Предлагаемое изобретение относится к биологически активным пептидам и белкам, способным ингибировать подвижность клеток, стимулированную хемокином фракталкином, а также к лекарственным средствам на их основе.
Известно, что хемокины являются основными регуляторными белками, по градиенту которых осуществляется рекрутирование и миграция лейкоцитов при воспалении. Кроме того, известно, что воспаление является неотъемлемым компонентом многих патологических состояний, таких как онкогенез, атеросклероз, аутоиммунные заболевания, болезнь Альцгеймера и др.
К семейству хемокинов принадлежат, в частности, относительно короткие белки - моноцитарный хемотаксический белок-1 (МСР-1) и фракталкин. Функция хемокинов осуществляется через специальные рецепторы, экспрессируемые клетками иммунной системы и клетками-мишенями действия хемокинов. Известно, что фракталкин в отличие от других хемокинов является мембраносвязанным хемокином, который включает в себя хемокиновый, трансмембранный и внутриклеточный домены. Хемокиновый домен заякорен в мембране через трансмембранный муцин-подобный стержень, соединенный с внутриклеточным доменом [1]. Заякоренный хемокиновый домен фракталкина высвобождается из клеточной мембраны протеолизом и переходит в растворимую форму, обозначаемую как растворимый фракталкин. В мембранной форме фракталкин действует как адгезионная молекула, в растворимой форме - как хемоаттрактант. Фракталкин экспрессируется дендритными клетками, эндотелиальными и гладкомышечными клетками и нейронами и связывается с рецептором, экспрессируемым клетками иммунной системы и глиальными клетками центральной нервной системы. В экспериментах на животных показано, что взаимодействие фракталкина с рецептором играет заметную роль в атерогенезе [2], ревматоидном артрите [3] и нефропатии [4]. Выявлена связь экспрессии фракталкина, фракталкинового рецептора и боли у пациентов, страдающих устойчивыми болями при панкреатите [5]. Действие фракталкина проявляется как в растворимой, так и в связанной форме в гомеостатических условиях и при воспалении. В настоящее время ингибирование связывания фракталкина с рецептором можно рассматривать в качестве одной из возможностей противовоспалительной терапии сердечно-сосудистых, ревматологических, панкреатических и неврологических заболеваний. Поэтому поиск и синтез ингибиторов такого рода представляется актуальным.
Установлено, что в патогенезе атеросклероза ведущая роль принадлежит хемокину МСР-1 (monocyte chemoattractant protein-1), который экспрессируется макрофагами, гладкомышечными и эндотелиальными клетками стенки сосуда. Известен С - концевой фрагмент 65-76 моноцитарного хемотаксического белка - 1 (МСР-1), ингибирующий МСР-1-зависимую миграцию моноцитов in vitro и in vivo [9]. Поскольку в экспериментах на животных установлена независимая роль МСР-1 и фракталкина в миграции моноцитов/макрофагов и их участия в формировании атеросклеротических бляшек [8], несомненный интерес в дополнение к антагонисту, описанному в [9], представляет разработка пептидных фрагментов структуры хемокинового домена фракталкина - антагонистов, способных ингибировать фракталкин-зависимую миграцию моноцитов.
В настоящее время в литературе представлены единичные примеры антагонистов фракталкина [6, 7]. Наиболее близким к заявляемому антагонисту является ингибитор миграции Т - лимфоцитов и естественных киллеров крови человека in vitro и моноцитов/макрофагов мыши in vivo [7]. Этот ингибитор (F1) представляет собой последовательность хемокинового домена фракталкина (1-77 аминокислотных остатков), модифицированную в N-концевой части добавлением остатка изолейцина в «нулевом» положении и заменой аминокислотных остатков QHHGVT на LDNGVS, либо эта последовательность, ковалентно связанная с Fc - фрагментом иммуноглобулина (F1-Ig). Ингибитор F1 получали с помощью химического синтеза или генной инженерии с использованием химерных фагмидных векторов. Из F1 затем получали конъюгат с Fc-фрагментом иммуноглобулина - F1-Ig. F1 (10 нг/мл) и F1-Ig (35 нг/мл) ингибировали миграцию CD8 (CD - cluster determination, кластеры дифференцировки - антигены клеточной поверхности) Т-лимфоцитов in vitro на 18% и 24% соответственно, а естественных киллеров - на 26% и 34%. Более значительные эффекты ингибирования (для естественных киллеров при F1 73% и при F1-Ig 69% и для Т-лимфоцитов при F1 49% и при F1-Ig 73%) зарегистрированы при 10-кратном увеличении концентрации антагонистов. Поскольку в настоящее время это единственные антагонисты фракталкина, ингибирующие миграцию Т-лимфоцитов и естественных киллеров крови человека in vitro, стимулируемую хекмокиновым доменом, они выбраны в качестве прототипа заявляемого изобретения.
Недостатками прототипа являются дороговизна и сложность синтеза, обусловленные длиной аминокислотной последовательности и методом синтеза (метод генной инженерии с использованием химерных фагмидных векторов), что предполагает многостадийную дорогостоящую очистку и трудности в стандартизации препарата. Кроме того, в случае предполагаемого применения подобных антагонистов в противовоспалительной терапии, белковые соединения могут вызывать аллергические осложнения.
Задачей настоящего изобретения явился поиск и синтез относительно коротких пептидов, лишенных побочных реакций, ингибирующих фракталкин-стимулированную миграцию моноцитарных клеток, причем синтез, очистка и идентификация таких пептидов должны быть значительно проще и дешевле по сравнению с синтезом, выделением и идентификацией белка-прототипа.
Поставленная задача решается путем синтеза пептидов формулы: H-PKEQWVKD-NH2 и H-DAMQHLDRQAAA-NH2.
Заявляемые пептиды содержат 8 и 12 аминокислотных остатков соответственно (для сравнения прототип - белок длиной в 78 аминокислотных остатков и его конъюгат с Fc-фрагментом иммуноглобулина). Синтез заявляемых пептидов осуществляется твердофазным способом с помощью Fmoc-технологии, что в настоящее время с учетом длины пептида является рутинной задачей. Очистка пептидов осуществляется путем обычной одностадийной ВЭЖХ. Все это делает получение заявляемых антагонистов значительно проще и дешевле, чем синтез белка-прототипа.
Неочевидность изобретения обусловлена тем, что поставленная задача решается путем синтеза пептидов последовательности 53-60 и 60-71 фракталкина, хотя из литературы неизвестно ни одного фрагмента этого белка, способного ингибировать фракталкин-стимулированную миграцию моноцитарных клеток. Таким образом, из литературных данных не следовало, что поиск пептидных ингибиторов фракталкин-стимулированной подвижности моноцитарных клеток среди фрагментов молекулы фракталкина может привести к желаемым результатам. Заявляемые пептиды нетоксичны, поскольку являются фрагментами эндогенного фракталкина.
Синтез заявляемых пептидов осуществляется по стандартной технологии пептидного синтеза на твердой фазе с применением наиболее современной Fmoc-(9-флуоренилметоксикарбонил)-технологии. В качестве нерастворимого носителя использовали сополимер стирола с 1% дивинилбензола с кислотолабильными якорными группами. Для блокирования функциональных групп боковых цепей аминокислот применяли следующие защиты: тpeт-бутильную для карбоксильных групп аспарагиновой и глутаминовой кислот; трет-бутилоксикарбонильную (Воc) - защиту для ε-аминогруппы лизина; 2,2,5,7,8-пентаметилхроман-6-сульфонильную (Pmc) - защиту для гуанидиновой функции аргинина; тритильную (Trt) - группу для карбоксамидной функции глутамина и имидазольного кольца гистидина. Пептидную цепь наращивали по одной аминокислоте, начиная синтез с С-конца. Для создания амидных связей использовали N,N'-диизопропилкарбодиимид в присутствии 1-гидроксибензотриазола (DIC/HOBt-метод). Для заключительного деблокирования и отщепления пептидов от носителя применяли трифторуксусную кислоту со скэвенджерами. Сырой продукт твердофазного синтеза очищали с использованием препаративной ВЭЖХ.
Пример 1. Синтез амида октапептида H-Pro-Lys-Glu-Gln-Trp-Val-Lys-Asp-NH2 (1), соответствующего последовательности 53-60
Список сокращений:
АА - аминокислота;
Вое - трет-бутилоксикарбонил;
But - трет-бутил;
DIC - N,N'-диизопропилкарбодиимид;
DCM - дихлорметан;
DMSO-d6 - дейтерированный диметилсульфоксид;
DTT - дитиотреитол;
Fmoc - 9-флуоренилметоксикарбонил;
НОВТ - 1-гидроксибензотриазол;
NMP - N-метилпирролидон;
4-MePip - 4 метилпиперидин;
Pmc - 2,2,5,7,8-пентаметилхроман-6-сульфонил;
TIBS - триизобутилсилан;
TFA - трифторуксусная кислота;
ВЭЖХ - высокоэффективная жидкостная хроматография.
В работе использованы производные L-аминокислот (Bachem, Швейцария), DIC, HOBt, TIBS, (Fluka, Швейцария). Для синтеза применяли N-метилпирролидон, дихлорметан, пиперидин, метанол и TFA (Applied Biosystems GmbH, Германия). Аналитическую ВЭЖХ проводили на хроматографе (Gilson, Франция), использовали колонку Kromasil C18, 4.6×250 мм, 5 мкм; буфер А: 0.1% водная трифторуксусная кислота, буфер Б: 80% ацетонитрил + 20% буфера А; градиент Б от 10 до 70% за 30 мин. Скорость потока 1 мл/мин, детекция при 220 нм. Структура полученных пептидов доказана спектрами 1Н-ЯМР и данными масс-спектрометрии. 1H-ЯМР-спектры снимали на спектрометре WM-500 (Bruker) 500 МГц (ФРГ) в DMSO-d6 при 300К, концентрация пептидов составляла 2-3 мг/мл. Химические сдвиги измерялись относительно тетраметилсилана. Масс-спектры регистрировали на приборе PC-Kompact MALDI (Kratos, Англия).
Для твердофазного синтеза использовали сополимер стирола с 1% дивинилбензола с 4-(2,4-диметоксифенил)-Fmoc-аминометилфенокси - якорной группой (Rink-amide-полимер) фирмы Nova BioChem, Германия, предназначенный для получения амидов пептидов, содержащий 0.56 ммоль/г аминогрупп. Синтез амида октапептида проводили с С-конца, ступенчато (присоединяя по одной аминокислоте), исходя из 0.45 г (0.25 ммоль) Rink-amide-полимера. Синтез проводили в автоматическом режиме на пептидном синтезаторе Applied Biosystems 431 А по стандартной программе для однократной конденсации Fmoc-аминокислот. (См. протокол твердофазного синтеза).
Протокол твердофазного синтеза
Заключительное деблокирование и отщепление октапептида от полимера проводили в одну стадию путем обработки соответствующего нонапептидилполимера смесью 10 мл TFA, 0.25 мл деионизованной воды и 0.25 мл TIBS, 0.25 г DTT в течение 2 ч. Затем полимер отфильтровывали, промывали 2×2 мл деблокирующей смеси, фильтрат упаривали и к остатку прибавляли сухой эфир. Осадок отфильтровывали, промывали DCM (3×3 мл) эфиром (3×5 мл), сушили в вакуум-эксикаторе. Сырой продукт твердофазного синтеза с содержанием основного вещества 75% очищали с помощью препаративной ВЭЖХ на приборе Beckman (США), используя колонку Диасорб С 16 13 ОТ (25×250 мм), размер частиц сорбента 10 мкм. В качестве элюентов использовали буфер А - 0.01 М раствор ацетата аммония и буфер Б - 80% ацетонитрила в воде. Элюцию проводили градиентом 0.5% в минуту буфера Б в буфере А от 100% буфера А со скоростью 10 мл/мин. Пептиды детектировали при длине волны 220 нм. Фракции, содержащие целевой продукт, объединяли, ацетонитрил упаривали и лиофилизовали. Выход пептида (1) составил 100 мг 58.2% (в расчете на стартовую аминокислоту, присоединенную к полимерному носителю, т.е. это суммарный выход на все стадии синтеза), чистота пептида - 98% (колонка Kromasil С 18, 4.6×250 мм, 5 мкм; буфер А: 0.1% водная трифторуксусная кислота, буфер Б: 80% ацетонитрил+20% буфера А; градиент Б от 10 до 70% за 30 мин; Rt - 12.43 мин); m/z: 1028.6 [М+Н]+, вычислено М=1027.17.
Пример 2. Синтез амида додекапептида H-Asp-Ala-Met-Gln-His-Leu-Asp-Arg-Gln-Ala-Ala-Ala-NH2 (2), соответствующего последовательности 60-71 хемокинового домена фракталкина
Твердофазный синтеза пептида (2) проведен на том же полимерном носителе и по тому же протоколу с соответствующим увеличением количества синтетических циклов, что и синтез пептида (1).
Заключительное отщепление защитных групп проводили одновременно с отщеплением целевого пептида от полимерного носителя действием трифторуксусной кислоты со скэвенджерами: TFA 10 мл, TIBS 0.25 мл, DTT 0.25 г, тиоанизол 0.5 мл, фенол 0.75 г, вода 0.25 мл в течение 2 ч. Полученный пептид очищали с помощью высокоэффективной жидкостной хроматографии на обращенной фазе, как описано в примере 1.
Выход пептида (2) составил 146 мг 62.9% (в расчете на стартовую аминокислоту, присоединенную к полимерному носителю, т.е. это суммарный выход на все стадии синтеза), чистота пептида - 97% (колонка Kromasil С 18, 4.6×250 мм, 5 мкм; буфер А: 0.1% водная трифторуксусная кислота, буфер Б: 80% ацетонитрил + 20% буфера А; градиент Б от 10 до 70% за 30 мин; Rt - 11.12 мин); m/z: 1325.46 [M+H]+, 1347.46
[M+Na]+, 1363.45 [М+К]+; вычислено М=1324.48.
Пример 3. Анализ влияния синтезированных пептидов на миграцию моноцитарных клеток in vitro
Исследования проводили в 48-луночной камере Бойдена с использованием фракции CD14+CD16+моноцитов периферической крови здоровых добровольцев. Мононуклеарные клетки выделяли из периферической крови путем центрифугирования в градиенте плотности Histopaque (ρ=1.077, Sigma). Для удаления из полученной популяции клеток нейтрофильных и эозинофильных гранулоцитов и NK-клеток в клеточную суспензию вносили конъюгированные с магнитными бусами FcR Blocking Reagent и Non-Monocyte Depletion Cocktail, состоящий из конъюгированных с магнитными бусами антител к CD15 и CD56 антигенам, в концентрациях, рекомендованных производителем (Miltenyi Biotec Inc., Germany). Затем клетки отмывали от несвязавшихся антител, после чего наносили на колонку, помещенную в магнитное поле, и собирали фракцию клеток, прошедшую через колонку. Для выделения CD16+ моноцитов клетки инкубировали с моноклональными антителами к CD16, конъюгированными с магнитными бусами, после чего клеточную суспензию наносили на колонку, помещенную в магнитное поле. В конечном итоге, освобождались от всех клеток, за исключением CD16+ моноцитов, которые задерживались на колонке. CD16+, моноциты снимали с колонки, отмывали в фосфатном буфере без Са2+ и Mg2+ и ресуспендировали в растворе Хэнкса с 0,5% бычьего сывороточного альбумина. Полученная суспензия клеток содержала не менее 65% CD16+ моноцитов, которые, по данным цитофлуориметрического анализа, коэкспрессировали CD14 и рецепторы фракталкина CX3CR1, т.е. имели CD14+CD16+CX3CR1+ фенотип. Количество жизнеспособных клеток, окрашенных трипановым синим, составляло не менее 98%. В лунки камеры Бойдена наносили по 105 клеток и камеру инкубировали 45 мин в CO2 - инкубаторе.
Полученные результаты представлены на фиг.1. Фракталкин (100 нг/мл) стимулировал миграцию моноцитов на 75% в сравнении со спонтанной миграцией клеток. Октапептид (53-60) (100 мкг/мл) достоверно ингибировал (на 70%) стимулированную фракталкином миграцию CD14+CD16+ моноцитов (Фиг.1) Додекапептид (60-71) (100 мкг/мл) также проявлял ингибирующие свойства - достоверно ингибировал (на 60%) стимулированную фракталкином миграцию CD14+CD16+ моноцитов (Фиг.1). Ингибирование миграции моноцитов указанными пептидами происходило также и при концентрации 1 мкг/мл. Следует отметить, что при использовании пептидного фрагмента хемокина МСР-1, ингибирующего миграцию моноцитов т vitro в аналогичной (100 мкг/мл) концентрации, противовоспалительный эффект in vivo достигается в значительно меньшей дозе - 35 мкг/кг [9].
Таким образом, впервые показано, что пептидные фрагменты 53-60 и 60-71 аминокислотной последовательности хемокинового домена фракталкина ингибируют фракталкин - зависимую миграцию CD14+CD16+ моноцитов человека на 70% и 60% в концентрации 100 мкг/мл. В прототипе подобные эффекты ингибирования для естественных киллеров при F1 (73%), при F1-Ig (69%) и для Т-лимфоцитов при F1 (49%) и при F1-Ig (73%) достигаются при 0,35 мкг/мл. Однако получение синтетических пептидных ингибиторов хемокинового домена фракталкина намного проще и менее затратно по сравнению с прототипом. Заявляемые пептиды нетоксичны, поскольку являются короткими фрагментами эндогенного белка человека.

| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИД, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ИНГИБИРОВАТЬ МИГРАЦИЮ МОНОЦИТАРНЫХ КЛЕТОК, СТИМУЛИРОВАННУЮ БЕЛКОМ МСР-1 | 2003 |
|
RU2260598C2 |
| Пептидное лекарственное средство | 2022 |
|
RU2827765C2 |
| Пептид, обладающий способностью ингибировать миграцию клеток, стимулированную белком TARC | 2016 |
|
RU2629198C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С ПРОЛОНГИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ ДОДЕКАПЕПТИДА ИНГРАМОН | 2022 |
|
RU2793124C1 |
| Антитела против эотаксина 2, которые распознают дополнительные связывающие CCR3 хемокины | 2015 |
|
RU2705255C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДОДЕКАПЕПТИДА И ТРИПЕПТИД ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2340626C1 |
| АМИД НОНАПЕПТИДА, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ПРЕДОТВРАЩАТЬ ПОВЫШЕНИЕ ПРОНИЦАЕМОСТИ ЭНДОТЕЛИЯ СОСУДОВ | 2009 |
|
RU2402565C1 |
| КОНСТРУКЦИИ АНТИТЕЛ И ХЕМОКИНОВ И ИХ ПРИМЕНЕНИЕ ПРИ ИММУНОЛОГИЧЕСКИХ НАРУШЕНИЯХ | 2001 |
|
RU2252786C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ПРОИЗВОДСТВА Т-КЛЕТОК | 2018 |
|
RU2793344C2 |
| СПОСОБ УСИЛЕНИЯ ИММУННОГО ОТВЕТА НА АНТИГЕН У МЛЕКОПИТАЮЩИХ | 2004 |
|
RU2341289C2 |
Изобретение относится к области биотехнологии, а именно к пептиду для ингибирования фракталкин-стимулированной миграции моноцитарных клеток. Пептид выбрают из H-PKEQWVKD-NH2 или
H-DAMQHLDRQAAA-NH2. Предложенное изобретение позволяет синтезировать пептид, лишенный побочных реакций, ингибирующий фракталкин-стимулированную миграцию моноцитарных клеток. 1 ил., 1 табл., 3 пр.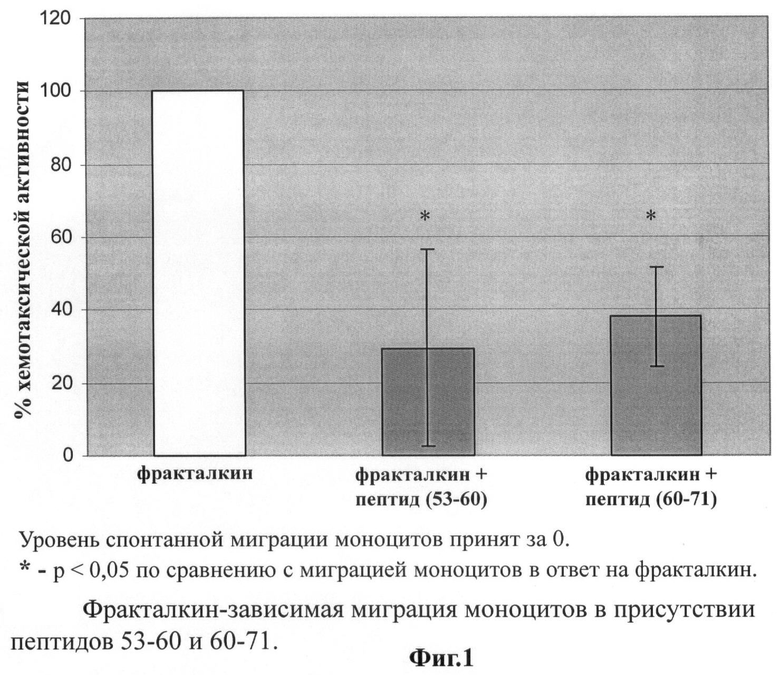
Пептид для ингибирования фракталкин-стимулированной миграции моноцитарных клеток, выбранный из H-PKEQWVKD-NH2 или Н-DAMQHLDRQAAA-NH2.
| DORGHAM К | |||
| еt al., An engineered CX3CR1 antagonist endowed with anti-inflammatory activity, J Leukoc BioL, 2009, v.86, no.4, pp.903-911 | |||
| INOUE A | |||
| еt al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| MARUYAMA K | |||
| еt al. | |||

