ПЕРЕКРЕСТНЫЕ ССЫЛКИ К РОДСТВЕННЫМ ЗАЯВКАМ
Данная заявка заявляет право на приоритет, вытекающее из предварительной патентной заявки №60/926403, зарегистрированной 25 апреля 2007 г., и предварительной патентной заявки №60/931449, зарегистрированной 22 мая 2007 г.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к прессованным твердым лекарственным формам, как, например, таблеткам и каплетам, и способам их получения. Конкретнее, изобретение относится к способам таблетирования и полученным с их помощью таблеткам лекарственных препаратов с низкой растворимостью в воде, как, например, тадалафил.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Во многих случаях при пероральном введении твердых лекарственных форм лекарственный препарат должен раствориться в водном желудочном соке, например в желудке больного, прежде чем препарат может проявить терапевтический эффект. Нерешенная проблема с прессованными твердыми лекарственными формами для перорального введения, такими как таблетки и каплеты (т.е. таблетки, имеющие форму капсулы), состоит в том, что скорость растворения некоторых лекарственных препаратов из лекарственной формы ограничивает их биологическую доступность. Эта проблема является сугубо банальной для лекарственных препаратов, которые представляют собой малые органические молекулы с низкой растворимостью в жидкостях, содержащих воду.
Имеется несколько путей, чтобы решить проблему, связанную с растворимостью плохо растворимых лекарственных препаратов. Например, сам лекарственный препарат может быть модифицирован. Физическая форма лекарственного препарата может варьироваться различными техническими приемами, чтобы оптимизировать скорость, при которой лекарственный препарат растворяется. Из этих технических приемов, имеющих наибольшее значение в данном изобретении, является уменьшение размера частиц. Скорость растворения твердого вещества часто может зависеть от площади поверхности, которая подвергается воздействию растворяющей среды; и поскольку площадь поверхности данной массы вещества обычно обратно пропорциональна размеру частиц вещества, то уменьшение размера частиц порошкообразного или гранулированного вещества может увеличить скорость его растворения.
Когда это эффективно, уменьшение размера частиц увеличивает скорость растворения измельченного твердого вещества при увеличении площади поверхности, которая подвергается воздействию растворяющей среды. Однако уменьшение размера частиц не всегда эффективно для увеличения скорости растворения лекарственного препарата, находящегося в прессованной твердой лекарственной форме. Во время процесса производства лекарственной формы многие гидрофобные лекарственные препараты имеют сильную склонность к агломерации в более крупные частицы, приводящей к полному уменьшению эффективной площади поверхности. Монография Remington (The Science and Practice of Pharmacy, 20th ed. 656, 657, A.R.Gennaro Ed., Lippincott Williams & Wilkins, Philadelphia 2000), которая включена в данное изобретение в качестве ссылки, содержит более полное обсуждение понятия "эффективная площадь поверхности" и влияния размера частиц на растворимость. Лекарственный препарат, который как будто был измельчен до мелкодисперсного состояния, будет иногда проявлять свойства по растворимости, которые характерны для частиц большего размера вследствие агломерации или подобного эффекта.
Имеются три известных способа производства прессованных твердых лекарственных форм: способ влажной грануляции, способ двойного прессования (также известный как сухая грануляция) и способ прямого прессования. В каждом из этих способов имеется стадия смешивания, которая может стимулировать агломерацию мелкоизмельченных частиц лекарственного препарата в более крупные, менее быстро растворяющиеся частицы.
В способе влажной грануляции смешиваются предварительно взвешенный лекарственный препарат и один или несколько других ингредиентов, как, например, разбавитель. Далее смесь смешивается с жидкостью, как, например, водой или этанолом, которые вызывают агломерацию частиц во влажную массу. Возможно, жидкость содержит связующее вещество. Влажную массу просеивают с получением гранул, которые впоследствии сушат. Сухие гранулы просеиваются с получением гранул заранее определенного размера. Затем гранулы обычно смешиваются с твердым смазывающим веществом и, возможно, с другими ингредиентами. Наконец, гранулы, содержащие смазывающее вещество и любые другие дополнительные гранулированные ингредиенты, прессуются в таблетку, на которую впоследствии может наноситься покрытие.
Способ двойного прессования или сухой грануляции имеет меньшее количество стадий, чем влажная грануляция, и не требуется контакта с жидкостью или сушка, что делает способ подходящим для технологии приготовления чувствительных к воде и нагреванию лекарственных препаратов. В способе двойного прессования лекарственный препарат и другие ингредиенты, как, например, смазывающее вещество, смешиваются и затем прессуются в первой стадии прессования. Имеются два удобных технологических приема первичного прессования. Один представляет собой уплотнение с помощью валкового пресса, в котором смесь подается между валками, которые прессуют смесь в пластинки; другой - представляет собой агломерацию, в которой смесь прессуется в агломерированную массу, которая имеет близкие к таблеткам формы, которые обычно больше, чем таблетки, предназначенные для человеческого потребления. Полученные пластинки или агломерированную массу далее превращают в гранулы, смешивают с твердым смазывающим веществом и прессуют во второй стадии прессования с получением конечной таблетки.
Способ прямого прессования является самым простым из трех известных способов получения прессованных твердых лекарственных форм. В способе прямого прессования лекарственный препарат и любые другие ингредиенты смешиваются вместе и непосредственно прессуются в конечную таблетку. Таблеточные ингредиенты должны иметь хорошие характеристики текучести и слипания (когезии), чтобы быть пригодными для прямого таблеточного прессования. Микрокристаллическая целлюлоза и лактоза представляют собой два обычно применяемых разбавителя в прямом таблеточном прессовании.
При разработке и производстве фармацевтических составов лекарственных препаратов с плохой растворимостью в воде, когда требуется его высокая биодоступность, обычно применяют комбинацию лекарственного препарата с малым размером частиц (как, например, мелкоизмельченные частицы лекарственного препарата) и соответствующего способа производства (например, один из вышеописанных способов). Способ мелкого измельчения, однако, может повлечь за собой большие издержки по времени и производительности процесса, и может также создать проблему, связанную с безопасностью, так как процесс мелкого измельчения может привести к тонкоизмельченному порошку лекарственного препарата.
Таким образом, любая новая композиция и/или производственный процесс, который обеспечит подходящие показатели растворения и биодоступности при использовании относительно больших частиц лекарственного препарата, может дать более безопасные и более экономически выгодные способы для производства твердых лекарственных форм.
Тадалафил, активный ингредиент в Циалисе® (Cialis®), был применен для лечения нарушения эректильной функции у мужчин. Инструкция по применению препарата Циалис® описывает данный продукт как миндалевидные таблетки с пленочным покрытием для перорального введения, содержащие тадалафил и следующие инертные ингредиенты: кроскармелозу натрия, гидроксипропилцеллюлозу, гипромеллозу, окись железа, лактозу моногидрат, стеарат магния, микрокристаллическую целлюлозу, лаурилсульфат натрия, тальк, двуокись титана и триацетин (см. http://pi.lilly.com/us/cialis-pi.pdf).
Тадалафил имеет химическое название - (6R-транс)-6-(1,3-бензодиоксол-5-ил)-2,3,6,7,12,12а-гексагидро-2-метил-пиразино[1',2':1,6]пиридо[3,4-b]индол-1,4-дион.
Структура тадалафила (CAS #171596-29-5) приведена ниже:
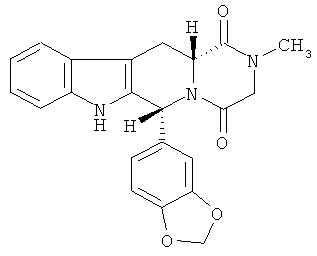
Тадалафил представляет собой твердое вещество, которое практически нерастворимо в воде и весьма незначительно растворимо в некоторых органических растворителях, таких как метанол, этанол и ацетон. В патенте США №6841167 сообщается, что растворимость тадалафила в воде составляет приблизительно 2 мкг в 1 мл воды при 25°C.
Были предприняты различные технические приемы в попытке преодолеть наблюдаемую плохую растворимость тадалафила в воде. В патенте США №6841167 сообщается о фармацевтическом составе, содержащем тадалафил в форме "свободного лекарственного препарата" вместе с разбавителем, смазывающим веществом, гидрофильным связующим веществом и разрыхлителем. При попытке приготовить составы тадалафила с возможно улучшенной биодоступностью предположительно созданы мягкие капсулы, содержащие тадалафил, суспендированный в фармацевтически приемлемом растворителе.
Другой технический прием, применяемый для улучшения растворимости, описан в патенте США №5985326, и включает приготовление составов с помощью "соосаждения" тадалафила, в котором "однородная смесь" тадалафила и носителя в несмешиваемом с водой растворителе и, возможно, с водой соосаждаются из "однородной смеси" с применением водной "среды соосаждения", в которой носитель практически нерастворим.
Патент США №6821975 внесен в «Оранжевую книгу» Федерального Управления США по контролю за продуктами и лекарствами (United States Federal Food and Drug Administration ("FDA")'s "Orange Book") для продукта Циалис® и очевидно предназначается для компании, которая продает на рынке Циалис®. Данный патент предположительно относится к "измельченной форме свободного лекарственного препарата» тадалафила, "содержащего частицы соединения, в котором по меньшей мере 90% частиц имеют размер менее чем приблизительно 40 микрон." Предпочтительно, когда по меньшей мере 90% частиц имеют размер менее 10 микрон.
Вышеописанные способы увеличения биодоступности тадалафила страдают недостатками в экономическом отношении и в смысле безопасности. Патент США №6821975 предлагает проведение мелкого измельчения, которое может быть трудоемким, а также могут возникнуть проблемы, связанные с безопасностью, вследствие полученного из него тонкоизмельченного порошка. Патент США №5985326 предлагает использовать значительные количества органического растворителя, который нежелателен для окружающей среды. Таким образом, способ, который использует большие частицы лекарственного препарата (с d(0.9) в 40 микрон или более) при сохранении нужной биодоступности, будет улучшать экономичность и безопасность процесса производства таблеток тадалафила.
Леви с сотр. (Levy et al.) сообщил о влиянии крахмала на скорость растворения салициловой кислоты из таблеток, полученных двойным прессованием (Levy, G. et al, J. Pharm. Sci. 1963, 52, 1047). Сообщалось, что увеличение содержания крахмала от 5 до 20% увеличивало скорость растворения салициловой кислоты в три раза. Это наблюдение приписывалось более быстрой распадаемости таблеток при более высоком содержании в них крахмала. В 1966 году Финхолт с сотр. (Finholt et al.) наблюдали, что тонкоизмельченные частицы крахмала, прибавленные к таблеткам фенобарбитала, увеличивали скорость растворения фенобарбитала из таблеток. Приходя к другому заключению, чем это было предложено Леви с сотр. (Levy et al.), Финхолт с сотр. (Finholt et al.) предположил, что крахмал действовал в качестве покрытия для кристаллов фенобарбитала и придавал ему гидрофильное свойство, которое улучшало контакт между частицами фенобарбитала и водной средой для растворения (Finholt, P. Medd Norsk Farm. Selsk. 1966, 28, 238).
Крахмал является обычной составной частью таблеток, где он применяется по ряду причин. Он обычно применяется, например, в качестве разбавителя, связующего вещества, разрыхлителя и вещества, способствующего скольжению. Разбавители увеличивают объем твердой фармацевтической композиции и могут делать фармацевтическую лекарственную форму, содержащую эту композицию, более удобной в обращении для больного и лица, осуществляющего уход за ним. Связующие вещества помогают связать активный ингредиент и другие составные части вместе, например, во время стадий грануляции и прессования. Разрыхлители ускоряют распад таблетки в желудке больного обычно путем проникновения воды в таблетку, вызывая ее набухание, тем самым приводя к распаду таблетки на более мелкие части (приводя к большей площади поверхности). Вещества, способствующие скольжению, улучшают сыпучесть порошкообразных композиций путем образования покрытия на поверхностях частиц. Согласно Справочнику по фармацевтическим наполнителям (Handbook of Pharmaceutical Excipients (4th Ed.603-604; Pharmaceutical Press: London 2003)), который полностью приведен в данном изобретении в качестве ссылки, крахмал, если он выполняет функции связующего вещества, обычно используется в количестве 5-15%. Если он функционирует в качестве разрыхлителя, то крахмал обычно прибавляется в количестве 3-15%. Количество разбавителя, которое предусматривается для конкретного применения, зависит от многих параметров и является весьма непостоянным. Однако, как отмечается в Справочнике, крахмал при использовании его в высоких концентрациях плохо прессуется и стремится увеличить хрупкость таблетки и защитного покрытия. Таким образом, применение высоких концентраций крахмала в качестве разбавителя часто ограничивается снижением прочности и повышением хрупкости (ухудшением стойкости к скалыванию), что происходит, когда количественное соотношение крахмала в составе увеличивается.
В публикации США №2006/0099252, которая включена в данное изобретение в качестве ссылки, описывается фармацевтический состав и способ его получения. Публикация описывает составы, содержащие высокие количества крахмала и способы получения таких составов; упомянутые способы включают смешивание активного соединения с крахмалом, прессование смеси в твердое тело, измельчение твердого тела в гранулы, увлажнение и сушку гранул и таблетирование высушенных гранул в твердый фармацевтический состав. В результате осуществления данного способа отмечалось значительное увеличение скорости растворения лекарственных препаратов с низкой растворимостью в воде.
В WO 2005/000296 описывается перорально доставляемая фармацевтическая композиция, содержащая лекарственный препарат с низкой растворимостью в воде и крахмальный клейстер с низкой вязкостью и/или показывающая многомодальное распределение частиц по размеру. Сообщалось, что состав, описанный в WO 2005/000296, показывает повышенную скорость растворения лекарственного препарата.
В публикации США №2008/0009502, которая приводится в данном изобретении в качестве ссылки, раскрывается твердый состав и способ его получения. Публикация раскрывает твердый состав, содержащий тадалафил и по меньшей мере один носитель. Предпочтительно, когда носитель представляет собой гидрофильный полимер, как например, повидон, гидроксипропилметилцеллюлозу и полиэтиленгликоль.
Весьма желательно получить прессованную твердую лекарственную форму для перорального введения с высокой скоростью растворения плохо растворимого лекарственного препарата при минимальном уменьшении размера его частиц.
Таким образом, данное изобретение стремится предложить улучшенные фармацевтические лекарственные формы для тадалафила, которые показывают высокие скорости растворения и которые не требуют уменьшения размера частиц лекарственного препарата (например, как при очень тонком измельчении).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Один из вариантов изобретения предлагает твердую фармацевтическую лекарственную форму, содержащую тадалафил и предпочтительно крахмал, с тем чтобы весовое соотношение крахмала к тадалафилу составляло приблизительно 4,5:1 или более, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равно 40 микронам.
Один вариант изобретения предлагает твердую фармацевтическую лекарственную форму, содержащую тадалафил и крахмал, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равно 40 микронам, в которой количество крахмала в твердой фармацевтической лекарственной форме предпочтительно составляет от приблизительно 45% до приблизительно 60% (вес.).
В предпочтительном варианте фармацевтическая лекарственная форма содержит связующее вещество. Предпочтительно, когда весовое соотношение крахмала к связующему веществу в твердой лекарственной форме составляет от приблизительно 1:1 до приблизительно 9:1.
В предпочтительном варианте фармацевтическая лекарственная форма показывает высокую скорость растворения. Предпочтительно, когда приблизительно 60% или более, а более предпочтительно, когда приблизительно 68% или более тадалафила высвобождается в течение 20 минут, если образец, содержащий 20 мг тадалафила, тестируется в среде по меньшей мере такой же эффективной, как 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°C, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об/мин. Предпочтительно, когда приблизительно 65% или более, а более предпочтительно, когда приблизительно 76% или более тадалафила высвобождается в течение 40 минут при аналогичных условиях.
В предпочтительном варианте фармацевтическая лекарственная форма показывает такую же скорость растворения, что и Циалис® (Cialis®) такой же дозы в тот же самый момент времени при том же условии тестирования. Предпочтительно, когда отношение скорости растворения лекарственной формы к Циалису® той же самой дозы составляет от 80% до 125% в тот же самый момент времени при том же условии тестирования.
В предпочтительном варианте фармацевтическая лекарственная форма биоэквивалентна Циалису® той же самой дозы. Предпочтительно, когда отношение Cmax лекарственной формы к Циалису® той же самой дозы составляет от 80% до 125%. Предпочтительно, когда отношение AUC лекарственной формы к Циалису® той же самой дозы составляет от 80% до 125%.
Один из вариантов изобретения предлагает способ приготовления вышеописанной твердой фармацевтической лекарственной формы, содержащей тадалафил и предпочтительно крахмал.
Один из вариантов изобретения предлагает способ приготовления твердой лекарственной формы тадалафила при помощи влажной грануляции или способа влажного двойного прессования.
Еще один вариант изобретения предлагает способ приготовления твердой лекарственной формы тадалафила, включающий:
а) смешивание твердых частиц тадалафила с гидрофильным веществом;
б) влажную грануляцию продукта стадии а) с жидкостью для грануляции; и
в) сушку продукта стадии б) с образованием высушенных гранул.
Предпочтительно, когда гидрофильным веществом в стадии а) является крахмал. Предпочтительно, когда способ дополнительно включает прессование гранул, полученных в последней стадии, с образованием прессованной твердой лекарственной формы.
Один из вариантов изобретения предлагает твердую фармацевтическую лекарственную форму, описанную в изобретении, для применения в медицине, предпочтительно для применения при лечении эректильной дисфункции у мужчин.
Один из вариантов изобретения предлагает применение твердой фармацевтической лекарственной формы, описанной в изобретении, в приготовлении медикамента для лечения эректильной дисфункции у мужчин.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
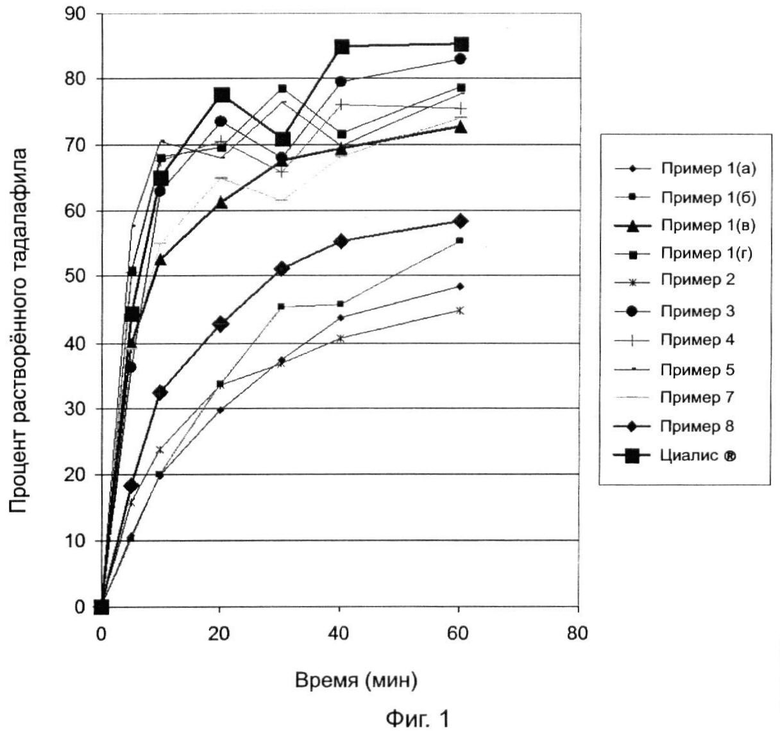
Фиг.1 представляет собой кривую, которая сравнивает скорости растворения тадалафила во времени для:
(1) (а) состава, содержащего тадалафил, смешанный с крахмалом;
(б) состава, содержащего тадалафил, смешанный с крахмалом и спрессованный в агломерированную массу;
(в) состав, содержащий тадалафил, смешанный с крахмалом, спрессованный в агломерированную массу и измельченный;
(г) состав, содержащий тадалафил, смешанный с крахмалом, спрессованный в агломерированную массу, измельченный и гранулированный с помощью влажной грануляции;
(2) состава 2 тадалафила, содержащего обычные наполнители и без крахмала;
(3) состава 3 тадалафила, содержащего крахмал и приготовленного согласно примеру 3;
(4) состава 4 тадалафила, содержащего малое количества крахмала и PVP и приготовленного согласно примеру 4;
(5) состава 5 тадалафила, содержащего крахмал и поверхностно-активное вещество и приготовленного согласно примеру 5;
(6) состава 7 тадалафила, содержащего крахмал и приготовленного согласно примеру 7;
(7) состава 8 тадалафила, приготовленного согласно примеру 8, который соответствует СОСТАВУ 5 из публикации США №2006/099252, за исключением того, что ралоксифен в СОСТАВЕ 5 заменен на тадалафил;
(8) Циалиса® (Cialis), 20 мг.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Как применяется в данном изобретении, термин "PVP" или "повидон" относится к поливинилпирролидону, термин "гидромеллоза" относится к гидроксипропилметилцеллюлозе, термин меглумин относится к N-метил-D-глюкамину и термин "Трис" относится к трис(гидроксиметил)амину.
Как применяется в данном изобретении, термин "инвертированный сахар" относится к эквимолярной смеси двух моносахаридов, глюкозы и фруктозы. Инвертированный сахар можно получить гидролизом сахарозы.
Как применяется в данном изобретении, термин "d(0.9)" относится к 90%-ному распределению частиц по размеру. d(0.9) представляет собой величину распределения, указывающую, что 90% частиц (по суммарному объему) имеют размер, отвечающий этой величине или меньшей, если распределение частиц по размеру измерено обычно принятым методом, как, например, лазерной дифракцией. Аппаратуру для измерения распределения частиц по размеру можно получить от различных источников, как например, от Malvern Instruments Ltd. (U.K.).
Как применяется в данном изобретении, если не указано особо, все проценты являются весовыми процентами, основанными на общем весе твердой лекарственной формы.
Как применяется в данном изобретении, продукт, названный Циалисом®, представляет собой пример серии №А149562.
Как применяется в данном изобретении, скорость растворения измеряется процентным содержанием тадалафила, растворенного в жидкой среде. Условие тестирования является следующим: образец, содержащий 20 мг тадалафила, тестируется в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°C, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об/мин.
Как применяется в данном изобретении, "подобная скорость растворения" как у Циалиса®, относится к отношению скорости растворения лекарственной формы к Циалису®, которая составляет от 80% до 125% для того же самого момента времени при том же условии тестирования для той же самой дозы.
Как применяется в данном изобретении, "биоэквивалентность" или "биоэквивалентный" означает, что продукт отвечает или должен отвечать требованию Управления США по контролю за продуктами и лекарствами (FDA) по биоэквивалентности. Согласно Федеральному уставу кодекса Соединенных Штатов (United States Code of Federal Regulation, Title 21, Vol.5, §320.1), "Биоэквивалентность означает отсутствие значительного отличия в скорости и степени, когда активный ингредиент или активный остаток в фармацевтических эквивалентах или фармацевтических вариантах становится доступным в месте действия лекарственного препарата при введении при одинаковой молярной дозе при одинаковых условиях в соответствующим образом обозначенном исследовании." Например, один из критериев, на который ссылается FDA, определяется в статистических методиках FDA для биоэквивалентных исследований с применением двух стандартных перекрестных методов лечения (Statistical Procedures for Bioequivalence Studies Using a Standard Two-Treatment Crossover Design (1992)), которые приведены в данном изобретении в качестве ссылки. Биоэквивалентность к Циалису® относится к 90%-ному доверительному интервалу отношения Cmax и/или отношения AUCmax, которое составляет от 80% до 125%.
Как применяется в данном изобретении, для тадалафила термин "Cmax" относится к максимальной концентрации тадалафила в плазме после приема внутрь дозы тадалафила; термин "Tmax" относится к времени после приема внутрь, когда достигается Cmax; термин "AUC0-t" относится к площади под кривой концентрации тадалафила в плазме от времени после приема внутрь. Кривая обычно обрывается, когда концентрация становится ниже предела обнаружения. Термин "AUC0-t" относится к AUC для времени «t» от момента приема внутрь. AUC0-∞ представляет общую площадь под кривой и является критерием общего воздействия тадалафила на больного. Если в исследование вовлекается большое количество субъектов, Cmax относится к среднему геометрическому Cmax большого количества субъектов, и AUC относится к среднему геометрическому AUC большого количества субъектов в исследовании.
Как применяется в данном изобретении, термин "отношение Cmax" относится к отношению Cmax тестируемой лекарственной формы к Cmax продукта сравнения Циалис® той же дозы. Отношение Cmax определяется как
Отношение Cmax=Cmax(тест.)/Cmax(сравн.)
Как применяется в данном изобретении, термин "отношение AUC" относится к отношению AUC тестируемого продукта к соответствующей AUC продукта сравнения Циалис® той же самой дозировки. Отношение AUC определяется как
Отношение AUC=AUC(тест)/AUC(сравн.)
Как применяется в данном изобретении, "состояние сытости" относится к состоянию субъекта, который съел рекомендуемую FDA еду или ее эквивалент с высоким содержанием жира (приблизительно 50% от общего калорийного содержания еды) и высокой калорийности (приблизительно от 800 до 1000 калорий). Предпочтительно, когда этот пробный завтрак будет составлять приблизительно 150, 250 и 500-600 калорий из белка, углевода и жира соответственно, как приведено в Руководстве FDA для промышленности: Исследования влияния пищи на биодоступность и биоэквивалентность в состоянии сытости (Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies (1992), которое приведено в данном изобретении в качестве ссылки.
Как применяется в данном изобретении, "состояние голода" относится к состоянию субъекта, который до приема внутрь лекарственной формы не ел по меньшей мере 10 часов, обычно в течение ночи. Предпочтительно, если пища не предусматривалась по меньшей мере в течение четырех часов после принятия дозы, как приведено в Руководстве FDA для промышленности: Исследования влияния пищи на биодоступность и биоэквивалентность в состоянии сытости (Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies (1992).
Один вариант, описанный в изобретении, предлагает твердую фармацевтическую лекарственную форму, содержащую тадалафил и крахмал, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равно 40 мкм, где весовое соотношение крахмала к тадалафилу составляет приблизительно 4,5:1 или более.
Другой вариант, описанный в изобретении, предлагает твердую фармацевтическую лекарственную форму, содержащую тадалафил и крахмал, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равно 40 мкм, в которой количество крахмала в твердой фармацевтической лекарственной форме составляет от приблизительно 45% до приблизительно 60% (вес.).
В предпочтительном варианте, описанном в изобретении, распределение частиц тадалафила по размеру таково, что d(0.9) больше или равно приблизительно 50 мкм, больше или равно приблизительно 60 мкм, или больше или равно приблизительно 70 мкм. Предпочтительно, когда распределение частиц тадалафила по размеру таково, что d(0.9) меньше или равно приблизительно 150 мкм или меньше или равен приблизительно 100 мкм.
Крахмал представляет собой встречающийся в природе полисахарид, который может быть получен из многих различных растительных источников, включая кукурузу, картофель, тапиоку, рис и пшеницу. Крахмал состоит из остатков амилозы и амилопектина. Крахмал коммерчески доступен от многочисленных производителей, как, например, Anheuser Busch, Starchem, AE Staley Mfg. Co., Matheson, Coleman & Bell, and Henkel Corp. Предпочтительным крахмалом для применения в данном изобретении является крахмальный клейстер, удовлетворяющий требованиям Официальной монографии национального фармацевтического справочника (Official Monograph of the National Formulary. United States Pharmacopeia & National Formulary 26/21 2843 (U.S. Pharmacopeial Convention, Inc.: Rockville, MD 2003)).
Предпочтительно, когда количество тадалафила в твердой фармацевтической лекарственной форме составляет от приблизительно 0,2% до приблизительно 20%, от приблизительно 2% до приблизительно 18%, от приблизительно 2,5% до приблизительно 10%, или от приблизительно 3% до приблизительно 8% (вес.).
В предпочтительном варианте весовое соотношение крахмала к тадалафилу в твердой лекарственной форме составляет приблизительно 4,5:1 или более, более предпочтительно приблизительно 5:1 или более, приблизительно 6:1 или более, приблизительно 10:1 или более, приблизительно 12:1 или более или приблизительно 15:1 или более.
В предпочтительном варианте весовое соотношение крахмала к тадалафилу в твердой лекарственной форме составляет приблизительно 600:1 или менее или приблизительно 100:1 или менее. Предпочтительно, когда весовое соотношение крахмала к тадалафилу составляет от приблизительно 4,5:1 до приблизительно 50:1, от приблизительно 4,5:1 до приблизительно 30:1, от приблизительно 10:1 до приблизительно 25:1, от приблизительно 10:1 до приблизительно 20:1, или от приблизительно 15:1 до приблизительно 20:1.
В предпочтительном варианте фармацевтическая лекарственная форма, описанная в изобретении, показывает высокую скорость растворения. Как применяется в данном изобретении, "высокая скорость растворения" приводит к высвобождению приблизительно 65% или более, предпочтительно приблизительно 76% или более тадалафила в течение 40 минут, когда образец, содержащий 20 мг тадалафила, тестируется в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°C, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об/мин.
Предпочтительно приблизительно 60% или более, более предпочтительно приблизительно 68% или более тадалафила высвобождается в течение 20 минут, когда образец, содержащий 20 мг тадалафила, тестируется в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°С, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об/мин.
В предпочтительном варианте фармацевтическая лекарственная форма показывает такую же скорость растворения как Циалис® той же дозировки в тот же самый момент времени при том же самом условии тестирования. Предпочтительно, когда отношение скорости растворения лекарственной формы к Циалису® той же самой дозировки в тот же самый момент времени при том же самом условии составляет от 80% до 125%, от 85% до 125%, от 90% и 125% или от 95% и 125%. Предпочтительно, когда момент времени составляет 20 минут или 40 минут. Условие тестирования является следующим: образец, содержащий 20 мг тадалафила, тестируется в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°С, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об/мин.
В предпочтительном варианте фармацевтическая твердая лекарственная форма, описанная в изобретении, биоэквивалентна Циалису® той же самой дозировки. Предпочтительно, когда концентрация тестируемой лекарственной формы в крови определяется Cmax и/или AUC.
Предпочтительно, когда соотношение Cmax фармацевтической твердой лекарственной формы к Циалису® той же самой дозировки составляет от 80% до 125%, от 85% до 125%, или от 90% до 125%. Вышеприведенные соотношения Cmax можно получить в состоянии сытости и/или в состоянии голодания.
Предпочтительно, когда отношение AUC фармацевтической твердой лекарственной формы к Циалису® той же самой дозировки составляет от 80% до 125%, от 85% до 125%, от 90% до 125%, или от 95% до 125%. Вышеприведенные отношения AUC можно получить в состоянии сытости и/или в состоянии голодания. Возможно вышеприведенные отношения AUC представляют отношения AUC0-t=71 ч. Возможно вышеприведенные отношения AUC представляют отношения AUC0-∞.
В одном варианте фармацевтическая лекарственная форма, описанная в изобретении, обеспечивает Cmax(сыт.) приблизительно 260 нг/мл или более, предпочтительно по меньшей мере приблизительно 300 нг/мл или более при дозировании 20 мг тадалафила. Предпочтительно, когда Cmax(сыт.) составляет менее чем приблизительно 450 нг/мл. Специалисту в данной области должно быть понятно, что величины Cmax будут изменяться в зависимости от введенной дозы.
В одном варианте фармацевтическая твердая лекарственная форма, описанная в изобретении, дает AUC0-∞(сыт.) приблизительно 6000 нг/мг или более, предпочтительно приблизительно 6800 нг/мг или более, более предпочтительно приблизительно 7200 нг/мг или более при дозировании 20 мг тадалафила. Предпочтительно, когда AUC0-∞(сыт.) составляет менее чем приблизительно 9400 нг/мг. Специалисту должно быть понятно, что величины AUC будут изменяться в зависимости от введенной дозы.
В одном варианте фармацевтическая твердая лекарственная форма, описанная в изобретении, дает Cmax(голод.) приблизительно 240 нг/мл или более, предпочтительно по меньшей мере приблизительно 260 нг/мл или более при дозировании 20 мг тадалафила. Предпочтительно, когда Cmax(голод) составляет менее чем приблизительно 400 нг/мл.
В одном варианте фармацевтическая твердая лекарственная форма, описанная в изобретении, дает AUC0-∞(голод.) приблизительно 6000 нг/мл или более, предпочтительно по меньшей мере приблизительно 6500 нг/мл или более при дозировании 20 мг тадалафила. Предпочтительно, когда AUC0-∞(сыт.) составляет менее чем приблизительно 9400 нг/мл.
В предпочтительном варианте фармацевтическая твердая лекарственная форма, описанная в изобретении, дополнительно содержит один или несколько фармацевтически приемлемых наполнителей, выбранных из группы, состоящей из разрыхлителей, поверхностно-активных веществ, связующих веществ, смазывающих веществ, разбавителей и веществ, обеспечивающих скольжение. Специалист в данной области с точки зрения данного изобретения сможет выбрать соответствующие наполнители. Примеры наполнителей можно найти в Справочнике фармацевтических наполнителей - Handbook of Pharmaceutical Excipients, 4th Ed.603-604; Pharmaceutical Press: London 2003.
Предпочтительно, когда наполнителем является связующее вещество. Прибавление связующего вещества может дополнительно увеличить как растворяющее действие крахмала, так и улучшить прессуемость. Примеры соответствующих связующих веществ включают крахмал, желатин, природные сахара, как, например, глюкозу, безводную лактозу, свободно сыпучую лактозу (free-flow lactose), бета-лактозу, сахаристое вещество из кукурузы, природные и синтетические смолы (камеди), как, например, аравийскую камедь, трагакант или альгинат натрия, карбоксиметилцеллюлозу и полиэтиленгликоль, но выбор ими не ограничивается.
Предпочтительно, когда связующее вещество выбрано из группы, состоящей из PVP, полиэтиленгликоля, сахаров, инвертированных сахаров, коллагена, альбумина, целлюлоз в неводных растворителях, полипропиленгликоля, сополимера полиоксиэтилена и полипропилена, сложного эфира полиэтиленгликоля, сложного эфира полиэтиленсорбитана, поли(этиленоксида), гидроксипропилметилцеллюлозы, гидроксипропилцеллюлозы, этилцеллюлозы, гидроксиэтилцеллюлозы, микрокристаллической целлюлозы и их смесей. Предпочтительно, когда связующее вещество выбрано из PVP, гидроксипропилметилцеллюлозы, гидроксипропилцеллюлозы, этилцеллюлозы и гидроксиэтилцеллюлозы. Более предпочтительно, когда связующим веществом является PVP. Предпочтительно, когда PVP выбран из по меньшей мере одного PVP с величиной вязкости К приблизительно 25 и PVP с величиной вязкости К приблизительно 30.
Предпочтительно, когда весовое соотношение крахмала к связующему веществу в твердой лекарственной форме составляет от приблизительно 1:1 до приблизительно 9:1. Более предпочтительно, когда весовое соотношение крахмала к связующему веществу составляет от приблизительно 3:1 до приблизительно 5:1.
В особенно предпочтительном варианте весовое соотношение крахмала к тадалафилу составляет от приблизительно 4,5:1 до приблизительно 50:1, и весовое соотношение крахмала к связующему веществу составляет от приблизительно 1:1 до приблизительно 9:1. Более предпочтительно, когда весовое соотношение крахмала к тадалафилу составляет от приблизительно 10:1 до приблизительно 25:1, и весовое соотношение крахмала к связующему веществу составляет от приблизительно 3:1 до приблизительно 5:1.
В предпочтительном варианте изобретения лекарственные формы, описанные в нем, дополнительно содержат один или несколько фармацевтически приемлемых наполнителей. Предпочтительно, когда один или несколько фармацевтически приемлемых наполнителей выбраны из группы, состоящей из разрыхлителей, поверхностно-активных веществ, связующих веществ, смазывающих веществ, разбавителей и веществ, способствующих скольжению. Предпочтительно, когда наполнитель включает поверхностно-активное вещество. Предпочтительно, когда поверхностно-активное вещество выбрано из группы, состоящей из меглумина, Трис, полоксамера, лаурилсульфата натрия и их смесей.
В предпочтительном варианте изобретения наполнитель включает смазывающее вещество. Примеры соответствующих смазывающих веществ включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и другие, но выбор ими не ограничивается.
В предпочтительном варианте изобретения наполнитель включает разрыхлитель. Предпочтительно, когда разрыхлитель представляет собой комбинацию бикарбоната натрия и винной кислоты.
Возможно, фармацевтическая лекарственная форма может также содержать один или несколько консервантов, стабилизаторов, красителей, ароматизирующих веществ, антиоксидантов и суспендирующих веществ. Примерами консервантов являются бензоат натрия, сорбиновая кислота и сложные эфиры п-гидроксибензойной кислоты, но выбор ими не ограничивается.
В предпочтительном варианте изобретения твердая лекарственная форма является прессованной твердой лекарственной формой. Предпочтительно, когда прессованная твердая лекарственная форма является таблеткой. В фармацевтической промышленности упругость таблеток к удару количественно определяется их твердостью и хрупкостью.
Твердость таблетки определяется измерением склонности таблетки к разрушению под действием приложенного давления. Установки для измерения твердости коммерчески доступны от целого ряда производителей, как, например, KRAEMER (UTS) Ltd. Предпочтительно, когда спрессованные твердые лекарственные формы, описанные в данном изобретении, имеют твердость по меньшей мере приблизительно 5 единиц боковой силы разрушения (Strong-Cobb units, "SCU"), как измерено с помощью системы KRAEMER (UTS). 1,4 SCU = 1 килограмм-силе. Предпочтительно, когда твердость составляет от приблизительно 5 до приблизительно 22, от приблизительно 7 до приблизительно 9, или от приблизительно 9 до приблизительно 22 SCU.
Предпочтительно, когда прессованные твердые лекарственные формы, описанные в данном изобретении, имеют хрупкость приблизительно 1% или менее, приблизительно 0,6% или менее, приблизительно 0,4% или менее, или приблизительно 0%. Хрупкость обычных таблеток измеряется процентом потери веса после обычного стандартного теста на хрупкость, известного каждому специалисту в данной области. Например, хрупкость можно измерить при стандартизированных условиях в соответствии с методом 1216 в USP/NF, взвесив определенное количество таблеток (обычно 20 или более), поместив их во вращающийся барабан из плексиглаза PLEXIGLAS, поднимая их во время повторных круговых вращений с помощью радиального рычага и затем удалением из через диаметр барабана. После повторных круговых вращений таблетки заново взвешивались и вычислялось процентное содержание "стертого" порошка и отломанных кусочков. Полагают, что для большинства продуктов приемлемо, когда максимальная средняя потеря веса от трех образцов составляет не более 1,0%.
В одном варианте изобретения предлагается твердая фармацевтическая лекарственная форма, содержащая тадалафил, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равно 40 мкм, в которой приблизительно 60% или более тадалафила растворяется в течение 20 минут, когда образец, содержащий 20 мг тадалафила, тестируется в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°C, с помощью описанной В американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об/мин.. Предпочтительно, когда приблизительно 68% или более тадалафила растворяется в течение 20 минут.
В одном варианте изобретения предлагается твердая фармацевтическая лекарственная форма, содержащая тадалафил, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равно 40 мкм, в которой приблизительно 65% или более тадалафила растворяется в течение 40 минут, когда образец, содержащий 20 мг тадалафила, тестируется в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющий pH 4,5 при 37°C, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об/мин. Предпочтительно, когда приблизительно 76% или более тадалафила растворяется в течение 40 минут.
В одном варианте изобретения предлагается способ приготовления любой из описанных выше твердых фармацевтических лекарственных форм, содержащих тадалафил.
В одном варианте изобретения предлагается способ приготовления твердой лекарственной формы тадалафила с помощью метода влажной грануляции или влажного двойного прессования. Метод влажного двойного прессования преимущественно применяется для получения прессованных твердых лекарственных форм с высоким содержанием мелкоизмельченного гидрофильного вещества, как, например, крахмала.
В производственном процессе может применяться несколько гидрофильных веществ. В способах, описанных в данном изобретении, относительно высокая концентрация крахмала в композиции оказывает заметное влияние на улучшение скорости растворения готовой лекарственной формы. Таким образом, предпочтительным гидрофильным наполнителем является крахмал.
Предпочтительный способ состоит из следующих стадий:
В первой стадии фармакологически активное соединение в измельченной форме смешивается с порошкообразным гидрофильным веществом, предпочтительно крахмалом, и возможно с поверхностно-активным веществом (например, меглумин, Трис или лаурилсульфат натрия). Вещества следует хорошо перемешать с получением однородной смеси. Количество фармакологически активного соединения и гидрофильного вещества, которое следует перемешать, будет определяться с учетом желаемого соотношения ингредиентов, содержания действующих веществ и размера готовой прессованной лекарственной формы.
Кроме активного соединения, гидрофильного вещества и возможного поверхностно-активного вещества, в этой стадии могут также смешиваться другие ингредиенты. Примеры включают смазывающие вещества, разрыхлители, связующие вещества и разбавители, но выбор ими не ограничивается. Смешивание можно осуществлять любым известным способом и с помощью любого оборудования, способного давать однородную порошкообразную смесь, как, например, V-образного смесителя (конусный смеситель), смесителя для порошков, мешалки с большими сдвигающими усилиями или гранулятора с псевдоожиженным слоем.
Возможно смесь от первой стадии прессуется в прочное твердое вещество, например агломерированную массу, слоистую структуру или пластину. Это можно выполнить с применением стандартных методик сухой грануляции, как, например, агломерации и уплотнения с помощью валковых прессов. В предпочтительном варианте прочное твердое вещество находится в форме агломерированной массы. В другом предпочтительном варианте прочное твердое вещество находится в форме прессованных пластин или уплотненных слоистых структур.
Предпочтительно, когда прочное твердое вещество измельчается в гранулы. Предпочтительно, когда стадия измельчения включает пропускание прочного твердого вещества через сито. Предпочтительно, когда стадия включает измельчение прочного твердого вещества с получением измельченного твердого вещества. Измельчение можно выполнить с помощью мельницы, как, например, мельницы Fitzpatrick, или просеивания. Предпочтительно, когда данная стадия дополнительно включает просеивание таким образом полученного измельченного твердого вещества.
Во второй стадии смесь или гранулы от первой стадии подвергаются влажной грануляции. Если продукт первой стадии представляет собой смесь, к смеси прибавляют жидкость для грануляции для образования гранул. Если продукт первой стадии представляет собой гранулы, гранулы увлажняются жидкостью для грануляции.
Предпочтительно, когда жидкость для грануляции выбрана из воды, C1-C4 спирта и их смесей. Более предпочтительно, когда жидкостью для грануляции является этанол.
В предпочтительном варианте гранулы второй стадии дополнительно содержат один или несколько фармацевтически приемлемых наполнителей. Примеры наполнителей включают разрыхлитель, связующее вещество, смазывающее вещество и вещество, способствующее скольжению, но выбор ими не ограничивается. Предпочтительно, когда наполнителем является связующее вещество. Возможно связующее вещество растворяется или диспергируется в жидкости для грануляции перед тем, как смесь или гранулы от первой стадии увлажняются жидкостью для грануляции.
Соответствующими связующими веществами являются, например, PVP, полиэтиленгликоль, сахара, инвертированные сахара, коллаген, альбумин, целлюлозы в неводных растворителях, поли(пропиленгликоль), сополимер полиоксиэтилена и полипропилена, сложный эфир полиэтиленгликоля, сложный эфир полиэтиленсорбитана, поли(этиленоксид), гидроксипропилцеллюлоза, этилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилметилцеллюлоза и микрокристаллическая целлюлоза. Предпочтительно, когда связующее вещество выбрано из PVP, гидроксипропилметилцеллюлозы, гидроксипропилцеллюлозы, этилцеллюлозы и гидроксиэтилцеллюлозы. Более предпочтительно, когда связующим веществом является PVP, более предпочтительно, когда PVP выбран из по меньшей мере одного PVP с величиной вязкости К, приблизительно 25 и PVP с величиной вязкости К, равной или приблизительно равной 30. В предпочтительном варианте связующим веществом является PVP и жидкость для грануляции представляет собой этанол. Предпочтительно, когда концентрация PVP в растворе составляет от приблизительно 40% до приблизительно 80%, более предпочтительно от приблизительно 55% до приблизительно 65% (вес.).
Возможно, могут прибавляться дополнительные твердые ингредиенты. Такие твердые ингредиенты включают наполнитель, подкисляющий агент, подщелачивающий агент, адсорбент, антиоксидант, буферное вещество, краситель, электролит, эмульгирующий (суспендирующий) агент, ароматизатор, душистое вещество, подсластитель, антиадгезив, связующее вещество, разбавитель, наполнитель, разрыхлитель, вещество, способствующее скольжению, смазывающее вещество, непрозрачную добавку и глянцеватель, но выбор ими не ограничивается. Соответствующие наполнители включают, например, двуосновный фосфат кальция, каолин, сахарозу, маннит, микрокристаллическую целлюлозу, порошкообразную целлюлозу, осажденный карбонат кальция, сорбит, крахмал, лактозу и их комбинации.
Рекомендации в отношении количества применяемой жидкости и типа и количества дополнительных ингредиентов, которые могут прибавляться во время этой стадии смачивания, можно получить с помощью ссылки на известные условия, применяемые в стандартных способах влажной грануляции. Таким образом, используется достаточное количество жидкости, так чтобы гранулы и любые другие твердые ингредиенты, включенные в данную стадию, были бы тщательно смочены, однако же не очень много, чтобы не оставалось значительного количества подвижной жидкости после того, как прибавлены все нерастворимые ингредиенты.
В третьей стадии влажные гранулы от второй стадии сушат. Сушку можно осуществлять с помощью любого стандартного оборудования для сушки, как, например, центробежной сушилки или сушилки в кипящем слое. Температура сушки будет отчасти зависеть от термолабильности активного ингредиента.
Возможно один или несколько фармацевтически приемлемых наполнителей смешиваются с высушенными гранулами. Например, если гранулы будут таблетироваться, возможно перед загрузкой гранул в сырьевой бункер таблеточной машины необходимо прибавить вещество, способствующее скольжению, и/или смазывающее вещество.
Возможно высушенные гранулы от третьей стадии измельчаются. Измельчение можно осуществить с помощью мельницы, как, например, мельницы Fitzpatrick, и/или путем просеивания.
Возможно, измельченные гранулы прессуются в прочное твердое вещество, например агломерированную массу, слоистую структуру или пластину. Это можно осуществить с помощью стандартных методик сухой грануляции, как, например, агломерации и уплотнения с помощью валковых прессов. Предпочтительно, когда прочное твердое вещество находится в форме агломерированной массы.
Предпочтительно, когда прочное твердое вещество измельчается в гранулы. Предпочтительно, когда стадия измельчения состоит из пропускания прочного твердого вещества через сито. Предпочтительно, когда данная стадия включает измельчение прочного твердого вещества в измельченное твердое вещество. Измельчение может осуществляться с помощью мельницы, как, например, мельницы Fitzpatrick, или путем просеивания. Предпочтительно, когда данная стадия дополнительно включает просеивание таким образом полученного измельченного твердого вещества.
В предпочтительном варианте гранулы от третьей стадии прессуются с образованием прессованной твердой лекарственной формы. Предпочтительно, когда прессованная твердая лекарственная форма является таблеткой. Возможно на таблетки наносится покрытие. Возможно таблетки с покрытием или без покрытия запаковываются обычным образом с соответствующей маркировкой, инструктирующей врачей и пациентов по их надлежащему применению.
Предпочтительно, когда твердость прессованной твердой лекарственной формы составляет по меньшей мере приблизительно 5, предпочтительно от приблизительно 5 до приблизительно 22, от приблизительно 7 до приблизительно 9, или от приблизительно 9 до приблизительно 22 единиц боковой силы разрушения, как измерено с помощью системы KRAEMER (UTS). Предпочтительно, когда прессованные твердые лекарственные формы, описанные в данном изобретении, имеют хрупкость (способность крошиться) приблизительно 1% или менее, приблизительно 0,6% или менее, приблизительно 0,4% или менее, или приблизительно 0%, как измерено с помощью описанного выше метода тестирования.
В соответствии с данным изобретением тадалафил может применяться для лечения болезни или состояния путем перорального введения таких лекарственных препаратов в лекарственных формах, описанных в изобретении. Таким образом, другой вариант изобретения предлагает способ лечения болезни или состояния, включающий пероральное введение пациенту твердой фармацевтической лекарственной формы, описанной в изобретении. В предпочтительном варианте болезнью является эректильная дисфункция у мужчин.
Один вариант изобретения предлагает твердую фармацевтическую лекарственную форму, описанную в изобретении, для применения в медицине.
Один вариант изобретения предлагает применение твердой фармацевтической лекарственной формы, описанной в изобретении, в приготовлении медикамента для лечения эректильной дисфункции у мужчин.
Следующие примеры составов являются только иллюстративными и никоим образом не предназначены для ограничения объема изобретения.
ПРИМЕРЫ
Общий
Если не указано особо, профили растворения получали путем тестирования образцов, содержащих 20 мг тадалафила в 1000 мл водного раствора, содержащего 0,14% (вес/вес) лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющего pH 4,5 при 37°С, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об/мин.
Если не указано особо, применяемый в примерах тадалафил имеет такое распределение частиц по размеру, что d(0.9) составляет от приблизительно 45 до приблизительно 58 мкм. Распределение частиц по размеру измерялось с помощью лазерной дифракции. Приборы для измерения распределения частиц по размеру от фирмы Malvern Instruments Ltd. (U.K.).
Если не указано особо, используемый в примерах крахмал является частично превращенным в клейстер кукурузным крахмалом (Starch 1500®, Colorcon, West Point, PA), используемый в примерах PVP, имеет величину вязкости К приблизительно 25 и/или приблизительно 30.
Твердость таблетки измерялась путем определения прочности на излом в поперечном направлении с помощью системы KRAEMER (UTS).
Хрупкость (способность крошиться) таблетки измерялась описанным выше методом.
Пример 1 (а-г)
Композиция, описанная в примере, готовилась из перечисленных в таблице 1 ингредиентов, следуя стадиям, как описано ниже. Образцы, взятые на различных стадиях процесса, тестировались на скорость растворения. Профили растворения образцов состава 1 приводятся ниже в таблице 9 и на Фиг.1.
Ингредиенты части I тщательно перемешивались (пример 1а);
Смесь прессовалась в агломерированную массу (пример 1б);
Агломерированная масса измельчалась (пример 1в);
К измельченной агломерированной массе прибавлялся раствор для грануляции (PVP, растворенный в этаноле);
Увлажненные гранулы, полученные в стадии 4, высушивались и измельчались в сухие гранулы (пример 1 г).
Сравнительный пример 2
Таблетка тадалафила с обычными наполнителями (микрокристаллическая целлюлоза и лактоза) производилась с помощью влажной грануляции. Ингредиенты в таблице 2 готовились влажной грануляцией и затем прессовались в таблетки весом 400 мг. Профиль растворения состава 2 приведен ниже в таблице 9 и Фиг.1.
Ингредиенты части I совместно превращались в измельченное состояние;
Ингредиенты части I и части II тщательно перемешивались;
Смесь гранулировалась прибавлением раствора для грануляции (полоксамер, растворенный в воде), затем гранулы сушились и измельчались;
Далее ингредиенты части II смешивались с гранулами от стадии 2;
Затем ингредиент стадии III смешивался с гранулами от стадии 2 в течение приблизительно 5 минут;
Содержащие смазку гранулы от стадии 4 прессовались в таблетки.
Пример 3 (агломерация + влажная грануляция; весовое соотношение крахмал: тадалафил = 17,5:1; весовое соотношение крахмал: PVP K-30 = 3,2:1)
Гранулы тадалафила готовились из ингредиентов, перечисленных в таблице 3, методом агломерации и влажной грануляции, как описано ниже. Профиль растворения состава 3 приведен ниже в таблице 9 и на Фиг.1.
Ингредиенты части I тщательно смешивались и прессовались в агломерированную массу;
Агломерированная масса измельчалась с получением гранул;
Гранулы увлажнялись раствором PVP в этаноле;
Влажные гранулы сушились и измельчались;
Ингредиенты части III перемешивались вместе с гранулами;
Далее ингредиент части IV смешивался с гранулами в течение приблизительно 5 минут;
Смесь стадии 6 прессовалась в таблетки.
Пример 3а (агломерация + влажная грануляция; весовое соотношение крахмал: тадалафил = 17,5:1; весовое соотношение крахмал: PVP K-30 = 3,2:1)
Гранулы тадалафила готовились из ингредиентов, перечисленных в таблице 3а, методом агломерации и влажной грануляции, как описано в примере 3. Твердость таблетки составляла 7-9 SCU. Хрупкость (способность крошиться) таблетки составляла 0%.
Пример 4: (Весовое соотношение крахмал: тадалафил = 12,5:1; весовое соотношение крахмал: PVP K-30 = 2,9:1)
Тадалафил перемешивался с крахмалом, прессовался в агломерированную массу, измельчался и подвергался влажной грануляции, следуя стадиям, как описано ниже. Профиль растворения состава 4 приведен ниже в таблице 9 и на Фиг.1.
Ингредиенты части I тщательно смешивались и прессовались в агломерированную массу;
Агломерированная масса измельчалась;
Гранулы подвергались влажной грануляции с раствором PVP в этаноле;
Влажные гранулы сушились и измельчались;
Ингредиенты части III перемешивались вместе с гранулами;
Гранулы прессовались в таблетки.
Пример 5 (Влажная грануляция перед агломерацией; весовое соотношение крахмал: тадалафил = 17,5:1; весовое соотношение крахмал: PVP K-30 = 2,9:1)
Гранулы тадалафила готовились из ингредиентов, перечисленных в таблице 5, с помощью метода влажной грануляции и агломерации, как описано ниже. Профиль растворения состава 5 приведен ниже в таблице 9 и на Фиг.1.
Ингредиенты части I тщательно смешивались;
Раствор для грануляции (PVP, растворенный в этаноле) прибавлялся к смеси с образованием гранул;
Гранулы сушились и измельчались с помощью ступки и пестика и просеивались через сито с величиной ячейки 30 меш;
Гранулы прессовались в агломерированную массу и измельчались;
Ингредиенты части III затем прибавлялись и тщательно перемешивались;
Гранулят прессовался в таблетки.
Пример 6 (Влажная грануляция без агломерации; весовое соотношение крахмал: тадалафил = 17,5:1; весовое соотношение крахмал: PVP K-30 = 2,9:1)
Гранулы тадалафила готовились из ингредиентов, перечисленных в таблице 6, с помощью метода влажной грануляции, как описано ниже.
Ингредиенты части I тщательно смешивались;
Раствор для грануляции (PVP, растворенный в этаноле) прибавлялся к смеси;
Гранулы сушились и измельчались с помощью ступки и пестика и затем просеивались через сито с величиной ячейки 30 меш;
Затем прибавлялись ингредиенты части III и тщательно перемешивались;
Гранулы прессовались в таблетки.
Пример 7 (Без агломерации; весовое соотношение крахмал: тадалафил = 5:1; весовое соотношение крахмал: PVP K-30 = 5:1)
Таблетки тадалафила, содержащие крахмал, производились с помощью влажной грануляции без агломерации, как описано ниже. Профиль растворения состава 7 приводится ниже в таблице 9 и на Фиг.1.
Ингредиенты части I тщательно смешивались;
Ингредиенты части II прибавлялись и смешивались со смесью части I;
Прибавлялся раствор для грануляции с последующим прибавлением раствора для грануляции II и перемешиванием;
После увлажнения гранулы сушились и измельчались;
Прибавлялись ингредиенты части III и хорошо перемешивались;
Ингредиент части IV прибавлялся и перемешивался.
Готовые гранулы прессовались в таблетки.
Сравнительный пример 8 (на основе состава 5 из публикации США 2006/0099252; соотношение крахмал: тадалафил = 4,4:1)
Гранулы тадалафила готовились из ингредиентов, перечисленных в таблице 8, с помощью метода агломерации и влажной грануляции, как описано ниже. Это пример, использующий 75% крахмала и весовое соотношение крахмал: тадалафил = 4,4:1. Профиль растворения состава 8 приведен ниже в таблице 9 и на Фиг.1.
Ингредиенты части I тщательно смешивались и прессовались в агломерированную массу;
Агломерированная масса измельчалась;
Гранулы подвергались влажной грануляции с раствором PVP в спирте;
Влажные гранулы сушились и измельчались;
Ингредиенты части III перемешивались вместе с гранулами;
Смесь стадии 5 прессовалась в таблетки.
Результаты по проверке растворимости
Профиль растворения составов, описанных в примерах, и продукта сравнения (% растворения)
Пример 9: Фармакокинетическое исследование
Перекрестное открытое рандоминизированное исследование с 12 субъектами проводилось в едином центре, с одинаковой дозой, тремя периодами, тремя последовательностями, по трем схемам лечения. Исследование предназначалось для сравнения относительной биодоступности двух составов тадалафила (примеры 3а и 7). Продуктом сравнения был Циалис®, 20 мг. В таблице 10 представлены фармакокинетические параметры тадалафила в состояниях сытости и голода.
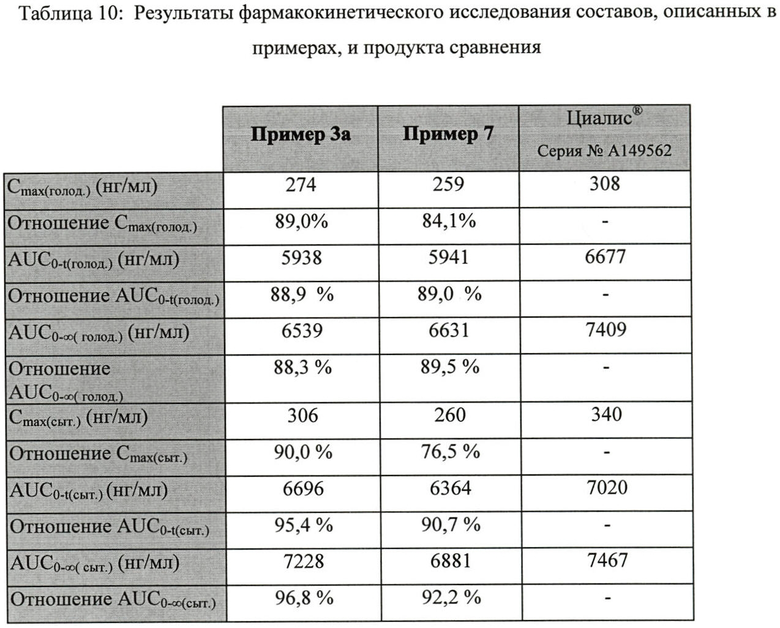
Все величины AUC0-t соответствовали t=71 час.
Все величины Cmax и AUC представляли среднее геометрическое значение для большого количества субъектов.
Хотя в данном изобретении были описаны определенные предпочтительные варианты, специалисту в данной области должно быть понятно, что изменения и модификации описанных вариантов могут осуществляться в пределах сущности и объема изобретения. Таким образом, предполагается, что изобретение ограничивается только до степени, требуемой прилагаемой формулой изобретения и применимыми прецедентными нормами.
Прессованная твердая фармацевтическая лекарственная форма содержит тадалафил и крахмал. Весовое соотношение крахмала к тадалафилу составляет приблизительно 5:1 или более. Распределение частиц тадалафила по размеру таково, что d(0,9) больше или равен 40 мкм. Твердость прессованной твердой лекарственной формы составляет 5 единиц боковой силы разрушения или более. Также описаны способы приготовления таблеток тадалафила. Лекарственная форма тадалафила по изобретению с частицами большого размера показывает высокую скорость растворения, когда приблизительно 60%, предпочтительно 68% или более тадалафила высвобождается в течение 20 минут при дозировке в 20 мг тадалафила. Лекарственная форма тадалафила применяется для лечения нарушения эректильной дисфункции у мужчин. 9 н. и 85 з.п. ф-лы, 1 ил., 10 табл., 9 пр.
1. Прессованная твердая фармацевтическая лекарственная форма, содержащая тадалафил и крахмал, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равно 40 мкм, при этом твердость прессованной твердой лекарственной формы составляет 5 единиц боковой силы разрушения или более, и весовое соотношение крахмала к тадалафилу составляет приблизительно 5:1 или более.
2. Лекарственная форма по п.1, в которой весовое соотношение крахмала к тадалафилу составляет приблизительно 6:1 или более.
3. Лекарственная форма по п.2, в которой весовое соотношение крахмала к тадалафилу составляет приблизительно 10:1 или более.
4. Лекарственная форма по п.3, в которой весовое соотношение крахмала к тадалафилу составляет приблизительно 12:1 или более.
5. Лекарственная форма по п.4, в которой весовое соотношение крахмала к тадалафилу составляет приблизительно 15:1 или более.
6. Лекарственная форма по п.1, в которой весовое соотношение крахмала к тадалафилу составляет от приблизительно 5:1 до приблизительно 50:1.
7. Лекарственная форма по п.6, в которой весовое соотношение крахмала к тадалафилу составляет от приблизительно 5:1 до приблизительно 30:1.
8. Лекарственная форма по п.7, в которой весовое соотношение крахмала к тадалафилу составляет от приблизительно 10:1 до приблизительно 25:1.
9. Лекарственная форма по п.8, в которой весовое соотношение крахмала к тадалафилу составляет от приблизительно 10:1 до приблизительно 20:1.
10. Лекарственная форма по п.9, в которой весовое соотношение крахмала к тадалафилу составляет от приблизительно 15:1 до приблизительно 20:1.
11. Лекарственная форма по п.1, в которой количество тадалафила составляет от 0,2% до приблизительно 20 вес.%.
12. Лекарственная форма по п.1, в которой крахмал представляет собой крахмальный клейстер.
13. Лекарственная форма по п.1, в которой приблизительно 60% или более тадалафила растворяется в течение 20 мин, если образец, содержащий 20 мг тадалафила, тестирован в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°С, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об/мин.
14. Лекарственная форма по п.1, в которой Cmax(голод.) составляет приблизительно 240 нг/мл или более при дозировке в 20 мг тадалафила.
15. Лекарственная форма по п.1, в которой AUC0-∞(сыт.) составляет приблизительно 6000 нг/мг или более при дозировке в 20 мг тадалафила.
16. Лекарственная форма по п.1, в которой AUC0-∞(голод.) составляет приблизительно 6000 нг/мл или более при дозировке в 20 мг тадалафила.
17. Лекарственная форма по п.1, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равен приблизительно 50 мкм.
18. Лекарственная форма по п.1, в которой распределение частиц тадалафила по размеру таково, что d(0.9) меньше или равно приблизительно 150 мкм.
19. Лекарственная форма по п.1, дополнительно содержащая один или несколько фармацевтически приемлемых наполнителей, выбранных из группы, состоящей из разрыхлителей, поверхностно-активных веществ, связующих веществ, смазывающих веществ, разбавителей и веществ, способствующих скольжению.
20. Лекарственная форма по п.1, дополнительно содержащая поверхностно-активное вещество.
21. Лекарственная форма по п.1, дополнительно содержащая смазывающее вещество.
22. Лекарственная форма по п.1, дополнительно содержащая разрыхлитель.
23. Лекарственная форма по п.1, дополнительно содержащая один или несколько консервантов, стабилизаторов, красителей, ароматизирующих веществ, антиоксидантов и суспендирующих веществ.
24. Прессованная твердая лекарственная форма по п.1, представляющая собой таблетку.
25. Способ приготовления твердой лекарственной формы по п.1, включающий применение метода влажного двойного прессования.
26. Способ приготовления твердой лекарственной формы по п.1, включающий применение метода влажной грануляции.
27. Способ приготовления твердой лекарственной формы по п.1, включающий:
а) смешивание измельченного тадалафила с крахмалом,
б) влажную грануляцию продукта стадии а) с жидкостью для грануляции, и
в) сушку продукта стадии б) с образованием высушенных гранул.
28. Твердая фармацевтическая лекарственная форма по п.1 для применения при лечении эректильной дисфункции у мужчин.
29. Прессованная твердая фармацевтическая лекарственная форма, содержащая тадалафил и крахмал, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равно 40 мкм, и в которой количество крахмала составляет от приблизительно 45% до приблизительно 60 вес.%, причем твердость прессованной твердой лекарственной формы составляет 5 единиц боковой силы разрушения или более.
30. Лекарственная форма по п.29, в которой количество тадалафила составляет от 0,2% до приблизительно 20 вес.%.
31. Лекарственная форма по п.30, в которой количество тадалафила составляет от приблизительно 2% до приблизительно 18 вес.%.
32. Лекарственная форма по п.31, в которой количество тадалафила составляет от приблизительно 2,5% до приблизительно 10 вес.%.
33. Лекарственная форма по п.32, в которой количество тадалафила составляет от приблизительно 3% до приблизительно 8 вес.%.
34. Лекарственная форма по п.29, в которой крахмал представляет собой крахмальный клейстер.
35. Лекарственная форма по п.29, в которой приблизительно 60% или более тадалафила растворяется в течение 20 мин, если образец, содержащий 20 мг тадалафила, тестирован в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°С, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об./мин.
36. Лекарственная форма по п.29, в которой приблизительно 68% или более тадалафила растворяется в течение 20 мин, если образец, содержащий 20 мг тадалафила, тестирован в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°C, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об./мин.
37. Лекарственная форма по п.29, в которой приблизительно 65% или более тадалафила растворяется в течение 40 мин, если образец, содержащий 20 мг тадалафила, тестирован в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°С, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об./мин.
38. Лекарственная форма по п.37, в которой приблизительно 76% или более тадалафила растворяется в течение 40 мин, если образец, содержащий 20 мг тадалафила, тестирован в 1000 мл водной среды, содержащей 0,14 вес.% лаурилсульфата натрия и 6 г/л NaH2PO4 и имеющей pH 4,5 при 37°C, с помощью описанной в американской фармакопее (USP) установки II с лопастями, вращающимися со скоростью 50 об./мин.
39. Лекарственная форма по п.29, в которой Cmax(голод.) составляет приблизительно 240 нг/мл или более при дозировке приблизительно 20 мг тадалафила.
40. Лекарственная форма по п.39, в которой Cmax(голод.) составляет приблизительно 260 нг/мл или более.
41. Лекарственная форма по п.40, в которой Cmax(сыт.) составляет приблизительно 385 нг/мл или менее.
42. Лекарственная форма по п.29, в которой AUC0-∞(сыт.) составляет приблизительно 6000 нг/мг или более при дозировке в 20 мг тадалафила.
43. Лекарственная форма по п.42, в которой AUC0-∞(сыт.) составляет приблизительно 6800 нг/мг или более.
44. Лекарственная форма по п.43, в которой AUC0-∞(сыт.) составляет приблизительно 7200 нг/мг или более.
45. Лекарственная форма по п.42, у которой AUC0-∞(сыт.) составляет приблизительно 9400 нг/мл или менее.
46. Лекарственная форма по п.29, в которой AUC0-∞(голод.) составляет приблизительно 6000 нг/мл или более при дозировке в 20 мг тадалафила.
47. Лекарственная форма по п.46, в которой AUC0-∞(голод.) составляет приблизительно 6500 нг/мл или более.
48. Лекарственная форма по п.46, в которой AUC0-∞(голод.) составляет приблизительно 9300 нг/мл или менее.
49. Лекарственная форма по п.29, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равен приблизительно 50 мкм.
50. Лекарственная форма по п.49, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равен приблизительно 60 мкм.
51. Лекарственная форма по п.50, в которой распределение частиц тадалафила по размеру таково, что d(0.9) больше или равен приблизительно 70 мкм.
52. Лекарственная форма по п.29, в которой распределение частиц тадалафила по размеру таково, что d(0.9) меньше или равен приблизительно 150 мкм.
53. Лекарственная форма по п.52, в которой распределение частиц тадалафила по размеру таково, что d(0.9) меньше или равен приблизительно 100 мкм.
54. Лекарственная форма по п.29, дополнительно содержащая один или несколько фармацевтически приемлемых наполнителей, выбранных из группы, состоящей из разрыхлителей, поверхностно-активных веществ, связующих веществ, смазывающих веществ, разбавителей и веществ, способствующих скольжению.
55. Лекарственная форма по п.54, в которой наполнителем является связующее вещество.
56. Лекарственная форма по п.55, в которой связующее вещество выбрано из группы, состоящей из поливинилпирролидона, гидроксипропилметилцеллюлозы, гидроксипропилцеллюлозы, этилцеллюлозы, гидроксиэтилцеллюлозы и их смесей.
57. Лекарственная форма по п.56, в которой связующим веществом является поливинилпирролидон.
58. Лекарственная форма по п.57, в которой поливинилпирролидон выбран из по меньшей мере одного поливинилпирролидона с величиной вязкости К приблизительно 25 и поливинилпирролидона с величиной вязкости К приблизительно 30.
59. Лекарственная форма по п.58, содержащая крахмал, причем весовое соотношение крахмала к связующему веществу в твердой лекарственной форме составляет от приблизительно 1:1 до приблизительно 9:1.
60. Лекарственная форма по п.59, в которой весовое соотношение крахмала к связующему веществу составляет от приблизительно 3:1 до приблизительно 5:1.
61. Лекарственная форма по п.29, дополнительно содержащая поверхностно-активное вещество.
62. Лекарственная форма по п.61, в которой поверхностно-активное вещество выбрано из группы, состоящей из меглумина, Трис, лаурилсульфата натрия и их смесей.
63. Лекарственная форма по п.29, дополнительно содержащая смазывающее вещество.
64. Лекарственная форма по п.63, в которой смазывающее вещество выбрано из группы, состоящей из олеата натрия, стеарата натрия, стеарата магния, бензоата натрия, ацетата натрия, хлорида натрия и их смесей.
65. Лекарственная форма по п.29, дополнительно содержащая разрыхлитель.
66. Лекарственная форма по п.65, в которой разрыхлитель представляет собой комбинацию бикарбоната натрия и винной кислоты.
67. Лекарственная форма по п.29, дополнительно содержащая один или несколько консервантов, стабилизаторов, красителей, ароматизирующих веществ, антиоксидантов и суспендирующих веществ.
68. Прессованная твердая лекарственная форма по п.29, представляющая собой таблетку.
69. Прессованная твердая лекарственная форма по п.68, твердость которой составляет от приблизительно 5 до приблизительно 22 единиц боковой силы разрушения.
70. Прессованная твердая лекарственная форма по п.69, твердость которой составляет от приблизительно 7 до приблизительно 9 единиц боковой силы разрушения.
71. Прессованная твердая лекарственная форма по п.69, твердость которой составляет от приблизительно 9 до приблизительно 22 единиц боковой силы разрушения.
72. Прессованная твердая лекарственная форма по п.66, хрупкость которой составляет приблизительно 1% или менее.
73. Прессованная твердая лекарственная форма по п.72, хрупкость которой составляет приблизительно 0,6% или менее.
74. Прессованная твердая лекарственная форма по п.73, хрупкость которой составляет приблизительно 0,4% или менее.
75. Прессованная твердая лекарственная форма по п.74, хрупкость которой составляет приблизительно 0%.
76. Способ приготовления твердой лекарственной формы по п.29, включающий применение метода влажного двойного прессования.
77. Способ приготовления твердой лекарственной формы по п.29, включающий применение метода влажной грануляции.
78. Способ приготовления твердой лекарственной формы по п.29, включающий:
а) смешивание измельченного тадалафила с крахмалом,
б) влажную грануляцию продукта стадии а) с жидкостью для грануляции, и
в) сушку продукта стадии б) с образованием высушенных гранул.
79. Способ по п.78, в котором в стадии а) поверхностно-активное вещество смешивают с тадалафилом и крахмалом.
80. Способ по п.78, дополнительно включающий прессование смеси от стадии а) в агломерированную массу.
81. Способ по п.80, в котором агломерированную массу измельчают в гранулы.
82. Способ по п.78, в котором жидкость для грануляции выбрана из воды, C1-C4 спирта и их смесей.
83. Способ по п.82, в котором жидкостью для грануляции является этанол.
84. Способ по п.78, в котором жидкость для грануляции дополнительно содержит связующее вещество.
85. Способ по п.84, в котором связующим веществом является поливинилпирролидон.
86. Способ по п.85, в котором концентрация поливинилпирролидона в растворе составляет от приблизительно 40% до приблизительно 80 вес.%.
87. Способ по п.86, в котором концентрация поливинилпирролидона в растворе составляет от приблизительно 55% до приблизительно 65 вес.%.
88. Способ по п.78, в котором высушенные гранулы от стадии в) измельчают.
89. Способ по п.88, дополнительно включающий прессование измельченных гранул в агломерированную массу.
90. Способ по п.89, в котором агломерированную массу измельчают в гранулы.
91. Способ по п.78, в котором гранулы от последней стадии прессуют с образованием прессованной твердой лекарственной формы.
92. Способ по п.91, в котором прессованной твердой лекарственной формой является таблетка.
93. Твердая фармацевтическая лекарственная форма по п.29 для применения при лечении эректильной дисфункции у мужчин.
94. Применение твердой фармацевтической лекарственной формы по п.29 в производстве медикамента для лечения эректильной дисфункции у мужчин.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| US 6821975 B1, 23.11.2004 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

