Изобретение относится к новым производным пиримидин нуклеозида, обладающим превосходной противоопухолевой активностью.
До настоящего времени в качестве коммерчески доступных противоопухолевых средств пиримидинового ряда, представляющих антагонистические средства метаболизма, известны такие, как 5-фтороурацил (Duschinsky. R., et al., J. Am. Chem. Soc. 79, 4559, (1957)), Tegafur (Hiller, SA. et al., Dokl. Akad. Nauk USSR, 176, 332, (1967)), UFT (Fujii, S., et al., Gann, 69, 763, (1978)), Carmofur (Hoshi, A., et al., Gann, 67, 725, (1976)), Doxyfluride (Cook, A.F., et al., J. Med. Chem., 22, 1330, (1979)), Cytarabine (Evance, J.S., et al., Proc. Soc. Exp. Bio. Med., 106, 350, (1961)), Ancytabine (Hoshi, A., et al., Gann, 63, 353, (1972)), Enocytabine (Aoshima, M., et al., Cancer Res., 36, 2726 (1976)) и т.д.
Среди мононуклеозидов пиримидина с цианогруппой в рибозной группировке известны только 3-цианотимин нуклеозид и 3-цианоурацил нуклеозид (Непроверенная патентная публикация Японии N Hei-2-83392, Hei-2-104586 и Hei-2-503002).
Настоящие изобретатели на протяжении длительного времени проводили длительные исследования с целью разработки абсолютно новых антиметаболитов, которые бы превосходили существующие противоопухолевые средства, описанные выше, для того чтобы найти те соединения, у которых цианогруппа введена в 2-положение сахарной части молекул пиримидинового ряда нуклеозидов, которые обладают сильной противоопухолевой активностью в отношении различных опухолевых систем и которые могут быть промежуточными продуктами при получении таких соединений, которые бы обладали сильной противоопухолевой активностью, и они завершили это изобретение.
Новые производные пиримидин нуклеозида, обладающие сильной противоопухолевой активностью, согласно настоящему изобретению имеют следующую общую формулу
или общую формулу
к ним относятся также фармакологически приемлемые нетоксичные соли соединений этих общих формул.
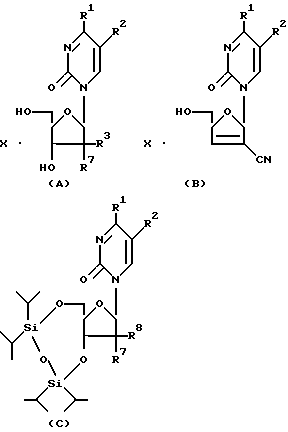
В вышеприведенных общих формулах (1) и (2) R1 - гидроксильная группа или аминогруппа, которая возможно имеет заместитель, выбранный из следующей группы A или B; R2 - атом водорода или алкильная группа, имеющая от 1 до 4 атомов углерода; R3 - атом водорода или гидроксильная группа; а R4 и R5 каждый - атом водорода или вместе могут образовать группу -R6R7Si-O-SiR6'R7' - (в которой R6, R7, R6' и R7' могут быть одинаковыми или отличаться один от другого и каждый представляет алкильную группу, имеющую от 1 до 4 атомов углерода).
(Группа A)
Представляет алифатический ацил, имеющий от 1 до 4 атомов углерода, и ароматический ацил, имеющий от 7 до 11 атомов углерода, и который может иметь заместитель в кольце.
(Группа B)
Представляет алкоксикарбонильную группу с C1-C4 алкилом, алкенилоксикарбонильную группу с C2-C4 алкенилом, аралкилоксикарбонильную группу, имеющую от 8 до 12 атомов углерода и которая может иметь заместитель в кольце.
Алифатический ацил, имеющий от 1 до 4 атомов углерода, в качестве заместителя R1, упомянутого выше, включает формил, ацетил, пропионил, бутирил и изопропионил, предпочтителен алифатический ацил, имеющий от 1 до 2 атомов углерода. Ароматический ацил, имеющий от 7 до 11 атомов углерода, включает бензоил, α-нафтоил, β-нафтоил, предпочтителен бензоил. Заместитель в ароматическом кольце включает алкил, имеющий от 1 до 4 атомов углерода, алкоксид, имеющий 1-4 атома углерода, предпочтительны метил, этил, метокси, этокси- и ацетильная группы. Алкильная группировка алкоксикарбонильной группы, имеющей C1-C4 алкильную группу, включает метил, этил, н-пропил, изопропил, н-бутил, изобутил и трет. -бутил, предпочтительны метил и трет.- бутил. Алкенильная группировка алкенилоксикарбонильной группы, имеющей C2-C4 алкенил, включает винил, аллил, изопропенил, 1-бутенил и 2-бутенил, предпочтителен аллил. Аралкильная группировка аралкилоксикарбонильной группы, имеющей от 8 до 12 атомов углерода, включает бензил, фенэтил, α-нафтилметил и β-нафтилметил, предпочтителен бензил. Заместитель в ароматическом кольце включает алкил, имеющий от 1 до 4 атомов углерода, алкоксил, имеющий от 1 до 4 атомов углерода и алифатическую ацилоксигруппу, имеющую от 1 до 4 атомов углерода, предпочтительны метил, этил, метокси, этокси и ацетоксигруппы.
R1, упомянутый выше, предпочтительно включает гидроксильную группу, аминогруппу, аминогруппу, замещенную C1-C2 алифатическим ацилом, аминогруппу, замещенную ароматическим ацилом, имеющим 7 атомов углерода, который может иметь заместитель в кольце, аминогруппу, замещенную алкоксикарбонильной группой, имеющей от 1 до 4 атомов углерода, аминогруппу, замещенную алкенилоксикарбонильной группой, имеющей C3 алкенил, аминогруппу, замещенную аралкилоксикарбонильной группой, имеющей 8 атомов углерода, которая может иметь заместитель в кольце, более предпочтительны гидроксильная группа, аминогруппа, аминогруппа, замещенная алифатическим ацилом, имеющим 1-2 атома углерода, аминогруппа, замещенная ароматическим ацилом, имеющим 7 атомов углерода, наиболее предпочтительны аминогруппа и гидроксильная группа.
R2 алкильная группа, имеющая 1-4 атома углерода, включает метил, этил, пропил, изопропил, бутил, изобутил и трет.-бутил, предпочтителен метил.
R2, упомянутый выше, предпочтительно включает атом водорода и метил.
R6, R6', R7 или R7'-алкильные группы, имеющие 1-4 атомов углерода, включают метил, этил, пропил, изопропил, бутил, изобутил и трет.-бутил, предпочтителен изопропил.
R4 и R5, упомянутые выше, каждый представляет атом водорода или вместе образуют тетраметилдисилоксидиильную группу, тетраэтилдисилоксидиильную группу, тетрапропилдисилоксидиильную группу, тетраизопропилдисилоксиильную группу, тетрабутилдисилоксдиильную группу, диэтилдиизопропилдисилоксдиильную группу, или дибутилдиизопропилдисилоксдиильную группу, предпочтительны атом водорода или тетраизопропилдисилоксдиильная группа, более предпочтителен атом водорода.
Фармакологически приемлемые нетоксичные соли соединений, имеющих вышеприведенные общие формулы (1) или (2) настоящего изобретения, могут быть представлены солями минеральных кислот, таких как хлориды, бромиды и сульфаты, органическими сульфонатами, такими как метансульфонат, бензолсульфонат, алифатическими карбоксилатами, такими как ацетат, пропионат, бутират и капроат, и ароматическими карбоксилатами, такими как бензоат.
Среди перечисленных солей соли минеральных кислот (в частности хлористоводородной кислоты) и алифатические карбоксилаты (в частности уксусной кислоты) предпочтительны.
В соединениях (1) и (2) можно предпочтительно упомянуть:
1) соединения, в которых R1 представляет гидроксильную группу или аминогруппу, которая может иметь заместитель, выбранный из следующей группы A' или B'; R2 представляет атом водорода или алкильную группу, имеющую 1-4 атомов углерода; R3 представляет атом водорода или гидроксильную группу; R4 и R5 каждый представляет атом водорода или они вместе образуют тетраизопропилдисилоксдиильную группу.
(Группа A') представляет алифатический ацил, имеющий 1-2 атома углерода и ароматический ацил, имеющий 7 атомов углерода, который может иметь заместитель в кольце.
(Группа B') представляет алкоксикарбонильную группу, имеющую C1-C4 алкил, алкенилоксикарбонильную группу, имеющую C3 алкенил и аралкилоксикарбонильную группу, имеющую 8 атомов углерода, которая может иметь заместитель в кольце;
2) соединения, в которых R1 представляет гидроксильную группу или аминогруппу, которая может иметь заместитель, выбранный из следующей группы A; R2 представляет атом водорода или метильную группу; R3 представляет атом водорода или гидроксильную группу; а R4 и R5 каждый представляет атом водорода.
(Группа A') представляет алифатический ацил, имеющий 1-2 атома углерода и ароматический ацил, имеющий 7 атомов углерода, который может иметь заместитель в кольце;
3) соединения, в которых R1 представляет гидроксильную группу или аминогруппу; R2 представляет атом водорода или метильную группу; R3 представляет атом водорода или гидроксильную группу; а R4 и R5 каждый представляет атом водорода.
Соединения (1) и (2) настоящего изобретения, как правило, могут быть представлены примерами, приведенными в табл. 1-3, но настоящее изобретение не ограничивается этими примерами.
Табл. 1-3 иллюстрируют соединения формулы A, соединения формулы B и соединения формулы C. В табл. 1-3 Et, Pr, t.Bu, Al, Bz, BzpMe, BzpOMe, By и ByOAc означают этильную группу, пропильную группу, трет.-бутильную группу, аллильную группу, ацетильную группу, бензоильную группу, пара-метилбензоильную группу, пара-метоксибензоильную группу и пара-ацетоксибензильную группу соответственно.
Соединения (1) и (2) настоящего изобретения могут быть получены при использования урацила или 5-низший алкилурацила, известного соединения (3), (M. Muraoka, A. Tanaka and T. Veda, Chem. Pharm. Bull, 18, 261, (1970)), следуя реакционным стадиям, изображенным на реакционных схемах 1- 4 (см. в конце описания). В схемах 1-4 R1 и R2 имеют значения, которые приведены выше, R4a и R5a вместе представляют группу формулы: -R6R7Si-O-SiR6'R7'-, в которой R6, R7, R6' и R7' имеют значения, определенные выше. R9 представляет алкокситиокарбонильную группу, имеющую C1-C4 алкил или арилокситиокарбонильную группу, имеющую C6-C10 арил. Алкил, имеющий 1-4 атома углерода, включает метил, этил, пропил, бутил и т.д., а арил, имеющий 6-10 атомов углерода, включает фенил, нафтил, и т.д. предпочтительны метил и соответственно фенил. R10 представляет триарилметильную группу, в которой арильная группировка может быть замещена и которая включает фенил, нафтил и т.д., предпочтителен фенил. Заместитель для арильной группировки включает алкильную группу, имеющую 1-4 атома углерода, такую как метил, этил, пропил и бутил, алкоксигруппу, имеющую 1-4 атома углерода, такую как метокси, этокси, пропокси и бутокси, и ацилокси группу, имеющую 2-4 атома углерода, такую как ацетокси, пропилокси и бутирилокси, предпочтительны метильная и метоксильная группы. X представляет атом галогена, предпочтителен хлор или бром. Более детально соответствующие реакционные стадии объяснены на схемах 1 - 4 (см. в конце текста).
Стадия 1.
Эта стадия относится к получению соединения (4) путем рибозилирования соединения (3).
Рибозилирование, как правило, осуществляют способами, обычно используемыми в данной области химии, например, следующим образом: (i) ртутную соль соединения (3), получаемую при добавлении спиртового раствора соли хлорида двухвалентной ртути к водному раствору гидроксида натрия и соединения (3), запускают в реакцию с известными соединением 2,3,5-три-O-бензоил-Д-рибозилхлоридом в бензоле. Метоксид натрия воздействует на полученное соединение, в метаноле при этом образуется соединение (4) (M.Muraoka, A.Tanaka and T. Ueda, Chem. Pharm. Bull, 18, 261, (1970)), а (ii) соединение (3) обрабатывают триметилсилилхлоридом в бензоле в присутствии органического амина, такого как триэтиламин, с целью получения бис(триметилсилил)урацила, который затем обрабатывают 2,3,5-три-О-бензоил-Д-рибозил хлоридом, а затем полученное соединение обрабатывают раствором метоксида натрия в метаноле для получения соединения (4) [T.Nishimura, B.Shimiru and I.Iwai, Chem. Pharm. Bull., 11, 1470, (1963)].
Стадия 2.
Эта стадия относится к получению соединения (5) за счет превращения карбонильной группировки в 4-положении соединения (4) в аминогруппу. Превращение в аминогруппу обычно проводят с использованием традиционных методов, известных специалистам в данной области, например, следующим образом:
(i) гексаметилдисилазан и сульфат аммония оставляют взаимодействовать с соединением (4) при нагревании в безводном формамиде с целью получения соединения (5) [Compiled by Townsend and Tipson, Nucleic Acid Chemistry, 227, (1978)].
(ii) Гидроксильные группы в положении 2'-, 3'-, и 5'-соединения (4) защищают путем ацилирования или бензоилирования. Тионилхлорид и безводный диметилформамид воздействуют в хлороформе, не содержащем спирта, на полученное соединение, а затем смесь обрабатывают метанольным раствором аммиака для получения соединения (5) [Compiled by Townsend and Tipson, Nucleic Acid Chemistry, 223, (1978)].
(iii) Гидроксильные группы в положениях 2'-, 3'- и 5' соединения (4) защищают путем ацилирования или бензоилирования, затем воздействуют на защищенное производное дифосфопентасульфидом в пиридине и получают 4-тиосоединение. На полученное соединение воздействуют низшим алкил йодидом, таким метил йодид и этил йодид, и гидроксидом щелочного металла, таким как гидроксид натрия, с целью получения 4-алкилтио соединения как промежуточного продукта. Далее 4-алкилтио соединение обрабатывают жидким аммиаком с целью получения соединения (5) [J.J. Fox, N. Miller and I.Wenpen, Journal of Medicinal Chemistry, 9, 101 (1966)].
Стадия 3.
В этой стадии соединение X-R6R7Si-O-SiR6'R7' воздействует на 3'- и 5'-положения соединения (4), которые в это же время защищаются, с целью получения соединения (5). Эта стадия проводится в соответствии с известным методом [M. J. Robins, J. S. Wilson, L. Sawyer and M.N.G. James, Can. J. Chem. 61, 1911, (1983)].
Поскольку используется растворитель, то предпочтительно упомянуть основной растворитель, такой как пиридин.
Реакцию проводят при температуре от -10 до 100oC, предпочтительно от 0 до 50oC.
Хотя длительность реакционного процесса варьируется в зависимости от температуры, при которой проводят реакцию, и от соединения, оно обычно составляет от 1 до 30 ч, предпочтительно от 1 до 5 ч.
После завершения реакции, например, отгоняют растворитель, и реакционную смесь выливают в воду, Образовавшуюся смесь экстрагируют растворителем, не смешивающимся с водой, таким как бензол, простой эфир и этилацетат, а затем растворитель отгоняют и получают соединение. Полученное таким образом соединение используют в последующей стадии. Если необходимо, соединение может быть очищено методом хроматографии или путем перекристаллизации.
Стадия 3'.
Эта стадия включает ацилирование аминогруппы в 4-положении соединения (5) и обработку таким образом ацилированного соединения X-R6R7Si-O-SiR6'R7' - X с одновременной защитой 3' и 5' - положений этого ацилированного соединения с целью получения соединения (6). 3'- и 5' - положения могут быть защищены таким же образом, как и в стадии (3).
Ацилирование аминогруппы в 4-положении осуществляют способом, как правило, обычно используемым специалистами в данной области. Например, в случае алифатического ацила или ароматического ацила реакционноспособное производное соответствующей карбоновой кислоты, такое как галоиданигидрид или ангидрид кислоты оставляют взаимодействовать или соответствующую карбоновую кислоту оставляют взаимодействовать в присутствии конденсирующего средства; или в случае алкоксикарбонила, алкенилоксикарбонила или аралкилоксикарбонила в реакцию запускают эфир галогеномуравьиной кислоты, имеющий соответствующую алкокси-, алкенилокси- или аралкилоксигруппу, или в реакцию запускают диалкилдикарбонат, диалкенил дикарбонат или диаралкил дикарбонат, имеющий соответствующий алкил, алкенил или аралкил.
В качестве галоидангидрида кислоты можно упомянуть, например, хлорангидриды или бромангидриды.
В качестве используемого конденсирующего средства могут быть, например, упомянуты N,N'-дициклогексилкарбодиимид (ДСС), 1,1-оксалилдиимидазол, 2,2-дипиридилдисульфид, N, N'-дисукцинимидил карбонат, N,N'-бис(2-оксо-3-оксазолидинил)-фосфинохлорид, N,N'-карбодиимидазол, N,N'-дисукцинимидил оксалат (DSO), N,N'-дифталимид оксалат (ДФО), N,N'-бис(норборненилсукцинимидил)оксалат (BNO), 1,1'-бис(бензотриазолил)оксалат (BBTO), 1,1'-бис(6-хлоробензотриазолил)оксалат (BCTO), 1,1'-бис(6-трифторометилбензотриазолил)оксалат (BTBO) и др.
Используемый растворитель не имеет, в частности ограничений, если только он не ингибирует реакцию, и включает ароматические углеводороды, такие как бензол, толуол и ксилол, простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан, и диметиловый эфир диэтиленгликоля, спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет. -бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, оксанол, циклогексанол, и метилцеллозольв, кетоны, такие как ацетон, метилэтилкетон, метилизобутил кетон, изофорон, и циклогексанон, нитрилы, такие как ацетонитрил, изобутиронитрил, амиды, такие как формамид, диметилформамид, диметилацетамид и гексаметилфосфотриамид, сульфоксиды, такие как диметилсульфоксид, сульфолан, и смеси на основе этих растворителей и воды, предпочтительны ароматические углеводороды, такие как бензол, толуол, ксилол, простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля, спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет. -бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол, и метилцеллозольв, кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон, и циклогексанон, нитрилы, такие как ацетонитрил, изобутиронитрил, амиды, такие как формамид, диметилформамид, диметилацетамид и гексаметилфосфотриамид, сульфоксиды, такие как диметилсульфоксид и сульфолан, и смешанные растворители этих органических растворителей и воды.
Реакцию проводят в температурном интервале от 0 до 150oC, предпочтительно от 0 до 100oC.
Хотя длительность реакции зависит от соединения, температуры, при которой проводится реакция и т.д., обычно она длится от 1 до 30 ч, предпочтительно, когда время реакции составляет от 2 до 5 ч.
После завершения реакции, например, отгоняют растворитель, а реакционную смесь выливают в воду. Образовавшуюся смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, простой эфир и этилацетат, и растворитель отгоняют от экстракта для получения соединения. Полученное таким образом соединение используют в последующей стадии как таковое. При необходимости соединение может быть выделено и очищено методами хроматографии или путем перекристаллизации.
Стадия 4.
Эта стадия, предназначенная для получения соединения (7), осуществляется путем обработки соединения (6) тиокарбонилирующим реагентом в инертном растворителе за счет тиокарбонилирования путем замещения гидроксильной группы в 2'-положении соединения (5).
Используемый растворитель не имеет ограничений, если он не ингибирует реакцию, и включает амиды, такие как диметилформамид и диметилацетамид, сульфоксиды, такие как диметилсульфоксид, и нитрилы, такие как ацетонитрил, предпочтителен ацетонитрил.
Если реагент, который используется для осуществления реакции, представляет собой тиокарбонилирующий реагент для гидроксильной группы, то он не имеет, в частности ограничений, и включает низший алкоксикарбонилгалид, такой как метокситиокарбонилхлорид, и этокситиокарбонилхлорид, а также арилтиокарбонилгалид, такой как фенокситиокарбонилхлорид и нафтокситиокарбонилхлорид.
Реакцию проводят при температуре от -20 до 50oC, предпочтительно от -10 до 10oC.
Хотя продолжительность реакции зависит от соединения, температуры проведения реакции и т.д., она обычно составляет от 1 до 30 ч, предпочтительно от 2 до 5 ч.
Для более эффективного проведения реакции могут быть использованы органические амины, такие как 4,4-диметиламинопиридин и триэтиламин.
После завершения реакции нужное соединение может быть выделено стандартными способами. Например, реакционную смесь выливают в воду, образовавшуюся смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, простой эфир и этилацетат, а затем растворитель отгоняют для получения нужного соединения. Полученное таким образом соединение обычно используют как таковое без дальнейшей обработки в последующей стадии. Если в этом есть необходимость, то соединение может быть очищено и выделено различными хроматографическими методами или путем перекристаллизации.
Стадия 5.
Эта стадия предназначена для получения соединения (1a), которое представляет собой соединение (1), в котором R4 и R5 вместе образуют группу формулы -R6R7Si-O-SiR6R7-; а R3 представляет атом водорода, и состоит в обработке соединения (7), полученного в стадии 4, восстанавливающим средством и реагентом, вводящим нитрильную группу, в инертном растворителе.
Используемый растворитель, в частности не ограничивается в выборе, если он не ингибирует реакцию, и включает алифатические углеводороды, такие как гексан, гептан, лигроин, петролейный эфир, ароматические углеводороды, такие как бензол, толуол и ксилол, простые эфир, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля, предпочтительны ароматические углеводороды, такие как бензол и толуол.
В качестве реагента, используемого для введения нитрильной группы, используют предпочтительно алкилизонитрилы, такие как трет.-изобутиронитрил, а восстанавливающий реагент предпочтительно включает гидрид триалкилолова, имеющий в алкильной группировке 1-4 атома углерода, например, трибутилолова гидрид.
Реакцию обычно проводят при температуре от 50 до 250oC, предпочтительно от 80 до 150oC. Хотя длительность реакции варьируется в зависимости от соединения, температуры реакции и т.д., оно обычно составляет от 30 мин до 12 ч, в предпочтительных случаях оно варьируется от 1 ч до 5 ч.
Для более эффективного проведения реакции в качестве катализатора может быть использован радикальный инициатор, такой как азобисизобутиронитрил.
Соединение (1a), полученное непосредственно после завершения реакции этой стадии, - это смесь соединений, в которых координация нитрильной группы представляет α-координацию и β-координацию соответственно. Такие соединения могут быть подвергнуты обработке методом абсорбционной или ионно-обменной хроматографии, с использованием различных носителей, таких как активированный уголь и силикагель, гель-фильтрации, с использованием колонки с Сефадексом, или перекристаллизации с использованием органического растворителя, такого как простой эфир, этилацетат и хлороформ, для разделения смеси на соответствующие соединения в зависимости от необходимых целей.
Стадия 6.
Эта стадия предназначена для получения соединения (8) и осуществляется путем окисления гидроксильной группы в 2-положении соединения (6) и может быть проведена и использованием известных методов (F. Hansske et al., Tetrahedron 40, 125, (1984)).
Используемый растворитель, в частности не ограничивается до тех пор, пока он не ингибирует реакцию и может растворять исходные вещества до некоторой степени, и он включает ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метилен хлорид, хлороформ; простые эфиры, такие как тетрагидрофуран, диоксан и диметоксиэтан; амиды, такие как диметилформамид, диметилацетамид и гексаметилфосфотриамид; сульфоксиды, такие как диметилсульфоксид, кетоны, такие как ацетон и метилэтилкетон; и нитрилы, такие как ацетонитрил; предпочтителен галогенированный углеводород, такой как метиленхлорид или хлороформ.
Реакцию проводят при комнатной температуре от 0 до 100oC, предпочтительно от 10 до 40oC.
Хотя продолжительность реакции зависит от соединения, температуры проведения реакции и т.д., она обычно составляет от 10 мин до 12 ч предпочтительно от 30 мин до 3 ч.
Между прочим, окисление, о котором шла речь выше, может быть ускорено при добавлении перемещающегося между слоями катализатора, такого как триэтилбензиламмонийхлорид и трибутилбензиламмоннийбромид.
Реакцию обычно проводят при температуре от 0 до 100oC, предпочтительно от 10 до 40oC.
Хотя время реакции варьируется в зависимости от соединения, температуры реакции и т.д., оно обычно составляет от 10 мин до 12 ч, предпочтительно от 30 мин до 3 ч.
Соединение (8), полученное в этой стадии, может быть собрано, разделено и очищено путем сочетания подходящих разнообразных методов. Например, реакционную смесь выливают в воду, образовавшуюся реакционную смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, простой эфир и этилацетат, а затем растворитель отгоняют от экстракта для получения соединения (8). Если в этом есть необходимость, то соединение может быть далее подвергнуто очистке, например, с использованием методов абсорбционной или ионно-обменной хроматографии с использованием различных носителей, таких как активированный уголь и силикагель, гель-фильтрации, с использованием колонки с Сефадексом, или перекристаллизации с использованием органического растворителя, такого как простой эфир, этилацетат и хлороформ.
Стадия 7.
Эта стадия предназначена для получения требуемого соединения (1б) и осуществляется путем обработки цианидом соединения (8) в инертном растворителе в присутствии основания.
Используемый растворитель, в частности не ограничивается до тех пор, пока не ингибирует реакцию, и включает смешанный растворитель на основе алифатического углеводорода, такого как гексан, гептан, лигроин и петролейный эфир и воды; смешанный растворитель на основе ароматического углеводорода, такого как бензол, толуол и ксилол и воды; смешанный растворитель на основе простого эфира, такого как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля и воды; предпочтителен смешанный растворитель на основе простого эфира и воды.
Основание, используемое в данной реакции, в частности не ограничивается, и здесь могут быть упомянуты органические основания и неорганические основания, включающие гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, и карбонаты щелочных металлов, такие как карбонат натрия и карбонат калия, предпочтителен бикарбонат щелочного металла.
Цианид, который используется в реакции, в частности не ограничивается до тех пор, пока он растворим в воде с образованием цианид иона, и предпочтительно включает цианиды щелочных металлов, такие как цианид натрия и цианид калия.
Реакцию обычно проводят при температуре от 0 до 100oC, предпочтительно в температурном интервале 10-40oC.
Хотя продолжительность реакции варьируется в зависимости от соединения, температуры проведения реакции и т.д., обычно она проводится в течение от 30 мин до 96 ч, предпочтительно от 5 до 24 ч.
Соединение (1б), полученное в этой стадии, может быть собрано, выделено и очищено путем сочетания различных методов. Например, реакционную смесь выливают в воду, образовавшуюся смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, простой эфир и этилацетат, а затем растворитель отгоняют из экстракта с получением требуемого соединения. Если в этом есть необходимость, то полученное таким образом соединение может быть далее подвергнуто обработке методами абсорбционной или ионно-обменной хроматографии, с использованием таких носителей, как активированный уголь и силикагель гель-фильтрации с использованием колонки с Сефадексом, или перекристаллизации с использованием органического растворителя, такого, как простой эфир, этилацетат и хлороформ.
Соединение (1б), выделенное непосредственно после завершения реакции, - это смесь соединений, в которых координация нитрильной группы - это α-координация и β-координация соответственною. Такие соединения могут быть подвергнуты в дальнейшем разделению методом адсорбционной или ионно-обменной хроматографии с использованием таких носителей, как активированный уголь и силикагель, гель-фильтрации с использованием колонки с Сефадексом или перекристаллизации с использованием такого органического растворителя, как простой эфир, этилацетат и хлороформ, для выделения из смеси соответствующих соединений в зависимости от требуемых целей.
Стадия 8.
Эта стадия предназначена для получения соединения (9) и осуществляется путем обработки соединения (1б) тиокарбонилирующим реагентом в инертном растворителе для проведения тиокарбонилирования путем замещения гидроксильной группы в 2'-положении соединения (1б), и может быть осуществлено так же, как это было осуществлено в стадии 4.
Стадия 9.
Эта стадия предназначена для получения соединения (1а) методом восстановительного элиминирования тиокарбонилоксигруппы в 2'-положении соединения (9).
Используемый растворитель, в частности не ограничивается до тех пор, пока он не ингибирует реакцию, и включает алифтические углеводороды, такие как гексан, гептан, лигроин и петройлейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля, а также ароматические углеводороды, такие как бензол и толуол; предпочтительны ароматические углеводороды, такие как бензол и толуол.
Используемый реагент включает предпочтительно гидриды триалкил олова, такие как трибутилолово гидрид.
Реакцию обычно проводят при температуре от 50 до 250oC, предпочтительно при температуре кипения используемого растворителя.
Продолжительность реакции обычно составляет от 30 мин до 10 ч, предпочтительно от 30 мин до 3 ч.
Для того чтобы более эффективно провести реакцию, в качестве катализатора может быть использован радикальный инициатор, такой как азобисизобутиронитрил.
Полученное таким образом требуемое соединение может быть собрано, выделено и очищено путем сочетания различных подходящих методов. Например, реакционную смесь выливают в воду, образовавшуюся смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, простой эфир и этилацетат, а затем растворитель отгоняют из экстракта для получения требуемого соединения. Если в этом есть необходимость, то полученное таким образом соединение может быть далее подвергнуто, например, адсорбционной или ионно-обменной хроматографии с использованием различных носителей, таких как активированный уголь и силикагель, гель-фильтрации с использованием колонки с Сефадексом или перекристаллизации с использованием такого растворителя, как эфир, этилацетат и хлороформ.
Соединение (1а), полученное непосредственно после реакции в этой стадии, - это смесь соединений, в которых координация нитрильной группы - это α-координация и β-координация соответственно. Такие соединения могут быть подвергнуты, например, адсорбционной или ионно-обменной хроматографии с использованием различных носителей, таких как активированный уголь и силикагель, гель-фильтрации с использованием колонки с Сефадексом или перекристаллизации с использованием органического растворителя, такого как простой эфир, этилацетат и хлороформ, для разделения этой смеси соединений на соответствующие отдельные соединения в зависимости от поставленной цели.
Стадии 10 и 12.
Эти стадии предназначены для получения нужных соединений (1с) и (1д) и осуществляются путем воздействия элиминирующего средства для R4a и R5a на соединения (1б) и (1а) в инертном растворителе для того, чтобы удалить заместитель при аминогруппе, если в этом есть необходимость.
Хотя растворитель, используемый в реакции элиминирования R4a и R5a, в частности не ограничивается, пока он не ингибирует реакцию, здесь следует упомянуть, что предпочтительны эфиры, такие как тетрагидрофуран и диоксан. Используемый реагент, в частности не ограничивается до тех пор, пока он обычно используется для элиминирования силильной группы, а здесь можно упомянуть один реагент, который образует анион фтора, такой как тетрабутиламмоний фторид.
Реакцию обычно проводят при температуре от 0 до 40oC, предпочтительно при комнатной температуре.
Хотя продолжительность реакции варьируется в зависимости от соединения, температуры, при которой проводится реакция, оно составляет от 10 мин до 24 ч, предпочтительно от 1 ч до 5 ч.
Полученное таким образом соединение может быть собрано, разделено и очищено при сочетании различных подходящих методов. Например, реакционную смесь выливают в воду, экстрагируют образовавшуюся смесь несмешивающимся с водой растворителем, таким как бензол, простой эфир и этилацетат, а затем растворитель отгоняют из экстракта для получения соединения (8). Если в этом есть необходимость, то полученное таким образом соединение может быть подвергнуто дальнейшей обработке, например, адсорбционной или ионно-обменной хроматографии с использованием различных носителей, таких как активированный уголь и силикагель, гель-фильтрации с использованием колонки с Сефадексом или перекристаллизации с использованием органического растворителя, такого как простой эфир, этилацетат и хлороформ.
Между прочим, в случае, когда R2 представляет замещенную аминогруппу, то она может иногда элиминироваться одновременно.
Стадии 11 и 13.
Эти стадии, предназначенные для получения соединений (1с) и (1д) настоящего изобретения, осуществляют под воздействием элиминирующего средства на защитную группу с отщеплением заместителя у R2 в инертном растворителе, и эта стадия выбирается, если в ней есть необходимость.
Хотя элиминирование защитной группировки варьируется в зависимости от защитной группировки, обычно оно осуществляется согласно известному специалистам в данной области следующему методу:
а) в том случае, когда защитная группировка представляет собой алифатическую ацильную группу, ароматический ацил или алкилоксикарбонил, то такие защитные группировки могут быть удалены при обработке кислотой в присутствии или отсутствии растворителя. В качестве используемой кислоты здесь можно упомянуть хлористоводородную кислоту, уксусную кислоту, серную кислоту, фосфорную кислоту и бромистоводородную кислоту, предпочтительна уксусная кислота.
В качестве используемого растворителя здесь можно упомянуть спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет.-бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, и октанол; ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как тетрагидрофуран и диоксан; и смешанный растворитель на основе органического растворителя и воды.
Реакцию проводят при температуре от 0 до 40oC, предпочтительно при комнатной температуре.
Хотя продолжительность реакции варьируется в зависимости от температуры реакции, она составляет от 10 мин до 24 ч, предпочтительно от 1 до 5 ч.
Полученное таким образом требуемое соединение может быть собрано, разделено и очищено путем сочетания различных подходящих методов. Обычно реакционную смесь перегоняют, а остаток подвергают, например, адсорбционной или ионно-обменной хроматографии с использованием различных носителей, таких как активированный уголь и силикагель, гель-фильтрации с использованием колонки с Сефадексом или перекристаллизации с использованием таких растворителей, как простой эфир, этилацетат и хлороформ;
б) в том случае, если защитная группировка представлена аралкилоксикарбонилом, то такая защитная группировка может быть элиминирована путем каталитического восстановления с использованием катализатора.
В качестве используемого растворителя здесь следует упомянуть спирты, такие как метанол, этанол, н-пропанол, изопропанол и н-бутанол, насыщенные углеводороды, такие как гексан и циклогексан, эфиры, такие как тетрагидрофуран и диоксан, и низшие жирные кислоты, такие как уксусная кислота и пропионовая кислота, предпочтительны метанол, этанол, уксусная кислота и пропионовая кислота.
В качестве используемого катализатора здесь можно предпочтительно упомянуть платину и палладий на углероде.
Реакцию проводят при температуре 0 - 40oC, предпочтительно ее проведение при комнатной температуре.
Хотя продолжительность реакции варьируется в зависимости от температуры реакции, она составляет от 10 мин до 24 ч, предпочтительно от 1 ч до 5 ч.
Полученное таким образом требуемое соединение может быть собрано, разделено и очищено путем использования различных подходящих методов. Обычно реакционную смесь перегоняют, а остаток подвергают, например, адсорбционной или ионно-обменной хроматографии с использованием различных носителей, таких, например, как активированный уголь и силикагель, гель-фильтрации с использованием колонки с Сефадексом, или перекристаллизации с использованием органического растворителя, такого как простой эфир, этилацетат и хлороформ.
Стадия 14.
Эта стадия предназначена для получения соединения (10) и осуществляется путем действия защитного реагента на соединение (1д) в инертном растворителе.
В качестве используемого растворителя здесь может быть упомянут основной растворитель, такой как пиридин и нейтральный растворитель, такой как бензол, толуол и простой эфир.
Хотя защитный реагент, который используется в данной реакции, в частности не ограничивается, пока он может специфически защищать только одну гидроксильную группу в 5'-положении, то удобно использовать для этих целей трифенилхлорметан, монометокситритилхлорид, диметокситритилхлорид и т.д.
Реакцию обычно проводят при температуре от 0 до 100oC, предпочтительно от -10 до 50oC.
Продолжительность реакции обычно составляет от 30 мин до 10 ч, предпочтительно от 1 ч до 5 ч.
При использовании нейтрального растворителя в качестве растворителя для более эффективного проведения реакции может быть использован органический амин, такой как триэтиламин.
Полученное таким образом соединение может быть собрано, разделено и очищено путем сочетания различных подходящих методов. Например, реакционную смесь выливают в воду, образовавшуюся смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, простой эфир и этилацетат, а затем растворитель отгоняют из экстракта для получения нужного соединения. Если в этом есть необходимость, то полученные таким образом соединения могут быть подвергнуты, например, адсорбционной или ионно-обменной хроматографии с использованием различных носителей, таких как активированный уголь и силикагель, гель-фильтрации с использованием колонки с Сефадексом или перекристаллизации с использованием органического растворителя, такого как простой эфир, этилацетат и хлороформ.
Стадия 15.
Эта стадия предназначена для получения соединения (11) и осуществляется при действии реагента, элиминирующего гидроксильную группу, на соединение (10) в инертном растворителе.
В качестве используемого растворителя можно упомянуть, например, алифатические углеводороды, такие как гексан, гептан, лигроин и петролинейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан, и диметиловый эфир диэтиленгликоля; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон, и циклогексанон; нитросоединения, такие как нитроэтан, и нитробензол; нитрилы, такие как ацетонитрил и изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид и гексаметилфосфотриамид, и сульфоксиды, такие как диметилсульфоксид и сульфолан; предпочтительны ароматические углеводороды, такие как бензол, толуол и ксилол.
В качестве используемого реагента может быть упомянуто соединение, имеющее тиокарбонильную группу, такое как тиокарбонилдиимидазол, фенокситиокарбонилхлорид.
Реакцию проводят при температуре от -10 до 50oC, предпочтительно при комнатной температуре.
Продолжительность реакции обычно составляет от 1 ч до 24 ч, предпочтительно от 3 до 10 ч.
Полученное таким образом нужное соединение может быть собрано, разделено и очищено путем сочетания подходящих различных методов. Например, реакционную смесь выливают в воду, образовавшуюся смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, простой эфир и этилацетат, а затем растворитель отгоняют из экстракта для получения нужного соединения. Если в этом есть необходимость, полученное таким образом соединение может быть подвергнуто адсорбционной или ионнообменной хроматографии с использованием различных носителей, таких как активированный уголь и силикагель, гель-фильтрации с использованием колонки с Сефадексом или перекристаллизации с использованием органического растворителя, такого как простой эфир, этилацетат и хлороформ.
Полученное таким образом требуемое соединение может быть собрано, разделено и очищено путем сочетания различных подходящих методов. Например, реакционную смесь выливают в воду, образовавшуюся смесь экстрагируют несмешивающимся с водой растворителем, таким как бензол, простой эфир и этилацетат, а затем растворитель отгоняют из экстракта для получения требуемого соединения. Если в этом есть необходимость, то полученное таким образом соединение может быть подвергнуто, например, адсорбционной или ионно-обменной хроматографии с использованием различных носителей, таких как активированный уголь и силикагель, гель-фильтрации с использованием колонки с Сефадексом или перекристаллизации с использованием органического растворителя, такого как простой эфир, этилацетат и хлороформ.
Стадия 16.
Эта стадия предназначена для получения требуемого соединения (2a) настоящего изобретения и осуществляется действием средства, снимающего защиту с гидроксильной группы соединения (11) в инертном растворителе.
Хотя элиминирование защитной группировки варьируется в зависимости от природы защитной группировки, обычно ее осуществляют по методу, который известен специалистам в данной области, в том случае, когда защитная группа представляет собой триарилметил, который является предпочтительной защитной группировкой, ее элиминирование проводят следующим образом.
Хотя используемый растворитель, в частности не ограничивается в этой реакции, он предпочтительно включает спирты, такие как метанол, этанол; простые эфиры, такие как тетрагидрофуран и диоксан; и смешанный растворитель на основе органического растворителя и воды.
В качестве реагента обычно используют кислоту. Кислота, в частности не ограничивается, пока используемая кислота представлена кислотой Бренстеда, и предпочтительно включает неорганическую кислоту, такую как хлористоводородная кислота, и серная кислота, и органическую кислоту, такую как уксусная кислота и пара-толуолсульфокислота. Дополнительно, сильная кислотная катионнообменная смола типа Dowex 50 может быть использована.
Реакцию обычно проводят при температуре от 0 до 50oC, предпочтительно при комнатной температуре.
Хотя продолжительность реакции зависит от исходных материалов типа кислот, которые используются в реакции и т.д., она обычно протекает от 10 мин до 18 ч, предпочтительно, когда время реакции составляет от 30 мин до 5 ч.
Полученное таким образом требуемое соединение может быть собрано, разделено и очищено при сочетании различных подходящих методов. Например, реакционную смесь выливают в воду, образовавшуюся смесь экстрагируют несмешивающимся с водой органическим растворителем, таким как бензол, эфир и этилацетат, а затем растворитель отгоняют из экстракта для получения требуемого соединения. Если в этом есть необходимость, то полученное таким образом соединение подвергают адсорбционной или ионно-обменной хроматографии, используя различные носители, такие как активированный уголь и силикагель, гель-фильтрацию, используют колонку с Сефадексом или перекристаллизацию, используя такой органический растворитель, как простой эфир, этилацетат и хлороформ.
Настоящее изобретение далее иллюстрируется примерами, сравнительными примерами и примерами приготовления лекарственных форм. TIPDS означает (1,1,3,3-тетраизопропилдисилокс-1,3-диил).
Пример 1. 1-[2'-Циано-3',5'-О-(1,1,3,3- тетраизопропилдисилокс-1,3-диил)-β-D-рибофуранозил]тимин.
В 15 мл смеси эфир-вода (2 : 1) растворили 997 мг 1-(3,5-0-TIPDS-β-D-эритро-пентофуран-2-урозил)тимина, к полученному раствору добавили 196 мг цианида натрия и 336 мг гидрокарбоната натрия, образовавшуюся смесь перемешивали при комнатной температуре в течение 36 ч. После завершения реакции к реакционной смеси добавили этилацетат и эту смесь промыли три раза водой. Слой этилацетата отделили и высушили над безводным сульфатом натрия, а растворитель удалили при упаривании. Остаток очистили методом колоночной хроматографии, использовав колонку с силикагелем (диаметр 2,4 см х 9,5 см). В качестве элюента использовали смесь этилацетата и гексана (1 : 2). После элюирования получили 1,03 г (97,5% выход) соединения, название которого дано в заголовке. Соединение было получено в виде пены белого цвета.
1H ЯМР (CDCl3) δ м.д.: 9.20 и 8.52 (1H, уш.с.); 7.43 и 7.36 (1H, д, J= 6.8 Гц); 6.22 и 6.00 (1H, с); 5.08 (1H, уш.с.); 4.32-3.94 (4H, м); 1.92 (3H, д, J=1.7 Гц); 1.12-1.07 (28H, м).
Пример 2. 1-(2'-Циано-2'-деокси-3',5'-О-TIPDS-β-D-арабинофуранозил)-тимин.
В 2 мл безводного ацетонитрила растворили 100 мг соединения примера 1 и 10 мг 4,4-диметиламинопиридина (обозначаемого здесь далее аббревиатурой ДМАР). К образовавшемуся раствору добавили 39 мкл феноксикарбонилхлорида и 40 мкл триэтиламина при 0oC и в атмосфере аргона, затем смесь перемешивали в течение 3 ч. После завершения реакции добавили этилацетат к реакционной смеси и смесь промыли три раза водой. Смесь высушили над сульфатом натрия и растворитель отогнали. Остаток очистили методом колоночной хроматографии, использовав колонку с силикагелем (диаметр колонки 1,6 и высота 10 см). В качестве элюента использовали смесь метанола и хлороформа (1:99), в результате после элюирования получили 71 мг (73,7%) соединения, название которого дано в заголовке.
1H ЯМР (CDCl3) δ м.д.: 7.36 (1H, д, J=1.2 Гц); 6.28 (1H, д, J=7.3 Гц); 4.67 (1H, дд, J=8.3, 9.3 Гц); 4.17 (1H, дд, J=2.2 Гц, 13.2 Гц); 4.04 (1H, дд, J=2.9, 13.2 Гц); 3.78 (1H, дд, J=2.2, 2.9 Гц); 3.58 (1H, дд, J=7.3, 9.3 Гц); 1.94 (1H, дд, J=1.2 Гц); 1.15-1.04 (28H, м).
Пример 3. 1-(2'-Циано-2'-деокси-β-D-арабинофуранозил)тимин.
При 0oC в токе азота в 3 мл безводного тетрагидрофурана (THF) растворили 178 мг соединения примера 2, к образовавшемуся раствору по каплям добавили 20 мкл уксусной кислоты и 0,70 мл раствора тетрабутиламмония в 1 M-THF, затем смесь перемешивали в течение 1,5 ч. После завершения реакции реакционную смесь сконцентрировали, очистили методом колоночной хроматографии, использовав колонку с силикагелем (диаметр колонки 1,8 и высота 8 см). В качестве элюента использовали смесь этанола и хлороформа (8-10 : 92-90) и кристаллизовали из эфира и этанола, в результате получили 27 мг соединения, название которого дано в заголовке. Соединение получено в виде кристаллов белого цвета.
1H ЯМР (CDCl3) δ м.д.: 11.49 (1H, с); 7.85 (1H, д, J=1.1 Гц); 6.25 (1H, д, J=0.6 Гц); 6.20 (1H, д, J=7.1 Гц); 5.30 (1H, т, J=4.9 Гц); 4.47 (1H, ддд, J= 6.0, 8.2, 8.8 Гц); 3.89 (1H, дд, J=7.1, 8.8 Гц); 3.47 (1H, ддд, J=2.2, 3.3, 8.2 Гц); 3.62 (1H, ддд, J=3.3, 4.9, 11.1 Гц); 1.78 (3H, д, J=1.1 Гц).
Пример 4. 1-(2'-Циано-2'-деокси-3',5'-О-TIPDS-β-D-арабинофуранозил)-N4-бензоилцитозин.
Повторили процедуру примера 1, использовав 294 мг N4-бензоил-1-(3,5-TIPDS-β-D-эритропентофуран-2-урозил)цитозина. Далее были повторены процедуры примера 2 с использованием сырого продукта, в результате получили 174 мг (49,1%) соединения, название которого дано в заголовке. Соединение было получено в виде твердого продукта желтовато-белого цвета.
1H-ЯМР (CDCl3) δ м.д. 8.89 (1H, уш.с); 8.11 (1H, д, J = 7.7 Гц); 7.93-7.45 (5H, м); 7.67 (1H, д, J=7.7 Гц); 6.36 (1H, д, J=6.6 Гц); 4.67 (1H, т, J=8.1 Гц); 4.18 (1H, дд, J=2.9, 13.2 Гц); 3.91 (1H, дд, J=2.9, 2.9, 8.1 Гц); 3.75 (1H, дд, J=6.6, 8.1 Гц); 1.15-1.04 (28H, м).
Пример 5. 1-(2'-Циано-2'-деокси-β-D-арабинофуранозил)-N4-бензоилцитозин.
Повторили процедуру примера 3, использовав 100 мг соединения примера 4, а кристаллизацию провели из метанола, в результате получили 25 мг озаглавленного соединения в виде кристаллов белого цвета.
1H-ЯМР (DMCO-d6) δ м.д.: 11.34 (1H, уш.с); 8.4 (1H, д, J=7.7 Гц); 8.00 (2H, м), 7.66-7.49 (3H, м), 7.42 (1H, д, J=7.7 Гц); 6.29 (1H, д, J=5.5 Гц); 6.25 (1H, д, J=7.1 Гц); 5.28 (1H, уш.с); 4.47 (1H, ддд, J=5.5, 7,1 Гц, 7.7 Гц); 3.94 (1H, дд, J=7.1, 7.7 Гц); 3.86 (1H, ддд, J=2.5, 3.8, 7.1 Гц); 3.79 (1H, уш.д, J=12.5 Гц); 3.65 (1H, уш.д, J=12.5 Гц).
Пример 6.1-(2'-Циано-2'-деокси-3', 5'-О-TIPDS-β-D-арабинофуранозил)-N4-ацетилцитозин.
Были повторены процедуры примера 4 с использованием 2 г N4-ацетил-1-(3,50-0-TIPDS-β-D-эритропентофуран-2-урозил)цитозина; кристаллы, полученные после упаривания растворителя, были собраны путем фильтрации смесью эфир/гексан, и после их очистки получили 703 мг соединения, название которого дано в заголовке. Соединение получено в виде кристаллов белого цвета.
1H-ЯМР (CDCl3) δ м. д.: 9.92 (1H, уш.с); 8.07 (1H, д, J=7.7 Гц); 7.55 (1H, д, J=7.7 Гц); 6.34 (H, д, J=7.0 Гц); 4.63 (1H, т, J=8.8 Гц); 4.18 (1H, дд, J= 2.4 Гц, 13.4 Гц); 4.06 (1H, дд, J=2.7 Гц, 13.4 Гц); 3.89 (1H, дд, J= 2.4, 2.7, 8.8 Гц); 3.72 (1H, дд, J=7.0, 8.8 Гц); 2.30 (3H, с), 1.13-1.03 (28H, м).
Пример 7. 1-(2'-Циано-2'-деокси-β-D-арабинофуранозил)-N4-ацетилцитозин.
Процедуры примера 3 были повторены с использованием 1,07 г соединения примера 6. После очистки кристаллов, полученных путем упаривания растворителя и собранных с помощью фильтрации смесью эфира и гексана, получили 480 мг соединения, название которого дано в заголовке. Соединение получено в виде кристаллов белого цвета.
1H-ЯМР (DMCO-d6) δ м.д.: 10.97 (1H, уш.с); 8.36 (1H, д, J=7.7 Гц); 7.26 (1H, д, J=7.7 Гц); 6.27 (1H, д, J=6.1 Гц); 6.22 (1H, д, J=7.1 Гц); 5.24 (1H, уш. с); 4.43 (1H, дд, J=6.1, 7.1, 7.1 Гц); 3.92 (1H, т, J=7.1 Гц); 3.84 (1H, ддд, J=2.8, 3.3, 7.1 Гц); 3.76 (1H, уш.д, J=12.1 Гц); 3.63 (1H, уш.д, J=12.1 Гц), 2.11 (3H, с).
Пример 8. 1-(2'-Циано-2'-деокси-β-D-арабинофуранозил)цитозин.
В 55 мл метанола растворили 100 мг соединения примера 7, к полученному раствору добавили 2,5 мл уксусной кислоты, затем кипятили с обратным холодильником на масляной бане в течение 5 дней. После завершения реакции растворитель упарили, а остаток очистили, пропустив через колонку с силикагелем ( ⊘ 1,8 x 7 см). В качестве элюента использовали смесь метанола и хлороформа (12-15 : 88-85) и далее очистку проводили методом ВЭЖХ (D-ODS-5,5% метанол-вода), а затем кристаллизовали из смеси этанол-эфир. В результате получили 29 мг соединения, название которого дано в заголовке. Соединение было получено в виде кристаллов белого цвета.
1H-ЯМР (DMCO-d6) δ м.д.: 7.83 (1H, д, J=7.1 Гц); 7.27 (2H, уш.с); 6.17 (1H, д, J=6.6 Гц); 6.15 (1H, д, J=7.1 Гц); 5.79 (1H, д, J=7.6 Гц); 5.14 (1H, т, J=4.9 Гц); 4.40 (1H, дд, J=6.6 Гц, 7.1, 7.7 Гц); 3.77 (1H, т, J=7.1 Гц); 3.74 (1H, ддд, J= 2.8, 4.5, 7.7 Гц); 3.73 (1H, ддд, J=2.8, 4.9, 12.6 Гц); 3.60 (1H, ддд, J=2.8, 4.9, 12.6 Гц).
Пример 9. 1-(2'-Циано-2'-деокси-β-D-арабинофуранозил)цитозин моногидрохлорид.
В 5 мл 3%-ной смеси хлористоводородная кислота-метанол растворили 40 мг соединения примера 8, затем смесь перемешивали в течение 50 мин при комнатной температуре. После завершения реакции провели кристаллизацию, используя смесь этанол-эфир. В результате получили 26 мг соединения, название которого дано в заголовке. Соединение получено в виде кристаллов белого цвета.
1H-ЯМР (DMCO-d6) δ м.д.: 8.5 (1H, с); 8.30 (1H, д, J=7.7 Гц); 6.21 (1H, д, J=7.2 Гц); 6.12 (1H, д, J=7.7 Гц); 4.43 (1H, дд, J=7.1 Гц, 7.7 Гц); 3.97 (1H, т, J=7.1 Гц); 3.83 (1H, ддд, J=2.8, 3.3, 7.7 Гц); 3.76 (1H, дд, J=2.8, 12.6 Гц); 3.62 (1H, дд, J=3.8 Гц, 12.6 Гц).
Пример 10. 1-(2'-Циано-2'-деокси-3',5'-О-TIPDS-β-D-рибофуранозил)тимин.
В 4 мл безводного толуола суспендировали 400 мг 3',5'-0-TIPDS-2'-0-фенокситиокарбонилтимидина и к полученной суспензии добавили 1,8 мл трет.-бутилизонитрила, затем последовало нагревание на масляной бане при 100oC в токе газа аргона. К полученной смеси по каплям добавили 4 мл раствора азобисизобутиронитрила (50 мг) и трибутилолово гидрида (0,25 мл) в толуоле, добавление велось в течение часа с помощью шприцевого насоса. Через три часа после завершения прикапывания к смеси добавили 0,25 мл трибутилолово гидрида и полученную смесь перемешивали в течение 19 ч, затем отогнали растворитель(и). Остаток очистили методом колоночной хроматографии, используя колонку с силикагелем ( ⊘ 2,2 x 8 см). В качестве элюента использовали хлороформ. В результате получили 70 мг соединения, название которого дано в заголовке. Соединение было получено в виде пены желтого цвета.
1H-ЯРМ (CDCl3) δ м.д.: 8.57 (1H, уш.с.); 7.38 (1H, д, J=1.1 Гц); 6.01 (1H, д, J=2.6 Гц); 4.22-4.01 (4H, м); 3.48 (1H, дд, J=2.6 Гц, 4.8 Гц); 1.90 (3H, д, J=1.1 Гц); 1.10-1.01 (28H, м).
Пример 11. 1-(2'-Циано-2'-деокси-β-D-рибофуранозил)тимин.
Процедуры примера 3 были повторены с использованием соединения примера 10 (70 мг) и после очистки твердого продукта, полученного в результате упаривания растворителя, собранного при фильтрации эфиром, получили 17 мг соединения, название которого дано в заголовке. Соединение было получено в виде твердого продукта желтовато-белого цвета.
1H-ЯРМ (DMCO-d6) δ м.д.: 11.49 (1H, уш.с.); 7.64 (1H, д, J=1.1 Гц); 6.32 (1H, д, J=5.5 Гц); 6.27 (1H, д, J=8.2 Гц); 5.22 (1H, уш.с.); 4.37 (1H, ддд, J= 2.8, 5.5, 5.5 Гц); 3.93 (1H, м); 3.75 (1H, дд, J=5.5, 8.2 Гц); 3.66-3.51 (2H, м); 1,78 (3H, д, J=1.1 Гц).
Пример 12. 1-(2'-Циано-2', 3'-деокси-2', 3'-дидегидро-β-D-рибофуранозил/тимин.
В 3 мл уксусной кислоты растворили 112 мл соединения сравнительного примера 3 и полученный раствор перемешивали при комнатной температуре в течение 1 ч. После завершения реакции растворитель упарили и остаток очистили над силикагелем в колонке диаметром 1,6 и высотой 8,5 см. В качестве элюента использовали смесь этанола и хлороформа (8:92). Растворитель упарили, и выпавшие кристаллы собрали путем фильтрации, используя смесь эфира и гексана. В результате получили 22 мг соединения, название которого дано в заголовке. Соединение было получено в виде кристаллов.
1H-ЯРМ (CDCl3) δ м.д.: 11.53 (1H, уш.с.); 7.81 (1H, д, J=1.1 Гц); 7.63 (1H, д, J= 1.1 Гц); 7.02 (1H, дд, J=1.7, 3.9 Гц); 5.33 (1H, т, J=4.9 Гц); 5.05 (1H, ддд, J= 2.8, 2.8, 3.9 Гц); 3.74 (1H, ддд, J=2.8, 4.9, 12.6 Гц); 3.67 (1H, ддд, J=2.8, 4.9, 12.6 Гц); 1.75 (3H, д, J=1.1 Гц).
Пример 13. 1-(2'-Циано-2',3'-дидеокси-2',3'-дидегидро-β-D-арабинофуранозил)-N4-ацетилцитозин.
Процедуры примера 12 были повторены с использованием 70 мг соединения сравнительного примера 4. После очистки путем пропускания через колонку с силикагелем (диаметр колонки 1,8 и высота 7 см) (в качестве элюента использовали смесь этанола и хлороформа 10:90) провели кристаллизацию из смеси этанола и этилацетата, в результате получили 14 мг соединения, название которого дано в заголовке. Соединение получено в виде кристаллов.
1H-ЯРМ (DMCO-d6) δ м. д.: 11.02 (1H, c); 8.33 (1H, д, J=7.1 Гц); 7.65 (1H, т, J= 7.1 Гц); 7.24 (1H, д, J=7.1 Гц); 7.12 (1H, дд, J=1.7, 3.3 Гц); 5.30 (1H, т, J=4.9 Гц); 5.12 (1H, м); 3.74 (1H, дд, J=3.3, 12.6 Гц); 3.67 (1H, дд, J=3.3, 12.6 Гц); 2.12 (3H, с).
Пример 14. N4-бензилоксикарбонилцитидин.
В пиридине, насколько это возможно, растворили 4,86 г цитидина, и полученный раствор подвергли дважды азеотропной перегонки для удаления содержащейся в нем влаги. В остатку добавили 100 мл пиридина и к полученной смеси добавили 12,6 мл триметилхлоросилана при охлаждении льдом, затем перемешивали смесь в течение 30 мин. К образовавшейся смеси по каплям добавили 49 мл карбобензоксихлорида (30-35% раствор в толуоле). После перемешивания смеси при комнатной температуре смесь оставили стоять на ночь. Затем к смеси добавили 40 мл воды и полученную смесь перемешивали в течение 1,5 ч. После добавления метиленхлорида отделили органический слой и промыли насыщенным раствором хлорида натрия. Затем органический слой высушили над безводным сульфатом магния и отогнали растворитель. Остаток подвергли азеотропной перегонке три раза с толуолом и этанолом. В результате получили 6,13 г соединения, название которого дано в заголовке. Соединение получено как кристаллический остаток.
1H-ЯРМ (270 МГц в DMCO-d6) δ м.д.: 8.40 (1H, д, J=7.3 Гц); 7.31-7.55 (5H, м); 7.02 (1H, д, J= 7.3 Гц); 5.77 (1H, д, J=2.4 Гц); 5.19 (2H, c); 3.88-3.99 (3H, м).
Пример 15. 3',5'-0-TIPDS-N4-бензилоксикарбонилцитидин.
В пиридине растворили 6,0 г соединения примера 14 и полученный раствор подвергли дважды азеотропной дистилляции для удаления влаги, содержащейся в этом соединении. Остаток растворили в 200 мл пиридина и затем туда добавили 5,09 мл 1,3-дихлоро-1,1,3,3-тетраизопропилдисилоксана, после чего смесь перемешивали при комнатной температуре. После того как смесь стояла в течение двух дней, растворители отогнали. Остаток растворили в метиленхлориде и раствор интенсивно промыли водой, 0,5 М хлористоводородной кислотой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия. Остаток высушили над безводным сульфатом натрия, растворитель упарили и в результате получили 10,23 г соединения, название которого дано в заголовке.
1H-ЯРМ (270 МГц в DMCO-d6) δ м.д.: 10.83 (1H, уш. с.); 8.12 (1H, д, J= 7.8 Гц); 7.32-7.43 (5H, м); 7.03 (1H, д, J=7.3 Гц); 5.59 (1H, с); 5.19 (1H, с); 3.91-4.24 (5H, м); 0.80-1.14 (28H, м).
Пример 16. N4-бензилоксикарбонил-1-(3,5-0-TIPDS-β-D-эритропентофуран-2-урозил)цитозин.
К 70 мл метиленхлорида добавили 13,16 г пиридиний дихромата, 3,31 мл уксусного ангидрида, 0,94 мл пиридина и 2,5 г целита, полученную смесь перемешивали в течение 40 мин. Отдельно растворили 7,23 г соединения примера 15 в 30 мл метиленхлорида и полученный раствор добавили к полученному выше раствору. После перемешивания полученной таким образом смеси при комнатной температуре в течение 5 ч к ней добавили этилацетат и отогнали метиленхлорид. Остаток растворили в этилацетате, а нерастворимую часть отфильтровали. После промывания фильтрата 1 M хлористоводородной кислотой, насыщенным водным раствором хлорида натрия, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия фильтрат высушили над безводным сульфатом магния. Растворитель отогнали, а остаток очистили методом хроматографии на силикагеле (в качестве элюента использовали смесь метиленхлорида и метанола 99:1) и в результате получили 2,84 г соединения, название которого дано в заголовке.
1H-ЯМР (270 МГц в DMCO-d6) δ м.д.: 10.98 (1H, уш.с.); 8.15 (1H, д, J=7.3 Гц); 7.32-7.44 (5H, м); 7.09 (1H, д, J=7.3 Гц); 5.49 (1H, с); 5.20 (2H, с); 5.07 (1H, д, J=8.3 Гц); 3.95-4.04 (3H, м); 0.92-1.12 (28H, м).
Пример 17. N4-бензилоксикарбонил-2'-циано-3',5'-О-TIPDS-β-D-арабино-фуранозилцитозин.
В 25 мл тетрагидрофурана растворили 2,71 г соединения примера 16 и к этому раствору добавили 13 мл воды, затем перемешали. К полученной смеси добавили 436 мг цианида натрия при охлаждении льдом. Затем добавили 740 мг гидрокарбоната натрия и перемешивали после этого при комнатной температуре в течение 7 ч. После этого смесь оставили стоять в холодильнике на ночь и затем добавили к ней 217 мг цианида натрия. Полученную смесь перемешивали при комнатной температуре в течение 8 ч, после этого отогнали растворители, а остаток растворили в этилацетате. Раствор промыли три раза насыщенным водным раствором хлорида натрия, а затем высушили над безводным сульфатом магния, после этого отогнали растворитель. Остаток очистили, пропустив через колонку с силикагелем. В качестве элюента использовали смесь метиленхлорида и метанола 99,25 : 0,75. Провели перекристаллизацию из ацетонитрила и в результате получили 392 мг соединения, название которого дано в заголовке.
1H-ЯМР (270 МГц в DMCO-d6) δ м.д.: 11.0 (1H, уш.с.); 8.05 (1H, д, J=7.3 Гц); 7.83 (1H, уш.с.); 7.33-7.44 (5H, м); 7.13 (1H, д, J=7.8 Гц); 5.93 (1H, с); 5.21 (2H, с); 3.93-4.33 (4H, м); 0.96-1.07 (28H, м),
Пример 18. N4-бензилоксикарбонил-2'-циано-деокси-3',5'-О-TIPDS-β-D-арабинофуранозилцитозин.
В 8 мл толуола растворили 0,40 мг соединения из сравнительного примера 5 и к полученному раствору добавили 13,4 мг α,α'-азобисизобутиронитрила и 0,20 мл трибутилолово гидрида в указанной последовательности и в токе газа азота, затем перемешивали при 100oC в течение 2 ч. Растворитель отогнали и остаток очистили над силикагелем (в качестве элюента использовали смесь метиленхлорида и метанола 99:1), в результате получили 202 мг соединения, название которого дано в заголовке.
1H-ЯМР (270 МГЦ в DMCO-d6) δ м.д. : 10.96 (1H, уш.с.); 8.02 (1H. д, J= 7.3 Гц); 7.32-7.44 (5H, м); 7.12 (1H, д, J=7.8 Гц); 6.20 (1H, д, J=7.8 Гц); 5.20 (2H, с); 3.91-4.72 (5H, м); 0.95-1.23 (28H, м).
Пример 19. N4-бензилоксикарбонил-2'-циано-2'-деокси-β-D-арабинофуранозилцитозин.
В 5 мл тетрагидрофурана растворили 192 мг соединения примера 18 и после того, как полученный раствор был охлажден на льду в токе газа азота, к нему добавили раствор 0,02 мл уксусной кислоты и 168 мг тетрабутиламмонийфторида, растворенного в 1,2 мл тетрагидрофурана, затем перемешивали на льду в течение 2 ч. Растворители отогнали и остаток очистили методом хроматографии на силикагеле (в качестве элюента использовали смесь метиленхлорида и метанола 95: 5), в результате получили 94 мг соединения, название которого дано в заголовке.
1H-ЯМР (270 МГц в DMCO-d6) δ м.д. : 10.92 (1H, уш.с.); 8.36 (1H, д, J= 7.3 Гц); 7.34-7.44 (5H, м); 7.11 (1H, д, J=7.8 Гц); 6.25 (1H, д, J=5.4 Гц); 5.20 (2H, с); 5.24 (1H, д, J=4.4 Гц); 4.43 (1H, кв. J=7.3 Гц, 12.7 Гц); 3.61-3.93 (4H, м),
Пример 20. 2'-Циано-2',3-дидеокси-2',3'-дидегидро-β-D-рибофуранозилцитозин.
Были повторены процедуры синтеза соединения примера 12 с использованием 300 мг соединения примера 8, в результате получили 60 мг соединения, название которого дано в заголовке.
1H-ЯМР (270 МГц в DMCO-d6) δ м.д. : 7.79 (1H, д, J=7.3 Гц); 7.55-7.56 (1H, м); 7.36 (2H, д, J=7.3 Гц); 7.07 (1H, дд, J=1.96, 3.90 Гц); 5.78 (1H, д, J=7.3 Гц); 5-18-5.21 (1H, м); 5.01-5.03 (1H, м); 3.65-3.70 (2H, м).
Сравнительный пример 1. 1-[2'-Циано-2'-деокси-5'-О-(4,4'-диметокситрифенилметил)-β-D-арабинофуранозил]тимин.
В 7 мл безводного пиридина растворили 267 мг соединения примера 3 и к полученному раствору добавили 508 мг 4,4-диметокситрифенилметил хлорида, затем все перемешивали при комнатной температуре в течение 1,5 ч в токе газа аргона. После завершения реакции растворитель отогнали и к остатку добавили 100 мл этилацетата. После промывания смеси в течение трех раз 50 мл воды и высушивания над безводным сульфатом натрия растворитель упарили. Остаток очистили методом хроматографии на колонке с силикагелем (диаметр колонки 1,8 и высота 8,5 см). В качестве элюента использовали смесь этанола с хлороформом (1-2 : 99-98). В результате получили 574 мг соединения, название которого дано в заголовке. Соединение получено в виде желтовато-белой пены.
1H-ЯМР (CDCl3) δ м. д. : 8.40 (1H, уш.с.); 7.50 (1H, д, J=1.2 Гц); 4.77-7.26 (9H, м); 6.90-6.80 (4H, м); 6.27 (1H, д, J=6.8 Гц); 4.74 (1H, д, J=6.8 Гц); 3.93 (1H, м); 3.79 (6H, с); 3.61 (1H, м); 3.30 (2H, м); 1.67 (1H, д, J=1.2 Гц).
Сравнительный пример 2. 1-[2'-Циано-2'-деокси-5'-О-(4,4-диметокситрифенилметил)-β-D-арабинофуранозил]-N4-ацетилцитозин.
Процедуры сравнительно примера 1 были повторены с использованием 194 мг соединения примера 7. В результате получили 326 мг соединения, название которого дано в заголовке. Соединение получено в виде пены желтовато-белого цвета.
1H-ЯМР (CDCl3) δ м. д. : 8.80 (1H, уш.с.); 8.19 (1H, д, J=7.6 Гц); 7.41-7.14 и 6.89-6.76 (14H, м); 6.30 (1H, д, J=6.1 Гц); 4.79 (2H, м); 4.08 (2H, м); 3.79 (6H, с); 3.56 (2H, м); 2.09 (3H, с).
Сравнительный пример 3. 1-[2'-Циано-2',3'-дидеокси-2',3'-дидегидро-5'-0-(4,4'-диметокситрифенилметил)-β-D-рибофуранозил]тимин.
В 3 мл безводного диметилформамида растворили 200 мг соединения сравнительного примера 1 и к полученному раствору добавили 94 мг тиокарбонилдиимидазола, затем последовало перемешивание в течение 13 ч при комнатной температуре и 40-минутное перемешивание в токе газа аргона. После завершения реакции добавили этилацетат к реакционной смеси и смесь промыли три раза водой и высушили над безводным сульфатом натрия, затем растворители отогнали. Остаток очистили методом хроматографии на колонке с силикагелем ( ⊘ 2 x 6,5 см). В качестве элюента использовали смесь этилацетата с гексаном от 1:1 до 2: 1). В результате получили 162 мг соединения, название которого дано в заголовке. Соединение было получено в виде карамели белого цвета.
1H-ЯМР (CDCl3) δ м.д.: 8.40 (1H, уш.с.); 7.45 (1H, д, J=1.1 Гц); 7.10 (1H, д, J= 1.8 Гц); 7.06 (1H, д, J=1.8 Гц, 4.0 Гц); 5.10 (1H, ддд, J=2.6, 3.3, 4.0 Гц); 4.12 (3H, с); 3.61 (1H, дд, J=2.6, 11.0 Гц); 3.45 (1H, дд, J= 3.3, 11.0 Гц); 1.90 (3H, д, J=1.1 Гц).
Сравнительный пример 4. 1-[2'-Циано-2',3'-дидеокси-2',3'-дидегидро-5'-О-(4,4-диметокситрифенилметил)-β-D-арабинофуранозил]-N4-ацетилцитозин.
Были повторены процедуры сравнительного примера 3 с использованием 326 мг соединения, полученного в сравнительном примере 2, и после проведения очистки, кристаллизации из эфира получили 163 мг соединения, название которого дано в заголовке. Соединение получено в виде кристаллов.
1H-ЯМР (DMCO-d6) δ м. д.: 9,22 (1H, c); 8,16 (1H, д, J = 7,3); 7,35 - 7,22 (9H, м); 6,95 (1H, дд, J = 1,8, 4,0 Гц); 6,90-6,84 (5H, м); 6,68 (1H, дт, J = 1.8 Гц); 5,08 (1H, ддд, J = 2.6, 2.9, 4.0 Гц); 3.82 (6H, с.); 3,71 (1H, дд, J=2,9, 11.7 Гц); 3.59 (1H, дд. J =2.6 Гц); 2.24 (3Н, с).
Сравнительный пример 5. N4-бензилоксикарбонил-2'-циано-2'-фенокситиокарбонил-3',5'-О-TIPDS-β-D-арабинофуранозил-цитозин.
В пиридине растворили 525 мг соединения примера 17 и после удаления влаги путем азеотропной перегонки остаток растворили в 5 мл метиленхлорида. К полученному раствору добавили 40 мг диметиламинопиридина, 0,17 мл фенилхлороформиата и 0,17 мл триэтиламина в указанной последовательности при охлаждении льдом в токе газа азота, а затем смесь перемешивали при охлаждении льдом в течение 4 ч. К образовавшейся смеси добавили метиленхлорид и после промывания полученной смеси насыщенным водным раствором хлорида натрия, 0,1 М раствором хлористоводородной кислоты и насыщенным раствором хлорида натрия и сушки над безводным сульфатом магния растворители отогнали. Остаток очистили методом хроматографии на силикагеле. В качестве элюента использовали смесь метиленхлорида с метанолом 99.5 : 0.5. В результате получили 0,46 г соединения, название которого дано в заголовке.
1H-ЯМР (270 Мгц в DMCO-d6) δ м.д.: 11.02 (1H, уш.с.); 8.00 (1H, д, J = 7.8 Гц); 7.31 -7.98 (1OH, м); 7.11 (1H, д, J =7.8 Гц); 6.29 (1H, с); 5.75 (1H, уш.с); 5.20 (2H, с); 3.98-4.17 (3H, м); 1.00 - 1.12 (28H, м).
Препаративный пример 1. Жесткие капсулы.
В каждую стандартную желатиновую жесткую капсулу типа колпачок - емкость загружали 100 мг порошкообразного комплекса примера 1, 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния. Приготовленные таким образом капсулы промывали и сушили для обеспечения твердокапсульных препаратов.
Препаративный пример 2. Таблетки.
Таблетки готовили следующим образом: смешивали 100 мг комплекса примера 1, 0,2 г коллоидальной двуокиси кремния, 5 мг стеарата магния, 275 мг микрокристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы. Полученную смесь таблетировали. При необходимости на таблетки может быть нанесено покрытие.
Препаративный пример 3. Инъекции.
В 10% по объему пропиленгликоля перемешали 1,5% по весу комплекса примера 1. Затем смесь довели до заранее определенного объема добавлением дистиллированной воды для инъекций, после этого провели стерилизацию.
Препаративный пример 4. Суспензии.
К 5 мл добавили при перемешивании 100 мг тонкоизмельченного комплекса примера 1, 100 мг карбоксиметилцеллюлозы натрия, 5 мг бензоата натрия, 1,0 г раствора сорбитола (фармакопея Японии) и 0,25 мл ванилина, затем провели гомогенизацию с целью получения суспензии.
Активность соединений. Описание методики испытания.
1. Оценка противоопухолевой активности in vitro.
Противоопухолевую активность in vitro оценивали, используя штамм раковых клеток человека. В качестве культуральной среды для раковых клеток использовали раствор RPMD 640, содержащий 10% иммобилизованной бычьей сыворотки и 50 г/мл канамицина. Раковые клетки (1•104 клеток/мл) иннокулировали в 1 мл культуральной жидкости, содержащей образцы соединений различной концентрации и культивировали в инкубаторе в атмосфере углекислого газа при 37oC в течение 72 ч. Жизнеспособность раковых клеток определяли по методу MTT, согласно которому определяют количество живых клеток в культуральных жидкостях, содержащих образец соединения, и соответственно несодержащих образец тестируемого соединения (слепой опыт), измеряя интенсивность видимого света (поглощение в видимой области спектра) с использованием для этих целей 3-(4,5-диметилтиазол-2-ил)-2,5-дифенил-тетразолиум бромида, который дает окрашивание, интенсивность которого растет пропорционально числу живых клеток. Степень противоопухолевой активности оценивали по ИК50 (концентрация мк/мл), необходимой для ингибирования пролиферации клеток на 50%. Значение ИК50 определяли из графической зависимости % пролиферации раковых клеток в образце, содержащем соединение настоящего изобретения (% относительно слепого опыта) от концентрации (в логарифмических координатах) соединения в образце. Результаты суммированы в табл. 4.
Соединения (1) и (2) настоящего изобретения проявляют сильную противоопухолевую активность в отношении Р388 клеток, трансплантированных мышам, а также в отношении различных форм рака человека. Они могут легко абсорбироваться организмом при пероральном приеме и обладают низкой токсичностью со слабым побочным действием. В соответствии с этим они весьма полезны при лечении или профилактике заболеваний, порождающих опухоли, как новый тип противоопухолевых средств на основе пиримидин нуклеозида. Кроме того, соединения (1) и (2) настоящего изобретения весьма полезны как полупродукт при производстве превосходных противоопухолевых средств. Производные пиримидин нуклеозида настоящего изобретения могут вводиться теплокровным животным, включая человека. Лекарственные формы включают препараты для внутривенных инъекций, подкожных инъекций, внутримышечных инъекций и суппозитории для парентерального введения, а также таблетки, капсулы, порошки и гранулы для перорального приема.
Хотя доза для взрослого организма варьируется в зависимости от заболевания, которое подлежит лечению, способа введения препарата, количества доз и периода лечения, препарат вводят в количестве от 0,01 до 5 г в сутки за один раз или проводят дробный прием препарата.
Кроме того, настоящее соединение может быть использовано в сочетании с другими потивоопухолевыми средствами, такими как препараты на основе нитрозомочевины, например, 5 Fu, AraC, ACNU и BCNU, цисплатина, дайномицин, адриамицин, митомицин С или этопозид. Помимо этого, настоящие производные пиримидин нуклеозида могут быть получены в нужных для введения выпускных формах традиционными способами. Таким образом, настоящее изобретение включает фармацевтические препараты и составы, содержащие фармацевтически приемлемые производные пиримидин нуклеозида.
Состав для инъекций выпускают в виде ампул для одноразового введения или во флаконах, содержащих дозу для многоразового введения. Составы могут содержать добавки, такие как суспендирующие средства, стабилизаторы и диспергаторы, и, как, правило, состав представляет собой порошок, который вновь растворяют перед использованием в соответствующем растворителе, таком как стерилизованная вода - водная среда, не содержащая пирогенных веществ. Такой состав может быть получен, например, путем растворения производного пиримидин нуклеозида в ацетоне, разлива по ампулам и сушкой вымораживанием после добавления воды. Далее, составы для перорального приема могут быть выпущены в форме таблеток, капсул, порошков, гранул и сиропов, содержащих подходящие количества производных пиримидин нуклеозида для приема.
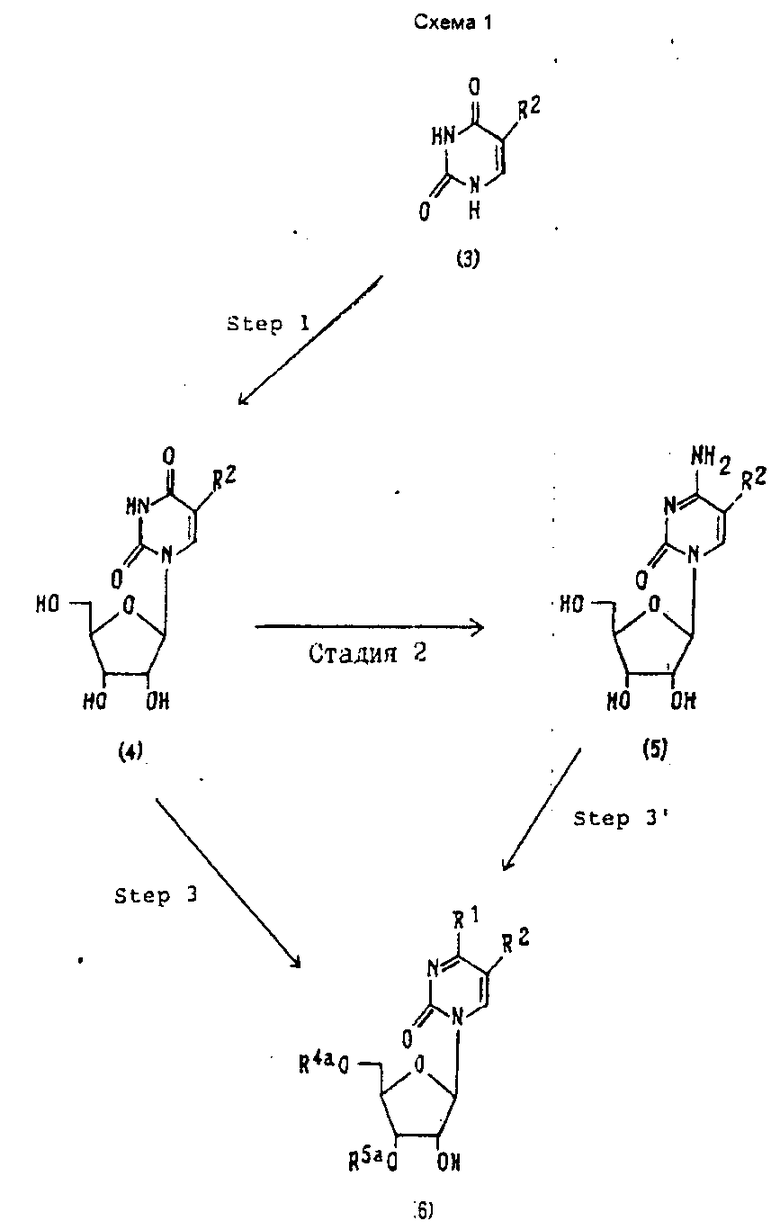





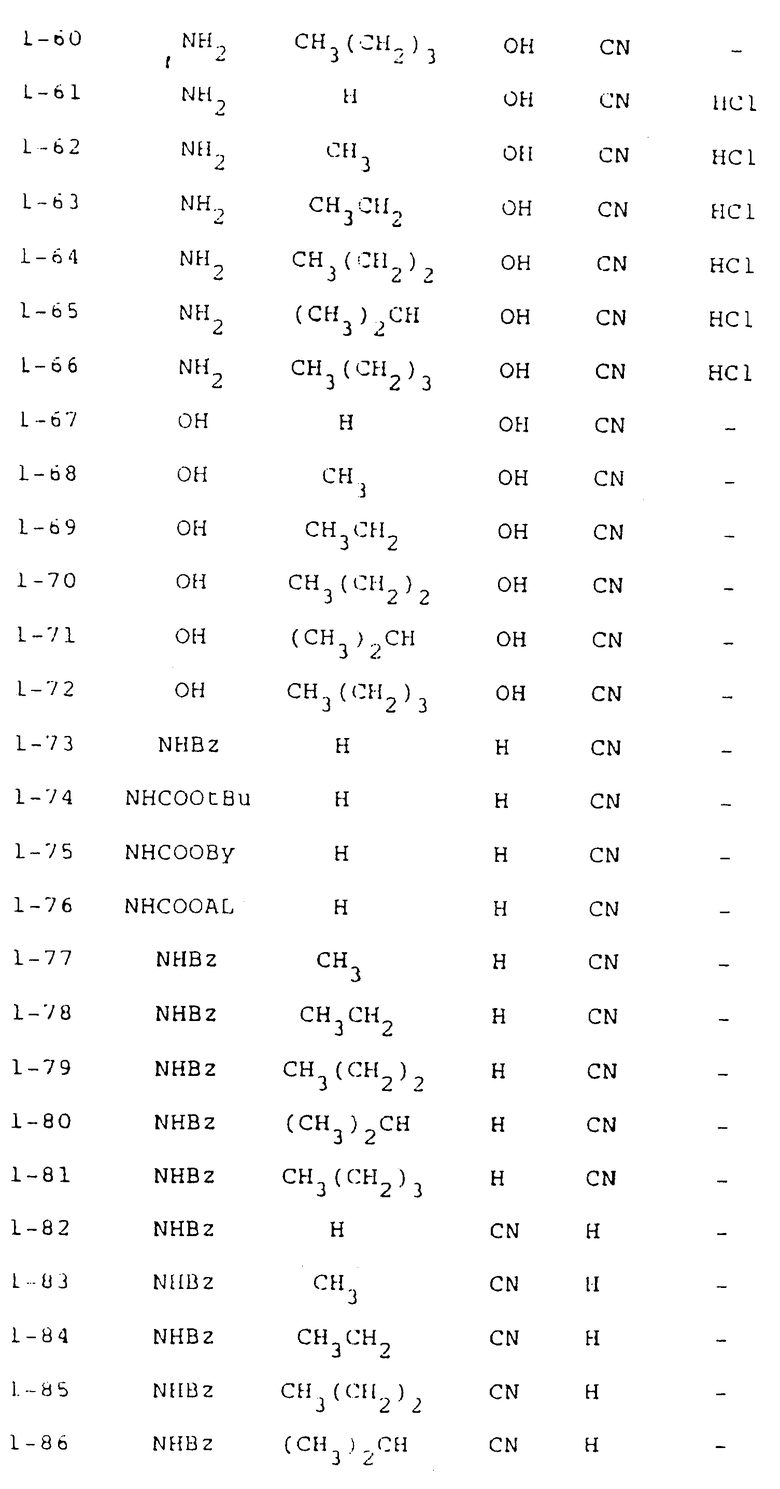






Предложены новые производные пиримидина, представленные общими формулами (1) и (2)
и соли этих соединений. В приведенных выше формулах R1 -гидроксильная группа или аминогруппа, которая может быть замещена ацильной группой: R2 - атом водорода или алкильная группа, имеющая 1-4 атома углерода; R3 - атом водорода или гидроксильная группа; а R4 и R5 - каждый атом водорода или вместе они могут образовывать группу - R6R7Si-O-Si-R6'R7'- (в которой R6, R7, R6' и R7' могут быть одинаковыми или отличаться друг от друга и каждый представляет C1-C4 алкил). Действие: заявленные соединения проявляют превосходную противоопухолевую активность или служат промежуточными соединениями в процессе получения полезных противоопухолевых средств. 7 с. и 4 з.п. ф-лы, 4 табл.

или их фармакологически приемлемые соли,
где R1 - OH, аминогруппа, возможно замещенная алифатическим C1 - C5-ацилом, бензоилом;
R2 - H, C1 - C4-алкил;
R3 - H, OH;
R4 - H;
R5 - H или R4 и R5 вместе образуют группу - R6R7Si-O-Si R6'R7', где R6, R7, R6', R7' одинаковые или различные, C1 - C4-алкил.
1-(2'-циано-β-D-2'-деокси-арабинофуранозил)цитозина,
1-(2'-циано-β-D-2'-деокси-арабинофуранозил)тимина,
1-(2'-циано-β-D-2'-деокси-рибофуранозил)цитозина,
1-(2'-циано-β-D-2'-деокси-арабинофуранозил)тимина,
1-(2'-циано-β-D-2'-деокси-рибофуранозил)тимина,
или их фармакологически приемлемые соли.
где R1 и R2 определены в п.1, R4а и R5a вместе образуют группу - R6R7-Si-O-Si-R6'R7', где R6, R7, R6', R7' определены в п.1, отличающийся тем, что проводят взаимодействие соединения общей формулы
R1, R2, R4a, R5a имеют указанные значения, а R9 - алкокси C1 - C4-тиокарбонильная группа с восстанавливающим и цианирующим агентом.
где R1 и R2 определены в п.1, R4а и R5a определены в п.7,
отличающийся тем, что проводят взаимодействие соединения общей формулы
где R1, R2, R4a и R5a имеют вышеуказанные значения с цианирующим агентом.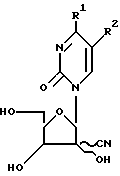
где R1 и R2 определены в п.1,
отличающийся тем, что проводят взаимодействие соединения общей формулы 1в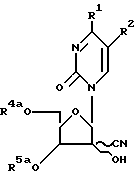
где R1 и R2 определены выше, а R4а и R5a определены в п.7 с деблокирующим агентом.
где R1 и R2 определены в п.1, R4а и R5a определены в п.7, отличающийся тем, что проводят взаимодействие соединения общей формулы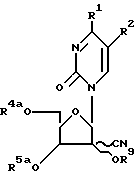
где R1, R2, R4a, R5a определены выше, R9-алкокситиокарбонил,
содержащая C1 - C4-алкил, с восстановителем.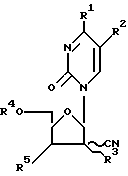
где R1 и R2 определены в п.1, а R3, R5 вместе образуют углерод-углеродную связь,
отличающийся тем, что проводят взаимодействие соединения общей формулы
где R1, R2, R3, R5 имеют указанные значения, а R10 - защитная группа с деблокирующим агентом.
| SU, 4616121, A 61 K 31/70, 10.04.90 | |||
| Chem.Pharm.Bull | |||
| Способ использования делительного аппарата ровничных (чесальных) машин, предназначенных для мериносовой шерсти, с целью переработки на них грубых шерстей | 1921 |
|
SU18A1 |

