Область техники
Настоящее изобретение относится к способам и композициям для предотвращения повреждения тканей в результате воспалительного действия.
Предпосылки изобретения
Настоящее изобретение было создано при помощи финансирования правительства США (NIH Grant ROL HL37942). Правительство США имеет определенные права на данное изобретение.
Воспалительная реакция служит для устранения вредных агентов из организма. Существует широкий спектр патогенных поражений, которые могут инициировать воспалительную реакцию, включая инфекцию, аллергены, аутоиммунные стимулы, иммунную реакцию на пересаженную ткань, вредные химические вещества и токсины, ишемию/реперфузию, гипоксию, механическую и термическую травму. Воспаление обычно оказывается весьма локализованным действием, служащим для удаления, смягчения разбавлением и удаления поражающего агента и поврежденной ткани. Реакция организма становится фактором заболевания в тех случаях, когда она приводит к нежелательному повреждению тканей хозяина в процессе устранения агента-мишени или ответа на травматическое поражение.
В качестве примеров, воспаление является компонентом патогенеза при некоторых сосудистых заболеваниях или поражениях. Подобные примеры включают ишемию/реперфузионное повреждение (N.G.Frangogiannis et al., in Myocardial Ischemia: Mechanisms, Reperfusion, Protection, M. Karmazyn, ed., Birkhuser Verlag (1996) at 236-284; H.S. Sharma et al., Med. of Inflamm., 6, 175 (1987), атеросклероз (R. Ross, Nature, 362, 801 (1993)), воспалительные аневризмы аорты (N. Girardi et al., Ann. Thor. Surg., 64, 251 (1997); D.I. Walker et al., Brit. J. Surg., 59, 609 (1972); R.L. Pennel et al., J. Vasc. Surg., 2, 859 (1985)), а также рестеноз после баллоноангиопластики (см. R. Ross, процитированный выше). Клетки, принимающие участие в воспалительном процессе, включают лейкоциты (т.е. клетки иммунной системы - нейтрофилы, эозинофилы, лимфоциты, моноциты, базофилы, макрофаги, дендритные и тучные клетки), сосудистый эндотелий, сосудистые клетки гладкой мускулатуры, фибробласты и миоциты.
Высвобождение воспалительных цитокинов, таких как фактор некроза опухоли альфа (TNFα), лейкоцитами является средством, при помощи которого иммунная система противостоит патогенным инвазиям, включая инфекции. TNFα стимулирует экспрессию и активацию факторов адгезии на лейкоциты и эндотелиальные клетки, примирует нейтрофилы на усиленный противовоспалительный ответ на вторичные стимулы, а также усиливает адгезивную окислительную активность нейтрофилов. См. Sharma et al., процитированный выше. Кроме того, макрофаги/дендритные клетки действуют как вспомогательные клетки, процессируя антиген для презентации лимфоцитам. Лимфоциты, в свою очередь, получают стимул к действию в качестве провоспалительных цитотоксичных клеток.
В целом, цитокины стимулируют нейтрофилы, усиливая окислительную (например, супероксид и вторичные продукты) и неокислительную (например, миелопероксидаза и другие ферменты) воспалительную активность. Нежелательное и избыточное высвобождение цитокинов может вызывать контрпродуктивное усиленное патогенное действие в результате высвобождения разрушающих ткань окислительных и неокислительных воспалительных продуктов (К. G. Tracey et al., J. Ехр. Med., 167, 1211 (1988); and. 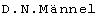 et al., Rev. Infect.Dis., 9 (suppl. 5), S 602-S 606 (1987)). Например, TNFα может индуцировать адгезию нейтрофилов к стенкам кровеносных сосудов, а затем их миграцию по сосуду к месту повреждения и высвобождение их окислительных и неокислительных продуктов воспаления.
et al., Rev. Infect.Dis., 9 (suppl. 5), S 602-S 606 (1987)). Например, TNFα может индуцировать адгезию нейтрофилов к стенкам кровеносных сосудов, а затем их миграцию по сосуду к месту повреждения и высвобождение их окислительных и неокислительных продуктов воспаления.
Несмотря на то, что моноциты медленно собираются в очагах воспаления, при благоприятных условиях они развиваются в долговременные, постоянные вспомогательные клетки и макрофаги. При стимуляции триггером воспаления моноциты/макрофаги также продуцируют и секретируют ряд цитокинов (включая TNFα), комплемент, липиды, реакционноспособные виды кислорода, протеазы и факторы роста, перестраивающие ткань и регулирующие окружающие ткань функции.
Например, была установлена патогенность воспалительных цитокинов при артрите (С.A.Dinarello, Semin. Immunol., 4, 133 (1992)); ишемии (A.Seekamp et al., Agents-Actions-Supp., 41, 137 (1993)); септическом шоке ( et al., Rev. Infect. Dis., 9 (suppl. 5), S602-S606 (1987)); астме (N.M. Cembrzynska et al., Am. Rev. Respir. Dis., 147, 291 (1993)); отторжении при трансплантации органов (D.K. Imagawa et al., Transplantation, 51, 57 (1991); рассеянном склерозе (Н.Р. Hartung, Ann. Neurol., 33, 591 (1993)); спиде (Т. Matsuyama et al., AIDS, 5, 1405 (1991) и ожоге глаз щелочью (F. Miyamoto et al., Opthalmic Res., 30, 168 (1997)). Кроме того, образование супероксида в лейкоцитах способствует репликации вируса иммунодефицита человека (ВИЧ) (S. Legrand-Poels et al., AIDS Res. Hum. Retroviruses, 6, 1389 (1990)).
Хорошо известно, что аденозин и некоторые его аналоги, неселективно активирующие подтипы рецепторов аденозина, снижают выработку нейтрофила воспалительных окислительных продуктов (B.N.Cronstein et al., Ann. N.Y.Acacd Sci., 451, 291 (1985); P.A.Roberts et al., Biochem. J., 227, 669 (1985); D.J.Schrier et al., J. Immunol., 137, 3284 (1986); В.N.Cronstein el al., Clinical Immunol and Immunopath., 42, 76 (1987); M.A.Iannone et al., in Topics and Perspective in Adenosine Research, E. Gerlach et al., cds., Springer-Verlag, Berlin, p. 286 (1987); S.T.McGarrity et al., J.Leukocyte Biol., 44, 411421 (1988); J.De La Harpe et al., J.Immunol, 143, 596 (1989); S.Т.McGarrity el al., J. Immunol., 142, 1986 (1989); and С.P.Nielson et al., Br.J. Pharmacol, 97, 882 (1989)). Например, было установлено, что аденозин ингибирует высвобождение супероксида из нейтрофилов, стимулируемых хемоатрактантами, такими как синтетическая имитация бактериальных пептидов, f-met-leu-phe (fMLP), а также компонента C5a комплемента (B.N.Cronstein et al., J. Immunol., 135, 1366 (1985). Аденозин может уменьшить сильно увеличенный окислительный выброс (очаг) PMN (нейтрофил), вначале примированный THF-α, а затем стимулированный вторым стимулом, таким как f-met-leu-phe (G.W. Sullivan et al., Clin. Res., 41, 172A (1993)). Кроме того, было установлено, что аденозин способен снижать уровень репликации ВИЧ в Т-клеточной линии (S. Sipka et al., Acta. Biochim. Biopys. Hung., 23, 75 (1998)). Однако не подтвержден тот факт, что in vivo аденозин обладает противовоспалительной активностью (G.S. Firestein et al., Clin. Res., 41, 170А (1993); and B.N.Cronstein et al., Clin. Res., 244A (1993)).
Было высказано предположение о том, что на нейтрофилах находится более одного подтипа рецептора аденозина, которые могут оказывать обратное действие на высвобождение супероксида (B.N.Cronstein et al., J. Clin. Invest., 85, 1150 (1990)). Существование рецептора А2А на нейтрофилах было первоначально установлено Van Calker et al. (D.Van Calker et al., Eur. J. Pharmacology, 206, 285 (1991).
Постепенно разрабатывались все более и более эффективные и/или селективные соединения в качестве агонистов А2A-рецепторов аденозина (АР) на основе анализов по связыванию радиолигандов и физиологических реакций. Вначале были разработаны соединения, не обладающие селективностью или обладающие невысокой селективностью по отношению к А2A-рецепторам, такие как сам аденозин или 5'-карбоксамиды аденозина, такие как 5'-N-этилкарбоксамидоаденозин (NECA) (B.N.Cronstein et al., J. Immunol., 135, 1366 (1985)). Позднее было установлено, что добавление 2-алкиламинозаместителей повышает эффективность и селективность, например, CV1808 и CGS21680 (M.F.Jarvis et al., J. Pharmacol. Ехр. Ther., 251, 888 (1989)). Производные 2-алкоксизамещенного аденозина, такие как WRC-0090, еще более эффективны и селективны как агонисты А2A-рецептора у коронарной артерии (М. Ueeda et al., J. Med. Chem., 34, 1334 (1991)). Было установлено, что производные 2-алкилгидразиноаденозина, например, SHA 211 (также называемый WRC-0474), также являются агонистами А2A-рецептора у коронарной артерии (К. Niiya et al., J. Med. Chem., 35, 4557 (1992)).
Существует одно описание сочетания относительно неспецифических аналогов аденозина: R-фенилизопропиладенозина(R-PIA) и 2-хлораденозина (CI-Ado) с ингибитором фосфодиэстеразы (PDE), приводящее к снижению окислительной активности нейтрофила (М.А.Iannone et al., Topics and Perspectives in Adenosine Research, E. Garlach et al., eds., Springer-Verlag, Berlin, pp. 286-298 (1987)). Однако аналоги R-PIA и CI-Ado действительно являются более эффективными активаторами A1-рецепторов аденозина, чем А2A-рецепторы аденозина, и, таким образом, вероятно, вызывают побочное действие вследствие активации А1-рецепторов на сердечной мышце и других тканях, вызывая "блокаду сердца".
R.A.Olsson et al. (патент США № 5278150) описывают селективные агонисты А2-рецептора аденозина формулы:
где Rib представляет рибозил, R1 может представлять Н, a R2 может представлять циклоалкил. Указано, что данные соединения могут быть использованы для лечения гипертензии, атеросклероза, а также в качестве сосудорасширяющих средств.
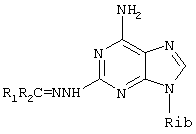
Olsson et al. (патент США № 5140015) описывают конкретные агонисты А2-рецептора аденозина формулы:
где C(X)BR2 может представлять CH2OH, a R1 может представлять алкил- или алкоксиалкил. Указано, что данные соединения могут быть использованы в качестве сосудорасширяющих средств или гипотензивных средств.
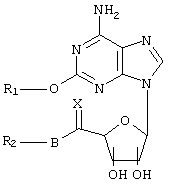
Linden et al. (патент США № 5877180) основываются на открытии того, что некоторые воспалительные заболевания, такие как артрит и астма, могут быть подвергнуты эффективному лечению путем введения соединений, которые являются селективными агонистами А2A-рецепторов аденозина, предпочтительно в сочетании с ингибитором фосфодиэстеразы, тип IУ. Вариант осуществления изобретения Linden et al. предусматривает способ лечения воспалительных заболеваний путем введения эффективного количества A2A-рецептора аденозина следущей формулы:
где R и X имеют значения, указанные в патенте.
Предпочтительный вариант изобретения (Linden et al.) предусматривает введение ингибитора фосфодиэстеразы (PDE), тип IУ, в сочетании с агонистом А2A-рецептора аденозина. Ингибитор Фосфодиэстеразы (PDE), тип IУ, включает рацемические и оптически активные 4-(полиалкоксифенил)-2-пирролидоны следующей формулы:
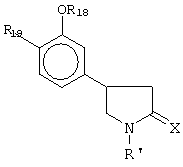
где R', R18, R19 и X имеют значения, указанные в патенте США №4193926. Ролипрам является примером подходящего ингибитора PDE, тип 1У, представленного вышеприведенной формулой.
G. Cristalli (патент США №5593975) описывает производные 2-арилэтинила, 2-циклоалкилэтинила или 2-гидроксиалкилэтинила, в которых остаток рибозида замещен карбоксиамино или замещенным карбоксиамино (R3HNC(О)-).
Производные 2-алкинилпурина описаны Miyasaka et al. (патент США № 4956345), при этом 2-алкинилгруппа замещена (С3-С16)алкилом. Указано, что соединения в соответствии с патентом США № 5593975 расширяют сосуды и ингибируют агрегацию тромбоцитов, таким образом, они могут быть использованы в качестве противоишемических, противоатеросклеротических и гипотензивных средств.
Однако сохраняется потребность в селективных агонистах А2-рецепторов аденозина для медицинского применения, оказывающих меньшее побочное действие.
Краткое описание изобретения
Настоящее изобретение включает соединения и способы их применения для лечения воспаления в ткани млекопитающих. Воспаление ткани может быть вызвано патологическими агентами либо физической, химической или тепловой травмой, либо травмой от медицинских процедур, таких как пересадка органов, тканей или клеток, ангиопластика (РСТА), воспаление после ишемии/реперфузии или имплантации. Данные соединения включают новый класс производных 2-алкиниладенозина, замещенных в положении этина замещенными фрагментами циклоалкила. Остаток рибозида предпочтительно замещен в положении 5'("X") N-алкил-(или циклоалкил)карбоксиамино ("аминокарбонил") фрагментом. Таким образом, настоящее изобретение предусматривает способ подавления воспалительной реакции у млекопитающих, таких как человек, а также защиту воспаленной ткани введением эффективного количества одного или нескольких соединений в соответствии с данным изобретением.
Соединения по данному изобретению имеют следующую общую формулу (I):
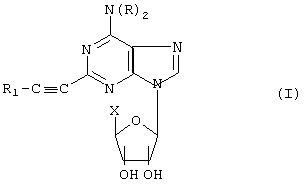
где (а) каждый из R независимо представляет водород, C1-C6алкил, С3-С7циклоалкил, фенил или фенил(C1-С3)алкил;
(b) X представляет -СН2ОН, -CO2R2, -OC(O)R2, -CH2OC(O)R2 или C(O)NRЗR4;
(c) каждый из R2, R3 и R4 отдельно представляет Н, C1-6алкил; C1-6-алкил, замещенный одним-тремя C1-6-алкокси, С3-7циклоалкилами, C1-6-алкилтио, галогенами, гидрокси, амино, моно (C1-6-алкил)амино, ди(C1-6-алкил)амино или С6-10-арилами, где арил может быть замещен одним-тремя галогенами, C1-6-алкилами, гидрокси, амино, моно(C1-6-алкил)амино или ди(C1-6-алкил)амино; C6-10-арил; или С6-10-арил, замещенный одним-тремя галогенами, гидрокси, амино, моно(C1-6-алкил) амино, ди(C1-6-алкил)амино или C1-6-алкилами;
(d) R1 представляет (X-(Z)-)n[(С3-С10)циклоалкил]-(Z')-, где Z и Z' независимо представляют (C1-C6)алкил, необязательно прерванный одним-тремя S или непероксидным О, либо отсутствуют, а n равно 1-3,
или их фармацевтически приемлемая соль.
Данное изобретение предусматривает соединение формулы I, которое может быть использовано для медикаментозного лечения, предпочтительно для лечения или защиты тканей от воспаления, такого как воспалительная реакция, а также использование соединения формулы I для изготовления лекарственных средств для лечения воспалительной реакции, вызванной патологическим состоянием или симптомом у млекопитающих, таких как человек, связанным с воспалением.
Несмотря на описание некоторых агонистов А2A-рецепторов аденозина в качестве сосудорасширяющих средств, которые могут быть непосредственно использованы для лечения гипертензии, тромба, атеросклероза и т.п., тканезащитное действие соединений формулы (I) не упоминается в известных источниках.
Данное изобретение также предусматривает применение указанных соединений с ингибиторами фосфодиэстеразы, тип IУ, для синергического снижения воспалительной реакции иммунных клеток.
Данное изобретение также предусматривает фармацевтическую композицию, включающую эффективное количество соединения формулы I или его фармацевтически приемлемой соли, в сочетании с фармацевтически приемлемым разбавителем или носителем и необязательно в сочетании с ингибитором фосфодиэстеразы (PDE), тип IУ. Композиция предпочтительно представлена в виде стандартной, дозированной лекарственной формы.
Кроме того, данное изобретение предусматривает терапевтический способ профилактики или лечения патологического состояния или симптома у млекопитающих, таких как человек, когда предполагается активность А2A-рецепторов аденозина и желателен агонизм указанной активности, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Предполагается, что активация А2A-рецепторов аденозина ингибирует воспаление, воздействуя на нейтрофилы, тучные клетки, моноциты/макрофаги, Т-клетки и/или эозинофилы. Ингибирование таких воспалительных клеток приводит к защите тканей после их поражения.
Воспалительные реакции, которые могут быть подвергнуты лечению (включая профилактическое) соединением формулы I необязательно с ингибитором PDE, тип IУ, включают воспаления, вызванные:
(a) аутоиммунной стимуляцией (аутоиммунные заболевания), такой как красная волчанка, рассеянный склероз, бесплодие, вызванное эндометриозом, сахарный диабет типа I, включая разрушение панкреатических островков, ведущее к диабету, и воспалительные последствия диабета, включая язвы на ногах, болезнь Крона, язвенный колит, воспалительное заболевание кишечника, остеопороз и ревматоидный артрит;
(b) аллергическими заболеваниями, такими как астма, сенная лихорадка, ринит, весенний конъюнктивит и другие эозинофилопосредованные состояния;
(c) кожными заболеваниями, такими как псориаз, контактный дерматит, экзема, инфекционные язвы кожи, открытые раны, целлюлит;
(d) инфекционными заболеваниями, включая сепсис, септический шок, энцефалит, инфекционный артрит, эндотоксический бактериально-токсический шок, грамотрицательный шок, реакция Яриша-Герксхаймера, опоясывающий лишай, токсический шок, церебральная малярия, бактериальный менингит, респираторный дистресс-синдром у взрослых (ARDS), болезнь Лайма, ВИЧ-инфекция (усиленная TNFa-ВИЧ-репликация, ингибирование TNFα-активности ингибитора обратной транскриптазы);
(e) изнуряющими болезнями: кахексия после рака и ВИЧ;
(f) трансплантацией органа, тканей или клеток (например, костного мозга, роговицы, почки, легких, печени, сердца, кожи, панкреатических островков), включая отторжение при трансплантации и болезнь "трансплантат против хозяина";
(g) побочным действием лекарственной терапии, включая побочное действие от лечения амфотерицином В, от иммуноподавляющей терапии, например лечение интерлейкином-2, побочное действие от лечения ОКТЗ, GM-CSF, циклоспорином, а также побочное действие лечения аминогликозидами, стоматита и мукозита, вызванное подавлением иммунитета;
(h) кардиососудистыми состояниями, включая заболевания кровообращения, индуцируемые или вызванные воспалительной реакцией, такими как ишемия, атеросклероз, заболевание периферических сосудов, рестеноз после ангиопластики, воспалительная аневризма аорты, васкулит, удар, повреждение спинного мозга, застойная сердечная недостаточность, геморрагический шок, ишемическое/реперфузионное повреждение, вазоспазм после субарахноидального кровоизлияния, вазоспазм после инсульта, плеврит, перикардит, а также сердечно-сосудистые осложнения диабета;
(i) диализом, включая перикардит, вызванный перитонеальным диализом;
(j) подагрой и
(k) химической или термической травмой, вызванной ожогами, кислотой, щелочью и т.п.
Особенно интересно и эффективно применение данных соединений для лечения воспалительных реакций, вызванных пересадкой органа, тканей или клеток, т.е. пересадка аллогенной или ксеногенной ткани реципиенту-млекопитающему, аутоиммунных заболеваний и воспалительных состояний, вызванных патологиями кровообращения и их лечением, включая ангиопластику, введение стента, шунта или имплантация. Неожиданно было обнаружено, что введение одного или нескольких соединений формулы (I) эффективно после возникновения воспалительной реакции, например, после поражения объекта патологией или травмой, инициирующей воспалительную реакцию.
Данное изобретение также включает способ измерения ответа или связывания соединения формулы I в или с обозначенными сайтами А2A-рецепторами аденозина, включающими указанные рецепторы, in vivo или in vitro, с количеством соединения формулы I, эффективным для связывания указанных рецепторов. Ткани или клетки, включающие связанные лигандом сайты рецепторов, могут быть использованы для измерения селективности тест-соединений на специфические подтипы рецептора, количества биоактивного соединения в крови или других физиологических жидкостях, либо могут быть использованы в качестве инструмента для идентификации потенциальных терапевтических агентов для лечения заболеваний или состояний, ассоциируемых с активацией сайта рецепторов, путем контакта указанных агентов с указанными комплексами лиганд-рецептор и измерения степени замещения лиганда и/или связывания агента, либо клеточной реакции на указанный агент (например, накопление цАМФ).
Подробное описание изобретения
Если не указано иначе, то определения имеют следующие значения. Галоид означает фтор, хлор, бром или иод. Алкил, алкокси, аралкил, алкиларил и т.д. означают как прямые, так и разветвленные алкильные группы, однако ссылка на отдельный радикал, такой как "пропил", подразумевает только радикал с прямой цепью, при этом изомер с разветвленной цепью, такой как "изопропил", указан конкретно. Арил включает фенильный радикал или орто-конденсированный бициклический, карбоциклический радикал, имеющий приблизительно от девяти до десяти кольцевых атомов, в которых по меньшей мере одно кольцо является ароматическим. Гетероарил включает радикал, присоединенный через углерод кольца моноциклического ароматического кольца, содержащего от пяти до шести кольцевых атомов, состоящих из углерода и от одного до четырех гетероатомов, каждый из которых выбран из группы, включающей непероксидный кислород, серу и N(X), где X отсутствует или представляет Н, О, (C1-С4)алкил, фенил или бензил, а также радикал орто-конденсированного бициклического гетероцикла, содержащего приблизительно от восьми до десяти полученных из него кольцевых атомов, особенно бензопроизводное или производное, полученное в результате его конденсации с пропиленовым, триметиленовым или тетраметиленовым дирадикалами.
Специалисту в данной области должно быть понятно, что соединения формулы (I) имеют несколько хиральных центров и могут быть выделены в оптически активных и рацемических формах. Фрагмент рибозида формулы (I) предпочтительно получают из D-рибозы, т.е. 3', 4'-гидроксильные группы являются альфа-группами по отношению к кольцу сахара, а 2'- и 5'-группы являются бета-группами (3R, 4S, 2R, 5S). Если две группы на циклогексильной группе находятся в положении 4, то они предпочтительно являются транс-группами. Некоторые соединения могут проявлять полиморфизм. Подразумевается, что настоящее изобретение включает любые рацемические, оптически активные, полиморфные или стереоизомерные формы либо их смеси соединений в соответствии с данным изобретением, обладающих описываемыми здесь полезными свойствами, при этом в данной области хорошо известно, как получать оптически активные формы (например, разложением рацемической формы перекристаллизацией или ферментами, синтезом из оптически активных исходных материалов, хиральным синтезом либо хроматографическим разделением с применением хиральной неподвижной фазы) и как определять активность агониста аденозина, применяя описываемые здесь тесты или другие подобные исследования, хорошо известные в данной области.
Конкретные и предпочтительные значения, указанные ниже для радикалов, заместителей и интервалов, служат только для иллюстрации; они не исключают другие определенные значения или другие значения, входящие в установленные интервалы для радикалов и заместителей.
Конкретно, (С1-С6)алкил может представлять метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, пентил, 3-пентил или гексил. В данном описании термин "циклоалкил" подразумевает бициклоалкил (норборнил, 2.2.2-бициклооктил и т.д.) и трициклоалкил (адамантил и т.д.), необязательно включающий один-два N, О или S. Циклоалкил также подразумевает (циклоалкил)алкил. Таким образом, (С3-С6)циклоалкил может представлять циклопропил, циклобутил, циклопентил или циклогексил; (С3-С6)циклоалкил(C1-C6)алкил может представлять циклопропилметил, циклобутилметил, циклопентилметил, циклогексилметил; 2-циклопропилэтил, 2-циклобутилэтил, 2-циклопентилэтил или 2-циклогексилэтил.
(C1-С6)алкокси может представлять метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, пентокси, 3-пентокси или гексилокси; (C2-C6)алкенил может представлять винил, аллил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 3-бутенил, 1-пентенил, 2-пентенил, 3-пентенил, 4-пентенил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил или 5-гексенил; (С2-С6)алкинил может представлять этинил, 1-пропинил, 2-пропинил, 1-бутинил, 2-бутинил, 3-бутинил, 1-пентинил, 2-пентинил, 3-пентинил, 4-пентинил, 1-гексинил, 2-гексинил, 3-гексинил, 4-гексинил или 5-гексинил; (C1-C6)алканоил может представлять ацетил, пропаноил или бутаноил; галоид(C1-C6)алкил может представлять иодметил, бромметил, хлорметил, фторметил, трифторметил, 2-хлорэтил, 2-фторэтил, 2,2,2-трифторэтил или пентафторэтил; гидрокси(C1-С6)алкил может представлять гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил, 1-гидроксипропил, 2-гидроксипропил, 3-гидроксипропил, 1-гидроксибутил, 4-гидроксибутил, 1-гидроксипентил, 5-гидроксипентил, 1-гидроксигексил или 6-гидроксигексил; (C1-C6)алкоксикарбонил (CO2R2) может представлять метоксикарбонил, этоксикарбонил, пропоксикарбонил, изопропоксикарбонил, бутоксикарбонил, пентоксикарбонил или гексилоксикарбонил; (C1-С6)алкилтио может представлять метилтио, этилтио, пропилтио, изопропилтио, бутилтио, изобутилтио, пентилтио или гексилтио; (С2-С6)алканоилокси может представлять ацетокси, пропаноилокси, бутаноилокси, изобутаноилокси, пентаноилокси или гексаноилокси; арил может представлять фенил, инденил или нафтил, а гетероарил может представлять фурил, имидазолил, триазолил, триазинил, оксазоил, изоксазоил, тиазолил, изотиазоил, пираксолил, пирролил, пиразинил, тетразолил, пуридил (или его N-оксид), тиентил, пиримидинил (или его N-оксид), индолил, изохинолил (или его N-оксид) либо хинолил (или его N-оксид).
Конкретным значением R является амино, монометиламино или циклопропиламино.
Конкретным значением R1 является карбокси- или (С1-С4)алкоксикарбонилциклогексил(C1-C4)алкил.
Конкретным значением R2 является Н или (C1-C4)алкил, т.е. метил или этил.
Конкретным значением R3 является Н, метил или фенил.
Конкретным значением R4 является Н, метил или фенил.
Конкретным значением Z является -СН2- или -СН2-СН2-.
Конкретным значением X является CO2R2, (C2-C5)алканоилметил или амидо.
Конкретным значением n является 1.
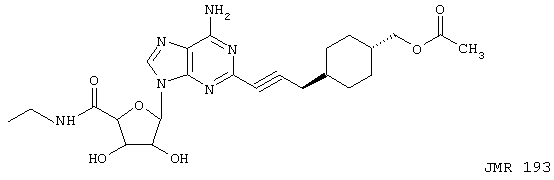
Предпочтительными соединениями формулы (I) являются такие соединения, в которых каждый из R представляет Н, X представляет этиламинокарбонил, а R1 представляет 4-карбоксициклогексилметил (DWH-146a), R1 представляет 4-метоксикарбонилциклогексилметил (DWH-146e) или R1 представляет 4-ацетоксиметилциклогексилметил (JMR-193). Они представлены ниже (DWH-146 (кислота) и метилэфир (e)), а также JMR-193.
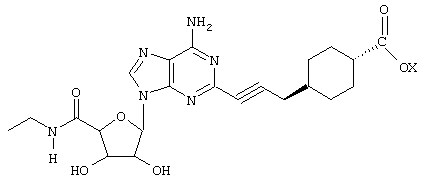
DWH-146 (кислота, X=Н; эфир, X=Me)
Синтез метил 4-[3-(6-амино-9-(5-[(этиламино)карбонил]-3,4-дигидрокситетрагидро-Z-фуранил-9Н-2-пуринил)-2-пропинил]-1-циклогексанкарбоксилата (DWH-146e) осуществляют перекрестным связыванием иодаденозинпроизводного (N-этил-1'-деокси-1'-(амино-2-иод-9Н-пурин-9-ил)-β-D-рибофурануорамид) с метил 4-(2-пропинил)-1-циклогексанкарбоксилатом, применяя катализатор Pd11. Синтез производного иодаденозина осуществляют из гуанозина. Гуанозин вначале обрабатывают уксусным ангидридом, который ацеталирует сахарные гидроксилы, а затем хлорируют положение 6 хлоридом тетраметиламмония и оксихлоридом фосфора. Иодирование положения 2 осуществляют в результате модифицированной реакции Сандмейера с последующим замещением 6-Cl и сахарных ацетатов аммиаком. Гидроксилы 2' и 3' защищают как ацетонид, а гидроксил 5' иодируют до кислоты с перманганатом калия. Снятие защиты с ацетонидов 2' и 3', этерификация Фишера 5' кислоты этанолом и превращение полученного сложного этилового эфира в этиламид этиламином дает N-этил-1'-деокси-1'-(амино-2-иод-9Н-пурин-9-ил)-β-D-рибофурануорамид.
Ацетилен (метил 4-(2-пропинил)-1-циклогексанкарбоксилат) синтезируют, исходя из транс-1,4-циклогександиметанола. Вначале транс-диол монотозилируют с последующим замещением тозилата анионом ацетилена. Гидроксил получаемых разновидностей гидроксилацетилена окисляют до кислоты реактивом Джонса с последующим метилированием (триметилсилил)диазометаном, получая 4-(3-пропинил)-1-циклогексанкарбоксилат.
Реакцию перекрестного связывания осуществляют в следующих, описанных выше условиях. К раствору N,N-диметилформамида (0,5 мл), ацетонитрила (1 мл), триэтиламина (0,25 мл) и N-этил-1'-деокси-1'-(амино-2-иод-9Н-пурин-9-ил)-β-D-рибофурануроамида (25 мг, 0,06 моль) добавляют бис(трифенилфосфин)палладий-дихлорид (1 мг, 2 мол.%) и иодид меди (I) (0,06 мг, 5 мол.%).
К полученной смеси добавляют метил 4-(2-пропинил)-1-циклогексанкарбоксилат (54 мг, 0,3 ммоль) и реакционную смесь перемешивают в атмосфере N2 в течение 16 часов. Растворитель удаляют в вакууме, а полученный остаток подвергают флэш-хроматографии в 20% метаноле в хлороформе (Rf=0,45), получая 19 мг (не совсем белое твердое вещество, т.пл. 125°С (разложение)) метил 4-[3-(6-амино-9-(5-[(этиламино)карбонил]-3,4-дигидрокситетрагидро-Z-фуранил)-9Н-2-пуринил]-2-пропинил-1-циклогексанкарбоксилата (DWH-146e).
DWH-146е и JMR193 намного более эффективны как ингибиторы в системах воспалительных моделей, чем сравнительное соединение, CGS21680 (2-[п-(карбоксиэтил)фенилэтиламино]-5'-N-этилкарбоксамидоаденозин). Например, DWH-146e приблизительно в 80 раз сильнее в отношении A2A-рецепторов и в 40 раз селективнее относительно А2A по сравнению с А3-рецепторами, чем CGS21680.
Примерами фармацевтически приемлемых солей являются органические аддитивные соли кислот, образуемые с кислотами, образующими физиологически приемлемый анион, например тозилат, метансульфонат, малат, ацетат, цитрат, малонат, тартрат, сукцинат, бензоат, аскорбат, α-кетоглутарат и α-глицерофосфат. Также могут быть образованы подходящие неорганические соли, включающие гидрохлориды, сульфаты, нитраты, бикарбонаты и карбонаты.
Фармацевтически приемлемые соли могут быть получены с использованием стандартных методов, хорошо известных в данной области, например взаимодействием достаточно основного соединения, такого как амин, с подходящей кислотой с получением физиологически приемлемого аниона. Могут быть также получены соли карбоновых кислот щелочного металла (например, натрий, калий или литий) или щелочноземельного металла (например, кальций).
Соединения формулы I могут быть приготовлены в виде фармацевтических композиций и введены хозяину-млекопитающему, такому как человек, в различных формах, учитывающих выбранный способ введения, т.е. пероральный или парентеральный, внутривенный, внутримышечный, местный или подкожный.
Таким образом, данные соединения могут быть введены системно, например, пероральным способом, в сочетании с фармацевтически приемлемым наполнителем, таким как инертный разбавитель или ассимилируемый съедобный носитель. Они могут быть заключены в капсулы с твердыми или мягкими желатиновыми оболочками, могут быть спрессованы в таблетки либо непосредственно подмешаны в еду пациента. При пероральном терапевтическом введении активное соединение может быть смешано с одним или несколькими формообразующими веществами и использовано в виде таблеток для проглатывания, защечных таблеток, пастилок, капсул, эликсиров, суспензий, сиропов, облаток и т.п. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Конечно, процентный состав композиций и препаратов может варьироваться и обычно составляет приблизительно от 2 до 60% от массы определенной дозированной лекарственной формы. Количество активного соединения в данных композициях для терапевтического применения таково, что обеспечивает эффективный уровень дозирования.
Таблетки, пастилки, пилюли, капсулы и т.п. также могут содержать следующие вещества: связующие, такие как трагакант, акация, кукурузный крахмал или желатин; эксципиенты, такие как дикальций фосфат; дезинтегрирующий агент, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота и т.п.; смазывающий агент, такой как стеарат магния, и подслащивающий агент, такой как сахароза, фруктоза, лактоза или аспартам, либо ароматизирующее вещество, такое как перечная мята, масло грушанки или вишневая отдушка. Если дозированная лекарственная форма имеет вид капсулы, она может содержать, помимо материалов вышеуказанного типа, жидкий носитель, такой как растительное масло или полиэтиленгликоль. Различные другие материалы могут присутствовать в виде оболочек или каким-либо другим способом модифицировать физический вид твердой дозированной лекарственной формы. Например, таблетки, пилюли или капсулы могут быть покрыты желатином, воском, шеллаком, сахаром и т.п. Сироп или эликсир может содержать активное соединение, сахарозу или фруктозу в качестве подслащивающего агента, метил и пропилпарабены в качестве консервантов, краситель и отдушку, такую как вишневое или апельсиновое ароматизирующее вещество. Безусловно, любой материал, применяемый при получении любой дозированной лекарственной формы, должен быть фармацевтически приемлемым и по существу нетоксичным в применяемых количествах. Кроме того, активное соединение может быть заключено в препараты и устройства с пролонгированным высвобождением.
Активное соединение также может быть введено внутривенно или внутрибрюшинно путем вливаний или инъекций. Растворы активного соединения или его солей могут быть получены в воде, необязательно смешанной с нетоксичным поверхностно-активным веществом. Дисперсии также могут быть получены в глицерине, жидких полиэтиленгликолях, триацетине, их смесях, а также в маслах. В обычных условиях хранения и применения данные препараты содержат консервант, предупреждающий рост микроорганизмов.
Фармацевтические лекарственные формы, предназначенные для инъекций или инфузий (вливаний), могут включать стерильные водные растворы, дисперсии или стерильные порошки, включающие активный ингредиент, предназначенные для незапланированного приготовления стерильных растворов или дисперсий для инъекций или вливаний, необязательно инкапсулированные в липосомы. Во всех случаях конечная лекарственная форма должна быть стерильной, жидкой и устойчивой в условиях изготовления и хранения. Жидкий носитель или наполнитель может представлять собой разбавитель или жидкую дисперсионную среду, включающую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль, жидкие полиэтиленгликоли и т.п.), растительные масла, нетоксичные глицериновые сложные эфиры, а также их подходящие смеси. Нужная текучесть может быть обеспечена, например, образованием липосом, использованием необходимого размера частиц при использовании дисперсий либо применением поверхностно-активных веществ. Действие микроорганизмов может быть предотвращено различными антибактериальными и противогрибковыми агентами, например парабенами, хлорбутанолом, фенолом, сорбиновой кислотой, тимеросалом и т.п. Во многих случаях предпочтительно включение изотонических агентов, например сахаров, буферов или хлорида натрия. Пролонгированная абсорбция композиций для инъекций может быть обеспечена применением в композициях агентов, замедляющих абсорбцию, например моностеарата алюминия и желатина.
Стерильные растворы для инъекций получают, вводя активное соединение в нужном количестве в соответствующий растворитель вместе с различными вышеуказанными ингредиентами с последующей, в соответствии с требованиями, стерилизацией фильтрованием. При использовании стерильных порошков для получения стерильных растворов для инъекций предпочтительными способами являются вакуумная сушка и сушка вымораживанием, обеспечивающие получение порошка активного ингредиента плюс любой дополнительный желаемый ингредиент, присутствующий в растворах, ранее подвергнутых стерилизации фильтрованием.
При местном введении данные соединения могут быть нанесены в чистом виде, т.е. когда они находятся в виде жидкостей. Однако обычно желательно наносить их на кожу в виде композиций или составов в сочетании с дерматологически приемлемым носителем, который может иметь твердую или жидкую форму.
Применимые твердые носители включают тонкоизмельченные твердые вещества, такие как тальк, глина, микрокристаллическая целлюлоза, двуокись кремния, окись алюминия и т.п. Применимые жидкие носители включают воду, спирты или гликоли либо смеси вода-спирт/гликоль, в которых данные соединения могут быть эффективно растворены или диспергированы, необязательно с помощью нетоксичных поверхностно-активных веществ. Чтобы оптимизировать свойства для определенного назначения, могут быть добавлены адъюванты, такие как ароматизаторы и дополнительные антимикробные агенты. Полученные жидкие композиции могут быть нанесены при помощи абсорбирующих подушечек, применяемых для пропитывания повязок и других перевязочных материалов, либо распылены на пораженный участок при помощи распылителей нагнетательного или аэрозольного типа.
Загустители, такие как синтетические полимеры, жирные кислоты, соли и сложные эфиры жирных кислот, жирные спирты, модифицированные целлюлозы или модифицированные минеральные материалы также могут быть использованы с жидкими носителями для получения наносимых паст, гелей, мазей, мыл и т.п., накладываемых непосредственно на кожу пациента.
Примеры дерматологических композиций, которые могут быть использованы для доставки соединений формулы I в кожу, описаны Jacquet et al. (патент США № 4608392), Geria (патент США № 4992478), Smith et al. (патент США № 4559157) и Wortzman (патент США № 4820508).
Эффективные дозы соединений формулы I могут быть определены в результате сравнения их эффективности in vitro и in vivo на животных моделях. Способы экстраполяции эффективных доз у мышей и других животных на человека известны в данной области; см., например, патент США № 4938949. Эффективные дозы ингибиторов PDE, тип IУ, известны в данной области. Например, см. патент США №5877180, кол.12.
В целом, концентрация соединения (соединений) формулы (I) в жидкой композиции, такой как лосьон, составляет приблизительно 0,1-25% мас., предпочтительно приблизительно 0,5-10% мас. Концентрация в полутвердой или твердой композиции, такой как гель или порошок, составляет приблизительно 0,1-5% мас., предпочтительно приблизительно 0,5-2,5% мас.
Количество соединения либо его активной соли или производного, необходимого для лечения, варьируется в зависимости не только от конкретной выбранной соли, но также и от способа введения, природы подвергаемого лечению состояния, возраста и состояния пациента, и в конечном счете определяется лечащим врачом или клиницистом.
Однако в целом подходящая доза находится в интервале приблизительно от 0,5 до 100 мкг/кг, например приблизительно от 10 до 75 мкг/кг массы тела в сутки, возможно от 3 до приблизительно 50 мкг на килограмм массы тела реципиента в сутки, предпочтительно в интервале от 6 до 90 мкг/кг/день, наиболее предпочтительно в интервале от 15 до 60 мкг/кг/день.
Данное соединение предпочтительно вводить в виде дозированной лекарственной формы, например, содержащей от 5 до 1000 мкг, целесообразно от 10 до 750 мкг, наиболее предпочтительно от 50 до 500 мкг активного ингредиента на дозированную лекарственную форму.
В идеале, активный ингредиент должен быть введен таким образом, чтобы достигнуть максимальной концентрации в плазме активного соединения, составляющей приблизительно от 0,1 до 10 нМ, предпочтительно приблизительно от 0,2 до 10 нМ, наиболее предпочтительно приблизительно от 0,5 до 5 нМ. Это может быть достигнуто, к примеру, в результате внутривенной инъекции 0,05-5% раствора активного ингредиента, необязательно в солевом растворе, либо перорального введения в виде болюса, содержащего приблизительно 1-100 мкг активного ингредиента. Желаемый уровень в крови можно поддерживать путем непрерывных вливаний, обеспечивающих приблизительно 0,01-5,0 мкг/кг/час, либо периодических вливаний, содержащих приблизительно 0,4-15 мкг/кг активного ингредиента (ингредиентов).
Желаемая доза может быть введена в виде разовой дозы или в виде разделенных доз, вводимых через соответствующие периоды времени, например, в виде двух, трех, четырех и более разделенных доз в сутки. В свою очередь разделенная доза может быть также разделена, например, на ряд дискретных введений (доз), таких как многократные ингаляции через инсуффлятор или закапывание нескольких капель в глаза. Например, желательно вводить данные композиции внутривенно в течение длительного периода времени после поражения, вызвавшего воспаление.
Способность соединения в соответствии с настоящим изобретением действовать в качестве агониста (или антагониста) А2А-рецептора аденозина может быть определена с использованием фармакологических моделей, хорошо известных в данной области, либо с применением описанных ниже тестов.
Далее настоящее изобретение описано со ссылкой на следующие подробные примеры, которые приведены для иллюстрации изобретения, а не для его ограничения. В спектрах ЯМР далее Гц - герц, с - синглет, д - дублет, т - триплет, м - мультиплет, уш - уширенный.
Пример 1
транс-(1-[4-[Гидроксиметил)циклогексил]метил)-4-метил-бензолсульфонат (5.2)
Гидрид натрия (1,69 г, 70 ммоль) добавляют к раствору 10 г (70 ммоль) [4-(гидроксиметил)циклогексил]метан-1-ола (5.1) в 700 мл тетрагидрофурана, перемешивают в течение 1 часа, затем добавляют п-толуолсульфонилхлорид (13,3 г, 70 ммоль) и реакционную смесь подвергают кипячению с обратным холодильником в течение 5 часов. Затем реакционную смесь охлаждают до 0°С и медленно гасят водой до исчезновения реакционноспособного гидрида. По окончании гашения гидрида реакционную смесь разбавляют простым эфиром (700 мл) и экстрагируют 2 раза 10% водным карбонатом калия (700 мл). Органическую часть сушат, применяя сульфат натрия, и растворитель удаляют при пониженном давлении. Продукт очищают хроматографией на колонке из силикагеля, элюируя смесью ацетон-дихлорметан (5:95) с получением соединения 5.2 (35%).
1Н ЯМР (300 МГц, CDCl3) δ 7.75 (д, J=8.3 Гц, 2Н), 7.32 (д, J=8.1 Гц, 2Н), 3.79 (д, J=6.35 Гц, 2Н), 3.39 (д, J=6.35 Гц, 2Н), 2.42 (с, 3Н), 1.75 (м, 4Н), 1.59 (м, 1Н), 1.37 (м, 1Н), 0.9 (м, 4Н). 13С ЯМР (300 МГц, CDCl3) δ 145.3, 133.4, 130.3, 130.3, 128.3, 128.3, 75.8, 68.5, 40.6, 37.8, 28.9, 28.9, 28.9, 28.9, 22.1.
Пример 2
(4-Проп-2-инилциклогексил)метан-1-ол (5.3)
Комплекс литийацетилид-этилендиамин (90%) (6,4 г, 70 ммоль) медленно добавляют к раствору 5.2 (3 г, 10 ммоль) в 40 мл диметилсульфоксида. Реакционной смеси дают возможность перемешиваться в течение 5 дней, а затем медленно гасят при 0°С водой. Смесь разбавляют простым эфиром (300 мл) и экстрагируют 3 раза насыщенным водным хлоридом аммония (200 мл). Органические вещества сушат сульфатом натрия. Растворитель удаляют при пониженном давлении и продукт очищают хроматографией на колонке из силикагеля, элюируя смесью этилацетат-гексаны (20:80) с получением соединения 5.3 (85%).
1Н ЯМР (300 МГц, CDCl3) δ 3.41 (д, J=6.5 Гц, 2Н), 2.07 (дд, J=2.5, 6.5 Гц, 2Н), 1.96-1.75 (м, 5Н), 1.41 (м, 2Н), 0,95 (м, 4). 13C ЯМР (300 МГц, CDCl3) δ 83.8, 69.6, 68.9, 40.7, 37.7, 32.3, 32.3, 29.6, 29.6, 26.5.
Пример 3
4-Проп-2-инилциклогексанкарбоновая кислота (5.4)
Раствор триоксида хрома (1,1 г, 11 ммоль) в 1,5 М серной кислоте (40 мл, 27 ммоль) поддерживают при температуре 0°С в течение более 2 часов, на протяжении которых добавляют соединение 5.3 (0,46 г, 3 ммоль) в 80 мл ацетона. Затем реакционную смесь перемешивают еще в течение 2 часов при комнатной температуре. Реакционную смесь разбавляют простым эфиром (200 мл) и 2 раза экстрагируют водой. Органические вещества сушат сульфатом натрия. Растворитель удаляют при пониженном давлении и продукт очищают хроматографией на колонке из силикагеля, элюируя смесью ацетон-дихлорметан (70:30) с получением соединения 5.4 (75%).
1H ЯМР (300 МГц, CDCl3) δ 2.24 (дт, J=3.66, 12.1 Гц, 1Н), 2.10 (дд, J=2.7, 6.5 Гц, 2Н), 2.04-1.89 (м, 5Н), 1.76 (д, J=2.3 Гц, 1Н), 1.43 (дкв, J=3.28, 13.1 Гц, 2Н), 1.03 (дкв, J=3.28, 13.1 Гц, 2Н). 13С ЯМР (300 МГц, CDCl3) δ 183.2, 83.3, 69.9, 43.4, 36.7, 31.8, 28.9, 26.3.
Пример 4
Метил 4-проп-2-инилциклогексанкарбоксилат (5.5)
Раствор (триметилсилил)диазометана (2,0 М) в гексанах (1 мл, 2 ммоль) добавляют к раствору соединения 5.4 (0,34 г, 2 ммоль) в 15 мл смеси метанол:дихлорметан (3:7). Растворители удаляют при пониженном давлении, получая 100% конверсию исходного материала в продукт.
1H ЯМР (300 МГц, CDCl3) δ 2.24 (дт, J=3.66, 12.1 Гц, 1Н), 2.10 (dд, J=2.7, 6.5 Гц, 2Н), 2.06 (дд, J=1.54, 6.54 Гц, 1Н), 2.00-1.89 (м, 3Н), 1.76 (д, J=2.3 Гц, 1Н). 1.43 (дкв, J=3.28, 13.1 Гц, 2Н), 1.03 (дкв, J=3.28, 13.1 Гц, 2Н). 13С ЯМР (300 МГц, CDCl3) δ 176.8, 83.3, 69.8, 51.9, 43.4, 36.7, 31.9, 29.2, 26.3.
Пример 5
[(2R,3R,4R,5R)-3,4-Диацетилокси-5-(2-амино-6-оксогидропурин-9-ил)оксолан-2-ил]метилацетат (6.2)
Суспензию 113 г (0,4 моль) сухого гуанозина (6.1), уксусного ангидрида (240 мл, 2,5 моль), сухого пиридина (120 мл) и сухого ДМФ (320 мл) нагревают в течение 3,75 час при 75°С, не позволяя температуре превысить 80°С. Прозрачный раствор затем переносят в 3 л колбу Эрленмейера, которую наполняют 2-пропанолом. После охлаждения раствора до комнатной температуры инициируют кристаллизацию и дают ей возможность протекать при 4°С в течение ночи. Белый твердый осадок фильтруют, промывают 2-пропанолом и перекристаллизовывают из него, получая соединение 6.2 (96%).
1Н ЯМР (300 МГц, CDCl3) δ 8.20 (с, 1Н, Н-8), 6.17 (д, J=5.41 Гц, 1Н, Н-1') 5.75 (т, J=5.39 Гц, 1H, H-2'), 5.56 (т, J=5.0, Н-3'), 4.41 (м, 3Н, Н-4', 5'), 2.14 (с, 3Н, Ас), 2.11 (с, 3Н, Ас), 2.10 (с, 3Н, Ас). 13С ЯМР (300 МГц, CD3OD) δ 171.0, 170.3, 170.2, 157.7, 154.8, 152.4, 136.7, 117.7, 85.5, 80.4, 73.0, 71.3, 64.0, 31.3, 21.2, 21.0.
Пример 6
[(2R,3R,4R,5R)-3,4-Диацетилокси-5-(2-амино-6-хлорпурин-9-ил)оксолан-2-ил]метилацетат (6.3)
В 1000 мл колбу помещают 80 г (0,195 моль) [(2R,3R,4R,5R)-3,4-диацетилокси-5-(2-амино-6-оксогидропурин-9-ил)оксолан-2-ил]метилацетата (6,2), тетраметиламмонийхлорид (44 г, 0,4 моль), безводный ацетонитрил (400 мл) и N,N-диметиланилин (25 мл). Колбу помещают на ледяную солевую баню и охлаждают до 2°С. К данному раствору по каплям добавляют POCl3 (107 мл, 1,15 моль) со скоростью, поддерживающей температуру ниже 5°С (45 минут). Затем колбу удаляют с ледяной бани, оборудуют конденсатором, помещают на масляную баню и дают возможность смеси кипеть с обратным холодильником в течение 10 минут, при этом цвет раствора меняется на красно-коричневый. Затем растворитель удаляют при пониженном давлении, получая маслянистый остаток, который переносят в мензурку, содержащую 1000 г льда и 400 мл CHCl3, и дают возможность смеси перемешиваться в течение 1,5 часов, до разложения оставшегося POCl3. Затем органическую фазу удаляют, водную фазу экстрагируют 3×50 мл CHCl3 и объединяют с органической фазой. Объединенные органические вещества затем подвергают обратной экстракции 50 мл воды с последующим перемешиванием с 200 мл насыщенного NaHCO3. После этого органическую фазу экстрагируют NaHCO3, до тех пор пока водный экстракт не станет нейтральным (2Х). Органическую фазу наконец экстрагируют насыщенным раствором соли, а затем сушат над MgSO4 в течение 16 часов. К раствору добавляют 800 мл 2-пропанола, после чего раствор концентрируют при пониженном давлении. К маслянистому твердому веществу добавляют 200 мл 2-пропанола и раствор помещают в холодильник на ночь. Кристаллический продукт фильтруют, промывают и дают возможность сохнуть в течение ночи, получая соединение 6.3 (77%).
1H ЯМР (300 МГц, CD3OD) δ 8.31 (с, 1Н, Н-8), 7.00 (с, 2Н, NH2) 6.06 (д, J=5.8 Гц, 1Н, Н-1'), 5.83 (т, J=6.16 Гц, 1Н, Н-2'), 5.67 (м, 1Н, Н-3'), 4.29 (м, 3Н, Н-4', 5'), 2.07 (с, 3Н, Ас), 1.99 (с, 3Н, Ас), 1.98 (с, 3Н, Ас). 13С ЯМР (300 МГц, CD3OD) δ 171.0, 170.4, 170.2, 160.8, 154.6, 150.8, 142.2, 124.5, 85.8, 80,6. 72.8, 71.2, 63.9, 21.4, 21.3, 21.1.
Пример 7
[(2R,3R,4R,5R)-3,4-Диацетилокси-5-(6-хлор-2-иодпурин-9-ил)оксолан-2-ил]метилацетат (6.4)
Изоамилнитрит (5 мл, 37 ммоль) добавляют к раствору 5,12 г (12 ммоль) [(2R,3R,4R,5R)-3,4-диацетилокси-5-(2-амино-6-хлорпурин-9-ил)оксалан-2-ил]метилацетата (6.3), I2 (3,04 г, 12 ммоль), CH2I2 (10 мл, 124 ммоль) и Cul (2,4 г, 12,6 ммоль) в ТГФ (60 мл). Смесь нагревают при кипячении с обратным холодильником в течение 45 минут, а затем дают ей возможность охладиться до комнатной температуры. К данному раствору добавляют 100 мл насыщенного Na2S2O3, устраняющего красноватый цвет, вызванный присутствием иода. Водный раствор экстрагируют 3Х хлороформом, который сливают вместе, сушат над MgSO4 и концентрируют при пониженном давлении. Затем продукт очищают на колонке из силикагеля, применяя CHCl3-МеОН (98:2) с получением [(2R,3R,4R,5R)-3,4-диацетилокси-5-(6-хлор-2-иодпурин-9-ил)оксолан-2-ил]метилацетата (6.4) (80% кристаллизуется из EtOH).
1H ЯМР (300 МГц, CDCl3) δ 8.20 (с, 1Н Н-8), 6.17 (д, J=5.41 Гц, 1Н, Н-1'), 5.75 (т, J=5.39 Гц, 1Н, H-2'), 5.56 (т, J=5.40 Гц, 1Н, H-3'), 4.38 (м, 3Н, Н-4', 5'), 2.14 (с, 1Н, Ас), 2.11 (с, 1 Н, Ас), 2.10 (с, 1Н, Ас).
Пример 8
(4S,2R,3R,5R)-2-(6-Амино-2-иодпурин-9-ил)-5-(гидроксиметил)оксолан-3,4-диол (6.5)
В колбу, содержащую 6,0 г (11,1 ммоль) [(2R,3R,4R,5R)-3,4-диацетилокси-5-(6-хлор-2-иодпурин-9-ил)оксолан-2-ил]метилацетата (6.4), добавляют 100 мл жидкого NH3 при -78°С и раствору дают возможность перемешиваться в течение 6 часов. После этого раствору дают возможность нагреться до комнатной температуры в течение ночи с одновременным выпариванием NH3, получая коричневое масло. Продукт кристаллизуют из горячего изопропанола, получая соединение 6.5 (80%), т.пл. 143-145°С, r.f.=0,6 в 20% МеОН/CHCl3.
1H ЯМР (300 МГц, DMSO-d6) δ 8.24 (с, 1Н), 7.68(c, 2H), 5.75(д, J=6.16, 1H), 5.42 (д, J=5.40 Гц, 1H), 5.16 (д, J=4.62 Гц, 1Н), 4.99 (т, J=5.39 Гц, 1Н), 4.67 (д, J=4.81 Гц, 1Н), 4.06 (д, J=3.37 Гц, 1Н), 3.89 (м, 1H), 3.54 (м, 2Н).
Пример 9
[(1R,2R,4R,5R)-4-(6-Амино-2-иодпурин-9-ил)-7,7-диметил-3,6,8-триоксабицикло[3.3.0]окт-2-ил]метан-1-ол (6.6)
К раствору 2,0 г (5,08 ммоль) (4S,2R,3R,5R)-2-(6-амино-2-иодпурин-9-ил)-5-(гидроксиметил)оксолан-3,4-диола (6.6) в 100 мл ацетона добавляют 9,6 г п-толуолсульфоновой кислоты и 5 мл диметоксипропана. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, на протяжении которого добавляют 15 г NaHCO3 а затем перемешивают еще в течение 3 часов. Остаток фильтруют и промывают 2Х EtOAc. Затем фильтрат концентрируют при пониженном давлении. Остаток подвергают хроматографии на колонке с силикагелем с МеОН-CHCl3 (1:99), получая соединение 6.6 (72%) в виде твердого вещества, т.пл. 185-187°С.
1H ЯМР (300 МГц, DMSO-d6) δ 8.22 (с, 1H, Н-8), 7.69 (с, 2Н, NH2), 6.00 (д, J=2.70 Гц, 1H, Н-1'), 5.21 (м, 1H, Н-2'), 5.07 (ушс, 1H, ОН), 4.88 (м, 1H, Н-3'), 4.13 (м, 1H, Н-4'), 3.47 (м, 2Н, Н-5'), 1.49 и 1.28 (с, 3Н, С(СН3)2).
Пример 10
(2S,1R,4R,5R)-4-{6-Амино-2-иодпурин-9-ил)-7,7-диметил-3,6,8-триоксабицикло[3.3.0]октан-2-карбоновая кислота (6.7)
К перемешиваемому раствору 1,6 г (3,7 ммоль) [(1R,2R,4R,5R)-4-(6-амино-2-иодпурин-9-ил)-7,7-диметил-3,6,8-триоксабицикло[3.3.0]окси-2-ил]метан-1-ола (6.6) в 200 мл H2O добавляют 0,60 г КОН и, по каплям, раствор 1,70 г (10,8 ммоль) KMnO4 в 50 мл Н2O. Смесь помещают в темное место при комнатной температуре на 225 час. Затем реакционную смесь охлаждают до 5-10°С и обесцвечивают раствором 4 мл 30% H2O2 в 16 мл воды, поддерживая при этом температуру ниже 10°С при помощи ледяной-солевой бани. Смесь фильтруют через целит, а фильтрат концентрируют при пониженном давлении приблизительно до 10 мл, затем подкисляют до рН 4 2 н. HCl. Полученный осадок отфильтровывают и промывают простым эфиром, получая после сушки соединение 6.7 (70%) в виде белого твердого вещества, т.пл. 187-190°С. 1H ЯМР (300 МГц, DMSO-d6) δ 8.11 (с, 1Н, Н-8), 7.62 (с, 2Н, NH2), 7.46 (с, 1Н, СООН), 6.22 (с, 1Н, Н-1'), 5.42 (д, J=5.71 Гц, 1Н, Н-2'), 5.34 (д, J=6.16 Гц, 1Н, Н-3'), 4.63 (с, 1Н, Н-4'), 1.46 и 1.30 (с, 3Н, С(СН3)2).
Пример 11
(2S,3S,4R,5R)-5-(6-Амино-2-иодпурин-9-ил)-3,4-дигидроксиоксолан-2-карбоновая кислота (6.8)
Раствор 1,72 г (3,85 ммоль) (2S,1R,4R,5R)-4-(6-амино-2-иодпурин-9-ил)-7,7-диметил-3,6,8-триоксабицикло[3.3.0]октан-2-карбоновой кислоты (6.7) в 80 мл 50% НСООН перемешивают при 80°С в течение 1,5 часов. Реакционную смесь выпаривают при пониженном давлении, растворяют в H2O и раствор вновь выпаривают. Этот процесс повторяют до тех пор, пока остаток не утратит запах муравьиной кислоты. В результате перекристаллизации из воды получают 1,33 г (85%) соединения 6.8 в виде белого твердого вещества, т.пл. 221-223°С, разл.
1H ЯМР (300 МГц, DMSO-d6) δ 8.31 (с, 1Н, Н-8), 7.68 (с, 2Н, NH2), 5.90 (д, J=6.55 Гц, 1Н, Н-1'), 4.42 (м, 1Н, 4-2'), 4.35 (д, J=2.31 Гц, 1H, 11-4'), 4.22 (м, 1H, Н-3').
Пример 12
[2S,3S,4R,5R)-5-(5-Амино-2-иодпурин-9-ил)-3,4-дигидроксиоксолан-2-ил]-N-этилкарбоксамид (6.9)
К охлажденному (5°С) и перемешиваемому раствору 1,29 г (3,17 ммоль) (2S,3S,4R,5R)-5-(6-амино-2-иодпурин-9-ил)-3,4-дигидроксиоксолан-2-карбоновой кислоты (6.8) в 150 мл абсолютного этанола по каплям добавляют 1,15 мл охлажденного льдом SOCl2. Смесь перемешивают при комнатной температуре в течение ночи, а затем ее рН доводят до 8 насыщенным водным NaHCO3. Смесь фильтруют, затем фильтрат концентрируют при пониженном давлении, получая белое твердое вещество, которое сушат, а затем повторно растворяют в 20 мл сухого этиламина при -20°С в течение 3 часов, а затем при комнатной температуре в течение ночи. Реакционную смесь разбавляют абсолютным этанолом, осажденный продукт отфильтровывают и промывают сухим простым эфиром, получая 530 мг (72%) 6.9 в виде чистого твердого вещества, т.пл. 232-234°С.
1H ЯМР (300 МГц, DMSO-d6) δ 8.34 (с, 1Н, Н-8), 8.12 (т, 1Н, NH), 7.73 (с, 2Н, NH2), 5.85, (д, J=6.93 Гц, 1Н, Н-1'), 4.54 (м, 1Н, Н-2'), 4.25 (д, J=1.92 Гц, 1Н, H-4'), 4.13 (м, 1Н, Н-3'), 3.28 (м, 2Н, СН2СН3), 1.00 (т, J=7.2 Гц, 3Н, СН2СН3).
Пример 13
Метил 4-(3-{9-[(4S,5S,2R,3R)-5-(N-этилкарбамоил)-3,4-дигидроксиоксолан-2-ил]-6-аминопурин-2-ил)}проп-2-инил)циклогексанкарбоксилат (DWH-146e)
К дегазированному раствору 25 мг (0,063 ммоль) [(2S,3S,4R,5R)-5-(6-амино-2-иодопурин-9-ил)-3,4-дигидроксиоксолан-2-ил]-N-этилкарбоксамида (6.9), 16,9 мг (0,094 ммоль) (5.5) и 0,75 Cul в 5 мл (каждого) триэтиламина (TEA) и ацетонитрила добавляют 15 мг Pd(PPh3)4. Раствор перемешивают в течение 24 часов при 70°С, после чего фильтруют через целит и подвергают хроматографии на силикагеле с МеОН-CHCl3 (5:95), получая соединение DWH-146e (24%).
Пример 14
(4-Проп-2-инилциклогексил)метилацетат (5.6)
Уксусный ангидрид (0,92 мл, 8,25 ммоль) и пиридин (0,2 мл, 2,5 ммоль) добавляют к раствору соединения 5.3 (250 мг, 1,65 ммоль) в 25 мл простого эфира. Реакционной смеси дают возможность перемешиваться при комнатной температуре в течение 24 час. К реакционной смеси добавляют воду, а органическую фазу далее экстрагируют 10% NaHCO3. Органический слой сушат MgSO4 и выпаривают. Остаток подвергают хроматографии на силикагеле с EtOAc-гексанами (5:95), получая соединение 5.6 (47%).
Пример 15
[4-(3-{9-(4S,5S,2R,3R)-5-(N-Этилкарбамоил)-3,4-дигидроксиоксолан-2-ил]-6-аминопурин-2-ил}проп-2-инил)циклогексил]метилацетат (JMR 193)
К дегазированному раствору 125 мг (0,29 ммоль) [(2S,3S,4R,5R)-5-(6-амино-2-иодпурин-9-ил)-3,4-дигидроксиоксолан-2-ил]-N-этилкарбоксамида (6.9), 150 мг (0,77 ммоль) соединения (5.6) и 1,0 мг Cul в 1,3 мл TEA и 4 мл ТМФ добавляют 25 мг Pd(PPh3)4. Раствор перемешивают в течение 72 часов при 60°С, после чего его фильтруют через целит и подвергают хроматографии на силикагеле с МеОН-CHCl3 (5:95), получая соединение JMR193 (10%).
Пример 16
[(2S,3S,4R,5R)-5-(6-Амино-2-{3-[4-(гидроксиметил)циклогексил]проп-2-инил}пурин-9-ил)-3,4-дигидроксиоксолан-2-ил]-N-этилкарбоксамид
А. (4-Проп-2-инилциклогексил)метан-1-ол
Комплекс литийацетилид-этилендиамин (90%) (6,4 г, 70 ммоль) медленно добавляют к раствору транс-[4-(гидроксиметил) циклогексил]метил 4-метилбензолсульфоната (3 г, 10 ммоль) в 40 мл диметилсульфоксида. Реакционной смеси дают возможность перемешиваться в течение 5 дней, а затем ее медленно гасят при 0°С водой. Данную смесь разбавляют простым эфиром (300 мл) и промывают 3 раза насыщенным водным хлоридом аммония (200 мл). Органические вещества сушат сульфатом натрия. Растворитель удаляют при пониженном давлении. Продукт очищают хроматографией на колонке из силикагеля, элюируя смесью этилацетат-гексаны (20:80) и получая продукт (85%).
1H ЯМР (300 МГц, CDCl3) d 3.41 [д, J=6.5 Гц, 2Н), 2.07 (дд, J=2.5, 6.5 Гц, 2Н), 1.96-1.75 (м, 5Н), 1.41 (м, 2Н), 0,95 (м, 4). 13С ЯМР (300 МГц, CDCl3) d 83.8, 69.6, 68.9, 40.7, 37.7, 32.3, 32.3, 29.6, 29.6, 26.5.
B. [(2S,3S,4R,5R)-5-(6-Амино-2-{3-[4-(гидроксиметил)циклогексил]проп-1-инил}пурин-9-ил)-3,4-дигидроксиоксолан-2-ил]-N-этилкарбоксамид (JMR2037)
Pd(PPh3)4, 10 мг, добавляют к дегазированному раствору 28 мг (0,065 ммоль) [(2S,3S,4R,5R)-5-(6-амино-2-иодопурин-9-ил)-3,4-дигидроксиоксолан-2-ил]-N-этилкарбоксамида, 30 мг (0,20 ммоль ) (4-проп-2-инилциклогексил)метан-1-ола, 1,0 мг Cul в 1 мл триэтиламина (TEA), 1 мл ДМФ и 1 мл ацетонитрила. Раствор перемешивают в течение 60 часов при комнатной температуре, после чего его фильтруют через целит и подвергают хроматографии на силикагеле с МеОН-CHCl3 (7:93), получая 5 мг (17%) соединения JMR2037. Указанное в заголовке соединение было подвергнуто описываемым здесь тестам на связывание, в результате которых было установлено, что оно связывается с рекомбинантными А2A-рецепторами человека, при этом Ki составляет 694±69 нМ.
Общий способ 4: получение 2-AAs (2-алкиниладенозинов)
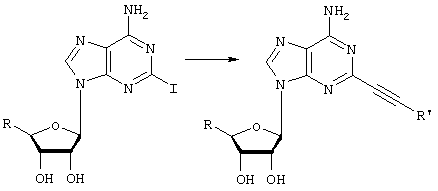
В высушенную на пламени 25 мл круглодонную колбу в атмосфере азота загружают 2-иодNECA (40 мг) и растворяют в 2 мл ДМФА. Затем добавляют соответствующий алкин (примерно 0,1 мл), после чего следует добавление 4 мл ацетонитрила и 0,1 мл ТЭА. Все три растворителя дегазируют азотом в течение по меньшей мере 24 часов. К данному раствору добавляют 5 мол.% Pd(PPh3)4 и 6 мол.% иодида меди. Желтоватый раствор перемешивают при комнатной температуре в течение 24 часов или до завершения реакции по данным ВЭЖХ. Если реакция за это время не завершится, добавляют дополнительный катализатор, Cul и ТЭА. После завершения реакции в вакууме удаляют растворители и красно-черный остаток переносят в небольшое количество ДМФА. Данный раствор добавляют на препаративную силикагелевую пластину для ТСХ (Analtech 1000 микрон, 20 см ×20 см) и элюируют вначале 120 мл смеси 40% гексан/СН2Cl2, а затем 40 мл МеОН. Собирают УФ-активную полосу (обычно желтая окраска) в средней части пластины, медленно промывают 4×25 мл 20% MeOH/CH2Cl2 и концентрируют. Затем данный продукт очищают ВЭЖХ с обращенной фазой.
Пример 17
4-{3-[6-Амино-9-(5-этилкарбамоил-3,4-дигидрокситетрагидрофуран-2-ил)-9Н-пурин-2-ил]проп-2-инил}циклогексанкарбоновая кислота (ATL146a)
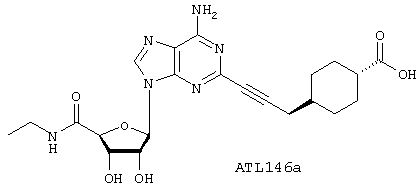
Взаимодействие ATL146e с пятью эквивалентами LiOH в смеси ТГФ/вода в течение 6 часов дает ATL146a (7 мг, 72%) в виде белого твердого вещества, которое кристаллизуют из смеси МеОН/вода (0,1% ТФУ) после очистки ВЭЖХ с обращенной фазой.
1H ЯМР (ДМСО-d6) δ 8,70 (с, 1Н), 8,41 (с, 1Н), 7,62 (с, 2Н), 5,89 (д, J=7,25 Гц, 1Н), 4,53 (м, 1Н), 4,27 (с, 1Н), 4,08 (д, J=3,6 Гц, 1Н), 2,29 (д, J=6,4 Гц, 2Н), 2,15-1,99 (м, 1Н), 1,92-1,76 (м, 4Н), 1,52-1,38 (м, 1H), 1,38-1,19 (м, 2Н), 1,02 (т, J=6,3 Гц, 3Н).
13С ЯМР (ДМСО-d6) 176,7, 169,2, 155,6, 148,9, 145,2, 141,6, 119,0, 87,7, 85,0, 84,6, 81,6, 73,1, 71,9, 43,2, 35,9, 33,3, 31,2, 28,3, 25,6, 15,0. МС высок. разреш. (бомбард. быстр, атомами) m/z 474,2196 [(М+Н)+ вычислено для C22H29N6O6 474,2182].
Пример 18
Этиламид 5-{6-амино-2-[3-(4-гидроксиметилциклогексил)проп-1-инилl]пурин-9-ил}-3,4-дигидрокситетрагидрофуран-2-карбоновой кислоты (JR2037)
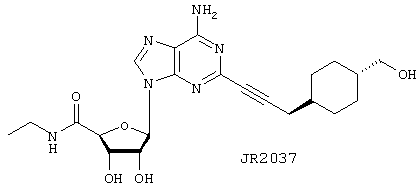
Взаимодействие (4-проп-2-инилциклогексил)метан-1-ола (5.3) (30 мг, 0,2 ммоль) с 2-иoдNECA (28 мг, 0,07 ммоль) при общих условиях, описанных выше, дает JR2037 (7 мг, 24%) в виде белого твердого вещества.
1H ЯМР (CD3OD) δ 8,22 (с, 1Н), 5,92 (д, J=7,7 Гц, 1H), 4,70-4,66 (дд, J=7,7 Гц, 4,8 Гц, 1Н), 4,40 (д, J=1,2 Гц, 1H), 4,25-4,23 (дд, J=4,8 Гц, 1,2 Гц, 1H), 3,51-3,37 (м, 2Н), 3,31 (д, J=6 Гц, 2Н), 2,30 (д, J=6,8 Гц, 2Н), 1,94-1,89 (м, 2Н), 1,83-1,78 (м, 2Н), 1,64-1,42 (м, 2Н), 1,12 (т, J=7,3 Гц, 3Н), 1,09-0,91 (м, 4Н).
13С ЯМР (CD3OD) δ 170,3, 155,4, 148,5, 146,0, 141,6, 118,8, 88,7, 85,5, 84,6, 80,6, 73,1, 71,3, 66,8, 39,6, 36,9, 33,3, 31,5, 28,6, 25,6, 13,5. МС высок. разреш. (бомбард. быстр, атомами) m/z 459,2373 [(М+H)+ вычисл. для С22Н31N6O5 459,2356].
Пример 19
Метиловый эфир 4-{3-[6-амино-9-(3,4-дигидрокси-5-гидроксиметилтетрагидрофуран-2-ил)-9Н-пурин-2-ил]проп-2-инил}циклогексанкарбоновой кислоты (JR2145)
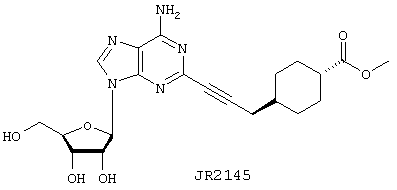
JR2145 синтезируют в соответствии с общим протоколом 4, используя алкин (5.5) и 10 мг (0,025 ммоль) 2-иодаденозина. Продукт выделяют в виде грязно-белого твердого вещества: выход 5,5 мг (48%).
1Н ЯМР (CD3OD) δ 8,35 (с, 1Н), 5,87 (д, J=6,5 Гц, 1H), 4,70-4,66 (дд, J=6,4 Гц, 5,6 Гц, 1H), 4,26 (дд, J=5,0 Гц, 2,3 Гц, 1H), 3,85 (дд, J=12,7 Гц, 2,3 Гц, 1H), 3,70 (дд, J=12,3 Гц, 2,7 Гц, 1H), 3,59 (с, 3Н), 2,31 (д, J=6,6 Гц, 2Н), 1,95 (м, 4Н), 1,61-1,20 (м, 6Н). APCI m/z (относит. интенсивн.) 446,3 (МН+, 100) 314,5 (30). МС высок. разреш. (бомбард. быстр. атомами) m/z 446,2040 [(М+Н)+ вычисл. для C21H29N5O6 446,2040].
Пример 20
4-{3-{6-Амино-9-(3,4-дигидрокси-5-гидроксиметилтетрагидрофуран-2-ил)-9Н-пурин-2-ил]проп-2-инил}циклогексилметиловый эфир уксусной кислоты (JR2147)
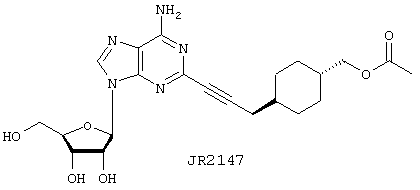
Указанное в заголовке соединение, JR2147, синтезируют в соответствии с общим протоколом 4, используя алкин (5.3) и 10 мг (0,025 ммоль) 2-иодаденозина. Продукт выделяют в виде грязно-белого твердого вещества: выход 4,8 мг (48%).
1H ЯМР (CD3OD) δ 8,25 (с, 1Н), 5,75 (д, J=6,5 Гц, 1Н), 4,70-4,66 (дд, J=8,1 Гц, 4,6 Гц, 1Н), 4,40 (д, J=1,2 Гц, 1Н), 4,25-4,23 (дд, J=4,6 Гц, 1,2 Гц, 1H), 3,83 (д, J=6,5, 2Н), 3,53-3,31 (м, 2Н), 2,29 (д, J=6,5 Гц, 2Н), 1,97 (с, 3Н), 1,93-1,89 (м, 2Н), 1,79-1,75 (м, 2Н), 1,64-1,42 (м, 2Н), 1,12 (т, J=7,3 Гц, 3Н), 1,09-0,91 (м, 4Н). APCI m/z (относит. интенсивн.) 460,3 (МН+, 100), 328,5 (35). МС высок. разреш. (бомбард. быстр. атомами) m/z 460,2193 [(М+Н)+ вычисл. для C22H30N5O6 460,2196].
Пример 21
2-Аминоэтиловый эфир 4-{3-[6-амино-9-(5-этилкарбамоил-3,4-дигидрокситетрагидрофуран-2-ил)-9Н-пурин-2-ил]проп-2-инил}циклогексанкарбоновой кислоты (JR3033)
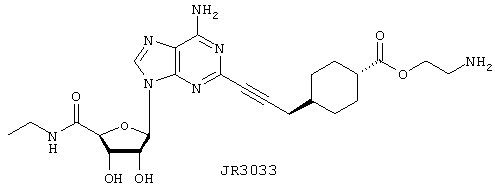
Указанное в заголовке соединение, JR3033, синтезируют, осуществляя взаимодействие чистой трифторуксусной кислоты в течение 2 ч с продуктом взаимодействия 2-иoдNECA и JR3115 в соответствии с общим способом 4. Продукт выделяют в виде соли с ТФУ: выход 9 мг (98%). APCI m/z (относит. интенсивн.) 516,4 (МН+, 100), 343,3(10).
Пример 22
Этиламид 5-{6-амино-2-[3-(4-этилкарбамоилциклогексил)проп-1-инил]пурин-9-ил}-3,4-дигидрокситетрагидрофуран-2-карбоновой кислоты (JR3037)
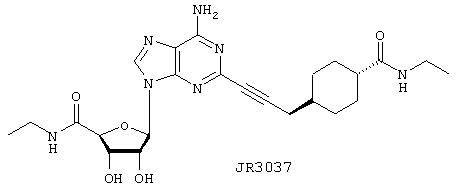
В запаянную трубку, содержащую 5 мл свежеперегнанного этиламина, добавляют 10 мг (0,02 ммоль) ATL146e. Колбу герметично закрывают и содержимое перемешивают при 60°С в течение 80 ч. После истечения данного времени реакция проходит примерно на 50% по данным ВЭЖХ. Сосуд охлаждают до 0°С, открывают и этиламин удаляют в вакууме, получая 4,5 мг (73%) JR3037 в виде белого твердого вещества и выделяют, регенерируя 4,0 мг исходного вещества после очистки остатка ВЭЖХ с обращенной фазой.
Спектры 1H ЯМР (CD3CD-d4) δ, 13С ЯМР (CD3CD-d4) δ соответствовали вышеуказанной структуре. APCI m/z (относит. интенсивн.) 500,8 (МН+, 100), 327,4 (3).
Пример 23
Этиламид 5-{6-амино-2-[3-(4-карбамоилциклогексил)проп-1-инил]пурин-9-ил}-3,4-дигидрокситетрагидрофуран-2-карбоновой кислоты (JR3055)
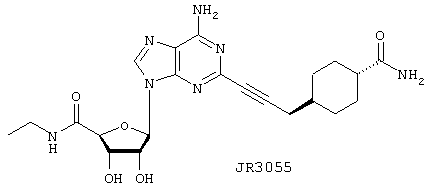
В герметичную трубку, содержащую 10 мл насыщенного раствора MeOH/NH3, добавляют 5 мг (0,01 ммоль) ATL146e. Колбу закрывают и содержимое перемешивают при 25°С в течение 48 ч. Сосуд охлаждают до 0°С, открывают и аммиак удаляют, барботируя N2 в течение 1 часа. Затем в вакууме удаляют остающийся растворитель, получая 4,0 мг (83%) JR3055 в виде белого твердого вещества после очистки остатка ВЭЖХ с обращенной фазой.
1H ЯМР (CD3OD-d6) δ 8,41 (с, 1Н), 5,98 (д, J=7,2 Гц, 1H), 4,65 (дд, J=7,3 Гц, 4,8 Гц, 1Н), 4,41 (д, J =2,0 Гц, 1Н), 4,28 (дд, J=4,6 Гц, 2,0 Гц, 1H), 3,35 (м, 2Н), 2,37 (д, J=6,4 Гц, 2Н), 2,10 (м, 1H), 1,90 (м, _Н), 1,53 (м, _Н), 1,23 (м, _Н), 1,12 (т, J=7,3 Гц, 3Н). 13С ЯМР (CD3OD-d4) δ соответствует вышеуказанной структуре. APCI m/z (относит, интенсивн.) 472,3 (МН+, 100), 299,4 (10).
Пример 24
Этиламид 5-{6-амино-2-[3-(4-метилкарбамоилциклогексил)проп-1-инил]пурин-9-ил}-3,4-дигидрокситетрагидрофуран-2-карбоновой кислоты (JB3065)

В герметичную трубку, содержащую 10 мл 2,0 М метиламина в метаноле, добавляют 16,5 мг (0,03 ммоль) ATL146e. Колбу закрывают и содержимое перемешивают при 70°С в течение 120 ч. Сосуд охлаждают до 0°С, открывают и растворитель удаляют в вакууме, получая 8,0 мг (48%) JR3065 в виде белого твердого вещества после очистки остатка ВЭЖХ с обращенной фазой. APCI m/z (относит. интенсивн.) 486,3 (МН+, 100), 313,4 (35).
Пример 25
1-(4-Проп-2-инилциклогексил)этанон (JR3115)
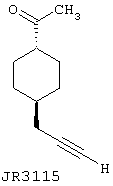
В 50 мл высушенную на пламени круглодонную колбу в атмосфере азота добавляют 840 мг (4,43 ммоль) Cul и 2 мл безводного ТГФ и раствор охлаждают до -40°С на бане ацетонитрил/сухой лед. К данному раствору частями в течение 30 минут добавляют 6,8 мл (9,50 ммоль) 1,4 М MeLi (в эфире) и содержимое перемешивают в течение общего времени, равного 2 часам. В отдельной колбе готовят хлорангидрид 4-проп-2-инилциклогексанкарбоновой кислоты (88), перемешивая 88 в 5 мл SOCl2 при 50°С в течение 3 часов и затем выпаривая тионилхлорид при пониженном давлении, получая желтое маслянистое вещество. Данный остаток переносят в 2 мл ТГФ и по каплям добавляют к раствору метилкупрата при -60°С. Колбу промывают дополнительными 2 мл ТГФ и данный раствор также добавляют по каплям. Реакционную смесь перемешивают в течение 40 минут и затем гасят метанолом, приводя к образованию пузырей, и реакционная смесь изменяет цвет от оранжевого до желтого. Реакционную смесь перемешивают в течение дополнительных 30 минут при комнатной температуре, после чего добавляют 5 мл EtOAc и 3 мл насыщенного раствора NH4Cl. После двух минут перемешивания добавляют 20 мл воды и 20 мл EtOAc и светло-зеленый органический слой отделяют от голубого водного слоя. Водный слой экстрагируют дополнительно 3×15 мл EtOAC, сушат и концентрируют до получения остатка, который подвергают хроматографии с использованием смеси EtOAc/гексан. APCI m/z (относит. интенсивн.) 165,1 (МН+,100).
Пример 26
Этиламид 5-(6-амино-2-{3-[4-(1-гидроксиэтил)циклогексил]проп-1-инил}пурин-9-ил)-3,4-дигидрокситетрагидрофуран-2-карбоновой кислоты
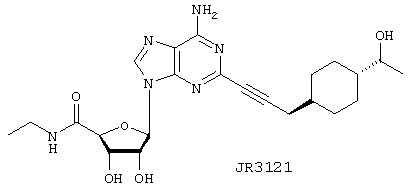
Указанное в заголовке соединение, JR3121, синтезируют, осуществляя взаимодействие NaBH4 в ТГФ с продуктом взаимодействия 2-иодNECA и JR3115 в соответствии с общим способом 4. Продукт выделяют в виде грязно-белого твердого вещества. Выход 7 мг (52%). APCI m/z (относит. интенсивн.) 473,4 (МН+, 100).
Пример 27
Изопропил 4-проп-2-инилциклогексанкарбоксилат
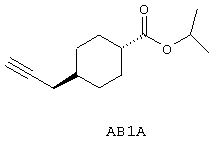
1-(3-Диметиламинопропил)-3-этилкарбодиимида гидрохлорид (103 мг, 0,54 ммоль) и диметиламинопиридин (12 мг, 0,098 ммоль) добавляют к раствору 4-проп-2-инилциклогексанкарбоновой кислоты (78 мг, 0,47 ммоль) в безводном метиленхлориде (8,0 мл). После перемешивания при комнатной температуре в течение 1,5 ч добавляют изопропиловый спирт (2,0 мл, 26 ммоль) и смесь перемешивают 19 ч при комнатной температуре. Добавляют воду (25 мл) и смесь экстрагируют метиленхлоридом (3×15 мл). Объединенные экстракты промывают водой (25 мл) и насыщенным раствором соли (20 мл), сушат над Na2SO4, фильтруют и выпаривают досуха при пониженном давлении. Неочищенный продукт очищают колоночной хроматографией, элюируют смесью гексан/эфир (95:5). Выход 76 мг, 78%.
1H ЯМР (CDCI3) δ 0,96-1,12, 1,19-1,25, 1,33-1,52, 1,86-2,00 (4 х м, 10Н, 9 х циклогексил, ацетилен), 1,20 (д, 6Н, -ОСН(СН3)2), 2,09 (дд, 2Н, -С6Н10СН2ССН), 2,16 (тт, 1Н, 1 х циклогексил), 4,97 (м, 1Н, -ОСН(СН3)2).
Пример 28
Изопропиловый эфир 4-{3-[6-амино-9-(5-этилкарбамоил-3,4-дигидрокситетрагидрофуран-2-ил)-9Н-пурин-2-ил]проп-2-инил}циклогексанкарбоновой кислоты
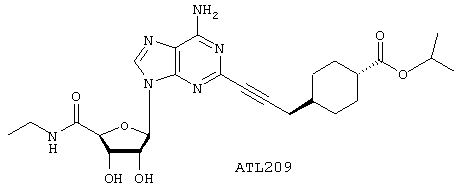
1H ЯМР (CD3OD) δ 1,13-1,26, 1,36-1,50, 1,54-3,67, 1,94-2,03 (4 х м, 18Н, циклогексил, 2 х -СН3, -NHCH2СН3), 2,36 (д, 2Н, -С6Н10СН2СС-), 3,45 (м, 2Н, -NHCH2СН3), 4,29 (дд, 1Н, 3'-Н), 4,45 (д, 1Н, 4'-Н), 4,72 (дд, 1Н, 2'-Н), 5,97 (д, 1Н, 1'-Н), 8,28 (с, 1Н, 8-Н). APCI m/z (относит. интенсивн.) 515,6 (MH+, 100).
Пример 29
трет-Бутил 4-проп-2-инилциклогексанкарбоксилат
Указанное в заголовке соединение, АВ1В, синтезируют, как описано для АВ1А, используя трет-бутиловый спирт вместо изопропилового спирта. Выход 56 мг, 56%.
1H ЯМР (CDCl3) δ 0,97-1,12, 1,18-1,23, 1,32-1,52, 1,85-2,00 (4 х м, 10Н, 9 х циклогексил, ацетилен), 1,43 (с, 9Н, 3 х -ОССН3), 2,05-2,16 (м, 3Н, -С6Н10СН2ССН, 1 х циклогексил).
Пример 30
трет-Бутиловый эфир 4-{3-[6-амино-9-(5-этилкарбамоил-З,4-дигидрокситетрагидрофуран-2-ил)-9Н-пурин-2-ил]проп-2-инил}циклогексанкарбоновой кислоты
Указанное в заголовке соединение, ATL211, получают в виде продукта взаимодействия 2-иoдNECA и JR3115 в соответствии с общим способом 4. Выход 27,5 мг, 63%.
1H ЯМР (CD3OD) δ 1,08-1,23, 1,33-1,49, 1,54-1,67, 1,92-2,03 (4 х м, 10Н, циклогексил), 1,17 (т, 3Н, -NHCH2СН3), 1,43 (с, 9Н, 3 х -СН3), 2,41 (д, 2Н, -С6Н10СН2СС-), 3,40 (м, 2Н, -NHCH2СН3), 4,32 (дд, 1Н, 3'-Н), 4,46 (д, 1Н, 4'-Н), 4,70 (дд, 1Н, 2'-Н), 6,03 (д, 1Н, 1'-Н), 8,46 (с, 1Н, 8-Н). APCI m/z (относит. интенсивн.) 529,6 (MH+,100).
Пример 31
Исследования по связыванию радиолиганда
Связывание с А2А-рецепторами определяют при помощи радиолиганда 125I-ZM241385. Фиг.2 показывает конкуренцию селективных агонистов за связывание с рекомбинантными А2A-рецепторами аденозина человека. Соединение DWH-146e в высшей степени селективно в отношении рекомбинантного A2A-подтипа человека (hA2A). Селективность в отношении А3-рецептора (не показано) менее внушительна, но тем не менее выше приблизительно в 50 раз. Соединение DWH146e приблизительно в 5 и 50 раз более эффективно, чем WRC0470 и CGS21680, соответственно (Фиг.1). Неожиданным и интересным открытием является то, что сложный эфир DWH-146e также приблизительно в 50 раз эффективнее, чем кислота DWH-146a (Фиг.1).
Пример 31а
Действие DWH-146e и JMR193 на окислительную активность нейтрофилов
А. Материалы.
f-met-leu-phe (fMLP), люминол, супероксид дисмутазы, цитохром С, фибриноген, аденозин дезаминазу и трипановый синий получают от Sigma Chemical. Гипак-фиколл приобретают у ICN (Aurora, ОН), а также у Cardinal Scientific (Santa Fe, NM) и Accurate Chemicals, и у Scientific (Westerbury, NY). Эндотоксин (липополисахарид; Е. coli K235) получают от List Biologicals (Campbell, CA). Сбалансированный солевой раствор Хенкса (HBSS) и набор для пробы с лизатом амебоцитов приобретают у Bio-Wittaker (Walkersville, MD). Сывороточный альбумин человека (HSA) получают от Cutter Biological (Elkhart, IN). Рекомбинантный фактор опухоли некроза альфа человека предоставляется Dianippon Pharmaceutical Co. Ltd. (Osaka, Japan). ZM241385 (4-(2-[7-амино-2-(2-фурил)[1,2,4]триазоло[2,3-а] [1,3,5]триазин-5-ил-амино]этил)фенол) был подарен Simon Poucher, Zeneca Pharmaceuticals, Cheshire, UK. Готовят маточные растворы (1 мМ и 10 мМ в ДМСО) и хранят их при -20°С.
В. Получение нейтрофилов человека.
Очищенные нейтрофилы (приблизительно 98% нейтрофилов и >95% жизнеспособных, как установлено методом исключения с применением трипанового синего, содержащие <1 тромбоцита на 5 нейтрофилов и <50 пг/мл эндотоксина (проба с лизатом амебоцитов), получают из нормальной гепаринизированной (10 ед/мл) венозной крови в результате одностадийной процедуры разделения на гипак-фиколле (A. Ferrante et al., J. Immunol. Meth., 36, 109 (1980) ).
С. Высвобождение при воспалении реактивного кислорода из примированных и стимулированных нейтрофилов человека.
Хемилюминесценция
Усиленная люминолом хемилюминесценция, мера измерения окислительной активности нейтрофилов, зависит как от выработки супероксида, так и от мобилизации миелопероксидазы, фермента лизосомных гранул. Свет излучается неустойчивым высокоэнергетическим кислородом, вырабатываемым активированными нейтрофилами. Очищенные нейтрофилы (5-10×105/мл) инкубируют в сбалансированном солевом растворе Хенкса, содержащем 0,1% сывороточного альбумина человека (1 мл) с DWH-146а, DWH-146e, CGS 21680 или JMR193 или без них, с ролипрамом или без него, и с фактором некроза опухоли альфа (1 ед/мл) или без него, в течение 30 минут при 37°С на встряхиваемой ледяной бане. Затем определяют усиленную люминолом (1×10-4 М), стимулированную f-met-leu-phe (1 мсМ) хемилюминесценцию на фотометре Chronolog® (Crono-log Corp., Havertown, PA) при 37°С в течение 2-4 минут. Хемилюминесценцию определяют как относительный максимум излучаемого света (=высоте кривой) по сравнению с таковым в образцах с фактором некроза опухоли альфа и без DWH, JMR или ролипрама.
D. Результаты
Как показано на Фиг.2, как JMR193, так и DWH-146e снижают окислительную активность нейтрофилов человека, стимулированную f-met-leu-phe и примированную фактором некроза опухоли альфа, при измерении усиленной люминолом хемилюминесценции, более эффективно, чем агонист А2А-рецептора аденозина CGS21680. На горизонтальной оси показаны концентрации CGS21680, DWH-146a, DWH-146e или JMR193 (log nM). На вертикальной оси показан результирующий пик активности нейтрофилов человека как относительное количество стимулированного высвобождения реактивного кислорода, измеряемое при помощи усиленной люминолом хемилюминесценции, по отношению к контрольным образцам, которые не были примированы фактором некроза опухоли альфа. Средние величины SEM (n=4-5 отдельных экспериментов).
Данные под горизонтальной осью на Фиг.2 показывают величину ЕС50 для снижения активности нейтрофилов человека (на основании данных Фиг.2). Средние величины SEM (n=4-5 отдельных экспериментов). *р<0,05, снижение IC50 по сравнению с CGS21680.
JMR193 и DWH-146e снижают окислительный выброс стимулированных нейтрофилов-EC50 менее 1 нМ (0,8 и 0,3 нМ, соответственно). Агонисты А2А-рецептора аденозина свободной кислоты DWH-146a и CGS21680, напротив, не так эффективно ингибируют окислительный выброс (53 и 9 нМ, соответственно; Фиг.2). Ингибирование DWH-146e окислительного выброса стимулированных нейтрофилов подавлялось селективным антагонистом A2A-AR ZM241385.
Как показано на Фиг.3, JMR193 (1 нм) с ролипрамом (100 нМ) синергически снижает стимулированное высвобождение реактивного кислорода. Нейтрофилы человека примируют фактором некроза опухоли альфа (1 ед/мл) и стимулируют f-met-leu-phe (1 мкМ). Вертикальная ось показывает процентную величину ингибирования окислительной активности, измеряемую усиленной люминолом хемилюминесценцией. Средние величины SEM (n=4) отдельным экспериментам. *р<0,05 синергизм между JMR193 и ролипрамом вместо аддитивной активности.
Как показано на Фиг.4, высокоселективный антагонист А2A-рецептора аденозина ZM241385 (100 нМ) (ZM) противодействует окислительной активности нейтрофилов человека, ингибируемой JMR193 (10 нМ), как следует из результатов усиленной люминолом хемилюминесценции. Средние величины SEM 4 отдельных экспериментов. *р=0,0004 ZM241385 противодействует ингибируемой JMR193 окислительной активности.
Е. [цАМФ]i нейтрофилов человека и адгезия нейтрофилов к биологической поверхности.
24-Луночный планшет для культивирования тканей покрывают фибриногеном человека (5 мг/мл в 1,5% бикарбонате натрия; 0,5 мл/лунку; Sigma Chemical) на ночь при 37°С в 5% СО2.
Нейтрофилы (3-4×106/0,5 мл/образец) инкубируют в лунке планшета с покрытием в течение 45 минут в 0,5 мл HBSS, содержащего 0,1% HSA и ADA (1 ед/мл) при наличии и отсутствии рекомбинантного человеческого TNFα (10 ед/мл), DWH-146e (3-300 нМ), ролипрама (300 нМ) и/или ZM241385 (100 нМ). После инкубирования в лунки добавляют 0,5 мл HCl (0,2 н.) и инкубируют еще в течение 45 минут, экстрагируя цАМФ. Затем образцы центрифугируют на микрофуге в течение 2 минут, удаляя дебрис клеток. Полумиллилитровые образцы замораживают для анализа на цАМФ (В. Brooker et al., Science, 194, 270 (1976)). Стенки дважды промывают нормальным солевым раствором, а оставшийся монослой гидролизуют 0,2 мл 0,2 н. NaOH, содержащей SDS, в течение 2 часов при комнатной температуре. Затем образцы белка замораживают (-70°С) для дальнейшего анализа белка с целью определения сравнительной адгезии PMN (К.Р. Stowell et al., Anal. Biochem., 85, 572 (1978)).
Результаты
DWH-146e (30-300 нМ) отдельно и синергически вместе с ролипрамом (300 нМ) повышает содержание цАМФ в нейтрофилах человека и вместе с ролипрамом синергически снижает адгезию нейтрофилов к покрытой фибриногеном поверхности (Фиг.5). Действию DWH-146e (300 нМ) + ролипрам (300 нМ) на продуцирование цАМФ нейтрофилами и адгезии противодействует селективный антагонист А2A-рецептора аденозина, ZM241385 (ZM; 100 нМ). Средняя величина SEM 5 отдельных экспериментов. *р<0,05 повышенное [цАМФ] в нейтрофилах по сравнению с отсутствием DWH-146e; **р<0,05 снижение адгезии нейтрофилов по сравнению с отсутствием DWH-146e.
F. Окислительная активность адгезивных нейтрофилов человека.
Способы
Применяя модифицированные способы из раздела Е, нейтрофилы (2×106/мл), полученные в результате разделения на гипак-фиколле инкубируют в течение 15 минут при 37°С в 0,45 мл сбалансированного солевого раствора Хенкса, содержащего 0,1% человеческого сывороточного альбумина и аденозиндезаминазы (1 ед/мл), ролипрама (300 нМ) и DWH-146e (3-300 нМ). После инкубирования добавляют цитохром С (120 мкМ) и каталазу (0,062 мг/мл) в присутствии и в отсутствие рекомбинантного фактора некроза опухоли альфа человека (1 ед/мл), а 200 мкл аликвоты клеточной суспензии немедленно переносят в парные ячейки 96-луночного матраса для культуры тканей, покрытого на ночь фибриногеном человека. Определяют оптическую плотность образцов при 500 нм против соответствующих образцов супероксид дисмутазы (200 ед/мл).
G. Результаты.
Как показано на Фиг.6, ингибирование стимулируемого фактором некроза опухоли альфа высвобождения супероксида адгезивными нейтрофилами человека на покрытой фибриногеном поверхности обеспечивают ролипрам (300 нМ) и DWH-146e. DWH-146e снижает окислительный выброс адгезивных нейтрофилов, а также синергетически снижает окислительный выброс в присутствии ролипрама, который сам по себе не влияет на окислительную активность нейтрофилов. На горизонтальной оси показаны концентрации DWH-146e в нМ, а на вертикальной оси - количество супероксида, высвобождаемого нейтрофилами, определяемое путем измерения снижения цитохрома С. Наблюдается заметный синергизм с DWH-146e и ингибитором PDE IV типа, ролипрамом, в снижении окислительной активности адгезивных нейтрофилов человека, стимулируемых фактором некроза опухоли альфа. Средние величины SEM повторов по 4-5 отдельным экспериментам. *р<0,05 снижение высвобождения супероксида по сравнению с тем, когда отсутствует DWH-146e; **р<0,05 снижение высвобождения супероксида по сравнению с наличием ролипрама и отсутствием DWH-146e.
Пример 32
Лечение ишемического/реперфузионного повреждения почек с применением DWH-146e
Чтобы определить, снижает ли активация А2A-рецептора аденозина, индуцируемая DWH-146e, креатинин плазмы через 24 и 48 часов после ишемического/реперфузионного повреждения у крыс, почки крыс подвергают 45-минутной ишемии и 24- или 48-часовой реперфузии. DWH-146e (0,004 мкг/кг/мин) или наполнитель начинают непрерывно вводить через мини-насос за 5 часов до ишемии/реперфузии. Как показано на Фиг.7, DWH-146e существенно снижает креатинин в плазме у 7/7 крыс (Р<0,05) и у 6/6 крыс, получавших DWH-146e (Р<0,001), через 24 и 48 часов соответственно.
Чтобы определить, является ли действие DWH-146e на снижение креатинина в плазме у крыс, подвергнутых ишемии/реперфузии, опосредованным А2A-рецептором, почки крыс подвергают 45-минутной ишемии, а затем 48-часовой реперфузии. DWH-146e (0,004 мкг/кг/мин) начинают непрерывно вводить через мини-насос за 5 часов до ишемии. Как показано на Фиг.8, улучшение почечной функции было полностью устранено антагонистом А2A ZM241385 (0,003 мкг/кг/мин - эквимолярная скорость доставки по сравнению с DWH-146e) (*Р<0,001 в случае наполнителя вместо DWH; **р<0,05 DWH против DWH/ZM. N=5 для наполнителя, DW; N=6 для DWH/ZM. ANOVA с последующей коррекцией Bonferroni).
DWH-146e при концентрации, не оказывающей гемодинамического действия, предотвращает отек почек, некроз и концентрацию эритроцитов во внутреннем мозговом веществе.
Защита от повреждения почек, оказываемая DWH-146е (0,01 мкг/кг/мин подкожно в течение 48 часов), коррелирует с резким ингибированием адгезии нейтрофилов к сосудистому эндотелию. Полагают, что ингибирование под действием DWH-146e взаимодействия между нейтрофилами и сосудистым эндотелием отвечает, по меньшей мере частично, за защиту почек от повреждения.
Для того, чтобы определить, снижает ли активизация A2A-AR количество нейтрофилов во внутреннем мозговом веществе крыс, подвергнутых ишемии/реперфузии с применением Neuroludica®, почку рассматривают при 100-кратном увеличении и делают вытяжку всей почки. Подсчитывают количество полиморфноядерных лейкоцитов, рассматривая срезы почки при 250-кратном увеличении. На почечные срезы накладывают оптические рамки, рассматриваемые под микроскопом, и подсчитывают все полиморфноядерные лейкоциты (PMN) внутри каждой рамки. Такая система гарантирует, что каждый PMN будет подсчитан только один раз. Как показано на Фиг.9, плотность нейтрофилов составляет 15,65/мм2 при использовании наполнителя и 3,02/мм2 при лечении с применением DWH-146e.
Пример 33
Действие DWH-146e на реперфузионное повреждение легких
А. Способы.
Применяют изолированную, перфузированную цельной кровью, вентилируемую модель легких кролика. Кролики-доноры подвергаются удалению легких после легочной артериальной инъекции PGEi1 и промывания Евро-Коллинз-консервантом, затем легкие сохраняют в течение 18 часов при 4°С. Группа I легких (n=9) служит контрольной группой. Группу II легких (n=9) реперфузируют цельной кровью, которую вначале пропускают через фильтр, удаляющий лейкоциты. В группе III (n=9) к реперфузату крови добавляют DWH-146e (25 мкг/кг) сразу же после реперфузии и вводят на протяжении реперфузионного периода (1 мкг/кг/мин). Все легкие подвергают реперфузии в течение 30 минут и записывают давление в легочной артерии (РАР), легочное сосудистое сопротивление (PVR), воздушную растяжимость (CPL) и артериальную оксигенацию. Записывают активность миелопероксидазы (МРО), чтобы определить объем секвестрации нейтрофилов, а также измеряют влажное/сухое весовое соотношение для определения отека легких.
В. Результаты.
Артериальная оксигенация в группах II и III существенно выше указанной оксигенации в группе I после 30 минут реперфузии (514,27±35,80 и 461,12±43,77 против 91,41±20,58 мм рт.ст., р<0,001. Как показано на Фиг.10, группа легких III проявляет нарастающее потребление pO2 на протяжении реперфузии. Отсутствие лейкоцитов в группе легких II улучшает артериальную оксигенацию в начале реперфузии. *р=0,004 (группа II против групп I и III); **р<0,001 (группы II и III против группы I).
Как показано на Фиг.11, средний PVR в группе II существенно ниже по сравнению с контрольными легкими (*р<0,001). PVR группы легких III существенно ниже, чем даже у легких, реперфузированных кровью без лейкоцитов (**р<0,001 против групп I и II). Легочное сосудистое сопротивление существенно ниже в группе III (22,783±357 дин·сек·см-5) по сравнению с обеими группами II и I (31,057±1743 и 36,911±2173 дин·сек·см-5, р<0,001). Воздушная растяжимость лучше в группах II и III по сравнению с группой I (1,68±0,08 и 1,68±0,05 против 1,36±0,13, р=0,03). Проницаемость микрососудов в группе III снижена до 106,82±17,09 по сравнению с 165,70±21,83 нг синего красителя Эванса/гм ткани в группе I (р=0,05). Как показано на Фиг.12, активность миелопероксидазы в группе III существенно ниже, чем в группе I (*р=0,03). МРО = миелопероксидаза. Активность миелопероксидазы в группе III составляет 39,88±4,87 по сравнению с 88,70±18,69 ΔOD/гм/мин в группе I (р=0,03), а активность миелопероксидазы в группе II составляет 56,06±7,46.
С. Выводы.
DWH-146e снижает секвестрацию нейтрофилов легких и резко улучшает функцию легочного трансплантата. Нейтрофилы являются важными компонентами воспалительного каскада реперфузионного повреждения, и их источником может быть как циркулирующая кровь, так и сам легочный имплантат. Селективное активирование А2A аденозина прекращает нейтрофилопосредованную воспалительную реакцию и уменьшает реперфузионное повреждение легких после трансплантации.
Результаты световой микроскопии показывают, что контрольные легкие в группе I имеют тяжелую инфильтрацию лейкоцитами и отек в альвеолярных пространствах после 18 часов ишемического хранения и 30 минут реперфузии. В группе II (легкие, подвергнутые реперфузии лишенной лейкоцитов кровью) и в группе III (легкие, получавшие DWH-146e во время реперфузии) такая инфильтрация была намного слабее.
Пример 34
Действие DWH-146e на образование неоинтимы после артериального повреждения
Активация лейкоцитов при высвобождении воспалительных цитокинов происходит после подкожного коронарного вмешательства и может играть некоторую роль в рестенозе. У мышей образование здоровой неоинтимы в присутствии интактной эндотелиальной выстилки происходит после лигирования общей сонной артерии. Применяя данную модель, мышей C57/BL6 рандомизируют во время лигирования сонной артерии для 7-дневного вливания через осмотический насос DWH-146e (n=7) или наполнителя (n=8).
На 14-й день после лигирования сонной артерии гистоморфометрия показывает существенное уменьшение площади неоинтимы (0,005±0,004 мм2 против 0,021±0,014 мм2, р=0,02), а также отношение площади неоинтимы к средней площади (0,13±0,07 против 0,64±0,44, р=0,01) у получавших лечение животных по сравнению с контролем. Средняя площадь в обеих группах почти одинакова (0,035±0,007 мм2 против 0,036±0,009 мм2, р=0,81). Такое ограничение роста неоинтимы продолжалось 28 дней. Фиг.13 суммирует действие DWH-146e по ингибированию роста неоинтимы на модели мышей LCCA. Данные эксперименты показывают, что на модели лигирования сонной артерии мыши пролонгированное стимулирование А2A (7 дней) с применением DWH-146e приводит к существенному снижению образования неоинтимы по меньшей мере в течение 21 дня, возможно, благодаря его действию на активирование и функцию лейкоцитов.
Пример 35
Ингибирование стимулированного эндотоксином высвобождения TNFα моноцитов человека
А. Материалы.
Гипак-фиколл приобретают у ICN (Aurora, ОН), Cardinal Scientific (Santa Fe, NM), Accurate Chemicals and Scientific (Westbury, NY). Эндотоксин (липополисахарид; E. coli 0111B4) получают от List Biologicals (Campbell, CA). Сбалансированный солевой раствор Хенкса (HBSS) и набор для проб с лизатом амебоцитов приобретают у BioWittaker (Walkersville, MD). Сывороточный альбумин человека (HSA) получают от Cutter Biological (Elkhart, IN). ZM241385 (4-(2-[7-амино-2-(2-фурил) [1,2,4]триазол[2,3-а][1,3,5]триазин-5-иламино]этил]фенол) был подарен Simon Poucher, Zeneca Pharmaceuticals, Cheshire, UK. Готовят маточные растворы (1 мМ и 10 мМ в ДМСО) и хранят их при -20°С.
В. Продуцирование TNFα очищенными адгезивными моноцитами человека.
Способы
Богатый моноцитами монослой (>65% моноцитов) получают, инкубируя 1 мл мононуклеарной фракции лейкоцитов (5×105/мл), полученной в результате разделения на гипак-фиколле (А. Ferrante et al., J. Immunol. Meth., 36, 109 (1980)) в лунках 24-луночного планшета для культивирования тканей (1 час; 37°С; 5% CO2). Неприкрепленные лейкоциты удаляют промыванием и культуральную среду (1 мл сбалансированного солевого раствора Хенкса, содержащего 0,1% сывороточного альбумина человека, аденозиндезаминазу [5 ед/мл] и 1% инактивированной нагреванием аутогенной сыворотки), добавляют в лунки, содержащие адгезивные мононуклеарные клетки. Как указано выше, добавляют следующие соединения: (1) эндотоксин (100 нг/мл) и селективный антагонист А2A AR ZM241385 (100 нМ), а также (2) селективные агонисты А2A-рецептора аденозина JMR193 (1-1000 нМ), DWH-146e (1-1000 нМ) и CGS21680 (10-1000 нМ). Затем образцы инкубируют в течение 4 часов (37°С; 5% CO2) и надосадочные слои собирают. Суспендированные клетки удаляют центрифугированием, а свободные от клеток образцы замораживают (-70°С). TNFα исследуют в свободных от клеток надосадочных жидкостях (n=6), применяя набор ELISA (Coulter/Immunotech, Miami, FL).
С. Результаты.
Как показано на Фиг.10, 14, агонисты A2A-рецептора аденозина снижают продуцирование TNFα эндотоксин-стимулированными адгезивными моноцитами. Селективный антагонист A2A AR ZM241385 (100 нМ) противостоит действию JMR193 на продуцирование TNFα (Фиг.15).
Таким образом, DWH-146e и JMR193 снижают LPS эндотоксин-стимулированное продуцирование TNFα моноцитами человека в результате действия механизма, зависящего от связывания агониста с А2A-рецепторами аденозина.
Пример 36
Активность DWH-146e в модели перитонита у мышей
Предварительные исследования экспериментального перитонита включают инъекцию зимозана (Zym) в качестве сильного стимула воспаления (Y. Zhang et al., Eur. J. Pharmacol., 313, 237 (1996)). Как показано на Фиг.16, после инъекции зимозана средняя концентрация лейкоцитов, определяемая при помощи гемоцитометра neubauer, составляет 7,325±1,893/мм3. Внутрибрюшинная инъекция DWH-146e в дозе, составляющей 2,5 мкг/кг, за час до зимозана ингибирует развитие перитонита, при атом средняя концентрация лейкоцитов ±SEM составляет 2,012±374/мм3 6 часов спустя (р<0,05). Таким образом, данные исследования показывают, что А2A AR является инструментом при опосредовании проникновения PMN в брюшину после введения зимозана.
Пример 37
Кардиозащита, опосредуемая противовоспалительным действием JMR193
Соединения в соответствии с настоящим изобретением исследуют, индуцируя миокардиальное "оглушение", вид поражения сердца, наступающего после повторных, временных периодов прекращения коронарного тока крови, путем повторной окклюзии поступления крови в коронарную артерию.
А. Действие четырех циклов окклюзии-реперфузии.
Левую переднюю нисходящую (LAD) коронарную артерию в группе собак изолируют и заключают в петлевой обтуратор. Поступление крови в LAD-артерию собак подвергают окклюзии 4 раза в течение 5 минут. После каждой окклюзии ток крови возобновляют на десять минут. Одной группе из шести собак после каждого периода окклюзии вливают раствор, содержащий соединение ацетата (JMR193), полученное в примере 15 (JMR193) (0,01 мкг/кг/мин). Второй группе из шести собак вливают раствор, содержащий наполнитель (носитель). После последнего цикла окклюзии-реперфузии сердечную функцию животных контролируют в течение 3 часов. Результаты представлены на Фиг.17 и 18. Фиг.17 показывает систолическую левую вентрикулярную (LV) реакцию утолщения у 6 контрольных собак. Сердечное утолщение снижается приблизительно на 50% через 3 часа после последней окклюзии. Фиг.18 показывает LV реакцию утолщения у 6 собак, получивших внутривенное вливание тест-соединения, JMR193 (0,01 мкг/кг/мин.), начиная с базового периода и на протяжении всего эксперимента. Сердечная функция в результате вливания JMR193 почти нормализовалась через 90 минут после реперфузии.
В. Действие десяти циклов окклюзии-реперфузии.
Две дополнительные группы собак подвергают десяти (а не 4) циклам окклюзии-реперфузии, при этом каждая окклюзия продолжается 5 минут, прерываемая 5 минутами реперфузии. В данном примере двум животным вливают раствор, содержащий соединение ацетата (JMR193), полученный в примере 15 (0,01 мкг/кг/мин), после каждого периода окклюзии. Другим трем животным вливают раствор, содержащий наполнитель (носитель). После последнего цикла окклюзии-реперфузии сердечную функцию животных контролируют в течение 3 часов.
Результаты представлены на Фиг.19 и 20. На Фиг.19 показана систолическая левая вентрикулярная реакция утолщения у 3 контрольных собак. Это более тяжелое сердечное поражение, чем в примере 23А, но результат LV-утолщение полностью отсутствует вскоре после реперфузии и остается неизменным в течение 3 часов. Фиг.20 показывает LV утолщение у 2 собак, получивших внутривенное вливание тест-соединения, JMR193 (0,01 мкг/кг/мин), начиная с базового периода и на протяжении всего эксперимента. По сравнению с контрольной группой, собаки, получавшие вливание JMR193, демонстрируют существенное и заметное улучшение сердечной функции сразу же после реперфузии, длившейся 3 часа.
С. Действие ацетатного соединения JMR193 на поглощение нейтрофилов во время окклюзии-реперфузии.
Некоторым животным вводят нейтрофилы, меченные радиоактивными изотопами. Нейтрофилы выделяют из крови собак, инкубируют с соединением, содержащим 99mTc, и вновь вводят собакам. Нейтрофилы, меченные 99mTc, вводят внутривенно в качестве маркера, чтобы определить степень воспаления в реперфузионной зоне после четырех циклов ишемии-реперфузии. Воспаление, вызванное циклами окклюзии-реперфузии, является причиной адгезии радиоактивных нейтрофилов, количество которых подсчитывают при помощи гамма-камеры. JMR193 ингибирует адгезию нейтрофилов. Результаты представлены на Фиг.21, где локализация нейтрофилов, меченных 99mTc у собак, получавших только наполнитель (сплошные полоски), выше, чем у собак, получавших JMR193 (заштрихованные полоски). Таким образом, снижение количества меченных радиоактивными изотопами нейтрофилов в центральной ишемической зоне, вызванное вливанием JMR193, иллюстрирует снижение (*) сердечного воспаления.
Исследования, описанные в примерах 23А и 23В, показывают, что сердечное воспаление играет существенную отрицательную роль, вызывая миокардиальное поражение. Кроме того, введение агониста А2A-рецептора аденозина, такого как, например, JMR193, либо предотвращает легкое повреждение (Фиг.17 и 18), либо существенно смягчает дисфункцию миокарда, сопровождающую тяжелое поражение (Фиг.19 и 20).
Все публикации, патенты и патентные документы приведены здесь в качестве ссылки, как если бы они отдельно перечислялись в качестве ссылок. Данное изобретение описано со ссылкой на различные конкретные и предпочтительные варианты и способы его осуществления. Однако подразумевается, что возможны многие варианты и модификации, если они не нарушают сущность и объем данного изобретения.
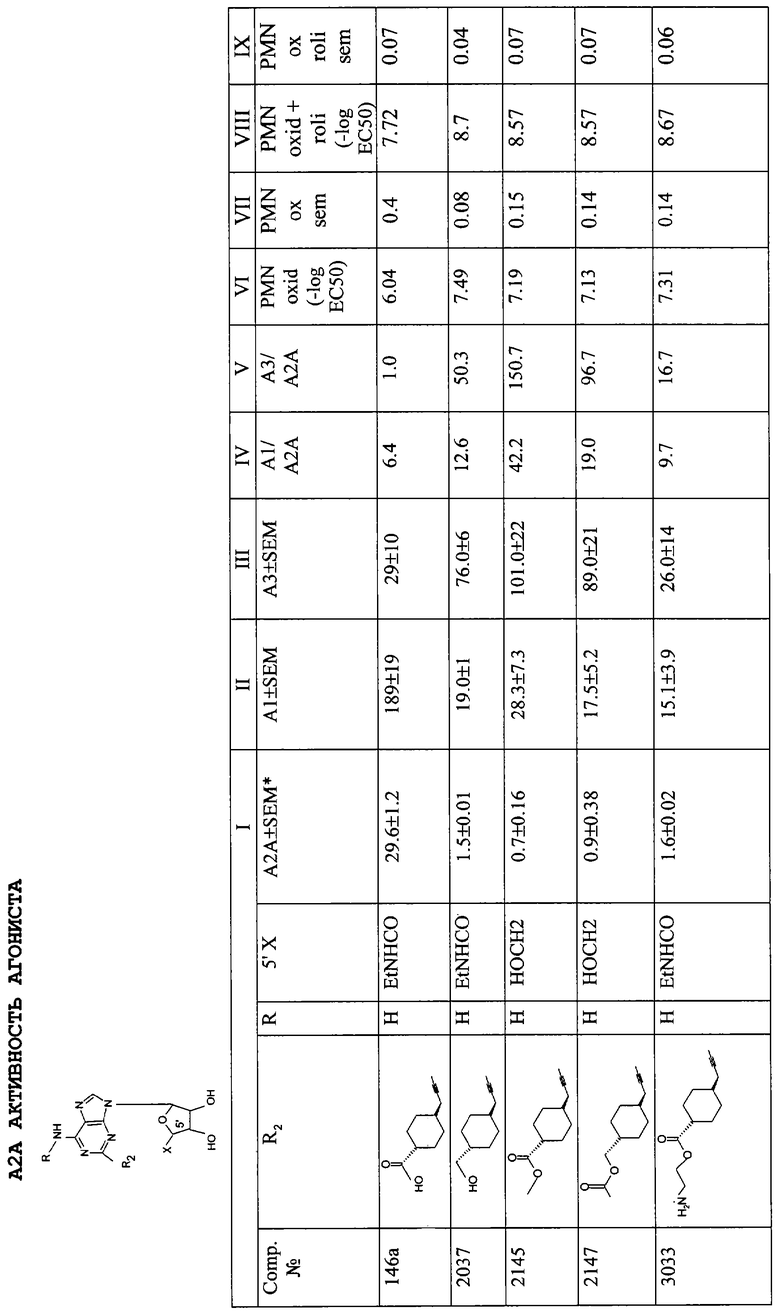
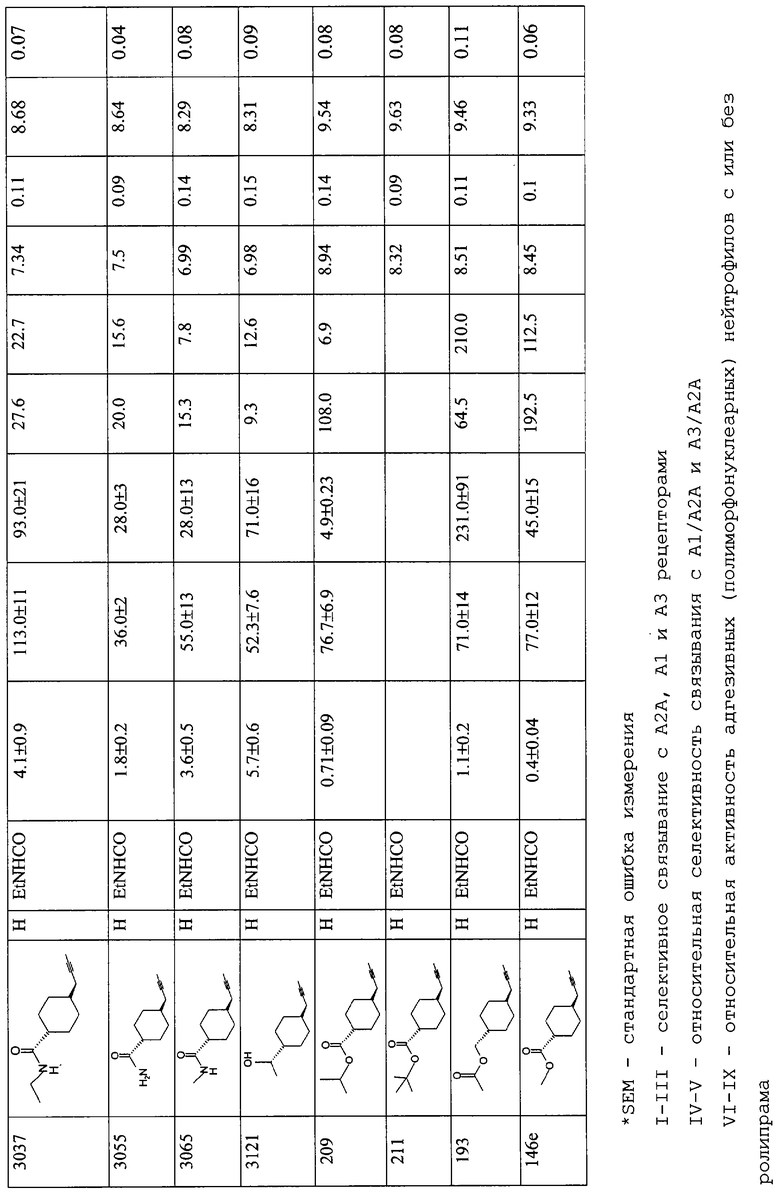
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ АРИЛПИПЕРИДИНИЛАЛКИНИЛАДЕНОЗИНЫ В КАЧЕСТВЕ AR АГОНИСТОВ | 2007 |
|
RU2442789C2 |
| ПРОИЗВОДНЫЕ 2-ПИРАЗИНОНА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ ИЛИ СОСТОЯНИЯ, ПРИ КОТОРЫХ ПОЛЕЗНО ИНГИБИРОВАНИЕ АКТИВНОСТИ НЕЙТРОФИЛЬНОЙ ЭЛАСТАЗЫ | 2007 |
|
RU2448098C2 |
| N-(2-АРИЛПРОПИОНИЛ)СУЛЬФОНАМИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ | 1999 |
|
RU2255084C2 |
| ПРОИЗВОДНЫЕ 2-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2005 |
|
RU2375347C2 |
| ОМЕГА-АМИНОАЛКИЛАМИДЫ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ ХЕМОТАКСИСА ПОЛИМОРФНОЯДЕРНЫХ И ОДНОЯДЕРНЫХ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2272024C2 |
| АМИДИНЫ И ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2375346C2 |
| СЕЛЕКТИВНЫЕ АНТАГОНИСТЫ АДЕНОЗИНОВЫХ A РЕЦЕПТОРОВ | 2007 |
|
RU2467009C2 |
| 2-АРИЛУКСУСНЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2356887C2 |
| (R)-4-(ГЕТЕРОАРИЛ)ФЕНИЛЭТИЛЬНЫЕ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2008 |
|
RU2475486C2 |
| 2-АРИЛПРОПИОНОВЫЕ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2002 |
|
RU2317075C2 |
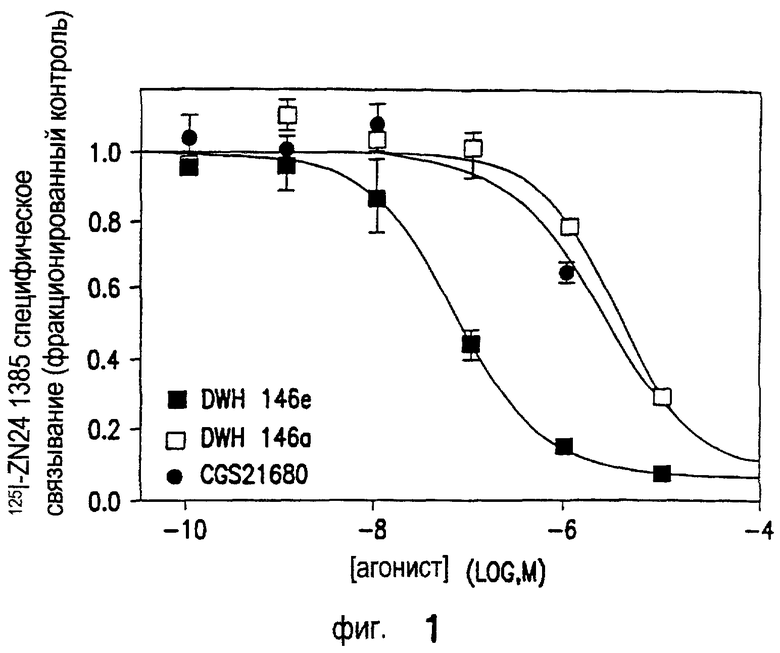
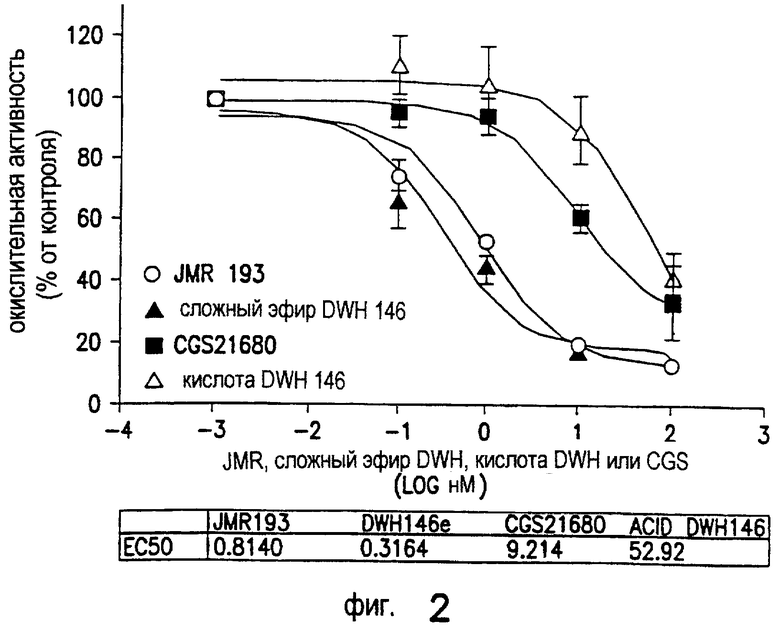
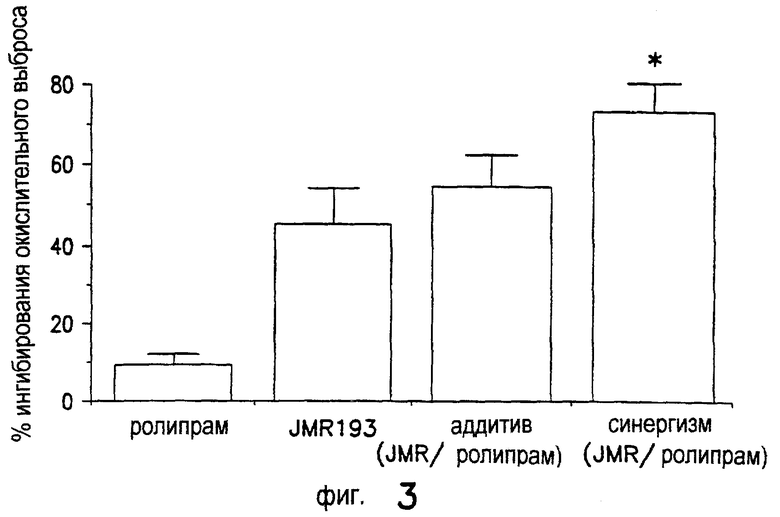
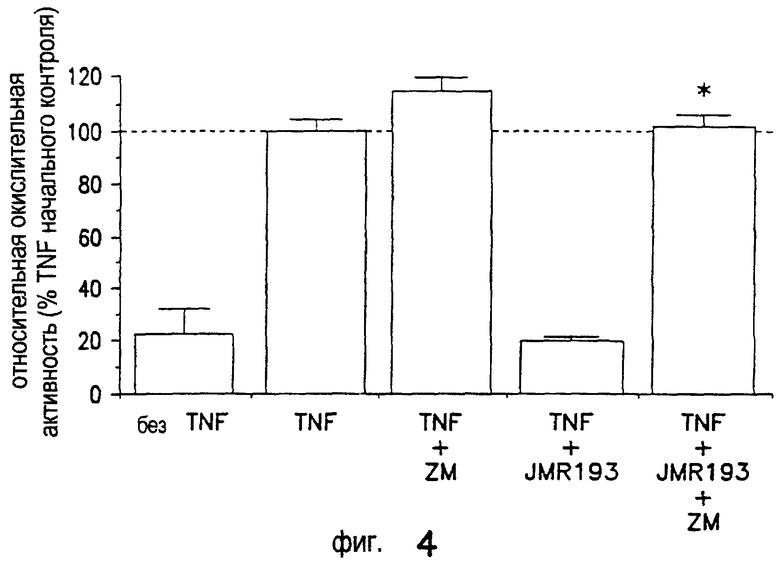
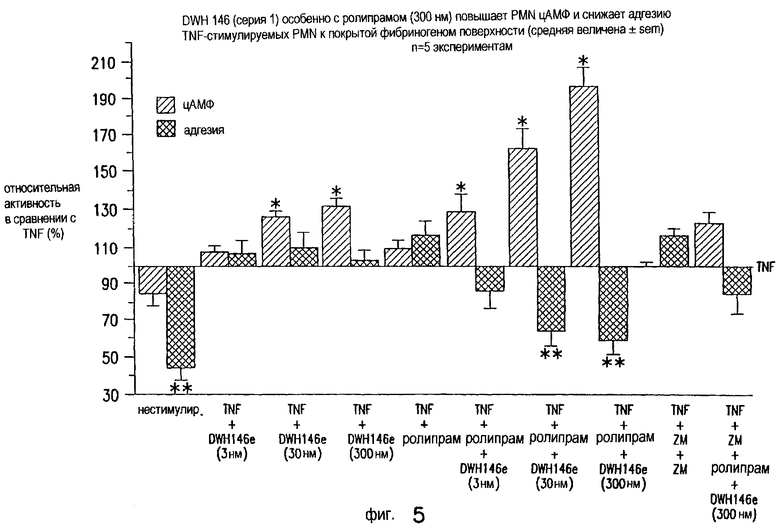
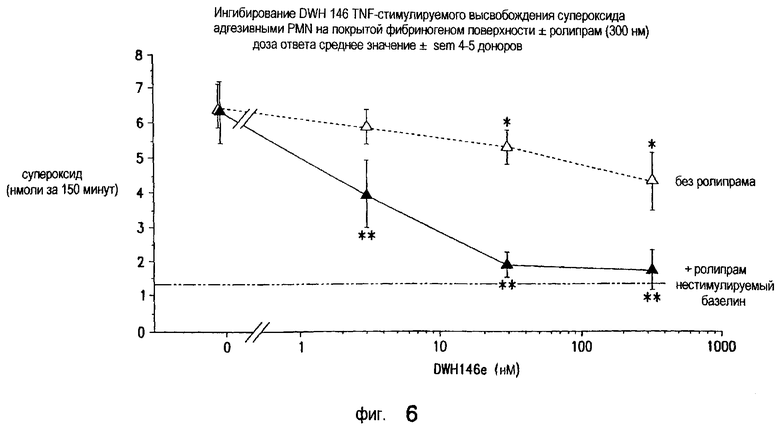


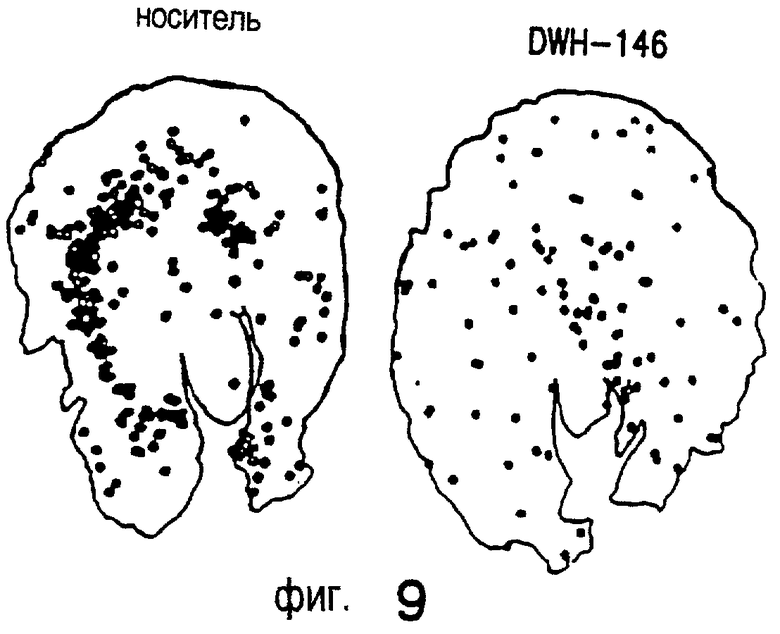
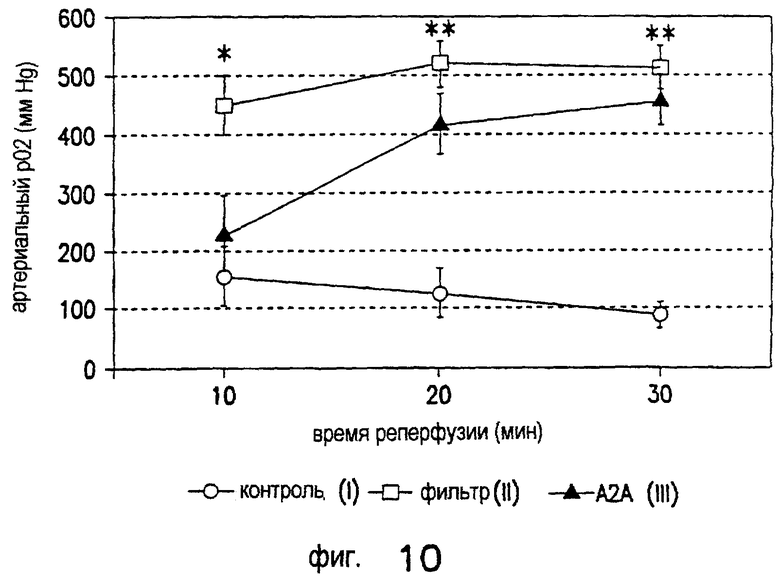
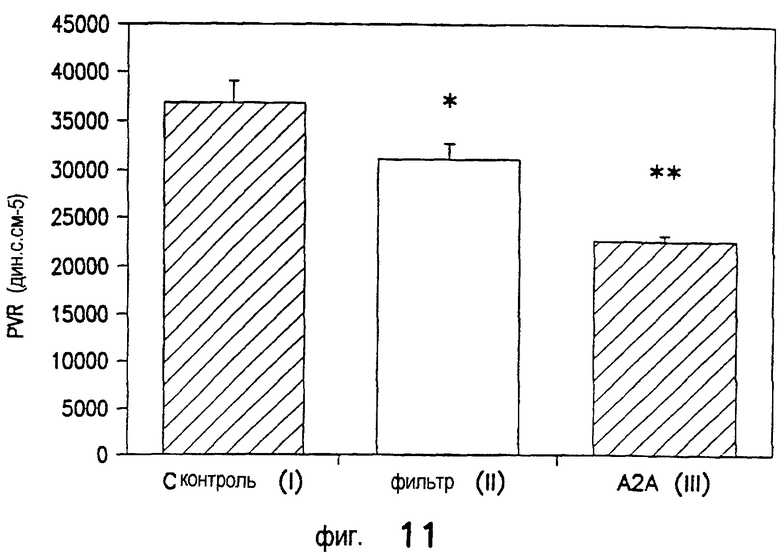
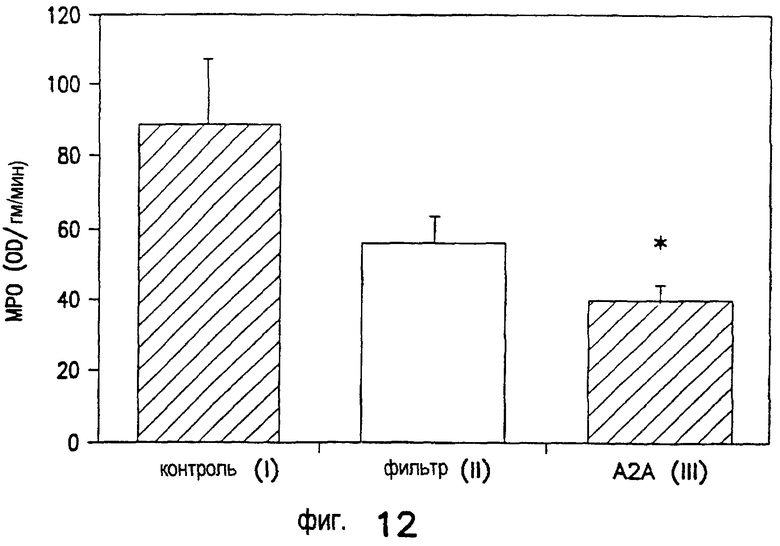
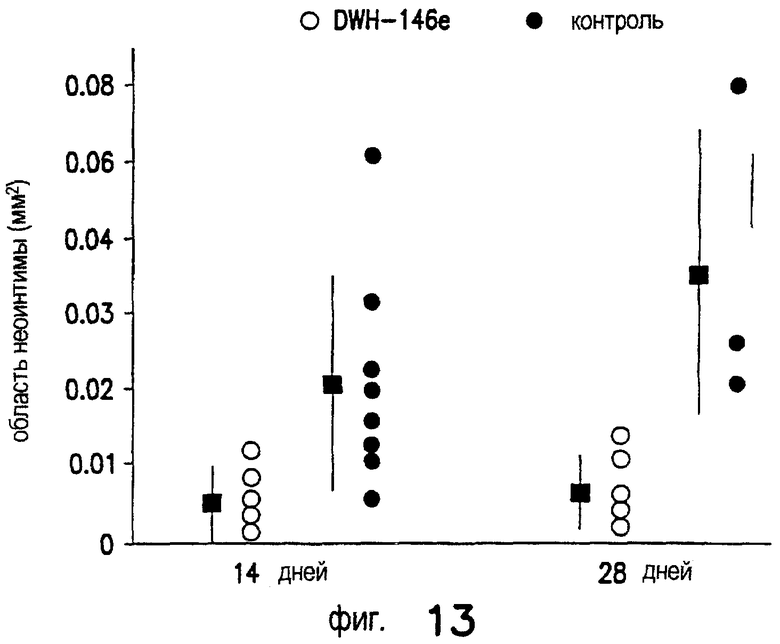
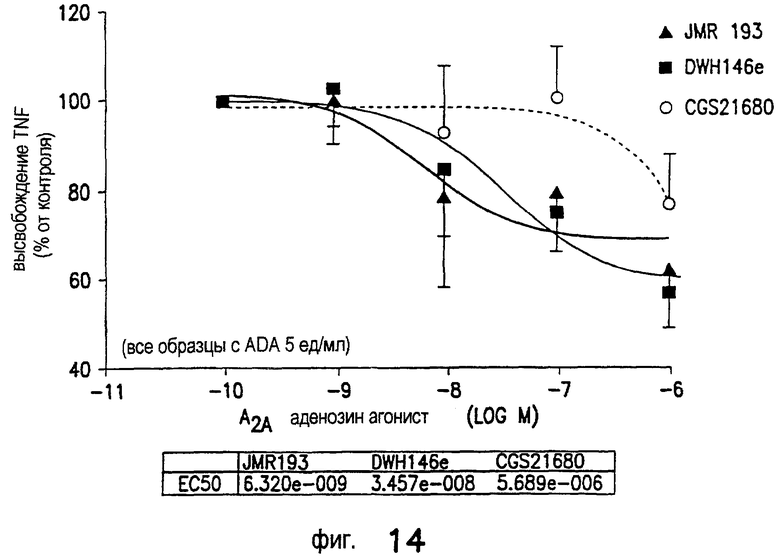
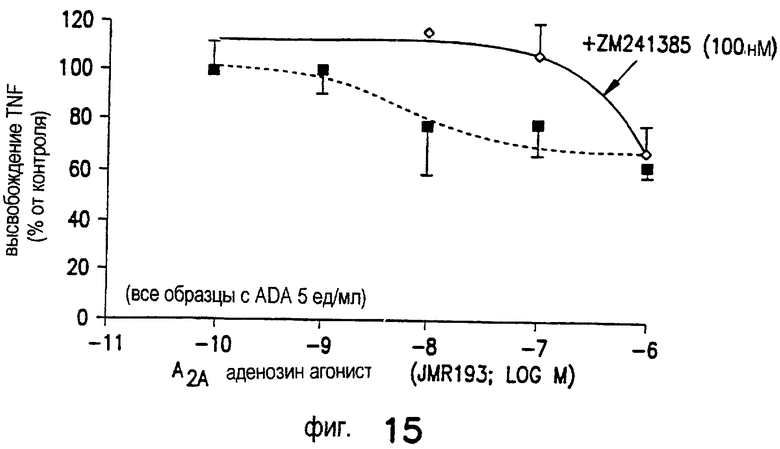
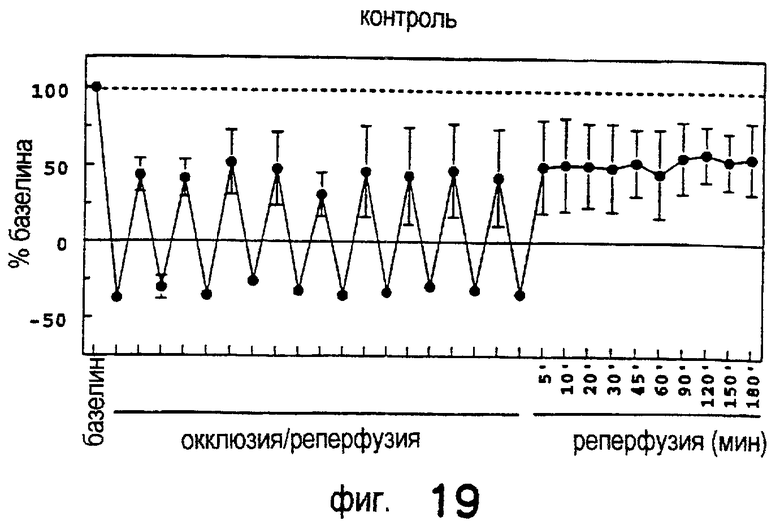
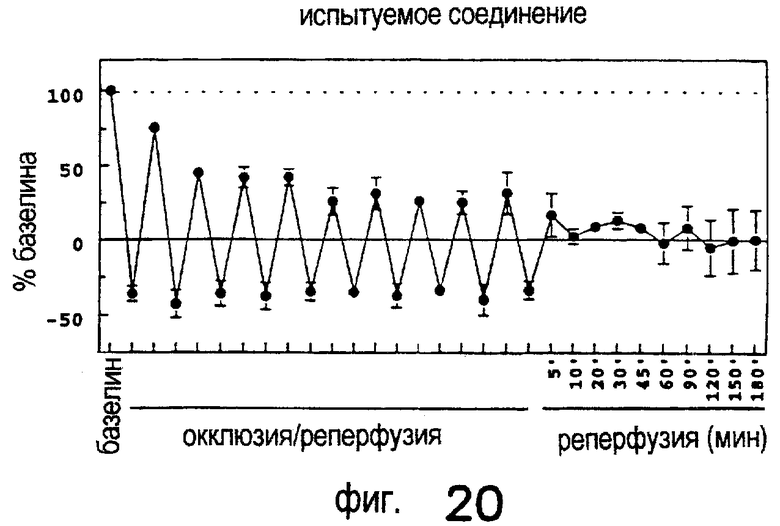
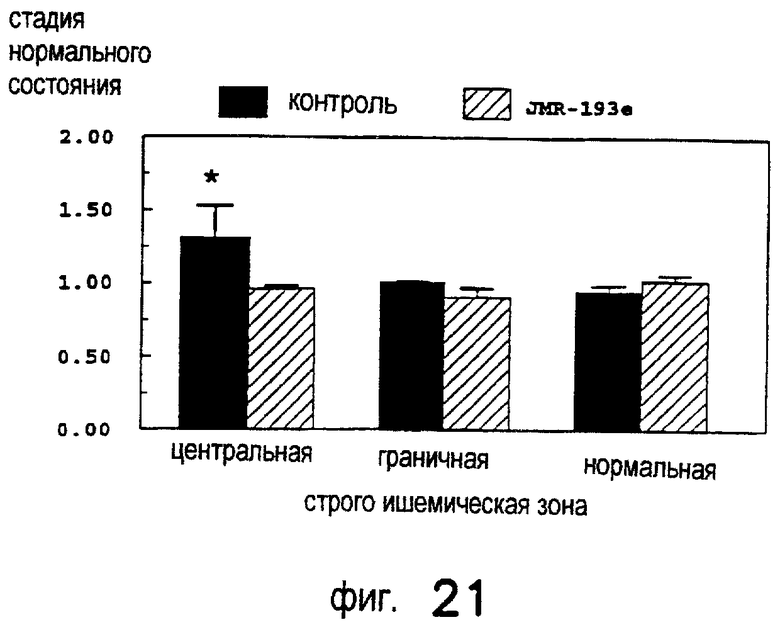
Изобретение относится к соединению формулы (I), где каждый из R независимо представляет водород, C1-C6алкил, С3-С7циклоалкил, фенил или фенил(C1-C3)алкил; Х и X' представляют -CH2OH, -CO2R2, -OC(O)R2, -CH2OC(O)R2 или C(O)NR3R4; R2, R3 и R4 независимо представляют Н, C1-6-алкил, необязательно замещенный одним-тремя C1-6-алкокси, C1-6-алкилтио, галогенами, гидрокси, амино, моно(C1-6-алкил)амино, ди(C1-6-алкил)амино; Z и Z' независимо представляют (C1-C6)алкил, необязательно прерванный одним-тремя атомами S или непероксидным О, либо отсутствуют; n=1-3, или его фармацевтически приемлемой соли. Соединения по изобретению являются агонистами А2A-рецепторов аденозина и могут использоваться для подавления воспалительной реакции или лечения воспаления. 55 з.п. ф-лы, 1 табл., 21 ил.

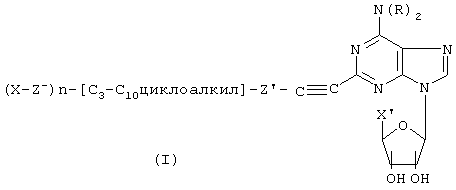
где (а) каждый из R независимо представляет водород, C1-C6алкил, С3-С7циклоалкил, фенил или фенил (С1-С3)алкил;
(b) Х и X' каждый независимо представляет -CH2OH, -CO2R2, -OC(O)R2, -СН2ОС(O)R2 или C(O)NR3R4;
(c) каждый из R2, R3 и R4 независимо представляет Н, C1-6-алкил; или C1-6-алкил, замещенный одной-тремя группами C1-6-алкокси, C1-6-алкилтио, галогена, гидрокси, амино, моно(C1-6-алкил)амино, ди(C1-6-алкил)амино;
(d) Z и Z' независимо представляют (С1-C6)алкил, необязательно прерванный одним-тремя атомами S или непероксидным О, либо отсутствуют, a n равно 1-3;
или его фармацевтически приемлемая соль.
| US 5593975 А, 14.01.1997 | |||
| Baraldi P.G., J | |||
| Med Chem., 1998, 41, р.3174-3185 | |||
| Mager P., Med Chem | |||
| Res., 1998, v.8, №6, p.277-290 | |||
| ПРОИЗВОДНЫЕ 2,6-ДИАМИНОПУРИН-β-D-РИБОФУРАНУРОНАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМСОСТАВ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ ЛЕЙКОЦИТОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ПОДАВЛЕНИЯ АКТИВНОСТИ ЛЕЙКОЦИТОВ | 1994 |
|
RU2129561C1 |

