Изобретение относится к соединениям, которые связываются и модулируют активность нейронных никотиновых ацетилхолиновых рецепторов, к способам получения таких соединений, к фармацевтическим композициям, содержащим такие соединения, и к способам применения таких соединений для лечения различных состояний и расстройств, включая связанные с дисфункцией центральной нервной системы (ЦНС).
Терапевтический потенциал соединений, которые нацелены на нейронные никотиновые рецепторы (NNR), известные также как никотиновые ацетилхолиновые рецепторы (nAChR), являлся предметом нескольких современных обзоров (см. Breining et al., Ann. Rep. Med. Chem. 40: 3 (2005), Hogg and Bertrand, Curr. Drug Targets: CNS Neurol. Disord. 3: 123 (2004), Suto and Zacharias, Expert Opin. Ther. Targets 8: 61 (2004), Dani et al., Bioorg. Med. Chem. Lett. 14: 1837 (2004), Bencherif and Schmitt, Curr. Drug Targets: CNS Neurol. Disord. 1: 349 (2002)). В число различных заболеваний, для лечения которых лиганды NNR были предложены в качестве терапевтического средства, входят когнитивные расстройства, включая болезнь Альцгеймера, синдром дефицита внимания и шизофрения (Newhouse et al., Curr. Opin. Pharmacol. 4: 36 (2004), Levin and Rezvani, Curr. Drug Targets: CNS Neurol. Disord. 1: 423 (2002), Graham et al., Curr. Drug Targets: CNS Neurol. Disord. 1: 387 (2002), Ripoll et al., Curr. Med. Res. Opin. 20(7): 1057 (2004), и McEvoy and Allen, Curr. Drug Targets: CNS Neurol. Disord. 1: 433 (2002)); боль и воспаление (Decker et al., Curr. Top. Med. Chem. 4(3): 369 (2004), Vincler, Expert Opin. Invest. Drugs 14(10): 1191 (2005), Jain, Curr. Opin. Inv. Drugs 5: 76 (2004), Miao et al., Neuroscience 123: 777 (2004)); депрессия и тревога (Shytle et al., Mol. Psychiatry 7: 525 (2002), Damaj et al., Mol. Pharmacol. 66: 675 (2004), Shytle et al., Depress. Anxiety 16: 89 (2002)); нейродегенерация (O'Neill et al., Curr. Drug Targets: CNS Neurol. Disord. 1: 399 (2002), Takata et al., J. Pharmacol. Exp. Ther. 306: 772 (2003), Marrero et al., J. Pharmacol. Exp. Ther. 309: 16 (2004)); болезнь Паркинсона (Jonnala and Buccafusco, J. Neurosci. Res. 66: 565 (2001)); наркомания (Dwoskin and Crooks, Biochem. Pharmacol. 63: 89 (2002), Coe et al., Bioorg. Med. Chem. Lett. 15(22): 4889 (2005)); ожирение (Li et al., Curr.Top.Med.Chem. 3: 899 (2003)); и синдром Туретта (Sacco et al., J. Psychopharmacol. 18(4):457 (2004), Young et al., Clin. Ther. 23(4):532 (2001)).
Ограничение применения некоторых никотиновых соединений связано с тем, что с ними связаны различные нежелательные побочные эффекты, например, вызванные стимулированием мышечных и ганглиозных рецепторов. По этой причине желательно иметь в распоряжении соединения, композиции и способы профилактики и/или лечения различных состояний или расстройств (например, расстройств ЦНС), включая облегчение симптомов таких расстройств, где соединения будут демонстрировать характерную для никотиновых соединений фармакологию с благоприятными эффектами (например, влиянием на деятельность ЦНС), но без значительных сопряженных побочных эффектов. Далее, весьма желательно разработать соединения, композиции и способы, которые будут оказывать влияние на деятельность ЦНС, при отсутствии значительного влияния на рецепторы тех подтипов, которые потенциально могут вызывать нежелательные побочные эффекты (например, заметную активность в сердечно-сосудистой системе и скелетных мышцах). Настоящее изобретение относится к таким соединениям, композициям и способам.
Настоящее изобретение относится к определенным амидным соединениям, которые могут быть получены из некоторых гетероарилсодержащих карбоновых кислот и определенных диазабициклоалканов, в частности 3,7-диазабицикло[3.3.0]октана и 3,7-диазабицикло[3.3.1]нонана. Такие амидные соединения с высокой аффинностью связывания в отношении обнаруженных в ЦНС NNR подтипа α4β2 и демонстрируют селективность в отношении NNR подтипа α4β2 по сравнению с NNR подтипа α7, также обнаруженными в ЦНС. Кроме того, настоящее изобретение относится к фармацевтически приемлемым солям, получаемым из данных соединений, и их фармацевтическим композициям, которые могут применяться для лечения и/или профилактики широкого спектра состояний и расстройств, и, в частности, расстройств, характеризуемых нарушением никотиновой холинэргической нейротрансмиссии или дегенерацией никотиновых холинэргических нейронов. Помимо этого, изобретение относится к способам лечения и/или профилактики расстройств, например расстройств ЦНС, а также лечения некоторых состояний (например, облегчения боли и воспаления) у млекопитающих, нуждающихся в таком лечении. Данные способы включают введение субъекту терапевтически эффективного количества указанных соединений (включая их соли) или фармацевтических композиций, содержащих указанные соединения. Далее, настоящее изобретение обеспечивает способ лечения расстройств, выбранных из группы, состоящей из возрастного ухудшения памяти, умеренных когнитивных нарушений, предстарческой деменции (раннего начала болезни Альцгеймера), старческой деменции (деменции типа Альцгеймера), деменции с тельцами Леви, сосудистой деменции, болезни Альцгеймера, инсульта, комплекса СПИД-деменция, синдрома дефицита внимания, гиперактивного расстройства с дефицитом внимания, дислексии, шизофрении, шизофрениформного расстройства и шизоаффективного расстройства. Кроме того, обеспечен способ лечения расстройств, выбранных из группы, состоящей из деменции типа Альцгеймера в степени от легкой до умеренной, синдрома дефицита внимания, легких когнитивных нарушений и возрастного ухудшения памяти.
Фармацевтические композиции содержат соединение по настоящему изобретению, которое при применении в эффективных количествах взаимодействует с соответствующими сайтами никотиновых рецепторов субъекта и благодаря этому действует как терапевтическое средство для лечения и профилактики широкого спектра состояний и расстройств. Описываемые фармацевтические композиции приносят терапевтическую пользу индивидуумам, страдающим указанными заболеваниями и демонстрирующим клинические проявления указанных заболеваний, в том отношении, что, в случае применения в эффективных количествах, могут: (i) демонстрировать никотиновую фармакологию и воздействовать на соответствующие сайты никотиновых рецепторов (например, действовать в качестве фармакологических агонистов, активируя никотиновые рецепторы) и/или (ii) вызывать секрецию нейротрансмиттеров и вследствие этого предотвращать и подавлять симптомы, связанные с указанными заболеваниями. Помимо этого, указанные соединения обладают потенциальной возможностью (i) увеличивать количество никотиновых холинэргических рецепторов в мозге пациента, (ii) демонстрировать нейропротективное действие и/или (iii) в случае применения в эффективных количествах не вызывать заметных вредных побочных эффектов (например, заметного увеличения кровяного давления и частоты сердцебиений, заметного неблагоприятного влияния на желудочно-кишечный тракт и заметного действия на скелетные мышцы). Считается, что фармацевтические композиции, содержащие соединения по настоящему изобретению, являются безопасными и эффективными при профилактике и лечении широкого спектра состояний и расстройств.
Указанные выше и другие аспекты настоящего изобретения детально изложены в подробном описании изобретения и примерах, приведенных ниже по тексту.
Фиг.1 представляет собой график, показывающий результаты исследования по распознаванию объектов крысами, которым перорально вводили N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан. Результаты показаны в виде зависимости коэффициента распознавания (%) от дозы (мг/кг).
Фиг.2 представляет собой график, показывающий результаты исследования по распознаванию объектов крысами, которым перорально вводили N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан. Результаты показаны в виде зависимости коэффициента распознавания (%) от дозы (мг/кг).
Ниже приведено подробное описание подтипа селективных соединений, фармацевтических композиций, содержащих указанные соединения, способов получения указанных соединений, а также способов лечения и/или профилактики с применением указанных соединений.
Более глубокое понимание описанных в настоящем описании соединений и способов будет достигнуто при обращении к следующим предпочтительным вариантам осуществления.
При определении объема настоящего изобретения будут применяться следующие определения.
В данном описании, если не указано иное, термин «алкил» включает алкильные группы как с линейными, так и с разветвленными цепями. В их число могут входить, но, не ограничиваясь ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил или изогексил. Таким образом, термин «C1-4алкил» охватывает алкильные группы, содержащие от 1 до 4 атомов углерода, включая, но, не ограничиваясь ими, метил, этил, н-пропил, изопропил или трет-бутил.
Как использовано в настоящем описании, если не указано иное, термин «циклоалкил» относится к необязательно замещенной, частично или полностью насыщенной моноциклической, бициклической или мостиковой углеводородной кольцевой системе. Термин «C3-8циклоалкил» может представлять собой, но, не ограничиваясь ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Как использовано в настоящем описании, гетероциклические радикалы являются 3-10-членными, включая один или несколько гетероатомов, выбранных из кислорода, серы и азота. Примеры подходящих гетероциклических фрагментов включают, но, не ограничиваясь ими, пиперидинил, морфолинил, пирролидинил, имидазолидинил, пиразолидинил, изотиазолидинил, тиазолидинил, изоксазолидинил, оксазолидинил, пиперазинил, оксанил (тетрагидропиранил) и оксоланил (тетрагидрофуранил).
Как использовано в настоящем описании, радикалы C1-6алкокси содержат от 1 до 6 атомов углерода в линейной или разветвленной цепи, а также включают C3-6циклоалкокси радикалы и алкокси радикалы, которые содержат C3-6циклоалкильные фрагменты. Примеры включают, не ограничиваясь указанными, метокси, этокси, пропокси, изопропокси, бутокси, трет-бутокси, изобутокси, циклопропилметокси, аллилокси или пропаргилокси.
Как использовано в настоящем описании, термин «ароматический» относится к 3-10, предпочтительно 5- или 6-членным ароматическим и гетероароматическим кольцам.
Как использовано в настоящем описании, термин «фрагменты, содержащие ароматические группы» относится к фрагментам, которые представляют собой или включают ароматические группы. Соответственно, в данное определение включены фенильная и бензильная группы, поскольку каждая из них является или включает ароматическую группу, в данное определение включены пиридинильная и пиримидинильная группы, поскольку обе являются гетероароматическими, т.е. подгруппой ароматических.
Как использовано в настоящем описании, арильные радикалы выбраны из фенила, нафтила и инденила.
Как использовано в настоящем описании, гетероарильные радикалы являются 3-10-членными, предпочтительно 5-6-членными, включая один или несколько гетероатомов, выбранных из кислорода, серы и азота. Примеры подходящих 5-членных кольцевых гетероарильных фрагментов включают фуранил, тиенил, пирролил, имидазолил, оксазолил, тиазолил, изоксазолил, изотиазолил, оксадиазолил, тетразолил, триазолил и пиразолил. Примеры подходящих 6-членных гетероарильных фрагментов включают пиридинил, пиримидинил, пиразинил и пиридазинил. Примеры 9-членных гетероарильных групп включают бензимидазолил, индолизинил, индолил, пуринил и индолинил. Примеры 10-членных гетероарильных групп включают хинолинил и изохинолинил.
Следует иметь в виду, что в тексте описания число и природа заместителей на кольцах соединений по настоящему изобретению должны быть выбраны таким образом, чтобы избежать стерически неприемлемых комбинаций.
Названия некоторых соединений по настоящему изобретению были генерированы с помощью компьютерной программы (ACDLabs 8.0/Name (IUPAC)).
Примеры подходящих фармацевтически приемлемых солей включают аддитивные соли неорганических кислот, например хлориды, бромиды, сульфаты, фосфаты и нитраты; аддитивные соли органических кислот, такие как ацетаты, галактараты, пропионаты, сукцинаты, лактаты, гликоляты, малаты, тартраты, цитраты, малеаты, фумараты, метансульфонаты, п-толуолсульфонаты и аскорбаты; соли с кислыми аминокислотами, такими как аспартаты и глутаматы; соли щелочных металлов, такие как соли натрия и соли калия; соли щелочноземельных металлов, такие как соли магния и соли кальция; аммониевые соли; соли органических оснований, такие как соли триметиламина, соли триэтиламина, соли пиридина, соли пиколина, соли дициклогексиламина и соли N,N'-дибензилэтилендиамина; а также соли с основными аминокислотами, такие как соли лизина и аргинина. В некоторых случаях указанные соли могут являться гидратами или сольватами, например производными этанола. Репрезентативные соли получают, как описано в патентах США № 5597919, на имя Dull и соавторов, 5616716, на имя Dull и соавторов, и 5663356, на имя Ruecroft и соавторов.
Соединения формулы (I) и их фармацевтически приемлемые соли могут существовать в сольватированной, например гидратированной, форме, а также в несольватированной форме, и настоящее изобретение охватывает все такие формы.
Как использовано в настоящем описании, термин «агонист» означает вещество, которое стимулирует связывающееся с ним соединение, как правило, рецептор. Стимулирование определяют в контексте конкретного исследования или оно может быть очевидным из литературы, в соответствии с приведенным в данном описании обсуждением, в котором осуществляется сравнение с фактором или веществом, которое принято считать «агонистом» или «антагонистом» конкретного партнера по связыванию, в основном, в сходных обстоятельствах, согласно оценке специалиста в данной области. Стимулирование можно определить как усиление конкретного эффекта или функции, которые вызываются взаимодействием агониста или частичного агониста с партнером по связыванию, и оно может включать аллостерические эффекты.
Как использовано в настоящем описании, «антагонист» представляет собой вещество, которое ингибирует связанное с ним соединение, как правило, рецептор. Ингибирование определяют в контексте конкретного исследования или оно может быть очевидным из литературы, в соответствии с приведенным в данном описании обсуждением, в котором осуществляется сравнение с фактором или веществом, которое принято считать «агонистом» или «антагонистом» конкретного партнера по связыванию, в основном, в сходных обстоятельствах, согласно оценке специалиста в данной области. Ингибирование можно определить как уменьшение конкретного эффекта или функции, которые вызываются взаимодействием антагониста с партнером по связыванию, и оно может включать аллостерические эффекты.
Как использовано в настоящем описании, термины «частичный агонист» или «частичный антагонист» относятся к веществу, которое вызывает соответственно стимулирование или ингибирование своего партнера по связыванию, уровень которого не является в полной мере или полностью агонистическим или антагонистическим, соответственно. Следует понимать, что стимулирование и, следовательно, ингибирование определяется природой любого вещества или категории веществ, определяемых как агонисты, антагонисты или частичные агонисты.
Как использовано в настоящем описании, термин «собственная активность» или «эффективность» относится к определенной степени биологической эффективности связывания с партнером по комплексу. В отношении фармакологии рецепторов, т.е. контекста, в котором следует определять собственную активность или эффективность, она будет зависеть от конкретных обстоятельств связывания с партнером по комплексу, и рассмотрение активности уместно в отношении конкретного биологического результата. Например, в некоторых ситуациях собственная активность может изменяться в зависимости от конкретно вовлеченной системы вторичного мессенджера. См. Hoyer, D. and Boddeke, H., Trends Pharmacol. Sci. 14(7): 270-5 (1993). Специалисту в данной области будет понятно, уместны ли такие специфические оценки в тех или иных конкретных обстоятельствах и как они могут соотноситься в контексте настоящего изобретения.
Как использовано в настоящем описании, модулирование рецептора включает агонизм, частичный агонизм, антагонизм, частичный антагонизм или обратный агонизм в отношении рецептора.
Как использовано в настоящем описании, нейротрансмиттеры, высвобождение которых опосредуется описанными в данном описании соединениями, включают, но, не ограничиваясь ими, ацетилхолин, дофамин, норэпинефрин, серотонин и глутамат, и описанные в данном описании соединения действуют в качестве модуляторов на NNR подтипа α4β2 ЦНС.
Соединения
Соединения по настоящему изобретению представляют собой амидные соединения, полученные из определенных гетероарильных карбоновых кислот и определенных диазабициклоалканов. Такие соединения могут быть представлены формулой (I):
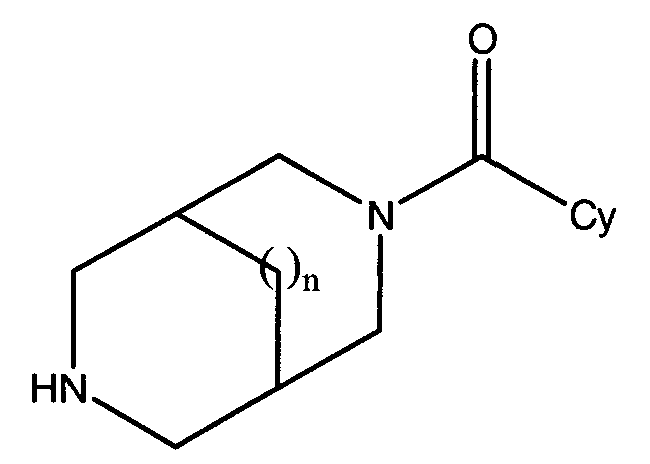
Формула (I)
где n имеет значения 0 или 1, и Cy представляет собой гетероарильную группу, выбранную из таких групп, как 2-фуранил, 3-фуранил, 2-оксазолил, 4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 1,3,4-оксадиазол-2-ил, 1,2,4-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 3-изотиазолил, 4-изотиазолил, 5-изотиазолил, 1,3,4-тиадиазол-2-ил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил и 4-пиридинил, где указанные гетероарильные группы необязательно замещены вплоть до 3 заместителями, отличными от водорода, независимо выбранными из C1-6алкила, замещенного C1-6алкила, C2-6алкенила, замещенного C2-6алкенила, C2-6алкинила, замещенного C2-6алкинила, C3-8гетероциклила, замещенного C3-8гетероциклила, C3-8циклоалкила, замещенного C3-8циклоалкила, C5-10арила, C5-10гетероарила, замещенного C5-10арила, замещенного C5-10гетероарила, C1-6алкил-C5-10арила, C1-6алкил-C5-10гетероарила, замещенного C1-6алкил-C5-10арила, замещенного C1-6алкил-C5-10гетероарила, C5-10арил-C1-6алкила, C5-10гетероарил-C1-6алкила, замещенного C5-10арил-C1-6алкила, замещенного C5-10гетероарил-C1-6алкила, галогена, -OR', -NR'R”, -CF3, -CN, -NO2, -C2R', -SR', -N3, -C(=O)NR'R”, -NR'C(=O)R”, -C(=O)R', -C(=O)OR', -OC(=O)R', -OC(=O)NR'R”, -NR'C(=O)OR”, -SO2R', -SO2NR'R” и -NR'SO2R”, где R' и R” независимо выбраны из водорода, C1-6алкила, C3-8циклоалкила, C3-8гетероциклила, C5-10арила, C5-10гетероарила или C5-10арил-C1-6алкила, или R' и R”, а также атомы, к которым они присоединены, вместе могут образовывать C3-8гетероциклическое кольцо, где термин «замещенный», примененный в отношении алкила, алкенила, алкинила, гетероциклила, циклоалкила, арила, гетероарила, алкиларила, алкилгетероарила, арилалкила и гетероарилалкила, относится к замещению одним или несколькими алкилами, арилами, гетероарилами, галогенами, группами -OR' и -NR'R”,
или их фармацевтически приемлемые соли.
Один из вариантов осуществления изобретения относится к соединениям формулы (I), где n имеет значения 0 или 1, и Cy представляет собой C5-10гетерооарильную группу, выбранную из таких групп, как 2-фуранил или 3-фуранил, 2-оксазолил, 4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 1,3,4-оксадиазол-2-ил, 1,2,4-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 3-изотиазолил, 4-изотиазолил, 5-изотиазолил, 1,3,4-тиадиазол-2-ил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил и 4-пиридинил, где указанные гетероарильные группы необязательно замещены вплоть до 3 заместителями, отличными от водорода, независимо выбранными из C1-6алкила, замещенного C1-6алкила, галогена и C2-6алкинила, замещенного фенилом.
В одном из вариантов осуществления n равно 0. В другом варианте осуществления n равно 1. В еще одном варианте осуществления Cy представляет собой 2-фуранил. В еще одном варианте осуществления Cy представляет собой 2-фуранил, замещенный галогеном. В одном из вариантов осуществления Cy представляет собой 2-фуранил, замещенный хлором. В еще одном варианте осуществления n равно 0, и Cy представляет собой 2-фуранил, необязательно замещенный галогеном. В одном из вариантов осуществления n равно 1, и Cy представляет собой 2-фуранил, необязательно замещенный галогеном. В еще одном варианте осуществления, 2-фуранил замещен по положению 5. В другом варианте осуществления R' и R” независимо выбраны из метила, этила, н-пропила, изопропила, н-бутила, изобутила, втор-бутила или трет-бутила. В другом варианте осуществления R' и R” независимо выбраны из фенила или бензила.
В некоторых случаях соединения по настоящему изобретению являются хиральными. Настоящее изобретение включает все энантиомерные или диастереомерные формы указанных соединений.
Примеры конкретных соединений по настоящему изобретению включают следующие соединения:
N-(фуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(3-метилфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(5-метилфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(3-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(3-бромфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(5-бромфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(4-фенилфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(5-(2-пиридинил)фуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(5-(фенилэтинил)фуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(фуран-3-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(оксазол-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(оксазол-4-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(оксазол-5-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(изоксазол-3-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(изоксазол-4-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(изоксазол-5-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(3-бромизоксазол-5-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(3-метоксиизоксазол-5-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(1,2,4-оксадиазол-3-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(1,2,4-оксадиазол-5-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(1,3,4-оксадиазол-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(тиазол-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(тиазол-4-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(тиазол-5-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(изотиазол-3-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(изотиазол-4-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(изотиазол-5-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(1,2,4-тиадиазол-3-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(1,2,4-тиадиазол-5-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(1,3,4-тиадиазол-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
N-(пиридин-4-илкарбонил)-3,7-диазабицикло[3.3.0]октан,
а также их фармацевтически приемлемые соли.
Примеры соединений по настоящему изобретению также включают следующие соединения:
N-(фуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(3-метилфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(5-метилфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(3-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(3-бромфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(5-бромфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(4-фенилфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(5-(2-пиридинил)фуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(5-(фенилэтинил)фуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(фуран-3-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(оксазол-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(оксазол-4-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(оксазол-5-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(изоксазол-3-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(изоксазол-4-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(изоксазол-5-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(3-бромизоксазол-5-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(3-метоксиизоксазол-5-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(1,2,4-оксадиазол-3-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(1,2,4-оксадиазол-5-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(1,3,4-оксадиазол-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(тиазол-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(тиазол-4-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(тиазол-5-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(изотиазол-3-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(изотиазол-4-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(изотиазол-5-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(1,2,4-тиадиазол-3-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(1,2,4-тиадиазол-5-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(1,3,4-тиадиазол-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
N-(пиридин-4-илкарбонил)-3,7-диазабицикло[3.3.1]нонан,
а также их фармацевтически приемлемые соли.
Один из вариантов осуществления относится к такому соединению, как N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан или его фармацевтически приемлемым солям. Другой вариант осуществления относится к такому соединению, как N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан или его фармацевтически приемлемым солям.
Получение соединений
Соединения по настоящему изобретению могут быть получены путем сочетания монозащищенного диазабицикла (т.е. диазабицикла, в котором одна из двух функциональных аминогрупп приведена в нереакционноспособное состояние путем преобразования в подходящее производное) с подходящим образом функционализированным хлорангидридом гетероарилсодержащей кислоты, группы или другим реакционноспособным производным такой карбоновой кислоты.
Существует большое число способов получения монозащищенных диазабициклов, применяемых для получения соединений по настоящему изобретению. Способы синтеза подходящим образом защищенного 3,7-диазабицикло[3.3.0]октана описаны в PCT WO 02/070523 Colon-Cruz и соавторами, а также в патентной заявке США 2006/0019985 Zhenkun и соавторами, в которых N-бензилмалеинимид конденсируют либо с параформальдегидом и N-бензилглицином, или с N-(метоксиметил)-N-(триметилсилилметил)бензиламином, с получением 3,7-дибензил-3,7-диазабицикло[3.3.0]октан-2,4-диона (известного также как 2,5-дибензилтетрагидропирроло[3,4-c]пиррол-1,3-дион). Последующее преобразование полученного промежуточного продукта может проходить несколькими путями. В одном случае обработка α-хлорэтилхлорформиатом приводит к получению 3-бензил-3,7-диазабицикло[3.3.0]октан-2,4-диона (известного также как 2-бензилтетрагидропирроло[3,4-c]пиррол-1,3-дион), который затем последовательно восстанавливают (используя комплекс боран-диметилсульфид), преобразовывают в N-(трет-бутоксикарбонильное) производное и гидрируют (для удаления второй бензильной группы). Это приводит к получению N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октана, который можно применять для сочетания с карбоновыми кислотами и их производными, для получения соединений по настоящему изобретению. Альтернативно, 3,7-дибензил-3,7-диазабицикло[3.3.0]октан-2,4-дион может быть восстановлен (литийалюминийгидридом), частично гидрирован (для удаления одной бензильной группы), преобразован в N-(трет-бутоксикарбонильное) производное и гидрирован (для удаления второй бензильной группы), с получением N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октана. Другие способы введения и удаления бензильной, трет-бутоксикарбонильной и других групп для защиты аминогруппы хорошо известны специалистам в данной области и подробно описаны в T.W.Greene and P.G. M.Wuts, Protective Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, New York (1999).
Альтернативный способ получения N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октана был описан в заявках на патент США 2004/0186107 Schrimpf и соавторами, и 2005/0101602 Basha и соавторами и включает конденсацию малеинимида и N-(метоксиметил)-N-(триметилсилилметил)бензиламина, с получением 7-бензил-3,7-диазабицикло[3.3.0]октан-2,4-диона (также известного как 5-бензилтетрагидропирроло[3,4-c]пиррол-1,3-дион). Последующая обработка восстанавливающим реагентом (например, литийалюминийгидридом) приводит к получению 3-бензил-3,7-диазабицикло[3.3.0]октана, свободная аминогруппа которого может быть защищена трет-бутоксикарбонильной группой, с последующим удалением бензильной защитной группы гидрогенолизом.
В качестве альтернативы малеинимидам в описанных реакциях конденсации могут быть использованы эфиры малеиновой кислоты. Так, например, согласно PCT WO 96/007656, на имя Schaus и соавторов, конденсация N-бензилглицина с параформальдегидом и диметилмалеатом приводит к получению диметилового эфира N-бензил-цис-3,4-пирролидиндикарбоновой кислоты. Полученное соединение может быть восстановлено, например, литийалюминийгидридом, с получением диола, который может быть затем подвергнут взаимодействию с метансульфонилхлоридом в присутствии триэтиламина, с получением соответствующего димезилата. Дальнейшая обработка аммиаком и нагревание приводит к получению N-бензил защищенного 3,7-диазабицикло[3.3.0]октана. Как указанно выше, полученное соединение может быть преобразовано в N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октан.
Для получения соединений по настоящему изобретению могут быть использованы подходящие производные 3,7-диазабицикло[3.3.1]нонана (биспидина). Одним из таких производных является N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.1]нонан, который можно получить целым рядом способов. Один из синтезов осуществляется через получение промежуточного N-бензил-N'-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.1]нонана, описанного Stead и соавторами в Org.Lett. 7: 4459 (2005). Так, реакция Манниха между N-(трет-бутоксикарбонил)пиперидин-4-оном, бензиламином и параформальдегидом приводит к получению N-бензил-N'-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.1]нонан-9-она, который затем можно последовательно обработать п-толуолсульфонгидразином и боргидридом натрия (для удаления карбонильного кислорода), с получением N-бензил-N'-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.1]нонана. Бензильную группу можно удалить, как описано выше, с получением N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.1]нонана. Альтернативный способ синтеза диазабицикло[3.3.1]нонанов, подходящих для преобразования либо в N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.1]нонан, либо другие монозамещенные производные, был описан Jeyaraman и Avila в Chem.Rev. 81(2):149-174(1981) и в патенте США 5468858, на имя Berlin и соавторов.
Одним из способов получения амидов по настоящему изобретению является сочетание N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октана или N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.1]нонана с подходящим образом функционализированными карбоновыми кислотами, с последующим удалением трет-бутоксикарбонильной защитной группы. Многие из таких карбоновых кислот коммерчески доступны, и другие могут быть легко получены по методикам, известным специалистам в данной области. Конденсация амина и карбоновой кислоты с образованием амида, как правило, требует использования походящего активирующего реагента, такого как, например, N,N'-дициклогексилкарбодиимид (DCC), гексафторфосфат (бензотриазол-1-илокси)трис(диметиламино)фосфония (BOP), гексафторфосфат (бензотриазол-1-илокси)трипирролидинофосфония (PyBOP), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-бис(тетраметилен)урония (HBPyU), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HBTU), тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU) или (1-этил-3-(3-диметиламинопропил)карбодиимид) (EDCI), с 1-гидроксибензотриазолом (HOBt). Другие активирующие реагенты хорошо известны специалисту в данной области (см., например, Kiso and Yajima, Peptides, pp 39-91, Academic Press, San Diego, CA (1995)).
Альтернативно, амидная связь может быть образована сочетанием монозамещенного диазабицикла с подходящим образом функционализированным хлорангидридом кислоты, который может быть коммерчески доступным или может быть получен из подходящим образом функционализированной карбоновой кислоты. Хлорангидрид кислоты может быть получен обработкой подходящей карбоновой кислоты, в числе прочих реагентов, тионилхлоридом или оксалилхлоридом.
После образования амида, удаление защитной группы (например, трет-бутоксикарбонильной группы) кислотой, водной или безводной, позволяет получить соединения по настоящему изобретению.
Специалисту в области органического синтеза будет понятно, что существует большое число способов получения соединения по настоящему изобретению, которые мечены радиоактивными изотопами и подходят для различных диагностических применений. Так, например, конденсация гетероароматической карбоновой кислоты, меченой 11C или 18F, с N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октаном или N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.1]нонаном с применением описанных выше способов, с последующим удалением трет-бутоксикарбонильной группы, приведет к получению соединения, подходящего для применения в позитронно-эмиссионной томографии.
Способы лечения
Соединения по настоящему изобретению являются модуляторами NNR подтипа α4β2, которые характерны для ЦНС, и могут применяться для профилактики и/или лечения различных состояний или расстройств, включая расстройства ЦНС, у субъектов, имеющих такие состояния или расстройства или восприимчивых к ним, путем модулирования α4β2 NNR. Указанные соединения обладают способностью селективно связываться с NNR подтипа α4β2 и проявлять никотиновую фармакологию (т.е. действовать в качестве агонистов, частичных агонистов, антагонистов и т.п.). Например, соединения по настоящему изобретению при введении в эффективных количествах пациентам, нуждающимся в этом, в определенной степени обеспечивают профилактику прогрессирования расстройств ЦНС (т.е. обеспечивают защитные эффекты), облегчение симптомов расстройств ЦНС и/или уменьшение вероятности повторного наступления расстройства ЦНС.
Соединения по настоящему изобретению могут применяться для лечения и/или профилактики таких типов состояний и расстройств, для которых в качестве терапевтических средств предлагались никотиновые соединения других типов. См., например, ссылки, приведенные выше в разделе описания предпосылок создания изобретения, а также Williams et al., Drug News Perspec. 7(4): 205 (1994), Arneric et al., CNS Drug Rev. 1(1): 1-26 (1995), Arneric et al., Exp. Opin. Invest. Drugs 5(1): 79-100 (1996), Bencherif et al., J. Pharmacol. Exp. Ther. 279: 1413 (1996), Lippiello et al., J. Pharmacol. Exp. Ther. 279: 1422 (1996), Damaj et al., J. Pharmacol. Exp. Ther. 291: 390 (1999); Chiari et al., Anesthesiology 91: 1447 (1999), Lavand'homme and Eisenbach, Anesthesiology 91: 1455 (1999), Holladay et al., J. Med. Chem. 40(28): 4169-94 (1997), Bannon et al., Science 279: 77 (1998) и патенты США №№ 5583140, на имя Bencherif и соавторов, 5597919, на имя Dull и соавторов, 5604231, на имя Smith и соавторов, и 5852041, на имя Cosfort и соавторов, содержание которых включено в настоящее описание посредством ссылки во всей их полноте.
Соединения по настоящему изобретению и их фармацевтические композиции полезны при лечении и/или профилактике ряда расстройств ЦНС, включая нейродегенеративные расстройства, нейропсихиатрические расстройства, неврологические расстройства и зависимости. Данные соединения и их фармацевтические композиции могут найти применение для лечения и/или профилактики когнитивных расстройств (возрастных или других), расстройств функции внимания и деменций (включая расстройства, вызванные инфекционными агентами или метаболическими нарушениями); для обеспечения нейрозащиты; для лечения конвульсий и множественных церебральных инфарктов; для лечения расстройств настроения, навязчивых желаний и склонности к пагубным привычкам; для обеспечения анальгезии; для регулирования воспаления (например, опосредованного цитокинами и ядерным фактором каппа B) и лечения воспалительных расстройств; для достижения облегчения боли; а также для лечения инфекций (в качестве противоинфекционных средств для лечения бактериальных, грибковых и вирусных инфекций). В число расстройств, заболеваний и состояний, для лечения и/или профилактики которых могут применяться соединения и фармацевтические композиции по настоящему изобретению, входят: возрастное ухудшение памяти, легкие когнитивные нарушения, предстарческая деменция (раннее начало болезни Альцгеймера), старческая деменция (деменция типа Альцгеймера), деменция с тельцами Леви, ВИЧ-деменция, сосудистая деменция, болезнь Альцгеймера, удар, комплекс СПИД-деменция, синдром дефицита внимания, гиперактивное расстройство с дефицитом внимания, дислексия, шизофрения, шизофрениформное расстройство, шизоаффективное расстройство, паркинсонизм, включая болезнь Паркинсона, болезнь Пика, хорея Хантингтона, поздняя дискинезия, гиперкинезия, прогрессирующий супрануклеарный паралич, болезнь Крейтцфельдта-Якоба, рассеянный склероз, боковой амиотрофический склероз, эпилепсия, мания, тревога, депрессия, панические расстройства, биполярные расстройства, генерализованное тревожное расстройство, обсессивно-компульсивное расстройство, приступы ярости, синдром Туретта, аутизм, пристрастие к наркотикам и алкоголю, влечение к табаку, ожирение, кахексия, псориаз, волчанка, острый холангит, афтозный стоматит, астма, язвенный колит, воспалительные заболевания кишечника, паучит, вирусный пневмонит и артрит (например, ревматоидный артрит и остеоартрит), эндотоксикоз, сепсис, атеросклероз, идиопатический легочный фиброз и неоплазии.
Преимущество лечения или профилактики заболеваний, расстройств и состояний соединениями по настоящему изобретению заключается в том, что оно проходит без заметных неблагоприятных побочных эффектов (например, значительного увеличения кровяного давления, частоты сердцебиений, значительного отрицательного влияния на желудочно-кишечный тракт и значительного воздействия на скелетные мышцы). Соединения по настоящему изобретению, при применении в эффективных количествах, могут модулировать активность NNR подтипа α4β2 без заметного взаимодействия с подтипами никотиновых рецепторов, которые характерны для ганглий человека (что демонстрируется их незначительной способностью вызывать никотиновую функцию в хромаффинной ткани надпочечников) или скелетной мускулатуры (что демонстрируется их незначительной способностью вызывать никотиновую функцию в клеточных препаратах, экспрессирующих никотиновые рецепторы мышечного типа). Так, например, данные соединения способны лечить и/или предотвращать заболевания, расстройства и состояния, не вызывая при этом значительных побочных эффектов, связанных с активностью ганглиозной и нейромышечной локализации. Таким образом, введение соединений по настоящему изобретению обеспечивает терапевтическое окно, в котором достигается лечение определенных заболеваний, расстройств и состояний, и удается избежать некоторых побочных эффектов. Т.е. эффективная доза такого соединения достаточна для получения желаемого воздействия на заболевание, расстройство или состояние, но недостаточна (т.е. не достигается достаточно высокий уровень) для появления нежелательных побочных эффектов.
Таким образом, настоящее изобретение относится к применению соединения формулы (I) или его терапевтически приемлемой соли в терапии (например, в лечении описанных выше заболеваний).
В еще одном аспекте настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственных средств, предназначенных для применения при лечении расстройств ЦНС (например, описанных выше расстройств, заболеваний или состояний).
В еще одном аспекте изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственных средств, предназначенных для применения при лечении деменции типа Альцгеймера в степени от легкой до умеренной, синдрома дефицита внимания, легких когнитивных нарушений и возрастного ухудшения памяти.
Диагностические применения
Соединения по настоящему изобретению могут быть использованы в диагностических композициях, например зондах, особенно если они модифицированы таким образом, что в них включены подходящие метки. Указанные метки могут использоваться, например, для определения относительного количества и/или функции конкретных рецепторов, особенно рецепторов подтипа α4β2. С этой целью соединения по настоящему изобретению наиболее предпочтительно метят атомами радиоактивных изотопов, такими как 11C, 18F, 76Br, 123I или 125I.
Вводимые соединения могут быть определены с применением обычных способов детектирования, подходящих для используемой метки. Примеры способов детектирования включают позиционную эмиссионную томографию (PET) и однофотонную эмиссионную компьютерную томографию (SPECT). Описанные выше радиоактивные метки полезны при получении изображений в PET (например, 11C, 18F или 76Br) и SPECT (например, 123I), при периоде полураспада примерно 20,4 минут для 11C, примерно 109 минут для 18F, примерно 13 часов для 123I и примерно 16 часов для 76Br. Для визуализации выбранных подтипов рецепторов при не насыщающих концентрациях желательна высокая удельная активность. Вводимые дозировки, как правило, находятся ниже диапазона токсичности и обеспечивают высококонтрастные изображения. Ожидается, что соединения можно будет вводить в нетоксичных количествах. Определение дозировки выполняют способом, известным специалисту в области получения изображений, с использованием радиоактивных меток. См., например, патент США № 5969144, на имя London и соавторов.
Соединения по настоящему изобретению можно вводить с применением известных методик. См., например, патент США № 5969144, на имя London и соавторов. Данные соединения можно вводить в составе композиций, которые содержат другие ингредиенты, например, ингредиенты таких типов, которые применимы при получении диагностических композиций. Соединения, полезные в соответствии с осуществлением настоящего изобретения, наиболее предпочтительно используют в формах, имеющих высокую чистоту. См. патент США № 5853696, на имя Elmalch и соавторов.
После того, как соединение введено субъекту (например, человеку), наличие указанного соединения в организме субъекта может быть отображено и количественно определено с помощью подходящих методик для индикации наличия, количества и функциональности выбранных подтипов NNR. Помимо людей, данные соединения также можно вводить животным, например мышам, крысам, собакам и обезьянам. Способы SPECT и PET могут быть реализованы с использованием любых подходящих методик и аппаратуры. См. Villemagne et al., In: Arneric et al. (Eds.) Neuronal Nicotinic Receptors: Pharmacology and Therapeutic Opportunities, 235-250 (1998) и патент США № 5853696, на имя Elmalch и соавторов, для ознакомления с типовыми методиками формирования изображения.
Радиоактивно меченные соединения с высокой аффинностью связывания в отношении определенных подтипов NNR (например, α4β2) и предпочтительно демонстрируют незначительное неспецифическое связывание с другими подтипами никотиновых холинэргических рецепторов (например, рецепторами таких подтипов, которые связаны с мышцами и ганглиями). Соединения по настоящему изобретению как таковые могут применяться в качестве средства для неинвазивного формирования отображения распределения некоторых подтипов никотиновых холинэргических рецепторов в организме субъекта, в частности в мозге, для диагностики различных заболеваний и расстройств ЦНС.
В одном из аспектов диагностические композиции по настоящему изобретению могут применяться в способе диагностики заболевания у субъекта, например у человека. Данный способ включает введение пациенту соединения, меченного обнаруживаемой меткой, как описано в настоящем описании, и детектирование связывания данного соединения с выбранными подтипами NNR (например, рецепторами подтипа α4β2). Специалисты в области применения способов диагностики, например PET и SPECT, могут применять описанные в данном описании радиоактивно меченые соединения для диагностики широкого спектра состояний и расстройств, включая состояния и расстройства, связанные с дисфункцией центральной и автономной нервных систем. Такие расстройства включают широкий спектр заболеваний и расстройств ЦНС, включая болезнь Альцгеймера, болезнь Паркинсона и шизофрению. Эти и другие конкретные заболевания и расстройства, которые можно исследовать приведенным способом, включают заболевания и расстройства, приведенные в патенте США № 5952339, на имя Bencherif и соавторов.
В другом аспекте описанные диагностические композиции могут применяться в способе, предназначенном для наблюдения за определенными подтипами никотиновых рецепторов в организме субъекта, например человека. Данный способ включает введение пациенту описанных в данном описании соединений, меченных обнаруживаемой меткой, и детектирование связывания данных соединений в отношении никотиновых рецепторов выбранного подтипа (например, рецепторов подтипа α4β2).
Фармацевтические композиции
Согласно одному из вариантов осуществления настоящего изобретения, предоставлена фармацевтическая композиция, содержащая в качестве активного ингредиента терапевтически эффективное количество соединения по настоящему изобретению в комбинации с одним или несколькими фармацевтически приемлемыми разбавителями, эксципиентами и/или инертными носителями.
Способ, которым вводятся соединения по настоящему изобретению, может меняться. Указанные композиции предпочтительно вводят перорально (например, в виде жидкой формы, содержащей растворитель, например водную, или неводную жидкость, или в твердом носителе). Предпочтительные композиции для перорального введения включают пилюли, таблетки, капсулы, каплеты, сиропы и растворы, включая твердые желатиновые капсулы и капсулы с высвобождением по времени. Композиции могут быть получены в виде дозированных форм, включающих одну дозу или содержащих активный ингредиент в количестве больше или меньше одной дозы. Предпочтительно композиции получают в жидкой или полутвердой форме. Могут применяться композиции, содержащие жидкий фармацевтически инертный носитель, такой как вода или другие фармацевтически совместимые жидкости или полутвердые вещества. Применение таких жидкостей и полутвердых веществ хорошо известно специалистам в данной области.
Соединения по настоящему изобретению можно также вводить с помощью инъекции, например, внутривенно, внутримышечно, подкожно, интраперитонеально, внутриартериально, интратекально и интрацеребровентрикулярно. Внутривенное введение является предпочтительным способом инъекции. Подходящие носители для инъекции хорошо известны специалисту в данной области и включают 5% растворы декстрозы, физиологический солевой раствор и забуференный фосфатом солевой раствор. Соединения могут вводиться также с помощью инфузии или инъекции (например, в виде суспензии или эмульсии в фармацевтически приемлемой жидкости или смеси жидкостей).
Композиции по настоящему изобретению можно вводить также с применением других способов, например ректального введения. Композиции, применимые для ректального введения, например суппозитории, хорошо известны специалистам в данной области. Соединения по настоящему изобретению можно вводить также путем ингаляции (например, в форме аэрозоля, либо назально, либо с применением средств доставки, описанных в патенте США № 4922901, на имя Brooks и соавторов, содержание которого включено в настоящее описание во всей своей полноте); местно (например, в форме лосьона); трансдермально (например, с применением трансдермального пластыря, используя технические решения, предлагаемые Novartis и Alza Corporation, или инъекцией порошка); или с помощью буккальной, сублингвальной или интраназальной абсорбции. Хотя имеется возможность вводить описываемые соединения в форме чистого активного соединения, предпочтительно использовать каждое соединение в форме фармацевтической композиции или состава для успешного и эффективного введения.
Примеры способов введения соединений по настоящему изобретению должны быть очевидны специалисту в данной области. Применимость композиций может зависеть от конкретной применяемой композиции и конкретного субъекта, получающего лечение. Например, композиции могут вводиться в форме таблеток, твердых желатиновых капсул или в форме капсул с отсроченным по времени высвобождением. Такие композиции могут содержать жидкий носитель, который может быть масляным, водным, эмульсионным или содержать определенные растворители, подходящие для данного способа введения.
Введение фармацевтических композиций, описанных в настоящем описании, может быть периодическим или производиться в постепенно меняющемся, непрерывном, постоянном или регулируемом режиме теплокровным животным (например, млекопитающим, таким как мыши, крысы, кошки, кролики, собаки, свиньи, коровы или обезьяны); но преимущественно их предпочтительно вводят людям. Кроме того, можно менять время дня, в которое осуществляется введение композиции, и число введений в течение дня.
Подходящей дозировкой соединений по настоящему изобретению является их количество, которое эффективно для предупреждения появления симптомов расстройства или для лечения некоторых симптомов расстройства, которым страдает пациент. Под «эффективным количеством», «терапевтическим количеством» или «эффективной дозой» понимается количество, достаточное для того, чтобы вызвать желаемые фармакологические или терапевтические эффекты, тем самым приводящие к эффективному предупреждению или лечению расстройства. Таким образом, при лечении расстройств ЦНС эффективное количество соединения представляет собой количество, достаточное для прохождения через барьер кровь-мозг субъекта, связывания с сайтами подходящих рецепторов в мозге субъекта и модулирования активности никотиновых рецепторов соответствующих подтипов (например, модулирования секреции нейротрансмиттеров, что приводит к эффективной профилактике или лечению расстройства). Профилактика расстройства проявляется в отсрочке наступления симптомов расстройства. Лечение расстройства проявляется в уменьшении симптомов, связанных с расстройством, или облегчении повторно наступающих симптомов расстройства.
Эффективная дозировка может изменяться в зависимости от таких факторов, как состояние пациента, тяжесть симптомов расстройства, а также способа введения фармацевтической композиции. Для пациентов из числа людей эффективная доза конкретных соединений, как правило, соответствует введению соединения в количестве, достаточном для модулирования относящихся к болезни рецепторов, с целью влияния на высвобождение нейротрансмиттеров (например, дофамина), но это количество должно быть недостаточным для того, чтобы оказать влияние на скелетные мышцы и ганглии в сколько-нибудь заметной степени. Эффективные дозы соединений будут, разумеется, различаться для разных пациентов, но в целом они включают количества, с которых начинается влияние на ЦНС или появляются другие желаемые терапевтические эффекты, но ниже того количества, при котором наблюдается влияние на мышцы и ганглии.
Как правило, для введения эффективной дозы соединений необходимо вводить их в количестве менее 5 мг/кг массы тела пациента. Часто соединения можно вводить в количестве от менее примерно 1 мг/кг массы тела пациента до менее примерно 100 мкг/кг массы тела пациента и иногда примерно от 10 мкг/кг до менее 100 мкг/кг массы тела пациента. Указанные выше эффективные дозировки, как правило, относятся к количеству активного средства, вводимому в виде одной дозы или в виде одной или нескольких доз в течение 24-часового периода. Для людей достижение эффективной дозы соединений по настоящему изобретению может потребовать введения соединения в количестве не менее примерно 1, но не более чем примерно 1000, и часто не более чем примерно 500 мг/24 ч/пациент.
Композиции, полезные в качестве диагностических, могут применяться, как указано в патентах США №№ 5853696, на имя Elmalch и соавторов и 5969144, на имя London и соавторов, содержание которых включено в настоящее описание посредством ссылки. Соединения по настоящему изобретению также можно вводить в составе композиций, которые содержат другие ингредиенты, например, ингредиенты таких типов, которые используются при создании диагностических композиций.
Следующие далее примеры предназначены для иллюстрации настоящего изобретения, и их не следует истолковывать как ограничение объема изобретения. В данных примерах все части и процентные доли приведены по массе, если не указано иное.
Биологические исследования
Пример 1: Связывание радиоактивных лигандов рецепторами nAChR ЦНС подтипа α4β2 nAChR
Получение мембран из коры головного мозга крыс:
Крыс (самок крыс Sprague-Dawley), массой 150-250 г, содержали в условиях 12 ч цикла свет/темнота при свободном доступе к воде и пище, производства PMI Nutrition International, Inc. Животным проводили анестезию 70% CO2 и затем обезглавливали. Мозг животных удаляли и помещали на охлажденную льдом площадку. Кору мозга удаляли и помещали в 20 объемов (масса:объем) ледяного препаративного буфера (137 мМ NaCl, 10,7 мМ KCl, 5,8 мМ KH2PO4, 8 мМ Na2HPO4, 20 мМ HEPES (свободная кислота), 5 мМ йодацетамид, 1,6 мМ EDTA, pH 7,4); добавляли PMSF, растворенный в метаноле до конечной концентрации 100 мкМ, и полученную суспензию гомогенизировали с использованием прибора Polytron. Гомогенат центрифугировали при 18000×g в течение 20 минут при 4°C и полученный осадок ресуспендировали в 20 объемах ледяной воды. Через 60 минут инкубирования на льду, вновь отделяли осадок центрифугированием при 18000×g в течение 20 минут при 4°C. Полученный конечный осадок ресуспендировали в 10 объемах буфера и сохраняли при -20°C.
Получение мембран из SH-EP1/человеческих клональных клеток α4β2:
Клеточный осадок из 40150 мм культуральных чашек объединяли и гомогенизировали с использованием прибора Polytron (Kinematica GmbH, Switzerland) в 20 миллилитрах ледяного препаративного буфера. Гомогенат центрифугировали при 48000 g в течение 20 минут при 4°C. Полученный осадок ресуспендировали в 20 мл ледяного препаративного буфера и сохраняли при -20°C.
В день проведения исследования замороженные мембраны оттаивали и центрифугировали при 48000×g в течение 20 минут. Супернатант отделяли декантацией и отбрасывали. Осадок ресуспендировали в забуференном фосфатном солевом растворе Dulbecco (PBS, Life Technologies) pH 7,4 и гомогенизировали с использованием прибора Polytron в течение 6 секунд. Концентрацию белка определяли с использованием комплекта для анализа белка Pierce BCA, с бычьим сывороточным альбумином в качестве стандарта (Pierce Chemical Company, Rockford, IL).
Препараты мембран (приблизительно 50 мкг для клеток человека и 200-300 мкг белка в случае крысиного α4β2) инкубировали в PBS (50 мкл и 100 мкл. соответственно) в присутствии конкурирующего соединения (от 0,01 нМ до 100 мкМ) и 5 нМ [3H]никотина в течение 2-3 часов на льду. Инкубирование прерывали быстрым фильтрованием на коллекторе для одновременной обработки нескольких образцов (Brandel, Gaithersburg, MD), используя фильтры GF/B, предварительно пропитанные 0,33% полиэтиленимином (масса/объем) для уменьшения неспецифического связывания. Ткань 3 раза промывали PBS, pH 7,4. К фильтрам, содержавшим промытую ткань, добавляли сцинтилляционную жидкость и давали достичь равновесия. Затем фильтры подсчитывали для определения связанной мембранами радиоактивности, используя методики подсчета сцинтилляции жидкостей (2200CA Tri-Carb LSC, Packard Instruments, 50% эффективность, или Wallac Trilux 1450 MicroBeta, 40% эффективность, Perkin Elmer).
Данные выражали в распадах в минуту (DPM). В каждом анализе определение данных для каждой точки повторяли 2-3 раза. Результаты по каждой точке усредняли и наносили на график зависимости логарифма концентрации лекарственного средства. Значение IC50, которое представляет собой концентрацию соединения, обеспечивающую 50% ингибирование связывания, определяли методом нелинейной регрессии наименьших квадратов. Значения Ki рассчитывали с применением уравнения Cheng-Prusoff (1973):
K i =IC 50 /(1+N/Kd),
где N означает концентрацию [3H]никотина, и Kd означает аффинность в отношении никотина (3 нМ, определено в отдельном эксперименте).
Подтип α7 nAChR
Крыс (самок крыс Sprague-Dawley), массой 150-250 г, содержали в условиях 12 ч цикла свет/темнота при свободном доступе к воде и пище, производства PMI Nutrition International, Inc. Животным проводили анестезию 70% CO2 и затем обезглавливали. Мозг животных удаляли и помещали на охлажденную льдом площадку. Гиппокамп удаляли и помещали в 10 объемов (масса:объем) ледяного препаративного буфера (137 мМ NaCl, 10,7 мМ KCl, 5,8 мМ KH2PO4, 8 мМ Na2HPO4, 20 мМ HEPES (свободная кислота), 5 мМ йодацетамид, 1,6 мМ EDTA, pH 7,4); добавляли PMSF, растворенный в метаноле до конечной концентрации 100 мкМ, и полученную суспензию гомогенизировали с использованием прибора Polytron. Гомогенат центрифугировали при 18000×g в течение 20 минут при 4°C и полученный осадок ресуспендировали в 20 объемах ледяной воды. После 60-мин инкубирования на льду, вновь отделяли осадок центрифугированием при 18000×g в течение 20 минут при 4°C. Полученный конечный осадок ресуспендировали в 10 объемах буфера и сохраняли при -20°C. В день исследования ткани оттаивали, центрифугировали при 18000×g в течение 20 минут и затем ресуспендировали в ледяном PBS (забуференный фосфатном солевой раствор Dulbecco, 138 мМ NaCl, 2,67 мМ KCl, 1,47 мМ KH2PO4, 8,1 мМ Na2HPO4, 0,9 мМ CaCl2, 0,5 мМ MgCl2, Invitrogen/Gibco, pH 7,4) до конечной концентрации примерно 2 мг белка/мл. Белок определяли способом Lowry et al., J. Biol. Chem. 193: 265(1951), используя бычий сывороточный альбумин в качестве стандарта.
Связывание [3H]MLA измеряли с применением модификации методики Davies et al., Neuropharmacol. 38: 679 (1999). [3H]MLA (с удельной активностью 25-35 Ки/ммоль) получали у Tocris. Связывание [3H]MLA определяли с помощью 2 ч инкубирования при 21°C. Инкубирование осуществляли в 48-луночных микротитровальных планшетах, содержавших примерно 200 мкг белка на лунку в конечном объеме инкубирования 300 мкл. Буфер для инкубирования представлял собой PBS, и конечная концентрация [3H]MLA составляла 5 нМ. Реакцию связывания прерывали путем фильтрования белка, содержавшего связанный лиганд, на фильтрах из стекловолокна (GF/B, Brandel) с использованием Brandel Tissue Harvester при комнатной температуре. Фильтры смачивали деионизированной водой, содержавшей 0,33% полиэтиленимина для уменьшения неспецифического связывания. Каждый фильтр промывали PBS (3×1 мл) при комнатной температуре. Неспецифическое связывание определяли путем внесения 50 мкМ нерадиоактивного MLA в выбранные лунки.
Ингибирование связывания [3H]MLA тестируемыми соединениями определяли путем введения тестируемых соединений в семи различных концентрациях в выбранные лунки. Введение каждой из концентраций повторяли трижды. Значения IC50 принимали равными концентрации соединений, при которой происходило 50% ингибирование специфического связывания [3H]MLA. Константы ингибирования (значения Ki), выраженные в нМ, рассчитывали исходя из значений IC50, используя методику Cheng et al., Biochem. Pharmacol. 22:3099-3108 (1973).
Пример 2: Определение выделения дофамина
Выделение дофамина измеряли с использованием стриарных синаптосом, полученных из мозга крысы, согласно методикам, описанным Rapier et al., в J. Neurochem. 54: 937(1990). Крыс (самки Sprague-Dawley) массой 150-250 г, содержали в условиях 12 ч цикла свет/темнота при свободном доступе к воде и пище, производства PMI Nutrition International, Inc. Животным проводили анестезию 70% CO2 и затем обезглавливали. Быстро удаляли мозг и иссекали стриатум. Ткани стриатума каждых 2 крыс объединяли и гомогенизировали в ледяном 0,32М растворе сахарозы (5 мл), содержавшем 5 мМ HEPES, pH 7,4, используя гомогенизатор стекло/стекло. Затем полученный препарат ткани центрифугировали при 1000×g в течение 10 минут. Осадок отбрасывали и супернатант центрифугировали при 12000×g в течение 20 минут. Полученный осадок ресуспендировали в перфузионном буфере, содержавшем ингибиторы моноаминоксидазы (128 мМ NaCl, 1,2 мМ KH2PO4, 2,4 мМ KCl, 3,2 мМ CaCl2, 1,2 мМ MgSO4, 25 мМ HEPES, 1 мМ аскорбиновую кислоту, 0,02 мМ паргилин HCl и 10 мМ глюкозу, pH 7,4) и центрифугировали в течение 15 мин при 25000×g. Полученный осадок ресуспендировали в перфузионном буфере (1,4 мл) для немедленного использования.
Суспензию синаптосом инкубировали в течение 10 минут при 37°C для восстановления метаболической активности. Добавляли [3H]дофамин ([3H]DA, удельная активность=28 Ки/ммоль, NEN Research Products) до конечной концентрации 0,1 мМ и суспензию инкубировали при 37°C еще 10 мин. Аликвоту ткани (50 мкл) и перфузионный буфер (100 мкл) помещали в супрафузионные камеры Brandel Suprafusion System (series 2500, Gaithersburg, MD). В камеры прокачивали перфузионный буфер (комнатной температуры) со скоростью 1,5 мл/мин в течение периода промывания, равного 16 мин. Затем в перфузионный поток вводили тестируемое соединение (10 мМ) или никотин (10 мМ) в течение 48 сек. На протяжении эксперимента из каждой камеры непрерывно отбирали фракции (каждая 24 с), для фиксации исходного выделения и максимального выделения, индуцированного агонистом, и для повторной фиксации базового уровня после введения агониста. Перфузионную жидкость собирали непосредственно в сцинтилляционные флаконы, и к ней добавляли сцинтилляционную жидкость. Количество выделившегося [3H]DA определяли подсчетом сцинтилляции. Для каждой камеры интегрированную площадь пика нормализовали относительно его исходного уровня.
Высвобождение выражали в процентах от высвобождения, полученного при действии равной концентрации L-никотина. В каждом исследовании эксперимент с каждым тестируемым соединением повторяли неоднократно, используя 2-3 камеры; результаты повторных экспериментов усредняли. Где это было целесообразно, строили кривые зависимости доза-ответ для тестируемых соединений. Определяли максимальную активацию для индивидуальных соединений (Emax) как процент от максимальной активации, вызванной L-никотином. Также определяли концентрацию соединения, приводившую к половине максимальной активации (EC50) потока специфических ионов.
Пример 3: Селективность по сравнению с периферическими nAChR
Взаимодействие с мышечным подтипом рецепторов nAChR человека
Активацию nAChR мышечных типов устанавливали на примере клональной линии клеток человека TE671/RD, которые получены из эмбриональной рабдомиосаркомы (Stratton et al., Carcinogen 10: 899 (1989)). Такие клетки экспрессируют рецепторы, которые имеют фармакологический (Lukas, J. Pharmacol. Exp. Ther. 251: 175 (1989)), электрофизиологический (Oswald et al., Neurosci.Lett. 96: 207(1989)) и молекулярно-биологический профили (Luther et al., J. Neurosci. 9: 1082(1989)), подобные nAChR мышечных типов.
Клетки TE671/RD поддерживали в фазе пролиферативного роста согласно стандартным протоколам (Bencherif et al., Mol. Cell. Neurosci. 2: 52 (1991) и Bencherif et al., J. Pharmacol. Exp. Ther. 257: 946 (1991)). Клетки культивировали в среде Игла, модифицированной Dulbecco (Gibco/BRL) с 10% лошадиной сывороткой (Gibco/BRL), 5% фетальной телячьей сывороткой (HyClone, Logan UT), 1 мМ пируватом натрия, 4 мМ L-глутамином и 50000 единицами пенициллина-стрептомицина (Irvine Scientific). Когда клетки достигли 80% конфлюентности, их помещали в 12-луночные планшеты из полистирола (Costar). Эксперименты проводили по достижении клетками 100% конфлюентности.
Деятельность никотиновых ацетилхолиновых рецепторов (nAChR) исследовали, используя выделение 86Rb+ согласно методике, описанной Lukas et al., Anal. Biochem. 175: 212 (1988). В день эксперимента осторожно удаляли ростовую среду из лунок и в каждую лунку добавляли ростовую среду, содержащую хлорид рубидия-86 (106 мкКи/мл). Клетки инкубировали при 37°C в течение минимум 3 ч. После периода поглощения изотопа клетками, избыток 86Rb+ удаляли и клетки дважды промывали не содержащим метки забуференным фосфатном солевым раствором Dulbecco (138 мМ NaCl, 2,67 мМ KCl, 1,47 мМ KH2PO4, 8,1 мМ Na2HPO4, 0,9 мМ CaCl2, 0,5 мМ MgCl2, Invitrogen/Gibco, pH 7,4), стараясь не взмутить осадок клеток. Затем клетки подвергали воздействию либо 100 мкМ тестируемого соединения, 100 мкМ L-никотина (Acros Organics) или чистого буфера в течение 4 мин. После такой обработки супернатант, содержавший высвободившийся 86Rb+, удаляли и переносили в сцинтилляционные сосуды. Добавляли сцинтилляционную жидкость, и измеряли высвободившуюся радиоактивность путем подсчета сцинтилляции жидкости.
В каждом из исследований каждый эксперимент повторяли по 2 раза и полученные результаты усредняли. Количество высвободившегося 86Rb+ сравнивали как с положительным контролем (100 мкМ L-никотин), так и с отрицательным контролем (чистый буфер) для определения высвобождения в процентах по отношению к L-никотину.
Когда это было целесообразно, строили кривые зависимости доза-ответ для тестируемых соединений. Определяли максимальную активацию для индивидуальных соединений (Emax) как процент от максимальной активации, вызванной L-никотином. Также определяли концентрацию соединения, приводившую к половине максимальной активации (EC50) потока специфических ионов.
Взаимодействие с ганглиозным подтипом рецепторов nAChR крысы
Активацию ганглиозных крысиных рецепторов nAChR определяли на примере клональной линии PC12 феохромоцитомы, которая является непрерывной клональной клеточной линией из ганглионарной пластинки, полученной из опухоли хромаффинной ткани надпочечника крысы. Данные клетки экспрессируют ганглионоподобные nAChR (см. Whiting et al., Nature 327: 515(1987); Lukas, J. Pharmacol. Exp. Ther. 251: 175 (1989); Whiting et al., Mol. Brain Res. 10: 61 (1990)).
Крысиные клетки PC12 поддерживали в фазе пролиферативного роста согласно стандартным протоколам (Bencherif et al., Mol. Cell. Neurosci. 2: 52 (1991) и Bencherif et al., J. Pharmacol. Exp. Ther. 257: 946 (1991)). Клетки культивировали в среде Игла, модифицированной Dulbecco (Gibco/BRL) с 10% лошадиной сывороткой (Gibco/BRL), 5% фетальной телячьей сывороткой (HyClone, Logan UT), 1 мМ пируватом натрия, 4 мМ L-глутамином и 50000 единицами пенициллина-стрептомицина (Irvine Scientific). Когда клетки достигли 80% конфлюентности, их помещали в 12-луночные планшеты Nunc (Nunclon) и покрывали 0,03% поли-L-лизином (Sigma, растворенным в 100 мМ борной кислоте). Эксперименты проводили по достижении клетками 80% конфлюентности.
Функцию никотиновых ацетилхолиновых рецепторов (nAChR) исследовали, используя выделение 86Rb+ согласно методике, описанной Lukas et al., Anal. Biochem. 175: 212 (1988). В день эксперимента осторожно удаляли ростовую среду из лунок и в каждую лунку добавляли ростовую среду, содержащую хлорид рубидия-86 (106 мкКи/мл). Клетки инкубировали при 37°C в течение минимум 3 ч. После периода поглощения изотопа клетками, избыток 86Rb+ удаляли и клетки дважды промывали не содержащим метки забуференным фосфатном солевым раствором Dulbecco (138 мМ NaCl, 2,67 мМ KCl, 1,47 мМ KH2PO4, 8,1 мМ Na2HPO4, 0,9 мМ CaCl2, 0,5 мМ MgCl2, Invitrogen/Gibco, pH 7,4), стараясь не взмутить осадок клеток. Затем клетки подвергали воздействию 100 мкМ тестируемого соединения, 100 мкМ никотина или чистого буфера в течение 4 мин. После такой обработки, супернатант, содержавший высвободившийся 86Rb+, удаляли и переносили в сцинтилляционные сосуды. Добавляли сцинтилляционную жидкость и измеряли высвободившуюся радиоактивность путем подсчета сцинтилляции жидкости.
В каждом из исследований каждый эксперимент повторяли по 2 раза и полученные результаты усредняли. Количество высвободившегося 86Rb+ сравнивали как с положительным контролем (100 мкМ никотин), так и с отрицательным контролем (чистый буфер) для определения высвобождения в процентах по отношению к L-никотину.
Когда это было целесообразно, строили кривые зависимости доза-ответ для тестируемых соединений. Определяли максимальную активацию для индивидуальных соединений (Emax) как процент от максимальной активации, вызванной L-никотином. Также определяли концентрацию соединения, приводившую к половине максимальной активации (EC50) потока специфических ионов.
Взаимодействие с ганглиозным подтипом рецепторов nAChR человека
Клеточная линия SH-SY5Y является непрерывной линией, полученной последовательным субклонированием родительской клеточной линии, а именно SK-N-SH, которая была первоначально получена из периферической нейробластомы человека. Клетки SH-SY5Y экспрессируют ганглионоподобные nAChR (Lukas et al., Mol. Cell. Neyrosci. 4:1 (1993)).
Человеческие клетки SH-SY5Y поддерживали в фазе пролиферативного роста согласно стандартным протоколам (Bencherif et al., Mol. Cell. Neurosci. 2: 52 (1991) и Bencherif et al., J. Pharmacol. Exp. Ther. 257:946 (1991)). Клетки культивировали в среде Игла, модифицированной Dulbecco (Gibco/BRL) с 10% лошадиной сывороткой (Gibco/BRL), 5% фетальной телячьей сывороткой (HyClone, Logan UT), 1мМ пируватом натрия, 4 мМ L-глутамином и 50000 единицами пенициллина-стрептомицина (Irvine Scientific). Когда клетки достигли 80% конфлюентности, их помещали в 12-луночные планшеты из полистирола (Costar). Эксперименты проводили по достижении клетками 100% конфлюентности.
Функцию никотиновых ацетилхолиновых рецепторов (nAChR) исследовали, используя выделение 86Rb+ согласно методике, описанной Lukas et al., Anal. Biochem. 175: 212 (1988). В день эксперимента осторожно удаляли ростовую среду из лунок и в каждую лунку добавляли ростовую среду, содержащую хлорид рубидия-86 (106 мкКи/мл). Клетки инкубировали при 37°C в течение минимум 3 ч. После периода поглощения изотопа клетками, избыток 86Rb+ удаляли и клетки дважды промывали не содержащим метки забуференным фосфатном солевым раствором Dulbecco (138 мМ NaCl, 2,67 мМ KCl, 1,47 мМ KH2PO4, 8,1 мМ Na2HPO4, 0,9 мМ CaCl2, 0,5 мМ MgCl2, Invitrogen/Gibco, pH 7,4), стараясь не взмутить осадок клеток. Затем клетки подвергали воздействию 100 мкМ тестируемого соединения, 100 мкМ никотина или чистого буфера в течение 4 мин. После такой обработки супернатант, содержавший высвободившийся 86Rb+, удаляли и переносили в сцинтилляционные сосуды. Добавляли сцинтилляционную жидкость и измеряли высвободившуюся радиоактивность путем подсчета сцинтилляции жидкости.
В каждом из исследований каждый эксперимент повторяли по 2 раза и полученные результаты усредняли. Количество высвободившегося 86Rb+ сравнивали как с положительным контролем (100 мкМ никотин), так и с отрицательным контролем (чистый буфер) для определения высвобождения в процентах по отношению к L-никотину.
Если это было целесообразно, строили кривые зависимости доза-ответ для тестируемых соединений. Определяли максимальную активацию для индивидуальных соединений (Emax) как процент от максимальной активации, вызванной L-никотином. Также определяли концентрацию соединения, приводившую к половине максимальной активации (EC50) потока специфических ионов.
Пример 4: Тест по распознаванию новых объектов (NOR)
Тест по распознаванию новых объектов (NOR) осуществляли согласно описанию Ennaceur and Delacour в Behav.Brain Res. 100: 85-92 (1988).
Примеры синтеза
Пример 5: Синтез N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октана
Пример 5 относится к синтезу N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октана, который получали, как описано в заявках на патент США 2004/0186107, на имя Schrimpf и соавторов, и 2005/0101602, на имя Basha и соавторов, по следующей методике:
5-бензилтетрагидропирроло[3,4-c]пиррол-1,3-дион (или 7-бензил-3,7-диазабицикло[3.3.0]октан-2,4-дион)
Трифторуксусную кислоту (ТФУК, 0,50 мл, 6,5 ммоль) в атмосфере азота добавляли к холодному (0°C) раствору малеинимида (6,27 г, 0,0646 моль) в дихлорметане (150 мл). По каплям в течение 45 минут добавляли раствор N-(метоксиметил)-N-(триметилсилилметил)бензиламина (20 г, 0,084 моль) в дихлорметане (100 мл). После того, как добавление было закончено, смесь медленно нагревали до комнатной температуры и перемешивали в течение 16 ч. Смесь концентрировали, полученный остаток растворяли в дихлорметане (200 мл) и промывали насыщенным водным раствором бикарбоната натрия (2×50 мл). Водный слой отделяли и экстрагировали дихлорметаном (2×75 мл). Объединенные дихлорметановые экстракты промывали насыщенным раствором соли (50 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали, получая 12,5 г (выход 83,9%) светло-желтого воскообразного твердого вещества (МС m/z 231 (M+H)).
2-бензилоктагидропирроло[3,4-c]пиррол (или 3-бензил-3,7-диазабицикло[3.3.0]октан)
Неочищенный 5-бензилтетрагидропирроло[3,4-c]пиррол-1,3-дион (4,9 г, 0,021 моль) растворяли в холодном (0°C) сухом тетрагидрофуране (ТГФ) (50 мл) в атмосфере азота и к непрерывно перемешиваемому раствору по каплям в течение 30 мин добавляли литийалюминийгидрид (63 мл 1М в ТГФ, 0,063 моль). Полученную смесь перемешивали при комнатной температуре в течение 30 мин и затем нагревали до кипения с обратным холодильником в течение 4 ч. Затем смесь охлаждали до 0°C и гасили медленным добавлением избытка твердого декагидрата сульфата натрия. Смесь нагревали до комнатной температуры и перемешивали в течение 16 ч. Твердые вещества удаляли фильтрованием, и остаток промывали этилацетатом (3×100 мл). Объединенные фильтраты концентрировали, получая 4,2 г (выход 99%) воскообразного вещества (МС m/z 203 (M+H)).
трет-бутиловый эфир 5-бензилгексагидропирроло[3,4-c]пиррол-2-карбоновой кислоты (или N-бензил-N'-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октан)
Неочищенный 2-бензилоктагидропирроло[3,4-c]пиррол (4,2 г, 0,021 моль) растворяли в ТГФ (50 мл). Добавляли дитрет-бутилдикарбонат (5,5 г, 0,025 моль) и водный насыщенный раствор NaHCO3 (10 мл) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили водой (10 мл) и добавляли этилацетат (30 мл). Водный слой экстрагировали этилацетатом (2×20 мл), объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали. Очистка колоночной хроматографией на силикагеле (1:1 гексан/этилацетат) приводила к получению 5,07 г (выход 79,8%) указанного в заголовке соединения (МС m/z 303 (M+H)).
Трет-бутиловый эфир гексагидропирроло[3,4-c]пиррол-2-карбоновой кислоты (или N-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.0]октан)
Трет-бутиловый эфир 5-бензилгексагидропирроло[3,4-c]пиррол-2-карбоновой кислоты (5,07 г, 0,0168 моль) растворяли в метаноле (50 мл) и в атмосфере азота добавляли 20% Pd(OH)2/C (влажный) (~2 г). Полученную смесь нагревали (45-50°C) и встряхивали в течение 2 ч в атмосфере водорода под давлением 40 фунт/кв.дюйм. Полученную смесь фильтровали и концентрировали, получая 3,49 г (выход 97,7%) указанного в заголовке соединения (МС m/z 213 (M+H)).
Пример 6: Синтез N-(фуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октана
Пример 6 относится к синтезу фуран-2-ил(гексагидропирроло[3,4-c]пиррол-2-ил)метанона (или N-(фуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октана), который получали по следующим методикам, являющимся иллюстративными для реакции сочетания, используемой для получения гетероатомных амидов 3,7-диазабицикло[3.3.0]октана:
Трифторацетат фуран-2-ил(гексагидропирроло[3,4-c]пиррол-2-ил)метанона (или трифторацетат N-(фуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октана)
Фуран-2-карбоновую кислоту (0,037 г, 0,33 ммоль) и триэтиламин (0,125 мл, 0,99 ммоль) смешивали в сухом дихлорметане (1 мл) и добавляли гексафторфосфат O-(бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU; 0,125 г, 0,33 ммоль). Добавляли раствор трет-бутилового эфира гексагидропирроло[3,4-c]пиррол-2-карбоновой кислоты (0,064 г, 0,30 ммоль) в дихлорметане (0,5 мл) и смесь перемешивали при комнатной температуре в течение ночи. Смесь встряхивали с 10% водным раствором гидроксида натрия и отделяли органический слой. Водный слой экстрагировали хлороформом (2×2 мл). Объединенные органические экстракты промывали водой (2×1 мл) и концентрировали. Полученный остаток растворяли в диметилформамиде (ДМФА) (0,3 мл) и очищали ВЭЖХ (градиент смеси ацетонитрил/вода). Фракции, содержащие желаемое вещество, объединяли и концентрировали, оставляя защищенный трет-бутоксикарбонилом продукт. Полученное вещество растворяли в смеси трифторуксусной кислоты (0,5 мл) и дихлорметана (0,5 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Летучие вещества удаляли на роторном испарителе, с последующим выдерживанием в глубоком вакууме, получая 77 мг масла (выход 80%) (1H ЯМР (d4-метанол, 300 МГц) 3,20 (м, 2H), 3,47-4,2 (м, 8H), 6,60 (т, 1H), 7,18 (д, 1H), 7,72 (д, 1H); МС m/z 207 (M+H)).
Пример 7: Синтез трифторацетата N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октана
Пример 7 относится к синтезу трифторацетата 5-хлорфуран-2-ил(гексагидропирроло[3,4-c]пиррол-2-ил)метанона (или трифторацетата N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октана), который получали по следующим методикам, являющимся иллюстративными для реакции сочетания, используемой для получения гетероароматических амидов 3,7-диазабицикло[3.3.0]октана:
5-хлорфуран-2-карбоновая кислота
Водный раствор гидроксида натрия (80 мл с концентрацией 10%) добавляли к раствору нитрата серебра (8,0 г, 47 ммоль) в воде (20 мл). Полученную суспензию перемешивали и медленно обрабатывали 30% водным раствором гидроксида аммония до получения прозрачного раствора. Добавляли раствор 5-хлорфуран-2-карбоксальдегида (3,0 г, 23 ммоль) (Aldrich Chemical) в метаноле (5 мл) и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь фильтровали и фильтрат промывали эфиром (100 мл). Затем водный фильтрат подкисляли (pH ~3) добавлением холодной 20% серной кислоты. Полученную смесь экстрагировали этилацетатом (3×100 мл). Экстракты промывали насыщенным водным раствором хлорида натрия (100 мл), сушили (безводным сульфатом натрия) и концентрировали в вакууме, получая 3,2 г (выход 95%) белого твердого вещества (т.пл. 178-179°C). Данная реакция может быть без труда проведена в большем объеме, и ее неоднократно проводили в масштабе >10 г.
Трифторацетат N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октана
Оксалихлорид (12,2 г, 95,8 ммоль), содержащий каплю ДМФА, по каплям добавляли к ледяному раствору 5-хлорфуран-2-карбоновой кислоты (6,25 г, 47,9 ммоль) в 200 мл дихлорметана. После окончания добавления удаляли баню со льдом и реакционную смесь нагревали до комнатной температуры в течение 1 часа. Затем удаляли летучие вещества в вакууме и остаток растворяли в ТГФ (50 мл). Затем добавляли полученный раствор хлорангидрида кислоты к перемешиваемому ледяному раствору трет-бутилового гексагидропирроло[3,4-c]пиррол-2-каарбоновой кислоты (10,2 г, 47,9 ммоль) и диизопропиламина (25 г, ~4 эквивалентов) в ТГФ (200 мл). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Затем летучие вещества удаляли в вакууме и остаток распределяли между водой (100 мл) и эфиром (300 мл). Эфирный слой и два эфирных экстракта (100 мл) водного слоя концентрировали на роторном испарителе. Остаток подвергали колоночной хроматографии на силикагеле, элюируя 0-60% градиентом этилацетата в гексане. Концентрирование выбранных фракций приводило к получению 13,9 г (выход 85,3%) бледно-желтого сиропа. Часть полученного вещества (12,9 г, 37,9 ммоль) растворяли в смеси дихлорметана и трифторуксусной кислоты (по 100 мл каждого). Полученную смесь перемешивали при комнатной температуре в течение 2 ч и затем концентрировали в вакууме. Остаток распределяли между хлороформом (200 мл) и 50% водным раствором карбоната калия (200 мл) и водный слой экстрагировали хлороформом (3×200 мл). Объединенные хлороформные слои сушили над безводным сульфатом натрия и концентрировали в вакууме, получая 8,66 г (выход 95%) бледно-желтого твердого вещества (1H ЯМР (d4-метанол, 300 МГц) 3,15-3,35 (м, 4H), 3,50-4,20 (м, 6H), 6,51 (д, 1H), 7,17 (д, 1H); МС m/z 241 (M+H)).
Пример 8: Синтез трет-бутилового эфира 3,7-диазабицикло[3.3.1]нонан-3-карбоновой кислоты
Пример 8 относится к синтезу трет-бутилового эфира 3,7-диазабицикло[3.3.1]нонан-3-карбоновой кислоты (или N-(третбутоксикарбонил)-3,7-диазабицикло[3.3.1]нонана), который получают по следующей методике:
трет-бутиловый эфир 7-бензил-3,7-диазабицикло[3.3.1]нонан-3-карбоновой кислоты (или N-бензил-N'-(трет-бутоксикарбонил)-3,7-диазабицикло[3.3.1]нонан)
Трет-бутиловый эфир 7-бензил-3,7-диазабицикло[3.3.1]нонан-3-карбоновой кислоты получали по методике, изложенной Stead et al., Org. Lett. 7(20): 4459(2005).
Трет-бутиловый эфир 3,7-диазабицикло[3.3.1]-3-карбоновой кислоты
Трет-бутиловый эфир 7-бензил-3,7-диазабицикло[3.3.1]нонан-3-карбоновой кислоты (0,49 г, 1,6 ммоль) растворяли в метаноле (20 мл) и в атмосфере азота добавляли 20% Pd(OH)2/C (влажный) (~2 г). Полученную смесь нагревали примерно до 50°C и встряхивали в течение 2 ч в атмосфере водорода при давлении 55 фунтов/кв.дюйм. Полученную смесь фильтровали и концентрировали, получая 0,32 г (выход 94%) указанного в заголовке соединения (МС m/z 227 (M+H)).
Пример 9: Синтез трифторацетата N-(фуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонана
Пример 9 относится к синтезу трифторацетата (3,7-диазабицикло[3.3.1]нон-3-ил)-фуран-2-илметанона (или трифторацетата N-(фуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонана), который получали по приведенной ниже методике, являющейся иллюстративной для реакции сочетания, используемой для синтеза гетероароматических амидов 3,7-диазабицикло[3.3.1]нонана:
Трифторацетат (3,7-диазабицикло[3.3.1]нон-3-ил)фуран-2-илметанона (или трифторацетата N-(фуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонана)
Фуран-2-карбоновую кислоту (0,032 г, 0,29 ммоль) и триэтиламин (0,121 мл, 0,870 ммоль) смешивали в сухом дихлорметане (1 мл) и добавляли HBTU (0,11 г, 0,29 ммоль). Добавляли раствор трет-бутилового эфира 3,7-диазабицикло[3.3.1]-3-карбоновой кислоты (0,059 г, 0,26 ммоль) в дихлорметане (0,5 мл) и смесь перемешивали при комнатной температуре в течение ночи. Смесь обрабатывали 10% водным раствором гидроксида натрия и экстрагировали хлороформом (2×2 мл). Объединенные органические экстракты промывали водой (2×1 мл) и концентрировали. Полученный остаток растворяли в диметилформамиде (ДМФА) (0,3 мл) и очищали ВЭЖХ (градиент смеси ацетонитрил/вода). Фракции, содержащие желаемое вещество, объединяли и концентрировали, оставляя защищенный трет-бутоксикарбонилом продукт. Полученное вещество растворяли в смеси трифторуксусной кислоты (0,5 мл) и дихлорметана (0,5 мл), и смесь перемешивали при комнатной температуре в течение 1 ч. Летучие вещества удаляли на роторном испарителе с последующим выдерживанием в глубоком вакууме, получая 36 мг масла (выход 41%) (1H ЯМР (d4-метанол, 300 МГц) 2,10 (ушир.с, 2H), 2,35 (ушир.с, 2H), 3,30-3,45 (м, 4H), 3,55 (м, 2H), 6,65 (м, 1H), 7,15 (д, 1H) и 7,75 (д, 1H); МС m/z 221 (M+H)).
Пример 10: Синтез трифторацетата N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонана
Пример 10 относится к синтезу трифторацетата (3,7-диазабицикло[3.3.1]нон-3-ил)-5-хлорфуран-2-илметанона (или трифторацетата N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонана), который получали способом, подобно описанному в примере 9, согласно следующей методике:
5-хлорфуран-2-карбоновую кислоту (0,96 г, 6,5 ммоль) смешивали с триэтиламином (2,9 мл, 21 ммоль) в сухом дихлорметане (10 мл) и добавляли HBTU (2,47 г, 65,1 ммоль). Добавляли раствор трет-бутилового эфира 3,7-диазабицикло[3.3.1]-3-карбоновой кислоты (1,5 г, 66 ммоль) в дихлорметане (5 мл), и смесь перемешивали при комнатной температуре в течение ночи. Смесь обрабатывали 10% водным раствором гидроксида натрия и экстрагировали хлороформом (2×20 мл). Объединенные органические экстракты промывали водой (2×10 мл) и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле, элюируя градиентом этилацетата в гексане, получая защищенный трет-бутоксикарбонилом продукт, в виде вязкого масла. Полученное вещество растворяли в смеси трифторуксусной кислоты (20 мл) и дихлорметана (20 мл), и смесь перемешивали при комнатной температуре в течение 1 ч. Летучие вещества удаляли на роторном испарителе, с последующим выдерживанием продукта в глубоком вакууме, получая 1,38 г (выход 57,5%) вязкого желтого масла (1H ЯМР (d4-метанол, 300 МГц) 2,00 (ушир.с, 2H), 2,15 (ушир.с, 2H), 3,15-3,35 (м, 6H), 4,25 (м, 2H), 6,53 (д, 1H) и 7,10 (д, 1H); МС m/z 255 (M+H)).
Пример 11: Таблицы спектральных данных и данных о связывании рецепторов
Описанные выше методики сочетания с образованием амидов применяли для получения соединений, показанных в таблицах 1 и 2. В некоторых случаях соединения синтезировали в количестве, достаточном для получения данных ядерного магнитного резонанса (ЯМР). В других случаях соединения получали в меньшем количестве в различных устройствах для параллельного синтеза, и такие соединения были охарактеризованы (структурно) только способом ЖХМС.
Крысиного α4β2
Человеческого α4β2
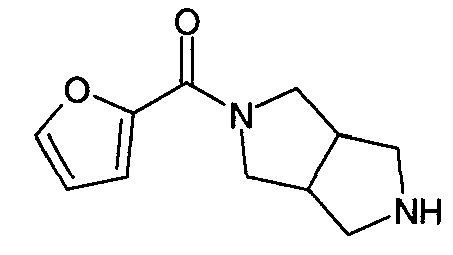

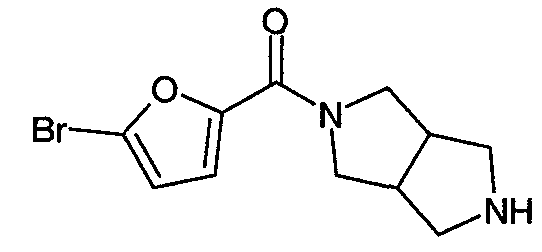

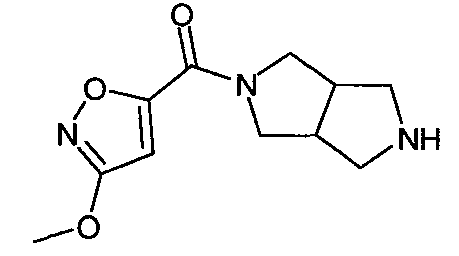
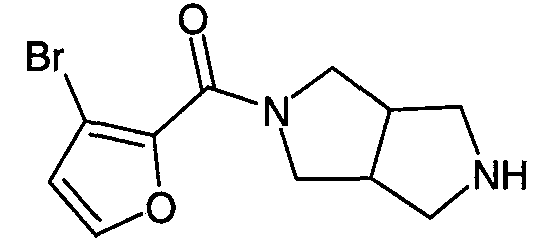
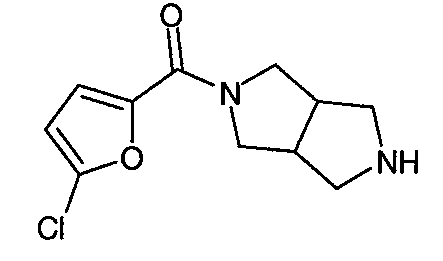
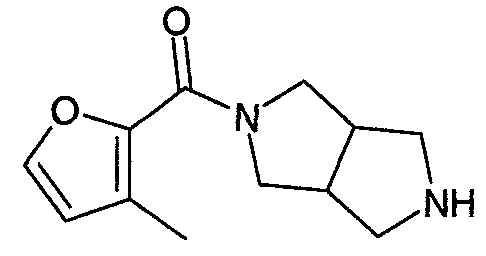
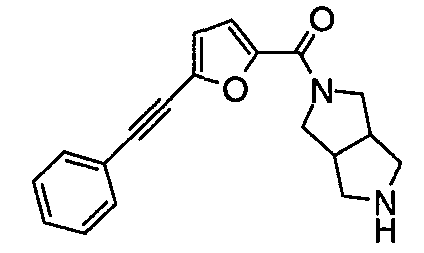

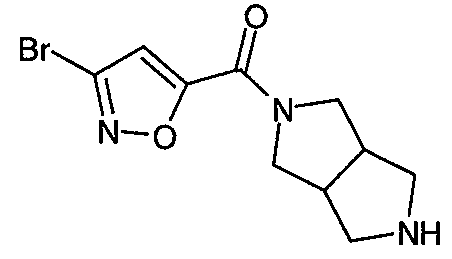
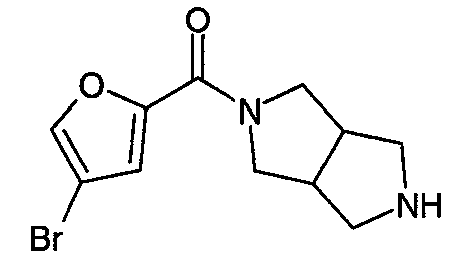
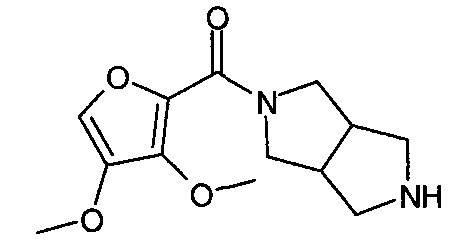
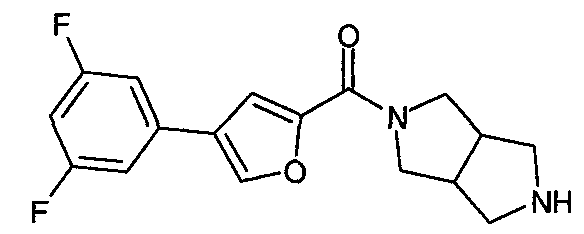
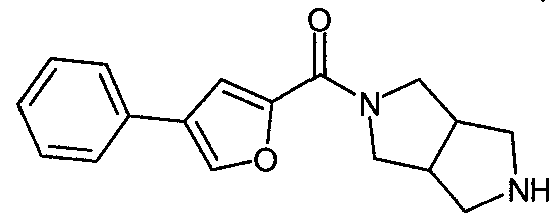
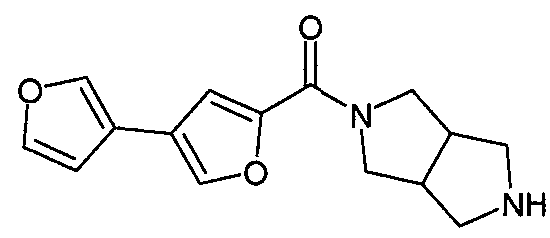
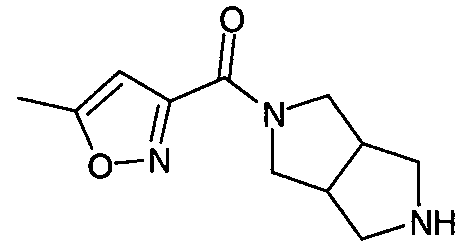




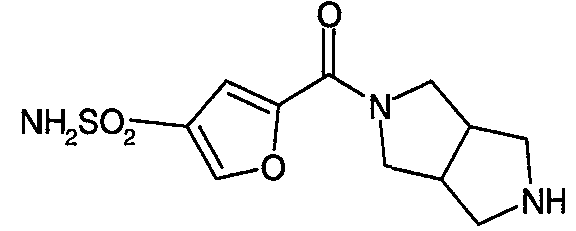
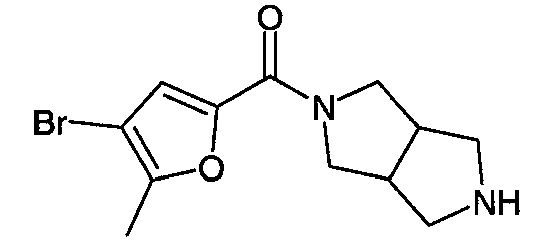

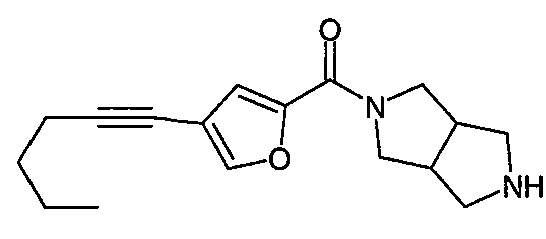

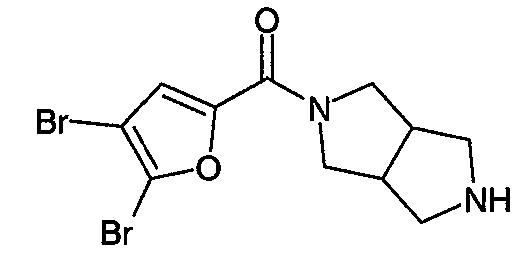

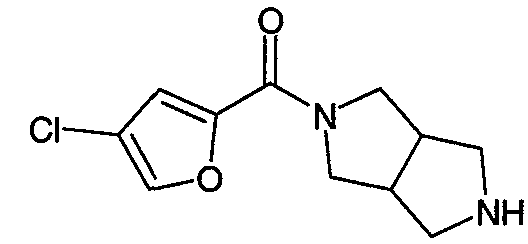

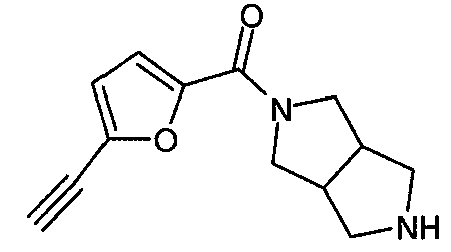

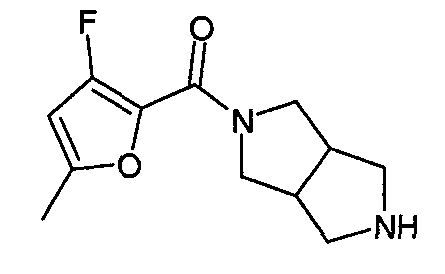
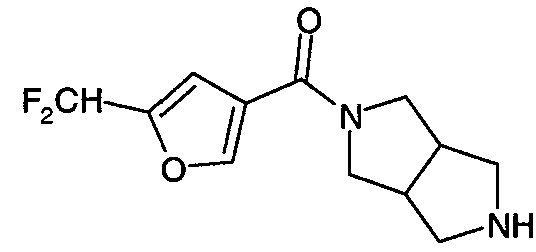

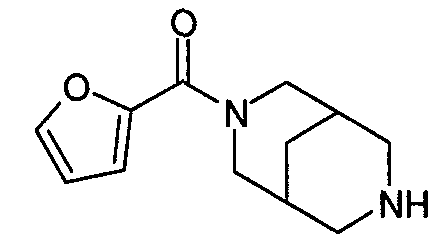
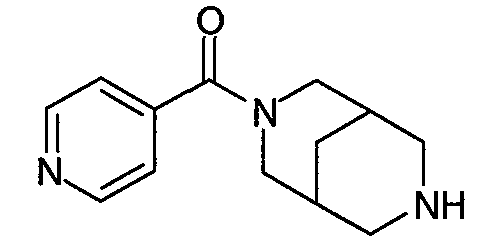

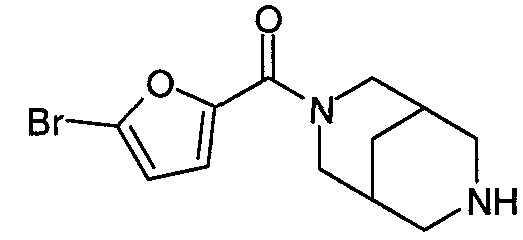


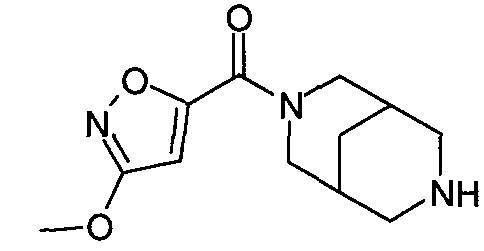

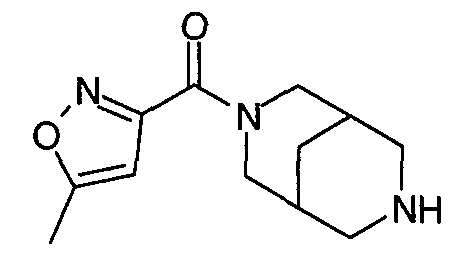

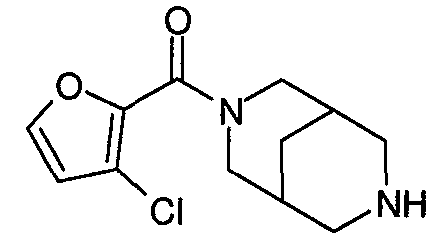


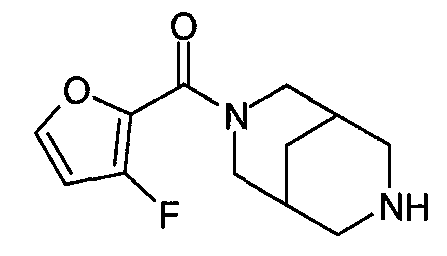



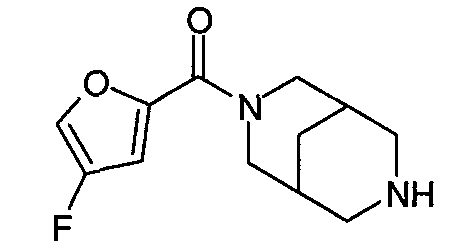

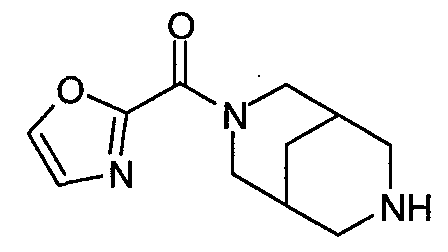

Краткое описание биологических данных
Соединения в таблицах 1 и 2, которые являются репрезентативными для настоящего изобретения, демонстрируют значения констант ингибирования (значения Ki) в отношении крысиных и человеческих рецепторов подтипа α4β2 в диапазоне от 1 нМ до 1000 нМ и от 1 нМ до 220 нМ, соответственно, что указывает на высокую аффинность в отношении рецепторов подтипа α4β2. Значения Ki для рецепторов подтипа α7 находятся в диапазоне от 1700 нМ до 210000 нМ (во многих случаях соединения не демонстрируют достаточного связывания с рецепторами подтипа α7 в ходе высокопроизводительного скрининга, чтобы гарантировать определение Ki). Те же самые соединения демонстрируют относительно небольшую функциональную активность в отношении рецепторов как человеческих мышечных подтипов (1-25% от максимальной реакции на никотин), так и человеческих ганглиозных рецепторов (1-20% от максимальной реакции на никотин).
Воздействие некоторых репрезентативных соединений оценивали в экспериментах на тест по распознаванию объектов, NOR. Так, например, оба соединения, а именно N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан (фиг.1) и N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.1]нонан (фиг.2) проявляли активность при распознавании объектов у крыс в количествах 0,1 мг/кг и 0,3 мг/кг соответственно. Этот факт доказывает эффективность (и потенциал) соединений по настоящему изобретению при лечении когнитивных расстройств, расстройств внимания и деменций, а также потенциал указанных соединений для лечения людей.
Изложенный выше материал является иллюстрацией настоящего изобретения и его не следует истолковывать как ограничение объема изобретения. Объем изобретения определен изложенной ниже формулой изобретения, причем в пункты формулы изобретения включены их эквиваленты. Все патенты и публикации, которые приведены в настоящем описании, включены посредством ссылок во всей своей полноте для использования в любых целях.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИДЫ ДИАЗАБИЦИКЛОАЛКАНОВ, СЕЛЕКТИВНЫЕ В ОТНОШЕНИИ АЦЕТИЛХОЛИНОВОГО ПОДТИПА НИКОТИНОВЫХ РЕЦЕПТОРОВ | 2007 |
|
RU2517693C2 |
| СЕЛЕКТИВНЫЕ К ПОДТИПУ РЕЦЕПТОРА АЗАБИЦИКЛОАЛКАНОВЫЕ ПРОИЗВОДНЫЕ | 2008 |
|
RU2417984C1 |
| N, N'-замещенные 3, 7-диазабицикло[3.3.1]нонаны, фармацевтические композиции на их основе и их применение | 2015 |
|
RU2613071C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2767857C2 |
| ПРОИЗВОДНЫЕ ДИАЗАГОМОАДАМАНТАНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2549551C2 |
| ОКСАДИАЗОЛЫ В КАЧЕСТВЕ АГОНИСТОВ МУСКАРИНОВОГО РЕЦЕПТОРА M1 И/ИЛИ M4 | 2019 |
|
RU2817018C2 |
| НЕКОНКУРЕНТНЫЕ АНТАГОНИСТЫ НИКОТИНОВЫХ РЕЦЕПТОРОВ | 2011 |
|
RU2582339C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2010 |
|
RU2531588C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 2006 |
|
RU2446171C2 |
| ЛЕЧЕНИЕ НЕВРОЛОГИЧЕСКИХ РАССТРОЙСТВ | 2017 |
|
RU2765868C2 |

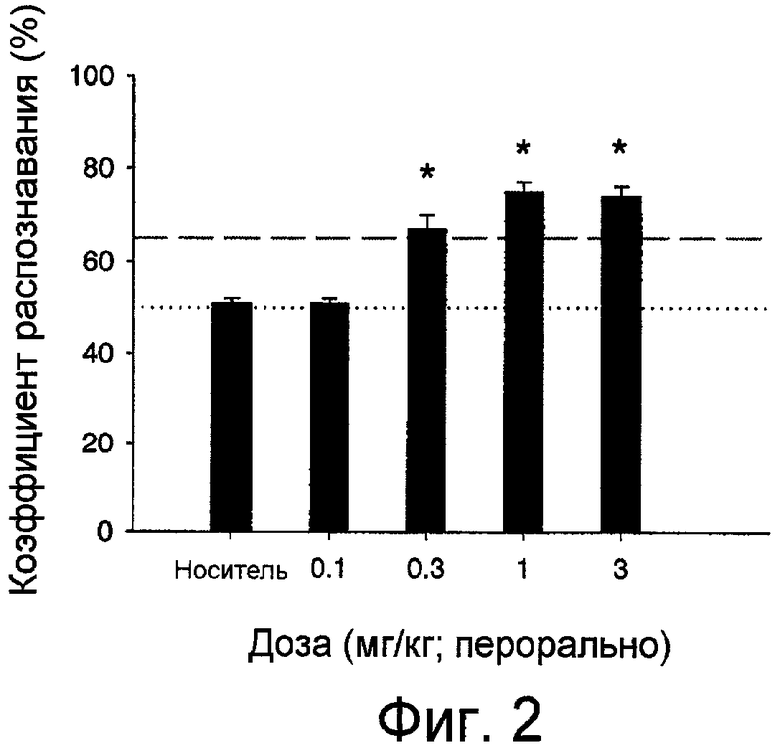
Изобретение относится к новому соединению, а именно N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октану или его фармацевтически приемлемым солям, фармацевтическим композициям для лечения состояний и расстройств, связанных с дисфункцией центральной нервной системы, содержащих это соединение, а также способам лечения. Данное соединение демонстрирует селективность в отношении нейронных никотиновых рецепторов подтипа α4β2 в центральной нервной системе (ЦНС) и связывается с ними с высокой аффинностью. Это соединение и композиции на его основе могут применяться для лечения и/или профилактики широкого спектра заболеваний и расстройств, в частности расстройств ЦНС. 6 н.п. ф-лы, 2 ил., 1 табл., 11 пр.
1. Соединение N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан или его фармацевтически приемлемые соли.
2. Способ лечения возрастного ухудшения памяти, слабого нарушения когнитивных способностей, предстарческой деменции, раннего начала болезни Альцгеймера, старческой деменции, деменции типа Альцгеймера, деменции с тельцами Леви, сосудистой деменции, болезни Альцгеймера, удара, комплекса СПИД-деменция, синдрома дефицита внимания, гиперактивного расстройства с дефицитом внимания, дислексии, шизофрении, шизофреноформного расстройства, нарушения познавательной способности при шизофрении и шизоаффективного расстройства, включающий введение N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан или его фармацевтически приемлемой соли.
3. Способ для лечения деменции типа Альцгеймера в степени от слабой до умеренной, умеренного нарушения когнитивных способностей или возрастного ухудшения памяти, включающий введение N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан или его фармацевтически приемлемой соли.
4. Способ лечения синдрома дефицита внимания или гиперактивности, включающий введение N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан или его фармацевтически приемлемой соли.
5. Способ лечения шизофрении, шизофреноформного расстройства, нарушения познавательной способности при шизофрении и шизоаффективного расстройства, включающий введение N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан или его фармацевтически приемлемой соли.
6. Фармацевтическая композиция для лечения состояний и расстройств, связанных с дисфункцией центральной нервной системы, содержащая в качестве активного ингредиента соединение N-(5-хлорфуран-2-илкарбонил)-3,7-диазабицикло[3.3.0]октан или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых разбавителей, эксципиентов или инертных носителей.
| WO 2004016616 А1, 26.02.2004 | |||
| WO 2004076453 А1, 10.09.2004 | |||
| US 20040186107 А, 32.09.2004 | |||
| US 20050101602 С2, 27.08.2005 | |||
| АЗАБИЦИКЛИЧЕСКИЕ СЛОЖНЫЕ ЭФИРЫ КАРБАМИНОВЫХ КИСЛОТ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2172739C2 |
| ДИАЗАБИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2124014C1 |

