Перекрестные ссылки на родственные заявки
Настоящая заявка испрашивает приоритет в соответствии со статьей 35 U.S.C. 119(e) согласно предварительной заявке на патент США No. 61/884,958, поданной 30 сентября 2013 г. и озаглавленной "Соединения, ингибирующие киназу", и в соответствии со статьей 35 U.S.C. 119(a) согласно китайской заявке на патент No. 201310485048.1, поданной 16 октября 2013 г. и озаглавленной "Ароматическое амидное производное, способ его получения и его применение в медицине". Полное содержание каждой из вышеуказанных заявок включено в настоящую заявку посредством ссылки.
Область и уровень техники
Настоящее изобретение относится к химическим соединениям, фармацевтическим композициям, содержащим эти соединения, и к применению их при лечении заболеваний. В частности, настоящее изобретение относится к применению замещенных никотинимидов в качестве необратимых ингибиторов тирозинкиназ, полезных при лечении заболеваний, опосредованных тирозинкиназой Брутона (ВТК), включая рак, воспалительные и аутоиммунные заболевания.
Тирозинкиназа ВТК представляет собой нерецепторную тирозинкиназу Тес-cемейства протеинкиназ, которая играет важную роль во многих путях сигнальной трансдукции, регулируя выживание, активацию, пролиферацию и дифференцировку В-лимфоцитов. ВТК сверхэкспрессируется и является активной при некоторых В-клеточных лейкозах. ВТК экспрессируется в злокачественных клетках у людей, страдающих В-клеточным острым лимфобластным лейкозом (ALL) из клеток-предшественников (ВСР), хроническим лимфоцитарным лейкозом (CLL) и неходжкинской лимфомой (NHL). ВТК является вышележащим активатором антиапоптотических сигнальных молекул и сетей, включая: передатчик сигнала и активатор транскрипции 5 (STAT5), фосфатидилинозитол (PI), 3 киназа/АK/мишень рапамицина млекопитающих (mTOP) путь и ядерный фактор каппа-В (NF-κB) D'Cruz, Osmond J., OncoTargets and Therapy 2013: 6, 161-176.
В PCT/CN2012/000971 De Man et. al. показано, что ВТК экспрессируется в В-клетках и миелоидных клетках, а также является терминальным ферментом сигнального пути антигенраспознающего В-клеточного рецептора (BCR). Мутации ВТК у человека приводят к агаммаглобулинемии, сцепленной с Х-хромосомой (XLA), иммунодефицитному состоянию, связанному с неспособностью генерировать зрелые В-клетки, что приводит к уменьшению иммуноглобулина в сыворотке крови. ВТК, таким образом, вовлекается в регуляцию продукции аутоантител при аутоиммунных заболеваниях. Кроме того, ВТК может играть определенную роль в терапии аутоиммунных заболеваний, характеризующуюся продуцированием провоспалительных цитокинов и хемокинов В-клетками вследствие участия ВТК в сигнальном пути BCR. В связи с вовлечением ВТК в регуляцию пролиферации и апоптоза В-клеток, ингибиторы ВТК могут быть использованы для лечения В-клеточных лимфом. Ингибирование ВТК имеет существенное значение, в частности, для В-клеточных лимфом, обусловленных постоянной активной передачей сигнала BCR. Davis et al., Nature, 463 (2010), 88-94.
Адаптивные иммунные ответы могут включать активацию В-лимфоцитов, и отсутствие активации В-лимфоцитов является признаком аутоиммунного заболевания. Лечение аутоиммунных заболеваний, таких как ревматоидный артрит (RA), с применением ритуксимаба, анти-CD20 терапии, показывает, что анти-В-клеточные терапии являются эффективными. Кроме того, лечение ритуксимабом, как было показано, снижает симптомы заболевания у пациентов с рецидивирующим рассеянным склерозом (RRMS) и с системной красной волчанкой (SLE). Соответственно, нацеливание на В-клетки иммунной системы является эффективным подходом для лечения аутоиммунных заболеваний.
Сущность изобретения
Настоящее изобретение включает некоторые замещенные соединения, описанные в настоящем документе, их фармацевтически приемлемые соли, сольваты и гидраты, получение соединений, промежуточных соединений, фармацевтических композиций и их составов и способов лечения заболеваний, включая раки, воспаление и аутоиммунные заболевания.
Настоящее изобретение включает соединения Формулы I и их фармацевтически приемлемые соли, как указано ниже и далее определено в настоящем описании:
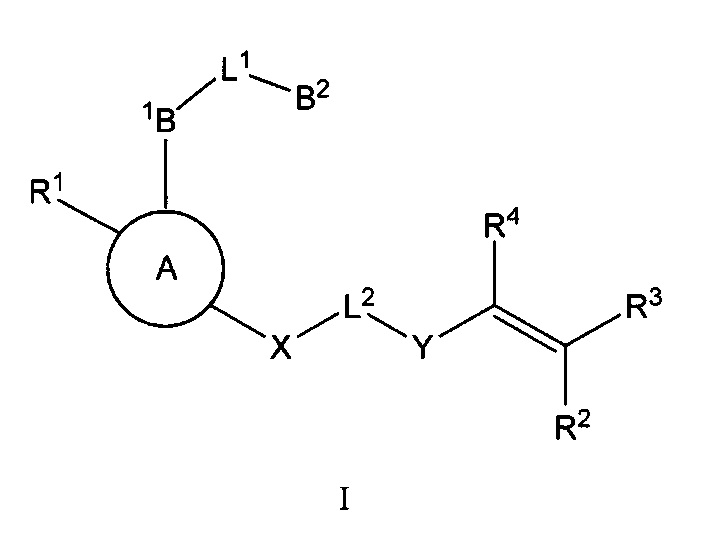
В некоторых аспектах соединения по настоящему изобретению являются необратимыми ингибиторами киназ, включая тирозинкиназу Брутона (ВТК). В некоторых аспектах соединения по настоящему изобретению являются селективными ингбиторами ВТК.
В некоторых аспектах изобретение относится к способам лечения пролиферативного заболевания, в частности, раков, условий, вызывающих воспаление, и аутоиммунных заболеваний, опосредованных, по меньшей мере, частично, ВТК, отдельно или в комбинации с другими видами терапии.
Соединения по настоящему изобретению и их фармацевтически приемлемые композиции могут быть использованы в качестве ингибиторов протеинкиназ. В некоторых вариантах осуществления эти соединения являются эффективными в качестве ингибиторов тирозинкиназы Брутона (ВТК). В некоторых аспектах изобретение включает фармацевтически приемлемые соли соединений Формулы I:
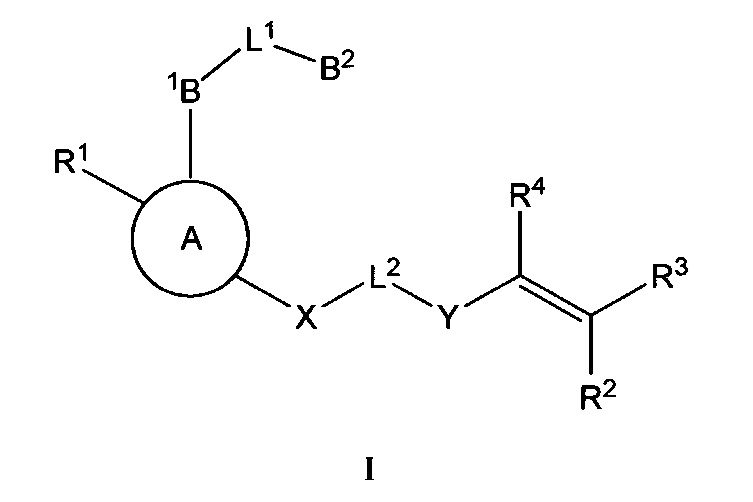
где:
А выбран из C3-12циклоалкила, C3-12гетероциклоалкила, C3-12арила или C3-12гетероарила, любой из которых необязательно замещен G1 заместителями;
B1 выбран из C3-12циклоалкил-C0-12алкил-, C3-12тероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G2 заместителями;
B2 выбран из C0-12алкила, C3-12циклоалкил-C3-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G3 заместителями;
L1 выбран из -C0-2алкил-, -CR5R6-, -C0-3алкил(R5)(OH), -C(О)-, -CH2O-, -OCH2-, -CF2-, -SCR5R6-, -CR5R6S-, -N(R5), -N(R5)C(O)-, -C(O)N(R5)-, -N(R5)C(O)N(R6)-, -O-, -S-, -S(O)m1-, -N(R5)S(O)m1- или -S(O)m1N(R5)-;
L2 выбран из -C0-4алкил-, -C(O)-, -N(R7)-, -N(R7)C(O)- или -N(R7)S(O)m2-;
X выбран из C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G4 заместителями;
Y выбран из -C(O)-, -N(R8)-, -N(R8)C(O)-, -S(O)m3- или -N(R8)S(O)m3-;
R1 выбран из -C(O)R9, -C(O)NR9R10, -C(O)OR9, C1-4алкинила, OR9, S(O)m4R9R10 или -CN;
R2, R3 и R4 каждый независимо выбран из C0-12алкила, -CN, галогена, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G5 заместителями;
R5, R6, R7, R8, R9 и R10 каждый независимо выбран из C0-12алкила, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G6 заместителями;
G1, G2, G3, G4, G5 и G6 каждый независимо выбран из одного или нескольких C0-12алкила, -C2-12алкенила, -C2-12алкинила, D, -CD3, -OCD3, галогена, -CN, -оксо-, -CF3, -OCF3, -OCHF2, -NO2, -B(OH)2, -P(О)C0-3 алкила, -PO(OR11)2, -PO(OR11)R12, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил-, гетероарил-C3-12гетероциклоалкил-, -C0-6алкилOR11, -OC(O)NR11R12, -C(O)OR11, -C(O)NR11R12, -C(O)R11, -NR11R12, -NR11C(O)R12, -NR13C(O)NR11R12, -S(O)m5R11 и -NR11S(O)m5R12, любой из которых необязательно замещен Q1 заместителями;
Q1 выбран из одного или нескольких C0-12алкил-, -C2-12алкенила, -C2-12алкинила, D, галогена, -CN, -CD3, -OCD3, -оксо-, -CF3, -OCF3, -OCHF2, -NO2, -B(ОН)2, -PO(OR14)2, -PO(OR14)R15, NR14R15, -C(O)NR14OH, -C0-6aлкилOR14, арил-C0-12алкил-, гетероарил-C0-12алкил-, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12циклоалкил-, гетероарил-C3-12циклоалкил-, C3-12гетероциклоалкил-C3-12циклоалкил-, C3-12циклоалкил-C3-12циклоалкил-, C3-12гетероциклоалкил-C3-12гетероциклоалкил-, арил-C3-12гетероциклоалкил-гетероарил-C3-12гетероциклоалкил-, -C(O)-C(O)NR14R15, -C(O)-C(O)OR14, -OC(O)R14, -NR14C(O)R15, -NR14S(O)m6R15, -(CR15R16)n1C(O)R14, -(CR15R16)n1C(O)OR14, (CR15R16)n1C(O)NR14R17, -(CR15R16)n1S(O)m6NR14R17, -(CR15R16)n1NR14R17, -(CR15R16)n1OR14, -(CR15R16)n1S(O)m6R14, -NR16C(O)NR14R15 и -NR16S(O)m6NR14R15, любой из которых необязательно замещен независимо выбранными Е1 заместителями;
Е1 выбран из одного или более C0-12алкил-, -C2-12алкенила, -C2-12алкинила, D, галогена, -CN, -оксо-, -CD3, -OCD3, -CF3, -OCF3, -OCHF2, -NO2, -B(ОН)2, -PO(OR18)2, -PO(OR18)19, -C(O)NR18OH, -C(O)NR18R19, -C0-12алкилOR18, арил-C0-12алкил-, гетероарил-C0-12алкил-, С3-12циклоалкил-C0-12алкил-, С3-12гетероциклоалкил-C0-12алкил-, арил-C0-12циклоалкил-, гетероарил-C3-12циклоалкил-, C3-12гетероциклоалкил-C3-12циклоалкил-, С3-12циклоалкил-C3-12циклоалкил-, С3-12гетероциклоалкил-C3-12гетероциклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C3-12гетероциклоалкил-, -C(O)-C(O)NR18R19, -C0-12aлкилC(O)OR18, -C(О)-C(O)OR18, -OC(O)R18, -NR18C(O)R19, -NR18C(O)OR19, -NR18S(O)m7R19, -(CR19R20)n2C(O)R18, -(CR19R20)n2C(O)OR18, -(CR19R20)n2C(O)NR18R21, -CCR19R20))m2(O)m7NR18R21, (CR19R20)n2NR18R21, -CCR19R20)n2OR18, -(CR19R20)m2(O)m7R18, -NR20C(O)NR18R19 и -NR20S(O)m7NR18R19 заместителей;
R11, R12, R13, R14, R15, R16, R17, R18, R19, R20 и R21 каждый независимо выбран из Н, C1-6алкил-, С3-8циклоалкил-C0-6алкил-, С3-8гетероциклоалкил-C0-6алкил-, арил-C0-6алкил-, арил-C3-8циклоалкил-, арил-C3-8гетероциклоалкил-, гетероарил-C1-6алкил-, гетероарил-C3-8циклоалкил- или гетероарил-C3-8гетероциклоалкил-;
R3 и R4, взятые совместно с атомами углерода, к которым они присоединены, образуют 3-12-членное частично насыщенное или ненасыщенное кольцо, где указанное кольцо необязательно включает один или более дополнительных гетероатомов, выбранных из O, N или S(O)m8;
m1, m2, m3, m4, m5, m6, m7, m8, n1 и n2 каждый независимо выбран из 0, 1 или 2; или их фармацевтически приемлемые соль, сольват или пролекарство.
В некоторых вариантах осуществления Формулы I соединения по настоящему изобретению представляют собой подкласс соединений Формулы I, имеющий Формулу Iа:
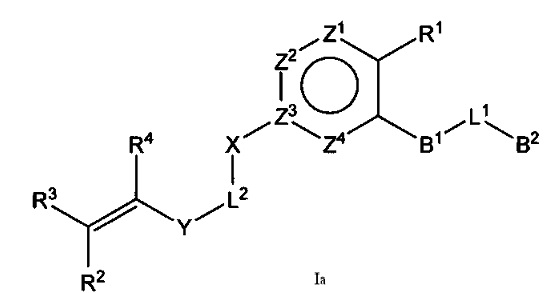
где:
B1 выбран из C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G2 заместителями;
B2 выбран из C0-12алкила, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12иклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G3 заместителями;
L1 выбран из -C0-2алкил-, -CR5R6-, -C0-3алкил(R5)(OH), -C(О)-, -CH2O-, -OCH2-, -CF2-, -SCR5R6-, -CR5R6S-, -N(R5), -N(R5)C(O)-, -C(O)N(R5)-, -N(R5)C(O)N(R6)-, -O-, -S-, -S(O)m1-, -N(R5)S(O)m1- или -S(O)m1N(R5)-;
L2 выбран из -C0-4алкил-, -C(O)-, -N(R7)-, -N(R7)C(O)- или -N(R7)S(O)m2-;
X выбран из C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G4 заместителями;
Y выбран из -C(O)-, -N(R8)-, -N(R8)C(O)-, -S(O)m3- или -N(R8)S(O)m3-;
Z1 представляет собой (CRa)0-1;
Z2 выбран из CRb, NRb, О или S;
Z3 выбран из C или N;
Z4 выбран из CRc, NRc, О или S;
R1 выбран из -C(O)R9, -C(O)NR9R10, -C(O)OR9, С1-4алкинила, OR9, S(O)m4R9R10 или -CN;
R2, R3, R4, Ra, Rb и Rc каждый независимо выбран из C0-12алкила, -CN, галогена, С3-12циклоалкил-C0-12алкил-, С3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G5 заместителями;
R5, R6, R7, R8, R9 и R10 каждый независимо выбран из C0-12алкила, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G6 заместителями;
G1, G2, G3, G4, G5 и G6 каждый независимо выбран из одного или нескольких С0-12алкила, -C2-12алкенила, -C2-12алкинила, D, -CD3, -OCD3, галогена, -CN, -оксо-, -CF3, -OCF3, -OCHF2, -NO2, -B(ОН)2, -P(О)C0-3алкила, -PO(OR11)2, -PO(OR11)R12, С3-12циклоалкил-C0-12алкил-, С3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил-, гетероарил-C3-12гетероциклоалкил-, -C0-6алкилOR11, -OC(O)NR11R12, -C(O)OR11, -C(O)NR11R12, -C(O)R11, -NR11R12, -NR11C(O)R12, -NR13C(O)NR11R12, -S(O)m5R11 и -NR11S(O)m5R12, любой из которых необязательно замещен независимо выбранными Q1 заместителями;
Q1 выбран из одного или нескольких C0-12алкил-, -C2-12алкенила, -C2-12алкинила, D, галогена, -CN, -CD3, -OCD3, -оксо-, -CF3, -OCF3, -OCHF2, -NO2, -B(ОН)2, -PO(OR14)2, -PO(OR14)R15, NR14R15, -C(O)NR14OH, -C0-6алкилOR14, арил-C0-12алкил-, гетероарил-C0-12алкил-, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12циклоалкил-, гетероарил-C3-12циклоалкил-, C3-12 гетероциклоалкил-C3-12циклоалкил-, C3-12циклоалкил-C3-12циклоалкил-, С3-12гетероциклоалкил-C3-12гетероциклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C3-12гетероциклоалкил-, -C(O)-C(O)NR14R15, -C(O)-C(O)OR14, -OC(O)R14, -NR14C(O)R15, -NR14S(O)m6R15, -(CR15R16)n1C(O)R14, -(CR15R16)n1C(O)OR14, (CR15R16)n1C(O)NR14R17, -(CR15R16)n1S(O)m6NR14R17, -(CR15R16)n1NR14R17, -(CR15R16)n1OR14, -(CR15R16)n1S(O)m6R14, -NR16C(O)NR14R15 и -NR16S(O)m6NR14R15, любой из которых необязательно замещен независимо выбранными Е1 заместителями;
Е1 выбран из одного или более C0-12алкил-, -C2-12алкенила, -C2-12алкинила, D, галогена, -CN, -оксо-, -CD3, -OCD3, -CF3, -OCF3, -OCHF2, -NO2, -B(OH)2, -PO(OR18)2, -PO(OR18)R19, -C(O)NR18OH, -C(O)NR18R19, -Co-l2aлкилOR18, арил-C0-12алкил-, гетероарил-C0-12алкил-, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12циклоалкил-гетероарил-C3-12циклоалкил-, C3-12гетероциклоалкил-C3-12циклоалкил-, С3-12циклоалкил-C3-12циклоалкил-, С3-12гетероциклоалкил-C3-12гетероциклоалкил-, арил-C3-12гетероциклоалкил-гетероарил-C3-12гетероциклоалкил-, -C(O)-C(O)NR18R19, -C0-12алкилС(О)OR18, -C(О)-C(O)OR18, -OC(O)R18, -NR18C(O)R19, -NR18C(O)OR19, -NR18S(O)m7R19, -(CR19R20)n2C(O)R18, -(CR19R20)n2C(O)OR18, -(CR19R20)n2C(O)NR18R21, -(CR19R20)n2S(O)m7NR18R21, (CR19R20)n2NR18R21, -(CR19R20)n2OR18, -(CR19R20))m2(O)m7R18, -NR20C(O)NR18R19 и -NR20S(O)m7NR18R19 заместителей;
R11, R12, R13, R14, R15,R16, R17, R18, R19, R20 и R21 каждый независимо выбран из Н, С1-6алкил-, C3-8циклоалкил-C0-6алкил-, C3-8гетероциклоалкил-C0-6алкил-, арил-C0-6алкил-, арил-C3-8циклоалкил-, арил-C3-8гетероциклоалкил-, гетероарил-C1-6алкил-, гетероарил-C3-8циклоалкил- или гетероарил-C3-8гетероциклоалкил-;
R3 и R4, взятые совместно с атомами углерода, к которым они присоединены, образуют 3-12-членное частично насыщенное или ненасыщенное кольцо, где указанное кольцо необязательно включает один или более дополнительных гетероатомов, выбранных из О, N или S(O)m8;
m1, m2, m3, m4, m5, m6, m1, m8, n1 и n2 каждый независимо выбран из 0, 1 или 2;
или их фармацевтически приемлемые соль, сольват или пролекарство.
В некоторых вариантах осуществления Формулы I соединения по настоящему изобретению представляют собой подкласс соединений Формулы I, выбранный из одной из Формул Ib-Ii:
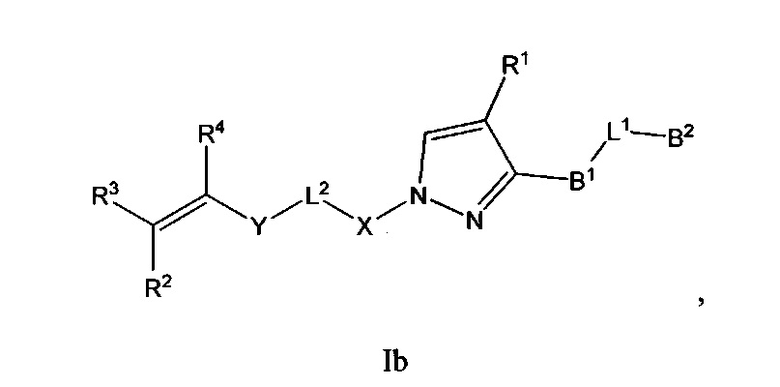
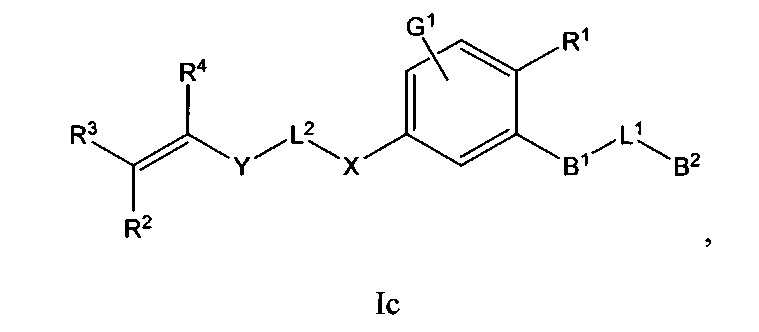
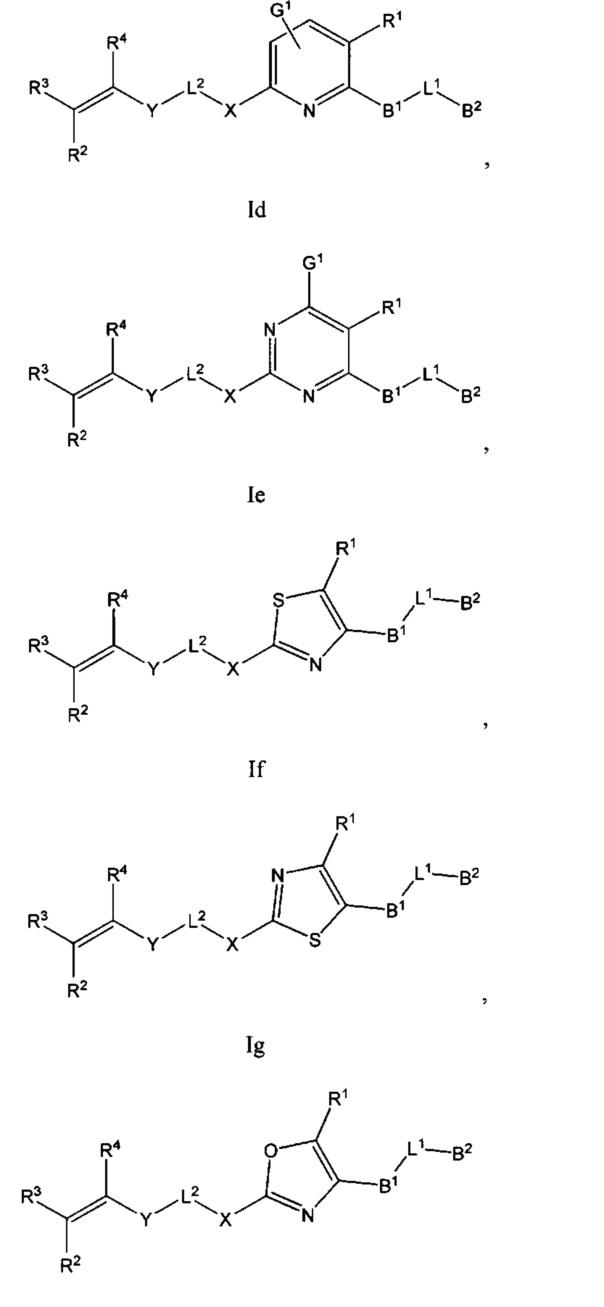 и
и
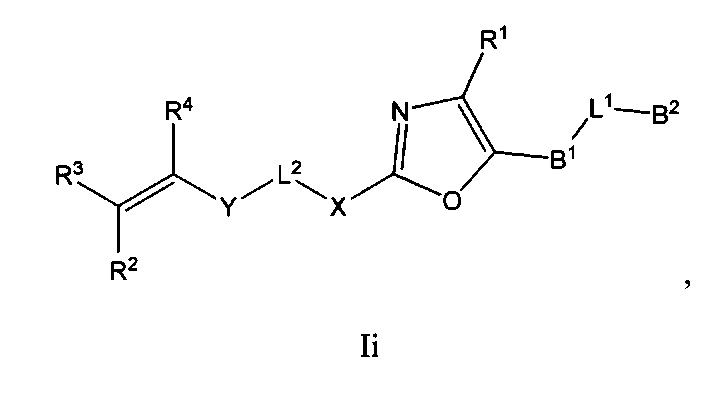
где:
B1 выбран из C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G2 заместителями;
B2 выбран из C0-12алкила, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12иклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G3 заместителями;
L1 выбран из -C0-2алкил-, -CR5R6-, -C0-3алкил(R5)(OH), -C(О)-, -CH2O-, -OCH2-, -CF2-, -SCR5R6-, -CR5R6S-, -N(R5), -N(R5)C(O)-, -C(O)N(R5)-, -N(R5)C(O)N(R6)-, -O-, -S-, -S(O)m1-, -N(R5)S(O)m1- или -S(O)m1N(R5)-;
L2 выбран из -C0-4алкил-, -C(O)-, -N(R7)-, -N(R7)C(O)- или -N(R7)S(O)m2-;
X выбран из C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G4 заместителями;
Y выбран из -C(O)-, -N(R8)-, -N(R8)C(O)-, -S(O)m3- или -N(R8)S(O)m3-;
R1 выбран из -C(O)R9, -C(O)NR9R10, -C(O)OR9, С1-4алкинила, OR9, S(O)m4R9R10 или -CN;
R2, R3 и R4 каждый независимо выбран из С0-12алкила, -CN, галогена, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12гетероциклоалкил-, любой из которых необязательно замещен G5 заместителями;
R5, R6, R7, R8, R9 и R10 каждый независимо выбран из С0-12алкила, С3-12циклоалкил-C0-12алкил-, C3-12 гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил- или гетероарил-C3-12 гетероциклоалкил-, любой из которых необязательно замещен G6 заместителями;
G1, G2, G3, G4, G5 и G6 каждый независимо выбран из одного или нескольких C0-12алкила, -C2-12алкенила, -C2-12алкинила, D, -CD3, -OCD3, галогена, -CN, -оксо-, -CF3, -OCF3, -OCHF2, -NO2, -B(ОН)2, -P(О)C0-3алкила, -PO(OR11)2, -PO(OR11)R12, C3-12циклоалкил-C0-12алкил-, С3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил-, гетероарил-C3-12гетероциклоалкил-, -C0-5алкилOR11, -OC(O)NR11R12, -C(O)OR11, -C(O)NR11R12, -C(O)R11, -NR11R12, -NR11C(O)R12, -NR13C(O)NR11R12, -S(O)m5R11 и -NR11S(O)m5R12, любой из которых необязательно замещен независимо выбранными Q1 заместителями;
Q1 выбран из одного или более C0-12алкил-, -C2-12алкенила, -C2-12алкинила, D, галогена, -CN, -CD3, -OCD3, -оксо-, -CF3, -OCF3, -OCHF2, -NO2, -B(ОН)2, -PO(OR14)2, -PO(OR14)R15, NR14R15, -C(O)NR14OH, -C0-6алкилOR14, арил-C0-12алкил-, гетероарил-C0-12алкил-, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12циклоалкил-, гетероарил-C3-12циклоалкил-, C3-12гетероциклоалкил-C3-12циклоалкил-, C3-12циклоалкил-C3-12циклоалкил-, С3-12гетероциклоалкил-C3-12гетероциклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C3-12гетероциклоалкил-, -C(O)-C(O)NR14R15, -C(O)-C(O)OR14, -OC(O)R14, -NR14C(O)R15, -NR14S(O)m6R15, -(CR15R16)n1C(O)R14, -(CR15R16)n1C(O)OR14, (CR15R16)n1C(O)NR14R17, -(CR15R,6)n1S(O)m6NR14R17, -(CR15R16)n1NR14R17, -(CR15R16)n1OR14, -(CR15R16)n1S(O)m6R14, -NR16C(O)NR14R15, -NR16S(O)m6NR14R15, любой из которых необязательно замещен независимо выбранными Е1 заместителями;
Е1 выбран из одного или более С0-12алкил-, -C2-12алкенила, -C2-12алкинила, D, галогена, -CN, -оксо-, -CD3, -OCD3, -CF3, -OCF3, -OCHF2, -NO2, -B(ОН)2, -PO(OR18)2, -PO(OR18)R19, -C(O)NR18OH, -C(O)NR18R19, -C0-12алкилOR18, арил-C0-12алкил-, гетероарил-C0-12алкил-, C12циклоалкил-C0-12алкил-, С3-12гетероциклоалкил-C0-12алкил-, арил-C0-12циклоалкил-, гетероарил-C3-12циклоалкил-, C3-12гетероциклоалкил-C3-12циклоалкил-, С3-12циклоалкил-C3-12циклоалкил-, С3-12гетероциклоалкил-C3-12гетероциклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C3-12гетероциклоалкил-, -C(O)-C(O)NR18R19, -C0-12aлкилC(O)OR18, -C(О)-C(O)OR18, -OC(O)R18, -NR18C(O)R19, -NR18C(O)OR19, -NR18S(O)m7R19, -(CR19R20)n2C(O)R18, -(CR19R20)n2C(O)OR18, -(CR19R20)n2C(O)NR18R21, -(CR19R20)n2S(O)m7NR18R21, (CR19R20)n2NR18R21, -(CR19R20)n2OR18, -(CR19R20))n2(O)m7R18, -NR20C(O)NR18R19 и -NR20S(O)m7NR18R19 заместителей;
R11, R12, R13, R14, R15, R16, R17, R18, R19, R20 и R21 каждый независимо выбран из Н, C1-6алкил-C3-8циклоалкил-C0-6алкил-, C3-8гетероциклоалкил-C0-6алкил-, арил-C0-6алкил-, арил-C3-8циклоалкил-, арил-C3-8гетероциклоалкил-, гетероарил-C1-6алкил-, гетероарил-C3-8циклоалкил- или гетероарил-C3-8гетероциклоалкил-;
R3 и R4, взятые совместно с атомами углерода, к которым они присоединены, образуют 3-12-членное частично насыщенное или ненасыщенное кольцо, где указанное кольцо необязательно включает один или более дополнительных гетероатомов, выбранных из O, N или S(O)m8;
m1, m2, m3, m4, m5, m6, m7, m8, n1, и n2 каждый независимо выбран из 0, 1 или 2;
или их фармацевтически приемлемые соль, сольват или пролекарство.
В некоторых вариантах осуществления Формул I и Ia-Ii В1 выбран из C4-8циклоалкил-C0-12алкил, C4-8гетероциклоалкил-C0-12алкил-, С4-8арил-C0-12алкил- или С4-8гетероарил-C0-12алкил-, любой из которых необязательно замещен G2 заместителями.
В некоторых вариантах осуществления Формул I и Ia-Ii В2 выбран из С4-8циклоалкил-C0-12алкил-, С4-8гетероциклоалкил-C0-12алкил-, С4-8арил-C0-12алкил- или С4-8гетероарил-C0-12алкил-, любой из которых необязательно замещен G3 заместителями.
В некоторых вариантах осуществления Формул I и Ia-Ii L1 выбран из -C0-2алкил-, -CR5R6-, -C0-3aлкил(R5)(OH)-, -C(O)-, -CH2O-, -ОСН2-, -CF2-, -N(R5)-, -N(R5)C(O)-, -C(O)N(R5)-, -О- или -S(O)m1-.
В некоторых вариантах осуществления Формул I и Ia-Ii L1 выбран из -C0-2алкил-, -CR5R6-, -C1-2aлкил(R5)(OH)-, -C(O)-, -CF2-, -N(R5)-, -N(R5)C(O)-, -C(O)N(R5)-, -О- или -S(O)m1-.
В некоторых вариантах осуществления Формул I и Ia-Ii L2 выбран из -C0-2алкил-, -C(О)- или -N(R7)-.
В некоторых вариантах осуществления Формул I и Ia-Ii X выбран из С4-8циклоалкил-C0-12алкил-, С4-8гетероциклоалкил-C0-12алкил-, С4-8арил-C0-12алкил- или С4-8гетероарил-C0-12алкил-.
В некоторых вариантах осуществления Формул I и Ia-Ii Y выбран из -C(О)-, -N(R8)-, -N(R8)C(O)- или -S(O)m3-.
В некоторых вариантах осуществления Формул I и Ia-Ii R1 выбран из -C(O)R9, -C(O)NR9R10, -C(O)OR9, С1-4алкинила или-CN.
В некоторых вариантах осуществления Формул I и Ia-Ii R2, R3 и R4 каждый независимо выбран из С0-12алкила, -CN, галогена, С3-6циклоалкил-C0-12алкил-, C3-6гетероциклоалкил-C0-12алкил-, любой из которых необязательно замещен G5 заместителями.
В некоторых вариантах осуществления Формул I и Ia-Ii G1, G2, G3, G4, G5 и G6 каждый независимо выбран из одного до трех C0-12алкила, -C2-12алкенила, -C2-12алкинила, D, -CD3, -OCD3, галогена, -CN, -оксо-, -CF3, -OCF3, -OCHF2, -NO2, -B(ОН)2, -P(О)C0-3алкила, -PO(OR11)2, -PO(OR11)R12, С3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил-, гетероарил-C3-12гетероциклоалкил-, -C0-6aлкилOR11,-OC(O)NR11R12, -C(O)OR11, -C(O)NR11R12, -C(O)R11, -NR11R12, -NR11C(O)R12, -NR13C(O)NR11R12, -S(O)m5R11 и -NR11S(O)m5R12, любой из которых необязательно замещен независимо выбранными Q1 заместителями.
В некоторых вариантах осуществления Формул I и Ia-Ii G1, G2, G3, G4, G5 и G6 каждый независимо выбран из от одного до двух C0-12алкила, -C2-12алкенила, -C2-12алкинила, D, -CD3, -OCD3, галогена, -CN, -оксо-, -CF3, -OCF3, -OCHF2, -NO2, -B(ОН)2, -P(О)C0-3алкила, -PO(OR11)2, -PO(OR11)R12, C3-12циклоалкил-C0-12алкил-, C3-12гетероциклоалкил-C0-12алкил-, арил-C0-12алкил-, арил-C3-12циклоалкил-, арил-C3-12гетероциклоалкил-, гетероарил-C0-12алкил-, гетероарил-C3-12циклоалкил-, гетероарил-C3-12гетероциклоалкил-, -C0-6aлкилOR11, -OC(O)NR11R12, -C(O)OR11, -C(O)NR11R12, -C(O)R11, -NR11R12, -NR11C(O)R12, -NR13C(O)NR11R12, -S(O)m5R11 и -NR11S(O)m5R12, любой из которых необязательно замещен независимо выбранными Q1 заместителями.
В некоторых аспектах настоящее изобретение относится к фармацевтической композиции, включающей соединение или соль любого из соединений Формулы I, объединенных с или без одного или более фармацевтическими носителями.
В некоторых аспектах настоящее изобретение относится к способу лечения, по меньшей мере, одного онкозаболевания, хронического воспаления и аутоиммунного заболевания, опосредованных, по меньшей мере, частично, ВТК, включающему введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения или соли соединения Формулы I.
В некоторых аспектах настоящее изобретение относится к способу лечения рака, хронического воспаления или аутоиммунного заболевания у млекопитающего, включающему введение терапевтически эффективного количества соединения Формулы I или его фармацевтически приемлемой соли млекопитающему, нуждающемуся в этом.
В некоторых аспектах настоящее изобретение относится к способу необратимого ингибирования тирозинкиназ, при этом указанный способ включает введение пациенту терапевтически эффективного количества ингибитора тирозинкиназы, включая соединение Формулы I.
В некоторых аспектах настоящее изобретение относится к способу необратимого ингибирования ВТК, при этом указанный способ включает введение пациенту терапевтически эффективного количества ингибитора ВТК, включая соединение Формулы I.
Способы, описанные в настоящем документе, включают введение субъекту, нуждающемуся в этом, композиции, содержащей терапевтически эффективное количество одного или более описанных здесь соединений, ингибирующих ВТК. Без привязки к какой-либо конкретной теории, разнообразные роли, которые играет сигнальная система ВТК в различных функциях гематопоэтических клеток, например, в активации В-клеточных рецепторов, позволяет предположить, что малые молекулы ВТК ингибиторов полезны для снижения риска или для лечения различных заболеваний, на которые влияют или которые влияют на многие типы клеток гематопоэтической клеточной линии, включая, например, аутоиммунные заболевания, гетероиммунные состояния или заболевания, воспалительные заболевания, рак (например, В-клеточные пролиферативные нарушения) и тромбоэмболические заболевания.
Кроме того, соединения, ингибирующие ВТК, описанные здесь, могут быть использованы для ингибирования небольшой подруппы других тирозинкиназ, которые гомологичны ВТК, поскольку они имеют остаток цистеина (включая остаток Cys 481), которые могут образовывать ковалентную связь с ингибитором. Таким образом, подмножество отличных от ВТК тирозинкиназ могут использоваться в качестве терапевтических мишеней при ряде нарушений здоровья.
Описанные здесь способы могут быть использованы для лечения аутоиммунного заболевания, которое включает, но не ограничиваясь ими, ревматоидный артрит, псориатический артрит, остеоартрит, болезнь Стилла, ювенильный артрит, волчанку, диабет (тип I и тип II), тяжелую псевдопаралитическую миастению gravis, тиреоидит Хашимото, атрофическую форму аутоиммунного тиреоидита (тироидит Орда), диффузный токсический зоб (болезнь Грейвса), синдром Шегрена, рассеянный склероз, синдром Гийена-Барре, острый рассеянный энцефаломиелит, болезнь Аддисона, опсо-миоклональный синдром, анкилозирующий спондилит, синдром антифосфолипидных антител, апластическую анемию, аутоиммунный гепатит, целиакию, синдром Гудпасчера, идиопатическую тромбоцитопению purpura, оптический неврит, склеродермию, первичный цирроз печени, неспецифический аортоартериит (болезнь Такаясу), временный артерит, тепловую аутоиммунную гемолитическую анемию, грануломатоз Вегенера, псориаз, общую алопецию, болезнь Бехчета, хроническую усталость, дисаутономию, эндометриоз, интерстициальный цистит, нейромиотонию, склеродермию или вульводинию.
Описанные здесь способы могут быть использованы для лечения гетероиммунных состояний или заболеваний, которые включают, но не ограничиваются ими, реакцию «трансплантат против хозяина», последствия трансплантации, трансфузии, анафилаксию, аллергии (например, аллергические реакции на пыльцу растений, латекс, лекарства, продукты питания, укусы насекомых, шерсть животных, перхоть животных, пылевых клещей или тараканов), гиперчувствительность I типа, аллергический конъюнктивит, аллергический ринит, аллергическую астму и атопический дерматит.
Описанные здесь способы могут быть использованы для лечения воспалительного заболевания, которое включает, но не ограничивается ими, астму, воспалительное заболевание кишечника, аппендицит, блефарит, бронхиолит, бронхит, бурсит, цервицит, холангит, холецистит, колит, конъюнктивит, цистит, дакриоаденит, дерматит, дерматомиозит, энцефалит, эндокардит, эндометрит, энтерит, энтероколит, эпикондилит, эпидидимит, фасциит, фиброз, гастрит, гастроэнтерит, гепатит, гнойный гидраденит, ларингит, мастит, менингит, миелит в сочетании с миокардитом, миозит, нефрит, оофорит, орхит, остеомиелит, отит, панкреатит, паротит, перикардит, перитонит, фарингит, плеврит, флебит, пневмонит, пневмонию, проктит, простатит, пиелонефрит, ринит, сальпингит, синусит, стоматит, синовит, тендинит, тонзиллит, увеит, вагинит, васкулит и вульвит.
Описанные здесь способы могут быть использованы для лечения рака, например, В-клеточных пролиферативных расстройств, которые включают в себя, но не ограничиваются ими, диффузную В-крупноклетчатую лимфому, фолликулярную лимфому, хроническую лимфоцитарную лимфому, хронический лимфоцитарный лейкоз, В-клеточный пролимфоцитарный лейкоз, лимфоплазмацитарную лимфому/макроглобулинемию Вальденстрема, лимфому маргинальной зоны селезенки, миелому клеток плазмы, плазмоцитому, экстранодальную В-клеточную лимфому маргинальной зоны, нодальную В-клеточную лимфому маргинальной зоны, мантийноклеточную лимфому, медиастинальную (тимусную) В-крупноклеточную лимфому, интраваскулярную В-крупноклеточную лимфому, первичную эффузионную лимфому, лимфому/лейкоз Беркитта и лимфогранулематоз.
Описанные здесь способы могут быть использованы для лечения тромбоэмболических нарушений, которые включают, но не ограничиваются ими, инфаркт миокарда, стенокардию (включая нестабильную стенокардию), реокклюзию или рестеноз после ангиопластики или аортокоронарного шунтирования, инсульт, транзиторную ишемию, периферические артериальные окклюзионные расстройства, эмболии легочной артерии и тромбоз глубоких вен.
В настоящем документе описан способ лечения гематологической злокачественной опухоли у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Гематологическое злокачественное новообразование представляет собой хронический лимфолейкоз (CLL), малую лимфолимфому (SLL), CLL высокого риска или не-CLL/SLL лимфому. В некоторых вариантах осуществления Гематологическое злокачественное новообразование представляет собой фолликулярную лимфому, диффузную крупноклеточную В-клеточную лимфому (DLBCL), мантийноклеточную лимфому, макроглобулинемию Вальденстрема, множественную миелому, лимфому маргинальной зоны, лимфому Беркитта, высокозлокачественную В-клеточную лимфому не-Беркитта или экстранодальную В-клеточную лимфому маргинальной зоны. В некоторых вариантах осуществления гематологическое злокачественное новообразование представляет собой острый или хронический миелогенный (или миелоидный) лейкоз, миелодиспластический синдром или острый лимфобластный лейкоз. В некоторых вариантах осуществления гематологическое злокачественное новообразование представляет собой рецидивирующую или рефракторную диффузную крупноклеточную В-клеточную лимфому (DLBCL), рецидивирующей или рефрактерной мантии клеточной лимфомы, рецидивирующую или рефракторную фолликулярную лимфому, рецидивирующую или рефракторную CLL; рецидивирующую или рефракторную SLL; рецидивирующую или рефракторную множественную миелому. В некоторых вариантах осуществления гематологическое злокачественное новообразование является гематологическим злокачественным новообразованием, которое классифицируется как с высокой степенью риска. В некоторых вариантах осуществления гематологическое злокачественное новообразование представляет собой CLL высокого риска или SLL высокого риска.
В-клеточные лимфопролиферативные заболевания (BCLDs) представляют собой новообразования крови и включают, в частности, неходжкинскую лимфому, множественную миелому и лейкоз. BCLDs могут возникать либо в лимфатической ткани (как в случае лимфомы), или в костном мозге (как и в случае лейкоза и миеломы), и все они связаны с неконтролируемым ростом лимфоцитов или лейкоцитов. Существует множество подтипов BCLD, например, хронический лимфолейкоз (CLL) и неходжкинская лимфома (NHL). Течение болезни и лечение BCLD зависит от подтипа BCLD; тем не менее, даже в пределах каждого подтипа клинические проявления, морфологические признаки и ответ на терапию являются гетерогенными.
Злокачественные лимфомы представляют собой неопластическую трансформацию клеток, которые находятся преимущественно в лимфоидных тканях. Две группы злокачественных лимфом представляют собой лимфому Ходжкина и неходжкинскую лимфому (NHL). Лимфомы этих двух типов инфильтрируют ретикулоэндотелиальные ткани. Однако они различаются происхождением опухолевой клетки, локализацией заболевания, наличием системных симптомов и ответом на лечение (Freedman et al., "Non-Hodgkin's Lymphomas" Chapter 134, Cancer Medicine, (одобренная публикация American Cancer Society, B.C. Decker Inc., Hamilton, Ontario, 2003).
Настоящее изобретение относится к способу лечения неходжкинской лимфомы у индивидуума, нуждающегося в этом, включающему: введение индивидууму композиции, содержащей терапетвическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Кроме того, настоящее изобретение относится к способу лечения рецидивирующей или рефрактерной неходжкинской лимфомы у индивидуума, нуждающегося в этом, включающему введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId. В некоторых вариантах осуществления неходжкинская лимфома представляет собой рецидивирующую или рефрактерную диффузную крупноклеточную В-клеточную лимфому (DLBCL), рецидивирующую или рефрактерную лимфому из клеток мантийной зоны или рецидивирующую или рефрактерную фолликулярную лимфому.
Неходжкинские лимфомы (NHL) представляют собой разнородную группу злокачественных опухолей, которые преимущественно имеют В-клеточное происхождение. NHL может развиваться в любых органах, ассоциированных с лимфатической системой, таких как селезенка, лимфатические узлы или миндалины, и может проявляться в любом возрасте. При NHL часто наблюдается увеличение лимфатических узлов, лихорадка и потеря веса. NHL классифицируется либо как В-клеточная, либо как Т-клеточная NHL. Лимфомы, связанные с лимфопролиферативными расстройствами после трансплантации костного мозга или стволовых клеток, обычно представляют собой В-клеточные NHL. В классификационной схеме рабочих препаратов (Working Formulation classification scheme), NHL по степени злокачественности была разделена на низкую, среднюю и высокую степень в зависимости от истории болезни (смотри "The Non-Hodgkin's Lymphoma Pathologic Classification Project," Cancer 49 (1982): 2112-2135). Лимфому с низкой степенью злокачественности часто называют индолентной лимфомой с медианой выживаемости от 5 до 10 лет (Horning and Rosenberg (1984) N. Engl. J. Med. 311: 1471-1475). Хотя химиотерапия может вызывать ремиссию, у большинства пациентов с индолентной лимфомой выздоровление наступает очень редко, и у большинства пациентов со временем наступает рецидив, требующий дальнейшей терапии. Лимфомы со средней и высокой степенью злокачественности представляют собой более агрессивные опухоли, однако пациенты имеют больше шансов на излечение с применением химиотерапии. Тем не менее, у значительной части этих пациентов будет наблюдаться рецидив и потребуется дальнейшее лечение.
Неограничивающий перечень В-клеточных NHL включает лимфому Беркитта (например, эндемическая лимфома Беркитта и спорадическая лимфома Беркитта), В-клеточную лимфому кожи, лимфому кожи из клеток маргинальной зоны (MZL), диффузную крупноклеточную В-клеточную лимфому (DLBCL), диффузную смешанную мелкоклеточную и крупноклеточную лимфому, диффузную мелкоклеточную с расщепленными ядрами лимфому, диффузную мелкоклеточную лимфоцитарную лимфому, экстранодальную В-клеточную лимфому из клеток маргинальной зоны, фолликулярную лимфому, фолликулярную мелкоклеточную с расщепленными ядрами лимфому (1 степень злокачественности), фолликулярную смешанную лимфому из мелких клеток с расщепленными ядрами и крупных клеток (2 степень злокачественности), фолликулярную крупноклеточную лимфому (3 степень злокачественности), интраваскулярную крупноклеточную В-клеточную лимфому, интраваскулярный лимфоматоз, крупноклеточную иммунобластную лимфому, крупноклеточную лимфому (LCL), лимфобластную лимфому, MALT-лимфому, лимфому из клеток мантийной зоны (MCL), иммунобластную крупноклеточную лимфому, лимфобластную лимфому из предшественников В-клеток, лимфому из клеток мантийной зоны, хронический лимфоцитарный лейкоз (CLL)/мелкоклеточную лимфоцитарную лимфому (SLL), экстранодальную В-клеточную лимфому из клеток маргинальной зоны, ассоциированную с лимфомой лимфоидной ткани слизистых оболочек (MALT), медиастинальную крупноклеточную В-клеточную лимфому, узловую В-клеточную лимфому из клеток маргинальной зоны, В-клеточную лимфому из клеток маргинальной зоны селезенки, первичную медиастинальную В-клеточную лимфому, лимфоплазмоцитарную лимфому, волосатоклеточный лейкоз, макроглобулинемию Вальденстрема и первичную лимфому центральной нервной системы (CNS). В пределах объема настоящего изобретения рассматриваются дополнительные неходжкинские лимфомы, которые легко обнаруживаются обычным специалистом в данной области.
Настоящее изобретение относится к способу лечения DLCBL у индивидуума, нуждающегося в этом, включающему введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Используемый в данном описании термин "диффузная крупноклеточная В-клеточная лимфома (DLCBL)" относится к новообразованию из В-лимфоцитов герминативного центра с диффузным характером роста и показателем высокой промежуточной пролиферации. DLCBL составляют примерно 30% всех лимфом и могут представлять собой несколько морфологических вариантов, включая центробластную, иммунобластную, богатую Т-клетками/гистиоцитами, анапластическую и лимфобластную лимфомы. Генетические тесты показали, что существуют различные подтипы DLCBL. Эти подтипы, по всей видимости, имеют разные прогнозы выздоровления и ответы на лечение. DLCBL может проявиться у людей любой возрастной группы, но встречается, главным образом, у пожилых людей (средний возраст - середина 60 лет).
Настоящее изобретение относится к способу лечения диффузной крупноклеточной В-клеточной лимфомы из активированных В-клеток подтипа (ABC-DLCBL) у индивидуума, нуждающегося в этом, включающему введение индивидууму необратимого ингибитора ВТК в количестве от 300 мг/сутки вплоть до и включая 1000 мг/сутки. Подтип ABC диффузной В-клеточной лимфомы (ABC-DLBCL), как полагают, возникает из В-клеток постгерминативного центра, которые подверглись генетическим мутациям (арестованные клетки) во время плазматической дифференцировки. Подтип ABC DLCBL (ABC-DLBCL) насчитывает около 30% общего количества диагнозов DLBCL. Этот подтип считается наименее излечимым из молекулярных подтипов DLBCL и, как таковой, обычно демонстрирует значительно сниженные показатели выживаемости пациентов с диагнозом ABC-DLCBL по сравнению с индивидуумами с другими типами DLCBL. ABC-DLBCL чаще всего ассоциируется с хромосомными транслокациями, дерегулирующими главный регулятор BCL6 герминативного центра, и с мутациями, инактивирующими ген PRDM1, который кодирует транскрипционный репрессор, необходимый для дифференцировки клеток плазмы.
Особенно значимый сигнальный путь в патогенезе ABC-DLCBL представляет собой путь, опосредованный комплексом транскрипционного ядерного фактора (NF)-κB. Семейство NF-кВ состоит из 5 членов (р50, р52, р65, c-rel и RelB), которые образуют гомо- и гетеродимеры, и функционируют как транскрипционные факторы, опосредующие различные процессы пролиферации и апоптоза, воспалительные и иммунные реакции, и имеют решающее значение для нормального развития и выживания В-клеток. NF-kB широко задействован в эукариотических клетках как регулятор генов, который контролирует пролиферацию клеток и выживание клеток. Как таковые, многие различные типы опухолей человека имеют разрегулированный NF-kB: то есть, NF-kB является конститутивно активным. Активный NF-kB включает экспрессию генов, которые поддерживают клеточную пролиферацию и защищают эти клетки от условий, которые в противном случае позволили бы им умереть через апоптоз.
Зависимость ABC DLCBL от NF-kB обусловлена вышележащим сигнальным путем IkВ киназы, приводящим к образованию комплекса белков CARD11, BCL10 и MALT1 (СВМ комплекса). Интерференция в СВМ пути гасит сигналы NF-kB в клетках ABC DLBCL и индуцирует апоптоз. Молекулярный базис конститутивной активности пути NF-kB является предметом проводимого в настоящее время исследования, однако ясно, что некоторые соматические изменения в геноме ABC DLCBL задействуют этот путь. Например, соматические мутации суперспирального домена CARD11 в DLCBL делают этот каркасный сигнальный белок способным спонтанно служить центром развития белок-белковых взаимодействий с MALT1 и BCL10, вызывая IKK активность и активацию NF-kB. Конститутивная активность сигнального пути В-клеточного рецептора вовлекается в активацию NF-kB в ABC DLCBL с диким типом CARD11, и это ассоциируется с мутациями в пределах цитоплазматических хвостов субъединиц В-клеточного рецептора CD79A и CD79B. Онкогенные активирующие мутации в сигнальном пути адаптерного белка MYD88 активируют NF-kB и усиливают активацию пути В-клеточного рецептора, поддерживая выживаемость клеток ABC DLBCL. В дополнение, инактивирующие мутации в негативном регуляторе А20 сигнального пути NF-kB встречаются почти исключительно в ABC DLCBL.
Действительно, генетические изменения, влияющие на многие компоненты сигнального пути NF-kB, недавно были идентифицированы у более чем 50% пациентов с ABC-DLBCL, где эти очаги поражения поддерживали конститутивную активацию NF-kB, способствуя тем самым росту лимфомы. К ним относятся мутации CARD 11 (~10% случаев), лимфоцит-специфического цитоплазматического поддерживающего белка, который вместе с MALT1 и BCL10 формирует сигнальный комплекс BCR, который ретранслирует сигналы от антигенных рецепторов к нисходящим медиаторам активации NF-kB. Еще большая доля случаев (-30%) несут биаллельные генетические повреждения, инактивирующие негативный регулятор А20 сигнального пути NF-kB. Кроме того, высокие уровни экспрессии генов-мишеней NF-kB наблюдались в образцах опухолей ABC-DLBCL. Смотри, например, U. Klein et al., (2008), Nature Reviews Immunology 8:22-23; R.E. Davis et al., (2001), Journal of Experimental Medicine 194: 1861-1874; G. Lentz et al., (2008), Science 319: 1676-1679; M. Compagno et al., (2009), Nature 459: 712-721; и L. Srinivasan et al., (2009), Cell 139: 573-586).
Настоящее изобретение относится к способу лечения фолликулярной лимфомы у индивидуума, нуждающегося в этом, включающему введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соеднения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Используемый в данном описании термин "фолликулярная лимфома" относится к любому из нескольких типов неходжкинских лимфом, в которых лимфоматозные клетки сгруппированны в узелки или фолликулы. Термин фолликулярная используется потому, что клетки в лимфатических узлах имеют тенденцию расти в виде круглой или узловатой формы. Средний возраст людей, страдающих такой лимфомой, составляет около 60 лет.
Настоящее изобретение относится к способу лечения хронического лимфоцитарного лейкоза (CLL) или мелкоклеточной лимфоцитарной лимфомы (SLL) у индивидуума, нуждающегося в этом, включающему введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Обычно считают, что CLL и SLL являются одной и той же болезнью с несколько разными проявлениями. Является ли лимфома CLL или SLL определяют в зависимости от места, где созревают раковые клетки. Когда раковые клетки находятся преимущественно в лимфатических узлах лимфатической системы, формируя структуры в виде лимской фасоли (в основном система крошечных сосудов, находящихся в организме), лимфома называется SLL. SLL составляет от около 5% до 10% от всех лимфом. Когда большая часть раковых клеток находится в крови и костном мозге, лимфома называется CLL.
Как CLL, так и SLL представляют собой медленно прогрессирующие болезни, хотя CLL, которая является гораздо более распространенной, как правило, прогрессирует медленнее. CLL и SLL лечатся одним и тем же способом. Они обычно не рассматриваются как излечимые посредством стандартной терапии, однако, в зависимости от стадии и скорости развития заболевания, большинство пациентов живут дольше, чем 10 лет. Иногда, с течением времени, эти медленно растущие лимфомы могут трансформироваться в лимфомы более агрессивного типа.
Хронический лимфоцитарный лейкоз (CLL) представляет собой наиболее распространенный тип лейкоза. По оценкам, 100,760 людей в Соединенных Штатах живут или находятся в стадии ремиссии CLL. Большинство (>75%) людей с впервые диагностированным CLL находятся в возрасте старше 50 лет. В настоящее время терапия CLL фокусируется в большей мере на контролировании этой болезни и ее симптомов, чем на непосредственном лечении. CLL лечится посредством химиотерапии, лучевой терапии, биологической терапии или трансплантации костного мозга. Симптомы заболевания иногда лечатся хирургически (спленэктомия, удаление увеличенной селезенки) или посредством радиационной терапии ("циторедукция" увеличенных лимфатических узлов). Хотя CLL в большинстве случаев прогрессирует медленно, болезнь обычно считается неизлечимой. Некоторые CLL классифицируются как состояния высокого онкогенного риска. Используемый в данном описании термин "CLL высокого онкогенного риска" означает, что CLL характеризуется, по меньшей мере, одним из следующих показателей: 1) 17р13-; 2) 1q22-; 3) немутировавший IgVH вместе с ZAP-70+ и/или CD38+; или 4) трисомия 12.
Лечение CLL обычно применяется, когда клинические симптомы или анализы крови пациента показывают, что болезнь прогрессировала до момента, когда она может повлиять на качество жизни пациента.
Мелкоклеточная лимфоцитарная лимфома (SLL) очень похожа на CLL, описанный выше, а также на рак В-клеток. В случае SLL атипичные лимфоциты поражают, главным образом, лимфатические узлы. Однако в случае CLL атипичные клетки поражают, главным образом, кровь и костный мозг. Селезенка может быть затронута при обоих состояниях. На SLL приходится около 1 случая заболевания из всех 25 случаев заболевания неходжкинской лимфомой. Болезнь может проявиться в любое время от юного возраста до глубокой старости, но редко возникает в возрасте моложе 50 лет. SLL рассматривается как индолентная лимфома. Это означает, что болезнь прогрессирует очень медленно, и пациенты, как правило, живут много лет после постановки диагноза. Однако для большинства пациентов диагноз часто ставится на поздних стадиях заболевания, и, несмотря на то, что SLL хорошо реагирует на разные химиотерапевтические препараты, она, в большинстве случаев, признается неизлечимой. Хотя некоторые виды рака, как правило, чаще наблюдаются у представителей какого-то одного или другого пола, случаи заболеваний и смертей вследствие SLL равномерно распределены между мужчинами и женщинами. Средний возраст на момент постановки диагноза составляет 60 лет.
Несмотря на то, что SLL представляет собой индолентную лимфому, она постоянно прогрессирует. Обычная картина этого заболевания представляет один из высоких показателей положительного клинического ответа на лучевую терапию и/или химиотерапию с последующим периодом ремиссии. Но спустя месяцы или годы после терапии наступает неизбежный рецидив. Повторное лечение снова приводит к ответу, но затем болезнь снова будет рецидивировать. Это означает, что, хотя краткосрочный прогноз SLL является неплохим, через какое-то время у многих пациентов развиваются фатальные осложнения этого рецидивирующего заболевания. Принимая во внимание возраст пациентов с диагнозом CLL и SLL, существует потребность для простого и эффективного лечения заболевания с минимальными побочными эффектами, которые не отражаются на качестве жизни пациента. Настоящее изобретение удовлетворяет эту давнюю потребность в данной области.
Объектом изобретения является способ лечения лимфомы из клеток мантийной зоны у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Используемый в данном описании термин "лимфома из клеток мантийной зоны" (MCL) относится к подтипу В-клеточной лимфомы по причине антиген-наивных CD5-позитивных В-клеток из прегерминативного центра в пределах мантийной зоны, которая окружает нормальный герминативный центр фолликул. Клетки MCL обычно сверхэкспрессируют циклин D1 в результате хромосомной транслокации t(11; 14) в ДНК. Более конкретно, имеет место хромосомная транслокация t(11, 14)(q13; q32). Только около 5% всех лимфом относятся к этому типу. Клетки MCL имеют размер от небольшого до среднего. Мужчины поражаются более часто. Средний возраст пациентов около 60 лет. Лимфома, когда она диагностируется, как правило, уже широко распространена в организме, включая лимфатические узлы, костный мозг и, очень часто, селезенку. Лимфома из клеток мантийной зоны представляет собой не очень быстро растущую лимфому, но трудно поддается лечению.
В некоторых вариантах осуществления настоящее изобретение представляет способ лечения В-клеточной лимфомы из клеток маргинальной зоны у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Используемый в данном изобретении термин "В-клеточная лимфома из клеток маргинальной зоны" относится к группе родственных В-клеточных новообразований, в которые вовлечены лимфоидные ткани маргинальной зоны, очаговой области за пределами мантийной зоны фолликула. Лимфомы маргинальной зоны составляют от около 5% до 10% от всех лимфом. Клетки в этих лимфомах под микроскопом выглядят маленькими. Существуют 3 основных типа лимфом маргинальной зоны, включая экстранодальную В-клеточную лимфому маргинальной зоны, нодальную В-клеточную лимфому маргинальной зоны и лимфому из клеток маргинальной зоны селезенки.
В некоторых вариантах осуществления настоящее изобретение представляет способ лечения MALT лимфомы у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIа-IId.
Термин "лимфома, ассоциированная с лимфоидной тканью слизистой оболочки (MALT)", как он использован здесь, относится к экстранодальным проявлениям лимфомы маргинальной зоны. Большинство MALT лимфом представляют собой лимфомы с низкой степенью злокачественности, хотя меньшая часть лимфом либо проявляется сначала как неходжкинская лимфома (NHL) со средней степенью злокачественности, либо развивается из формы с низкой степенью злокачественности. В большинстве случаев MALT лимфомы локализуются в желудке, и примерно 70% MALT лимфом желудка ассоциированы с хеликобактерной инфекцией. Было идентифицировано несколько цитогенетических патологий, при этом наиболее распространенной является трисомия 3 или t(11; 18). Многие из этих других MALT лимфом были связаны также с бактериальными или вирусными инфекциями. Средний возраст больных с лимфомой MALT составляет около 60.
В некоторых вариантах осуществления настоящее изобретение представляет способ лечения нодальной В-клеточной лимфомы маргинальной зоны у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Термин "нодальная В-клеточная лимфома маргинальной зоны" относится к индолентной В-клеточной лимфоме, которая обнаруживается, по большей части, в лимфатических узлах. Заболевание встречается редко и составляет только 1% от всех неходжкинских лимфом (NHL). Она наиболее часто диагностируется у пациентов пожилого возраста, при этом женщины более восприимчивы, чем мужчины. Заболевание классифицируется как лимфома маргинальной зоны, поскольку мутация возникает в В-клетках маргинальной зоны. Из-за локализации ее в лимфатических узлах, это заболевание также классифицируется как нодальная лимфома.
В некоторых вариантах осуществления настоящее изобретение представляет способ лечения В-клеточной лимфомы маргинальной зоны селезенки у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Термин "В-клеточная лимфома из клеток маргинальной зоны селезенки" относится к конкретной мелкоклеточной В-клеточной лимфоме с низкой степенью злокачественности, которая включена в классификацию Всемирной организации здравоохранения. Характерными особенностями лимфомы являются спленомегалия, умеренные лимфоциты с морфологией "ворсинчатых клеток", интрасинусоидальный характер вовлечения различных органов, особенно костного мозга, а также относительно индолентное течение болезни. Прогрессирование опухоли с увеличением бластных форм и агрессивное развитие болезни наблюдаются у меньшинства пациентов. Молекулярные и цитогенетические исследования показали неоднородные результаты, вероятно, из-за отсутствия стандартизованных диагностических критериев.
В некоторых вариантах осуществления настоящее изобретение представляет способ лечения лимфомы Беркитта у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Термин "лимфома Беркитта" относится к типу неходжкинской лимфомы (NHL), которая обычно наблюдается у детей. Она представляет собой высоко агрессивный тип В-клеточной лимфомы, которая во многих случаях возникает и включает кроме лимфатических узлов другие части тела. Несмотря на то, что это быстрорастущая опухоль, лимфома Беркитта часто является излечимой посредством современных методов интенсивной терапии. Существуют два основных типа лимфом Беркитта - спорадический и эндемический варианты: эндемическая лимфома Беркитта и спорадическая лимфома Беркитта.
Эндемическая лимфома Беркитта поражает детей в гораздо большей мере, чем взрослых, и в 95% случаев связана с вирусной инфекцией Эпштейна-Барр (EBV). Она наблюдается главным образом в экваториальной Африке, где около половины всех случаев детского рака представляют собой лимфому Беркитта. Она характеристически имеет высокую вероятность вовлечения челюстной кости, довольно отличительную особенность, которая редко встречается при спорадической лимфоме Беркитта. Она также обычно включает брюшную полость.
Спорадическая лимфома Беркитта представляет собой разновидность лимфомы Беркитта, которая выявляется в разных странах мира, в том числе в Европе и Америке. Здесь так же этому заболеванию в основном подвержены дети. Связь с вирусом Эпштейна-Барр (EBV) не такая сильная, как в случае эндемической разновидности, хотя прямые доказательства инфекции EBV присутствуют у одного из пяти пациентов. В сравнении с частотой вовлечения лимфатических узлов при спорадической форме поражение в большей степени локализовано в органах брюшной полости, в особенности, этому подвержены дети, более чем в 90% всех случаев. Поражение костного мозга является более распространенным при спорадической форме.
В некоторых вариантах осуществления настоящее изобретение представляет способ лечения макроглобулинемии Вальденстрема (WM) у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Термин "макроглобулинемия Вальденстрема", также известный как лимфоплазматическая лимфома, представляет рак с вовлечением подтипа белых кровяных клеток, называемых лимфоцитами. Заболевание характеризуется неконтролируемой клональной пролиферацией терминально дифференцированных В-лимфоцитов. Оно также характеризуется клетками лимфомы, секретирующими антитело, называемое иммуноглобулин М (IgM). Антитела IgM циркулируют в крови в больших количествах и приводят к сгущению крови. Это может привести к снижению кровотока во многих органах, что может вызвать проблемы со зрением (из-за плохой циркуляции крови в кровеносных сосудах в задней части глаз) и неврологические проблемы (такие, как головная боль, головокружение и спутанность сознания), вызванные плохим кровотоком в головном мозге. Другие симптомы могут включать чувство усталости и слабости и склонность к легкому возникновению кровотечения. Предрасполагающий этиологический фактор до конца не изучен, но ряд факторов риска был идентифицирован, в том числе локус 6р21.3 на хромосоме 6. Существует 2-3-кратное увеличение риска развития WM у людей с личной историей аутоиммунных заболеваний, обусловленных выработкой аутоиммунных антител, и особенно повышенные риски ассоциированы с гепатитом, вирусом иммунодефицита человека и риккетсиозом.
В некоторых вариантах осуществления настоящее изобретение представляет способ лечения миеломы у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIa-IId.
Множественная миелома, также известная как ММ, миелома, плазмаклеточная миелома или как болезнь Калера (позднее Отто Калера), представляет собой рак белых кровяных клеток, известных как плазматические клетки. Тип В-клеток, плазматические клетки, являются важной частью иммунной системы, ответственной за продуцирование антител у людей и других позвоночных. Они производятся в костном мозге и транспортируются посредством лимфатической системы.
В некоторых вариантах осуществления настоящее изобретение представляет способ лечения лейкоза у индивидуума, нуждающегося в этом, включающий введение индивидууму композиции, содержащей терапевтическое количество, по меньшей мере, одного соединения, имеющего структуру Формул I, Ia-Ii или IIа-IId.
Лейкоз представляет собой рак крови или костного мозга, характеризующийся аномальным увеличением числа клеток крови, как правило, лейкоцитов (белых клеток крови). Лейкоз является широким термином, охватывающим целый спектр заболеваний. Первым разделением по типам является его острая и хроническая формы: (i) острый лейкоз характеризуется быстрым увеличением количества незрелых клеток крови. Такое скопление делает костный мозг неспособным производить здоровые клетки крови. При остром лейкозе требуется немедленное лечение в связи с быстрым прогрессированием и накоплением злокачественных клеток, которые затем выходят в кровоток и распространяются по другим органам тела. Острые формы лейкоза являются наиболее распространенными формами лейкоза у детей; (и) хронический лейкоз отличается чрезмерным увеличением количества относительно зрелых, но все-таки аномальных белых клеток крови. Имея обычно несколько месяцев или лет для развития, клетки производятся гораздо с более высокой скоростью, чем нормальные клетки, в результате чего образуется много аномальных белых кровяных телец в крови. Хронический лейкоз в основном наблюдается у пожилых людей, но теоретически может проявиться в любой возрастной группе. Кроме того, эти заболевания подразделяются соответственно по виду клеток крови, которые подвергаются воздействию. В соответствии с этим фактором лейкозы делятся на лимфобластный или лимфоцитарный лейкозы и миелоидный или миелогенный лейкозы: (i) при лимфобластном или лимфоцитарном лейкозах раковое преобразование имеет место в том типе клеток костного мозга, в которых обычно происходит образование лимфоцитов, представляющих собой клетки иммунной системы, борющиеся с инфекцией; (ii) при миелоидном или миелогенном лейкозах раковые изменения происходят в том типе клеток костного мозга, в которых обычно происходит образование эритроцитов, некоторых других белых клеток и тромбоцитов.
В рамках этих основных категорий есть несколько подкатегорий, включая, но не ограничиваясь ими, острый лимфобластный лейкоз (ALL), острый миелоидный лейкоз (AML), хронический миелоидный лейкоз (CML) и лейкоз ворсистых клеток (HCL).
Симптомы, диагностические тесты и прогностические тесты для каждого из вышеупомянутых патологических состояний известны в данной области. Смотри, например, Harrison's Principles of Internal Medicine®," 16th ed., 2004, The McGraw-Hill Companies, Inc. Dey et al. (2006), Cytojournal 3 (24) и классификацию лимфом "Revised European American Lymphoma" (REAL) (смотри, например, интернет-Cайт, поддержанный Национальным институтом рака).
Для разработки диапазона терапевтически эффективных доз соединений, ингибирующих ВТК для лечения любого из указанных выше заболеваний, обычно используется ряд моделей на животных.
Например, дозирование соединений, ингибирующих ВТК для лечения аутоиммунного заболевания, можно определить на модели ревматоидного артрита у мышей. В этой модели артрит стимулировали у мышей Balb/c путем введения антител к коллагену и липополисахарида. Смотри Nandakumar et al. (2003), Am. J. Pathol 163: 1827-1837.
В другом примере дозирование ингибиторов Btk для лечения В-клеточного пролиферативного расстройства можно исследовать, например, с помощью модели ксенотрансплантата человек-мышь, в которой клетки В-клеточной лимфомы человека (например, клетки Ramos) имплантированы иммунодефицитным мышам (например, бестимусным "nude" мышам), как описано, например, в Pagel et al. (2005), Clin Cancer Res 11 (13): 4857-4866.
Известны также модели с использованием животных для лечения тромбоэмболических заболеваний.
Терапевтическая эффективность соединения для одного из вышеуказанных заболеваний может быть оптимизирована в процессе лечения. Например, субъект, проходящий курс лечения, может пройти диагностическую оценку для установления корреляции между снижением выраженности симптомов заболевания или патологии и ингибированием активности ВТК, достигаемым при введении конкретной дозы ингибитора ВТК. Клеточные анализы, известные в данной области, могут быть использованы для определения in vivo активности ВТК в присутствии или в отсутствие ингибитора ВТК. Например, поскольку активированная ВТК фосфорилируется по тирозину 223 (Y223) и тирозину 551 (Y551), фосфоспецифическое иммуноцитохимическое окрашивание PY 223 или P-Y551-положительных клеток может быть использовано для обнаружения или количественного определения активизации ВТК в популяции клетки (например, с помощью анализа окрашенных и неокрашенных клеток путем FACS). Смотри, например, Nisitani et al. (1999), Proc. Natl. Acad. Sci, USA 96: 2221-2226. Таким образом, количество соединения ингибитора ВТК, которое вводится субъекту, может быть увеличено или уменьшено по мере необходимости так, чтобы поддерживать оптимальный уровень ингибирования ВТК для лечения болезненного состояния субъекта.
Соединения, описанные в настоящем изобретении, необратимо ингибируют ВТК и могут быть использованы для лечения млекопитающих, страдающих от патологических состояний или заболеваний, зависимых от тирозинкиназы Брутона или опосредованных тирозинкиназой Брутона, в том числе, но не ограничиваясь этим, рака, аутоиммунных и других воспалительных заболеваний. Соединения, описанные в настоящем изобретении, показали эффективность при самых различных заболеваниях и патологических состояниях, описанных в настоящем изобретении.
Еще один аспект заключается в использовании соединений Формулы I, Ia-Ii или IIa-IId или их фармацевтически приемлемой соли для получения лекарственного средства для применения при лечении хронических В-клеточных расстройств, в которых Т-клетки играют важную роль.
Еще в другом аспекте изобретения соединения Формул I, Ia-Ii или IIa-IId используются для изготовления лекарственного средства для применения при лечении ВТК-опосредованных заболеваний или патологических состояний. Это включает, но не ограничивается, лечение В-клеточной лимфомы, развивающейся в результате постоянной активации сигнального пути В-клеточного рецептора.
ВТК опосредованные расстройства или ВТК опосредованные патологические состояния, описываемые здесь, означают любое болезненное состояние или другое ухудшенное состояние, при котором центральную роль играют В-клетки, тучные клетки, миелоидные клетки, остеокласты. Эти заболевания включают, но не ограничиваются ими, иммунные, аутоиммунные и воспалительные заболевания, аллергии, инфекционные заболевания, резорбционные костные расстройства и пролиферативные заболевания.
Иммунные, аутоиммунные и воспалительные заболевания, которые можно лечить или предупреждать с помощью соединений Формул I, Ia-Ii или IIa-IId, дополнительно включают ревматические заболевания (например, инфекционный артрит, прогрессирующий хронический артрит, деформирующий артрит, травматический артрит, подагрический артрит, остеопороз, синдром Рейтера, полихондрит, острый синовит и спондилит), гломерулонефрит (с нефротическим синдромом или без него), аутоиммунные гематологические заболевания (например, гемолитическая анемия, апластическая анемия, идиопатическая тромбоцитопения и нейтропения) и аутоиммунные воспалительные заболевания кишечника (например, язвенный колит и болезнь Крона), болезнь трансплантат против хозяина, отторжение аллотрансплантата, хронический тиреоидит, склеродермию, первичный билиарный цирроз печени, системную красную волчанку, контактный дерматит, экзему, солнечные ожоги кожи, хроническую почечную недостаточность, синдром Стивенса-Джонсона, воспалительную боль, идиопатическую спру, кахексию, саркоидоз, кератоконъюнктивит, отит, пародонтоз, легочный интерстициальный фиброз, пневмокониоз, синдром легочной недостаточности, легочную эмфизему, пневмосклероз, силикоз, хроническое воспалительное заболевание легких (например, хроническое обструктивное легочное заболевание) и другие воспалительные или обструктивные заболевания дыхательных путей.
Аллергии, которые можно лечить или предупреждать, включают, среди прочего, аллергии на пищевые продукты, пищевые добавки, яды насекомых, пылевых клещей, на пыльцу, на животные продукты и контактные аллергены, гиперчувствительность типа I, аллергическую астму, аллергический конъюнктивит.
Инфекционные заболевания, которые можно лечить или предупреждать, включают, среди прочего, сепсис, септический шок, эндотоксический шок, сепсис, вызываемый грамотрицательными бактериями, дизентерию, менингит, церебральную малярию, пневмонию, туберкулез, вирусный миокардит, вирусный гепатит (гепатит А, гепатит В и гепатит С), ВИЧ-инфекции, ретинит, вызванный цитомегаловирусом, грипп, герпес, инфекции, ассоциированные с тяжелыми ожогами, миалгию, вызванную инфекциями, кахексию, вторичную по отношению к инфекциям, и ветеринарные инфекции, вызываемые вирусами, такими как лентивирус, артритический козий вирус, меди-висна вирус, кошачий вирус иммунодефицита, бычий вирус иммунодефицита или собачий вирус иммунодефицита.
В некоторых вариантах осуществления изобретения соединения Формулы I присутствуют как вещество по существу в чистом виде.
В некоторых вариантах осуществления изобретения соединения Формулы I выбраны из любого из приведенных в настоящем описании Примеров или их фармацевтически приемлемых солей.
Каждое определение вышеприведенной переменной включает любое ее подмножество, и соединения Формулы I включают любую комбинацию таких переменных или подмножеств переменных.
Настоящее изобретение включает соединения и их соли, их физические формы, получение соединений, используемых промежуточных соединений и фармацевтические композиции и их лекарственные формы и рецептуры.
Соединения по настоящему изобретению и термин "соединение" в формуле изобретения включают любые фармацевтически приемлемые соли или сольваты, и любые аморфные или кристаллические формы, или таутомеры, независимо от того, перечислены ли конкретно они в контексте.
Настоящее изобретение включает все изомеры соединений. Соединения могут иметь один или несколько асимметричных атомов углерода и могут существовать в виде двух или более стереоизомеров. В случае, когда соединение по настоящему изобретению содержит алкенильную или алкениленовую группу, возможно образование геометрических цис/транс (или Z/E) изомеров. В случае, если соединение содержит, например, кето или оксим группу или ароматический фрагмент молекулы, может наблюдаться таутомерная изомерия ('таутомерия'). Индивидуальное соединение может проявлять более чем один тип изомерии.
Настоящее изобретение включает любые стереоизомеры, даже если не указано конкретно, по отдельности, а также их смеси, геометрические изомеры и фармацевтически приемлемые соли, в том числе соединения, проявляющие более чем один тип изомерии. В случае, когда соединение или стереоцентр описано или показано без определенной стереохимии, оно должно приниматься охватывающим все возможные отдельные изомеры, конфигурации и их смеси. Таким образом, образец вещества, содержащий смесь стереоизомеров, будет охвачен описанием или стереоизомеров, или описанием без определенной стереохимии.
Предполагаются также любые цис/транс изомеры или таутомеры описанных соединений. Когда таутомер соединения Формулы I существует, соединение Формулы I по настоящему изобретению включает любые возможные таутомеры и их фармацевтически приемлемые соли и их смеси, за исключением тех случаев, где конкретно указано иное.
Подробное описание изобретения
В настоящей заявке описаны соединения Формулы I, которые включают соединения Формул Ia-Ii и IIa-IId, и композиции и рецептуры, содержащие такие соединения, и способы применения и получения таких соединений. Эти соединения являются полезными для лечения заболеваний или состояний, модулированных, по меньшей мере, частично, ВТК.
В варианте осуществления описано соединение в соответствии с Формулой I и вышеизложенными вариантами осуществления, где соединение Формулы I представлено как соединение Формулы IIа:
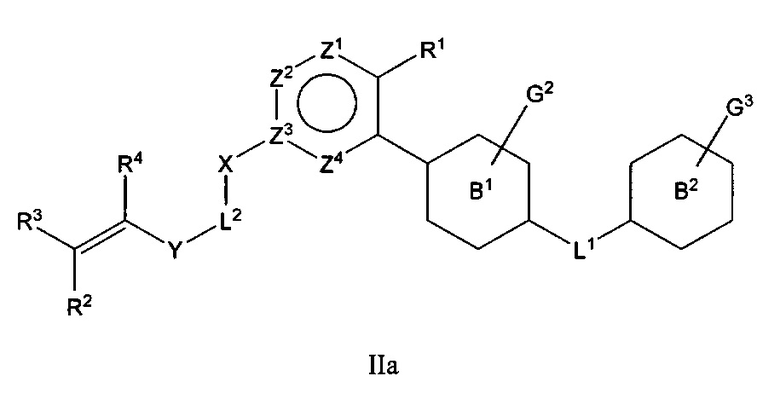
где R1-R4, G2, G3, L1, L2, X, Y и Z1-Z4 являются такими, как описано ранее для соединения Формулы I, и В1 и В2 независимо выбраны из С6циклоалкила, C6гетероциклоалкила, C6арила или С6гетероарила.
В варианте осуществления описано соединение в соответствии с Формулой I и вышеизложенными вариантами осуществления, где соединение Формулы I представлено как соединение Формулы IIb:
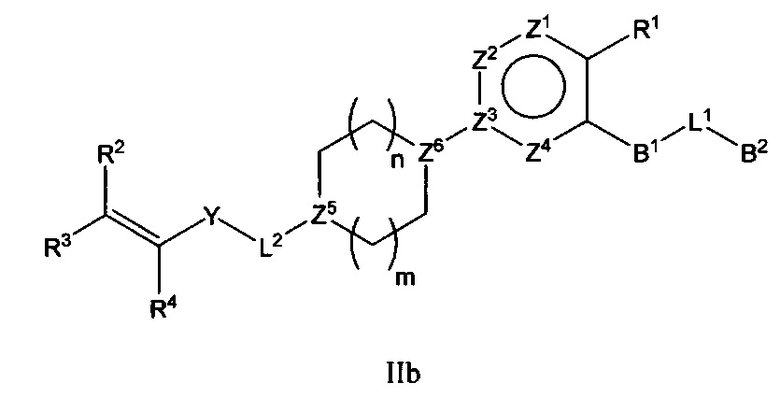
где R1-R4, В1, В2, L1, L2, Y и Z1-Z4 являются такими, как описано ранее для соединения Формулы I, Z5 и Z6 каждый независимо выбран из C(Ra) или N, где Ra представляет собой алкил или Н, и n и m каждый независимо выбран из 0, 1 или 2.
В варианте осуществления описано соединение в соответствии с Формулой I и вышеизложенными вариантами осуществления, где соединение Формулы I представлено как соединение Формулы IIс:
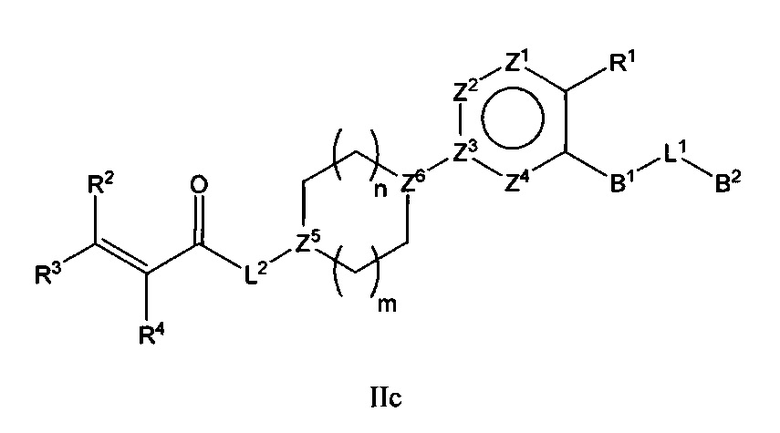
где R1-R4, В1, В2, L1, L2, Y и Z1-Z4 являются такими, как описано ранее для соединения Формулы I, Z5 и Z6 каждый независимо выбран из С или N, и n и m каждый независимо выбран из 0, 1 или 2.
В варианте осуществления описано соединение в соответствии с Формулой I и вышеизложенными вариантами осуществления, где соединение Формулы I представлено как соединение Формулы IId:
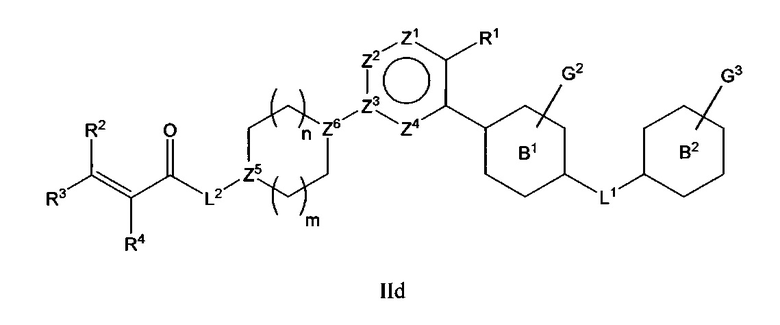
где R1-R4, L1, L2, G2, G3 и Z1-Z4 являются такими, как описано ранее для соединения Формулы I, Z и Z каждый независимо выбран из C или N, В1 и В2 независимо выбраны из C6циклоалкила, Сбгетероциклоалкила, С6арила или С6гетероарила, и n и m каждый независимо выбран из 0, 1 или 2.
Настоящее изобретение включает соединения, промежуточные соединения, примеры и способы синтеза, описанные в данном документе. Соединения Формулы I получают в соответствии со схемами реакций, описанных в настоящей заявке. Если не указано иное, то заместители в схемах определены, как указано выше.
Способы синтеза:
Соединения по настоящему изобретению включают промежуточные соединения, примеры и способы синтеза, описанные в настоящей заявке. Способы синтеза, описанные здесь, как правило, предваряются их соответствующими схемами синтеза. В случае, когда способ для промежуточного соединения или примера относится к аналогичному способу для аналогичного промежуточного соединения или примера, такая ссылка включает способ для такого же аналогичного промежуточного соединения или примера, связанной с ним схемы синтеза, а также способы и схемы, используемые для синтеза аналогичных промежуточного соединения или примера.
Соединения Формул I, Ia-Ii и IIa-d могут быть получены способами, описанными ниже, вместе со способами синтеза, известными в области органической химии, или путем модификаций и дериватизаций, которые известны специалистам в данной области техники. Исходные вещества, используемые в настоящем документе, являются коммерчески доступными или могут быть получены стандартными способами, известными в данной области [такими, как способы, описанные в стандартных справочниках, как, например, Compendium of Organic Synthetic Methods, Vol. I-VI (Wiley-Interscience) или Comprehensive Organic Transformations, by R.C. Larock (Wiley-Interscience)]. Предпочтительные способы включают, но не ограничиваются ими, те, которые описаны ниже.
При проведении любой из следующих последовательностей синтеза может быть необходимым и/или желательным защитить чувствительные или реакционноспособные группы на любой из рассматриваемых молекул. Это может быть достигнуто с помощью обычных защитных групп, таких, как те, которые описаны в Т.W. Greene, Protective Groups in Organic Chemistry, John Wiley & Sons, 1981; T.W. Greene и P.G.M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1991 и T.W. Greene и P.G. M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1999, которые включены в настоящее описание посредством ссылки.
Соединения Формул I, Ia-Ii и IIa-D, или их фармацевтически приемлемые соли могут быть получены в соответствии со схемами реакций, описанных ниже, и используя обычную квалификацию в данной области техники. Если не указано иное, то заместители в схемах определены, как указано выше. Выделение и очистка продуктов осуществляется с помощью стандартных методик, которые известны среднему специалисту в области химии.
Когда указывается общий или приводимый в качестве примера способ синтеза, специалист в данной области может легко определить соответствующие реагенты, если они не указаны, основываясь на общих или типовых способах. Некоторые из общих способов приведены в качестве примеров для общего способа получения соединений. Специалист в данной области техники может легко адаптировать такие способы для синтеза других конкретных соединений. Изображение незамещенной позиции в структурах, показанное или указанное в общих способах, представлено для удобства и не исключает возможности замены, как описано где-либо в настоящем документе. В случае определенных групп, которые могут присутствовать либо в качестве групп в общих способах, либо в качестве не показанных необязательных заместителей, следует обратиться к описаниям в остальной части этого документа, включая формулу изобретения и подробное описание.
Общий способ синтеза соединений Формул I, Ia-Ii и IIa-d показан на Общей Схеме ниже.
Общая Схема:
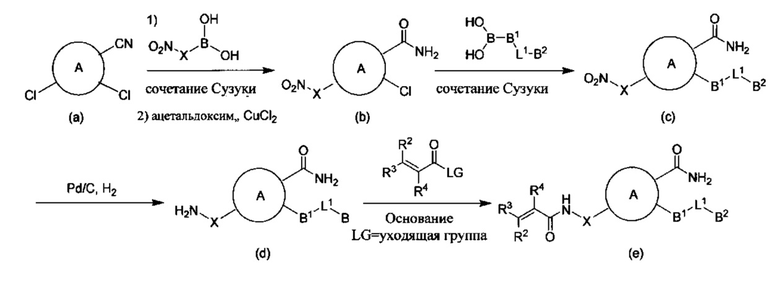
В случаях, когда A, В1, В2, L1, X, R2, R3 и R4 являются такими, как определено выше для соединения Формул I, Ia-Ii и IIa-d, и LG представляет собой подходящую уходящую группу, такую как трифлат, мезилат, тозилат, HATU, Cl, Br или I.
В типичном способе получения соединения Формул I, Ia-Ii и IIa-D соединение Формулы (а) вступало в реакцию с подходящей бороновой кислотой в условиях сочетания Сузуки. Подходящие условия включают, но не ограничиваются ими, обработку соединений Формулы (а) подходящим основанием, таким как CS2CO3 или K2CO3, и подходящим палладиевым катализатором, таким как Pd(dppf)Cl2 DCM. Подходящие растворители для использования в приведенном выше способе синтеза включают 1,4-диоксан, воду, DME и их смеси. Смесь дегазировали азотом шесть раз и нагревали до кипения с обратным холодильником в течение около 16 ч в атмосфере азота с получением соединения Формулы (b). Приведенный выше способ осуществляли при температурах между около 50° и около 150°. Предпочтительно, реакцию проводили при температуре около 100°. Указанный выше способ, предпочтительно, проводили при или около атмосферном давлении, хотя, при желании, могут быть использованы более высокие или более низкие степени давления. Главным образом, предпочтительно использовать эквимолярные количества реагентов, хотя, при желании, могут быть использованы большие или меньшие количества.
Соединение Формулы (b) подвергали взаимодействию с подходящей бороновой кислотой в условиях реакции сочетания Сузуки. Подходящие условия включают, но не ограничиваются ими, обработку соединений Формулы (b) подходящим основанием, таким как CS2CO3 или K2СО3, и подходящим палладиевым катализатором, таким как Pd(dppf)Cl2 DCM или Pd(PPh3)4. Подходящие растворители для использования в приведенном выше способе синтеза включают 1,4-диоксан, воду, DME и их смеси. Смесь дегазировали азотом шесть раз и нагревали до кипения с обратным холодильником в течение около 5 часов в атмосфере азота с получением соединения Формулы (с). Приведенный выше способ осуществляли при температурах между около 40° и около 120°. Предпочтительно, реакцию проводили при температуре около 90°. Указанный выше способ, предпочтительно, проводили при или около атмосферном давлении, хотя, при желании, могут быть использованы более высокие или более низкие степени давления. Главным образом, предпочтительно использовать эквимолярные количества реагентов, хотя, при желании, могут быть использованы большие или меньшие количества.
Соединение Формулы (с) подвергали взаимодействию в атмосфере водорода в присутствии палладия на угле, предпочтительно, дегазируя смесь водородом около 6 раз. Подходящие растворители для использования в приведенном выше способе синтеза включают этилацетат и метанол. Описанный выше способ проводили при температурах между около 10° и около 60°, или, предпочтительно, при температуре окружающей среды с получением соединения Формулы (d). Указанный выше способ, предпочтительно, проводили при или около атмосферном давлении, хотя, при желании, могут быть использованы более высокие или более низкие степени давления. Главным образом, предпочтительно использовать эквимолярные количества реагентов, хотя, при желании, могут быть использованы большие или меньшие количества.
Соединение Формулы (d) подвергали взаимодействию с подходящим акрилоилхлоридом с подходящим основанием. Подходящие основания включают органические основания, такие как TEA или DIPEA. Приведенный выше способ осуществляли при температурах между около -10° и около температуры окружающей среды, или, предпочтительно, при 0° с получением соединения Формулы (d). Указанный выше способ, предпочтительно, проводили при или около атмосферном давлении, хотя, при желании, могут быть использованы более высокие или более низкие степени давления. Главным образом, предпочтительно использовать эквимолярные количества реагентов, хотя, при желании, могут быть использованы большие или меньшие количества.
Примеры
Способы получения и Промежуточные соединения
Схема 1
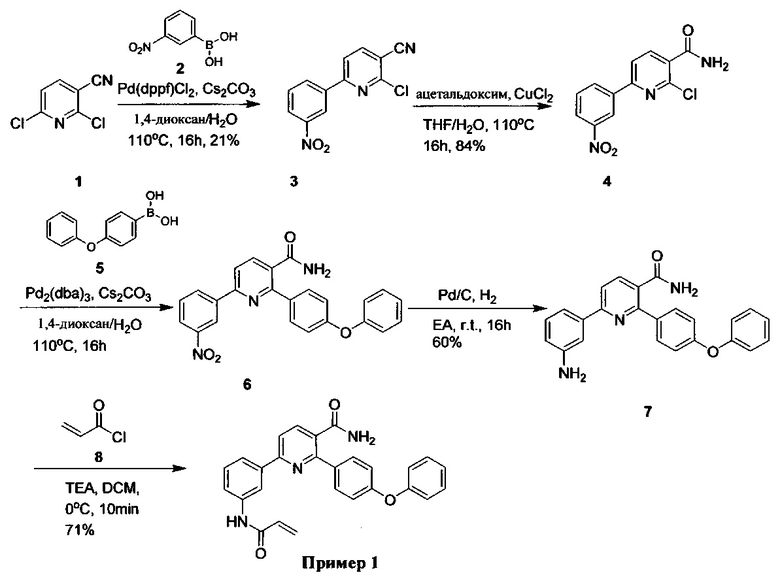
2-Хлор-6-(3-нитрофенил)никотинонитрил (3). К раствору 3-нитрофенилбороновой кислоты 2 (5.19 г, 30 ммоль), CS2CO3 (19.56 г, 60 ммоль) и 2,6-дихлорникотинонитрила 1 (5.51 г, 33 ммоль) в сухом 1,4-диоксане (100 мл) добавляли Pd(dppf)Cl2⋅DCM (2.4 г, 3.0 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до кипения с обратным холодильником в течение 16 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали и остаток очищали с помощью флэш-хроматографии, элюируя от 20:1 до 3:1 РЕ/ЕА с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (1.6 г, 21%).
2-Хлор-6-(3-нитрофенил)никотинамид (4). К раствору 2-хлор-6-(3-нитрофенил)никотинонитрила 3 (259 мг, 1.0 ммоль) и ацетальдоксима (88 мг, 1.5 ммоль) в тетрагидрофуране (5 мл) и воде (5 мл) добавляли CuCl2 (15 мг, 0.1 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали в течение 16 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали и сырой продукт очищали с помощью флэш-хроматографии, элюируя смесью 20:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (240 мг, 84%). MS (ESI): m/z=277.9 [М+Н]+.
6-(3-Нитрофенил)-2-(4-феноксифенил)никотинамид (6). К раствору 2-хлор-6-(3-нитрофенил)никотинамида 4 (240 мг, 0.87 ммоль), CS2CO3 (567 мг, 1.74 ммоль) и бифенил-4-илбороновой кислоты 5 (203 мг, 0.95 ммоль) в 1,4-диоксане (10 мл) и воде (2.5 мл) добавляли Pd2(dba)3 (80 мг, 0.09 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до кипения с обратным холодильником в течение 16 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя 150:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (110 мг, сырое).
6-(3-Аминофенил)-2-(4-феноксифенил)никотинамид (7). К раствору 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида 6 (110 мг, 0.27 ммоль) в этилацетате (5 мл) добавляли Pd/C (10 мг) в атмосфере водорода, и смесь дегазировали водородом 6 раз, затем перемешивали в течение 16 ч при температуре окружающей среды в атмосфере водорода. Раствор фильтровали, и фильтрат упаривали до сырого продукта в виде красного масла (61 мг, 60%). MS (ESI): m/z=382.1 [М+Н]+.
Пример 1
6-(3-Акриламидофенил)-2-(4-феноксифенил)никотинамид
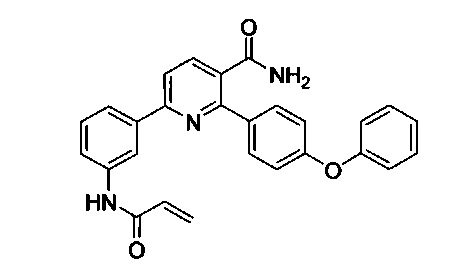
К раствору 6-(3-аминофенил)-2-(4-феноксифенил)никотинамида 7 (38 мг, 0.1 ммоль) в DCM (5 мл) добавляли TEA (0.05 мл, 0.4 ммоль) и акрилоилхлорид 8 (9 мг, 0.1 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (30 мг, 71%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, DMSO) δ 10.36 (s, 1H), 8.43 (s, 1H), 7.72-7.97 (m, 7H), 7.49 (s, 1H), 7.33-7.52 (m, 3H), 7.24(t, J=6.9 Гц, 1H), 6.9-7.13 (m, 4H), 6.45 (dd, J=16.9, 10.0 Гц, 1H), 6.30 (d, J=16.9 Гц, 1H), 5.79 (d, J =10.5 Гц, 1H), MS (ESI, методика A): m/z=436.0 [M+H]+, tR=1.553 мин, ВЭЖХ: 97.5% (214 нм), 98.0% (254 нм).
Схема 2
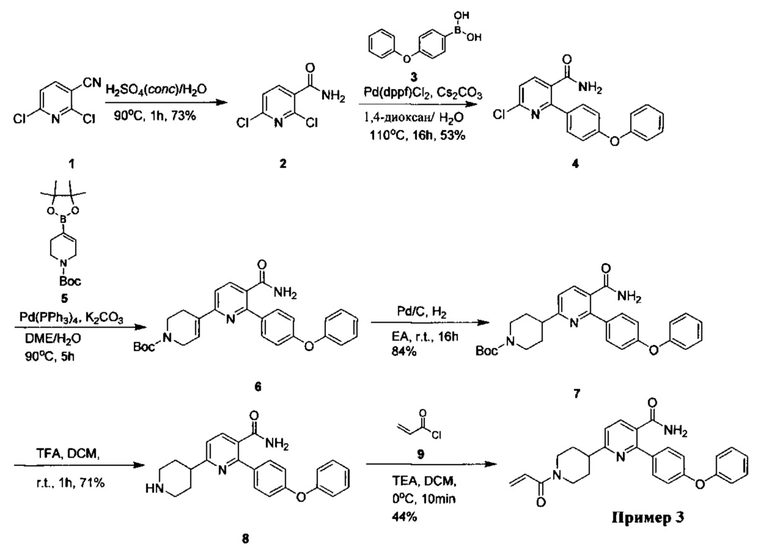
2,6-Дихлорникотинамид (2). К 2,6-дихлорникотинонитрилу 1 (1.73 г, 10 ммоль) добавляли концентрированную H2SO4 (10 мл) и воду (2 мл). Смесь нагревали до 90° и перемешивали в течение 1 ч. После охлаждения до комнатной температуры раствор выливали в ледяную воду, затем доводили до значения рН=8 аммиачной водой. Осадок отфильтровывали, промывали водой (20 мл) и высушивали под вакуумом с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (1.4 г, 73%). MS (ESI): m/z=191.1 [М+Н]+.
6-Хлор-2-(4-феноксифенил)никотинамид (4). К раствору 2,6-дихлорникотинамида 2 (668 мг, 3.5 ммоль), CS2CO3 (1.14 г, 7 ммоль) и 4-феноксифенилбороновой кислоты 3 (749 мг, 3.5 ммоль) в 1,4-диоксане (30 мл) добавляли Pd(dppf)Cl2⋅DCM (285 мг, 0.35 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до кипения с обратным холодильником в течение 16 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и остаток очищали с помощью флэш-хроматографии, элюируя от 20:1 до 3:1 смесью петролейный эфир/этилацетат (РЕ/ЕА), с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (611 мг, 53%). MS (ESI): m/z=325.0 [М+Н]+.
трет-Бутил-4-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат (6). К раствору 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридина 5 (232 мг, 0.75 ммоль), K2CO3 (207 мг, 1.50 ммоль) и 6-хлор-2-(4-феноксифенил)никотинамида 4 (162 мг, 0.50 ммоль) в 1,2-диметоксиэтане (5 мл) и воде (1 мл) добавляли Pd(PPh3)4 (115 мг, 0.10 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали в течение 5 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя 70:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (232 мг, сырое).
трет-Бутил-4-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пиперидин-1-карбоксилат (7). К раствору 4-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата 6 (232 мг, 0.75 ммоль) в этилацетате (5 мл) добавляли Pd/C (10 мг) в атмосфере водорода, и смесь дегазировали водородом 6 раз, затем перемешивали в течение 16 ч при температуре окружающей среды в атмосфере водорода. Раствор фильтровали, и фильтрат упаривали до сырого продукта в виде твердого вещества коричневого цвета (215 мг, 60% за две стадии). MS (ESI): m/z=474.1 [М+Н]+.
2-(4-Феноксифенил)-6-(пиперидин-4-ил)никотинамид (Пример 2) (8). К раствору трет-бутил-4-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пиперидин-1 -карбоксилата 7 (215 мг, 0.45 ммоль) в сухом дихлорметане (6 мл) добавляли TFA (2 мл), и полученную в результате смесь перемешивали в течение 1 ч при температуре окружающей среды. Растворитель удаляли, и остаток разделяли между насыщенным водным бикарбонатом натрия (30 мл) и этилацетатом (20 мл). Органическую фазу отделяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Сырой остаток очищали с помощью флэш-хроматографии, элюируя 5:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (120 мг, 71%). MS (ESI): m/z=374.2 [М+Н]+.
Пример 3
6-(1-Акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамид
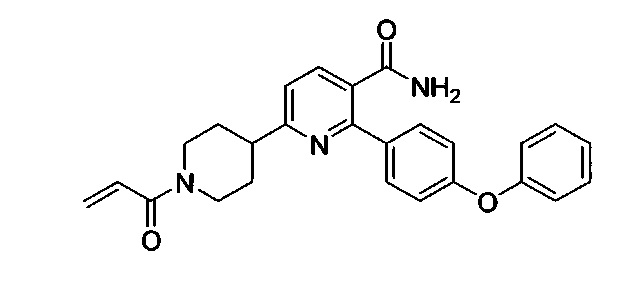
К раствору 2-(4-феноксифенил)-6-(пиперидин-4-ил)никотинамида 8 (26 мг, 0.07 ммоль) в DCM (5 мл) добавляли TEA (0.05 мл, 0.4 ммоль) и акрилоилхлорид 9 (7 мг, 0.07 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (13 мг, 44%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 7.90 (d, J=8.0 Гц, 1Н), 7.71-7.62 (m, 2H), 7.42-7.31 (m, 2H), 7.21-7.11 (m, 2H), 7.10-7.01 (m, 4H), 6.60 (dd, J=16.8, 10.5 Гц, 1H), 6.26 (dd, J=16.8, 2.0 Гц, 1H), 5.84 (s, 1H), 5.68 (dd, J=10.5, 2.0 Гц, 1H), 5.54 (s, 1H), 4.79 (d, J=13.3 Гц, 1H), 4.13 (d, J=12.8 Гц, 1H), 3.17 (t, J=12.3 Гц, 1H), 3.39 (t, J=11.7 Гц, 1H), 2.76 (t, J=11.7 Гц, 1H), 2.03 (br, 2H), 1.80 (ddd, J=25.5, 12.5, 4.2 Гц, 2H). MS (ESI, методика A): m/z=428.0 [M+H]+, tR=1.480 мин. ВЭЖХ: 96.7% (214 нм), 99.3% (254 нм).
Схема 3
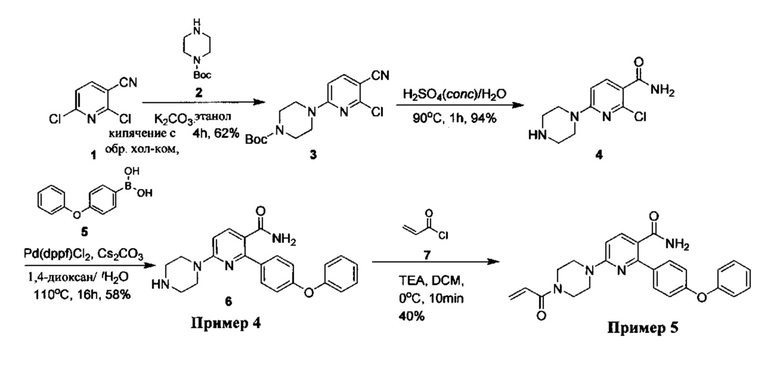
трет-Бутил-4-(6-хлор-5-цианопиридин-2-ил)пиперазин-1-карбоксилат (3). К раствору 2,6-дихлорникотинонитрила 1 (519 мг, 3.0 ммоль) и трет-бугип пиперазин-1-карбоксилата 2 (558 мг, 3.0 ммоль) в этаноле (15 мл) добавляли K2CO3 (636 мг, 6.0 ммоль), и полученный в результате раствор нагревали до кипения с обратным холодильником в течение 4 ч. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя от 5:1 до 2:1 РЕ/ЕА, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (600 мг, 62%). MS (ESI): m/z=345.1 [M+Na]+.
6-Хлор-2-(4-феноксифенил)никотинамид (4). К трет-бутил-(6-хлор-5-цианопиридин-2-ил)пиперазин-1-карбоксилату 3 (600 мг, 10 ммоль) добавляли H2SO4 (конц., 5 мл) и воду (1 мл). Смесь нагревали до 90° и перемешивали в течение 1 ч. После охлаждения до комнатной температуры раствор выливали в ледяную воду, затем доводили до значения рН=8 аммиачной водой. Осадок отфильтровывали, промывали водой (20 мл) и высушивали под вакуумом с получением указанного в заголовке соединения в виде твердого вещества не совсем белого цвета (420 мг, 94%). MS (ESI): m/z=241.0 [М+Н]+.
2-(4-Феноксифенил)-6-(пиперазин-1-ил)никотинамид (Пример 4) (6). К раствору 6-хлор-2-(4-феноксифенил)никотинамида 4 (420 мг, 1.75 ммоль), K2CO3 (483 мг, 3.5 ммоль) и 4-феноксифенилбороновой кислоты 5 (374 мг, 1.75 ммоль) в 1,4-диоксане (15 мл) и воде (3 мл) добавляли Pd(PPh3)4 (231 мг, 0.20 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали в течение 5 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя 70:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (260 мг, 58%). MS (ESI): m/z=375.0 [М+Н]+.
Пример 5
6-(4-Акрилоилпиперазин-1-ил)-2-(4-феноксифенил)никотинамид
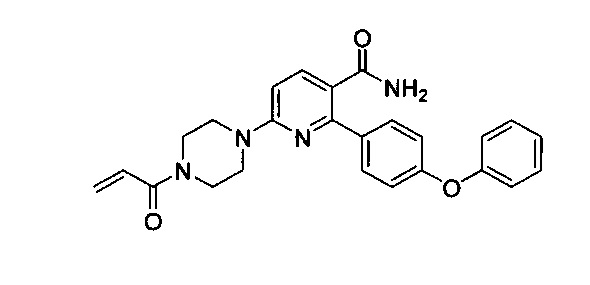
К раствору 2-(4-феноксифенил)-6-(пиперазин-1-ил)никотинамида 6 (59 мг, 0.16 ммоль) в DCM (5 мл) добавляли TEA (0.05 мл, 0.4 ммоль) и акрилоилхлорид 7 (14 мг, 0.16 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (27 мг, 40%) в виде твердого вещества светло-желтого цвета. 1Н ЯМR (400 МГц, CDCl3) δ 8.04 (d, J=8.7 Гц, 1Н), 7.64 (d, J=8.2 Гц, 2H), 7.40 (t, J=7.6 Гц, 2H), 7.18 (t, J=7.3 Гц, 1H), 7.08 (dd, J=12.0, 8.4 Гц, 4H), 6.66 (d, J=8.5 Гц, 1H), 6.64-6.56 (m, 1H), 6.37 (d, J=16.7 Гц, 1H), 5.77 (d, J=10.6 Гц, 1H), 5.57 (s, 1H), 5.33 (s, 1H), 3.81 (s, 4H), 3.73 (s, 4H). MS (ESI, методика A): m/z=429.2 [M+H]+, tR=1.454 мин, ВЭЖХ: 97.5% (214 нм), 98.0% (254 нм).
Схема 4
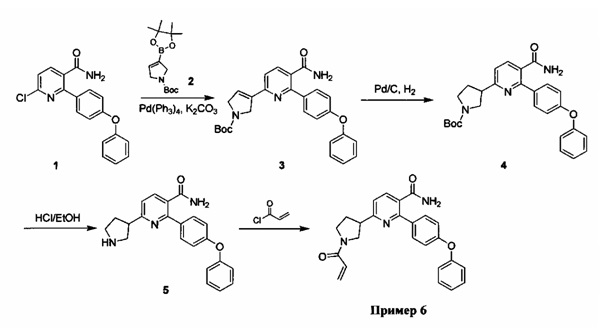
6-Хлор-2-(4-феноксифенил)никотинамид (1). 6-Хлор-2-(4-феноксифенил)никотинамид 1 синтезировали с использованием способа, аналогичного описанному выше, с получением твердого вещества желтого цвета (611 мг, 53%). MS (ESI): m/z=325.0 [М+Н]+.
трет-Бутил-3-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)-2,5-дигидро-1Н-пиррол-1-карбоксилат (3). Указанное в заголовке соединение синтезировали с использованием способа, аналогичного описанному при получении трет-бутил-4-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата, в виде твердого вещества белого цвета (300 мг, сырое). MS (ESI): m/z=458.2 [М+Н]+.
трет-Бутил-3-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пирролидин-1-карбоксилат (4). Указанное в заголовке соединение синтезировали с использованием способа, аналогичного описанному при получении трет-бутил-4-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пиперидин-1-карбоксилата, в виде твердого вещества белого цвета (240 мг, 45% за две стадии). MS (ESI): m/z=460.1 [М+Н]+.
2-(4-Феноксифенил)-6-(пирролидин-3-ил)никотинамид (5). Указанное в заголовке соединение синтезировали с использованием способа, аналогичного описанному при получении 2-(4-феноксифенил)-6-(пиперидин-4-ил)никотинамида, в виде твердого вещества белого цвета (190 мг, сырое). MS (ESI): m/z=360.1 [М+Н]+.
Пример 6
6-(1-Акрилоилпирролидин-3-ил)-2-(4-феноксифенил)никотинамид
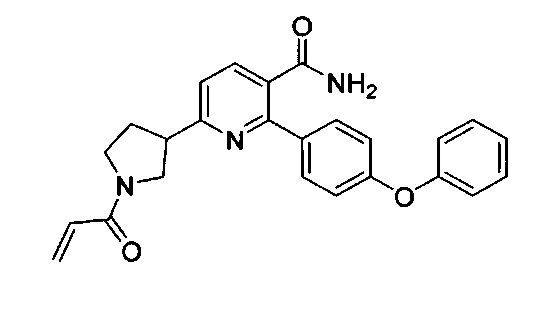
Указанное в заголовке соединение синтезировали с использованием способа, аналогичного описанному в Примере 3, в виде твердого вещества белого цвета (18 мг, 29%). 1Н ЯМР (300 МГц, CD3OD) δ 7.85 (d, J=7.9 Гц, 1Н), 7.72 (d, J=8.8 Гц, 2Н), 7.37 (m, 3Н), 7.15 (t, J=7.4 Гц, 1H), 7.02 (m, 4Н), 6.65 (dd, J=16 J, 10.6 Гц, 1Н), 6.28 (m, 1Н), 5.74 (dd, J=9.5, 7.4 Гц, 1H), 3.98 (m, 2H), 3.69 (m, 3H), 2.34 (dtd, J=30.9, 12.6, 8.0 Гц, 2H). MS (ESI, методика A): m/z=414.2 [M+H]+, tR=1.421 мин. ВЭЖХ: 100% (214 нм), 100% (254 нм).
Схема 5
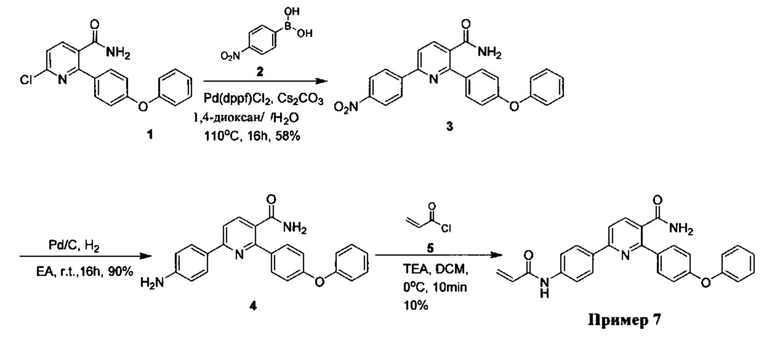
трет-Бутил-4-(6-хлор-5-цианопиридин-2-ил)пиперазин-1-карбоксилат (3). К раствору 6-хлор-2-(4-феноксифенил)никотинамида 1 (130 мг, 0.4 ммоль) и 4-нитрофенилбороновой кислоты 2 (67 мг, 0.4 ммоль) в DME (10 мл) / воде (3 мл) добавляли K2CO3 (110 мг, 0.8 ммоль) и Pd(dppf)Cl2 (33 мг, 0.04 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали в течение 5 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя 70:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (60 мг, сырое). MS (ESI): m/z=412.1 [М+Н]+.
6-(4-Аминофенил)-2-(4-феноксифенил)никотинамид (4). К раствору трет-бутил-4-(6-хлор-5-цианопиридин-2-ил)пиперазин-1-карбоксилата 3 (60 мг, 0.15 ммоль) в этилацетате (5 мл) добавляли Pd/C (6 мг) в атмосфере водорода, и смесь дегазировали водородом 6 раз, затем перемешивали в течение 16 ч при температуре окружающей среды в атмосфере водорода. Раствор фильтровали, и фильтрат упаривали до сырого продукта в виде твердого вещества коричневого цвета (42 мг, 90%). MS (ESI): m/z=382.0 [М+Н]+.
Пример 7
6-(4-Акриламидофенил)-2-(4-феноксифенил)никотинамид
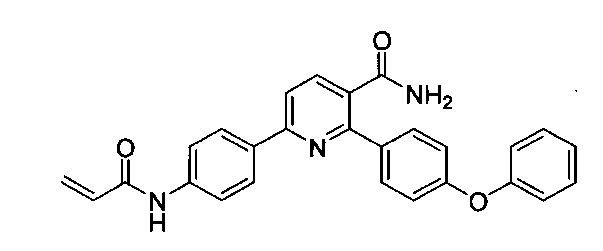
К раствору 6-(4-аминофенил)-2-(4-феноксифенил)никотинамида 4 (42 мг, 0.11 ммоль) в DCM (5 мл) добавляли TEA (0.05 мл, 0.4 ммоль) и акрилоилхлорид 5 (10 мг, 0.11 ммоль) при 0°.Смесь перемешивали при 0° в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (2.5 мг, 10%) в виде твердого вещества белого цвета. 1Н ЯМР (300 МГц, CD3OD) δ 8.17-8.11 (m, 1H), 7.98-7.92 (m, 1Н), 7.90-7.84 (m, 1Н), 7.81 (s, 3Н), 7.69-7.60 (m, 2Н), 7.58-7.52 (m, 1Н), 7.42-7.34 (m, 2Н), 7.18-7.12 (m, 1H), 7.10-7.00 (m, 3Н), 6.47-6.38 (m, 2Н), 5.82-5.76 (m, 1H). MS (ESI, методика A): m/z=435.9 [М+Н]+, tR=1.540 мин, ВЭЖХ: 97.5% (214 нм), 98.7% (254 нм).
Схема 6
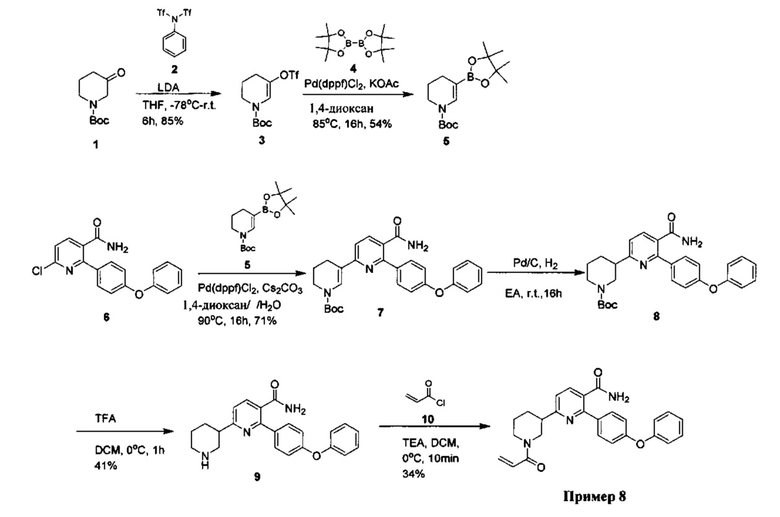
трет-Бутил-5-(трифторметилсульфонилокси)-3,4-дигидропиридин-1(2Н)-карбоксилат (3). К раствору трет-бутил-3-оксопиперидин-1-карбоксилата 1 (495 мг, 5.0 ммоль) в тетрагидрофуране (10 мл) в трехгорлой колбе добавляли диизопропиламид лития (2.0 М, 2.5 мл, 5.0 ммоль) при -78°. После перемешивания в течение 2 ч при -78° добавляли раствор трифтор-N-фенил-N-(трифторметилсульфонил)метансульфонамида 2 (1.8 г, 5.0 ммоль) в тетрагидрофуране (5 мл), и раствор перемешивали в течение еще 30 мин при этой же температуре, затем нагревали до комнатной температуры и перемешивали в течение 1 ч. Добавляли воду (30 мл), чтобы остановить реакцию, и раствор экстрагировали этилацетатом (3×30 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя петролейным эфиром с получением указанного в заголовке соединения в виде коричневого масла (1.4 г, 85%).
трет-Бутил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидропиридин-1(2Н)-карбоксилат (5). К раствору 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) 4 (559 мг, 2.2 ммоль), КОАс (588 мг, 6.0 ммоль) и трет-бутил-5-(трифторметилсульфонилокси)-3,4-дигидропиридин-1(2Н)-карбоксилата 3 (662 мг, 2.0 ммоль) в сухом 1,4-диоксане (10 мл) добавляли Pd(dppf)Cl2⋅DCM (326 мг, 0.4 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 85° и перемешивали на протяжении ночи в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя от 100:1 до 20:1 РЕ/ЕА, с получением указанного в заголовке соединения в виде коричневого масла (336 мг, 54%).
трет-Бутил-5-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)-3,4-дигидропиридин-1(2Н)-карбоксилат (7). К раствору трет-бутил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидропиридин-1(2Н)-карбоксилата 5 (260 мг, 0.84 ммоль), CS2CO3 (456 мг, 1.4 ммоль) и 6-хлор-2-(4-феноксифенил)никотинамида 6 (227 мг, 0.7 ммоль) в 1,4-диоксане (10 мл) добавляли Pd(dppf)Cl2⋅DCM (57 мг, 0.07 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали на протяжении ночи в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя от 100:1 до 20:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (240 мг, 71%). MS (ESI): m/z=472.2 [М+Н]+.
трет-Бутил-3-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пиперидин-1-карбоксилат (8). К раствору трет-бутил-5-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)-3,4-дигидропиридин-1(2Н)-карбоксилата 7 (240 мг, 0.5 ммоль) в этилацетате (5 мл) добавляли Pd/C (24 мг) в атмосфере водорода, и смесь дегазировали водородом 6 раз, затем перемешивали в течение 16 ч при температуре окружающей среды в атмосфере водорода. Раствор фильтровали, и фильтрат упаривали до сырого продукта в виде твердого вещества коричневого цвета (230 мг, сырое). MS (ESI): m/z=474.2 [М+Н]+.
2-(4-Феноксифенил)-6-(пиперидин-3-ил)никотинамид (9). К раствору трет-бутил-3-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пиперидин-1-карбоксилата 8 (150 мг, 0.32 ммоль) в сухом дихлорметане (6 мл) добавляли TFA (2 мл), и полученную в результате смесь перемешивали в течение 1 ч при температуре окружающей среды. Растворитель удаляли, и остаток разделяли между насыщенным водным бикарбонатом натрия (30 мл) и этилацетатом (20 мл). Органическую фазу отделяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Сырой остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя 5:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (77 мг, 71%). MS (ESI): m/z=374.2 [М+Н]+.
Пример 8
6-(1-Акрилоилпиперидин-3-ил)-2-(4-феноксифенил)никотинамид
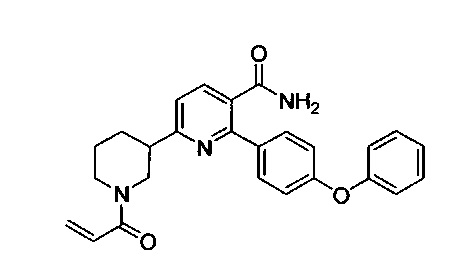
К раствору 2-(4-феноксифенил)-6-(пиперидин-3-ил)никотинамида 9 (19 мг, 0.05 ммоль) в DCM (5 мл) добавляли TEA (0.05 мл, 0.4 ммоль) и акрилоилхлорид 10 (5 мг, 0.05 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (7 мг, 34%) в виде твердого вещества белого цвета. 1Н ЯМР (300 МГц, CDCl3) δ 7.97 (d, J=8.9 Гц, 1Н), 7.69 (d, J=8.1 Гц, 2Н), 7.38 (t, J=7.8 Гц, 2Н), 7.24-7.15 (m, 2Н), 7.12-7.01 (m, 4Н), 6.62 (dd, J=16.6, 10.6 Гц, 1Н), 6.28 (d, J=16.4 Гц, 1Н), 5.75-5.60 (m, 2Н), 5.46 (s, 1Н), 4.89-4.55 (m, 1Н), 4.30-3.90 (m, 1Н), 3.50 (t, J=12.2 Гц, 0.5Н), 3.24-3.06 (m, 1H), 2.99 (d, J=10,7 Гц, 1Н), 2.80 (t, J=12.2 Гц, 0.5Н), 2.13 (s, 1Н), 1.87 (d, J=13.4 Гц, 2H), 1.64 (s, 1H). MS (ESI, методика A): m/z=428.0 [M+H]+, tR=1.526 мин, ВЭЖХ: 93.3% (214 нм), 99.8% (254 нм).
Схема 7
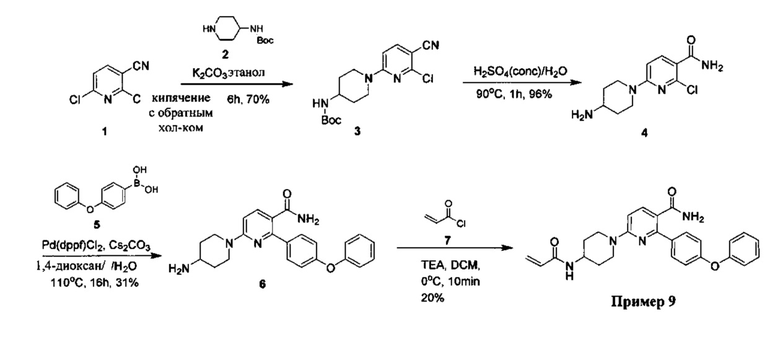
трет-Бутил-1-(6-хлор-5-цианопиридин-2-ил)пиперидин-4-илкарбамат (3). К раствору 2,6-дихлорникотинонитрила 1 (346 мг, 2.0 ммоль) в этаноле (10 мл) добавляли трет-бутил пиперидин-4-илкарбамат 2 (400 мг, 2.0 ммоль) и K2CO3 (552 мг, 4.0 ммоль). Смесь нагревали до кипения с обратным холодильником и перемешивали в течение 4 ч. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя от 20:1 до 5:1 РЕ/ЕА, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (470 мг, 70%). MS (ESI, методика A): m/z=337.2 [М+Н]+.
6-(4-Аминопиперидин-1-ил)-2-хлорникотинамид (4). К трет-бутил-1-(6-хлор-5-цианопиридин-2-ил)пиперидин-4-илкарбамату 3 (470 мг, 1.4 ммоль) добавляли H2SO4 (конц., 5 мл) и воду (1 мл). Смесь нагревали до 90° и перемешивали в течение 1 ч. После охлаждения до комнатной температуры раствор выливали в ледяную воду, затем доводили до значения рН=8 аммиачной водой. Осадок отфильтровывали, промывали водой (10 мл) и высушивали под вакуумом с получением указанного в заголовке соединения в виде твердого вещества белого цвета (340 мг, 96%). MS (ESI): m/z=255.0 [М+Н]+.
6-(4-Аминопиперидин-1-ил)-2-(4-феноксифенил)никотинамид (6). К раствору 6-(4-аминопиперидин-1-ил)-2-хлорникотинамида 4 (51 мг, 0.2 ммоль), K2CO3 (55 мг, 0.4 ммоль) и 4-феноксифенилбороновой кислоты 5 (43 мг, 0.2 ммоль) в 1,4-диоксане (15 мл) и воде (3 мл) добавляли Pd(dppf)Cl2 (18 мг, 0.02 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали в течение 5 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 70:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (24 мг, 31%). MS (ESI): m/z=389.2 [М+Н]+.
Пример 9
6-(4-Акриламидопиперидин-1-ил)-2-(4-феноксифенил)никотинамид
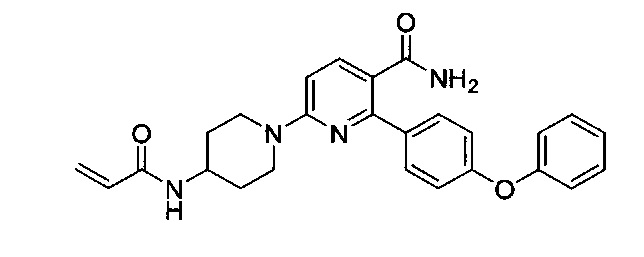
К раствору 6-(4-аминопиперидин-1-ил)-2-(4-феноксифенил)никотинамида 6 (62 мг, 0.16 ммоль) в DCM (5 мл) добавляли TEA (0.05 мл, 0.4 ммоль) и акрилоилхлорид 7 (15 мг, 0.16 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (14 мг, 20%) в виде твердого вещества белого цвета. 1Н ЯМР (300 МГц, CDCl3) δ 7.98 (d, J=8.8 Гц, 1Н), 7.62 (d, J=8.5 Гц, 2Н), 7.38 (t, J=7.8 Гц, 2Н), 7.16 (t, J=7.4 Гц, 1H), 7.12-7.00 (m, 4Н), 6.65 (d, J=8.9 Гц, 1Н), 6.31 (d,J=16.9 Гц, 1H), 6.07 (dd, J=16.9, 10.2 Гц, 1Н), 5.66 (d, J=10.2 Гц, 1Н), 5.52-5.48 (m, 1Н), 5.28 (s, 1Н), 4.44 (d, J=13.4 Гц, 2Н), 4.24-4.08 (m, 1Н), 3.08 (t, J=11.9 Гц, 2Н), 2.08 (d, J=10.7 Гц, 2Н), 1.62 (s, 1Н), 1.47 (dt, J=11.3, 6.1 Гц, 2H). MS (ESI, методика A): m/z=443.2 [M+H]+, tR=1.434 мин, ВЭЖХ: 99.1% (214 нм), 99.3% (254 нм).
Схема 8
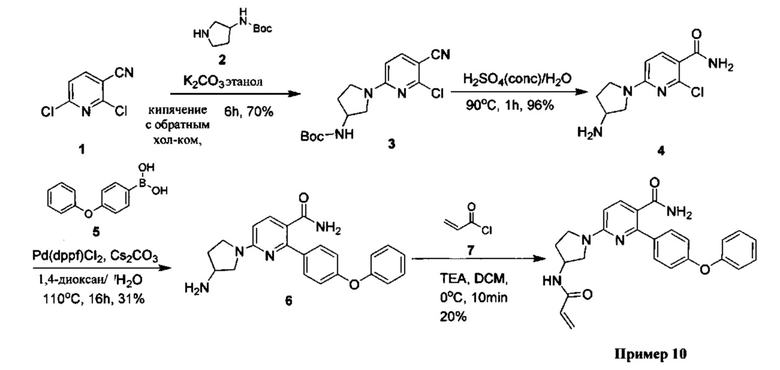
трет-Бутил-1-(6-хлор-5-цианопиридин-2-ил)пирролидин-3-илкарбамат (3). К раствору 2,6-дихлорникотинонитрила 1 (346 мг, 2.0 ммоль) в этаноле (10 мл) добавляли трет-бутил пиперидин-4-илкарбамат 2 (372 мг, 2.0 ммоль) и K2CO3 (552 мг, 4.0 ммоль). Смесь нагревали до кипения с обратным холодильником и перемешивали в течение 4 ч. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя от 20:1 до 5:1 РЕ/ЕА, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (365 мг, 57%). MS (ESI, методика A): m/z=345.1 [М+Na]+.
6-(4-Аминопиперидин-1-ил)-2-хлорникотинамид (4). К трет-бутил-1-(6-хлор-5-цианопиридин-2-ил)пирролидин-3-илкарбамату 3 (357 мг, 1.1 ммоль) добавляли H2SO4 (конц., 5 мл) и воду (1 мл). Смесь нагревали до 90° и перемешивали в течение 1 ч. После охлаждения до комнатной температуры раствор выливали в ледяную воду, затем доводили до значения рН=8 аммиачной водой. Осадок отфильтровывали, промывали водой (10 мл) и высушивали под вакуумом с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (192 мг, 72%). MS (ESI, методика A): m/z=241.0 [М+Н]+.
6-(3-Аминопирролидин-1-ил)-2-(4-феноксифенил)никотинамид (6). К раствору 6-(4-аминопиперидин-1-ил)-2-хлорникотинамида 4 (192 мг, 0.79 ммоль), K2CO3 (220 мг, 1.6 ммоль) и 4-феноксифенилбороновой кислоты 5 (171 мг, 0.8 ммоль) в 1,4-диоксане (15 мл) и воде (3 мл) добавляли Pd(dppf)Cl2 (69 мг, 0.08 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали в течение 5 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя 70:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (92 мг, 31%). MS (ESI): m/z=375.2 [М+Н]+.
Пример 10
6-(3-Акриламидопирролидин-1-ил)-2-(4-феноксифенил)никотинамид
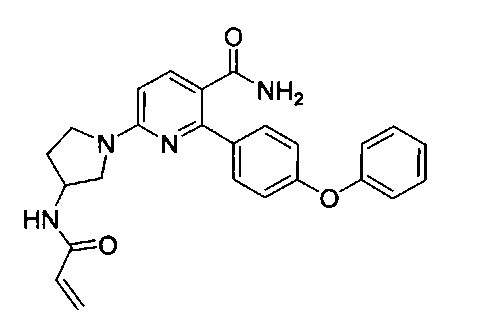
К раствору 6-(3-аминопирролидин-1-ил)-2-(4-феноксифенил)никотинамида 6 (92 мг, 0.25 ммоль) в DCM (5 мл) добавляли TEA (0.14 мл, 1.0 ммоль) и акрилоилхлорид 7 (23 мг, 0.25 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (58 мг, 54%) в виде твердого вещества белого цвета. 1Н ЯМР (300 МГц, CDCl3) δ 7.95 (d, J=8.7 Гц, 1Н), 7.62 (d, J=8.5 Гц, 2Н), 7.38 (t, J=7.8 Гц, 2Н), 7.16 (t, J=7.4 Гц, 1Н), 7.05 (dd, J=12.6, 8.3 Гц, 4H), 6.40-625 (m, 1H), 6.10 (dd, J=16.8, 10.2 Гц, 2H), 5.67 (d, J=10.2 Гц, 1H), 5.48 (s, 1H), 5.31 (s, 1H), 4.71 (d, J=4.4 Гц, 1H), 3.85 (dd, J=11.2, 6.0 Гц, 1H), 3.75-340 (m, 4H), 2.40-2.25 (m, 1H), 2.10-1.90 (m, 1H). MS (ESI, методика A): m/z=429.0 [M+H]+, tR=1.443 мин, ВЭЖХ: 100% (214 нм), 100% (254 нм).
Пример 11
1-(1-Акрилоилпиперидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
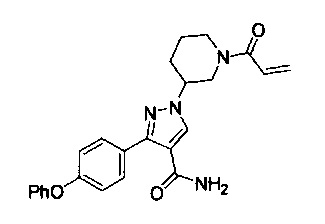
Смотри схемы Примера 34 и связанные с ними экспериментальные методики и данные, описанные ниже.
Пример 12
1-(1-Акрилоилпиперидин-4-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
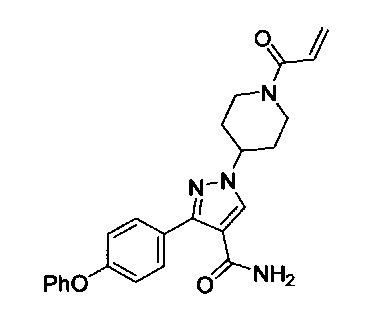
Смотри схемы Примера 34 и связанные с ними экспериментальные методики и данные, описанные ниже.
Пример 13
1-(1-Акрилоилпирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
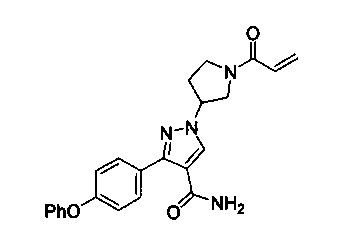
Смотри схемы Примера 34 и связанные с ними экспериментальные методики и данные, описанные ниже.
Пример 14
1-(Азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
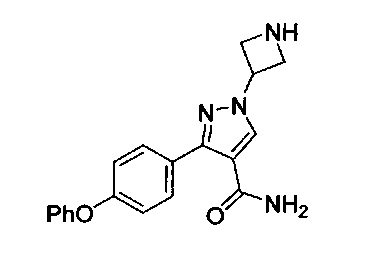
Смотри схемы Примера 34 и связанные с ними экспериментальные методики и данные, описанные ниже.
Схема 9
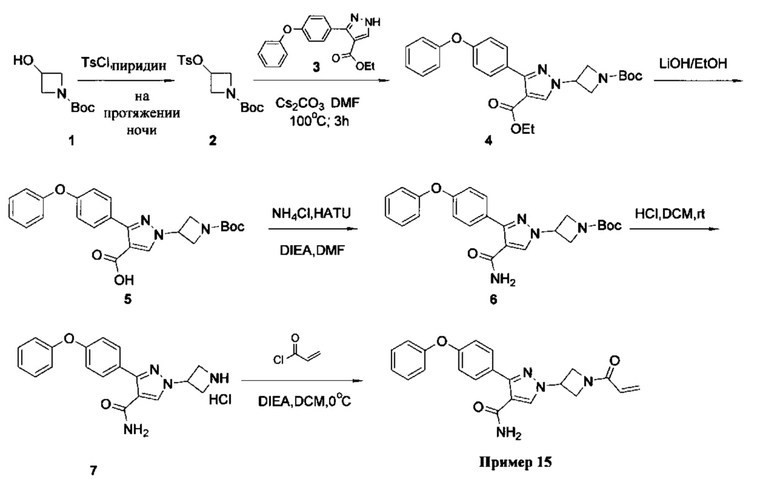
трет-Бутил-3-(тозилокси)азетидин-1-карбоксилат (2). К раствору трет-бутил-3-гидроксиазетидин-1-карбоксилата 1 (2.0 г, 11.5 ммоль) в пиридине (25 мл) добавляли TsCl (2.64 г, 13.8 ммоль), и полученный в результате раствор перемешивали в течение 16 ч при комнатной температуре. Растворитель выпаривали, и сырой остаток очищали на колонке с силикагелем, элюируя смесью 5:1 петролейный эфир/этилацетат, с получением указанного в заголовке соединения (3.2 г, 85%) в виде бесцветного масла. MS (ESI): m/z=350.0 [M+Na]+.
Этил-1-(1-(трет-бутоксикарбонил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксилат (4). К раствору трет-бутил-3-(тозилокси)азетидин-1-карбоксилата 2 (1.3 г, 3.88 ммоль) и CsCO3 (2.12 г, 6.48 ммоль) в DMF (25 мл) добавляли этил 3-(4-феноксифенил)-1Н-пиразол-4-карбоксилат 3 (1.0 г, 3.24 ммоль), и полученный в результате раствор перемешивали в течение 16 ч при 100°. Растворитель выпаривали, и сырой остаток очищали на колонке с силикагелем, элюируя смесью 3:1 петролейный эфир/этилацетат, с получением указанного в заголовке соединения (1.39 г, 93%) в виде твердого вещества белого цвета. MS (ESI): m/z=464.0 [М+Н]+.
1-(1-(трет-Бутоксикарбонил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоновая кислота (5). К раствору этил 1-(1-(трет-бутоксикарбонил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксилата 4 (1.39 г, 3.0 ммоль) в EtOH (20 мл) и воде (2 мл) добавляли LiOH (630 мг, 15.0 ммоль), и полученный в результате раствор перемешивали в течение 16 ч при комнатной температуре. Растворитель выпаривали, и остаток растворяли в воде (5 мл), и полученный в результате раствор подкисляли 2 н. соляной кислотой до рН=6. Осадок отфильтровывали, промывали водой (15 мл) и высушивали под вакуумом с получением указанного в заголовке соединения (1.2 г, 92%) в виде твердого вещества белого цвета. MS (ESI): m/z=436.0 [М+Н]+.
трет-Бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)азетидин-1-карбоксилат (6). К раствору 1-(1-(трет-бутоксикарбонил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоновой кислоты 5 (800 мг, 1.84 ммоль), NH4Cl (120 мг, 2.21 ммоль) и HATU (1.04 мг, 2.76 ммоль) в сухом DMF (10 мл) добавляли DIPEA (957 мг, 7.36 ммоль), и полученный в результате раствор перемешивали на протяжении ночи при комнатной температуре. После завершения реакции раствор концентрировали, разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке с силикагелем, элюируя 40:1 DCM/MeOH, с получением указанного в заголовке соединения (700 мг, 82%) в виде твердого вещества белого цвета. MS (ESI): m/z=435.0 [М+Н]+.
1-(Азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (7). К раствору 3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)азетидин-1-карбоксилата 6 (700 мг, 1.61 ммоль) в DCM (20 мл) добавляли конц. HCl (5 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После завершения реакции раствор концентрировали с получением указанного в заголовке соединения (600 мг, 100%) в виде твердого вещества белого цвета. MS (ESI): m/z=335.0 [М+Н]+.
Пример 15
1-(1-Акрилоилазетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
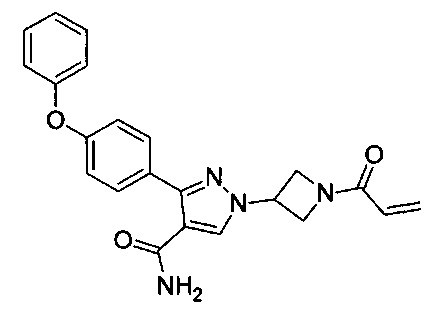
К раствору 1-(азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида 7 (300 мг, 0.81 ммоль) в сухом DCM (15 мл) добавляли DIEA (316 мг, 2.43 ммоль) и акрилоил хлорид (88 мг, 0.97 ммоль) при 0°, и полученный в результате раствор перемешивали при 0° в течение 10 мин. Добавляли воду (10 мл), чтобы остановить реакцию. Смесь разбавляли DCM (20 мл), и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (ACN-H2O=20-70, 0.1% FA) с получением указанного в заголовке соединения (160 мг, 51%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, DMSO) δ 8.38 (s, 1Н), 7.81 (d, J=8.7 Гц, 2Н), 7.47-7.38 (m, 3Н), 7.20-7.14 (m, 1Н), 7.11-6.99 (m, 5Н), 6.43-6.34 (m, 1Н), 6.20-6.12 (m, 1Н), 5.76-5.70 (m, 1Н), 5.38-5.29 (m, 1Н), 4.78-4.68 (m, 1Н), 4.59-4.49 (m, 1Н), 4.48-4.40 (m, 1Н), 4.32-4.22 (m, 1H). MS (ESI, методика A): m/z=389.1 [М+Н]+, tR=1.374 мин. ВЭЖХ: 99.7% (214 нм), 99.7% (254 нм).
Пример 16
1-(4-Акриламидофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
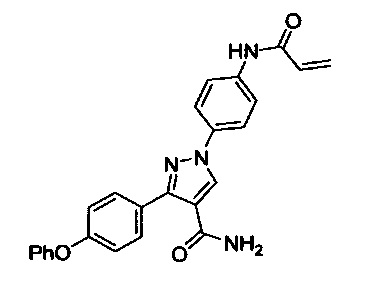
Смотри схемы Примера 34 и связанные с ними экспериментальные методики и данные, описанные ниже.
Пример 17
1-(3-Аминофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
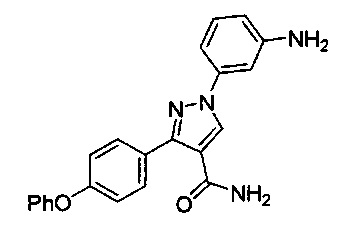
Смотри схемы Примера 34 и связанные с ними экспериментальные методики и данные, описанные ниже.
Пример 18
1-(3-Акриламидофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
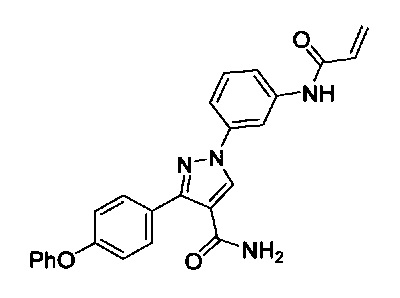
Смотри схемы Примера 34 и связанные с ними экспериментальные методики и данные, описанные ниже.
Пример 19
(S)-1-(1-акрилоилпирролидин-3-ил)-3-(4-феноксифенил)-1H-пиразол-4-карбоксамид
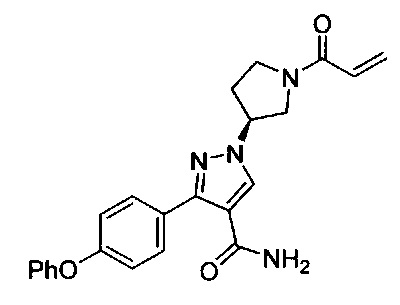
Смотри схемы Примера 34 и связанные с ними экспериментальные методики и данные, описанные ниже.
Пример 20
(R)-1-(1-акрилоилпирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
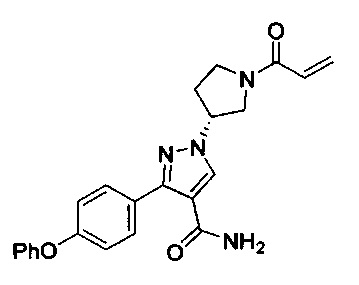
Смотри схемы Примера 34 и связанные с ними экспериментальные методики и данные, описанные ниже.
Схема 10
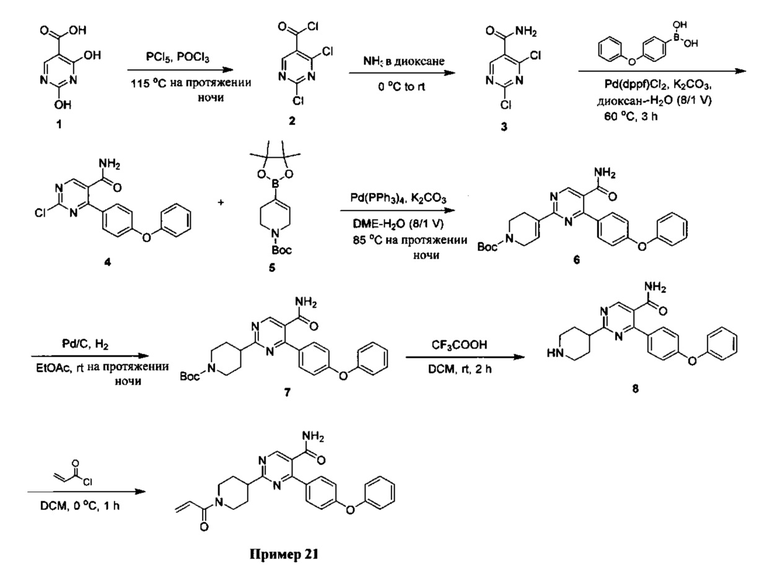
2,4-Дихлорпиримидин-5-карбонилхлорид (2). К раствору 2,4-дигидроксипиримидин-5-карбоновой кислоты (5.0 г, 32.0 ммоль) в POCl3 (50 мл) добавляли PCl5 (23.9 г, 115.2 ммоль), и полученный в результате раствор нагревали до 115° и перемешивали в течение 12 ч. Растворитель выпаривали, и сырой остаток разбавляли этилацетатом (100 мл). Твердое вещество отфильтровывали, и фильтрат концентрировали с получением указанного в заголовке соединения в виде коричневого масла (6.2 г, 92%).
2,4-Дихлорпиримидин-5-карбоксамид (3). К раствору 2,4-дихлорпиримидин-5-карбонилхлорида (6.2 г, 29.3 ммоль) в 1,4-диоксане (50 мл) добавляли по каплям аммиак (1.0 г, 58.3 ммоль) в 1,4-диоксане (50 мл) при 0°, и полученный в результате раствор перемешивали в течение 12 ч при температуре окружающей среды. Смесь разбавляли этилацетатом (50 мл) и промывали водой (2×80 мл) и рассолом (2×80 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения в виде твердого вещества белого цвета (4.5 г, 80%). MS (ESI): m/z=191.8 [М+Н]+.
2-Хлор-4-(4-феноксифенил)пиримидин-5-карбоксамид (4). К раствору 4-феноксифенилбороновой кислоты (1.0 г, 4.7 ммоль), K2CO3 (1.4 г, 10.4 ммоль) и 2,4-дихлорпиримидин-5-карбоксамида (1.0 г, 5.2 ммоль) в 1,4-диоксане (24 мл) и воде (3 мл) добавляли Pd(dppf)Cl2 (380 мг, 0.52 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 60° и перемешивали в течение 1 ч в атмосфере азота. После охлаждения до комнатной температуры раствор выливали в воду (50 мл), и затем экстрагировали этилацетатом (3×40 мл). Объединенные органические слои высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали с помощью колоночной хроматографии, элюируя смесью 100:1 дихлорметан/метанол с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (410 мг, 24%). MS (ESI): m/z=325.9 [М+Н]+.
трет-Бутил-4-(5-карбамоил-4-(4-феноксифенил)пиримидин-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат (6). К раствору 2-хлор-4-(4-феноксифенил)пиримидин-5-карбоксамида 4 (410 мг, 1.26 ммоль), K2CO3 (521.6 мг, 3.78 ммоль) и трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата 5 (583 мг, 1.89 ммоль) в DME (30 мл) и воде (5 мл) добавляли Pd(PPh3)4 (146 мг, 0.126 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз и затем нагревали до 85° и перемешивали в течение 12 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя смесью 80:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (420 мг, 71%). MS (ESI): m/z=472.8 [М+Н]+.
трет-Бутил-4-(5-карбамоил-4-(4-феноксифенил)пиримидин-2-ил)пиперидин-1-карбоксилат (7). К раствору трет-бутил-4-(5-карбамоил-4-(4-феноксифенил)пиримидин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата 6 (100 мг, 0.212 ммоль) в этилацетате (5 мл) добавляли Pd/C (20 мг) в атмосфере водорода, и смесь дегазировали водородом 6 раз, затем перемешивали в течение 16 ч при температуре окружающей среды в атмосфере водорода. Раствор фильтровали, и фильтрат упаривали до сырого продукта в виде твердого вещества коричневого цвета (96 мг). MS (ESI): m/z=474.8 [М+Н]+.
4-(4-Феноксифенил)-2-(пиперидин-4-ил)пиримидин-5-карбоксамид (8). К раствору трет-бутил-4-(5-карбамоил-4-(4-феноксифенил)пиримидин-2-ил)пиперидин-1-карбоксилата 7 (96 мг, сырое) в сухом дихлорметане (2 мл) добавляли TFA (2 мл), и полученную в результате смесь перемешивали в течение 1 ч при температуре окружающей среды. Растворитель удаляли, и остаток разделяли между насыщенным водным бикарбонатом натрия (30 мл) и этилацетатом (20 мл). Органическую фазу отделяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Сырой остаток очищали с помощью флэш-хроматографии, элюируя смесью 5:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (70 мг, 88% за две стадии). MS (ESI): m/z=374.9 [М+Н]+.
Пример 21
2-(1-Акрилоилпиперидин-4-ил)-4-(4-феноксифенил)пиримидин-5-карбоксамид
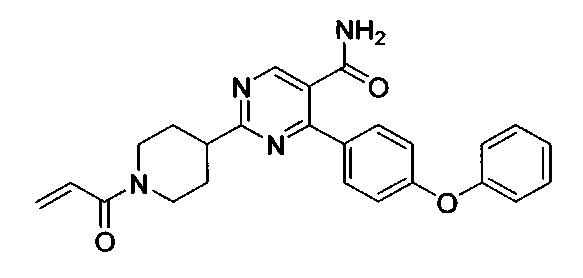
К раствору 4-(4-феноксифенил)-2-(пиперидин-4-ил)пиримидин-5-карбоксамида 8 (70 мг, 0.187 ммоль) в DCM (3 мл) добавляли TEA (56.7 мг, 0.56 ммоль) и акрилоилхлорид (25.3 мг, 0.28 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 мин. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (50 мг, 63%) в виде твердого вещества светло-желтого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 8.95 (s, 1Н), 7.82 (d, J=8.8 Гц, 2H), 7.49-7.36 (m, 2H), 7.27-7.17 (m, 1H), 7.16-7.04 (m, 4H), 6.64 (dd, J=16.8, 10.6 Гц, 1H), 6.29 (dd, J=16.8, 1.9 Гц, 1H), 5.94 (s, 1H), 5.77 (s, 1H), 5.71 (dd, J=10.6, 1.9 Гц, 1H), 4.76 (d, J=12.5 Гц, 1H), 4.14 (d, J=14.1 Гц, 1H), 3.37-3.19 (m, 2H), 2.89 (t, J=11.5 Гц, 1H), 2.16 (d, J=11.0 Гц, 2H), 1.97 (d, J=9.9 Гц, 2H). MS (ESI, методика F): m/z=428.8 [M+H]+, tR=1.407 мин, ВЭЖХ: 99.8% (214 нм), 99.6% (254 нм).
Схема 11
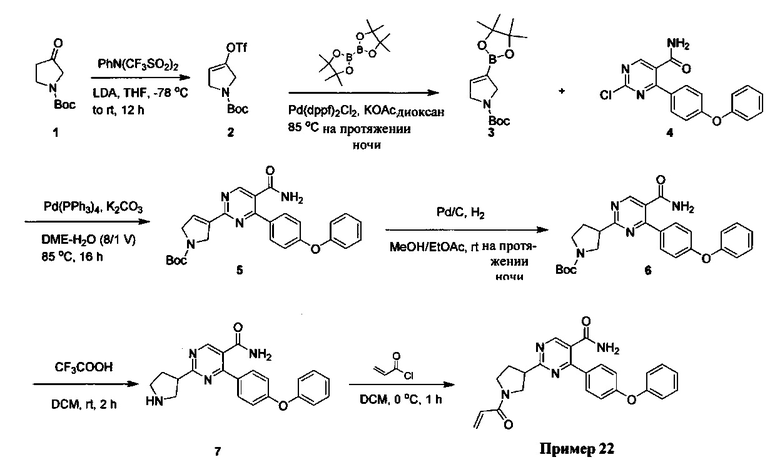
1-(трет-Бутоксикарбонил)-2,5-дигидро-1Н-пиррол-3-ил-трифторметансульфонат (2). К раствору трет-бутил-3-оксопирролидин-1-карбоксилата 1 (5.0 г, 27.0 ммоль) в тетрагидрофуране (130 мл) в трехгорлой колбе добавляли диизопропиламид лития (2.0 М, 16.2 мл, 32.4 ммоль) при -78°. После перемешивания в течение 2 ч при -78° добавляли раствор трифтор-N-фенил-N-(трифторметилсульфонил)метансульфонамида (10.1 г, 28.4 ммоль) в тетрагидрофуране (20 мл), и раствор перемешивали в течение еще 30 мин при этой же температуре, затем нагревали до комнатной температуры и перемешивали в течение 1 ч. Добавляли воду (30 мл), чтобы остановить реакцию, и раствор экстрагировали этилацетатом (3×200 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя петролейным эфиром, с получением указанного в заголовке соединения в виде коричневого масла (4.2 г, 49%). MS (ESI): m/z=318.1 [М+Н]+.
трет-Бутил-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2H-пиррол-1(5H)-карбоксилат (3). К раствору бис(пинаколато)диборона (4.0 г, 15.9 ммоль), КОАс (2.59 г, 26.4 ммоль) и 1-(трет-бутоксикарбонил)-2,5-дигидро-1H-пиррол-3-ил трифторметансульфоната 2 (4.2 г, 13.2 ммоль) в сухом 1,4-диоксане (10 мл) добавляли Pd(dppf)Cl2 (965 мг, 1.32 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 85° и перемешивали на протяжении ночи в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя от 100:1 до 20:1 петролейным эфиром/этилацетатом с получением указанного в заголовке соединения в виде коричневого масла (1.2 г, 31%). MS (ESI): m/z=296.1 [M+H]+.
трет-Бутил-3-(5-карбамоил-4-(4-феноксифенил)пиримидин-2-ил)-2H-пиррол-1(5H)-карбоксилат (5). К раствору трет-бутил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2H-пиррол-1(5H)-карбоксилата 3 (136 мг, 0.46 ммоль), K2CO3 (127 мг, 0.92 ммоль) и 2-хлор-4-(4-феноксифенил)пиримидин-5-карбоксамида 4 (100 мг, 0.31 ммоль) в 1,4-диоксане (15 мл) и воде (3 мл) добавляли Pd(PPh3)4 (35.5 мг, 0.031 ммоль) в атмосфере азота. Смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали в течение 12 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя смесью 70:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (90 мг, 64%). MS (ESI): m/z=458.8 [М+Н]+.
трет-Бутил-3-(5-карбамоил-4-(4-феноксифенил)пиримидин-2-ил)пирролидин-1-карбоксилат (6). К раствору трет-бутил-3-(5-карбамоил-4-(4-феноксифенил)пиримидин-2-ил)-2H-пиррол-1(5H)-карбоксилата 5 (90 мг, 0.20 ммоль) в этилацетате (3 мл) и МеОН (3 мл) добавляли Pd/C (20 мг) в атмосфере водорода, и смесь дегазировали водородом 6 раз, затем перемешивали в течение 16 ч при температуре окружающей среды в атмосфере водорода. Раствор фильтровали, и фильтрат упаривали до сырого продукта в виде твердого вещества коричневого цвета (80 мг, 88%). MS (ESI): m/z=461.1 [М+Н]+.
4-(4-Феноксифенил)-2-(пирролидин-3-ил)пиримидин-5-карбоксамид (7). К раствору трет-бутил-3-(5-карбамоил-4-(4-феноксифенил)пиримидин-2-ил)пирролидин-1-карбоксилата 6 (80 мг, 0.174 ммоль) в сухом дихлорметане (2 мл) добавляли TFA (2 мл), и полученную в результате смесь перемешивали в течение 1 ч при температуре окружающей среды. Растворитель удаляли, и остаток разделяли между насыщенным водным бикарбонатом натрия (30 мл) и этилацетатом (20 мл). Органическую фазу отделяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения в виде твердого вещества белого цвета (70 мг, сырое). MS (ESI): m/z=360.8 [М+Н]+.
Пример 22
2-(1-Акрилоилпирролидин-3-ил)-4-(4-феноксифенил)пиримидин-5-карбоксамид
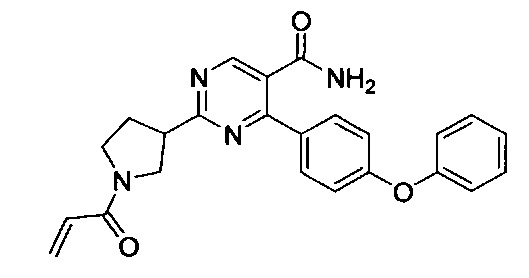
К раствору 4-(4-феноксифенил)-2-(пирролидин-3-ил)пиримидин-5-карбоксамида 7 (70 мг, сырое) в DCM (5 мл) добавляли TEA (59 мг, 0.58 ммоль) и акрилоилхлорид (26 мг, 0.29 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 мин. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (20 мг, 25%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 8.95 (d, J=6.2 Гц, 1Н), 7.82 (d, J=6.6 Гц, 2H), 7.42 (t, J=7.8 Гц, 2H), 7.22 (t, J=7.4 Гц, 1H), 7.10 (t, J=7.9 Гц, 4H), 6.55-6.47 (m, 1H), 6.39 (d, J=16.8, Гц, 1H), 5.84 (s, 2H), 5.71 (d, J=10.1 Гц, 1H), 4.14-3.96 (m, 2H), 3.94-3.78 (m, 2H), 3.76-3.62 (m, 1H), 2.55-2.36 (m, 2H). MS (ESI, методика F): m/z=414.8 [M+H]+, tR=1.382 мин, ВЭЖХ: 99.0% (214 нм), 98.4% (254 нм).
Схема 12
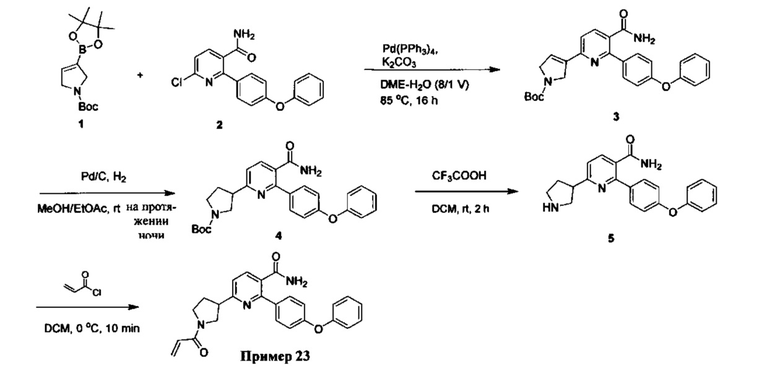
трет-Бутил-3-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)-2H-пиррол-1(5H)-карбоксилат (3). К раствору трет-бутил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2H-пиррол-1(5H)-карбоксилата 1 (136 мг, 0.0.46 ммоль), K2CO3 (127.5 мг, 0.92 ммоль) и 6-хлор-2-(4-феноксифенил)никотинамида 2 (100 мг, 0.31 ммоль) в 1,2-диметоксиэтане (5 мл) и воде (1 мл) добавляли Pd(PPh3)4 (35.6 мг, 0.031 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 85° и перемешивали в течение 12 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя смесью 50:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (84 мг, 60%). MS (ESI): m/z=458.1 [М+Н]+.
трет-Бутил-5-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пирролидин-1-карбоксилат (4). К раствору трет-бутил-5-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)-2H-пиррол-1(5H)-карбоксилата 3 (84 мг, 0.18 ммоль) в этилацетате (5 мл) и МеОН (5 мл) добавляли Pd/C (40 мг) в атмосфере водорода, и смесь дегазировали водородом 6 раз, затем перемешивали в течение 12 ч при температуре окружающей среды в атмосфере водорода. Раствор фильтровали, и фильтрат упаривали до сырого продукта в виде твердого вещества коричневого цвета (80 мг, 95%). MS (ESI): m/z=459.8 [М+Н]+.
2-(4-Феноксифенил)-6-(пирролидин-3-ил)пиридин-3-карбоксамид (5). К раствору трет-бутил-3-(5-карбамоил-6-(4-феноксифенил)пиридин-2-ил)пирролидин-1-карбоксилата 4 (80 мг, 0.17 ммоль) в сухом дихлорметане (2 мл) добавляли TFA (2 мл), и полученную в результате смесь перемешивали в течение 1 ч при температуре окружающей среды. Растворитель удаляли, и остаток разделяли между насыщенным водным бикарбонатом натрия (30 мл) и этилацетатом (20 мл). Органическую фазу отделяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения в виде твердого вещества белого цвета (70 мг, сырое). MS (ESI): m/z=359.9 [М+Н]+.
Пример 23
6-(1-Акрилоилпирролидин-3-ил)-2-(4-феноксифенил)пиридин-3-карбоксамид
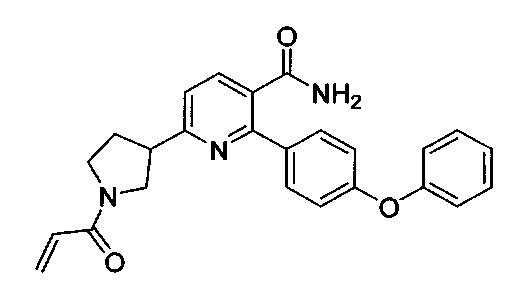
К раствору 2-(4-феноксифенил)-6-(пирролидин-3-ил)пиридин-3-карбоксамида 5 (70 мг, сырое) в DCM (5 мл) добавляли TEA (59 мг, 0.58 ммоль) и акрилоилхлорид (26.4 мг, 0.29 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 мин. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (18 мг, 25% за две стадии) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 8.06 (s, 1H), 7.72 (s, 2H), 7.41 (t, J=7.8 Гц, 2H), 7.28-7.24 (m, 1H), 7.19 (t, J=7.4 Гц, 1H), 7.11-7.09 (m, 4H), 6.55-6.47 (m, 1H), 6.41 (d, J=16.8, Гц, 1H), 5.71 (d, J=10.1 Гц, 1H), 5.65 (br, 1H), 5.45 (br, 1H), 4.14-3.96 (m, 1H), 3.94-3.78 (m, 2H), 3.76-3.62 (m, 2H), 2.55-2.36 (m, 2H). MS (ESI, методика F): m/z=413.8 [M+H]+, tR=1.393 мин, ВЭЖХ: 97.3% (214 нм), 99.5% (254 нм).
Схема 13
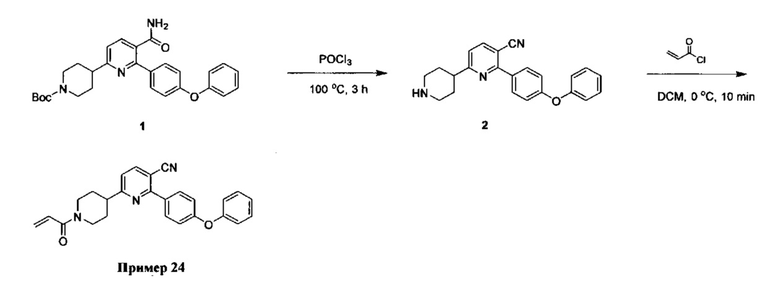
2-(4-Феноксифенил)-6-(пиперидин-4-ил)пиридин-3-карбонитрил (2). Раствор трет-бутил-4-(5-карбамоил-4-(4-феноксифенил)пиримидин-2-ил) пиперидин-1-карбоксилата (100 мг, 0.21 ммоль) в POCl3 (3 мл) нагревали до 100° и перемешивали в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь выливали в воду (3 мл), и подщелачивали насыщенным водным NaHCO3 до pH=10, затем раствор экстрагировали этилацетатом (3×60 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Сырой остаток очищали с помощью препаративной TLC, элюируя смесью 8:1 DCM/MeOH, с получением указанного в заголовке соединения в виде коричневого масла (10 мг, 13%). MS (ESI): m/z=356.1 [М+Н]+.
Пример 24
6-(1-Акрилоилпиперидин-4-ил)-2-(4-феноксифенил)пиридин-3-карбонитрил
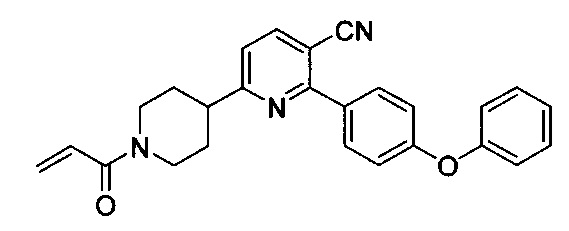
К раствору 2-(4-феноксифенил)-6-(пиперидин-4-ил)пиридин-3-карбонитрила 2 (10 мг, 0.028 ммоль) в DCM (2 мл) добавляли TEA (8.5 мг, 0.084 ммоль) и акрилоилхлорид (3.8 мг, 0.042 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 мин. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (13 мг, 44%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 8.01 (d, J=8.1 Гц, 1H), 7.97 (dd, J=9.2, 2.3 Гц, 2H), 7.41 (t, J=7.9 Гц, 2H), 7.24-7.17 (m, 2H), 7.16-7.09 (m, 4H), 6.64 (dd, J=16.8, 10.6 Гц, 1H), 6.32 (dd, J=16.8, 1.8 Гц, 1H), 5.73 (dd, J=10.6, 1.9 Гц, 1H), 4.85-4.79 (m, 1H), 4.21-4.16 (m, 1H), 3.25-4.18 (m, 1H), 3.12 (tt, J=12.0, 3.8 Гц, 1H), 2.84-2.78 (m, 1H), 2.09-2.06 (m, 2H), 1.93-1.82 (m, 2H). MS (ESI, методика F): m/z=409.8 [M+H]+, tR=1.669 мин, ВЭЖХ: 98.7% (214 нм), 98.8% (254 нм).
Схема 14
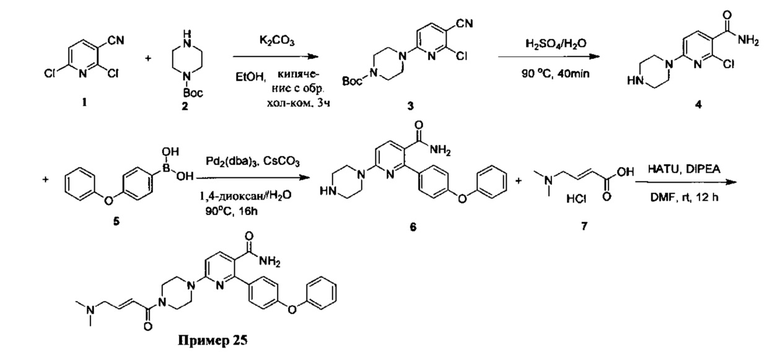
трет-Бутил-4-(6-хлор-5-цианопиридин-2-ил)пиперазин-1-карбоксилат (3). К раствору 2,6-дихлорникотинонитрила 1 (1.0 г, 5.78 ммоль) и трет-бутил-пиперазин-1-карбоксилата 2 (1.08 г, 5.78 ммоль) в этаноле (20 мл) добавляли K2CO3 (636 мг, 6.0 ммоль). Полученный в результате раствор нагревали до кипения с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя от 5:1 до 2:1 РЕ/ЕА, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (800 мг, 43%). MS (ESI): m/z=345.1 [M+Na]+.
6-Хлор-2-(4-феноксифенил)никотинамид (4). К трет-бутил-(6-хлор-5-цианопиридин-2-ил)пиперазин-1-карбоксилату 3(800 мг, 2.5 ммоль) добавляли конц. H2SO4 (5 мл) и воду (1 мл). Смесь нагревали до 90° и перемешивали в течение 40 мин. После охлаждения до комнатной температуры раствор выливали в ледяную воду, затем доводили до значения pH=8 с помощью водного аммиака. Осадок отфильтровывали, промывали водой (20 мл) и высушивали под вакуумом с получением указанного в заголовке соединения в виде твердого вещества не совсем белого цвета (580 мг, 96%). MS (ESI): m/z=241.1 [М+Н]+.
2-(4-Феноксифенил)-6-(пиперазин-1-ил)никотинамид (6). К раствору 6-хлор-2-(4-феноксифенил)никотинамида 4 (580 мг, 2.41 ммоль), CS2CO3 (2.35 г, 7.23 ммоль) и 4-феноксифенилбороновой кислоты 5 (619 мг, 2.89 ммоль) в 1,4-диоксане (15 мл) и воде (3 мл) добавляли Pd2(dbа)3 (220 мг, 0.24 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали в течение 12 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя смесью 10:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (480 мг, 52%). MS (ESI): m/z=375.1 [М+Н]+.
Пример 25
(Е)-6-(4-(4-(диметиламино)бут-2-еноил)пиперазин-1-ил)-2-(4-феноксифенил)никотинамид
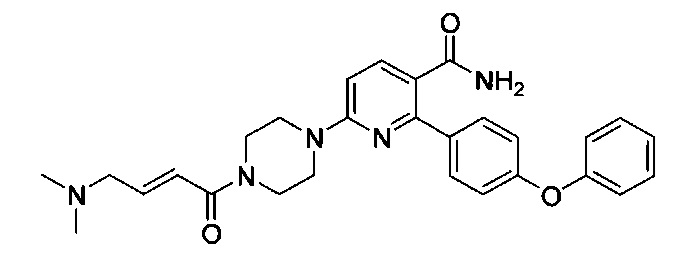
Смесь 2-(4-феноксифенил)-6-(пиперазин-1-ил)никотинамида 6 (100 мг, 0.27 ммоль), (Е)-4-(диметиламино)бут-2-еноевой кислоты гидрохлорида 7 (53 мг, 0.32 ммоль), HATU (152 мг, 0.4 ммоль) и DIPEA (172.5 мг, 1.34 ммоль) в DMF (10 мл) перемешивали при комнатной температуре в течение 12 ч. Раствор выливали в воду (50 мл), и затем экстрагировали этилацетатом (3×50 мл). Объединенные органические слои высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью препаративной TLC, элюируя смесью 10:1 DCM/MeOH, с получением указанного в заголовке соединения (30 мг, 23%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, MeOD) δ 7.78 (d, J=8.7 Гц, 1H), 7.69 (d, J=8.6 Гц, 2Н), 7.40 (t, J=7.9 Гц, 2Н), 7.17 (d, J=7.4 Гц, 1Н), 7.04 (dd, J=15.3, 8.3 Гц, 4Н), 6.93 (d, J=15.3 Гц, 1Н), 6.82 (d, J=8.7 Гц, 1Н), 6.77 (m, 1H), 3.89-3.68 (m, 10Н), 2.76 (s, 6Н). MS (ESI, методика F): m/z=485.9 [М+Н]+, tR=1.249 мин, ВЭЖХ: 93.3% (214 нм), 93.0% (254 нм).
Схема 15
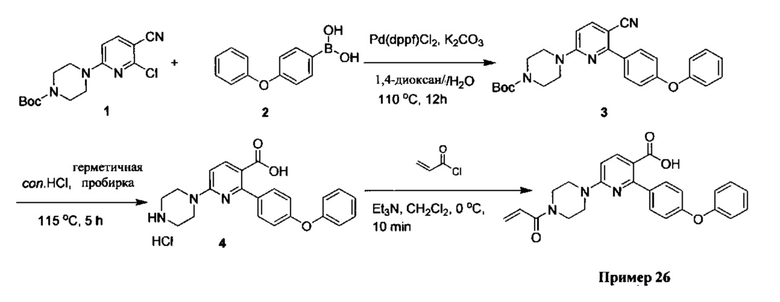
2-(4-Феноксифенил)-6-(пиперазин-1-ил)никотинамид (3). К раствору трет-бутил-4-(6-хлор-5-цианопиридин-2-ил)пиперазин-1-карбоксилата 1 (300 мг, 0.93 ммоль), K2CO3 (385 мг, 2.79 ммоль) и 4-феноксифенилбороновой кислоты 2 (297 мг, 1.39 ммоль) в 1,4-диоксане (12 мл) и воде (2 мл) добавляли Pd(dppf)Cl2 (68 мг, 0.093 ммоль) в атмосфере азота. Смесь дегазировали азотом 6 раз, затем нагревали до 90° и перемешивали в течение 12 ч в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью флэш-хроматографии, элюируя смесью 20:1 DCM/MeOH, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (260 мг, 58%). MS (ESI): m/z=457.1 [М+Н]+.
2-(4-Феноксифенил)-6-(пиперазин-1-ил)никотиновой кислоты гидрохлорид (4). В 20 мл герметичную пробирку помещали раствор 2-(4-феноксифенил)-6-(пиперазин-1-ил)никотинамида 3 (140 мг, 0.31 ммоль) в конц. HCl (5 мл). Смесь нагревали до 115° и перемешивали в течение 4 ч. После охлаждения до комнатной температуры раствор концентрировали под вакуумом с получением указанного в заголовке соединения в виде твердого вещества не совсем белого цвета (120 мг, 95%). MS (ESI): m/z=376.1 [М+Н]+.
Пример 26
6-(4-Акрилоилпиперазин-1-ил)-2-(4-феноксифенил)пиридин-3-карбоновая кислота
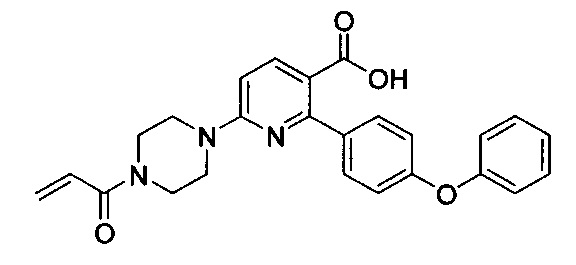
К раствору 2-(4-феноксифенил)-6-(пиперазин-1-ил)никотиновой кислоты гидрохлорида 4 (120 мг, 0.32 ммоль) в DCM (5 мл) добавляли TEA (162 мг, 1.6 ммоль) и акрилоилхлорид (35 мг, 0.38 ммоль) при 0°. Смесь перемешивали при 0° в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 20:1 DCM/MeOH, с получением указанного в заголовке соединения (15 мг, 11%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 8.17 (d, J=8.9 Гц, 1Н), 7.55 (d, J=8.3 Гц, 2H), 7.38 (t, J=7.7 Гц, 2H), 7.16 (t, J=7.4 Гц, 1H), 7.11 (d, J=8.0 Гц, 2H), 7.03 (d, J=8.3 Гц, 2H), 6.65-6.55(m, 2H), 6.38 (d, J=16.7 Гц, 1H), 5.78 (d, J=10.6 Гц, 1H), 3.86-375 (m, 8H). MS (ESI, методика F): m/z=429.8 [M+H]+, tR=1.477 мин, ВЭЖХ: 93.5% (214 нм), 93.0% (254 нм).
Схема 16
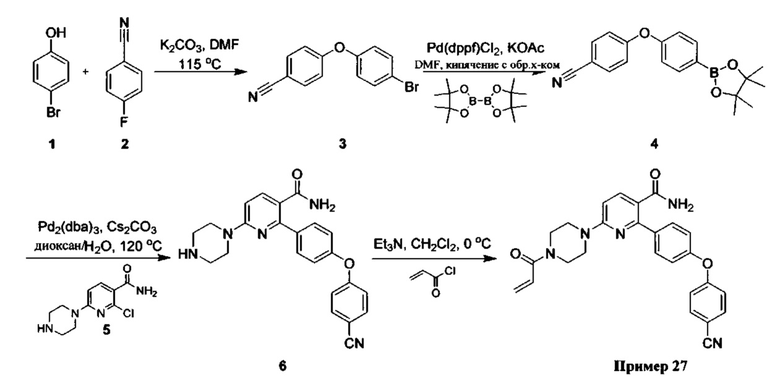
4-(4-Бромфенокси)бензонитрил (3). Смесь 4-бромфенола 1 (0.50 г, 2.9 ммоль), 4-фторбензонитрила 2 (0.28 г, 2.31 ммоль), K2CO3 (0.80 г, 5.8 ммоль) и DMF (4 мл) перемешивали при 115° в течение 16 ч. После охлаждения до комнатной температуры смесь концентрировали под вакуумом. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью 100:1 петролейный эфир/EtOAc с получением указанного в заголовке соединения (0.59 г, 74%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, DMSO-d6) δ 7.86 (d, J=8.7 Гц, 2Н), 7.64 (d, J=6.0 Гц, 2Н), 7.13 (dd, J=5.1, 4.7 Гц, 4Н).
4-(4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)бензонитрил (4). Смесь 4-(4-бромфенокси)бензонитрила 3 (0.59 г, 2.15 ммоль), бис(пинаколато)диборона (1.09 г, 4.3 ммоль), КОАс (0.63 г, 6.45 ммоль), Pd(dppf)Cl2 (0.157 г, 0.215 ммоль) и DMF (3.5 мл) дегазировали N2 6 раз и затем перемешивали при кипении с обратным холодильником в течение 16 ч. После охлаждения до комнатной температуры смесь концентрировали под вакуумом. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью 50:1 петролейный эфир/EtOAc, с получением указанного в заголовке соединения (0.55 г, 79%) в виде твердого вещества белого цвета.
2-(4-(4-Цианофенокси)фенил)-6-(пиперазин-1-ил)никотинамид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 2-(4-(гидрокси(фенил)метил)фенил)-6-(пиперидин-4-ил)никотинамида (смотри Схему 48) в виде твердого вещества желтого цвета (0.15 г, 37%). MS (ESI): m/z=400.1 [М+Н]+.
Пример 27
6-(4-Акрилоилпиперазин-1-ил)-2-(4-(4-цианофенокси)фенил)никотинамид
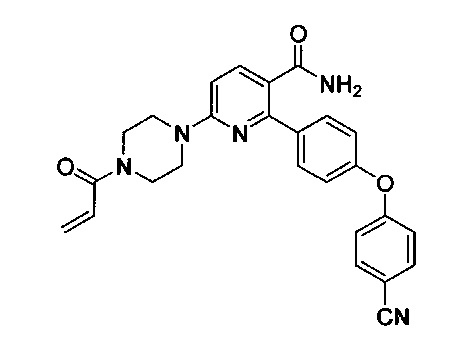
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (40 мг, 23%). 1Н ЯМР (400 МГц, DMSO-d6) δ 7.88 (d, J=8.7 Гц, 2Н), 7.73 (d,J=8.6 Гц, 2Н), 7.68 (d, J=8.7 Гц, 1Н), 7.59 (s, 1Н), 7.26 (s, 1H), 7.21-7.14 (m, 4Н), 6.86 (dd, J=17.8, 9.3 Гц, 2Н), 6.16 (dd, J=16.7, 2.2 Гц, 1H), 5.73 (dd, J=10.4, 2.2 Гц, 1Н), 3.66-3.67 (m, 8Н). MS (ESI, методика A): m/z=454.1 [М+Н]+, tR=1.685 (мин). ВЭЖХ: 97.1% (214 нм), 99.7% (254 нм).
Схема 17
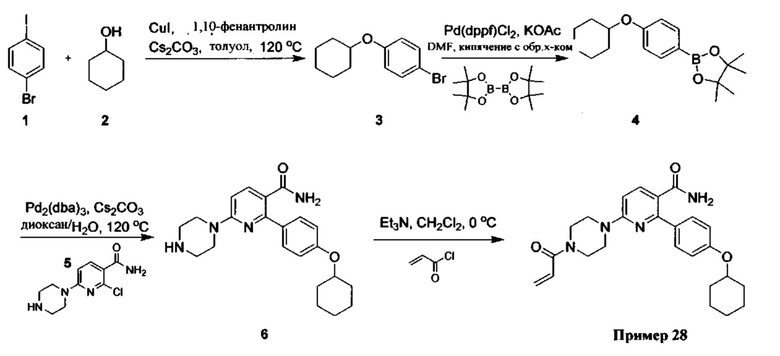
1-Бром-4-(циклогексилокси)бензол (3). Смесь 1-бром-4-иодбензола 1 (2.83 г, 10 ммоль), циклогексанола 2 (5.0 г, 50 ммоль), CuI (0.381 г, 2.0 ммоль), 1,10-фенантролина (0.793 г, 4.0 ммоль), CS2CO3 (8.15 г, 25 ммоль) и толуола (5 мл) перемешивали при 120° в герметичной пробирке в атмосфере N2 в течение 16 ч. После охлаждения до комнатной температуры смесь фильтровали через слой целита. Фильтрат высушивали над Na2SO4, концентрировали под вакуумом и очищали с помощью хроматографии на силикагеле, элюируя смесью 20:1 петролейный эфир/EtOAc с получением указанного в заголовке соединения (1.17 г, 46%) в виде бесцветного масла. 1Н ЯМР (400 МГц, CDCl3) δ 7.40-7.33 (m, 2Н), 6.83-6.77 (m, 2Н), 4.26-4.16 (m, 1Н), 2.04-1.92 (m, 2Н), 1.87-1.75 (m, 2Н), 1.65-1.47 (m, 3Н), 1.45-1.25 (m, 3Н).
2-(4-(Циклогексилокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)бензонитрила (смотри Схему 16) в виде коричневого масла (1.04 г, 75%). 1Н ЯМР (400 МГц, CDCl3) δ 7.75 (d, J=8.5 Гц, 2Н), 6.91 (d, J=8.5 Гц, 2Н), 4.40-4.25 (m, 1Н), 2.09-1.94 (m, 2Н), 1.89-1.76 (m, 2Н), 1.66-1.49 (m, 4Н), 1.48-1.23 (m, 14Н).
2-(4-(Циклогексилокси)фенил)-6-(пиперазин-1-ил)никотинамид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида (смотри Схему 1) в виде коричневой смолы (0.157 г, 41%). MS (ESI): m/z=381.1 [М+Н]+.
Пример 28
6-(4-Акрилоилпиперазин-1-ил)-2-(4-(циклогексилокси)фенил)никотинамид
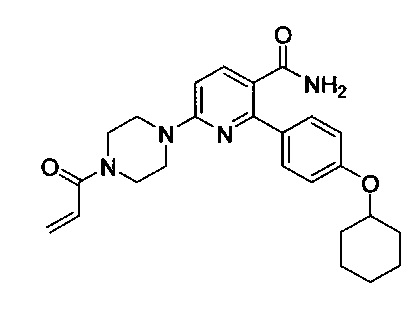
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (61 мг, 34%). 1Н ЯМР (400 МГц, CD3OD) δ 7.76 (d, J=8.7 Гц, 1Н), 7.63 (d, J=8.7 Гц, 2Н), 6.96 (d, J=8.7 Гц, 2Н), 6.87-6.76 (m, 2Н), 6.27 (dd, J=16.8, 1.8 Гц, 1Н), 5.80 (dd, J=10.6, 1.8 Гц, 1Н), 4.43-4.35 (m, 1Н), 3.79-3.75 (m, 8Н), 2.07-1.96 (m, 2Н), 1.89-1.78 (m, 2Н), 1.63-1.34 (m, 6Н). MS (ESI, методика A): m/z=435.2 [М+Н]+, tR=1.800 (мин). ВЭЖХ: 98.2% (214 нм), 98.5% (254 нм).
Схема 18
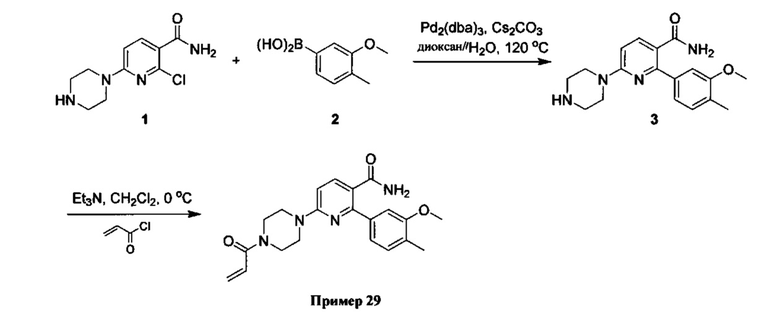
2-(3-Метокси-4-метилфенил)-6-(пиперазин-1-ил)никотинамид (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 2-(4-(гидрокси(фенил)метил)фенил)-6-(пиперидин-4-ил)никотинамида (смотри Схему 48) в виде твердого вещества коричневого цвета (0.238 г, 58%). MS (ESI): m/z=327.1 [М+Н]+.
Пример 29
6-(4-Акрилоилпиперазин-1-ил)-2-(3-метокси-4-метилфенил)никотинамид
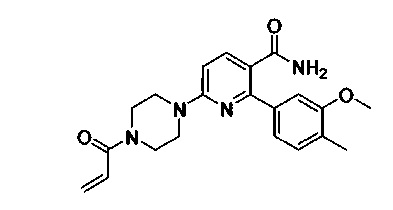
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (115 мг, 43%). 1Н ЯМР (400 МГц, CDCl3) δ 7.77 (d, J=8.7 Гц, 1Н), 7.23 (s, 1H), 7.21-7.15 (m, 2Н), 6.88-6.78 (m, 2Н), 6.26 (dd, J=16.8, 1.9 Гц, 1Н), 5.80 (dd, J=10.6, 1.9 Гц, 1Н), 3.88 (s, 3Н), 3.75-3.78 (m, 8Н), 2.24 (s, 3Н). MS (ESI, методика A): m/z=381.1 [М+Н]+, tR=1.513 (мин). ВЭЖХ: 95.9% (214 нм), 98.7% (254 нм).
Схема 19
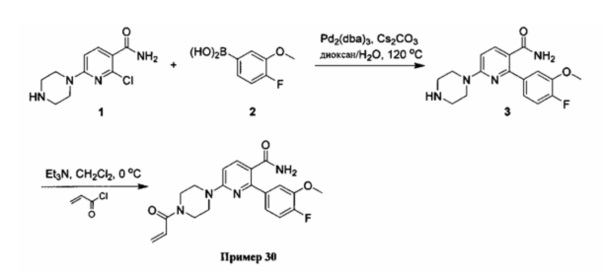
2-(4-Фтор-3-метоксифенил)-6-(пиперазин-1-ил)никотинамид (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида (смотри Схему 1) в виде коричневой смолы (0.264 г, 72%). MS (ESI): m/z=331.1 [М+Н]+.
Пример 30
6-(4-Акрилоилпиперазин-1-ил)-2-(4-фтор-3-метоксифенил)никотинамид
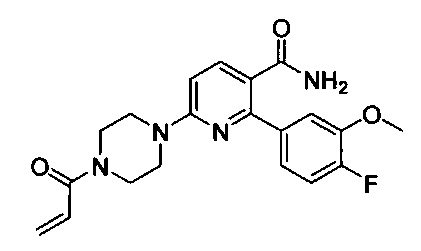
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (45 мг, 15%). 1Н ЯМР (400 МГц, CD3OD) δ 7.78 (d, J=8.7 Гц, 1Н), 7.41 (dd, J=8.3, 2.0 Гц, 1Н), 7.28-7.22 (m, 1Н), 7.16-7.12 (m, =1Н), 6.88-6.77 (m, 2Н), 6.27 (dd, J=16.8, 1.9 Гц, 1Н), 5.80 (dd, J=10.6, 1.9 Гц, 1Н), 3.93 (s, 3Н), 3.87-3.71 (m, 8Н). MS (ESI, методика A): m/z=385.1 [М+Н]+, tR=1.527 (мин). ВЭЖХ: 99.6% (214 нм), 99.7% (254 нм).
Схема 20
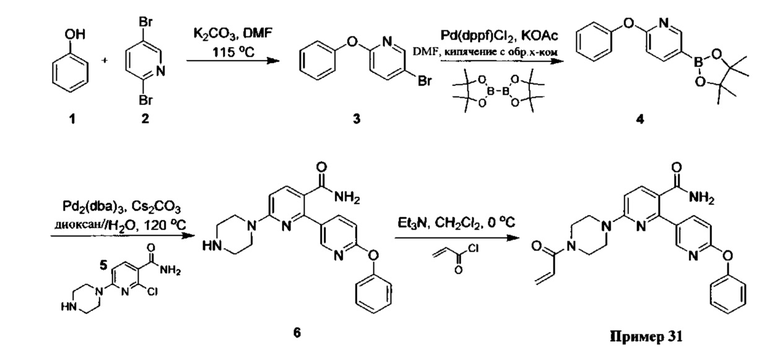
5-Бром-2-феноксипиридин (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 4-(4-бромфенокси)бензонитрила (смотри Схему 16) в виде твердого вещества желтого цвета (3.85 г, 81%).
2-Фенокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)бензонитрила (смотри Схему 16) в виде твердого вещества желтого цвета (0.536 г, 90%).
6'-Фенокси-6-(пиперазин-1-ил)-2,3'-бипиридин-3-карбоксамид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида (смотри Схему 1) в виде коричневой смолы (0.14 г, 55%). MS (ESI): m/z=376.1 [М+Н]+.
Пример 31
6-(4-Акрилоилпиперазин-1-ил)-6'-фенокси-2,3'-бипиридин-3-карбоксамид
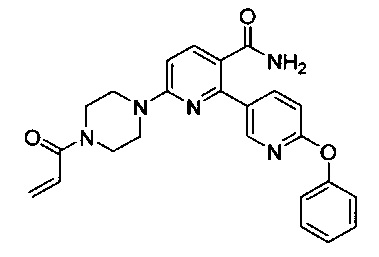
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (50 мг, 31%). 1Н ЯМР (400 МГц, CD3OD) δ 8.43 (d, J=2.4 Гц, 1Н), 8.10 (dd, J=8.6, 2.4 Гц, 1Н), 7.80 (d, J=8.7 Гц, 1Н), 7.49-7.41 (m, 2Н), 7.29-7.22 (m, 1Н), 7.16 (d, J=7.9 Гц, 2Н), 6.97 (d, J=8.6 Гц, 1Н), 6.88-6.77 (m, 2Н), 6.27 (dd, J=16.8, 1.8 Гц, 1Н), 5.80 (dd, J=10.6, 1.8 Гц, 1H), 3.87-3.69 (m, 8Н). MS (ESI, методика A): m/z=430.1 [М+Н]+, tR=1.620 (мин). ВЭЖХ: 95.1% (214 нм), 96.2% (254 нм).
Схема 21
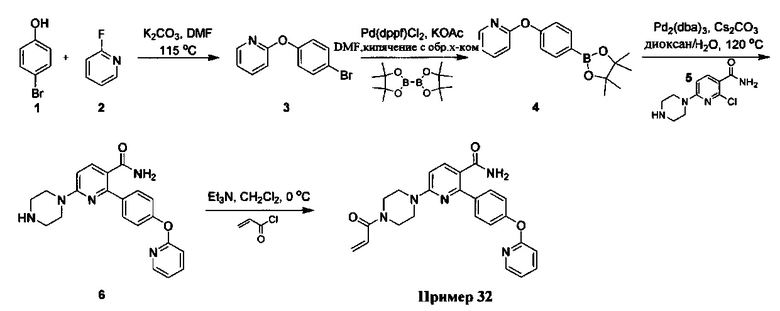
2-(4-Бромфенокси)пиридин (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 4-(4-бромфенокси)бензонитрила (смотри Схему 16) в виде твердого вещества желтого цвета (2.83 г, 57%). MS (ESI): m/z=250.1 [М+Н]+.
2-(4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)пиридин (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)бензонитрила (смотри Схему 16) в виде твердого вещества коричневого цвета (0.882 г, 74%). MS (ESI): m/z=298.1 [М+Н]+.
6-(Пиперазин-1-ил)-2-(4-(пиридин-2-илокси)фенил)никотинамид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 2-(4-(гидрокси(фенил)метил)фенил)-6-(пиперидин-4-ил)никотинамида (смотри Схему 48) в виде коричневой смолы (0.198 г, 53%). MS (ESI): m/z=376.0 [М+Н]+.
Пример 32
6-(4-Акрилоилпиперазин-1-ил)-2-(4-(пиридин-2-илокси)фенил)никотинамид
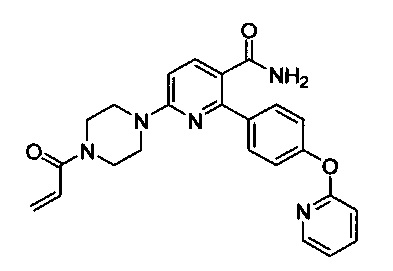
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (41 мг, 18%). 1Н ЯМР (400 МГц, CD3OD) δ 8.18 (dd, J=5.0, 1.4 Гц, 1H), 7.86 (ddd, J=8.4, 7.3, 2.0 Гц, 1H), 7.82-7.74 (m, 3Н), 7.20-7.14 (m, 3Н), 7.01 (d, J=8.3 Гц, 1H), 6.88-6.79 (m, 2Н), 6.27 (dd, J=16.8, 1.9 Гц, 1Н), 5.80 (dd, J=10.6, 1.9 Гц, 1Н), 3.86-3.71 (m, 8Н). MS (ESI, методика A): m/z=430.1 [М+Н]+, tR=1.560 (мин). ВЭЖХ: 100% (214 нм), 100% (254 нм).
Схема 22
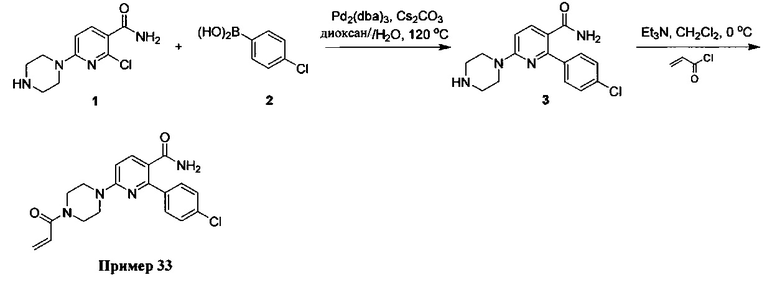
2-(4-Хлорфенил)-6-(пиперазин-1-ил)никотинамид (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 2-(4-(гидрокси(фенил)метил)фенил)-6-(пиперидин-4-ил)никотинамида (смотри Схему 48) в виде коричневой смолы (0.23 г, 72%).
Пример 33
6-(4-Акрилоилпиперазин-1-ил)-2-(4-хлорфенил)никотинамид
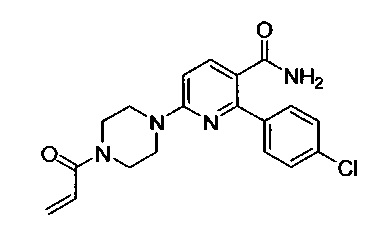
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (50 мг, 18%). 1Н ЯМР (400 МГц, CD3OD) δ 7.78 (d, J=8.7 Гц, 1H), 7.72-7.64 (m, 2Н), 7.45-7.39 (m, 2Н), 6.88-6.76 (m, 2Н), 6.27 (dd, J=16.8, 1.9 Гц, 1H), 5.80 (dd, J=10.6, 1.9 Гц, 1H), 3.87-3.69 (m, 8Н). MS (ESI, методика A): m/z=371.0 [М+Н]+, tR=1.645 (мин). ВЭЖХ: 95.1% (214 нм), 95.3% (254 нм).
Схема 23
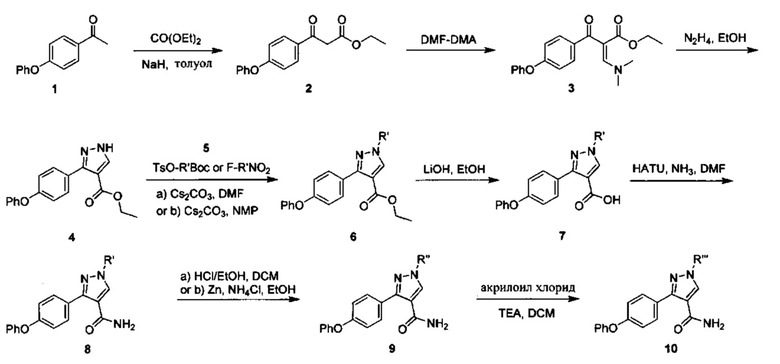
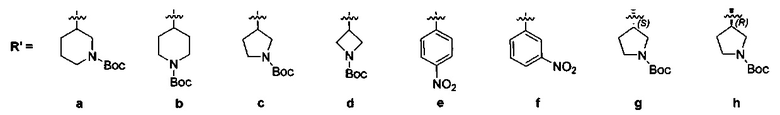
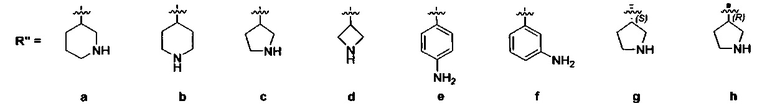
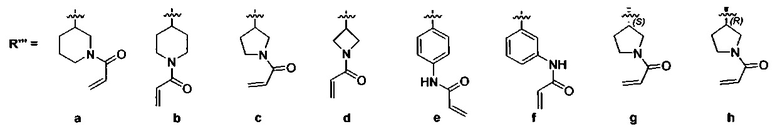
Этил 3-оксо-3-(4-феноксифенил)пропаноат (2). К раствору диэтилкарбоната (14 г, 120 ммоль) в толуоле (100 мл) добавляли NaH (60%, 4.7 г, 12 ммоль) при 0°, и полученный в результате раствор нагревали до 90°, затем добавляли раствор 1-(4-феноксифенил)этанона 1 (10 г, 47 ммоль) в толуоле (50 мл) по каплям на протяжении 30 мин, и раствор нагревали до кипения с обратным холодильником в течение 20 мин. После охлаждения до комнатной температуры добавляли АсОН/Н2O (55 мл/275 мл). Толуол удаляли под вакуумом, и сырой остаток разбавляли водой (500 мл), затем смесь экстрагировали дихлорметаном (4×800 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 9:1 РЕ/ЕА, с получением указанного в заголовке соединения в виде коричневого масла (11.5 г, 79%). MS (ESI): m/z=285.0 [М+Н]+.
Этил 3-(диметиламино)-2-(4-феноксибензоил)акрилат (3). Раствор этил 3-оксо-3-(4-феноксифенил)пропаноата 2 (9.6 г, 34 ммоль) в DMA-DMF (100 мл) перемешивали при 100° в течение 1 ч. Полученный в результате раствор концентрировали под высоким вакуумом с получением указанного в заголовке соединения (12 г, 100%) в виде коричневого густого масла. MS (ESI): m/z=340.1 [М+Н]+.
Этил 3-(4-феноксифенил)-1Н-пиразол-4-карбоксилат (4). К раствору этил 3-(диметиламино)-2-(4-феноксибензоил)акрилата 3 (6 г, 17 ммоль) в этаноле (30 мл) добавляли гидрат гидрозина (875 мг, 17 ммоль), и полученный в результате раствор нагревали до 85° и перемешивали в течение 1 ч. Растворитель выпаривали, и сырой остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 4:1 РЕ/ЕА, с получением указанного в заголовке соединения в виде коричневого масла (4.3 г, 78%). MS (ESI): m/z=309.0 [М+Н]+.
Этил-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксилат (6 с). CS2CO3 (1.08 г, 3.3 ммоль) добавляли к раствору этил 3-(4-феноксифенил)-1Н-пиразол-4-карбоксилата 4 (500 мг, 1.6 ммоль) и трет-бутил-3-(тозилокси)пирролидин-1-карбоксилата 5 с (1.1 г, 3.2 ммоль) в DMF (20 мл), и полученную в результате смесь перемешивали при 100° в течение 16 ч. Реакционную смесь концентрировали под высоким вакуумом, и сырой остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 20:1 DCM/EA, с получением указанного в заголовке соединения (780 мг, 100%) в виде бесцветного густого масла. MS (ESI): m/z=478.1 [М+Н]+.
Аналогичный способ использовали для получения следующих соединений: 6а, 6b, 6d, 6g, 6h.
трет-Бутил-3-(4-(этоксикарбонил)-3-(4-феноксифенил)-1Н-пиразол-1-ил)пиперидин-1-карбоксилат (6а), густое бесцветное масло, 340 мг, 36%. MS (ESI): m/z=492.1 [М+Н]+.
трет-Бутил-4-(4-(этоксикарбонил)-3-(4-феноксифенил)-1Н-пиразол-1-ил)пиперидин-1-карбоксилат (6b), густое масло, 557 мг, 71%. MS (ESI): m/z=492.1 [М+Н]+.
Этил-1-(1-(трет-бутоксикарбонил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксилат (6d), твердое вещество белого цвета, 1.7 г, 61%. MS (ESI): m/z=464.1 [М+Н]+.
(S)-Этил-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4- карбоксилат (6g), бесцветное масло, 1.5 г, 99%. MS (ESI): m/z=478.1 [М+Н]+.
(R)-Этил-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксилат (6h), желтое масло, 2 г, 100%. MS (ESI): m/z=478.1 [M+H]+.
Этил 1-(4-нитрофенил)-3-(4-феноксифенил)-1H-пиразол-4-карбоксилат (6е). К раствору этил 3-(4-феноксифенил)-1H-пиразол-4-карбоксилата 4 (300 мг, 1.0 ммоль) и 1-фтор-4-нитробензола 5е (140 мг, 1.0 ммоль) в 1-метил-2-пирролидоне (8 мл) добавляли карбонат цезия (950 мг, 3.0 ммоль), и полученный в результате раствор нагревали до 130° и перемешивали в течение 1 ч. После охлаждения до температуры окружающей среды растворитель выпаривали, и сырой остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 4:1 РЕ/ЕА, с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (290 мг, 68%). MS (ESI): m/z=430.1 [М+Н]+.
Аналогичные способы использовали для получения соединения 6f:
Этил 1-(3-нитрофенил)-3-(4-феноксифенил)-1H-пиразол-4-карбоксилат (6f), твердое вещество желтого цвета, 290 мг, 40%. MS (ESI): m/z=430.1 [М+Н]+.
1-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоновая кислота (7с). К раствору этил 1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксилата 6с (390 мг, 0.8 ммоль) в EtOH (8 мл) добавляли 1 н. LiOH/H2O (4 мл), и реакционную смесь перемешивали при 75° в течение 1 ч, затем к раствору добавляли 1 н. НО/H2O до рН=4-5, концентрировали с получением указанного в заголовке соединения (690 мг, сырое) в виде твердого вещества белого цвета, содержащего соль LiCl. MS (ESI): m/z=450.1 [M+H]+.
Аналогичный способ использовали для получения следующих соединений: 7а, 7b, 7d, 7е, 7f, 7g,7h.
1-(1-(трет-Бутоксикарбонил)пиперидин-3-ил)-3-(4-феноксифенил)-1H-пиразол-4-карбоновая кислота (7а), твердое вещество белого цвета, 310 мг, 100%. MS (ESI): m/z=464.1 [М+Н]+.
1-(1-(трет-Бутоксикарбонил)пиперидин-4-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоновая кислота (7b), твердое вещество желтого цвета, 525 мг, 99%. MS (ESI): m/z=464.1 [М+Н]+.
1-(1-(трет-Бутоксикарбонил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоновая кислота (7d), твердое вещество желтого цвета, 300 мг, 80%. MS (ESI): m/z=436.1 [M+H]+.
1-(4-Нитрофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоновая кислота (7е), твердое вещество белого цвета, 280 мг, 99%. MS (ESI): m/z=402.1 [М+Н]+.
1-(3-Нитрофенил)-3-(4-феноксифенил)-1H-пиразол-4-карбоновая кислота (7f), желтое масло, 260 мг, 100%. MS (ESI): m/z=402.1 [М+Н]+.
(S)-1-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоновая кислота (7g), пена желтого цвета, 1.2 г, 99%. MS (ESI): m/z=450.1 [М+Н]+.
(R)-1-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоновая кислота (7h), желтое масло, 1.2 г, 99%. MS (ESI): m/z=450.1 [М+Н]+.
Трет-бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пирролидин-1-карбоксилат (8с). К раствору 1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоновой кислоты 7с (630 мг, сырое, 0.67 ммоль) в DMF (20 мл) добавляли HATU (510 мг, 1.33 ммоль) и перемешивали при комнатной температуре в течение 30 минут, полученный в результате раствор барботировали газообразным NH3 в течение 10 минут. После перемешивания при комнатной температуре в течение 1 ч раствор концентрировали под высоким вакуумом, и сырой остаток разбавляли DCM (50 мл), промывали водой (3×20 мл), высушивали над Na2SO4, концентрировали с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 100:1 DCM/MeOH, с получением указанного в заголовке соединения (120 мг, 33%) в виде бесцветной пены. MS (ESI): m/z=471.2 [М+Na]+.
Аналогичный способ использовали для получения следующих соединений: 8а, 8b, 8d, 8е, 8f, 8g, 8h.
трет-Бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пиперидин-1-карбоксилат (8а), бесцветное масло, 310 мг, 100%. MS (ESI): m/z=463.1 [М+Н]+.
трет-Бутил-4-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пиперидин-1-карбоксилат (8b), твердое вещество желтого цвета, 320 мг, 63%. MS (ESI): m/z=463.1 [М+Н]+.
трет-Бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)азетидин-1-карбоксилат (8d), твердое вещество желтого цвета, 300 мг, 99%. MS (ESI): m/z=431.1 [М+Н]+.
1-(4-Нитрофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (8е), твердое вещество желтого цвета, 230 мг, 99%. MS (ESI): m/z=401.1 [М+Н]+.
1-(3-Нитрофенил)-3-(4-феноксифенил)-1H-пиразол-4-карбоксамид (8f), твердое вещество желтого цвета, 260 мг, 100%. MS (ESI): m/z=401.0 [М+Н]+.
(S)-трет-Бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пирролидин-1-карбоксилат (8g), твердое вещество белого цвета, 1.2 г, 100%. MS (ESI): m/z=449.0 [М+Н]+.
(R)-трет-Бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пирролидин-1-карбоксилат (8h), твердое вещество белого цвета, 960 мг, 80%. MS (ESI): m/z=449.0 [М+Н]+.
3-(4-Феноксифенил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид. К раствору трет-бутил-3-(4-карбамоил-3-(4-феноксифенил)-1 Н-пиразол-1 -ил)пирролидин-1 -карбоксилата 8 с (120 мг, 0.27 ммоль) в DCM (10 мл) добавляли 6 н. HCl/EtOH (5 мл, 30 ммоль), перемешивали при комнатной температуре в течение 1 ч, полученный в результате раствор концентрировали под высоким вакуумом, и сырой остаток очищали с помощью препаративной ВЭЖХ с получением указанного в заголовке соединения (86 мг, 91%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CD3OD): δ 8.20 (s, 1Н), 7.75 (d, J=8.6 Гц, 2H), 7.43-7.36 (m, 2H), 7.20-7.13 (m, 1H), 7.08-7.00 (m, 4H), 5.32-5.27 (m, 1H), 3.87-3.72 (m, 3H), 3.59-3.50 (m, 1H), 2.67-2.56 (m, 1H), 2.52-2.43 (m, 1H). MS (ESI, методика A): m/z=349.1 [M+H]+, tR=1.242 мин. ВЭЖХ: 99.3% (214 нм), 99.5% (254 нм).
Аналогичный способ использовали для получения следующих соединений: 9а, 9b, 9d, 9g, 9е и 9f.
3-(4-Феноксифенил)-1-(пиперидин-3-ил)-1Н-пиразол-4-карбоксамид (9а), твердое вещество белого цвета, 240 мг, 99%. 1Н ЯМР (400 МГц, CD3OD): δ 8.23 (s, 1Н), 7.73 (d, J=8.6 Гц, 2H), 7.48-7.31 (m, 2H), 7.21-7.12 (m, 1H), 7.12-6.97 (m, 4H), 4.76-4.65 (m, 1H), 3.74-3.57 (m, 2H), 3.43-3.36 (m, 1H), 3.28-3.15 (m, 1H), 2.39-2.02 (m, 3H), 2.01-1.84 (m, 1H). MS (ESI, методика A): m/z=363.2 [M+H]+, tR=1.255 мин. ВЭЖХ: 100% (214 нм), 100% (254 нм).
3-(4-Феноксифенил)-1-(пиперидин-4-ил)-1Н-пиразол-4-карбоксамид (9b), твердое вещество светло-желтого цвета, 3.1 мг, 40%. 1Н ЯМР (300 МГц, CD3OD): δ 8.18 (s, 1Н), 7.67 (d, J=8.7 Гц, 2H), 7.43-7.31 (m, 2H), 7.19-7.09 (m, 1H), 7.07-6.95 (m, 4H), 4.65-4.55 (m, 1H), 3.64-3.54 (m, 2H), 3.29-3.17(m, 2H), 2.42-2.23 (m, 4H). MS (ESI, методика A): m/z=363.0 [M+H]+, tR=1.288 мин. ВЭЖХ: 100% (214 нм), 100% (254 нм).
1-(Азетидин-3-ил)-3-(4-феноксифенил)-1H-пиразол-4-карбоксамид (Пример 14) (9d), твердое вещество желтого цвета, 300 мг, 100%. 1Н ЯМР (300 МГц, CD3OD): δ 8.20-8.14 (m, 1Н), 7.82-7.66 (m, 2H), 7.44-7.30 (m, 2H), 7.18-7.11 (m, 1H), 7.07-6.96 (m, 4H), 5.52-5.40 (m, 1H), 4.64-4.55 (m, 4H). MS (ESI, методика A): m/z=335.1 [M+H]+, tR=1.235-1.255 мин. ВЭЖХ: 100% (214 нм), 100% (254 нм).
(S)-3-(4-феноксифенил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид (9g), твердое вещество белого цвета, 90 мг, 83%. 1Н ЯМР (400 МГц, CD3OD): δ 8.25 (s, 1Н), 7.80-7.68 (m, 2H), 7.45-7.33 (m, 2H), 7.20-7.11 (m, 1H), 7.11-6.98 (m, 4H), 5.38-5.25 (m, 1H), 3.88-3.69 (m, 3H), 3.62-3.49 (m, 1H), 2.68-2.54 (m, 1H), 2.54-2.40 (m, 1H). MS (ESI, методика A): m/z=349.1 [M+H]+, tR=1.219 мин. ВЭЖХ: 100% (214 нм), 100% (254 нм).
(R)-3-(4-феноксифенил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид (9h), желтое масло, 100 мг, 100%. MS (ESI, методика A): m/z=349.1 [М+Н]+.
1-(4-Аминофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (9е). К раствору 1-(4-нитрофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида 8е (200 мг, 0.50 ммоль) в этаноле (8 мл) добавляли насыщенный водный NH4Cl (4 мл) с последующим добавлением цинковой пыли (260 мг, 4.0 ммоль) порциями на протяжении 5 мин, затем полученный в результате раствор перемешивали в течение 10 ч при температуре окружающей среды. Смесь фильтровали, и фильтрат разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (150 мг, 81%). 1Н ЯМР (400 МГц, CD3OD): δ 8.47 (s, 1Н), 7.78 (d, J=8.7 Гц, 2Н), 7.51 (d, J=8.7 Гц, 2Н), 7.44-7.36 (m, 2Н), 7.16 (s, 1Н), 7.19-7.13 (m, 4Н), 6.83 (d, J=8.7 Гц, 2Н). MS (ESI, методика A): m/z=371.1 [М+Н]+, tR=1.468 мин. ВЭЖХ: 97.8% (214 нм), 98.0% (254 нм).
Аналогичные способы использовали для получения соединения 8f.
1-(3-Аминофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (8f), твердое вещество белого цвета, 105 мг, 47%. 1Н ЯМР (400 МГц, CD3OD): δ 8.60 (s, 1Н), 7.81 (d, J=8.6 Гц, 2H), 7.44-7.36 (m, 2H), 7.28-7.03 (m, 8H), 6.75 (d, J=7.9 Гц, 1H). MS (ESI, методика A): m/z=371.1 [M+H]+, tR=1.507 мин. ВЭЖХ: 96.9% (214 нм), 98.2% (254 нм).
1-(1-Акрилоилпирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (Пример 13) (10с). К раствору 3-(4-феноксифенил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида 9с (43 мг, 0.12 ммоль) и TEA (50 мг, 0.5 ммоль) в DCM (10 мл) добавляли акрилоилхлорид (22 мг, 0.24 ммоль) и перемешивали при 75° в течение 1 ч, полученное в результате вещество концентрировали и очищали с помощью препаративной TLC, элюируя смесью 10:1 DCM/MeOH, с получением указанного в заголовке соединения (17 мг, 35%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CD3OD): δ 8.18 (d, J=8.7 Гц, 1Н), 7.70 (d, J=8.6 Гц, 2H), 7.43-7.34 (m, 2H), 7.18-7.08 (m, 1H), 7.03 (dd, J=15.5, 8.3 Гц, 4H), 6.72-6.58 (m, 1H), 6.32 (dd, J=15.9, 4.0 Гц, 1H), 5.83-5.73 (m, 1H), 5.18-5.04 (m, 1H), 4.21-3.65 (m, 4H), 2.63-2.45 (m, 2H). MS (ESI, методика A): m/z=403.1 [M+H]+, tR=1.414 мин. ВЭЖХ: 95.3% (214 нм), 95.3% (254 нм).
Аналогичный способ использовали для получения следующих соединений: 10а, 10b, 10d, 10е, 10f, 10g и 10h.
1-(1-Акрилоилпиперидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (Пример 11) (10a). Твердое вещество белого цвета, 39 мг, 23%. 1Н ЯМР (400 МГц, CD3OD) δ 8.20 (d, J=5.4 Гц, 1Н), 7.71 (d, J=8.3 Гц, 2H), 7.44-7.35 (m, 2H), 7.19-7.12 (m, 1H), 7.04 (dd, J=12.3, 8.4 Гц, 4H), 6.89-6.73 (m, 1H), 6.3-6.12 (m, 1H), 5.76 (dd, J=25.5, 10.7 Гц, 1H), 4.71 (d, J=11.5 Гц, 0.5 H), 4.45-4.17 (m, 2H), 4.08 (d, J=14.3 Гц, 0.5 H), 3.85-3.73 (m, 0.5 H), 3.45-3.34 (m, 1H), 3.27-3.14 (m, 0.5 H), 2.39-2.18 (m, 2H), 2.06-1.92 (m, 1H), 1.71-1.61 (m, 1H). MS (ESI, методика A): m/z=417.2 [M+H]+, tR=1.468 мин. ВЭЖХ: 97.7% (214 нм), 98.7% (254 нм).
1-(1-Акрилоилпиперидин-4-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (Пример 12) (10b). Твердое вещество белого цвета, 4.7 мг, 20.8%. 1Н ЯМР (400 МГц, CD3OD) δ 8.19 (s, 1Н), 7.69 (d, J=8.5 Гц, 2H), 7.45-7.33 (m, 2H), 7.25-7.12 (m, 1H), 7.04 (dd, J=12.2, 8.5 Гц, 4H), 6.84 (dd, J=16.8, 10.7 Гц, 1H), 6.24 (dd, J=16.8, 1.5 Гц, 1H), 5.79 (dd, J=10.7, 1.5 Гц, 1H), 4.77-4.64 (m, 1H), 4.61-4.46 (m, 1H), 4.35-4.23 (m, 1H), 3.38 (d, J=13.0 Гц, 1H), 3.03-2,92 (m, 1H), 2.34-2.17 (m, 2H), 2.12-1.96 (m, 2H). MS (ESI, методика A): m/z=417.1 [M+H]+. tR=1.421 мин. ВЭЖХ: 97.5% (214 нм), 97.8% (254 нм).
1-(1-Акрилоилазетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (10d). Твердое вещество белого цвета, 19 мг, 18%. 1Н ЯМР (300 МГц, CD3OD) δ 8.21 (s, 1Н), 7.71 (d, J=9.1 Гц, 2H), 7.42-7.31 (m, 2H), 7.20-7.09 (m, 1H), 7.06-6.94 (m, 4H), 6.46-6.22 (m, 2H), 5.78 (dd, J=9.9, 2.3 Гц, 1H), 5.33 (td, J=8.0, 4.0 Гц, 1H), 4.83-4.65 (m, 2H), 4.62-4.41 (m, 2H). MS (ESI, методика A): m/z=389.1 [M+H]+, tR=l.388-1.401 мин. ВЭЖХ: 95.1% (214 нм), 96.1% (254 нм).
1-(4-Акриламидофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (Пример 16) (10е). Твердое вещество белого цвета, 52 мг, 46%. 1Н ЯМР (400 МГц, CD3OD) δ 8.67 (s, 1Н), 7.89-7.78 (m, 6H), 7.44-7.36 (m, 2H), 7.20-7.12 (m, 1H), 7.11-7.02 (m, 4H), 6.52-6.37 (m, 2H), 5.82 (dd, J=9.4, 2.4 Гц, 1H). MS (ESI, методика A): m/z=425.2 [M+H]+, tR=1.521 мин. ВЭЖХ: 97.3% (214 нм), 97.0% (254 нм).
1-(3-Акриламидофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (Пример 18) (10f). Твердое вещество белого цвета, 24 мг, 24%. 1Н ЯМР (400 МГц, CD3OD) δ 8.70 (s, 1Н), 8.35 (s, 1H), 7.84 (d, J=8.7 Гц, 2H), 7.60 (d, J=8.1 Гц, 2H), 7.54-7.48 (m, 1H), 7.44-7.36 (m, 2H), 7.11-7.03 (m, 1H), 7.20-7.13(m, 4H), 6.53-6.38(m, 2H), 5.83 (dd, J=9.2, 2.6 Гц, 1H). MS (ESI, методика A): m/z=425.1 [M+H]+, tR=1.547 (мин). ВЭЖХ: 97.6% (214 нм), 98.6% (254 нм).
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (Пример 20) (10g). Твердое вещество серого цвета, 120 мг, 27%. 1Н ЯМР (400 МГц, CD3OD) δ 8.21 (d, J=6.6 Гц, 1H), 7.73-7.64 (m, 2H), 7.43-7.34 (m, 2H), 7.20-7.10 (m, 1H), 7.10-6.96 (m, 4H), 6.75-6.55 (m, 1H), 6.32 (ddd, J=16.8, 4.8, 1.9 Гц, 1H), 5.86-5.72 (m, 1H), 5.21-5.03 (m, 1H), 4.22-4.07 (m, 1H), 4.05-3.67 (m, 3H), 2.64-2.43 (m, 2H). MS (ESI, методика A): m/z=403.2 [M+H]+, tR=1.421 мин. ВЭЖХ: 99.0% (214 нм), 99.0% (254 нм).
(R)-1-(1-Акрилоилпирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид (Пример 19) (10h). Твердое вещество белого цвета, 20 мг, 6%. 1Н ЯМР (300 МГц, CD3OD): δ 8.16 (d, J=5.9 Гц, 1Н), 7.67 (d, J=8.7 Гц, 2H), 7.42-7.30 (m, 2H), 7.17-6.92 (m, 5H), 6.71-6.56 (m, 1H), 6.34-6.25 (m, 1H), 5.80-5.72 (m, 1H), 5.18-5.02 (m, 1H), 4.20-4.05 (m, 1H), 4.02-3.65 (m, 3H), 2.62-2.45 (m, 2H). MS (ESI, методика A): m/z=403.1 [M+H]+, tR=1.409 мин. ВЭЖХ: 95.0% (214 нм), 97.0% (254 нм).
Схема 24
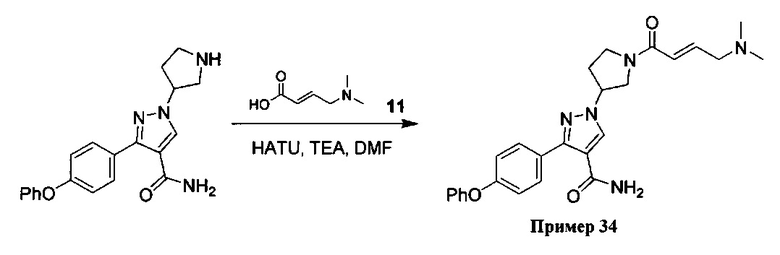
Пример 34
1-(1-(4-(Диметиламино)бут-2-еноил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
К раствору 4-(диметиламино)бут-2-еноевой кислоты 11 (22 мг, 0.17 ммоль) и HATU (98 мг, 0.26 ммоль) в DMF (10 мл) добавляли 3-(4-феноксифенил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид 9 с (Схема 23) (60 мг, 0.17 ммоль) и DIPEA (22 мг, 0.5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Раствор концентрировали под высоким вакуумом, и остаток разбавляли DCM (10 мл), промывали водой (3×10 мл), высушивали над Na2SO4, концентрировали с получением остатка, который очищали с помощью препаративной TLC, элюируя смесью 10:1 DCM/MeOH, с получением указанного в заголовке соединения (26 мг, 37%) в виде твердого вещества коричневого цвета. 1Н ЯМР (300 МГц, CD3OD): δ 8.20 (d, J=9.0 Гц, 1Н), 7.73-7.56 (m, 2Н), 7.41-7.27 (m, 2Н), 7.11 (t, J=7.4 Гц, 1H), 7.05-6.91 (m, 4Н), 6.88-6.66 (m, 2Н), 5.22-5.02 (m, 1Н), 4.24-4.06 (m, 1Н), 4.05-3.76 (m, 4.5Н), 3.76-3.61 (m, 0.5Н), 2.86 (d, J=6.1 Гц, 6Н), 2.66-2.39 (m, 2Н). MS (ESI, методика A): m/z=459.8 [М+Н]+, tR=1.244 мин. ВЭЖХ: 97.5% (214 нм), 98.2% (254 нм).
Схема 92
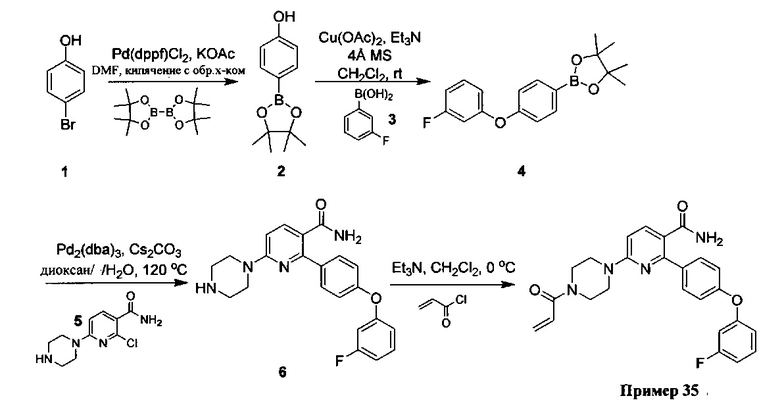
4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)фенол (2). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)бензонитрила (смотри Схему 16) в виде твердого вещества белого цвета (10.2 г, 80%).
2-(4-(3-Фторфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (4). Смесь 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенол 2 (1.0 г, 4.5 ммоль), 3-фторфенилбороновой кислоты 3 (0.70 г, 5.0 ммоль), Cu(ОАс)2 (1.0 г, 5.0 ммоль), Et3N (2.75 г, 27.3 ммоль), 4А молекулярных сит (1 г) и CH2Cl2 (15 мл) перемешивали при комнатной температуре в течение 16 ч, затем смесь концентрировали под вакуумом, и остаток очищали с помощью хроматографии на силикагеле, элюируя смесью 20:1 петролейный эфир/EtOAc, с получением указанного в заголовке соединения (0.45 г, 32%) в виде твердого вещества белого цвета.
2-(4-(3-Фторфенокси)фенил)-6-(пиперазин-1-ил)никотинамид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида (смотри Схему 1) в виде твердого вещества белого цвета (0.15 г, 41%). MS (ESI): m/z=393.1 [М+Н]+.
Пример 35
6-(4-Акрилоилпиперазин-1-ил)-2-(4-(3-фторфенокси)фенил)никотинамид
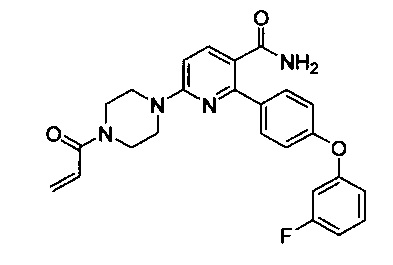
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (50 мг, 27%). 1Н ЯМР (400 МГц, CD3OD) δ 7.79 (d, J=8.7 Гц, 1Н), 7.77-7.70 (m, 2Н), 7.42-7.33 (m, 1Н), 7.13-7.06 (m, 2Н), 6.93-6.75 (m, 5Н), 6.27 (dd, J=16.8, 1.9 Гц, 1Н), 5.80 (dd, J=10.6, 1.9 Гц, 1Н), 3.84-3.72 (m, 8Н). MS (ESI, методика A): m/z=447.1 [М+Н]+, tR=1.746 (мин). ВЭЖХ: 95.9% (214 нм), 96.4% (254 нм).
Схема 26
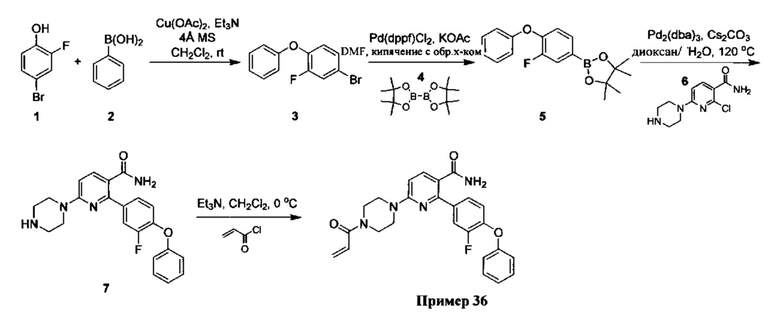
4-Бром-2-фтор-1-феноксибензол (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 2-(4-(3-фторфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (смотри Схему 25) в виде масла желтого цвета (1.5 г, 21%). MS (ESI): m/z=267.1 [М+Н]+.
2-(3-Фтор-4-феноксифенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)бензонитрила (смотри Схему 16) в виде масла желтого цвета (1.04 г, 73%).
2-(3-Фтор-4-феноксифенил)-6-(пиперазин-1-ил)никотинамид (7). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида (смотри Схему 1) в виде коричневой смолы (0.194 г, 49%). MS (ESI): m/z=393.1 [М+Н]+.
Пример 36
6-(4-Акрилоилпиперазин-1-ил)-2-(3-фтор-4-феноксифенил)никотинамид
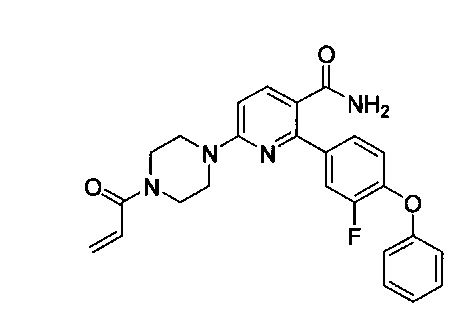
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (38 мг, 17%). 1Н ЯМР (400 МГц, DMSO-d6) δ (m, 2Н), 7.59 (d,J=11.4 Гц, 1Н), 7.48 (d, J=8.2 Гц, 1Н), 7.42 (t, J=7.7 Гц, 2Н), 7.35-7.28 (m, 1H), 7.24-7.15 (m, 2Н), 7.04 (d, J=7.9 Гц, 2Н), 6.95-6.81 (m, 2Н), 6.16 (d, J=16.6 Гц, 1Н), 5.73 (d, J=10.2 Гц, 1Н), 3.79-3.50 (m, 8Н). MS (ESI, методика A): m/z=447.1 [М+Н]+, tR=1.753 (мин). ВЭЖХ: 98.6% (214 нм), 98.4% (254 нм).
Схема 95
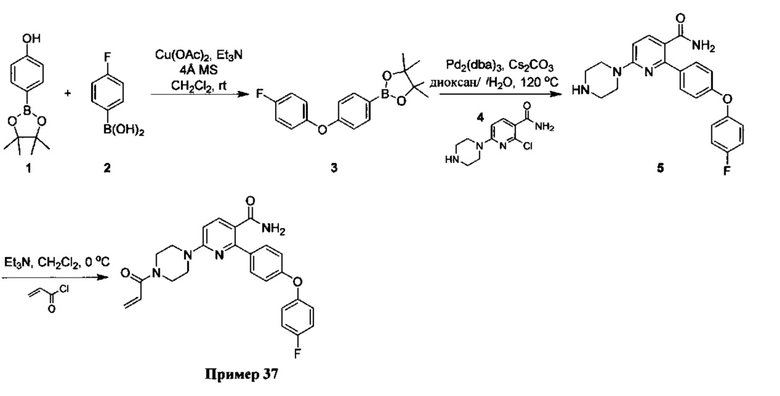
2-(4-(4-Фторфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 2-(4-(3-фторфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (смотри Схему 25) в виде твердого вещества белого цвета (0.45 г, 32%).
2-(4-(4-Фторфенокси)фенил)-6-(пиперазин-1-ил)никотинамид (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида (смотри Схему 1) в виде коричневой смолы (0.18 г, 41%).
Пример 37
6-(4-Акрилоилпиперазин-1-ил)-2-(4-(4-фторфенокси)фенил)никотинамид
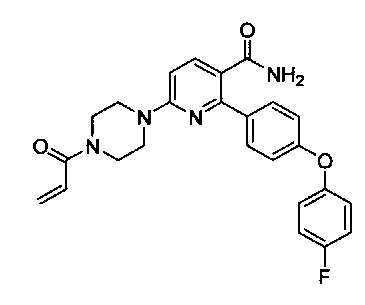
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (80 мг, 44%). 1H ЯМР (400 МГц, CD3OD) δ 7.77 (d, J=8.7 Гц, 1Н), 7.69 (d, J=8.7 Гц, 2Н), 7.17-7.05 (m, 4Н), 7.00 (d, J=8.7 Гц, 2Н), 6.87-6.76 (m, 2Н), 6.26 (dd, J=16.8, 1.8 Гц, 1Н), 5.80 (dd, J=10.6, 1.8 Гц, 1H), 3.87-3.71 (m, 8H). MS (ESI, методика A): m/z=447.0 [M+H]+, tR=1.739 (мин). ВЭЖХ: 99.8% (214 нм), 100% (254 нм).
Схема 28
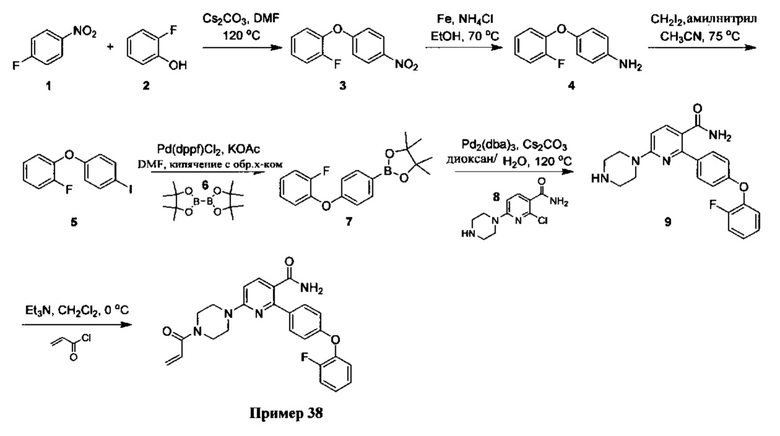
1-Фтор-2-(4-нитрофенокси)бензол (3). Смесь 1-фтор-4-нитробензола 1 (10.0 г, 70.8 ммоль), 2-фторфенола 2 (9.54 г, 85.0 ммоль), CS2CO3 (34.6 г, 106 ммоль) и DMF (80 мл) нагревали до 120° и перемешивали в течение 2 ч. После охлаждения до комнатной температуры, смесь разбавляли водой (150 мл) и экстрагировали EtOАс (200 мл×2). Объединенную органическую фазу промывали рассолом (100 мл×3), высушивали над Na2SO4 и фильтровали. Фильтрат концентрировали под вакуумом. Остаток перекристаллизовывали из МеОН (20 мл×2) с получением указанного в заголовке соединения в виде твердого вещества светло-желтого цвета (14.6 г, 88%).
4-(2-Фторфенокси)бензоламин (4). К смеси 1-фтор-2-(4-нитрофенокси)бензола 3 (16.2 г, 69.3 ммоль), насыщенного водного раствора NH4Cl (30 мл) и EtOH (150 мл) медленно добавляли железный порошок (19.4 г, 347 ммоль), и затем полученную в результате смесь нагревали до 70° в течение 3.5 ч. После охлаждения до комнатной температуры смесь фильтровали. Фильтрат концентрировали под вакуумом. Остаток растворяли в EtOAc (160 мл) и последовательно промывали водой (100 мл×2) и рассолом (100 мл×2). Органическую фазу высушивали над Na2SO4, фильтровали и концентрировали под вакуумом с получением указанного в заголовке соединения (13.6 г, 96%) в виде твердого вещества желтого цвета. 1Н ЯМР (400 МГц, DMSO-d6) δ 7.35-7.28 (m, 1Н), 7.14-7.00 (m, 2Н), 6.93-6.85 (m, 1Н), 6.80-6.73 (m, 2Н), 6.64-6.56 (m, 2Н), 4.99 (brs, 2Н).
1-Фтор-2-(4-иодофенокси)бензол (5). Смесь 4-(2-фторфенокси)бензоламина 4 (7.94 г, 39.1 ммоль), CH2I2 (29.0 г, 136.8 ммоль) и CH3CN (120 мл) нагревали до 55° и затем добавляли амилнитрит (11.6 г, 97.7 ммоль). Полученную в результате смесь нагревали до 75° в течение 3.5 ч. После охлаждения до комнатной температуры, смесь разбавляли EtOAc и затем последовательно промывали 10% Na2S2O3 и рассолом. Органическую фазу высушивали над Na2SO4, фильтровали, концентрировали под вакуумом и очищали с помощью хроматографии на силикагеле, элюируя петролейным эфиром, с получением указанного в заголовке соединения (4.12 г, 51%) в виде светло-желтого масла. 1Н ЯМР (400 МГц, DMSO) δ 7.78-7.63 (m, 2Н), 7.44-7.37 (m, 1Н), 7.34-7.14 (m, 3Н), 6.87-6.74 (m, 2Н).
2-(4-(2-Фторфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (7). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)бензонитрила (смотри Схему 16) в виде масла желтого цвета (0.40 г, 13%).
2-(4-(2-Фторфенокси)фенил)-6-(пиперазин-1-ил)никотинамид (9). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида (смотри Схему 1) в виде твердого вещества белого цвета (0.16 г, 31%). MS (ESI): m/z=393.1 [М+Н]+.
Пример 38
6-(4-Акрилоилпиперазин-1-ил)-2-(4-(2-фторфенокси)фенил)никотинамид
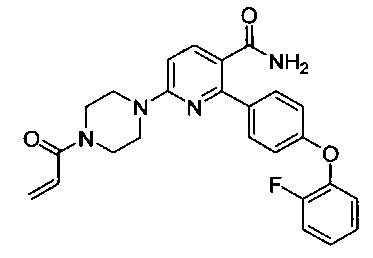
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (28 мг, 24%). 1Н ЯМР (400МГц, CD3OD) δ 7.77 (d, J=8.7 Гц, 1H), 7.71-7.68 (m, 2Н), 7.34-7.13 (m, 4Н), 6.99 (d, J=8.7 Гц, 2H), 6.88-6.79 (m, 2Н), 6.26 (dd, J=16.8, 1.9 Гц, 1H), 5.80 (dd, J=10.6, 1.9 Гц, 1H), 3.89-3.71 (m, 8H). MS (ESI, методика A): m/z=447.1 [M+H]+, tR=1.723 (мин). ВЭЖХ: 98.6% (214 нм), 98.8% (254 нм).
Схема 29
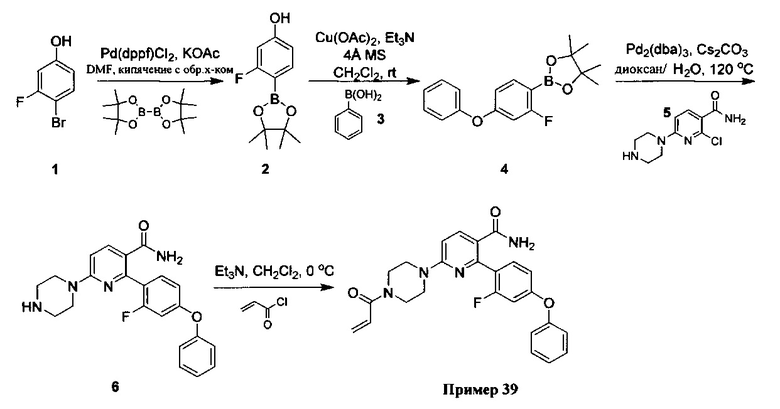
3-Фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенол (2). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенола (смотри Схему 25) в виде твердого вещества белого цвета (4.1 г, 66%). MS (ESI): m/z=239.2 [М+Н]+.
2-(2-Фтор-4-феноксифенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 2-(4-(3-фторфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (смотри Схему 25) в виде твердого вещества белого цвета (2.1 г, 39%).
2-(2-Фтор-4-феноксифенил)-6-(пиперазин-1-ил)никотинамид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида (смотри Схему 1) в виде твердого вещества коричневого цвета (0.105 г, 21%). MS (ESI): m/z=393.1 [М+Н]+.
Пример 39
6-(4-Акрилоилпиперазин-1-ил)-2-(2-фтор-4-феноксифенил)никотинамид
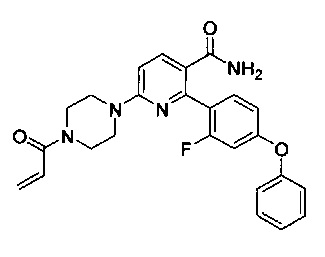
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (78 мг, 65%). 1Н ЯМP(400 МГц, MeOD) δ 7.78 (d, J=8.7 Гц, 1Н), 7.59 (dd, J=11.8, 2.0 Гц, 1H), 7.48 (d, J=9.2 Гц, 1H), 7.44-7.33 (m, 2H), 7.17-7.06 (m, 2H), 7.05-6.99 (m, 2H), 6.90-6.79 (m, 2H), 6.27 (dd, J=16.8, 1.8 Гц, 1H), 5.80 (dd, J=10.6, 1.8 Гц, 1H), 3.86-3.70 (m, 8H). MS (ESI, методика A): m/z=447.0 [M+H]+, tR=1.770 (мин). ВЭЖХ: 96.5% (214 нм), 96.9% (254 нм).
Схема 30
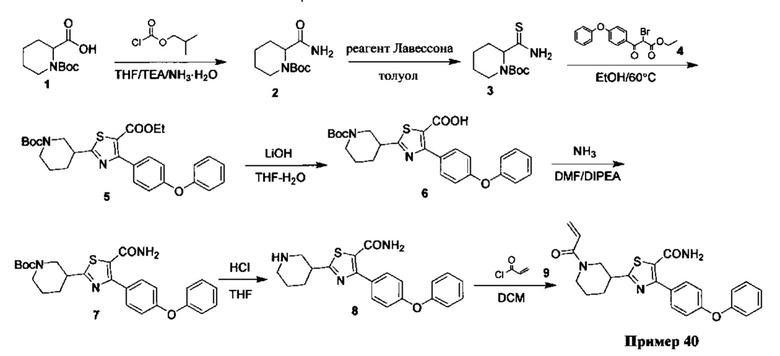
трет-Бутил-2-карбамоилпиперидин-1-карбоксилат (2). К раствору 1-(трет-бутоксикарбонил)пиперидин-2-карбоновой кислоты 1 (5.0 г, 21.8 ммоль) в сухом THF (100 мл) добавляли TEA (3.30 г, 32.7 ммоль), изобутил карбонохлоридат (3.27 г, 23.99 ммоль) при 0°C, и полученный в результате раствор перемешивали в течение 20 мин. Добавляли NH3-H2O и перемешивали при комнатной температуре в течение 2 ч. Смесь разбавляли этилацетатом (30 мл) и промывали насыщенным водным Na2CO3 (2×20 мл) и насыщенной водной лимонной кислотой (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением сырого продукта (3.6 г, 72%) в виде твердого вещества белого цвета. МS (ESI): m/z=173. [M-55]+.
трет-Бутил-2-карбамотиоилпиперидин-1-карбоксилат (3). К раствору трет-бутил-2-карбамоилпиперидин-1-карбоксилата 2 (2.0 г, 8.77 ммоль) в сухом толуоле (20 мл) добавляли реагент Лавессона (2.13 г, 5.26 ммоль) в атмосфере N2. Полученный в результате раствор перемешивали при 80°C в течение 16 ч. Смесь разбавляли этилацетатом (30 мл), и промывали насыщенным водным (sat. aq.) Na2CO3 (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 4:1 РЕ/ЕА, с получением указанного в заголовке соединения (330 мг, 15%) в виде твердого вещества белого цвета. MS (ESI): m/z=245.2 [М+Н]+.
Этил-2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоксилат (5). Раствор трет-бутил-2-карбамотиоилпиперидин-1-карбоксилата 3 (60 мг, 0.239 ммоль) и этил 2-бром-3-оксо-3-(4-феноксифенил)пропаноата 4 (250 мг, 0.7 ммоль) в EtOH (10 мл) перемешивали в течение 2 ч при 60°C. Смесь разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 40:1 РЕ/ЕА, с получением указанного в заголовке соединения (120 мг, 34%) в виде бесцветного масла. MS (ESI): m/z=509.3 [М+Н]+.
2-(1-(трет-Бутоксикарбонил)пиперидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоновая кислота (6). Раствор этил 2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоксилата 5 (120 мг, 0.263 ммоль) в THF-H2O (1/1, 20 мл) и добавленный LiOH (30 мг, 0.71 ммоль) перемешивали при комнатной температуре в течение 13 ч. Смесь выпаривали и разбавляли водой (5 мл), и раствор подкисляли 2 н. соляной кислотой до pH=4. Остаток экстрагировали этилацетатом (ЕА) (3×30 мл), Высушивали и концентрировали с получением сырого соединения (ПО мг, 100%) в виде жидкости светло-красного цвета. MS (ESI): m/z=481.0 [М+Н]+.
трет-Бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пиперидин-1-карбоксилат (7). Смесь 2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоновой кислоты 6 (110 мг, 0.229 ммоль), HATU (113 мг, 0.297 ммоль), DIPEA (88 мг, 0.687 ммоль) и сухого DMF (10 мл) барботировали NH3 в течение 20 мин, и полученный в результате раствор перемешивали в течение 1 ч при температуре окружающей среды. Смесь разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью препаративной TLC с 3:2 РЕ/ЕА с получением указанного в заголовке соединения (100 мг, 75%) в виде бесцветного масла. MS (ESI): m/z=480.1 [М+Н]+.
4-(4-Феноксифенил)-2-(пиперидин-3-ил)тиазол-5-карбоксамид (8). К раствору трет-бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пиперидин-1-карбоксилата 7 (100 мг, 0.21 ммоль) в DCM (5 мл) добавляли TFA (2.5 мл) при температуре окружающей среды. Смесь перемешивали в течение 1 ч. Смесь разбавляли водой (2×150 мл), экстрагировали этилацетатом (2×20 мл) и промывали рассолом (2×150 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (60 мг, 76%) в виде коричневого масла. MS (ESI): m/z=380.0 [М+Н]+.
Пример 40
2-(1-Акрилоилпиперидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоксамид
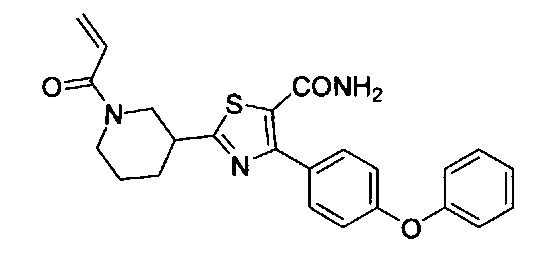
К раствору 4-(4-феноксифенил)-2-(пиперидин-3-ил)тиазол-5-карбоксамида 8 (60 мг, 0.155 ммоль) в сухом дихлорметане (10 мл) добавляли TEA (19 мг, 0.21 ммоль) и акрилоилхлорид 9 (32 мг, 0.316 ммоль), и полученный в результате раствор перемешивали при комнатной температуре в течение 1 ч. Добавляли воду (10 мл), чтобы остановить реакцию. Смесь разбавляли этилацетатом (30 мл), промывали рассолом (2×20 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением остатка, который очищали с помощью препаративной TLC с 17:1 DCM/MeOH с получением указанного в заголовке соединения (33 мг, 50%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, DMSO) δ 7.76 (d, J=8.5 Гц, 2Н), 7.76 (brs, 1Н),7.70 (brs, 1Н), 7.44 (t, J=8.0 Гц, 2Н), 7.19 (t, J=7.4 Гц, 1Н), 7.08 (t, J=8.6 Гц, 4Н), 6.92-6.82 (m, 1Н), 6.14-6.04 (m, 1Н), 5.73-5.64 (m, 1Н), 4.65-4.62 (m, 0.5Н), 4.15-4.04 (m, 0.5Н), 4.02-4.00 (m, 1H), 3.59-3.56 (m, 0.5Н), 3.26-3.14 (m, 1.5Н), 3.09-2.98 (m, 1Н), 2.22-2.16 (m, 1Н), 1.90-1.74 (m, 2Н), 1.58-1.48 (m, 1Н). MS (ESI, методика A): m/z=433.8 [М+Н]+, tR=1.488 мин. ВЭЖХ: 99.1% (214 нм), 99.2% (254 нм).
Схема 31
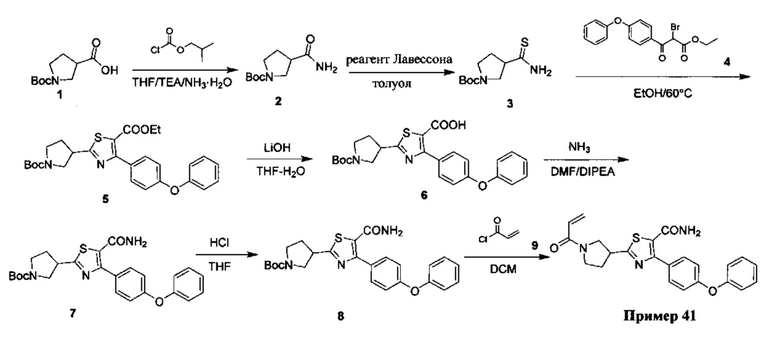
трет-Бутил-3-карбамоилпирролидин-1-карбоксилат (2). К раствору 1-(трет-бутоксикарбонил)пирролидин-3-карбоновой кислоты 1 (5 г, 23.23 ммоль) в сухом THF (100 мл) добавляли TEA (4.69 г, 46.46 ммоль), изобутилкарбонохлоридат (3.8 г, 27.87 ммоль) при 0°C, и полученный в результате раствор перемешивали в течение 20 мин. Добавляли NH3-Н2О и перемешивали при комнатной температуре в течение 2 ч. Смесь разбавляли этилацетатом (30 мл), и промывали насыщенным (sat.) Na2CO3 (2×20 мл) и насыщенной лимонной кислотой (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением сырого продукта (2.39 г, 49%) в виде твердого вещества желтого цвета. MS (ESI): m/z=159.0 [М+Н]+.
трет-Бутил-3-карбамотиоилпирролидин-1-карбоксилат (3). К раствору трет-бутил-3-карбамоилпирролидин-1-карбоксилата 2 (0.76 г, 3.55 ммоль) в сухом толуоле (20 мл) добавляли реагент Лавессона (0.71 г, 1.77 ммоль) в атмосфере N2. Полученный в результате раствор перемешивали при 80°C в течение 16 ч. Смесь разбавляли этилацетатом (30 мл) и промывали насыщенным Na2CO3 (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 6:1 РЕ/ЕА, с получением указанного в заголовке соединения (230 мг, 28%) в виде коричневого масла. MS (ESI): m/z=175.2 [М+Н]+.
Этил 2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоксилат (5). Раствор трет-бутил-3-карбамотиоилпирролидин-1-карбоксилата 3 (360 мг, 0.1 ммоль) и этил 2-бром-3-оксо-3-(4-феноксифенил)пропаноата 4 (230 мг, 0.1 ммоль) в EtOH (10 мл) перемешивали в течение 2 ч при 60°C. Смесь разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 30:1 РЕ/ЕА, с получением указанного в заголовке соединения (100 мг, 20%) в виде бесцветного масла. MS (ESI): m/z=495.3[М+H]+.
2-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоновая кислота (6). Раствор этил 2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоксилата 5 (100 мг, 0.2 ммоль) в THF-H2O (1/1, 20 мл) с добавленным LiOH (84 мг, 2 ммоль) перемешивали при комнатной температуре в течение 13 ч. Смесь выпаривали и разбавляли водой (5 мл), и раствор подкисляли 2 н. соляной кислотой до pH=4. Остаток экстрагировали ЕА (3×30 мл). Высушивали и выпаривали с получением сырого соединения (95 мг, 100%) в виде красного масла. MS (ESI): m/z=411.0 [М+Н]+.
трет-Бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пирролидин-1-карбоксилат (7). Смесь 2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоновой кислоты 6 (950 мг, 0.2 ммоль), НАTU (116 мг, 0.3 ммоль), DIPEA (103 мг, 0.8 ммоль) и сухого DMF (10 мл) барботировали NH3 в течение 20 мин, и полученный в результате раствор перемешивали в течение 1 ч при температуре окружающей среды. Смесь разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью препаративной TLC с 2:1 РЕ/ЕА с получением указанного в заголовке соединения (70 мг, 75%) в виде бесцветного масла. MS (ESI): m/z=466.1 [М+Н]+.
трет-Бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пирролидин-1-карбоксилат (8). К раствору трет-бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пирролидин-1-карбоксилата 7 (70 мг, 0.15 ммоль) в DCM (5 мл) добавляли TFA (2.5 мл) при температуре окружающей среды. Смесь перемешивали в течение 1 ч. Смесь разбавляли водой (2×150 мл) и экстрагировали этилацетатом (2×20 мл) и промывали рассолом (2×150 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (65 мг, 100%) в виде коричневого масла. MS (ESI): m/z=366.1 [М+Н]+.
Пример 41
2-(1-Акрилоилпирролидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоксамид
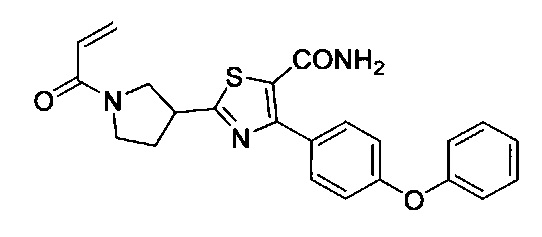
К трет-бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пирролидин-1-карбоксилату 8 (65 мг, 0.15 ммоль) в сухом дихлорметане (10 мл) добавляли TEA (50 мг, 0.45 ммоль) и акрилоилхлорид 9 (20.4 мг, 0.225 ммоль), и полученный в результате раствор перемешивали при комнатной температуре в течение 1 ч. Добавляли воду (10 мл), чтобы остановить реакцию. Смесь разбавляли этилацетатом (30 мл), промывали рассолом (2×20 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением остатка, который очищали с помощью препаративной TLC с 1:1 РЕ/ЕА с получением указанного в заголовке соединения (22 мг, 35%) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, DMSO) δ 7.76 (d, J=8.0 Гц, 2Н), 7.76 (brs, 1Н),7.70 (brs, 1Н), 7.44 (t, J=8.0 Гц, 2Н), 7.19 (t, J=1А Гц, 1Н), 7.07 (t, J=8.6 Гц, 4H), 6.65-6.59 (m, 1H), 6.19-6.15 (m, 1H), 5.72-5.68 (m, 1H), 4.11-4.07 (m, 0.5H), 3.99-3.95 (m, 0.5H), 3.91-3.78 (m, 2H), 3.70-3.62 (m, 1.5H), 3.50-3.47 (m, 0.5H), 2.44-2.36 (m, 1H), 2.28-2.25 (m, 0.5H), 2.18-2.13 (m, 0.5H). MS (ESI, методика A): m/z=419.8 [M+H]+, tR=1.429 мин. ВЭЖХ: 97.2% (214 нм), 97.5% (254 нм).
Схема 105
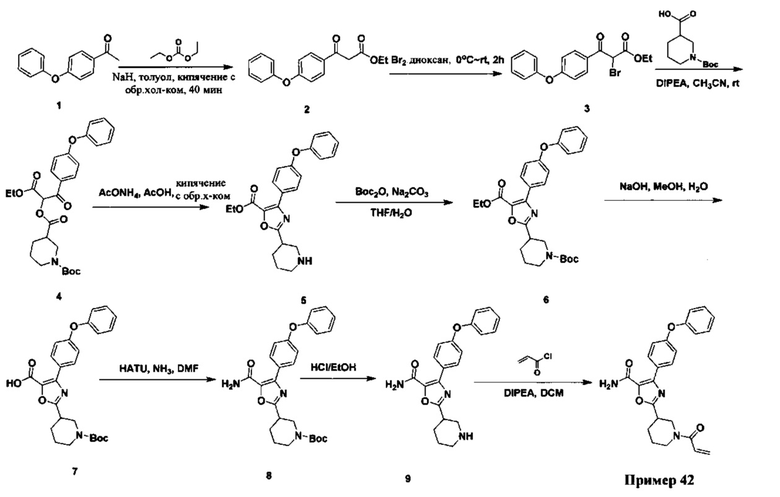
Этил 3-оксо-3-(4-феноксифенил)пропаноат (2). К смеси диэтилкарбоната (14 г, 120 ммоль) и NaH (4.8 г, 120 ммоль) в толуоле (100 мл) добавляли 1-(4-феноксифенил)этанон 1 (10 г, 47 ммоль) по каплям в течение 20 мин. Полученную в результате смесь нагревали до кипения с обратным холодильником в течение 40 мин. После охлаждения до комнатной температуры реакцию останавливали АсОН/H2O (6 мл/30 мл), затем разбавляли ЕА (100 мл). Органический слой отделяли, промывали рассолом, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток наносили на колонку с силикагелем, элюируя смесью 20:1 РЕ/ЕА, с получением указанного в заголовке соединения (7.5 г, 56%) в виде масла желтого цвета. MS (ESI): m/z=285.1 [М+Н]+.
Этил 2-бром-3-оксо-3-(4-феноксифенил)пропаноат (3). К смеси этил 3-оксо-3-(4-феноксифенил)пропаноата 2 (6.55 г, 23.1 ммоль) в диоксане (100 мл) добавляли Br2 (3.69 г, 23.1 ммоль) по каплям при 0°C в атмосфере N2. Полученную в результате смесь перемешивали при комнатной температуре в течение 1 ч. Летучую фазу удаляли при пониженном давлении. В результате этого получали указанное в заголовке сырое соединение (9.5 г, избыток) в виде масла желтого цвета. MS (ESI): m/z=363.0/365.0 [М+Н]+.
1-трет-Бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-ил)пиперидин-1,3-дикарбоксилат (4). К смеси этил 2-бром-3-оксо-3-(4-феноксифенил)пропаноата 3 (1.0 г, 2.75 ммоль) в CH3CN (20 мл) добавляли DIEA (0.39 г, 3.0 ммоль) по каплям. Полученную в результате смесь перемешивали при комнатной температуре на протяжении ночи. Летучую фазу удаляли при пониженном давлении. Остаток наносили на колонку с силикагелем, элюируя смесью 3:1 РЕ/ЕА, с получением указанного в заголовке соединения (1.15 г, 82%) в виде масла желтого цвета. MS (ESI): m/z=412.1 [М+Н-Вос]+.
Этил 4-(4-феноксифенил)-2-(пиперидин-3-ил)оксазол-5-карбоксилат (5). Смесь 1-трет-бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-ил)пиперидин-1,3-дикарбоксилата 4 (1.15 г, 2.25 ммоль) и AcONH4 (0.86 г, 11.23 ммоль) в АсОН (20 мл) перемешивали при 120°C в течение 2 ч. Летучую фазу удаляли при пониженном давлении. Остаток растворяли в ЕА (50 мл), который промывали насыщенным NaHCO3, рассолом, высушивали над сульфатом натрия, фильтровали и концентрировали. В результате этого получали указанное в заголовке сырое соединение (0.85 г, сырое) в виде масла желтого цвета. MS (ESI): m/z=393.1 [М+Н]+.
Этил-2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-4-(4-феноксифенил)оксазол-5-карбоксилат (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксилата (смотри Схему 37) в виде масла бледно-желтого цвета (0.35 г, 33%, две стадии). MS (ESI): m/z=437.0 [М+Н-56]+.
2-(1-(трет-Бутоксикарбонил)пиперидин-3-ил)-4-(4-феноксифенил)оксазол-5-карбоновая кислота (7). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-4'-феноксибифенил-2-карбоновой кислоты (смотри Схему 45) в виде масла желтого цвета (350 мг, избыток). MS (ESI): m/z=465.1 [М+Н]+.
трет-Бутил-3-(5-карбамоил-4-(4-феноксифенил)оксазол-2-ил)пиперидин-1-карбоксилат (8). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пиррол и дин-1-карбоксилата (смотри Схему 23) в виде твердого вещества бледно-желтого цвета (250 мг, избыток). MS (ESI): m/z=408.0 [М+Н-56]+.
4-(4-Феноксифенил)-2-(пиперидин-3-ил)оксазол-5-карбоксамид гидрохлорид (9). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде твердого вещества белого цвета (150 мг, избыток). MS (ESI): m/z=364.1 [М+Н]+.
Пример 42
2-(1-Акрилоилпиперидин-3-ил)-4-(4-феноксифенил)оксазол-5-карбоксамид
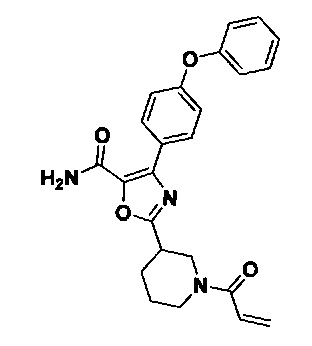
Указанное в заголовке соединение получали с использованием способа, аналогичного описанному ниже в Примере 43, в виде твердого вещества белого цвета (60 мг, 45%, четыре стадии). 1Н ЯМР (300 МГц, DMSO) δ 8.26 (d, J=9.0 Гц, 2Н), 7.92 (s, 1H), 7.67 (s, 1Н), 7.53-7.29 (m, 2Н), 7.25-7.13 (m, 1Н), 7.11-6.99 (m, 4Н), 6.85 (dd, J=16.7, 10.5 Гц, 1Н), 6.11 (dd, J=16.7, 2.4 Гц, 1Н), 5.68 (dd, J=10.4, 2.4 Гц, 1Н), 4.39-4.31 (m, 1H), 4.12-4.04 (m, 1Н), 3.33-3.14 (m, 2Н), 3.02-2.89 (m, 1Н), 2.14-2.06 (m, 2Н), 1.90-1.65 (m, 2Н). MS (ESI, методика A): m/z=418.1 [М+Н]+, tR=1.488 мин. ВЭЖХ: 97% (214 нм), 97% (254 нм).
Схема 33
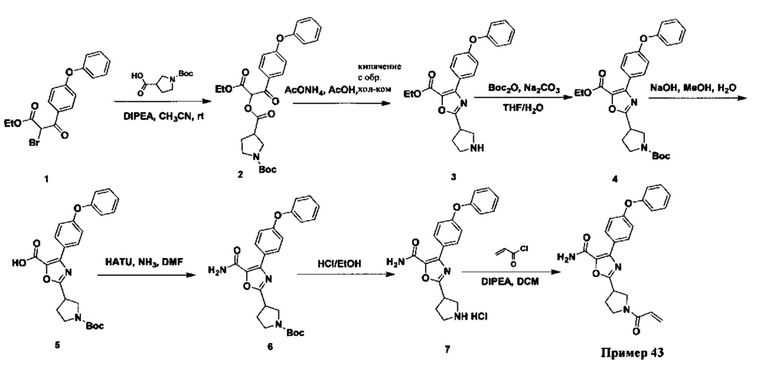
1-трет-Бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-ил)пирролидин-1,3-дикарбоксилат (2). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-трет-бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-ил)пиперидин-1,3-дикарбоксилата (смотри Схему 32) в виде масла бледно-желтого цвета (1.1 г, 80%). MS (ESI): m/z=441.8 [М+Н-56]+.
Этил 4-(4-феноксифенил)-2-(пирролидин-3-ил)оксазол-5-карбоксилат (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 4-(4-феноксифенил)-2-(пиперидин-3-ил)оксазол-5-карбоксилата (смотри Схему 32) в виде масла желтого цвета (0.80 г, сырое). MS (ESI): m/z=420.1 [М+Н+41]+.
Этил 2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-4-(4-феноксифенил)оксазол-5-карбоксилат (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксилата (смотри Схему 37) в виде масла бледно-желтого цвета (0.35 г, 33%, две стадии). MS (ESI): m/z=423.1 [М+Н-56]+.
2-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-4-(4-феноксифенил)оксазол-5-карбоновая кислота (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-4'-феноксибифенил-2-карбоновой кислоты (смотри Схему 45) в виде масла желтого цвета (350 мг, избыток). MS (ESI): m/z=395.0 [М+Н-56]+.
трет-Бутил-3-(5-карбамоил-4-(4-феноксифенил)оксазол-2-ил)пирролидин-1-карбоксилат (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пирролидин-1-карбоксилата (смотри Схему 23) в виде твердого вещества бледно-желтого цвета (250 мг, избыток). MS (ESI): m/z=394.1 [М+Н-56]+.
4-(4-Феноксифенил)-2-(пирролидин-3-ил)оксазол-5-карбоксамид гидрохлорид (7). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде твердого вещества белого цвета (150 мг, избыток). MS (ESI): m/z=350.1 [М+Н]+.
Пример 43
2-(1-Акрилоилпирролидин-3-ил)-4-(4-феноксифенил)оксазол-5-карбоксамид
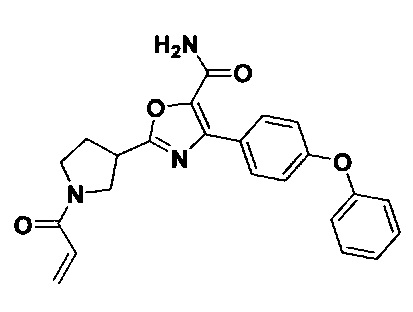
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 15 в виде твердого вещества белого цвета (45 мг, 34%, четыре стадии). 1Н ЯМР (400 МГц, DMSO) δ 8.27 (d, J=8.8 Гц, 2Н), 7.94 (d, J=8.0 Гц, 1Н), 7.70 (d, J=8.0 Гц, 1Н), 7.43 (t, J=7.9 Гц, 2Н), 7.19 (t, J=7.9 Гц, 1Н), 7.11-7.02 (m, 4Н), 6.69-6.55 (m, 1H), 6.19 (d, J=18.0 Гц, 1Н), 5.70 (d, J=10.4 Гц, 1H), 4.08-3.96 (m, 1H), 3.90-3.65 (m, 3H), 3.64-3.55 (m, 0.5Н), 3.53-3.45 (m, 0.5Н), 2.45-2.37 (m, 1Н), 2.36-2.27 (m, 1H). MS (ESI, методика A): m/z=404.1 [М+Н]+, tR=1.542 мин. ВЭЖХ: 98% (214 нм), 98% (254 нм).
Схема 34
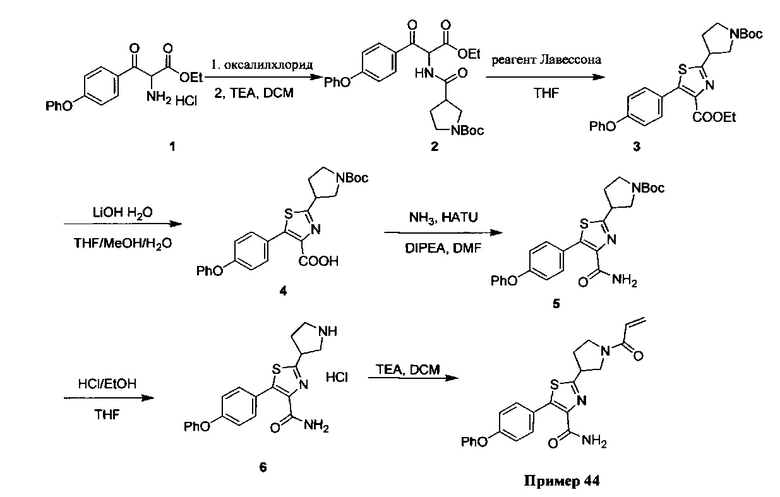
трет-Бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-илкарбамоил)пирролидин-1-карбоксилат (2). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-илкарбамоил)пиперидин-1-карбоксилата (смотри Схему 35) в виде масла желтого цвета (0.35 г, 30%). MS (ESI): m/z=440.7 [М-55]+.
Этил 2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-5-(4-феноксифенил)тиазол-4-карбоксилат (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-5-(4-феноксифенил)тиазол-4-карбоксилата (смотри Схему 35) в виде масла желтого цвета (0.12 г, 61%). MS (ESI): m/z=494.7 [М+Н]+.
2-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-5-(4-феноксифенил)тиазол-4-карбоновая кислота (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоновой кислоты (смотри Схему 46) в виде бесцветного масла (110 мг, 97%). MS (ESI): m/z=488.7 [М+Na]+.
трет-Бутил-3-(4-карбамоил-5-(4-феноксифенил)тиазол-2-ил)пирролидин-1-карбоксилат (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-Бутил-4-карбамоилпиперидин-1-карбоксилата (смотри Схему 42) в виде бесцветного масла (95 мг, 87%). MS (ESI): m/z=487.7 [М+Na]+.
5-(4-Феноксифенил)-2-(пирролидин-3-ил)тиазол-4-карбоксамид гидрохлорид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде бесцветного масла (90 мг, 100%). MS (ESI): m/z=365.8 [М+Н]+.
Пример 44
2-(1-Акрилоилпирролидин-3-ил)-5-(4-феноксифенил)тиазол-4-карбоксамид
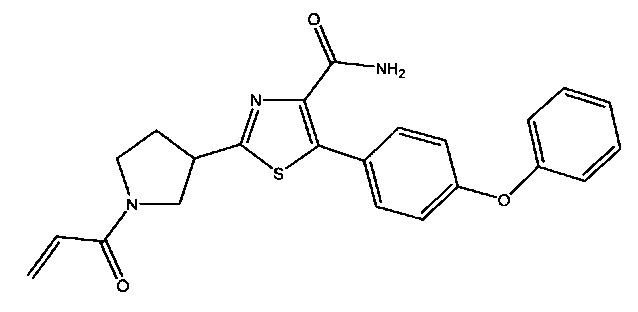
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 3 в виде твердого вещества белого цвета (30 мг, 30%). 1Н ЯМР (300 МГц, CD3OD) δ 7.52 (d, J=9.0 Гц, 2Н), 7.37 (d, J=9.0 Гц, 2Н), 7.15 (t, J=7.4 Гц, 1Н), 7.06-7.02 (m, 2Н), 6.99-6.94 (m, 2Н), 6.64 (ddd, J=16.9, 10.4, 7.2 Гц, 1Н), 6.29 (dt, J=16.8, 1.9 Гц, 1Н), 5.76 (dt, J=10.4, 1.7 Гц, 1Н), 4.19-4.05 (m, 0.5Н), 4.03-3.82 (m, 3Н), 3.83-3.71 (m, 1Н), 3.65-3.55 (m, 0.5Н), 2.61-2.23 (m, 2Н). MS (ESI, методика F): m/z=419.7 [М+Н]+, tR=1.487 мин. ВЭЖХ: 99.3% (214 нм), 98.7% (254 нм).
Схема 35
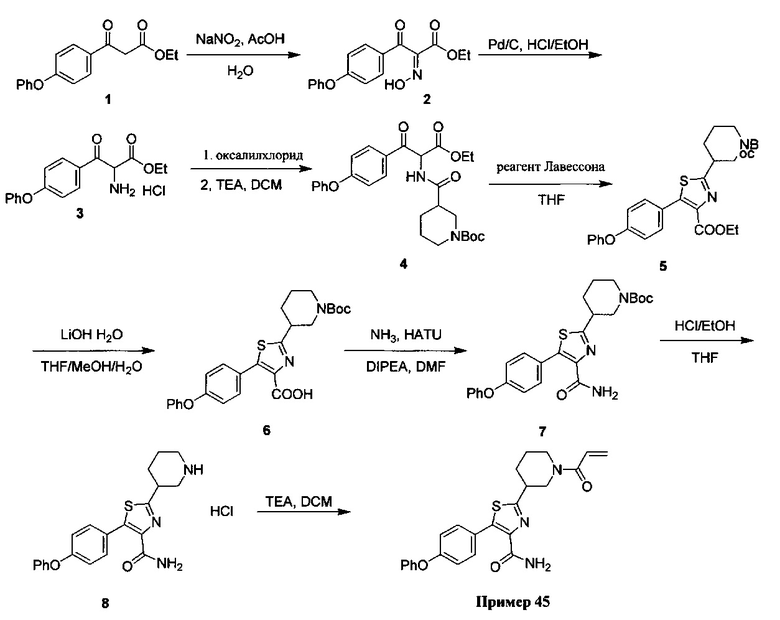
(Е)-этил 2-(гидроксиимино)-3-оксо-3-(4-феноксифенил)пропаноат (2). К раствору этил 3-оксо-3-(4-феноксифенил)пропаноата 1 (1 г, 3.52 ммоль) в АсОН (15 мл) добавляли раствор NaNO2 (0.364 г, 5.28 ммоль) в воде (10 мл) при 0°С. Смесь перемешивали при температуре окружающей среды в течение 15 ч. После окончания смесь экстрагировали этилацетатом (2×20 мл). Органические слои промывали водой (2×10 мл) и рассолом (2×10 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения (1.1 г, 100%) в виде твердого вещества желтого цвета. MS (ESI): m/z=313.8 [М+Н]+.
Этил 2-амино-3-оксо-3-(4-феноксифенил)пропаноат гидрохлорид (3). К раствору (Е)-этил 2-(гидроксиимино)-3-оксо-3-(4-феноксифенил)пропаноата 2 (6 г, 19.15 ммоль) в МеОН (50 мл) добавляли Pd/C (10%, 500 мг) и HCl/EtOH (33%, 20 мл) в атмосфере водорода, и полученный в результате раствор перемешивали в течение 24 ч при температуре окружающей среды, затем фильтровали, и фильтрат концентрировали. Сырой продукт очищали с помощью препаративной ВЭЖХ, элюируя смесью CH3CN/H2O (содержащей 0.5% TFA) от 10:90 до 60:40 c получением указанного в заголовке соединения (3,5 г, 61%) в виде масла желтого цвета. MS (ESI): m/z=299.9 [M+H]+.
трет-Бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-илкарбамоил)пиперидин-1-карбоксилат (4). К раствору 1-(трет-бутоксикарбонил)пиперидин-3-карбоновой кислоты (0.5 г, 2.18 ммоль) в DCM (10 мл) добавляли оксалилхлорид (0.415 г, 3.27 ммоль) и 5 капель DMF. Смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали с получением сырой смеси. К раствору этил 2-амино-3-оксо-3-(4-феноксифенил)пропаноат гидрохлорида 3 (0.73 г, 2.18 ммоль) в DCM (20 мл) добавляли TEA (0.66 г, 6.54 ммоль) и раствор полученной выше смеси в DCM (10 мл). Полученный в результате раствор перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь промывали рассолом (2×10 мл), высушивали над Na2SO4, фильтровали и концентрировали с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 1:1 РЕ/ЕА, с получением указанного в заголовке соединения (0.16 г, 14%) в виде масла желтого цвета. MS (ESI): m/z=454.7 [М-55]+.
Этил 2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-5-(4-феноксифенил)тиазол-4-карбоксилат (5). К раствору трет-бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-илкарбамоил)пиперидин-1-карбоксилата 4 (0.16 г, 0.31 ммоль) в THF (20 мл) добавляли реагент Лавессона (0.127 г, 0.31 ммоль). Смесь дегазировали N2 3 раза, затем нагревали до кипения с обратным холодильником и перемешивали в течение 2 ч. После окончания смесь экстрагировали этилацетатом (2×20 мл). Органические слои промывали насыщенным NaHCO3 (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 2:1 РЕ/ЕА, с получением указанного в заголовке соединения (90 мг, 57%) в виде масла желтого цвета. MS (ESI): m/z=508.8 [М+Н]+.
2-(1-(трет-Бутоксикарбонил)пиперидин-3-ил)-5-(4-феноксифенил)тиазол-4-карбоновая кислота (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоновой кислоты (смотри Схему 46) в виде твердого вещества белого цвета (80 мг, 94%). MS (ESI): m/z=480.8 [М+Н]+.
трет-Бутил-3-(4-карбамоил-5-(4-феноксифенил)тиазол-2-ил)пиперидин-1-карбоксилат (7). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-4-карбамоилпиперидин-1-карбоксилата (смотри Схему 42) в виде бесцветного масла (40 мг, 50%). MS (ESI): m/z=479.7 [М+Н]+.
5-(4-Феноксифенил)-2-(пиперидин-3-ил)тиазол-4-карбоксамид гидрохлорид (8). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде масла желтого цвета (35 мг, 100%). MS (ESI): m/z=379.8[М+Н]+.
Пример 45
2-(1-Акрилоилпиперидин-3-ил)-5-(4-феноксифенил)тиазол-4-карбоксамид
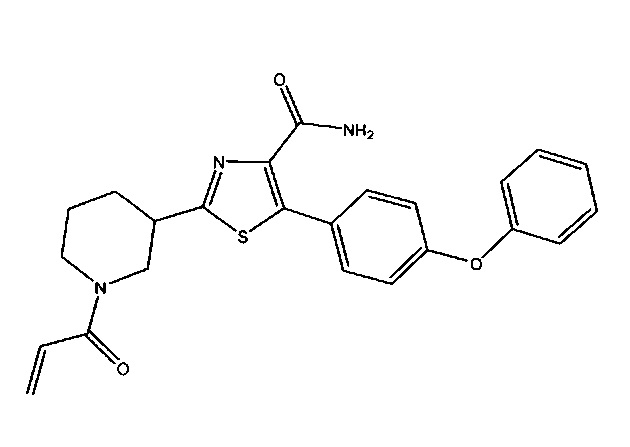
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 3 в виде твердого вещества белого цвета (10 мг, 27%). 1Н ЯМР (300 МГц, CD3OD) δ 7.56-7.48 (m, 2Н), 7.44-7.32 (m, 2Н), 7.15 (t, J=7.4 Гц, 1Н), 7.03 (d, J=0.9 Гц, 2Н), 6.99-6.94 (m, 2Н), 6.84 (ddd, J=23.2, 16.7, 10.6 Гц, 1H), 6.21 (dd, J=16.8, 1.9 Гц, 1Н), 5.75 (d, J=10.7 Гц, 1Н), 4.62 (d, J=11.6 Гц, 0.5Н), 4.27 (d, J=13.2 Гц, 0.5Н), 4.15 (d, J=12.8 Гц, 0.5H), 4.00 (d, J=13.6 Гц, 0.5H), 3.72 (dd, J=13.5, 8.9 Гц, 0.5H), 3.42-3.33 (m, 1H), 3.29-3.15 (m, 1.5H), 2.35-2.23 (m, 1H), 2.08-1.85 (m, 2H), 1.75-1.55 (m, 1H). MS (ESI, методика F): m/z=433.8 [M+H]+, tR=1.557 мин. ВЭЖХ: 96.7% (214 нм), 98.0% (254 нм).
Схема 115
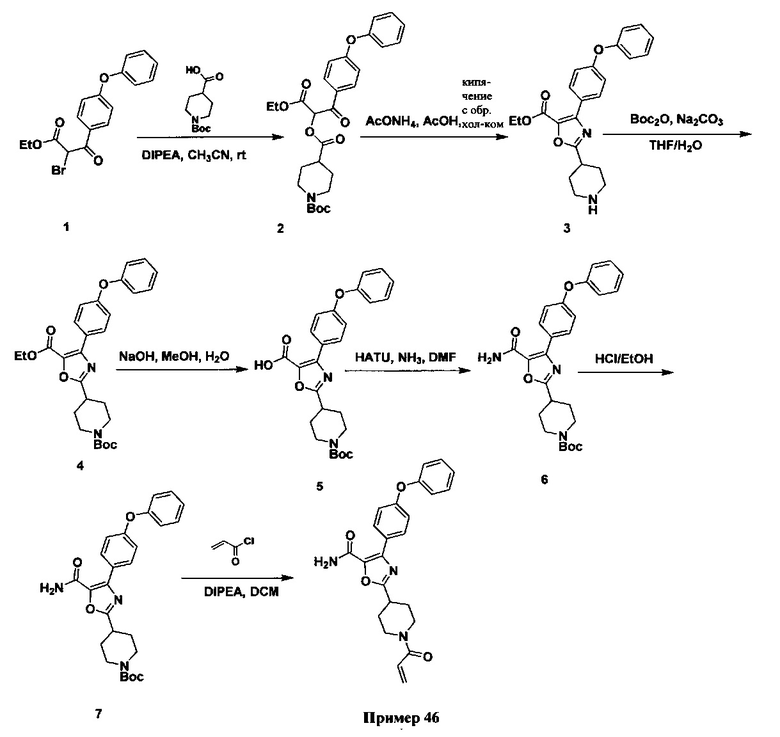
1-трет-Бутил-4-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-ил)пиперидин-1,4-дикарбоксилат (2). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-трет-бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-ил)пиперидин-1,3-дикарбоксилата (смотри Схему 32) в виде масла бледно-желтого цвета (0.85 г, 60%). MS (ESI): m/z=456.1 [М+Н-56]+.
Этил 4-(4-феноксифенил)-2-(пиперидин-4-ил)оксазол-5-карбоксилат (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 4-(4-феноксифенил)-2-(пиперидин-3-ил)оксазол-5-карбоксилата (смотри Схему 32) в виде масла желтого цвета (0.80 г, сырое). MS (ESI): m/z=393.2 [М+Н]+.
Этил-2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-4-(4-феноксифенил)оксазол-5-карбоксилат (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксилата (смотри Схему 37) в виде масла бледно-желтого цвета (0.85 г, 68%, две стадии). MS (ESI): m/z=437.2 [М+Н-56]+.
2-(1-(трет-Бутоксикарбонил)пиперидин-4-ил)-4-(4-феноксифенил)оксазол-5-карбоновая кислота (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-4'-феноксибифенил-2-карбоновой кислоты (смотри Схему 45) в виде масла желтого цвета (0.8 г, 99%). MS (ESI): m/z=409.1 [М+Н-56]+.
трет-Бутил-4-(5-карбамоил-4-(4-феноксифенил)оксазол-2-ил)пиперидин-1-карбоксилат (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пирролидин-1-карбоксилата (смотри Схему 23) в виде твердого вещества бледно-желтого цвета (390 мг, избыток). MS (ESI): m/z=408.0 [М+Н-56]+.
4-(4-Феноксифенил)-2-(пиперидин-4-ил)оксазол-5-карбоксамид гидрохлорид (7). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде твердого вещества белого цвета (400 мг, избыток). MS (ESI): m/z=364.1 [М+Н]+.
Пример 46
2-(1-Акрилоилпиперидин-4-ил)-4-(4-феноксифенил)оксазол-5-карбоксамид
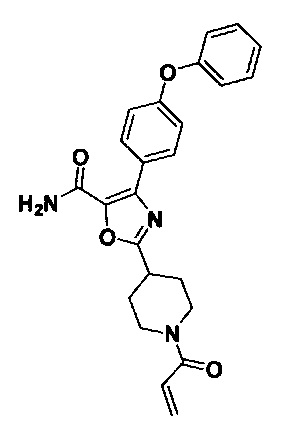
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 15 в виде твердого вещества белого цвета (45 мг, 34%, три стадии). 1Н ЯМР (300 МГц, DMSO) δ 8.26 (d, J=9.0 Гц, 2Н), 7.92 (s, 1Н), 7.674 (s, 1H), 7.53-7.29 (m, 2Н), 7.25-7.13 (m, 1Н), 7.11-6.99 (m, 4Н), 6.85 (dd, J=16.7, 10.5 Гц, 1Н), 6.11 (dd, J=16.7, 2.4 Гц, 1Н), 5.68 (dd, J=10.4, 2.4 Гц, 1H), 4.39-4.31 (m, 1Н), 4.12-4.04 (m, 1H), 3.33-3.14 (m, 2Н), 3.02-2.89 (m, 1H), 2.14-2.06 (m, 2Н), 1.90-1.65 (m, 2Н). MS (ESI, методика A): m/z=418.1 [М+Н]+, tR=1.489 мин. ВЭЖХ: 97% (214 нм), 97% (254 нм).
Схема 37
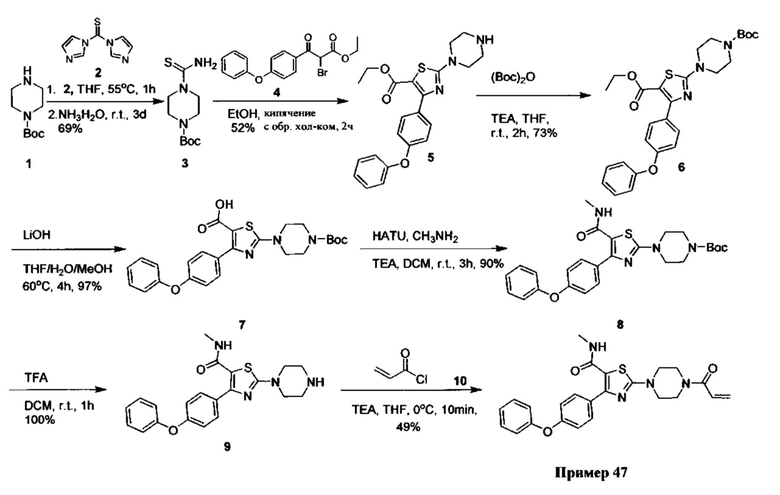
трет-Бутил-4-карбамотиоилпиперазин-1-карбоксилат (3). К раствору ди(1Н-имидазол-1-ил)метантиона 2 (2.14 г, 12 ммоль) в безводном THF (30 мл) добавляли трет-бутил пиперазин-1-карбоксилат 1 (1.86 г, 10 ммоль) при температуре окружающей среды. Смесь оставляли перемешиваться при температуре окружающей среды в течение 2 ч, затем смесь нагревали при 55°C в течение 1 ч. Смесь концентрировали под вакуумом до около половины объема. К оставшейся реакционной смеси добавляли 2 М раствор аммиака в метаноле (20 мл) и оставляли перемешиваться при температуре окружающей среды в течение 3 дней. Растворитель удаляли, и остаток очищали с помощью хроматографии, элюируя смесью 50:1 DCM/MeOH, с получением указанного в заголовке соединения (1.7 г. 69%) в виде твердого вещества белого цвета. МS (ESI): m/z=246.1 [M+H]+.
Этил 4-(4-феноксифенил)-2-(пиперазин-1-ил)тиазол-5-карбоксилат (5). К раствору этил 2-бром-3-оксо-3-(4-феноксифенил)пропаноата 4 (362 мг, 1 ммоль) в этаноле (10 мл) добавляли трет-бутил-4-карбамотиоилпиперазин-1-карбоксилат 3 (245 мг, 1 ммоль) при температуре окружающей среды. Смесь затем нагревали до кипения с обратным холодильником и перемешивали в течение 2 ч. После охлаждения до температуры окружающей среды растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя от 40:1 до 20:1 смесью DCM/MeOH, с получением указанного в заголовке соединения (214 мг, 52%) в виде твердого вещества коричневого цвета. MS (ESI): m/z=410.1 [М+Н]+.
Этил 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксилат (6). К соединению этил 4-(4-феноксифенил)-2-(пиперазин-1-ил)тиазол-5-карбоксилат 5 (214 мг, 0.51 ммоль) добавляли ди-трет-бутилдикарбонат (134 мг, 0.6 ммоль) и TEA (0.2 мл, 1.5 ммоль) при температуре окружающей среды. Смесь затем перемешивали при температуре окружающей среды в течение 2 ч. Растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 50:1 DCM/MeOH, с получением указанного в заголовке соединения (190 мг, 73%) в виде твердого вещества коричневого цвета. MS (ESI): m/z=510.1 [М+Н]+.
2-(4-(трет-Бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоновая кислота (7). К раствору этил 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксилата 6 (190 мг, 0.37 ммоль) в THF (50 мл)/H2O (1 мл)/МеОН (3 мл) добавляли гидроксид лития (30 мг, 0.74 ммоль) при температуре окружающей среды. Смесь затем нагревали до 60°C и перемешивали в течение 2 ч. После охлаждения до температуры окружающей среды растворитель удаляли, и остаток очищали с помощью хроматографии, элюируя смесью 10:1 DCM/MeOH, с получением указанного в заголовке соединения (173 мг, 97%) в виде твердого вещества белого цвета. MS (ESI): m/z=482.0 [М+Н]+.
трет-Бутил-4-(5-(метилкарбамоил)-4-(4-феноксифенил)тиазол-2-ил)пиперазин-1-карбоксилат (8). К раствору 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоновой кислоты 7 (124 мг, 0.26 ммоль) в DCM (10 мл) добавляли HATU (117 мг, 0.3 ммоль) и метанамин (0.26 мл, 0.26 ммоль) при температуре окружающей среды. Смесь перемешивали при температуре окружающей среды в течение 1 ч. Растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 1:5 ЕА/РЕ, с получением указанного в заголовке соединения (115 мг, 90%) в виде бесцветного масла. MS (ESI): m/z=495.2 [М+Н]+.
N-метил-4-(4-феноксифенил)-2-(пиперазин-1-ил)тиазол-5-карбоксамид (9). К раствору трет-бутил-4-(5-(метилкарбамоил)-4-(4-феноксифенил)тиазол-2-ил)пиперазин-1-карбоксилата 8 (115 мг, 0.23 ммоль) в DCM (5 мл) добавляли TFA (1 мл) при температуре окружающей среды. Смесь перемешивали в течение 1 ч при температуре окружающей среды. Растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 10:1 DCM/MeOH, с получением указанного в заголовке соединения (117 мг, 100%) в виде бесцветного масла.
Пример 47
2-(4-Акрилоилпиперазин-1-ил)-N-метил-4-(4-феноксифенил)тиазол-5-карбоксамид
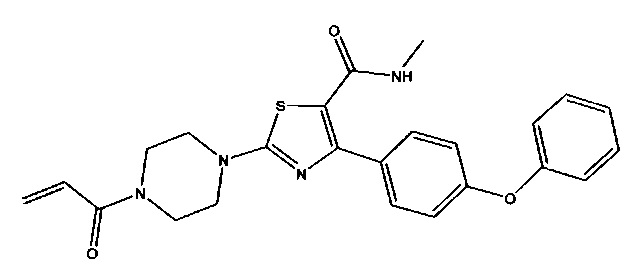
К раствору N-метил-4-(4-феноксифенил)-2-(пиперазин-1-ил)тиазол-5-карбоксамида 9 (117 мг, 0.23 ммоль) в DCM (5 мл) добавляли TEA (0.1 мл, 0.7 ммоль) и акрилоилхлорид 10 (0.21 мг, 0.23 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя от 50:1 до 20:1 смесью DCM/MeOH, с получением указанного в заголовке соединения (50 мг, 49%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 7.57 (d, J=8.6 Гц, 2Н), 7.40 (t, J=7.9 Гц, 2Н), 7.19 (t, J=7.4 Гц, 1Н), 7.09 (d, J=8.4 Гц, 4Н), 6.60 (dd, J=16.8, 10.5 Гц, 1Н), 6.37 (d, J=16.7 Гц, 1Н), 5.79 (d, J=11.8 Гц, 1Н), 5.59 (d, J=4.1 Гц, 1Н), 3.78 (d, J=48.5 Гц, 4Н), 3.62 (s, 4Н), 2.80 (d, J=4.8 Гц, 3Н), MS (ESI, методика A): m/z=449.1 [М+Н]+, tR=1.528 мин. ВЭЖХ: 96.9% (214 нм), 96.9% (254 нм).
Схема 120
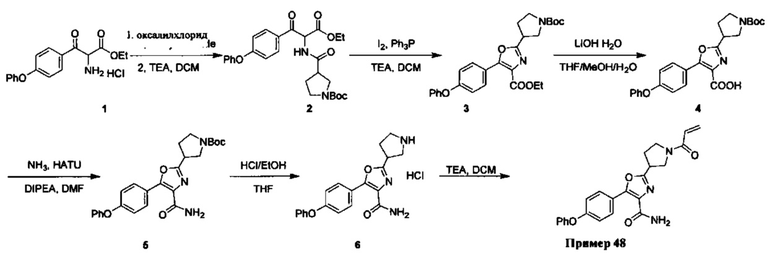
трет-Бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-илкарбамоил)пирролидин-1-карбоксилат (2). Указанное в заголовке соединение получали, используя способ, аналогичный описанному в Общей Схеме и Примере 15 в виде масла желтого цвета (0.35 г, 30%). MS (ESI): m/z=440.7 [М-55]+.
Этил-2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-5-(4-феноксифенил)оксазол-4-карбоксилат (3). К раствору Ph3P (159 мг, 0.604 ммоль) в DCM (20 мл) добавляли I2 (153 мг, 0.604 ммоль). Полученную в результате смесь дегазировали N2 3 раза и перемешивали при температуре окружающей среды в течение 2 ч. Добавляли TEA (122 мг, 1.208 ммоль) и перемешивали в течение 10 мин, затем добавляли раствор трет-бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-илкарбамоил)пирролидин-1-карбоксилата 2 (150 мг, 0.302 ммоль) и перемешивали в течение 15 ч. Смесь разбавляли этилацетатом (50 мл) и промывали насыщенным Na2S2O3 (20 мл), рассолом (3×20 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 1:1 РЕ/ЕА, с получением указанного в заголовке соединения (0.11 г, 76%) в виде бесцветного масла. MS (ESI): m/z=422.8 [М-55]+.
2-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-5-(4-феноксифенил)оксазол-4-карбоновая кислота (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоновой кислоты (смотри Схему 46) в виде масла желтого цвета (110 мг, 100%). MS (ESI): m/z=394.8 [М-55]+.
трет-Бутил-3-(4-карбамоил-5-(4-феноксифенил)оксазол-2-ил)пирролидин-1-карбоксилат (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-4-карбамоилпиперидин-1-карбоксилата (смотри Схему 42) в виде бесцветного масла (95 мг, 92%). MS (ESI): m/z=393.8 [М-55]+.
5-(4-Феноксифенил)-2-(пирролидин-3-ил)оксазол-4-карбоксамид гидрохлорид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде твердого вещества белого цвета (75 мг, 92%). MS (ESI): m/z=349.9 [М+Н]+.
Пример 48
2-(1-Акрилоилпирролидин-3-ил)-5-(4-феноксифенил)оксазол-4-карбоксамид
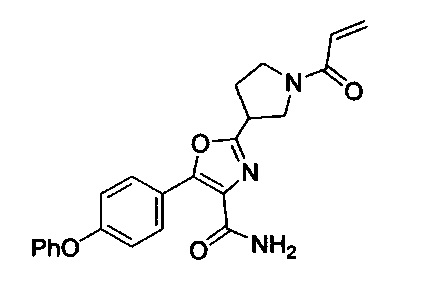
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1 в виде твердого вещества не совсем белого цвета (50 мг, 64%). 1Н ЯМР (400 МГц, DMSO) δ 8.27-8.22 (m, 2Н), 7.61 (s, 1Н), 7.58 (s, 1H), 7.45 (t, J=7.9 Гц, 2Н), 7.21 (t, J=7.3 Гц, 1Н), 7.14-7.06 (m, 4Н), 6.67-6.58 (m, 1Н), 6.16 (d, J=16.7 Гц, 1H), 5.70 (d, J=10.4 Гц, 1H), 4.05-4.01 (m, 0.5Н), 3.97-3.92 (m, 0.5Н), 3.90-3.59 (m, 3.5Н), 3.53-3.45 (m, 0.5Н), 2.45-2.21 (m, 2Н). MS (ESI, методика A): m/z=404.1 [М+Н]+, tR=1.484 мин. ВЭЖХ: 99.5% (214 нм), 99.3% (254 нм).
Схема 122
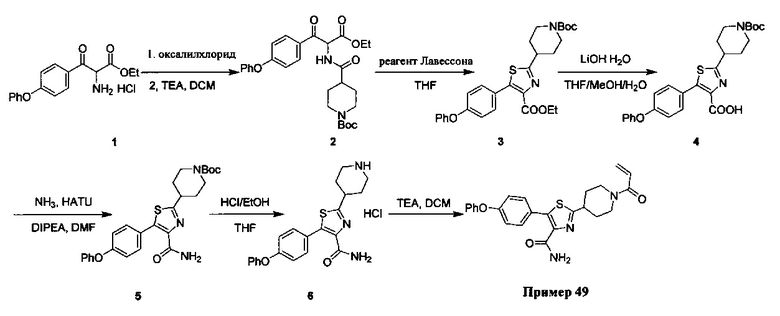
трет-Бутил-4-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-илкарбамоил)пиперидин-1-карбоксилат (2). Указанное в заголовке соединение получали, используя способ, аналогичный описанному в Общей Схеме и Примере 15 в виде бесцветного масла (0.18 г, 16%). MS (ESI): m/z=454.8[М-55]+.
Этил 2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-5-(4-феноксифенил)тиазол-4-карбоксилат (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-2-карбамотиоилпиперидин-1-карбоксилата (смотри Схему 30) в виде бесцветного масла (0.12 г, 67%). MS (ESI): m/z=508.8 [М+Н]+.
2-(1-(трет-Бутоксикарбонил)пиперидин-4-ил)-5-(4-феноксифенил)тиазол-4-карбоновая кислота (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоновой кислоты (смотри Схему 46) в виде масла желтого цвета (120 мг, 100%). MS (ESI): m/z=480.8 [М+Н]+.
трет-Бутил-4-(4-карбамоил-5-(4-феноксифенил)тиазол-2-ил)пиперидин-1-карбоксилат (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пиперидин-1-карбоксилата (смотри Схему 30) в виде бесцветного масла (100 мг, 88%). MS (ESI): m/z=479.8 [М+Н]+.
5-(4-Феноксифенил)-2-(пиперидин-4-ил)тиазол-4-карбоксамид гидрохлорид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде твердого вещества белого цвета (80 мг, 92%). MS (ESI): m/z=379.8 [М+Н]+.
Пример 49
2-(1-Акрилоилпиперидин-4-ил)-5-(4-феноксифенил)тиазол-4-карбоксамид
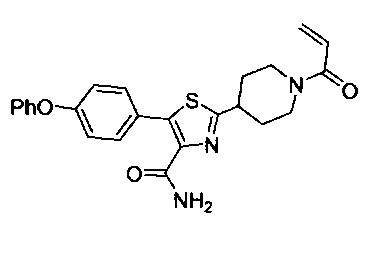
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 51 в виде твердого вещества не совсем белого цвета (20 мг, 24%). 1Н ЯМР (400 МГц, DMSO) δ 7.68 (s, 1Н), 7.56 (d, J=8.7 Гц, 2Н), 7.50 (s, 1Н), 7.47-7.42 (m, 2Н), 7.21 (t, J=7.4 Гц, 1Н), 7.10 (d, J=7.7 Гц, 2Н), 7.00 (d, J=8.7 Гц, 2Н), 6.85 (dd, J=16.7, 10.5 Гц, 1H), 6.12 (dd, J=16.7, 2.4 Гц, 1Н), 5.69 (dd, J=10.5, 2.4 Гц, 1Н), 4.49 (d, J=12.8 Гц, 1Н), 4.15 (d, J=12.8 Гц, 1Н), 3.33-3.19 (m, 2Н), 2.88 (d, J=11.5 Гц, 1Н), 2.12 (d, J=12.8 Гц, 2Н), 1.73-1.55 (m, 2Н). MS (ESI, методика A): m/z=434.0 [М+Н]+, tR=1.606 мин. ВЭЖХ: 95.0% (214 нм), 96.6% (254 нм).
Схема 40
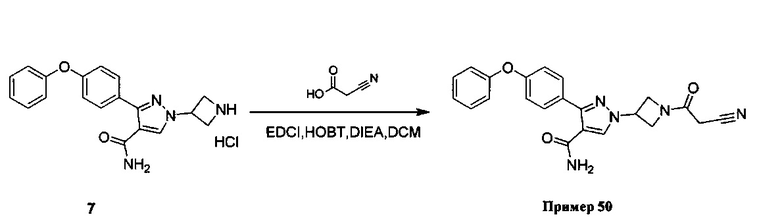
Пример 50
1-(1-(2-Цианоацетил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
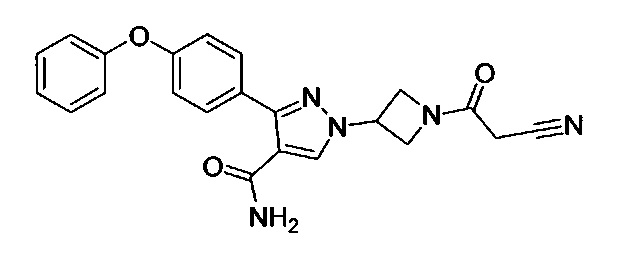
К раствору 1-(азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида 7 (смотри схему Примера 15, соответствующие данные и методики) (900 мг, 2.4 ммоль), 2-цианоуксусной кислоты (178 мг, 2.1 ммоль), EDCI (534 мг, 2.8 ммоль) и НОВТ (64 мг, 0.48 ммоль) в сухом DCM (20 мл) добавляли DIPEA (936 мг, 7.2 ммоль) при 0°C, затем раствор оставляли медленно нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 1 ч. После завершения реакции раствор промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке с силикагелем, элюируя смесью 30:1 DCM/MeOH, с получением указанного в заголовке соединения (250 мг, 26%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 8.18 (s, 1Н), 7.61-7.50 (m, 2H), 7.43-7.32 (m, 2H), 7.20-7.13 (m, 1H), 7.12-7.03 (m, 4H), 5.65 (s, 2H), 5.24-5.14 (m, 1H), 4.87-4.73 (m, 2H), 4.62-4.49 (m, 2H), 3.35 (s, 2H). MS (ESI, методика A): m/z=402.1 [M+H]+, tR=1.361 мин. ВЭЖХ: 98.0% (214 нм), 98.6% (254 нм).
Схема 125
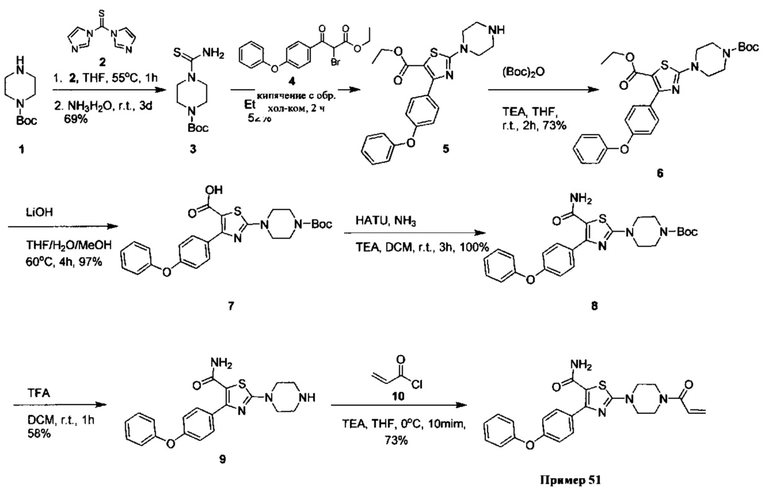
трет-Бутил-4-карбамотиоилпиперазин-1-карбоксилат (3). К раствору ди(1Н-имидазол-1-ил)метантиона 2 (2.14 г, 12 ммоль) в безводном THF (30 мл) добавляли трет-бутил пиперазин-1-карбоксилат 1 (1.86 г, 10 ммоль) при температуре окружающей среды. Смесь оставляли перемешиваться при температуре окружающей среды в течение 2 ч, и затем смесь нагревали до 55°C в течение 1 ч. Смесь концентрировали под вакуумом до около половины объема. К оставшейся реакционной смеси добавляли 2 М аммиак в метаноле (20 мл) и оставляли перемешиваться при температуре окружающей среды в течение 3 дней. Растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 50:1 DCM/MeOH, с получением указанного в заголовке соединения (1.7 г, 69%) в виде твердого вещества белого цвета. MS (ESI): m/z=246.1 [М+Н]+.
Этил 4-(4-феноксифенил)-2-(пиперазин-1-ил)тиазол-5-карбоксилат (5). К раствору этил 2-бром-3-оксо-3-(4-феноксифенил)пропаноата 4 (362 мг, 1 ммоль) в этаноле (10 мл) добавляли трет-бутил-4-карбамотиоилпиперазин-1-карбоксилат 3 (245 мг, 1 ммоль) при температуре окружающей среды. Смесь затем нагревали до кипения с обратным холодильником и перемешивали в течение 2 ч. После охлаждения до температуры окружающей среды растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя от 40:1 до 20:1 DCM/MeOH, с получением указанного в заголовке соединения (214 мг, 52%) в виде твердого вещества коричневого цвета. MS (ESI): m/z=410.1 [М+Н]+.
Этил-2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксилат (6). К соединению этил 4-(4-феноксифенил)-2-(пиперазин-1-ил)тиазол-5-карбоксилат 5 (214 мг, 0.51 ммоль) добавляли ди-трет-бутил-дикарбонат (134 мг, 0.6 ммоль) и TEA (0.2 мл, 1.5 ммоль) при температуре окружающей среды. Смесь затем перемешивали при температуре окружающей среды в течение 2 ч. Растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 50:1 DCM/MeOH, с получением указанного в заголовке соединения (190 мг, 73%) в виде твердого вещества коричневого цвета. MS (ESI): m/z=510.1 [М+Н]+.
2-(4-(трет-Бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоновая кислота (7). К раствору этил 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксилата 6 (190 мг, 0.37 ммоль) в THF (50 мл)/H2O (1 мл)/МеОН (3 мл) добавляли гидроксид лития (30 мг, 0.74 ммоль) при температуре окружающей среды. Смесь затем нагревали до 60°C и перемешивали в течение 2 ч. После охлаждения до температуры окружающей среды растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 10:1 DCM/MeOH, с получением указанного в заголовке соединения (173 мг, 97%) в виде твердого вещества белого цвета. MS (ESI): m/z=482.0 [М+Н]+.
трет-Бутил-4-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пиперазин-1-карбоксилат (8). К раствору 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоновой кислоты 7 (173 мг, 4.6 ммоль) в DCM (10 мл) добавляли HATU (164 мг, 0.43 ммоль) при температуре окружающей среды. Смесь дегазировали три раза газообразным аммиаком и перемешивали при температуре окружающей среды в атмосфере аммиака в течение 6 ч. Растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 30:1 DCM/MeOH, с получением указанного в заголовке соединения (170 мг, 100%) в виде твердого вещества белого цвета. MS (ESI): m/z=481.1 [М+Н]+.
4-(4-Феноксифенил)-2-(пиперазин-1-ил)тиазол-5-карбоксамид (9). Раствор трет-бутил-4-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пиперазин-1-карбоксилата 8 (170 мг, 0.35 ммоль) в DCM (5 мл) и TFA (1 мл) при температуре окружающей среды перемешивали в течение 1 ч. Растворитель удаляли, и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя от 20:1 до 10:1 смесью DCM/MeOH, с получением указанного в заголовке соединения (110 мг, 58%) в виде твердого вещества белого цвета.
Пример 51
2-(4-Акрилоилпиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксамид
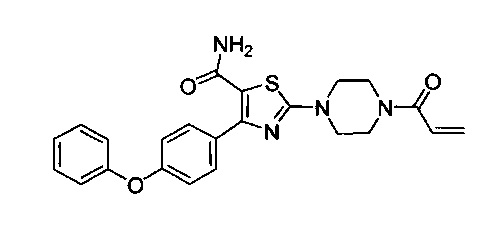
К раствору 4-(4-феноксифенил)-2-(пиперазин-1-ил)тиазол-5-карбоксамида 9 (110 мг, 0.22 ммоль) в DCM (5 мл) добавляли TEA (0.1 мл, 0.66 ммоль) и акрилоилхлорид 10 (0.20 мг, 0.22 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 10 минут. Растворитель удаляли, и остаток очищали с помощью препаративной TLC, элюируя смесью 25:1 DCM/MeOH, с получением указанного в заголовке соединения (70 мг, 73%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, MeOD) δ 7.66-7.61 (m, 2Н), 7.37-7.43 (m, 2Н), 7.18 (t, J=7.4 Гц, 1H), 7.10-7.04 (m, 4H), 6.83 (dd, J=16.8, 10.6 Гц, 1H), 6.27 (dd, J=16.8, 1.8 Гц, 1H), 5.81 (dd,J=10.6, 1.9 Гц, 1H), 3.87-3.78 (m, 4H), 3.69-3.61 (m, 4H). MS (ESI, методика A): m/z=435.0 [M+H]+, tR=1.539 мин, ВЭЖХ: 95.5% (214 нм), 96.4% (254 нм).
Схема 128
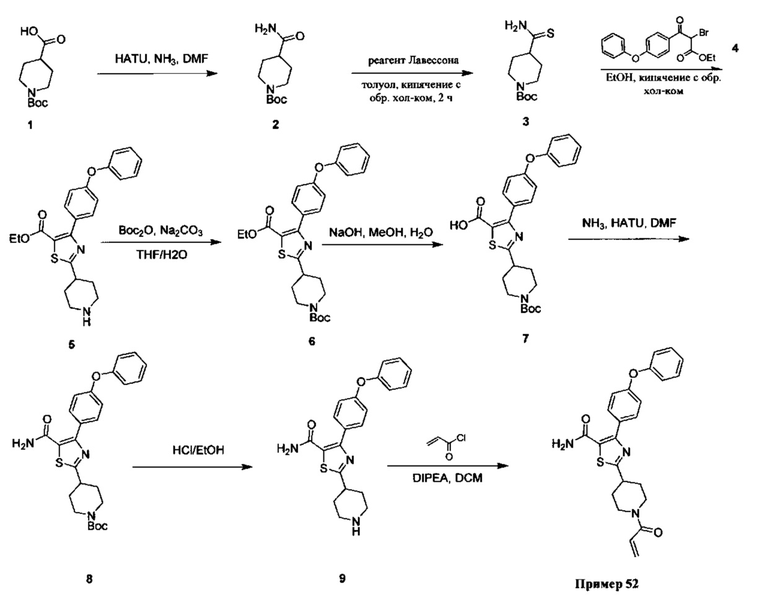
трет-Бутил-4-карбамоилпиперидин-1-карбоксилат (2). К смеси 1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоты 1 (2.29 г, 10 ммоль), DIEA (3.87 г, 30 ммоль) и HATU (4.18 г, 11 ммоль) в DMF (50 мл) добавляли NH3, барботируя в течение 20 мин. Полученную в результате смесь перемешивали при комнатной температуре на протяжении ночи. Смесь разбавляли водой (200 мл) и экстрагировали ЕА (2×50 мл). Органические слои объединяли, промывали рассолом, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток наносили на колонку с силикагелем, элюируя ЕА, с получением указанного в заголовке соединения (1.3 г, 56%) в виде твердого вещества белого цвета. MS (ESI): m/z=173.2 [М+Н-56]+.
трет-Бутил-4-карбамотиоилпиперидин-1-карбоксилат (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения циклопентанкарботиоамида (смотри Схему 30) в виде твердого вещества бледно-желтого цвета (0.35 г, 29%). MS (ESI): m/z=245.2 [М+Н]+.
Этил 4-(4-феноксифенил)-2-(пиперидин-4-ил)тиазол-5-карбоксилат (5). Неочищенное указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 2-циклопентил-4-(4-феноксифенил)тиазол-5-карбоксилата (смотри Схему А-2) в виде масла желтого цвета (0.90 г, избыток). MS (ESI): m/z=408.8 [М+Н]+.
Этил 4-(4-феноксифенил)-2-(пиперидин-4-ил)тиазол-5-карбоксилат (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксилата (смотри Схему 37) в виде твердого вещества желтого цвета (0.30 г, 30%, две стадии). MS (ESI): m/z=509.1 [М+Н]+.
2-(1-(трет-Бутоксикарбонил)пиперидин-4-ил)-4-(4-феноксифенил)тиазол-5-карбоновая кислота (7). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-4'-феноксибифенил-2-карбоновой кислоты (смотри Схему 45) в виде масла желтого цвета (250 мг, избыток). MS (ESI): m/z=481.0 [М+Н]+.
трет-Бутил-4-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пиперидин-1-карбоксилат (8). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пирролидин-1-карбоксилата (смотри Схему 23) в виде твердого вещества бледно-желтого цвета (250 мг, избыток). MS (ESI): m/z=480.1 [М+Н]+.
4-(4-Феноксифенил)-2-(пиперидин-4-ил)тиазол-5-карбоксамид (9). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде твердого вещества белого цвета (300 мг, избыток). MS (ESI): m/z=380.1 [М+Н]+.
Пример 52
2-(1-Акрилоилпиперидин-4-ил)-4-(4-феноксифенил)тиазол-5-карбоксамид
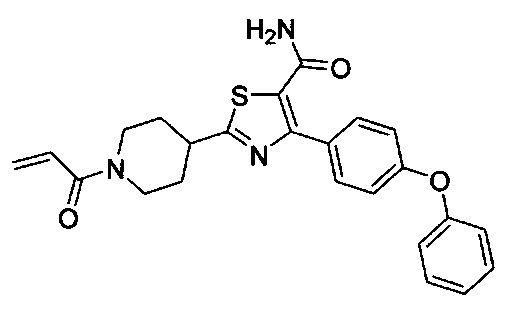
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 15, в виде твердого вещества белого цвета (50 мг, 23%, четыре стадии). 1Н ЯМР (300 МГц, DMSO) δ 7.76-7.69 (m, 4Н), 7.42 (t, J=7.4 Гц, 2Н), 7.19 (t, J=7.4 Гц, 1Н), 7.09-7.00 (m, 4Н), 6.84 (dd, J=16.7, 10.4 Гц, 1Н), 6.10 (dd, J=16.7, 2.4 Гц, 1Н), 5.68 (dd, J=10.4, 2.4 Гц, 1Н), 4.52-4.41 (m, 1Н), 4.19-4.08 (m, 1Н), 3.32-3.15 (m, 2Н), 2.90-2.78 (m, 1Н), 2.14-2.06 (m, 2Н), 1.73-1.46 (m, 2Н). MS (ESI, методика A): m/z=434.0 [М+Н]+, tR=1.540 мин. ВЭЖХ: 96% (214 нм), 96% (254 нм).
Схема 43
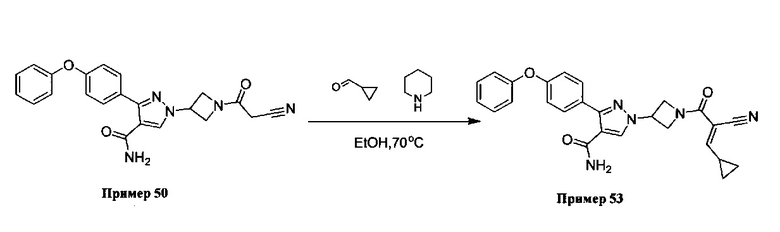
Пример 53
(Е)-1-(1-(2-циано-3-циклопропилакрилоил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
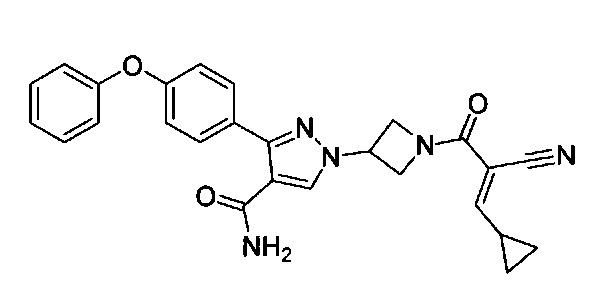
К раствору 1-(1-(2-цианоацетил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида (100 мг, 0.25 ммоль) и пиперидина (2 капли) в EtOH (10 мл) добавляли циклопропанкарбальдегид (70 мг, 1.0 ммоль), затем раствор перемешивали при 70°C в течение 1 ч. После завершения реакции раствор концентрировали и очищали с помощью препаративной TLC смесью 10:1 DCM/MeOH с получением сырого продукта (70 мг) и затем очищали с помощью препаративной ВЭЖХ (ACN/H2O=42%, 0.1% FA) с получением указанного в заголовке соединения (15 мг, 13%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 8.16 (s, 1Н), 7.64-7.57 (m, 2Н), 7.45-7.37 (m, 2Н), 7.22-7.16 (m, 1Н), 7.15-7.03 (m, 5Н), 5.60 (s, 2Н), 5.23-5.13 (m, 1H), 5.11-4.95 (m, 2Н), 4.68-4.54 (m, 2Н), 2.16-2.05 (m, 1Н), 1.33-1.29 (m, 2Н), 1.02-0.95 (m, 2Н). MS (ESI, методика A): m/z=454.1 [М+Н]+, tR=1.485 мин. ВЭЖХ: 100% (214 нм), 100% (254 нм).
Схема 44
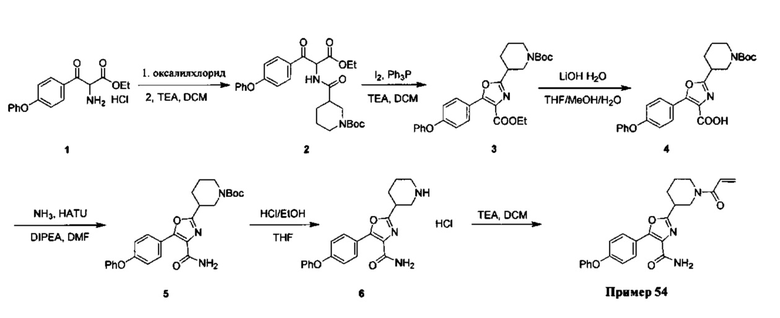
трет-Бутил-3-(1-этокси-1,3-диоксо-3-(4-феноксифенил)пропан-2-илкарбамоил)пиперидин-1-карбоксилат (2). Указанное в заголовке соединение получали, используя способ, аналогичный описанному в Общей Схеме и Примере 15, в виде масла желтого цвета (0.4 г, 36%). MS (ESI): m/z=454.8 [М-55]+.
Этил-2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-5-(4-феноксифенил)оксазол-4-карбоксилат (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 2-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-5-(4-феноксифенил)оксазол-4-карбоксилата (смотри Схему 44) в виде масла желтого цвета (0.25 г, 65%). MS (ESI): m/z=492.8 [М+Н]+.
2-(1-(трет-Бутоксикарбонил)пиперидин-3-ил)-5-(4-феноксифенил)оксазол-4-карбоновая кислота (4). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоновой кислоты (смотри Схему 46) в виде масла желтого цвета (0.25 г, 100%). MS (ESI): m/z=464.8 [М+Н]+.
трет-Бутил-3-(4-карбамоил-5-(4-феноксифенил)оксазол-2-ил)пиперидин-1-карбоксилат (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пиперидин-1-карбоксилата (смотри Схему 30) в виде масла желтого цвета (0.2 г, 84%). MS (ESI): m/z=407.8 [М-55]+.
5-(4-Феноксифенил)-2-(пиперидин-3-ил)оксазол-4-карбоксамид гидрохлорид (6). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде коричневого масла (180 мг, 100%). MS (ESI): m/z=363.8[М+Н]+.
Пример 54
2-(1-Акрилоилпиперидин-3-ил)-5-(4-феноксифенил)оксазол-4-карбоксамид
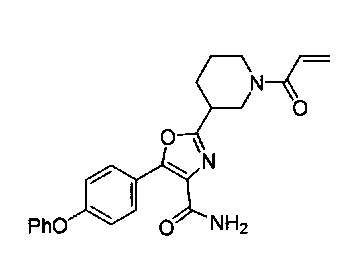
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества желтого цвета (70 мг, 37%). 1Н ЯМР (400 МГц, DMSO) δ 8.24 (d, J=7.2 Гц, 2Н), 7.57 (s, 1.5Н), 7.50 (s, 0.5Н), 7.45 (t, J=7.9 Гц, 2H), 7.21 (t, J=7.4 Гц, 1H), 7.11 (d, J=3.1 Гц, 2H), 7.08 (d, J=4.2 Гц, 2H), 6.96-6.78 (m, 1H), 6.13 (t, J=18 Гц, 1H), 5.75-5.57 (m, 1Н), 4.51 (d, J=11.6 Гц, 0.5H), 4.08 (d, J=12 Гц, 0.5H), 3.93 (d, J=13.2 Гц, 0.5H), 3.85-3.73 (m, 0.8H), 3.34-3.20 (m, 2H), 3.10-2.97 (m, 0.7H), 2.27-2.09 (m, 1H), 2.07-1.64 (m, 2H), 1.60-1.43 (m, 1H). MS (ESI, методика F): m/z=418.1 [M+H]+, tR=1.679 (мин). ВЭЖХ: 100% (214 нм), 100% (254 нм).
Схема 45
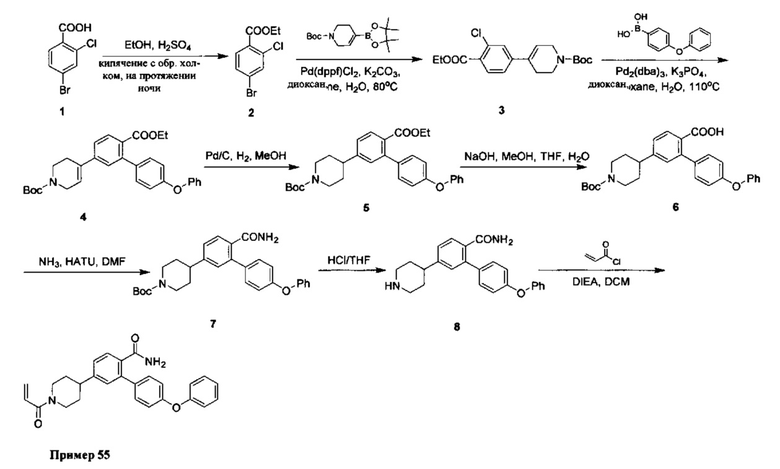
Этил 4-бром-2-хлорбензоат (2). К смеси 4-бром-2-хлорбензойной кислоты 1 (1.9 г, 8.1 ммоль) в EtOH (50 мл) осторожно добавляли H2SO4 (5 мл) по каплям при 0°C. Полученную в результате смесь нагревали до кипения с обратным холодильником на протяжении ночи. Летучую фазу удаляли при пониженном давлении. Остаток разбавляли ЕА (100 мл) и промывали водой (2×50 мл) и рассолом, высушивали над сульфатом натрия, фильтровали и концентрировали. В результате получали указанное в заголовке соединение (2.0 г, 95%) в виде коричневого масла. MS (ESI): m/z=263.0/265.0 [М+Н]+.
трет-Бутил-4-(3-хлор-4-(этоксикарбонил)фенил)-5,6-дигидропиридин-1(2Н)-карбоксилат (3). Смесь этил 4-бром-2-хлорбензоата 2 (0.53 г, 2.0 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата (0.74 г, 2.4 ммоль), карбоната калия (0.83 г, 6.0 ммоль) и Pd(dppf)Cl2 (0.15 г, 0.2 ммоль) в диоксане/H2O (20 мл/5 мл) перемешивали при 80°C в течение 4 ч в атмосфере N2. Летучую фазу удаляли при пониженном давлении. Остаток наносили на колонку с силикагелем, элюируя смесью 6:1 РЕ/ЕА, с получением указанного в заголовке соединения (0.7 г, 96%) в виде масла желтого цвета. MS (ESI): m/z=310.1 [М+Н-56]+.
трет-Бутил-4-(6-(этоксикарбонил)-4'-феноксибифенил-3-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат (4). Смесь трет-бутил-4-(3-хлор-4-(этоксикарбонил)фенил)-5,6-дигидропиридин-1(2Н)-карбоксилата 3 (200 мг, 0.66 ммоль), 4-феноксифенилбороновой кислоты (172 мг, 0.80 ммоль), K3PO4 (414 мг, 2.0 ммоль), трициклогексилфосфина (40 мг, 0.14 ммоль) и Pd2(dbа)3 (64 мг, 0.07 ммоль) в диоксане/H2O (15 мл/3 мл) перемешивали при 110°C на протяжении ночи в атмосфере N2. Летучую фазу удаляли при пониженном давлении. Остаток наносили на колонку с силикагелем, элюируя смесью 6:1 РЕ/ЕА, с получением указанного в заголовке соединения (200 мг, 61%) в виде масла желтого цвета. MS (ESI): m/z=500.0 [М+Н]+.
трет-Бутил-4-(6-(этоксикарбонил)-4'-феноксибифенил-3-ил)пиперидин-1-карбоксилат (5). Смесь 4-(6-(этоксикарбонил)-4'-феноксибифенил-3-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата 4 (180 мг, 0.36 ммоль) и Pd/C (влажность 10%, 18 мг) в МеОН (10 мл) перемешивали при комнатной температуре в атмосфере Н2 в течение 5 ч. Твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. В результате получали указанное в заголовке соединение (180 мг, 99%) в виде бесцветного масла, которое сразу использовали на следующей стадии. MS (ESI): m/z=524.0 [М+Na]+.
5-(1-(трет-Бутоксикарбонил)пиперидин-4-ил)-4'-феноксибифенил-2-карбоновая кислота (6). Смесь трет-бутил-4-(6-(этоксикарбонил)-4'-феноксибифенил-3-ил)пиперидин-1-карбоксилата 5 (180 мг, 0.36 ммоль) и NaOH (72 мг, 1.8 ммоль) в THF/MeOH/H2O (5 мл/5 мл/5 мл) перемешивали при комнатной температуре на протяжении ночи. Летучую фазу удаляли при пониженном давлении. Значение pH доводили до 3 с помощью HCl (1 н.), затем экстрагировали ЕА (2×30 мл). Органические слои объединяли, промывали рассолом, высушивали над сульфатом натрия, фильтровали и концентрировали. В результате получали указанное в заголовке соединение (180 мг, избыток) в виде бесцветного масла, которое сразу использовали на следующей стадии. MS (ESI): m/z=374.1 [М+Н-Вос]+.
трет-Бутил-4-(6-карбамоил-4'-феноксибифенил-3-ил)пиперидин-1-карбоксилат (7). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(4-карбамоил-3-(4-феноксифенил)-1Н-пиразол-1-ил)пирролидин-1-карбоксилата (смотри Схему 23) в виде твердого вещества бледно-желтого цвета (200 мг, избыток). MS (ESI): m/z=416.8 [М+Н-56]+.
трет-Бутил-4-(6-карбамоил-4'-феноксибифенил-3-ил)пиперидин-1-карбоксилат (8). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде твердого вещества белого цвета (200 мг, избыток). MS (ESI): m/z=373.1 [М+Н]+.
Пример 55
5-(1-Акрилоилпиперидин-4-ил)-4'-феноксибифенил-2-карбоксамид
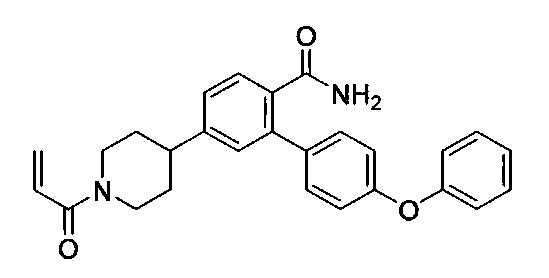
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 61, в виде твердого вещества белого цвета (70 мг, 20%). 'Н ЯМР (300 МГц, CDCl3) δ 7.75 (d, J=7.9 Гц, 1H), 7.44-7.32 (m, 4Н), 7.23-7.28 (m, 1Н), 7.20-7.12 (m, 2Н), 7.11-6.99 (m, 4Н), 6.62 (dd, J=16.5 Гц, 10.5 Гц, 1Н), 6.31 (dd, J=16.5 Гц, 1.8 Гц, 1Н), 5.73 (brs, 1Н), 5.71 (dd, J=10.5 Гц, 1.8 Гц, 1H), 5.35 (brs, 1Н), 4.99-4.74 (m, 1Н), 4.24-4.03 (m, 1Н), 3.30-3.05 (m, 1Н), 2.97-2.58 (m, 2Н), 2.04-1.88 (m, 2Н), 1.83-1.58 (m, 2Н). MS (ESI, методика A): m/z=427.1 [М+Н]+, tR=1.563 мин. ВЭЖХ: 99% (214 нм), 99% (254 нм).
Схема 136
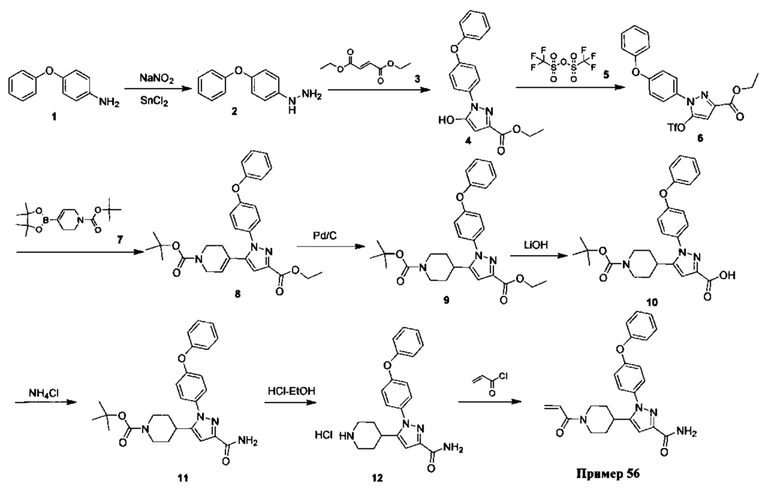
(4-Феноксифенил)гидразин (2). К раствору 4-феноксибензоламина 1 (1.0 г, 5.4 ммоль) в HCl (100 мл) добавляли NaNO2 (700 мг, 10.1 ммоль) при 0°C, затем смесь перемешивали при 0°C в течение 1 ч. К смеси добавляли раствор SnCl2 (5.0 г, 22.1 ммоль) в HCl (100 мл), и полученную в результате смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли водный NaOH (3 н.), чтобы довести pH до 10, и смесь экстрагировали ЕА (3×50 мл), объединенную органическую фазу промывали рассолом (20 мл), высушивали над Na2SO4. Растворитель удаляли при пониженном давлении с получением сырого указанного в заголовке соединения (567 мг, 56%) в виде твердого вещества желтого цвета, которое использовали на следующей стадии без дополнительной очистки. MS (ESI): m/z=201.0 [М+Н]+.
Этил 5-гидрокси-1-(4-феноксифенил)-1Н-пиразол-3-карбоксилат (4). К раствору диэтилфумарата 3 (260 мг, 1.5 ммоль) в EtOH (25 мл) добавляли (4-феноксифенил)гидразин 2 (254 мг, 1.3 ммоль), и полученный в результате раствор перемешивали на протяжении ночи при 80°C. Растворитель выпаривали под вакуумом, и сырой остаток разбавляли водой (30 мл). Раствор экстрагировали этилацетатом (3×30 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 20:1 DCM/MeOH, с получением указанного в заголовке соединения (174 мг, 42%) в виде твердого вещества желтого цвета. MS (ESI): m/z=325.1 [М+Н]+.
Этил 1-(4-феноксифенил)-5-(трифторметилсульфонилокси)-1Н-пиразол-3-карбоксилат (6). К раствору этил 5-гидрокси-1-(4-феноксифенил)-1Н-пиразол-3-карбоксилата 4 (170 мг, 0.5 ммоль) в DCM (20 мл) добавляли трифторметансульфоновый ангидрид 5 (295 мг, 1.0 ммоль) и перемешивали в течение 1 ч при -30°C в атмосфере N2. Смесь выливали в воду (60 мл), и раствор экстрагировали этилацетатом (3×30 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 5:1 РЕ/ЕА, с получением указанного в заголовке соединения (210 мг, 92%) в виде твердого вещества желтого цвета. MS (ESI): m/z=457.0 [М+Н]+.
трет-Бутил-4-(3-(этоксикарбонил)-1-(4-феноксифенил)-1Н-пиразол-5-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат (8). К раствору этил 1-(4-феноксифенил)-5-(трифторметилсульфонилокси)-1H-пиразол-3-карбоксилата 6 (210 мг, 0.5 ммоль), K2СО3 (552 мг, 4.0 ммоль) и трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1-(2Н)-карбоксилата 7 (201 мг, 0.6 ммоль) в диоксане/H2O (10:1, 20 мл) добавляли Pd(dppf)Cl2 (327 мг, 0.2 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90°C и перемешивали на протяжении ночи в атмосфере азота. После охлаждения до комнатной температуры растворитель выпаривали, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 20:1 DCM/MeOH, с получением указанного в заголовке соединения (95 мг, 42%) в виде твердого вещества желтого цвета. MS (ESI): m/z=490.2[М+Н]+.
трет-Бутил-4-(3-(этоксикарбонил)-1-(4-феноксифенил)-1Н-пиразол-5-ил)пиперидин-1-карбоксилат (9). Смесь трет-бутил-4-(3-(этоксикарбонил)-1-(4-феноксифенил)-1Н-пиразол-5-ил)-5,6-дигидропиридин-1-(2Н)-карбоксилата 8 (95 мг, 0.2 ммоль), Pd/C (10% палладий на угле, 56.5% воды, 48 мг) и МеОН (20 мл) дегазировали Н2 6 раз и затем перемешивали в атмосфере Н2 при комнатной температуре в течение 1 ч. Затем смесь фильтровали, и фильтрат концентрировали под вакуумом с получением указанного в заголовке соединения (56 мг, 58%) в виде бесцветного масла. MS (ESI): m/z=492.2 [М+Н]+.
5-(1-(трет-Бутоксикарбонил)пиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоновая кислота (10). К раствору трет-бутил-4-(3-(этоксикарбонил)-1-(4-феноксифенил)-1Н-пиразол-5-ил)пиперидин-1-карбоксилата 9 (166 мг, 0.3 ммоль) в THF/MeOH/H2O (20:20:10 мл) добавляли LiOH (60 мг, 1.4 ммоль), и полученный в результате раствор перемешивали в течение 15 ч при температуре окружающей среды. Смесь концентрировали, остаток разбавляли водой (10 мл), и раствор подкисляли лимонной кислотой до pH=4, экстрагировали этилацетатом (3×20 мл), промывали рассолом (20 мл), высушивали над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения (120 мг, 76%) в виде твердого вещества коричневого цвета. MS (ESI): m/z=464.2 [М+Н]+
трет-Бутил-4-(3-карбамоил-1-(4-феноксифенил)-1Н-пиразол-5-ил)пиперидин-1-карбоксилат (11). К раствору 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоновой кислоты 10 (250 мг, 0.59 ммоль) и HATU (147 мг, 0.38 ммоль) в сухом N,N-диметилформамиде (25 мл) добавляли DIPEA (67 мг, 0.51 ммоль), и полученный в результате раствор перемешивали в течение 20 мин при температуре окружающей среды, затем добавляли NH4Cl (28 мг, 0.51 ммоль) и перемешивали на протяжении ночи. Смесь разбавляли этилацетатом (50 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 20:1 DCM/MeOH, с получением указанного в заголовке соединения (100 мг, 84%) в виде твердого вещества желтого цвета. MS (ESI): m/z=463.2 [М+Н]+.
1-(4-Феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорид (12). К раствору трет-бутил-4-(3-карбамоил-1-(4-феноксифенил)-1Н-пиразол-5-ил)пиперидин-1-карбоксилата 11 (100 мг, 0.21 ммоль) в EtOH (20 мл) добавляли HCl/EtOH (33%, 2 мл) при температуре окружающей среды. Смесь перемешивали в течение 12 ч при комнатной температуре. Смесь концентрировали с получением указанного в заголовке соединения (70 мг, 89%) в виде масла желтого цвета. MS (ESI): m/z=363.1 [М+Н]+.
Пример 56
5-(1-Акрилоилпиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоксамид
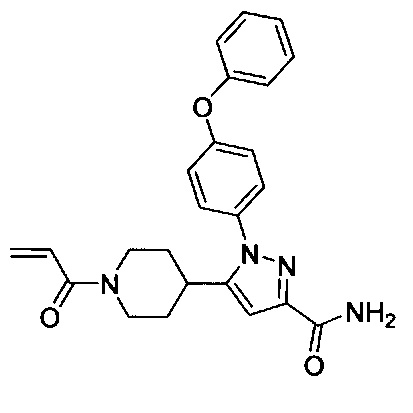
К раствору 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида 12 (110 мг, 0.3 ммоль), DIPEA (117 мг, 0.9 ммоль) и СН2Сl2 (10 мл) добавляли по каплям акрилоилхлорид (28 мг, 0.3 ммоль), и затем реакционную смесь перемешивали при 0°C в течение 1 ч. Смесь разбавляли СН2Сl2 (20 мл) и промывали насыщенным водным раствором NaHCO3 (20 мл). Водную фазу повторно экстрагировали EtOАс (20 мл). Объединенную органическую фазу высушивали над Na2SO4, концентрировали под вакуумом и очищали с помощью препаративной TLC, хроматографируя смесью 20:1 СН2Сl2/МеОН, с получением указанного в заголовке соединения (66 мг, 52%) в виде масла желтого цвета. 1Н ЯМР (300 МГц, CD3OD) δ 7.52-7.37 (m, 4Н), 7.24-7.06 (m, 5Н), 6.84-6.68 (m, 2Н), 6.21 (d, J=2.0 Гц, 0.5Н), 6.15 (d, J=2.0 Гц, 0.5Н), 5.73 (dd, J=10.6, 2.0 Гц, 1H), 4.61 (d, J=12.8 Гц, 1H), 4.15 (d, J=14.0 Гц, 1H), 3.04 (tt, J=8.0, 7.5 Гц, 2H), 2.77-2.59 (m, 1H), 2.03-1.81 (m, 2H), 1.72-1.46 (m, 2H). MS (ESI, методика A): m/z=417.1 [M+H]+, tR=1.547 мин. ВЭЖХ: 98.2% (214 нм), 98.8% (254 нм).
Схема 140
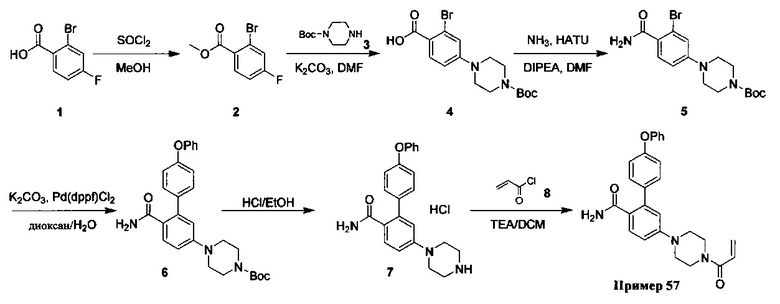
Метил 2-бром-4-фторбензоат (2). К смеси 2-бром-4-фторбензойной кислоты 1 (2.19 г, 10 ммоль) в МеОН (20 мл) добавляли SOCl2 (0.5 мл) по каплям при комнатной температуре. Полученную в результате смесь перемешивали при 60°C в течение 10 ч. Смесь концентрировали с получением указанного в заголовке соединения (2 г, 86%) в виде коричневого масла. MS (ESI): m/z=232.8 [М+Н]+.
2-Бром-4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)бензойная кислота (4). К раствору метил 2-бром-4-фторбензоата 2 (1.8 г, 7.72 ммоль) в DMSO (30 мл) добавляли трет-бутил пиперазин-1-карбоксилат 3 (1.726 г, 9.27 ммоль) и K2CO3 (4.26 г, 30.9 ммоль). Смесь перемешивали при 120°C в течение 5 ч. После окончания смесь экстрагировали этилацетатом (2×50 мл) и промывали водой (50 мл) и рассолом (2×50 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 10:1 РЕ/ЕА, с получением указанного в заголовке соединения (1.1 г, 36%) в виде масла желтого цвета. MS (ESI): m/z=328.7 [М-55]+.
трет-Бутил-4-(3-бром-4-карбамоилфенил)пиперазин-1-карбоксилат (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пирролидин-1-карбоксилата (смотри Схему 31) в виде твердого вещества желтого цвета (0.8 г, 73%). MS (ESI): m/z=327.7 [М -55]+.
трет-Бутил-4-(6-карбамоил-4'-феноксибифенил-3-ил)пиперазин-1-карбоксилат (6). К смеси трет-бутил-4-(3-бром-4-карбамоилфенил)пиперазин-1-карбоксилата 5 (192 мг, 0.5 ммоль), 4-феноксифенилбороновой кислоты (102 мг, 0.5 ммоль) и K2CO3 (207 мг, 1.5 ммоль) в диоксане/воде (20/4 мл) добавляли Pd(dppf)2Cl2 (36.55 мг, 0.05 ммоль). Смесь дегазировали N2 3 раза, затем нагревали до 100°C и перемешивали в течение 16 ч. После охлаждения до комнатной температуры к смеси добавляли этилацетат (30 мл), промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 30:1 DCM/MeOH, с получением указанного в заголовке соединения (50 мг, 21%) в виде твердого вещества желтого цвета. MS (ESI): m/z=473.8 [М+Н]+.
4'-Фенокси-5-(пиперазин-1-ил)бифенил-2-карбоксамид гидрохлорид (7). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-(4-феноксифенил)-5-(пиперидин-4-ил)-1Н-пиразол-3-карбоксамид гидрохлорида (смотри Схему 46) в виде твердого вещества желтого цвета (40 мг, 92%). MS (ESI): m/z=374.1 [М+Н]+.
Пример 57
5-(4-Акрилоилпиперазин-1-ил)-4'-феноксибифенил-2-карбоксамид
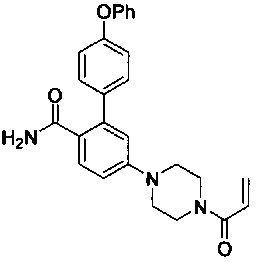
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества не совсем белого цвета (15 мг, 36%). 1Н ЯМР (400 МГц, DMSO) δ 7.52-7.30 (m, 6Н), 7.25-6.93 (m, 7Н), 6.91-6.82 (m, 2Н), 6.15 (d, J=16.7 Гц, 1Н), 5.73 (d, J=9.9 Гц, 1H), 3.70 (d, J=12.0 Гц, 4Н), 3.27 (s, 4Н). MS (ESI, методика A): m/z=428.1 [М+Н]+, tR=1.525 мин. ВЭЖХ: 96.4% (214 нм), 99.3% (254 нм).
Схема 142
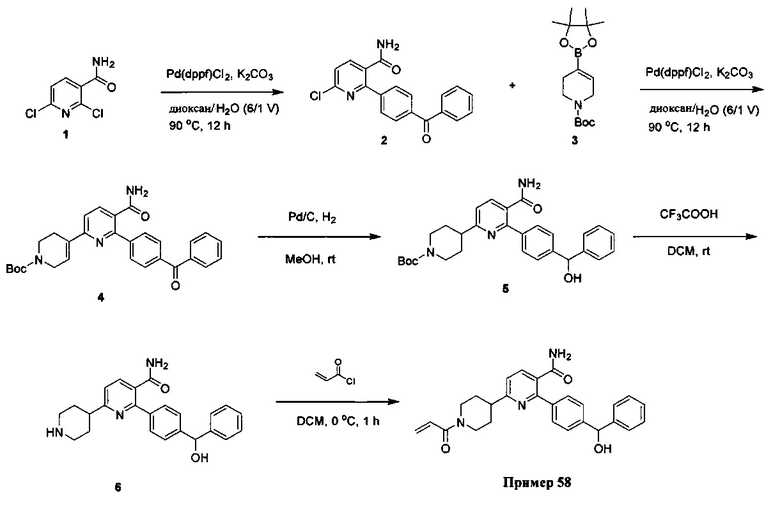
2-(4-Бензоилфенил)-6-хлорникотинамид (2). К раствору 4-феноксифенилбороновой кислоты (650 мг, 2.88 ммоль), K2CO3 (1.08 г, 7.86 ммоль) и 2,6-дихлорникотинамида (500 мг, 2.62 ммоль) в 1,4-диоксане (25 мл) и воде (4 мл) добавляли Pd(dppf)Cl2 (192 мг, 0.262 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 60°C и перемешивали в течение 12 ч в атмосфере азота. После охлаждения до комнатной температуры раствор выливали в воду (50 мл), и затем экстрагировали этилацетатом (2×50 мл). Объединенные органические слои высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали с помощью колоночной хроматографии, элюируя смесью 80:1 дихлорметан/метанол, с получением указанного в заголовке соединения в виде коричневого масла (868 мг, 98%). MS (ESI): m/z=336.9 [М+Н]+.
трет-Бутил-4-(6-(4-бензоилфенил)-5-карбамоилпиридин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат (4). К раствору трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата (1.2 г, 3.87 ммоль), K2CO3 (1.07 г, 7.74 ммоль) и 2-(4-бензоилфенил)-6-хлорникотинамида 2 (868 мг, 2.58 ммоль) в 1,4-диоксане (24 мл) и воде (3 мл) добавляли Pd(dppf)Cl2 (188 мг, 0.258 ммоль) в атмосфере азота, и смесь дегазировали азотом 6 раз, затем нагревали до 90°C и перемешивали в течение 12 ч в атмосфере азота. После охлаждения до комнатной температуры раствор выливали в воду (50 мл) и затем экстрагировали этилацетатом (3×40 мл). Объединенные органические слои высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт очищали с помощью колоночной хроматографии, элюируя смесью 80:1 дихлорметан/метанол, с получением указанного в заголовке соединения в виде коричневого масла (800 мг, 64%). MS (ESI): m/z=484.0 [М+Н]+.
трет-Бутил-4-(5-карбамоил-6-(4-(гидрокси(фенил)метил)фенил)пиридин-2-ил)пиперидин-1-карбоксилат (5). К раствору трет-бутил-4-(6-(4-бензоилфенил)-5-карбамоилпиридин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата 4 (800 мг, 1.65 ммоль) в МеОН (15 мл) добавляли Pd/C (150 мг) в атмосфере водорода, и смесь дегазировали водородом 6 раз, затем перемешивали в течение 16 ч при температуре окружающей среды в атмосфере водорода. Раствор фильтровали, и фильтрат упаривали с получением сырого продукта в виде твердого вещества белого цвета (500 мг, 62%). MS (ESI): m/z=488.1 [М+Н]+.
2-(4-(Гидрокси(фенил)метил)фенил)-6-(пиперидин-4-ил)никотинамид (6). К раствору трет-бутил-4-(5-карбамоил-6-(4-(гидрокси(фенил)метил)фенил)пиридин-2-ил)пиперидин-1-карбоксилата 5 (500 мг, 1.03 ммоль) в сухом дихлорметане (5 мл) добавляли TFA (2 мл), и полученную в результате смесь перемешивали в течение 1 ч при температуре окружающей среды. Растворитель удаляли, и остаток разделяли между насыщенным водным бикарбонатом натрия (30 мл) и этилацетатом (20 мл). Органическую фазу отделяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения в виде твердого вещества белого цвета (350 мг, 88%). MS (ESI): m/z=388.1 [М+Н]+.
Пример 58
6-(1-Акрилоилпиперидин-4-ил)-2-(4-(гидрокси(фенил)метил)фенил)никотинамид
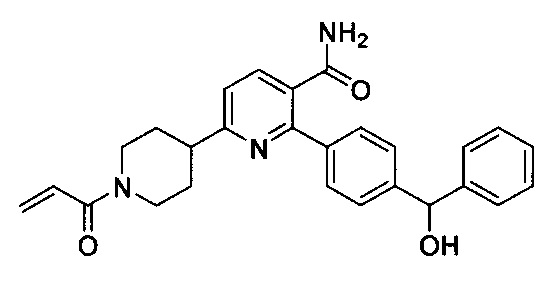
К раствору 2-(4-(гидрокси(фенил)метил)фенил)-6-(пиперидин-4-ил) никотинамида 6 (350 мг, 0.90 ммоль) в DCM (10 мл) добавляли TEA (182 мг, 1.8 ммоль) и акрилоилхлорид (90 мг, 0.99 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 10 мин. Растворитель удаляли, и остаток очищали с помощью препаративной ВЭЖХ с получением указанного в заголовке соединения (150 мг, 38%) в виде твердого вещества белого цвета. 1Н ЯМР (300 МГц, CDCl3) δ 8.03 (d, J=7.9 Гц, 1Н), 7.65 (d, J=7.1 Гц, 2H), 7.56-7.15 (m, 9H), 6.67-6.49 (m, 1H), 6.27 (d, J=16.6 Гц, 1H), 5.88 (s, 1H), 5.68 (d, J=10.1 Гц, 1H), 5.52 (d, J=27.1 Гц, 2H), 4.79-4.72 (m, 1H), 4.11-4.06 (m, 1H), 3.18-3.10 (m, 2H), 2.80-2.74 (m, 1H), 2.08-1.98 (m, 2H), 1.78 (d, J=10.5 Гц, 2H). MS (ESI, методика F): m/z=441.8 [M+H]+, tR=1.268 мин, ВЭЖХ: 100.0% (214 нм), 100.0% (254 нм).
Схема 49
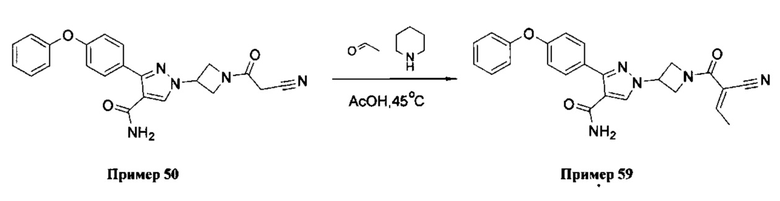
Пример 59
(Е)-1-(1-(2-цианобут-2-еноил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
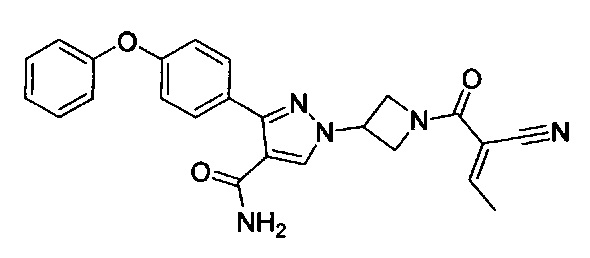
К раствору 1-(1-(2-цианоацетил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида (150 мг, 0.37 ммоль) и пиперидина (3 капли) в АсОН (10 мл) добавляли ацетальдегид (99 мг, 0.74 ммоль), затем перемешивали при 45°C на протяжении ночи. После завершения реакции к раствору добавляли насыщенный NaOH до pH=6-7. Затем раствор экстрагировали DCM (2×20 мл). Органический слой объединяли и концентрировали под вакуумом досуха. Остаток очищали с помощью препаративной ВЭЖХ (МеОН-H2O=60-70, 0.1% FA) с получением указанного в заголовке соединения (40 мг, 26%) в виде твердого вещества белого цвета. 1Н ЯМР (300 МГц, CDCl3) δ 8.15 (s, 1Н), 7.79-7.68 (m, 1H), 7.62-7.54 (m, 2H), 7.43-7.34 (m, 2H), 7.21-7.13 (m, 1H), 7.13-7.04 (m, 4H), 5.62 (s, 2H), 5.26-5.10 (m, 1H), 5.08-4.79 (m, 2H), 4.71-4.51 (m, 2H), 3.51-3.36 (m, 1H), 2.26-2.16 (m, 2H). MS (ESI, методика A): m/z=428.1 [M+H]+, tR=1.438 мин. ВЭЖХ: 98.4% (214 нм), 98.4% (254 нм).
Схема 50
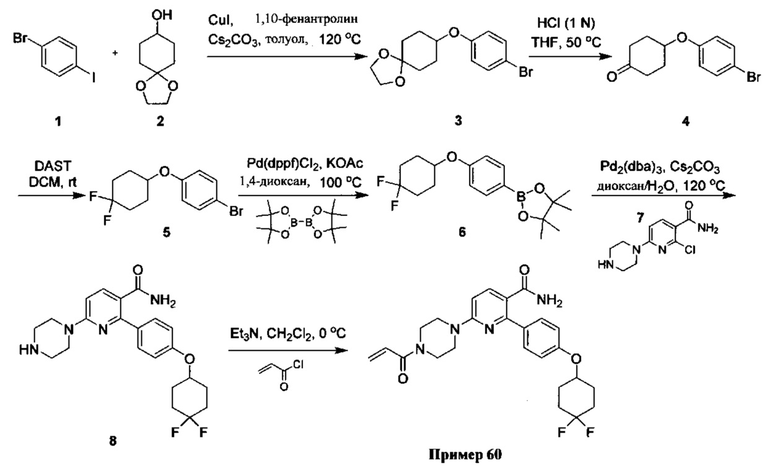
8-(4-Бромфенокси)-1,4-диоксаспиро[4.5]декан (3). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 1-бром-4-(циклогексилокси)бензола (смотри Схему 17) в виде бесцветного масла (0.272 г, 87%).
4-(4-Бромфенокси)циклогексанон (4). Раствор 8-(4-бромфенокси)-1,4-диоксаспиро[4.5]декана 3 (0.272 г, 0.87 ммоль) в THF (5 мл) обрабатывали HCl (1 н., 5 мл) при 50°C в течение 1 ч, затем смесь нейтрализовали NaHCO3 и экстрагировали EtOAc (5 мл×3). Объединенную органическую фазу высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 5:1 петролейный эфир/EtOAc, с получением указанного в заголовке соединения (147 мг, 63%) в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3) δ 7.46-7.37 (m, 2Н), 6.91-6.83 (m, 2Н), 4.68 (s, 1Н), 2.75-2.64 (m, 2Н), 2.43-2.25 (m, 4Н), 2.15-2.08 (s, 2Н).
1-Бром-4-((4,4-дифторциклогексил)окси)бензол (5). К раствору 4-(4-бромфенокси)циклогексанона 4 (0.538 г, 2.0 ммоль) в сухом DCM (5 мл) добавляли раствор DAST (0.654 г, 4.0 ммоль) в сухом DCM (5 мл) при 0°C, и полученную в результате смесь перемешивали при комнатной температуре в течение 4 ч. Реакцию останавливали ледяной водой (5 мл), подщелачивали насыщенным NaHCO3 до pH=8 и экстрагировали EtOAc (10 мл×3). Объединенную органическую фазу высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 10:1 петролейный эфир/EtOAc, с получением указанного в заголовке соединения (0.547 г, 94%) в виде масла желтого цвета.
2-(4-((4,4-Дифторциклогексил)окси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (6). Смесь 1-бром-4-((4,4-дифторциклогексил)окси)бензола 5 (0.328 г, 1.13 ммоль), бис(пинаколато)диборона (0.429 г, 1.69 ммоль), Pd(dppf)Cl2 (0.168 г, 0.23 ммоль), KOAc (0.323 г, 3.29 ммоль) и сухого диоксана (3 мл) перемешивали при 100°C в течение 1 ч. После охлаждения до комнатной температуры смесь выливали в воду (10 мл) и экстрагировали EtOAc (15 мл×3). Объединенную органическую фазу высушивали нал Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя РЕ, с получением указанного в заголовке соединения (0.258 г, 68%) в виде твердого вещества белого цвета.
2-(4-((4,4-Дифторциклогексил)окси)фенил)-6-(пиперазин-1-ил)никотинамид (8). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 6-(3-нитрофенил)-2-(4-феноксифенил)никотинамида (смотри Схему 1) в виде твердого вещества белого цвета (0.10 г, 31%). MS (ESI): m/z=417.1 [М+Н]+.
Пример 60
6-(4-Акрилоилпиперазин-1-ил)-2-(4-((4,4-дифторциклогексил)окси)фенил)никотинамид
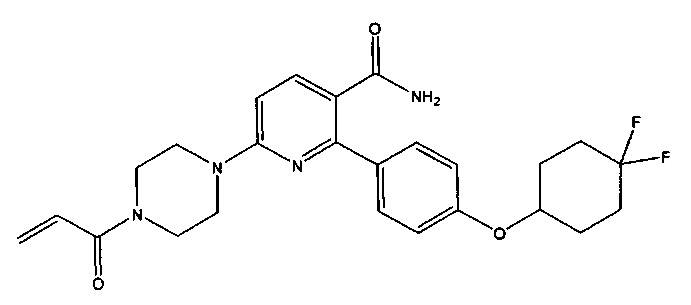
Указанное в заголовке соединение получали способом, аналогичным описанному для получения Примера 1, в виде твердого вещества белого цвета (6 мг, 11%). 1Н ЯМР (400 МГц, DMSO-d6) δ 7.88 (d, J=8.7 Гц, 2Н), 7.73 (d, J=8.6 Гц, 2Н), 7.68 (d, J=8.7 Гц, 1H), 7.59 (s, 1H), 7.26 (s, 1H), 7.21-7.14 (m, 4H), 6.86 (dd, J=17.8, 9.3 Гц, 2H), 6.16 (dd, J=16.7, 2.2 Гц, 1H), 5.73 (dd, J=10.4, 2.2 Гц, 1H), 3.66-3.67 (m, 8H). MS (ESI, методика A): m/z=471.2 [M+H]+, tR=1.753 (мин). ВЭЖХ: 99.2% (214 нм), 99.2% (254 нм).
Схема 51
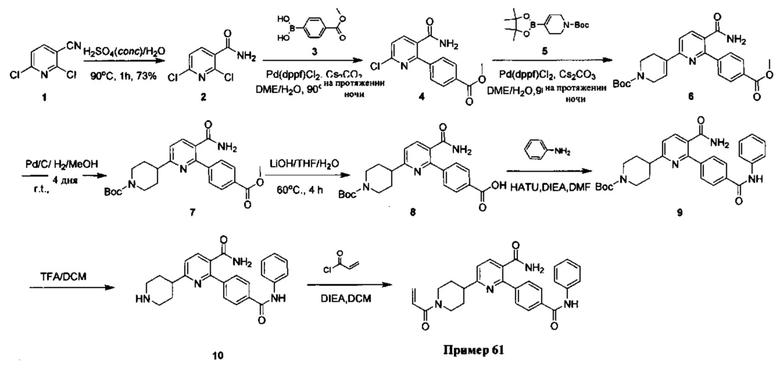
2,6-Дихлорникотинамид (2). Указанное в заголовке соединение синтезировали с использованием способа, аналогичного описанному для получения 2,6-дихлорникотинамида (смотри Схему 51) (9.5 г, 81%) в виде твердого вещества коричневого цвета. MS (ESI): m/z=191.0 [М+Н]+.
Метил 4-(3-карбамоил-6-хлорпиридин-2-ил)бензоат (4). К раствору 2,6-дихлорникотинамида 2 (1.91 г, 10.0 ммоль), 4-(метоксикарбонил)фенилбороновой кислоты 3 (1.8 г, 10.0 ммоль) и Pd(dppf)Cl2 (816 мг, 1.0 ммоль) в DME/H2O (20 мл/2 мл) добавляли CS2CO3 (6.5 г, 20.0 ммоль). Полученный в результате раствор дегазировали N2 6 раз и перемешивали на протяжении ночи при 90°C под защитой N2. После завершения реакции раствор концентрировали, разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке с силикагелем, разбавляя от 100:1 до 60:1 DCM/MeOH, с получением указанного в заголовке соединения (1.95 г, 67%) в виде твердого вещества белого цвета. MS (ESI): m/z=291.1 [М+Н]+.
трет-Бутил-4-(5-карбамоил-6-(4-(метоксикарбонил)фенил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат (6). К раствору метил 4-(3-карбамоил-6-хлорпиридин-2-ил)бензоата 4 (170 мг, 0.59 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата 5 (200 мг, 0.65 ммоль) и Pd(dppf)Cl2 (50 мг, 0.06 ммоль) в DME/H2O (20 мл/2 мл) добавляли CS2CO3 (390 мг, 1.2 ммоль), полученный в результате раствор дегазировали N2 6 раз и перемешивали на протяжении ночи при 90°C в атмосфере N2. После завершения реакции раствор концентрировали, разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке с силикагелем, разбавляя от 50:1 до 10:1 DCM/MeOH, с получением указанного в заголовке соединения (145 мг, 56%) в виде коричневого масла. MS (ESI): m/z=438.2 [М+Н]+.
трет-Бутил-4-(5-карбамоил-6-(4-(метоксикарбонил)фенил)пиридин-2-ил)пиперидин-1-карбоксилат (7). К раствору трет-бутил-4-(5-карбамоил-6-(4-(метоксикарбонил)фенил)пиридин-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата 6 (145 мг, 0.33 ммоль) в МеОН (10 мл) добавляли Pd/C (15 мг) и дегазировали Н2 6 раз и перемешивали на протяжении ночи при комнатной температуре в атмосфере H2. После завершения реакции раствор фильтровали, и фильтрат концентрировали с получением указанного в заголовке соединения (100 мг, 69%) в виде коричневого масла. MS (ESI): m/z=440.2 [М+Н]+.
4-(6-(1-(трет-Бутоксикарбонил)пиперидин-4-ил)-3-карбамоилпиридин-2-ил)бензойная кислота (8) К раствору трет-бутил-4-(5-карбамоил-6-(4-(метоксикарбонил)фенил)пиридин-2-ил)пиперидин-1-карбоксилата 7 (460 мг, 1.05 ммоль) в THF/H2O (20 мл/2 мл) добавляли LiOH (84 мг, 2.1 ммоль), полученный в результате раствор перемешивали на протяжении ночи при комнатной температуре. После завершения реакции раствор концентрировали, добавляли воду (10 мл) и подкисляли HCl (конц., 1 мл) до pH=5-6, разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (390 мг, 87%) в виде твердого вещества желтого цвета. MS (ESI): m/z=426.2 [М+Н]+.
трет-Бутил-4-(5-карбамоил-6-(4-(фенилкарбамоил)фенил)пиридин-2-ил)пиперидин-1-карбоксилат (9). К раствору 4-(6-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-3-карбамоилпиридин-2-ил)бензойной кислоты 8 (390 мг, 0.917 ммоль), анилина (102 мг, 1.101 ммоль) и HATU (418 мг, 1.101 ммоль) в сухом DMF (10 мл) добавляли DIEA (355 мг, 2.751 ммоль), полученный в результате раствор перемешивали на протяжении ночи при комнатной температуре. Смесь разбавляли этилацетатом (30 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью препаративной TLC смесью 20:1 DCM/MeOH с получением указанного в заголовке соединения (120 мг, 27%) в виде твердого вещества желтого цвета. MS (ESI): m/z=501.2 [М+Н]+.
2-(4-(Фенилкарбамоил)фенил)-6-(пиперидин-4-ил)никотинамид (10) К раствору трет-бутил-4-(5-карбамоил-6-(4-(фенилкарбамоил)фенил)пиридин-2-ил)пиперидин-1-карбоксилата 9 (120 мг, 0.24 ммоль) в сухом DCM (10 мл) добавляли TFA (3 мл), полученный в результате раствор перемешивали на протяжении ночи при комнатной температуре. Смесь промывали NaHCO3/H2O (3×20 мл) и рассолом (3×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток концентрировали с получением указанного в заголовке соединения (100 мг, 99%) в виде твердого вещества желтого цвета. MS (ESI): m/z=401.2 [М+Н]+.
Пример 61
6-(1-Акрилоилпиперидин-4-ил)-2-(4-(фенилкарбамоил)фенил)никотинамид
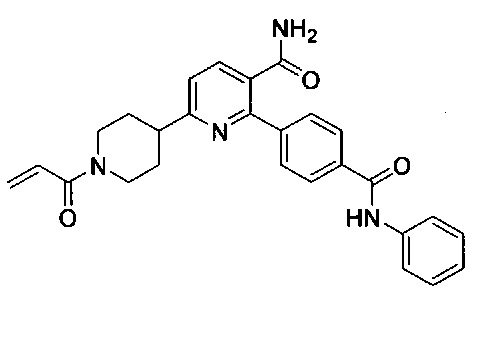
К раствору 2-(4-(фенилкарбамоил)фенил)-6-(пиперидин-4-ил)никотинамида 10 (100 мг, 0.24 ммоль) в сухом DCM (15 мл) добавляли DIEA (95 мг, 0.72 ммоль) и акрилоилхлорид (35 мг, 0.36 ммоль) при 0°C, и полученный в результате раствор перемешивали при 0°C в течение 10 мин. Добавляли воду (10 мл), чтобы остановить реакцию. Смесь разбавляли DCM (20 мл) и промывали водой (2×20 мл) и рассолом (2×20 мл). Органический слой высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (ACN-H2O=30-90, 0.1% FA) с получением указанного в заголовке соединения (31 мг, 29%) в виде твердого вещества белого цвета. 1Н ЯМР (300 МГц, CDCl3) δ 8.59 (s, 1Н), 8.04-7.58 (m, 3), 7.75-7.54 (m, 4Н), 7.37-7.27 (m, 2Н), 7.21-7.08 (m, 2Н), 6.65-6.49 (m, 1Н), 6.29-6.10 (m, 3), 5.74-5.63 (m, 1Н), 4.79-4,63 (m, 1Н), 4.17-3.99 (m, 1H), 3.27-2.96 (m, 2Н), 2.83-2.66 (m, 1Н), 2.07-1.88 (m, 2Н), 1.83-1.60 (m, 2Н). MS (ESI): m/z=455.2 [М+Н]+, tR=1.316 мин. ВЭЖХ: 95.3% (214 нм), 93.0% (254 нм).
Схема 52
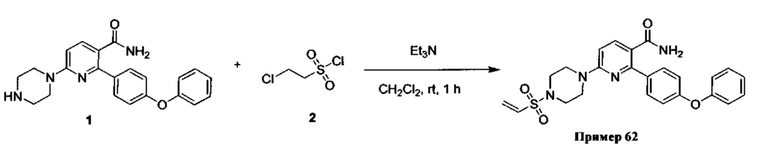
Пример 62
2-(4-Феноксифенил)-6-(4-(винилсульфонил)пиперазин-1-ил)никотинамид
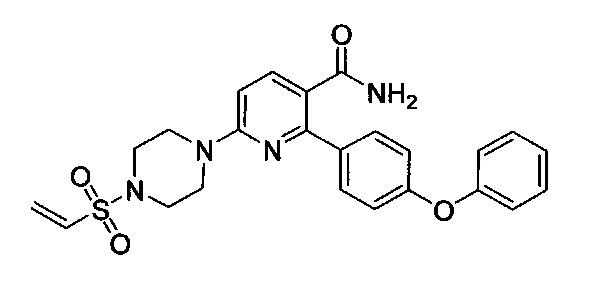
К раствору 2-(4-феноксифенил)-6-(пиперазин-1-ил)никотинамида 1 (200 мг, 0.53 ммоль) в DCM (8 мл) добавляли TEA (161 мг, 1.59 ммоль) и 2-хлорэтансульфонил хлорид (131 мг, 0.80 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1 ч. Раствор выливали в воду (50 мл), и затем экстрагировали CH2Cl г (2×50 мл). Объединенные органические слои высушивали над безводным сульфатом натрия, фильтровали и концентрировали, и остаток очищали с помощью препаративной TLC, элюируя смесью 100:1 DCM/MeOH, с получением указанного в заголовке соединения (17 мг, 6.9%) в виде твердого вещества белого цвета. 1Н ЯМР (300 МГц, CDCl3) δ 8.01 (d, J=8.8 Гц, 1H), 7.59 (d, J=8.6 Гц, 2Н), 7.43-7.30 (m, 2Н), 7.15 (t, J=7.4 Гц, 1Н), 7.09-7.01 (m, 4H), 6.64 (d, J=8.8 Гц, 1H), 6.42 (dd, J=16.6, 9.7 Гц, 1H), 6.26 (d, J=16.5 Гц, 1H), 6.06 (d, J=9.7 Гц, 1H), 5.46 (br, 1H), 5.28 (br, 1H), 3.87-3.75 (m, 4H), 3.32-3.18 (m, 4H). MS (ESI, методика F): m/z=465.1.0 [M+H]+, tR=1.546 мин, ВЭЖХ: 99.5% (214 нм), 98.3% (254 нм).
Схема 53
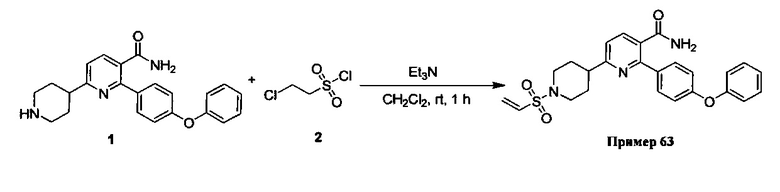
Пример 63
2-(4-Феноксифенил)-6-(1-(винилсульфонил)пиперидин-4-ил)никотинамид
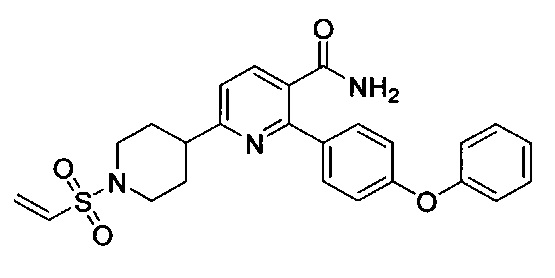
К раствору 2-(4-феноксифенил)-6-(пиперидин-4-ил)никотинамида 1 (200 мг, 0.54 ммоль) в DCM (8 мл) добавляли TEA (164 мг, 1.62 ммоль) и 2-хлорэтансульфонил хлорид (131 мг, 0.80 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1 ч. Раствор выливали в воду (50 мл), и затем экстрагировали этилацетатом (2×50 мл). Объединенные органические слои высушивали над безводным сульфатом натрия, фильтровали и концентрировали, и остаток очищали с помощью препаративной ВЭЖХ с получением указанного в заголовке соединения (40 мг, 16%) в виде твердого вещества белого цвета. 1Н ЯМР (300 МГц, CDCl3) δ 8.03 (d, J=8.0 Гц, 1H), 7.71-7.61 (m, 2Н), 7.43-7.31 (m, 2Н), 7.17 (dd, J=15.6, 7.8 Гц, 2Н), 7.10-7.01 (m, 4Н), 6.46 (dd, J=16.6, 9.8 Гц, 1Н), 6.26 (d, J=16.6 Гц, 1H), 6.04 (d, J=9.9 Гц, 1H), 5.60 (b, 1H), 5.41 (b, 1H), 3.94-3.82 (m, 2H), 2.97-2.86 (m, 1H), 2.80-2.71 (m, 2H), 2.11-1.92 (m, 4H). MS (ESI, методика F): m/z=464.0 [M+H]+, tR=1.566 мин, ВЭЖХ: 99.3% (214 нм), 100.0% (254 нм).
Следующие примеры могут быть синтезированы в соответствии с методиками, приведенными ниже, и с привлечением обычного специалиста в данной области техники. Эти примеры, как полагают, будут полезны в качестве ингибиторов ВТК исходя из биологической активности соединений, описанных выше.
Пример 64
1-(1-Акрилоилпиперидин-3-ил)-3-(4-(фенилкарбамоил)фенил)-1Н-пиразол-4-карбоксамид
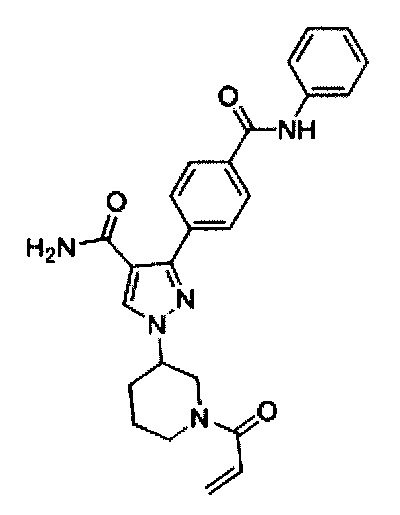
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примере 15.
Пример 65
1-(1-Акрилоилпиперидин-3-ил)-3-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)-1Н-пиразол-4-карбоксамид
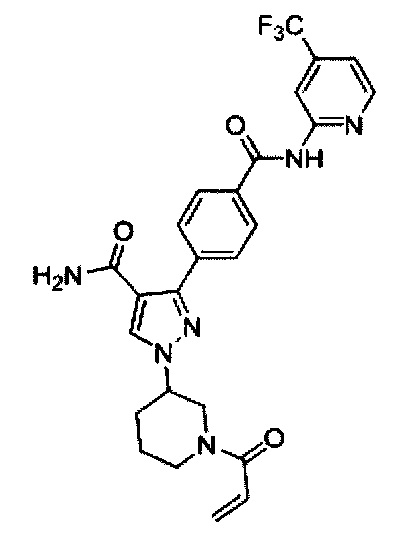
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примере 15.
Пример 66
6-(4-Акриламидофенил)-2-(4-(фенилкарбамоил)фенил)никотинамид
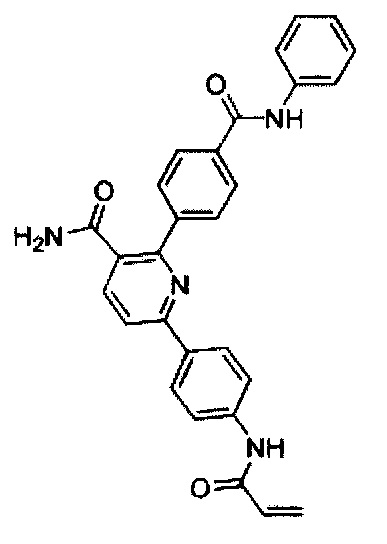
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примере 16.
Пример 67
6-(4-Акриламидофенил)-2-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)никотинамид
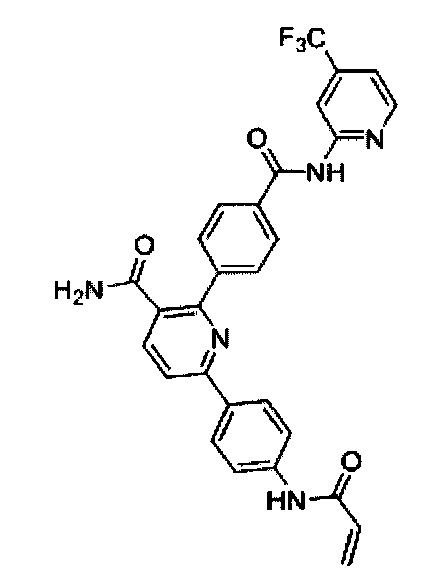
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примере 16.
Пример 68
1-(1-Акрилоилпиперидин-3-ил)-3-(4-(гидрокси(фенил)метил)фенил)-1Н-пиразол-4-карбоксамид
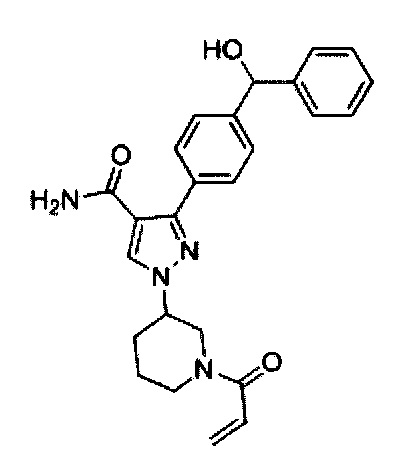
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примерах 15 и 58.
Пример 69
1-(1-Акрилоилпиперидин-3-ил)-3-(4-(метил(фенил)амино)фенил)-1Н-пиразол-4-карбоксамид
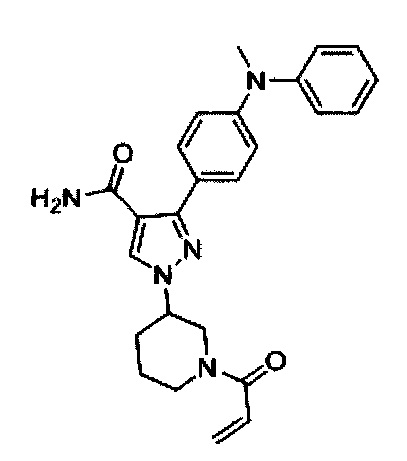
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примере 15.
Пример 70
6-(4-Акриламидофенил)-2-(4-(1-гидрокси-1-фенилэтил)фенил)никотинамид
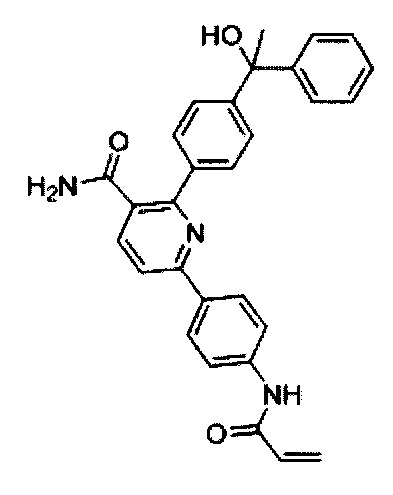
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примерах 16 и 58.
Пример 71
6-(4-Акриламидофенил)-2-(4-(дифтор(фенил)метил)фенил)никотинамид
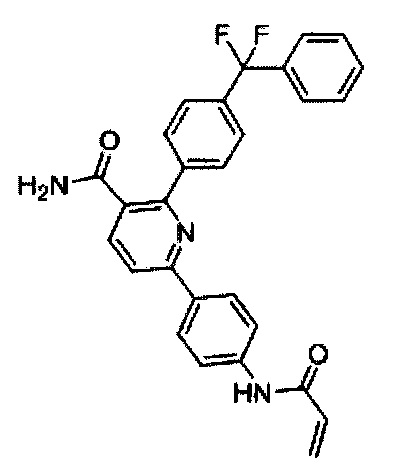
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примере 16.
Пример 72
1-(1-Акрилоилпиперидин-3-ил)-3-(4-(фенилсульфонил)фенил)-1Н-пиразол-4-карбоксамид
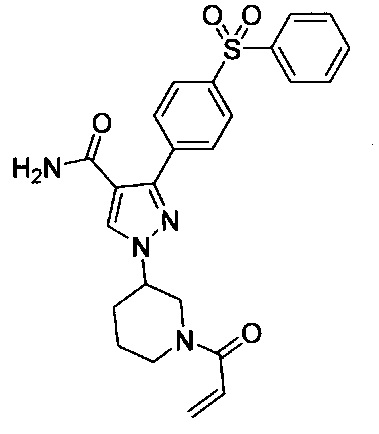
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примере 15.
Пример 73
6-(4-Акриламидофенил)-2-(4-(фенилсульфонил)фенил)никотинамид
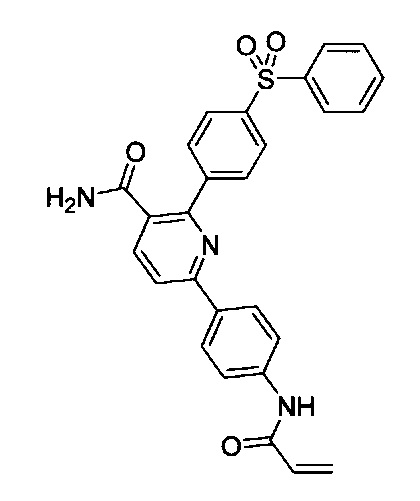
Указанное в заголовке соединение может быть получено с использованием способа, аналогичного описанному в Общей Схеме и Примере 16.
Схема A-1
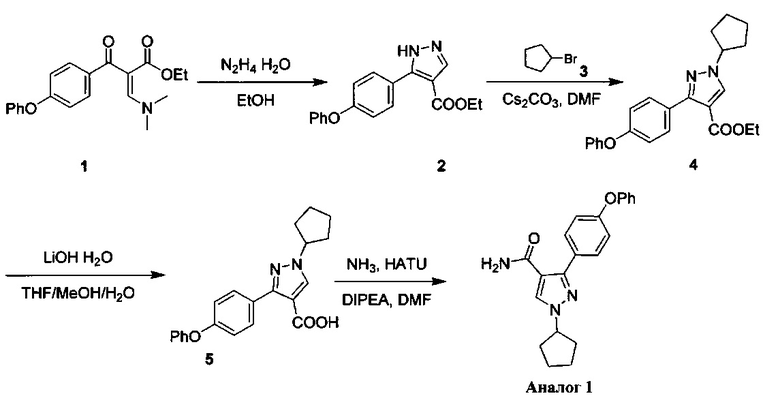
Этил 5-(4-феноксифенил)-1Н-пиразол-4-карбоксилат (2). Указанное в заголовке соединение получали способом, аналогичным описанному для получения этил 3-(4-феноксифенил)-1Н-пиразол-4-карбоксилата (смотри Схему 23) в виде масла желтого цвета (0.3 г, 97%). MS (ESI): m/z=308.8 [М+Н]+.
Этил 1-циклопентил-3-(4-феноксифенил)-1Н-пиразол-4-карбоксилат (4). К раствору этил 5-(4-феноксифенил)-1Н-пиразол-4-карбоксилата 2 (150 мг, 0.486 ммоль) в DMF (10 мл) добавляли бромциклопентан 3 (87 мг, 0.584 ммоль) и CS2CO3 (475.5 мг, 1.46 ммоль). Смесь перемешивали при 100С в течение 1 ч. После окончания смесь экстрагировали этилацетатом (2×30 мл) и промывали водой (2×10 мл) и рассолом (3×10 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения (0.13 г, 71%) в виде масла желтого цвета. MS (ESI): m/z=376.9 [М+Н]+.
1-Циклопентил-3-(4-феноксифенил)-1H-пиразол-4-карбоновая кислота (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоновой кислоты (смотри Схему 46) в виде масла желтого цвета (0.12 г, 100%). MS (ESI): m/z=348.8 [М+Н]+.
Аналог 1
1-Циклопентил-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамид
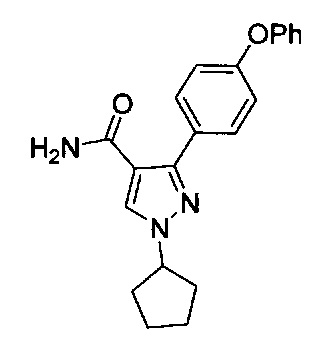
Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-3-(5-карбамоил-4-(4-феноксифенил)тиазол-2-ил)пирролидин-1-карбоксилата (смотри Схему 31) в виде твердого вещества не совсем белого цвета (70 мг, 59%). 1Н ЯМР (300 МГц, DMSO) δ 8.23 (s, 1Н), 7.79 (d, J=8.5 Гц, 2Н), 7.41 (t, J=7.0 Гц, 3Н), 7.15 (t, J=7.1 Гц, 1H), 7.05 (d, J=7.6 Гц, 2Н), 7.00 (s, 2Н), 6.98 (d, J=1.4 Гц, 1Н), 4.78-4.62 (m, 1H), 2.19-1.55 (m, 8Н). MS (ESI, методика F): m/z=347.9 [М+Н]+, tR=1.589 (мин). ВЭЖХ: 97.6% (214 нм), 98.0% (254 нм).
Схема А-2
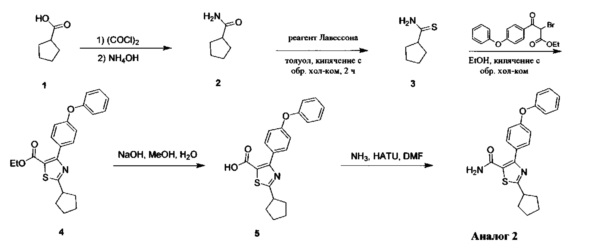
Циклопентанкарбоксамид (2). К смеси циклопентанкарбоновой кислоты 1 (2.85 г, 25 ммоль) и DMF (3 капли) в DCM (60 мл) осторожно добавляли (СОСl)2 (3.2 г, 25 ммоль) по каплям при 0°C в атмосфере N2. Полученную в результате смесь перемешивали при комнатной температуре в течение 2 ч, затем к этой смеси добавляли по каплям NH4OH (5 мл) при 0°C. После добавления смесь перемешивали при комнатной температуре в течение еще 1 ч, затем разбавляли DCM (50 мл). Смесь промывали водой (50 мл), рассолом, высушивали над сульфатом натрия, фильтровали и концентрировали. В результате получали указанное в заголовке соединение (1.8 г, 64%) в виде твердого вещества белого цвета. MS (ESI): m/z=114 [M+H]+.
Циклопентанкарботиоамид (3). Смесь циклопентанкарбоксамида 2 (1.8 г, 15.9 ммоль) и реагента Лавессона (3.2 г, 8.0 ммоль) в толуоле (40 мл) перемешивали при 80°C в течение 2 ч в атмосфере N2. Реакцию останавливали насыщенным NaHCO3 (50 мл), затем экстрагировали ЕА (2×40 мл). Органические слои объединяли, промывали рассолом, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток наносили на колонку с силикагелем, элюируя смесью 3:1 РЕ/ЕА, с получением указанного в заголовке соединения (0.25 г, 12%) в виде твердого вещества желтого цвета. MS (ESI): m/z=130.1 [М+Н-56]+.
Этил 2-циклопентил-4-(4-феноксифенил)тиазол-5-карбоксилат (4). Смесь циклопентанкарботиоамида 3 (150 мг, 1.16 ммоль) и этил 2-бром-3-оксо-3-(4-феноксифенил)пропаноата (422 мг, 1.16 ммоль) в ETOH (15 мл) нагревали до кипения с обратным холодильником в течение 2 ч. Летучую фазу удаляли при пониженном давлении. Остаток растворяли в ЕА (50 мл), который промывали насыщенным NaHCO3 (30 мл), рассолом, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток наносили на колонку с силикагелем, элюируя смесью 4:1 РЕ/ЕА, с получением указанного в заголовке соединения (200 мг, 43%) в виде масла бледно-желтого цвета. MS (ESI): m/z=394.1 [М+Н]+.
2-Циклопентил-4-(4-феноксифенил)тиазол-5-карбоновая кислота (5). Указанное в заголовке соединение получали способом, аналогичным описанному для получения 5-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-4'-феноксибифенил-2-карбоновой кислоты (смотри Схему 45) в виде твердого вещества бледно-желтого цвета (190 мг, 99%). MS (ESI): m/z=366.1 [М+Н]+.
Аналог 2
2-Циклопентил-4-(4-феноксифенил)тиазол-5-карбоксамид
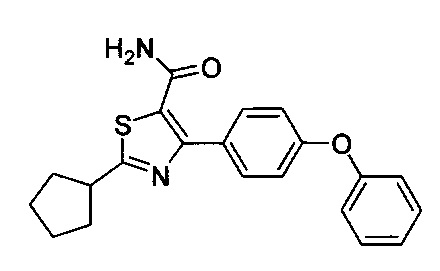
Указанное в заголовке соединение получали способом, аналогичным описанному для получения трет-бутил-4-(6-карбамоил-4'-феноксибифенил-3-ил)пиперидин-1-карбоксилата (смотри Схему 45) в виде твердого вещества белого цвета (70 мг, 38%). 1Н ЯМР (300 МГц, CDCl3) δ 7.66-7.56 (m, 2Н), 7.43-7.33 (m, 2Н), 7.17 (t, J=7.4 Гц, 1Н), 7.12-7.00 (m, 4H), 5.73 (brs, 2H), 3.52-3.35 (m, 1H), 2.32-2.16 (m, 2H), 1.90-1.77 (m, 4H), 1.76-1.66 (m, 2H). MS (ESI, методика A): m/z=365.1 [M+H]+, tR=1.780 мин. ВЭЖХ: 97% (214 нм), 96% (254 нм).
Аналитические условия
Если не указано иное, все растворители, химические вещества и реагенты были получены из коммерческих источников и использовались без дополнительной очистки. Спектры 1Н ЯМР были получены в CDCl3, DMSO-d6, CD3OD или ацетоне-d6 при 25°C при 300 МГц или 400 МГц на OXFORD (Varian) с химическим сдвигом (δ, ppm) по отношению к TMS в качестве внутреннего стандарта. Хроматограммы и спектры ВЭЖХ-МС получали с помощью системы 1200-6110 Agilent. Препаративную ВЭЖХ проводили на приборе Gilson GX-281 (Gilson) и Р230 Preparative Gradient System (градиент: 95% воды, 5% ацетонитрила, градиент 30-50 мин до 25% воды, 75% ацетонитрила). Использовалась микроволновая система СЕМ Discover SP.
Биологические свойства: Определение IC50 ингибиторов ВТК в биохимическом анализе активности киназы ADP-Glo.
Активность описанных в настоящем документе Примеров и Аналогов в качестве ингибиторов ВТК продемонстрирована и подтверждена фармакологическими анализами in vitro. Активность, которой обладают соединения, может быть продемонстрирована in vivo. Специалистам в данной области техники будет понятно, что могут быть использованы различные форматы анализа для определения активности соединений, описанных в настоящем документе.
Материалы:
ADP-Glo™ метод анализа киназной активности (cat. V9102, 10000 проб), компоненты:
- 1×50 мл реагент ADP-Glo™,
- 1×100 мл буфер для определения киназы,
- 1×субстрат для определения киназы (лиофилизированный),
- 1×5 мл Ultra Pure ATP, 10 мМ,
- 1×5 мл ADP, 10 мМ.
Реагенты и планшет:
Tris. HCl (Sigma cat. 154563), MgCl2 (Sigma cat. M1028), MnCl2 (Sigma, M3634), BSA (Sigma cat. 05470), субстрат ВТК (Signalchem, P61-58), DTT (Sigma, D0632), DMSO (Sigma, S5879), фермент ВТК (1.5 мг/мл, степень чистоты 75%, 90 нг/мкл, получен собственными силами), 384-луночный аналитический планшет (cat. 3674).
Условия анализа:
- Концентрация фермента: 8 нг/5 мкл,
- Концентрация АТР: 50 мкМ,
- Концентрация субстрата (пептид): 0.2 мг/мл,
- Композиция реакционного буфера: 40 мМ Tris-HCl pH7.5, 10 мМ MgCl2, 2 мМ MnCl2, 0.1 мг/мл BSA, 0.05 мМ DTT,
- Концентрация тестируемого соединения: DMSO ≤0.5%.
Методики:
Получение градиентного раствора дозированной формы соединения:
3-Кратное серийное разведение тестируемого соединения было сделано для 10 градиентных точек (100, 33.33, 11.11, 3.70, 1.23, 0.41, 0,14, 0.046, 0.015, 0.005 мкМ) в 100% DMSO. Промежуточное разбавление было сделано путем добавления 2 мкл разбавленного соединения в 78 мкл буфера для анализа (содержащего 40 мМ Tris. HCl, pH 7.5, 10 мМ MgCl2, 2 мМ MnCl, 0.1 мг/мл BSA, 0.05 мМ DTT), получая конечную концентрацию соединения (1000, 333.33, 111.11, 37.04, 12.35, 4.12, 1.37, 0.46, 0.15, 0.05 нМ) и концентрацию DMSO 0.5% в процентах.
Протокол анализа киназы ADP-Glo для тестирования ингибиторов ВТК:
В первую очередь для анализа были приготовлены 1х и 2х буферы. Киназу ВТК разбавляли 1х буфером для анализа, и субстрат разбавляли 2х буфером для анализа. 1 мкл разведенного соединения переносили в 384-луночный аналитический планшет, затем добавляли 2.0 мкл раствора фермента и центрифугировали при 2000 об/мин в течение 1 мин. Эту смесь инкубировали при 24°C в течение 30 минут. В аналитический планшет добавляли 2 мкл смеси пептидного субстрата/АТР, чтобы начать реакцию. Смесь тщательно перемешивали, и затем 384-луночный планшет центрифугировали и инкубировали при 24°C в течение 60 минут. Добавляли 5.0 мкл реагента ADP-Glo, чтобы остановить активность киназы и удалить неизрасходованный АТР, и планшет тщательно перемешивали и инкубировали при 24°C в течение 40 мин. Затем добавляли 10.0 мкл реагента для определения киназы, и планшет центрифугировали и затем выдерживали при 24°C в течение 30 минут. Сигнал люминесценции был прочитан на планшет-ридере Envision.
Анализ полученных данных:
% Ингибирования соединений при каждой различной концентрации вычисляется по формуле (1):
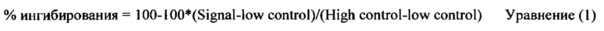
Значения IC50 соединений рассчитывали из 4-параметрической модели, используя Уравнение (2):
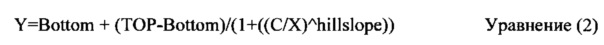
Bottom - самое низкое значение эффекта, который наблюдался,
ТОР - самое высокое значение эффекта, который наблюдался,
Hillslope - коэффициент Хилла, характеризующий наклон кривой.
В Уравнении 2 Y представляет % ингибирования, X представляет собой значение логарифма концентрации тестируемого соединения. IC50 представляет собой концентрацию соединения, когда была достигнута половина максимального ингибирования.
Все данные были проанализированы с помощью программного обеспечения IDBS XLfit 5 (ID Business Solutions Ltd., UK). Данные IC50 представлены в Таблице 1.
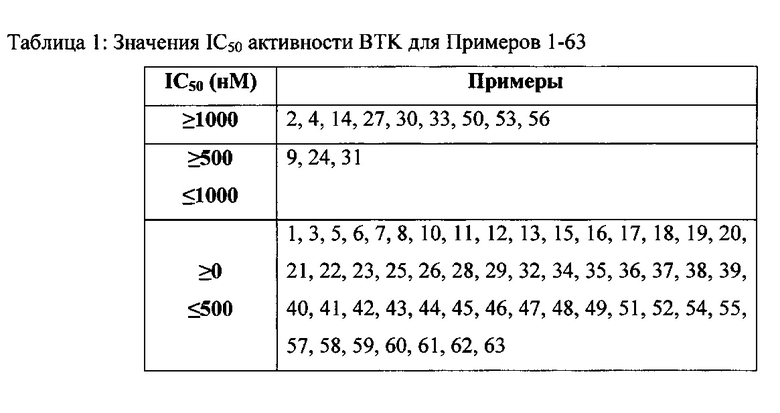
В Таблице 2 приведено сравнение Примеров 3 и 5 с Аналогами 1 и 2 по показателю активности ВТК in vitro. Примеры 3 и 5 демонстрируют примерно 100-кратное увеличение активности по сравнению с Аналогами 1 и 2 при прямом сравнительном исследовании.

Примеры 3 и 5 также демонстрируют 10х-100х различия в активности между ВТК и Src и, таким образом, являются селективными для ВТК относительно Src.
Кроме того, определяли селективность Примеров 3 и 5. В Таблице 3 приведены биохимические данные in vitro, показывающие, что Примеры 3 и 5 являются селективными для ВТК относительно Src.
Таблица 3: Данные селективности для Примеров 3 и 5
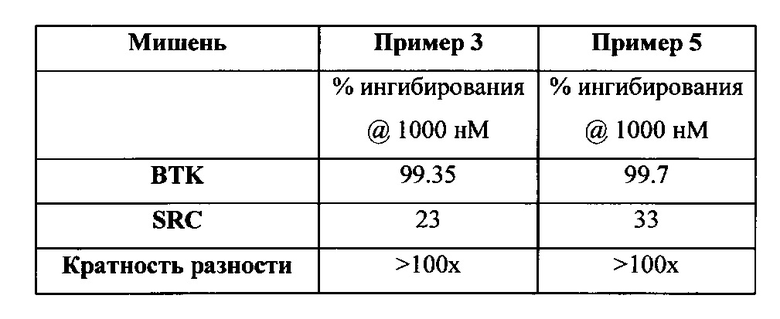
Для получения данных, представленных в Таблице 3, меченые киназой штаммы Т7-фага выращивали параллельно в 24-луночных планшетах, применяя в качестве хозяина Е. coli, полученную из штамма BL21. E.coli выращивали до фазы логарифмического роста и инфицировали фагом Т7 из замороженного стокового раствора (множественность заражения ~0.4), и инкубировали при встряхивании при 32°C до лизиса (90-150 минут). Лизаты центрифугировали (6000 g) и отфильтровывали (0.2 мкм) для удаления остатков клеток. Оставшиеся киназы продуцировали в клетках НЕК-293 и затем вводили ДНК-метки для детектирования методом количественного ПЦР. Покрытые стрептавидином магнитные бусины обрабатывали биотинилированными низкомолекулярными лигандами в течение 30 минут при комнатной температуре, получая аффинные смолы для испытаний с киназами. Бусины с присоединенными лигандами блокировали избытком биотина и промывали блокирующим буфером (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 мМ DTT) для удаления несвязанного лиганда и уменьшения неспецифичного связывания фага. Реакции связывания проводили комбинированием киназ, аффинных бусин со связанными лигандами и тестируемых соединений в 1х буфере для связывания (20% SeaBlock, 0.17х PBS, 0.05% Tween 20, 6 мМ DTT).
Тестируемые соединения готовили в виде 40х стоковых растворов в 100% DMSO и разбавляли непосредственно в растворе для анализа. Все реакции проводили в полипропиленовых 384-луночных планшетах в конечном объеме 0.04 мл. Аналитические планшеты инкубировали при комнатной температуре при встряхивании в течение 1 часа, и аффинные бусины промывали отмывочным буфером (1x PBS, 0.05% Tween 20). Затем бусины заново суспендировали в элюирующем буфере (1x PBS, 0.05% Tween 20, 0.5 мМ небиотинилированного аффинного лиганда) и инкубировали при комнатной температуре при встряхивании в течение 30 минут. Концентрацию киназы в элюатах определяли методом количественного ПЦР.
Соединение(я) подвергали скринингу при 1000 нМ, и результаты теста первого уровня связывающего взаимодействия представлены в Таблице 3 как "% Ctrl", где более низкие числа показывают более сильное действие. % Ctrl вычисляется согласно Уравнению (1) ниже:

где:
тестируемое соединение = Пример
негативный контроль = DMSO (100% Ctrl)
позитивный контроль = контрольное соединение (0% Ctrl)
Дополнительные данные биологической селективности Примеров 3 и 5 в отношении каждого члена семейства протеинкиназ SRC, а также EGFR, приведены в Таблице 4 ниже. Данные, приведенные в Таблице 4, были получены с использованием той же методики, которая описана выше для данных, приведенных в Таблице 3.
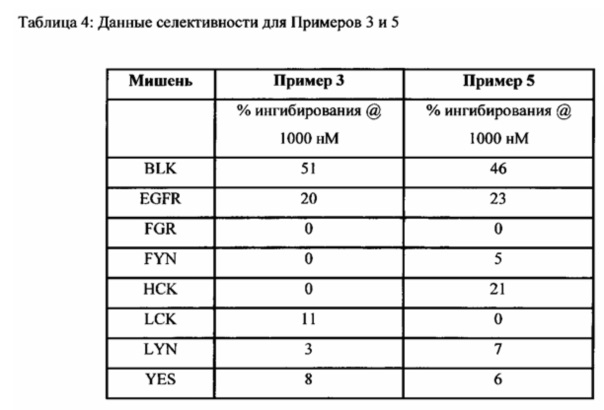
Композиции:
Настоящее изобретение включает фармацевтические композиции, содержащие соединение или его фармацевтически приемлемую соль по настоящему изобретению, которое разработано для желательного способа введения без или с одним или более фармацевтически приемлемыми и пригодными носителями.
Соединения также могут быть включены в фармацевтические композиции в комбинации с одним или более другими терапевтически активными соединениями.
Фармацевтические композиции по настоящему изобретению содержат соединение по изобретению (или его фармацевтически приемлемую соль) в качестве активного ингредиента, необязательно, фармацевтически приемлемый носитель (носители) и, необязательно, другие терапевтические ингредиенты или вспомогательные вещества. Композиции включают композиции, подходящие для перорального, ректального, местного и парентерального (включая подкожное, внутримышечное и внутривенное) введения, хотя наиболее подходящий путь в любом данном случае будет зависеть от конкретного пациента и природы и тяжести состояний, при которых вводится активный ингредиент. Фармацевтические композиции могут быть в целях удобства представлены в единичной стандартной лекарственной форме и получены любым из способов, хорошо известных в области фармации.
Соединения по настоящему изобретению могут быть объединены в качестве активного ингредиента в однородной смеси с фармацевтическим носителем в соответствии с общепринятыми способами получения фармацевтических композиций. Носитель может иметь разнообразные формы в зависимости от формы препарата, требуемого для введения, например, пероральной или парентеральной (включая внутривенную). Таким образом, фармацевтические композиции по настоящему изобретению могут быть представлены в виде дискретных единиц, подходящих для перорального введения, таких как капсулы, облатки или таблетки, каждая содержащая заранее определенное количество активного ингредиента. Кроме того, композиции могут быть представлены в виде порошка, в виде гранул, в виде раствора, в виде суспензии в водной жидкости, в виде неводной жидкости, в виде эмульсии масло-в-воде или в виде жидкой эмульсии вода-в-масле. Помимо общих лекарственных форм, изложенных выше, соединение, представленное Формулой I или его фармацевтически приемлемой солью, также может быть введено с помощью способов контролируемого высвобождения и/или доставки лекарственного средства. Композиции могут быть получены с помощью любого из способов фармации. В общем, такие способы включают стадию приведения в контакт активного ингредиента с носителем, который содержит один или более необходимых ингредиентов. В общем, композиции получают путем равномерного и тщательного смешивания активного ингредиента с жидкими носителями, или тонко измельченными твердыми носителями, или ими обоими. Затем продукту можно в целях удобства придать желаемую форму.
Используемый фармацевтический носитель может быть, например, твердым веществом, жидкостью или газообразным. Примеры твердых носителей включают лактозу, каолин, сахарозу, тальк, желатин, агар, пектин, аравийскую камедь, стеарат магния и стеариновую кислоту. Примерами жидких носителей являются сахарный сироп, арахисовое масло, оливковое масло и вода. Примеры газообразных носителей включают диоксид углерода и азот.
Таблетка, содержащая композицию по настоящему изобретению, может быть получена путем прессования или формования, необязательно с одним или несколькими дополнительными ингредиентами или адъювантами. Прессованные таблетки могут быть получены прессованием в соответствующей машине активного ингредиента в свободно текучей форме, такой как порошок или гранулы, необязательно смешанного со связующим веществом, смазывающим веществом, инертным разбавителем, поверхностно-активным или диспергирующим веществом. Формованные таблетки могут быть изготовлены формованием в соответствующей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Каждая таблетка, предпочтительно, содержит от около 0,05 мг до около 5 г активного ингредиента, и каждая облатка или капсула, предпочтительно, содержит от около 0,05 мг до около 5 г активного ингредиента.
Композиция, предназначенная для перорального введения человеку, может содержать от около 0,5 мг до около 5 г активного вещества, смешанного с соответствующим и подходящим количеством материала-носителя, которое может варьироваться от около 5 до около 95 процентов от общей массы композиции. Стандартные лекарственные формы обычно содержат от около 1 мг до около 2 г активного ингредиента, обычно 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 800 мг или 1000 мг.
Для изготовления лекарственного препарата соединения по настоящему изобретению могут быть предоставлены с высокой степенью чистоты, по меньшей мере, около 90%, 95% или 98% по весу.
Фармацевтические композиции по настоящему изобретению, пригодные для парентерального введения, могут быть получены в виде растворов или суспензий активных соединений в воде.
Может быть включено подходящее поверхностно-активное вещество, такое как, например, гидроксипропилцеллюлоза. Также могут быть приготовлены дисперсии в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Кроме того, может быть включен консервант для предотвращения негативного роста микроорганизмов.
Фармацевтические композиции по настоящему изобретению, пригодные для применения в виде инъекций, включают стерильные водные растворы или дисперсии. Кроме того, композиции могут быть в форме стерильных порошков для экстемпорального приготовления таких стерильных инъекционных растворов или дисперсий. Во всех случаях конечная инъецируемая форма должна быть стерильной и должна быть фактически жидкой для легкого введения через шприц. Фармацевтические композиции должны быть стабильными в условиях производства и хранения; при этом, предпочтительно, должны быть защищены от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), растительные масла и их подходящие смеси.
Фармацевтические композиции по настоящему изобретению могут быть в форме, подходящей для местного применения, такой как, например, аэрозоль, крем, мазь, лосьон, присыпка или тому подобное. Кроме того, композиции могут быть в форме, пригодной для использования в трансдермальных устройствах. Эти композиции могут быть получены с использованием соединения, представленного Формулой I по настоящему изобретению, или его фармацевтически приемлемой соли с помощью общепринятых технологических методов. В качестве примера, крем или мазь готовят путем смешивания гидрофильного вещества и воды совместно с от около 5% масс. до около 10% масс. соединения для приготовления крема или мази, имеющих желаемую консистенцию.
Фармацевтические композиции по настоящему изобретению могут быть в форме, подходящей для ректального введения, где носитель представляет собой твердое вещество. Предпочтительно, чтобы смесь формировалась в суппозитории со стандартной дозой. Подходящие носители включают масло какао и другие вещества, обычно используемые в данной области техники. Суппозитории могут быть легко получены сначала смешиванием композиции с размягченным или расплавленным носителем(ями) с последующим охлаждением и формованием.
В дополнение к вышеупомянутым ингредиентам-носителям, фармацевтические композиции, описанные выше, могут включать, в случае необходимости, один или более дополнительных ингредиентов, таких как разбавители, буферы, ароматизирующие вещества, связующие, поверхностно-активные вещества, загустители, смазывающие вещества, консерванты (в том числе антиоксиданты), и тому подобное. Кроме того, могут быть включены другие вспомогательные вещества для придания композиции изотоничности с кровью предполагаемого реципиента.
Композиции, содержащие соединение, описываемое Формулой I, или его фармацевтически приемлемые соли, также могут быть получены в порошкообразной или жидкой концентрированной форме.
Области применения:
Соединения по настоящему изобретению ингибируют активность ВТК у животных, включая людей, и пригодны для лечения и/или профилактики различных заболеваний и состояний, таких как рак, воспаление, фиброзные заболевания и аутоиммунные заболевания, которые вызваны, опосредованы и/или передаются ВТК.
В частности, соединения по настоящему изобретению и композиции на их основе являются ингибиторами ВТК и полезны при лечении состояний, модулированных, по меньшей мере, частично, ВТК. В некоторых аспектах настоящее изобретение включает способ лечения рака, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения рака, опосредованного, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли Формулы I.
В некоторых аспектах настоящее изобретение включает способ лечения или способ изготовления лекарственного средства для лечения рака, такого, как описанные в настоящем документе, который опосредован, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения хоминга лимфоцитов и воспаления, опосредованных, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли Формулы I.
В некоторых аспектах настоящее изобретение включает способ лечения или способ изготовления лекарственного средства для лечения хоминга лимфоцитов и воспаления, таких, как описанные в настоящем документе, которые опосредованы, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения невропатической боли, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения нейропатической боли, опосредованной, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли Формулы I.
В некоторых аспектах настоящее изобретение включает способ лечения или способ изготовления лекарственного средства для лечения нейропатической боли, такой, как описанная в настоящем документе, которая опосредована, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения фиброзных заболеваний, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения фиброзных заболеваний, опосредованных, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли Формулы I.
В некоторых аспектах настоящее изобретение включает способ лечения или способ изготовления лекарственного средства для лечения фиброзных заболеваний, таких, как описанные в настоящем документе, которые опосредованы, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения тромбоза, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения тромбоза, опосредованного, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли Формулы I.
В некоторых аспектах настоящее изобретение включает способ лечения или способ изготовления лекарственного средства для лечения тромбоза, такого, как описанный в настоящем документе, которое опосредовано, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения холестатического зуда, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
В некоторых аспектах настоящее изобретение включает способ лечения холестатического зуда, опосредованного, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли Формулы I.
В некоторых аспектах настоящее изобретение включает способ лечения или способ изготовления лекарственного средства для лечения холестатического зуда, такого, как описанные в настоящем документе, которые опосредовано, по меньшей мере, частично, ВТК, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению.
Соединения Формулы I по настоящему изобретению могут быть использованы для лечения различных видов онкозаболеваний, включая, но не ограничиваясь ими, солидные опухоли, саркому, фибросаркому, остеому, меланому, ретинобластому, рабдомиосаркому, глиобластому, нейробластому, тератокарциному, гемопоэтическое злокачественное новообразование и злокачественный асцит. Более конкретно, виды онкозаболеваний включают, но не ограничиваются ими, рак легких, рак мочевого пузыря, рак поджелудочной железы, рак почки, рак желудка, рак молочной железы, рак толстой кишки, рак предстательной железы (включая метастазы в кости), гепатоклеточную карциному, рак яичников, плоскоклеточную карциному пищевода, меланому, анапластическую крупноклеточную лимфому и воспалительную миофибробластическую опухоль и глиобластому.
В некоторых аспектах вышеуказанные способы используются для лечения одного или более видов рака мочевого пузыря, толстой и прямой кишки, немелкоклеточного рака легкого, молочной железы или поджелудочной железы. В некоторых аспектах вышеуказанные способы используются для лечения одного или более видов рака яичников, желудка, шеи и головы, предстательной железы, гепатоклеточного рака, почек, глиомы или саркомы.
В некоторых аспектах настоящее изобретение включает способ, в том числе вышеуказанные способы, где соединение применяется для ингибирования клеточного эпителиально-мезенхимального перехода (ЕМТ).
В некоторых аспектах способ дополнительно включает введение, по меньшей мере, дополнительного активного вещества. В некоторых аспектах изобретение включает способ лечения рака, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения или соли по изобретению, где, по меньшей мере, одно дополнительное активное противораковое вещество используется как часть способа.
В некоторых аспектах настоящее изобретение включает способ лечения заболевания, описанного в настоящем документе, опосредованного, по меньшей мере, частично, ВТК, включающий назначение млекопитающему, нуждающемуся в этом, терапевтически эффективной схемы приема лекарства, содержащего соединение или соль Формулы I и, по меньшей мере, одно дополнительное активное вещество. Как правило, уровни доз порядка от около 0,01 мг/кг до около 150 мг/кг массы тела в день являются эффективными при лечении указанных выше состояний, или, в качестве альтернативы, от около 0,5 мг до около 7 г на каждого пациента в день. Например, воспаление, рак, псориаз, аллергия/астма, состояние и заболевания иммунной системы, состояние и заболевания центральной нервной системы (CNS) могут эффективно лечиться путем введения от около 0,01 до 50 мг соединения на килограмм массы тела в день или, альтернативно, от около 0,5 мг до около 3,5 г на каждого пациента в день.
Является понятным, однако, что конкретная величина дозы для любого конкретного пациента будет зависеть от целого ряда факторов, включая возраст, массу тела, общее состояние здоровья, пол, диету, время введения, путь введения, скорость выведения, комбинацию лекарственных средств и тяжесть конкретного заболевания, подвергающегося терапии.
Общие определения и сокращения:
За исключением случаев, когда указано иное, применяются следующие общие условные обозначения и определения. Если иное здесь не указано, формулировка и термины должны быть даны в их самой широкой обоснованной интерпретации, как понятно специалисту в данной области техники. Любые приведенные примеры не являются ограничивающими. Любые заголовки разделов или подразделов в настоящем документе даны для удобства читателя и/или формального соответствия и не являются ограничивающими.
Описание соединения в настоящем документе является открытым и охватывает любой материал или композицию, содержащую описанное соединение (например, композицию, содержащую рацемическую смесь, таутомеры, эпимеры, стереоизомеры, неочищенные смеси и т.д.). Поскольку соль, сольват или гидрат, полиморф, или другой комплекс соединения включает само это соединение, описание соединения охватывает вещества, содержащие такие формы. Изотопно меченые соединения также включены, кроме специально исключенных случаев. Например, водород не ограничивается водородом, содержащим ноль нейтронов. Например, дейтерий именуется в настоящем документе как "D" и означает атом водорода, имеющий один нейтрон.
Термин "активное вещество" означает соединение по изобретению в любой форме - соли, полиморфа, кристалла, сольвата или гидрата.
Термин "замещенный" и заместители, содержащиеся в формулах в настоящем документе, относятся к замещению одного или более водородных радикалов в данной структуре заданным радикалом, или, если не указано, к замене любым химически возможным радикалом. Когда более чем одно положение в данной структуре может быть замещено более чем одним заместителем, выбранным из указанных групп, то заместители могут быть или одинаковыми, или различными в каждом положении (независимо выбранном), если не указано иное. В некоторых случаях две позиции в данной структуре могут быть замещены одним общим заместителем. Понятно, что химически невозможные или с высокой степенью нестабильные конфигурации нежелательны или не предполагаются для использования, как будет понятно специалисту в данной области.
В описаниях и формуле изобретения, где объект изобретения (например, замещение в заданном молекулярном положении) излагается как выбранный из группы возможностей, перечисление специально предназначено для включения любого подмножества из указанной группы. В случае нескольких различных позиций или заместителей любая комбинация группы или различных подмножеств также предполагается.
Если не указано иное, заместитель, дирадикал или другая группа, упомянутая в настоящем документе, могут быть связаны в любой подходящей позиции с рассматриваемой подвергаемой воздействию молекулой. Например, термин "индолил" включает 1-индолил, 2-индолил, 3-индолил и т.д.
Правило для описания содержания углерода в некоторых фрагментах представляет собой "(Сa-b)" или "Са-Cb", означающее, что фрагмент может содержать любое число от "а" до "b" атомов углерода. Соалкил означает одинарную ковалентную химическую связь, когда он является связывающим фрагментом, и водород, когда он является концевым фрагментом. Аналогичным образом, "х-y" может указывать на функциональную группу, содержащую от x до у атомов, например, 5.6-гетероциклоалкил означает гетероциклоалкил, имеющий либо пять, либо шесть кольцевых членов. "Сх-y" может быть использован для определения числа атомов углерода в группе. Например, "С0-12алкил" означает алкил, имеющий 0-12 атомов углерода, где С0алкил означает одинарную ковалентную химическую связь в случае связывающей группы и означает атом водорода в случае концевой группы. С0-12алкил включает различные альтернативные варианты, в том числе, но не ограничиваясь ими, C1-12алкил, C2-12алкил, C3-12алкил, С4-12алкил, С5-12алкил, С6-12алкил, С7-12алкил, C8-12алкил, C9-12алкил, C10-12алкил, C11-12алкил, C1-11алкил, С1-10алкил, С1-9алкил, С1-8алкил, С1-7алкил, С1-6алкил, С1-5алкил, С1-4алкил, C1-3алкил, С1-2алкил, C1алкил и С0алкил. С0-12алкил дополнительно включает любую комбинацию "а" и "b" и/или "x" и "y" число атомов углерода, включая, но не ограничиваясь ими, С2-12алкил, С3-11алкил, С4-10алкил, С5-9алкил, C6-8алкил и C7алкил.
Термин "отсутствует" в контексте настоящего документа для описания структурной переменной (например, "-R- отсутствует") означает, что дирадикал R не имеет атомов, и только представляет собой связь между другими соседними атомами, если не указано иное. Если не указано иное (например, посредством соединительного "-"), присоединения фрагментов наименования соединения находятся в крайнем правом положении описанного фрагмента. То есть, наименование заместителя начинается с концевого фрагмента, продолжается любыми мостиковыми фрагментами и заканчивается соединительным фрагментом. Например, "гетероарилтиоС1-4алкил" представляет собой гетероарильную группу, присоединенную через тиосеру к С1-4алкилу, где алкил присоединен к химической группе, несущей заместитель.
Термин "алифатический" означает любой углеводородный фрагмент и может содержать линейные, разветвленные и циклические части, и может быть насыщенным или ненасыщенным.
Термин "алкил" означает любую насыщенную углеводородную группу, которая является линейной или разветвленной. Примеры алкильных групп включают метил, этил, пропил, 2-пропил, н-бутил, изо-бутил, трет-бутил, пентил, и тому подобное.
Термин "алкенил" означает любую этиленненасыщенную линейную или разветвленную углеводородную группу. Типичные примеры включают, но не ограничиваются ими, этенил, 1-пропенил, 2-пропенил, 1-, 2- или 3-бутенил, и тому подобное.
Термин "алкинил" означает любую ацетиленненасыщенную линейную или разветвленную углеводородную группу. Типичные примеры включают, но не ограничиваются ими, этинил, 1-пропинил, 2-пропинил, 1-, 2- или 3-бутинил, и тому подобное.
Термин "алкокси" означает -O-алкил, -O-алкенил или -O-алкенил. "Галоалкокси" означает -O-(галоалкил) группу. Типичные примеры включают, но не ограничиваются ими, трифторметокси, трибромметокси, и тому подобное.
"Галоалкил" означает алкил, предпочтительно, низший алкил, который замещен одним или более одинаковыми или различными атомами галогена.
"Гидроксиалкил" означает алкил, предпочтительно, низший алкил, который замещен одним, двумя или тремя гидроксильными группами; например, гидроксиметил, 1 или 2-гидроксиэтил, 1,2-, 1,3- или 2,3-дигидроксипропил, и тому подобное.
Термин "алканоил" означает -C(O)-алкил, -C(O)-алкенил или -C(O)-алкинил.
"Алкилтио" означает -S-(алкил) или -S-(незамещенный циклоалкил) группу. Типичные примеры включают, но не ограничиваются ими, метилтио, этилтио, пропилтио, бутилтио, циклопропилтио, циклобутилтио, циклопентилтио, циклогексилтио, и тому подобное.
Термин "циклический" означает любую кольцевую систему с или без гетероатомов (N, О или S(O)0-2), и которая может быть насыщенной, частично насыщенной или ненасыщенной. Кольцевые системы могут быть мостиковыми, и могут включать конденсированные кольца. Размер кольцевых систем может быть описан с помощью таких терминов, как "x-yциклическая", что означает циклическую кольцевую систему, которая может содержать от x до y атомов в кольце. Например, термин "9-10карбоциклический" означает 5,6 или 6,6 конденсированную бициклическую карбоциклическую кольцевую систему, которая может быть насыщенной, ненасыщенной или ароматической. Это также означает фенил, конденсированный с одной 5- или 6-членной насыщенной или ненасыщенной карбоциклической группой. Неограничивающие примеры таких групп включают нафтил, 1,2,3,4 тетрагидронафтил, инденил, инданил, и тому подобное.
Термин "карбоциклический" означает циклический кольцевой фрагмент, содержащий только атомы углерода в кольце(ах) без учета ароматичности. Термин 3-10-членный карбоциклический означает химически возможные моноциклические и конденсированные бициклические карбоциклы, имеющие от 3 до 10 атомов в кольце. Аналогичным образом, термин 4-6-членный карбоциклический означает моноциклические карбоциклические кольцевые фрагменты, имеющие от 4 до 6 атомов углерода в кольце, и термин 9-10-членный карбоциклический означает конденсированные бициклические карбоциклические кольцевые фрагменты, имеющие от 9 до 10 атомов углерода в кольце.
Термин "циклоалкил" означает неароматический 3-12 углеродный моноциклический, бициклический или полициклический алифатический кольцевой фрагмент. Циклоалкильной группой может быть циклоалкил, полициклоалкил, мостик или спироалкил. Одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженную систему пи-электронов. Примеры, без ограничения, циклоалкильных групп представляют собой циклопропан, циклобутан, циклопентан, циклопентен, циклогексан, циклогексадиен, адамантан, циклогептан, циклогептатриен, и тому подобное.
Термин "ненасыщенный карбоциклический" означает любую циклоалкильную группу, содержащую, по меньшей мере, одну двойную или тройную связь. Термин "циклоалкенил" означает циклоалкильную группу, имеющую, по меньшей мере, одну двойную связь в кольцевом фрагменте.
Термины "бициклоалкил" и "полициклоалкил" означают структуру, состоящую из двух или более циклоалкильных фрагментов, которые имеют два или более общих атомов. Если циклоалкильные фрагменты имеют строго два общих атома, они, как говорят, являются "конденсированными". Примеры включают, но не ограничиваются ими, бицикло[3.1.0]гексил, пергидронафтил, и тому подобное. Если циклоалкильные фрагменты имеют более двух общих атомов, они называются "мостиковыми". Примеры включают, но не ограничиваются ими, бицикло[2.2.1]гептил ("норборнил"), бицикло[2.2.2]октил, и тому подобное.
Термин "спироалкил" означает структуру, состоящую из двух циклоалкильных фрагментов, которые имеют строго один общий атом. Примеры включают, но не ограничиваются ими, спиро[4.5]децил, спиро[2,3]гексил, и тому подобное.
Термин "ароматический" означает планарные кольцевые фрагменты, содержащие 4n+2 пи электронов, где n представляет собой целое число.
Термин "арил" означает ароматические фрагменты, содержащие только атомы углерода в своей кольцевой системе. Неограничивающие примеры включают фенил, нафтил и антраценил. Термины "арил-алкил", или "арилалкил", или "аралкил" относятся к любому алкилу, который образует связанную мостиком часть с концевым арилом.
Термин "аралкил" означает алкил, который замещен арильной группой, как определено выше; например, -CH2 фенил, -(CH2)2фенил, -(СН2)3фенил, CH3CH(CH3)CH2фенил, и тому подобное, и их производные.
Термин "гетероциклический" означает циклический кольцевой фрагмент, содержащий, по меньшей мере, один гетероатом (N, O или S(O)0-2), включая гетероарил, гетероциклоалкил, в том числе ненасыщенные гетероциклические кольца.
Термин "гетероциклоалкил" означает неароматический моноциклический, бициклический или полициклический гетероциклический кольцевой фрагмент из от 3 до 12 кольцевых атомов, содержащий, по меньшей мере, одно кольцо, имеющее один или более гетероатомов. Такие кольца могут также иметь одну или более двойных связей. Тем не менее, кольца не имеют полностью сопряженную систему пи-электронов. Примеры, без ограничения, гетероциклоалкильных колец включают азетидин, оксетан, тетрагидрофуран, тетрагидропиран, оксепан, оксокан, тиетан, тиазолидин, оксазолидин, оксазетидин, пиразолидин, изоксазолидин, изотиазолидин, тетрагидротиофен, тетрагидротиопиран, тиепан, тиокан, азетидин, пирролидин, пиперидин, N-метилпиперидин, азепан, 1,4-диазапан, азокан, [1,3]диоксан, оксазолидин, пиперазин, гомопиперазин, морфолин, тиоморфолин, 1,2,3,6-тетрагидропиридин, и тому подобное. Другие примеры гетероциклоалкильных колец включают окисленные формы серосодержащих колец. Таким образом, тетрагидротиофен-1-оксид, тетрагидротиофен-1,1-диоксид, тиоморфолин-1-оксид, тиоморфолин-1,1-диоксид, тетрагидротиопиран-1-оксид, тетрагидротиопиран-1,1-диоксид, тиазолидин-1-оксид и тиазолидин-1,1-диоксид также считаются гетероциклоалкильными кольцами. Термин "гетероциклоалкил" также включает конденсированные кольцевые системы и может включать карбоциклическое кольцо, которое является частично или полностью ненасыщенным, такое как бензольное кольцо, с образованием бензоконденсированных гетероциклоалкильных колец. Например, 3,4-дигидро-1,4-бензодиоксин, тетрагидрохинолин, тетрагидроизохинолин, и тому подобное. Термин "гетероциклоалкил" также включает гетеробициклоалкил, гетерополициклоалкил или гетероспироалкил, которые являются бициклоалкильной, полициклоалкильной или спироалкильной группами, в которых один или более атом(ов) углерода замещены одним или более гетероатомами, выбранными из O, N и S. Например, 2-окса-спиро[3.3]гептан, 2,7-диазаспиро[4,5]декан, 6-окса-2-тиа-спиро[3,4]октан, октагидропирроло[1,2-а]пиразин, 7-аза-бицикло[2.2.1]гептан, 2-окса-бицикло[2.2.2]октан, и тому подобное, представляют собой такие гетероциклоалкилы.
Примеры насыщенных гетероциклических групп включают, но не ограничиваются ими, оксиранил, тиаранил, азиридинил, оксетанил, тиатанил, азетидинил, тетрагидрофуранил, тетрагидротиофенил, пирролидинил, тетрагидропиранил, тетрагидротиопиранил, пиперидинил, 1,4-диоксанил, 1,4-оксатианил, морфолинил, 1,4-дитианил, пиперазинил, 1,4-азатианил, оксепанил, тиепанил, азепанил, 1,4-диоксепанил, 1,4-оксатиепанил, 1,4-оксаазепанил, 1,4-дитиепанил, 1,4-тиеазепанил, 1,4-диазепанил.
Неарильные гетероциклические группы включают насыщенные и ненасыщенные системы и могут включать группы, имеющие только 4 атома в своей кольцевой системе. Такие гетероциклические группы включают бензо-конденсированные кольцевые системы и кольцевые системы, замещенные одним или более оксо- фрагментами. Описание кольцевой серы понимается включающим сульфид, сульфоксид или сульфон, где это возможно. Гетероциклические группы также включают частично ненасыщенные или полностью насыщенные 4-10-членные кольцевые системы, например, одиночные кольца размером от 4 до 8 атомов и бициклические кольцевые системы, в том числе ароматические 6-членные арильные или гетероарильные кольца, конденсированные с неароматическим кольцом. Кроме того, включены 4-6-членные кольцевые системы ("4-6-членный гетероцикл"), которые включают 5-6-членные гетероарилы, и включают группы, такие как азетидинил и пиперидинил. Гетероциклы могут быть присоединены через гетероатом, где такое возможно. Например, группа, образованная из пиррола, может быть пиррол-1-ил (N-присоединенная) или пиррол-3-ил (С-присоединенная). Другие гетероциклы включают имидазо(4,5-b)пиридин-3-ил и бензоимидазол-1-ил.
Примеры гетероциклических групп включают пирролидинил, тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 1,2,3,6-тетрагидропиридинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, 3Н-индолил, хинолизинил, и тому подобное.
Термин "ненасыщенный гетероциклический" означает гетероциклоалкильную группу, содержащую, по меньшей мере, одну ненасыщенную связь. Термин "гетеробициклоалкил" означает бициклоалкильную структуру, в которой, по меньшей мере, один атом углерода замещен гетероатомом. Термин "гетероспироалкил" означает спироалкильную структуру, в которой, по меньшей мере, один атом углерода замещен гетероатомом.
Примеры частично ненасыщенных гетероалициклических групп включают, но не ограничиваются ими: 3,4-дигидро-2Н-пиранил, 5,6-дигидро-2Н-пиранил, 2Н-пиранил, 1,2,3,4-тетрагидропиридинил и 1,2,5,6-тетрагидропиридинил.
Термины "гетероарил" или "гетарил" означают моноциклический, бициклический или полициклический ароматический гетероциклический кольцевой фрагмент, содержащий 5-12 атомов. Примеры таких гетероарильных колец включают, но не ограничиваются ими, фурил, тиенил, пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридил, пиридазинил, пиримидинил, пиразинил и триазинил. Термины "гетероарил" также включают гетероарильные кольца с конденсированными карбоциклическими кольцевыми системами, которые являются частично или полностью ненасыщенными, такие как бензольное кольцо, с образованием бензоконденсированного гетероарила. Например, бензоимидазол, бензоксазол, бензотиазол, бензофуран, хинолин, изохинолин, хиноксалин, индазол, имидазо[1,2-а]пиридин, 3-метил-2-оксо-2,3-дигидробензо[d]оксазол-5-ил, 2-метил-2Н-индазол-5-ил, 3-метилимидазо[1,5-а]пиридин, 2-метил-1Н-бензо[d]имидазол, 1Н-пирроло[2,3-b]пиридин, 3,4-дигидро-2Н-бензо[b][1,4]оксазин, 2-оксо-2,3-дигидро-бензо[d]оксазол, 3-оксо-3,4-дигидро-2Н-бензо[b][1,4]оксазин, 2,3-дигидро-бензо[b][1,4]диоксин, 2-метил-[1,2,4]триазол[1,5-а]пиридин, и тому подобное. Кроме того, термины "гетероарил" включает конденсированные 5-6, 5-5, 6-6 кольцевые системы, необязательно обладающие одним атомом азота в месте соединения колец. Примеры таких гетарильных колец включают, но не ограничиваются ими, пирролопиримидинил, имидазо[1,2-а]пиридинил, имидазо[2,1-b]тиазолил, имидазо[4,5-b]пиридин, пирроло[2,1-f][1,2,4]триазинил, и тому подобное. Гетероарильные группы могут быть присоединены к другим группам через их атомы углерода или гетероатом(ы), если это применимо. Например, пиррол может быть присоединен через атом азота или любой из атомов углерода.
Гетероарилы включают, например, 5- и 6-членные моноциклические группы, такие как пиразинил и пиридинил, и 9- и 10-членные конденсированные бициклические кольцевые фрагменты, такие как хинолинил. Другие примеры гетероарила включают хинолин-4-ил, 7-метокси-хинолин-4-ил, пиридин-4-ил, пиридин-3-ил и пиридин-2-ил. Другие примеры гетероарила включают пиридинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фуранил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил, фуропиридинил, и тому подобное. Примеры 5-6-членных гетероарилов включают тиофенил, изоксазолил, 1,2,3-триазолил, 1,2,3-оксадиазолил, 1,2,3-тиадиазолил, 1,2,4-триазолил, 1,3,4-оксадиазолил, 1,3,4-тиадиазолил, 1,2,5-оксадиазолил, 1,2,5-тиадиазолил, пиридил, пиридазинил, пиримидинил, пиразинил, 1,2,4-оксадиазолил, 1,2,5-триазинил, 1,3,5-триазинил, 6-оксо-1,6-дигидропиридин, и тому подобное.
"Гетероаралкильная" группа означает алкил, предпочтительно, низший алкил, который замещен гетероарильной группой; например, -CH2-пиридинил, -(CH2)2пиримидинил, -(СН2)3имидазолил, и тому подобное, и их производные.
Фармацевтически приемлемым гетероарилом является такой, который достаточно устойчив для присоединения к соединению по настоящему изобретению, составления фармацевтической композиции, и затем введения пациенту, нуждающемуся в этом.
Примеры моноциклических гетероарильных групп включают, но не ограничиваются ими: пирролил, фуранил, тиофенил, пиразолил, имидазолил, изоксазолил, оксазолил, изотиазолил, тиазолил, 1,2,3-триазолил, 1,3,4-триазолил, 1-окса-2,3-диазолил, 1-окса-2,4-диазолил, 1-окса-2,5-диазолил, 1-окса-3,4-диазолил, 1-тиа-2,3-диазолил, 1-тиа-2,4-диазолил, 1-тиа-2,5-диазолил, 1-тиа-3,4-диазолил, тетразолил, пиридинил, пиридазинил, пиримидинил, пиразинил.
Примеры конденсированных кольцевых гетероарильных групп включают, но не ограничиваются ими: бензодуранил, бензотиофенил, индолил, бензимидазолил, индазолил, бензотриазолил, пирроло[2,3-b]пиридинил, пирроло[2,3-с]пиридинил, пирроло[3,2-с]пиридинил, пирроло[3,2-b]пиридинил, имидазо[4,5-b]пиридинил, имидазо[4,5-с]пиридинил, пиразоло[4,3-d]пиридинил, пиразоло[4,3-с]пиридинил, пиразоло[3,4-с]пиридинил, пиразоло[3,4-b]пиридинил, изоиндолил, индазолил, пуринил, индолинил, имидазо[1,2-а]пиридинил, имидазо[1,5-а]пиридинил, пиразоло[1,5-а]пиридинил, пирроло[1,2-b] пиридазинил, имидазо[1,2-с]пиримидинил, хинолинил, изохинолинил, циннолинил, азахиназолин, хиноксалинил, фталазинил, 1,6-нафтиридинил, 1,7-нафтиридинил, 1,8- нафтиридинил, 1,5-нафтиридинил, 2,6-нафтиридинил, 2,7-нафтиридинил, пиридо[3,2-d]пиримидинил, пиридо[4,3-d]пиримидинил, пиридо[3,4-d]пиримидинил, пиридо[2,3-d]пиримидинил, пиридо[2,3-b]пиразинил, пиридо[3,4-b]пиразинил, пиримидо[5,4-d]пиримидинил, пиримидо[2,3-b]пиразинил, пиримидо[4,5-d]пиримидинил.
"Арилтио" означает -S-арильную или -S-гетероарильную группу, как определено в данном описании. Типичные примеры включают, но не ограничиваются ими, фенилтио, пиридилтио, фуранилтио, тиенилтио, пиримидинилтио, и тому подобное, и их производные.
Термин "9-10-членный гетероцикл" означает конденсированный 5,6 или 6,6 бициклический гетероциклический кольцевой фрагмент, который может быть насыщенным, ненасыщенным или ароматическим. Термин "9-10-членный конденсированный бициклический гетероциклический" также означает фенил, конденсированный с одной 5- или 6-членной гетероциклической группой. Примеры включают бензофуранил, бензотиофенил, индолил, бензоксазолил, 3Н-имидазо[4,5-с]пиридинил, дигидрофтазинил, 1Н-имидазо[4,5-с]пиридин-1-ил, имидазо[4,5-b]пиридил, 1,3-бензо[1,3]диоксолил, 2Н-хроманил, изохроманил, 5-оксо-2,3-дигидро-5Н-[1,3]тиазоло[3,2-а]пиримидил, 1,3-бензотиазолил, 1,4,5,6-тетрагидропиридазил, 1,2,3,4,7,8-гексагидроптеридинил, 2-тиоксо-2,3,6,9-тетрагидро-1Н-пурин-8-ил, 3,7-дигидро-1Н-пурин-8-ил, 3,4-дигидропиримидин-1-ил, 2,3-дигидро-1,4-бензодиоксинил, бензо[1,3]диоксолил, 2Н-хроменил, хроманил, 3,4-дигидрофталазинил, 2,3-дигидро-1Н-индолил, 1,3-дигидро-2Н-изоиндол-2-ил, 2,4,7-триоксо-1,2,3,4,7,8-гексагидроптеридинил, тиено[3,2-d]пиримидинил, 4-оксо-4,7-дигидро-3Н-пирроло[2,3-d]пиримидинил, 1,3-диметил-6-оксо-2-тиоксо-2,3,6,9-тетрагидро-1Н-пуринил, 1,2-дигидроизохинолинил, 2-оксо-1,3-бензоксазолил, 2,3-дигидро-5Н-1,3-тиазоло-[3,2-а]пиримидинил, 5,6,7,8-тетрагидрохиназолинил, 4-оксохроманил, 1,3-бензотиазолил, бензимидазолил, бензотриазолил, пуринил, фурилпиридил, тиофенилпиримидил, тиофенилпиридил, пирролилпиридил, оксазолилпиридил, тиазолилпиридил, 3,4-дигидропиримидин-1-ил, имидазолилпиридил, хинолинил, изохинолинил, хиназолинил, хиноксалинил, нафтиридинил, пиразолил[3,4]пиридин, 1,2-дигидроизохинолинил, циннолинил, 2,3-дигидро-бензо[1,4]диоксин-4-ил, 4,5,6,7-тетрагидро-бензо[b]-тиофенил-2-ил, 1,8-нафтиридинил, 1,5- нафтиридинил, 1,6-нафтиридинил, 1,7- нафтиридинил, 3,4-дигидро-2Н-1,4-бензотиазин, 4,8-дигидрокси-хинолинил, 1-оксо-1,2-дигидро-изохинолинил, 4-фенил-[1,2,3]тиадиазолил, и тому подобное.
Термин "арилокси" означает -O-арильную или -O-гетероарильную группу, как определено в данном описании. Типичные примеры включают, но не ограничиваются ими, фенокси, пиридинилокси, фуранилокси, тиенилокси, пиримидинилокси, пиразинилокси, и тому подобное, а также их производные.
Термин "оксо" означает соединение, содержащее карбонильную группу. Специалист в данной области понимает, что группе "оксо" необходима вторая связь с атомом, к которому присоединен оксо.
Термин "гало" или "галоген" означает фтор, хлор, бром или йод.
Термин "ацил" означает -C(O)R группу, где R может быть выбран из неограничивающей группы, включающей водород или необязательно замещенный низший алкил, тригалометил, незамещенный циклоалкил, арил или другой подходящий заместитель.
"Тиоацил" или "тиокарбонил" обозначает группу -C(S)R'' с R, как определено выше.
Термин "защитная группа" означает подходящую химическую группу, которая может быть присоединена к функциональной группе и удалена на более поздней стадии, чтобы открыть сохраненную функциональную группу. Примеры подходящих защитных групп для различных функциональных групп описаны в Т.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2d Ed., John Wiley and Sons (1991 и более поздние издания); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); и L. Paquette, ed. Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995). Термин "гидроксильная защитная группа", как он использован здесь, если не указано иное, включает Ac, CBZ и различные гидроксизащитные группы, хорошо знакомые специалистам в данной области, в том числе группы, указанные в Greene.
Термин "линейная структура" означает фрагмент, имеющий заместители, которые не циклизуются с образованием кольцевой системы. Типичный пример включает, но не ограничивается им, соединение, включая -NRXRY, где любые атомы "RX" и любые атомы "RY" не соединяются с образованием кольца.
Используемый в данном описании термин "фармацевтически приемлемая соль" означает такие соли, которые сохраняют биологическую эффективность и свойства исходного соединения, и являются безопасными или нетоксичными. Термин "фармацевтически приемлемая соль(и)" известен в данной области техники и включает соли кислотных или основных групп, которые могут присутствовать в соединениях и получены или являются результом фармацевтически приемлемых оснований или кислот.
Термин "фармацевтическая композиция" означает активное соединение в любой форме, подходящей для эффективного введения субъекту, например, смеси соединения и, по меньшей мере, одного фармацевтически приемлемого носителя.
Используемый в описании термин "физиологически/фармацевтически приемлемый носитель" означает носитель или разбавитель, который не вызывает значительного раздражения в организме и не подавляет биологическую активность и свойства вводимого соединения.
Термин "фармацевтически приемлемый наполнитель" означает инертное вещество, добавляемое к фармацевтической композиции для дополнительного облегчения введения соединения. Примеры, без ограничения, наполнителей включают карбонат кальция, фосфат кальция, различные сахара и типы крахмала, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли.
Термины "лечить", "терапия" и "лечение" означают улучшение состояния, облегчение, подавление развития, или частично или полностью предотвращение расстройства или состояния, к которому этот термин применяется, или одного или более симптомов такого расстройства или состояния. "Предотвращение" означает лечение до возникновения инфекции.
"Терапевтически эффективное количество" означает такое количество вводимого соединения, которое ослабит до некоторой степени один или более симптомов расстройства, подлежащего лечению, или приведет к замедлению развития или, по меньшей мере, частичному купированию состояния.
ЯМР - ядерный магнитный резонанс
MDP(S) - очистка ВЭЖХ (система)
LC/MS - жидкостная хроматомасс-спектрометрия
LDA - диизопропиламид лития
tret-BuOH - трет-бутанол
АсОН - уксусная кислота
CDI - 1,1'-карбонилдиимидазол
DCE - 1,1-дихлорэтан
DCM - дихлорметан
DMF - диметилформамид
THF - тетрагидрофуран
МеОН - метанол
EtOH - этанол
EtOAc - этилацетат
MeCN - ацетонитрил
DMSO - диметилсульфоксид
Boc - трет-бутилоксикарбонил
DME - 1,2-диметоксиэтан
DMF - N,N-диметилформамид
DIPEA/DIEA - диизопропилэтиламин
PS-DIEA - диизопропилэтиламин на полимерной подложке
PS-PPh3-Pd - Pd(PPh3)4 на полимерной подложке
LAH - алюмогидрид лития
EDC - 1-(3-диметиламинопропил)-3-этилкарбодиимид
HATU - 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксид гексафторфосфат
HOBt - 1-гидроксибензотриазол
DMAP - 4-диметиламинопиридин
SEM-C1 - 2-(триметилсилил)этоксиметил хлорид
TBTU - O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат
TEMPO - 2,2,6,6-тетраметилпиперидин-1-оксил
TFA (А) - трифторуксусная кислота (ангидрид)
TLC - тонкослойная хроматография
TMSCN - триметилсилилцианид
Min - минута(ы)
NMO - N-метилморфолин N-оксид
h - час(ы)
d - день(дни)
RT, R.T., r.t., r.t или rt - комнатная температура
tR - время удерживания
Conc. - концентрированная
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2013 |
|
RU2652117C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ИНГИБИТОР, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ ПРОИЗВОДНОЕ ПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2807611C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
| ПРОИЗВОДНОЕ N-(АЗААРИЛ)ЦИКЛОЛАКТАМ-1-КАРБОКСАМИДА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2765785C2 |
| ФЕНОКСИПИРИДИНИЛАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ PDE4-ОПОСРЕДОВАННЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2009 |
|
RU2509077C2 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АМИЛОИДОМ ИЛИ АМИЛОИДПОДОБНЫМИ БЕЛКАМИ | 2007 |
|
RU2469026C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ОПОСРЕДОВАНИЯ АКТИВНОСТИ ТИРОЗИНКИНАЗЫ 2 | 2020 |
|
RU2826012C2 |
Изобретение относится к группе конкретных соединений, указанных в формуле изобретения, или их фармацевтически приемлемым солям. Также предложены соединение формулы IId или его фармацевтически приемлемая соль, фармацевтическая композиция, применение соединений и способ необратимого ингибирования тирозинкиназы Брутона. Предложенные соединения являются эффективными в качестве ингибиторов тирозинкиназы Брутона и могут быть использованы для лечения рака, хронического воспаления и аутоиммунного заболевания, которые опосредованы, по меньшей мере, частично, тирозинкиназой Брутона. 6 н. и 4 з.п. ф-лы, 4 табл., 73 пр.
1. Соединение, выбранное из группы, состоящей из:
6-(3-акриламидофенил)-2-(4-феноксифенил)никотинамида
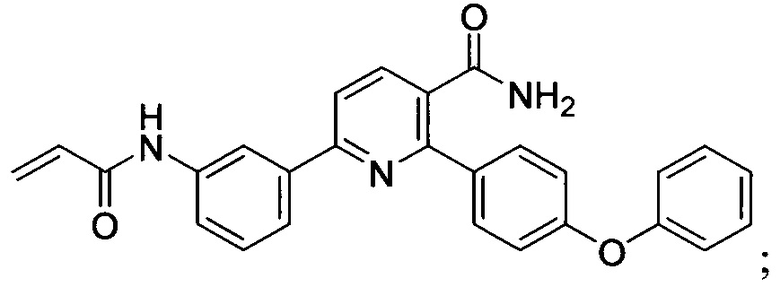
2-(4-феноксифенил)-6-(пиперидин-4-ил)никотинамида
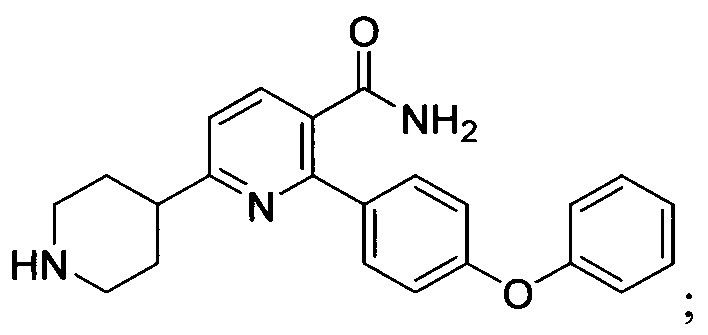
6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида
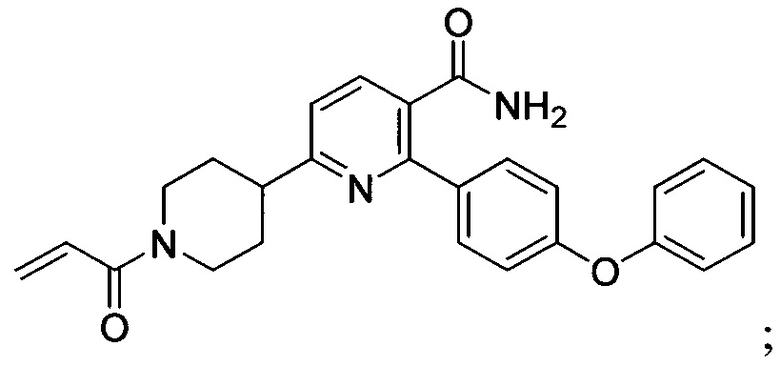
2-(4-феноксифенил)-6-(пиперазин-1-ил)никотинамида
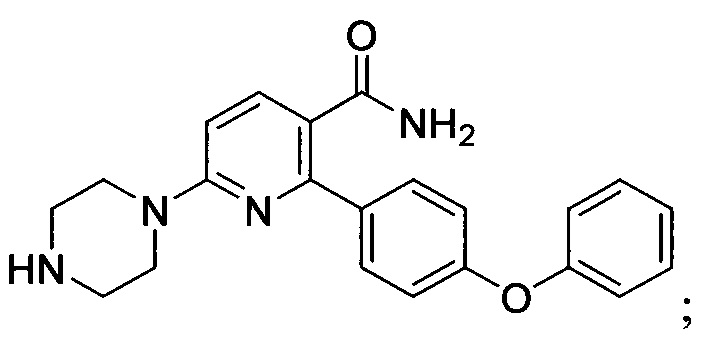
6-(4-акрилоилпиперазин-1-ил)-2-(4-феноксифенил)никотинамида
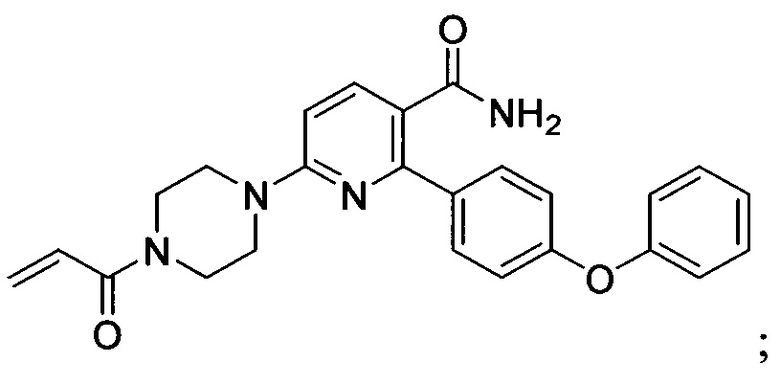
6-(1-акрилоилпирролидин-3-ил)-2-(4-феноксифенил)никотинамида
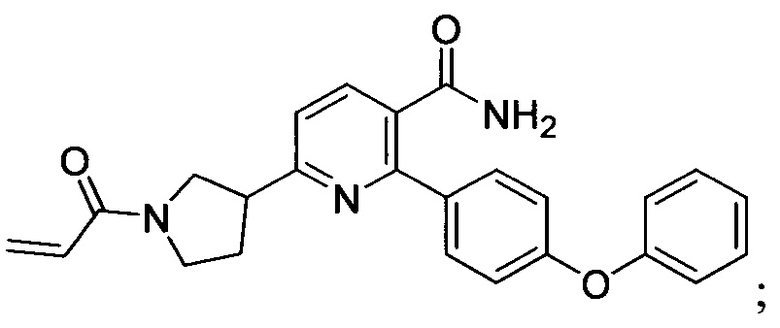
6-(4-акриламидофенил)-2-(4-феноксифенил)никотинамида
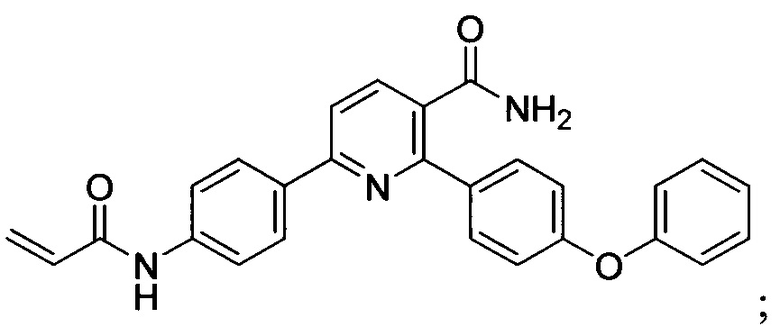
6-(1-акрилоилпиперидин-3-ил)-2-(4-феноксифенил)никотинамида
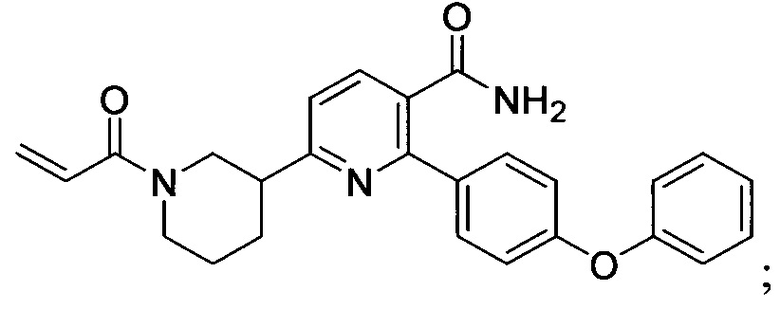
6-(4-акриламидопиперидин-1-ил)-2-(4-феноксифенил)никотинамида
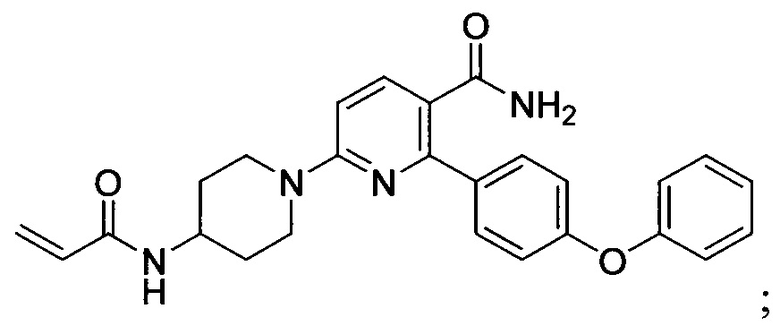
6-(3-акриламидопирролидин-1-ил)-2-(4-феноксифенил)никотинамида
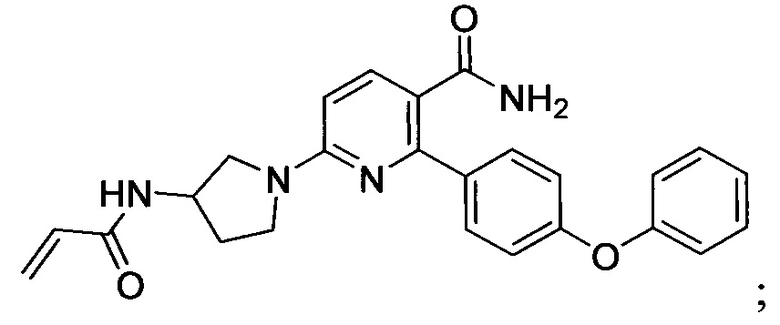
1-(1-акрилоилпиперидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
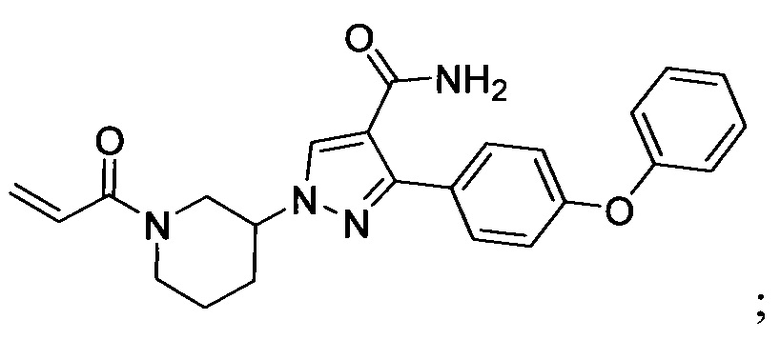
1-(1-акрилоилпиперидин-4-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
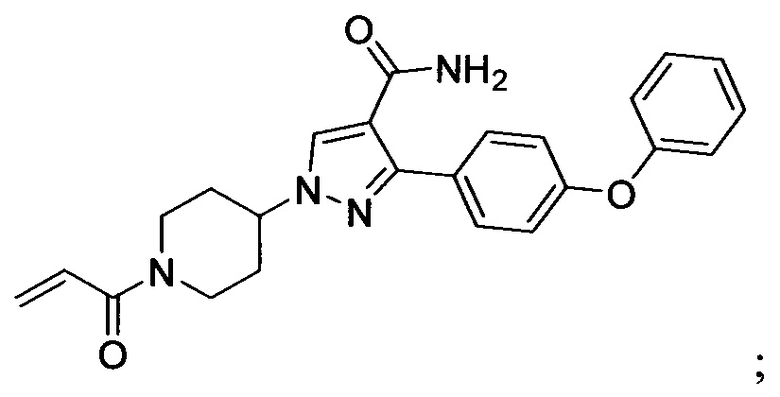
1-(1-акрилоилпирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
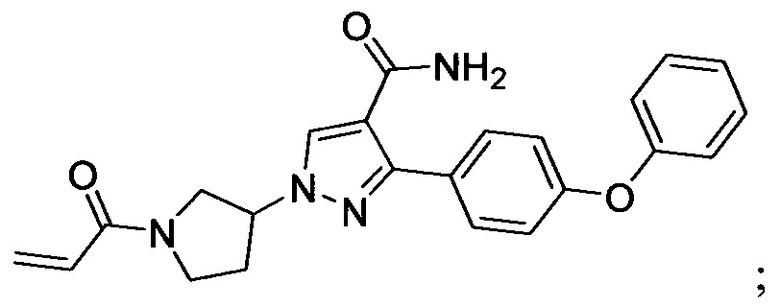
1-(азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
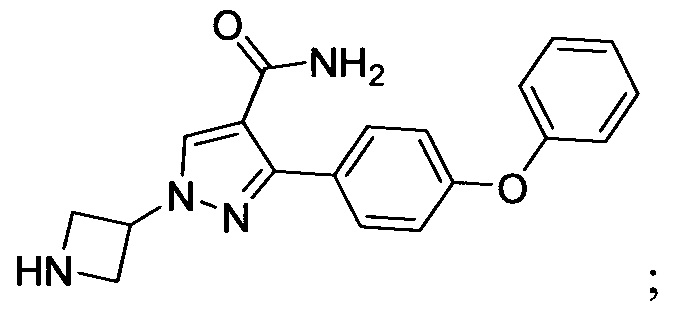
1-(1-акрилоилазетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
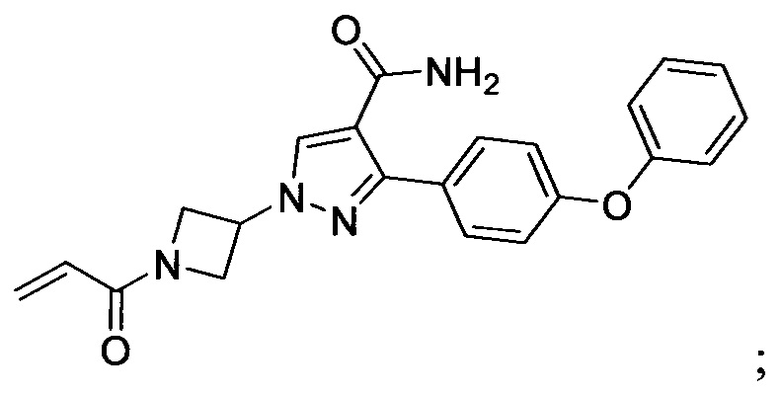
1-(4-акриламидофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида 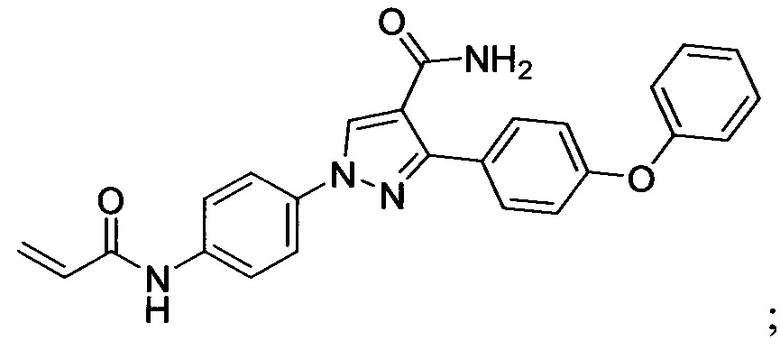
1-(3-аминофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
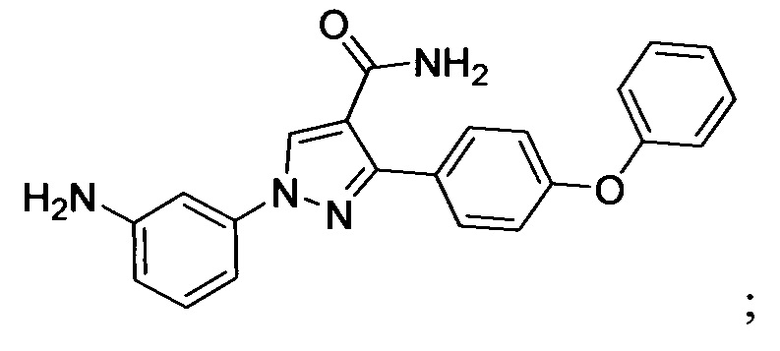
1-(3-акриламидофенил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
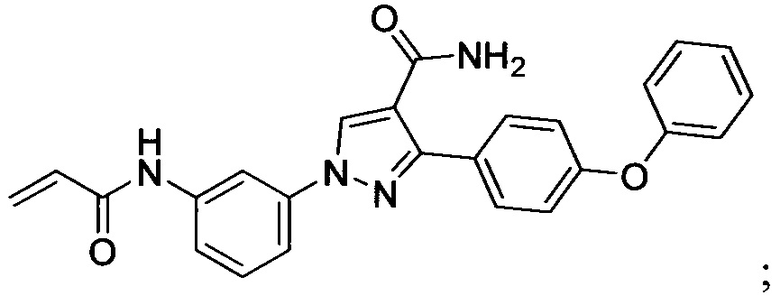
(S)-1-(1-акрилоилпирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
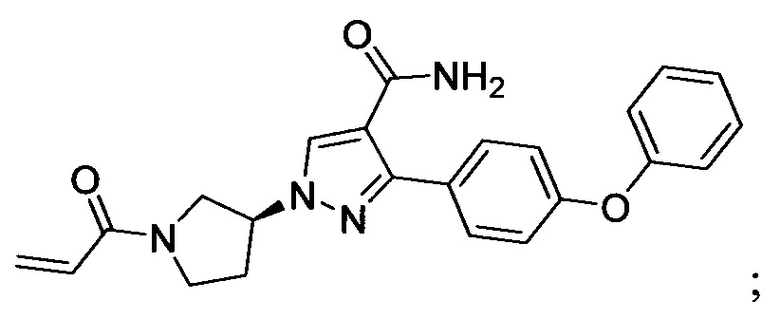
(R)-1-(1-акрилоилпирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
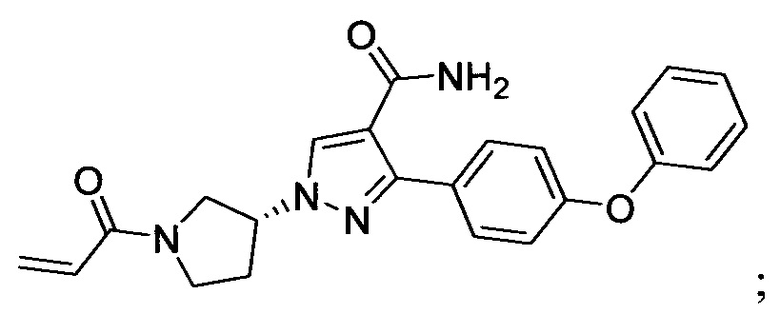
2-(1-акрилоилпиперидин-4-ил)-4-(4-феноксифенил)пиримидин-5-карбоксамида
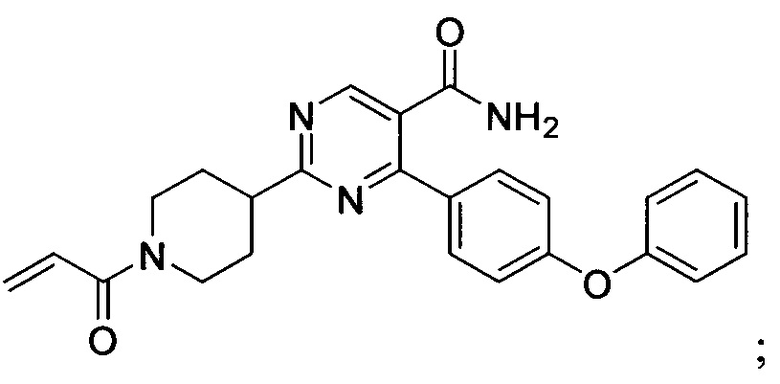
2-(1-акрилоилпирролидин-3-ил)-4-(4-феноксифенил)пиримидин-5-карбоксамида
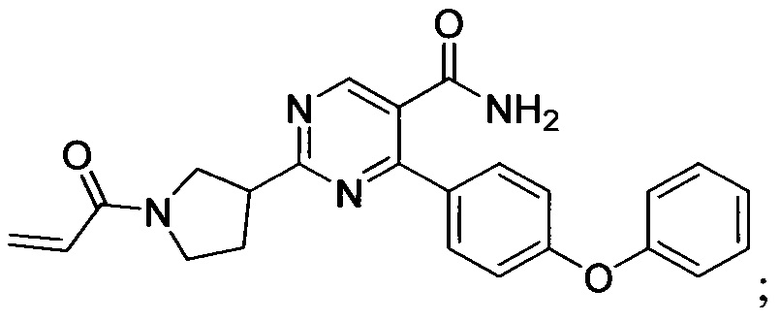
6-(1-акрилоилпирролидин-3-ил)-2-(4-феноксифенил)пиридин-3-карбоксамида
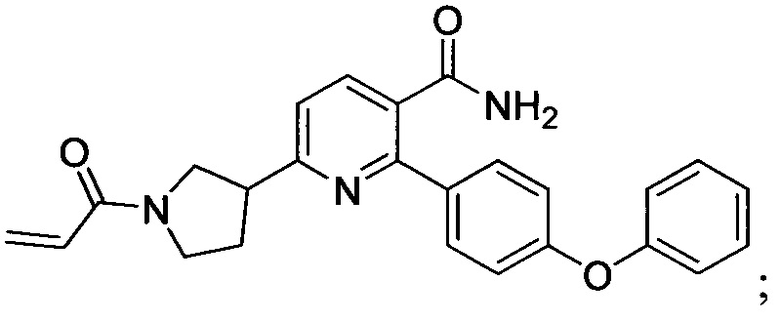
6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)пиридин-3-карбонитрила
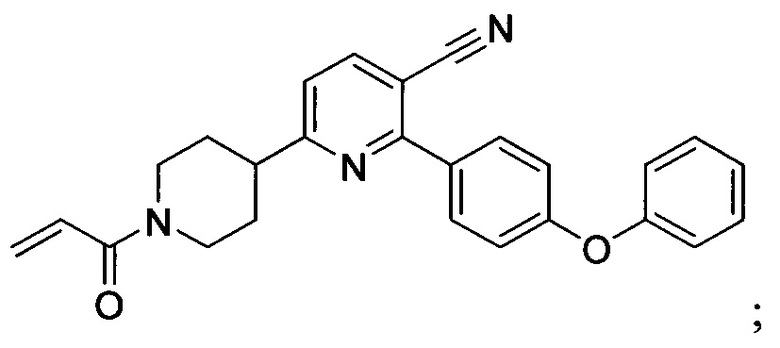
(Е)-6-(4-(4-(диметиламино)бут-2-еноил)пиперазин-1-ил)-2-(4-феноксифенил)никотинамида
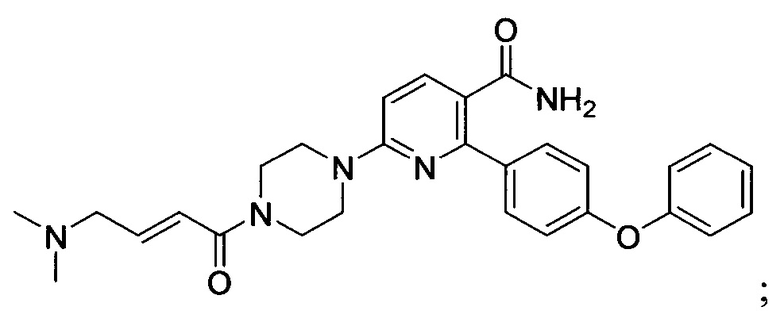
6-(4-акрилоилпиперазин-1-ил)-2-(4-феноксифенил)пиридин-3-карбоновой кислоты
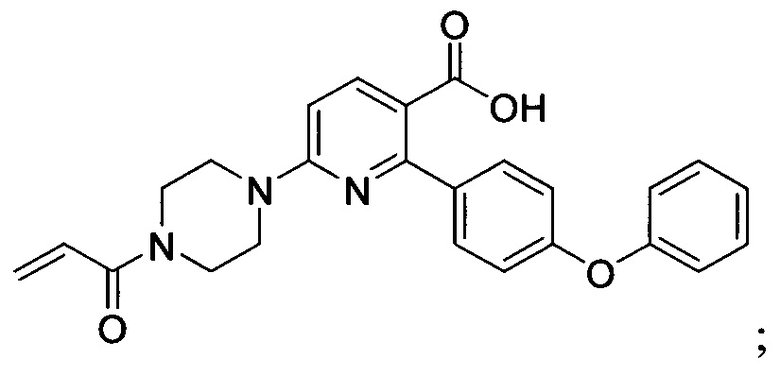
6-(4-акрилоилпиперазин-1-ил)-2-(4-(4-цианофенокси)фенил)никотинамида
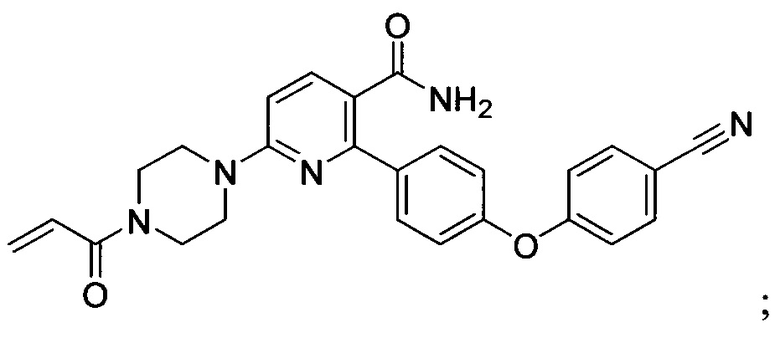
6-(4-акрилоилпиперазин-1-ил)-2-(4-(циклогексилокси)фенил)никотинамида
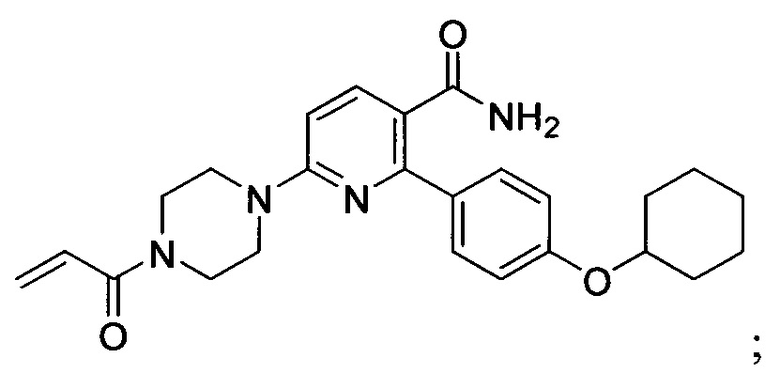
6-(4-акрилоилпиперазин-1-ил)-2-(3-метокси-4-метилфенил)никотинамида
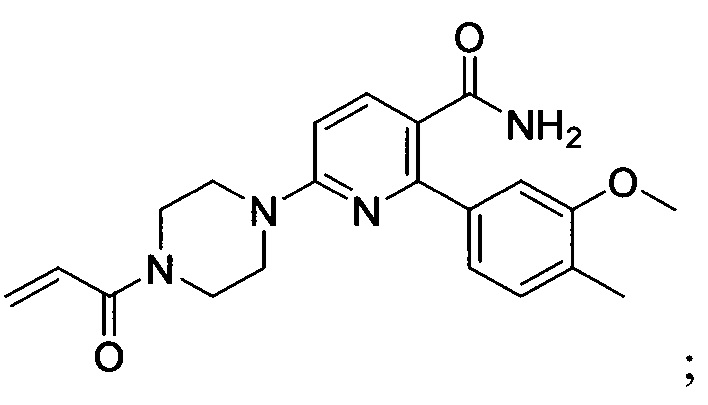
6-(4-акрилоилпиперазин-1-ил)-2-(4-фтор-3-метоксифенил)никотинамида
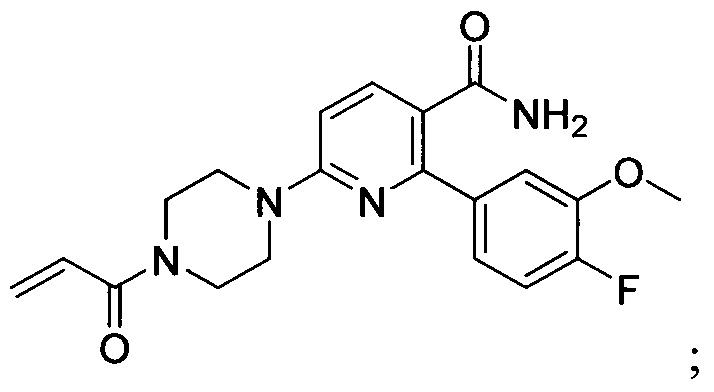
6-(4-акрилоилпиперазин-1-ил)-6'-фенокси-2,3'-бипиридин-3-карбоксамида
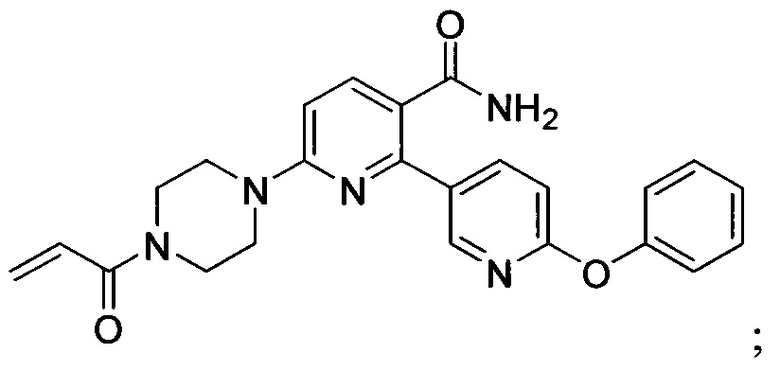
6-(4-акрилоилпиперазин-1-ил)-2-(4-(пиридин-2-илокси)фенил)никотинамида
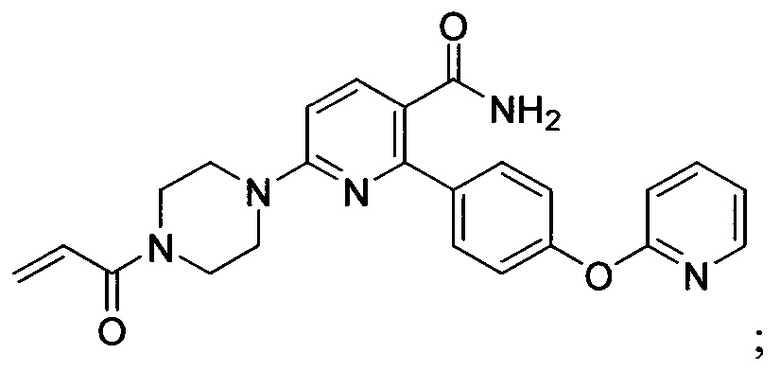
6-(4-акрилоилпиперазин-1-ил)-2-(4-хлорфенил)никотинамида
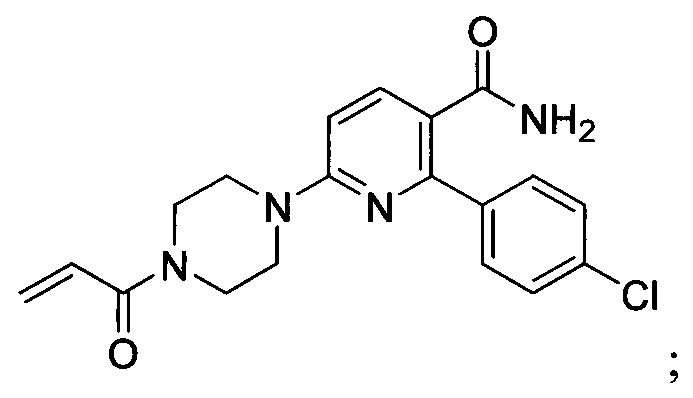
1-(1-(4-(диметиламино)бут-2-еноил)пирролидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
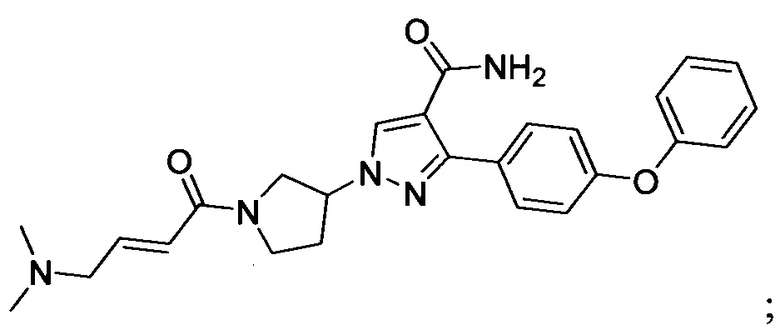
6-(4-акрилоилпиперазин-1-ил)-2-(4-(3-фторфенокси)фенил)никотинамида
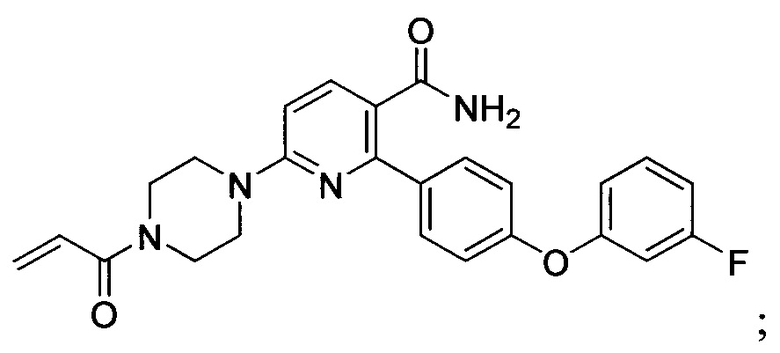
6-(4-акрилоилпиперазин-1-ил)-2-(3-фтор-4-феноксифенил)никотинамида
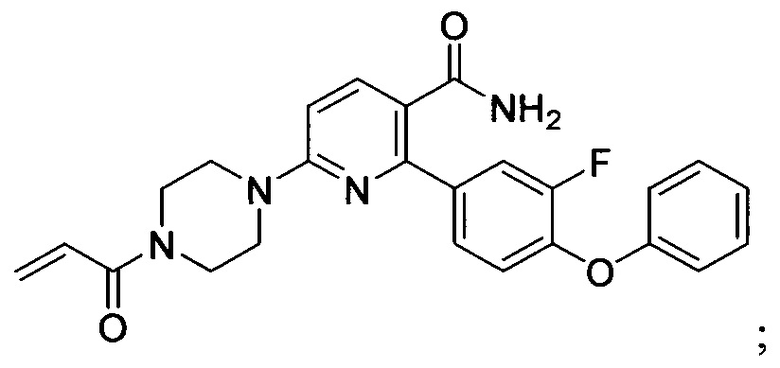
6-(4-акрилоилпиперазин-1-ил)-2-(4-(4-фторфенокси)фенил)никотинамида
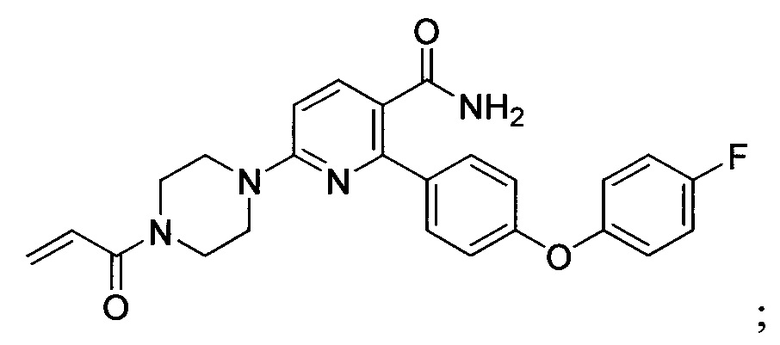
6-(4-акрилоилпиперазин-1-ил)-2-(4-(2-фторфенокси)фенил)никотинамида
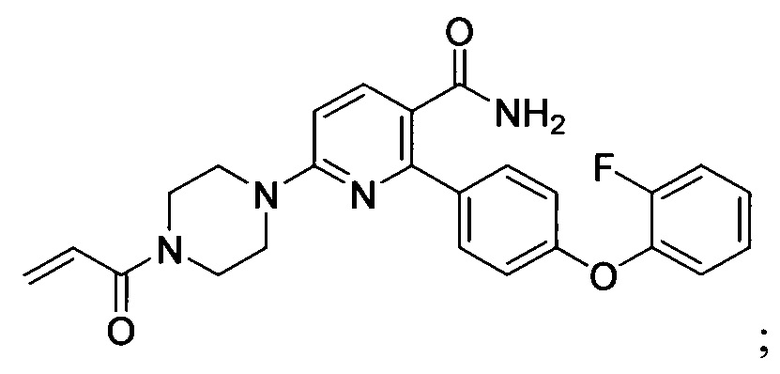
6-(4-акрилоилпиперазин-1-ил)-2-(2-фтор-4-феноксифенил)никотинамида
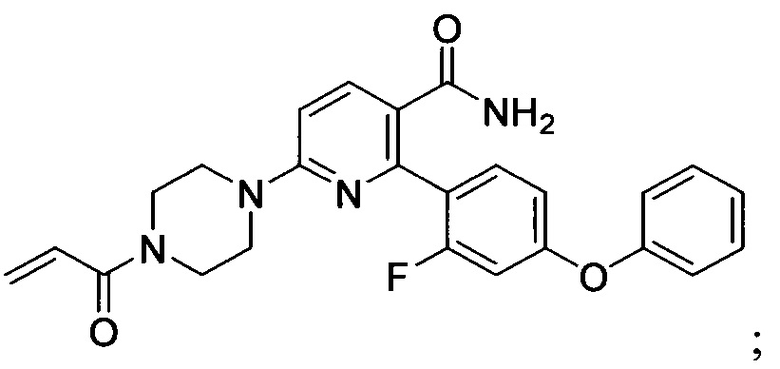
2-(1-акрилоилпиперидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоксамида
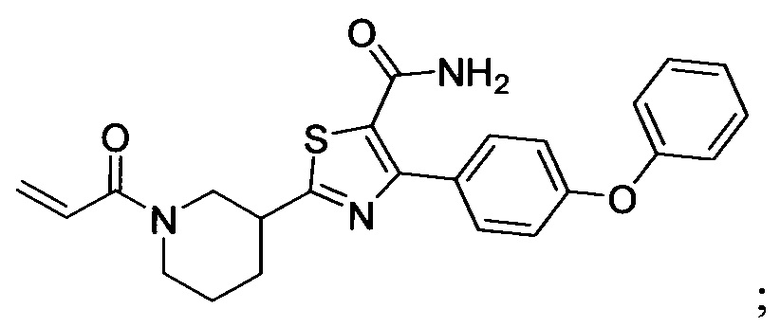
2-(1-акрилоилпирролидин-3-ил)-4-(4-феноксифенил)тиазол-5-карбоксамида
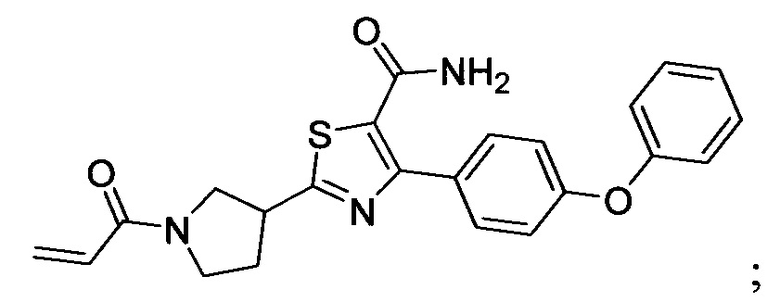
2-(1-акрилоилпиперидин-3-ил)-4-(4-феноксифенил)оксазол-5-карбоксамида
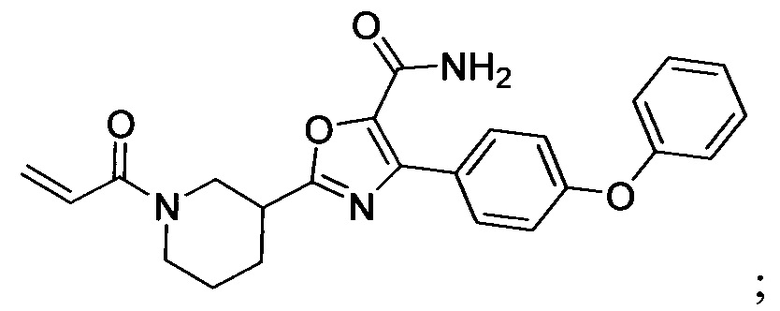
2-(1-акрилоилпирролидин-3-ил)-4-(4-феноксифенил)оксазол-5-карбоксамида
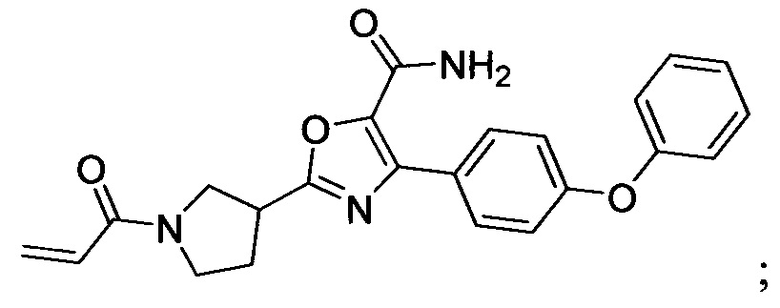
2-(1-акрилоилпирролидин-3-ил)-5-(4-феноксифенил)тиазол-4-карбоксамида
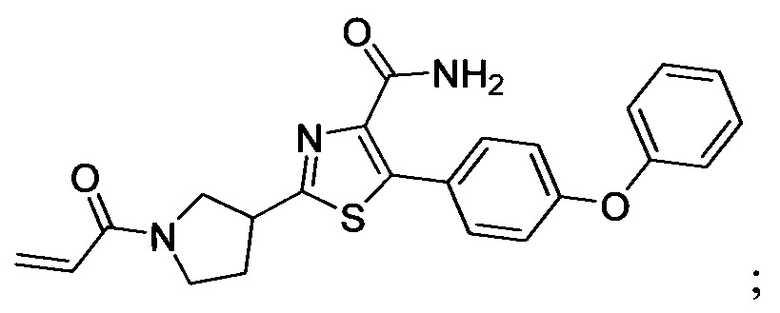
2-(1-акрилоилпиперидин-3-ил)-5-(4-феноксифенил)тиазол-4-карбоксамида
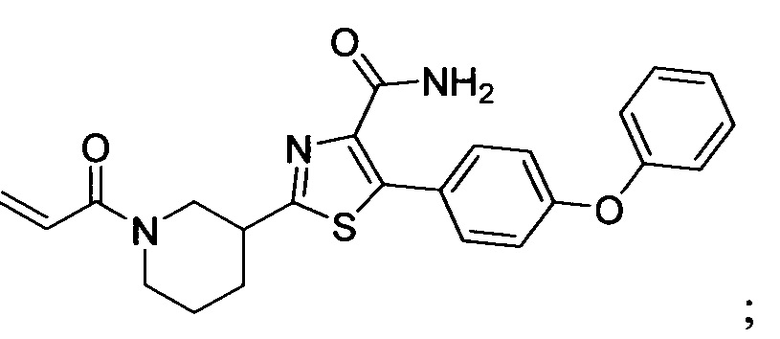
2-(1-акрилоилпиперидин-4-ил)-4-(4-феноксифенил)оксазол-5-карбоксамида
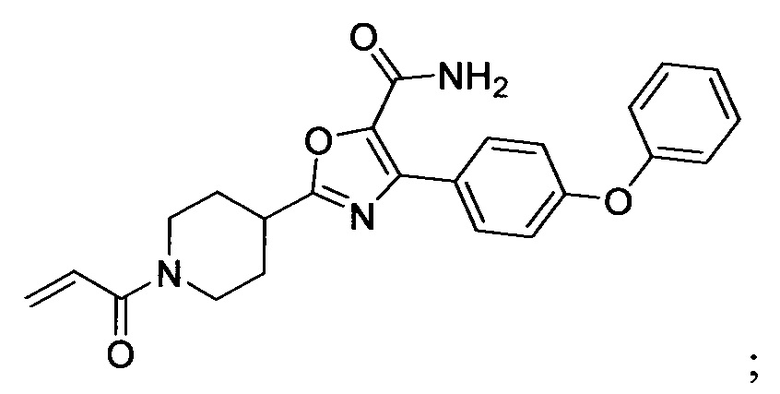
2-(4-акрилоилпиперазин-1-ил)-N-метил-4-(4-феноксифенил)тиазол-5-карбоксамида
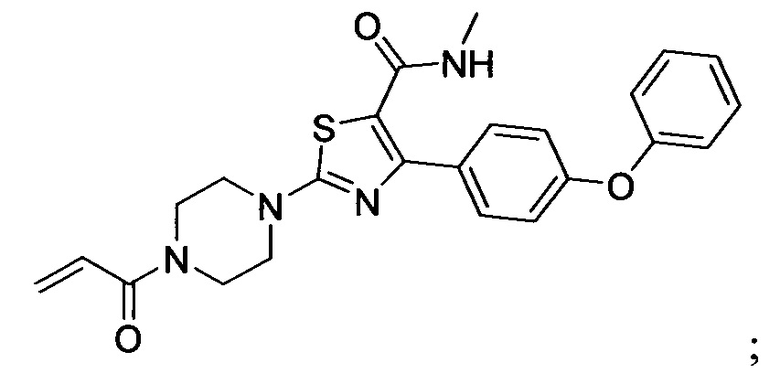
2-(1-акрилоилпирролидин-3-ил)-5-(4-феноксифенил)оксазол-4-карбоксамида
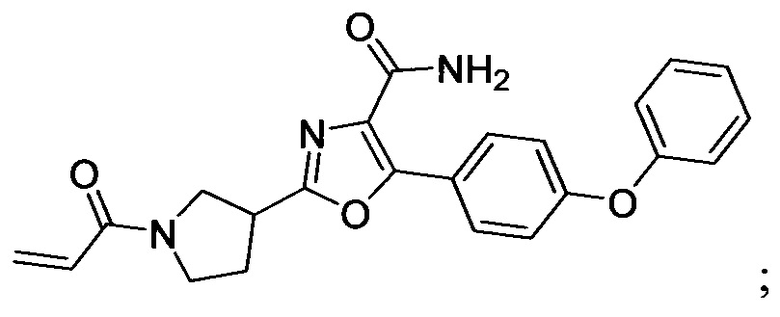
2-(1-акрилоилпиперидин-4-ил)-5-(4-феноксифенил)тиазол-4-карбоксамида
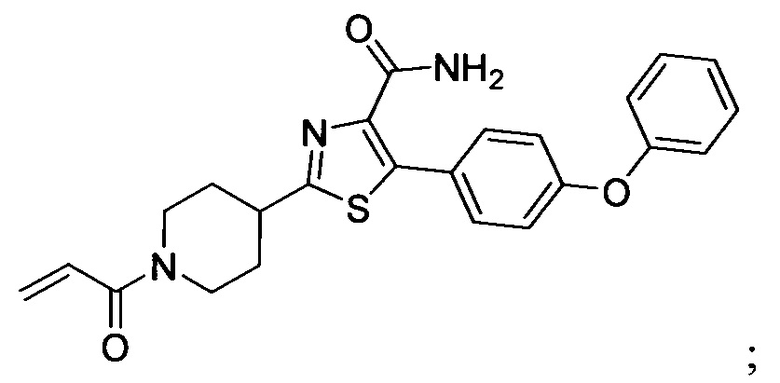
2-(4-акрилоилпиперазин-1-ил)-4-(4-феноксифенил)тиазол-5-карбоксамида
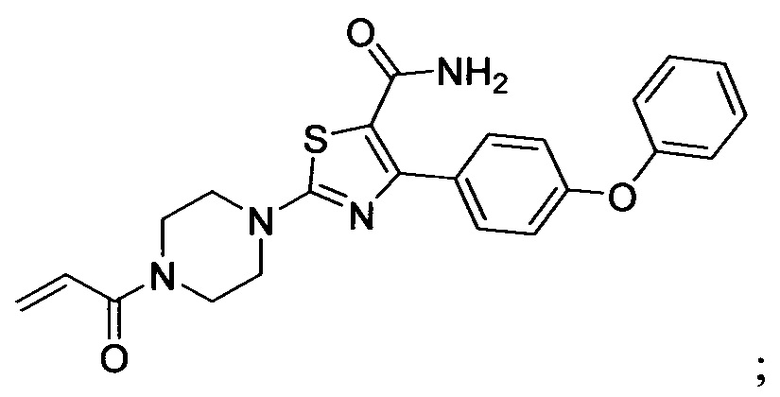
2-(1-акрилоилпиперидин-4-ил)-4-(4-феноксифенил)тиазол-5-карбоксамида
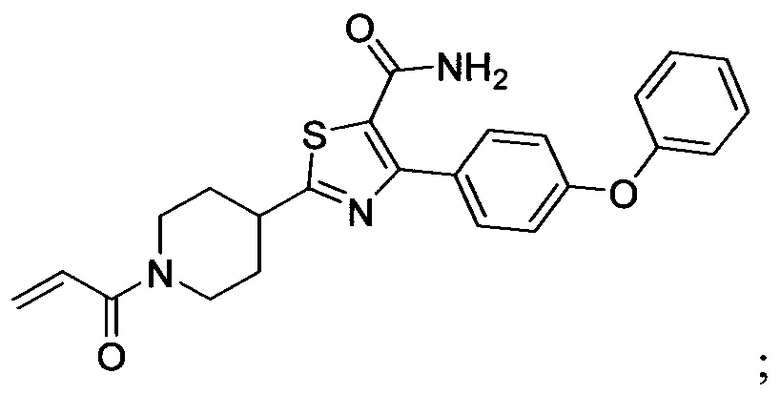
(Е)-1-(1-(2-циано-3-циклопропилакрилоил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
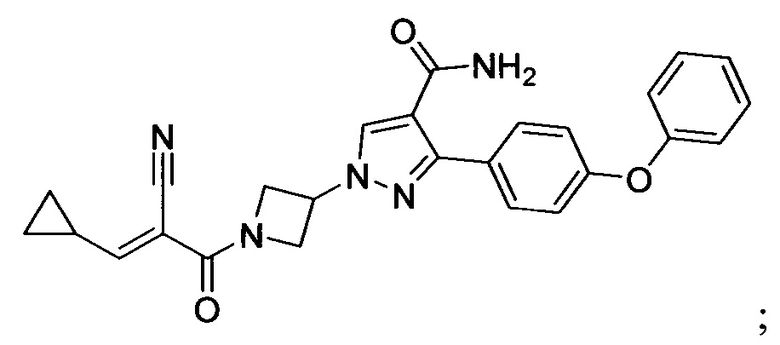
2-(1-акрилоилпиперидин-3-ил)-5-(4-феноксифенил)оксазол-4-карбоксамида
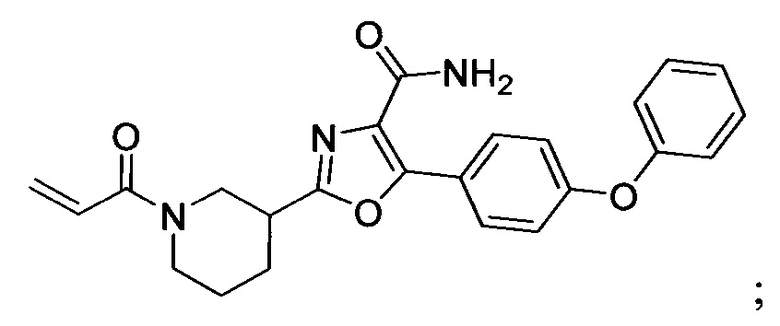
5-(1-акрилоилпиперидин-4-ил)-4'-феноксибифенил-2-карбоксамида
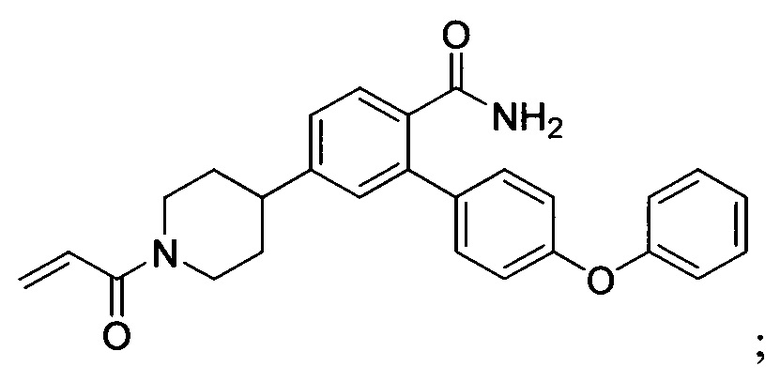
5-(1-акрилоилпиперидин-4-ил)-1-(4-феноксифенил)-1Н-пиразол-3-карбоксамида
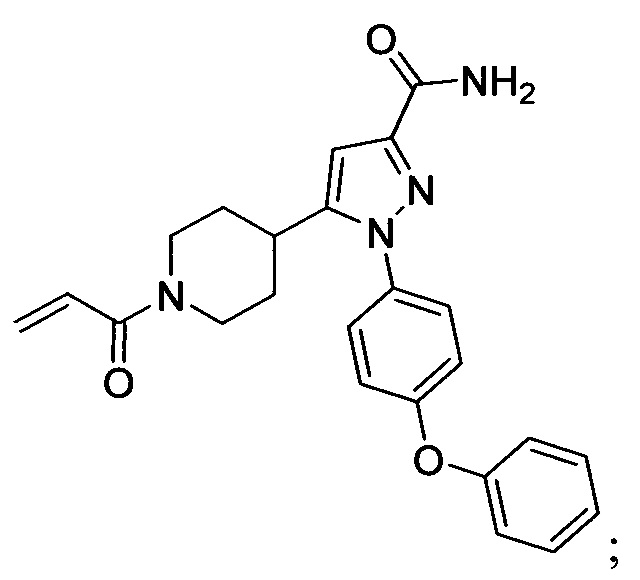
5-(4-акрилоилпиперазин-1-ил)-4'-феноксибифенил-2-карбоксамида
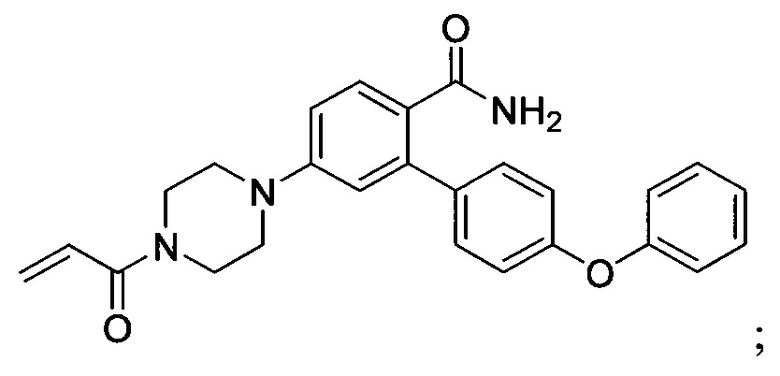
6- (1-акрилоилпиперидин-4-ил)-2-(4-(гидрокси(фенил)метил)фенил)никотинамида
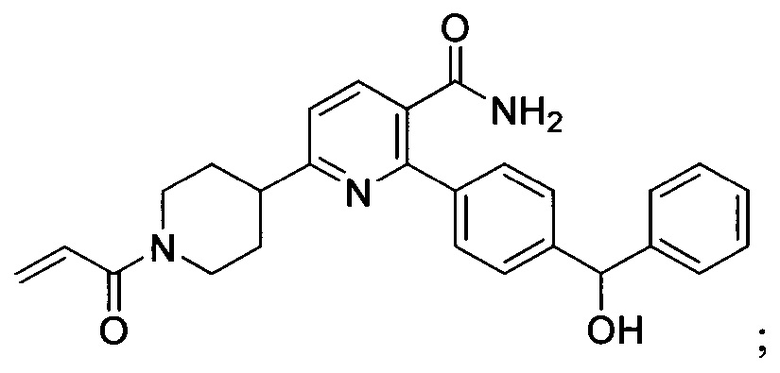
(Е)-1-(1-(2-цианобут-2-еноил)азетидин-3-ил)-3-(4-феноксифенил)-1Н-пиразол-4-карбоксамида
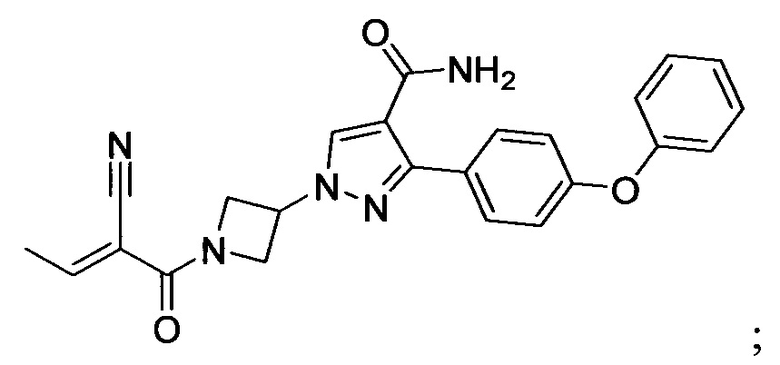
6-(4-акрилоилпиперазин-1-ил)-2-(4-((4,4-дифторциклогексил)окси)фенил)никотинамида
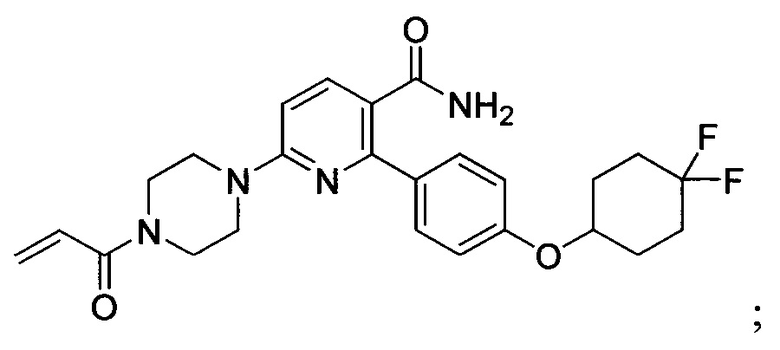
6-(1-акрилоилпиперидин-4-ил)-2-(4-(фенилкарбамоил)фенил)никотинамида
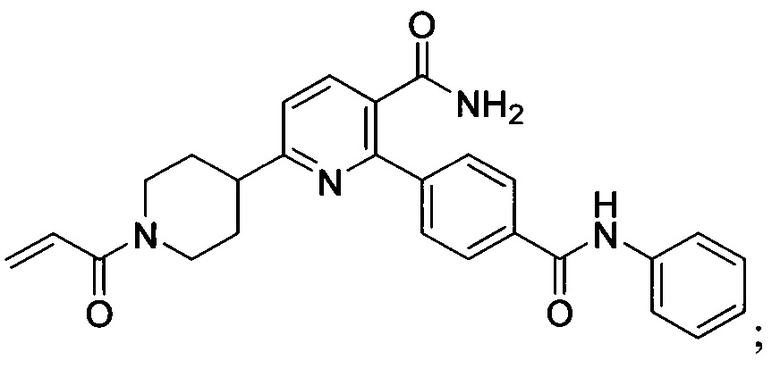
2-(4-феноксифенил)-6-(4-(винилсульфонил)пиперазин-1-ил)никотинамида
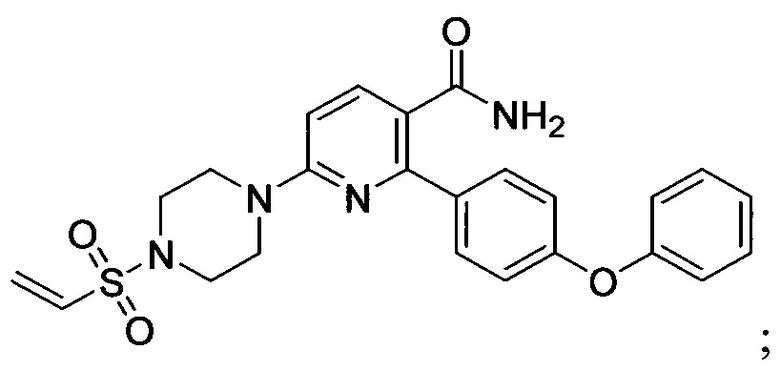
2-(4-феноксифенил)-6-(1-(винилсульфонил)пиперидин-4-ил)никотинамида
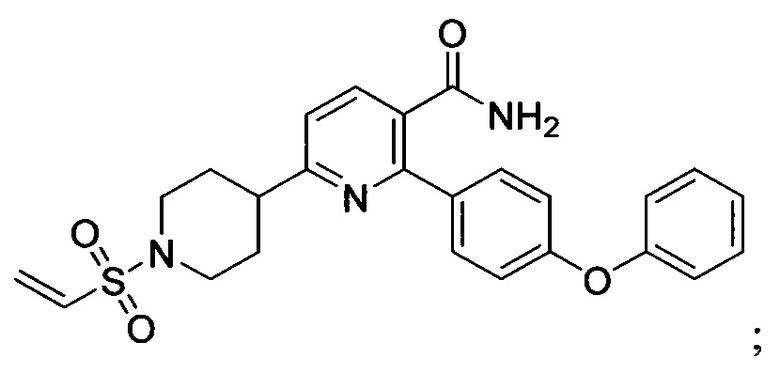
1-(1-акрилоилпиперидин-3-ил)-3-(4-(фенилкарбамоил)фенил)-1Н-пиразол-4-карбоксамида
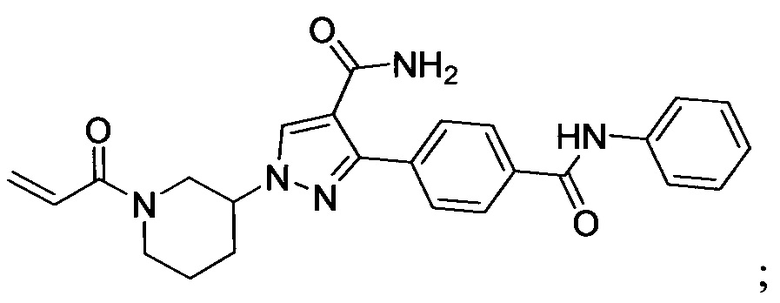
1-(1-акрилоилпиперидин-3-ил)-3-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)-1Н-пиразол-4-карбоксамида
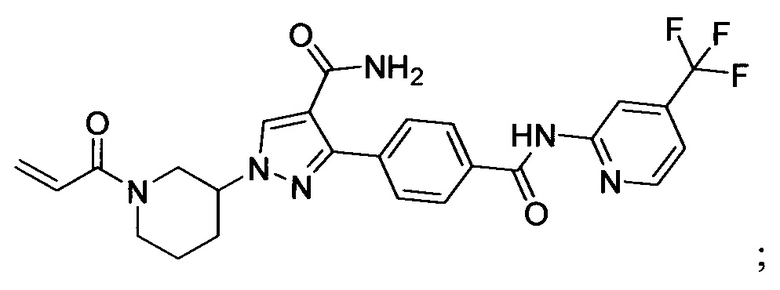
6-(4-акриламидофенил)-2-(4-(фенилкарбамоил)фенил) никотинамида
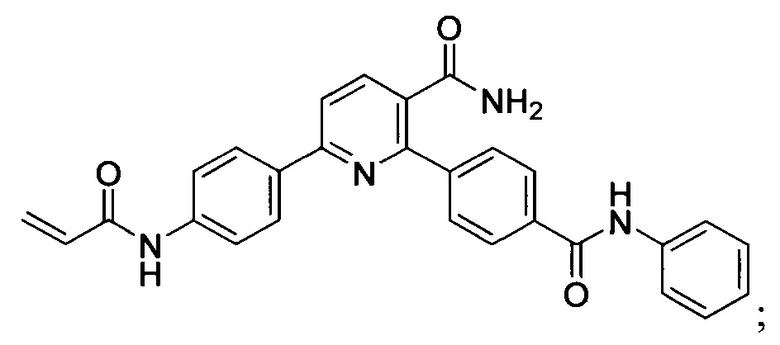
6-(4-акриламидофенил)-2-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)никотинамида
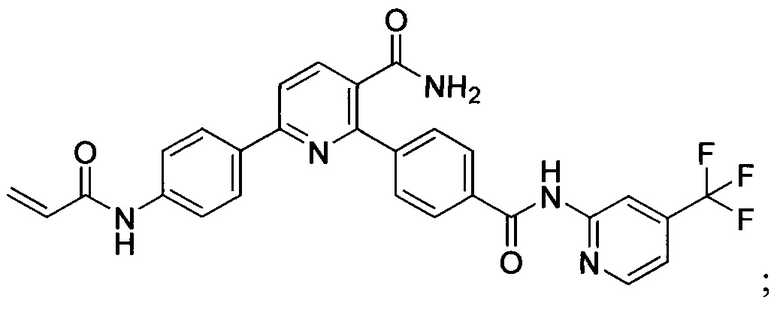
1-(1-акрилоилпиперидин-3-ил)-3-(4-(гидрокси(фенил)метил)фенил)-1Н-пиразол-4-карбоксамида
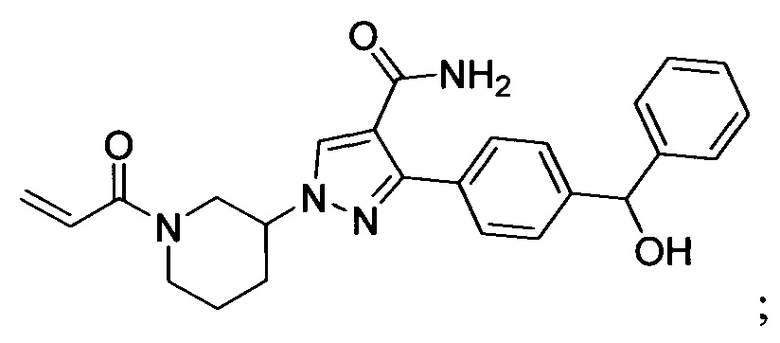
1-(1-акрилоилпиперидин-3-ил)-3-(4-(метил(фенил)амино)фенил)-1Н-пиразол-4-карбоксамида
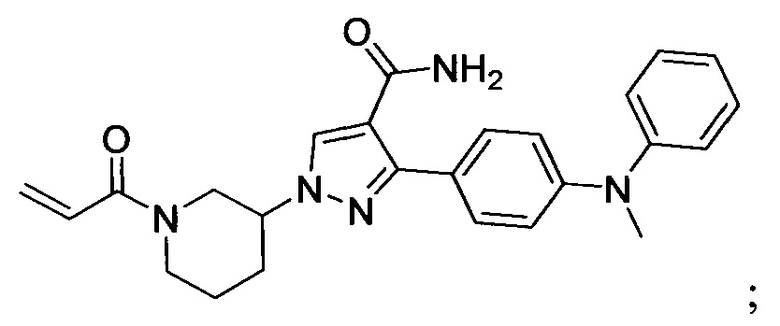
6-(4-акриламидофенил)-2-(4-(1-гидрокси-1-фенилэтил)фенил)никотинамида
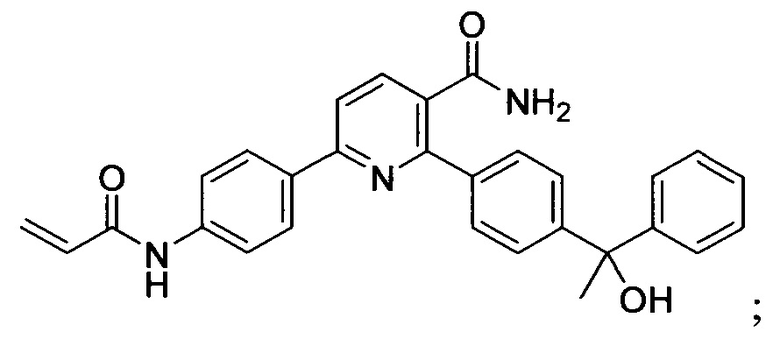
6-(4-акриламидофенил)-2-(4-(дифтор(фенил)метил)фенил)никотинамида
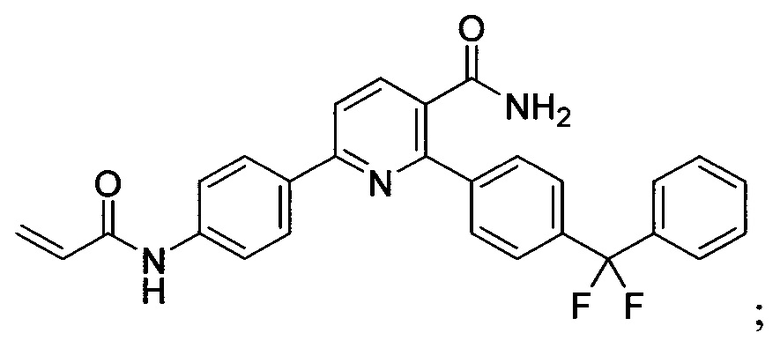
1-(1-акрилоилпиперидин-3-ил)-3-(4-(фенилсульфонил)фенил)-1Н-пиразол-4-карбоксамида
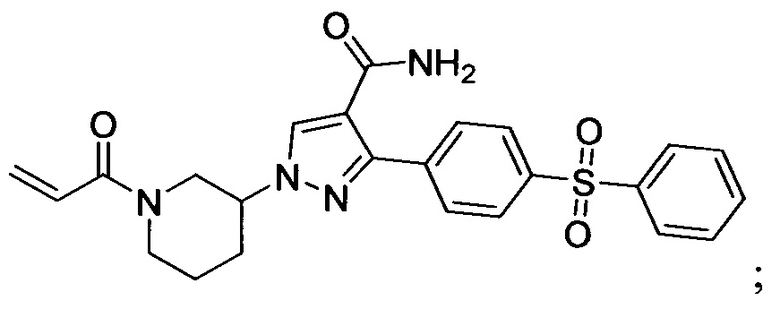
6-(4-акриламидофенил)-2-(4-(фенилсульфонил)фенил)-никотинамида
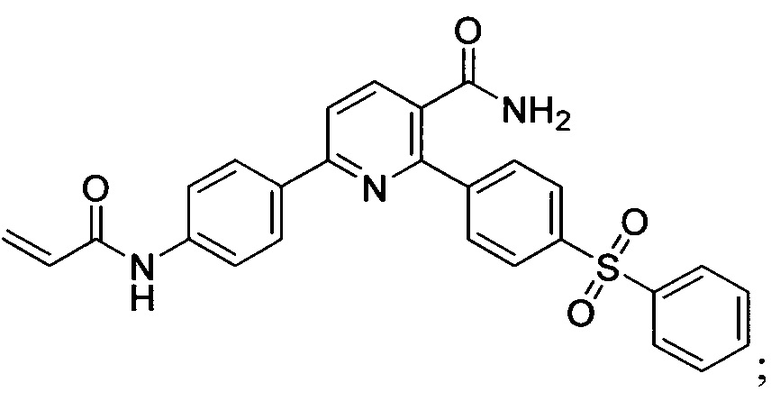
или его фармацевтически приемлемая соль.
2. Соединение, выбранное из группы, состоящей из:
6-(3-акриламидофенил)-2-(4-феноксифенил)никотинамида
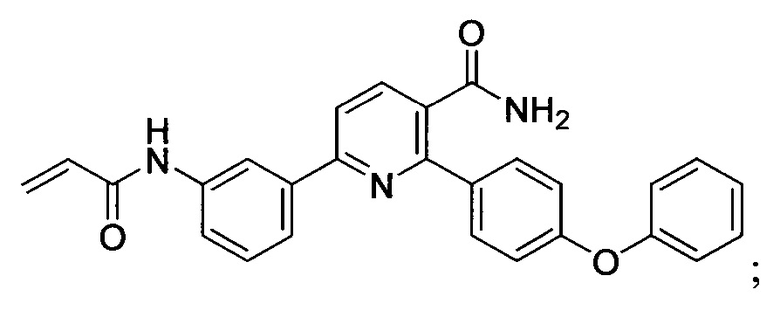
6-(1-акрилоилпиперидин-4-ил)-2-(4-феноксифенил)никотинамида
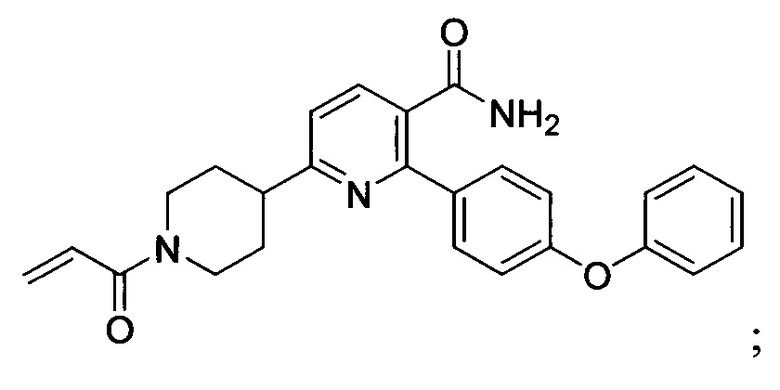
6-(4-акрилоилпиперазин-1-ил)-2-(4-феноксифенил)никотинамида
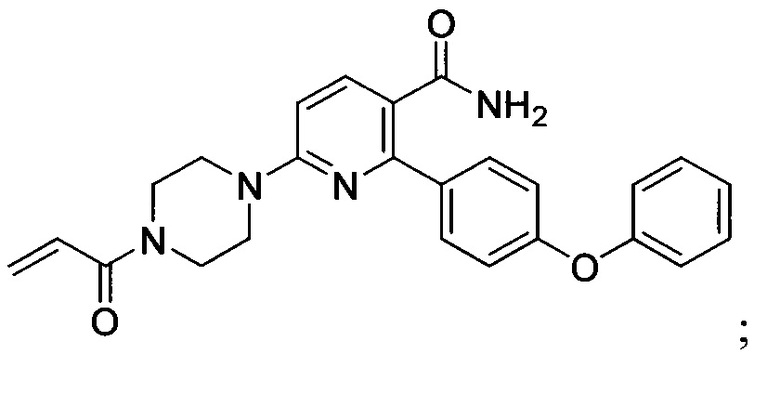
6-(1-акрилоилпирролидин-3-ил)-2-(4-феноксифенил)никотинамида;
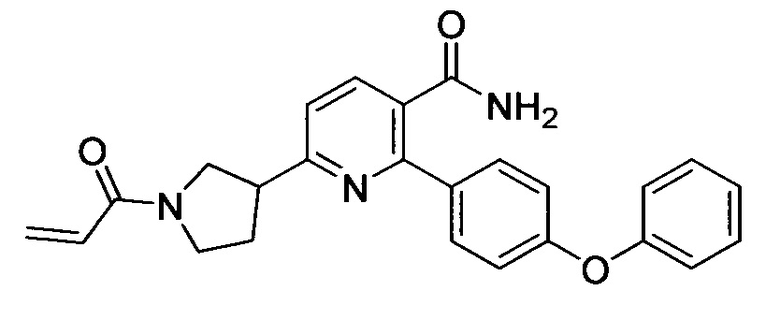
6-(4-акриламидофенил)-2-(4-феноксифенил)никотинамида
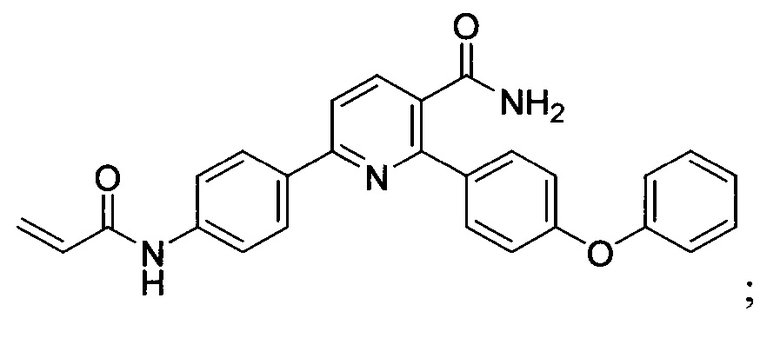
6-(1-акрилоилпиперидин-3-ил)-2-(4-феноксифенил)никотинамида
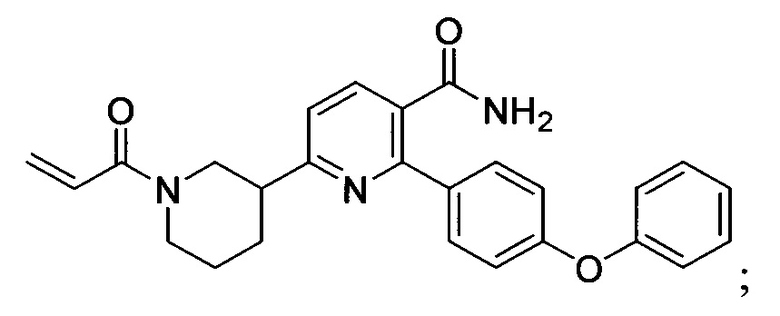
6-(4-акриламидопиперидин-1-ил)-2-(4-феноксифенил)никотинамида
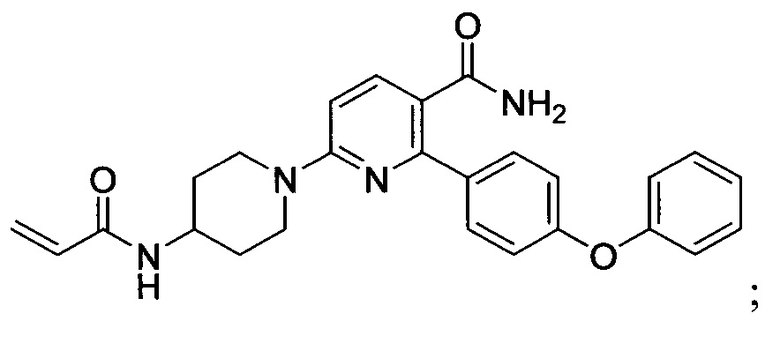
6-(3-акриламидопирролидин-1-ил)-2-(4-феноксифенил)никотинамида
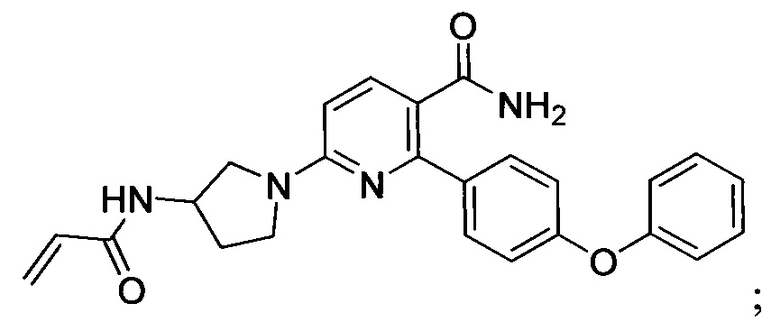
6-(1-акрилоилпирролидин-3-ил)-2-(4-феноксифенил)пиридин-3-карбоксамида;
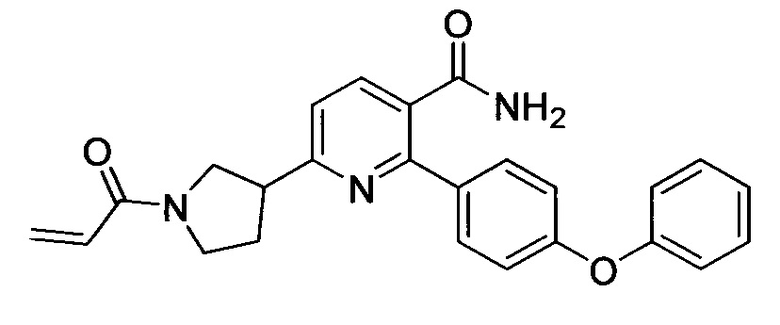
(Е)-6-(4-(4-(диметиламино)бут-2-еноил)пиперазин-1-ил)-2-(4-феноксифенил)никотинамида
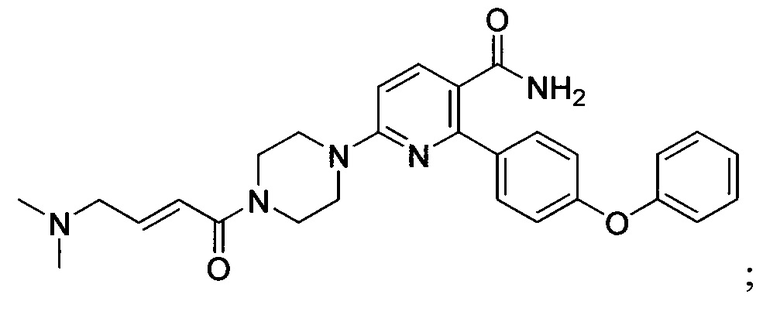
6-(4-акрилоилпиперазин-1-ил)-2-(4-(4-цианофенокси)фенил)никотинамида
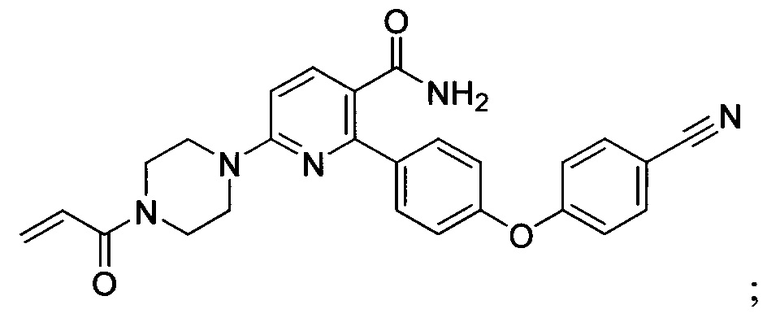
6-(4-акрилоилпиперазин-1-ил)-2-(4-(циклогексилокси)фенил)никотинамида
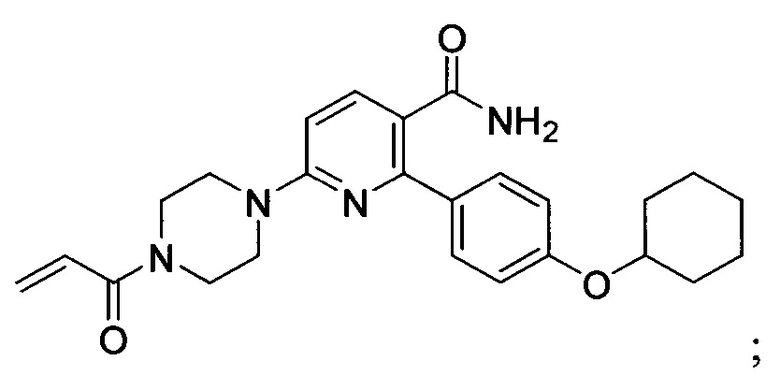
6-(4-акрилоилпиперазин-1-ил)-2-(3-метокси-4-метилфенил)никотинамида
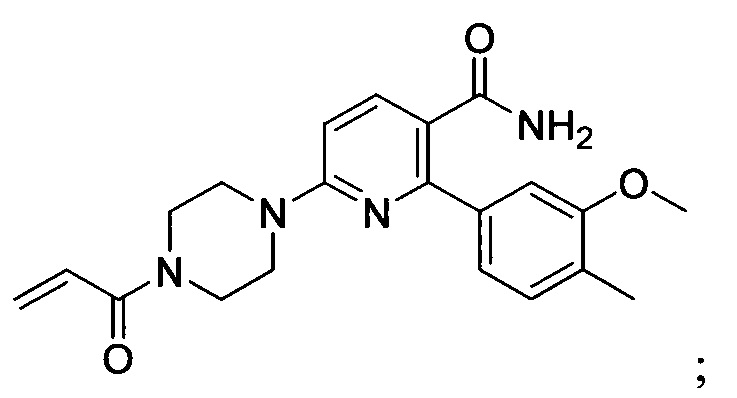
6-(4-акрилоилпиперазин-1-ил)-2-(4-фтор-3-метоксифенил)никотинамида
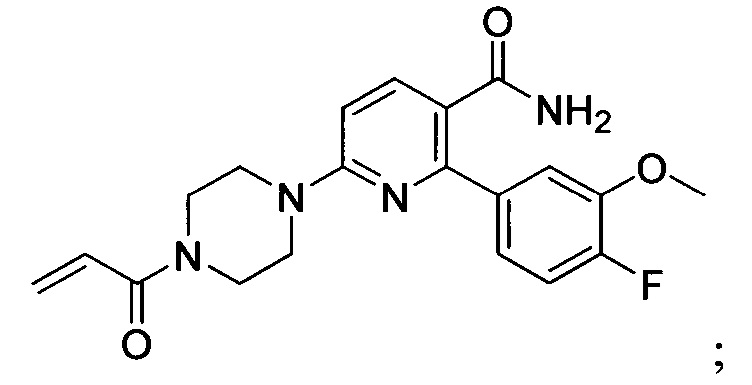
6-(4-акрилоилпиперазин-1-ил)-6'-фенокси-2,3'-бипиридин-3-карбоксамида
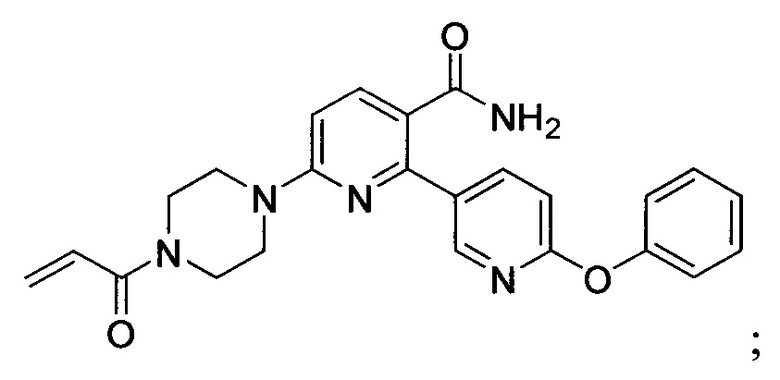
6-(4-акрилоилпиперазин-1-ил)-2-(4-(пиридин-2-илокси)фенил)никотинамида
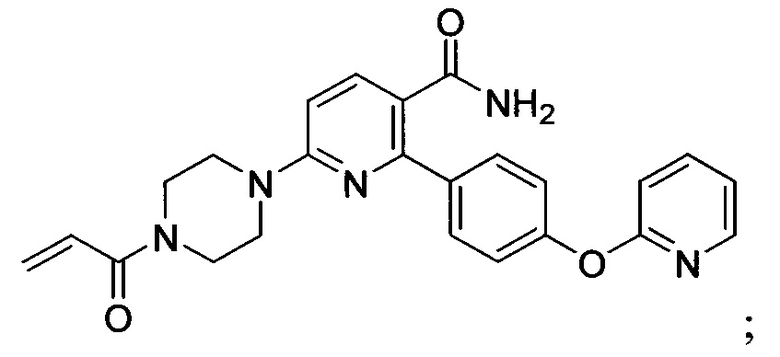
6-(4-акрилоилпиперазин-1-ил)-2-(4-хлорфенил)никотинамида
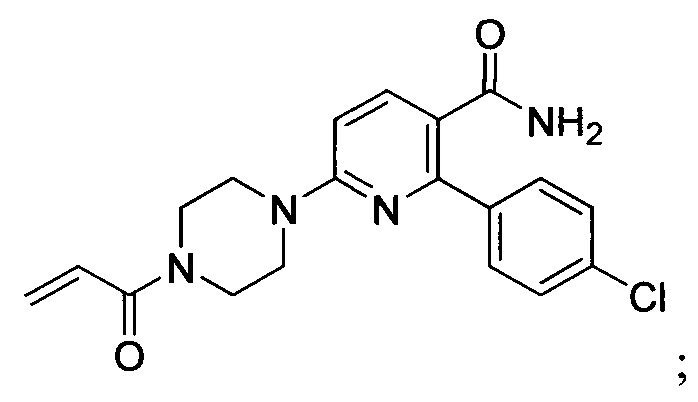
6-(4-акрилоилпиперазин-1-ил)-2-(4-(3-фторфенокси)фенил)никотинамида
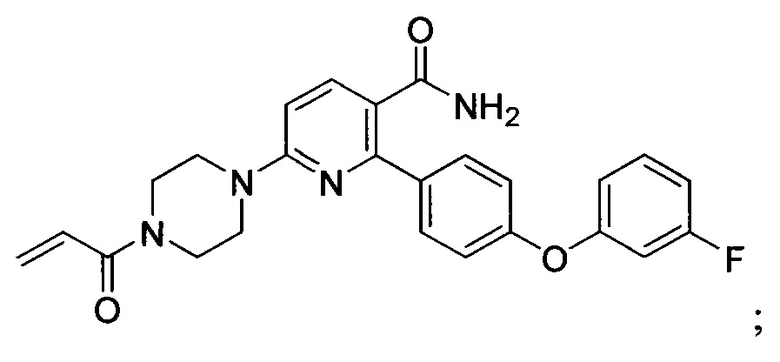
6-(4-акрилоилпиперазин-1-ил)-2-(3-фтор-4-феноксифенил)никотинамида
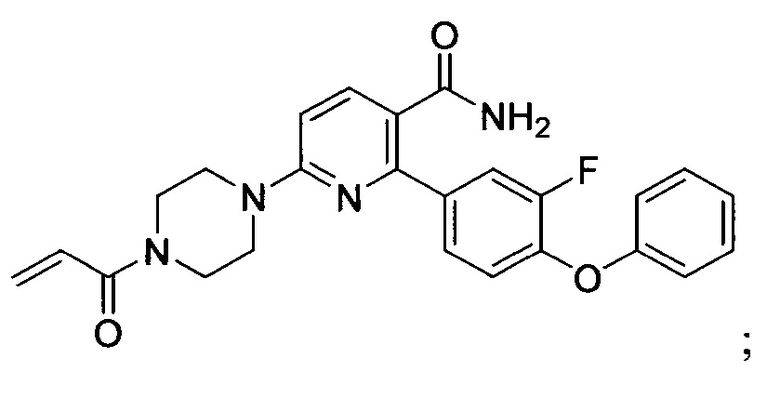
6-(4-акрилоилпиперазин-1-ил)-2-(4-(4-фторфенокси)фенил)никотинамида
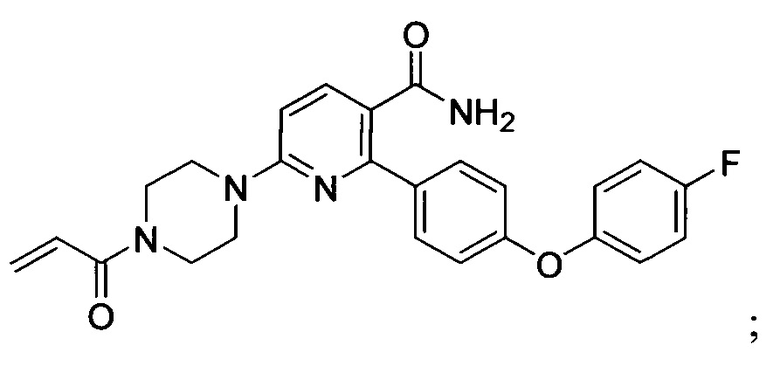
6-(4-акрилоилпиперазин-1-ил)-2-(4-(2-фторфенокси)фенил)никотинамида
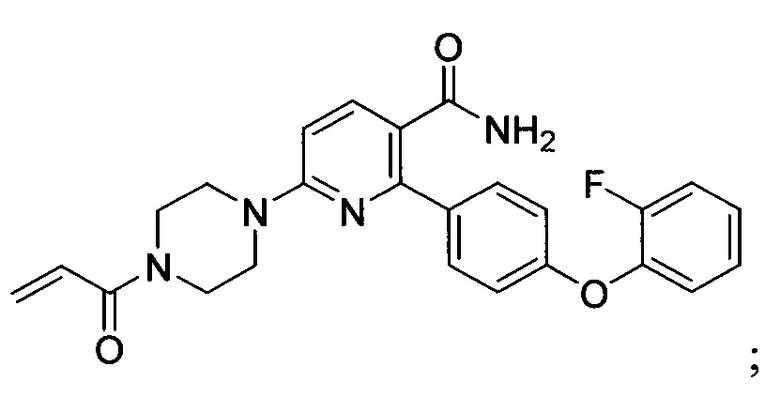
6-(4-акрилоилпиперазин-1-ил)-2-(2-фтор-4-феноксифенил)никотинамида
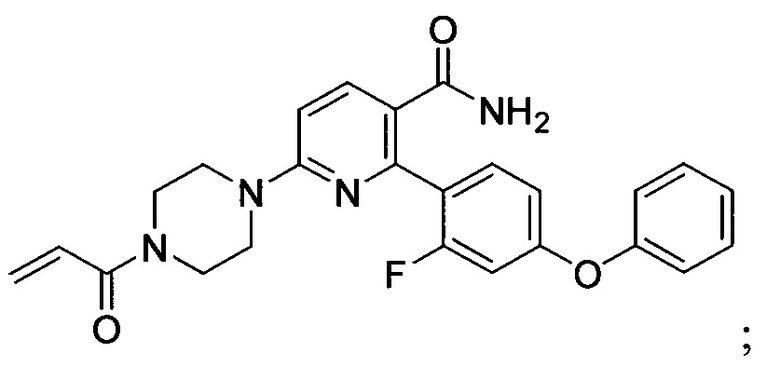
5-(1-акрилоилпиперидин-4-ил)-4'-феноксибифенил-2-карбоксамида
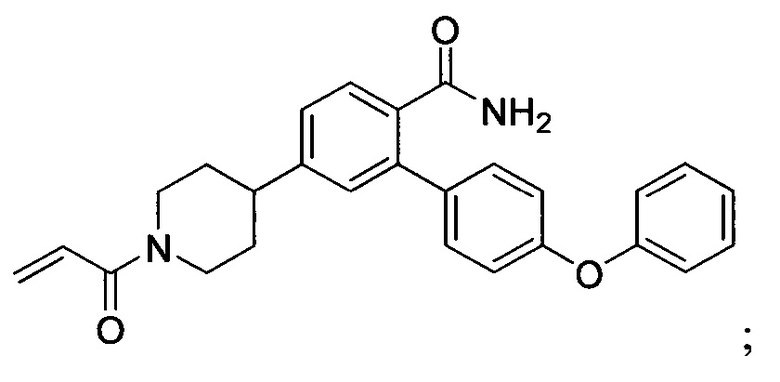
5-(4-акрилоилпиперазин-1-ил)-4'-феноксибифенил-2-карбоксамида
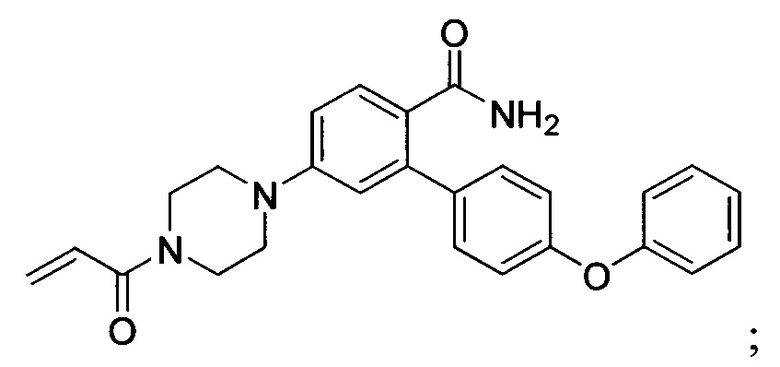
6-(1-акрилоилпиперидин-4-ил)-2-(4-(гидрокси(фенил)метил)фенил)никотинамида
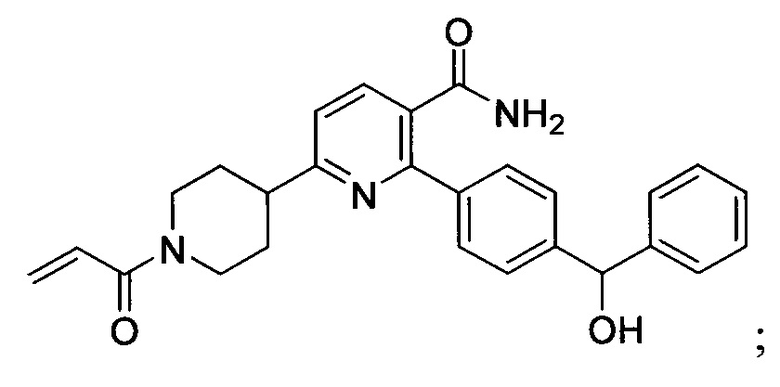
6-(4-акрилоилпиперазин-1-ил)-2-(4-((4,4-дифторциклогексил)окси)фенил)никотинамида
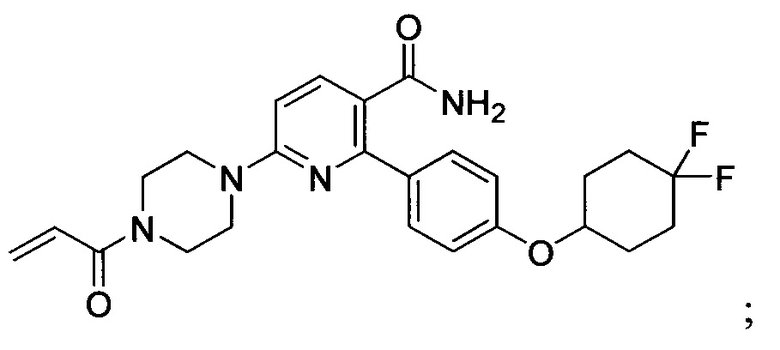
6-(1-акрилоилпиперидин-4-ил)-2-(4-(фенилкарбамоил)фенил)никотинамида
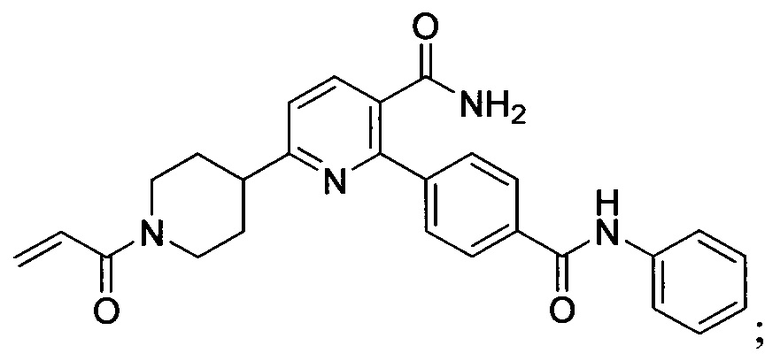
2-(4-феноксифенил)-6-(4-(винилсульфонил)пиперазин-1-ил)никотинамида
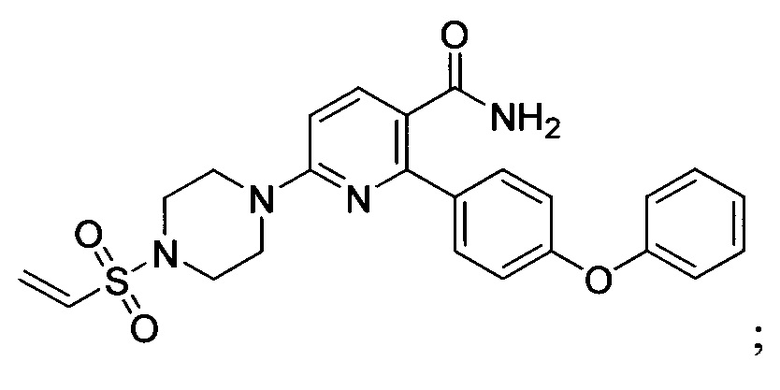
2-(4-феноксифенил)-6-(1-(винилсульфонил)пиперидин-4-ил)никотинамида
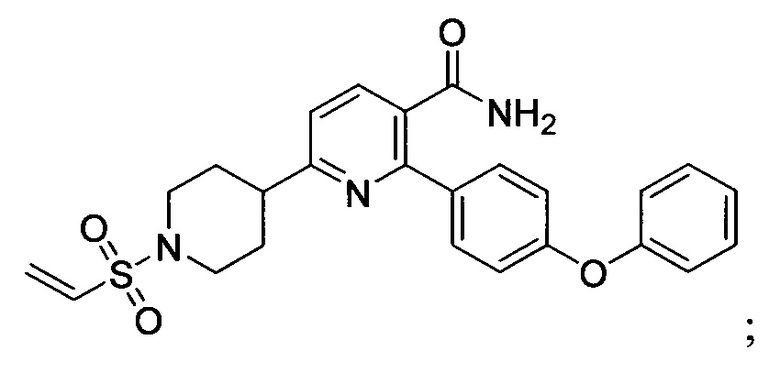
6-(4-акриламидофенил)-2-(4-(фенилкарбамоил)фенил)никотинамида
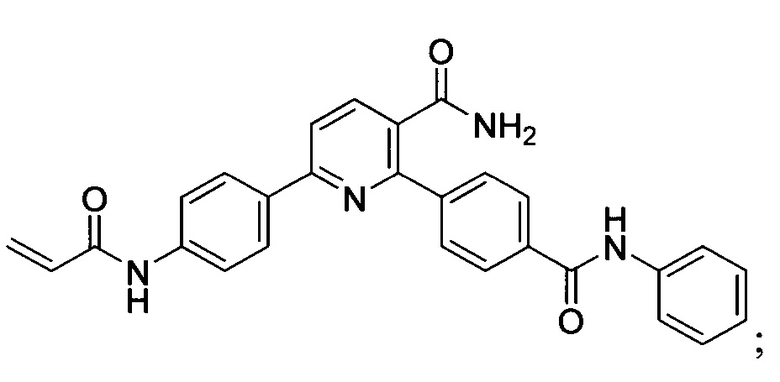
6-(4-акриламидофенил)-2-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)никотинамида
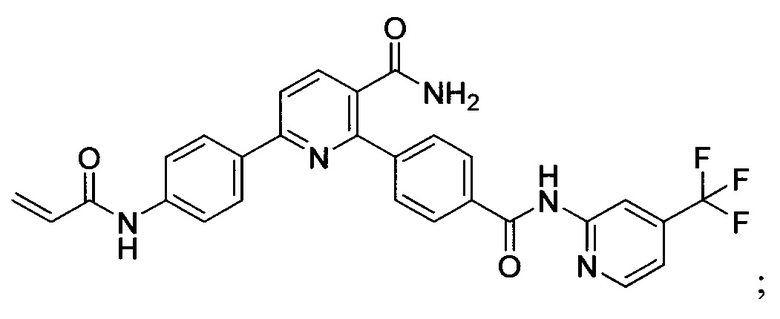
6-(4-акриламидофенил)-2-(4-(1-гидрокси-1-фенилэтил)фенил)никотинамида
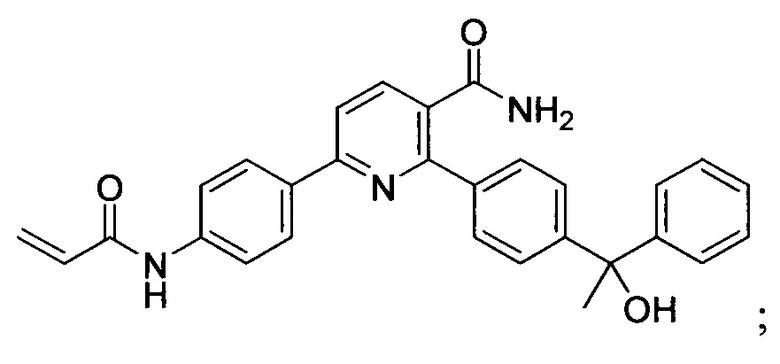
6-(4-акриламидофенил)-2-(4-(дифтор(фенил)метил)фенил)никотинамида
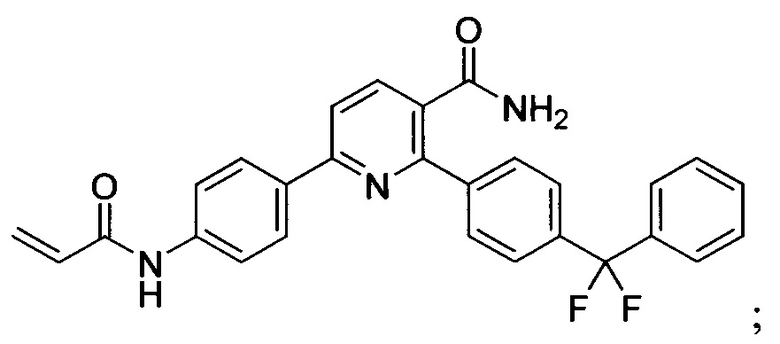
1-(1-акрилоилпиперидин-3-ил)-3-(4-(фенилсульфонил)фенил)-1Н-пиразол-4-карбоксамида
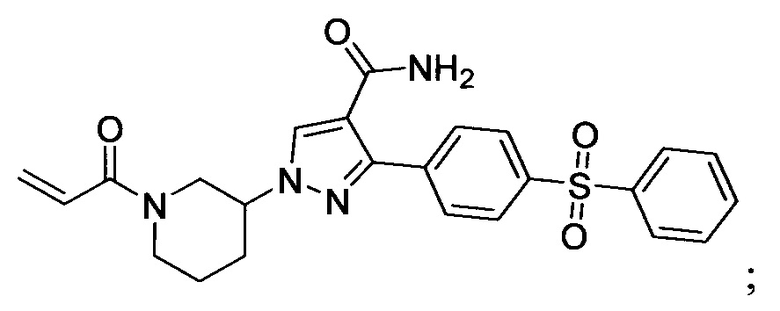
6-(4-акриламидофенил)-2-(4-(фенилсульфонил)фенил)никотинамида
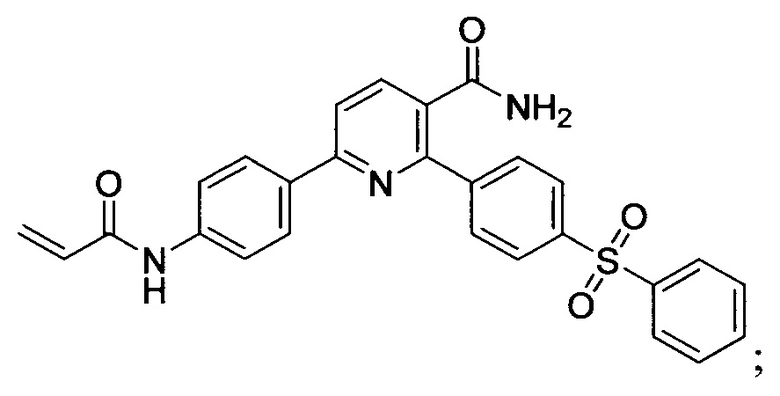
или его фармацевтически приемлемая соль.
3. Соединение по п. 2, представляющее собой
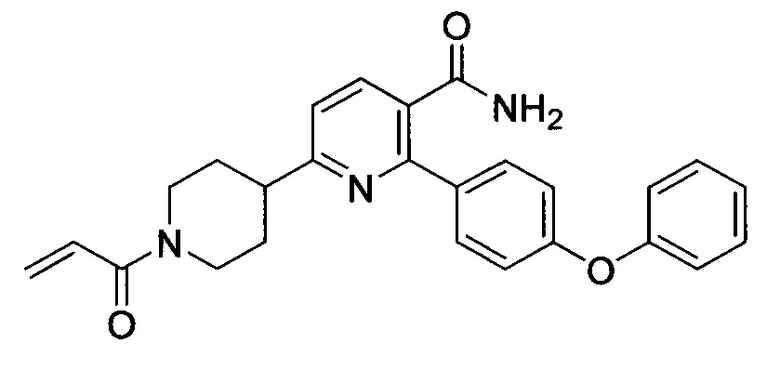
или
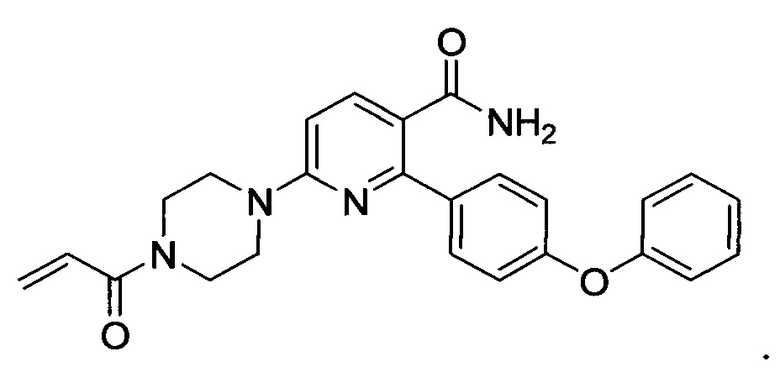
4. Соединение, имеющее Формулу IId, или его фармацевтически приемлемая соль:
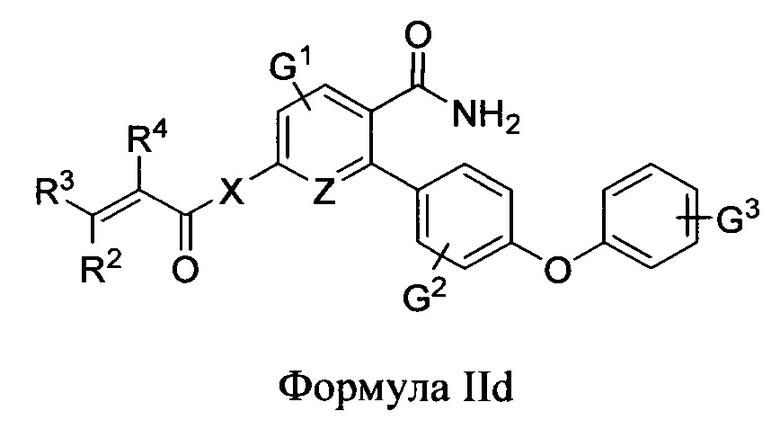
где X представляет собой 
Z представляет собой С или N;
G1, G2 и G3 независимо выбраны из группы, состоящей из одного или более Н, D (дейтерий), C1-12 алкила, галогена, CN и CF3;
R2 и R3 каждый независимо выбраны из группы, состоящей из Н, D, C1-12 алкила, галогена, CN и CF3, причем C1-12 алкил необязательно замещен группой NR11R12;
R4 выбран из группы, состоящей из Н, D, C1-12 алкила, галогена, CN и CF3; и
R11 и R12 независимо представляют собой Н или C1-6 алкил.
5. Соединение по п. 4, в котором
X представляет собой 
6. Соединение по п. 5, в котором G1, G2 и G3 независимо выбраны из группы, состоящей из одного или более Н и галогена.
7. Соединение по п. 4, в котором R2 и R3 представляют собой Н или C1-12 алкил, причем C1-12 алкил необязательно замещен группой NR11R12, и R11 и R12 представляют собой Н или СН3.
8. Фармацевтическая композиция для лечения заболевания, опосредованного тирозинкиназой Брутона (BTK), содержащая соединение по любому из пп. 1-7 или его фармацевтически приемлемую соль.
9. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-7 для лечения, по меньшей мере, одного из рака, хронического воспаления и аутоиммунного заболевания, которые опосредованы, по меньшей мере, частично, BTK.
10. Способ необратимого ингибирования BTK, включающий введение пациенту терапевтически эффективного количества ингибитора BTK, содержащего соединение по любому из пп. 1-7.
| WO 2009158571 A1, 30.12.2009 | |||
| WO 2012170976 A2, 13.12.2012 | |||
| WO 2008033834 A1, 20.03.2008 | |||
| WO 2006099075 A2, 21.09.2006 | |||
| EA 200901313 A1, 30.04.2010. |

