ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка притязает на приоритет на основании предварительной заявки 60/850834, поданной 11 октября 2006 года. Указанная заявка включена в данное описание в виде ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым ингибиторам киназ и модулирующим соединениям, применимым для лечения различных заболеваний. Более конкретно, изобретение относится к таким соединениям, способам лечения заболеваний и способам синтеза соединений. Предпочтительно соединения применимы для модулирования киназной активности C-Abl, c-Kit, VEGFR, PDGFR, Flt-3, c-MET, семейства HER, семейства киназ Raf и их патологических полиморфных форм.
УРОВЕНЬ ТЕХНИКИ
Несколько представителей семейства протеинкиназ, как было ясно показано, вовлечены в патогенез различных пролиферативных и миелопролиферативных заболеваний и, следовательно, представляют собой важные мишени для лечения таких заболеваний. Некоторые из пролиферативных заболеваний, имеющих отношение к настоящему изобретению, включают рак, ревматоидный артрит, атеросклероз и ретинопатии. Важными примерами киназ, которые, как было показано, вызывают или вносят вклад в патогенез таких заболеваний, являются киназа C-Abl и онкогенный слитый белок bcr-киназа Abl; киназа c-Kit, c-MET, семейство HER, PDGF-рецепторная киназа, VEGF-рецепторная киназа, киназа Flt-3 и семейство киназ Raf.
Киназа C-Abl является важной нерецепторной тирозинкиназой, вовлеченной в сигнальную трансдукцию в клетке. Такая повсеместно экспрессируемая киназа - при активации факторами, которые действуют выше в каскаде передачи сигнала, включая факторы роста, окислительный стресс, стимуляцию интегрином и ионизирующую радиацию - локализуется в плазматической мембране клетки, клеточном ядре и других клеточных компартментах, включая актиновый цитоскелет (Van Etten, Trends Cell Biol. (1999) 9; 179). Существует две нормальные изоформы киназы Abl: Abl-1A и Abl-1B. N-концевая половина киназы c-Abl имеет важное значение для аутоингибирования каталитической активности киназного домена (Pluk et al., Cell (2002) 108: 247). Недавно раскрыты подробные аспекты механизма такого аутоингибирования (Nagar et al., Cell (2003) 112: 859). Было показано, что N-концевой миристоил-аминокислотный остаток Abl-1B внутри молекулы занимает гидрофобный карман, образованный альфа-спиралями в C-части киназного домена. Такое внутримолекулярное связывание индуцирует образование новой области связывания для внутримолекулярной стыковки домена SH2 и домена SH3 с киназным доменом, тем самым нарушая и ингибируя каталитическую активность киназы. Таким образом, сложная внутримолекулярная негативная регуляция киназной активности обусловлена такими N-концевыми областями киназы c-Abl. Аномальная обусловленная нарушенной регуляцией форма c-Abl образуется в результате события хромосомной транслокации, называемой филадельфийской хромосомой (P.C. Nowell et al., Science (1960) 132: 1497; J.D. Rowley, Nature (1973) 243: 290). Такая аномальная хромосомная транслокация приводит к аберрантному генному слиянию между геном киназы Abl и геном области локализации сайта инициации реаранжировки (BCR), таким образом кодируя аномальный белок, называемый bcr-Abl (G.Q. Daley et al., Science (1990) 247: 824; M.L. Gishizky et al., Proc. Natl. Acad. Sci. USA (1993) 90: 3755; S. Li et al., J. Exp. Med. (1999) 189: 1399). Слитый белок bcr-Abl не содержит регуляторного сайта миристоилирования (B. Nagar et al., Cell (2003) 112: 859) и в результате функционирует как онкобелок, который вызывает хронический миелоидный лейкоз (CML). CML является злокачественным заболеванием, поражающим плюрипотентные гематопоэтические стволовые клетки. p210-форма bcr-Abl наблюдается у 95% пациентов с CML и у 20% пациентов с острым лимфоцитарным лейкозом. Также описана форма p185 и показано, что она связана с причиной развития острого лимфоцитарного лейкоза у 10% пациентов.
Показано, что большинство низкомолекулярных ингибиторов киназ, о которых сообщается в литературе, связываются с одним из трех путей. Большая часть известных ингибиторов взаимодействуют с доменом связывания ATP активного центра и проявляют свои эффекты, конкурируя с ATP за занимаемое положение. Другие ингибиторы, как было показано, связываются с отдельной гидрофобной областью белка, известной как карман с конформацией «погружения DFG внутрь», а другие, как было показано, связываются как с ATP-доменом, так и с карманом с конформацией «погружения DFG внутрь». Примеры специфичных ингибиторов Raf-киназ можно найти в Lowinger et al., Current Pharmaceutical Design (2002) 8: 2269-2278; Dumas, J. et al., Current Opinion in Drug Discovery and Development (2004) 7: 600-616; Dumas, J. et al., WO 2003068223 A1 (2003); Dumas, J., et al., WO 9932455 A1 (1999) и Wan, P.T.C., et al., Cell (2004) 116: 855-867.
Физиологически киназы регулируются общим механизмом активации/дезактивации, при котором специфичная последовательность петли активации киназного белка связывается в специфичном кармане на том же самом белке, который называют карманом управления переключением (подробности см. в WO 200380110049). Такое связывание происходит в том случае, когда специфичные аминокислотные остатки петли активации модифицируются, например, в результате фосфорилирования, окисления или нитрозилирования. Связывание петли активации в кармане переключения приводит к конформационному изменению белка с образованием активной формы (Huse, M. and Kuriyan, J. Cell (109) 275-282).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Соединения согласно настоящему изобретению находят применение при лечении гиперпролиферативных заболеваний, рака млекопитающих и особенно рака человека, включая, без ограничения, злокачественные меланомы, меланомы, глиобластомы, рак яичника, рак поджелудочной железы, рак простаты, рак легкого, рак молочной железы, рак почек, карциномы шейки матки, метастазы первичного солидного рака щитовидной железы во вторичных опухолевых очагах, миелопролиферативные заболевания, хронический миелогенный лейкоз, острый лимфоцитарный лейкоз, другие миелопролиферативные расстройства, папиллярную карциному щитовидной железы, немелкоклеточный рак легкого, мезотелиому, гиперэозинофильный синдром, стромальные опухоли желудочно-кишечного тракта, рак ободочной кишки, глазные болезни, характеризующиеся гиперпролиферацией, приводящие к слепоте, включая различные ретинопатии, например диабетическую ретинопатию и связанную с возрастом дегенерацию желтого пятна, ревматоидный артрит, астму, хроническое обструктивное легочное заболевание, воспаление у человека, ревматоидный спондилит, остеоартрит, астму, подагрический артрит, сепсис, септический шок, эндотоксический шок, грамотрицательный сепсис, синдром токсического шока, респираторный дистресс-синдром взрослых, удар, реперфузионное повреждение, травму нервной системы, ишемию нервной системы, псориаз, рестеноз, хроническое обструктивное легочное заболевание, заболевания, обусловленные резорбцией костей, реакцию «трансплантат против хозяина», болезнь Крона, язвенный колит, воспалительное заболевание кишечника, изжогу и их сочетания, заболевание, вызванное киназой c-Abl, ее онкогенными формами, ее аберрантными формами в виде слитых белков и ее полиморфами, заболевание, вызванное киназой Raf, ее онкогенными формами, ее аберрантными формами в виде слитых белков и ее полиморфами, киназой c-Kit, ее онкогенными формами, ее аберрантными формами в виде слитых белков и ее полиморфами, киназой Flt-3, ее онкогенными формами, ее аберрантными формами в виде слитых белков и ее полиморфами, киназой VEGFR, ее онкогенными формами, ее аберрантными формами в виде слитых белков и ее полиморфами, киназой PDGFR, ее онкогенными формами, ее аберрантными формами в виде слитых белков и ее полиморфами, киназой c-MET, ее онкогенными формами, ее аберрантными формами в виде слитых белков и ее полиморфами, и заболевание, вызванное киназой HER, ее онкогенными формами, ее аберрантными формами в виде слитых белков и ее полиморфами.
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ
Следующее описание относится к различным соединениям и их остаткам.
Карбоциклил относится к углеродным циклам, выбранным из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила, циклогептанила, циклооктанила, норборанила, норборенила, бицикло[2.2.2]октанила и бицикло[2.2.2]октенила.
Галоген относится к фтору, хлору, брому и йоду.
Арил относится к моноциклическим или конденсированным бициклическим кольцевым системам, характеризуемым делокализованными π-электронами (ароматичностью), которые являются общими для атомов углерода в цикле, по меньшей мере, одного карбоциклического кольца; предпочтительные арильные циклы выбраны из фенила, нафтила, тетрагидронафтила, инденила и инданила.
Гетероарил относится к моноциклическим или конденсированным бициклическим кольцевым системам, характеризуемым делокализованными π-электронами (ароматичностью), которые являются общими для атомов углерода или гетероатомов в цикле, включая азот, кислород или серу, по меньшей мере, одного карбоциклического или гетероциклического кольца; при этом гетероарильные циклы выбраны, без ограничения, из пирролила, фурила, тиенила, оксазолила, тиазолила, изоксазолила, изотиазолила, имидазолила, пиразолила, оксадиазолила, тиадиазолила, триазолила, тетразолила, пиридинила, пиримидинила, пиразинила, пиридазинила, триазинила, индолила, индолинила, изоиндолила, изоиндолинила, индазолила, бензофуранила, бензотиенила, бензотиазолила, бензотиазолонила, бензоксазолила, бензоксазолонила, бензизоксазолила, бензизотиазолила, бензимидазолила, бензимидазолонила, бензтриазолила, имидазопиридинила, пиразолопиридинила, имидазолонопиридинила, тиазолопиридинила, тиазолонопиридинила, оксазолопиридинила, оксазолонопиридинила, изоксазолопиридинила, изотиазолопиридинила, триазолопиридинила, имидазопиримидинила, пиразолопиримидинила, имидазолонопиримидинила, тиазолопиримидинила, тиазолонопиримидинила, оксазолопиримидинила, оксазолонопиримидинила, изоксазолопиримидинила, изотиазолопиримидинила, триазолопиримидинила, дигидропуринонила, пирролопиримидинила, пуринила, пиразолопиримидинила, фталимидила, фталимидинила, пиразинилпиридинила, пиридинопиримидинила, пиримидинопиримидинила, циннолинила, хиноксалинила, хиназолинила, хинолинила, изохинолинила, фталазинила, бензодиоксила, бензизотиазолин-1,1,3-трионила, дигидрохинолинила, тетрагидрохинолинила, дигидроизохинолила, тетрагидроизохинолинила, бензоазепинила, бензодиазепинила, бензоксапинила и бензоксазепинила.
Гетероциклил относится к моноциклическим кольцам, содержащим атомы углерода и гетероатомы, выбранные из кислорода, азота или серы, и в которых нет делокализованных π-электронов (ароматичности), общих для атомов углерода или гетероатомов в цикле; при этом гетероциклические кольца включают, без ограничения, оксетанил, азетадинил, тетрагидрофуранил, пирролидинил, оксазолинил, оксазолидинил, тиазолинил, тиазолидинил, пиранил, тиопиранил, тетрагидропиранил, диоксалинил, пиперидинил, морфолинил, тиоморфолинил, тиоморфолинил-S-оксид, тиоморфолинил-S-диоксид, пиперазинил, азепинил, оксепинил, диазепинил, тропанил и гомотропанил.
Полиарил относится к двум или более моноциклическим или конденсированным арильным бициклическим кольцевым системам, характеризуемым делокализованными π-электронами (ароматичностью), общими для атомов углерода в цикле, по меньшей мере, одного карбоциклического кольца, при этом кольца, входящие в них, необязательно связаны вместе.
Полигетероарил относится к двум или более моноциклическим или конденсированным бициклическим системам, характеризуемым делокализованными π-электронами (ароматичностью), общими для атомов углерода или гетероатомов в цикле, включая азот, кислород или серу, по меньшей мере, одного карбоциклического или гетероциклического кольца, в которых кольца, входящие в них, необязательно связаны вместе, при этом, по меньшей мере, одно из моноциклических или конденсированных бициклических колец полигетероарильной системы выбрано из гетероарила, который в широком смысле определен выше, и другие кольца выбраны из любого арила, гетероарила или гетероциклила, которые в широком смысле определены выше.
Полигетероциклил относится к двум или более моноциклическим или конденсированным бициклическим кольцевым системам, содержащим атомы углерода и гетероатомы, выбранные из кислорода, азота или серы, в которых нет делокализованных π-электронов (ароматичности), общих для атомов углерода или гетероатомов в цикле, и входящие в них кольца необязательно связаны, при этом по меньшей мере одно из моноциклических или конденсированных бициклических колец полигетероарильной системы выбрано из гетероциклила, который в широком смысле определен выше, а другие циклы выбраны из любого арила, гетероарила или гетероциклила, которые в широком смысле определены выше.
Низший алкил относится к C1-C6-алкилам с линейной или разветвленной цепью.
Термин «замещенный», относящийся к остатку, обозначает, что с остатком может быть связан дополнительный заместитель в любом допустимом положении на остатке.
Термин «соли» охватывает фармацевтически приемлемые соли, обычно используемые в форме солей щелочных металлов и свободных жирных кислот и в форме аддитивных солей свободных оснований. Природа соли не является решающей при условии, что она является фармацевтически приемлемой. Подходящие фармацевтически приемлемые кислотно-аддитивные соли могут быть получены из неорганической кислоты или из органической кислоты. Примерами таких неорганических кислот являются хлористоводородная, бромистоводородная, йодистоводородная, азотная, угольная, серная и фосфорная. Подходящие органические кислоты могут быть выбраны из алифатических, циклоалифатических, ароматических, арилалифатических и содержащих гетероциклил карбоновых кислот и сульфоновых кислот, примерами которых являются муравьиная, уксусная, пропионовая, янтарная, гликолевая, глюконовая, молочная, яблочная, винная, лимонная, аскорбиновая, глюкуроновая, малеиновая, фумаровая, пировиноградная, аспарагиновая, глутаминовая, бензойная, антраниловая, метилсульфоновая, стеариновая, салициловая, пара-гидроксибензойная, фенилуксусная, миндальная, эмбоновая (памовая), метансульфоновая, этансульфоновая, 2-гидроксиэтансульфоновая, бензолсульфоновая, пантотеновая, толуолсульфоновая, 2-гидроксиэтансульфоновая, сульфаниловая, циклогексиламиносульфоновая, альгиновая, 3-гидроксимасляная, галактаровая и галактуроновая кислота. Подходящие фармацевтически приемлемые соли соединений согласно изобретению, содержащих группу свободной кислоты, включают соли металлов и органические соли. Более предпочтительные соли металлов включают, без ограничения, подходящие соли щелочных металлов (группы Ia), соли щелочноземельных металлов (группы IIa) и других физиологически приемлемых металлов. Могут быть получены соли алюминия, кальция, лития, магния, калия, натрия и цинка. Предпочтительные органические соли могут быть получены из первичных аминов, вторичных аминов, третичных аминов и солей четвертичного аммония, включая в частности, трометамин, диэтиламин, тетра-N-метиламмоний, N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглюмин (N-метилглюкамин) и прокаин.
Термин «пролекарство» относится к производным активных соединений, которые in vivo превращаются в активную форму. Например, форма карбоновой кислоты активного лекарственного средства может быть этерифицирована с образованием пролекарства, и сложный эфир затем подвергается превращению in vivo и опять превращается в форму карбоновой кислоты. См. Ettmayer et al., J. Med. Chem, 2004, 47(10), 2393-2404 и Lorenzi et al., J. Pharm. Exp. Therapeutics, 2005, 883-8900.
1. Первый аспект изобретения - соединения, способы, получение и аддукты
Изобретение относится к соединениям формулы Ia:
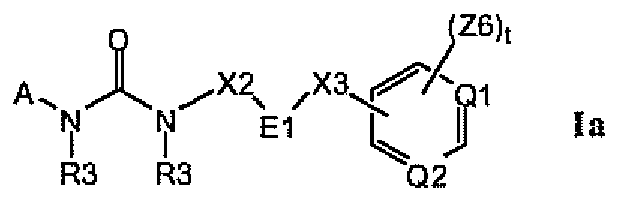
где Q1 и Q2, каждый отдельно и независимо, выбран из группы, состоящей из N и C-Z6, при условии что оба Q1 и Q2 одновременно не означают C-Z6;
E1 выбран из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила, пирролидинила, пиперидинила, фенила, тиенила, оксазолила, тиазолила, изоксазолила, изотиазолила, пирролила, пиразолила, оксадиазолила, тиадиазолила, фурила, имидазолила, пиридила, пиримидинила и нафтила, и где цикл E1 замещен одним или несколькими остатками R16, и где цикл E1 замещен одним или несколькими остатками R18;
где A выбран из группы, состоящей из фенила, C3-C8-карбоциклила, пирролила, фурила, тиенила, оксазолила, тиазолила, изоксазолила, изотиазолила, имидазолила, пиразолила, оксадиазолила, тиадиазолила, триазолила, тетразолила, пиразинила, пиридазинила, триазинила, пиридинила, пиримидинила и G4;
G1 означает гетероарил, выбранный из группы, состоящей из пирролила, фурила, тиенила, оксазолила, тиазолила, изоксазолила, изотиазолила, имидазолила, пиразолила, оксадиазолила, тиадиазолила, триазолила, тетразолила, пиразинила, пиридазинила, триазинила, пиридинила и пиримидинила;
G2 означает конденсированный бициклический гетероарил, выбранный из группы, состоящей из индолила, индолинила, изоиндолила, изоиндолинила, индазолила, бензофуранила, бензотиенила, бензотиазолила, бензотиазолонила, бензоксазолила, бензоксазолонила, бензизоксазолила, бензизотиазолила, бензимидазолила, бензимидазолонила, бензтриазолила, имидазопиридинила, пиразолопиридинила, имидазолонопиридинила, тиазолопиридинила, тиазолонопиридинила, оксазолопиридинила, оксазолонопиридинила, изоксазолопиридинила, изотиазолопиридинила, триазолопиридинила, имидазопиримидинила, пиразолопиримидинила, имидазолонопиримидинила, тиазолопиримидинила, тиазолонопиримидинила, оксазолопиримидинила, оксазолонопиримидинила, изоксазолопиримидинила, изотиазолопиримидинила, триазолопиримидинила, дигидропуринонила, пирролопиримидинила, пуринила, пиразолопиримидинила, фталимидила, фталимидинила, пиразинилпиридинила, пиридинопиримидинила, пиримидинопиримидинила, циннолинила, хиноксалинила, хиназолинила, хинолинила, изохинолинила, фталазинила, бензодиоксила, бензизотиазолин-1,1,3-трионила, дигидрохинолинила, тетрагидрохинолинила, дигидроизохинолила, тетрагидроизохинолинила, бензоазепинила, бензодиазепинила, бензоксапинила и бензоксазепинила;
G3 означает неконденсированный бициклический гетероарил, выбранный из группы, состоящей из пиридилпиримидинила, пиримидинилпиримидинила, оксазолилпиримидинила, тиазолилпиримидинила, имидазолилпиримидинила, изоксазолилпиримидинила, изотиазолилпиримидинила, пиразолилпиримидинила, триазолилпиримидинила, оксадиазолилпиримидинила, тиадиазолилпиримидинила, морфолинилпиримидинила, диоксотиоморфолинилпиримидинила и тиоморфолинилпиримидинила;
G4 означает гетероциклил, выбранный из группы, состоящей из оксетанила, азетадинила, тетрагидрофуранила, пирролидинила, оксазолинила, оксазолидинила, имидазолонила, пиранила, тиопиранила, тетрагидропиранила, диоксалинила, пиперидинила, морфолинила, тиоморфолинила, тиоморфолинил-S-оксида, тиоморфолинил-S-диоксида, пиперазинила, азепинила, оксепинила, диазепинила, тропанила и гомотропанила;
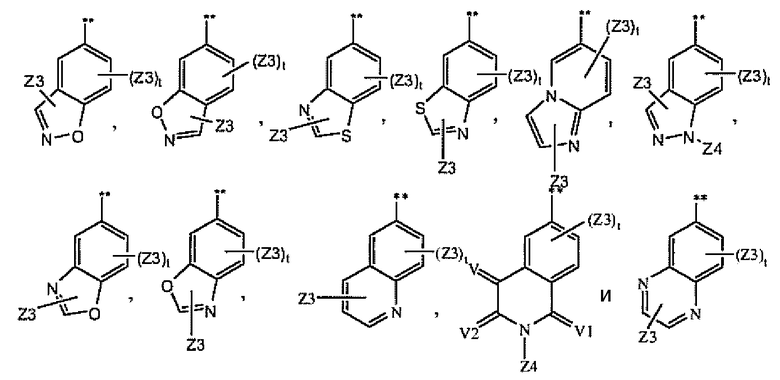
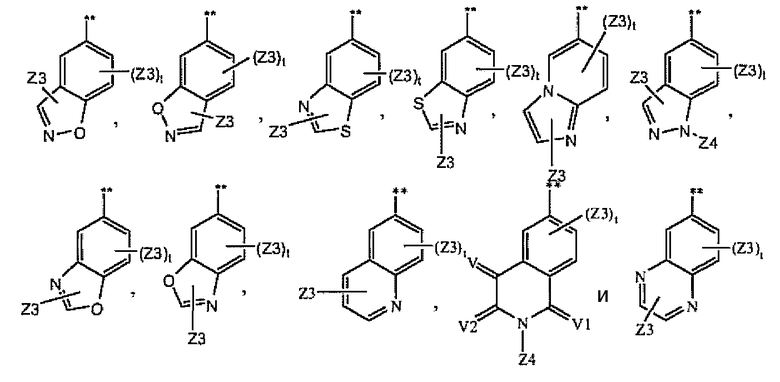
цикл A замещен в любом замещаемом положении одним остатком A1, где A1 выбран из группы, состоящей из A2, A3 и A4;
A2 выбран из группы, состоящей из
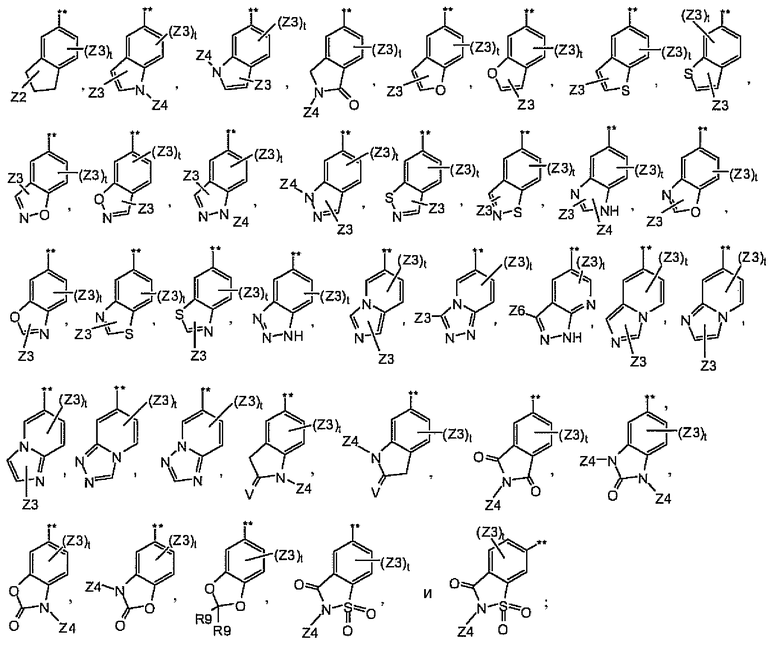
A3 выбран из группы, состоящей из
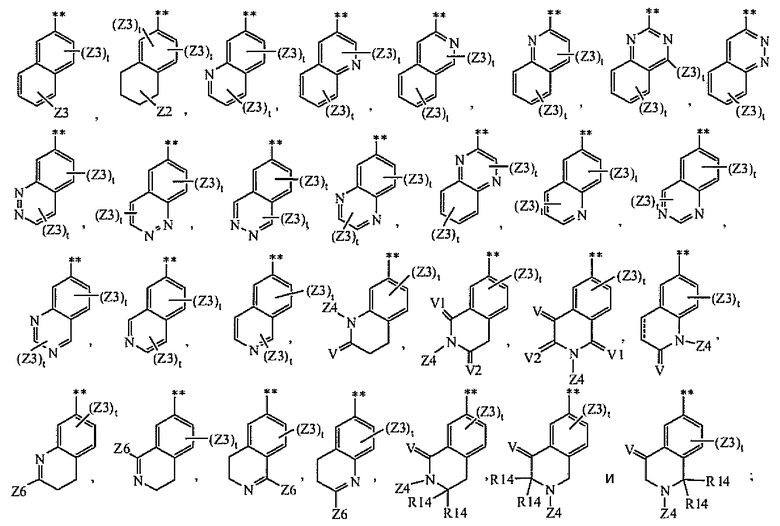
A4 выбран из группы, состоящей из
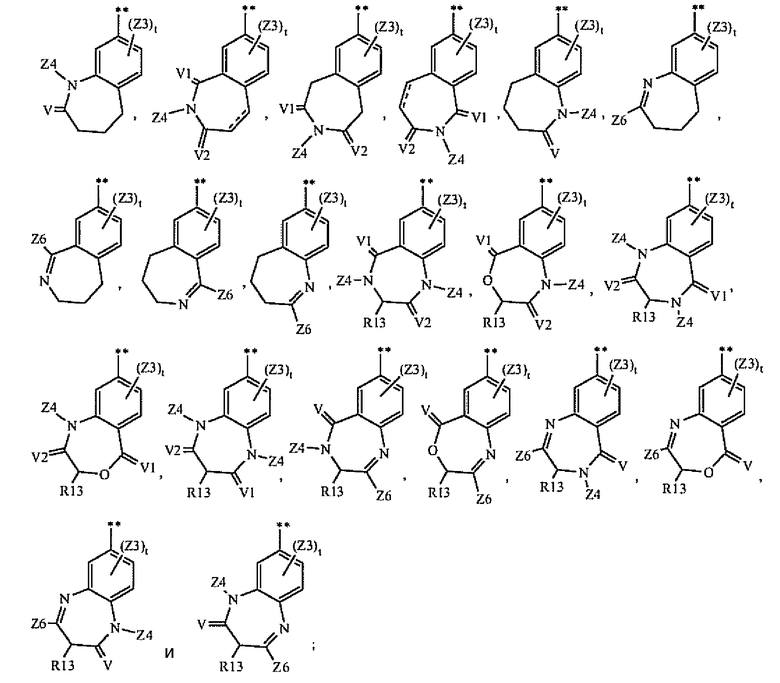
и где символ (**) означает точку связывания с циклом A формулы Ia;
и где «----» означает либо насыщенную, либо ненасыщенную связь;
цикл A необязательно замещен одним или несколькими остатками R2;
X2 выбран из группы, состоящей из C1-C6-алкила, разветвленного C2-C6-алкила и прямой связи, где E1 непосредственно связан с группой NR3 формулы Ia;
X3 выбран из группы, состоящей из -C(=O)-, -O-, -O-(CH2)n-, -S-(CH2)n, -NR3-(CH2)n-, -O-(CH2)q-O-, -O-(CH2)q-NR3-, -N(R3)-(CH2)q-N(R3)-, -(CH2)n-N(R4)-C(-O)-, -(CH2)n-N(R4)-C(=O)(CH2)n-, -(CH2)n-C(=O)N(R4)-, -(CH2)p-, C2-C5-алкенила, C2-C5-алкинила и C3-C6-циклоалкила, и где атомы углерода в остатках -(CH2)n-, -(CH2)q-, -(CH2)p-, C2-C5-алкенила и C2-C5-алкинила X3 могут быть дополнительно замещены одним или несколькими C1-C6-алкилами;
V, V1 и V2, каждый независимо и соответственно выбран из группы, состоящей из O и H2;
каждый Z2 независимо и отдельно выбран из группы, состоящей из водорода, арила, C1-C6-алкила, C3-C8-карбоциклила, гидроксила, гидрокси-C1-C6-алкила-, цианогруппы, (R3)2N-, (R4)2N-, (R4)2NC1-C6-алкила-, (R4)2NC2-C6-алкил-N(R4)-(CH2)n-, (R4)2NC2- C6-алкил-O(CH2)n-, (R3)2NC(O)-, (R4)2NC(O)-, (R4)2NC(O)C1-C6-алкила-, карбоксила, карбокси-C1-C6-алкила-, C1-C6-алкоксикарбонила-, C1-C6-алкоксикарбонил-C1-C6-алкила-, (R3)2NSO2-, (R4)2NSO2-, -SO2R5, -SO2R8, -(CH2)nN(R4)C(O)R8, -C(O)R8, =O, =NOH, =N(OR6), -(CH2)nG1, -(CH2)nG4, -(CH2)nO(CH2)nG1, -(CH2)nO(CH2)nG4, -(CH2)nNR3(CH2)n-арила, -(CH2)nNR3(CH2)nG1, -(CH2)nNR3(CH2)nG4, -(CH2)nNHC(O)NHS(O)2R8, -(CH2)nNHS(O)2NHC(O)R8, -C(O)NHS(O)2R8, -(CH2)NHC(O)(CH2)nR5, -(CH2)nNHS(O)2R5, -(CH2)nC(O)NH(CH2)qR5, (CH2)nC(O)R5, -(CH2)nOC(O)R5 и -(CH2)nR5;
в том случае когда Z2 содержит остаток алкила или алкилена, такие остатки могут быть дополнительно замещены одним или несколькими C1-C6-алкилами;
каждый Z3 независимо и отдельно выбран из группы, состоящей из H, C1-C6-алкила, разветвленного C3-C7-алкила, C3-C8-карбоциклила, галогена, фтор-C1-C6-алкила, где остаток алкила может быть частично или полностью фторирован, цианогруппы, гидроксила, метоксигруппы, оксогруппы, (R3)2NC(O)-, (R4)2NC(O)-, -N(R4)C(O)R8, (R3)2NSO2-, (R4)2NSO2-, -N(R4)SO2R5, - N(R4)SO2R8, -(CH2)nN(R3)2, -(CH2)nN(R4)2, -O(CH2)qN(R4)2, -O(CH2)q-O-C1-C6-алкила-, -N(R3)(CH2)q-O-C1-C6-алкила, -N(R3)(CH2)qN(R4)2, -O(CH2)qR5, -NR3(CH2)qR5, - C(O)R5, -C(O)R8, -R5 и нитрогруппы;
в том случае когда Z3 содержит остаток алкила или алкилена, такие остатки могут быть дополнительно замещены одним или несколькими C1-C6-алкилами;
каждый Z4 независимо и отдельно выбран из группы, состоящей из H, C1-C6-алкила, гидрокси-C2-C6-алкила, C1-C6-алкокси-C2-C6-алкила-, (R4)2N-C2-C6-алкила-, (R4)2N-C2-C6-алкила-N(R4)-C2-C6-алкила-, (R4)2N-C2-C6-алкил-O-C2-C6-алкила-, (R4)2NC(O)-C1-C6-алкила-, карбокси-C1-C6-алкила-, C1-C6-алкоксикарбонил-C1-C6-алкила-, -C2-C6-алкилN(R4)C(O)R8, R8-C(=NR3)-, -SO2R8, -COR8, -(CH2)nG1, -(CH2)nG4, -(CH2)q-O(CH2)nG1, -(CH2)qO(CH2)nG4, -(CH2)qNR3(CH2)nG1, -(CH2)qNR3(CH2)nG4, -(CH2)qNHC(O)(CH2)nR5, -(CH2)qC(O)NH(CH2)qR5, -(CH2)qC(O)R5, -(CH2)qOC(O)R5, -(CH2)qR5, -(CH2)qNR4(CH2)qR5 и -(CH2)qO(CH2)qR5;
в том случае когда Z4 содержит остаток алкила или алкилена, такие остатки могут быть дополнительно замещены одним или несколькими C1-C6-алкилами;
каждый Z6 независимо и отдельно выбран из группы, состоящей из H, C1-C6-алкила, разветвленного C3-C7-алкила, гидроксила, гидрокси-C1-C6-алкила, разветвленного гидрокси-C2-C6-алкила-, C1-C6-алкоксигруппы, C1-C6-алкокси-C1-C6-алкила-, разветвленного C1-C6-алкокси-C2-C6-алкила-, разветвленной C2-C6-алкоксигруппы-, C1-C6-алкилтиогруппы, (R3)2N-, -N(R3)COR8, (R4)2N-, -R5, - N(R4)C(O)R8, -N(R3)SO2R6, -C(O)N(R3)2, -C(O)N(R4)2, -C(O)R5, -SO2NHR4, галогена, фтор-C1-C6-алкила, где алкил полностью или частично фторирован, цианогруппы, фтор-C1-C6-алкоксигруппы, где алкил полностью или частично фторирован, -O(CH2)qN(R4)2, - N(R3)(CH2)qN(R4)2, -O(CH2)qO-C1-C6-алкила, -O(CH2)qN(R4)2, -N(R3)(CH2)qO-DC1-C6-алкила, -N(R3)(CH2)qN(R4)2, -O(CH2)qR5 и -N(R3)(CH2)qR5, -(NR3)rR17, -(O)rR17, -(S)rR17, -(CH2)nR17, -(CH2)nG1, -(CH2)nG4, -(CH2)qO(CH2)nG1, -(CH2)qO(CH2)nG4, -(CH2)qN(R3)(CH2)nG1 и -(CH2)qNR3(CH2)nG4;
каждый R2 выбран из группы, состоящей из Z3-замещенного арила, Z3-замещенного G1, Z3-замещенного G4, C1-C6-алкила, разветвленного C3-C8-алкила, R19-замещенного C3-C8-карбоциклила, гидроксил-C1-C6-алкила, разветвленного гидроксил-C3-C6-алкила-, гидроксилзамещенного C3-C8-карбоциклила-, циано-C1-C6-алкила-, цианозамещенного разветвленного C3-C6-алкила-, цианозамещенного C3-C8-карбоциклила-, (R4)2NC(O)C1-C6-алкила-, (R4)2NC(O)-замещенного разветвленного C3-C6-алкила-, (R4)2NC(O)-замещенного C3-C8-карбоциклила-, фтор-C1-C6-алкила, где алкил полностью или частично фторирован, галогена, цианогруппы, C1-C6-алкоксигруппы и фтор-C1-C6-алкоксигруппы, где алкильная группа полностью или частично фторирована;
каждый R3 независимо и отдельно выбран из группы, состоящей из H, C1-C6-алкила, разветвленного C3-C7-алкила, C3-C7-циклоалкила и Z3-замещенного фенила;
каждый R4 независимо и отдельно выбран из группы, состоящей из H, C1-C6-алкила, гидрокси-C1-C6-алкила-, дигидрокси-C1-C6-алкила-, C1-C6-алкокси-C1-C6-алкила-, разветвленного C3-C7-алкила-, разветвленного гидрокси-C1-C6-алкила-, разветвленного C1-C6-алкокси-C1-C6-алкила-, разветвленного дигидрокси-C2-C6-алкила-, -(CH2)pN(R7)2, -(CH2)pR5, -(CH2)pC(O)N(R7)2, -(CH2)nC(O)R5, -(CH2)nC(O)OR3, C3-C8-карбоциклила, гидроксилзамещенного C3-C8-карбоциклила-, алкоксизамещенного C3-C8-карбоциклила-, дигидроксилзамещенного C3-C8-карбоциклила- и -(CH2)nR17;
каждый R5 независимо и отдельно выбран из группы, состоящей из
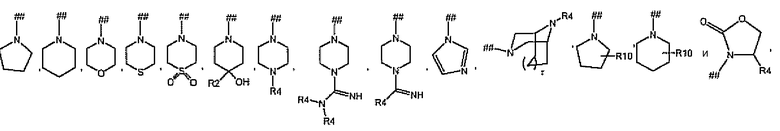
и где символ (##) означает точку присоединения остатка R5;
каждый R6 независимо и отдельно выбран из группы, состоящей из C1-C6-алкила, разветвленного C3-C7-алкила, C3-C8-карбоциклила, фенила, G1 и G4;
каждый R7 независимо и отдельно выбран из группы, состоящей из H, C1-C6-алкила, гидрокси-C2-C6-алкила-, дигидрокси-C2-C6-алкила-, C2-C6-алкокси-C2-C6-алкила-, разветвленного C3-C7-алкила-, разветвленного гидрокси-C2-C6-алкила-, разветвленного C2-C6-алкокси-C2-C6-алкила-, разветвленного дигидрокси-C2-C6-алкила-, -(CH2)qR5, -(CH2)nC(O)R5, -(CH2)nC(O)OR3, C3-C8-карбоциклила, гидроксилзамещенного C3-C8-карбоциклила-, алкоксизамещенного C3-C8-карбоциклила-, дигидроксизамещенного C3-C8-карбоциклила и -(CH2)nR17;
каждый R8 независимо и отдельно выбран из группы, состоящей из C1-C6-алкила, разветвленного C3-C7-алкила, фтор-C1-C6-алкила, где остаток алкила частично или полностью фторирован, C3-C8-карбоциклила, Z3-замещенного фенила-, Z3-замещенного фенил-C1-C6-алкила-, Z3-замещенного G1-, Z3-замещенного G1-C1-C6-алкила-, Z2-замещенного G4-, Z2-замещенного G4-C1-C6-алкила-, OH, C1-C6-алкоксигруппы, N(R3)2, N(R4)2 и R5;
каждый R9 независимо и отдельно выбран из группы, состоящей из H, F, C1-C6-алкила, разветвленного C3-C7-алкила, C3-C7-циклоалкила, фенила, фенил-C1-C6-алкила-, -(CH2)nG1 и -(CH2)nG4;
каждый R10 независимо и отдельно выбран из группы, состоящей из CO2H, CO2C1-C6-алкила, -C(O)N(R4)2, OH, C1-C6-алкоксигруппы и -N(R4)2;
каждый R13 независимо и отдельно выбран из группы, состоящей из H, C1-C6-алкила, разветвленного C3-C7-алкила, карбоциклила, гидрокси-C2-C7-алкила, C1-C6-алкокси-C2-C7-алкила-, (R4)2NC(O)-, (R4)2NC(O)C1-C6-алкила-, карбокси-C1-C6-алкила-, C1-C6-алкоксикарбонила-, C1-C6-алкоксикарбонил-C1-C6-алкила-, (R4)2N-C2-C6-алкила-, (R4)2N-C2-C6-алкил-N(R4)(CH2)q-, R5-C2-C6-алкил-N(R4)(CH2)q-, (R4)2N-C2-C6-алкил-O(CH2)q-, R5-C2-C6-алкил-O(CH2)q-, -(CH2)qN(R4)C(O)R8, арила, арил-C1-C6-алкила, арилокси-C2-C6-алкила-, ариламино-C2-C6-алкила-, C1-C6-алкоксикарбонил-C1-C6-алкила-, -C2-C6-алкилN(R4)C(O)R8, R8C(=NR3)-, -SO2R8, -COR8, -(CH2)nG1, -(CH2)n-G4, -(CH2)nO(CH2)nG1, -(CH2)nO(CH2)nG4, -(CH2)nN(R3)(CH2)nG1 и (CH2)nN(R3)(CH2)nG4;
каждый R14 независимо и соответственно выбран из группы, состоящей из H, C1-C6-алкила, разветвленного C3-C6-алкила и C3-C7-карбоциклила;
каждый R16 независимо и отдельно выбран из группы, состоящей из C1-C6-алкила, разветвленного C3-C7-алкила, C3-C8-карбоциклила, галогена, фтор-C1-C6-алкила, где остаток алкила может быть частично или полностью фторирован, цианогруппы, гидроксила, C1-C6-алкоксигруппы, фтор-C1-C6-алкоксигруппы, где остаток алкила может быть частично или полностью фторирован, -N(R3)2, -N(R4)2 и нитрогруппы;
каждый R17 выбран из группы, состоящей из фенила, нафтила, пирролила, фурила, тиенила, оксазолила, тиазолила, изоксазолила, изотиазолила, имидазолила, пиразолила, оксадиазолила, тиадиазолила, триазолила, тетразолила, пиразинила, пиридазинила, триазинила, оксетанила, азетадинила, тетрагидрофуранила, оксазолинила, оксазолидинила, пиранила, тиопиранила, тетрагидропиранила, диоксалинила, азепинила, оксепинила, диазепинила, пирролидинила и пиперидинила;
где R17 может быть дополнительно замещен одним или несколькими остатками Z2, Z3 или Z4;
R18 независимо и отдельно выбран из группы, состоящей из водорода, C1-C6-алкила, разветвленного C3-C7-алкила, C3-C8-карбоциклила, галогена, фтор-C1-C6-алкила, где остаток алкила может быть частично или полностью фторирован, цианогруппы, гидроксила, C1-C6-алкоксигруппы, фтор-C1-C6-алкоксигруппы, где остаток алкила может быть частично или полностью фторирован, -N(R3)2, -N(R4)2, C2-C3-алкинила и нитрогруппы;
R19 означает H или C1-C6-алкил;
где два остатка R3 или R4, независимо и отдельно выбранные из группы, состоящей из C1-C6-алкила и разветвленного C3-C6-алкила, гидроксиалкила и алкоксиалкила, и связанные с одним и тем же атомом азота, могут быть соединены с образованием C3-C7-гетероциклического кольца;
и n равно 0-6; p равно 1-4; q равно 2-6; r равно 0 или 1; t равно 1-3, v равно 1 или 2;
при условии, что соединения формулы Ia не могут представлять собой
1.1. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-1b:
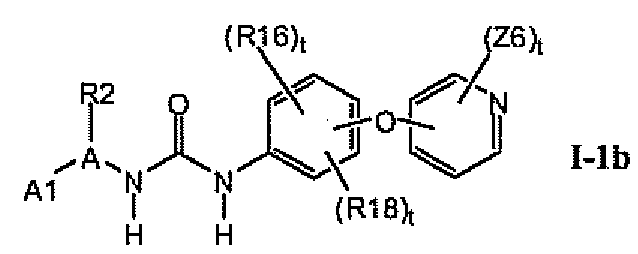
где цикл A представляет собой пиразолил.
1.1.1. Соединения формулы I-1b, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-1b указанные соединения имеют структуры формулы I-1c:
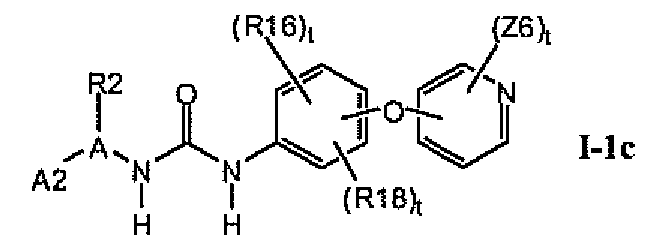
1.1.2. Соединения формулы Ib, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-1b указанные соединения имеют структуры формулы I-1d:
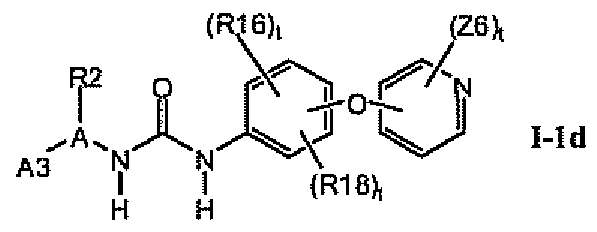
1.1.3. Соединения формулы I-1b, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-1b указанные соединения имеют структуры формулы I-1e
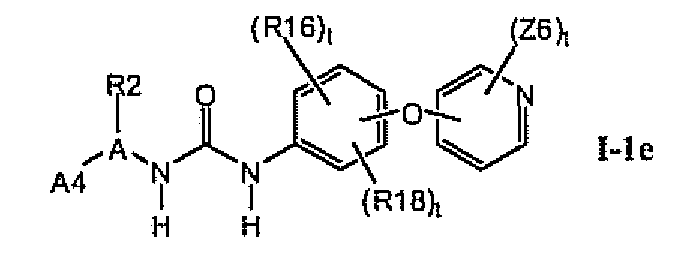
1.1.4. Более предпочтительные соединения в разделе 1.1
В предпочтительном варианте соединений из раздела 1.1 указанные соединения имеют структуры формулы I-1f:
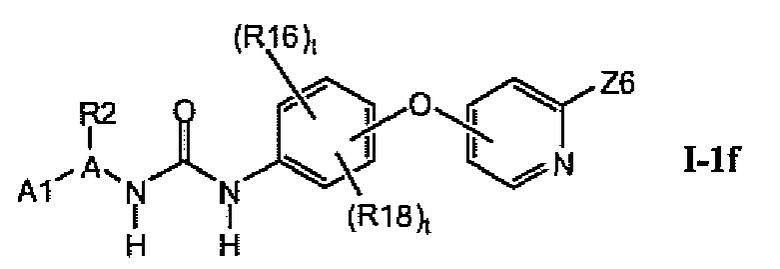
1.1.5. Соединения из раздела 1.1.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.1.4 указанные соединения имеют структуры формулы I-1g:
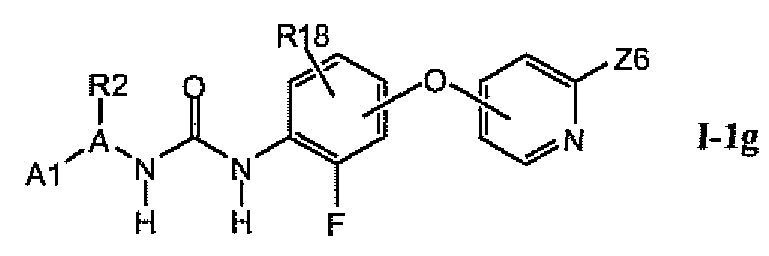
1.1.6. Соединения из раздела 1.1.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.1.5 указанные соединения имеют структуры формулы I-1h:
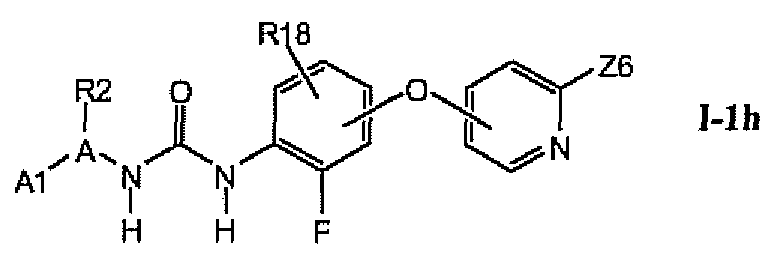
где A1 выбран из группы, состоящей из
1.1.7. Соединения из раздела 1.1.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.1.5 указанные соединения имеют структуры формулы I-1i:
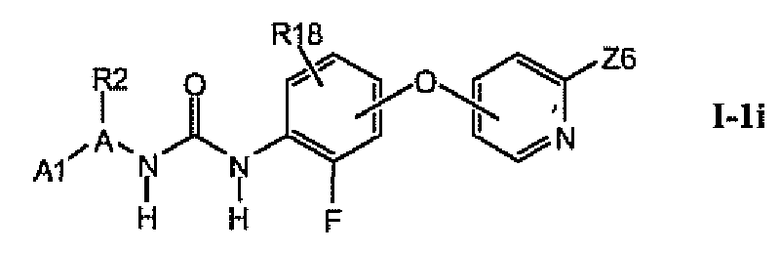
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.2. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-2a:
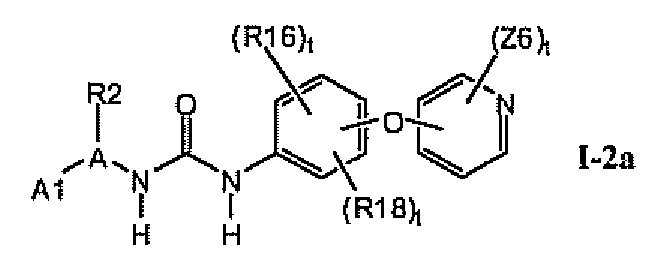
где цикл A представляет собой изоксазолил.
1.2.1. Соединения формулы I-2a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-2a указанные соединения имеют структуры формулы I-2b:
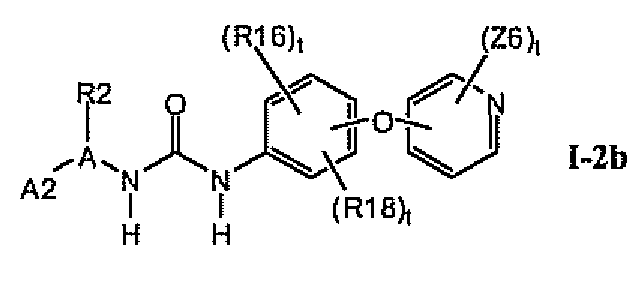
1.2.2. Соединения формулы I-2a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-2a указанные соединения имеют структуры формулы I-2c:
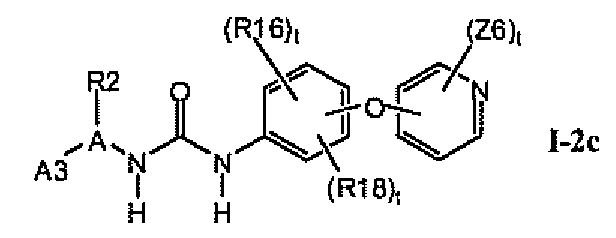
1.2.3. Соединения формулы I-2a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-2a указанные соединения имеют структуры формулы I-2d:
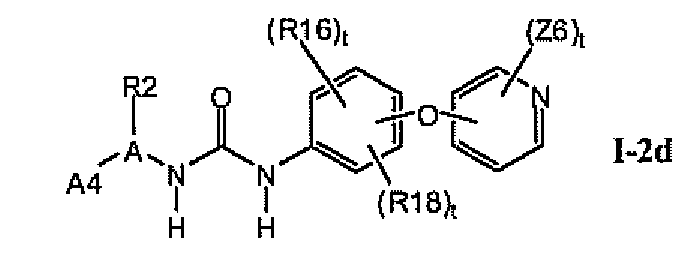
1.2.4. Более предпочтительные соединения в разделе 1.2
В предпочтительном варианте соединений из раздела 1.2 указанные соединения имеют структуры формулы I-2e:
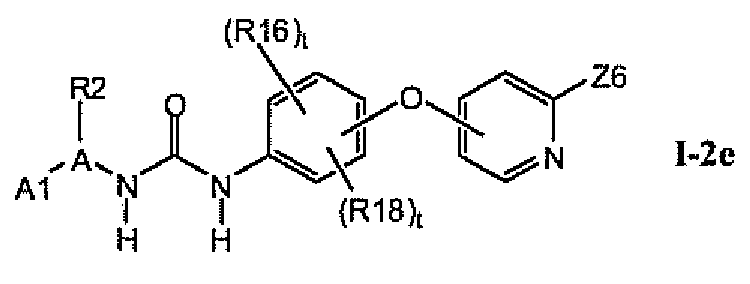
1.2.5. Соединения из раздела 1.2.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.2.4 указанные соединения имеют структуры формулы I-2f:
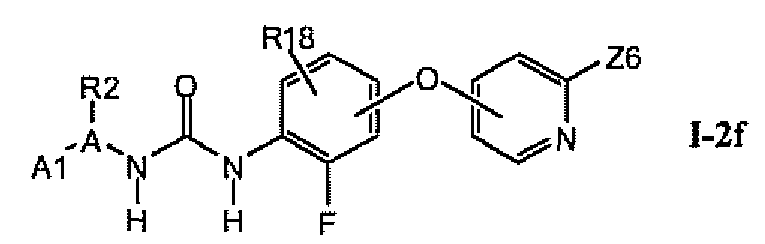
1.2.6. Соединения из раздела 1.2.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.2.5 указанные соединения имеют структуры формулы I-2g:
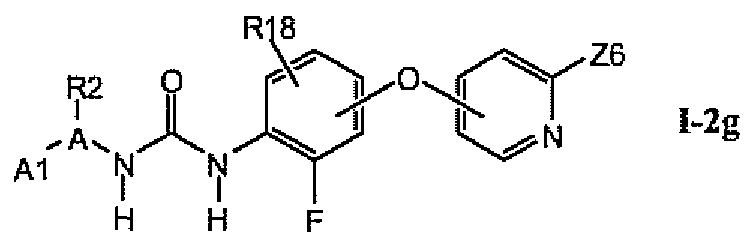
где A1 выбран из группы, состоящей из
1.2.7 Соединения из раздела 1.2.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.2.5 указанные соединения имеют структуры формулы I-2h:
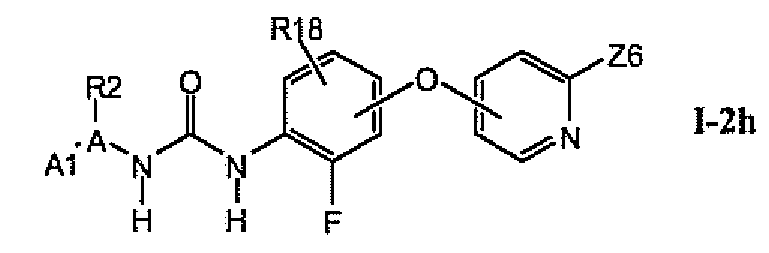
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.3. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-3a:
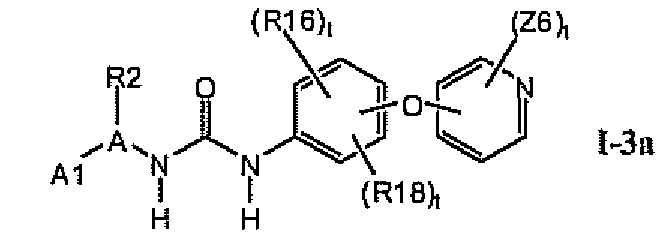
где цикл A представляет собой тиенил.
1.3.1. Соединения формулы I-3a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-3a указанные соединения имеют структуры формулы I-3b:
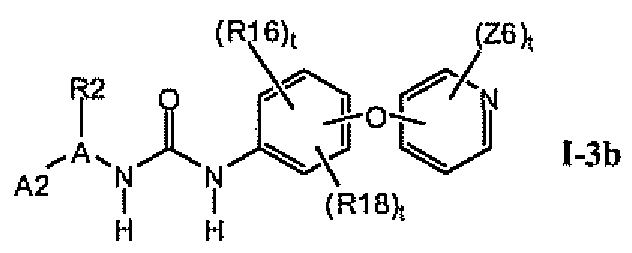
1.3.2. Соединения формулы Ix, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-3a указанные соединения имеют структуры формулы I-3c:
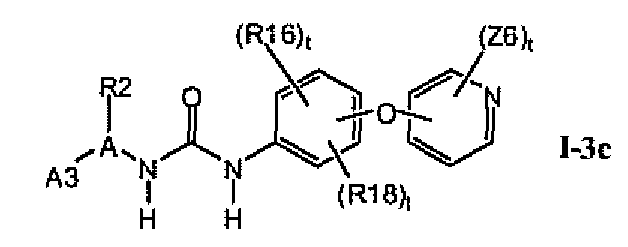
1.3.3. Соединения формулы I-3a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-3a указанные соединения имеют структуры формулы I-3d:
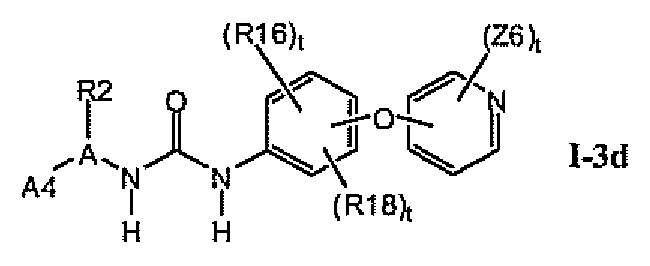
1.3.4. Более предпочтительные соединения из раздела 1.3
В предпочтительном варианте соединений из раздела 1.3 указанные соединения имеют структуры формулы I-3e:
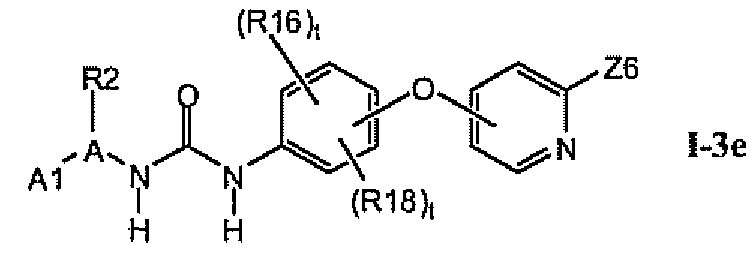
1.3.5. Соединения из раздела 1.3.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.3.4 указанные соединения имеют структуры формулы 1-3f:
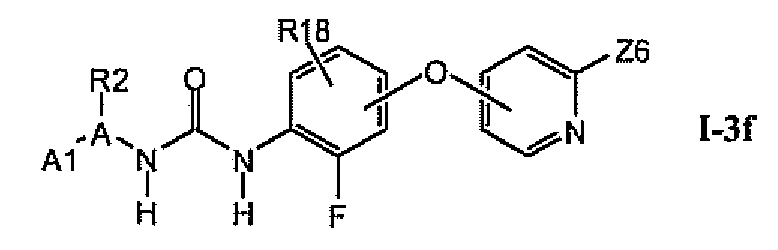
1.3.6. Соединения из раздела 1.3.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.3.5 указанные соединения имеют структуры формулы I-3g:
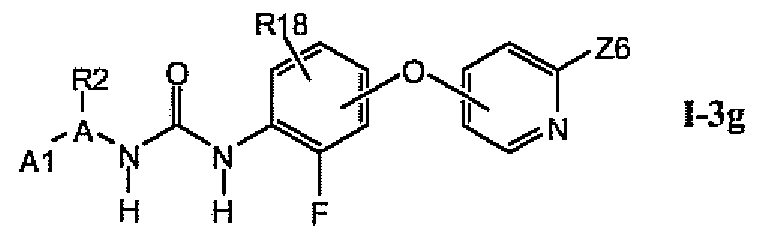
где A1 выбран из группы, состоящей из
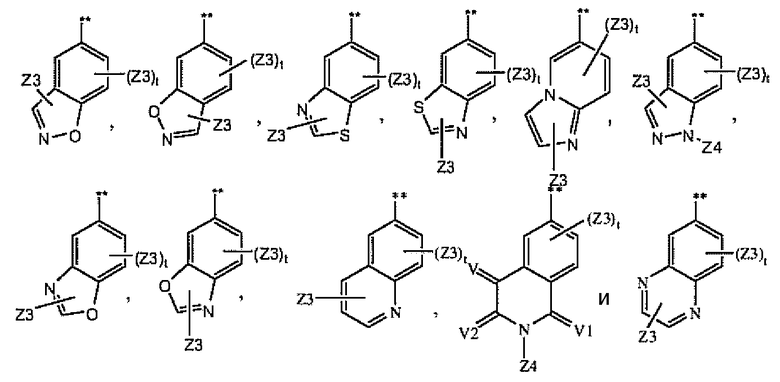
1.3.7. Соединения из раздела 1.3.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.3.5 указанные соединения имеют структуры формулы I-3h:
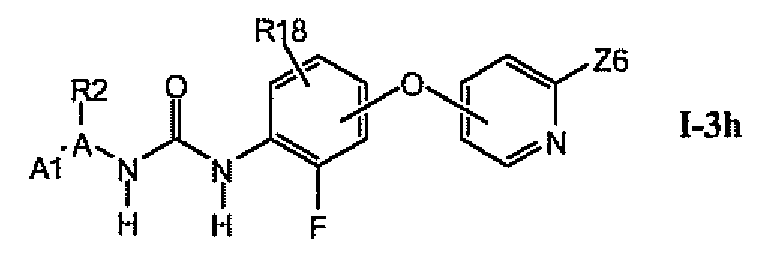
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.4 Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-4a:
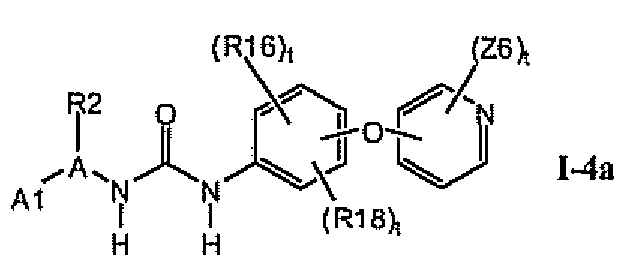
где цикл A представляет собой фурил.
1.4.1. Соединения формулы Iii, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-4a указанные соединения имеют структуры формулы I-4b:
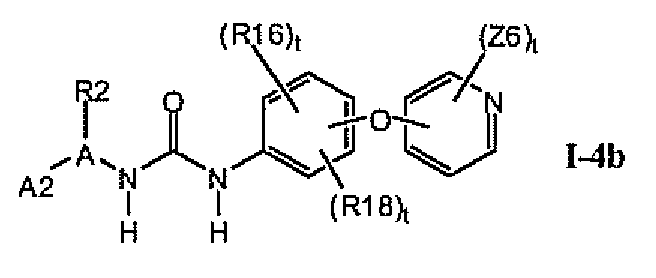
1.4.2. Соединения формулы Iii, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-4a указанные соединения имеют структуры формулы I-4c:
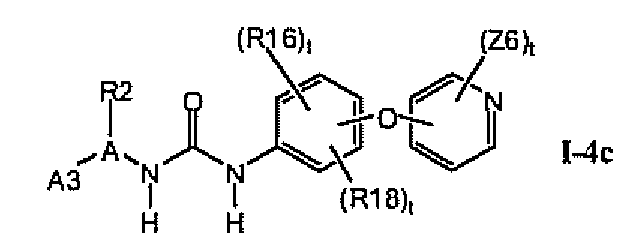
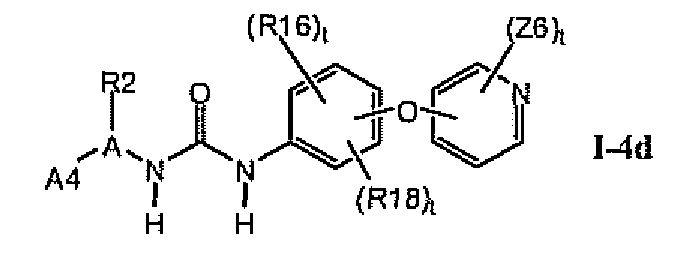
1.4.3. Соединения формулы Im, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-4a указанные соединения имеют структуры формулы I-4d:
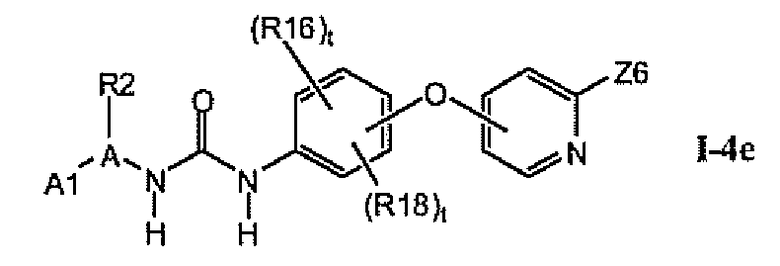
1.4.4. Более предпочтительные соединения из раздела 1.4
В предпочтительном варианте соединений из раздела 1.4 указанные соединения имеют структуры формулы I-4e:
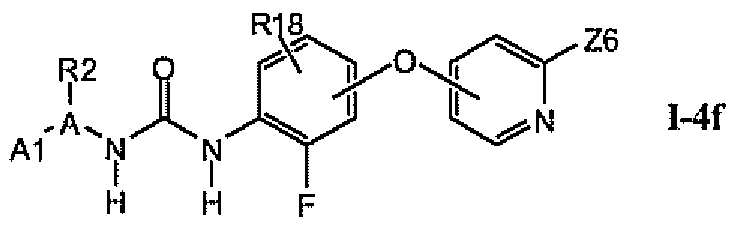
1.4.5. Соединения из раздела 1.4.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.4.4 указанные соединения имеют структуры формулы I-4f:
1.4.6. Соединения из раздела 1.4.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.4.5 указанные соединения имеют структуры формулы I-4g:
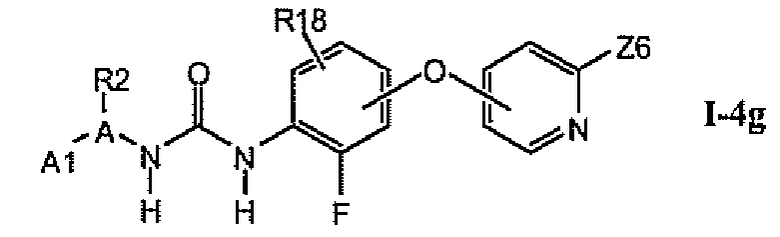
где A1 выбран из группы, состоящей из
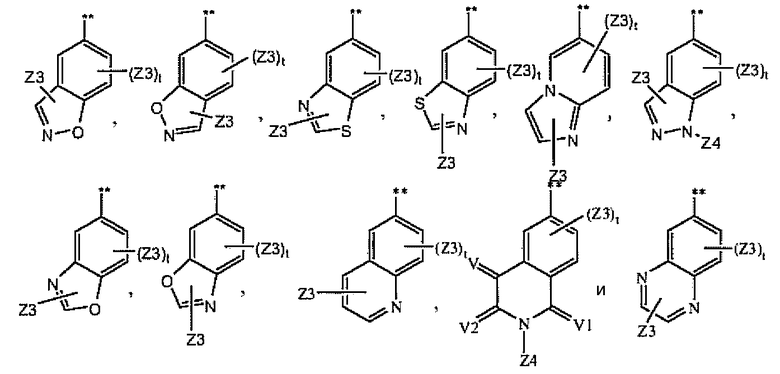
1.4.7. Соединения из раздела 1.4.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.4.5 указанные соединения имеют структуры формулы I-4h:
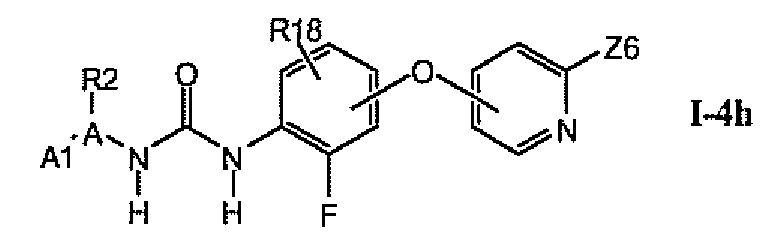
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.5 Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-5a:
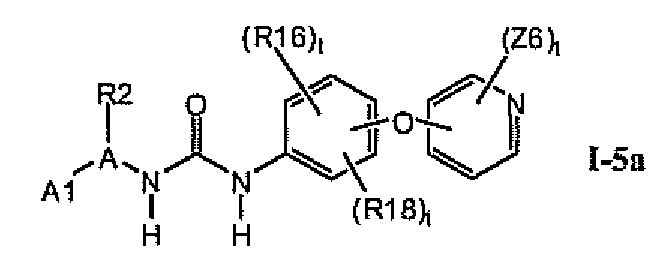
где цикл A представляет собой пирролил.
1.5.1. Соединения формулы I-5a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-5a указанные соединения имеют структуры формулы I-5b:
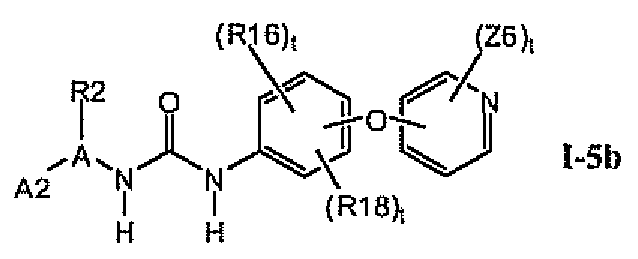
1.5.2. Соединения формулы I-5a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-5a указанные соединения имеют структуры формулы I-5c:
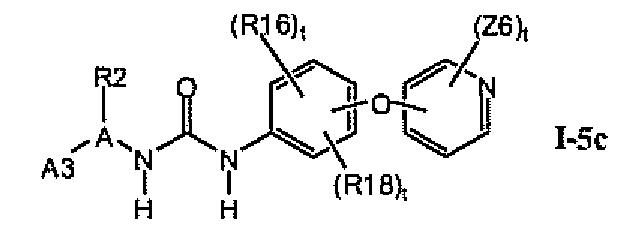
1.5.3. Соединения формулы I-5a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы 1-5a указанные соединения имеют структуры формулы I-5d:
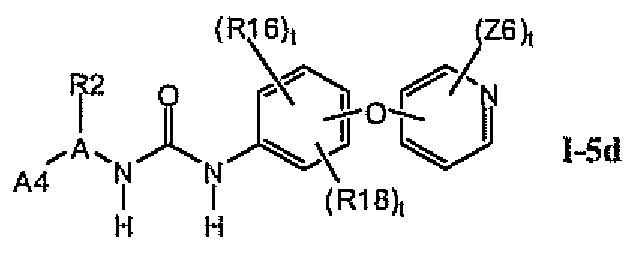
1.5.4. Более предпочтительные соединения из раздела 1.5
В предпочтительном варианте соединений из раздела 1.5 указанные соединения имеют структуры формулы I-5e:
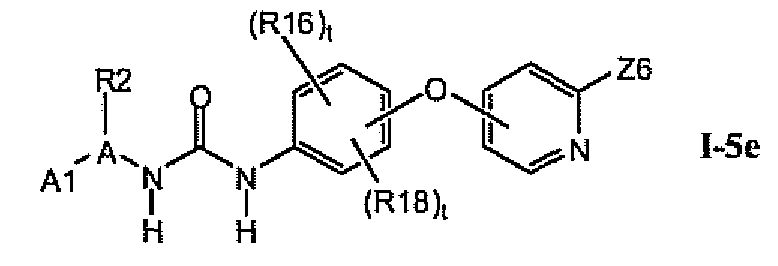
1.5.5. Соединения из раздела 1.5.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.5.4 указанные соединения имеют структуры формулы 1-5f:
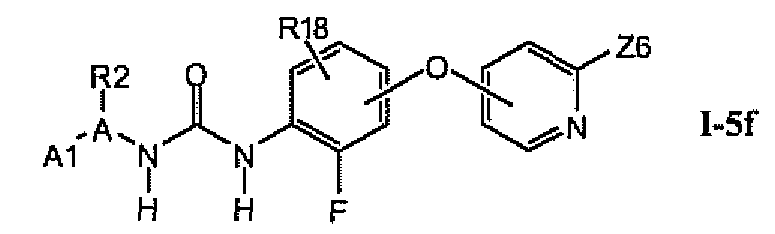
1.5.6. Соединения из раздела 1.5.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.5.5 указанные соединения имеют структуры формулы I-5g:
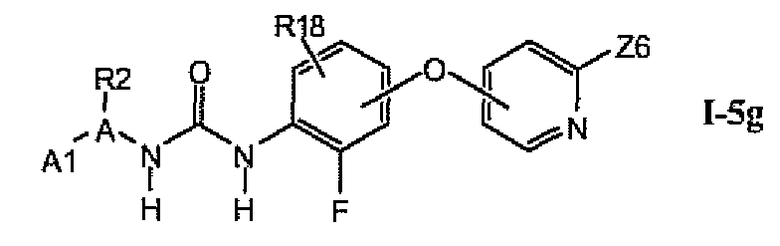
где A1 выбран из группы, состоящей из
1.5.7. Соединения из раздела 1.5.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.5.5 указанные соединения имеют структуры формулы I-5h:
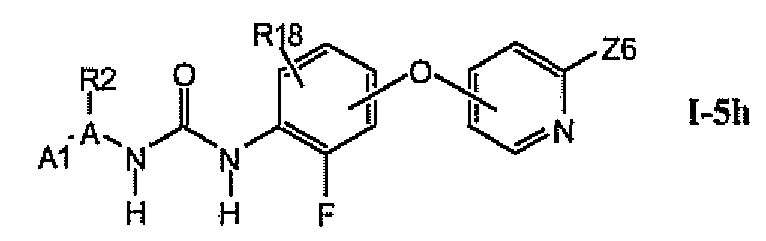
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.6. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-6a:
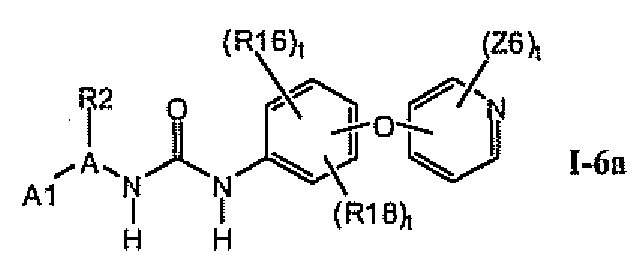
где цикл A представляет собой имидазолил.
1.6.1. Соединения формулы I-6a, которые иллюстрируют предпочтительные остатки A1
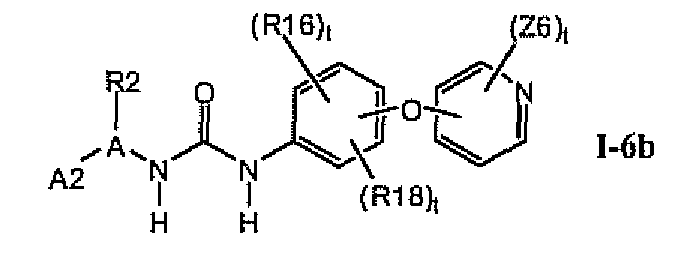
В предпочтительном варианте соединений формулы 1-6a указанные соединения имеют структуры формулы I-6b:
1.6.2. Соединения формулы I-6a, которые иллюстрируют предпочтительные остатки A1
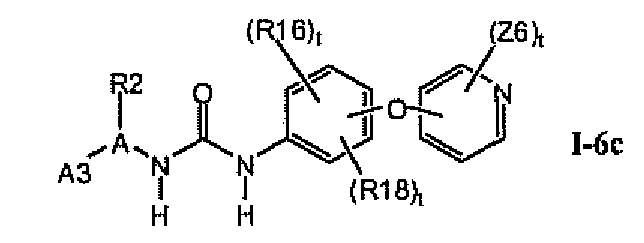
В предпочтительном варианте соединений формулы I-6a указанные соединения имеют структуры формулы I-6c:
1.6.3 Соединения формулы I-6a, которые иллюстрируют предпочтительные остатки A1
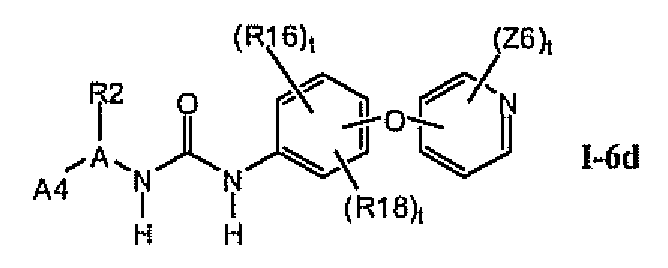
В предпочтительном варианте соединений формулы I-6a указанные соединения имеют структуры формулы I-6d:
1.6.4. Более предпочтительные соединения из раздела 1.6
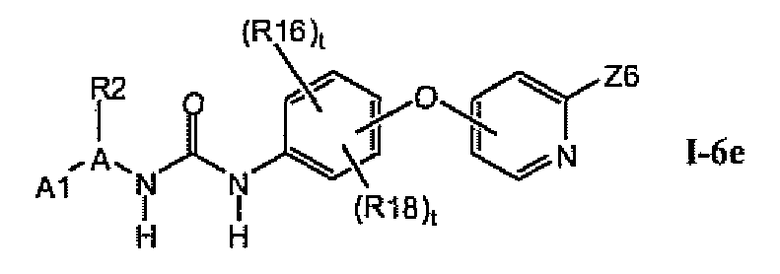
В предпочтительном варианте соединений из раздела 1.6 указанные соединения имеют структуры формулы I-6e:
1.6.5. Соединения из раздела 1.6.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.6.4 указанные соединения имеют структуры формулы I-6f:
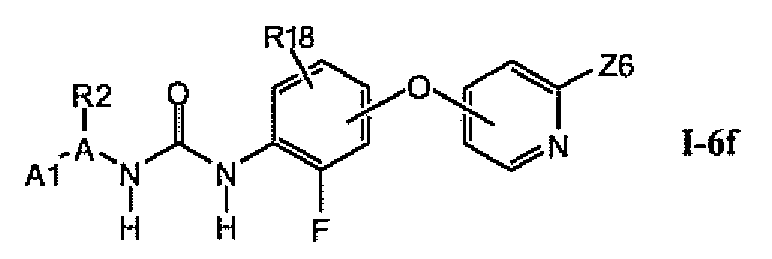
1.6.6. Соединения из раздела 1.6.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.6.5 указанные соединения имеют структуры формулы I-6g:
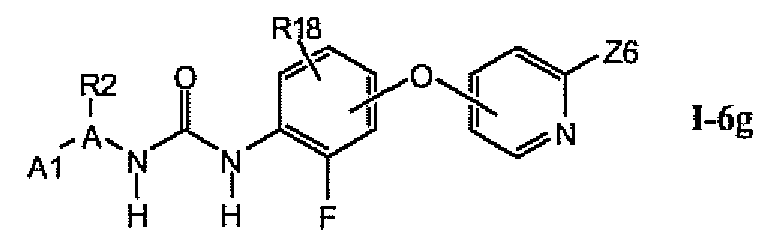
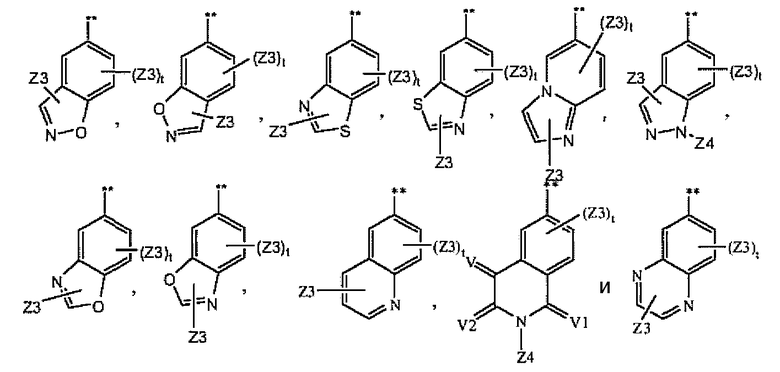
где A1 выбран из группы, состоящей из
1.6.7. Соединения из раздела 1.6.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.6.5 указанные соединения имеют структуры формулы I-6h:
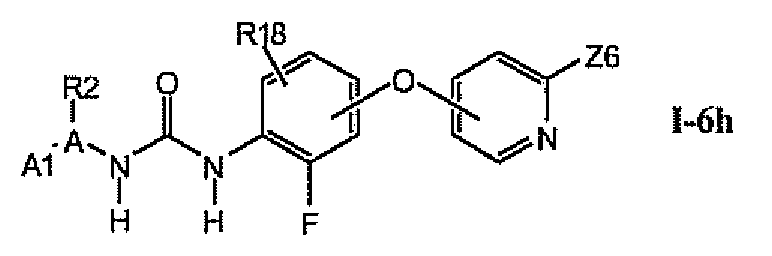
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.7. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-7a:
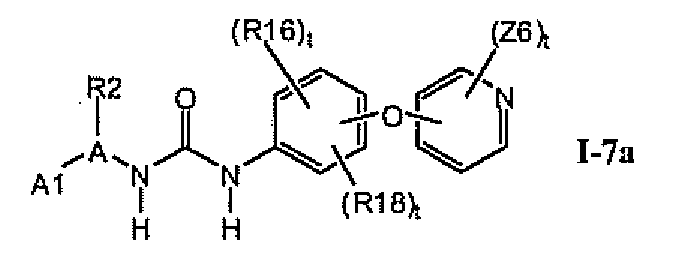
где цикл A представляет собой тиазолил.
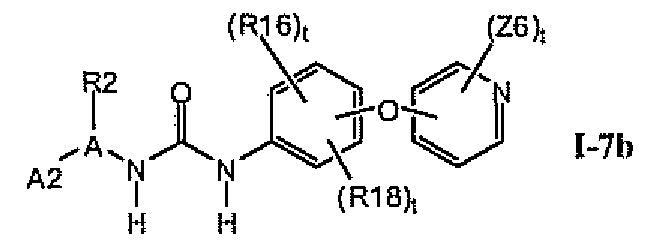
1.7.1. Соединения формулы I-7a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-7a указанные соединения имеют структуры формулы I-7b:
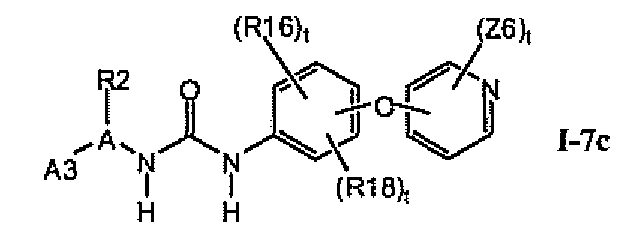
1.7.2. Соединения формулы I-7a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-7a указанные соединения имеют структуры формулы I-7c:
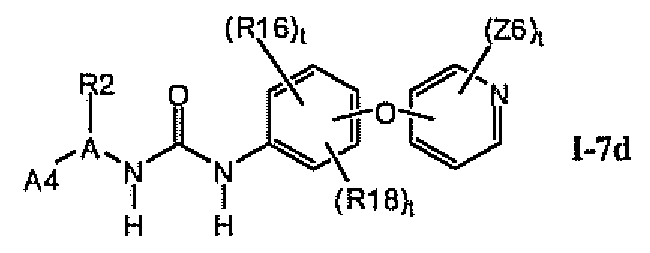
1.7.3. Соединения формулы I-7a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-7a указанные соединения имеют структуры формулы I-7d:
1.7.4. Более предпочтительные соединения из раздела 1.7
В предпочтительном варианте соединений из раздела 1.7 указанные соединения имеют структуры формулы I-7e:
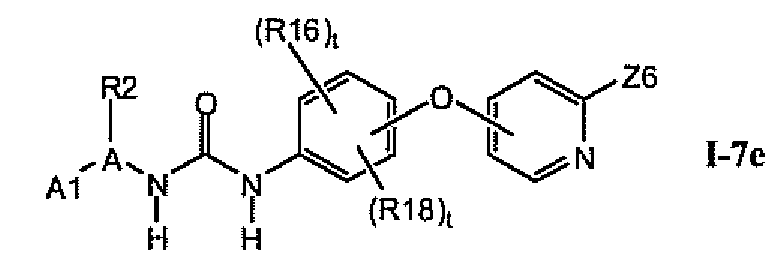
1.7.5. Соединения из раздела 1.7.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.7.4 указанные соединения имеют структуры формулы I-7f:
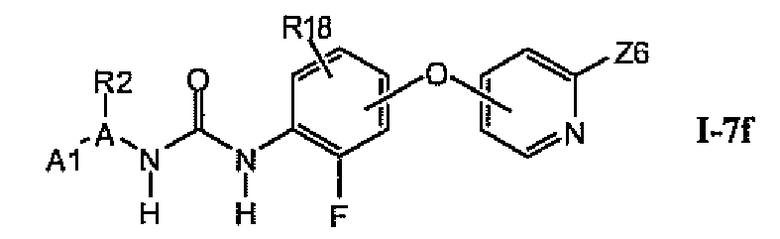
1.7.6. Соединения из раздела 1.7.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.7.5 указанные соединения имеют структуры формулы I-7g:
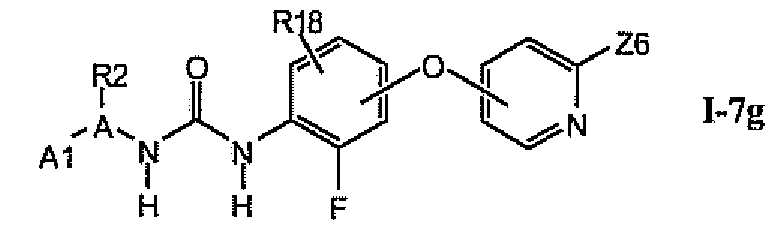
где A1 выбран из группы, состоящей из
1.7.7. Соединения из раздела 1.7.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.7.5 указанные соединения имеют структуры формулы I-7h:
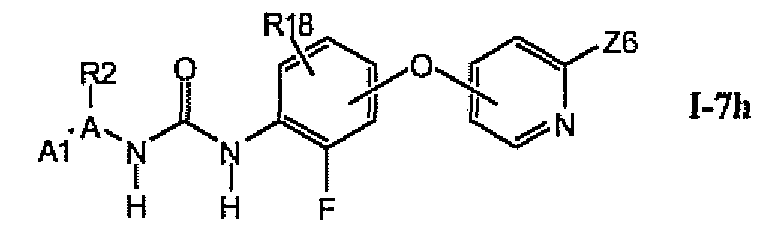
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.8. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-8a:
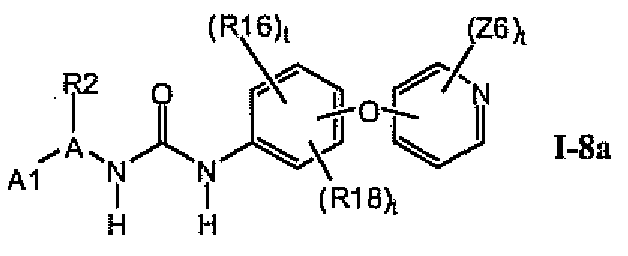
где цикл A представляет собой оксазолил.
1.8.1. Соединения формулы I-8a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-8a указанные соединения имеют структуры формулы I-8b:
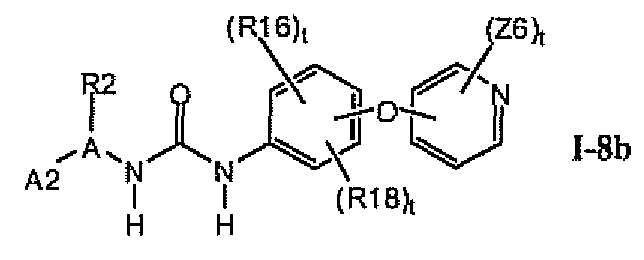
1.8.2. Соединения формулы I-8a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-8a, указанные соединения имеют структуры формулы I-8c:
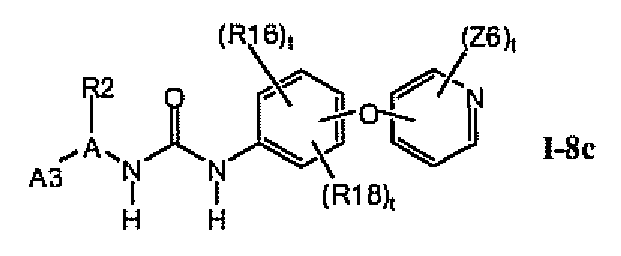
1.8.3. Соединения формулы I-8a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-8a указанные соединения имеют структуры формулы I-8d:
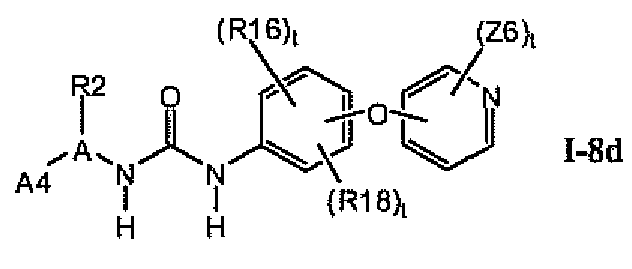
1.8.4. Более предпочтительные соединения из раздела 1.8
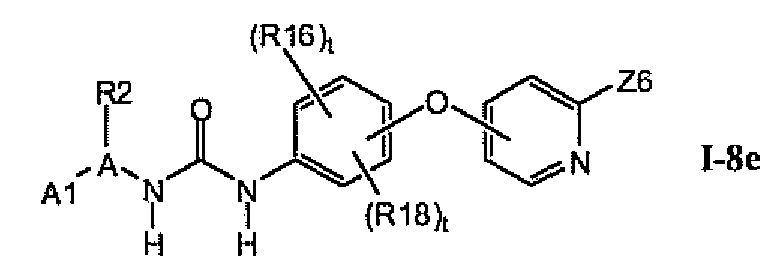
В предпочтительном варианте соединений из раздела 1.8 указанные соединения имеют структуры формулы I-8e:
1.8.5. Соединения из раздела 1.8.4 с предпочтительными остатками R16
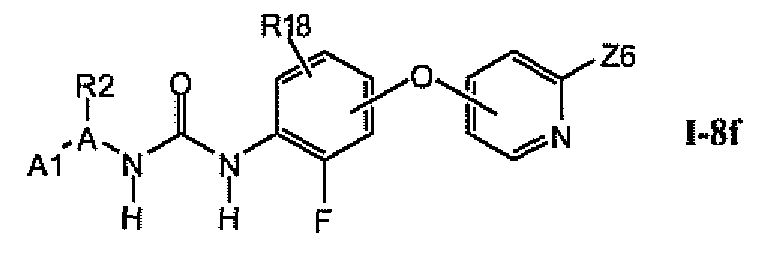
В предпочтительном варианте соединений из раздела 1.8.4 указанные соединения имеют структуры формулы I-8f:
1.8.6. Соединения из раздела 1.8.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.8.5 указанные соединения имеют структуры формулы I-8g:
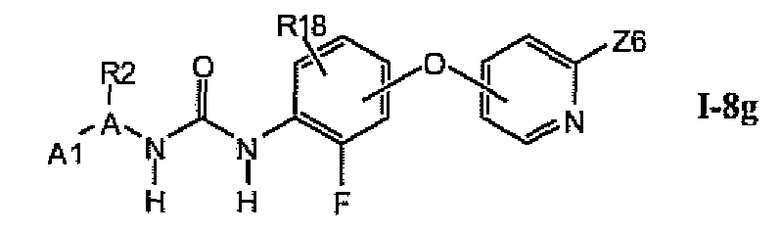
где A1 выбран из группы, состоящей из
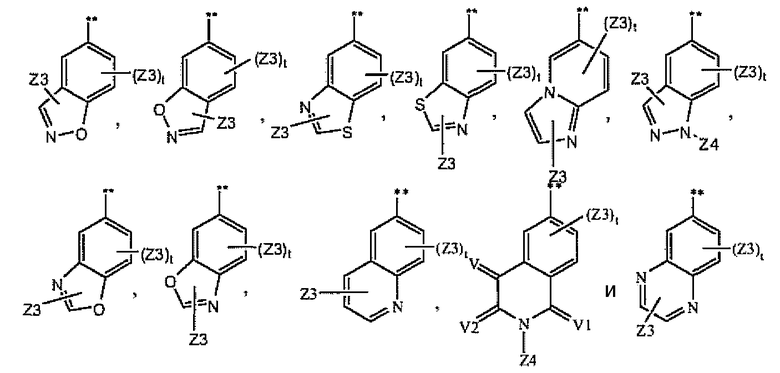
1.8.7. Соединения из раздела 1.8.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.8.5 указанные соединения имеют структуры формулы I-8h:
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
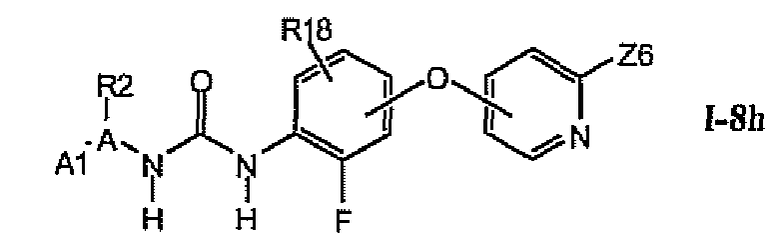
1.9. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-9a:
где цикл A представляет собой изотиазолил.
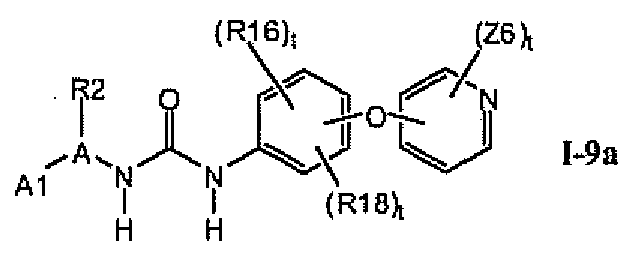
1.9.1. Соединения формулы I-9a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-9a указанные соединения имеют структуры формулы I-9b:
1.9.2. Соединения формулы I-9a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-9a указанные соединения имеют структуры формулы I-9c:
1.9.3. Соединения формулы I-9a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-9a указанные соединения имеют структуры формулы I-9d:
1.9.4. Более предпочтительные соединения из раздела 1.9
В предпочтительном варианте соединений из раздела 1.9 указанные соединения имеют структуры формулы I-9e:
1.9.5. Соединения из раздела 1.9.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.9.4 указанные соединения имеют структуры формулы 1-9f:
1.9.6. Соединения из раздела 1.9.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.9.5 указанные соединения имеют структуры формулы I-9g:
где A1 выбран из группы, состоящей из
1.9.7. Соединения из раздела 1.9.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.9.5 указанные соединения имеют структуры формулы I-9h:
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.10. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-10a:
где цикл A представляет собой фенил.
1.10.1. Соединения формулы I-10a, которые иллюстрируют предпочтительные остатки A1
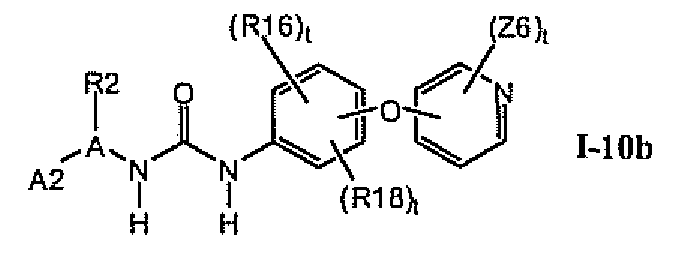
В предпочтительном варианте соединений формулы I-10a указанные соединения имеют структуры формулы I-10b:
1.10.2. Соединения формулы I-10a, которые иллюстрируют предпочтительные остатки A1
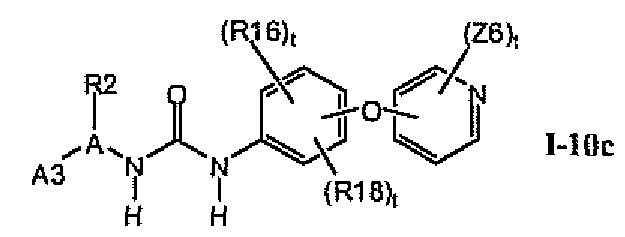
В предпочтительном варианте соединений формулы I-10a указанные соединения имеют структуры формулы I-10c:
1.10.3. Соединения формулы I-10a, которые иллюстрируют предпочтительные остатки A1
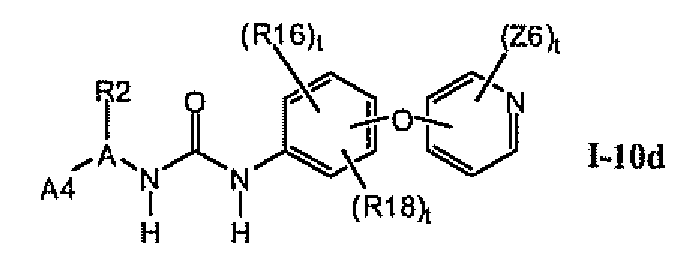
В предпочтительном варианте соединений формулы I-10a указанные соединения имеют структуры формулы I-10d:
1.10.4. Более предпочтительные соединения из раздела 1.10
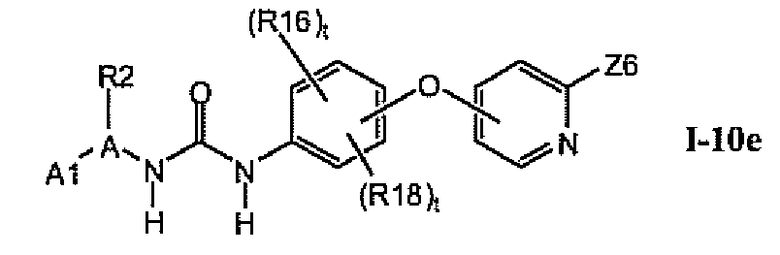
В предпочтительном варианте соединений из раздела 1.10 указанные соединения имеют структуры формулы I-10e:
1.10.5. Соединения из раздела 1.10.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.10.4 указанные соединения имеют структуры формулы I-10f:
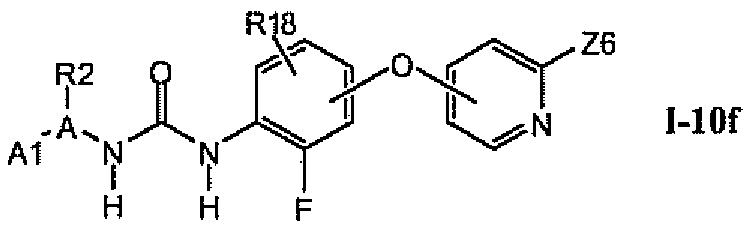
1.10.6. Соединения из раздела 1.10.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.10.5 указанные соединения имеют структуры формулы I-10g:
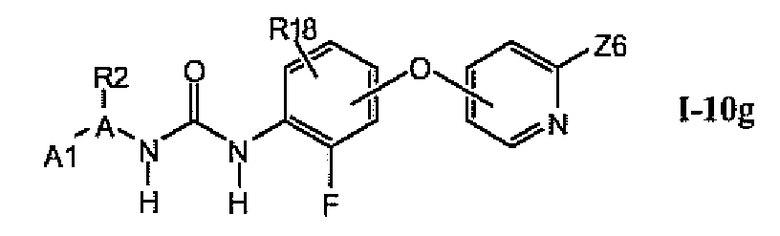
где A1 выбран из группы, состоящей из
1.10.7. Соединения из раздела 1.10.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.10.5 указанные соединения имеют структуры формулы I-10h:
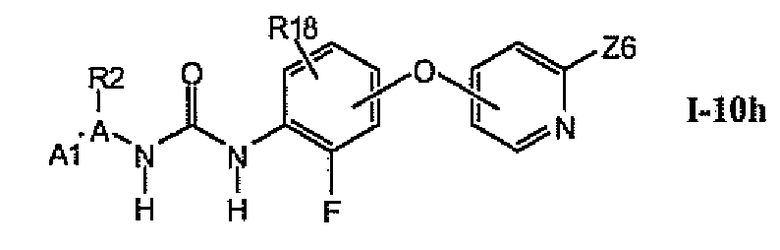
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.11. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-11a:
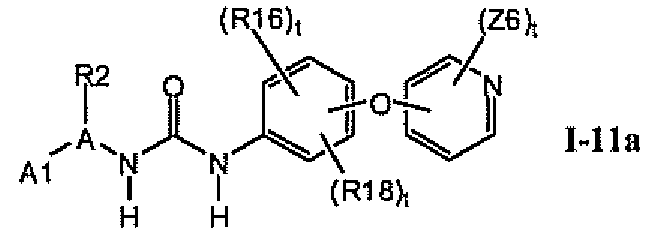
где цикл A представляет собой пиримидинил.
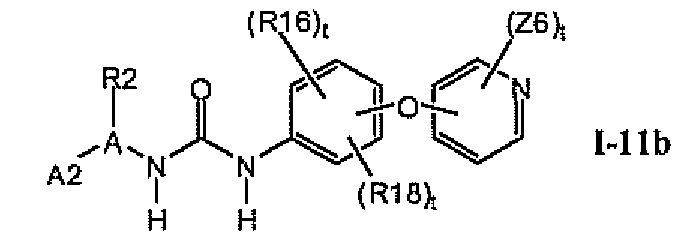
1.11.1. Соединения формулы I-11a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-11a указанные соединения имеют структуры формулы I-11b:
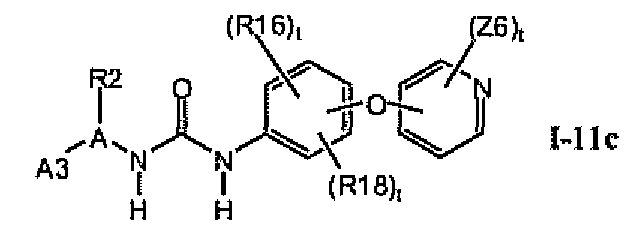
1.11.2. Соединения формулы I-11a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-11a указанные соединения имеют структуры формулы I-11c:
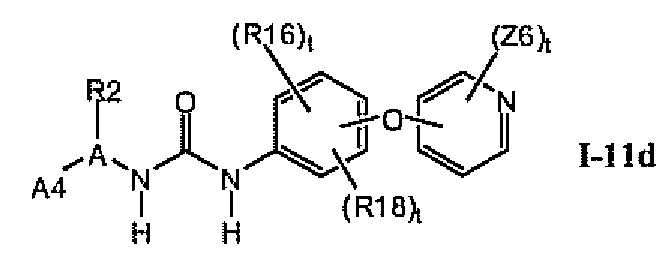
1.11.3. Соединения формулы I-11a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-11a, указанные соединения имеют структуры формулы 1-11d:
1.11.4. Более предпочтительные соединения из раздела 1.11
В предпочтительном варианте соединений из раздела 1.11 указанные соединения имеют структуры формулы I-11e:
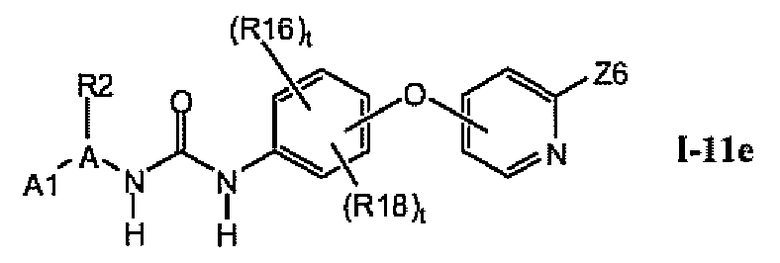
1.11.5. Соединения из раздела 1.11.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.11.4 указанные соединения имеют структуры формулы I-11f:
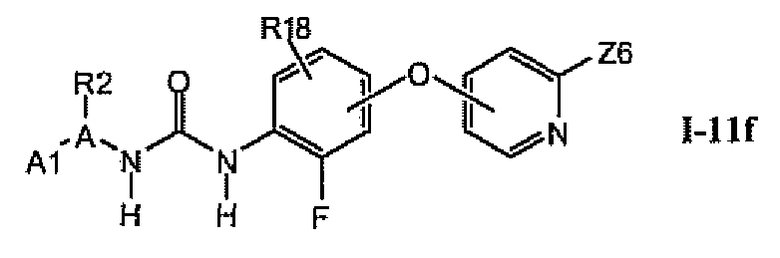
1.11.6. Соединения из раздела 1.11.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.11.5 указанные соединения имеют структуры формулы I-11g:
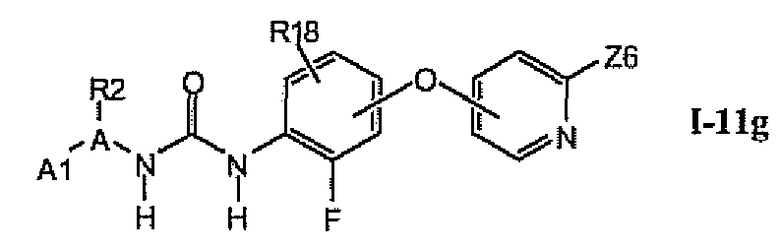
где A1 выбран из группы, состоящей из
1.11.7. Соединения из раздела 1.11.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.11.5 указанные соединения имеют структуры формулы I-11h:
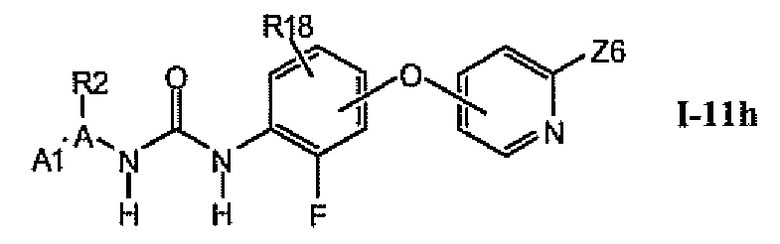
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.12. Соединения формулы Ia, которые иллюстрируют предпочтительные остатки A и X2-E1
В предпочтительном варианте соединений формулы Ia указанные соединения имеют структуры формулы I-12a:
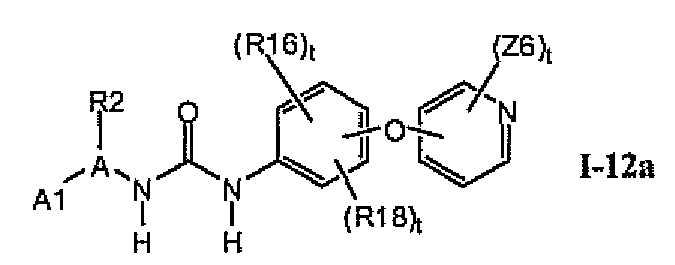
где цикл A представляет собой пиридинил.
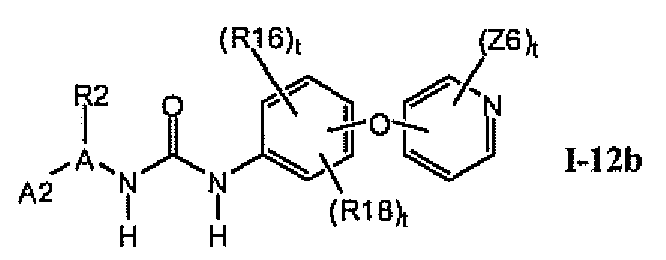
1.12.1. Соединения формулы I-12a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-12a указанные соединения имеют структуры формулы I-12b:
1.12.2. Соединения формулы I-12a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-12a указанные соединения имеют структуры формулы I-12c:
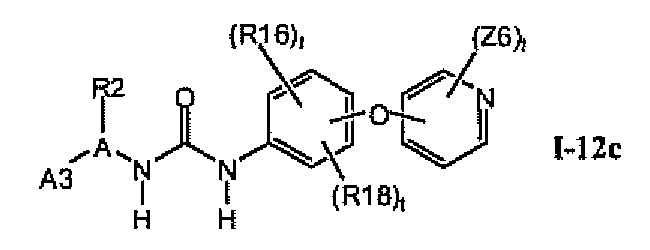
1.12.3. Соединения формулы I-12a, которые иллюстрируют предпочтительные остатки A1
В предпочтительном варианте соединений формулы I-12a указанные соединения имеют структуры формулы I-12d:
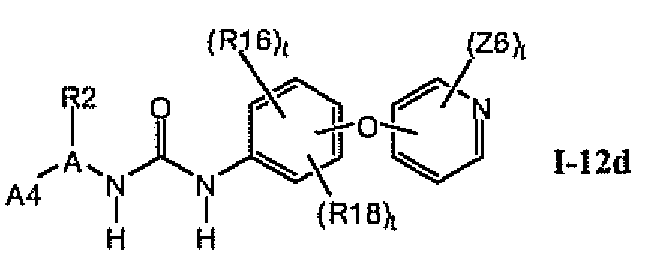
1.12.4. Более предпочтительные соединения из раздела 1.12
В предпочтительном варианте соединений из раздела 1.12 указанные соединения имеют структуры формулы I-12e:
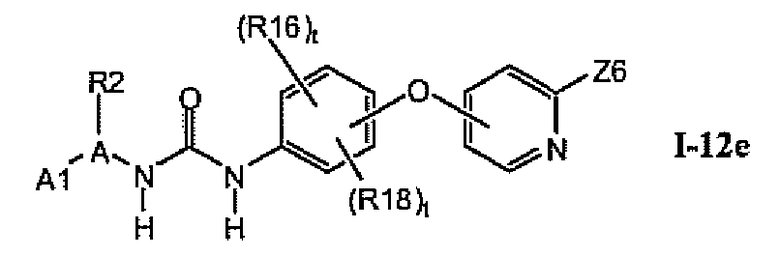
1.12.5. Соединения из раздела 1.12.4 с предпочтительными остатками R16
В предпочтительном варианте соединений из раздела 1.12.4 указанные соединения имеют структуры формулы I-12f:
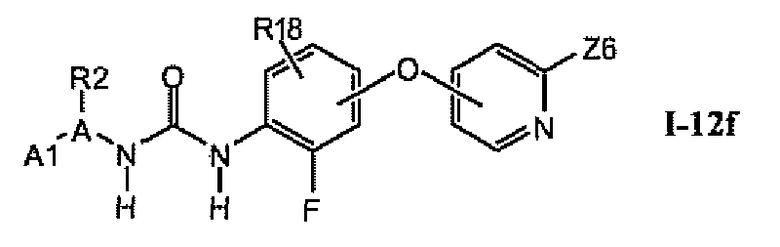
1.12.6. Соединения из раздела 1.12.5 с более предпочтительными остатками A1
В более предпочтительном варианте соединений из раздела 1.12.5 указанные соединения имеют структуры формулы I-12g:
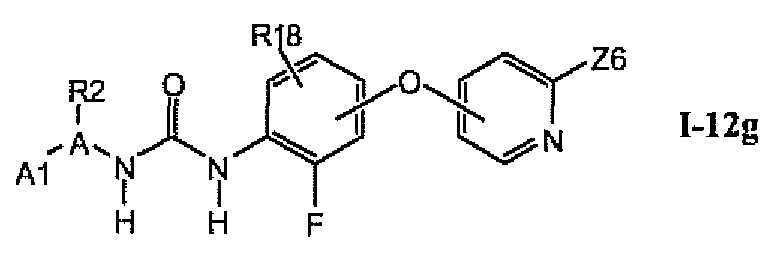
где A1 выбран из группы, состоящей из
1.12.7. Соединения из раздела 1.12.5 с более предпочтительными остатками Z6
В более предпочтительном варианте соединений из раздела 1.12.5 указанные соединения имеют структуры формулы I-12h:
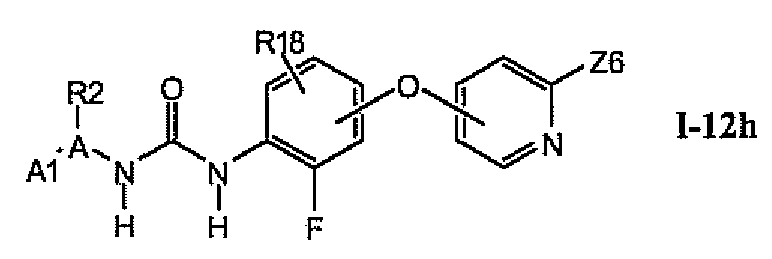
где Z6 означает -C(O)NHR4, -NHR4 или R19-замещенный пиразол.
1.13 Способы
1.13a Способы модулирования белков
Изобретение относится к способам модулирования киназной активности различных киназ, например киназы C-Abl, киназы bcr-Abl, Flt-3, c-Kit, PDGFR, VEGFR, c-MET, семейства киназ HER и семейства киназ Raf. Киназы могут представлять собой киназы дикого типа, их онкогенные формы, их аберрантные формы в виде слитых белков или полиморфы любой из указанных выше форм. Способ включает стадию осуществления контакта киназы определенного вида с соединениями согласно изобретению и особенно соединениями, указанными в разделах 1.1-1.12. Виды киназ могут быть активированными или инактивированными, и такие виды могут быть модулированы фосфорилированием, сульфатированием, ацилированием жирными кислотами, гликозилированием, нитрозилированием, цистинилированием (т.е. проксимальные остатки цистеина в киназах взаимодействуют друг с другом с образованием дисульфидной связи) или окислением. Киназная активность может быть выбрана из группы, состоящей из катализа реакций переноса фосфора, ингибирования фосфорилирования, окисления или нитрозилирования указанной киназы другим ферментом, усиления дефосфорилирования, восстановления или денитрозилирования указанной киназы другим ферментом, клеточной локализации киназы и привлечения других белков в сигнальные комплексы посредством модулирования конформации киназы.
1.13b Способы лечения
Способы согласно изобретению также включают лечение индивидуумов, страдающих от состояния, выбранного из группы, состоящей из рака и гиперпролиферативных заболеваний. Такие способы включают введение таким индивидуумам соединений согласно изобретению и особенно соединений, описанных в разделах 1.1-1.12, при этом указанные заболевания включают, без ограничения, заболевание, вызванное киназой c-Abl, ее онкогенными формами, ее аберрантными формами в виде слитых белков и ее полиморфами, хронический миелогенный лейкоз, острый лимфоцитарный лейкоз, другие миелопролиферативные расстройства, стромальные опухоли желудочно-кишечного тракта, связанную с возрастом дегенерацию желтого пятна, гиперэозинофильный синдром, глиобластомы, рак яичника, рак поджелудочной железы, рак простаты, рак легкого, рак молочной железы, рак почек, карциномы шейки матки, метастазы первичной солидной опухоли во вторичных очагах, глазные болезни, характеризующиеся гиперпролиферацией, приводящие к слепоте, включая различные ретинопатии, например диабетическую ретинопатию и связанную с возрастом дегенерацию желтого пятна, ревматоидный артрит, меланомы, рак ободочной кишки, рак щитовидной железы, заболевание, вызванное мутацией в киназном пути RAS-RAF-MEK-ERK-MAP, воспаление у человека, ревматоидный спондилит, остеоартрит, астму, подагрический артрит, сепсис, септический шок, эндотоксический шок, грамотрицательный сепсис, синдром токсического шока, респираторный дистресс-синдром взрослых, удар, реперфузионное повреждение, травму нервной системы, ишемию нервной системы, псориаз, рестеноз, хроническое обструктивное легочное заболевание, заболевания, обусловленные резорбцией костей, реакцию «трансплантат против хозяина», болезнь Крона, язвенный колит, воспалительное заболевание кишечника, изжогу и их сочетания. Способ введения не является решающим и может быть выбран из группы, состоящей из перорального, парентерального, ингаляционного и подкожного введения.
1.14 Фармацевтические композиции
Соединения согласно изобретению, особенно соединения, описанные в разделах 1.1-1.12, могут составлять часть фармацевтической композиции при объединении одного или нескольких таких соединений с фармацевтически приемлемым носителем. Кроме того, композиции могут содержать вспомогательное вещество, выбранное из группы, состоящей из адъювантов, эксципиентов, разбавителей и стабилизаторов.
2. Синтез соединений согласно настоящему изобретению
Соединения согласно изобретению можно получить с использованием способов и инструкций, приведенных в заявке WO 2006/071940, зарегистрированной 23 декабря 2005 года, включенной в виде ссылки, и используя общие способы синтеза, показанные на схемах ниже и в прилагаемых примерах.
Как показано на схеме 1, мочевины общей формулы 1 могут быть легко получены при взаимодействии аминов общей формулы 2 с изоцианатами 3 или заменителями изоцианатов 4 (трихлорэтилкарбаматами) или 5 (изопропенилкарбаматами). Предпочтительные условия для получения соединений общей формулы I включают нагревание раствора 4 или 5 с 2 в присутствии третичного основания, такого как диизопропилэтиламин, триэтиламин или N-метилпирролидин, в растворителе, таком как диметилформамид, диметилсульфоксид, тетрагидрофуран или 1,4-диоксан, при температуре от 50 до 100°C в течение периода времени в диапазоне от 1 часа до 2 суток.
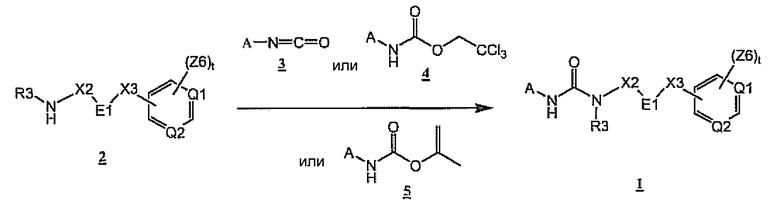
Схема 1
Как показано на схеме 2, изоцианаты 3 могут быть получены из аминов A-NH2 6 по реакции с фосгеном или эквивалентом фосгена, таким как дифосген, трифосген или N,N-дикарбонилимидазол. Трихлорэтилкарбаматы 4 и изопропенилкарбаматы 5 легко получают из аминов A-NH2 (6) в результате ацилирования трихлорэтилхлорформиатом или изопропенилхлорформиатом в стандартным условиях, известных специалистам в данной области. Предпочтительные условия для получения 4 и 5 включают обработку соединения 6 подходящим хлорформиатом в присутствии пиридина в апротонном растворителе, таком как дихлорметан, или в присутствии водного гидроксида или карбоната в двухфазной системе растворителей вода/этилацетат.
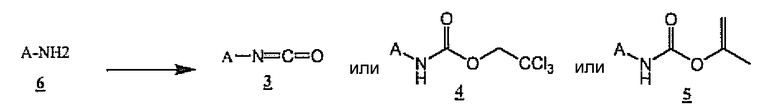
Схема 2
Кроме того, соединения формулы 1 также могут быть получены из карбоновых кислот 7 благодаря образованию in situ промежуточных ацилазидов (перегруппировка Курциуса), как показано на схеме 3. Предпочтительные условия реакции, показанной на схеме 3, включают смешивание кислоты 7 с амином 2 и дифенилфосфорилазидом в растворителе, таком как 1,4-диоксан или диметилформамид, в присутствии основания, такого как триэтиламин, и повышение температуры реакции примерно до 80-120°C, чтобы осуществить перегруппировку Курциуса
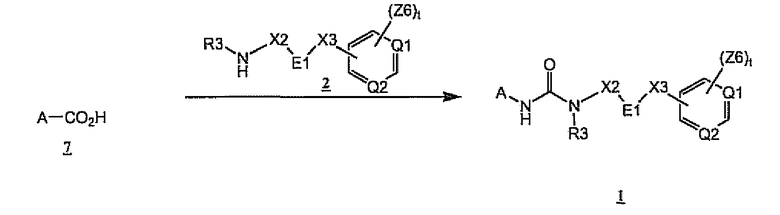
Схема 3
Существует множество способов получения аминов A-NH2 6 и кислот A-CO2H 7, зависящих от природы остатка A. Многие такие способы подробно описаны в публикации WO 2006/071940, которая включена в виде ссылки в данное описание. Предпочтительные способы синтеза изображены на следующих схемах в качестве неограничивающих примеров, где A означает 1-замещенный пиразол (необязательно замещенный R2) или A и A1 связаны C-C-связью.
Как показано на схеме 4, A1-замещенные пиразоламины 10 (предпочтительный аспект A-NH2 6, схема 2) получают в результате конденсации гидразинов 8 и бета-кетонитрилов 9. Предпочтительным условием для такого превращения является нагревание в этанольном растворе HCl. Гидразины 8, в свою очередь, могут быть получены диазотированием аминов 11 с последующим восстановлением или альтернативно в результате гидролиза гидразонов 13, полученных опосредованным палладием связыванием бензофенонгидразона с соединениями формулы A1-X 12, где X означает галоген или остаток трифлата.
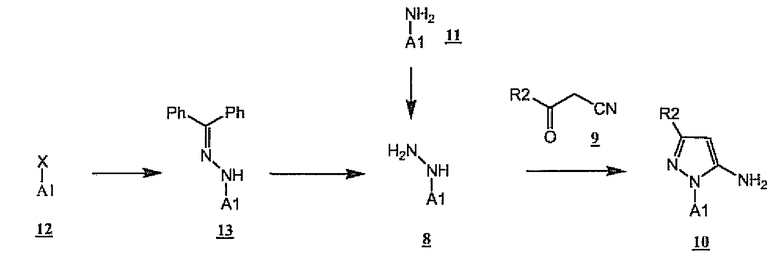
Схема 4
Неограничивающий пример реакции, изображенной на схеме 4, проиллюстрирован получением соединения 19 (схема 5 и прилагаемые примеры). Таким образом, коммерчески доступный 6-гидроксихинолин 14 может быть превращен в трифторметансульфонат 15 обработкой ангидридом трифторметансульфоновой кислоты и пиридином. Взаимодействие 15 с бензофенонгидразоном в присутствии палладиевого катализатора, предпочтительно катализатора, содержащего лиганд бис(дифенилфосфино)ферроцен, дает гидразон 16. Взаимодействие 16 с этанольным раствором HCl при кипячении с обратным холодильником дает гидразин 17, который можно объединить с кетонитрилами общей формулы 18 при дополнительном нагревании в этанольном растворе HCl, получая хинолинпиразоламины формулы 19. В другом аспекте указанной последовательности синтеза гидразон 16 может быть превращен непосредственно в пиразол 19 в результате прямого взаимодействия с кетонитрилом 18 при нагревании в этанольном растворе HCl.
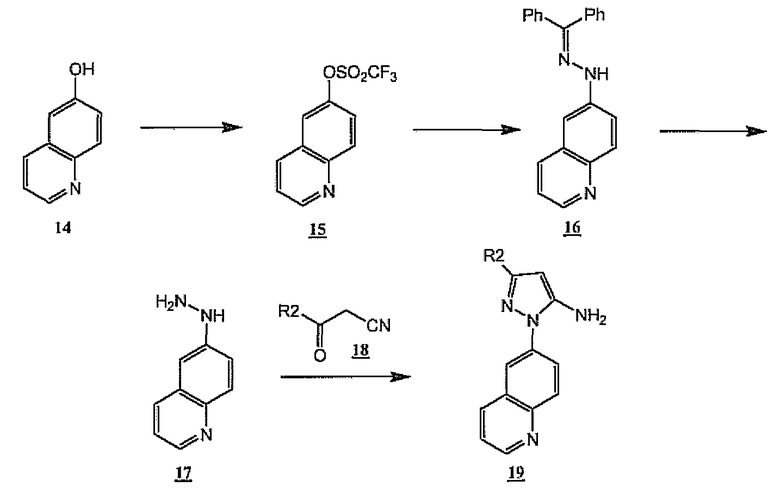
Схема 5
Другой предпочтительный способ конструирования A1-замещенных пиразолов проиллюстрирован общим способом получения кислоты пиразола 22 (схема 6), варианта A-CO2H 7 (схема 3). Как показано на схеме 6, объединение сложного эфира пиразол-5-карбоновой кислоты 20 с A1-X 12, где X означает галогенид, трифторметансульфонат или бороновую кислоту, подходящие для прямого катализируемого переходным металлом связывания с пиразолами 20, дает A1-замещенные сложные эфиры пиразола 21. Предпочтительные условия для таких превращений включают смешивание бороновой кислоты 11 [X-B(OH)2] и сложных эфиров 20 в дихлорметане с ацетатом меди и пиридином в присутствии измельченных молекулярных сит с нагреванием или без нагревания. Предпочтительные сложные эфиры для такого превращения включают этиловый, трет-бутиловый и бензиловый сложные эфиры. Сложные эфиры 21, в свою очередь, могут быть превращены в кислоты 22 в стандартных условиях, известных специалистам в данной области, таких как условия омыления, кислотного гидролиза или гидрирования.
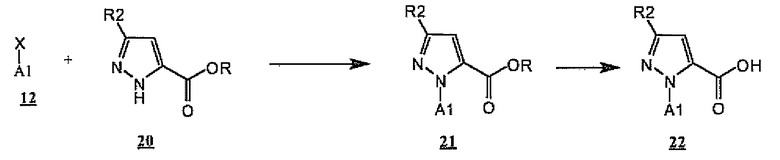
Схема 6
Синтез промежуточных продуктов, применимых для конструирования соединений формулы 1, где A и A1 связаны C-C-связью, показан на схеме 7. В данном случае катализируемые палладием реакции (например, реакции Сузуки или Стилле) A1-X 12 с дополнительным компонентом 23 или 24 дают соединения 25 или 26, примеры общих промежуточных продуктов A-NH2 6 или A-CO2H 7 соответственно. В такой последовательности реакций синтеза X-группы на реагентах 12 и 23 или 24 представляют собой остатки, которые подвергаются катализируемым переходными металлами реакциям кросс-сочетания, такие как остатки галогенидов или трифлатов и бороновых кислот или сложных эфиров, станнанов, силанов, цинкорганических или других металлоорганических остатков, известных специалистам в данной области в качестве подходящих субстратов для таких целей. X-группы на схеме 7 представляют собой взаимодействующие остатки для процессов кросс-сочетания, так что в том случае, когда A1-X 12 является галогенидом или трифлатом, A-X 23 или A-X 24 могут быть взаимодействующими металлоорганическими соединениями, такими как станнан или тому подобные или бороновая кислота или сложный эфир. Подобным образом, если A1-X 12 является металлоорганическим реагентом или бороновой кислотой или сложным эфиром, A-X может быть галогенидом или трифлатом.
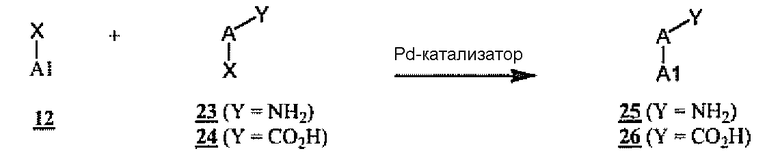
Схема 7
На схеме 7, как будет понятно специалистам в данной области, показано, что существуют дополнительные эквиваленты для Y-групп 23 и 24, которые могут быть использованы в синтезе взаимозаменяемо с NH2 и CO2H в случае добавления дополнительных стадий превращения. Например, Y-группа 23 также может представлять собой защищенную аминогруппу, например N-Boc, или заменитель аминогруппы, такой как нитрогруппа, которая может приводить к образованию соединений формулы 25 после кислотного гидролиза или восстановления соответственно. Подобным образом, будет понятно, что Y-группа 24 также может быть группой сложного эфира или нитрила, которая может быть гидролизована до кислоты формулы 26 с использованием стандартных способов синтеза.
Неограничивающий пример способа, изображенного на схеме 7, проиллюстрирован получением соединения 29, являющегося примером общего промежуточного продукта A-NH2 6, указанного выше. Таким образом, коммерчески доступную хинолин-6-бороновую кислоту 27 можно объединить с коммерчески доступным 5-фтор-2-йоданилином 28 в присутствии палладиевого катализатора с получением соединения 29, являющегося примером общего промежуточного продукта A-NH2 6, указанного выше.
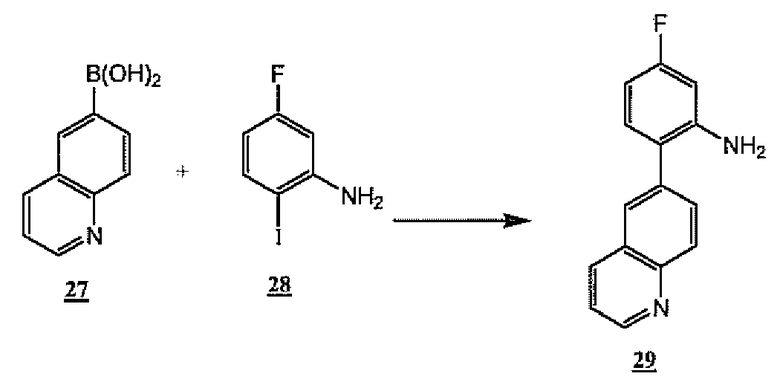
Схема 8
Амины 2 (схемы 1 и 3 выше), применимые для изобретения, могут быть синтезированы согласно способам, общеизвестным специалистам в данной области. Неограничивающие примеры проиллюстрированы на следующих далее схемах. Общее получение ариламина 32, примера амина 2, указанного выше, приведено на схеме 9. Таким образом, хлорпиридины формулы 31 подвергают взаимодействию с фенолами формулы 30 в присутствии основания, такого как трет-бутоксид калия. Реакции обычно проводят при температурах от 0°C до 150°C в растворителях, таких как диметилацетамид, диметилформамид или диметилсульфоксид. Некоторые неограничивающие примеры общего способа, изображенного на схеме 9, показаны на схемах 10-12 ниже.
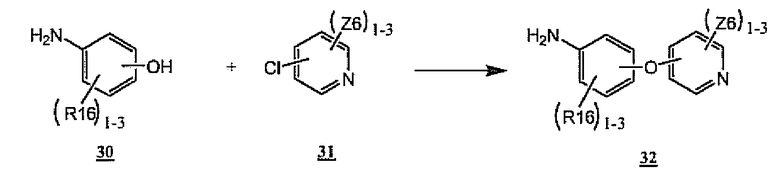
Схема 9
На схеме 10 коммерчески доступный 3-фтор-4-аминофенол подвергают взаимодействию с трет-бутоксидом калия и хлорпиридинами 34 или 35 с получением аминоэфиров 36 и 37 соответственно. Предпочтительным растворителем для такого превращения является диметилацетамид при температуре от 80 до 100°C.
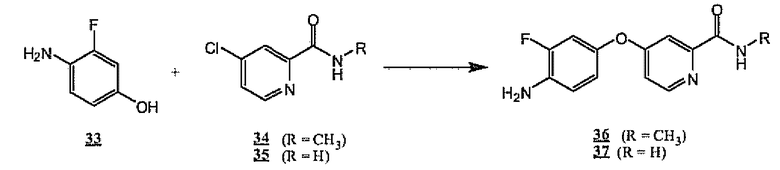
Схема 10
Подобным образом коммерчески доступный 2-метил-4-аминофенол 38 объединяют с хлорпиридинами 34 и 35, получая аминоэфиры 39 и 40 соответственно (схема 11).
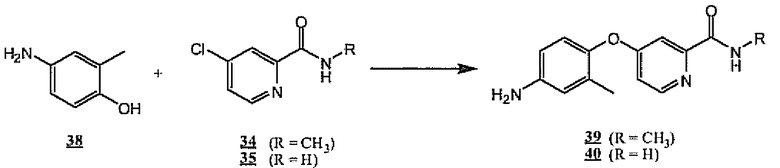
Схема 11
Схема 12 иллюстрирует получение метазамещенных пиридил- аминоэфиров 47 и 48, являющихся примерами общих промежуточных продуктов 2, указанных выше. Как показано на схеме 12, коммерчески доступный 2-хлор-4-фторфенол 41 обрабатывают метилхлорформиатом, получая карбонат 42. Затем нитрование в стандартных условиях дает аддукт 43. Гидролиз карбоната дает фенол 44. Сопутствующее восстановление обоих остатков нитрогруппы и хлора дает аминофенол 45. Последовательная обработка фенола 45 трет-бутоксидом калия и 3,5-дихлорпиридином и нагревание в диметилацетамиде дает соединение 47. Удаление атома хлора 47 гидрированием дает амин формулы 48, являющийся вариантом общего амина 2.
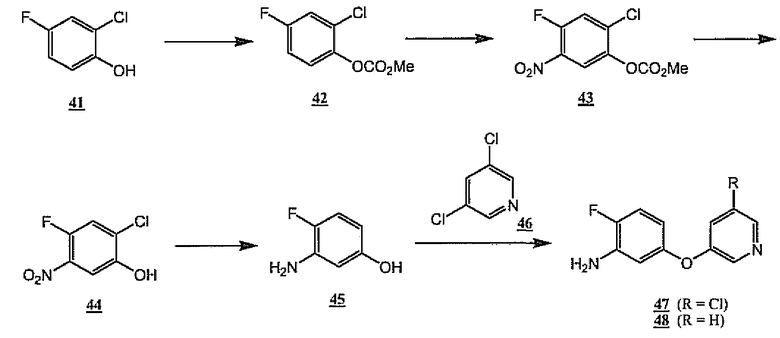
Схема 12
Амины общей формулы 2 также могут быть получены общим способом, показанным на схеме 13. Таким образом, галогенпиридин 49 (X означает галоген) или галогенпиримидин 50 (X означает галоген) может быть превращен в Z6-замещенный пиридин 51 или Z6-замещенный пиримидин 52 соответственно. Существует несколько способов для осуществления такого превращения, зависящих от природы Z6. Когда остаток Z6 связан с Q-содержащим циклом через атом азота Z6, предпочтительные способы включают нагревание соединений формулы 49 или 50 с избытком амина Z6-H, либо неразбавленным, либо в растворителе, таком как N-метилпирролидинон, ДМФА, ДМСО или спиртовой растворитель, при температурах в диапазоне от комнатной температуры до 200°C. В случае арил- и гетероариламинов Z6-H дополнительные предпочтительные способы включают нагревание соединений 49 или 50 с избытком амина Z6-H и кислотным катализатором (например, TsOH, HCl, HOAc или т.п.) в подходящем растворителе, таком как ДМФА, ДМСО или спиртовой растворитель. Дополнительные предпочтительные способы в случае арил- и гетероариламинов Z6-H включают объединение Z6-H с соединениями 49 или 50 в присутствии переходного металла в качестве катализатора, такого как палладиевый катализатор, в подходящем растворителе, подобном 1,4-диоксану или ДМФА, с нагреванием при необходимости. Когда остаток Z6 связан с Q-содержащим циклом через атом кислорода или атом серы Z6, предпочтительные способы включают нагревание 49-50 со спиртом или тиолом Z6-H в присутствии сильного основания (например, NaH или трет-бутоксид калия), либо неразбавленного, используя Z6-H в качестве растворителя, либо в полярном растворителе, таком как ДМФА или ДМСО, при температурах в диапазоне от комнатной температуры до 200°C. Когда остаток Z6 связан с Q-содержащим циклом через атом углерода Z6, предпочтительные способы включают осуществление контакта соединений 49 или 50 с соединением формулы Z6-M в присутствии палладиевого катализатора, где M означает вид группы, которая принимает участие в катализируемых переходными металлами реакциях кросс-сочетания. Примеры подходящих M-групп включают, без ограничения, бороновые кислоты, бороновые сложные эфиры, цинк, триалкилолово, силикон, магний, литий и алюминий. Необязательно превращения, показанные на схеме 13, могут быть осуществлены при нагревании в микроволновой печи. Специалистам в данной области будет понятно, что введенные остатки Z6, показанные на схеме 13, могут содержать необязательные защитные группы, которые могут быть удалены при последующих превращениях (не показано). Некоторые неограничивающие примеры способа, изображенного на схеме 13, показаны на схемах 14 и 15 ниже.
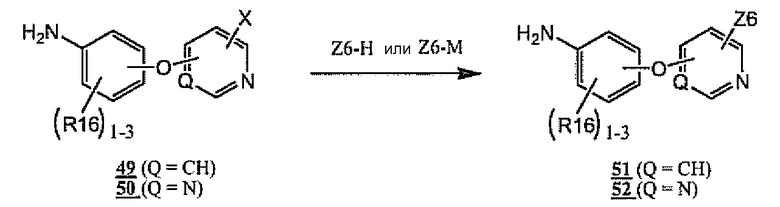
Схема 13
На схеме 14 фенол 33 и 2,4-дихлорпиридин (51) объединяют, используя общую схему 9, получая хлорпиридин 52. Дальнейшее взаимодействие хлорпиридина 52 с N-метилпиразолборонатом 53 в присутствии тетракис(трифенилфосфин)палладия дает 54, который является примером общего амина 2.
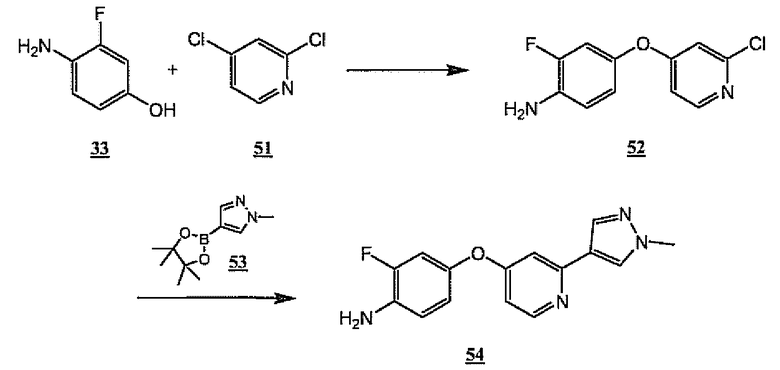
Схема 14
На схеме 15 показано получение аминопиридина 55 из хлорпиридина 52 общим способом, показанным на схеме 13. Предпочтительные условия для такого превращения включают осуществление контакта хлорпиридина 52 с изопропиламином в N-метилпирролидиноне с нагреванием в микроволновой печи.
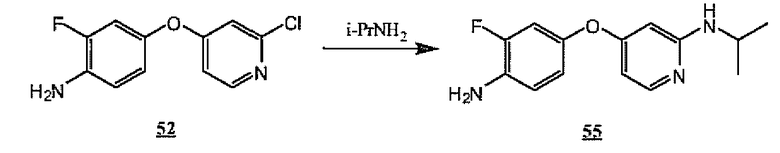
Схема 15
Схема 16 иллюстрирует альтернативное получение соединений общей формулы 1, представленное в виде получения мочевины 61. В том случае, когда общий амин 2 является первичным (R3 = H), амин 2 может быть превращен в изопропенилкарбамат 56, трихлорэтилкарбамат 57, или 4-нитрофенилкарбамат 58 в результате взаимодействия с изопропенилхлорформиатом, трихлорэтилхлорформиатом или 4-нитрофенилхлорформиатом соответственно. Альтернативно по аналогии со схемой 2 амин 2 (R3 = H) может быть превращен в отдельный изоцианат 59. По аналогии со схемой 1 взаимодействие карбаматов 56-58 или изоцианата 59 с R3-замещенным амином 60 дает мочевину 61, являющуюся примером соединения общей формулы 1.
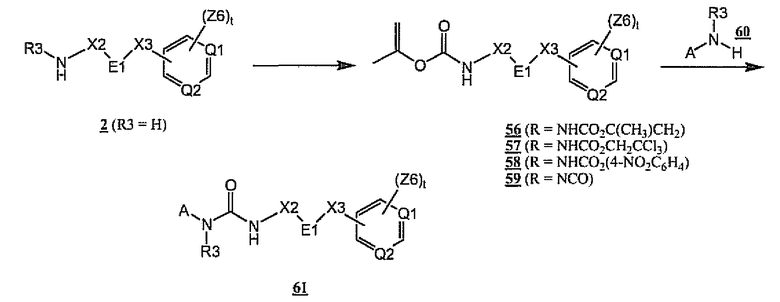
Схема 16
Дополнительная подгруппа мочевин общей формулы 1 может быть получена, как показано на схеме 17. В тех случаях, когда R3 не является H, монозамещенные мочевины 1 или 61 затем могут быть необязательно превращены в бис-R3-замещенные мочевины 62 (формула 1). Таким образом, как показано на схеме 17, воздействие на 1 или 61 алкилгалогенидами или циклоалкилгалогенидами в присутствии основания, например карбоната калия, гидрида натрия или трет-бутоксида калия, в подходящем растворителе, таком как ДМФА, дает мочевины 62, где вновь введенной группой R3 является алкил или циклоалкил. Альтернативно воздействие на мочевины 1 или 61 ацетатом меди(II) и Z3-замещенными фенилбороновыми кислотами [см. Chan et al., Tetrahedron Lett. 2003, 44, 3863-3865; Chan et al., Tetrahedron Lett. 1998, 39, 2933-2936; Chan, D.M.T. Tetrahedron Lett, 1996, 37, 9013-9016] дает аналогичные бис-R3-замещенные мочевины, в которых вновь введенной группой R3 является Z3-замещенный фенил.
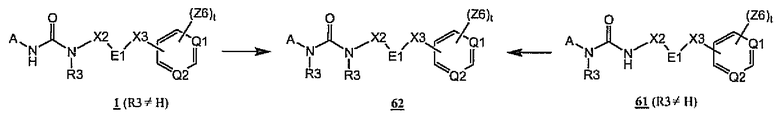
Схема 17
Общие амины A-NH2 (6), в которых A-цикл представляет собой изоксазол, могут быть получены способами, описанными на схеме 18. Многие примеры R2-замещенных аминоизоксазолов 64 и 65 являются коммерчески доступными. Они также могут быть получены из общих кетонитрильных промежуточных продуктов 63 в результате конденсации с гидроксиламином либо в кислых, либо в щелочных условиях, которые описаны в литературе (Takase, et al. Heterocycles, (1991), 32, pp.1153-1158). Бромирование изоксазолов 64 или 65 с использованием стандартных условий (см. Sircar, et al. J. Org. Chem. (1985), 50, pp.5723-7; Carr, et al. J. Med. Chem. (1977), 20, pp.934-9; Chan et al., US 5514691) дает бромизоксазолы 66 и 67 соответственно. По аналогии со схемами 7 и 8 соединения 66 и 67 могут быть превращены в A1-содержащие аминоизоксазолы 68 и 69, являющиеся примерами общего амина 6 и 25, в результате опосредованного палладием связывания с реагентами формулы A1-M (70), где остаток «M» в A1-M является остатком, который принимает участие в катализируемых переходными металлами реакциях кросс-сочетания, таким как остаток бороновой кислоты или боронового сложного эфира, станнана, силана, цинкорганический остаток или другой металлоорганический остаток, которые известны специалистам в данной области в качестве подходящего субстрата для таких целей. С использованием общих способов, показанных на схемах 1 и 2, амины 68 и 69 могут быть превращены в мочевины общей формулы 1. Специалистам в данной области будет понятно, что A1-остаток 68-70 может содержать защитные группы, которые могут быть удалены до или после превращения в мочевины формулы 1 в подходящих для удаления защиты условиях. Кроме того, может быть понятно, что аминогруппа в 64-69 необязательно может быть защищена подходящей защитной группой (такой как трет-бутилкарбамат), если требуется для проведения стадий бромирования или сочетания в присутствии палладия.
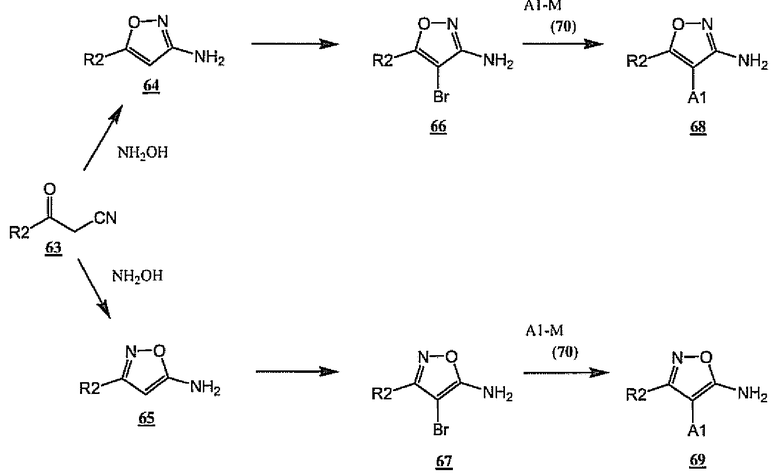
Схема 18
По аналогии со схемой 18 амины 73 и 74, являющиеся примерами общих аминов A-NH2 (6), в которых A-цикл представляет собой изотиазол, могут быть получены, как показано на схеме 19, в результате взаимодействия бромизотиазолов 71 и 72 и A1-M (70). Требуемые изотиазолы 71 и 72 могут быть получены способами, описанными в литературе (см. Hegde, V., WO 94/21647 (1994); Hackler, et. al. J. Heterocyclic Chem. (1989), 26, рр.1575-8). С использованием общих способов, показанных на схемах 1 и 2, амины 73 и 74 могут быть превращены в мочевины общей формулы 1.
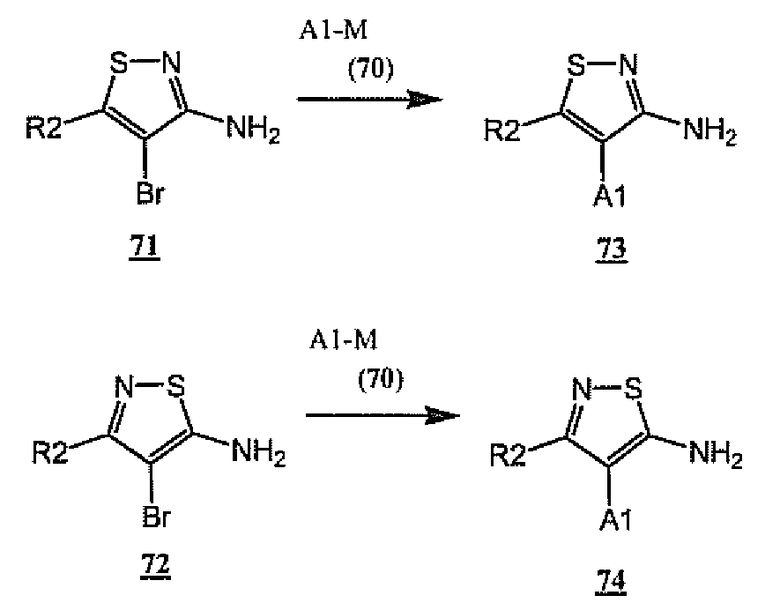
Схема 19
2.1. Примеры
Общий способ A: К перемешиваемому раствору карбоновой кислоты (0,50 ммоль, 1,00 экв.) и DPPA (0,75 ммоль, 1,50 экв.) в 1,4-диоксане (5,0 мл) при комнатной температуре добавляли Et3N (1,5 ммоль, 3,00 экв.). После перемешивания в течение 30 минут при комнатной температуре добавляли соответствующий амин (0,76 ммоль, 1,50 экв.) в диоксане и смесь нагревали при 95-100°C. Спустя 2 часа реакционную смесь после завершения реакции охлаждали до комнатной температуры, разбавляли насыщенным раствором соли и экстрагировали EtOAc (2×). Объединенные органические фазы промывали 3М HCl (1×), насыщенным раствором NaHCO3 (2×) и насыщенным раствором соли (1×), сушили (MgSO4), фильтровали и упаривали, получая сырой продукт, который очищали флэш-хроматографией на колонке, получая целевую мочевину.
Пример A1: 4-Амино-2-фторфенол (1,13 г, 8,9 ммоль) и соединение из примера A22 (1,5 г, 8,9 ммоль) объединяли способом согласно примеру A2, получая 4-(4-амино-2-фторфенокси)-N-метилпиколинамид (300 мг, выход 13%).
1H-ЯМР (ДМСО-d6) δ 8,78 (д, J=4,8 Гц, 1H), 8,47 (д, J=5,4 Гц, 1H), 7,32 (д, J=2,4 Гц, 1H), 7,11 (м, 1H), 7,01 (т, J=9,0 Гц, 1H), 6,51 (дд, J=13,2, 2,4 Гц, 1H), 6,42 (дд, J=8,4, 1,6 Гц, 1H), 5,51 (ушир.с, 2H), 2,76 (д, J=4,8 Гц, 3H); MC (ESI) m/z: 262,1 (М+H+).
Пример A2: Раствор 4-амино-3-фторфенола (2,00 г, 15,7 ммоль) в безводном DMA (32 мл) дегазировали, откачивая воздух из свободного пространства над раствором и снова заполняя аргоном (повторяли 3 раза). Раствор обрабатывали трет-бутоксидом калия (2,12 г, 18,9 ммоль) и полученную в результате смесь недолго обрабатывали ультразвуком, чтобы перевести все твердые вещества в объем растворителя, и перемешивали при комнатной температуре в течение 30 минут. Добавляли соединения из примера A22 (2,68 г, 15,7 ммоль). Реакционную смесь дегазировали второй раз и реакционную смесь нагревали до 100°C в течение ночи в атмосфере аргона. Реакционную смесь вливали в этилацетат (400 мл) и промывали водой (3×100 мл) и насыщенным раствором соли (2×100 мл). Объединенные водные фазы экстрагировали EtOAc (100 мл). Объединенные органические фазы сушили (MgSO4), концентрировали в вакууме до получения коричневого масла и очищали хроматографией на силикагеле, получая 4-(4-амино-3-фторфенокси)-N-метилпиколинамид (3,18 г, выход 77%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,76 (м, 1H), 8,48 (д, J=5,7 Гц, 1H), 7,36 (д, J=2,6 Гц, 1H), 7,10 (дд, J=5,7, 2,6 Гц, 1H), 7,02 (дд, J=11,8, 2,6 Гц, 1H), 6,86 (т, J=9,8 Гц, 1H), 6,79 (дд, J=8,9, 2,5 Гц, 1H), 5,23 (с, 2H), 2,79 (д, J=4,9 Гц, 3H); MC (ESI) m/z: 262,0 (М+H+).
Пример A3: В NMP (15 мл) помещали 3-амино-4-хлорфенол (1,70 г, 11,8 ммоль) и трет-бутоксид калия (1,40 г, 12,4 ммоль) и смесь перемешивали в течение ночи при комнатной температуре. Темный раствор обрабатывали 3,5-дифторпиридином (2,73 г, 23,7 ммоль) и порошкообразным карбонатом калия (818 мг, 5,92 ммоль) и затем смесь нагревали до 80°C и перемешивали в течение 24 часов. Полученную черную смесь охлаждали до комнатной температуры, разбавляли насыщенным раствором соли (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные этилацетатные экстракты промывали насыщенным раствором бикарбоната натрия (50 мл), водой (50 мл) и насыщенным раствором соли (50 мл), сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на колонке, получая 2-хлор-5-(5-фторпиридин-3-илокси)бензоламин в виде вязкого масла, которое использовали без дополнительной очистки.
1H-ЯМР (ДМСО-d6): δ 5,57 (ушир.с, 2H), 6,26-6,30 (дд, 1H), 6,50 (с, 1H), 7,19-7,22 (м, 1H), 7,45-7,50 (м, 1H), 8,26 (с, 1H), 8,39 (с, 1H); MC (ESI) m/z: 239,0 (М+H+).
Пример A4: Смесь соединения из примера A10 (4,6 г, 19,3 ммоль) и 10% Pd(OH)2/C (0,5 г, 0,35 ммоль) в EtOH (50 мл) перемешивали в атмосфере H2 при комнатной температуре в течение 3 часов. Смесь фильтровали через целит и промывали EtOH. Фильтрат концентрировали, получая 2-фтор-5-(пиридин-3-илокси)анилин (3,5 г, выход 88%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,53 (д, J=2,4 Гц, 1H), 8,48 (д, J=3,9 Гц, 1H), 7,80-7,69 (м, 2H), 7,05 (дд, J=11,1, 8,7 Гц, 1H), 6,53 (дд, J=7,5, 3,0 Гц, 1H), 6,28 (дт, J=8,7, 3,3 Гц, 1H); MC (ESI) m/z: 205,3 (М+H+).
Пример A5: К раствору 2,4-дифторфенола (2 г, 15,4 ммоль) в CH2Cl2 (20 мл) добавляли триэтиламин (3,21 мл, 23 ммоль) и этилхлорформиат (1,77 мл, 18,4 ммоль) при 0°C. После перемешивания смеси в течение 1 часа при комнатной температуре добавляли насыщенный раствор NaHCO3 раствор (30 мл), органический слой отделяли и водный слой экстрагировали CH2Cl2 (1×25 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали, получая 2,4-дифторфенилэтилкарбонат (3,11 г, выход 100%) в виде жидкости.
К раствору 2,4-дифторфенилэтилкарбоната (3,1 г, 16 ммоль) в серной кислоте (10 мл) медленно добавляли дымящую HNO3 (0,78 мл, 19 ммоль), поддерживая внутреннюю температуру около 0°C. Через 15 минут добавляли ледяную воду (70 мл), продукт экстрагировали этилацетатом (2×50 мл), объединенные органические слои промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали, получая нитропродукт в виде густого сиропа. Указанный нитропродукт растворяли в метаноле (20 мл) и к полученному раствору добавляли твердый NaHCO3 (4,0 г, 47 ммоль) и полученную смесь перемешивали в течение 16 часов при комнатной температуре. Смесь фильтровали и фильтрат концентрировали. Полученное твердое вещество растворяли в воде (20 мл) и подкисляли 3М раствором HCl до pH~5. Продукт экстрагировали CH2Cl2 (3×25 мл), объединенные органические слои промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали, получая 2,4-дифтор-5-нитрофенол (2,34 г, выход 84%).
1H ЯМР (400 МГц, ацетон-d6) δ 9,59 (с, 1H), 7,78 (т, J=7,2 Гц, 1H), 7,45 (т, J=10,4 Гц, 1H); MC (ESI) m/z: 176,0 (М+H+).
К суспензии 2,4-дифтор-5-нитрофенола (1,01 г, 5,77 ммоль) в EtOAc добавляли гидроксид палладия (0,08 г, 0,57 ммоль) и полученную взвесь перемешивали в атмосфере водорода в течение 6 часов. Смесь фильтровали через подушку целита, промывая EtOAc (2×10 мл) и фильтрат концентрировали, получая 5-амино-2,4-дифторфенол (0,8 г, выход 96%) в виде твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,28 (с, 1H), 6,91 (т, J=7,2 Гц, 1H), 6,35 (т, J=8,8 Гц, 1H), 4,84 (ушир.с, 2H); MC (ESI) m/z: 146,0 (М+H+).
К раствору 5-амино-2,4-дифторфенола (0,3 г, 2,07 ммоль) в ДМСО (2 мл) добавляли трет-бутоксид калия (0,23 г, 2,07 ммоль) при комнатной температуре. После перемешивания в течение 1 часа добавляли 3,5-дихлорпиридин (0,37 г, 2,5 ммоль) и карбонат калия (0,14 г, 1 ммоль) и смесь нагревали до 190°C в течение 1 часа в микроволновом реакторе. Добавляли воду (30 мл) и продукт экстрагировали EtOAc (2×35 мл) и объединенные органические слои промывали насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией (EtOAc/гексан), получая 5-(5-хлорпиридин-3-илокси)-2,4-дифторбензоламин (0,35 г, выход 66%) в виде твердого вещества.
1H ЯМР (400 МГц, ацетон-d6) δ 8,33-8,30 (м, 2H), 7,44 (т, J=2,4 Гц, 1H), 7,13 (т, J=10,8 Гц, 1H), 6,78 (т, J=8,4 Гц, 1H), 4,85 (ушир.с, 2H); MC (ESI) m/z: 257,0 (М+H+).
К раствору 5-(5-хлорпиридин-3-илокси)-2,4-дифторбензоламина (0,35 г, 1,4 ммоль) в 1М растворе HCl (10 мл) добавляли Pd/C (0,015 г) и смесь встряхивали в аппарате Парра в атмосфере водорода (40 psi, 0,279 МПа) в течение 24 часов. Смесь фильтровали через целит и фильтровальную подушку промывали водой (2×5 мл) и фильтрат концентрировали на лиофилизаторе, получая гидрохлорид. Полученное соединение нейтрализовали насыщенным водным раствором NaHCO3, свободный амин экстрагировали в EtOAc (2×35 мл) и объединенные органические слои промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали, получая 2,4-дифтор-5-(пиридин-3-илокси)бензоламин (0,19 г, выход 63%) в виде твердого вещества.
1H ЯМР (400 МГц, ацетон-d6) δ 8,33-8,30 (м, 2H), 7,37-7,29 (м, 2H), 7,09 (т, J=10,4 Гц, 1H), 6,70 (т, J=8,4 Гц, 1H), 4,78 (ушир.с, 2H); MC (ESI) m/z: 223,0 (М+H+).
Пример A6: Раствор 4-амино-о-крезола (0,301 г, 2,44 ммоль) в безводном диметилацетамиде (6 мл) дегазировали в вакууме и обрабатывали трет-бутоксидом калия (0,33 г, 2,93 ммоль) в атмосфере аргона. Реакционную смесь недолго обрабатывали ультразвуком, чтобы суспендировать все твердые вещества в объеме жидкости. Затем реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли соединение из примера A22 (0,417 г, 2,44 ммоль) и полученную смесь нагревали до 100°C в течение ночи. Охлажденную реакционную смесь распределяли между этилацетатом (50 мл) и водой (20 мл). Затем органический слой промывали водой (3×20 мл) и насыщенным раствором соли (2×20 мл). Объединенные водные фазы экстрагировали этилацетатом (2×20 мл). Объединенные органические фазы сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле (EtOAc/гексаны), получая 4-(4-амино-2-метилфенокси)-N-метилпиколинамид (530 мг, выход 84%) в виде желтой пены.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,75 (м, 1H), 8,45 (дд, J=4,6, 0,5 Гц, 1H), 7,27 (дд, J=2,6, 0,4 Гц, 1H), 7,04 (дд, J=5,5, 2,6 Гц, 1H), 6,78 (д, J=8,5 Гц, 1H), 6,53 (д, J=2,3 Гц, 1H), 6,48 (дд, J=8,6, 2,5 Гц, 1H), 5,10 (с, 2H), 2,78 (д, J=5,0 Гц, 3H), 1,93 (с, 3H); MC (ESI) m/z: 258,0 (М+H+).
Пример A7: Используя способ, аналогичный способу, описанному в примере A2, 4-амино-3-фторфенол (14 г, 0,11 ммоль) и соединение из примера A25 (16 г, 0,10 ммоль) объединяли, получая 4-(4-амино-3-фторфенокси)пиколинамид (8,8 г, выход 36%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,46 (д, J=5,7 Гц, 1H), 8,09 (ушир.с, 1H), 7,68 (ушир.с, 1H), 7,34 (д, J=2,4 Гц, 1H), 7,10 (дд, J=5,6, 2,6 Гц, 1H), 7,01 (дд, J=5,7, 2,4 Гц, 1H), 6,84 (т, J=9,0 Гц, 1H), 6,77 (дд, J=5,7, 2,4 Гц, 1H), 5,22 (с, 2H); MC (ESI) m/z: 248,1 (М+H+).
Пример A8: Раствор соединения из примера A23 (2,0 г, 8,4 ммоль) в 2-аминоэтаноле (6,0 мл) нагревали до 150°C в течение 3 часов. Растворитель удаляли при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем, получая 2-(4-(4-амино-3-фторфенокси)пиридин-2-иламино)этанол (1,2 г, выход 54%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,78 (д, J=5,6 Гц, 1H), 6,85 (дд, J=12,0, 2,4 Гц, 1H), 6,78 (т, J=8,8 Гц, 1H), 6,67 (дд, J=8,8, 2,0 Гц, 1H), 6,44 (т, J=5,2 Гц, 1H), 6,06 (дд, J=6,0, 2,4 Гц, 1H), 5,80 (д, J=2,0 Гц, 1H), 5,08 (с, 2H), 4,68 (ушир.с, 1H), 3,43 (м, 2H), 3,25-3,20 (м, 2H); MC (ESI) m/z: (М+H+) 264,1.
Пример A9: Раствор соединения из примера A23 (4,0 г, 16,8 ммоль) и N,O-диметилгидроксиламин·HCl (3,3 г, 34 ммоль) объединяли в 1,4-диоксане (50 мл) и реакционную смесь нагревали в течение ночи при 110°C. Реакционную смесь концентрировали в вакууме, нейтрализовали 3М NaOH и экстрагировали EtOAc (3×). Объединенные органические фазы промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали в вакууме, получая 4-(4-амино-3-фторфенокси)-N-метокси-N-метилпиридин-2-амин (4,4 г, выход 99%).
1H ЯМР (ДМСО-d6) δ 8,06 (д, J=5,2 Гц, 1H), 6,95 (дд, J=12,4, 2,8 Гц, 1H), 6,83 (дд, J=8,8, 8,4 Гц, 1H), 6,75 (дд, J=8,4, 2,4 Гц, 1H), 6,43 (д, J=2,4 Гц, 1H), 6,37 (дд, J=5,6, 2,4 Гц, 1H), 5,16 (с, 2H), 3,61 (с, 3H), 3,14 (с, 3H); MC (ESI) m/z: 264,2 (М+H+).
Смесь 2-фтор-4-(2-(метокси(метил)амино)пиридин-4-илокси)анилин (2,0 г, 7,6 ммоль) и 10% Pd/C (200 мг, 0,18 ммоль) в MeOH (15 мл) перемешивали в атмосфере H2 (50 psi, 0,349 МПа) при комнатной температуре в течение 48 часов. Смесь фильтровали через целит и осадок на фильтре промывали MeOH. Фильтрат концентрировали, получая 4-(4-амино-3-фторфенокси)-N-метилпиридин-2-амин (1,2 г, выход 68%).
1H ЯМР (ДМСО-d6) δ 7,86 (д, J=6,3 Гц, 1H), 6,82-6,69 (м, 3H), 6,18 (дд, J=6,0, 2,1 Гц, 1H), 5,84 (д, J=2,1 Гц, 1H), 5,41 (ушир.с, 1H), 3,62 (с, 2H), 2,84 (д, J=3,0 Гц, 3H); MC (ESI) m/z: 234,2 (М+H+).
Пример A10: Раствор соединения из примера A24 (0,95 г, 7,47 ммоль) и трет-бутоксида калия (0,92 г, 8,2 ммоль) в диметилацетамиде (2,0 мл) дегазировали в вакууме и снова заполняли N2 (4×) и затем перемешивали в течение 30 минут. Добавляли 3,5-дихлорпиридин и полученный раствор нагревали до 80°C в течение ночи. Смесь фильтровали и фильтрат концентрировали в вакууме и очищали хроматографией на силикагеле, получая 5-(5-хлорпиридин-3-илокси)-2-фторанилин (0,5 г, выход 28%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,37 (с, 1H), 8,29 (с, 1H), 7,51 (с, 1H), 7,00 (дд, J=10,8, 8,8 Гц, 1H), 6,46 (дд, J=7,6, 2,8 Гц, 1H), 6,22 (м, 1H), 5,38 (с, 2H); MC (ESI) m/z: 239,2 (М+H+).
Пример A11: Смесь соединения из примера A8 (0,263 г, 1,0 ммоль), имидазола (0,0749 г, 1,1 ммоль) и TBSCl (0,181 г, 1,2 ммоль) в ДМФА (10 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Остаток гасили H2O (10 мл) и pH доводили до ~8, используя NaHCO3. Водный раствор экстрагировали EtOAc (3×20 мл) и объединенные органические слои сушили (MgSO4), концентрировали в вакууме и очищали хроматографией, получая 4-(4-амино-3-фторфенокси)-N-(2-(трет-бутилдиметилсилилокси)этил)пиридин-2-амин (0,252 г, выход 67%) в виде светло-желтого масла. МС (ESI) m/z: 378,3 (M+H+).
Пример A12: К раствору соединения из примера A17 (7,5 г, 32,5 ммоль) в EtOH (60 мл) добавляли 1,0М водный раствор NaOH (10 мл, 100 ммоль). Полученную смесь нагревали при 85°C в течение ночи. Основную часть этанола удаляли в вакууме и концентрат разбавляли водой (50 мл) и промывали этилацетатом. Водный слой подкисляли до pH 1-2 добавлением 3М HCl. Кислый раствор экстрагировали EtOAc (3×200 мл) и экстракты промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали в вакууме, получая 5-(3-амино-4-фторфенокси)пиколиновую кислоту (6,2 г, выход 77%).
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,40 (д, J=2,7 Гц, 1H), 8,01 (д, J=8,4 Гц, 1H), 7,38 (дд, J=8,7, 2,7 Гц, 1H), 7,03 (дд, J=11,4, 8,7 Гц, 1H), 6,50 (дд, J=7,5, 3,0 Гц, 1H), 6,26 (м, 1H), 5,39 (ушир.с, 2H); MC (ESI) m/z: 249,1 (М+H+).
5-(3-амино-4-фторфенокси)пиколиновую кислоту (0,14 г, 0,56 ммоль) растворяли в ТГФ (3 мл) и перемешивали при 0°C в течение 5 мин. По каплям к реакционной смеси добавляли 1М раствор борана (3,4 мл) при 0°C в течение периода времени, составляющего 30 минут. Баню со льдом убирали и перемешивание продолжали при комнатной температуре в течение 7 часов. Реакционную смесь охлаждали на бане со льдом и обрабатывали 3М HCl (5 мл). Раствор нагревали в течение 1 часа при 50°C. Раствор промывали EtOAc (2×) и водный слой охлаждали на бане со льдом и нейтрализовали 3М NaOH. Раствор экстрагировали EtOAc (3×), объединенные органические слои промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали в вакууме, получая (5-(3-амино-4-фторфенокси)пиридин-2-ил)метанол (0,13 г, выход 98%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,24 (д, J=2,8 Гц, 1H), 7,46 (д, J=8,8 Гц, 1H), 7,40 (дд, J=2,8, 8,4 Гц, 1H), 6,99 (дд, J=8,8, 11,2 Гц, 1H), 6,40 (дд, J=2,8, 7,6 Гц, 1H), 6,15 (дт, J=3,2, 8,8 Гц, 1H), 5,40 (т, J=5,6 Гц, 1H), 5,33 (с, 2H), 4,54 (д, J=6,0 Гц, 2H); MC (ESI) m/z: 235,0 (М+H+).
Пример A13: NaH (100 мг, 3,3 ммоль) медленно добавляли к раствору соединения из примера A12 (0,50 г, 2,1 ммоль) в безводном ТГФ (50 мл) при 0°C. Через 30 минут добавляли CS2 (0,49 г, 6,4 ммоль) и реакционную смесь перемешивали при 0°C в течение 1 часа. Добавляли метилиодид (2,4 г, 17 ммоль) при 0°C и реакционной смеси давали возможность нагреться до комнатной температуры в течение ночи. Растворитель удаляли при пониженном давлении, получая сырой продукт. Неочищенный O-(5-(3-амино-4-фторфенокси)пиридин-2-ил)метил-S-метилкарбонодитиоат (0,69 г, 2,1 ммоль) растворяли в толуоле (5 мл) и добавляли гидрид трибутилолова (1 мл) и AIBN (50 мг). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Растворитель удаляли при пониженном давлении и остаток фильтровали и промывали CH2Cl2. Фильтрат упаривали и остаток очищали хроматографией на колонке с силикагелем, получая 2-фтор-5-(6-метилпиридин-3-илокси)бензоламин (0,26 г, выход 56%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,20 (д, J=2,8 Гц, 1H), 7,30 (дд, J=2,8 и 8,4 Гц, 1H), 7,25 (д, J=8,4 Гц, 1H), 6,97 (дд, J=8,8, 11,6 Гц, 1H), 6,38 (дд, J=3,2, 7,6 Гц, 1H), 6,13 (дт, J=3,2, 8,8 Гц, 1H), 5,31 (с, 1H), 2,44 (с, 3H); MC (ESI) m/z: 219,0 (М+H+).
Пример A14: Раствор 4-амино-3-фторфенола (0,20 г, 1,6 ммоль) в 4 мл безводного DMA обрабатывали трет-бутоксидом калия (0,24 г, 1,9 ммоль). Полученный темно-красный раствор перемешивали при комнатной температуре в течение 1 часа в закрытом крышкой флаконе. Добавляли 4-хлор-2-метоксипиридин (0,26 г, 1,6 ммоль) и реакционную смесь нагревали в течение ночи при 100°C. Добавляли воду (50 мл) и раствор экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на колонке с силикагелем, получая 2-фтор-4-(2-метоксипиридин-4-илокси)бензоламин (0,20 г, выход 58%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,02 (д, J=6,0 Гц, 1H), 6,95 (дд, J=2,8, 12,0 Гц, 1H), 6,82 (дд, J=8,4, 8,8 Гц, 1H), 6,73 (дд, J=2,0, 8,4 Гц, 1H), 6,54 (дд, J=2,4, 6,0 Гц, 1H), 6,10 (д, J=2,4 Гц, 1H), 5,17 (с, 1H), 3,81 (с, 3H); MC (ESI) m/z: 235,0 (М+H+).
Пример A15: В закрытый тефлоновой крышкой флакон загружали 4-амино-3-фторфенол (0,291 г, 2,29 ммоль) и безводный ДМФА (2,3 мл). Полученный раствор дегазировали в вакууме и снова заполняли аргоном (3×). Содержимое флакона обрабатывали трет-бутоксидом натрия (0,27 г, 2,41 ммоль) в атмосфере аргона и быстро закрывали крышкой. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После добавления 4-хлорпиколинонитрила (0,317 г, 2,29 ммоль) и K2CO3 (0,174 г, 1,26 ммоль) флакон снова дегазировали и нагревали на масляной бане при 90°C в течение ночи. Реакционную смесь разбавляли EtOAc (60 мл) и промывали насыщенным раствором соли (25 мл). Водную фазу снова экстрагировали EtOAc (50 мл). Объединенные органические слои промывали насыщенным раствором соли (25 мл), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией, получая 4-(4-амино-3-фторфенокси)пиколинонитрил (0,162 г, выход 31%) в виде бесцветного масла.
1H ЯМР (ДМСО-d6) δ 8,56 (д, J=5,6 Гц, 1H), 7,62 (д, J=2,0 Гц, 1H), 7,14 (дд, J=6,0, 2,8 Гц, 1H), 7,03 (дд, J=11,6, 2,4 Гц, 1H), 6,88-6,77 (м, 2H), 5,25 (с, 2H); MC (ESI) m/z: 230,0 (М+H+).
Пример A16: Раствор 5-амино-2-хлор-4-фторфенола (100 мг, 0,619 ммоль) в дегазированном диметилацетамиде (2 мл) обрабатывали трет-бутоксидом калия (83 мг, 0,743 ммоль) и 5-хлор-2-цианопиридином (86 мг, 0,619 ммоль). Полученную смесь нагревали до 80°C в течение ночи, затем охлаждали до комнатной температуры и разбавляли водой (10 мл). Смесь экстрагировали EtOAc (30 мл). Органическую фазу промывали водой (3×30 мл) и насыщенным раствором соли (30 мл), сушили (Na2SO4) и концентрировали в вакууме, получая 5-(5-амино-2-хлор-4-фторфенокси)пиколинонитрил в виде темного масла, которое использовали без дополнительной очистки. МС (ESI) m/z: 264,0 (M+H+).
Пример A17: Раствор 3-амино-4-фторфенол (5,6 г, 44 ммоль) в диметилацетамиде (60 мл) дегазировали в вакууме и обрабатывали трет-бутоксидом калия (5,3 г, 47 ммоль). Полученный раствор перемешивали в течение 30 минут. 5-Бромпиридин-2-карбонитрил (6,6 г, 36 ммоль) добавляли одной порцией и смесь нагревали при 80°C в течение ночи. Растворитель удаляли в вакууме и остаток очищали хроматографией на силикагеле, получая 5-(3-амино-4-фторфенокси)пиколинонитрил (3,5 г, выход 44%).
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,47 (д, J=3,0 Гц, 1H), 7,98 (д, J=8,4 Гц, 1H), 7,44 (дд, J=8,8, 2,7 Гц, 1H), 7,06 (т, J=9,2 Гц, 1H), 6,52 (д, J=7,6 Гц, 1H), 6,28 (м, 1H), 5,44 (ушир.с, 2H); MC (ESI) m/z: 230,0 (М+H+).
Пример A18: В DMA (10 мл) помещали 3-амино-4-фторфенол (500 мг, 3,93 ммоль), трет-бутоксид калия (441 мг, 3,93 ммоль) и 4-хлор-2-(метилтио)пиримидин (632 мг, 3,93 ммоль). Смесь нагревали до 50°C и перемешивали в течение ночи. Смесь охлаждали до комнатной температуры и разбавляли водой (30 мл), экстрагировали этилацетатом (2×25 мл) и объединенные органические фазы промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали, получая темное масло. Масло очищали хроматографией на колонке, получая 2-фтор-5-(2-(метилтио)пиримидин-4-илокси)бензоламин (841 мг, выход 85%) в виде масла, которое использовали без дополнительной очистки. МС (ESI) m/z: 252,0 (M+H+).
Пример A19: Раствор пиридин-3-бороновой кислоты (0,68 г, 5,5 ммоль) и 2-метил-5-нитрофенола (0,85 г, 5,5 ммоль) в ДХМ (10 мл) обрабатывали пиридином (1,00 мл, 12,4 ммоль), ацетатом меди (1,5 г, 8,3 ммоль) и порошкообразными молекулярными ситами 4Å (330 мг). Реакционную смесь перемешивали в течение 7 дней при комнатной температуре на открытом воздухе. Смесь вливали в воду (50 мл) и экстрагировали ДХМ (2×50 мл). Объединенные органические фазы промывали насыщенным водным раствором NaHCO3 (25 мл), водой (25 мл), насыщенным раствором NH4Cl (2×25 мл) и насыщенным раствором соли (25 мл), сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая 3-(2-метил-5-нитрофенокси)пиридин (81 мг, выход 6%).
1H ЯМР (400 МГц, CDCl3) δ 8,48 (дд, J=4,6, 1,0 Гц, 1H), 8,43 (д, J=2,4 Гц, 1H), 7,99 (дд, J=8,0, 2,0 Гц, 1H), 7,70 (д, J=2,4 Гц, 1H), 7,46 (д, J=8,4 Гц, 1H), 7,39-7,30 (м, 2H), 2,42 (с, 3H); MC (ESI) m/z: 231,0 (М+H+).
Раствор 3-(2-метил-5-нитрофенокси)пиридина (80 мг, 0,35 ммоль) и 10% Pd/C (50% wet, 165 мг, 0,08 ммоль) в метаноле (4 мл) обрабатывали муравьиной кислотой (89%, 1 мл, 35 ммоль) и полученный раствор перемешивали при комнатной температуре. Через 1 час реакционную смесь фильтровали через целит и осадок на фильтре промывали метанолом. Фильтраты концентрировали в вакууме, разбавляли 40 мл водного раствора с pH 12 и экстрагировали этилацетатом (3×25 мл). Экстракты сушили (Na2SO4) и концентрировали в вакууме, получая 4-метил-3-(пиридин-3-илокси)бензоламин (58 мг, выход 83%).
1H ЯМР (400 МГц, CDCl3) δ 8,36 (м, 2H), 8,32 (дд, J=4,6, 1,4 Гц, 1H), 7,26-7,18 (м, 3H), 7,05 (д, J=8,0 Гц, 1H), 6,49 (дд, J=8,8, 2,4 Гц, 1H), 6,29 (д, J=2,4 Гц, 1H), 2,11 (с, 3H); MC (ESI) m/z: 201,0 (М+H+).
Пример A20: В DMA (8 мл) помещали 3-амино-4-фторфенол (281 мг, 2,21 ммоль), трет-бутоксид калия (248 мг, 2,21 ммоль) и 5-бром-2-(трифторметил)пиридин (500 мг, 2,21 ммоль). Смесь нагревали до 75°C в течение ночи, затем охлаждали до комнатной температуры и разбавляли водой (75 мл). Смесь экстрагировали этилацетатом (2×40 мл) и объединенные органические фазы промывали насыщенным раствором соли (40 мл), сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на колонке, получая 2-фтор-5-(6-(трифторметил)пиридин-3-илокси)бензоламин (161 мг, выход 26%) в виде масла, которое использовали без дополнительной очистки. МС (ESI) m/z: 273,0 (M+H+).
Пример A21: В ДМФА (5 мл) помещали 5-(3-амино-4-фторфенокси)пиколиновую кислоту из примера A12 (500 мг, 2,01 ммоль), 2,0М раствор метиламина/ТГФ (10 мл, 20,1 ммоль) и HOBt (324 мг, 2,12 ммоль). К смеси добавляли гидрохлорид N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (772 мг, 4,03 ммоль) и раствор перемешивали в течение ночи при комнатной температуре. Раствор обрабатывали, дополнительно добавляя эквивалент гидрохлорида N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (775 мг), и нагревали до 40°C, затем охлаждали до комнатной температуры и перемешивали в течение ночи. Раствор разбавляли этилацетатом (30 мл) и промывали водой (30 мл), насыщенным раствором соли (30 мл), сушили (Na2SO4) и концентрировали в вакууме, получая 5-(3-амино-4-фторфенокси)-N-метилпиколинамид (530 мг, выход 101%) в виде густого масла, которое использовали без дополнительной очистки. МС (ESI) m/z: 262,0 (M+H+).
Пример A22: К перемешиваемому безводному ДМФА (25 мл) медленно добавляли SOCl2 (125 мл) с такой скоростью, при которой температура реакции поддерживалась при 40-50°C. Порциями в течение 30 минут добавляли пиридин-2-карбоновую кислоту (25 г, 0,2 моль) и полученную смесь кипятили с обратным холодильником в течение 16 часов, за это время происходило осаждение желтого твердого вещества. После охлаждения до комнатной температуры смесь разбавляли толуолом (80 мл) и концентрировали. Указанный процесс повторяли три раза. Полученный сухой остаток промывали толуолом и сушили при пониженном давлении, получая 4-хлорпиридин-2-карбонилхлорид (27,6 г, выход 79%), который использовали на следующей стадии без очистки.
К раствору 4-хлорпиридин-2-карбонилхлорида (27,6 г, 0,16 моль) в безводном ТГФ (100 мл) при 0°C по каплям добавляли раствор MeNH2 в EtOH. Полученную смесь перемешивали при 3°C в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении, получая твердое вещество, которое суспендировали в EtOAc и фильтровали. Фильтрат промывали насыщенным раствором соли (2×100 мл), сушили и концентрировали, получая 4-хлор-N-метилпиколинамид (16,4 г, выход 60%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,78 (ушир.с, 1H), 8,55 (д, J=5,2 Гц, 1H), 7,97 (д, J=2,0 Гц, 1H), 7,66 (м, 1H), 2,82 (д, J=4,8 Гц, 3H); MC (ESI) m/z: 171,0 (М+H+).
Пример A23: Используя способ, аналогичный способу, описанному в примере A2, 2,4-дихлорпиридин (8,0 г, 54 ммоль) и 3-фтор-4-аминофенол (8,0 г, 62,9 ммоль) объединяли, получая 4-(2-хлорпиридин-4-илокси)-2-фторфениламин (11 г, выход 86%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,24 (д, J=5,7 Гц, 1H), 7,00 (дд, J=9,0, 2,7 Гц, 1H), 6,89-6,73 (м, 4H), 5,21 (ушир.с, 2H); MC (ESI) m/z: 239,2 (М+H+).
Пример A24: Метилхлорформиат (77,3 г, 0,82 моль) добавляли по каплям к охлажденному до -10°C раствору 2-хлор-4-фторфенола (100 г, 0,68 моль) и гидроксида натрия (32,8 г, 0,82 моль) в воде (550 мл). После завершения добавления осажденное твердое вещество собирали фильтрованием и промывали водой, получая 2-хлор-4-фторфенилметилкарбонат (110 г, выход 79%).
1H ЯМР (300 МГц, ДМСО-d6) δ 7,62 (дд, J=8,1, 2,7 Гц, 1H), 7,50 (дд, J=9,0, 5,4 Гц, 1H), 7,30 (тд, J=8,1, 3,0 Гц, 1H), 3,86 (с, 3H); MC (ESI) m/z: 205,2 (М+H+).
К суспензии 2-хлор-4-фторфенилметилкарбоната (110 г, 0,54 моль) в концентрированной H2SO4 (50 мл) медленно добавляли смесь, состоящую из концентрированной H2SO4 (40 мл) и дымящей HNO3 (40,8 мл, 0,89 моль). Полученную смесь перемешивали в течение 30 минут при 0°C. Реакционную смесь вливали в ледяную воду и осажденное твердое вещество собирали фильтрованием и промывали водой, получая 2-хлор-4-фтор-5-нитрофенилметилкарбонат (120 г, выход 90%).
1H ЯМР (400 МГц, ДМСО-d6): δ 8,45 (д, J=7,2 Гц, 1H), 8,12 (д, J=10,8 Гц, 1H), 3,89 (с, 3H); MC (ESI) m/z: 250,1 (М+H+).
Смесь 2-хлор-4-фтор-5-нитрофенилметилкарбоната (120 г, 0,48 моль) и гидроксида натрия (22,7 г, 0,57 моль) в воде (300 мл) кипятили с обратным холодильником в течение 4 часов. Нерастворимые твердые вещества удаляли фильтрованием и фильтрат подкисляли разбавленной HCl. Осажденное твердое вещество собирали фильтрованием и промывали водой, получая 2-хлор-4-фтор-5-нитрофенол (90 г, выход 98%).
1H ЯМР (400 МГц, ДМСО-d6) δ 11,18 (с, 1H), 8,10 (д, J=10,4 Гц, 1H), 7,62 (д, J=7,2 Гц, 1H); MC (ESI) m/z: 192,1 (М+H+).
2-Хлор-4-фтор-5-нитрофенол (85 г, 0,45 моль) и 10% Pd/C (25 г, 0,023 моль) объединяли в EtOH и гидрировали (50 psi, 0,349 МПа) в течение 12 часов. Реакционную смесь фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле, получая 3-амино-4-фторфенол (40 г, выход 70%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,87 (с, 1H), 6,70 (дд, J=11,2, 8,8 Гц, 1H), 6,14 (дд, J=7,8, 2,4 Гц, 1H), 5,84 (м, 1H), 4,92 (с, 2H); MC (ESI) m/z: 128,2 (М+H+).
Пример A25: 4-Хлорпиколинамид получали, используя способ, аналогичный способу, описанному в примере A22, заменяя MeNH2 на NH3.
1H ЯМР (300 МГц, ДМСО-d6) δ 8,59 (д, J=5,2 Гц, 1H), 8,18 (ушир.с, 1H), 8,00 (д, J=2,0 Гц, 1H), 7,79 (ушир.с, 1H), 7,72 (дд, J=5,2, 2,0 Гц, 1H); MC (ESI) m/z: 157,0 (М+H+).
Пример A26: Используя способ, аналогичный способу, описанному в примере A2, 2-фтор-4-аминофенол (2,6 г, 24 ммоль) и 2,4-дихлорпиридин (2,88 г, 20 моль) объединяли, получая 4-(2-хлорпиридин-4-илокси)-3-фторфениламин (3,2 г, выход 67%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,25 (д, J=5,6 Гц, 1H), 6,99 (м, 1H), 6,90 (м, 2H), 6,50 (д, J=1,6 Гц, 1H), 6,41 (д, J=10,4 Гц, 1H), 5,51 (с, 2H); MC (ESI) m/z: 239,1 (М+H+).
Смесь 4-(2-хлорпиридин-4-илокси)-3-фторфениламина (2,0 г, 8,4 ммоль) и бензилметиламина (20 мл) нагревали до 200°C в течение ночи в стальном баллоне. Реакционную смесь концентрировали в вакууме и очищали хроматографией на силикагеле, получая 4-(4-амино-2-фторфенокси)-N-бензил-N-метилпиридин-2-амин (1,0 г, выход 37%). МС (ESI) m/z: 324,2 (M+H+).
К раствору 4-(4-амино-2-фторфенокси)-N-бензил-N-метилпиридин-2-амина (1,0 г, 3,1 ммоль) в MeOH (10 мл) добавляли 10% Pd/C (0,25 г, 0,23 ммоль). Реакционную смесь перемешивали в атмосфере H2 (50 psi, 0,349 МПа) при 75°C в течение 12 часов. Реакционную смесь фильтровали, концентрировали при пониженном давлении и очищали обращенно-фазовой препаративной ВЭЖХ, получая 4-(4-амино-2-фторфенокси)-N-метилпиридин-2-амин (560 мг, выход 78%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,80 (д, J=5,6 Гц, 1H), 6,90 (т, J=9,0 Гц, 1H), 6,40-6,45 (м, 3H), 6,06 (дд, J=8,0, 2,8 Гц, 1H), 5,73 (д, J=2,8 Гц, 1H), 5,37 (с, 2H), 2,68 (д, J=4,8 Гц, 3H); MC (ESI) m/z: (М+H+): 234,2.
Пример A27: Соединение из примера A23 (0,597 г, 2,5 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (0,728 г, 3,75 ммоль), Cs2CO3 (3,10 г, 9,5 ммоль) и Pd(PPh3)4 (0,289 г, 0,25 ммоль) объединяли в ДМФА/H2O (20 мл). Реакционную смесь дегазировали, покрывали N2 и нагревали при 90°C в течение ночи. После завершения реакции смесь разбавляли H2O (5 мл) и экстрагировали EtOAc (3×50 мл). Объединенные органические фазы промывали насыщенным раствором соли (20 мл), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией, получая 4-(2-(1H-пиразол-4-ил)пиридин-4-илокси)-2-фторбензоламин (0,56 г, 83%) в виде светло-желтого твердого вещества.
1H ЯМР (400 Гц, ДМСО-d6) δ 13,01 (с, 1H), 8,38 (д, J=5,6 Гц, 1H), 8,35 (с, 1H), 8,06 (с, 1H), 7,29 (д, J=2,4 Гц, 1H), 7,03 (дд, J=11,6, 2,4 Гц, 1H), 6,89 (т, J=8,8 Гц, 1H), 6,84 (м, J=8,4 Гц, 1H), 6,60 (м, 1H), 5,20 (с, 2H); MC (ESI) m/z: 271,0 (М+H+).
Пример A28: Раствор соединения из примера A23 (3 г, 12,6 ммоль), 1-метил-3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-пиразола (5,2 г, 25,2 ммоль) и Na2CO3 (2,7 г, 25,2 ммоль) в DME (18 мл) и воде (6 мл) барботировали азотом в течение 20 минут. Добавляли Pd(PPh3)4 (729 мг, 0,63 ммоль) и полученную смесь нагревали до 100°C в течение 16 часов. Растворитель удаляли при пониженном давлении и сырой продукт суспендировали в воде и экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая 2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)анилин (2 г, выход 56%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,31 (д, J=5,7 Гц, 1H), 8,21 (с, 1H), 7,92 (с, 1H), 7,12 (с, J=2,4 Гц, 1H), 6,96 (м, 1H), 6,85-6,72 (м, 2H), 6,56 (м, 1H), 5,15 (с, 2H), 3,84 (с, 3H); MC (ESI) m/z: 285,0 (М+H+).
Пример A29: По аналогии с примером A2 4-амино-3-фторфенол (0,12 г, 0,53 ммоль), трет-бутоксид калия (0,080 г, 0,71 ммоль) и трет-бутил-4-хлорпиколинат (159 мг, 0,53 ммоль) объединяли, получая трет-бутил-4-(4-амино-3-фторфенокси)пиколинат (151 мг, выход 67%). МС (ESI) m/z: 305,0 (M+H+).
К раствору LiAlH4 (699 мг, 18,4 ммоль) в ТГФ (15 мл) добавляли трет-бутил-4-(4-амино-3-фторфенокси)пиколинат (1,4 г, 4,6 ммоль) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 2 часов. Реакционную смесь гасили 10% водным раствором NaOH (4 мл), полученную суспензию фильтровали и фильтрат экстрагировали EtOAc (3×30 мл), получая (4-(4-амино-3-фторфенокси)пиридин-2-ил)метанол (700 мг, выход 70%). МС (ESI) m/z: 235,1 (M+H+).
Раствор (4-(4-амино-3-фторфенокси)пиридин-2-ил)метанола (750 мг, 3,2 ммоль) и Et3N (821 мг, 8 ммоль) в ДМФА (10 мл) при 0°C обрабатывали трет-бутилдиметилсилилхлоридом (624 мг, 4,16 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 4 часов. Растворитель удаляли в вакууме и остаток очищали хроматографией на колонке с силикагелем, получая 4-(2-((трет-бутилдиметилсилилокси)метил)пиридин-4-илокси)-2-фторбензоламин (370 мг, выход 33%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,32 (д, J=5,6 Гц, 1H), 7,02 (с, 1H), 6,67-6,82 (м, 4H), 4,76 (с, 2H), 3,71 (с, 2H), 0,89 (с, 9H), 0,07 (с, 6H); MC (ESI) m/z: 349,2 (М+H+).
Пример A30: Соединение из примера A23 (1 г, 4,2 ммоль) и этил(4-метоксибензил)амин (10 мл) объединяли и нагревали до 200°C в течение 30 часов. Реакционный раствор вливали в смесь HOAc/вода (20%, об./об.) и экстрагировали EtOAc (3×100 мл). Объединенные органические фазы промывали насыщенным раствором соли (3×50 мл) и насыщенным раствором NaHCO3 (2×100 мл), сушили (NaSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая [4-(4-амино-3-фторфенокси)пиридин-2-ил]этил-(4-метоксибензил)амин (1,2 г, выход 78%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,90 (д, J=5,6 Гц, 1H), 7,07 (д, J=8,4 Гц, 2H), 6,82 (д, J=8,0 Гц, 2H), 6,74 (м, 2H), 6,63 (д, J=7,2 Гц, 1H), 6,02 (д, J=4,0 Гц, 1H), 5,90 (с, 1H), 5,09 (с, 2H), 4,53 (с, 2H), 3,67 (с, 3H), 3,44 (м, 2H), 1,00 (т, J=6,8 Гц, 3H); MC (ESI) m/z: 368,2 (М+H+).
Трифторуксусную кислоту (10 мл) добавляли к раствору [4-(4-амино-3-фторфенокси)пиридин-2-ил]этил-(4-метоксибензил)амина (1,2 г, 3,27 ммоль) в CH2Cl2 (50 мл) и полученный раствор нагревали до 40°C в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток обрабатывали HCl (5 мл, 12М, 60 ммоль) и водой (50 мл). Раствор промывали EtOAc (4×50 мл). Водный слой обрабатывали NaHCO3 вплоть до pH 8 и затем экстрагировали EtOAc (3×50 мл). Объединенные экстракты промывали насыщенным раствором соли (3×50 мл), сушили (NaSO4) и концентрировали в вакууме, получая 4-(4-амино-3-фторфенокси)-N-этилпиридин-2-амин (0,45 г, выход 67%).
1H ЯМР (300 МГц, ДМСО-d6) δ 7,79 (д, J=5,7 Гц, 1H), 6,85 (дд, J=11,7, 2,4 Гц, 1H), 6,78 (т, J=8,7 Гц, 1H), 6,67 (дд, J=8,7, 2,4 Гц, 1H), 6,39 (м, 1H), 6,05 (дд, J=5,7, 2,1 Гц, 1H), 5,72 (д, J=2,1 Гц, 1H), 5,09 (с, 2H), 3,15 (м, 2H), 1,03 (т, J=7,2 Гц, 3H); MC (ESI) m/z: 248,2 (М+H+).
Пример A31: К раствору соединения из примера A23 (0,30 г, 1,3 ммоль) в NMP (5 мл) добавляли изопропиламин (0,54 мл, 6,3 ммоль) и нагревали микроволновым излучением при 200°C в течение 6 часов. Добавляли воду и раствор экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли, сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на колонке с силикагелем (EtOAc/гексан: EtOAc: MeOH/CH2Cl2), получая 4-(4-амино-3-фторфенокси)-N-изопропилпиридин-2-амин (0,16 г, выход 49%). МС (ESI) m/z: 262,2 (M+H+).
Пример A32: Раствор 3,5-динитробензонитрила (5 г, 25,9 моль), 5-хлорпиридин-3-ола (3,35 г, 25,9 моль) и K2CO3 (7,2 г, 52 моль) в ДМФА (150 мл) нагревали при 100°C в течение ночи. Смесь концентрировали в вакууме и остаток вливали в воду. Водный слой экстрагировали этилацетатом (3×150 мл) и объединенные органические фазы промывали насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая 3-(5-хлорпиридин-3-илокси)-5-нитробензонитрил (3,1 г, выход 44%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,56 (с, 1H), 8,51 (с, 1H), 8,47 (с, 1H), 8,22 (с, 1H), 8,19 (с, 1H), 7,87 (с, 1H).
Порошок железа (6,3 г, 112 ммоль) добавляли к смеси 3-(5-хлорпиридин-3-илокси)-5-нитробензонитрила (3,1 г, 11,2 моль) в уксусной кислоте (100 мл) и реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли воду (200 мл) и смесь нейтрализовали до pH 7 насыщенным раствором Na2CO3 и экстрагировали EtOAc (3×150 мл). Объединенные органические фазы промывали насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали на силикагеле, получая 3-амино-5-(5-хлорпиридин-3-илокси)бензонитрил (1,92 г, выход 71%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,53 (д, J=1,6 Гц, 1H), 8,44 (д, J=2,4 Гц, 1H), 7,80 (т, J=2,4 Гц, 1H), 6,77 (с, 1H), 6,72 (д, J=1,6 Гц, 1H), 6,56 (д, J=2,0 Гц, 1H), 5,92 (с, 2H); MC (ESI) m/z: 246,2 [M+H]+.
Пример A33: 3,5-динитробензонитрил (3 г, 16 ммоль), 6-метилпиридин-3-ол (1,7 г, 16 ммоль) и K2CO3 (4,3 г, 31 ммоль) растворяли в ДМФА и нагревали до 110°C в течение ночи. Реакционную смесь вливали в воду и смесь экстрагировали EtOAc. Объединенные органические фазы промывали насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая 3-(6-метилпиридин-3-илокси)-5-нитробензонитрил (3 г, выход 76%).
1H ЯМР (400 МГц, ДМСО) δ 8,50 (с, 1H), 8,38 (с, 1H), 8,08 (с, 1H), 8,01 (с, 1H), 7,59-7,56 (д, J=10 Гц, 1H), 7,38-7,36 (д, J=8,4 Гц, 1H), 1,98 (с, 3H); MC (ESI) m/z: 256,3 [M+H]+.
Смесь 3-(6-метилпиридин-3-илокси)-5-нитробензонитрила (3 г, 0,012 моль) и порошка железа в уксусной кислоте (200 мл) перемешивали при комнатной температуре в течение 6 часов. Добавляли H2O и смесь доводили до pH 7 насыщенным раствором Na2CO3. Водный слой экстрагировали EtOAc и объединенные органические фазы промывали насыщенным раствором соли, сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая 3-амино-5-(6-метилпиридин-3-илокси)бензонитрил (2 г, выход 76%).
1H ЯМР (400 МГц, ДМСО) δ 8,25 (с, 1H), 7,42 (д, J=10 Гц, 1H), 7,30 (д, J=8,4 Гц, 1H), 6,62 (с, 1H), 6,51 (с, 1H), 6,38 (с, 1H), 5,78 (с, 2H), 2,49 (с, 3H); MC (ESI) m/z: 226,2 [M+H]+.
Пример A34: 3,5-Динитробензонитрил (1,50 г, 7,77 ммоль) добавляли к взвеси пиридин-3-ола (739 мг, 7,77 ммоль) и карбоната калия (10,7 г, 77,7 ммоль) в ДМФА (15 мл), смесь нагревали до 60°C и перемешивали в течение ночи. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом (50 мл) и водой (100 мл). Органическую фазу отделяли, промывали насыщенным раствором бикарбоната натрия (50 мл) и насыщенным раствором соли (50 мл), сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией (колонка Si-40, этилацетат/гексаны), получая светло-желтое твердое вещество, идентифицированное как 3-нитро-5-(пиридин-3-илокси)бензонитрил (1,31 г, выход 69%). МС (ESI) m/z: 242,0 (M+H+).
Раствор 3-нитро-5-(пиридин-3-илокси)бензонитрила (1,31 г, 9,42 ммоль) и дигидрата хлорида олова(II) (6,13 г, 27,2 ммоль) в этаноле (20 мл) нагревали до 70°C в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь вливали в смесь лед/вода (100 мл). Водную смесь подщелачивали (pH ~8) гидроксидом натрия, разбавляли этилацетатом (50 мл) и фильтровали через бумагу, чтобы удалить большую часть солей. Полученный раствор экстрагировали этилацетатом (2×75 мл) и объединенные органические фазы промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали в вакууме, получая светло-желтое твердое вещество, идентифицированное как 3-амино-5-(пиридин-3-илокси)бензонитрил (660 мг, выход 57%). МС (ESI) m/z: 212,0 (M+H+).
Пример A35: Используя способ, аналогичный способу, описанному в примере A3, 3-амино-4-фторфенол (491 мг, 3,86 ммоль) и 4-хлорпиримидин-2-амин (500 мг, 3,86 ммоль) объединяли, получая 4-(3-амино-4-фторфенокси)пиримидин-2-амин (509 мг, выход 59%). МС (ESI) m/z: 221,0 (M+H+).
Пример A36: Раствор 1,3-дифтор-2-метилбензола (15 г, 0,12 моль) в H2SO4 (100 мл) по каплям обрабатывали HNO3 (65%, 11,4 г, 0,12 моль) при -10°C. Полученную смесь перемешивали в течение примерно 30 минут. Смесь вливали в ледяную воду и экстрагировали EtOAc (3×200 мл). Объединенные органические фазы промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали в вакууме, получая 1,3-дифтор-2-метил-4-нитробензол (16 г, выход 78%).
1H ЯМР (400 МГц, CDCl3) δ 7,80 (м, 1H), 6,8-7,1 (м, 1H), 2,30 (с, 3H).
1,3-дифтор-2-метил-4-нитробензол (16 г, 0,092 моль), бензиловый спирт (10 г, 0,092 моль) и K2CO3 (25,3 г, 0,18 моль) объединяли в ДМФА (250 мл) и нагревали до 100°C в течение ночи. Смесь вливали в воду и экстрагировали EtOAc (3×200 мл). Объединенные органические фазы промывали насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на колонке с силикагелем, получая 1-бензилокси-3-фтор-2-метил-4-нитробензол (8 г, выход 33%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,04 (т, J=8,8 Гц, 1H), 7,30-7,46 (м, 5 H), 7,08 (д, J=9,2 Гц, 1H), 5,28 (с, 2H), 2,13 (с, 3H).
1-Бензилокси-3-фтор-2-метил-4-нитробензол (8 г, 0,031 моль) и 10% Pd-C (1 г) объединяли в метаноле (100 мл) и смесь перемешивали в атмосфере H2 (1 атм) в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме, получая 4-амино-3-фтор-2-метилфенол (4,2 г, выход 96%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,61 (с, 1H), 6,42 (т, J=8,4 Гц, 1H), 7,11 (д, J=8,4 Гц, 1H), 4,28 (с, 2H), 1,96 (с, 3H); MC (ESI) m/z: 142,1 [M+H]+.
Трет-бутоксид калия (3,5 г, 0,031 моль) добавляли к раствору 4-амино-3-фтор-2-метилфенола (4,2 г, 0,03 моль) в DMAc и полученную смесь перемешивали в течение 30 минут при комнатной температуре. К полученной смеси добавляли раствор 2,4-дихлорпиридина (4,38 г, 0,03 моль) в DMAc и смесь нагревали при 100°C в течение ночи. Реакционную смесь концентрировали в вакууме и остаток растворяли в этилацетате (200 мл) и фильтровали через силикагель, промывая EtOAc. Фильтрат концентрировали и очищали хроматографией на силикагеле, получая 4-(2-хлорпиридин-4-илокси)-2-фтор-3-метилбензоламин (3,2 г, выход 42%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,21 (д, J=6,0 Гц, 1H), 6,84 (с, 1H), 6,81 (дд, J=5,6, 2,4 Гц, 1H), 6,67 (м, 2H), 5,12 (с, 2H), 1,91 (с, 3H); MC (ESI) m/z 253,1 [M+H]+.
4-(2-Хлорпиридин-4-илокси)-2-фтор-3-метилбензоламин (1,0 г, 3,3 ммоль), 1-метил-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1H-пиразол (1 г, 4,8 ммоль), Na2CO3 (0,84 г, 6,6 ммоль) и Pd(PPh3)4 (0,25 г, 0,2 ммоль) объединяли в DME (75 мл) и воде (25 мл). Смесь барботировали азотом в течение 15 минут и кипятили с обратным холодильником в течение ночи. Реакционную смесь экстрагировали EtOAc (3×100 мл) и объединенные органические фазы промывали насыщенным раствором соли, концентрировали в вакууме и очищали хроматографией на силикагеле, получая 2-фтор-3-метил-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)анилин (0,74 г, выход 75%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,27 (д, J=6,0 Гц, 1H), 8,18 (с, 1H), 7,90 (с, 1H), 7,07 (с, 1H), 6,63 (м, 2H), 6,45 (дд, J=5,6, 2,4 Гц, 1H), 5,06 (с, 2H), 3,82 (с, 3H), 1,95 (с, 3H); MC (ESI) m/z: 299,2 [M+H]+.
Пример A37: Раствор 1,2,3-трифтор-4-нитробензола (30 г, 0,17 моль) и бензилового спирта (18,4 г, 0,17 моль) в ДМФА (300 мл) обрабатывали K2CO3 (35 г, 0,25 моль) и полученную смесь перемешивали при комнатной температуре в течение 8 часов. Добавляли воду (300 мл) и смесь экстрагировали EtOAc (3×500 мл). Объединенные органические фазы промывали насыщенным раствором соли, сушили (MgSO4), концентрировали в вакууме и подвергали хроматографии на силикагеле, получая 1-бензилокси-2,3-дифтор-4-нитробензол (16 г, выход 36%).
1H ЯМР (400 МГц, ДМСО-d6): δ 8,06 (м, 1H), 7,49-7,30 (м, 6H), 5,37 (с, 2H).
Смесь 1-бензилокси-2,3-дифтор-4-нитробензола (14 г, 52,8 ммоль) и Pd/C (10%, 1,4 г) в MeOH (200 мл) перемешивали в атмосфере водорода (30 psi, 0,210 МПа) в течение 2 часов. Катализатор удаляли фильтрованием и фильтрат концентрировали в вакууме, получая 4-амино-2,3-дифторфенол (7 г, выход 92%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,05 (с, 1H), 6,45 (т, J=8,8 Гц, 1H), 6,34 (т, J=9,2 Гц, 1H), 4,67 (с, 2H).
Используя способ, аналогичный способу, описанному в примере A2, 4-амино-2,3-дифторфенол (6 г, 41,4 ммоль), трет-бутоксид калия (4.9 г, 43,5 ммоль) и 2,4-дихлорпиридин (6,1 г, 41,4 ммоль) объединяли, получая 4-(2-хлорпиридин-4-илокси)-2,3-дифторфениламин (7 г, выход 66%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,27 (д, J=6,0 Гц, 1H), 7,05 (с, 1H), 6,95 (м, 1H), 6,92 (д, J=8,8 Гц, 1H), 6,62 (д, J=8,8 Гц, 1H), 5,60 (с, 2H).
Пример A38: Раствор соединения из примера A37 (2 г, 7,8 ммоль), 1-метил-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1H-пиразола (1,6 г, 7,8 ммоль) и Na2CO3 (1,65 мг, 15,6 ммоль) в DME (12 мл) и H2O (4 мл) барботировали азотом в течение 20 минут. Pd(PPh3)4 (450 мг, 0,4 ммоль) добавляли и полученную смесь нагревали до 70°C в атмосфере азота в течение 16 часов. Растворитель удаляли при пониженном давлении и сырой продукт суспендировали в воде и экстрагировали EtOAc (3×10 мл). Органический слой промывали насыщенным раствором соли, сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на колонке с силикагелем, получая 2,3-дифтор-4-[2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси]фениламин (1,3 г, выход 55%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,40 (д, J=6,0 Гц, 1H), 8,32 (с, 1H), 8,02 (с, 1H), 7,26 (с, 1H), 6,96 (т, J=8,8 Гц, 1H), 6,70-6,67 (м, 2H), 5,62 (с, 2H), 3,92 (с, 3H); MC (ESI) m/z: 303,2[M+H]+.
Пример A39: Соединение из примера A23 (2,0 г, 8,4 ммоль) и 4-метоксибензиламин (50 мл) объединяли в стальном баллоне и нагревали до 160°C в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении и очищали обращенно-фазовой препаративной ВЭЖХ, получая N-(4-метоксибензил)-4-(4-амино-3-фторфенокси)пиридин-2-амин (1,0 г, выход 35%).
Раствор N-(4-метоксибензил)-4-(4-амино-3-фторфенокси)пиридин-2-амина (500 мг, 1,47 ммоль) в CH2Cl2 (10 мл) обрабатывали нитратом аммония-церия(IV) (1,64 г, 2,99 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь промывали водой, концентрировали в вакууме и очищали хроматографией на силикагеле, получая 4-(4-амино-3-фторфенокси)пиридин-2-амин (250 мг, выход 77%).
1H ЯМР (300 МГц, ДМСО-d6) δ 7,73 (д, J=6,0 Гц, 1H), 6,88 (дд, J=9,0, 2,0 Гц, 1H), 6,80 (т, J=8,7 Гц, 1H), 6,68 (м, 1H), 6,06 (дд, J=4,5, 1,8 Гц, 1H), 5,84 (с, 2H), 5,75 (д, J=1,5 Гц, 1H), 5,08 (с, 2H); MC (ESI) m/z: 220,3 (М+H+).
Пример A40: Раствор 4-амино-2-метилфенола (4,25 г, 34,5 ммоль) в диметилацетамиде (50 мл) дегазировали в вакууме и помещали под слой аргона. Добавляли трет-бутоксид калия (5,0 г, 44,6 ммоль) и реакционную смесь второй раз дегазировали и перемешивали при комнатной температуре в атмосфере аргона в течение 30 минут. Добавляли 2,4-дихлорпиридин (4,6 г, 31,3 ммоль) и смесь нагревали до 100°C в течение ночи. Растворитель удаляли при пониженном давлении и остаток очищали хроматографией на силикагеле, получая 4-(2-хлорпиридин-4-илокси)-3-метилбензоламин (4,5 г, выход 56%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,21 (д, J=5,2 Гц, 1H), 6,75-6,80 (м, 3H), 6,45-6,50 (м, 2H), 5,15 (с, 2H), 1,92 (с, 3H); MC (ESI) m/z: 235,1 (М+H+).
Раствор 4-(2-хлорпиридин-4-илокси)-3-метилбензоламина (595 мг, 2,54 ммоль), 1-метил-4-(4,4,5,5-тетраметил)-[1,3,2]диоксаборолан-2-ил)-4H-пиразола (790 мг, 3,80 ммоль) и Cs2CO3 (2,53 г, 7,77 ммоль) в 10 мл ДМФА (10 мл) и воде (3 мл) дегазировали в вакууме и помещали в атмосферу азота. Добавляли Pd(PPh3)4 (295 мг, 0,26 ммоль) и реакционную смесь нагревали до 90°C в течение ночи. Реакционную смесь разбавляли EtOAc (30 мл) и промывали водой (2×10 мл) и насыщенным раствором соли (2×10 мл). Водную часть экстрагировали EtOAc (2×15 мл) и объединенные органические фазы промывали насыщенным раствором соли (10 мл), концентрировали в вакууме и очищали на силикагеле, получая 3-метил-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)бензоламин в виде бледно-желтой пены (627 мг, выход 88%).
1H ЯМР (400 МГц, ДМСО-d6): δ 8,27 (д, J=6,0 Гц, 1H), 8,18 (с, 1H), 7,90 (д, J=0,7 Гц, 1H), 7,07 (д, J=2,2 Гц, 1H), 6,74 (д, J=8,6 Гц, 1H), 6,49 (д, J=2,5 Гц, 1H), 6,46-6,40 (м, 2H), 5,02 (с, 2H), 3,84 (с, 3H), 1,94 (с, 3H); MC (ESI) m/z: 281,2 (М+H+).
Пример A41: 4-Хлор-2-метилсульфанилпиримидин (1,4 г, 8,8 ммоль), 4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1H-пиразол (2,0 г, 10,3 ммоль), Na2CO3 (2,8 г, 26,4) и Pd(PPh3)4 (500 мг, 0,43 ммоль) объединяли в растворителе, состоящем из смеси толуол/EtOH/H2O (4/4/1,20 мл). Смесь дегазировали, используя вакуум, и снова заполняли свободное пространство над смесью аргоном. Реакционную смесь нагревали в течение ночи при 100°C. Нерастворимую часть фильтровали и фильтрат концентрировали и очищали хроматографией на силикагеле, получая 2-(метилтио)-4-(1H-пиразол-4-ил)пиримидин (1,2 г, выход 71%).
1H ЯМР (400 МГц, CDCl3) δ 8,45 (д, J=6,4 Гц, 1H), 8,24 (с, 1H), 7,23 (с, 1H), 7,05 (д, J=6,4 Гц, 1H), 2,51 (с, 3H).
К раствору 2-(метилтио)-4-(1H-пиразол-4-ил)пиримидин (200 мг, 1 ммоль) в дихлорметане (3 мл) и H2O (1 мл) добавляли 4-метоксибензилхлорид (200 мг, 1,28 ммоль) при 0°C. Смесь перемешивали при комнатной температуре в течение ночи. Органический слой отделяли, промывали насыщенным раствором соли и концентрировали в вакууме, получая неочищенный 4-(1-(4-метоксибензил)-1H-пиразол-4-ил)-2-(метилтио)пиримидин.
1H ЯМР (300 МГц, ДМСО-d6) δ 8,58 (с, 1H), 8,50 (д, J=5,4 Гц, 1H), 8,16 (с, 1H), 7,40 (д, J=5,4 Гц, 1H), 7,27 (д, J=8,4 Гц, 2H), 7,22 (д, J=8,4 Гц, 2H), 5,30 (с, 2H), 3,72 (с, 3H), 2,51 (с, 3H); MC (ESI) m/z: 313 (М+H+).
К раствору 4-(1-(4-метоксибензил)-1H-пиразол-4-ил)-2-(метилтио)пиримидин (200 мг, 0,64 ммоль) в дихлорметане добавляли m-CPBA (220 мг, 1,28 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Добавляли воду, органический слой отделяли и водный слой экстрагировали дихлорметаном. Объединенные органические фазы промывали насыщенным раствором соли и концентрировали в вакууме. Остаток объединяли с 3-амино-4-фторфенолом (165 мг, 1,28 ммоль) и K2CO3 (176 мг, 1,28 ммоль) в ДМФА (5 мл) и полученную смесь нагревали при 90°C в течение ночи. После фильтрования и концентрирования остаток очищали хроматографией на колонке с силикагелем, получая 5-(4-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиримидин-2-илокси)-2-фторбензоламин (210 мг, выход 84%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,50 (с, 1H), 8,44 (д, J=5,4 Гц, 1H), 8,10 (с, 1H), 7,42 (д, J=5,4 Гц, 1H), 7,25 (д, J=8,4 Гц, 2H), 6,98 (т, J=9,6 Гц, 1H), 6,91 (д, J=8,4 Гц, 2H), 6,52 (дд, J=2,7, 8,7 Гц, 1H), 6,28 (м, 1H), 5,30 (ушир.с, 2H), 5,26 (с, 2H), 3,72 (с, 3H); MC (ESI) m/z: 392,2 (М+H+).
К раствору 5-(4-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиримидин-2-илокси)-2-фторбензоламин (50 мг, 0,13 ммоль) в дихлорметане (3 мл) добавляли ТФУ (0,3 мл) при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Растворитель удаляли в вакууме, остаток промывали эфиром и обрабатывали насыщенным раствором аммиака. Твердые вещества собирали фильтрованием и сушили в вакууме, получая 5-(4-(1H-пиразол-4-ил)пиримидин-2-илокси)-2-фторбензоламин (15 мг, выход 43%).
1H ЯМР (300 МГц, MeOD) δ 8,44 (д, J=5,1 Гц, 1H), 8,23 (ушир.с, 2H), 7,40 (д, J=5,4, 1H), 7,02 (дд, J=10,8, 8,7 Гц, 1H), 6,73 (дд, J=2,7, 7,2 Гц, 1H), 6,50 (м, 1H); MC (ESI) m/z: 272,2 (М+H+).
Пример A42. Используя способ, аналогичный способу, описанному в примере A3, 3-амино-4-фторфенол (0,127 г, 1,0 ммоль) и 5-бром-2-нитропиридин (0,203 г, 1,0 ммоль) объединяли, получая 2-фтор-5-(6-нитропиридин-3-илокси)бензоламин (0,098 г, выход 39%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,36 (д, J=2,8 Гц, 1H), 8,30 (д, J=8,8 Гц, 1H), 7,56 (дд, J=8,8, 2,8 Гц, 1H), 7,07 (м, 1H), 6,53 (дд, J=7,6, 3,2 Гц, 1H), 6,31 (с, 1H), 5,48 (с, 2H); MC (ESI) m/z: 250,0 (М+H+).
Пример B1: К перемешиваемому раствору бензил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,991 г, 2,52 ммоль, 1,00 экв.) в ТГФ (10 мл) и H2O (2,5 мл) добавляли NaIO4 (1,62 г, 7,56 ммоль, 3,00 экв.). Полученную суспензию перемешивали при 25°C в течение 30 минут и затем обрабатывали 3М HCl (1,68 мл, 5,04 ммоль, 2,0 экв.). Смесь перемешивали в течение 2,5 часов. Надосадочную жидкость сливали с твердых веществ, которые промывали ТГФ. Объединенные органические фазы промывали насыщенным раствором соли (2×), сушили (MgSO4) и концентрировали в вакууме, получая неочищенную 2-(бензилоксикарбонил)-1,2,3,4-тетрагидроизохинолин-6-илбороновую кислоту (0,640 г, выход 82%) в виде пены, которую как таковую использовали для следующей реакции.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,68-7,58 (м, 2H), 7,45-7,29 (м, 6H), 7,17 (м, 1H), 5,13 (с, 2H), 4,62-4,56 (ушир.м, 2H), 3,65 (ушир.с, 2H), 2,86 (т, 2H, J=5,60 Гц); MC (ESI) m/z: 312,0 (М+H+).
К перемешиваемой суспензии 2-(бензилоксикарбонил)-1,2,3,4-тетрагидроизохинолин-6-илбороновой кислоты (0,640 г, 2,06 ммоль, 1,00 экв.) и молекулярных сит 4Å МС (0,64 г) в CH2Cl2 (20 мл) добавляли пиридин (0,168 мл, 2,06 ммоль, 1,00 экв.), затем этил 3-трет-бутил-1H-пиразол-5-карбоксилат (0,404 г, 2,06 ммоль, 1,00 экв.) и Cu(OAc)2 (0,374 г, 2,06 ммоль, 1,00 экв.). Полученную сине-зеленую смесь перемешивали при 25°C. Через 40 часов смесь разбавляли H2O и сливали с молекулярных сит. Слои разделяли и органическую фазу промывали H2O (2×). Объединенные водные фазы экстрагировали CH2Cl2 (1×). Объединенные органические фазы сушили (MgSO4), концентрировали в вакууме и очищали флэш-хроматографией (EtOAc/гексаны), получая бензил-6-(3-трет-бутил-5-(этоксикарбонил)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,46 г, выход 48%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,41-7,28 (м, 5H), 7,24-7,20 (м, 3H), 6,96 (с, 1H), 5,15 (с, 2H), 4,67 (ушир.м, 2H), 4,17 (кв, 2H, J=7,2 Гц), 3,66 (ушир.с, 2H), 2,86 (т, 2H, J=6,0 Гц), 1,29 (с, 9H), 1,18 (т, 3H, J=7,2 Гц); MC (ESI) m/z: 462,3 (М+H+).
К перемешиваемому раствору бензил-6-(3-трет-бутил-5-(этоксикарбонил)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (0,160 г, 0,347 ммоль) в смеси 1:1:1 ТГФ/EtOH/H2O (3 мл) при 22°C добавляли LiOH·H2O (0,0727 г, 1,73 ммоль). Через 3 часа, когда реакция была завершена, реакционную смесь подкисляли (pH 2-3), используя 1М HCl, и экстрагировали EtOAc (3×). Объединенные органические фазы промывали насыщенным раствором соли (2×), сушили (MgSO4), фильтровали и упаривали, получая 1-(2-(бензилоксикарбонил)-1,2,3,4-тетрагидроизохинолин-6-ил)-3-трет-бутил-1H-пиразол-5-карбоновую кислоту (0,16 г, выход 106%) в виде масла, которое использовали без дополнительной очистки.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,41-7,31 (м, 5H), 7,328-7,20 (м, 3H), 6,91 (с, 1H), 5,15 (с, 2H), 4,65 (ушир.м, 2H), 3,66 (ушир.с, 2H), 2,86 (т, 2H, J=6,0 Гц), 1,29 (с, 9H); MC (ESI) m/z: 434,2 (М+H+).
Пример B2: Этил-3-трет-бутил-1-(2-(трифторметилсульфонилокси)хинолин-6-ил)-1H-пиразол-5-карбоксилат (см. WO 2006/071940A2, 0,380 г, 0,806 ммоль), MeNH2·HCl (0,109 г, 1,61 ммоль) и Et3N (0,449 мл, 3,22 ммоль) объединяли в ДМФА (8 мл) и перемешивали при комнатной температуре в течение ночи. Добавляли дополнительные порции MeNH2·HCl (0,109 г, 1,61 ммоль) и Et3N (0,449 мл, 3,22 ммоль) и реакционную смесь перемешивали еще в течение 4 часов при комнатной температуре и в течение 3 часов при 60°C. После завершения реакции смесь разбавляли насыщенным раствором соли и экстрагировали EtOAc. Экстракты промывали насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая этил-3-трет-бутил-1-(2-(метиламино)хинолин-6-ил)-1H-пиразол-5-карбоксилат (240 мг, выход 85%).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,90 (д, J=9,2 Гц, 1H), 7,68 (д, J=2,8 Гц, 1H), 7,53 (д, J=9,2 Гц, 1H), 7,46 (дд, J=8,8, 2,0 Гц, 1H), 7,17 (кв, J=4,8 Гц, 1H), 6,98 (с, 1H), 6,80 (д, J=8,8 Гц, 1H), 4,16 (кв, J=7,2 Гц, 2H), 2,92 (д, J=4,8 Гц, 3H), 1,32 (с, 9H), 1,13 (т, J=7,2 Гц, 3H); MC (ESI) m/z: 353,2 (М+H+).
LiOH·H2O (0,143 г, 3,40 ммоль) добавляли к раствору этил-3-трет-бутил-1-(2-(метиламино)хинолин-6-ил)-1H-пиразол-5-карбоксилата (0,240 г, 0,68 ммоль) в смеси вода/ТГФ/EtOH (1:1:1, 9 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре, разбавляли 3М HCl и экстрагировали EtOAc и ТГФ. Объединенные органические фазы промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали в вакууме, получая 3-трет-бутил-1-(2-(метиламино)хинолин-6-ил)-1H-пиразол-5-карбоновую кислоту (0,22 г, выход 100%).
1H-ЯМР (ДМСО-d6) δ 7,90 (д, J=9,2 Гц, 1H), 7,66 (д, J=2,4 Гц, 1H), 7,52 (д, J=8,8 Гц, 1H), 7,46 (дд, J=9,2, 2,8 Гц, 1H), 7,14 (м, 1H), 6,88 (ушир.с, 1H), 6,79 (д, J=9,2 Гц, 1H), 2,92 (д, J=4,8 Гц, 3H), 1,31 (с, 9H); MC (ESI) m/z: 325,2 (М+H+).
Пример B3: Раствор ангидрида трифторметансульфоновой кислоты (42,8 г, 0,15 моль) в CH2Cl2 (100 мл) по каплям добавляли к охлажденному до 0°C раствору 6-гидроксихинолина (20,00 г, 0,138 моль) и пиридина (23 г, 0,277 моль) в CH2Cl2 (500 мл). Охлаждающую баню убирали и полученный раствор перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь промывали водой (3×300 мл) и органическую фазу сушили (MgSO4) и концентрировали в вакууме, получая неочищенный хинолин-6-илтрифторметансульфонат (40 г, выход >100%) в виде масла.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,00 (д, 1H, J=2,8 Гц), 8,50 (д, 1H, J=8,0 Гц), 8,21 (д, J=2,8 Гц, 1H), 8,18 (д, J=9,2 Гц, 1H), 7,80 (м, 1H), 7,64 (м, 1H); MC (ESI) m/z: 277,9 (М+H+).
К суспензии хинолин-6-илтрифторметансульфоната (40 г, 0,14 моль), гидразона бензофенона (35,6 г, 0,18 моль), карбоната цезия (74 г, 0,23 моль) и 1,1'-бис(дифенилфосфино)ферроцена (2,5 г, 4,5 ммоль) в дегазированном толуоле (1 л) добавляли ацетат палладия (0,013 г, 0,058 ммоль). Полученную смесь нагревали до 90°C в атмосфере азота. Через 16 часов смесь концентрировали в вакууме и остаток очищали хроматографией на колонке с силикагелем (EtOAc/петролейный эфир), получая 1-(дифенилметилен)-2-(хинолин-6-ил)гидразин (32 г, выход 68,6%).
1H-ЯМР (300 МГц, ДМСО-d6) δ 9,22 (с, 1H), 8,58 (т, J=1,8 Гц, 1H), 8,13 (д, J=3,6 Гц, 1H), 7,80 (д, J=3,6 Гц, 1H), 7,61 (д, J=3,9 Гц, 1H), 7,59-7,51 (м, 4H), 7,50 (д, J=3,6 Гц, 2H), 7,33-7,39 (м, 6H); MC (ESI) m/z: 324 (М+H+).
Раствор 1-(дифенилметилен)-2-(хинолин-6-ил)гидразина (32 г, 99 ммоль) и 4,4-диметил-3-оксопентанонитрила (26 г, 0,15 моль) в этаноле (500 мл) обрабатывали концентрированной HCl (80 мл, 12н., 0,96 моль) и смесь кипятили с обратным холодильником в течение ночи. Охлажденную реакционную смесь концентрировали в вакууме и остаток промывали Et2O, чтобы удалить дифенилкетон. Неочищенный продукт растворяли в EtOAc и нейтрализовали (pH 8) насыщенным раствором Na2CO3. Органический слой сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая 5-трет-бутил-2-хинолин-6-ил-2H-пиразол-3-иламин (23 г, выход 87%).
1H-ЯМР (300 МГц, DMCO-d6) δ 8,86 (м, 1H), 8,39 (д, J=5,7 Гц, 1H), 8,11-8,02 (м, 3H), 7,54 (м, 1H), 5,46 (с, 1H), 5,42 (ушир.с, 2H), 1,23 (с, 9H); MC (ESI) m/z: 267,2 (М+H+).
К охлажденному раствору (-10°C) 5-трет-бутил-2-хинолин-6-ил-2H-пиразол-3-иламина (8,00 г, 30 ммоль) в 100 мл CH2Cl2 добавляли пиридин (8,0 мл, 99 ммоль) и DMAP (100 мг), затем добавляли раствор трихлорэтилхлорформиата (8,9 мл, 42 ммоль) в 30 мл CH2Cl2 в течение периода времени, составляющего 20 минут. После перемешивания в течение 1 часа добавляли воду (100 мл), перемешивание продолжали еще в течение 10 минут и органический слой отделяли. Органический слой промывали насыщенным раствором соли, сушили и темно-коричневый остаток, полученный после удаления растворителя, кристаллизовали из ацетонитрила, получая 2,2,2-трихлорэтил-3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-илкарбамат в виде белого твердого вещества (8,23 г, выход 62%).
1H ЯМР (ДМСО-d6) δ 10,15 (ушир.с, 1H), 8,93 (м, 1H), 8,41 (д, J=8 Гц, 1H), 8,11 (м, 2H), 7,90 (дд, J=8, 2 Гц, 1H), 7,60 (дд, J=6,4, 4,2 Гц, 1H), 6,39 (с, 1H), 4,85 (с, 2H), 1,32 (с, 9H); MC (ESI) m/z: 442 (М+H+).
Пример B4: Хинолин-6-илбороновую кислоту (0,34 г, 2,0 ммоль) растворяли в CH2Cl2 (30 мл) и пиридине (1 мл) с молекулярными ситами (активированные 4Å) и перемешивали при комнатной температуре в течение 6 часов. Добавляли этил-3-трет-бутил-1H-пиразол-5-карбоксилат (0,39 г, 2,0 ммоль) и ацетат меди(II) (0,36 г, 2,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 суток на открытом воздухе. Реакционную смесь фильтровали через подушку целита, фильтрат концентрировали в вакууме и очищали хроматографией на силикагеле, получая этил-3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-карбоксилат (0,21 г, выход 33%). МС (ESI) m/z: 324,0 (M+H+).
Гидроксид лития (62 мг, 2,6 ммоль) добавляли к раствору этил-3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-карбоксилата (0,21 г, 0,65 ммоль) в смеси диоксан-H2O-EtOH (1:1:1, 6 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Раствор концентрировали и остаток растворяли в H2O (2 мл). Добавляли 3М HCl и осадок собирали фильтрованием и промывали водой. Твердое вещество сушили в вакууме, получая 3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-карбоновую кислоту (0,18 г, выход 94%) в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,96 (дд, J=2,0, 4,0 Гц, 1H), 8,47 (дд, J=1,2, 8,4 Гц, 1H), 8,09 (м, 1H), 8,06 (с, 1H), 7,82 (дд, J=2,8, 9,2 Гц, 1H), 7,61 (дд, J=4,8, 8,8 Гц, 1H), 7,01 (с, 1H), 1,33 (с, 9H); MC (ESI) m/z: 296,0 (М+H+).
Пример B5: Гидрохлорид этилового эфира [3-(5-амино-3-трет-бутилпиразол-1-ил)нафталин-1-ил]уксусной кислоты (см. WO 2006/071940, 1,60 г, 4,55 ммоль) обрабатывали аммиаком в метаноле (7 М, 13 мл, 91 ммоль) и реакционную смесь нагревали в герметично закрытой пробирке в течение 6 суток. Растворитель удаляли в вакууме и остаток подвергали хроматографии, получая 2-(3-(5-амино-3-трет-бутил-1H-пиразол-1-ил)нафталин-1-ил)ацетамид (610 мг, выход 41%). МС (ESI) m/z: 323,3 (M+H+).
К смеси насыщенного раствора бикарбоната натрия (20 мл), этилацетата (20 мл) и 2-(3-(5-амино-3-трет-бутил-1H-пиразол-1-ил)нафталин-1-ил)ацетамида (300 мг, 0,931 ммоль) добавляли Troc-Cl (296 мг, 1,40 ммоль). Смесь энергично перемешивали в течение ночи. Смесь разбавляли этилацетатом (30 мл) и органическую фазу отделяли, промывали 5% лимонной кислотой (30 мл) и насыщенным раствором соли (30 мл), сушили (Na2SO4) и концентрировали в вакууме, получая твердое вещество, которое растирали в этилацетате и фильтровали, получая 2,2,2-трихлорэтил-1-(4-(2-амино-2-оксоэтил)нафталин-2-ил)-3-трет-бутил-1H-пиразол-5-илкарбамат (241 мг, выход 52%). МС (ESI) m/z: 499,0 (M+H+).
Пример B6: К перемешиваемой суспензии трет-бутил 5-(5-амино-3-трет-бутил-1H-пиразол-1-ил)-1H-индазол-1-карбоксилата (см. WO 2006/071940 A2, 0,250 г, 0,70 ммоль) и Troc-Cl (0,10 мл, 0,74 ммоль) в EtOAc (7 мл) при комнатной температуре добавляли насыщенный раствор NaHCO3 (2,9 мл, 2,1 ммоль). Через 3 часа, когда реакция была завершена, реакционную смесь разбавляли гексанами (35 мл) и фильтровали. Твердое вещество тщательно промывали гексанами и сушили получая трет-бутил-5-(3-трет-бутил-5-((2,2,2-трихлорэтокси)карбонил)-1H-пиразол-1-ил)-1H-индазол-1-карбоксилат (0,36 г, выход 97%). МС (ESI) m/z: 532,0 (M+H+).
Пример B7: К перемешиваемому раствору трет-бутил-6-(5-амино-3-трет-бутил-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (см. WO 2006/071940A2, 0,075 г, 0,20 ммоль) и Troc-Cl (0,028 мл, 0,21 ммоль) в EtOAc (2 мл) добавляли насыщенный раствор NaHCO3 (0,82 мл, 0,61 ммоль). Полученный двухфазный раствор перемешивали при комнатной температуре в течение ночи. Слои разделяли и водную фазу экстрагировали EtOAc (2×). Объединенные органические фазы промывали насыщенным раствором соли (1×), сушили (MgSO4) и концентрировали в вакууме, получая неочищенный трет-бутил-6-(3-трет-бутил-5-((2,2,2-трихлорэтокси)карбонил)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,110 г, выход 100%).
1H ЯМР (ДМСО-d6) δ 9,93 (ушир.с, 1H), 7,29-7,24 (м, 2H), 6,83-6,80 (м, 1H), 6,27 (с, 1H), 4,85 (с, 2H), 4,52 (ушир.с, 2H), 3,57-3,53 (м, 2H), 2,82-2,79 (м, 2H), 1,44 (с, 9H), 1,27 (с, 9H); MC (ESI) m/z: 545,0 (М+H+).
Пример B8: Раствор трет-бутил-5-(5-амино-3-трет-бутил-1H-пиразол-1-ил)-1H-индазол-1-карбоксилата (см. WO 2006/071940 A2, 0,64 г, 1,80 ммоль) в EtOAc (6 мл) обрабатывали 1 М водным раствором NaOH (2,7 мл). К перемешиваемой двухфазной реакционной смеси при 0°C по каплям в течение 1 минуты добавляли изопропенилхлорформиат (0,26 мл). Реакционную смесь перемешивали в течение 4 часов при комнатной температуре. Реакционную смесь разбавляли EtOAc (20 мл). Органический слой промывали H2O (2×10 мл), насыщенным раствором соли (10 мл), сушили (MgSO4) и концентрировали, получая трет-бутил-5-(3-трет-бутил-5-((проп-1-ен-2-илокси)карбониламино)-1H-пиразол-1-ил)-1H-индазол-1-карбоксилат (0,69 г, выход 87%) в виде светло-желтой пены.
1H ЯМР (ДМСО-d6) δ 9,77 (с, 1H), 8,52 (с, 1H), 8,17 (д, J=9 Гц, 1H), 7,97 (д, J=2 Гц, 1H), 7,74 (дд, J=9, 2 Гц, 1H), 6,34 (с, 1H), 4,7 (м, 2H), 1,80 (с, 3H), 1,67 (с, 9H), 1,30 (с, 9H); MC (ESI) m/z: 440,2 (М+H+).
Пример B9: Используя способ, аналогичный способу, описанному в примере B3, 6-(2-(дифенилметилен)гидразинил)хинолин (4,0 г, 12,3 ммоль) и 4-метил-3-оксопентанонитрил (1,5 г, 13,5 ммоль) объединяли, получая 3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-амин. (1,1 г, выход 36%).
1H ЯМР (400 МГц, CDCl3) δ 8,93 (дд, J=4,4, 1,6 Гц, 1H), 8,21-8,18 (м, 2H), 8,05-8,02 (м, 2H), 7,44 (дд, J=8,4, 4,4 Гц, 1H), 5,56 (с, 1H), 3,85 (ушир.с, 2H), 2,97 (м, 1H), 1,31 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 253,2 (М+H+).
Используя способ, аналогичный способу, описанному в примере B3, 3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-амин (0,378 г, 1,5 ммоль) превращали в 2,2,2-трихлорэтил-3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-илкарбамат (0,391 г, выход 61%). МС (ESI) m/z: 427,0 (M+H+).
Пример B10: Используя способ, аналогичный способу, описанному в примере B3, 6-(2-дифенилметилен)гидразинил)хинолин (4,0 г, 12,3 ммоль) и 3-оксопентанонитрил (1,3 г, 1,1 экв.) объединяли, получая 5-этил-2-хинолин-6-ил-2H-пиразол-3-иламин (2,5 г, выход 85%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,87 (дд, J=7,8, 1,8 Гц, 1H), 8,39 (дд, J=8,4, 1,5 Гц, 1H), 8,12 (с, 1H), 8,06-8,03 (м, 2H), 7,54 (дд, J=8,4, 1,2 Гц, 1H), 5,46 (ушир.с, 2H), 5,40 (с, 1H), 2,49 (кв, J=7,5 Гц, 2H), 1,16 (т, J=7,5 Гц, 3H); MC (ESI) m/z: 239,2 (М+H+).
Используя способ, аналогичный способу, описанному в примере B3, 5-этил-2-хинолин-6-ил-2H-пиразол-3-иламин (0,378 г, 1,5 ммоль) превращали в 2,2,2-трихлорэтил-3-этил-1-(хинолин-6-ил)-1H-пиразол-5-илкарбамат (0,287 г, выход 41%) в виде белой пены. МС (ESI) m/z: 413,0 (M+H+).
Пример B11: Используя способ, аналогичный способу, описанному в примере B3, 6-(2-(дифенилметилен)гидразинил)хинолин (5,0 г, 15,5 ммоль) и 4,4,4-трифтор-3-оксобутиронитрил (2,3 г, 16,8 ммоль) объединяли, получая 2-хинолин-6-ил-5-трифторметил-2H-пиразол-3-иламин (2,3 г, выход 53%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,95 (дд, J=1,5, 4,2 Гц, 1H), 8,47 (д, J=7,2 Гц, 1H), 8,22 (д, J=2,4 Гц, 1H), 8,14 (д, J=9,3 Гц, 1H), 7,97 (дд, J=2,4, 9,0 Гц, 1H), 7,60 (дд, J=7,2, 4,2 Гц, 1H), 5,96 (ушир.с, 2H), 5,85 (с, 1H); MC (ESI) m/z: 279,2 (М+H+).
Используя способ, аналогичный способу, описанному в примере B3, 2-хинолин-6-ил-5-трифторметил-2H-пиразол-3-иламин (0,47 г, 1,7 ммоль) превращали в 2,2,2-трихлорэтил-1-(хинолин-6-ил)-3-(трифторметил)-1H-пиразол-5-илкарбамат (0,333 г, выход 43%). МС (ESI) m/z: 453,0 (M+H+).
Пример B12: Используя способ, аналогичный способу, описанному в примере B3, 6-(2-(дифенилметилен)гидразинил)хинолин (5,0 г, 15,5 ммоль) и 3-циклопентил-3-оксопропаннитрил (3,0 г, 1,1 экв.) объединяли, получая 3-циклопентил-1-(хинолин-6-ил)-1H-пиразол-5-амин (2,3 г, выход 53%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,87 (м, 1H), 8,38 (дд, J=1,5, 8,4 Гц, 1H), 8,10 (с, 1H), 8,04-8,02 (м, 2H), 7,55 (дд, J=4,2, 8,1 Гц, 1H), 5,41 (ушир.с, 2H), 5,38 (с, 1H), 2,90 (м, 1H), 1,85-1,96 (м, 2H), 1,53-1,70 (м, 6H); MC (ESI) m/z: 279,3 (М+H+).
Используя способ, аналогичный способу, описанному в примере B3, 3-циклопентил-1-(хинолин-6-ил)-1H-пиразол-5-амин (0,418 г, 1,5 ммоль) превращали в 2,2,2-трихлорэтил-3-циклопентил-1-(хинолин-6-ил)-1H-пиразол-5-илкарбамат (0,394 г, выход 58%). МС (ESI) m/z: 453,0 (M+H+).
Пример B13: Используя способ, аналогичный способу, описанному в примере B3, 6-(2-(дифенилметилен)гидразинил)хинолин (4,0 г, 12,3 ммоль) и 3-циклобутил-3-оксопропионитрил (1,7 г, 1,1 экв.) объединяли, получая 5-циклобутил-2-хинолин-6-ил-2H-пиразол-3-иламин (1,3 г, выход 40%).
1H ЯМР (300 МГц, CDCl3) δ 8,92 (дд, J=4,5, 1,2 Гц, 1H), 8,16-8,20 (м, 2H), 8,00-8,04 (м, 2H), 7,43 (дд, J=8,4, 1,2 Гц, 1H), 5,64 (с, 1H), 3,83 (ушир.с, 2H), 3,53 (м, 1H), 2,40-2,20 (м, 4H), 2,08-1,92 (м, 2H); MC (ESI) m/z: 265,1 (М+H+).
Используя способ, аналогичный способу, описанному в примере B3, 5-циклобутил-2-хинолин-6-ил-2H-пиразол-3-иламин (0,396 г, 1,5 ммоль) превращали в 2,2,2-трихлорэтил-3-циклобутил-1-(хинолин-6-ил)-1H-пиразол-5-илкарбамат (0,412 г, выход 63%). МС (ESI) m/z: 439,0 (M+H+).
Пример B14: Дегазированную смесь этил 5-хлор-2-йодбензоата (0,621 г, 2,00 ммоль), Pd(PPh3)4 (0,116 мг, 0,1 ммоль), хинолин-6-илбороновой кислоты (0,381 г, 2,2 ммоль), K2CO3 (0,553 г, 4,0 ммоль), диметоксиэтана (20 мл) и воды (5 мл) кипятили с обратным холодильником в течение ночи. Растворители удаляли при пониженном давлении. Остаток разбавляли насыщенным раствором NH4Cl (15 мл) и экстрагировали EtOAc (3×30 мл). Объединенные органические слои сушили (MgSO4), концентрировали в вакууме и очищали хроматографией, получая этил-5-хлор-2-(хинолин-6-ил)бензоат (0,244 г, выход 39%) в виде бесцветного масла. МС (ESI) m/z: 312,0 (M+H+).
К перемешиваемому раствору этил-5-хлор-2-(хинолин-6-ил)бензоата (0,244 г, 0,78 ммоль) в смеси 1:1:1 ТГФ/EtOH/H2O (21 мл) при комнатной температуре добавляли LiOH·H2O (0,164 г, 3,91 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и остаток разбавляли H2O (10 мл). Водный раствор подкисляли до pH~4, используя 3М HCl, и экстрагировали EtOAc (3×30 мл). Объединенные органические слои промывали насыщенным раствором соли (20 мл), сушили (MgSO4) и концентрировали, получая 5-хлор-2-(хинолин-6-ил)бензойную кислоту (0,201 г, выход 91%) в виде белого твердого вещества. МС (ESI) m/z: 284,0 (M+H+).
К перемешиваемому раствору 5-хлор-2-(хинолин-6-ил)бензойной кислоты (0,201 г, 0,708 ммоль) и TEA (0,148 мл, 1,06 ммоль) в 1,4-диоксане (10 мл) при комнатной температуре добавляли DPPA (0,191 мл, 0,244 ммоль). После перемешивания в течение 30 минут при комнатной температуре добавляли 2,2,2-трихлорэтанол (0,680 мл, 7,08 ммоль) и реакционную смесь перемешивали с нагреванием при 100°C в течение 2 часов. После завершения реакции реакционную смесь разбавляли насыщенным раствором соли (10 мл) и экстрагировали EtOAc (3×25 мл). Объединенные органические фазы промывали 5% лимонной кислотой (10 мл), насыщенным раствором NaHCO3 (10 мл) и насыщенным раствором соли (10 мл), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией, получая 2,2,2-трихлорэтил-5-хлор-2-(хинолин-6-ил)фенилкарбамат (0,25 г, выход 82%) в виде белого твердого вещества. МС (ESI) m/z: 431,0 (M+H+).
Пример B15: 2,2,2-Трихлорэтил-4-хлор-2-(хинолин-6-ил)фенилкарбамат получали из этил-4-хлор-2-йодбензоата, используя способ, аналогичный способу, описанному в примере B14. МС (ESI) m/z: 431,0 (M+H+).
Пример B16: Смесь 5-нитро-1H-индазола (50 г, 0,31 моль) и 10% Pd/C (5,0 г) в MeOH (400 мл) нагревали в атмосфере H2 (30 psi, 0,210 МПа) в течение ночи. После фильтрования смеси фильтрат концентрировали, получая 1H-индазол-5-иламин в виде желтого твердого вещества (40 г, выход 97%).
1H ЯМР (300 МГц, ДМСО-d6) δ 12,50 (ушир.с, 1H), 7,70 (с, 1H), 7,22 (д, J=6,6 Гц, 1H), 6,77 (д, J=6,6 Гц, 1H), 6,74 (с, 1H), 4,72 (ушир.с, 1H); MC (ESI) m/z: 134,2 (М+H+).
К раствору 1H-индазол-5-иламина (8,0 г, 60,1 ммоль) в концентрированной HCl (20 мл, 240 ммоль) добавляли водный раствор (50 мл) NaNO2 (4,2 г, 60,1 ммоль) при 0°C и полученную смесь перемешивали в течение 1 часа. Затем добавляли раствор SnCl2·2H2O (27 г, 120 ммоль) в концентрированной HCl (30 мл) при 0°C. Реакционную смесь перемешивали еще в течение 2 часов при комнатной температуре. Добавляли раствор 4-метил-3-оксопентанонитрила (8,0 г, 1,1 экв.) в этаноле (50 мл) и полученную смесь кипятили с обратным холодильником в течение ночи. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на силикагеле, получая 2-(1H-индазол-5-ил)-5-изопропил-2H-пиразол-3-иламин (8,5 г, выход 59%, две стадии).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,09 (с, 1H), 7,82 (с, 1H), 7,57 (д, J=6,6 Гц, 1H), 7,51 (д, J=6,6 Гц, 1H), 5,31 (с, 1H), 5,12 (с, 2H), 2,74 (м, 1H), 1,15 (д, J=5,1 Гц, 6H); MC (ESI) m/z: 242,3 (М+H+).
Перемешиваемый раствор 2-(1H-индазол-5-ил)-5-изопропил-2H-пиразол-3-иламина (8,0 г, 33 ммоль) в диоксане (80 мл)/10% NaOH (30 мл) обрабатывали (Boc)2O (8,6 г, 39,4 ммоль). Полученную смесь перемешивали в течение 3 часов и затем экстрагировали ДХМ (3×100 мл). Органический слой концентрировали в вакууме и остаток очищали хроматографией на силикагеле, получая трет-бутиловый эфир 5-(5-амино-3-изопропилпиразол-1-ил)индазол-1-карбоновой кислоты (6,8 г, 47%) в виде белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8,43 (с, 1H), 8,10 (д, J=9,3 Гц, 1H), 8,00 (ушир.с, 1H), 7,82 (д, J=9,3 Гц, 1H), 5,36 (с, 1H), 5,29 (ушир.с, 2H), 2,76 (м, 1H), 1,64 (с, 9H), 1,16 (д, J=7,2 Гц, 6H), MC (ESI) m/z: 442,2 (М+H+).
Раствор трет-бутил-5-(5-амино-3-изопропил-1H-пиразол-1-ил)-1H-индазол-1-карбоксилат (1,50 г) в EtOAc (15 мл) обрабатывали 1 М водным раствором NaOH (6,8 мл). К перемешиваемой двухфазной реакционной смеси при 0°C по каплям добавляли изопропенилхлорформиат (0,64 мл) в течение 1 минуты. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли EtOAc (100 мл), промывали H2O (2×30 мл), насыщенным раствором соли (30 мл), сушили (MgSO4) и концентрировали, получая трет-бутил-5-(3-изопропил-5-((проп-1-ен-2-илокси)карбониламино)-1H-пиразол-1-ил)-1H-индазол-1-карбоксилат (1,90 г, выход 99%) в виде белой пены. МС (ESI) m/z: 425,8 (M+H+).
Пример B17: Используя способ, аналогичный способу, описанному в примере B16, 1H-индазол-5-иламин (5,0 г, 37,5 ммоль) и 3-оксопентанонитрил (4,0 г, 1,1 экв.) объединяли и очищали хроматографией на силикагеле, получая 5-этил-2-(1H-индазол-5-ил)-2H-пиразол-3-иламин (5,2 г, выход 61%, две стадии).
1H ЯМР (300 МГц, DMCO-d6) δ 8,04 (с, 1H), 7,58 (с, 1H), 7,57 (д, J=6,6 Гц, 1H), 7,50 (д, J=6,6 Гц, 1H), 5,30 (с, 1H), 5,13 (ушир.с, 2H), 2,47 (кв, J=6,9 Гц, 2H), 1,14 (т, J=6,9 Гц, 3H); MC (ESI) m/z: 228,3 (М+H+).
Используя способ, аналогичный способу, описанному в примере B16, 5-этил-2-(1H-индазол-5-ил)-2H-пиразол-3-иламин (5,0 г, 22 ммоль) превращали в трет-бутиловый эфир 5-(5-амино-3-этилпиразол-1-ил)индазол-1-карбоновой кислоты (3,0 г, выход 42%) в виде белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6): δ 8,42 (с, 1H), 8,09 (д, J=6,6 Гц, 1H), 7,98 (с, 1H), 7,81 (д, J=6,6 Гц, 1H), 5,35 (с, 1H), 5,29 (ушир.с, 2H), 2,44
Трет-бутил-5-(5-амино-3-этил-1H-пиразол-1-ил)-1H-индазол-1-карбоксилат (0,50 г) превращали в трет-бутил-5-(3-этил-5-((проп-1-ен-2-илокси)карбониламино)-1H-пиразол-1-ил)-1H-индазол-1-карбоксилат (0,55 г, выход 88%), используя способ, аналогичный способу, описанному в примере 16. МС (ESI) m/z: 412,3 (M+H+).
Пример B18: Раствор N-бензгидрилиден-N'-хинолин-6-илгидразина (32 г, 0,099 моль) в EtOH (500 мл) обрабатывали концентрированной HCl (80 мл, 0,96 ммоль). После перемешивания в течение 10 минут добавляли этиловый эфир 5,5-диметил-2,4-диоксогексановой кислоты (26 г, 0,15 моль) и смесь нагревали до 80°C в течение ночи. Реакционную смесь концентрировали в вакууме, получая остаток, который промывали Et2O, получая гидрохлорид этил-5-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-3-карбоксилат (40 г, 0,11 моль, выход 112%). МС (ESI) m/z: 324,1 (M+H+).
Суспензию гидрохлорида этил-5-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-3-карбоксилата (32 г, 0,089 моль) в ТГФ (300 мл) обрабатывали водным раствором LiOH (2 N, 100 мл, 0,20 ммоль) и полученную смесь нагревали до 40°C в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении и оставшийся водный слой промывали EtOAc. Водную фазу подкисляли до pH 3 и полученный осадок собирали фильтрованием, промывали холодным эфиром и сушили в вакууме, получая 5-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-3-карбоновую кислоту (21 г, выход 71%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,03 (м, 1H), 8,50 (д, J=8,7 Гц, 1H), 8,20 (д, J=2,4 Гц, 1H), 8,15 (д, J=8,8 Гц, 1H), 7,79 (дд, J=8,7 Гц, 2,4 Гц, 1H), 7,67 (дд, J=8,4, 4,4 Гц, 1H), 6,68 (с, 1H), 1,17 (с, 9H); MC (ESI) m/z: 296,3 (М+H+).
Пример B19: Раствор нитрита натрия (502 мг, 7,27 ммоль) в H2O (8 мл) по каплям добавляли к хорошо перемешиваемой охлажденной до 0°C смеси 2-метилхинолин-6-амина (1,00 г, 6,32 ммоль) в концентрированной HCl (10 мл). Полученную смесь перемешивали при 0°C в течение 1 часа. Добавляли дигидрат хлорида олова(II) (6,13 г, 27,2 ммоль) в концентрированной HCl (8 мл) и перемешивание продолжали при 0°C в течение 1 часа и затем при комнатной температуре в течение 2 часов. Добавляли этанол (60 мл) и 4,4-диметил-3-оксопентанонитрил (1,03 г, 8,22 ммоль) и смесь кипятили с обратным холодильником в течение ночи. После завершения реакции реакционную смесь концентрировали в вакууме и разбавляли этилацетатом (100 мл). Смесь охлаждали на бане лед/вода и подщелачивали (pH~8), используя твердый гидроксид натрия. Раствор фильтровали через целит и осадок на фильтре промывали водой (50 мл) и этилацетатом (100 мл). Органическую фазу отделяли, промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали, получая пену. Пену перемешивали в эфире (50 мл) и оставляли стоять на несколько часов. Полученное твердое веществе собирали фильтрованием и сушили в вакууме, получая 3-трет-бутил-1-(2-метилхинолин-6-ил)-1H-пиразол-5-амин (428 мг, выход 24%). МС (ESI) m/z: 281,2 (M+H+).
Раствор 3-трет-бутил-1-(2-метилхинолин-6-ил)-1H-пиразол-5-амина (420 мг, 1,50 ммоль) в CH2Cl2 (15 мл) обрабатывали пиридином (592 мг, 7,49 ммоль) и TROC-Cl (333 мг, 1,57 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, затем промывали 5% лимонной кислотой (2×20 мл), насыщенным водным раствором NaHCO3 (20 мл) и насыщенным раствором соли (20 мл). Органическую фазу сушили (Na2SO4) и концентрировали, получая смесь 2,2,2-трихлорэтил-3-трет-бутил-1-(2-метилхинолин-6-ил)-1H-пиразол-5-илкарбамата (выход 73%), с 16% примеси бис-Troc-аддукта. Смесь использовали без дополнительной очистки. МС (ESI) m/z: 456,5 (M+H+).
Пример B20: Используя способ, аналогичный способу, описанному в примере B4, имидазо[1,2-a]пиридин-6-илбороновую кислоту (0,200 г, 1,23 ммоль) и этил-3-трет-бутил-1H-пиразол-5-карбоксилат (0,267 г, 1,36 ммоль) объединяли, получая этил-3-трет-бутил-1-(H-имидазо[1,2-a]пиридин-6-ил)-1H-пиразол-5-карбоксилат (0,0355 г, выход 9%) в виде бесцветного масла. МС (ESI) m/z: 313,2 (M+H+).
Используя способ, аналогичный способу, описанному в примере B4, этил-3-трет-бутил-1-(H-имидазо[1,2-a]пиридин-6-ил)-1H-пиразол-5-карбоксилат (0,071 г, 0,23 ммоль) превращали в 3-трет-бутил-1-(H-имидазо[1,2-a]пиридин-6-ил)-1H-пиразол-5-карбоновую кислоту (0,0643 г, выход 99%) в виде белого твердого вещества. МС (ESI) m/z: 285,0 (M+H+).
Пример B21: Используя способ, аналогичный способу, описанному в примере B4, имидазо[1,2-a]пиридин-6-илбороновую кислоту (0,500 г, 3,09 ммоль) и этил-3-изопропил-1H-пиразол-5-карбоксилат (0,619 г, 3,40 ммоль) объединяли, получая этил-3-изопропил-1-(H-имидазо[1,2-a]пиридин-6-ил)-1H-пиразол-5-карбоксилат (0,098 г, выход 11%) в виде бесцветного масла. МС (ESI) m/z: 299,3 (M+H+).
Используя способ, аналогичный способу, описанному в примере B4, этил-3-изопропил-1-(H-имидазо[1,2-a]пиридин-6-ил)-1H-пиразол-5-карбоксилат (0,098 г, 0,33 ммоль) превращали в 3-изопропил-1-(H-имидазо[1,2-a]пиридин-6-ил)-1H-пиразол-5-карбоновую кислоту (0,087 г, выход 98%) в виде белого твердого вещества. МС (ESI) m/z: 271,0 (M+H+).
Пример B22: К перемешиваемой суспензии 6-аминобензотиазола (0,500 г, 3,33 ммоль) в концентрированной HCl (5 мл) при 0-5°C добавляли раствор NaNO2 (0,276 г, 3,99 ммоль) в H2O (5 мл). Смесь перемешивали при 0-5°C в течение 75 минут вплоть до получения прозрачного желтого раствора. Затем добавляли раствор SnCl2·2H2O (2,76 г, 13,3 ммоль) в концентрированной HCl (5 мл). После завершения добавления суспензию перемешивали при комнатной температуре в течение 2 часов. Добавляли 4-метил-3-оксопентанонитрил (0,444 г, 3,99 ммоль) и EtOH (50 мл) и реакционную смесь перемешивали с нагреванием при 75°C. Через 18 часов, когда реакция была завершена, реакционную смесь охлаждали до комнатной температуры и концентрировали до получения водного остатка. Остаток полностью охлаждали на льду и сильно подщелачивали (pH 12-13) добавлением 6М NaOH. Еще холодную смесь экстрагировали EtOAc (2×). Объединенные органические фазы промывали H2O (2×), насыщенным раствором соли (1×), сушили (MgSO4), фильтровали и упаривали, получая неочищенный 1-(бензо[d]тиазол-6-ил)-3-изопропил-1H-пиразол-5-амин (0,8 г, выход 93%) в виде масла, которое использовали как таковое для следующей реакции.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,36 (с, 1H), 8,30 (д, J=2,4 Гц, 1H); 8,10 (д, J=8,8 Гц, 1H), 7,74 (дд, J=2,4 и 8,8 Гц, 1H), 5,36 (с, 1H), 5,33 (ушир.с, 2H), 2,76 (септет, J=6,8 Гц, 1H), 1,17 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 259,0 (М+H+).
К перемешиваемому раствору 1-(бензо[d]тиазол-6-ил)-3-изопропил-1H-пиразол-5-амина (0,80 г, 3,1 ммоль) и пиридина (0,51 мл, 6,2 ммоль) в CH2Cl2 (30 мл) при комнатной температуре добавляли Troc-Cl (0,51 мл, 3,7 ммоль). Через 2 часа, когда реакция была завершена, реакционную смесь промывали 10% CuSO4 (2×), H2O (1×), насыщенным раствором соли (1×), сушили (MgSO4), упаривали и очищали флэш-хроматографией на колонке (EtOAc/гексаны), получая 2,2,2-трихлорэтил-1-(бензо[d]тиазол-6-ил)-3-изопропил-1H-пиразол-5-илкарбамат (0,31 г, выход 23%) в виде масла. МС (ESI) m/z: 433,0 (M+H+), 435,0 (M+2+H+).
Пример B23: 1-Метил-5-нитро-1H-бензо[d]имидазол (полученный, как описано в WO 2005/092899; 1,14 г, 6,43 ммоль) в EtOH (50 мл) перемешивали в атмосфере H2 (1 атм) при комнатной температуре в присутствии 10% Pd/C (50% масс. H2O, 1,37 г, 0,643 ммоль). Через 18 часов, когда реакция была завершена, реакционную смесь фильтровали через целит, промывали EtOH. Объединенные фильтраты концентрировали, получая неочищенный 1-метил-1H-бензо[d]имидазол-5-амин (1,02 г, выход 108%) в виде темно-оранжевого масла, которое использовали как таковое для следующей реакции.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,87 (с, 1H), 7,17 (д, J=8,4 Гц, 1H), 6,75 (д, J=2,0 Гц, 1H), 6,59 (дд, J=2,0 и 8,4 Гц, 1H), 4,73 (ушир.с, 2H), 3,69 (с, 3H); MC (ESI) m/z: 148,0 (М+H+).
Используя способ, аналогичный способу, описанному в примере B22, 1-метил-1H-бензо[d]имидазол-5-амин (0,50 г, 3,4 ммоль), NaNO2 (0,28 г, 4,1 ммоль), SnCl2·2H2O (2,8 г, 14 ммоль) и 4-метил-3-оксопентанонитрил (0,45 г, 4,1 ммоль) объединяли, получая неочищенный 3-изопропил-1-(1-метил-1H-бензо[d]имидазол-5-ил)-1H-пиразол-5-амин (0,63 г, выход 73%) в виде пены, которую использовали как таковую для следующей реакции.
1H ЯМР (400 МГц, ДМСО-d6): δ 8,22 (с, 1H), 7,72 (дд, J=0,40 и 1,2 Гц, 1H), 7,60 (дд, J=0,40 и 8,4 Гц, 1H), 7,42 (дд, J=2,0 и 8,4 Гц, 1H), 5,32 (с, 1H), 5,08 (ушир.с, 2H), 3,85 (с, 3H), 2,75 (септет, J=6,8 Гц, 1H), 1,16 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 250,0 (М+H+).
Используя способ, аналогичный способу, описанному в примере B22, 3-изопропил-1-(1-метил-1H-бензо[d]имидазол-5-ил)-1H-пиразол-5-амин (0,63 г, 2,5 ммоль) превращали в 2,2,2-трихлорэтил-3-изопропил-1-(1-метил-1H-бензо[d]имидазол-5-ил)-1H-пиразол-5-илкарбамат (0,5 г, выход 47%) и выделяли в виде масла.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,86 (ушир.с, 1H), 8,24 (с, 1H), 7,67 (ушир.с, 1H), 7,62 (д, J=8,4 Гц, 1H), 7,36 (дд, J=2,0 и 8,4 Гц, 1H), 6,23 (с, 1H), 4,81 (с, 2H), 3,85 (с, 3H), 2,90 (септет, J=6,8 Гц, 1H), 1,22 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 430,0 (М+H+), 432,0 (M+2+H+).
Пример B24: К перемешиваемому раствору 1-(2-(бензилоксикарбонил)-1,2,3,4-тетрагидроизохинолин-6-ил)-3-трет-бутил-1H-пиразол-5-карбоновой кислоты из примера B1 (0,320 г, 0,738 ммоль, 1,0 экв.) и TEA (0,118 мл, 0,849 ммоль, 1,15 экв.) в 1,4-диоксане (7,5 мл) при 20°C добавляли DPPA (0,183 мл, 0,849 ммоль, 1,15 экв.). Через 30 минут добавляли 2,2,2-трихлорэтанол (1,0 мл, 10,4 ммоль, 14 экв.) и реакционную смесь перемешивали при нагревании до 100°C. Через 4 часа, когда реакция была завершена, реакционную смесь разбавляли насыщенным раствором соли и экстрагировали EtOAc (2×). Объединенные органические фазы промывали 5% лимонной кислотой (1×), насыщенным раствором NaHCO3 (1×) и насыщенным раствором соли (1×), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая бензил 6-(3-трет-бутил-5-((2,2,2-трихлорэтокси)карбонил)амино-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,260 г, выход 61%) в виде масла. МС (ESI) m/z: 579,0 (M+H+), 581,0 (M+2+H+).
Пример B25: Используя способ, описанный в примере B26, 3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-амин из примера B9 (1,00 г, 4,0 ммоль), бис(триметилсилил)амид лития (1,0M в ТГФ, 7,9 мл, 7,9 ммоль) и изопропенилхлорформиат (0,48 мл, 4,4 ммоль) объединяли, получая проп-1-ен-2-ил-3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-илкарбамат (0,85 г, выход 65%). МС (ESI) m/z: 337,2 (M+H+).
Пример B26: Раствор 5-трет-бутил-2-хинолин-6-ил-2H-пиразол-3-иламина из примера B3 (1,00 г, 3,8 ммоль) в ТГФ (20 мл) охлаждали до -78°C и обрабатывали бис(триметилсилил)амидом лития (1,0М в ТГФ, 7,5 мл, 7,5 ммоль). Полученную смесь перемешивали при -78°C в течение 30 минут. Добавляли изопропенилхлорформиат (0,45 мл, 0,41 ммоль) и перемешивание продолжали при -78°C в течение 30 минут. Реакционную смесь гасили при -78°C водным раствором HCl (2н., 4 мл, 8 ммоль), нагревали до комнатной температуры и распределяли между водой (200 мл) и EtOAc (200 мл). Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая проп-1-ен-2-ил-3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-илкарбамат (0,5 г, выход 38%). МС (ESI) m/z: 351,2 (M+H+).
Пример B27: 4-Фтор-3-нитрофенилбороновую кислоту (0,9 г, 4,9 ммоль) растворяли в CH2Cl2 (10 мл) и пиридине (1 мл) с молекулярными ситами (активированные 4Å) и сушили в течение 6 часов. Смесь 4-фтор-3-нитрофенилбороновой кислоты, трет-бутил-3-изопропил-1H-пиразол-5-карбоксилата (1,0 г, 4,9 ммоль), ацетата меди(II) (0,88 г, 4,9 ммоль) и молекулярных сит (4Å, активированные, порошок) перемешивали при комнатной температуре в течение 7 суток на открытом воздухе. Реакционную смесь фильтровали через подушку целита. Фильтрат концентрировали в вакууме и очищали хроматографией на колонке с силикагелем (EtOAc/гексан), получая трет-бутил-1-(4-фтор-3-нитрофенил)-3-изопропил-1H-пиразол-5-карбоксилат (0,74 г, выход 44%). МС (ESI) m/z: 350,3 (M+H+).
К раствору трет-бутил-1-(4-фтор-3-нитрофенил)-3-изопропил-1H-пиразол-5-карбоксилата (0,74 г, 2,1 ммоль) в смеси ТГФ/вода (12 мл) добавляли LiOH (300 мг, 13 ммоль) и H2O2 (30% масс., 0,96 мл). Реакционную смесь нагревали в течение ночи при 60°C. Добавляли раствор Na2S2O3 до тех пор, пока тест на пероксид (йодкрахмальная бумага) не становился отрицательным. Добавляли уксусную кислоту вплоть до значения pH 4-5. Раствор экстрагировали EtOAc и органический слой промывали насыщенным раствором соли, сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на колонке с силикагелем (EtOAc/гексаны), получая трет-бутил-1-(4-гидрокси-3-нитрофенил)-3-изопропил-1H-пиразол-5-карбоксилат (0,27 г, выход 37%). МС (ESI) m/z: 348,3 (M+H+).
К раствору трет-бутил-1-(4-гидрокси-3-нитрофенил)-3-изопропил-1H-пиразол-5-карбоксилата (0,27 г, 0,78 ммоль) в смеси этилацетат/метанол (1:1, 10 мл) добавляли палладий-на-угле (30 мг) и смесь гидрировали (50 psi, 0,349 МПа) в течение ночи в аппарате Парра. Раствор фильтровали и промывали метанолом. Объединенные фильтраты концентрировали, получая трет-бутил-1-(3-амино-4-гидроксифенил)-3-изопропил-1H-пиразол-5-карбоксилат. Неочищенный трет-бутил-1-(3-амино-4-гидроксифенил)-3-изопропил-1H-пиразол-5-карбоксилат обрабатывали 25% ТФУ в CH2Cl2 (2 мл) и перемешивали в течение ночи при комнатной температуре. Растворитель выпаривали, получая 1-(бензо[d]оксазол-5-ил)-3-трет-бутил-1H-пиразол-5-карбоновую кислоту. К раствору 1-(бензо[d]оксазол-5-ил)-3-трет-бутил-1H-пиразол-5-карбоновой кислоты в ксиленах (3 мл) добавляли триэтилортоформиат (0,16 мл, 0,96 ммоль) и каталитическое количество PPTS. Реакционную смесь нагревали при 140°C в течение 4 часов. Растворитель выпаривали и остаток обрабатывали метиленхлоридом при перемешивании в течение 1 часа. Полученное твердое вещество отфильтровывали и промывали метиленхлоридом, получая 1-(бензо[d]оксазол-5-ил)-3-изопропил-1H-пиразол-5-карбоновую кислоту (0,1 г, выход 45%: для трех стадий). МС (ESI) m/z: 272,0 (M+H+).
Пример B28: В толуол (8 мл) помещали 1-(дифенилметилен)гидразин (1,00 г, 5,10 ммоль), ацетат палладия (10,4 мг, 0,0464 ммоль) и 2-(дифенилфосфино)-1-(2-(дифенилфосфино)нафталин-1-ил)нафталин (44 мг, 0,0696 ммоль) и реакционную смесь перемешивали при 100°C в атмосфере Ar в течение 5 минут и затем охлаждали до комнатной температуры. К полученному темно-фиолетовому раствору добавляли 6-бромхиноксалин (970 мг, 4,64 ммоль), трет-бутоксид натрия (624 мг, 6,50 ммоль) и толуол (2 мл). Реакционную смесь помещали в атмосферу Ar и нагревали до 100°C в течение 5 часов, охлаждали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли эфиром (50 мл) и водой (30 мл) и фильтровали через подушку целита. Подушку промывали эфиром (20 мл) и водой (20 мл). Объединенные органические слои промывали насыщенным раствором соли (50 мл), сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией (этилацетат/гексаны), получая 1-(дифенилметилен)-2-(хиноксалин-6-ил)гидразин (305 мг, выход 20%) в виде ярко-желтой пены.
1H ЯМР (300 МГц, ДМСО-d6) δ 7,35-7,41 (м, 5H), 7,51-7,53 (м, 2H), 7,58-7,65 (м, 3H), 7,75 (с, 1H), 7,89 (с, 2H), 8,61 (с, 1H), 8,74 (с, 1H), 9,60 (с, 1H); MC (ESI) m/z: 325,0 (М+H+).
В этанол (10 мл) помещали 1-(дифенилметилен)-2-(хиноксалин-6-ил)гидразин (300 мг, 0,925 ммоль), пивалоилацетонитрил (156 мг, 1,25 ммоль) и гидрат пара-толуолсульфоновой кислоты (704 мг, 3,70 ммоль). Реакционную смесь кипятили с обратным холодильником и перемешивали в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (50 мл) и насыщенным раствором бикарбоната натрия (50 мл). Органическую фазу отделяли, промывали 1н. NaOH (30 мл) и насыщенным раствором соли (30 мл), сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией (колонка Si-25, этилацетат/гексаны), получая желто-коричневую пену, идентифицированную как 3-трет-бутил-1-(хиноксалин-6-ил)-1H-пиразол-5-амин (57 мг, выход 23%). МС (ESI) m/z: 268,2 (M+H+).
Пример B29: К раствору фенэтиламина (60,5 г, 0,5 моль) и Na2CO3 (63,6 г, 0,6 моль) в EtOAc/HO (800 мл, 4:1) по каплям добавляли этилхлорформиат (65,1 г, 0,6 моль) при 0°C в течение 1 часа. Смесь нагревали до комнатной температуры и перемешивали еще в течение 1 часа. Органическую фазу отделяли и водный слой экстрагировали EtOAc. Объединенные органические фазы промывали H2O и насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали флэш-хроматографией, получая этилфенэтилкарбамат (90,2 г).
1H ЯМР (400 МГц, CDCl3) δ 7,32-7,18 (м, 5H), 4,73 (ушир.с, 1H), 4,14-4,08 (кв, J=6,8 Гц, 2H), 3,44-3,43 (м, 2H), 2,83-2,79 (т, J=6,8 Гц, 2H), 1,26-1,21 (т, J=6,8 Гц, 3H).
Суспензию этилфенэтилкарбамата (77,2 г, 40 ммоль) в полифосфорной кислоте (300 мл) нагревали до 140-160°C и перемешивали в течение 2,5 часов. Реакционную смесь охлаждали до комнатной температуры, осторожно вливали в смесь льда с H2O и перемешивали в течение 1 часа. Водный раствор экстрагировали EtOAc (3×300 мл). Объединенные органические фазы промывали H2O, 5% K2CO3 и насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали флэш-хроматографией, получая 3,4-дигидро-2H-изохинолин-1-он (24 г).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,91 (ушир.с, 1H), 7,83 (д, J=7,5 Гц, 1H), 7,43 (т, J=7,5 Гц, 1H), 7,33-7,25 (м, 2H), 3,37-3,32 (м, 2H), 2,87 (т, J=6,6 Гц, 2H).
К смеси концентрированной HNO3 и концентрированной H2SO4 (200 мл, 1:1), охлажденной на бане, содержащей смесь льда и соли, по каплям добавляли 4-дигидро-2H-изохинолин-1-он (15 г, 0,102 моль) в течение 15 минут. После перемешивания в течение 2 часов полученную смесь вливали в смесь льда и H2O и перемешивали в течение 30 минут. Осадок отфильтровывали, промывали H2O и сушили на воздухе, получая 7-нитро-3,4-дигидро-2H-изохинолин-1-он (13 г).
1H ЯМР (300 МГц, ДМСО-d6) δ 8,53 (д, J=2,4 Гц, 1H), 8,31 (д, J=2,4 Гц, 1H), 8,29 (д, J=2,4 Гц, 1H), 7,62 (д, J=8,4 Гц, 1H), 3,44-3,39 (м, 2H), 3,04 (т, J=6,6 Гц, 2H).
Суспензию 7-нитро-3,4-дигидро-2H-изохинолин-1-она (11,6 г, 60 ммоль) и 10% Pd/C (1,2 г,) в MeOH перемешивали в течение ночи при комнатной температуре в атмосфере H2 (40 psi, 0,279 МПа). Смесь фильтровали через целит и промывали MeOH. Фильтрат упаривали в вакууме, получая 8,2 г 7-амино-3,4-дигидро-2H-изохинолин-1-он, который использовали без дополнительной очистки.
К суспензии 7-амино-3,4-дигидро-2H-изохинолин-1-она (8,1 г, 50 ммоль) в концентрированной HCl (100 мл) на бане, содержащей лед и H2O, по каплям добавляли раствор NaNO2 (3,45 г, 50 ммоль) в H2O с такой скоростью, чтобы температура реакционной смеси никогда не превышала 5°C. Через 30 минут по каплям добавляли раствор SnCl2·2H2O (22,5 г, 0,1 моль) в концентрированной HCl (150 мл). Полученную смесь перемешивали еще в течение 2 часов при 0°C. Осадок собирали отсасыванием, промывали эфиром, получая 7-гидразино-3,4-дигидро-2H-изохинолин-1-он (8,3 г), который использовали для следующей реакции без дополнительной очистки.
Смесь 7-гидразино-3,4-дигидро-2H-изохинолин-1-она (8,0 г, 37,6 ммоль) и 4,4-диметил-3-оксопентанонитрила (5,64 г, 45 ммоль) в EtOH (100 мл) и концентрированной HCl (10 мл) кипятили с обратным холодильником в течение ночи. После удаления растворителя остаток промывали эфиром, получая гидрохлорид 7-(5-амино-3-трет-бутилпиразол-1-ил)-3,4-дигидро-2H-изохинолин-1-она в виде желтого твердого вещества (11,5 г, выход 96%), который использовали без дополнительной очистки.
К раствору гидрохлорида 7-(5-амино-3-трет-бутилпиразол-1-ил)-3,4-дигидро-2H-изохинолин-1-она (0,5 г, 1,76 ммоль) в CH2Cl2 (25 мл) добавляли пиридин (0,22 мл) и трихлорэтилхлорформиат (0,27 мл) при 0°C и смесь перемешивали в течение ночи при комнатной температуре. ЖХ-МС показала, что реакция не была завершена. Добавляли пиридин (0,25 мл) и TROC-Cl (0,25 мл) и затем смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли CH2Cl2, органический слой промывали 3М HCl и насыщенным раствором соли, сушили (Na2SO4) и концентрировали в вакууме. Остаток растворяли в EtOAc и добавляли гексан. Твердое вещество отфильтровывали, получая 2,2,2-трихлорэтил-3-трет-бутил-1-(1-оксо-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиразол-5-илкарбамат (0,46 г, выход 57%). МС (ESI) m/z: 458,0 (M+H+).
Пример B30: К раствору гидрохлорида 7-(5-амино-3-трет-бутилпиразол-1-ил)-3,4-дигидро-2H-изохинолин-1-она из примера B29 (20 г, 0,070 моль) в ТГФ (400 мл) порциями добавляли LAH (15 г, 0,395 моль) при 0-5°C. Полученную смесь кипятили с обратным холодильником в течение ночи, затем добавляли 10% раствор NaOH. После перемешивания в течение 1 часа при комнатной температуре добавляли Boc2O (23 г, 0,106 моль) и раствор перемешивали в течение ночи. После фильтрования фильтрат концентрировали, получая сырой продукт, который очищали обращенно-фазовой хроматографией, получая трет-бутиловый эфир 7-(5-амино-3-трет-бутилпиразол-1-ил)-3,4-дигидро-1H-изохинолин-2-карбоновой кислоты (12 г, выход 75%).
1H ЯМР (300 МГц, ДМСО-d6) δ 7,32 (с, 1H), 7,29 (д, J=2,7 Гц, 1H), 7,18 (д, J=8,4 Гц, 1H), 5,32 (с, 1H), 5,15 (с, 1H), 4,51 (с, 2H), 3,52 (т, J=5,6 Гц, 2H), 2,75 (т, J=5,6 Гц, 2H), 1,40 (с, 9H), 1,17 (с, 9H); MC (ESI) m/z: 371 (М+H+).
К перемешиваемому раствору трет-бутил-7-(5-амино-3-трет-бутил-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (0,50 г, 1,35 ммоль) и Troc-Cl (0,19 мл, 1,38 ммоль) в EtOAc (15 мл) добавляли насыщенный раствор NaHCO3 (2,75 мл, 2,02 ммоль). Полученную двухфазную смесь перемешивали при комнатной температуре в течение 5 часов. Слои разделяли и органический слой промывали насыщенным раствором NaHCO3 (1×) и насыщенным раствором соли (1×), сушили (Na2SO4) и концентрировали в вакууме, получая трет-бутил-7-(3-трет-бутил-5-((2,2,2-трихлорэтокси)карбонил)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,69 г, выход 94%). МС (ESI) m/z: 545,0 (M+H+).
Пример 1: Раствор соединения из примера B3 (7,0 г, 15,8 ммоль), соединения из примера A2 (4,14 г, 15,8 ммоль) и DIEA (4,5 г, 34,9 ммоль) в ДМСО (70 мл) нагревали на масляной бане при 70°C в течение 8 часов. Реакционную смесь вливали в воду (500 мл), перемешивали в течение ночи и твердые вещества отделяли фильтрованием. Последующая кристаллизация неочищенного продукта из толуола и ацетона давала 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину в виде белого кристаллического твердого вещества (4,06 г, выход 46%).
1H ЯМР (ДМСО-d6) δ 8,90 (м, 2H), 8,79 (м, 1H), 8,52 (м, 2H), 8,2 (м, 3H), 7,96 (дд, J=9, 2 Гц, 1H), 7,63 (дд, J=8, 4 Гц, 1H), 7,40 (ушир.с, 1H), 7,30(дд, J=3, 12 Гц, 1H), 7,17 (м, 1H), 7,05 (д, J=9 Гц, 1H), 6,50 (с, 1H), 2,80 (д, J=5 Гц), 1,32 (с, 9H); MC (ESI) m/z: 554 (М+H+).
Свободное основание обрабатывали 0,1М HCl, получая бис гидрохлорид 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины в виде бледно-желтого рыхлого твердого вещества (2,40 г).
1H ЯМР (ДМСО-d6) δ 9,56 (с, 1H), 9,26 (м, 2H), 9,10 (д, J=8 Гц, 1H), 8,85 (м, 1H), 8,55 (м, 2H), 8,46 (д, J=9 Гц, 1H), 8,33 (дд, J=9, 2 Гц, 1H), 8,11 (т, J=9 Гц, 1H), 8,03 (дд, дд, J=9, 2 Гц, 1H), 7,46 (д, J=3 Гц, 1H), 7,30 (дд, J=3, 12 Гц, 1H), 7,20 (дд, J=3, 6 Гц, 1H), 7,04 (ушир.д, J=7 Гц, 1H), 6,49 (с, 1H), 2,80 (д, J=4,5 Гц), 1,33 (с, 9H).
Пример 2: Соединение из примера B1 (142 мг, 0,33 ммоль) и Et3N (0,15 мл, 0,72 ммоль) объединяли в диоксане (3 мл). Добавляли DPPA (0,13 мл, 0,59 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 90 минут. Добавляли соединение из примера A2 (94 мг, 0,36 ммоль) и полученную смесь нагревали до 95°C в течение 4 часов. Реакционную смесь концентрировали в вакууме и очищали хроматографией на силикагеле, получая бензил 6-(3-трет-бутил-5-(3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)уреидо)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (95 мг, выход 42%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,00 (ушир.с, 1H), 8,84 (с, 1H), 8,79 (кв, J=4,8 Гц, 1H), 8,52 (д, J=5,6 Гц, 1H), 8,20 (т, J=9,2 Гц, 1H), 7,40-7,28 (м, 10H), 7,17 (дд, J=5,6, 2,8 Гц, 1H), 7,05 (м, 1H), 6,40 (с, 1H), 5,14 (с, 2H), 4,66 (м, 2H), 3,68 (м, 2H), 2,91 (т, J=5,6 Гц, 2H), 2,79 (д, J=4,8 Гц, 3H), 1,27 (с, 9H); MC (ESI) m/z: 692,2 (М+H+).
Раствор бензил-6-(3-трет-бутил-5-(3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)уреидо)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (93 мг, 0,13 ммоль) в метаноле (3 мл) обрабатывали 10% Pd/C (50% влаги, 74 мг, 0,03 ммоль) и муравьиной кислотой (88%, 0,60 мл, 14 ммоль). Полученную реакционную смесь перемешивали в течение 90 минут и фильтровали через целит, промывая метанолом. Фильтрат концентрировали в вакууме и очищали на силикагеле, получая 1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину (42 мг, выход 56%). Продукт обрабатывали водным раствором HCl (0,1М, 0,75 мл), получая гидрохлорид 1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,38 (ушир.с, 2H), 9,10 (д, J=1,8 Гц, 1H), 9,05 (с, 1H), 8,80 (м, 1H), 8,53 (д, J=5,4 Гц, 1H), 8,15 (т, J=9,1 Гц, 1H), 7,46-7,34 (м, 4H), 7,32 (дд, J=11,6, 2,8 Гц, 1H), 7,18 (м, 1H), 7,05 (м, 1H), 6,39 (с, 1H), 4,33 (ушир.с, 2H), 3,40 (2H закрыт H2O), 3,09 (т, J=6,0 Гц, 2H), 2,79 (д, J=5,0 Гц, 3H), 1,28 (с, 9H); MC (ESI) m/z: 558,3 (М+H+).
Пример 3: Используя общий способ A, соединение из примера B4 (80 мг, 0,27 ммоль), из примера A1 (0,18 г, 0,68 ммоль), триэтиламин (30 мг, 0,30 ммоль) и DPPA (82 мг, 0,30 ммоль) объединяли, получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(3-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину, которую обрабатывали 3М HCl/EtOAc, получая ее HCl-соль (125 мг, выход 78%).
1H ЯМР (400 МГц, ДМСО-d6): δ 9,79 (ушир.м, 1H), 9,16 (ушир.м, 1H), 9,05 (ушир.м, 1H), 8,93 (ушир.м, 1H), 8,79 (ушир.м, 1H), 8,53 (д, J=5,6 Гц, 1H), 8,42 (ушир.м, 1H), 8,33 (ушир.м, 1H), 8,22 (ушир.м, 1H), 7,91 (ушир.м, 1H), 7,68 (дд, J=2,4 и 14,4 Гц, 1H), 7,37 (д, J=2,4 Гц, 1H), 7,34 (т, J=9,2 Гц, 1H), 7,19 (ушир.м, 1H), 6,49 (с, 1H), 2,79 (д, J=5,2 Гц, 3H), 1,31 (с, 9H); MC (ESI) m/z: 554,2 (М+H+).
Пример 4: К раствору соединения из примера B8 (0,132 г, 0,30 ммоль) в ТГФ (1,0 мл) добавляли соединение из примера A2 (0,083 г, 0,315 ммоль) и 1-метилпирролидин (2,6 мг, 0,03 ммоль). Смесь нагревали при 55°C в течение ночи. Растворитель удаляли и остаток растворяли в MeOH (4,5 мл), к которому добавляли 3 М HCl/EtOAc (1,3 мл, 3,8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи с последующим нагреванием при 55°C в течение 3 часов. Реакционную смесь концентрировали досуха, разбавляли насыщенным раствором NaHCO3 (7 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали насыщенным раствором NaHCO3 (7 мл), H2O (7 мл) и насыщенным раствором соли (7 мл), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией, получая 1-(3-трет-бутил-1-(1H-индазол-5-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину (80 мг, выход 49%) в виде белого твердого вещества. Продукт превращали в соответствующую HCl-соль в результате взаимодействия с HCl (4,0 М в диоксане, 1,0 экв.).
1H ЯМР (ДМСО-d6) δ 9,17 (с, 1H), 9,13 (с, 1H), 8,99 (м, 1H), 8,56 (д, J=5,6 Гц, 1H), 8,23-8,18 (м, 2H), 7,96 (д, J=2,0 Гц, 1H), 7,72 (д, J=8,8 Гц, 1H), 7,58 (д, J=2,4 Гц, 1H), 7,49 (дд, J=8,8, 1,6 Гц, 1H), 7,32 (дд, J=11,6, 2,8 Гц, 1H), 7,24 (дд, J=6,0, 3,0 Гц, 1H), 7,07 (дд, J=8,8, 1,6 Гц, 1H), 6,47 (с, 1H), 2,81 (д, J=4,8 Гц, 3H), 1,30 (с, 9H); MC (ESI) m/z: 543,2 (М+H+).
Пример 5: Используя общий способ A, соединение из примера B4 (80 мг, 0,27 ммоль) и соединение из примера A6 (99 мг, 0,38 ммоль) объединяли, получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(3-метил-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину (149 мг, выход 99%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,08 (с, 1H), 8,97 (дд, J=4,1, 1,2 Гц, 1H), 8,77 (кв, J=4,6 Гц, 1H), 8,62 (с, 1H), 8,51-8,48 (м, 2H), 8,20-8,16 (м, 2H), 7,97 (д, J=8,9, 2,0 Гц, 1H), 7,63 (дд, J=8,5, 4,2 Гц, 1H), 7,46 (д, J=2,4 Гц, 1H), 7,32 (дд, J=8,7, 2,5 Гц, 1H), 7,27 (д, J=2,6 Гц, 1H), 7,08 (м, 1H), 7,06 (д, J=8,7 Гц, 1H), 6,47 (с, 1H), 2,78 (д, J=4,6 Гц, 3H), 2,04 (с, 3H), 1,33 (с, 9H); MC (ESI) m/z: 550,2 (М+H+).
Пример 6: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (0,19 г, 0,43 ммоль) и соединение из примера A7 (0,11 г, 0,43 ммоль) объединяли, получая гидрохлорид 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)мочевины (0,160 г, выход 64%).
1H ЯМР (ДМСО-d6) δ 9,55 (с, 1H), 9,27-9,24 (м, 2H), 9,10 (д, J=8,8 Гц, 1H), 8,56-8,54 (м, 2H), 8,46 (д, J=9,2 Гц, 1H), 8,32 (дд, J=9,6, 2,4 Гц, 1H), 8,27 (с, 1H), 8,13 (т, J=9,2 Гц, 1H), 8,04 (дд, J=8,4, 5,2 Гц, 1H), 7,85 (с, 1H), 7,52 (д, J=2,4 Гц, 1H), 7,32 (дд, J=11,6, 2,4 Гц, 1H), 7,24 (дд, J=6,0, 2,8 Гц, 1H), 7,05 (дкв, J=8,8, 1,2 Гц, 1H), 6,50 (с, 1H), 1,33 (с, 9H); MC (ESI) m/z: 540,3 (М+H+).
Пример 7: Соединение из примера B3 (0,12 г, 0,27 ммоль), соединение из примера A9 (63 мг, 0,27 ммоль) и DIEA (77 мг, 0,60 ммоль) объединяли в ДМСО (1 мл) и нагревали в течение ночи при 50-55°C. Добавляли воду (50 мл) и смесь экстрагировали EtOAc (3×100 мл), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на колонке с силикагелем (EtOAc/гексан), получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метиламино)пиридин-4-илокси)фенил)мочевину. Твердое вещество обрабатывали 0,100М HCl (2 экв.), получая гидрохлорид 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метиламино)пиридин-4-илокси)фенил)мочевины (52 мг, выход 32%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,23 (ушир.с, 1H), 9,17 (ушир.с, 1H), 9,06 (ушир.м, 1H), 8,66 (ушир.м, 1H), 8,53 (ушир.с, 1H), 8,0-8,3 (м, 4H), 7,92 (д, J=6,8 Гц, 1H), 7,74 (м, 1H), 7,35 (дд, J=2,8 и 11,6 Гц, 1H), 7,07 (м, 1H), 6,62 (д, J=6,4 Гц, 1H), 6,48 (с, 1H), 6,18 (ушир.с, 1H), 2,88 (д, J=4,8 Гц, 2H), 1,32 (с, 9H); LC-MC (EI) m/z: 526,2 (М+H+).
Пример 8: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B6 (0,178 г, 0,335 ммоль), соединение из примера A10 (0,0840 г, 0,352 ммоль) и DIEA (0,0701 мл, 0,402 ммоль) объединяли, очищали флэш-хроматографией на колонке (EtOAc/гексаны) и очищали второй раз флэш-хроматографией на колонке (EtOAc/CH2Cl2), получая трет-бутил-5-(3-трет-бутил-5-(3-(5-(5-хлорпиридин-3-илокси)-2-фторфенил)уреидо)-1H-пиразол-1-ил)-1H-индазол-1-карбоксилат (0,0486 г, выход 23%) в виде твердого вещества.
1H ЯМР (400 МГц, ацетон-d6) δ 8,52 (ушир.д, 1H, J=2,8 Гц), 8,46 (с, 1H), 8,37 (д, 1H, J=2,0 Гц), 8,35-8,32 (м, 2H), 8,24 (дт, 1H, J=0,8 и 8,8 Гц), 8,818 (дд, 1H, J=2,8 и 6,8 Гц), 7,22 (дд, 1H, J=8,8 и 10,8 Гц), 6,81 (ддд, 1H, J=3,2, 4,0 и 8,8 Гц), 1,73 (с, 9H), 1,34 (с, 9H); MC (ESI) m/z: 620,2 (М+H+).
Вещество, полученное на предыдущей стадии (0,0486 г, 0,078 ммоль), и 4М HCl в диоксане (5,0 мл) объединяли при комнатной температуре. Добавляли небольшое количество MeOH, получая гомогенный раствор. Смесь нагревали в течение ночи при 40°C. После завершения реакции реакционную смесь концентрировали в вакууме, растворяли в MeCN/H2O, замораживали и подвергали лиофилизации, получая 1-(3-трет-бутил-1-(1H-индазол-5-ил)-1H-пиразол-5-ил)-3-(5-(5-хлорпиридин-3-илокси)-2-фторфенил)мочевину (0,0475 г, выход 103%) в виде бис-HCl-соли.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,14 (с, 1H), 8,95 (с, 1H), 8,43-8,42 (м, 1H), 8,34-8,33 (м, 1H), 8,20 (с, 1H), 8,00-7,97 (м, 1H), 7,88-7,87 (м, 1H), 7,70-7,67 (м, 1H), 7,60-7,59 (м, 1H), 7,45-7,42 (м, 1H), 7,32-7,27 (м, 1H), 6,81-6,77 (м, 1H), 6,38 (с, 1H), 1,27 (с, 9H); MC (ESI) m/z: 520,2 (М+H+).
Пример 9: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B7 (0,300 г, 0,550 ммоль), соединение из примера A10 (0,138 г, 0,577 ммоль) и DIEA (0,115 мл, 0,659 ммоль) объединяли и очищали флэш-хроматографией на колонке (EtOAc/гексаны), получая трет-бутил-6-(3-трет-бутил-5-(3-(5-(5-хлорпиридин-3-илокси)-2-фторфенил)уреидо)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,090 г, выход 26%) в виде пленки.
1H ЯМР (400 МГц, ацетон-d6) δ 8,50 (ушир.с, 1H), 8,36 (с, 1H), 8,35-8,32 (м, 2H), 8,19-8,16 (м, 1H), 7,47-7,46 (м, 1H), 7,38-7,36 (м, 2H), 7,31-7,29 (м, 1H), 7,27-7,22 (м, 1H), 6,83-6,79 (м, 1H), 6,46 (с, 1H), 4,63 (ушир.с, 2H), 3,68-3,65 (м, 2H), 2,89-2,86 (м, 2H), 1,50 (с, 9H), 1,32 (с, 9H); MC (ESI) m/z: 635,2 (М+H+).
Вещество, полученное в предыдущей реакции (0,090 г, 0,14 ммоль, 1,00 экв.), и 4М HCl в диоксане (5,00 мл) объединяли при 22°C. Добавляли небольшое количество MeOH, чтобы сделать смесь гомогенной. Через 2,5 часа, когда реакция была завершена, реакционную смесь концентрировали в вакууме, растворяли в MeCN/H2O, замораживали и подвергали лиофилизации, получая 1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)-3-(5-(5-хлорпиридин-3-илокси)-2-фторфенил)мочевину (76 мг, выход 89%) в виде бис-HCl-соли.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,51 (ушир.с, 2H), 9,26 (ушир.с, 1H), 9,22 (с, 1H), 8,42-8,41 (м, 1H), 8,33-8,32 (м, 1H), 7,95-7,92 (м, 1H), 7,60-7,59 (м, 1H), 7,42-7,29 (м, 4H), 6,82-6,78 (м, 1H), 6,34 (с, 1H), 4,32-4,30 (м, 2H), 3,39-3,35 (м, 2H), 3,10-3,06 (м, 2H), 1,26 (с, 9H); MC (ESI) m/z: 535,2 (М+H+).
Пример 10: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (0,150 г, 0,351 ммоль) и соединение из примера A2 (0,101 г, 0,386 ммоль) объединяли, получая гидрохлорид 1-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (0,126 г, выход 62%).
1H ЯМР (ДМСО-d6) δ 9,36 (с, 1H), 9,18-9,15 (м, 2H), 8,92 (д, J=8,4 Гц, 1H), 8,85-8,80 (м, 1H), 8,53 (д, J=5,6 Гц, 1H), 8,44 (д, J=2,4 Гц, 1H), 8,36 (д, J=9,2 Гц, 1H), 8,22 (дд, J=9,2, 2,4 Гц, 1H), 8,14 (т, J=9,2 Гц, 1H), 7,92 (дд, J=8,4, 4,8 Гц, 1H), 7,42 (д, J=2,4 Гц, 1H), 7,31 (дд, J=11,6, 2,8 Гц, 1H), 7,19 (дд, J=5,6, 2,8 Гц, 1H), 7,04 (дд, J=8,8, 2,0 Гц, 1H), 6,45 (с, 1H), 2,96 (м, 1H), 2,79 (д, J=4,8 Гц, 3H), 1,28 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 540,3 (М+H+).
Пример 11: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B10 (0,15 г, 0,363 ммоль) и соединение из примера A2 (0,100 г, 0,38 ммоль) объединяли, получая гидрохлорид 1-(3-этил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины (0,120 г, выход 58%).
1H ЯМР (ДМСО-d6) δ 9,42 (с, 1H), 9,21-9,18 (м, 2H), 8,96 (д, J=8,4 Гц, 1H), 8,87-8,82 (м, 1H), 8,53 (д, J=5,6 Гц, 1H), 8,48 (д, J=1,6 Гц, 1H), 8,38 (д, J=9,2 Гц, 1H), 8,25 (дд, J=9,2, 1,6 Гц, 1H), 8,14 (т, J=8,8 Гц, 1H), 7,95 (дд, J=8,0, 4,8 Гц, 1H), 7,43 (д, J=2,0 Гц, 1H), 7,31 (дд, J=12,0, 2,4 Гц, 1H), 7,19 (дд, J=5,2, 2,0 Гц, 1H), 7,05 (дт, J=8,8, 1,6 Гц, 1H), 6,44 (с, 1H), 2,79 (д, J=4,8 Гц, 3H), 2,64 (кв, J=7,6 Гц, 2H), 1,25 (т, J=7,6 Гц, 3H); MC (ESI) m/z: 526,2 (М+H+).
Пример 12: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (0,195 г, 0,441 ммоль), соединение из примера A10 (0,111 г, 0,464 ммоль) и DIEA (0,0923 мл, 0,530 ммоль) объединяли и очищали сначала флэш-хроматографией на колонке (EtOAc/гексаны) и затем обращенно-фазовой хроматографией (MeCN (w/0,1% ТФУ)/H2O (w/0,1% ТФУ)), получая водный раствор ТФУ-соли требуемого продукта. Водный остаток обрабатывали насыщенным раствором NaHCO3 (pH 8) и экстрагировали EtOAc (3×). Объединенные органические фазы промывали насыщенным раствором соли (1×), сушили (MgSO4) и упаривали, получая продукт (0,0258 г, выход 11%) в виде свободного основания. Свободное основание обрабатывали сертифицированной 0,1н. HCl (0,97 мл, 2,0 экв.), получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(5-(5-хлорпиридин-3-илокси)-2-фторфенил)мочевину (0,0262 г, выход 10%) в виде бис-HCl-соли.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,33 (с, 1H), 9,22-9,21 (м, 1H), 9,14-9,13 (м, 1H), 8,83-8,81 (м, 1H), 8,42-8,41 (м, 1H), 8,36 (ушир.с, 1H), 8,33-8,29 (м, 2H), 8,15-8,12 (м, 1H), 7,94-7,91 (м, 1H), 7,88-7,84 (м, 1H), 7,59-7,57 (м, 1H), 7,34-7,28 (м, 1H), 6,82-6,78 (м, 1H), 6,46 (с, 1H), 1,30 (с, 9H); MC (ESI) m/z: 531,0 (М+H+).
Пример 13: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (100 мг, 0,226 ммоль), DIEA (73 мг, 0,566 ммоль) и соединение из примера A18 (63 мг, 0,25 ммоль) объединяли, получая гидрохлорид 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(2-(метилтио)пиримидин-4-илокси)фенил)мочевины (61 мг, выход 50%).
1H-ЯМР (ДМСО-d6) δ 1,30 (с, 9H), 2,50 (с, 3H), 6,47 (с, 1H), 6,76 (д, 1H), 6,86-6,90 (м, 1H), 7,29-7,34 (м, 1H), 7,92-7,98 (м, 2H), 8,20-8,23 (м, 1H), 8,37 (д, 1H), 8,44 (с, 1H), 8,50 (д, 1H), 8,95 (д, 1H), 9,19-9,20 (м, 1H), 9,28 (с, 1H), 9,46 (с, 1H); MC (ESI) m/z: 544,2 (М+H+).
Пример 14: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (0,10 г, 0,23 ммоль), соединение из примера A12 (53 мг, 0,23 ммоль) и DIEA (64 мг, 0,50 ммоль) объединяли и очищали обращенно-фазовой хроматографией на колонке, получая ТФУ-соль 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(6-(гидроксиметил)пиридин-3-илокси)фенил)мочевины. Остаток растворяли в 3М HCl и упаривали вместе с изопропиловым спиртом (3×). К остатку добавляли EtOAc и твердое вещество отфильтровывали, промывали EtOAc и сушили в вакууме, получая HCl-соль 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(6-(гидроксиметил)пиридин-3-илокси)фенил)мочевины (40 мг, выход 34%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,15 (ушир.м, 1H), 9,05 (ушир.м, 1H), 8,63 (ушир.м, 1H), 8,32 (ушир.м, 1H), 8,23 (ушир.м, 2H), 8,03 (м, 1H), 7,90 (м, 1H), 7,73 (ушир.м, 1H), 7,56 (м, 2H), 7,28 (дд, J=9,2, 12,4 Гц, 1H), 6,74 (м, 1H), 6,44 (с, 1H), 4,60 (м, 2H), 1,30 (с, 9H); MC (ESI) m/z: 527,2 (М+H+).
Пример 15: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (0,120 г, 0,281 ммоль) и соединение из примера A7 (0,0763 г, 0,309 ммоль) объединяли, получая гидрохлорид 1-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (0,101 г, выход 65%).
1H ЯМР (ДМСО-d6) δ 9,23 (с, 1H), 9,11-9,08 (м, 2H), 8,77 (д, J=4,8 Гц, 1H), 8,53 (д, J=6,0 Гц, 1H), 8,35 (д, J=2,0 Гц, 1H), 8,29 (д,J=8,8 Гц, 1H), 8,18-8,11 (м, 3H), 7,84-7,80 (м, 1H), 7,75 (с, 1H), 7,43 (д, J=2,4 Гц, 1H), 7,31 (дд, J=11,6, 2,4 Гц, 1H), 7,20 (дд, J=6,0, 2,4 Гц, 1H), 7,05 (дд, J=9,6, 2,8 Гц, 1H), 6,45 (с, 1H); MC (ESI) m/z: 526,2 (М+H+).
Пример 16: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (85 мг, 0,19 ммоль), соединение из примера A13 (42 мг, 0,19 ммоль) и DIEA (55 мг, 0,42 ммоль) объединяли в ДМСО (1 мл) и нагревали в течение ночи при 50-55°C. Добавляли воду (50 мл) и смесь экстрагировали EtOAc (3×100 мл), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на колонке с силикагелем, получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(6-метилпиридин-3-илокси)фенил)мочевину.
Продукт обрабатывали 0,10 М водным раствором HCl, получая HCl-соль 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(6-метилпиридин-3-илокси)фенил)мочевины (56 мг, выход 52%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,38 (ушир.с, 1H), 9,27 (д, J=2,4 Гц, 1H), 9,11 (дд, J=1,6 и 4,8 Гц, 1H), 8,77 (д, J=8,0 Гц, 1H), 8,50 (д, J=3,2 Гц, 1H), 8,34 (д, J=2,4 Гц, 1H), 8,29 (д, J=9,2 Гц, 1H), 8,11 (дд, J=2,4 и 9,2 Гц, 1H), 7,94 (дд, J=3,2 и 6,8 Гц, 1H), 7,83 (м, 2H), 7,68 (д, J=8,8 Гц, 1H), 7,32 (дд, J=9,2, 10,8 Гц, 1H), 6,79 (м, 1H), 6,44 (с, 1H), 2,61 (с, 3H), 1,30 (с, 9H); MC (ESI) m/z: 511,2 (М+H+).
Пример 17: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (213 мг, 0,50 ммоль), соединение из примера A6 (145 мг, 0,56 ммоль) и DIEA (0,09 мл, 0,517 ммоль) объединяли в ДМФА (2 мл), получая 1-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(3-метил-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину (194 мг, выход 73%).
1H ЯМР (400 МГц, ДМСО-d6): δ 9,07 (с, 1H), 8,97 (дд, J=4,2, 1,8 Гц, 1H), 8,76 (кв, J=4,9 Гц, 1H), 8,64 (с, 1H), 8,51-8,48 (м, 2H), 8,19-8,16 (м, 2H), 7,97 (дд, J=9,0, 2,4 Гц, 1H), 7,63 (дд, J=8,3, 4,2 Гц, 1H), 7,45 (д, J=2,4 Гц, 1H), 7,33 (дд, J=8,9, 2,6 Гц, 1H), 7,28 (д, J=2,6 Гц, 1H), 7,10-7,04 (м, 2H), 6,43 (с, 1H), 2,95 (м, 1H), 2,78 (д, J=4,9 Гц, 3H), 2,04 (с, 3H), 1,28 (д, J=6,7 Гц, 6H); MC (ESI) m/z: 536,2 (М+H+).
Пример 18: mCPBA (1,07 г ~70%, 4,34 ммоль) добавляли к раствору соединения из примера A18 (545 мг, 2,17 ммоль) в CH2Cl2 (15 мл) и раствор перемешивали при комнатной температуре. Смесь промывали насыщенным раствором бикарбоната натрия (3×20 мл) и насыщенным раствором соли (30 мл), сушили (Na2SO4) и концентрировали в вакууме, получая 0,65 г желто-коричневой пены, которая, как было доказано, является смесью сульфоксида и сульфона, которую можно использовать как таковую. В смесь 2,0 N метиламин/ТГФ (22 мл) помещали неочищенную смесь сульфоксида/сульфона (0,61 г, 2,2 ммоль) и перемешивали в течение ночи при 40°C. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом (25 мл), промывали 5% лимонной кислотой (25 мл), насыщенным раствором бикарбоната натрия (25 мл) и насыщенным раствором соли (25 мл), сушили (Na2SO4), концентрировали в вакууме и очищали обращенно-фазовой хроматографией, получая соль трифторуксусной кислоты 4-(3-амино-4-фторфенокси)-N-метилпиримидин-2-амина (301 мг, выход 60%). МС (ESI) m/z: 235,0 (M+H+).
В ДМСО (2 мл) помещали соединение из примера B3 (159 мг, 0,359 ммоль), DIEA (139 мг, 1,08 ммоль) и соль трифторуксусной кислоты 4-(3-амино-4-фторфенокси)-N-метилпиримидин-2-амина (150 мг, 0,431 ммоль). Смесь нагревали до 50°C в течение ночи, затем разбавляли этилацетатом (25 мл), промывали 5% лимонной кислотой (50 мл), насыщенным раствором бикарбоната натрия (50 мл) и насыщенным раствором соли (50 мл), сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на колонке, получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(2-(метиламино)пиримидин-4-илокси)фенил)мочевину (93 мг, выход 49%).
1H-ЯМР (ДМСО-d6) δ 1,31 (с, 9H), 2,54-2,86 (ушир.д, 3H), 6,46 (с, 1H), 6,57-6,61 (ушир.м, 1H), 6,91-6,93 (ушир.м, 1H), 7,32-7,37 (м, 1H), 7,94-8,05 (м, 2H), 8,23-8,33 (м, 2H), 8,40 (д, 1H), 8,48 (с, 1H), 8,98 (д, 1H), 9,19-9,21 (м, 1H), 9,43-9,47 (ушир.м, 1H), 9,68-9,73 (ушир.м, 1H); MC (ESI) m/z: 527,2 (М+H+).
Пример 19: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (85 мг, 0,20 ммоль), соединение из примера A9 (46 мг, 0,20 ммоль) и DIEA (57 мг, 0,44 ммоль) объединяли в ДМСО (1 мл), получая 1-(2-фтор-4-(2-(метиламино)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину. Продукт обрабатывали 0,100 М водным раствором HCl, получая HCl-соль 1-(2-фтор-4-(2-(метиламино)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (52 мг, выход 48%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,17 (с, 1H), 9,14 (ушир.с, 1H), 8,98 (дд, J=1,2 и 4,0 Гц, 1H), 8,50 (д, J=8,4 Гц, 1H), 8,42 (ушир.с, 1H), 8,20 (д, J=2,8 Гц, 1H), 8,17 (д, J=9,2 Гц, 1H), 7,97 (дд, J=2,4 и 9,2 Гц, 1H), 7,91 (д, J=7,2 Гц, 1H), 7,64 (дд, J=4,0 и 8,4 Гц, 1H), 7,34 (дд, J=2,4 и 11,6 Гц, 1H), 7,07 (дд, J=1,2 и 8,8 Гц, 1H), 6,60 (д, J=6,4 Гц, 1H), 6,43 (с, 1H), 6,17 (ушир.с, 1H), 2,95 (м, 1H), 2,87 (д, J=4,4 Гц, 3H), 1,27 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 512,3 (М+H+).
Пример 20: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B10 (0,13 г, 0,314 ммоль), соединение из примера A7 (0,086 г, 0,346 ммоль) и DIEA (0,12 мл, 0,69 ммоль) растворяли в ДМСО (1,5 мл) и смесь нагревали при 55°C в течение ночи, получая 1-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)-3-(3-этил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,088 г, выход 55%). Продукт превращали в соответствующую HCl-соль в результате взаимодействия с HCl (4,0М HCl/диоксан, 1,0 экв.).
1H ЯМР (ДМСО-d6) δ 9,37 (с, 1H), 9,18-9,15 (м, 2H), 8,90 (д, J=8,0 Гц, 1H), 8,54 (д, J=5,6 Гц, 1H), 8,43 (с, 1H), 8,21 (д, J=8,8 Гц, 1H), 8,22-8,12 (м, 3H), 7,91 (м, 1H), 7,78 (с, 1H), 7,45 (д, J=1,6 Гц, 1H), 7,31 (дд, J=12, 2,0 Гц, 1H), 7,21 (дд, J=5,2, 1,4 Гц, 1H), 7,05 (д, J=9,2 Гц, 1H), 6,44 (с, 1H), 2,64 (кв, J=7,6 Гц, 2H), 1,25 (т, J=7,2 Гц, 3H); MC (ESI) m/z: 512,3 (М+H+).
Пример 21: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (198 мг, 373 ммоль), DIEA (121 мг, 0,933 ммоль) и соединение из примера A21 (117 мг, 0,448 ммоль) объединяли, получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(6-(метилкарбамоил)пиридин-3-илокси)фенил)мочевину (140 мг, выход 67%) в качестве гидрохлорида.
1H-ЯМР (ДМСО-d6) δ 1,30 (с, 9H), 2,81 (д, 3H), 6,45 (с, 1H), 6,81-6,83 (м, 1H), 7,30-7,35 (м, 1H), 7,43-7,46 (м, 1H), 7,91-8,02 (м, 3H), 8,19-8,21 (м, 1H), 8,34-8,43 (м, 3H), 8,65-8,66 (м, 1H), 8,91 (д, 1H), 9,17-9,19 (м, 1H), 9,28 (ушир.с, 1H), 9,44 (с, 1H); MC (ESI) m/z: 554,2 (М+H+).
Пример 22: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B14 (0,125 г, 0,291 ммоль) и соединение из примера A7 (0,079 г, 0,320 ммоль) объединяли, получая гидрохлорид 1-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)-3-(5-хлор-2-(хинолин-6-ил)фенил)мочевины (0,070 г, выход 43%).
1H ЯМР (ДМСО-d6) δ 9,20 (д, J=3,6 Гц, 1H), 9,04 (д, J=1,6 Гц, 1H), 8,92 (д, J=8,0 Гц, 1H), 8,54-8,52 (м, 2H), 8,36 (д, J=9,2 Гц, 1H), 8,32 (д, J=1,6 Гц, 1H), 8,23 (т, J=8,8 Гц, 1H), 8,18-8,17 (м, 2H), 8,02 (дд, J=8,4, 1,6 Гц, 1H), 7,93-7,90 (м, 1H), 7,76 (с, 1H), 7,43-7,39 (м, 2H), 7,31-7,26 (м, 2H), 7,20 (дд, J=5,6, 2,4 Гц, 1H), 7,06 (дд, J=8,8, 1,2 Гц, 1H); MC (ESI) m/z: 528,0 (М+H+).
Пример 23: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (35 мг, 0,02 ммоль), соединение из примера A14 (47 мг, 0,20 ммоль) и DIEA объединяли в ДМСО и нагревали в течение ночи при 60°C, получая HCl-соль 1-(2-фтор-4-(2-метоксипиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (54 мг, выход 49%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,35 (ушир.с, 1H), 9,13 (ушир.с, 1H), 8,85 (д, J=2,0 Гц, 1H), 8,74 (с, 1H), 8,35 (дд, J=1,6 и 8,4 Гц, 1H), 8,25 (м, 1H), 7,90 (с, 1H), 7,74 (д, J=8,4 Гц, 1H), 7,71 (ушир.с, 1H), 7,29 (м, 2H), 6,46 (с, 1H), 4,31 (кв, J=7,2 Гц, 2H), 2,66 (с, 3H), 1,29 (с, 9H), 1,22 (т, J=7,2 Гц, 3H); MC (ESI) m/z: 556,3 (М+H+).
Пример 24: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B19 (150 мг, 0,329 ммоль) и соединение из примера A2 (94 мг, 0,362 ммоль) объединяли, получая гидрохлорид 1-(3-трет-бутил-1-(2-метилхинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины (113 мг, выход 60%).
1H-ЯМР (ДМСО-d6) δ 1,33 (с, 9H), 2,79 (д, 3H), 3,00 (с, 3H), 6,49 (с, 1H), 7,02-7,04 (м, 1H), 7,19-7,20 (м, 1H), 7,30 (д, 1H), 7,45 (с, 1H), 8,01 (д, 1H), 8,07-8,09 (м, 1H), 8,34-8,37 (м, 1H), 8,50-8,57 (м, 3H), 8,85-8,87 (м, 1H), 9,10 (д, 1H), 9,29 (с, 1H), 9,61 (с, 1H); MC (ESI) m/z: 568,2 (М+H+).
Пример 25: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (120 мг, 0,28 ммоль), соединение из примера A20 (80 мг, 0,29 ммоль) и DIEA (110 мг, 0,84 ммоль) объединяли, получая гидрохлорид 1-(2-фтор-5-(6-(трифторметил)пиридин-3-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (62 мг, выход 40%).
1H-ЯМР (ДМСО-d6) δ 1,25 (д, 6H), 2,93 (пентет, 1H), 6,41 (с, 1H), 6,85-6,88 (м, 1H), 7,32-7,37 (м, 1H), 7,51-7,54 (м, 1H), 7,87-7,90 (м, 2H), 7,96-7,98 (м, 1H), 8,16-8,18 (м, 1H), 8,33 (д, 1H), 8,40 (с, 1H), 8,52 (с, 1H), 8,87 (д, 1H), 9,15-9,16 (м, 1H), 9,28 (с, 1H), 9,42 (с, 1H); MC (ESI) m/z: 551,2 (М+H+).
Пример 26: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (0,200 г, 0,468 ммоль) и соединение из примера A15 (0,113 г, 0,491 ммоль) объединяли, получая 1-(4-(2-цианопиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,238 г, 100%). МС (ESI) m/z: 508,3 (M+H+).
1-(4-(2-Цианопиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,108 г, 0,221 ммоль) и N-ацетилцистеин (0,072 г, 0,441 ммоль) растворяли в MeOH (0,3 мл). Добавляли ацетат аммония (0,041 г, 0,0,529 ммоль) и реакционную смесь нагревали при 60°C в атмосфере N2 в течение ночи. Реакционную смесь после завершения реакции разбавляли H2O (10 мл), подщелачивали, используя K2CO3, экстрагировали EtOAc (2×30 мл) и ТГФ (20 мл). Объединенные органические слои промывали насыщенным раствором соли (20 мл), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией, получая 1-(4-(2-карбамидоилпиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,019 г, выход 17%) в виде белого твердого вещества. Продукт превращали в соответствующую HCl-соль в результате взаимодействия с HCl (4,0 М HCl/диоксан, 1,0 экв.).
1H ЯМР (ДМСО-d6) δ 9,57 (с, 2H), 9,36-9,34 (м, 2H), 9,20 (д, J=1,2 Гц, 1H), 9,09 (дд, J=4,4, 1,2 Гц, 1H), 8,74 (д, J=8,0 Гц, 1H), 8,68 (д, J=5,2 Гц, 1H), 8,35 (д, J=2,0 Гц, 1H), 8,28 (д, J=9,2 Гц, 1H), 8,18-8,10 (м, 2H), 7,92 (д, J=2,4 Гц, 1H), 7,80 (дд, J=8,4, 4,8 Гц, 1H), 7,32-7,26 (м, 2H), 7,05 (дд, J=8,8, 1,2 Гц, 1H), 6,44 (с, 1H), 2,97-2,93 (м, 1H), 1,28 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 525,3 (М+H+).
Пример 27: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B7 (159 мг, 0,291 ммоль), DIEA (45 мг, 0,35 ммоль) и соединение из примера A34 (74 мг, 0,35 ммоль) объединяли, получая трет-бутил-6-(3-трет-бутил-5-(3-(3-циано-5-(пиридин-3-илокси)фенил)уреидо)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (83 мг, выход 47%). МС (ESI) m/z: 608,3 (M+H+).
В CH2Cl2 (8 мл) помещали трет-бутил-6-(3-трет-бутил-5-(3-(3-циано-5-(пиридин-3-илокси)фенил)уреидо)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (83 мг, 0,14 ммоль). Проводили барботирование реакционной смеси, используя HCl (газообразный), вплоть до насыщения раствора и затем раствор перемешивали при комнатной температуре в течение 4 часов. Концентрирование в вакууме давало твердое вещество, которое растирали в эфире (10 мл). Твердое вещество собирали фильтрованием, промывали эфиром (2 мл) и сушили, получая гидрохлорид 1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)-3-(3-циано-5-(пиридин-3-илокси)фенил)мочевины (69 мг, выход 93%).
1H ЯМР (300 МГц, ДМСО-d6) δ 1,26 (с, 9H), 3,06-3,09 (м, 2H), 3,35-3,40 (м, 2H), 4,28-4,30 (м, 2H), 6,33 (с, 1H), 7,23-7,24 (м, 1H), 7,31-7,34 (м, 1H), 7,39-7,47 (м, 4H), 7,63-7,67 (м, 2H), 7,77-7,78 (м, 1H), 8,52-8,54 (м, 1H), 8,59 (м, 1H), 8,93 (с, 1H), 9,42-9,43 (м, 2H), 10,16 (с, 1H); MC (ESI) m/z: 527,2 (М+H+).
Пример 28: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера A35 (95 мг, 0,428 ммоль), DIEA (158 мг, 1,22 ммоль) и соединение из примера B3 (180 мг, 0,407 ммоль) объединяли, получая гидрохлорид 1-(5-(2-аминопиримидин-4-илокси)-2-фторфенил)-3-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (102 мг, выход 48%).
1H ЯМР (300 МГц, ДМСО-d6) δ 1,31 (с, 9H), 6,46 (с, 1H), 6,65 (д, J=6,8 Гц, 1H), 6,91-6,94 (м, 1H), 7,32-7,37 (м, 1H), 7,91-7,94 (м, 1H), 7,97-8,00 (м, 1H), 8,20-8,23 (м, 1H), 8,31-8,33 (м, 1H), 8,36-8,39 (м, 1H), 8,45-8,46 (м, 1H), 8,92-8,94 (м, 1H), 9,18 (м, 1H), 9,45 (м, 1H), 9,66 (с, 1H), NH2 отсутствует; MC (ESI) m/z: 513,3 (М+H+).
Пример 29: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (0,200 г, 0,468 ммоль) и соединение из примера A15 (0,113 г, 0,491 ммоль) в присутствии DIEA (0,179 мл, 0,103 ммоль) объединяли, получая 1-(4-(2-цианопиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,238 г, 100%) в виде бесцветного масла. Продукт превращали в соответствующую HCl-соль в результате взаимодействия с HCl (4,0 М в диоксане, 1,0 экв.).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (с, 1H), 9,09-9,08 (м, 2H), 8,73 (д, J=8,0 Гц, 1H), 8,60 (д, J=6,0 Гц, 1H), 8,32 (д, J=2,4 Гц, 1H), 8,27 (д, J=8,8 Гц, 1H), 8,16 (т, J=9,2 Гц, 1H), 8,10 (дд, J=9,2, 2,4 Гц, 1H), 7,80 (дд, J=8,0, 4,4 Гц, 1H), 7,72 (д, J=2,8 Гц, 1H), 7,31 (дд, J=11,6, 2,8 Гц, 1H), 7,23 (дд, J=5,6, 2,8 Гц, 1H), 7,05 (дд, J=9,2, 2,8 Гц, 1H), 6,45 (с, 1H), 2,95 (м, 1H), 1,27 (д, J=7,2 Гц, 6H); MC (ESI) m/z: 508,3 (М+H+).
Пример 30: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (0,2 г, 0,453 ммоль) и соединение из примера A29 (0,158 г, 0,453 ммоль) объединяли в ДМСО (4 мл) при 70°C в присутствии DIEA (0,176 г, 1,36 ммоль), получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(4-(2-((трет-бутилдиметилсилилокси)метил)пиридин-4-илокси)-2-фторфенил)мочевину (0,12 г, выход 43%).
1H ЯМР (400 МГц, CDCl3) δ 9,02 (ушир.с, 1H), 8,86 (д, J=8,5 Гц, 1H), 7,65 (м, 3H), 7,27 (дд, J=8, 4,4 Гц, 1H), 6,99 (с, 1H), 6,89 (ушир.д, J=9,0 Гц, 1H), 6,73 (дд, J=12, 2,5 Гц, 1H), 6,65 (с, 1H), 6,60 (м, 1H), 4,71 (с, 2H), 1,36 (с, 9H), 0,85 (с, 9H), 0,05 (с, 6H); MC (ESI) m/z: 641,3 (М+H+).
Раствор 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(4-(2-((трет-бутилдиметилсилилокси)метил)пиридин-4-илокси)-2-фторфенил)мочевины (0,12 г, 0,19 ммоль) в ТГФ (2 мл) обрабатывали TBAF (1,0 мл, 1,0 М раствор в ТГФ) при комнатной температуре в течение 1 часа. Добавляли воду (10 мл) и отделившееся твердое вещество отфильтровывали, промывали водой и сушили, получая десилилированный продукт 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(гидроксиметил)пиридин-4-илокси)фенил)мочевину в виде белого твердого вещества (0,090 г, выход 91%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,01 (ушир.с, 1H), 8,97 (дд, J=4,2, 1,6 Гц, 2H), 8,50 (ушир.д, J=8,3 Гц, 1H), 8,36 (д, J=5,5 Гц, 2H), 8,18 (м, 2H), 7,97 (дд, J=9, 2 Гц, 1H), 7,63 (дд, J=9, 4,4 Гц, 1H), 7,22 (дд, J=12, 2,5 Гц, 1H), 6,99 (м, 1H), 6,93 (д, J=2,5 Гц, 1H), 6,82 (дд, J=5,7, 2,5 Гц, 1H), 6,48 (с, 1H), 5,40 (т, J=6 Гц, 1H), 4,50 (д, J=8 Гц, 2H), 1,32 (с, 9H); MC (ESI) m/z: 527,2 (М+H+).
Свободное основание превращали в гидрохлорид.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,31 (ушир.с, 1H), 9,23 (м, 1H), 9,07 (дд, J=4,2, 1,6 Гц, 1H), 8,70 (ушир.д, J=8,3 Гц, 1H), 8,65 (д, J=6,8 Гц, 2H), 8,32 (д, J=2 Гц, 1H), 8,27 (д, J=9 Гц, 1H), 8,22 (д, J=9 Гц, 1H), 8,09 (дд, J=9, 2,3 Гц, 1H), 7,75 (дд, J=8, 4,5 Гц, 1H), 7,43-7,37 (м, 2H), 7,34 (д, J=2,8 Гц, 1H), 7,12 (м, 1H), 6,48 (с, 1H), 4,77 (с, 2H), 1,32 (с, 9H); MC (ESI) m/z: 527,2 (М+H+).
Пример 31: Используя способ, аналогичный способу, описанному в примере 4, соединение из примера B25 (0,30 г, 0,89 ммоль) и соединение из примера A31 (0,26 г, 0,98 ммоль) в присутствии N-метилпирролидина (каталитическое количество) объединяли, получая 1-(2-фтор-4-(2-(изопропиламино)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,26 г, выход 54%). Продукт обрабатывали метансульфоновой кислотой, получая мезилат 1-(2-фтор-4-(2-(изопропиламино)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (260 мг, выход 88%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,03 (м, 1H), 9,01 (с, 1H), 8,96 (дд, J=1,6 и 4,0 Гц, 1H), 8,49 (ушир.д, J=8,4 Гц, 1H), 8,33 (ушир.м, 1H), 8,17 (м, 2H), 7,95 (дд, J=2,8 и 9,2 Гц, 1H), 7,87 (д, J=7,6 Гц, 1H), 7,63 (д, J=4,4 и 8,4 Гц, 1H), 7,33 (дд, J=2,8 и 11,6 Гц, 1H), 7,06 (м, 1H), 6,61 (дд, J=2,4 и 7,2 Гц, 1H), 6,41 (с, 1H), 6,09 (ушир.с, 1H), 3,81 (м, 1H), 2,91 (м, 1H), 2,30 (с, 3H), 1,25 (д, J=6,8 Гц, 6H), 1,13 (д, J=6,0 Гц, 6H); MC (ESI) m/z: 540,3 (М+H+).
Пример 32: Используя общий способ A, соединение из примера B20 (0,0643 г, 0,226 ммоль) и соединение из примера A7 (0,168 г, 0,678 ммоль) объединяли, получая 1-(3-трет-бутил-1-(H-имидазо[1,2-a]пиридин-6-ил)-1H-пиразол-5-ил)-3-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)мочевину (0,071 г, 59%) в виде белого твердого вещества. Продукт превращали в соответствующую HCl-соль посредством взаимодействия с HCl (4,0 М в диоксане, 1,0 экв.).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,48 (с, 1H), 9,33 (д, J=0,8 Гц, 1H), 9,13 (д, J=1,6 Гц, 1H), 8,53 (д, J=5,2 Гц, 1H), 8,41 (д, J=2,4 Гц, 1H), 8,26 (д, J=2,0 Гц, 1H), 8,17-8,09 (м, 4H), 7,72 (с, 1H), 7,39 (д, J=2,4 Гц, 1H), 7,32 (дд, J=12,0, 2,8 Гц, 1H), 7,20 (дд, J=5,6, 2,8 Гц, 1H), 7,05 (дд, J=9,2, 1,6 Гц, 1H), 6,49 (с, 1H), 1,32 (с, 9H); MC (ESI) m/z: 529,3 (М+H+).
Пример 33: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (100 мг, 0,23 ммоль) и соединение из примера A12 (55 мг, 0,23 ммоль) в присутствии DIEA (90 мкл, 0,51 ммоль) объединяли, получая 1-(2-фтор-5-(6-(гидроксиметил)пиридин-3-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (30 мг, выход 25%). Продукт обрабатывали метансульфоновой кислотой, получая мезилат 1-(2-фтор-5-(6-(гидроксиметил)пиридин-3-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (23 мг, выход 65%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,11 (ушир.с, 1H), 9,10 (м, 1H), 9,06 (м, 1H), 8,65 (д, J=8,4 Гц, 1H), 8,34 (с, 1H), 8,25 (д, J=1,6 Гц, 1H), 8,21 (д, J=9,2 Гц, 1H), 8,03 (дд, J=2,4 и 9,2 Гц, 1H), 7,91 (дд, J=2,8 и 6,4 Гц, 1H), 7,75 (дд, J=4,8 и 8,4 Гц, 1H), 7,58 (с, 1H), 7,30 (м, 1H), 6,75 (м, 1H), 6,40 (с, 1H), 4,61 (с, 2H), 2,92 (м, 1H), 2,32 (с, 3H), 1,25 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 513,3 (М+Н+).
Пример 34: Используя способ, аналогичный способу, описанному в примере B19 для стадии 2, соединение из примера A2 (1,00 г, 3,83 ммоль) и 2,2,2-трихлорэтилкарбонхлоридат (1,30 г, 6,12 ммоль) объединяли, получая 2,2,2-трихлорэтил-2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенилкарбамат. МС (ESI) m/z: 436,0, 438,0 (M+H).
Раствор соединения из примера B28 (57 мг, 0,213 ммоль), 2,2,2-трихлорэтил-2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенилкарбамата (102 мг, 0,235 ммоль) и DIEA (110 мг, 0,853 ммоль) в ДМСО (1,5 мл) нагревали до 60°C в течение ночи. Затем раствор обрабатывали дополнительным количеством 2,2,2-трихлорэтил-2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенилкарбамата (~200 мг), нагревали до 60°C в течение ночи. Реакционную смесь разбавляли этилацетатом (25 мл) и 5% лимонной кислотой (20 мл). Органическую фазу отделяли, промывали насыщенным раствором бикарбоната натрия (20 мл) и насыщенным раствором соли (20 мл), сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией (колонка Si-25, MeOH/EtOAc), получая продукт с примесями. Повторная очистка обращенно-фазовой хроматографией (колонка C18-25, CH3CN/H2O) давала остаток, который обрабатывали 1н. гидроксидом натрия (3 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические фазы сушили (Na2SO4), концентрировали в вакууме и обрабатывали смесью 4н. HCl/диоксан (0,1 мл), получая гидрохлорид 1-(3-трет-бутил-1-(хиноксалин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины (14 мг, выход 12%).
1H ЯМР (300 МГц, ДМСО-d6) δ 1,31 (с, 9H), 2,77 (д, 3H), 6,47 (с, 1H), 7,00-7,05 (м, 1H), 7,15-7,18 (м, 1H), 7,26-7,28 (м, 1H), 7,39 (м, 1H), 7,65 (м, 1H), 8,08-8,13 (м, 2H), 8,21-8,25 (м, 2H), 8,50 (м, 1H), 8,78 (м, 1H), 8,97-9,03 (м, 3H), 9,13 (с, 1H); MC (ESI) m/z: 555,2 (М+H+).
Пример 35: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (0,145 г, 0,339 ммоль) и соединение из примера A27 (0,087 г, 0,323 ммоль) в присутствии DIEA (0,124 мл, 0,710 ммоль) объединяли, получая 1-(4-(2-(1H-пиразол-4-ил)пиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,112 г, 63%) в виде белой пены. Продукт превращали в соответствующий мезилат в результате взаимодействия с MsOH (1,0 экв.).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,10-9,03 (м, 3H), 8,63-8,52 (м, 4H), 8,26-8,20 (м, 2H), 8,03 (д, J=3,6 Гц, 1H), 7,78-7,70 (м, 2H), 7,40 (д, J=10,8 Гц, 1H), 7,14-7,09 (м, 2H), 6,44 (с, 1H), 2,95 (м, 1H), 2,33 (с, 3H), 1,27 (д, J=7,2 Гц, 6H); MC (ESI) m/z: 549,3 (М+H+).
Пример 36: Соединение из примера B22 (0,310 г, 0,715 ммоль), соединение из примера A2 (0,187 г, 0,715 ммоль) и DIEA (0,274 мл, 1,57 ммоль) объединяли в ДМСО (3 мл) и перемешивали при 70°C. Через 18 часов, когда реакция была завершена, реакционную смесь охлаждали до комнатной температуры, разбавляли насыщенным раствором соли и экстрагировали EtOAc (3×). Объединенные органические фазы промывали насыщенным раствором соли (2×), сушили (MgSO4), упаривали и очищали флэш-хроматографией на колонке (EtOAc/гексаны), получая свободное основание (84,1 мг, выход 22%). Полученное таким образом свободное основание обрабатывали сертифицированной 0,1н. HCl (3,1 мл, 2,0 экв.), получая 1-(1-(бензо[d]тиазол-6-ил)-3-изопропил-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину (45 мг) в виде бис-HCl-соли.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,49 (с, 1H), 9,00 (с, 2H), 8,81 (кв, J=4,8 Гц, 1H), 8,52 (д, J=5,6 Гц, 1H), 8,39 (д, J=1,6 Гц, 1H), 8,24 (д, J=8,80 Гц, 1H), 8,19 (т, J=9,2 Гц, 1H), 7,70 (дд, J=2,4 и 8,8 Гц, 1H), 7,42 (д, J=2,4 Гц), 7,31 (дд, J=3,2 и 12,0 Гц, 1H), 7,18 (дд, J=2,8 и 6,0 Гц, 1H), 7,06 (ддд, J=1,2, 2,8 и 8,8 Гц, 1H), 6,42 (с, 1H), 2,92 (септет, J=7,2 Гц, 1H), 2,79 (д, J=4,8 Гц, 3H), 1,26 (д, J=7,2 Гц, 6H); MC (ESI) m/z: 546,3 (М+H+).
Пример 37: Соединение из примера B23 (0,200 г, 0,464 ммоль), соединение из примера A2 (0,121 г, 0,464 ммоль) и i-Pr2NEt (0,178 мл, 1,02 ммоль) объединяли в ДМСО (2 мл) и перемешивали, нагревая при 70°C. Через 18 часов, когда реакция была завершена, реакционную смесь охлаждали до комнатной температуры, разбавляли насыщенным раствором соли и экстрагировали EtOAc (3×). Объединенные органические фазы промывали насыщенным раствором соли (2×), сушили (MgSO4), концентрировали в вакууме и очищали флэш-хроматографией на колонке (EtOAc/гексаны - EtOAc - ТГФ), получая продукт с примесями. Продукт очищали второй раз обращенно-фазовой хроматографией (MeCN (w/0,1% ТФУ)/H2O (w/0,1% ТФУ)), получая требуемый продукт (110 мг, выход 36%) в виде ТФУ-соли после лиофилизации. Полученную таким образом ТФУ-соль растворяли в ТГФ и подвергали круговому встряхиванию с MP-карбонатной смолой (110 мг) в течение 2 часов. Надосадочную жидкость сливали и шарики промывали ТГФ (2×). Объединенные слитые надосадки концентрировали, разбавляли MeCN/H2O и затем обрабатывали сертифицированной 0,1н. HCl (3,3 мл, 2,0 экв.), получая 1-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(1-метил-1H-бензо[d]имидазол-5-ил)-1H-пиразол-5-ил)мочевину (31 мг) в виде бис-HCl-соли.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,46 (ушир.с, 1H), 9,11 (с, 1H), 9,07 (с, 1H), 8,76 (ушир.кв, J=4,8 Гц, 1H), 8,50 (д, J=5,6 Гц, 1H), 8,11 (т, J=9,2 Гц, 1H), 8,06 (д, J=8,8 Гц), 7,98 (д, J=2,0 Гц, 1H), 7,78 (м, 1H), 7,37 (д, J=2,8 Гц, 1H), 7,28 (дд, J=2,4 и 11,2 Гц, 1H), 7,16 (дд, J=2,4 и 5,6 Гц, 1H), 7,02 (ддд, J=1,2, 2,8 и 8,8 Гц, 1H), 6,38 (с, 1H), 4,08 (с, 3H), 2,92 (септет, J=6,8 Гц, 1H), 2,76 (д, J=4,8 Гц, 3H), 1,24 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 543,2 (М+H+).
Пример 38: Используя общий способ A, соединение из примера B21 (0,0054 г, 0,20 ммоль) и соединение из примера A2 (0,16 г, 0,60 ммоль) объединяли, получая 1-(1-(H-имидазо[1,2-a]пиридин-6-ил)-3-изопропил-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину (0,045 г, выход 43%) в виде белого твердого вещества. Продукт превращали в соответствующий мезилат в результате взаимодействия с MsOH (1,0 экв.).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (м, 1H), 8,49 (д, J=6,0 Гц, 1H), 8,33 (д, J=2,0 Гц, 1H), 8,24 (дд, J=9,6, 3,0 Гц, 1H), 7,15 (д, J=2,0 Гц, 1H), 8,08 (д, J=10,0 Гц, 1H), 8,01 (т, J=8,8 Гц, 1H), 7,53 (д, J=3,5 Гц, 1H), 7,12 (дд, J=6,0, 3,0 Гц, 1H), 7,06 (дд, J=11,6, 2,8 Гц, 1H), 6,96 (м, 1H), 6,45 (с, 1H), 3,01 (м, 1H), 2,94 (с, 3H), 2,70 (с, 3H), 1,33 (д, J=6,4 Гц, 6H); MC (ESI) m/z: 529,3 (М+H+).
Пример 39: Используя общий способ A, соединение из примера B21 (0,030 г, 0,11 ммоль) и соединение из примера A7 (0,082 г, 0,33 ммоль) объединяли, получая 1-(1-(H-имидазо[1,2-a]пиридин-6-ил)-3-изопропил-1H-пиразол-5-ил)-3-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)мочевину (0,0245 г, выход 43%) в виде белого твердого вещества. Продукт превращали в соответствующую HCl-соль в результате взаимодействия с HCl (4,0 М в диоксане, 1,0 экв.).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,26 (д, J=0,8 Гц, 1H), 8,69 (д, J=6,4 Гц, 1H), 8,38 (д, J=1,6 Гц, 1H), 8,26 (дд, J=9,6, 1,2 Гц, 1H), 8,20-8,11 (м, 3H), 7,96 (с, 1H), 7,48 (д, J=5,6 Гц, 1H), 7,23 (дд, J=11,6, 2,8 Гц, 1H), 7,10 (д, J=9,2 Гц, 1H), 6,51 (с, 1H), 3,03 (м, 1H), 1,37 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 515,2 (М+H+).
Пример 40: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера A39 (63 мг, 0,29 ммоль) и соединение из примера B9 (122 мг, 0,29 ммоль) объединяли, получая 1-(4-(2-аминопиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину с примесью 2,2,2-трихлорэтанола (56 мг, выход 28%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,99-8,96 (м, 2H), 8,93 (д, J=1,5 Гц, 1H), 8,49 (м, 1H), 8,19-8,16 (м, 2H), 8,10 (т, J=9,2 Гц, 1H), 7,95 (дд, J=9,1, 2,3 Гц, 1H), 7,80 (д, J=5,8 Гц, 1H), 7,63 (дд, J=8,3, 4,0 Гц, 1H), 7,15 (дд, J=11,8, 2,8 Гц, 1H), 6,95 (м, 1H), 6,44 (с, 1H), 6,13 (дд, J=5,9, 2,2 Гц, 1H), 5,94 (с, 2H), 5,82 (д, J=2,0 Гц, 1H), 2,94 (м, 1H), 1,27 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 498,2 (М+H+).
Раствор указанной выше 1-(4-(2-аминопиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (44 мг, 0,061 ммоль, теоретически) и пиридина (0,30 мл, 3,7 ммоль) в CH2Cl2 (1 мл) обрабатывали уксусным ангидридом (0,040 мл, 0,39 ммоль). Реакционную смесь перемешивали в течение 60 часов и затем распределяли между EtOAc и 2М водным раствором Na2CO3. Органический слой промывали водой и насыщенным раствором соли. Водные фазы снова экстрагировали EtOAc. Объединенные органические фазы сушили (Na2SO4), концентрировали в вакууме и очищали обращенно-фазовой хроматографией, получая 1-(4-(2-ацетамидопиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (25 мг, выход 76%).
1H ЯМР (400 МГц, ДМСО-d6) δ 10,53 (с, 1H), 9,01 (с, 1H), 8,96-8,94 (м, 2H), 8,49 (м, 1H), 8,18-8,11 (м, 4H), 7,95 (дд, J=8,8, 2,4 Гц, 1H), 7,64-7,59 (м, 2H), 7,21 (дд, J=11,8, 2,7 Гц, 1H), 6,98 (м, 1H), 6,65 (дд, J=5,8, 2,4 Гц, 1H), 6,43 (с, 1H), 2,93 (м, 1H), 2,03 (с, 3H), 1,26 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 540,3 (М+H+).
Пример 41: Используя способ, аналогичный способу, описанному в примере 4, соединение из примера B25 (100 мг, 0,30 ммоль) и соединение из примера A30 (74 мг, 0,30 ммоль) в присутствии N-метилпирролидина (каталитическое количество) объединяли, получая 1-(4-(2-(этиламино)пиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (70 мг, выход 45%). Продукт обрабатывали метансульфоновой кислотой, получая мезилат 1-(4-(2-(этиламино)пиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевины (71 мг, выход 87%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,02 (м, 1H), 9,01 (с, 1H), 8,97 (дд, J=1,6 и 4,0 Гц, 1H), 8,49 (ушир.д, J=8,4 Гц, 1H), 8,37 (ушир.с, 1H), 8,17 (м, 2H), 7,95 (дд, J=2,4 и 8,8 Гц, 1H), 7,88 (д, J=7,2 Гц, 1H), 7,63 (д, J=4,4 и 8,4 Гц, 1H), 7,33 (дд, J=2,8 и 11,6 Гц, 1H), 7,06 (м, 1H), 6,61 (дд, J=2,0 и 7,2 Гц, 1H), 6,41 (с, 1H), 6,13 (ушир.с, 1H), 3,23 (м, 2H), 2,92 (м, 1H), 2,28 (с, 3H), 1,25 (д, J=6,8 Гц, 6H), 1,13 (т, J=7,2 Гц, 3H); MC (ESI) m/z: 526,2 (М+H+).
Пример 42: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (295 мг, 0,69 ммоль) и соединение из примера A40 (214 мг, 0,763 ммоль) объединяли в ДМФА (3 мл), получая 1-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(3-метил-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)мочевину (278 мг, выход 72%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,00 (с, 1H), 8,94 (дд, J=4,2, 1,6 Гц, 1H), 8,59 (с, 1H), 8,45 (дд, J=8,6, 1,0 Гц, 1H), 8,29 (д, J=6,0 Гц, 1H), 8,20 (с, 1H), 8,15-8,13 (м, 2H), 7,94 (дд, J=9,1, 2,4 Гц, 1H), 7,91 (с, 1H), 7,60 (дд, J=8,5, 4,1 Гц, 1H), 7,40 (д, J=2,3 Гц, 1H), 7,27 (дд, J=8,6, 2,4 Гц, 1H), 7,11 (д, J=2,2 Гц, 1H), 6,99 (д, J=8,8 Гц, 1H), 6,45 (дд, J=5,7, 2,4 Гц, 1H), 6,39 (с, 1H), 3,83 (с, 3H), 2,92 (м, 1H), 2,05 (с, 3H), 1,25 (д, J=6,9 Гц, 6H); MC (ESI) m/z: 559,2 (М+H+).
Пример 43: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (0,711 г, 1,66 ммоль) и соединение из примера A28 (0,450 г, 1,58 ммоль) в присутствии DIEA (0,61 мл, 3,48 ммоль) объединяли, получая 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,431 г, выход 48%) в виде белого твердого вещества. Продукт превращали в соответствующий мезилат в результате взаимодействия с MsOH (1,0 экв.).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,08-9,04 (м, 3H), 8,66 (д, J=8,8 Гц, 1H), 8,57-8,54 (м, 2H), 8,26-8,16 (м, 4H), 8,05 (дд, J=9,2, 2,4 Гц, 1H), 7,75 (кв, J=4,4 Гц, 1H), 7,64 (с, 1H), 7,37 (дд, J=11,6, 2,0 Гц, 1H), 7,12-7,08 (м, 2H), 6,41 (с, 1H), 3,90 (с, 3H), 2,92 (м, 1H), 2,33 (с, 3H), 1,24 (д, J=7,2 Гц, 6H); MC (ESI) m/z: 563,3 (М+H+).
Пример 44: Используя способ, аналогичный способу, описанному в примере 4, соединение из примера B26 (100 мг, 0,29 ммоль) и соединение из примера A31 (75 мг, 0,29 ммоль) в присутствии N-метилпирролидина (каталитическое количество) объединяли, получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(изопропиламино)пиридин-4-илокси)фенил)мочевину (59 мг, выход 32%). Продукт обрабатывали метансульфоновой кислотой, получая мезилат 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(изопропиламино)пиридин-4-илокси)фенил)мочевины (63 мг, выход 93%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,03 (м, 1H), 9,00 (с, 1H), 8,98 (м, 1H), 8,54 (ушир.д, J=8,4 Гц, 1H), 8,35 (ушир.м, 1H), 8,17 (м, 2H), 7,97 (дд, J=2,4 и 9,2 Гц, 1H), 7,86 (д, J=7,2 Гц, 1H), 7,66 (д, J=4,4 и 8,4 Гц, 1H), 7,33 (дд, J=2,8 и 11,6 Гц, 1H), 7,05 (м, 1H), 6,61 (дд, J=2,4 и 6,8 Гц, 1H), 6,45 (с, 1H), 6,08 (ушир.с, 1H), 3,81 (м, 1H), 2,29 (с, 3H), 1,29 (с, 9H), 1,13 (д, J=6,0 Гц, 6H); MC (ESI) m/z: 554,2 (М+H+).
Пример 45: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B10 (0,060 г, 0,15 ммоль) и соединение из примера A28 (0,041 г, 0,15 ммоль) в присутствии DIEA (0,056 мл, 0,32 ммоль) объединяли, получая 1-(3-этил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)мочевину (47,6 мг, выход 60%) в виде белой пены. Продукт превращали в соответствующий мезилат в результате взаимодействия с MsOH (1,0 экв.).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,03-8,95 (м, 3H), 8,55-8,48 (м, 3H), 8,19-8,13 (м, 3H), 7,95 (дд, J=9,2, 2,4 Гц, 1H), 7,64 (дд, J=8,4, 4,4 Гц, 1H), 7,55 (с, 1H), 7,32 (дд, J=12,0, 2,8 Гц, 1H), 7,07-7,01 (м, 2H), 6,36 (с, 1H), 3,86 (с, 3H), 2,56 (кв, J=7,2 Гц, 2H), 2,25 (с, 3H), 1,18 (т, J=7,6 Гц, 3H); MC (ESI) m/z: 549,3 (М+H+).
Пример 46: Используя общий способ A, соединение из примера B27 (77 мг, 0,28 ммоль) и соединение из примера A2 (150 мг, 0,57 ммоль) в присутствии DPPA (67 мкл, 0,31 ммоль) и Et3N (44 мкл, 0,31 ммоль) объединяли, получая 1-(1-(бензо[d]оксазол-5-ил)-3-изопропил-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину (105 мг, выход 70%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,96 (д, J=2,0 Гц, 1H), 8,88 (с, 1H), 8,86 (с, 1H), 8,77 (кв, J=4,8 Гц, 1H), 8,49 (д, J=6,0 Гц, 1H), 8,16 (т, J=9,2 Гц, 1H), 7,94 (дд, J=3,2 и 5,2 Гц, 1H), 7,57 (дд, J=2, и 8,8 Гц, 1H), 7,38 (д, J=2,8 Гц, 1H), 7,28 (дд, J=2,4 и 11,6 Гц, 1H), 7,14 (дд, J=2,8 и 5,6 Гц, 1H), 7,03 (м, 1H), 6,37 (с, 1H), 2,76 (д, J=4,8 Гц, 3H), 1,23 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 530,2 (М+H+).
Пример 47: К суспензии 5-амино-2-фторбензонитрила (1,00 г, 7,38 ммоль) в концентрированной HCl (15 мл) при 0°C медленно в течение 15 минут добавляли раствор NaNO2 (0,64 г, 9,28 ммоль) в воде (15 мл). Полученную смесь перемешивали в течение 90 минут при 0°C. Раствор, содержащий SnCl2·2H2O (3,37 г, 14,9 ммоль), концентрированную HCl (5 мл) и воду (5 мл) добавляли по каплям в течение 20 минут. Смесь перемешивали в течение 2 часов при 0°C и экстрагировали EtOAc (4×25 мл). Водную часть охлаждали на бане со льдом и осторожно обрабатывали 70 мл 3М NaOH (70 мл) до конечного значения pH 5. Водную часть экстрагировали EtOAc (2×50 мл). Все органические фазы объединяли и концентрировали в вакууме, получая коричневое масло (2,58 г), которое объединяли с пивалоилацетонитрилом (1,00 г, 8,0 ммоль) в изопропаноле (15 мл). Полученный раствор кипятили с обратным холодильником в течение 28 часов. Реакционную смесь концентрировали в вакууме, разбавляли EtOAc (30 мл) и промывали водой (20 мл), насыщенным водным раствором NaHCO3 (20 мл), водой (20 мл) и насыщенным раствором соли (20 мл). Водную фазу дополнительно экстрагировали EtOAc (2×20 мл). Объединенные органические фазы сушили (MgSO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая 5-(5-амино-3-трет-бутил-1H-пиразол-1-ил)-2-фторбензонитрил (1,24 г, выход 65%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,05 (м, 1H), 7,97 (м, 1H), 7,61 (т, J=9,0 Гц, 1H), 5,43 (с, 1H), 5,42 (с, 2H); MC (ESI) m/z: 259,3 (М+H+).
Раствор 5-(5-амино-3-трет-бутил-1H-пиразол-1-ил)-2-фторбензонитрила (86 мг, 0,33 ммоль) и оксима ацетона (37 мг, 0,50 ммоль) в DMAc (1 мл) обрабатывали трет-бутоксидом калия (56 мг, 0,50 ммоль). Реакционную смесь перемешивали 45 минут при комнатной температуре. Смесь разбавляли EtOAc (30 мл), промывали водой (10 мл) и насыщенным раствором соли (2×10 мл), сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на силикагеле, получая O-2-циано-4-(5-амино-3-трет-бутил-1H-пиразол-1-ил)фенилоксим пропан-2-она (47 мг, выход 45%).
1H ЯМР (400 МГц, ацетон-d6) δ 7,93-7,89 (м, 2H), 7,63 (дд, J=8,8, 0,8 Гц, 1H), 5,52 (с, 1H), 4,87 (с, 2H), 2,17 (с, 3H), 2,08 (c, 3H), 1,26 (с, 9H); MC (ESI) m/z: 312,3 (М+H+).
Раствор O-2-циано-4-(5-амино-3-трет-бутил-1H-пиразол-1-ил)фенилоксима пропан-2-она (47 мг, 0,15 ммоль) в этилацетате (5 мл) обрабатывали 2М водным раствором Na2CO3 (0,67 мл) и изопропенилхлорформиата (0,050 мл, 0,46 ммоль). Реакционную смесь перемешивали при комнатной температуре. Через 2 часа добавляли дополнительное количество изопропенилхлорформиата (0,1 мл, 0,92 ммоль). Через 1 час дополнительно добавляли изопропенилхлорформиат (0,1 мл, 0,92 ммоль) и 2М водный раствор Na2CO3 (0,5 мл, 1 ммоль). Еще через час реакционную смесь разбавляли EtOAc (10 мл), промывали водой (10 мл) и насыщенным раствором соли (10 мл), сушили (MgSO4) и концентрировали в вакууме, получая изопропенилкарбамат O-2-циано-4-(5-амино-3-трет-бутил-1H-пиразол-1-ил)фенилоксима пропан-2-она (62 мг, выход 58%), который использовали без дополнительной очистки. МС (ESI) m/z: 396,2 (M+H+).
Изопропенилкарбамат с предыдущей стадии (60 мг, 0,15 ммоль), пример A2 (40 мг, 0,15 ммоль) и N-метилпирролидин (1 мг, 0,015 ммоль) объединяли в ТГФ (1 мл) и нагревали до 55°C в течение ночи. Реакционную смесь концентрировали и подвергали хроматографии, получая соответствующую мочевину (97 мг, выход >100%) в виде темной пены. МС (ESI) m/z: 599,2 (M+H+).
Описанную выше мочевину растворяли в этаноле и обрабатывали 3 М водным раствором HCl (0,5 мл). Через 24 часа добавляли еще 0,5 мл 3 М водного раствора HCl и перемешивание продолжали в течение 3 суток. Реакционную смесь распределяли между 2 М водным раствором Na2CO3 и EtOAc. Органический слой промывали насыщенным водным раствором NaHCO3, водой и насыщенным раствором соли, сушили (Na2SO4), концентрировали в вакууме и очищали хроматографией на силикагеле и перекристаллизовывали из ацетона, получая 1-(1-(3-аминобензо[d]изоксазол-5-ил)-3-трет-бутил-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину (33 мг, выход 39% за 2 стадии).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,93 (д, J=2,2 Гц, 1H), 8,86 (с, 1H), 7,77 (кв, J=4,8 Гц, 1H), 8,50 (д, J=5,4 Гц, 1H), 8,20 (т, J=9,3 Гц, 1H), 7,99 (д, J=1,2 Гц, 1H), 7,64-7,59 (м, 2H), 7,37 (д, J=2,4 Гц, 1H), 7,29 (дд, J=11,9, 2,6 Гц, 1H), 7,15 (дд, J=5,6, 2,6 Гц, 1H), 7,03 (м, 1H), 6,55 (с, 2H), 6,41 (с, 1H), 2,77 (д, J=4,7 Гц, 3H), 1,27 (с, 9H); MC (ESI) m/z: 559,2 (М+H+).
Пример 48: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B9 (0,175 г, 0,41 ммоль) и соединение из примера A42 (0,097 г, 0,389 ммоль) объединяли, получая 1-(2-фтор-5-(6-нитропиридин-3-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,129 г, выход 63%) в виде светло-желтого масла.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,94 (дд, J=4,4, 2,0 Гц, 1H), 8,48 (д, J=8,4 Гц, 1H), 8,31 (д, J=8,8 Гц, 1H), 8,26 (д, J=2,8 Гц, 1H), 8,20 (д, J=8,8 Гц, 1H), 8,11 (д, J=2,4 Гц, 1H), 8,00 (м, 1H), 7,91 (дд, J=9,2, 2,4 Гц, 1H), 7,63 (м, 1H), 7,58 (дд, J=8,8, 2,8 Гц, 1H), 7,22 (м, 1H), 6,84 (м, 1H), 6,46 (с, 1H), 2,98 (м, 1H), 1,30 (д, J=7,2 Гц, 6H); MC (ESI) m/z: 528,3 (М+H+).
1-(2-фтор-5-(6-нитропиридин-3-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,129 г, 0,245 ммоль) растворяли в MeOH (2,0 мл), добавляли NH4Cl (0,131 г, 2,45 ммоль) и цинковую пыль (0,160 г, 2,45 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь фильтровали через целит и промывали метанолом (30 мл) и EtOAc (50 мл). Фильтрат концентрировали в вакууме, распределяли между EtOAc (30 мл) и водой (20 мл). Отделенную органическую фазу промывали насыщенным раствором соли (10 мл), сушили (MgSO4) и концентрировали, получая 1-(5-(6-аминопиридин-3-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,0495 г, выход 41%) в виде белой пены. МС (ESI) m/z: 498,2 (M+H+).
1-(5-(6-аминопиридин-3-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,0495 г, 0,099 ммоль) растворяли в ДХМ (1,0 мл), добавляли пиридин (0,49 мл, 6,0 ммоль) и уксусный ангидрид (0,066 мл, 0,65 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Завершенную реакцию гасили 2 М раствором NaHCO3 (12 мл) и экстрагировали EtOAc (25 мл). Органический слой промывали H2O (15 мл) и насыщенным раствором соли (10 мл), сушили (MgSO4), концентрировали в вакууме и очищали хроматографией, получая 1-(5-(6-ацетамидопиридин-3-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (0,0234 г, выход 44%) в виде желтой пены. Продукт превращали в соответствующий мезилат в результате взаимодействия с MsOH (1,0 экв.).
1H ЯМР (400 МГц, ДМСО-d6) δ 10,54 (с, 1H), 9,09 (с, 1H), 9,07-9,04 (м, 2H), 8,65 (д, J=8,0 Гц, 1H), 8,25 (д, J=2,0 Гц, 1H), 8,21 (д, J=8,8 Гц, 1H), 8,11-8,07 (м, 2H), 8,02 (дд, J=8,8, 2,4 Гц, 1H), 7,85 (м, 1H), 7,75 (м, 1H), 4,48 (дд, J=8,8, 3,2 Гц, 1H), 7,24 (м, 1H), 6,67 (м, 1H), 6,40 (с, 1H), 2,92 (м, 1H), 2,31 (с, 3H), 2,08 (с, 3H), 1,24 (д, J=7,2 Гц, 6H); MC (ESI) m/z: 540,0 (М+H+).
Пример 49: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B24 (150 мг, 0,26 ммоль) и соединение из примера A28 (74 мг, 0,26 ммоль) в присутствии DIEA (90 мкл, 0,52 ммоль) объединяли, получая бензил-6-(3-трет-бутил-5-(3-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)уреидо)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (100 мг, выход 56%).
К раствору бензил-6-(3-трет-бутил-5-(3-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)уреидо)-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (100 мг, 0,14 ммоль) в метаноле/EtOAc (1:1, 10 мл) добавляли 10% Pd/C. Раствор перемешивали в течение ночи в атмосфере H2 (1 атм) при комнатной температуре. Раствор фильтровали и концентрировали в вакууме, получая 1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)мочевину (73 мг, выход 90%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,00 (ушир.с, 1H), 8,02 (м, 1H), 8,35 (д, J=5,6 Гц, 1H), 8,25 (с, 1H), 8,15 (дт, J=2,4 и 8,8 Гц, 1H), 7,95 (с, 1H), 7,1-7,3 (м, 3H), 7,99 (м, 1H), 6,65 (м, 1H), 6,36 (д, J=2,8 Гц, 1H), 3,95 (м, 1H), 3,84 (с, 3H), 3,53 (м, 1H), 3,01 (м, 1H), 2,88 (м, 1H), 2,79 (м, 1H), 2,60 (м, 1H), 1,25 (с, 9H); MC (ESI) m/z: 581,3 (М+H+).
Пример 50: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B29 (0,20 г, 0,43 ммоль) и соединение из примера A27 (118 мг, 0,43 ммоль) объединяли, получая 1-(4-(2-(1H-пиразол-4-ил)пиридин-4-илокси)-2-фторфенил)-3-(3-трет-бутил-1-(1-оксо-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиразол-5-ил)мочевину (123 мг, выход 47%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,88 (ушир.с, 1H), 8,83 (с, 1H), 8,33 (д, J=5,6 Гц, 1H), 8,10 (д, J=8,8 Гц, 1H), 8,07 (м, 2H), 7,85 (д, J=2,0 Гц, 1H), 7,57 (дд, J=2,4 и 8,0 Гц, 1H), 7,42 (д, J=8,0 Гц, 1H), 7,31 (ушир.с, 1H), 7,18 (дд, J=2,4 и 12,0 Гц, 1H), 6,95 (м, 1H), 6,65 (м, 1H), 6,33 (с, 1H), 3,35 (м, 2H), 2,91 (м, 2H), 1,22 (с, 9H); MC (ESI) m/z: 581,3 (М+H+).
Пример 51: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B30 (0,20 г, 0,37 ммоль) и соединение из примера A27 (100 мг, 0,37 ммоль) объединяли, получая трет-бутил-7-(5-(3-(4-(2-(1H-пиразол-4-ил)пиридин-4-илокси)-2-фторфенил)уреидо)-3-трет-бутил-1H-пиразол-1-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (130 мг, выход 53%), который обрабатывали смесью 4,0 М HCl/диоксан (2 мл) и перемешивали при комнатной температуре в течение 4 часов. Твердое вещество отфильтровывали, промывали этилацетатом и сушили в вакууме, получая HCl-соль 1-(4-(2-(1H-пиразол-4-ил)пиридин-4-илокси)-2-фторфенил)-3-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиразол-5-ил)мочевины (120 мг, выход 96%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,51 (ушир.с, 2H), 9,27 (ушир.с, 1H), 9,21 (ушир.с, 1H), 8,69 (ушир.с, 2H), 8,54 (д, J=7,2 Гц, 1H), 8,22 (т, J=9,2 Гц, 1H), 7,84 (м, 1H), 7,3-7,5 (м, 4H), 7,13 (м, 1H), 7,10 (дд, J=2,4 и 6,4 Гц, 1H), 6,37 (с, 1H), 4,38 (м, 2H), 3,38 (м, 2H), 3,05 (м, 2H), 1,28 (с, 9H); MC (ESI) m/z: 567,3 (М+H).
Пример 52: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера A36 (110 мг, 0,363 ммоль) и соединение из примера B10 (150 мг, 0,363 ммоль) объединяли и очищали хроматографией (колонка Si-25, метанол/этилацетат), получая 1-(2,3-дифтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(3-этил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину в виде белой пены (66 мг, выход 32%).
1H ЯМР (400 МГц, диметилсульфоксид-d6) δ 1,27 (т, 3H), 2,65 (кв, 2H), 3,89 (с, 3H), 6,46 (с, 1H), 6,74-6,76 (м, 1H), 7,22 (т, 1H), 7,29 (с, 1H), 7,65-7,68 (с, 1H), 7,97-8,02 (м, 3H), 8,20-8,22 (м, 2H), 8,31 (с, 1H), 8,40-8,42 (м, 1H), 8,50-8,53 (м, 1H), 9,00-9,01 (м, 1H), 9,11 (с, 1H), 9,19 (с, 1H); MC (ESI) m/z: 567,0 (М+H+).
Пример 53: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера A38 (108 мг, 0,363 ммоль) и соединение из примера B10 (150 мг, 0,363 ммоль) объединяли и очищали хроматографией (колонка Si-25, метанол/этилацетат), получая 1-(3-этил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-3-метил-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)мочевину в виде белой пены (78 мг, выход 38%).
1H ЯМР (400 МГц, диметилсульфоксид-d6) δ 1,29 (т, 3H), 2,09 (с, 3H), 2,67 (кв, 2H), 3,91 (с, 3H), 6,47 (с, 1H), 6,59-6,61 (м, 1H), 7,00-7,02 (м, 1H), 7,22 (с, 1H), 7,67-7,70 (м, 1H), 7,99-8,10 (м, 3H), 8,22-8,24 (м, 2H), 8,30 (с, 1H), 8,39 (д, 1H), 8,53-8,55 (м, 1H), 9,00-9,03 (м, 2H), 9,10 (с, 1H); MC (ESI) m/z: 563,3 (М+H+).
Пример 54: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (0,10 г, 0,23 ммоль) и соединение из примера A32 (56 мг, 0,23 ммоль) в присутствии DIEA (68 мкл) объединяли, получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(3-(5-хлорпиридин-3-илокси)-5-цианофенил)мочевину (39 мг, выход 32%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,47 (с, 1H), 8,98 (дд, J=2,0 и 4,4 Гц, 1H), 8,82 (с, 1H), 8,53 (д, J=2,0 Гц, 1H), 8,49 (м, 1H), 8,45 (д, J=2,4 Гц, 1H), 8,17 (м, 2H), 7,97 (дд, J=2,8 и 9,2 Гц, 1H), 7,84 (т, J=2,0 Гц, 1H), 7,70 (т, J=1,6 Гц, 1H), 7,65 (дд, J=4,0 и 8,0 Гц, 1H), 7,45 (т, J=2,0 Гц, 1H), 7,31 (м, 1H), 6,48 (с, 1H), 2,50 (с, 3H), 1,34 (с, 9H); MC (ESI) m/z: 538,0 (М+H+).
Пример 55: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера B3 (0,10 г, 0,23 ммоль) и соединение из примера A33 (51 мг, 0,23 ммоль) в присутствии DIEA (68 мкл) объединяли, получая 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(3-циано-5-(6-метилпиридин-3-илокси)фенил)мочевину (31 мг, выход 27%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,43 (с, 1H), 8,98 (дд, J=2,0 и 4,4 Гц, 1H), 8,74 (с, 1H), 8,48 (м, 1H), 8,33 (д, J=2,8 Гц, 1H), 8,16 (м, 2H), 7,96 (дд, J=2,8 и 9,2 Гц, 1H), 7,63 (м, 2H), 7,50 (дд, J=2,8 и 8,0 Гц, 1H), 7,34 (д, J=8,4 Гц, 1H), 7,29 (т, J=2,0 Гц, 1H), 7,17 (м, 1H), 6,46 (с, 1H), 2,50 (с, 3H), 1,33 (с, 9H); MC (ESI) m/z: 518,0 (М+H+).
Пример 56: Используя способ, аналогичный способу, описанному в примере 1, соединение из примера A41 (15 мг, 0,055 ммоль) и соединение из примера B9 (24 мг, 0,056 ммоль) объединяли, получая 1-(5-(4-(1H-пиразол-4-ил)пиримидин-2-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевину (9 мг, выход 29%).
1H ЯМР (400 МГц, ДМСО-d6) δ 13,36 (с, 1H), 9,09 (с, 1H), 9,07 (с, 1H), 8,95 (м, 1H), 8,50-8,45 (м, 2H), 8,17-8,12 (м, 2H), 8,01 (дд, J=6,8, 2,9 Гц, 1H), 7,92 (дд, J=9,0, 2,1 Гц, 1H), 7,61 (дд, J=8,2, 4,1 Гц, 1H), 7,51 (д, J=5,0 Гц, 1H), 7,27 (дд, J=11,0, 8,9 Гц, 1H), 6,85 (м, 1H), 6,40 (с, 1H), 2,89 (м, 1H), 1,22 (д, J=6,8 Гц, 6H); MC (ESI) m/z: 550,2 (М+H+).
Следующие примеры получали способами, описанными на схемах 1-17, используя общий способ A, согласно описанным выше примерам и способами, описанными в WO 2006/071940, зарегистрированной 23 декабря 2005 года, включенной в виде ссылки:
1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)-3-(3-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(2-(метиламино)хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевина, 1-(1-(4-(2-амино-2-оксоэтил)нафталин-2-ил)-3-трет-бутил-1H-пиразол-5-ил)-3-(2-хлор-5-(5-фторпиридин-3-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(пиридин-3-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2,4-дифтор-5-(пиридин-3-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)-3-(2,4-дифтор-5-(пиридин-3-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(1H-индазол-5-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(пиридин-3-илокси)фенил)мочевина, 1-(5-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-3-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(2-гидроксиэтиламино)пиридин-4-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(4-хлор-5-(6-цианопиридин-3-илокси)-2-фторфенил)мочевина, 1-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)-3-(1-(хинолин-6-ил)-3-(трифторметил)-1H-пиразол-5-ил)мочевина, 1-(3-циклопентил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевина, 1-(3-циклобутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(5-(6-цианопиридин-3-илокси)-2-фторфенил)мочевина, 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(3-фтор-4-(2-(метиламино)пиридин-4-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)-3-(4-метил-3-(пиридин-3-илокси)фенил)мочевина, 1-(2-фтор-5-(6-метилпиридин-3-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(3-этил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метиламино)пиридин-4-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(1H-индазол-5-ил)-1H-пиразол-5-ил)-3-(2-фтор-5-(2-(метиламино)пиримидин-4-илокси)фенил)мочевина, 1-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)-3-(4-хлор-2-(хинолин-6-ил)фенил)мочевина, 1-(1-(1H-индазол-5-ил)-3-изопропил-1H-пиразол-5-ил)-3-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)мочевина, 1-(3-трет-бутил-1-(2-метилхинолин-6-ил)-1H-пиразол-5-ил)-3-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)мочевина, 1-(4-(2-карбамоилпиридин-4-илокси)-3-метилфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(2-метилхинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(3-метил-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевина, 1-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(2-метилхинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(4-(2-(диметиламино)пиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(3-метил-4-(2-(метиламино)пиридин-4-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(4-(2-карбамоилпиридин-4-илокси)-3-метилфенил)мочевина, 1-(5-(2-аминопиримидин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(2-метилхинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(2-фтор-4-(2-(метиламино)пиримидин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(2-фтор-5-(6-(метилкарбамоил)пиридин-3-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(2-метил-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевина, 1-(3-трет-бутил-1-(2-метилхинолин-6-ил)-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метиламино)пиридин-4-илокси)фенил)мочевина, 1-(4-(2-(1H-пиразол-4-ил)пиридин-4-илокси)-2-фторфенил)-3-(3-метил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хиноксалин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хиноксалин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(1-(бензо[d]оксазол-5-ил)-3-трет-бутил-1H-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевина, 1-(4-(2-(1H-пиразол-4-ил)пиридин-4-илокси)-3-метилфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(4-(2-(1H-пиразол-4-ил)пиридин-4-илокси)-2-фторфенил)-3-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(3-фтор-4-(2-(изопропиламино)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)-3-(4-(2-(изопропиламино)пиридин-4-илокси)-3-метилфенил)мочевина, 1-(4-(2-(циклопентиламино)пиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина и 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(3-метил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина.
Следующие примеры получали способами, описанными на схемах 1-17, общим способом A, согласно описанными выше примерам и способами, описанными в WO 2006/071940, зарегистрированной 23 декабря 2005 года, включенной в виде ссылки: 1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-5-ил)-3-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-илокси)фенил)мочевина, 1-(5-(4-(1H-пиразол-4-ил)пиримидин-2-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(2-фтор-4-метил-5-(4-(1-метил-1H-пиразол-4-ил)пиримидин-2-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(2-фтор-5-(4-(1-метил-1H-пиразол-4-ил)пиримидин-2-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(1-изопропил-3-(хинолин-6-ил)-1H-пиразол-4-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(1-изопропил-4-(хинолин-6-ил)-1H-пиррол-3-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(1-изопропил-5-метил-3-(хинолин-6-ил)-1H-пиразол-4-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(2-изопропил-5-(хинолин-6-ил)оксазол-4-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(2-изопропил-5-(хинолин-6-ил)тиазол-4-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(5-изопропил-2-(хинолин-6-ил)фуран-3-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(5-изопропил-2-(хинолин-6-ил)тиофен-3-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(4-изопропил-1-(хинолин-6-ил)-1H-имидазол-2-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(5-изопропил-2-(хинолин-6-ил)-1H-пиррол-3-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(4-изопропил-1-(хинолин-6-ил)-1H-пиррол-2-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(5-метил-2-(хинолин-6-ил)пиридин-3-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(1-изопропил-3-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиразол-4-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(1-изопропил-4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиррол-3-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(2-изопропил-5-(1,2,3,4-тетрагидроизохинолин-6-ил)оксазол-4-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(2-изопропил-5-(1,2,3,4-тетрагидроизохинолин-6-ил)тиазол-4-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(5-изопропил-2-(1,2,3,4-тетрагидроизохинолин-6-ил)фуран-3-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(5-изопропил-2-(1,2,3,4-тетрагидроизохинолин-6-ил)тиофен-3-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(4-3(4-изопропил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-имидазол-2-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(5-изопропил-2-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиррол-3-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(4-изопропил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-пиррол-2-ил)мочевина, 1-(2-фтор-4-(2-(1-метил-1H-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(5-метил-2-(1,2,3,4-тетрагидроизохинолин-6-ил)пиридин-3-ил)мочевина, 4-(3-фтор-4-(3-(1-изопропил-3-(хинолин-6-ил)-1H-пиразол-4-ил)уреидо)фенокси)-N-метилпиколинамид, 4-(3-фтор-4-(3-(1-изопропил-4-(хинолин-6-ил)-1H-пиррол-3-ил)уреидо)фенокси)-N-метилпиколинамид, 4-(3-фтор-4-(3-(2-изопропил-5-(хинолин-6-ил)оксазол-4-ил)уреидо)фенокси)-N-метилпиколинамид, 4-(3-фтор-4-(3-(2-изопропил-5-(хинолин-6-ил)тиазол-4-ил)уреидо)фенокси)-N-метилпиколинамид, 4-(3-фтор-4-(3-(5-изопропил-2-(хинолин-6-ил)тиофен-3-ил)уреидо)фенокси)-N-метилпиколинамид, 4-(3-фтор-4-(3-(4-изопропил-1-(хинолин-6-ил)-1H-имидазол-2-ил)уреидо)фенокси)-N-метилпиколинамид, 4-(3-фтор-4-(3-(5-изопропил-2-(хинолин-6-ил)-1H-пиррол-3-ил)уреидо)фенокси)-N-метилпиколинамид, 4-(3-фтор-4-(3-(4-изопропил-1-(хинолин-6-ил)-1H-пиррол-2-ил)уреидо)фенокси)-N-метилпиколинамид, 4-(3-фтор-4-(3-(5-метил-2-(хинолин-6-ил)пиридин-3-ил)уреидо)фенокси)-N-метилпиколинамид, 4-(3-фтор-4-(3-(5-изопропил-2-(хинолин-6-ил)фуран-3-ил)уреидо)фенокси)-N-метилпиколинамид, 1-(5-(4-(1H-пиразол-4-ил)пиримидин-2-илокси)-2-фтор-4-метилфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(2-фтор-5-(4-(1-метил-1H-пиразол-4-ил)пиримидин-2-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(5-(4-(1H-пиразол-4-ил)пиримидин-2-илокси)-2-фтор-4-метилфенил)-3-(1-(бензо[d]оксазол-5-ил)-3-изопропил-1H-пиразол-5-ил)мочевина, 1-(2-фтор-4-метил-5-(4-(1-метил-1H-пиразол-4-ил)пиримидин-2-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1H-пиразол-5-ил)мочевина, 1-(1-(бензо[d]оксазол-5-ил)-3-изопропил-1H-пиразол-5-ил)-3-(2-фтор-5-(4-(1-метил-1H-пиразол-4-ил)пиримидин-2-илокси)фенил)мочевина, 1-(5-(4-(1H-пиразол-4-ил)пиримидин-2-илокси)-2-фтор-4-метилфенил)-3-(1-(имидазо[1,2-a]пиридин-6-ил)-3-изопропил-1H-пиразол-5-ил)мочевина, 1-(2-фтор-5-(4-(1-метил-1H-пиразол-4-ил)пиримидин-2-илокси)фенил)-3-(1-(имидазо[1,2-a]пиридин-6-ил)-3-изопропил-1H-пиразол-5-ил)мочевина.
Раздел 3
Анализ киназы Abl (SEQ ID NO: 1)
Активность киназы Abl (SEQ ID NO: 1) определяли по продукции АДФ в результате киназной реакции посредством связывания с системой пируваткиназа/лактатдегидрогеназа (например, Schindler, et al., Science (2000) 289, 1938-1942). В данном анализе окисление NADH (соответственно снижение при A340 нм) непрерывно регистрировали спектрофотометрически. Реакционная смесь (100 мкл) содержала киназу Abl (1 нМ Abl из deCode Genetics), пептидный субстрат (EAIYAAPFAKKK (SEQ ID NO: 3), 0,2 мМ), MgCl2 (10 мМ), пируваткиназу (4 единицы), лактатдегидрогеназу (0,7 единиц), фосфоенолпируват (1 мМ) и NADH (0,28 мМ) в 90 мМ трис-буфере, содержащем 0,2% октилглюкозид и 3,5% ДМСО, pH 7,5. Тестируемые соединения инкубировали с Abl (SEQ ID NO: 1) и другими реагентами при 30°C в течение 2 часов перед добавлением АТФ (500 мкМ) для начала реакции. Поглощение при 340 нм регистрировали непрерывно в течение 2 часов при 30°C на считывающем устройстве для планшетов Polarstar Optima (BMG). Скорость реакции вычисляли, используя временной интервал от 1,0 до 2,0 часов. Ингибирование в процентах определяли в результате сравнения скорости реакции со скоростью реакции в контроле (т.е. в отсутствие тестируемого соединения). Значения IC50 вычисляли на основании ряда значений ингибирования в процентах, определенных в диапазоне концентраций ингибитора, используя стандартные компьютерные программы, которые имеются в пакете программ GraphPad Prism.
Анализ киназы pAbl
Активность киназы pAbl (SEQ ID NO: 1) определяли по продукции АДФ в результате киназной реакции посредством связывания с системой пируваткиназа/лактатдегидрогеназа (например, Schindler, et al., Science (2000) 259, 1938-1942). В данном анализе, окисление NADH (соответственно снижение при A340 нм) непрерывно регистрировали спектрофотометрически. Реакционная смесь (100 мкл) содержала киназу pAbl (2 нМ pAbl из deCode Genetics), пептидный субстрат (EAIYAAPFAKKK (SEQ ID NO: 3), 0,2 мМ), MgCl2 (10 мМ), пируваткиназу (4 единицы), лактатдегидрогеназу (0,7 единиц), фосфоенолпируват (1 мМ) и NADH (0,28 мМ) в 90 мМ трис-буфере, содержащем 0,2% октилглюкозид и 3,5% ДМСО, pH 7,5. Тестируемые соединения инкубировали с pAbl (SEQ ID NO: 1) и другими реагентами при 30°C в течение 2 часов перед добавлением АТФ (500 мкМ) для начала реакции. Поглощение при 340 нм регистрировали непрерывно в течение 2 часов при 30°C на считывающем устройстве для планшетов Polarstar Optima (BMG). Скорость реакции вычисляли, используя временной интервал от 1,0 до 2,0 часов. Ингибирование в процентах определяли в результате сравнения скорости реакции со скоростью реакции в контроле (т.е. в отсутствие тестируемого соединения). Значения IC50 вычисляли на основании ряда значений ингибирования в процентах, определенных в диапазоне концентраций ингибитора, используя стандартные компьютерные программы, которые имеются в пакете программ GraphPad Prism. pAbl получали в виде фосфорилированной формы фермента, используемой в анализе Abl (см. выше).
Анализ киназы Abl(T315I) (SEQ ID NO: 2)
Активность киназы Abl(T315I) (SEQ ID NO: 2) определяли по продукции АДФ в результате киназной реакции посредством связывания с системой пируваткиназа/лактатдегидрогеназа (например, Schindler, et al. Science (2000) 259, 1938-1942). В данном анализе окисление NADH (соответственно снижение при A340 нм) непрерывно регистрировали спектрофотометрически. Реакционная смесь (100 мкл) содержала киназу Abl(T315I) (SEQ ID NO: 2) (6 нМ Abl(T315I) из deCode Genetics), пептидный субстрат (EAIYAAPFAKKK (SEQ ID NO: 3), 0,2 мМ), MgCl2 (10 мМ), пируваткиназу (4 единицы), лактатдегидрогеназу (0,7 единиц), фосфоенолпируват (1 мМ) и NADH (0,28 мМ) в 90 мМ трис-буфере, содержащем 0,2% октилглюкозид и 3,5% ДМСО, pH 7,5. Тестируемые соединения инкубировали с Abl(T315I) и другими реагентами при 30°C в течение 2 часов перед добавлением АТФ (500 мкМ) для начала реакции. Поглощение при 340 нм регистрировали непрерывно в течение 2 часов при 30°C на считывающем устройстве для планшетов Polarstar Optima (BMG). Скорость реакции вычисляли, используя временной интервал от 1,0 до 2,0 часов. Ингибирование в процентах определяли в результате сравнения скорости реакции со скоростью реакции в контроле (т.е. в отсутствие тестируемого соединения). Значения IC50 вычисляли на основании ряда значений ингибирования в процентах, определенных в диапазоне концентраций ингибитора, используя стандартные компьютерные программы, которые имеются в пакете программ GraphPad Prism.
Киназа Abl (SEQ ID NO: 1)

Киназа Abl(T315I) (SEQ ID NO: 2)

Культивирование клеток
Клетки BaF3 (исходные или трансфицированные следующим образом: bcr-Abl дикого типа или точечные мутанты bcr-Abl T315I, E255K, Y253F, M351T) получали от профессора Richard Van Etten (New England Medical Center, Boston, MA). Коротко, клетки выращивали в RPMI 1640 с добавлением 10% охарактеризованной фетальной сыворотки теленка (HyClone, Logan, UT) при 37°C, 5% CO2, 95% влажности. Клеткам давали возможность размножаться вплоть до 80% насыщения, в этой точке их субкультивировали или собирали для анализа.
Анализ пролиферации клеток
Серийное разведение тестируемого соединения распределяли в 96-луночном планшете с черным прозрачным дном (Corning, Corning, NY). В случае каждой линии клеток в лунку вносили по 3000 клеток в полной среде роста. Планшеты инкубировали в течение 72 часов при 37°C, 5% CO2, 95% влажности. В конце периода инкубации в каждую лунку добавляли реагент Cell Titer Blue (Promega, Madison, WI) и осуществляли инкубацию еще в течение 4,5 часов при 37°C, 5% CO2, 95% влажности. Затем планшеты считывали на BMG Fluostar Optima (BMG, Durham, NC), используя возбуждение при 544 нм и эмиссию при 612 нм. Данные анализировали, используя компьютерную программу Prism (Graphpad, San Diego, CA), чтобы получить значения IC50.
Суммарные биологические данные. Биохимические значения IC 50 для соединений формулы Ia
В общем, соединения 1-56, раскрытые в настоящем описании, имели ингибирующую активность >50% в концентрации 0,1-2 мкМ против киназы Abl и киназы Abl-T315I.
Суммарные биологические данные. Значения IC 50 , полученные на целых клетках, для соединений формулы Ia
В общем, соединения 1-56, раскрытые в настоящем описании, в концентрации 1-10 мкМ ингибировали более чем на 50% пролиферацию клеток BaF/3, несущих bcr-Abl дикого типа или точечные мутанты bcr-Abl, включая T315I, E255K, Y253F и M351T.
Раздел 4 - Важные структурные сравнения в соответствии с биологической активностью
В WO 2006/071940 A2 описаны ингибиторы киназ, включая киназу C-Abl, киназу B-Raf, c-MET, киназу VEGF и семейство HER, в которых центральный фенильный цикл не замещен. Пример таких ингибиторов показан ниже, где центральный фенильный цикл не замещен (R16 и R18 = H). Соединения A, B и C, обсуждаемые ниже, взяты из публикации WO 2006/071940A2.
Репрезентативные ключевые структуры
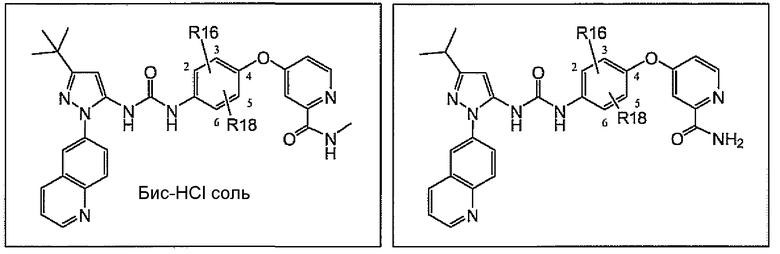
Неожиданно было обнаружено, что ингибиторы, которые содержат другие заместители R16, отличные от H, имеют более высокую активность, измеряемую по ингибированию киназы in vitro, а также измеряемую по антипролиферативной эффективности на целых клетках in vivo при использовании раковых клеток. В качестве иллюстрации в таблице 1 показано, что соединение из примера 1 согласно настоящему изобретению, содержащее остаток 2-F в качестве заместителя R16, в 5,5 раза более эффективно по отношению к фосфорилированной киназе Abl (p-Abl), чем незамещенное соединение A, содержащее R16 = H. Соединение из примера 1 в 6,3 раза более эффективно, чем соединение A против мутанта T315I киназы Abl, клинического изолята онкогенной киназы Abl, найденного у пациентов с хроническим миелогенным лейкозом, которые резистентны к лечению имеющимися в настоящее время терапевтическим средствами, включая Gleevec® (M.E. Gorre et al., Science (2001) 293: 876; S. Branford et al., Blood (2002) 99: 3472; N. von Bubnoff et al., Lancet (2002) 359: 487) и дазатиниб (N.P. Shah et al., Science (2004) 305: 399). Соединение из примера 5, содержащее остаток 3-метила в качестве R16-заместителя, в 4 раза более эффективно против киназы p-Abl, чем незамещенное (R16 = H) соединение A. Соединение из примера 15, содержащее остаток 2-F в качестве заместителя R16, в 8 раз более эффективно против нефосфорилированной киназы Abl (u-Abl), чем незамещенное (R16 = H) соединение B (из WO 2006/071940A2). Соединение из примера 15 в 14 раз более эффективно, чем соединение B против киназы p-Abl и в 18 раз более эффективно, чем соединение B против мутанта T315I киназы Abl.
Структуры соединений из примера 4 (R16=2-F, R18=H) и соединения C (R16, R18=H)
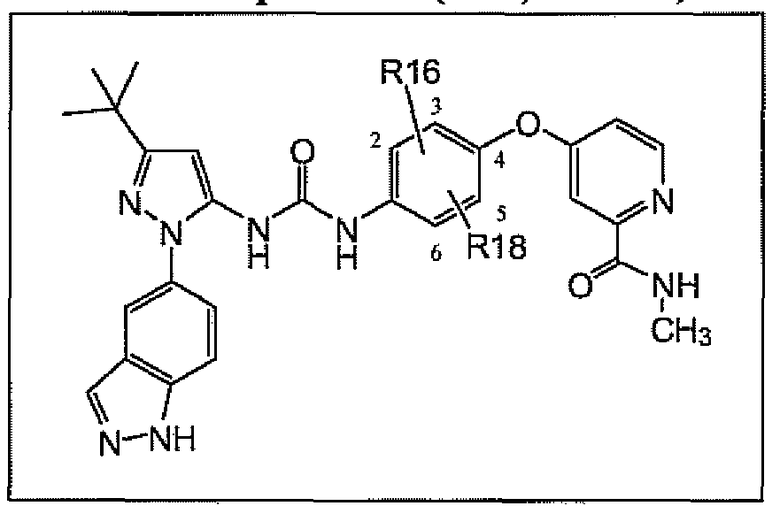
Указанная закономерность также очевидна в случае других аналогов, родственных указанным выше. Как показано в таблице 1, содержащее индазолил соединение из примера 4, которое содержит остаток 2-F в качестве заместителя R16, в 2,2 раза более эффективно, чем незамещенное (R16 = H) соединение C по отношению к киназе u-Abl, в 18 раз более эффективно, чем соединение C против киназы p-Abl и в 13 раз более эффективно, чем соединение C против мутанта T315I киназы Abl.
Указанное неожиданное повышение эффективности против таких киназ также обнаружено в анализах целых клеток, в которых измеряли эффективности таких ингибиторов киназ Abl в блокировании пролиферации клеток, содержащих онкогенные формы киназы Abl: киназы в виде слитого белка bcr-Abl (C.L. Sawyers, New England Journal of Medicine (1999) 340: 1330; S. Faderl et al., New England Journal of Medicine (1999) 341: 164; J.B. Konopka et al., Proceeding of the National Academy of Sciences USA (1985) 82: 1810). В таблице 2 показана повышенная эффективность замещенных R16-содержащих соединений из примеров 1, 5 и 15 по сравнению с их незамещенными аналогами, соединениями A и B. R16-замещенные аналоги в 2,6-4,5 раза более эффективны, чем незамещенные аналоги в клетках BaF3, экспрессирующих онкогенную киназу bcr-abl, в 1,5-3,5 раза более эффективны в клетках BaF3, экспрессирующих мутантную онкогенную форму T315I киназы bcr-abl, в 3,5-7,2 раза более эффективны, чем в клетках BaF3, экспрессирующих мутантную онкогенную форму Y253F киназы bcr-abl, в 4,4-6 раз более эффективны в клетках BaF3, экспрессирующих мутантную онкогенную форму E255K киназу bcr-abl и в 3,2-4,2 раза более эффективны в клетках BaF3, экспрессирующих мутантную онкогенную форму M351T киназы bcr-abl. Пять указанных форм киназы bcr-abl являются онкогенными и являются причиной хронического миелогенного лейкоза человека. Кроме того, четыре мутантных формы киназы bcr-abl резистентны к имеющемуся в настоящее время ингибитору bcr-abl Gleevec®.
bcr-abl
IC50
IC50
IC50
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗАБИЦИКЛОСОЕДИНЕНИЕ И ЕГО СОЛЬ | 2010 |
|
RU2565078C2 |
| НОВОЕ ПРОИЗВОДНОЕ КУМАРИНА, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2428420C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2013 |
|
RU2637944C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА | 2012 |
|
RU2632915C2 |
| ПИРРОЛО[2,3-С]ПИРИДИНЫ В КАЧЕСТВЕ ВИЗУАЛИЗИРУЮЩИХ АГЕНТОВ ДЛЯ НЕЙРОФИБРИЛЛЯРНЫХ КЛУБКОВ | 2015 |
|
RU2695373C2 |
| НОВЫЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ И ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ (1) | 2005 |
|
RU2330021C2 |
| ПИРРОЛО[2,3-С]ПИРИДИНЫ В КАЧЕСТВЕ ВИЗУАЛИЗИРУЮЩИХ АГЕНТОВ ДЛЯ НЕЙРОФИБРИЛЛЯРНЫХ КЛУБКОВ | 2015 |
|
RU2788916C2 |
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| НОВОЕ ПРОИЗВОДНОЕ ТЕТРАГИДРОИЗОХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ПРИМЕНЕНИЕ ЕГО И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРЕДОТВРАЩЕНИЯ ЗАБОЛЕВАНИЯ | 2009 |
|
RU2474575C2 |



Настоящее изобретение относится к новым соединениям
Соединение формулы Ia:
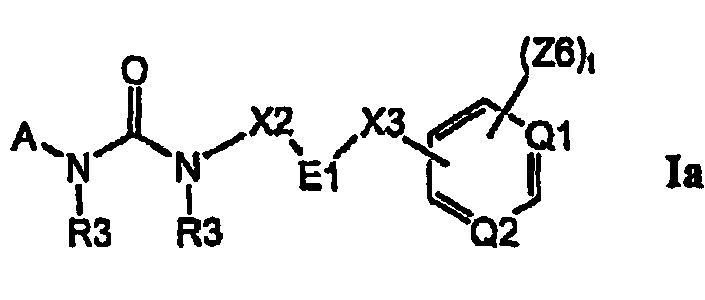
где Q1 и Q2, каждый отдельно и независимо, выбран из группы, состоящей из N и C-Z6, при условии, что оба Q1 и Q2 одновременно не являются C-Z6; Е1 представляет собой фенил, и где цикл Е1 замещен одним или несколькими остатками R16, и где цикл Е1 замещен одним или несколькими остатками R18; где А выбран из группы, состоящей из фенила и пиразолила; G1 означает пиразолил; цикл А замещен в любом замещаемом положении одним остатком А1, где А1 выбран из группы, состоящей из А2 и A3; А2 и A3 таких, как указано в п.1 формулы изобретения, цикл А необязательно замещен одним или несколькими остатками R2; Х2 представляет собой прямую связь, где Е1 непосредственно связан с группой NR3 в формуле Ia; Х3 представляет собой -O-; V и V2 представляют собой Н2, и V1 независимо выбран из группы, состоящей из H2 или О; каждый Z2 независимо и отдельно выбран из группы, состоящей из водорода, C1-С6-алкила; каждый Z3 независимо и отдельно выбран из группы, состоящей из Н, C1-С6-алкила, разветвленного С3-С7-алкила и -(CH2)nN(R4)2; каждый Z4 представляет собой Н; каждый Z6 независимо и отдельно выбран из группы, состоящей из Н, C1-С6-алкила, разветвленного С3-С7-алкила, гидроксила, гидрокси-С3-С6-алкила, разветвленного гидрокси-С2-С6-алкила-, (R3)2N-, (R4)2N-, -N(R3)C(O)R8, C(O)N(R4)2, галогена, -(CH2)nG1 и -R17; каждый R2 выбран из группы, состоящей из C1-С6-алкила и разветвленного С3-С8-алкила; каждый R3 представляет собой Н; каждый R4 представляет собой Н, C1-С6-алкил или разветвленный С3-С7-алкил; каждый R8 независимо и отдельно выбран из группы, состоящей из C1-С6-алкила и разветвленного С3-С7-алкила; каждый R16 независимо и отдельно выбран из группы, состоящей из C1-С6-алкила, разветвленного С3-С7-алкила, галогена и цианогруппы; каждый R17 представляет собой пиразолил; где R17 может быть дополнительно замещен одним или несколькими остатками Z2 или Z3; R18 представляет собой водород и n равно 0-6; р равно 1-4; q равно 2-6; r равно 0 или 1; t равно 1-3, v равно 1 или 2. Кроме того, изобретение описывает способ модулирования киназной активности дикого типа, фармацевтическую композицию, предназначенную для лечения гиперпролиферативных заболеваний и рака у млекопитающих, также описаны способы лечения рака у пациента. Технический результат: получены и описаны новые соединения, которые могут найти свое применение в модулировании киназной активности. 7 н. и 13 з.п., 56 прим., 2 табл.
1. Соединение формулы Ia:
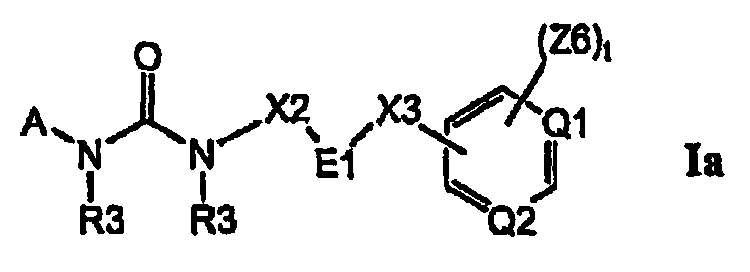
где Q1 и Q2, каждый отдельно и независимо, выбран из группы, состоящей из N и C-Z6, при условии, что оба Q1 и Q2 одновременно не являются С-Z6;
Е1 представляет собой фенил, и где цикл Е1 замещен одним или несколькими остатками R16, и где цикл Е1 замещен одним или несколькими остатками R18;
где А выбран из группы, состоящей из фенила и пиразолила;
G1 означает пиразолил;
цикл А замещен в любом замещаемом положении одним остатком А1, где
А1 выбран из группы, состоящей из А2 и A3;
А2 выбран из группы, состоящей из
A3 выбран из группы, состоящей из
и где символ (**) означает точку присоединения к циклу А в формуле Ia;
и где «----» означает либо насыщенную, либо ненасыщенную связь;
цикл А необязательно замещен одним или несколькими остатками R2;
Х2 представляет собой прямую связь, где Е1 непосредственно связан с группой NR3 в формуле Ia;
Х3 представляет собой -O-;
V и V2 представляют собой Н2, и VI независимо выбран из группы, состоящей из Н2 или О;
каждый Z2 независимо и отдельно выбран из группы, состоящей из водорода, C1-С6-алкила;
каждый Z3 независимо и отдельно выбран из группы, состоящей из Н, С1-С6-алкила, разветвленного С3-С7-алкила и - (CH2)nN(R4)2;
каждый Z4 представляет собой Н;
каждый Z6 независимо и отдельно выбран из группы, состоящей из Н, C1-С6-алкила, разветвленного С3-С7-алкила, гидроксила, гидрокси-C1-С6-алкила, разветвленного гидрокси-С2-С6-алкила-, (R3)2N-, (R4)2N-, -N(R3)C(O)R8, C(O)N(R4)2, галогена, -(CH2)nG1 и -R17;
каждый R2 выбран из группы, состоящей из C1-С6-алкила и разветвленного С3-С8-алкила;
каждый R3 представляет собой Н;
каждый R4 представляет собой Н, C1-С6-алкил или разветвленный С3-С7-алкил;
каждый R8 независимо и отдельно выбран из группы, состоящей из С1-С6-алкила и разветвленного С3-С7-алкила;
каждый R16 независимо и отдельно выбран из группы, состоящей из С1-С6-алкила, разветвленного С3-С7-алкила, галогена и цианогруппы;
каждый R17 представляет собой пиразолил;
где R17 может быть дополнительно замещен одним или несколькими остатками Z2 или Z3;
R18 представляет собой водород;
и n равно 0-6; р равно 1-4; q равно 2-6; r равно 0 или 1; t равно 1-3, v равно 1 или 2.
2. Соединение по п.1, имеющее формулу Ib
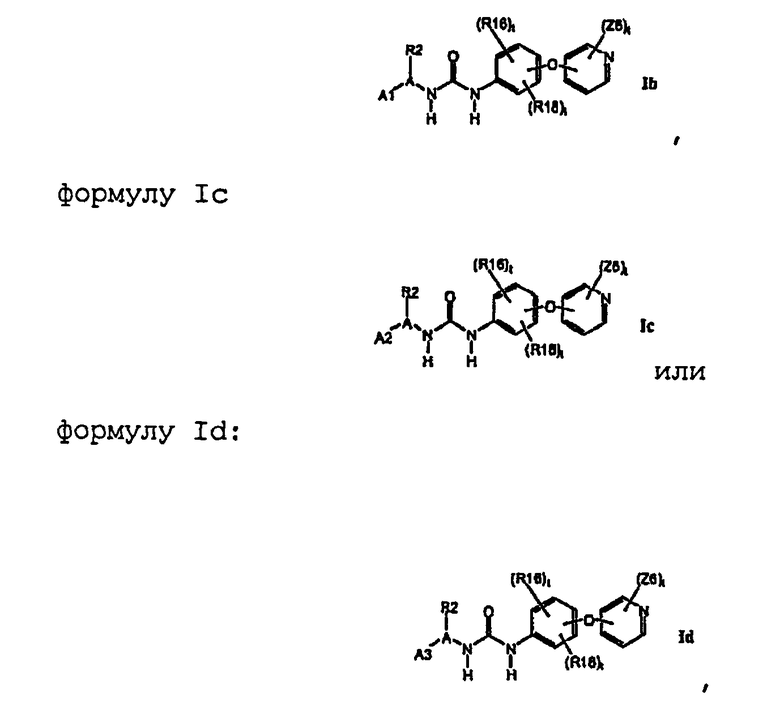
где А означает пиразолил.
3. Соединение по п.1, имеющее формулу Ill:
где А означает фенил.
4. Соединение, выбранное из группы, состоящей из:
1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины,
1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(3-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины,
1-(3-трет-бутил-1-(1Н-индазол-5-ил)-1Н-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины,
1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины,
1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(3-метил-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины,
1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)мочевины,
1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-4-(2-(метиламино)пиридин-4-илокси)фенил)мочевины,
1-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)мочевины,
1-(3-этил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины,
1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-5-(6-(гидроксиметил)пиридин-3-илокси)фенил)мочевины,
1-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)мочевины,
1-(3-изопропил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(3-метил-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевины,
1-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)-3-(3-этил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)мочевины,
1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-5-(6-(метилкарбамоил)пиридин-3-илокси)фенил)мочевины,
1-(2-фтор-4-(2-(1-метил-1Н-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)мочевины,
1-(3-этил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-4-(2-(1-метил-1Н-пиразол-4-ил)пиридин-4-илокси)фенил)мочевины,
1-(3-трет-бутил-1-(1,2,3,4-тетрагидроизохинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-4-(2-(1-метил-1Н-пиразол-4-ил)пиридин-4-илокси)фенил)мочевины,
1-(4-(2-(1Н-пиразол-4-ил)пиридин-4-илокси)-2-фторфенил)-3-(3-трет-бутил-1-(1-оксо-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиразол-5-ил)мочевины,
1-(4-(2-(1Н-пиразол-4-ил)пиридин-4-илокси)-2-фторфенил)-3-(3-третбутил-1-(1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиразол-5-ил)мочевины,
1-(3-этил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-3-метил-4-(2-(1-метил-1Н-пиразол-4-ил)пиридин-4-илокси)фенил)мочевины и
1-(2,3-дифтор-4-(2-(1-метил-1Н-пиразол-4-ил)пиридин-4-илокси)фенил)-3-(3-этил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)мочевины.
5. Соединение по п.4, которое представляет собой 1-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)-3-(3-изопропил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)мочевину, или его фармацевтически приемлемые соли или таутомеры.
6. Соединение по п.4, которое представляет собой 1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(3-метил-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину, или его фармацевтически приемлемые соли или таутомеры.
7. Соединение по п.4, которое представляет собой 1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)мочевину, или его фармацевтически приемлемые соли или таутомеры.
8. Соединение по п.4, которое представляет собой 1-(3-трет-бутил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)-3-(4-(2-карбамоилпиридин-4-илокси)-2-фторфенил)мочевину, или его фармацевтически приемлемые соли или таутомеры.
9. Соединение по п.4, которое представляет собой 1-(2-фтор-4-(2-(метилкарбамоил)пиридин-4-илокси)фенил)-3-(3-изопропил-1-(хинолин-6-ил)-1Н-пиразол-5-ил)мочевину, или его фармацевтически приемлемые соли или таутомеры.
10. Способ модулирования киназной активности киназы дикого типа, ее онкогенных форм, ее аберрантных форм в виде слитых белков и полиморфов любой из указанных выше форм, включающий стадию осуществления контакта указанной киназы с соединением по любому из пп.1-9.
11. Фармацевтическая композиция, содержащая соединение по любому из пп.1-9, или его соль, предназначенное для лечения гиперпролиферативных заболеваний и раковых заболеваний млекопитающих, вместе с фармацевтически приемлемым носителем, включающим вспомогательное вещество, выбранное из группы, состоящей из адъювантов, эксципиентов, разбавителей и стабилизаторов.
12. Фармацевтическая композиция по п.11, содержащая соединение по п.4 или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
13. Фармацевтическая композиция по п.11, содержащая соединение по п.5 или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
14. Фармацевтическая композиция по п.11, содержащая соединение по п.6 или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
15. Фармацевтическая композиция по п.11, содержащая соединение по п.7 или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
16. Фармацевтическая композиция по п.11, содержащая соединение по п.8 или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
17. Фармацевтическая композиция по п.11, содержащая соединение по п.9 или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем.
18. Способ лечения пациента, страдающего от состояния, выбранного из группы, состоящей из рака, гиперпролиферативных заболеваний, вторичного развития рака в результате метастазов и заболеваний, характеризуемых ангиогенезом, включающий введение указанному пациенту соединения по любому из пп.1-9.
19. Способ лечения пациента, страдающего от хронического миелогенного лейкоза, острого лимфоцитарного лейкоза, других миелопролиферативных расстройств, стромальных опухолей желудочно-кишечного тракта, гиперэозинофильного синдрома, глиобластом, рака яичника, рака поджелудочной железы, рака простаты, рака легкого, рака молочной железы, рака почек, карцином шейки матки, метастазов первичной солидной опухоли во вторичных очагах, глазных болезней, характеризующихся гиперпролиферацией, приводящих к слепоте, ретинопатий, диабетической ретинопатией и связанной с возрастом дегенерации желтого пятна, ревматоидного артрита, меланом, рака ободочной кишки, рака щитовидной железы, и их сочетаний, включающий стадию введения указанному пациенту соединения по любому из пп.1-9.
20. Способ введения млекопитающему соединения по любому из пп.1-9, выбранный из группы, состоящей из перорального, парентерального, ингаляционного и подкожного введения.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 99124188 A, 27.01.1998. | |||

