Область техники, к которой относится изобретение
[0001]
Перекрестная ссылка на родственную заявку
По настоящей заявке испрашивается приоритет заявки на патент Японии № 2015-070927, зарегистрированной 31 марта 2015 года, содержание которой включено в настоящее изобретение путем ссылки на нее.
Настоящее изобретение относится к новым кристаллам 3,5-дизамещенного бензолалкинильного соединения, которые являются стабильными, характеризуются высокой способностью к всасыванию при пероральном введении и которые применяются в качестве противоопухолевого средства.
Уровень техники
[0002]
Обычно необходимо, чтобы фармацевтические композиции для перорального введения характеризовались не только стабильностью активного ингредиента, но и высокой способностью к всасыванию при пероральном введении и, кроме того, важно, чтобы такие композиции можно было получать в промышленном масштабе крупными партиями.
[0003]
Кристаллы могут существовать в форме полиморфов, которые содержат одну и ту же молекулу, но имеют различные расположения молекул. Известно, что такие полиморфы характеризуются отличающимися пиками на порошковых рентгенограммах (при рентгеноструктурном анализе). Кроме того, известно, что такие кристаллические полиморфы проявляют различную растворимость, способность к всасыванию при пероральном введении, стабильность и другие подобные свойства. Поэтому, при создании лекарственных средств необходимо находить оптимальные кристаллические формы с учетом различных факторов.
[0004]
В настоящее время, в качестве противоопухолевых средств известен ряд ингибиторов FGFR (рецептора фактора роста фибробластов), и в патентных документах 1, 2 и 3 описан (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-он (в дальнейшем именуемый в этом изобретении как "соединение 1") в качестве соединения, которое обладает высокой ингибирующей активностью в отношении FGFR и которое проявляет противоопухолевую активность.
[0005]
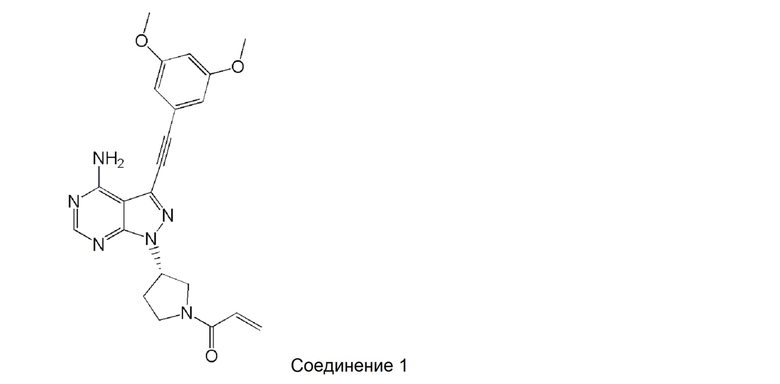
[0006]
Однако, ни в одном из патентных документов 1, 2 или 3 не раскрываются или не предлагаются кристаллы соединения 1, и не описаны их стабильность, способность к всасыванию при пероральном введении и получение кристаллов методом кристаллизации.
Список противопоставленных материалов
Патентные документы
[0007]
Патентный документ 1: WO2013/108809
Патентный документ 2: WO2015/008844
Патентный документ 3: WO2015/008839
Сущность изобретения
Техническая задача
[0008]
Задачей настоящего изобретения является получение кристаллов соединения 1, которое применяют в качестве противоопухолевого средства и которое описано в патентном документе 1, при этом кристаллы являются стабильными, характеризуются способностью к всасыванию при пероральном введении и пригодны для получения в промышленном масштабе крупными партиями. Задачей настоящего изобретения является также и разработка способа кристаллизации соединения 1.
Решение задачи
[0009]
Авторы настоящего изобретения провели обширные исследования и обнаружили, что соединение 1 имеет три кристаллических формы (кристаллическая форма I, кристаллическая форма II, кристаллическая форма III). Авторы настоящего изобретения обнаружили, что среди этих форм кристаллическая форма II характеризуется высокой стабильностью, проявляет высокую способность к всасыванию при пероральном введении, обладает высокой кристалличностью, имеет высокую химическую чистоту, и она пригодна для получения в промышленном масштабе крупными партиями с однородным распределением частиц по размерам, в результате чего авторами и было создано изобретение. Они также обнаружили, что кристаллическая форма II может быть получена путем добавления конкретного растворителя к соединению 1 с целью его кристаллизации. Авторы изобретения также обнаружили, что кристаллическая форма I соединения 1 характеризуется высокой стабильностью и высокой способностью к всасыванию при пероральном введении.
[0010]
А именно, в настоящем изобретении предлагаются следующие пункты.
Пункт 1.
Кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, где кристаллы характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, три характеристических пика при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°.
Пункт 2.
Кристаллы по пункту 1, которые характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, пять характеристических пика при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°.
Пункт 3.
Кристаллы по пункту 1 или 2, которые характеризуются порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°) 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°.
Пункт 4.
Кристаллы по любому одному из пунктов 1-3, которые имеют химическую чистоту 99,0% или более.
Пункт 5.
Кристаллы по любому одному из пунктов 1-4, которые характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно при 166°C при исследовании методом дифференциальной сканирующей калориметрии.
Пункт 6.
Фармацевтическая композиция, включающая кристаллы по любому одному из пунктов 1-5.
Пункт 7.
Фармацевтическая композиция для перорального введения, где композиция включает кристаллы по любому одному из пунктов 1-5.
Пункт 8.
Кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, где кристаллы характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, семь характеристических пика при углах дифракции (2θ±0,2°), выбранных из 13,5°, 17,9°, 19,5°, 20,6°, 22,0°, 22,6°, 23,3°, 23,7° и 24,2°.
Пункт 9.
Кристаллы по пункту 8, которые характеризуются порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°) 13,5°, 17,9°, 19,5°, 20,6°, 22,0°, 22,6°, 23,3°, 23,7° и 24,2°.
Пункт 10.
Кристаллы по пункту 8 или 9, которые характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно при 169°C при исследовании методом дифференциальной сканирующей калориметрии.
Пункт 11.
Фармацевтическая композиция, включающая кристаллы по любому одному из пунктов 8-10.
Пункт 12.
Фармацевтическая композиция для перорального введения, где композиция включает кристаллы по любому одному из пунктов 8-10.
[0011]
Пункт 13.
Кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, где кристаллы получают способом, включающим
стадию (1) добавления (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она в один или более растворителей, выбранных из группы, состоящей из воды, C1-4 спиртов, C3-5 эфиров алифатических карбоновых кислот, C3-6 кетонов, C2-5 апротонных полярных органических растворителей и смесей этих растворителей, и
стадию (2) перемешивания растворителя, к которому был добавлен (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-он на стадии (1) для кристаллизации (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она.
Пункт 14.
Кристаллы по пункту 13, которые характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, три характеристических пика при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°.
Пункт 15.
Кристаллы по пункту 13 или 14, которые имеют химическую чистоту 99,0% или более.
Пункт 16.
Кристаллы по любому одному из пунктов 13-15, которые характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно при 166°C при исследовании методом дифференциальной сканирующей калориметрии.
Пункт 17.
Способ кристаллизации (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, где способ включает
стадию (1) добавления (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она в один или более растворителей, выбранных из группы, состоящей из воды, C1-4 спиртов, C3-5 эфиров алифатических карбоновых кислот, C3-6 кетонов, C2-5 апротонных полярных органических растворителей и смесей этих растворителей, и
стадию (2) перемешивания растворителя, к которому был добавлен (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]-пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-он на стадии (1) для кристаллизации (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она.
[0012]
Пункт 18.
Способ кристаллизации по пункту 17, где кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, полученные на стадии (2), характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, три характеристических пика при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°.
Пункт 19.
Способ кристаллизации по пункту 17 или 18, где кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]-пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, полученные на стадии (2), имеют химическую чистоту 99,0% или более.
Пункт 20.
Способ кристаллизации по любому одному из пунктов 17-19, где кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло-3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, полученные на стадии (2), характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно при 166°C при исследовании методом дифференциальной сканирующей калориметрии.
Пункт 21.
Способ уменьшения образования отложений кристаллов (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она на поверхностях оборудования, где способ включает
стадию (1) добавления (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она в растворитель, выбранный из группы, состоящей из воды, C1-4 спиртов, C3-5 эфиров алифатических карбоновых кислот, C3-6 кетонов, C2-5 апротонных полярных органических растворителей и смесей этих растворителей, и
стадию (2) перемешивания растворителя, к которому был добавлен (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-он на стадии (1) с целью кристаллизации (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)-этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она.
[0013]
Пункт 22.
Способ уменьшения образования отложений кристаллов на поверхностях оборудования по пункту 21, где кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, полученные на стадии (2), характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, три характеристических пика при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°.
Пункт 23.
Способ уменьшения образования отложений кристаллов на поверхностях оборудования по пункту 21 или 22, где кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, полученные на стадии (2), имеют химическую чистоту 99,0% или более.
Пункт 24.
Способ уменьшения образования отложений кристаллов на поверхностях оборудования по любому одному из пунктов 21-23, где кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]-пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, полученные на стадии (2), характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно при 166°C при исследовании методом дифференциальной сканирующей калориметрии.
Пункт 25.
Кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, где кристаллы получают способом, включающим
стадию (1) добавления (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она в растворитель, содержащий C7-10 углеводород, C2-8 эфир, C6-10 эфир алифатической карбоновой кислоты или смесь C7-10 углеводорода и C3-5 эфира алифатической карбоновой кислоты, и
стадию (2) перемешивания растворителя, к которому был добавлен (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]-пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-он на стадии (1) с целью кристаллизации (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она.
Пункт 26.
Кристаллы по пункту 25, которые характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, семь характеристических пика при углах дифракции (2θ±0,2°), выбранных из 13,5°, 17,9°, 19,5°, 20,6°, 22,0°, 22,6°, 23,3°, 23,7° и 24,2°.
Пункт 27.
Кристаллы по пункту 25 или 26, которые характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно при 169°C при исследовании методом дифференциальной сканирующей калориметрии.
[0014]
Пункт 28.
Способ кристаллизации (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, где способ включает
стадию (1) добавления (S)-1-(3-(4-амино-3-((3,5-диметокси-фенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она в растворитель, содержащий C5-10 углеводород, C2-8 эфир, C6-10 эфир алифатической карбоновой кислоты или смесь C5-10 углеводорода и C3-5 эфира алифатической карбоновой кислоты, и
стадию (2) перемешивания растворителя, к которому был добавлен (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]-пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-он на стадии (1) с целью кристаллизации (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она.
Пункт 29.
Способ кристаллизации по пункту 28, где кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, полученные на стадии (2), характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, семь характеристических пика при углах дифракции (2θ±0,2°), выбранных из 13,5°, 17,9°, 19,5°, 20,6°, 22,0°, 22,6°, 23,3°, 23,7° и 24,2°.
Пункт 30.
Кристаллы по пункту 28 или 29, где кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, полученные на стадии (2), характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно при 169°C при исследовании методом дифференциальной сканирующей калориметрии.
Положительные эффекты изобретения
[0015]
Кристаллическая форма II соединения 1 настоящего изобретения характеризуется высокой стабильностью, проявляет высокую способность к всасыванию при пероральном введении, обладает высокой кристалличностью, имеет высокую химическую чистоту, и она пригодна для получения в промышленном масштабе крупными партиями с однородным распределением частиц по размерам. В связи с чем, кристаллическая форма II может применяться в качестве перорально вводимого лекарственного средства. Кроме того, кристаллическая форма I соединения 1 характеризуется высокой стабильностью, высокой способностью к всасыванию при пероральном введении, обладает высокой кристалличностью и имеет высокую химическую чистоту. В связи с чем, кристаллическая форма I может применяться в качестве перорально вводимого лекарственного средства.
Краткое описание чертежей
[0016]
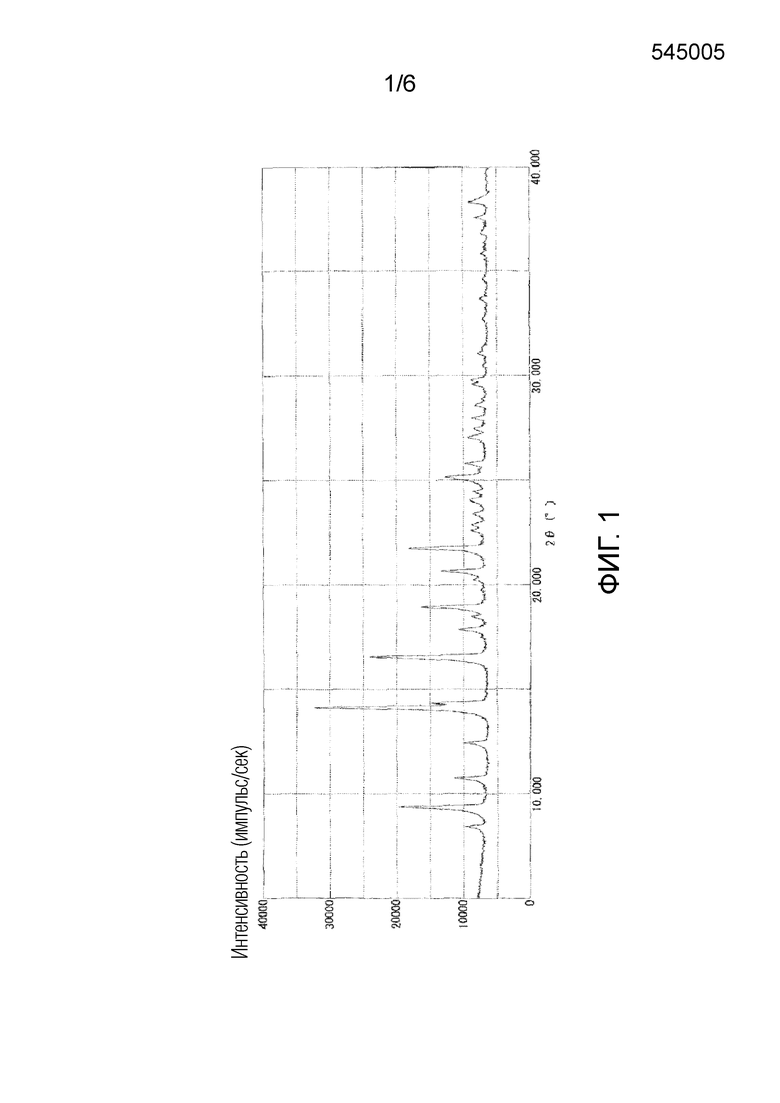
На фигуре 1 приведена порошковая рентгенограмма кристаллической формы II соединения 1 (вертикальная ось: интенсивность (число импульсов в секунду), горизонтальная ось: угол дифракции (2θ±0,2°)).
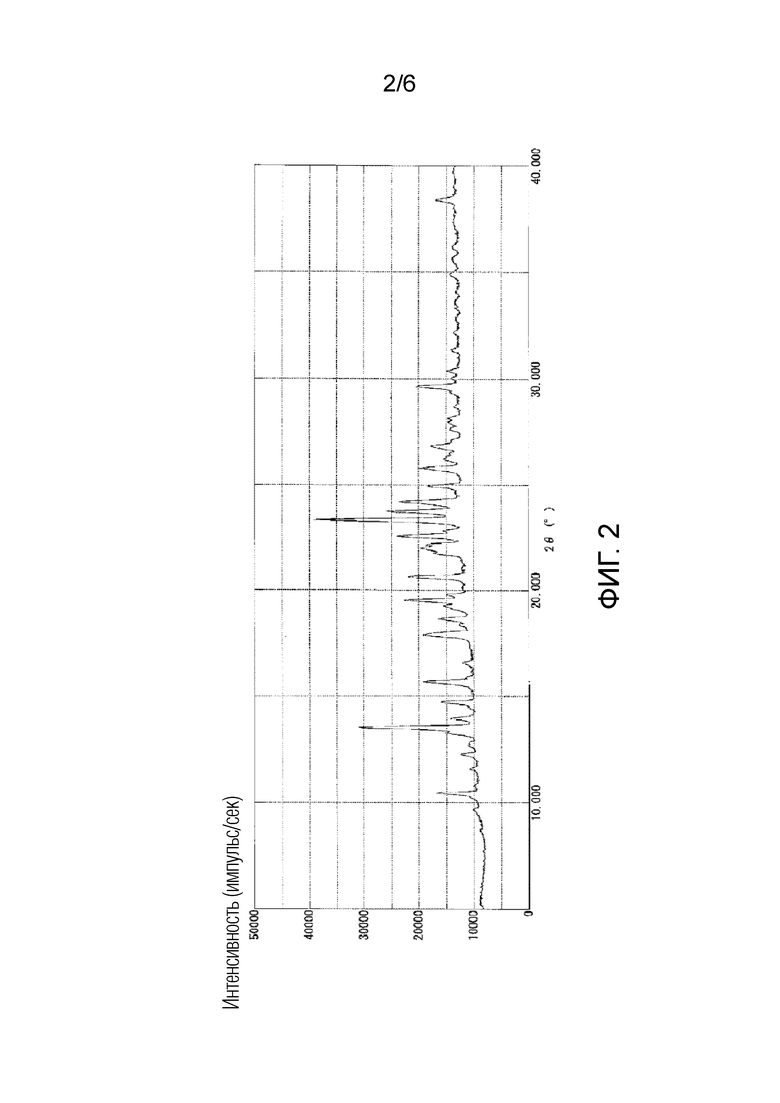
На фигуре 2 приведена порошковая рентгенограмма кристаллической формы I соединения 1 (вертикальная ось: интенсивность (число импульсов в секунду), горизонтальная ось: угол дифракции (2θ±0,2°)).
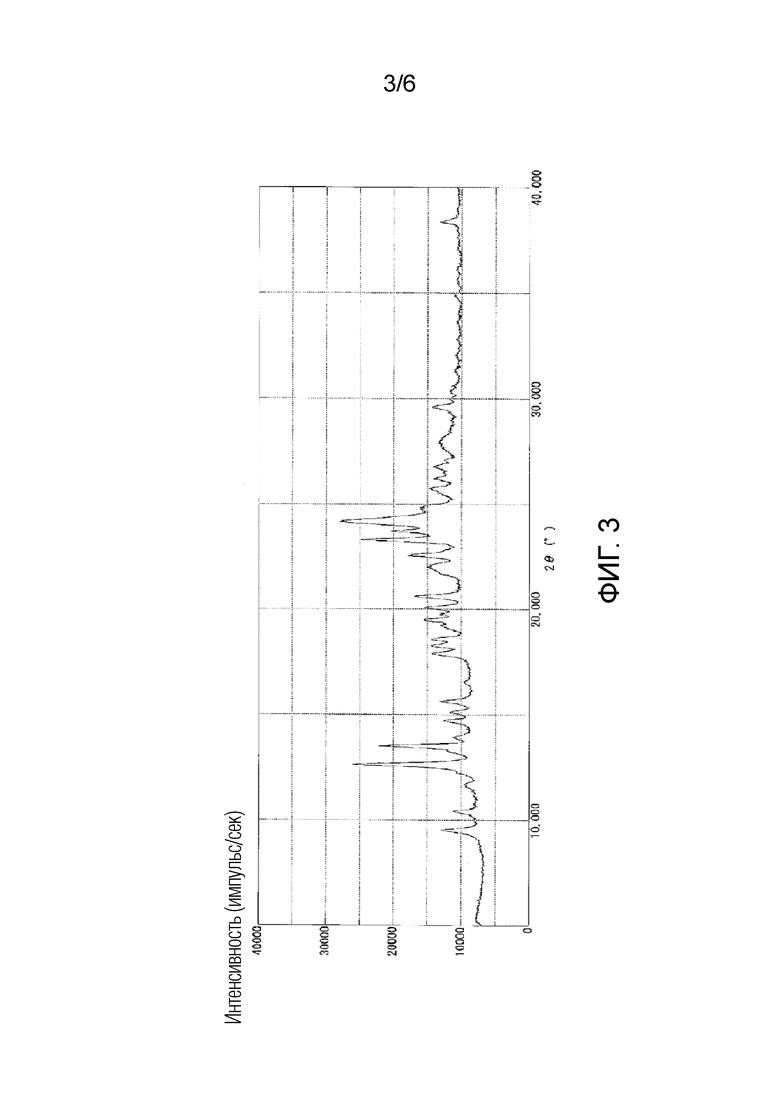
На фигуре 3 приведена порошковая рентгенограмма кристаллической формы III соединения 1 (вертикальная ось: интенсивность (число импульсов в секунду), горизонтальная ось: угол дифракции (2θ±0,2°)).
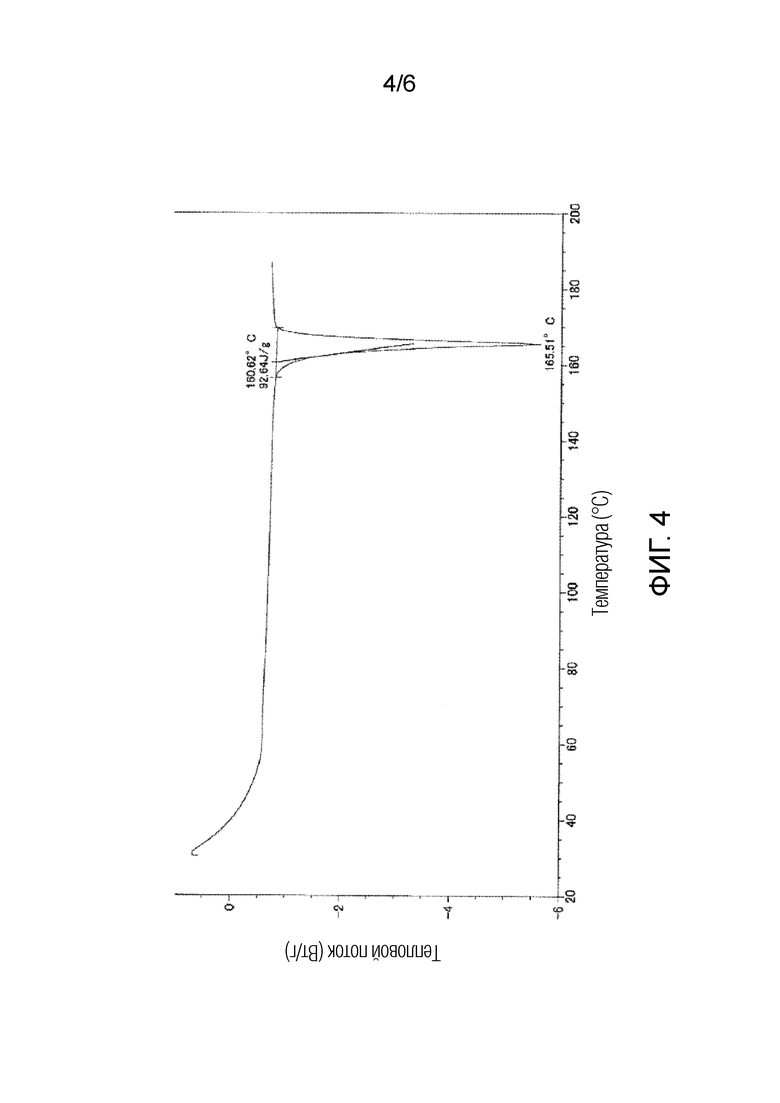
На фигуре 4 приведена кривая дифференциальной сканирующей калориметрии (DSC) для кристаллической формы II соединения 1.
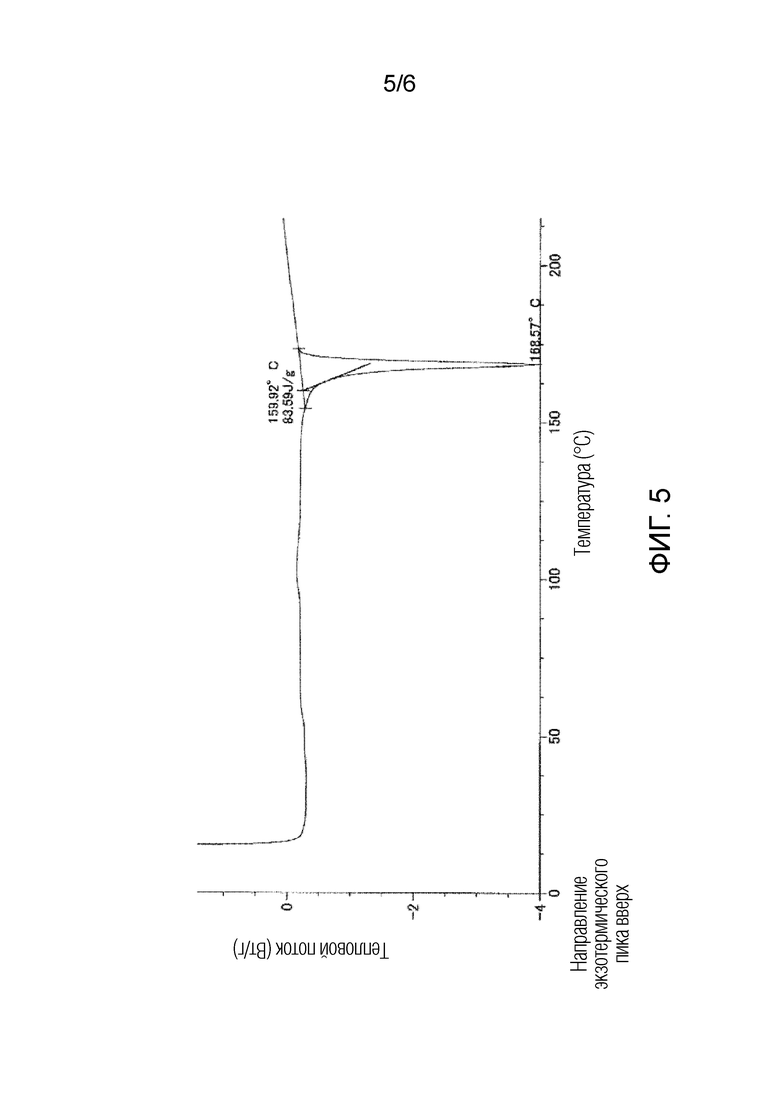
На фигуре 5 приведена кривая дифференциальной сканирующей калориметрии (DSC) для кристаллической формы I соединения 1.
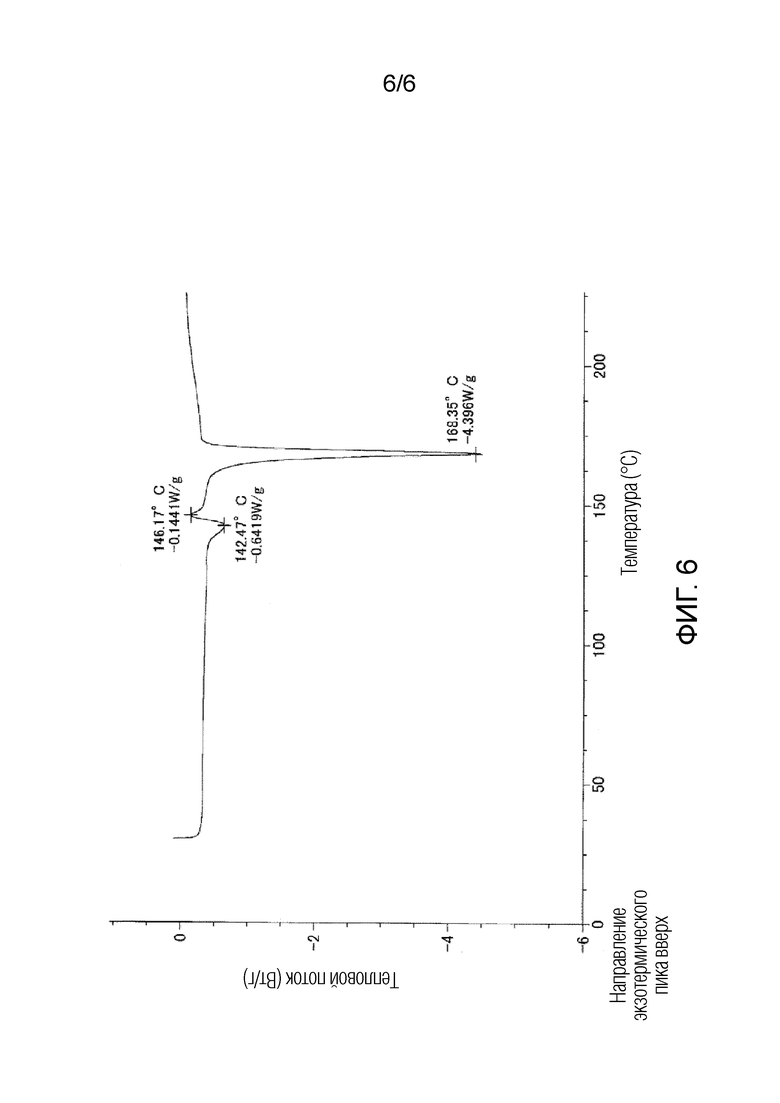
На фигуре 6 приведена кривая дифференциальной сканирующей калориметрии (DSC) для кристаллической формы III соединения 1.
Описание вариантов осуществления изобретения
[0017]
Соединение 1 по настоящему изобретению может быть синтезировано с помощью метода, раскрытого в патентном документе 1.
[0018]
Кристаллы относятся к твердому веществу, в котором атомы или молекулы расположены строго повторяющимся образом, и они отличаются от аморфного твердого вещества, которое не имеет повторяющихся фрагментов. Кристаллы и аморфные твердые вещества могут быть исследованы такими методами, как порошковый рентгеноструктурный анализ (XRD), дифференциальная сканирующая колориметрия (DSC), термогравиметрия-дифференциальный термический анализ (TG-DTA) или ИК-спектроскопия (IR).
[0019]
Кристаллы могут существовать в форме полиморфов, которые содержат одну и ту же молекулу, но имеют различные расположения молекул. Известно, что такие полиморфы характеризуются отличающимися пиками на порошковых рентгенограммах (при рентгеноструктурном анализе). Кроме того, известно, что такие кристаллические полиморфы проявляют различную растворимость, способность к всасыванию при пероральном введении, стабильность и другие подобные свойства. Поэтому, при создании лекарственных средств необходимо находить оптимальные кристаллические формы с учетом различных факторов.
[0020]
Авторы настоящего изобретения провели обширные исследования и обнаружили, что соединение 1 имеет три кристаллических формы (кристаллическую форму I, кристаллическую форму II и кристаллическую форму III).
[0021]
Кристаллическая форма III может быть получена в результате использования смеси этилацетата и гексана. Однако, исследование методом дифференциальной сканирующей калориметрии (DSC) показало, что кристаллическая форма III характеризуется эндотермическим пиком или экзотермическим пиком приблизительно при 145°C, что указывает на более низкую стабильность кристаллической формы III по сравнению со стабильностью кристаллической формы I или кристаллической формы II и на возможность изменения кристаллической формы, например, в процессе получения кристаллов или в процессе приготовления лекарственного средства. Поэтому, кристаллическая форма III считается неподходящей кристаллической формой кристаллов для лекарственных средств, так как лекарственные средства должны быть стабильными.
[0022]
В отличие от этого, при исследовании методом дифференциальной сканирующей калориметрии кристаллической формы I не были обнаружены эндотермический пик или экзотермический пик, что указывало на стабильность кристаллической формы I, на малую вероятность изменения кристаллической формы в процессе приготовления лекарственного средства и при других подобных манипуляциях, и что кристаллическая форма I является химически очень стабильной. Кроме того, в силу своей чрезвычайно высокой способности к всасыванию при пероральном введении, кристаллическая форма I подходит в качестве кристаллической формы для лекарственных средств, которые должны быть стабильными и обладать высокой способностью к всасыванию при пероральном введении.
[0023]
При исследовании методом дифференциальной сканирующей калориметрии кристаллической формы II также не были обнаружены эндотермический пик или экзотермический пик, что указывало на стабильность кристаллической формы II, на малую вероятность изменения кристаллической формы в процессе приготовления лекарственного средства и при других подобных манипуляциях, и что кристаллическая форма II является химически очень стабильной. Кроме того, кристаллическая форма II не прилипает прочно к оборудованию, такому как реактор и перемешивающая лопасть, при осаждении ее в растворителе, и она пригодна для получения в промышленном масштабе крупными партиями. Кристаллическая форма II является также подходящей для эффективного получения соединения 1 с чрезвычайно высокой химической чистотой. В связи с этим, кристаллическая форма II подходит в качестве кристаллической формы для лекарственных средств, для которых требуется устойчивое получение стабильной и высокочистой кристаллической формы в больших объемах.
[0024]
Кристаллическая форма I может представлять собой любые кристаллы при условии, что кристаллическая форма I содержит кристаллическую форму I соединения 1. Кристаллическая форма II может представлять собой любые кристаллы при условии, что кристаллическая форма II содержит кристаллическую форму II соединения 1. Кристаллическая форма I или кристаллическая форма II могут представлять собой монокристаллы кристаллической формы I или кристаллической формы II, или полиморфную смесь, которая содержит другие кристаллы. В частности, предпочтительно, чтобы 90 масс.% или более кристаллов составляла кристаллическая форма I или кристаллическая форма II, и более предпочтительно, 95 масс.% или более кристаллов составляла кристаллическая форма I или кристаллическая форма II, и особенно предпочтительно, 99 масс.% или более кристаллов составляла кристаллическая форма I или кристаллическая форма II.
[0025]
В этом изобретении, термин "химическая чистота" относится к чистоте, определяемой методом высокоэффективной жидкостной хроматографии, и химическая чистота соединения 1 обозначает чистоту, определенную для соединения I методом высокоэффективной жидкостной хроматографии. Может быть соответствующим образом определена длина волны детектора, используемого при определении чистоты. В частности, предпочтительно, чтобы химическая чистота кристаллов соединения 1 составляла 95,0% или более, более предпочтительно, 98,0% или более, и особенно предпочтительно, 99,0% или более.
[0026]
Кристаллическая форма I и кристаллическая форма II по настоящему изобретению каждая включает формы с различными габитусами кристаллов (то есть, с различной внешней формой) вследствие различного роста поверхности кристалла. Так, например, кристаллическая форма I и кристаллическая форма II каждая включает кристаллы, которые характеризуются различными относительными интенсивностями пиков, даже если наборы пиков при угле дифракции 2θ кристаллической формы I или кристаллической форма II, определенных методом рентгеноструктурного анализа, являются одинаковыми. Используемый в изобретении термин "относительная интенсивность" обозначает величину площади каждого пика относительно площади самого большого пика (принимаемого за 100) из числа пиков при угле дифракции 2θ на порошковой рентгенограмме.
[0027]
Ошибка определения пиков при угле дифракции 2θ на порошковой рентгенограмме в настоящем изобретении составляет приблизительно ±0,2°. Наличие этой ошибки обусловлено используемыми при измерении приборами, установкой образца, методами анализа данных и так далее. Так, например, значения, полученные для кристаллов по настоящему изобретению методом рентгеноструктурного анализа, включают ошибку ±0,2° при угле дифракции 2θ.
[0028]
Эндотермический пик (самая высокая величина пика), измеренный методом дифференциальной сканирующей калориметрии (DSC) может изменяться в зависимости от скорости увеличения температуры в минуту depending, массы образца, чистоты образца и других факторов. В этом изобретении, термин "приблизительно при" означает ±5,0°C.
[0029]
Кристаллическая форма II по настоящему изобретению может быть получена путем добавления соединения 1 в конкретный растворитель и перемешивания смеси с целью кристаллизации соединения 1. Так, например, в настоящем изобретении предлагается способ кристаллизации для получения кристаллической формы II, где способ включает:
стадию (1) добавления соединения 1 в растворитель; и
стадию (2) перемешивания растворителя, к которому было добавлено соединение 1 на стадии (1) с целью кристаллизации соединения 1.
Этот способ может быть также назван иначе как способ уменьшения образования отложений кристаллов соединения 1 на поверхностях оборудования, где способ включает:
стадию (1) добавления соединения 1 в растворитель; и
стадию (2) перемешивания растворителя, к которому было добавлено соединение 1 на стадии (1) с целью кристаллизации соединения 1, с получением в результате кристаллической формы II.
[0030]
Растворители, используемые для кристаллизации с получением кристаллической формы I по настоящему изобретению, включают C7-10 углеводороды, C2-8 эфиры, C6-10 эфиры алифатических карбоновых кислот и смеси C7-10 углеводородов и C3-5 эфиров алифатических карбоновых кислот.
[0031]
C7-10 углеводороды обозначают углеводороды, имеющие от 7 до 10 углеродных атомов, и их примеры включают гептан и декан, при этом гептан является предпочтительным.
[0032]
C2-8 эфиры обозначают эфиры, имеющие от 2 до 8 углеродных атомов, и их примеры включают диэтиловый эфир, третбутилметиловый эфир, циклопентилметиловый эфир и тетрагидрофуран, при этом третбутилметиловый эфир является предпочтительным.
[0033]
C6-10 эфиры алифатических карбоновых кислот обозначают эфиры алифатических карбоновых кислот, имеющие от 6 до 10 углеродных атомов в эфирах в целом, и их примеры включают бутилацетат, пентилацетат, гексилацетат, октилацетат и бутилпропионат, при этом бутилацетат является предпочтительным.
[0034]
C3-5 эфиры алифатических карбоновых кислот обозначают эфиры алифатических карбоновых кислот, имеющие от 3 до 5 углеродных атомов в эфирах в целом, и их примеры включают метилацетат, этилацетат, пропилацетат, изопропилацетат, метилпропионат и этилпропионат, при этом этилацетат является предпочтительным.
[0035]
Растворители, используемые для кристаллизации с получением кристаллической формы I по настоящему изобретению, включают растворители, выбранные из группы, состоящей из C7-10 углеводородов, C2-8 эфиров, C6-10 эфиров алифатических карбоновых кислот, смеси C7-10 углеводороды-C3-5 эфиры алифатических карбоновых кислот и смесей этих растворителей с гептаном, третбутилметиловым эфиром, бутилацетатом, и смесь гептан-этилацетат является предпочтительной.
[0036]
Растворители, используемые для получения кристаллической формы II по настоящему изобретению, включают растворители, выбранные из группы, состоящей из воды, C1-4 спиртов, C3-5 эфиров алифатических карбоновых кислот, C3-6 кетонов, C2-5 апротонных полярных органических растворителей и смесей этих растворителей.
[0037]
C1-4 спирты обозначают спирты, имеющие от 1 до 4 углеродных атомов, и их примеры включают метанол, этанол, н-пропанол, изопропанол, н-бутанол и третбутанол, при этом этанол и изопропанол являются предпочтительными.
[0038]
C3-5 эфиры алифатических карбоновых кислот обозначают упомянутые выше эфиры алифатических карбоновых кислот, и этилацетат является предпочтительным.
[0039]
C3-6 кетоны обозначают кетоны, имеющие от 3 до 6 углеродных атомов в кетонах в целом, и их примеры включают ацетон, метилэтилкетон, метилизобутилкетон и циклогексанон, при этом ацетон и метилэтилкетон являются предпочтительными.
[0040]
C2-5 апротонные полярные органические растворители включают ацетонитрил, N-метил-2-пирролидон, N,N-диметилформамид, N,N-диметилацетамид и диметилсульфоксид.
[0041]
Растворители, используемые для кристаллизации с получением кристаллической формы II по настоящему изобретению, включают растворители, выбранные из группы, состоящей из воды, C1-4 спиртов, C3-5 эфиров алифатических карбоновых кислот, C3-6 кетонов, C2-5 апротонных полярных органических растворителей и смесей этих растворителей с растворителями, выбранными из группы, состоящей из воды, C1-4 спиртов, C3-5 эфиров алифатических карбоновых кислот, C3-6 кетонов, и смеси этих растворителей являются предпочтительными. Более предпочтительные растворители включают этанол, изопропанол, ацетон, метилэтилкетон, этилацетат и смесь вода-этанол. Особенно предпочтительным растворителем является смесь вода-этанол. При использовании смеси вода-C1-4 спирт, отношение C1-4 спирта к воде может быть подобрано так, что C1-4 спирт обычно присутствует в количестве от 0,01 до 100 частей по массе, предпочтительно, от 0,1 до 50 частей по массе, и более предпочтительно, от 1 до 30 частей по массе, на одну часть по массе воды.
[0042]
Количество растворителя, добавляемое к кристаллической форме I или кристаллической форме II по настоящему изобретению, с точки зрения достижения высокого выхода кристаллов, должно быть больше массы соединения 1 от 1 до 100 раз (по отношению объема к массе), предпочтительно, от 2 до 50 раз (по отношению объема к массе), и более предпочтительно, от 4 до 30 раз (по отношению объема к массе).
[0043]
Температура проведения кристаллизации для получения кристаллической формы I или кристаллической формы II по настоящему изобретению может быть соответствующим образом определена, исходя из используемого растворителя, в диапазоне от 0°C до температуры кипения растворителя. Необязательно, чтобы температура при проведении кристаллизации оставалась на протяжении всего процесса одинаковой, и может осуществляться нагревание или охлаждение в диапазоне от 0°C до температуры кипения растворителя. Используемый в изобретении термин "нагревание" подразумевает поддержание температуры растворителя при 40°C или выше, и "охлаждение" подразумевает поддержание температуры растворителя ниже 15°C.
[0044]
Перемешивание при проведении кристаллизации с получением кристаллической формы I или кристаллической формы II по настоящему изобретению может быть осуществлено соответствующим образом путем использования мешалки, перемешивающей лопасти, магнитной мешалки или других мешалок, в зависимости от количества растворителя и размера термического реактора и других подобных факторов. Скорость перемешивания обычно составляет от 1 до 600 об/мин, и предпочтительно, от 10 до 300 об/мин.
[0045]
Предпочтительно, чтобы время перемешивания при проведении кристаллизации с получением кристаллической формы I или кристаллическая формы II по настоящему изобретению равнялось или было больше предварительно установленного периода времени, способствующего осуществлению кристаллизации и достижению высокого выхода кристаллов, и предпочтительно, чтобы время перемешивания было меньше предварительно установленного периода времени для уменьшения разложения кристаллов, приводящего к снижению выхода. Время перемешивание составляет от 1 минуты до 120 часов, предпочтительно, от 1 часа до 72 часов, и более предпочтительно, от 3 часов до 48 часов.
[0046]
Уменьшение образования отложений кристаллической формы II по настоящему изобретению на поверхностях оборудования при проведении кристаллизации означает уменьшение количества кристаллов, остающихся в реакторе, до менее чем 20% от теоретического выхода; предпочтительно, чтобы это количество составляло менее 10%, и более предпочтительно, менее 5% от теоретического выхода.
[0047]
Кристаллическая форма I или кристаллическая форма II по настоящему изобретению, осажденные в растворители, могут быть выделены и очищены хорошо известными методами разделения и очистки, такими как фильтрация, промывка органическим растворителем и сушка при пониженном давлении. Используемые для промывки органические растворители включают описанные выше растворители, и предпочтительно, чтобы этими органическими растворителями были этанол, изопропанол, ацетон, метилэтилкетон, этилацетат и смесь вода-этанол. Пониженное давление при сушке составляет 0,01 МПа или менее, и предпочтительно, 0,005 МПа или менее. Температура при проведении сушки при пониженном давлении составляет от 0 до 200°C, и предпочтительно, от 25 до 100°C.
[0048]
При проведении кристаллизации по настоящему изобретению, может быть добавлена кристаллическая форма I или кристаллическая форма II в качестве затравочных кристаллов. Количество добавляемых затравочных кристаллов составляет от 0,1 до 10 масс.%, и предпочтительно, от 1 до 3 масс.% от теоретического выхода кристаллизуемого соединения 1.
[0049]
Полученная таким образом кристаллическая форма I соединения 1 характеризуется порошковой рентгенограммой, содержащей, по меньшей мере, семь пиков при углах дифракции (2θ±0,2°), выбранных из 13,5°, 17,9°, 19,5°, 20,6°, 22,0°, 22,6°, 23,3°, 23,7° и 24,2°. Более предпочтительно, чтобы кристаллическая форма I соединения 1 характеризовалась порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°) 13,5°, 17,9°, 19,5°, 20,6°, 22,0°, 22,6°, 23,3°, 23,7° и 24,2°, которая приведена на фигуре 2. В типичном варианте осуществления, кристаллическая форма I соединения 1 характеризуется эндотермическим пиком (самой высокой величиной пика), например, приблизительно в диапазоне от 164 до 174°C, более предпочтительно, приблизительно при 169°C, как это показано по результатам дифференциальной сканирующей калориметрии (DSC) на фигуре 5.
[0050]
Полученная таким образом кристаллическая форма I соединения 1 характеризуется порошковой рентгенограммой, содержащей, по меньшей мере, семь пиков при углах дифракции (2θ±0,2°), выбранных из 13,5°, 17,9°, 19,5°, 20,6°, 22,0°, 22,6°, 23,3°, 23,7° и 24,2°, и характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно в диапазоне от 164 до 174°C при исследовании методом дифференциальной сканирующей калориметрии (DSC). Более предпочтительно, чтобы кристаллическая форма I соединения 1 характеризовалась порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°) 13,5°, 17,9°, 19,5°, 20,6°, 22,0°, 22,6°, 23,3°, 23,7° и 24,2°, которая приведена на фигуре 2, и характеризовалась эндотермическим пиком (самой высокой величиной пика) приблизительно при 169°C при исследовании методом дифференциальной сканирующей калориметрии (DSC).
[0051]
Полученная таким образом кристаллическая форма II соединения 1 характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, три пика при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°. Более предпочтительно, чтобы кристаллическая форма II соединения 1 характеризовалась порошковой рентгенограммой, содержащей, по меньшей мере, пять пиков при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°. Еще более предпочтительно, чтобы кристаллическая форма II соединения 1 характеризовалась порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°) 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°, которая приведена на фигуре 1. В типичном варианте осуществления, кристаллическая форма II соединения 1 характеризуется эндотермическим пиком (самой высокой величиной пика), например, приблизительно в диапазоне от 161 до 171°C, и более предпочтительно, приблизительно при 166°C, как это показано по результатам дифференциальной сканирующей калориметрии (DSC) на фигуре 4.
[0052]
Кристаллическая форма II по настоящему изобретению характеризуются порошковой рентгенограммой, содержащей, по меньшей мере, три пика при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°, и характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно в диапазоне от 161 до 171°C при исследовании методом дифференциальной сканирующей калориметрии (DSC). Более предпочтительно, чтобы кристаллическая форма II характеризовалась порошковой рентгенограммой, содержащей, по меньшей мере, пять пиков при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°, и характеризовалась эндотермическим пиком (самой высокой величиной пика) приблизительно при 166°C при исследовании методом дифференциальной сканирующей калориметрии (DSC). Еще более предпочтительно, чтобы кристаллическая форма II характеризовалась порошковой рентгенограммой, содержащей, по меньшей мере, семь пиков при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°, и характеризовалась эндотермическим пиком (самой высокой величиной пика) приблизительно при 166°C при исследовании методом дифференциальной сканирующей калориметрии (DSC).
[0053]
Так как соединение 1 обладает высокой ингибирующей активностью в отношении FGFR, его кристаллическая форма I и кристаллическая форма II по настоящему изобретению обе применяются в качестве противоопухолевого средства. Примеры типов раков-мишеней включают, но этим не ограничивая, рак головы и шеи, рак желудочно-кишечного тракта (например, рак пищевода, рак желудка, желудочно-кишечную стромальную опухоль, рак двенадцатиперстной кишки), рак печени, рак желчных путей (например, рак желчного пузыря и рак желчного протока), рак поджелудочной железы, рак тонкого кишечника, рак толстого кишечника (например, колоректальный рак, рак толстой кишки и рак прямой кишки), рак легкого, рак молочной железы, рак яичников, рак матки (например, рак шейки матки и рак эндометрия), рак почки, рак мочевого пузыря, рак предстательной железы, рак уротелия, саркомы костей и мягких тканей, рак крови (например, B-клеточную лимфому, хронический лимфолейкоз, периферическую Т-клеточную лимфому, миелодиспластический синдром, острый миелолейкоз и острый лимфолейкоз), множественную миелому, рак кожи и мезотелиому.
[0054]
При использовании кристаллической формы I или кристаллической формы II по настоящему изобретению в качестве лекарственного средства, к кристаллической форме I или кристаллической форме II может быть необязательно добавлен фармацевтический носитель для приготовления соответствующей лекарственной формы, в зависимости от цели профилактики или лечения. Примеры лекарственной формы включают пероральные лекарственные средства, инъецируемые средства, суппозитории, мази и пластыри, при этом пероральные лекарственные средства являются предпочтительными. Эти лекарственные формы могут быть приготовлены методами, которые хорошо известны специалистам в области фармацевтики.
[0055]
Используемый фармацевтический носитель включает целый ряд органических или неорганических веществ, обычно используемых в фармацевтике в качестве носителей для лекарственных средств; и носитель добавляют в виде наполнителя, связующего, разрыхлителя, скользящего вещества или окрашивающего вещества к твердым лекарственным средствам, или в виде растворителя, солюбилизирующего средства, суспендирующего средства, изотонического вещества, буфера, или успокаивающего средства к жидким лекарственным средствам. Могут быть также необязательно введены добавки к фармацевтическим препаратам, такие как консерванты, антиоксиданты, окрашивающие вещества, подсластители и стабилизаторы.
[0056]
Пероральные твердые лекарственные средства могут быть приготовлены путем добавления вспомогательного вещества, необязательно вместе с наполнителей, связующим, разрыхлителем, скользящим веществом, окрашивающим веществом, веществом, исправляющим вкус лекарственного средства или ароматизатором, и другими веществами к кристаллической форме I или кристаллической форме II по настоящему изобретению с получением таблеток, таблеток, покрытых оболочкой, гранул, порошков, капсул или других подобных лекарственных форм обычным методом.
[0057]
Инъецируемые средства могут быть приготовлены путем добавления регулятора pH, буфера, стабилизатора, изотонического средства, местного анестетика и других веществ к кристаллической форме I или кристаллической форме II по настоящему изобретению и переработки смеси в подкожное, внутримышечное или внутривенное инъецируемое средство обычным методом.
[0058]
Количество кристаллической формы I или кристаллической формы II по настоящему изобретению, добавляемое в каждую лекарственную форму с разовой дозой изменяется в зависимости от симптомов у пациента, которому вводят лекарственное средство или лекарственную форму. Однако, требуемое количество для лекарственной формы с разовой дозой обычно составляет от 0,05 до 1000 мг для пероральных лекарственных средств, от 0,01 до 500 мг для инъецируемых средств и от 1 до 1000 мг для суппозиториев.
[0059]
Суточная доза лекарственного средства в описанных выше лекарственных формах изменяется в зависимости от симптомов, массы тела, возраста, пола пациента и других факторов; и она не может быть одинаковой для любых случаев. Однако, доза в расчете на содержание кристаллической формы I или кристаллической формы II по настоящему изобретению обычно составляет от 0,05 до 5000 мг, и предпочтительно, от 0,1 до 1000 мг для взрослого человека (с массой тела 50 кг) в сутки, и лекарственное средство предпочтительно вводить в форме одной дозы или в форме двух или трех разделенных доз в сутки.
Примеры
[0060]
Далее настоящее изобретение описывается более подробно с помощью примеров, однако, настоящее изобретение не ограничивается этими примерами. Несмотря на то, что настоящее изобретение достаточно подробно описано в примерах, тем не менее, любой специалист в этой области мог бы добавить к нему различные изменения и/или модификации. За исключением тех случаев, когда такие изменения и/или модификации выходят за пределы объема настоящего изобретения, настоящее изобретение охватывает такие изменения и/или модификации.
Используемые в примерах реагенты являются продуктами, производимыми промышленностью, если специально не указано иное.
[0061]
Проведение порошкового рентгеноструктурного анализа (XRD)
Исследование испытуемого вещества методом порошкового рентгеноструктурного анализа проводили при следующих условиях после несильного измельчения некоторого количества испытуемого вещества в агатовой ступке, если в этом была необходимость.
Прибор: RINT-ULTIMA+2100 фирмы Rigaku Corporation
Мишень: Cu
Выход рентгеновского излучения: 40 мА, 40 кВ
Диапазон сканирования: от 5,0 до 40,0°
Шаг сканирования: 0,010°
Скорость сканирования: 5,00°C/мин.
Щель расходимости: 1/2°
Щель рассеивания: 3,00 мм
Приемная щель: 13,00 мм
Эксплуатация прибора и сбор данных осуществлялись в соответствии с методами и методиками, приведенными в инструкции к прибору.
[0062]
Проведение дифференциальной сканирующей калориметрии (DSC)
Анализ методом дифференциальной сканирующей калориметрии проводили при следующих условиях.
Прибор: TA Instruments Q1000
Образец: приблизительно 1 мг
Кювета для образца: из алюминия
Скорость повышения температуры: 10°C/мин
Атмосфера: азот
Расход азота: 50 мл/мин
Эксплуатация прибора и сбор данных осуществлялись в соответствии с методами и методиками, приведенными в инструкции к прибору.
[0063]
Высокоэффективная жидкостная хроматография
Высокоэффективную жидкостную хроматографию проводили при следующих условиях.
[0064]
Прибор: система для жидкостной хроматографии серии 1200 (фирмы Agilent Technologies)
Образец: 0,1 мг/мл 0,1% водный раствор фосфорной кислоты-раствор ацетонитрила (1:1)
Подвижная фаза A: 0,1% водный раствор фосфорной кислоты
Подвижная фаза B: ацетонитрил
Колонка: Ascentis Express C18, 4,6×150 мм, S=2,7 мкм
Длина волны измерения: 210 нм
Эксплуатация прибора и сбор данных осуществлялись в соответствии с методами и методиками, приведенными в инструкции к прибору.
Кроме того, высокоэффективную жидкостную хроматографию проводили также при следующих условиях.
[0065]
Прибор: ACQUITY SQD, Quadrupole (фирмы Waters)
Образец: 0,1 мг/мл раствор ацетониторила
Подвижная фаза A: 0,1% водный раствор муравьиной кислоты
Подвижная фаза B: 0,1% муравьиная кислота-ацетонитрил
Колонка: YMC-Triart C18, 2,0×50 мм, 1,9 мкм (YMC)
Длина волны измерения: 254 нм
Эксплуатация прибора и сбор данных осуществлялись в соответствии с методами и методиками, приведенными в инструкции к прибору.
[0066]
Пример 1. Получение кристаллической форма II (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она
Этанол (9 мл) и воду (1 мл) добавляли к соединению 1 (1,00 г), полученному методом, раскрытом в патентном документе 1, и смесь перемешивали при 75°C в течение 5 минут. Затем температуру понижали до комнатной температуры, и смесь перемешивали в течение 26 часов, затем фильтровали осадок, получая в результате кристаллическую форму II соединения 1 (771 мг, выход 77%).
[0067]
Как показано на фигуре 1, кристаллическая форма II характеризовалась порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°) 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°. Как показано на фигуре 4, кристаллическая форма II характеризовалась эндотермическим пиком (самой высокой величиной пика) приблизительно при 166°C при исследовании методом дифференциальной сканирующей калориметрии (DSC).
[0068]
Пример 2. Получение кристаллической формы I соединения 1
Третбутилметиловый эфир (1 мл) добавляли к соединению 1 (50 мг), полученному методом, раскрытом в патентном документе 1, и смесь перемешивали при комнатной температуре в течение 20 часов, получая в результате кристаллическую форму I соединения 1 (28 мг, выход 56%).
[0069]
Как показано на фигуре 2, кристаллическая форма I характеризовалась порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°) 13,5°, 17,9°, 19,5°, 20,6°, 22,0°, 22,6°, 23,3°, 23,7° и 24,2°. Как показано на фигуре 5, кристаллическая форма I характеризовалась эндотермическим пиком (самой высокой величиной пика) приблизительно при 170°C при исследовании методом дифференциальной сканирующей калориметрии (DSC).
[0070]
Сравнительный пример 1. Кристаллическая форма III соединения 1
Из соединения 1 (1,91 г), полученного методом, раскрытом в патентном документе 1, получали кристаллическую форму III соединения 1 (821 мг, выход 43%) таким же образом, как в примере 1, используя смесь этилацетата и н-гексана.
[0071]
Как показано на фигуре 3, кристаллическая форма III соединения 1 характеризовалась порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°) 9,5°, 12,6°, 13,5°, 20,1°, 20,6°, 22,5°, 23,3°, 23,7° и 24,2°. Как показано на фигуре 6, кристаллическая форма III соединения 1 характеризовалась эндотермическим пиком (самой высокой величиной пика) приблизительно при 140°C и 170°C при исследовании методом дифференциальной сканирующей калориметрии (DSC).
[0072]
Пример испытания 1. Стабильность в твердом состоянии кристаллической формы II соединения 1
Кристаллическую форму I и кристаллическую форму II соединения 1 выдерживали при 40°C, при 40°C (при влажности 75%) или при 60°C в течение 1 месяца. После чего определяли их химическую чистоту методом высокоэффективной жидкостной хроматографии, и изменение химической чистоты под воздействием каждого из условий составляло 0,1% или менее. При исследовании методом дифференциальной сканирующей калориметрии (DSC) кристаллов примеров 1 и 2 и сравнительного примера 1, в отличие от кристаллической формы III соединения 1, описанной в сравнительном примере 1, в случае кристаллической формы I и кристаллической формы II соединения 1 пик не обнаруживался, что позволяет предположить наличие фазового перехода при повышении температуры. Эти результаты указывают на то, что кристаллическая форма I и кристаллическая форма II соединения 1 характеризуются высокой стабильностью в твердом состоянии.
[0073]
Пример испытания 2. Способность к всасыванию при пероральном введении кристаллической формы II соединения 1
Кристаллическую форму I и кристаллическую форму II соединения 1 каждую суспендировали в 0,5% водном растворе гидроксипропил- метилцеллюлозы (HPMC) и перорально вводили мышам линии BALB/c в дозе 50 мг/кг. Через 0,5, 1, 2, 4 и 6 часов после введения, брали кровь у каждой мыши из ретро-орбитального синуса и определяли концентрацию соединения 1 в плазме. Результаты приведены в таблице 1. Способность как кристаллической формы I, так и кристаллической формы II соединения 1 к всасыванию при пероральном введении была высокой, при этом способность кристаллической формы I к всасыванию при пероральном введении была выше. Было также подтверждено, что в результате способности как кристаллической формы I, так и кристаллической формы II к всасыванию при пероральном введении достигалась концентрация, достаточная для обеспечения лечебного эффекта.
[0074]
Таблица 1
мкM·час
[0075]
Пример испытания 3. Сравнение химической чистоты кристаллической формы I и кристаллической формы II соединения 1, полученных из одной и той же партии соединения 1
Неочищенное соединение 1 (50 мг, химическая чистота 98,6%), полученное методом, раскрытым в патентном документе 1, добавляли в 1 мл ацетона, и смесь перемешивали при комнатной температуре в течение 20 часов, затем осадок фильтровали, получая в результате кристаллическую форму II соединения 1.
[0076]
Аналогично, неочищенное соединение 1, полученное, как описано выше, добавляли в этилацетат, и смесь перемешивали при комнатной температуре в течение 20 часов, затем осадок фильтровали, получая в результате кристаллическую форму II соединения 1.
[0077]
Неочищенное соединение 1, полученное, как описано выше, добавляли в третбутилметиловый эфир, и смесь перемешивали при комнатной температуре в течение 20 часов, затем осадок фильтровали, получая в результате кристаллическую форму I соединения 1.
[0078]
В таблице 2 приведена химическая чистота неочищенного соединения 1 и кристаллической формы II и кристаллической формы I соединения 1, полученных их неочищенного соединения 1 путем использования соответствующих растворителей. Обычно можно ожидать, что перекристаллизация повышает химическую чистоту, и эти результаты показывают, что кристаллическая форма II представляет собой кристаллы, из которых могут быть эффективно удалены примеси. Так как руководство ICH-Q3A Международного совета по гармонизации технических требований к лекарственным средствам для медицинского применения (Япония, США и Европа) указывает, что присутствие примесей в количестве 0,03% или более в лекарственном веществе должно регулироваться соответствующими государственными органами, результаты примеров испытаний говорят о возможности практического использования.
[0079]
Таблица 2
форма II
[0080]
Пример испытания 4. Сравнение образования отложений кристаллов на поверхностях оборудования для кристаллической формы I и кристаллической формы II соединения 1
Неочищенное соединение 1, полученное методом, раскрытом в патентном документе 1 (получали 767 мг с теоретическим выходом) и смесь этилацетата (30 мл) и гептана (24 мл) загружали в реактор и нагревали до температуры кипения растворителя с обратным холодильником в течение 1,5 часов. После охлаждения, фильтровали только осадок, диспергированный в растворителе в реакторе, с получением кристаллической формы I соединения 1 (290 мг, выход 38%). Отдельно собирали осадок, прилипший к реактору и другому оборудованию (образовавшееся отложение) и получали кристаллическую форму I соединения 1 (312 мг, выход 41%).
[0081]
Из неочищенной формы 1 получали таким же образом кристаллическую форму II, используя смесь воды с этанолом, ацетоном или этилацетатом, но образовавшееся отложение кристаллической формы II составляло менее 5%.
[0082]
Эти результаты показали, что образование отложений, которое происходило при получении кристаллической формы I соединения 1 составляло приблизительно 40% от выхода, что указывает на то, что образование отложений может приводить к снижению выхода или нарушению нормальной работы технологического оборудования в случае промышленного производства. Вместе с тем, в случае кристаллической формы II, такой проблемы, как образования отложений на поверхностях оборудования, не возникало, и кристаллическая форма II может применяться для промышленного производства крупными партиями.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВАЯ СОЛЬ КОНДЕНСИРОВАННОГО ПИРИМИДИНОВОГО СОЕДИНЕНИЯ И ЕЕ КРИСТАЛЛ | 2016 |
|
RU2702660C2 |
| СОЛЬ И КРИСТАЛЛИЧЕСКАЯ ФОРМА СОЕДИНЕНИЯ, ОБЛАДАЮЩЕГО АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К РЕЦЕПТОРУ S1P | 2020 |
|
RU2833357C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПИРИДО[3,4-D]ПИРИМИДИНОВОГО ПРОИЗВОДНОГО | 2017 |
|
RU2793759C2 |
| КРИСТАЛЛЫ АЗАБИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ | 2016 |
|
RU2697521C2 |
| КРИСТАЛЛ ПРОИЗВОДНОГО БЕНЗОКСАЗОЛА | 2019 |
|
RU2787767C2 |
| НОВЫЕ КРИСТАЛЛЫ УРАЦИЛЬНОГО СОЕДИНЕНИЯ | 2016 |
|
RU2686722C1 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА ТРИАЗОЛО (4,5-d) ПИРИМИДИНА | 2005 |
|
RU2418802C2 |
| ПРОТИВООПУХОЛЕВОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ПРЕРЫВИСТОГО ВВЕДЕНИЯ ИНГИБИТОРА FGFR | 2014 |
|
RU2664118C2 |
| ПРОТИВОРАКОВАЯ ТЕРАПИЯ С ПРИМЕНЕНИЕМ СОЕДИНЕНИЙ 3,5-ДИЗАМЕЩЕННОГО БЕНЗОЛАЛКИНИЛА И ПЕМБРОЛИЗУМАБА | 2020 |
|
RU2822064C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СВОБОДНОГО ОСНОВАНИЯ | 2014 |
|
RU2675851C2 |
Изобретение относится к новой кристаллической форме (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]-пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, который обладает противоопухолевым действием. Кристаллическая форма характеризуется порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°, и эндотермическим пиком при приблизительно 166оС, полученным методом дифференциальной сканирующей калориметрии. 3 н. и 2 з.п. ф-лы, 6 ил., 2 табл., 6 пр.
1. Кристаллы (S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразоло[3,4-d]пиримидин-1-ил)-1-пирролидинил)-2-пропен-1-она, где кристаллы характеризуются порошковой рентгенограммой, содержащей характеристические пики при углах дифракции (2θ±0,2°), выбранных из 9,5°, 14,3°, 16,7°, 19,1°, 20,8°, 21,9° и 25,2°.
2. Кристаллы по п. 1, которые имеют химическую чистоту 99,0% или более.
3. Кристаллы по п. 1 или 2, которые характеризуются эндотермическим пиком (самой высокой величиной пика) приблизительно при 166°C при исследовании методом дифференциальной сканирующей калориметрии.
4. Фармацевтическая композиция, обладающая противоопухолевой активностью, включающая эффективное количество кристаллов по любому одному из пп. 1-3.
5. Фармацевтическая композиция для перорального введения, обладающая противоопухолевой активностью, включающая эффективное количество кристаллов по любому одному из пп. 1-3.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| RU 2009127883А1, 27.01.2011 | |||
| BAVIN Mike, Polimorphism in Process Development, Chemistry & Idustry, 1989, 16, pp | |||
| Приспособление для получения световых декораций на прозрачном экране | 1920 |
|
SU527A1 |
| STEPHEN BYRN et al., Pharmaceutical Solids : A Strategic Approach to Regulatory Considerations, Pharmaceutical Research, 1995, 12(7), pp.945-954 | |||
| Yuki Kagobutsu Kessho Sakusei, Hadbook -Centri to Know-how-, Maruzen Co., Ltd, 2008, pp.57-84 | |||
| Mitsuru HASHIDA, Keiko Toyo Seisai no Sekkei to Hyoka, Kabushiki Kaisha Yakugya Jihosha,1995, pp.76-79, 171-172. | |||

