По настоящей заявке испрашивается приоритет по предварительной заявке на патент США №60/763,712, поданной 31 января 2006 года, которая полностью включена в настоящую заявку путем ссылки.
Область техники, к которой относится изобретение
Настоящее изобретение относится к ингибиторам киназ, фармацевтическим композициям, содержащим такие ингибиторы, и способам получения указанных ингибиторов. Ингибиторы киназ по настоящему изобретению применимы для лечения воспаления, остеоартрита, ревматоидного артрита, рака, аутоиммунных заболеваний и других заболеваний, опосредованных цитокинами.
Предшествующий уровень техники
Ряд хронических и острых воспалительных заболеваний связан с перепроизводством провоспалительных цитокинов. Такие цитокины включают (но не ограничиваются ими) фактор некроза опухоли альфа (TNF-α), интерлейкин 1 бета (IL-1β), интерлейкин 8 (IL-8) и интерлейкин 6 (IL-6). Ревматоидный артрит (RA) представляет собой хроническое заболевание, начало которого и прогрессирование видимого разрушения костей и суставов, наблюдаемое при этом расстраивающем здоровье заболевании, непосредственно связаны с TNF-α и IL-1β. Терапевтические виды лечения RA, недавно получившие одобрение, включают растворимый рецептор TNF-α (ENBREL™) и антагонист рецептора IL-1 (ANAKINRA™). Эти виды лечения работают путем блокировки способности соответствующих цитокинов связываться с естественными рецепторами. В настоящее время исследуются альтернативные способы лечения опосредованных цитокинами заболеваний. Один из таких методов включает ингибирование сигнального пути, который регулирует синтез и продуцирование провоспалительных цитокинов, таких как р38.
Цитокин Р38 (также известный как CSBP или RK) представляет собой серин/треонин-митоген-активированную протеинкиназу (МАРК), которая, как было показано, регулирует провоспалительные цитокины. Сначала Р38 МАРК была идентифицирована как киназа, которая становится тирозинфосфорилированной в моноцитах мышей после лечения липосахаридом (LPS). Связь между р38 МАРК и ответной реакцией клеток на цитокины была впервые установлена Саклатвалой с сотр. (Saklatvala et al., Cell, 1994, 78:1039-1049), которые показали, что IL-1 активирует каскад протеинкиназы, что приводит к фосфорилированию небольшого белка теплового шока, Hsp27, вероятно, активированной митоген-акктивированным белком протеинкиназой 2 (МАРКАР киназа-2). Анализ последовательностей пептидов, полученных из очищенной киназы, показывает, что она родственна р38 МАРК, активированной LPS в моноцитах мыши (Han, J., et al., Science, 1994, 265:808-811). В то же время показано, что сама р38 МАРК активируется киназой в обратном направлении в ответ на множество клеточных стрессов, включающих УФ-облучение и осмотический шок, и была подтверждена идентичность киназы, которая непосредственно фосфорилирует Hsp27, как МАРКАР киназа-2 (Rouse, J., et al., Cell, 1994, 78:1027-1037). Впоследствии было показано, что р38 МАРК является молекулярной мишенью ряда производных пиридинилимидазола, которые ингибируют продупирование TNF из LPS-стимулированных моноцитов человека (Lee, J., et al., Nature, 372:739-746). Это было главным открытием, которое привело к разработке ряда селективных ингибиторов р38 МАРК и прояснению ее роли в цитокиновой передаче сигнала.
Теперь известно, что разнообразные формы р38 МАРК (α, β, γ, δ), каждая из которых кодирована отдельным геном, образуют часть каскада киназы, участвующего в реакции клеток на целый ряд стимулов, включающих осмотический стресс, УФ-свет и опосредованные цитокинами явления. Считается, что эти четыре изоформы р38 регулируют различные аспекты внутриклеточной передачи сигнала. Активация р38 представляет собой часть каскада передачи сигнала, которая приводит к синтезу и продуцированию провоспалительных цитокинов, таких как TNF-α. Р38 функционирует путем фосфорилирования субстратов в прямом направлении, которое включает другие киназы и факторы транскрипции. Показано, что агенты, которые ингибируют р38 МАРК, блокируют продуцирование цитокинов, включающих TNF-α; IL-6, IL-8 и IL-1β, но не ограниченных ими, в моделях in vitro и in vivo (Adams, J.L., et al., Progress in Medicinal Chemistry, 2001, 38:1-60).
Abl (известный также как Ableson) представляет собой тирозинкиназу, которая экспрессируется в гемопоэтических клетках и участвует в развитии различных «жидких опухолей», включающих хронический миелоидный лейкоз (CML) и острый лимфобластный лейкоз (ALL). Трансформация является результатом транслокации хромосомы, известной под названием филадельфийская хромосома. Это приводит к существенно активированной химере между Ableson и областью локализации сайта инициации реаранжировки (BCR) - Abl-BCR белку. Препарат GLEEVEC®, известный также под названием иматиниб (новартис), представляет собой сильный ингибитор Abl и в настоящее время используется для лечения пациентов с CML (N. Engl. J. Med., 2001, 344:1031-1037). Упомянутое лекарственное средство становится стандартом лечения при этом смертельном заболевании, и его также проверяют на целом ряде других злокачественных новообразований, включающих гастроинтестинальные стромальные опухоли (GIST).
Имеется подтверждение того, что фибробласты реагируют на белок фактора роста TGF-β, стимулируя Аbl-путь, и приводят к морфологическим изменениям, свидетельствующим о фиброзе; следовательно, Аbl мог бы играть роль в патогенезе фибротических заболеваний, подобных идиопатическому фиброзу легких. Леоф с сотр. (Leof et al., J. Clin. Invest., 2004, 114(9) 1308-1316) показали доклиническую эффективность препарата GLEEVEC® на модели опосредуемого блеомицином фиброза легких у мышей. В настоящее время препарат GLEEVEC® оценивают на пациентах с фиброзом легких.
Цитокин ТЕК (известный также под названием Tie-2) представляет собой еще один рецептор тирозинкиназы, экспрессируемый только на эндотелиальных клетках, которые, как было показано, играют роль в ангиогенезе. Связывание фактора ангиопоэтина-1 приводит в результате к аутофосфорилированию киназного домена ТЕК и процессу трансдукции сигнала, который, по-видимому, опосредует взаимодействие эндотелиальных клеток с периэндотелиальными поддерживающими клетками, тем самым способствуя развитию вновь образованных кровеносных сосудов. С другой стороны, фактор ангиопоэтин-2, по-видимому, противодействует действию ангиопоэтина-1 на ТЕК и нарушает ангиогенез (Maisonpierre et al., Science, 1997, 277:55-60). Цитокин Tie2 подвергается позитивной регуляции в ангиогенных сосудах опухоли (Trogan, E. Br. J. Cancer, 1998, 77:51-56), и существует доказательство того, что он может играть поддерживающую роль в гемопоэтических видах рака (L.Naldini et al., Cancer Cell, 2005, 8:211-226; Suda, T. et al., Cell, 2004, 118:149-161). Кроме его возможной роли в развитии рака, ангиогенез, возможно, также связан с заболеваниями, подобными ревматоидному артриту (RA), псориазу, и с равитием патологий, вызываемых воспалением. Образование паннуса, разрушительного легиона, ответственного за прогрессирование артрита, частично управляется образованием новых кровеносных сосудов, а патологическая роль Tie2 продемонстрирована недавно в работе (Lin, С.et al., Arthritis и Rheumatism, 2005, 52(5):1585-1594) на артритных моделях RA, индуцированного коллагеном у мышей. Следовательно, ингибирование Tie2 могло бы дать полезный эффект в отношении пролиферативных и воспалительных заболеваний.
Несколько других киназ связаны с прогрессированием пролиферативных заболеваний, таких как рак. Среди них многочисленные белки семейства Src, как показано, играют ту же роль, что Src и, возможно, предоставляют параллельные пути передачи сигнала во время неконтролируемой клеточной пролиферации. Основные примеры включают тирозинкиназы Lyn, Fyn, Lck и Hck. Lyn и Hck причастны к развитию острого лимфобластного лейкоза В-клеток (B-ALL), гемопоэтического рака В-клеток (Li, S. et al. Nat Genet., 2004 36(5):453-461).
Семейство рецепторов тирозинкиназы Eph (эритропоэтин продуцирующая гепатомная амплифицированная последовательность) связывает эфрины, которые управляют многочисленными клеточными процессами. По-видимому, Ephs играют роль модулирования адгезии, моторики и инвазивности опухолевых клеток, и некоторые доказательства демонстрируют активную роль Ephs и эфринов в образовании новых кровеносных сосудов во время патологических процессов. Рецепторы Eph A сверхпродуцируются в сосудистой сети опухолей легких, почек и желудка, и показано, что доминантно-негативные растворимые белки EphA2 или A3 модулируют ангиогенез и прогрессирование опухолей in vivo (Lackmarm M. et al., IUBMB Life, 2005, 57(6):421-31).
Эндотелиальные факторы роста сосудов и родственные им рецепторы, например KDR (VEGFR2) и FLT1 (VEGFRR1), являются основными регуляторами ангиогенеза. Белковое терапевтическое средство AVASTIN®, как было показано, является перспективным для лечения рака прямой кишки и действует посредством VEGFR-пути. SUTENT/SU11248 (сунитиниб малеат) (Pfizer) представляет собой сильнодействующий ингибитор KDR, он показал многообещающие результаты против карцином желудочно-кишечного тракта (GIST) и ренальных клеток.
Показано, что периферические моноциты крови (РВМС) экспрессируют и секретируют провоспалительные цитокины при стимуляции липополисахаридом (LPS) in vitro. Ингибиторы Р38 эффективно блокируют указанное действие, когда РВМС предварительно обрабатывают такими соединениями до стимуляции LPS (Lee, J.C., et al., Int. J. Immunopharmacol., 1988, 10:835-843). Эффективность ингибиторов р38 на моделях воспалительных заболеваний на животных стимулировала исследования основных механизмов, которые могли бы объяснить действие упомянутых ингибиторов. Роль р38 в реакции клеток на IL-1 и TNF исследовалась на ряде клеточных систем, имеющих отношение к воспалительной реакции с использованием пиридинилимидазольного ингибитора, таких как эндотелиальные клетки и IL-8 (Hashimoto, S., et al., J. Pharmacol. Exp. Ther., 2001, 293:370-375), фибробласты и IL-6/GM-CSF/PGE2 (Beyaert, R., et al., EMBO J., 1996, 15:1914-1923), нейтрофилы и IL-8 (Albanyan, E. A., et al.. Infect. Immun., 2000, 68:2053-2060) макрофаги и IL-1 (Caivano, M. и Cohen, P., J. Immunol., 2000, 164:3018-3025) и гладкомышечные клетки и RANTES (Maruoka, S., et al., Am. J. Respir. Crit. Care Med., 1999, 161:659-668). Деструктивные эффекты многих болезненных состояний вызваны сверхпродуцированием провоспалительных цитокинов. Способность ингибиторов р38 регулировать это сверхпродуцирование делает их превосходными кандидатами в качестве средств, модифицирующих заболевания.
Известные ингибиторы р38 МАРК проявляют активность на целом ряде моделей широко известных заболеваний. Ингибиторы р38 МАРК показывают положительные эффекты на ряде стандартных моделей воспаления у животных, включающих индуцированный коллагеном артрит у крыс (Jackson, J.R., et al., J. Pharmacol. Exp. Ther., 1998, 284:687-692); индуцированный адъювантом артрит у крыс (Badger, A. M., et al., Arthritis Rheum., 2000, 43:175-183; Badger, A.M., et al., J. Pharmacol. Exp. Ther., (1996) 279:1453-1461) и индуцированный каррагенаном отек лап у мышей (Nishikori, Т., et al., Eur. J. Pharm., 2002, 451:327-333). Молекулы, которые блокируют функцию р38, как показано, эффективны в отношении ингибирования резорбции костей, воспаления и других иммунных и обусловленных воспалением патологий на упомянутых моделях у животных.
Следовательно, безопасный и эффективный ингибитор киназ предоставил бы средства для лечения изнурительных заболеваний, которые можно регулировать модуляцией одной или нескольких киназ.
Публикация международной патентной заявки WO 2004/078116 раскрывает некоторые соединения в качестве ингибиторов киназ. Среди упомянутых соединений находятся некоторые N1-замещенные производные индазола, имеющие заместитель в положении 5, который содержит пиразол-5-илмочевинную группу. Примеры таких соединений включают соединения примеров 94 и 138, в которых N1 заместитель представляет собой соответственно метильную группу и 2-гидрокси-2-метилпропильную группу.
В настоящее время обнаружено, что соединения, обладающие наиболее желательными свойствами, можно получить, выбрав первичную спиртовую группу -СН2СН2OН, в качестве заместителя N1 и конкретный заместитель в положении 5, содержащий 3-трет-бутил-1-п-толил-1Н-пиразол-5-ильную группу.
Сущность изобретения
Настоящее изобретение относится к соединениям, которые ингибируют одну или несколько киназ и опосредованные киназами явления, такие как ингибирование продуцирования цитокинов, ангиогенез или клеточная пролиферация. Такие соединения находят применение в качестве терапевтических средств для заболеваний, которые можно лечить путем ингибирования сигнальных путей киназ.
Подробное описание изобретения
Один объект изобретения относится к соединению формулы I:
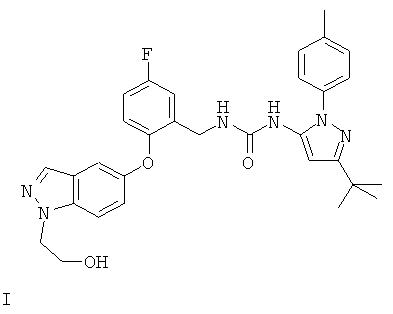
или его фармацевтически приемлемой соли.
Соединение может также быть описано химическим названием 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-(5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)бензил)мочевина.
Обнаружено, что соединение формулы I обладает сильной активностью против некоторых киназ. Кроме того, наблюдается, что растворимость соответствующего соединения, в котором заместитель N1 замещен сложноэфирной фосфатной группой формулы -СН2СН2OРО3Н, на несколько порядков выше, чем растворимость соединения формулы I. Таким образом, соединение формулы I обладает уникальной возможностью для создания растворимых пролекарств. Более конкретно, как показано далее данными испытаний, соединение формулы I является существенно более сильнодействующим ингибитором Аbl и Tie2, чем соединения примеров 94 и 138 из WO 2004/078116. Кроме того, соединения примеров 94 и 138 из WO 2004/078116 не содержат первичную спиртовую группу, которая может быть дериватизирована для получения пролекарства.
Кроме соединения формулы I, изобретение также включает фармацевтически приемлемые соли соединения и сольваты соединения и его фармацевтически приемлемые соли.
Термин «фармацевтически приемлемый» указывает, что вещество или композиция совместима химически и/или токсикологически с другими ингредиентами, составляющими рецептуру, и/или с млекопитающими, которые подвергаются лечению ею.
Термин «сольват» относится к ассоциации или комплексу одной или нескольких молекул растворителя и соединения по изобретению. Примеры растворителей, которые образуют сольваты, включают воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин, но не ограничиваются ими. Термин «гидрат» относится к комплексу, в котором молекулой растворителя является вода.
Еще один объект настоящего изобретения относится к применению соединения формулы I в изготовлении лекарственного средства для лечения опосредованного киназами состояния у млекопитающего.
В еще одном объекте изобретение относится к способу лечения или профилактики опосредованного киназами состояния, включающему введение соединения формулы I в количестве, эффективном для лечения или профилактики опосредованного киназами состояния.
Изобретение также относится к пролекарствам соединения формулы I.
Термин «пролекарство» относится к соединению, которое можно превращать в физиологических условиях или путем сольволиза в конкретное соединение или в соль такого соединения.
Свободная гидроксигруппа соединения по изобретению может давать производное в качестве пролекарства путем превращения гидроксигруппы в такую группу, как (но не ограничивается перечисленными ниже) сложноэфирнофосфатная phosphate ester, полусукцинатная, диметиламиноацетатная или фосфорилоксиметилоксикарбонильная группа, как изложено в монографии (Advanced Drug Delivery Reviews, 1996, 19, 115). Карбаматные пролекарства гидроксигрупп также включены, как и карбонатные пролекарства, сульфонатные сложные эфиры и сульфатные сложные эфиры гидроксигрупп. Дериватизация гидроксигрупп с получением таких производных, как (ацилокси)метиловые и (ацилокси)этиловые простые эфиры, в которых ацильная группа может быть алкиловым сложным эфиром, необязательно замещенным группами, включающими эфирную, аминную и карбоксильную функциональные группы, но не ограниченными ими, или ацильная группа, представляющая собой сложный эфир аминокислоты, как описано выше, также включена. Пролекарства такого типа описаны в работе (J. Med. Chem., 1996, 39, 10). Более конкретные примеры включают замену атома водорода спиртовой группы такой группой, как (С1-С6)алканоилоксиметил, 1-((С1-С6)алканоилокси)этил, 1-метил-1-((С1-С6)алканоилокси)этил, (С1-С6)алкоксикарбонилоксиметил, N-(C1-С6)алкоксикарбониламинометил, сукциноил, (С1-С6)алканоил, α-амино(С1-С4)алканоил, арилацил и α-аминоацил или α-аминоацил-α-аминоацил, где каждая α-аминоацильная группа независимо выбрана из встречающихся в природе L-аминокислот, Р(O)(ОН)2, -Р(О)(O(С1-С6)алкил)2 или гликозил (радикал, возникающий при удалении гидроксильной группы полуацетальной формы углевода).
Таким образом, еще один объект настоящего изобретения относится к применению пролекарства соединения формулы I в изготовлении лекарственного средства для лечения соединением по пункту 1 состояния, опосредованного киназами у млекопитающего. В одном варианте осуществления изобретения пролекарство представляет собой фосфатное пролекарство.
Еще один объект настоящего изобретения относится к способу лечения или профилактики опосредованного киназами состояния у млекопитающего соединением формулы I, включающему введение млекопитающему пролекарства соединения формулы I в количестве, эффективном для лечения или профилактики упомянутого опосредованного киназами состояния. В одном варианте осуществления изобретения пролекарство представляет собой фосфатное пролекарство.
В соответствии с еще одним объектом изобретения настоящее изобретение относится к 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фтор-фенокси)-1Н-индазол-1-ил)этилдигидрофосфату или его фармацевтически приемлемой соли.
Не желая связывать себя теорией, полагают, что фосфатный сложный эфир действует как пролекарство для соответствующего первичного спирта.
Как описано выше, было обнаружено, что фосфатный сложный эфир соединения формулы I обладает особенно хорошей растворимостью.
Термин «фармацевтически приемлемая соль», если не указано иное, включает соли, которые сохраняют биологическую эффективность соответствующей свободной кислоты или основания конкретного соединения и не являются нежелательными биологически или каким-либо иным образом. Соединение по изобретению может содержать достаточно кислую группу, достаточно основную группу или функциональные группы обоих типов и соответственно реагировать с любым числом неорганических или органических оснований или кислот с образованием фармацевтически приемлемой соли. Примеры фармацевтически приемлемых солей включают соли, полученные реакцией соединений по настоящему изобретению с неорганической или органической кислотой или с неорганическим основанием. Такие соли, включают сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, йодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, γ-гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты и манделаты, но не ограничиваются ими. Так как единичные соединения по изобретению могут включать более одного кислотного или основного остатка, соединения по настоящему изобретению могут содержать моно-, ди- или три соли в одном соединении.
Если соединение по изобретению представляет собой основание, желательная фармацевтически приемлемая соль может быть получена любым подходящим способом, имеющимся в данной области техники, например обработкой свободного основания кислым соединением, например неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидильная кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота или подобные.
Если соединение по изобретению представляет собой кислоту, желательная фармацевтически приемлемая соль может быть получена любым подходящим способом, имеющимся в данной области техники, например обработкой свободной кислоты неорганическим или органическим основанием. Примеры подходящих неорганических солей включают соли, образованные щелочными и щелочноземельными металлами, такими как литий, натрий, калий, барий и кальций. Примеры подходящих солей органических оснований включают, например, соли аммония, дибензиламмония, бензиламмония, 2-гидроксиэтиламмония, бис(2-гидроксиэтил)аммония, фенилэтилбензиламина, дибензилэтилендиамина и им подобные соли. Другие соли кислотных остатков могут включать, например, соли, образованные с прокаином, хинином и N-метилглюкозамином, плюс соли, образованные с основными аминокислотами, такими как глицин, орнитин, гистидин, фенилглицин, лизин и аргинин.
Соединения формулы I включают также и другие свои соли, которые необязательно являются фармацевтически приемлемыми солями и которые могут использоваться в качестве промежуточных соединений для получения и/или очистки соединений формулы I и/или для разделения энантиомеров соединений формулы I.
Изобретение также охватывает меченные изотопами соединения по настоящему изобретению, которые идентичны соединениям, перечисленным здесь, но за исключением того, что один или несколько атомов заменены атомом, имеющим атомную массу или атомное число, которые отличаются от атомной массы или атомного числа, обычно встречающихся в природе. Меченные изотопами соединения по настоящему изобретению можно обычно получать по следующим методикам, аналогичным методикам, раскрываемым на схемах и/или в примерах, приведенных ниже, путем замены реагента, не содержащего изотопной метки, реагентом, меченным изотопом.
Синтез соединений по изобретению
Соединения по настоящему изобретению можно получать синтетическими путями, которые включают способы, аналогичные способам, хорошо известным в химии, или как описано в публикации международной патентной заявки WO 2004/078116, в частности, учитывая описание, которое содержится здесь. Исходные вещества обычно доступны из коммерческих источников, таких как Aldrich Chemicals (Milwaukee, WI), или легко получаются способами, которые хорошо известны специалистам в данной области техники (например, получаются способами, как правило, описанными в справочных изданиях (Louis F. Fieser и Mary Fieser, Reagents for Organic Synthesis, v.1-19, Wiley, N.Y. (1967-1999 ed.) или (Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая дополнения (доступны также через online базу данных Beilstein).
Еще один объект настоящего изобретения относится к способу получения соединения формулы I или его фармацевтически приемлемой соли, который включает:
(а) сочетание соединения формулы II
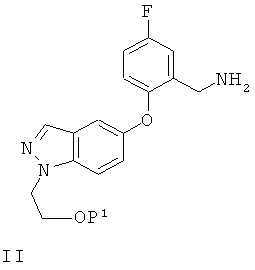
или его соли, в которых Р1 представляет собой атом водорода или группу, защищающую гидроксил, с соединением формулы III
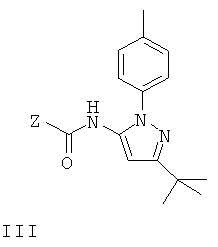
в котором Z представляет собой уходящую группу, или с соответствующим изоцианатом; или
(b) восстановление соединения формулы IV
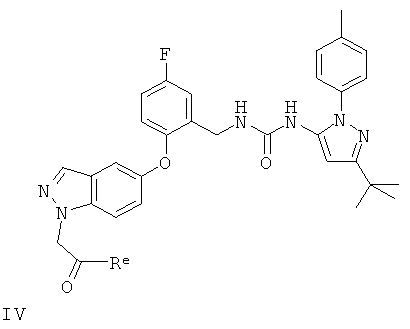
в котором Re представляет собой атом водорода или спиртовый остаток;
с последующим удалением любой защитной группы и, если требуется, образованием фармацевтически приемлемой соли.
Примеры удобных групп, защищающих гидроксил, обозначенных Р1, включают циклические полукетали, такие как тетрагидро-2Н-пиран-2-ил.
Уходящая группа, обозначенная Z, может представлять собой, например, незамещенную или замещенную гидрокарбилоксигруппу, например галоген(1-6С)алкоксигруппу, такую как 2,2,2-трихлорэтокси, алкенилоксигруппу, такую как CH2=O(CH3)O-, или арилоксигруппу, необязательно замещенную, например, одной или несколькими группами, выбранными из F, Cl, Вr и NO2. Конкретные значения для необязательно замещенной арилоксигруппы включают фенокси, 4-хлорфенокси, 4-бромфенокси, 4-фторфенокси, 4-нитрофенокси и 2-нитрофенокси. В конкретном варианте осуществления изобретения Z представляет собой фенокси.
Показано, что соединение II можно успешно выделять с хорошим выходом и высокой степенью чистоты без стадии хроматографии, когда Z в соединении III представляет собой необязательно замещенную арилоксигруппу, такую как феноксигруппа.
Сочетание соединения формулы (II) с соединением формулы (III), когда Z представляет собой необязательно замещенную феноксигруппу, удобно проводить при температуре от 0 до 100°С, и более конкретно при температуре окружающей среды. Подходящие растворители включают апротонные растворители, такие как простые эфиры (например, тетрагидрофуран или п-диоксан), ДМФА, ДМСО или ацетонитрил. Реакцию сочетания удобно проводить в присутствии основания, такого как третичный амин (например, триэтиламина или DMA).
Конкретные значения Re, когда он обозначает остаток спирта, включают (1-6С)алкоксигруппы, такие как этокси.
Соединения формул (II) и (IV), как полагают, являются новыми и относятся к дополнительным объектам изобретения.
Соединения формулы (III), где Z представляет собой необязательно замещенную арилоксигруппу, как полагают, также являются новыми и относятся к дополнительным объектам изобретения.
Еще одним объектом настоящего изобретения является способ получения фосфатного пролекарства соединения формулы I, т.е. 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этилдигидрофосфата или его фармацевтически приемлемой соли, который включает фосфорилирование 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-(5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)бензил)мочевины или ее соли.
Фосфорилирование удобно осуществлять взаимодействием спирта с диалкил- или диарилдиалкилфосфинамидитом, таким как ди-трет-бутил диизопропилфосфинамидит или дифенилдиизопропилфосфинамидит, с последующим удалением алкильных или арильных групп на фосфатном продукте гидролизом или каталитическим гидрированием.
Для пояснения на схемах 1 и 2 и в примерах иллюстрируются способы получения соединений по настоящему изобретению, а также ключевых интермедиатов. Специалисты в данной области поймут, что для синтеза соединений по настоящему изобретению можно использовать другие синтетические пути. Хотя конкретные исходные вещества и реагенты описаны в схемах и рассматриваются ниже, их можно легко заменить другими исходными веществами и реагентами, чтобы обеспечить получение целого ряда производных и/или условия реакции. Кроме того, многие из соединений, полученных способами, описанными ниже, могут быть дополнительно модифицированы с учетом данного описания с помощью традиционной химии, хорошо известной специалистам.
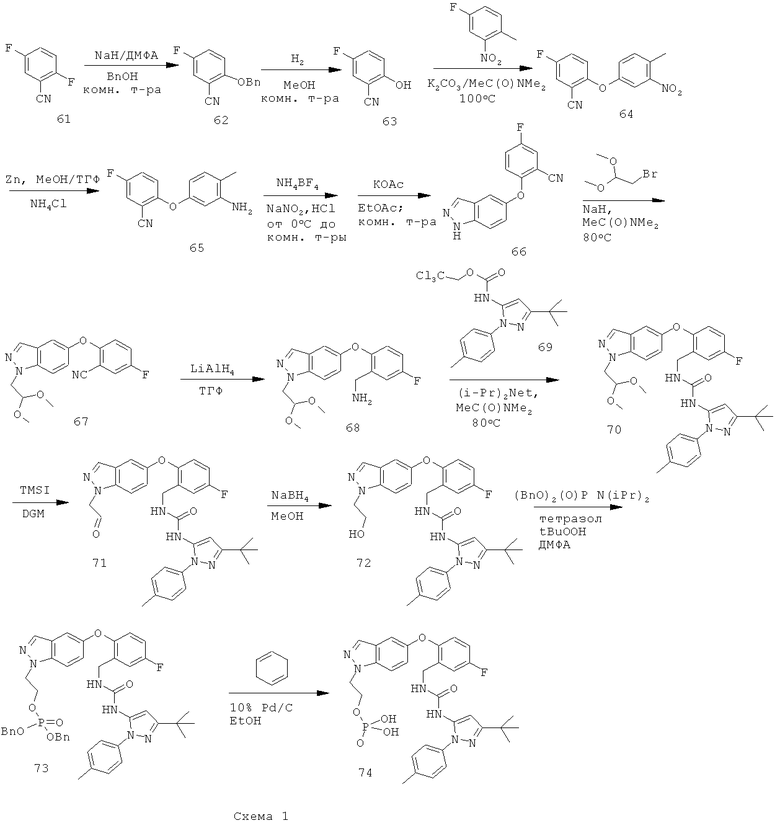
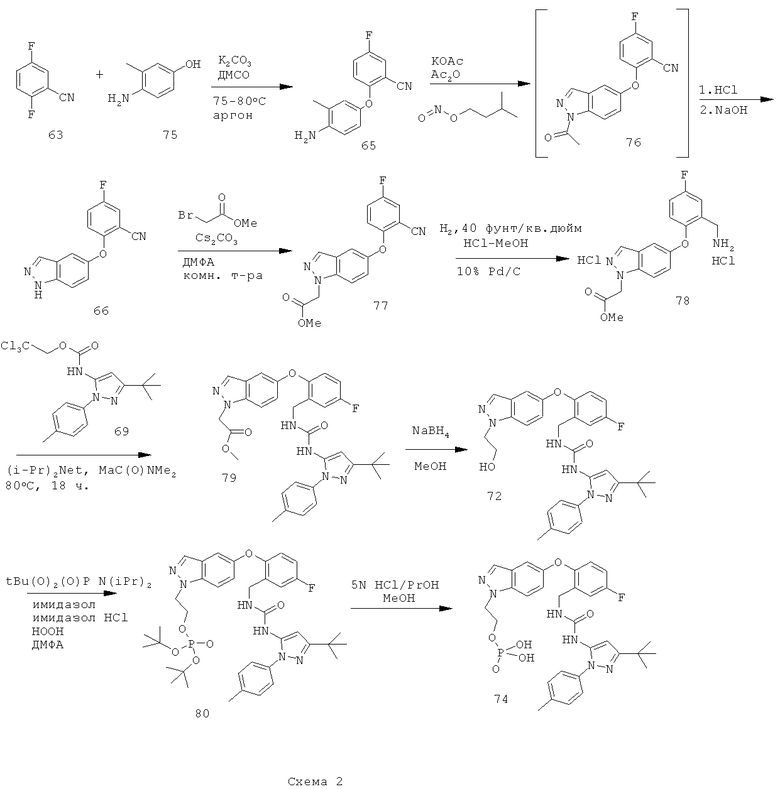
Восстановление нитрильной группы в соединении 77, как показано на схеме 2, требует высоких концентраций кислоты (например, НСl или уксусной кислоты), чтобы свести к минимуму образование димера. Однако более высокие концентрации кислоты могут вызвать гидролиз сложноэфирного производного 78 в соответствующую кислоту. Схема 3 иллюстрирует усовершенствованный способ получения соединения формулы I, который уменьшает образование примесей димера и кислоты.
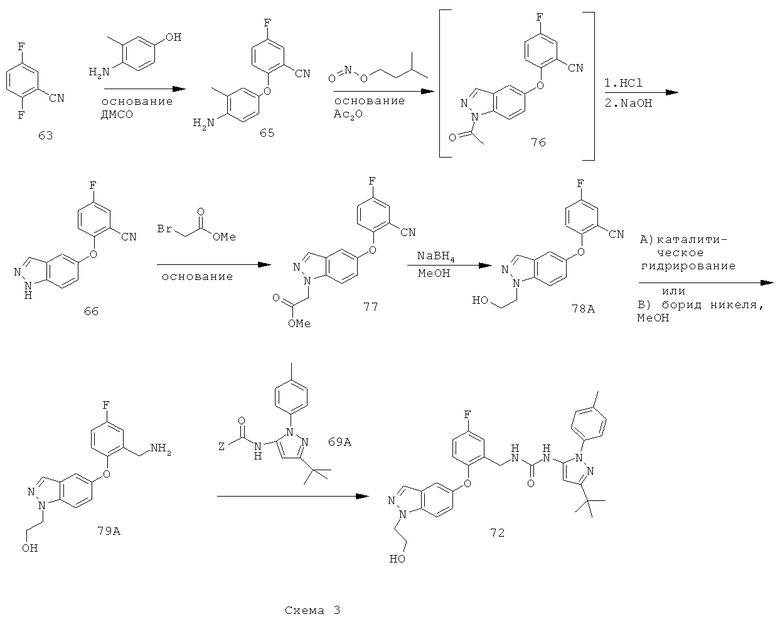
Схема 3 иллюстрирует способ восстановления сложноэфирной группы соединения 77 в соответствующую спиртовую группу перед проведением восстановления нитрильной группы, чтобы получить соединение 79А, не содержащее примесей кислот и с минимальным содержанием примесей димера.
Более конкретно, соединение 77 можно восстанавливать по стадиям: сначала сложноэфирную группу восстанавливают натрийборгидридом в подходящем растворителе, чтобы получить соответствующее спиртовое производное 78А. Затем восстанавливают нитрильную группу соединения 78А в стандартных условиях каталитического гидрирования или действием борида никеля в подходящем органическом растворителе и получают соответствующее метиламиносоединение 79А.
Путь, показанный на схеме 3, обеспечивает несколько преимуществ, по сравнению с путями, показанными на схеме 2, для образования соединения формулы I. Используя пути, показанные на схеме 3, восстановление нитрильной группы можно осуществлять при более высоких концентрациях НСl, что благоприятствует снижению количества примеси образующегося димера. Кроме того, путь, показанный на схеме 3, отменяет использование натрийборгидрида на конечной стадии, что позволяет избежать дополнительных стадий очистки для удаления остаточных борных примесей из конечного продукта. Таким образом, путем исключения стадии восстановления боргидридом натрия ранее в последовательности реакций, остаточный бор можно легко удалить во время промежуточных стадий последовательности. Кроме того, реакцию сочетания аминоспирта 79А с соединением 69А можно проводить при более низких температурах, повышая таким образом чистоту продукта 72. Соответственно, путь, показанный на схеме 3, дает возможность улучшить производительность синтеза соединения формулы I и в результате обеспечивает эффективный синтетический путь получения соединения формулы I, который является более приспособляемым для крупномасштабного производства или более подходящим для такого производства.
Соответственно, предоставляется также способ получения соединения формулы (II), включающий:
(1) восстановление соединения формулы (V)
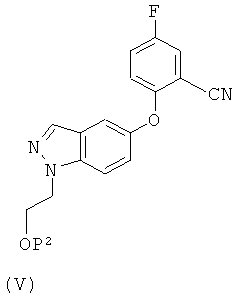
в котором Р2 представляет собой водород или группу, защищающую гидроксил, в условиях каталитического гидрирования или в присутствии борида никеля.
Касательно стадии (i): катализаторы гидрирования включают любой подходящий палладиевый катализатор, такой как Pd(OH)2 или палладий, нанесенный на уголь. Гидрирование происходит в кислых условиях (таких, как при прибавлении кислоты, например НС1 или уксусной кислоты) или при добавлении аммиака. Каталитическое гидрирование можно проводить в любой подходящей системе растворителей, таких как спирт (например, метанол, этанол, изопропанол), сложный эфир (например, этилацетат) или простой эфир (например, ТГФ). Смешанные растворители, например спирт и ТГФ, также подходят для стадии гидрирования. Давление водорода может составлять от 25 до 100 фунт/кв. дюйм, например 40 фунт/кв. дюйм. Восстановление обычно проводят при температурах от 20 до 100°С.
Касательно стадии (i): борид никеля можно получать in situ из соли переходного металла, предпочтительно соли Ni(II), и натрийборгидрида. В предпочтительном варианте осуществления изобретения борид никеля получают из хлорида никеля (II) и натрийборгидрида. Реакцию удобно проводить в подходящем растворителе, таком как спирт (например, метанол, этанол, изопропанол). Восстановление обычно проводят при температуре окружающей среды.
Соединение формулы (V) может быть получено восстановлением соединения формулы (VI)
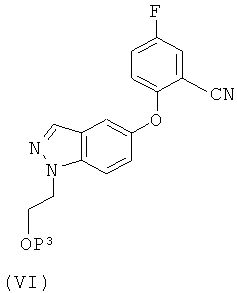
где Р3 такой, как определено для Р2, используя условия восстановления для любого сложного эфира (например, натрийборгидрид) в подходящем растворителе, таком как спирт (например, метанол, этанол, изопропанол).
При получении соединений по настоящему изобретению может быть необходима защита отдельных функциональных групп (например, первичных или вторичных аминогрупп, спиртов и т.п.) промежуточных соединений. Необходимость такой защиты будет варьироваться в зависимости от природы функциональной группы и условий способов получения. Например, подходящие аминозащитные группы (NH-Pg) включают ацетил, трифторацетил, трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBz) и 9-флуоренилметиленоксикарбонил (Fmoc). Удобные защитные группы для гидроксила включают тетрагидро-2Н-пиран-2-ильную, бензильную, триалкилсилильную и ацетальную. Необходимость такой защиты легко определяет специалист в данной области. Для общего описания защитных групп и их применения см. (Т.W.Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991).
СПОСОБЫ ЛЕЧЕНИЯ
Соединения по изобретению можно использовать для лечения заболеваний, опосредованных модуляцией или регуляцией протеинкиназ. Соответственно, еще один объект изобретения относится к способам лечения или профилактики заболеваний или состояний, описанных здесь, путем введения млекопитающему, например человеку, терапевтически эффективного количества соединения по настоящему изобретению или его сольвата, метаболита или фармацевтически приемлемой соли в количестве, эффективном для лечения или профилактики указанного заболевания. В одном варианте осуществления изобретения способ включает введение млекопитающему соединения по настоящему изобретению в количестве, эффективном для ингибирования одной или нескольких киназ.
Термин «эффективное количество» относится к количеству соединения, которое при введении млекопитающему, нуждающемуся в таком лечении, является достаточным, чтобы осуществить лечение заболевания, опосредованного активностью одной или нескольких протеинкиназ, таких как р38 МАРК, и связанных с ними явлений, опосредованных киназами, таких как продуцирование цитокинов. Так, например, терапевтически эффективное количество соединения по настоящему изобретению, или его соли, или его активного метаболита представляет собой количество, достаточное для модуляции, регуляции или ингибирования активности одной или нескольких протеинкиназ таким образом, чтобы уменьшить или облегчить болезненное состояние, которое опосредовано такой активностью.
Термин «лечение» предназначен для обозначения по меньшей мере облегчения болезненного состояния у млекопитающего, например человека, на которое влияет, по меньшей мере частично, активность одной или нескольких протеинкиназ. Термины «лечить» и «лечение» относятся как к терапевтическому лечению, так и к профилактическим или превентивным мерам, где объект должен предотвратить или замедлить (уменьшить) нежелательное физиологическое изменение или нарушение. Для целей настоящего изобретения полезные или желательные клинические результаты включают (но не ограничиваются ими) облегчение симптомов, уменьшение степени развития заболевания, стабилизированное (т.е. не ухудшающееся) состояние болезни, задержка или замедление прогрессирования заболевания, улучшение или облегчение болезненного состояния и ремиссию (или частичную, или полную), или определяемую, или неопределяемую. «Лечение» может также означать продление времени жизни по сравнению с временем жизни, ожидаемым при отсутствии лечения. Выражение «те, кто нуждается в лечении» включает тех, кто уже страдает рассматриваемым заболеванием или состоянием, а также тех, кто подвержен такому заболеванию или состоянию, или тех, у кого необходимо предотвратить такое заболевание или состояние.
Количество соединения по настоящему изобретению, вводимое млекопитающему, будет варьироваться в зависимости от ряда факторов, таких как конкретное соединение, болезненное состояние и его тяжесть, особенность (например, масса тела) млекопитающего, нуждающегося в лечении, но может быть определено по стандартной методике специалистом в данной области.
Термин «млекопитающее», используемый здесь, относится к теплокровному животному, которое страдает заболеванием, описанным здесь, или для которого существует риск развития такого заболевания, и включает (но не ограничивается перечисленными ниже) морских свинок, собак, кошек, крыс, мышей, хомяков и приматов, в том числе людей.
Один объект настоящего изобретения относится к соединениям по настоящему изобретению или их фармацевтическим солям, которые могут входить в состав фармацевтических композиций для введения животным или людям для лечения или профилактики опосредованного киназами состояния. Термин «опосредованное киназами состояние», используемый здесь, обозначает любое заболевание или другое опасное состояние, в котором, как известно, играет роль р38, и включает состояния, которые, как известно, вызваны сверхпродуцированием IL-1, TNF, IL-6 или IL-8. Такие состояния включают (но не ограничиваются перечисленными) воспалительные заболевания, аутоиммунные заболевания, деструктивные заболевания костей, пролиферативные заболевания, инфекционные заболевания, вирусные заболевания, фиброзные заболевания и нейродегенеративные заболевания.
Воспалительные заболевания, которые можно лечить или предотвращать, включают острый панкреатит, хронический панкреатит, астму, аллергии и синдром дыхательной недостаточности у взрослых, но не ограничиваются ими.
Аутоиммунные заболевания, которые можно лечить или предотвращать, включают (но не ограничиваются перечисленными ниже) гломералонефрит, ревматоидный артрит, системную красную волчанку, склеродерму, хронический тиреоидит, базедову болезнь, аутоиммунный гастрит, инсулинозависимый сахарный диабет (тип I), аутоиммунную гемолитическую анемию, аутоиммунную нейтропению, тромбоцитопению, атопический дерматит, хронический активный гепатит, астенический бульбарный паралич, рассеянный склероз, воспалительные заболевания кишечника, язвенный колит, болезнь Крона, псориаз или реакцию «трансплантат против хозяина».
Деструктивные заболевания костей, которые можно лечить или предотвращать, включают (но не ограничиваются перечисленными ниже) остеопороз, остеоартрит и множественные, связанные с миеломой, заболевания костей.
Фиброзные заболевания, которые можно лечить или предотвращать, включают (но не ограничиваются перечисленными ниже) идиопатический фиброз легких, фиброз почек и печени.
Пролиферативные заболевания, которые можно лечить или предотвращать, включают (но не ограничиваются перечисленными ниже) острый миелогенный лейкоз, хронический миелогенный лейкоз, хроническую меланому, саркому Капоши, миелодиспластический синдром, множественную миелому, астроцитому, рак кости, рак мозга, рак молочной железы, колоректальный рак, рак желудка, глиому, глиобластому, многоформный рак головы и шеи, гематологический рак, нарушения кроветворения, интерстициальные легочные процессы, лимфоцитарный лейкоз, меланому, миелоидный лейкоз, немелкоклеточный рак легких, рак яичников, рак предстательной железы, саркому, рак кожи, мелкоклеточный рак легких и рак желудка. Другие пациенты, которых можно лечить, включают тех, кто подвергается трансплантации костного мозга.
Инфекционные заболевания, которые можно лечить или предотвращать, включают (но не ограничиваются перечисленными ниже) сепсис, септический шок и шигеллез.
Вирусные заболевания, которые можно лечить или предотвращать, включают (но не ограничиваются перечисленными ниже) острую инфекцию гепатита (включая гепатит А, гепатит В и гепатит С), ВИЧ-инфекция и CMV-ретинит.
Дегенеративные состояния или заболевания, которые можно лечить или предотвращать соединениями по настоящему изобретению, включают (но не ограничиваются перечисленными ниже) болезнь Альцгеймера, болезнь Паркинсона, ишемию головного мозга и другие нейродегенеративные заболевания.
Термин «опосредованные киназами состояния» включает также ишемию/реперфузию при инсульте, сердечные приступы, ишемию миокарда, гипоксию органов, сосудистую гиперплазию, гипертрофию сердца и индуцированную тромбином агрегацию тромбоцитов.
Кроме того, ингибиторы киназ по настоящему изобретению применимы также для ингибирования экспрессии индуцируемых провоспалительных протеинов, таких как простагландинэндопероксидсинтаза-2 (PGHS-2), также называемая циклооксигеназой-2 (СОХ-2). Следовательно, другие «опосредованные киназами состояния» включают (но не ограничиваются ими) отек, отсутствие болевой чувствительности, лихорадку и боль, такую как нервно-мышечную боль, головную боль, боль при раке, зубную боль и боль при артрите.
Состояния и заболевания, которые можно лечить и предотвращать ингибиторами киназ по настоящему изобретению, можно сгруппировать удобным образом по цитокину (например, IL-1, TNF, IL-6, IL-8), который, как полагают, является ответственным за заболевание.
Таким образом, опосредованное IL-1 заболевание или состояние включает ревматоидный артрит, остеоартрит, инсульт, эндотоксемию и/или синдром токсического шока, воспалительную реакцию, индуцированную эндотоксином, воспалительное заболевание кишечника, туберкулез, атеросклероз, дегенерацию мышц, кахексию, псориатический артрит, синдром Рейтера, подагру, травматический артрит, краснушный артрит, острый синовит, диабет, панкреатическое заболевание β-клеток и болезнь Альцгеймера.
Заболевания или состояния, опосредованные TNF, включают (но не ограничены ими) ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие виды артритов, сепсис, септический шок, эндотоксический шок, грамотрицательный сепсис, синдром токсического шока, синдром дыхательной недостаточности у взрослых, церебральную малярию, хроническое воспалительное заболевание легких, силикоз, саркоидоз легких, резорбцию костей, повреждение при реперфузии, реакцию «трансплантат против хозяина», отторжение аллотрансплантатов, лихорадку и боли в мышцах, обусловленные инфекцией, кахексию, вторичную в отношении инфекции, СПИД, СПИД-ассоциированный комплекс или злокачественность, образование келоидов, образование рубцов на тканях, болезнь Крона, язвенный колит или пирезис. Заболевания, опосредованные TNF, включают также вирусные инфекции, такие как ВИЧ, CMV (вирус мозаики цветной капусты), грипп и герпес; и ветеринарные вирусные инфекции, такие как лентивирусные инфекции, включающие вирус конской инфекционной анемии, козлиный вирус артрита, висна-вирус или меди-вирус, но не ограниченные ими; или ретровирусные инфекции, включающие кошачий вирус иммунодефицита, бычий вирус иммунодефицита или собачий вирус иммунодефицита.
Заболевания или состояния, опосредованные IL-8, включают заболевания, характеризующиеся выраженным нейтрофильным инфильтратом, такие как псориаз, воспалительные заболевания кишечника, астма, реперфузионные повреждения сердца и почек, синдром дыхательной недостаточности у взрослых, тромбоз и гломерулонефрит, но не ограничиваются ими.
Кроме того, соединения по настоящему изобретению могут быть использованы местно для лечения или профилактики состояний, вызванных или усиленных IL-1 или TNF. Такие состояния включают (но не ограничиваются ими) воспаленные суставы, экзему, псориаз, воспалительные кожные заболевания, такие как солнечный ожог, воспалительные заболевания глаз, такие как конъюнктивит, пирезис, боль и другие заболевания, связанные с воспалением.
Хотя соединения по настоящему изобретению в первую очередь имеют значение как терапевтические средства для применения на теплокровных животных (включая человека), они также применимы во всех случаях, когда требуется ингибировать действие цитокинов. Следовательно, они применимы в качестве фармакологических стандартов для использования при разработке новых биологических тестов и при поиске новых фармакологических средств.
Размер дозы соединения по настоящему изобретению для лечебных или профилактических целей будет, разумеется, варьироваться в зависимости от природы и тяжести заболевания, возраста и пола животного или пациента и пути введения в соответствии с известными принципами медицины.
Еще один аспект настоящего изобретения относится к соединению по изобретению для применения в качестве лекарственного средства для лечения заболеваний или состояний, описанных здесь, у млекопитающего, например человека, страдающего таким заболеванием или состоянием. Изобретение также относится к применению соединения по изобретению для получения лекарственного средства для лечения заболеваний и состояний, описанных в настоящей заявке, у теплокровных животных, таких как млекопитающие, например люди, страдающие такими заболеваниями.
Комбинированная терапия
Соединения по настоящему изобретению можно использовать в сочетании с другими лекарственными средствами и видами терапии, используемыми в лечении болезненных состояний, для которых полезно ингибирование киназ и цитокинов, ассоциированных с ними, таких как IL-1, TNF, IL-6 или IL-8. Доза второго лекарственного средства может быть выбрана должным образом на основании дозы, используемой в клинике. Соотношение соединения по изобретению и второго лекарственного средства можно правильно определить при соответствии субъекту введения, пути введения, заболеванию-мишени, клиническому состоянию, комбинации и другим факторам. В случаях, где субъектом введения является, например, человек, второе лекарственное средство можно использовать в количестве от 0,01 до 100 массовых частей на массовую часть соединения по настоящему изобретению.
Второе лекарственное средство рецептуры фармацевтической комбинации или схемы приема обладает, например, дополнительными видами активности в отношении соединения по настоящему изобретению, такими, что они не влияют неблагоприятно друг на друга. Такие лекарственные средства соответственно входят в состав комбинаций в количествах, которые эффективны для подразумеваемой цели. Соответственно, еще одним объектом настоящего изобретения является композиция, содержащая соединение по настоящему изобретению в сочетании со вторым лекарственным средством, таким, как описано здесь.
Соединение по настоящему изобретению и дополнительное фармацевтически лекарственное средство (или средства) можно вводить вместе в единой фармацевтической композиции или отдельно, и, когда их вводят отдельно, введение может происходить одновременно или последовательно в любом порядке. Такие последовательные введения могут происходить быстро друг за другом или через некоторый интервал времени.
Количества соединения по настоящему изобретению и второго лекарственного средства (или средств) и соответствующее время введения выбирают так, чтобы добиться желательного комбинированного терапевтического эффекта.
Комбинированная терапия может приводить к «синергизму» и демонстрировать «синергический» эффект, т.е. эффект, который достигается в результате совместного использования активных ингредиентов, больше суммы эффектов, получаемых при использовании соединений по отдельности. Синергический эффект может достигаться, когда соединение по настоящему изобретению и второе лекарственное средство представляют собой: (1) входящие вместе в одну рецептуру и введенные или доставленные одновременно в объединенной стандартной лекарственной форме; (2) доставляемые путем чередования или параллельного введения в виде отдельных рецептур или (3) по некоторым другим схемам. При альтернативной терапии синергический эффект может достигаться, когда соединение по настоящему изобретению и второе лекарственное средство вводят или доставляют последовательно, например, различными инъекциями из отдельных шприцев. Например, во время альтернативной терапии эффективную дозу каждого активного ингредиента можно вводить последовательно, т.е. сериями, тогда как при комбинированной терапии эффективные дозы двух или более активных ингредиентов вводят вместе.
Например, благодаря способности ингибировать цитокины, соединения по настоящему изобретению важны для лечения некоторых воспалительных и невоспалительных заболеваний, которые в настоящее время лечат ингибирующим циклооксигеназу нестероидным противовоспалительным лекарственным средством (NSAID), таким как индометацин, кеторолак, ацетилсалициловая кислота, ибупрофен, сулиндак, толметин и пироксикам. Совместное введение соединения по настоящему изобретению и NSAID может привести в результате к уменьшению количества второго лекарственного средства, необходимого для получения терапевтического эффекта, и, следовательно, снижается вероятность вредных побочных эффектов NSAID, таких как воздействие на желудочно-кишечный тракт. Таким образом, согласно дополнительному признаку изобретения оно относится к фармацевтической композиции, которая содержит соединение по настоящему изобретению или его фармацевтически приемлемую соль в комбинации или в смеси с ингибирующим циклооксигеназу нестероидным противовоспалительным средством и фармацевтически приемлемым разбавителем или носителем.
Соединения по настоящему изобретению можно также использовать для лечения таких состояний, как ревматоидный артрит, в сочетании со средствами против артрита, такими как золото, метотрексат, стероиды и пенициллинамин, и таких состояний, как остеоартрит, в сочетании со стероидами.
Соединения по настоящему изобретению можно также использовать для лечения разрушающих здоровье заболеваний, например остеоартрита, в сочетании с хондропротективными, антиразрушительными и/или репаративными средствами, такими как диацерин, рецептуры с гиалуроновой кислотой, такие как Hyalan, румалон, артепарон, и соли глюкозамина, такие как антрил.
Соединения по настоящему изобретению можно также использовать для лечения астмы в сочетании с антиастматическими средствами, такими как бронходилаторы и антагонисты лейкотриена.
Введение соединений по изобретению
Соединения по настоящему изобретению можно вводить любым путем, подходящим для состояния, подвергаемого лечению. Подходящие пути включают пероральный, парентеральный (включая подкожный, внутримышечный, внутривенный, внутриартериальный, интрадермальный, интратекальный и эпидуральный), трансдермальный, ректальный, назальный, местный (включая буккальный и сублингвальный), вагинальный, интраперитонеальный, внутрилегочный и интраназальный. Следует понимать, что используемый путь может изменяться, например, в зависимости от состояния реципиента. В тех случаях когда соединение вводят перорально, оно может входить в состав пилюли, капсулы, таблетки и т.п. с фармацевтически приемлемым носителем или эксципиентом. Для парентерального введения соединения может быть приготовлена стандартная лекарственная форма для инъекций, в состав которой входит соединение и фармацевтически приемлемая парентеральная среда, как подробно описано ниже.
Фармацевтические рецептуры
Для использования соединения по настоящему изобретению для терапевтического лечения (включая профилактику) млекопитающих, включая человека, обычно его используют в виде фармацевтической композиции в соответствии со стандартной фармацевтической практикой. Согласно этому объекту изобретение относится к фармацевтической композиции, содержащей соединение по настоящему изобретению вместе с фармацевтически приемлемым разбавителем или носителем.
Фармацевтические композиции по изобретению составляют, дозируют и вводят с использованием количеств, концентраций, схем, курса, среды и пути, которые согласуются с надлежащей медицинской практикой. Факторы для рассмотрения в данном контексте включают конкретное заболевание, подвергаемое лечению, млекопитающее, подвергаемое лечению, клиническое состояние конкретного пациента, причину заболевания, место доставки лекарственного средства, способ введения, схему введения и другие факторы, известные лечащему врачу. Терапевтически эффективное количество соединения для введения следует определять с учетом всех этих факторов, и оно представляет собой минимальное количество соединения, необходимое для предотвращения, улучшения или лечения заболевания. Соединение по настоящему изобретению обычно вводят в состав фармацевтических лекарственных форм, чтобы обеспечить легко контролируемую дозу лекарственного средства и дать возможность пациенту следовать предписанной схеме.
Композиция для применения по данному изобретению предпочтительно является стерильной. В частности, рецептуры, используемые для введения in vivo, должны быть стерильными. Такую стерилизацию легко осуществить, например, фильтрацией через стерильные мембраны для фильтрации. Соединение обычно можно хранить в виде твердой композиции, лиофилизованной рецептуры или в виде водного раствора.
Фармацевтические композиции соединений по настоящему изобретению могут быть приготовлены для различных путей и типов введения. Например, соединение по настоящему изобретению, имеющее нужную степень чистоты, необязательно можно смешивать с фармацевтически приемлемыми разбавителями, носителями, эксципиентами или стабилизаторами (Remington's Pharmaceutical Sciences (1980) 16th edition, Osol, A. Ed.) в виде лиофилизованного состава, молотого порошка или водного раствора. Приготовление композиции можно проводить путем смешивания соединения при температуре окружающей среды при соответствующем рН и при требуемой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, которые нетоксичны для реципиентов в используемых дозах и концентрациях. Величина рН композиции зависит в основном от конкретного применения и концентрации соединения, но может изменяться в пределах, например, от около 3 до около 8. Композиция в ацетатном буфере при рН5 представляет собой подходящий вариант осуществления изобретения. Композиции можно готовить, используя обычные процедуры растворения и смешивания. Например, нерасфасованное лекарственное вещество (т.е. соединение по настоящему изобретению или стабилизированная форма соединения (например, комплекс с циклодекстриновым производным или другим известным комплексообразующим агентом)) растворяют в подходящем растворителе в присутствии одного или нескольких эксципиентов.
Конкретный используемый носитель, разбавитель или эксципиент зависит от средства и цели, для которых применяют соединение по настоящему изобретению.
Растворители обычно выбирают, базируясь на растворителях, признанных специалистами в данной области как безопасные (GRAS) для введения млекопитающим. Вообще безопасные растворители представляют собой нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы в воде или могут смешиваться с водой. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, PEG 400, PEG 300) и т.п. и их смеси. Приемлемые разбавители, носители, эксципиенты и стабилизаторы являются нетоксичными по отношению к реципиентам в используемых дозах и концентрациях и включают буферы, такие как фосфат, цитрат и другие органические кислоты; антиоксиданты, включающие аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмонийхлорид, гексаметионинхлорид, бензалконийхлорид, бензетонийхлорид, фенол, бутиловый или бензиловый спирт, алкилпарабены, такие как метил- или пропилпарабен, катехол, резорцин, циклогексанол, 3-пентанол; и м-крезол); полипептиды с низкой молекулярной массой (меньше чем примерно 10 остатков); белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включающие глюкозу, маннозу или декстрины; хелатообразующие агенты, такие как EDTA; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы металлов (например, комплексы Zn-белок); и/или неионогенные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (PEG). Рецептуры могут также включать один или несколько стабилизирующих агентов, поверхностно-активных веществ, увлажняющих агентов, смазывающих веществ, эмульгаторов, суспендирующих агентов, консервантов, антиоксидантов, веществ для придания непрозрачности, веществ, способствующих скольжению, технологических добавок, окрашивающих веществ, подслащивающих веществ, ароматизирующих добавок, вкусовых добавок и других известных добавок для обеспечения красивого внешнего вида лекарственного средства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или облегчения изготовления фармацевтического продукта (т.е. лекарственного средства). Активные фармацевтические ингредиенты можно также помещать в микрокапсулы, полученные, например, методом коацервации или межфазной полимеризацией, например микрокапсулы из гидроксиметилцеллюлозы или желатина и поли((метилметакрилата) соответственно; в коллоидные системы для доставки лекарственных средств (например, липосомы, микросферы альбумина, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие методики описаны в (Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980)). «Липосома» представляет собой небольшой пузырек, состоящий из липидов различных типов, фосфолипидов и/или поверхностно-активных веществ, которые применимы для доставки лекарственного средства млекопитающему. Компоненты липосомы обычно упорядочены в двухслойное образование, аналогичное размещению липидов в биологических мембранах.
Можно изготавливать препараты соединения по настоящему изобретению с замедленным высвобождением. Подходящие примеры препаратов с замедленным высвобождением включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащих соединение по настоящему изобретению, причем матрицы имеют форму профилированных изделий, например пленок или микрокапсул. Примеры матриц с замедленным высвобождением включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт)), полилактиды (патент США №3,773,919), сополимеры L-глутаминовой кислоты и гамма-этил-L-глутамата, не способный разлагаться этиленвинилацетат, способные разлагаться сополимеры молочной кислоты и гликолевой кислоты, такие как LUPRON DEPOT™ (микросферы для инъекций, состоящие из сополимера молочной кислоты и гликолевой кислоты и лепролид ацетата) и поли-D-(-)-3-гидроксимасляная кислота.
Фармацевтическая композиция по настоящему изобретению может быть в виде стерильного препарата для инъекций, такого как стерильная водная или масляная суспензия для инъекций. Такую суспензию можно приготовить в соответствии с известными в данной области способами, используя подходящие суспендирующие и увлажняющие агенты, которые были упомянуты выше. Стерильный препарат для инъекций может также представлять собой стерильный раствор или суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как раствор в 1,3-бутандиоле, или может быть получен в виде лиофилизированного порошка. Приемлемыми средами и растворителями, которые можно использовать, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды можно обычно использовать стерильные нелетучие масла. Для такой цели можно использовать любое безвкусное нелетучее масло, включая синтетические моно- и диглицериды. Кроме того, для получения препаратов для инъекций можно использовать жирные кислоты, такие как олеиновая кислота.
Фармацевтические композиции по настоящему изобретению, подходящие для парентерального введения включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые делают композицию изотонической в отношении крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители.
Композиции по изобретению могут также быть приготовлены в форме, подходящей для перорального применения (например, в виде таблеток, леденцов, твердых и мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков и гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения ингаляцией (например, в виде тонкоизмельченного порошка или жидкого аэрозоля), для введения инсуффляцией (например, в виде тонкоизмельченного порошка).
Подходящие фармацевтически приемлемые эксципиенты для рецептуры таблеток включают, например, инертные разбавители, такие как лактоза, карбонат натрия, фосфат кальция или карбонат кальция, гранулирующие агенты, разрыхлители, такие как кукурузный крахмал или альгиновая кислота; связующие агенты, такие как крахмал; смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк; консерванты, такие как этил- или пропил-п-гидроксибензоат, и антиоксиданты, такие как аскорбиновая кислота. Композиции в виде таблеток могут быть без нанесенного покрытия или с нанесенным покрытием или для модифицирования их распадаемости и последующего всасывания активного ингредиента в желудочно-кишечном тракте, или для улучшения их стабильности и/или внешнего вида в любом случае используют широко распространенные вещества для покрытий и методики, известные в данной области техники.
Композиции для перорального применения могут быть изготовлены в виде твердых желатиновых капсул, в которых активный ингредиент смешивают с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешивают с водой или маслом, таким как арахисовое масло, жидкий парафин или оливковое масло.
Водные суспензии обычно содержат активный ингредиент в виде токоизмельченного порошка вместе с одним или несколькими суспендирующими агентами, такими как натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и акациевая камедь; диспергирующие и увлажняющие агенты, такие как лецитин или продукты конденсации алкиленоксидов с жирными кислотами (например, полиоксиэтиленстеарат), или продукты конденсации оксида этилена с неполными сложными эфирами, производными жирных кислот и гексита, такие как полиоксиэтиленсорбитолмоноолеат, или продукты конденсации оксида этилена с неполными сложными эфирами, производными жирных кислот и ангидридов гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов (таких как этил- или пропил-п-гидроксибензоат), антиоксидантов (таких как аскорбиновая кислота), окрашивающих веществ, вкусовых добавок и/или подслащивающих веществ (таких как сахароза, сахарин или аспартам).
Масляные суспензии могут быть приготовлены путем суспендирования активного ингредиента в растительном масле (таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло) или в минеральном масле (таком как жидкий парафин). Масляные суспензии могут также содержать загуститель, такой как пчелиный воск, тяжелый парафин или цетиловый спирт. Для получения приятного на вкус перорального препарата могут быть прибавлены подслащивающие вещества, такие как упомянуты выше, и вкусовые добавки. Эти композиции можно сохранять прибавлением антиоксидантов, таких как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для получения водной суспензии путем прибавления воды обычно содержат активный ингредиент вместе с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или несколькими консервантами. Подходящие диспергирующие или увлажняющие агенты и суспендирующие агенты являются примерами агентов, уже упомянутых выше. Дополнительные эксципиенты, такие как подслащивающие вещества, вкусовые добавки и красящие вещества, также могут присутствовать.
Фармацевтические композиции по изобретению могут также быть в виде эмульсий масло-в-воде. Масляная фаза может быть растительным маслом, таким как оливковое масло или арахисовое масло, или минеральным маслом, таким как, например, жидкий парафин или смесь любых из них. Подходящими эмульгаторами могут быть, например, встречающиеся в природе смолы, такие как акациевая камедь и трагакантовая камедь, встречающиеся в природе фосфатиды, такие как соевые бобы, лецитин, сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гексита (например, сорбитаноноолеат), и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, такие как полиоксиэтиленсорбитанмоноолеат. Эмульсии могут также содержать подслащивающие вещества, вкусовые добавки и консерванты.
Сиропы и эликсиры могут быть изготовлены с подслащивающими веществами, такими как глицерин, пропиленгликоль, сорбит, аспартам или сахароза, и могут также содержать смягчающее средство, консервант, вкусовые добавки и/или красящие вещества.
Рецептуры суппозиториев могут быть приготовлены путем смешивания активного ингредиента с подходящим, не вызывающим раздражения эксципиентом, который является твердым при обычной температуре, но становится жидким при ректальной температуре и, следовательно, плавится в прямой кишке, высвобождая лекарственное средство. Подходящие эксципиенты включают, например, масло какао и полиэтиленгликоли. Рецептуры, подходящие для вагинального введения, могут существовать в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев, содержащих, кроме активного ингредиента, такие подходящие носители, которые известны в данной области техники.
Рецептуры для местного введения, такие как кремы, мази, гели и водные или масляные растворы или суспензии, можно, как правило, получать путем составления смеси активного ингредиента с обычным приемлемым для местного применения носителем или разбавителем по общепринятым методикам, хорошо известным в данной области техники.
Композиции для трансдемального введения могут быть изготовлены в виде трансдермальных кожных пластырей, которые хорошо известны специалистам средней квалификации в данной области техники.
Композиции для введения путем инсуффляции могут быть приготовлены в виде тонкоизмельченного порошка, содержащего частицы, средний диаметр которых составляет, например, 30 мкм или намного меньше, причем порошок содержит или один активный ингредиент или разбавлен одним или несколькими физиологически приемлемыми носителями, такими как лактоза. Порошок для инсуффляции затем удобно сохранять в капсуле, содержащей, например, 1-50 мг активного ингредиента для применения с турбоингалятором, таким как используют для инсуффляции известного средства хромогликата натрия.
Композиции для введения ингаляцией могут быть в виде традиционного аэрозоля, находящегося под давлением, устроенного таким образом, чтобы распылять активный ингредиент либо в виде аэрозоля, содержащего тонкоизмельченное твердое вещество, либо в виде жидких капель. Можно использовать традиционные пропелланты для аэрозоля, такие как летучие фторированные углеводороды или углеводороды, и аэрозольное устройство предусматривает удобное распыление отмеренного количества активного ингредиента.
Фармацевтическая композиция (или рецептура) для применения может быть упакована различными путями в зависимости от способа, используемого для введения лекарственного средства. Например, готовое изделие может включать контейнер, в котором находится фармацевтическая композиция в соответствующей форме. Подходящие контейнеры хорошо известны специалистам и включают такие виды как флаконы (пластмассовые и стеклянные), саше, ампулы, пластиковые мешки, металлические цилиндры и т.п. Контейнер может также включать монтаж, исключающий несанкционированный доступ, для предотвращения неосторожного доступа к содержимому упаковки. Кроме того, на контейнере имеется этикетка с описанием содержимого контейнера. Этикетка может также включать соответствующие предупреждения. Композиции могут также быть упакованы в контейнеры для одной дозы и контейнеры для нескольких доз, например запаянные ампулы и сосуды, и могут храниться в лиофилизированном состоянии, что требует только прибавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием. Экстемпоральные инъекционные растворы и суспензии приготавливают из стерильных порошков, гранул и таблеток ранее описанного типа. Предпочтительными стандартными лекарственными формами являются такие, которые содержат дневную дозу или стандартную дневную субдозу активного ингредиента, как изложено выше, или соответствующую ее часть.
Кроме того, изобретение относится к композициям для ветеринарии, содержащим по меньшей мере один активный ингредиент, как определено выше, вместе с носителем для ветеринарии. Носители для ветеринарии представляют собой вещества, применимые для введения композиции, и могут быть твердыми, жидкими или газообразными веществами, которые в остальном инертны или приемлемы для ветеринарии и совместимы с активным ингредиентом. Упомянутые ветеринарные композиции можно вводить парентерально, перорально или любым другим требуемым путем.
Количество соединения по настоящему изобретению, которое объединяют с одним или несколькими эксципиентами для получения единой лекарственной формы, необходимо варьировать в зависимости от субъекта, подвергаемого лечению, тяжести заболевания или состояния, скорости введения, используемого соединения и указаний лечащего врача. В одном варианте осуществления изобретения подходящее количество соединения по настоящему изобретению вводят млекопитающему, нуждающемуся в этом. В одном из вариантов осуществления имеет место введение в количестве от около 0,001 мг/кг массы тела до около 60 мг/кг массы тела в день. В еще одном варианте осуществления изобретения имеет место введение в количестве 0,5 мг/кг массы тела до около 40 мг/кг массы тела в день. В некоторых примерах дозировки ниже нижнего предела вышеуказанного интервала могут быть более подходящими, хотя в некоторых случаях могут использоваться еще более высокие дозы, причем они не вызывают никакого вредного побочного действия, при условии, что такие более высокие дозы сначала разделяют на несколько небольших доз для введения в течение дня. Для получения дополнительной информации о путях введения и схемах дозировки см. Chapter 25.3 in Volume 5 of Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990, которая включена в данную заявку путем ссылки.
Готовые изделия
Еще один вариант осуществления настоящего изобретения относится к промышленному изделию или «набору», содержащему вещества, применимые для лечения заболеваний, описанных выше. В одном варианте осуществления изобретения набор включает контейнер, содержащий соединение по настоящему изобретению. Подходящие контейнеры включают, например, флаконы, сосуды, шприцы, блистерные упаковки и т.п. Контейнер может быть изготовлен из разных материалов, таких как стекло или пластмасса. В контейнере может храниться соединение по настоящему изобретению или его композиция, которые эффективны для лечения заболевания и могут иметь стерильный порт доступа (например, контейнер может представлять собой мешок для раствора для внутривенного вливания или сосуд с пробкой, которая поддается прокалыванию гиподермальной инъекционной иглой).
Набор может также дополнительно содержать на контейнере или связанные с контейнером этикетку или вкладыш в упаковку. Термин «вкладыш в упаковку» относится к инструкциям, обычно включаемым в упаковки для продажи лечебных продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предупреждениях, касающихся применения таких лечебных продуктов. В одном варианте осуществления изобретения на этикетке или во вкладышах указано, что композицию, содержащую соединение по настоящему изобретению, можно использовать для лечения заболевания или нарушения, опосредованного киназами. На этикетке или вкладыше может быть также указано, что композицию можно также использовать для лечения других заболеваний.
В некоторых вариантах осуществления изобретения наборы подходят для доставки твердых пероральных форм соединения по настоящему изобретению, таких как таблетки или капсулы. Такой набор предпочтительно включает ряд стандартных доз. Такие наборы могут включать карту с дозами, расположенными в порядке их намечаемого использования. Примером такого набора является «блистерная упаковка». Блистерные упаковки хорошо известны в упаковочной промышленности и широко используются для упаковки фармацевтических стандартных лекарственных форм. При желании может быть предоставлено дополнительное напоминание, например, в виде чисел, номеров, букв или других отметок или вкладыша-календаря, в котором обозначены те дни в схеме лечения, в которые следует вводить дозу.
Согласно еще одному варианту осуществления изобретения набор может содержать (а) первый контейнер с соединением по настоящему изобретению, которое в нем содержится; и (b) второй контейнер со второй фармацевтической рецептурой, содержащейся в нем, где вторая фармацевтическая рецептура содержит второе соединение, применимое для лечения опосредованного киназами заболевания или нарушения. Альтернативно или дополнительно набор может включать третий контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), физиологический раствор, забуференный фосфатом, раствор Рингера и раствор декстрозы. Набор может дополнительно включать другие вещества, желательные с коммерческой и пользовательской точки зрения, в том числе другие буферы, разбавители, фильтры, иглы и шприцы.
Набор может дополнительно содержать инструкции по введению соединения по настоящему изобретению и, если она имеется, по введению второй фармацевтической рецептуры. Например, если набор содержит первую композицию соединения по настоящему изобретению и вторую фармацевтическую рецептуру, он может дополнительно содержать инструкции для одновременного, последовательного или раздельного введения первой и второй фармацевтических композиций пациенту, который в этом нуждается.
В некоторых других вариантах осуществления изобретения, где набор содержит композицию по изобретению и вторую фармацевтическую рецептуру, набор может включать контейнер для размещения композиций по отдельности, такой как разделенный флакон или разделенный пакет из фольги, однако отдельные композиции могут также находиться в едином, не разделенном контейнере. В некоторых вариантах осуществления изобретения набор содержит инструкции по введению отдельных компонентов. Форма набора особенно предпочтительна в тех случаях, когда отдельные компоненты предпочтительно вводят в различных лекарственных формах (например, перорально и парентерально), при различных интервалах доз или когда требуется титрование отдельных компонентов комбинации по указанию лечащего врача.
Биологическая активность
Ингибирование р38 МАРК
Можно оценивать активность соединения по настоящему изобретению для ингибирования р38 МАРК in vitro, in vivo или на линии клеток. Анализы in vitro включают анализы, которые определяют ингибирование или активности киназ, или активности АТФазы, активированной р38 МАРК. Альтернативные in vitro анализы выражают количественно способность ингибитора связываться с р38 МАРК, которая может быть измерена либо введением радиоактивной метки в ингибитор до связывания, выделением комплекса ингибитор/р38 МАРК и определением количества связанной радиоактивной метки, либо экспериментом с конкуренцией, в котором новые ингибиторы инкубируются с р38 МАРК, связанной с известными радиолигандами. Вышеупомянутые и другие анализы, применимые in vitro и на клеточных культурах, хорошо известны специалистам в данной области.
Анализы ингибирующего действия соединений по настоящему изобретению на культурах клеток могут быть использованы для определения количеств TNF-α:, IL-1, IL-6 или IL-8, продуцируемых в цельной крови или ее клеточных фракциях, в клетках, обработанных ингибитором, по сравнению с клетками, обработанными отрицательным контролем. Уровень указанных цитокинов можно определять с помощью коммерчески доступных ELISA или как описано ниже в разделе «Биологические примеры».
Сведения, подтверждающие возможность осуществления изобретения
Для иллюстрации изобретения включены следующие примеры. Однако следует понимать, что изобретение не ограничивается упомянутыми примерами, они только подразумевают предполагаемый способ практического осуществления изобретения.
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Биологическую активность соединений по настоящему изобретению доказывают с помощью следующих ниже анализов in vitro. В примерах С-М используют следующие соединения: соединение 72: 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-(5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)бензил)мочевина (примеры 1 и 3); и соединение 74: 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этилдигидрофосфат (примеры 2 и 4).
Пример А
Биохимический анализ р38
Активность р38 анализируют при комнатной температуре в 100 мкл реакционной смеси, содержащей 5 нМ активированного фермента р38α и 1 мкМ ATF-2 (активирующий слитый белок фактора транскрипции 2) в качестве субстрата в 25 мМ HEPES (рН 7,4), 100 мкМ ванадата, 1 мкМ DTT, 10 мкМ MgCl2 и 10 мкМ [γ-33Р]-АТФ (~0,1 мкСi Р33/реакцию). Реакцию останавливают через 30-40 минут путем прибавления 25% ТСА, выдерживают в течение 5 минут и затем переносят непосредственно на мембранный фильтр GF-B. Фильтр дважды промывают в течение 30 секунд 0,5% фосфорной кислотой, используя автоматический харвестер (Tomtec Mach III Automated Harvestor). После промывки вакуум поддерживают в течение 30 секунд, чтобы высушить фильтр. Прибавляют приблизительно 30 мкл сцинтиллятора на лунку фильтра и затем считывают в жидкостном сцинтилляционном счетчике (Packard TopCount HTS).
Пример В
Анализ с использованием РВМС
Способность соединений по настоящему изобретению ингибировать продуцирование TNF-α оценивают, используя мононуклеарные клетки периферической крови человека («РВМС»), которые синтезируют и выделяют TNF-α при стимуляции липополисахаридом.
Растворы испытываемых соединений готовят 5-кратными серийными разведениями в ДМСО, которые затем разводят, получая 5х исходные растворы разведением MEM, 2% термоинактивированной фетальной телячьей сывороткой («FBS»), 20 мкМ HEPES, 2 мкМ L-глутамина и 1% пенициллин/стрептомицином.
Клетки РВМС выделяют из крови человека следующим образом. Образцы цельной крови отбирают у людей-добровольцев в Vacutainer™ CPT от Becton Dickinson. Пробирки перемешивают и центрифугируют при комнатной температуре (18-25°С) горизонтально в течение минимум 15 минут при 1500-1800 RCF (относительная центробежная сила). Для каждого донора светлые слои кровяного сгустка помещают в отдельную пробирку и промывают дважды физиологическим раствором, забуференным фосфатом («PBS»). Дебрис повторно суспендируют в MEM, 2% термоинактивированной фетальной телячьей сыворотке («FBS»), 20 мМ HEPES, 2 мМ L-глутамина и 1% пенициллин/стрептомицина. Общее число клеток определяют гемоцитометром и суспензию клеток доводят до 2×106 клеток/мл.
В каждую лунку 96-луночного культурального планшета прибавляют 0,1 мл суспензии клеток. Прибавляют тест-раствор соединения (30 мкл) и клетки инкубируют в инкубаторе при 37°С/5% СO2 в течение 1 часа и затем в каждую лунку прибавляют 20 мкл 7,5 нг/мл липополисахарида (LPS Е. Coli K-235) и клетки возвращают в инкубатор 37°С/5% СO2 на 16-20 часов. Клетки центрифугируют в течение 15 минут при 1100 RCF. Приблизительно 0,12 мл супернатанта переносят в чистый 96-луночный полипропиленовый планшет. Образцы или анализируют немедленно, или хранят при -80°С до готовности для анализа. Уровни TNF-α определяют в каждом образце, используя ELISA анализ человеческого TNF-α, такой, как описано ниже.
Уровни TNF-α определяют, используя следующий анализ. Готовят планшеты, покрытые TNF-альфа антителами, прибавляя 150 мкл 2 мкг/мл очищенных анти-TNF-α мышиных моноклональных IgG в карбонат-бикарбонатном буфере (BupH™ Carbonate-Bicarbonate Buffer Pack) в лунки 96-луночного планшета Immulon 4 (Immulon 4 ELISA Flat Bottom Plate; Dynex, номер в каталоге 011-010-3855) и инкубируют в течение ночи при 2-8°С. Раствор для покрытия удаляют, прибавляют 200 мкл блокирующего буфера (20 мМ HEPES рН 7,4, 150 мМ NaCl, 2% BSA) и планшеты хранят при 2-8°С до готовности использовать. Стандартную кривую рекомбинантной TNF-α человека по 10 точкам получают 1:2 сериальным разведением в разбавителе образцов (20 мМ HEPES, рН 7,4, 150 мМ NaCl, 2 мМ MgCl2, 1% BSA) с самой высокой концентрацией 6000 пг/мл.
Блокирующий раствор удаляют из TNF-α ELISA планшетов 5-кратным промыванием 300 мкл «промывочного буфера» (20 мМ HEPES, рН 7,4, 150 мМ NaCl, 2 мМ MgCl2, 0,02% Tween-20). Во все лунки прибавляют 50 мкл разбавителя образцов и затем прибавляют или 50 мкл стандартного раствора TNF-α, или супернатант тестируемого соединения во все лунки. Планшет инкубируют при комнатной температуре в течение 1 часа при встряхивании (300 об/мин). Планшет промывают пять раз 300 мкл «промывочного буфера». Прибавляют 100 мкл 0,2 мкг/мл биотинилированных козьих антител против TNF-α человека в «разбавителе антител» (20 мМ HEPES, рН 7,4, 150 мМ NaCl, 2 мМ MgCl2, 1% BSA, 0,02% Твин-20) на лунку, и планшет инкубируют при комнатной температуре в течение 1 часа при встряхивании (300 об/мин). Планшет промывают пять раз 300 мкл «промывочного буфера» на лунку. Прибавляют 100 мкл 0,02 мкг/мл стрептавидин-щелочной фосфатазы в разбавителе антител на лунку и планшет инкубируют при комнатной температуре в течение одного часа при встряхивании (300 об/мин). Планшет промывают пять раз 300 мкл «промывочного буфера» на лунку. Прибавляют в каждую лунку 200 мкл 1 мг/мл pNPP (п-нитрофенилфосфата) в диэтаноламинном буфере с 0,5 мМ MgCl2 и планшет инкубируют в течение 35-45 минут при комнатной температуре при встряхивании (300 об/мин). Ход реакции контролируют определением оптической плотности: когда верхний стандарт достигает OD от 2,0 до 3,0, в каждую лунку прибавляют 50 мкл 2N NaOH. Оптическую плотность в каждой лунке определяют в течение 30 минут, используя ридер титрационного микропланшета, установленный на 405 нм. Данные анализируют в схеме XL обработки 4-параметрической кривой.
В вышеописанных анализах используют следующие реагенты. Физиологический раствор, забуференный фосфатом Дульбекко без кальция или магния (Gibco, каталожный номер 14190); минимальная эссенциальная среда Игла (MEM; Gibco, каталожный номер 11090); пенициллин-стрептомицин (Gibco, каталожный номер 15140); L-глутамин, 200 мМ (Gibco, каталожный номер 25030); HEPES, 1М (Gibco, каталожный номер 15630); фетальная телячья сыворотка («FBS»; HyClone, каталожный номер SH30070,03); липополисахариды из Escherichia coli K-235 («LPS»; Sigma, каталожный номер L2018); анти-TNF-α, очищенные моноклональные мышиные антитела IgG (R&D SysteMC, каталожный номер МАВ210); ВupН™ карбонат-бикарбонатный буфер Pack (Pierce, каталожный номер 28382); HEPES (FW 238,3; Sigma, каталожный номер Н3575); NaCl (Sigma, каталожный номер S7653); альбумин бычьей сыворотки («BSA»; Jackson ImmunoReseach, каталожный номер 001-000-162); полиоксиэтилен (20) сорбитанмонолаурат (Sigma, каталожный номер Р2287); гексагидрат хлорида магния (Sigma, каталожный номер М2670); рекомбинантный человеческий TNF-α (R&D SysteMC, каталожный номер 210ТА010); биотинилированный очищенный козий IgG с TNF-α аффинностью (R&D SysteMC, каталожный номер BAF210); стрептавидин-щелочная фосфатаза (Jackson ImmunoResearch, каталожный номер 016-050-084); буфер диэтаноламинного субстрата (Pierce, каталожный номер 34064); п-нитрофенилфосфат (Sigma, каталожный номер N2765).
Пример С
Активность соединения 72 против различных киназ
Исследовали активность соединения 72 в отношении р38α в лабораторном анализе р38α радиоактивной киназы с АТФ-2 в качестве субстрата. Селективность соединения 72 определяют путем анализа активности 202 протеинкиназ в присутствии 1 мкМ соединения 72, затем проводят определения IC50 для тех киназ, которые ингибируются на>80% в первичных испытаниях (концентрация АТФ при Km) по установленным протоколам.
Как показано в таблице 1, соединение 72 представляет собой сильнодействующий ингибитор р38α цитоплазматической тирозинкиназы Абельсона (Abl), Абельсон-родственного гена (Arg) и тирозинкиназы рецептора Tie-2. Наряду с указанными видами активности, соединение 72 является умеренно активным против резистентной к иматинибу формы BCR-Abl T315I, src-семейства тирозинкиназ Hck, Lyn и Fyn, и тирозинкиназ рецептора FGFR1 (рецептор 1 фактора роста фибробласта; FGFR1) и VEGFR2 (рецептор 2 сосудистого эндотелиального фактора роста; KDR).
Соединения примеров 94 и 138 из WO 2004/078116 также были испытаны для сравнения.
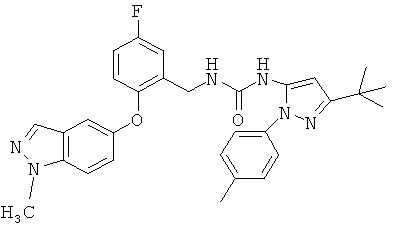
Пример 94
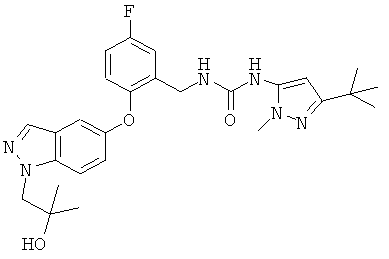
Пример 138
Результаты представлены ниже в таблице 1а.
Результаты показывают, что соединение 72 является значительно более сильным ингибитором Аbl и Tie2, чем соединения примеров 94 и 138 из WO 2004/078116.
Пример D
Активность соединения 72 против пути р38 в клетках
Клетки HeLa обрабатывают соединением 72 в течение 60 минут перед стимуляцией анизомицином (1 мкг/мл) в течение 60 минут, чтобы индуцировать активацию пути р38. Клетки метят антителами против фосфорилированной формы HSP27 (Ser78) и GAPDH в качестве контроля нормализации. Клетки отображаются и выражаются количественно с помощью прибора для визуализации LI-COR Aeriustm.
Результаты показывают, что соединение 72 представляет собой сильный ингибитор пути р38 в клетках с выраженной IC50 2 нМ. В функциональном анализе клеток это соединение испытывают также на его способность ингибировать продуцирование TNF-α в цельной крови, обработанной бактериальным липополисахаридом (LPS), чтобы вызвать продуцирование цитокинов (как определено ELISA), генерируя выраженную IC50 30 нМ.
Пример Е
Активность соединения 72 против рецептора Tie2 на клетках
Для эксперимента по зависимости от времени HUVEC освобождают от сыворотки в течение 2 часов, затем стимулируют 200 нг/мл агониста рецептора Tie2 ангиопоэтина-1 (Ang-1). Клетки собирают в указанные моменты времени и подвергают иммуноблоттингу с антителами против фосфо-АКТ, фосфо-Erk, фосфо-НSР27, фосфо-р38 и GAPDH. Сигналы выражают количественно и нормализуют к GAPDH, чтобы оценить степень индукции в результате активации рецептора Tie2. Результаты демонстрируют в виде иммуноблоттинга и графика, показывающего зависимость от времени активации путей Erk, АКТ и р38 в ответ на агонист Ang-1 рецептора Tie2 в эндотелиальных клетках пупочной вены человека (HUVEC). Результаты показывают, что только пути АКТ и Erk активируются в ответ на активацию рецептора Tie2.
Для исследования активации Tie2 HUVEC освобождают от сыворотки в течение 2 часов, затем обрабатывают различными концентрациями соединения 72 в течение 60 минут перед стимуляцией Ang-1 (200 нг/мл) в течение 10 минут. Tie2 затем очищают иммуноаффинным методом и подвергают иммуноблоттингу (IP) с антителами против фосфотирозина (рТуr) и против Tie2, чтобы определить степень аутофосфорилирования Tie2. Лизаты подвергают иммуноблоттингу с антителами против-фосфо-АКТ (Ser473) соответственно. Сигналы выражают количественно и нормализуют к GAPDH как к контролю нагрузки. Результаты показаны в иммуноблоте и на графике. Обработка соединением 72 приводит к зависимому от дозы ингибированию фосфорилирования как Tie2, так и его эффекторов в прямом направлении Erk и АКТ с IC50, составляющей меньше 33 нМ для первого и меньше 11 нМ для последнего.
Пример F
Активность соединения 72 против BCR-Abl в клетках
Клеточную линию BCR-Аbl-положительного хронического миелоидного лейкоза К562 обрабатывают различными концентрациями иматиниба или соединения 72, как указано, в течение 72 часов при 37°С с 5% СO2. Затем измеряют жизнеспособность клеток, используя CellTiter Blue (Promega), чтобы определить значения IC50 для каждого соединения. Значения IC50, которые были определены, составляют 278 нМ и 62 нМ для иматиниба и соединения 72 соответственно.
Получают экстракты клеток К562, обработанных различными концентрациями иматиниба или соединения 72 в течение 2 часов при 37°С, 5% СO2, и подвергают иммуноблоттингу для аутофосфорилированных Abl (p-Abl, Tyr245) и Аbl субстратов STAT5 (pSTAT5, Tyr694) и CrkL (pCrkL, Tyr207). Результаты показывают, что соединение 72 является более чем в четыре раза более сильным ингибитором пролиферации в линии клеток К562, стимулированной BCR-Abl. Измеряют результаты иммуноблоттинга экстрактов К562, обработанных pBCR-Аbl, pSTAT5, pCrkL или GAPDH при следующих концентрациях иматиниба или соединения 72: 5000, 1000, 333, 111, 37 и 0 нМ. Данный анализ показывает, что как соединение 72, так и иматиниб способны ингибировать аутофосфорилирование BCR-Abl, а также ее субстратов STAT5 и CrkL. Соединение 72, согласно данным по пролиферации, является более сильным ингибитором фосфорилирования вышеупомянутых субстратов.
Пример G
Физические свойства, метаболический профиль и показатели безопасности соединений 72 и 74
1. Определение растворимости
Результаты по определению растворимости показаны в таблице 2.
Для сравнения: растворимости соединений примеров 94 и 138 из WO 2004/078116, как было показано, составляют менее 0,001 мг/мл при рН 6,5 и 7,4.
2. Анализы ингибирования CYP (Флуоресцентный анализ, ЖХ-МС)
Значения IC50 для CYP3A4, CYP2C19 и CYP2C9 получают путем измерения отношения образования метаболита конкретного субстрата к внутреннему стандарту по сравнению с контролем методом жидкостной хроматографии/масс-спектрометрии (ЖХ/МС), используя смешанные микросомы печени человека (n=1). Значения IC50 для CYP2D6 и CYP1A2 получают путем измерения относительной флуоресценции по сравнению с контролем коммерчески доступных флуоресцентных зондов Р450-изоформы и рекомбинантного белка (n=1). Результаты показаны в таблице 3.
Тестостерон: ~10 мкМ
3. Анализы off-target токсичности
Соединение 72 анализируют на связывание (анализ замены лиганда) при 10 мкМ против следующего набора из 28 рецепторов, ионных каналов и транспортеров.
Из 28 испытанных молекулярных форм только натриевый канал (сайт 2), как было показано, в значительной степени взаимодействует (замещение более 50%) с соединением.
4. Анализы для оценки безопасности
Анализ проводят, используя протокол пэтч-кламп. Анализ AMES (анализ бактериальной обратной мутации) проводят в соответствии с протоколом. Соединение 72 испытывают при 0,5-5000 мкг/тест-планшет с/без активацией/активации S9. Микроядерные анализы in vitro и in vivo проводят в соответствии с протоколом. Результаты показаны в таблице 4.
5. Исследование переносимости
Мышам (штамм CD-1, n=5 в каждой исследуемой группе) вводят определенными дозами соединение 72 (в виде соединения 74) при 10, 30 или 100 мг/кг BID в течение 14 дней. Оценивают потерю массы во время проведения исследования. Кровь удаляют в последний день для клинических исследований (альбумин, щелочная фосфатаза, ALT, амилаза, BUN, кальций, холестерин, креатинин, глюкоза, фосфат, общий билирубин, ТР, глобулин). Результаты показаны в таблице 5. Случаев смертности, связанных с введением соединения, не наблюдалось. В данном исследовании даже при 100 мг/кг не отмечалось токсичности, ограничивающей дозу (DLT), поэтому не определена максимально переносимая доза (MTD).
DLT не наблюдается
MTD не достигается
Пример Н
Фармакокинетика соединения 72 в грызунах и приматах
Некомпартментные (независимые от модели) способы используют для анализа кривых концентрация плазмы-время, полученных для всех видов животных, как описано ниже. Ниже приводятся значения клиренса (С1), рассчитанные в процентах отношений экстракции (ER%), объема распределения (Vz), времени до достижения ½-максимальной концентрации (t1/2) и воздействия (AUC, площадь под кривой) для мышей, крыс и обезьян соответственно.
1. Мыши: Фармакокинетику соединения 72 изучают после внутривенного (IV, 1 мг/кг) или перорального введения болюса (РО, 10 мг/кг) мышам-самкам Balb/c (n=3/момент времени). Образцы крови получают через 15 и 30 минут и 1, 2, 4, 8 и 24 часа после перорального введения и через 15 и 30 минут и 1, 2,4, и 8 часа после внутривенного введения IV.
2. Крысы: Фармакокинетику соединения 72 изучают на крысах-самцах линии Sprague-Dawley (n=3/момент времени) после внутривенного (IV, 1 мг/кг) или перорального введения болюса (РО, 10 мг/кг). Образцы крови отбирают в те же моменты времени, какие перечислены для исследования фармококинетики на мышах.
3. Обезьяны: Фармакокинетику соединения 72 изучают на обезьянах-самцах Cynomologus (n=3/момент времени) после внутривенного IV (2 мг/кг) или перорального введения болюса (РО, 10 мг/кг). Образцы крови отбирают в таких же точках времени, какие перечислены для исследования факмакокинетики на мышах.
Пример I
Соединение 72 на модели индуцированного коллагеном артрита (CIA) у крыс
В данном исследовании соединение 72 вводят определенными дозами в виде его пролекарства (соединения 74). Крысам-самкам Lewis (массой 200 г, n=8 в каждой группе, подвергаемой лечению) вводят инъекцию коллагена типа II в неполном адъюванте Фрейнда (инъекция 0,6 мг коллагена, дорсальная поверхность день 0 и день 6). Животным вводят: (1) среду: 5 мл/кг, РО, BID×7; (2) соединение 72: 0,3, 1, 3 или 10 мг/кг, РО, BID×7; или (3) ENBREL®: 10 мг/кг, SC, дни 1 и 4. ENBREL® служит в качестве положительного контроля. Соединения вводят на 11-18 день. Диаметр лодыжки измеряют на 11-й день и в каждый последующий день (фигура 70). В исследовании определяют массу задней лапы и ее микроскопическую гистологию. Удаляют обе задние лапы и колени, задние лапы взвешивают и затем помещают в формалин вместе с коленями. После 1-2 дней в фиксаже и 4-5 дней в декальцинаторе суставы лодыжек и колени обрабатывают, заделывают, разделяют на части и окрашивают толуидиновым синим. Баллы от 0 до 5 дают для воспаления, образования паннуса и резорбции кости лодыжкам и коленям.
Результаты показывают, что соединение 72 дозозависимым образом улучшает все параметры, связанные с данной моделью заболевания. Для диаметра лодыжки и гистопатологии соединение 72 имело явную ED50 1,5 и 3,0 мг/кг соответственно. В дозе 10 мг/кг соединение 72 вызывало регрессию заболевания к концу исследования.
Пример J
Соединение 72 ингибирует ангиогенез in vivo
В данном исследовании соединение 72 вводят определенными дозами в виде его пролекарства (соединения 74). Мышам-самкам (массой 19-22 г, n=4 в каждой группе, подвергаемой лечению) имплантируют подкожно 500 мкл восстановленного матригеля фактора роста, содержащего 1500 нг/мл bFGF или 0 нг/мл bFGF в качестве отрицательного контроля. Испытываемые соединения вводят перорально через зонд в указанных дозах и по указанным схемам. Животным вводят: (1) среду: 30 мМ фосфатного буфера, 10 мл/кг, PO, BID×7; (2) соединение 72: 10, 30 или 100 мг/кг активного лекарственного средства, РО, BID×7; или (3) SU11248: (сутент) 40 мг/кг, РО, QD×7. Сутент представляет собой ингибитор KDR и в данном исследовании служит в качестве положительного лекарственного контроля. Животных умертвляют на 7-й день. Матригель собирают, взвешивают и гомогенизируют в физиологическом растворе, забуференном фосфатом, рН 7,4. Образцы центрифугируют и полученный лизат анализируют на концентрацию гемоглобина, используя колориметрическую реакцию. Ингибирование ангиогенеза, индуцированного bFGF, соединением 72 зависит от дозы. При дозе 100 мг/кг ангиогенез ингибируется в значительной степени (на 72%) (р<0,05, тест множественного сравнения Даннета).
Пример К
Соединение 72 ингибирует опухоли, управляемые BCR-Abl
В данном исследовании соединение 72 вводят определенными дозами в виде его пролекарства (соединения 74). Мышам-самкам Scid-beige (17-20 г, n=8 в каждой группе, подвергаемой лечению) имплантируют подкожно клетки 107 К562 с правой стороны. Испытываемые соединения вводят перорально через зонд в указанных дозах и по указанным схемам, когда опухоли достигают размера 200±50 мм3. Животным вводят:
(1) среду: 20% Trappsol, 10 мл/кг, РО, BID×7; (2) соединение 74: 30 или 100 мг/кг активного лекарственного средства, РО, BID или QD×7; или (3) иматиниб (GLEEVEC®): 400 мг/кг, РО, BID×7. Иматиниб представляет собой известный ингибитор Abl и в данном исследовании служит в качестве положительного лекарственного контроля. В данном исследовании контролируют размер опухоли и массу тела. Результаты показаны в таблице 6.
Соединение 74 было эффективным на данной модели. Введение 30 мг/кг BID или 100 мг/кг QD приводит к остановке роста опухоли, и имеет место 94% регрессия опухоли, а при увеличении дозы до 100 мг/кг BID у 5 или 8 животных имел место полный ответ (100% регрессия опухоли). Соединение 72 хорошо переносится, вызывая потерю массы тела менее 10% в ходе лечения.
Пример L
Соединение 72 на модели множественной миеломы
В данном исследовании соединение 72 вводят определенными дозами в виде его пролекарства (соединение 74). Мышам-самкам линии Scid-beige (массой 17-20 г, n=8 в каждой группе) имплантируют подкожно клетки 15×106 RPMI 8226 с правой стороны или со средой (20% Trappsol, 10 мл/кг, PO, BID×14), или с соединением 74 (100 мг/кг активного лекарственного средства, РО, BID×14). Испытываемые соединения вводят перорально принудительно через зонд в указанных дозах и по указанным схемам, когда опухоли достигают размера 200±50 мм3. Во время исследования контролируют размер опухолей и массу тела, и соединение 72 оценивают по его способности ингибировать рост (% TGI=(1-Т/С)×100). Результаты показаны в таблице 7. Соединение 72 (вводят определенными дозами в виде его пролекарства соединения 74) оказывает статистически значимое действие на рост опухоли в ходе данного исследования. Эти данные позволяют предположить, что действие соединения 72, наблюдаемое в этом исследовании, является дополнением к его способности ингибировать р38 или не зависит от нее. Соединение 72 является хорошо переносимым, вызывая в ходе лечения потерю массы тела менее 10%.
Клетки множественной миеломы RPMI 8226 обрабатывают различными концентрациями протеосомального ингибитора MG-132 (служит в качестве положительного контроля) или соединения 72 в течение 72 часов при 37°С, 5% СO2. Затем измеряют жизнеспособность клеток, используя CellTiter Blue (Promega) для определения значений IC50 для каждого соединения (Таблица 8). Обработка соединением 72 не является анти-пролиферативной в указанном интервале концентраций. MG-132 дает значение IC50, которое находится в предыдущих интервалах лабораторных испытаний (Таблица 8). Эти данные указывают, что TGI эффект, наблюдаемый для соединения 72 in vivo, вероятно, обусловлен эффектами вне опухоли. Такая роль согласуется с опубликованными сообщениями, предполагающими, что активность как р38, так и Tie2 может быть решающей для роста/выживания опухолей этого типа in vivo.
Пример М
Активность соединения 72 против VEGFR2 на клетках
HUVEC освобождают от сыворотки крови в течение 2 часов, затем обрабатывают различными концентрациями соединения 72 или контрольного ингибитора VEGFR2 ((Z)-3-[(2,4-диметил-3-(этоксикарбонил)пиррол-5-ил)метилиденил]индолин-2-она) в течение 60 минут перед стимуляцией 20 нг/мл VEGF в течение 5 минут. Готовят лизаты различных обработок и подвергают иммуноблоттингу антителами, специфичными для VEGFR2, фосфорилированными на Tyr1175. Полученные блотты для соединения 72 и контрольного ингибитора VEGFR1 затем отображают и выражают количественно, с использованием прибора для визуализации LI-COR Aeriustm. Сигнал фосфо-VEGFR2 нормализуют к GAPDH в качестве контроля нагрузки. Указанные величины затем используют для генерирования значений IC50 для соединения 72 и контрольного ингибитора VEGFR2 VEGFR1, используя схему обработки 4-параметрической кривой.
Соединение 72 ингибирует аутофосфорилирование сосудистого эндотелиального рецептора типа II (VEGFR2) в HUVEC. Результаты показывают, что обработка соединением 72 приводит к дозозависимому ингибированию аутофосфорилирования VEGFR2 на тирозиновом остатке 1175 с IC50 48 нМ.
ПРЕПАРАТИВНЫЕ ПРИМЕРЫ
В примерах, описанных ниже, все температуры приведены в градусах Цельсия, если не указано иное. Реагенты приобретали у коммерческих поставщиков, таких как Aldrich Chemical Company, Lancaster, TCI или Maybridge, и использовали без дополнительной очистки, если не указано иное. Тетрагидрофуран (ТГФ), N,N-диметилформамид (ДМФА), дихлорметан (DCM), толуол, диоксан и 1,2-дифторэтан приобретали от Aldrich в герметично закрытых флаконах, и используют в том виде, в котором поставлены.
Реакции, описанные ниже, обычно проводят при положительном давлении азота или аргона или с осушительной трубкой (если не указано иное) в безводных растворителях, и реакционные колбы обычно снабжены резиновой перегородкой для введения субстратов и реагентов с помощью шприца. Оборудование из стекла высушивают в печи и/или при нагревании.
Для колоночной хроматографии используют систему Biotage (производитель: Dyax Corporation) с колонкой, заполненной силикагелем, или картридж SepPak с силикагелем (Waters).
Спектры 1Н ЯМР регистрируют на приборе Varian, работающем при 400 МГц. Спектры 1Н ЯМР получают для растворов в CDCl3 (приводят в м.д.), используя хлороформ в качестве эталона (7,25 м.д.). При необходимости используют другие растворители для ЯМР. При указании мультиплетности пиков используют следующие сокращения: с (синглет), д (дублет), т (триплет), м (мультиплет), уш (уширенный), дд (дублет дублетов), дт (дублет триплетов). Константы спин-спинового взаимодействия, если указаны, приводятся в герцах (Гц).
Пример 1
Получение 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-(5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)бензил)мочевины (72). Способ 1
Синтез соединения (72) по способу 1 показан на схеме 1, стадии А-J.
Стадия А. Получение 2-(бензилокси)-5-фторбензонитрила 62. В 4-горлую круглодонную колбу объемом 12 л, снабженную механической мешалкой и термометром и содержащую суспензию гидрида натрия (74 г, 1,82 моль) в ДМФА (1,95 л), в атмосфере азота прибавляют бензиловый спирт (142 г, 1,3 моль) через капельную воронку медленно в течение 30 минут. Смесь перемешивают на ледяной бане (0°С) в течение 120 минут и прибавляют раствор 2,5-дифторбензонитрила (180 г, 1,3 моль) в ДМФА (650 мл) через капельную воронку в течение 35 минут. Полученную смесь нагревают до комнатной температуры. Через 2 часа смесь охлаждают на ледяной бане (4°С) и гасят насыщенным раствором NH4Cl (130 мл), затем прибавляют воду (2,6 л×3). Взвесь перемешивают при комнатной температуре в течение ночи, осадок отфильтровывают и промывают водой (650 мл×3). Твердое вещество сушат в вакууме в течение 2 дней, получая 292 г (99,3%) требуемого соединения. 1Н ЯМР (400 МГц, ДМСО-d6) δ □,□8 (дд, 1Н, J=3,2 Гц, J=8,2 Гц), 7,5-7,76 (м, 1Н), 7,35 медленно 7,48 (м, 6Н), 5,28 (с, 2Н).
Стадия В. Получение 5-фтор-2-гидроксибензонитрила 63. Сухой 10% Pd/C (2,48 г) помещают в 5-литровую колбу в атмосфере N2. Раствор 2-(бензилокси)-5-фторбензонитрила (148 г, 651 ммоль) в метаноле (2,34 л) медленно прибавляют в атмосфере N2. Воздух из колбы удаляют в вакууме и в колбу три раза загружают Н2 (из баллона). Смесь перемешивают при комнатной температуре в течение 1 часа. Вышеуказанный процесс повторяют еще два раза, чтобы довести реакцию до завершения. Смесь перемешивают при комнатной температуре дополнительно 2 часа. Смесь фильтруют через слой целита в атмосфере N2 и промывают EtOAc (150 мл×2). Отходы палладия промывают водой (50 мл) и отбрасывают.Фильтраты концентрируют, получая 94,6 г (106%) требуемого соединения в виде желтого твердого вещества. МС (APCI-) m/z 137 (M-1). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,07 (уш, 1Н), 7,58 (дд, 1Н, J=3,2 Гц, J=8,2 Гц), 7,43 (м, 1Н), 6,91-7,03 (дд, 1Н, J=4,5 Гц, J=9,2 Гц).
Стадия С.Получение 5-фтор-2-(4-метил-3-нитрофенокси)бензонитрила 64. В 3-горлой колбе объемом 5 л, снабженной механической мешалкой, обратным холодильником и датчиком температуры J-Kem, раствор 5-фтор-2-нитротолуола (158 г, 1,02 моль) в N,N-диметилацетамиде (500 мл) прибавляют к смеси 5-фтор-2-гидроксибензонитрила (147 г, 1,07 моль) и карбоната калия (163 г, 1,2 моль) в N,N-диметилацетамиде (573 мл) при комнатной температуре. Смесь нагревают до 100°С. Через 8 часов реакционную смесь охлаждают до комнатной температуры и прибавляют воду (3400 мл) на ледяной бане. Осадок отфильтровывают, промывают водой (1073 мл×2), сушат в вакууме (40°С) и получают требуемое соединение (264 г, 90%) в виде коричневого твердого вещества. МС (APCI-) m/z 272 (M-1). 1Н ЯМР (400 МГц, CDCl3) δ 8,10 (д, 1Н, J=9 Гц), 7,41-7,48 (м, 1Н), 7,38 (м, 1Н), 7,07-7,14 (м, 1Н), 6,88-7,10 (м, 1Н), 2,63 (с, 3Н).
Стадия D. Получение 2-(3-амино-4-метилфенокси)-5-бензонитрила 65. В 4-горлую колбу объемом 12 л, снабженную механической мешалкой, обратным холодильником, и датчиком температуры J-Kem, помещают 2-(3-амино-4-метилфенокси)-5-бензонитрил (247 г, 907 ммоль) и Zn пыль (297 г, 4,5 моль) и суспендируют в 2,2 л смеси МеОН/ТГФ (1:1). Медленно прибавляют насыщенный раствор NH4Cl (1,6 л) в течение 40 минут через капельную воронку (поддерживая температуру внутри ниже 60°С). Реакционную смесь охлаждают до комнатной температуры и перемешивают при комнатной температуре в течение 1 часа. Цинковую пыль удаляют фильтрованием и слой на фильтре промывают ЕtOАс (450 мл×3). Прибавляют насыщенный водный раствор NH4OAc (1500 мл, приготовленный из 2 кг NH4OAc и 4 л Н2O) и Н20 (1500 мл×2). Фильтраты концентрируют на роторном испарителе до тех пор, пока не останется в основном вода. Рыжевато-коричневое твердое вещество отфильтровывают, промывают Н2O (1500 мл×4) и сушат в вакууме, получая 211,5 г (96%) требуемого соединения. МС (APCI+) m/z 243 (М+1). 1H ЯМР (400 МГц, CDCl3) δ 7,38 (м, 1Н), 7,18 (м, 1Н), 6,65-6,85 (м, 5Н), 3,60 (уш, 2Н), 2,17 (с, 3Н).
Стадия Е. Получение 2-(1Н-индазол-5-илокси)-5-фторбензонитрила 66. В 6-литровой круглодонной колбе, снабженной механической мешалкой и датчиком температуры J-Kem, холодный раствор (0°С) 2-(3-амино-4-метилфенокси)-5-бензонитрила (209 г, 862 ммоль) и NH4BF4 в смеси уксусной кислоты (1,75 л) и воды (862 мл) обрабатывают по каплям концентрированной НСl (359 мл) в течение 2 минут. Взвесь перемешивают на ледяной бане в течение 30 минут и прибавляют нитрит натрия. Реакционную смесь перемешивают при 0°С в течение 1 часа и затем нагревают до комнатной температуры. После перемешивания при комнатной температуре в течение 3 часов растворители удаляют на роторном испарителе и азеотропной перегонкой с толуолом (200 мл×2). Остаток сушат в вакууме в течение ночи. Твердое вещество суспендируют в ацетоне (200 мл) и EtOAc (200 мл) и удаляют растворители на роторном испарителе. Остаток суспендируют в EtOAc (200 мл) и растворитель удаляют на роторном испарителе. Остаток суспендируют в EtOAc (1145 мл) и прибавляют одной порцией ацетат калия (254 г, 2,6 моль). Смесь перемешивают при комнатной температуре в течение 20 часов и фильтруют. Слой на фильтре промывают EtOAc (500 мл×6). Объединенные фильтраты концентрируют. Остаток суспендируют в ЕtOАс (500 мл) и гептане (500 мл). Отфильтровывают желтое твердое вещество (141,3 г). Маточный раствор концентрируют и перекристаллизовывают из смеси МТВЕ/гексан (100 мл/100 мл), получая 20,1 г. Второй маточный раствор концентрируют и растирают в смеси МТВЕ/гептан (100 мл/100 мл), получая дополнительные 14,28 г. Объединенные порции дают 176 г (выход 81%) требуемого соединения. МС (APCI+) m/z 254 (М+1). 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,38 (уш, 1H), 8,07 (с, 1Н), 7,94 (дд, 1Н, J=3,1 Гц, J=8,2 Гц), 7,64 (д, 1Н, J=9 Гц), 7,54 (м, 2Н), 7,21 (дд, 1Н, J=9 Гц), 6,96 (дд, 1Н, J=4,3 Гц, J=9,3 Гц).
Стадия F. Получение 2-(1-(2,2-диметоксиэтил)-1Н-индазол-5-илокси)-5-фторбензонитрила 67. 2-(1Н-Индазол-5-илокси)-5-фторбензонитрил (5,000 г, 19,7 ммоль) суспендируют в N,N-диметилацетамиде (50 мл) и порциями прибавляют гидрид натрия (0,684 г, 25,7 ммоль) при комнатной температуре. Прибавляют по каплям 2-бром-1,1-диметоксиэтан (3,49 мл, 29,6 ммоль) и реакционную смесь нагревают при 80°С в течение 18 часов. Реакционную смесь охлаждают до комнатной температуры, концентрируют при пониженном давлении и получают сырое вещество, которое распределяют между ЕtOАс и насыщенным водным раствором NH4Cl. Объединенные органические слои промывают водой (3х), насыщенным раствором хлорида натрия, сушат (MgSO4) и затем концентрируют при пониженном давлении, получая сырое вещество. Сырой продукт очищают колоночной флэш-хроматографией (элюент - 25% EtOAc/смесь гексанов) и получают 3,88 г (57,6%) требуемого соединения. Изомер N-2 не выделяют. МС (APCI+) m/z 342 (М+1). 1Н ЯМР (400 МГц, CDCl3) δ 7,98 (с, 1Н), 7,54 (д, 1Н, J=9 Гц), 7,36 (м, 2Н), 7,16 (м, 2Н), 6,82 (дд, 1Н, J=4,3 Гц, J=9,3 Гц), 4,76 (т, 1Н, J=5,3 Гц), 4,49 (д, 2Н, J=5,3 Гц), 3,41 (с, 6Н).
Стадия G. Получение (2-(1-(2,2-диметоксиэтил)-1Н-индазол-5-илокси)-5-фторфенил)метанамина 68. 2-(1-(2,2-Диметоксиэтил)-1Н-индазол-5-илокси)-5-фторбензонитрил (3,80 г, 11,13 ммоль) в ТГФ (100 мл) прибавляют по каплям при перемешивании и 0°С к суспензии литийалюминийгидрида (0,6338 г, 16,70 ммоль) в ТГФ (100 мл). Температуру реакционной смеси (0°С) поддерживают до завершения прибавления исходного вещества. Реакционную смесь нагревают до комнатной температуры и перемешивают до тех пор, пока полностью не израсходуется исходное вещество (по данным ВЭЖХ-анализа). Реакционную смесь прибавляют по каплям к перемешиваемому при комнатной температуре насыщенному водному раствору соли Рошеля. Двухфазную смесь интенсивно перемешивают в течение 30 минут до растворения солей алюминия. Слои разделяют, и водный слой экстрагируют ЕtOAc. Объединенные органические слои сушат (MgSO4), концентрируют при пониженном давлении и получают 3,73 г (97%) сырого продукта, который используют без дополнительной очистки. МС (APCI+) m/z 346 (М+1). 1Н ЯМР (400 МГц, CDCl3) δ 7,90 (с, 1Н), 7,48 (д, 1Н, J=9 Гц), 6,9-7,15 (м, 5Н), 4,76 (т, 1Н, J=5,3 Гц), 4,49 (д, 2Н, J=5,3 Гц), 3,88 (с, 2Н), 3,36 (с, 6Н).
Стадия Н. Получение 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-(2-(1-(2,2-диметоксиэтил)-1Н-индазол-5-илокси)-5-фторбензил)мочевины 70. (2-(1-(2,2-Диметоксиэтил)-1Н-индазол-5-илокси)-5-фторфенил)метанамин (2,00 г, 5,791 ммоль), 2,2,2-трихлорэтил-3-трет-бутил-1-п-толил-1Н-пиразол-5-илкарбамат (3,516 г, 8,686 ммоль) и N,N-диизопропилэтиламин (3,026 мл, 17,37 ммоль) суспендируют в N,N-диметилацетамиде (50 мл) и нагревают при 80°С в течение ночи. Реакционную смесь концентрируют при пониженном давлении и остаток разбавляют EtOAc и насыщенным водным раствором NH4Cl. Органический слой промывают водой (3х), насыщенным раствором хлорида натрия, сушат (MgSO4) и затем концентрируют при пониженном давлении, получая сырое вещество, которое очищают колоносной флэш-хроматографией (элюент 35-45% EtOAc/смесь гексанов). После выпаривания растворителя получают требуемый продукт в виде бледно-желтой пены (2,57 г, 74%). МС (APCI+) m/z 602 (M+1). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,88 (с, 1Н), 7,43 (д, 1Н, J=9 Гц), 6,72-7,30 (м, 9Н), 6,18 (с, 1Н), 6,01 (уш, 1Н), 5,47 (т, 1Н, J=6,1 Гц), 4,76 (т, 1Н, J=5,3 Гц), 4,44 (т, 4Н, J=5,6 Гц), 3,38 (с, 6Н), 2,37 (с, ЗН), 1,30 (с, 9Н).
Стадия I. Получение 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-(5-фтор-2-(1-(2-оксоэтил)-1Н-индазол-5-илокси)бензил)мочевины 71. 1-(3-трет-Бутил-1-п-толил-1Н-пиразол-5-ил)-3-((2-(1-(2,2-диметоксиэтил)-1 Н-индазол-5-илокси)-5-фторфенил)метил)-мочевину (0,500 г, 0,8324 ммоль) растворяют в дихлорметане при комнатной температуре и к перемешиваемому раствору прибавляют по каплям йодтриметилсилан (0,3554 мл, 2,497 ммоль). Реакционную смесь контролируют ВЭЖХ-анализом и, когда исходное вещество не определяется, смесь разбавляют дихлорметаном и промывают 10% водным раствором Na2S2O4, насыщенным водным раствором NaHCO3, насыщенным раствором хлорида натрия, сушат (MgSO4) и концентрируют при пониженном давлении, получая 536 мг (количественный выход) сырого продукта, который используют без дополнительной очистки. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,76 (с, 1Н), 7,97 (с, 1Н). 6,78- 7,30 (м, 10Н), 6,17 (с, 1Н), 5,99 (уш, 1Н), 5,44 (кв, 1Н, J=6,1 Гц), 4,46 (м, 4Н), 2,35 (с, 3Н), 1,34 (с, 9Н).
Стадия J. Получение 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-(5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)бензил)мочевины 72. 1-(3-трет-Бутил-1-п-толил-1Н-пиразол-5-ил)-3-((5-фтор-2-(1-(2-оксоэтил)-1Н-индазол-5-илокси)фенил)метил)-мочевину (2,179 г, 3,929 ммоль) суспендируют в МеОН (40 мл) и обрабатывают натрийборгидридом (0,7432 г, 19,64 ммоль) порциями при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре до тех пор, пока не наблюдается полное превращение исходного вещества в продукт (ВЭЖХ-анализ). Реакционную смесь концентрируют при пониженном давлении, затем разбавляют насыщенным водным раствором NH4Cl и экстрагируют дихлорметаном. Органические экстракты сушат (MgSO4), концентрируют при пониженном давлении, получая сырой продукт, который очищают колоночной флэш-хроматографией (элюент 2-6%-2-6% i-PrOH/DCM) и получают 1,21 г (55%) требуемого соединения. МС (APCI+) m/z 557 (M+1). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,29 (с, 1H), 7,97 (с, 1Н), 7,68 (д, 1Н, J=9,1 Гц), 7,35 (с, 1Н), 6,85-7,4 (м, 8Н), 6,24 (с, 1Н), 4,87 (т, 1Н, J=5,4 Гц), 4,43 (т, 1Н, J=5,6 Гц), 4,28 (д, 1Н, J=5,9 Гц), 3,80 (кв, 1Н, J=5,5 Гц), 2,35 (с, 3Н), 1,25 (с, 9Н).
Пример 2
Получение 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этил дигидрофосфата (74). Способ 1
Синтез соединения (74) по способу 1, показанному на схеме 1, стадии К-L.
Стадия К. Получение дибензил-2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-5-ил)этилфосфата 73. 1-(3-трет-Бутил-1-п-толил-1Н-пиразол-5-ил)-3-((5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)фенил)метил)-мочевину (0,357 г, 0,6414 ммоль), дифенил-диизопропил фосформидит (0,2970 мл, 0,9620 ммоль) и 2Н-тетразол (0,07189 г, 1,026 ммоль) суспендируют в ДМФА (10 мл) и перемешивают при комнатной температуре до тех пор, пока ТСХ-анализ (10:1 DCM-Et2O) не покажет, что исходное вещество полностью израсходовано. Прибавляют трет-бутилгидропероксид (0,4439 мл, 3,207 ммоль) и затем реакцию контролируют ВЭЖХ-анализом. После завершения реакции прибавляют 10% водный раствор Na2S2O3 и реакционную смесь перемешивают в течение 10 минут. Реакционную смесь выливают в насыщенный водный раствор NaHCO3 и экстрагируют EtOAc. Объединенные органические экстракты промывают водой (2 х) и насыщенным раствором хлорида натрия, сушат (MgSO4) и концентрируют при пониженном давлении, получая сырой продукт, который очищают колоночной флэш-хроматографией (элюент: 30-40% EtOAc/смесь гексанов) и получают 458 мг (87%) требуемого соединения. МС (APCI+) m/z 817 (M+1). 1Н ЯМР (400 МГц, CDCl3) δ 7,85 (с, 1Н), 6,7-7,35 (м, 10Н), 6,18 (с, 1Н), 6,16 (с, 1Н), 4,62 (с, 1Н), 5,36 (т, 1Н, J=6 Гц), 4,81 (д, 4Н, J=8,2 Гц), 4,54 (т, 2Н, J=5,1 Гц), 4,39 (м, 4Н), 2,35 (с, 3Н), 1,25 (с, 9Н).
Стадия L. Получение 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этилдигидрофосфата 74. Дибензил-2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этилфосфат (1,920 г, 2,35 ммоль) суспендируют в ЕtOН (0,05 М, 50 мл) и прибавляют циклогекса-1,4-диен (3,56 мл, 82,14 ммоль) и 10% Pd/C (20% масс., 0,384 г). Реакционную смесь кипятят с обратным холодильником в течение ночи. Реакционную смесь фильтруют через бумагу GF и промывают МеОН. Органические слои концентрируют при пониженном давлении, получая сырой продукт, который растворяют в МеОН и пропускают через фильтр Gellman acrodisk (0,45 um) для удаления следов загрязнения палладием. После выпаривания растворителя получившуюся смолу растирают в CH3CN, отфильтровывают образовавшиеся твердые вещества, сушат в высоком вакууме в течение 3 часов и получают 1,21 г (89%) требуемого соединения. МС (APCI+) m/z 637 (М+1). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,30 (с, 1Н), 8,00 (с, 1Н), 7,71 (д, 1Н, J=9 Гц), 6,88-7,37 (м, 10Н), 6,24 (с, 1Н), 4,61 (уш, 1Н), 4,28 (д, 2Н, J=5,7 Гц), 4,19 (дд, 2Н, J=5,2 Гц), 2,35 (с, 3Н), 1,25 (с, 9Н).
Пример 3
Получение 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-((5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)фенил)метил)мочевины (72). Способ 2
Альтернативный способ синтеза соединения (72) по способу 2 показан на схеме 2, стадии А-F.
Стадия А. Получение 2-(4-амино-3-метилфенокси)-5-фторбензонитрила 65. В 4-горлой колбе объемом 3 л, которая после вакуумирования заполнена аргоном, 2,5-дифторбензонитрил (295,3 мл, 812,0 ммоль) и 4-амино-3-метилфенол (100,0 г, 812,0 ммоль) растворяют в сухом ДМСО (2,75 М) при быстром перемешивании при комнатной температуре. Раствор вакуумируют/заполняют аргоном (3 х). Прибавляют карбонат калия (185,2 г, 1340 ммоль) (~325 меш). Реакционную смесь вакуумируют/заполняют аргоном (3 х) и нагревают при 80°С (датчик внутри) в течение 16 часов. ТСХ указывает на полную конверсию. Реакционную смесь охлаждают до комнатной температуры и медленно выливают в 2 л ледяной воды при быстром перемешивании. Остаток в круглодонной колбе неоднократно вымывают водой и выливают в ледяную воду до тех пор, пока общий объем не достигнет 3 л и все твердые вещества не окажутся в ледяной воде. Суспензию быстро перемешивают в течение 2 часов, пока она не нагреется до комнатной температуры. Коричневые твердые вещества отфильтровывают, промывают водой (3 л), сушат на воздухе, сушат, используя латексную пластину, и сушат в высоком вакууме при 40°С в течение 44 часов, получая 194 г (99%) требуемого продукта в виде рыжевато-коричневого твердого вещества. МС (APCI+) m/z 243 (М+1). 1Н ЯМР (400 МГц, CDCl3) δ 7,38 (м, 1Н), 7,18 (м, 1Н), 6,65-6,85 (м, 5Н), 3,60 (уш, 2Н), 2,17 (с, 3Н).
Стадия В. Получение 2-(1Н-индазол-5-илокси)-5-фторбензонитрила 66. В одногорлой колбе объемом 500 мл, содержащей большой якорь магнитной мешалки, 2-(4-амино-3-метилфенокси)-5-фторбензонитрил (274,5 мл, 219,6 ммоль) растворяют в хлороформе (0,8 М) и обрабатывают ацетатом калия (25,86 г, 263,5 ммоль) при комнатной температуре. Реакционную смесь охлаждают до 0°С и обрабатывают уксусным ангидридом (62,28 мл, 658,8 ммоль), прибавляя его через воронку в течение 5 минут. Реакционную колбу снабжают обратным холодильником, обрабатывают дополнительной порцией хлороформа (100 мл) и нагревают до 40°С. Прибавляют по каплям через капельную воронку изоамилнитрит (58,74 мл, 439,2 ммоль). Воронку промывают хлороформом (60 мл+40 мл), удаляют из колбы и заменяют стеклянной пробкой. Реакционную смесь нагревают и кипятят в течение 20 часов, охлаждают до комнатной температуры и концентрируют в вакууме до коричневого твердого вещества. После высушивания в высоком вакууме в течение 30 минут коричневое твердое вещество суспендируют в 1 л воды, энергично перемешивают 15 минут и отфильтровывают. Полученное оранжево-коричневое твердое вещество (83 г) помещают в 400 мл МеОН и обрабатывают 1,0 М хлористоводородной кислотой (241,6 мл, 241,6 ммоль) при комнатной температуре. Реакционную колбу снабжают обратным холодильником и нагревают до 70°С в течение 20 часов. Реакционную смесь охлаждают до 0°С и прибавляют 1,0 М раствор гидроксида натрия (252,6 мл, 252,6 ммоль), затем воду (250 мл). Полученную желтую суспензию перемешивают на ледяной бане 10 минут и затем дают возможность осесть. Суспензию фильтруют, промывают водой (1 л), сушат на воздухе, сушат, используя латексную пластину, и сушат в высоком вакууме при 40°С в течение 36 часов, получая 50,7 г (91%) требуемого продукта в виде легкосыпучего рыжевато-коричневого твердого вещества. МС (APCI+) m/z 254 (M+l). 1H ЯМР (400 МГц, ДМСО-d6) δ 13,38 (уш, 1H), 8,07 (с, 1H), 7,94 (дд, 1H, J=3,1 Гц, J=8,2 Гц), 7,64 (д, 1H, J=9 Гц), 7,54 (м, 2Н), 7,21 (дд, 1Н, J=9 Гц), 6,96 (дд, 1H, J=4,3 Гц, J=9,3 Гц).
Стадия С. Получение метил-2-(5-(2-циано-4-фторфенокси)-1Н-индазол-1-ил)ацетата 77. 2-(1Н-Индазол-5-илокси)-5-фторбензонитрил (40,7 г, 160,7 ммоль) растворяют в 400 мл ДМФА и раствор помещают в водяную баню (комнатная температура). Прибавляют карбонат цезия (157,1 г, 482,2 ммоль). После выдерживания на водяной бане в течение 30 минут прибавляют по каплям метил-2-бромацетат (33,47 мл, 353,6 ммоль). Смесь перемешивают при комнатной температуре 4 часа. К смеси прибавляют воду (400 мл, слабоэкзотермическая реакция) и всю смесь переносят в 2-литровую делительную воронку (с 400 мл ЕtOАс). Слои разделяют и водный слой экстрагируют ЕtOАс (3×200 мл). Объединенные экстракты промывают водой и насыщенным раствором хлорида натрия и слои разделяют. Органический слой сушат над MgSO4, фильтруют через слой целита, концентрируют при пониженном давлении и сушат в высоком вакууме в течение 1 часа, получая 66,3 г рыжевато-коричневого твердого вещества. Твердое вещество растворяют в 400 мл горячего ЕtOАс и обрабатывают древесным углем в течение 10 минут при кипячении. Смесь фильтруют через слой целлита и концентрируют при пониженном давлении. Нерастворимые вещества удаляют фильтрованием. Твердое вещество растворяют в 300 мл горячего ЕtOАс и прибавляют 300 мл смеси гексанов. Смесь охлаждают до комнатной температуры. Светло-коричневое твердое вещество выделяют фильтрованием, 32,24 г (61,7%). МС (APCI+) m/z 326 (M+1) was detected. 1H ЯМР (400 МГц, CDCl3) δ 8,03,1Н), 7,41-7,36 (м, 3Н), 7,22-7,16 (м, 2Н), 6,81 (дд, J=4,29 Гц, 9,37 Гц, 1Н), 5,19 (с, 2Н), 3,79 (с, 3Н).
Стадия D: Получение дигидрохлорида метил-2-(5-(2-(аминометил)-4-фторфенокси)-1Н-индазол-1-ил)ацетата 78. Метил-2-(5-(2-циано-4-фторфенокси)-1Н-индазол-1-ил)ацетат (10,00 г, 30,74 ммоль) помещают в сосуд Парра для гидрированид на 2500 мл. Прибавляют метанол (0,1 М, 310 мл) и концентрированную НСl (25,62 мл, 307,4 ммоль). Прибавляют 10% Pd/C (2,0 г, тип Degussa) и сосуд продувают газообразным водородом (3 раза, вакуумирование не проводят). Сосуд загружают газообразным водородом до давления 40 фунт/кв.дюйм и оставляют при встряхивании на 18 часов. К смеси прибавляют дополнительно 1,0 г 10% Pd/C и продолжают гидрирование в сосуде Парра еще 18 часов. Смесь фильтруют через слой целита и концентрируют при пониженном давлении (температуру бани поддерживают ниже 20°С). Остаток подвергают азеотропной перегонке с МеОН (3×100 мл). Температуру бани поднимают до 40°С после третьей азеотропной перегонки. Остаток (масло) сушат в высоком вакууме в течение 2 часов, получая требуемый продукт в виде желтого твердого вещества (11,6 г, 93,8%). МС (APCI+) m/z 330 (M+1). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,08 (с, 1Н), 6,84-7,76 (м, 6Н), 5,43 (с, 2Н), 4,8 (уш, 2Н), 4,105 (кв, 2Н, J=5,6 Гц), 3,68 (с, 3Н).
Стадия Е. Получение метил-2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)ацетата 79. Раствор дигидрохлорида метил-2-(5-(2-(аминометил)-4-фторфенокси)-1Н-индазол-1-ил)ацетата (17,5 г, 43,51 ммоль) в диметилацетамиде (0,5 М, 88 мл) обрабатывают 2,2,2-трихлорэтил-3-трет-бутил-1-п-толил-1Н-пиразол-5-илкарбаматом (17,96 г, 44,38 ммоль), затем диизопропилэтиламином (30,3 мл, 174 ммоль) при комнатной температуре. Смесь нагревают при 80°С в течение ночи. Смесь концентрируют в вакууме и остаток распределяют между ЕtOАс насыщенным водным раствором NH4Cl. Органический слой отделяют и промывают насыщенным раствором хлорида натрия, фильтруют через бумагу 1PS, упаривают в вакууме до коричневой пены. Пену растирают в эфире и твердые вещества, выпавшие в осадок, отфильтровывают, получая 22,55 г (89%) требуемого соединения. МС (APCI+) m/z 585 (M+1). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,3 (с, 1Н), 8,07 (с, 1Н), 7,68 (д, 1Н, J=8,9 Гц), 6,85-7,37 (м, 8Н), 6,24 (с, 1Н), 5,4 (с, 2Н), 4,28 (д, 2Н, J=5,8 Гц), 3,68 (с, 3Н), 2,35 (с, 3Н), 1,25 (с, 9Н).
Стадия F. Получение 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-((5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)фенил)метил)мочевины 72. Раствор метил-2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)ацетата (4,6 г, 7,87 ммоль) в метаноле (0,1 М, 80 мл) обрабатывают натрийборгидридом (893 мг, 23,6 ммоль) порциями при комнатной температуре. Смесь перемешивают при комнатной температуре в течение 18 часов, растворитель выпаривают в вакууме и получают темно-желтый остаток. Остаток распределяют между дихлорметаном и насыщенным водным раствором NH4Cl. Органический слой фильтруют через бумагу 1PS, упаривают в вакууме и очищают с помощью системы Biotage, элюируя 2-10% i-PrOH/DCM. Нужные фракции упаривают в вакууме, получая белую пену (2,97 г, 68%). МС (APCI+) m/z 557 (М+1). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,29 (с, 1Н), 7,97 (с, 1Н), 7,68 (д, 1Н, J=9,1 Гц), 7,35 (с, 1Н), 6,85-7,4 (м, 8Н), 6,24 (с, 1Н), 4,87 (т, 1Н, J=5,4 Гц), 4,43 (т, 1Н, J=5,6 Гц), 4,28 (д, 1Н, J=5,9 Гц), 3,80 (кв, 1Н, J=5,5 Гц), 2,35 (с, 3Н), 1,25 (с, 9Н).
Пример 4
Получение 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этил дигидрофосфата (74). Способ 2
Альтернативный способ для синтеза соединения (72) по способу 2 показан на схеме 2, стадии G-Н.
Стадия G. Получение ди-трет-бутил 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этилфосфата 80. Раствор 1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-((5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)фенил)метил)мочевины (1,5 г, 2,69 ммоль) в ДМФА (0,2М, 15 мл) обрабатывают имидазолом (183 мг, 2,69 ммоль) и гидрохлоридом имидазола (423 мг, 4,04 ммоль) при комнатной температуре. После полного растворения прибавляют ди-трет-бутил-диизопропилфосфорамидит (1,28 мл, 4,04 ммоль). Перемешивание продолжают при комнатной температуре в течение 18 часов. Медленно прибавляют пероксид водорода (50% водный, 0,535 мл, 9,43 ммоль) при комнатной температуре медленно и продолжают перемешивание в течение 1 часа. Реакция завершается (как показывают ГХ и ТСХ). Остаток распределяют между дихлорметаном и 10% водным раствором Na2S2O3. Водный слой экстрагируют ЕtOАс (2 х). Объединенные органические экстракты промывают насыщенным раствором хлорида натрия, фильтруют через бумагу 1PS, упаривают в вакууме, получая желтое масло. Масло очищают пропусканием через слой силикагеля, элюируя DCM, 3-5% MeOH/DCM. Нужные фракции упаривают в вакууме и получают требуемый продукт в виде белой пены (1,59 г, 79%). МС (APCI+) m/z 749 (М+). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,29 (с, 1Н), 8,01 (с, 1Н), 7,73 (д, 1Н, J=9,1 Гц), 6,79-7,35 (м, 8Н), 6,23 (с, 1Н), 4,62 (т, 2Н, J=4,7 Гц), 4,26 (м, 4Н), 2,35 (с, 3Н), 1,43, с, 9Н), 1,25 (с, 18Н).
Стадия Н. Получение 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этилдигидрофосфата 74. Раствор ди-трет-бутил-2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этилфосфата (3,66 г, 4,89 ммоль) в ЕtOН (0,2 М, 24 мл) обрабатывают 5 N HCl/i-PrOH (2,93 мл, 14,7 ммоль) при комнатной температуре. Перемешивание при комнатной температуре продолжают в течение ночи. Растворитель выпаривают в вакууме и остаток растворителя довыпаривают из толуола и эфира (2х). Пену бежевого цвета растворяют в метаноле и медленно выливают в стакан, содержащий воду. Выпавшие в осадок твердые вещества отфильтровывают и получают 2,83 г (91%) требуемого продукта. МС (APCI+) m/z 637 (M+1). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,30 (с, 1H), 8,00 (с, 1H), 7,71 (д, 1H, J=9 Гц), 6,88-7,37 (м, 10Н), 6,24 (с, 1H), 4,61 (уш, 1H), 4,28 (д, 2Н, J=5,7 Гц), 4,19 (дд, 2Н, J=5,2 Гц), 2,35 (с, 3Н), 1,25 (с, 9Н).
Пример 5
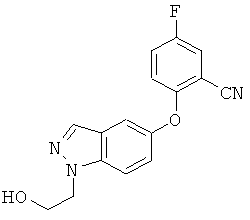
5-Фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)бензонитрил (78А)
К раствору метил-2-(5-(2-циано-4-фторфенокси)-1Н-индазол-1-ил)ацетата 77 (полученного, как описано в примере 3) (60,0 г, 184,4 ммоль) в МеОН (370 мл) прибавляют NaBH4 (24,42 г) пятью порциями по 5 г за 90 минут. Температура поднимается с 25°С до 44,7°С при последнем прибавлении NaBH4. Смесь концентрируют в вакууме. К концентрату прибавляют насыщенный раствор NH4Cl (600 мл) и ЕtOАс (600 мл) и смесь энергично перемешивают в течение 1 часа. Слои разделяют и водный слой экстрагируют ЕtOАс (2×100 мл). Объединенные органические слои промывают насыщенным раствором NH4Cl (2×100 мл), водой (200 мл), насыщенным раствором хлорида натрия (500 мл), сушат над MgSO4, фильтруют через бумагу GF/F, концентрируют в ярко-оранжевое твердое вещество. Неочищенное твердое вещество растворяют в СН2Сl2 (1 л) и прибавляют при перемешивании смесь гексанов (2,25 л). Образуется светло-коричневое твердое вещество; после перемешивания в течение 30 мин твердое вещество собирают и сушат в высоком вакууме, получая 45,5 г.Фильтрат концентрируют при пониженном давлении, остаток повторно растворяют в СН2Сl2 (100 мл) и прибавляют смесь гексанов (200 мл). Полученное твердое вещество (6,2 г) выделяют фильтрованием. Объединяют две порции, получая 51,7 г (94,3%) соединения 78А. 1Н ЯМР (400 МГц, CDC13) δ 7,99 (с, 1Н), 7,49 (д, 1Н), 7,39-7,36 (м, 2Н), 7,21-7,16 (м, 2Н), 6,90 (дд, 1Н), 4,49 (т, 2Н), 4,17-4,13 (м, 2Н), 2,81 (т, 1Н).
Пример 6
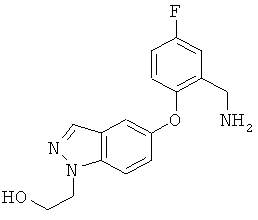
2-(5-(2-(Аминометил)-4-фторфенокси)-1Н-индазол-1-ил)этанол (79А)
(Способ А)
Во встряхиваемый сосуд Парра объемом 250 мл помещают 5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)бензонитрил 78А (полученный, как описано в примере 5) (80,0 г, 269,1 ммоль), Pd(OH)2 (32 г, 40% загрузка), ЕtOН (5 л) и концентрированную НСl (224 мл, 2691 ммоль). Сосуд закупоривают, продувают водородом, выпускают водород и подают водород до давления 180 фунт/кв.дюйм. Внутреннюю температуру понижают до 18°С и после перемешивания в течение 23 часов считают реакцию завершенной (ВЭЖХ). Реакционную смесь удаляют и всю последовательность повторяют два раза - с 80 г исходного вещества и затем с 53,2 г исходного вещества. Объединенные реакционные смеси (из 4 реакций) фильтруют через GF/F и фильтрат концентрируют при пониженном давлении. Остаток растворяют в 1 N НСl (3,0 л) и промывают ЕtOс (3×2 л). К водному слою прибавляют ЕtOАс (2 л) и фазы энергично перемешивают в течение 10 минут и затем органический слой отбрасывают. Эту стадию повторяют еще два раза и водный слой переносят в охлажденную колбу (ледяная баня). К кислой водной смеси прибавляют гранулы NaOH (40 г×11) таким образом, чтобы температура не поднималась выше 35°С. Как только рН становится ~14, смесь переносят в делительную воронку и экстрагируют СН2Сl2 (4×1 л). Объединенные экстракты промывают водой (2×1 л) и насыщенным раствором хлорида натрия (2×1 л). Экстракт сушат над MgSO4 (~250 г), фильтруют через GF/F, концентрируют при пониженном давлении. Концентрат растворяют в СН2Сl2 (3,5 л) и прибавляют смесь гексанов (4 л). После прибавления первых 2 л смеси гексанов образуется коричневое твердое вещество. После перемешивания в течение 1 часа твердое вещество отделяют фильтрованием и промывают на фильтре 500 мл смеси СН2Сl2/смесь гексанов (1:2), сушат в вакууме и получают 165,1 г (55,6%) соединения 79А. 1Н ЯМР (400 МГц, CDCl3) δ 7,90 (с, 1Н), 7,42 (д, 1Н), 7,17-7,12 (м, 3Н), 6,87 (тд, 1Н), 6,80 (дд, 1Н), 4,46 (т, 2Н), 4,12 (т, 2Н), 3,86 с (2Н), 1,87 (уш.с, 1Н).
Пример 7
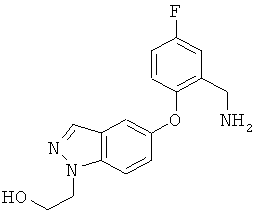
2-(5-(2-(Аминометил)-4-фторфенокси)-1Н-индазол-1-ил)этанол (79А)
(Способ В)
К раствору (5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)бензонитрила (1,0 г, 3,36 ммоль) и NiCl2·6Н2O (148 мг, 0,336 ммоль) в МеОН (30 мл) прибавляют NaBH4 (636 мг, 16,8 ммоль) порциями (2-3) и реакционную смесь перемешивают в течение 3 часов. Медленно прибавляют насыщенный раствор Na2CO3 (15 мл) и смесь перемешивают 75 минут, в ходе которых образуется мелкодисперсное твердое вещество. Реакционную смесь фильтруют через GF/F и слой на фильтре промывают МеОН (20 мл). Фильтрат концентрируют в вакууме до примерно 15 мл, прибавляют изопропилацетат (20 мл) и смесь концентрируют. К концентрату прибавляют изопропилацетат (20 мл) и смесь перемешивают 5 минут. Фазы разделяют и органический слой промывают 10% раствором хлорида натрия (20 мл). Прибавляют лимонную кислоту (0,1 М, 20 мл) и фазы энергично перемешивают. Органический слой удаляют и водный слой промывают изопропилацетатом (20 мл) и ЕtOАс (20 мл). К водному слою прибавляют изопропилацетат (20 мл) и 50% раствор NaOH (1 мл). Фазы энергично перемешивают и водный слой отбрасывают. Органический слой промывают 10% раствором хлорида натрия (20 мл), сушат над Na2SO4 и концентрируют, получая 850 мг (85%).
Пример 8
Фенил-3-трет-бутил-1-п-толил-1Н-пиразол-5-илкарбамат
К раствору 3-трет-бутил-1-п-толил-1H-пиразол-5 -амина (102,9 г, 448,9 ммоль) в ЕtOАс (1 л) при 10,9°С прибавляют раствор NaOH (2,5 М, 269 мл), двухфазная реакционная смесь нагревается до 15,9°С, и затем температура стабилизируется. Прибавляют фенилхлорформиат (98,4 г, 156,5 ммоль) одной порцией, температура поднимается до 25,5°С, а затем стабилизируется. Ледяную баню убирают и контролируют реакцию с помощью ВЭЖХ. Через 1 час прибавляют дополнительные 25 мл 2,5 М раствора NaOH. Реакционную смесь перемешивают в течение ночи и затем разбавляют ЕtOАс (200 мл). Органический слой промывают насыщенным раствором хлорида натрия (1 л), сушат над Na2SO4 (200 г), концентрируют до объема примерно 400-500 мл. Концентрированный раствор нагревают до 60°С. После растворения твердых веществ медленно прибавляют гептан (2 л), при этом образуются твердые вещества. После перемешивания в течение 30 минут отфильтровывают твердое вещество, слой на фильтре промывают 400-500 мл гептана и сушат в вакуумной печи при температуре окружающей среды, получая 156,9 г (95,0%) указанного в заголовке соединения. 1Н ЯМР (400 МГц, CDCl3) δ 7.39 - 7,31 (м, 6Н), 7,26-7,225 (м, 2Н), 7,13 (уш.с, 2Н), 6,96 (уш.с, 1Н), 6,47 (уш.с, 1Н), 2,42 (с, 3Н), 1,34(с, 9Н).
Пример 9
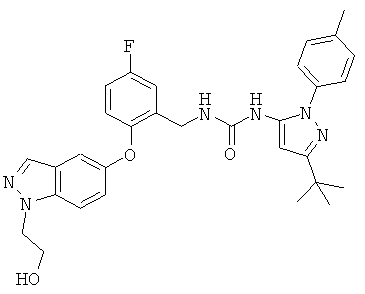
1-(3-трет-Бутил-1-п-толил-1Н-пиразол-5-ил)-3-(5-фтор-2-(1-(2-гидроксиэтил)-1Н-индазол-5-илокси)бензил)мочевина (72)
К раствору 2-(5-(2-(аминометил)-4-фторфенокси)-1Н-индазол-1-ил)этанола (71,55 г, 237 ммоль) в DMA (350 мл), температура которого 4,1°С, прибавляют раствор фенил-3-трет-бутил-1-п-толил-1Н-пиразол-5-илкарбамата (80,5 г, 230 ммоль) в DMA (350 мл). Температура поднимается до 18,4°С и стабилизируется. Ледяную баню удаляют и реакционную смесь перемешивают в течение 2 часов. Прибавляют дополнительную порцию 2-(5-(2-(аминометил)-4-фторфенокси)-1Н-индазол-1-ил)этанола и реакционную смесь перемешивают в течение 2 часов. Реакционную смесь распределяют между изопропилацетатом (1,6 л) и водой (2,4 л). Водный слой отбрасывают, органический слой промывают водой 0,5 N NaOH (2×2,4 л) и затем насыщенным раствором хлорида натрия (1×2,4 л). Органический слой сушат над Na2SO4 (300 г), концентрируют и получают 110,8 г (86,6%).
Предшествующее описание рассматривается только в качестве иллюстрации принципов изобретения. Более того, так как многочисленные модификации и изменения будут легко очевидны для специалистов в данной области техники, нежелательно ограничивать изобретение точным толкованием и способом, показанными, как описано выше. Соответственно, все приемлемые модификации и эквиваленты можно рассматривать как попадающие в объем притязаний изобретения, как определено представленной ниже формулой изобретения.
Слова «содержать», «содержащий», «включать», «включающий» и «включает» при использовании в настоящем описании и нижеследующей формуле изобретения предназначены для того, чтобы определить наличие установленных признаков, целых чисел, компонентов или стадий, но они не мешают наличию или добавлению одного или нескольких других признаков, целых чисел, компонентов, стадий или их групп.
Изобретение относится к соединению формулы I и его пролекарству, представляющему собой 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1 Н-индазол-1-ил)этилдигидрофосфат, а также к их фармацевтически приемлемым солям и фармацевтическим композициям на их основе. Указанные соединения являются ингибиторами киназ. Объектами изобретения также являются способы получения соединения формулы I, промежуточные соединения для его получения и способы лечения опосредованного киназами состояния у млекопитающего. В частности, указанные соединения могут применяться при лечении воспалительных заболеваний, аутоиммунных заболеваний, деструктивных заболеваний костей, пролиферативных заболеваний, инфекционных заболеваний, вирусных заболеваний или нейродегенеративных заболеваний. 10 н. и 5 з.п. ф-лы, 8 табл., 13 биологических пр., 9 препаративных пр.
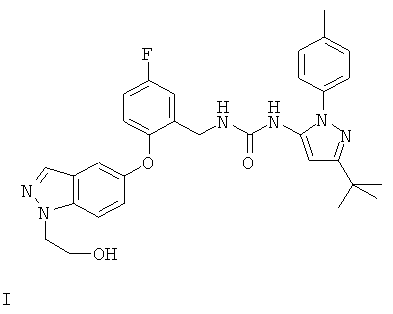
1. Соединение формулы I
или его фармацевтически приемлемая соль.
2. Способ получения соединения по п.1, который включает связывание соединения формулы II
или его соли, где Р1 представляет собой атом водорода с соединением формулы III
где Z представляет собой незамещенную или замещенную гидрокарбилоксигруппу, например галоген(1-6С)алкоксигруппу, такую как 2,2,2-трихлорэтокси, алкенилоксигруппу, такую как СР2=С(СН3)O-или необязательно замещенную арилоксигруппу.
3. Фармацевтическая композиция для лечения опосредованного киназами состояния, содержащая соединение по п.1 и фармацевтически приемлемый носитель или разбавитель.
4. Соединение по п.1 в качестве лекарственного средства для лечения опосредованного киназами состояния.
5. Способ лечения опосредованного киназами состояния у млекопитающего, включающий введение указанному млекопитающему соединения по п.1.
6. Способ по п.5, в котором указанное опосредованное киназами состояние представляет собой воспалительное заболевание, аутоиммунное заболевание, деструктивное заболевание костей, пролиферативное заболевание, инфекционное заболевание, вирусное заболевание или нейродегенеративное заболевание.
7. Соединение формулы II
или его соль.
8. Соединение формулы IV
где Re представляет собой атом водорода или (1-6С)алкоксигруппу, такую как этокси.
9. Способ лечения опосредованного киназами состояния у млекопитающего, включающий введение указанному млекопитающему пролекарства соединения по п.1, в котором пролекарство представляет собой 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этилдигидрофосфат или его фармацевтически приемлемую соль.
10. Способ по п.9, в котором указанное опосредованное киназами состояние представляет собой воспалительное заболевание, аутоиммунное заболевание, деструктивное заболевание костей, пролиферативное заболевание, инфекционное заболевание, вирусное заболевание или нейродегенеративное заболевание.
11. Соединение, представляющее собой 2-(5-(2-((3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)метил)-4-фторфенокси)-1Н-индазол-1-ил)этилдигидрофосфат или его фармацевтически приемлемую соль.
12. Фармацевтическая композиция для лечения опосредованного киназами состояния, содержащая соединение по п.11 и фармацевтически приемлемый носитель или разбавитель.
13. Способ по п.2, где Z представляет собой арилоксигруппу, необязательно замещенную одной или несколькими группами, выбранными из F, Cl, Вr и NO2.
14. Способ по п.2, где указанное соединение формулы (II) получено способом, включающим восстановление соединения формулы (V)
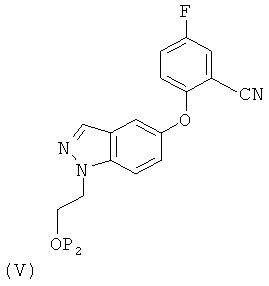
где P представляет собой водород в условиях каталитического гидрирования или в присутствии борида никеля.
15. Способ получения соединения по п.1, который включает восстановление соединения формулы IV
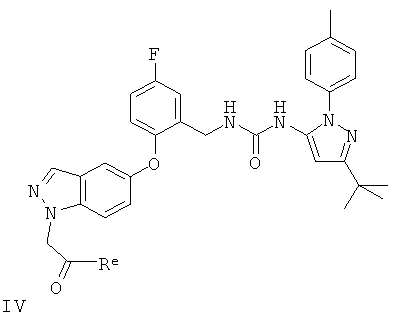
где Re представляет собой атом водорода или (1-6С)алкоксигруппу, такую как этокси и, если требуется, образованием фармацевтически приемлемой соли.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ АГЕНТОВ | 2000 |
|
RU2242474C2 |

