ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям, к фармацевтическим композициям, содержащим указанные соединения, и к применению указанных соединений в терапии. Настоящее изобретение также относится к способам получения указанных соединений и к новым промежуточным соединениям, используемым для их получения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Чувство боли у млекопитающих имеет место вследствие активации периферических терминалей специализированной популяции сенсорных нейронов, известных как болевые рецепторы. Капсаицин, активный ингредиент в жгучих перцах, вызывает продолжительную активацию болевых рецепторов и также вызывает зависимое от дозы чувство боли у людей. Клонирование ванилоидного рецептора 1 (VR1 или TRPV1) показало, что VR1 является молекулярной мишенью для капсаицина и его аналогов (Caterina, M.J., Schumacher, М.А., et al. Nature (1997), v.389, p.816-824). Функциональные исследования с использованием VR1 свидетельствуют о том, что он активируется также вредным повышением температуры тела, закислением ткани и другими воспалительными медиаторами (Tominaga, М., Caterina, M.J. et al. Neuron (1998), v.21, p.531-543). Экспрессия VR1 также регулируется после повреждения периферического нерва по типу, который приводит к невропатической боли. Эти свойства VR1 делают его чрезвычайно подходящей мишенью для боли и для заболеваний, вовлекающих воспаление. Несмотря на то что агонисты рецептора VR1 могут действовать в качестве анальгетиков посредством разрушения болевых рецепторов, применение таких агонистов, как капсаицин и его аналоги, ограничено из-за их жгучести, нейротоксичности и индукции гипотермии. Вместо этого, агенты, которые блокируют активность VR1, должны оказаться более полезными. Антагонисты могли бы сохранить аналгетические свойства, но избежать побочных эффектов от жгучести и нейротоксичности.
Соединения с ингибиторной активностью в отношении VR1, как полагают, могут иметь потенциальное применение для лечения и/или профилактики расстройств, таких как боль, особенно боль воспалительного или травматического происхождения, таких как артрит, ишемия, рак, фибромиалгия, боль в пояснице и послеоперационная боль (Walker et al. J Pharmacol Exp Ther. (2003) Jan; 304(1):56-62). Помимо этого, потенциальными болевыми состояниями, которые можно лечить с помощью ингибирования VR1, являются висцеральные боли, такие как хроническая тазовая боль, цистит, синдром раздраженного кишечника (IBS), панкреатит и тому подобное, а также невропатическая боль, такая как ишиалгия, диабетическая невропатия, ВИЧ-невропатия, рассеянный склероз и тому подобное (Walker et al., там же, Rashid et al. J Pharmacol Exp Ther. (2003) Mar; 304(3):940-8). Эти соединения, как полагают, также потенциально полезны для воспалительных расстройств, таких как астма, кашель и воспалительное заболевание кишечника (IBD) (Hwang and Oh Curr Opin Pharmacol (2002) Jun; 2(3):235-42). Соединения с блокирующей активностью в отношении VR1 также полезны против зуда и кожных заболеваний, таких как псориаз, и против гастроэзофагеальной рефлюксной болезни (GERD), рвоты, рака, недержания мочи и гиперактивного мочевого пузыря (Yiangou et al. BJU Int (2001) Jun; 87(9):774-9, Szallasi Am J Clin Pathol (2002) 118:110-21). Ингибиторы VR1 могут иметь потенциальное применение также для лечения и/или профилактики эффектов воздействия на активаторы VR1, такие как капсаицин или слезоточивый газ, кислоты или нагревание (Szallasi, там же).
Другое потенциальное применение относится к лечению устойчивости к активаторам VR1.
Ингибиторы VR1 могут быть также полезны в лечении интерстициального цистита и боли, связанной с интерстициальным циститом.
В WO 2004/100865 раскрыты соединения, демонстрирующие ингибиторную активность в отношении ванилоидного рецептора 1 (VR1).
ОПРЕДЕЛЕНИЯ:
Используемые в данной заявке следующие термины имеют следующие значения:
Термин "(+, -)" будет означать рацемическую смесь такого соединения.
Термин "алкил", используемый отдельно или в виде суффикса или префикса, относится к гидрокарбильным радикалам с прямой или разветвленной цепью, содержащим от 1 до примерно 12 атомов углерода.
Термин "алкилен", используемый отдельно или в виде суффикса или префикса, относится к двухвалентным углеводородным радикалам с прямой или разветвленной цепью, содержащим от 1 до примерно 12 атомов углерода, которые служат для соединения двух структур вместе.
Термин "алкенил", используемый отдельно или в виде суффикса или префикса, относится к одновалентным углеводородным радикалам с прямой или разветвленной цепью, имеющим по меньшей мере одну углерод-углеродную двойную связь и содержащим по меньшей мере 2 и вплоть до примерно 12 атомов углерода.
Термин "алкинил", используемый отдельно или в виде суффикса или префикса, относится к одновалентным углеводородным радикалам с прямой или разветвленной цепью, имеющим по меньшей мере одну углерод-углеродную тройную связь и содержащим по меньшей мере 2 и вплоть до примерно 12 атомов углерода.
Термин "амин" или "амино" относится к радикалам общей формулы -NRR', где R и R' независимо выбраны из водорода или алкильного радикала.
Термин "ароматический" относится к гидрокарбильным радикалам, имеющим одно или более полиненасыщенных углеродных колец, имеющих ароматический характер (например, 4n+2 делокализованных электронов) и содержащих от 6 вплоть до примерно 14 атомов углерода.
Термин "арил", используемый отдельно или в виде суффикса или префикса, относится к углеводородному радикалу, имеющему одно или более полиненасыщенных углеродных колец, имеющих ароматический характер (например, 4n+2 делокализованных электрона) и содержащих от 5 вплоть до примерно 14 атомов углерода, где радикал расположен на атоме углерода ароматического кольца.
Термин "циклоалкил", используемый отдельно или в виде суффикса или префикса, относится к одновалентному, содержащему кольцо углеводородному радикалу, содержащему по меньшей мере от 3 вплоть до примерно 12 атомов углерода.
Термин "гетероциклоалкил" относится к насыщенному или ненасыщенному циклоалкилу, в котором по меньшей мере один кольцевой атом углерода (и любые связанные атомы водорода) независимо заменены на по меньшей мере один гетероатом, выбранный из О и N. Такие циклоалкилы включают в себя, но не ограничиваются этим, такие группы, как морфолинил, пиперидинил, пиперазинил и пирролидинил.
Термин "галогено" или "галоген" относится к радикалам фтора, хлора, брома и йода.
Термин "гетероцикл" или "гетероциклический" или "гетероциклическая группировка" относится к содержащим кольцо одновалентным и двухвалентным радикалам, имеющим один или более гетероатомов, независимо выбранных из N, О, Р и S, в виде части кольцевой структуры, и содержащим по меньшей мере от 3 и вплоть до примерно 20 атомов в кольцах, предпочтительно 5- и 6-членных кольцах. Гетероциклические группировки могут быть насыщенными или ненасыщенными, содержащими одну или более двойных связей, и гетероциклические группировки могут содержать более чем одно кольцо.
Термин "гетероарил" относится к гетероциклическим одновалентным и двухвалентным радикалам, имеющим ароматический характер.
Гетероциклические группировки включают в себя, например, моноциклические группировки, такие как: азиридин, оксиран, тииран, азетидин, оксетан, тиэтан, пирролидин, пирролин, имидазолидин, пиразолидин, диоксолан, сульфолан, 2,3-дигидрофуран, 2,5-дигидрофуран, тетрагидрофуран, тиофан, тиофен, пиперидин, 1,2,3,6-тетрагидропиридин, пиперазин, морфолин, тиоморфолин, пиран, тиопиран, 2,3-дигидропиран, тетрагидропиран, 1,4-дигидропиридин, 1,4-диоксан, 1,3-диоксан, диоксан, гомопиперидин, 2,3,4,7-тетрагидро-1H-азепин гомопиперазин, 1,3-диоксепан, 4,7-дигидро-1,3-диоксепин и гексаметиленоксид. Кроме того, гетероциклические группировки включают в себя гетероарильные кольца, такие как: пиридил, пиразинил, пиримидинил, пиридазинил, тиенил, фурил, пирролил, имидазолил, тиазолил, оксазолил, пиразолил, изотиазолил, изоксазолил, 1,2,3-триазолил, тетразолил, 1,2,3-тиадиазолил, 1,2,3-оксадиазолил, 1,2,4-триазолил, 1,2,4-тиадиазолил, 1,2,4-оксадиазолил, 1,3,4-триазолил, 1,3,4-тиадиазолил и 1,3,4-оксадиазолил. Кроме того, гетероциклические группировки охватывают полициклические группировки, такие как: индол, индолин, хинолин, тетрагидрохинолин, изохинолин, тетрагидроизохинолин, 1,4-бензодиоксан, кумарин, дигидрокумарин, бензофуран, 2,3-дигидробензофуран, 1,2-бензизоксазол, бензотиофен, бензоксазол, бензтиазол, бензимидазол, бензтриазол, тиоксантин, карбазол, карболин, акридин, пиролизидин и хинолизидин.
Помимо полициклических гетероциклов, описанных выше, гетероциклические группировки включают в себя полициклические гетероциклические группировки, в которых кольцевая конденсация между двумя или более кольцами включает более чем одну связь, общую для обоих колец, и более чем два атома, общих для двух колец. Примеры таких мостиковых гетероциклов включают в себя хинуклидин, диазабицикло[2.2.1]гептан и 7-оксабицикло[2.2.1]гептан.
Термин "гидрокарбильный" относится к любой структуре, содержащей только атомы углерода и водорода, вплоть до 14 атомов углерода.
Термин "млекопитающее" включает в себя любое из множества теплокровных позвоночных животных класса Mammalia, включая, но не ограничиваясь этим, людей, обычно характеризующихся волосяным покровом на коже.
Термин "пациент" относится к субъекту, который получает медицинское внимание, помощь или лечение.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Одним воплощением изобретения является соединение формулы I:
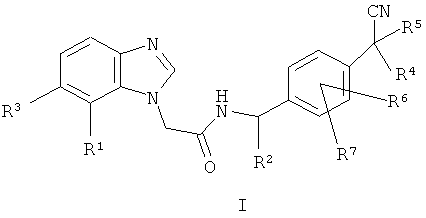
где
R1 выбран из CN, галогена, С(=O)СН3;
R2 выбран из метила или Н;
R3 выбран из Н или галогена;
каждый из R4 и R5 независимо выбран из метила или этила, или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-6-членную циклоалкильную или 5- или 6-членную гетероциклоалкильную группу;
каждый из R6 и R7 независимо выбран из Н, галогена, метила или этила;
или его фармацевтически приемлемая соль;
где соединение формулы I не является следующим:
N-[4-(1-циано-1-метилэтил)бензил]-2-(6,7-дифтор-1Н-бензимидазол-1-ил)-ацетамид;
2-(7-хлор-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-3-фторбензил]ацетамид;
(+)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклогексил)фенил]этил}ацетамид;
(+)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(+,-)-2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}ацетамид;
(+)-2-(7-ацетил-1H-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид;
(+)-N-{1-[4-(1-цианоциклогексил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(+)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид;
(+)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(+)-N-{1-[4-(1-цианоциклобутил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(R)(+)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(R)(+)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид;
(+)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид;
(+)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид;
(R)(+)-N-{1-[4-(1-циано-1-этилпропил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R3 выбран из хлора или фтора.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, и R3 выбран из хлора или фтора.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R4 и R5 независимо выбраны из метила или этила.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, и R4 и R5 независимо выбраны из метила или этила.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R3 выбран из хлора или фтора, и R4 и R5 независимо выбраны из метила или этила.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, R3 выбран из хлора или фтора, и R4 и R5 независимо выбраны из метила или этила.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-, 4- или 6-членную циклоалкильную или 5- или 6-членную гетероциклоалкильную группу.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, и R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-, 4- или 6-членную циклоалкильную или 5- или 6-членную гетероциклоалкильную группу.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R3 выбран из хлора или фтора, и R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-, 4- или 6-членную циклоалкильную или 5- или 6-членную гетероциклоалкильную группу.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, R3 выбран из хлора или фтора, и R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-, 4- или 6-членную циклоалкильную или 5- или 6-членную гетероциклоалкильную группу.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где каждый из R6 и R7 независимо выбран из фтора или хлора.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где каждый из R6 и R7 независимо выбран из Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, и каждый из R6 и R7 независимо выбран из Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R3 выбран из хлора или фтора, и каждый из R6 и R7 независимо выбран из Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, R3 выбран из хлора или фтора, и каждый из R6 и R7 независимо выбран из Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R4 и R5 независимо выбраны из метила или этила, и каждый из R6 и R7 независимо выбран из Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, R4 и R5 независимо выбраны из метила или этила, и каждый из R6 и R7 независимо выбран из Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R3 выбран из хлора или фтора, R4 и R5 независимо выбраны из метила или этила, и каждый из R6 и R7 представляет собой Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, R3 выбран из хлора или фтора, R4 и R5 независимо выбраны из метила или этила, и каждый из R6 и R7 представляет собой Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-, 4- или 6-членную циклоалкильную или 5- или 6-членную гетероциклоалкильную группу, и каждый из R6 и R7 представляет собой Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-, 4- или 6-членную циклоалкильную или 5- или 6-членную гетероциклоалкильную группу, и каждый из R6 и R7 представляет собой Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R3 выбран из хлора или фтора, R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-, 4- или 6-членную циклоалкильную или 5- или 6-членную гетероциклоалкильную группу, и каждый из R6 и R7 представляет собой Н.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 независимо выбран из хлора или фтора, R3 выбран из хлора или фтора, R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-, 4- или 6-членную циклоалкильную или 5- или 6-членную гетероциклоалкильную группу, и каждый из R6 и R7 представляет собой Н.
Одним воплощением изобретения является соединение, выбранное из:
(S)(-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида;
(S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамида;
(S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(7-хлор-1Н-бензимидазол-1-ил)ацетамида;
(S)(-)-N-{1-[4-(1-циано-1-этилпропил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[4-(1-цианоциклобутил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[4-(1-цианоциклогексил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклогексил)фенил]этил}ацетамида;
(-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклобутил)фенил]этил}ацетамида;
(-)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида;
(S)(-)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(-)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-2-(7-хлор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-3-фторбензил]ацетамида;
(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(-)-N-{1-[4-(4-цианотетрагидро-2Н-тиопиран-4-ил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[3-хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[3-хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(7-циано-1Н-бензимидазол-1-ил)ацетамида;
(-)-2-(6,7-дифтор-1H-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(S)(-)-2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)-N-{[4-(1-циано-1-метилэтил)фенил]этил}ацетамида;
и его фармацевтически приемлемая соль.
Одним воплощением изобретения является соединение, выбранное из:
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(7-хлор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-циано-1-этилпропил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-цианоциклобутил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-цианоциклогексил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклогексил)фенил]этил}ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклопропил)фенил]этил}ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклобутил)фенил]этил}ацетамида;
(+,-)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида;
(+,-)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(+,-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида;
(+,-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(+,-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(+,-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(+,-)-N-{1-[4-(4-цианотетрагидро-2Н-тиопиран-4-ил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
N-[4-(1-циано-1-метилэтил)-2-метилбензил]-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
2-(7-циано-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-2-метилбензил]ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[3-хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[3-хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(7-циано-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-2-(7,6-дифтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)-N-{[4-(1-циано-1-метилэтил)-2-метилбензил]ацетамида;
и его фармацевтически приемлемые соли.
Одним воплощением изобретения является соединение (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид и его фармацевтически приемлемая соль.
Одним воплощением изобретения является соединение (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид и его фармацевтически приемлемая соль.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль для применения в лечении хронических ноцицептивных болевых расстройств у млекопитающих.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль для применения в лечении остеоартрита.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль для применения в лечении хронического тендинита.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль для применения в лечении тазовой боли.
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль для применения в лечении периферической невропатии (главным образом PHN (постгерпетической невралгии)).
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль для применения в лечении гастроэзофагеальной рефлюксной болезни (GERD).
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль для применения в лечении синдрома раздраженного кишечника (IBS).
Одним воплощением изобретения является соединение формулы I или его фармацевтически приемлемая соль для применения в лечении гиперактивного мочевого пузыря.
Одним воплощением изобретения является способ лечения ноцицептивных болевых расстройств, включающий введение эффективного количества соединения формулы I или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения ноцицептивных болевых расстройств, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения ноцицептивных болевых расстройств, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения хронических ноцицептивных болевых расстройств, включающий введение эффективного количества соединения формулы I или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения хронических ноцицептивных болевых расстройств, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения хронических ноцицептивных болевых расстройств, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения остеоартрита, включающий введение эффективного количества соединения формулы I или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения остеоартрита, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения остеоартрита, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения хронического тендинита, включающий введение эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения хронического тендинита, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения хронического тендинита, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения хронического тендинита, включающий введение эффективного количества соединения формулы I или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения хронического тендинита, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения хронического тендинита, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения тазовой боли, включающий введение эффективного количества соединения формулы I или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения тазовой боли, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения тазовой боли, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метил этил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения периферической невропатии (главным образом PHN), включающий введение эффективного количества соединения формулы I или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения периферической невропатии (главным образом PHN), включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения периферической невропатии (главным образом PHN), включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения гастроэзофагеальной рефлюксной болезни (GERD), включающий введение эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения гастроэзофагеальной рефлюксной болезни (GERD), включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения гастроэзофагеальной рефлюксной болезни (GERD), включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения синдрома раздраженного кишечника (IBS), включающий введение эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения синдрома раздраженного кишечника (IBS), включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения синдрома раздраженного кишечника (IBS), включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения гиперактивного мочевого пузыря, включающий введение эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения гиперактивного мочевого пузыря, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является способ лечения гиперактивного мочевого пузыря, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
Одним воплощением изобретения является фармацевтическая композиция, содержащая соединение формулы 1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Одним воплощением изобретения является фармацевтическая композиция, содержащая соединение (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Одним воплощением изобретения является фармацевтическая композиция, содержащая соединение (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Признаки и преимущества изобретения могут быть более легко поняты средними специалистами в данной области техники при прочтении следующего подробного описания. Следует принимать во внимание, что некоторые признаки изобретения, которые для ясности описаны выше и ниже в контексте отдельных воплощений, могут быть также объединены с образованием единого воплощения. И наоборот, различные признаки изобретения, которые для краткости описаны в контексте единого воплощения, могут быть объединены с образованием их подкомбинаций.
Некоторые соединения по изобретению имеют хиральный центр. Такие формы могут быть фракционированы хиральной хроматографией, и правовращающие фракционированные соединения обладают большей антагонистической активностью, чем левовращающие. Не желая быть связанными какой-либо теорией, в настоящее время считают, что (+)-изомеры представляют собой (R)-энантиомеры, а (-)-изомеры представляют собой (S)-энантиомеры. Таким образом, хотя правовращающие, (D) или (+) или (R), и левовращающие, (L) или (-) или (S)-соединения, представляют собой соединения по изобретению, конкретные соединения по изобретению представляют собой левовращающие, (S) или (-), соединения.
Знак заявленного вращения наблюдают для длины волны натрия, измеренной при 22°С стандартным способом в растворителях и концентрациях, в которых, как полагают, межмолекулярные ассоциации не происходят.
Недавно подтвердили, что хиральный (-)-2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрил, полученный фракционированием соответствующей рацемической смеси, имеет (S)-конфигурацию. Также подтвердили, что этот (-)-амин является хиральным исходным веществом, дающим в результате (-)-активные конечные соединения, заявленные в данной заявке. Поскольку никакой инверсии аминного хирального стереогенного центра, как наблюдали, не происходило при реакции сочетания, предполагают, что конфигурация (-)-активных конечных соединений (полученных с этим конкретным хиральным амином) также представляет собой (S)-конфигурацию.
Для хиральных конечных (-)-активных соединений, полученных с использованием различных, но сходных бензиловых аминов, описанных выше, в значительной степени предполагают, что хиральный центр имеет такую же (S)-конфигурацию, однако могут быть исключения из этого общего утверждения.
Для подтверждения хиральной структуры 2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила проводили анализы. Результаты анализов инфракрасного колебательного кругового дихроизма (VCD), объединенные с молекулярно-механическими расчетами и расчетами методом теории функционала плотности предсказанных VCD-спектров, соответствовали предложенным конфигурациям.
Конкретные соединения, описанные в данной заявке, иллюстрируют, но не ограничивают изобретение, другие соединения в объеме изобретения будут очевидны специалистам в данной области техники при рассмотрении процессов, способов и соединений, описанных в данной заявке.
Соединения, предложенные в данной заявке, полезны в форме свободного основания, но могут быть также предложены в форме фармацевтически приемлемой соли и/или в форме фармацевтически приемлемого гидрата. Например, фармацевтически приемлемая соль соединений формулы I включает в себя соли, полученные из минеральных кислот, таких как, например: метансульфокислота, этансульфокислота, соляная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, йодистоводородная кислота, азотистая кислота и фосфористая кислота. Фармацевтически приемлемая соль может быть также получена с органическими кислотами, включающими алифатические моно- и дикарбоксилаты и ароматические кислоты.
Другие фармацевтически приемлемые соли соединений по настоящему изобретению включают в себя, например, сульфат, пиросульфат, бисульфат, бисульфит, нитрат и фосфат.
Соединения формулы I могут быть получены способами, известными в химических областях для получения структурно аналогичных соединений. Соответственно, соединения по данному изобретению могут быть получены с использованием процедур, известных в литературе, начиная с известных соединений или легко получаемых промежуточных соединений.
В данной заявке предложены синтетические способы получения соединений-предшественников или применения в практических аспектах настоящего изобретения.
Специалистам в данной области техники будет понятно, что некоторые соединения по настоящему изобретению содержат, например, асимметрично замещенный углерод и, соответственно, могут существовать и быть выделены в оптически активных и рацемических формах. Некоторые соединения могут демонстрировать полиморфизм, таким образом, следует понимать, что настоящее изобретение охватывает рацемические, оптически активные, полиморфные или стереоизомерные формы или их смеси, причем эти формы обладают свойствами, полезными в лечении расстройств, описанных ниже. Получение оптически активных форм хорошо известно в данной области техники (например, посредством разделения рацемических форм методами перекристаллизации, синтеза из оптически активных исходных веществ, хирального синтеза или хроматографического разделения с использованием хиральной стационарной фазы).
Соединения формулы I представляют собой антагонисты VR-1. Соединения формулы I и их фармацевтически приемлемые соли могут быть также использованы в способе лечения боли, острой боли, хронической боли, ноцицептивной боли, острой ноцицептивной боли, хронической ноцицептивной боли, невропатической боли, острой невропатической боли, хронической невропатической боли, воспалительной боли, острой воспалительной боли, хронической воспалительной боли. Лечение таких расстройств включает введение теплокровному животному, предпочтительно млекопитающему, более предпочтительно человеку, нуждающемуся в таком лечении, эффективного количества соединения формулы I или фармацевтически приемлемой соли указанного соединения.
Кроме того, предложено применение соединения формулы I в лечении остеоартрита, хронического тендинита, тазовой боли и периферической невропатии (главным образом PHN), гастроэзофагеальной рефлюксной болезни (GERD), синдрома раздраженного кишечника (IBS) и гиперактивного мочевого пузыря.
Кроме того, предложено применение соединения формулы I в получении лекарственного средства для лечения расстройства, такого как боль, у теплокровного животного, предпочтительно млекопитающего, более предпочтительно человека, страдающего таким расстройством.
Кроме того, в изобретении предложена фармацевтическая композиция, подходящая для лечения вышеописанных расстройств, включающая введение теплокровному животному, имеющему такое расстройство, эффективного количества фармацевтической композиции соединения формулы I или фармацевтически приемлемой соли.
В изобретении также предложена фармацевтическая композиция, содержащая соединение формулы I, как определено в данной заявке, или фармацевтически приемлемую соль, в комбинации с фармацевтически приемлемым носителем.
По меньшей мере одно соединение, описанное в данной заявке, демонстрирует антагонистическую активность в отношении VR-1 в анализе, описанном в данной заявке, лучшую, чем примерно 1 мкМ. Обнаружили, что выбранные соединения по настоящему изобретению являются активными антагонистами с активностью, меньшей, чем примерно 100 нМ.
Соединения, описанные в данной заявке, могут быть предложены или доставлены в форме, подходящей для перорального применения, например, в виде таблетки, лепешки, твердой и мягкой капсулы, водного раствора, масляного раствора, эмульсии и суспензии. Соединения также могут быть предложены для местного введения, например, в виде крема, мази, геля, спрея или водных растворов, масляных растворов, эмульсий или суспензий. Соединения, описанные в данной заявке, могут быть также предложены в форме, подходящей для назального введения, например, в виде назального спрея, капель в нос или сухого порошка. Композиции также можно вводить во влагалище или прямую кишку в форме суппозитория. Соединения, описанные в данной заявке, также можно вводить парентерально, например, путем внутривенной, интравезикулярной, подкожной или внутримышечной инъекции или инфузии. Соединения можно вводить путем инсуффляции (например, в виде высокодисперсного порошка). Соединения можно также вводить чрескожно или сублингвально.
Соединения по изобретению соответственно могут быть получены традиционными методами с использованием традиционных фармацевтических эксципиентов, хорошо известных в данной области техники. Таким образом, композиции, предназначенные для перорального применения, могут содержать, например, один или более красителей, подсластителей, ароматизаторов и/или консервантов.
Количество активного ингредиента, который объединяют с одним или более эксципиентами для получения единой лекарственной формы, будет при необходимости варьировать в зависимости от пациента, которого лечат, и конкретного пути введения. Размер дозы соединения формулы I для терапевтических или профилактических целей, естественно, будет варьировать в соответствии с природой и тяжестью состояний, возрастом и полом животного или пациента и пути введения, в соответствии с хорошо известными принципами медицины. Известны различные анализы и тесты in vivo для определения полезности соединений при расстройствах, указанных выше, и особенно в качестве антагонистов рецепторов VR-1.
Соединение формулы I или его фармацевтически приемлемую соль, сольват или гидролизующийся in vivo сложный эфир, или фармацевтическую композицию или препарат, содержащие соединение формулы I, вводят параллельно, одновременно, последовательно или отдельно от другого(их) соединения или соединений, выбранных из следующих:
(1) средств лечения невропатической боли, включающих, например, габапентин, лидодерм, прегаблин и эквиваленты, включающие, но не ограничивающиеся этим, их фармацевтически приемлемую соль и фармацевтически активный(е) изомер(ы) и метаболит(ы);
(2) средств лечения ноцицептивной боли, включающих, например, целекоксиб, эторикоксиб, лумиракоксиб, рофекоксиб, валдекоксиб, диклофенак, локсопрофен, напроксен, парацетамол и эквиваленты, включающие, но не ограничивающиеся этим, их фармацевтически приемлемую соль и фармацевтически активный(е) изомер(ы) и метаболит(ы);
(3) средств лечения недержания мочи, включающих, например, дарифенацин, флавоксат, оксибутинин, пропиверин, робалзотан, солифенацин, тиспий, толтеродин и эквиваленты, включающие, но не ограничивающиеся этим, их фармацевтически приемлемую соль и фармацевтически активный(е) изомер(ы) и метаболит(ы).
В таких комбинациях продуктов используют соединения по данному изобретению в дозовом диапазоне, описанном в данной заявке, и другой фармацевтически активный агент в одобренных дозовых диапазонах и/или дозировке, описанных в опубликованной ссылке.
Способы получения
В другом аспекте настоящего изобретения предложены способы получения соединений формулы I или их солей, сольватов или сольватированных солей.
В нижеследующем описании таких способов следует понимать, что, при необходимости, подходящие защитные группы будут добавляться и впоследствии удаляться из различных реагентов и промежуточных соединений таким образом, который будет легко понятен специалисту в области органического синтеза. Традиционные способы использования таких защитных групп, а также примеры подходящих защитных групп описаны, например, в "Protective Groups in Organic Synthesis", T.W.Green, P.G.M.Wuts, Wiley-Interscience, New York, (1999). Ссылки и описания других подходящих реакций описаны в учебниках по органической химии, например, в "Advanced Organic Chemistry", March, 4th ed. McGraw Hill (1992) или в "Organic Synthesis", Smith, McGraw Hill, (1994). Для типичных примеров гетероциклической химии см., например, "Heterocyclic Chemistry", J.A.Joule, K.Mills, G.F.Smith, 3rd ed. Chapman and Hall (1995), p.189-224 и "Heterocyclic Chemistry", T.L.Gilchrist, 2nd ed. Longman Scientific and Technical (1992), p.248-282.
Термин "комнатная температура" и "температура окружающей среды" будет означать, если не указано иное, температуру от 16 до 25°С.
Сокращения
Экспериментальные способы:
Все исходные вещества имеются в продаже или описаны в литературе. Спектры 1Н-ЯМР регистрируют на Variant при 400 МГц. Масс-спектры регистрируют на (LC-MS; LC: Agilent 1100, Waters ESI-MS, колонка Phenomenex Synergi Polar (4 мкм) 30×2 мм, скорость потока: 1,75 мл/мин, подвижная фаза: А = вода (0,05% TFA) В = MECN (0,05% TFA), градиент: 5-95%, время градиента: 2,25 мин). Конечные соединения анализируют на LCMS Agilent 1100 (MS: Agilent APPI-MSD, скорость потока: 3,5 мл/мин, колонка: Zorbax SB (1,8 мкм) 4,6×30 мм, температура колонки: 70°С, подвижная фаза: А = вода (0,05% TFA) В = MECN (0,05% TFA), градиент: 5-95%, время градиента: 4,5 мин). Энантиомеры каждого продукта могут быть разделены с использованием колонок Chiralcel OD или AD от Chiral Technologies Inc.
Конечные продукты названы путем преобразования рацемического изображения молекулы в название IUPAC с использованием лабораторного программного обеспечения ACD. Энантиомерную характеристику в начале каждого названия [(+), (-), (+,-), R, S] добавляют в зависимости от того, что известно о соединении в это время.
Схема 1: Синтез (7-хлор-1Н-бензимидазол-1-ил)уксусной кислоты

Стадия а) промежуточное соединение 1
Синтез 2-[(2-хлор-6-нитрофенил)амино]этанола
2,3-Дихлорнитроанилин (300 г, 1,56 моль) смешивают с этанолом (600 мл) и этаноламином (282 мл, 4,68 моль). Смесь нагревают при температуре дефлегмации в течение 20 ч, затем охлаждают и концентрируют в вакууме. Для удаления гидрохлоридной соли этаноламина неочищенный продукт растворяют в 3,5 л AcOEt и 1 л воды. Водную фазу удаляют и органическую фазу дважды промывают 700 мл воды и рассолом. После сушки над безводным сульфатом магния раствор фильтруют и упаривают в вакууме с получением желаемого продукта (336 г, 99%) в виде оранжевого масла.
Стадия б) промежуточное соединение 2
Синтез 2-[(2-хлор-6-аминофенил)амино]этанола
К раствору 2-[(2-хлор-6-нитрофенил)амино]этанола (120 г, 0,554 моль) в метаноле (1,5 л) при 60°С добавляют раствор Na2S2O4 (85%, 318 г, 1,55 моль) в воде (1,12 л) в течение 20 мин. Полученную суспензию перемешивают при 60°С в течение дополнительных 20 мин. Обесцвеченную смесь оставляют охлаждаться и концентрируют в вакууме. В ледяную баню добавляют 800 мл 1,5 М раствора NaOH и смесь экстрагируют три раза 500 мл AcOEt. Органическую фазу промывают рассолом и сушат над сульфатом магния. Растворители выпаривают с получением желаемого продукта (72,4 г, 70%).
Стадия в) промежуточное соединение 3
Синтез 2-(7-хлор-1Н-бензимидазол-1-ил)этанола
2-[(2-Хлор-6-аминофенил)амино]этанол (72,4 г, 0,388 моль) растворяют в муравьиной кислоте (350 мл) и перемешивают при температуре дефлегмации в течение 1 часа. Реакционную смесь концентрируют досуха при пониженном давлении с получением темного твердого вещества, затем к остатку добавляют 500 мл 2 н. HCl и смесь нагревают при температуре дефлегмации в течение 30 мин. Раствор охлаждают на льду и добавляют 50%-ный раствор NaOH до щелочного значения и полученную суспензию фильтруют в вакууме, и полученное твердое вещество сушат с получением желаемого продукта (70,8 г, 93%).
Стадия г) промежуточное соединение 4
Синтез 2-(7-хлор-1Н-бензимидазол-1-ил)уксусной кислоты
2-(7-Хлор-1Н-бензимидазол-1-ил)этанол (50 г, 0,254 моль) растворяют в 1 л ацетонитрила и натрий-фосфатном буфере (750 мл, рН 6,7) и смесь нагревают до 40°С, в течение 3 часов добавляют TEMPO (2,9 г 18,5 ммоль), затем твердый NaClO2 (119 г, 85%, 1,06 моль). Одновременно добавляют раствор NaOCl (1,65 М, 40 мл) до тех пор, пока реакционная смесь не становится темно-коричневой. Смесь оставляют перемешиваться в течение 16 часов при 45°С. Избыток окислителя гасят (в ледяной бане) твердым Na2SO3 (100 г), который добавляют до полного обесцвечивания реакционной смеси. На этой стадии образуется осадок. Это твердое вещество, которое содержит 2-(7-хлор-1Н-бензимидазол-1-ил)уксусную кислоту и минеральный продукт, фильтруют и растворяют в 500 мл воды. Полученный раствор затем подкисляют до рН 2 с помощью 6 н. HCl. Осадок фильтруют и промывают водой с получением 3,76 г желаемого продукта. Водную фазу из реакционной смеси подкисляют 6 н. HCl до рН 2 и твердое вещество, которое образуется, фильтруют и промывают водой с получением 41,83 г желаемого продукта. Раствор ацетонитрила, полученный из органической фазы, концентрируют с получением суспензии неочищенного продукта в воде, которую очищают путем ее растворения в 50%-ном растворе NaOH. Водный раствор затем промывают AcOEt и осаждают с помощью 6 н. HCl до рН 6 с получением 1,76 г желаемого продукта с общим выходом 47,35 г (88%) желаемого продукта.
Схема 2: Синтез (6,7-Дифтор-1Н-бензимидазол-1-ил)уксусной кислоты

Схема 2: синтез (6,7-дифтор-1H-бензимидазол-1-ил)уксусной кислоты.
Стадия а) промежуточное соединение 5
Синтез 2-[(2,3-дифтор-6-нитрофенил)амино]этанола
Раствор 1,2,3-трифтор-4-нитробензола (5,0 г, 28,2 ммоль) и этаноламина (1,72 г, 28,2 ммоль) в 100 мл этанола перемешивают в течение ночи при комнатной температуре, затем при 70°С в течение 5 часов. Реакционную смесь концентрируют досуха и очищают с помощью флэш-хроматографии на силикагеле, используя градиент от 80/20 до 20/80 гептан/этилацетат, с получением оранжевого твердого вещества. Выход (3,8 г, 62%). 1Н-ЯМР (400 МГц, CDCl3) δ млн-1 1.67 (t, J=5.08 Гц, 1Н), 3.77-3.83 (m, 2Н), 3.88-3.94 (m, 2Н), 6.51 (ddd, J=9.77, 8.59, 7.03 Гц, 1Н), 8.02 (ddd, J=9.77, 5.66, 2.34 Гц, 1Н), 8.21 (s, 1Н).
Стадия б) промежуточное соединение 6
Синтез 2-[(6-амино-2,3-дифторфенил)амино]этанола
К раствору 2-[(2,3-дифтор-6-нитрофенил)амино]этанола (3,8 г, 17,4 ммоль) в 70 мл этилацетата и 30 мл этанола добавляют 10% Pd/C (380 мг). Реакционную смесь встряхивают при давлении водорода 50 фунтов на кв.дюйм (344,75 кПа) в течение 3 часов. Давление периодически доводят до 50 фунтов на кв.дюйм (344,75 кПа). Реакционную смесь фильтруют через целит, промывают этанолом и концентрируют. Полученное вещество используют без дополнительной очистки на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3) δ млн-1 3.17-3.27 (m, 2Н), 3.68-3.78 (m, 2Н), 6.38 (ddd, J=8.89, 4.69, 2.05 Гц, 1Н), 6.61-6.70 (m, 1Н).
Стадия в) промежуточное соединение 7
Синтез 2-(6,7-дифтор-1Н-бензимидазол-1-ил)этанола
Раствор 2-[(6-амино-2,3-дифторфенил)амино]этанола в 100 мл муравьиной кислоты нагревают до 100°С в течение 2 часов. Реакционную смесь концентрируют досуха, вливают в 100 мл 2 н. NH3 в этаноле и перемешивают в течение 2,5 ч. Реакционную смесь концентрируют и вливают в этилацетат. Полученный осадок собирают фильтрацией и промывают холодным этилацетатом. Маточную жидкость концентрируют и очищают с помощью флэш-хроматографии на силикагеле, используя этилацетат/гептан. Общий выход 3,2 г или 93% для двух стадий на основе 3,8 г 2-[(2,3-дифтор-6-нитрофенил)амино]этанола.
Стадия в) промежуточное соединение 8
Синтез (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислоты.
2-(6,7-Дифтор-1Н-бензимидазол-1-ил)этанол (2,96 г, 15 ммоль) вливают в 75 мл MeCN и натрий-фосфатный буфер (56 мл, 0,67 М, рН 6,8) и смесь нагревают до 42°С. Добавляют TEMPO (165 мг, 1,05 моль) с последующим одновременным добавлением по каплям раствора NaClO2 (3,38 г, 80% чистоты, 30 ммоль в 15 мл воды) и раствора отбеливателя (350 мкл 6% NaOCl в 7,5 мл воды) в течение 1,5 часов. Через 48 ч добавляют такие же количества NaClO2 и отбеливателя. После дополнительных 24 часов добавляют TEMPO (165 мг, 1,05 моль) и реакционную смесь перемешивают в течение 72 ч. Потемневшую реакционную смесь оставляют охлаждаться до комнатной температуры с последующим добавлением по каплям 30 мл насыщенного раствора Na2SO3 (экзотермическая). Реакционная смесь становится почти бесцветной. Используя 2 н. NaOH, рН повышают до 9,2 и реакционную смесь экстрагируют 4 раза этилацетатом. рН затем снижают до 3,8 с помощью 2 н. HCl и раствор оставляют стоять в течение 48 часов. Выделяют 1,98 г белого кристаллического вещества. Маточную жидкость уменьшают до половины объема и оставляют стоять. Собирают еще 260 мг (Общий выход 2,23 г, 70%).
1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 5.19 (s, 2Н), 7.25 (ddd, J=1.62, 8.89, 7.62 Гц, 1Н), 7.49 (ddd, J=8.94, 3.86, 1.07 Гц, 1Н), 8.13-8.28 (m, 1Н), 13.38 (s, 1Н).
Схема 3: Синтез (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты
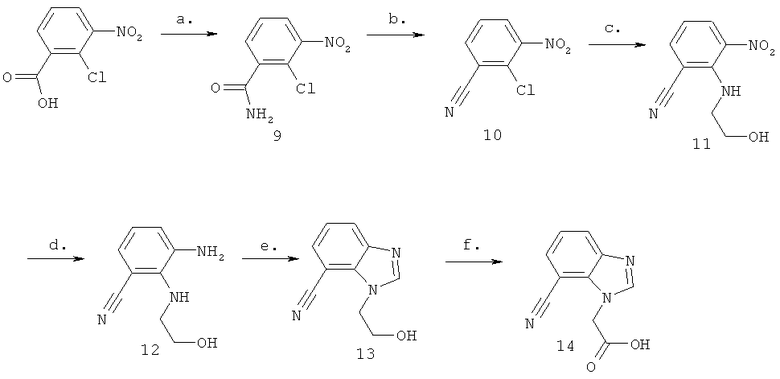
Стадия а) промежуточное соединение 9
Синтез 2-хлор-3-нитробензамида
2-Хлор-3-нитробензойную кислоту (100 г, 0,496 г) нагревают до температуры дефлегмации в чистом тионилхлориде в течение 2,5 часов с перемешиванием (выделяется газ). После охлаждения тионилхлорид упаривают досуха. Полученное твердое вещество растворяют в 150 мл дихлорметана, охлаждают в ледяной бане и добавляют 400 мл 28%-ного гидроксида аммония в течение 1 часа (реакция является экзотермической). Затем добавляют 100 мл воды для облегчения осаждения. Образовавшийся осадок фильтруют, промывают водой и сушат в течение 16 часов над Р2О5 в вакууме с получением желаемого продукта (83,2 г, 83%) в виде бледно-желтого рыхлого твердого вещества.
1Н-ЯМР (300 МГц, ДМСО-d6) δ млн1 7.61 (t, J=7.93 Гц, 1Н), 7.72 (dd, J=7.63, 1.47 Гц, 1Н), 8.04 (dd, J=7.94, 1.47 Гц, 1Н).
Стадия б) промежуточное соединение 10
Синтез 2-хлор-3-нитробензонитрила
2-Хлор-3-нитробензамид (83 г, 0,413 моль, хорошо высушенный) добавляют к кипящему раствору дегидратирующего агента*, и эту смесь затем оставляют при этой температуре на 4 часа и при комнатной температуре на 16 часов. Смесь гасят льдом, добавляют 400 мл воды для облегчения разделения фаз и водную фазу отбрасывают. Органическую фазу промывают водой и рассолом и затем сушат над безводным Na2SO4. Раствор фильтруют и концентрируют с получением желаемого продукта (74,4 г, 99%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ млн1 7.76 (t, J=7.93 Гц, 1Н), 8.27 (dd, J=7.93 1.47 Гц, 1Н), 8.36 (dd, J=8.22 1.47 Гц, 1Н).
* Получение триметилсилилполифосфата (дегидратирующий агент):
Р2О5 (254 г; 1,79 моль) в 1 л безводного дихлорметана перемешивают при температуре дефлегмации и добавляют 330 мл гексаметилдисилоксана (1,54 моль) в течение 1 часа с помощью капельной воронки (реакция является экзотермической). Реакционную смесь затем оставляют перемешиваться при этой температуре в течение 1 часа.
Стадия в) промежуточное соединение 11
Синтез 2-[(2-гидроксиэтил)амино]-3-нитробензонитрила
2-Хлор-3-нитробензонитрил (74 г, 0,408 моль) смешивают с этанолом (370 мл) и этаноламином (57 мл). Смесь перемешивают в течение 16 ч при комнатной температуре. Для завершения реакции реакционную смесь подвергают дефлегмации в течение 2 часов. После охлаждения смесь концентрируют в вакууме; продукт осаждается в виде красного твердого вещества. Для удаления гидрохлоридной соли этаноламина суспензию растирают с 500 мл воды и фильтруют в вакууме. Твердое вещество промывают этанолом и эфиром, затем сушат получением желаемого продукта (75 г, 89%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ млн-1 3.55-3.60 (m, 2Н), 3.69-3,74 (m, 2Н), 6.75 (dd, J=7.63, 8.52 Гц, 2Н), 7.90 (dd, J=7.63, 1.76 Гц, 2Н), 8.27 (dd, J=8.52, 1.76 Гц, 1Н), 3.35 m, 1Н).
Стадия г) промежуточное соединение 12
Синтез 3-амино-2-[(2-гидроксиэтил)амино]бензонитрила
Метанол (500 мл) и 5% Pd/активированный уголь (влажный, 3,45 г) добавляют к 2-[(2-гидроксиэтил)амино]-3-нитробензонитрилу (69 г, 0,333 моль). Суспензию встряхивают в аппарате Парра при давлении водорода 20 фунтов на кв. дюйм (137,9 кПа) в течение 1 часа. Смесь затем фильтруют на целите и упаривают досуха с получением желаемого вещества (62,7 г). Этот продукт используют на следующей стадии без дополнительной очистки.
1Н-ЯМР (300 МГц, MeOD) δ млн-1 3.41 (t, J=5.43 Гц, 2Н), 3.70 (t, J=5.43 Гц, 2Н), 6.75 (t, J=7.71 Гц, 1Н), 6.87 (dd, J=7.71, 1.61 Гц, 1Н), 6.92 (dd, J=7.71, 1.61 Гц, 1Н).
Стадия д) промежуточное соединение 13
Синтез 1-(2-гидроксиэтил)-1Н-бензимидазол-7-карбонитрила
3-Амино-2-[(2-гидроксиэтил)амино]бензонитрил (38 г, неочищенный) растворяют в муравьиной кислоте (150 мл) и перемешивают при температуре дефлегмации в течение 1 часа. Реакционную смесь концентрируют досуха при пониженном давлении с получением темного твердого вещества. Это твердое вещество растворяют в 200 мл метанола с нагреванием и, пока оно еще горячее, добавляют 60 мл триэтиламина и нагревают с обратным холодильником в течение 1 часа. Смесь концентрируют в вакууме и осадок фильтруют и промывают водой, затем сушат получением желаемого соединения (27 г, 70% от промежуточного соединения 11).
1Н-ЯМР (300 МГц, ДМСО-d6) δ млн1 3.79 (dt, J=5.14 Гц, 2Н), 4.51 (t, J=5.14 Гц, 2Н), 5.04 (t, J=5.14 Гц, 1Н), 7.34 (dd, J=7.63, 0.77 Гц, 1Н), 7.74 (dd, J=7.63, 0.77 Гц), 8.02 (dd, J=7.73, 0.77 Гц), 8.36 (s, 1Н).
Стадия е) промежуточное соединение 14
Синтез (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты
1-(2-Гидроксиэтил)-1Н-бензимидазол-7-карбонитрил (61,4 г, 0,328 моль) растворяют в ацетонитриле (1,2 л) и натрий-фосфатном буфере (930 мл, рН 6,8) и смесь нагревают до 40°С. Добавляют TEMPO (3,6 г 22,7 моль), затем твердый NaClO2 (148,3 г, 85%, 1,31 моль) в течение 3 часов. Одновременно добавляют раствор NaOCl (1,65 М, 50 мл) до тех пор, пока реакционная смесь не станет темно-коричневой. Смесь оставляют перемешиваться в течение 16 часов при 45°С. Избыток окислителя гасят (в ледяной бане) твердым Na2SO3, который добавляют до полного обесцвечивания реакционной смеси.
На этой стадии образуется осадок. Это твердое вещество содержит желаемые продукты и продукты в виде минеральных солей, его фильтруют и растворяют в 500 мл воды. Полученный раствор затем подкисляют до рН 2 с помощью 6 н. HCl. Осадок фильтруют и промывают водой с получением желаемого продукта (12,7 г, 19%). Водную фазу, полученную из реакционной смеси, подкисляют 6 н. HCl до рН 2, и твердое вещество, которое образуется, фильтруют и промывают водой и сушат с получением желаемого продукта (43,0 г, 65%) конечного продукта. Ацетонитрил из органической фазы упаривают с получением суспензии неочищенного продукта в воде, которую очищают путем ее растворения в 50%-ном растворе NaOH, промывкой AcOEt и осаждением с помощью 6 н. HCl до рН 6 с получением желаемого продукта (4,2 г, 6%), (59,9 г, объединенные выходы 90%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ млн-1 5.31 (s, 2Н), 7.36 (t, J=7.78 Гц, 1Н), 7.74 (dd, J=1.03, 7.78 Гц, 1Н), 8.03 (dd, J=1.03, 7.78 Гц, 1Н), 8.37 (s, 1Н).
Схема 4: Синтез (7-ацетил-1Н-бензимидазол-1-ил)уксусной кислоты
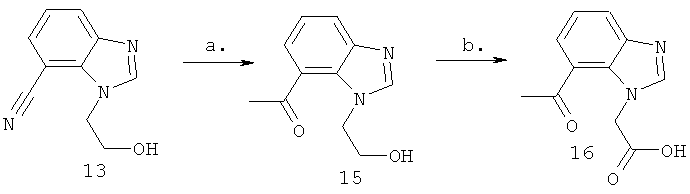
Стадия а) промежуточное соединение 15
Синтез 1-[1-(2-гидроксиэтил)-1Н-бензимидазол-7-ил]этанона.
Раствор 1-(2-гидроксиэтил)-1Н-бензимидазол-7-карбонитрила (0,29 г, 1,5 ммоль) в сухом THF (6,2 мл) охлаждают до -78°С и медленно добавляют MeLi (5,8 мл, 9,3 ммоль). После добавления реакционной смеси позволяют нагреться до температуры окружающей среды и оставляют в таком состоянии на 30 мин. Температуру снова снижают до -78°С и медленно добавляют воду (4 мл). После нагревания реакционную смесь подкисляют до рН 4 и нагревают при 50°С в течение 30 мин. Растворители удаляют при пониженном давлении и остаток разделяют между этилацетатом и водн. NaHCO3. Органический экстракт затем промывают водой и рассолом, сушат над Na2SO4 и концентрируют. Очистку осуществляют на флэш-колонке с силикагелем с использованием смеси этилацетат-метанол в качестве элюента с получением желаемого продукта (0,25 г, 80%).
1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 2.67 (s, 3H), 3.51 (q, J=5.1 Гц, 2Н), 4.41 (t, J=5.3 Гц, 2Н), 4.77 (t, J=5.1 Гц, 1Н), 7.29 (t, J=7.8 Гц, 1Н), 7.78 (dd, J=7.6, 1.0 Гц, 1Н), 7.88 (dd, J=8.1, 1.0 Гц, 1Н), 8.20 (s, 1Н).
Стадия б) промежуточное соединение 16
Синтез (7-ацетил-1Н-бензимидазол-1-ил)уксусной кислоты.
1-[1-(2-Гидроксиэтил)-1Н-бензимидазол-7-ил]этанон (0,30 г, 1,47 ммоль) окисляют до желаемой кислоты в соответствии со способом, описанным для синтеза (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты (стадия е, промежуточное соединение 14), с получением желаемого продукта (0,24 г, 75%).
1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 2.64 (s, 3H), 5.34 (s, 2Н), 7.46 (t, J=7.81 Гц, 1Н), 7.90-7.99 (m, J=6.84, 6.84 Гц, 2Н), 8.56 (s, 1Н).
Схема 5: Синтез (7-фтор-1Н-бензимидазол-1-ил)уксусной кислоты
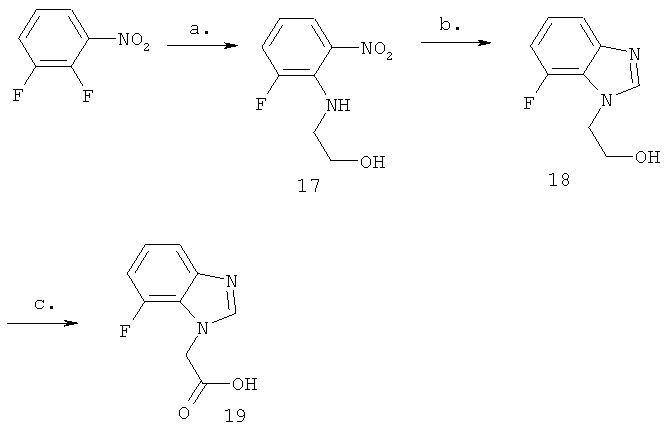
Стадия а) промежуточное соединение 17
Синтез 2-[(2-фтор-6-нитрофенил)амино]этанола
2,3-Дифторнитробензол (15 г, 94,3 ммоль) растворяют в 200 мл этанола. Добавляют этаноламин (11,4 мл, 188,7 ммоль, 2 экв.) и смесь перемешивают при комнатной температуре в течение ночи (реакция завершается по данным TLC). Этанол упаривают и полученный остаток растворяют в этилацетате, промывают водой (для удаления избытка этаноламина), сушат над сульфатом магния, фильтруют и упаривают досуха с получением желаемого продукта в виде темно-оранжевого масла (18,3 г, 97%). Это неочищенное вещество используют на следующей стадии без дополнительной очистки.
1Н-ЯМР (400 МГц, CD3OD) δ 3.61-3.68 (m, 2Н), 3.69-3.76 (m, 2Н), 4.88 (s, 2Н), 6.61-6.67 (m, 1Н), 7.27 (ddd, J=14,16, 7.91, 1.56 Гц, 1Н), 7.91 (dt, J=8.69, 1.51 Гц, 1Н). MS [М+Н], вычислено: 201, найдено: 201.
Стадия б) промежуточное соединение 18
Синтез 2-(7-фтор-1H-бензимидазол-1-ил)этанола
2-[(2-Фтор-6-нитрофенил)амино]этанол (18,3 г, 91,5 ммоль) растворяют в 90 мл муравьиной кислоты, добавляют к суспензии 10% Pd-C (300 мг) в 10 мл муравьиной кислоты. Смесь встряхивают в аппарате Парра при атмосферном давлении Н2 в течение ночи. Реакционную смесь фильтруют через целит, растворитель упаривают в вакууме и полученный остаток растворяют в 2 М NH3 в этаноле. Этот раствор перемешивают при комнатной температуре в течение 1 ч (для гидролиза аддукта муравьиной кислоты). Образуется осадок. Эту смесь упаривают досуха и очищают с помощью колоночной хроматографии (SiO2, DCM/MeOH, от 10:1 до 5:1) с получением желаемого продукта в виде белого твердого вещества (10,5 г, 64%) TLC: DCM/MeOH, 5:1, Rf=0,23.
1Н-ЯМР (400 МГц, CD3OD) δ 3.86-3.95 (m, 2Н), 4.46 (t, J=5.27 Гц, 2Н), 7.03 (dd, J=11.72, 8.01 Гц, 1Н), 7.21 (td, J=8.11, 4.88 Гц, 1Н), 7.47 (d, J=7.81 Гц, 1Н), 8.13 (s, 1Н). MS [М+Н], вычислено: 181, найдено: 181.
Стадия в) промежуточное соединение 19
Синтез (7-фтор-1Н-бензимидазол-1-ил)уксусной кислоты
2-(7-Фтор-1Н-бензимидазол-1-ил)этанол (706 мг, 3,92 ммоль) суспендируют в 20 мл ацетонитрила и 15 мл 1 М натрий-фосфатного буфера (рН 6,5). Смесь нагревают до 35°С. Добавляют TEMPO (43 мг, 0,27 ммоль), затем NaClO2 (80%, 887 мг, 7,84 ммоль), растворенный в 4 мл воды и разбавленном отбеливателе (2 мл 0,4%-ного водного раствора). После добавления отбеливателя реакционная смесь становится красно-коричневой. Для завершения реакции, при необходимости, добавляют больше TEMPO (22 мг), NaClO2 (440 мг в 2 мл воды) и разбавленного отбеливателя (1 мл) и смесь перемешивают в течение 6 часов при 35°С. После охлаждения при комнатной температуре реакционную смесь гасят путем добавления насыщенного водного Na2SO3 (5 мл). Красно-коричневая окраска исчезает. рН доводят до 8-9 путем добавления 2 М NaOH и смесь промывают этилацетатом (2х). Органический слой удаляют и водную фазу подкисляют 1 М HCl (до рН 3). Желаемый продукт кристаллизуется в водной фазе в виде белого твердого вещества (537 мг, 70%). TLC: дихлорметан/метанол, 10:1+5% триэтиламин, Rf=0,33 (s.m.: Rf=0,56).
1Н-ЯМР (400 МГц, CD3OD) δ 5.19 (s, 2Н); 7.05 (dd, J=11.52, 8.20 Гц, 1Н); 7.24 (td, J=8.15, 4.98 Гц, 1Н); 7.49 (d, J=8.20 Гц, 1Н); 8.19 (s, 1Н). MS [М+Н], вычислено: 196, найдено: 195.
Схема 6: Синтез 2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила
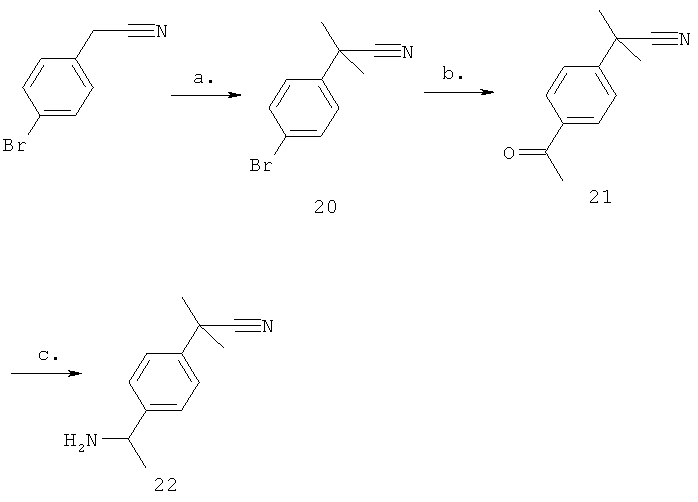
Стадия а) промежуточное соединение 20
Синтез 2-(4-бромфенил)-2-метилпропаннитрила.
Получение 2-(4-бромфенил)-2-метилпропаннитрила осуществляют, как описано в J. Med. Chem. (1995), no 38, page 1608-1628. Гидрид натрия (60%-ная суспензия в масле, 6,66 г, 166,3 ммоль) добавляют несколькими порциями в течение 1 часа к 2-(4-бромфенил)ацетонитрилу (10 г, 51,0 ммоль), растворенному в безводном DMF и метилйодиде (14,838 г, 102,0 ммоль) при 0°С. Этот раствор превращается в вязкую и коричнево-оранжевую пасту. Его оставляют перемешиваться с медленным нагреванием до комнатной температуры (18 ч). Органический раствор распределяют между водой и этилацетатом, разделяют, сушат над безводным сульфатом натрия и фильтруют. Раствор концентрируют при пониженном давлении и полученный неочищенный продукт очищают на силикагеле, используя градиент 0-20% этилацетата в гексане, с получением желаемого соединения (4,9 г, 42%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 1.71 (s, 6Н), 7.35 (d, J=8.79 Гц, 2Н), 7.52 (d, J=8.79 Гц, 2Н).
Стадия б) промежуточное соединение 21
Синтез 2-(4-ацетилфенил)-2-метилпропаннитрила.
2-(4-Бромфенил)-2-метилпропаннитрил (1 г, 4,46 ммоль) растворяют в безводном THF (75 мл), раствор охлаждают до -100°С в бане с диэтиловым эфиром и жидким азотом, добавляют 2 М н-бутиллитий в с-гексане (4,0 мл, 8,0 ммоль), и эту реакционную смесь, перемешиваемую в течение 10 мин, затем добавляют N-метокси-N-метилацетамид (1,6 г, 15,6 ммоль), и реакционную смесь затем оставляют медленно нагреваться до комнатной температуры. После обработки (промывки кислотным рассолом) и концентрирования неочищенную смесь очищают на силикагеле, используя градиент 0-50% этилацетата в гексане, с получением желаемого соединения (660 мг, 78%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 1.76 (s, 6Н), 2.62 (s, 3H), 7.59 (d, J=8.79 Гц, 2Н), 7.99 (d, J=8.79 Гц, 2Н).
Стадия в) промежуточное соединение 22
Синтез 2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила.
Получение 2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила осуществляют в соответствии с общим способом, описанным в Tetrahedron (2004), по 60, page 1463-1471. Ацетофенон, 2-(4-ацетилфенил)-2-метилпропаннитрил (600 мг, 3,21 ммоль) растворяют в 28%-ном растворе аммиака в этаноле (20,0 мл). Добавляют изопропилат титана (1,82 г, 6,42 ммоль) и эту реакционную смесь затем оставляют перемешиваться в течение 18 ч при комнатной температуре. Боргидрид натрия добавляют двумя порциями, затем оставляют перемешиваться в течение 3 ч. Бесцветный раствор медленно становится серым, затем белым, добавляют воду и оксид титана удаляют фильтрацией. Органический раствор распределяют между водой и этилацетатом, разделяют, сушат над безводным сульфатом натрия и фильтруют. Раствор концентрируют при пониженном давлении. Полученный остаток растворяют в диэтиловом эфире, фильтруют и добавляют HCl в эфире, полученный осадок фильтруют, затем сушат с получением желаемого продукта в виде HCl соли (500 мг, 69%) в виде желтого твердого вещества.
1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.61 (d, J=6.90 Гц, 3H), 1.70 (s, 6Н), 4.46 (q, J=6.90 Гц, 1Н), 7.49 (dt, J=8.64, 2.34, 2.10 Гц, 2Н), 7.60 (dt, J=8.64, 2.10 Гц, 2Н). MS [М+Н] вычислено: 189,1, найдено: 189,3.
Схема 7: Синтез 2-(4-(1-аминоэтил)-2-фторфенил)-2-метилпропаннитрила

Стадия а) промежуточное соединение 23
Синтез 2-(4-бром-2-фторфенил)-2-метилпропаннитрила
2-(4-Бром-2-фторфенил)-2-метилпропаннитрил получают из (4-бром-2-фторфенил)ацетонитрила (10 г, 51,0 ммоль) с использованием способа, описанного выше для промежуточного соединения 20, с получением желаемого продукта (11 г, 89%) в виде неочищенного бледно-желтого масла, которое не требует дополнительной очистки.
1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 1.77 (d, J=0.78 Гц, 6Н), 7.24-7.40 (m, 3H).
Стадия б) промежуточное соединение 24
Синтез 2-(4-ацетил-2-фторфенил)-2-метилпропаннитрила
2-(4-Ацетил-2-фторфенил)-2-метилпропаннитрил получают из (4-бром-2-фторфенил)ацетонитрила (5,0 г, 21,0 ммоль) с использованием способа, описанного для промежуточного соединения 21, с получением желаемого продукта (4,15 г, 99%) в виде неочищенного желтого масла, которое не очищают дополнительно после обработки.
1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 1.79-1.81 (m, 6Н), 2.58 (s, 3H), 7.60 (t, J=7.91 Гц, 1Н), 7.65 (dd, J=12.40, 1.46 Гц, 1Н), 7.72 (dd, J=8.11, 1.86 Гц, 1Н).
Стадия в) промежуточное соединение 25
Синтез 2-(4-(1-аминоэтил)-2-фторфенил)-2-метилпропаннитрила
2-(4-(1-Аминоэтил)-2-фторфенил)-2-метилпропаннитрил получают из неочищенного (4-ацетил-2-фторфенил)ацетонитрила (2,4 г, 11,7 ммоль) с использованием способа, описанного для промежуточного соединения 22, с получением желаемого продукта в виде HCl соли (2,1 г, 67%) в виде неочищенного бледно-желтого масла, которое не требует дополнительной очистки после обработки.
1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.19 (d, J=6.44 Гц, 3H), 1.67 (s, 6Н), 1.95 (s, 3H) 3.95 (q, J=6.44 Гц, 1Н), 7.19 (dd, J=8.11, 1.66 Гц, 1Н), 7.26 (dd, J=13.48, 1.56 Гц, 1Н), 7.34 (t, J=8.30 Гц, 1Н). MS [М+Н] вычислено: 207,13, найдено: 207,15.
Схема 8: Синтез 2-{4-(аминометил)-2-фторфенил)-2-метилпропаннитрила
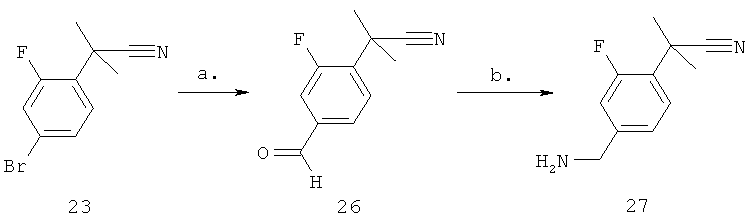
Стадия а) промежуточное соединение 26
Синтез 2-(4-формил-2-фторфенил)-2-метилпропаннитрила
2-(4-Формил-2-фторфенил)-2-метилпропаннитрил получают из (4-бром-2-фторфенил)ацетонитрила (5,2 г, 21,5 ммоль) и N-метокси-N-метилформамида (3,8 г, 43,0 ммоль) с использованием способа, аналогичного способу, описанному для промежуточного соединения 21, с получением желаемого продукта, который используют непосредственно на следующей стадии.
1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн1 1.82 (s, 3H), 1.81 (s, 3H), 7.59 (dd, J=11.62, 1.07 Гц, 1Н), 7.65-7.74 (m, 2Н), 9.97 (d, J=1.95 Гц, 1Н).
Стадия б) промежуточное соединение 27
Синтез 2-(4-(аминометил)-2-фторфенил)-2-метилпропаннитрила
2-(4-(1-Аминоэтил)-2-фторфенил)-2-метилпропаннитрил получают из неочищенного (4-ацетил-2-фторфенил)ацетонитрила (неочищенный 26) с помощью общего способа, описанного для промежуточного соединения 22, с получением желаемого продукта в виде HCl соли (1,2 г, 16% для стадий а и б) в виде неочищенного бледно-желтого масла, которое не требует дополнительной очистки после обработки.
1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ 1.77 (s, 6Н), 4.13 (s, 2Н), 7.31 (d, J=10.55 Гц, 2Н), 7.58 (t, J=8.11 Гц, 1Н). MS [М+Н] вычислено: 193,1, найдено: 193,3.
Схема 9: Синтез 1-[4-(1-аминоэтил)фенил]циклобутанкарбонитрила
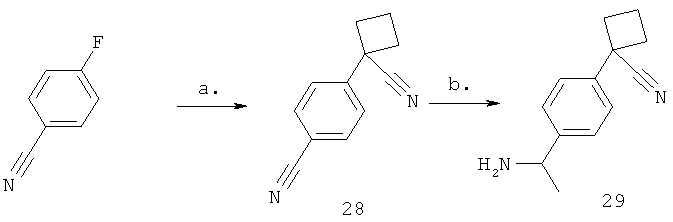
Стадия а) промежуточное соединение 28
Синтез 4-(1-цианоциклобутил)бензонитрила
Твердый KHMDS (3,48 г, 17,5 ммоль) растворяют в THF (20,0 мл) и охлаждают до 0°С. Добавляют циклобутанкарбонитрил (1,42 г, 17,5 ммоль) и полученный раствор перемешивают в течение 40 минут. Добавляют раствор 4-фторбензонитрила (2,12 г, 17,5 ммоль) в THF (10,0 мл) и смесь перемешивают в течение 2 часов при 0°С. К реакционной смеси добавляют 1 н. HCl (50,0 мл) и водную фазу экстрагируют EtOAc (4×40,0 мл). Объединенные органические фазы сушат над MgSO4, фильтруют и концентрируют на роторном испарителе. Продукт очищают с помощью флэш-хроматографии (CombiFlash), элюируя смесью гептанов и EtOAc (от 0% EtOAc до 40% EtOAc), с получением желаемого продукта (1,76 г, 9,67 ммоль, 55%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.92-2.12 (m, 1Н), 2.19-2.37 (m, 1Н), 2.57-2.69 (m, 2Н), 2.71-2.82 (m, 2Н), 7.67 (d, J=8.59 Гц, 2Н), 7.91 (d, J=8.79 Гц, 2Н).
Стадия б) промежуточное соединение 29
Синтез 1-[4-(1-аминоэтил)фенил]циклобутанкарбонитрила
4-(Цианоциклобутил)бензонитрил (335 мг, 1,84 ммоль) смешивают с THF (10,0 мл) и охлаждают до -78°С в газообразном N2. Добавляют MeLi (1,15 мл, 1,84 ммоль, 1,60 М в Et2O) и смесь перемешивают при -78°С в течение 15 минут. Добавляют смесь NaBH4 (70,0 мг, 1,84 ммоль) в МеОН (10,0 мл) и раствор нагревают до 0°С в течение 1 часа. Добавляют 1 н. HCl (40,0 мл) и раствор концентрируют досуха на роторном испарителе. Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Synergi Gemini (5 мкм) 21,2×50 мм (высокий рН), подвижная фаза: А = вода (10 мМ NH4CO3) В = MECN, (95,0 мг, 0,475 ммоль, 26%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.23 (d, J=6.64 Гц, 3H), 1.90-2.06 (m, 1Н), 2.16-2.35 (m, 1Н), 2.52-2.64 (m, 2Н), 2.64-2.78 (m, 2Н), 4.00 (q, J=6.64 Гц, 1Н), 7.36 (d, J=8.59 Гц, 2Н), 7.42 (d, J=8.20 Гц, 2Н).
Схема 10: Синтез 1-[4-(1-аминоэтил)фенил]-циклопропилкарбонитрила

Стадия а) промежуточное соединение 30
Синтез 4-(1-цианоциклопропил)бензонитрила
Твердый KHMDS (6,82 г, 34,3 ммоль) растворяют в THF (60,0 мл) и охлаждают до -40°С. Добавляют циклопропанкарбонитрил (2,30 г, 34,3 ммоль) и полученный раствор перемешивают в течение 30 минут. Добавляют раствор 4-фторбензонитрила (4,15 г, 34,3 ммоль) в THF (20,0 мл) и смесь перемешивают в течение 20 минут при -40°С, затем в течение 2 часов при комнатной температуре. Добавляют насыщенный раствор NaHCO3 (50,0 мл) и водную фазу экстрагируют EtOAc (4×40,0 мл). Объединенные органические фазы сушат над MgSO4, фильтруют и концентрируют на роторном испарителе. Продукт очищают с помощью флэш-хроматографии (CombiFlash), элюируя смесью гептанов и EtOAc (от 0% EtOAc до 70% EtOAc) (743 мг, 4,42 ммоль, 13%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.60-1.67 (m, 2Н), 1.84-1.90 (m, 2Н), 7.49 (d, J=8.59 Гц, 2Н), 7.85 (d, J=8.79 Гц, 2Н).
Стадия б) промежуточное соединение 31
Синтез 1-[4-(1-аминоэтил)фенил]циклопропилкарбонитрила
4-(Цианоциклопропил)бензонитрил (132 мг, 0,786 ммоль) смешивают с THF (10,0 мл) и охлаждают до -78°С в газообразном N2. Добавляют MeLi (0,639 мл, 1,02 ммоль, 1,60 М в Et2O) и смесь перемешивают при -78°С в течение 60 минут. Добавляют смесь NaBH4 (39,0 мг, 1,02 ммоль) в МеОН (10,0 мл) и раствор нагревают до 0°С в течение 1 часа. Добавляют 1 н. HCl (40,0 мл) и раствор концентрируют досуха на роторном испарителе. Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Synergi Gemini (5 мкм) 21,2×50 мм (высокий рН), подвижная фаза: А = вода (10 мМ NH4CO3) В = MECN, (23,0 мг, 0,0623 ммоль, 14%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.32 (d, J=6.64 Гц, 3H), 1.42-1.51 (m, 2Н), 1.66-1.77 (m, 2Н), 4.09-4.20 (m, J=6.84 Гц, 1Н45), 7.30 (d, J=8.59 Гц, 2Н), 7.42 (d, J=8.20 Гц, 1Н).
Схема 11: Синтез 1-[4-(1-аминоэтил)фенил]-циклогексилкарбонитрила

Стадия а) промежуточное соединение 32
Синтез 4-(1-цианоциклогексил)бензонитрила
Твердый KHMDS (4,36 г, 22,0 ммоль) растворяют в THF (80,0 мл) и охлаждают до 0°С. Добавляют циклогексанкарбонитрил (2,38 г, 22,0 ммоль) и полученный раствор перемешивают в течение 40 минут. Добавляют раствор 4-фторбензонитрила (1,33 г, 10,95 ммоль) в THF (10,0 мл) и смесь перемешивают в течение 2 часов при 0°С и в течение 10 часов при комнатной температуре. Добавляют 1 н. HCl (50,0 мл) и водную фазу экстрагируют EtOAc (4×50,0 мл). Объединенные органические фазы сушат над MgSO4, фильтруют и концентрируют на роторном испарителе. Продукт очищают с помощью флэш-хроматографии (CombiFlash), элюируя смесью гептанов и EtOAc (от 0% EtOAc до 30% EtOAc) (1,55 г, 7,38 ммоль, 67%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.23-1.38 (m, 1Н), 1.52-1.68 (m, 2Н), 1.68-1.78 (m, 1Н), 1.78-1.93 (m, 4Н), 2.00-2.11 (m, 2Н), 7.75 (d, J=8.79 Гц, 2Н), 7.91 (d, J=8.79 Гц, 2Н).
Стадия б) промежуточное соединение 33
Синтез 1-[4-(1-аминоэтил)фенил]циклогексилкарбонитрила
4-(Цианоциклогексил)бензонитрил (1,55 г, 7,38 ммоль) смешивают с THF (40,0 мл) и охлаждают до -78°С в газообразном N2. Добавляют MeLi (9,23 мл, 14,8 ммоль, 1,60 М в Et2O) и смесь перемешивают при -78°С в течение 60 минут. Добавляют смесь NaBH4 (558 мг, 14,8 ммоль) в МеОН (40,0 мл) и раствор нагревают до 0°С в течение 2 часов. Добавляют 1 н. HCl (50,0 мл) и раствор концентрируют досуха на роторном испарителе. Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Synergi Gemini (5 мкм) 21,2×50 мм (высокий рН), подвижная фаза: А = вода (10 мМ NH4CO3) В = MECN, (510 мг, 2,24 ммоль, 30%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.22 (d, J=6.64 Гц, 3H), 1.25-1.34 (m, 1Н), 1.52-1.68 (m, 2Н), 1.69-1.76 (m, 1Н), 1.76-1.88 (m, 4Н), 1.99-2.07 (m, 2Н), 3.96 (q, J=6.51 Гц, 1Н), 7.37-7.45 (m, 4Н).
Схема 12: Синтез (7-хлор-6-фтор-1Н-бензимидазол-1-ил)уксусной кислоты.

Стадия а) промежуточное соединение 34
2-[(2-Хлор-3-фтор-6-нитрофенил)амино]этанол.
2-Хлор-1,3-дифтор-4-нитробензол (5,37 г, 27,7 ммоль), этаноламин (1,69 г, 27,7 ммоль) и Et3N (2,80 г, 27,7 ммоль) перемешивают в EtOH (40,0 мл) при комнатной температуре в течение 2 часов. Затем растворитель упаривают и полученный остаток суспендируют в EtOAc (50,0 мл) и промывают 0,5 н. NaOH (50,0 мл). Водную фазу экстрагируют 4 раза EtOAc (4×50,0 мл). Объединенные органические фазы сушат с MgSO4, фильтруют и концентрируют. Продукт очищают с помощью флэш-хроматографии на силикагеле, элюируя смесями гептана и EtOAc (5,53 г, 85%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 3.39 (dd, J=10.55, 5.27 Гц, 2Н), 3.53 (t, J=5.47 Гц, 2Н), 4.89 (s, 1Н), 6.89 (dd, J=9.37, 8.01 Гц, 1Н), 7.19 (t, J=4.69 Гц, 1Н), 8.01 (dd, J=9.47, 5.96 Гц, 1Н).
Стадия б) промежуточное соединение 35
2-[(6-Амино-2-хлор-3-фторфенил)амино]этанол.
2-[(2-Хлор-3-фтор-6-нитрофенил)амино]этанол (5,52 г, 22,7 ммоль) растворяют в МеОН (40,0 мл). К первому раствору добавляют предварительно смешанный раствор Na2S2O4 (13,8 г, 79,5 ммоль) в воде (40,0 мл). Полученный раствор перемешивают в течение 5 минут при 60°С, затем в течение 2 часов при комнатной температуре. Растворители выпаривают и полученный остаток суспендируют в насыщенном растворе NaHCO3(40,0 мл). Водную фазу экстрагируют 4 раза EtOAc (4×40,0 мл). Объединенные органические фазы сушат с MgSO4, фильтруют и концентрируют. Продукт является достаточно чистым по данным 1Н-ЯМР (2,07 г, 45%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 2.99 (m, 2Н), 3.32 (s, 1Н), 3.48 (t, J=5.57 Гц, 2Н), 4.09 (s, 1Н), 4.73-4.95 (m, 2Н), 6.55 (dd, J=8.79, 5.66 Гц, 1Н), 6.71 (t, J=8.89 Гц, 1Н).
Стадия в) промежуточное соединение 36
2-(7-Хлор-6-фтор-1Н-бензимидазол-1-ил)этанол.
2-[(6-Амино-2-хлор-3-фторфенил)амино]этанол (2,07 г, 10,2 ммоль) растворяют в муравьиной кислоте (50,0 мл) и полученный раствор нагревают до 100°С в течение 1 часа. Раствор охлаждают до комнатной температуры и затем упаривают досуха. Остаток суспендируют в NH3 (50,0 мл, 2 н. в EtOH) и перемешивают в течение 1 часа. Раствор концентрируют досуха и остаток суспендируют в EtOAc (50,0 мл). Органическую фазу промывают 2 н. NaOH (50,0 мл) и полученную водную фазу экстрагируют 4 раза EtOAc (4×50,0 мл). Объединенные органические фазы сушат с MgSO4, фильтруют и концентрируют. Продукт растворяют в EtOAc, фильтруют и перекристаллизовывают из EtOAc (895 мг, 41%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 3.74 (t, J=5.27 Гц, 2Н), 4.52 (t, J=5.47 Гц, 2Н), 4.99 (s, 1Н), 7.25 (dd, J=10.16, 8.79 Гц, 1Н), 7.64 (dd, J=8.79, 4.49 Гц, 1Н), 8.21 (s, 1Н).
Стадия г) промежуточное соединение 37
(7-Хлор-6-фтор-1Н-бензимидазол-1-ил)уксусная кислота.
2-(7-Хлор-6-фтор-1Н-бензимидазол-1-ил)этанол (550 мг, 2,57 ммоль) растворяют в АсОН (20,0 мл). Добавляют по каплям реагент Джонсаа (3,31 мл, 3,00 ммоль, 0,907 М) и раствор перемешивают в течение 1 часа при комнатной температуре. Раствор разбавляют iPrOH (50,0 мл) и растворители выпаривают. К раствору добавляют 6 н. NaOH до рН 11. Водную фазу промывают EtOAc (50,0 мл) и органическую фазу экстрагируют 3 раза водой (3×50,0 мл). Объединенные водные фазы подкисляют 12 н. HCl до рН 3. Водную фазу затем экстрагируют 4 раза EtOAc (4×50,0 мл). Объединенные органические фазы сушат с MgSO4, фильтруют и концентрируют. Продукт является достаточно чистым по данным 1Н-ЯМР (236 мг, 40%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 5.18 (s, 2Н), 7.24 (dd, J=10.16, 8.79 Гц, 1Н), 7.64 (dd, J=8.79, 4.49 Гц, 1Н), 8.13-8.30 (m, 1Н).
а Реагент Джонса получают путем смешивания 997 мг CrO3 в 1,00 мл H2SO4 и 10,0 мл воды.
Альтернативное получение (7-хлор-6-фтор-1Н-бензимидазо-1-ил)уксусной кислоты
Стадия 1. Синтез N-(2-хлор-3-фтор-6-нитрофенил)глицинметилового эфира

В трехгорлую круглодонную колбу, снабженную барботером азота и термометром, загружают K2CO3 (40,0 г, 289,3 ммоль), глицинметилового эфира гидрохлорид (17,4 г, 138,8 ммоль) и 2-пропанол (200 мл). Полученную смесь перемешивают при комнатной температуре в течение одного часа. 3-Хлор-2,4-дифторнитробензол (22,4 г, 115,7 ммоль, 1 экв.) загружают в реакционную смесь, которая становится суспензией желтого цвета. Эту суспензию перемешивают при комнатной температуре в течение 18 ч. Протекание реакции контролируют с помощью 1Н-ЯМР спектроскопии.
После подтверждения завершения реакции смесь разбавляют iPrOAc (560 мл) и к вышеуказанной смеси по каплям добавляют 1 М HCl (225 мл) (контроль выделения CO2) при энергичном перемешивании. Темно-желтая суспензия превращается в прозрачный и бесцветный двухфазный раствор. Органический слой отделяют и промывают 1 М HCl (3×100 мл) и затем сушат над MgSO4. Осушитель фильтруют и фильтрат упаривают досуха на роторном испарителе с получением неочищенного продукта в виде желтого твердого вещества (29,1 г), которое по данным 1Н-ЯМР является очень чистым.
1Н-ЯМР (400 МГц, CDCl3) δ: 8.08 (ddd, 1Н), 6.7 (m, 1Н), 4.35 (s, 2Н), 3.8 (s, 3H).
Реакционную смесь подвергают мониторингу путем отбора аликвоты реакционной смеси в iPrOAc, гашения ее 1 н. HCl и затем отделения и упаривания органического слоя досуха и анализа полученного желтого твердого вещества с помощью 1Н-ЯМР.
Стадия 2. Превращение N-(2-хлор-3-фтор-6-нитрофенил)глицинметилового эфира в (7-хлор-6-фтор-1Н-бензимидазо-1-ил)уксусной кислоты гидрохлорид
А.

В колбу Парра для гидрирования загружают N-(2-хлор-3-фтор-6-нитрофенил)глицинметиловый эфир (15 г, 60,8 ммоль) с муравьиной кислотой (150 мл). Полученную смесь нагревают до примерно 50°С с образованием прозрачного раствора, который охлаждают до комнатной температуры. К этому раствору добавляют 5% Pd-C (0,6 г, 4 мас.%) и реакционную смесь гидрируют при 40 фунтах на кв.дюйм (275,8 кПа) в течение 3 ч. Завершение реакции подтверждают с помощью 1Н-ЯМР.
Также осуществляют идентичную реакцию с использованием 12,3 г (49,8 ммоль) N-(2-хлор-3-фтор-6-нитрофенил)глицинметилового эфира, 125 мл муравьиной кислоты и 0,5 г катализатора.
После подтверждения завершения реакции реакционную смесь после двух вышеописанных экспериментов фильтруют через воронку из пористого стекла над слоем целита. Осадок на фильтре промывают горячей муравьиной кислотой (60°С) до тех пор, пока фильтрат не станет бесцветным. Фильтрат упаривают досуха при пониженном давлении и полученный коричневый осадок сушат в вакуумной печи при 60°С в течение 3 ч с получением коричневого твердого вещества (24,2 г). Гидрирование приводит к образованию смеси продуктов, которую превращают в бензимидазол следующим образом.
Это полученное неочищенное вещество растворяют в муравьиной кислоте (242 мл) и конц. HCl (242 мл). Полученную смесь нагревают до температуры дефлегмации и выдерживают в течение 2 ч. После подтверждения завершения реакции с помощью 1Н-ЯМР реакционную смесь упаривают досуха на роторном испарителе. Полученный твердый остаток растворяют в 300 мл MeCN с образованием суспензии, которую снова концентрируют досуха. Полученный коричневый твердый остаток растирают с 300 мл MeCN при комнатной температуре в течение одного часа. Твердое вещество собирают фильтрацией и сушат в вакуумной печи при 60°С в течение 18 ч с получением желаемого продукта (23,2 г, 79%) в виде гидрохлоридной соли. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 9.25 (s, 1Н), 7.83 (dd, 1Н), 7.52 (t, 1Н), 5.5 (s, 2Н).
Стадия 3. Выделение желаемой (7-хлор-6-фтор-1Н-бензимидазол-1-ил)уксусной кислоты.
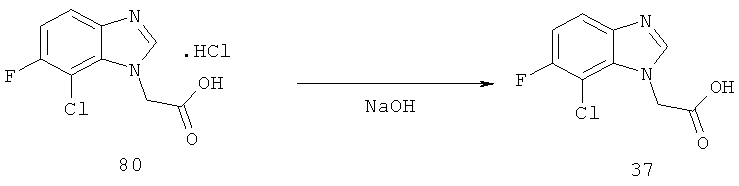
Гидрохлоридную соль желаемой кислоты (5 г) суспендируют в воде (25 мл). При умеренном перемешивании к этой суспензии по каплям добавляют 2 М NaOH до тех пор, пока все не растворится, с получением прозрачного коричневого раствора. Этот водный раствор промывают EtOAc (2×25 мл). Водный слой отделяют и подкисляют путем добавления по каплям 1 М HCl до тех пор, пока рН реакционной смеси не достигнет примерно 4,2. Образуется суспензия, которую охлаждают до 0-5°С, погружая в баню со льдом и водой и выдерживая в течение одного часа при умеренном перемешивании. Твердое вещество собирают фильтрацией и осадок на фильтре сушат на воздухе в течение выходных с получением желаемого продукта 37 в виде светло-коричневого твердого вещества (2,5 г, 60%).
Схема 13: Синтез (6-хлор-7-фтор-1Н-бензимидазол-1-ил)уксусной кислоты.

Стадия а) промежуточное соединение 38
2-(6-Хлор-7-фтор-1H-бензимидазол-1-ил)этилформат
1,3-Дихлор-2-фтор-4-нитробензол (9,70 г, 46,2 ммоль), этаноламин (7,05 г, 231,0 ммоль) перемешивают в EtOH (25,0 мл) при 60°С в течение 24 часов. Затем растворитель упаривают и полученный остаток растворяют в МеОН (150,0 мл). К первому раствору добавляют предварительно смешанный раствор Na2S2O4 (23,7 г, 136,4 ммоль) в воде (100 мл). Полученный раствор перемешивают в течение 30 минут при 60°С. Растворители выпаривают и полученный остаток суспендируют в насыщенном растворе NaHCO3 (40,0 мл). Водную фазу экстрагируют 4 раза EtOAc (4×100,0 мл). Объединенные органические фазы фильтруют и концентрируют. Полученное неочищенное вещество нагревают при 100°С в муравьиной кислоте (60,0 мл) в течение 3 часов, затем перемешивают при комнатной температуре в течение 18 ч. Реакционную смесь концентрируют при пониженном давлении, к полученному остатку добавляют конц. раствор бикарбоната натрия (10 мл) и экстрагируют этилацетатом (3×100 мл). Объединенные органические фазы сушат с MgSO4, фильтруют и концентрируют. Полученное твердое вещество растирают с метанолом с получением ожидаемого продукта 2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)этилформата (2,2 г, 19%). 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 4.49-4.62 (m, 2Н), 4.64-4.77 (m, 2Н), 7.31 (dd, J=8.59, 7.03 Гц, 1Н), 7.46 (d, J=8.59 Гц, 1Н), 8.02 (s, 1Н), 8.23 (s, 1Н).
Стадия б) промежуточное соединение 39
2-(6-Хлор-7-фтор-1Н-бензимидазол-1-ил)этанол.
2-(6-Хлор-7-фтор-1Н-бензимидазол-1-ил)этилформат (1,1 г, 4,53 ммоль) растворяют в МеОН (50,0 мл) с триэтиламином (5,0 мл) и перемешивают при комнатной температуре в течение 18 часов. Реакционную смесь затем концентрируют с получением ожидаемого продукта 2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)этанола (0,95 г, 98%) в виде белого порошка. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 3.90 (t, J=4.69 Гц, 2Н), 4.47 (t, J=5.08 Гц, 2Н), 7.30 (dd, J=8.79, 6.84 Гц, 1Н, 7.45 (dd, J=8.59, 0.78 Гц, 1Н), 8.16-8.19 (m, 1Н).
Стадия в) промежуточное соединение 40
(6-Хлор-7-фтор-1Н-бензимидазол-1-ил)уксусная кислота.
2-(6-Хлор-7-фтор-1Н-бензимидазол-1-ил)этанол (800 мг, 3,74 ммоль) вливают в 40 мл MeCN и натрий-фосфатный буфер (30 мл, 0,67 М, рН 6,8) и смесь нагревают до 36°С. Добавляют TEMPO (174 мг, 1,12 моль) с последующим одновременным добавлением по каплям раствора NaClO2 (201 мг, 80% чистоты, 2,23 ммоль в 15 мл воды) и раствора отбеливателя (500 мкл 6%-ного раствора NaOCl) в течение 2 часов. Через 24 часа реакционную смесь оставляют охлаждаться до комнатной температуры, затем добавляют по каплям насыщенный раствор Na2SO3. Используя конц. HCl, рН снижают до 1 и осаждается белый порошок, который фильтруют и промывают несколькими каплями дистиллированной воды. Полученное твердое вещество сушат в вакууме с получением ожидаемого продукта (7-хлор-6-фтор-1Н-бензимидазол-1-ил)уксусной кислоты (540 мг, 63%) в виде белого порошка. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 4.08 (s, 2Н), 6.50 (dd, J=8.59, 7.03 Гц, 1Н), 6.66 (d, J=8.59 Гц, 1Н), 7.37 (s, 1Н).
Схема 14: Синтез 4-[4-(1-аминоэтил)-3-метилфенил]тетрагидро-2Н-тиопиран-4-карбонитрила
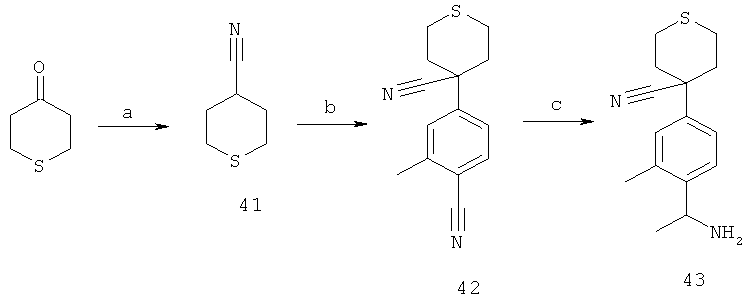
Стадия а) промежуточное соединение 41
Тетрагидро-2H-тиопиран-4-карбонитрил
Смесь тетрагидро-4H-тиопиран-4-она (8,0 г, 68,9 ммоль), тозилметилизоцианата (14,76 г, 75,6 ммоль) и 1,2-диметоксиэтана (400 мл) перемешивают в атмосфере азота при 0°С в ледяной бане, при этом медленно добавляя раствор t-BuOK в THF (1,0 М, 15,1 ммоль) через шприц. Смеси позволяют нагреться до комнатной температуры с перемешиванием в течение 5 часов. Содержимое опять охлаждают до 0°С в ледяной бане и добавляют воду (10,0 мл) для гашения реакции. Растворитель удаляют на роторном испарителе и в сосуд добавляют EtOAc (100 мл) и насыщенный водный NaHCO3 (100 мл). Фазы разделяют и органическую фазу промывают рассолом, сушат с Na2SO4 и концентрируют до остатка, который очищают с помощью колоночной хроматографии на силикагеле (EtOAc/гексаны) с получением продукта (3,5 г, 27,6 ммоль, 41%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.79-1.94 (m, 2Н), 1.97-2.14 (m, 2Н), 2.51-2.73 (m, 4Н), 2.96-3.10 (m, 1Н).
Стадия б) промежуточное соединение 42
4-(4-Циано-3-метилфенил)тетрагидро-2Н-тиопиран-4-карбонитрил
Раствор тетрагидро-2Н-тиопиран-4-карбонитрила (197 мг, 1,55 ммоль) и THF (2,0 мл) медленно добавляют через шприц к смеси KHMDS (324 мг, 1,63 ммоль) в THF (3,0 мл), перемешивая при -78°С в атмосфере азота. Температуру поддерживают при -78°С в течение 30 мин, затем в сосуд медленно добавляют смесь 4-фтор-2-метилбензонитрила (222 мг, 1,64 ммоль) в THF (1,0 мл). Смеси позволяют нагреться до комнатной температуры и перемешивают в течение 3 часов, затем гасят насыщенным водным NH4Cl. Растворитель удаляют на роторном испарителе и добавляют EtOAc (10,0 мл) и воду (10,0 мл). Фазы разделяют и органическую фазу промывают рассолом, сушат с Na2SO4 и концентрируют с получением продукта (252 мг, 1,04 ммоль, 67%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.84-1.95 (m, 2Н), 2.03-2.14 (m, 2Н), 2.51 (s, 3H), 2.57-2.72 (m, 4Н), 7.23-7.31 (m, 1Н), 7.41 (dd, J=9.96, 1.76 Гц, 1Н), 7.89 (dd, J=8.59, 5.86 Гц, 1Н).
Стадия в) промежуточное соединение 43
4-[4-(1-Аминоэтил)-3-метилфенил]тетрагидро-2Н-тиопиран-4-карбонитрил
4-(4-Циано-3-метилфенил)тетрагидро-2Н-тиопиран-4-карбонитрил (983 мг, 4,06 ммоль) перемешивают в THF (40,0 мл) при -78°С в атмосфере азота по мере того, как в сосуд через шприц медленно добавляют 1,6 М раствор метиллития в Et2O. Содержимое перемешивают в течение 3 часов при -20°С, затем гасят МеОН (40,0 мл) и нагревают до комнатной температуры. Медленно добавляют NaBH4 (460 мг, 12,2 ммоль) при 0°С в ледяной бане и смесь нагревают до комнатной температуры и перемешивают в течение 16 часов. Добавляют 1,0 н. водную HCl до рН 3,0 и растворитель удаляют на роторном испарителе. К остатку добавляют концентрированную водную HCl (3,0 мл) и смесь перемешивают в течение 16 часов. Содержимое нейтрализуют 1,0 М водным NaOH и добавляют EtOAc (40,0 мл). Слои разделяют и органический слой сушат с Na2SO4 и концентрируют до остатка, который очищают колоночной хроматографией на силикагеле (10% МеОН в DCM) (253 мг, 1,05 ммоль, 26%).
Схема 15: Синтез 2-[4-(1-аминоэтил)-3-метилфенил]-2-метилпропаннитрила

Стадия а) промежуточное соединение 44
(4-Бром-3-метилфенил)метанол
Метил-4-бром-3-метилбензоат (17,0 г, 74,2 ммоль) растворяют в безводном THF (100 мл). Раствор охлаждают до 0°С и к смеси добавляют 2 М раствор алюмогидрида лития в THF (40 мл). Раствор оставляют перемешиваться в течение 30 мин при этой температуре. Затем в реакционную смесь добавляют по каплям холодный раствор HCl до растворения комплекса алюминия. Желаемый продукт экстрагируют этилацетатом, органическую фазу промывают рассолом (200 мл), сушат с MgSO4 и концентрируют в вакууме с получением ожидаемого продукта (4-бром-3-метилфенил)метанола (14,4 г, 97%) в виде прозрачного желтого масла, чистого согласно протонному ЯМР, и используют в таком виде на следующей стадии. 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 2,40 (s, 3H), 4.61 (s, 2Н), 6.89-7.10 (m, J=8.20, 1.56, 0.59 Гц, 1Н), 7.23 (d, J=2.34 Гц, 1Н), 7.50 (d, J=8.20 Гц, 1Н).
Стадия б) промежуточное соединение 45
1-Бром-4-(бромметил)-2-метилбензол
(4-Бром-3-метилфенил)метанол (14,4 г, 71,6 ммоль) растворяют в безводном CH2Cl2 (150 мл) и добавляют CBr4 (26,1 г, 79,0 ммоль). Реакционную смесь охлаждают до 0°С и маленькими порциями добавляют PPh3 (20,7 г, 79,0 ммоль). Реакционную смесь перемешивают в течение 2 ч и образующийся оксид трифенилфосфина отфильтровывают, и растворитель удаляют в вакууме. Полученное полутвердое вещество фильтруют через слой силикагеля и промывают смесью гексан/EtOAc (9:1) с получением ожидаемого продукта 1-бром-4-(бромметил)-2-метилбензола в виде прозрачного масла, загрязненного бромоформом, которое используют непосредственно на следующей стадии. 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 4.39-4.44 (m, 3H), 7.07 (dd, J=8.20, 2.34 Гц, 1Н), 7.26 (t, J=1.17 Гц, 1Н), 7.49 (d, J=8.20 Гц, 1Н).
Стадия в) промежуточное соединение 46
2-(4-Бром-3-метилфенил)ацетонитрил
1-Бром-4-(бромметил)-2-метилбензол (неочищенный 45 со стадии b, 68,2 ммоль), растворенный в CH2Cl2 (500 мл), смешивают и перемешивают с цианидом калия (24 г, 364 ммоль) и N-тетра(н-бутил)аммония бромидом (1,2 г, 3,64 ммоль) в дистиллированной воде (500 мл). Эту реакционную смесь энергично перемешивают в течение 6 ч. Органическую фазу отделяют, сушат над MgSO4, фильтруют и концентрируют. Полученный остаток очищают на силикагеле, используя градиент от 0 до 30% этилацетата в гептане, с получением ожидаемого продукта 2-(4-бром-3-метилфенил)ацетонитрила (12,4 г, 87% после двух стадий) в виде чистого желтого масла. 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 2.41 (s, 3H), 3.68 (s, 2Н), 7.01 (dd, J=8.50, 2.05 Гц, 1Н), 7.21 (d, J=1.37 Гц, 1Н), 7.53 (d, J=8.20 Гц, 1Н).
Стадия г) промежуточное соединение 47
2-(4-Бром-3-метилфенил)-2-метилпропаннитрил
К перемешиваемому раствору 2-(4-бром-3-метилфенил)ацетонитрила (11,2 г, 53,3 ммоль) в безводном DMF (125 мл) добавляют метилйодид (13,2 мл, 213 ммоль). Раствор охлаждают до 0°С и маленькими порциями добавляют гидрид натрия (60%-ная суспензия в масле, 3,84 г, 160 ммоль) в течение 20 мин. Реакционную смесь затем оставляют перемешиваться и медленно нагревают до комнатной температуры в течение 18 ч. Затем при 0°С медленно добавляют воду (500 мл), затем экстрагируют этилацетатом, содержащим 10% гексанов. Органический слой отделяют, сушат с MgSO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта 2-(4-бром-3-метилфенил)-2-метилпропаннитрила (12,6 г, 99%) в виде прозрачного желтого масла, которое используют на следующей стадии без дополнительной очистки. 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 1.71 (s, 6Н), 2.43 (s, 3H), 7.14 (dd, J=8.40, 2.34 Гц, 1Н), 7.34 (d, J=2.54 Гц, 1Н) 7.53, (d, J=8.40 Гц, 1Н).
Стадия д) промежуточное соединение 48
2-(4-Ацетил-3-метилфенил)-2-метилпропаннитрил
К перемешиваемому раствору 2-(4-бром-3-метилфенил)-2-метилпропаннитрила (5,0 г, 21,0 ммоль) в безводном THF (75 мл) при -100°С добавляют н-бутиллитий (2 M в с-гексане) (21 мл, 42 ммоль). Эту реакционную смесь перемешивают при этой температуре 5 мин, затем добавляют N-метокси-N-метилацетамид (4,33 г, 42 ммоль). Затем раствор нагревают до комнатной температуры в течение 1 ч. Затем медленно добавляют кислотный рассол (30 мл рассола, 15 мл 3%-ной водн. HCl) и раствор экстрагируют EtOAc (3×100 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают на силикагеле, используя градиент от 0 до 30% EtOAc в гептане, с получением ожидаемого продукта 2-(4-ацетил-2-метилфенил)-2-метилпропаннитрила (1,0 г, 24%). 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 1.71 (s, 6Н) 2.53 (s, 3H), 2.56 (s, 3H), 7.30-7.38 (m, 2Н), 7.70 (d, J=8.01 Гц, 1Н).
Стадия е) промежуточное соединение 49
2-[4-(1-Аминоэтил)-3-метилфенил]-2-метилпропаннитрил
К перемешиваемому раствору 2-(4-ацетил-3-метилфенил)-2-метилпропаннитрила (1,0 г, 4,97 ммоль) в 7 М аммиаке в МеОН (40 мл) добавляют изопропилат титана (IV) (3,0 г, 9,95 ммоль). Реакционную смесь оставляют перемешиваться в течение 24 ч при комнатной температуре. После охлаждения до 0°С добавляют боргидрид натрия (1,0 г, 19,9 ммоль) и реакционную смесь перемешивают и оставляют нагреваться до комнатной температуры 1 ч. Затем добавляют конц. гидроксид аммония (15 мл) и оксид титана удаляют фильтрацией и промывают этилацетатом. Фильтрат экстрагируют EtOAc (2×100 мл) и объединенные органические слои сушат над MgSO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта 2-[4-(1-аминоэтил)-3-метилфенил]-2-метилпропаннитрила (1,0 г, 99%) в виде прозрачного желтого масла. 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн1 1.33 (d, J=6.64 Гц, 3H), 1.44 (s, 2Н) 1.70 (s, 6Н) 2.36 (s, 3H), 4.34 (q, J=6.44 Гц, 1Н) 7.22 (d, J=1.95 Гц, 1Н), 7.28 (dd, J=8.20, 2.15 Гц, 1Н), 7.48 (d, J=8.20 Гц, 1Н).
Схема 16: Синтез 2-[4-(1-аминоэтил)-2-метилфенил]-2-метилпропаннитрила

Стадия а) промежуточное соединение 50
(4-Бром-2-метилфенил)метанол
4-Бром-2-метилбензойную кислоту (11,1 г, 51,6 ммоль) растворяют в безводном THF (11,2 мл). Раствор охлаждают до 0-5°С и в смесь добавляют 1 М раствор ВН3 в THF (103 мл). Раствор оставляют перемешиваться в течение 3 ч при комнатной температуре. Затем добавляют холодную воду (20 мл) и реакционную смесь промывают насыщенным раствором NaHCO3 (120 мл). Водную фазу экстрагируют диэтиловым эфиром (3×300 мл) и объединенные органические фазы промывают рассолом (200 мл), сушат с MgSO4 и концентрируют в вакууме. Полученное масло очищают флэш-хроматографией, элюируя смесью гексан/EtOAc от 95:5 до 70:30, с получением ожидаемого продукта (4-бром-2-метилфенил)метанола (10,4 г, 100%) в виде прозрачного масла.
1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.36-7.30 (2Н, m), 7.25-7.21 (1Н, m), 4.65 (2Н, s), 2.32 (3H, s).
Стадия б) промежуточное соединение 51
1-Бром-4-(бромметил)-3-метилбензол
(4-Бром-2-метилфенил)метанол (10,4 г, 51,6 ммоль) растворяют в безводном CH2Cl2 (150 мл) и добавляют СВr4 (18,8 г, 56,8 ммоль). Реакционную смесь охлаждают до 0-5°С и добавляют PPh3 (14,9 г, 56,8 ммоль). Реакционную смесь перемешивают в течение ночи, затем добавляют гексан/EtOAc (9:1) (250 мл) при энергичном перемешивании. Оксид трифенилфосфина, который образуется во время реакции, отфильтровывают и фильтраты концентрируют в вакууме. Полученное масло очищают через слой силикагеля смесью гексан/EtOAc (8:2). Растворитель удаляют на роторном испарителе, а бромоформ удаляют вакуумной дистилляцией (15 мм рт.ст., т.к.: 40-50°С) с получением ожидаемого продукта 1-бром-4-(бромметил)-3-метилбензола (13,23 г, 97%) в виде желтого масла. 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.37-7.27 (2Н, m), 7.17 (1Н, d, J=8,1 Гц), 4.45 (2Н, s), 2.39 (3H, s).
Стадия в) промежуточное соединение 52
2-(4-Бром-2-метилфенил)ацетонитрил
1-Бром-4-(бромметил)-3-метилбензол (13,2 г, 50,1 ммоль) растворяют в DMF (65 мл). Реакционную смесь охлаждают до 0-5°С и добавляют NaCN (3,66 г, 74,6 ммоль), затем воду (8 мл). Реакционную смесь перемешивают в течение ночи при комнатной температуре и добавляют воду (170 мл), затем насыщ. NaHCO3 (130 мл) и гексан/Et2O (2:1) (150 мл). Органическую фазу отделяют и водную фазу экстрагируют смесью гексан/Et2O (2:1) (3×150 мл). Объединенные органические фазы промывают водой (170 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта 2-(4-бром-2-метилфенил)ацетонитрила (9,76 г, 93%) в виде оранжевого масла. 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.40-7.32 (2Н, m), 7.23 (2Н, d, J=8,2 Гц), 3.61 (2Н, s), 2.32 (3H, s).
Стадия г) промежуточное соединение 53
2-(4-Бром-2-метилфенил)-2-метилпропаннитрил
К перемешиваемому раствору 2-(4-бром-2-метилфенил)ацетонитрила (3,93 г, 18,8 ммоль) в безводном DMF (35 мл) добавляют метилйодид (2,45 мл, 39,4 ммоль). Раствор охлаждают до 0°С и добавляют гидрид натрия (60%-ная суспензия в масле, 1,47 г, 61,1 ммоль) 3 равными порциями в течение 20 мин. Реакционную смесь затем оставляют перемешиваться и медленно нагревают до комнатной температуры в течение 18 ч. Раствор превращается в густую коричнево-оранжевую пасту. Затем при 0°С медленно добавляют воду (50 мл) и раствор экстрагируют раствором гексан/Et2O, 2:1 (3×50 мл). Органический слой промывают рассолом, сушат с MgSO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (9:1), с получением ожидаемого продукта 2-(4-бром-2-метилфенил)-2-метилпропаннитрила (3,1 г, 66%) в виде прозрачного желтоватого масла. 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.40-7.30 (2Н, m), 7.16 (2Н, d, J=8.5 Гц), 2.63 (3H, s), 1.77 (6Н, s).
Стадия д) промежуточное соединение 54
2-(4-Ацетил-2-метилфенил)-2-метилпропаннитрил
К перемешиваемому раствору 2-(4-бром-2-метилфенил)-2-метилпропаннитрила (3,1 г, 13,0 ммоль) в безводном THF (75 мл) при -78°С добавляют н-бутиллитий (2 M в с-гексане) (7,16 мл, 14,3 ммоль). Эту реакционную смесь перемешивают при этой температуре в течение 10 мин, затем добавляют N-метокси-N-метилацетамид (2,77 мл, 26,0 ммоль) и затем раствор нагревают до комнатной температуры в течение 1 ч. Затем медленно добавляют кислотный рассол (30 мл рассола, 15 мл 3%-ной водн. HCl) и раствор экстрагируют EtOAc (3×100 мл). Органический слой сушат с MgSO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью флэш-хроматографии с использованием градиента от 5 до 20% EtOAc в гексане с получением ожидаемого продукта 2-(4-ацетил-2-метилфенил)-2-метилпропаннитрила (1,77 г, 68%) в виде желтоватого масла. 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.84-7.74 (2Н, m), 7.43-7.40 (1Н, m), 2.70 (3H, s), 2.60 (3H, s), 1.81 (6Н, s).
Стадия е) промежуточное соединение 55
2-[4-(1-Аминоэтил)-2-метилфенил]-2-метилпропаннитрил
К перемешиваемому раствору 2-(4-ацетил-2-метилфенил)-2-метилпропаннитрила (1,77 г, 8,80 ммоль) в 7 М аммиаке в МеОН (45 мл) добавляют свежедистиллированный изопропилат титана (IV) (5,20 мл, 17,6 ммоль). Реакционную смесь оставляют перемешиваться в течение 18 ч при комнатной температуре. После охлаждения до 0°С добавляют боргидрид натрия (500 мг, 13,2 ммоль) и реакционную смесь перемешивают при 0°С до тех пор, пока газ не будет больше выделяться, и затем при комнатной температуре в течение 3 ч. Затем добавляют воду (25 мл) и оксид титана удаляют фильтрацией на воронке Бюхнера. Фильтрат экстрагируют EtOAc (3×100 мл) (добавляют рассол (10 мл) для облегчения разделения между обоими слоями) и объединенные органические слои концентрируют при пониженном давлении. Полученный неочищенный продукт (который содержит воду) растворяют в Et2O (75 мл), промывают рассолом, сушат над MgSO4 и фильтруют. К хорошо перемешиваемому фильтрату медленно добавляют 5-6 н. HCl в 2-пропаноле (2,2 мл). Полученный белый осадок фильтруют на воронке Бюхнера и сушат в вакууме с получением ожидаемого продукта 2-[4-(1-аминоэтил)-2-метилфенил]-2-метилпропаннитрила (1,58 г, 75%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CD3OD): δ 7.55 (1Н, d, J=8,0 Гц), 7.44-7.37 (2Н, m), 4.97 (3H, s), 4.52 (1Н, q, J=6.9 Гц), 2.75 (3H, s), 1.86 (6Н, s), 1.69 (3H, d, J=6.9 Гц).
Схема 17: Синтез 2-[4-(1-аминоэтил)-2-хлорфенил]-2-метилпропаннитрила
Стадия а) промежуточное соединение 56
(4-Бром-2-хлорфенил)метанол
К перемешиваемому раствору 4-бром-2-хлорбензойной кислоты (4,27 г, 18,1 ммоль) в тетрагидрофуране (39 мл) при 0°С добавляют боран-тетрагидрофурановый комплекс (1 M в THF) (36,3 мл, 36,3 ммоль). Реакционную смесь перемешивают в течение 16 ч при комнатной температуре. При 0°С медленно добавляют воду, затем также медленно добавляют водн. насыщ. NaHCO3. Полученный раствор экстрагируют EtOAc (3×50 мл). Объединенный органический слой промывают рассолом, сушат над безводным MgSO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью флэш-хроматографии (элюент: гексаны/EtOAc от 85:15 до 70:30) с получением ожидаемого продукта (4-бром-2-хлорфенил)метанола (4,34 г, 108%). 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.53 (1Н, d, J=1.8 Гц), 7.43 (1Н, dd, J=8.2, 1.8 Гц), 7.38 (1Н, d, J=8.2 Гц), 4.74 (2Н, d, J=6.2 Гц), 1,90 (1Н, t, J=6.3 Гц).
Стадия б) промежуточное соединение 57
4-Бром-1-(бромметил)-2-хлорбензол
К перемешиваемому раствору (4-бром-2-хлорфенил)метанола (4,34 г, 19,6 ммоль) в дихлорметане (98 мл) при 0°С добавляют тетрабромид углерода (6,5 г, 19,6 ммоль) и трифенилфосфин (5,14 г, 19,6 ммоль). Реакционную смесь перемешивают в течение 16 ч при комнатной температуре. Затем растворитель удаляют и неочищенное твердое вещество суспендируют в смеси гексаны/EtOAc, 9:1 (100 мл), и фильтруют через слой силикагеля. Слой промывают смесью гексаны/EtOAc, 9:1 (100 мл), и фильтрат концентрируют в вакууме с получением ожидаемого продукта 4-бром-1-(бромметил)-2-хлорбензола (7,19 г, 129%), загрязненного бромоформом. 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.57 (1Н, d, J=2.0 Гц), 7.39 (1Н, dd, J=8.2, 2.0 Гц), 7.30 (1Н, d, J=8.2 Гц), 4.53 (2Н, s).
Стадия в) промежуточное соединение 58
2-(4-Бром-2-хлорфенил)ацетонитрил
К перемешиваемому раствору 4-бром-1-(бромметил)-2-хлорбензола (7,19 г, 25,3 ммоль) в дихлорметане (60 мл) и воде (60 мл) добавляют бромид тетрабутиламмония (0,82 г, 2,53 ммоль). Затем добавляют цианид калия (4,94 г, 75,8 ммоль) в воде (60 мл). Полученный раствор перемешивают в течение 4 ч при комнатной температуре, и он быстро становится оранжевым. Затем добавляют насыщенный водн. раствор NaHCO3 и смесь экстрагируют CH2Cl2 (3×100 мл). Объединенный органический слой промывают рассолом, сушат над безводным MgSO4 и фильтруют через слой силикагеля. Слой промывают CH2Cl2 и фильтрат концентрируют при пониженном давлении с получением ожидаемого продукта 2-(4-бром-2-хлорфенил)ацетонитрила (5,38 г, 92%), загрязненного бромоформом. 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.60 (1Н, d, J=1.9 Гц), 7.47 (1Н, dd, J=8.3, 1.9 Гц), 7.39 (1Н, d, J=8.3 Гц), 3.79 (2Н, s).
Стадия г) промежуточное соединение 59
2-(4-Бром-2-хлорфенил)-2-метилпропаннитрил
К перемешиваемому раствору 2-(4-бром-2-хлорфенил)ацетонитрила (1,11 г, 4,82 ммоль) в безводном DMF (7,6 мл) добавляют метилйодид (0,63 мл, 10,1 ммоль). Раствор охлаждают до 0°С и добавляют гидрид натрия (60%-ная суспензия в масле, 0,63 г, 15,7 ммоль) 5 порциями в течение 1 ч. Реакционную смесь затем оставляют перемешиваться с медленным нагреванием до комнатной температуры в течение 18 ч. Раствор превращается в густую коричнево-оранжевую пасту. Затем медленно добавляют воду (20 мл) и раствор экстрагируют раствором гексан/Et2O, 3:1 (3×25 мл). Органический слой промывают рассолом, сушат над MgSO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью флэш-хроматографии (элюент: гексан/EtOAc от 9:1 до 8:2) с получением ожидаемого продукта 2-(4-бром-2-хлорфенил)-2-метилпропаннитрила (0,79 г, 63%). 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.61 (1Н, d, J=2.1 Гц), 7.43 (1Н, dd, J=8.5, 2.1 Гц), 7.34 (1Н, d, J=8.6 Гц), 1.85 (6Н, s).
Стадия д) промежуточное соединение 60
2-(4-Ацетил-2-хлорфенил)-2-метилпропаннитрил
К перемешиваемому раствору 2-(4-бром-2-хлорфенил)-2-метилпропаннитрила (2,85 г, 11,0 ммоль) в безводном THF (60 мл) при -78°С добавляют м-бутиллитий (2 M в с-гексане) (6,1 мл, 12,1 ммоль). Реакционную смесь перемешивают при этой температуре в течение 10 мин, затем добавляют неразбавленный N-метокси-N-метилацетамид (2,34 мл, 22,0 ммоль) и раствор перемешивают в течение 10 мин при -78°С и затем нагревают до комнатной температуры в течение еще одного часа. Затем медленно добавляют кислотный рассол (60 мл) и раствор экстрагируют EtOAc (3×60 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью флэш-хроматографии (элюент: гексан/EtOAc от 1:0 до 7:3) с получением ожидаемого продукта 2-(4-ацетил-2-хлорфенил)-2-метилпропаннитрила (1,57 г, 64%) в виде желтого масла. 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.96 (1Н, d, J=1.9 Гц), 7.81 (1Н, dd, J=8.3, 1.9 Гц), 7.55 (1Н, d, J=8.3 Гц), 2.56 (3H, s), 1.85 (6Н, s).
Стадия е), промежуточное соединение 61
2-(4-(1-Аминоэтил)-2-хлорфенил)-2-метилпропаннитрила гидрохлорид
К перемешиваемому раствору 2-(4-ацетил-2-хлорфенил)-2-метилпропаннитрила (1,57 г, 7,08 ммоль) в 7 М аммиаке в растворе метанола (35 мл) добавляют свежедистиллированный изопропилат титана (IV) (4,2 мл, 14,2 ммоль). Реакционную смесь оставляют перемешиваться в течение 18 ч при комнатной температуре. После охлаждения до 0°С добавляют боргидрид натрия (0,40 г, 10,6 ммоль) и реакционную смесь перемешивают при 0°С до тех пор, пока не прекратится выделение газа, и затем при комнатной температуре в течение 4 ч. Затем добавляют воду (40 мл) и оксид титана удаляют фильтрацией на воронке Бюхнера. Фильтрат экстрагируют EtOAc (3×50 мл) и объединенные органические слои концентрируют при пониженном давлении. Полученный остаток растворяют в Et2O (200 мл), промывают рассолом, сушат над безводным MgSO4 и фильтруют. К этому раствору затем добавляют 5 н. HCl в 2-пропаноле (1,8 мл, 8,85 ммоль). Полученный осадок фильтруют на воронке Бюхнера и сушат с получением желаемого 2-(4-(1-аминоэтил)-2-хлорфенил)-2-метилпропаннитрила гидрохлорида (1,17 г, 64%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CD3OD): δ 7.69 (1Н, d, J=10.7 Гц), 7.68 (1Н, s), 7.52 (1Н, dd, J=8.3, 2.0 Гц), 4.53 (1Н, q, J=6.8 Гц), 1.90 (6Н, s), 1.66 (3H, d, J=6.9 Гц). 13С-ЯМР (75 МГц, CD3OD): δ 142.5, 140.2, 136.0, 132.0, 130.1, 127.9, 52.0, 37.8, 28.4, 21.3.
Схема 18: Синтез 2-[4-(аминометил)фенил]-2-метилпропаннитрила
Стадия а) промежуточное соединение 62
4-(1-Циано-1-метилэтил)бензонитрил
2-Метил-2-(4-метилфенил)пропаннитрил (3,51 г, 15,7 ммоль) растворяют в сухом DMF (10 мл). Добавляют Zn(CN)2 (2,02 г, 17,2 ммоль) и Pd(PPh3)4 (906 мг, 5 мол.%) и смесь нагревают при 100°С в течение 3 часов. Реакционную смесь разбавляют водой и твердое вещество извлекают фильтрацией. Твердый остаток растворяют в метаноле, фильтруют для удаления нерастворимых примесей, концентрируют в вакууме и очищают с помощью MPLC с прямой фазой (SiO2, от гексана до EtOAc) с получением 4-(1-циано-1-метилэтил)бензонитрила (2,28 г, 13,4 ммоль, 85%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.70 (s, 6Н), 7.73 (dt, J=8.64, 2.03 Гц, 2Н), 7.92 (dt, J=8.69, 2.00 Гц, 2Н).
Стадия б) промежуточное соединение 63
2-[4-(Аминометил)фенил]-2-метилпропаннитрил
4-(1-Циано-1-метилэтил)бензонитрил (2,28 г, 13,41 ммоль) растворяют в сухом THF и раствор охлаждают до 0°С. Добавляют Red-Al (85%-ный раствор в толуоле, 2,62 мл, 13,41 ммоль гидрида) и реакционную смесь перемешивают при 0°С в течение 4 часов. Смесь гасят метанолом и концентрируют в вакууме. Остаток растворяют в CH2Cl2, промывают 2 порциями воды, сушат над сульфатом магния, фильтруют и упаривают досуха. Неочищенный продукт очищают с помощью MPLC с прямой фазой (SiO2, от DCM до DCM/MeOH, 3:1) с получением 2-[4-(аминометил)фенил]-2-метилпропаннитрила (900 мг, 5,17 ммоль, 39%) в виде бесцветного масла. 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.68 (s, 6Н), 4.00-4.05 (m, 2Н), 7.55 (q, J=8.59 Гц, 4Н), 8.35 (уширенный s, 2Н).
Схема 19: Синтез 2-[4-(аминометил)-3-метилфенил]-2-метилпропаннитрила
Стадия а) промежуточное соединение 64
2-(4-Формил-3-метилфенил)-2-метилпропаннитрил
2-(4-Бром-3-метилфенил)-2-метилпропаннитрил (3,31 г, 13,9 ммоль) растворяют в сухом THF (80 мл). Смесь охлаждают до -78°С и добавляют трет-BuLi (1,7 М в пентане, 18 мл, 30,6 ммоль). Через 30 минут реакционную смесь гасят DMF (10 мл), летучие компоненты выпаривают и остаток очищают с помощью MPLC с прямой фазой (SiO2, от гептана до гептан/EtOAc, 3:1) с получением 2-(4-формил-3-метилфенил)-2-метилпропаннитрила (1,89 г, 10,2 ммоль, 73%) в виде бесцветной жидкости. 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.70 (s, 6Н), 2.64 (s, 3H), 7.49 (s, 1Н), 7.55 (dd, J=8.01, 2.15 Гц, 1Н), 7.86 (d, J=7.81 Гц, 1Н), 10.22 (s, 1Н).
Стадия б) промежуточное соединение 65
2-[4-(Аминометил)-3-метилфенил]-2-метилпропаннитрил
2-(4-Формил-3-метилфенил)-2-метилпропаннитрил (1,89 г, 10,2 ммоль) растворяют в 50 мл 7 М NH3 в МеОН. Добавляют Ti(OiPr)4 (6 ммоль, 20 ммоль) и смесь перемешивают в течение 3 часов при комнатной температуре. Добавляют NaBH4 (0,6 г, 15 ммоль) и реакционную смесь оставляют при комнатной температуре в течение ночи. Смесь гасят путем добавления 25 мл 2 М NH4OH. Полученный осадок отфильтровывают и промывают EtOAc. Фазы разделяют, органическую фазу сушат над сульфатом магния, фильтруют и упаривают досуха с получением 2-[4-(аминометил)-3-метилфенил]-2-метилпропаннитрила (1,325 г, 7,09 ммоль, 87%). Неочищенный продукт используют без дополнительной очистки. 1Н-ЯМР (400 МГц, ДМСО-D6) δ 1.64 (s, 6Н, первый ротамер), 1.65 (s, 6Н, второй ротамер), 2.28 (s, 3H, первый ротамер), 2.29 (s, 3H, второй ротамер), 3.32 (broad s, 2Н), 3.67 (s, 2Н, первый ротамер), 3.68 (s, 2Н, второй ротамер), 7.24-7.27 (m, 1Н) 7.28 (s, 1Н), 7.36-7.40 (m, 1Н).
Схема 20: Отделение (S)-(-)-2-[4-(аминометил)-3-метилфенил]-2-метилпропаннитрила от (R)-(+)-2-[4-(аминометил)-3-метилфенил]-2-метилпропаннитрила
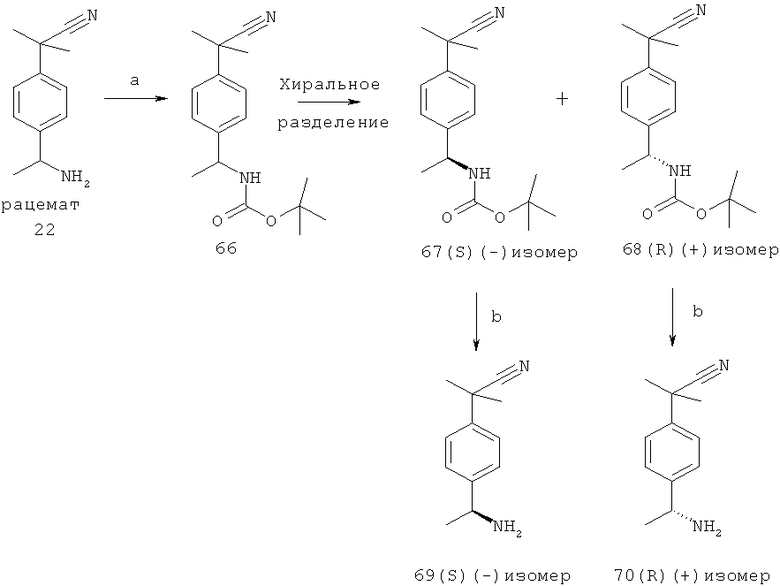
Стадия а) промежуточное соединение 66, 67 и 68.
трет-Бутил-N-[1-[4-(2-цианопропан-2-ил)фенил]этил]карбамат
К перемешиваемому раствору рацемического 2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила гидрохлорида (1 г, 3,97 ммоль) в THF (12 мл) добавляют Et3N (2,22 мл, 15,9 ммоль). Добавляют по каплям ди-трет-бутилдикарбонат (0,87 г, 3,97 ммоль) в THF (2 мл) и реакционную смесь перемешивают при комнатной температуре в течение 18 ч. Полученную смесь фильтруют через слой силикагеля и промывают раствором EtOAc и гексана, 1:1. Фильтрат концентрируют при пониженном давлении и неочищенный продукт оставляют стоять до тех пор, пока не затвердеет. Его очищают перекристаллизацией в 30 мл гексана с получением желаемого соединения трет-бутил-N-[1-[4-(2-цианопропан-2-ил)фенил]этил]-карбамата (0,87 г, 68%) в виде белого твердого вещеста.
1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.44-7.41 (2Н, m), 7.33-7.30 (2Н, m), 4.80 (2Н, s), 1.71 (6Н, s), 1.44 (3H, s) 1.42 (9Н, s).
Два энантиомера 67 и 68 разделяют на хиральной колонке CHIRALPAK ® AY ® с использованием смеси элюента гексан:IРА (изопропанол) (9:1) при 25°С с УФ-детектором, установленным на 220 нм. Два энантиомера имели значения измерений оптического вращения [α]D=-76,2° (1,36, МеОН) для изомера 67 и [α]D=+78,7° (с=1,20, МеОН) для изомера 68.
Стадия б) промежуточное соединение 69 (такой же способ осуществляют для получения 70 из 68).
S(-)2-[4-(1-Аминоэтил)фенил]-2-метилпропаннитрил
4 М хлористый водород (34,7 мл, 138,70 ммоль) в диоксане добавляют к раствору (-)трет-бутил-N-[1-[4-(2-цианопропан-2-ил)фенил]этил]карбамата 67 (8,00 г, 27,74 ммоль) в тетрагидрофуране (45 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 2 часов и растворитель концентрируют досуха. Остаток суспендируют в EtOAc (45 мл) и перемешивают в течение 1 часа. Твердое вещество собирают и сушат на воздухе с получением HCl соли (S)-2-(4-(1-аминоэтил)фенил)-2-метилпропаннитрила (6,08 г, 97%) в виде белого твердого вещества. Вещество суспендируют в МТВЕ (80 мл) и добавляют 2 М NaOH (40 мл). Смесь осторожно перемешивают в течение 1 часа и отделяют органическую фазу. Водную фазу экстрагируют МТВЕ (40 мл). Объединенные органические слои сушат над безводным MgSO4 и растворитель концентрируют с получением ожидаемого продукта S(-)-2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила (4,88 г, 93%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 1.39 (d, J=6.64 Гц, 3H), 1.53-1.63 (m, 2Н), 1.68-1.76 (m, 6Н), 4.14 (q, J=6.64 Гц, 1Н), 7.34-7.40 (m, 2Н), 7.41-7.46 (m, 2Н). MS (ESI, ионизиция электрораспылением) (М+Н)+ 188,9. [α]D=-21,0° (1.44, EtOH).
Для другого изомера 70, полученного описанным выше способом, [α]D=+19,0° (с=0,39, EtOH)
Подтверждение абсолютной конфигурации промежуточного соединения 69 и 70 с использованием Колебательного Кругового Дихроизма (VCD).
Краткое описание.
Этот метод предполагает вычисление VCD-спектров чистых энантиомеров, для которых должна быть определена абсолютная конфигурация. Эти вычисленные спектры затем сравнивают с экспериментальными VCD-спектрами, полученными из хиральных веществ. Сравнивая специфические спектральные характеристики, составляют подтверждение абсолютной конфигурации энантиомеров.
Компьютерные спектральные модели.
Поиск методом молекулярной механики Монте-Карло низкоэнергетических конформеров для 69 проводили с использованием MacroModel в графическом интерфейсе Maestro (Schrödinger Inc.). 12 идентифицированных конформеров с наименьшей энергией используют в качестве исходных точек и минимизируют с использованием функциональной теории плотности (DFT) в Gaussian03.
Для каждого из конформеров определяли оптимизированные структуры, частоты/интенсивности гармонических колебаний, вращательные силы VCD и свободные энергии при STP (стандартные температура и давление) (включая энергию нулевых колебаний). В этих расчетах использовали обобщенное градиентное приближение (GGA) B3LYP обменно-корреляционных функционалов плотности. В частности, GGA представляет собой комбинацию обменного функционала Бекке (3-параметрический HF/DFT гибридный обменный функционал [В3]) {Becke, A.D.J. Chem. Phys. 93, 98, 5648} с динамическим корреляционным функционалом Ли-Янга-Парра (LYP) {Lee, С; Yang, W.; Parr, R.G. Phys. Rev. В 1988, 37, 785}. Использовали базисный набор 6-31G* {Hariharan, P.C.; Pople, J.A. Theor. Chim. Acta, 1973, 28, 213}.
Инфракрасные и VCD-спектры для каждого конформера получают из исходящих файлов Gaussian 03 с использованием одной из множества программ для подгонки лоренцевых форм спектральных линий (ширина линии 5 см-1) к вычисленным спектрам. Таким образом, могут быть сделаны прямые сравнения между смоделированными и экспериментальными спектрами.
Эксперимент
30 мг 69 и 70, соответственно, растворяли в 0,28 мл d6-ДМСО. Растворы отдельно загружали в 0,1 мм BaF2 инфракрасную ячейку для анализа 4 см-1 разрешения с использованием 5-часового двухисточникового протокола сканирования VCD. Все анализы проводили с использованием прибора ChiralR™ Bio Tools, Inc. Прибор включал один набор фотоупругих модуляторов для поляризационной модуляции при 37,024 кГц с λ/4 запаздыванием (оптимизировано для получения спектральной области с центром около 1400 см-1). Использовали усиление синхронизации с постоянной времени 30 мкс и фильтром верхних частот 20 кГц и фильтром низких частот 4 кГц.
Результаты
Сравнение инфракрасных спектров колебательного кругового дихроизма (VCD) с предсказанными VCD-спектрами (полученными в результате расчетов методом молекулярной механики и функциональной теории плотности) показывает, что структура согласуется с предложенной S-конфигурацией для промежуточного соединения 69 и R-конфигурацией для промежуточного соединения 70.
Схема 21: Энантиоселективный синтез S(-)2-[4-(аминометил)-3-метилфенил]-2-метилпропаннитрила

Стадия а) промежуточное соединение 71
N-{(S,S)-1-[4-(1-Циано-1-метилэтил)фенил]этил}-2-метилпропан-2-сульфинамид.
В 500 мл 3-горлую круглодонную колбу, снабженную термометром, впускным отверстием для азота, резиновой прокладкой и магнитной мешалкой, загружают 8,13 г (36,93 ммоль, предположительно содержащего 85 мас.% только кетона) арилметилкетона и (S)-(-)-трет-бутилсульфинамид (4,47 г, 1 экв.). Установку продувают азотом. Добавляют безводный THF (82 мл, 10 мас. частей неочищенной массы) и полученный раствор умеренно перемешивают в атмосфере азота. К этому раствору через шприц добавляют Ti(OEt)4 (15,32 мл, 2 экв.). Полученный темно-желтый раствор нагревают в атмосфере азота до слабой дефлегмации при умеренном перемешивании и выдерживают в течение примерно 24 часов (наблюдается небольшое вспенивание реакционной смеси). Анализ с помощью 1Н-ЯМР показывает, что реакция завершилась.
Сначала реакционную смесь охлаждают до температуры окружающей среды и затем до 0-5°С путем погружения в водяную баню со льдом. К этой смеси добавляют боргидрид натрия (2,1 г, 1,5 экв.) (внимание: наблюдается умеренное вспенивание и экзотермичность от 3 до 10°С). Полученную реакционную смесь перемешивают при 0-5°С в течение еще 5 часов и затем оставляют нагреваться до температуры окружающей среды и перемешивают в течение ночи (примерно 14 часов). Отбирают образец для IPC 1Н-ЯМР, который показывает, что весь кетимин израсходован.
Смесь охлаждают до 0-5°С.Постепенно добавляют ацетон (16,3 мл, примерно 6 экв.) в течение 15-20 минут для расходования оставшегося гидрида (внимание: наблюдается экзотермичность от примерно 3 до 15°С). После того как добавление завершено, реакционную смесь перемешивают в течение 15 минут. Загружают МеОН (2 мл) для проверки присутствия остаточного гидрида - выделение водорода не наблюдается. Смесь оставляют нагреваться до комнатной температуры и переносят в 500-мл колбу Эрленмейера. Круглодонную колбу промывают 160 мл МТВЕ. Полученную смесь энергично перемешивают в течение 5 минут. По частям загружают рассол (10 мл) в течение 2-3 минут для индукции осаждения титанового реагента. Образовавшуюся суспензию энергично перемешивают в течение 30 минут и затем фильтруют через толстый слой песка. Слой песка промывают 3×30 мл МТВЕ. Объединенный фильтрат перемешивают с 150 мл рассола в течение следующего 1 часа с получением мутной двухфазной смеси, которую переносят в разделительную воронку. Нижний водный слой удаляют и верхний органический слой фильтруют через слой целита. Фильтрат упаривают досуха с получением ожидаемого продукта N-{(S,S)-1-[4-(1-циано-1-метилэтил)фенил]этил}-2-метилпропан-2-сульфинамида (11,1 г) в виде желтого сиропа. Предполагается, что это вещество является 100% чистым и его используют непосредственно на следующей стадии без дополнительной очистки.
Как сообщалось Эллманом (Ellman et al. Tetrahedron, 40 (1999) 6709-6712), неочищенный продукт состоит из смеси 2 диастереомеров с преобладанием одного. Соотношение этих двух изомеров означает степень асимметрии, вызванной хиральной сульфинильной группой. Обычно соотношение, наблюдающееся для этого уменьшения, варьируется от 92:8 до 95:5, как показано HPLC и 1Н-ЯМР анализами неочищенной смеси продуктов.
1Н-ЯМР 400 МГц (ХЛОРОФОРМ-D) δ 1.26 (s, 9Н), 1.53 (d, 3H, J=8 Гц), 1.75 (s, 6Н), 4.52-4.62 (m, 1Н), 7.40 (d, 2Н, J=8 Гц), 7.48 (d, 2Н, J=8 Гц).
Стадия б) промежуточное соединение 72
В 250-мл круглодонную колбу, снабженную магнитной мешалкой, содержащую замещенный сульфинамид (11,1 г, 1 экв., предполагается, что является 100% чистым), полученный на предыдущей стадии, добавляют диоксан (28,3 мл). Полученный раствор охлаждают до 0-5°С в водяной бане со льдом, энергично перемешивая. В этот раствор через шприц загружают HCl в диоксане (28,3 мл, 3 экв.) одной порцией. Белое твердое вещество начинает немедленно осаждаться из раствора. Полученную суспензию перемешивают при 0-5°С (температура бани) в течение следующих 10 минут с получением густой белой суспензии. На этой стадии охлаждающую баню удаляют. Небольшой образец реакционной смеси отбирают и анализируют с помощью тонкослойной хроматографии. Весь исходный сульфинамид израсходован.
Смесь разбавляют EtOAc (28,3 мл) и перемешивают в течение следующих 5 минут при комнатной температуре. Белое твердое вещество затем собирают вакуумной фильтрацией. Колбу промывают 2×10 мл EtOAc. Осадок на фильтре промывают еще 2×10 мл EtOAc и затем сушат в токе азота. Осадок на фильтре затем сушат при 40-45°С в вакуумной печи в течение ночи с получением 4,28 г кристаллического белого твердого вещества. Анализ этого вещества с помощью 1Н-ЯМР спектроскопии показал присутствие только желаемого амина гидрохлорида.
Согласно наблюдению авторов изобретения оптическая чистота этого хирального амина, которая непосредственно коррелирует с соотношением двух диастереомерных сульфинамидных предшественников, варьируется от 84 до 90% е.е. (энантиомерный избыток). Эти количества подтверждают, что конечная стадия HCl обработки не влияет на оптическую чистоту амина.
1Н-ЯМР 400 МГц (ХЛОРОФОРМ-D) δ 1.20 (d, 3H, J=8 Гц), 1.70 (s, 3H), 1.71 (s, 3H), 3.73 (АВ q, 1Н, J=14,6 Гц), 4.50 (br. s, 1Н), 7.17 (d, 2Н, J=8 Гц), 7.21-7.31 (m, 5Н), 7.41 (d, 2Н, J=8 Гц).
HCl соль амина (3,12 г, 13,89 ммоль, 1 экв.) растворяют в 26 мл воды в 100-мл круглодонной колбе с мягким перемешиванием магнитной мешалкой. В этот раствор добавляют 2 М водный гидроксид натрия (7,6 мл, 1,1 экв.) для доведения его результирующего рН до 13-14, что приводит к образованию легкой эмульсии. В эту эмульсию загружают МТВЕ (26 мл) и полученную смесь энергично перемешивают. Верхний органический слой собирают и водный слой разбавляют рассолом (15 мл), затем экстрагируют еще 26 мл МТВЕ. Органические слои объединяют, промывают рассолом (15 мл) и затем сушат над безводным сульфатом натрия. В результате удаления высушивающего агента фильтрацией и выпаривания фильтрата досуха получили амин в виде бледно-желтого легкого масла.
Масло, полученное выше, растворяют в ацетоне (26 мл) и перемешивают в атмосфере азота. В амин загружают раствор (+)-миндальной кислоты (2 г, 0,95 экв. относительно используемой HCl соли амина) в ацетоне (15 мл), полученный в отдельном сосуде. Данный сосуд затем промывают ацетоном (5 мл). Осаждение кристаллического твердого вещества (соли) происходило быстро с получением тяжелой белой суспензии. Эту суспензию перемешивают в течение 15 минут и затем охлаждают до 0-5°С и перемешивают в течение еще 30 минут. Белое твердое вещество собирают вакуумной фильтрацией и промывают ацетоном (2×10 мл). Осадок на фильтре сушат в вакуумной печи при 40-45°С в течение ночи (примерно 16 часов) с получением 3,67 г (77%) соли (+)-миндальной кислоты в виде кристаллического белого твердого вещества.
1Н-ЯМР спектроскопический анализ выделенного выше продукта в дейтерированном хлороформе показал, что присутствует только один энантиомер амина.
В 2-л круглодонную колбу, снабженную подвесной мешалкой, загружают соль миндальной кислоты (50,8 г, 149,3 ммоль, 1 экв.) и МТВЕ (1 л, 6,5 частей) с получением суспензии. К этой суспензии при умеренном перемешивании добавляют раствор NaOH (1 М, 672 мл, 672 ммоль, 4,5 экв.). Полученную двухфазную смесь энергично перемешивают при комнатной температуре в течение 30 минут и затем переносят в разделительную воронку. Слои разделяют. Водный слой разбавляют 500 мл рассола и экстрагируют МТВЕ (2×250 мл). Объединенные органические фазы сушат над MgSO4. В результате удаления высушивающего агента с последующим выпариваем фильтрата досуха получили свободный амин в виде светло-желтого масла (27,7 г, 99%).
1Н-ЯМР 400 МГц (ХЛОРОФОРМ-D) δ 1.20 (d, 3H, J=8 Гц), 1.70 (s, 3H), 1.71 (s, 3H), 3.73 (АВ квартет, 1Н, J=14, 6 Гц), 4.50 (br. s, 1Н), 7.17 (d, 2Н, J=8 Гц), 7.21-7.31 (m, 5Н), 7.41 (d, 2Н, J=8 Гц).
Схема 22: Энантиоселективный синтез (-)1-[4-[1-аминоэтил]фенил]-циклобутан-1-карбонитрила

Стадия а) промежуточное соединение 73
1-(4-Бромфенил)циклобутанкарбонитрил
К перемешиваемой суспензии гидрида натрия (60%-ная суспензия в масле, 12,24 г, 255 ммоль) в безводном ДМСО (240 мл) добавляют по каплям 2-(4-бромфенил)ацетонитрил (20,0 г, 102 ммоль), растворенный в безводном ДМСО (40 мл). Через 45 мин реакционную смесь охлаждают до 0°С и медленно добавляют 1,3-дибромпропан (30,9 г, 15,5 мл, 153 ммоль), растворенный в безводном ДМСО (40 мл), для поддержания температуры ниже 45°С. Реакционную смесь перемешивают в течение ночи при комнатной температуре и вливают в холодную воду (1,2 л). Продукт экстрагируют CH2Cl2 (6×100 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении. Продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан: EtOAc (от 100% до 95:5), с получением ожидаемого продукта 1-(4-бромфенил)циклобутанкарбонитрила (9,9 г, 41%) в виде желтоватого масла. 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D): δ 7.57-7.49 (2Н, m), 7.33-7.24 (2Н, m), 2.89-2.77 (2Н, m), 2.66-2.34 (3H, m), 2.15-1.99 (1Н, m).
Стадия б) промежуточное соединение 74
1-(4-Ацетилфенил)циклобутанкарбонитрил
К перемешиваемому раствору 1-(4-бромфенил)циклобутанкарбонитрила (2,8 г, 11,9 ммоль) в безводном THF (70 мл) при -78°С добавляют н-бутиллитий (2 M в с-гексане) (7,44 мл, 14,9 ммоль). Реакционную смесь перемешивают при этой температуре в течение 10 мин, затем добавляют N-метокси-N-метилацетамид (2,50 мл, 23,8 ммоль) и раствор затем оставляют нагреваться до комнатной температуры в течение 1 ч. Затем медленно добавляют смесь рассола (30 мл) и 1 н. HCl (15 мл) и раствор экстрагируют EtOAc (3×100 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью флэш-хроматографии с использованием градиента от 0 до 20% EtOAc в гексане с получением 1-(4-ацетилфенил)циклобутанкарбонитрила (1,35 г, 57%) в виде желтоватого масла. 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-D) δ 8.03- (2Н, m), 7.56-7.50 (2Н, m), 2.94-2.81 (2Н, m), 2.71-2.57 (2Н, m), 2.62 (3H, s), 2.57-2.40 (1Н, m), 2.18-2.05 (1Н, m).
Стадия в) промежуточное соединение 75
(-)-1-[4-[1-Аминоэтил]фенил]циклобутан-1-карбонитрил
В 1-(4-ацетилфенил)циклобутанкарбонитрил (8,6 г, 43,2 ммоль) и безводный THF (85 мл) с (S)-(-)-трет-бутилсульфинамидом (5,7 г, 1,1 экв.) добавляют тетраэтилат титана (18 мл, 2 экв.). Полученную смесь нагревают до температуры дефлегмации в атмосфере азота и выдерживают при температуре дефлегмации в течение 18 часов. Аликвоту отбирают и анализируют в отношении завершения реакции.
Смесь охлаждают до комнатной температуры и затем до 0-5°С. В реакционную смесь небольшими порциями в течение 10 минут добавляют боргидрид натрия (2,4 г, 1,5 эквивалента). Полученную смесь оставляют нагреваться до комнатной температуры в течение 1 часа 3 часов.
Реакционную смесь гасят путем добавления ацетона (19 мл, 6 экв.) и полученную смесь разбавляют рассолом (25 мл). При умеренном перемешивании добавляют МТВЕ (80 мл) и перемешивание продолжают в течение примерно 15 минут. Полученную двухфазную смесь фильтруют через керамическую воронку, упакованную целитом. Органический слой собирают и сушат над безводным сульфатом магния. В результате упаривания этого органического раствора досуха при пониженном давлении получили желаемый замещенный хиральный сульфинамид в виде вязкого сиропа (13,3 г). Это неочищенное вещество используют без дополнительной очистки.
Неочищенный хиральный сульфинамид (13,3 г), полученный выше, растворяют в диоксане (130 мл) с умеренным перемешиванием в круглодонной колбе. В полученную смесь загружают 4 М HCl в диоксане (32,8 мл, 3 экв.) и перемешивание продолжают при комнатной температуре в течение ночи (примерно 16 часов). Реакционную смесь упаривают досуха на роторном испарителе и полученный остаток растирают с EtOAc (100 мл) в течение 1 часа. Белое твердое вещество фильтруют и промывают. В результате сушки осадка на фильтре в вакуумной печи в течение ночи получили ожидаемый хиральный амин (-)-1-[4-[1-аминоэтил]фенил]циклобутан-1-карбонитрил (5,0 г) в его гидрохлоридной форме в виде белого твердого вещества. Данные 1Н-ЯМР (400 МГц, ДМСО-d6) δ 1.52 (3H, d, J=8 Гц), 1.94-2.07 (1Н, m), 2.20-2.35 (1Н, m), 2.55-2.67 (2Н, m), 2.70-2.80 (2Н, m), 4.44 (1Н, q, J=6.8 Гц), 7.52 (2Н, J=8.4 Гц), 7.61 (2Н, d, J=8.4 Гц), 8.40-8.90 (3H, m).
Перед реакцией сочетания для получения конечных хиральных примеров получают свободное основание промежуточного соединения 75 с использованием способа, изложенного на стадии b, описанной при получении промежуточного соединения 72.
Схема 23: Энантиоселективный синтез (-)2-[4-[1-аминоэтил]-3-метилфенил]-2-метилпропаннитрила

К 2-(4-ацетил-3-метилфенил)-2-метилпропаннитрилу (1,47 г, приблизительный анализ 80% => 5,8 ммоль) и безводному THF (12 мл, 10 частей) с (S)-(-)-трет-бутилсульфинамидом (0,7 г, 1 экв.) добавляют тетраэтилат титана (2,4 мл, 2 экв.). Полученную смесь нагревают до температуры дефлегмации в атмосфере азота и выдерживают в течение 22 часов (в течение ночи) с последующим перемешиванием при комнатной температуре в течение выходных. Анализ реакционной смеси показал, что реакция завершилась.
При умеренном перемешивании реакционную смесь охлаждают до 0-5°С (водяная баня со льдом). В реакционную смесь небольшими порциями в течение 10 минут добавляют боргидрид натрия (0,35 г, 1,5 эквивалента). Полученную смесь перемешивают в течение 3 часов при 0-5°С и затем позволяют ей медленно нагреваться до комнатной температуры. Перемешивание при комнатной температуре продолжают в течение еще 1 часа.
После охлаждения смеси до 0-5°С реакционную смесь гасят путем добавления ацетона (2 мл, 6 экв.) и полученную смесь разбавляют рассолом (4 мл). При умеренном перемешивании добавляют МТВЕ (20 мл) и перемешивание продолжают в течение 10 минут. Эту водно-органическую смесь фильтруют через слой целита. Органическую фазу собирают и сушат над безводным сульфатом магния. В результате упаривания этого органического раствора досуха при пониженном давлении получили желаемый замещенный хиральный сульфинамид в виде желтого вязкого сиропа (1,88 г). Спектр 1Н-ЯМР этого вещества показывает присутствие желаемого замещенного сульфинамида в виде основного продукта вместе с другими примесями. Это полученное неочищенное вещество, как полагают, является 100% чистым и его используют без дополнительной очистки.
Полученный выше неочищенный хиральный сульфинамид (0,28 г, 0,4 ммоль) растворяют в диоксане (3 мл) с умеренным перемешиванием в круглодонной колбе. В образовавшийся раствор загружают 4 М HCl в диоксане (0,7 мл, 3 экв.) и полученную смесь перемешивают при комнатной температуре в течение 30 минут с получением тяжелой белой суспензии. Эту суспензию упаривают досуха на роторном испарителе и полученный остаток растирают с EtOAc (10 мл) в течение 10 минут. Белое твердое вещество собирают фильтрацией и затем промывают EtOAc. В результате естественной сушки осадка на фильтре при комнатной температуре получают гидрохлорид желаемого хирального амина (-)2-[4-[1-аминоэтил]-3-метилфенил]-2-метилпропаннитрила в виде белого порошка (95 мг).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 1.48 (3H, d, J=8 Гц), 1.68 (6Н, s), 2.39 (3H, s), 4.53 (1Н, ABq, J=16,8 Гц), 7.39 (1Н, d, J=2 Гц), 7.44 (1Н, dd, J=8,2 Гц), 7,64 (1Н, d, J=8 Гц), 6.8-9 (неразрешенный m, обмен на D2O).
Схема 24: Синтез 2-[4-(1-аминоэтил)фенил]-2-этилбутаннитрила

Стадия а) промежуточное соединение 77
4-(1-Циано-1-этилпропил)бензонитрил.
4-Цианометилбензонитрил (1,02 г, 7,18 ммоль) и этилйодид (2,25 г, 14,4 ммоль) добавляют к DMF (20,0 мл) и полученный раствор охлаждают до 0°С. Порциями добавляют NaH (576 мг, 14,4 ммоль). Смесь нагревают до комнатной температуры и затем перемешивают в течение 3 часов. Добавляют воду (100 мл) и водную фазу экстрагируют EtOAc (4 раза по 50,0 мл). Объединенные органические фазы сушат с MgSO4, фильтруют и концентрируют на роторном испарителе. Продукт очищают с помощью Combi-Flash хроматографии на силикагеле, элюируя смесями гептана и EtOAc (от 100/0 до 70/30), (936 мг, 5,51 ммоль, 77%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 0.77 (t, J=7.42 Гц, 6Н), 1.89-2.14 (m, 4Н), 7.64 (d, J=8.40 Гц, 2Н), 7.92 (d, J=8.59 Гц, 2Н).
Стадия б) промежуточное соединение 78
2-[4-(1-Аминоэтил)фенил]-2-этилбутаннитрил.
4-(1-Циано-1-этилпропил)бензонитрил (936 мг, 5,51 ммоль) добавляют к THF (20,0 мл) и полученный раствор охлаждают до -78°С. Добавляют MeLi (3,44 мл, 5,51 ммоль) и раствор перемешивают в течение 20 минут. NaBH4 (208 мг, 5,51 ммоль) смешивают с МеОН (20,0 мл) и добавляют к первому раствору. Полученный раствор нагревают до комнатной температуры и перемешивают в течение 1 часа. Добавляют 1 н. HCl (50,0 мл), затем 1 н. NaOH (60,0 мл). Водную фазу экстрагируют EtOAc (4 раза по 50,0 мл) и объединенные органические фазы сушат с MgSO4, фильтруют и концентрируют на роторном испарителе. Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (10 мМ NH4CO3), В = MeCN (528 мг, 2,44 ммоль, 44%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 0.77 (t, J=7.32 Гц, 6Н), 1.22 (d, J=6.45 Гц, 3H), 1.82-2.07 (m, 4Н), 3.97 (q, J=6.64 Гц, 1Н), 7.32 (d, J=8.40 Гц, 2Н), 7.40 (d, J=8.20 Гц, 2Н).
Пример 1:

(R)(+) и (S)(-)-2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид.
2-[4-(1-Аминоэтил)фенил]-2-метилпропаннитрила гидрохлорид, полученный согласно схеме 6 (557 мг, 2,49 ммоль), (7-циано-1Н-бензимидазол-1-ил)уксусную кислоту (500 мг, 2,49 ммоль), полученную согласно схеме 3, и DMAP (601 мг, 4,97 ммоль) смешивают в DMF (10,0 мл). Добавляют HATU (945 мг, 2,49 ммоль) и смесь перемешивают в течение 18 часов. Реакционную смесь фильтруют и затем очищают на HPLCMS: Waters prep LCMS, скорость потока: 27 мл/мин, колонка: SynerSi (4 мкм) Polar RP, 21,2×50 мм, подвижная фаза: А = вода (0,05% TFA) В = MeCN, используемый градиент от 40% до 70% В в А, в течение 10 мин. Фракции объединяют, добавляют 1 н. NaOH и желаемое соединение экстрагируют этилацетатом. Объединенные органические фракции концентрируют при пониженном давлении с получением продукта (610 мг, 1,64 ммоль, 66%). 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.50 (d, J=7.03 Гц, 3H), 1.68 (s, 6Н) 4.98-5.09 (m, 1Н), 5.33 (s, 2Н) 7.37-7.43 (m, 3H), 7.46 (d, J=8.20 Гц, 2Н), 7.68 (d, J=7.62 Гц, 1Н), 7.97 (dd, J=8.20, 0.98 Гц, 1Н), 8.28 (s, 1Н), 8.89 (d, J=7.62 Гц, 1Н). MS [М+Н], вычислено: 372,2, найдено: 372,3.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson, Inc., скорость потока: 18 мл/мин, колонка®: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика [α]D=-156 (с=8,2, МеОН) и [α]D=+136 (с=13,4, МеОН).
Пример 2:

(R)(+) и (S)(-)-N-{1-[4-(1-Циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид.
N-{1-[4-(1-Циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид синтезируют согласно примеру 1 путем смешивания (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислоты (500 мг, 2,36 ммоль), полученной согласно схеме 2, 2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила гидрохлорида (525 мг, 2,36 ммоль), полученного согласно схеме 6, DMAP (570 мг, 4,71 ммоль) и HATU (896 мг, 2,36 ммоль) в DMF (10,0 мл). Продукт очищают на HPLCMS: Waters prep LCMS, 27 мл/мин, колонка: SynerSi (4 мкм) Polar RP, 21,2×50 мм, подвижная фаза: А = вода (0,05% TFA) В = MeCN, используемый градиент от 40% до 60% В в А, в течение 10 мин. Фракции объединяют, добавляют 1 н. NaOH, и желаемое соединение экстрагируют этилацетатом. Объединенные органические фракции концентрируют при пониженном давлении с получением продукта (450 мг, 2,36 ммоль, 50%). 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.49 (d, J=7.03 Гц, 3H), 1.68 (s, 6Н), 5.03 (m, 1Н), 5.20 (m, 2Н), 7.30 (dt, J=11.23, 8.98, 7.32 Гц, 1Н), 7.35-7.42 (m, 2Н), 7.44-7.54 (m, 3H), 8.56 (s, 1Н). MS [М+Н], вычислено: 383,2, найдено: 383,3.
HRMS(ESI+) вычислено для C21H21F2N4O 383,16779 [М+Н]+, найдено 383,16782.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson, Inc., скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = i-PrOH (0,1% DEA), изократика 80% А, 30% В. [α]D=-160 (с=5,4, МеОН) и [α]D=+164 (с=6,0, МеОН).
Пример 3
(R)(+) и (S)(-)-N-{1-[4-(1-Циано-1-метилэтил)фенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамид.
N-{1-[4-(1-Циано-1-метилэтил)фенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамид синтезируют согласно примеру 1 путем смешивания (7-фтор-1Н-бензимидазол-1-ил)уксусной кислоты (200 мг, 1,03 ммоль), полученной согласно схеме 5, 2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила гидрохлорида (231 мг, 1,03 ммоль), полученного согласно схеме 6, DMAP (249 мг, 2,06 ммоль) и HATU (391 мг, 1,03 ммоль) в DMF (4,0 мл). Продукт очищают на HPLCMS: Waters prep LCMS, 27 мл/мин, колонка: SynerSi (4 мкм) Polar RP, 21,2×50 мм, подвижная фаза: А = вода (0,05% TFA) В = MeCN, используемый градиент от 30% до 50% В в А, в течение 10 мин. Фракции объединяют, добавляют 1 н. NaOH, и желаемое соединение экстрагируют этилацетатом. Объединенные органические фракции концентрируют при пониженном давлении с получением продукта (226 мг, 0,621 ммоль, 60%). 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.48 (d, J=7.03 Гц, 3H), 1.69 (s, 6Н), 5.03 (q, J=6.90 Гц, 4Н), 5.09 (d, J=16.99 Гц, 1Н), 5.14 (d, J=16.99 Гц, 1Н), 7.00 (dd, J=11.43, 8.11 Гц, 1Н), 7.20 (td, J=8.11, 4.88 Гц, 1Н), 7.37 (d, J=8.20 Гц, 2Н), 7.43-7.50 (m, 3H), 8.10 (s, 1Н); MS [М+Н], вычислено: 365,2, найдено: 365,3.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, Inc., скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 4:
(R) (+) и (S)(-)-N-{1-[4-(1-Циано-1-метилэтил)фенил]этил}-2-(7-хлор-1H-бензимидазол-1-ил)ацетамид.
N-{1-[4-(1-Циано-1-метилэтил)фенил]этил}-2-(7-хлор-1H-бензимидазол-1-ил)ацетамид синтезируют согласно примеру 1 путем смешивания (7-хлор-1H-бензимидазол-1-ил)уксусной кислоты (100 мг, 0,48 ммоль), полученной согласно схеме 1, 2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила гидрохлорида (107 мг, 0,48 ммоль), полученного согласно схеме 6, DMAP (115 мг, 0,95 ммоль) и HATU (181 мг, 0,48 ммоль) в DMF (2,0 мл). Продукт очищают на HPLCMS: Waters prep LCMS, 27 мл/мин, колонка: SynerSi (4 мкм) Polar RP, 21,2×50 мм, подвижная фаза: А = вода (0,05% TFA) В = MeCN, используемый градиент от 40% до 70% В в А, в течение 10 мин. Фракции объединяют и сушат сублимацией с получением желаемого продукта в виде соли TFA (50 мг, 0,100 ммоль, 21%). 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.48 (d, J=7.03 Гц, 3H), 1.69 (s, 6Н) 5.03 (q, J=6.71 Гц, 1Н), 5.21-5.34 (m, 2Н) 7.16-7.28 (m, 2Н), 7.38 (d, 2Н) 7.46 (d, 2Н), 7.59 (dd, J=7.52, 1.27 Гц, 1Н), 8.13 (s, 1Н); MS [М+Н], вычислено: 381,1, найдено: 381,3.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, Inc., скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 5:
(+) и (-)-N-{1-[4-(1-Циано-1-этилпропил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид.
2-[4-(1-Аминоэтил)фенил]-2-этилбутаннитрил (520 мг, 2,41 ммоль), полученный согласно схеме 24, (6,7-дифтор-1Н-бензимидазол-1-ил)уксусную кислоту (510 мг, 2,41 ммоль), полученную согласно схеме 2, и Et3N (731 мг, 7,22 ммоль, 1,00 мл) смешивают в MeCN (15,0 мл). Добавляют HATU (915 мг, 2,41 ммоль) и смесь перемешивают в течение 2 часов. Добавляют 1 н. NaOH (40,0 мл) и водную фазу экстрагируют EtOAc (4 раза по 40,0 мл). Объединенные органические фазы сушат с MgSO4, фильтруют и концентрируют на роторном испарителе. Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (10 мМ NH4CO3) В = MeCN, (804 мг, 1,96 ммоль, 81%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 0.77 (t, J=7.32 Гц, 6Н), 1.40 (d, J=7.03 Гц, 3H), 1.83-2.13 (m, 4Н)4.96 (ddd, J=14.65, 7.23, 7.03 Гц, 1Н) 5.08 (s, 2Н), 7.21 (ddd, J=11.57, 8.93, 7.62 Гц, 1Н), 7.35-7.41 (m, 4Н), 7.45 (ddd, J=8.84, 3.86, 0.78 Гц, 1Н), 8.19-8.22 (m, 1Н), 8.83 (d, J=8.01 Гц, 1Н); MS [М+Н], вычислено: 411,0, найдено: 411,3.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel AD 21×250 мм (20 мкм), подвижная фаза: А = гексан (0,1% DEA) В = i-PrOH (0,1% DEA), αD=-149 (с=3,2, МеОН) и αD=+178 (с=3,4, МеОН).
Пример 6
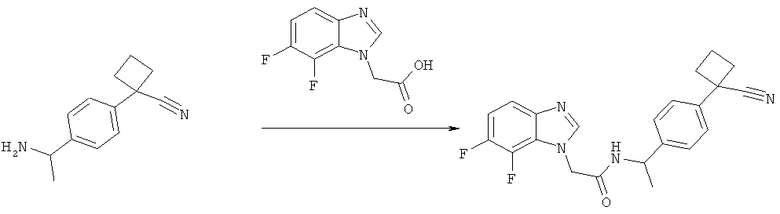
(+) и (-)-N-{1-[4-(1-Цианоциклобутил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид.
N-{1-[4-(1-Цианоциклобутил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид синтезируют согласно примеру 5 путем смешивания (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислоты (212 мг, 1,00 ммоль), полученной согласно схеме 2, 1-[4-(1-аминоэтил)фенил]циклобутанкарбонитрила (200 мг, 1,00 ммоль), полученного согласно схеме 9, Et3N (304 мг, 3,00 ммоль, 0,418 мл) и HATU (280 мг, 1,00 ммоль) в MeCN (10,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (10 мМ NH4CO3) В = MeCN, (220 мг, 0,558 ммоль, 56%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.39 (d, J=7.03 Гц, 3H), 1.91-2.05 (m, 1Н), 2.16-2.34 (m, 1Н), 2.53-2.64 (m, 2Н), 2.66-2.78 (m, 2Н), 4.87-5.01 (m, 1Н), 5.07 (s, 2Н), 7.11-7.31 (m, 1Н), 7.33-7.49 (m, 5Н), 8.19 (s, 1Н), 8.84 (d, J=7.81 Гц, 1Н); MS [М+Н], вычислено: 395,0, найдено: 395,2.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), αD=-135 (с=0,57, МеОН) и αD=+199 (с=4,1, МеОН).
Пример 7
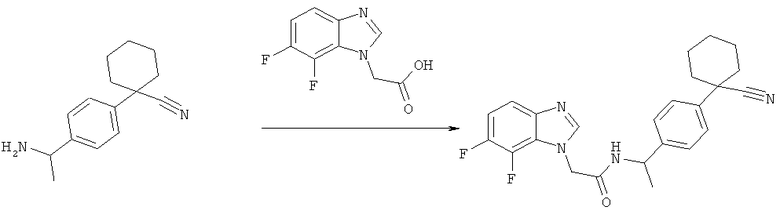
(+) и (-)-N-{1-[4-(1-Цианоциклогексил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид.
N-{1-[4-(1-Цианоциклогексил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид синтезируют согласно примеру 5 путем смешивания (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислоты (233 мг, 1,10 ммоль), полученной согласно схеме 2, 1-[4-(1-аминоэтил)фенил]циклогексанкарбонитрила (250 мг, 1,10 ммоль), полученного согласно схеме 11, Et3N (111 мг, 1,10 ммоль, 0,153 мл) и HATU (418 мг, 1,10 ммоль) в MeCN (10,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (10 мМ NH4CO3) В = MeCN, (336 мг, 0,796 ммоль, 72%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.38 (d, J=7.03 Гц, 3H), 1.23-1.35 (m, 1Н), 1.51-1.69 (m, 2Н), 1.69-1.78 (m, 1Н), 1.77-1.89 (m, 4Н), 1.99-2.09 (m, 2Н), 4.87-5.00 (m, 1Н), 5.07 (s, 2Н), 7.14-7.28 (m, 1Н), 7.37 (d, J=8.40 Гц, 2Н), 7.42-7.53 (m, 3H), 8,19 (s, 1Н), 8,83 (d, J=7,81 Гц, 1Н); MS [М+Н], вычислено: 424,0, найдено: 423,3.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), αD=-151 (с=5,6, МеОН) и αD=+194 (с=2,8, МеОН).
Пример 8
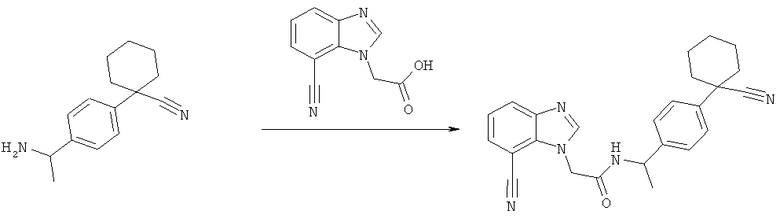
(+) и (-)-2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклогексил)-фенил]этил}ацетамид.
2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклогексил)фенил]-этил}ацетамид синтезируют согласно примеру 5 путем смешивания (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты (216 мг, 1,08 ммоль), полученной согласно схеме 3, 1-[4-(1-аминоэтил)фенил]циклогексанкарбонитрила (246 мг, 1,08 ммоль), полученного согласно схеме 11, Et3N (328 мг, 3,24 ммоль, 0,452 мл) и HATU (411 мг, 1,08 ммоль) в MeCN (10,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (10 мМ NH4CO3) В = MeCN, (443 мг, 1,08 ммоль, количественно). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн1 1.22-1.36 (m, 1Н), 1.39 (d, J=6.84 Гц, 3H) 1.53-1.68 (m, 2Н), 1.68-1.77 (m, 1Н), 1.77-1.89 (m, 4Н), 1.98-2.08 (m, 2Н), 4.88-5.02 (m, 1Н), 5.24 (dd, J=24.61, 17.77 Гц, 2Н), 7.35 (t, J=7.91 Гц, 1Н), 7.40 (d, J=8.40 Гц, 2Н), 7.46 (d, J=8.40 Гц, 2Н), 7.72 (d, J=7.62 Гц, 1Н), 8.01 (dd, J=8.20, 0.78 Гц, 1Н), 8.35 (s, 1Н), 8.85 (d, J=7.81 Гц, 1Н); MS [М+Н], вычислено: 412,0, найдено: 412,3.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), αD=-208 (с=2,2, МеОН) и αD=+190 (с=2,4, МеОН).
Пример 9
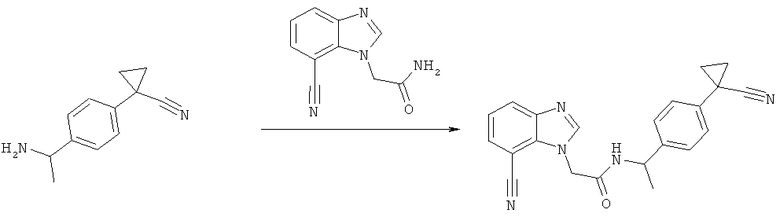
(+) и (-)-2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклопропил)-фенил]этил}ацетамид.
2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклопропил)фенил]-этил}ацетамид синтезируют согласно примеру 5 путем смешивания (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты (86,5 мг, 0,430 ммоль), полученной согласно схеме 3, 1-[4-(1-аминоэтил)фенил]циклопропанкарбонитрила (80,0 мг, 0,430 ммоль), полученного согласно схеме 10, Et3N (131 мг, 1,29 ммоль, 0,180 мл) и HATU (163 мг, 0,430 ммоль) в MeCN (10,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Synergi Polar (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,05% TFA) В = MeCN, (23,0 мг, 0,0623 ммоль, 14%). 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-D) δ млн-1 1.35 (dd, J=7.81, 5.47 Гц, 2Н), 1.49 (d, J=7.03 Гц, 3H), 1.68 (dd, J=7.42, 5.08 Гц, 2Н), 5.00-5.13 (m, 3H), 6.65 (d, J=7.42 Гц, 1Н), 7.22 (d, J=8.59 Гц, 2Н), 7.29 (d, J=8.20 Гц, 2Н), 7.33 (d, J=7.81 Гц, 1Н), 7.59 (d, J=7.62 Гц, 1Н), 7.96-8.06 (m, 2Н); MS [М+Н], вычислено: 370,0, найдено: 370,0.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 10
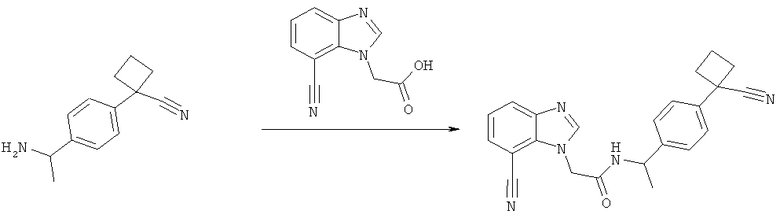
(+) и (-)-2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклобутил)-фенил]этил}ацетамид.
2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклобутил)фенил]этил}-ацетамид синтезируют согласно примеру 5 путем смешивания (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты (95,0 мг, 0,475 ммоль), полученной согласно схеме 3, 1-[4-(1-аминоэтил)фенил]циклобутанкарбонитрила (95 мг, 0,475 ммоль), полученного согласно схеме 9, Et3N (144 мг, 1,43 ммоль, 0,199 мл) и HATU (181 мг, 0,475 ммоль) в MeCN (10,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (10 мМ NH4CO3) В = MeCN, (34,0 мг, 0,0888 ммоль, 19%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.40 (d, J=7.03 Гц, 3H), 1.89-2.05 (m, 1Н), 2.15-2.34 (m, 1Н), 2.52-2.66 (m, 2Н), 2.66-2.80 (m, 2Н), 4.88-5.01 (m, 1Н), 5.25 (dd, J=22.66, 17.58 Гц, 2Н), 7.31-7.46 (m, 5Н), 7.71 (dd, J=7.62, 0.78 Гц, 1Н), 8.01 (dd, J=8.01, 0.78 Гц, 1Н), 8.36 (s, 1Н), 8.87 (d, J=7.62 Гц, 1Н); MS [М+Н], вычислено: 384,0, найдено: 384,2.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 11

(R)(+) и (3)(-)-2-(7-Ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид.
2-(7-Ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]-этил}ацетамид синтезируют согласно примеру 5 путем смешивания 7-ацетил-1Н-бензимидазол-1-илуксусной кислоты (90,0 мг, 0,413 ммоль), полученной согласно схеме 4, 2-[4-(1-аминоэтил)фенил]-2-метилпропаннитрила (77,6 мг, 0,413 ммоль), полученного согласно схеме 6, и Et3N (125 мг, 1,24 ммоль, 0,173 мл) и HATU (157 мг, 0,413 ммоль) в сухом MeCN (10,0 мл). Продукт очищают с помощью обращенно-фазной HPLC: препаративные насосы Gilson; скорость потока: 30 мл/мин; колонка: Synergi Polar (4 мкм) 21,2×50 мм; подвижная фаза: А = вода (0,05% TFA) В = MeCN (49,5 мг, 0,128 ммоль, 31,0%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.35 (d, J=7.03 Гц, 3H), 1.66 (s, 6Н), 2.38 (s, 3H), 4.74-4.87 (m, J=7.23, 7.23 Гц, 1Н), 5.14 (s, 2Н), 7.26 (t, J=7.81 Гц, 1Н), 7.35 (d, J=8.40 Гц, 2Н), 7.45 (d, J=8.40 Гц, 2Н), 7.71 (d, J=7.62 Гц, 1Н), 7.85 (d, J=8.01 Гц, 1Н), 8.21 (s, 1Н), 8.65 (d, J=7.81 Гц, 1Н); MS [М+Н] вычислено: 389,19, найдено: 389,09.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson; скорость потока: 18 мл/мин; колонка: Chiralcel AD 21×250 мм (20 мкм); подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA); αD=+171 (с=2, МеОН) и αD=-162 (с=2, МеОН).
Пример 12

(+) и (-)-2-(7-Ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид.
2-(7-Ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид синтезируют согласно примеру 5 путем смешивания HATU (179 мг, 0,472 ммоль), 7-ацетил-1Н-бензимидазол-1-илуксусной кислоты (103 мг, 0,472 ммоль), полученной согласно схеме 4, 2-[4-(1-аминоэтил)-2-фторфенил]-2-метилпропаннитрила (97,0 мг, 0,472 ммоль), полученного согласно схеме 7, и Et3N (143 мг, 1,42 ммоль, 0,200 мл) в сухом MeCN (10,0 мл). Продукт очищают с помощью обращенно-фазной HPLC: препаративные насосы Gilson; скорость потока: 30 мл/мин; колонка: Gemini (5 мкм) 21,2×50 мм; подвижная фаза: А=10,0 мМ NH4HCO3 в H2O В=MeCN (52,4 мг, 0,129 ммоль, 27,0%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.36 (d, J=7.03 Гц, 3H), 1.70 (s, 6Н), 2.40 (s, 3H), 4.72-4.90 (m, 1Н), 5.17 (dd, J=21.48, 17.19 Гц, 2Н), 7.18 (d, J=7.81 Гц, 1Н), 7.21-7.32 (m, 2Н), 7.40 (t, J=8.30 Гц, 1Н), 7.72 (d, J=7.62 Гц, 1Н), 7.86 (d, J=7.81 Гц, 1Н), 8.22 (s, 1Н), 8.72 (d, J=7.42 Гц, 1Н); MS [М+Н] вычислено: 407,18, найдено: 407,17.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson; скорость потока: 18 мл/мин; колонка: Chiralcel AD 21×250 мм (20 мкм); подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA); αD=+166 (с=2, МеОН) и αD=-159 (с=1,60, МеОН).
Пример 13:
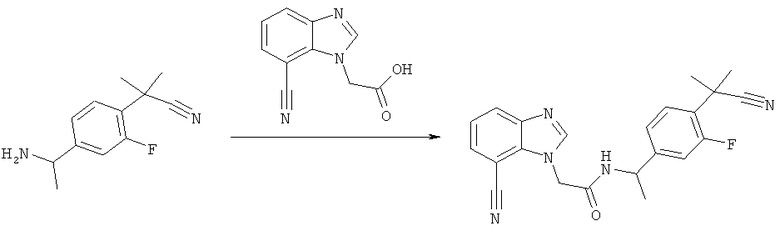
(+) и (-)-2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид.
2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид синтезируют согласно примеру 1 путем смешивания 2-[4-(1-аминоэтил)-2-фторфенил]-2-метилпропаннитрила гидрохлорида (260 мг, 0,94 ммоль), полученного согласно схеме 7, (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты (200 мг, 0,94 ммоль), полученной согласно схеме 3, и DMAP (228 мг, 1,89 ммоль, 1,00 мл) и HATU (358 мг, 2,49 ммоль) в DMF (5,0 мл). Продукт очищают с помощью HPLC: Waters prep LCMS, скорость потока: 27 мл/мин, колонка: SynerSi (4 мкм) Polar RP, 21,2×50 мм, подвижная фаза: А = вода (0,05% TFA) В = MeCN, используемый градиент от 40% до 60% В в А, в течение 10 мин. Фракции объединяют, добавляют 1 н. NaOH, и желаемое соединение экстрагируют этилацетатом. Объединенные органические фракции концентрируют при пониженном давлении с получением продукта (69 мг, 1,77 ммоль, 19%). 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.50 (d, J=7.03 Гц, 3H), 1.72-1.78 (m, 6Н), 5.02 (q, J=6.97 Гц, 1Н), 5.31 (d, J=17.58 Гц, 1Н), 5.36 (d, J=17.58 Гц, 1Н), 7.17-7.25 (m, 2Н), 7.39 (dd, J=8.20, 7.62 Гц, 1Н), 7.43 (t, J=8.40 Гц, 1Н), 7.67 (dd, J=7.62, 0.78 Гц, 1Н), 7.97 (dd, J=8.20, 0.98 Гц, 1Н), 8.26 (s, 1Н); MS [М+Н], вычислено: 390,2, найдено: 390,3.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel® AD 21×250 мм (20 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 70% А: 30% В изократика αD=-173 (с=5,5, МеОН) и αD=+180 (с=5,7, МеОН).
Пример 14

N-{1-[4-(1-Циано-1-метилэтил)-3-фторфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид
Амид синтезируют в соответствии с модифицированным примером 1 путем смешивания (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислоты (781 мг, 3,68 ммоль), полученной согласно схеме 2, 2-[4-(1-аминоэтил)-2-фторфенил]-2-метилпропаннитрила гидрохлорида (1,07 мг, 4,41 ммоль), полученного согласно схеме 7, Et3N (1,23 мл, 8,82 ммоль) и HATU (1,68 г, 4,41 ммоль) в DMF (15,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,075% TFA) В = MeCN (0,075% TFA), (565 мг в виде соли TFA, 1,10 ммоль, 30%); 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.38 (d, J=6.84 Гц, 3H), 1.71 (s, 6Н), 4.87-5.00 (m, J=7.23, 7.23 Гц, 1Н), 5.09 (s, 2Н), 7.16-7.29 (m, 3H), 7.36-7.51 (m, 2Н), 8.19 (s, 1Н), 8.86 (d, J=7.62 Гц, 1Н). MS [М+Н], вычислено: 401,4, найдено: 401.3.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralpak OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = iPrOH (0,1% DEA).
Пример 15

2-(7-Хлор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид
Амид синтезируют согласно примеру 1 путем смешивания (7-хлор-1Н-бензимидазол-1-ил)уксусной кислоты (521 мг, 2,48 ммоль), полученной согласно схеме 1, 2-[4-(1-аминоэтил)-2-фторфенил]-2-метилпропаннитрила гидрохлорида 721 мг, 2,97 ммоль), полученного согласно схеме 7, Et3N (824 мкл, 5,95 ммоль) и HATU (1,13 г, 2,97 ммоль) в DMF (10,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,075% TFA) В = MeCN (0,075% TFA), (421 мг в виде соли TFA, 0,82 ммоль, 33%); 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.38 (d, J=7.03 Гц, 3H), 1.64-1.76 (m, 6Н), 4.88-4.98 (m, 1Н), 5.27 (s, 2Н), 7.18-7.28 (m, 3H), 7.28-7.33 (m, 1Н), 7.41 (t, J=8.30 Гц, 1Н), 7.65 (d, J=7.81 Гц, 1Н), 8.47 (s, 1Н), 8.85 (d, J=7.62 Гц, 1Н). MS [М+Н], вычислено: 399,9, найдено: 399,3.
Энантиомеры разделяют с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralpak OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = iPrOH (0,1% DEA).
Пример 16:

2-(7-Хлор-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-3-фторбензил]ацетамид.
2-(7-Хлор-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-3-фторбензил]ацетамид синтезируют согласно примеру 14 с (7-хлор-1Н-бензимидазол-1-ил)уксусной кислотой (150 мг, 0,712 ммоль), полученной согласно схеме 1, и 2-[4-(аминометил)-2-фторфенил]-2-метилпропаннитрила гидрохлоридом (196 мг, 0,855 ммоль), полученным согласно схеме 8, Et3N (238 мкл, 1,71 ммоль) и HATU (325 мг, 0,855 ммоль) в DMF (3,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,075% TFA) В = MeCN (0,075% TFA). Выход = 14 мг в виде соли TFA (0,035 ммоль, 5%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.67 (s, 6Н), 4.30 (d, J=5.86 Гц, 2Н), 5.24 (s, 2Н), 7.10-7.22 (m, 3H), 7.23-7.28 (m, 1Н), 7.38 (t, J=8.40 Гц, 1Н), 7.62 (d, J=7.81 Гц, 1Н), 8.31 (s, 1Н), 8.81 (s, 1Н). MS [М+Н], вычислено: 385,1, найдено: 385,0.
Пример 17:
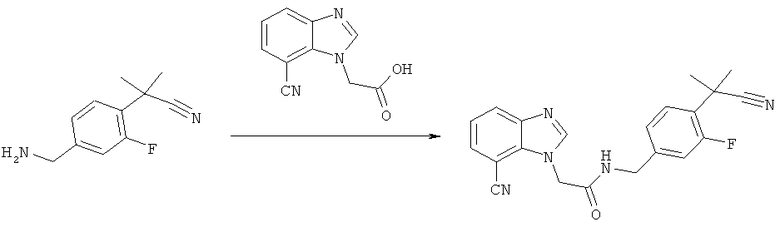
2-(7-Циано-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-3-фторбензил]ацетамид.
2-(7-Циано-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-3-фторбензил]ацетамид синтезируют согласно примеру 14 с (7-циано-1Н-бензимидазол-1-ил)уксусной кислотой (150 мг, 0,746 ммоль), полученной согласно схеме 3, 2-[4-(аминометил)-2-фторфенил]-2-метилпропаннитрила гидрохлоридом (205 мг, 0,896 ммоль), полученным согласно схеме 8, Et3N (250 мкл, 1,79 ммоль) и HATU (341 мг, 0,896 ммоль) в DMF (3,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,075% TFA) В = MeCN (0,075% TFA). Выход = 21 мг в виде соли TFA (0,043 ммоль, 6%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.62-1.71 (m, 6Н), 4.32 (d, J=5.86 Гц, 2Н), 5.26 (s, 2Н), 7.13-7.25 (m, 2Н), 7.29-7.41 (m, 2Н), 7.71 (d, J=7.42 Гц, 1Н), 8.00 (d, J=8.01 Гц, 1Н), 8.36 (s, 1Н), 8.84-8.93 (m, 1Н). MS [М+Н], вычислено: 376,2, найдено: 376,0.
Пример 18:
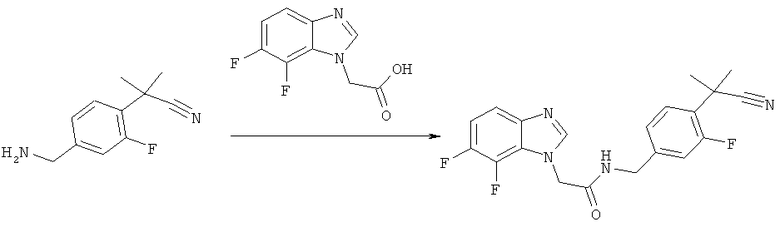
N-[4-(1-Циано-1-метилэтил)-3-фторбензил]-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид.
N-[4-(1-Циано-1-метилэтил)-3-фторбензил]-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид синтезируют согласно примеру 14 с (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислотой (150 мг, 0,707 ммоль), полученной согласно схеме 2, 2-[4-(аминометил)-2-фторфенил]-2-метилпропаннитрила гидрохлоридом (194 мг, 0,848 ммоль), полученным согласно схеме 8, Et3N (237 мкл, 1,70 ммоль) и HATU (323 мг, 0,848 ммоль) в DMF (3,0 мл). Продукт очищают с помощью HPLC: препаративные насосы Gilson, скорость потока: 30 мл/мин, колонка: Gemini (5 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,075% TFA) В = MeCN (0,075% TFA). Выход = 25 мг в виде соли TFA, 0,05 ммоль, 7%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.74-1.87 (m, 6Н), 4.43 (d, J=5.66 Гц, 2Н), 5.22 (s, 2Н), 7.19-7.40 (m, 2Н), 7.45-7.61 (m, 2Н), 8.34 (s, 1Н), 8.91-9.04 (m, 1Н). MS [М+Н], вычислено: 387,1, найдено: 387,0.
Пример 19:
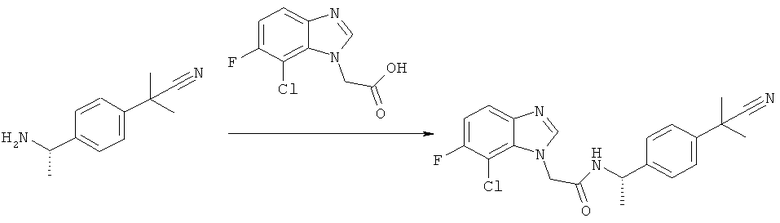
(S)(-)-2-(7-Хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид.
Смесь (7-хлор-6-фтор-1Н-бензимидазол-1-ил)уксусной кислоты (1,00 г, 4,39 ммоль), полученной согласно схеме 12, (S)(-)-2-{4-[1-аминоэтил]фенил}-2-метилпропаннитрила (825 мг 4,39 ммоль), полученного согласно схеме 20 или 21, и Et3N (1,33 г, 13,2 ммоль, 1,84 мл) перемешивают в MeCN (50,0 мл). Добавляют HATU (1,67 г, 4,39 ммоль) и после 2 часов перемешивания смесь разбавляют 1 н. NaOH (100 мл) и затем экстрагируют 4 раза EtOAc (4×75,0 мл). Органические фазы объединяют и сушат над Na2SO4. Смесь фильтруют и концентрируют. Продукт очищают с помощью преп. насоса Gilson, скорость потока: 30 мл/мин, колонка: Gemini™ (5 мкм) 21,2×50 мм, подвижная фаза: А=10 мМ бикарбоната аммония, В=MeCN (1,01 г, 58%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.39 (d, J=7.03 Гц, 3H), 1.66 (s, 6Н), 4.92 (квинт., J=7.23 Гц, 1Н), 5.19 (s, 2Н), 7.23 (dd, J=10.16, 8.98 Гц, 1Н), 7.37 (d, J=8.20 Гц, 2Н), 7.47 (d, J=8.59 Гц, 2Н), 7.63 (dd, J=8.79, 4.49 Гц, 1Н), 8.21 (s, 1Н), 8.81 (d, J=7.81 Гц, 1Н). MS [М+Н], вычислено: 399,1, найдено: 399,2. [α]D=-165 (с=1,02 МЕТАНОЛ).
HRMS(ESI+) вычислено для C21H21ClFN4O 399,13824 [М+Н]+, найдено 399,13832.
Пример 20:

(+) и (-) 2-(7-Хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилзтил)-3-фторфенил]этил}ацетамид.
Смесь (7-хлор-6-фтор-1Н-бензимидазол-1-ил)уксусной кислоты (210 мг, 0,921 ммоль), полученной согласно схеме 12, 2-{4-[1-аминоэтил]-2-фторфенил}-2-метилпропаннитрила (190 мг 0,921 ммоль), полученного согласно схеме 7, и Et3N (280 мг, 2,76 ммоль, 0,39 мл) перемешивают в MeCN (10,0 мл). Добавляют HATU (350 мг, 0,921 ммоль) и после 2 часов перемешивания смесь разбавляют 1 н. NaOH (40,0 мл) и затем экстрагируют 4 раза EtOAc (4×40,0 мл). Органические фазы объединяют и сушат над MgSO4. Смесь фильтруют и концентрируют. Продукт очищают с помощью преп. насоса Gilson, скорость потока: 30 мл/мин, колонка: Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN (206 мг, 54%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.38 (d, J=6.84 Гц, 3H), 1.71 (s, 6Н), 4.85-5.00 (m, J=7.13, 7.13 Гц, 1Н), 5.21 (s, 2Н), 7.15-7.31 (m, 3H), 7.42 (t, J=8.30 Гц, 1Н), 7.63 (dd, J=8.89, 4.39 Гц, 1Н), 8.21 (s, 1Н), 8.84 (d, J=7.81 Гц, 1Н). Энантиомеры разделяют на хиральный колонке AD, элюируя 30% EtOH (0,1% DIEA) и 70% гексанами (0,1% DIEA). MS [М+Н], вычислено: 417,1, найдено: 417,3, [α]D=+154 (с=13, МеОН) и [α]D=-155 (с=15, МеОН).
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 21:

(+) и (-)-2-(7-Хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамид.
К раствору (7-хлор-6-фтор-1Н-бензимидазол-1-ил)уксусной кислоты (100 мг, 0,44 ммоль), полученной согласно схеме 12, 2-{4-[1-аминоэтил]-2-метилфенил}-2-метилпропаннитрила (88,5 мг 0,44 ммоль), полученного согласно схеме 16, и DMAP (106 мг, 0,88 ммоль), перемешиваемому в безводном DMF (2,0 мл), добавляют HATU (166 мг, 0,44 ммоль). После 18 часов перемешивания смесь разбавляют метанолом (200 мкл). Раствор очищают непосредственно на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN с использованием кратковременного от 40 до 60% В 10 мин градиентного способа. Чистые фракции объединяют, TFA нейтрализуют 4 н. раствором NaOH и затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта (+) и (-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}-ацетамида (290 мг, 53%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.48 (d, J=7.03 Гц, 3H), 1.76 (s, 6Н), 2.61 (s, 3H), 4.99 (q, J=7.03 Гц, 1Н) 5.19-5.36 (m, 2Н), 7.13-7.25 (m, 3H), 7.33 (d, J=8.20 Гц, 1Н), 7.59 (dd, J=8.98, 4.30 Гц, 1Н), 8.14 (s, 1Н). MS [М+Н], вычислено: 413,2, найдено: 413,3.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 22:
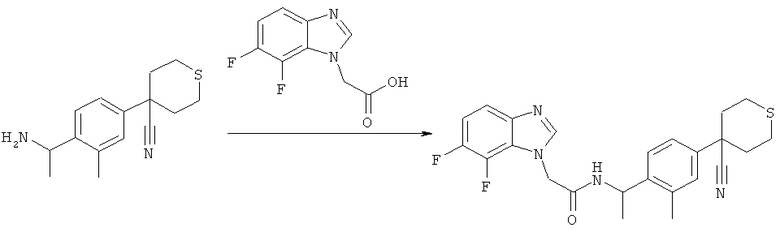
(+) и (-) N-{1-[4-(4-Цианотетрагидро-2Н-тиопиран-4-ил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид
Амид синтезируют согласно примеру 1 путем смешивания (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислоты (96 мг, 0,45 ммоль), полученной согласно схеме 2, 4-[4-(1-аминоэтил)-3-метилфенил]тетрагидро-2Н-тиопиран-4-карбонитрила (129 мг, 0,50 ммоль), полученного согласно схеме 14, Et3N (0,69 мкл, 0,50 ммоль) и HATU (189 мг, 0,50 ммоль) в MECN (3,0 мл). Соединение очищают с помощью колоночной хроматографии на силикагеле (10% МеОН в DCM) (121 мг, 59%). 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.35 (d, J=7.03 Гц, 3H), 2.05-2.16 (m, 2Н), 2.26-2.35 (m, 5Н), 2.72-2.81 (m, J=14.06 Гц, 2Н), 2.88-2.99 (m, J=12.50, 12.50 Гц, 2Н), 5.01-5.11 (m, 3H), 7.16-7.26 (m, 1Н), 7.30 (s, 1Н), 7.33-7.38 (m, 1Н), 7.39-7.48 (m, 2Н), 8.18 (s, 1Н), 8.84 (d, J=7.42 Гц, 1Н). MS [М+Н], вычислено: 455,5, найдено: 455,3.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 23:

N-[4-(1-Циано-1-метилэтил)бензил]-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид
(6,7-Дифтор-1Н-бензимидазол-1-ил)уксусную кислоту (42 мг, 0,2 ммоль), полученную согласно схеме 2, растворяют в CH2Cl2 (2 мл). Добавляют триэтиламин (83 мкл, 0,6 ммоль), затем пивалоилхлорид (25 мкл, 0,2 ммоль). Через 30 минут добавляют 2-[4-(аминометил)фенил]-2-метилпропаннитрил (35 мг, 0,2 ммоль), полученный согласно схеме 18, растворенный в 1 мл CH2Cl2. Смесь перемешивают в течение ночи при комнатной температуре, концентрируют в вакууме и очищают на HPLCMS: Waters prep LCMS, 27 мл/мин, колонка: X-Bridge Prep С18 OBD, 30×50 мм, размер частиц 5 мкм, подвижная фаза: А = вода (10 мМ NH4CO3) В = MeCN, используемый градиент от 40% до 60% В в А, в течение 10 мин. Фракции объединяют и сушат сублимацией с получением желаемого продукта (17 мг, 0,05 ммоль, 25%). MS (ESI) (М+1) 383,3. 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.65 (s, 6Н), 2.30 (s, 3H) 4.29 (d, J=5.47 Гц, 2Н), 5.09 (s, 2Н), 7.19-7.31 (m, 3H), 7.32-7.35 (m, 1Н), 7.47 (dd, J=8.98, 3.91 Гц, 1Н), 8.22 (8, 1Н), 8.66-8.74 (m, 1Н).
Пример 24:

N-[4-(1-Циано-1-метилэтил)-2-метилбензил]-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид
(6,7-Дифтор-1Н-бензимидазол-1-ил)уксусную кислоту (91 мг, 0,43 ммоль), полученную согласно схеме 2, растворяют в DCM (3 мл). Добавляют DIPEA (0,1 мл), затем пивалоилхлорид (53 мкл, 0,43 ммоль). Через 1 час добавляют 2-[4-(аминометил)-3-метилфенил]-2-метилпропаннитрил (80 мг, 0,43 ммоль), полученный согласно схеме 19. Смесь перемешивают в течение ночи при комнатной температуре, концентрируют в вакууме и очищают на HPLCMS: Waters prep LCMS, 27 мл/мин, колонка: X-Bridge Prep C18 OBD, 30×50 мм, размер частиц 5 мкм, подвижная фаза: А = вода (10 мМ NH4CO3) В = MeCN, используемый градиент от 30% до 50% В в А, в течение 10 мин. Фракции объединяют и сушат сублимацией с получением желаемого продукта (59 мг, 0,15 ммоль, 35%). MS (ESI) (М+1) 369,2. 1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.67 (s, 6Н), 4.33 (d, J=5.86 Гц, 2Н), 5.10 (s, 2Н), 7.23 (ddd, J=11.62, 8.89, 7.62 Гц, 1Н), 7.32 (d, J=8.59 Гц, 2Н), 7.44-7.51 (m, 3H), 8.21 (s, 1Н), 8.80 (t, J=5.86 Гц, 1Н).
Пример 25:
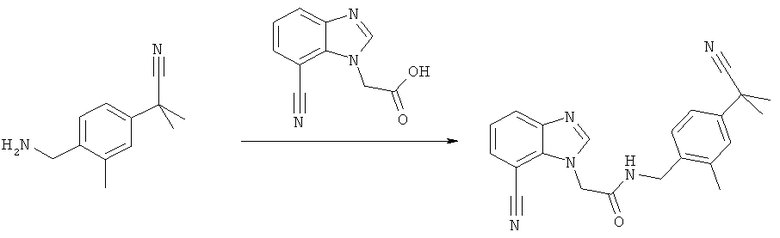
2-(7-Циано-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-2-метилбензил]ацетамид
(7-Циано-1Н-бензимидазол-1-ил)уксусную кислоту (86 мг, 0,43 ммоль), полученную согласно схеме 3, растворяют в DCM (3 мл). Добавляют DIPEA (0,1 мл), затем пивалоилхлорид (53 мкл, 0,43 ммоль). Через 1 час добавляют 2-[4-(аминометил)-3-метилфенил]-2-метилпропаннитрил (80 мг, 0,43 ммоль), полученный согласно схеме 19. Смесь перемешивают в течение ночи при комнатной температуре, концентрируют в вакууме и очищают на HPLCMS: Waters prep LCMS, 27 мл/мин, X-Bridge Prep C18 OBD, 30×50 мм, размер частиц 5 мкм, подвижная фаза: А = вода (10 мМ NH4CO3) В = MeCN, используемый градиент от 30% до 50% В в А, в течение 10 мин. Фракции объединяют и сушат сублимацией получением желаемого продукта (47 мг, 0,13 ммоль, 30%). MS (ESI) (М+1) 372,3.1Н-ЯМР (400 МГц, ДМСО-D6) δ млн-1 1.65 (s, 6Н), 2.31 (s, 3H), 4.29 (d, J=5.08 Гц, 2Н), 5.26 (s, 2Н), 7.25-7.29 (m, 1Н), 7.30-7.40 (m, 3H), 7.73 (dd, J=7.62, 0.98 Гц, 1Н), 8.03 (dd, J=8.20, 0.78 Гц, 1Н), 8.38 (s, 1Н), 8.74 (t, J=5.27 Гц, 1Н).
Пример 26:
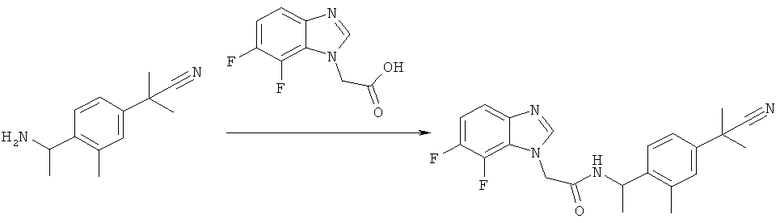
(+) и (-)-N-{1-[4-(1-Циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид
К раствору (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислоты (250 мг, 1,18 ммоль), полученной согласно схеме 2, 2-[4-(1-аминоэтил)-3-метилфенил]-2-метилпропаннитрила (202 мг 1,18 ммоль), полученного согласно схеме 15, и DMAP (285 мг, 2,35 ммоль), перемешиваемому в безводном DMF (4,0 мл), добавляют HATU (448 мг, 1,18 ммоль). После 18 часов перемешивания смесь разбавляют 1 н. NaOH (40,0 мл) и затем экстрагируют 4 раза EtOAc (4×40,0 мл). Органические фазы объединяют и сушат над MgSO4. Смесь фильтруют и концентрируют. Продукт очищают на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,01 М аммоний-карбонатный буфер), В = MeCN с использованием кратковременного от 40 до 60% В 10 мин градиентного способа при высоком рН. Чистые фракции объединяют, затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида (350 мг, 75%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.44 (d, J=6.84 Гц, 3H), 1.66 (s, 6Н), 2.36 (s, 3H), 5.06 (d, J=16.99 Гц, 1Н), 5.11 (d, J=16.99 Гц, 1Н), 5.20 (q, J=7.03 Гц, 1Н), 7.14 (ddd, J=11.38, 8.84, 7.52 Гц, 1Н), 7.26 (d, J=2.15 Гц, 1Н), 7.32 (dd, J=6.05, 2.15 Гц, 1Н), 7.37-7.43 (m, 2Н), 8.09 (s, 1Н). MS [М+Н], вычислено: 397,2, найдено: 397,3. Энантиомеры разделяют на хиральной колонке OD с использованием метанола и CO2 в качестве подвижной фазы.
Альтернативно, хиральный (-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид может быть получен непосредственно с использованием хирального промежуточного соединения (-)2-[4-[1-аминоэтил]-3-метилфенил]-2-метилпропаннитрила, полученного, как описано на схеме 23, и соответствующей кислоты выше.
Пример 27:
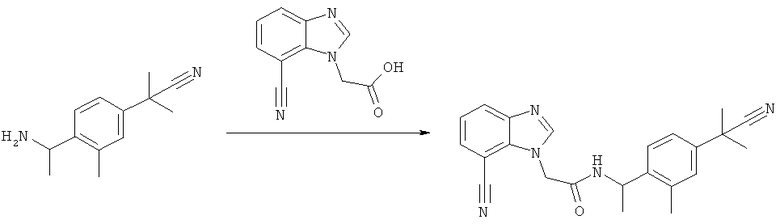
(+) и (-)-2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}ацетамид
К раствору (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты (250 мг, 1,24 ммоль), полученной согласно схеме 3, 2-[4-(1-аминоэтил)-3-метилфенил]-2-метилпропаннитрила (251 мг 1,24 ммоль), полученного согласно схеме 15, и DMAP (150 мг, 1,24 ммоль), перемешиваемому в безводном DMF (4,0 мл), добавляют HATU (473 мг, 1,24 ммоль). После 18 часов перемешивания смесь разбавляют метанолом (200 мкл). Раствор очищают непосредственно на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN с использованием кратковременного от 40 до 60% В 10 мин градиентного способа. Чистые фракции объединяют, TFA нейтрализуют 4 н. раствором NaOH и затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта (+) и (-)-2-(7-циано-1H-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}ацетамида (206 мг, 43%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.45 (d, J=6.84 Гц, 3Н), 1.66 (s, 6H), 2.37 (s, 3Н), 5.20 (q, J=6.84 Гц, 1H), 5.30 (s, 2H), 7.25 (d, J=1.95 Гц, 1H), 7.32 (dd, J=8.20, 2.15 Гц, 1H), 7.38 (dd, J=8.11, 7.71 Гц, 1Н), 7.44 (d, J=8.20 Гц, 1H), 7.67 (dd, J=7.42, 0.78 Гц, 1H), 7.96 (dd, J=8.20, 0.98 Гц, 1H), 8,24 (s, 1H). MS [M+H], вычислено: 386,2, найдено: 386,2.
Альтернативно, хиральный (-)-2-(7-циано-1H-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}ацетамид может быть получен непосредственно с использованием хирального промежуточного соединения (-)2-[4-[1-аминоэтил]-3-метилфенил]-2-метилпропаннитрила, полученного, как описано на схеме 23, и соответствующей кислоты выше.
Пример 28:

(+) и (-)-N-{1-[4-(1-Циано-1-метилэтил)-2-метилфенил]этил}-2-(7-фтор-1H-бензимидазол-1-ил)ацетамид
К раствору (7-фтор-1H-бензимидазол-1-ил)уксусной кислоты (250 мг, 1,29 ммоль), полученной согласно схеме 5, 2-[4-(1-аминоэтил)-3-метилфенил]-2-метилпропаннитрила (260 мг 1,29 ммоль), полученного согласно схеме 15, и DMAP (156 мг, 1,29 ммоль), перемешиваемому в безводном DMF (4,0 мл), добавляют HATU (490 мг, 1,29 ммоль). После 18 часов перемешивания смесь разбавляют метанолом (200 мкл). Раствор очищают непосредственно на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN с использованием кратковременного от 40 до 60% В 10 мин градиентного способа. Чистые фракции объединяют, TFA нейтрализуют 4 н. раствором NaOH и затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта (+) и (-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамида (306 мг, 63%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.44 (d, J=6.84 Гц, 3H), 1.66 (s, 6Н) 2.35 (s, 3H), 5.02-5.14 (m, 2Н), 5.19 (q, J=7.03 Гц, 1Н), 6.99 (dd, J=11.52, 7.42 Гц, 1Н), 7.19 (dt, J=8.15, 4.98 Гц, 1Н), 7.26 (d, J=2.15 Гц, 1Н), 7.32 (dd, J=8.01, 2.34 Гц, 3H), 7.39 (d, J=8.20 Гц, 1Н), 7.45 (d, J=7.62 Гц, 1Н), 8.09 (s, 1Н). MS [М+Н], вычислено: 379,2, найдено: 379,2.
Альтернативно, хиральный (-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамид может быть получен непосредственно с использованием хирального промежуточного соединения (-)2-[4-[1-аминоэтил]-3-метилфенил]-2-метилпропаннитрила, полученного, как описано на схеме 23, и соответствующей кислоты выше.
Пример 29:

(+) и (-)-N-{1-[3-Хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид
К раствору (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислоты (285 мг, 1,35 ммоль), полученной согласно схеме 2, 2-[4-(1-аминоэтил)-2-хлорфенил]-2-метилпропаннитрила (300 мг 1,35 ммоль), полученного согласно схеме 17, и DMAP (325 мг, 2,69 ммоль), перемешиваемому в безводном DMF (3,0 мл), добавляют HATU (511 мг, 1,35 ммоль). После 18 часов перемешивания смесь разбавляют метанолом (200 мкл). Раствор очищают непосредственно на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN с использованием кратковременного от 40 до 60% В 10 мин градиентного способа. Чистые фракции объединяют, TFA нейтрализуют 4 н. раствором NaOH и затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта (+) и (-)-N-{1-[3-хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида (300 мг, 55%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.50 (d, J=7.03 Гц, 3H), 1.83 (s, 6Н) 5.02 (q, J=7.03 Гц, 1Н), 5.14 (s, 2Н), 7.17 (ddd, J=11.33, 8.98, 7.42 Гц, 1Н), 7.33 (dd, J=8.20, 1.95 Гц, 1Н), 7.43 (ddd, J=8.89, 3.81, 1.37 Гц, 1Н), 7.47 (d, J=2.34 Гц, 1Н), 7.51 (d, J=8.20 Гц, 1Н), 8.13 (s, 1Н). MS [М+Н], вычислено: 379,1, найдено: 379,3.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 30:

(+) и (-)-N-{1-[3-Хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(7-циано-1Н-бензимидазол-1-ил)ацетамид
К раствору (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты (271 мг, 1,35 ммоль), полученной согласно схеме 3, 2-[4-(1-аминоэтил)-2-хлорфенил]-2-метилпропаннитрила (300 мг 1,35 ммоль), полученного согласно схеме 17, и DMAP (325 мг, 2,69 ммоль), перемешиваемому в безводном DMF (3,0 мл), добавляют HATU (511 мг, 1,35 ммоль). После 18 часов перемешивания смесь разбавляют метанолом (200 мкл). Раствор очищают непосредственно на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN с использованием кратковременного от 40 до 60% В 10 мин градиентного способа. Чистые фракции объединяют, TFA нейтрализуют 4 н. раствором NaOH и затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта (+) и (-)-N-{1-[3-хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(7-циано-1Н-бензимидазол-1-ил)ацетамида (290 мг, 53%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.50 (d, J=7.03 Гц, 3H), 1.83 (s, 6Н) 5.02 (q, J=7.03 Гц, 1Н), 5.14 (s, 2Н) 7.17 (ddd, J=11.33, 8.98, 7.42 Гц, 1Н), 7.33 (dd, J=8.20, 1.95 Гц, 1Н), 7.43 (ddd, J=8.89, 3.81, 1.37 Гц, 1Н), 7.47 (d, J=2.34 Гц, 1Н), 7.51 (d, J=8.20 Гц, 1Н), 8.13 (s, 1Н). MS [М+Н], вычислено: 417,1, найдено: 417,3.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 31:

(+) и (-)-2-(6,7-Дифтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамид.
К раствору (6,7-дифтор-1Н-бензимидазол-1-ил)уксусной кислоты (500 мг, 2,36 ммоль), полученной согласно схеме 2, 2-{4-[1-аминоэтил]-2-метилфенил}-2-метилпропаннитрила (477 мг, 2,36 ммоль), полученного согласно схеме 16, и DMAP (570 мг, 4,71 ммоль), перемешиваемому в безводном DMF (4,0 мл), добавляют HATU (896 мг, 2,36 ммоль). После 18 часов перемешивания смесь разбавляют EtOAc (100 мл). Органическую фазу промывают в дистиллированной воде, сушат над безводным MgSO4. Смесь фильтруют и концентрируют. Продукт очищают на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN с использованием кратковременного от 40 до 60% В 10 мин градиентного способа. Чистые фракции объединяют, TFA нейтрализуют 4 н. раствором NaOH и затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта (+) и (-)-2-(6,7-дифтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида (627 мг, 67%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.48 (d, J=7.03 Гц, 3H), 1.75 (s, 6Н), 2.61 (s, 3H), 4.99 (q, J=7.03 Гц, 1Н), 5.12 (s, 2Н), 7.12-7.25 (m, 3H), 7.33 (d, J=8.20 Гц, 1Н), 7.42 (ddd, J=8.89, 3.61, 1.17 Гц, 1Н), 8.13 (s, 1Н). MS [М+Н], вычислено: 397,2, найдено: 397,2.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 32:

(+) и (-)-2-(7-Циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамид.
К раствору (7-циано-1Н-бензимидазол-1-ил)уксусной кислоты (100 мг, 0,50 ммоль), полученной согласно схеме 3, 2-{4-[1-аминоэтил]-2-метилфенил}-2-метилпропаннитрила (101 мг, 0,50 ммоль), полученного согласно схеме 16, и DMAP (120 мг, 1,00 ммоль), перемешиваемому в безводном DMF (2,0 мл), добавляют HATU (189 мг, 0,50 ммоль). После 18 часов перемешивания смесь разбавляют метанолом (200 мкл). Раствор очищают непосредственно на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN с использованием кратковременного от 40 до 60% В 10 мин градиентного способа. Чистые фракции объединяют, TFA нейтрализуют 4 н. раствором NaOH и затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта (+) и (-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида (143 мг, 75%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.49 (d, J=7.03 Гц, 3H), 1.75 (s, 6Н), 2.61 (s, 3H), 4.99 (q, J=7.03 Гц, 1Н), 5.28-5.40 (m, 2Н), 7.21-7.29 (m, 2Н), 7.33 (d, J=8.20 Гц, 1Н), 7.37-7.44 (m, 1Н), 7.69 (dd, J=7.42, 0.78 Гц, 1Н), 7.98 (dd, J=8.20, 1.17 Гц, 1Н), 8.27 (s, 1Н). MS [М+Н], вычислено: 386,2, найдено: 386,2.
Полагают, что энантиомеры могут быть разделены с помощью хиральной HPLC: препаративные насосы Gilson, скорость потока: 18 мл/мин, колонка: Chiralcel OD 21×250 мм (5 мкм), подвижная фаза: А = гексан (0,1% DEA) В = EtOH (0,1% DEA), 80% А: 20% В изократика.
Пример 33:
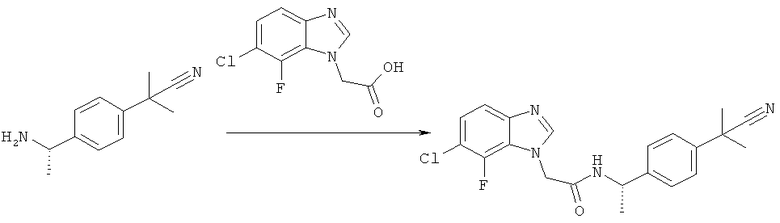
(S)-(-)2-(6-Хлор-7-фтор-1Н-бензимидазол-1-ил)-N-{[4-(1-циано-1-метилэтил)фенил]этил}ацетамид.
К раствору (6-хлор-7-фтор-1Н-бензимидазол-1-ил)уксусной кислоты (100 мг, 0,44 ммоль), полученной согласно схеме 13, (S) (-)-2-{4-[1-аминоэтил]фенил}-2-метилпропаннитрила (88,5 мг 0,44 ммоль), полученного согласно схеме 20 или 21, и DMAP (106 мг, 0,88 ммоль), перемешиваемому в безводном DMF (2,0 мл), добавляют HATU (166 мг, 0,44 ммоль). После 18 часов перемешивания смесь разбавляют метанолом (200 мкл). Раствор очищают непосредственно на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN с использованием кратковременного от 40 до 60% В 10 мин градиентного способа. Чистые фракции объединяют, TFA нейтрализуют 4 н. раствором NaOH и затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта (S)-(-)-2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида (290 мг, 53%) в виде прозрачного твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.50 (d, J=7.03 Гц, 3H), 1.69 (s, 6Н), 5.03 (q, J=7.01 Гц, 1Н), 5.07-5.18 (m, 2Н), 7.29 (dd, J=9.37, 6.64 Гц, 3H), 7.39 (d, J=8.59 Гц, 2Н), 7.44 (d, J=9.37 Гц, 1Н), 7.48 (d, J=8.59 Гц, 2Н), 8.14 (s, 1H). MS [М+Н], вычислено: 399,1, найдено: 399,2, [α]D=-138° (С=0,5 МЕТАНОЛ).
Пример 34:

2-(6-Хлор-7-фтор-1Н-бензимидазол-1-ил)-N-{[4-(1-циано-1-метилэтил)-2-метилбензил]ацетамид.
К раствору (6-хлор-7-фтор-1Н-бензимидазол-1-ил)уксусной кислоты (85 мг, 0,37 ммоль), полученной согласно схеме 13, 2-[4-(аминометил)-3-метилфенил]-2-метилпропаннитрила (70 мг 0,37 ммоль), полученного согласно схеме 19, и DMAP (90 мг, 0,74 ммоль), перемешиваемому в безводном DMF (2,0 мл), добавляют HATU (142 мг, 0,37 ммоль). После 18 часов перемешивания смесь разбавляют метанолом (200 мкл). Раствор очищают непосредственно на препаративной системе LCMS, снабженной колонкой Synergi Polar (4 мкм) 21,2×50 мм, подвижная фаза: А = вода (0,1% TFA), В = MeCN с использованием кратковременного от 30 до 60% В 10 мин градиентного способа. Чистые фракции объединяют, TFA нейтрализуют 4 н. раствором NaOH и затем экстрагируют этилацетатом, который отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением ожидаемого продукта 2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)-N-{[4-(1-циано-1-метилэтил)-2-метилбензил]ацетамида (18 мг, 12%) в виде прозрачного твердого вещества. 1Н-ЯМР (400 МГц, МЕТАНОЛ-D4) δ млн-1 1.46 (d, J=7.03 Гц, 3H), 1.68 (s, 6H), 2.38 (s, 3H), 5.05-5.16 (m, 2H), 5.21 (q, J=6.90 Гц, 1H), 7.25-7.31 (m, 2H), 7.35 (dd, J=8.98, 1.95 Гц, 1H), 7.41 (d, J=8.20 Гц, 1H), 7.44 (d, J=8.98 Гц, 1H), 8.14 (s, 1H). MS [М+Н], вычислено: 413,2, найдено: 413,3.
Фармакология:
Биологическая оценка TRPV1 человека, FLIPR™ анализ мобилизации кальция.
Активность соединений по настоящему изобретению (IC50) измеряют с помощью анализа визуализации с использованием 384-луночного планшета, который контролирует индуцированный лекарственным средством уровень внутриклеточного Са2 в целых клетках. Активацию hTRPV1 (человеческий рецептор V1 для кратковременного потенциала (human Transient Potential Receptor V1), соответствует инвентарному номеру: ААМ89472 со следующей модификацией: вместо фенилаланина в положении 589 присутствует лейцин), рецепторов, экспрессирующихся в клетках НЕК T-Rex (эмбриональная почка человека, тетрациклин-регулируемые клетки), количественно определяли в устройстве Molecular Devices FLIPR II™ по увеличению флюоресцентного сигнала. Ингибирование hTRPV1 соединениями определяют по уменьшению флюоресцентного сигнала в ответ на активацию 20 нМ капсаицином. hVR1-индуцибельные клетки HEK T-Rex выращивают в 1Х среде Игла в модификации Дульбекко с добавками (DMEM, Wisent, 319-005-CL) с 10% эмбриональной бычьей сыворотки (Wisent, 090850), 2 мМ L-глутамина (Wisent, 609-065-EL), 5 мкг/мл бластицидина S HCL (Invitrogen R-210-01) и 350 мкл/мл зеоцина (Invitrogen R-250-05). Клетки сеют в 384-луночный черный планшет, покрытый полилизином (falcon, BD) в концентрации 10000 клеток/лунку/50 мкл на 16 часов или 5500 клеток/лунку/50 мкл на 48 часов во влажном инкубаторе (5% СО2 и 37°С) в среде DMEM без селективного агента. Клетки HEK T-Rex hVR1 индуцировали 0,1 мкг/мл тетрациклина (Invitrogen, 550205) за 16 часов до эксперимента. В день эксперимента среды удаляют из планшетов с клетками путем переворачивания. В каждую лунку добавляют загрузочный раствор 30 мкл сбалансированного солевого раствора Хэнкса, 1 мМ CaCl2 и 5 мМ глюкозы, рН 7,4 (Wisent, 311-520-VL,) с индикаторным красителем кальция FLUO-4 AM 4 мкМ (Molecular Probes F14202) и Pluronic F-127 0,004% (Invitrogen P3000MP), используя раскапыватель Labsystems multidrop. Планшеты инкубируют при 37°С в течение 30-40 минут до начала эксперимента. Инкубацию завершают путем промывки клеток 4 раза в буфере для анализа с использованием Skatron Embla (Moleculare Devices corp), оставляя остаточный объем 25 мкл буфера/лунку. Планшеты с клетками затем переносят в FLIPR, готовые к добавлениям соединения.
В день эксперимента капсаицин и соединения разбавляют в трехкратном концентрационном диапазоне (10-точечное последовательное разведение) для добавления с помощью FLIPR. Для всех анализов кальция берутся фоновые показания в течение 10 секунд, затем добавляют 12,5 мкл соединения, получая общий объем лунки 37,5 мкл. Данные собирают каждую секунду для 60 изображений и затем каждые 10 секунд для 23 изображений перед добавлением агониста в течение общего периода времени 300 секунд. Перед добавлением агониста берут второе фоновое показание в течение 10 секунд, затем добавляют 12,5 мкл агониста или буфера, получая конечный объем 50 мкл. После стимуляции агонистом FLIPR продолжает собирать данные каждую секунду для 60 изображений и затем каждые 10 секунд для 21 изображения в течение общего периода времени 280 секунд. Излучение флюоресценции считывают с использованием фильтра 1 (излучение 520-545 нм) с помощью CCD-камеры на FLIPR.
Соединения, обладающие антагонистическими свойствами в отношении hVR1, будут ингибировать увеличение внутриклеточного кальция в ответ на добавление капсаицина, что приводит, таким образом, к уменьшению флюоресцентного сигнала. Данные экспортируют с помощью программы FLIPR в виде суммы флюоресценции, рассчитанной под кривой после добавления капсаицина. Данные анализируют с использованием сигмоидальных соответствий нелинейной программы вычерчивания кривых по точкам (XLfit версия 5.0.6 от ID Business Solutions Limited, Guildford, UK). Для каждого соединения получают данные в отношении максимального ингибирования, экстремума-наклона и IC50.
Были получены следующие результаты.
Список сокращений:
Примеры фармацевтических композиций
Соединение по изобретению просеивают и смешивают с лактозой, кукурузным крахмалом и предварительно желатинизированным кукурузным крахмалом. В очищенной воде растворяют полисорбат 80. К смеси добавляют подходящие объемы полисорбата 80 и смесь гранулируют. После сушки гранулы просеивают и смешивают со стеаратом магния. Затем гранулы прессуют в таблетки.
Исходные компоненты отвешивают и затем просеивают. Действующее вещество (1) перемешивают с исходными компонентами (2) и (3) в течение 30 минут и затем измельчают. Полученную смесь перемешивают с компонентами (4) и (5) в течение 15 минут. Затем полученной смесью заполняют твердые желатиновые капсулы подходящего размера, которые затем упаковывают надлежащим образом.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| 2, 5-ДИОКСОИМИДАЗОЛИДИН-4-ИЛ-МЕТИЛКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2003 |
|
RU2326117C2 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| ИМИДАЗОПИРИДИНИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2020 |
|
RU2833052C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ МИМЕТИКОВ ТРО | 2006 |
|
RU2385865C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2298550C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| МОДУЛЯТОРЫ АКТИВНОСТИ НЕС1 И СПОСОБЫ ДЛЯ НИХ | 2011 |
|
RU2576036C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2419608C2 |
Описываются новые производные бензимидазола общей формулы I
где R1=CN, галоген или С(=O)СН3; R2 означает метил или Н; R3=Н или галоген; R4 и R5 независимо означают метил или этил, или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют С3-6циклоалкил или 5-, 6-членный гетероциклоалкил; R6 и R7 независимо означают Н, галоген, метил или этил; или их фармацевтически приемлемые соли, фармацевтические композиции, содержащие эти соединения, и их применение в терапии. Данные соединения могут применяться при лечении остеоартрита, хронического тендинита, тазовой боли и периферической невропатии, гастроэзофагеальной рефлюксной болезни, синдрома раздраженного кишечника и сверхактивного мочевого пузыря. 29 н. и 10 з.п. ф-лы, 34 прим.
1. Соединение формулы
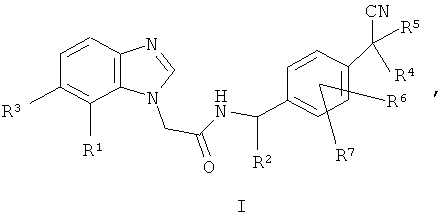
где R1 выбран из CN, галогена или С(=O)СН3;
R2 выбран из метила или Н;
R3 выбран из Н или галогена;
каждый из R4 и R5 независимо выбран из метила или этила, или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-6-членную циклоалкильную или 5-или 6-членную гетероциклоалкильную группу;
каждый из R6 и R7 независимо выбран из Н, галогена, метила или этила;
или его фармацевтически приемлемая соль;
где соединение формулы I не является следующим:
N-[4-(1-циано-1-метилэтил)бензил]-2-(6,7-дифтор-1Н-бензимидазол-1-ил)-ацетамид;
2-(7-хлор-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-3-фторбензил]ацетамид;
(+)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклогексил)фенил]этил}ацетамид;
(+)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(+,-)-2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}ацетамид;
(+)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид;
(+)-N-{1-[4-(1-цианоциклогексил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(+)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид;
(+)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(+)-N-{1-[4-(1-цианоциклобутил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(R)(+)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид;
(R)(+)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид;
(+)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид;
(+)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамид;
(R)(+)-N-{1-[4-(1-циано-1-этилпропил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид.
2. Соединение, выбранное из:
(S)(-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида;
(S)(-)-N-{1-[4-(1-циано-1-этилпропил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[4-(1-цианоциклобутил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[4-(1-цианоциклогексил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклогексил)-фенил]этил}ацетамида;
(-)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-фенил]этил}ацетамида;
(S)(-)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(-)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-2-(7-хлор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-3-фторбензил]ацетамида;
(-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамида;
(S)(-)-2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)-N-{[4-(1-циано-1-метилэтил)фенил]этил}ацетамида;
или его фармацевтически приемлемая соль.
3. Соединение, выбранное из:
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-фенил]этил}ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(7-хлор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-циано-1-этилпропил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-цианоциклобутил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-цианоциклогексил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклогексил)-фенил]этил}ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклопропил)-фенил]этил}ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-цианоциклобутил)-фенил]этил}ацетамида;
(+,-)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-фенил]этил}ацетамида;
(+,-)-2-(7-ацетил-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(+,-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида;
(+,-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-фторфенил]этил}ацетамида;
(+,-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(+,-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(+,-)-N-{1-[4-(4-цианотетрагидро-2Н-тиопиран-4-ил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
N-[4-(1-циано-1-метилэтил)-2-метилбензил]-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
2-(7-циано-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-2-метилбензил]ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[4-(1-циано-1-метилэтил)-2-метилфенил]этил}-2-(7-фтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[3-хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-N-{1-[3-хлор-4-(1-циано-1-метилэтил)фенил]этил}-2-(7-циано-1Н-бензимидазол-1-ил)ацетамида;
(+,-)-2-(7,6-дифтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
(+,-)-2-(7-циано-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)-3-метилфенил]этил}ацетамида;
2-(6-хлор-7-фтор-1Н-бензимидазол-1-ил)-N-[4-(1-циано-1-метилэтил)-2-метилбензил] ацетамида;
или его фармацевтически приемлемая соль.
4. Соединение (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамид или его фармацевтически приемлемая соль.
5. Соединение (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид или его фармацевтически приемлемая соль.
6. Соединение по п.1 для применения в лечении ноцицептивных болевых расстройств у млекопитающего.
7. Соединение по п.1 для применения в лечении хронических ноцицептивных болевых расстройств у млекопитающего.
8. Соединение по п.1 или его фармацевтически приемлемая соль для применения в лечении остеоартрита.
9. Соединение по п.1 или его фармацевтически приемлемая соль для применения в лечении тендинита.
10. Соединение по п.1 или его фармацевтически приемлемая соль для применения в лечении хронического тендинита.
11. Соединение по п.1 или его фармацевтически приемлемая соль для применения в лечении тазовой боли.
12. Соединение по п.1 или его фармацевтически приемлемая соль для применения в лечении периферической невропатии (главным образом PHN (постгерпетической невралгии)).
13. Соединение по п.1 или его фармацевтически приемлемая соль для применения в лечении гастроэзофагеальной рефлюксной болезни (GERD).
14. Соединение по п.1 или его фармацевтически приемлемая соль для применения в лечении синдрома раздраженного кишечника (IBS).
15. Соединение по п.1 или его фармацевтически приемлемая соль для применения в лечении гиперактивного мочевого пузыря.
16. Способ лечения ноцицептивных болевых расстройств, включающий введение эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
17. Способ лечения ноцицептивных болевых расстройств, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1H-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
18. Способ лечения ноцицептивных болевых расстройств, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
19. Способ лечения хронических ноцицептивных болевых расстройств, включающий введение эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
20. Способ лечения хронических ноцицептивных болевых расстройств, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1 метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
21. Способ лечения хронических ноцицептивных болевых расстройств, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
22. Способ лечения остеоартрита, включающий введение эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
23. Способ лечения остеоартрита, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
24. Способ лечения остеоартрита, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
25. Способ лечения тендинита, включающий введение эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
26. Способ лечения тендинита, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
27. Способ лечения тендинита, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
28. Способ лечения хронического тендинита, включающий введение эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
29. Способ лечения хронического тендинита, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
30. Способ лечения хронического тендинита, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
31. Способ лечения тазовой боли, включающий введение эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
32. Способ лечения тазовой боли, включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
33. Способ лечения тазовой боли, включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
34. Способ лечения периферической невропатии (главным образом PHN), включающий введение эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
35. Способ лечения периферической невропатии (главным образом PHN), включающий введение эффективного количества соединения (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
36. Способ лечения периферической невропатии (главным образом PHN), включающий введение эффективного количества соединения (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамида или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
37. Фармацевтическая композиция, обладающая ингибиторным действием в отношении ванилоидного рецептора 1 (VR1), содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
38. Фармацевтическая композиция, обладающая ингибиторным действием в отношении ванилоидного рецептора 1 (VR1), содержащая соединение (S)(-)-2-(7-хлор-6-фтор-1Н-бензимидазол-1-ил)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-ацетамид или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
39. Фармацевтическая композиция, обладающая ингибиторным действием в отношении ванилоидного рецептора 1 (VR1), содержащая соединение (S)(-)-N-{1-[4-(1-циано-1-метилэтил)фенил]этил}-2-(6,7-дифтор-1Н-бензимидазол-1-ил)ацетамид или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
| WO 23006033620 A1, 30.03.2006 | |||
| ГРУЗОВАЯ ТЕЛЕЖКА ПОДВЕСНОГО ТОЛКАЮЩЕГОКОНВЕЙЕРА | 0 |
|
SU250031A1 |
| RU 2005136529, 10.06.2006. | |||

