ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] В этой заявке испрашивается приоритет предварительной заявки на патент США № 62/752685, зарегистрированной 30 октября 2018 года, полное содержание которой включено в настоящее изобретение путем ссылки на нее.
УРОВЕНЬ ТЕХНИКИ
[0002] Обеспечение тканей кислородом на необходимом уровне является непременным условием для поддержания функции и физиологии клеток млекопитающих. Дефицит обеспечения тканей кислородом является характерным для ряда патофизиологических состояний, при которых кровоток недостаточен для обеспечения соответствующей оксигенации. Гипоксическая (с низким содержанием кислорода) тканевая среда активирует сигнальный каскад, который запускает индукцию или репрессию транскрипции множества генов, участвующих в таких событиях, как ангиогенез (неоваскуляризация), метаболизм глюкозы и выживание/гибель клеток. Ключ к этому транскрипционному ответу на гипоксическое воздействие лежит в факторах транскрипции, а именно, факторах, индуцируемых гипоксией (HIF). Факторы HIF являются дисрегулируемыми при множестве типов рака в результате действия зависимых и независимых от гипоксии механизмов, и экспрессию факторов, индуцируемых гипоксией (HIF), связывают с неблагоприятным прогнозом для пациента.
[0003] Факторы HIF состоят из чувствительной к кислороду субъединицы HIFα и конститутивно экспрессируемой субъединицы ΗIFβ. Когда факторы HIF активируются, субъединицы HIFα и ΗIFβ собирают функциональный гетеродимер (субъединица α гетеродимеризуется с субъединицей β). Как HIFα, так и ΗIFβ имеют две идентичные структурные характеристики: основную спираль-петлю-спираль (bHLH) и домены PAS (PAS представляет собой аббревиатуру, относящуюся к первым белкам, PER, ARNT, SIM, в которых был идентифицирован этот мотив). Существует три субъединицы HIFα человека (HIF-1α, HIF-2α и HIF-3α), чувствительные к кислороду. Среди трех субъединиц, HIF-1α экспрессируется наиболее повсеместно и индуцируется низкими концентрациями кислорода во многих типах клеток и тканей. HIF-2α очень похож на HIF-1α как по структуре, так и по функциям, но проявляет более ограниченную клеточную и тканеспецифичную экспрессию, а также может дифференцированно регулироваться ядерной транслокацией. HIF-3α также проявляет консервативность с HIF-1α и HIF-2α в доменах HLH и PAS. HIF-1β (также называемый ядерным транслокатором арилуглеводородного рецептора), партнер по димеризации субъединиц HIFα, конститутивно экспрессируется во всех типах клеток и не регулируется концентрацией кислорода.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0004] В конкретных аспектах, в настоящем изобретении предлагается твердая дисперсия, включающая соединение формулы (I):
Формула (I).
В некоторых вариантах осуществления, твердая дисперсия дополнительно включает фармацевтически приемлемый полимер. Полимер может включать гидрофобные области и гидрофильные области. В некоторых вариантах осуществления, полимер выбирают из сложных эфиров целлюлозы; простых эфиров целлюлозы; полиалкиленоксидов; поливинилхлоридов; поливиниловых спиртов; полиакрилатов; полиметакрилатов; гомополимеров и сополимеров N-виниллактамов, полиакриламидов и винилацетатов; графт-сополимеров полиэтиленгликоля, поливинилкапролактама и поливинилацетата; олигосахаридов; полисахаридов; и их смесей. В некоторых вариантах осуществления, полимер представляет собой сложный эфир целлюлозы или простой эфир целлюлозы. В некоторых вариантах осуществления, полимер выбирают из метилцеллюлозы, этилцеллюлозы, гидроксиэтилцеллюлозы, ацетатфталата целлюлозы (CAP), гидроксипропилметилцеллюлозы (HPMC), гидроксипропилцеллюлозы, фталата гидроксипропилметилцеллюлозы (HPMCP), ацетатсукцината гидроксипропилметилцеллюлозы (HPMCAS), метилового эфира полиэтиленгликоля, графт-сополимера полиэтиленгликоль-поливинилацетат-поливинилкапролактам (Soluplus), полиэтиленгликоля 6000 (PEG 6000), поливинилпирролидона (PVP), сополимера поливинилпирролидона и винилацетата (PVP-VA), сополимера этилакрилата, метилметакрилата и триметиламмониоэтилметакрилата хлорида 1:2:0.1 (например, Eudragit RS 100), сополимера метакриловой кислоты типа В (например, Eudragit S 100), сополимера метакриловой кислоты типа В с поливинилацетатфталатом (например, Sureteric), блок-сополимера полиоксиэтилен-полиоксипропилен (например, Pluronic F-68) и полиоксиэтилена (20) сорбитан моноолеата (Tween 80). В некоторых вариантах осуществления, полимер выбирают из HPMCAS, CAP и графт-сополимера полиэтиленгликоль-поливинилацетат-поливинилкапролактам (например, Soluplus). В некоторых вариантах осуществления, полимер выбирают из HPMCAS-L, HPMCAS-M и HPMCAS-H, например, HPMCAS-H.
[0005] В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве от 1% до 50% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, полимер присутствует в количестве от 50% до 99% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве от 15% до 35% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, полимер присутствует в количестве от 65% до 85% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве от 22,5% до 27,5% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, полимер присутствует в количестве от 72,5% до 77,5% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве приблизительно 25% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, полимер присутствует в количестве приблизительно 75% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, массовое отношение соединения формулы (I) к полимеру составляет от 1:99 до 1:1, например, от 15:85 до 35:65. В некоторых вариантах осуществления, массовое отношение соединения формулы (I) к полимеру составляет от 22,5:77,5 до 27,5:72,5, например, приблизительно 25:75.
[0006] В некоторых вариантах осуществления, твердая дисперсия является, по существу, некристаллической. В некоторых вариантах осуществления, твердая дисперсия является аморфной. В некоторых вариантах осуществления, твердая дисперсия характеризуется температурой стеклования (Tg) от 80 до 100°C, например, от 82 до 92°C. В некоторых вариантах осуществления, твердая дисперсия характеризуется температурой стеклования (Tg) приблизительно 87°C, например, 87±3°C. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 2% примесей по массе. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 2% примесей по массе после трех месяцев хранения при комнатной температуре. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 2% воды по массе. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 2% воды после трех месяцев хранения при комнатной температуре. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 5000 ppm ацетона. В некоторых вариантах осуществления, энантиомерная чистота соединения формулы (I) составляет, по меньшей мере, 95%, например, по меньшей мере 99%.
[0007] В некоторых вариантах осуществления, распределение частиц по размерам твердой дисперсии характеризуется величиной d10 менее чем 6 мкм. В некоторых вариантах осуществления, распределение частиц по размерам твердой дисперсии характеризуется величиной d50 менее чем 18 мкм. В некоторых вариантах осуществления, распределение частиц по размерам твердой дисперсии характеризуется величиной d90 менее чем 45 мкм. В некоторых вариантах осуществления, твердая дисперсия характеризуется насыпной плотностью, по меньшей мере, 0,20 г/мл. В некоторых вариантах осуществления, твердая дисперсия характеризуется насыпной плотностью после уплотнения, по меньшей мере, 0,35 г/мл. В некоторых вариантах осуществления, распределение частиц по размерам твердой дисперсии характеризуется величиной d10 менее чем 15 мкм. В некоторых вариантах осуществления, распределение частиц по размерам твердой дисперсии характеризуется величиной d50 менее чем 45 мкм. В некоторых вариантах осуществления, распределение частиц по размерам твердой дисперсии характеризуется величиной d90 менее чем 90 мкм. В некоторых вариантах осуществления, твердая дисперсия характеризуется насыпной плотностью, по меньшей мере, 0,15 г/мл. В некоторых вариантах осуществления, твердая дисперсия характеризуется насыпной плотностью после уплотнения, по меньшей мере, 0,30 г/мл. В некоторых вариантах осуществления, твердая дисперсия характеризуется величиной Cmax GB (максимальной концентрацией в буфере, моделирующем желудочный сок), по меньшей мере, 300 мкгА/мл. В некоторых вариантах осуществления, твердая дисперсия характеризуется величиной Cmax FaSSIF (максимальной концентрацией в биорелевантной среде, моделирующей кишечный сок натощак и после еды), по меньшей мере 400 мкгА/мл. В некоторых вариантах осуществления, твердая дисперсия характеризуется величиной AUC FaSSIF (площади под фармакокинетической кривой в биорелевантной среде, моделирующей кишечный сок натощак и после еды), по меньшей мере, 40000 мкгА/мл. В некоторых вариантах осуществления, твердая дисперсия характеризуется величиной AUC FaSSIF, по меньшей мере, 85000 мкгА/мл. В некоторых вариантах осуществления, твердую дисперсию получают путем сушки методом распыления. В некоторых вариантах осуществления, твердую дисперсию получают путем плавления, испарения растворителя, распылительной сушки, слияния, замешивания, совместного измельчения, лиофилизации, экструзией горячего расплава, агломерации в расплаве или сверхкритической флюидной технологии.
[0008] В конкретных аспектах, в настоящем изобретении предлагается аморфная твердая дисперсия, включающая в расчете на суммарную массу твердой дисперсии:
(a) от 22,5% до 27,5% соединения формулы (I):
Формула (I)
и
(b) от 72,5% до 77,5% HPMCAS.
[0009] В конкретных аспектах, в настоящем изобретении предлагается фармацевтическая композиция, включающая описанную в изобретении твердую дисперсию и фармацевтически приемлемое вспомогательное вещество. В некоторых вариантах осуществления, фармацевтическая композиция представляет собой капсулу или таблетку. В некоторых вариантах осуществления, фармацевтическую композицию приготавливают для перорального введения. В некоторых вариантах осуществления, фармацевтически приемлемое вспомогательное вещество включает связующее вещество, наполнитель, разрыхлитель, смазывающее вещество, скользящее вещество или их комбинацию. В некоторых вариантах осуществления, твердая дисперсия присутствует в количестве от 15% до 50% от массы фармацевтической композиции. В некоторых вариантах осуществления, фармацевтическая композиция включает, в расчете на суммарную массу фармацевтической композиции: (a) от 15% до 50% твердой дисперсии; (b) от 20% до 50% связующего вещества; (c) от 20% до 40% наполнителя; (d) от 1,0% до 5,0% разрыхлителя; и (e) от 0,25% до 1,25% смазывающего вещества. В некоторых вариантах осуществления, связующее вещество представляет собой микрокристаллическую целлюлозу. В некоторых вариантах осуществления, наполнитель представляет собой маннит. В некоторых вариантах осуществления, разрыхлитель представляет собой кроскармеллозу натрия. В некоторых вариантах осуществления, смазывающее вещество представляет собой стеарат магния. В некоторых вариантах осуществления, фармацевтическая композиция дополнительно включает, в расчете на суммарную массу фармацевтической композиции, от 0,1% до 1,25% скользящего вещества, где, необязательно, скользящее вещество представляет собой коллоидный диоксид кремния. В некоторых вариантах осуществления, фармацевтическая композиция дополнительно включает покрытие, где, необязательно, покрытие представляет собой покрытие на основе поливинилового спирта. В конкретных вариантах осуществления, покрытие на основе поливинилового спирта в фармацевтической композиции дополнительно включает полиэтиленгликоль. В конкретных вариантах осуществления, фармацевтическая композиция имеет покрытие, которое представляет собой OpaDry II.
[0010] В конкретных аспектах, в настоящем изобретении предлагается упакованная твердая дисперсия, включающая описанную в изобретении твердую дисперсии и осушающее вещество. В некоторых вариантах осуществления, осушающее вещество представляет собой SiO2. В некоторых вариантах осуществления, упаковка включает контейнер с низкой проницаемостью для паров воды.
[0011] В конкретных аспектах, в настоящем изобретении предлагается способ лечения болезни фон Гиппеля - Линдау (VHL), включающий введение субъекту, нуждающемуся в этом, эффективного количества описанной в изобретении твердой дисперсии или фармацевтической композиции. В некоторых вариантах осуществления, субъект также страдает от гемангиобластомы, феохромоцитомы, панкреатической нейроэндокринной опухоли или почечно-клеточной карциномы, например, от почечно-клеточной карциномы. В настоящем изобретении также предлагается способ лечения почечно-клеточной карциномы, включающий введение субъекту, нуждающемуся в этом, эффективного количества описанной в изобретении твердой дисперсии или фармацевтической композиции. В некоторых вариантах осуществления, почечно-клеточная карцинома представляет собой светлоклеточную почечно-клеточную карциному.
[0012] В конкретных аспектах, в настоящем изобретении предлагается способ лечения HIF-2α-опосредованного заболевания или состояния, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества описанной в изобретении твердой дисперсии или фармацевтической композиции. В некоторых вариантах осуществления, заболевание или состояние представляет собой рак. В некоторых вариантах осуществления, заболевание или состояние выбирают из почечно-клеточной карциномы, болезни фон Гиппеля - Линдау, легочной артериальной гипертензии, глиобластомы и колита. В настоящем изобретении также предлагается способ ингибирования HIF-2α, включающий контактирование HIF-2α с эффективным количеством описанной в изобретении твердой дисперсии или фармацевтической композиции. Описанные в изобретении способы могут дополнительно включать введение второго терапевтического средства.
[0013] В конкретных аспектах, в настоящем изобретении предлагается способ приготовления описанной в изобретении твердой дисперсии, включающий: (a) получение раствора соединения формулы (I) и полимера в растворителе; и (b) и удаление растворителя с получением твердой дисперсии. В некоторых вариантах осуществления, растворитель включает ацетон, метилэтилкетон, тетрагидрофуран, воду или их комбинацию. В некоторых вариантах осуществления, растворитель включает ацетон. В некоторых вариантах осуществления, растворитель включает до 5% воды. В некоторых вариантах осуществления, растворитель удаляют лиофилизацией или распылительной сушкой. В некоторых вариантах осуществления, растворитель удаляют распылительной сушкой. Способ может дополнительно включать сушку твердой дисперсии в полочной сушилке, в которой удаляют остаточный растворитель. В некоторых вариантах осуществления, раствор включает от 8% до 14% твердых веществ по массе.
[0014] В конкретных аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает (a) твердую дисперсию, включающую соединение формулы (I); и (b) одно или более фармацевтически приемлемых вспомогательных веществ. В некоторых вариантах осуществления, твердая лекарственная форма представляет собой капсулу или таблетку. В некоторых вариантах осуществления, твердая лекарственная форма представляет собой таблетку. В некоторых вариантах осуществления, одно или более фармацевтически приемлемых вспомогательных веществ включают связующее вещество, наполнитель, разрыхлитель и смазывающее вещество. В некоторых вариантах осуществления, твердая дисперсия присутствует в количестве от 15% до 50% от массы твердой лекарственной формы. В некоторых вариантах осуществления, твердая дисперсия включает фармацевтически приемлемый полимер. В некоторых вариантах осуществления, фармацевтически приемлемый полимер представляет собой HPMCAS. В некоторых вариантах осуществления, полимер присутствует в количестве от 15% до 35% от массы твердой лекарственной формы. В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве от 1% до 15% от массы твердой лекарственной формы. Твердая лекарственная форма может включать от 5 мг до 100 мг соединения формулы (I), например, приблизительно 10 мг соединения формулы (I) или приблизительно 40 мг соединения формулы (I).
[0015] Твердая лекарственная форма по настоящему изобретению может включать связующее вещество в количестве от 20% до 50% от массы твердой лекарственной формы, где, необязательно, связующее вещество представляет собой микрокристаллическую целлюлозу. Твердая лекарственная форма может включать наполнитель в количестве от 20% до 40% от массы твердой лекарственной формы. В некоторых вариантах осуществления, твердая лекарственная форма включает интрагранулярный наполнитель и экстрагранулярный наполнитель, где интрагранулярный наполнитель присутствует в количестве от 12% до 22% от массы твердой лекарственной формы, и где экстрагранулярный наполнитель присутствует в количестве от 8% до 18% от массы твердой лекарственной формы. В некоторых вариантах осуществления, наполнитель представляет собой маннит. Твердая лекарственная форма может включать разрыхлитель в количестве от 1,0% до 5,0% от массы твердой лекарственной формы. В некоторых вариантах осуществления, твердая лекарственная форма включает интрагранулярный разрыхлитель и экстрагранулярный разрыхлитель, где интрагранулярный разрыхлитель присутствует в количестве от 0,9% до 3,0% от массы твердой лекарственной формы, и где экстрагранулярный разрыхлитель присутствует в количестве от 0,1% до 2,0% от массы твердой лекарственной формы. В некоторых вариантах осуществления, разрыхлитель представляет собой кроскармеллозу натрия. Твердая лекарственная форма может включать смазывающее вещество в количестве от 0,25% до 1,25% от массы твердой лекарственной формы. В некоторых вариантах осуществления, твердая лекарственная форма включает интрагранулярное смазывающее вещество и экстрагранулярное смазывающее вещество, где интрагранулярное смазывающее вещество присутствует в количестве от 0,15% до 0,75% от массы твердой лекарственной формы, и где экстрагранулярное смазывающее вещество присутствует в количестве от 0,10% до 0,50% от массы твердой лекарственной формы. В некоторых вариантах осуществления, смазывающее вещество представляет собой стеарат магния. В некоторых вариантах осуществления, твердая лекарственная форма включает скользящее вещество в количестве от 0,10% до 1,25% от массы твердой лекарственной формы. В некоторых вариантах осуществления, скользящее вещество представляет собой коллоидный диоксид кремния, например, CabOSil. В некоторых вариантах осуществления, твердая лекарственная форма включает покрытие. В некоторых вариантах осуществления, покрытие представляет собой покрытие на основе PVA, например, OpaDry II.
[0016] Твердая лекарственная форма по настоящему изобретению может характеризоваться твердостью от 5 до 25 KP (единиц твердости по Кнупу). В некоторых вариантах осуществления, масса твердой лекарственной формы составляет от 50 до 750 мг, например, приблизительно 125 мг или приблизительно 500 мг. В некоторых вариантах осуществления, твердая лекарственная форма включает менее чем 2% примесей по массе. В некоторых вариантах осуществления, твердая лекарственная форма включает менее чем 2% примесей по массе после шести месяцев хранения при комнатной температуре. В некоторых вариантах осуществления, твердая лекарственная форма включает менее чем 3% воды по массе. В некоторых вариантах осуществления, твердая лекарственная форма включает менее чем 3% воды по массе после шести месяцев хранения при комнатной температуре. В некоторых вариантах осуществления, твердая лекарственная форма характеризуется временем распадаемости от 1 до 5 минут.
[0017] В конкретных аспектах, в настоящем изобретении предлагается упакованная твердая лекарственная форма, включающая описанную в изобретении твердую лекарственную форму и осушающее вещество. В некоторых вариантах осуществления, осушающее вещество представляет собой SiO2. В некоторых вариантах осуществления, упаковка включает контейнер с низкой проницаемостью для паров воды. Упакованная лекарственная форма может дополнительно включать хлопковый, вискозный или полиэфирный жгут. В настоящем изобретении также предлагается набор, включающий описанную в изобретении твердую лекарственную форму и инструкции по введению твердой лекарственной формы субъекту, нуждающемуся в этом.
[0018] В конкретных аспектах, в настоящем изобретении предлагается способ лечения болезни фон Гиппеля - Линдау (VHL), включающий введение субъекту, нуждающемуся в этом, описанной в изобретении твердой лекарственной формы. В некоторых вариантах осуществления, субъект также страдает гемангиобластомой, феохромоцитомой, панкреатической нейроэндокринной опухолью или почечно-клеточной карциномой, например, почечно-клеточной карциномой. В настоящем изобретении также предлагается способ лечения почечно-клеточной карциномы, включающий введение субъекту, нуждающемуся в этом, описанной в изобретении твердой лекарственной формы. В некоторых вариантах осуществления, почечно-клеточная карцинома представляет собой светлоклеточную почечно-клеточную карциному.
[0019] В конкретных аспектах, в настоящем изобретении предлагается способ лечения HIF-2α-опосредованного заболевания или состояния, включающий введение субъекту, нуждающемуся в этом, описанной в изобретении твердой лекарственной формы. В некоторых вариантах осуществления, заболевание или состояние представляет собой рак. В некоторых вариантах осуществления, заболевание или состояние выбирают из почечно-клеточной карциномы, болезни фон Гиппеля - Линдау, легочной артериальной гипертензии, глиобластомы и колита. В настоящем изобретении также предлагается способ ингибирования HIF-2α, включающий контактирование HIF-2α с описанной в изобретении твердой лекарственной формой. Любой из заявленных способов может дополнительно включать введение второго терапевтического средства.
[0020] В конкретных аспектах, в настоящем изобретении предлагается способ приготовления описанной в изобретении твердой лекарственной формы, включающий: (a) смешение соединения формулы (I) и одного или более фармацевтически приемлемых вспомогательных веществ с формированием нарезанных гранул; и (b) прессование гранул путем приложения усилия прессования от 5 кН до 20 кН. В настоящем изобретении также предлагается способ приготовления описанной в изобретении твердой лекарственной формы, включающий: (a) перемешивание соединения формулы (I), связующего вещества, наполнителя, разрыхлителя и смазывающего вещества с формированием перемешенной смеси; (b) грануляцию перемешенной смеси, необязательно, с использованием роликового пресса с формированием гранулированной смеси; (c) смешение второго наполнителя, второго разрыхлителя и второго смазывающего вещества с гранулированной смесью с формированием смеси для таблетирования; и (d) прессование смеси для таблетирования в таблетку, где наполнитель и второй наполнитель являются одинаковыми или разными; разрыхлитель и второй разрыхлитель являются одинаковыми или разными; и смазывающее вещество и второе смазывающее вещество являются одинаковыми или разными. В некоторых вариантах осуществления, способ дополнительно включает смешение скользящего вещества с гранулированной смесью (c). В некоторых вариантах осуществления, способ дополнительно включает нанесение на таблетку слоя покрытия, такого как OpaDry II.
ВКЛЮЧЕНИЕ В ИЗОБРЕТЕНИЯ СВЕДЕНИЙ ПУТЕМ ССЫЛКИ
[0021] Содержание всех публикаций, патентов и заявок на патенты, упомянутых в данном изобретении, включено в настоящее изобретение путем ссылок на них в той же степени, как если бы содержание каждой отдельная публикации, патента или заявки на патент было бы специально и индивидуально включено путем ссылки на нее.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0022] На фигуре 1 приведена порошковая рентгенограмма кристаллического соединения формулы (I).
[0023] На фигуре 2 показано наложение дифрактограмм семи твердых дисперсий соединения формулы (I), кристаллического соединения формулы (I), и высушенного распылением соединения формулы (I). Последние две дифрактограммы характеризуются четко различающимися пиками.
[0024] На фигуре 3 показано наложение дифрактограмм трех твердых дисперсий соединения формулы (I) и кристаллического соединения формулы (I).
[0025] На фигуре 4 приведены полученные методом сканирующей растровой электронной микроскопии (SEM) изображения твердых дисперсий соединения формулы (I) и HPMCAS-H (слева), CAP (посередине) или SOLUPLUS (справа) при 5000-кратном увеличении.
[0026] На фигуре 5 показано три серии наложений порошковых рентгенограмм твердых дисперсий соединения формулы (I) и кристаллического соединения формулы (I). В каждой серии, верхняя уширенная кривая представляет твердую дисперсию, хранившуюся на воздухе в течение 4 недель при 40°C и 75% RH, средняя уширенная кривая представляет твердую дисперсию, хранившуюся без доступа воздуха в течение 4 недель при тех же самых условиях, и нижняя кривая представляет твердую дисперсию до хранения (t=0). Кривая, содержащая четко различающиеся пики, представляет кристаллическое соединение формулы (I).
[0027] На фигуре 6 приведена порошковая рентгенограмма твердой дисперсии 1:3 соединение формулы (I):HPMCAS-H.
[0028] На фигуре 7 приведены полученные методом сканирующей растровой электронной микроскопии (SEM) изображения твердой дисперсии 1:3 соединение формулы (I):HPMCAS-H при 500-кратном увеличении (слева) и 5000-кратном увеличении (справа).
[0029] На фигуре 8 представлена кривая распределения частиц по размерам для твердой дисперсии.
[0030] На фигуре 9 приведены кривые высокоэффективной жидкостной хроматографии (HPLC) холостого образца (нижняя кривая), соединения формулы (I) (средняя кривая) и твердой дисперсии соединения формулы (I) и HPMCAS-H (верхняя кривая).
[0031] На фигуре 10 представлены профили модулированной дифференциальной сканирующей калориметрии (mDSC) твердой дисперсии 1:3 соединение формулы (I):HPMCAS-H через 24 часа после воздействия среды с 33% влажностью (верхняя кривая), 50% влажностью (средняя кривая) или 75% влажностью (нижняя кривая).
[0032] На фигуре 11 приведены порошковые рентгенограммы влажных твердых дисперсий, выдерживаемых при комнатной температуре в течение 1 дня, 3 дней, 5 дней или 7 дней (кривые сверху вниз, соответственно).
[0033] На фигуре 12 приведена порошковая рентгенограмма твердой дисперсии 1:3 соединение формулы (I):HPMCAS-H.
[0034] На фигуре 13 представлена кривая распределения частиц по размерам для твердой дисперсии.
[0035] На фигуре 14 изображена блок-схема процесса приготовления твердой лекарственной формы.
[0036] На фигуре 15 приведен профиль растворимости для таблеток 10 мг и 40 мг.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0037] При серийном производстве подходящих для применения в медицине потенциальных лекарственных средств, необходимо, как правило, следовать принципам надлежащей практике организации производства. В изобретении предлагаются способы приготовления фармацевтических композиций, включающих соединение формулы (I):
Формула (I).
[0038] Предлагаемые в изобретении композиции обладают предпочтительными физическими свойствами, которые могут обеспечивать положительные эффекты с точки зрения переработки, приготовления, стабильности, биодоступности, хранения и транспортировки, наряду с другими важными фармацевтическими характеристиками. Описанные в изобретении способы позволяют осуществить серийное производство в соответствии с принципами надлежащей практике организации производства (GMP).
[0039] Если не указано иное, то используемое в изобретении соединение, называемое в изобретении 3-(((1S,2S,3R)-2,3-дифтор-1-гидрокси-7-(метилсульфонил)-2,3-дигидро-1H-инден-4-ил)окси)-5-фторбензонитрилом, соответствует соединению формулы (I), изображенной ниже.
Формула (I).
[0040] Если не указано иное, то все используемые в изобретении технические и научные термины имеют значения, которые являются общепринятыми для любого специалиста в области, к которой относится настоящее изобретение.
[0041] Используемая в описании изобретения и пунктах формулы изобретения форма единственного числа включает в себя также множественное число, если из контекста в явном виде не следует иное.
[0042] "Необязательный" или "необязательно" означает, что последовательно описываемое событие или обстоятельство может происходить или может не происходить, и что описание включает случаи, когда событие или обстоятельство происходит, и случаи, при которых оно не происходит. Например, "необязательно замещенный арил" означает, что арильная группа может быть замещенной или не замещенной, и что описание включает как замещенные арильные группы, так и арильные группы, не имеющие замещения.
[0043] Соединение формулы I также включает кристаллические и аморфные формы соединения, фармацевтически приемлемых солей и активных метаболитов этих соединений, имеющих такой же тип активности, в том числе, например, сокристаллы, полиморфы, псевдополиморфы, сольваты, гидраты, несольватированные полиморфы (в том числе ангидраты), конформационные полиморфы, и аморфные формы соединения, а также их смеси.
[0044] Используемый в изобретении термин "сокристалл" относится к твердой фазе (которая может быть или может не быть кристаллической), в которой два или более различных молекулярных и/или ионных компонентов (обычно в стехиометрическом отношении) удерживаются вместе за счет неионных взаимодействий, включающих, но этим не ограничивая, водородную связь, диполь-дипольные взаимодействия, диполь-квадрупольные взаимодействия или силы дисперсионного взаимодействия (силы Ван-дер-Ваальса). Не происходит передача протона между неодинаковыми компонентами, и твердая фаза не представляет собой ни простую соль, ни сольват. Обсуждение сокристаллов можно найти, например, в публикации S. Aitipamula et al., Crystal Growth and Design. 2012, 12 (5), pp. 2147-2152.
[0045] Используемый в изобретении термин "твердая форма" относится к соединению, которое преимущественно не находится в жидком или газообразном состоянии. Используемый в изобретении термин "твердая форма" включает в себя полутвердые вещества. Твердые формы могут быть кристаллическими, аморфными, частично кристаллическими, частично аморфными, или их смесями. "Однокомпонентная" твердая форма, включающая соединение формулы (I), состоит, главным образом, из соединения формулы (I). "Многокомпонентная" твердая форма, включающая соединение формулы (I), включает один или более дополнительных компонентов, таких как ионы и/или молекулы, внутри твердой формы. Например, в конкретных вариантах осуществления, аморфная многокомпонентная твердая форма, включающая соединение формулы (I), включает один или более полимеров и соединение формулы (I), диспергированное в твердой матрице, которая включает полимер (полимеры).
[0046] Термин "кристаллический", когда его используют для описания вещества, соединения, компонента, продукта или формы, означает, что вещество, соединение, компонент, продукт или форма является по существу кристаллическим, что устанавливается, например, методом рентгеноструктурного анализа.
[0047] Термин "кристаллическая форма" относится к кристаллическими модификациям, включающим данное вещество, в том числе к однокомпонентным кристаллическим формам, и включающим, но этим не ограничивая, полиморфы, сольваты, гидраты, сокристаллы, другие молекулярные комплексы, соли, сольваты солей, гидраты солей, сокристаллы солей, и другие молекулярные комплексы солей, и их полиморфы. В некоторых вариантах осуществления, кристаллическая форма вещества может практически не содержать аморфных форм и/или других кристаллических форм. В других вариантах осуществления, кристаллическая форма вещества может содержать менее чем приблизительно 1%, 2%, 3%, 4%, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45% или 50% одной или более аморфных форм и/или других кристаллических форм по массе. Кристаллические формы вещества могут быть получены целым рядом методов. Такие методы включают, но этим не ограничивая, перекристаллизацию из расплава, охлаждение расплава, перекристаллизацию из растворителя, перекристаллизацию в ограниченном пространстве, таком как, например, в нанопорах или капиллярах, перекристаллизацию на поверхностях или шаблонах, таких как, например, на полимерах, перекристаллизацию в присутствии добавок, таких как, например, сокристаллические противомолекулы, десольватацию, дегидратацию, быстрое испарение, быстрое охлаждение, медленное охлаждение, диффузию из паровой фазы, сублимацию, измельчение и измельчение с добавкой небольшого количества растворителя.
[0048] Термины "полиморф" и "полиморфная форма" относятся к двум или более кристаллическим формам, которые состоят, по существу, из одной и той же молекулы, одних и тех же молекул или ионов. Различные полиморфы могут иметь различные физические свойства, такие как, например, температуры плавления, теплоты плавления, растворимости, скорости растворения и/или колебательные спектры, вследствие различной группировки или конформации молекул или ионов в кристаллической решетке. Различия в физических свойствах, проявляемые полиморфами, могут влиять на фармацевтические параметры, такие как стабильность свойств при хранении, прессуемость и плотность (важные при приготовлении и производстве), и скорость растворения (важный фактор для биодоступности). Различия в стабильности могут быть следствием изменений реакционной способности (например, дифференциальное окисление, в результате чего лекарственная форма теряет окраску более быстро, когда она состоит из одного полиморфа, чем когда она состоит из другого полиморфа) или механических изменений (например, таблетки крошатся при хранении в виду того, что полиморф с благоприятными кинетическими характеристиками превращается в более термодинамически стабильный полиморф), или в результате и того и другого (например, таблетки из одного полиморфа более подвержены разрушению при высокой влажности). В результате различий в растворимости/растворении, в экстремальном случае, некоторые полиморфные переходы могут приводить к потере активности или, в другом экстремальном случае, к проявлению токсичности. Кроме того, физические свойства кристалла могут играть важную роль в процессе технологической обработки, например, один полиморф может характеризоваться большей вероятностью образования сольватов или возникновением проблем при фильтровании и при отмывке водой от примесей (например, форма частиц и распределение частиц по размерам могут быть различными для разных полиморфов).
[0049] Термины "аморфный" и "аморфная форма" применяют в изобретении для описания вещества, компонента или продукта, которые не являются, по существу, кристаллическими, что устанавливается методом рентгеноструктурного анализа. В конкретных вариантах осуществления, аморфная форма вещества может практически не содержать кристаллические формы. В других вариантах осуществления, аморфная форма вещества может содержать менее чем приблизительно 1%, 2%, 3%, 4%, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45% или 50% одной или более кристаллических форм по массе. В других вариантах осуществления, аморфная форма вещества может включать дополнительные компоненты или ингредиенты (например, добавку, полимер или вспомогательное вещество, которые могут способствовать дополнительной стабилизации аморфной формы). В некоторых вариантах осуществления, аморфная форма может представлять собой твердый раствор. Аморфные формы вещества могут быть получены целом рядом методов. Такие методы включают, но этим не ограничивая, нагревание, охлаждение расплава, быстрое охлаждение расплава, испарение растворителя, быстрое испарение растворителя, десольватацию, сублимацию, измельчение, размол в шаровой мельнице, криоизмельчение, распылительную сушку и лиофилизацию.
[0050] Если не указано иное, то термин "твердая дисперсия" относится к твердому состоянию, которое включает, по меньшей мере, два образующих компонента, где один образующий компонент гомогенно диспергирован в значительной степени равномерно в другом образующем компоненте или компонентах. Твердая дисперсия включает твердые или стеклообразные растворы, то есть, дисперсия образующих ее компонентов является такой композицией, которая по своему характеру является химически и физически гомогенной. В одном варианте осуществления, первый образующий компонент представляет собой активный фармацевтический ингредиент (API), такой как соединение формулы (I), а второй образующий компонент представляет собой матрицу, которая включает полимер, где API диспергирован в значительной степени равномерно внутри матрицы (в полимере). API может присутствовать в аморфном состоянии или в мелкокристаллической диспергированной форме. Кроме того, API может находиться в виде смеси аморфной и кристаллической форм. Твердая дисперсия может включать более чем два образующих компонента. Например, два или более API могут быть диспергированы внутри матрицы, и матрица может включать два или более полимеров. Без ограничения, твердые дисперсии могут быть отнесены с физической точки зрения к эвтектической смеси, твердому раствору, стеклянному раствору или суспензии, аморфному осадку в стеклообразном или кристаллическом носителе, комплексу, комплексному образованию или комбинации различных систем. Кроме того, твердые дисперсии могут быть приготовлены с использованием различных методов, известных специалистам в данной области, например, путем совместного растворения API и полимера в растворителе, затем использования распылительной сушки, распылительного отверждения, испарения, отверждения или воздействия микроволнового излучения, путем смешения и прямого прессования, механического смешения при повышенной температуре, но ниже температуры плавления, влажного гранулирования, экструзии с последующей сферонизацией, смешения в расплаве, экструзии горячего расплава и путем использования других подобных методов. "Твердая матрица" относится к матрице, которая представляет собой твердое вещество.
[0051] Термин "полимер" относится к соединению, включающему повторяющиеся структурные звенья (мономеры), соединенные ковалентными химическими связями. Полимеры могут быть дополнительно дериватизированы, сшиты, привиты или подвергнуты блокировке концевых групп. Неограничивающие примеры полимеров включают сополимеры, терполимеры, четвертичные полимеры и гомологи. Термин "сополимер" относится к полимеру, состоящему, по существу, из двух или более различных типов повторяющихся структурных звеньев (мономеров).
[0052] Термины "около" и "приблизительно", применительно к количеству, относятся к количеству, которое отклоняется от указанного количества на 30%, например, на 20%, 15%, 10% или 5%.
[0053] Описанные в изобретении соединения могут иметь природный изотопный состав, или один или более из атомов могут быть искусственным образом обогащены конкретны изотопом, имеющим такой же атомный номер, но атомную массу или массовое число, отличающиеся от атомной массы или массового числа, преимущественно обнаруживаемые в природе. Все изотопные варианты соединений по настоящему изобретению, будь то радиоактивные или нерадиоактивные, включены в объем настоящего изобретения. Например, водород имеет три природных изотопа, обозначаемых как 1H (протий), 2H (дейтерий) и 3H (тритий). Протий является самым распространенным изотопом в природе. Обогащение дейтерием может давать конкретные терапевтические преимущества, такие как повышенный in vivo период полувыведения и/или повышенная экспозиция, или может давать соединение, применяемое для исследования in vivo путей выведения из организма и метаболизма лекарственного средства. Обогащенные изотопами соединения могут быть приготовлены традиционными методами, хорошо известными специалистам в данной области.
[0054] "Изомеры" представляют собой различные соединения, которые имеют одинаковую молекулярная формула. "Стереоизомеры" представляют собой изомеры, которые отличаются только расположением атомов в пространстве. "Энантиомеры" представляют собой пару стереоизомеров, чьи зеркальные изображения не налагаются друг на друга. Смесь 1:1 пары энантиомеров называют "рацемической смесью". Термин "(±)" используют в соответствующих случаях для обозначения рацемической смеси. "Диастереоизомеры" или "диастереомеры" представляют собой стереоизомеры, которые имеют, по меньшей мере, два асимметрических атома, которые не являются зеркальными изображениями друг друга. Абсолютную стереохимию указывают в соответствии с номенклатурой Кана - Ингольда - Прелога. Когда соединение является чистым энантиомером, стереохимия на каждом хиральном углероде может быть указана или как R, или как S. Выделенные индивидуальные соединения, чья абсолютная конфигурация является неизвестной, могут быть обозначены (+) или (-), в зависимости от направления (вращения вправо или лево), в котором они вращают плоскополяризованный свет при длине волны D-линии натрия. Конкретные описанные в изобретении соединения содержат один или более центров асимметрии и могут образовывать вследствие этого энантиомеры, диастереомеры и другие стереоизомерные формы, центры асимметрии которых могут быть определены, в терминах абсолютной стереохимии, как (R)- или (S)-. Предполагается, что настоящие химические структурные элементы, фармацевтические композиции и способы включают все эти возможные стереоизомеры, в том числе рацемические смеси, оптически чистые формы, смеси диастереомеров и промежуточные смеси. Оптически активные (R)- и (S)-изомеры могут быть получены с использованием хиральных синтонов или хиральных реагентов или разделены с использованием традиционных методов. Оптическая активность соединения может быть проанализирована любым подходящим методом, в том числе, но этим не ограничивая, хиральной хроматографией и поляриметрией, и может быть определена степень преобладания одного стереоизомера над другим изомеров.
[0055] При необходимости, могут быть проведены выделение и очистка описанных в изобретении химических структурных элементов и промежуточных соединений любым подходящим методом разделения или очистки, таким как, например, фильтрация, экстракция, кристаллизация, колоночная хроматография, тонкослойная хроматография или хроматография в толстом слое, или комбинация этих методов. Конкретные иллюстрации подходящих методов разделения и выделения можно найти в приведенных ниже примерах. Однако, могут быть также использованы другие эквивалентные методы разделения и выделения.
[0056] Термин "соль" или "фармацевтически приемлемая соль" относится к солям, образованным целым рядом хорошо известных органических и неорганических противоионов. Фармацевтически приемлемые соли присоединения кислоты могут быть образованы с неорганическими кислотами и органическими кислотами. Неорганические кислоты, из которых могут быть образованы соли, включают, например, хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и другие подобные неорганические кислоты. Органические кислоты, из которых могут быть образованы соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, пировиноградную кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, коричную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, п-толуолсульфоновую кислоту, салициловую кислоту и другие подобные кислоты. Фармацевтически приемлемые соли присоединения основания могут быть образованы с неорганическими и органическими основаниями. Соли, которые могут быть образованы из неорганических оснований, включают, например, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия, и другие подобные соли. Соли, которые могут быть образованы из органических оснований, включают, например, соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе природных замещенных аминов, циклических аминов, катионообменных смол, и другие подобные соли, в частности, такие как соли изопропиламина, триметиламина, диэтиламина, триэтиламина, трипропиламина и этаноламина. В некоторых вариантах осуществления, фармацевтически приемлемую соль присоединения основания выбирают из солей аммония, калия, натрия, кальция и магния.
[0057] Термин "фармацевтически приемлемое вспомогательное вещество" включает, без ограничение, любой адъювант, носитель, вспомогательное вещество, связующее вещество, наполнитель, разрыхлитель, смазывающее вещество, скользящее вещество, подсластитель, разбавитель, консервант, краситель, окрашивающее вещество, интенсификатор вкусоароматических свойств, поверхностно-активное вещество, смачивающие средство, диспергирующее вещество, суспендирующее вещество, стабилизатор, изотоническое средство, растворитель или эмульгатор, которые одобрены Управлением по надзору за качеством пищевых продуктов и медикаментов США для применения на людях или домашних животных.
[0058] Термин "эффективное количество" или "терапевтически эффективное количество" относится к такому количеству описанного в изобретении соединения, которое является достаточным для осуществления предполагаемого действия, включающего, но этим не ограничивая, описанное ниже лечение заболевания. Терапевтически эффективное количество может варьировать в зависимости от предполагаемого применения лечения (in vivo) или от субъекта и болезненного состояния, подвергаемого лечению, например, от массы тела и возраста субъекта, тяжести болезненного состояния, способа введения и других подобных факторов, которые могут быть легко приняты определены любым обыкновенным специалистом в данной области. Термин также применяется в отношении дозы, которая должна индуцировать конкретную ответную реакцию в клетках-мишенях, например, снижение адгезии тромбоцитов и/или миграции клеток. Конкретная доза будет варьировать в зависимости от выбранных конкретных соединений, применяемого режима дозирования, возможности введения в комбинации с другими соединения, моментов времени введения, ткани, в которую вводят дозу, и физической системы доставки, с помощью которой осуществляется ее перенос.
[0059] Используемый в изобретении термин "лечение" относится к подходу для достижения положительных или требуемых результатов в отношении заболевания, нарушения или медицинского состояния, включая, но этим не ограничивая, терапевтический положительный эффект и/или профилактический положительный эффект. Под терапевтическим положительным эффектов подразумевают ликвидацию или уменьшение интенсивности лежащего в основе нарушения, подвергаемого лечению. Кроме того, терапевтический положительный эффект достигается путем ликвидации или уменьшения интенсивности одного или более из физиологических симптомов, ассоциированных с лежащим в основе нарушением, вследствие чего у субъекта наблюдается улучшение, несмотря на то что субъект может все еще страдать от лежащего в основе нарушения. В конкретных вариантах осуществления, для осуществления профилактического положительного эффекта, композиции вводят субъекту с повышенным риском развития конкретного заболевания, или субъекту, сообщающему о наличии у него одного или более физиологических симптомов заболевания, несмотря на то что это заболевание у него еще не диагностировано.
[0060] Используемый в изобретении в качестве термина "терапевтический эффект" включает в себя описанные выше терапевтический положительный эффект и/или профилактический положительный эффект. Профилактический эффект включает отсрочку или исключение возникновения заболевания или состояния, отсрочку или исключение появления симптомов заболевания или состояния, замедление, остановку или устранение прогрессирования заболевания или состояния, или любую их комбинацию.
[0061] Используемый в изобретении термин "совместное введение", "введение в комбинации с" и их грамматические эквиваленты включают в себя введение двух или более лекарственных средств животному, в том числе людям, для того чтобы оба лекарственных средства и/или их метаболиты присутствовали в организме субъекта в одно и то же время. Совместное введение включает одновременное введение в раздельных композициях, введение в различные моменты времени в различных композициях, или введение в композиции, в которой присутствуют оба лекарственных средства.
[0062] Термины "антагонист" и "ингибитор" используются взаимозаменяемо, и они относятся к соединению, обладающему способностью ингибировать биологическую функцию (например, активность, экспрессию, связывание, белок-белковое взаимодействие) белка-мишени или фермента-мишени (например, HIF-2α). Соответственно, термины "антагонист" и "ингибитор" определяются в контексте биологической роли белка-мишени. В то время как предпочтительные антагонисты в изобретении специфично взаимодействуют (например, связываются) с мишенью, тем не менее, соединения, которые ингибируют биологическую активность белка-мишени путем взаимодействия с другими членами пути сигнальной трансдукции, членом которого является белок-мишень, также конкретно включены в это определение. Предпочтительную биологическую активность, ингибируемую антагонистом, ассоциирует с воспалением.
[0063] Используемый в изобретении термин "агонист" относится к соединению, обладающему способность инициировать или усиливать биологическую функцию белка-мишени путем ингибирования или активности или экспрессии белка-мишени. Соответственно, термин "агонист" определяются в контексте биологической роли полипептида-мишени. В то время как предпочтительные агонисты в изобретении специфично взаимодействуют (например, связываются) с мишенью, тем не менее, соединения, которые инициируют или усиливают биологическую активность полипептида-мишени путем взаимодействия с другими членами пути сигнальной трансдукции, членом которого является полипептид-мишень, также конкретно включены в это определение.
[0064] "Сигнальная трансдукция" представляет собой процесс, при котором стимулирующие или ингибирующие сигналы передаются в клетку и внутри клетки, для того чтобы вызвать внутриклеточный ответ. Модулятор пути сигнальной трансдукции относится к соединению, которое модулирует активность одного или более клеточных белков, картированных на одном и том же специфическом пути сигнальной трансдукции. Модулятор может усиливать (агонист) или подавлять (антагонист) активность сигнальной молекулы.
[0065] Используемый в изобретении термин "гетеродимеризация" относится к комплексу, образованному в результате нековалентного связывания HIF-2α с ΗIF-1β (ARNT). Гетеродимеризация HIF-2α с ΗIF-1β (ARNT) требуется для HIF-2α ДНК связывания и транскрипционной активности, и она опосредуется HLH и PAS-B доменами. Транскрипционная активность, следующая за гетеродимеризацией HIF-2α с HIF-1β (ARNT), может воздействовать на пять групп генов-мишеней, включающих ангиогенные факторы, переносчиков глюкозы и гликолитические ферменты, факторы стволовых клеток, факторы выживания и факторы инвазии.
[0066] Термин "HIF-2α" относится к мономерному белку, который содержит три консервативных структурированных домена: основная спираль-петля-спираль (bHLH) и два Per-ARNT-Sim (PAS) домена, обозначаемых PAS-A и PAS-B, в дополнение к C-концевым регуляторным областям. В научной литературе для "HIF-2α" альтернативно также используют несколько других названий, чаще всего эндотелиальный PAS домен-содержащий белок 1 (EPAS-1), который кодируется геном EPAS1. Альтернативные названия включают основная спираль-петля-спираль-PAS белок (MOP2). В качестве представителя bHLH/PAS семейства факторов транскрипции, "HIF-2α" образует комплекс активного гетеродимерного фактора транскрипции путем связывания с ARNT (также известного как HIF-1β) белком в результате нековалентных взаимодействий.
[0067] Термин "полость HIF-2α PAS-B домена" относится к внутренней полости внутри PAS-B домена HIF-2α. Кристаллическая структура PAS-B домена может содержать большую (приблизительно 290 Å) полость в ее ядре. Однако, боковые цепи аминокислоты в структуре раствора являются динамическими. Например, эти боковые цепи могут проявлять тенденцию к более глубокому внедрению в ядро, и могут съеживать полость до 1 или 2 меньших полостей или даже могут расширять полость. Полость выстлана аминокислотными остатками, включающими PHE-244, SER-246, HIS-248, MET-252, PHE-254, ALA-277, PHE-280, TYR-281, MET-289, SER-292, HIS-293, LEU-296, VAL- 302, VAL-303, SER-304, TYR-307, MET-309, LEU-319, THR-321, GLN-322, GLY-323, ILE-337, CYS-339 и ASN-341 HIF-2α PAS-B домена. Система нумерации позаимствована из известных структур, представленных в базе данных белковых структур (PDB) Научно-исследовательского сотрудничества в области структурной биоинформатики (RCSB) с PDB кодом 3H7W. Другие системы нумерации в PDB также могли бы быть использованы для определения этих же представленных выше аминокислот, которые выстилают полость.
[0068] Термин "пролиферация клеток" относится к явлению, в силу которого изменилось число клеток в результате их деления. Этот термин также включает в себя клеточный рост, в результате которого изменилась морфология клеток (например, увеличение размера) в соответствии с пролиферативным сигналом.
[0069] Термин "селективное ингибирование" или "селективно ингибирует" относится к способности биологически активного средства преимущественно снижать целевую сигнальную активность по сравнению с нецелевой сигнальной активностью, путем прямого или косвенного взаимодействия с целью.
[0070] "Субъект" относится к животному, такому как млекопитающее, например к человеку. Описанные в изобретении способы могут применяться как при лечении людей, так и в ветеринарной практике. В некоторых вариантах осуществления, субъектом является млекопитающее, и в некоторых вариантах осуществления, субъектом является человек. "Млекопитающее" включает людей, и как домашних животных, таких как лабораторные животные и домашние животные (например, кошки, собаки, свиньи, крупный рогатый скот, овцы, козы, лошади, кролики), так и недомашних животных, таких как дикие животные и другие подобные животные.
[0071] Термин "in vivo" относится к событию, которое происходит в организме субъекта.
[0072] Термин "in vitro" относится к событию, которое происходит вне организма субъекта. Например, in vitro анализ включает в себя любой анализ, проводимый вне организма субъекта. In vitro анализы включают в себя клеточные анализы, при которых используют живые или фиксированные клетки. In vitro анализы также включают в себя бесклеточный анализ, при котором не используют интактные клетки.
[0073] Подразумевается, что изобретение также включает в себя in vivo метаболические продукты раскрытых соединений. Такие продукты могут образовываться в результате, например, протекания реакций окисления, восстановления, гидролиза, амидирования, этерификации и других подобных реакций вводимого соединения, главным образом, вследствие ферментативных процессов. Соответственно, изобретение включает соединения, образующиеся в результате применения способа, включающего введение соединения по настоящему изобретению млекопитающему в течение периода времени, достаточного для образования его метаболического продукта. Такие продукты обычно идентифицируют путем введения меченного радиоактивным изотопом соединения по изобретению в поддающейся обнаружению дозе животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, обеспечения достаточного времени для протекания метаболизма и выделения продуктов его превращения из мочи, крови или других биологических образцов.
[0074] Термин "OpaDry II" относится к водному пленочному покрытию, производимому фирмой Colorcon, со спецификациями, приведенными в таблице ниже:
классификация
[0075] Используемые в изобретении названия химических соединений и структурные диаграммы представляют собой модифицированную форму номенклатурной системы IUPAC (Международного союза теоретической и прикладной химии) с использованием программного обеспечения ChemDraw Professional 15.1 или приложения mol2nam программного обеспечения OpenEye Scientific Software. Для используемых в изобретении сложных химических названий, замещающую группу обычно называют перед группой, к которой она присоединяется. Например, циклопропилэтил включает этиловую цепь с циклопропильным заместителем. Кроме описанных ниже случаев, в изобретении указываются все связи в химических структурных диаграммах, за исключением всех связей на некоторых углеродных атомах, которые, как предполагается, связаны с достаточным количеством атомов водорода для полного исчерпания свободных валентностей.
Твердые дисперсии
[0076] В конкретных аспектах, в настоящем изобретении предлагается твердая дисперсия, включающая соединение формулы (I):
Формула (I),
или его фармацевтически приемлемую соль.
[0077] В некоторых вариантах осуществления, твердая дисперсия дополнительно включает фармацевтически приемлемый полимер. Соединение формулы (I) может быть диспергировано в твердой матрице, которая включает полимер. В некоторых вариантах осуществления, полимер включает гидрофобные области и гидрофильные области. В некоторых вариантах осуществления, полимер представляет собой водорастворимый полимер.
[0078] Подходящие полимеры для применения в заявленных твердых дисперсиях включают, но этим не ограничивая, сложные эфиры целлюлозы; простые эфиры целлюлозы; полиалкиленоксиды; полиакрилаты; полиметакрилаты; гомополимеры и сополимеры N-виниллактамов, полиакриламиды, полимеры винилацетата; графт-сополимеры полиэтиленгликоля, поливинилкапролактам и поливинилацетат; олигосахариды; полисахариды; и их смеси.
[0079] В некоторых вариантах осуществления, полимер представляет собой полимер простого эфира целлюлозы. Например, полимер может быть выбран из метилцеллюлозы, этилцеллюлозы, (гидроксиалкил)целлюлозы (например, гидроксиэтилцеллюлозы (HEC) или гидроксипропилцеллюлозы (HPC)), или (гидроксиалкил)алкил-целлюлозы (например, гидроксипропилметилцеллюлозы (HPMC или гидроксипропилметилцеллюлозы)). В некоторых вариантах осуществления, полимер представляет собой метилцеллюлозу. В некоторых вариантах осуществления, полимер представляет собой этилцеллюлозу. В некоторых вариантах осуществления, полимер представляет собой HEC. В некоторых вариантах осуществления, полимер представляет собой HPC. В некоторых вариантах осуществления, полимер представляет собой HPMC.
[0080] В некоторых вариантах осуществления, полимер представляет собой полимер сложного эфира целлюлозы. В некоторых вариантах осуществления, полимер представляет собой фталат целлюлозы (например, ацетатфталат целлюлозы или фталат гидроксипропилметилцеллюлозы (HPMCP)) или сукцинат целлюлозы (например, сукцинат гидроксипропилметилцеллюлозы (HPMCS) или ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS)). В некоторых вариантах осуществления, полимер представляет собой HPMCP. В некоторых вариантах осуществления, полимер представляет собой HPMCAS.
[0081] Описанные в изобретении целлюлозные полимеры могут быть дополнительно определены путем указания конкретной марки. Например, полимер HPMC может быть выбран из HPMC E3, HPMC E5, HPMC E6, HPMC E15, HPMC K3, HPMC A4 или HPMC A15. В некоторых вариантах осуществления, HPMCAS представляет собой HPMCAS-L, HPMCAS-M, HPMCAS-H, HPMCAS-LF, HPMCAS-MF, HPMCAS-HF, HPMCAS-LG, HPMCAS-MG или HPMCAS-HG. В некоторых вариантах осуществления, HPMCP представляет собой HPMCP 50 или HPMCP 55. В некоторых вариантах осуществления, полимер представляет собой HPMCP. В некоторых вариантах осуществления, полимер представляет собой HPMCP 50. В некоторых вариантах осуществления, полимер представляет собой HPMCAS-L, HPMCAS-M или HPMCAS-H. В некоторых вариантах осуществления, полимер представляет собой HPMCAS-H. В некоторых вариантах осуществления, полимер представляет собой HPMCAS-HG.
[0082] В некоторых вариантах осуществления, полимер представляет собой полиалкиленоксид. В некоторых вариантах осуществления, полимер представляет собой высокомолекулярный полиалкиленоксид. В некоторых вариантах осуществления, полимер представляет собой полиэтиленоксид (PEG или PEO) или сополимеры этиленоксида и пропиленоксида (полоксамеры). Подходящие PEG включают, без ограничения, PEG 400, PEG 600, PEG 1450, PEG 3350, PEG 4000, PEG 6000, PEG 8000, PEG 20000 и их смеси. Подходящие полоксамеры включают, без ограничения, полоксамер 124, полоксамер 188, полоксамер 237, полоксамер 338, полоксамер 407 и их смеси. В одном варианте осуществления, полимер представляет собой метиловый эфир полиэтиленгликоля. В некоторых вариантах осуществления, полимер представляет собой полиэтиленгликоль 6000 (PEG 6000).
[0083] В некоторых вариантах осуществления, полимер представляет собой полиакрилат или полиметакрилат. В некоторых вариантах осуществления, полимер представляет собой сополимер метакриловая кислота/этилакрилат, сополимер метакриловая кислота/метилметакрилат, сополимер бутилметакрилат/2-диметиламиноэтилметакрилат, полигидроксиалкилакрилаты или полигидроксиалкилметакрилаты. Подходящие полиакрилаты или полиметакрилаты включают, без ограничения, те, которые продают под торговой маркой EudragitTM фирмы Rohm GmbH, например, Eudragit RS 100, Eudragit L 100, Eudragit L 100-55, и Eudragit S 100, эквивалентные им продукты других производителей, и их смеси. В одном варианте осуществления, полимер представляет собой Eudragit RS 100. В некоторых вариантах осуществления, полимер представляет собой Eudragit S 100.
[0084] В некоторых вариантах осуществления, полимер представляет собой гомополимер или сополимер N-виниллактамов. В некоторых вариантах осуществления, полимер представляет собой гомополимер или сополимер N-винилпирролидона. В некоторых вариантах осуществления, полимер представляет собой гомополимер поливинилпирролидона (PVP или повидон) или сополимер (например, сополимер, включающий мономеры N-винилпирролидона и винилацетата (коповидон) или N-винилпирролидона и винилпропионата). Подходящие повидоны включают, без ограничения, повидоны, имеющие K-величину (меру измерения вязкости водного раствора повидона) приблизительно 12, приблизительно 15, приблизительно 17, приблизительно 25, приблизительно 30 или приблизительно 90, и их смеси. В некоторых вариантах осуществления, полимер представляет собой поливинилпирролидон K30. В некоторых вариантах осуществления, полимер представляет собой поли(1-винилпирролидон-ко-винилацетат).
[0085] В некоторых вариантах осуществления, полимер представляет собой полиакриламид. В некоторых вариантах осуществления, полимер представляет собой полимер винилацетата (например, сополимеры винилацетата и кротоновой кислоты, поливинилацетат, поливиниловый спирт частично гидролизованный поливинилацетат). В некоторых вариантах осуществления, полимер представляет собой граф-сополимер полиэтиленгликоля, поливинилкапролактама и поливинилацетата (например, SOLUPLUSTM фирмы BASF или эквивалентный продукт). В некоторых вариантах осуществления, полимер представляет собой олиго- или полисахарид (например, каррагинаны, галактоманнаны или ксантановую камедь).
[0086] В некоторых вариантах осуществления, полимер выбирают из метилцеллюлозы, этилцеллюлозы, гидроксиэтилцеллюлозы, ацетатфталата целлюлозы (CAP), гидроксипропилметилцеллюлозы (HPMC), гидроксипропилцеллюлозы, фталата гидроксипропилметил-целлюлозы (HPMCP), ацетатсукцината гидроксипропилметилцеллюлозы (HPMCAS), метилового эфира полиэтиленгликоля, графт-сополимера полиэтиленгликоль-поливинилацетат-поливинилкапролактам (например, SOLUPLUS), полиэтиленгликоля 6000 (PEG 6000), поливинилпирролидона (PVP), сополимера поливинилпирролидона и винилацетата (PVP-VA), сополимера этилакрилата, метилметакрилата и триметиламмониоэтилметакрилата хлорида 1:2:0,1 (Eudragit RS 100), сополимера метакриловой кислоты типа В (Eudragit S 100), Sureteric, Pluronic F-68 и полиоксиэтилен (20) сорбитан моноолеата (Tween 80). Предпочтительно, чтобы полимер выбирали из HPMCAS, CAP и SOLUPLUS, например, из HPMCAS. В некоторых вариантах осуществления, полимер выбирают из HPMCAS марки L (HPMCAS-L), HPMCAS марки M (HPMCAS-M) и HPMCAS марки H (HPMCAS-H). В некоторых вариантах осуществления, полимер представляет собой HPMCAS-H.
[0087] В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве от 1% до 50% в расчете на массу твердой дисперсии, например, от 1% до 45%, от 1% до 40%, от 1% до 35%, от 1% до 30%, от 5% до 50%, от 5% до 45%, от 5% до 40%, от 5% до 35%, от 5% до 30%, от 10% до 50%, от 10% до 45%, от 10% до 40%, от 10% до 35%, от 10% до 30%, от 15% до 50%, от 15% до 45%, от 15% до 40%, от 15% до 35%, от 15% до 30%, от 20% до 50%, от 20% до 45%, от 20% до 40%, от 20% до 35%, от 20% до 30%, от 22,5% до 50%, от 22,5% до 45%, от 22,5% до 40%, от 22,5% до 35%, от 22,5% до 30%, от 22,5% до 27,5%, от 25% до 50%, от 25% до 45%, от 25% до 40%, от 25% до 35%, от 25% до 30%, или от 25% до 27,5% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве, по меньшей мере 1% в расчете на массу твердой дисперсии, например, по меньшей мере 2%, по меньшей мере 3%, по меньшей мере 4%, по меньшей мере 5%, по меньшей мере 7,5%, по меньшей мере 10%, по меньшей мере 12,5%, по меньшей мере 15%, по меньшей мере 17,5%, по меньшей мере 20%, по меньшей мере 22,5%, по меньшей мере 25%, по меньшей мере 27,5%, по меньшей мере 30%, по меньшей мере 32,5%, по меньшей мере 35%, по меньшей мере 37,5%, по меньшей мере 40%, по меньшей мере 42,5%, по меньшей мере 45%, по меньшей мере 47,5%, или, по меньшей мере, 50% в расчете на массу твердой дисперсии.
[0088] В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве приблизительно 1%, приблизительно 2%, приблизительно 3%, приблизительно 4%, приблизительно 5%, приблизительно 7,5%, приблизительно 10%, приблизительно 12,5%, приблизительно 15%, приблизительно 17,5%, приблизительно 20%, приблизительно 22,5%, приблизительно 25%, приблизительно 27,5%, приблизительно 30%, приблизительно 35%, приблизительно 40%, приблизительно 45%, или приблизительно 50% в расчете на массу твердой дисперсии.
[0089] В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве от 1% до 50% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве от 15% до 35% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве, по меньшей мере, от 22,5% до 27,5% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве приблизительно 25% в расчете на массу твердой дисперсии.
[0090] В некоторых вариантах осуществления, полимер присутствует в количестве от 50% до 99% в расчете на массу твердой дисперсии, например, от 55% до 99%, от 60% до 99%, от 65% до 99%, от 70% до 99%, от 50% до 95%, от 55% до 95%, от 60% до 95%, от 65% до 95%, от 70% до 95%, от 50% до 90%, от 55% до 90%, от 60% до 90%, от 65% до 90%, от 70% до 90%, от 50% до 85%, от 55% до 85%, от 60% до 85%, от 65% до 85%, от 70% до 85%, от 50% до 80%, от 55% до 80%, от 60% до 80%, от 65% до 80%, от 70% до 80%, от 50% до 77,5%, от 52,5% до 77,5%, от 55% до 77,5%, от 57,5% до 77,5%, от 62,5% до 77,5%, от 65% до 77,5%, от 67,5 до 77,5%, от 70% до 77,5%, от 72,5% до 77,5%, от 50% до 75%, от 52,5% до 75%, от 55% до 75%, от 57,5% до 75%, от 60% до 75%, от 62,5% до 75%, от 65% до 75%, от 67,5 до 75%, от 70% до 75%, или от 72,5% до 75% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, полимер присутствует в количестве, по меньшей мере, 50% в расчете на массу твердой дисперсии, например, по меньшей мере, 55%, по меньшей мере, 57,5%, по меньшей мере, 60%, по меньшей мере, 62,5%, по меньшей мере, 65%, по меньшей мере, 67,5%, по меньшей мере, 70,5%, по меньшей мере, 72,5%, или, по меньшей мере, 75% в расчете на массу твердой дисперсии.
[0091] В некоторых вариантах осуществления, полимер присутствует в количестве приблизительно 60%, приблизительно 62.5%, приблизительно 65%, приблизительно 67,5%, приблизительно 70%, приблизительно 72,5%, приблизительно 75%, приблизительно 77,5%, приблизительно 80%, приблизительно 82,5%, приблизительно 85%, приблизительно 87,5%, or приблизительно 90% в расчете на массу твердой дисперсии.
[0092] В некоторых вариантах осуществления, полимер присутствует в количестве от 50% до 99% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, полимер присутствует в количестве от 65% до 85% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, полимер присутствует в количестве, по меньшей мере, от 72,5% до 77,5% в расчете на массу твердой дисперсии. В некоторых вариантах осуществления, полимер присутствует в количестве приблизительно 75% в расчете на массу твердой дисперсии.
[0093] В некоторых вариантах осуществления, массовое отношение соединения формулы (I) к полимеру составляет от 1:99 до 1:1, например, от 5:95 до 1:1, от 10:90 до 1:1, от 15:85 до 1:1, от 20:80 до 1:1, от 25:75 до 1:1, от 1:99 до 35:65, от 5:95 до 35:65, от 10:90 до 35:65, от 15:85 до 35:65, от 20:80 до 35:65, от 25:75 до 35:65, от 1:99 до 27,5:72.5, от 5:95 до 27,5:72,5, от 10:90 до 27,5:72,5, от 15:85 до 27,5:72,5, от 20:80 до 27,5:72,5, или от 22,5:77,5 до 27,5:72,5. В некоторых вариантах осуществления, массовое отношение соединения формулы (I) к полимеру составляет от 1:99 до 1:1. В некоторых вариантах осуществления, массовое отношение соединения формулы (I) к полимеру составляет от 15:85 до 35:65. В некоторых вариантах осуществления, массовое отношение соединения формулы (I) к полимеру составляет от 22,5:77,5, до 27,5:72,5. В некоторых вариантах осуществления, массовое отношение соединения формулы (I) к полимеру составляет приблизительно 25:75.
[0094] Описанная в изобретении твердая дисперсия может быть охарактеризована порошковым рентгеноструктурным анализом (XRPD), дифференциальной сканирующей калориметрией (DSC), модулированной дифференциальной сканирующей калориметрией (mDSC), сканирующей электронной микроскопией (SEM), титрованием воды по Карлу Фишеру (KF), остаточным растворителем, размером частиц, насыпной плотностью, насыпной плотностью после уплотнения, хиральной высокоэффективной жидкостной хроматографией (HPLC) и ахиральной HPLC.
[0095] Порошковый рентгеноструктурный анализ (XRPD) может быть использован для оценки кристалличности твердой дисперсии, например, для выявления того, является ли твердая дисперсия кристаллической, полукристаллической или аморфной. В некоторых вариантах осуществления, твердая дисперсия является, по существу, некристаллической. В некоторых вариантах осуществления, не более чем приблизительно на 20%, не более чем приблизительно на 15%, не более чем приблизительно на 10%, не более чем приблизительно на 5%, не более чем приблизительно на 4%, не более чем приблизительно на 3%, не более чем приблизительно на 2%, или не более чем приблизительно на 1% твердая дисперсия является кристаллической, что определяется порошковым рентгеноструктурным анализов. В некоторых вариантах осуществления, твердая дисперсия является аморфной. Порошковая рентгенограмма аморфной твердой дисперсии обычно характеризуется уширенным фоновым сигналом, то есть на рентгенограмме отсутствуют дискретные пики, которые можно было бы обнаружить в случае кристаллического материала. Предпочтительно, если порошковый рентгеноструктурный анализ (XRPD) проводят на рентгеновском дифрактометре Bruker D2 Phaser с использованием метода сканирования θ/2θ, при котором величину напряжения устанавливают на 30 кВ и величину тока на 10 мА при вращении 15 об/мин, с держателем образца с нулевым фоном, при ширине щели 1,0 мм и при ширине призмы, установленной на 1,0 мм.
[0096] Температура стеклования (Tg) и температура плавления (Tm) твердой дисперсии может быть измерена любым подходящим методом, например, mDSC. В некоторых вариантах осуществления, твердая дисперсия характеризуется Tg от 70 до 110°C, например, от 75 до 105°C, от 80 до 100°C, от 80 до 95°C, от 82 до 92°C, или от 83 до 89°C. В некоторых вариантах осуществления, твердая дисперсия характеризуется величиной Tg при приблизительно 87°C, например, 87,0±2,0°C. Предпочтительно, когда анализ mDSC проводят на приборе TA Q2000 с системой охлаждения RCS 90 в диапазоне температур от 0 до 250°C при скорости нагревания 1,5°C в минуту, где режим сканирования модулируют при частоте 60 секунд и амплитуде 1°C. В некоторых вариантах осуществления, твердая дисперсия характеризуется величиной Tg от 60 до 145°C, например, от 65 до 140°C, от 70 до 135°C, от 75 до 130°C, от 77,5 до 120°C, от 80 до 110°C, от 82,5 до 100°C, или от 85 до 90°C.
[0097] Изображение, полученное методом сканирующей электронной микроскопии (SEM), может быть использовано для визуальной характеристики морфологии твердой дисперсии. В некоторых вариантах осуществления, твердая дисперсия включает целые и разрушенные сферы. Поверхности сфер могут быть гладкими. В некоторых вариантах осуществления, предпочтительно, когда в изображении твердой дисперсии, полученном методом сканирующей электронной микроскопии (SEM), не обнаруживается кристаллический материал. Предпочтительно, когда метод сканирующей электронной микроскопии (SEM) проводят с использованием сканирующего электронного микроскопа FEI Quanta 200 SEM с системой для автоматического напыления материала мишени Au/Pd Polaron Autocoater E5200 при напряжении 15 кВ, размере пятна 3,0 мА, токе накала 2,52 A и токе эмиссии 96 мкА.
[0098] Титрование воды по Карлу Фишеру (KF) может быть использовано для определения содержания воды в твердой дисперсии. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 2% воды по массе. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 2% воды после трех месяцев хранения при комнатной температуре. Твердая дисперсия по настоящему изобретению может быть гигроскопичной. В некоторых вариантах осуществления, воздействие на твердую дисперсию влажности, например 33%, 50% или 75% влажности, снижает величину Tg твердой дисперсии. В некоторых вариантах осуществления, величина Tg твердой дисперсии снижается на 10% или более при воздействии 33% относительной влажности в течение 24 часов. Не привлекая для обоснования какую-либо конкретную теорию, тем не менее можно предположить, что влияние влажности на температуру стеклования может указывать на то, что при комнатной температуре подвижность молекул возможна, когда влажность составляет более чем 33%, что может обеспечивать соединению формулы (I) способность образовывать центры кристаллизации и/или кристаллизоваться. Предпочтительно, когда твердую дисперсию по настоящему изобретению хранят при условиях, при которых поддерживаются уровни относительной влажности менее чем 50%, например, менее чем 33%. В некоторых вариантах осуществления, твердую дисперсию расфасовывают вместе с осушающим веществом, таким как SiO2. В некоторых вариантах осуществления, твердую дисперсию расфасовывают в контейнер с низкой проницаемостью для паров воды, такой как пакет из полиэтилентерефталата или полиэтилена низкой плотности, необязательно, в ящик из полиэтилена высокой плотности. В некоторых вариантах осуществления, твердую дисперсию расфасовывают и хранят вместе с осушающим веществом в контейнере с низкой проницаемостью для паров воды.
[0099] Уровни остаточного растворителя в твердой дисперсии по настоящему изобретению могут быть определены методом парофазной газовой хроматографии (GCHS). Например, для анализа образца может быть использован газовый хроматограф Agilent 6890A/7694 GC/HS, оборудованный колонкой JW Scientific DB-624 30 м x 0,32 мм, 1,8 мкм, со следующими параметрами: температура образца 105°C; температура петли 110°C; температура линии транспортировки 115°C; время цикла газовой хроматографии 45 мин; время установления равновесия в виале 30 мин; размер петлевого дозатора 1 мл; время создания давления в виале 20 сек; давление газа-носителя 0,05 МПа; и давление в виале 0,10 МПа. В некоторых вариантах осуществления, твердая дисперсия включает остаточный растворитель. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 25000 ppm ацетона. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 5000 ppm ацетона.
[0100] Метод рассеяния лазерного излучения может быть применен на порошке, таком как раскрытая в изобретении твердая дисперсия, для определения распределения частиц по размеру. Например, для анализа образца может быть использован анализатор размера сухих частиц Malvern Aero S, имеющий высоту бункера 3 мм при давлении 0,07 и скорости подачи 40%. Предпочтительно, когда образцы подвергают анализу в течение 10 секунд в трех параллельных опытах. Для анализа с несферическим алгоритмом и поглощением образца от 0,1 до 15% могут быть использованы показатель преломления 1,681 и плотность 0,5 г/мл. В некоторых вариантах осуществления, распределение частиц по размеру регистрируются с использованием средних значений d10, d50 и d90, где 10% частиц в твердой дисперсии по массе имеют диаметр меньше величины d10, 50% частиц в твердой дисперсии по массе имеют диаметр меньше величины d50, и 90% частиц в твердой дисперсии по массе имеют диаметр меньше величины d90. В некоторых вариантах осуществления, распределение частиц по размерам твердой дисперсии характеризуется величиной d10 менее чем 16 мкм, например, менее чем 15 мкм, менее чем 14 мкм, менее чем 12 мкм, менее чем 10 мкм, менее чем 8 мкм или менее чем приблизительно 6 мкм. В некоторых вариантах осуществления, распределение частиц по размерам твердой дисперсии характеризуется величиной d50 менее чем 50 мкм, например, менее чем 45 мкм, менее чем 40 мкм, менее чем 35 мкм, менее чем 30 мкм, менее чем 28 мкм, менее чем 25 мкм, менее чем 23 мкм или менее чем приблизительно 18 мкм. В некоторых вариантах осуществления, распределение частиц по размерам твердой дисперсии характеризуется величиной d90 менее чем 100 мкм, например, менее чем 90 мкм, менее чем 80 мкм, менее чем 70 мкм, менее чем 65 мкм, менее чем 55 мкм или менее чем приблизительно 45 мкм.
[0101] Насыпная плотность твердой дисперсии по настоящему изобретению может быть определена путем измерения массы и объема образца твердой дисперсии. В некоторых вариантах осуществления, твердая дисперсия характеризуется насыпной плотностью, по меньшей мере, 0,10 г/мл, например, по меньшей мере, 0,15 г/мл, по меньшей мере, 0,20 г/мл, по меньшей мере, 0,25 г/мл, или, по меньшей мере, 0,30 г/мл.
[0102] Насыпная плотность после уплотнения твердой дисперсии по настоящему изобретению может быть определена путем измерения массы и объема образца твердой дисперсии после механического уплотнения образца до тех пор, пока не будет обнаружено небольшое дополнительное изменение или отсутствие дополнительного изменения объема. В некоторых вариантах осуществления, твердая дисперсия характеризуется насыпной плотностью после уплотнения, по меньшей мере, 0,20 г/мл, например, по меньшей мере, 0,25 г/мл, по меньшей мере, 0,30 г/мл, по меньшей мере, 0,35 г/мл, по меньшей мере, 0,40 г/мл, по меньшей мере, 0,45 г/мл, или, по меньшей мере, 0,50 г/мл.
[0103] Чистота твердой дисперсии по настоящему изобретению может быть оценена методом высокоэффективной жидкостной хроматографии (HPLC). Например, может быть использован хроматограф Agilent 1220, оборудованный колонкой Kinetex, C18, 4,6×100 мм, 2,6 мкм, с использованием следующих параметров: подвижная фаза A, 0,1% муравьиной кислоты в воде; подвижная фаза B, 0,1% муравьиной кислоты в ацетонитриле; градиент 95% A (0 мин), 60% A (6 мин), 5% A (12 мин), 95% A (15,1 мин), с достижением равновесия с подвижной фазой B; расход, 0,8 мл/мин; температура колонки 40°C; температура образца комнатная температура; вводимый объем 5 мкл; длина волны детектирования 240 нм; и время записи хроматограммы 18,5 мин. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 5% примесей по массе, например, менее чем 2% примесей по массе. В некоторых вариантах осуществления, твердая дисперсия включает менее чем 2% примесей по массе после трех месяцев хранения при комнатной температуре. В некоторых вариантах осуществления, энантиомерная чистота соединения формулы (I) составляет, по меньшей мере, 95%. В некоторых вариантах осуществления, энантиомерная чистота соединения формулы (I) составляет, по меньшей мере, 99%. В некоторых вариантах осуществления, энантиомерная чистота соединения формулы (I) составляет, по меньшей мере, 99,5%.
[0104] Характеристика растворения твердой дисперсии может быть получена путем проведения испытания на растворимость в условиях недостаточного разбавления в результате измерения растворимости соединения формулы (I) в биорелевантной среде, моделирующей кишечный сок (FaSSIF), после 30 минут воздействия среды с низким значением pH (SGF). Из профиля растворимости может быть получена максимальная концентрация соединения формулы (I) в SGF (Cmax GB) и SIF (Cmax FaSSIF), а также площадь под кривой после прохождения твердой дисперсии через желудок (AUC FaSSIF).
[0105] В некоторых вариантах осуществления, величина Cmax GB твердой дисперсии составляет, по меньшей мере, 100 мкгА/мл, например, по меньшей мере, 150, по меньшей мере, 200, по меньшей мере, 250, по меньшей мере, 300, по меньшей мере, 350, по меньшей мере, 400, по меньшей мере, 450, по меньшей мере, 500, или, по меньшей мере, 550 мкгА/мл. Предпочтительно, чтобы величина Cmax GB составляла, по меньшей мере 300, мкгА/мл. В некоторых вариантах осуществления, величина Cmax GB составляет от 100 до 600 мкгА/мл, например, от 150 до 600, от 200 до 550, от 250 до 500, от 300 до 450, от 350 до 450, от 400 до 450, или от 375 до 425 мкгА/мл. Предпочтительно, когда величина Cmax GB составляет от 375 до 425 мкгА/мл. В некоторых вариантах осуществления, величина Cmax GB составляет приблизительно 409 мкгА/мл, например, 410±10 мкгА/мл.
[0106] В некоторых вариантах осуществления, величина Cmax FaSSIF твердой дисперсии составляет, по меньшей мере, 50 мкгА/мл, например, по меньшей мере, 100, по меньшей мере, 150, по меньшей мере, 200, по меньшей мере, 250, по меньшей мере, 300, по меньшей мере, 350, по меньшей мере, 400, по меньшей мере, 450, по меньшей мере, 500, по меньшей мере, 550, по меньшей мере, 600, по меньшей мере, 650, по меньшей мере, 700, по меньшей мере, 750, или, по меньшей мере, 800 мкгА/мл. Предпочтительно, чтобы величина Cmax FaSSIF составляла не менее 300 мкгА/мл. В некоторых вариантах осуществления, величина Cmax FaSSIF составляет от 50 до 900 мкгА/мл, например, ОТ 150 ДО 800, от 250 до 700, от 350 до 600, или от 350 до 575 мкгА/мл. Предпочтительно, чтобы величина Cmax FaSSIF составляла от 350 до 575 мкгА/мл. В некоторых вариантах осуществления, величина Cmax FaSSIF составляет приблизительно 545 мкгА/мл, например, 545±50 мкгА/мл, или приблизительно 435 мкгА/мл, например, 435±50 мкгА/мл.
[0107] В некоторых вариантах осуществления, величина AUC FaSSIF твердой дисперсии составляет, по меньшей мере, 10000 мкгА/мл, например, по меньшей мере, 20000, по меньшей мере, 30000, по меньшей мере, 40000, по меньшей мере, 50000, по меньшей мере, 60000, по меньшей мере, 70000, по меньшей мере, 80000, по меньшей мере, 90000, или, по меньшей мере, 100000 мкгА/мл. Предпочтительно, чтобы величина AUC FaSSIF составляла не менее 40000 мкгА/мл. В некоторых вариантах осуществления, величина AUC FaSSIF составляет от 10000 до 200000 мкгА/мл, например, от 10000 до 175000, от 15000 до 150000, от 20000 до 125000, или от 25000 до 100000 мкгА/мл. В некоторых вариантах осуществления, величина AUC FaSSIF составляет приблизительно 90000 мкгА/мл, например, 90000±1000 мкгА/мл. В некоторых вариантах осуществления, величина AUC FaSSIF составляет приблизительно 65000 мкгА/мл, например, 65000±1000 мкгА/мл.
[0108] В некоторых вариантах осуществления, степень увеличения AUC твердой дисперсии составляет, по меньшей мере, 3,0, например, по меньшей мере, 5,0, по меньшей мере, 10,0, по меньшей мере, 15,0, по меньшей мере, 20,0, по меньшей мере, 25,0, по меньшей мере, 30,0, по меньшей мере, 35,0, или, по меньшей мере, 40,0, где степень увеличения AUC представляет собой отношение величины AUC FaSSIF твердой дисперсии к величине AUC FaSSIF соединения формулы (I). Предпочтительно, когда степень увеличения AUC составляет, по меньшей мере, 10. В некоторых вариантах осуществления, степень увеличения AUC составляет от 3,0 до 50,0, например, от 5,0 до 45,0, от 10,0 до 40,0, от 15,0 до 40,0, от 20,0 до 40,0, или от 25,0 до 45,0. В некоторых вариантах осуществления, степень увеличения AUC составляет приблизительно 35,0, например, 35,0±3,0. В некоторых вариантах осуществления, степень увеличения AUC составляет приблизительно 25,0, например, 25,0±3,0.
[0109] В некоторых аспектах, в настоящем изобретении предлагается аморфная твердая дисперсия, включающая, по массе относительно суммарной массы твердой дисперсии:
(a) от 22,5% до 27,5% соединения формулы (I):
Формула (I);
и
(b) от 72,5% до 77,5% HPMCAS.
[0110] В некоторых аспектах, в настоящем изобретении предлагается аморфная твердая дисперсия, включающая, по массе относительно суммарной массы твердой дисперсии:
(a) от 20% до 30% соединения формулы (I):
Формула (I);
и
(b) от 70% до 80% полимера, выбранного из сложных эфиров целлюлозы и простых эфиров целлюлозы,
где твердая дисперсия характеризуется температурой стеклования (Tg) от 80 до 100°C, и где твердая дисперсия включает менее чем 2% воды по массе.
[0111] В некоторых аспектах, в настоящем изобретении предлагается аморфная твердая дисперсия, включающая, по массе относительно суммарной массы твердой дисперсии:
(a) от 1% до 50% соединения формулы (I):
Формула (I);
и
(b) от 50% до 99% полимера, выбранного из HPMCAS, CAP и SOLUPLUS.
[0112] В некоторых аспектах, в настоящем изобретении предлагается аморфная твердая дисперсия, включающая, по массе относительно суммарной массы твердой дисперсии:
(a) от 10% до 30% соединения формулы (I):
Формула (I);
и
(b) от 70% до 90% HPMCAS,
где твердая дисперсия включает менее чем 2% воды по массе.
[0113] В некоторых аспектах, в настоящем изобретении предлагается аморфная твердая дисперсия, включающая, по массе относительно суммарной массы твердой дисперсии:
(a) от 22,5% до 27,5% соединения формулы (I):
Формула (I);
и
(b) от 72,5% до 77,5% полимера, выбранного из HPMCAS-L, HPMCAS-M и HPMCAS-H,
где энантиомерная чистота соединения формулы (I) составляет, по меньшей мере, 95%.
[0114] В некоторых вариантах осуществления, в изобретении предлагается твердая дисперсия, включающая приблизительно 25% по массе соединения формулы (I), диспергированного в твердой матрице, которая включает HPMCAS. В некоторых вариантах осуществления, предлагается твердая дисперсия в форме порошка. В некоторых вариантах осуществления, предлагается твердая дисперсия в форме гранул. В некоторых вариантах осуществления, предлагается твердая дисперсия в форме пленки. В некоторых вариантах осуществления, твердая дисперсия остается аморфной после хранения либо при 25°C и 60% RH, либо при 40°C и 75% RH, в течение 4 недель. В некоторых вариантах осуществления, твердая дисперсия остается аморфной после хранения либо при 2-8°C, 25°C и 60% RH, либо при 40°C и 75% RH, в течение 3 месяцев. В некоторых вариантах осуществления, твердая дисперсия является умеренно гигроскопичной. В некоторых вариантах осуществления, твердая дисперсия характеризуется повышенной растворимостью соединения формулы (I) в водной среде по сравнению с чистым соединением формулы (I). В некоторых вариантах осуществления, водной средой является вода, смоделированный желудочный сок (SGF), смоделированная интестинальная жидкость (FaSSIF) или 5% раствор глицерина в воде.
[0115] Не привлекая для обоснования какую-либо конкретную теорию, тем не менее можно утверждать, что предложенные в изобретении твердые дисперсии характеризуются физическими свойствами, например, стабильностью, растворимостью и/или скоростью растворения, которые позволяют применять эти твердые дисперсии в клинических и терапевтических лекарственных формах. В некоторых вариантах осуществления, предложенные в изобретении конкретные твердые дисперсии подходят для применения в твердой лекарственной форме для перорального введения.
Кристаллическая форма
[0116] Кристаллические формы соединения формулы (I) могут использоваться, по меньшей мере, в производстве фармацевтических композиций.
[0117] В одном варианте осуществления предлагается кристаллическая форма соединения формулы (I):
Формула (I),
где кристаллическая форма характеризуется порошковой рентгенограммой, приведенной на фигуре 1.
[0118] В одном варианте осуществления предлагается кристаллическая форма соединения формулы (I):
Формула (I),
где кристаллическая форма характеризуется величиной Tm приблизительно 209°C.
[0119] В одном варианте осуществления предлагается кристаллическая форма соединения формулы (I):
Формула (I),
где кристаллическая форма характеризуется величиной Tm приблизительно 80°C.
[0120] В одном варианте осуществления предлагается кристаллическая форма соединения формулы (I):
Формула (I),
где кристаллическая форма характеризуется порошковой рентгенограммой, имеющей, по меньшей мере, один 2Θ пик, выбранный из 7,6, 15,0, 19,6, 22,9, 25,8 или 30,2.
[0121] В одном варианте осуществления предлагается кристаллическая форма соединения формулы (I):
Формула (I),
где кристаллическая форма характеризуется порошковой рентгенограммой, имеющей, по меньшей мере, два 2Θ пика, выбранных из 7,6, 15,0, 19,6, 22,9, 25,8 или 30,2.
[0122] В одном варианте осуществления предлагается кристаллическая форма соединения формулы (I):
Формула (I),
где кристаллическая форма характеризуется порошковой рентгенограммой, имеющей, по меньшей мере, три 2Θ пика, выбранных из 7,6, 15,0, 19,6, 22,9, 25,8 или 30,2.
[0123] В одном варианте осуществления предлагается кристаллическая форма соединения формулы (I):
Формула (I),
где кристаллическая форма характеризуется порошковой рентгенограммой, имеющей, по меньшей мере, четыре 2Θ пика, выбранных из 7,6, 15,0, 19,6, 22,9, 25,8 или 30,2.
[0124] В одном варианте осуществления предлагается кристаллическая форма соединения формулы (I):
Формула (I),
где кристаллическая форма характеризуется порошковой рентгенограммой, имеющей, по меньшей мере, пять 2Θ пиков, выбранных из 7,6, 15,0, 19,6, 22,9, 25,8 или 30,2.
Способы приготовления твердой дисперсии
[0125] В некоторых аспектах, в настоящем изобретении предлагается способ приготовления описанной в изобретении твердой дисперсии, включающий (a) приготовление раствора соединения формулы (I) и полимера в растворителе; и (b) удаление растворителя с получением твердой дисперсии. Полимер может быть выбран из любого описанного в изобретении подходящего полимера, например, HPMCAS.
[0126] В некоторых вариантах осуществления, растворитель включает ацетон, метилэтилкетон, тетрагидрофуран, воду или их комбинацию. В некоторых вариантах осуществления, растворитель включает ацетон. Растворитель может включать до 5% воды. В некоторых вариантах осуществления, растворитель включает ацетон и до 5% воды. В некоторых вариантах осуществления, растворитель представляет собой от 95 до 99% ацетона и от 1 до 5% воды по объему. В некоторых вариантах осуществления, растворитель представляет собой от 90 до 99% ацетона и от 1 до 10% воды по объему.
[0127] В некоторых вариантах осуществления, растворитель удаляют методом лиофилизации или распылительной сушки. В некоторых вариантах осуществления, твердую дисперсию получают путем плавления, испарения растворителя, распылительной сушки, слияния, замешивания, совместного измельчения, лиофилизации, экструзией горячего расплава, агломерации в расплаве или сверхкритической флюидной технологии. Предпочтительно, когда растворитель удаляют путем распылительной сушки. Удаление растворителя может дополнительно включать сушку твердой дисперсии в полочной сушилке для удаления остаточного растворителя, который остается после распылительной сушки.
[0128] В некоторых вариантах осуществления, раствор включает от 5% до 20% твердых веществ по массе, например, от 8% до 14% твердых веществ по массе. В некоторых вариантах осуществления, раствор включает приблизительно 8% твердых веществ по массе, например, 8±2% твердых веществ.
[0129] В некоторых вариантах осуществления, приготовление (a) включает добавление соединения формулы (I) в растворитель (например, ацетон) и смешение полученной смеси до тех пор, пока соединение формулы (I) полностью не растворяется, или до тех пор, пока не образуется прозрачный раствор. Приготовление (a) может дополнительно включать добавление подходящего полимера (например, HPMCAS-H) в раствор соединения формулы (I) в ацетоне, и затем смешение. В некоторых вариантах осуществления, удаление (b) включает распылительную сушку раствора соединения формулы (I) и полимера в растворителе. Распылительная сушка может быть осуществлена в любой подходящей сушилке, например, в установке для распылительной сушки SPX Anhydro MicraSpray MS-150, оборудованной двухлоточной форсункой. В некоторых вариантах осуществления, распыление осуществляют в схеме с замкнутым контуром. Давление распыления в распылительной сушке может составлять от 0,20 до 0,35 МПа, например, от 0,25 до 0,28 МПа. В некоторых вариантах осуществления, норма распыления составляет от 5,0 до 20,0 кг/час, например, от 7,5 до 12,5 кг/час. В некоторых вариантах осуществления, температура на входе является повышенной, например, от 65 до 85°C. Температура на выходе может быть также повышенной, например, от 40 до 45°C. В некоторых вариантах осуществления, расход сушильного газа составляет от 150 до 200 кг/час. Температура в конденсаторе установки для распылительной сушки находится обычно ниже температуры замерзания, например, от -30 до -10°C.
[0130] Для удаления остаточного растворителя, остающегося после распылительной сушки, необязательно используют вторичную сушку. Вторичная сушка может включать полочную сушку, например, в полочной сушилке Despatch. В некоторых вариантах осуществления, твердую дисперсию загружают на полки полочной сушилки до глубины приблизительно 2,54 см. Температура сушки является обычно повышенной, например, от 30 до 50°C. В некоторых вариантах осуществления, вторичная сушка имеет продолжительность от 1 до 24 часов, например, от 6 до 18 часов, от 8 до 12 часов, или приблизительно 10 часов.
[0131] Вторичное извлечение необязательно используют для извлечения остаточной твердой дисперсии из распылительной форсунки, главной камеры и выходного отверстия после распылительной сушки. В некоторых вариантах осуществления, остаточная твердая дисперсия из распылительной форсунки, главной камеры и/или выходного отверстия может быть собрана и добавлена к основному материалу. Добавление остаточной твердой дисперсии, собранной из одной или более распылительных форсунок, главной камеры и выходного отверстия распылительной сушилки, к основному материалу может увеличивать суммарный выход твердой дисперсии, по меньшей мере, на 10%, например, по меньшей мере, на 15%, по меньшей мере, на 20%, по меньшей мере, на 25%, по меньшей мере, на 30%, по меньшей мере, на 35%, или, по меньшей мере, на 40%.
[0132] В некоторых аспектах, в настоящем изобретении предлагается способ приготовления раскрытой в изобретении твердой дисперсии, включающий (a) приготовление раствора соединения формулы (I) и HPMCAS в растворителе, где раствор включает от суммарно от 6% до 15% твердых веществ по массе; и (b) удаление растворителя с получением твердой дисперсии. Полимер может быть выбран из любого описанного в изобретении подходящего полимера, например, HPMCAS.
Фармацевтические композиции
[0133] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает: (a) твердую дисперсию, включающую соединение формулы (I); и (b) одно или более фармацевтически приемлемых вспомогательных веществ.
[0134] Твердая лекарственная форма может представлять собой капсулу или таблетку. В некоторых вариантах осуществления, твердая лекарственная форма представляет собой таблетку. В некоторых вариантах осуществления, одно или фармацевтически приемлемых вспомогательных веществ включают связующее вещество, наполнитель, разрыхлитель и смазывающее вещество. В некоторых вариантах осуществления, одно или фармацевтически приемлемых вспомогательных веществ дополнительно включают скользящее вещество и покрытие. В некоторых вариантах осуществления, одно или фармацевтически приемлемых вспомогательных веществ включают связующее вещество, наполнитель, разрыхлитель, смазывающее вещество, скользящее вещество и покрытие. В некоторых вариантах осуществления, твердая дисперсия присутствует в количестве от 15% до 50% от массы твердой лекарственной формы.
[0135] В некоторых вариантах осуществления, твердая дисперсия включает фармацевтически приемлемый полимер, например, HPMCAS. В некоторых вариантах осуществления, полимер присутствует в количестве от 15% до 35% от массы твердой лекарственной формы. В некоторых вариантах осуществления, соединение формулы (I) присутствует в количестве от 1% до 15% от массы твердой лекарственной формы. В некоторых вариантах осуществления, твердая лекарственная форма включает от 5 мг до 100 мг соединения формулы (I), например, 10 мг или 40 мг соединения формулы (I).
[0136] В некоторых вариантах осуществления, твердая лекарственная форма включает связующее вещество в количестве от 20% до 50% от массы твердой лекарственной формы. Связующее вещество может представлять собой микрокристаллическую целлюлозу. В некоторых вариантах осуществления, твердая лекарственная форма включает наполнитель в количестве от 20% до 40% от массы твердой лекарственной формы. Наполнитель может представлять собой маннит. В некоторых вариантах осуществления, твердая лекарственная форма включает интрагранулярный наполнитель и экстрагранулярный наполнитель, где, необязательно, интрагранулярный наполнитель присутствует в количестве от 12% до 22% от массы твердой лекарственной формы, и экстрагранулярный наполнитель присутствует в количестве от 8% до 18% от массы твердой лекарственной формы.
[0137] В некоторых вариантах осуществления, твердая лекарственная форма включает разрыхлитель в количестве от 1,0% до 5,0% от массы твердой лекарственной формы. Твердая лекарственная форма может включать интрагранулярный разрыхлитель и экстрагранулярный разрыхлитель, где, необязательно, интрагранулярный разрыхлитель присутствует в количестве от 0,9% до 3,0% от массы твердой лекарственной формы, и экстрагранулярный разрыхлитель присутствует в количестве от 0,1% до 2,0% от массы твердой лекарственной формы. В некоторых вариантах осуществления, разрыхлитель представляет собой кроскармеллозу натрия.
[0138] В некоторых вариантах осуществления, твердая лекарственная форма включает смазывающее вещество в количестве от 0,25% до 1,25% от массы твердой лекарственной формы. Твердая лекарственная форма может включать интрагранулярное смазывающее вещество и экстрагранулярное смазывающее вещество, где, необязательно, интрагранулярное смазывающее вещество присутствует в количестве от 0,15% до 0,75% от массы твердой лекарственной формы, и экстрагранулярное смазывающее вещество присутствует в количестве от 0,10% до 0,50% от массы твердой лекарственной формы. В некоторых вариантах осуществления, смазывающее вещество представляет собой стеарат магния.
[0139] В некоторых вариантах осуществления, твердая лекарственная форма включает скользящее вещество в количестве от 0,1% до 1,25% от массы твердой лекарственной формы. Твердая лекарственная форма может включать интрагранулярное скользящее вещество и экстрагранулярное скользящее вещество, где, необязательно, интрагранулярное скользящее вещество присутствует в количестве от 0,05% до 0,75% от массы твердой лекарственной формы, и экстрагранулярное скользящее вещество присутствует в количестве от 0,05% до 0,50% от массы твердой лекарственной формы. В некоторых вариантах осуществления, твердая лекарственная форма включает экстрагранулярное скользящее вещество в количестве от 0,2% до 0,5% от массы твердой лекарственной формы. В некоторых вариантах осуществления, скользящее вещество представляет собой коллоидный диоксид кремния, например, Cab-O-Sil.
[0140] В некоторых вариантах осуществления, твердая лекарственная форма включает покрытие. В некоторых вариантах осуществления, покрытие представляет собой покрытие на основе PVA, такое как OpaDry II. В некоторых вариантах осуществления, покрытие окрашено пигментом, где, необязательно, покрытие окрашено в голубой цвет. Твердая лекарственная форма может включать покрытие в количестве от 1% до 5% от массы твердой лекарственной формы, например, приблизительно 3% по массе.
[0141] В некоторых вариантах осуществления, твердость твердой лекарственная форма составляет от 5 до 25 KP (единиц твердости по Кнупу). Масса твердой лекарственной формы может составлять от 50 до 750 мг, например, приблизительно 125 мг или приблизительно 500 мг. В некоторых вариантах осуществления, твердая лекарственная форма включает менее чем 2% примесей по массе.
[0142] В некоторых вариантах осуществления, твердая лекарственная форма включает менее чем 3% воды по массе. В некоторых вариантах осуществления, твердая лекарственная форма включает менее чем 3% воды по массе после шести месяцев хранения при комнатной температуре. В некоторых вариантах осуществления, твердая лекарственная форма характеризуется временем распадаемости от 1 до 5 минут.
[0143] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает: (a) твердую дисперсию, включающую соединение формулы (I), и полимер, выбранный из HPMCAS, CAP и SOLUPLUS; и (b) одно или более фармацевтически приемлемых вспомогательных веществ.
[0144] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает: (a) твердую дисперсию, включающую от 5 до 100 мг соединения формулы (I); и (b) одно или более фармацевтически приемлемых вспомогательных веществ.
[0145] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает, в расчете на суммарную массу фармацевтической композиции:
(a) от 15% до 50% твердой дисперсии, включающей соединение формулы (I);
(b) от 20% до 50% связующего вещества;
(c) от 20% до 40% наполнителя;
(d) от 1,0% до 5,0% разрыхлителя; и
(e) от 0,25% до 1,25% смазывающего вещества.
[0146] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает, в расчете на суммарную массу фармацевтической композиции:
(a) от 15% до 50% твердой дисперсии, включающей соединение формулы (I) и HMPCAS;
(b) от 20% до 50% микрокристаллической целлюлозы;
(c) от 20% до 40% маннита;
(d) от 1,0% до 5,0% кроскармеллозы натрия; и
(e) от 0,25% до 1,25% стеарата магния.
[0147] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает, в расчете на суммарную массу фармацевтической композиции:
(a) от 15% до 50% твердой дисперсии, включающей соединение формулы (I);
(b) от 20% до 50% связующего вещества;
(c) от 20% до 40% наполнителя;
(d) от 1,0% до 5,0% разрыхлителя;
(e) от 0,25% до 1,25% смазывающего вещества;
(f) от 0,1% до 1,25% скользящего вещества; и
(g) от 1% до 5% покрытия.
[0148] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает, в расчете на суммарную массу фармацевтической композиции:
(a) от 15% до 50% твердой дисперсии, включающей соединение формулы (I) и HMPCAS;
(b) от 20% до 50% микрокристаллической целлюлозы;
(c) от 20% до 40% маннита;
(d) от 1,0% до 5,0% кроскармеллозы натрия;
(e) от 0,25% до 1,25% стеарата магния;
(f) от 0,1% до 1,25% коллоидного диоксида кремния; и
(g) необязательно, покрытие на основе PVA.
[0149] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает: (a) твердую дисперсию, включающую соединение формулы (I); и (b) одно или более фармацевтически приемлемых вспомогательных веществ, где масса твердой лекарственной формы составляет от 50 до 750 мг.
[0150] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает: (a) твердую дисперсию, включающую соединение формулы (I); (b) одно или более фармацевтически приемлемых вспомогательных веществ; и (c) менее чем 2% примесей по массе.
[0151] В некоторых аспектах, в настоящем изобретении предлагается фармацевтическая твердая лекарственная форма для перорального введения соединения формулы (I),
Формула (I),
где твердая лекарственная форма включает: (a) твердую дисперсию, включающую соединение формулы (I); (b) одно или более фармацевтически приемлемых вспомогательных веществ; и (c) менее чем 3% воды по массе после шести месяцев хранения при комнатной температуре.
[0152] Соединение или твердая дисперсия по настоящему изобретению могут быть приготовлены в любой подходящей лекарственной форме. Фармацевтическая композиция по настоящему изобретению обычно содержит активный ингредиент (например, соединение формулы (I)), и одно или более фармацевтически приемлемых вспомогательных веществ и/или носителей, включая, но этим не ограничивая, инертные твердые наполнители и наполнители, связующие вещества, разбавители, разрыхлители, смазывающие вещества, скользящие вещества, покрытия, стерильные водные растворы и различные органические растворители, усилители проницаемости, солюбилизаторы и адъюванты. Соединение или твердая дисперсия по настоящему изобретению могут быть приготовлены в любой подходящей лекарственной форме. В некоторых вариантах осуществления, фармацевтически приемлемое вспомогательное вещество выбирают из микрокристаллической целлюлозы, маннита, кроскармеллозы натрия и стеарата магния. В некоторых вариантах осуществления, фармацевтически приемлемое вспомогательное вещество выбирают из микрокристаллической целлюлозы, маннита, кроскармеллозы натрия, стеарата магния и Cab-O-Sil. В некоторых вариантах осуществления, в настоящем изобретении предлагается фармацевтическая композиция, включающая соединение формулы (I) и фармацевтически приемлемое вспомогательное вещество, выбранное из микрокристаллической целлюлозы, маннита, кроскармеллозы натрия и стеарата магния. В некоторых вариантах осуществления, в настоящем изобретении предлагается фармацевтическая композиция, включающая соединение формулы (I) и фармацевтически приемлемое вспомогательное вещество, выбранное из микрокристаллической целлюлозы, маннита, кроскармеллозы натрия, стеарата магния и коллоидного диоксида кремния. В некоторых вариантах осуществления, в настоящем изобретении предлагается фармацевтическая композиция, включающая соединение формулы (I), микрокристаллическую целлюлозу, маннит, кроскармеллозу натрия и стеарат магния. В некоторых вариантах осуществления, в настоящем изобретении предлагается фармацевтическая композиция, включающая соединение формулы (I), микрокристаллическую целлюлозу, маннит, кроскармеллозу натрия, стеарат магния, Cab-O-Sil и покрытие на основе PVA.
[0153] Лекарственные формы могут быть приготовлены в любой подходящей форме, которая может зависеть от способа введения. В некоторых вариантах осуществления, описанная в изобретении фармацевтическая композиция может быть приготовлена в лекарственной форме для введения субъекту. В некоторых вариантах осуществления, фармацевтическую композицию приготавливают для перорального, внутривенного, интраартериального, аэрозольного, парентерального, буккального, местного, трансдермального, ректального, внутримышечного, подкожного, внутрикостного, интраназального, внутрилегочного, трансмукозального, ингаляционного и/или интраперитонеального введения. В некоторых вариантах осуществления, лекарственную форму приготавливают для перорального введения. Например, фармацевтическая композиция может быть приготовлена в форме пилюли, таблетки, капсулы, ингаляционного препарата, жидкой суспензии, жидкой эмульсии, геля или порошка. Предпочтительно, чтобы фармацевтическую композицию приготавливали в форме пилюли, таблетки или капсулы. В некоторых вариантах осуществления, фармацевтическая композиция может быть приготовлена в качестве разовой дозы в жидкой, гелеобразной, полужидкой, полутвердой, пенообразной или твердой форме.
[0154] Количество каждого вводимого соединения будет зависеть от подвергаемого лечению млекопитающего, тяжести нарушения или состояния, скорости введения, фармакокинетики соединения и индивидуального суждения лечащего врача. Однако, эффективная доза может находиться в диапазоне от приблизительно 0,001 до приблизительно 100 мг на кг массы тела в сутки, в форме разовой дозы или разделенных доз. В некоторых вариантах осуществления, доза находится в диапазоне от приблизительно 0,1 мг/кг до приблизительно 1,0 мг/кг массы тела в сутки. В некоторых случаях, уровни доз ниже нижнего предела упомянутого выше диапазона могут быть более чем достаточными, в то время как в других случаях, могут быть использованы значительно более высокие дозы, не вызывая какого-либо опасного побочного эффекта, например, путем разделения таких более высоких доз на несколько низких доз для введения в течение дня.
[0155] В некоторых вариантах осуществления, в изобретении предлагается фармацевтическая композиция, включающая количество соединения формулы (I), приготовленная для введения субъекту, нуждающемуся в этом. В некоторых вариантах осуществления, фармацевтическая композиция включает приблизительно 0,1-1000 мг соединения формулы (I), например, 0,1-500 мг, 1-250 мг, 5-125 мг, 5-100 мг, 5-75 мг, или 5-50 мг соединения формулы (I). В некоторых вариантах осуществления, фармацевтическая композиция включает приблизительно или более чем приблизительно 1 мг, 2 мг, 3 мг, 4 мг, 5 мг, 6 мг, 7 мг, 8 мг, 9 мг, 10 мг, 15 мг, 20 мг, 25 мг, 30 мг, 35 мг, 40 мг, 45 мг, 50 мг, 55 мг, 60 мг, 65 мг, 70 мг, 75 мг, 80 мг, 85 мг, 90 мг, 95 мг или 100 мг соединения формулы (I). В некоторых вариантах осуществления, фармацевтическая композиция включает 1-100 мг соединения формулы (I) в разовой дозе, например, 5-50 мг соединения формулы (I). В некоторых вариантах осуществления, фармацевтическая композиция включает приблизительно 10 мг соединения формулы (I). В некоторых вариантах осуществления, фармацевтическая композиция включает приблизительно 40 мг соединения формулы (I).
[0156] В некоторых вариантах осуществления, терапевтически эффективное количество соединения формулы (I), которое может представлять собой суточное количество, вводимое в течение периода проведения лечения, позволяет в достаточной степени достигать любого одного или более терапевтических эффектов, описанных в изобретении. В качестве примера, терапевтически эффективное количество может находиться в диапазоне приблизительно 0,001-100 мг/кг массы тела, 0,001-50 мг/кг массы тела, 0,01-50 мг/кг массы тела, 0,01-25 мг/кг массы тела, 0,01-10 мг/кг массы тела, 0,01-5 мг/кг массы тела, 0,01-4 мг/кг массы тела, 0,01-3 мг/кг массы тела, 0,01-2 мг/кг массы тела, 0,01-1 мг/кг массы тела, 0,01-0,75 мг/кг массы тела, 0,1-5 мг/кг массы тела, 0,1-4 мг/кг массы тела, 0,1-3 мг/кг массы тела, 0,1-2 мг/кг массы тела, 0,1-1 мг/кг массы тела, или 0,1-0,75 мг/кг массы тела соединения формулы (I). В некоторых вариантах осуществления, терапевтическое количество может составлять приблизительно или более чем приблизительно 0,001 мг/кг массы тела, 0,01 мг/кг массы тела, 0,1 мг/кг массы тела, 0,2 мг/кг массы тела, 0,3 мг/кг массы тела, 0,4 мг/кг массы тела, 0,5 мг/кг массы тела, 0,6 мг/кг массы тела, 0,7 мг/кг массы тела, 0,8 мг/кг массы тела, 0,9 мг/кг массы тела, 1 мг/кг массы тела, 1,5 мг/кг массы тела, 2 мг/кг массы тела, 3 мг/кг массы тела, 4 мг/кг массы тела, 5 мг/кг массы тела, 6 мг/кг массы тела, 7 мг/кг массы тела, 8 мг/кг массы тела, 9 мг/кг массы тела, or 10 мг/кг массы тела соединения формулы (I). В некоторых вариантах осуществления, эффективное количество составляет, по меньшей мере, приблизительно 0,01 мг/кг массы тела соединения формулы (I). В некоторых вариантах осуществления, эффективное количество составляет количество в диапазоне приблизительно 0,01-10 мг/кг массы тела соединения формулы (I).
[0157] В некоторых вариантах осуществления, предлагается композиция в одной или более разовых дозах. Например, композиция может быть введена в 1, 2, 3, 4, 5, 6, 7, 14, 30, 60 или более дозах. Такое количество может вводиться каждый день, например, в индивидуальных дозах, вводимых один, два или три, или более раз в сутки. Однако, не следует считать, что заявленные в изобретении суточные дозы требуют введения суточной дозы каждый день. Например, если одно из лекарственных средств применяется соответствующим образом в форме с замедленным высвобождением, то две или более величин суточной дозы могут быть введены с меньшей частотой, например, в форме депо от одного раза в два дня до одного раза в месяц или даже более продолжительно. Как правило, для субъекта наиболее удобно, чтобы ингибитор, соединение формулы (I), вводили один раз в сутки, например, утром, вечером или в течение дня.
[0158] Разовые дозы могут быть введены одновременно или последовательно. Композиция может быть введена в течение длительного периода лечения. В качестве иллюстрации, период лечения может составлять, по меньшей мере, приблизительно один месяц, например, по меньшей мере, приблизительно, 3 месяца, по меньшей мере, приблизительно 6 месяцев или, по меньшей мере, приблизительно 1 год. В некоторых случаях, введение может продолжаться на протяжении практически всей оставшейся жизни субъекта.
[0159] Фармацевтическая композиция для перорального введения. В некоторых вариантах осуществления, в изобретении предлагается фармацевтическая композиция для перорального введения, содержащая, по меньшей мере, одно соединение по настоящему изобретению и фармацевтическое вспомогательное вещество, подходящее для перорального введения. Композиция может находиться в форме твердого вещества, жидкости, геля, полужидкого вещества или полутвердого вещества. В некоторых вариантах осуществления, композиция находится в форме твердого вещества. В некоторых вариантах осуществления, композиция дополнительно включает второе лекарственное средство.
[0160] В некоторых вариантах осуществления, в изобретении предлагается твердая фармацевтическая композиция для перорального введения, содержащая: (i) соединение формулы (I); и (ii) фармацевтическое вспомогательное вещество, подходящее для перорального введения. В некоторых вариантах осуществления, композиция дополнительно содержит: (iii) третье лекарственное средство или даже четвертое лекарственное средство. В некоторых вариантах осуществления, каждое соединение или лекарственное средство присутствует в терапевтически эффективном количестве. В других вариантах осуществления, одно или более соединений или лекарственных средств присутствуют в субтерапевтическом количестве, и соединения или лекарственные средства действуют синергетически с образованием терапевтически эффективной фармацевтической композиции.
[0161] Фармацевтические композиции по изобретению, подходящие для перорального введения, могут быть представлены в виде дискретных лекарственных форм, таких как твердые или мягкие капсулы, крахмальные капсулы, пастилки, леденцы или таблетки, или в форме жидкостей или спрей-аэрозолей, содержащих заданное количество активного ингредиента, в форме порошка или в гранулах, в форме раствора или суспензии в водной или неводной жидкости, в форме эмульсии типа "масло в воде" или жидкой эмульсии типа "вода в масле", или в форме диспергируемых порошков или гранул, или в форме сиропов или эликсиров. Предпочтительно, когда фармацевтическую композицию для перорального введения приготавливают в форме таблетки. Такие лекарственные формы могут быть приготовлены любым из используемых в фармацевтике методов, который обычно включает стадию смешения активного ингредиента (активных ингредиентов) со вспомогательным веществом. Обычно, композицию приготавливают путем равномерного и тщательного смешения активного ингредиента (активных ингредиентов) с жидкими вспомогательными веществами или мелкодисперсными твердыми вспомогательными веществами, или и с теми, и с другими, и затем, в случае необходимости, формования продукта в требуемую форму. Например, таблетка может быть приготовлена путем прессования или формования, необязательно, с одним или более фармацевтически приемлемыми вспомогательными веществами. Прессованные таблетки могут быть приготовлены путем прессования в соответствующей установке активного ингредиента (активных ингредиентов) в сыпучей форме, такой как порошок или гранулы, необязательно, смешанного с вспомогательным веществом, таким как, но этим не ограничивая, связующее вещество, наполнитель, разрыхлитель, скользящее вещество и/или смазывающее вещество. Формованные таблетки могут быть изготовлены путем формования в соответствующей установке смеси порошкообразного соединения, увлажненного с помощью инертного жидкого разбавителя.
[0162] Настоящее изобретение дополнительно включает в себя безводные фармацевтические композиции и лекарственные формы, включающие активный ингредиент, так как вода может способствовать разложению некоторых соединений. Например, воду могут добавлять (например, 5%) с целью моделирования длительного хранения для определения таких характеристик, как срок годности или стабильность лекарственных форм в течение времени. Безводные фармацевтические композиции и лекарственные формы по изобретению могут быть приготовлены с использованием безводных или содержащих низкую концентрацию влаги ингредиентов и условий с низким содержанием влаги или с низкой влажностью. Фармацевтические композиции и лекарственные формы по изобретению, которые содержат лактозу, могут быть приготовлены безводными, если ожидается реальный контакт с влагой и/или влажностью в процессе производства, расфасовки и/или хранения. Безводная фармацевтическая композиция может быть приготовлена и может храниться таким образом, что сохраняется ее безводный характер. Соответственно, безводные композиции могут быть расфасованы с использованием материалов, по поводу которых известно, что они предотвращают возможность воздействия воды, вследствие чего они могут быть включены в подходящие формуляры наборов. Примеры подходящих упаковок включают, но этим не ограничивая, контейнеры с низкой проницаемостью для паров воды, герметические упаковки из фольги, пластмассы или других подобных материалов, контейнеры с разовой дозой, блистерные упаковки и контурные упаковки.
[0163] Активный ингредиент может быть объединен в однородную смесь с фармацевтическим вспомогательным веществом методами фармацевтического компаундирования. Вспомогательное вещество может принимать разнообразные формы в зависимости от формы препарата, требуемой для введения. При приготовлении композиции для пероральной лекарственной формы, в качестве вспомогательных веществ может быть использована любая подходящая фармацевтическая среда, такая как, например, вода, гликоли, масла, спирты, ароматизирующие средства, консерванты, окрашивающие вещества и другие подобные вещества, в случае пероральных жидких препаратов (таких как суспензии, растворы и эликсиры) или аэрозолей; или могут быть использованы вспомогательные вещества, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие средства, смазывающие вещества, скользящие вещества, связующие вещества и разрыхлители в случае пероральных твердых препаратов, в некоторых вариантах осуществления без использования лактозы. При необходимости, на таблетки может быть нанесено покрытие стандартными методами водного или неводного нанесения.
[0164] Связующие вещества, подходящие для использования в заявленных фармацевтических композиций и лекарственных формах, включают, но этим не ограничивая, кукурузный крахмал, картофельный крахмал или другие крахмалы, желатин, природные и синтетические камеди, такие как аравийская камедь, альгинат натрия, альгиновую кислоту, другие альгинаты, порошкообразную трагакантовую камедь, гуаровую камедь, целлюлозу и ее производные (например, этилцеллюлозу, ацетат целлюлозы, карбоксиметилцеллюлозу кальция, карбоксиметилцеллюлозу натрия), поливинилпирролидон, метилцеллюлозу, прежелатинизированный крахмал, гидроксипропил- метилцеллюлозу, микрокристаллическую целлюлозу, и их смеси. В некоторых вариантах осуществления, связующее вещество представляет собой микрокристаллическую целлюлозу. В некоторых вариантах осуществления, микрокристаллическую целлюлозу выбирают из микрокристаллической целлюлозы PH101, микрокристаллической целлюлозы PH105, микрокристаллической целлюлозы PH112, микрокристаллической целлюлозы PH113, микрокристаллической целлюлозы PH200, микрокристаллической целлюлозы PH301 и микрокристаллической целлюлозы PH302. В некоторых вариантах осуществления, связующее вещество представляет собой микрокристаллическую целлюлозу PH101 или микрокристаллическую целлюлозу PH105. В некоторых вариантах осуществления, связующее вещество is микрокристаллическая целлюлоза PH101. В некоторых вариантах осуществления, связующее вещество представляет собой микрокристаллическую целлюлозу PH105. В некоторых вариантах осуществления, связующее вещество присутствует в количестве, по меньшей мере, 0,5 мг, по меньшей мере, 1 мг, по меньшей мере, 5 мг, по меньшей мере, 10 мг, по меньшей мере, 20 мг, по меньшей мере, 30 мг, по меньшей мере, 50 мг, по меньшей мере, 100 мг, по меньшей мере, 150 мг, по меньшей мере, 200 мг, по меньшей мере, 300 мг, или, по меньшей мере, 500 мг.
[0165] Примеры подходящих наполнителей для использования в раскрытых в изобретении фармацевтических композиций и лекарственных формах включают, но этим не ограничивая, тальк, карбонат кальция (например, гранулы или порошок), микрокристаллическую целлюлозу, порошок целлюлозы, декстраты, каолин, маннит, кремниевую кислоту, сорбит, крахмал, прежелатинизированный крахмал, и их смеси. В некоторых вариантах осуществления, наполнитель представляет собой маннит. В некоторых вариантах осуществления, маннит выбирают из маннита M100, маннита M200 и маннита M300. В некоторых вариантах осуществления, наполнитель представляет собой маннит M100. В некоторых вариантах осуществления, наполнитель присутствует в количестве, по меньшей мере, 0,5 мг, по меньшей мере, 1 мг, по меньшей мере, 5 мг, по меньшей мере, 10 мг, по меньшей мере, 20 мг, по меньшей мере, 30 мг, по меньшей мере, 50 мг, по меньшей мере, 100 мг, по меньшей мере, 150 мг, по меньшей мере, 200 мг, по меньшей мере, 300 мг, или, по меньшей мере, 500 мг.
[0166] Разрыхлитель могут использоваться в композиции по изобретению для приготовления таблеток, которые распадаются при воздействии водной окружающей среды. Использование слишком большого количества разрыхлителя позволяет получать таблетки, которые могут распадаться в бутылке. Использование слишком малого количества разрыхлителя может быть недостаточным для протекания распада и может изменять скорость и степень высвобождения активного ингредиента (активных ингредиентов) из лекарственной формы. Для приготовления лекарственных форм раскрытых в изобретении соединений может быть использовано соответствующее количество разрыхлителя, которое не является ни слишком малым, ни слишком большим, для того чтобы негативно влиять на изменение высвобождения активного ингредиента (активных ингредиентов). Используемое количество разрыхлителя может варьировать в зависимости от типа лекарственной формы и способа введения. В фармацевтической композиции может быть использовано от приблизительно 0,5 до приблизительно 15 массовых процентов разрыхлителя, например, от приблизительно 1,0 до приблизительно 5,0 массовых процентов разрыхлитель. Разрыхлители, которые могут быть использованы для приготовления фармацевтической композиции и лекарственных форм по изобретению, включают, но этим не ограничивая, агар-агар, альгиновую кислоту, карбонат кальция, микрокристаллическую целлюлозу, кроскармеллозу натрия, кросповидон, полакрилин калия, натрия крахмала гликолят, картофельный или кукурузный крахмал, другие крахмалы, прежелатинизированный крахмал, другие крахмалы, глины, другие альгины, другие целлюлозы, камеди или их смеси. В некоторых вариантах осуществления, разрыхлитель представляет собой кроскармеллозу натрия.
[0167] Смазывающие вещества, которые могут быть использованы для приготовления фармацевтических композиций и лекарственных форм по изобретению, включают, но этим не ограничивая, стеарат кальция, стеарат магния, минеральное масло, легкое минеральное масло, глицерин, сорбит, маннит, полиэтиленгликоль, другие гликоли, стеариновую кислоту, лаурилсульфат натрия, тальк, гидрированное растительное масло (например, арахисовое масло, хлопковое масло, подсолнечное масло, сезамовое масло, оливковое масло, кукурузное масло и соевое масло), стеарат цинка, этилолеат, этиллаурат, агар-агар, или их смеси. Дополнительные смазывающие вещества включают, например, коллоидный силикагель, коагулированный аэрозоль синтетического диоксида кремния, или их смеси. Смазывающее вещество может быть необязательно добавлено в количестве менее чем приблизительно 1 массового процента от массы фармацевтической композиции. В некоторых вариантах осуществления, смазывающее вещество представляет собой стеарат магния. В некоторых вариантах осуществления, смазывающее вещество присутствует в количестве, по меньшей мере, 0,1 мг, по меньшей мере, 0,2 мг, по меньшей мере, 0,3 мг, по меньшей мере, 0,4 мг, по меньшей мере, 0,5 мг, по меньшей мере, 0,6 мг, по меньшей мере, 0,7 мг, по меньшей мере, 0,8 мг, по меньшей мере, 0,9 мг, по меньшей мере, 1 мг, или, по меньшей мере, 5 мг.
[0168] Скользящие вещества, которые могут быть использованы для приготовления фармацевтических композиций и лекарственных форм по изобретению, включают, но этим не ограничивая, коллоидный диоксид кремния, CAB-O-SIL® (Cabot Co. of Boston, MA), не содержащий асбеста тальк, стеарат магния, крахмал и тальк. В некоторых вариантах осуществления, скользящее вещество присутствует в количестве, по меньшей мере, 0,1 мг, по меньшей мере, 0,2 мг, по меньшей мере, 0,3 мг, по меньшей мере, 0,4 мг, по меньшей мере, 0,5 мг, по меньшей мере, 0,6 мг, по меньшей мере, 0,7 мг, по меньшей мере, 0,8 мг, по меньшей мере, 0,9 мг, по меньшей мере, 1 мг, по меньшей мере, 1,1 мг, по меньшей мере, 1,2 мг, по меньшей мере, 1,3 мг, по меньшей мере, 1,4 мг, по меньшей мере, 1,5 мг, по меньшей мере, 2 мг, по меньшей мере, 3 мг, по меньшей мере, 4 мг, или, по меньшей мере, 5 мг.
[0169] Когда требуются водные суспензии и/или эликсиры для перорального введения, активный ингредиент в них может быть объединен с различными подсластителями или ароматизаторами, окрашивающим веществом или красителями и, по желанию, эмульгаторами и/или суспендирующими средствами, вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и различные их комбинации.
[0170] Таблетки могут быть без нанесенного на них слоя или с нанесенным слоем с помощью известных методов для отсрочки распада и абсорбции в желудочно-кишечном тракте и вследствие этого обеспечения пролонгированного действия в течение более длительного периода времени. Например, может быть использован обеспечивающий задержку во времени материал, такой как глицерилмоностеарат или глицерилдистеарат. Покрытие может дополнительно предотвращать или замедлять сорбцию влаги. Лекарственные формы для перорального применения могут быть также представлены твердыми желатиновыми капсулами, в которых активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или мягкими желатиновыми капсулами, в которых активный ингредиент смешан с водной или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом. Энтеросолюбильные покрытия включают, но этим не ограничивая, CAP, акрилатные полимеры, HPMCP, HPMC, PVAP, PVA, жирные кислоты, жиры, фенилсалицилат, воски, шеллак, аммонизированный шеллак и ацетатфталаты целлюлозы. Неэнтеросолюбильные покрытия включают, но этим не ограничивая, HPMC, метилгидроксиэтилцеллюлозы (MHEC), EC, HPC, повидон, карбоксиметилцеллюлозу натрия (SCMC), PEG и акрилатные полимеры. Таблетки в сахарной оболочке представляют собой прессованные таблетки, покрытые слоем сахара, который позволяет избавиться от неприятных вкусов и запахов и защитить таблетки от окисления. Таблетки с пленочным покрытием представляют собой прессованные таблетки, которые покрыты тонким слоем или пленкой водорастворимого материала. Пленочные покрытия включают, но этим не ограничивая, гидроксиэтилцеллюлозу, карбоксиметил-целлюлозу натрия, полиэтиленгликоль 4000 и ацетатфталат целлюлозы. Пленочное покрытие придает таблеткам те же самые свойства, что и сахарное покрытие. В некоторых вариантах осуществления, покрытие представляет собой покрытие на основе PVA, такое как OpaDry II. В некоторых вариантах осуществления, покрытие окрашено пигментом. Например, покрытие может включать голубой, зеленый, красный, пурпурный, оранжевый, серебряный и/или желтый пигменты. В некоторых вариантах осуществления, покрытие представляет собой OpaDry II, например, OpaDry II голубое. В некоторых вариантах осуществления, покрытие присутствует в количестве, по меньшей мере, 0,5 мг, по меньшей мере, 1 мг, по меньшей мере, 2,5 мг, по меньшей мере, 5 мг, по меньшей мере, 7,5 мг, по меньшей мере, 10 мг, по меньшей мере, 12,5 мг, по меньшей мере, 15 мг, по меньшей мере, 17,5 мг, по меньшей мере, 20 мг, по меньшей мере, 25 мг, или, по меньшей мере, 30 мг.
[0171] Поверхностно-активное вещество, которое может быть использовано для приготовления фармацевтической композиции и лекарственных форм по изобретению включает, но этим не ограничивая, гидрофильные поверхностно-активные вещества, липофильные поверхностно-активные вещества и их смеси. А именно, может быть использована смесь гидрофильных поверхностно-активных веществ, может быть использована смесь липофильных поверхностно-активных веществ или может быть использована смесь, по меньшей мере, одного гидрофильного поверхностно-активного вещества и, по меньшей мере, одного липофильного поверхностно-активного вещества.
[0172] Подходящее гидрофильное поверхностно-активное вещество может, как правило, иметь величину HLB, по меньшей мере, 10, в то время как подходящие липофильные поверхностно-активные вещества могут, как правило, иметь величину HLB приблизительно 10 или менее чем приблизительно 10. Эмпирическим параметром, используемым для характеристики относительной гидрофильности и гидрофобности неионогенных амфифильных соединений, является гидрофильно-липофильный баланс (величина "HLB"). Поверхностно-активные вещества с более низкими величинами HLB являются более липофильными или гидрофобными и имеют более высокую растворимость в маслах, в то время как поверхностно-активные вещества с более высокими величинами HLB являются более гидрофильными и имеют более высокую растворимость в водных растворах. Гидрофильными поверхностно-активными веществами обычно считают те соединения, которые имеют величину HLB больше чем приблизительно 10, а также анионные, катионные или цвиттер-ионные соединения, для которых шкалу HLB обычно не применяют. Аналогично, липофильными (то есть, гидрофобными) поверхностно-активными веществами являются соединения, которые имеют величину HLB равную или меньшую чем приблизительно 10. Однако, величина HLB поверхностно-активного вещества является всего лишь грубым показателем, обычно используемым для оценки возможности получения промышленных, фармацевтических и косметических эмульсий.
[0173] Гидрофильные поверхностно-активные вещества могут быть либо или ионогенными, или неионогенными. Подходящие ионогенные поверхностно-активные вещества включают, но этим не ограничивая, соли алкиламмония; соли фусидовой кислоты; производные аминокислот, олигопептидов и полипептидов с жирной кислотой; глицеридные производные аминокислот, олигопептидов и полипептидов; лецитины и гидрированные лецитины; лизолецитины и гидрированные лизолецитины; фосфолипиды и их производные; лизофосфолипиды и их производные; соли эфиров карнитина с жирной кислотой; соли алкилсульфатов; соли жирных кислот; докузат натрия; ацилактилаты; моно- и диацетилированные эфиры винной кислоты моно- и диглицеридов; сукцинилированные моно- и диглицериды; эфиры лимонной кислоты моно- и диглицеридов; и их смеси.
[0174] Внутри упомянутой выше группы, ионогенные поверхностно-активные вещества включают, например, лецитины, лизолецитин, фосфолипиды, лизофосфолипиды и их производные; соли эфиров карнитина с жирной кислотой; соли алкилсульфатов; соли жирных кислот; докузат натрия; ацилактилаты; моно- и диацетилированные эфиры винной кислоты моно- и диглицеридов; сукцинилированные моно- и диглицериды; эфиры лимонной кислоты моно- и диглицеридов; и их смеси.
[0175] Ионогенные поверхностно-активные вещества могут находиться в ионизированных формах лецитина, лизолецитина, фосфатидилхолина, фосфатидилэтаноламина, фосфатидилглицерина, фосфатидной кислоты, фосфатидилсерина, лизофосфатидилхолина, лизофосфатидилэтаноламина, лизофосфатидилглицерина, лизофосфатидной кислоты, лизофосфатидилсерина, PEG-фосфатидилэтаноламина, PVP-фосфатидилэтаноламина, эфиров молочной кислоты жирных кислот, стеароил-2-лактилата, стеароиллактилата, сукцинилированных моноглицеридов, моно/диацетилированных эфиров винной кислоты моно/диглицеридов, эфиров лимонной кислоты моно/диглицеридов, холилсаркозина, капроата, каприлата, капрата, лаурата, миристата, пальмитата, олеата, рицинолеата, линолеата, линолената, стеарата, лаурилсульфата, терацилсульфата, докузата, лауроилкарнитинов, пальмитоилкарнитинов, миристоилкарнитинов, и их солей и их смесей.
[0176] Гидрофильные неионогенные поверхностно-активные вещества могут включать, но этим не ограничивая, алкилглюкозиды; алкилмальтозиды; алкилтиоглюкозиды; лаурилмакроголглицериды; полиоксиалкиленалкиловые эфиры, такие как полиэтиленгликоль-алкиловые эфиры; полиоксиалкиленалкилфенолы, такие как, такие как полиэтиленгликольалкилфенолы; эфиры полиоксиалкиленалкилфенолов и жирных кислот, такие как моноэфиры полиэтиленгликолей и жирных кислот и диэфиры полиэтиленгликолей и жирных кислот; эфиры полиэтиленгликольглицеринов и жирных кислот; эфиры полиглицеринов и жирных кислот; эфиры полиоксиалкиленсорбитанов и жирных кислот, такие как эфиры полиэтиленгликольсорбитанов и жирных кислот; гидрофильные продукты переэтерификации полиола, по меньшей мере, с одним представителем группы глицеридов, растительных масел, гидрированных растительных масел, жирных кислот и стеролов; полиоксиэтиленстеролы, их производные и аналоги; полиоксиэтилированные витамины и их производные; блок-сополимеры полиоксиэтилен-полиоксипропилен; и их смеси; эфиры полиэтиленгликольсорбитанов и жирных кислот и гидрофильные продукты переэтерификации полиола, по меньшей мере, с одним представителем группы триглицеридов, растительных масел и гидрированных растительных масел. Полиол может представлять собой глицерин, этиленгликоль, полиэтиленгликоль, сорбит, пропиленгликоль, пентаэритрит или сахарид.
[0177] Другие гидрофильные неионогенные поверхностно-активные вещества включают, без ограничения, PEG-10 лаурат, PEG-12 лаурат, PEG-20 лаурат, PEG-32 лаурат, PEG-32 дилаурат, PEG-12 олеат, PEG-15 олеат, PEG-20 олеат, PEG-20 диолеат, PEG-32 олеат, PEG-200 олеат, PEG-400 олеат, PEG-15 стеарат, PEG-32 дистеарат, PEG-40 стеарат, PEG-100 стеарат, PEG-20 дилаурат, PEG-25 глицерил- триолеат, PEG-32 диолеат, PEG-20 глицериллаурат, PEG-30 глицерил- лаурат, PEG-20 глицерилстеарат, PEG-20 глицерилолеат, PEG-30 глицерилолеат, PEG-30 глицериллаурат, PEG-40 глицериллаурат, PEG-40 косточковое пальмовое масло, PEG-50 гидрированное касторовое масло, PEG-40 касторовое масло, PEG-35 касторовое масло, PEG-60 касторовое масло, PEG-40 гидрированное касторовое масло, PEG-60 гидрированное касторовое масло, PEG-60 кукурузное масло, PEG-6 капрат/каприлатглицериды, PEG-8 капрат/каприлатглицериды, полиглицерил-10 лаурат, PEG-30 холестерин, PEG-25 фитостерол, PEG-30 соя стерол, PEG-20 триолеат, PEG-40 сорбитан олеат, PEG-80 сорбитан лаурат, полисорбат 20, полисорбат 80, POE-9 лауриловый эфир, POE-23 лауриловый эфир, POE-10 олеиловый эфир, POE-20 олеиловый эфир, POE-20 стеариловый эфир, токоферил PEG-100 сукцинат, PEG-24 холестерин, полиглицерил-10 олеат, Tween 40, Tween 60, сахарозы моностеарат, сахарозы монолаурат, сахарозы монопальмитат, PEG 10-100 нонилфенольные соединения, PEG 15-100 октилфенольные соединения и полоксамеры.
[0178] Подходящие липофильные поверхностно-активные вещества включают, например, только жирные спирты; эфиры глицерина и жирных кислот; ацетилированные эфиры глицерина и жирных кислот; эфиры низших спиртов и жирных кислот; эфиры пропиленгликоля и жирных кислот; эфиры сорбитана и жирных кислот; эфиры полиэтиленгликольсорбитана и жирных кислот; стеролы и производные стерола; производные полиоксиэтилированных стеролов и стеролов; полиэтиленгликольалкиловые эфиры; сложные эфира сахаров; простые эфира сахаров; производные молочной кислоты и моно- и диглицеридов; гидрофобные продукты переэтерификации полиола, по меньшей мере, с одним представителем группы глицеридов, растительных масел, гидрированных растительных масел, жирных кислот и стероидов; производные жирорастворимых витаминов/витаминов; и их смеси. Внутри этой группы, предпочтительные липофильные поверхностно-активные вещества включают эфиры глицерина и жирных кислот, эфиры пропиленгликоля и жирных кислот, и их смеси, или предпочтительными являются гидрофобные продукты переэтерификации полиола, по меньшей мере, с одним представителем группы растительных масел, гидрированных растительных масел и триглицеридов.
[0179] В одном варианте осуществления, композиция может включать солюбилизатор для обеспечения хорошей солюбилизации и/или растворения соединения по настоящему изобретению и для минимизации осаждения соединения по настоящему изобретению. Это может представлять особую важность для композиции для неперорального применения, например, композиции для инъекции. Солюбилизатор может быть также добавлен для повышения растворимости гидрофильного лекарственного средства и/или других компонентов, таких как поверхностно-активные вещества, или для поддержания композиции в форме стабильного или гомогенного раствора или дисперсии.
[0180] Примеры подходящих солюбилизаторов включают, но этим не ограничивая, следующие вещества: спирты и полиолы, такие как этанол, изопропанол, бутанол, бензиловый спирт, этиленгликоль, пропиленгликоль, бутандиолы и их изомеры, глицерин, пентаэритрит, сорбит, маннит, транскутол, диметилизосорбид, полиэтиленгликоль, полипропиленгликоль, поливиниловый спирт, гидроксипропил- метилцеллюлоза и другие производные целлюлозы, циклодекстрины и производные циклодекстринов; эфиры полиэтиленгликолей со средней молекулярной массой от приблизительно 200 до приблизительно 6000, такие как эфир тетрагидрофурфурилового спирта и PEG (гликофурол) или метокси PEG; амиды и другие азотсодержащие соединения, такие как 2-пирролидон, 2-пиперидон, ε-капролактам, N-алкилпирролидон, N-гидроксиалкилпирролидон, N-алкилпиперидон, N-алкилкапролактам, диметилацетамид и поливинилпирролидон; эфиры, такие как этилпропионат, трибутилцитрат, ацетилтриэтилцитрат, ацетилтрибутилцитрат, триэтилцитрат, этилолеат, этилкаприлат, этилбутират, триацетин, пропиленгликоля моноацетат, пропиленгликоля диацетат, ε-капролактон и его изомеры, δ-валеролактон и его изомеры, β-бутиролактон и его изомеры; и другие известные в данной области солюбилизаторы, такие как диметилацетамид, диметилизосорбид, N-метилпирролидоны, монооктаноин, моноэтиловый эфир диэтиленгликоля и вода.
[0181] Могут быть также использованы смеси солюбилизаторов. Примеры включают, но этим не ограничивая, триацетин, триэтилцитрат, этилолеат, этилкаприлат, диметилацетамид, N-метилпирролидон, N-гидроксиэтилпирролидон, поливинилпирролидон, гидроксипропил метилцеллюлозу, гидроксипропилциклодекстрины, этанол, полиэтиленгликоль 200-100, гликофурол, транскутол, пропиленгликоль и диметилизосорбид. Особенно предпочтительные солюбилизаторы включают сорбит, глицерин, триацетин, этиловый спирт, PEG-400, гликофурол и пропиленгликоль.
[0182] На количество солюбилизатора, которое может быть введено, не накладывают особых ограничений. Количество данного солюбилизатора может ограничиваться биологически приемлемым количеством, которое может быть легко определено любым специалистом в данной области. В некоторых случаях, предпочтительно включать количества солюбилизаторов намного больше, чем их биологически приемлемые количества, например, для максимизации концентрации лекарственного средства, при этом избыток солюбилизатора удаляют перед введением композиции пациенту с использованием традиционных методов, таких как дистилляция или испарения. Если солюбилизатор присутствует, то он может находиться при массовом отношении 10%, 25%, 50%, 100% или до приблизительно 200% по массе, в расчете на общую массу лекарственного средства и других вспомогательных средств. При необходимости, могут также использоваться очень малые количества солюбилизатора, например, 5%, 2%, 1% или еще меньше. Как правило, солюбилизатор может присутствовать в количестве от приблизительно 1% до приблизительно 100%, чаще всего, от приблизительно 5% до приблизительно 25% по массе.
[0183] Композиция может дополнительно включать одну или более фармацевтически приемлемых добавок и вспомогательных веществ. Такие добавки и вспомогательные вещества включают, без ограничения, вещества для устранения клейкости, антивспениватели, буферные вещества, полимеры, антиоксиданты, консерванты, хелатообразующие вещества, модуляторы вязкости, модификаторы тоничности, ароматизаторы, окрашивающие вещества, ароматизаторы, замутняющие средства, суспендирующие вещества, связующие вещества, наполнители, пластификаторы, смазывающие вещества, и их смеси.
[0184] Кроме того, в композицию могут быть введены кислота или основание с целью облегчения технологии изготовления, повышения стабильности или по другим причинам. Примеры фармацевтически приемлемых оснований включают аминокислоты, эфиры аминокислот, гидроксид аммония, гидроксид калия, гидроксид натрия, гидрокарбонат натрия, гидроксид алюминия, карбонат кальция, гидроксид магния, алюмосиликат магния, синтетический алюмосиликат, синтетический гидрокальцит, гидроксид магния алюминия, диизопропилэтиламин, этаноламин, этилендиамин, триэтаноламин, триэтиламин, триизопропаноламин, триметиламин, трис(гидроксиметил)аминометан (TRIS) и другие подобные основания. Кроме того, подходящими основаниями являются основания, которые представляют собой соли фармацевтически приемлемой кислоты, такой как уксусная кислота, акриловая кислота, адипиновая кислота, альгиновая кислота, алкансульфоновая кислота, аминокислоты, аскорбиновая кислота, бензойная кислота, борная кислота, масляная кислота, угольная кислота, лимонная кислота, жирные кислоты, муравьиная кислота, фумаровая кислота, глюконовая кислота, гидрохиносульфоновая кислота, изоаскорбиновая кислота, молочная кислота, малеиновая кислота, щавелевая кислота, para-бромфенилсульфоновая кислота, пропионовая кислота, п-толуолсульфоновая кислота, салициловая кислота, стеариновая кислота, янтарная кислота, дубильная кислота, винная кислота, тиогликолевая кислота, толуолсульфоновая кислота, мочевая кислота, и другие подобные кислоты. Могут быть также использованы соли многоосновных кислот, такие как фосфат натрия, двузамещенный фосфорнокислый натрий и однозамещенный фосфорнокислый натрий. Когда основание представляет собой соль, катионом может являться любой подходящий и фармацевтически приемлемый катион, такой как аммоний, щелочные металлы, щелочноземельные металлы и другие подобные катионы. Примеры могут включать, но этим не ограничивая, натрий, калий, литий, магний, кальций и аммоний.
[0185] Подходящими кислотами являются фармацевтически приемлемые органические или неорганические кислоты. Примеры подходящих неорганических кислот включают хлористоводородную кислоту, бромистоводородную кислоту, иодистоводородную кислоту, серную кислоту, азотную кислоту, борную кислоту, фосфорную кислоту, и другие подобные кислоты. Примеры подходящих органических кислот включают уксусную кислоту, акриловую кислоту, адипиновую кислоту, альгиновую кислоту, алкансульфоновые кислоты, аминокислоты, аскорбиновую кислоту, бензойную кислоту, борную кислоту, масляную кислоту, угольную кислоту, лимонную кислоту, жирные кислоты, муравьиную кислоту, фумаровую кислоту, глюконовую кислоту, гидрохиносульфоновую кислоту, изоаскорбиновую кислоту, молочную кислоту, малеиновую кислоту, метансульфоновую кислоту, щавелевую кислоту, пара-бромфенилсульфоновую кислоту, пропионовую кислоту, п-толуолсульфоновую кислоту, салициловую кислоту, стеариновую кислоту, янтарную кислоту, дубильную кислоту, винную кислоту, тиогликолевую кислоту, толуолсульфоновую кислоту, мочевую кислоту и другие подобные кислоты.
[0186] Когда способ по изобретению включает комбинированную терапию, например, где вторичное лекарственное средство вводят совместно с соединением формулы (I), лекарственные средства могут быть введены раздельно, одновременно, или в различные моменты времени в течение дня, или они могут быть введены в одной композиции. При комбинированных терапиях по изобретению, каждое лекарственное средство может быть введено методом, обеспечивающим "немедленное высвобождение" или методом, обеспечивающим "контролируемое высвобождение". Когда дополнительное активное средство представляет собой, например, кортикостероид, любая лекарственная форма, содержащая оба активных средства, например, и соединение формулы (I) и кортикостероид, может обеспечивать немедленное высвобождение или контролируемое высвобождение кортикостероида, и или немедленное высвобождение, или контролируемое высвобождение соединения формулы (I). В других лекарственных формах по настоящему изобретению, два или более дополнительных активных средств, которые могут относится или могут не относится к одному и тому же классу лекарственных средств, могут присутствовать в комбинации вместе с соединением формулы (I). В таком случае, эффективное количество одного или каждого присутствующего индивидуального дополнительного активного средства может, как правило, быть уменьшено по сравнению с его количеством, которое потребовалось бы в случае использования только одного добавляемого лекарственного средства.
[0187] В некоторых вариантах осуществления, предлагаемую в изобретении твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию, или предлагаемую в изобретении композицию, включающую твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию, вводят перорально, парентерально, местно или мукозально. В некоторых вариантах осуществления, предлагаемую в изобретении твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию, или предлагаемую в изобретении композицию, включающую твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию, вводят перорально.
[0188] В некоторых вариантах осуществления, предлагаемую в изобретении твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию, или предлагаемую в изобретении композицию, включающую твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию, вводят при частоте дозирования один, два, три или четыре раза в сутки. В некоторых вариантах осуществления, предлагаемая в изобретении твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция, или предлагаемая в изобретении композиция, включающая твердую дисперсию, твердая лекарственная форма или фармацевтическая композиция, включает соединение формулы (I) в количестве от приблизительно 0,1 до приблизительно 100 мг, от приблизительно 0,5 до приблизительно 75 мг, от приблизительно 5 до приблизительно 50 мг, от приблизительно 5 мг до приблизительно 45 мг, от приблизительно 5 до приблизительно 15 мг, или от приблизительно 35 до приблизительно 45 мг. В некоторых вариантах осуществления, в изобретении предлагается лекарственная форма с разовой дозой, применяемая для перорального введения человеку, включающая количество, равное или большее чем приблизительно 1, 2, 3, 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90 или 100 мг, соединения формулы (I) и фармацевтически приемлемое вспомогательное вещество. В некоторых вариантах осуществления, количество активного ингредиента составляет приблизительно 10 мг. В некоторых вариантах осуществления, количество активного ингредиента составляет приблизительно 40 мг.
[0189] В некоторых вариантах осуществления, второе активное средство вводят один или два раза в сутки, один раз в два дня, один раз в неделю, один раз в две недели, или один раз в три недели, в количестве от приблизительно 1 до приблизительно 1000 мг, от приблизительно 5 до приблизительно 500 мг, от приблизительно 10 до приблизительно 350 мг, или от приблизительно 50 до приблизительно 200 мг. В некоторых вариантах осуществления, второе активное средство вводят перорально или внутривенно. Конкретное количество второго активного средства будет зависеть от используемого конкретного средства, подвергаемого лечению или контролю типа заболевания, тяжести и стадии заболевания, и количества (количеств) предлагаемых в изобретении соединений и любых необязательных дополнительных активных средств, одновременно вводимых пациенту.
[0190] Как уже было указано в изобретении выше, настоящее изобретение также включает в себя способ снижения, лечения и/или предотвращения неблагоприятных или нежелательных эффектов, связанных с проведением традиционной терапии, включающей, но этим не ограничивая, хирургическое вмешательство, химиотерапию, лучевую терапию, гормональную терапию, биологическую терапию и иммунотерапию. Предлагаемая в изобретении твердая дисперсия, включающая соединение формулы (I) и другие активные ингредиенты, может быть введена пациенту до, во время или после возникновения неблагоприятного эффекта, связанного с проведением традиционной терапии.
Способы приготовления фармацевтической композиции
[0191] В конкретных аспектах, в настоящем изобретении предлагается способ приготовления твердой лекарственной формы, включающий: (a) смешение соединения формулы (I) и одного или более фармацевтически приемлемых вспомогательных веществ с получением измельченных гранул; и (b) прессование гранул путем приложения усилия прессования от 5 кН до 20 кН.
[0192] В конкретных аспектах, в настоящем изобретении предлагается способ приготовления твердой лекарственной формы, включающий: (a) смешение соединения формулы (I), связующего вещества, наполнителя, разрыхлителя и смазывающего вещества, с получением в результате этого перемешанной смеси; (b) гранулирование перемешанной смеси, необязательно, с использованием роликового пресса, с получением в результате этого гранулированной смеси; (c) смешение второго наполнителя, второго разрыхлителя и второго смазывающего вещества с гранулированной смесью, с получением в результате этого смеси для таблетирования; и (d) прессование смеси для таблетирования в таблетку, где наполнитель и второй наполнитель являются одинаковыми или разными; разрыхлитель и второй разрыхлитель являются одинаковыми или разными; и смазывающее вещество и второе смазывающее вещество являются одинаковыми или разными. В некоторых вариантах осуществления, связующее вещество представляет собой микрокристаллическую целлюлозу, наполнитель представляет собой маннит, разрыхлитель представляет собой кроскармеллозу натрия и смазывающее вещество представляет собой стеарат магния. В некоторых вариантах осуществления, второй наполнитель представляет собой маннит, второй разрыхлитель представляет собой кроскармеллозу натрия, и второе смазывающее вещество представляет собой стеарат магния. Способ может дополнительно включать смешение скользящего вещества с гранулированной смесью. В некоторых вариантах осуществления, скользящее вещество представляет собой коллоидный диоксид кремния, такой как Cab-O-Sil. В некоторых вариантах осуществления, способ дополнительно включает нанесение покрытия на таблетку. Покрытие может представлять собой покрытие на основе PVA, такое как OpaDry II.
[0193] В некоторых вариантах осуществления, способ включает предварительное нанесение покрытия из связующего вещества на корпус смесителя, где, необязательно, связующее вещество представляет собой микрокристаллическую целлюлозу. Предварительное нанесение покрытия на корпус смесителя может включать перемешивание связующего вещества, такого как связующее вещество для смешения, в течение, по меньшей мере, 1 минуты. Метод может дополнительно включать смешение интрагранулярных ингредиентов в смесителе с нанесенным покрытием, где, необязательно, интрагранулярные ингредиенты включают соединение формулы (I), связующее вещество, наполнитель, разрыхлитель и, необязательно, смазывающее вещество. Смешение может включать перемешивание в течение, по меньшей мере, 5 минут, необязательно, при скорости вращения 15 об/мин или выше. В некоторых вариантах осуществления, способ дополнительно включает измельчение перемешиваемых интрагранулярных ингредиентов. Способ может дополнительно включать смешение измельченных интрагранулярных ингредиентов в смесителе, например, перемешивание в течение, по меньшей мере, 10 минут при скорости вращения 15 об/мин. В некоторых вариантах осуществления, смазывающее вещество, такое как стеарат магния, добавляют в смеситель и смешивают с измельченными и перемешенными интрагранулярными ингредиентами. Эта смесь может быть подвергнута гранулированию, необязательно, с использованием роликового пресса. Необязательно, способ дополнительно включает пропускание гранулированного материала через мельницу. Способ может дополнительно включать перенесение измельченных гранул в смеситель и смешение с экстрагранулярными ингредиентами, где, необязательно, экстрагранулярные ингредиенты включают наполнитель и разрыхлитель. В некоторых вариантах осуществления, способ дополнительно включает добавление смазывающего вещества, такого как стеарат магния, к смеси и смешение с получением смеси для таблетирования. В некоторых вариантах осуществления, способ дополнительно включает добавление скользящего вещества, такого как коллоидный диоксид кремния, к смеси и смешение с получением смеси для таблетирования. Необязательно, смесь для таблетирования подвергают уплотнению с использованием роликового уплотнителя. В некоторых вариантах осуществления, способ включает прессование смесь для таблетирования в таблетку. В некоторых вариантах осуществления, таблетки имеют овальную форму. В некоторых вариантах осуществления, обеспечиваемое прессом усилие прессования составляет от 5 до 20 кН, например, приблизительно 7,5 кН или приблизительно 15 кН. В некоторых вариантах осуществления, таблетка состоит из приблизительно 500 мг смеси для таблетирования, например, 500±25 мг. В некоторых вариантах осуществления, таблетка состоит из приблизительно 125 мг смеси для таблетирования, например 125±10 мг.
Расфасованные композиции и наборы
[0194] В конкретных аспектах, в настоящем изобретении предлагается расфасованная твердая дисперсия, включающая описанную в изобретении твердую дисперсию и осушающее вещество. Подходящие осушающие вещества включают силикагель, бентонитовую глину или молекулярные сита. В некоторых вариантах осуществления, упаковка включает контейнер с низкой проницаемостью для паров воды, такой как бутылка из полиэтилена высокой плотности (HDPE), пакет из полиэтилена низкой плотности (LDPE), двойные пакеты из полиэтилена низкой плотности (LDPE), горловины которых затягивают с помощью жгута, бутылка из полиэтилена высокой плотности (HDPE), закрываемая завинчивающейся пробкой, бочка из полиэтилена низкой плотности (LDPE), или их комбинация.
[0195] В конкретных аспектах, в настоящем изобретении предлагается расфасованная твердая лекарственная форма, включающая описанную в изобретении твердую лекарственную форму и осушающее вещество. Подходящие осушающие вещества включают силикагель, Sorb-it®, Clariant, бентонитовую глину или молекулярные сита. В некоторых вариантах осуществления, упаковка включает контейнер с низкой проницаемостью для паров воды, такой как бутылка из полиэтилена высокой плотности (HDPE), пакет из полиэтилена низкой плотности (LDPE), двойные пакеты из полиэтилена низкой плотности (LDPE), горловины которых затягивают с помощью жгута, бутылка из полиэтилена высокой плотности (HDPE), закрываемая завинчивающейся пробкой, бочка из полиэтилена низкой плотности (LDPE), двухсторонние блистерные упаковки из фольги, двухсторонние блистерные упаковки из фольги холодного формования, двухсторонние блистерные упаковки из алюминиевой фольги, двухсторонние блистерные упаковки из алюминиевой фольги холодного формования, блистерные упаковки из поливинилхлорида (PVC), блистерные упаковки из поливинилиденхлорида (PVDC), блистерные упаковки из политетрафторэтилена (Aclar), блистерные упаковки из полихлортрифторэтилена (PCTFE), пластиковая бутыль, многослойная бутылка, или их комбинация. Расфасованная твердая лекарственная форма может дополнительно включать хлопковый, вискозный или полиэфирный жгут. В некоторых вариантах осуществления, расфасованная твердая лекарственная форма включает бутылку, такую как бутылка из полиэтилена высокой плотности (HDPE), которая содержит твердую лекарственную форму, осушающее вещество и, необязательно, хлопковый, вискозный или полиэфирный жгут. В некоторых вариантах осуществления, расфасованная твердая лекарственная форма включает бутылку, такую как бутылка из полиэтилена высокой плотности (HDPE), которая содержит твердую лекарственную форму, осушающее вещество и, необязательно, хлопковый, вискозный или полиэфирный жгут.
[0196] В конкретных аспектах, в настоящем изобретении предлагается набор, включающий описанную в изобретении твердую лекарственную форму и инструкции по введению твердой лекарственной формы субъекту, нуждающемуся в этом. В некоторых вариантах осуществления, набор включает твердую лекарственную форму, включающую соединение формулы (I), расфасованную в контейнер с низкой проницаемостью для паров воды с осушающим веществом. Необязательно, но этикетка может находиться на самом контейнере или связана с ним. Например, этикетка находится на самом контейнере, когда буквы, цифры или другие символы, образующие этикетку, прикреплены, отлиты или вытравлены на самом контейнере, и этикетка связана с контейнером, когда она присутствует в таре или чехле, таком как коробка, в которых также хранится контейнер, например, в виде листка-вкладыша в упаковке. Кроме того, этикетка может использоваться для указания того, что содержимое следует использовать для конкретного терапевтического применения. В некоторых вариантах осуществления, этикетка включает инструкции по применению содержимого, например, в описанных в изобретении способах. В некоторых вариантах осуществления, предлагаемая в изобретении фармацевтическая композиция представлена в упаковке или контейнере, которые содержат одну или несколько лекарственных форм с разовой дозой, включающих соединение формулы (I). Упаковка может содержать металлическую или пластиковую фольгу, например, представлять собой блистерную упаковку. К упаковке или контейнеру могут прилагаться инструкции по введению лекарственной формы с разовой дозой. В некоторых вариантах осуществления, к упаковке или контейнеру прилагается уведомление в форме, установленной государственным органом, регулирующим производство, применение или продажу фармацевтических препаратов, в котором отражено одобрение агентством применения формы лекарственного средства на людях или животных. Такое уведомление, например, может представлять собой информацию об одобрении Управлением по контролю качества продовольствия и медикаментов США применения в медицинской практике лекарственных средств, или листок-вкладыш об одобрении продукта. В некоторых вариантах осуществления, композиции, включающие соединение формулы (I), приготавливают, помещают в соответствующий контейнер и обеспечивают этикеткой, содержащей информацию по поводу лечения указанного состояния.
Способы лечения
[0197] В одном аспекте, в настоящем изобретении предлагается способ лечения пролиферативного нарушения у субъекта, нуждающегося в этом, включающий введение указанному субъекту предложенных в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции. В некоторых вариантах осуществления, пролиферативное нарушение представляет собой онкологическое заболевание. В некоторых дополнительных вариантах осуществления, указанное онкологическое заболевание представляет собой рак, выбранный из группы, состоящей из рака легких, плоскоклеточного рака головы и шеи, рака поджелудочной железы, рака молочной железы, рака яичников, почечно-клеточной карциномы, рака предстательной железы, нейроэндокринного рака, рака желудка, рака мочевого пузыря и рака толстой кишки. В некоторых дополнительных вариантах осуществления, онкологическое заболевание представляет собой почечно-клеточную карциному. В конкретных аспектах, в настоящем изобретении предлагается способ лечения почечно-клеточной карциномы, включающий введение субъекту, нуждающемуся в этом, эффективного количества предлагаемых в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции. В некоторых вариантах осуществления, почечно-клеточная карцинома представляет собой светлоклеточную почечно-клеточную карциному.
[0198] В некоторых вариантах осуществления, в настоящем изобретении предлагается способ лечения онкологического заболевания, где соединение формулы (I) является эффективным в одном или более случаях при ингибировании пролиферации раковых клеток, ингибировании метастазирования раковых клеток, уничтожении раковых клеток и снижении тяжести или частоты возникновения симптомов, связанных с присутствием раковых клеток. В некоторых других вариантах осуществления, указанный способ включает введение в раковые клетки предлагаемых в изобретении терапевтически эффективного количества твердой дисперсии, твердой лекарственной формы или фармацевтической композиции. В некоторых вариантах осуществления, введение осуществляется in vitro. В других вариантах осуществления, введение осуществляется in vivo.
[0199] В некоторых вариантах осуществления, в настоящем изобретении предлагается способ лечения болезни фон Гиппеля-Линдау (VHL), включающий введение субъекту, нуждающемуся в этом, предлагаемые в изобретении твердую дисперсию, твердую лекарственную форму или фармацевтическую композиции. В некоторых вариантах осуществления, субъект также страдает от гемангиобластомы, феохромоцитомы, панкреатической нейроэндокринной опухоли или почечно-клеточной карциномы. В некоторых вариантах осуществления, субъект страдает от почечно-клеточной карциномы. Болезнь фон Гиппеля-Линдау (VHL) представляет собой аутосомно-доминантный синдром, который создает у пациентов предпосылки развития не только рака почки (пожизненный риск ~70%), но также гемангиобластом, феохромоцитомы и панкреатических нейроэндокринных опухолей. Болезнь фон Гиппеля-Линдау (VHL) приводит к опухолям с постоянно активными белками HIF-α, большинство из которых зависят от активности HIF-2α (Maher, et al. Eur. J. Hum. Genet. 19: 617-623, 2011). HIF-2α связывали с раком сетчатки глаза, раком надпочечной железы и раком поджелудочной железы как через болезнь фон Гиппеля-Линдау (VHL), так и через активацию мутаций. Недавно, были идентифицированы мутации HIF-2α, при которых белковый продукт экспрессии мутантного гена приобретает новые и патологические функции, при эритроцитозе и параганглиоме с полицитемией (Zhuang, et al. NEJM 367: 922-930, 2012; Percy, et al. NEJM 358: 162-168, 2008; and Percy, et al. Am. J. Hematol. 87: 439-442, 2012). А именно, было показано, что ряд известных продуктов HIF-2α генов-мишеней (например, VEGF, PDGF, и циклин D1) играют ключевые роли при раке почек, печени, толстой кишки, легких и головного мозга. Более того, терапии, нацеленные на один из ключевых продуктов HIF-2α регулируемого гена, VEGF, уже одобрены для лечения этих типов рака.
[0200] Используемый в изобретении термин "терапевтически эффективное количество" твердой дисперсии, твердой лекарственной формы или фармацевтической композиции относится к количеству соединения формулы (I), достаточному для осуществления предполагаемого применения, включающего, но этим не ограничивая, лечения заболевания, определяемого в изобретении. Кроме того, в заявленных способах предполагается применение субтерапевтического количества соединения формулы (I) для лечения конкретного болезненного состояния.
[0201] Вводимое количество твердой дисперсии, твердой лекарственной формы или фармацевтической композиции может варьировать в зависимости от предполагаемого применения (in vitro или in vivo), или от субъекта и болезненного состояния, подвергаемого лечению, например, массы и возраста субъекта, тяжести болезненного состояния, способа введения и других подобных факторов, которые могут быть легко определены любым обычным специалистом в данной области.
[0202] Измерение ингибирования биологических эффектов HIF-2α может включать проведение анализа биологического образца, такого как образец, взятый у субъекта. В зависимости от проводимого анализа, может быть выбран любой из целого ряда биологических образцов. Примеры биологических образцов включают, но этим не ограничивая, образцы крови (например, плазмы или сыворотки крови), образцы конденсата выдыхаемого воздуха, жидкость бронхоальвеолярного лаважа, образцы мокроты, образцы мочи и образцы тканей.
[0203] Субъект, подвергаемый лечению с применением твердой дисперсии, твердой лекарственной формы или фармацевтической композиции по настоящему изобретению, может находиться под постоянным контролем с целью определения эффективности лечения, и схема лечения может быть скорректирована с учетом физиологических реакций субъекта на лечение. Например, если ингибирование биологического эффекта в результате ингибирования HIF-2α будет выше или ниже порогового значения, то количество или частота дозирования могут быть, соответственно, уменьшены или увеличены. Способы могут дополнительно включать продолжение проведения терапии, если терапия определена как эффективная. Способы могут включать поддержание, постепенное уменьшение, уменьшение вводимого количества соединения или прекращение введения соединения при проведении терапии, если терапия определена как эффективная. Способы могут включать увеличение вводимого количества соединения при проведении терапии, если установлено, что она неэффективна. В качестве варианта, способы могут включать прекращение проведении терапии, если установлено, что она неэффективна. В некоторых вариантах осуществления, лечение с применением соединения формулы (I) прекращают, если ингибирование биологического эффекта выше или ниже порогового значения, например, при отсутствии ответа или при нежелательной реакции. Биологический эффект может заключаться в изменении любого из множества физиологических показателей.
[0204] Обычно, ингибитор HIF-2α, такой как соединение формулы (I), представляет собой соединение, которое ингибирует один или несколько биологических эффектов HIF-2α. Примеры биологических эффектов HIF-2α включают, но этим не ограничивая, гетеродимеризацию HIF-2α в HIF-1β, экспрессию целевого гена HIF-2α, экспрессию гена VEGF и секрецию белка VEGF. В некоторых вариантах осуществления, ингибитор HIF-2α является селективным в отношении HIF-2α, так что ингибитор ингибирует гетеродимеризацию HIF-2α в HIF-1β, но не гетеродимеризацию HIF-1α в HIF-1β. Такие биологические эффекты могут подавляться примерно на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или более.
[0205] Индуцируемые гипоксией факторы (HIF), такие как HIF-2α, являются факторами транскрипции, которые реагируют на изменения количества доступного кислорода в клеточной среде (например, на снижение количества кислорода или гипоксию). Сигнальный каскад HIF опосредует влияние гипоксии, состояния низкой концентрации кислорода, воздействующей клетку. Гипоксия часто препятствует дифференцировке клеток. Однако гипоксия способствует образованию кровеносных сосудов и имеет важное значение для формирования сосудистой системы у эмбрионов и злокачественных опухолей. Гипоксия в ранах также способствует миграции кератиноцитов и восстановлению эпителия. Соединение формулы (I) может быть введено в количестве, эффективном для снижения любого одного или нескольких таких эффектов активности HIF-2α.
[0206] Активность HIF-2α может быть ингибирована путем ингибирования гетеродимеризации HIF-2α в HIF-1β (ARNT), например, с помощью соединения формулы (I). Доступен целый ряд методов измерения димеризации HIF-2α. В некоторых вариантах осуществления, ингибитор HIF-2α связывается с полостью домена PAS-B HIF-2α.
[0207] Ингибирование гетеродимеризации HIF-2α в HIF-1β (ARNT) может быть также определено по снижению экспрессии мРНК гена-мишени HIF-2α. Количественное определение мРНК может быть выполнено с использованием технологии ПЦР в реальном времени. (Wong, et al , "Real-time PCR for mRNA quantitation", 2005. BioTechniques 39, 1: 1-1.). Еще одним методом определения ингибирования гетеродимеризации HIF-2α в HIF-1β (ARNT) является коиммунопреципитация.
[0208] Как уже было описано выше в изобретении, HIF-2α представляет собой фактор транскрипции, который играет важную роль в регуляции экспрессии генов-мишеней. Неограничивающие примеры генов-мишеней HIF-2α включают HMOX1, SFTPA1, CXCR4, PAI1, BDNF, hTERT, ATP7A и VEGF. Например, HIF-2α является активатором VEGF. Дополнительные неограничивающие примеры генов-мишеней HIF-2α включают HMOX1, EPO, CXCR4, PAI1, CCND1, CLUT1, IL6 и VEGF. Соединение формулы (I) может быть введено в количестве, эффективном для снижения экспрессии любого одного или нескольких генов, индуцированных активностью HIF-2α. Доступен целый ряд методов для определения уровней экспрессии генов, которые включают обнаружение продуктов транскрипции генов (полинуклеотидов) и продуктов трансляции (полипептидов). Например, экспрессия гена может быть обнаружена и количественно определена на уровне ДНК, РНК или мРНК. Различные методы, которые используются для количественной оценки мРНК, включают методы гибридизации in situ, методы флуоресцентной гибридизации in situ, репортерные гены, анализы защиты от РНКаз, нозерн-блоттинг, обратную транскрипцию (RT)-PCR, серийный анализ экспрессии генов (SAGE), ДНК-микрочип, мозаичный микрочип и секвенирование РНК. Примеры методов обнаружения полинуклеотидов включают, но этим не ограничивая, селективное колориметрическое обнаружение полинуклеотидов на основе зависимых от расстояния оптических свойств наночастиц золота и определение в фазе раствора полинуклеотидов с использованием взаимодействующих флуоресцентных меток, и конкурентную гибридизацию. Примеры обнаружения белков включают, но этим не ограничивая микроскопию и иммуноокрашивание белков, иммунопреципитацию белков, иммуноэлектрофорез, вестерн-блоттинг, анализ бицинхониновой кислоты (BCA), спектрофотометрию, масс-спектрофотометрию и ферментный анализ.
[0209] В некоторых вариантах осуществления, ингибирование HIF-2α характеризуется снижением экспрессии гена VEGF. Это снижение может быть измерено любым из целого ряда методов, например, описанным в изобретении методом. В качестве дополнительного примера, уровень экспрессии мРНК VEGF может быть измерен с помощью количественной ПЦР (QT-PCR), микрочипа, секвенирования РНК и наноструны. В качестве другого примера, для измерения уровня секреции белка VEGF может быть использован иммуноферментный анализ (ELISA).
[0210] В конкретных аспектах, в настоящем изобретении предлагается способ лечения HIF-2α-опосредованного заболевания или состояния, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества описанных в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции. В некоторых вариантах осуществления, заболевание или состояние представляет собой рак. В некоторых вариантах осуществления, заболевание или состояние выбирают из почечно-клеточной карциномы, болезни фон Гиппеля - Линдау, легочной артериальной гипертензии, глиобластомы и колита.
[0211] В некоторых вариантах осуществления, в изобретении предлагается способ ингибирования HIF-2α, включающий контактирование HIF-2α с эффективным количеством описанных в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции. В некоторых других вариантах осуществления, заявляемые способы могут применяться для лечения болезненного состояния, ассоциированного с HIF-2α. Предполагаемое болезненное состояние может представлять собой любое болезненное состояние, которое прямо или косвенно является результатом аномальной активности или аномального уровня экспрессии HIF-2α. В некоторых вариантах осуществления, болезненное состояние представляет собой пролиферативное нарушение, такое как описанное в изобретении пролиферативное нарушение, включая, но этим не ограничивая, рак. Роль HIF-2α в образование опухолей и прогрессировании опухоли отмечается при многих типах рака у человека. Постоянно активный HIF-2α может быть следствием ненормального состояния при болезни фон Гиппеля-Линдау (VHL) или низкой концентрации кислорода в раковых клетках. Быстро растущие опухоли являются обычно гипоксическими вследствие недостаточной васкуляризации, состояния, которое активирует HIF-2α для поддержания выживаемости и пролиферации опухолевых клеток. Постоянная активация HIF-2α проявляется в качестве отличительного признака при различных типах рака у человека, и следовательно, положительный терапевтический эффект могут оказывать лекарственные средства, которые целенаправленно воздействуют на HIF-2α.
[0212] Данные, представленные в приведенных ниже примерах, иллюстрируют противораковое действие соединения формулы (I). Таким образом, заявляемый способ может применяться, в частности, для лечения пролиферативного нарушения, такого как неопластическое состояние. Неограничивающие примеры таких состояний включают, но этим не ограничивая, следующие неопластические состояния: акантома, ацинарно-клеточная аденокарцинома, акустическая невринома, акральная лентигинозная меланома, акроспирома, острый эозинофильный лейкоз, острый лимфобластный лейкоз, острый мегакариобластный лейкоз, острый моноцитарный лейкоз, острый миелобластный лейкоз с созреванием, острый миелоидный дендритноклеточный лейкоз, острый миелолейкоз, острый промиелоцитарный лейкоз, адамантинома, аденокарцинома, аденокистозная карцинома, аденома, аденоматоидная одонтогенная опухоль, адренокортикальная карцинома, Т-клеточный лейкоз взрослых, агрессивный NK-клеточный лейкоз, СПИД-ассоциированные типы рака, СПИД-ассоциированная лимфома, альвеолярная саркома мягких тканей, амелобластическая фиброма, рак анального канала, анапластическая крупноклеточная лимфома, анапластический рак щитовидной железы, ангиоиммунобластная Т-клеточная лимфома, ангиомиолипома, ангиосаркома, рак аппендикса, астроцитома, атипическая тератоидно-рабдоидная опухоль, базальноклеточная карцинома, базальноподобная карцинома, B-клеточный лейкоз, B-клеточная лимфома, карцинома протока Беллини, рак желчных путей, рак мочевого пузыря, бластома, рак костей, костная опухоль, глиома ствола головного мозга, опухоль головного мозга, рак молочной железы, опухоль Бреннера, бронхиальная опухоль, бронхиолоальвеолярная карцинома, бурая опухоль, лимфома Беркитта, карциноидная опухоль, карцинома, карциносаркома, болезнь Кастлемена, эмбриональная опухоль центральной нервной системы, астроцитома мозжечка, церебральная астроцитома, рак шейки матки, холангиокарцинома, хондрома, хондросаркома, хордома, хориокарцинома, папиллома хориоидного сплетения, хронический лимфолейкоз, хронический моноцитарный лейкоз, хронический миелолейкоз, хроническое миелопролиферативное нарушение, хронический нейтрофильный лейкоз, светлоклеточная почечно-клеточная карцинома, светлоклеточная опухоль, рак толстой кишки, колоректальный рак, краниофарингиома, кожная Т-клеточная лимфома, возвышающаяся дерматофибросаркома, дермоидная киста, десмопластическая мелкокруглоклеточная опухоль, диффузная крупноклеточная В-клеточная лимфома, дизэмбриопластическая нейроэпителиальная опухоль, эмбриональная карцинома, опухоль эндодермального синуса, эндометриальный рак, рак эндометрия матки, эндометриоидная опухоль, T-клеточная лимфома, ассоциированная с энтеропатией, эпендимобластома, эпендимома, эпителиоидная саркома, эритролейкоз, рак пищевода, эстезионейробластома, саркома Юинга, экстракраниальная герминогенная опухоль, внегонадная герминогенная опухоль, рак внепеченочных желчных протоков, экстрамаммиллярный рак Педжета, рак фаллопиевой трубы, фиброма, фибросаркома, фолликулярная лимфома, фолликулярный рак щитовидной железы, рак желчного пузыря, ганглиоглиома, ганглионеврома, рак желудка, лимфома желудка, рак желудочно-кишечного тракта, карциноидная опухоль желудочно-кишечного тракта, гастроинтестинальная стромальная опухоль, эмбрионально-клеточная опухоль, герминома, хориокарцинома матки, гестационная трофобластическая опухоль, гигантоклеточная остеобластома, мультиформная глиобластома, глиома, глиоматоз головного мозга, гломическая опухоль, глюкагонома, гонадобластома, гранулезоклеточная опухоль, волосатоклеточный лейкоз, рак головы и шеи, рак сердца, гемангиобластома, гемангиоперицитома, гемангиосаркома, гематологическая злокачественная опухоль, гепатоцеллюлярная карцинома, Т-клеточная лимфома печени и селезенки, лимфома Ходжкина, гипофарингеальный рак, гипоталамическая глиома, воспалительный рак молочной железы, внутриглазная меланома, островково-клеточная карцинома, ювенильный миеломоноцитарный лейкоз, саркома Капоши, рак почки, опухоль Клацкина, опухоль Крукенберга, рак гортани, меланома типа злокачественного лентиго, лейкоз, рак губы и полости рта, липосаркома, рак легких, лютеома, лимфангиома, лимфангиосаркома, лимфоэпителиома, лимфолейкоз, лимфома, макроглобулинемия, злокачественная фиброзная гистиоцитома, злокачественная глиома, злокачественная мезотелиома, злокачественная опухоль оболочек периферических нервов, злокачественная рабдоидная опухоль, злокачественная тритоновая опухоль, солодовая лимфома, лимфома из клеток зоны мантии, тучноклеточный лейкоз, опухоль зародышевых клеток средостения, опухоль средостения, медуллярный рак щитовидной железы, медуллобластома, медуллоэпителиома, меланома, менингиома, карцинома из клеток Меркеля, мезотелиома, метастатический плоскоклеточный рак шеи скрытой первичной локализации, метастатическая уротелиальная карцинома, смешанные опухоли Мюллерова, моноцитарный лейкоз, рак ротовой полости, муцинозная опухоль, синдром множественной эндокринной неоплазии, множественная миелома, фунгоидный микоз, миелодиспластическая болезнь, миелолейкоз, миелоидная саркома, миелопролиферативное заболевание, миксома, рак носовой полости, рак носоглотки, неоплазия, невринома, нейробластома, нейрофиброма, неврома, узловая меланома, неходжкинская лимфома, немеланоцитарный рак кожи, немелкоклеточный рак легких, глазная онкология, олигоастроцитома, олигодендроглиома, онкоцитома, менингиома оболочки зрительного нерва, рак ротовой полости, рак ротоглотки, остеосаркома, рак яичников, эпителиальный рак яичников, опухоль зародышевых клеток яичников, опухоль яичников с низкой степенью злокачественности, опухоль Панкоста, рак поджелудочной железы, папиллярный рак щитовидной железы, папилломатоз, параганглиома, рак придаточных пазух носа, рак паращитовидной железы, рак полового члена, периваскулярная эпителиоидно-клеточная опухоль, рак глотки, феохромоцитома, опухоль паренхимы шишковидной железы промежуточной дифференцировки, пинеобластома, питуицитома, аденома гипофиза, опухоль гипофиза, новообразование плазматических клеток, плевролегочная бластома, полиэмбриома, предшественник Т-лимфобластной лимфомы, примитивная нейроэктодермальная опухоль, рак предстательной железы, псевдомиксома брюшной полости, рак прямой кишки, почечно-клеточная карцинома, ретинобластома, рабдомиома, рабдомиосаркома, преобразование Рихтера, крестцово-копчиковая тератома, рак слюнной железы, саркома, шванноматоз, карцинома сальной железы, опухолевый метастаз, семинома, серозная опухоль, опухоль из клеток Сертоли-Лейдига, опухоль полового тяжа-стромы, синдром Сезари, перстневидно-клеточная карцинома, рак кожи, голубая мелкокруглоклеточная опухоль, мелкоклеточная карцинома, мелкоклеточный рак легких, мелкоклеточная лимфома, рак тонкого кишечника, саркома мягких тканей, соматостатинома, рак трубочистов, опухоль позвоночника, лимфома маргинальной зоны селезенки, плоскоклеточная карцинома, рак желудка, поверхностная распространяющаяся меланома, супратенториальная примитивная нейроэктодермальная опухоль, поверхностная эпителиально-стромальная опухоль, синовиальная саркома, Т-клеточный острый лимфобластный лейкоз, Т-клеточный лейкоз из больших гранулярных лимфоцитов, Т-клеточный лейкоз, Т-клеточная лимфома, Т-клеточный пролимфоцитарный лейкоз, тератома, лимфатический рак в терминальной стадии, рак яичка, текома, рак горла, рак тимуса, тимома, рак щитовидной железы, переходно-клеточный рак почечной лоханки и мочеточника, переходно-клеточная карцинома, урахальный рак, рак уретры, урогенитальное новообразование, саркома матки, увеальная меланома, рак влагалища, синдром Вернера-Моррисона, бородавчатая карцинома, глиома зрительного пути, рак вульвы, макроглобулинемия Вальденстрема, опухоль Уортина, опухоль Вильмса или любая их комбинация.
[0213] В некоторых вариантах осуществления, способы введения описанных в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции применяют для лечения рака надпочечной железы, рака крови, рака костного мозга, рака головного мозга, рака молочной железы, рака шейки матки, рака толстой кишки, рака головы и шеи, рака почки, рака печени, рака легких, рака яичника, рака поджелудочной железы, рака плазматических клеток, рака прямой кишки, рака сетчатки глаза, рака кожи, рака позвоночника, рака горла или любой их комбинации.
[0214] В некоторых вариантах осуществления настоящего изобретения рассматривается подвергаемый обследованию человек, например, субъект, у которого диагностировано состояние пролиферативного нарушения или риск его развития или возникновения. В других конкретных вариантах осуществления рассматриваются субъекты, не являющиеся людьми, например низшие приматы, такие как макака, шимпанзе, горилла, зеленая мартышка, орангутан, бабуин или другой низший примат, в том числе такие субъекты, не являющиеся людьми, по поводу которых известно, что они могут быть использованы в качестве моделей при проведении доклинических исследований. В других конкретных вариантах осуществления рассматривается субъект, не являющийся человеком, который является млекопитающим, например, мышью, крысой, кроликом, свиньей, овцой, лошадью, крупным рогатым скотом, козой, песчанкой, хомяком, морской свинкой или другим млекопитающим. Также предполагаются другие варианты осуществления, в которых субъектом или биологическим источником может быть позвоночное животное, не являющееся млекопитающим, например, другое высшее позвоночное животное или виды пернатых, амфибий или рептилий, или другой субъект или биологический источник. В конкретных вариантах осуществления настоящего изобретения используют трансгенное животное. Трансгенное животное представляет собой животное, не являющееся человеком, у которого одна или более клеток в организме включает нуклеиновую кислоту, которая является неэндогенной (то есть гетерологичной) и присутствует как экстрахромосомный элемент в части его клетки или стабильно интегрирована в его ДНК зародышевой линии (то есть в геномную последовательность большинства или всех его клеток).
[0215] В некоторых вариантах осуществления, терапевтическую эффективность измеряют по результату лечения пролиферативного нарушения, такого как рак. В целом терапевтическая эффективность способов и композиций по изобретению применительно к лечению пролиферативного нарушения (например, рака, доброкачественного или злокачественного) может быть измерена степенью, в которой способы и композиции способствуют ингибированию пролиферации опухолевых клеток, ингибированию васкуляризации опухоли, уничтожению опухолевых клеток, снижению скорости роста опухоли и/или уменьшению размера, по меньшей мере, одной опухоли. В изобретении обсуждаются несколько параметров, которые следует учитывать при определении терапевтической эффективности. Соответствующая комбинация параметров для конкретной ситуации может быть установлена лечащим врачом. Положительный эффект от применения способа по изобретению при лечении рака (например, уменьшение размера опухоли или уничтожение раковых клеток) может быть установлен с использованием любого подходящего метода, такого как методы, которые в настоящее время используются в клинике для отслеживания размера опухоли и прогрессирования рака. Предпочтительно, когда в качестве первичного параметра эффективности, используемого для оценки лечения рака способом и композициями по настоящему изобретению, применяют уменьшение размера опухоли. Размер опухоли может быть рассчитан любым подходящим методом, например, измерением геометрических размеров или оценкой объема опухоли с использованием доступного компьютерного программного обеспечения, такого как программа FreeFlight, разработанная в Wake Forest University, которая позволяет точно оценить объем опухоли. Размер опухоли может быть определен путем визуализации опухоли с использованием, например, компьютерной томографии (CT), ультразвука, однофотонной эмиссионной компьютерной томографии (SPECT), спиральной компьютерной томографии, магнитной резонансной томография (MRI), фотографий и других подобных методов. В вариантах осуществления, в которых опухоль удаляют хирургическим путем после завершения терапевтического лечения, присутствие опухолевой ткани и размер опухоли могут быть определены путем общего анализа ткани, подлежащей хирургическому удалению, и/или путем патологического анализа удаленной ткани.
[0216] В некоторых вариантах осуществления, в результате применения способа и композиций по изобретению рост опухоли стабилизируют (то есть, одна или более опухолей не увеличиваются в размере более чем на 1%, 5%, 10%, 15% или 20%, и/или не метастазируют). В некоторых вариантах осуществления, опухоль стабилизируют в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более недель. В некоторых вариантах осуществления, опухоль стабилизируют в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более месяцев. В некоторых вариантах осуществления, опухоль стабилизируют в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более лет. Предпочтительно, чтобы способ по изобретению позволял уменьшать размер опухоли, по меньшей мере, приблизительно на 5% (например, по меньшей мере, приблизительно на 10%, 15%, 20% или 25%). Более предпочтительно, если размер опухоли уменьшается, по меньшей мере, приблизительно на 30% (например, по меньшей мере, приблизительного на 35%, 40%, 45%, 50%, 55%, 60% или 65%). Еще более предпочтительно, если размер опухоли уменьшается, по меньшей мере, приблизительно на 70% (например, по меньшей мере, приблизительно на 75%, 80%, 85%, 90% или 95%). И наиболее предпочтительно, если опухоль полностью ликвидируется или ее размеры уменьшаются до величины ниже уровня обнаружения. В некоторых вариантах осуществления, у субъекта не обнаруживается наличие опухоли (например, при ремиссии) в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более недель после лечения. В некоторых вариантах осуществления, у субъекта не обнаруживается наличие опухоли в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более месяцев после лечения. В некоторых вариантах осуществления, у субъекта не обнаруживается наличие опухоли в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более лет после лечения.
[0217] В некоторых вариантах осуществления, эффективность способов по настоящему изобретению по снижению размера опухоли может быть определена путем измерения процента некротической (то есть, мертвой) ткани в хирургически удаленной опухоли после завершения терапевтического лечения. В некоторых дополнительных вариантах осуществления, лечение является терапевтически эффективным, если процент некроза в удаленной ткани составляет более чем приблизительно 20% (например, по меньшей мере, приблизительно 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100%), более предпочтительно, приблизительно 90% или более (например, приблизительно 90%, 95% или 100%). Наиболее предпочтительно, если процент некроза в удаленной ткани составляет 100%, то есть, опухолевая ткань отсутствует или не поддается обнаружению.
[0218] Эффективность способов по настоящему изобретению может быть определена с использованием ряда вторичных параметров. Примеры вторичных параметров включают, но этим не ограничивая, обнаружение новых опухолей, обнаружение опухолевых антигенов или маркеров (например, CEA, PSA или CA-125), биопсию, снижение стадии хирургического вмешательства (то есть, превращение стадии хирургического вмешательства из стадии неоперабельный опухоли в стадию операбельной опухоли), сканограммы позитронно-эмиссионной томографии (PET), выживаемость, выживаемость без признаков прогрессирования заболевания, время до прогрессирования заболевания, оценки качества жизни, такие как "Оценка положительного клинического результата" (Clinical Benefit Response Assessment), и другие подобные параметры, каждый из которых может указывать на общее прогрессирование (или ремиссию) рака у человека. Биопсия особенно подходит для выявления факта уничтожения раковых клеток в ткани. Радиоиммуноанализ (RAID) применяют для определения местонахождения и стадии развития опухолей, используя уровни маркеров в сыворотке крови (антигенов), продуцируемых опухолями и/или ассоциированных с опухолями ("опухолевые маркеры" или "опухолеассоциированные антигены"), и уровни этих маркеров могут использоваться в качестве диагностического подхода перед лечением, диагностического индикатора возникновения рецидива после лечения и показателя терапевтической эффективности после лечения. Примеры опухолевых маркеров или опухолеассоциированных антигенов, которые могут быть оценены в качестве показателей терапевтической эффективности, включают, но этим не ограничивая, карциноэмбриональный антиген (CEA), простатический специфический антиген (PSA), CA-125, CA19-9, молекулы ганглиозида (например, GM2, GD2, and GD3), MART-1, белки теплового шока (например, gp96), сиалил Tn (STn), тирозиназа, MUC-1, HER-2/neu, c-erb-B2, KSA, PSMA, p53, RAS, EGF-R, VEGF, MAGE и gp100. Известны и другие опухолеассоциированные антигены. Радиоиммуноанализ в сочетании с системами эндоскопического обнаружения также позволяет эффективно различать незначительные опухоли в окружающей ткани (смотрите, например, патентный документ U.S. Pat. No. 4932412).
[0219] В некоторых вариантах осуществления, положительный эффект лечение рака у пациента с применением способа по изобретению подтверждают одним или более из следующих результатов: (a) полное исчезновение опухоли (то есть, полная ремиссия), (b) уменьшение размера опухоли приблизительно на величину от 25% до приблизительно 50% в течение, по меньшей мере, четырех недель после завершения проведения терапии по сравнению с размером опухоли до лечения, (c) уменьшение, по меньшей мере, приблизительно на 50% размера опухоли в течение, по меньшей мере, четырех недель после завершения проведения терапии по сравнению с размером опухоли до проведения терапии, и (d) снижение, по меньшей мере, на 2% (например, снижение приблизительно на 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% или 90%) уровня специфического опухолеассоциированного антигена через приблизительно 4-12 недель после завершения проведения терапии по сравнению с уровнем опухолеассоциированного антигена до проведения терапии. Несмотря на то, что снижение уровня опухолеассоциированного антигена, по меньшей мере, на 2% является предпочтительным, тем не менее, любое снижение уровня опухолеассоциированного антигена является доказательством положительного эффекта лечения рака у пациента с применением способа по изобретению. Например, применительно к неоперабельному местно распространенному раку поджелудочной железы, положительный эффект лечения может быть подтвержден снижением, по меньшей мере, на 10% уровня опухолеассоциированного антигена CA19-9 через 4-12 недель после завершения проведения терапии по сравнению с уровнем опухолеассоциированного антигена CA19-9 до проведения терапии. Аналогично, применительно к местно распространенному раку прямой кишки, положительный эффект лечения может быть подтвержден снижением, по меньшей мере, на 10% уровня опухолеассоциированного антигена CEA через 4-12 недель после завершения проведения терапии по сравнению с уровнем опухолеассоциированного антигена CEA до проведения терапии.
[0220] Что касается оценок качества жизни, например, с помощью критерия положительного клинического результата (Clinical Benefit Response Criteria), то терапевтический положительный эффект лечения в соответствии с изобретением может быть доказан путем использования таких показателей, как интенсивность боли, потребление болеутоляющих средств и/или оценка в баллах по шкале Карновского. В качестве варианта или дополнительно, положительный эффект при лечении рака у пациента, доказывается (a) снижением, по меньшей мере, на 50% (например, снижением, по меньшей мере, на 60%, 70%, 80%, 90% или 100%) интенсивности боли, в соответствии с полученной от пациента информацией, например, в течение любых последующих четырех недель через 12 недель после завершения проведения лечения, по сравнению с интенсивностью боли, в соответствии с полученной от пациента информацией, до проведения лечения, (b) снижением, по меньшей мере, на 50% (например, снижением, по меньшей мере, на 60%, 70%, 80%, 90% или 100%) потребления болеутоляющих средств, в соответствии с полученной от пациента информацией, например, в течение любых последующих четырех недель через 12 недель после завершения проведения лечения, по сравнению с потреблением болеутоляющих средств, в соответствии с полученной от пациента информацией, до проведения лечения, и/или (c) увеличением, по меньшей мере, на 20 баллов (например, увеличением, по меньшей мере, на 30 баллов, 50 баллов, 70 баллов или 90 баллов) оценки в баллах по шкале Карновского, в соответствии с полученной от пациента информацией, например, в течение любых последующих четырех недель через 12 недель после завершения проведения лечения, по сравнению с оценкой в баллах по шкале Карновского, в соответствии с полученной от пациента информацией, до проведения лечения.
[0221] Положительный эффект лечения пролиферативного нарушения (например, рака, либо доброкачественного, либо злокачественного) у пациента при желании может быть доказан одним или более (в любой комбинации) из упомянутых выше результатов, хотя об эффективности лечения могут свидетельствовать и альтернативные или дополнительные результаты упоминаемых тестов и/или других тестов.
[0222] В некоторых вариантах осуществления, размер опухоли уменьшают в результате применения способа по изобретению, предпочтительно, без проявления значительных неблагоприятных явлений у субъекта. Неблагоприятные явления классифицируют или "ранжируют" в соответствии с Программой оценки терапии рака (Cancer Therapy Evaluation Program (CTEP)) Национального института рака США (National Cancer Institute (NCI)), при этом оценка 0 соответствует минимальным неблагоприятным побочным эффектам, а оценка 4 соответствует наиболее тяжелым неблагоприятным явлениям. Желательно, чтобы способ по изобретению ассоциировался с минимальными неблагоприятными явлениями, например, неблагоприятными явлениями с оценкой 0, оценкой 1 или оценкой 2, по шкале оценок CTEP/NCI. Однако, как было указано в изобретении выше, несмотря на то, что уменьшение размера опухоли является предпочтительным событием, тем не менее, это не является необходимым, поскольку фактический размер опухоли может не уменьшиться, несмотря на уничтожение опухолевых клеток. Для достижения терапевтического эффекта является достаточным уничтожение раковых клеток. Точно так же, для достижения терапевтического эффекта является достаточным любое уменьшение размер опухоли.
[0223] Обнаружение, регулярный контроль и ранжирование различных типов рака у человека дополнительно описаны в документе Cancer Facts and Figures 2001, American Cancer Society, New York, N.Y., и патентном документе International Patent Application WO 01/24684. Соответственно, лечащий врач может использовать стандартные тесты для определения эффективности различных вариантов осуществления способа по изобретению при лечении рака. Однако, при оценке эффективности лечения, лечащий врач может также учитывать, помимо размера опухоли и ее распространения, качество жизни и выживаемость пациента.
[0224] В одном аспекте, в изобретении предлагается способ лечения легочной артериальной гипертензии (PAH). В одном варианте осуществления, способ включает введение эффективного количества описанных в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции указанному субъекту. Легочная артериальная гипертензия (PAH) представляет собой опасное для жизни и прогрессирующее заболевание различного происхождения, характеризующееся ремоделированием легочных сосудов, что вызывает повышение легочного сосудистого сопротивления и легочного артериального давления, что чаще всего приводит к правосторонней сердечной недостаточности. Наиболее распространенным симптомом на момент первичного осмотра является одышка, с пониженной способностью переносить физическую нагрузку в качестве характерного признака заболевания. Легочная артериальная гипертензия (PAH) включает идиопатическую легочную артериальную гипертензию, наследственную легочную артериальную гипертензию (например BMPR2, ALK1, эндоглин), обусловленную действием лекарственного средства легочную артериальную гипертензию, обусловленную действием токсического вещества легочную артериальную гипертензию, и легочную артериальную гипертензию, ассоциированную с другим состоянием (например, заболеваниями соединительной ткани, ВИЧ-инфекцией, портальной гипертензией, врожденными пороками сердца, шистосомозом или хронической гемолитической анемией). Идиопатическая легочная артериальная гипертензия возникает при отсутствии известных факторов риска и является наиболее распространенной формой заболевания. Состояние легочной артериальной гипертензии, подвергаемое лечению в соответствии с настоящим изобретением, может включать любое из этих состояний легочной артериальной гипертензии.
[0225] В целом, медиана периода выживаемости при легочной артериальной гипертензии после постановки диагноза, от начальной регистрации Национальными институтами здравоохранения США (U.S. National Institutes of Health) с совокупными сведениями о состоянии больного после установления диагноза и выписки из стационара, составляет менее 3 лет для 194 не подвергавшихся лечению пациентов с идиопатической или наследственной легочной артериальной гипертензией (ранее называвшейся первичной легочной гипертензией) со средним возрастом 36 лет. В настоящее время средняя выживаемость после постановки диагноза у взрослых оценивается в 5-7 лет с таким же неблагоприятным общим прогнозом и для детей. В некоторых вариантах осуществления, твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию вводят в количестве, эффективном для увеличения средней выживаемости среди подвергающейся лечению популяции субъектов, например, приблизительно на 0,5, 1, 2, 3, 4, 5 , 6, 7, 8, 9, 10, 15 или более лет.
[0226] Патология легочной артериальной гипертензии может включать патологические изменения внутренней оболочки, среды и адвентициальных слоев сосудистой стенки. Как эндотелиальные клетки, так и гладкомышечные клетки стенок сосудов характеризуются аномальным ростом с избыточной клеточной пролиферацией и устойчивостью к апоптозу. Эти аномалии резидентных сосудистых клеток в сочетании с воспалением, избыточным сужением сосудов и тромбозом in situ могут способствовать физическому сужению дистальных отделов легочных артериол. Не приводя в качестве обоснования какую-либо теорию, тем не менее можно предположить, что это сужение может вызывать увеличение сопротивления легочных сосудов, что приводит к хроническому и прогрессирующему повышению легочного артериального давления. В некоторых случаях, легочная артериальная гипертензия может быть вызвана гипоксией. В некоторых вариантах осуществления, твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию вводят в количестве, эффективном для отсрочки, снижения частоты возникновения или тяжести одного или нескольких таких симптомов, связанных с легочной артериальной гипертензией.
[0227] Лечение легочной артериальной гипертензии включает лечение субъекта, у которого диагностировано наличие легочной артериальной гипертензии, а также профилактику легочной артериальной гипертензии. В некоторых вариантах осуществления, количество твердой дисперсии, твердой лекарственной формы или фармацевтической композиции, вводимое субъекту (либо в разовой дозе, либо в нескольких разделенных дозах), является эффективным для отсрочки начала или снижения частоты проявления одного или нескольких симптомов легочной артериальной гипертензии. Отсрочка или снижение заболеваемости может относиться к референтной популяции, такой как популяция субъектов с легочной артериальной гипертензией, не подвергавшихся лечению, или субъектов с легочной артериальной гипертензией, но подвергавшихся лечению другим лекарственным средством. Отсрочка или снижение частоты проявления одного или нескольких симптомов легочной артериальной гипертензии может представлять собой уменьшение приблизительно на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или более. В некоторых вариантах осуществления, отсрочку или снижение частоты заболеваемости измеряют увеличением времени прогрессирования заболевания, например, временем между проявлением одного или нескольких первых симптомов и проявлением одного или нескольких вторых симптомов. Отсрочка может составлять приблизительно дни, недели, месяцы или годы (например, 1, 2, 3, 4, 5, 6, 7 или более дней; 1, 2, 3, 4, 5, 6, 7, 8 или более недель; 1, 2, 3, 4, 5, 6 или более месяцев; или 1, 2, 3, 4, 5 или более лет). В случае профилактики, субъектом может быть человек с риском развития легочной артериальной гипертензии.
[0228] В одном варианте осуществления, количество твердой дисперсии, твердой лекарственной формы или фармацевтической композиции, вводимое субъекту (либо в разовой дозе, либо в нескольких разделенных дозах) является эффективным при (a) стабилизации легочного артериального давления (PAP) по сравнению с тенденцией поведения PAP у субъекта до проведения лечения, или (b) снижении PAP относительно начального уровня PAP у субъекта; где PAP представляет собой систолическое легочное артериальное давление (SPAP) или диастолическое легочное артериальное давление (DPAP). Известен целый ряд методов измерения легочного артериального давления (PAP). Например, величина PAP может быть измерена путем введения катетера в легочную артерию. Обычно, средняя величина давления составляет 9-18 мм.рт.ст., и заклинивающее давление, измеренное в левом предсердии, может составлять 6-12 мм.рт.ст. Заклинивающее давление может быть повышенным при левосторонней сердечной недостаточности, стенозе митрального клапана и других состояниях, таких как серповидноклеточная анемия.
[0229] Величина легочного артериального давления у субъекта может быть стабилизирована на уровне, существующим на момент начала лечения, например, на протяжении дней, недель, месяцев или лет. В некоторых вариантах осуществления, исходная величина уровня легочного артериального давления у субъекта может быть снижена приблизительно на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50% или более. В некоторых вариантах осуществления, скорость увеличения величины легочного артериального давления может быть снижена приблизительно на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или более.
[0230] Количество твердой дисперсии, твердой лекарственной формы или фармацевтической композиции, вводимое субъекту (либо в разовой дозе, либо в нескольких разделенных дозах), может быть эффективным при уменьшении гипертрофии правого желудочка, измеряемой индексом Фултона, по сравнению с популяцией субъектов с легочной артериальной гипертензией, не подвергавшихся лечению. Индекс Фултона представляет собой отношение массы правого желудочка к суммарной массе левого желудочка и перегородки (RV / (LV+S)). Масса может быть измерена непосредственно (например, при исследовании на животных) или может быть рассчитана путем экстраполяции данных по измерению объемов живого субъекта (например, путем определения по эхокардиограмме). Снижение может быть рассчитано относительно популяции субъектов с легочной артериальной гипертензией, не подвергавшихся лечению. Например, среднее уменьшение индекса Фултона среди популяции субъектов с легочной артериальной гипертензией, подвергавшихся лечению с помощью соединения формулы (I), по сравнению с популяцией субъектов с легочной артериальной гипертензией, не подвергавшихся лечению, может составлять приблизительно 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50% или более. В некоторых вариантах осуществления, снижение индекса Фултона составляет, по меньшей мере, приблизительно 20%.
[0231] В некоторых вариантах осуществления, количество твердой дисперсии, твердой лекарственной формы или фармацевтической композиции, вводимое субъекту (либо в разовой дозе, либо в нескольких разделенных дозах), может быть эффективным при уменьшении величины физиологического показателя легочной артериальной гипертензии. Уменьшение может быть измерено в течение указанного периода времени, например, уровень уменьшения на протяжении дней (например, на протяжении приблизительно 1, 2, 3, 4, 5, 6,7 или более дней), недель (например, на протяжении 1, 2, 3, 4, 5, 6 или более недель), месяце (например, на протяжении 1, 2, 3, 4, 5, 6, 8, 10 или более месяцев) или на протяжении более длительного периода (например, например, на протяжении 1, 2, 3, 4, 5 или более лет) после начала проведения лечения (проведение которого может продолжаться на протяжении всего периода). Может быть измерен любой из целого ряда физиологических показателей, таких как сопротивление легочных сосудов, легочное артериальное давление, системное артериальное давление, насыщение смешанной венозной крови кислородом, насыщение артериальной крови кислородом, сердечный индекс, давление заклинивания легочных капилляров, давление в правом предсердии, тест на преодоление пешком дистанции за 6 минут, уровень натрийуретического пептида головного мозга и диффузионная способность легких. Например, твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция может присутствовать в количестве, эффективном для (a) снижения одного или более из физиологических показателей: сопротивление легочных сосудов, легочное артериальное давление, системное артериальное давление, давление заклинивания легочных капилляров, сердечный индекс, давление в правом предсердии, и уровень натрийуретического пептида головного мозга; и/или для (b) повышения одного или более из физиологических показателей: насыщение смешанной венозной крови кислородом, насыщение артериальной крови кислородом, сердечный индекс, тест на преодоление пешком дистанции за 6 минут и диффузионная способность легких. Количество твердой дисперсии, твердой лекарственной формы или фармацевтической композиции может быть эффективным при увеличении времени, необходимого до достижения летальной дозы, увеличении времени до наступления клинического ухудшения, снижении частоты возникновения правосторонней сердечной недостаточности, или при промотировании благоприятного изменения функционального класса по классификации Всемирной организации здравоохранения (ВОЗ).
[0232] Одним физиологическим показателем легочной артериальной гипертензии является сопротивление легочных сосудов (PVR). Исходный или референсный уровень PVR может составлять 200 дин·сек/см5 или более, 240 дин·сек/см5 или более, 300 дин·сек/см5 или более, 400 дин·сек/см5 или более, 500 дин·сек/см5 или более, 600 дин·сек/см5 или более, 700 дин·сек/см5 или более, или 800 дин·сек/см5 или более. В некоторых вариантах осуществления, предлагаемое в изобретении лечение является эффективным, если после начала лечения конечный уровень PVR у субъекта снижается относительно исходного или референсного уровня PVR на 70 дин·сек/см5 или более, 100 дин·сек/см5 или более, 130 дин·сек/см5 или более, или 160 дин·сек/см5 или более.
[0233] Другим физиологическим показателем легочной артериальной гипертензии является легочное артериальное давление (PAP). Исходный или референсный уровень PAP может составлять 20 мм.рт.ст. или более, 25 мм.рт.ст. или более, 30 мм.рт.ст. или более, 35 мм.рт.ст. или более, 40 мм.рт.ст. или более, 45 мм.рт.ст. или более, 50 мм.рт.ст. или более, 60 мм.рт.ст. или более, или 70 мм.рт.ст. или более. В некоторых вариантах осуществления, предлагаемое в изобретении лечение является эффективным, если после начала лечения конечный уровень PAP у субъекта снижается относительно исходного или референсного уровня PAP на 0,5 мм.рт.ст. или более, 1 мм.рт.ст. или более, 1,5 мм.рт.ст. или более, 5 мм.рт.ст. или более, 10 мм.рт.ст. или более, 20 мм.рт.ст. или более, 30 мм.рт.ст. или более, 40 мм.рт.ст. или более, или 50 мм.рт.ст.
[0234] Еще одним физиологическим показателем легочной артериальной гипертензии является сердечный индекс (CI). Исходный или референсный уровень CI может составлять 5 л/мин/м2 или ниже, 2,5 л/мин/м2 или ниже, 2 л/мин/м2 или ниже, 1,5 л/мин/м2 или ниже, или 1 л/мин/м2 или ниже. В некоторых вариантах осуществления, предлагаемое в изобретении лечение является эффективным, если после начала лечения конечный уровень CI повышается относительно исходного или референсного уровня CI на 0,1 или более, 0,2 или более, 0,3 или более, 0,4 или более, 0,5 или более, 1 или более, или 2 или более.
[0235] Еще одним физиологическим показателем легочной артериальной гипертензии является давление заклинивания легочных капилляров (PCWP). Исходный или референсный уровень PCWP может составлять 36 мм.рт.ст. или менее, 24 мм.рт.ст. или менее, 18 мм.рт.ст. или менее, 10 мм.рт.ст., или 5 мм.рт.ст. или менее. В некоторых вариантах осуществления, предлагаемое в изобретении лечение является эффективным, если после начала лечения конечный уровень PCWP повышается относительно исходного или референсного уровня PCWP на 0,2 мм.рт.ст. или более, 0,3 мм.рт.ст. или более, 0,4 мм.рт.ст. или более, 0,5 мм.рт.ст. или более, 0,6 мм.рт.ст. или более, 1 мм.рт.ст. или более, или 5 мм.рт.ст. или более.
[0236] Еще одним физиологическим показателем легочной артериальной гипертензии является давление в правом предсердии (RAP). Исходный или референсный уровень RAP может составлять 4 мм.рт.ст. или более, 6 мм.рт.ст. или более, 8 мм.рт.ст. или более, 10 мм.рт.ст. или более, 12 мм.рт.ст. или более, 16 мм.рт.ст. или более, 20 мм.рт.ст. или более, or 25 мм.рт.ст. или более. В некоторых вариантах осуществления, предлагаемое в изобретении лечение является эффективным, если после начала лечения конечный уровень RAP у субъекта снижается относительно исходного или референсного уровня RAP на 5 мм.рт.ст. или более, 2,5 мм.рт.ст. или более, 1 мм.рт.ст. или более, 0,5 мм.рт.ст. или более, или 0,2 мм.рт.ст. или более.
[0237] Еще одним физиологическим показателем легочной артериальной гипертензии является тест на преодоление пешком дистанции за 6 минут (6 MWD). Исходный или референсный уровень для теста 6 MWD может составлять 50 м или менее, 100 м или менее, 200 м или менее, 300 м или менее, 400 м или менее, или 500 м или менее. В некоторых вариантах осуществления, предлагаемое в изобретении лечение является эффективным, если после начала лечения конечный уровень для теста 6 MWD у субъекта повышается относительно исходного или референсного уровня для теста 6 MWD на 10 м или более, 15 м или более, 20 м или более, 25 м или более, 30 м или более, или 50 м или более; или если конечный уровень для теста 6 MWD у субъекта повышается на 3% или более, 4% или более, 5% или более, 10% или более, или 20% или более от исходного уровня.
[0238] Еще одним физиологическим показателем легочной артериальной гипертензии является уровень натрийуретического пептида головного мозга (BNP). Исходный или референсный уровень BNP может составлять 60 пг/мл или выше, 80 пг/мл или выше, 100 пг/мл или выше, 120 пг/мл или выше, 140 пг/мл или выше, 200 пг/мл или выше, 500 пг/мл или выше, или 1000 пг/мл или выше. В некоторых вариантах осуществления, предлагаемое в изобретении лечение является эффективным, если после начала лечения конечный уровень BNP у субъекта снижается относительно исходного или референсного уровня BNP. Например, конечный уровень BNP у субъекта может снижаться на 1 пг/мл или более, 2 пг/мл или более, 5 пг/мл или более, 10 пг/мл или более, 20 пг/мл или более, 100 пг/мл или более, 500 пг/мл или более, или 1000 пг/мл или более.
[0239] В качестве физиологического показателя легочной артериальной гипертензии может быть также использована диффузионная способность легких по монооксиду углерода (DLCO) или диффузионная способность легких по CO. Исходный или референсный уровень DLCO может составлять 90% или менее, 80% или менее, 70% или менее, 50% или менее, 45% или менее, или 40% или менее. В некоторых вариантах осуществления, предлагаемое в изобретении лечение является эффективным, если после начала лечения конечный уровень DLCO повышается относительно исходного уровня. Например, конечный уровень DLCO может повышаться относительно исходного или референсного уровня DLCO на 1% или более, 5% или, 10% или более, 15% или более, 20% или более, или 50% или более.
[0240] В качестве параметра для определения эффективности в популяции из одного или более субъектов может быть использован средний уровень выживаемости. Референсный средний уровень выживаемости может составлять 95% или ниже, 93% или ниже, 90% или ниже, 86% или ниже, 82% или ниже, или 78% или ниже. Средний уровень выживаемости может представлять собой средний уровень выживаемости в течение 1 года. В некоторых вариантах осуществления, предлагаемое в изобретении лечение является эффективным в популяции из одного или более субъектов, если после начала лечения, средний уровень выживаемости повышается. Например, средний уровень выживаемости может повышаться относительно референсного среднего уровень выживаемости на 1% или более, 2% или более, 5% или более, 10% или более, или 20% или более.
[0241] В одном аспекте, в изобретении предлагается способ лечения глиобластома (в том числе мультиформной глиобластомы, или GBM) у субъекта, нуждающегося в этом. В одном варианте осуществления, способ включает введение эффективного количества описанных в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции указанному субъекту. Глиомы представляют собой гетерогенную опухоль, обычно состоящую из опухолевых клеток и стволовых раковых клеток глиомы (GSC), которые способствуют самообновлению, пролиферации и выживанию. Эти популяции стволовых клеток могут преимущественно экспрессировать гены, регулируемые HIF-2α, такие как VEGF, Oct4, Glut-1, NOTCH, и простатическую кислую фосфатазу (PAP). Популяции раковых клеток глиомы (GSC) имеют тенденцию находиться в периваскулярной нише с целью поддержания функции сосудов и роста опухоли, а также устойчивости к воздействию радиации. Эффекты агрессивного роста глиомы и подобных стволовым клеткам фенотипов могут быть опосредованы путями сигнальной трансдукции выше HIF, такими как PI3K/AKT/mTOR, и общим повышением активности HIF-2α. Гистопатологические особенности мультиформной глиобластомы (GBM) включают инфильтративную инвазию в паренхиму головного мозга, значительные очаги некроза палисадной ткани и обширные характерные участки микроваскулярной пролиферации. В некоторых вариантах осуществления, твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию вводят в количестве, эффективном для отсрочки прогрессирования, снижения частоты проявления или снижения степени одной или нескольких из этих характеристик, связанных с глиобластомой, или других характеристик, описанных в изобретении. В некоторых вариантах осуществления, твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию вводят (либо в разовой дозе, либо в нескольких раздельных дозах) в количестве, эффективном для увеличения средней выживаемости среди подвергающейся лечению популяции субъектов, например, приблизительно на 0,5 , 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15 или более лет.
[0242] Лечение глиобластомы включает лечение субъекта, у которого диагностировано наличие глиобластомы, а также предотвращение возникновения глиобластомы, например, у субъекта с повышенным риском развития глиобластомы. В некоторых вариантах осуществления, количество твердой дисперсии, твердой лекарственной формы или фармацевтической композиции, вводимое субъекту (либо в разовой дозе, либо в нескольких раздельных дозах) является эффективным в случаях одном или более ингибирования клеток глиобластомы, ингибирования метастазирования клеток глиобластомы, уничтожения клеток глиобластомы, снижения размера опухоли и снижения тяжести или частоты проявления симптомов, ассоциированных с присутствием клеток глиобластомы. Степень проявления одного или более из этих терапевтических эффектов может составлять приблизительно 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или более. В некоторых вариантах осуществления, терапевтическую эффективность измеряют увеличением времени прогрессирования заболевания, например, временем между проявлением одного или нескольких первых симптомов и проявлением одного или нескольких вторых симптомов, или отсрочкой между двумя или более возникновениями этих же симптомов. Отсрочка может составлять приблизительно дни, недели, месяцы или годы (например, 1, 2, 3, 4, 5, 6, 7 или более дней; 1, 2, 3, 4, 5, 6, 7, 8 или более недель; 1, 2, 3, 4, 5, 6 или более месяцев; или 1, 2, 3, 4, 5 или более лет). В случае профилактики, субъектом может быть индивидуум с риском развития глиобластомы, например, субъект в состоянии ремиссии, имеющий случаи заболевания глиобластомой в семье и/или имеющий некоторую другую предрасположенность. Степень терапевтической эффективности может быть оценена относительно исходного состояния субъекта (например, относительно исходного размера опухоли, скорости роста, степени метастазирования, тяжести и частоты возникновения одного или более симптомов), или относительно референсной популяции (например, популяции, не подвергавшейся лечению, популяции, подвергавшейся лечению другим лекарственным средством).
[0243] Эффективность лечения глиобластомы может быть установлена любым подходящим методом, таким как методы, используемые в настоящее время в клинике для контроля за размером опухоли и прогрессированием рака. Измерение размера опухоли является одним из методов определения, замедлился ли рост опухоли, приостановлен ли рост опухоли, или произошло обратное развитие опухоли (например, в случае уничтожения клеток глиобластомы). Размер опухоли может быть рассчитан любым подходящим методом, например, измерением геометрических размеров или оценкой объема опухоли с использованием доступного компьютерного программного обеспечения, такого как программа FreeFlight, разработанная в Wake Forest University, которая позволяет точно оценить объем опухоли. Размер опухоли может быть определен путем визуализации опухоли с использованием, например, компьютерной томографии (CT), ультразвука, однофотонной эмиссионной компьютерной томографии (SPECT), спиральной компьютерной томографии, магнитной резонансной томография (MRI), фотографий и других подобных методов. В вариантах осуществления, в которых опухоль удаляют хирургическим путем после завершения терапевтического лечения, присутствие опухолевой ткани и размер опухоли могут быть определены путем общего анализа ткани, подлежащей хирургическому удалению, и/или путем патологического анализа удаленной ткани.
[0244] Желательно, чтобы в результате лечения рост опухоли был стабилизирован (то есть, одна или более опухолей не увеличиваются в размере более чем на 1%, 5%, 10%, 15% или 20%, и/или не метастазируют). В некоторых вариантах осуществления, опухоль стабилизируют в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более недель. В некоторых вариантах осуществления, опухоль стабилизируют в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более месяцев. В некоторых вариантах осуществления, опухоль стабилизируют в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более лет. Предпочтительно, чтобы способ по изобретению позволял уменьшать размер опухоли, по меньшей мере, приблизительно на 5% (например, по меньшей мере, приблизительно на 10%, 15%, 20% или 25%). Более предпочтительно, если размер опухоли уменьшается, по меньшей мере, приблизительно на 30% (например, по меньшей мере, приблизительного на 35%, 40%, 45%, 50%, 55%, 60% или 65%). Еще более предпочтительно, если размер опухоли уменьшается, по меньшей мере, приблизительно на 70% (например, по меньшей мере, приблизительно на 75%, 80%, 85%, 90% или 95%). И наиболее предпочтительно, если опухоль полностью ликвидируется или ее размеры уменьшаются до величины ниже уровня обнаружения. В некоторых вариантах осуществления, у субъекта не обнаруживается наличие опухоли (например, при ремиссии) в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более недель после лечения. В некоторых вариантах осуществления, у субъекта не обнаруживается наличие опухоли в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более месяцев после лечения. В некоторых вариантах осуществления, у субъекта не обнаруживается наличие опухоли в течение, по меньшей мере, приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более лет после лечения. Когда опухоль подвергают хирургическому удалению после завершения терапевтического лечения, эффективность способа по изобретению при уменьшении размера опухоли размер опухоли может быть определена путем измерения процента удаленной ткани, которая является некротической. В некоторых вариантах осуществления, a лечение является терапевтически эффективным, если процент некроза в удаленной ткани составляет более чем приблизительно 20% (например, по меньшей мере, приблизительно 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100%), более предпочтительно, приблизительно 90% или более (например, приблизительно 90%, 95% или 100%). Наиболее предпочтительно, если процент некроза в удаленной ткани составляет 100%, то есть, опухолевая ткань отсутствует или не поддается обнаружению. В некоторых вариантах осуществления, скорость роста глиобластомы снижают приблизительно на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или более, включая полную остановку роста глиобластомы (например, снижают скорость изменения размера опухоли). В некоторых вариантах осуществления, уничтожают приблизительно более 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или более выявленных клеток глиобластомы (например, в виде опухолевой массы).
[0245] Для определения терапевтической эффективности может быть использован целый ряд вторичных параметров. Примеры вторичных параметров включают, но этим не ограничивая, обнаружение новых опухолей, обнаружение опухолевых антигенов или маркеров (например, CEA, PSA или CA-125), биопсию, снижение стадии хирургического вмешательства (то есть, превращение стадии хирургического вмешательства из стадии неоперабельный опухоли в стадию операбельной опухоли), сканограммы позитронно-эмиссионной томографии (PET), выживаемость, выживаемость без признаков прогрессирования заболевания, время до прогрессирования заболевания, оценки качества жизни, такие как "Оценка положительного клинического результата" (Clinical Benefit Response Assessment), и другие подобные параметры, каждый из которых может указывать на общее прогрессирование (или ремиссию) рака у человека. Биопсия особенно подходит для выявления факта уничтожения раковых клеток в ткани. Радиоиммуноанализ (RAID) применяют для определения местонахождения и стадии развития опухолей, используя уровни маркеров в сыворотке крови (антигенов), продуцируемых опухолями и/или ассоциированных с опухолями ("опухолевые маркеры" или "опухолеассоциированные антигены"), и уровни этих маркеров могут использоваться в качестве диагностического подхода перед лечением, диагностического индикатора возникновения рецидива после лечения и показателя терапевтической эффективности после лечения. Примеры маркеров глиобластомы которые могут быть оценены в качестве показателей терапевтической эффективности, включают, но этим не ограничивая, ABCC3, GPNMB, NNMT, и SEC61γ (смотрите, например, патентный документ US7115265).
[0246] Дополнительный пример метода оценки эффективности лечения, экспрессия биомаркеров заболевания, например, глиобластомы, может быть подвергнута сравнению между субъектом, имеющим заболевание или с повышенным риском возникновения заболевания, с экспрессией тех же самых биомаркеров у субъекта в динамике по времени. В некоторых случаях, экспрессия одного и того же набора биомаркеров может быть подвергнута сравнению между субъектом, имеющим заболевание или с повышенным риском возникновения заболевания, и одним или более здоровыми субъектами. При оценке исхода заболевания или эффекта лечения, популяция пациентов, каждый из которых страдает от заболевания, может находиться под наблюдением врача в течение определенного периода времени. Уровни экспрессии биомаркеров могут быть установлены путем оценки экспрессии биомаркера в образце, взятом у одного пациента, оценки экспрессии биомаркера дополнительных образцов, взятых у того же самого пациента позже через некоторое время, и сравнения экспрессии биомаркера в последних образцах с исходным образцом или образцами. Этот метод может быть использован в случае биомаркеров, которые указывают, например, на прогрессирование или усугубление заболевания, отсутствие эффективности применяемой схемы лечения, на ремиссию заболевания или эффективность применяемой схемы лечения. Кроме того, эффективность лечения субъекта может быть оценена целым рядом методов, включающих, но этим не ограничивая, физикальное обследование, биопсию или любой из многочисленных методов визуализации, такой как ультразвуковая эхография, компьютерная томография, магниторезонансная визуализация, магнитно-резонансная спектроскопия или позитронная эмиссионная томография.
[0247] Глиобластома ассоциируется с целым рядом симптомов. Типичные симптомы заболевания включают судорожный припадок, тошноту и рвоту, головную боль, потерю памяти и гемипарез, и прогрессирующее нарушение памяти, личностное расстройство или неврологическое расстройство вследствие вовлечения в патологический процесс височной и лобной доли головного мозга. В некоторых случаях, опухоль может быстро вызвать появление симптомов, но, в редких случаях, развитие опухоли может протекать бессимптомно до тех пор, пока она не достигнет огромных размеров. Терапевтическая эффективность может быть измерена величиной уменьшения любого одного или более из этих симптомов, например, величиной уменьшения частоты возникновения или тяжести симптомов. В некоторых вариантах осуществления, уменьшение симптомов составляет приблизительно 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или более, измеренное с использованием соответствующей шкалы.
[0248] Что касается оценок качества жизни, например, с помощью критерия положительного клинического результата (Clinical Benefit Response Criteria), то терапевтический положительный эффект лечения в соответствии с изобретением может быть доказан путем использования таких показателей, как интенсивность боли, потребление болеутоляющих средств и/или оценка в баллах общего состояния по шкале Карновского. Оценка в баллах общего состояния по шкале Карновского позволяет классифицировать пациентов в соответствии с их функциональным нарушением. Балльная шкала оценки общего состояния Карновского имеет диапазон 0-100 баллов. Обычно, более низкое количество баллов по шкале Карновского предвещает неблагоприятный прогноз по выживаемости. В некоторых вариантах осуществления, альтернативно или дополнительно, положительный эффект при лечении рака у пациента, доказывается (a) снижением, по меньшей мере, на 50% (например, снижением, по меньшей мере, на 60%, 70%, 80%, 90% или 100%) интенсивности боли, в соответствии с полученной от пациента информацией, например, в течение любых последующих четырех недель через 12 недель после завершения проведения лечения, по сравнению с интенсивностью боли, в соответствии с полученной от пациента информацией, до проведения лечения, (b) снижением, по меньшей мере, на 50% (например, снижением, по меньшей мере, на 60%, 70%, 80%, 90% или 100%) потребления болеутоляющих средств, в соответствии с полученной от пациента информацией, например, в течение любых последующих четырех недель через 12 недель после завершения проведения лечения, по сравнению с потреблением болеутоляющих средств, в соответствии с полученной от пациента информацией, до проведения лечения, и/или (c) увеличением, по меньшей мере, на 20 баллов (например, увеличением, по меньшей мере, на 30 баллов, 50 баллов, 70 баллов или 90 баллов) оценки в баллах общего состояния по шкале Карновского, в соответствии с полученной от пациента информацией, например, в течение любых последующих четырех недель через 12 недель после завершения проведения лечения, по сравнению с оценкой в баллах общего состояния по шкале Карновского, в соответствии с полученной от пациента информацией, до проведения лечения.
[0249] В одном аспекте, в настоящем изобретении предлагается способ лечения воспалительного заболевания системы органов пищеварения у субъекта, нуждающегося в этом. В некоторых вариантах осуществления, способ включает введение субъекту эффективного количества предлагаемых в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции. В одном аспекте, в настоящем изобретении предлагается способ уменьшения воспаления системы органов пищеварения у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества предлагаемых в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции. В некоторых вариантах осуществления, твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию вводят в количестве, эффективном для отсрочки прогрессирования, снижения частоты возникновения воспаления или воспалительного заболевания или уменьшения степени одной или более характеристик, связанных с воспалением или воспалительным заболеванием. В некоторых вариантах осуществления, твердую дисперсию, твердую лекарственную форму или фармацевтическую композицию вводят, либо в разовой дозе, либо в нескольких разделенных дозах, в количестве, эффективном для достижения ремиссии воспаления или воспалительного заболевания.
[0250] Система органов пищеварения состоит из желудочно-кишечного тракта и вспомогательных органов пищеварения, включающих язык, слюнные железы, пищевод, желудок, поджелудочную железу, печень, желчный пузырь, тонкий кишечник, толстый кишечник, толстую кишку, задний проход и прямую кишку. Некоторые варианты осуществления настоящего изобретения относится конкретно к нижнему отделу желудочно-кишечного тракта, включающего тонкий кишечник и толстый кишечник. Термин "тонкий кишечник" включает в себя двенадцатиперстную кишку, тонкую кишку и подвздошную кишку, и термин " толстый кишечник " включает слепую кишку, аппендикс, толстая кишка, восходящую ободочную кишку, правый печеночный угол, поперечную ободочную кишку, левый селезеночный угол, нисходящую ободочную кишку, сигмовидную ободочную кишку, прямую кишку, анальный канал и заднепроходное отверстие.
[0251] В настоящем изобретении предлагается как способы уменьшения воспаления системы органов пищеварения, так и способы лечения воспалительного заболевания системы органов пищеварения. Используемый в изобретении термин "воспаление" относится к собирательному термину для местной аккумуляции жидкостей, белков плазмы крови, и/или лейкоцитов, инициируемой аутоиммунным ответом, телесным повреждением, инфекцией, сосудистым заболеванием, воздействием химического вещества, радиоактивного излучения или локальным иммунным ответом. Обычно, воспаление характеризуется одним или более признаками, включающими, например, покраснение кожи, боль, повышение температуры тела, опухание и/или потерю функции. Воспаление может быть связано с хроническими (продолжительными) воспалительными заболеваниями или нарушениями или с острыми (непродолжительными) воспалительными заболеваниями или нарушениями.
[0252] При осуществлении на практике любого из заявляемых способов, твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция может уменьшать воспаление системы органов пищеварения, например, воспаление одного или более из органов, таких как язык, слюнные железы, пищевод, желудок, поджелудочная железа, печень, желчный пузырь, тонкий кишечник, толстый кишечник, толстая кишка, заднепроходное отверстие и прямая кишка. В некоторых вариантах осуществления, твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция уменьшает воспаление нижнего отдела желудочно-кишечного тракта, например, воспаление тонкого кишечника, толстого кишечника или толстой кишки. В некоторых примерах вариантов осуществления, твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция уменьшает воспаление толстой кишки. Воспаление может проявляться в форме энтерита, гастрита, гастроэнтерита, колита, энтероколита, дуоденита, еюнита, илеита, проктита или аппендицита. Воспаление может быть острым или хроническим. В некоторых предпочтительных вариантах осуществления, воспаление классифицируют как колит.
[0253] При физиологических условиях, желудочно-кишечный тракт может характеризоваться очень высоким значением кислородного градиента. Хроническое воспалительное заболевание кишечника обычно характеризуется активным потреблением O2 рекрутированными воспалительными клетками, включающими макрофаги, дендритные клетки и нейтрофилы. Возникающий дисбаланс между потреблением и снабжением кислородом вызывает тяжелую форму воспалительной гипоксии слизистой оболочки кишечника. Эта "воспалительная гипоксия" обычно приводит к повышению уровней факторов, индуцируемых гипоксией, 1α и 2α (HIF-1α и HIF-2α) в интестинальном эпителии субъектов, страдающих от воспаления системы органов пищеварения, в том числе субъектов, страдающих от воспалительного заболевания кишечника, и в экспериментальных моделях колита на мышах. При воспалительном заболевании кишечника, было показано, что HIF-1α и HIF-2α играют противоположные роли в прогрессировании заболевания. Было показано, что повышение экспрессии HIF-1α играет защитную роль при воспалительном заболевании кишечника, в том числе для выживания эпителиальных клеток, индукции некоторых белков защитного барьера и белков плотных контактов, увеличения уровня противомикробного β-дефензина и предотвращения избыточного иммунного ответа в результате повышенной регуляции CD39/CD73 и T регуляторных клеток. Продолжительная экспрессия HIF-2α может приводить к устойчивому самопроизвольному интестинальному воспалению и повреждению при воспалительном заболевании кишечника, включая апоптоз эпителиальных клеток, дисрегуляцию белков защитного барьера и белков плотных контактов, повышение уровня провоспалительных цитокинов и избыточный иммунный ответ. Было показано, что HIF-2α регулирует рекрутмент функцию миелоидных клеток, в том числе нейтрофилов и макрофагов, способствуя прогрессированию воспаления и воспалению, опосредованному раком толстой кишки. Недавно стало известно, что HIF-1α является фактором, способствующим воспалению в экспериментальной модели колита при нокдауне в клеточных линиях миелоидных клеток.
[0254] Не привлекая в качестве обоснования какую-либо конкретную теорию, авторы настоящего изобретения, тем не менее, выдвинули гипотезу о том, что экспрессия HIF-2α в эпителиальных и миелоидных клетках толстой кишки является главным фактором, способствующим инициированию, прогрессированию и сохранению хронического воспаления. Предполагается, что блокада HIF-2α должна уменьшать или ингибировать рекрутмент типов воспалительных клеток и также их провоспалительных продуктов, приводя к ремиссии или предотвращению воспаления системы органов пищеварения, в частности, у субъектов, страдающих болезнью Крона и язвенным колитом.
[0255] Субъект с воспалением системы органов пищеварения может страдать от воспалительного заболевания кишечника, болезни Крона, колита, целиакии, эозинофильной энтеропатии или аппендицита. Используемый в изобретении термин "воспалительное заболевание кишечника" относится к патологии, характеризующейся воспалительным состоянием толстого и/или тонкого кишечника. Болезнь Крона и колит представляют собой два типа воспалительного заболевания кишечника.
[0256] В некоторых вариантах осуществления, воспалительное заболевание кишечника включает колит, например, язвенный колит. "Колит" представляет собой воспаление толстого кишечника. Колит может быть острым или хроническим. Используемый в изобретении термин "колит" включает язвенный колит, микроскопический колит, лимфатический колит, коллагенозный колит, диверсионный колит, химический колит, ишемический колит, инфекционный колит, панколит, левосторонний колит, обширный колит, сегментарный колит, микроскопический колит, индуцированный ионизирующим излучением колит, индуцированный лекарственными препаратами колит и проктит. "Язвенный колит" представляет собой хроническое воспалительное заболевание, поражающее толстую кишку. Он характеризуется воспалением слизистой оболочки толстой кишки. Симптомы могут проявляться в форме от умеренной до тяжелой и могут включать кровь в экскрементах, диарею, кровавую диарею, императивный позыв на дефекацию, ложные болезненные позывы на дефекацию, недержание, утомление, повышенную частоту дефекации, выделение слизи, ночные дефекации, неприятные ощущения в животе, лихорадку, потерю массы тела, парадоксальную констипацию, анемию и абдоминальную болезненность. Язвенный колит является перемежающимся заболеванием, при котором у большинства пациентов течение болезни характеризуется периодами рецидива и периодами облегчения с периодическими внезапными обострениями болезни. Термины "внезапное обострение болезни" или "рецидив" относятся к усилению симптомов язвенного колита, например, увеличению частоты дефекации, усилению кровотечение из прямой кишки и/или атипичному виду слизистой оболочки, обнаруживаемому при эндоскопии. Несмотря на то, что симптомы язвенного колита могут ослабевать без хирургического вмешательства, тем не менее, заболевание обычно требует лечения для достижения состояния ремиссии.
[0257] Используемый в изобретении термин "активный" язвенный колит относится к язвенному колиту, который является биологически активным. Пациенты с активным заболеванием могут иметь клинические проявления и обнаруживать один или более признаков или симптомов язвенного колита, например, кровотечение из прямой кишки, увеличение частоты дефекации, воспаление слизистой оболочки или не соответствующие норме результаты лабораторных анализов (например, повышенные значения скорости оседания эритроцитов (ESR) или С-реактивного белка (CRP), или пониженный гемоглобин). "Рефракторный" язвенный колит в отношении конкретной терапии обозначает язвенный колит, который является активным или который рецидивирует или периодическими внезапно обостряется не смотря на проведение лечения с применением этой терапии.
[0258] Используемый в изобретении термин "болезнь Крона" относится к типу воспалительного заболевания кишечника, характеризующемуся воспалением слизистой оболочки желудочно-кишечного тракта. Симптомы могут включать диарею, боль в животе, лихорадку, утомление, кровь в экскрементах и потерю массы тела.
[0259] При подозрении наличия у пациента язвенного колита, первичный диагноз обычно основывается на комбинации симптомов, данных эндоскопии и гистологии. Диагностирование обычно включает анализ кала, анализ мочи и тесты на анемию, железодефицит, лейкоцитоз и/или тромбоцитоз. В зависимости от тяжести заболевания, могут иметь повышенные значения маркеры воспаления, такие как скорость оседания эритроцитов (ESR) и C-реактивный белок. Однако, обычно считается, что только эндоскопия с биопсией является методом для однозначной постановки диагноза язвенного колита. Данные эндоскопии, которые подтверждают диагноз язвенного колита, могут включать эритему, потерю нормального сосудистого рисунка, эрозии, кровотечение, зернистость, рыхлость, изъязвления и псевдополипы. Во время проведения эндоскопии может быть также проведена биопсия для дифференцирования язвенного колита от болезни Крона. Биопсийные образцы исследует на искажение архитектуры крипт кишечника, воспаление крипт, укорачивание крипт, повышение числа лимфоцитов и плазматических клеток в собственной пластинке, абсцессы крипта, истощение муцина и геморрагию или воспаление в собственной пластинке.
[0260] Язвенный колит может поражать часть толстой кишки, или практически полностью всю толстую кишку. Язвенный колит может представлять собой проктит, в случае которого язвенный колит ограничивается заднепроходным отверстием и слизистой оболочкой прямой кишки. Язвенный колит может представлять собой левосторонний колит, в случае которого колит ограничивается частью толстой кишки, наиболее удаленной от селезеночного изгиба ободочной кишки, более конкретно, язвенный колит, который простирается от прямой кишки и до селезеночного изгиба ободочной кишки. Язвенный колит может представлять собой обширный колит, в случае которого поражается практически вся толстая кишка. Соответственно, в некоторых вариантах осуществления, в настоящем изобретении предлагается способ уменьшения воспаления у субъекта, страдающего от язвенного колита, включая проктит, левосторонний колит и обширный колит.
[0261] Язвенный колит обычно дополнительно характеризуется формой тяжести заболевания, например, ремиссией, легкой, средней или тяжелой формой язвенного колита. Способы по настоящему изобретению могут применяться для лечения легкой, средней или тяжелой форм язвенного колита, или язвенного колита, который находится в стадии ремиссии. Например, эффективное количество предлагаемых в изобретении твердой дисперсии, твердой лекарственной формы или фармацевтической композиции может быть введено субъекту, страдающему легкой формой язвенного колита. Твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция могут быть введены субъекту, страдающему язвенным колитом средней формой тяжести. Твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция могут быть введены субъекту, страдающему язвенным колитом в тяжелой форме. Твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция могут быть введены субъекту, страдающему язвенным колитом легкой или средней формы тяжести. Твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция могут быть введены субъекту, страдающему язвенным колитом в средней или тяжелой формы. Твердая дисперсия, твердая лекарственная форма или фармацевтическая композиция могут быть введены субъекту, страдающему язвенным колитом в стадии ремиссии.
[0262] Существуют многочисленные показатели для оценки формы тяжести язвенного колита, включающие индекс Мейо (Mayo score), индекс Лихтигера (Lichtiger score) и простой клинический индекс активности колита (Simple Clinical Colitis Activity Index). Эти показатели обычно служат одним из факторов в обобщенном эндоскопическом показателе, например, обобщенный показатель индекса Мейо или эндоскопического индекса тяжести язвенного колита (Ulcerative Colitis Endoscopic Index of Severity). Типичные гистологические классификации включают гистопатологический индекс Робартса (Robarts Histopathology index) и индекс Нэнсу (Nancy index). Для оценки тяжести заболевания может быть использован комбинированный критерий, включающий один или более из этих показателей, влияние заболевания на качество жизни субъекта, поддающиеся измерению маркеры активности и степени заболевания и общего течения заболевания, такие как внекишечные проявления болезни, интестинальное поражение и частота внезапных обострений болезни.
[0263] В некоторых вариантах осуществления, описанный в изобретении способ дополнительно включает введение второго терапевтического средства. Второе средство может быть введено внутривенно или подкожно. В некоторых вариантах осуществления, второе средство вводят один или два раза в сутки, один раз в два дня, один раз в неделю, один раз в две недели или один раз в три недели. Второе средство может быть введено в количестве от приблизительно 1 до приблизительно 1000 мг, от приблизительно 5 до приблизительно 500 мг, от приблизительно 10 до приблизительно 350 мг, или от приблизительно 50 до приблизительно 200 мг. В одном варианте осуществления, второе активное средство вводят перорально один или два раза в сутки, один раз в два дня, один раз в неделю, один раз в две недели или один раз в три недели, в количестве от приблизительно 1 до приблизительно 1000 мг, от приблизительно 5 до приблизительно 500 мг, от приблизительно 10 до приблизительно 350 мг, от приблизительно 10 до приблизительно 200 мг, от приблизительно 10 до приблизительно 100 мг, или от приблизительно 20 до приблизительно 50 мг. Конкретное количество второго активного средства будет зависеть от конкретного используемого средства, типа заболевания, подвергаемого лечению или ведению, тяжести и стадии заболевания, и количества соединения формулы (I), и любых необязательных дополнительных активных средств, одновременно вводимых пациенту.
ПРИМЕРЫ
[0264] Следующие далее примеры приведены с целью иллюстрации различных вариантов осуществления изобретения и не коим образом не ограничивают настоящее изобретение. Настоящие примеры вместе с описанными в изобретении способами являются иллюстрациями предпочтительных вариантов осуществления, и их не следует рассматривать в качестве ограничений объема изобретения. Для специалистов в данной области является очевидным, что в изобретение могут быть внесены изменения и предложены другие варианты применения, которые не противоречат сущности изобретения, определяемой объемом формулы изобретения.
Методы характеризации
Порошковый рентгеноструктурный анализ проводили на рентгеновском дифрактометре Bruker D2 Phaser с использованием метода сканирования θ/2θ, при котором величину напряжения устанавливали на 30 кВ и величину тока на 10 мА при вращении 15 об/мин, с держателем образца с нулевым фоном, при ширине щели 1,0 мм и при ширине призмы, установленной на 1,0 мм.
Модулированная дифференциальная сканирующая калориметрия
[0266] Модулированную дифференциальную сканирующую калориметрию (MDSC) проводили на дифференциальном сканирующем калориметре TA Instruments Q2000 с системой охлаждения Refrigerated Cooling System 90. MDSC использовали для определения температуры стеклования (Tg) и температуры плавления (Tm), если эти превращения происходили. Использовали температурный диапазон 0-250°C при скорости нагревания 1,5°C в минуту. Режим сканирования модулировали при частоте 60 секунд и амплитуде 1°C.
Парофазная газовая хроматография
[0267] Уровни остаточного растворителя определяли методом парофазной газовой хроматографии (GCHS). Например, для анализа образца использовали газовый хроматограф Agilent 6890A/7694 GC/HS, оборудованный колонкой JW Scientific DB-624 30 м x 0,32 мм, 1,8 мкм, со следующими параметрами: температура образца 105°C; температура петли 110°C; температура линии транспортировки 115°C; время цикла газовой хроматографии 45 мин; время установления равновесия в виале 30 мин; размер петлевого дозатора 1 мл; время создания давления в виале 20 сек; давление газа-носителя 0,05 МПа; и давление в виале 0,10 МПа.
Высокоэффективная жидкостная хроматография
[0268] Высокоэффективную жидкостную хроматографию (HPLC) проводили на хроматографе Agilent 1220, оборудованном колонкой Kinetex, C18, 4,6×100 мм, 2,6 мкм, с использованием следующих параметров: подвижная фаза A, 0,1% муравьиной кислоты в воде; подвижная фаза B, 0,1% муравьиной кислоты в ацетонитриле; градиент 95% A (0 мин), 60% A (6 мин), 5% A (12 мин), 95% A (15,1 мин), с достижением равновесия с подвижной фазой B; расход, 0,8 мл/мин; температура колонки 40°C; температура образца комнатная температура; вводимый объем 5 мкл; длина волны детектирования 240 нм; и время записи хроматограммы 18,5 мин.
Сканирующая электронная микроскопия
[0269] Получение изображения методом сканирующей электронной микроскопии (SEM) проводили на сканирующем электронном микроскопе FEI Quanta 200 SEM с системой для автоматического напыления материала мишени Au/Pd Polaron Autocoater E5200 при напряжении 15 кВ, размере пятна 3,0 мА, токе накала 2,52 A и токе эмиссии 96 мкА.
[0270] Пример 1. Характеризация 3-(((1S,2S,3R)-2,3-дифтор-1-гидрокси-7-(метилсульфонил)-2,3-дигидро-1H-инден-4-ил)окси)-5-фторбензонитрила.
[0271] Температуру плавления (Tm) и температуру стеклования (Tg) 3-(((1S,2S,3R)-2,3-дифтор-1-гидрокси-7-(метилсульфонил)-2,3-дигидро-1H-инден-4-ил)окси)-5-фторбензонитрила, соединения формулы (I), определяли методом mDSC и обнаруживали, что они составляют 209,2°C и 79,9°C, соответственно. Рентгенограмму соединения формулы (I) получали методом XRPD (фигура 1), и хроматограмма указывала на то, что соединение является кристаллическим веществом.
[0272] Растворимость соединения формулы (I) определяли дважды в ацетоне и в смесях ацетон/H2O. Первое измерение проводили при комнатной температуре в течение 30 минут при смешении, которое заканчивалось центрифугированием в центрифужных пробирках. Наблюдаемая растворимость представлена в таблице 1.
Таблица 1
[0273] Второе измерение проводили для дополнительной оценки комбинаций ацетон/H2O при увеличении содержании воды с шагом 1% H2O. Растворимость, представленная в таблице 2, наблюдалась после приведения образцов в состояние равновесия в течение 48 часов при комнатной температуре.
Таблица 2
(масс.%)
[0274] Пример 2. Скрининг твердых дисперсий.
[0275] Приготавливали семь композиций полимерных дисперсий, включающих соединение формулы (I), и подвергали их распылительной сушке из смеси 95:5 ацетон:H2O при соотношении соединения формулы (I) к полимеру 25:75 в соответствии с параметрами, приведенными в таблице 3.
Таблица 3
[0276] Для удаления остаточного растворителя после начальной распылительной сушки использовали процесс вторичной сушки в полочной сушилке. В этой операции, "влажную" твердую дисперсию нагревали до 40°C и выдерживали на полке конвекционного сушильного шкафа в течение 26 часов. Стадия вторичной обработки при повышенной температуре позволяла удалить остаточный ацетон из твердой дисперсии. Для измерения остаточного растворителя, остающегося в твердой дисперсии после вторичной сушки использовали анализ методом парофазной газовой хроматографии (GC). Определяли остаточное содержание ацетона в нескольких композициях на предмет того, чтобы его содержание было намного ниже предельного значения для ацетона 5000 ppm, установленного Международной конференцией по гармонизации (International Conference on Harmonization (ICH)). Суммарный выход, измеренные величины Tg и содержание остаточного ацетона в каждой из твердых дисперсий приведены в таблице 4.
Таблица 4
Tg (°C)
соединение формулы (I):
HPMCAS-M
соединение формулы (I):
HPMCAS-H
соединение формулы (I):
PVP-VA
соединение формулы (I):
HPMCP HP-55
соединение формулы (I):
SOLUPLUS
соединение формулы (I):
CAP
соединение формулы (I):
Eudragit S100
LOQ: нижняя граница определяемых концентраций
[0277] Термический анализ твердых дисперсий методом модулированной дифференциальной сканирующей калориметрии (mDSC) показал, что все дисперсии имели единственную температуру стеклования Tg, что указывало на тщательно перемешанную аморфную твердую дисперсию с высокой степенью гомогенности. Значения Tg твердых дисперсий находились в диапазоне от 70°C до 140°C (таблица 4). Эти относительно высокие значения Tg указывают на высокую физическую стабильность, например, склонность соединения формулы (I) к перекристаллизации в процессе длительного хранения была низкой. Твердые дисперсии хранили при температурах ниже Tg для обеспечения длительной физической стабильности, в результате чего подвижность соединения формулы (I) в дисперсии света была низкой. Раствор, содержащий 100% соединения формулы (I) в ацетоне, подвергали распылительной сушке и извлекали в кристаллической форме, которая подтверждалось отсутствием Tg и порошковой рентгенограммой. Характеризация методом рентгеноструктурного анализа (XRPD) указывала на то, что твердая дисперсия соответствовала аморфной структуре вследствие отсутствия наблюдаемых кристаллических пиков на дифрактограммах твердой дисперсии (фигура 2).
[0278] Морфологию поверхности частиц твердых дисперсий оценивали методом сканирующей электронной микроскопии. Было обнаружено, что твердые дисперсии включают целые и разрушенные сферы с гладкими поверхностями. В образцах твердых дисперсий не обнаруживалось присутствие кристаллического материала.
[0279] Характеристику растворения твердых дисперсий получали путем проведения испытания на растворимость в условиях недостаточного разбавления. Испытание на растворимость использовали для измерения пересыщения лекарственного средства, превышающего суммарную растворимость кристаллического соединение формулы (I) в биорелевантной среде, моделирующей кишечный сок (FaSSIF), после 30 минут воздействия среды с низким значением pH (SGF). При проведении испытания, образцы переносили из SGF [теоретическая Cmax=2000 мкгА/мл] в FaSSIF [теоретическая Cmax=1000 мкгА/мл].
[0280] Наблюдаемые величины Cmax GB, Cmax FaSSIF, AUC FaSSIF, и увеличения AUC приведены в таблице 5. Твердые дисперсии на основе HPMCAS-H, CAP и SOLUPLUS обеспечивали приблизительно 30-кратное усиление солюбилизации по сравнению с кристаллическим лекарственным средством (AUCтвердая дисперсия/AUCсоединение формулы (I)), как это показано в таблице 5. Все три твердых дисперсии обеспечивали существенное увеличение Cmax, одновременно при этом сохраняя пределы перенасыщения в течение, по меньшей мере, 180 минут воздействия FaSSIF. Эти три твердые дисперсии демонстрируют высокий результат при проведении теста на растворение при прохождении через желудок, и ожидалось, что эти твердые дисперсии значительно повысят биодоступность соединения формулы (I) по сравнению с кристаллической формой лекарственного средства.
Таблица 5
GB (мкгА/мл)
AUC
PVP-VA
Eudragit S100
HPMCAS-M
HPMCP HP-55
SOLUPLUS
CAP
HPMCAS-H
[0281] Пример 3. Дополнительный скрининг твердых дисперсий.
[0282] Приготавливали три композиции твердой дисперсии для in vivo испытания, включающие соединение формулы (I) в соотношении 25:75 с HPMCAS-H, CAP или SOLUPLUS. Все композиции подвергали распылительной сушке с использованием таких же параметров, как в примере 2. Для удаления остаточного растворителя после начальной распылительной сушки использовали процесс вторичной сушки в полочной сушилке. В этой операции, "влажную" твердую дисперсию нагревали до 40°C и выдерживали на полке конвекционного сушильного шкафа в течение 24 часов. Стадия вторичной обработки при повышенной температуре позволяла удалить остаточный ацетон из твердой дисперсии. Для измерения остаточного растворителя, остающегося в твердой дисперсии после вторичной сушки использовали анализ методом парофазной газовой хроматографии (GCHS). Определяли остаточное содержание ацетона во всех трех композициях на предмет того, чтобы его содержание было намного ниже предельного значения для ацетона 5000 ppm, установленного Международной конференцией по гармонизации (International Conference on Harmonization (ICH)), причем композиции с HPMCAS-H и SOLUPLUS содержали остаточный ацетон ниже чем LOQ испытания (<200 ppm). Композицию с CAP сушили в течение еще 24 часов и определяли, что после повторного испытания содержание остаточного ацетона было < LOQ. В таблице 6 приведены результаты по содержанию остаточного растворителя для двух композиций. После вторичной сушки проводили анализ методом HPLC и анализ чистоты, результаты которых подтвердили, что процессе приготовления композиций не происходило внесение каких-либо примесей или разложения соединения формулы (I).
Таблица 6
соединение формулы(I)
: HPMCAS-H
соединение формулы(I)
: CAP
соединение формулы (I)
: SOLUPLUS
LOQ: нижняя граница определяемых концентраций
[0283] Получали значения Cmax GB, Cmax FaSSIF и AUC FaSSIF, которые представлены в таблице 7. Твердые дисперсии с HPMCAS-H, CAP и SOLUPLUS обеспечивали от 10-кратного до 30-кратного повышения солюбилизации лекарственного средства по сравнению с кристаллическим лекарственным средством (AUCтвердая дисперсия/AUCсоединение формулы (I)). Все три твердых дисперсии обеспечивали существенное увеличение Cmax, одновременно при этом сохраняя пределы перенасыщения в течение, по меньшей мере, 180 минут воздействия FaSSIF. Эти три твердые дисперсии демонстрировали высокий результат при проведении теста на растворение при прохождении через желудок, и ожидалось, что эти твердые дисперсии значительно повысят биодоступность соединения формулы (I) по сравнению с кристаллической формой лекарственного средства.
Таблица 7
(мкгА/мл*мин)
AUC
[0284] Термический анализ твердых дисперсий методом модулированной дифференциальной сканирующей калориметрии (mDSC) показал, что все дисперсии имели единственную температуру стеклования Tg, что указывало на тщательно перемешанную аморфную твердую дисперсию с высокой степенью гомогенности. Относительно высокие значения Tg указывают на высокую физическую стабильность, например, склонность соединения формулы (I) к перекристаллизации в процессе длительного хранения является низкой. Твердые дисперсии хранили при температурах ниже Tg для обеспечения длительной физической стабильности, в результате чего подвижность соединения формулы (I) в дисперсии света была низкой.
[0285] Характеризация твердого состояния методом рентгеноструктурного анализа (XRPD) указывало на то, что твердые дисперсии представляли собой аморфные дисперсии, так как в дифрактограммах твердых дисперсий не обнаруживали кристаллических пиков (фигура 3).
[0286] Морфологию поверхности частиц твердых дисперсий оценивали методом сканирующей электронной микроскопии. На фигуре 4 приведены изображения твердых дисперсий соединения формулы (I), полученные методом сканирующей электронной микроскопии, слева направо для HPMCAS-H, CAP или SOLUPLUS, при увеличении в 5000 раз. Было обнаружено, что твердые дисперсии включают целые и разрушенные сферы с гладкими поверхностями.
[0287] Пример 4. Дозирование в форме суспензий.
[0288] Оценивали стабильность суспензий твердых дисперсий при концентрациях в суспензии 10 мг соединения формулы (I) в 1 мл раствора, содержащего 1 масс.% смеси 1:1 метилцеллюлоза:Tween 80. Проводили мониторинг характеристики каждой суспензии твердой дисперсии через 4 и 24 часа после ее приготовления, используя испытание на растворимость в SGF/FaSSIF в качестве меры сравнения.
[0289] Наблюдали стабильную характеристику растворения в течение 4 часов (при хранении при комнатной температуре ~ 21°C с перемешиванием при 750 об/мин) для суспензий композиций твердых дисперсий, приготовленных при 10 мгА/мл в стандартном носителе, что является достаточным временем для in vivo дозирования после разбавления суспензий. Профили стабильности и растворения суспензий для композиций твердых дисперсии, выдерживаемых в течение 4 часов или 24 часов, приведены в таблицах 8 и 9, соответственно. Испытания на растворимость проводили при концентрации 2,0 мгА/мл в SGF, и затем при переносе в FaSSIF снижали до 1,0 мгА/мл. Предполагается, что суспензии твердых дисперсии могут храниться до 4 часов перед их дозированием.
Таблица 8
(мкгА/мл)
(мкгА/мл)
(мкгА/мл*мин)
соединение формулы (I)
: HPMCAS-H
соединение формулы (I)
: CAP
соединение формулы (I)
: SOLUPLUS
Таблица 9
(мкгА/мл)
(мкгА/мл)
(мкгА/мл*мин)
соединение формулы (I)
: HPMCAS-H
соединение формулы (I)
: CAP
соединение формулы (I)
: SOLUPLUS
[0290] Пример 5. Стабильность твердых дисперсий.
[0291] Для быстрой оценки физической и химической стабильности композиций твердых дисперсий соединения формулы (I), дисперсии подвергали старению в течение 4 недель при 25°C/60% RH и при 40°C/75% RH, как на открытом воздухе, так и в закрытой упаковке с осушающим веществом. Твердые дисперсии оценивали на изменения аморфного физического состояния методом порошкового рентгеноструктурного анализа (XRPD), химической чистоты методом высокоэффективной жидкостной хроматографии (HPLC) и морфологии частиц методом сканирующей электронной микроскопии (SEM).
[0292] Порошковый рентгеноструктурный анализ (XRPD) состаренных образцов твердых дисперсий показал, что через 4 недели все композиции твердых дисперсий оставались аморфными при отсутствии поддающегося обнаружению кристаллического материала, как показано на фигуре 5. Морфологию поверхности частиц состаренных твердых дисперсий характеризовали методом сканирующей электронной микроскопии. Изображения, полученные методом сканирующей электронной микроскопии, показали, что твердые дисперсии состояли из целых и разрушенных сфер с гладкими поверхностями, за исключением композиции SOLUPLUS при соотношении 40/75 на открытом воздухе, которая выглядела как стеклообразные агломерированные сферы, но все равно оставалась аморфной, что подтверждалось порошковым рентгеноструктурным анализом (XRPD). Анализ чистоты состаренных твердых дисперсий проводили на композициях с HPMCAS-H и CAP через две и четыре недели при соотношении 40/75 на открытом воздухе и в закрытых условиях. Никаких существенных примесей не было обнаружено.
[0293] Пример 6. Композиции с липидным носителем.
[0294] Приготавливали пять композиций соединения формулы (I) с липидными носителями. Желатиновые капсулы размера 0 заполняли композициями при 60°C. Каждая капсула имела суммарную массу 500±50 мг и содержала 5,0±0,3 масс.% соединения формулы (I) и приблизительно 95 масс.% липидного носителя, выбранного из: (1) 1:4 TPGS-1000:Gelucire 44/14; (2) 2:3 TPGS-1000:Gelucire 44/14; (3) 1:4 TPGS-1000:Gelucire 50/13; (4) 2:3 TPGS-1000:Gelucire 50/13 и (5) 1:1:1 TPGS-1000:Gelucire 44/14:Gelucire 50/13 (Gelucire представляет собой смеси моно, ди и триглицеридов с эфирами полиэтиленгликоля жирных кислот.
[0295] Для каждой композиции рассчитывали величины Cmax GB, Cmax FaSSIF и AUC FaSSIF, которые представлены в таблице 10. Композиция на основе 2:3 TPGS-1000:Gelucire 44/14 обеспечивала 4 кратное увеличение солюбилизации лекарственного средства по сравнению с кристаллическим лекарственным средством.
Таблица 10
(мкгА/мл)
(мкгА/мл)
(мкгА/мл*мин)
формулы (I)
[0296] Приготавливали приблизительно 35 капсул, содержащих 25 мгА (25 мг соединения формулы (I)) в 2:3 TPGS-1000:Gelucire 44/14 для in vivo и ускоренного испытания стабильности. Проводили исследование методом высокоэффективной жидкостной хроматографии (HPLC) и анализ чистоты. Не обнаруживали признаков разложения соединения формулы (I) в присутствии или отсутствии вспомогательных веществ капсул.
[0297] Липидные капсулы подвергали старению в течение 4 недель при 25°C/60% RH (в закрытых условиях) и при 40°C/75% RH (на открытом воздухе или в закрытых упаковках). Липидные капсулы оценивали на изменения химической чистоты методом высокоэффективной жидкостной хроматографии (HPLC). Анализ чистоты состаренных липидных капсул проводили через две и четыре недели при 40/75 как на открытом воздухе, так и в закрытых условиях, в присутствии и отсутствии вспомогательных веществ капсул. Не обнаруживали связанного с соединение формулы (I) разложения через четыре недели ни при каких условиях. Пик, относящийся к желатину, возрастал от 0,30% суммарной площади пиков до 0,9-1,3% суммарной площади пиков через четыре недели для всех конфигураций, хотя пик, относящийся к TPGS-1000, оставался постоянным на протяжении всего исследования.
[0298] Пример 7. Твердые дисперсии с HPMCAS-H.
[0299] В емкость объемом 75 л с пневматической мешалкой загружали ацетон (23,0 кг), а затем соединение формулы (I) (0,5 кг). Полученную суспензию перемешивали в течение 6 минут при комнатной температуре, после чего получали прозрачный раствор, что указывало на полное растворение соединения формулы (I). Затем добавляли полимер HPMCAS-H (1,5 кг), и полученный раствор перемешивали в течение 14 часов при комнатной температуре. Ацетон удаляли путем распылительной сушки раствора, используя установку для распылительной сушки SPX Anhydro MicraSpray MS-150, снабженную двухлоточной форсункой. Распыление осуществляли в конфигурации с замкнутым контуром с использованием следующих параметров: форсунка - система распыления с двумя лотками; растворитель - ацетон; общее содержание сухих веществ - 2,0 кг; состав раствора - сухих веществ 8%, общее масса раствора - 25,0 кг; давление распыления - 0,25-0,28 МПа; скорость распыления - 10,0 кг/час; температура на входе - 74,9-77,0°C; температура на выходе - 42,0-42,9°С; расход сушильного газа - 174 кг/час; температура конденсатора - от -17,6 до -17,1°C.
[0300] Для удаления остаточного ацетона, оставшегося после распылительной сушки, проводили вторичную сушку в полочной сушилке Despatch объемом 113,3 л. Глубина слоя твердой дисперсии на полках составляла приблизительно 2,54 см, и заданная температура сушилки составляла 40°C. Было обнаружено, что при 40°C происходит быстрое удаление ацетона из твердой дисперсии, и полное высыхание происходит примерно через 5 часов после загрузки в сушилку. Высушенная твердая дисперсия соответствует пределу для ацетона 5000 ppm, установленному Международной конференция по гармонизации (ICH Option 1).
[0301] После завершения распылительной сушки, было обнаружено, что твердая дисперсия содержала статический заряд. В результате, в сушильной камере МС-150 наблюдалось некоторое скопление твердой дисперсии, хотя материал при этом оставался рыхлым и не слипался. Суммарный выход составлял только 64%, вероятно, из-за потерь в камере. Минимальное количество твердой дисперсии (~ 100 г) обнаруживалось внутри пылеуловительной камеры с матерчатыми фильтрами, на стенках основной камеры оседало ~ 400 г, и небольшое оседание обнаруживалось на стенках выпускной технологической трубы (~ 25 г).
[0302] Оценивали время выдерживания как для распыляемого раствора, так и для влажной дисперсии, перед вторичной сушкой. Общее количество примесей, обнаруженных методом ВЭЖХ, для выдерживаемого распыляемого раствора и выдерживаемой влажной твердой дисперсии, представлено в таблице 11. Как распыляемый раствор, так и влажную твердую дисперсию, выдерживали при комнатной температуре в течение времени, указанного в таблице. Данные по стабильности распыляемого раствора показали рост примесей приблизительно на 0,05% в день. Данные по стабильности влажной твердой дисперсии показали рост примесей приблизительно на 0,02% в день.
Таблица 11
[0303] Высушенную твердую дисперсию охарактеризовывали методами порошкового рентгеноструктурного анализа (XRPD), модулированной дифференциальной сканирующей калориметрии (mDSC), сканирующей электронной микроскопии (SEM), титрованием воды по Карлу Фишера (KF), определением остаточного растворителя, размера частиц, насыпной плотности/насыпной плотности после уплотнения и высокоэффективной жидкостной хроматографией (HPLC) (хиральной и ахиральной). На фигуре 6 приведена порошковая рентгенограмма твердой дисперсии. На дифрактограмме отсутствуют кристаллические пики, что указывает на то, что твердая дисперсия является по природе своей аморфной. Измерение массовых процентов воды и остаточного растворителя показало, что твердая дисперсия содержала 0,75 массовых процента H2O, и содержание остаточного ацетона составляла величину ниже LOQ (<200 ppm).
[0304] Температуру стеклования твердой дисперсии измеряли методом модулированной дифференциальной сканирующей калориметрии (mDSC). Полученные данные показывают, что Tg составляет приблизительно 86°C. Наблюдался только один тепловой эффект (Tg), что позволяет предположить, что в результате распылительной сушки получали гомогенный стекловидный раствор. Анализ методом сканирующей электронной микроскопии (фигура 7) показал при увеличении в 500 раз (слева) и в 5000 раз (справа), что морфология твердой дисперсии состояла из целых и разрушенных сфер с гладкими поверхностями. Присутствие кристаллического материала в образце не обнаруживалось.
[0305] Как показано на фигуре 8, для определения распределения частиц по размерам было проведено исследование порошка твердой дисперсии методом рассеяния лазерного излучения. Использовали анализатор размера сухих частиц Malvern Aero S с высотой бункера 3 мм при давлении 0,7 бар и скорости подачи 40%. Образцы анализировали в трех экземплярах в течение 10 секунд. Для анализа использовали показатель преломления 1,681 и плотность 0,5 г/мл с применением несферического алгоритма. Поглощение света образцом составляло от 0,1 до 15%. Определения распределения частиц по размерам показало, что твердая дисперсия характеризуется мономодальным распределением и что средняя величина d10 составляет 4,9 мкм, средняя величина d50 составляет 15,6 мкм, средняя величина d90 составляет 41,5 мкм.
[0306] Кроме того, измеряли насыпную плотность и насыпную плотность после уплотнения. Было обнаружено, что насыпная плотность составляет 0,25 г/мл, а насыпная плотность после уплотнения составляет 0,41 г/мл. Рассчитывали сыпучесть с выражением результата через индекс Карра, равный 44,35, и отношение Хауснера, равное 1,80.
[0307] Проводили анализ твердой дисперсии методом высокоэффективной жидкостной хроматографии (HPLC), и было обнаружено, что твердая дисперсия содержит 24,6% соединения формулы (I). На фигуре 9 представлены хроматограммы твердой дисперсии (верхняя хроматограмма), соединения формулы (I) (средняя хроматограмма) и холостого опыта (нижняя хроматограмма). По данным, полученным методом HPLC, соединение формулы (I) и твердая дисперсия характеризовались минимальными различиями в содержании примесей. Кроме того, проводили анализ твердой дисперсии на хиральную чистоту, который показал, что энантиомерная чистота соединения формулы (I) составляет 99,8%.
[0308] Изучали влияние относительной влажности на величину Tg для твердой дисперсии. Твердую дисперсию подвергали воздействию 33%, 50% или 75% влажности в течение 24 часов. После воздействия, образцы помещали в герметично закрытые тигли для анализа методом дифференциальной сканирующей калориметрии. Термограммы каждого образца приведены на фигуре 10. Дисперсии характеризовались падением величины Tg при увеличении относительной влажности, по-видимому, вследствие гигроскопичности материала. Было показано, что при воздействии 33% и 75% относительной влажности, величина Tg падала до 71°C и 51°C, соответственно. Факт влияния влажности (содержания воды) на температуру стеклования твердой дисперсии позволяет предположить, что молекулярная подвижность теоретически возможна при комнатной температуре (25ºC), когда влажность составляет > 33%, что делает возможным для соединения формулы (I) образовывать зародыши кристаллизации и соответственно кристаллизоваться. Твердую дисперсию расфасовывали в упаковку для длительного хранения, в которой поддерживалась относительная влажность <33%.
[0309] Адсорбцию воды твердой дисперсией определяли методом динамической сорбции паров (DVS). Для определения адсорбции/десорбции воды, дисперсии подвергали воздействию линейно изменяющейся влажности от 5 до 95% и затем уменьшающейся влажности от 95 до 5%. Результаты анализа методом DVS показывают, что твердая дисперсия является относительно негигроскопичной и адсорбирует до 5,5 масс.% H2O при относительной влажности 90%.
[0310] Образец остающегося материала, собранный из камеры распылительной сушилки, был проанализирован на морфологию частиц методом сканирующей электронной микроскопии (SEM ), на аморфный характер методом порошкового рентгеноструктурного анализа (XRPD) и на наличие примесей методом высокоэффективной жидкостной хроматографии (HPLC). Все испытания показали эквивалентность остающегося в камере материала и собираемой в результате распылительной сушки твердой дисперсии, что указывало на то, что остающийся в камере материал и собираемая в результате распылительной сушки твердая дисперсия могут быть объединены.
[0311] Твердую дисперсию расфасовывали в двойные пакеты из полиэтилена низкой плотности (LDPE) и помещали в бочку из полиэтилена высокой плотности (HDPE). Партии по 2 г твердой дисперсии расфасовывали в двойные пакеты из полиэтилена низкой плотности, горловины которых затягивают с помощью жгута. Упакованные в пакеты образцы хранили в бутылях из полиэтилена высокой плотности объемом 75 см3, закрытых завинчивающимися крышками. Затем проводили испытания стабильности упакованных твердых дисперсий в условиях, соответствующих рекомендациям Международной конференции по гармонизации технических требований к регистрации лекарственных препаратов для медицинского применения (ICH). После трех месяцев хранения при 2-8°C, 25°C/60% RH или 40°C/75% RH не наблюдалось значимых изменений внешнего вида, результатов анализа, родственных веществ, аморфного характера или результатов термического анализа. При 40°C/75% RH, влажность увеличилась с 0,75% при T0 до 2,42% через 3 месяца. Предполагается, что добавление осушающего вещества в упаковку должно предотвратить это увеличение влажности. Данные о стабильности, собранные для твердой дисперсии, сведены в таблице 12.
Таблица 12
2-8°C
25°C, 60% RH
40°C, 75% RH
[0312] Пример 8. Приготовление твердых дисперсий.
[0313] Соединение формулы (I) (2,25 кг) смешивали с ацетоном (103,5 кг) в течение 15 минут при комнатной температуре до тех пор, пока не образовывался прозрачный раствор. Добавляли полимер HPMCAS-H (6,75 кг), и полученную смесь перемешивали в течение 8 часов и выдерживали в условиях окружающей среды в течение ночи. Полученный раствор сушили распылением с использованием распылительной сушилки Anhydro MS-150, снабженной распылительными системами с двухлоточной форсункой. Следующие параметры распыления использовали и контролировали на протяжении всего процесса распылительной сушки: температура на входе - 76,0±20,0°C; температура на выходе - 42,0±5,0°С; температура чиллера - -30,0°C; температура конденсатора - -17,0±10,0°С; скорость подачи раствора - 10,0±2,0 кг/час; расход сушильного газа - 170,0±20,0 кг/час; давление распыления - 0,25±0,05 МПа; давление в камере - 34 см.вод.ст.; ΔP в циклоне - 150 мм.вод.ст.; и ΔP в пылеуловительной камере с тканевыми фильтрами - 70 мм.вод.ст. После распылительной сушки, твердую дисперсию загружали на лотки и сушили при 40°C в течение приблизительно 14 часов. Суммарно извлекали 8,2 кг сухой твердой дисперсии, 1,3 кг из которых было собрано из остающейся в камере твердой дисперсии, суммарный выход составил 92%. Твердую дисперсию расфасовывали в двойные пакеты из полиэтилена низкой плотности (LDPE) толщиной 102 мкм вместе с двадцатью пакетами по 5 г осушающего вещества силикагеля, помещенными между пакетами, и затем помещали в бочку из полиэтилена высокой плотности (HDPE) объемом 57 л и хранили при 2-8°C.
[0314] Твердую дисперсию расфасовывали партиями по 2 грамма в двойные пакеты из полиэтилена низкой плотности (LDPE), горловины которых затягивали с помощью жгута, с 0,5 граммами осушающего вещества, добавленными между пакетами из полиэтилена низкой плотности. Каждый образец хранили в бутылях из полиэтилена высокой плотности (HDPE) объемом 75 см3, закрытых завинчивающимися крышками. Образцы хранили в течение трех месяцев при 2-8°C, при 25°C и относительной влажности 25% или при 40°C и относительной влажности 75%, затем проводили оценку стабильности твердой дисперсии. Никаких значительных изменений, кроме небольшого увеличения результата анализа на содержание воды, не наблюдалось ни при каких условиях с нулевого момента времени. При проведении порошкового рентгеноструктурного анализа (XRPD) кристаллизации не обнаруживали.
[0315] Эту партию твердой дисперсии оценивали путем сравнения с характеристиками, одобренными соответствующими государственными органами. Анализ включал внешний вид, чистоту, определяемую методом HPLC, хиральную чистоту, определяемую методом HPLC, содержание воды по методу Карла Фишера (KF), содержание остаточного растворителя, данные порошкового рентгеноструктурного анализа (XRPD) и данные модулированной дифференциальной сканирующей калориметрии. Результаты этой оценки представлены в таблице 13.
Таблица 13
2-8°C
25°C, 60% RH
40°C, 75% RH
[0316] Пример 9. Оценка загрузки твердых веществ при распылительной сушке.
[0317] Оценивали вязкость раствора и склонность к осаждению композиций соединения формулы (I) и HPMCAS-H в различных композициях растворителей. Как показано в таблице 14, было обнаружено, что вязкость в ацетоне возрастает по мере увеличения загрузки твердых веществ. Раствор 97:3 ацетон/H2O характеризовался такой же вязкостью, что и чистый ацетон при загрузке 14 масс.% твердых веществ. Эти величины указывают, что каждая из этих композиций может использоваться в качестве распыляемых растворов с применением стандартных насосов для перекачки растворов.
Таблица 14
[0318] Оценивали визуально склонность к осаждению трех растворов. Никакого осаждения соединения формулы (I) или полимера не обнаруживалось через три дня при комнатной температуре. Через три дня распыляемый раствор выглядел слегка мутным из-за HPMCAS-H и увеличения вязкости по мере растворения полимера.
[0319] Приготавливали композицию соединения формулы (I) с химически чистым ацетатсукцинатом гидроксипропилметилцеллюлозы (HPMCAS-H) в соотношении 1:3 (то есть, при 25% загрузке лекарственного средства). Проводили распылительную сушку в масштабе 100 г с использованием 12 масс.% твердых веществ в смеси 97/3 ацетон/H2O, и проводили вторичную сушку с использованием конвекционной полочной сушилки. Характеризацию твердой дисперсии проводили с использованием методов XRPD, SEM, mSDC, GC-HS, ахиральной ВЭЖХ и измерения размера частиц. Кроме того, была проведена оценка стабильности распыляемого раствора и стабильности влажной твердой дисперсии.
[0320] В колбу с магнитной мешалкой загружали 97/3 ацетон/H2O (733,3 г), и затем соединение формулы (I) (25 г). После перемешивания при комнатной температуре, получали прозрачный раствор, что указывало на полное растворение соединения формулы (I). Затем к раствору добавляли полимер HPMCAS-H (75 г) и перемешивали при комнатной температуре до получения прозрачного раствора.
[0321] Распылительную сушку для удаления растворителя ацетон/H2O с получением аморфной твердой дисперсии проводили на установке для распылительной сушки Buchi B-290, снабженной двухлоточной форсункой. Распыление проводили с рециркуляцией с использованием высокоэффективного циклона.
[0322] Выход влажной твердой дисперсии составлял 93% (без учета образцов раствора, взятых для проведения анализа на стабильность). Для удаления остаточного ацетона и воды, оставшихся после распылительной сушки, проводили вторичную сушку в полочной сушилке. Заданная температура сушилки составляла 40°C, и твердую дисперсию сушили в течение 24 часов. После сушки получали сухую твердую дисперсию с выходом 79% без учета образцов раствора, взятых для проведения анализа на содержание остаточного растворителя.
[0323] Твердая дисперсия характеризовалась быстрым удалением ацетона при 40°C. Сушку завершали приблизительно через 5 часов после загрузки в сушилку. Твердая дисперсия соответствовала пределу 5000 ppm для ацетона, рекомендуемого Международной конференцией по гармонизации технических требований к регистрации лекарственных препаратов для медицинского применения (ICH Option 1). Содержание воды после сушки в течение 24 часов составляло 0,74 масс.%.
[0324] Во процессе проведения распылительной сушки собирали дополнительную информацию о твердой дисперсии, для того чтобы определить время выдерживания как для распыляемого раствора, так и для влажной твердой дисперсии, перед вторичной сушкой. В таблице 15 приведены сумма примесей в распыляемом растворе и во влажной твердой дисперсии после выдерживания при комнатной температуре в течение 7 дней. Влажная твердая дисперсия содержала ~ 21 000 ppm ацетона.
Таблица 15
[0325] Данные по стабильности распыляемого раствора не показали изменения содержания примесей. На основании этих данных был сделан вывод о том, что распыляемый раствор ацетон/H2O можно использовать (то есть использовать распылительную сушку) через семь (7) дней после его приготовления при выдерживании при комнатной температуре. Данные по стабильности влажной твердой дисперсии не показали роста содержания примесей в течение семи (7) дней. На основании этих данных был сделан вывод о том, что влажную твердую дисперсию можно при необходимости выдерживать в течение нескольких дней при комнатной температуре.
[0326] Проводили также оценку вероятности кристаллизации влажной твердой дисперсии методами порошкового рентгеноструктурного анализа (XRPD, и модулированной дифференциальной сканирующей калориметрии (mDSC) . На фигуре 11 представлена порошковая рентгенограмма твердой дисперсии после хранения в течение семи дней при комнатной температуре. Как следует из порошковой рентгенограммы, влажная твердая дисперсия оставалась аморфной после 7 дней хранения без признаков кристаллизации. Как указано в таблице 15, было обнаружено, что в течение периода выдерживания величина Tg находилась в диапазоне от 58 до 62°C, и никаких событий кристаллизации не наблюдалось.
[0327] Высушенную твердую дисперсию охарактеризовывали методами порошкового рентгеноструктурного анализа (XRPD), модулированной дифференциальной сканирующей калориметрии (mDSC), сканирующей электронной микроскопии (SEM), определения воды по Карлу Фишеру (KF), определения остаточного растворителя, размера частиц и ахиральной высокоэффективной жидкостной хроматографии (HPLC). На фигуре 12 приведена порошковая рентгенограмма твердой дисперсии. На дифракционной картине отсутствуют кристаллические пики, и, поэтому, твердая дисперсия является по своей природе аморфной.
[0328] Измерение массовых процентов воды по Карлу Фишеру (KF) и остаточного растворителя показало, что твердая дисперсия содержала 0,75 массовых процента H2O, и содержание остаточного ацетона составляла величину ниже LOQ (<200 ppm).
[0329] Методом модулированной дифференциальной сканирующей калориметрии (mDSC) было обнаружено, что величина Tg твердой дисперсии составляет приблизительно 84°C. Наблюдался только один тепловой эффект (Tg), что позволяет предположить, что в результате распылительной сушки получали гомогенный стекловидный раствор. Анализ методом сканирующей электронной микроскопии (SEM) показал, что морфология твердой дисперсии состояла из целых и разрушенных сфер с гладкими поверхностями. Присутствие кристаллического материала в образце не обнаруживалось.
[0330] Как показано на фигуре 13, для определения распределения частиц по размерам было проведено исследование порошка твердой дисперсии методом рассеяния лазерного излучения. Использовали анализатор размера сухих частиц Malvern Aero S с высотой бункера 3 мм при давлении 0,7 бар и скорости подачи 40%. Образцы анализировали в трех экземплярах в течение 10 секунд. Для анализа использовали показатель преломления 1,681 и плотность 0,5 г/мл с применением несферического алгоритма. Поглощение света образцом составляло от 0,1 до 15%. Определения распределения частиц по размерам показало, что твердая дисперсия характеризуется мономодальным распределением и что средняя величина d10 составляет 1,9 мкм, средняя величина d50 составляет 9,8 мкм, и средняя величина d90 составляет 26,6 мкм. При исследовании распределения, диапазон размера частиц ограничивали частицами с размером не более 200 мкм, в силу того, что агломераты крупных частиц с размером ~ 1 мм не диспергировались в анализаторе размера частиц.
[0331] Методом высокоэффективной жидкостной хроматографии (HPLC) была определена концентрация соединения формулы (I) в твердой дисперсии, которая составляла 25,6%. Соединение формулы (I) и твердая дисперсия характеризовались минимальным различием в содержании примесей (0,90% и 0,92%, соответственно), определенном методом высокоэффективной жидкостной хроматографии (HPLC).
[0332] Пример 10. Приготовление таблеток.
[0333] Приготавливали партию твердой дисперсии массой 80 г, содержащей смесь 1:3 соединение формулы (I) : HPMCAS-H, в соответствии с общими методами, описанными в примерах 3 и 8. Приготавливали три вида таблетки лекарственного препарата из этой твердой дисперсии на прессе с одним пуансоном с использованием технологических приемов сухой грануляции, измельчения, смешивания и прессования. Таблетки изготавливали при 32,00% загрузки твердой дисперсии (8% загрузки активного вещества) с получением таблеток с 40 мг активного вещества, прессованных с помощью модифицированной капсульной оснастки (~ 16,499 мм x 8,498 мм) при суммарной массе 500 мг. В таблицах 16, 17 и 18 приведены композиции трех видов таблетки.
Таблица 16
HPMCAS-H
Таблица 17
HPMCAS-H
Таблица 18
HPMCAS-H
[0334] Каждый состав сначала прессовали до целевой твердой фракции (~ 0,7) плоских гранул размером приблизительно 13 мм. Гранулы измельчали через сито с размером отверстий 30 меш. Добавляли экстрагранулярные вспомогательные вещества, и полученную смесь прессовали в таблетки. Использовали величину давление приблизительно 90 МПа для достижения целевой твердости 15 килопонд (КП) для композиций, приведенных в таблица 16 и 18. Аналогичную твердость достигали для композиции, приведенной в таблице 17 при использовании усилия прессования приблизительного 150 МПа. Распадаемость таблеток, приготовленных из композиций, приведенных в таблицах 16 и 18, происходила менее чем за 1 минуту, между тем, как время распадаемости таблеток, приготовленных из композиции, представленной в таблице 17, составляло приблизительно 1,5 минуты.
[0335] Приготавливали дополнительные таблетки с твердостью приблизительно 10 КП, и подвергали их испытанию на растворение с использованием прибора с лопастями (прибора II), рекомендуемого Фармакопей США, в 0,1 н. HCl (900 мл) при 37°C. Скорость вращения лопастей поддерживали на уровне 75 об/мин в течение первых 60 минут, затем увеличивали до 250 об/мин в течение последних 15 минут испытания. В каждый момент времени отбирали образцы по 10 мл и фильтровали их через полиэтиленовый фильтр с размером пор 10 мкм, затем разбавляли и проводили анализ методом ВЭЖХ. Испытания на растворение не показали различий в профилях высвобождения между этими тремя композициями.
[0336] Пример 11. Приготовление и характеризация таблеток.
[0337] Приготавливали дополнительные таблетки с использованием композиций, описанных в таблицах 16 и 18, для оценки их стабильности, фармакокинетики (PK) и растворения в среде, с выбранными оптимальными характеристиками. Использовали общий метод приготовления, описанный в примере 10. Прессованные таблетки визуально были белого цвета. Использовали фракцию твердых гранул 0,7 и целевую твердость таблеток 14 KP (прочность на разрыв 1,61 MPa). После прессования, оценивали содержание действующего вещества и сумму примесей в таблетках методом высокоэффективной жидкостной хроматографии (HPLC) (таблица 19).
Таблица 19
[0338] Ожидаемое содержание действующего вещества таблеток подтверждали методом высокоэффективной жидкостной хроматографии (HPLC). Не было обнаружено заметного различия между композициями в сумме примесей.
[0339] Проводили испытание растворимости кристаллического соединения формулы (I), а также твердой дисперсии 1:3 соединение формулы (I) : HPMCAS-H, в типичной среде для растворения при pH 2 и рН 6,8. Рассчитывали фактор разбавления, используя объем 900 мл среды наряду с растворимостью и дозой действующего вещества в таблетка, и эти данные приведены в таблице 20.
Таблица 20
*потенциальные резко выделяющиеся значения
[0340] Оценивали фармакокинетику соединения формулы (I) в двух различных композициях таблеток (таблица 21), композиции таблеток 1 (приведенной в таблице 16) и композиции таблеток 3 (приведенной в таблице 18). Каждую композицию вводили группе самцов собак натощак (n=4/группа) (то есть, каждая собака получала разовую пероральную дозу, содержащую 40 мг соединения формулы (I)). Через определенные интервалы времени после введения таблеток, собирали образцы крови для анализа концентраций соединения формулы (I) в плазме с течением времени. В целях сравнения, фармакокинетику для композиции таблетки 1 и 3 сравнивали с фармакокинетикой для твердой дисперсии (1 : 3 соединение формулы (I) : HPMCAS-H), вводимой в форме суспензии.
Таблица 21
[0341] Оценки средних значений фармакокинетических параметров приведены в таблице 21. Для сравнения между группами использовали величину AUC0-t (площадь под кривой зависимости концентрации в плазме от времени, от нулевого момента времени до последнего момента времени сбора данных) соединения формулы (I). Нормализованные по дозе средние значения AUC0-t были аналогичны композиции суспензии 1 : 3 соединение формулы (I) : HPMCAS-H (в пределах 20%), 10,8±3,2 и 8,9±1,4 (мкг*ч/мл)/(мг/кг) для композиции таблетки 1 и композиции таблетки 3, соответственно. Было обнаружено, что нормализованное по дозе среднее значение Cmax в плазме одинаково для композиций двух таблеток и немного выше, чем для композиции суспензии. Наблюдаемые значения tmax позволяют предположить, что композиция таблетки 1 всасывается быстрее, чем композиция таблетки 3.
[0342] Композиции таблеток 1 и 3 помещали в условия для ускоренного исследования стабильности (40°C/75% RH) на открытом воздухе и проводили испытание на химическую чистоту и содержание действующего вещества через 2 и 4 недели. В условиях для проведения ускоренного исследования, для композиции таблетки 1 наблюдалось снижение содержание действующего вещества таблетки с 99,3% до 96,7%, в то время как сумма примесей увеличивалось с 1,24% в нулевой момент времени до 2,02% через 4 недели. Для композиции таблетки 3 снижение содержание действующего вещества в таблетке составляло от 100,1% до 96,2%, в то время сумма примесей увеличилось с 1,25% в нулевой момент времени до 1,77% через 4 недели.
[0343] Пример 12. Приготовление и характеризация композиции таблеток.
[0344] Заранее приготавливали композицию таблетки 1, 40 мг активного вещества, с фракцией твердых веществ 0,70 и твердостью таблетки 14,1 КП, прочностью на разрыв 1,61 МПа (смотрите примеры 10 и 11). Эта таблетка характеризовалась временем распадаемости 33 секунды. Приготавливали дополнительные таблетки с пониженным содержанием твердой фракции (гранул) 0,6 для обеспечения дополнительной прессуемости таблетки. Затем гранулы прессовали до достижения твердости таблеток в диапазоне от 11,6 до 39,7 KP (прочности на разрыв 1,2-4,7 МПа), и измеряли распадаемость. Время распадаемости для этих таблеток составляло приблизительно 30 секунд. Затем композицию модифицировали путем удаления всех экстрагранулярных компонентов, ac-di-sol и маннита, которые заменяли на микрокристаллическую целлюлозу (MCC). Затем таблетку прессовали до достижения твердости 20,7 КП (прочности на разрыв 2,1 МПа), и было обнаружено, что распадаемость составляла 27 секунд. Сама по себе интрагранулярная смесь также была подвергнута прессованию до достижения твердости 17,9 КП (прочности на разрыв 2,1 МПа). Время распадаемости самой по себе интрагранулярной смеси составляло 26 секунд.
[0345] Поскольку небольшие изменения в композиции не приводили желаемому результату, и прессование самой по себе интрагранулярной смеси приводило к очень быстрой распадаемости, внимание было обращено на исследование влияния замены сорта микрокристаллической целлюлозы (MCC) PH101 на PH105 на время распадаемости. Это замена могла бы обеспечить более точное соответствие между размером частиц твердой дисперсии и основной добавкой для прессования, микрокристаллической целлюлозой (MCC), и, в результате, обеспечить более тесный контакт с частицами, позволяя увеличить связывание во время вальцевания.
[0346] Затем таблетки с содержанием активного вещества 40 мг, в которых микрокристаллическую целлюлозу сорта PH101 в композиции таблетки 1 заменяли на микрокристаллическую целлюлозу сорта PH105, прессовали (из гранул с твердой фракцией 0,59) до твердости 27 КП. Было обнаружено, что полученные таблетки характеризуются временем распадаемости 1:40 мин и истираемостью 0,03%. С целью увеличении времени распадаемости, таблетки с содержанием активного вещества 10 мг также подвергали прессованию, и было обнаружено, что они характеризуются твердостью 10 КП и временем распадаемости 2:21 мин.
[0347] Приготавливали обычную смесь композиции таблетки 4, имеющую состав, представленный в таблице 22, затем прессовали в таблетки с содержанием активного вещества 10 мг и 40 мг . Выбранная для прессования оснастка представляла собой стандартную вогнутую овальную форму с диаметром 6,4 мм для таблетки с содержанием активного вещества 10 мг и модифицированную овальную форму 7 мм x 14 мм ля таблетки с содержанием активного вещества 40 мг. Приготавливали обычную смесь в количестве приблизительно 1,25 кг с использованием 480 г твердой дисперсии. Эту обычную смесь разделяли на порции для прессования, как таблеток с содержанием активного вещества 10 мг, так и таблеток с содержанием активного вещества 40 мг. Композиция таблеток с содержанием активного вещества 10 мг и 40 мг приведена в таблице 22. Композиция таблеток с нанесенным покрытием с содержанием активного вещества 40 мг приведена в таблице 23.
Таблица 22
10 мг
(мг)
40 мг
(мг)
HPMCAS-H
Таблица 23
40 мг
(мг)
[0348] Основное оборудование, используемое для гранулирования и приготовления таблеток, представляло собой бункерный смеситель с технологической емкостью объемом 5 л, сито 40 меш, мельницу Quadro Comil U5, роликовый пресс Vector TFC Lab-Micro и ротационный пресс для таблетирования Piccola-B. Вкратце, процесс гранулирования и приготовления таблеток включает смешивание, предотвращение комкообразования и роликовое прессование интрагранулярных компонентов, смешивание и предотвращение комкообразования экстрагранулярных компонентов, и прессование таблеток с содержанием активного вещества 10 и 40 мг. Блок-схема процесса приведена на фигуре 14.
[0349] Более подробно, технологический процесс включал следующие стадии: (1) на внутреннюю поверхность технологической емкости смесителя предварительно наносили слой микрокристаллической целлюлозы (в течение 1 мин при 20 об/мин); (2) в смесителе с нанесенным слоем покрытия смешивали интрагранулярные ингредиенты (в течение 5 мин при 20 об/мин); (3) измельчали интрагранулярные ингредиенты (Screen 032R, 4000 об/мин); (4) смешивали интрагранулярные ингредиенты в смесители (в течение 15 мин при 20 об/мин); (5) в смеситель добавляли просеянный через сито 40 меш стеарат магния, и перемешивали (в течение 4 мин при 20 об/мин); (6) смесь гранулировали с использованием роликового пресса (скорость шнека 40 об/мин, скорость ролика 3 об/мин, давление 10 МПа); (7) сухой гранулированный материал пропускали через мельницу (Screen 050G, 2000 об/мин); (8) измельченные гранулы переносили в смеситель, добавляли экстрагранулярные ингредиенты, за исключением стеарата магния, и перемешивали (в течение 10 мин при 20 об/мин); (9) смесь пластифицировали путем добавлением просеянного через сито 40 меш стеарата магния, затем перемешивали (в течение 4 мин при 20 об/мин); (10) прессовали таблетки (с содержанием активного вещества 10 мг и 40 мг) с использованием таблеточного пресса с соответствующими приспособлениями (в процессе прессования контролировали массу, толщину и твердость).
[0350] Приготавливали обычную смесь в количестве 1,25 кг с использованием вальцевания для достижения насыпной плотности на ленты приблизительно 0,8 г/мл (~ 0,6 твердой фракции). Обработку проводили при комнатной температуре (приблизительно 25,2°C) и относительной влажности 26%. Измеряли, помимо размера частиц гранул после измельчения, насыпную плотность и насыпную плотность после уплотнения (таблица 24).
Таблица 24
[0351] Интрагранулярная смесь характеризовалась самой низкой насыпной плотностью и самой высокой величиной отношения Хауснера. После гранулирования, насыпная плотность повышалось до 0,5 г/мл, а отношение Хауснера снижалось, что свидетельствовало об улучшении сыпучести. Добавление экстрагранулярных наполнителей не изменяло объемных характеристик гранул, но немного улучшало сыпучесть, на что указывало более низкая величина отношения Хауснера.
[0352] Распределение частиц по размерам показало, что большая часть гранул (48,7%) задерживалась на ситах с размером отверстий в диапазоне 250-595 мкм, при этом основную массу остального материала собирали в поддоне. Обнаруживали 31% мелких частиц (частиц с размером менее 74 мкм). Суммарный выход составлял 96,1%, всего было собрано 1,2 кг конечной смеси.
[0353] Гранулы разделяли для прессования таблеток с содержанием активного вещества 10 и 40 мг. Таблетки с содержанием активного вещества 40 мг изготавливали с использованием 949,95 г гранул. Описание инструментальной оснастки, а также число позиций, заданная масса содержимого и твердость таблетки приведены в таблице 25.
Таблица 25
(таблетка с содержанием активного вещества 40 мг)
(таблетка с содержанием активного вещества 10 мг)
[0354] Пресс Piccola эксплуатировали со скоростью 20 об/мин при скорости вращения лопастей 5 об/мин. Усилие прессования доводили до 15 кН с предварительным усилием прессования 480-600 Н. Было обнаружено, что сила выталкивания во время прессования составляла 181 Н. В процессе прессования было получено 1736 таблеток с выходом 91,4%. Таблетки в процессе прессования характеризовались диапазоном твердости от 16,9 до 22,8 КП, при этом большая часть таблеток имела твердость приблизительно 20 КП. Толщина таблеток составляла приблизительно 5,7-5,8 мм, а масса таблеток составляла в среднем 500 мг. Испытания на распадаемость показали в среднем время распадаемости 1:37, а истираемость составляла 0,14%.
[0355] Остальные гранулы, приблизительно 257,05 г, использовали для прессования таблеток с содержанием активного вещества 10 мг. Пресс Piccola эксплуатировали со скоростью 20 об/мин при скорости вращения лопастей 4 об/мин. Усилие прессования доводили до 7,5 кН с предварительным усилием прессования 280 Н. Было обнаружено, что сила выталкивания во время прессования составляла 80-89 Н. Всего в процессе прессования было получено 1561 таблетка с выходом 75,9%. Таблетки в процессе прессования характеризовались диапазоном твердости от 8,8 до 10,5 КП, при этом большая часть таблеток имела твердость приблизительно 10 КП. Толщина таблеток составляла приблизительно 3,9 мм, а масса таблеток составляла в среднем 126 мг. Испытания на распадаемость показали в среднем время распадаемости 2:04, а истираемость составляла 0,01%.
[0356] Таблетки охарактеризовывали по внешнему виду, путем идентификации методом высокоэффективной жидкостной хроматографии (HPLC), путем проведения анализа примесей, однородности состава, содержания воды и растворимости. Результаты испытаний представлены в таблице 26.
Таблица 26
[0357] Внешний вид таблеток был белым без видимых вкраплений или пятен, и содержание действующего вещества по данным анализа методом HPLC составляло почти 100%. Сумма примесей была такой же, как и в твердой дисперсии, а однородность состава соответствовала критериям Фармакопеи США (USP). Содержание воды в двух таблетках было одинаковым. Анализ растворения проводили в среде 0,01 н HCl (pH 2) с 0,5% лаурилсульфата натрия (SLS) (900 мл) при 37°C при скорости вращения лопастей мешалки 75 об/мин. Сбор данных проводили через 5, 15, 30, 45 и 60 минут. В каждый момент времени отбирали образец объемом 10 мл и фильтровали через полиэтиленовый фильтр с размером пор 10 мкм, затем разбавляли и проводили анализ методом HPLC. На фигуре 15 представлены кривые растворения для таблеток с содержанием действующего вещества 10 и 40 мг .
[0358] Полное растворение наблюдалось через 15 минут для таблеток с обеими дозами действующего вещества. Стандартное отклонение для всех временных точек составляло 1-3%, и остаточное стандартное отклонение (RSD) также варьировало в диапазоне 1-3%, указывая на то, что растворение было очень схожим для этих таблеток. Профиль растворения соответствует критериям USP/FDA для лекарственных форм с немедленным высвобождением для плохо растворимых в воде лекарственных средств, в случае которых 85% растворение достигается через 30 и/или 45 минут.
[0359] Не расфасованные таблетки упаковывали в двойные пакеты из полиэтилена низкой плотности (LDPE) с осушающим веществом SiO2 между пакетами, и затем помещали в бочку из полиэтилена высокой плотности (HDPE).
[0360] Пример 13. Фармакокинетические исследования таблеток.
[0361] Фармакокинетику при введении таблеток с содержанием действующего вещества 40 мг, описанных в примере 12, оценивали на самцах собак как натощак, так и при приеме корма. Композицию вводили группе самцов собак натощак (n=4/группа) и группе самцов собак, принимавших корм (n=4/группа), при этом каждая собака получала разовую пероральную дозу 40 мг. Через определенные интервалы времени после введения дозы, собирали образцы крови для проведения анализа концентраций в плазме соединения формулы (I) с течением времени. Средние оценки фармакокинетических параметров приведены в таблице 27.
Таблица 27
[0362] При оценке фармакокинетики введения таблетки при приеме пищи и натощак, нормализованные по дозе средние значения AUC0-t были одинаковыми между группами (в пределах 20%), составляя 8,6 и 9,4 (мкг*ч/мл)/(мг/кг), соответственно. Вариабельность воздействия в группе между животными при состоянии сытости была меньше, чем когда таблетку вводили натощак. Нормализованное по дозе среднее значение Cmax в плазме оказалась немного выше в случае введения таблеток натощак. Наблюдаемые значения tmax позволяют предположить, что композиция таблетки быстрее всасывалась в состоянии натощак, при этом состояние натощак характеризуется быстрым значением tmax и немного более высоким значением Cmax. Значения AUC0-t были аналогичными, что указывало на отсутствие значительного влияния присутствия пищи в желудке на фармакокинетику при введении композиции таблетки.
[0363] Пример 14. Стабильность таблеток.
[0364] Оценивали стабильность как таблеток с содержанием активного вещества 10 мг, так и содержанием активного средства 40 мг, описанных в примере 12. Таблетки с содержанием активного вещества 10 мг хранили во флаконах из полиэтилена высокой плотности (HDPE) объемом 30 см3 (30 таблеток в одном флаконе) с 0,5 г осушающего вещества SiO2 и 9 г чистого полиэфирного жгута. Таблетки с содержанием активного вещества 40 мг хранили во флаконах из полиэтилена высокой плотности (HDPE) объемом 60 см3 (30 таблеток в одном флаконе) с 0,5 г осушающего вещества SiO2 и 9 г чистого полиэфирного жгута. Все флаконы герметизировали с помощью фольгированной термозапечатываемой пробки. Флаконы помещали на шесть месяцев в климатические камеры при 2-8°C, 25°C/60% RH и 40°C/75% RH. Краткая сводка результатов хранения таблеток с содержанием активного вещества 10 и 40 мг через 6 месяцев представлена в таблице 28 и таблице 29, соответственно.
Таблица 28
Таблица 29
[0365] Никаких значительных различий в физических или химических свойствах таблеток в течение шести месяцев не обнаруживалась даже в условиях проведения ускоренного испытания на стабильность при хранении.
[0366] Пример 15. Сцинтилляционный анализ сближения (SPA) HIF-2α
[0367] Используемый для анализа общий объем составлял около 100 мкл со следующими компонентами: 2 мкл соединения в 100% DMSO, 88 мкл буфера с белком и зондом и 10 мкл шариков SPA. Соединение разводили в эталонном планшете, включающем 10 точек дозозависимого ответа с 3-кратным разведением соединения от 100 мкМ до 5 нМ. Анализы проводили на 96-луночном планшете, в котором один столбец, обозначенный как контроль с высоким сигналом, содержал DMSO без соединения, а другой столбец, обозначенный как контроль с низким сигналом, не содержал белка. Перед посевом в отсутствии соединения, приготавливали буферный раствор, состоящий из 25 мМ TRIS pH 7,5 (Sigma), 150 мМ NaCl (Sigma), 15% глицерина (Sigma), 0,15% BSA (Sigma), 0,001% Tween-20 (Sigma), 150 нМ N-(3-хлорфенил-4,6-t2)-4-нитробензо[c][1,2,5]оксадиазол-5-амина и 100 нМ HIF-2α домена HIS TAG-PASB, и приводили его в равновесие в течение 30 минут. Затем соединения, которые должны были быть протестированы, помещали в 96-луночный планшет Isoplate-96 SPA с белым прозрачным дном (Perkin Elmer). К соединениям добавляли 88 мкл буферного раствора, затем планшет, покрытый пластиковой крышкой и алюминиевой фольгой, помещали в шейкер и приводили в равновесие в течение 1 часа. После достижения равновесия, в каждую лунку планшета добавляли 10 мкл 2 мг/мл раствора SPA-шариков, меченых с помощью YSi Cu His (Perkin Elmer), накрывали крышкой и приводили в равновесие в течение еще 2 часов. Затем планшеты вынимали из шейкера, помещали в жидкостной сцинтилляционный счетчик 1450 LSC и люминесцентный счетчик MicroBeta Trilux (Perkin Elmer) для измерения степени смещения зонда. Определяли процент ингибирования и рассчитывали значения IC50 с использованием системы Dotmatics по следующему уравнению: % ингибирования = [(контроль высокого сигнала - образец)/( контроль высокого сигнала - контроль низкого сигнала)] x 100. Было обнаружено, что соединение формулы (I) имело величину IC50 менее 50 нМ в анализе SPA.
[0368] Пример 16. Иммуноферментный анализ (ELISA) VEGF
[0369] В первый день исследования, около 7500 клеток линии 786-O в 180 мкл среды для культивирования высевали в каждую лунку 96-луночного планшета с белым прозрачным дном (07-200-566, Fisher Scientific). Через четыре часа, проводили последовательные 10-кратные разведения исходных растворов соединения в среде для культивирования из 500-кратных исходных растворов DMSO, и 20 мкл этих 10-кратно разбавленных исходных растворов соединения добавляли в каждую лунку для получения следующих конечных концентраций (мкМ): 20, 6,67, 2,22, 0,74, 0,25, 0,082, 0,027, 0,009, 0,003, 0,001 и 0. Каждую концентрацию соединения добавляли в планшет в двух экземплярах. Примерно через 20 часов, среду удаляли отсасыванием, и в каждую лунку добавляли 180 мкл среды для культивирования. В каждую лунку добавляли около 20 мкл свежеприготовленных 10-кратно разбавленных исходных растворов соединения. Приблизительно через 24 часа, среду для культивирования клеток удаляли, и определяли концентрацию VEGF с использованием набора для проведения анализа ELISA, приобретенного у компании R&D systems, в соответствии с методом, предложенным компанией-производителем. Величину ЕС50 рассчитывали с помощью программного обеспечения GraphPad Prism, используя уравнение доза-ответ-ингибирование (четыре параметра). Планшет с посеянными клетками затем подвергали люминесцентному анализу жизнеспособности клеток CellTiter-Glo (Promega), путем добавления в каждую лунку 50 мкл реагента Celltiter Glo и встряхивания планшета в течение 8 минут при 550 об/мин (Thermomixer R, Eppendorf), затем сигнал люминесценции сразу же считывали в планшет-ридере (задержка 3 секунды, время интегрирования 0,5 секунды/лунка, многоканальный микропланшетный ридер Synergy 2). Было обнаружено, что соединение формулы (I) имеет величину ЕС50 менее 50 нМ в анализе VEGF ELISA.
[0370] Пример 17. Исследования на основе анализа люцифиразной активности
[0371] Получали клетки единичного клона 786-O-Hif-Luc путем инфицирования клеток линии 786-O (ATCC® CRL-1932TM) доступным для использования лентивирусом, который доставляет ген люциферазы, управляемый множеством чувствительных к HIF элементов (репортер Cignal Lenti HIF (luc): CLS- 007L, Qiagen) при величине показателя множественности заражения (MOI) 25, в течение 24 часов. К клеткам добавляли свежую среду (среду Игла, модифицированную Дульбекко (DMEM, D5796, Sigma), дополненную 10% FBS (F6178, Sigma), 100 единицами пенициллина и 100 мкг стрептомицина/мл (P4333, Sigma)) в течение еще 24 часов. Затем отбирали пул инфицированных клеток относительно 2 мкг/мл пуромицина (P8833, Sigma) в течение 10 дней с последующим ограниченным разведением для отбора единичных клонов. Клоны подвергали испытанию на их ответ на воздействие ингибиторов HIF-2, и те, клоны, которые характеризовались самым большим динамическим диапазоном (786-0-Hif-Luc), расширяли и использовали для анализа люциферазы. Для анализа люциферазы около 7500 клеток 786-O-Hif-Luc в 90 мкл среды для культивирования высевали в каждую лунку 96-луночного белого непрозрачного планшета (08-771-26, Fisher Scientific) за день до проведения обработки.
[0372] В день проведения обработки, производили последовательные 10-кратные разведения исходных растворов соединений в среде для культивирования из 500-кратных исходных растворов DMSO, и 10 мкл 10-кратно разбавленных исходных растворов добавляли в каждую лунку для получения следующих конечных концентраций (мкМ): 20, 6,67, 2,22, 0,74, 0,25, 0,08, 0,027, 0,009, 0,003, 0,001 и 0. Каждую концентрацию тестировали в трех экземплярах. Приблизительно через 24 часа, определяли активность люциферазы с использованием реагента ONE-Glo Luciferase Assay Reagent (E6110, Promega) в соответствии с методикой, рекомендованной фирмой-производителем. Величину EC50 рассчитывали с использованием программного обеспечения Dotmatics. Было обнаружено, что соединение формулы (I) имеет величину ЕС50 менее 50 нМ в анализе активности люциферазы.
[0373] Пример 18. Скрининг параметров масштабирования процесса получения высушенной распылением дисперсии (SDD).
[0374] Приготавливали раствор смеси соединение формулы (I) : HPMCAS-H в ацетоне в емкости объемом 50 л с верхним расположением мешалки и смешивали все ингредиенты до тех пор, пока визуально не обнаруживалось образование прозрачного раствора. Композиции раствора и время смешения приведены ниже:
[0375] Получение высушенной распылением дисперсии (SDD) соединения формулы (I) исследовали путем изучения различных технологических параметров и полученных объемных характеристик приготовленного выше раствора. На распылительной сушилке с незамкнутым контуром с производительностью 200 кг/час по осушающему газу исследовали следующие технологические диапазоны: температура на выходе: 35-46°C; скорость подачи раствора: 340-455 г/мин; расчетное относительное насыщение на выходе: 7-11%. В зависимости от различных термодинамических условий и размеров используемых форсунок, обнаруживали полученные в результате диапазоны объемных характеристик: диаметр частиц D (v 0,5): 33-67 мкм; насыпная плотность: 0,16-0,20 г/мл
[0376] Демонстрационная партия
Пример условий перед получением демонстрационной партии представлял собой соотношение L:G, равное 0,121, и температуру на выходе 42°C. Эти условия позволяли получать высушенную распылением дисперсию (SDD) с приемлемой стабильностью, характеристиками и выходом. Для получения высушенной распылением дисперсии 25% соединения формулы (I) : HPMCAS-H были выбраны следующие условия распылительной сушки
[0377] На основе кривой сушки, построенной для демонстрационной партии, были выбраны следующие условия вторичной сушки с использованием конвекционной полочной сушилки.
[0378] Несмотря на то, что в изобретении были продемонстрированы и описаны предпочтительные варианты осуществления настоящего изобретения, тем не менее, для специалистов в данной области является очевидным, что такие варианты осуществления приводятся только в качестве иллюстрации. Для специалистов в данной области очевидно возможность существования многочисленных вариантов, изменений и замен без отклонения от сущности изобретения. Следует иметь в виду, что при применении изобретения на практике могут быть использованы различные альтернативы описанным в изобретении вариантам осуществления изобретения. Предполагается, что представленная далее формула изобретения определяет объем изобретения, и что этот объем изобретения охватывает способы и структуры и их эквиваленты.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ СТАБИЛИЗАЦИИ ИНДУЦИРУЕМОГО ГИПОКСИЕЙ ФАКТОРА-2 АЛЬФА КАК СПОСОБ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2602498C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ СОЕДИНЕНИЯ ПИРАЗОЛА, ДИСПЕРГИРОВАННОГО В МАТРИЦЕ ПОЛИМЕРА | 2020 |
|
RU2826604C1 |
| УЛУЧШЕННЫЙ ТЕРАПЕВТИЧЕСКИЙ ИНДЕКС ИНГИБИТОРОВ ПРОТИВ ИММУННОЙ КОНТРОЛЬНОЙ ТОЧКИ С ИСПОЛЬЗОВАНИЕМ КОМБИНИРОВАННОЙ ТЕРАПИИ, ВКЛЮЧАЮЩЕЙ ЭКСТРАКТ PHY906, ЭКСТРАКТ Scutellaria baicalensis Georgi (S) ИЛИ СОЕДИНЕНИЕ ИЗ ТАКИХ ЭКСТРАКТОВ | 2017 |
|
RU2753523C2 |
| АНТАГОНИСТ РЕЦЕПТОРА CRF1, ЕГО ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ И ТВЕРДЫЕ ФОРМЫ ДЛЯ ЛЕЧЕНИЯ ВРОЖДЕННОЙ ГИПЕРПЛАЗИИ НАДПОЧЕЧНИКОВ | 2019 |
|
RU2824490C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ВВЕДЕНИЕ | 2013 |
|
RU2802442C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ВВЕДЕНИЯ | 2013 |
|
RU2692779C2 |
| СПОСОБЫ УСИЛЕНИЯ СТАБИЛИЗАЦИИ ИНДУЦИРУЕМОГО ГИПОКСИЕЙ ФАКТОРА-1 АЛЬФА | 2010 |
|
RU2521251C2 |
| СПОСОБ УМЕНЬШЕНИЯ МУЛЬТИЛЕКАРСТВЕННОЙ РЕЗИСТЕНТНОСТИ С ИСПОЛЬЗОВАНИЕМ ТРИПИРОФОСФАТА ИНОЗИТА | 2010 |
|
RU2563127C2 |
| НАНОЧАСТИЦА, СОДЕРЖАЩАЯ РАПАМИЦИН И АЛЬБУМИН, В КАЧЕСТВЕ ПРОТИВОРАКОВОГО АГЕНТА | 2008 |
|
RU2483714C2 |
| Фармацевтические комбинации | 2017 |
|
RU2759669C2 |

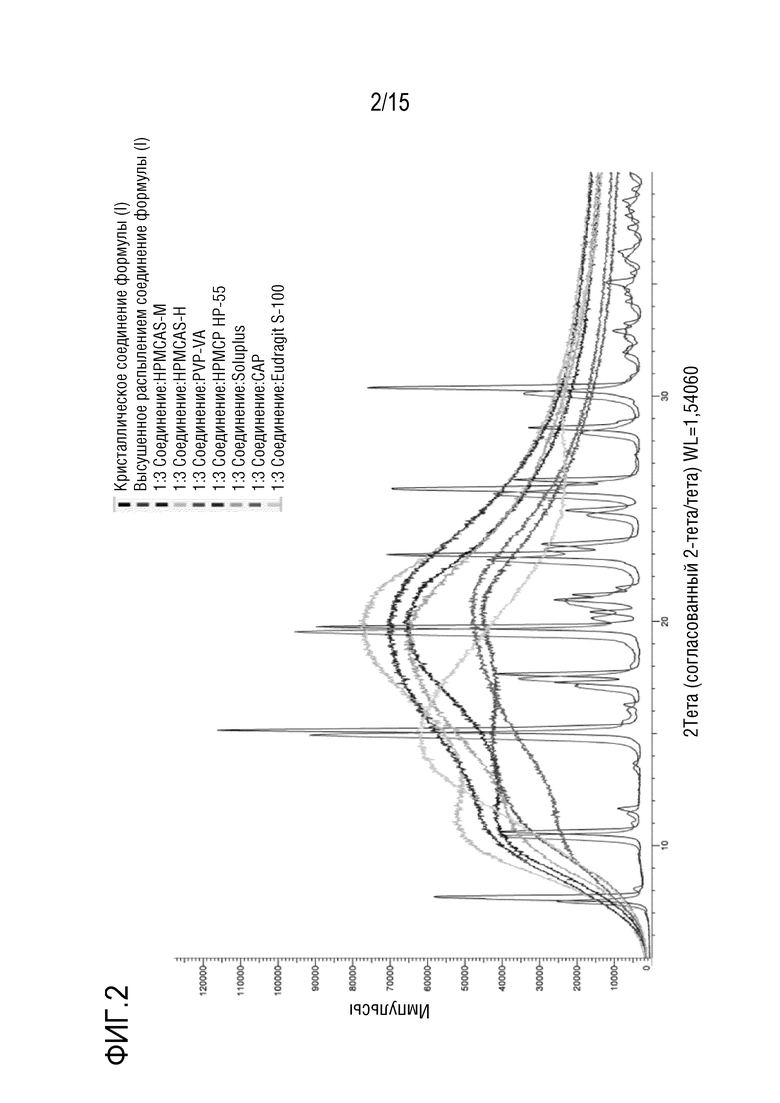
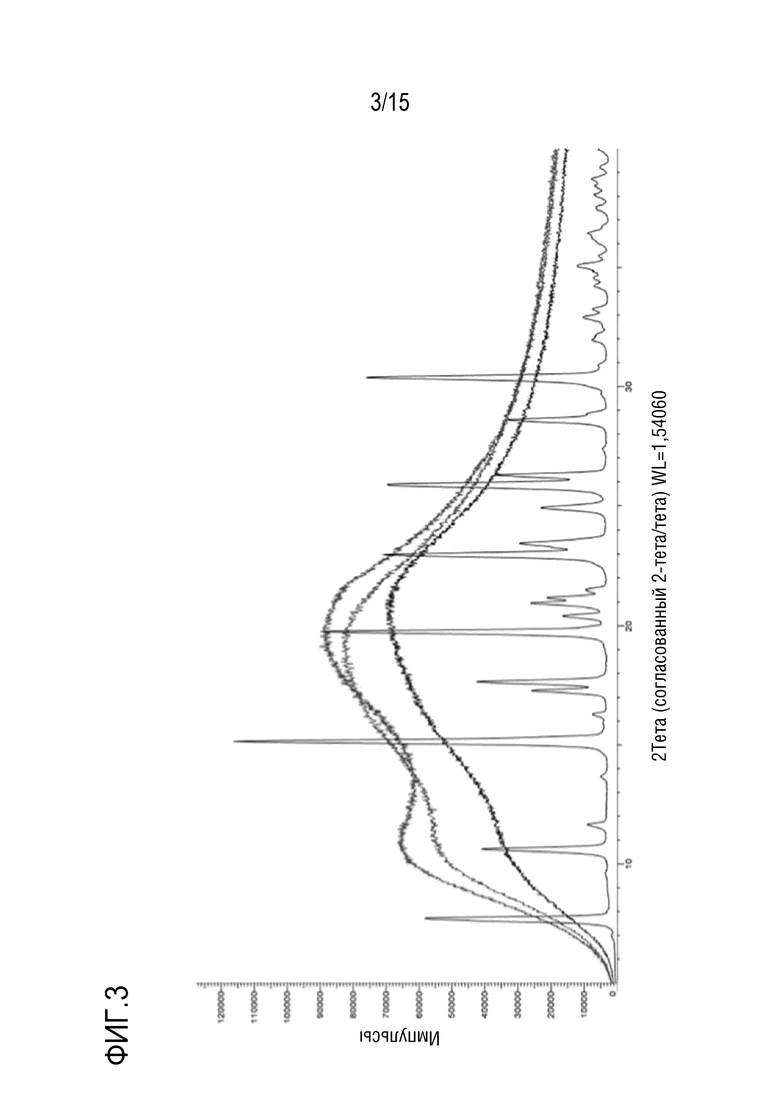

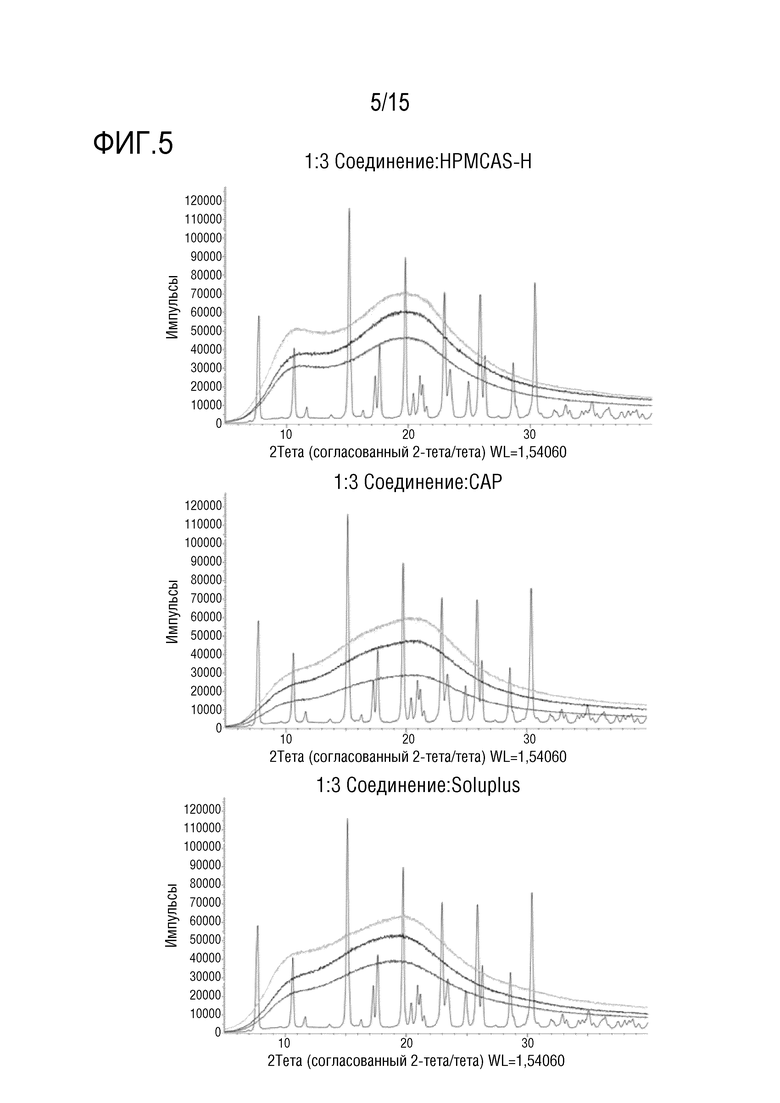
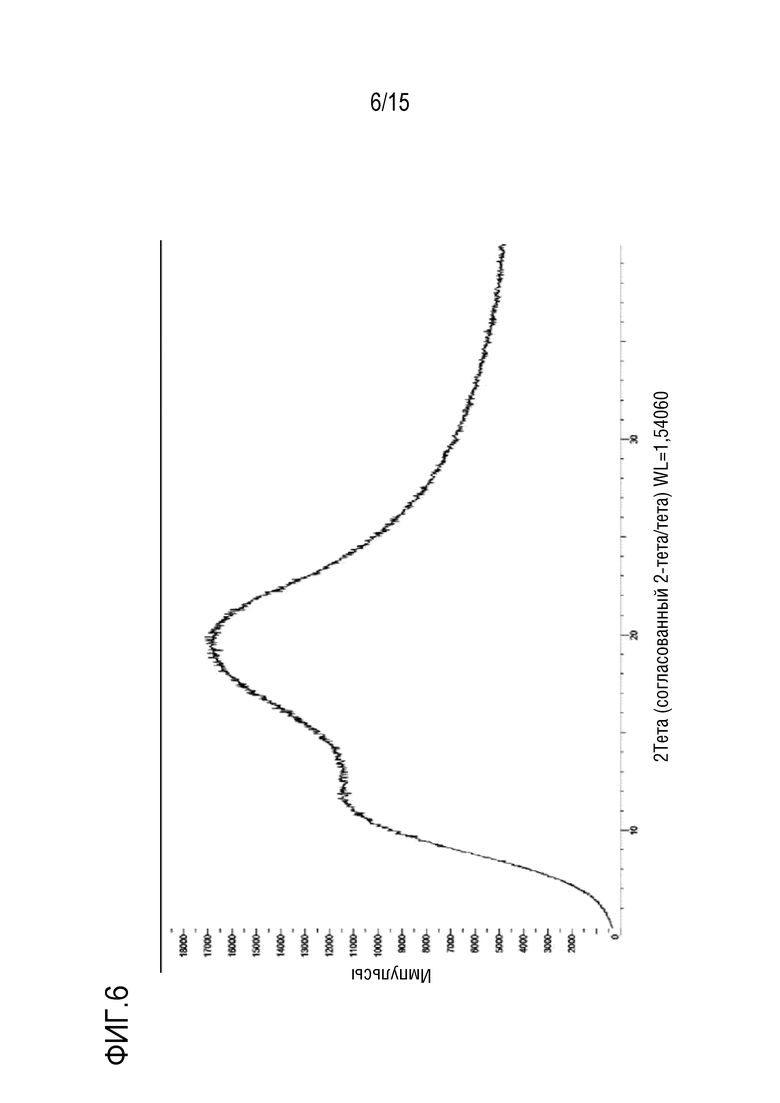
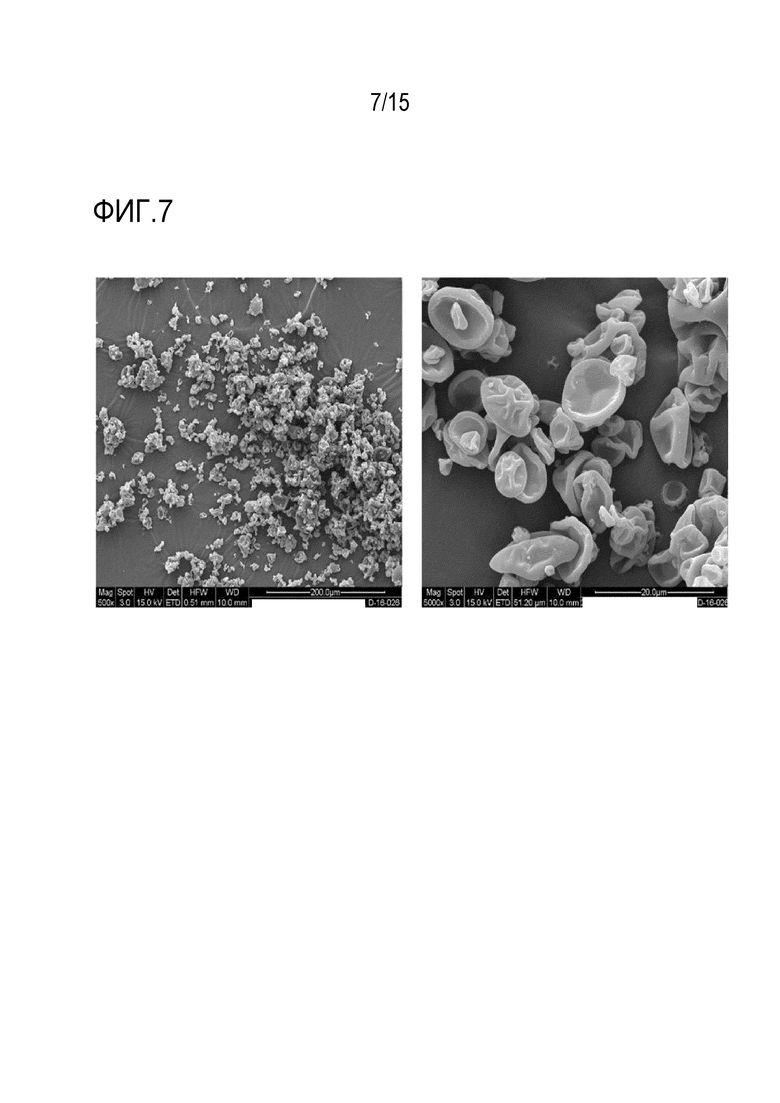

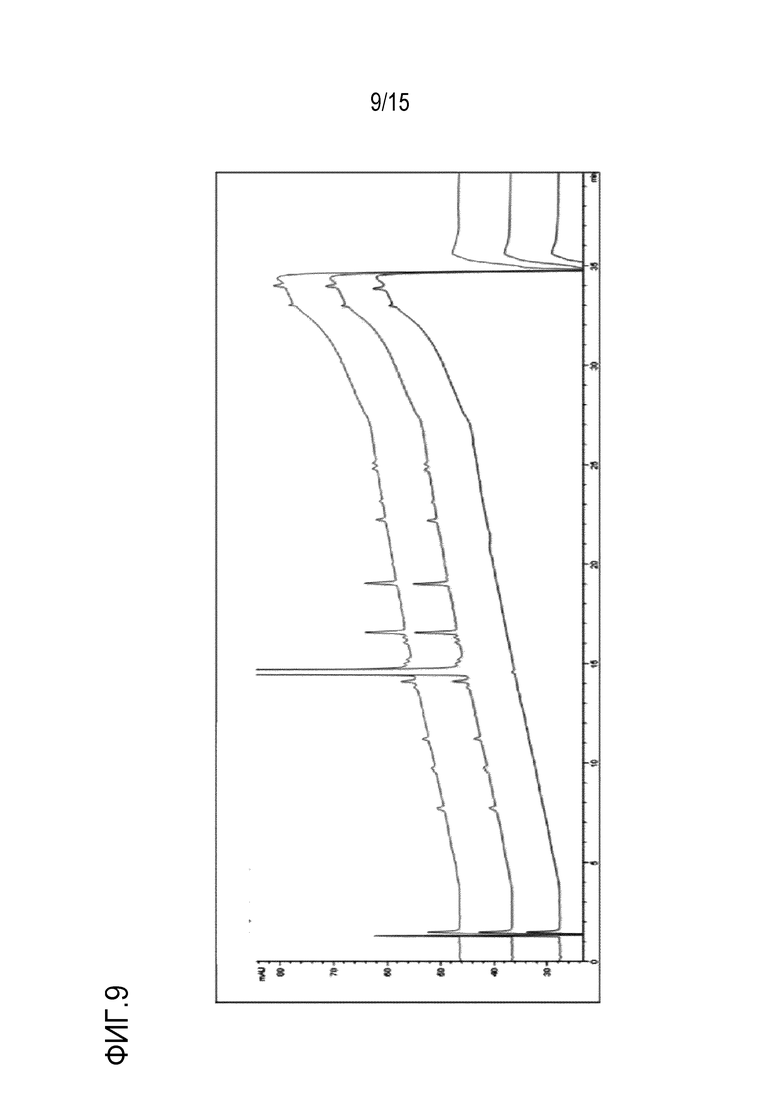
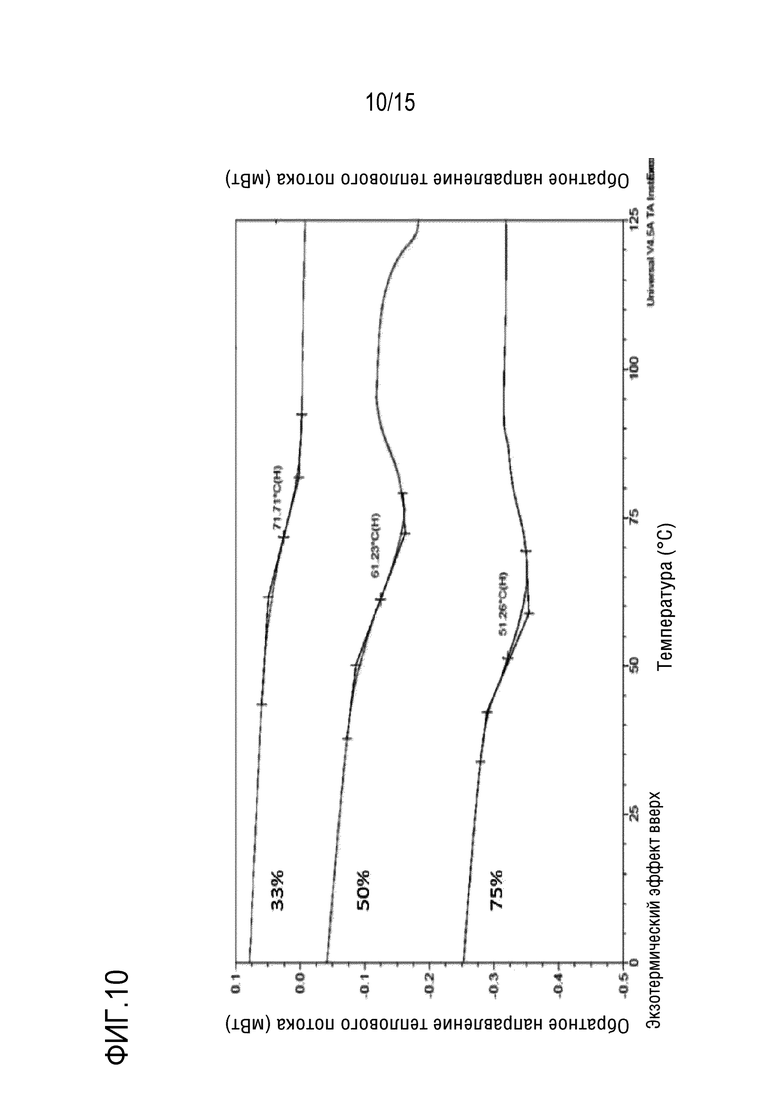

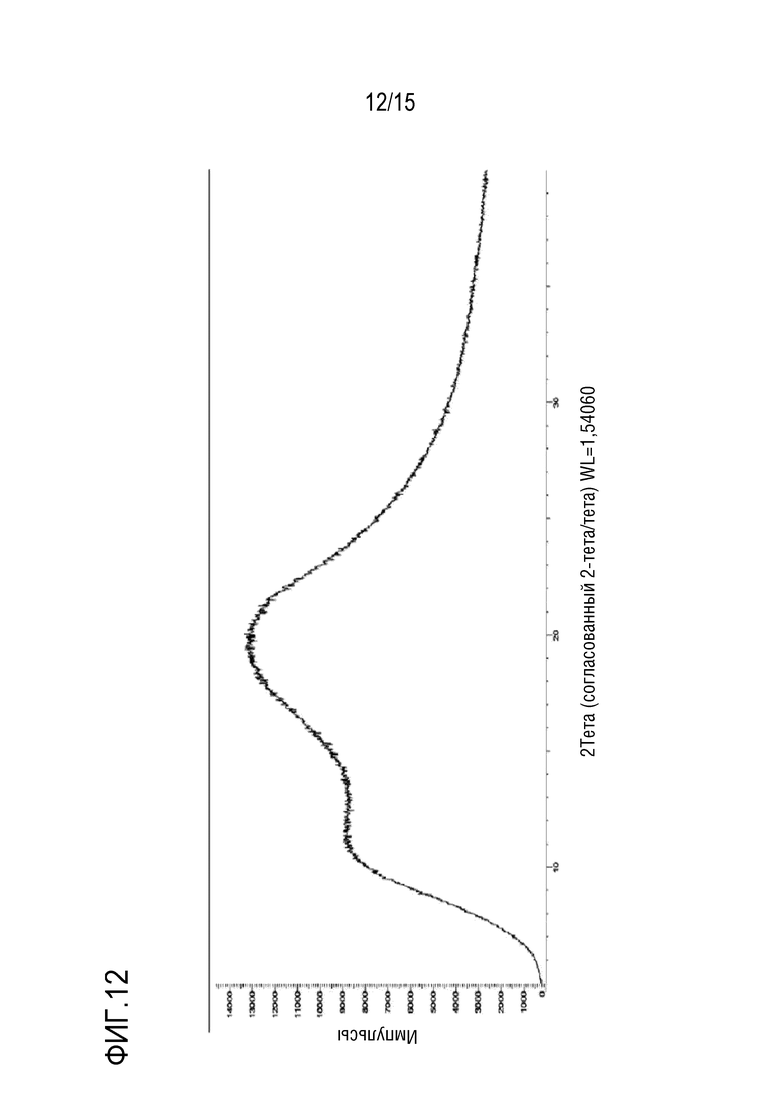
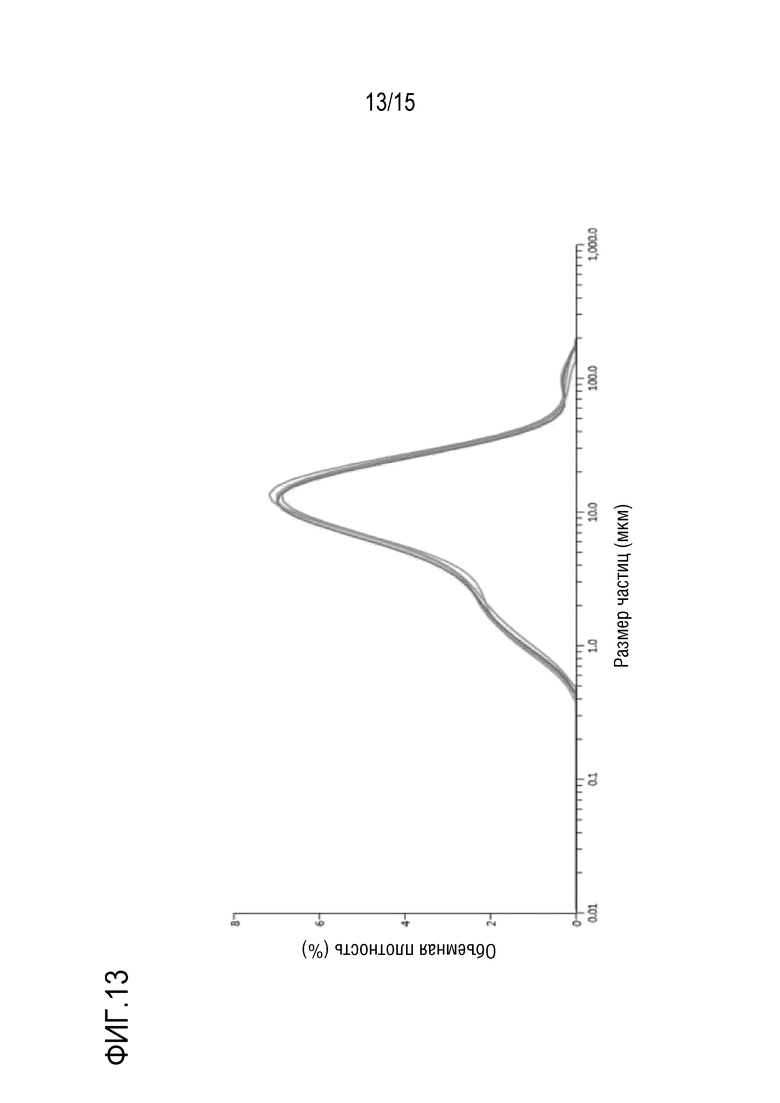
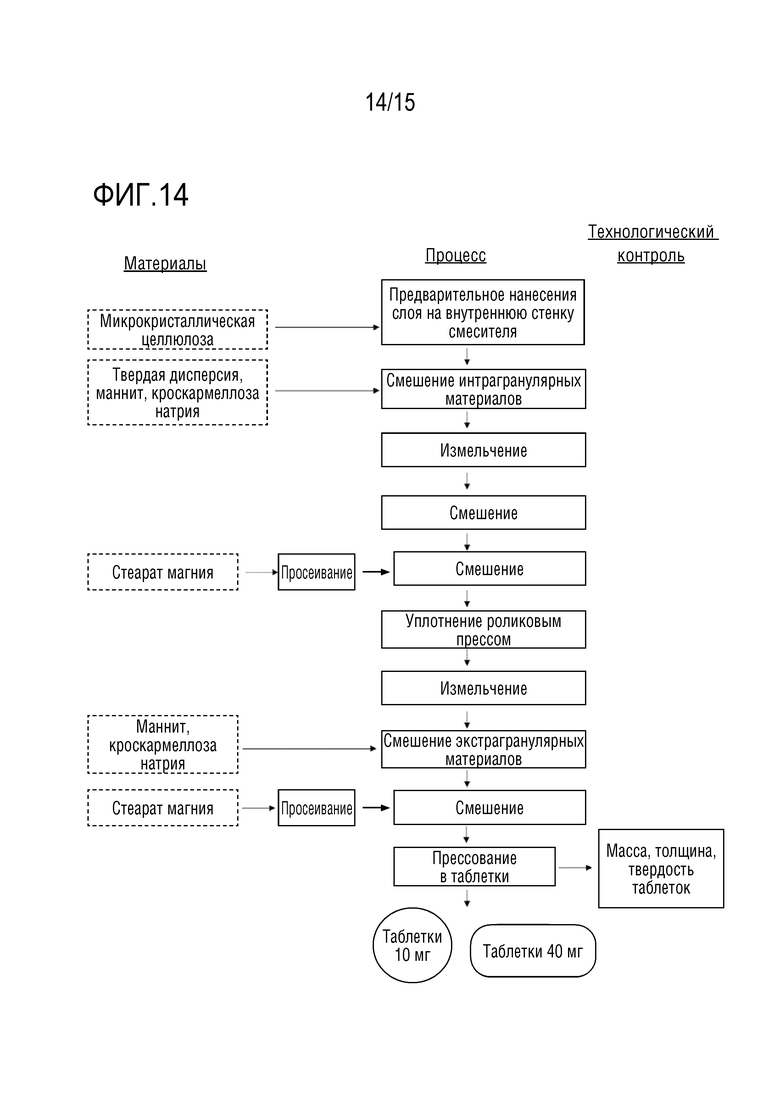
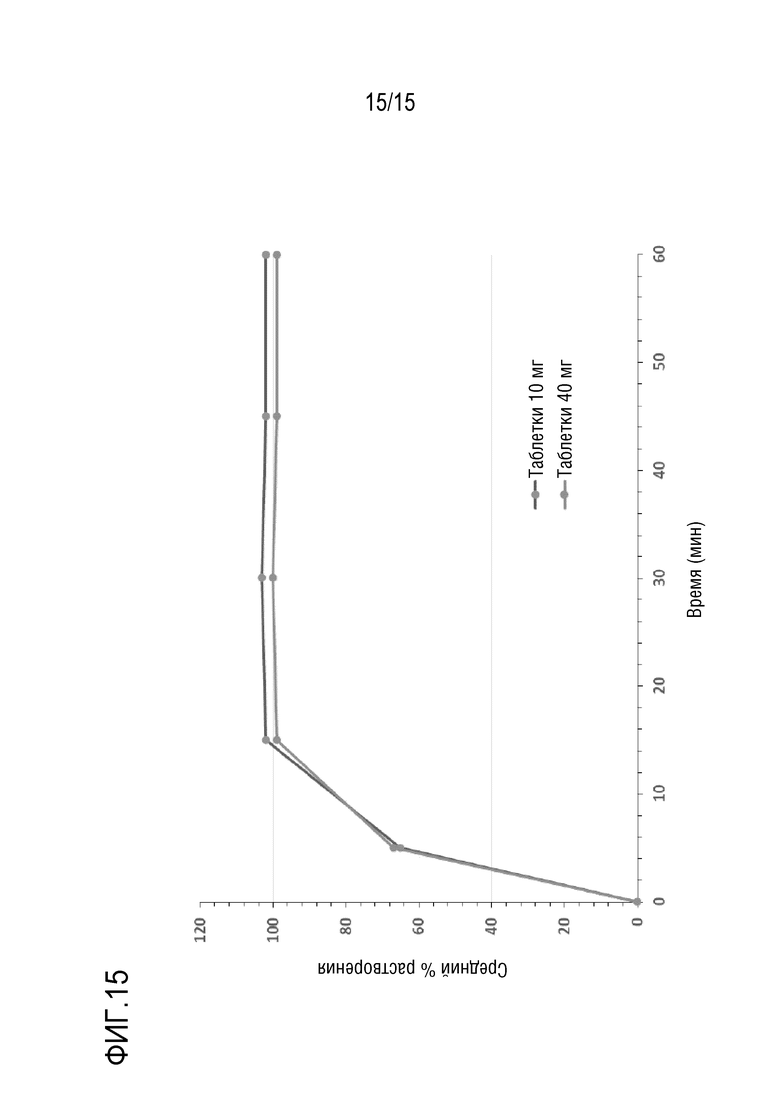
Группа изобретений относится к фармацевтической твердой лекарственной форме, ингибирующей HIF-2α, для перорального введения соединения формулы (I)
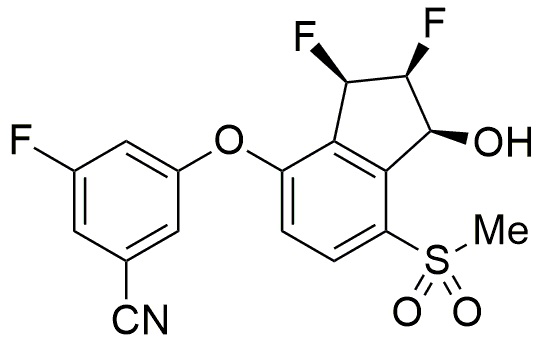
Формула (I),
а также способам лечения, включающим введение указной лекарственной формы. Твердая лекарственная форма включает твердую дисперсию, включающую соединение формулы (I) и одно или более фармацевтически приемлемых вспомогательных веществ. При этом твердая дисперсия включает фармацевтически приемлемый полимер, выбранный из группы, состоящей из ацетатсукцината гидроксипропилметилцеллюлозы (HPMCAS), ацетатфталата целлюлозы (CAP) и графт-сополимера полиэтиленгликоль-поливинилацетат-поливинилкапролактам, а твердая лекарственная форма представляет собой капсулу или таблетку. Также заявлен способ лечения болезни фон Гиппеля-Линдау (VHL) и способ лечения почечно-клеточной карциномы. Изобретение обеспечивает повышение стабильности, биодоступности и хранения фармацевтической лекарственной формы. 3 н. и 15 з.п. ф-лы, 15 ил., 33 табл., 18 пр.
1. Фармацевтическая твердая лекарственная форма, ингибирующая HIF-2α, для перорального введения соединения формулы (I)
Формула (I),
где твердая лекарственная форма включает твердую дисперсию, включающую соединение формулы (I) и одно или более фармацевтически приемлемых вспомогательных веществ, где твердая дисперсия включает фармацевтически приемлемый полимер, выбранный из группы, состоящей из ацетатсукцината гидроксипропилметилцеллюлозы (HPMCAS), ацетатфталата целлюлозы (CAP) и графт-сополимера полиэтиленгликоль-поливинилацетат-поливинилкапролактам;
и твердая лекарственная форма представляет собой капсулу или таблетку.
2. Твердая лекарственная форма по п. 1, где твердая лекарственная форма представляет собой таблетку.
3. Твердая лекарственная форма по п. 1 или 2, где одно или более фармацевтически приемлемых вспомогательных веществ включают связующее вещество, наполнитель, разрыхлитель и смазывающее вещество.
4. Твердая лекарственная форма по п. 3, где одно или более фармацевтически приемлемых вспомогательных веществ дополнительно включают скользящее вещество.
5. Твердая лекарственная форма по любому одному из пп. 1-4, где твердая дисперсия присутствует в количестве от 15 до 50% от массы твердой лекарственной формы.
6. Твердая лекарственная форма по любому одному из пп. 1-5, где соединение формулы (I) присутствует в твердой дисперсии в количестве от 15 до 35% в расчете на массу твердой дисперсии.
7. Твердая лекарственная форма по любому одному из пп. 1-5, где фармацевтически приемлемый полимер присутствует в твердой дисперсии в количестве от 65 до 85% в расчете на массу твердой дисперсии.
8. Твердая лекарственная форма по п. 7, где фармацевтически приемлемый полимер представляет собой HPMCAS.
9. Твердая лекарственная форма по п. 6 или 8, где фармацевтически приемлемый полимер присутствует в количестве от 15 до 35% от массы твердой лекарственной формы.
10. Твердая лекарственная форма по любому одному из пп. 1-9, где соединение формулы (I) присутствует в количестве от 1 до 15% от массы твердой лекарственной формы.
11. Твердая лекарственная форма по любому одному из пп. 1-9, включающая от 5 до 100 мг соединения формулы (I).
12. Твердая лекарственная форма по любому одному из пп. 1-11, включающая приблизительно 10 мг соединения формулы (I).
13. Твердая лекарственная форма по любому одному из пп. 1-11, включающая приблизительно 40 мг соединения формулы (I).
14. Способ лечения болезни фон Гиппеля-Линдау (VHL), включающий введение субъекту, нуждающемуся в этом, твердой лекарственной формы по любому одному из пп. 1-13.
15. Способ по п. 14, где субъект также страдает от гемангиобластомы, феохромоцитомы, панкреатической нейроэндокринной опухоли или почечно-клеточной карциномы.
16. Способ по п. 14, где субъект страдает от почечно-клеточной карциномы.
17. Способ лечения почечно-клеточной карциномы, включающий введение субъекту, нуждающемуся в этом, твердой лекарственной формы по любому одному из пп. 1-13.
18. Способ по п. 15 или 16, где почечно-клеточная карцинома представляет собой светлоклеточную почечно-клеточную карциному.
| WO 2016145045 А1, 15.09.2016 | |||
| WO 2015035223 A1, 12.03.2015 | |||
| US 20090143423 A1, 04.06.2009 | |||
| "Краткий курс молекулярной фармакологии" под ред | |||
| П.В.Сергеева, М., 1975 | |||
| Л.Е | |||
| Холодов и др | |||
| "Клиническая фармакокинетика", М., "Медицина", 1985 | |||
| И.М | |||
| Перцев, Фармацевтические и медико-биологические аспекты лекарств, Т.1, 1999. |

