Настоящая заявка испрашивает приоритет на основании заявки Индии №610/СНЕ/2006, поданной 3 апреля 2006, и предварительной заявки США №60/801437, поданной 18 мая 2006, которые приведены здесь посредством ссылки.
Область изобретения
Настоящее изобретение относится к новым соединениям, представленным формулой I, их производным, аналогам, таутомерным формам, стереоизомерам, биоизостерам, диастереомерам, полиморфам, фармацевтически приемлемым солям, сольватам и содержащим их фармацевтически приемлемым композициям, которые применяют при лечении диабета типа II и диабетических осложнений, а также для лечения дислипидемии, гиперхолестеринемии, ожирения и гипергликемии.
Более конкретно, настоящее изобретение относится к соединениям формулы I, которые преимущественно являются ингибиторами серинпротеазы, в особенности ингибиторами дипептидилпептидазы, более конкретно ингибиторами дипептидилпептидазы IV, а также к их производным, аналогам, таутомерным формам, стереоизомерам, биоизостерам, диастереомерам, полиморфам, фармацевтически приемлемым солям и сольватам. Кроме того, настоящее изобретение относится к фармацевтически приемлемым композициям, содержащим вышеуказанные соединения.
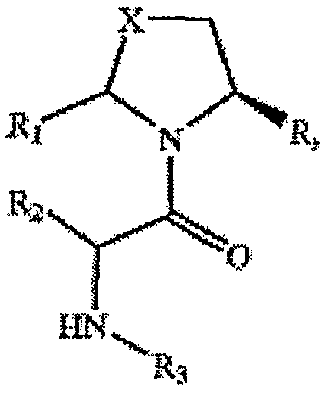
Соединения настоящего изобретения представлены формулой I

его производными, аналогами, таутомерными формами, стереоизомерами, биоизостерами, диастереомерами, полиморфами, фармацевтически приемлемыми солями и фармацевтически приемлемыми сольватами, где
Х представляет собой СН2, CHF, CF2, CHCl, CHOH, CHOCH3, NH, NCOCH3, CHPh, O или S,
Y представляет собой CN,
R1 и R5 выбраны из водорода, С1-С4алкила и гидрокси,
R2 выбран из водорода, С1-С4алкила, замещенного алкила, С1-4алкоксиС1-4алкила, С1-4гидроксиалкила, R5NHC1-4алкила и R5NHC(NH)NHC1-4алкила,
R3 выбран из водорода и С1-С4алкила,
R4 выбран из водорода, С1-С4алкила, замещенного алкила, С1-С4алкокси, С1-С4алканоилокси, гидрокси, амино, нитро, С2-С6алкенила, ацила и галогена,
n представляет собой 1 или 2,
m представляет собой 0, 1 или 2,
R представляет собой R11, R12 или R13, где
R11 включает в себя, по меньшей мере, одну из групп, выбранных из нижеследующих а), b) или с), где необязательно замещенные циклоалкильные, гетероциклильные и гетероарильные группы связаны с норадамантильной частью либо непосредственно, либо через смежный метилен или этилен, либо С-С связью, либо C-N связью.
а) Циклоалкильная группа, которая необязательно замещена С1-С4алкилом, диалкилом или оксо, предпочтительно С4-С7 кольцевая система, более предпочтительно С5-С6 кольцевая система, которая может быть дополнительно функционализирована или замещена с высокой степенью замещения. Примерами возможных циклоалкильных групп являются циклопентан, циклогексан, циклопентандион, циклогександион и необязательные заместители включают в себя С1-С4алкил, диалкил и оксо.
b) Необязательно замещенная гетероарильная группа, предпочтительно, 5-10-членная кольцевая система, в которой гетероарильное кольцо представляет собой моноциклическую, ароматическую кольцевую систему или бициклическую ароматическую кольцевую систему, включающую в себя один, два или более гетероатомов, выбранных из азота, серы и кислорода. Возможные гетероарильные группы включают в себя, но не ограничиваются перечисленными, тетразол, триазол, пиразол, имидазол, оксадиазол, пиридин, пиримидин, индол, фуран, бензофуран, бензимидазол, индазол, тиофен и бензотиофен, и заместители в гетероарильном кольце, которые могут быть одинаковыми или различными, выбраны из R6 и R7, где R6 представляет собой водород, С1-С4алкил, С2-С4алкенил, гидрокси, гидроксиалкил, алкиламино, галогеналкил, амино, ацил, СООR9 или СОR9 и R7 выбран из группы, состоящей из водорода, гидрокси, галогена, амино, нитро, С1-С8 алкила, С2-С4 алкенила, СООR9, CONR8R9, COR9, NHCOOR8, NHS(O)2R8, NHS(O)R8, NHS(O)2NHR8, NR8COOR9, NR8COR9, NR8S(O)2R9, NR8CONR8R9, NR8C(S)NR8R9, NHC(O)NH(S)(O)2R8, OSO2R8, OCONR8R9, SO2R8, SOR8, SR8, SO2NR8R9 и S(O)3OR8. Когда присутствуют R6 и R7 у смежных атомов углерода кольцевой системы, они могут вместе образовывать шестичленное ароматическое кольцо, такое как фенил, или гетероциклическое кольцо, такое как пиридин с дополнительными заместителями, такими как амино, гидрокси, алкил, алкилсульфонил, алкилтио, алкилсульфинил, карбокси или оксо.
с) Гетероциклическая группа, необязательно замещенная С1-С3алкилом, диалкилом или оксогруппами, где гетероциклическая кольцевая система представляет собой 4-10-членную моно- или бициклическую кольцевую систему с одним или несколькими гетероатомами, выбранными из группы, состоящей из азота, серы и кислорода, где гетероатомы также могут присутствовать как функциональные группы, такие как N-оксиды, сульфоксиды и сульфодиоксиды, где гетероциклическая кольцевая система может содержать одну или две двойные связи и где моноциклическое гетероциклическое кольцо может быть необязательно конденсировано с гетероарильным, арильным или циклоалкильным кольцом, необязательно замещенным С1-С5алкилом, галогенами, гидрокси, амино, нитро, галогеналкилом, алкиламино, карбокси, NH(CO)R8, NHS(O)2R8, NHC(O)NHR9, NHSOR8, NHS(O)2NHR8, NR8COOR9, NR8COR9, NR8S(O)2R9, NR8CONR8R9, NR8C(S)NR8R9 или NHC(O)NHS(O)2R8. Примеры таких гетероциклических кольцевых радикалов включают в себя, но не ограничиваются перечисленными, имидазолидинон, изотиазолидин-1,1-диоксид, пирролидин, пирролидиндион, оксопирролидин, изоксазолидиндион, изоиндолдион, морфолин, тиоморфолин, тиоморфолин-1,1-диоксид, тиофен-1,1-диоксид, тиазолидиндион, пиперидин, пиперазин, тетрагидропиримидинон, [1,2]-тиазинан-1,1-диоксид, тетрагидротиофен-1,1-диоксид, пиперидинон и тетрагидротиопиран-1,1-диоксид.
R12 выбран из водорода, галогена, галогеналкила, гидрокси, карбокси, нитро, амино, циано, алкилсульфинила, алкилсульфонила, алкилтио, амидинила, алкокси, алкоксикарбониламино, уреидо, тиоуреидо, алканоила, алканоилокси, алканоиламино, карбамоила, гуанидила, необязательно замещенного С1-С8алкила или С2-С6алкенила.
R13 представляет собой необязательно замещенный арил, где заместители могут быть одинаковыми или различными и включают в себя, по меньшей мере одну из групп, выбранных из
а) водорода;
b) С1-С8алкила, С2-С6алкенила, галогена, алкилгалогена, алкокси, алкилсульфонила, алкилсульфинила, алкокси, алканоила, алканоилокси, ациламино, карбониламино, гуанидила, нитро, амино, СООR9, R8NHC(O)R9, COR9, CONR8R9, NHC(O)OR8, NHC(O)R8, NHC(O)NR8R9, NHC(O)NR8R9, NHS(O)2R8, NHS(O)R8, NHS(O)2NHR8, NHS(O)2NHC(O)R8, NR8COOR9, NR8COR9, NR8S(O)2R9, NR8CONR8R9, NR8C(S)NR8R9, NHC(O)NHS(O)2R8, S(O)2R8, SOR8, SR8, S(O)2NR8R9, OCF3, OS(O)2R8 или OCONR8R9;
c) насыщенной, частично насыщенной или ненасыщенной моно- или бициклической гетероциклической кольцевой системы, необязательно замещенной одной или несколькими группами, выбранными из С1-С3алкила, С2-С6алкенила, диалкила и оксо, где гетероциклическая кольцевая система представляет собой 4-10-членное кольцо с одним или несколькими гетероатомами, выбранными из группы, состоящей из азота, серы и кислорода, где гетероатомы могут также присутствовать в качестве функциональных групп, таких как N-оксиды, сульфоксиды и сульфодиоксиды. Примеры таких гетероциклических кольцевых радикалов включают в себя, но не ограничиваются перечисленными, пиридин, пиримидин, имидазолидинон, имидазолидинтион, индазол, индол, изоиндол, хиназолин, хинолин, изохинолин, цинналон, изотиазолидин-1,1-диоксид, пирролидинон, 2-пиперидинон, тетрагидропиримидинон, азитидинон и тиазан-1,1-диоксид.
R8, R9 и R10-группы, которые необязательно замещены галогеновыми, гидрокси, алкокси, циано, нитро, алкильными, ацильными, ацилкоси, гидроксиалкильными, амино, алкилтио или тиоалкильными группами, которые могут быть одинаковыми или различными, представляют собой отдельно выбранные из водорода, необязательно замещенного С1-С8алкила, арила, арилалкила, алкоксикарбонила и арилалкоксикарбонила. Когда R8 и R9 присутствуют вместе у атома азота, то они могут образовать 5- или 6-членную, частично ненасыщенную или ненасыщенную циклическую систему, содержащую атомы углерода, по меньшей мере, один атом азота и необязательно один или несколько других гетероатомов, выбранных из кислорода, серы и азота.
Настоящее изобретение также относится к способам получения соединений формулы I, их производных, аналогов, таутомерных форм, стереоизомеров, биоизостеров, диастереомеров, полиморфов, фармацевтически приемлемых солей, и сольватов.
Настоящее изобретение также относится к новым промежуточным соединениям, способам их получения, их применению при получении соединений формулы I и их производных, аналогов, таутомерных форм, стереоизомеров, биоизостеров, диастереомеров, полиморфов, фармацевтически приемлемых солей и сольватов.
Уровень техники изобретения
Диабет характеризуется повышенными уровнями глюкозы в плазме или гипергликемией в состоянии натощак или после введения глюкозы во время перорального теста на толерантность глюкозы. Имеются два типа диабета. Диабет 1 типа, обычно диагностируемый у детей и молодых людей, ранее был известен как ювенильный диабет. При диабете 1 типа организм не продуцирует инсулин. Диабет типа 2 является наиболее распространенной формой диабета. При диабете 2 типа организм либо не продуцирует достаточно инсулина, либо клетки игнорируют инсулин. Пациенты с диабетом 2 типа подвергаются повышенному риску макрососудистых и микрососудистых осложнений, включающих заболевание коронарной артерии, инсульт, гипертензию, нефропатию, заболевание периферических сосудов, нейропатию и ретинопатию.
Существенная, быстро увеличивающаяся часть популяции людей поражена диабетом 2 типа, заболеванием, характеризующимся повышенными уровнями глюкозы в крови и относительной недостаточностью инсулина. Недавние исследования обнаружили, что активность двух сильнодействующих стимуляторов секреции инсулина, GLP-1 и GIP быстро анулируется серинпептидазой дипептидилпептидазой IV (DPP-IV). DPP-IV является членом семейства серинпептидаз. CD26 или GPPIV представляет собой мембрана-ассоциированную пептидазу из 766 аминокислот, которые широко распределены во многих тканях. DPP-4 также существует как растворимая форма, циркулирующая в плазме, и существенная активность, подобная DPP-4, детектирована в плазме из организма человека и грызунов. Главной биологической активностью CD26 (DPP-IV) является его ферментативная функция. DPP-IV предпочитает субстраты с аминоконцевым пролином или аланином в положении 2, но может также расщеплять субстраты с непредпочтительными аминокислотами в положении 2. Структура GIP, GLP-1 и GLP-2 обнаруживает в высокой степени сохраненный аланин в положении 2, превращая эти пептиды в идеальные предполагаемые субстраты для аминопептидазы дипептидилпептидазы 4 (DPP-4). Eur. J. Biochem. 1993, 214(3), 829-35.
Взглянув на доступные курсы лечения диабета 2 типа, которые по существу не изменяются многие годы, становится ясно, что все они имеют свои собственные ограничения. Имеются много фармакологических стратегий для достижения этих целей. Первыми из этих рядов являются ингибиторы альфа глюкозидазы, такие как акарбоза и миглитол, которые действуют путем создания препятствия при воздействии альфа-глюкозидазы, присутствующей в щеточной каемке малого кишечника. Результатом этого ингибирования является уменьшение усвоения пищи и последующая абсорбция глюкозы в системном кровообращении. Уменьшение поглощения глюкозы позволяет панкреатическим бета-клеткам более эффективно регулировать секрецию инсулина. Преимуществом применения ингибиторов альфа-глюкозидазы является то, что они функционируют в кишечнике локально и не обладают большим системным воздействием. Гипогликемия обычно не возникает при применении ингибиторов альфа-глюкозидазы, но они являются эффективными для уменьшения уровней глюкозы натощак в плазме (FPG) и уровней гликозилированного гемоглобина (HbAIc). Распространенными неблагоприятными побочными действиями этих ингибиторов являются абдоминальное вздутие и дискомфорт, диарея и метеоризм.
Препараты сульфонилмочевины и классы меглитинидов пероральных гипогликемических лекарственных средств называются эндогенными стимуляторами секреции инсулина, потому что они индуцируют высвобождение из поджелудочной железы эндогенного инсулина. Вследствие того, что эти лекарственные средства могут индуцировать отчетливую гипогликемию, курс лечения инициируют самой низкой возможной дозой, осторожно управляют до получения в результате дозы в FPG 110-140 мг/декалитр. Препараты сульфонилмочевины действуют путем связывания и ингибированием АТР-зависимого калиевого канала поджелудочной железы, который обычно вовлечен в опосредованную глюкозой секрецию инсулина. Сульфонилкарбамиды оказывают незначительное воздействие на циркуляцию триглицеридов, липопротеинов или холестерина. Несульфонилкарбамидные стимуляторы секреции инсулина являются как быстро, так и кратковременно действующими. Однако меглитиниды (несульфонилкарбамидные стимуляторы секреции инсулина) оказывают воздействия на калиевую проводимость. Подобно сульфонилкарбамидам меглитиниды не оказывают непосредственных воздействий на уровни циркуляции липидов в плазме.
Бигуаниды снижают уровни глюкозы в сыворотке увеличением опосредованной инсулином супрессии продуцирования глюкозы в печени и увеличением стимулированного инсулином поглощения глюкозы скелетными мышцами. Метформин является членом этого класса и обычно наиболее широко рекомендуемым инсулин-сенсибилизирующим лекарственным средством при клиническом применении. Введение метформина не приводит к увеличению высвобождения инсулина из поджелудочной железы и как таковой риск гипогликемии является минимальным. Вследствие того, что главным местом действия метформина является печень, его применение может быть противопоказано пациентам с дисфункцией печени. В случае молодых женщин с диабетом 2 типа, применение метформина весьма рекомендуется для уменьшения заболеваемости, а также и возможности синдрома поликистоза яичников. Однако два бигуанида, фенформин и метформин могут вызывать лактоцидоз и тошноту/диарею.
Семейство ядерных рецепторов, активируемых пролифератором пероксисом (РРАR), подвергали особенно тщательному исследованию в области диабета и в результате появились тиазолидиндионы как класс терапевтических средств. Первое поколение тиазолидиндионов было агонистами гамма рецепторов PPAR и было способно уменьшать резистентность к инсулину. Однако одним неблагоприятным действием, связанным с агонистами гамма рецепторов PPAR, является увеличение массы тела. Недавно созданы молекулы, которые активируют альфа PPAR. Этот класс способен снижать уровни триглицеридов и также способен повышать чувствительность к инсулину и в результате созданы двойные агонисты альфа PPAR/гамма PPAR с предлагаемыми полезными действиями выше, чем у существующих предпочтительных гамма- и альфа PPAR лекарственных средств при лечении диабета 2 типа. Но проблемы безопасности замедляют введение этих лекарственных средств.
Одним из вызывающих интерес для разработки классов агентов являются агонисты GLP-1. Первичные метаболические ответные реакции на высвобождение GLP-1 из энтероэндокринных L-клеток кишки представляют собой ингибирование секреции глюкагона и усиление зависимого от глюкозы высвобождения инсулина из поджелудочной железы, оба эффекта приводят к снижению гликемического движения. Но гормональное действие GLP-1 быстро заканчивается в результате ферментативного расщепления DPP IV. Недавние клинические данные показали, что либо инфузия GLP-1, либо ингибирование DPP IV может приводить к драматическим понижениям концентраций глюкозы в плазме, понижениям HbAIc и улучшению функции бета-клеток поджелудочной железы. Следовательно, обе представленные возможные цели для предотвращения гипергликемии связаны с диабетом и нарушением инсулиновой функции. Существует как польза, так и бесполезные воздействия в современных терапевтических методах нацеливающего действия GLP-1 у пациентов с диабетом. Современное применение миметиков GLP-1 и/или агонистов (GLP-1R) рецепторов GLP-1 сосредоточены на пептидах или модифицированных пептидах и их требуется вводить инъекцией, которая приводит к проблемам с комплаентностью пациента.
По другому, новому механизму лечения диабета 2 типа действуют ингибиторы дипептидилпептидазы-IV. Дипептидилпептидаза IV представляет собой многофункциональный белок, вовлеченный в расщепление гормонов внутренней секреции, следовательно, служащий для регулирования гомеостаза глюкозы и, в результате, рассматриваемый как мишень для управляющего воздействия на диабет 2 типа. Полезное действие дипептидилпептидазы IV при лечении диабета 2 типа основано на том факте, что дипептидилпептидаза IV in vivo легко инактивирует GLP-1 и GIP. Они представляют собой инкретины, которые продуцируются, когда происходит потребление пищи. Эти инкретины стимулируют продуцирование инсулина. Ингибирование дипептидилпептидазы IV приводит к пониженной инактивации инкретинов, и, в свою очередь, повышенной эффективности инкретинов в стимулировании продуцирования инсулина поджелудочной железой. Следовательно, ингибирование дипептидилпептидазы IV приводит к повышенному уровню сывороточного инсулина. Интересным наблюдением является то, что инкретины продуцируются только, когда происходит потребление пищи. Поэтому не предполагают, что ингибирование дипептидилпептидазы IV увеличит уровень инсулина между приемами пищи, что может привести к гипогликемии. Следовательно, предполагают, что ингибирование дипептидилпептидазы увеличит уровень инсулина без повышения риска гипогликемии. Исследованные ингибиторы дипептидилпептидазы IV имеют преимущество над другими новыми терапевтическими методами, поскольку их можно вводить перорально. Комплаентность у пациентов является более высокой при перорально доставляемых лекарственных средствах, чем при доставке, которая требует инъекции. Поэтому ингибиторы дипептидилпептидазы IV представляют собой перспективные новые подходы для лечения диабета типа 2, которые функционируют, по меньшей мере частично, как непрямые стимуляторы секреции инсулина. Механизм и применение ингибиторов DPPIV при различных заболеваниях хорошо объясняется в патентах предшествующего уровня техники, подобных WO 2005/033106 и введенных здесь посредством ссылки во всей своей полноте.
Предшествующий уровень техники
В международной патентной заявке WO 00/34241 описаны соединения общей формулы
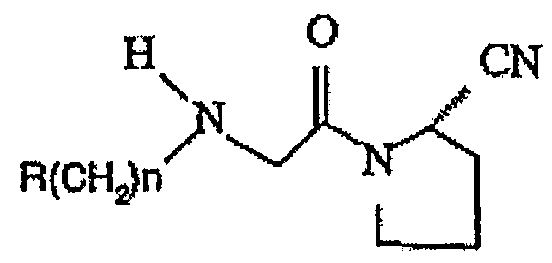
где R представляет собой замещенный адамантил.
В международной патентной заявке WO 03/04498 описаны соединения общей формулы
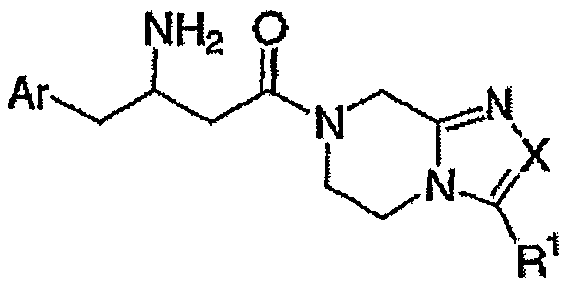
В заявке США US 2005/038020 описаны соединения общей формулы
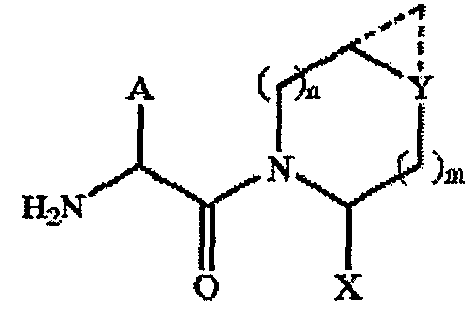
где А представляет собой необязательно замещенную адамантильную группу.
В международной патентной заявке WO 2006/090244 описаны соединения формулы
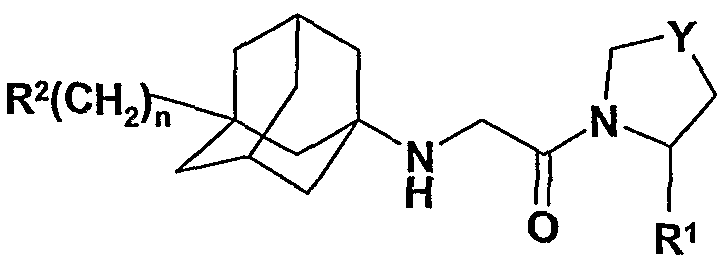
где n представляет собой 0, 1, 2 или 3.
R2 представляет собой замещенный алкил, замещенный или незамещенный алкенил, замещенный или незамещенный алкинил, замещенный или незамещенный циклоалкил, замещенный или незамещенный циклоалкилалкил, замещенный или незамещенный циклоалкенил, замещенный или незамещенный циклоалкенилалкил, замещенный или незамещенный арил, замещенный или незамещенный арилалкил, замещенный или незамещенный гетероарил, замещенную или незамещенную гетероциклическую группа, замещенный или незамещенный гетероциклилалкил, замещенный или незамещенный гетероарилалкил, -NR3R4, -NH-S(O)m-R3, -NH-CR3R4, C(O)-R5, -C(O)O-R3,
-C(O)NR3R4, -S(O)m-, NR3R4, нитро, циано, формил, ацетил, галоген, -SRa или защитную группу.
В международной патентной заявке WO 2005/021536 описаны cоединения формулы
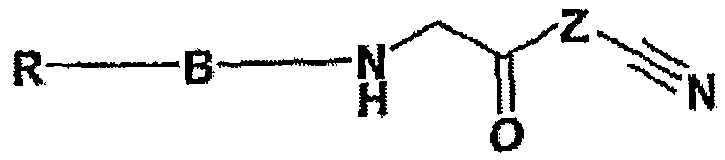
где один из возможных заместителей В представляет собой адамантиламин.
Один из репрезентативных примеров представляет собой
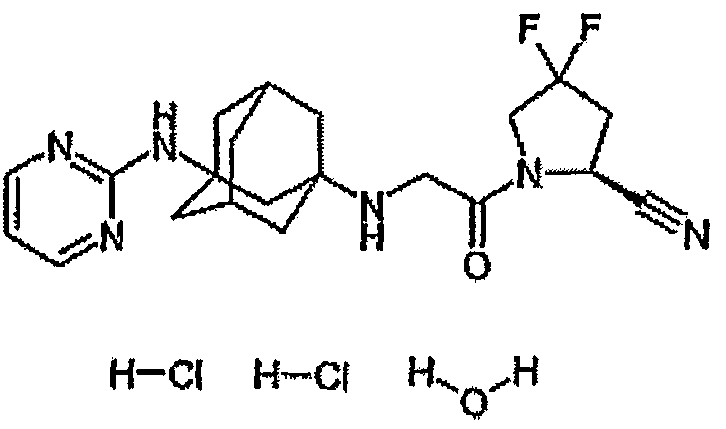
В международной патентной заявке WO 2006/012395 описаны соединения формулы
в качестве ингибиторов пептидазы. Один из репрезентативных примеров представляет собой
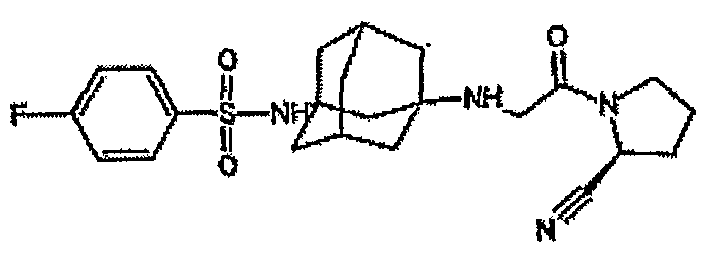
В международной патентной заявке WO 2005095339 описаны соединения формулы
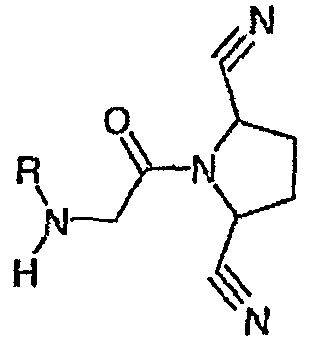
в качестве ингибиторов DPPIV.
В US 20050215784 в качестве ингибиторов DPPIV описаны соединения формулы
Хотя имеется немного ингибиторов DPPIV в различных стадиях клинических испытаний, такие, как в примерах, указанных выше (LAF-237, MK-0431, BMS-477118, GSK23A), все еще имеется необходимость в новых соединениях в этой области и целью настоящего изобретения является получение новых норадамантилцианопирролидиновых соединений, представленных формулой I, которые обладают активностью ингибиторов DPPIV, а также способы их получения.
Сущность изобретения
Цель настоящего изобретения представляет собой предложение новых соединений формулы I, обладающих активностью как ингибиторы серинпротеазы, конкретно активностью как ингибиторы дипептидилпептидазы IV, для снижения уровней глюкозы в крови, уровней липидов, уровней холестерина и уменьшения массы тела, против диабета 2 типа и диабетических осложнений. Поэтому главной целью настоящего изобретения является получение новых норадамантилцианопирролидиновых соединений, представленных формулой I, их производных, аналогов, таутомерных форм, стереоизомеров, биоизостеров, диастереомеров, полиморфов, фармацевтически приемлемых солей, сольватов и содержащих их фармацевтически приемлемых композиций.
По другому аспекту настоящего изобретения предложен способ получения норадамантилцианопирролидиновых соединений формулы I, их производных, аналогов, таутомерных форм, стереоизомеров, биоизостеров, диастереомеров, полиморфов, фармацевтически приемлемых солей и фармацевтически приемлемых сольватов.
По другому аспекту настоящего изобретения предложены новые промежуточные соединения, способы их получения и применение таких промежуточных соединений в способах получения указанных норадамантилцианопирролидиновых соединений, представленных формулой I и их производных, аналогов, таутомерных форм, стереоизомеров, полиморфов, биоизостеров, диастереомеров, фармацевтически приемлемых солей и сольватов.
По другому аспекту настоящего изобретения предложены фармацевтические композиции, содержащие соединения настоящего изобретения, представленные формулой I, их производные, аналоги, таутомерные формы, стереоизомеры, биоизостеры, диастереомеры, полиморфы, соли, сольваты или их смеси в сочетании с подходящими носителями, растворителями, разбавителями и другими средами, обычно применяемыми при получении таких композиций.
Подробное описание изобретения
Настоящее изобретение предлагает соединения, представленные формулой I,
их производные, аналоги, таутомерные формы, стереоизомеры, биоизостеры, диастереомеры, полиморфы, фармацевтически приемлемые соли и их фармацевтически приемлемые сольваты, где
Х представляет собой CH2, CHF, CF2, CHCl, CHOH, CHOCH3, NH, NCOCH3, CHPh, O или S,
Y представляет собой CN,
R1 и R5 выбраны из водорода, С1-С4алкила и гидрокси,
R2 выбран из водорода, С1-С4алкила, замещенного алкила, С1-4алкоксиС1-4алкила, С1-4гидроксиалкила, R5NHC1-4алкила и R5NHC(NH)NHC1-4алкила,
R3 выбран из водорода и С1-С4алкила,
R4 выбран из водорода, С1-4алкила, замещенного алкила, С1-4алкокси, С1-4алканоилокси, гидрокси, амино, нитро, С2-С6алкенила, ацила и галогена,
n представляет собой 1 или 2,
m представляет собой 0, 1 или 2,
R представляет собой R11, R12 или R13, где
R11 включает в себя, по меньшей мере одну из групп, выбранных из а), b) или с), где необязательно замещенные циклоалкильные, гетероциклильные и гетероарильные группы связаны с норадамантильной частью, либо непосредственно, либо через смежный метилен или этилен, либо связью С-С, либо связью C-N.
а) Циклоалкильная группа, которая необязательно замещена С1-С4алкилом, диалкилом или оксо, предпочтительно С4-С7 кольцевая система, более предпочтительно С5-С6 кольцевая система, которая может быть дополнительно функционализирована или замещена с высокой степенью замещения. Примеры возможных циклоалкильных групп представляют собой циклопентан, циклогексан, циклопентандион, циклогександион и возможные заместители включают в себя С1-С4алкил, диалкил и оксо.
b) Необязательно замещенная гетероарильная группа, предпочтительно 5-10-членная кольцевая система, в которой гетероарильное кольцо представляет собой моноциклическую ароматическую кольцевую систему или бициклическую ароматическую кольцевую систему, включающую в себя один, два или более гетероатомов, выбранных из азота, серы и кислорода. Возможные гетероарильные группы включают в себя, но не ограничиваются перечисленными, тетразол, триазол, пиразол, имидазол, оксадиазол, пиридин, пиримидин, индол, фуран, бензофуран, бензимидазол, индазол, тиофен и банзотиофен, и заместители у гетероарильного кольца, которые могут быть одинаковыми или различными, выбраны из R6 и R7, где R6 представляет собой водород, С1-С4алкил, С2-С4алкенил, гидрокси, гидроксиалкил, алкиламино, галогеналкил, амино, ацил, СООR9 или COR9 и R7 выбран из группы, состоящей из водорода, гидрокси, галогена, амино, нитро, С1-С8алкила, С2-С4алкенила, СООR9, CONR8R9, COR9, NHCOOR8, NHS(O)2R8, NHS(O)R8, NHS(O)2NHR8, NR8COOR9, NR8COR9, NR8S(O)2R9, NR8CONR8R9, NR8C(S)NR8R9, NHC(O)NHS(O)2R8, OSO2R8, OCONR8R9, SO2R8, SOR8, SR8, SO2NR8R9 и S(O)2OR8. Когда R6 и R7 присутствуют у смежных атомов углерода кольцевой системы, они вместе могут образовывать шестичленное ароматическое кольцо, такое как фенил, или гетероциклическое кольцо такое как пиридин, с дополнительными заместителями, такими как амино, гидрокси, алкил, алкилсульфонил, алкилтио, алкилсульфинил, карбокси или оксо.
Дополнительные предпочтительные гетероарильные группы включают в себя
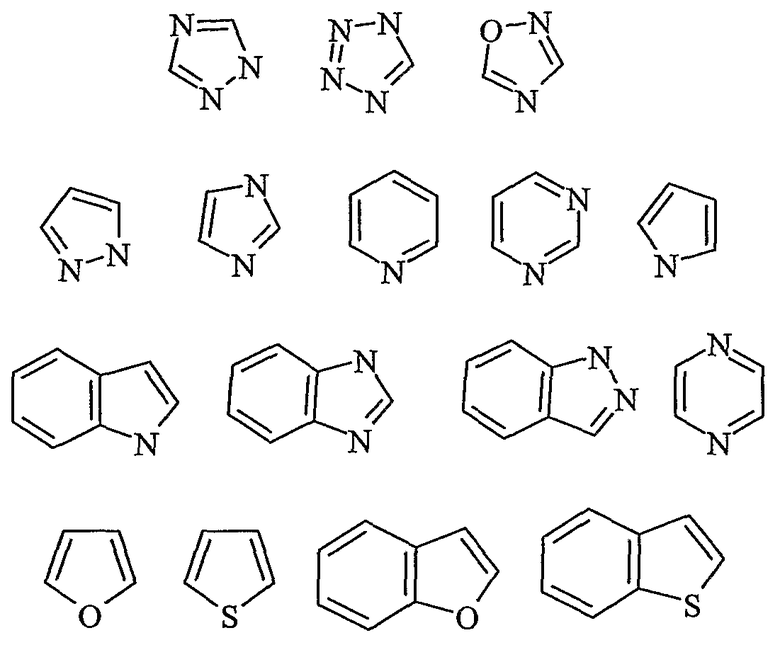
эти примеры не ограничивают настоящее изобретение.
с) Гетероциклическая группа необязательно замещена С1-С3алкильными, диалкильными и оксогруппами, где гетероциклическая кольцевая система представляет собой 4-10-членную моно- или бициклическую кольцевую систему с одним или несколькими гетероатомами, выбранными из группы, состоящей из азота, серы и кислорода, где гетероатомы также могут присутствовать как функциональные группы, такие как N-оксиды, сульфоксиды и сульфодиоксиды, где гетероциклическая кольцевая система может содержать одну или две двойные связи и где моноциклическое гетероциклическое кольцо может быть необязательно конденсировано с гетероарильным, арильным или циклоалкильным кольцом, необязательно замещенным С1-С5алкилом, галогенами, гидрокси, амино, нитро, галогеналкилом, алкиламино, карбокси, NH(CO)R8, NHS(O)2R8, NHC(O)NHR9, NHSOR8, NHS(O)2NHR8, NR8COOR9, NR8COR9, NR8S(O)2R9, NR8CONR8R9, NR8C(S)NR8R9 или NHC(O)NHS(O)2R8. Необязательные заместители гетероциклических кольцевых систем включают в себя С1-С3алкильные, диалкильные и оксогруппы. Примеры таких гетероциклических радикалов включают в себя, но не ограничиваются перечисленными, имидазолидинон, изотиазолидин-1,1-диоксид, пирролидин, пирролидиндион, оксопирролидин, изоксазолидиндион, изоиндолдион, морфолин, тиоморфолин, тиоморфолин-1,1-диоксид, тиофен-1,1-диоксид, тиазолидиндион, пиперидин, пиперазин, тетрагидропиримидинон, [1,2]-тиазинан-1,1-диоксид, тетрагидротиофен-1,1-диоксид, пиперидинон и тетрагидротиопиран-1,1-диоксид.
Дополнительные предпочтительные гетероциклические группы включают в себя
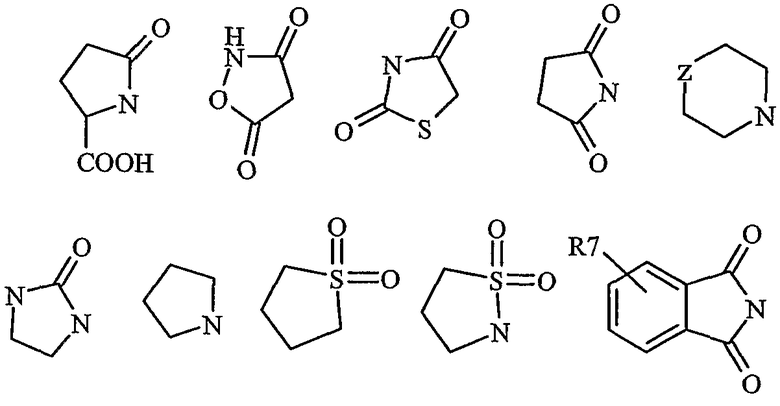
где R7 представляет собой, как указано выше по тексту, и Z представляет собой CH2, O, S, SO2, NH, NR6 или СН(ОН). Эти примеры не ограничивают настоящее изобретение.
R12 выбран из водорода, галогена, галогеналкила, гидрокси, карбокси, нитро, амино, циано, алкилсульфинила, алкилсульфонила, алкилтио, амидинила, алкокси, алкоксикарбониламино, уреидо, тиоуреидо, алканоила, алканоилокси, алканоиламино, карбамоила, гуанидила, необязательно замещенного С1-С8алкила и С2-С6алкенила.
R13 представляет собой необязательно замещенный арил, где заместители могут быть одинаковыми или различными, и включают в себя, по меньшей мере, одну из групп, выбранных из
а) водорода;
b) С1-С8алкила, С2-С6алкенила, галогена, алкилгалогена, алкокси, алкилсульфонила, алкилсульфинила, алкокси, алканоила, алканоилокси, ациламино, карбониламино, гуанидила, нитро, амино, СООR9, R8NHC(O)R9, COR9, CONR8R9, NHC(O)OR8, NHC(O)R8, NHC(O)NR8R9, NHC(O)NR8R9, NHS(O)2R8, NHS(O)R8, NHS(O)2NHR8, NHS(O)2NHC(O)R8, NR8COOR9, NR8COR9, NR8S(O)2R9, NR8CONR8R9, NR8C(S)NR8R9, NHC(O)NHS(O)2R8, S(O)2R8, SOR8, SR8, S(O)2NR8R9, OCF3, OS(O)2R8 или OC(O)NR8R9;
c) насыщенная, частично насыщенная или ненасыщенная моно- или бициклическая гетероциклическая кольцевая система, необязательно замещенная одной или несколькими группами, выбранными из С1-С3алкила, С2-С6алкенила, диалкила и оксо, где гетероциклическая кольцевая система представляет собой 4-10-членное кольцо с одним или несколькими гетероатомами, выбранными из группы, состоящей из азота, серы и кислорода, где гетероатомы также могут присутствовать в качестве функциональных групп, таких как N-оксиды, сульфоксиды, сульфодиоксиды. Примеры таких гетероциклических кольцевых радикалов включают в себя, но не органичиваются перечисленными, пиридин, пиримидин, имидазолидинон, имидазолидинтион, индазол, индол, изоиндол, хиназолин, хинолин, изохинолин, цинналон, изотиазолидин-1,1-диоксид, пирролидинон, 2-пиперидинон, тетрагидропиримидинон, азитидинон и тиазан-1,1-диоксид.
Дополнительно предпочтительные гетероциклические группы включают в себя
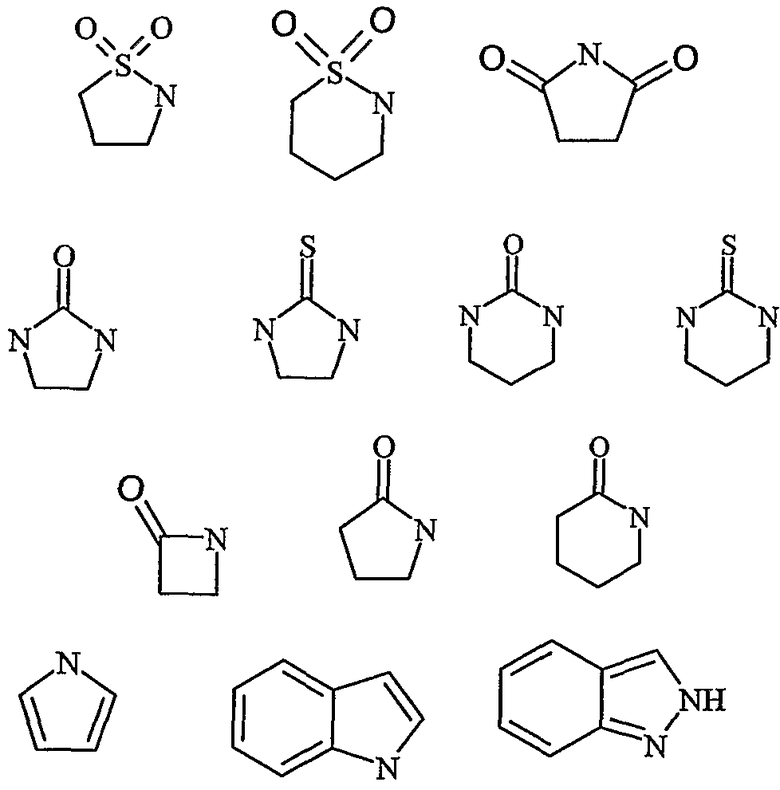
эти примеры не ограничивают настоящее изобретение.
Группы R8, R9 и R10, которые необязательно замещены группами галогена, гидрокси, алкокси, циано, нитро, алкила, ацила, ацилокси, гидроксиалкила, амино, алкилтио, или тиоалкила, могут быть одинаковыми или различными, и в отдельности выбраны из водорода, необязательно замещенного С1-С8алкила, арила, арилалкила, алкоксикарбонила и арилалкоксикарбонила. Когда R8 и R9 присутствуют вместе у атома азота, они могут образовать 5- или 6-членную насыщенную, частично ненасыщенную или ненасыщенную циклическую систему, содержащую атомы углерода, по меньшей мере один атом азота и необязательно один или несколько других гетероатомов, выбранных из кислорода, серы и азота.
В результате соединения изобретения получали либо в свободной форме, либо в виде их соли, если присутствуют солеобразующие группы. Соединения настоящего изобретения можно превратить в фармацевтически приемлемые соли взаимодействием их с соответствующими кислотами или основаниями.
Некоторые соединения формулы I настоящего изобретения могут содержать один или несколько хиральных центров и настоящее изобретение включает в себя выделенные стереоизомеры, их смеси, а также соответствующие рацематы.
Перечисление терминов в списке ниже по тексту представляет собой обозначение различных терминов, применяемых для описания данного изобретения.
Термин «алкил» относится к насыщенной, имеющей нормальное или разветвленное строение алифатической углеводородной цепи, которая необязательно может быть замещена с высокой степенью замещения. Примеры «алкила» включают в себя, но не ограничиваются перечисленным, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, н-пентил и изобутил. Заместители могут быть выбраны из галогенов, гидрокси, алкокси, ацила, амино и нитро. Если не указано иное, например, выражение «Сx-Cyалкил», которое относится к алкильной группе со специфическим числом атомов углерода, в полной спецификации термин «алкильная группа» относится к С1-С8-группе. Подобную терминологию применяют также к другим предпочтительным группам.
Термин «алкенил», применяемый здесь, либо в отдельности, либо в сочетании с другими радикалами, обозначает имеющую нормальное или разветвленное строение С2-С6 алифатическую углеводородную цепь, содержащую одну или несколько углерод-углеродных двойных связей, которая может быть необязательно замещенной с высокой степенью замещения. Термин «алкенил» включает в себя диены и триены, имеющие цепи нормального и разветвленного строения, и включает в себя такие группы, как винил, аллил, 2-бутенил, 3-бутенил, 2-пентенил, 3-пентенил, 4-пентенил, 2-гексенил, 3-гексенил, 4-гексенил, 5-гексенил, 2-гептенил, 3-гептенил, 4-гептенил, 5-гептенил и 6-гептенил.
Термин «ацил» относится к -C(O)Ra-группе, где Ra представляет собой имеющий нормальное или разветвленное строение С1-С4алкил или арил.
Применяемый здесь термин «ациламино» представлен -NHC(O)Ra-группой, где Ra имеет значения, как указано выше по тексту, и примеры представляют собой CH3CONH, C2H5CONH, C3H7CONH, C6H5CONH.
Термин «алканоилокси» относится к -ОС(О)Ra-группе, где Ra представляет собой С1-С4алкил нормального или разветвленного строения, как указано выше по тексту; примеры представляют собой ацетилокси и пропанилокси.
Термин «алканоил» относится к -С(O)Ra-группе, где Ra представляет собой С1-С4алкил нормального или разветвленного строения, как указано выше по тексту; примеры представляют собой ацетил или пропаноил.
Термин «алканоиламино» относится к -NH-C(O)Ra-группе, где Ra представляет собой С1-С4алкил нормального или разветвленного строения, как указано выше по тексту; примеры представляют собой СН3СОNH- и C2H5CONH-.
Термин «алкокси» относится к -ORa-группе, где Ra представляет собой алкил, как указано здесь. Репрезентативные примеры включают в себя, но не ограничиваются перечисленным, метокси и этокси.
Термин «алкоксикарбонил» относится к -C(O)ORa-группе, где Ra представляет собой алкил, как указано здесь.
Термин «алкоксикарбониламино» относится к -NHC(O)ОRa-группе, где Ra представляет собой алкил, как указано здесь.
Термин «алкиламино» относится к -N(Ra)2-группе, где один Ra представляет собой алкил, а другой Ra представляет собой независимо Н или алкил, как указано здесь.
Термин «алкилсульфинил» относится к -S(O)Ra-группе, где Ra представляет собой алкил, как указано здесь.
Термин «алкилсульфонил» относится к -S(O)2Ra-группе, где Ra представляет собой алкил, как указано здесь.
Термин «алкилтио» относится к -SRa-группе, где Ra представляет собой алкил, как указано здесь. Репрезентативные примеры включают в себя, но не ограничиваются перечисленным, -S-CH3, -S-CH2CH3.
Термин «алкилгалоген» относится к RaX-группе, где Ra представляет собой алкил, как указано выше по тексту, и Х представляет собой атом галогена, выбранный из фтора, хлора, брома и иода.
Термин «галоген» относится к атому фтора, хлора, брома или иода.
Термин «гидроксиалкил» относится к RaOH-группе, где Ra представляет собой алкил, как указано здесь, и репрезентативные примеры включают в себя, но не ограничиваются перечисленным, гидроксиметил, гидроксиэтил и гидроксипропил.
Термин «арил» относится к ароматической кольцевой системе с пятью-десятью атомами углерода, которая может быть моноциклической или бициклической и ненасыщенной или частично насыщенной, и один или несколько атомов углерода могут быть необязательно заменены одним или несколькими гетероатомами, выбранными из N, O и S. Термин «арил» включает в себя кольцо(а), необязательно замещенное с высокой степенью замещения, и заместители могут включать в себя алкил, алкилен, диалкил и оксо.
Термин «арилалкил» относится к Ar-Ra-группе, где Ar и Ra имеют значения, как указано выше по тексту.
Термин «арилалкоксикарбонил» относится к -С(О)ОRaAr-группе, где Ar и Ra имеют значения, как указано выше по тексту.
Термин «гетероарил» относится к моноциклической ароматической кольцевой системе или конденсированной бициклической ароматической кольцевой системе, включающей в себя два или более ароматических кольца, предпочтительно два кольца. Эти гетероарильные кольца содержат один или несколько гетероатомов, таких как азот, сера и кислород, где функциональные группы, такие как N-оксиды, сульфоксиды и диоксиды, допустимы в качестве замены гетероатома. Термин «гетероарил» включает в себя необязательно замещенные кольцевые системы. Примеры гетероарильных групп включают в себя фуран, тиофен, пиррол, имидазол, пиразол, триазол, тетразол, тиазол, оксазол, изоксазол, оксадиазол, тиадиазол, изотиазол, пиридин, пиридазин, пиразин, пиримидин, хинолин, изохинолин, бензофуран, бензотиофен, индол, индазол и их замещенные варианты.
Термин «гетероциклил» относится к 3-15-членному кольцу, которое является либо насыщенным, либо имеет одну или несколько двойных связей. Такие гетероциклические кольца содержат один или несколько гетероатомов, таких как атомы азота, серы и/или кислорода, где функциональные группы, такие как N-оксиды, сульфоксиды и диоксиды, представляют собой допустимые замены гетероатома. Такое кольцо может быть необязательно конденсировано с одним или несколькими другими гетероциклическими кольцами (кольцом), арильными кольцами (кольцом) или циклоалкильными кольцами (кольцом).
Термин «стереоизомеры» относится к некоторым, описанным здесь, соединениям, которые содержат один или несколько хиральных центров, или иначе, могут быть способны существовать в виде нескольких стереоизомеров. Объем настоящего изобретения включает в себя чистые стереоизомеры, смеси стереоизомеров такие, как очищенные энантиомеры/диастереомеры или смеси, обогащенные энантиомерами/диастереомерами, и рацематы.
Термин «биоизостеры» относится к соединениям или группам, которые обладают близкими молекулярными конфигурациями и объемами, приблизительно с одинаковым распределением электронов, и которые проявляют аналогичные физические свойства, такие как гидрофобность. Биоизостерные соединения действуют аналогично биохимически ассоциированным системам как агонисты или антагонисты, и тем самым порождают биологические свойства, которые являются близкими друг другу.
Термин «фармацевтически приемлемые соли» включает в себя соли, полученные из неорганических оснований, таких как Li, Na, K, Ca, Mg, Fe, Cu, Zn, Al и Mn, соли органических оснований, таких как N,N-диацетилэтилендиамин, 2-диметиламиноэтанол, изопропиламин, морфолин, пиперазин, пиперидин, прокаин, диэтиламин, триэтиламин, триметиламин, трипропиламин, трометамин, адамантиламин, диэтаноламин, этилендиамин, N,N-бензилфенилэтиламин, гидроксид холина, дициклогексиламин, метформин, бензиламин, фенилэтиламин, диалкиламин, триалкиламин, тиамин, аминопиримидин, аминопиридин, пурин, пиримидин, и спермидин, хиральные основания, подобные алкилфениламину, глицину и фенилглицину, соли природных аминокислот, таких как глицин, аланин, валин, лейцин, изолейцин, норлейцин, тирозин, цистеин, цистин, метионин, пролин, гидроксипролин, гистидин, орнитин, лизин, аргинин, серин, треонин, и фенилаланин, аминокислоты неприродного происхождения, такие как D-изомеры или замещенные аминокислоты, соли с кислотной группой аминокислот, таких как аспарагиновая кислота и глутаминовая кислота, гуанидин, замещенный гуанидин, где заместители выбраны из нитро, амино, алкила, алкенила, алкинила, аммония или замещенных солей аммония. Соли могут включать в себя аддитивные соли с кислотой, такие как сульфаты, нитраты, фосфаты, перхлораты, бораты, гидрогалогениды, выбранные из HCl, HBr, HI, ацетаты, тартраты, малеаты, цитраты, сукцинаты, пальмоаты, метансульфонаты, бензоаты, салицилаты, гидроксинафтоаты, бензолсульфонаты, аскорбаты, глицерофосфаты и кетоглутараты.
Термин «фармацевтически приемлемые сольваты» обозначает аддукты и сокристаллаты, такие как гидраты или сольваты, включающие в себя другие растворители, например спирты.
Термин «соединения изобретения» или «настоящего изобретения» относится к соединениям настоящего изобретения, представленным формулой I, как указано здесь, их производным, аналогам, таутомерным формам, стереоизомерам, биоизостерам, диастереомерам, полиморфам, а также их фармацевтически приемлемым солям и сольватам.
Термин «подходящие фармацевтически приемлемые носители» включают в себя твердые наполнители или разбавители, и стерильные водные или органические растворы. Активный ингредиент будет присутствовать в таких фармацевтических композициях в количествах, достаточных для обеспечения требуемого действия, как описано выше по тексту. Поэтому для перорального применения соединения можно объединять с подходящим твердым, жидким носителем или разбавителем, чтобы сформировать капсулы, таблетки, порошки, сиропы, растворы, суспензии и т.д. Фармацевтические композиции могут содержать дополнительные компоненты, такие как корригенты, подсластители и эксципиенты.
Предпочтительные соединения настоящего изобретения представлены формулой Ia и общей структурой, приведенной ниже по тексту. Помимо этого предпочтительные варианты осуществления представлены их производными, аналогами, таутомерными формами, стереоизомерами, биоизостерами, диастереомерами, полиморфами, фармацевтически приемлемыми солями и их фармацевтически приемлемыми сольватами.

где n представляет собой 1 или 2
m представляет собой 0, 1 или 2
Х представляет собой CH2, CHF или S
Y представляет собой CN
R3 независимо выбран из водорода и С1-С4алкила
R4 представляет собой водород, С1-С4алкил, замещенный алкил, С1-С4алкокси, С1-С4алканоилокси, гидрокси, амино, нитро, С2-С6алкенил, ацил или галоген
R представляет собой R11, R12 или R13 и R11, R12, R13 имеют значения, как указано выше по тексту.
Предпочтительные соединения настоящего изобретения включают в себя следующие соединения:
(2S)-1-[1H-1,2,4-триазол-1-илметил(трицикло[3,3,1,03,7]нон-3-иламино)ацетил]пирролидин-2-карбонитрил
(2S,4S)-4-фтор-1-{N-[2-(1H-1,2,4-триазол-1-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
(2S,4R)-4-фтор-1-{N-[2-(1H-1,2,4-триазол-1-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
(4R)-3-{N-[2-(1H-1,2,4-триазол-1-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}-1,3-тиазолидин-4-карбонитрил
(2S)-1-{N-[2-(1H-тетразол-1-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
(2S)-1-{N-[2-[(4-метилпиперазин-1-ил)метил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-{N-[2-[(4-метилпиперазин-1-ил)метил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила
(2S)-1-{N-[2-(тиоморфолин-4-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-{N-[2-(тиоморфолин-4-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила
(2S)-1-{N-[2-[(1,1-диоксидоизотиазолидин-2-ил)метил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
(2S)-1-{N-[2-[(2,4-диоксо-1,3-тиазолидин-3-ил)метил]гексагидро-2,5-метанпентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
(2S)-1-{N-[2-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
((2S)-1-{N-[2-(1,2,4-оксадиазол-3-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-ил)ацетонитрил
(2S)-1-{N-[2-[4-(1,1-диоксидоизотиазолидин-2-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
(2S,4S)-1-{N-[2-[4-(1,1-диоксидоизотиазолидин-2-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}-4-фторпирролидин-2-карбонитрил
(2S,4R)-1-{N-[2-[4-(1,1-диоксидоизотиазолидин-2-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}-4-фторпирролидин-2-карбонитрил
(4R)-3-{N-[2-[4-(1,1-диоксидоизотиазолидин-2-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}-1,3-тиазолидин-4-карбонитрил
(2S)-1-{N-[2-[4-(2-оксопирролидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
(2S,4S)-4-фтор-1-{N-[2-[4-(2-оксопирролидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
(4R)-3-{N-[2-[4-(2-оксопирролидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}-1,3-тиазолидин-4-карбонитрил
(2S)-1-{N-[2-[4-(1H-пиррол-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
(2S)-1-{N-[2-[4-(2-оксоимидазолидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
((2S)-1-{N-[2-[4-(3-метил-2-оксоимидазолидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-ил)ацетонитрил
(2S)-1-[(трицикло[3,3,1,03,7]нон-3-иламино)ацетил]пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-[(трицикло[3,3,1,03,7]нон-3-иламино)ацетил]пирролидин-2-карбонитрила
(2S)-1-{[(1-гидрокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-{[(1-гидрокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрила
(2S)-1-{[(1-метокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-{[(1-метокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрила
(2S)-1-{[(1-этокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-{[(1-этокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрила
хлористоводородная соль (2S)-1-{[(1-аминотрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрила
(2S)-1-[N-(2-фторгексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-[N-(2-фторгексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрила
(2S)-1-{N-[2-(2-оксопирролидин-1-ил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-{N-[2-(2-оксопирролидин-1-ил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила
(2S)-1-{N-[2-(1,1-диоксидоизотиазолидин-2-ил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-{N-[2-(1,1-диоксидоизотиазолидин-2-ил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила
(2S)-1-[N-(2-фенилгексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрил
хлористоводородная соль (2S)-1-[N-(2-фенилгексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрила
(2S)-1-{N-[2-(4-нитрофенил)гексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрил
(2S)-1-{N-[2-(4-аминофенил)гексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрил
По другому аспекту настоящего изобретения предложены способы получения описанных выше по тексту соединений общей формулы I.
Схема I
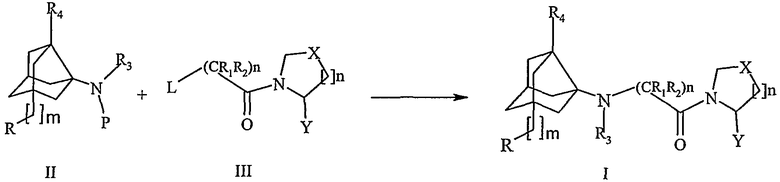
Где
Р представляет собой Н или защитную группу,
L представляет собой удаляемую группу, выбранную из группы, состоящей из атомов галогенов, тозилатов, мезилатов и трифлатов
R, R1, R2, R3, R4, n, m, X и Y имеют значения, как указано выше по тексту.
Соединения формулы I получают согласно следующим стадиям, включающим в себя:
а. Взаимодействие одного эквивалента соединения формулы III приблизительно с 1-5 эквивалентами аминного соединения формулы II в свободной форме или в виде соли или в виде соединения, где амин защищен, в присутствии одного или нескольких оснований, таких как (но не ограничиваясь перечисленными) гидриды щелочных металлов, подобные NaH и KH; органолитиевые соединения, такие как CH3Li и BuLi; алкоксиды, подобные NaOMe, NaOEt, и KOtBu; третичные амины, подобные триэтиламину и DBU; карбонаты, подобные карбонату калия, бикарбонату калия, карбонату натрия и карбонату цезия.
b. Указанное взаимодействие осуществляли при температуре в интервале приблизительно от -5ºС до 120ºС в инертном растворителе, таком как дихлорметан, диметилформамид, тетрагидрофуран, ацетонитрил, ДМСО и т.д.
с. Указанное взаимодействие осуществляли в течение от 0,5 до 72 часов, предпочтительно в течение от 0,5 до 60 часов.
d. Выделение образовавшегося соединения формулы I.
Соединения формулы III можно получать способами, представленными в современном уровне техники (WO 2003/002553, WO 00/034241, WO 98/19998, US 2005/0038020, Bioorganic and Medicinal Chemistry Letters 1996, 6,1163-66, Journal of Medicinal Chemistry 2003, 46, 2774-2789).
Промежуточное соединение II можно получить путем последовательных реакций, которые суммированы на схеме II и III.
Схема II
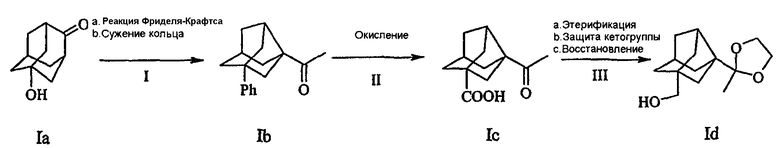
Схема II описана следующими стадиями, включающими в себя:
1а. Осуществление взаимодействия бензола и 5-гидроксиадамантаноном по реакции Фриделя-Крафтса в присутствии трифлатной кислоты в течение периода приблизительно 1-4 ч при температуре кипячения с обратным холодильником с получением 5-фениладамантанона.
1b. Сужение кольца адамантанона до норадамантанона происходило в 2 стадии, сначала происходило превращение в 2-метил-5-фениладамантан-2-ол по реакции Гриньяра, где реагент Гриньяра выбран из группы, включающей в себя метилметаллгалогениды, подобные CH3MgCl, CH3MgBr, CH3MgI с последующим сужением кольца в присутствии окисляющего агента в растворителе, подобном воде, ТГФ, бензолу, или сочетании указанных растворителей, с последующей обработкой основанием в протонном растворителе, таком как вода, спирты с образованием Ib. Предпочтительные спирты представляют собой С1-С4 спирты и основание выбирают из NaOH и КОН. Окисляющий агент можно выбрать из моноксида хлора, гипохлоритов, подобных NaOCl, или тетраацетата свинца, в присутствии иода.
II. Фенильную группу соединения Ib превращали в карбоксильную группу в присутствии окисляющего агента, подобного NaIO4/RuCl3 при температуре окружающей среды с образованием Ic.
III. Карбоксильную группу соединения Ic превращали в сложный алкиловый эфир, кетогруппу защищали 1,2-диолом, таким как 1,2-этандиол или 1,3-диолом, таким как 1,3-пропандиол, в присутствии органических растворителей, подобных бензолу или толуолу, при температуре кипячения с обратным холодильником, с применением кислотного катализатора, такого как р-TSA, CSA и эфират BF3. На следующей стадии сложный эфир восстанавливали в спирт восстанавливающим агентом, таким как LiAlH4, NaBH4 и DIBAl-H в инертном растворителе, подобном ТГФ, простому эфиру или их смесям при температуре приблизительно 0ºС с образованием Id.
Схема III
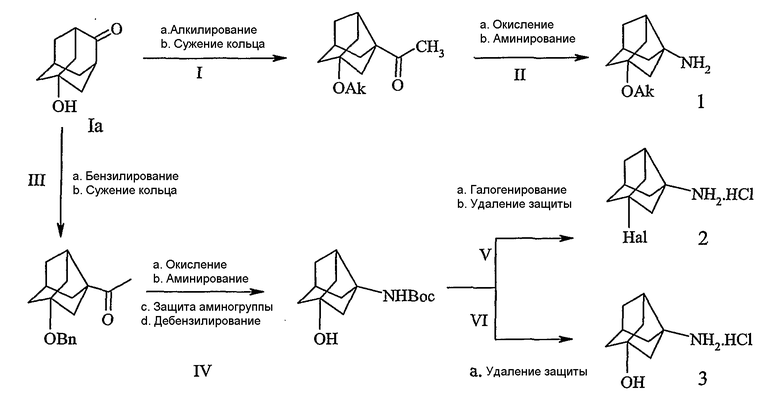
Схема III описана следующими стадиями:
Ia. Гидроксигруппу Ia в положении 5 алкилировали алкилирующим агентом, подобным алкилгалогенидам (таким, как CH3I, CH3Br или изопропилбромид) с применением сильных оснований, таких как NaH, KH или NaOCH3 с образованием 5-гидроксиадамантанона, с последующим сужением кольца.
Ib. 5-Алкоксиадамантанон превращали в 5-алкоксинорадамантилэтанон путем превращения его в 2-метил-5-алкоксиадамантан-2-ол по реакции Гриньяра с последующим сужением кольца адамантанона в норадамантан в присутствии окисляющего агента в растворителе, подобном воде, ТГФ, бензолу, или сочетании таких растворителей, с последующей обработкой основанием в протонном растворителе, таком как вода, спирты или их смеси, с образованием 5-алкоксинорадамантилэтанона.
IIa. Этаноновую группу в соединении, полученном на стадии 1, превращали в карбоновую кислоту обработкой окисляющим агентом, подобным гипобромитам, гипохлоритам (таким, как NaOBr, NaOCl), приблизительно при 0ºС с последующей стадией аминирования.
IIb. Кислоту превращали в аминную функцию обработкой азидом, подобным NaN3 или nBu4NN3 в кислотных условиях в присутствии растворителей, подобных CHCl3, CH2Cl2, CH3CN при температуре приблизительно от 35 до 50ºС с получением алкоксинорадамантиламинов (1).
IIIa. По другому пути синтеза гидроксильную группу соединения Ia бензилировали взаимодействием с бензилгалогенидами, подобно бензилбромиду, в растворителе (например, ТГФ, ДМФА, NMP) приблизительно при 0ºС в течение приблизительно 10-18 час с образованием бензилоксиадамантанона, с последующим сужением кольца с образованием бензилоксинорадамантилэтанона.
IV. Бензилоксинорадамантилэтанон, полученный на стадии III, превращали в соответствующую кислоту посредством окисляющего агента, с последующей стадией аминирования (b). Полученный таким образом амин защищали общепринятыми группами для защиты амина, подобными Boc, Cbz или Fmoc с последующим дебензилированием (d). Дебензилирование осуществляли в атмосфере H2 c катализатором Pd/C в протонных растворителях, подобных метанолу, этанолу или IPA при комнатной температуре в течение 1-3 ч с образованием гидроксинорадамантиламина, где аминогруппа защищена.
V. Галогенированием гидроксильной группы с последующим удалением защиты амина соединения, полученного на стадии IVd, получали галогензамещенный норадамантиламин в форме соли 2.
VI. Удаление защиты амина соединения, полученного на стадии IVd, получали гидроксизамещенный норадамантиламин в форме соли 3.
Полученные таким образом промежуточные соединения 1, 2, 3 подвергали реакции с хлорацетилцианопирролидинами с образованием соответствующих конечных соединений, в соответствии со схемой I.
Схема IV
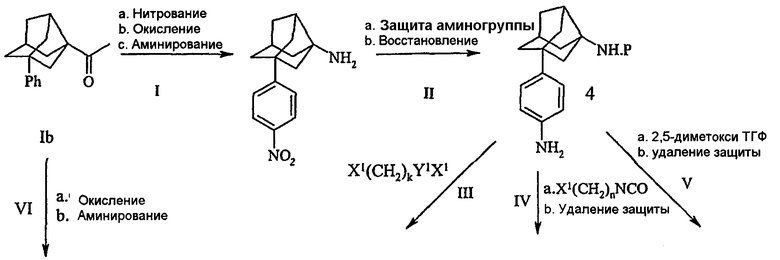
Х1 представляет собой галоген; Y1 представляет собой SO2, CO; P представляет собой Boc, CBz, Fmoc; k представляет собой 1, 2, 3.
Схема IV описана следующим образом.
Ia. Фенильную группу соединения Ib (полученного по схеме I) нитровали нитрующей смесью по общепринятой методике HNO3/H2SO4 приблизительно при 0ºС.
Ib. Нитрофенилнорадамантилэтанон, полученный на стадии Ia, окисляли для превращения этанона в соответствующую кислоту окисляющим агентом с последующей стадией аминирования.
Ic. Превращение кислоты в амин выполняли обработкой азидом, подобным NaN3 или n-BuNN3, в кислотных условиях в присутствии растворителей, подобных CHCl3, CH2Cl2 или ацетонитрилу при температуре приблизительно от 35 до 50ºС.
IIa. Аминогруппу в соединении, полученном на стадии I, защищали общепринятыми для амина защитными группами, подобными Boc, Cbz или Fmoc.
IIb. Нитрогруппу восстанавливали в амин восстанавливающими агентами, такими как Pd/C, Pd(OAc)2, Zn/формиат аммония или Fe/NH4Cl и т.д. в присутствии растворителей, таких как сложные эфиры (например, этилацетат), спирты (например, метанол, этанол), ТГФ, вода или их сочетание с образованием фениламиносоединения (4).
III. Фениламиносоединение (4), полученное на стадии II, подвергали взаимодействию с X1(CH2)nY1X1, где Х1 представляет собой галогенную группу, выбранную из F, Cl, Br, I, Y1 представляет собой SO2 или СО, в присутствии органического основания, подобного триэтиламину или DBU, и инертного растворителя, подобного ТГФ, CH2Cl2, ацетонитрилу или ДМФ с последующей циклизацией в присутствии гидроксидов щелочных металлов, таких как LiOH, NaOH или КОН в воде с межфазным катализатором, таким как тетраалкиламмонийгалогениды (например, тетрабутиламмонийиодид) и удалением защитной группы амина с получением соответствующего гетероциклилзамещенного фенилнорадамантиламинного соединения (5).
IV. В другом способе соединение (2), полученное на стадии II, подвергали взаимодействию с X1(CH2)NCO в присутствии органических оснований, подобных триэтиламину или DBU, растворителя, подобного ТГФ, CH2Cl2, ацетонитрилу, ДМФ или их смесям, с последующей циклизацией с получением соответствующего гетероциклилзамещенного соединения, которое путем дальнейшего удаления защиты амина образовало соответствующие гетероциклилзамещенные фенилнорадамантиламинные соединения (6).
V. В другом способе фениламиносоединение (2), полученное на стадии II, подвергали взаимодействию с 2,5-диметокситетрагидрофураном в ледяной уксусной кислоте при температуре кипячения с обратным холодильником с образованием замещенных пиррольных соединений, которые после удаления защиты амина образовали соответствующие пирролзамещенные фенилнорадамантиламинные соединения (7).
VIa. Этаноновую группу фенилнорадамантилэтанона (Ib) (полученного по схеме II) превращали в кислотную группу с помощью окисляющего агента с последующей стадией аминирования.
VIb. Превращением кислоты в амин с помощью азидов, как указано выше по тексту, получали соответствующие фенилзамещенные норадамантиламинные соединения (8).
Полученные промежуточные аминные соединения 4, 5, 6, 7, 8 подвергали взаимодействию с хлорацетилцианопирролидинами с образованием соответствующих конечных соединений, согласно схеме I.
Cхема V
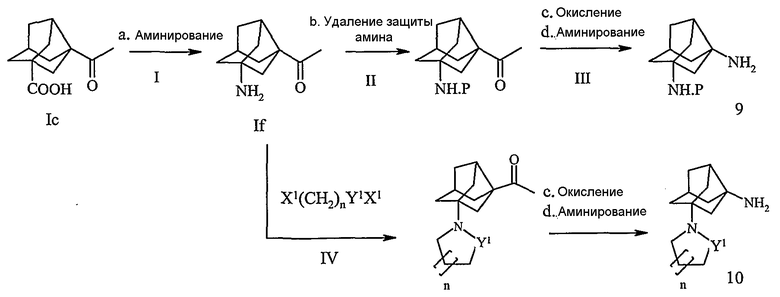
Схема V описана следующим образом:
Ia. Группу карбоновой кислоты соединения Ic (полученного по схеме II) превращали в амин обработкой азидом, подобным NaN3 в кислотной среде или дифенилфосфорилазидом в присутствии органических оснований, подобных триэтиламину, растворителей, подобных бензолу или толуолу при температуре кипячения с обратным холодильником.
IIb. Аминную группу соединения If, полученного на стадии I, защищали общепринятой для амина защитной группой, подобной Вос, Cbz или Fmoc, с последующей стадией окисления.
IIIc. Окисление соединения, полученного на стадии II, осуществляли окисляющими агентами с получением производного карбоновой кислоты.
IIId. Такую группу карбоновой кислоты соединения, полученного на стадии IIIc, превращали в амин обработкой азидом, например NaN3, в кислотной среде или дифенилфосфорилазидом в присутствии органических оснований, подобных триэтиламину, в растворителе, подобном бензолу или толуолу, при температуре кипячения с обратным холодильником, с последующим гидролизом гидроксидом металла, такого как КОН, NaOH, LiOH, в воде при комнатной температуре с образованием соответствующего замещенного амином норадамантиламина (9).
IV. Аминосоединение (If), полученное на стадии I, подвергали взаимодействию с Х1(CH2)nY1X1, где Х1 представляет собой галоген и Y1 представляет собой -SO2- или
-СО-, в присутствии органических оснований, подобных триэтиламину, и растворителя, подобного ТГФ или СН2Cl2, с последующей циклизацией с применением гидроксидов щелочных металлов, в воде, с межфазным катализатором, таким как тетрабутиламмонийиодид, с получением соответствующих гетероциклилзамещенных соединений с этаноновой группой, которую после стадий окисления и аминирования, как указано на схемах IIIc и IIId, превращали в соответствующие замещенные гетероциклом норадамантиламины (10).
Полученные аминные промежуточные соединения 9 и 10 подвергали взаимодействию с хлорацетилцианопирролидинами с образованием соответствующих конечных соединений, в соответствии со схемой I.
Схема VI:

Схема VI описана следующим образом:
I. Для получения соединения Ie гидроксильную группу
соединения Id превращали в удаляемую группу L, например, мезилированием, толизированием или галогенированием в присутствии органического основания, подобного триэтиламину, N,N-диизопропилэтиламину, пиридину, NMP или N-метилморфолину при температуре приблизительно 0ºС.
II. Отщепляемую группу L Ie замещали группой R11 в присутствии основания и растворителя, при температуре приблизительно 80-120ºС с образованием Ii. Основания выбирали из карбонатов или бикарбонатов щелочных металлов, подобных Na2CO3, NaHCO3, K2CO3 или KHCO3. Растворители выбирали из диметилформамида, ДМСО, NMP или подобного растворителя.
IIIa. Удаляли защитную группу у защищенной кетогруппы соединения Ii, полученного на стадии II, например, взаимодействием с p-TSA в ацетоне при температуре кипячения с обратным холодильником, с образованием этанонового соединения.
IIIb. Этаноновое соединение окисляли в соответствующую карбоновую кислоту в присутствии окисляющего агента, который можно выбрать из моноксида хлора, гипохлоритов, подобных NaOCl или тетраацетата свинца, в присутствии иода.
IIIc. Карбоновую кислоту превращали в амин взаимодействием с азидом, подобным NaN3 в кислотных условиях, в присутствии растворителей, подобных CHCl3, CH2Cl2 или ацетонитрилу, приблизительно при 35-45ºС с образованием замещенного R11 норадамантиламина (11).
IV. В другом способе гидроксильную группу Id, которую получали по схеме II, бензилировали взаимодействием с бензилгалогенидом, подобным бензилбромиду, в растворителях, подобных ТГФ, ДМФА или NMP приблизительно при 0ºС в течение приблизительно 10-18 ч с образованием имеющего защищенную кетогруппу бензилоксинорадамантана (Ig).
Va. У соединения Ig удаляли защиту кетогруппы с получением этанона, как описано в стадии IIIa.
Vb. Этаноновую группу бензилоксинорадамантилэтанонового соединения окисляли с получением карбоновой кислоты, как описано на стадии IIIb.
Vc. Карбоксильную группу соединения, полученного на стадии Vb, превращали в амин взаимодействием с дифенилфосфорилазидом, в присутствии органического основания, подобного триэтиламину, растворителях, подобных бензолу или толуолу, при температуре кипячения с обратным холодильником, с последующим гидролизом гидроксидами металлов, таких как КОН, NaOH или LiOH в воде при комнатной температуре с образованием бензилоксинорадамантиламина (Ih).
Via. Аминогруппу соединения Ih, полученного на стадии Vc, защищали общепринятыми для амина защитными группами, например, такими, как описано ранее, с последующей стадией дебензилирования.
VIb. Дебензилирование осуществляли в атмосфере Н2 с Pd/C в протонных растворителях, подобных метанолу, этанолу или IPA, при комнатной температуре в течение 1-3 ч с образованием соединения Ij.
VIIa. Гидроксильную группу соединения Ij мезилировали, тозилировали или галогенировали с образованием удаляемой группы, в присутствии органических оснований, подобных триэтиламину, N,N-диизопропилэтиламину, пиридину, N-метилпиперидину или N-метилморфолину при температуре окружающей среды.
VIIb. Cоединение, полученное на стадии VIIa, подвергали взаимодействию с R11 группой в присутствии основания и растворителя при температуре в интервале приблизительно 80-120ºС. Основания выбирали из карбонатов или бикарбонатов щелочных металлов, подобных Na2CO3, NaHCO3, K2CO3 и KHCO3. Растворители выбирали из ДМФА, ДМСО, NMP и других.
VIIc. У защищенной аминогруппы замещенного R11 соединения, полученного на стадии VIIb, удаляли защиту общепринятыми способами, например обработкой сухим HCl в растворителях, подобных EtOAc, простому эфиру или 1,4-диоксана при температуре от 0ºС до комнатной температуры с образованием R11-замещенного норадамантиламина в виде хлористоводородной соли (12).
VIIIa. В другом способе гидроксильную группу Ij подвергали взаимодействию с R11-группой в присутствии трифенилфосфина, диизопропилазодикарбоксилата, и органического растворителя, подобного бензолу, толуолу или ТГФ, при температуре приблизительно от 20ºС приблизительно до 110ºС в течение приблизительно 2-6 часов с образованием R11-замещенного соединения.
VIIIb. Защитную группу аминогруппы R11-замещенного соединения, полученного на стадии VIIIa, удаляли обработкой трифторуксусной кислотой в растворителе, подобном CH2Cl2 или СНCl3 приблизительно при 0ºС с образованием R11-замещенного норадамантанамина, как, например, соль с TFA (13). Полученные промежуточные соединения 11, 12 и 13 подвергали взаимодействию с хлорацетилцианопирролидинами с образованием соответствующих конечных соединений, в соответствии со схемой I.
Схема VII:
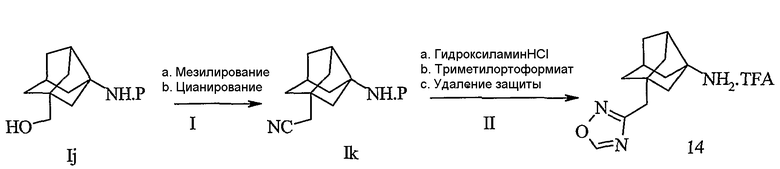
Схема VII описана следующим образом.
Ia. Для образования удаляемой группы L гидроксигруппу соединения Ij мезилировали, тозилировали или галогенировали в присутствии органических оснований, подобных триэтиламину, N,N-диизопропилэтиламину, пиридину, N-метилпиперидину или N-метилморфолину при температуре окружающей среды. Соединение, полученное из Ia, подвергали взаимодействию с цианирующим агентом, подобным NaCN, в присутствии апротонного растворителя, подобного ДМФА, при 100-110ºС в течение приблизительно 12-15 ч с получением соединения (Ik).
IIa. Соединение Ik подвергали взаимодействию с гидрохлоридом гидроксиламина с образованием амидоксима, который затем подвергали взаимодействию с триметилортоформиатом в присутствии каталитического количества камфорсульфокислоты. В конце после удаления защиты получали соединение 14.
Полученное промежуточное соединение 14 подвергали взаимодействию с хлорацетилцианопирролидинами с образованием соответствующих конечных соединений, в соответствии со схемой I.
Понятно, что в любой из вышеуказанных схем любую реакционноспособную группу в молекуле субстрата можно защитить в соответствии с любой общепринятой методикой, известной в предшествующем уровне техники. Подходящие защитные группы включают в себя тетраарилбутилдиметилсилил, метоксиметил, трифенилметил, бензилоксикарбонил, ТНР и т.д. для защиты гидроксильных или фенольных гидроксигрупп; N-Вос, N-Cbz, N-Fmoc и бензофенонимин для защиты амино или анилиногрупп; ацетальную защиту для альдегидов, кетальную защиту для кетонов. Способы образования и удаления таких защитных групп, которые известны в области техники, зависят от молекулы, которую защищают.
Также частью изобретения является то, что при осуществлении изобретения, везде, где присутствует отщепляемая группа, ее можно выбрать из группы, включающей в себя атомы галогенов (подобных хлору, брому), о-толуолсульфонил, о-метилсульфонил.
Стереоизомеры соединений, в соответствии с данным изобретением, можно получать с применением реагентов в отдельной энантиомерной форме в способе, везде, где возможно, или выполнением реакции в присутствии реагентов или катализаторов в форме отдельного энантиомера, или разделением смеси стереоизомеров общепринятыми способами. Некоторые из предпочтительных способов включают в себя применение микробного разделения, разделения диастереомерных солей, образованных с хиральными кислотами, такими как манделовая кислота, камфорсульфоновая кислота, винная кислота или молочная кислота, везде, где применимо, или хиральными основаниями, такими как бруцин, хининовые алкалоиды или их производные.
Фармацевтически приемлемые соли можно получать взаимодействием соединения формулы I приблизительно с 1-5 эквивалентами оснований, таких как гидроксиды щелочных металлов, алкоксиды щелочных металлов, гидроксид кальция или гидроксид магния, в протонных или апротонных растворителях, подобных метанолу, этанолу, пропанолу, IPA, простому эфиру, ТГФ, диоксану и т.д. В альтернативном случае, везде, где применимо, аддитивные соли с кислотой получали обработкой кислотами, такими как галогенводородные кислоты, подобные HCl, HBr; азотная кислота, серная кислота, фосфорная кислота, п-толуолсульфоновая кислота, метансульфоновая кислота, уксусная кислота или лимонная кислота, в растворителях, включающих в себя по меньшей мере один, выбранный из этилацетата, простых эфиров, спиртов, ацетона, ТГФ, диоксана или их смесей.
Различные полиморфы соединения общей формулы I настоящего изобретения можно получать кристаллизацией соединения формулы I при различных условиях. Например, с применением обычно применяемых растворителей или их смесей для перекристаллизации, кристаллизации при различных температурных интервалах, различных методов охлаждения, таких как от очень быстрого до очень медленного охлаждения во время кристаллизации, выдерживанием соединения при температуре окружающей среды, нагреванием или плавлением его с последующим постепенным охлаждением и т.д. Присутствие полиморфов можно определить одним или несколькими методами, подобными ЯМР-спектроскопии с твердым зондом, DSC, TGA, рентгенографии или ИК-спектроскопии.
В другом варианте осуществления настоящего изобретения соединения можно очищать с применением таких методов, как кристаллизация из растворителей, таких как пентан, простой диэтиловый эфир, простой изопропиловый эфир, хлороформ, дихлорметан, этилацетат, ацетон, метанол, этанол, изопропанол, вода или их сочетания, или соединение I можно очищать колоночной хроматографией с применением оксида алюминия или силикагеля и элюированием колонки такими растворителями, как гексан, петролейный эфир, дихлорметан, хлороформ, этилацетат, ацетон, метанол или их сочетания.
Настоящее изобретение также предлагает фармацевтические композиции, содержащие соединения, как указано выше, их производные, аналоги, таутомерные формы, стереоизомеры, биоизостеры, полиморфы, энантиомеры, диастереомеры, фармацевтически приемлемые соли или фармацевтически приемлемые сольваты в сочетании с подходящими фармацевтически приемлемыми носителями или разбавителями. Фармацевтические композиции в соответствии с настоящим изобретением применимы в качестве антидиабетических средств, гиполипидемических средств или антигиперхолестеринемических средств.
Подходящие фармацевтически приемлемые носители включают в себя твердые наполнители, разбавители и стерильные водные или органические растворы. Активный ингредиент будет присутствовать в таких фармацевтических композициях в количествах, достаточных для обеспечения требуемого эффекта, как указано выше по тексту. Поэтому для перорального введения соединения можно объединять с подходящим твердым или жидким носителем или разбавителем для образования, например, капсул, таблеток, порошков, сиропов, растворов или суспензий. Фармацевтические композиции могут, если требуется, содержать дополнительные компоненты, такие как корригенты, подсластители или эксципиенты.
Путем введения может быть пероральный, назальный, легочный, трансбуккальный, подкожный, внутрикожный, чрескожный или парентеральный, например ректальный, депо, подкожный, внутривенный, внутриуретральный, внутримышечный, интраназальный, в виде глазного раствора или мази, которые эффективно транспортируют активное соединение настоящего изобретения, которое ингибирует ферментативную активность DPPIV в соответствующем или требуемом месте действия. Для перорального введения, если применяют твердый носитель, препарат может быть в форме таблетки, или может быть помещен в твердую желатиновую капсулу в форме порошка или гранулы, или он может быть в форме пастилки или леденца. Если применяют жидкий носитель, то препарат может быть в форме сиропа, эмульсии, твердой желатиновой капсулы или стерильной инъецируемой жидкости, такой как водная или неводная жидкая суспензия или раствор. Для назального введения применяют в качестве аэрозольной аппликации жидкий носитель, конкретно водный носитель. Для парентеральной аппликации подходящие композиции представляют собой инъецируемые растворы или суспензии, предпочтительно водные растворы.
Дополнительный аспект настоящего изобретения представляет собой применение соединений изобретения в качестве фармацевтической композиции в терапевтически эффективном количестве для лечения метаболических нарушений, снижения уровня глюкозы в крови, для лечения диабета типа II, для лечения ухудшения переносимости глюкозы, для лечения снижения уровня глюкозы натощак, для лечения ожирения, для предупреждения гипергликемии, для лечения дислипидемии, гиперхолестеринемии, гиполипидемии или для уменьшения массы тела или диабетических осложнений, включающих заболевание коронарной артерии, инсульт, гипертензию, нефропатию, заболевание периферических сосудов, нейропатию или ретинопатию.
Соединения настоящего изобретения являются эффективными во всем широком интервале доз. Например, при лечении людей дозы могут быть приблизительно от 0,05 до 1000 мг и предпочтительно приблизительно от 0,1 до 500 мг в день. Точная доза зависит от способа введения, от терапевтических требований, формы, в которой вводят активный ингредиент, и пациента, подвергаемого лечению, массы тела субъекта для лечения и предпочтения и квалификации врача в палате. Здесь в качестве субъекта рассматривают человека.
Изобретение также включает в себя пролекарственные производные соединений изобретения, которые после введения подвергаются химическому превращению посредством метаболических процессов перед превращением в активные фармакологические вещества. Обычно такие пролекарственные производные будут представлять собой функциональные производные соединений изобретения, которое легко превращается in vivo в соединения изобретения.
Изобретение также включает в себя активные метаболиты соединений настоящего изобретения.
Испытание активности дипептидилпептидазы IV
Ингибирование протеолитической активности DPP-IV испытывали с помощью гидролиза Ala-Pro-7-амино-4-трифторметилкумарина (Ala-Pro-AFC) и последующего флуориметрического количественного определения выделенного AFC. Для испытания применяли человеческий рекомбинантный DPP-IV (экспрессированный в клетках Sf9 насекомых). Тестируемые соединения растворяли в диметилсульфоксиде (ДМСО). Обычно фермент (приблизительно 20 нг/мл в 100 мМ буфера Tris-HCl, рН 8,0) предварительно инкубировали в отсутствии (1% ДМСО) и в присутствии различных концентраций тестируемых соединений в течение 15 мин при 37ºС. Реакцию инициировали добавлением 20 мкМ Ala-Pro-AFC и дополнительно инкубировали в течение 30 мин при 37ºС. Выделяемое количество AFC измеряли спектрофотометром с длинами волн возбуждения и испускания при 400 нм и 510 нм соответственно. Результаты выражали в процентах ингибирования активности фермента. В исследование всегда включали ссылочный стандарт (известный ингибитор DPP-IV).
Обнаружено, что соединения настоящего изобретения ингибируют индуцированную DPPIV флуоресценцию с константами ингибирования в интервале приблизительно от 0,5 нМ до 500 нМ. В предпочтительной области, соединения настоящего изобретения ингибировали индуцируемую DPPIV флуоресценцию с константой ингибирования приблизительно от 0,1 нМ до 300 нМ и в более предпочтительной области соединения настоящего изобретения ингибировали индуцированную DPPIV флуоресценцию с константами ингибирования приблизительно от 1 нМ до 120 нМ.
Как показано в таблице ниже по тексту, соединения примеров вызывают сильное ингибирование DPP-IV.
Следующие примеры предоставляют возможность специалисту в области техники для практического осуществления изобретения и представляют собой только иллюстрацию изобретения, но не ограничивают объем изобретения.
Получение промежуточных соединений
Препарат 1
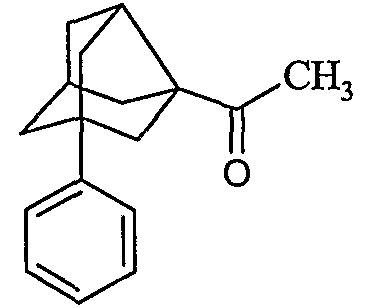
1-(1-Фенилтрицикло[3,3,1,03,7]нон-3-ил)этанон
Стадия I: Адамантанон (12 г, 80 ммоль) добавляли при перемешивании к азотной кислоте (98%, 100 мл) при температуре бани со льдом в течение 15 минут. Реакционную смесь перемешивали при комнатной температуре в течение 72 ч и затем нагревали до 60ºС в течение 2 ч до тех пор, пока не испарится большая часть диоксида азота. Избыток азотной кислоты отгоняли при пониженном давлении. Легкое желтое масло затвердевало после охлаждения (аддукт NO3 с гидроксикетоном). Добавляли воду (40 мл) и конц. H2SO4 (98%, 15 мл). Полученный прозрачный желтый раствор нагревали на паровой бане в вытяжном шкафу (в токе азота) в течение 1 ч. Затем раствор охлаждали и экстрагировали смесью 2:1 н-гексана и простого диэтилового эфира для удаления непрореагировавшего адамантанона (1,0 г). Кислотный слой нейтрализовали 30% вод. раствором NaOH и пока раствор был теплым, экстрагировали хлороформом. Экстракты объединяли, промывали насыщенным раствором соли и концентрировали в вакууме. Неочищенный продукт растворяли в CH2Cl2 (15 мл) и добавляли гексан до прекращения образования осадка. Твердое вещество выделяли фильтрованием и сушили с получением 5-гидроксиадамантан-2-она. Выход: 9,0 г (70%). Твердое вещество. Т.пл.: 278,8-300ºС (разложение) m/z (M+1) 167; 1Н ЯМР (CDCl3) 300 МГц δ 2,70-2,55 (м, 2Н), 2,36-2,32 (м, 1Н), 2,11-1,93 (м, 10Н). 13С ЯМР (CDCl3) 75 МГц δ 217,0, 66,7, 46,7, 46,6, 44,7 (2С), 43,8, 37,9 (2С), 29,5.
Стадия II: К перемешиваемому раствору соединения 5-гидроксиадамантан-2-она (10,0 г, 60,2 ммоль) в бензоле (180 мл) добавляли трифторметансульфоновую кислоту (5,3 мл, 60,2 ммоль) в течение периода 30 минут при комнатной температуре. После перемешивания реакционной смеси в течение 5 минут при комн. темп. смесь кипятили с обратным холодильником в течение 4 ч. Реакционную смесь охлаждали до 0º и добавляли насыщенный водный раствор NaHCO3 (76 мл) в течение периода 30 минут. Два слоя разделяли, водный слой экстрагировали простым эфиром и объединенный слой промывали водой и насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением 5-фениладамантан-2-она (10,5 г) в виде белого твердого вещества с выходом 80%. Т.пл.: 53,8-60,9ºС. m/z (M+1) 227; ИК (см-1): 2921, 2853, 1717, 1446, 1060, 758, 698. 1Н ЯМР (CDCl3) 300 МГц δ 7,37-7,30 (м, 4Н), 7,26-7,19 (м, 1Н), 2,70-2,63 (м, 2Н), 2,30-2,0 (м, 11Н). 13С ЯМР (CDCl3) 75 МГц δ 217,8, 147,8, 128,3 (2С), 126,1, 124,6 (2С), 46,9 (2С), 44,3 (2С), 41,9, 35,9, 38,4 (2С), 28,0.
Стадия III: Только что полученный метилмагнийиодид в простом эфире (1 М, 85 мл) добавляли через трубку к 5-фениладамантан-2-ону (9,6 г, 42,5 ммоль), полученному на стадии II, в ТГФ (85 мл) при 0ºС. После перемешивания при 0ºС в течение 0,5 ч реакционную смесь гасили добавлением насыщенного водного раствора NH4Cl. Органический слой отделяли и водный слой экстрагировали простым диизопропиловым эфиром. Объединенные органические слои промывали водой и насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением 2-метил-5-фениладамантан-2-ола (9,9 г) в виде не совсем белого твердого вещества с выходом 97%. Т.пл.: 98-100,4ºС. m/z (M+23) 265; 1Н ЯМР (CDCl3) 300 МГц δ 7,42-7,28 (м, 4Н), 7,24-7,18 (м, 1Н), 2,47-2,26 (м, 2Н), 2,14-2,09 (м, 3Н), 1,96-1,70 (м, 6Н), 1,61-1,57 (м, 2Н), 1,42 (с, 3Н). 13С ЯМР (CDCl3) 75 МГц δ 150,0, 128,1 (2С), 125,7, 124,8 (2С), 73,3, 44,0 (2С), 40,5 (2С), 39,6 (2С), 35,9, 32,0, 27,6.
Стадия IV: 2-Метил-5-фениладамантан-2-ол (20 г, 82,6 ммоль), полученный на стадии III (86 г, 355,4 ммоль), растворенный в смеси АсОН (76,3 мл) и ТГФ (360 мл), добавляли по каплям через присоединенную воронку к охлажденному на ледяной бане раствору NaOCl (4%, 3,5 л) в течение периода 15 минут. Добавляли твердый n-Bu4NI (13,1 г, 35,6 ммоль) и реакционную смесь перемешивали в течение 1,5 ч. Два слоя разделяли, водный слой экстрагировали простым диизопропиловым эфиром и объединенный органический слой промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Остаток растворяли в метаноле (165 мл), добавляли КОН (39,8 г, 300 ммоль) и смесь кипятили с обратным холодильником в течение 1 ч. Растворитель выпаривали при пониженном давлении и неочищенный продукт очищали колоночной хроматографией с получением соединения 1-(1-фенилтрицикло[3,3,1,03,7]нон-3-ил)этанон (50,0 г) с выходом 59% в виде вязкой жидкости. M/z (M+1) 241; ИК (см-1): 2924, 2867, 1697, 1445, 1356, 1223, 757, 699. 1Н ЯМР (CDCl3) 300 МГц δ 7,38-7,19 (м, 5Н), 2,86-2,80 (м, 1Н), 2,59-2,50 (м, 1Н), 2,32-2,25 (м, 1Н), 2,23 (с, 3Н), 2,10-1,99 (м, 4Н), 1,90-1,79 (м, 4Н), 1,78-1,71 (м, 1Н). 13С ЯМР (CDCl3) 75 МГц δ 211,7, 146,7, 128,2 (2С), 125,7 (2С), 124,7, 61,9, 50,2, 49,0, 47,9, 45,7, 42,4, 42,3, 37,6, 26,4.
Препарат 2
[3-(2-Метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метилметансульфонат
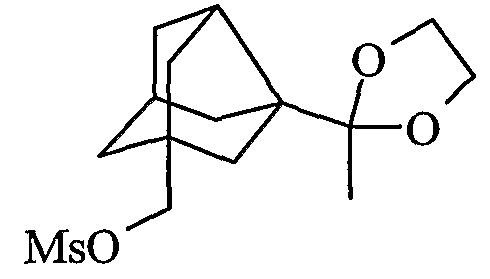
Стадия I: К перемешиваемой смеси 1-(фенилтрицикло[3,3,1,03,7]нон-3-ил)этанона (8,0 г, 33,3 ммоль), который получен, как указано ранее (препарат I), четыреххлористого углерода (66 мл), ацетонитрила (66 мл) и воды (100 мл), охлажденной до 0ºС, добавляли периодат натрия (31,9 г, 149 ммоль) и гидрат хлорида рутения (III) (0,44 г, 1,7 ммоль). Реакционную смесь постепенно нагревали до окружающей температуры и перемешивали в течение 2 ч. Реакционную смесь разбавляли простым диизопропиловым эфиром (100 мл) и перемешивали в течение 15 мин до осаждения черного вещества RuO2. Затем реакционную смесь фильтровали через слой целита и органический слой экстрагировали 1н раствором NaOH (3×25 мл). Органический слой сушили над Na2SO4 и растворитель выпаривали в вакууме с получением непрореагировавшего исходного вещества (3,04 г, 12,67 ммоль). Водный слой подкисляли конц. НСl и экстрагировали EtOAc. Объединенные органические слои промывали водой, насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 3-ацетилтрицикло[3,3,1,03,7]нонан-1-карбоновой кислоты (4,0 г) в виде не совсем белого твердого вещества с выходом 57%. Т.пл.: 90-95,0ºС. m/z (M+1) 209. ИК см-1 2935, 1694, 1413, 1357, 974, 746. 1Н ЯМР (CDCl3) 300 МГц, δ 2,77-2,73 (м, 1Н), 2,54-2,48 (м, 1Н), 2,44-2,34 (м, 1Н), 2,20 (с, 3Н), 2,18-2,09 (м, 1Н), 7,38-7,19 (м, 5Н), 2,86-2,80 (м, 1Н), 2,59-2,50 (м, 1Н), 2,32-2,25 (м, 1Н), 2,23 (с, 3Н), 2,18-2,09 (м, 1Н), 2,06-1,74 (м, 7Н), 1,73-1,61 (м, 1Н). 13С ЯМР (CDCl3) 75 МГц δ 211,0, 181,3, 61,5, 50,3, 47,9, 45,9, 45,6, 42,6, 41,9, 36,8, 35,6, 26,4.
Стадия II: К 3-ацетилтрицикло[3,3,1,03,7]нонан-1-карбоновой кислоте (2,4 г, 11,4 ммоль), полученной на стадии I, в МеОН (48 мл), охлажденной до температуры бани со льдом, добавляли ацетилхлорид (1,64 мл, 22,8 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 2 ч. Летучие продукты удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением метил-3-ацетилтрицикло[3,3,1,03,7]нонан-1-карбоксилата (2,4 г) с выходом 93% в виде вязкой жидкости. M/z (M+1) 223. ИК см-1 2953, 1728, 1698, 1461, 1234, 1078, 755. 1Н ЯМР (CDCl3) 300 МГц δ 3,66 (с, 3Н), 2,75-2,70 (м, 1Н), 2,53-2,46 (м, 1Н), 2,40-2,32 (м, 1Н), 2,19 (с, 3Н), 2,15-2,05 (м, 1Н), 2,03-1,60 (м, 8Н). 13С ЯМР (CDCl3) 75 МГц δ 210,9, 175,3, 61,4, 53,4, 51,6, 50,5, 48,0, 45,9, 45,6, 42,6, 41,9, 36,8, 35,9, 26,4.
Стадия III: Смесь метил-3-ацетилтрицикло[3,3,1,03,7]нонан-1-карбоксилата (2,0 г, 8,9 ммоль), полученного на стадии II, 1,2-этандиола (8,9 мл), р-TSA (47 мг, 5 мол.%) и бензола (36 мл) кипятили с обратным холодильником с применением устройства Дина-Старка в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли 10% вод. NaHCO3 (36 мл) и разделяли два слоя. Водный слой экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель выпаривали при пониженном давлении с получением метил-3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нонан-1-карбоксилата (2,2 г) с выходом 93% в виде вязкой жидкости. M/z (M+1) 267. ИК см-1 2954, 1731, 1698, 1460, 1236, 1046, 752. 1Н ЯМР (CDCl3) 300 МГц δ 4,07-3,94 (м, 4Н), 3,66 (с, 3Н), 2,44-2,36 (м, 2Н), 2,27-2,18 (м, 1Н), 2,12-2,02 (м, 1Н), 1,94-1,81 (м, 8Н), 1,30 (с, 3Н).
Стадия IV: Метил-3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нонан-1-карбоксилат (2,3 г, 8,64 ммоль), полученный на стадии III, в ТГФ (15 мл) медленно добавляли в атмосфере N2 через капельную воронку к суспензии LiAlH4 (0,32 г, 8,64 ммоль) в простом эфире (15 мл) при температуре бани со льдом. Реакционную смесь перемешивали в течение 30 минут перед гашением реакции добавлением насыщенного водного раствора NH4Cl (9 мл), за которым следовал 1н раствор NaOH (9 мл), и реакционную смесь перемешивали при комнатной температуре в течение 15 мин перед фильтрованием смеси через слой целита. Органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель выпаривали при пониженном давлении с получением [3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метанола (1,9 г) с выходом 94% в виде вязкой жидкости. M/z (М+1) 269; ИК см-1 3436, 2936, 1634, 1459, 1373, 1309, 1047, 757. 1Н ЯМР (CDCl3) 300 МГц δ 4,15-3,80 (м, 4Н), 3,53 (с, 3Н), 2,50-2,30 (м, 2Н), 1,90-1,30 (м, 10Н), 1,29 (с, 3Н).
Стадия V: К раствору [3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метанола (2,0 г, 8,47 ммоль), полученного на стадии IV, в ТГФ (35 мл) при температуре бани со льдом, последовательно добавляли триэтиламин (3,5 мл, 25,4 ммоль), DMAP (52 мг, 0,42 ммоль) и метансульфонилхлорид (0,97 мл, 12,7 ммоль). После перемешивания реакционной смеси при такой же температуре в течение 0,5 ч, смесь разбавляли водой и экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением названного соединения [3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метилметансульфоната (2,4 г) с выходом 90% в виде вязкой жидкости. m/z (M+1) 317. ИК см-1 2954, 1461, 1356, 1216, 1174, 1048, 756.
1 ЯМР (CDCl3) 300 МГц δ 4,15-3,90 (м, 4Н), 3,00 (с, 3Н), 2,44-2,36 (м, 2Н), 1,89-1,79 (м, 2Н), 1,78-1,42 (м, 8Н), 1,29 (с, 3Н). 13С ЯМР (CDCl3) 75 МГц δ 112,1, 76,3, 64,95, 64,9, 57,3, 46,6, 45,3, 44,9, 44,5, 42,9, 39,8, 36,9, 36,7, 36,3, 20.
Препарат 3
{3-[(трет-бутоксикарбонил)амино]трицикло[3,3,1,03,7]нон-1-ил}метилметансульфонат
Стадия I: К суспензии NaH (60% дисперсия в медицинском масле, 3,36 г, 21 ммоль) в ТГФ (84 мл), охлажденной до температуры бани со льдом, добавляли раствор [3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метанола (10,0 г, 42,0 ммоль) (который получали как указано ранее (препарат 2), на стадии IV) в ТГФ (84 мл) через шприц в течение периода 30 минут. После перемешивания реакционной смеси в течение 30 минут при комнатной температуре, последовательно добавляли nBu4NI (0,37 г, 1,0 ммоль); с последующим добавлением бензилбромида (5,0 мл, 42,0 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч до обнаруживаемого методом ТСХ завершения реакции. После охлаждения реакционной смеси до температуры бани со льдом избыток NaH гасили добавлением насыщенного водного раствора NH4Cl. Два слоя разделяли и водный слой экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением неочищенной массы вещества, которое очищали колоночной хроматографией с получением 2-[1-(бензилоксиметил)трицикло[3,3,1,03,7]нон-3-ил]-2-метил-1,3-диоксолана (11,0 г) в виде вязкой жидкости с выходом 80%. м/z (М+1) 329; 1Н ЯМР (CDCl3) 300 МГц δ 7,38-7,22 (м, 5Н), 4,50 (с, 2Н), 4,02-3,92 (м, 4Н), 3,24 (с, 2Н), 2,38-2,30 (м, 2Н), 1,88-1,71 (м, 2Н), 1,70-1,60 (м, 4Н), 1,55-1,39 (м, 4Н), 1,27 (с, 3Н).
Стадия II: Смесь 2-[1-(бензилоксиметил)трицикло[3,3,1,03,7]нон-3-ил]-2-метил-1,3-диоксолана (2,6 г, 7,9 ммоль), п-толуолсульфоновой кислоты (0,3 г, 1,6 ммоль) и ацетона (31,6 мл) кипятили с обратным холодильником в течение 4 ч. Летучие вещества удаляли в вакууме и остаток разбавляли EtOAc, промывали 10% вод. NaHCO3 и насыщенным раствором соли, сушили над Na2SO4 и растворитель выпаривали при пониженном давлении с получением 1-{1-[(бензилокси)метил]трицикло[3,3,1,03,7]нон-3-ил}этанона (2,1 г) в виде вязкой жидкости с выходом 93%. м/z (M+1) 285; 1Н ЯМР (CDCl3) 300 МГц δ 7,40-7,22 (м, 5Н), 4,50 (с, 2Н), 3,27 (с, 2Н), 2,70-2,62 (м, 1Н), 2,47-2,40 (м, 1Н), 2,16 (с, 2Н), 2,0-1,90 (м, 2Н), 1,81-1,56 (м, 7Н), 1,50 (дд, J=3,0, 11,0 Гц, 1Н).
Стадия III: К смеси NaOH (30,6 г, 765 ммоль), Н2О (255 мл) и 1,4-диоксана (51 мл) при температуре бани со льдом добавляли Br2 (15,2 мл, 285,6 ммоль) и перемешивали в течение 15 минут. Такой раствор гипобромита добавляли по каплям к перемешиваемому раствору 1-{1-[(бензилокси)метил]трицикло[3,3,1,03,7]нон-3-ил}этанона (14,5 г, 51,0 ммоль) в 1,4-диоксане (51 мл) при температуре бани со льдом. Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 1 ч. Реакционную смесь охлаждали до температуры бани со льдом и гасили добавлением АсОН (46,7 мл, 765 ммоль). Реакционную смесь разбавляли водой, экстрагировали EtOAc, объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4, растворитель удаляли при пониженном давлении с получением 1-{1-[(бензилокси)метил]трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (8,0 г) в виде белого твердого вещества с выходом 55%. m/z (М-1), 285; 1Н ЯМР (CDCl3) 300 МГц δ 7,40-7,22 (м, 5Н), 4,50 (с, 2Н), 3,27 (с, 2Н), 2,70-2,62 (м, 1Н), 3,47-3,38 (м, 1Н), 2,16 (с, 2Н), 2,12-1,98 (м, 2Н), 1,90-1,55 (м, 7Н), 1,50 (дд, J=3,0, 11,0 Гц, 1Н).
Стадия IV: К перемешиваемой смеси 1-[(бензилокси)метил]трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (1,8 г, 6,3 ммоль), полученной на стадии III, триэтиламина (2,6 мл, 18,9 ммоль) и толуола (25 мл) при температуре бани со льдом добавляли дифенилфосфорилазид (1,5 мл, 6,93 ммоль). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение одного часа и затем кипятили с обратным холодильником в течение 4 ч. После завершения реакции реакционную смесь переносили в делительную воронку и промывали водой. Органический слой перемешивали с вод. раствором КОН (50% масс./об., 12,6 мл) и nBu4NI (120 мг, 0,32 ммоль) в течение 2 ч. Реакционную смесь охлаждали до температуры бани со льдом, подкисляли 2н KHSO4 до рН 2, экстрагировали простым эфиром, водный слой подщелачивали водным раствором NaOH (50% масс./об.) и экстрагировали хлороформом. Объединенные органические слои сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением 1-[(бензилокси)метил]трицикло[3,3,1,03,7]нонан-3-амина (0,82 г) в виде вязкой жидкости с выходом 51%. m/z (М+1) 258; 1Н ЯМР (CDCl3) 300 МГц δ 7,37-7,24 (м, 5Н), 4,50 (с, 2Н), 3,24 (с, 2Н), 2,37-2,29 (м, 1Н), 2,0-1,81 (м, 3Н), 1,78-1,70 (м, 1Н), 1,70-1,45 (м, 7Н), 1,40 (дд, J=2,9, 10,7 Гц, 1Н).
Стадия V: К перемешиваемому раствору 1-[(бензилокси)метил]трицикло[3,3,1,03,7]нонан-3-амина (0,82 г, 3,2 ммоль), полученного на стадии IV, в дихлорметане (13 мл) при температуре бани со льдом добавляли Et3N (0,67 мл, 4,8 ммоль) и ди-трет-бутилдикарбонат (0,77 г, 3,5 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 1 ч летучие вещества удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением трет-бутил[1-(бензилоксиметил)трицикло[3,3,1,03,7]нон-3-ил]карбамата в виде вязкой жидкости (1,0 г) с выходом 88%. m/z (М+1) 358; 1Н ЯМР (CDCl3) 300 МГц δ 7,37-7,24 (м, 5Н), 4,49 (с, 2Н), 3,25 (с, 2Н), 2,48-2,38 (м, 1Н), 2,37-2,30 (м, 1Н), 2,10-1,87 (м, 2Н), 1,82-1,67 (м, 2Н), 1,67-1,39 (м, 6Н), 1,44 (с, 9Н).
Стадия VI: К перемешиваемой смеси трет-бутил[1-(бензилоксиметил)трицикло[3,3,1,03,7]нон-3-ил]карбамата (1,0 г, 2,8 ммоль), полученного на стадии V, в МеОН (11 мл), добавляли Pd/C (10%, 0,2 г). Затем подавали из баллона под давлением Н2 в течение 2 ч. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением трет-бутил[1-(гидроксиметил)трицикло[3,3,1,03,7]нон-3-ил]карбамата в виде вязкой жидкости (0,7 г) с выходом 95%. m/z (M+1) 268; 1Н ЯМР (CDCl3) 300 МГц δ 4,74 (с (ушир), 1Н), 3,45 (с, 2Н), 2,50-2,40 (м, 1Н), 2,40-2,33 (м, 1Н), 2,05-1,87 (м, 4Н), 1,80 (дд, J=2,4, 10,3 Гц, 1Н), 1,65 (дд, J=3,0, 10,0 Гц, 1Н), 1,54-1,40 (м, 3Н), 1,45 (с, 9Н), 1,37 (дд, J=3,1, 10,0 Гц, 1Н).
Стадия VII: К перемешиваемому раствору трет-бутил[1-(гидроксиметил)трицикло[3,3,1,03,7]нон-3-ил]карбамата (0,65 г, 2,45 ммоль), полученного на стадии VI, в ТГФ (10 мл) при температуре бани со льдом последовательно добавляли Et3N (1,0 мл, 7,35 ммоль), DMAP (20 мг, 0,16 ммоль) и метансульфонилхлорид (0,29 мл, 3,7 ммоль). После перемешивания при такой же температуре в течение 0,5 ч реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением указанного соединения {3-[(трет-бутоксикарбонил)амино]трицикло[3,3,1,03,7]нон-1-ил}метилметансульфоната в виде вязкой жидкости (0,84 г) с выходом 100%. м/z (М+1) 358; 1Н ЯМР (CDCl3) 300 МГц δ 4,76 (с (ушир), 1Н), 4,02 (с, 2Н), 3,01 (с, 2Н), 2,99 (с, 3Н), 2,54-2,44 (м, 1Н), 2,42-2,35 (м, 1Н), 2,20-1,87 (м, 4Н), 1,82 (дд, J=2,2, 10,5 Гц, 1Н), 1,76-1,58 (м, 4Н), 1,57-1,48 (м, 1Н), 1,45 (с, 9Н).
Препарат 4
Бензил-[2-(4-аминофенил)гексагидро-2,5-метанпентален-3а(1Н)-ил]карбамат

Стадия I: Cмесь для нитрования (1 мл) [смесь для нитрования получали смешиванием 10,5 г азотной кислоты (d 1,375 при 22ºС), 180,0 г конц. серной кислоты (d 1,84 при 22ºС) и 16 г Н2О] добавляли по каплям к перемешиваемому раствору 1-(1-фенилтрицикло[3,3,1,03,7]нон-3-ил)этанона (240 мг, 1,0 ммоль), полученного, как указано ранее (препарат 1), в нитрометане (4 мл) при 0ºС. После перемешивания в течение 2 ч реакционную смесь выливали в охлажденную льдом воду и экстрагировали EtOAc, объединенные органические слои промывали насыщенным раствором соли и сушили над Na2SO4. Растворитель выпаривали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 1-[1-(4-нитрофенил)трицикло[3,3,1,03,7]нон-3-ил]этанона в виде не совсем белого твердого вещества (250 мг) с выходом 88%. Т.пл.: 61,3-66,6ºС, m/z (М+1) 286; ИК см-1 2953, 1697, 1598, 1519, 1217, 1110, 852, 768. 1Н ЯМР (CDCl3) 300 МГц δ 8,17 (д, J=8,7 Гц, 2Н), 7,43 (д, J=8,7 Гц, 2Н), 2,87-2,80, (м, 1Н), 2,61-2,55 (м, 1Н), 2,34-2,26 (м, 1Н), 2,23 (с, 3Н), 2,13-1,98 (м, 4Н), 1,90-1,71 (м, 5Н).
Стадия II: К перемешиваемому раствору NaOH (6,3 г, 158,0 ммоль), Н2О (54,0 мл) и 1,4-диоксана (7 мл) при температуре бани со льдом добавляли Br2 (3,2 мл, 59,0 ммоль) и перемешивали в течение 5 минут. Образованный раствор гипобромита добавляли по каплям к перемешиваемому раствору 1-[1-(4-нитрофенил)трицикло[3,3,1,03,7]нон-3-ил]этанона (3,0 г, 10,53 ммоль), полученного на стадии I в 1,4-диоксане (14 мл) при температуре бани со льдом. Реакционную смесь постепенно нагревали до комнатной температуры и через 1 час смесь охлаждали до температуры бани со льдом и подкисляли конц. HCl, разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-(4-нитрофенил)трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (2,27 г) с выходом 75%, в виде не совсем белого твердого вещества; Т.пл.: 145-150ºС, m/z (М-1) 286; ИК см-1 3437, 2945, 1693, 1596, 1511, 1408, 1352, 946, 839, 749. 1Н ЯМР (CDCl3) 300 МГц δ 8,16 (д, J=8,8 Гц, 2Н), 7,42 (д, J=8,8 Гц, 2Н), 2,96-2,88 (м, 1Н), 2,59-2,52 (м, 1Н), 2,44-2,33 (м, 1Н), 2,23-2,03 (м, 4Н), 2,02-1,68 (м, 5Н).
Стадия III: К перемешиваемому раствору 1-(4-нитрофенил)трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (3,0 г, 10,45 ммоль), полученной на стадии II, в CHCl3 (21 мл) добавляли конц. H2SO4 (4,2 мл, 78,9 ммоль), затем порциями добавляли твердый NaN3 так, чтобы температура реакционной смеси не повышалась выше 40ºС. Реакционную смесь нагревали до 45ºС и после перемешивания в течение 2 ч снова охлаждали до температуры бани со льдом, разбавляли водой и экстрагировали EtOAc. Водный слой подщелачивали 50% раствором NaOH и экстрагировали CHCl3. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-(4-нитрофенил)трицикло[3,3,1,03,7]нонан-3-амина в виде не совсем белого твердого вещества (2,0 г) с выходом 74%. Т.пл.: 201,0-205,9ºС, m/z (M+1) 259; ИК см-1 3435, 2941, 1643, 1596, 1518, 1400, 1349, 1013, 750. 1Н ЯМР (CDCl3) 300 МГц δ 8,14 (д, J=8,8 Гц, 2Н), 7,39 (д, J=8,8 Гц, 2Н), 3,70-3,60 (д (ушир), 2Н), 2,62-2,50 (м, 2Н), 2,38-2,23 (м, 3Н), 2,15-2,0 (м, 3Н), 1,95-1,60 (м, 4Н).
Стадия IV: К перемешиваемой смеси 1-(4-нитрофенил)трицикло[3,3,1,03,7]нонан-3-амина (2,0 г, 8,0 ммоль), полученного на стадии III, К2СО3 (3,5 г, 24 ммоль) в ТГФ (80 мл), охлажденной до температуры бани со льдом, добавляли бензилхлорформиат (50% масс./об. в толуоле, 2,2 мл, 12 ммоль). После перемешивания при комной температуре в течение 2 ч реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением бензил-[2-(4-нитрофенил)гексагидро-2,5-метанпентален-3а(1Н)-ил]карбамата в виде не совсем белого твердого вещества (2,1 г) с выходом 70%. Т.пл.: 104,1-105,9ºС; m/z (М+1) 393; ИК см-1 3440, 2952, 1714, 1518, 1349, 1216, 757; 1Н ЯМР (CDCl3) 300 МГц δ 8,12 (д, J=8,8 Гц, 2Н), 7,50-7,27 (м, 7Н), 5,20-5,0 (с (ушир), 2Н), 2,65-2,55 (м, 1Н), 2,50-1,55 (м, 11Н). 13С ЯМР (CDCl3) 75 МГц δ 155,1, 154,2, 146,1, 136,4, 131,1, 128,5 (2С), 128,1 (2С), 126,6 (2С), 123,4 (2С), 66,3, 64,4, 49,2, 48,0, 47,5, 43,8, 42,1, 40,8, 37,0.
Стадия V: К перемешиваемому раствору бензил[2-(4-нитрофенил)гексагидро-2,5-метанпентален-3а(1Н)-ил]карбамата (1,0 г, 2,55 ммоль), который получен на стадии IV в смеси 1:2:4 воды, ТГФ и этанола, соответственно (10 мл) добавляли твердый NH4Cl (0,5 г, 9,3 ммоль) и порошок Fe (0,5 г, 9,0 ммоль). Реакционную смесь нагревали при температуре кипячения с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Фильтрат выпаривали при пониженном давлении, и остаток разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением бензил-[2-(4-аминофенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата в виде не совсем белого твердого вещества (0,75 г) с выходом 81%. m/z (M+1) 363; ИК см-1 3441, 2950, 1715, 1517, 1346, 1216, 756. 1Н ЯМР (CDCl3) 300 МГц δ 7,45-7,05 (м, 9Н), 5,20-5,0 (с (ушир), 2Н), 2,65-2,50 (м, 1Н), 2,50-2,40 (м, 1Н), 2,36-1,57 (м, 10Н).
Препарат 5
(2S,4S)-1-(хлорацетил)-4-фторпирролидин-2-карбонитрил
Стадия I: К перемешиваемому раствору (2S,4R)-4-гидроксипирролидин-2-карбоновой кислоты (13,1 г, 0,1 моль) в метаноле (400 мл), охлажденному до 0ºС, добавляли ацетилхлорид (14,3 мл, 0,2 моль) в течение периода 30 мин. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 ч. Летучие вещества удаляли при пониженном давлении и остаток некоторое время растирали с простым эфиром с получением хлористоводородной соли (2S,4R)-4-гидроксипирролидин-2-карбоксилата в виде белого порошка (14,5 г) с выходом 100%. m/z (M+1) 145; 1Н ЯМР (ДМСО-d6) 300 МГц δ 5,7-5,5 (с (ушир), 1Н), 4,55-4,35 (м, 2Н), 3,76 (с, 3Н), 3,35 (д, J=12 Гц, 1Н), 3,10 (д, J=12 Гц, 1Н), 2,25-2,0 (м, 2Н).
Стадия II: К перемешиваемой суспензии хлористоводородной соли, полученной на стадии I (14,5 г, 0,1 моль), в СН2Cl2 (400 мл), охлажденной до 0ºС, добавляли Et3N (28 мл, 0,2 моль), DMAP (0,61 г, 5 ммоль) и ангидрид Вос (27,5 мл, 0,12 моль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 2 ч. Затем растворитель удаляли в вакууме и добавляли простой эфир к оставшемуся твердому веществу. Твердое вещество фильтровали через воронку с фильтром из спекшегося стекла и тщательно промывали простым эфиром. Фильтрат упаривали при пониженном давлении. Остаток растворяли в СН2Cl2 и промывали насыщ. NaCl и насыщ. NaHCO3 c последующим высушиванием над безводным Na2SO4. Растворитель выпаривали при пониженном давлении с получением светло-желтого масла, которое затвердевало в высоком вакууме. Полученное твердое вещество некоторое время растирали с гексаном. Твердое вещество сушили в высоком вакууме с получением 1-трет-бутил-2-метил-(2S,4R)-4-гидроксипирролидин-1,2-дикарбоксилата в виде белого твердого вещества (24,5 г) с выходом 100%. (М+1) 245; 1Н ЯМР (CDCl3) 300 МГц δ 4,54-4,37 (м, 2Н), 3,75 (с, 3Н), 3,70-3,40 (м, 2Н), 2,48-2,21 (м, 2Н), 2,13-2,0 (м, 1Н), 1,46 (с, 3Н), 1,41 (с, 6Н).
Стадия III: К перемешиваемому раствору соединения, полученного на стадии II (24,5 г, 0,1 моль), в 1,2-дихлорэтане (300 мл), охлажденному до -10ºС, добавляли диэтиламиносульфотрифторид (19,7 мл, 0,15 моль) в течение периода 30 минут. Реакционную смесь перемешивали при этой температуре в течение 1 ч, затем при комнатной температуре в течение 16 ч. Реакционную смесь гасили добавлением смеси измельченного льда (300 г) и твердого NaHCO3 (25,2 г, 0,3 моль). Два слоя разделяли и водный слой экстрагировали дихлорметаном. Объединенный органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-трет-бутил-2-метил-(2S,4S)-4-фторпирролидин-1,2-дикарбоксилата в виде вязкой жидкости (24,7 г) с выходом 100%. [α]D-53,3, (с, 1,0, CDCl3). m/z (M+1) 248; 1Н ЯМР (CDCl3) 300 МГц δ 5,20 (ддд, J=3,8, 3,8, 49,1 Гц, 1Н), 4,55 (д, J=9,5 Гц, ½ Н), 4,42 (d, J=9,5 Гц, ½ Н), 3,90-3,55 (м, 2Н), 3,75 (с, 3Н), 2,55-2,20 (м, 2Н), 1,46 (с, 3Н), 1,41 (с, 6Н).
Стадия IV: К перемешиваемому раствору 1-трет-бутил-2-метил-(2S,4S)-4-фторпирролидин-1,2-дикарбоксилата (24,7 г, 0,1 моль), полученного на стадии III, в ТГФ (200 мл), охлажденному до 0ºС, добавляли раствор LiOH (3,6 г, 0,15 моль) в воде (200 мл) в течение периода 30 мин. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 ч до обнаружения методом ТСХ завершения реакции. Реакционную смесь разбавляли водой, простым эфиром и два слоя разделяли. Водный слой подкисляли конц. HCl и экстрагировали EtOAc. Объединенный органический слой промывали водой и насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением (2S,4S)-1-(трет-бутоксикарбонил)-4-фторпирролидин-2-карбоновой кислоты в виде не совсем белого твердого вещества (21 г) с выходом 90%. [α]D-65,7 (с, 1,0, CDCl3); m/z (M-1) 232; 1Н ЯМР (CDCl3) 300 МГц δ 5,22 (ддд, J=3,8, 3,8, 52,3 Гц, 1Н), 4,60-4,40 (м, 1Н), 3,95-3,50 (м, 2Н), 2,78-2,15 (м, 2Н), 1,6-1,35 (м, 9Н).
Стадия V: К перемешиваемому раствору кислоты, полученной на стадии IV (17,3 г, 0,074 моль), в ацетонитриле (220 мл) при комнатной температуре добавляли пиридин (6,6 мл, 0,082 моль), ангидрид Вос (20 мл, 0,089 моль). Через 1 ч добавляли твердый NH4HCO3 (9,4 г, 0,12 моль) и реакционную смесь перемешивали в течение 12 ч. Реакционную смесь разбавляли EtOAc и промывали смесью (1:1 об./об.) насыщенного раствора соли и 1н HCl. Органический слой сушили над безводным Na2SO4 и растворитель удаляли в вакууме с получением трет-бутил-(2S,4S)-2-(аминокарбонил)-4-фторпирролидин-1-карбоксилата (17 г) в виде вязкой жидкости. Вещество применяли для следующей реакции без дополнительной очистки. m/z (M+1) 233; 1Н ЯМР (CDCl3) 300 МГц δ 6,70-6,60 (с (ушир), ½ Н), 6,30-6,10 (с (ушир), ½ Н), 5,50-5,40 (с (ушир), 1Н), 5,22 (ддд, J=3,4, 3,4, 52,0 Гц, 1Н), 4,50-4,30 (м, 1Н), 3,95-3,50 (м, 2Н), 2,90-2,10 (м, 2Н), 1,48 (с, 9Н).
Стадия VI: К перемешиваемому раствору амида, полученного на предыдущей стадии (17,9 г, 0,077 моль) в EtОAc (35 мл) при 0ºС добавляли сухой HCl в EtОAc (4н, 225 мл) в течение периода 30 мин. После перемешивания при 0ºС в течение 1 ч летучие вещества удаляли при пониженном давлении и остаток некоторое время растирали с простым эфиром с получением хлористоводородной соли (2S,4S)-4-фторпирролидин-2-карбоксамида в виде не совсем белого порошка (12 г) с выходом 92%. m/z (M+1) 133; 1H ЯМР (ДМСО-d6) 300 МГц δ 10,60-10,30 (с (ушир), ½ Н), 8,90-8,60 (с (ушир), ½ Н), 8,10 (с (ушир), 1Н), 7,72 (с (ушир), 1Н), 5,38 (ддд, J=3,7, 3,7, 52,4 Гц, 1Н), 4,32 (д, J=10,5 Гц, 1Н), 4,28 (д, J=10,4 Гц, 1Н), 3,64-3,29 (м, 2Н), 2,73-2,50 (м, 1Н), 2,41-2,24 (м, 1Н).
Стадия VII: К перемешиваемой суспензии хлористоводородной соли, которая получена на стадии VI (12 г, 0,071 моль) в дихлорметане (140 мл), охлажденной до 0ºС, добавляли Et3N (30 мл, 0,213 моль), хлорацетилхлорид (8,1 мл, 0,107 моль). Реакционную смесь постепенно нагревали до комн. темп. и перемешивали в течение 1 ч. Реакционную массу фильтровали через воронку с фильтром из спекшегося стекла, промывали слой соли простым эфиром и фильтрат выпаривали в вакууме с получением неочищенного вещества (2S,4S)-1-(хлорацетил)-4-фторпирролидин-2-карбоксамида (14,8 г) как вязкую жидкость в виде смеси ротомеров 3:1. m/z (М+1) 209; 1Н ЯМР (ДМСО-d6) 300 МГц δ 7,26 (с (ушир), ½ Н), 7,04 (с (ушир), ½ Н), 5,34 (д, J=52,5 Гц, 0,8 Н), 5,25 (д, J=53,0 Гц, 0,2Н), 4,58-4,30 (м, 3Н), 3,90-3,50 (м, 2Н), 2,60-2,20 (м, 2Н).
Стадия VIII: К перемешиваемому раствору соединения, полученного на стадии VII (14,7 г, 0,07 моль) в сухом ТГФ (140 мл) в атмосфере N2 при 0ºС добавляли ангидрид трифторуксусной кислоты (15 мл, 0,107 моль). Реакционную смесь постепенно нагревали до комн. темп. и перемешивали в течение 1 ч. Добавляли воду и два слоя разделяли. Водный слой экстрагировали EtOAc и объединенный органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении с получением неочищенного вещества, которое очищали колоночной хроматографией с получением (2S,4S)-1-(хлорацетил)-4-фторпирролидин-2-карбонитрила в виде желтовато-коричневого твердого вещества (8,7 г) с выходом 64% (смесь 3:1 двух ротомеров). [α]D-51,0 (с, 1,0, СНСl3); ИК см-1 3031, 3007, 2962, 2241, 1679, 1407, 1280, 1225, 1076, 860; m/z (M+1), 191; 1Н ЯМР (СDCl3) 300 МГц δ 5,45 (ддд, J=3,4, 3,4, 51,3 Гц, 0,8 Н), 5,37 (ддд, J=3,4, 3,4, 51,0 Гц, 0,2 Н), 5,06 (д, J=8,9 Гц, 0,2 Н), 4,95 (д, 9,3, Гц, 0,8 Н), 4,30-3,55 (м, 2 Н), 4,06 (с, 2Н), 2,65-2,55 (м, 1Н), 2,50-2,25 (м, 1 Н).
Препарат 6
(2S,4R)-1-(2-хлорацетил)-4-фторпирролидин-2-карбонитрил
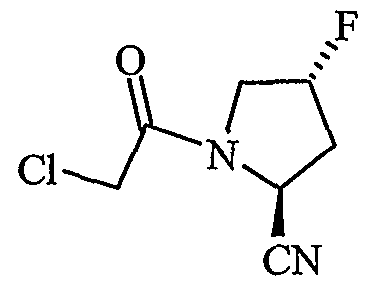
Соединение (2S,4R)-1-(2-хлорацетил)-4-фторпирролидин-2-карбонитрил синтезировали из (2S,4S)-4-гидроксипирролидин-2-карбоновой кислоты с применением такой же последовательности стадий и методик, как указано выше по тексту для (2S,4S)-1-(2-хлорацетил)-4-фторпирролидин-2-карбонитрила, исходя из (2S,4R)-4-гидроксипирролидин-2-карбоновой кислоты. (2S,4R)-1-(2-Хлорацетил)-4-фторпирролидин-2-карбонитрил: твердое вещество, смесь 4:1 двух ротамеров; m/z (М+1) 191; 1Н ЯМР (CDCl3) 300 МГц δ 5,38 (д (ушир), J=51,3 Гц, 0,8Н), 5,33 (д (т), J=51,0 Гц, 0,2Н), 5,02 (д, J=8,5 Гц, 0,2Н), 4,72 (д, 8,5 Гц, 0,8Н), 4,40-3,30 (м, 2Н), 4,06 (с, 2Н), 3,0-2,65 (м, 1Н), 2,62-2,40 (м, 1Н).
Препарат 7
(4R)-3-(хлорацетил)-1,3-тиазолидин-4-карбонитрил
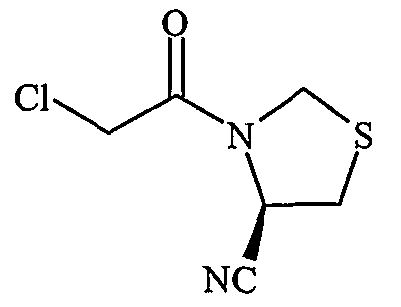
Стадия I: К перемешиваемому водному раствору 40% формальдегида (40 мл) при 0ºС добавляли порциями твердый L-цистеин (12,1 г, 0,1 моль) в течение периода 30 минут. Реакционную смесь перемешивали в течение 4 ч, после чего реакционную смесь фильтровали через воронку с фильтром из спекшегося стекла. Твердые вещества промывали абсолютным этанолом, затем простым диэтиловым эфиром. Твердые вещества сушили в высоком вакууме с получением (4R)-1,3-тиазолидин-4-карбоновой кислоты (12,5 г) с выходом 94%. Т.пл. 215-217ºС; m/z (M+1) 134; ИК см-1 3429, 3049, 2357, 1629, 1463, 1383, 1343, 1014, 862; 1Н ЯМР (D2O) 300 МГц δ 4,40-4,30 (м, 2Н), 4,30-4,22 (м, 1Н), 3,40-3,18 (м, 2Н).
Стадия II: К перемешиваемой смеси соединения, полученного на стадии I (13,3 г, 0,1 моль), в ацетонитриле (400 мл), охлажденной до 0ºС, добавляли пиридин (20,1 мл, 0,22 моль) и ангидрид Вос (58 мл, 0,24 моль). После перемешивания реакционной смеси в течение 1 ч при комнатной температуре добавляли твердый NH4HCO3 (11,8 г, 0,15 моль) и реакционную смесь перемешивали в течение следующих 2 ч. Реакционную смесь распределяли между этилацетатом и смесью 2н НСl и насыщенного раствора соли 1:1. Водный слой экстрагировали этилацетатом и объединенные органические слои сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением трет-бутил-(4R)-4-(аминокарбонил)-1,3-тиазолидин-3-карбоксилата (23 г) в виде смолистой жидкости с количественным выходом. Вещество применяли в следующей реакции без дополнительной очистки. m/z (M+1) 233; ИК см-1 3334, 2978, 2932, 1682, 1392, 1163, 763; 1Н ЯМР (CDCl3) 300 МГц δ 5,70-5,50 (с (ушир), 1Н), 4,80-4,60 (м, 2Н), 4,48-4,30 (м, 1Н), 3,50-3,10 (м, 2Н).
Стадия III: К перемешиваемому раствору трет-бутил-(4R)-4-(аминокарбонил)-1,3-тиазолидин-3-карбоксилата (23 г, 0,1 моль) в этилацетате (50 мл) при 0ºС добавляли сухой HCl в этилацетате (3,5 н, 250 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 ч, летучие вещества удаляли при пониженном давлении и остаток растирали некоторое время с этилацетатом с получением хлористоводородной соли (4R)-1,3-тиазолидин-4-карбоксамида в виде не совсем белого порошка (16,5 г) с количественным выходом. Т.пл. 201,8-203,9ºС; m/z (M+1) 133; ИК см-1 3387, 3250, 3189, 2857, 1706, 1675, 1612, 1371, 1123, 894; 1Н ЯМР (ДМСО-d6) 300 МГц, δ 10,30-9,60 (с (ушир), 1/2Н), 8,10 (с (ушир), 1/2Н), 7,77 (с (ушир), 1Н), 4,40 (т, J=7,0 Гц, 1Н), 4,31 (д, J=9,6 Гц, 1Н), 4,25 (д, J=9,6 Гц, 1Н), 3,50-3,30 (м, 1Н), 3,15 (дд, J=7,0, 11,7 Гц, 1Н).
Стадия IV: К перемешиваемой суспензии хлористоводородной соли, полученной на стадии III (16,5 г, 0,1 моль), в дихлорметане (200 мл), охлажденной до 0ºС, добавляли Et3N (41 мл, 0,3 моль) и хлорацетилхлорид (8,8 мл, 0,11 моль). Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 1 ч. Реакционную массу фильтровали через воронку с фильтром из спекшегося стекла, слой соли промывали простым эфиром и фильтрат выпаривали в вакууме с получением неочищенного соединения (4R)-3-(хлорацетил)-1,3-тиазолидин-4-карбоксамида (20,8 г) в виде вязкой жидкости, который применяли на стедующей стадии без дополнительной очистки. m/z (M+1) 209; ИК см-1 3418, 2925, 1736, 1667, 1416, 1219, 771; 1Н ЯМР (CDCl3) 300 МГц δ 6,60-6,30 (с (ушир), 1/2Н), 6,0-5,65 (с (ушир), ½ Н), 5,02 (дд, J=3,6, 6,9 Гц, 0,8Н), 4,88-4,75 (м, 0,4Н), 4,70 (д, J=8,8 Гц, 0,8Н), 4,62 (д, J=8,8 Гц, 0,8Н), 4,51 (д, J=10,0 Гц, 0,2Н), 4,16 (с, 1,6Н), 4,11 (с, 0,4Н), 3,56 (дд, J=3,4, 11,8 Гц, 0,8Н), 3,44 (дд, J=4,7, 8,0 Гц, 0,4Н), 3,15 (дд, J=7,1, 11,8 Гц, 0,8Н), 1,28 (с, 1,6Н), 1,25 (с, 7,4Н).
Стадия V: К перемешиваемому раствору соединения, полученного на стадии IV (20,7 г, 0,1 моль), в сухом ТГФ (200 мл) в атмосфере N2 при 0ºС добавляли ангидрид трифторуксусной кислоты (21 мл, 0,15 моль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 1 ч. Реакционную смесь разбавляли водой и разделяли два слоя. Водный слой экстрагировали EtOAc и объединенные органические слои промывали насыщенным раствором соли и сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении и остаток очищали колоночной хроматографией с получением (4R)-3-(хлорацетил)-1,3-тиазолидин-4-карбонитрила (10,0 г) в виде не совсем белого твердого вещества с выходом 53%. [α]D -147,24 (с, 0,5, CHCl3); Т.пл. 85,9-87,3ºС; m/z (М+1) 191; ИК см-1 2982, 2936, 2245, 1679, 1666, 1393, 1284, 1261, 984, 788; 1Н (CDCl3) 300 МГц δ 5,29 (т, J=4,3 Гц), 4,72 (д, J=8,8 Гц, 1Н), 4,66 (д, J=8,8 Гц, 1Н), 3,45-3,30 (м, 2Н).
Препарат 8
Трет-Бутил(3-аминотрицикло[3,3,1,03,7]нон-1-ил)карбамат
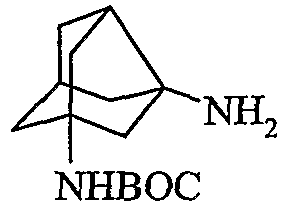
Стадия I: К раствору карбоновой кислоты, полученной на стадии I (препарат 2) (2,7 г, 12,9 ммоль), в толуоле (52 мл) при температуре бани со льдом добавляли Et3N (5,8 мл, 38,7 ммоль) и дифенилфосфорилазид (3,3 мл, 15,5 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры, перемешивали в течение одного часа и кипятили с обратным холодильником в течение 4 ч. После охлаждения до комнатной температуры реакционную смесь переносили в делительную воронку и промывали один раз водой. Органический слой переносили в круглодонную колбу, охлаждали до температуры бани со льдом и добавляли водн. раствор КОН (50% масс./об., 26 мл) и nBu4NI (476 мг, 1,29 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь охлаждали до температуры бани со льдом, подкисляли до рН 2 конц. HCl, экстрагировали один раз простым эфиром, водный слой подщелачивали водн. раствором NaOH (50% масс./об.) и экстрагировали хлороформом. Объединенные органические слои сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением аминосоединения 1-(1-аминотрицикло[3,3,1,03,7]нон-3-ил]этанона в виде вязкой жидкости (1,3 г) с выходом 56%. m/z (M+1) 180; 1Н ЯМР (CDCl3) 300 МГц δ 2,68-2,61 (м, 1Н), 2,49-2,43 (м, 1Н), 2,17 (с, 3Н), 2,05 (ддд, J=2,3, 2,3, 10,6 Гц, 1Н), 1,95-1,78 (м, 3Н), 1,78-1,43 (м, 6Н).
Стадия II: К перемешиваемому раствору аминосоединения, полученного на стадии I (1,3 г, 7,26 ммоль), в дихлорметане (29 мл) при температуре бани со льдом Et3N (2 мл, 14,5 ммоль), добавляли ангидрид Вос (2,1 мл, 8,7 ммоль) и DMAP (44 мг, 0,36 ммоль). После перемешивания в течение 1 ч при комн. темп. растворитель удаляли при пониженном давлении и неочищенную реакционную массу очищали колоночной хроматографией с получением трет-бутил-(3-ацетилтрицикло[3,3,1,03,7]нон-1-ил)карбамата (1,8 г) в виде вязкой жидкости с выходом 90%. m/z (M+1) 280; 1Н ЯМР (CDCl3) 300 МГц δ 4,73 (с (ушир), 1Н), 2,72-2,64 (м, 1Н), 2,17 (с, 3Н), 2,10-1,78 (м, 8Н), 1,78-1,68 (м, 2Н), 1,43 (с, 9Н).
Стадия III: К смеси NaOH (1,32 г, 33,0 ммоль), Н2О (8,8 мл) и 1,4-диоксана (2 мл) при температуре бани со льдом добавляли Br2 (0,6 мл, 12,3 ммоль) и перемешивали в течение 5 минут. Полученный раствор гипобромита добавляли по каплям к перемешиваемому раствору соединения, полученного на стадии II (0,6 г, 2,2 ммоль) в 1,4-диоксане (2,4 мл) приблизительно при 10ºС. Реакционную смесь постепенно нагревали до комн. темп., перемешивали в течение 1 ч, затем охлаждали до 0ºС и гасили добавлением уксусной кислоты (2 мл, 36,3 ммоль). Смесь разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-[(трет-бутоксикарбонил)амино]трицикло[3,3,1,03,7]-нонан-3-карбоновой кислоты (0,54 г) в виде вязкой жидкости с выходом 87%. m/z (M-1) 280; 1Н ЯМР (CDCl3) 300 МГц δ 4,75 (с (ушир), 1Н), 2,78-2,60 (м, 1Н), 2,47-2,40 (м, 1Н), 2,33-2,22 (м, 1Н), 2,18-1,72 (м, 8Н), 1,62-1,53 (м, 1Н), 1,43 (с, 9Н).
Стадия IV: К раствору кислоты (0,55 г, 1,95 ммоль), полученной на стадии III, в толуоле (8 мл) при температуре бани со льдом добавляли Et3N (1,2 мл, 8,8 ммоль) и дифенилфосфорилазид (0,5 мл, 2,3 ммоль). Реакционную смесь постепенно нагревали до комн. темп., перемешивали в течение одного часа и затем нагревали при кипячении с обратным холодильником в течение 4 ч. После охлаждения до комнатной температуры реакционную смесь переносили в делительную воронку и промывали один раз водой. Органический слой переносили в круглодонную колбу, охлажденную до температуры бани со льдом и добавляли водн. раствор КОН (50% масс./об., 4 мл) и nBu4NI (10 мг, 0,02 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь охлаждали до температуры бани со льдом, подкисляли до рН 2 конц. HCl, экстрагировали один раз простым эфиром и водный слой подщелачивали водн. раствором NaOH (50% масс./об.) и экстрагировали хлороформом. Объединенные органические слои сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением названного соединения трет-бутил(3-аминотрицикло[3,3,1,03,7]нон-1-ил)карбамата в виде вязкой жидкости (0,3 г) с выходом 60%. m/z (M+1) 253; 1Н ЯМР (CDCl3) 300 МГц δ 2,35-2,28 (м, 1Н), 2,20-1,80 (м, 5Н), 1,78-1,53 (м, 5Н), 1,52-1,47 (м, 1Н), 1,43 (с, 9Н).
Препарат 9
1-(3-Аминотрицикло[3,3,1,03,7]нон-1-ил)пирролидин-2-он
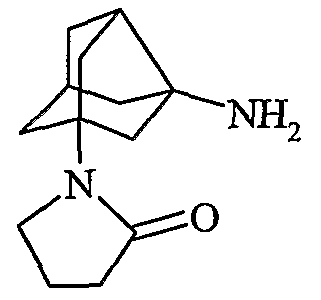
Стадия I: К перемешиваемому раствору соединения, полученного, как указано ранее (стадия I, препарат 8) (0,9 г, 5,0 ммоль), в ТГФ (20 мл) при 0ºС добавляли Et3N (2,1 мл, 15 ммоль) и 4-хлорбутироилхлорид (1,02 г, 7,5 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение 1 ч добавляли по каплям водный раствор NaOH (50%, 10 мл), с последующим добавлением n-Bu4NI (182 мг, 10 мол.%). После перемешивания в течение 16 ч реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное соединение очищали колоночной хроматографией с получением 1-(3-ацетилтрицикло[3,3,1,03,7]нон-1-ил)пирролидин-2-она в виде вязкой жидкости (1,0 г) с выходом 81%. m/z (M+1) 248; 1Н ЯМР (CDCl3) 300 МГц δ 3,42 (т, J=6,5 Гц, 2Н), 2,72-2,66 (м, 1Н), 2,51-2,40 (м, 2Н), 2,32 (т, J=7,8 Гц, 2Н), 2,28-2,12 (м, 2Н), 2,18 (с, 3Н), 2,10-1,87 (м, 5Н), 1,81-1,69 (м, 2Н), 1,67-1,60 (м, 2Н).
Стадия II: К перемешиваемой смеси NaOH (2,4 г, 60,6 ммоль), Н2О (16 мл) и 1,4-диоксана (4 мл) при температуре бани со льдом добавляли Br2 (1,13 мл, 22,6 ммоль) и перемешивали в течение 5 минут. Полученный раствор гипобромита добавляли по каплям к перемешиваемому раствору соединения, полученного на стадии I (1,0 г, 4,04 ммоль), в 1,4-диоксане (18 мл) при 10ºС. Температуру реакционной смеси повышали до комнатной температуры и реакционную смесь перемешивали в течение 1 ч. Затем смесь охлаждали до температуры бани со льдом и гасили добавлением уксусной кислоты (3,9 мл, 65,7 ммоль). Реакционную смесь разбавляли водой, экстрагировали EtOAc и объединенные органические слои промывали насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением 1-(2-оксопирролидин-1-ил)трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты в виде вязкой жидкости (1,3 г) с выходом 100%. m/z (M+1) 250; 1Н ЯМР (CDCl3) 300 МГц δ 3,42 (т, J=7,0 Гц, 2Н), 2,78-2,71 (м, 1Н), 2,47-2,23 (м, 6Н), 2,08-1,90 (м, 6Н), 1,85-1,72 (м, 2Н), 1,62 (дд, J=2,4 11,0 Гц, 1Н).
Стадия III: К перемешиваемому раствору кислоты, полученной на стадии II (0,5 г, 2,0 ммоль) в CHCl3 (21 мл) при комнатной температуре добавляли конц. H2SO4 (1,0 мл, 20 ммоль) с последующим добавлением порциями NaN3 (0,39 г, 6,0 ммоль) в течение периода 30 мин, таким образом, чтобы температура реакционной смеси не повышалась выше 40ºС. Реакционную смесь нагревали до 45ºС и перемешивали в течение 2 ч, затем охлаждали до температуры бани со льдом, разбавляли водой и экстрагировали EtOAc. Водный слой подщелачивали добавлением 50% раствора NaOH и экстрагировали CHCl3. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-(3-аминотрицикло[3,3,1,03,7]нон-1-ил)пирролидин-2-она в виде не совсем белого твердого вещества (0,25 г) с выходом 74%. m/z (M+1) 221; 1Н ЯМР (CDCl3) 300 МГц δ 3,41 (т, J=7,0 Гц, 2Н), 2,46 (дд, J=2,4, 10,3 Гц, 1Н), 2,38-2,28 (м, 1Н), 2,30 (т, J=8,3 Гц, 2Н), 2,16-1,82 (м, 9Н), 1,78 (дд, J=2,6, 10,8 Гц, 1Н), 1,66-1,58 (м, 1Н), 1,51 (дд, J=2,5, 10,8 Гц, 1Н).
Препарат 10
1-(1,1-Диоксидоизотиазолидин-2-ил)трицикло[3,3,1,03,7]нонан-3-амин
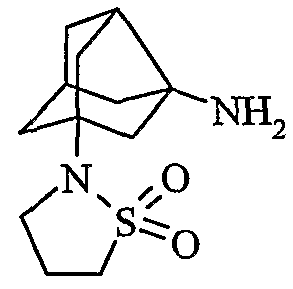
Стадия I: К перемешиваемому раствору соединения, полученного, как указано ранее (стадия I, препарат 8) (1,0 г, 5,6 ммоль) в ТГФ (23 мл) при 0ºС добавляли Et3N (1,2 мл, 8,4 ммоль), с последующим добавлением 4-хлорбутирилхлорида (1,02 г, 7,5 ммоль). После перемешивания реакционной смеси при комн. темп. в течение 1 ч добавляли водный раствор NaOH (50% масс./об., 11 мл); с последующим добавлением n-Bu4NI (182 мг, 0,56 ммоль). Реакционную смесь перемешивали в течение 16 ч, разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением 1-[1-(1,1-диоксидоизотиазолидин-2-ил)трицикло[3,3,1,03,7]нон-3-ил]этанона в виде вязкой жидкости (1,0 г) с выходом 61%. m/z (M+1) 284; 1Н ЯМР (CDCl3) 300 МГц δ 3,37 (т, J=6,6 Гц, 2Н), 3,16 (т, J=7,5 Гц, 2Н), 2,76-2,69 (м, 1Н), 2,53-2,47 (м, 1Н), 2,39-2,26 (м, 3Н), 2,18 (с, 3Н), 2,22-2,07 (м, 4Н), 2,20 (дд, J=3,2, 11,0 Гц, 1Н), 1,96-1,89 (м, 1Н), 1,78-1,69 (м, 2Н), 1,61 (дд, J=2,7 11,0 Гц, 1Н).
Стадия II: К перемешиваемой смеси NaOH (2,1 г, 53,0 ммоль), Н2О (14 мл) и 1,4-диоксана (4 мл) при температуре бани со льдом добавляли Br2 (1,0 мл, 19,8 ммоль) и смесь перемешивали в течение 5 минут. Полученный таким образом раствор гипобромита добавляли по каплям к перемешиваемому раствору соединения, полученного на стадии I (1,0 г, 3,53 ммоль) в 1,4-диоксане (7 мл) при 10ºС. Температуру реакционной смеси постепенно повышали до комнатной температуры и реакционную смесь перемешивали в течение 1 ч, затем смесь охлаждали до температуры бани со льдом и гасили добавлением АсОН (3,9 мл, 65,7 ммоль). Реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-(1,1-диоксидоизотиазолидин-2-ил)трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты в виде вязкой жидкости (0,9 г) с выходом 90%. m/z (M+1) 286; 1Н ЯМР (CDCl3) 300 МГц δ 3,37 (т, J=6,6 Гц, 2Н), 3,16 (т, J=7,8 Гц, 2Н), 2,84-2,76 (м, 1Н), 2,52-2,41 (м, 2Н), 2,38-2,26 (м, 2Н), 2,25-1,98 (м, 6Н), 1,85-1,72 (м, 2Н), 1,61 (дд, J=2,2, 11,3 Гц, 1Н).
Стадия III: К перемешиваемому раствору кислоты, полученной на стадии II (0,29 г, 1,0 ммоль) в CHCl3 (5 мл) при комнатной температуре добавляли конц. H2SO4 (0,53 мл, 10 ммоль) с последующим добавлением порциями NaN3 (0,2 г, 3,0 ммоль) в течение периода 30 мин; поддерживая в то же время температуру ниже 40ºС. Реакционную смесь нагревали до 45ºС и перемешивали в течение 2 ч. Реакционную смесь охлаждали до температуры бани со льдом, разбавляли водой и экстрагировали EtOAc. Водный слой подщелачивали добавлением 50% раствора NaOH и экстрагировали CHCl3. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-(1,1-диоксидоизотиазолидин-2-ил)трицикло[3,3,1,03,7]нонан-3-амина в виде вязкой жидкости (0,17 г) с выходом 66%. m/z (M+1) 257; 1Н ЯМР (CDCl3) 300 МГц δ 3,36 (т, J=6,6 Гц, 2Н), 3,15 (т, J=7,5 Гц, 2Н), 2,40-2,24 (м, 4Н), 2,18 (дд, J=3,0, 10,4 Гц, 1Н), 2,13-1,83 (м, 6Н), 1,76 (дд, J=2,4, 10,8 Гц, 1Н), 1,65-1,58 (м, 1Н), 1,48 (дд, J=2,4, 10,8 Гц, 1Н).
Препарат 11
Трет-бутил-(1-гидрокситрицикло[3,3,1,03,7]нон-3-ил)карбамат
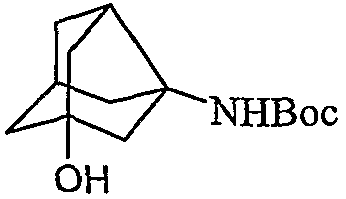
Стадия I: К суспензии NaH (60% дисперсия в медицинском масле, 1,92 г, 80 ммоль) в ТГФ (80 мл), охлажденной до температуры бани со льдом, добавляли 4-гидроксиадамантанон (6,64 г, 40 ммоль), растворенный в ТГФ (80 мл), через шприц в течение периода 15 минут. После перемешивания реакционной смеси в течение 30 мин, добавляли nBu4NI (1,4 г, 4 ммоль) с последующим добавлением бензилбромида (5,26 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч до обнаружения методом ТСХ исчезновения гидроксиадамантанона. Избыток NaH гасили добавлением насыщ. водн. pаствора NH4Cl к реакционной смеси, охлажденной льдом. Два слоя разделяли и водный слой экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением неочищенной реакционной массы, которую очищали колоночной хроматографией с получением 5-(бензилокси)адамантан-2-она в виде смолистой жидкости (7,93 г) с выходом 77%. m/z (M+1) 257; 1H ЯМР (CDCl3) 300 МГц δ 7,60-7,20 (м, 5Н), 4,51 (с, 2Н), 2,72-2,63 (м, 2Н), 2,40-2,36 (м, 1Н), 2,35-1,92 (м, 10Н).
Стадия II: Свежеприготовленный метилмагнийиодид в простом эфире (0,5М, 114 мл) добавляли через трубку к соединению, полученному на стадии I (7,3 г, 28,5 ммоль) в ТГФ (57 мл при 0ºС). После перемешивания в течение 0,5 ч реакционную смесь гасили добавлением насыщ. водн. раствора NH4Cl. Органический слой отделяли и водный слой экстрагировали простым изопропиловым эфиром. Объединенный органический слой промывали водой и насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением 5-(бензилокси)-2-метиладамантан-2-ола в виде смолистой жидкости (смесь 4:6 изомеров α и β) (7,5 г) с выходом 95%. m/z (M+1) 273; 1Н ЯМР (CDCl3) 300 МГц δ 7,42-7,20 (м, 5Н), 4,51 (с, 0,8Н), 4,48 (с, 1,2Н), 2,40-1,30 (м, 13Н), 1,40 (с, 1,2Н), 1,35 (с, 1,8Н).
Стадия III: Соединение, полученное на стадии II (7,5 г, 27,5 ммоль), растворяли в смеси АсОН (5,5 мл) и ТГФ (28 мл) и добавляли по каплям через присоединенную воронку к охлажденному льдом раствору NaOCl (4%, 275 мл) в течение периода 15 минут. Добавляли nBu4NI (210 мг, 2 моль%) и реакционную смесь перемешивали в течение 1,5 ч. Реакционную смесь выливали в делительную воронку и разделяли два слоя. Водный слой экстрагировали простым диизопропиловым эфиром и объединенный органический слой промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество кипятили с обратным холодильником с метанольным раствором KOH (3,0 г КОН в 55 мл МеОН) в течение 1 ч. Растворитель выпаривали при пониженном давлении и вещество очищали колоночной хроматографией с получением метилкетон-1-[1-(бензилокси)трицикло[3,3,1,03,7]нон-3-ил]этанона в виде смолистой жидкости (4,84 г), с выходом 65%. m/z (M+1) 271; 1Н ЯМР (CDCl3) 300 Мгц 7,40-7,20 (м, 5Н), 4,52 (с, 2Н), 2,70-2,64 (м, 1Н), 2,58-2,52 (м, 1Н), 2,33-2,27 (м, 1Н), 2,18 (с, 3Н), 2,11-1,88 (м, 5Н), 1,78-1,58 (м, 4Н).
Стадия IV: К смеси NaOH (10,8 г, 270 ммоль), Н2О (72 мл) и 1,4-диоксана (20 мл) при температуре бани со льдом добавляли Br2 (5,2 мл, 100,8 ммоль) и перемешивали в течение 5 минут. Раствор гипобромита добавляли по каплям к раствору соединения, полученного на стадии III (4,84 г, 18 ммоль) в 1,4-диоксане (18 мл), выдерживая при температуре бани со льдом. Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 1 ч, после чего охлаждали до температуры бани со льдом и гасили добавлением АсОН (3,9 мл, 65,7 ммоль), разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-(бензилокси)трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты в виде смолистой жидкости (4,1 г) с выходом 83%. m/z (M+1) 273; 1Н ЯМР (CDCl3) 300 МГц δ 7,40-7,20 (м, 5Н), 4,52 (с, 2Н), 2,78-2,70 (м, 1Н), 2,58-2,48 (м, 1Н), 2,45-2,37 (м, 1Н), 2,18-1,50 (м, 9Н).
Стадия V: К раствору кислоты, полученной на стадии IV (1,36 г, 5 ммоль) в толуоле (20 мл) при температуре бани со льдом добавляли Et3N (2,1 мл, 15 ммоль) и дифенилфосфорилазид (DPPA, 1,3 мл, 6 ммоль). Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение одного часа, после чего температуру повышали до температуры кипячения с обратным холодильником в течение 4 ч. После охлаждения до комнатной температуры смесь переносили в делительную воронку и промывали один раз водой. Органический слой переносили в круглодонную колбу, охлаждали до температуры бани со льдом и добавляли водн. раствор КОН (50% масс./об., 10 мл) и nBu4NI (92 мг, 0,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь охлаждали до температуры бани со льдом, подкисляли до рН 2 конц. HCl, экстрагировали один раз простым эфиром и водный слой подщелачивали водн. раствором NaOH (50% масс./об.) и экстрагировали хлороформом. Объединенный органический слой сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением чистого амина 1-(бензилокси)трицикло[3,3,1,03,7]нонан-3-амина в виде смолистой жидкости (614 мг) с выходом 80%. m/z (M+1) 244; 1Н ЯМР (CDCl3) 300 МГц δ 2,40-2,20 (м, 5Н), 2,70-2,48 (м, 3Н), 2,40 (с (ушир), 1Н), 2,27-2,16 (м, 1Н), 2,10-1,40 (м, 9Н).
Стадия VI: К раствору аминосоединения, полученного на стадии V (590 мг, 2,4 ммоль), в дихлорметане (10 мл) при температуре бани со льдом добавляли Et3N (0,5 мл, 3,6 ммоль) с последующим добавлением ангидрида Вос (654 мг, 3,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли при пониженном давлении и неочищенную реакционную массу очищали колоночной хроматографией с применением в качестве элюента смеси EtOAc/гексаны с получением производного Вос трет-бутил-(1-бензилокситрицикло[3,3,1,03,7]нон-3-ил)карбамата в виде смолистой жидкости (740 мг) с выходом 90%. m/z (M+1) 343; 1H ЯМР (CDCl3) 300 МГц δ 7,40-7,20 (м, 5Н), 4,80-4,70 (с (ушир), 1Н), 4,51 (с, 2Н), 2,50-2,25 (м, 4Н), 2,0-1,75 (м, 6Н), 1,65-1,50 (м, 2Н), 1,46 (с, 9Н).
Стадия VII: Смесь соединения, полученного на стадии VI (730 г, 2,1 ммоль) и Pd(OH)2/C (20% влажн., 150 мг) в МеОН (9 мл) перемешивали в атмосфере Н2 при комнатной температуре в течение 2 ч. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением трет-бутил-(1-гидрокситрицикло[3,3,1,03,7]нон-3-ил)карбамата в виде не совсем белого твердого вещества (520 мг) с выходом 97%. M/z (M+1) 254; 1H ЯМР (CDCl3) 300 МГц δ 4,72 (с (ушир), 1Н), 2,48-2,35 (м, 2Н), 2,32-2,18 (м, 2Н), 1,93-1,70 (м, 6Н), 1,55-1,35 (м, 2Н), 1,45 (с, 9Н).
Пример 1
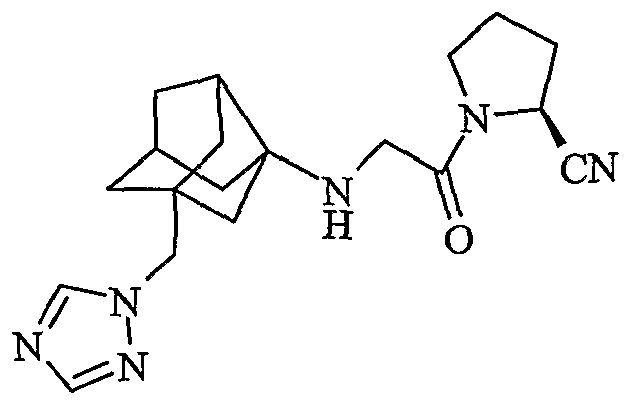
Стадия I: Перемешиваемую смесь [3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метилметансульфоната (2,4 г, 7,5 ммоль) [который получен, как указано ранее (препарат 2)], К2СО3 (4,5 г, 34,2 ммоль) и 1,2,4-триазола (1,5 г, 22,5 ммоль) в ДМФА (30 мл) нагревали до 110ºС в течение 5 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением 1-{[3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метил}-1Н-1,2,4-триазола (1,82 г) в виде вязкой жидкости с выходом 85%. m/z (M+1) 290; ИК см-1 2932, 1668, 1506, 1441, 1373, 1311, 1211, 1140, 1047, 876, 753. 1Н ЯМР (CDCl3) 300 МГц δ 7,99 (с, 1Н), 7,93 (с, 1Н), 4,07 (с, 2Н), 4,03-3,92 (м, 4Н), 2,42-2,32 (м, 2Н), 1,86-1,79 (м, 1Н), 1,74-1,65 (м, 2Н), 1,52-1,37 (м, 8Н), 1,26 (с, 3Н).
Стадия II: Перемешиваемый раствор 1-{[3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метил}-1Н-1,2,4-триазола (2,6 г, 9,09 ммоль), полученный на стадии I, и п-толуолсульфоновой кислоты (0,16 г) в ацетоне (36 мл) кипятили с обратным холодильником в течение 4 ч. Летучие соединения удаляли при пониженном давлении и остаток разбавляли этилацетатом, промывали 10% водн. раствором NaHCO3 и насыщенным раствором соли, сушили над Na2SO4 и растворитель выпаривали при пониженном давлении с получением 1-[1-(1H-1,2,4-триазол-1-илметил)трицикло[3,3,1,03,7]нон-3-ил]этанона (2,2 г, выход 86%) в виде вязкой жидкости. m/z (M+1) 246; 1Н ЯМР (CDCl3) 300 МГц δ 7,99 (с, 1Н), 7,93 (с, 1Н), 4,10 (с, 2Н), 2,75-2,65 (м, 1Н), 2,49-2,42 (м, 1Н), 2,16 (с, 3Н), 2,0-1,45 (м, 10Н), 13С ЯМР (CDCl3) 75 МГц δ 211,0, 151,6, 143,6, 61,6, 56,9, 48,2, 45,9, 45,7, 42,6, 41,9, 37,3, 36,6, 26,3.
Стадия III: К перемешиваемому раствору NaOH (1,75 г, 43,8 ммоль), Н2О (14,6 мл) и 1,4-диоксана (2 мл) при температуре бани со льдом добавляли Br2 (0,8 мл, 16,4 ммоль) и смесь перемешивали в течение 5 минут. Полученный раствор гипобромита добавляли по каплям к перемешиваемому раствору 1-[1-(1H-1,2,4-триазол-1-илметил)трицикло[3,3,1,03,7]нон-3-ил]этанона (0,7 г, 2,92 ммоль) в 1,4-диоксане (4 мл) при температуре бани со льдом. Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 1 ч. Затем смесь охлаждали до температуры бани со льдом и гасили добавлением АсОН (3,9 мл, 65,7 ммоль). Неочищенную реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-(1Н-1,2,4-триазол-1-илметил)трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (0,54 г) с выходом 75% в виде не совсем белого твердого вещества. Т.пл.: 230-235ºС. m/z (M+1) 248. ИК см-1 3436, 3102, 2924, 2511, 1937, 1689, 1523, 1308, 1137, 979, 732. 1Н ЯМР (CD3OD) 300 МГц δ 8,43 (с, 1Н), 7,9 (с, 1Н), 4,17 (с, 2Н), 2,70-2,62 (м, 1Н), 2,40-2,33 (м, 1Н), 2,06-1,95 (м, 2Н), 1,84-1,43 (м, 8Н).
Стадия IV: К перемешиваемой суспензии 1-(1Н-1,2,4-триазол-1-илметил)трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (0,13 г, 0,52 ммоль), полученной на стадии III, в CHCl3 (2,6 мл) добавляли конц. H2SO4 (0,25 мл, 5,2 ммоль). К полученному гомогенному раствору добавляли порциями NaN3 (0,1 г, 1,56 ммоль) в течение периода 30 минут, поддерживая температуру реакционной смеси ниже 40ºС. После перемешивания реакционной смеси в течение 2 ч при комн. темп. реакционную смесь охлаждали до температуры бани со льдом, разбавляли водой и экстрагировали EtOAc. Водный слой подщелачивали добавлением 50% водн. раствора NaOH и экстрагировали CHCl3. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-(1Н-1,2,4-триазол-1-илметил)трицикло[3,3,1,03,7]нонан-3-амина (0,08 г) в виде вязкой жидкости с выходом 70%. m/z (M+1) 219; 1Н ЯМР (CDCl3) 300 МГц δ 7,98 (с, 1Н), 7,93 (с, 1Н), 4,07 (с, 2Н), 2,38-2,30 (м, 1Н), 2,06-1,98 (м, 1Н), 1,96-1,80 (м, 2Н), 1,72-1,57 (м, 4Н), 1,55-1,36 (м, 4Н).
Стадия V: К перемешиваемой смеси 1-(1Н-1,2,4-триазол-1-илметил)трицикло[3,3,1,03,7]нонан-3-амина (0,06 г, 0,38 ммоль) и К2СО3 (0,13 г, 0,96 ммоль) в ДМСО (1 мл) при температуре бани со льдом добавляли соединение (2S)-1-(хлорацетил)пирролидин-2-карбонитрил (0,07 г, 0,32 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ), реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением производного триазола (2S)-1-[1H-1,2,4-триазол-1-илметил(трицикло[3,3,1,03,7]нон-3-иламино)ацетил]пирролидин-2-карбонитрила (0,07 г) с выходом 60% в виде не совсем белого твердого вещества. Т.пл.: 230-235ºС. m/z (M+1) 355. ИК см-1 3429, 2929, 2224, 1658, 1511, 1426, 1330, 1276, 1141, 1018, 747. 1Н ЯМР (CDCl3) 300 МГц δ 8,0 (с, 1Н), 7,9 (с, 1Н), 4,78 (ушир. д, J=6,3 Гц, 1Н), 4,09 (с, 2Н), 3,65-3,35 (м, 4Н), 2,40-2,05 (м, 8Н), 1,80-1,62 (м, 4Н), 1,58-1,32 (м, 4Н).
Пример 2
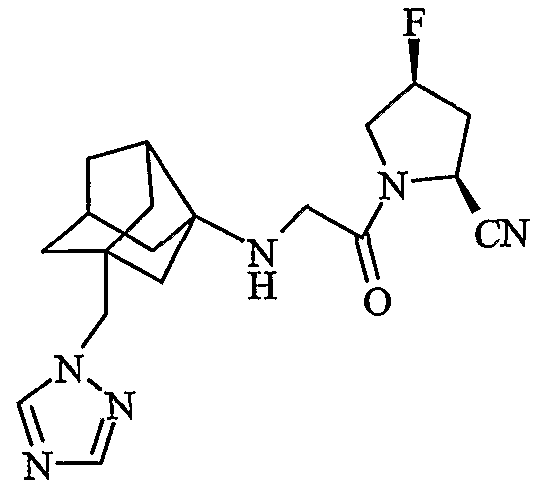
К перемешиваемой смеси триазоламина, который получен на стадии IV примера 1 (0,65 г, 3 ммоль) и К2СО3 (1,24 г, 9 ммоль) в ДМСО (12 мл) при температуре бани со льдом добавляли (2S,4S)-1-(хлорацетил)-4-фторпирролидин-2-карбонитрил (0,57 г, 3 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S,4S)-4-фтор-1-{N-[2-(1Н-1,2,4-триазол-1-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого порошка (0,4 г) с выходом 36%. ИК см-1 3444, 2951, 1672, 1518, 1416, 1301, 1135, 935; m/z (M+1) 373; 1Н ЯМР (CDCl3) 300 МГц δ 8,0 (с, 1Н), 7,92 (с, 1Н), 5,44 (ддд, J=3,4, 3,4, 51,2 Гц, 0,8Н), 5,35 (ддд, J=3,4, 3,4, 51,3 Гц, 0,2Н), 5,04 (т, J=8,8 Гц, 0,2Н), 4,95 (д, 9,2 Гц, 0,8Н), 4,10 (с, 2Н), 4,05-3,51 (м, 2,4Н), 3,4 (с (ушир), 1,6Н), 2,82-2,62 (м, 1Н), 2,45-2,16 (м, 5Н), 1,91-1,69 (м, 4Н), 1,57-1,38 (м, 4Н).
Пример 3
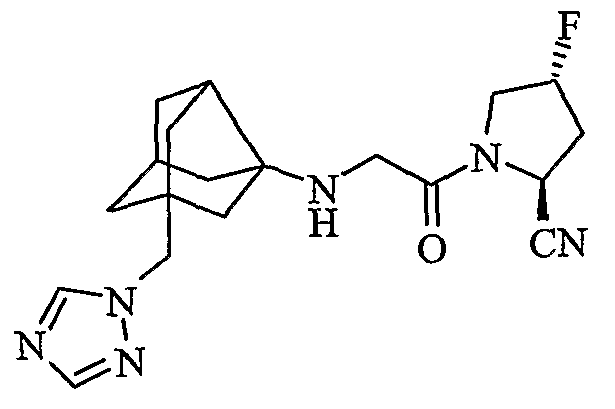
К перемешиваемой смеси триазоламина, который получен на стадии IV в примере 1 (0,4 г, 1,83 ммоль) и К2СО3 (0,4 г, 2,8 ммоль) в ДМСО (4 мл) при температуре бани со льдом добавляли (2S,4R)-1-(хлорацетил)-4-фторпирролидин-2-карбонитрил (0,35 г, 1,83 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S,4R)-4-фтор-1-{N-[2-(1H-1,2,4-триазол-1-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде смолистой жидкости (0,21 г) в виде смеси двух ротамеров с выходом 25%. m/z (M+1) 373; 1Н ЯМР (CDCl3) 300 МГц δ 8,0 (с, 1Н), 7,92 (с, 1Н), 5,35 (д (ушир), J=51,5 Гц, 0,8Н), 5,30 (д (ушир), J=51,3 Гц, 0,2Н), 4,97 (т, J=8,4 Гц, 0,2Н), 4,80 (т, 8,4 Гц, 0,8Н), 4,20-3,32 (м, 4Н), 4,10 (с, 2Н), 2,87-2,40 (м, 1Н), 2,40-2,33 (м, 1Н), 2,25-2,15 (м, 1Н), 1,98-1,38 (м, 11Н).
Пример 4
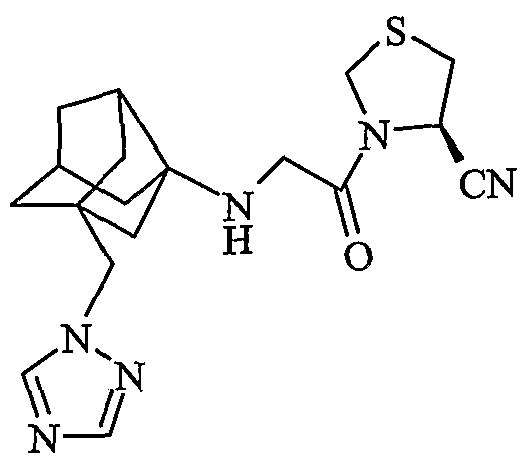
К перемешиваемой смеси триазоламина, который получен на стадии IV примера 1 (0,27 г, 1,05 ммоль) и К2СО3 (0,58 г, 4,2 ммоль) в ДМСО (4 мл) при температуре бани со льдом добавляли (4R)-3-(хлорацетил)-1,3-тиазолидин-4-карбонитрил (0,2 г, 1,05 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (4R)-3-{N-[2-(1H-1,2,4-триазол-1-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}-1,3-тиазолидин-4-карбонитрила в виде светложелтого твердого вещества (0,18 г) с выходом 46%. m/z (М+1) 373; 1Н ЯМР (CDCl3) 300 МГц δ 7,99 (с, 1Н), 7,92 (с, 1Н), 5,32 (т, J=4,1 Гц, 1Н), 4,70-4,55 (м, 2Н), 4,08 (с, 2Н), 3,62-3,48 (м, 2Н), 3,40-3,26 (м, 2Н), 2,42-2,35 (м, 1Н), 2,25-2,15 (м, 1Н), 1,90-1,39 (м, 10Н).
Пример 5
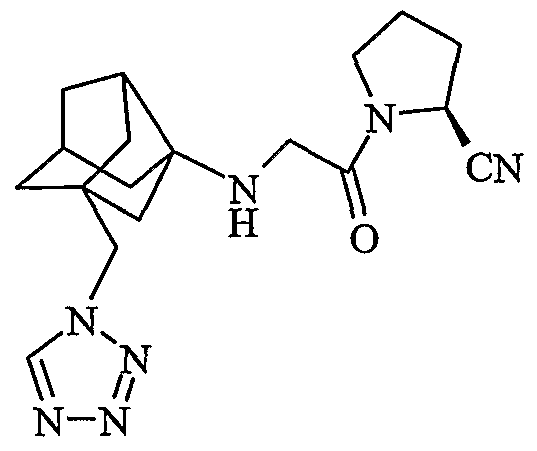
Cтадия I: Смесь {3-[(трет-бутоксикарбонил)амино]-трицикло[3,3,1,03,7]нон-1-ил}метилметансульфонатов, которые получены, как указано ранее (препарат 3) (0,8 г, 2,3 ммоль), К2СО3 (0,95 г, 6,9 ммоль), тетразола (0,24 г, 3,45 ммоль) и ДМФА (10,0 мл) нагревали до 110ºС в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением трет-бутил[1-(1Н-тетразол-1-илметил)трицикло[3,3,1,03,7]нон-3-ил]карбамата в виде вязкой жидкости (0,2 г) с выходом 27%. m/z (M+1) 320; 1Н ЯМР (CDCl3) 300 МГц δ 8,48 (с, 1Н), 4,72 (с (ушир), 1Н), 4,56 (с, 2Н), 2,52-2,43 (м, 1Н), 2,37-2,30 (м, 1Н), 2,20-1,80 (м, 4Н), 1,72 (ддд, J=2,7, 12,3, 15,4 Гц, 2Н), 1,56-1,38 (м, 4Н), 1,44 (с, 9Н).
Стадия II: К перемешиваемому раствору соединения, полученного на стадии I выше по тексту (0,2 г, 0,62 ммоль) в EtOAc (2,0 мл), охлажденному до температуры бани со льдом, добавляли раствор сухого HCl в EtOAc (3н, 3 мл). Реакционную смесь перемешивали при такой же температуре в течение 2 ч и летучие соединения удаляли при пониженном давлении с получением неочищенного вещества, которое растирали некоторое время с простым эфиром с получением чистой хлористоводородной соли 1-(1Н-тетразол-1-илметил)трицикло[3,3,1,03,7]нонан-3-амина (160 мг) с выходом 100%. m/z (М+1) 220; 1Н ЯМР (CD3OD) 300 МГц δ 8,72 (с,1Н), 4,70 (с, 2Н), 2,52-2,43 (м, 1Н), 2,42-2,37 (м, 1Н), 2,0-1,80 (м, 5Н), 1,70-1,50 (м, 5Н).
Стадия III: К перемешиваемому раствору хлористоводородной соли, полученной на стадии II (0,162 г, 0,62 ммоль), в ДМСО (2,5 мл) при комнатной температуре в атмосфере азота добавляли (2S)-1-(хлорацетил)пирролидин-2-карбонитрил (0,11 г, 0,62 ммоль) и К2СО3 (0,34 г, 2,48 ммоль). После перемешивания в течение 3 ч реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-(1H-тетразол-1-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,09 г) с выходом 40%. m/z (M+1) 355; 1Н ЯМР (CDCl3) 300 МГц δ 8,49 (с, 1Н), 4,77 (д, J=6,7 Гц, 1Н), 4,56 (с, 2Н), 3,64-3,38 (м, 2Н), 3,40 (с, 2Н), 2,40-2,05 (м, 6Н), 1,80-1,65 (м, 5Н), 1,60-1,40 (м, 5Н).
Пример 5А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в примере 5 (36 мг, 0,1 ммоль), в метаноле (2 мл), охлажденному до 0ºС, добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие вещества удаляли при пониженном давлении и остаток растирали некоторое время с простым эфиром с получением не совсем белой хлористоводородной соли (2S)-1-{N-[2-(1H-тетразол-1-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила (38 мг).
Пример 6
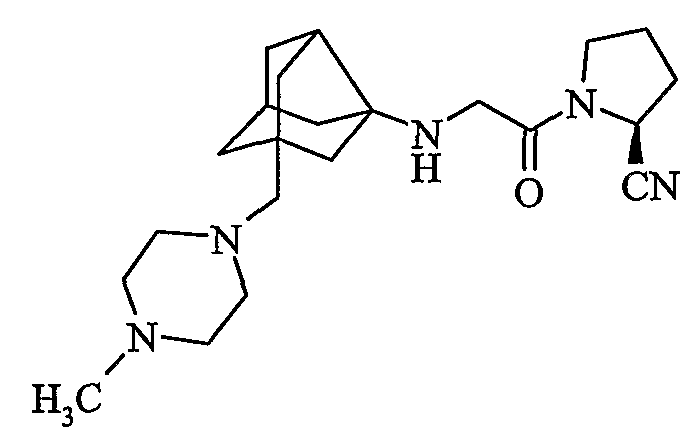
Стадия I: Смесь {3-[(трет-бутоксикарбонил)амино]-трицикло[3,3,1,03,7]нон-1-ил}метилметансульфонатов, которые получены, как указано ранее (препарат 3) (0,85 г, 2,4 ммоль), К2СО3 (1,0 г, 7,2 ммоль), N-метилпиперазина (0,37 мл, 3,6 ммоль) и ДМФА (10,0 мл) нагревали до 110ºС в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением трет-бутил[1-(4-метилпиперазин-1-ил)метил]трицикло[3,3,1,03,7]нон-3-ил]карбамата в виде вязкой жидкости (0,39 г) с выходом 46%. m/z (M+1) 350; 1Н ЯМР (CDCl3) 300 МГц δ 4,72 (с (ушир), 1Н), 2,55-2,25 (м, 10Н), 2,48 (с, 3Н), 2,38 (с, 2Н), 2,0-1,85 (м, 4Н), 1,80-1,72 (м, 1Н), 1,70-1,40 (м, 4Н), 1,45 (с, 9Н), 1,35-1,22 (м, 1Н).
Стадия II: К перемешиваемому раствору соединения, полученного в соответствии со стадией I (0,38 г, 1,09 ммоль) в EtOAc (4,0 мл), охлажденному до температуры бани со льдом, добавляли раствор сухого HCl в EtOAc (3н, 6 мл). Реакционную смесь перемешивали при такой же температуре в течение 2 ч и летучие вещества удаляли при пониженном давлении с получением неочищенных веществ, которые растирали некоторое время с простым диэтиловым эфиром с получением хлористоводородной соли 1-[(4-метилпиперазин-1-ил)метил]трицикло[3,3,1,03,7]нонан-3-амина (330 мг) с выходом 85%.
Стадия III: К перемешиваемому раствору гидрохлорида (0,33 г, 0,92 ммоль), полученного на стадии II, в ДМСО (3,7 мл) при комнатной температуре в атмосфере азота добавляли (2S)-1-(хлорацетил)пирролидин-2-карбонитрил (0,16 г, 0,92 ммоль) и К2СО3 (0,76 г, 5,53 ммоль). После перемешивания в течение 3 ч реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-[(4-метилпиперазин-1-ил)метил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,12 г) с выходом 47%. m/z (M+1) 386; 1Н ЯМР (CDCl3+CD3OD) 300 МГц δ 4,78 (д, J=5,7 Гц, 1Н), 3,85-3,50 (м, 4Н), 3,30-3,10 (м, 5Н), 2,85-2,70 (м, 8Н), 2,52-2,45 (м, 2Н), 2,38-2,20 (м, 4Н), 2,10-1,90 (м, 4Н), 1,70-1,40 (м, 5Н), 1,40-1,27 (м, 1Н).
Пример 6А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в примере 7 (39 мг, 0,1 ммоль) в метаноле (2 мл), охлажденному до 0ºС, добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие соединения удаляли при пониженном давлении и остаток растирали некоторое время с простым диэтиловым эфиром с получением хлористоводородной соли (2S)-1-{N-[2-[(4-метилпиперазин-1-ил)метил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (41 мг).
Пример 7
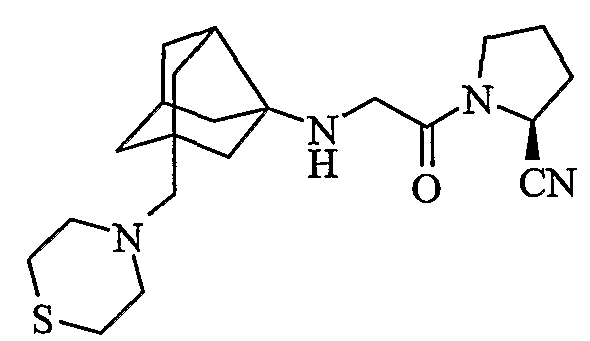
Стадия I: Смесь {3-[(трет-бутоксикарбонил)амино]-трицикло[3,3,1,03,7]нон-1-ил}метилметансульфонатов, которые получали, как указано ранее (препарат 3) (0,85 г, 2,4 ммоль), К2СО3 (1,0 г, 7,2 ммоль), тиоморфолина (0,4 мл, 3,6 ммоль) и ДМФА (10,0 мл) нагревали до 110ºС в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением трет-бутил[1-(тиоморфолин-4-илметил)трицикло[3,3,1,03,7]нон-3-ил]карбамата в виде вязкой жидкости (0,18 г) с выходом 21%. m/z (M+1) 353; 1Н ЯМР (CDCl3) 300 МГц δ 4,72 (с (ушир), 2,75-2,66 (м, 4Н), 2,65-2,58 (м, 4Н), 2,40-2,28 (м, 2Н), 2,17 (с, 2Н), 2,0-1,85 (м, 4Н), 1,80-1,72 (м, 1Н), 1,62-1,50 (м, 4Н), 1,45 (с, 9Н), 1,35-1,20 (м, 1Н).
Стадия II: К перемешиваемому раствору соединения (0,22 г, 0,63 ммоль), полученного на стадии I, в EtOAc (2,0 мл), охлажденному до температуры бани со льдом, добавляли раствор сухого HCl в EtOAc (3н, 4 мл). Реакционную смесь перемешивали при такой же температуре в течение 2 ч и растворитель удаляли при пониженном давлении с получением неочищенного вещества, которое растирали некоторое время с простым диэтиловым эфиром с получением хлористоводородной соли 1-(тиоморфолин-4-илметил)трицикло[3,3,1,03,7]нонан-3-амина (180 мг) с выходом 88%.
Стадия III: К перемешиваемому раствору хлористоводородной соли (0,17 г, 0,51 ммоль), полученной на стадии II (выше по тексту) в ДМСО (2,0 мл) при комнатной температуре в атмосфере азота добавляли (2S)-1-(хлорацетил)пирролидин-2-карбонитрил (0,09 г, 0,52 ммоль) и К2СО3 (0,35 г, 2,55 ммоль). После перемешивания в течение 3 ч реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-тиоморфолин-4-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,07 г) с выходом 35%. m/z (M+1) 389; 1H ЯМР (CDCl3) 300 МГц δ 4,83-4,75 (м, 1Н), 3,90-3,45 (м, 4Н), 2,85-2,70 (м, 4Н), 2,70-2,60 (м, 4Н), 2,48-2,35 (м, 2Н), 2,35-2,15 (м, 4Н), 2,0-1,75 (м, 4Н), 1,68-1,40 (м, 5Н), 1,38-1,25 (м, 1Н).
Пример 7А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в соответствии с примером 7 (39 мг, 0,1 ммоль), в метаноле (2 мл), охлажденному до 0ºС, добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие вещества удаляли при пониженном давлении и остаток растирали некоторое время с простым диэтиловым эфиром с получением хлористоводородной соли (2S)-1-{N-[2-тиоморфолин-4-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (41 мг).
Пример 8
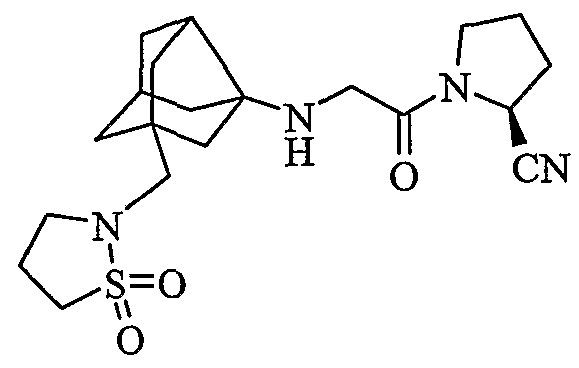
Стадия I: Перемешиваемую смесь [3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метилметансульфоната, который получен, как указано ранее (препарат 2) (1,0 г, 2,9 ммоль), К2СО3 (1,16 г, 8,7 ммоль) и изотиазолидин-1,1-диоксида (0,53 г, 4,35 ммоль) в ДМФА (12,0 мл) нагревали при 110ºС в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением 2-{[3-(2-метил-1,3-диоксолан-2-ил)трицикло[3,3,1,03,7]нон-1-ил]метил}изотиазолидин-1,1-диоксид в виде вязкой жидкости (0,69 г) с выходом 70%. m/z (M+1) 342; 1Н ЯМР (CDCl3) 300 МГц δ 4,04-3,92 (м, 4Н), 3,30 (т, J=6,8 Гц, 2Н), 3,10 (т, J=7,4 Гц, 2Н), 2,94 (д, J=14,6 Гц, 1Н), 2,87 (д, J=14,6 Гц, 1Н), 2,40-2,27 (м, 4Н), 1,88-1,72 (м, 2Н), 1,72-1,55 (м, 4Н), 1,55-1,38 (м, 4Н), 1,27 (с, 3Н).
Стадия II: Перемешиваемый раствор соединения, полученного на стадии I (0,68 г, 2,0 ммоль), и п-толуолсульфокислоты (38 мг, 0,2 ммоль) в ацетоне (8 мл) кипятили с обратным холодильником в течение 4 ч. Реакционную смесь разбавляли EtOAc и промывали 10% водн. NaHCO3 и насыщенным раствором соли, сушили над Na2SO4 и растворитель выпаривали при пониженном давлении с получением 1-{1-[1,1-диоксидоизотиазолидин-2-ил)метил]трицикло[3,3,1,03,7]нон-3-ил}этанона (0,55 г) с выходом 92% в виде вязкой жидкости.
Стадия III: К перемешиваемой смеси NaOH (1,2 г, 27,8 ммоль), Н2О (8 мл) и 1,4-диоксана (1 мл) при температуре бани со льдом добавляли Br2 (0,56 мл, 10,4 ммоль) и смесь перемешивали в течение 15 минут. Полученный раствор гипобромита добавляли по каплям к перемешиваемому раствору соединения, полученного на стадии II (0,55 г, 1,85 ммоль) в 1,4 диоксане (3 мл) при температуре бани со льдом. Реакционную смесь постепенно нагревали до комнатной температуры и затем перемешивали в течение 1 ч; смесь охлаждали до температуры бани со льдом и гасили добавлением АсОН (1,7 мл, 27,8 ммоль). Реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-[(1,1-диоксидоизотиазолидин-2-ил)метил]трицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (0,39 г) с выходом 70%. m/z (M-1) 298; 1Н ЯМР (CDCl3) 300 МГц δ 3,32 (т, J=6,8 Гц, 2Н), 3,10 (т, J=7,4 Гц, 2Н), 2,95 (с, 2Н), 2,80-2,73 (м, 1Н), 2,43-2,41 (м, 1Н), 2,40-2,29 (м, 2Н), 2,10-2,0 (м, 2Н), 1,82-1,72 (м, 3Н), 1,67-1,54 (м, 4Н), 1,46 (дд, J=3,2 11,0 Гц, 1Н).
Стадия IV: К перемешиваемому раствору кислоты, полученной на стадии III (0,39 г, 1,31 ммоль) в CHCl3 (7 мл) добавляли конц. H2SO4 (1,4 мл, 26 ммоль). Медленно, порциями добавляли твердый NaN3 (0,26 г, 3,93 ммоль), поддерживая температуру реакции ниже 40ºС. Реакционную смесь перемешивали при комн. темп. в течение 2 ч, затем охлаждали до температуры бани со льдом, разбавляли водой и экстрагировали EtOAc. Водный слой подщелачивали добавлением 50% раствора NaOH и экстрагировали СНCl3. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4, растворитель выпаривали при пониженном давлении с получением 1-[(1,1-диоксидоизотиазолидин-2-ил)метил]трицикло[3,3,1,03,7]нонан-3-амина (0,26 г) в виде вязкой жидкости с выходом 74%. m/z (M+1) 271; 1Н ЯМР (CDCl3) 300 МГц δ 3,32 (т, J=6,8 Гц, 2Н), 3,10 (т, J=7,4 Гц, 2Н), 2,92 (с, 2Н), 2,40-2,28 (м, 3Н), 2,01-1,83 (м, 3Н), 1,75-1,52 (м, 6Н), 1,50-1,43 (м, 1Н), 1,39-1,32 (м, 1Н).
Стадия V: К перемешиваемой смеси амина, полученного на стадии IV (0,26 г, 0,96 ммоль), и К2СО3 (0,42 г, 2,9 ммоль) в ДМСО (4,0 мл) при температуре бани со льдом в атмосфере азота добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,17 г, 1,0 ммоль). После перемешивания в течение 3 ч при комн. темп. реакционную смесь разбавляли EtOAc, промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель выпаривали при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-[(1,1-диоксидоизотиазолидин-2-ил)метил]гексагидро-2,5-метанпентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,17 г) с выходом 43%. m/z (M+1) 407; 1Н ЯМР (CDCl3) 300 МГц δ 4,87-4,76 (м, 1Н), 3,70-3,40 (м, 2Н), 3,44 (с, 2Н), 3,32 (м, J=6,7 Гц, 2Н), 3,10 (т, J=7,5 Гц, 2Н), 2,93 (с, 2Н), 2,41-2,10 (м, 8Н), 1,90-1,46 (м, 7Н), 1,43-1,35 (м, 1Н).
Пример 9
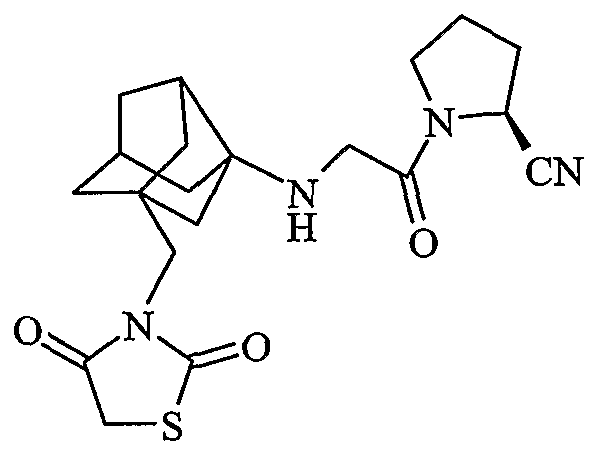
Стадия I: Смесь соединения, полученного, как указано ранее (препарат 3) (0,9 г, 2,6 ммоль), К2СО3 (1,1 г, 7,8 ммоль) и тиазолидин-2,4-диона (0,47 г, 4,0 ммоль) в ДМФА (10,5 мл) нагревали при 110ºС в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением 3{3-[(трет-бутоксикарбонил)амино]трицикло[3,3,1,03,7]нон-1-ил}метил-1,3-тиазолидин-2,4-диона в виде вязкой жидкости (0,28 г) с выходом 25%. m/z (M+1) 367; 1Н ЯМР (CDCl3) 300 МГц δ 4,72 (с (ушир), 1Н), 3,96 (с, 2Н), 3,57 (с, 2Н), 2,46-2,38 (м, 1Н), 2,38-2,30 (м, 1Н), 2,02-1,85 (м, 4Н), 1,80-1,70 (м, 1Н), 1,63 (дд, J=2,9, 10,5 Гц, 1Н), 1,52-1,40 (м, 3Н), 1,44 (с, 9Н), 1,37 (дд, J=3,0, 11,0 Гц, 1Н).
Стадия II: К перемешиваемому раствору соединения, полученного на стадии I (0,2 г, 0,54 ммоль) в EtOAc (2,0 мл), охлажденному до температуры бани со льдом, добавляли раствор сухого HCl в EtOAc (3н, 3 мл). Реакционную смесь перемешивали при такой же температуре в течение 2 ч и растворитель удаляли при пониженном давлении с получением неочищенного вещества, которое растирали некоторое время с простым диэтиловым эфиром с получением хлористоводородной соли (165 мг) с выходом 100%. M/z (M+1) 267; 1Н ЯМР (CD3OD) 300 МГц δ 4,10 (с, 2Н), 3,60 (дд, J=9,0, 11,0 Гц, 2Н), 2,48-2,42 (м, 1Н), 2,34 (ддд, J=1,6, 6,9, 8,5 Гц, 1Н), 1,88-1,68 (м, 6Н), 1,68-1,53 (м, 3Н), 1,50 (дд, J=2,0, 11,4 Гц, 1Н).
Стадия III: К перемешиваемому раствору хлористоводородной соли, полученной на стадии II (0,165 г, 0,54 ммоль) в ДМСО (2,2 мл) при комнатной температуре в атмосфере азота постепенно добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,1 г, 0,54 ммоль) и К2СО3 (0,23 г, 1,62 ммоль). После перемешивания в течение 3 ч реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-[(2,4-диоксо-1,3-тиазолидин-3-ил)метил]гексагидро-2,5-метанпентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,11 г) с выходом 50%. m/z (M+1) 358; 1Н ЯМР (CDCl3) 300 МГц δ 4,77 (д, J=7,4 Гц, 1Н), 3,96 (с, 2Н), 3,75-3,38 (м, 2Н), 3,56 (с, 2Н), 3,41 (с, 2Н), 2,40-2,10 (м, 6Н), 1,90-1,57 (м, 10Н), 1,57-1,33 (м, 4Н).
Пример 10
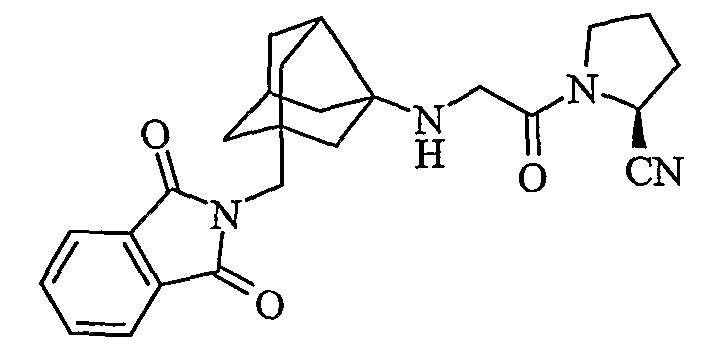
Стадия I: К перемешиваемому раствору трет-бутил[1-(гидроксиметил)трицикло[3,3,1,03,7]нон-3-ил]карбамата (который получен на стадии VI (препарат 3)) (0,67 г, 2,5 ммоль) в толуоле (10 мл) добавляли фталимид (0,52 г, 3,5 ммоль), трифенилфосфин (1,05 г, 4,0 ммоль) и диизопропилазодикарбоксилат (0,8 мл, 4,0 ммоль). Реакционную смесь нагревали при 90ºС в течение 4 ч. Летучие вещества удаляли при пониженном давлении и остаток очищали колоночной хроматографией с получением трет-бутил[2-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата (0,45 г) в виде вязкой жидкости с выходом 46%. m/z (M+1) 397; 1Н ЯМР (CDCl3) 300 МГц δ 7,90-7,80 (м, 2Н), 7,77-7,68 (м, 2Н), 4,75 (с (ушир), 1Н), 3,60 (с, 2Н), 2,47-2,35 (м, 1Н), 2,36-2,29 (м, 1Н), 2,02-1,84 (м, 4Н), 1,77-1,65 (м, 2Н), 1,64-1,35 (м, 5Н), 1,41 (с, 9Н), 1,34-1,24 (м, 1Н).
Стадия II: К перемешиваемому раствору соединения, полученного на стадии I (0,45 г, 1,12 ммоль) в дихлорметане (1,1 мл) при 0ºС добавляли трифторуксусную кислоту (1,1 мл). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 1 ч. Летучие вещества удаляли в вакууме и остаток растирали некоторое время с простым диэтиловым эфиром с получением 2-[(3-аминотрицикло[3,3,1,03,7]нон-1-ил)метил]-1Н-изоиндол-1,3(2Н)-дион (0,4 г) в виде соли с трифторуксусной кислотой с выходом 86%. m/z (M+1) 297; 1Н ЯМР (ДМСО-d6) 300 МГц δ 8,05 (с (ушир), 2Н), 7,92-7,80 (м, 4Н), 3,51 (с, 2Н), 2,38-2,30 (м, 1Н), 2,30-2,22 (м, 1Н), 1,85-1,66 (м, 6Н), 1,60-1,50 (м, 3Н), 1,45-1,38 (м, 1Н).
Стадия III: К перемешиваемой смеси соединения, полученного на стадии II (0,4 г, 0,98 ммоль), и К2СО3 (0,54 г, 3,92 ммоль) в ДМСО (4,0 мл) при температуре бани со льдом в атмосфере азота добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,17 г, 1,0 ммоль). После перемешивания при комн. темп. в течение 3 ч реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (S)-1-{N-[2-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,18 г) с выходом 43%. m/z (M+1) 433; 1Н ЯМР (CDCl3) 300 МГц δ 7,90-7,80 (м, 2Н), 7,75-7,65 (м, 2Н), 4,86-4,72 (м, 1Н), 3,72-3,38 (м, 2Н), 3,60 (с, 2Н), 3,40 (с, 2Н), 2,40-2,05 (м, 7Н), 1,90-1,78 (м, 2Н), 1,70-1,40 (м, 3Н).
Пример 11
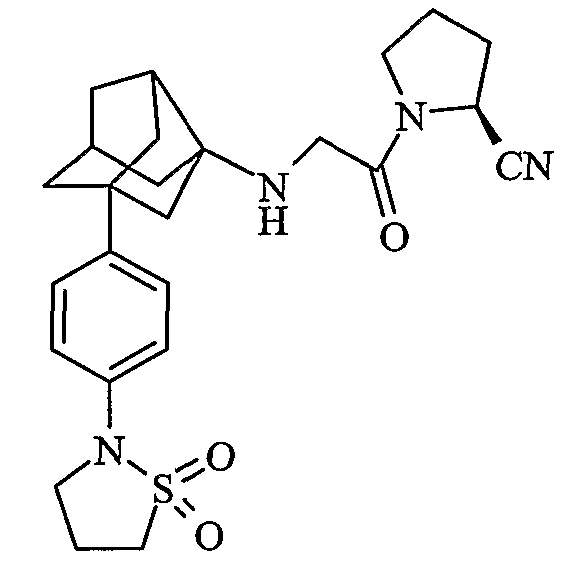
Стадия I: К перемешиваемому раствору бензил-[2-(4-аминофенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата (1,1 г, 3,03 ммоль), полученного, как указано ранее (препарат 4), в ТГФ (30 мл) при 0ºС добавляли Et3N (0,66 мл, 4,6 ммоль) и 3-хлорпропансульфонилхлорид (0,42 мл, 3,3 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Добавляли водный раствор NaOH (50% масс./об., 6 мл) с последующим добавлением n-Bu4NI (56 мг, 0,15 ммоль). После перемешивания в течение 16 ч реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением бензил-[2-(4-(1,1-диоксидоизотиазолидин-2-ил)фенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата в виде не совсем белого твердого вещества (0,93 г) с выходом 66%. Т.пл.: 215,6-219,2ºС, m/z (M+1) 467; ИК см-1 3441, 2953, 1716, 1516, 1314, 1215, 770. 1Н ЯМР (CDCl3) 300 МГц δ 7,40-7,16 (м, 9Н), 5,13-5,0 (ушир.с, 2Н), 3,75 (т, J=6,6 Гц, 2Н), 3,35 (т, J=7,5 Гц, 2Н), 2,59-2,38 (м, 4Н), 2,30-1,60 (м, 10Н).
Стадия II: Смесь бензил-[2-(4-(1,1-диоксидоизотиазолидин-2-ил)фенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата (0,8 г, 1,7 ммоль), полученного на стадии I, Pd/C (10%, 0,4 г) в МеОН (17 мл) перемешивали при комнатной температуре в атмосфере Н2 в течение 2 ч. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением 1-[4-(1,1-диоксидоизотиазолидин-2-ил)фенил]трицикло[3,3,1,03,7]нонан-3-амина в виде не совсем белого твердого вещества (0,49 г) с выходом 85%. m/z (M+1) 333; ИК см-1 3418, 1652, 1137, 772. 1Н ЯМР (CDCl3) 300 МГц, δ 7,31 (д, J=8,8 Гц, 2Н), 7,22 (д, J=8,8 Гц, 2Н), 3,78 (т, J=6,6 Гц, 2Н), 3,43 (т, J=7,4 Гц, 2Н), 2,58-2,47 (м, 3Н), 2,40-2,34 (м, 1Н), 2,27-2,11 (м, 2Н), 2,10-1,95 (м, 4Н), 1,93-1,70 (м, 4Н).
Стадия III: К перемешиваемой смеси 1-[4-(1,1-диоксидизотиазолидин-2-ил)фенил]трицикло[3,3,1,03,7]нонан-3-амина (0,4 г, 1,2 ммоль) и К2СО3 (0,48 г, 3,6 ммоль) в ДМСО (4,8 мл) при температуре бани со льдом добавляли (2S)-1-(хлорацетил)пирролидин-2-карбонитрил (0,25 г, 1,44 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-[4-(1,1-диоксидизотиазолидин-2-ил)фенил]гексагидро-2,5-метанопентален-3а(Н)-ил]глицил}пирролидин-2-карбонитрила в виде белого твердого вещества (0,28 г) с выходом 50%. Т.пл. 214-216ºС, m/z (M+1) 469; ИК-1 3436, 2932, 2240, 1658, 1517, 1414, 1308, 1137, 952, 740. 1Н ЯМР (300 МГц, CD3OD) δ: 7,35 (д, J=8,5 Гц, 2Н), 7,23 (д, J=8,5 Гц, 2Н), 4,85 (т, J=5,4 Гц, 1Н), 4,15-3,95 (м, 2Н), 3,81-3,70 (м, 3Н), 3,60-3,50 (м, 1Н), 3,42 (т, J=7,4 Гц, 2Н), 2,65-2,45 (м, 4Н), 2,40-1,75 (м, 14Н).
Пример 12
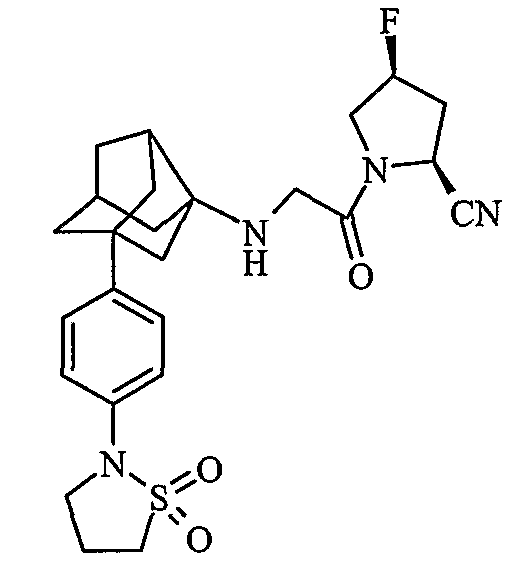
К перемешиваемой смеси 1-[4-(1,1-диоксидизотиазолидин-2-ил)фенил]трицикло[3,3,1,03,7]нонан-3-амина (который получен на стадии II примера 8) (0,17 г, 0,5 ммоль) и К2СО3 (0,21 г, 1,5 ммоль) в ДМСО (2 мл) при температуре бани со льдом добавляли (2S,4S)-1-хлорацетил)-4-фторпирролидин-2-карбонитрил (0,1 г, 0,5 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S,4S)-1-{N-[2-[4-(1,1-диоксидизотиазолидин-2-ил)фенил]гексагидро-2,5-метанопентален-3а(Н)-ил]глицил}-4-фторпирролидин-2-карбонитрила в виде не совсем белого порошка (0,09 г) с выходом 37% в форме смеси 3:1 двух ротомеров. ИК см-1 3438, 2953, 2776, 1673, 1518, 1427, 1301, 1135, 1078, 955; m/z (M+1) 487; 1Н ЯМР (CDCl3) 300 МГц δ 7,25 (д, J=6,2 Гц, 2Н), 7,20 (д, J=8,6 Гц, 2Н), 5,42 (д (ушир), J=51,0 Гц, 0,8Н), 5,34 (д (ушир), J=50,4 Гц, 0,2Н), 5,15 (д, J=10,2 Гц, 0,2Н), 4,98 (д, J=7,4 Гц, 0,8Н), 4,0-3,50 (м, 4Н), 3,76 (т, J=6,5 Гц, 2Н), 3,35 (т, J=6,5 Гц, 2Н), 2,80-2,28 (м, 6Н), 2,15-1,58 (м, 10Н).
Пример 13
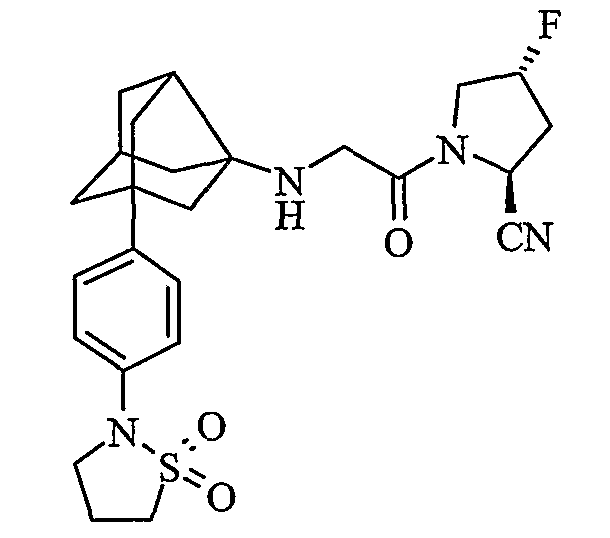
К перемешиваемой смеси 1-[4-(1,1-диоксидизотиазолидин-2-ил)фенил]трицикло[3,3,1,03,7]нонан-3-амина, который получен на стадии II примера 8 (0,22 г, 0,7 ммоль) и К2СО3 (0,3 г, 2,1 ммоль) в ДМСО (3,4 мл) при температуре бани со льдом добавляли (2S,4R)-1-(хлорацетил)-4-фторпирролидин-2-карбонитрил (0,13 г, 2,1 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S,4R)-1-{N-[2-[4-(1,1-диоксидоизотиазолидин-2-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}-4-фторпирролидин-2-карбонитрила в виде не совсем белого порошка (0,14 г) в форме смеси 3:1 двух ротамеров, с выходом 43%. m/z (M+1) 487; 1H ЯМР (CDCl3) 300 МГц δ 7,25 (д, J=7,9 Гц, 2Н), 7,20 (д, J=8,7 Гц, 2Н), 5,35 (д (ушир), J=51,4 Гц, 0,8Н), 5,28 (д (ушир), J=50,7 Гц, 0,2Н), 5,02 (т, J=7,2 Гц, 0,2Н), 4,80 (т, J=8,4 Гц, 0,8Н), 4,02-3,41 (м, 4Н), 3,76 (т, J=6,6 Гц, 2H), 3,37 (т, J=7,4 Гц, 2Н), 2,85-2,25 (м, 6Н), 2,15-1,55 (м, 10Н).
Пример 14
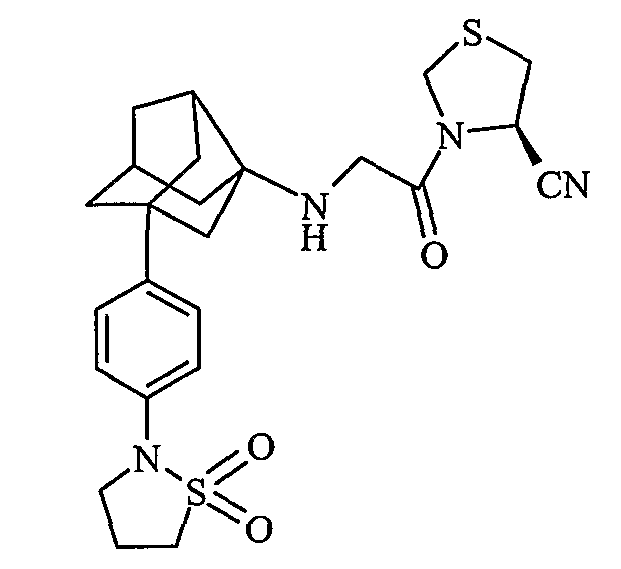
К перемешиваемой смеси 1-[4-(1,1-диоксидоизотиазолидин-2-ил)фенил]трицикло[3,3,1,03,7]нонан-3-амина, который получен на стадии II примера 8 (0,25 г, 1,1 ммоль), и К2СО3 (0,46 г, 3,3 ммоль) в ДМСО (5 мл) при температуре бани со льдом добавляли (4R)-3-(хлорацетил)-1,3-тиазолидин-4-карбонитрил (0,2 г, 1,1 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (4R)-3-{N-[2-[4-(1,1-диоксидоизотиазолидин-2-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}-1,3-тиазолидин-4-карбонитрила в виде не совсем белого порошка (0,1 г) с выходом 40%. m/z (M+1) 487; ИК см-1 3444, 2952, 2873, 1672, 1518, 1416, 1301, 1135, 953; 1Н ЯМР (CDCl3) 300 Мгц δ 7,25 (д, J=6,2 Гц, 2Н), 7,20 (д, J=8,6 Гц, 2Н), 5,38-5,48 (м, 1Н), 4,72-4,51 (м, 2Н), 3,75 (т, J=6,6 Гц, 2Н), 3,70-3,55 (м, 1Н), 3,36 (т, J=7,4 Гц, 2Н), 3,32-3,20 (м, 1Н), 2,60-2,40 (м, 4Н), 2,20-1,58 (м, 10Н).
Пример 15
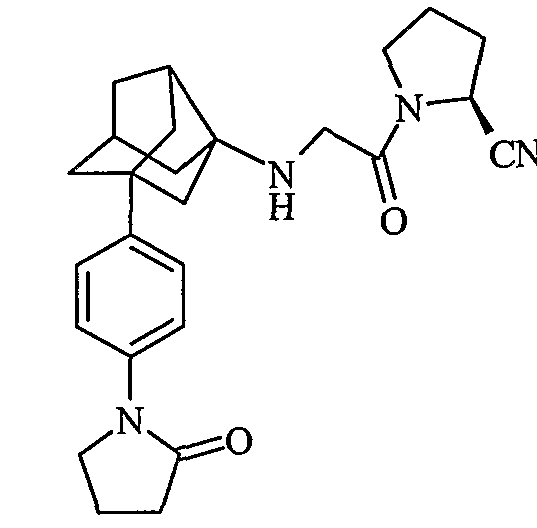
Стадия I: К перемешиваемому раствору бензил-[2-(4-аминофенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата (1,1 г, 3,03 ммоль), полученного, как указано ранее (препарат 4), в ТГФ (30 мл) при 0ºС последовательно добавляли Et3N (0,66 мл, 4,6 ммоль) и 4-хлорбутирилхлорид (0,37 мл, 3,3 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Добавляли водный раствор NaOH (50% масс./об., 6 мл) с последующим добавлением n-Bu4NI (56 мг, 0,15 ммоль) и реакционную смесь перемешивали в течение 16 ч. Реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4, растворитель удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением бензил-[2-[4-(2-оксопирролидин-1-ил)фенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата в виде не совсем белого твердого вещества (0,9 г, выход 69%). m/z (M+1) 431; 1Н ЯМР (CDCl3) 300 МГц δ 7,55 (д, J=8,6 Гц, 2Н), 7,42-7,20 (м, 7Н), 5,15-5,0 (ушир. с, 2Н), 3,87 (т, J=7,0 Гц, 2Н), 2,67-2,53 (м, 3Н), 2,48-2,42 (м, 1Н), 2,35-1,55 (м, 12Н).
Стадия II: Cмесь бензил-[2-[4-(2-оксопирролидин-1-ил)фенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата (0,73 г, 1,7 ммоль), полученного на стадии I, и Pd/C (10%, 0,4 г) в МеОН (17 мл) перемешивали при комнатной температуре в атмосфере Н2 в течение 2 ч. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением 1-[4-(3-аминотрицикло[3,3,1,03,7]нон-1-ил)фенил]пирролидин-2-она в виде не совсем белого твердого вещества (0,43 г, 85% выход). m/z (M+1) 297; 1Н ЯМР (CDCl3) 300 МГц δ 7,52 (д, J=8,6 Гц, 2Н), 7,25 (д, J=8,6 Гц, 2Н), 3,86 (т, J=6,9 Гц, 2Н), 2,62 (т, J=7,9 Гц, 2Н), 2,52-2,42 (м, 1Н), 2,32-1,60 (м, 13Н).
Стадия III: К перемешиваемому раствору 1-[4-(3-аминотрицикло[3,3,1,03,7]нон-1-ил)фенил]пирролидин-2-она, полученного на стадии II (0,36 г, 1,2 ммоль), в ДМСО (4,8 мл) при температуре бани со льдом в атмосфере азота добавляли К2СО3 (0,48 г, 3,6 ммоль) с последующим добавлением (2S)-1-(хлорацетил)пирролидин-2-карбонитрила (0,21 г, 1,2 ммоль). После перемешивания при комн. темп. в течение 3 ч реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением названного соединения (2S)-1-{N-[2-[4-(2-оксопирролидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде белого твердого вещества (0,26 г) с выходом 50%. Т.пл.: 254-256ºС; m/z (M+1) 433; 1Н ЯМР (300 МГц, CDCl3) δ: 7,54 (д, J=8,5 Гц, 2Н), 7,23 (д, J=8,5 Гц, 2Н), 4,30-4,20 (м, 1Н), 4,06-4,18 (м, 2Н), 3,87 (т, J=7,0 Гц, 1Н), 3,80-3,50 (м, 4Н), 2,64 (т, J=7,8 Гц, 2Н), 2,58-1,60 (м, 18Н).
Пример 16

К перемешиваемой смеси лактамамина, 1-[4-(3-аминотрицикло[3,3,1,03,7]нон-1-ил)фенил]пирролидин-2-она, который получен на стадии II примера 12 (0,15 г, 0,5 ммоль), и К2СО3 (0,21 г, 1,5 ммоль) в ДМСО (2 мл) при температуре бани со льдом добавляли (2S,4S)-1-(хлорацетил-4-фторпирролидин-2-карбонитрил (0,1 г, 0,5 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S,4S)-4-фтор-1-{N-[2-[4-(2-оксопирролидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого порошка (0,11 г) с выходом 49%. m/z (M+1) 451; 1Н ЯМР (CDCl3) 300 МГц δ 7,51 (д, J=8,5 Гц, 2Н), 7,25 (д, J=8,5 Гц, 2Н), 5,43 (ддд, J=3,1, 3,1, 51,3 Гц, 1Н), 5,12 (д, J=8,7 Гц, 0,2Н), 4,97 (д, J=9,5 H), 4,05-3,62 (м, 2Н), 3,85 (т, J=6,9 Гц, 2Н), 3,55-3,40 (м, 2Н), 2,81-2,61 (м, 1Н), 2,60 (т, J=8,2 Гц, 2Н), 2,49-2,40 (м, 1Н), 2,32-2,25 (м, 1Н), 2,21-2,05 (м, 4Н), 2,0-1,60 (м, 9Н).
Пример 17
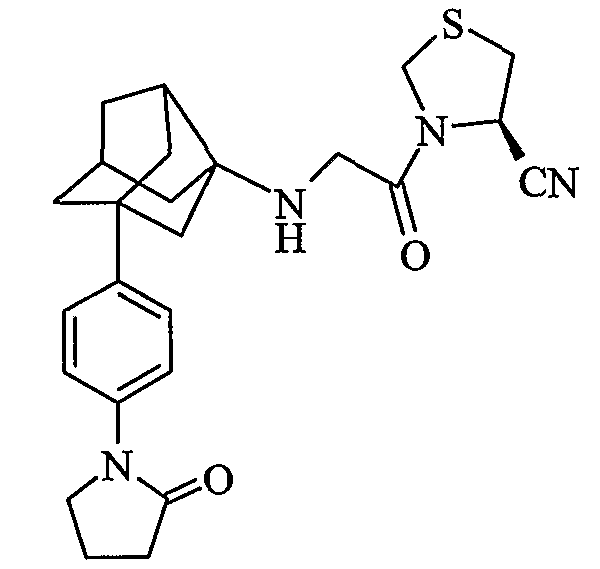
К перемешиваемой смеси лактамамина, полученного на стадии II примера 12 (0,15 г, 0,5 ммоль), и К2СО3 (0,21 г, 1,5 ммоль) в ДМСО (2 мл) при температуре бани со льдом добавляли (4R)-3-(хлорацетил)-1,3-тиазолидин-4-карбонитрил (0,1 г, 1,1 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (4R)-3-{N-[2-[4-(2-оксопирролидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}-1,3-тиазолидин-4-карбонитрила в виде не совсем белого порошка (0,09 г) с выходом 40%. m/z (M+1) 451; 1Н ЯМР (CDCl3) 300 МГц δ 7,51 (д, J=6,7 Гц, 2Н), 7,24 (д, J=7,0 Гц, 2Н), 5,34 (т, J=4,1 Гц, 1Н), 4,65 (д, J=7,5 Гц, 1Н), 4,60 (д, J=7,5 Гц, 1Н), 3,85 (т, J=6,9 Гц, 2Н), 3,70-3,55 м, 2Н), 3,40-3,25 (м, 2Н), 2,60 (т, J=7,8 Гц, 2Н), 2,49-2,42 (м, 1Н), 2,33-2,25 (м, 1Н), 2,21-2,02 (м, 4Н), 2,0-1,6 (м, 8Н).
Пример 18
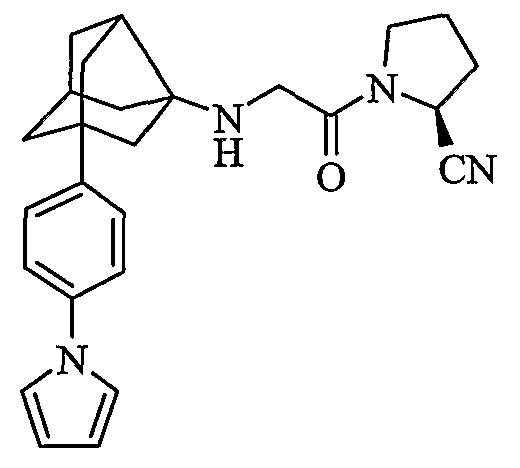
Стадия I: К перемешиваемому раствору бензил-[2-(4-аминофенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата, полученного, как указано ранее (препарат 4) (1,09 г, 3,0 ммоль), в ледяной уксусной кислоте (12 мл) добавляли 2,5-диметокситетрагидрофуран (0,44 г, 3,3 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 1 ч. Смесь разбавляли этилацетатом, промывали водой, 10% водн. NaHCO3 и насыщенным раствором соли. Объединенный органический слой сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением бензил-[2-[4-(1H-пиррол-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата (0,8 г) с выходом 65% в виде вязкой жидкости. m/z (M+1) 413; 1Н ЯМР (CDCl3) 300 МГц δ 7,50-7,20 (м, 9Н), 7,08-7,04 (м, 2Н), 6,37-6,30 (м, 2Н), 5,10 (с, 2Н), 2,65-2,52 (м, 1Н), 2,50-2,42 (м, 1Н), 2,40-2,12 (м, 2Н), 2,10-1,70 (м, 6Н), 1,70-1,52 (м, 2Н).
Стадия II: К перемешиваемому раствору соединения, полученного на стадии I (0,8 г, 1,94 ммоль) в МеОН (20 мл) добавляли Pd/C (10%, 0,1 г). Реакционную смесь перемешивали при комн. темп. в течение 2 ч под давлением Н2, подаваемого из баллона. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением 2-[4-(1H-пиррол-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-амина в виде вязкой жидкости (0,45 г) с выходом 83%. m/z (M+1) 279; 1Н ЯМР (CD3ОD) 300 МГц δ 7,40-7,25 (м, 4Н), 7,20-7,05 (м, 2Н), 6,30-6,20 (м, 2Н), 2,57-2,46 (м, 1Н), 2,46-2,38 (м, 1Н), 2,30-2,13 (м, 2Н), 2,13-1,81 (м, 7Н), 1,80-1,70 (м, 1Н).
Стадия III: К перемешиваемой смеси соединения, полученного на стадии II (0,41 г, 1,5 ммоль), и К2СО3 (0,62 г, 4,5 ммоль) в ДМСО (6 мл) при температуре бани со льдом в атмосфере N2 добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,26 г, 1,5 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-[4-(1Н-пиррол-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества, (0,2 г) с выходом 32%. m/z (M+1) 415; 1H ЯМР (CDCl3) 300 МГц δ 7,38-7,26 (м, 4Н), 7,10-7,05 (м, 2Н), 6,36-6,32 (м, 2Н), 4,84-4,78 (м, 1Н), 3,75-3,40 (м, 2Н), 3,50 (с, 2Н), 2,50-2,42 (м, 1Н), 2,38-2,28 (м, 2Н), 2,25-2,0 (м, 6Н), 2,0-1,70 (м, 7Н).
Пример 19
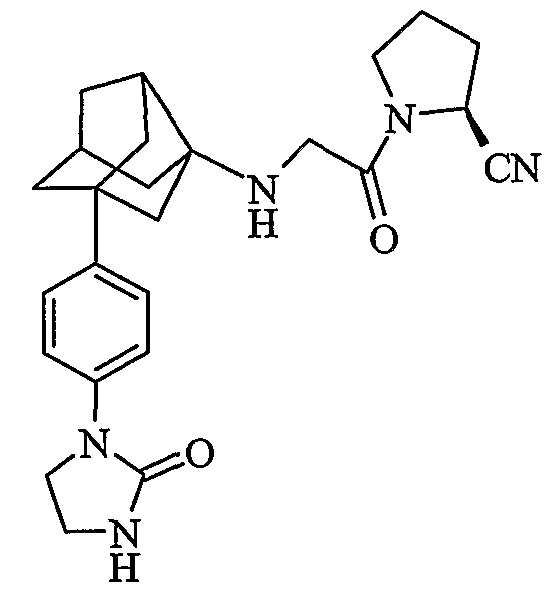
Стадия I: К перемешиваемому раствору бензил-[2-(4-аминофенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата, полученного, как указано ранее (препарат 4) (1,09 г, 3,0 ммоль), в ТГФ (12 мл) при 0ºС добавляли Et3N (0,65 мл, 4,5 ммоль), с последующим добавлением 2-хлорэтилизоцианата (0,3 мл, 3,3 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Добавляли водный раствор NaOH (50% масс./об., 6 мл) с последующим добавлением n-Bu4NI (55 мг, 0,15 ммоль) и реакционную смесь перемешивали в течение 16 ч. Два слоя разделяли и водный слой экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель выпаривали при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением бензил-[2-[4-(2-оксоимидазолидин-1-ил]гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата (0,7 г) в виде не совсем белого твердого вещества с выходом 54%. m/z (M+1) 432; 1Н ЯМР (CDCl3) 300 МГц δ 7,44 (д, J=8,8 Гц, 2Н), 7,40-7,30 (м, 5Н), 7,21 (д, J=8,8 Гц, 2Н), 5,09 (с, 2Н), 3,92 (т, J=7,4 Гц, 3Н), 3,55 (т, J=8,5 Гц, 2Н), 2,59-2,50 (м, 1Н), 2,44-2,38 (м, 1Н), 2,30-2,11 (м, 3Н), 2,11-1,90 (м, 3Н), 1,89-1,66 (м, 2Н), 1,64-1,46 (м, 2Н).
Стадия II: К перемешиваемому раствору соединения, полученного на стадии I (0,65 г, 1,5 ммоль), в МеОН (15 мл) добавляли Pd/C (10%, 0,1 г). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч под давлением Н2, подаваемого из баллона. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением 1-[4-(3-аминотрицикло[3,3,1,03,7]нон-1-ил)фенил]имидазолидин-2-она (0,4 г) в виде не совсем белого твердого вещества с выходом 90%. Т.пл.: 215-220ºС; m/z (M+1) 298; ИК см-1 3412, 3245, 2955, 1687, 1518, 1485, 1263, 805; 1Н ЯМР (CDCl3+ДМСО-d6) 300 МГц δ 8,26 (с (ушир), 2Н), 7,46 (д, J=8,7 Гц, 2Н), 7,16 (д, J=8,7 Гц, 2Н), 6,70 (с (ушир), 1Н), 3,86 (т, J=7,3 Гц, 2Н), 3,48 (т, J=7,3 Гц, 2Н), 2,50-2,45 (м, 1Н), 2,22-2,07 (м, 3Н), 2,02-1,63 (м, 8Н).
Стадия III: К перемешиваемой смеси соединения, полученного на стадии II (0,4 г, 1,35 ммоль), и К2СО3 (0,56 г, 4,05 ммоль) в ДМСО (6 мл) при температуре бани со льдом в атмосфере N2 добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,23 г, 1,35 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель выпаривали при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-[4-(2-оксоимидазолидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,21 г) с выходом 36%. m/z (M+1) 434; 1Н ЯМР (CDCl3) 300 МГц δ 7,45 (д, J=8,8 Гц, 3Н), 7,23 (д, J=8,8 Гц, 2Н), 4,86-4,76 (м, 1Н), 4,67 (с (ушир), 1Н), 3,93 (т, J=7,4 Гц, 2Н), 3,75-3,40 (м, 4Н), 3,48 (с, 2Н), 3,46-2,38 (м, 1Н), 2,36-2,23 (м, 2Н), 2,23-1,70 (м, 12Н), 1,65-1,57 (м, 1Н).
Пример 20
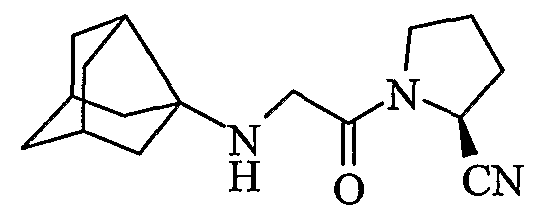
Стадия I: К перемешиваемой смеси трицикло[3,3,1,03,7]нонан-3-амина (0,28 г, 2,0 ммоль) и К2СО3 (0,83 г, 6,0 ммоль) в ДМСО (8 мл) при температуре бани со льдом в атмосфере N2 добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,34 г, 2,0 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-[(трицикло[3,3,1,03,7]нон-3-иламино)ацетил]пирролидин-2-карбонитрила в виде вязкой жидкости (0,23 г) с выходом 42%. m/z (M+1) 274; 1H ЯМР (CDCl3) 300 МГц δ 4,87-4,76 (м, 1Н), 3,71-3,40 (м, 2Н), 3,41 (с, 2Н), 2,35-2,05 (с, 7Н), 1,92-1,74 (м, 6Н), 1,68-1,48 (м, 4Н).
Пример 19А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного по примеру 18 (27 мг, 0,1 ммоль) в метаноле (2 мл), охлажденному до 0ºС, добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие соединения удаляли при пониженном давлении и остаток растирали некоторое время с простым диэтиловым эфиром с получением хлористоводородной соли (2S)-1-[(трицикло[3,3,1,03,7]нон-3-иламино)ацетил]пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (31 мг). m/z (M+1) 274; 1Н ЯМР (ДМСО-d6) 300 МГц δ 9,44 (с (ушир), 2Н), 4,86 (дд, J=4,4 7,0 Гц, 1Н), 4,10-3,88 (м, 2Н), 3,78-3,65 (м, 1Н), 3,60-3,47 (м, 1Н), 2,48-2,41 (м, 1Н), 2,34-2,27 (м, 2Н), 2,26-2,17 (м, 2Н), 2,10-1,99 (м, 2Н), 1,99-1,83 (м, 6Н), 1,63-1,43 (м, 4Н).
Пример 21
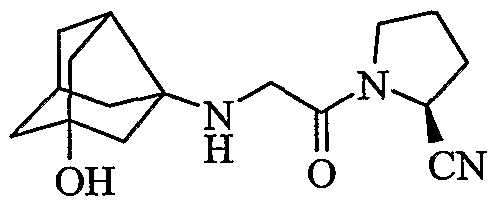
Стадия I: К перемешиваемому раствору трет-бутил(1-гидрокситрицикло[3,3,1,03,7]нон-3-ил)карбамата, который получен как указано ранее (препарат 11) (500 мг, 1,97 ммоль) в EtOAc (5 мл), охлажденному до температуры бани со льдом, добавляли раствор сухого HCl в EtOAc (4н, 5 мл). Затем реакционную смесь перемешивали в течение 2 ч, летучие вещества удаляли при пониженном давлении. Неочищенное вещество растирали некоторое время с простым диэтиловым эфиром с получением гидрохлорида 3-аминотрицикло[3,3,1,03,7]нонан-1-ола (280 мг) с выходом 75%. m/z (M+1) 154; 1Н ЯМР (CD3OD) 300 МГц δ 2,51-2,44 (м, 1Н), 2,34 (ддд, J=2,0, 7,0, 8,9 Гц, 1Н), 2,17-2,04 (м, 2Н), 1,94-1,71 (м, 6Н), 1,60-1,53 (м, 2Н).
Стадия II: К перемешиваемой смеси соединения, полученного на стадии I (280 мг, 1,5 ммоль), и К2СО3 (820 мг, 6 ммоль) в ДМСО (6 мл) при температуре бани со льдом в атмосфере N2 добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (205 мг, 1,2 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{[(1-гидрокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрила в виде вязкой жидкости (150 мг) с выходом 35%. m/z (M+1) 291; 1Н ЯМР (CDCl3) 300 МГц δ 4,78 (д, J=7,0 Гц, 1Н), 3,75-3,38 (м, 2Н), 3,43 (с, 2Н), 2,45-2,37 (м, 1Н), 2,35-1,95 (м, 8Н), 1,90-1,60 (м, 6Н), 1,53-1,40 (м, 2Н).
Пример 22
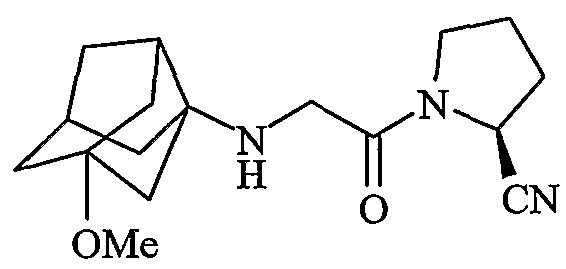
Стадия I: К суспензии NaH (60% дисперсия в медицинском масле, 0,96 г, 24 ммоль) в ТГФ (40 мл), охлажденной до температуры бини со льдом, добавляли через шприц гидроксиадамантанон (3,32 г, 20 ммоль), растворенный в ТГФ (40 мл), в течение периода 15 минут. После перемешивания реакционной смеси в течение 30 мин добавляли иодметан (1,38 мл, 22 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч до тех пор, пока методом ТСХ не обнаруживали завершение реакции. Избыток NaH гасили добавлением насыщенного водн. раствора NH4Cl к охлаждаемой льдом реакционной смеси. Два слоя разделяли и водный слой экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением неочищенной реакционной массы, которую очищали колоночной хроматографией с получением 5-метоксиадамантан-2-она (3,0 г) с выходом 83%. m/z (M+1) 181; 1Н ЯМР (СDCl3) 300 МГц δ 3,26 (с, 3Н), 2,67-2,61 (м, 2Н), 2,39-2,33 (м, 1Н), 2,20-1,93 (м, 10Н).
Стадия II: Только что полученный метилмагнийиодид в простом эфире (1М, 32 мл) через трубку добавляли к 5-метоксиадамантан-2-ону (3,0 г, 16 ммоль) в ТГФ (32 мл) при 0ºС. После перемешивания смеси при 0ºС в течение 0,5 ч, реакционную смесь гасили добавлением насыщенного водн. раствора NH4Cl. Органический слой отделяли и водный слой экстрагировали простым диэтиловым эфиром. Объединенный органический слой промывали водой и насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением 5-метокси-2-метиладамантан-2-ола в виде смеси аномеров 1:1 (выход 3,0 г, 96%). m/z (M+23 219; 1Н ЯМР (CDCl3) 300 МГц δ 3,24 (с, 1½ Н), 3,23 (с, 1½ Н), 2,68-2,62 (м, 1Н), 2,40-1,55 (м, 10Н), 1,53-1,35 (м, 2Н), 1,38 (с, 3Н).
Стадия III: 5-Метокси-2-метиладамантан-2-ол (3,0 г, 15 ммоль), растворенный в смеси АсОН (3,0 мл) и ТГФ (15 мл (15 мл) добавляли по каплям с помощью капельной воронки к охлажденному на бане со льдом раствору NaOCl (4%, 150 мл) в течение периода 15 минут. Добавляли n-Bu4NI (0,55 г, 1,5 ммоль) и реакционную смесь перемешивали в течение 1,5 ч. Реакционную смесь разделяли на два слоя. Водный слой экстрагировали простым диизопропиловым эфиром и объединенный органический слой промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель выпаривали при пониженном давлении с получением гипохлорита, который разбавляли метанолом (30 мл) и добавляли твердый КОН (1,68 г, 30 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 1 ч. Летучие вещества удаляли и неочищенное вещество разбавляли простым эфиром, промывали водой, насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали в вакууме. Неочищенное вещество очищали колоночной хроматографией с получением 1-(1-метокситрицикло[3,3,1,03,7]нон-3-ил)этанона (1,92 г) с выходом 66%. m/z (M+1) 195; 1Н ЯМР (CDCl3) 300 МГц δ 3,29 (с, 3Н), 2,69-2,62 (м, 1Н), 2,57-2,50 (м, 1Н), 2,26-2,16 (м, 1Н), 2,18 (с, 3Н), 2,10-1,61 (м, 7Н), 1,59-1,52 (м, 2Н).
Стадия IV: К смеси NaOH (0,58 г, 147 ммоль), Н2О (40,0 мл) и 1,4-диоксана (10 мл) при температуре бани со льдом добавляли Br2 (2,8 мл, 55,0 ммоль) и перемешивали в течение 5 минут. Полученный раствор гипобромита добавляли по каплям к перемешиваемому раствору 1-(1-метокситрицикло[3,3,1,03,7]нон-3-ил)этанона, полученного на стадии III (1,9 г, 9,8 ммоль) в 1,4 диоксане (10 мл) при температуре бани со льдом. Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 1 ч. Реакционную смесь охлаждали до температуры бани со льдом и добавляли АсОН (3,9 мл, 65,7 ммоль). Реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-метокситрицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (1,3 г) с выходом 72%. m/z (M+1) 197; 1Н ЯМР (CDCl3) 300 МГц δ 3,30 (с, 3Н), 2,78-2,70 (м, 1Н), 2,57-2,48 (м, 1Н), 2,38-2,28 (м, 1Н), 2,11-2,0 (м, 2Н), 1,86-1,69 (м, 5Н), 1,59-1,51 (м, 2Н).
Стадия V: К суспензии 1-метокситрицикло[3,3,1,03,7]нонан-3-карбоновой кислоты, полученной на стадии IV (0,39 г, 2,0 ммоль), в CHCl3 (10 мл) добавляли конц. H2SO4 (1,0 мл, 20 ммоль). Добавляли порциями твердый NaN3 (0,39 г, 6,0 ммоль), поддерживая температуру реакционной смеси ниже 40ºС. После перемешивания при комнатной температуре в течение 2 ч реакционную смесь охлаждали до температуры бани со льдом, разбавляли водой и экстрагировали EtOAc. Водный слой подщелачивали добавлением 50% раствора NaOH и экстрагировали CHCl3. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-метокситрицикло[3,3,1,03,7]нонан-3-амина (0,23 г) с выходом 69%. m/z (M+1) 168; 1Н ЯМР (CDCl3) 300 МГц δ 3,27 (с, 3Н), 2,40-2,34 (м, 1Н), 2,16-2,08 (м, 1Н), 1,97 (ддд, J=2,0, 6,8, 8,7 Гц, 1Н), 1,92-1,53 (м, 7Н), 1,51-1,38 (м, 2Н).
Стадия VI: К перемешиваемому раствору аминосоединения, полученного на стадии V (0,16 г, 0,95 ммоль) в ДМСО (4,0 мл) добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,17 г, 0,96 ммоль) и К2СО3 (0,4 г, 2,9 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. Реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{[(1-метокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрила (0,12 г) с выходом 41%. m/z (M+1) 304; 1Н ЯМР (CDCl3) 300 МГц δ 4,82-4,75 (м, 1Н), 3,75-3,40 (м, 4Н), 3,27 (с, 3Н), 2,47-2,40 (м, 1Н), 2,35-1,68 (13Н), 1,57-1,41 (м, 2Н).
Пример 16А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в примере 16 (30 мг, 0,1 ммоль) в метаноле (2 мл), охлажденному до 0ºС, добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие вещества удаляли при пониженном давлении и остаток растирали некоторое время с простым диэтиловым эфиром с получением хлористоводородной соли (2S)-1-{[1-метокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (30 мг).
Пример 23
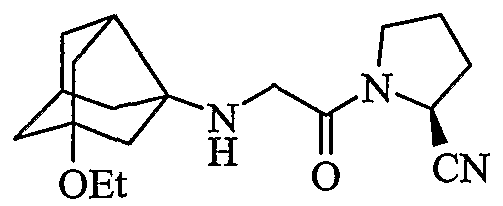
Способ такой же, как описан для метоксисоединения, полученного в примере 16, отличающийся тем, что на первой стадии применяли этилирующий агент n-Bu4NI и этилбромид, в остальном способ, аналогичный указанному выше.
Стадия I: К суспензии NaH (60% дисперсия в медицинском масле, 0,96 г, 24 ммоль) в ТГФ (40 мл), охлажденной до температуры бани со льдом, добавляли гидроксиадамантанон (3,32 г, 20 ммоль), растворенный в ТГФ (40 мл) через шприц в течение периода 15 минут. Реакционную смесь перемешивали в течение 30 мин, затем добавляли n-Bu4NI (0,74 г, 2 ммоль) и этилбромид (1,6 мл, 22 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч. После охлаждения реакционной смеси до 0ºС избыток NaH гасили добавлением насыщ. водн. раствора NH4Cl. Два слоя разделяли, и водный слой экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением неочищенной реакционной массы, которую очищали колоночной хроматографией с получением 5-этоксиадамантан-2-она (3,0 г) с выходом 77%. M/z (M+1) 195; 1Н ЯМР (CDCl3) 300 МГц δ 3,47 (кв, J=7,0 Гц, 2Н), 2,67-2,60 (м, 2Н), 2,38-2,30 (м, 1Н), 2,13-1,78 (м, 10Н), 1,17 (т, J=7,0 Гц, 3Н).
Стадия II: 5-Этокси-2-метиладамантан-2-ол получали из 5-этоксиадамантан-2-она в виде смеси аномеров 1:1 (1,6 г) с выходом 99%. m/z (M+23) 233; 1H ЯМР (CDCl3) 300 МГц δ 3,51-3,41 (м, 2Н), 2,31-2,23 (м, 1Н), 2,18-2,05 (м, 2Н), 2,02-1,80 (м, 4Н), 1,79-1,47 (м, 7Н), 1,46-1,32 (м, 4Н), 1,16 (т, J=6,5 Гц, 3Н).
Стадия III: 1-(1-Этокситрицикло[3,3,1,0 3,7 ]нон-3-ил)этанон получали из 5-этокси-2-метиладамантан-2-ола с выходом 66%. m/z (М+1) 209; 1Н ЯМР (CDCl3) 300 МГц δ 3,49 (кв, J=7,1 Гц, 2Н), 2,69-2,61 (м, 1Н), 2,56-2,48 (м, 1Н), 2,22-2,18 (м, 1Н), 2,17 (с, 3Н), 2,02-1,84 (м, 4Н), 1,78-1,50 (м, 5Н), 1,18 (т, J=7,1 Гц, 3Н).
Стадия IV: 1-Этокситрицикло[3,3,1,0 3,7 ]нонан-3-карбоновую кислоту получали из 1-(1-этокситрицикло[3,3,1,03,7]нон-3-ил)этанона с выходом 68%. m/z (М+1) 195; 1Н ЯМР (CDCl3) 300 МГц δ 3,49 (кв, J=6,7 Гц, 2Н), 2,75-2,68 (м, 1Н), 2,52-2,47 (м, 1Н), 2,36-2,28 (м, 1Н), 2,10-1,95 (м, 2Н), 1,83 (с, 1,86-1,68 (м, 5Н), 1,54 (ддд, J=3,0, 10,9, 13,6 Гц, 2Н), 1,18 (т, J=6,7 Гц, 3Н).
Стадия V: 1-Этокситрицикло[3,3,1,0 3,7 ]нонан-3-амин получали из 1-этокситрицикло[3,3,1,03,7]нонан-3-карбоновой кислоты с выходом 53%. m/z (М+1) 182; 1Н ЯМР (CDCl3) 300 МГц δ 3,49 (кв, J=7,0 Гц, 2Н), 2,39-2,32 (м, 1Н), 2,16-2,07 (м, 1Н), 1,99-1,92 (м, 1Н), 1,92-1,75 (м, 5Н), 1,71-1,38 (м, 4Н), 1,17 (т, J=7,0 Гц, 3Н).
Стадия VI: Реакцией взаимодействия амина, полученного на стадии V, и цианопирролидинового соединения получали (2S)-1-{[1-этокситрицикло[3,3,1,0 3,7 ]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрил с выходом 41%. m/z (М+1) 318; 1Н ЯМР (CDCl3) 300 МГц δ 4,81-4,75 (м, 1Н), 3,75-3,40 (м, 6Н), 2,45-2,37 (м, 1Н), 2,36-2,13 (м, 4Н), 2,12-1,95 (м, 2Н), 1,88-1,68 (м, 6Н), 1,56 (ддд, J=3,0, 3,0, 10,5 Гц, 1Н), 1,49-1,41 (м, 1Н), 1,17 (т, J=7,0 Гц, 3Н).
Пример 17А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в примере 17 (32 мг, 0,1 ммоль), в метаноле (2 мл), охлажденному до 0ºС, добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие соединения удаляли при пониженном давлении и остаток растирали некоторое время с простым диэтиловым эфиром с получением хлористоводородной соли (2S)-1-{[(1-этокситрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (30 мг). m/z (М+1) 318; 1Н ЯМР (DMSO-d6) 300 МГц δ 9,41 (с (ушир), 2Н), 4,86 (дд, J=4,6, 6,8 Гц, 1Н), 4,10-3,90 (м, 2Н), 3,75-3,65 (м, 1Н), 3,57-3,25 (м, 3Н), 2,48-2,38 (м, 2Н), 2,30-2,17 (м, 2Н), 2,16-2,0 (м, 4Н), 1,90-1,14 (м, 6Н), 1,53-1,40 (м, 2Н), 1,06 (т, J=7,0 Гц, 3Н).
Пример 24
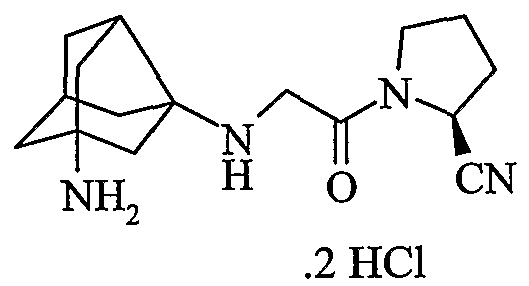
Стадия I: К перемешиваемой смеси соединения, полученного, как указано ранее (препарат 8) (0,3 г, 1,2 ммоль), и К2СО3 (0,5 г, 3,6 ммоль) в ДМСО (5 мл) при температуре бани со льдом в атмосфере N2 добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,14 г, 0,83 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением трет-бутил-(2S)-1-{[(1-аминотрицикло[3,3,1,03,7]нон-3-ил)карбамат]ацетил}пирролидин-2-карбонитрила в виде вязкой жидкости (0,18 г) с выходом 40%. m/z (М+1) 389; 1Н ЯМР (CDCl3) 300 МГц δ 4,79 (д, J=7,5 Гц, 1Н), 4,70 (с (ушир), 1Н), 3,70-3,40 (м, 2Н), 3,46 (с, 2Н), 2,40-2,32 (м, 1Н), 2,32-1,90 (м, 6Н), 1,90-1,63 (м, 5Н), 1,55-1,43 (м, 1Н).
Стадия II: К перемешиваемому раствору соединения, полученного на стадии I (0,04 г, 0,1 ммоль), в EtOAc (2 мл), охлажденному до температуры бани со льдом, добавляли раствор сухого HCl в EtOAc (3н, 2 мл). Реакционную смесь перемешивали в течение 2 ч и растворитель удаляли при пониженном давлении с получением неочищенного вещества, которое растирали некоторое время с простым диэтиловым эфиром с получением соли дигидрохлорид (2S)-1-{[(1-аминотрицикло[3,3,1,03,7]нон-3-ил)амино]ацетил}пирролидин-2-карбонитрила (0,02 г) с выходом 69%. m/z (М+1) 289; 1Н ЯМР (DMSO-d6) 300 МГц δ 9,69 (с (ушир), 2Н), 8,57 (с (ушир), 3Н), 4,86 (дд, J=4,4, 7,0 Гц, 1Н), 4,16-3,90 (м, 2Н), 3,78-3,65 (м, 1Н), 3,65-3,50 (м, 1Н), 2,60-2,55 (м, 1Н), 2,50-2,43 (м, 1Н), 2,35-2,18 (м, 4Н), 2,13-1,98 (м, 2Н), 2,10-2,0 (м, 3Н), 1,96-1,70 (м, 5Н), 1,68-1,60 (м, 1Н), 1,50-1,43 (м, 2Н).
Пример 25

Стадия I: К перемешиваемому раствору трет-бутил(1-гидрокситрицикло[3,3,1,03,7]нон-3-ил)карбамата, полученного как указано ранее (препарат 11) (1,1 г, 4,34 ммоль), в дихлорметане, охлажденному до -15ºС в атмосфере N2 добавляли диэтиламиносульфотрифторид (0,85 мл, 6,51 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 ч и впоследствии перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь охлаждали до температуры бани со льдом и гасили добавлением смеси измельченного льда и твердого NaHCO3 (1,1 г, 13 ммоль). Два слоя разделяли и водный слой экстрагировали хлороформом. Объединенный органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением трет-бутил(1-фтортрицикло[3,3,1,03,7]нон-3-ил)карбамата (0,26 г) в виде вязкой жидкости с выходом 24%. m/z (М-1) 254; 1Н ЯМР (CDCl3) 300 МГц δ 2,55-2,40 (м, 1Н), 2,0-1,75 (м, 2Н), 1,70-1,40 (м, 9Н), 1,57 (с, 6Н), 1,45 (с, 3Н).
Стадия II: К перемешиваемому раствору соединения, полученного на стадии I (0,25 г, 0,98 ммоль), в EtOAc (1 мл), охлажденному до температуры бани со льдом, добавляли раствор сухого HCl в EtOAc (3н, 3 мл). После перемешивания реакционной смеси в течение 2 ч летучие вещества удаляли при пониженном давлении. Неочищенное вещество растирали некоторое время с простым эфиром с получением хлористоводородной соли 1-фтортрицикло[3,3,1,03,7]нонан-3-амина (0,19 г) с выходом 99%. m/z (М+1) 156; 1Н ЯМР (ДМСО-d6) 300 МГц δ 8,45 (с (ушир), 2Н), 2,47-2,18 (м, 4Н), 1,96-1,72 (м, 6Н), 1,63 (дд, J=2,6, 9,6 Гц, 1Н), 1,50-1,42 (м, 1Н).
Стадия III: К перемешиваемой смеси хлористоводородной соли, полученной на стадии II (0,19 г, 0,98 ммоль), и К2СО3 (0,53 г, 4,0 ммоль) в ДМСО (4,0 мл) при температуре бани со льдом в атмосфере азота добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,17 г, 1,0 ммоль). После перемешивания при комн. темп. в течение 3 ч реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-[N-(2-фторгексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,11 г) с выходом 38%. m/z (М+1) 292; 1Н ЯМР (CDCl3) 300 МГц δ 4,80-4,73 (м, 1Н), 3,77-3,32 (м, 2Н), 3,52 (с, 2Н), 2,50-2,42 (м, 1Н), 2,87-2,08 (м, 6Н), 1,50-1,42 (м, 1Н).
Пример 23А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в примере 23 (29 мг, 0,1 ммоль) в метаноле (2 мл), охлажденному до 0ºС, добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие вещества удаляли при пониженном давлении и остаток некоторое время растирали с простым эфиром с получением хлористоводородной соли (2S)-1-[N-(2-фторгексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (30 мг).
Пример 26

Стадия I: К перемешиваемой смеси 1-(3-аминотрицикло[3,3,1,03,7]нон-1-ил)пирролидин-2-она, полученного, как указано ранее (препарат 9) (0,25 г, 1,1 ммоль), и К2СО3 (0,46 г, 3,3 ммоль) в ДМСО (5 мл) при температуре бани со льдом добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,2 г, 1,1 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-(2-оксопирролидин-1-ил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде вязкой жидкости (0,16 г) с выходом 40. m/z (М+1) 357; 1Н ЯМР (CDCl3) 300 МГц δ 4,78 (д, J=7,4 Гц, 1Н), 3,70-3,38 (м, 2Н), 3,46 (с, 2Н), 3,41 (т, J=7,0 Гц, 2Н), 2,60 (дд, J=4,0, 10,2 Гц, 1Н), 2,40-1,75 (м, 17Н), 1,72-1,62 (м, 1Н), 1,58-1,50 (м, 1Н).
Пример 20А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в примере 19 (36 мг, 0,1 ммоль) в метаноле (2 мл), охлажденному до 0ºС добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие вещества удаляли при пониженном давлении и остаток некоторое время растирали с простым эфиром с получением хлористоводородной соли (2S)-1-{N-[2-(2-оксопирролидин-1-ил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (38 мг).
Пример 27
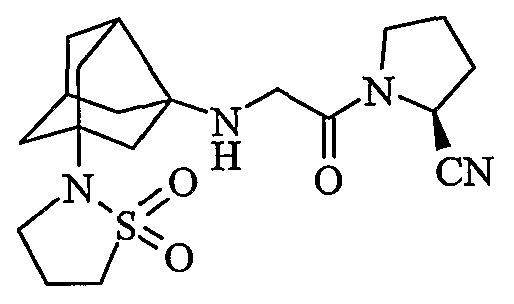
Стадия I: К перемешиваемой смеси 1-(1,1-диоксидоизотиазолидин-2-ил)трицикло[3,3,1,03,7]нонан-3-амина, полученного, как указано ранее (препарат 10) (0,17 г, 0,66 ммоль), и К2СО3 (0,28 г, 2,0 ммоль) в ДМСО (2,6 мл) при температуре бани со льдом добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,13 г, 0,66 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2(1,1-диоксидоизотиазолидин-2-ил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде вязкой жидкости (0,1 г) с выходом 40%. m/z (М+1) 393; 1Н ЯМР (CDCl3) 300 МГц δ 4,80-4,75 (м, 1Н), 3,70-3,37 (м, 4Н), 3,36 (т, J=6,6 Гц, 2Н), 3,16 (т, J=7,4 Гц, 2Н), 2,43-2,37 (м, 1Н), 2,35-2,13 (м, 9Н), 2,05-1,99 (м, 1Н), 1,95-1,75 (м, 5Н), 1,70-1,63 (м, 1Н), 1,54-1,47 (м, 1Н).
Пример 21А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в примере 20 (39 мг, 0,1 ммоль) в метаноле (2 мл), охлажденному до 0ºС, добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие вещества удаляли при пониженном давлении и остаток некоторое время растирали с простым диэтиловым эфиром с получением хлористоводородной соли (2S)-1-{N-[2-(1,1-диоксидоизотиазолидин-2-ил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (41 мг). m/z (М+1) 393; 1Н ЯМР (ДМСО-d6) 300 МГц δ 9,60 (с (ушир), 2Н), 4,86 (дд, J=4,4, 7,0 Гц, 1Н), 4,12-3,96 (м, 2Н), 3,59-3,47 (м, 2Н), 3,28 (т, J=6,8 Гц, 2Н), 3,19 (т, J=7,5 Гц, 2Н), 2,43-2,37 (м, 1Н), 2,31-2,13 (м, 8Н), 2,10-1,97 (м, 3Н), 1,96-1,76 (м, 5Н), 1,50-1,43 (м, 1Н).
Пример 28
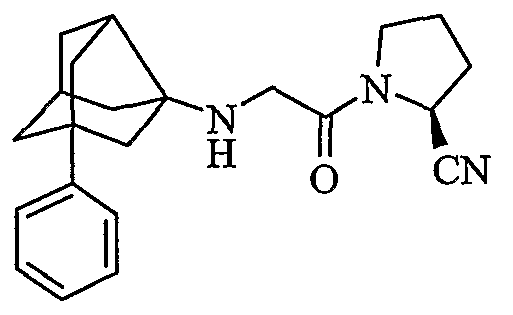
Стадия I: К перемешиваемой смеси NaOH (2,4 г, 60,0 ммоль), Н2О (16 мл) и 1,4-диоксана (2 мл) при температуре бани со льдом добавляли Br2 (0,56 мл, 10,4 ммоль) и перемешивали в течение 15 минут. Полученный раствор гипобромита добавляли по каплям к перемешиваемому раствору 1-(1-фенилтрицикло[3,3,1,03,7]нон-3-ил)этанона, полученного, как указано ранее (препарат I), (1,0 г, 4,0 ммоль) в 1,4-диоксане (6 мл), при температуре бани со льдом. После перемешивания при комн. темп. в течение 1 ч реакционную смесь снова охлаждали до температуры бани со льдом и гасили добавлением АсОН (3,6 мл, 60 ммоль). Реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением 1-фенилтрицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (0,62 г) с выходом 64%. m/z (М+1) 243; 1Н ЯМР δ 7,40-7,25 (м, 4Н), 7,25-7,18 (м, 1Н), 2,90-2,86 (м, 1Н), 2,54-2,48 (м, 1Н), 2,38 (ддд, J=2,0, 2,0, 11,0 Гц, 1Н), 2,20-2,05 (м, 4Н), 1,98-1,78 (м, 5Н), 1,70 (дд, J=2,8, 11,5 Гц, 1Н).
Стадия II: К перемешиваемому раствору 1-фенилтрицикло[3,3,1,03,7]нонан-3-карбоновой кислоты (0,6 г, 2,48 ммоль), полученной на стадии I, и триэтиламина (1,0 мл, 7,44 ммоль) в толуоле (10 мл) в атмосфере N2 при температуре бани со льдом добавляли дифенилфосфорилазид (0,64 мл, 3,0 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч, затем кипятили с обратным холодильником в течение 4 ч. Реакционную смесь охлаждали до комн. темп., промывали водой и перемешивали с водным раствором КОН (50% масс./об., 5,0 мл) и nBu4NI (92 мг, 0,25 ммоль) в течение 2 ч при комн. темп. Реакционную смесь охлаждали до температуры бани со льдом, подкисляли конц. HCl до рН 2, экстрагировали простым диэтиловым эфиром. Водный слой подщелачивали водн. раствором NaOH (50% масс./об.) и экстрагировали хлороформом. Объединенный органический слой сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением 1-фенилтрицикло[3.3.1.03,7]нонан-3-амина (0,33 г) в виде вязкой жидкости с выходом 62%. m/z (М+1) 214; 1Н ЯМР (CDCl3) 300 МГц δ 7,40-7,22 (м, 4Н), 7,22-7,15 (м, 1Н), 2,42-2,35 (м, 1Н), 2,20-2,05 (м, 2Н), 2,02-1,52 (м, 9Н).
Стадия III: К перемешиваемой смеси соединения, полученного на стадии II (0,2 г, 0,94 ммоль), и К2СО3 (0,39 г, 2,8 ммоль) в ДМСО (4,0 мл) при температуре бани со льдом в атмосфере азота добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,16 г, 0,94 ммоль). После перемешивания в течение 3 ч при комн. темп. реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-[N-(2-фенилгексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,16 г) с выходом 48%. m/z (М+1) 350; 1Н ЯМР (CDCl3) 300 МГц δ 7,35-7,16 (м, 5Н), 4,85-4,76 (м, 1Н), 3,76-3,40 (м, 2Н), 3,50 (с, 2Н), 2,48-2,41 (м, 1Н), 2,36-2,27 (м, 2Н), 2,23-2,0 (м, 6Н), 1,94-1,68 (м, 4Н), 1,66-1,60 (м, 1Н).
Пример 26А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в примере 26 (35 мг, 0,1 ммоль) в метаноле (2 мл), охлажденному до 0ºС, добавляли TMS-Cl (25 мкл, 0,2 ммоль). Через 30 минут летучие вещества удаляли при пониженном давлении и остаток растирали некоторое время с простым диэтиловым эфиром с получением хлористоводородной соли (2S)-1-[N-(2-фенилгексагидро-2,5-метанопентален-3а(1Н)-ил)глицил]пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (38 мг).
Пример 29
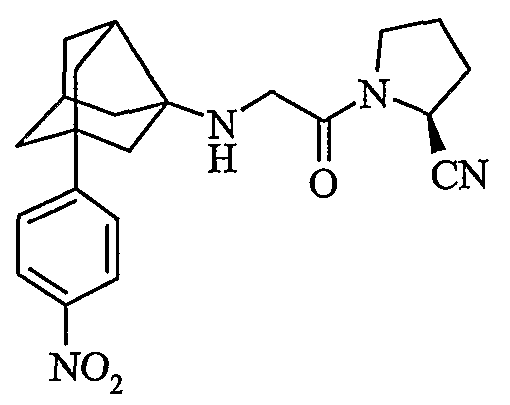
К перемешиваемой смеси 1-(4-нитрофенил)трицикло[3,3,1,03,7]нонан-3-амина, который получен, как указано ранее (стадия III, препарат 4) (0,77 г, 3,0 ммоль), и К2СО3 (1,25 г, 9,0 ммоль) в ДМСО (12 мл) при температуре бани со льдом в атмосфере N2 добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,51 г, 3,0 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением (2S)-1-{N-[2-(4-нитрофенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,5 г) с выходом 42%. m/z (M+1) 395; 1Н ЯМР (CDCl3) 300 МГц δ 8,14 (д, J=8,9 Гц, 2Н), 7,41 (д, J=8,9 Гц, 2Н), 4,83-4,73 (м, 1Н), 3,78-3,40 (м, 2Н), 3,48 (с, 2Н), 2,51-2,45 (м, 1Н), 2,37-2,06 (м, 6Н), 2,02-1,60 (м, 9Н).
Пример 30
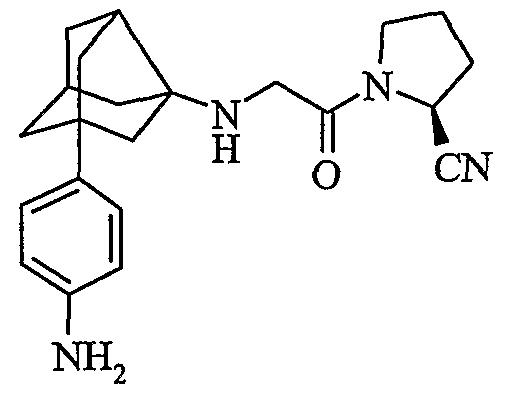
К перемешиваемому раствору соединения, полученного в примере 29 (0,2 г, 0,51 ммоль), в смеси воды, ТГФ и этанола 1:2:4 (7 мл) соответственно последовательно добавляли твердый NH4Cl (0,1 г, 1,87 ммоль) и порошок Fe (0,1 г, 1,78 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 2ч. После охлаждения до комнатной температуры реакционную смесь фильтровали через небольшой слой целита и промывали слой EtOAc. Фильтрат упаривали при пониженном давлении и остаток разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали водой, сушили над безводным Na2SO4 и растворитель выпаривали при пониженном давлении с получением (2S)-1-{N-[2-(4-аминофенил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила в виде не совсем белого твердого вещества (0,09 г) с выходом 50%. m/z (М+1) 365; 1Н ЯМР (CDCl3) 300 МГц δ 7,05 (д, J=8,5 Гц, 2Н), 6,63 (д, J=8,5 Гц, 2Н), 4,90-4,75 (м, 1Н), 3,80-3,40 (м, 4Н), 2,45-1,70 (м, 15Н), 1,65-1,57 (м, 1Н).
Пример 31
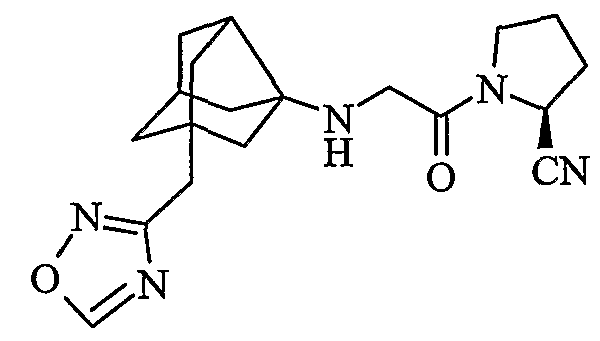
Стадия I: Перемешиваемую смесь соединения, полученного, как указано ранее (препарат 3) (1,1 г, 3,18 ммоль), цианида натрия (0,164 г, 3,5 ммоль) в ДМФА (7,0 мл) нагревали до 110ºС в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением трет-бутил[1-(цианометил)трицикло[3,3,1,03,7]нон-3-ил]карбамата в виде вязкой жидкости (0,3 г) с выходом 34%. m/z (M+1) 277; 1H ЯМР (CDCl3) 300 МГц δ 4,73 (ушир. с, 1Н), 2,60-2,35 (м, 2Н), 2,31 (с, 2Н), 2,20-1,40 (с, 10Н), 1,45 (с, 9Н).
Стадия II: К перемешиваемому раствору соединения, полученного на стадии I (0,25 г, 0,9 ммоль) в смеси этанола и воды 1:1 (9 мл) при комнатной температуре добавляли гидрохлорид гидроксиламина (0,32 г, 4,5 ммоль), с последующим добавлением твердого Na2CO3 (0,57 г, 5,4 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 12 ч. Летучие вещества удаляли при пониженном давлении и остаток растворяли в этилацетате и промывали водой. Органический слой сушили над безводным Na2SO4 и упаривали при пониженном давлении. Остаток растворяли в триметилортоформиате (0,2 мл) и добавляли каталитическое количество камфорсульфоновой кислоты. Реакционную смесь кипятили с обратным холодильником в течение 4 ч. Летучие вещества удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением трет-бутил[1-(1,2,4-оксадиазол-3-илметил)трицикло[3.3.1.03,7]нон-3-ил]карбамата (0,15 г) с выходом 51% в виде вязкой жидкости. m/z (М+1) 320; 1Н ЯМР (CDCl3) 300 МГц δ 8,62 (с, 1Н), 4,71 (ушир. с, 1Н), 2,79 (с, 2Н), 2,48-2,38 (м, 1Н), 2,35-2,28 (м, 1Н), 2,10-1,88 (м, 4Н), 1,74 (дд, J=2,8, 10,5 Гц, 2Н), 1,65-1,38 (м, 4Н), 1,43 (с, (Н),
Стадия III: К перемешиваемому раствору соединения, полученного на стадии II (0,15 г, 0,47 ммоль) в CH2Cl2 (2 мл), охлажденному до 0ºС, добавляли трифторуксусную кислоту (0,5 мл). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 1 ч. Летучие вещества удаляли при пониженном давлении и остаток некоторое время растирали с простым эфиром с получением трифторуксуснокислой соли 1-(1,2,4-оксадиазол-3-илметил)трицикло[3,3,1,03,7]нонан-3-амина в виде белого порошка (0,12 г) с выходом 77%. m/z (М+1) 220; 1Н ЯМР (ДМСО-d6) 300 МГц δ 9,53 (с, 1Н), 8,09 (ушир. с, 3Н), 2,78 (с, 2Н), 2,38-2,25 (м, 2Н), 1,88-1,70 (м, 6Н), 1,60-1,42 (м, 4Н).
Стадия IV: К перемешиваемому раствору соединения, полученного на стадии III (0,12 г, 0,36 ммоль), в ДМСО (1,5 мл) при комнатной температуре в атмосфере азота добавляли (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,06 г, 0,36 ммоль) и К2СО3 (0,2 г, 1,44 ммоль). После перемешивания в течение 3 ч реакционную смесь разбавляли EtOAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением ((2S)-1-{N-[2-(1,2,4-оксадиазол-3-илметил)гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-ил)ацетонитрила в виде не совсем белого твердого вещества (0,05 г) с выходом 40%. m/z (М+1) 356; 1Н ЯМР (CDCl3) 300 МГц δ 8,64 (с, 1Н), 4,84-4,75 (м, 1Н), 3,75-3,40 (м, 2Н), 3,49 (с, 2Н), 2,81 (с, 2Н), 2,40-2,10 (м, 6Н), 1,92-1,70 (м, 6Н), 1,68-1,48 (м, 4Н).
Пример 31А: Хлористоводородная соль: К перемешиваемому раствору соединения, полученного в примере 31 (20 мг, 0,056 ммоль) в метаноле (1 мл), охлажденному до 0º, добавляли TMS-Cl (15 мкл, 0,12 ммоль). Через 30 минут летучие вещества удаляли при пониженном давлении и остаток некоторое время растирали с простым эфиром с получением хлористоводородной соли соединения в виде не совсем белого твердого вещества (21 мг).
Пример 32
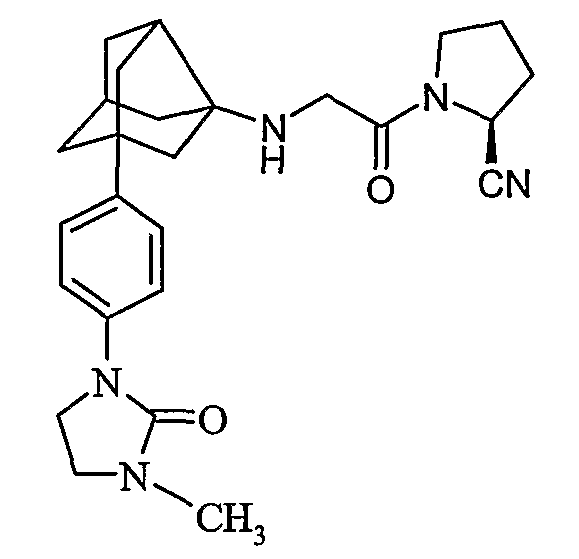
Стадия I: К перемешиваемой суспензии NaH (50% дисперсия в медицинском масле, 50 мг, 1,05 ммоль) в ТГФ (3 мл), охлажденной до 0ºС в атмосфере N2 добавляли по каплям с помощью шприца раствор соединения, полученного на стадии I примерa 19 (0,3 г, 0,7 ммоль) в ТГФ (4 мл). После перемешивания при комнатной температуре в течение 30 минут реакционную смесь снова охлаждали до температуры бани со льдом и добавляли MeI (0,1 мл, 1,5 ммоль). Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 2 ч. Избыток NaH гасили добавлением водн. раствора NH4Cl после охлаждения реакционной смеси до температуры бани со льдом. Два слоя разделяли и водный слой экстрагировали EtOAc. Объединенный органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением трет-бутил[2-[4-(3-метил-2-оксоимидазолидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]карбамата (250 мг) в виде не совсем белого твердого вещества с выходом 80%. m/z (М+1) 446; 1Н ЯМР (CDCl3) 300 МГц δ 7,48 (д, J=8,7 Гц, 2Н), 7,43-7,30 (м, 5Н), 7,28-7,18 (м, 2Н), 5,17-5,03 (м, 3Н), 3,80 (т, J=7,2 Гц, 2Н), 3,47 (т, J=7,2 Гц, 2Н), 2,91 (с, 3Н), 2,61-2,52 (м, 1Н), 2,48-2,40 (м, 1Н), 2,32-2,55 (м, 10Н).
Стадия II: К перемешиваемому раствору соединения, полученного на стадии I (0,25 г, 0,56 ммоль) в смеси CH2Cl2 и МеОН 1:1 (10 мл) добавляли Pd/C (10% масс/масс, 0,1 г) и перемешивали при комнатной температуре в атмосфере Н2 в течение 2 ч. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением 1-[4-(3а-аминогексагидро-2,5-метанопентален-2(1Н)-ил)фенил]-3-метилимидазолидин-2-она (0,15 г) в виде не совсем белого твердого вещества с выходом 86%. Т.пл. 227-230ºС; m/z (М+1) 312; ИК см-1 3416, 3249, 2964, 1682, 1519, 1485, 1261, 810; 1Н ЯМР (CDCl3+ДМСО-d6) 300 МГц δ 8,38 (ушир. с, 2Н), 7,47 (д, J=8,6 Гц, 2Н), 7,19, (д, J=8,6 Гц, 2Н), 3,76 (т, J=7,4 Гц, 2Н), 3,43 (т, J=7,4 Гц, 2Н), 2,77 (с, 3Н), 2,48-2,40 (м, 2Н), 2,22-2,14 (м, 1Н), 2,10-2,0 (м, 2Н), 2,0-1,86 (м, 4Н), 1,80-1,60 (м, 3Н).
Стадия III: К перемешиваемой смеси соединения, полученного на стадии II (0,15 г, 0,48 ммоль), и К2СО3 (0,21 г, 1,5 ммоль) в ДМСО (2 мл) при температуре бани со льдом добавляли соединение (S)-1-(2-хлорацетил)пирролидин-2-карбонитрил (0,09 г, 0,5 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3 ч. После завершения реакции (контролируемой методом ТСХ) реакционную смесь разбавляли EtAc и промывали водой и насыщенным раствором соли, сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией с получением ((2S)-1-{N-[2-[4-(3-метил-2-оксоимидазолидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-ил)ацетонитрила в виде не совсем белого твердого вещества (0,09 г) с выходом 42%. m/z (М+1) 449; 1Н ЯМР (CDCl3) 300 МГц δ 7,47 (д, J=8,6 Гц, 2Н), 7,22 (т, J=8,6 Гц, 2Н), 4,85-4,76 (м, 1Н), 3,79 (т, J=7,3 Гц, 2Н), 3,75-3,40 (м, 4Н), 3,48 (с, 2Н), 2,89 (с, 3Н), 2,46-2,39 (м, 1Н), 2,36-2,15 (м, 3Н), 2,12-1,58 (м, 12Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2010 |
|
RU2574410C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HDM2 | 2013 |
|
RU2690663C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| КОНДЕНСИРОВАННЫЕ КОЛЬЦЕВЫЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ TRK | 2015 |
|
RU2708674C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| Бициклические конденсированные гетероарильные или арильные соединения в качестве модуляторов IRAK4 | 2016 |
|
RU2684324C1 |
Настоящее изобретение относится к новому соединению, представленному формулой I, и его фармацевтически приемлемой соли, где Х представляет собой CH2, CHF или S, Y представляет собой CN, R1, R2, R3 и R4 представляют собой водород, n представляет собой 1, m представляет собой 0 или 1, R представляет собой R11, R12 или R13, где R11 включает по меньшей мере одну из групп, выбранных из нижеследующих b) или с), где необязательно замещенная гетероциклильная и гетероарильная группы связаны с норадамантильной частью либо непосредственно, либо через метиленовую смежную группу, либо С-С связью, либо C-N связью; b) замещенная 5-членная гетероарильная группа, в которой гетероарильное кольцо представляет собой моноциклическую ароматическую кольцевую систему, включающую два или более гетероатомов, выбранных из азота и кислорода; с) гетероциклильная группа необязательно замещенная C1-С3алкильной или оксогруппами, где гетероциклическая кольцевая система представляет собой 5-9-членную моно- или бициклическую кольцевую систему с одним или более гетероатомами, выбранными из группы, состоящей из азота и серы, где гетероатомы также могут присутствовать в качестве функциональных групп, где гетероциклическая кольцевая система может содержать одну или две двойные связи, и где моноциклическое гетероциклическое кольцо может быть конденсировано с фенильным кольцом, R12 выбран из водорода, галогена, гидрокси, амино и С1-С4алкокси; R13 представляет собой замещенный фенил, где заместители, которые могут быть одинаковыми или различными, включают по меньшей мере одну из групп, выбранных из а) водорода; b) нитро, амино; с) насыщенной или ненасыщенной моноциклической гетероциклической кольцевой системы, необязательно замещенной одной или несколькими группами, выбранными из C1-С3алкила и оксо, где гетероциклическая кольцевая система представляет собой 5-членное кольцо с одним или несколькими гетероатомами, выбранными из группы, состоящей из азота и серы, где гетероатомы также могут присутствовать в качестве функциональных групп. Настоящее изобретение также относится к фармацевтической композиции, обладающей ингибитующей активностью дипептидилпептидазы IV, способам получения нового соединения формулы I и применению при лечении диабета типа II и диабетических осложнений и также для лечения дислипидемии, гиперхолестеринемии, ожирения и гипергликемии. Технический результат - новые ингибиторы дипептидилпептидазы IV. 6 н. и 4 з.п. ф-лы, 1 табл.

1. Соединение формулы I,
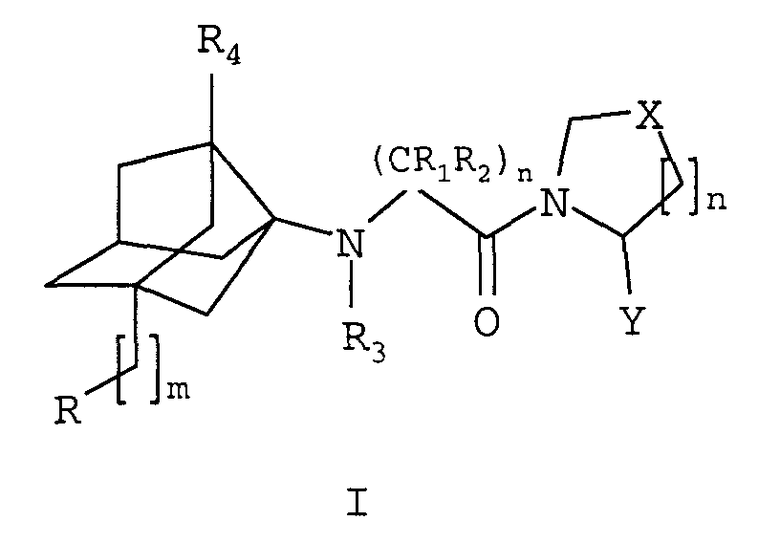
его фармацевтически приемлемые соли, где
Х представляет собой CH2, CHF или S,
Y представляет собой CN,
R1, R2, R3 и R4 представляют собой водород,
n представляет собой 1,
m представляет собой 0 или 1,
R представляет собой R11, R12 или R13, где
R11 включает по меньшей мере одну из групп, выбранных из нижеследующих b) или с), где необязательно замещенная гетероциклильная и гетероарильная группы связаны с норадамантильной частью либо непосредственно, либо через метиленовую смежную группу, либо С-С связью, либо C-N связью;
b) замещенная 5-членная гетероарильная группа, в которой гетероарильное кольцо представляет собой моноциклическую ароматическую кольцевую систему, включающую два или более гетероатомов, выбранных из азота и кислорода;
с) гетероциклильная группа необязательно замещенная C1-С3алкильной или оксогруппами, где гетероциклическая кольцевая система представляет собой 5-9-членную моно- или бициклическую кольцевую систему с одним или более гетероатомами, выбранными из группы, состоящей из азота и серы, где гетероатомы также могут присутствовать в качестве функциональных групп,
где гетероциклическая кольцевая система может содержать одну или две двойные связи, и
где моноциклическое гетероциклическое кольцо может быть конденсировано с фенильным кольцом,
R12 выбран из водорода, галогена, гидрокси, амино и С1-С4алкокси;
R13 представляет собой замещенный фенил, где заместители, которые могут быть одинаковыми или различными, включают по меньшей мере одну из групп, выбранных из
a)водорода;
b) нитро, амино;
c) насыщенная или ненасыщенная моноциклическая гетероциклическая кольцевая система необязательно замещенная одной или несколькими группами, выбранными из C1-С3алкила и оксо, где гетероциклическая кольцевая система представляет собой 5-членное кольцо с одним или несколькими гетероатомами, выбранными из группы, состоящей из азота и серы,
где гетероатомы также могут присутствовать в качестве функциональных групп.
2. Соединение по п.1, где
R представляет собой R11,
m представляет собой 1,
R11 имеет значения как указано в п.1.
3. Соединение по п.1, где
R представляет собой R11 или R12,
m представляет собой 0,
R11 и R12 имеют значения как указано в п.1.
4. Соединение по п.1, где
R представляет собой R13,
m представляет собой 0,
R13 имеет значения как указано в п.1.
5. Соединение, выбранное из группы, состоящей из
(2S)-1-[1H-1,2,4-тpиaзoл-1-илмeтил-(тpициклo[3,3,1,03,7]нoн-3-иламино)ацетил]пирролидин-2-карбонитрила,
(2S)-1-{N-[2-[(1,1-диоксидоизотиазолидин-2-ил)метил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила,
(2S)-1-{N-[2-[4-(2-оксопирролидин-1-ил)фенил]гексагидро-2,5-метанопентален-3а(1Н)-ил]глицил}пирролидин-2-карбонитрила.
6. Способ получения соединения формулы I
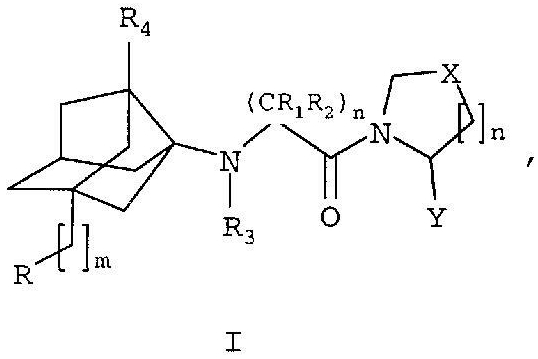
включающий связывание соединения формулы II, которое находится в свободной форме, в форме соли или в защищенной форме, с соединением формулы III
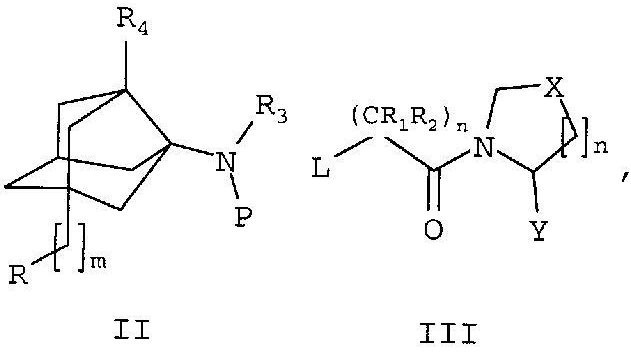
где Р представляет собой водород или защитную группу, выбранную из трет-бутоксикарбонила (ВОС) и карбобензилокси (Cbz), L представляет собой удаляемую группу, выбранную из галогена, O-толуолсульфонила и O-метилсульфонила, и R, R1, R2, R3, R4, X, Y, n и m имеют значения как указано в п.1.
7. Способ по п.6, где реакцию связывания осуществляют в инертном растворителе, выбранном из дихлорметана, диметилформамида, ДМСО, тетрагидрофурана и ацетонитрила, и в присутствии основания, выбранного из третичных аминов, карбонатов, гидроксидов и их смесей.
8. Промежуточное соединение, выбранное из
1-(1Н-1,2,4-триазол-1-илметил)трицикло[3,3,1,03,7]нонан-3-амина,
1-[(1,1-диоксидоизотиазолидин-2-ил)метил]трицикло[3,3,1,03,7]нонан-3-амина,
1-[4-(3-аминотрицикло[3,3,1,03,7]нон-1-ил)фенил]пирролидин-2-она.
9. Фармацевтическая композиция, обладающая ингибирующей активностью DPP-IV, включающая а) соединение формулы I, его фармацевтически приемлемую соль по любому одному из пп.1-6, и b) по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
10. Применение соединения формулы I, его фармацевтически приемлемой соли, по любому одному из пп.1-6 для получения фармацевтической композиции для лечения и/или предупреждения диабета типа II, для лечения метаболических нарушений, снижения уровня глюкозы в крови, ухудшения переносимости глюкозы, снижения уровня глюкозы натощак, дислипидемии, гиперхолестеринемии, гиполипидемии или ожирения, для предупреждения гипергликемии или для уменьшения массы тела или диабетических осложнений, включающих заболевание коронарной артерии, инсульт, гипертензию, нефропатию, заболевание периферических сосудов, нейропатию или ретинопатию.
| N-ЗАМЕЩЕННЫЕ 2-ЦИАНОПИРРОЛИДИНЫ | 1997 |
|
RU2180901C2 |
| N-ЗАМЕЩЕННЫЕ 2-ЦИАНПИРРОЛИДИНЫ | 1999 |
|
RU2251544C2 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

