Область техники
Настоящее изобретение относится к сложноэфирному соединению аминокислоты и к композициям, содержащим сложноэфирное соединение аминокислоты. Изобретение также относится к применению соединения или композиции для ингибирования фермента р38 МАП-киназы. Кроме того, изобретение относится к кислоте, полученной посредством гидролиза сложноэфирной группы соединения по настоящему изобретению.
Предшествующий уровень техники
Неадекватная активация лейкоцитов, в том числе - моноцитов, макрофагов и нейтрофилов, ведущая к продукции повышенных уровней цитокинов, таких как ФНО-α (фактор некроза опухолей альфа), ИЛ 1-β (интерлейкин 1-β) и ИЛ-8 (интерлейкин-8), является особенностью патогенеза нескольких воспалительных заболеваний, включая ревматоидный артрит, язвенный колит, болезнь Крона, хроническую обструктивную болезнь легких (ХОБЛ), астму и псориаз, и клеточно-пролиферативных заболеваний с воспалительным компонентом. Продукция цитокинов воспалительными клетками является результатом реакции на разнообразные внешние стимулы, приводящие к активации ряда внутриклеточных сигнальных механизмов. Среди них выделяется суперсемейство митоген-активируемых протеинкиназ (МАПК), состоящее из высоко консервативных сигнальных киназ, которые регулируют рост клеток, их дифференциацию и реакции на стресс. Клетки млекопитающих содержат по меньшей мере три семейства МАПК: регулируемые внеклеточными сигналами (ERK; от англ.: extracellular signal-regulated kinases) р42/44 МАПК, с-JunNH2-терминальные киназы (JNK; от англ.: c-Jun NH2-terminal kinases) и р38 МАПК (также называемая p38a/Mpk2/RK/SAPK2a/CSBP1/2). р38МАПК была впервые клонирована после ее идентификации как киназы, которая фосфорилирована по остатку тирозина, после стимуляции моноцитов липополисахаридом (ЛПС) [Han et al., Science 1994, 265, 808]. Были описаны дополнительные гомологи р38 МАПК, которые включают р38β [Jiang et al., J. Biol. Chem., 1996, 271, 17920], p38γ [Li et al., Biochem. Biophys. Res. Commun., 1996, 228, 334] и р38δ [Jiang et al., J. Biol. Chem., 1997, 272, 30122]. Если p38α и p38β экспрессируются повсеместно, то экспрессия р38γ прежде всего ограничена скелетными мышцами, а р38δ преимущественно экспрессируется в легких и почках.
Выделение цитокинов иммунными клетками организма-хозяина и реакция лейкоцитов на цитокины и другие провоспалительные стрессы в различной степени регулируются р38 МАПК [Cuendaetal., FEBSLett., 1995, 364, 229-233]. В других типах клеток р38 МАПК регулирует стрессорные реакции, такие как продукция ИЛ-8 бронхиальными эпителиальными клетками, стимулируемая ФНО-α, и положительная регуляция молекулы клеточной адгезии ICAM-1 в эндотелиальных клетках, стимулированных ЛПС. После активации посредством двойного фосфорилирования TGY мотива киназами МКК3 и МКК6 с двойной специфичностью р38 МАПК оказывает свои эффекты за счет фосфорилирования факторов транскрипции и других киназ. Активированная МАП-киназой протеинкиназа-2 (МАРКАР-К2) идентифицирована как мишень для р38 фосфорилирования. Продемонстрировано, что мыши [Kotlyarov et al., Nat. Cell Biol., 1999, 1, 94-97], у которых отсутствовала MAPKAP-K2, выделяли сниженные уровни ФНО-α, ИЛ-1β, ИЛ-6, ИЛ-10 и ИФН-γ в ответ на эндотоксический шок, опосредованный ЛПС/галактозамином. Регуляция уровней этих цитокинов и ЦОГ-2 осуществляется на уровне мРНК. Уровни ФНО-α регулируются через контроль трансляции обогащенными аденилат-уридилатными последовательностями элементами 3'-нетранслируемых областей (3'-НТО) мРНК ФНО-α, причем МАРКАР-К2 сигналы усиливают трансляцию мРНК ФНО-α. МАРКАР-К2 сигналы приводят к повышению стабильности мРНК для ЦОГ-2, ИЛ-6 и макрофагального воспалительного белка. МАРКАР-К2 определяет локализацию в клетке р38 МАПК и преобразование сигналов р38 МАПК, поскольку содержит сигнал ядерной локализации на карбоксильном конце и сигнал ядерного экспорта в качестве части аутоингибиторного домена [Engeletal., EMBOJ., 1998, 17, 3363-3371]. В стрессированных клетках МАРКАР-К2 и р38 МАПК мигрируют в цитоплазму из ядра, эта миграция происходит только в том случае, если р38 МАПК каталитически активна. Считается, что это явление запускается за счет экспозиции сигнала ядерного экспорта МАРКАР-К2 в результате фосфорилирования протеинкиназой р38 МАПК [Meng et al., J. Boil. Chem., 2002, 277, 37401-37405]. Кроме того, p38 МАПК прямо или косвенно приводит к фосфорилированию нескольких факторов транскрипции, которые считают опосредующими воспаление, в том числе ATF1/2 (факторы активации транскрипции ½), CHOP-10/GADD-153 (ген 153, индуцируемый остановкой роста и повреждением ДНК), SAP-1 (вспомогательный белок-1 фактора ответа сыворотки) и MEF2C (фактор-усилитель миоцитов 2) [Fosteretal., DrugNewsPerspect., 2000, 13, 488-497].
В нескольких случаях было продемонстрировано, что ингибирование активности р38 МАПК мелкими молекулами можно использовать для лечения нескольких болезненных состояний, опосредованных неадекватной продукцией цитокинов, включая ревматоидный артрит, ХОБЛ, астму и ишемию мозга. Эта возможность была предметом нескольких обзоров [Salituro et al., Current Medicinal Chemistry, 1999, 6, 807-823 и Kumar et al., Nature Reviews Drug Discovery, 2003, 2, 717-726].
Показано, что ингибиторы p38 МАПК эффективны в моделях ревматоидного артрита у животных, таких как коллаген-индуцированный артрит у крыс [Revesz et al., Biorg. Med. Chem. Lett., 2000, 10, 1261-1364] и адъювант-индуцированный артрит у крыс [Wadsworth et al., J. Pharmacol. Exp. Ther., 1999, 291, 1685-1691]. В моделях повреждения легких, вызванного панкреатитом, у мышей предварительное лечение ингибитором р38 МАПК снижало выделение ФНО-α в дыхательных путях и отек легких [Denham et al., Crit. Care Ved., 2000, 29, 628 и Yang et al., Surgery, 1999, 126, 216]. Ингибирование p38 МАПК перед нагрузкой овальбумином (ОВА) у мышей, сенсибилизированных к ОВА, снижало аккумуляцию цитокинов и воспалительных клеток в дыхательных путях в модели аллергического воспаления дыхательных путей [Underwood et al., J. Pharmacol. Exp. Ther., 2000, 293, 281]. Повышенную активность p38 МАП-киназы наблюдали у пациентов, страдающих воспалительным заболеванием кишечника [Waetzig et al., J. Immunol., 2002, 168, 5432-5351]. Показано, что ингибиторы р38 МАПК эффективны в моделях гипертрофии сердца [Behr et al., Circulation, 2001, 104, 1292-1298] и фокальной церебральной ишемии [Baron et al., J. Pharmacol. Exp. Ther., 2001, 296, 312-321].
В публикациях WO 2007/129040 и WO 2009/060160 раскрыты сложные эфиры альфа-аминокислот, которые являются ингибиторами р38 МАП-киназы. В раскрытых соединениях сложноэфирная группа является сложноэфирной группой, которая может быть гидролизована одним или более внутриклеточными эстеразными ферментами до карбоксильной группы, а заместители у атома углерода сложного эфира альфа-аминокислоты образуют боковую цепь, которая является боковой цепью природной или неприродной альфа-аминокислоты.
Заявлено, что раскрытые соединения являются сильными и селективными ингибиторами р38 МАПК (р38α, β, γ и δ) и их изоформ и сплайс-вариантов, в частности - р38α, р38β и р38β2. Поэтому эти соединения могут быть использованы в медицине, например - для лечения и профилактики иммунных и воспалительных заболеваний, описанных в данной публикации. Соединения характеризуются наличием в молекуле аминокислотного мотива или мотива сложного эфира аминокислоты -NH-CHR1R2, который может быть гидролизован внутриклеточной карбоксилэстеразой. Соединения, содержащие липофильный мотив сложного эфира аминокислоты, проникают через клеточную мембрану и гидролизуются до кислоты внутриклеточными карбоксилэстеразами. Полярный продукт гидролиза аккумулируется в клетке, так как он не может легко проникать через клеточную мембрану. Соответственно, активность соединений в отношении р38 МАП-киназы продлевается и усиливается внутри клетки. Соединения по этому изобретению относятся к ингибиторам р38 МАП-киназы, охватываемым раскрытием в Международной заявке на патент WO 03076405, но отличаются от них тем, что они содержат мотив сложного эфира аминокислоты, указанный выше.
В публикации WO 2007/129040 также раскрыто то, что соединения, к которым она относится, включают соединения, избирательно накапливающиеся в макрофагах. Известно, что макрофаги играют ключевую роль в воспалительных заболеваниях за счет выделения цитокинов, в частности - ФНОα и ИЛ-1 (van Roon et al., Arthritis and Rheumatism, 2003, 1229-1238). При ревматоидном артрите они вносят основной вклад в поддержание воспаления суставов и деструкцию суставов. Макрофаги участвуют также в росте и развитии опухолей (Naldini and Carraro, Curr. Drug Targets Inflamm. Allergy, 2005, 3-8). Поэтому агенты, которые избирательно нацелены на пролиферацию макрофагальных клеток, могут иметь значение для лечения рака и аутоиммунных заболеваний. Можно ожидать, что нацеленность на специфические типы клеток приведет к снижению побочных эффектов. Способ, которым эстеразный мотив присоединен к ингибитору р38 киназы, определяет, будет ли он гидролизован, и, соответственно, будет ли он накапливаться в различных типах клеток. Более конкретно, макрофаги содержат карбоксилэстеразу hCE-1 человека, тогда как другие клетки ее не содержат. В соответствии с пунктом 1 формулы изобретения из публикации WO 2007/129040, если азот эстеразного мотива R1CH(R2)NH- не связан непосредственно с с карбонильной группой (-С(=O)-), то есть, если Y не является -С(=O), -С(=O)O- или -C(=O)NR3-радикалом, то сложный эфир будет гидролизоваться только hCE-1, и поэтому ингибиторы будут накапливаться только в макрофагах. В данной публикации, если не указано «моноцит» или «моноциты», термин «макрофаг» или «макрофаги» будет использован для обозначения макрофагов (включая опухолеассоциированные макрофаги) и/или моноцитов.
В публикации WO 2009/060160 раскрыта группа специфических соединений, попадающих в объем изобретения WO 2007/129040, но конкретно не идентифицированных и не проиллюстрированных примерами. Соединения обладают свойством избирательности к макрофагам, обсуждавшимся выше.
Сущность изобретения
Авторы настоящего изобретения обнаружили, что соединение трет-бутил-N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланинат неожиданно хорошо ингибирует активность р38 МАП-киназы. Испытания показали, что значение IC50 для ингибирования сложным эфиром ФНО-α в крови человека значительно ниже, чем можно было ожидать, исходя из значений IC50 для ингибирования сложным эфиром ФНО-α в крови человека, полученных для родственных соединений. Кроме того, испытания, проведенные авторами настоящего изобретения, продемонстрировали, что биодоступность соединения по настоящему изобретению значительно выше, чем можно было предсказать, исходя из сходных по структуре известных соединений.
Соответственно, настоящее изобретение обеспечивает соединение, которое является трет-бутил-N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланинатом или его фармацевтически приемлемой солью, гидратом или сольватом.
Настоящее изобретение также обеспечивает фармацевтическую композицию, содержащую соединение совместно с одним или более фармацевтически приемлемыми носителями и/или наполнителями.
В другом аспекте изобретение обеспечивает соединение, определенное выше, или композицию, определенную выше, для применения в способе воздействия на организм человека или животного посредством лечения.
Настоящее изобретение также обеспечивает соединение, определенное выше, или композицию, определенную выше, для применения для ингибирования активности фермента р38 МАП-киназы in vitro или in vivo. Кроме того, обеспечены соединение, определенное выше, или композиция, определенная выше, для применения для профилактики или лечения аутоиммунного или воспалительного заболевания. Также обеспечены соединение, определенное выше, или композиция, определенная выше, для применения для лечения клеточно-пролиферативного заболевания.
В другом аспекте изобретение обеспечивает способ ингибирования активности фермента р38 МАП-киназы, который включает обеспечение контакта фермента с количеством соединения, определенного выше, или композиции, определенной выше, эффективным для такого ингибирования. Также обеспечен способ лечения или профилактики аутоиммунного или воспалительного заболевания у субъекта, который включает введение указанному субъекту эффективного количества соединения, определенного выше, или композиции, определенной выше. Также обеспечен способ лечения клеточно-пролиферативного заболевания у субъекта, который включает введение указанному субъекту эффективного количества соединения, определенного выше, или композиции, определенной выше. Лечение может включать облегчение симптомов или снижение инцидентности клеточно-пролиферативного заболевания.
В следующем аспекте изобретение обеспечивает применение соединения, определенного выше, или композиции, определенной выше, для производства лекарственного средства для ингибирования активности фермента р38 МАП-киназы. Далее обеспечено применение соединения, определенного выше, или композиции, определенной выше, для производства лекарственного средства для профилактики или лечения аутоиммунного или воспалительного заболевания. Также обеспечено применение соединения, определенного выше, или композиции, определенной выше, для производства лекарственного средства для лечения клеточно-пролиферативного заболевания.
Изобретение также обеспечивает средство для ингибирования активности фермента р38 МАП-киназы, содержащее соединение, определенное выше, или композицию, определенную выше, в качестве активного ингредиента. Это средство в характерном случае предназначено для профилактики или лечения аутоиммунного или воспалительного заболевания. Альтернативно оно может быть предназначено для лечения клеточно-пролиферативного заболевания.
Изобретение также обеспечивает кислоту, которая является N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланином.
Сведения, подтверждающие возможность осуществления изобретения
Настоящее изобретение обеспечивает соединение, которое является трет-бутил-N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланинатом.
Соединение по настоящему изобретению может быть получено в форме соли, гидрата или сольвата. Поэтому изобретение также обеспечивает соль, гидрат или сольват соединения по настоящему изобретению. В характерном случае соль является фармацевтически приемлемой солью.
При использовании в контексте настоящего изобретения термин «фармацевтически приемлемая соль» относится к соли фармацевтически приемлемой кислоты или фармацевтически приемлемого основания. Фармацевтически приемлемые кислоты включают неорганические кислоты, такие как хлористоводородная, серная, фосфорная, дифосфорная, бромистоводородная или азотная кислоты, и органические кислоты, такие как лимонная, салициловая, глутаминовая, молочная, фумаровая, малеиновая, яблочная, аскорбиновая, янтарная, винная, бензойная, уксусная, метансульфоновая, этансульфоновая, бензолсульфоновая или п-толуолсульфоновая кислоты. Фармацевтически приемлемые основания включают гидроксиды щелочных металлов (например, натрия или калия) и щелочноземельных металлов (например, кальция, бария или магния) и органические основания, такие как алкиламины, аралкиламины и гетероциклические амины. Примерами подходящих органических оснований являются, но не ограничиваются этим, N-метил-D-глюкамин, холин, трис(гидроксиметил)аминометан, L-аргинин, L-лизин, N-этилпиперидин, дибензиламин. Обзор подходящих солей см. в руководстве Handbook of Pharmaceutical Salts: Properties, Selection and Use авторов Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Подходящие соли соединений по настоящему изобретению включают соли, указанные выше в качестве примеров фармацевтически приемлемых солей.
Термин «сольват» при использовании в контексте настоящего изобретения описывает молекулярный комплекс, содержащий соединение по настоящему изобретению и стехиометрическое количество молекул одного или более фармацевтически приемлемого растворителя, например - этанола. Термин «гидрат» в контексте настоящего изобретения используют, если указанным растворителем является вода.
Во избежание противоречий следует указать, что соединение по настоящему изобретению может быть использовано в любой таутомерной форме.
Соединение по настоящему изобретению содержит хиральный центр. В характерном случае соединение находится в форме производного L-аланината (как изображено в Примере 1). Однако соединение может существовать как производное D-аланината или как смесь D- и L-форм. Если присутствует смесь, то предпочтительно по меньшей мере 90%, 95% или 99% соединения по настоящему изобретению присутствует в ней в форме производного L-аланината.
Далее обсуждаются подходящая схема и способ получения соединения по настоящему изобретению со ссылкой на приведенный ниже раздел «Описание примеров осуществления изобретения».
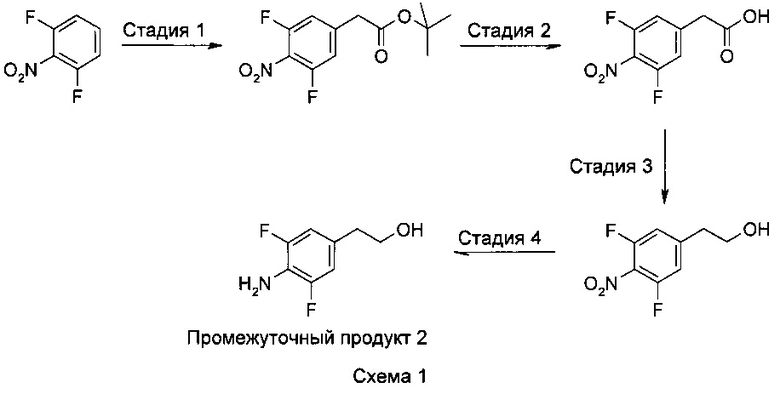
Исходными материалами в характерном случае являются 4-хлорфенил-3-(2,4-дифторфенил)-3-оксопропанимидотиоата гидрохлорид и 2-(4-амино-3,5-дифторфенил)этанол. 2-(4-амино-3,5-дифторфенил)этанол может быть получен с использованием следующей схемы, которая аналогична схеме 1 из раздела «Описание примеров осуществления изобретения»:
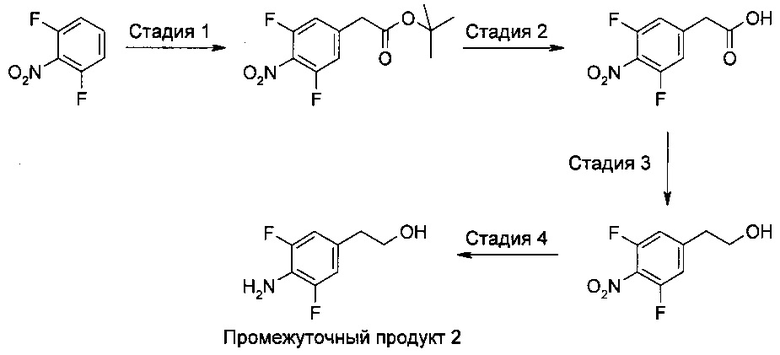
Дифторнитробензол коммерчески доступен. Стадия 1 требует присоединения трет-бутилацетатной группы к фенильному кольцу в пара-положении по отношению к нитрогруппе. Стадия 2 требует гидролиза сложноэфирной группы с получением соответствующей кислоты. Кислота восстанавливается до первичного спирта на Стадии 3. На Стадии 4 нитрогруппа восстанавливается до амина.
4-хлорфенил-3-(2,4-дифторфенил)-3-оксопропанимидотиоата гидрохлорид может быть получен с использованием экспериментальных процедур, описанных в публикации WO 2003076405.
Затем может быть синтезировано соединение трет-бутил-N-(2-{4-(6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил)-L-аланинат с использованием следующей схемы, которая аналогична Схеме 2 в разделе «Описание примеров осуществления изобретения».
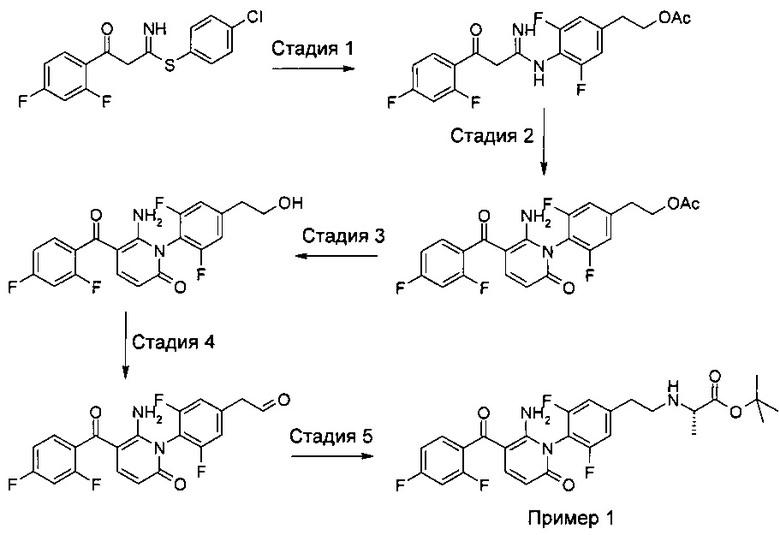
На Стадии 1 2-(4-амино-3,5-дифторфенил)этанол и 4-хлорфенил-3-(2,4-дифторфенил)-3-оксопропанимидотиоата гидрохлорид реагируют друг с другом с образованием 2-(4-{[3-(2,4-дифторфенил)-3-оксопропанимидоил]амино}-3,5-дифторфенил)этилацетата. На Стадии 2 добавляют пропиоловую кислоту с получением 2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этилацетата. На Стадии 3 ацетатную группу гидролизуют с получением спирта, и на Стадии 4 полученную спиртовую группу окисляют до альдегида. Затем на Стадии 5 получают соединение по настоящему изобретению посредством добавления трет-бутил-L-аланината гидрохлорида. Трет-бутил-L-аланината гидрохлорид коммерчески доступен.
Изобретение также обеспечивает фармацевтическую композицию, содержащую соединение по настоящему изобретению совместно с одним или более фармацевтически приемлемыми носителями и/или наполнителями. Фармацевтическая композиция в характерном случае содержит до 85 масс. % соединения по настоящему изобретению. Более предпочтительно она содержит до 50 масс. % соединения по настоящему изобретению. Предпочтительные фармацевтические композиции являются стерильными и не содержат пирогенов.
Соединение по настоящему изобретению может быть введено в различных лекарственных формах. Так, оно может быть введено перорально, например - в форме таблеток, пастилок, капсул, леденцов, водных или масляных суспензий, диспергируемых порошков или гранул. Соединения по настоящему изобретению также могут быть введены парентерально - подкожно, внутривенно, внутримышечно, внутригрудинно, чрескожно или с использованием инфузионных техник. В зависимости от используемого носителя и концентрации лекарственное средство может быть суспензировано или растворено в носителе. Предпочтительно, в носителе могут быть растворены адъюванты, такие как местный анестетик, консервант и буферный агент. Соединения по настоящему изобретению также могут быть введены в форме суппозиториев. Соединения по настоящему изобретению могут быть введены посредством ингаляции в форме аэрозоля с использованием ингалятора или небулайзера.
Соединение по настоящему изобретению в характерном случае входит в состав композиции для введения совместно с фармацевтически приемлемым носителем или разбавителем. Например, твердые формы для орального введения могут содержать, совместно с активным соединением, солюбилизирующие агенты, например - циклодекстрины или модифицированные циклодекстрины; разбавители, например - лактозу, декстрозу, сахарозу, целлюлозу, кукурузный крахмал или картофельный крахмал; смазывающие агенты, например - диоксид кремния, тальк, стеариновую кислоту, стеараты магния или кальция и/или полиэтиленгликоли; связующие агенты, например - крахмалы, аравийскую камедь, трагакантовую камедь, желатин, сироп, гуммиарабик, сорбитол, метилцеллюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; дезагрегирующие агенты, например - крахмал, альгиновую кислоту, альгинаты или натрия крахмала гликоляты; пенообразующие смеси; красители; подсластители; смачивающие агенты, такие как лецитин, полисорбаты, лаурилсульфаты; и, в целом, нетоксичные и фармакологические неактивные вещества, используемые в фармацевтических композициях. Такие фармацевтические композиции могут быть получены известными способами, например - посредством смешивания, гранулирования, таблетирования, нанесения сахарного покрытия или нанесения пленочного покрытия.
Жидкие дисперсии для перорального введения могут быть растворами, сиропами, эмульсиями и суспензиями. Жидкие композиции могут содержать стандартные добавки, такие как суспензирующие агенты, например - сорбитол, сироп, метилцеллюлозу, сироп глюкозы, желатин, гидрогенизированные пищевые жиры; эмульгаторы, например - лецитин, сорбитана моноолеат или гуммиарабик; неводные разбавители (которые могут включать пищевые масла), например - миндальное масло, фракционированное кокосовое масло, жирные сложные эфиры, такие как глицерин, пропиленгликоль или этиловый спирт; консерванты, например - метил- или пропил-п-гидроксибензоат или сорбиновую кислоту, и, по желанию, стандартные вкусовые добавки или красители. Растворы могут содержать солюбилизирующие агенты, например - циклодекстрины или модифицированные циклодекстрины. Сиропы могут содержать в качестве носителей, например, сахарозу или сахарозу с глицерином и/или маннитол и/или сорбитол.
Суспензии и эмульсии могут содержать в качестве носителя, например, природную камедь, агар, натрия альгинат, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензии или растворы для внутримышечных инъекций могут содержать, совместно с активным соединением, например, стерильную воду, оливковое масло, этилолеат, гликоли, например - пропиленгликоль; солюбилизирующие агенты, например - циклодекстрины или модифицированные циклодекстрины, и, при желании, подходящее количество лидокаина гидрохлорида.
Растворы для внутривенного введения или инфузий могут содержать в качестве носителя, например, стерильную воду и солюбилизирующие агенты, например - циклодекстрины или модифицированные циклодекстрины, или, предпочтительно, они могут находиться в форме стерильных водных изотонических солевых растворов.
Для местного нанесения на кожу лекарственное средство может быть приготовлено в форме крема, лосьона или мази. Композиции в форме крема или мази, которые могут быть использованы в качестве лекарственного средства, являются стандартными композициями, хорошо известными в данной области техники, например, такими, как описано в стандартных руководствах по фармацевтике, таких как Британская фармакопея.
Для местного применения посредством ингаляции лекарственное средство может быть приготовлено для доставки в форме аэрозоля, например - с помощью струйных распылителей под давлением или ультразвуковых распылителей или, предпочтительно, в форме дозированных аэрозолей, распыляемых газом-вытеснителем, или посредством введения микронизированных порошков без газа-вытеснителя, например - ингаляционных капсул или других систем доставки «сухих порошков». Вспомогательные вещества, такие как, например, газы-вытеснители (например, Frigen в случае дозированных аэрозолей), поверхностно-активные вещества, эмульгаторы, стабилизаторы, консерванты, вкусовые добавки и наполнители (например, лактоза в случае порошковых ингаляторов) могут присутствовать в таких ингаляционных композициях. Для целей ингаляции доступно большое количество аппаратов, с помощью которых можно генерировать и вводить аэрозоли с оптимальным размером частиц с использованием техники ингаляции, подходящей для пациента. Кроме того, для использования адаптеров (спейсерных устройств, расширителей) и грушевидных контейнеров (например, Nebulator®, Volumatic®) и автоматических устройств, выделяющих аэрозольный спрей (Autohaler®), для дозированных аэрозолей, в частности - в случае порошковых ингаляторов, доступно много технических решений (например, Diskhaler®, Rotadisk®, Turbohaler® или ингаляторы, например, такие, как описано в Европейской патентной заявке ЕР 0505321).
Для местного применения в глазах лекарственное средство может быть приготовлено в форме раствора или суспензии в подходящем стерильном водном или неводном носителе. Также могут быть включены добавки, например - буферы, такие как натрия метабисульфит или динатрия эдетат; консерванты, в том числе бактерицидные и фунгицидные агенты, такие как фенилртути ацетат или нитрат, бензалкония хлорид или хлоргексидин, и загустители, такие как гипромеллоза.
Субъекту вводят терапевтически эффективное количество соединения по настоящему изобретению. Следует понимать, что специфический уровень дозы для каждого конкретного субъекта будет зависеть от многих факторов, включая активность специфического использованного соединения, возраст, массу тела, общее состояние здоровья, пол, диету, время введения, путь введения, скорость экскреции, комбинацию лекарственных средств и степень тяжести конкретного заболевания, подвергающегося лечению. Оптимальные уровни дозы и частоту дозирования обычно определяют в клиническом испытании.
Характерная дневная доза достигает 50 мг на кг массы тела, например - от 0,001 мг/кг массы тела до 50 мг/кг массы тела, в соответствии с активностью специфического соединения, возраста, массы тела и состояния субъекта, подлежащего лечению, типа и степени тяжести болезни и частоты и пути введения. Предпочтительно дневные уровни дозы лежат в диапазоне от 0,5 мг до 2 г, более предпочтительно - от 0,1 мг до 10 мг. Соединение по настоящему изобретению обычно вводят пациенту в нетоксичном количестве.
Изобретение также обеспечивает соединение, определенное в данной публикации, или композицию, определенную в данной публикации, для использования в способе воздействия на организм человека или животного посредством лечения.
Обнаружено, что соединения и композиции по настоящему изобретению ингибируют активность фермента р38 МАП-киназы. Поэтому соединения и композиции можно использовать для профилактики и лечения болезней и состояний, модулируемых активностью р38 МАП-киназы. Болезни и состояния, модулируемые активностью р38 МАП-киназы, включают клеточно-пролиферативные заболевания, такие как рак и псориаз, полиглутаминовое заболевание, такое как болезнь Хантингтона, нейродегенеративное заболевание, такое как болезнь Альцгеймера, аутоиммунное заболевание, такое как ревматоидный артрит, диабет, гематологическое заболевание, воспалительное заболевание, сердечно-сосудистое заболевание, атеросклероз и воспалительные осложнения инфекции. Конкретными примерами являются клеточно-пролиферативное заболевание, аутоиммунное заболевание и воспалительное заболевание.
Аутоиммунное заболевание часто имеет воспалительный компонент. Такие состояния включают острую диссеминированную универсальную алопецию, АНЦА-позитивные заболевания (АНЦА = антитела к цитоплазме нейтрофилов), болезнь Бехчета, болезнь Шагаса, синдром хронической усталости, дизавтономия, энцефаломиелит, анкилозирующий спондилит, апластическая анемия, гнойный гидраденит, аутоиммунный гепатит, аутоиммунный оофорит, целиакия, воспалительное заболевание кишечника, болезнь Крона, сахарный диабет 1 типа, синдром Фанкони, гигантоклеточный артериит, гломерулонефрит, синдром Гудпасчера, болезнь Грейвса, синдром Гийена-Барре, болезнь Хашимото, пурпура Геноха-Шенлейна, болезнь Кавасаки, системная красная волчанка, микроскопический колит, микроскопический полиартериит, смешанное заболевание соединительной ткани, рассеянный склероз, миастения гравис, опсоклонус-миоклонус синдром, неврит зрительного нерва, тиреоидит Орда, пемфигус, узловой периартериит, синдром Рейтера, синдром Шегрена, височный артериит, гранулематоз Вегенера, «теплый тип» аутоиммунной гемолитической анемии, интерстициальный цистит, болезнь Лайма, очаговая склеродермия, псориаз, саркоидоз, склеродермия, язвенный колит и витилиго.
Другие воспалительные состояния, которые можно предотвратить или лечить соединениями и композициями по настоящему изобретению, включают, например, аппендицит, дерматит, дерматомиозит, эндокардит, фиброзит, гингивит, глоссит, гепатит, гнойный гидраденит, ирит, ларингит, мастит, миокардит, нефрит, отит, панкреатит, паротит, перикардит, перитонит, фарингит, плеврит, пневмонит, простатит, пиелонефрит и стоматит, отторжение трансплантата (включая такие органы, как почки, печень, сердце, легкие, поджелудочная железа (например, островковые клетки), костный мозг, роговица, тонкий кишечник, кожные аллографты, кожные гомографты и ксенографты клапанов сердца, сывороточная болезнь и болезнь «трансплантат против хозяина»), острый панкреатит, хронический панкреатит, острый респираторный дистресс-синдром, синдром Сезари, врожденная гиперплазия надпочечников, негнойный тиреоидит, гиперкальциемия, связанная с раком, пемфигус, буллезный герпетиформный дерматит, тяжелая мультиформная эритема, эксфолиативный дерматит, себорейный дерматит, сезонный или круглогодичный аллергический ринит, бронхиальная астма, контактный дерматит, атопический дерматит, реакции гиперчувствительности к лекарствам, аллергический конъюнктивит, кератит, герпес зостер офтальмологический, ирит и иридоциклит, хориоретинит, неврит зрительного нерва, симптоматический саркоидоз, химиотерапия скоротечного или диссеминированного туберкулеза легких, идиопатическая тромбоцитопеническая пурпура взрослых, вторичная тромбоцитопения взрослых, приобретенная (аутоиммунная) гемолитическая анемия, лейкемия и лимфома у взрослых, острая лейкемия у детей, региональный энтерит, аутоиммунный васкулит, рассеянный склероз, хроническая обструктивная болезнь легких, отторжение трансплантата солидных органов, сепсис, первичный билиарный цирроз печени и первичный склерозирующий холангит.
Соединения и композиции по настоящему изобретению можно использовать для профилактики и лечения воспалительных и аутоиммунных болезней и состояний. Воспалительные и аутоиммунные болезни и состояния, которые можно лечить с использованием соединений и композиций по настоящему изобретению включают ревматоидный артрит, псориатический артрит, диабет 1 типа, астму, воспалительное заболевание кишечника, системную красную волчанку и сопровождаемые воспалением инфекционные состояния (например, сепсис), псориаз, болезнь Крона, язвенный колит, хроническую обструктивную болезнь легких, рассеянный склероз, атопический дерматит и болезнь «трансплантат против хозяина».
Соединения и композиции по настоящему изобретению также можно использовать для лечения клеточно-пролиферативного заболевания, например -рака. Примерами раков, которые можно лечить, являются рак молочной железы, рак яичников, рак легкого, рак поджелудочной железы, рак печени, рак толстого кишечника, рак почки, лимфома и меланома. Например, раками, которые можно лечить, являются рак молочной железы, рак яичников, рак поджелудочной железы, рак легкого, рак толстого кишечника, рак почки, лимфома или меланома.
Изобретение также относится к кислоте, которая является:
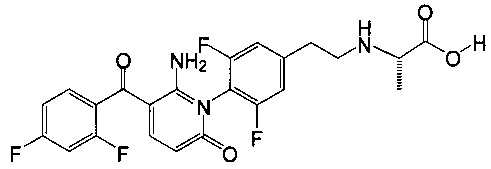
N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланином.
Кислота может быть получена посредством гидролиза соединения по настоящему изобретению.
Как обсуждалось выше, соединение по настоящему изобретению является селективным по отношению к макрофагам. Соответственно, сложноэфирная группа соединения по настоящему изобретению может быть гидролизована клетками, содержащими карбоксилэстеразу hCE-1 человека, и не может быть гидролизована клетками, содержащими hCE-2 или hCE-3. Поэтому кислота образуется внутри клеток, содержащих hCE-1, в процессе гидролиза сложноэфирной группы соединения по настоящему изобретению, и кислота избирательно накапливается в таких клетках.
Настоящее изобретение дополнительно проиллюстрировано приведенными ниже Примерами его осуществления.
Описание примеров осуществления изобретения
Соединение по настоящему изобретению может быть получено в соответствии со следующим Примером.
Аббревиатуры
КДИ = карбонилдиимидазол
ДХМ = дихлорметан
ДМФ = диметилформамид
EtOAc = этилацетат
HCl = соляная кислота
ЖХМС = высокоэффективная жидкостная хроматография/масс-спектрометрия
МеОН = метанол
MgSO4 = сульфат магния
Na2CO3 = карбонат натрия
NaHCO3 = гидрокарбонат натрия
ЯМР = ядерный магнитный резонанс
ТАБН = триацетоксиборогидрид натрия
ТГФ = тетрагидрофуран
г = грамм(ы)
мг = миллиграмм(ы)
мл = миллилитр(ы)
ммоль = миллимоль (миллимоли)
Коммерчески доступные реагенты и растворители (ЖХМС качества) использовали без дополнительной очистки. Растворители удаляли с использованием ротационного испарителя производства компании Buchi. Микроволновое излучение генерировали с использованием микроволнового синтезатора Biotage Initiator™ Eight. Очистку соединений с помощью флэш-хроматографической колонки выполняли с использованием силикагеля, размер частиц 40-63 мкм (230-400 меш), полученного из компании Fluorochem.
1Н ЯМР-спектры регистрировали на спектрометре AV 300 МГц производства компании Bruker в дейтерированных растворителях. Химические сдвиги (d) выражали в частях на миллион. Анализ посредством тонкослойной хроматографии (ТСХ) выполняли с использованием пластин Кизельгель 60 F254 (производства компании Merck) и визуализировали с использованием УФ-излучения.
Аналитическую ВЭЖХ/МС выполняли на системе Agilent НР1100 LC с использованием обратнофазных колонок Luna С18 (3 мм, 50 × 4,6 мм), градиент 5-95% В (А = вода / 0,1% муравьиная кислота, В = ацетонитрил / 0,1% муравьиная кислота) в течение 2,25 мин, объемная скорость = 2,25 мл/мин. УФ-спектры регистрировали при длинах волн 220 нм и 254 нм с использованием детектора G1315 DAD. Массовые спектры получали в диапазоне m/z от 150 до 800 с использованием детектора LC/MSD SL G1956 В. Данные интегрировали и представляли с использованием программного обеспечения ChemStation и ChemStation Data Browser.
Промежуточные продукты
Промежуточный продукт 1: 4-хлорфенил-3-(2,4-дифторфенил)-3-оксопропанимидотиоата гидрохлорид
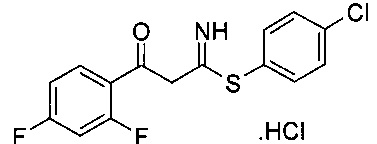
Промежуточный продукт 1 можно получить с использованием экспериментальных процедур, описанных в публикации WO 2003076405.
Промежуточный продукт 2: 2-(4-амино-3,5-дифторфенил)этанол
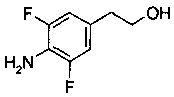
Промежуточный продукт 2 был синтезирован с использованием пути, изображенного на Схеме 1 ниже.
Стадия 1 - трет-бутил-(3,5-дифтор-4-нитрофенил)ацетат
Раствор дифторнитробензола (24,96 г, 157 ммоль) и трет-бутил-хлорацетат (38,0 мл, 267 ммоль) в безводном ДМФ (200 мл) добавили по каплям в течение часа к холодной (-35°C) суспензии калия трет-бутоксида (61,61 г, 549 ммоль) в безводном ДМФ (200 мл) под азотом. Реакционную смесь перемешивали при -35°C в течение 1,5 часов, погасили 2N HCl (240 мл) и экстрагировали гептаном (4×200 мл). Объединенные органические экстракты промыли водой (3×200 мл), рассолом (200 мл), высушили (MgSO4), профильтровали и сконцентрировали при пониженном давлении с получением желтого масла. Очистка посредством колоночной хроматографии (10% EtOAc в гептане) дала желтое масло (37,64 г). Другие две загрузки (10,00 г и 23,45 г дифторнитробензола) дали 14,30 г и 31,39 г продукта, соответственно. 1Н ЯМР всех трех загрузок продемонстрировал смесь желаемого соединения и небольших количеств неидентифицированных загрязнений. 3 загрузки объединили и использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (300 МГц, CDCl3) 7,05 (2Н, d, J=8,5 Гц), 3,56 (2Н, s), 1,46 (9Н, s).
Стадия 2 - (3,5-дифтор-4-нитрофенил)уксусная кислота
Трифторуксусную кислоту (150 мл) по каплям в течение 20 минут добавили к холодному (0°C) раствору трет-бутил-(3,5-дифтор-4-нитрофенил)ацетата (83,33 г, 305 ммоль) в ДХМ (300 мл). После завершения добавления реакционной смеси позволили нагреться до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь сконцентрировали при пониженном давлении с получением липкого коричневого твердого вещества. Растирание с гептаном дало титульное соединение в форме желтого твердого вещества (53,29 г, 67%-ный выход после двух стадий).
1Н ЯМР (300 МГц, CDCl3) 7,08 (2Н, d, J=8,5 Гц), 3,74 (2Н, s), -CO2H не виден.
Стадия 3 - 2-(3,5-дифтор-4-нитрофенил)этанол
Борандиметилсульфидный комплекс (35 мл, 368 ммоль) по каплям в течение 20 минут добавили к холодному (0°C) раствору (3,5-дифтор-4-нитрофенил)уксусной кислоты (53,29 г, 245 ммоль) в безводном ТГФ (500 мл) под азотом. После завершения добавления реакционной смеси позволили нагреться до комнатной температуры, перемешивали в течение 16 часов, охладили до 0°C, осторожно погасили МеОН (300 мл) и сконцентрировали при пониженном давлении с получением коричневого масла. Очистка посредством сухой флэш-хроматографии (60-80% EtOAc в гептане) дала титульное соединение в форме оранжевого масла (38,90 г, выход 78%).
1Н ЯМР (300 МГц, CDCl3) 7,01 (2Н, d, J=8,7 Гц), 3,93 (2Н, t, J=6,2 Гц), 2,92 (2Н, t, J=6,2 Гц), 2,34 (1Н, br s).
Стадия 4 - 2-(4-амино-3,5-дифторфенил)этанол
2-(3,5-дифтор-4-нитрофенил)этанол (38,90 г, 191 ммоль) растворили в EtOAc (250 мл). Из химического реактора удалили воздух и три раза заполнили его азотом. Добавили палладий на углероде (10 масс. %, 4,00 г), удалили из реактора газ и три раза заполнили азотом. Затем из реактора удалили газ, заполнили его водородом и соединили с баллоном, содержащим водород. После перемешивания при комнатной температуре под водородом в течение 15 часов снова заполнили баллон водородом и перемешивали смесь в течение дополнительных 25 часов. Реакционную смесь профильтровали через Celite® и сконцентрировали фильтрат при пониженном давлении с получением коричневого масла. Очистка посредством сухой флэш-хроматографии (50% EtOAc в гептане) дала титульное соединение в форме белого твердого вещества (20,70 г, выход 62%).
1Н ЯМР (300 МГц, CDCl3) 6,73-6,70 (2Н, m), 3,81 (2Н, t, J=6,4 Гц), 2,75 (2Н, t, J=6,4 Гц), -ОH и -NH2 не видны.
Пример 1
Трет-бутил-N-(2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил)-L-аланинат
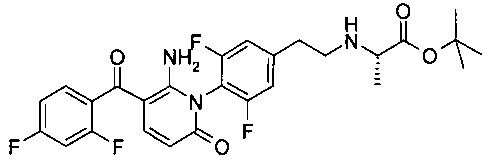
Пример 1 был синтезирован с использованием пути, изображенного на Схеме 2 ниже.
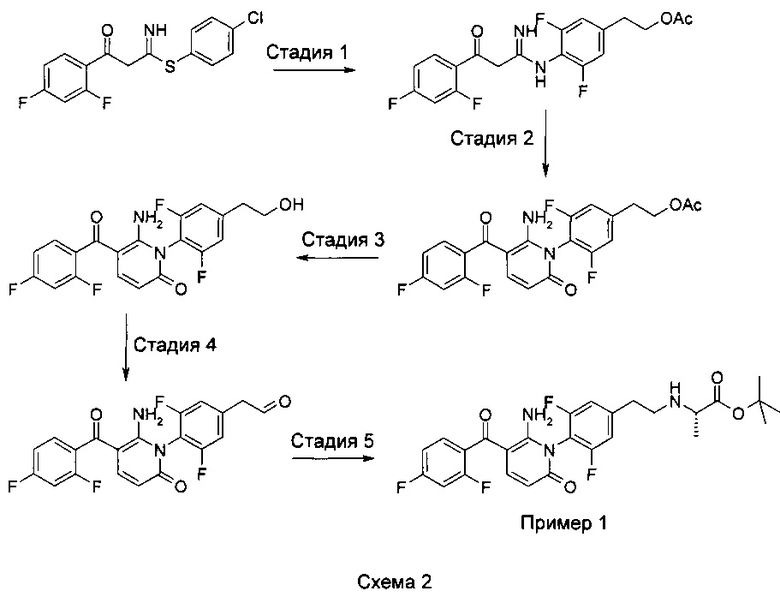
Стадия 1 - 2-(4-{[3-(2,4-дифторфенил)-3-оксопропанимидоил]амино}-3,5-дифторфенил)этилацетат
2-(4-амино-3,5-дифторфенил)этанол (20,71 г, 120 ммоль) добавили к раствору 4-хлорфенил-3-(2,4-дифторфенил)-3-оксопропанимидотиоата гидрохлорида (41,26 г, 114 ммоль) в ледяной уксусной кислоте (400 мл). Реакционную смесь перемешивали при 80°C в течение 2,5 часов и добавили уксусный ангидрид (21 мл, 228 ммоль). После дополнительных 45 минут при 80°C реакционной смеси дали остыть до комнатной температуры и сконцентрировали при пониженном давлении с получением коричневого масла. Растирание с EtOAc дало бежевое твердое вещество, которое промыли диэтиловым эфиром. Твердое вещество перенесли в насыщенный водный раствор NaHCO3 и интенсивно перемешивали в течение 30 минут. Твердое вещество собрали посредством фильтрации, промыли водой и высушили при пониженном давлении с получением титульного соединения в форме бежевого твердого вещества (23,36 г, выход 52%). ЖХМС: m/z 397 [М+Н]+.
Стадия 2 - 2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этилацетат
Пропиоловую кислоту (5,4 мл, 88 ммоль) добавили по каплям в течение 5 минут к холодному (0°C) раствору КДИ (14,27 г, 88 ммоль) в безводном ТГФ (400 мл) под азотом. После завершения добавления реакционной смеси дали нагреться до комнатной температуры и перемешивали в течение одного часа. Добавили раствор 2-(4-{[3-(2,4-дифторфенил)-3-оксопропанимидоил]амино}-3,5-дифторфенил)этилацетата (23,26 г, 59 ммоль) в безводном ТГФ (200 мл) и реакционную смесь перемешивали с обратным холодильником в течение 6,5 часов. Реакционной смеси дали остыть до комнатной температуры и оставили на 16,5 часов. Пропиоловую кислоту (5,4 мл, 88 ммоль), КДИ (14,27 г, 88 ммоль) и ТГФ (200 мл) обработали так, как описано выше, и добавили к реакционной смеси, которую затем перемешивали с обратным холодильником в течение дополнительных 6 часов. Затем реакционной смеси дали остыть до комнатной температуры и сконцентрировали при пониженном давлении с получением коричневого масла. Очистка посредством сухой флэш-хроматографии (5% МеОН в ДХМ) дала темно-коричневое твердое вещество, которое дополнительно очистили посредством растирания с EtOAc с получением титульного соединения в форме желтого твердого вещества (7,45 г, выход 28%).
ЖХМС: m/z 449 [М+Н]+ и 471 [M+Na]+
Стадия 3 - 6-амино-5-(2,4-дифторбензоил)-1-[2,6-дифтор-4-(2-гидроксиэтил)фенил]пиридин-2(1Н)-он
2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этилацетат (7,45 г, 17 ммоль) суспензировали в 6N HCl (80 мл) и реакционную смесь нагревали с обратным холодильником в течение 21,5 часов. Твердое вещество собрали посредством фильтрации, поместили в насыщенный водный раствор NaHCO3 (200 мл) и интенсивно перемешивали в течение 30 минут. Твердое вещество собрали посредством фильтрации, промыли водой и высушили в вакуумной печи (40°C) с получением титульного соединения в форме бежевого твердого вещества.
ЖХМС: m/z 407 [М+Н]+ и 429 [M+Na]+.
1Н ЯМР (300 МГц, ДМСО-d6) 7,57 (1Н, td, J=6,6, 8,3 Гц), 7,41 (1Н, td, J=2,4, 9,7 Гц), 7,37-7,29 (3Н, m), 7,23 (1Н, td, J=2,3, 8,5 Гц), 5,74 (1Н, d, J=9,8 Гц), 4,78 (1Н, t, J=5,1 Гц), 3,76-3,70 (2Н, m), 2,86 (2Н, t, J=6,7 Гц), -NH2 не виден.
Стадия 4 - {4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}-ацетальдегид
Перйодинан Десса-Мартина (1,03 г, 2,4 ммоль) добавили к суспензии 6-амино-5-(2,4-дифторбензоил)-1-[2,6-дифтор-4-(2-гидроксиэтил)фенил]пиридин-2(1Н)-она (823 мг, 2,0 ммоль) в ДХМ (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, погасили насыщенным водным раствором NaHCO3 (10 мл) и насыщенным водным раствором тиосульфата натрия (10 мл) и интенсивно перемешивали в течение 30 минут. Водную фазу отделили и дополнительно экстрагировали ДХМ (2×20 мл). Объединенные органические экстракты высушили (MgSO4), профильтровали и сконцентрировали при пониженном давлении с получением титульного соединения в форме светло-коричневого твердого вещества (819 мг). Это вещество использовали без дополнительной очистки на следующей стадии.
Стадия 5 - трет-бутил-N-(2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил)-L-аланинат
Трет-бутил-L-аланината гидрохлорид (552 мг, 3,0 ммоль) и ТАБН (1,29 г, 6,1 ммоль) добавили к раствору {4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}-ацетальдегида (819 мг, 2,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3,5 часов, погасили насыщенным водным раствором Na2CO3 (20 мл) и интенсивно перемешивали в течение 20 минут. Водную фазу отделили и дополнительно экстрагировали EtOAc (2×20 мл). Объединенные органические экстракты промыли рассолом (20 мл), высушили (MgSO4), профильтровали и сконцентрировали при пониженном давлении с получением желтого масла. Очистка посредством колоночной хроматографии (5% МеОН в ДХМ) дала титульное соединение в форме светло-желтого твердого вещества (492 мг, выход из двух стадий 78%).
ЖХМС: степень чистоты 98%, m/z 534 [М+Н]+.
1Н ЯМР (300 МГц, ДМСО-d6) 7,58 (1Н, td, J=6,8, 8,3 Гц), 7,41 (1Н, td, J=2,3, 9,8 Гц), 7,37-7,30 (3Н, m), 7,23 (1Н, td, J=2,3, 8,5 Гц), 5,74 (1Н, d, J=9,8 Гц), 3,20 (1Н, d, J=7,0 Гц), 2,89-2,70 (4Н, m), 1,42 (9Н, s), 1,16 (3Н, d, J=7,0 Гц), -NH2 и -NH- не видны.
Измерение биологической активности
р38 МАП-киназная активность
Способность соединений ингибировать активность р38 МАП-киназы была измерена в анализе, выполненном компанией Upstate (Данди, Соединенное Королевство). В конечном реакционном объеме, равном 25 мкл, р38 МАП-киназу альфа (5-10 мЕд) инкубировали с 25 мМ трис-буфера с рН 7,5, 0,002 мМ ЭГТА, 0,33 мг/мл основного белка миелина, 10 мМ ацетата Мg и [g-33р-АТФ] (удельная активность, равная примерно 500 циклов в минуту/пмоль, концентрация по потребности). Реакцию инициировали добавлением смеси МgАТФ. После инкубации в течение 40 минут при комнатной температуре реакцию остановили добавлением 5 мкл 3%-ного раствора фосфорной кислоты. Затем 10 мкл реакционной смеси нанесли на плоский фильтр Р30 и промыли три раза по 5 минут в 75 мМ растворе фосфорной кислоты и один раз в метаноле перед сушкой и подсчетом сцинтилляций.
Парные экспериментальные точки получены из серийных разведений маточного раствора в ДМСО с интервалом, равным 1/3 логарифмической единицы. Из максимальной концентрации, равной 10 мкМ, получено девять разведений и добавлен холостой раствор «без соединения». Стандартный радиометрический анализ способом связывания с фильтром выполнен при концентрации АТФ, равной или близкой к Km. Данные, полученные посредством подсчета сцинтилляций, получены и подвергнуты анализу степени согласия с использованием программного обеспечения Prism. Из полученной кривой определена и зарегистрирована концентрация, обеспечивающая 50%-ное ингибирование.
Стимуляция клеток ТНР-1 липополисахаридом (ЛПС)
Клетки ТНР-1 нанесли в объеме, равном 100 мкл, с плотностью 4×104 клеток/лунку в лунки подготовленных 96-луночных планшетов для тканевых культур с V-образным дном и инкубировали при 37°C в 5% CO2 в течение 16 часов. Через 2 часа после добавления ингибитора в 100 мкл среды для культивирования тканей клетки стимулировали ЛПС (Е. coli, штамм 005:В5, Sigma) в конечной концентрации, равной 1 мкг/мл, и инкубировали при 37°C в 5% CO2 в течение 6 часов. Концентрации ФНО-α измерили в бесклеточных супернатантах посредством твердофазного иммуноферментного сэндвич-анализа (R&D Systems №QTA00B).
Стимуляция цельной крови человека липополисахаридом
Цельную кровь получили посредством венозной пункции с использованием гепаринизированных вакутейнеров (Becton Dickinson) и разбавили равным объемом среды для культивирования тканей RPMI1640 (Sigma). По 100 мкл нанесли в лунки подготовленных 96-луночных планшетов для тканевых культур с V-образным дном. Через 2 часа после добавления ингибитора в 100 мкл среды RPMI1640 кровь стимулировали ЛПС (E. coli, штамм 005:В5, Sigma) в конечной концентрации, равной 100 нг/мл, и инкубировали при 37°C в 5% CO2 в течение 6 часов. Концентрации ФНО-α измерили в бесклеточных супернатантах посредством твердофазного иммуноферментного сэндвич-анализа (R&D Systems №QTA00B).
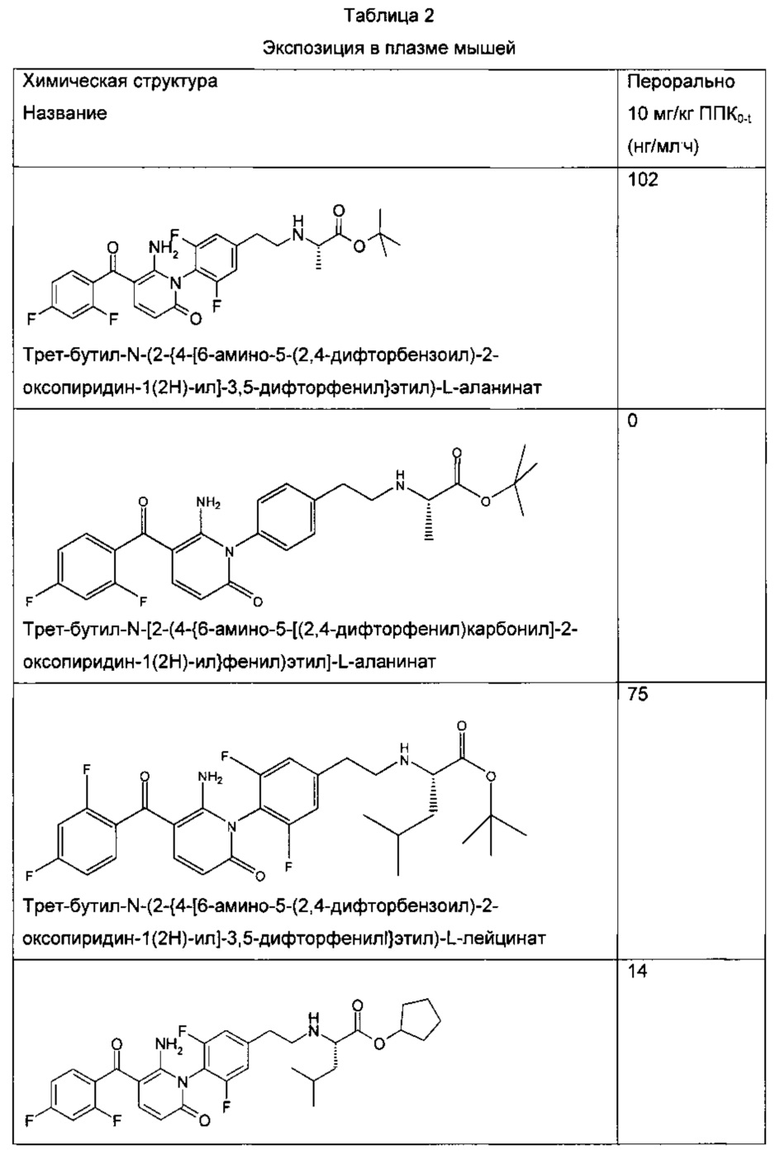
Экспозиция в плазме мышей
Из соединений приготовили композиции в водном растворе, содержавшем 8% ДМСО и 92% 11,25%-ного гидроксипропил-β-циклодекстрина, с использованием следующей процедуры: соединения полностью растворили в 100%-ном ДМСО, а затем добавили раствор гидроксипропил-β-циклодекстрина. Образовавшийся мелкий осадок повторно растворили посредством добавления водного раствора HCI и довели рН до 4 водным раствором гидроксида натрия.
Каждое соединение вводили орально в дозе, равной 10 мг/кг, при общем объеме дозы, равном 5 мл/кг, самцам мышей CD1 (25-20 г). В каждый момент времени использовали трех мышей. Пробы крови были взяты в следующие моменты времени: через 5, 15, 30, 60, 120, 240 и 360 минут посредством терминальной пункции сердца под анестезией галотаном/изофлураном. Пробы крови собрали в предварительно охлажденные пробирки, содержавшие NaF/ЭДТА, и перемешали. Пробы центрифугировали при 7-7,5 g в течение 2 минут. Плазму отсосали и заморозили.
Образцы плазмы приготовили посредством осаждения белка с использованием трех объемов ацетонитрила, содержавших внутренний стандарт. Супернатанты проанализировали посредством ЖХМС (Sciex API 3000, бинарный насос НР1100, СЕС PAL). Хроматография была основана на колонке Acentis С8 (50×2,1 мм) и мобильной фазе, содержавшей 5-95% ацетонитрила в воде / 0,1% муравьиной кислоте.
Экспозицию (площадь под кривой) рассчитали по графику зависимости концентрации в плазме от времени с использованием PK Solutions 2.0 (Summit Research Services, Монтроз, Колорадо).
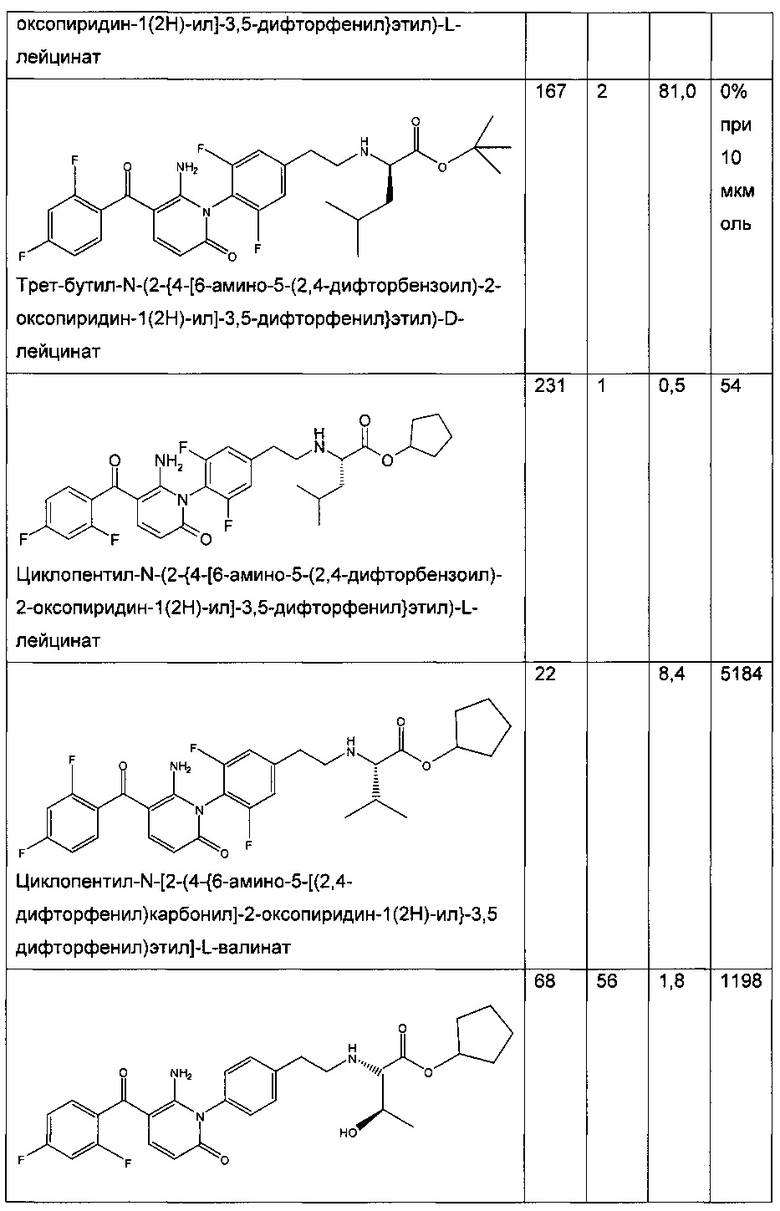
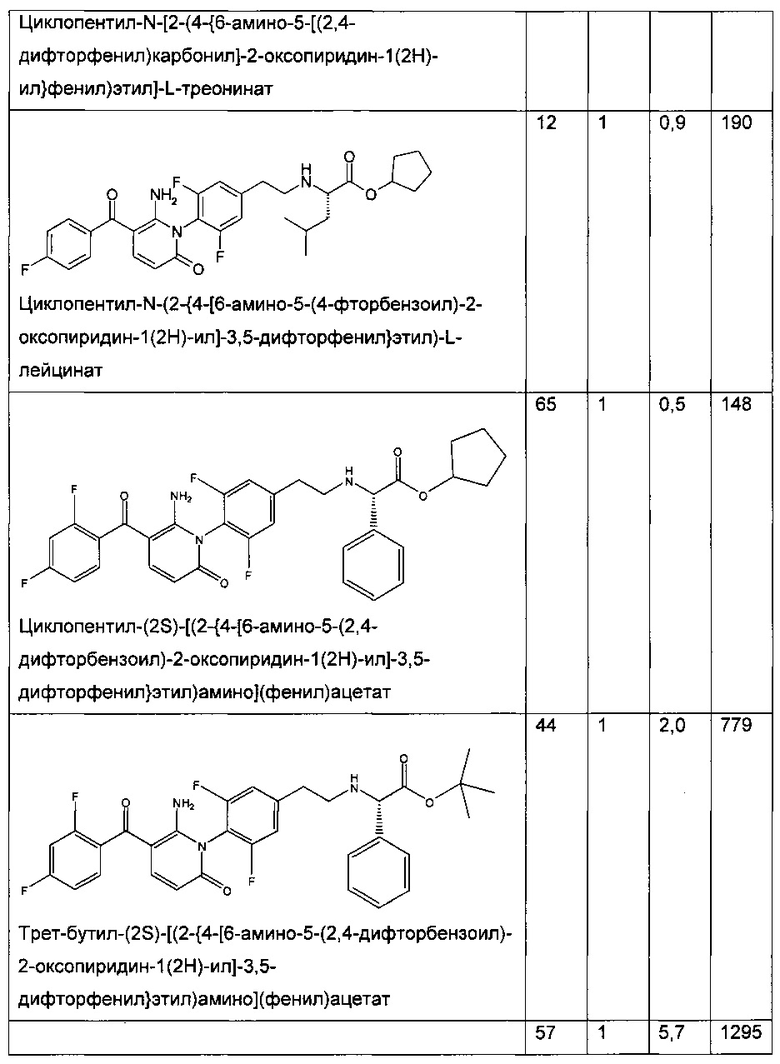
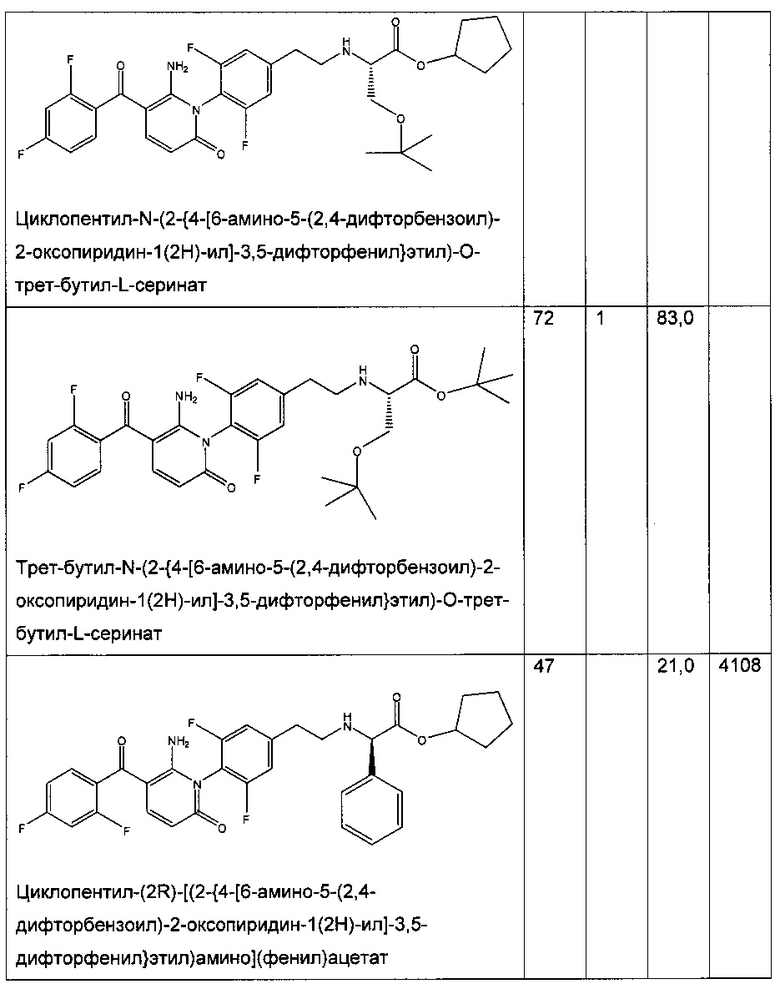
Таблица 1
Биологическая активность соединения по настоящему изобретению и структурно родственных соединений
В колонках A-D приведены следующие данные:
А - анализ влияния сложного эфира на фермент (р38 киназа A (Invitrogen)), IC50 (нМ);
В - анализ влияния кислоты на фермент (р38 киназа A (Invitrogen)), IC50 (нМ);
С - ингибирование ФНО-альфа сложным эфиром (клетки ТНР-1), IC50 (нМ); и
D - ингибирование ФНО-альфа сложным эфиром (цельная кровь человека), IC50 (нМ).
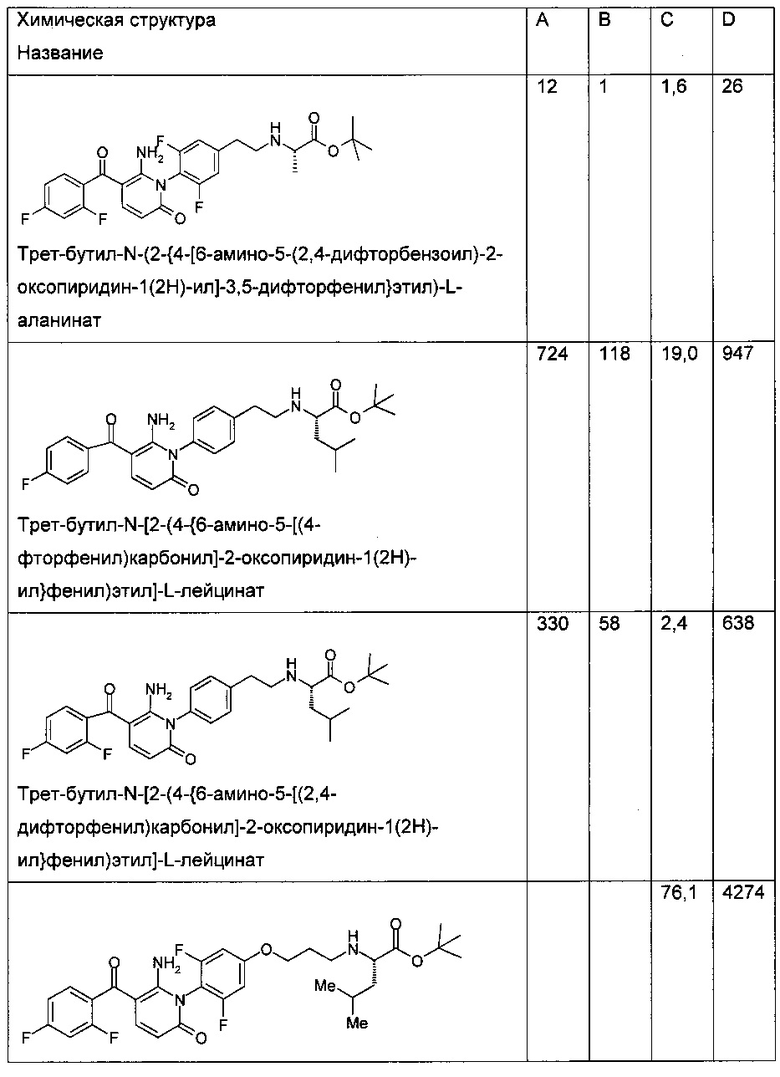
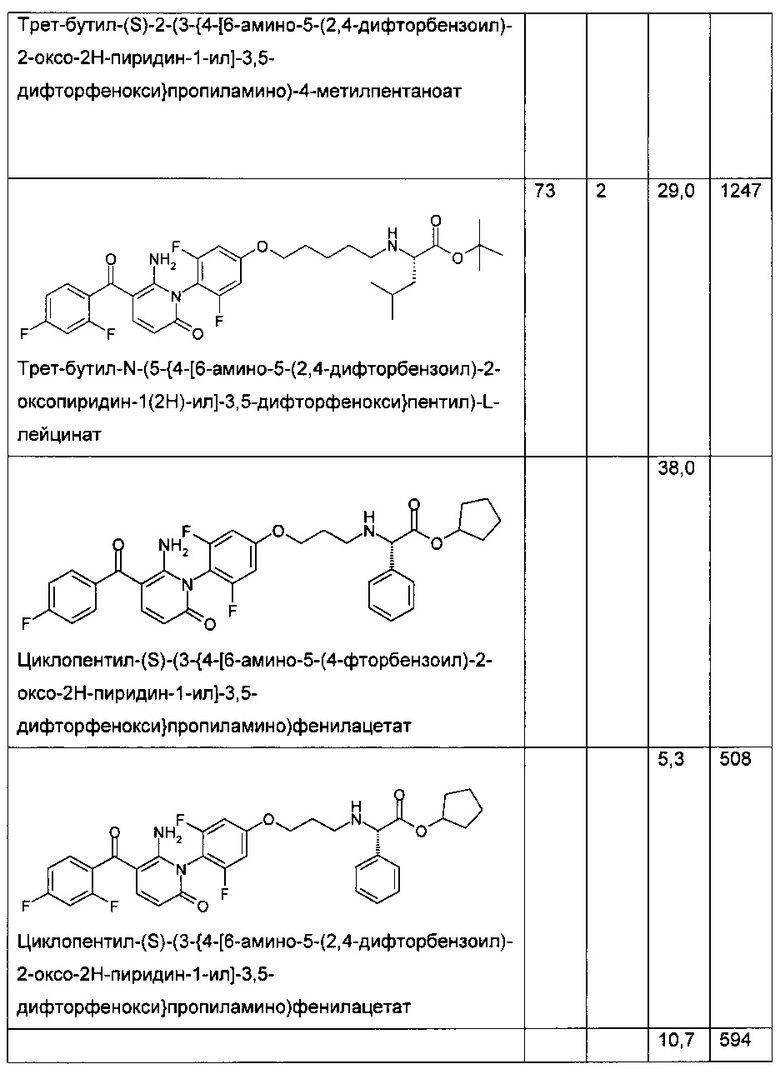
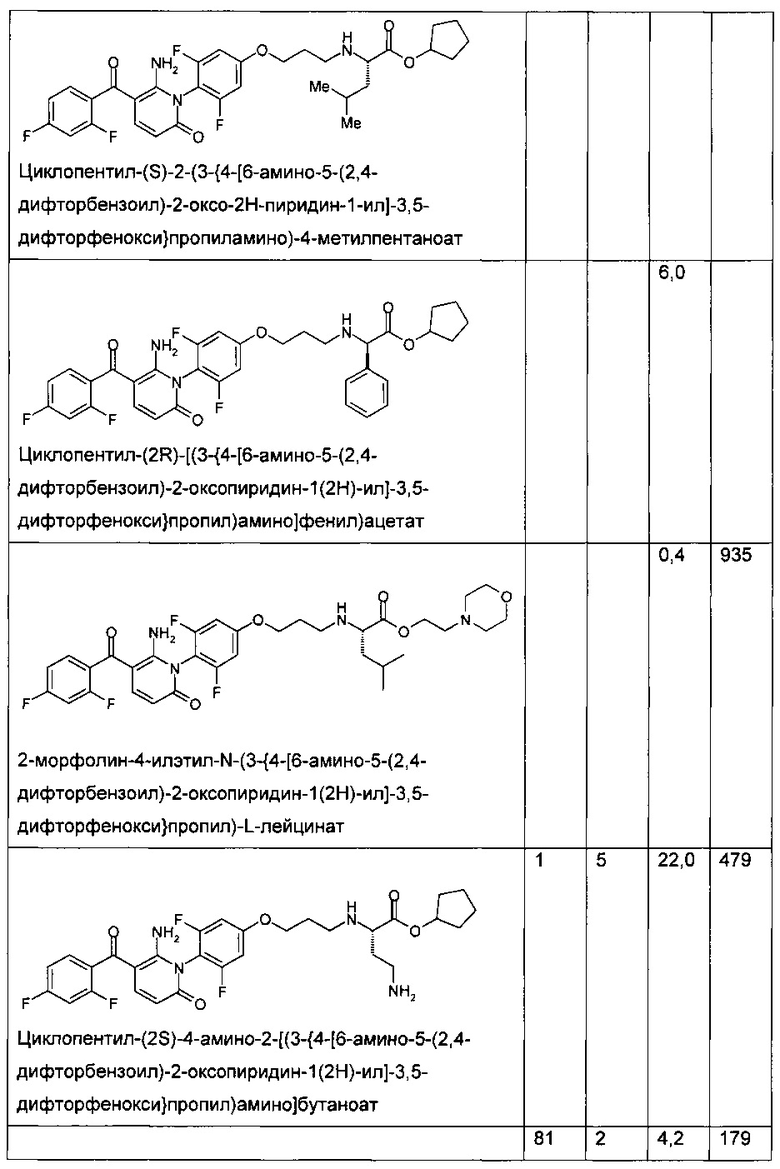
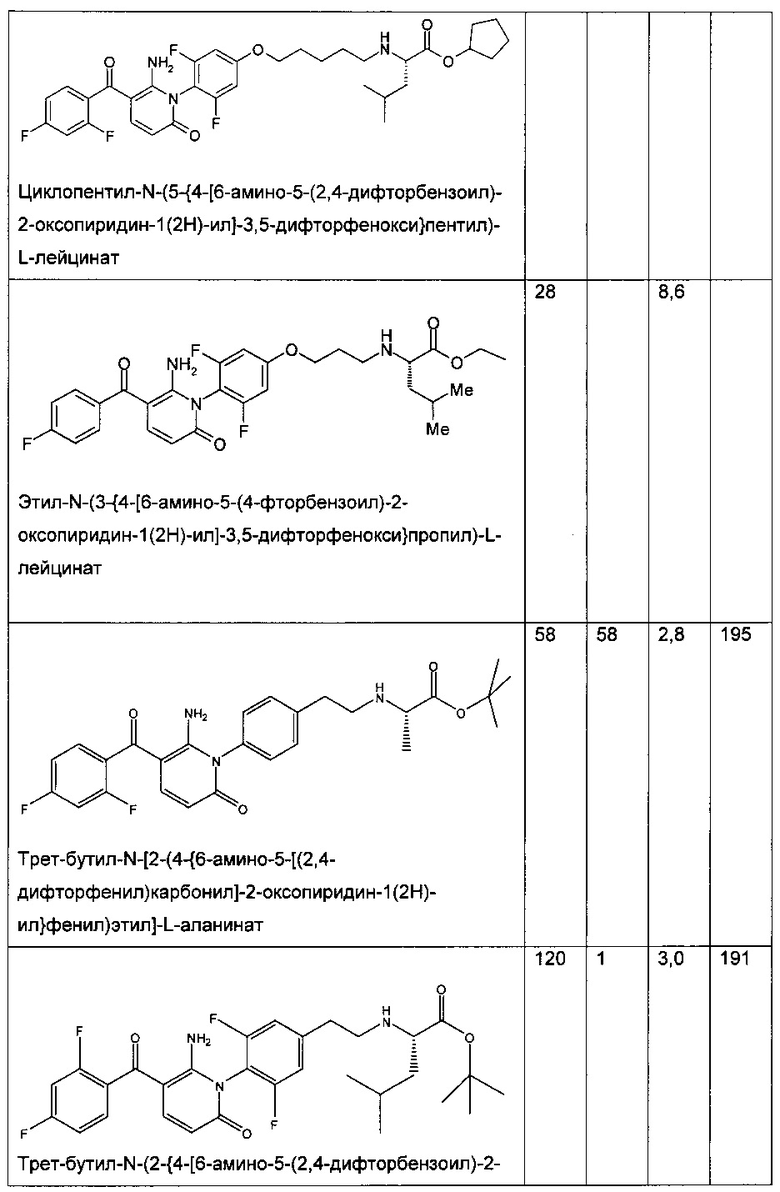

Выводы
Таблица 1 демонстрирует, что соединение по настоящему изобретению является хорошим ингибитором р38 МАП-киназы. Кроме того, значения IC50 для ингибирования сложным эфиром ФНО-α в клетках ТНР-1 и крови человека являются очень низкими. В частности, IC50 для ингибирования сложным эфиром ФНО-α в крови человека значительно ниже, чем можно было ожидать, исходя из значений IC50 для ингибирования сложным эфиром ФНО-α в крови человека, полученных для родственных соединений.
Данные по ППК (площади под кривой), представленные в Таблице 2, демонстрируют, что биодоступность соединения по настоящему изобретению значительно выше, чем у структурно сходных известных соединений.
Изобретение относится к соединению, которое является трет-бутил-N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланинатом или его фармацевтически приемлемой солью и обладает способностью ингибирования активности фермента р38 МАП-киназы (митоген-активируемой протеинкиназы). Изобретение относится также к фармацевтической композиции, содержащей указанное соединение, и способу ингибирования фермента р38 МАП-киназы. 3 н. и 4 з.п. ф-лы, 2 табл., 1 пр.
1. Соединение, которое является трет-бутил-N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланинатом или его фармацевтически приемлемой солью.
2. Соединение по п.1, которое является трет-бутил-N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланинатом.
3. Соединение по п.1, которое является:
(а) этансульфоновой солью трет-бутил-N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланината или
(б) метансульфоновой солью трет-бутил-N-[2-{4-[6-амино-5-(2,4-дифторбензоил)-2-оксопиридин-1(2Н)-ил]-3,5-дифторфенил}этил]-L-аланината.
4. Фармацевтическая композиция для ингибирования фермента р38 МАП-киназы, содержащая эффективное количество соединения по пп.1, 2 или 3 совместно с одним или более фармацевтически приемлемыми носителями и/или наполнителями.
5. Способ ингибирования фермента р38 МАП-киназы, отличающийся тем, что он включает обеспечение контакта фермента с количеством соединения, определенного в пп.1, 2 или 3, или композиции, определенной в п.4, эффективным в отношении этого ингибирования.
6. Способ по п.5, отличающийся тем, что он предназначен для лечения, облегчения симптомов или снижения инцидентности рака у субъекта, отличающийся тем, что он включает введение субъекту эффективного количества соединения, определенного в пп.1, 2 или 3, или композиции, определенной в п.4.
7. Способ по п.6, отличающийся тем, что он предназначен для лечения, облегчения симптомов или снижения инцидентности рака молочной железы, рака яичников, рака поджелудочной железы, рака легкого, рака толстого кишечника, рака почки, лимфомы или меланомы.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| ПРОИЗВОДНЫЕ НИКОТИНАМИДА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2003 |
|
RU2309951C2 |

