ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к области фармацевтических препаратов и конкретно относится к твердой дисперсии, содержащей (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-он, и способу ее получения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
В-клеточная лимфома является одной из распространенных злокачественных опухолей, влияющих на здоровье человека, и на ее долю приходится от 70 до 80 % злокачественных лимфом. Уровень заболеваемости В-клеточной лимфомой увеличивается с каждым годом. Это одна из самых распространенных злокачественных опухолей в Китае. На возникновение В-клеточной лимфомы влияют различные факторы, включая генетический фактор, биологический фактор, физический и химический фактор. В-клеточная лимфома характеризуется плохим прогнозом, и выживаемость пациентов все еще низка при современных схемах лечения.
Ревматоидный артрит (РА) является хроническим аутоиммунным заболеванием, которое в основном поражает фасеточные суставы. Патологическими признаками ревматоидного артрита являются в основном инфильтрация воспалительных клеток, гиперплазия и гипертрофия синовиальной ткани, а также повреждение костей. Этиология и патогенез до конца не выяснены. Частота ревматоидного артрита высока, приблизительно от 0,5 до 1 % у взрослых.
При В-клеточной лимфоме ингибирование активности тирозинкиназы Брутона, ВТК, эффективно ингибирует пролиферацию и выживание опухолевых клеток. Типичное лекарственное средство, ибрутиниб, было одобрено Управлением по контролю за пищевыми продуктами и лекарственными средствами (FDA) США для лечения лимфомы из клеток мантийной зоны и хронического лимфолейкоза, и клинически показало превосходную эффективность.
При ревматоидном артрите ингибирование активности BTK может ингибировать активность факторов транскрипции, таких как NF-κB, тем самым ингибируя высвобождение воспалительных факторов и уменьшая симптомы воспаления. Низкомолекулярный ингибитор BTK HM61713, разработанный Hanmi Pharmaceuticals, демонстрирует превосходную доклиническую эффективность в отношении артрита, и в настоящее время проходит фазу I клинических испытаний.
В WO2013184572 раскрыт пероральный препарат ибрутиниба, содержащая ибрутиниб, разбавитель, разрыхлитель, поверхностно-активное вещество и смазывающее вещество, для соответствия требованиям растворения, стабильности и биодоступности для получения лекарственного средства ибрутиниба.
В WO2016007185 раскрыто соединение формулы I, а именно (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-он, который по структуре аналогичен ибрутинибу. Соединение формулы I имеет такие особенности, как хорошая специфичность к мишени, высокая селективность к киназе и высокая пероральная биодоступность. Ожидается, что он уменьшит или устранит клинически нежелательную лекарственную реакцию ибрутиниба и будет иметь терапевтические преимущества в таких областях, как В-клеточная лимфома и ревматоидный артрит.
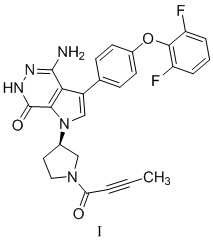 .
.
Существует ряд проблем для фармацевтически активных соединений с низкой растворимостью при получении высококачественной фармацевтической композиции или препарата, такого как таблетки, гранулы и порошок. Исследователям предстоит изучить и найти решение возникающим проблемам.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении активный ингредиент (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-он или его фармацевтически приемлемую соль диспергируют в структуре материала, содержащего лекарственное средство, с помощью растворителя способом твердой дисперсии с получением кодисперсной системы лекарственного средства и материала носителя, что решает проблему его плохой растворимости.
Настоящее изобретение обеспечивает твердую дисперсию (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-она или его фармацевтически приемлемой соли, содержащую (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-он или его фармацевтически приемлемую соль в виде активного ингредиента и материал носителя, где материал носителя выбирают из группы, состоящей из ацетат сукцината гидроксипропилметилцеллюлозы и фталата гидроксипропилметилцеллюлозы.
В некоторых вариантах осуществления, когда массовое соотношение материала носителя к активному ингредиенту, применяемому в твердой дисперсии по настоящему изобретению, составляет по меньшей мере 0,5:1 или более, однородная дисперсная система активного ингредиента и носителя может быть получена способом получения в эксперименте, кристаллическое состояние активного ингредиента изменяется до аморфного, растворимость и абсорбция лекарственного средства улучшаются, и лекарственное средство обладает быстрым началом действия и высокой биодоступностью после перорального введения. Сама твердая дисперсия является стабильной и не проявляет явления старения при ускоренном режиме в течение 6 месяцев, и нет значительного изменения в различных оценочных показателях.
В твердой дисперсии по настоящему изобретению массовое соотношение материала носителя к активному ингредиенту или его фармацевтически приемлемой соли может варьироваться в широких пределах, как минимум 0,5:1. В настоящем изобретении, чем выше содержание материала носителя, тем легче изменить активный ингредиент из кристаллической формы в аморфную форму и тем выше соответствующая биодоступность твердой дисперсии. Принимая во внимание баланс между содержанием лекарственного средства и биодоступностью, массовое соотношение материала носителя к активному ингредиенту или его фармацевтически приемлемой соли в настоящем изобретении может составлять от 0,5:1 до 4:1. В некоторых вариантах осуществления массовое соотношение может составлять 0,5:1, 0,6:1, 0,7:1, 0,8:1, 0,9:1, 1:1, 1,1:1, 1,2:1, 1,3:1, 1,4:1, 1,5:1, 1,6:1, 1,7:1, 1,8:1, 1,9:1, 2:1, 2,2:1, 2,4:1, 2,6:1, 2,8:1, 3:1, 3,2:1, 3,4:1 3,6:1, 3,8:1 и 4:1, предпочтительно от 0,8:1 до 3:1 и более предпочтительно от 1:1 до 2:1.
В некоторых вариантах осуществления твердая дисперсия по настоящему изобретению состоит из активного ингредиента (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-она или его фармацевтически приемлемой соли и материала носителя, где материал носителя выбран из группы, состоящей из ацетат сукцината гидроксипропилметилцеллюлозы и фталата гидроксипропилметилцеллюлозы.
Твердая дисперсия по настоящему изобретению может быть получена общеизвестными способами получения, такими как способ плавления, способ с применением растворителя и способ плавления растворителем. Другие способы получения включают получение эвтектической смеси способом измельчения по принципу совместного растворения и получение твердого поверхностного адсорбируемого вещества путем растворения лекарственного средства в органическом растворителе, который должен быть диспергирован и адсорбирован на инертном материале.
Способ с применением растворителя по настоящему изобретению представляет собой способ соосаждения, где лекарственное средство и носитель растворяют вместе в органическом растворителе, или лекарственное средство и носитель растворяют в указанном порядке в растворителе с последующим тщательным их перемешиванием, или материал носителя суспендируют и диспергируют в органическом растворителе активного ингредиента или его фармацевтически приемлемой соли, а затем растворитель удаляют для получения твердой дисперсии. Способ удаления растворителя известен или может быть определен специалистами в данной области техники и может представлять собой способ добавления по каплям высокополярного органического раствора в низкополярный растворитель для осаждения твердого вещества, а также способ сушки распылением или сушки в условиях пониженного давления.
В соответствии со способом плавления согласно настоящему изобретению лекарственное средство и носитель тщательно смешивают и нагревают до расплавления, или носитель нагревают до расплавления с последующим добавлением лекарственного средства для растворения при перемешивании, и затем расплав быстро охлаждают до твердого вещества при энергичном перемешивании или непосредственно вливают в капсулу и затем охлаждают.
Согласно способу плавления растворителем по настоящему изобретению лекарственное средство растворяют в небольшом количестве органического растворителя, тщательно смешивают с расплавленным носителем, выпаривают для удаления органического растворителя и охлаждают до получения твердого вещества.
Способ получения твердой дисперсии по настоящему изобретению представляет собой предпочтительно способ с применением растворителя (также известный как способ соосаждения), включающий этапы растворения материала носителя и активного ингредиента или его фармацевтически приемлемой соли в органическом растворителе или, суспендирования и диспергирования материала носителя в растворе активного ингредиента или его фармацевтически приемлемой соли в органическом растворителе, а затем удаление органического растворителя с получением твердой дисперсии.
Кроме того, способ удаления органического растворителя известен или может быть определен специалистами в данной области техники и может представлять собой способ добавления по каплям высокополярного органического раствора к низкополярному растворителю или воде для осаждения твердого вещества (а именно способ осаждения растворителем), а также способ сушки распылением или сушки в условиях пониженного давления.
В некоторых вариантах осуществления твердую дисперсию по настоящему изобретению получают путем растворения вместе активного ингредиента или его фармацевтически приемлемой соли и материала носителя ацетат сукцината гидроксипропилметилцеллюлозы в первом органическом растворителе, а затем добавления по каплям полученного раствора ко второму растворителю. Скорость добавления по каплям составляет предпочтительно от 1 до 100 г/мин, более предпочтительно от 2 до 50 г/мин и, в частности, 2, 6, 10, 14, 16, 20, 24, 28, 32, 36, 40, 44, 48 или 50 г/мин. Кроме того, вышеуказанный способ получения композиции может включать любую один этап фильтрации, промывки и сушки, для того, чтобы гарантировать, что количество остаточного растворителя в полученной твердой дисперсии составляет менее 120 млн-1, для соответствия требованиям получения лекарственного средства из твердой дисперсии.
Кроме того, первый растворитель представляет собой высокополярный органический растворитель, который известен или может быть определен специалистами в данной области техники, и включает, но не ограничивается ими, сульфоновый растворитель, такой как диметилсульфоксид, амидный растворитель, такой как N,N-диметилформамид и N,N-диметилацетамид, кетоновый растворитель, такой как ацетон, галогенированный углеводородный растворитель, такой как тетрахлорметан, спиртовой растворитель, такой как этанол и метанол, и, предпочтительно по меньшей мере один из диметилсульфоксида, N,N-диметилформамида, N,N-диметилацетамида, этанола и метанола.
Второй растворитель представляет собой менее полярный растворитель (также известный как низкополярный растворитель, который смешивается с первым растворителем, и после смешивания с первым органическим растворителем растворимость активного ингредиента или его фармацевтически приемлемой соли в системе снижается) и включает, но не ограничивается ими, алкановый растворитель, такой как н-гексан и простой петролейный эфир, спиртовой растворитель, такой как этанол и метанол, фурановый растворитель, такой как тетрагидрофуран, эфирный растворитель, такой как диэтиловый эфир и дипропиловый эфир, и воду или кислый водный раствор, и предпочтительно по меньшей мере один из метанола, этанола, воды и кислого водного раствора.
В одном варианте осуществления активный ингредиент или его фармацевтически приемлемая соль и материал носителя растворяют вместе по меньшей мере в одном органическом растворителе, выбранном из группы, состоящей из диметилсульфоксида, N,N-диметилформамида и N,N-диметилацетамида, и затем полученный раствор по каплям добавляют в воду. Скорость добавления по каплям составляет предпочтительно от 1 до 100 г/мин, более предпочтительно от 2 до 50 г/мин и, в частности, 2, 6, 10, 14, 16, 20, 24, 28, 32, 36, 40, 44, 48 или 50 г/мин
Твердая дисперсия по настоящему изобретению может быть дополнительно обработана в твердый препарат, такой как таблетка, пилюля, гранула, капсула и тому подобное. Количество активного ингредиента или его фармацевтически приемлемой соли составляет от 8 до 40 % и может составлять 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35 или 40 мас.% и предпочтительно от 15 до 25 мас.% твердого препарата.
В некоторых вариантах осуществления количество (масса или вес) активного ингредиента или его фармацевтически приемлемой соли по настоящему изобретению составляет от 10 до 500 мг и может составлять 200 мг, 190 мг, 180 мг, 170 мг, 160 мг, 150 мг, 140 мг, 130 мг, 120 мг, 110 мг, 100 мг, 95 мг, 75 мг, 50 мг, 25 мг, 15 мг и 10 мг и предпочтительно 200 мг, 100 мг или 25 мг. Кроме того, твердый препарат также содержит фармацевтически приемлемый эксципиент, который хорошо известен или может быть определен специалистами в данной области техники, и включает, но не ограничивается ими, по меньшей мере один из разрыхлителя, наполнителя, связующего вещества и смазывающего вещества.
В некоторых вариантах осуществления в среде, 0,15 % водном растворе додецилсульфата натрия (SDS), скорость растворения (%) активного ингредиента в твердом препарате по настоящему изобретению составляет 85 % или более и может быть большей или равной 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 и 100 % и предпочтительно 90 % или более в течение 45 мин, кроме того, скорость растворения (%) активного ингредиента в твердом препарате составляет 70 % или более и может быть большей или равной 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94 и 95 % в течение 15 мин. Твердый препарат быстро и полностью растворяется и обладает хорошей биодоступностью. Способ получения твердого препарата прост и подходит для крупномасштабного производства.
В некоторых вариантах осуществления твердый препарат по настоящему изобретению стабилен в течение по меньшей мере 3 месяцев, по меньшей мере 6 месяцев, по меньшей мере 9 месяцев, по меньшей мере 12 месяцев, по меньшей мере 18 месяцев или по меньшей мере 24 месяцев при 25 °C/60 % относительной влажности. В некоторых вариантах осуществления фармацевтическая композиция является стабильной в течение по меньшей мере 1 месяца, по меньшей мере 2 месяцев, по меньшей мере 3 месяцев, даже 6 месяцев или дольше при 40 °C/75 % относительной влажности.
В некоторых вариантах осуществления твердая дисперсия по настоящему изобретению является стабильной в течение по меньшей мере 3 месяцев, по меньшей мере 6 месяцев, по меньшей мере 9 месяцев, по меньшей мере 12 месяцев, по меньшей мере 18 месяцев или по меньшей мере 24 месяцев при 25 °C/60 % относительной влажности. В некоторых вариантах осуществления фармацевтическая композиция является стабильной в течение, по меньшей мере 1 месяца, по меньшей мере 2 месяцев, по меньшей мере, 3 месяцев, даже 6 месяцев или дольше при 40 °C/75 % относительной влажности.
Наполнитель обеспечивает объем для получения таблетки практичного размера, пригодного для обработки. Наполнитель также может вносить вклад в способ и улучшать физические свойства твердого препарата, такие как текучесть, прессуемость и прочность на раздавливание твердого препарата. Наполнитель по настоящему изобретению известен или может быть определен специалистами в данной области техники и включает, но не ограничивается ими, по меньшей мере один из декстрина, лактозы, сахарозы, гидрофосфата кальция, крахмала, безводного гидрофосфата кальция, гидрофосфата кальция, микрокристаллической целлюлозы и маннита. Предпочтительно количество наполнителя составляет от 30 до 90 мас.%, и более предпочтительно от 35 до 60 мас.% по отношению к массе твердого препарата. В одном варианте осуществления количество наполнителя может составлять 35, 38, 40, 42, 45, 47, 50, 52, 55, 58 и 60 мас.% по отношению к массе твердого препарата.
Разрыхлитель по настоящему изобретению известен или может быть определен специалистами в данной области техники и включает, но не ограничивается ими, по меньшей мере один из кроскармеллозы натрия, кросповидона, натрия карбоксиметилкрахмала, крахмала, прежелатинизированного крахмала и альгиновой кислоты. Предпочтительно количество разрыхлителя составляет от 1 до 20 мас.% по отношению к массе твердого препарата. В одном варианте осуществления количество разрыхлителя может составлять 1,0, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5, 8, 8,5, 9, 9,5, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 и 20 мас.% и предпочтительно от 5 до 15 мас.% по отношению к массе твердого препарата.
Связующее вещество по настоящему изобретению известно или может быть определено специалистами в данной области техники и включает, но не ограничивается ими, по меньшей мере один из поливинилпирролидона, крахмала, метилцеллюлозы, карбоксицеллюлозы, гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы и альгината, и предпочтительно по меньшей мере один из поливинилпирролидона (торговое наименование K30) и гидроксипропилцеллюлозы. Более предпочтительно количество связующего вещества составляет от 0,5 до 10 мас.% по отношению к массе твердого препарата. В одном варианте осуществления количество связующего вещества может составлять 0,5, 0,6, 0,7, 0,8, 0,9, 1, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5 8, 8,5, 9, 9,5 и 10 мас.% по отношению к массе твердого препарата.
Смазывающее вещество по настоящему изобретению известно или может быть определено специалистами в данной области техники и включает, но не ограничивается ими, по меньшей мере один из стеарата магния, стеариновой кислоты, пальмитиновой кислоты, стеарата кальция, талька, карнаубского воска и стеарилфумарата натрия. Предпочтительно, количество смазывающего вещества по настоящему изобретению составляет от 0,1 до 5 мас.% по отношению к массе твердого препарата. В одном варианте осуществления количество смазывающего вещества может составлять 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9%, 1%, 1,5%, 2%, 2,5%, 3%, 3,5%, 4%, 4,5 и 5 мас.% и предпочтительно от 0,1 до 2 мас.% по отношению к массе твердого препарата.
В предпочтительном варианте осуществления твердый препарат по настоящему изобретению содержит:
1) от 10 мг до 500 мг активного ингредиента или его фармацевтически приемлемой соли,
2) от 5 до 15 мас.% разрыхлителя,
3) от 30 до 90 мас.% наполнителя,
4) от 0,5 до 10 мас.% связующего вещества, и
5) от 0,1 до 5 мас.% смазывающего вещества.
Кроме того, материал носителя в твердом препарате представляет собой ацетат сукцинат гидроксипропилметилцеллюлозы.
Скорость растворения твердого препарата по настоящему изобретению определяли в соответствии со вторым способом (способ с применением лопастной мешалки) тестирования скорости растворения, описанным в общем правиле тома IV Китайской Фармакопеи, издание 2015 года. Тестирование на растворение композиции по настоящему изобретению проводили с применением 0,15 % водного раствора додецилсульфата натрия (SDS), предпочтительно 1000 мл, в качестве среды для растворения при 37 ± 0,5 °С и скорости лопастной мешалки 50 об/мин.
Скорость растворения твердой дисперсии по настоящему изобретению определяли в соответствии со вторым способом (способ с применением лопастной мешалки) тестирования скорости растворения, описанным в общем правиле тома IV Китайской Фармакопеи, издание 2015 года. Тестирование на растворение проводили с применением 1000 мл 0,15 % водного раствора SDS в качестве среды для растворения при 37 ± 0,5 °C и скорости лопастной мешалки 75 об/мин.
Настоящее изобретение также обеспечивает твердый препарат, содержащую активный ингредиент (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-он или его фармацевтически приемлемую соль и материал носителя, где в среде 0,15 % водном растворе додецилсульфата натрия (SDS), скорость растворения (%) активного ингредиента в твердом препарате составляет 85 % или более и может быть большей или равной 85, 86, 87, 88, 89, 90, 91, 92, 93, 94 95, 96, 97, 98, 99 и 100 % и предпочтительно 90 % или более в течение 45 мин, кроме того, скорость растворения (%) активного ингредиента в твердоом препарате составляет 70 % или более и может быть большей или равной 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94 и 95 % в течение 15 мин. В некоторых вариантах осуществления активный ингредиент или его фармацевтически приемлемая соль и материал носителя в твердой дисперсии находятся в форме твердой дисперсии, где материал носителя выбран из группы, состоящей из ацетат сукцината гидроксипропилметилцеллюлозы и фталата гидроксипропилметилцеллюлозы, и предпочтительно ацетат сукцината гидроксипропилметилцеллюлозы.
Способ получения твердого препарата, содержащего твердую дисперсию (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-она по настоящему изобретению является следующим: твердую дисперсию измельчают в порошок, тщательно смешивают с наполнителем и/или разрыхлителем, необходимым для формования твердого препарата, смешивают со связующим веществом и подвергают влажному гранулированию или сухому гранулированию, затем полученные гранулы сушат, просеивают через сито, измельчают, тщательно смешивают со смазывающим веществом и получают в виде пилюль или гранул или прессуют в таблетки или помещают в капсулы, или твердая дисперсия также может быть смешана с подходящими вспомогательными веществами и непосредственно помещена в капсулы или спрессована в таблетки, полученные гранулы или необработанные таблетки или капсулы могут быть дополнительно покрыты оболочкой если это необходимо.
Активный ингредиент (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7H-пирроло[2,3-d]пиридазин-7-он по настоящему изобретению может реагировать с кислотой с получением фармацевтически приемлемой соли. Кислота известна или может быть определена специалистами в данной области техники и включает, но не ограничивается ими, соляную кислоту, метансульфоновую кислоту, фумаровую кислоту, трифторуксусную кислоту и фосфорную кислоту.
Термин "по отношению к массе твердого препарата" в контексте настоящего изобретения означает, что расчет диапазонов количества применяемого активного ингредиента или других видов фармацевтических вспомогательных веществ основан на массе ядра таблетки без покрывающего агента, в Примере 1 приведено подробное описание.
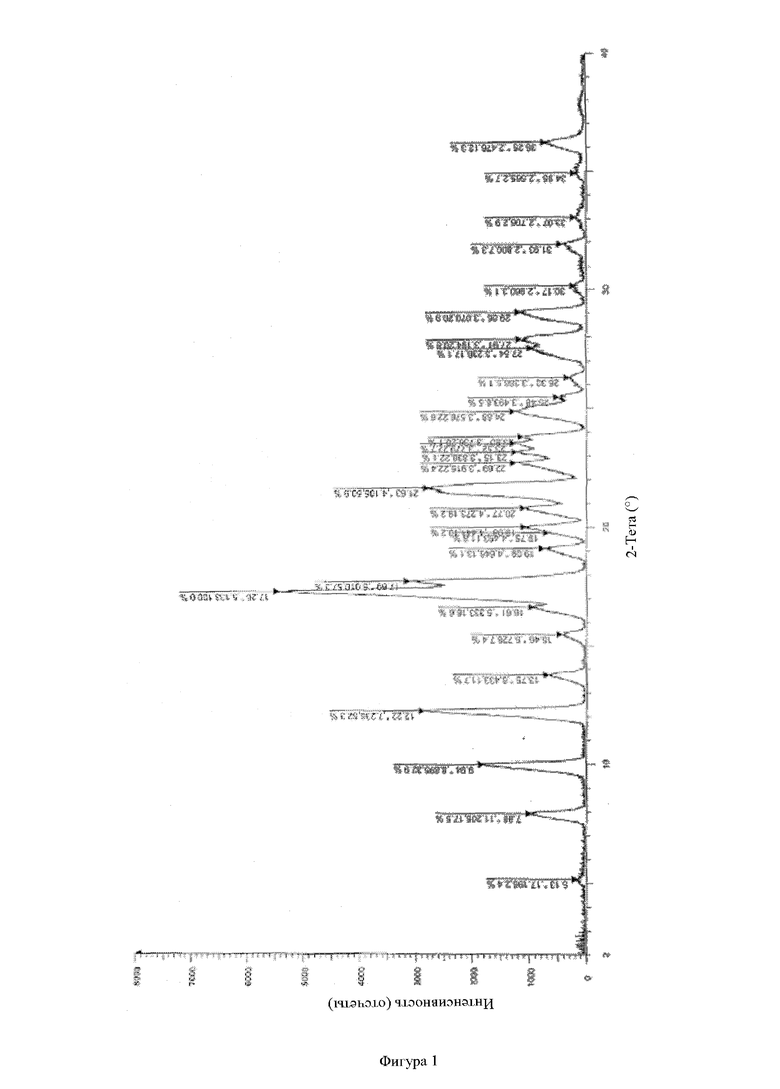
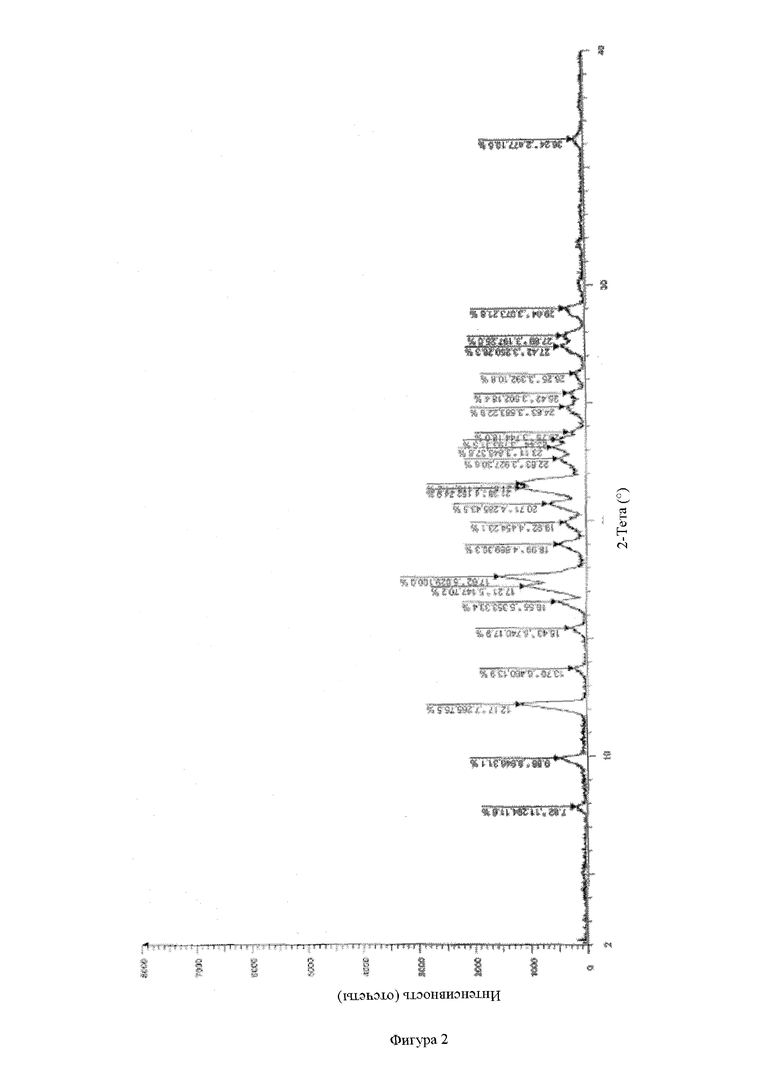
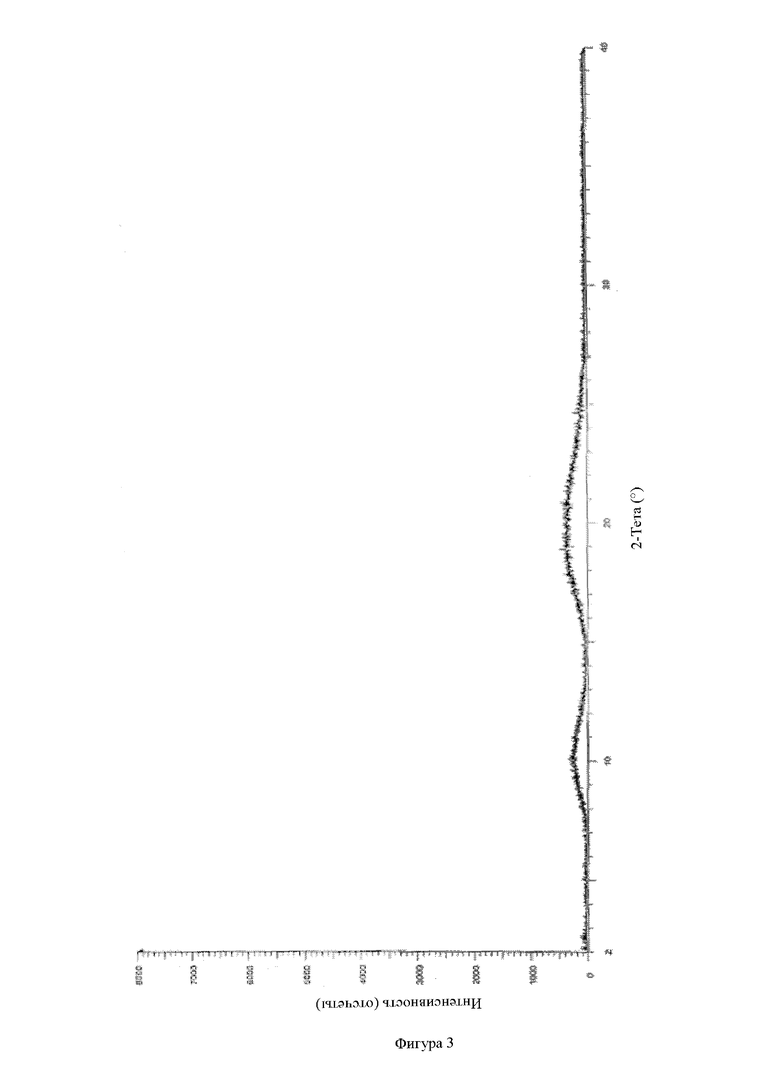
Типичными приемлемыми критериями стабильности настоящего изобретения являются следующие: согласно тестированию с помощью ВЭЖХ увеличение общего содержания примесей обычно составляет не более приблизительно 1 %, предпочтительно не более 0,5 %, и может составлять 0,01 %, 0,02 %, 0,03 %, 0,04 %, 0,05 %, 0,06 %, 0,07 %, 0,08 %, 0,09 %, 0,1 %, 0,2 %, 0,3 %, 0,4 % и 0,5 %, или/и общее содержание примесей составляет не более 0,5 % и может составлять 0,01 %, 0,02 %, 0,03 %, 0,04 %, 0,05 %, 0,06 %, 0,07 %, 0,08 %, 0,09 %, 0,1 %, 0,2 %, 0,3 %, 0,4 % и 0,5 %, или/и в соответствии с рентгеновской порошковой дифрактометрией, физическая форма активного ингредиента в твердой дисперсии/твердом препарате остается аморфной без явления старения.
Тестирование с помощью рентгеновской порошковой дифрактометрии по настоящему изобретению проводили на комплексном многофункциональном рентгеновском дифрактометре Rigaku UltimaIV. Конкретная информация для обнаружения: Cu анод (40 кВ, 40 мА), Cu-Kα1 луч (λ = 1,5418 Å), скорость сканирования 20 °/мин, диапазон сканирования (диапазон 2q): 3~45°, размер шага сканирования 0,02, ширина щели 0,01.
Условия обнаружения ВЭЖХ по настоящему изобретению: в качестве наполнителя применяли диоксид кремния, связанный октадецилсиланом (колонка Waters Symmetry C18), в качестве подвижной фазы и элюента применяли 0,01 моль/л калиевого дигидрофосфатного буферного раствора и ацетонитрил, длина волны обнаружения составляет 210 нм.
Фармацевтические вспомогательные вещества и реагенты, такие как ацетат сукцинат гидроксипропилметилцеллюлозы, имеются в продаже. (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-он (соединение А) или его фармацевтически приемлемая соль могут быть получены в соответствии со способом, описанным в примере 93 в WO2016007185.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Вышеуказанные и другие объекты и признаки настоящего изобретения будут очевидными со ссылкой на следующие графические материалы:
Фигура 1: рентгенодифракционный спектр соединения А активного фармацевтического ингредиента (АФИ).
Фигура 2: рентгенодифракционный спектр физической смеси соединения А АФИ и носителя ацетат сукцината гидроксипропилметилцеллюлозы.
Фигура 3: рентгенодифракционный спектр носителя ацетат сукцината гидроксипропилметилцеллюлозы.
Фигура 4: рентгенодифракционный спектр твердой дисперсии экспериментального примера 3.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет дополнительно подробно описано со ссылкой на следующие примеры и экспериментальные примеры. Указанные примеры и экспериментальные примеры предназначены только для иллюстративных целей и не должны рассматриваться как ограничивающие объем настоящего изобретения.
Пример 1: Получение твердых дисперсий
Твердые дисперсии получали с применением (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-она (называемого соединением А) и различных видов материалов носителей. Конкретные препараты приведены в таблице 1:
Таблица 1
Способ получения (способ соосаждения):
Соединение А и материал носителя взвешивают в соответствии с препаратами и полностью растворяют в N,N-диметилацетамиде (ДМФА). Рекомендуемое количество очищенной воды взвешивают в соответствии с отношением N,N-диметилацетамида к очищенной воде 1:15 (г/г). Раствор, содержащий соединение А и материал носителя, добавляют по каплям к воде со скоростью потока 30 г/мин, и белый хлопьевидный осадок осаждают, фильтруют, промывают и высушивают для получения твердой дисперсии.
Тестирование на растворение
Скорости растворения смесей АФИ и носителя из экспериментальных примеров 1-3 определяют в соответствии со вторым способом (способ с применением лопастной мешалки) тестирования скорости растворения, описанным в общем правиле тома IV Китайской Фармакопеи, издание 2015 года. Тестирование на растворение проводят с применением 1000 мл 0,15 % водного раствора SDS в качестве среды для растворения при 37 ± 0,5 °С и скорости лопастной мешалки 75 об/мин.
Таблица 2
Результаты показали, что при применении Eudragit L100-55 в качестве материала носителя, растворение было медленным и неполным, скорость растворения составляла всего лишь приблизительно 70 % в течение 45 минут, что влияет в некоторой степени на биодоступность соединения A после получения в виде лекарственного средства, при применении фталата гидроксипропилметилцеллюлозы или ацетат сукцината гидроксипропилметилцеллюлозы, особенно ацетат сукцината гидроксипропилметилцеллюлозы в качестве материала носителя, характеристики растворения значительно улучшались.
Исследование стабильности
Твердую дисперсию экспериментального примера 3 помещали при 25 °С/60 % относительной влажности и 40 °С/75 % относительной влажности соответственно, для исследования стабильности при длительном хранении. Данные представлены следующим образом:
Таблица 3
Пример 2
Соединение А и ацетат сукцинат гидроксипропилметилцеллюлозы (массовое соотношение: 1:1) получают в виде твердой дисперсии, которую измельчают в порошок для соответствия желаемому размеру частиц. Рекомендуемое количество твердой дисперсии, лактозы и микрокристаллической целлюлозы взвешивают в соответствии с разработанным препаратом и добавляют кроскармеллозу натрия, кросповидон, натрий карбоксиметилкрахмал или низкозамещенную гидроксипропилцеллюлозу в качестве разрыхлителя соответственно. Смесь выливают в грануляционную емкость, тщательно перемешивают и добавляют гидроксипропилцеллюлозу в качестве связующего вещества для получения гранул. Влажный и мягкий материал подвергаю мокрому размолу и высушивают, а затем сухие гранулы (содержание воды менее 3 %) подвергают сухому размолу. Добавляют рекомендуемое количество стеарата магния и тщательно перемешивают с гранулами. Полученные общие смешанные гранулы прессуют в таблетки. Конкретные соотношения в препарате приведены в таблице 4.
Таблица 4
Тестирование на растворение
Скорости растворения таблеток экспериментальных примеров с 4 по 7 определяют в соответствии со вторым способом (способ с применением лопастной мешалки) тестирования скорости растворения, описанным в общем правиле тома IV Китайской Фармакопеи, издание 2015 года. Тестирование на растворение проводят с применением 1000 мл 0,15 % водного раствора SDS в качестве среды для растворения при 37 ± 0,5 °С и скорости лопастной мешалки 50 об/мин.
Таблица 5
Результаты показали, что растворение экспериментальных примеров 5 и 7 было медленным, скорость растворения составляла всего приблизительно 50 % в течение 45 минут, и указанные твердые препараты не могли быстро высвобождать фармацевтически активный ингредиент. Напротив, экспериментальные примеры 1 и 3 показали лучшие характеристики растворения, и скорость растворения составляла 90,0 % в течение 45 минут.
Пример 3
Соединение А и ацетат сукцинат гидроксипропилметилцеллюлозы (массовое соотношение: 1:1) получают в виде твердой дисперсии способом соосаждения, которую затем измельчают в порошок. Рекомендуемое количество твердой дисперсии, лактозы, микрокристаллической целлюлозы и кроскармеллозы натрия взвешивают в соответствии с разработанным препаратом. Смесь выливают в грануляционную емкость, тщательно перемешивают и добавляют соответственно гидроксипропилметилцеллюлозу, поливинилпирролидон, предварительно желатинизированный крахмал или гидроксипропилцеллюлозу в качестве связующего вещества для получения гранул. Влажный и мягкий материал подвергают мокрому размолу и высушивают, а затем сухие гранулы (содержание воды менее 3 %) подвергали сухому размолу. Добавляют рекомендуемое количество стеарата магния и тщательно перемешивают с гранулами. Полученные общие смешанные гранулы прессуют в таблетки. Конкретные соотношения в препарате приведены в таблице 6.
Таблица 6
Тестирование на растворение
Скорости растворения таблеток в экспериментальных примерах 4 и с 8 по 11 определяют в соответствии со вторым способом (способ с применением лопастной мешалки) тестирования скорости растворения, описанным в общем правиле тома IV Китайской Фармакопеи, издание 2015 г. Тестирование на растворение проводят с применением 1000 мл 0,15 % водного раствора SDS в качестве среды для растворения при 37 ± 0,5 °С и скорости лопастной мешалки 50 об/мин.
Таблица 7
Результаты показали, что растворение в экспериментальном примере 9 было медленным и неполным, и скорость растворения составляла всего лишь приблизительно 85 % в течение 45 минут. Напротив, экспериментальные примеры с 8 по 11 демонстрировали лучшие характеристики растворения, и скорость растворения составляла 93,0 % в течение 45 минут.
Пример 4
Соединение А и ацетат сукцинат гидроксипропилметилцеллюлозы получают в виде твердой дисперсии способом соосаждения, которую затем измельчают в порошок. Рекомендуемое количество твердой дисперсии, лактозы, микрокристаллической целлюлозы и кроскармеллозы натрия взвешивают в соответствии с разработанным препаратом. Смесь выливают в грануляционную емкость, тщательно перемешивают и добавляют поливинилпирролидон в качестве связующего вещества для получения гранул. Влажный и мягкий материал подвергают мокрому размолу и высушивают, а затем сухие гранулы (содержание воды менее 3 %) подвергают сухому размолу. Добавляют экстрагранулярные вспомогательные вещества и тщательно смешивают с гранулами. Полученные общие смешанные гранулы прессуют в таблетки. Конкретные соотношения в препарате приведены в таблице 8.
Таблица 8
Тестирование на растворение
Скорости растворения таблеток в экспериментальных примерах с 12 по 15 определяют в соответствии со вторым способом (способ с применением лопастной мешалки) тестирования скорости растворения, описанным в общем правиле тома IV Китайской Фармакопеи, издание 2015 года. Тестирование на растворение проводят с применением 1000 мл 0,15 % водного раствора SDS в качестве среды для растворения при 37 ± 0,5 °С и скорости лопастной мешалки 50 об/мин.
Таблица 9
Результаты показывают, что при применении Eudragit L100-55 в качестве материала носителя, растворение было медленным и неполным, скорость растворения составляла всего лишь приблизительно 65 % в течение 45 минут, что влияет на биодоступность соединения A, при применении в качестве материала носителя ацетат сукцината гидроксипропилметилцеллюлозы, скорость растворения значительно улучшалась.
Исследование стабильности
Таблетку из экспериментального примера 14 помещают при 25 °С/60 % относительной влажности и 40 °С/75 % относительной влажности соответственно, для исследования стабильности при длительном хранении. Данные представлены далее:
Таблица 10
Пример 5: Фармакокинетические (ФК) исследования на животных
12 собак породы бигль распределяют в две группы (6 собак породы бигль на группу, половина самцы и половина самки). Собаки породы бигль голодали более 12 часов до начала эксперимента, а корм им предоставляют через 4 часа после введения лекарственного средства в день эксперимента. Воду во время эксперимента животным предоставляют без ограничения. Двум группам животных перорально вводят кристаллическую форму соединения A и твердую дисперсию соединения A соответственно (отношение соединения A к ацетат сукцинату гидроксипропилметилцеллюлозы составляло 1:1, согласно препарату экспериментального примера 3) при дозе введения 30 мг/кг. 0,6 мл крови берут из вен до введения и через 0,25, 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 12, 24 и 72 ч после введения. Кровь хранят в пробирке с антикоагулянтом этилендиаминтетрауксусной кислотой (ЭДТА) и центрифугируют в течение 10 минут при 3500 об/мин (4 °С) для отделения плазмы крови. Плазму хранят при минус 70 °С.
Концентрацию АФИ в плазме крови и вводимом растворе определяют способом жидкостной хроматографии с тандемной масс-спектрометрией (ЖХ/МС/МС). Фармакокинетические параметры собак породы бигль после введения лекарственного средства рассчитывают по полученной концентрации в плазме с применением некомпартментной модели программного обеспечения Phoenix WinNonlin 6.4. Результаты показаны в таблице 11.
Таблица 11
(нг/мл)
Из результатов в таблице 11 видно, что после того, как соединение А было получено в виде твердой дисперсии, абсорбция АФИ in vivo была значительно лучше, чем у кристаллической формы соединения A, что указывает на то, что твердая дисперсия, полученная с применением ацетат сукцината гидроксипропилметилцеллюлозы, может значительно увеличить биодоступность соединения A после получения в виде лекарственного средства и абсорбцию АФИ in vivo.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТВЕРДАЯ ДИСПЕРСИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2020 |
|
RU2816913C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛО[2,3-D]ПИРИДАЗИН-4-ОНЫ И ПИРАЗОЛО[3,4-D]ПИРИДАЗИН-4-ОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2017 |
|
RU2749038C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА ТИРОЗИНКИНАЗЫ БРУТОНА И СПОСОБ ЕЁ ПОЛУЧЕНИЯ | 2016 |
|
RU2728827C2 |
| АМИНОПИРИДАЗИНОНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2674701C2 |
| СОСТАВЫ ТВЕРДЫХ ДОЗИРОВАННЫХ ЛЕКАРСТВЕННЫХ ФОРМ АНТАГОНИСТА ОРЕКСИНОВОГО РЕЦЕПТОРА | 2013 |
|
RU2759837C2 |
| СОСТАВЫ ТВЕРДЫХ ДОЗИРОВАННЫХ ЛЕКАРСТВЕННЫХ ФОРМ АНТАГОНИСТА ОРЕКСИНОВОГО РЕЦЕПТОРА | 2013 |
|
RU2699358C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ГЕПАТИТА С | 2013 |
|
RU2665365C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2015 |
|
RU2671494C2 |
| ТВЁРДЫЕ ДИСПЕРСИИ | 2016 |
|
RU2694832C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ СОЕДИНЕНИЯ ПИРАЗОЛА, ДИСПЕРГИРОВАННОГО В МАТРИЦЕ ПОЛИМЕРА | 2020 |
|
RU2826604C1 |

Группа изобретений относится к области фармацевтики и медицины, а именно: к твердой дисперсии для лечения или предупреждения В-клеточной лимфомы и ревматоидного артрита, содержащей активный ингредиент (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-он или его фармацевтически приемлемую соль и материал носителя, где материал носителя выбран из группы, состоящей из ацетат сукцината гидроксипропилметилцеллюлозы и фталата гидроксипропилметилцеллюлозы, где массовое соотношение материала носителя к активному ингредиенту составляет от 1:1 до 2:1; к способу получения указанной твердой дисперсии, включающему этапы растворения вместе материала носителя и активного ингредиента или его фармацевтически приемлемой соли в органическом растворителе или суспендирования и диспергирования материала носителя в растворе активного ингредиента или его фармацевтически приемлемой соли в органическом растворителе, и затем удаления органического растворителя; и к твердому препарату для лечения или предупреждения В-клеточной лимфомы и ревматоидного артрита, содержащему указанную твердую дисперсию. Группа изобретений обеспечивает повышение растворимости активного ингредиента. 3 н. и 3 з.п. ф-лы, 4 ил., 11 табл., 5 пр.
1. Твердая дисперсия для лечения или предупреждения В-клеточной лимфомы и ревматоидного артрита, содержащая активный ингредиент (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-он или его фармацевтически приемлемую соль и материал носителя, где материал носителя выбран из группы, состоящей из ацетат сукцината гидроксипропилметилцеллюлозы и фталата гидроксипропилметилцеллюлозы, где массовое соотношение материала носителя к активному ингредиенту составляет от 1:1 до 2:1.
2. Твердая дисперсия по п. 1, где материал носителя представляет собой ацетат сукцината гидроксипропилметилцеллюлозы.
3. Твердая дисперсия по п. 1 или 2, состоящая из активного ингредиента (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1,6-дигидро-7Н-пирроло[2,3-d]пиридазин-7-она или его фармацевтически приемлемой соли и материала носителя.
4. Способ получения твердой дисперсии по любому из пп. 1-3, включающий этапы растворения вместе материала носителя и активного ингредиента или его фармацевтически приемлемой соли в органическом растворителе или суспендирования и диспергирования материала носителя в растворе активного ингредиента или его фармацевтически приемлемой соли в органическом растворителе, и затем удаления органического растворителя для получения твердой дисперсии.
5. Способ по п. 4, характеризующийся тем, что органический растворитель удаляют способом осаждения растворителем.
6. Твердый препарат для лечения или предупреждения В-клеточной лимфомы и ревматоидного артрита, содержащий твердую дисперсию по любому из пп. 1-3 и характеризующийся тем, что содержит:
1) от 10 до 500 мг активного ингредиента или его фармацевтически приемлемой соли,
2) от 5 до 15 мас.% разрыхлителя,
3) от 30 до 90 мас.% наполнителя,
4) от 0,5 до 10 мас.% связующего вещества, и
5) от 0,1 до 5 мас.% смазывающего вещества.
| WO 2016007185 A1, 14.01.2016 | |||
| ПЕРЦЕВ И.М | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Т | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| - Харьков: УкрФА | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| Телефонная трансляция | 1922 |
|
SU464A1 |
| WO 2016022942 A1, 11.02.2016 | |||
| WO 2015018521 A1, 12.02.2015 | |||
| EA 201591276 A1, 29.01.2016 | |||
| Междувагонное предохранительное ограждение для трамвая | 1928 |
|
SU15715A1 |

