Данная заявка испрашивает приоритет по предварительной патентной заявке США серии № 60/801857, поданной 18 мая 2006 года, и по предварительной патентной заявке США серии № 60/765114, поданной 3 февраля 2006 года, полное содержание которых включено тем самым в качестве ссылки.
Область изобретения
Настоящее изобретение относится к способу широкомасштабного получения агониста A2A-аденозинового рецептора, и также относится к полиморфам данного соединения и способам выделения определенного полиморфа.
Уровень техники
Аденозин является встречающимся в природе нуклеозидом, который проявляет свои биологические свойства посредством взаимодействия с семейством аденозиновых рецепторов, известных как A1, A2A, A2B и A3, которые регулируют важные физиологические процессы. Одним из биологических эффектов аденозина является действие в качестве коронарного вазодилятатора; данный эффект является следствием взаимодействия с A2A-аденозиновым рецептором. Показано, что данное свойство аденозина применимо в качестве вспомогательного средства для рентгено- и томографии сердца, когда коронарные артерии расширяют перед введением контрастного вещества (например, талий 201), и таким образом, при просмотре снимков, полученных таким способом, можно определить наличие или отсутствие заболевания коронарных артерий. Преимущество данной технологии заключается в том, что она позволяет избежать наболее традиционного способа индуцирования коронарного расширения сосудов посредством физической нагрузки на беговой дорожке, что является крайне нежелательным для пациента с коронарным заболеванием.
Однако введение аденозина имеет несколько недостатков. Аденозин имеет очень короткий период полураспада в организме человека (менее чем 10 секунд), и также обладает всеми эффектами, связанными с агонистами A1, A2A, A2B, и A3 рецепторов. Таким образом, применение избирательного агониста A2A-аденозинового рецептора могло бы обеспечить лучший способ продуцирования коронарной вазодилятации, в частности агониста с более длинным периодом полураспада и меньшим количеством побочных эффектов или их отсутствием.
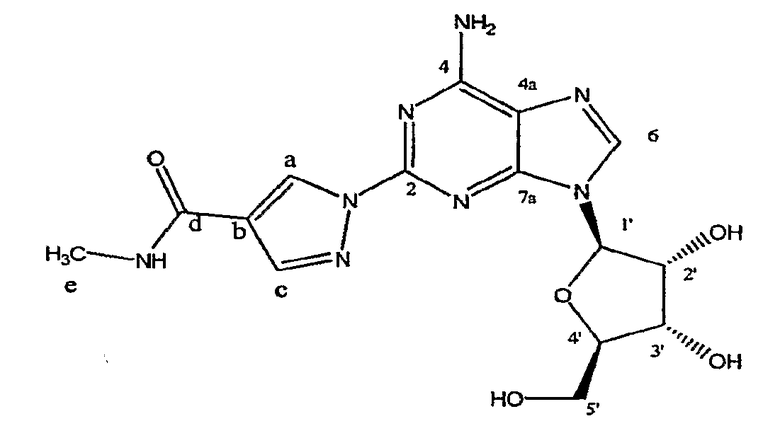
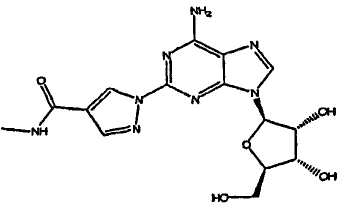
Класс соединений, обладающих данными необходимыми свойствами, был описан в патенте США № 6403567, полное описание которого, таким образом, включено в качестве ссылки. В частности, показано, что соединение, описанное в данном патенте, (l-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамид, является высокоизбирательным агонистом A2A-аденозинового рецептора, и в настоящий момент проходит клинические испытания, как коронарный вазодилятатор, применяемый в сердечной графии.
Принимая во внимание повышенный интерес в данном и похожих соединениях, необходим поиск новых способов синтеза, которые обеспечивают удобный способ получения больших количеств вещества с хорошим выходом и высокой степенью очистки. Патент, который описывает интересующее соединение (патент США № 6403567) предусматривает несколько способов получения данного соединения. Однако, хотя данные способы подходят для синтеза малых количеств, все синтетические способы, описанные в патенте, используют защитные группы, которые нежелательны для широкомасштабного синтеза.
Дополнительно, было открыто, что получаемый продукт, который является (l-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамидом) существует, по крайней мере, в трех различных кристаллических формах, наиболее устойчивая из которых является моногидратом. Данный полиморф стабилен при стрессовых условиях относительной влажности, вплоть до его температуры плавления. Таким образом, желательно, чтобы конечный продукт, создаваемый посредством нового синтеза, получали в качестве стабильного моногидрата.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Таким образом, задачей данного изобретения является создание подходящего способа широкомасштабного получения
(l-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида и его полиморфов, предпочтительно в виде его моногидрата. Таким образом, в первом аспекте, изобретение относится к получению соединения формулы I
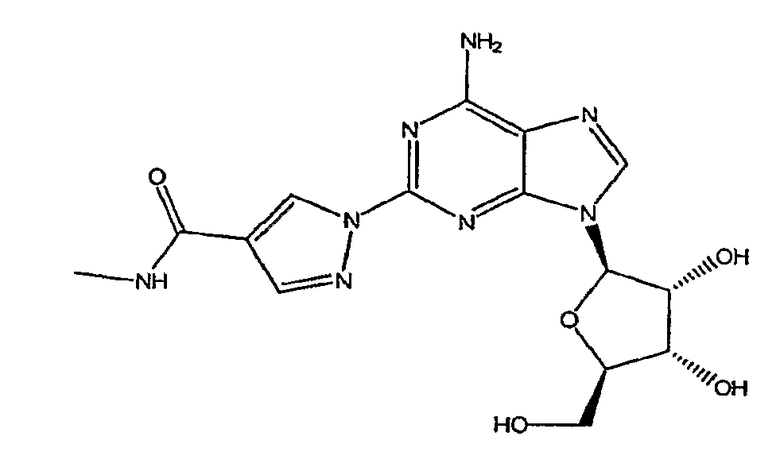
Формула I,
включающего
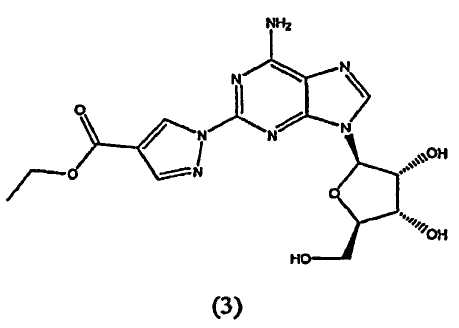
взаимодействие соединения формулы (3)
с метиламином.
В одном из вариантов осуществления способа химическую реакцию осуществляют в водном растворе метиламина, первоначально при температуре приблизительно 0-5°C, с последующим нагреванием до приблизительно 50-70°C. Альтернативно, химическую реакцию осуществляют как указано выше, но в герметичном реакционном реакторе под давлением.
Во втором варианте осуществления способа продукт выделяют в виде чистого моногидрата посредством растворения продукта в растворителе, например в диметилсульфоксиде, с добавлением чистой воды, фильтрованием суспензии, таким образом образованную, промывкой содержимого фильтра водой, затем этанолом, и высушивании твердого осадка, который остается, в вакууме при температуре, не превышающей 40°C.
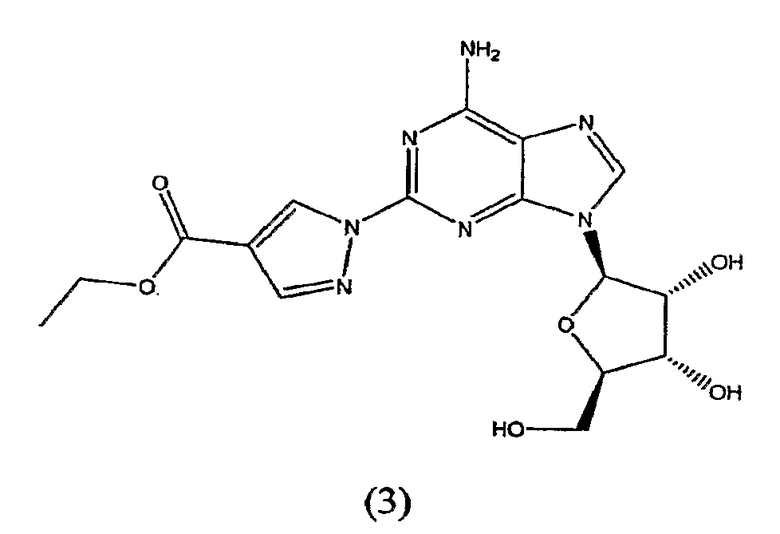
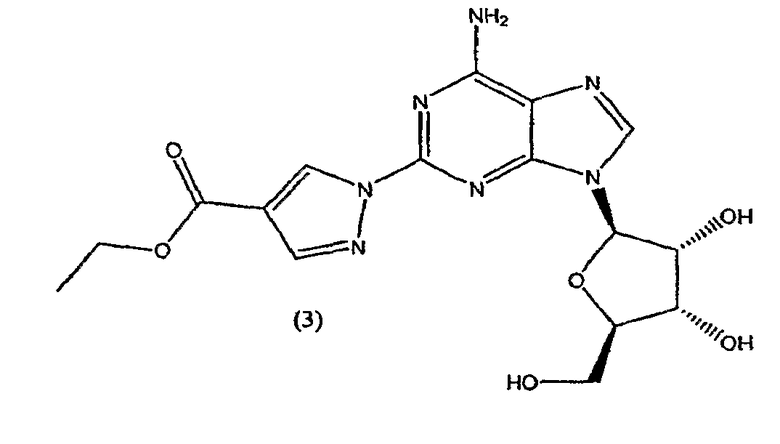
Во втором аспекте изобретение относится к получению соединения формулы (3)
включающего
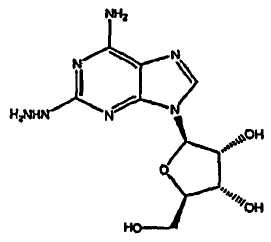
взаимодействие соединения формулы (2)
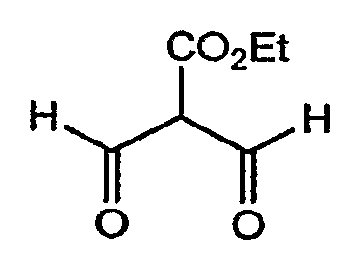
с этил-2-формил-3-оксопропионатом.
В одном из вариантов осуществления способа реакцию осуществляют в этаноле, при температуре приблизительно 80°C, с приблизительно 1,1 молярными эквивалентами этилового эфира 2-формил-3-оксопропионовой кислоты.
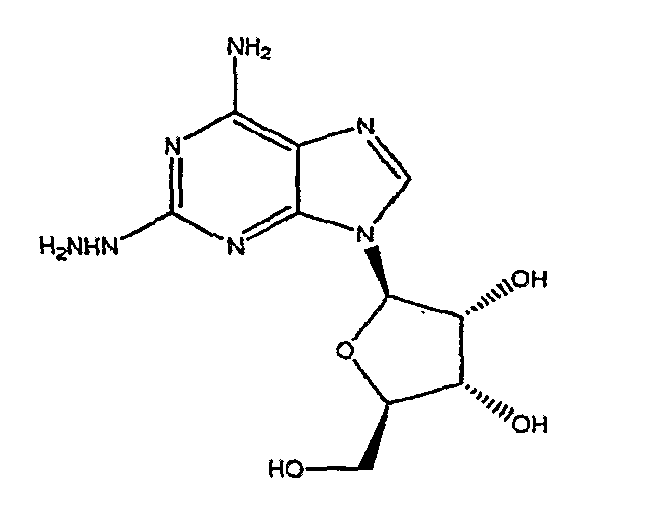
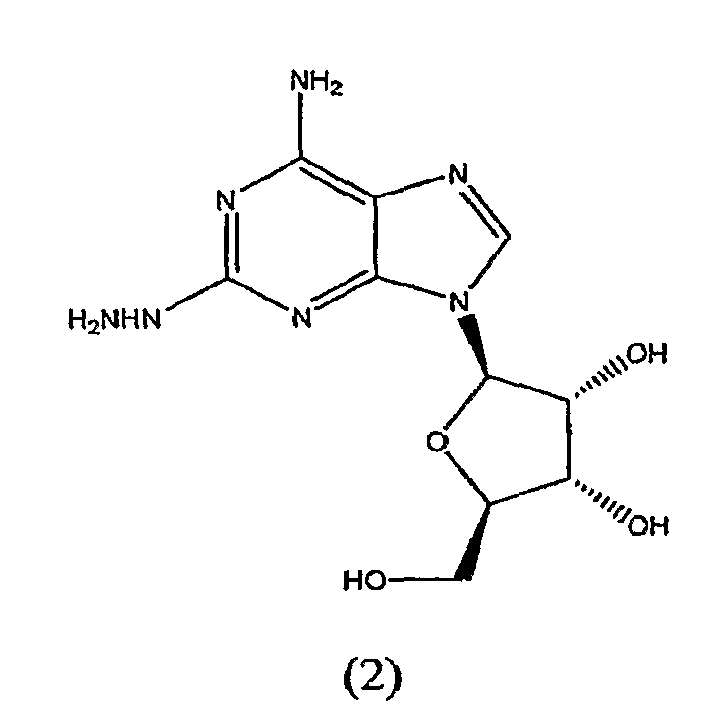
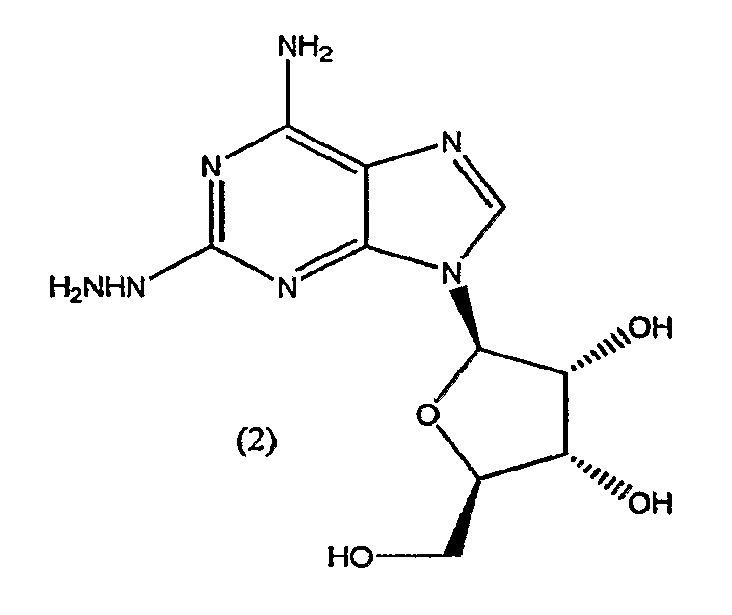
В третьем аспекте изобретение относится к получению соединения формулы (2)
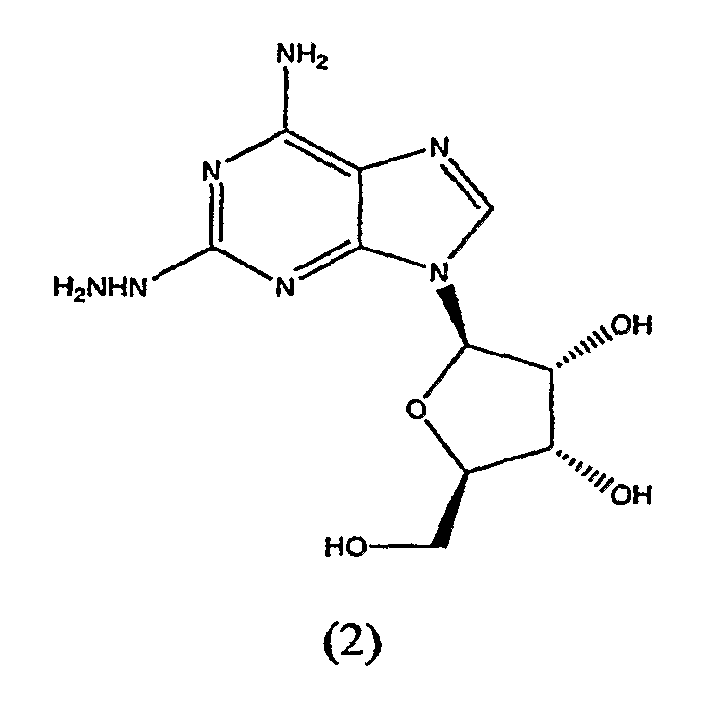
включающего
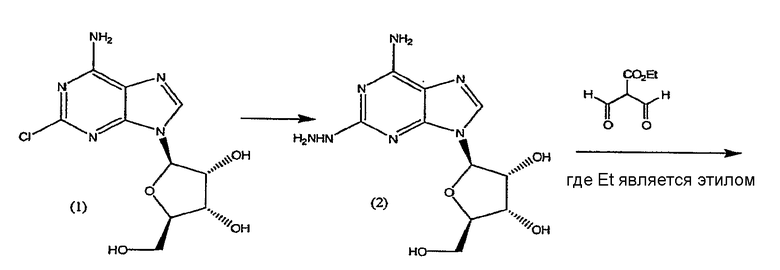
взаимодействие соединения формулы (1)
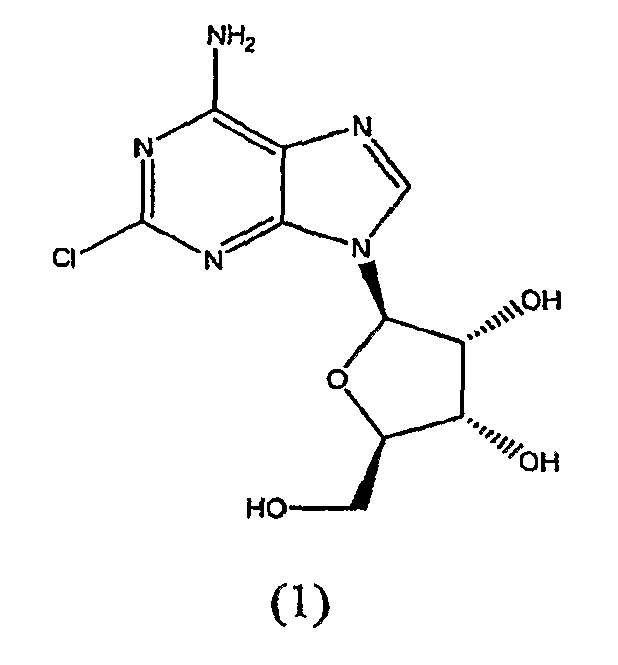
с гидразином.
Вышеописанный синтез пригоден для широкомасштабного синтеза необходимого продукта, который обеспечивает хороший выход, хотя в конечном продукте видна одна небольшая примесь. Показано, что данная примесь является неизменным промежуточным соединением формулы (2); которое представлено следующей структурной формулой:

Хотя данную примесь можно удалить из конечного продукта путем кристаллизации, было решено продолжить поиск альтернативного синтеза, который имел бы все преимущества выше описанного синтеза, но не образовывал соединения формулы (2) в виде примеси в конечном продукте.
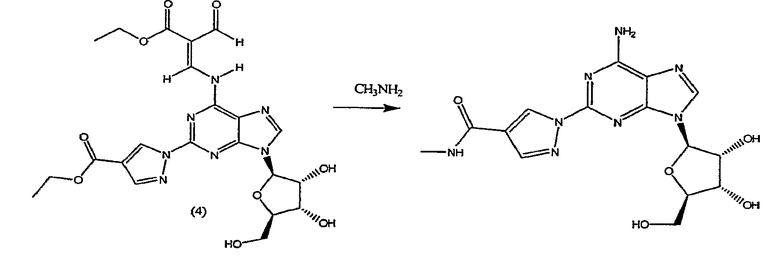
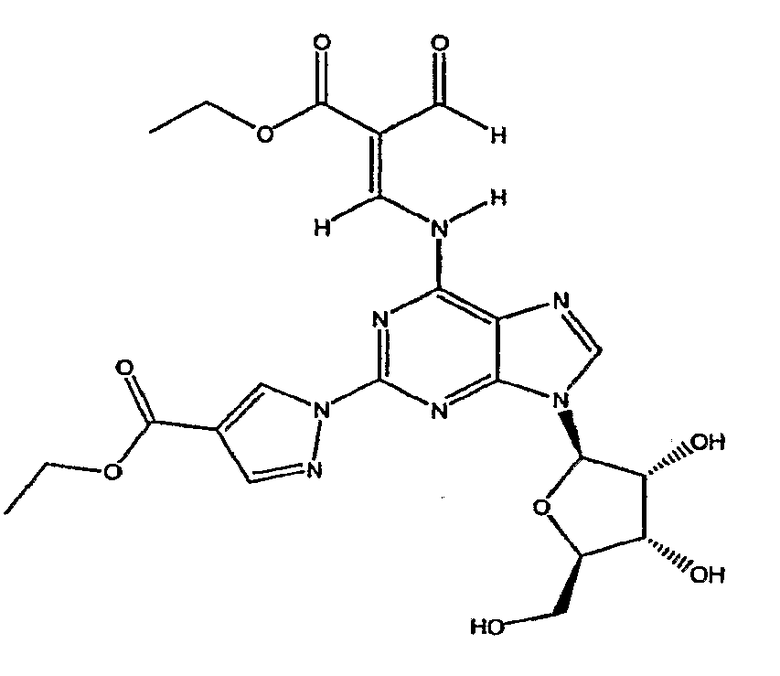
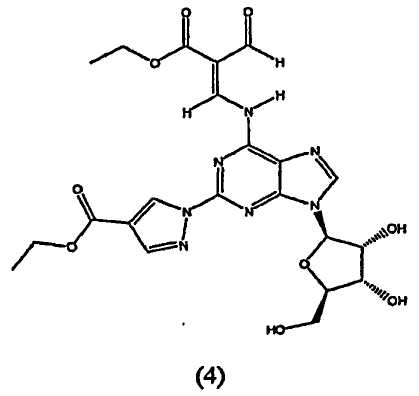
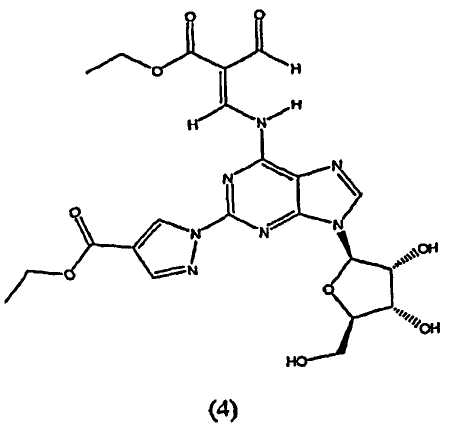
Таким образом, в четвертом аспекте изобретение относится к способу получение (l-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида посредством взаимодействия соединения формулы (4)
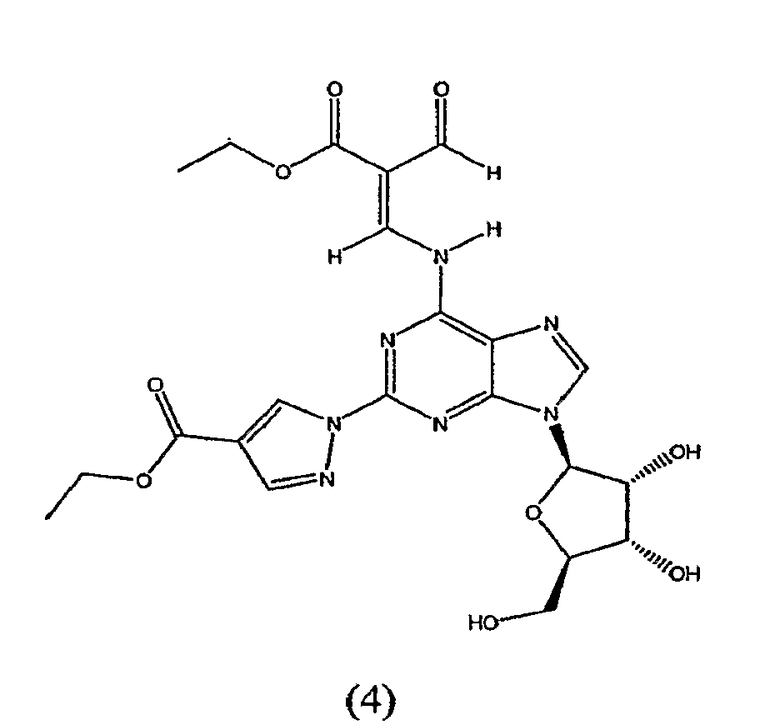
с метиламином.
В одном из вариантов осуществления способа химическую реакцию осуществляют в водном растворе метиламина, первоначально при температуре приблизительно 0-5°C, за которой следует нагревание приблизительно до 50-70°C. Альтернативно, химическую реакцию осуществляют как указано выше, но в герметичном реакторе под давлением.
Во втором варианте осуществления продукт выделяют в виде чистого моногидрата посредством растворения продукта в растворителе, например в диметилсульфоксиде, с добавлением чистой воды, фильтрованием суспензии, таким образом образованную, промывкой содержимого фильтра водой, затем этанолом, и высушиванием твердого осадка, который остается, в вакууме при температуре, не превышающей 40°C.
В пятом аспекте изобретение относится к способу получения соединения формулы (4)
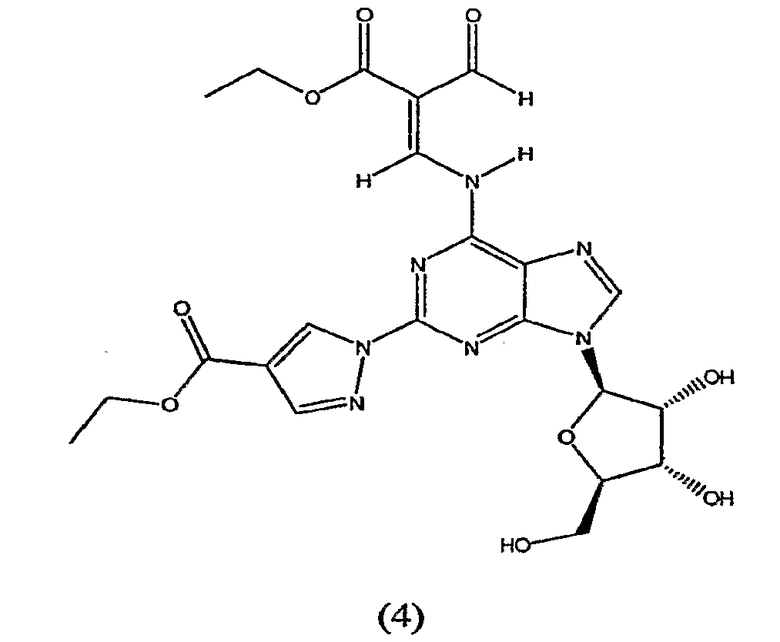
включающего взаимодействие соединения формулы (2)
с избытком этилового эфира 2-формил-3-оксопропионовой кислоты, предпочтительно приблизительно с 2-10 кратном избытком, более предпочтительно с 5-10 кратном избытком.
В одном из вариантов осуществления реакцию осуществляют в этаноле, при температуре приблизительно 80°C. Этиловый эфир 2-формил-3-оксопропионовой кислоты присутствует в 5-10 кратном избытке.
Определения и общие параметры
Фиг.1 является 1H ЯМР спектром моногидрата (l-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}-пиразол-4-ил)-N-метилкарбоксамида (Форма A).
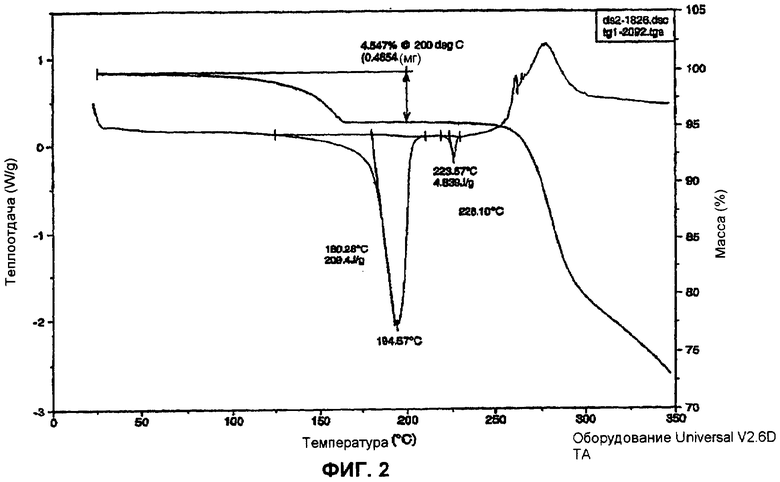
Фиг.2 изображает термический анализ моногидрата (l-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}-пиразол-4-ил)-N-метилкарбоксамида.
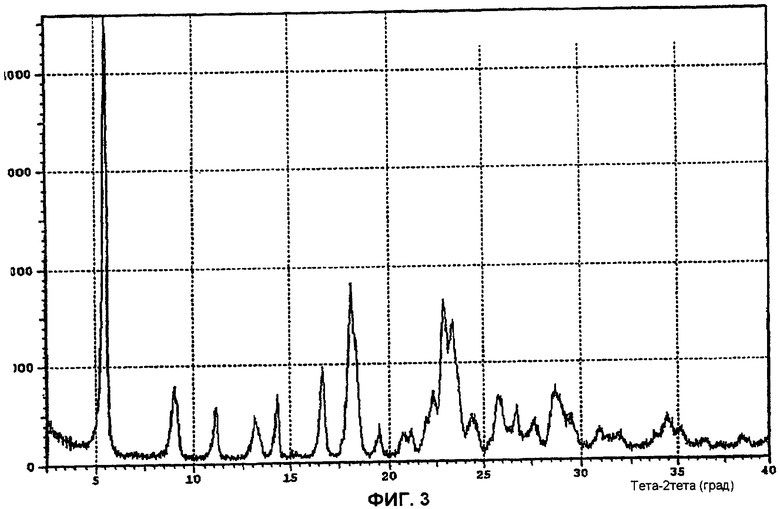
Фи.3 показывает дифракционную рентгенограмму моногидрата (l-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}-пиразол-4-ил)-N-метилкарбоксамида.
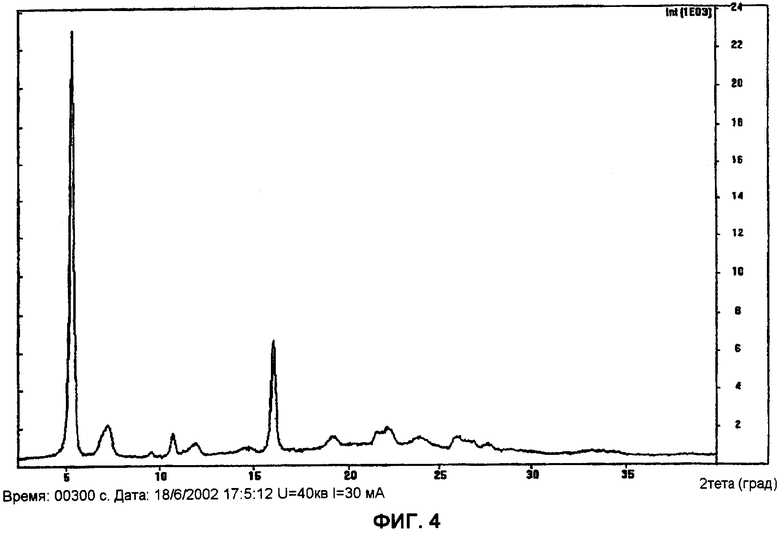
Фиг.4 показывает дифракционную рентгенограмму моногидрата (l-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}-пиразол-4-ил)-N-метилкарбоксамида формы B.
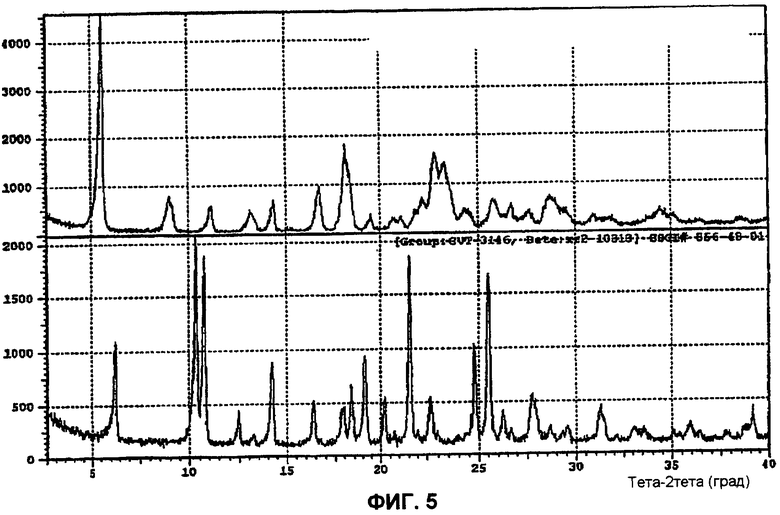
Фиг.5 показывает дифракционную рентгенограмму моногидрата (l-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}-пиразол-4-ил)-N-метилкарбоксамида формы C по сравнению с формой A.
Применяемые в настоящем описании следующие слова и фразы, как правило, имеют значения, которые объяснены ниже, за исключением ситуаций, если в контексте, в котором они употреблены, указано иначе.
"Необязательный" или "необязательно" означает, что дальнейшее описанное событие или обстоятельство может происходить или не происходить, и, что описание включает в себя примеры, где указанное событие или обстоятельство происходит и примеры, где оно не происходит.
Термин "терапевтически эффективное количество" относится к количеству соединения формулы I, которое достаточно для эффективного воздействия, как определено ниже, когда осуществляют введение млекопитающему, которому необходимо данное воздействие. Терапевтически эффективное количество в значительной степени зависит от субъекта и состояния заболевания, которое лечат, массы и возраста субъекта, тяжести состояния заболевания, способа введения и т.п., и может быть легко определено специалистом в данной области.
Термин "терапия" или "терапевтический" означает любое лечение заболевания у млекопитающих, включающее в себя:
(i) предотвращение заболевания, которое обеспечивает неразвитие клинических симптомов заболевания;
(ii) ингибирование заболевания, которое обеспечивает задержку развития клинических симптомов; и/или
(iii) излечивание заболевания, которое обеспечивает регрессию клинических симптомов.
Как применяют в настоящем документе, "фармацевтически пригодный носитель" включает в себя все без исключения растворители, диспергенты, покрытия, антибактериальные и антигрибковые вещества, изотонические вещества и вещества, препятствующие абсорбции и т.п. Применение подобных сред и веществ в фармацевтически активных субстанциях хорошо известно в данной области. За исключением случаев, если какое-либо подходящее вещество или среда несовместимы с активным ингредиентом, предполагается их применение в терапевтических композициях. Дополнительные активные ингредиенты также могут быть включены в композиции.
Подразумевают, что термин "полиморф" включает в себя некристаллические формы и сольваты (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида.
Было установлено, что данное соединение устойчиво при существовании, по крайней мере, в трех различных кристаллических формах, упомянутые в настоящем документе как Форма A, Форма B, Форма C и в аморфном состоянии.
Форма A: Данный полиморф можно получить посредством кристаллизации (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида из протонных растворителей, например этанола или смеси этанол/вода, или из полярного растворителя, например диметилсульфоксид/вода. Показано, что Форма A является моногидратом и наиболее устойчива из различных полиморфов при температуре окружающей среды. Она устойчива в стрессовых условиях относительной влажности вплоть до ее температуры плавления.
Форма B: Данный полиморф получают посредством испарения в вакууме раствора (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида в трифторэтаноле при температуре окружающей среды. Рентгеновский анализ данных кристаллов отчетливо различался от любых других полиморфов (см. фиг.4), но было трудно определить его строение, т.к. рентгеновский анализ давал неупорядоченные размытые пики, и полиморф содержал различное количество воды. Было установлено, что получение данного полиморфа достоверно трудно воспроизводимо.
Форма C: Данный полиморф получают посредством суспендирования (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида в ацетонитриле в течение длительного периода времени при 60°C. Рентгеновский анализ данных кристаллов отчетливо различался от любых других полиморфов (см. фиг.5). Показано, что полиморф C является неустойчивым гидратом, который при повышенных температурах, десольватируется в нестабильную форму.
Аморфное вещество: Данный полиморф получают посредством нагревания полиморфа Формы A при температуре до 200°C. Данный полиморф неустойчив в присутствии атмосферной влажности, формируя неустойчивые гидраты.
Способы анализа Форм A, B, C и Аморфного вещества
Рентгеновская дифракция порошка
Анализ рентгеновской дифракции порошка (XRPD) выполняли на рентгеновском порошковом дифрактометре Shimadzu XRD-6000 с использованием Cu Kα излучения. Прибор был оборудован тонко сфокусированной рентгеновской трубкой, и напряжение, и сила тока были установлены на 40 кВ и 40 мА соответственно. Дивергенция и рассеивающие щели были установлены на 1" и принимающая щель быль установлена на 0,15 мм. Дифрагированное излучение анализировали посредством NaI сцинтилляционного детектора. Непрерывное сканирование тета-два тета проводили при 3°/мин (0,4 с/0,02° шаг) от 2,5-40° 2θ. Для проверки настройки прибора использовали кремниевый стандарт. Данные собирали и анализировали с использованием программного обеспечения XRD-6000 в.4.1.
Анализ рентгеновской дифракции порошка (XRPD) также выполняли с применением дифрактометра Inel XRG-3000, оборудованного детектором CPS (нелинейный позиционно-чувствительный) с 28 ранжированием 120°. Калибровку прибора выполняли с помощью кремниевого стандартного образца. Напряжение и сила тока в трубке были установлены на 40 кВ и 30 мА соответственно. Монохроматическую щель установили на 5 мм по 80 мкм. Образцы поместили в алюминиевый держатель образцов с силиконовой вставкой или в стеклянные капилляры для XRPD. Каждый капилляр устанавливали на крышке гониометра, который двигался для обеспечения вращения капилляров в течение сбора данных. Данные в режиме реального времени собирали с использованием Cu Kα излучения при разрешении 0,03° 2θ. Как правило, данные собирали в течение периода 300 секунд. Графики XRPD образцы показывали только экспериментальные точки в пределе 2,5-40° 2θ.
Термический анализ
Термогравиметрический (TG) анализ выполняли на оборудовании TA 2050 или на 2950 термогравиметрическом анализаторе. Калибровочными стандартами были никель и Алюмель™. Образцы поместили в алюминиевый противень для образцов, вставили в TG нагревательную установку и тщательно уравновешивали. Образцы нагревали в азоте при условиях 10°C/мин до 300 или 350°C. Если не указано иначе, массы образцов уравновешивали при 25°C в TGA духовых шкафах перед анализом.
Анализы дифференциальной сканирующей калориметрии (DSC) выполняли на оборудовании TA дифференциального сканирующего калориметра 2920. Тщательно уравновешенные образцы помещали в сжатые емкости или в герметически закрытые емкости, которые содержали поры, чтобы можно было снизить давление. Каждый образец нагревали в азоте при условиях 10°C/мин до 300 или 350°C. Металл индий применяли в качестве калибровочного стандарта. Температуры записывали в области максимума теплового перехода.
Инфракрасная спектроскопия
Инфракрасную спектроскопию осуществляли на инфракрасном спектрофотометре преобразования Фурье (FT-IR) Magna 860® (Nicolet Instrument Corp.), оборудованного средним/дальним ИК источником Ever-Glo, расширенным уровнем расщепителя пучка калия бромида и дейтерированным триглицинсульфатным детектором (DTGS). Если не указано иначе, для отбора проб применяли прибор диффузного отражения Spectra-Tech, Inc. (Коллектор™). Каждый спектр представлял собой 256 взаимодополняющих считываний в спектральном разрешении 4 см-1. Получение образца для соединения состояло из помещения образца в микрокапсулу и выравнивания вещества по плоскости матового стекла. Установление исходного уровня проводили путем регулирования зеркала в рабочем положении. Спектр представлял собой отношение однолучевого показателя образца к однолучевому показателю исходного уровня. Калибровка длин волн в приборе осуществляли с применением полистирола.
ЯМР Спектроскопия
Спектр фазового раствора 1H ЯМР проводили при температуре окружающей среды на спектрометре AM-250 модели Bruker, работающем при 5,87 T (рабочая частота: 1H=250 МГц). Данные временного интервала получали с применением ширины импульса 7,5 ps и времени экспозиции 1,6834 секунды при спектральном окне 5000 Гц. Было собрано в общей сложности 16384 экспериментальных точек. Снижение времени выдержки 5 секунд применяли между переходными состояниями. Каждый набор данных, как правило, состоял из 128 усредненных переходных состояний. Спектр обрабатывали с применением программного обеспечения GRAMS 132 Al, версии 6,00. Сигнал свободной индукции (FID) обнуляли четыре раза числом координат данных и экпоненциально умножали на фактор расширения спектральной линии 0,61 Гц перед преобразованием Фурье. Спектры 1Η сравнивали с тетраметилсиланом (0 м.д.), который был добавлен в качестве внутреннего стандарта.
Альтернативно, анализ ЯМР выполняли, как описано в Примере 4.
Анализ сорбции/десорбции паров воды
Данные сорбции/десорбции паров воды собирали на паровом сорбционном анализаторе VTI SGA-100. Данные сорбции и десорбции собирали в пределах от 5% до 95% относительной влажности (RK) при 10% интервалах RH при продувании азотом. В качестве калибровочных стандартов применяли хлорид натрия (NaCl) и поливинилпирролидон (PVP). Критерии равновесия, применяемые для анализа, были менее чем 0,0100% изменения массы в течение 5 минут, с максимальным временем равновесия 180 минут, если критерий массы не соответствовал. Графические данные с учетом содержания начальной влажности не корректировали.
Номенклатура
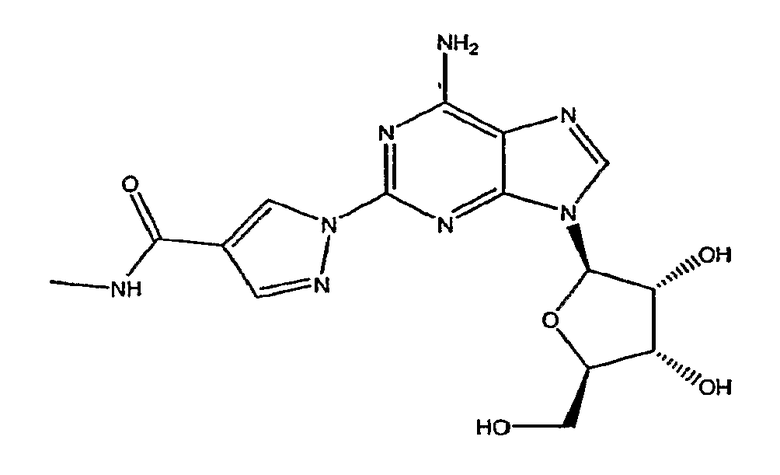
Структура соединения (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида является следующей:

Синтез (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида
Один способ для широкомасштабного синтеза (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида представлен в реакционной схеме I.
СХЕМА РЕАКЦИИ I
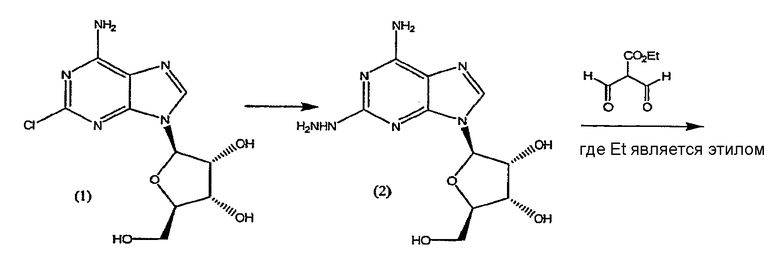
Стадия 1 - Получение формулы (2)
Соединение формулы (2) получают из соединения формулы (1) посредством реакции с гидразином моногидратом в отсутствии растворителя. Реакцию проводят при температуре приблизительно 40°C плюс/минус 5°C. Когда реакция завершена, продукт формулы (2) выделяют путем смешивания с протонным растворителем, в котором соединение формулы (2) обладает ограниченной растворимостью, например, с этанолом или изопропанолом. Смесь перемешивают в течение приблизительно 1-5 часов, и затем фильтруют. Твердый осадок очищают путем смешивания с водой, фильтрования и промывания водой, затем изопропанолом и высушивают под вакуумом, и переходят на следующую стадию без очистки.
Стадия 2 - Получение формулы (3)
Соединение формулы (2) затем переводят в соединение формулы (3) с помощью реакции с приблизительно 1-1,2 молярным эквивалентом этиловым эфиром 2-формил-3-оксопропионовой кислоты.
Реакцию проводят в протонном растворителе, предпочтительно этаноле, при температуре кипения с обратным холодильником в течение приблизительно 2-4 часов. После охлаждения до приблизительно 0°C твердый осадок отфильтровывают, промывают холодным этанолом и высушивают при пониженном давлении. Продукт формулы (3) переводят на следующую стадию без очистки.
Стадия 3 - Получение конечного продукта
Конечный продукт получают из соединения формулы (3) с помощью реакции с метиламином, предпочтительно с водным метиламином. Реакцию проводят при комнатной температуре, в течение приблизительно 4 часов. Продукт формулы I выделяют с помощью обычных способов, например с помощью фильтрования, промывки осадка холодным этанолом и высушивания при пониженном давлении.
Получение исходных веществ
(4S,2R,3R,5R)-2-(6-амино-2-хлорпурин-9-ил)-5-(гидроксиметил)оксолан-3,4-диол применяют в качестве исходного вещества на стадии 1. Данное соединение является коммерчески доступным.
Этиловый эфир 2-формил-3-оксопропионовой кислоты применяют в качестве исходного вещества на стадии 2. Он является коммерчески доступным, или может быть получен, как представлено на реакционной схеме II.
СХЕМА РЕАКЦИИ II
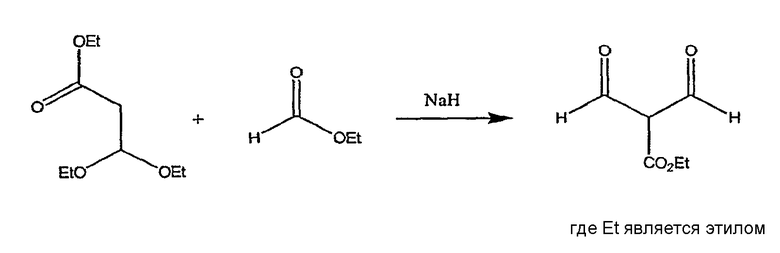
Этиловый эфир 3,3-диэтоксипропионовой кислоты взаимодействует с этилформиатом в присутствии сильного основания, предпочтительно гидрида натрия. Реакцию проводят приблизительно при 0-5°C, приблизительно в течение 24 часов. Продукт выделяют обычными способами, например посредством добавления воды и экстракции примесей обычными растворителями, например трет-бутилметиловым эфиром, подкисления водной фазы, например, соляной кислотой, с последующей экстракцией растворителем, таким как дихлорометан, и удаления растворителя из высушенного экстракта при пониженном давлении.
Предпочтительный способ широкомасштабного синтеза (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида представлен в реакционной схеме III.
Реакционная схема III
Стадия 1 - Получение формулы (2)
Соединение формулы (2) получают из соединения формулы (1) с помощью реакции с гидразином моногидратом в отсутствии растворителя. Реакцию проводят при температуре приблизительно 45-55°C плюс/минус 5°C. Когда реакция окончена, продукт формулы (2) выделяют путем смешивания с протонным растворителем, в котором соединение формулы (2) обладает ограниченной растворимостью, например с этанолом или изопропанолом. Смесь перемешивают в течение приблизительно 1-5 часов и затем фильтруют. Твердый осадок очищают путем смешивания с водой, фильтрования и промывкой водой, затем этанолом или изопропанолом и сушкой под вакуумом, и переходят на следующую стадию без очистки.
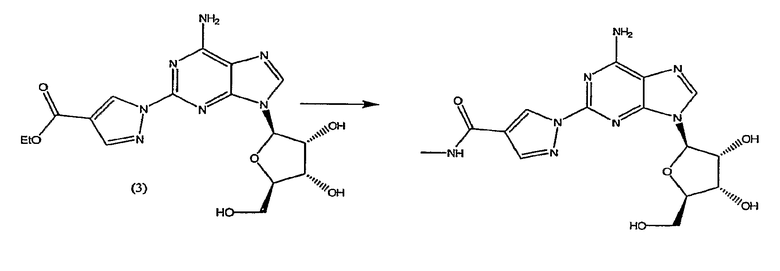
Стадия 2 - Получение формулы (4)
Соединение формулы (2) затем переводят в соединение формулы (4) с помощью реакции с избытком этиловым эфиром 2-формил-3-оксопропионовой кислоты, например 2-10 кратным избытком, предпочтительно приблизительно 5-10 кратным избытком. Реакцию проводят в протонном растворителе, например этаноле, при температуре кипения с обратным холодильником в течение приблизительно 2-4 часов. После охлаждения до приблизительно 0°C, твердый осадок отфильтровывают, промывают холодным этанолом и высушивают при пониженном давлении, и продукт формулы (4) переводят на следующую стадию без очистки.
Соединение формулы (4) изображено в виде алкенового производного (2E), т.к. он является основным изомером, образующимся в данной реакции. Однако следует отметить, что в данной реакции также может быть образовано значительное количество алкенового производного (2Z), структурной формулы
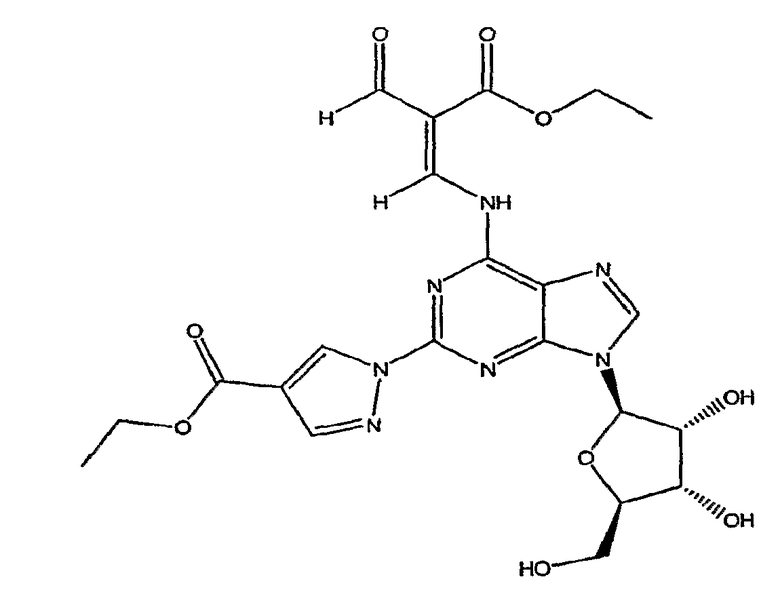
названное как этил-(2Z)-3-({9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)-оксолан-2-ил]-2-[4-(этоксикарбонил)пиразолил]пуринн-6-ил}амино)-2-формилпроп-2-еноат.
Таким образом, хотя соединение формулы (4) представлено только как алкеновое производное (2E), предполагается, что термин "соединение формулы (4)" означает, что соединение представляет собой только изомер (2E) или соединение, основную часть которого составляет изомер (2E), а также присутствует небольшая часть изомера (2Z). Превращение соединения формулы (4) в конечный продукт посредством реакции с метиламином, как описано в стадии 3, проводят одним и тем же способом, присутствует ли соединение формулы (4) в виде изомера (2E) или в виде смеси изомера (2E) и изомера (2Z).
Стадия 3 - Получение конечного продукта
Конечный продукт получают из соединения формулы (4) с помощью реакции с метиламином, предпочтительно с водным метиламином. Первоначально реакцию проводят приблизительно при 0-5°C в течение 8 часов, предпочтительно в реакторе под давлением, с последующим повышением температуры до 50-60°C в течение приблизительно 1 часа, и поддержание температуры в течение 15-30 мин. Продукт выделяют с помощью обычных способов, например охлаждением до 0-5°C и оставляя в вакууме приблизительно на 1 час, удаляя таким образом метиламин. Вакуум убирают, и оставшееся содержимое оставляют при 0-5°C в течение, по меньшей мере, 30 минут, с последующим фильтрованием. Осадок, полученный таким образом, промывают водой, затем этанолом и высушивают при пониженном давлении.
Данный процесс обеспечивает (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамид в виде его моногидрата. Данный полиморф можно дополнительно очистить посредством растворения в диметилсульфоксиде, фильтрования любых твердых примесей из раствора и осаждением моногидрата из раствора посредством добавления воды.
ПРИМЕР 1
Получение этилового эфира 2-формил-3-оксопропионовой кислоты
Трех- или четырехгорловую круглодонную колбу, оборудованную магнитной мешалкой, термопарой, цифровым термометром, подводом и выводом газа и дополнительной трубой, продули аргоном. Этиловый эфир 3,3-диэтоксипропионовой кислоты (64,5 г) в тетрагидрофуране загрузили в дополнительную воронку. Гидрид натрия (21,2 г 60% коллоидного раствора) загрузили в реакционную колбу вслед за тетрагидрофураном. Содержимое колбы охладили до 0-5°C на ледяной бане и добавили этилформиат (257 г). Смесь охладили до 0-5°C и содержимое дополнительной воронки добавили по каплям, поддерживая внутреннюю температуру менее чем 5°C. Ледяную баню удалили и содержимому позволили нагреться до температуры окружающей среды. Расход этилового эфира 3,3-диэтоксипропионовой кислоты контролировали с помощью TLC анализа. Реакцию останавливали посредством добавления ледяной воды (10,6 объемов) и экстрагировали три раза метил-трет-бутиловым эфиром (5,4 объема каждый) и удаляли органические слои. В водную фазу добавили концентрированную соляную кислоту до pH от 1 до 1,5. Подкисленный водный слой экстрагировали три раза с дихлорметаном и соединенные органические слои высушили с помощью сульфата натрия. Растворитель удалили при пониженном давлении, и осадок перегоняли в вакууме с получением этилового эфира 2-формил-3-оксопропионовой кислоты, 27,92 г, с 70% выходом.
ПРИМЕР 2
A. Получение 2-гидразиноаденозина (2)
Реакционную колбу, оборудованную механической мешалкой, подводом и выводом газа и термопарой, продули аргоном. Добавили гемигидрат 2-хлороаденозина (53,1 г), с последующим добавлением моногидрата гидразина (134 г). Смесь перемешивали при нагревании до 40-45°C в течение 2 часов. Ход реакции наблюдали с помощью TLC анализа. Когда реакция закончилась, нагревательный элемент убрали и добавили этанол (800 мл). Смесь перемешивали в течение 2 часов при температуре окружающей среды, затем осадок собрали посредством фильтрования. Отфильтрованный осадок промыли этанолом и высушивали при пониженном давлении в течение 30 минут. Осадок перенесли в чистую реакционную колбу, оборудованную механической мешалкой, и добавили воду (300 мл). Суспензию перемешивали при комнатной температуре в течение 18 часов, и осадок выделили с помощью фильтрации. Отфильтрованный осадок промыли ледяной водой (300 мл), затем промыли охлажденным льдом этанолом (300 мл). Осадок высушивали при пониженном давлении с получением 2-гидразинаденозина (41,38 г, 81,4% выхода, 99,3% чистого продукта).
B. Альтернативное получение 2-гидразиноаденозина (2)
Реакционный сосуд, содержащий гидрат гидразина (258 г, 250 мл), нагрели до 40-50°C. К теплой смеси порциями добавили гемигидрат хлораденозина (100 г), поддерживая температуру между 45 и 55°C. Температуру поддерживали на данном уровне в течение двух часов и затем добавили деионизированную воду (500 мл) в течение 30 минут, поддерживая температуру при 45-55°C. Затем смесь постепенно охлаждали до 0-5°C в течение 3 часов, затем перемешивали при данной температуре в течение следующих 30 минут. Осадок затем отфильтровывали и промывали холодной (2-5°C) деионизированной водой (200 мл), затем промыли этанолом (400 мл). Осадок высушили под вакуумом в течение 12 часов для получения 2-гидразинаденозина.
ПРИМЕР 3
Получение этил-1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-карбоксилата (3)
Этиловый эфир 2-формил-3-оксопропионовой кислоты (23,93 г, 0,17 моль) поместили в реакционную колбу, оборудованную механической мешалкой, подводом и выводом газа и обратным холодильником. В реакционную колбу добавили 2-пропанол, затем добавили 2-гидразинаденозин (44,45 г, 0,15 моль). Смесь нагревали до температуры кипения с обратным холодильником при перемешивании в течение 2-4 часов, при этом оценивая ход реакции посредством TLC анализа. Когда решали, что реакция закончилась, нагревательный элемент убрали и смесь охлаждали до комнатной температуры. Суспензию охлаждали при помешивании на ледяной бане в течение от 1,5 до 2 часов. Осадок выделяли посредством вакуумной фильтрации и промывали охлажденным льдом 2-пропанолом. Продукт, этил-1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-карбоксилат, высушивали при пониженном давлении до постоянной массы. Выход 54,29 г, чистый продукт (по HPLC) 96,6%.
ПРИМЕР 4
Получение 1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида

Смесь этил-1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-карбоксилата (46,4 г) и метиламина (40% в воде, 600 мл) перемешивали при температуре окружающей среды в течение приблизительно 4 часов, при этом оценивая ход реакции посредством TLC анализа. Большую часть избыточности метиламина было удалено при пониженном давлении, и оставшуюся смесь охлаждали при 0°C в течение 2 часов. Твердое вещество отфильтровали, промывали охлажденным льдом этанолом с крепостью 200, и высушивали при пониженном давлении с получением 1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-карбоксамида в виде его моногидрата, 36,6 г, чистый продукт 99,6%.
Структура вещества была подтверждена 1H ЯМР (см. фиг.1 и ниже). Термический анализ (см. фиг.2) представил результаты, соответствующие наличию одной молекулы воды. Были получены профили рентгеновской дифракции порошка (фиг.3).
1H и 13C ЯМР спектры получили следующим способом. Два образца вещества, полученные выше, взвесили и растворили в d6-DMSO - 5,3 мг использовали для 1H спектра, и 20,8 мг использовали для 13C спектра. Все спектры получали при температуре окружающей среды на спектрометре JEOL Eclipse* 400, производящего 400 МГц для 1H и 100 МГц для 13C.
Очистка моногидрата 1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-карбоксамида
Раствор моногидрата 1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-карбоксамида (100 г) в диметилсульфоксиде (300 мл) фильтровали через 0,6-0,8 микронные префильтры и 0,2 микронный префильтр для удаления любых твердых примесей. Отфильтрованное вещество затем медленно в течение периода 1 час добавляли к деионизированной воде (1 литр) с помешиванием, и, таким образом, помешивая суспензию в течение не менее чем 1 ч. Твердый осадок отфильтровали, промывали деионизированной водой (2×1 литр) и высушили в вакууме в течение не менее чем 1 час. Высушенный продукт затем смешивали до получения суспензии с деионизированной водой (1,5 литра) в течение не менее чем 2 часа, отфильтровали и промывали деионизированной водой (1 литр), затем промывали абсолютным этанолом (750 мл). Очищенный продукт затем высушили под вакуумом при температуре не более чем 40°C в течение не менее чем 12 часов с получением моногидрата 1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-карбоксамида, свободного от какой-либо примеси 2-гидразинаденозина.
ПРИМЕР 5
Получение этил-(2Е)-3-({9-[(4S , 2R , 3R,5R)-3,4-дигидрокси-5-(гидроксиметил)-оксолан-2-ил]-2-[4(этоксикарбонил)пиразолил]пурин-6-ил}амино)-2-формилпроп-2-еноата.
Смесь 2-гидразинаденозина (100 г, 0,34 моль), этилового эфира 2-формил-3-оксопропионовой кислоты (242 г, 1,7 моль) и абсолютного этанола загружали в реакционную колбу, и смесь нагревали до кипения с обратным холодильником в течение 2 часов. Когда решали, что реакция закончилась, нагревательный элемент убрали и смесь постепенно охлаждали до 5-10°C в течение 3 часов. Суспензию перемешивали в течение 30 минут при данной температуре, и фильтровали смесь. Осадок промывали холодным (5-10°C) абсолютным этанолом и затем высушивали в вакууме при температуре, не превышающей 40°C с получением этил-(2Е)-3-({9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)-оксолан-2-ил]-2-[4(этоксикарбонил)пиразолил]пурин-6-ил}амино)-2-формилпроп-2-еноата.
Элементный анализ дал следующие результаты: C, 48,75%; H, 4,86%; N, 18,05%; O, 27,57. Теоретические: C, 49,72%; H, 4,74%; N, 18,45%; O, 27,09. В пределах погрешностей эксперимента анализ соответствует гемигидрату ожидаемого продукта (C, 48,89%; H, 4,81%; N, 18,1%; O, 28,12).
1H и 13C ЯМР спектры были получены следующим способом. 20,2 мг соединения формулы (4) растворили в ~0,75 мл DMSO-d6, и спектры получали при температуре окружающей среды на спектрометре JEOL ECX-400 NMR, производящего 400 МГц для 1H и 100 МГц для 13C. Химические сдвиги были по отношению к растворителю DMSO, 2,50 м.д. для 1H и 39,5 м.д. для 13C.
РЕЗУЛЬТАТЫ
Химические сдвиги 1H и 13C перечислены в Таблице 1. Два изомера с соотношением ~60/30 были обнаружены и в спектре 1H и в спектре 13C, помеченные в таблице как основной и побочный.
Подтверждено, что соединение формулы (4) является смесью двух следующих изомеров:
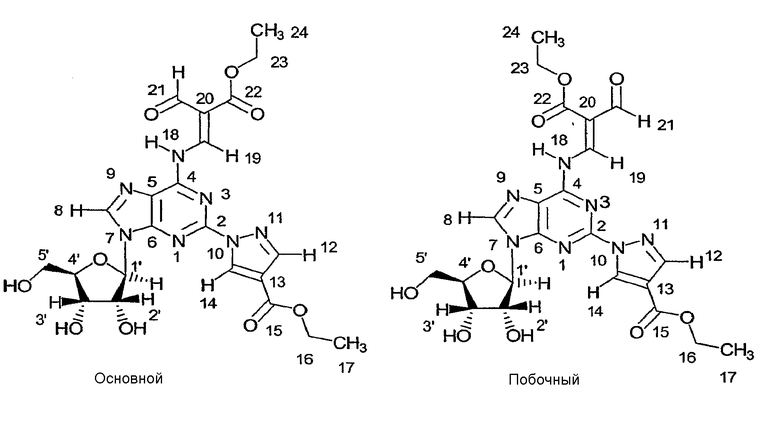
ПРИМЕР 6
Получение 1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида из соединения (4)
40% Водный раствор метиламина (1300 мл) помещали в реактор под давлением, охладили до 0-5°C и добавили продукт Примера 5 этил-(2Е)-3-({9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)-оксолан-2-ил]-2-[4-(этоксикарбонил)пиразолил]пурин-6-ил}амино)-2-формилпроп-2-еноат (100 г). Смесь перемешивали при 0-5°C в течение, по меньшей мере, 8 часов, контролируя реакцию до ее завершения. Когда реакция была завершена, смесь нагрели, поддерживая температуру между 50 и 60°C в течение 1 часа, и затем охладили менее чем до 30°C в течение периода времени 1 час. Когда температура стала ниже 30°C, смесь дегазировали посредством давления 100-150 мм Hg, позволяя температуре опуститься до 0-5°C. Смесь перемешивали при 0-5°C в течение, по меньшей мере, 1 часа, поддерживая давление 100-150 мм Hg. Затем вакуум заменяли на азот, поддерживая температуру 0-5°C в течение не менее 30 минут. Затем твердый продукт отфильтровали, промыли водой (3×500 мл), затем абсолютным этанолом (625 мл). Продукт высушили под вакуумом, не позволяя температуре превысить 40°C, для получения 1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида в виде его моногидрата.
1H и 13C ЯМР спектры получили следующим способом. Два образца вещества, полученные выше, взвесили и растворили в d6-DMSO - 5,3 мг использовали для 1H спектра, и 20,8 мг использовали для 13C спектра. Все спектры получали при температуре окружающей среды на спектрометре JEOL Eclipse* 400, производящего 400 МГц для 1H и 100 МГц для 13C.
Элементный анализ дал следующие результаты: C, 43,96%; H, 4,94%; N, 27,94. Теоретические: C, 44,12%; H, 4,94%; N, 27,44%; O, 27,09. Анализ соответствует моногидрату в пределах погрешностей эксперимента.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВИЗУАЛИЗАЦИЯ ПЕРФУЗИИ МИОКАРДА С ИСПОЛЬЗОВАНИЕМ АГОНИСТОВ АДЕНОЗИНОВЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2346693C2 |
| СПОСОБЫ И КОМПОЗИЦИИ, ПОВЫШАЮЩИЕ ПЕРЕНОСИМОСТЬ ПАЦИЕНТОМ МЕТОДОВ ВИЗУАЛИЗАЦИИ МИОКАРДА | 2007 |
|
RU2459626C2 |
| АГОНИСТЫ А3 РЕЦЕПТОРОВ АДЕНОЗИНА | 2002 |
|
RU2298557C2 |
| ЧАСТИЧНЫЕ И ПОЛНЫЕ АГОНИСТЫ АДЕНОЗИНОВЫХ РЕЦЕПТОРОВ A | 2003 |
|
RU2340623C2 |
| АГОНИСТЫ TLR7 | 2019 |
|
RU2817014C2 |
| ЧАСТИЧНЫЕ И ПОЛНЫЕ АГОНИСТЫ АДЕНОЗИНОВЫХ РЕЦЕПТОРОВ А | 2003 |
|
RU2317994C2 |
| ПРОИЗВОДНЫЕ 2-АЛКИНИЛАДЕНОЗИНА ДЛЯ БОРЬБЫ С ВОСПАЛИТЕЛЬНОЙ РЕАКЦИЕЙ | 2000 |
|
RU2258071C2 |
| ПРОИЗВОДНЫЕ АДЕНОЗИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ КОРРЕКЦИИ ЭЛЕКТРИЧЕСКИХ НАРУШЕНИЙ В СЕРДЦЕ МЛЕКОПИТАЮЩЕГО | 1997 |
|
RU2172320C2 |
| Способ химической обработки нефтедобывающих скважин или трубопроводов и основа реагента для его осуществления | 2024 |
|
RU2830667C1 |
| НОВОЕ ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕЕ, В ЧАСТНОСТИ, АГЕНТ ДЛЯ ПРЕДОТВРАЩЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ОПУХОЛЕЙ, И ТОМУ ПОДОБНОЕ, НА ОСНОВЕ ИНГИБИТОРНОГО ВОЗДЕЙСТВИЯ НА NAE | 2015 |
|
RU2658008C2 |

Настоящее изобретение относится к способам широкомасштабного получения агониста А2A-аденозинового рецептора, в частности моногидрата (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида
 . Заявлены также способы получения промежуточных продуктов, используемых для получения указанного выше моногидрата, и непосредственно сам моногидрат (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида. Технический результат - обеспечение новых удобных способов получения 1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида, которые обеспечивают получение больших количеств целевого вещества с хорошим выходом и высокой степенью очистки. 5 н. и 10 з.п. ф-лы, 6 пр., 5 ил.
. Заявлены также способы получения промежуточных продуктов, используемых для получения указанного выше моногидрата, и непосредственно сам моногидрат (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида. Технический результат - обеспечение новых удобных способов получения 1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида, которые обеспечивают получение больших количеств целевого вещества с хорошим выходом и высокой степенью очистки. 5 н. и 10 з.п. ф-лы, 6 пр., 5 ил.
1. Способ широкомасштабного получения моногидрата (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида

включающий взаимодействие соединения формулы (3)
с водным раствором метиламина при начальной температуре приблизительно 0-5°С с последующим нагреванием до приблизительно 50-70°С, и реакцию проводят в герметичном реакторе под давлением.
2. Способ по п.1, дополнительно включающий выделение конечного продукта (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида в виде чистого моногидрата посредством:
(a) растворения продукта в растворителе,
(b) добавления очищенной воды,
(c) фильтрования суспензии, полученной таким образом,
(d) промывания содержимого фильтра водой с последующей промывкой этанолом и
(e) высушивания твердого осадка, который остается, под вакуумом при температуре, не превышающей 40°С.
3. Способ по п.2, где растворителем, применяемым на стадии (а), является диметилсульфоксид.
4. Способ получения соединения формулы (3)
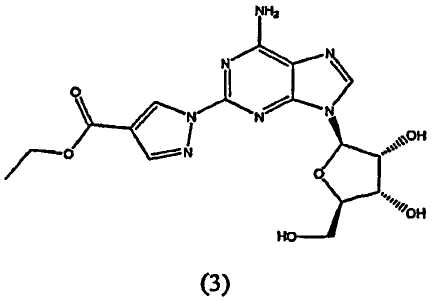
включающий взаимодействие соединения формулы (2)
с приблизительно 1,1 молярным эквивалентом этилового эфира 2-формил-3-оксопропионовой кислоты в растворителе.
5. Способ по п.4, где растворителем является этанол.
6. Способ по п.5, где реакцию проводят при температуре приблизительно 80°С.
7. Способ получения моногидрата (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида путем взаимодействия соединения формулы (4)
с водным раствором метиламина при начальной температуре приблизительно 0-5°С с последующим нагреванием до приблизительно 50-70°С в герметичном реакторе под давлением.
8. Способ по п.7, дополнительно включающий выделение конечного продукта (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида в виде чистого моногидрата посредством:
(a) растворения продукта по п.7 в растворителе,
(b) добавления очищенной воды,
(c) фильтрования суспензии, полученной таким образом,
(d) промывания содержимого фильтра водой с последующей промывкой этанолом и
(e) высушивания твердого осадка, который остается, под вакуумом при температуре, не превышающей 40°С.
9. Способ по п.8, где растворителем, применяемым на стадии (а), является диметилсульфоксид.
10. Способ получения соединения формулы (4)
включающий взаимодействие соединения формулы (2)
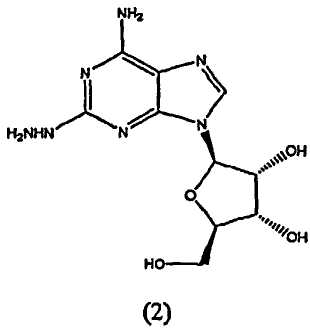
с приблизительно 2-10-кратным молярным избытком этилового эфира 2-формил-3-оксопропионовой кислоты в растворителе.
11. Способ по п.10, где растворителем является этанол.
12. Способ по п.11, где реакцию проводят при температуре приблизительно 80°С.
13. Способ по п.10, где приблизительно 5-10-кратный молярный избыток этилового эфира 2-формил-3-оксопропионовой кислоты взаимодействует с соединением формулы (2).
14. Моногидрат (1-{9-[(4S,2R,3R,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6-аминопурин-2-ил}пиразол-4-ил)-N-метилкарбоксамида, который находится в кристаллической форме.
15. Моногидрат по п.14, где кристаллическая форма характеризуется следующим Н ЯМР спектром:
| ПРИСПОСОБЛЕНИЕ ДЛЯ ДЕМОНТАЖА ШАРИКОПОДШИПНИКОВ | 1949 |
|
SU78779A1 |
| Niiya К et al, Journal of Medicinal Chemistry, 1992, 35(24), CC.4557-4561 | |||
| Киноэкран | 1925 |
|
SU4861A1 |

