ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к способу получения композиции поликарбоновой кислоты. Один из аспектов настоящего изобретения относится к частичному окислению ароматического соединения (например, пара-ксилола) с получением сырой ароматической дикарбоновой кислоты (например, сырой терефталевой кислоты), которая затем может быть подвергнута очистке и выделению. Другой аспект изобретения относится к усовершенствованной реакторной системе, которая обеспечивает более эффективный и экономичный процесс окисления.
УРОВЕНЬ ТЕХНИКИ
Реакции окисления используются в ряде существующих промышленных процессов. Например, жидкофазное окисление в настоящее время используют для окисления альдегидов до кислот (например, пропионового альдегида до пропионовой кислоты), окисление циклогексана до адипиновой кислоты и окисление алкилароматических соединений до спиртов, кислот или дикислот. Особенно значимым промышленным процессом окисления последней категории (окисление алкилароматических соединений) является жидкофазное каталитическое окисление пара-ксилола до терефталевой кислоты. Терефталевая кислота является важным соединением, находящим целый ряд применений. Основное применение терефталевой кислоты состоит в применении в качестве исходного сырья при производстве полиэтилентерефталата (ПЭТ, РЕТ). ПЭТ является хорошо известным пластиком, используемым в больших количествах во всем мире для производства изделий, таких как бутылки, волокна и упаковочный материал.
В типичном процессе жидкофазного окисления, включая частичное окисление пара-ксилола до терефталевой кислоты, жидкофазный поток сырья и газофазный поток окислителя вводят в реактор и получают в реакторе многофазную реакционную среду. Жидкофазный поток сырья, введенный в реактор, содержит, по меньшей мере, одно способное к окислению органическое соединение (например, пара-ксилол), тогда как газофазный поток окислителя содержит молекулярный кислород. По меньшей мере, часть молекулярного кислорода, введенного в реактор в виде газа, растворяется в жидкой фазе реакционной среды, что обеспечивает доступность кислорода для жидкофазной реакции. Если жидкая фаза многофазной реакционной среды содержит недостаточную концентрацию молекулярного кислорода (например, если некоторые части реакционной среды являются «обедненными по кислороду»), нежелательные побочные реакции могут приводить к образованию примесей и/или целевые реакции могут быть замедлены по скорости. Если жидкая фаза многофазной реакционной среды содержит слишком мало способного к окислению соединения, скорость реакции может быть неприемлемо медленной. Кроме того, если жидкая фаза реакционной среды содержит избыточную концентрацию способного к окислению соединения, дополнительные нежелательные побочные реакции могут вызывать образование примесей.
Обычные реакторы жидкофазного окисления оборудованы средствами смешения для перемешивания многофазной реакционной среды, находящейся в них. Перемешивание реакционной среды предусмотрено в целях стимулирования растворения молекулярного кислорода в жидкой фазе реакционной среды, поддержания относительно равномерных концентраций растворенного кислорода в жидкой фазе реакционной среды и поддержания относительно равномерных концентраций способного к окислению органического соединения в жидкой фазе реакционной среды.
Перемешивание реакционной среды, подвергающейся жидкофазному окислению, часто достигается с помощью механических средств перемешивания в емкостях, таких как, например, непрерывные реакторы смешения (НРС, CSTR). Хотя НРС могут обеспечить тщательное перемешивание реакционной среды, НРС имеют ряд недостатков. Например, НРС имеют относительно высокую капитальную стоимость, так как для них требуются дорогие двигатели, гидростатические подшипники и приводные валы и/или сложные перемешивающие механизмы. Кроме того, вращающиеся и/или колеблющиеся механические компоненты обычных НРС требуют регулярного технического обслуживания. Затраты труда и время отключения, связанные с таким техническим обслуживанием, увеличивают эксплуатационные расходы для НРС. Однако даже при регулярном техническом обслуживании системы механического перемешивания, используемые в НРС, склонны к механическим поломкам и могут потребовать замены в течение относительно короткого периода времени.
Реакторы по типу барботажной колонны дают привлекательную альтернативу НРС и другим реакторам окисления с механическим перемешиванием. Реакторы по типу барботажной колонны обеспечивают перемешивание реакционной среды без применения дорогого и ненадежного механического оборудования. Реакторы по типу барботажной колонны обычно включают удлиненную вертикальную реакционную зону, внутри которой находится реакционная среда. Перемешивание реакционной среды в реакционной зоне обеспечивается преимущественно за счет естественной плавучести пузырьков газа, поднимающихся вверх через жидкую фазу реакционной среды. Перемешивание за счет естественной плавучести, создаваемое в реакторах по типу барботажной колонны, снижает капитальные и эксплуатационные расходы относительно реакторов с механическим перемешиванием. Кроме того, по существу отсутствие движущихся механических деталей, связанных с реакторами по типу барботажной колонны, обеспечивает систему окисления, которая менее склонна к механическим поломкам, чем реакторы с механическим перемешиванием.
Когда жидкофазное частичное окисление пара-ксилола проводят в обычных реакторах окисления (НРС или в барботажной колонне), продукт, выводимый из реактора, как правило, представляет собой суспензию, содержащую сырую терефталевую кислоту (СТК, СТА) и маточную жидкость. СТК имеет относительно высокий уровень примесей (например, 4-карбоксибезальдегида, пара-толуиловой кислоты, флуоренонов и других окрашенных соединений), что делает ее неприемлемой в качестве сырья для производства ПЭТ. Следовательно, СТК, произведенная в обычных реакторах окисления, как правило, подвергается процессу очистки, который превращает СТК в очищенную терефталевую кислоту (ОТК, РТА), приемлемую для производства ПЭТ.
Один из типичных процессов очистки для превращения СТК в ОТК включает следующие стадии: (1) замена маточной жидкости суспензии, содержащей СТК, водой; (2) нагревание суспензии СТК/вода для растворения СТК в воде; (3) каталитическое гидрирование раствора СТК/вода для превращения примесей в более желаемые и/или легко отделяемые соединения; (4) осаждение полученной ОТК из раствора гидрирования с помощью множества стадий кристаллизации и (5) выделение кристаллизованной ОТК из остальных жидкостей. Хотя этот тип обычного процесса очистки является эффективным, он может быть очень дорогим. Отдельные факторы, вносящие вклад в повышение стоимости обычных методов очистки СТК, включают, например, тепловую энергию, требуемую для стимулирования растворения СТК в воде, катализатор, необходимый для гидрирования, поток водорода, требуемый для гидрирования, потерю выхода, вызванную гидрированием некоторого количества терефталевой кислоты, и множество сосудов, необходимых для многоступенчатой кристаллизации. Следовательно, было бы желательно разработать систему окисления, способную производить СТК, которая могла бы быть очищена без необходимости стимулированного нагреванием растворения в воде, гидрирования и/или многоступенчатой кристаллизации.
ЦЕЛИ ИЗОБРЕТЕНИЯ
Таким образом, целью настоящего изобретения является разработка более эффективной и экономичной системы жидкофазного окисления.
Другая цель изобретения состоит в разработке более эффективного и экономичного реактора и процесса жидкофазного каталитического частичного окисления пара-ксилола до терефталевой кислоты.
Еще одной целью настоящего изобретения является создание реактора по типу барботажной колонны, который способствует улучшенному протеканию реакций жидкофазного окисления с пониженным образованием примесей.
Еще одна цель настоящего изобретения состоит в разработке более эффективной и экономичной системы для производства чистой терефталевой кислоты (ЧТК, РТА) путем жидкофазного окисления пара-ксилола с получением сырой терефталевой кислоты (СТК) и затем очистки СТК до ЧТК.
Другая цель настоящего изобретения состоит в создании реактора по типу барботажной колонны для окисления пара-ксилола и производства СТК, способной подвергаться очистке без необходимости стимулируемого нагреванием растворения СТК в воде, гидрирования растворенной СТК и/или многоступенчатой кристаллизации гидрированной СТК.
Следует отметить, что объем настоящего изобретения, который определен в прилагаемой формуле изобретения, не ограничен способами и оборудованием, которые способны реализовать все перечисленные выше цели. Более того, объем заявленного изобретения может охватывать ряд систем, которые не достигают всех или каких-либо из перечисленных выше целей. Другие цели и преимущества настоящего изобретения будут легко очевидны специалисту в данной области техники при рассмотрении следующего подробного описания и сопровождающих чертежей.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Один из вариантов осуществления настоящего изобретения относится к способу производства композиции поликарбоновой кислоты, и этот способ включает следующие стадии: (а) проведение окисления многофазной реакционной среды в реакторе первичного окисления, в результате чего получают первую суспензию; и (b) проведение дополнительного окисления, по меньшей мере, части первой суспензии в реакторе вторичного окисления, где реактор вторичного окисления представляет собой реактор по типу барботажной колонны.
Другой вариант осуществления настоящего изобретения относится к реакторной системе. Реакторная система включает реактор первичного окисления и реактор вторичного окисления. Реактор первичного окисления определяет границы первого впускного отверстия и первого выпускного отверстия. Реактор вторичного окисления представляет собой реактор по типу барботажной колонны, который определяет второе впускное отверстие и второе выпускное отверстие. Первое выпускное отверстие связано передачей потока жидкости со вторым впускным отверстием.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Предпочтительные варианты осуществления настоящего изобретения описаны более подробно ниже со ссылкой на прилагаемые чертежи, где
ФИГ. 1 представляет собой вид сбоку реактора окисления, выполненного в соответствии с одним из вариантов осуществления настоящего изобретения, в частности, иллюстрирующий введение потоков сырья, окислителя и флегмы в реактор, присутствие многофазной реакционной среды в реакторе и выведение газа и суспензии из верхней части и нижней части реактора соответственно;
ФИГ. 2 представляет собой увеличенный вид сбоку в разрезе нижней части реактора по типу барботажной колонны, полученный вдоль линии 2-2 на ФИГ. 3, в частности, иллюстрирующий расположение и конфигурацию барботера окислителя, используемого для введения потока окислителя в реактор;
ФИГ. 3 представляет собой вид сверху барботера окислителя ФИГ. 2, в частности, иллюстрирующий, что в верхней части барботера отсутствуют отверстия для подачи окислителя;
ФИГ. 4 представляет собой вид снизу барботера окислителя ФИГ. 2, в частности, иллюстрирующий конфигурацию отверстий для подачи окислителя в нижней части барботера;
ФИГ. 5 представляет собой вид сбоку в разрезе барботера окислителя, полученный вдоль линии 5-5 на ФИГ. 3, в частности, иллюстрирующий ориентацию отверстий для подачи окислителя в нижней части барботера окислителя;
ФИГ. 6 представляет собой увеличенный вид сбоку в разрезе нижней части реактора по типу барботажной колонны, в частности, иллюстрирующий систему для введения потока сырья в реактор в многочисленных, разделенных вертикальными промежутками местоположениях;
ФИГ. 7 представляет собой вид сверху, полученный вдоль линии 7-7 на ФИГ. 6, в частности, иллюстрирующий, как система введения сырья, показанная на ФИГ. 6, распределяет поток сырья в предпочтительной радиальной зоне сырья (ЗС, FZ) и более чем одном азимутальном квадранте (Q1, Q2, Q3, Q4);
ФИГ. 8 представляет собой вид сверху в разрезе, аналогичный ФИГ. 7, но иллюстрирующий альтернативные средства для подачи потока сырья в реактор с использованием байонетных трубок, каждая из которых имеет множество небольших отверстий для сырья;
ФИГ. 9 представляет собой изометрический вид альтернативной системы для введения потока сырья в реакционную зону в многочисленных, разделенных вертикальными промежутками местоположениях без необходимости множества врезок в сосуд, в частности, иллюстрирующий, что система распределения сырья, по меньшей мере, частично может опираться на барботер окислителя;
ФИГ. 10 представляет собой вид сбоку системы распределения сырья с одной врезкой и барботера окислителя, представленных на ФИГ. 9;
ФИГ. 11 представляет собой вид сверху в разрезе, полученный по линии 11-11 на ФИГ. 10, и дополнительно иллюстрирует систему распределения сырья с одной врезкой, опирающуюся на барботер окислителя;
ФИГ. 12 представляет собой вид сбоку реактора по типу барботажной колонны, оборудованного внутренней и внешней реакционными емкостями;
ФИГ. 13 представляет собой увеличенный вид в разрезе реактора по типу барботажной колонны ФИГ. 12, полученный по линии 13-13, в частности, иллюстрирующий относительную ориентацию внутренней и внешней реакционных емкостей;
ФИГ. 14 представляет собой вид сбоку альтернативного реактора по типу барботажной колонны, оборудованного внутренней и внешней реакционными емкостями, в частности, иллюстрирующий, что внутренняя реакционная емкость имеет ступенчатый диаметр;
ФИГ. 15 представляет собой вид сбоку реактора по типу барботажной колонны, оборудованного внешним реактором вторичного окисления, который принимает суспензию от боковой фракции в реакторе первичного окисления;
ФИГ. 16 представляет собой вид сбоку реактора по типу барботажной колонны, оборудованного сквозным внешним реактором вторичного окисления, который принимает суспензию из увеличенного отверстия в боковой части реактора первичного окисления;
ФИГ. 17а представляет собой схематичный вид сбоку реактора по типу барботажной колонны, оборудованного внутренней структурой для усиления гидродинамики в реакторе;
ФИГ. 17b представляет собой вид в разрезе реактора ФИГ. 17а, полученный по линии 17b-17b на ФИГ. 17а;
ФИГ. 18а представляет собой схематичный вид сбоку реактора по типу барботажной колонны, оборудованного первой альтернативной внутренней структурой для усиления гидродинамики реактора;
ФИГ. 18b представляет собой вид в разрезе реактора ФИГ. 18а, полученный по линии 18b-18b на ФИГ. 18а;
ФИГ. 19а представляет собой схематичный вид сбоку реактора по типу барботажной колонны, оборудованного второй альтернативной внутренней структурой для усиления гидродинамики реактора;
ФИГ. 19b представляет собой вид в разрезе реактора ФИГ. 19а, полученный по линии 19b-19b на ФИГ. 19а;
ФИГ. 20а представляет собой схематичный вид сбоку реактора по типу барботажной колонны, оборудованного третьей альтернативной внутренней структурой для усиления гидродинамики реактора;
ФИГ. 20b представляет собой вид в разрезе реактора ФИГ. 20а, полученный по линии 20b-20b на ФИГ. 20а;
ФИГ. 21а представляет собой схематичный вид сбоку реактора по типу барботажной колонны, оборудованного четвертой альтернативной внутренней структурой для усиления гидродинамики реактора;
ФИГ. 21b представляет собой вид в разрезе реактора ФИГ. 21а, полученный по линии 21b-21b на ФИГ. 21а;
ФИГ. 22а представляет собой схематичный вид сбоку реактора по типу барботажной колонны, оборудованного пятой альтернативной внутренней структурой для усиления гидродинамики реактора;
ФИГ. 22b представляет собой вид в разрезе реактора ФИГ. 22а, полученный по линии 22b-22b на ФИГ. 22а;
ФИГ. 23а представляет собой схематичный вид сбоку реактора по типу барботажной колонны, оборудованного шестой альтернативной внутренней структурой для усиления гидродинамики реактора;
ФИГ. 23b представляет собой вид в разрезе реактора ФИГ. 23а, полученный по линии 23b-23b на ФИГ. 23а;
ФИГ. 24а представляет собой схематичный вид сбоку реактора по типу барботажной колонны, оборудованного седьмой альтернативной внутренней структурой для усиления гидродинамики реактора;
ФИГ. 24b представляет собой вид в разрезе реактора ФИГ. 24а, полученный по линии 24b-24b на ФИГ. 24а;
ФИГ. 25а представляет собой схематичный вид реактора по типу барботажной колонны со ступенчатым диаметром с усиливающей гидродинамику внутренней структурой;
ФИГ. 25b представляет собой вид в разрезе реактора ФИГ. 25а, полученный по линии 25b-25b на ФИГ. 25а;
ФИГ. 26 представляет собой вид сбоку реактора по типу барботажной колонны, содержащего многофазную реакционную среду, в частности, иллюстрирующий реакционную среду, которая теоретически распределена на 30 горизонтальных тонких слоев равного объема, чтобы количественно определить некоторые градиенты в реакционной среде;
ФИГ. 27 представляет собой вид сбоку реактора по типу барботажной колонны, содержащего многофазную реакционную среду, в частности, иллюстрирующий первый и второй дискретные 20-процентные сплошные объемы реакционной среды, которые имеют по существу различные концентрации кислорода и/или скорости потребления кислорода;
ФИГ. 28А и 28В представляют собой увеличенные виды частиц сырой терефталевой кислоты (СТК), произведенных в соответствии с одним из вариантов осуществления настоящего изобретения, в частности, иллюстрирующие, что каждая частица СТК представляет собой частицу низкой плотности с высокой площадью поверхности, состоящую из множества свободно связанных субчастиц СТК;
ФИГ. 29А и 29В представляют собой увеличенные виды полученной обычным образом СТК, особенно иллюстрирующие, что частица обычной СТК имеет увеличенный размер, более высокую плотность и более низкую площадь поверхности, чем частица заявляемой СТК ФИГ. 28А и 28В;
ФИГ. 30 показывает упрощенную схему технологических потоков процесса предшествующего уровня техники для производства очищенной терефталевой кислоты (ОТК) и
ФИГ. 31 показывает упрощенную схему технологических потоков процесса производства ОТК в соответствии с одним из вариантов осуществления настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Один из вариантов осуществления настоящего изобретения относится к жидкофазному частичному окислению способного к окислению соединения. Такое окисление предпочтительно проводят в жидкой фазе многофазной реакционной среды, находящейся в одном или в нескольких реакторах с перемешиванием. Подходящими реакторами с перемешиванием являются, например, реакторы с пузырьковым перемешиванием (например, реакторы по типу барботажной колонны), реакторы с механическим перемешиванием (например, непрерывные реакторы смешения) и реакторы с проточным перемешиванием (например, инжекторные реакторы). В одном из вариантов настоящего изобретения жидкофазное окисление проводят с использованием, по меньшей мере, одного реактора по типу барботажной колонны.
Используемое в данном случае определение «реактор по типу барботажной колонны» будет означать реактор для облегчения химических реакций в многофазной реакционной среде, где перемешивание реакционной смеси обеспечивается преимущественно за счет движения по направлению вверх пузырьков газа через реакционную среду. Используемое в данном случае определение «перемешивание» будет означать работу, распределенную в реакционной среде, которая вызывает потоки жидкости и/или перемешивание. Используемые в данном случае определения «большинство», «преимущественно» и «особенно» будут означать более чем 50%. Используемое в данном случае определение «механическое перемешивание» будет означать перемешивание реакционной среды, созданное физическим движением жесткого(их) или гибкого(их) элемента(ов) против или внутри реакционной среды. Например, механическое перемешивание может быть обеспечено за счет вращения, колебания и/или вибрации внутренних мешалок, лопастей, вибраторов или акустических диафрагм, расположенных в реакционной среде. Используемое в данном случае определение «перемешивание потоком» будет означать перемешивание реакционной среды, вызванное за счет высокой скорости инжекции и/или рециркулирования одной или нескольких текучих сред в реакционной среде. Например, перемешивание потоком может быть обеспечено с помощью насадок, эжекторов и/или эдукторов.
В предпочтительном варианте осуществления настоящего изобретения менее чем приблизительно 40% перемешивания реакционной среды в реакторе по типу барботажной колонны во время окисления обеспечивается с помощью механического перемешивания и/или перемешивания потоком, более предпочтительно менее чем приблизительно 20% перемешивания обеспечивается с помощью механического перемешивания и/или перемешивания потоком, и наиболее предпочтительно менее чем приблизительно 5% перемешивания обеспечивается с помощью механического перемешивания и/или перемешивания потоком. Предпочтительно количество механического перемешивания и/или перемешивания потоком, воздействующего на многофазную реакционную среду во время окисления, составляет менее чем приблизительно 3 киловатта на кубический метр реакционной среды, более предпочтительно менее чем приблизительно 2 киловатта на кубический метр и наиболее предпочтительно менее чем приблизительно 1 киловатт на кубический метр.
На ФИГ. 1 предпочтительный реактор по типу барботажной колонны 20 показан как состоящий из оболочки емкости 22, которая имеет реакционную секцию 24 и секцию отделения 26. Реакционная секция 24 определяет границы реакционной зоны 28, тогда как секция отделения 26 определяет границы зоны разделения 30. Преимущественно жидкофазный поток сырья вводят в реакционную зону 28 через впускное отверстие для сырья 32а,b,c,d. Преимущественно газофазный поток окислителя вводят в реакционную зону 28 через барботер окислителя 34, расположенный в нижней части реакционной зоны 28. Жидкофазный поток сырья и газофазный поток окислителя вместе образуют многофазную реакционную среду 36 в пределах реакционной зоны 28. Многофазная реакционная среда 36 содержит жидкую фазу и газовую фазу. Более предпочтительно многофазная реакционная среда 36 содержит трехфазную среду, имеющую твердофазный, жидкофазный и газофазный компоненты. Твердофазный компонент реакционной среды 36 предпочтительно выпадает в осадок внутри реакционной зоны 28 в результате реакции окисления, проводимой в жидкой фазе реакционной среды 36. Реактор по типу барботажной колонны 20 включает выпускное отверстие для суспензии 38, расположенное около днища реакционной зоны 28, и выпускное отверстие для газа 40, расположенное около верха зоны отделения 30. Суспензионный выходящий поток, содержащий жидкофазный и твердофазный компоненты реакционной среды 36, выводят из реакционной зоны 28 через выпускное отверстие для суспензии 38, тогда как преимущественно газообразный выходящий поток выводят из зоны отделения 30 через выпускное отверстие для газа 40.
Жидкофазный поток сырья, введенный в реактор по типу барботажной колонны 20 через впускное отверстие для сырья 32а,b,c,d, предпочтительно содержит способное к окислению соединение, растворитель и каталитическую систему.
Способное к окислению соединение, присутствующее в жидкофазном потоке сырья, предпочтительно содержит, по меньшей мере, одну углеводородную группу. Более предпочтительно способное к окислению соединение является ароматическим соединением. Еще более предпочтительно способное к окислению соединение является ароматическим соединением, по меньшей мере, с одной прикрепленной углеводородной группой, или, по меньшей мере, с одной прикрепленной замещенной углеводородной группой, или, по меньшей мере, с одним прикрепленным гетероатомом, или, по меньшей мере, с одной прикрепленной функцией карбоновой кислоты (-СООН). Даже более предпочтительно способное к окислению соединение является ароматическим соединением, по меньшей мере, с одной прикрепленной углеводородной группой, или, по меньшей мере, с одной прикрепленной замещенной углеводородной группой, причем каждая из прикрепленных групп содержит от 1 до 5 атомов углерода. Еще более предпочтительно способное к окислению соединение является ароматическим соединением, содержащим именно две прикрепленные группы, причем каждая из прикрепленных групп содержит именно один атом углерода и содержит метильные группы и/или замещенные метильные группы и/или, самое большее, одну группу карбоновой кислоты. Даже еще более предпочтительно способное к окислению соединение является пара-ксилолом, мета-ксилолом, пара-толуиловым альдегидом, мета-толуиловым альдегидом, пара-толуиловой кислотой, мета-толуиловой кислотой и/или ацетальдегидом. Наиболее предпочтительно способное к окислению соединение представляет собой пара-ксилол.
«Углеводородная группа», как это определено в описании, представляет собой, по меньшей мере, один атом углерода, который связан только с атомами водорода или с другими атомами углерода. «Замещенная углеводородная группа», как это определено в описании, представляет собой, по меньшей мере, один атом углерода, связанный, по меньшей мере, с одним гетероатомом и, по меньшей мере, с одним атомом водорода. «Гетероатомы», как это определено в данном случае, представляют собой все атомы, отличные от атомов углерода и водорода. Ароматические соединения, которые определены в описании, содержат ароматическое кольцо, предпочтительно имеющее, по меньшей мере, 6 атомов углерода, даже более предпочтительно имеющее только атомы углерода как часть ароматического кольца. Подходящими примерами таких ароматических колец являются, но не ограничиваются ими, бензол, бифенил, терфенил, нафталин и другие конденсированные ароматические кольца на основе углерода.
Если способное к окислению соединение, присутствующее в жидкофазном потоке сырья, является обычным твердым соединением (то есть является твердым при стандартной температуре и стандартном давлении), предпочтительно, чтобы способное к окислению соединение было по существу растворено в растворителе при введении в реакционную зону 28. Предпочтительно, чтобы точка кипения способного к окислению соединения при атмосферном давлении составляла, по меньшей мере, приблизительно 50ºС. Более предпочтительно температура кипения способного к окислению соединения находится в интервале приблизительно от 80 до 400ºС и наиболее предпочтительно в интервале от 125 до 155ºС. Количество способного к окислению соединения, присутствующее в жидкофазном потоке сырья, предпочтительно находится в интервале приблизительно от 2 до 40% масс., более предпочтительно в интервале приблизительно от 4 до 20% масс. и наиболее предпочтительно в интервале приблизительно от 6 до 15% масс.
В настоящее время отмечено, что способное к окислению соединение, присутствующее в жидкофазном сырье, может включать комбинацию двух или более различных способных к окислению химикатов. Эти два или более различных химических материалов могут быть поданы объединенными в жидкофазном потоке сырья или могут быть поданы отдельно в многочисленных потоках сырья. Например, способное к окислению соединение, содержащее пара-ксилол, мета-ксилол, пара-толуиловый альдегид, пара-толуиловую кислоту и ацетальдегид, может быть подано в реактор через одно впускное отверстие или через множество отдельных впускных отверстий.
Растворитель, присутствующий в жидкофазном потоке сырья, включает кислотный компонент и водный компонент. Растворитель предпочтительно присутствует в жидкофазном потоке сырья в концентрации в интервале приблизительно от 60 до 98% масс., более предпочтительно в интервале приблизительно от 80 до 96% масс. и наиболее предпочтительно в интервале от 85 до 94% масс. Кислотный компонент растворителя предпочтительно представляет собой преимущественно низкомолекулярную монокарбоновую кислоту, содержащую 1-6 атомов углерода, более предпочтительно 2 атома углерода. Наиболее предпочтительно кислотный компонент растворителя представляет собой преимущественно уксусную кислоту. Предпочтительно кислотный компонент составляет, по меньшей мере, приблизительно до 75% масс. растворителя, более предпочтительно, по меньшей мере, приблизительно 80% масс. растворителя и наиболее предпочтительно от 85 до 98% масс. растворителя, причем остаток составляет главным образом вода. Растворитель, введенный в реактор по типу барботажной колонны 20, может включать незначительные количества примесей, таких как, например, пара-толуиловый альдегид, терефталевый альдегид, 4-карбоксибензальдегид (4-КБА, 4-СВА), бензойная кислота, пара-толуиловая кислота, пара-толуиловый альдегид, альфа-бром-пара-толуиловая кислота, изофталевая кислота, фталевая кислота, тримеллитовая кислота, полиароматические соединения и/или суспендированные материалы в виде частиц. Предпочтительно, чтобы суммарное количество примесей в растворителе, введенном в реактор по типу барботажной колонны 20, составляло менее чем приблизительно 3% масс.
Каталитическая система, присутствующая в жидкофазном потоке сырья, предпочтительно является гомогенной, жидкофазной каталитической системой, способной активировать окисление (включая частичное окисление) способного к окислению соединения. Более предпочтительно каталитическая система содержит, по меньшей мере, один поливалентный переходный металл. Еще более предпочтительно поливалентный переходный металл представляет собой кобальт. Даже более предпочтительно каталитическая система включает кобальт и бром. Наиболее предпочтительно каталитическая система включает кобальт, бром и марганец.
Когда кобальт присутствует в каталитической системе, предпочтительно, чтобы количество кобальта, присутствующего в жидкофазном потоке сырья, было таковым, чтобы концентрация кобальта в жидкой фазе реакционной среды 36 поддерживалась в интервале приблизительно от 300 до 6000 массовых частей на миллион (масс.ч./млн), более предпочтительно в интервале приблизительно от 700 до 4200 масс.ч./млн, и наиболее предпочтительно в интервале от 1200 до 3000 масс.ч./млн. Когда в каталитической системе присутствует бром, предпочтительно, чтобы количество брома, присутствующего в жидкофазном потоке сырья, было таковым, чтобы концентрация брома в жидкой фазе реакционной среды 36 поддерживалась в интервале приблизительно от 300 до 5000 масс.ч./млн, более предпочтительно в интервале приблизительно от 600 до 4000 масс.ч./млн и наиболее предпочтительно в интервале от 900 до 3000 масс.ч./млн. Когда в каталитической системе присутствует марганец, предпочтительно, чтобы количество марганца, присутствующего в жидкофазном потоке сырья, было таковым, чтобы концентрация марганца в жидкой фазе реакционной среды 36 поддерживалась в интервале приблизительно от 20 до 1000 масс.ч/млн, более предпочтительно в интервале приблизительно от 40 до 500 масс.ч/млн, и наиболее предпочтительно в интервале от 50 до 200 масс.ч/млн.
Концентрации кобальта, брома и/или марганца в жидкой фазе реакционной среды 36, приведенные выше, выражены из расчета на усредненные по времени и усредненные по объему значения. Используемое в данном случае понятие «усредненное по времени» означает среднее значение, по меньшей мере, для 10 измерений, полученных в одинаковых условиях в течение непрерывного периода времени, по меньшей мере, 100 сек. Используемое в данном случае понятие «усредненное по объему» означает среднее значение, по меньшей мере, для 10 измерений, полученных в однородном 3-мерном пространстве по всему определенному объему.
Массовое отношение кобальта к брому (Со:Br) в каталитической системе, введенной в реакционную зону 28, предпочтительно находится в интервале приблизительно от 0,25:1 до 4:1, более предпочтительно в интервале приблизительно от 0,5:1 до 3:1 и наиболее предпочтительно в интервале от 0,75:1 до 2:1. Массовое отношение кобальта к марганцу (Со:Mn) в каталитической системе, введенной в реакционную зону 28, предпочтительно находится в интервале приблизительно от 0,3:1 до 40:1, более предпочтительно в интервале приблизительно от 5:1 до 30:1 и наиболее предпочтительно в интервале от 10:1 до 25:1.
Жидкофазный поток сырья, введенный в реактор по типу барботажной колонны 20, может включать небольшие количества примесей, таких как, например, толуол, этилбензол, пара-толуиловый альдегид, терефталевый альдегид, 4-карбоксибензальдегид (4-КБА, 4-СВА), бензойная кислота, пара-толуиловая кислота, пара-толуиловый альдегид, альфа-бром-пара-толуиловая кислота, изофталевая кислота, фталевая кислота, тримеллитовая кислота, полиароматические соединения и/или суспендированные материалы в виде частиц. Когда реактор по типу барботажной колонны 20 используют для производства терефталевой кислоты, мета-ксилол и орто-ксилол также считаются примесями. Предпочтительно, чтобы суммарное количество примесей в жидкофазном сырье, введенном в реактор по типу барботажной колонны 20, составляло менее чем приблизительно 3% масс.
Хотя ФИГ. 1 иллюстрирует вариант осуществления изобретения, где способное к окислению соединение, растворитель и каталитическая система смешаны вместе и вводятся в реактор по типу барботажной колонны 20 в виде единственного потока сырья, в альтернативном варианте осуществления изобретения способное к окислению соединение, растворитель и каталитическая система могут быть введены в реактор по типу барботажной колонны 20 по отдельности. Например, допустимо подавать поток пара-ксилола в реактор по типу барботажной колонны 20 через впускное отверстие, отдельное от впускного(ых) отверстия(ий) для растворителя и катализатора.
Преимущественно газофазный поток окислителя, введенный в реактор по типу барботажной колонны 20 через барботер окислителя 34, содержит молекулярный кислород (О2). Предпочтительно поток окислителя содержит в интервале приблизительно от 5 до 40% мол. молекулярного кислорода, более предпочтительно в интервале приблизительно от 15 до 30% мол. молекулярного кислорода и наиболее предпочтительно в интервале от 18 до 24% мол. молекулярного кислорода. Предпочтительно, чтобы остальное количество потока окислителя составлял главным образом газ или газы, такие как азот, которые инертны к окислению. Более предпочтительно поток окислителя состоит по существу из молекулярного кислорода и азота. Наиболее предпочтительно поток окислителя представляет собой сухой воздух, который содержит приблизительно 21% мол. молекулярного кислорода и приблизительно от 78 до 81% мол. азота. В альтернативном варианте настоящего изобретения поток окислителя может содержать по существу чистый кислород.
На ФИГ. 1 реактор по типу барботажной колонны 20 предпочтительно оборудован распределителем флегмы 42, расположенным выше верхней поверхности 44 реакционной среды 36. Распределитель флегмы 42 работает так, чтобы вводить капли преимущественно жидкофазного потока флегмы в зону отделения 30 с помощью любого средства образования капель, известного в данной области техники. Более предпочтительно распределитель флегмы 42 дает распыл капель, направленный вниз по направлению к верхней поверхности 44 реакционной среды 36. Предпочтительно этот направленный вниз распыл капель оказывает воздействие (то есть, затрагивает и влияет), по меньшей мере, приблизительно на 50% площади максимального горизонтального поперечного сечения зоны отделения 30. Более предпочтительно распыл капель оказывает воздействие, по меньшей мере, на 75% площади максимального горизонтального поперечного сечения зоны отделения 30. Наиболее предпочтительно распыл капель оказывает воздействие, по меньшей мере, на 90% площади максимального горизонтального поперечного сечения зоны отделения 30. Этот направленный вниз распыл жидкой флегмы может способствовать предупреждению образования пены на верхней поверхности или выше верхней поверхности 44 реакционной среды 36 и также может способствовать отделению любых капель жидкости или суспензии, захваченных перемещающимся вверх газом, который протекает в направлении выпускного отверстия для газа 40. Кроме того, жидкая флегма может служить для снижения количества материала в форме частиц и потенциального осаждения соединений (например, растворенной бензойной кислоты, пара-толуиловой кислоты, 4-КБА, терефталевой кислоты и каталитических солей металла), присутствующих в газообразном выходящем потоке, выводимом из зоны отделения 30 через выпускное отверстие для газа 40. Кроме того, введение капель флегмы в зону отделения 30 за счет перегонки может быть подогнано к композиции газообразного выходящего потока, выводимого через выпускное отверстие для газа 40.
Поток жидкой флегмы, введенный в реактор по типу барботажной колонны 20 через распределитель флегмы 42, предпочтительно имеет приблизительно тот же состав, что и компонент растворителя жидкофазного потока сырья, введенного в реактор по типу барботажной колонны 20 через впускное отверстие для сырья 32а,b,c,d. Следовательно, предпочтительно, чтобы поток жидкой флегмы содержал кислотный компонент и воду. Кислотный компонент потока флегмы предпочтительно представляет собой низкомолекулярную монокарбоновую кислоту, содержащую 1-6 атомов углерода, более предпочтительно 2 атома углерода. Наиболее предпочтительно кислотный компонент потока флегмы представляет собой уксусную кислоту. Предпочтительно кислотный компонент составляет, по меньшей мере, приблизительно до 75% масс. потока флегмы, более предпочтительно, по меньшей мере, приблизительно 80% масс. потока флегмы и наиболее предпочтительно от 85 до 98% масс. потока флегмы, причем остальное количество составляет вода. Так как поток флегмы, как правило, по существу имеет тот же состав, что и растворитель в жидкофазном потоке сырья, когда в описании дается ссылка на «весь растворитель», введенный в реактор, такой «весь растворитель» будет включать как поток флегмы, так и растворитель как часть потока сырья.
Во время жидкофазного окисления в реакторе по типу барботажной колонны 20, предпочтительно, чтобы потоки сырья, окислителя и флегмы по существу непрерывно вводились в реакционную зону 28, тогда как выходящие потоки газа и суспензии по существу непрерывно выводились из реакционной зоны 28. Используемое в данном случае определение «по существу непрерывно» означает период в течение, по меньшей мере, 10 час, прерываемый менее чем на 10 минут. Во время окисления предпочтительно, чтобы способное к окислению соединение (например, пара-ксилол) по существу непрерывно вводилось в реакционную зону 28 при скорости, по меньшей мере, приблизительно 8000 килограмм в час, более предпочтительно при скорости в интервале приблизительно от 15000 до 200000 кг/час, еще более предпочтительно в интервале приблизительно от 22000 до 150000 кг/час и наиболее предпочтительно в интервале приблизительно от 30000 до 100000 кг/час. Хотя, как правило, предпочтительно, чтобы расходы поступающих потоков сырья, окислителя и флегмы были по существу стационарными, надо отметить, что один из вариантов осуществления настоящего изобретения предполагает пульсирование поступающего потока сырья, окислителя и/или флегмы, чтобы улучшить смешение и массовую передачу. Когда поступающий поток сырья, окислителя и/или флегмы вводят в пульсирующем режиме, предпочтительно, чтобы их расходы менялись в пределах приблизительно от 0 до 500% от стационарных расходов, указанных в настоящем изобретении, более предпочтительно в пределах приблизительно от 30 до 200% от стационарных расходов, указанных в настоящем изобретении, наиболее предпочтительно в пределах от 80 до 120% от стационарных расходов, указанных в настоящем изобретении.
Средняя объемная скорость реакции (СОС, STR) в реакторе по типу барботажной колонны 20 определяется как масса способного к окислению соединения, поданная на единицу объема реакционной среды 36 в единицу времени (например, килограмм пара-ксилола, поданного на кубический метр в час). При обычном применении количество способного к окислению соединения, не превращенного в продукт, перед расчетом СОС, как правило, необходимо вычитать из количества способного к окислению соединения в потоке сырья. Однако конверсия и выход обычно являются высокими для многих способных к окислению соединений, предпочтительных в данном случае (например, для пара-ксилола), и удобно определять это понятие так, как указано выше. По причине капитальных затрат и рабочих материалов, наряду с другими, в целом предпочтительно, чтобы реакция проводилась с высокой СОС. Однако проведение реакции с все более и более высокой СОС может оказывать влияние на количество или выход частичного окисления. Реактор по типу барботажной колонны 20 особенно полезен, когда СОС способного к окислению соединения (например, пара-ксилола) находится в интервале приблизительно от 25 до 400 килограмм на кубический метр в час, более предпочтительно в интервале приблизительно от 30 до 250 кг/м3·час, еще более предпочтительно приблизительно от 35 до 150 кг/м3·час и наиболее предпочтительно от 40 до 100 кг/м3·час.
СОС кислорода в реакторе по типу барботажной колонны 20 определяют как массу молекулярного кислорода, потребленную на единицу объема реакционной среды 36 в единицу времени (например, килограмм молекулярного кислорода на кубический метр в час). По причине капитальных затрат и расхода растворителя на окисление, наряду с другими, в целом предпочтительно, чтобы реакция проводилась с высокой СОС кислорода. Однако проведение реакции с более и более высокой СОС кислорода снижает количество или выход частичного окисления. Без привязки к какой-либо теории, это, по-видимому, связано со скоростью переноса молекулярного кислорода из газовой фазы в жидкую фазу у площади межфазной поверхности и оттуда в объем жидкости. Слишком высокая СОС кислорода, вероятно, приводит к слишком низкому содержанию растворенного кислорода в объеме жидкой фазы реакционной среды.
Обобщенно-усредненную СОС кислорода определяют в данном случае как весь кислород, потребленный во всем объеме реакционной среды 36 в единицу времени (например, килограмм молекулярного кислорода, потребленного на кубический метр в час). Реактор по типу барботажной колонны 20 особенно полезен, когда обобщенно-усредненная СОС кислорода находится в интервале приблизительно от 25 до 400 килограмм на кубический метр в час, более предпочтительно в интервале приблизительно от 30 до 250 кг/м3·час, еще более предпочтительно приблизительно от 35 до 150 кг/м3·час и наиболее предпочтительно от 40 до 100 кг/м3·час.
Во время окисления в реакторе по типу барботажной колонны 20 предпочтительно, чтобы отношение удельного массового расхода всего растворителя (как из потока сырья, так и из потока флегмы) к удельному массовому расходу способного к окислению соединения, входящего в реакционную зону 28, поддерживалось в интервале приблизительно от 2:1 до 50:1, более предпочтительно в интервале приблизительно от 5:1 до 40:1 и наиболее предпочтительно в интервале приблизительно от 7,5:1 до 25:1. Предпочтительно отношение удельного массового расхода всего растворителя, введенного в виде части потока сырья, к удельному массовому расходу растворителя, введенного в виде части потока флегмы, поддерживается в интервале приблизительно от 0,5:1 до отсутствия какого-либо потока флегмы, более предпочтительно в интервале приблизительно от 0,5:1 до 4:1, еще более предпочтительно в интервале приблизительно от 1:1 до 2:1 и наиболее предпочтительно в интервале от 1,25:1 до 1,5:1.
Во время жидкофазного окисления в реакторе по типу барботажной колонны 20 предпочтительно, чтобы поток окислителя вводился в реактор по типу барботажной колонны 20 в количестве, которое обеспечивает молекулярный кислород слегка выше стехиометрической потребности в кислороде. Количество избыточного молекулярного кислорода, требуемое для наиболее хороших результатов с конкретным способным к окислению соединением, оказывает влияние на общую экономику жидкофазного окисления. Во время жидкофазного окисления в реакторе по типу барботажной колонны 20 предпочтительно, чтобы отношение удельного массового расхода окислителя к удельному массовому расходу способного к окислению органического соединения (например, пара-ксилола), поступающего в реактор 20, поддерживалось в интервале приблизительно от 0,5:1 до 20:1, более предпочтительно в интервале приблизительно от 1:1 до 10:1 и более предпочтительно в интервале от 2:1 до 6:1.
На ФИГ. 1 потоки сырья, окислителя и флегмы, введенные в реактор по типу барботажной колонны 20, вместе образуют, по меньшей мере, часть многофазной реакционной среды 36. Реакционная среда 36 предпочтительно является трехфазной средой, содержащей твердую фазу, жидкую фазу и газовую фазу. Как упоминалось выше, окисление способного к окислению соединения (например, пара-ксилола) преимущественно имеет место в жидкой фазе реакционной среды 36. То есть жидкая фаза реакционной среды 36 содержит растворенный кислород и способное к окислению соединение. Экзотермическая природа реакции окисления, которая имеет место в ректоре по типу барботажной колонны 20, заставляет часть растворителя (например, уксусной кислоты и воды), введенного через впускное отверстие для сырья 32а,b,c,d, кипеть/испаряться. Следовательно, газовая фаза реакционной среды 36 в реакторе 20 образуется преимущественно из испарившегося растворителя и нерастворенной, непрореагировавшей части потока окислителя.
В некоторых реакторах окисления предшествующего уровня техники используются теплообменные трубки/пластины, чтобы нагревать или охлаждать реакционную среду. Однако такие теплообменные структуры могут быть нежелательны в заявляемом реакторе и в процессе, описанном в данном случае. Следовательно, предпочтительно, чтобы реактор по типу барботажной колонны 20 по существу не включал поверхности, которые вступают в контакт с реакционной средой 36, и показывал усредненный по времени удельный тепловой поток больше чем 30000 ватт на квадратный метр. Кроме того, предпочтительно, чтобы менее чем приблизительно 50% усредненной по времени теплоты реакции реакционной среды 36 отводилось с помощью теплообменных поверхностей, более предпочтительно менее чем приблизительно 30% теплоты реакции отводилось с помощью теплообменных поверхностей и наиболее предпочтительно менее чем приблизительно 10% теплоты реакции отводилось с помощью теплообменных поверхностей.
Концентрация растворенного кислорода в жидкой фазе реакционной среды 36 представляет собой динамическое равновесие между скоростью массовой передачи от газовой фазы и скоростью потребления в ходе реакции в пределах жидкой фазы (то есть оно не устанавливается просто за счет парциального давления молекулярного кислорода в поддерживающей газовой фазе, хотя это и является одним из факторов в восполнении доли растворенного кислорода и действительно оказывает влияние на ограничивающую верхнюю концентрацию растворенного кислорода). Количество растворенного кислорода меняется локально, являясь более высоким около межфазной поверхности пузырьков. В общем случае количество растворенного кислорода зависит от соотношения факторов подачи и потребления в различных областях реакционной среды 36. По времени количество растворенного кислорода зависит от равномерности смешения газа и жидкости относительно скоростей химического потребления. При желании уровнять соответствующим образом подачу и потребление растворенного кислорода в жидкой фазе реакционной среды 36 предпочтительно, чтобы усредненная по времени и усредненная по объему концентрация кислорода в жидкой фазе реакционной среды 36 поддерживалась выше приблизительно 1 мол. ч/млн, более предпочтительно в интервале приблизительно от 4 до 1000 мол. ч./млн, еще более предпочтительно в интервале приблизительно от 8 до 500 мол. ч./млн и наиболее предпочтительно в интервале от 12 до 120 мол. ч./млн.
Реакция жидкофазного окисления, проводимая в реакторе по типу барботажной колонны 20, предпочтительно является реакцией с осаждением, которая дает твердые вещества. Более предпочтительно жидкофазное окисление, проводимое в реакторе по типу барботажной колонны 20, заставляет, по меньшей мере, приблизительно 10% масс. способного к окислению соединения (например, пара-ксилола), вводимого в реакционную зону 28, образовывать в реакционной среде 36 твердое соединение (например, частицы сырой терефталевой кислоты). Еще более предпочтительно жидкофазное окисление заставляет, по меньшей мере, приблизительно 50% масс. способного к окислению соединения давать в реакционной среде 36 твердое вещество. Наиболее предпочтительно жидкофазное окисление заставляет, по меньшей мере, приблизительно 90% масс. способного к окислению соединения давать в реакционной среде 36 твердое вещество. Предпочтительно, чтобы суммарное количество твердых веществ в реакционной среде 36 было больше чем приблизительно 3% масс. из расчета на усреднение по времени и усреднение по объему. Более предпочтительно суммарное количество твердых веществ в реакционной среде 36 поддерживается в интервале приблизительно от 5 до 40% масс., еще более предпочтительно в интервале приблизительно от 10 до 35% масс. и наиболее предпочтительно в интервале от 15 до 30% масс. Предпочтительно, чтобы существенная часть продукта окисления (например, терефталевой кислоты), произведенного в реакторе по типу барботажной колонны 20, присутствовала в реакционной среде 36 в виде твердых веществ, в противоположность остальному количеству, растворенному в жидкой фазе реакционной среды 36. Количество твердой фазы продукта окисления, присутствующее в реакционной среде 36, предпочтительно составляет, по меньшей мере, приблизительно 25% масс. из расчета на весь продукт окисления (твердая и жидкая фаза) в реакционной среде 36, более предпочтительно, по меньшей мере, приблизительно 75% масс. из расчета на весь продукт окисления в реакционной среде 36 и наиболее предпочтительно, по меньшей мере, 95% масс. из расчета на весь продукт окисления в реакционной среде 36. Численные интервалы, приведенные выше для количества твердых веществ в реакционной среде 36, по существу применимы для стационарного режима работы барботажной колонны 20 в течение по существу непрерывного периода времени, без запуска, остановки или операций оптимизации реактора типа барботажной колонны 20. Количество твердых веществ в реакционной среде 36 определяется с помощью гравиметрического метода. В указанном гравиметрическом методе соответствующую часть суспензии выводят из реакционной среды и взвешивают. При условиях, которые эффективно сохраняют общее твердофазное распределение, присутствующее в реакционной среде, свободную жидкость удаляют из части твердых веществ путем осаждения или фильтрования, эффективно без потери осажденных твердых веществ и при менее чем приблизительно 10% начальной массы жидкости, остающейся с частью твердых веществ. Оставшуюся жидкость на твердых веществах упаривают досуха, эффективно без сублимации твердых веществ. Оставшуюся часть твердых веществ взвешивают. Отношение массы части твердых веществ к массе исходной части суспензии представляет собой долю твердых веществ, обычно выражаемую в процентах.
Реакция с осаждением, проводимая в реакторе по типу барботажной колонны 20, может вызвать образование загрязнения (то есть накопление твердых веществ) на поверхности некоторых жестких структур, которые контактируют с реакционной средой 36. Следовательно, в одном из вариантов осуществления настоящего изобретения предпочтительно, чтобы реактор по типу барботажной колонны 20 по существу не включал внутренних теплообменных, перемешивающих или отклоняющих поток структур в реакционной зоне 28, так как такие структуры будут способствовать образованию загрязнений. Если внутренние структуры присутствуют в реакционной зоне 28, желательно исключить внутренние структуры, имеющие внешние поверхности, которые имеют значительную площадь обращенной наверх плоской поверхности, которая весьма склонна к образованию загрязнений. Таким образом, если любые внутренние структуры присутствуют в реакционной зоне 28, предпочтительно, чтобы менее чем приблизительно 20% всей площади обращенной наверх подвергающейся воздействию внешней поверхности таких внутренних структур образовывало по существу плоские поверхности, наклоненные менее чем приблизительно на 15 градусов от горизонтали. Внутренние структуры с конфигурацией такого типа называют как имеющие «незагрязняющуюся» конфигурацию.
Что касается ФИГ. 1, то физическая конфигурация реактора по типу барботажной колонны 20 помогает обеспечить оптимизированное окисление способного к окислению соединения (например, пара-ксилола) с минимальным образованием примесей. Предпочтительно, чтобы удлиненная реакционная секция 24 оболочки емкости 22 включала по существу основной цилиндрический корпус 46 и дно 48. Верхний конец реакционной зоны 28 определен горизонтальной плоскостью 50, простирающейся через верхнюю часть основного цилиндрического корпуса 46. Нижний конец 52 реакционной зоны 28 определен с помощью самой нижней внутренней поверхностью дна 48. Как правило, нижний конец 52 реакционной зоны 28 располагается рядом с отверстием для выпускного отверстия для суспензии 38. Следовательно, удлиненная реакционная зона 28, определенная внутри реактора по типу барботажной колонны 20, имеет максимальную длину «L», измеренную от верхнего конца 50 до нижнего конца 52 реакционной зоны 28 вдоль оси удлиненного основного цилиндрического корпуса 46. Длина «L» реакционной зоны 28 предпочтительно находится в интервале приблизительно от 10 до 100 метров, более предпочтительно в интервале приблизительно от 20 до 75 метров и наиболее предпочтительно в интервале от 25 до 50 метров. Реакционная зона 28 имеет максимальный диаметр (ширину) «D», который, как правило, равен максимальному внутреннему диаметру основного цилиндрического корпуса 46. Максимальный диаметр «D» реакционной зоны 28 предпочтительно находится в интервале приблизительно от 1 до 12 метров, более предпочтительно в интервале приблизительно от 2 до 10 метров, еще более предпочтительно в интервале приблизительно от 3,1 до 9 метров и наиболее предпочтительно в интервале от 4 до 8 метров. В предпочтительном варианте осуществления настоящего изобретения реакционная зона 28 имеет отношение длины к диаметру «L:D» в интервале приблизительно от 6:1 до 30:1. Еще более предпочтительно реакционная зона 28 имеет отношение «L:D» в интервале приблизительно от 8:1 до 20:1. Наиболее предпочтительно реакционная зона 28 имеет отношение «L:D» в интервале от 9:1 до 15:1.
Как обсуждалось выше, реакционная зона 28 реактора по типу барботажной колонны 20 принимает многофазную среду 36. Реакционная среда 36 имеет нижний конец, совпадающий с нижним концом 52 реакционной зоны 28, и верхний конец, расположенный у верхней поверхности 44. Верхняя поверхность 44 реакционной среды 36 определена вдоль горизонтальной плоскости, которая пересекает реакционную зону 28 в вертикальном положении, где содержимое реакционной зоны 28 переходит от сплошного газофазного состояния к сплошному жидкофазному состоянию. Верхняя поверхность 44 предпочтительно расположена при вертикальном расположении, где локальное усредненное по времени удерживание газа тонким горизонтальным слоем содержимого реакционной зоны 28 составляет 0,9.
Реакционная среда 36 имеет максимальную высоту «Н», измеряемую между ее верхним и нижним концами. Максимальная ширина «W» реакционной среды 36, как правило, равна максимальному диаметру «D» основного цилиндрического корпуса 46. Во время жидкофазного окисления в реакторе по типу барботажной колонны 20 предпочтительно, чтобы высота «Н» поддерживалась приблизительно на 60-120% от «L», более предпочтительно приблизительно от 80 до 110% от «L», наиболее предпочтительно приблизительно от 85 до 100% от «L». В предпочтительном варианте осуществления настоящего изобретения реакционная среда 36 имеет отношение высоты к ширине «H:W» больше чем приблизительно 3:1. Более предпочтительно реакционная среда 36 имеет отношение «H:W» в интервале приблизительно от 7:1 до 25:1. Еще более предпочтительно реакционная среда 36 имеет отношение высоты к ширине «H:W» в интервале приблизительно от 8:1 до 20:1. Наиболее предпочтительно реакционная среда 36 имеет отношение высоты к ширине «H:W» в интервале приблизительно от 9:1 до 15:1. В одном из вариантов изобретения L=H и D=W так, что различные размеры или отношения, предложенные для L и D, также применимы к Н и W, и наоборот.
Относительно высокие отношения L:D и H:W, предложенные в соответствии с вариантом осуществления настоящего изобретения, могут вносить вклад в некоторые значимые преимущества заявляемой системы. Как обсуждается более подробно ниже, установлено, что более высокие отношения L:D и H:W, а также некоторые другие признаки, обсуждающиеся ниже, могут стимулировать возникновение благоприятных вертикальных градиентов концентраций молекулярного кислорода и/или способного к окислению соединения (например, пара-ксилола) в реакционной среде 36. В отличие от общепринятого мнения, которое отдает предпочтение хорошо перемешанной реакционной среде с относительно однородными концентрациями повсюду, установлено, что вертикальное ступенчатое измерение концентраций кислорода и/или способного к окислению соединения способствует более эффективной и экономичной реакции окисления. Минимизация концентраций кислорода и способного к окислению соединения около верхней части реакционной среды 36 может содействовать исключению потерь непрореагировавшего кислорода и непрореагировавшего способного к окислению соединения через верхнее выпускное отверстие для газа 40. Однако если концентрации способного к окислению соединения и непрореагировавшего кислорода невелики по всей реакционной среде 36, то скорость и/или селективность окисления снижаются. Следовательно, предпочтительно, чтобы концентрации молекулярного кислорода и/или способного к окислению соединения были значительно более высокими около нижней части реакционной среды 36, чем около верхней части реакционной среды 36.
Кроме того, высокие отношения L:D и H:W приводят к тому, что давление в нижней части реакционной среды 36 значительно выше, чем давление в верхней части реакционной среды 36. Такой вертикальный градиент давления является результатом высоты и плотности реакционной среды 36. Одно из преимуществ вертикального градиента давления состоит в том, что повышенное давление в нижней части емкости приводит к большей растворимости кислорода и большему массовому переносу, чем достигалось бы в противном случае при сравнимых температурах и головном давлении в неглубоких реакторах. Следовательно, реакция окисления может быть проведена при более низких температурах, чем можно было бы ожидать в случае менее глубоких реакторов. Когда реактор по типу барботажной колонны 20 используется для частичного окисления пара-ксилола до сырой терефталевой кислоты (СТК), способность работать при более низких температурах с таким же или более хорошим массовым переносом кислорода имеет ряд преимуществ. Например, низкотемпературное окисление пара-ксилола снижает количество растворителя, сжигаемого во время реакции. Как обсуждается более подробно ниже, низкотемпературное окисление также способствует образованию небольших, имеющих высокую площадь поверхности, свободно связанных, легко растворяемых частиц СТК, которые могут быть подвергнуты более экономичным методикам очистки, чем большие, имеющие низкую площадь поверхности, плотные частицы СТК, производимые с помощью обычных высокотемпературных процессов окисления.
Во время окисления в реакторе 20 предпочтительно, чтобы усредненная по времени и усредненная по объему температура реакционной среды 36 поддерживалась в интервале приблизительно от 125 до 200ºС, более предпочтительно в интервале приблизительно от 140 до 180ºС и наиболее предпочтительно в интервале от 150 до 170ºС. Головное давление выше реакционной среды 36 предпочтительно поддерживается в интервале приблизительно от 1 до 20 абсолютных бар (абс. бар), более предпочтительно в интервале приблизительно от 2 до 12 абс. бар и наиболее предпочтительно в интервале от 4 до 8 абс. бар. Предпочтительно разница давлений между верхом реакционной среды 36 и низом реакционной среды 36 находится в интервале приблизительно от 0,4 до 5 бар, более предпочтительно разница давлений находится в интервале приблизительно от 0,7 до 3 бар и наиболее предпочтительно разница давлений составляет от 1 до 2 бар. Хотя в общем случае предпочтительно, чтобы головное давление выше реакционной среды 36 поддерживалось при относительно постоянном значении, один из вариантов настоящего изобретения подразумевает пульсирующее головное давление, чтобы способствовать улучшенному смешению и/или массовому переносу в реакционной среде 36. Когда головное давление пульсирует, предпочтительно, чтобы импульсные давления находились в интервале приблизительно от 60 до 140% от головного давления в стационарном состоянии, указанного в настоящем изобретении, более предпочтительно приблизительно от 85 до 115% от головного давления в стационарном состоянии, указанного в настоящем изобретении, и наиболее предпочтительно от 95 до 105% от головного давления в стационарном состоянии, указанного в настоящем изобретении.
Другое преимущество высокого отношения «L:D» реакционной зоны 28 состоит в том, что оно может вносить вклад в повышение средней приведенной скорости реакционной среды 36. Определение «приведенная скорость» и «приведенная скорость газа», используемое в данном случае по отношению к реакционной среде 36, означает объемную скорость потока газовой фазы реакционной среды 36 при подъеме в реакторе, поделенную на площадь горизонтального поперечного сечения реактора на этой отметке высоты. Повышенная приведенная скорость, обеспечиваемая высоким отношением L:D реакционной зоны 28, может способствовать локальному смешению и повышению удерживания газа реакционной среды 36. Усредненные по времени приведенные скорости реакционной среды 36 на одной четвертой высоты, на половине высоты и/или на трех четвертях высоты реакционной среды 36 предпочтительно составляют приблизительно больше чем 0,3 метра в секунду, более предпочтительно находятся в интервале приблизительно от 0,8 до 5 м/сек, еще более предпочтительно находятся в интервале приблизительно от 0,9 до 4 м/сек и наиболее предпочтительно в интервале приблизительно от 1 до 3 м/сек.
На ФИГ. 1 секция отделения 26 реактора по типу барботажной колонны 20 представляет собой просто расширенную часть оболочки емкости 22, расположенную непосредственно над реакционной секцией 24. Секция отделения 26 снижает скорость протекающей по направлению вверх газовой фазы в реакторе по типу барботажной колонны 20, когда газовая фаза поднимается над верхней поверхностью 44 реакционной среды 36 и достигает выпускного отверстия для газа 40. Такое снижение скорости подъема вверх газовой фазы способствует легкому удалению захваченных жидкостей и/или твердых веществ в протекающей наверх газовой фазе и, следовательно, снижает нежелательные потери компонентов, присутствующих в жидкой фазе реакционной среды 36.
Секция отделения 26 предпочтительно в большинстве случаев включает переходную стенку в форме усеченного конуса 54, цилиндрическую широкую боковую стенку 56 и верхнюю головную часть 58. Узкий нижний конец переходной стенки 54 соединен с верхом основного цилиндрического корпуса 46 реакционной секции 24. Широкий верхний конец переходной стенки 54 соединен с низом широкой боковой стенки 56. Предпочтительно, чтобы переходная стенка 54 простиралась вверх и наружу от ее узкого нижнего конца под углом в интервале приблизительно от 10 до 70 градусов от вертикали, более предпочтительно в интервале приблизительно от 15 до 50 градусов от вертикали и наиболее предпочтительно в интервале приблизительно от 15 до 45 градусов от вертикали. Широкая боковая стенка 56 имеет максимальный диаметр «Х», который, как правило, больше чем максимальный диаметр «D» реакционной секции 24, хотя когда верхняя часть реакционной секции 24 имеет меньший диаметр, чем весь максимальный диаметр реакционной секции 24, то Х может быть фактически меньше D. В предпочтительном варианте осуществления настоящего изобретения отношение диаметра широкой боковой стенки 56 к максимальному диаметру реакционной секции 24 «X:D» находится в интервале приблизительно от 0,8:1 до 4:1, наиболее предпочтительно в интервале от 1,1:1 до 2:1. Верхняя головная часть 58 соединена с верхом широкой боковой стенки 56. Верхняя головная часть 58 предпочтительно представляет собой эллиптический головной элемент, ограничивающий центральное отверстие, которое дает возможность газу покидать зону отделения 30 через выпускное отверстие для газа 40. С другой стороны, верхняя головная часть 58 может иметь любую конфигурацию, в том числе коническую. Зона отделения 30 имеет максимальную высоту «Y», которую измеряют от верха 50 реакционной зоны 28 до самой верхней части зоны отделения 30. Отношение длины реакционной зоны 28 к высоте зоны отделения 30 «L:Y» предпочтительно находится в интервале приблизительно от 2:1 до 24:1, более предпочтительно в интервале приблизительно от 3:1 до 20:1 и наиболее предпочтительно в интервале от 4:1 до 16:1.
Что касается ФИГ. 1-5, то местоположение и конфигурация барботера окислителя 34 будут обсуждены более подробно. ФИГ. 2 и 3 показывают, что барботер окислителя 34 может включать кольцевой элемент 60 и пару трубопроводов для поступления окислителя 64а,b. Обычно такие трубопроводы для поступления окислителя 64а,b могут входить в емкость на отметке высоты выше кольцевого элемента 60 и затем поворачивать вниз, как показано на ФИГ. 2. С другой стороны, трубопровод для поступления окислителя может входить в емкость ниже кольцевого элемента 60 или приблизительно на той же горизонтальной плоскости, что и кольцевой элемент 60. Каждый трубопровод для поступления окислителя 64а,b включает первый конец, соединенный с соответствующим впускным отверстием для окислителя 66а,b, образованным в оболочке емкости 22, и второй конец, подвижно соединенный с кольцевым элементом 60. Кольцевой элемент 60 предпочтительно образован из трубопроводов, более предпочтительно из множества секций прямых трубопроводов и наиболее предпочтительно из множества секций прямых трубок, жестко присоединенных одна к другой с образованием трубчатого многоугольного кольца. Предпочтительно кольцевой элемент 60 образован, по меньшей мере, из трех секций прямых труб, более предпочтительно из 6-10 секций труб и наиболее предпочтительно из 8 секций труб. Соответственно, когда кольцевой элемент 60 образован из 8 трубчатых секций, он имеет конфигурацию восьмиугольника. Предпочтительно, чтобы секции труб, которые образуют трубопроводы для поступления окислителя 64а,b и кольцевой элемент 60, имели номинальный диаметр больше чем приблизительно 0,1 метр, более предпочтительно в интервале приблизительно от 0,2 до 2 м и наиболее предпочтительно в интервале от 0,25 до 1 м. Как, по-видимому, наиболее хорошо показано на ФИГ.3, предпочтительно, чтобы по существу не было отверстий, образованных в верхней части кольца барботера 60.
Как, по-видимому, наиболее хорошо показано на ФИГ.4 и 5, нижняя часть кольца барботера окислителя 60 имеет множество отверстий для окислителя 68. Отверстия для окислителя 68 предпочтительно имеют такую конфигурацию, что, по меньшей мере, приблизительно 1% общей площади пропускного сечения, ограниченной отверстиями для окислителя 68, расположено ниже центральной линии 64 (ФИГ. 5) кольцевого элемента 60, где центральная линия 64 расположена на высоте объемного центра тяжести кольцевого элемента 60. Более предпочтительно, по меньшей мере, приблизительно 5% общей площади пропускного сечения, очерченной всеми отверстиями для окислителя 68, расположено ниже центральной линии 64, причем, по меньшей мере, приблизительно 2% общей площади пропускного сечения определено отверстиями 68, которые выпускают поток окислителя преимущественно по направлению вниз в пределах приблизительно 30 градусов от вертикали. Еще более предпочтительно, по меньшей мере, приблизительно 20% общей площади пропускного сечения, очерченной всеми отверстиями для окислителя 68, расположено ниже центральной линии 64, причем, по меньшей мере, приблизительно 10% общей площади пропускного сечения определено отверстиями 68, которые выпускают поток окислителя преимущественно по направлению вниз в пределах приблизительно 30 градусов от вертикали. Наиболее предпочтительно, по меньшей мере, приблизительно 75% общей площади пропускного сечения, очерченной всеми отверстиями для окислителя 68, расположено ниже центральной линии 64, причем, по меньшей мере, приблизительно 40% общей площади пропускного сечения определено отверстиями 68, которые выпускают поток окислителя преимущественно по направлению вниз в пределах приблизительно 30 градусов от вертикали. Доля общей площади пропускного сечения, очерченной всеми отверстиями для окислителя 68, которые расположены выше центральной линии 64, предпочтительно составляет меньше чем приблизительно 75%, более предпочтительно меньше чем приблизительно 50%, еще более предпочтительно меньше чем приблизительно 25% и наиболее предпочтительно меньше чем 5%.
Как показано на ФИГ. 4 и 5, отверстия для окислителя 68 включают направленные вниз отверстия 68а и скошенные отверстия 68b. Направленные вниз отверстия 68а имеют такую конфигурацию, чтобы выпускать поток окислителя преимущественно вниз под углом в пределах приблизительно 30 градусов от вертикали, более предпочтительно в пределах приблизительно 15 градусов от вертикали и наиболее предпочтительно в пределах приблизительно 5 градусов по вертикали. Что касается ФИГ. 5, то скошенные отверстия 68b имеют такую конфигурацию, чтобы выпускать поток окислителя преимущественно в сторону и вниз под углом «А», который находится в интервале приблизительно от 15 до 75 градусов от вертикали, более предпочтительно угол А находится в интервале приблизительно от 30 до 60 градусов от вертикали и наиболее предпочтительно угол А находится в интервале от 40 до 50 градусов от вертикали.
Предпочтительно, чтобы по существу все отверстия для окислителя 68 имели приблизительно одинаковый диаметр. Диаметр отверстий для окислителя 68 предпочтительно находится в интервале приблизительно от 2 до 300 миллиметров, более предпочтительно в интервале приблизительно от 4 до 120 мм и наиболее предпочтительно в интервале от 8 до 60 мм. Суммарное число отверстий для окислителя 68 в кольцевом элементе 60 выбирают так, чтобы оно соответствовало критерию низкого перепада давлений, подробно рассмотренному ниже. Предпочтительно суммарное число отверстий для окислителя 68, образованное в кольцевом элементе 60, составляет, по меньшей мере, приблизительно 10, более предпочтительно суммарное число отверстий для окислителя 68 находится в интервале приблизительно от 20 до 200 и наиболее предпочтительно суммарное число отверстий для окислителя 68 находится в интервале приблизительно от 40 до 100.
Хотя ФИГ. 1-5 иллюстрируют вполне конкретную конфигурацию для барботера окислителя 34, нужно отметить, что целый ряд конфигураций барботера окислителя может быть использован для достижения описанных в настоящем изобретении преимуществ. Например, нет необходимости в том, чтобы барботер окислителя имел восьмиугольную конфигурацию кольцевого элемента, показанную на ФИГ. 1-5. Более того, допустимо, чтобы барботер окислителя образовывал любую конфигурацию проточного(ых) трубопровода(ов), в которой используют множество расположенных на расстоянии отверстий для выпуска потока окислителя. Размер, число и направление выпуска отверстий для окислителя в проточном трубопроводе предпочтительно находятся в пределах интервалов, представленных выше. Кроме того, барботер окислителя предпочтительно конфигурирован так, чтобы обеспечить азимутальное и радиальное распределение молекулярного кислорода, описанное выше.
Независимо от конкретной конфигурации барботера окислителя 34, предпочтительно, чтобы барботер окислителя был физически конфигурирован и ориентирован таким образом, чтобы свести до минимума перепад давлений, связанный с выпуском потока окислителя из проточного(ых) трубопровода(ов) через отверстия для окислителя и в реакционную зону. Такой перепад давлений рассчитывают в виде усредненного по времени статического давления потока окислителя внутри проточного трубопровода у выходных отверстий для окислителя 66а,b минус усредненное по времени статическое давление в реакционной зоне на отметке высоты, где одна половина потока окислителя введена выше этого вертикального местоположения, а другая половина потока окислителя введена ниже этого вертикального местоположения. В предпочтительном варианте настоящего изобретения усредненный по времени перепад давлений, связанный с выпуском потока окислителя из барботера окислителя, составляет менее чем приблизительно 0,3 мегапаскаля (МПа), более предпочтительно менее чем приблизительно 0,2 МПа, еще более предпочтительно менее чем приблизительно 0,1 МПа и наиболее предпочтительно менее чем 0,05 МПа.
Необязательно для барботера окислителя 34 может быть предусмотрена непрерывная или периодическая промывка с помощью жидкости (например, уксусной кислоты, воды и/или пара-ксилола), чтобы предупредить загрязнение барботера окислителя твердыми веществами. При использовании такой промывки жидкостью предпочтительно, чтобы эффективное количество жидкости (то есть не только небольшое количество жидких капель, которое может естественно присутствовать в потоке окислителя) пропускалось через барботер окислителя и выходило из отверстий для окислителя, по меньшей мере, в течение одного периода больше одной минуты каждый день. Когда жидкость непрерывно или периодически выпускают из барботера окислителя 34, предпочтительно, чтобы усредненное по времени отношение удельного массового расхода жидкости через барботер окислителя к удельному массовому расходу молекулярного кислорода через барботер окислителя находилось в интервале приблизительно от 0,05:1 до 30:1, или в интервале приблизительно от 0,1:1 до 2:1, или даже в интервале от 0,2:1 до 1:1.
Во многих обычных реакторах по типу барботажной колонны, содержащих многофазную реакционную среду, по существу вся реакционная среда, расположенная ниже барботера окислителя (или другого механизма для введения потока окислителя в реакционную зону), имеет очень низкое значение удерживания газа. Как известно в данной области техники, «удерживание газа» просто представляет собой объемную долю многофазной среды, которая находится в газообразном состоянии. Зоны низкого удерживания газа в среде могут также быть названы «неаэрируемыми» зонами. Во многих обычных реакторах по типу барботажной колонны значительная часть общего объема реакционной среды расположена ниже барботера окислителя (или другого механизма для введения потока окислителя в реакционную зону). Следовательно, значительная часть реакционной среды, находящейся у днища обычных реакторов по типу барботажной колонны, является неаэрируемой.
Установлено, что сведение до минимума количества неаэрируемых зон в реакционной среде, подвергаемой окислению в реакторе по типу барботажной колонны, может свести до минимума образование некоторых нежелательных примесей. Неаэрируемые зоны реакционной среды содержат относительно немного пузырьков окислителя. Такой низкий объем пузырьков окислителя снижает количество молекулярного кислорода, доступного для растворения в жидкой фазе реакционной среды. Следовательно, жидкая фаза в неаэрируемой зоне реакционной среды имеет относительно низкую концентрацию молекулярного кислорода. Такие обедненные по кислороду, неаэрируемые зоны реакционной среды имеют тенденцию стимулировать нежелательные побочные реакции, а не целевую реакцию окисления. Например, когда пара-ксилол частично окисляют с образованием терефталевой кислоты, недостаточная доступность кислорода в жидкой фазе реакционной среды может вызвать образование нежелательно высоких количеств бензойной кислоты и сопряженных ароматических колец, в особенности включающих весьма нежелательные окрашенные молекулы, известные как флуореноны и антрахиноны.
В соответствии с одним из вариантов осуществления настоящего изобретения жидкофазное окисление проводят в реакторе по типу барботажной колонны, конфигурированном и ориентированном таким образом, чтобы объемная доля реакционной среды с низкими значениями удерживания газа была минимизирована. Такая минимизация неаэрируемых зон может быть количественно оценена путем теоретического распределения всего объема реакционной среды на 2000 дискретных горизонтальных тонких слоев одинакового объема. За исключением самого высокого и самого низкого горизонтальных тонких слоев, каждый горизонтальный тонкий слой имеет дискретный объем, ограниченный по его сторонам боковой стенкой реактора и ограниченный сверху и снизу воображаемыми горизонтальными плоскостями. Самый высокий горизонтальный тонкий слой ограничен снизу воображаемой горизонтальной плоскостью, а сверху верхней поверхностью реакционной среды. Самый нижний горизонтальный тонкий слой ограничен сверху воображаемой горизонтальной плоскостью, а снизу нижним концом емкости. После теоретического распределения реакционной среды на 2000 дискретных горизонтальных тонких слоев равного объема, может быть определено усредненное по времени и усредненное по объему удерживание газа каждого горизонтального тонкого слоя. Когда используется такой метод определения количества неаэрируемых зон, предпочтительно, чтобы число горизонтальных тонких слоев, имеющих усредненное по времени и усредненное по объему удерживание газа менее чем 0,1, составляло менее чем 30, более предпочтительно менее чем 15, еще более предпочтительно менее чем 6, даже более предпочтительно менее чем 4 и наиболее предпочтительно менее чем 2. Предпочтительно, чтобы число горизонтальных тонких слоев, имеющих удерживание газа менее чем 0,2, составляло менее чем 80, более предпочтительно менее чем 40, еще более предпочтительно менее чем 20, даже более предпочтительно менее чем 12 и наиболее предпочтительно менее чем 5. Предпочтительно, чтобы число горизонтальных тонких слоев, имеющих удерживание газа менее чем 0,3, составляло менее чем 120, более предпочтительно менее чем 80, еще более предпочтительно менее чем 40, даже более предпочтительно менее чем 20 и наиболее предпочтительно менее чем 15.
Что касается ФИГ. 1 и 2, то установлено, что расположение барботера окислителя 34 ниже в реакционной зоне 28, обеспечивает несколько преимуществ, в том числе уменьшение количества неаэрируемых зон в реакционной среде 36. С учетом высоты «Н» реакционной среды 36, длины «L» реакционной зоны 28 и максимального диаметра «D» реакционной зоны 28, предпочтительно, чтобы большая часть (то есть >50% масс.) потока окислителя была введена в реакционную зону 28 в пределах приблизительно 0,025Н, 0,022L и/или 0,25D от нижнего конца 52 реакционной зоны 28. Более предпочтительно большую часть потока окислителя вводят в реакционную зону 28 в пределах приблизительно 0,02Н, 0,018L и/или 0,2D от нижнего конца 52 реакционной зоны 28. Наиболее предпочтительно большую часть потока окислителя вводят в реакционную зону 28 в пределах приблизительно 0,015Н, 0,013L и/или 0,15D от нижнего конца 52 реакционной зоны 28.
В варианте осуществления изобретения, представленном на ФИГ.2, вертикальное расстояние «Y1» между нижним концом 52 реакционной зоны 28 и выходным отверстием верхних отверстий для окислителя 68 барботера окислителя 34 составляет менее чем приблизительно 0,25Н, 0,022L и/или 0,25D, так что по существу весь поток окислителя поступает в реакционную зону 28 в пределах приблизительно 0,25Н, 0,022L и/или 0,25D от нижнего конца 52 реакционной зоны 28. Более предпочтительно Y1 составляет менее чем 0,02Н, 0,018L и/или 0,2D. Наиболее предпочтительно Y1 составляет менее чем 0,015Н, 0,013L и/или 0,15D, но более чем 0,005Н, 0,004L и/или 0,06D. ФИГ. 2 иллюстрирует касательную линию 72 в местоположении, где нижний край основного цилиндрического корпуса 46 оболочки емкости 22 соединен с верхним краем эллиптической нижней головной части 48 оболочки емкости 22. С другой стороны, нижняя головная часть 48 может иметь любую конфигурацию, в том числе коническую, и касательная линия также будет определяться по нижнему краю основного цилиндрического корпуса 46. Вертикальное расстояние «Y2» между касательной линией 72 и верхом барботера окислителя 34 предпочтительно составляет, по меньшей мере, приблизительно 0,0012Н, 0,001L и/или 0,001D; более предпочтительно, по меньшей мере, приблизительно 0,005Н, 0,004L и/или 0,05D и наиболее предпочтительно, по меньшей мере, 0,01Н, 0,008L и/или 0,1D. Вертикальное расстояние «Y3» между нижним концом 52 реакционной зоны 28 и выходным отверстием нижних отверстий для окислителя 70 барботера окислителя 34 предпочтительно составляет менее чем приблизительно 0,015Н, 0,013L и/или 0,15D; более предпочтительно менее чем приблизительно 0,012Н, 0,01L и/или 0,1D и наиболее предпочтительно менее чем приблизительно 0,01Н, 0,008L и/или 0,075D, но более чем 0,003Н, 0,002L и/или 0,025D.
Помимо преимуществ, обеспечиваемых за счет минимизации неаэрируемых зон (то есть зон с низким удерживанием газа) в реакционной среде 36, установлено, что окисление может быть улучшено за счет максимального повышения удерживания газа всей реакционной среды 36. Реакционная среда 36 предпочтительно имеет усредненное по времени и усредненное по объему удерживание газа, по меньшей мере, приблизительно 0,4, более предпочтительно в интервале приблизительно от 0,6 до 0,9 и наиболее предпочтительно в интервале от 0,65 до 0,85. Некоторые физические и технологические характеристики реактора по типу барботажной колонны 20 вносят вклад в высокое удерживание газа, обсужденное выше. Например, в случае заданного размера реактора и скорости потока окислителя высокое отношение L:D реакционной зоны 28 дает меньший диаметр; это повышает приведенную скорость в реакционной среде 36, что, в свою очередь, повышает удерживание газа. Кроме того, фактический диаметр барботажной колонны и отношение L:D, как известно, оказывают влияние на среднее удерживание газа, даже в случае заданной постоянной приведенной скорости. Кроме того, минимизация неаэрируемых зон, особенно в нижней части реакционной зоны 28, вносит вклад в повышенное значение удерживания газа. Кроме того, головное давление и механическая конфигурация реактора по типу барботажной колонны могут оказывать воздействие на технологическую стабильность при высоких приведенных скоростях и значениях удерживания газа, раскрытых в настоящем изобретении.
Что касается ФИГ. 1, то установлено, что улучшенное распределение способного к окислению соединения (например, пара-ксилола) в реакционной среде 36 может быть обеспечено за счет введения жидкофазного потока сырья в реакционную зону 28 во множестве разделенных вертикальными промежутками местоположениях. Предпочтительно жидкофазный поток сырья вводят в реакционную зону 28, по меньшей мере, через 3 отверстия для сырья, более предпочтительно, по меньшей мере, через 4 отверстия для сырья. Как используется в данном случае, определение «отверстия для сырья» обозначают отверстия, где жидкофазный поток сырья выпускают в реакционную зону 28 для смешения с реакционной средой 36. Предпочтительно, чтобы, по меньшей мере, 2 отверстия для сырья были отделены друг от друга вертикальным промежутком, по меньшей мере, приблизительно на 0,5D, более предпочтительно, по меньшей мере, приблизительно на 1,5D и наиболее предпочтительно, по меньшей мере, приблизительно на 3D. Однако предпочтительно, чтобы самое высокое отверстие для сырья было отделено от самого низкого отверстия для сырья вертикальным промежутком не более чем приблизительно 0,75Н, 0,65L и/или 8D; более предпочтительно не более чем приблизительно 0,5Н, 0,4L и/или 5D и наиболее предпочтительно не более чем приблизительно 0,4Н, 0,35L и/или 4D.
Хотя желательно вводить жидкофазный поток сырья во множестве вертикальных местоположениях, также установлено, что обеспечивается улучшенное распределение способного к окислению соединения в реакционной среде 36, если основная часть жидкофазного потока сырья вводится в нижнюю половину реакционной среды 36 и/или реакционной зоны 28. Предпочтительно, по меньшей мере, приблизительно 75% масс. жидкофазного потока сырья вводится в нижнюю половину реакционной среды 36 и/или реакционной зоны 28. Наиболее предпочтительно, по меньшей мере, 90% масс. жидкофазного потока сырья вводится в нижнюю половину реакционной среды 36 и/или реакционной зоны 28. Кроме того, предпочтительно, чтобы, по меньшей мере, приблизительно 30% масс. жидкофазного потока сырья было введено в реакционную зону 28 в пределах приблизительно 1,5D от самого низкого вертикального местоположения, где поток окислителя вводится в реакционную зону 28. Такое самое низкое вертикальное местоположение, где поток окислителя вводится в реакционную зону 28, как правило, находится внизу барботера окислителя; однако ряд альтернативных конфигураций для введения потока окислителя в реакционную зону 28 подразумевается предпочтительным вариантом осуществления настоящего изобретения. Предпочтительно, по меньшей мере, приблизительно 50% масс. жидкофазного сырья вводится в пределах приблизительно 2,5D от самого низкого вертикального местоположения, где поток окислителя вводится в реакционную зону 28. Предпочтительно, по меньшей мере, приблизительно 75% масс. жидкофазного сырья вводится в пределах приблизительно 5D от самого низкого вертикального местоположения, где поток окислителя вводится в реакционную зону 28.
Каждое отверстие для сырья определяет площадь пропускного сечения, через которую выпускается сырье. Предпочтительно, чтобы, по меньшей мере, приблизительно 30% общей площади пропускного сечения всех впускных отверстий для сырья было расположено в пределах приблизительно 1,5D от самого низкого вертикального местоположения, где поток окислителя вводится в реакционную зону 28. Предпочтительно, по меньшей мере, приблизительно 50% общей площади пропускного сечения всех впускных отверстий для сырья располагается в пределах приблизительно 2,5D от самого низкого вертикального местоположения, где поток окислителя вводится в реакционную зону 28. Предпочтительно, по меньшей мере, приблизительно 75% общей площади пропускного сечения всех впускных отверстий для сырья располагается в пределах приблизительно 5D от самого низкого вертикального местоположения, где поток окислителя вводится в реакционную зону 28.
Что касается ФИГ. 1, то в одном из вариантов осуществления настоящего изобретения впускные отверстия для сырья 32а,b,c,d просто представляют собой ряд вертикально размещенных отверстий вдоль одной из стенок оболочки емкости 22. Такие отверстия для сырья имеют по существу одинаковые диаметры, которые составляют менее чем приблизительно 7 сантиметров, более предпочтительно находятся в интервале приблизительно от 0,25 до 5 см и наиболее предпочтительно в интервале от 0,4 до 2 см. Реактор по типу барботажной колонны 20 предпочтительно оборудован системой контроля расхода жидкофазного потока сырья из каждого отверстия для сырья. Такие системы контроля расхода предпочтительно включают индивидуальный дроссель 74а,b,c,d для каждого соответствующего впускного отверстия для сырья 32а,b,c,d. Кроме того, предпочтительно, чтобы реактор по типу барботажной колонны 20 был оборудован системой контроля расхода, которая дает возможность вводить, по меньшей мере, часть жидкофазного потока сырья в реакционную зону 28 при повышенной приведенной скорости ввода, которая составляет, по меньшей мере, приблизительно 2 метра в секунду, более предпочтительно, по меньшей мере, приблизительно 5 м/сек, еще более предпочтительно, по меньшей мере, приблизительно 6 м/сек и наиболее предпочтительно находится в интервале приблизительно от 8 до 20 м/сек. Как используется в данном случае, определение «приведенная скорость ввода» означает усредненный по времени объемный расход потока сырья из отверстия для сырья, поделенный на площадь отверстия для сырья. Предпочтительно, по меньшей мере, приблизительно 50% масс. потока сырья вводится в реакционную зону 28 при повышенной приведенной скорости ввода. Наиболее предпочтительно по существу весь поток сырья вводят в реакционную зону 28 при повышенной приведенной скорости ввода.
На ФИГ. 6 и 7 показана альтернативная система введения жидкофазного потока сырья в реакционную зону 28. В этом варианте осуществления изобретения поток сырья вводят в реакционную зону 28 на четырех различных отметках высоты. Каждая отметка высоты оборудована соответствующей системой распределения сырья 76а,b,c,d. Каждая система распределения сырья 76 включает основной трубопровод для сырья 78 и коллектор 80. Каждый коллектор 80 снабжен, по меньшей мере, двумя выходными отверстиями 82, 84, соединенными с соответствующими вставными трубопроводами 86, 88, каждый из которых простирается в реакционную зону 28 оболочки емкости 22. Каждый вставной трубопровод 86, 88 предоставляет соответствующее отверстие для сырья 87, 89 для выпуска потока сырья в реакционную зону 28. Отверстия для сырья 87, 89 предпочтительно имеют по существу одинаковые диаметры менее чем приблизительно 7 см, более предпочтительно в интервале приблизительно от 0,25 до 5 см и наиболее предпочтительно в интервале от 0,4 до 2 см. Предпочтительно, чтобы отверстия для сырья 87, 89 каждой системы распределения сырья 76а,b,c,d находились диаметрально напротив друг друга с тем, чтобы вводить поток сырья в реакционную зону 28 в противоположных направлениях. Кроме того, предпочтительно, чтобы расположенные диаметрально напротив друг друга отверстия для сырья 86, 88 соседних систем распределения сырья 76 были ориентированы на 90 градусов вращения относительного друг друга. При работе жидкофазный поток сырья запускается в основной трубопровод для сырья 78 и затем поступает в коллектор 80. Коллектор 80 распределяет поток сырья равномерно для одновременного введения на противоположных сторонах реактора 20 через отверстия для сырья 87, 89.
ФИГ. 8 иллюстрирует другую конфигурацию, где каждая система распределения сырья 76 оборудована байонетными трубками 90, 92, а не вставными трубопроводами 86, 88 (показанными на ФИГ. 7). Байонетные трубки 90, 92 простираются в реакционную зону 28 и имеют множество небольших отверстий для сырья 94, 96 для выпуска жидкофазного сырья в реакционную зону 28. Предпочтительно, чтобы небольшие отверстия для сырья 94, 96 байонетных трубок 90, 92 имели по существу одинаковый диаметр менее чем приблизительно 50 мм, более предпочтительно приблизительно от 2 до 25 мм и наиболее предпочтительно от 4 до 15 мм.
ФИГ. 9-11 иллюстрируют альтернативную систему распределения сырья 100. Система распределения сырья 100 вводит жидкофазный поток сырья во множестве разделенных вертикальными промежутками и разделенных продольными промежутками местоположениях без необходимости многократных врезок в боковую стенку реактора по типу барботажной колонны 20. Система введения сырья 100 обычно включает один входной трубопровод 102, коллектор 104, множество установленных вертикально распределительных трубок 106, продольный опорный механизм 108 и вертикальный опорный механизм 110. Входной трубопровод 102 пронизывает боковую стенку основного корпуса 46 оболочки емкости 22. Входной трубопровод 102 подвижно соединен с коллектором 104. Коллектор 104 распределяет поток сырья, принятый из входного трубопровода 102, равномерно по установленным вертикально распределительным трубкам 106. Каждая распределительная трубка 106 имеет множество разделенных вертикальными промежутками отверстий для сырья 112а,b,c,d для выпуска потока сырья в реакционную зону 28. Продольный опорный механизм 108 соединен с каждой распределительной трубкой 106 и подавляет относительное продольное перемещение распределительных трубок 106. Вертикальный опорный механизм 110 предпочтительно соединен с продольным опорным механизмом 108 и с верхней частью барботера окислителя 34. Вертикальный опорный механизм 110 по существу подавляет вертикальное перемещение распределительных трубок 106 в реакционной зоне 28. Предпочтительно, чтобы отверстия для сырья 112 имели по существу одинаковые диаметры менее чем приблизительно 50 мм, более предпочтительно приблизительно от 2 до 25 мм и наиболее предпочтительно от 4 до 15 мм. Вертикальные промежутки для отверстий для сырья 112 системы распределения 110, показанной на ФИГ. 9-11, могут быть по существу такими же, как описано выше для системы распределения сырья на ФИГ. 1. Необязательно отверстия для сырья могут представлять собой вытянутые насадки, а не простые прорези. Необязательно одно или несколько устройств для отклонения потока могут находиться снаружи проточного трубопровода и на траектории движения жидкостей, выходящих из него в реакционную среду. Необязательно отверстие около нижней части проточного трубопровода может быть выбрано по размеру так, чтобы проводить очистку от твердых веществ внутри системы распределения жидкофазного сырья, или непрерывно или периодически. Необязательно механические устройства, такие как откидные узлы, обратные клапаны, регуляторы избытка потока, клапаны экономайзера и т.д., могут быть использованы или для предотвращения попадания твердых веществ во время эксплуатационных нарушений, или для выгрузки накопившихся твердых веществ из системы распределения жидкофазного сырья.
Установлено, что схемы потоков реакционной среды во многих реакторах по типу барботажной колонны могут давать неравномерное азимутальное распределение способного к окислению соединения в реакционной среде, особенно когда способное к окислению соединение вводится преимущественно вдоль одной стороны реакционной среды. Используемое в данном случае определение «азимутальное» означает угол или промежуток около вертикальной оси удлинения реакционной зоны. Используемое в данном случае определение «вертикально» означает в пределах 45º от вертикали. В одном из вариантов осуществления настоящего изобретения поток сырья, содержащий способное к окислению соединение (например, пара-ксилол), вводят в реакционную зону через множество расположенных с азимутальными промежутками отверстий для сырья. Такие отверстия для сырья с азимутальными промежутками могут предупреждать появление областей с исключительно высокими или исключительно низкими концентрациями способного к окислению соединения в реакционной среде. Различные системы введения сырья, представленные на ФИГ. 6-11, являются примерами систем, которые обеспечивают подходящее азимутальное расположение отверстий для сырья.
Что касается ФИГ. 7, то для того, чтобы количественно оценить азимутально распределенное введение жидкофазного потока сырья в реакционную среду, реакционную среду можно теоретически распределить на четыре вертикальных азимутальных квадранта «Q1, Q2, Q3, Q4» приблизительно одинакового объема. Такие азимутальные квадранты «Q1, Q2, Q3, Q4» определяются парой воображаемых пересекающихся перпендикулярных вертикальных плоскостей «Р1, Р2», проходящих выше максимального вертикального размера и максимального радиального размера реакционной среды. Когда реакционная среда находится в цилиндрической емкости, линия пересечения воображаемых пересекающихся вертикальных плоскостей Р1, Р2 будет приблизительно совпадать с вертикальной центральной линией цилиндра, и каждый из азимутальных квадрантов Q1, Q2, Q3, Q4 будет, как правило, представлять собой клиновидный вертикальный объем, имеющий высоту, равную высоте реакционной среды. Предпочтительно, чтобы значительная часть способного к окислению соединения выпускалась в реакционную среду через отверстия для сырья, расположенные, по меньшей мере, в двух различных азимутальных квадрантах.
В предпочтительном варианте осуществления изобретения не более чем приблизительно 80% масс. способного к окислению соединения выпускается в реакционную среду через отверстия для сырья, которые могут быть расположены в одном азимутальном квадранте. Более предпочтительно не более чем 60% масс. способного к окислению соединения выпускается в реакционную среду через отверстия для сырья, которые могут быть расположены в одном азимутальном квадранте. Наиболее предпочтительно не более чем 40% масс. способного к окислению соединения выпускается в реакционную среду через отверстия для сырья, которые могут быть расположены в одном азимутальном квадранте. Такие параметры азимутального распределения способного к окислению соединения измеряют, когда азимутальные квадранты азимутально ориентированы так, что максимально возможное количество способного к окислению соединения выпускается в один из азимутальных квадрантов. Например, если весь поток сырья выпускается в реакционную среду через два отверстия для сырья, которые азимутально отделены друг от друга на 89 градусов, в целях определения азимутального распределения в четырех азимутальных квадрантах 100% масс. потока сырья выпускают в реакционную среду в одном азимутальном квадранте, так как азимутальные квадранты могут быть азимутально ориентированы таким образом, что оба отверстия для сырья располагаются в одном азимутальном квадранте.
Помимо преимуществ, связанных с соответствующим азимутальным расположением отверстий для сырья, также установлено, что может иметь значение соответствующее радиальное расположение отверстий для сырья в реакторе по типу барботажной колонны. Предпочтительно, чтобы значительная часть способного к окислению соединения, введенного в реакционную среду, выпускалась через отверстия для сырья, которые отделены радиальными промежутками по направлению внутрь от боковой стенки емкости. Следовательно, в одном из вариантов осуществления настоящего изобретения значительная часть способного к окислению соединения входит в реакционную зону через отверстия для сырья, расположенные в «предпочтительной радиальной зоне сырья», которая отделена промежутком по направлению внутрь от вертикальных боковых стенок, определяющих реакционную зону.
Что касается ФИГ. 7, то предпочтительная радиальная зона сырья «FZ» может иметь форму теоретического вертикального цилиндра, центрированного в реакционной зоне 28 и имеющего внешний диаметр «Dо», равный 0,9D, где «D» представляет собой диаметр реакционной зоны 28. Следовательно, внешнее кольцевое пространство «ОА», имеющее толщину 0,05D, определяет границу между предпочтительной радиальной зоной сырья FZ и внутренней стороной боковой стенки, определяющей реакционную зону 28. Предпочтительно, чтобы незначительное количество способного к окислению соединения вводилось в реакционную зону 28 через отверстия для сырья, расположенные в таком внешнем кольцевом пространстве ОА, или не вводилось вообще.
В другом варианте осуществления изобретения предпочтительно, чтобы незначительное количество способного к окислению соединения вводилось в центр реакционной зоны 28 или не вводилось вообще. Следовательно, как показано на ФИГ. 8, предпочтительная радиальная зона FZ может иметь форму теоретического вертикального кольцевого пространства, центрированного в реакционной зоне, которое имеет внешний диаметр DO, равный 0,9D, и имеет внутренний диаметр D1, равный 0,2D. Таким образом, в этом варианте изобретения внутренний цилиндр IC, имеющий диаметр 0,2D, «вырезан из» центра предпочтительной радиальной зоны сырья FZ. Предпочтительно, чтобы незначительное количество способного к окислению соединения вводилось в реакционную зону 28 через отверстия для сырья, расположенные в таком внутреннем цилиндре IC, или не вводилось вообще.
В предпочтительном варианте осуществления настоящего изобретения значительная часть способного к окислению соединения вводится в реакционную среду 36 через отверстия для сырья, расположенные в предпочтительной радиальной зоне сырья, независимо от того, имеет ли предпочтительная радиальная зона сырья цилиндрическую или кольцевую форму, описанную выше. Более предпочтительно, по меньшей мере, приблизительно 25% масс. способного к окислению соединения выпускается в реакционную среду 36 через отверстия для сырья, расположенные в предпочтительной радиальной зоне сырья. Еще более предпочтительно, по меньшей мере, приблизительно 50% масс. способного к окислению соединения выпускается в реакционную среду 36 через отверстия для сырья, расположенные в предпочтительной радиальной зоне сырья. Наиболее предпочтительно, по меньшей мере, приблизительно 75% масс. способного к окислению соединения выпускается в реакционную среду 36 через отверстия для сырья, расположенные в предпочтительной радиальной зоне сырья.
Хотя теоретические азимутальные квадранты и теоретическая предпочтительная зона сырья, показанные на ФИГ. 7 и 8, описаны со ссылкой на распределение жидкофазного потока сырья, также установлено, что соответствующее азимутальное и радиальное распределение газофазного потока окислителя также может создавать некоторые преимущества. Следовательно, в одном из вариантов настоящего изобретения описание азимутального и радиального распределения жидкофазного потока сырья, представленное выше, также применимо к модели, по которой газофазный поток окислителя вводят в реакционную зону 36.
На ФИГ. 12 и 13 показан альтернативный реактор по типу барботажной колонны 200, имеющий конфигурацию «реактор в реакторе». Реактор по типу барботажной колонны 200 включает внешний реактор 202 и внутренний реактор 204, причем внутренний реактор 204, по меньшей мере, частично расположен во внешнем реакторе 202. В предпочтительном варианте осуществления как внешний реактор, так и внутренний реактор 202 и 204 представляют собой реакторы по типу барботажной колонны. Предпочтительно внешний реактор 202 включает внешнюю реакционную емкость 206 и внешний барботер окислителя 208, тогда как внутренний реактор 204 включает внутреннюю реакционную емкость 210 и внутренний барботер окислителя 212.
Хотя ФИГ. 12 и 13 показывают внутреннюю реакционную емкость 210 как полностью расположенную во внешней реакционной емкости 206, допустимо, чтобы внутренняя реакционная емкость 210 только частично была расположена во внешней реакционной емкости 206. Однако предпочтительно, чтобы, по меньшей мере, приблизительно 50, 90, 95 или 100% высоты внутренней реакционной емкости 210 было расположено во внешней реакционной емкости 206. Более того, предпочтительно, чтобы часть каждой реакционной емкости была поднята выше части другой реакционной емкости, по меньшей мере, приблизительно на 0,01, 0,2, 1 или 2 максимальных диаметра внешней реакционной емкости.
В предпочтительном варианте осуществления настоящего изобретения каждая внешняя и внутренняя реакционные емкости 206 и 210 включают соответствующую расположенную вертикально боковую стенку, имеющую обычно конфигурацию цилиндра. Предпочтительно, расположенные вертикально боковые стенки внешней и внутренней реакционных емкостей 206 и 210 по существу являются концентрическими и определяют кольцевое пространство между ними. Внутренняя реакционная емкость 210 поддерживается вертикально к внешней реакционной емкости 206, предпочтительно главным образом с помощью вертикальных опор между нижними частями соответствующих емкостей. Кроме того, внутренняя реакционная емкость 210 может поддерживаться внешней реакционной емкостью 206 с помощью множества продольных опорных элементов 214, простирающихся между расположенной вертикально боковой стенкой внешней и внутренней реакционных емкостей 206 и 210. Предпочтительно такие продольные опорные элементы 214 имеют незагрязняющуюся конфигурацию с минимальной обращенной вверх плоской поверхностью, как описано ранее.
Хотя предпочтительно, чтобы расположенная вертикально боковая стенка внутренней реакционной емкости 210 была по существу цилиндрической, допустимо, чтобы некоторые части расположенной вертикально боковой стенки внутренней реакционной емкости 210 образовывали вогнутую поверхность относительно примыкающей части второй реакционной зоны 218. Предпочтительно любая часть расположенной вертикально боковой стенки внутренней реакционной емкости 210, которая вогнута относительно примыкающей части второй реакционной зоны 218, составляла менее чем приблизительно 25, 10, 5 или 0,1% от общей площади поверхности расположенной вертикально боковой стенки внутренней реакционной емкости 210. Предпочтительно отношение максимальной высоты вертикальной боковой стенки внутренней реакционной емкости 210 к максимальной высоте вертикальной боковой стенки внешней реакционной емкости 206 находится в интервале приблизительно от 0,1:1 до 0,9:1, более предпочтительно в интервале приблизительно от 0,2:1 до 0,8:1 и наиболее предпочтительно в интервале приблизительно от 0,3:1 до 0,7:1.
Внешняя реакционная емкость 206 определяет здесь первую реакционную зону 216, тогда как внутренняя реакционная емкость 210 определяет здесь вторую реакционную зону 218. Предпочтительно внешняя и внутренняя реакционные емкости 206 и 210 расположены в линию вертикально так, чтобы объемный центр тяжести второй реакционной зоны 218 был горизонтально смещен от объемного центра тяжести первой реакционной зоны 216, по меньшей мере, приблизительно на 0,4, 0,2, 0,1 или 0,01 части от максимального горизонтального диаметра первой реакционной зоны 216. Предпочтительно отношение площадей максимального горизонтального поперечного сечения первой реакционной зоны 216 и второй реакционной зоны 218 находится в интервале приблизительно от 0,01:1 до 0,75:1, более предпочтительно в интервале приблизительно от 0,03:1 до 0,5:1 и наиболее предпочтительно в интервале от 0,05:1 до 0,3:1. Предпочтительно отношение площади горизонтального поперечного сечения второй реакционной зоны 218 к площади горизонтального поперечного сечения кольцевого пространства, очерченного между внешней и внутренней реакционными емкостями 206 и 210, составляет, по меньшей мере, приблизительно 0,2:1, более предпочтительно находится в интервале приблизительно от 0,05:1 до 2:1 и наиболее предпочтительно в интервале приблизительно от 0,1:1 до 1:1, где площадь поперечного сечения измеряют на ¼ высоты, ½ высоты и/или ¾ высоты второй реакционной зоны 218. Предпочтительно, по меньшей мере, приблизительно 50, 70, 90 или 100% объема второй реакционной зоны 218 расположено во внешней реакционной емкости 206. Предпочтительно отношение объема первой реакционной зоны 216 к объему второй реакционной зоны 218 находится в интервале приблизительно от 1:1 до 100:1, более предпочтительно в интервале приблизительно от 4:1 до 50:1 и наиболее предпочтительно в интервале от 8:1 до 30:1. Предпочтительно первая реакционная зона 216 имеет отношение максимальной вертикальной высоты к максимальному горизонтальному диаметру в интервале приблизительно от 3:1 до 30:1, более предпочтительно приблизительно от 6:1 до 20:1 и наиболее предпочтительно в интервале от 9:1 до 15:1. Предпочтительно вторая реакционная зона 218 имеет отношение максимальной вертикальной высоты к максимальному горизонтальному диаметру в интервале приблизительно от 0,3:1 до 100:1, более предпочтительно в интервале приблизительно от 1:1 до 50:1 и наиболее предпочтительно в интервале от 3:1 до 30:1. Предпочтительно максимальный горизонтальный диаметр второй реакционной зоны 218 находится в интервале приблизительно от 0,1 до 5 метров, более предпочтительно в интервале приблизительно от 0,3 до 4 м и наиболее предпочтительно в интервале от 1 до 3 м. Предпочтительно максимальная вертикальная высота второй реакционной зоны 218 находится в интервале приблизительно от 1 до 100 м, более предпочтительно в интервале приблизительно от 3 до 50 м и наиболее предпочтительно в интервале от 10 до 30 м. Предпочтительно отношение максимального горизонтального диаметра второй реакционной зоны 218 к максимальному горизонтальному диаметру первой реакционной зоны 216 находится в интервале приблизительно от 0,05:1 до 0,8:1, более предпочтительно в интервале приблизительно от 0,1:1 до 0,6:1 и наиболее предпочтительно в интервале от 0,2:1 до 0,5:1. Предпочтительно отношение максимальной вертикальной высоты второй реакционной зоны 218 к максимальной вертикальной высоте первой реакционной зоны 216 находится в интервале приблизительно от 0,03:1 до 1:1, более предпочтительно в интервале приблизительно от 0,1:1 до 0,9:1 и наиболее предпочтительно в интервале от 0,3:1 до 0,8:1. Любые параметры (например, высота, ширина, площадь, объем, относительное горизонтальное размещение и относительное вертикальное размещение), определенные здесь для внешней реакционной емкости 206 и составных частей, также считаются применимыми к первой реакционной зоне 216, определяемой внешней реакционной емкостью 206, и наоборот. Кроме того, любые параметры, определенные здесь для внутренней реакционной емкости 210 и составных частей, также считаются применимыми ко второй реакционной зоне 218, определяемой внутренней реакционной емкостью 210, и наоборот.
Во время работы реактора по типу барботажной колонны 200 многофазная реакционная среда 220 вначале подвергается окислению в первой реакционной зоне 216, а затем подвергается окислению во второй реакционной зоне 218. Следовательно, при нормальной работе первая часть реакционной среды 220а располагается в первой реакционной зоне 216, тогда как вторая часть реакционной среды 220b располагается во второй реакционной зоне 218. После обработки во второй реакционной зоне 218 суспензионную фазу (то есть жидкую и твердую фазы) реакционной среды 220b выводят из второй реакционной зоны 218 и выгружают из реактора по типу барботажной колонны 200 через выпускное отверстие для суспензии 222 для последующей переработки ниже по технологическому потоку.
Внутренний реактор 204 предпочтительно имеет, по меньшей мере, одно внутреннее отверстие для газа, которое позволяет впускать дополнительный молекулярный кислород во вторую реакционную зону 218. Предпочтительно множество внутренних отверстий для газа определено внутренним барботером окислителя 212. Описания для барботера окислителя 34 на ФИГ. 1-5 также применимы к внутреннему барботеру окислителя 212 с точки зрения размеров и конфигураций трубопровода, размера и конфигурации отверстий, рабочего перепада давлений и жидкостных промывок. Заметным отличием является то, что предпочтительно располагать барботер окислителя 212 относительно выше, чтобы использовать нижнюю часть внутренней реакционной емкости 210 в виде зоны деаэрации. Например, варианты осуществления, описанные в настоящем изобретении для окисления пара-ксилола с образованием ТФК (ТРА), обеспечивают значительно пониженные пространственно-временные скорости реакции около нижней части второй реакционной зоны 218, и это ослабляет влияние деаэрации на образование примесей. Внутренняя реакционная емкость 210 имеет максимальную высоту «Hi». Предпочтительно, чтобы, по меньшей мере, приблизительно 50, 75, 95 или 100% общей площади пропускного сечения, определенной всеми внутренними отверстиями для газа, находилось на расстоянии, по меньшей мере, 0,05Hi, 0,1Hi или 0,25Hi от верха внутренней реакционной емкости 210. Также предпочтительно, чтобы, по меньшей мере, приблизительно 50, 75, 95 или 100% общей площади пропускного сечения, определенной всеми внутренними отверстиями для газа, находилось на расстоянии, по меньшей мере, приблизительно 0,5Hi, 0,25Hi или 0,1Hi выше днища внутренней реакционной емкости 210. Предпочтительно, чтобы, по меньшей мере, приблизительно 50, 75, 95 или 100% общей площади пропускного сечения, определенной всеми внутренними отверстиями для газа, находилось на расстоянии, по меньшей мере, 1, 5 или 10 м от верха внутренней реакционной емкости 210 и на расстоянии, по меньшей мере, приблизительно на 0,5, 1 или 2 м от днища внутренней реакционной емкости 210. Предпочтительно, чтобы, по меньшей мере, приблизительно 50, 75, 95 или 100% общей площади пропускного сечения, определенной всеми внутренними отверстиями для газа, соединялось непосредственно со второй реакционной зоной 218 и не соединялось непосредственно с первой реакционной зоной 216. Как используется в данном случае, определение «площадь пропускного сечения» означает минимальную площадь поверхности (плоскую или неплоскую), которая будет закрывать отверстие.
В общем случае модель, по которой потоки сырья, окислителя и флегмы вводят во внешний реактор 202, и модель, по которой работает внешний реактор 202, по существу являются такими же, что описаны выше для реактора по типу барботажной колонны 20 на ФИГ. 1-11. Однако одно различие между внешним реактором 202 (ФИГ. 12 и 13) и реактором по типу барботажной колонны 20 (ФИГ. 1-11) состоит в том, что внешний реактор 202 не включает выпускное отверстие, которое позволяет суспензионную фазу реакционной среды 220а непосредственно выпускать из внешней реакционной емкости 206 для переработки вниз по технологическому потоку. Точнее, реактор по типу барботажной колонны 200 требует, чтобы суспензионная фаза реакционной среды 220а перед выгрузкой из реактора по типу барботажной колонны 200 вначале проходила через внутренний реактор 204. Как упоминалось выше, во второй реакционной зоне 218 внутреннего реактора 204 реакционная среда 220b подвергается дополнительному окислению, чтобы способствовать очистке жидкой и/или твердой фаз реакционной среды 220b.
В способе, где пара-ксилол подают в реакционную зону 216, жидкая фаза реакционной среды 220а, которая выходит из первой реакционной зоны 216 и входит во вторую реакционную зону 218, как правило, содержит, по меньшей мере, некоторое количество пара-толуиловой кислоты. Предпочтительно, чтобы значительная часть пара-толуиловой кислоты, поступающей во вторую реакционную зону, была окислена во второй реакционной зоне 218. Следовательно, предпочтительно, чтобы усредненная по времени концентрация пара-толуиловой кислоты в жидкой фазе реакционной среды 220b, выходящей из второй реакционной зоны 218, была меньше чем усредненная по времени концентрация пара-толуиловой кислоты в жидкой фазе реакционной среды 220а/b, входящей во вторую реакционную зону 218. Предпочтительно, чтобы усредненная по времени концентрация пара-толуиловой кислоты в жидкой фазе реакционной среды 220b, выходящей из второй реакционной зоны 218, составляла менее чем приблизительно 50, 10 или 5% от усредненной по времени концентрации пара-толуиловой кислоты в жидкой фазе реакционной среды 220а/b, входящей во вторую реакционную зону 218. Предпочтительно усредненная по времени концентрация пара-толуиловой кислоты в жидкой фазе реакционной среды 220а/b, входящей во вторую реакционную зону 218, составляет, по меньшей мере, приблизительно 250 масс. ч./млн, более предпочтительно находится в интервале приблизительно от 500 до 6000 масс. ч./млн и наиболее предпочтительно в интервале от 1000 до 4000 масс. ч./млн. Предпочтительно усредненная по времени концентрация пара-толуиловой кислоты в жидкой фазе реакционной среды 220b, выходящей из второй реакционной зоны 218, составляет менее чем приблизительно 1000, 250 или 50 масс. ч./млн.
Внутренняя реакционная емкость 210 оборудована, по меньшей мере, одним прямым отверстием, которое позволяет реакционной среде 220а/b проходить непосредственно между реакционной зоной 216 и второй реакционной зоной 218. Предпочтительно, чтобы по существу все прямые отверстия во внутренней реакционной емкости 210 были расположены около верха внутренней реакционной емкости 210. Предпочтительно, по меньшей мере, приблизительно 50, 75, 90 или 100% общей площади пропускного сечения, определенного всеми прямыми отверстиями, находится на расстоянии менее чем приблизительно 0,5Нi, 0,25Нi или 0,1Нi от верха внутренней реакционной емкости 210. Предпочтительно менее чем приблизительно 50, 25, 10 или 1% общей площади пропускного сечения, определенного всеми прямыми отверстиями во внутренней реакционной емкости 210, находится на расстоянии более чем приблизительно 0,5Нi, 0,25Нi или 0,1Нi от верха внутренней реакционной емкости 210. Наиболее предпочтительно прямое отверстие, определенное внутренней реакционной емкостью 210, является единственным верхним отверстием 224, расположенным на самом верхнем конце внутренней реакционной емкости 210. Отношение площади пропускного сечения верхнего отверстия 224 к площади максимального горизонтального поперечного сечения второй реакционной зоны 218 предпочтительно составляет, по меньшей мере, 0,1:1, 0,2:1 или 0,5:1.
При нормальной работе реактора по типу барботажной колонны 200 реакционная среда 220 проходит из первой реакционной зоны 216, через прямое(ые) отверстие(ия) (например, верхнее отверстие 224) во внутреннюю реакционную емкость 210 и во вторую реакционную зону 218. Во второй реакционной зоне 218 суспензионная фаза реакционной среды 220b перемещается обычно в направлении вниз через вторую реакционную зону 218, тогда как газовая фаза реакционной среды 220b перемещается обычно по направлению вверх. Предпочтительно внутренняя реакционная емкость 210 определяет, по меньшей мере, одно выгружающее отверстие, которое позволяет суспензионной фазе выходить из второй реакционной зоны 218. Суспензионная фаза, выходящая из выгружающего отверстия внутренней реакционной емкости 210, затем выходит из реактора по типу барботажной колонны 200 через выпускное отверстие для суспензии 222. Предпочтительно выгружающее отверстие располагается на днище внутренней реакционной емкости 210 или рядом с ним. Предпочтительно, по меньшей мере, приблизительно 50, 75, 90 или 100% общей площади пропускного сечения, определенного всеми выгружающими отверстиями во внутренней реакционной емкости 210, располагается в пределах приблизительно 0,5Нi, 0,25Нi или 0,1Нi, от днища внутренней реакционной емкости 210.
Так как реакционная среда 220b подвергается обработке во второй реакционной зоне 218 внутреннего реактора 204, предпочтительно понижать удерживание газа реакционной среды 220b, пока суспензионная фаза реакционной среды 220b протекает по направлению вниз через вторую реакционную зону 218. Предпочтительно отношение усредненного по времени удерживания газа реакционной среды 220а/b, поступающей во вторую реакционную зону 218, к удерживанию газа реакционной среды 220b, выходящей из второй реакционной зоны 218, составляет, по меньшей мере, приблизительно 2:1, 10:1 или 25:1. Предпочтительно усредненное по времени удерживание газа реакционной среды 220а/b, выходящей из второй реакционной зоны 218, находится в интервале приблизительно от 0,4 до 0,9, более предпочтительно в интервале приблизительно от 0,5 до 0,8 и наиболее предпочтительно в интервале от 0,55 до 0,7. Предпочтительно усредненное по времени удерживание газа реакционной среды 220b, выходящей из второй реакционной зоны 218, составляет менее чем приблизительно 0,1, 0,05 или 0,02. Предпочтительно отношение усредненного по времени удерживания газа реакционной среды 220а в первой реакционной зоне 216 к удерживанию газа реакционной среды 220b во второй реакционной зоне 218 составляет больше чем приблизительно 1:1, более предпочтительно находится в интервале приблизительно от 1,25:1 до 5:1 и наиболее предпочтительно в интервале от 1,5:1 до 4:1, где величина удерживания газа измерена на любой высоте первой и второй реакционных зон 216 и 218, на любых соответствующих высотах первой и второй реакционных зон 216 и 218, на ¼ высоты первой и/или второй реакционных зон 216 и 218, на ½ высоты первой и/или второй реакционных зон 216 и 218, на ¾ высоты первой и/или второй реакционных зон 216 и 218, и/или представляет собой среднее значение по всем высотам первой и/или второй реакционных зон 216 и 218. Предпочтительно усредненное по времени удерживание газа части реакционной среды 220а в первой реакционной зоне 216 находится в интервале приблизительно от 0,4 до 0,9, более предпочтительно в интервале приблизительно от 0,5 до 0,8 и наиболее предпочтительно в интервале от 0,55 до 0,70, где удерживание газа измерено на любой высоте первой реакционной зоны 216, на ¼ высоты первой реакционной зоны 216, на ½ высоты первой реакционной зоны 216, на ¾ высоты первой реакционной зоны 216, и/или представляет собой среднее по всей высоте первой реакционной зоны 216. Предпочтительно усредненное по времени удерживание газа части реакционной среды 220b во второй реакционной зоне 218 находится в интервале приблизительно от 0,01 до 0,6, более предпочтительно в интервале приблизительно от 0,03 до 0,3 и наиболее предпочтительно в интервале от 0,08 до 0,2, где удерживание газа измерено на любой высоте второй реакционной зоны 218, на ¼ высоты второй реакционной зоны 218, на ½ высоты второй реакционной зоны 218, на ¾ высоты второй реакционной зоны 218, и/или представляет собой среднее по всей высоте второй реакционной зоны 218.
Температура реакционной среды 220 предпочтительно приблизительно равна температуре в первой и во второй реакционных зонах 216 и 218. Предпочтительно такая температура находится в интервале приблизительно от 125 до 200ºС, более предпочтительно в интервале приблизительно от 140 до 180ºС и наиболее предпочтительно в интервале от 150 до 170ºС. Однако возникают разницы температур в пределах первой реакционной зоны 216, которая является такой же, которая описана со ссылкой на ФИГ. 28. Предпочтительно разницы температур такого же порядка также существуют в пределах второй реакционной зоны 218, а также между первой реакционной зоной 216 и второй реакционной зоной 218. Такие дополнительные температурные градиенты связаны с химической реакцией, протекающей во второй реакционной зоне 218, с введением дополнительного окислителя во вторую реакционную зону 218 и статическими давлениями, существующими во второй реакционной зоне 218, по сравнению с давлениями в первой реакционной зоне 216. Как описывалось выше, удерживание пузырьков предпочтительно выше в первой реакционной зоне 216, чем во второй реакционной зоне 218. Следовательно, на отметках высоты ниже верхнего отверстия 224 статическое давление в реакционной зоне 216 больше, чем во второй реакционной зоне 218. Величина такой разницы давлений зависит от величины плотности жидкости или суспензии и от разницы в удерживании пузырьков между двумя реакционными зонами. Величина этой разницы давлений растет на отметках высоты еще ниже верхнего отверстия 224.
В одном из вариантов осуществления настоящего изобретения часть способного к окислению соединения (например, пара-ксилола), подаваемого в реактор по типу барботажной колонны 200, вводится непосредственно во вторую реакционную зону 218 внутреннего реактора 204. Однако предпочтительно, по меньшей мере, приблизительно 90, 95, 99 или 100% мол. всего способного к окислению соединения, поданного в реактор по типу барботажной колонны 200, вводить в первую реакционную зону 216 (а не во вторую реакционную зону 218). Предпочтительно мольное отношение количества способного к окислению соединения, введенного в первую реакционную зону 216, к количеству способного к окислению соединения, введенного во вторую реакционную зону 218, составляет, по меньшей мере, 2:1, 4:1 или 8:1.
Хотя ФИГ. 12 и 13 показывают конфигурацию, когда часть всего молекулярного кислорода, подаваемого в реактор по типу барботажной колонны 200, вводится во вторую реакционную зону 218 внутреннего реактора 204 через внутренний барботер окислителя 212, предпочтительно, чтобы основная часть всего молекулярного кислорода, подаваемого в реактор по типу барботажной колонны 200, вводилась в первую реакционную зону 216, причем остальное количество вводится во вторую реакционную зону 218. Предпочтительно, по меньшей мере, приблизительно 70, 90, 95 или 98% мол. всего молекулярного кислорода, подаваемого в реактор по типу барботажной колонны 200, вводится в первую реакционную зону 216. Предпочтительно мольное отношение количества молекулярного кислорода, введенного в первую реакционную зону 216, к количеству молекулярного кислорода, введенного во вторую реакционную зону 218, составляет, по меньшей мере, приблизительно 2:1, более предпочтительно находится в интервале приблизительно от 4:1 до 200:1, наиболее предпочтительно в интервале от 10:1 до 100:1. Хотя возможно, чтобы некоторое количество растворителя и/или способного к окислению соединения (например, пара-ксилола) было подано непосредственно во вторую реакционную зону 218, предпочтительно, чтобы менее чем приблизительно 10, 5 или 1% масс. всего количества растворителя и/или способного к окислению соединения, подаваемого в реактор по типу барботажной колонны 200, было подано непосредственно во вторую реакционную зону 218.
Объем, время пребывания и объемная скорость среды 220а в первой реакционной зоне 216 внешней реакционной емкости 206 предпочтительно значительно выше объема, времени пребывания и объемной скорости реакционной среды 220b во второй реакционной зоне 218 внутренней реакционной емкости 210. Следовательно, основная часть способного к окислению соединения (например, пара-ксилола), подаваемого в реактор по типу барботажной колонны 200, предпочтительно окисляется в первой реакционной зоне 216. Предпочтительно, по меньшей мере, приблизительно 80, 90 или 95% масс. всего способного к окислению соединения, которое окисляется в реакторе по типу барботажной колонны 200, окисляется в первой реакционной зоне 216. Предпочтительно, чтобы усредненная по времени приведенная скорость газа реакционной среды 220а в первой реакционной зоне 216 составляла, по меньшей мере, приблизительно 0,2, 0,4, 0,8 или 1 метр в секунду, где приведенная скорость газа измерена на любой высоте первой реакционной зоны 216, на ¼ высоты первой реакционной зоны 216, на ½ высоты первой реакционной зоны 216, на ¾ высоты первой реакционной зоны 216, и/или представляет собой среднее по всей высоте первой реакционной зоны 216.
Хотя реакционная среда 220b во второй реакционной зоне 218 может иметь такую же приведенную скорость газа, что и реакционная среда 220а в первой реакционной зоне 216, предпочтительно, чтобы усредненная по времени приведенная скорость газа реакционной среды 220b во второй реакционной зоне 218 была меньше усредненной по времени и усредненной по объему приведенной скорости газа реакционной среды 220b во второй реакционной зоне 218. Эта пониженная приведенная скорость газа во второй реакционной зоне 218 делается возможной, например, за счет пониженного потребления молекулярного кислорода во второй реакционной зоне 218 по сравнению с первой реакционной зоной 216. Предпочтительно отношение усредненной по времени приведенной скорости реакционной среды 220а в первой реакционной зоне 216 к усредненной по времени приведенной скорости реакционной среды 220b во второй реакционной зоне 218 составляет, по меньшей мере, приблизительно 1,25:1, 2:1 или 5:1, где приведенные скорости газа измерены на любой высоте первой и второй реакционных зон 216 и 218, на любых соответствующих высотах первой и второй реакционных зон 216 и 218, на ¼ высоты первой и/или второй реакционных зон 216 и 218, на ½ высоты первой и/или второй реакционных зон 216 и 218, на ¾ высоты первой и/или второй реакционных зон 216 и 218, и/или представляет собой среднее значение по всем высотам первой и/или второй реакционных зон 216 и 218. Предпочтительно усредненная по времени и усредненная по объему приведенная скорость газа реакционной среды 220b во второй реакционной зоне 218 составляет менее чем приблизительно 0,2, 0,1 или 0,06 метра в сек, где приведенная скорость газа измерена на любой высоте второй реакционной зоны 218, на ¼ высоты второй реакционной зоны 218, на ½ высоты второй реакционной зоны 218, на ¾ высоты второй реакционной зоны 218, и/или представляет собой среднее по всей высоте второй реакционной зоны 218. С такими низкими приведенными скоростями газа может быть получен нисходящий поток суспензионной фазы реакционной среды 220b во второй реакционной зоне 218, чтобы направленно перейти к поршневому режиму двухфазного потока. Например, во время окисления пара-ксилола до ТФК относительные вертикальные градиенты концентрации в жидкой фазе пара-толуиловой кислоты могут быть намного больше во второй реакционной зоне 218, чем в первой реакционной зоне 216. И это несмотря на то, что вторая реакционная зона 218 представляет собой барботажную колонну, имеющую аксиальное смещение жидкой и суспензионной композиций. Усредненная по времени приведенная скорость суспензионной фазы (твердое вещество + жидкость) и жидкой фазы реакционной среды 220b во второй реакционной зоне 218 предпочтительно составляет меньше чем приблизительно 0,2, 0,1 или 0,06 м/сек, где приведенная скорость измерена на любой высоте второй реакционной зоны 218, на ¼ высоты второй реакционной зоны 218, на ½ высоты второй реакционной зоны 218, на ¾ высоты второй реакционной зоны 218, и/или представляет собой среднее по всей высоте второй реакционной зоны 218.
В одном из вариантов осуществления настоящего изобретения реактор по типу барботажной колонны 200 работает таким образом, чтобы обеспечить осаждение твердых веществ во внутреннем реакторе 204. Если осаждение твердых веществ желательно, предпочтительно, чтобы усредненная по времени и усредненная по объему приведенная скорость газа реакционной среды 220b во второй реакционной зоне составляла менее чем приблизительно 0,05, 0,03 или 0,01 м/сек. Кроме того, если осаждение твердых веществ желательно, предпочтительно, чтобы усредненная по времени и усредненная по объему приведенная скорость суспензионной фазы и жидкой фазы реакционной среды 220b во второй реакционной зоне 218 составляла менее чем приблизительно 0,01, 0,005 или 0,001 м/сек.
Хотя допустимо, чтобы некоторая часть суспензионной фазы, выходящей из внутреннего реактора 204, непосредственно была рециркулирована назад в первую реакционную зону 216 без дополнительной обработки в нисходящем потоке, предпочтительно, чтобы прямое рециркулирование реакционной среды 220b от нижних отметок высоты второй реакционной зоны 218 в первую реакционную зону 216 было минимизировано. Предпочтительно масса реакционной среды 220b (твердой, жидкой и газовой фаз), выходящей из нижних 25% объема второй реакционной зоны 218 и непосредственно рециркулируемой назад в первую реакционную зону 216 без дополнительной обработки в нисходящем потоке, составляет менее чем 10, 1 или 0,1 части от массы (твердой, жидкой и газовой фаз) реакционной среды 220b, выходящей из второй реакционной зоны 218 и затем подвергнутой обработке в нисходящем потоке. Предпочтительно масса реакционной среды 220b, выходящей из нижних 50% объема второй реакционной зоны 218 и непосредственно рециркулируемой назад в первую реакционную зону 216 без дополнительной обработки в нисходящем потоке, составляет менее чем 20, 2 или 0,2 части от массы реакционной среды 220b, выходящей из второй реакционной зоны 218 и затем подвергнутой обработке в нисходящем потоке. Предпочтительно менее чем приблизительно 50, 75 или 90% масс. жидкой фазы реакционной среды 220b, выходящей из второй реакционной зоны 218 через отверстия в нижних 90, 60, 50 или 5% объема второй реакционной зоны 218, вводится в первую реакционную зону 216 в течение 60, 20, 5 или 1 мин после выхода из второй реакционной зоны 218. Предпочтительно жидкая фаза реакционной среды 220b, расположенная во второй реакционной зоне 218, имеет усредненное по массе время пребывания во второй реакционной зоне 218, по меньшей мере, приблизительно 1 мин, более предпочтительно находится в интервале приблизительно от 2 до 60 мин и наиболее предпочтительно в интервале от 5 до 30 мин. Предпочтительно менее чем приблизительно 50, 75 или 90% масс. жидкой фазы реакционной среды 220а/b, введенной во вторую реакционную зону 218, входит во вторую реакционную зону 218 в нижние 90, 60 или 30% объема второй реакционной зоны 218. Предпочтительно менее чем приблизительно 50, 75 или 90% масс. от общего объема жидкой фазы реакционной среды 220a/b, введенной в виде жидкофазного потока в первую реакционную зону 216, поступает в первую реакционную зону 216 в течение 60, 20, 5 или 1 мин после выведения из второй реакционной зоны 218 через выпускное отверстие для суспензии 222. Предпочтительно, по меньшей мере, приблизительно 75, 90, 95 или 99% масс. жидкой фазы реакционной среды 220b, выведенной из второй реакционной зоны 218, выходит из второй реакционной зоны 218 через отверстия в нижних 90, 60, 30 или 5% объема второй реакционной зоны 218.
Конструкция реактора по типу барботажной колонны в конфигурации «реактор в реакторе» 200 может быть изменена различным образом без отклонения от объема настоящего изобретения. Например, внутренняя реакционная емкость 210 может иметь большую высоту, чем внешняя реакционная емкость 206, если внутренняя реакционная емкость 210 простирается ниже нижнего конца внешней реакционной емкости 206. Внешняя и внутренняя реакционные емкости 206 и 210 могут быть цилиндрическими, как показано, или могут иметь любую форму. Внешняя и внутренняя реакционные емкости 206 и 210 не должны быть симметричными по оси, аксиально вертикальными или концентрическими. Газовая фаза, выходящая из внутреннего реактора 204, может быть направлена наружу из реактора по типу барботажной колонны 200 без объединения с реакционной средой 220a в первой реакционной зоне 216. Однако с точки зрения безопасности от воспламенения желательно ограничивать объемы захваченных газовых карманов менее чем приблизительно до 10, 2 или 1 м3. Кроме того, нет необходимости, чтобы суспензионная фаза, выходящая из внутреннего реактора 204, выходила через единственное отверстие для суспензии в днище внутренней реакционной емкости 210. Суспензионная фаза может выходить из реактора по типу барботажной колонны 200 через боковое выпускное отверстие в находящейся под давлением боковой стенке внешнего реактора 202.
На ФИГ. 14 представлен реактор по типу барботажной колонны 300, имеющий конфигурацию «реактор в реакторе» и конфигурацию ступенчатого диаметра. Реактор по типу барботажной колонны 300 включает внешний реактор 302 и внутренний реактор 304. Внешний реактор 302 представляет собой внешнюю реакционную емкость 306, имеющую широкую нижнюю секцию 306а и узкую верхнюю секцию 306b. Предпочтительно диаметр узкой верхней секции 306b меньше диаметра широкой нижней секции 306a. За исключением конфигурации ступенчатого диаметра внешней реакционной емкости реактор по типу барботажной колонны 300 на ФИГ. 14 предпочтительно конфигурирован и работает по существу аналогично реактору по типу барботажной колонны 200 на ФИГ. 12 и 13, описанному выше.
На ФИГ. 15 представлена реакторная система 400, включающая реактор первичного окисления 402 и реактор вторичного окисления 404. Реактор первичного окисления 402 предпочтительно конфигурирован и работает по существу аналогично внешнему реактору 202 на ФИГ. 12 и 13. Вторичный реактор окисления 404 предпочтительно конфигурирован и работает по существу аналогично внутреннему реактору 204 на ФИГ. 12 и 13. Однако основное различие между реакторной системой 400 на ФИГ. 15 и реактором по типу барботажной колонны 200 на ФИГ. 12 и 13 состоит в том, что реактор вторичного окисления 404 реакторной системы 400 расположен вне реактора первичного окисления 402. В реакторной системе 400 на ФИГ. 15 впускной трубопровод 405 используется для перемещения части реакционной среды 420 из реактора первичного окисления 402 в реактор вторичного окисления 404. Кроме того, выпускной трубопровод 407 используется для перемещения головных газов из верха реактора вторичного окисления 404 в реактор первичного окисления 402.
При нормальной работе реакционной системы 400 реакционная среда 420 вначале подвергается окислению в первичной реакционной зоне 416 реактора первичного окисления 402. Реакционная среда 420а затем выводится из первичной реакционной зоны 416 и перемещается во вторичную реакционную зону 418 через трубопровод 405. Во вторичной реакционной зоне 418 жидкая и/или твердая фазы реакционной среды 420b подвергаются дополнительному окислению. Предпочтительно, чтобы, по меньшей мере, приблизительно 50, 75, 95 или 99% масс. жидкой фазы и/или твердой фазы выводилось из первичной реакционной зоны 416 для переработки во вторичной реакционной зоне 416. Головные газы выходят из верхнего выпускного отверстия для газа реактора вторичного окисления 404 и перемещаются назад в реактор первичного окисления 402 через трубопровод 407. Суспензионная фаза реакционной среды 420b выходит из нижнего выпускного отверстия суспензии 422 реактора вторичного окисления 404 и затем подвергается дополнительной обработке в нисходящем потоке.
Впускной трубопровод 405 может быть присоединен к реактору первичного окисления 402 на любой высоте. Хотя на ФИГ. 15 не показано, реакционная среда 420 может быть механически подана насосом во вторичную реакционную зону 418, если это желательно. Однако более предпочтительно использовать гидростатический напор (силу тяжести), чтобы передавать реакционную среду 420 из первичной реакционной зоны 416 через впускной трубопровод 405 и во вторичную реакционную зону 418. Соответственно предпочтительно, чтобы впускной трубопровод 405 был соединен на одном конце с верхними 50, 30, 20 или 10% всей высоты и/или всего объема первичной реакционной зоны 416. Предпочтительно другой конец впускного трубопровода 405 соединяется с верхними 30, 20, 10 или 5% всей высоты и/или всего объема вторичной реакционной зоны 418. Предпочтительно впускной трубопровод 405 является горизонтальным и/или наклоненным вниз от реактора первичного окисления 402 в направлении реактора вторичного окисления 404. Выпускной трубопровод 407 может быть присоединен к любой отметке высоты в реакторе вторичного окисления 404, но предпочтительно, чтобы выпускной трубопровод 407 соединялся с реактором вторичного окисления 404 выше отметки высоты присоединения впускного трубопровода 405. Более предпочтительно выпускной трубопровод 407 присоединяется к верху реактора вторичного окисления 404. Выпускной трубопровод 407 предпочтительно присоединяется к реактору первичного окисления 402 выше отметки высоты присоединения впускного трубопровода 405. Более предпочтительно выпускной трубопровод 407 присоединяется к верхним 30, 20, 10 или 5% общей высоты и/или общего объема первичной реакционной зоны 416. Предпочтительно выпускной трубопровод 407 является горизонтальным и/или наклоненным вниз от реактора вторичного окисления 404 в направлении реактора первичного окисления 402. Хотя на ФИГ. 15 не показано, выпускной трубопровод 407 также может быть прикреплен непосредственно к выпускному трубопроводу для газа, который выводит газообразный выходящий поток из верха реактора первичного окисления 402. Верхнее пространство вторичной реакционной зоны 416 может быть выше или ниже верхнего пространства первичной реакционной зоны 418. Более предпочтительно верхняя протяженность первичной реакционной зоны 416 находится в пределах от 10 м выше и до 50 м ниже, от 2 до 40 м ниже, от 5 до 30 м ниже верхней протяженности вторичной реакционной зоны 418. Нижнее выпускное отверстие для суспензии 422 может выходить от любой отметки высоты реактора вторичного окисления 404, но предпочтительно, чтобы нижнее выпускное отверстие для суспензии 422 было соединено с реактором вторичного окисления 404 ниже отметки высоты присоединения впускного трубопровода 405. Точка присоединения нижнего выпускного отверстия для суспензии 422 более предпочтительно с большим расстоянием отделена по высоте от точки присоединения впускного трубопровода 405, причем два присоединения разделены, по меньшей мере, приблизительно 50, 70, 90 или 95% высоты вторичной реакционной зоны 418. Более предпочтительно нижнее выпускное отверстие для суспензии 422 присоединено к днищу реактора вторичного окисления 404, как показано на ФИГ. 15. Нижняя протяженность вторичной реакционной зоны 418 может быть установлена на высоте выше или ниже нижней протяженности первичной реакционной зоны 416. Более предпочтительно нижняя протяженность первичной реакционной зоны 416 находится на высоте в пределах 40, 20, 5 или 2 м выше или ниже нижней протяженности вторичной реакционной зоны 418.
Параметры (например, высота, ширина, площадь, объем, относительное горизонтальное размещение и относительное вертикальное размещение), определенные здесь для реактора первичного окисления 402 и составных частей, также считаются применимыми к первичной реакционной зоне 416, определяемой реактором первичного окисления 402, и наоборот. Любые параметры, определенные в настоящем изобретении для реактора вторичного окисления 404 и составных частей, также считаются применимыми к вторичной реакционной зоне 418, определяемой реактором вторичного окисления 404, и наоборот.
Как упоминалось выше, предпочтительно, чтобы реактор вторичного окисления 404 был расположен вне реактора первичного окисления 402. Предпочтительно реактор вторичного окисления 404 располагается вдоль стороны реактора первичного окисления 402 (то есть, по меньшей мере, часть реакторов первичного и вторичного окисления 402 и 404 делят общую отметку высоты). Первичная реакционная зона 416 реактора первичного окисления 402 имеет максимальный диаметр «Dp». Объемный центр тяжести вторичной реакционной зоны 418 предпочтительно по горизонтали отделен промежутком от объемного центра тяжести первичной реакционной зоны 416, по меньшей мере, приблизительно на 0,5Dp, 0,75Dp или 1,0Dp и на менее чем приблизительно 30Dp, 10Dp или 3Dp.
На ФИГ. 16 показана реакторная система 500, включающая реактор первичного окисления 502 и реактор вторичного окисления 504. Реактор первичного окисления определяет в данном случае зону первичного окисления 516, тогда как реактор вторичного окисления 504 определяет зону вторичного окисления 518. Каждая реакционная зона 516 и 518 принимает часть реакционной среды 520.
Конфигурация и работа реакторной системы 500 (ФИГ. 16) предпочтительно аналогичны конфигурации и работе реакторной системы 400 (ФИГ. 15). Однако в реакторной системе 500 установленная вертикально боковая стенка реактора первичного окисления 502 определяет границы, по меньшей мере, одного увеличенного отверстия 505, которое позволяет передавать реакционную среду 520 из первичной реакционной зоны 516 во вторичную реакционную зону 518, при этом одновременно дает возможность передавать отделенную газовую фазу из вторичной реакционной зоны 518 в первичную реакционную зону 516. Предпочтительно площадь пропускного сечения увеличенного отверстия 505, поделенная на площадь максимального горизонтального поперечного сечения вертикальной части вторичной реакционной зоны 518, находится в интервале приблизительно от 0,01 до 2, от 0,02 до 0,5 или от 0,04 до 0,2. Первичная реакционная зона 516 реактора первичного окисления 502 имеет максимальную высоту «Нр». Предпочтительно, чтобы центр размещения увеличенного отверстия 505 был вертикально отделен промежутком, по меньшей мере, приблизительно 0,1Нр, 0,2Нр или 0,3Нр от верха и/или днища первичной реакционной зоны 516.
На ФИГ. 17-25 показан ряд реакторов по типу барботажной колонны, оборудованных внутренними структурами, имеющими различные конфигурации. Установлено, что применение одной или нескольких внутренних структур, окруженных реакционной средой, неожиданно модифицирует сквозное перемешивание реакционной среды. Внутренняя структура определяет статическую зону, имеющую пониженную турбулентность по сравнению с турбулентностью реакционной среды, окружающей статическую зону.
Как показано на ФИГ. 17-25, внутренняя структура может принимать различные формы. В частности, ФИГ. 17 иллюстрирует реактор по типу барботажной колонны 600, в котором используют в целом цилиндрическую внутреннюю структуру 602, чтобы определить статическую зону. Внутренняя структура 602 по существу центрирована в основной реакционной зоне реактора по типу барботажной колонны 600 и отделена вертикальным промежутком от верхнего и нижнего концов основной реакционной зоны. ФИГ. 18 иллюстрирует реактор по типу барботажной колонны 610, в котором используют в целом цилиндрическую внутреннюю структуру 612, аналогичную внутренней структуре 602 на ФИГ. 17. Однако внутренняя структура 612 на ФИГ. 18 не центрирована в основной реакционной зоне реактора по типу барботажной колонны 610. Более того, объемный центр тяжести статической зоны, определенной внутренней структурой 612, горизонтально смещен от объемного центра тяжести основной реакционной зоны. Кроме того, днище внутренней структуры 612 расположено около нижней касательной линии реактора по типу барботажной колонны 610. ФИГ. 19 иллюстрирует реактор по типу барботажной колонны 620, в котором используется в целом цилиндрическая внутренняя структура 622, которая выше внутренней структуры 602 и 612 на ФИГ. 17 и 18. Кроме того, объемный центр тяжести статической зоны внутренней структуры 622 смещен от объемного центра основной реакционной зоны реактора по типу барботажной колонны 620. ФИГ. 20 иллюстрирует реактор по типу барботажной колонны 630, в котором используется внутренняя структура, включающая обычно цилиндрическую верхнюю часть 632 и обычно цилиндрическую нижнюю часть 634. Нижняя часть 634 внутренней структуры имеет более узкий диаметр, чем верхняя часть 632. ФИГ. 21 иллюстрирует реактор по типу барботажной колонны 640, в котором используется внутренняя структура, включающая обычно цилиндрическую нижнюю часть 642 и обычно цилиндрическую верхнюю часть 644. Верхняя часть 644 внутренней структуры имеет более узкий диаметр, чем нижняя часть 642. ФИГ. 22 иллюстрирует реактор по типу барботажной колонны 650, в котором используется первая, вторая и третья отдельные внутренние структуры 652, 654 и 656. Внутренние структуры 652, 654 и 656 отделены вертикальными промежутками друг от друга. Объемные центры тяжести статических зон, определенных первой и третьей внутренними структурами 652 и 656, горизонтально выровнены с объемным центром тяжести основной реакционной зоны реактора по типу барботажной колонны 650. Однако объемный центр тяжести статической зоны, определенной внутренней структурой 654, горизонтально смещен от объемного центра тяжести основной реакционной зоны реактора по типу барботажной колонны 650. ФИГ. 23 иллюстрирует реактор по типу барботажной колонны 660, в котором используется пара расположенных «бок о бок» первой и второй внутренних структур 662 и 664. Объемные центры тяжести статических зон, определенных первой и второй внутренними структурами 662 и 664, горизонтально отделены промежутком друг от друга и горизонтально отделены промежутком от объемного центра тяжести основной реакционной зоны реактора по типу барботажной колонны 660. Кроме того, первая и вторая внутренняя структуры 662 и 664 имеют конфигурацию «бок о бок» так, что, по меньшей мере, часть первой и второй внутренних структур 662 и 664 разделяют общую отметку высоты. ФИГ. 24 иллюстрирует реактор по типу барботажной колонны 670, в котором обычно используется призматическая внутренняя структура 672. В частности, внутренняя структура 672 имеет обычно треугольное горизонтальное поперечное сечение. ФИГ. 25 иллюстрирует реактор по типу барботажной колонны 680, в котором используется обычно цилиндрическая внутренняя структура 682, которая меньше внутренней структуры 602 на ФИГ. 17. Однако внешняя реакционная емкость реактора по типу барботажной колонны 680 имеет ступенчатый диаметр, образованный узкой нижней секцией 682 и широкой верхней секцией 684.
Как показано на ФИГ. 17-25, внутренняя структура, используемая в соответствии с одним из вариантов осуществления настоящего изобретения, может иметь ряд конфигураций и может быть расположена в разных положениях в пределах основной реакционной зоны реактора по типу барботажной колонны. Кроме того, внутренняя структура и статическая зона, определенная ею, могут быть получены из множества различных материалов. В одном из вариантов осуществления настоящего изобретения внутренняя структура полностью закрыта так, что никакой окружающей реакционной среды не поступает во внутреннюю структуру. Такая закрытая внутренняя структура может быть полой или сплошной. В другом варианте осуществления настоящего изобретения внутренняя структура включает одно или несколько отверстий, которые дают возможность реакционной среде поступать в статическую зону, определенную внутренней структурой. Однако так как одна из задач статической зоны состоит в том, чтобы создать зону пониженной турбулентности относительно турбулентности реакционной среды, окружающей внутреннюю структуру, предпочтительно, чтобы внутренняя структура не позволяла значительному количеству реакционной среды быстро протекать через внутреннюю структуру.
Конкретная конфигурация и технологические параметры реактора по типу барботажной колонны, оборудованного одной или несколькими внутренними структурами, будут описаны более подробно. Предпочтительно внутренняя структура расположена полностью внутри внешней реакционной емкости реактора по типу барботажной колонны; однако допустимо, чтобы, по меньшей мере, часть внутренней структуры выступала за пределы внешней реакционной емкости реактора по типу барботажной колонны. Как упоминалось выше, во время работы реактора по типу барботажной колонны, внутренняя структура определяет, по меньшей мере, одну статическую зону в пределах реактора по типу барботажной колонны. Основная реакционная зона реактора по типу барботажной колонны и статическая зона имеют отдельные объемы (то есть не перекрывают одна другую). Основная реакционная зона реактора по типу барботажной колонны определена внутри внешней реакционной емкости реактора по типу барботажной колонны, но снаружи внутренней структуры.
Как упоминалось выше, статическая зона, определенная внутренней структурой, представляет собой объем, который имеет пониженную турбулентность относительно турбулентности примыкающей реакционной среды в основной реакционной зоне. Предпочтительно, чтобы, по меньшей мере, приблизительно 90, 95, 98 или 99,9% объема статической зоны было заполнено материалом, отличным от реакционной среды, и/или было заполнено частью реакционной среды, имеющей по существу пониженную турбулентность в сравнении с турбулентностью реакционной среды, расположенной по соседству с внутренней структурой. Если статическая зона включает любую часть реакционной среды, предпочтительно, чтобы часть реакционной среды, находящаяся в статической зоне, имела усредненное по массе время пребывания в статической зоне, по меньшей мере, приблизительно 2, 8, 30 или 120 мин. Если статическая зона включает любую часть реакционной среды, предпочтительно, чтобы усредненное по времени удерживание газа реакционной среды в статической зоне было менее чем приблизительно 0,2, 0,1, 0,5 или 0,01, где удерживание газа измерено на любой отметке высоты статической зоны, на ¼ высоты статической зоны, на ½ высоты статической зоны, на ¾ высоты статической зоны, и/или представляет собой среднее по всей высоте статической зоны. Предпочтительно, чтобы усредненное по времени удерживание газа реакционной среды в реакционной зоне находилось в интервале приблизительно от 0,2 до 0,9, более предпочтительно приблизительно от 0,5 до 0,8 и наиболее предпочтительно от 0,55 до 0,7, где удерживание газа измерено на любой отметке высоты реакционной зоны, на ¼ высоты реакционной зоны, на ½ высоты реакционной зоны, на ¾ высоты реакционной зоны, и/или представляет собой среднее по всей высоте реакционной зоны. Если статическая зона включает любую часть реакционной среды, предпочтительно, чтобы усредненная по времени приведенная скорость газа реакционной среды в статической зоне была менее чем приблизительно 0,4, 0,2, 0,1 или 0,05 м/сек, где приведенная скорость газа измерена на любой отметке высоты статической зоны, на ¼ высоты статической зоны, на ½ высоты статической зоны, на ¾ высоты статической зоны, и/или представляет собой среднее по всей высоте статической зоны. Предпочтительно, чтобы усредненная по времени приведенная скорость газа реакционной среды в реакционной зоне, составляла, по меньшей мере, приблизительно 0,2, 0,4, 0,8 или 1 м/сек, где приведенная скорость газа измерена на любой отметке высоты реакционной зоны, на ¼ высоты реакционной зоны, на ½ высоты реакционной зоны, на ¾ высоты реакционной зоны, и/или представляет собой среднее по всей высоте реакционной зоны. Если статическая зона включает любую часть реакционной среды, предпочтительно, чтобы усредненная по времени приведенная скорость жидкой фазы реакционной среды в статической зоне была менее чем приблизительно 0,04, 0,01 или 0,004 м/сек, где приведенная скорость жидкой фазы измерена на любой отметке высоты статической зоны, на ¼ высоты статической зоны, на ½ высоты статической зоны, на ¾ высоты статической зоны, и/или представляет собой среднее по всей высоте статической зоны. Предпочтительно, чтобы усредненная по времени приведенная скорость жидкой фазы реакционной среды в реакционной зоне составляла менее чем приблизительно 0,1, 0,04 или 0,01 м/сек, где приведенная скорость жидкой фазы измерена на любой отметке высоты реакционной зоны, на ¼ высоты реакционной зоны, на ½ высоты реакционной зоны, на ¾ высоты реакционной зоны, и/или представляет собой среднее по всей высоте реакционной зоны. Любые параметры (например, высота, ширина, площадь, объем, относительное горизонтальное размещение и относительное вертикальное размещение), определенные здесь для внутренней структуры, также считаются применимыми к статической зоне, определенной внутренней структурой, и наоборот.
Предпочтительно, чтобы размер статической зоны, определенной внутренней структурой, был таким, чтобы статическая зона имела, по меньшей мере, одно местоположение, которое отделено промежутком от реакционной зоны, по меньшей мере, приблизительно на 0,05 часть от максимального горизонтального диаметра реакционной зоны или приблизительно на 0,2 м, иногда больше. Предпочтительно статическая зона имеет, по меньшей мере, одно местоположение, которое отделено промежутком от реакционной зоны, по меньшей мере, приблизительно на 0,4, 0,7 или 1,0 м. Предпочтительно статическая зона имеет, по меньшей мере, одно местоположение, которое отделено промежутком от реакционной зоны, по меньшей мере, приблизительно на 0,1, 0,2 или 0,3 части от максимального горизонтального диаметра реакционной зоны. Статическая зона предпочтительно имеет, по меньшей мере, два местоположения, которые отделены друг от друга расстоянием по вертикали, которое составляет, по меньшей мере, приблизительно 0,5, 1,2 или 4 части от максимального горизонтального диаметра реакционной зоны. Предпочтительно указанные два вертикально разделенные промежутком местоположения в статической зоне также, каждое, отделены от реакционной зоны, по меньшей мере, приблизительно на 0,05, 0,1, 0,2 или 0,3 части от максимального горизонтального диаметра реакционной зоны. Предпочтительно указанные два вертикально разделенные промежутком местоположения в статической зоне отделены друг от друга вертикальным промежутком, по меньшей мере, приблизительно 1, 3, 10 или 20 м, и каждое также отделено от реакционной зоны, по меньшей мере, приблизительно на 0,1, 04, 0,7 или 1 м. Предпочтительно объем статической зоны находится в интервале приблизительно от 1 до 50% от объема основной реакционной зоны, более предпочтительно в интервале приблизительно от 2 до 25% от объема основной реакционной зоны и наиболее предпочтительно в интервале от 4 до 15% от объема основной реакционной зоны.
Внешняя реакционная емкость реактора по типу барботажной колонны включает, как правило, цилиндрическую вертикальную внешнюю боковую стенку. Предпочтительно внутренняя структура включает цилиндрическую вертикальную боковую стенку, которая находится на расстоянии по направлению внутрь от внешней боковой стенки. Предпочтительно внутренняя структура не является частью теплообменника. Следовательно, предпочтительно, чтобы усредненный по времени удельный тепловой поток через установленные вертикально внутренние боковые стенки внутренней структуры составлял менее чем приблизительно 100, 15, 3 или 0,3 киловатта на квадратный метр. Кольцевое пространство, заполненное реакционной средой, предпочтительно ограничено между внутренней и внешней боковыми стенками. Внутренняя структура поддерживается вертикально к внешней емкости, предпочтительно с помощью установленных вертикально опор между нижними частями внутренней структуры и нижней частью внешней реакционной емкости. Кроме того, внутренняя структура предпочтительно поддерживается внешней реакционной емкостью за счет множества незагрязняющихся продольных поддерживающих элементов, простирающихся по направлению внутрь от внешней боковой стенки к внутренней боковой стенке. Предпочтительно площадь горизонтального поперечного сечения статической зоны на ¼ высоты, ½ высоты и/или ¾ высоты статической зоны составляет, по меньшей мере, приблизительно 2, от 5 до 75 или от 10 до 30% от площади горизонтального поперечного сечения кольцевого пространства на соответствующей отметке высоты. Предпочтительно максимальная высота внешней вертикальной боковой стенки находится в интервале приблизительно от 10 до 90% от максимальной высоты внутренней вертикальной боковой стенки, более предпочтительно в интервале приблизительно от 20 до 80% от максимальной высоты внешней вертикальной боковой стенки и наиболее более предпочтительно в интервале приблизительно от 30 до 70% от максимальной высоты внешней вертикальной боковой стенки. Хотя предпочтительно, чтобы внутренняя боковая стенка имела, как правило, цилиндрическую конфигурацию, допустимо, чтобы часть внутренней боковой стенки была вогнутой относительно примыкающей части статической зоны. Когда внутренняя боковая стенка включает вогнутую часть, предпочтительно, чтобы такая вогнутая часть образовывала менее чем приблизительно 25, 10, 5 или 0,1% от общей обращенной наружу площади поверхности, представленной внутренней боковой стенкой. Предпочтительно отношение общей площади поверхности внутренней структуры, которая находится в непосредственном контакте с реакционной средой, к общему объему реакционной зоны составляет менее чем приблизительно 1, 0,5, 0,3 или 0,15 квадратного метра на кубический метр. Предпочтительно, чтобы объемный центр тяжести статической зоны был горизонтально смещен от объемного центра тяжести основной реакционной зоны менее чем приблизительно на 0,4, 0,2, 0,1 или 0,01 части от максимального горизонтального диаметра основной реакционной зоны.
Когда реактор по типу барботажной колонны включает больше, чем одну внутреннюю структуру, определяющую больше, чем одну статическую зону, предпочтительно, чтобы статические зоны были расположены вертикально в ряд так, чтобы объемный центр тяжести всех статических зон, учитываемых вместе, был горизонтально смещен от объемного центра тяжести реакционной зоны приблизительно менее чем на 0,4, 0,2, 0,1 или 0,01 части от максимального горизонтального диаметра основной реакционной зоны. Кроме того, когда множество статических зон образовано внутри основной реакционной зоны, предпочтительно, чтобы количество отдельных статических зон, имеющих объем больше чем 0,2% от объема основной реакционной зоны, составляло менее чем приблизительно 100, 10, 5 или 2.
Внешняя реакционная емкость реактора по типу барботажной колонны предпочтительно имеет отношение максимальной вертикальной высоты к максимальному горизонтальному диаметру в интервале приблизительно от 3:1 до 30:1, более предпочтительно в интервале приблизительно от 6:1 до 20:1 и наиболее предпочтительно в интервале от 9:1 до 15:1. Внутренняя структура предпочтительно имеет отношение максимальной вертикальной высоты к максимальному горизонтальному диаметру в интервале приблизительно от 0,3:1 до 100:1, более предпочтительно в интервале приблизительно от 1:1 до 50:1 и наиболее предпочтительно в интервале от 3:1 до 30:1. Предпочтительно, чтобы максимальный горизонтальный диаметр внутренней структуры находился в интервале приблизительно от 0,1 до 5 м, более предпочтительно в интервале приблизительно от 0,3 до 4 м и наиболее предпочтительно в интервале от 1 до 3 м. Предпочтительно максимальная вертикальная высота внутренней структуры находится в интервале приблизительно от 1 до 100 м, более предпочтительно в интервале приблизительно от 3 до 50 м и наиболее предпочтительно в интервале от 10 до 50 м. Предпочтительно максимальный горизонтальный диаметр внутренней структуры находится в интервале приблизительно от 5 до 80, более предпочтительно приблизительно от 10 до 60, наиболее предпочтительно от 20 до 50% от максимального горизонтального диаметра внешней реакционной емкости. Предпочтительно максимальная вертикальная высота внутренней структуры 602 находится в интервале приблизительно от 3 до 100% от максимальной вертикальной высоты внешней реакционной емкости, более предпочтительно в интервале приблизительно от 10 до 90% от максимальной вертикальной высоты внешней реакционной емкости и наиболее предпочтительно в интервале от 30 до 80% от максимальной вертикальной высоты внешней реакционной емкости. Любые параметры (например, высота, ширина, площадь, объем, относительное горизонтальное размещение и относительное вертикальное размещение), определенные здесь для внешней реакционной емкости и структурных частей, также считаются применимыми к реакционной зоне, определенной внешней реакционной емкостью, и наоборот.
В одном из вариантов осуществления изобретения внутренняя структура полностью изолирует статическую зону от реакционной зоны. В другом варианте изобретения внутренняя структура определяет одно или несколько прямых отверстий, что обеспечивает возможность прямой передачи жидкости между статической зоной и реакционной зоной. Когда внутренняя структура определяет такие прямые отверстия, предпочтительно, чтобы максимальный диаметр самого маленького из прямых отверстий был меньше чем приблизительно 0,3, 0,2, 0,1 или 0,05 части от максимального горизонтального диаметра основной реакционной зоны. Когда внутренняя структура определяет такие прямые отверстия, предпочтительно, чтобы максимальный диаметр самого большого из прямых отверстий был меньше чем приблизительно 0,4, 0,3, 0,2 или 0,1 части от максимального горизонтального диаметра основной реакционной зоны. Когда внутренняя структура определяет такие прямые отверстия, предпочтительно, чтобы совокупная площадь пропускного сечения, определяемая всеми прямыми отверстиями, была меньше чем приблизительно 0,4, 0,3 или 0,2 части от площади максимального горизонтального поперечного сечения основной реакционной зоны. Внутренняя структура имеет максимальную высоту (Hi). Когда внутренняя структура определяет одно или несколько прямых отверстий, предпочтительно, чтобы менее чем приблизительно 50, 25 или 10% от совокупной площади пропускного сечения, определенной всеми прямыми отверстиями, находилось на расстоянии приблизительно более чем 0,5Нi, 0,25Нi или 0,1Нi от верха внутренней структуры. Когда в реакторе по типу барботажной колонны используется множество внутренних структур для формирования множества статических зон, допустимо, чтобы две или несколько статических зон включали взаимосвязанные отверстия и/или трубопроводы, которые обеспечивают передачу жидкости между статическими зонами. Предпочтительно максимальный диаметр самого маленького из любых взаимосвязанных отверстий и/или трубопроводов составляет менее чем приблизительно 0,3, 0,2, 0,1 или 0,05 части от максимального горизонтального диаметра основной реакционной зоны.
Как упоминалось выше, некоторые физические и рабочие характеристики реакторов по типу барботажной колонны, описанных выше с помощью ФИГ. 1-25, создают вертикальные градиенты давления, температуры и концентраций реагентов (то есть кислорода и способного к окислению соединения) перерабатываемой реакционной среды. Как обсуждалось выше, такие вертикальные градиенты могут обеспечить более эффективный и экономичный процесс окисления по сравнению с обычными процессами окисления, которые отдают предпочтение хорошо перемешиваемой реакционной среде с относительно равномерным повсюду давлением, температурой и концентрацией реагентов. Вертикальные градиенты кислорода, способного к окислению соединения (например, пара-ксилола) и температуры, которые делают возможным использование системы окисления в соответствии с вариантом осуществления настоящего изобретения, будут описаны более детально.
Что касается ФИГ. 26, то для того чтобы количественно определить градиенты концентрации реагентов в реакционной среде во время окисления в реакторе по типу барботажной колонны, весь объем реакционной среды может быть теоретически распределен на 30 дискретных горизонтальных тонких слоев равного объема. ФИГ. 26 иллюстрирует принцип деления реакционной среды на 30 дискретных горизонтальных тонких слоев равного объема. За исключением самого верхнего и самого нижнего горизонтальных тонких слоев, каждый горизонтальный тонкий слой представляет собой дискретный объем, ограниченный вверху и внизу воображаемыми горизонтальными плоскостями и ограниченный по его сторонам стенкой реактора. Самый верхний горизонтальный тонкий слой ограничен снизу воображаемой горизонтальной плоскостью, а сверху верхней поверхностью реакционной среды. Самый нижний горизонтальный тонкий слой ограничен вверху воображаемой горизонтальной плоскостью, а снизу днищем оболочки емкости. После распределения реакционной среды на 30 дискретных горизонтальных тонких слоев равного объема можно определить усредненную по времени и усредненную по объему концентрацию каждого горизонтального тонкого слоя. Отдельный горизонтальный тонкий слой, имеющий максимальную концентрацию из всех 30 горизонтальных тонких слоев, может быть идентифицирован как «С-макс горизонтальный тонкий слой». Отдельный горизонтальный тонкий слой, расположенный выше С-макс горизонтального слоя и имеющий минимальную концентрацию из всех горизонтальных тонких слоев, расположенных выше С-макс горизонтального тонкого слоя, может быть идентифицирован как «С-мин горизонтальный тонкий слой». Вертикальный градиент концентрации затем может быть рассчитан как отношение концентрации в С-макс горизонтальном тонком слое к концентрации в С-мин горизонтальном тонком слое.
Что касается количественного определения градиента концентрации кислорода, когда реакционную среду теоретически распределяют на 30 дискретных горизонтальных тонких слоев равного объема, О2-макс горизонтальный тонкий слой определяют как слой, имеющий максимальную концентрацию кислорода из всех 30 горизонтальных тонких слоев, а О2-мин горизонтальный тонкий слой определяют как слой, имеющий минимальную концентрацию кислорода из горизонтальных тонких слоев, расположенных выше О2-макс горизонтального тонкого слоя. Концентрации кислорода в горизонтальных тонких слоях измеряют в газовой фазе реакционной среды, исходя из влажного усредненного по времени и усредненного по объему мольного состава. Предпочтительно, чтобы отношение концентрации кислорода О2-макс горизонтального тонкого слоя к концентрации кислорода О2-мин горизонтального тонкого слоя находилось в интервале приблизительно от 2:1 до 25:1, более предпочтительно в интервале приблизительно от 3:1 до 15:1 и наиболее предпочтительно в интервале от 4:1 до 10:1.
Как правило, О2-макс горизонтальный тонкий слой будет располагаться около нижней части реакционной среды, тогда как О2-мин горизонтальный тонкий слой будет располагаться около верхней части реакционной среды. Предпочтительно О2-мин горизонтальный тонкий слой представляет собой один из 5 самых верхних горизонтальных тонких слоев из 30 дискретных горизонтальных тонких слоев. Наиболее предпочтительно О2-мин горизонтальный тонкий слой представляет собой один самый верхний слой из 30 дискретных горизонтальных тонких слоев, как показано на ФИГ. 26. Предпочтительно О2-макс горизонтальный тонкий слой представляет собой один из 10 самых нижних горизонтальных тонких слоев из 30 дискретных горизонтальных тонких слоев. Наиболее предпочтительно О2-макс горизонтальный тонкий слой представляет собой один из 5 самых нижних горизонтальных тонких слоев из 30 дискретных горизонтальных тонких слоев. Например, на ФИГ. 26 показан О2-макс горизонтальный тонкий слой в виде третьего горизонтального тонкого слоя от днища реактора. Предпочтительно, чтобы вертикальный промежуток между О2-мин и О2-макс горизонтальными тонкими слоями составлял, по меньшей мере, приблизительно 2W, более предпочтительно, по меньшей мере, приблизительно 4W и наиболее предпочтительно, по меньшей мере, 6W. Предпочтительно, чтобы вертикальный промежуток между О2-мин и О2-макс горизонтальными тонкими слоями составлял, по меньшей мере, приблизительно 0,2Н, более предпочтительно, по меньшей мере, приблизительно 0,4Н и наиболее предпочтительно, по меньшей мере, 0,6Н.
Усредненная по времени и усредненная по объему концентрация кислорода, из расчета на влажный кислород, О2-мин горизонтального тонкого слоя предпочтительно находится в интервале приблизительно от 0,1 до 3% мол., более предпочтительно в интервале приблизительно от 0,3 до 2% мол. и наиболее предпочтительно в интервале от 0,5 до 1,5% мол. Усредненная по времени и усредненная по объему концентрация кислорода, из расчета на влажный кислород, О2-макс горизонтального тонкого слоя предпочтительно находится в интервале приблизительно от 4 до 20% мол., более предпочтительно в интервале приблизительно от 5 до 15% мол. и наиболее предпочтительно в интервале от 6 до 12% мол. Усредненная по времени концентрация кислорода, исходя из сухого кислорода, в газообразном выходящем потоке, выпускаемом из реактора через выпускное отверстие для газа, предпочтительно находится в интервале приблизительно от 0,5 до 9% мол., более предпочтительно в интервале от 1 до 7% мол. и наиболее предпочтительно в интервале от 1,5 до 5% мол.
Так как концентрация кислорода падает так значительно по направлению к верху реакционной среды, предпочтительно, чтобы потребление кислорода было уменьшено в верхней части реакционной среды. Такое уменьшенное потребление около верха реакционной среды может быть осуществлено за счет создания вертикального градиента концентраций способного к окислению соединения (например, пара-ксилола), где минимальная концентрация способного к окислению соединения находится около верха реакционной среды.
Что касается количественного определения градиента концентраций способного к окислению соединения (например, пара-ксилола), то реакционную среду теоретически распределяют на 30 дискретных горизонтальных тонких слоев равного объема, ОС-макс горизонтальный тонкий слой идентифицируют как слой, имеющий максимальную концентрацию способного к окислению соединения из всех 30 горизонтальных слоев, а ОС-мин горизонтальный тонкий слой идентифицируют как слой, имеющий минимальную концентрацию способного к окислению соединения из горизонтальных слоев, расположенных выше ОС-макс горизонтального тонкого слоя. Концентрации способного к окислению соединения в горизонтальных тонких слоях измеряют в жидкой фазе из расчета на усредненную по времени и усредненную по объему массовую долю. Предпочтительно, чтобы отношение концентрации способного к окислению соединения ОС-макс горизонтального тонкого слоя к концентрации способного к окислению соединения ОС-мин горизонтального тонкого слоя составляло больше чем приблизительно 5:1, более предпочтительно больше чем приблизительно 10:1, еще более предпочтительно больше чем приблизительно 20:1 и наиболее предпочтительно находилось в интервале от 40:1 до 1000:1.
Как правило, ОС-макс горизонтальный тонкий слой будет располагаться около нижней части реакционной среды, тогда как ОС-мин горизонтальный тонкий слой будет располагаться около верхней части реакционной среды. Предпочтительно ОС-мин горизонтальный тонкий слой представляет собой один из 5 самых верхних горизонтальных тонких слоев из 30 дискретных горизонтальных тонких слоев. Наиболее предпочтительно ОС-мин горизонтальный тонкий слой представляет собой самый верхний слой из 30 дискретных горизонтальных тонких слоев, как показано на ФИГ. 26. Предпочтительно ОС-макс горизонтальный тонкий слой представляет собой один из 10 самых нижних горизонтальных тонких слоев из 30 дискретных горизонтальных тонких слоев. Наиболее предпочтительно ОС-макс горизонтальный тонкий слой представляет собой один из 5 самых нижних горизонтальных тонких слоев из 30 дискретных горизонтальных тонких слоев. Например, на ФИГ. 26 показан ОС-макс горизонтальный тонкий слой в виде пятого от днища реактора горизонтального тонкого слоя. Предпочтительно, чтобы вертикальный промежуток между ОС-мин и ОС-макс горизонтальными тонкими слоями составлял, по меньшей мере, приблизительно 2W, где W является максимальной шириной реакционной среды. Более предпочтительно вертикальный промежуток между ОС-мин и ОС-макс горизонтальными тонкими слоями составляет, по меньшей мере, приблизительно 4W и наиболее предпочтительно, по меньшей мере, 6W. При данной высоте Н реакционной среды предпочтительно, чтобы вертикальный промежуток между ОС-мин и ОС-макс горизонтальными тонкими слоями составлял, по меньшей мере, приблизительно 0,2Н, более предпочтительно, по меньшей мере, приблизительно 0,4Н и наиболее предпочтительно, по меньшей мере, 0,6Н.
Усредненная по времени и усредненная по объему концентрация способного к окислению соединения (например, пара-ксилола) в жидкой фазе ОС-мин горизонтального тонкого слоя составляет предпочтительно менее чем приблизительно 5000 масс.ч./млн, более предпочтительно менее чем приблизительно 2000 масс.ч./млн, еще более предпочтительно менее чем приблизительно 400 масс.ч./млн и наиболее предпочтительно находится в интервале от 1 до 100 масс.ч./млн. Усредненная по времени и усредненная по объему концентрация способного к окислению соединения в жидкой фазе ОС-макс горизонтального тонкого слоя предпочтительно находится в интервале приблизительно от 100 до 10000 масс.ч./млн, более предпочтительно в интервале приблизительно от 200 до 5000 масс.ч./млн и наиболее предпочтительно в интервале от 500 до 3000 масс.ч./млн.
Хотя предпочтительно, чтобы реактор по типу барботажной колонны создавал вертикальные градиенты концентраций способного к окислению соединения, также предпочтительно, чтобы объемный процент реакционной среды, имеющей концентрацию способного к окислению соединения в жидкой фазе выше 1000 масс.ч./млн, был минимизирован. Предпочтительно усредненный по времени объемный процент реакционной среды, имеющей концентрацию способного к окислению соединения в жидкой фазе свыше 1000 масс.ч./млн, составляет менее чем приблизительно 9%, более предпочтительно менее чем приблизительно 6%, и наиболее предпочтительно менее чем 3%. Предпочтительно усредненный по времени объемный процент реакционной среды, имеющей концентрацию способного к окислению соединения в жидкой фазе свыше 2500 масс.ч./млн, составляет менее чем приблизительно 1,5%, более предпочтительно менее чем приблизительно 1% и наиболее предпочтительно менее чем 0,5%. Предпочтительно усредненный по времени объемный процент реакционной среды, имеющей концентрацию способного к окислению соединения в жидкой фазе свыше 10000 масс.ч./млн, составляет менее чем приблизительно 0,3%, более предпочтительно менее чем приблизительно 0,1% и наиболее предпочтительно менее чем 0,03%. Предпочтительно усредненный по времени объемный процент реакционной среды, имеющей концентрацию способного к окислению соединения в жидкой фазе свыше 25000 масс.ч./млн, составляет менее чем приблизительно 0,03%, более предпочтительно менее чем приблизительно 0,015% и наиболее предпочтительно менее чем 0,007%. Установлено, что не требуется, чтобы объем реакционной среды, имеющей повышенные уровни способного к окислению соединения, находился в одном сплошном объеме. Во многих случаях хаотичная схема потоков в реакционной емкости по типу барботажной колонны дает одновременно две или более сплошных, но изолированных частей реакционной среды, имеющей повышенные уровни способного к окислению соединения. При каждом времени, используемом при усреднении по времени, все такие сплошные, но изолированные объемы больше чем 0,0001% об. от всей реакционной среды, складывают вместе, чтобы определить общий объем, имеющий повышенные уровни концентраций способного к окислению соединения в жидкой фазе.
Помимо градиентов концентраций кислорода и способного к окислению соединения, обсуждавшихся выше, предпочтительно, чтобы в реакционной среде существовал температурный градиент. Если рассмотреть снова ФИГ. 26, то этот температурный градиент можно количественно определить способом, аналогичным способу определения градиентов концентраций, путем распределения реакционной среды на 30 дискретных горизонтальных тонких слоев равного объема и измерения усредненной по времени и усредненной по объему температуры каждого тонкого слоя. Затем горизонтальный тонкий слой с самой низкой температурой из наиболее низких 15 горизонтальных тонких слоев может быть идентифицирован как Т-мин горизонтальный тонкий слой, а горизонтальный тонкий слой, расположенный выше Т-мин горизонтального тонкого слоя и имеющий максимальную температуру из всех тонких слоев выше Т-мин горизонтального тонкого слоя, может быть идентифицирован как «Т-макс горизонтальный тонкий слой». Предпочтительно, чтобы температура Т-макс горизонтального тонкого слоя была, по меньшей мере, на 1ºC выше, чем температура Т-мин горизонтального тонкого слоя. Более предпочтительно температура Т-макс горизонтального тонкого слоя находится в интервале приблизительно от 1,25 до 12ºС выше, чем температура Т-мин горизонтального тонкого слоя. Наиболее предпочтительно температура Т-макс горизонтального тонкого слоя находится в интервале приблизительно от 2 до 8ºС выше, чем температура Т-мин горизонтального тонкого слоя. Температура Т-макс горизонтального тонкого слоя предпочтительно находится в интервале приблизительно от 125 до 200ºС, более предпочтительно в интервале приблизительно от 140 до 180ºС и наиболее предпочтительно в интервале от 150 до 170ºС.
Как правило, Т-макс горизонтальный тонкий слой будет располагаться около центра реакционной среды, тогда как Т-мин горизонтальный тонкий слой будет располагаться около нижней части реакционной среды. Предпочтительно Т-мин горизонтальный тонкий слой представляет собой один из 10 самых нижних горизонтальных тонких слоев из 15 самых нижних горизонтальных тонких слоев. Наиболее предпочтительно Т-мин горизонтальный тонкий слой представляет собой один из 5 самых нижних горизонтальных тонких слоев из 15 самых нижних горизонтальных тонких слоев. Например, ФИГ. 26 показывает Т-мин горизонтальный тонкий слой как второй от днища реактора горизонтальный тонкий слой. Предпочтительно Т-макс горизонтальный тонкий слой представляет собой один из 20 средних горизонтальных тонких слоев из 30 дискретных горизонтальных тонких слоев. Наиболее предпочтительно Т-мин горизонтальный тонкий слой представляет собой один из 14 средних горизонтальных тонких слоев из 30 дискретных горизонтальных тонких слоев. Например, на ФИГ. 26 показан Т-макс горизонтальный тонкий слой в виде двадцатого от днища реактора горизонтального тонкого слоя (то есть одного из 10 средних горизонтальных тонких слоев). Предпочтительно, чтобы вертикальный промежуток между Т-мин и Т-макс горизонтальными тонкими слоями составлял, по меньшей мере, приблизительно 2W, более предпочтительно, по меньшей мере, приблизительно 4W и наиболее предпочтительно, по меньшей мере, 6W. Предпочтительно, чтобы вертикальный промежуток между Т-мин и Т-макс горизонтальными тонкими слоями составлял, по меньшей мере, приблизительно 0,2Н, более предпочтительно, по меньшей мере, приблизительно 0,4Н и наиболее предпочтительно, по меньшей мере, 0,6Н.
Как описано выше, когда в реакционной зоне существует вертикальный температурный градиент, он может обеспечивать преимущества выведения реакционной среды в поднятом по высоте местоположении, где температура реакционной среды является наиболее высокой, особенно когда выведенный продукт подвергается дополнительной обработке по направлению потока при более высоких температурах. Следовательно, когда реакционную среду 36 выводят из реакционной зоны через одно или несколько поднятых по высоте выпускных отверстий, как показано на ФИГ. 15 и 16, предпочтительно, чтобы поднятое(ые) по высоте выпускное(ые) отверстие(я) располагало(и)сь около Т-макс горизонтального тонкого слоя. Предпочтительно поднятое по высоте выпускное отверстие располагается в пределах 10 горизонтальных тонких слоев Т-макс горизонтального тонкого слоя, более предпочтительно в пределах 5 горизонтальных тонких слоев Т-макс горизонтального тонкого слоя и наиболее предпочтительно в пределах 2 горизонтальных тонких слоев Т-макс горизонтального тонкого слоя.
Следует отметить, что много заявленных признаков, описанных в настоящем изобретении, может быть использовано в составных реакторных системах окисления - не только для системы, в которой используется один реактор окисления. Кроме того, некоторые заявленные признаки, описанные в настоящем изобретении, могут быть использованы в реакторах окисления с механическим перемешиванием и/или с перемешиванием потоком - не только в реакторах с использованием пузырькового перемешивания (то есть в реакторах по типу барботажной колонны). Например, раскрыты некоторые преимущества, связанные со ступенчатой/переменной концентрацией кислорода и/или скоростью потребления кислорода по всей реакционной среде. Преимущества, реализуемые за счет ступенчатой концентрации/поглощения кислорода в реакционной среде, могут быть достигнуты независимо от того, содержится ли весь объем реакционной среды в одной емкости или во множестве емкостей. Кроме того, преимущества, реализуемые за счет ступенчатой концентрации/потребления кислорода в реакционной среде, могут быть достигнуты независимо от того, является(ются) ли реакционная(ые) емкость(и) механически перемешиваемой(ыми), перемешиваемой(ыми) потоком и/или перемешиваемой(ыми) пузырьками.
Один из методов количественной оценки ступенчатого изменения концентрации кислорода и/или скорости потребления кислорода в реакционной среде состоит в сравнении двух или нескольких дискретных 20%-ных сплошных объемов реакционной среды. Такие 20%-ные дискретные сплошные объемы не требуют ограничения какой-либо конкретной формой. Однако каждый 20%-ный сплошной объем должен быть образован непрерывным объемом реакционной среды (то есть каждый объем является «сплошным»), и 20%-ные сплошные объемы не должны перекрываться друг с другом (то есть объемы являются «дискретными»). Такие дискретные 20%-ные сплошные объемы могут быть расположены в одном и том же реакторе (ФИГ. 29) или во множестве реакторов. На ФИГ. 27 реактор по типу барботажной колонны показан как содержащий реакционную среду, которая включает первый дискретный 20%-ный сплошной объем 37 и второй дискретный 20%-ный сплошной объем 39.
Ступенчатое изменение доступности кислорода в реакционной среде может быть оценено количественно со ссылкой на 20%-ный сплошной объем реакционной среды, имеющий наиболее обогащенную мольную долю кислорода в газовой фазе, и со ссылкой на 20%-ный сплошной объем реакционной фазы, имеющий наиболее обедненную мольную долю кислорода в газовой фазе. В газовой фазе дискретного 20%-ного сплошного объема реакционной среды, содержащего самую высокую концентрацию кислорода в газовой фазе, усредненная по времени и усредненная по объему концентрация кислорода из расчета на влажный кислород предпочтительно находится в интервале приблизительно от 3 до 18% мол., более предпочтительно в интервале приблизительно от 3,5 до 14% мол. и наиболее предпочтительно в интервале от 4 до 10% мол. В газовой фазе дискретного 20%-ного сплошного объема реакционной среды, содержащего самую низкую концентрацию кислорода в газовой фазе, усредненная по времени и усредненная по объему концентрация кислорода из расчета на влажный кислород, предпочтительно находится в интервале приблизительно от 0,3 до 5% мол., более предпочтительно в интервале приблизительно от 0,6 до 4% мол. и наиболее предпочтительно в интервале от 0,9 до 3% мол. Кроме того, отношение усредненной по времени и усредненной по объему концентрации кислорода из расчета на влажный кислород в наиболее обогащенном 20%-ном сплошном объеме реакционной среды относительно наиболее обедненного 20%-ного сплошного объема реакционной среды предпочтительно находится в интервале приблизительно от 1,5:1 до 20:1, более предпочтительно в интервале приблизительно от 2:1 до 12:1 и наиболее предпочтительно в интервале от 3:1 до 9:1.
Ступенчатое изменение скорости потребления кислорода в реакционной среде может быть количественно оценено в значениях СОС кислорода (кислород-STR), описанной ранее. Понятие «СОС кислорода» ранее описано в общем смысле (то есть из прогноза среднего значения СОС кислорода всей реакционной среды); однако СОС кислорода также может быть рассмотрена в локальном смысле (то есть для части реакционной среды), чтобы количественно оценить ступенчатое изменение потребления кислорода по всей реакционной среде.
Установлено, что весьма полезно вызывать изменение СОС кислорода по всей реакционной среде в общей согласованности с желаемыми градиентами, раскрытыми в настоящем изобретении, относящимися к давлению в реакционной среде и мольной доле молекулярного кислорода в газовой фазе реакционной среды. Следовательно, предпочтительно, чтобы отношение СОС кислорода первого дискретного 20%-ного сплошного объема реакционной среды к СОС кислорода второго дискретного 20%-ного сплошного объема реакционной среды находилось в интервале приблизительно от 1,5:1 до 20:1, более предпочтительно в интервале приблизительно от 2:1 до 12:1 и наиболее предпочтительно в интервале от 3:1 до 9:1. В одном из вариантов изобретения «первый дискретный 20%-ный сплошной объем реакционной среды» располагается ближе, чем «второй дискретный 20%-ный сплошной объем реакционной среды» к местоположению, где молекулярный кислород изначально вводится в реакционную среду. Такие большие градиенты СОС кислорода желательны, независимо от того, находится ли реакционная среда частичного окисления в реакторе окисления по типу барботажной колонны или в любом другом типе реакционной емкости, где созданы градиенты давления и/или мольной доли молекулярного кислорода в газовой фазе в реакционной среде (например, в сосуде с механическим перемешиванием, имеющем множество вертикально расположенных зон перемешивания, достигнутых за счет использования множества лопастей, имеющих сильный радиальный поток, возможно дополненных горизонтальными дефлекторными устройствами, с потоком кислорода, поднимающимся, как правило, по направлению вверх от подачи около нижней части реакционной емкости, несмотря на то, что значительное обратное смешение потока окислителя может иметь место в пределах каждой вертикально расположенной зоны перемешивания и что некоторое обратное смешение потока окислителя может иметь место между примыкающими вертикально расположенными зонами перемешивания). То есть, когда существует градиент давления и/или мольной доли молекулярного кислорода в газовой фазе реакционной среды, заявители установили, что желательно создавать аналогичный градиент химического потребления растворенного кислорода с помощью раскрытых в изобретении средств.
Предпочтительным средством, вызывающим локальное измерение СОС кислорода, является контролирование местоположений подачи способного к окислению соединения и контролирование перемешивания жидкой фазы реакционной среды, чтобы контролировать градиенты концентрации способного к окислению соединения в соответствии с описанием настоящего изобретения. Другим полезным средством, вызывающим локальное изменение СОС кислорода, является изменение реакционной активности за счет создания локального колебания температуры и за счет изменения локальной смеси компонентов катализатора и растворителя (например, путем введения дополнительного газа, чтобы вызвать охлаждение испарением в определенной части реакционной среды; и путем добавления потока растворителя, содержащего более высокое количество воды, чтобы понизить активность в конкретной части реакционной среды).
Когда реактор окисления имеет конфигурацию «реактор в реакторе», которая описана выше со ссылкой на ФИГ. 12-14, предпочтительно, чтобы градиенты концентраций, градиенты температур и градиенты СОС кислорода, описанные в настоящем изобретении со ссылкой на ФИГ. 26 и 27, были применимы к части реакционной среды, расположенной внутри внешнего реактора и снаружи внутреннего реактора (например, к реакционной среде 220а на ФИГ. 12).
Что касается ФИГ. 1-27, то окисление предпочтительно проводят в реакторе по типу барботажной колонны в условиях, которые сильно отличаются в соответствии с предпочтительными вариантами осуществления, описанными в настоящем изобретении, от условий обычных реакторов окисления. При использовании реактора по типу барботажной колонны для проведения жидкофазного частичного окисления пара-ксилола до сырой терефталевой кислоты (СТК) в соответствии с предпочтительными вариантами осуществления, описанными в изобретении, пространственные профили локальной реакционной интенсивности, локальной интенсивности испарения и локальной температуры, объединенные со схемами потоков жидкости в пределах реакционной зоны, и предпочтительные относительно низкие температуры окисления вносят вклад в образование частиц СТК, имеющих уникальные и полезные свойства.
На ФИГ. 28А и 28В представлены базовые частицы СТК, произведенные в соответствии с одним из вариантов настоящего изобретения. На ФИГ. 28А показаны базовые частицы СТК при увеличении в 500 раз, тогда как ФИГ. 28В детализирует изображение на одной из базовых частиц СТК и показывает эту частицу при увеличении в 2000 раз. Как, по-видимому, наиболее хорошо представлено на ФИГ. 28В, каждая базовая частица СТК, как правило, образована из большого числа маленьких, агломерированных субчастиц СТК, образуя в результате базовую частицу СТК с относительно высокой площадью поверхности, высокой пористостью, низкой плотностью и хорошей растворимостью. Если не оговорено особо, различные свойства заявляемой СТК, описанные ниже, измерены с использованием типичного образца СТК, где типичный образец весит, по меньшей мере, 1 грамм и/или образован, по меньшей мере, из 10000 отдельных частиц СТК. Базовые частицы СТК, как правило, имеют средний размер частиц в интервале приблизительно от 20 до 150 микрон, более предпочтительно в интервале приблизительно от 30 до 120 микрон и наиболее предпочтительно в интервале от 40 до 90 микрон. Субчастицы СТК обычно имеют средний размер частиц в интервале приблизительно от 0,5 до 30 микрон, более предпочтительно в интервале приблизительно от 1 до 15 микрон и наиболее предпочтительно в интервале от 2 до 5 микрон. Относительно высокая площадь поверхности базовых частиц СТК, показанных на ФИГ. 28А и 28В, может быть количественно оценена с использованием метода измерения площади поверхности по Браунауэру-Эмметту-Теллеру (БЭТ). Предпочтительно базовые частицы СТК имеют среднюю площадь поверхности по БЭТ, по меньшей мере, приблизительно 0,6 квадратного метра на грамм (м2/г). Более предпочтительно базовые частицы СТК имеют среднюю площадь поверхности по БЭТ в интервале приблизительно от 0,8 до 4 м2/г. Наиболее предпочтительно базовые частицы СТК имеют среднюю площадь поверхности по БЭТ в интервале приблизительно от 0,9 до 2 м2/г. Физические свойства (например, размер частиц, площадь поверхности по БЭТ, пористость и растворимость) базовых частиц СТК, полученных с помощью оптимизированного процесса окисления предпочтительного варианта осуществления настоящего изобретения, позволяют проводить очистку частиц СТК с помощью более эффективных и/или экономичных способов, которые описаны далее более подробно со ссылкой на ФИГ. 31.
Значения среднего размера частиц, представленные выше, определены с использованием микроскопии в поляризованном свете и анализа изображения. Оборудование, используемое при анализе размера частиц, включает оптический микроскоп Nikon E800 c объективом 4х Plan Flour N.A. 0.13, цифровую камеру Spot RTTM и персональный компьютер с программным обеспечением для анализа изображения Image Pro PlusTM V4.5.0.19. Метод анализа размера частиц включает следующие основные стадии: (1) диспергирование порошков СТК в минеральном масле; (2) приготовление системы предметное стекло/покровное стекло для дисперсии; (3) изучение предметного стекла с использованием микроскопа в поляризованном свете (условия перекрещивающихся поляр - частицы проявляются в виде ярких объектов на темном фоне); (4) фиксация различных изображений для каждого образца препарата (размер поля=3×2,25 мм; размер пикселя = 1,84 мкм/пиксель); (5) проведение анализа изображения с помощью программного обеспечения Image Pro PlusTM; (6) перенос измерений частиц в сводную таблицу и (7) проведение статистического описания в сводной таблице. Стадия (5) «проведение анализа изображения с помощью программного обеспечения Image Pro PlusTM» включает подстадии: (а) установка порога изображения для определения белых частиц на черном фоне; (b) создание бинарного изображения; (с) прохождение однопроходного открытого светофильтра, чтобы убрать шум пикселя; (d) измерение всех частиц в изображении и (е) фиксация среднего диаметра, измеренного для каждой частицы. Программное обеспечение Image Pro PlusTM определяет средний диаметр отдельных частиц в виде среднечисленной длины диаметров частиц, измеренных при интервалах 2 градуса и проходящих через центр тяжести частицы. Стадия 7 «проведение статистического описания в сводной таблице» включает расчет объемно-взвешенного среднего размера частиц следующим образом. Объем каждой из n частиц в образце рассчитывают, как если бы она была сферической, с использованием pi/6*di^3; умножают объем каждой частицы на ее диаметр, чтобы найти pi/6*di^4; суммируют для всех частиц в образце значения pi/6*di^4; суммируют объемы всех частиц в образце и рассчитывают объемно-взвешенный диаметр частицы как сумму для всех n частиц в образце (pi/6*di^4), поделенную на сумму всех n частиц в образце (pi/6*di^3). Как используется в данном случае, «средний размер частиц» относится к объемно-взвешенному среднему размеру частиц, определенному в соответствии с описанным выше методом испытания; его также называют как D(4,3).
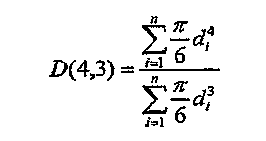
Кроме того, стадия (7) включает выявление размеров частиц, для которых различные фракции всего объема образца имеют меньший размер. Например, D(v,0,1) представляет собой размер частиц, для которого 10% всего объема образца являются более мелкими и 90% являются более крупными; D(v,0,5) представляет собой размер частиц, для которого одна половина всего объема образца является более крупной и другая половина является более мелкой; D(v,0,9) представляет собой размер частиц, для которого 90% всего объема образца являются более мелкими; и т.д. Кроме того, стадия (7) включает расчет значения D(v,0,9) минус D(v,0,1), что в данном случае определяется как «разброс частиц по размерам»; и стадия (7) включает расчет величины разброса частиц по размерам, поделенной на D(4,3), что определяется в данном случае как «относительный разброс частиц по размерам».
Кроме того, предпочтительно, чтобы D(v,0,1) частиц СТК, измеренный, как описано выше, находился в интервале приблизительно от 5 до 65 микрон, более предпочтительно в интервале приблизительно от 15 до 55 мкм и наиболее предпочтительно в интервале от 25 до 45 мкм. Предпочтительно, чтобы D(v,0,5) частиц СТК, измеренный, как описано выше, находился в интервале приблизительно от 10 до 90 мкм, более предпочтительно в интервале приблизительно от 20 до 80 мкм и наиболее предпочтительно в интервале от 30 до 70 мкм. Предпочтительно, чтобы D(v,0,9) частиц СТК, измеренный, как описано выше, находился в интервале приблизительно от 30 до 150 мкм, более предпочтительно в интервале приблизительно от 40 до 130 мкм и наиболее предпочтительно в интервале от 50 до 110 мкм. Предпочтительно, чтобы относительный разброс частиц по размерам находился в интервале приблизительно от 0,5 до 2,0, более предпочтительно в интервале приблизительно от 0,6 до 1,5 и наиболее предпочтительно в интервале от 0,7 до 1,3.
Значения площади поверхности по БЭТ, представленные выше, измерены на приборе Micromeritics ASAP2000 (Micromeritics Instrument Corporation of Norcross, GA). На первой стадии процесса измерения взвешивают 2-4 грамма образца частиц и сушат в вакууме при 50ºС. Затем образец помещают на коллектор для анализа газа и охлаждают до 77ºК. Измеряют изотерму адсорбции азота при 5 минимальных равновесных давлениях путем воздействия на образец известных объемов азота и измерения падения давления. Равновесные давления находятся приблизительно в интервале Р/Ро=0,01-0,20, где Р представляет собой равновесное давление и Ро представляет собой давление паров жидкого азота при 77К. Полученную изотерму затем изображают графически в соответствии со следующим уравнением БЭТ:
где Va представляет собой объем газа, адсорбированного образцом при Р, Vm представляет собой объем газа, требуемый для покрытия всей поверхности образца одним слоем газа, и С представляет собой константу. Из полученного графика определяют Vm и С. Затем Vm переводят в площадь поверхности с использованием площади поперечного сечения азота при 77К с помощью выражения:
где σ представляет собой площадь поперечного сечения азота при 77К, Т равна 77К, а R представляет собой газовую постоянную.
Как упоминалось выше, СТК, полученная в соответствии с одним из вариантов настоящего изобретения, показывает превосходные характеристики растворимости относительно СТК, полученной с помощью других процессов. Такая повышенная степень растворения позволяет проводить очистку заявляемой СТК более эффективно и/или с помощью более эффективных процессов очистки. Следующее описание относится к способу, которым может быть количественно оценена скорость растворения СТК.
Скорость растворения известного количества твердых веществ в известном количестве растворителя при перемешивании смеси может быть определена по различным методикам. Используемый в данном случае метод, называемый «испытанием рассчитанного по времени растворения», описан ниже. В течение всего испытания рассчитанного по времени растворения используется давление окружающей среды приблизительно 0,1 МПа. В течение всего испытания рассчитанного по времени растворения используется температура окружающей среды приблизительно 22ºС. Кроме того, твердые вещества, растворитель и все оборудование для растворения полностью уравновешивают термически при этой температуре до начала испытания, и во время проведения растворения отсутствует заметное нагревание или охлаждение химического стакана и его содержимого. Порцию растворителя, свежего тетрагидрофурана аналитического качества для ВЭЖХ (степень чистоты >99,9), далее ТГФ, составляющую 250 г, помещают в чистый высокий стеклянный химический стакан KIMAX объемом 400 мл (Kimble® номер партии 14020, Kimble/Kontes, Vineland, NJ), который является неизолированным, гладкостенным и имеет по существу цилиндрическую форму. Покрытую тефлоном магнитную мешалку (VWR номер партии 58948-230, приблизительно длиной 1 дюйм и диаметром 3/8 дюйма с восьмиугольным поперечным сечением, VWR International, West Chester, PA 19380) помещают в химический стакан, где она естественным образом оседает на дно. Образец перемешивают с использованием многоточечной на 15 точек магнитной мешалки Variomag® (H&P Labortechnik AG, Oberschleissheim, Germany) при настройке 800 об/мин. Такое перемешивание начинают не более чем за 5 минут до добавления твердых веществ и продолжают постоянно в течение, по меньшей мере, 30 минут после добавления твердых веществ. Твердый образец сырой или очищенной ТФК в виде частиц в количестве 250 мг взвешивают в весовой чашке, к которой образец не прилипает. При начальном времени, обозначенном как t=0, взвешенные твердые вещества вносят все одной порцией в перемешиваемый ТГФ и одновременно запускают таймер. По существу ТГФ очень быстро смачивает твердые вещества и образует разбавленную, хорошо перемешиваемую суспензию в течение 5 секунд. Затем отбирают образцы этой смеси при следующих промежутках времени, измеренных в минутах от t=0: 0,08, 0,25, 0,50, 0,75, 1,00, 1,50, 2,00, 2,50, 3,00, 4,00, 5,00, 6,00, 8,00, 10,00, 15,00 и 30,00. Каждый небольшой образец отбирают из разбавленной, хорошо перемешиваемой смеси с использованием нового одноразового шприца (Becton, Dickinson and Co., 5 мл, REF 30163, Franklin Lakes, NJ 07417). Сразу же после отбора из химического стакана приблизительно 2 мл прозрачного жидкого образца быстро выгружают через новый, неиспользованный шприцевой фильтр (диаметр 25 мм, 0,45 мкм, Gelman GHP Acrodisc GF®, Pall Corporation, East Hills, NY 11548) в новый помеченный стеклянный сосуд для образцов. Продолжительность каждого наполнения шприца, установки фильтра и выгрузки в сосуд для образцов составляет менее чем приблизительно 5 сек, и этот промежуток уместно начинать и заканчивать в пределах приблизительно 3 секунд от любой стороны каждого выбранного времени отбора образцов. В пределах приблизительно пяти минут от каждого наполнения сосуды для образцов закрывают крышкой и хранят при приблизительно постоянной температуре до проведения последующих химических анализов. После отбора последнего образца через 30 минут после t=0 все шесть образцов анализируют на количество растворенной ТФК с использованием метода ВЭЖХ-DAD, который в целом описан в данном описании далее. Однако в рассматриваемом тесте калибровочные стандарты и представленные результаты в обоих случаях основаны на миллиграммах растворенной ТФК на грамм растворителя, ТГФ (далее «ч/млн в ТГФ). Например, если все 250 мг твердых веществ представляют собой очень чистую ТФК и если это все количество полностью растворяется в 250 г ТГФ до отбора соответствующего образца, точная измеренная концентрация будет составлять 1000 ч./млн в ТГФ.
Когда СТК в соответствии с настоящим изобретением подвергают испытанию на рассчитанное по времени растворение, описанному выше, предпочтительно, чтобы образец, взятый через одну минуту после t=0, растворялся до концентрации, по меньшей мере, приблизительно 500 ч./млн в ТГФ, более предпочтительно, по меньшей мере, 600 ч./млн в ТГФ. Для образца, взятого через две минуты после t=0, предпочтительно, чтобы СТК в соответствии с настоящим изобретением растворялась до концентрации, по меньшей мере, приблизительно 700 ч./млн в ТГФ, более предпочтительно, по меньшей мере, 750 ч./млн в ТГФ. Для образца, взятого через четыре минуты после t=0, предпочтительно, чтобы СТК в соответствии с настоящим изобретением растворялась до концентрации, по меньшей мере, приблизительно 840 ч./млн в ТГФ, более предпочтительно, по меньшей мере, 880 ч/млн в ТГФ.
Установлено, что относительно простая модель развития по отрицательной экспоненте может быть использована для описания временной зависимости всего набора данных для полного испытания на рассчитанное по времени растворение, несмотря на сложность образцов в форме частиц и процесса растворения. Форма уравнения, называемого далее «моделью рассчитанного по времени растворения», является следующей:
S=A+B*(1-exp(-C*t)),
где t - время в минутах;
S - растворимость в ч./млн в ТГФ при времени t;
exp - экспоненциальная функция в основании натурального логарифма 2;
А, В - неизменные константы в ч./млн в ТГФ, где А относится, главным образом, к быстрому растворению более мелких частиц за очень короткое время и где сумма А+В относится, главным образом, к общему количеству растворенного вещества в конце определенного периода испытания; и
С - неизменная временная константа в обратных минутах.
Неизменные константы корректируют, чтобы свести до минимума сумму квадратов ошибок между точками фактических данных и соответствующих модельных значений, и этот метод обычно называют соответствием по «методу наименьших квадратов». Предпочтительным пакетом программ для этой регрессии данных является JMP Release 5.1.2 (SAS Institute Inc., JMP Software, SAS Campus Drive, Cary NC 27513).
Когда СТК в соответствии с настоящим изобретением испытывают с помощью испытания на рассчитанное по времени растворение и аппроксимируют к модели рассчитанного по времени растворения, как описано выше, предпочтительно, чтобы СТК имела временную константу «С» больше чем приблизительно 0,5 обратной минуты, более предпочтительно больше чем приблизительно 0,6 обратной минуты и наиболее предпочтительно больше чем 0,7 обратной минуты.
На ФИГ. 29А и 29В представлена частица обычной СТК, полученная с помощью обычного высокотемпературного процесса окисления в непрерывном реакторе с перемешиванием (НРП). На ФИГ. 29А показана частица обычной СТК при 500-кратном увеличении, тогда как на ФИГ. 29В изображение увеличено, и частица СТК показана при увеличении в 2000 раз. Визуальное сравнение частиц заявленной СТК, представленных на ФИГ. 28А и 29В, и частиц обычной СТК, представленных на ФИГ. 29А и 29В, показывает, что частица обычной СТК имеет более высокую плотность, более низкую площадь поверхности, более низкую пористость и больший размер частиц, чем частицы заявленной СТК. Действительно, обычная СТК, представленная на ФИГ. 29А и 29В, имеет средний размер частиц приблизительно 205 мкм и площадь поверхности по БЭТ приблизительно 0,57 м2/г.
ФИГ. 30 иллюстрирует обычный процесс производства очищенной терефталевой кислоты (ОТК). В обычном процессе получения ОТК пара-ксилол частично окисляют в реакторе высокотемпературного окисления 700 с механическим перемешиванием. Суспензию, содержащую СТК, выводят из реактора 700 и затем очищают в системе очистки 702. Продукт, ОТК, системы очистки 702 вводят в систему разделения 706 для отделения и сушки частиц ОТК. Система очистки 702 вносит значительную часть в затраты, связанные с производством частиц ОТК обычными способами. Система очистки 702, как правило, включает систему добавления воды/обмена 708, систему растворения 710, систему гидрирования 712 и три отдельных кристаллизационных емкости 704а,b,c. В системе добавления воды/обмена 708 значительная часть маточной жидкости заменяется водой. После добавления воды суспензию вода/СТК вводят в систему растворения 710, где смесь вода/СТК нагревают до полного растворения частиц СТК в воде. После растворения СТК раствор СТК в воде подвергают гидрированию в системе гидрирования 712. Гидрированный выходящий поток из системы гидрирования 712 затем подвергают трем стадиям кристаллизации в кристаллизационных емкостях 704а,b,c, после чего выделяют ОТК в системе отделения 706.
ФИГ. 31 иллюстрирует усовершенствованный процесс производства ОТК, в котором используется реакторная система окисления, включающая реактор первичного окисления 800а и реактор вторичного окисления 800b. В конфигурации, представленной на ФИГ. 31, исходную суспензию получают из реактора первичного окисления 800а и затем подвергают очистке в системе очистки 802, в которой реактор вторичного окисления 800b является частью. Исходная суспензия, выведенная из реактора первичного окисления 800а, предпочтительно содержит частицы твердой СТК и жидкий маточный раствор. Как правило, исходная суспензия содержит в интервале приблизительно от 10 до 50% масс. частиц твердой СТК, причем остальное количество составляет жидкий маточный раствор. Частицы твердой СТК, присутствующие в исходной суспензии, выведенной из реактора первичного окисления 800а, как правило, содержат, по меньшей мере, приблизительно 400 масс.ч./млн 4-карбоксибензальдегида (4-КБА), более типично, по меньшей мере, приблизительно 800 масс.ч./млн 4-КБА и наиболее типично в интервале приблизительно от 1000 до 15000 масс.ч./млн 4-КБА.
Система очистки 802 принимает начальную суспензию, выведенную из реактора первичного окисления 800а, и снижает концентрацию 4-КБА и других примесей, присутствующих в СТК. В системе очистки 802 получают более чистую/очищенную суспензию и эту суспензию подвергают отделению и сушке в системе отделения 804, в результате получая частицы более чистой твердой терефталевой кислоты, содержащие менее чем приблизительно 400 масс.ч./млн 4-КБА, более предпочтительно менее чем приблизительно 250 масс.ч./млн 4-КБА и наиболее предпочтительно в интервале приблизительно от 10 до 200 масс.ч./млн 4-КБА.
Система очистки 802 включает реактор вторичного окисления 800b, систему обмена жидкости 806, автоклав 808 и один кристаллизатор 810. В реакторе вторичного окисления 800b исходную суспензию подвергают окислению при температуре и давлении, которые приблизительно равны температуре и давлению в реакторе первичного окисления 800а. В системе обмена жидкости 806, по меньшей мере, 50% масс. маточной жидкости, присутствующей в суспензии, выведенной из реактора вторичного окисления 800b, заменяют свежим растворителем, чтобы в результате дает суспензию с растворителем, содержащую частицы СТК и растворитель. Суспензию с обмененным растворителем, выходящую из системы обмена жидкости 806, вводят в автоклав 808. В автоклаве 808 проводят дополнительную реакцию окисления при немного более высоких температурах, чем температуры, используемые в реакторе первичного окисления 800а.
Как описывалось выше, высокая площадь поверхности, небольшой размер частиц и низкая плотность частиц СТК, полученных в реакторе первичного окисления 800а, делают некоторые примеси, захваченные в частицы СТК, доступными для окисления в автоклаве 808 без необходимости полного растворения частиц СТК в автоклаве 808. Следовательно, температура в автоклаве 808 может быть ниже, чем во многих аналогичных процессах данной области техники. Дополнительное окисление, проводимое в автоклаве 808, предпочтительно снижает концентрацию 4-КБА в СТК, по меньшей мере, на 200 масс.ч./млн, более предпочтительно, по меньшей мере, приблизительно на 400 масс.ч./млн, и наиболее предпочтительно снижение находится в интервале от 600 до 6000 масс.ч./млн. Предпочтительно температура сжигания в автоклаве 808, по меньшей мере, приблизительно на 10ºС выше, чем температура первичного окисления в реакторе 800а, более предпочтительно приблизительно на 20-80ºС выше, чем температура первичного окисления в реакторе 800а и наиболее предпочтительно на 30-50ºС выше, чем температура первичного окисления в реакторе 800а. Температура сжигания предпочтительно находится в интервале приблизительно от 160 до 240ºС, более предпочтительно в интервале приблизительно от 180 до 220ºС и наиболее предпочтительно в интервале приблизительно от 190 до 210ºС. Для очищенного продукта из автоклава 808 необходима только одна стадия кристаллизации в кристаллизаторе 810 перед выделением в системе отделения 804. Подходящие методики вторичного окисления/сжигания описаны более подробно в патентной заявке США № 2005/0065373, полное описание которой включено точно в качестве ссылки.
Терефталевая кислота (например, ОТК), произведенная с помощью системы, представленной на ФИГ. 31, предпочтительно образует частицы, имеющие средний размер частиц, по меньшей мере, приблизительно 40 мкм, более предпочтительно в интервале приблизительно от 50 до 2000 мкм наиболее предпочтительно в интервале приблизительно от 60 до 200 мкм. Частицы ОТК предпочтительно имеют среднюю площадь поверхности по БЭТ менее чем приблизительно 0,25 м2/г, более предпочтительно в интервале приблизительно от 0,005 до 0,2 м2/г и наиболее предпочтительно в интервале от 0,01 до 0,18 м2/г. ОТК, полученная с помощью системы, представленной на ФИГ. 31, приемлема для использования в качестве сырья для производства ПЭТ. Как правило, ПЭТ получают путем этерификации терефталевой кислоты этиленгликолем с последующей поликонденсацией. Предпочтительно терефталевая кислота, произведенная с помощью вариантов осуществления настоящего изобретения, применяется в качестве сырья для процесса производства ПЭТ в трубчатом реакторе, описанном в патентной заявке США с регистрационным № 10/013318, направленной на рассмотрение 7 декабря 2001, полное описание которой включено в качестве ссылки.
Частицы ОТК с предпочтительной морфологией, описанные в данном изобретении, особенно могут быть использованы в описанном выше процессе окислительного сжигания для снижения содержания 4-КБА. Кроме того, такие предпочтительные частицы СТК обеспечивают преимущества в широком интервале других последующих процессов, в которых имеет место растворение и/или химическое взаимодействие частиц. Такие дополнительные последующие обработки включают, но не ограничиваются ими, реакцию, по меньшей мере, одного гидроксилсодержащего соединения с образованием сложноэфирных соединения, в особенности реакцию СТК с метанолом с образованием диметилтерефталата и примесных сложных эфиров; реакцию, по меньшей мере, с одним диолом с образованием сложноэфирных мономерных и/или полимерных соединений, в особенности реакцию СТК с этиленгликолем с образованием полиэтилентерефталата (ПЭТ); и полное или частичное растворение в растворителях, включая, но не ограничиваясь ими, воду, уксусную кислоту и N-метил-2-пирролидон, которые могут включать дополнительную обработку, в том числе, но без ограничения, повторное осаждение более чистой терефталевой кислоты и/или селективную химическую реакцию карбонильных групп, отличных от групп карбоновой кислоты. Особенно это относится к значительному растворению СТК в растворителе, содержащем воду, в сочетании с частичным гидрированием, что снижает количество альдегидов, в особенности 4-КБА, флуоренонов, фенонов и/или антрахинонов.
В соответствии с одним из вариантов осуществления настоящего изобретения предлагается способ частичного окисления способного к окислению ароматического соединения до одного или нескольких типов ароматических карбоновых кислот, где чистота части растворителя в сырье (то есть «загрузки растворителя») и чистота части способного к окислению соединения в сырье (то есть «загрузки способного к окислению соединения») контролируется в пределах некоторых интервалов, конкретно определенных ниже. Наряду с другими вариантами настоящего изобретения это дает возможность контролировать чистоту жидкой фазы и, если они присутствуют, твердой фазы и объединенной суспензионной фазы (то есть твердое вещество плюс жидкость) реакционной среды в пределах определенных предпочтительных интервалов, описанных ниже.
Что касается загрузки растворителя, то известно окисление способного(ых) к окислению ароматического(их) соединения(й) для получения ароматической карбоновой кислоты, где загрузка растворителя, введенного в реакционную среду, представляет собой смесь аналитически чистой уксусной кислоты и воды, как это часто используется в лабораторных условиях и в опытных установках. Аналогично, известно проведение окисления способного к окислению ароматического соединения до ароматической карбоновой кислоты, где растворитель, покидающий реакционную среду, отделяют от произведенной ароматической карбоновой кислоты и затем рециркулируют назад в реакционную среду в качестве загрузки растворителя, главным образом по причине стоимости производства. Такое рециркулирование растворителя со временем приводит к накоплению некоторых примесей сырья и побочных продуктов процесса в рециркулированном растворителе. В данной области техники известны различные средства, которые способствуют очистке рециркулированного растворителя перед повторным введением в реакционную среду. Как правило, более высокая степень очистки рециркулированного растворителя приводит к значительно более высокой стоимости производства, чем в случае более низкой степени очистки с помощью аналогичных средств. Один из вариантов осуществления настоящего изобретения относится к осмыслению и определению предпочтительных интервалов для большого числа примесей в загрузке растворителя, многие из которых прежде считались в значительной степени допустимыми, чтобы найти оптимальный баланс между суммарной стоимостью производства и общей чистотой продукта.
«Загрузка рециркулированного растворителя» определяется в данном случае как загрузка растворителя, который ранее составлял часть реакционной среды, подвергнутой окислению в зоне/реакторе окисления, и вышел из зоны/реактора окисления в виде части сырого жидкого и/или суспензионного продукта. Например, загрузка рециркулированного растворителя в реакционную среду частичного окисления при окислении пара-ксилола с образованием ТФК представляет собой растворитель, который первоначально составлял часть реакционной среды частичного окисления, был удален из реакционной среды в виде жидкой фазы суспензии ТФК, отделен от большей части твердой ТФК и затем возвращен в реакционную среду частичного окисления. Как описано выше, такая загрузка рециркулированного растворителя имеет склонность к накоплению всех сортов нежелательных примесей, если не предусмотрены конкретные дополнительные технологические стадии для очистки растворителя при значительных капитальных и эксплуатационных затратах. По экономическим причинам предпочтительно, чтобы, по меньшей мере, приблизительно 20% масс. загрузки растворителя в реакционную среду настоящего изобретения представляло собой рециркулированный растворитель, более предпочтительно, по меньшей мере, приблизительно 40% масс., еще более предпочтительно, по меньшей мере, приблизительно 80% масс. и наиболее предпочтительно, по меньшей мере, приблизительно 90% масс. На основании запаса растворителя и продолжительности рабочего цикла в промышленной установке предпочтительно, чтобы части рециркулированного растворителя проходили через реакционную среду, по меньшей мере, один раз за день работы, более предпочтительно, по меньшей мере, один раз в день в течение, по меньшей мере, семи последовательных дней работы и наиболее предпочтительно, по меньшей мере, один раз в день в течение, по меньшей мере, 30 последовательных дней работы.
На основании реакционной активности и с учетом металлических примесей, оставшихся в продукте окисления, установлено, что концентрации выбранных многовалентных металлов в загрузке рециркулированного растворителя предпочтительно находятся в пределах интервалов, конкретно указанных непосредственно ниже. Концентрация железа в рециркулированном растворителе предпочтительно находится ниже приблизительно 150 масс.ч./млн, более предпочтительно ниже приблизительно 40 масс.ч./млн и наиболее предпочтительно в интервале от 0 до 8 масс.ч./млн. Концентрация никеля в рециркулированном растворителе предпочтительно находится ниже приблизительно 150 масс.ч./млн, более предпочтительно ниже приблизительно 40 масс.ч./млн и наиболее предпочтительно в интервале от 0 до 8 масс.ч./млн. Концентрация хрома в рециркулированном растворителе предпочтительно находится ниже приблизительно 150 масс.ч./млн, более предпочтительно ниже приблизительно 40 масс.ч./млн и наиболее предпочтительно в интервале от 0 до 8 масс.ч./млн. Концентрация молибдена в рециркулированном растворителе предпочтительно находится ниже приблизительно 75 масс.ч./млн, более предпочтительно ниже приблизительно 20 масс.ч./млн и наиболее предпочтительно в интервале от 0 до 4 масс.ч./млн. Концентрация титана в рециркулированном растворителе предпочтительно находится ниже приблизительно 75 масс.ч./млн, более предпочтительно ниже приблизительно 20 масс.ч./млн и наиболее предпочтительно в интервале от 0 до 4 масс.ч./млн. Концентрация меди в рециркулированном растворителе предпочтительно находится ниже приблизительно 20 масс.ч./млн, более предпочтительно ниже приблизительно 4 масс.ч./млн и наиболее предпочтительно в интервале от 0 до 1 масс.ч./млн. Другие металлические примеси, как правило, также присутствуют в рециркулированном растворителе, и обычно их концентрация меняется на более низких уровнях относительно одного или нескольких перечисленных выше металлов. Контролирование перечисленных выше металлов в предпочтительных интервалах будет удерживать другие металлические примеси на приемлемых уровнях.
Такие металлы могут появляться в виде примесей в любом поступающем в процесс сырье (например, в поступающем способном к окислению соединении, в растворителе, в окислителе и в каталитических соединениях). С другой стороны, металлы могут появляться как продукты коррозии любой из установок процесса, контактирующей с реакционной средой и/или контактирующей с рециркулированным растворителем. Средства контролирования металлов в раскрытых интервалах концентраций включают соответствующие описание и мониторинг чистоты различного сырья и применение подходящих материалов конструкций, в том числе, но без ограничения ими, многих промышленных сортов титана и нержавеющей стали, в том числе сортов, известных как нержавеющие стали дуплекс-процесса и высоко молибденовые нержавеющие стали.
Заявители также раскрывают предпочтительные интервалы для выбранных ароматических соединений в рециркулированном растворителе. Эти интервалы включают как осажденные, так и растворенные ароматические соединения в рециркулированном растворителе.
Неожиданно, но даже осажденный продукт (например, ТФК) частичного окисления пара-ксилола является загрязнителем, который необходимо контролировать в рециркулированном растворителе. Так как существуют неожиданно предпочтительные интервалы для уровней твердых веществ в реакционной среде, любой осажденный продукт в загрузке растворителя сразу вычитается из количества способного к окислению соединения, которое может быть подано совместно. Более того, подача осажденной твердой ТФК в рециркулированном растворителе при повышенных уровнях, как установлено, отрицательно влияет на свойства частиц, образованных в среде окисления с осаждением, что приводит к нежелательным свойствам в операциях в нисходящем потоке (например, на фильтрацию продукта, промывку растворителем, окислительное сжигание сырого продукта, растворение сырого продукта для последующей обработки и т.д.). Другим нежелательным свойством осажденных твердых веществ в загрузке рециркулированного растворителя является то, что они часто содержат очень высокие уровни осажденных примесей по сравнению с концентрациями примесей в объеме твердых веществ в суспензиях ТФК, из которых получена большая часть рециркулированного растворителя. Возможно, повышенные уровни примесей, наблюдаемые в твердых веществах, суспендированных в рециркулированном растворителе, могут быть связаны со временем зародышеобразования для осаждения некоторых примесей из рециркулированного растворителя и/или с охлаждением рециркулированного растворителя, или преднамеренно или вследствие внешних потерь. Например, концентрации интенсивно окрашенного и нежелательного 2,6-дикарбоксифлуоренона наблюдаются на значительно более высоких уровнях в твердых веществах, присутствующих в рециркулированном растворителе при 80ºС, чем в твердых веществах ТФК, отделенной от рециркулированного растворителя при 160ºС. Аналогично, концентрации изофталевой кислоты наблюдаются на значительно более высоких уровнях в твердых веществах, присутствующих в рециркулированном растворителе, по сравнению с уровнями, наблюдаемыми в твердых веществах ТФК из реакционной среды. Путь, по которому конкретные осажденные примеси вовлекаются в рециркулированный растворитель при повторном введении в реакционную среду, как оказывается, меняется. Это зависит, по-видимому, от относительной растворимости примеси в жидкой фазе реакционной среды, по-видимому, от того, как осажденная примесь напластована в пределах осажденных твердых веществ и, по-видимому, от локальной скорости осаждения ТФК, где твердое вещество изначально повторно поступает в реакционную среду. Таким образом, найдено полезным контролировать уровень некоторых примесей в рециркулированном растворителе, как описано ниже, без учета того, присутствуют ли эти примеси в рециркулированном растворителе в растворенной форме или вовлечены в материал в виде частиц, находящихся в нем.
Количество осажденных твердых веществ, присутствующих в рециркулированном растворителе, определяют гравиметрическим методом следующим образом. Типичный образец выводят из подачи растворителя в реакционную среду, пока растворитель протекает по трубопроводу к реакционной среде. Полезный размер образца составляет приблизительно 100 г, отобранные в стеклянный контейнер, имеющий приблизительно 250 мл внутреннего объема. Перед стравливанием до атмосферного давления, но, при непрерывном протекании в направлении контейнера для образца, рециркулированный растворитель охлаждают менее чем до 100ºС; такое охлаждение проводят для того, чтобы ограничить испарение растворителя в течение короткого интервала перед герметичным закрытием стеклянного контейнера. После забора образца при атмосферном давлении стеклянный контейнер сразу же герметично закрывают. Затем образцу дают остыть приблизительно до 20ºС при температуре окружающего воздуха приблизительно 20ºС и без принудительной конвенции. По достижении 20ºС образец выдерживают в таких условиях, по меньшей мере, приблизительно 2 часа. Затем герметично закрытый контейнер энергично встряхивают до достижения равномерного распределения твердых веществ. Сразу после этого в контейнер для образцов вносят магнитную мешалку и вращают при достаточной скорости, чтобы эффективно поддерживать равномерное распределение твердых веществ. Пипеткой отбирают аликвоту объемом 10 мл перемешанной жидкости с суспендированными твердыми веществами и взвешивают. Затем объем жидкой фазы из этой аликвоты отделяют вакуумной фильтрацией, все еще приблизительно при 20ºС и без потерь твердых веществ. Влажные твердые вещества, отфильтрованные из этой аликвоты, затем сушат без возгонки твердых веществ, и такие высушенные твердые вещества взвешивают. Отношение массы высушенных твердых веществ к массе исходной аликвоты суспензии представляет собой фракцию твердых веществ, как правило, выражаемую в процентах, и в данном случае эту фракцию называют количеством «твердых веществ, осажденных при 20ºС» в загрузке растворителя.
Заявители установили, что ароматические соединения, растворенные в жидкой фазе реакционной среды и содержащие ароматические карбоновые кислоты, не имеющие неароматических углеводородных групп (например, изофталевую кислоту, бензойную кислоту, фталевую кислоту, 2,5,4'-трикарбоксибифенил), неожиданно являются наносящими вред компонентами. Хотя такие соединения имеют значительно пониженную химическую активность в рассматриваемой реакционной среде по сравнению со способными к окислению соединениями, имеющими неароматические углеводородные группы, установлено, что эти соединения, тем не менее, подвергаются множеству вредных реакций. Следовательно, полезно контролировать содержание этих соединений в предпочтительных интервалах в жидкой фазе реакционной среды. Это приводит к предпочтительным интервалам выбранных соединений в загрузке рециркулированного растворителя, а также к предпочтительным интервалам выбранных предшественников в загрузке способного к окислению ароматического соединения.
Например, при жидкофазном частичном окислении пара-ксилола до терефталевой кислоты (ТФК) заявители установили, что интенсивно окрашенная и нежелательная примесь 2,7-дикарбоксифлуоренон (2,7-ДКФ, 2,7-DCF) фактически не определима в реакционной среде, и продукт отводят, когда мета-замещенные ароматические соединения присутствуют в реакционной среде на очень низких уровнях. Установлено, что когда примесь изофталевой кислоты присутствует в загрузке растворителя в повышенных количествах, образование 2,7-ДКФ повышается почти прямо пропорционально. Установлено также, что когда примесь мета-ксилола присутствует в загрузке пара-ксилола, образование 2,7-ДКФ также повышается почти прямо пропорционально. Кроме того, установлено, что даже если загрузка растворителя и загрузка способного к окислению соединения освобождены от мета-замещенных ароматических соединений, некоторое количество изофталевой кислоты образуется во время типичного частичного окисления очень чистого пара-ксилола, особенно, когда бензойная кислота присутствует в жидкой фазе реакционной среды. Такая самообразованная изофталевая кислота вследствие ее более высокой растворимости, чем у ТФК, в растворителе, состоящем из уксусной кислоты и воды, со временем может накапливаться в промышленных установках, использующих рециркулированный растворитель. Следовательно, количество изофталевой кислоты в загрузке растворителя, количество мета-ксилола в загрузке способного к окислению соединения и скорость самообразования изофталевой кислоты в пределах реакционной среды, все соответствующим образом рассматриваются в балансе друг с другом и в балансе с любыми реакциями, которые потребляют изофталевую кислоту. Изофталевая кислота, как установлено, подвергается другим неэкономичным реакциям, помимо образования 2,7-ДКФ, которые описаны ниже. Кроме того, установлено, что существуют другие объекты для рассмотрения при установлении подходящих интервалов для мета-замещенных ароматических образцов при частичном окислении пара-ксилола до ТФК. Другие интенсивно окрашенные и нежелательные примеси, такие как 2,6-дикарбоксифлуоренон (2,6-ДКФ), как оказывается, в значительной степени связаны с растворенными пара-замещенными ароматическими образцами, которые всегда присутствуют в загрузке пара-ксилола при жидкофазном окислении. Следовательно, подавление образования 2,7-ДКФ лучше всего рассматривать с учетом уровня других произведенных окрашенных примесей.
Например, при жидкофазном частичном окислении пара-ксилола до ТФК установлено, что образование тримеллитовой кислоты повышается по мере того, как в реакционной среде повышаются уровни изофталевой кислоты и фталевой кислоты. Тримеллитовая кислота представляет собой трифункциональную карбоновую кислоту, приводящую к разветвлению полимерных цепочек при производстве ПЭТ из ТФК. Во многих вариантах применения ПЭТ степень разветвления необходимо контролировать на низких уровнях и, следовательно, в очищенной ТФК тримеллитовую кислоту необходимо контролировать на низких уровнях. Помимо образования тримеллитовой кислоты присутствие в реакционной среде мета-замещенных и орто-замещенных образцов также приводит к появлению других трикарбоновых кислот (например, 1,3,5-трикарбоксибензола). Кроме того, повышенное присутствие трикарбоновых кислот в реакционной среде повышает образование тетракарбоновых кислот (например, 1,2,4,5-тетракарбоксибензола). Контролирование суммарного производства всех ароматических карбоновых кислот, имеющих более двух карбоксильных групп, является одним из факторов при установлении предпочтительных уровней мета-замещенных и орто-замещенных образцов в загрузке рециркулированного растворителя, в загрузке способного к окислению соединения и в реакционной среде в соответствии с настоящим изобретением.
Например, при жидкофазном частичном окислении пара-ксилола до ТФК установлено, что повышенные уровни в жидкой фазе реакционной среды некоторых растворенных ароматических кислот, не имеющих неароматических углеводородных групп, непосредственно приводят к повышенному производству монооксида углерода и диоксида углерода. Такое повышенное производство оксидов углерода означает потерю выхода как окислителя, так и способного к окислению соединения, причем последнее из-за многих одновременно получаемых ароматических карбоновых кислот, которые, с одной стороны, могут рассматриваться как примеси, а с другой стороны, также имеют промышленное значение. Следовательно, соответствующее удаление относительно растворимых карбоновых кислот, не имеющих неароматических углеводородных групп, из рециркулируемого растворителя имеет экономическое значение при предотвращении потерь выхода способного к окислению соединения и окислителя, помимо подавления образования высоко нежелательных примесей, таких как различные флуореноны и тримеллитовая кислота.
Например, при жидкофазном частичном окислении пара-ксилола до ТФК установлено, что образование 2,5,4'-трикарбоксибифенила кажется неизбежным. 2,5,4'-Трикарбоксибифенил является ароматической трикарбоновой кислотой, образованной путем сочетания двух ароматических колец, вероятно, за счет сочетания растворенных пара-замещенных ароматических образцов с арильным радикалом, вероятно, арильным радикалом, образованным в результате декарбоксилирования или декарбонилирования пара-замещенных ароматических образцов. К счастью, 2,5,4'-трикарбоксибифенил обычно образуется в более низких количествах, чем тримеллитовая кислота, и обычно не приводит к значительно повышенным проблемам разветвления полимерных молекул при производстве ПЭТ. Однако заявители установили, что повышенные уровни 2,5,4'-трикарбоксибифенила в реакционной среде, включающей окисление алкилароматических соединений в соответствии с предпочтительными вариантами осуществления настоящего изобретения, приводят к повышенным уровням интенсивно окрашенного и нежелательного 2,6-ДКФ. Повышенные количества 2,6-ДКФ, по-видимому, образуются из 2,5,4'-трикарбоксибифенила за счет раскрытия цикла с потерей молекулы воды, хотя точный механизм реакции достоверно не известен. Если слишком большому количеству 2,5,4'-трикарбоксибифенила, более растворимома в растворителе, содержащем уксусную кислоту и воду, чем ТФК, позволить накапливаться в рециркулированном растворителе, степень конверсии до 2,6-ДКФ может стать неприемлемо большой.
Например, при жидкофазном частичном окислении пара-ксилола до ТФК установлено, что ароматические карбоновые кислоты, не имеющие неароматических углеводородных групп (например, изофталевая кислота), обычно приводят к умеренному подавлению химической активности реакционной среды, когда они присутствуют в жидкой фазе в достаточной концентрации.
Например, при жидкофазном частичном окислении пара-ксилола до ТФК установлено, что осаждение очень часто является неидеальным (то есть неравновесным) относительно соответствующих концентраций различных химических образцов в твердой фазе и в жидкой фазе. Вероятно, это обусловлено тем, что скорость осаждения является очень высокой при объемных реакционных скоростях, предпочтительных в данном случае, что приводит к неидеальному соосаждению примесей или даже к окклюзии. Следовательно, когда желательно ограничить концентрацию некоторых примесей (например, тримеллитовой кислоты и 2,6-ДКФ) в сырой ТФК, из-за конфигурации работы установки в нисходящем направлении, предпочтительно контролировать их концентрацию в загрузке растворителя, а также скорость их образования в реакционной среде.
Например, установлено, что бензофеноновые соединения (например, 4,4'-дикарбоксибензофенон и 2,5,4'-трикарбоксибензофенон), полученные во время частичного окисления пара-ксилола, оказывают нежелательное влияние на реакционную среду ПЭТ, даже хотя в ТФК бензофеноновые соединения не являются сами по себе интенсивно окрашенными, как флуореноны и антрахиноны. Соответственно, желательно ограничить присутствие бензофенонов и выбранных предшественников в загрузке рециркулированного растворителя и в загрузке способного к окислению соединения. Кроме того, установлено, что присутствие повышенных уровней бензойной кислоты независимо от того, находится ли она в рециркулированном растворителе или образуется в реакционной среде, приводит к повышенным скоростям производства 4,4'-дикарбоксибензофенона.
В процессе изучения заявители установили и количественно оценили неожиданный порядок реакций для ароматических соединений, не имеющих неароматические углеводородные группы, которые присутствуют при жидкофазном частичном окислении пара-ксилола до ТФК. При анализе только одного случая бензойной кислоты заявители установили, что повышенные уровни бензойной кислоты в реакционной среде некоторых вариантов осуществления настоящего изобретения приводят к значительно повышенному производству интенсивно окрашенной и нежелательной 9-флуоренон-2-карбоновой кислоты, к значительно повышенным уровням 4,4'-дикарбоксибифенила, к повышенным уровням 4,4'-дикарбоксибензофенона, к умеренному подавлению химической активности в направлении целевого окисления пара-ксилола, а также к повышенным уровням диоксидов углерода и сопутствующим потерям выхода. Установлено, что повышенные уровни бензойной кислоты в реакционной среде также приводят к повышенному производству изофталевой кислоты и фталевой кислоты, уровни которых желательно контролировать в низких интервалах в соответствии с аналогичными аспектами данного изобретения. Число и значение реакций с вовлечением бензойной кислоты является, вероятно, даже более неожиданными, так как некоторые современные исследователи рассматривают использование бензойной кислоты вместо уксусной кислоты в качестве основного компонента растворителя (см., например, патент США № 6562997). Кроме того, наблюдается, что бензойная кислота самообразуется при окислении пара-ксилола при скоростях, которые достаточно значительны относительно скоростей ее образования из примесей, таких как толуол и этилбензол, обычно находимых в загрузке способного к окислению соединения, содержащей промышленно чистый пара-ксилол.
С другой стороны, заявители раскрыли небольшое значение дополнительного регулирования композиции рециркулированного растворителя относительно присутствия способного к окислению ароматического соединения и относительно реакций ароматических промежуточных соединений, которые содержат неароматические углеводородные группы и относительно растворимы в рециркулированном растворителе. В общем случае такие соединения или подаются в реакционную среду, или образуются в реакционной среде при значительно более высоких скоростях, чем их количество, присутствующее в рециркулированном растворителе; а скорость потребления этих соединений в реакционной среде является достаточно высокой, удерживая одну или несколько неароматических углеводородных групп, чтобы ограничить их накопление в рециркулированном растворителе. Например, во время частичного окисления пара-ксилола в многофазной среде пара-ксилол испаряется в ограниченной степени наряду с большими количествами растворителя. Когда такой испарившийся растворитель выходит из реактора в виде части выходящего газа и конденсируется для выделения в виде рециркулированного растворителя, значительная часть испарившегося пара-ксилола также конденсируется в нем. Нет необходимости ограничивать концентрацию такого пара-ксилола в рециркулированном растворителе. Например, если растворитель отделяют от твердых веществ при выходе суспензии из реакционной среды окисления пара-ксилола, этот выделенный растворитель будет содержать концентрацию растворенной пара-толуиловой кислоты, аналогичную концентрации в точке удаления из реакционной среды. Хотя может быть важным ограничение фиксированной концентрации пара-толуиловой кислоты в жидкой фазе реакционной среды (см. ниже), нет необходимости регулировать отдельно пара-толуиловую кислоту в этой порции рециркулированного растворителя ввиду ее относительно хорошей растворимости и ее низкого удельного массового расхода относительно образования пара-толуиловой кислоты в пределах реакционной среды. Аналогично выявлены незначительные основания для ограничения концентраций в рециркулированном растворителе ароматических соединений с метильными заместителями (например, толуиловых кислот), ароматических альдегидов (например, терефталевого альдегида), ароматических соединений с гидроксиметильными заместителями (например, 4-гидроксиметилбензойной кислоты) и бромированных ароматических соединений, содержащих, по меньшей мере, одну неароматическую углеводородную группу (например, альфа-бром-пара-толуиловую кислоту) ниже концентраций, по сути найденных в жидкой фазе, выходящей из реакционной среды, существующей при частичном окислении ксилола в соответствии с предпочтительными вариантами осуществления настоящего изобретения. Неожиданно заявители установили, что также нет необходимости контролировать в рециркулированном растворителе концентрацию выбранных фенолов, по сути произведенных во время частичного окисления ксилола, ввиду того, что эти соединения образуются и разрушаются в пределах реакционной среды со скоростями, значительно более высокими, чем их присутствие в рециркулированном растворителе. Например, установлено, что 4-гидроксибензойная кислота оказывает относительно небольшое влияние на химическую активность в предпочтительных вариантах настоящего изобретения при совместной подаче со скоростями свыше 2 граммов 4-гидроксибензойной кислоты на 1 килограмм пара-ксилола, значительно более высокой, чем естественное присутствие в рециркулированном растворителе, несмотря на то, другие авторы указывают на нее как на сильный яд в аналогичной реакционной среде (см., например, W. Partenheimer, Catalysis Today 23, (1995), p. 81).
Таким образом, существует множество реакций и многочисленные рассуждения при установлении предпочтительных интервалов различных ароматических примесей в загрузке растворителя, как это показано в настоящее время. Такие результаты представлены в значениях совокупной средневзвешенной композиции всех потоков растворителя, которые подаются в реакционную среду в течение установленного периода времени, предпочтительно в течение одного дня, более предпочтительно одного часа и наиболее предпочтительно в течение 1 минуты. Например, если одна загрузка растворителя протекает по существу непрерывно с композицией 40 масс.ч./млн изофталевой кислоты с расходом 7 кг/мин, вторая загрузка растворителя протекает по существу непрерывно с композицией 2000 масс.ч./млн изофталевой кислоты с расходом 10 кг/мин, и не существует других потоков загрузки растворителя, поступающих в реакционную среду, то совокупная средневзвешенная композиция загрузки растворителя рассчитывается как (40*7+2000*10)/(7+10)=1,193 масс.ч./млн изофталевой кислоты. Следует отметить, что масса загрузки любого способного к окислению соединения или загрузки любого окислителя, которые, возможно, объединены с загрузкой растворителя до поступления в реакционную среду, не учитываются при расчете совокупной средневзвешенной композиции загрузки растворителя.
В таблице 1, представленной ниже, приведены предпочтительные значения для некоторых компонентов в загрузке растворителя, введенного в реакционную среду. Компонентами загрузки растворителя, которые приведены в таблице 1, являются следующие соединения: 4-карбоксибензальдегид (4-КБА), 4,4'-дикарбоксистильбен (4,4'-ДКС, 4,4'-DCS), 2,6-дикарбоксиантрахинон (2,6-ДКА, 2,6-DCA), 2,6-дикарбоксифлуоренон (2,6-ДКФ), 2,7-дикарбоксифлуоренон (2,7-ДКФ), 3,5-дикарбоксифлуоренон (3,5-ДКФ), 9-флуоренон-2-карбоновая кислота (9F-2КК, 9F-2CA), 9-флуоренон-4-карбоновая кислота (9F-4КК, 9F-4CA), все флуореноны, в том числе другие флуореноны, не перечисленные отдельно (все флуореноны), 4,4'-дикарбоксибифенил (4,4'-ДКБ), 2,5,4'-трикарбоксибифенил (2,5,4'-ТКБ, 2,5,4'-ТСВ), фталевая кислота (ФК, РА), изофталевая кислота (ИФК, IPA), бензойная кислота (БК, ВА), тримеллитовая кислота (ТМК, ТМА), 2,6-дикарбоксибензокумарин (2,6-ДКБК, 2,6-DCBC), 4,4'-дикарбоксибензил (4,4'-ДКБЗ, 4,4'-DCBZ), 4,4'-дикарбоксибензофенон (4,4'-ДКБФ, 4,4'-DCBР), 2,5,4'-трикарбоксибензофенон (2,5,4'-ТКБФ, 2,5,4'-ТСВР), терефталевая кислота (ТФК), осажденные при 20ºС твердые вещества и все ароматические карбоновые кислоты, не имеющие неароматических углеводородных групп. В представленной ниже таблице 1 приведены предпочтительные количества указанных примесей в СТК, произведенной в соответствии с вариантом осуществления настоящего изобретения.
Компоненты загрузки растворителя, введенной в реакционную смесь
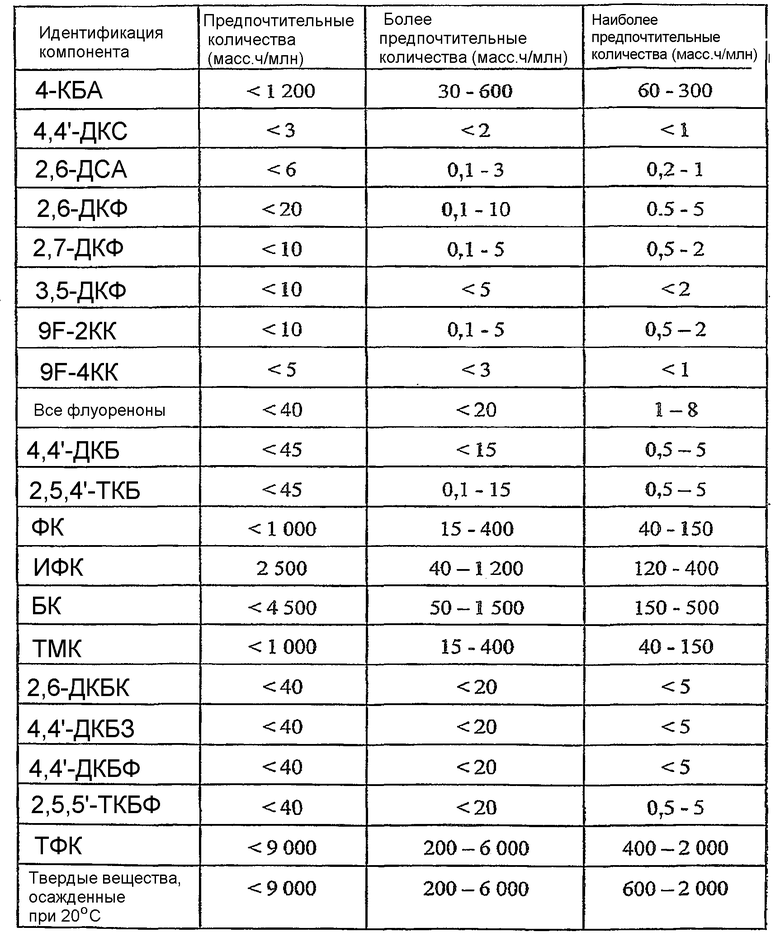
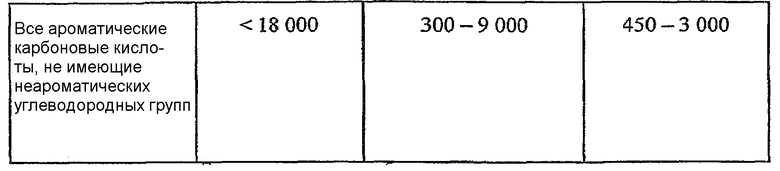
Многие другие ароматические примеси, как правило, также присутствуют в рециркулированном растворителе, причем обычно их количества меняются даже на более низких уровнях и/или в пропорции к одному или нескольким раскрытым ароматическим соединениям. Способы контролирования раскрытых ароматических соединений в предпочтительных интервалах будут обычно удерживать другие ароматические примеси на подходящих уровнях.
Когда бром используется в реакционной среде, большие количества ионных и органических форм брома, как известно, находятся в динамическом равновесии. Такие различные формы брома имеют разные характеристики стабильности при выходе из реакционной среды и при прохождении через различные технологические установки, относящиеся к рециркулированному растворителю. Например, альфа-бром-пара-толуиловая кислота может продолжать существовать как таковая при некоторых условиях или может быстро гидролизоваться при других условиях с образованием 4-гидроксиметилбензойной кислоты и бромистого водорода. В настоящем изобретении предпочтительно, чтобы, по меньшей мере, приблизительно 40% масс., более предпочтительно, чтобы, по меньшей мере, приблизительно 60% масс. и наиболее предпочтительно, чтобы, по меньшей мере, приблизительно 80% масс. общей массы брома, присутствующего в совокупной загрузке растворителя в реакционную среду, находилось в одной или в нескольких из следующих химических форм: ионный бром, альфа-бром-пара-толуиловая кислота и бромуксусная кислота.
Хотя важность и значение контролирования совокупной средневзвешенной чистоты загрузки растворителя в пределах раскрытых, желаемых интервалов настоящего изобретения до настоящего времени не были обнаружены и/или раскрыты, подходящие средства контроля чистоты загрузки растворителя могут быть собраны из различных методов, уже известных в данной области техники. Во-первых, любой растворитель, испаренный из реакционной среды, как правило, имеет подходящую чистоту, при условии, что жидкость или твердые вещества из реакционной среды не захвачены испаренным растворителем. Подача флегмы из капель растворителя в пространство отделения выходящего газа выше реакционной среды, как это описано в данном случае, соответствующим образом ограничивает такой захват; и из такого выходящего газа может быть сконденсирован рециркулированный растворитель подходящей чистоты относительно ароматического соединения. Во-вторых, более трудная и дорогая очистка загрузки рециркулированного растворителя связана с растворителем, отобранным из реакционной среды в жидкой форме, и с растворителем, который впоследствии контактирует с жидкой и/или твердой фазами реакционной среды, выведенной из реакционной емкости (например, рециркулированный растворитель, полученный с фильтра, на котором твердые вещества концентрировались и/или промывались, рециркулированный растворитель, полученный с центрифуги, на которой твердые вещества концентрировались и/или промывались, рециркулированный растворитель, отобранный с операции кристаллизации и т.д.). Однако в данной области также хорошо известны средства для эффективного проведения необходимой очистки таких потоков рециркулированных растворителей с использованием одного или нескольких открытий предшествующего уровня техники. Что касается контролирования осажденных твердых веществ в рециркулированном растворителе в пределах специально определенных интервалов, то подходящими средствами контроля являются, но не ограничиваются ими, гравиметрическая седиментация, механическая фильтрация с использованием фильтровальной ткани на ротационных ленточных фильтрах и ротационных барабанных фильтрах, механическая фильтрация с использованием стационарной фильтрующей среды в емкостях, работающих под давлением, гидроциклоны и центрифуги. Что касается контролирования растворенных ароматических образцов в рециркулированном растворителе в пределах специально определенных интервалов, то контролирующие средства включают, но не ограничиваются ими, средства, описанные в патенте США № 4939297 и в патентной заявке США, номер публикации 2005-0038288, которые включены в качестве ссылки. Однако ни одно из предшествующих изобретений не раскрывает и не описывает предпочтительные уровни чистоты совокупной загрузки растворителя, раскрытые в данном описании. Скорее, указанные изобретения предшествующего уровня предлагают только средства очистки выбранных и частичных потоков рециркулированного растворителя без раскрытия настоящего изобретения, оптимальных значений композиции совокупной средневзвешенной загрузки растворителя в реакционную среду.
Если вернуться к чистоте загрузки способного к окислению соединения, то известно, что некоторые уровни изофталевой кислоты, фталевой кислоты и бензойной кислоты присутствуют и в низких концентрациях приемлемы в очищенной ТФК, используемой для производства полимеров. Более того, известно, что эти образцы имеют относительно более высокую растворимость во многих растворителях и могут быть успешно удалены из очищенной ТФК с помощью процессов кристаллизации. Однако из варианта осуществления изобретения, описанного в данном изобретении, известно, что контролирование уровня некоторых относительно растворимых ароматических образцов, в особенности включающих изофталевую кислоту, фталевую кислоту и бензойную кислоту, в жидкой фазе реакционной среды неожиданно имеет большое значение для контролирования уровня полициклических и окрашенных ароматических соединений, образовавшихся в реакционной среде, для контролирования соединений с более чем 2 карбоксильными кислотными функциями в молекуле, для контролирования реакционной активности в пределах реакционной среды частичного окисления и для контролирования потерь выхода окислителя и ароматического соединения.
В данной области техники известно, что изофталевая кислота, фталевая кислота и бензойная кислота образуются в реакционной среде следующим образом. Примеси мета-ксилола в загрузке окисляются с хорошей конверсией и дают ИФК. Примеси орто-ксилола в загрузке окисляются с хорошей конверсией и дают фталевую кислоту. Примеси этилбензола и толуола в загрузке окисляются с хорошей конверсией и дают бензойную кислоту. Однако выявлено, что значительные количества изофталевой кислоты, фталевой кислоты и бензойной кислоты также образуются в пределах реакционной среды, содержащей пара-ксилол, способами, отличными от окисления мета-ксилола, орто-ксилола, этилбензола и толуола. Эти другие внутренние химические пути, вероятно, включают декарбонилирование, декарбоксилирование, перегруппировку переходных состояний и присоединение метильного и карбонильного радикалов к ароматическим кольцам.
При определении предпочтительных интервалов примесей в загрузке способного к окислению соединения многие факторы являются существенными. Любая примесь в загрузке, по-видимому, приводит к прямой потере выхода и к затратам на очистку продукта, если требования по чистоте окисленного продукта являются достаточно жесткими (например, в реакционной среде частичного окисления пара-ксилола толуол и этилбензол, обычно обнаруживаемые в пара-ксилоле промышленной чистоты, дают бензойную кислоту, и эту бензойную кислоту преимущественно удаляют из большей части промышленной ТФК). Когда продукт частичного окисления примесей загрузки выпадает в осадок при дополнительных реакциях, факторы, отличные от простой потери выхода и удаления, становятся значимыми при рассмотрении, какое увеличение стоимости очистки сырья вытекает из этого (например, в реакционной среде частичного окисления пара-ксилола этилбензол приводит к бензойной кислоте, а бензойная кислота затем дает интенсивно окрашенную 9-флуоренон-2-карбоновую кислоту, изофталевую кислоту, фталевую кислоту и повышенное количество оксидов углерода, наряду с другими). Когда реакционная среда сама генерирует дополнительные количества примеси за счет химических механизмов, непосредственно не связанных с примесями загрузки, анализ становится еще более сложным (например, в реакционной среде частичного окисления пара-ксилола бензойная кислота также самообразуется из самого пара-ксилола). Кроме того, обработка в направлении нисходящего потока сырого продукта окисления может оказывать влияние на рассмотрение предпочтительной чистоты сырья. Например, стоимость удаления до приемлемых уровней прямой примеси (бензойной кислоты) и стоимость удаления последующих примесей (изофталевой кислоты, фталевой кислоты, 9-флуоренон-2-карбоновой кислоты и т.д.) могут быть одинаковыми, могут отличаться друг от друга и могут отличаться от требований по удалению большинства не имеющих отношения примесей (например, продукта неполного окисления 4-КБА при окислении пара-ксилола до ТФК).
Раскрытые ниже интервалы чистоты загрузки для пара-ксилола являются предпочтительными, где пара-ксилол подают с растворителем и окислителем в реакционную среду для частичного окисления с получением ТФК. Эти интервалы являются более предпочтительными в процессе производства ТФК, включающем стадии последующего окисления для удаления из реакционной среды примесей, отличных от окислителя и растворителя (например, металлов катализатора). Такие интервалы все еще остаются более предпочтительными в процессах производства ТФК, в которых дополнительный 4-КБА удаляют из СТК (например, путем конверсии СТК до диметилтерефталата плюс примесные сложные эфиры и последующего удаления метилового эфира 4-КБА перегонкой, с помощью способов окислительного сжигания для превращения 4-КБА в ТФК, с помощью методов гидрирования для превращения 4-КБА в пара-толуиловую кислоту, которая затем отделяется методами дробной кристаллизации). Эти интервалы являются наиболее предпочтительными в процессах производства ТФК, в которых дополнительный 4-КБА удаляют из СТК с помощью способов окислительного сжигания для превращения 4-КБА в ТФК.
Используя новые знания предпочтительных интервалов рециркулирующих ароматических соединений и относительных количеств ароматических соединений, образованных непосредственно при окислении примесей загрузки, по сравнению с другими, не имеющими отношения химическими путями, выявлены улучшенные интервалы примесей для загрязненного пара-ксилола, который подают в процесс частичного окисления для получения ТФК. В таблице 2, приведенной ниже, представлены предпочтительные значения для количества мета-ксилола, орто-ксилола и этилбензола+толуола в загрузке пара-ксилола, выраженные в массовых частях на миллион пара-ксилола.
Компоненты загрязненной загрузки пара-ксилола
Специалисту в данной области техники теперь будет понятно, что указанные выше примеси в загрязненном пара-ксилоле могут оказывать наиболее сильное влияние на реакционную среду после накопления продуктов их частичного окисления в рециркулированном растворителе. Например, подача верхнего количества из наиболее предпочтительного интервала мета-ксилола, 400 масс.ч./млн, будет сразу же давать приблизительно 200 масс.ч./млн изофталевой кислоты в жидкой фазе реакционной среды при работе приблизительно с 33% масс. твердых веществ в реакционной среде. Это сравнимо с вводом из верхнего количества наиболее предпочтительного интервала для изофталевой кислоты в рециркулированном растворителе, 400 масс.ч./млн, которое после проведения типичного испарения растворителя для охлаждения реакционной среды, достигает приблизительно 1200 масс.ч./млн изофталевой кислоты в жидкой фазе реакционной среды. Следовательно, именно накопление продуктов частичного окисления в течение времени в рециркулированном растворителе является наиболее вероятным влиянием примесей мета-ксилола, орто-ксилола, этилбензола и толуола в загрузке загрязненного пара-ксилола. Соответственно, приведенные выше интервалы примесей в подаче загрязненного пара-ксилола предпочтительно поддерживать, по меньшей мере, полдня каждый день работы любой реакционной среды частичного окисления в конкретной промышленной установке, более предпочтительно, по меньшей мере, три четверти дня каждый день в течение, по меньшей мере, семи последовательных дней работы и наиболее предпочтительно, когда усредненная по массе композиция загрузки загрязненного пара-ксилола находится в пределах предпочтительных интервалов, по меньшей мере, в течение 30 последовательных дней работы.
Средства для получения загрязненного пара-ксилола предпочтительной чистоты уже известны в данной области техники и к ним относятся, но без ограничения, перегонка, методы дробной кристаллизации при температурах ниже температуры окружающей среды и методы на основе молекулярных сит с использованием селективной по размерам пор адсорбции. Однако предпочтительные интервалы степени чистоты, определенные в настоящем изобретении, на их высокой границе являются более затратными и дорогими, чем обычно имеет место при промышленных подачах пара-ксилола; и, к тому же, при низкой границе предпочтительные интервалы исключают чрезмерно дорогую очистку пара-ксилола для подачи в среду частичного окисления за счет обнаружения и раскрытия, где объединенное влияние самообразования примеси из самого пара-ксилола и уничтожающие примесь реакции в пределах реакционной среды становятся более важным, чем скорости подачи примесей в загрязненном пара-ксилоле.
Когда содержащий ксилол поток сырья содержит выбранные примеси, такие как этилбензол и/или толуол, окисление таких примесей может давать бензойную кислоту. Как используется в данном случае, определение «образованная из примесей бензойная кислота» будет означать бензойную кислоту, полученную из любого источника, отличного от ксилола, во время окисления ксилола.
Как установлено в данном изобретении, часть бензойной кислоты, выработанной во время окисления ксилола, получена из самого ксилола. Такая выработка бензойной кислоты из ксилола, очевидно, добавляется к любой части выработки бензойной кислоты, которая может быть образованной из примесей бензойной кислотой. Не привязываясь к какой-либо теории, полагают, что бензойная кислота получена из ксилола в реакционной среде, когда различные продукты промежуточного окисления ксилола самопроизвольно подвергаются декарбонилированию (потере монооксида углерода) или декарбоксилированию (потере диоксида углерода), в результате чего образуются арильные радикалы. Такие арильные радикалы затем могут отнимать атом водорода от одного из многих доступных источников в реакционной среде и давать самообразовавшуюся бензойную кислоту. Независимо от химического механизма определение «самообразовавшаяся бензойная кислота», используемое в данном случае, будет означать бензойную кислоту, полученную из ксилола при окислении ксилола.
Как обсуждалось выше, когда пара-ксилол окисляется с образованием терефталевой кислоты (ТФК), выработка самообразовавшейся бензойной кислоты приводит к потерям выхода по пара-ксилолу и к потерям выхода по окислителю. Кроме того, присутствие самообразовавшейся бензойной кислоты в жидкой фазе реакционной среды согласуется с ростом многих нежелательных побочных реакций, в особенности включающих образование интенсивно окрашенных соединений, называемых монокарбоксифлуоренонами. Самообразовавшаяся бензойная кислота также вносит вклад в нежелательное накопление бензойной кислоты в рециркулированном растворителе, что дополнительно повышает концентрацию бензойной кислоты в жидкой фазе реакционной среды. Следовательно, выработку самообразовавшейся бензойной кислоты желательно свести до минимума, но она также соответствующим образом одновременно учитывается с образованной из примесей бензойной кислотой, с факторами, влияющими на потребление бензойной кислоты, с факторами, относящимися к другим вопросам селективности реакции, и с суммарной экономикой процесса.
Установлено, что самообразовавшуюся бензойную кислоту можно контролировать на низких уровнях путем соответствующего выбора, например, температуры, распределения ксилола и доступности кислорода в пределах реакционной среды во время окисления. Без привязки к какой-либо теории, более низкие температуры и улучшенная доступность кислорода, оказывается, подавляют скорости декарбонилирования и/или декарбоксилирования, исключая, таким образом, аспект потери выхода из-за самообразовавшейся бензойной кислоты. Достаточная доступность, оказывается, направляет арильные радикалы на образование других менее опасных продуктов, в частности гидроксибензойных кислот. Распределение ксилола в реакционной среде также может влиять на баланс между конверсией арильного радикала до бензойной кислоты или до гидроксибензойных кислот. Независимо от химических механизмов, заявители раскрыли реакционные условия, которые, хотя и достаточно мягкие, чтобы снизить выработку бензойной кислоты, являются достаточно жесткими, чтобы окислить высокую долю произведенных гидроксибензойных кислот до монооксида углерода и/или диоксида углерода, которые легко удаляются из продукта окисления.
В предпочтительном варианте осуществления настоящего изобретения реактор окисления конфигурирован и работает таким образом, что образование самообразовавшейся бензойной кислоты является минимальным и окисление гидроксибензойных кислот до монооксида углерода и/или диоксида углерода сведено до минимума. Когда реактор окислении используется для окисления пара-ксилола до терефталевой кислоты, предпочтительно, чтобы пара-ксилол составлял, по меньшей мере, приблизительно 50% масс. от всех ксилолов в потоке сырья, введенного в реактор. Более предпочтительно, пара-ксилол составляет, по меньшей мере, приблизительно 75% масс. от всего ксилола в потоке сырья. Еще более предпочтительно, пара-ксилол составляет, по меньшей мере, приблизительно 95% масс. от всего ксилола в потоке сырья. Наиболее предпочтительно, пара-ксилол составляет по существу все количество ксилола в потоке сырья.
Когда реактор используется для окисления пара-ксилола до терефталевой кислоты, предпочтительно, чтобы скорость производства терефталевой кислоты была максимальной, тогда как скорость производства самообразовавшейся бензойной кислоты была минимальной. Предпочтительно, отношение скорости производства (по массе) терефталевой кислоты к скорости производства (по массе) самообразовавшейся бензойной кислоты составляет, по меньшей мере, приблизительно 500:1, более предпочтительно, по меньшей мере, приблизительно 1000:1 и наиболее предпочтительно, по меньшей мере, 1500:1. Как будет показано ниже, скорость производства самообразовавшейся бензойной кислоты предпочтительно измеряют, когда концентрация бензойной кислоты в жидкой фазе реакционной среды ниже 2000 масс.ч./млн, более предпочтительно ниже 1000 масс.ч./млн и наиболее предпочтительно ниже 500 масс.ч./млн, так как такие низкие концентрации подавляют до приемлемо низких скоростей реакции, по которым бензойная кислота превращается в другие соединения.
При объединении самообразовавшейся бензойной кислоты и образовавшейся из примесей бензойной кислоты отношение скорости производства (по массе) терефталевой кислоты к скорости производства (по массе) всей (самообразовавшейся и образовавшейся из примесей) бензойной кислоты предпочтительно составляет, по меньшей мере, приблизительно 400:1, более предпочтительно, по меньшей мере, приблизительно 700:1 и наиболее предпочтительно, по меньшей мере, 1100:1. Как будет показано ниже, суммированную скорость производства самообразовавшейся бензойной кислоты плюс образованной из примесей бензойной кислоты, предпочтительно измеряют, когда концентрация бензойной кислоты в жидкой фазе реакционной среды ниже 500 масс.ч./млн, так как такие низкие концентрации подавляют до приемлемо низких скоростей реакции, по которым бензойная кислота превращается в другие соединения.
Как показано в данной работе, повышенные концентрации бензойной кислоты в жидкой фазе реакционной среды приводят к повышенному образованию многих других ароматических соединений, некоторые из которых являются вредными примесями в ТФК; и, как показано в настоящем изобретении, повышенные концентрации бензойной кислоты в жидкой фазе реакционной среды приводят к повышенному образованию газообразных оксидов углерода, образование которых означает потерю выхода по окислителю и по ароматическим соединениям и/или растворителю. Более того, в настоящее время установлено, что значительная часть такого повышенного образования других ароматических соединений и оксидов углерода берет начало из реакций, в которых происходит превращение некоторых молекул самой бензойной кислоты, в отличие от бензойной кислоты, катализирующей другие реакции без ее собственного потребления. Соответственно, «чистое образование бензойной кислоты» определяется в данном случае как усредненная по времени масса всей бензойной кислоты, выходящей из реакционной среды, минус усредненная по времени масса всей бензойной кислоты, поступающей в реакционную среду, в течение одного и того же периода времени. Такое чистое образование бензойной кислоты часто является положительным, являясь результатом скоростей образования образованной из примесей бензойной кислоты и самообразовавшейся бензойной кислоты. Однако установлено, что скорость превращения бензойной кислоты до оксидов углерода и до некоторых других соединений, оказывается, растет приблизительно линейно по мере повышения концентрации бензойной кислоты в жидкой фазе реакционной среды, измеренной, когда другие реакционные условия, в том числе температура, доступность кислорода, СОС и реакционная активность, соответствующим образом поддерживаются постоянными. Следовательно, когда концентрация бензойной кислоты в жидкой фазе реакционной среды является достаточно высокой, вероятно из-за повышенной концентрации бензойной кислоты в рециркулированном растворителе, конверсия молекул бензойной кислоты в другие соединения, в том числе в оксиды углерода, может стать равной или больше, чем химическое образование новых молекул бензойной кислоты. В этом случае чистое образование бензойной кислоты может балансировать около нуля или даже быть отрицательным. Установлено, что когда чистое образование бензойной кислоты является положительным, отношение скорости производства (по массе) терефталевой кислоты в реакционной среде к скорости чистого образования бензойной кислоты в реакционной среде составляет предпочтительно больше чем приблизительно 700:1, более предпочтительно приблизительно больше чем 1100:1 и наиболее предпочтительно приблизительно 4000:1. Заявители установили, что когда чистое образование бензойной кислоты является отрицательным, отношение скорости производства (по массе) терефталевой кислоты в реакционной среде к скорости чистого образования бензойной кислоты в реакционной среде составляет предпочтительно больше чем приблизительно 200:(-1), более предпочтительно приблизительно больше чем 1000:(-1) и наиболее предпочтительно приблизительно 5000:(-1).
Заявители также раскрывают предпочтительные интервалы для композиции суспензии (жидкость + твердые вещества), выводимой из реакционной среды, и для части твердой СТК в суспензии. Композиции предпочтительной суспензии и предпочтительной СТК неожиданно являются более широкими и полезными. Например, очищенная ТФК, произведенная из такой предпочтительной СТК путем окислительного сжигания, имеет достаточно низкое содержание всех примесей и окрашенных примесей, поэтому очищенная ТФК приемлема без гидрирования дополнительного количества 4-КБА и/или окрашенных примесей для широкого спектра применений в ПЭТ волокнах и ПЭТ упаковках. Например, композиция предпочтительной суспензии дает жидкую фазу реакционной среды, которая имеет относительно низкую концентрацию важных примесей, и это существенно снижает образование других даже более нежелательных примесей, как это описано в настоящем изобретении. Кроме того, композиция предпочтительной суспензии очень способствует последующей обработке жидкости из суспензии, чтобы она становилась приемлемым чистым рециркулированным растворителем в соответствии с другими вариантами осуществления настоящего изобретения.
СТК, произведенная в соответствии с одним из вариантов осуществления настоящего изобретения, содержит меньше примесей установленных типов, чем СТК, произведенная с помощью обычных процессов и оборудования, особенно тех, в которых используется рециркулированный растворитель. Примеси, которые могут присутствовать в СТК, включают следующие соединения: 4-карбоксибензальдегид (4-КБА), 4,4'-дикарбоксистильбен (4,4'-ДКС, 4,4'-DCS), 2,6-дикарбоксиантрахинон (2,6-ДКА, 2,6-DCA), 2,6-дикарбоксифлуоренон (2,6-ДКФ), 2,7-дикарбоксифлуоренон (2,7-ДКФ), 3,5-дикарбоксифлуоренон (3,5-ДКФ), 9-флуоренон-2-карбоновая кислота (9F-2КК, 9F-2CA), 9-флуоренон-4-карбоновая кислота (9F-4КК, 9F-4CA), все флуореноны, включающие другие флуореноны, не перечисленные отдельно (все флуореноны), 4,4'-дикарбоксибифенил (4,4'-ДКБ), 2,5,4'-трикарбоксибифенил (2,5,4'-ТКБ, 2,5,4'-ТСВ), фталевая кислота (ФК, РА), изофталевая кислота (ИФК, IPA), бензойная кислота (БК, ВА), тримеллитовая кислота (ТМК, ТМА), пара-толуиловая кислота (ПТК, РТАС), 2,6-дикарбоксибензокумарин (2,6-ДКБК, 2,6-DCBC), 4,4'-дикарбоксибензил (4,4'-ДКБЗ, 4,4'-DCBZ), 4,4'-дикарбоксибензофенон (4,4'-ДКБФ, 4,4'-DCBР), 2,5,4'-трикарбоксибензофенон (2,5,4'-ТКБФ, 2,5,4'-ТСВР). В представленной ниже таблице 3 показаны предпочтительные количества этих примесей в СТК, произведенной в соответствии с вариантом осуществления настоящего изобретения.
Примеси в СТК
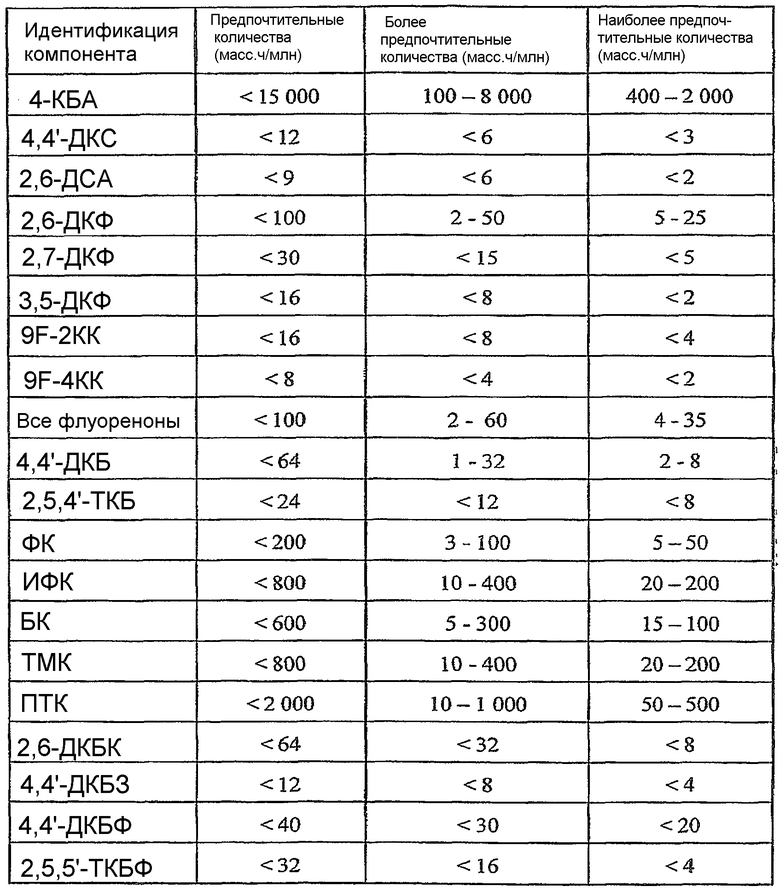
Кроме того, предпочтительно, чтобы СТК, произведенная в соответствии с вариантом осуществления настоящего изобретения, имела пониженное содержание окрашенных компонентов по сравнению с СТК, произведенной с помощью обычных процессов и оборудования, особенно тех, в которых используется рециркулированный растворитель. Следовательно, предпочтительно, чтобы СТК, произведенная в соответствии с одним из вариантов осуществления настоящего изобретения, имела процент пропускания при 340 нанометрах (нм), по меньшей мере, приблизительно 25%, более предпочтительно, по меньшей мере, приблизительно 50% и наиболее предпочтительно, по меньшей мере, 60%. Также предпочтительно, чтобы СТК, произведенная в соответствии с одним из вариантов осуществления настоящего изобретения, имела процент пропускания при 400 нм, по меньшей мере, приблизительно 88%, более предпочтительно, по меньшей мере, приблизительно 90% и наиболее предпочтительно, по меньшей мере, 92%.
Тест на процент пропускания дает меру измерения количества окрашенных, поглощающих свет примесей, присутствующих в ТФК или СТК. Используемый в данном случае тест относится к измерениям, выполненным на части раствора, приготовленного путем растворения 2,00 г сухой твердой ТФК или СТК в 20,0 мл диметилсульфоксида (ДМСО) аналитической чистоты или выше. Часть этого раствора помещают в полумикропроточную ячейку Hellma, PN 176.700, которая изготовлена из кварца и имеет длину пути 1,0 см и объем 0,39 мл (Hellma USA, 80 Skyline Drive, Plainview, NY 11803). Используют спектрофотометр Agilent 8453 Diode Array Spectrophotometer для измерения пропускания различных длин волн света через заполненную проточную ячейку (Agilent Technologies, 395 Page Mill Road, Palo Alto, CA 94303). После соответствующей коррекции поглощения относительно фона, в том числе, но без ограничения, ячейки и используемого растворителя данные процента пропускания, характеризующие долю падающего света, которая проходит через раствор, записывают непосредственно с помощью прибора. Значения процента пропускания при длинах волн 340 и 400 нм особенно полезны для отделения чистой ТФК от многих примесей, обычно находимых в ней.
Предпочтительные интервалы различных ароматических примесей в суспензионной фазе (твердые вещества + жидкость) реакционной среды представлены ниже в таблице 4.
Примеси в суспензии
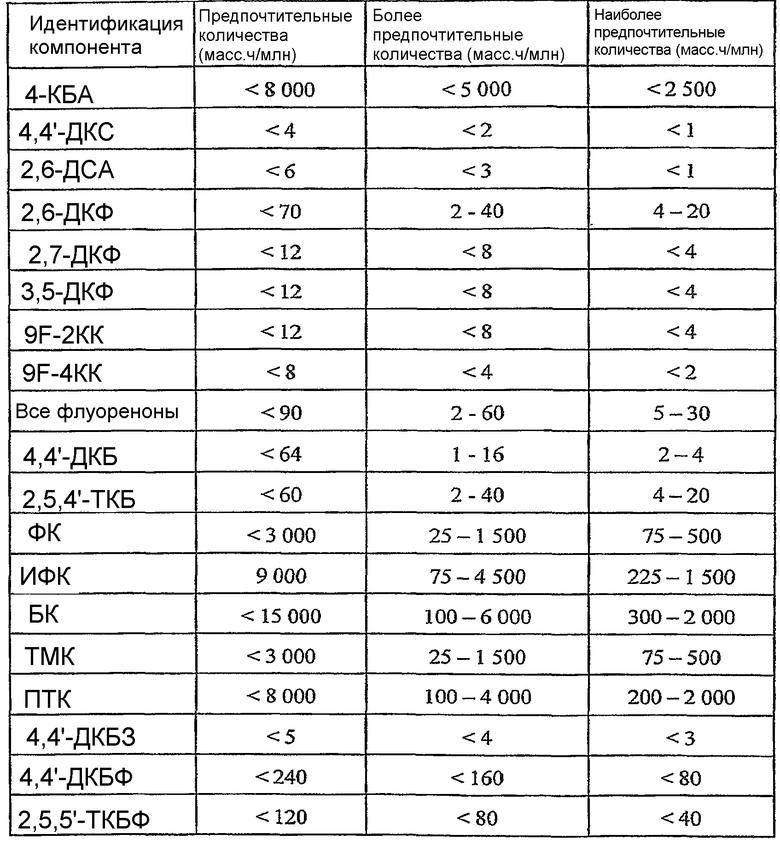
Такие предпочтительные композиции суспензии означают предпочтительную композицию жидкой фазы реакционной среды, исключая при этом экспериментальные проблемы, связанные с осаждением дополнительных компонентов жидкой фазы из реакционной среды в компоненты твердой фазы во время отбора образцов из реакционной среды, разделения жидкостей и твердых веществ и смещения аналитических условий.
Многие другие ароматические примеси, как правило, также присутствуют в суспензионной фазе реакционной среды и в СТК реакционной среды, и их количество обычно меняется при более низких уровнях и/или пропорционально по отношению к одному или нескольким раскрытым ароматическим соединениям. Контролирование раскрытых ароматических соединений в предпочтительных интервалах будет поддерживать другие ароматические примеси на приемлемых уровнях. Такие предпочтительные композиции суспензионной фазы в реакционной среде и в твердой СТК, отобранной непосредственно из суспензии, дают возможность работать с вариантами осуществления настоящего изобретения, описанными для частичного окисления пара-ксилола до ТФК.
Измерение концентрации компонентов, присутствующих на низком уровне, в растворителе, рециркулированном растворителе, СТК, суспензии из реакционной среды и ТФК проводятся с использованием методов жидкостной хроматографии. Далее описано два взаимозаменяемых варианта осуществления.
Способ, называемый ВЭЖХ-DAD, включает высокоэффективную жидкостную хроматографию (ВЭЖХ) в сочетании с детектированием с диодной матрицей, чтобы обеспечить разделение и количественную оценку различных видов молекул в данном образце. Прибор, используемый при проведении таких измерений, представляет собой модель 1100 ВЭЖХ, снабженную DAD (детектором с диодной матрицей), предлагаемым Agilent Technologies (Palo Alto, CA), хотя другие подходящие приборы также коммерчески доступны и от других поставщиков. Как известно в данной области техники, и время элюирования, и отклик детектора калибруют с использованием известных соединений, присутствующих в известных количествах, соединений и количеств, которые соответствуют соединениям и количествам, находящимся в неизвестных образцах.
Способ, называемый ВЭЖХ-МС, включает высокоэффективную жидкостную хроматографию (ВЭЖХ) в сочетании с масс-спектрометрией (МС), чтобы обеспечить разделение, идентификацию и количественную оценку различных видов молекул в данном образце. Прибор, используемый при проведении таких измерений, представляет собой Alliance HPLC и ZQ MS, предлагаемый Waters Corp. (Milford, MA), хотя другие подходящие приборы также коммерчески доступны и от других поставщиков. Как известно в данной области техники, и время элюирования, и масс-спектрометрический отклик калибруют с использованием известных соединений, присутствующих в известных количествах, соединений и количеств, которые соответствуют соединениям и количествам, находящимся в неизвестных образцах.
Другой вариант осуществления настоящего изобретения относится к частичному окислению ароматического, способного к окислению соединения при соответствующем уравновешивании подавления вредных ароматических примесей, с одной стороны, относительно производства монооксида углерода и диоксида углерода (вместе - оксиды углерода (СОх)), с другой стороны. Такие оксиды углерода, как правило, выходят из реакционной емкости в виде уходящего газа, и они означают деструктивную потерю растворителя и способного к окислению соединения, в том числе, конечно, и, в конечном итоге, предпочтительных окисленных производных (например, уксусной кислоты, пара-ксилола и ТФК). Заявителями выявлены более низкие границы для образования оксидов углерода, ниже которых, как оказывается, высокое образование вредных примесей, которые описаны ниже, и уровень низкой конверсии являются неизбежно слишком плохими для экономической применимости. Также раскрыты верхние границы диоксидов углерода, выше которых образование диоксидов углерода продолжает расти с небольшой дополнительной величиной, обеспеченной снижением образования вредных ароматических примесей.
Заявители также установили, что снижение жидкофазных концентраций загрузки способного к окислению ароматического соединения и ароматических промежуточных образцов в реакционной среде приводит к более низким скоростям образования вредных примесей во время реакции частичного окисления способного к окислению ароматического соединения. Такие вредные примеси включают сопряженные ароматические кольца и/или ароматические молекулы, содержащие больше желаемого числа число групп карбоновых кислот (например, при окислении пара-ксилола вредными примесями являются 2,6-дикарбоксиантрахинон, 2,6-дикарбоксифлуоренон, тримеллитовая кислота, 2,5,4'-трикарбоксибифенил и 2,5,4'-бензофенон). Ароматические промежуточные образцы включают ароматические соединения, перешедшие из загрузки способного к окислению соединения и все еще сохраняющие неароматические углеводородные группы (например, при окислении пара-ксилола ароматическими промежуточными образцами являются пара-толуиловый альдегид, терефталевый альдегид, пара-толуиловая кислота, 4-КБА, 4-гидроксиметилбензойная кислота и альфа-бром-пара-толуиловая кислота). Загрузка способного к окислению ароматического соединения и ароматические промежуточные образцы, сохраняющие неароматические углеводородные группы, когда они присутствуют в жидкой фазе реакционной среды, как оказывается, приводят к вредным примесям способом, аналогичным способу, уже описанному для растворенных ароматических образцов, не имеющих неароматических углеводородных групп (например, изофталевая кислота).
Противопоставив такую необходимость в более высокой реакционной активности для подавления образования вредных ароматических примесей во время частичного окисления способных к окислению ароматических соединений, заявители установили, что нежелательным сопутствующим результатом является повышенное производство оксидов углерода. Важно отметить, что такие оксиды углерода означают потерю выхода по способному к окислению соединения и окислителя, а не только растворителя. Понятно, что значительная, а иногда и принципиальная фракция, оксидов углерода поступает от способного к окислению соединения и его производных, а не от растворителя, и часто способное к окислению соединение стоит дороже из расчета на единицу углерода, чем растворитель. Кроме того, важно отметить, что желаемый продукт карбоновая кислота (например, ТФК) также подвергается избыточному окислению до оксидов углерода, когда присутствует в жидкой фазе реакционной среды.
Также важно отметить, что настоящее изобретение относится к реакциям в жидкой фазе реакционной среды и к концентрациям реагентов в ней. Это противоречит некоторым изобретениям предшествующего уровня техники, которые относятся непосредственно к образованию осажденного твердого вещества из ароматических соединений, сохраняющих неароматические углеводородные группы. Конкретно, в случае частичного окисления пара-ксилола до ТФК некоторые изобретения предшествующего уровня техники относятся к количеству 4-КБА, осажденного в твердой фазе СТК. Однако заявители настоящего изобретения раскрывают изменение более чем от двух к одному для отношения 4-КБА в твердой фазе к 4-КБА в жидкой фазе при использовании некоторого перечня температуры, давления, катализатора, композиции растворителя и объемной скорости реакции пара-ксилола, в зависимости от того, проводится ли частичное окисление в хорошо перемешиваемом автоклаве или в реакционной среде со ступенчатым изменением концентраций кислорода и пара-ксилола в соответствии с настоящим изобретением. Кроме того, заявители наблюдали, что отношение 4-КБА в твердой фазе к 4-КБА в жидкой фазе также может меняться в пределах от двух к одному или в хорошо перемешиваемой или ступенчатой реакционной среде, в зависимости от объемной скорости реакции пара-ксилола при другом аналогичном перечне температуры, давления, катализатора и композиции растворителя. Кроме того, 4-КБА в твердой фазе СТК, оказывается, не вносит вклад в образование вредных примесей, и 4-КБА в твердой фазе может быть выделен и окислен до ТФК и с высоким выходом (например, окислительным сжиганием суспензии СТК, как это описано в данной работе); тогда как удаление вредных примесей является значительно более трудным и дорогим, чем удаление твердофазного 4-КБА, а образование оксидов углерода означает постоянную потерю выхода. Таким образом, важно отличать, что данный аспект настоящего изобретения относится к композициям жидкой фазы в реакционной среде.
Независимо от того, является ли источником растворитель или способное к окислению соединение, заявители установили, что при конверсиях, приемлемых при промышленном применении, выработка оксидов углерода тесно соотносится с уровнем общей реакционной активности, несмотря на широкое изменение в определенной комбинации температуры металлов, галогенов, температуры, кислотности реакционной среды, измеренной по рН, концентрации воды, используемой, чтобы получить определенный уровень общей реакционной активности. Установлено, что при частичном окислении ксилола полезно повышать уровень общей реакционной активности с использованием концентрации в жидкой фазе толуиловой кислоты на средней высоте реакционной среды, внизу реакционной среды и вверху реакционной среды.
Таким образом, отсюда вытекает важность одновременного выравнивания, чтобы свести до минимума образование вредных примесей за счет повышения реакционной активности и чтобы также свести до минимума образование оксидов углерода за счет понижения реакционной активности. То есть если общее производство оксидов углерода подавляется слишком мало, то образуются избыточные уровни вредных примесей, и наоборот.
Кроме того, заявители установили, что растворимость и относительная реакционная способность целевой карбоновой кислоты (например, ТФК) и присутствие других растворенных ароматических образцов, не имеющих неароматических углеводородных групп, дает очень важную точку опоры для такого выравнивания оксидов углерода относительно вредных примесей. Целевой продукт карбоновая кислота, как правило, растворена в жидкой фазе реакционной среды, даже когда присутствует в твердой форме. Например, при температурах в предпочтительном интервале ТФК растворяется в реакционной среде, содержащей уксусную кислоту и воду, в количествах, находящихся в интервале приблизительно от одной тысячной масс.ч./млн до свыше 1% масс., причем растворимость повышается с ростом температуры. Хотя существуют различия в скоростях реакций в направлении образования различных вредных примесей из загрузки способного к окислению ароматического соединения (например, пара-ксилола), из ароматических промежуточных соединений (например, пара-толуиловой кислоты), из целевого продукта ароматической карбоновой кислоты (например, ТФК) и из ароматических образцов, не имеющих неароматических углеводородных групп (например, изофталевой кислоты), присутствие и реакционная способность двух последних групп определяют область убывающих ответов относительно дополнительного подавления первых двух групп, способного к окислению ароматического соединения и ароматических промежуточных реакционных соединений. Например, при частичном окислении пара-ксилола до ТФК, если растворенная ТФК достигает до 7000 масс.ч./млн в жидкой фазе реакционной среды при данных условиях, растворенная бензойная кислота достигает до 8000 масс.ч./млн, растворенная изофталевая кислота достигает до 6000 масс.ч./млн и растворенная фталевая кислота достигает до 2000 масс.ч./млн, то важность снижения уровня всех вредных соединений начинает убывать по мере повышения реакционной активности для подавления жидкофазной концентрации пара-толуиловой кислоты и 4-КБА ниже подобных уровней. То есть присутствие и концентрация в жидкой фазе реакционной среды ароматических образцов, не имеющих ароматических углеводородных групп, меняется очень незначительно за счет повышения реакционной активности, и их присутствие служит для расширения по направлению вверх области убывающих ответов для снижения концентрации реакционных промежуточных соединений, чтобы подавить образование вредных примесей.
Следовательно, один из вариантов осуществления настоящего изобретения предлагает предпочтительные интервалы оксидов углерода (монооксида углерода и диоксида углерода), ограниченных на нижнем конце низкой реакционной активностью и чрезмерным образованием вредных примесей, а на верхнем конце избыточными потерями углерода, но на уровнях, более низких, чем ранее раскрыты и описаны как полезные при промышленной реализации. Соответственно, образование оксидов углерода предпочтительно контролировать следующим образом. Отношение молей всех произведенных оксидов углерода к молям поданного способного к окислению ароматического соединения предпочтительно находится в интервале приблизительно от 0,02:1 до 0,25:1, более предпочтительно в интервале приблизительно от 0,04:1 до 0,22:1, еще более предпочтительно в интервале приблизительно от 0,05:1 до 0,19:1 и наиболее предпочтительно в интервале от 0,06:1 до 0,15:1. Отношение молей образованного диоксида углерода к молям поданного способного к окислению ароматического соединения предпочтительно находится в интервале приблизительно от 0,01:1 до 0,21:1, более предпочтительно в интервале приблизительно от 0,03:1 до 0,19:1, еще более предпочтительно в интервале приблизительно от 0,04:1 до 0,16:1 и наиболее предпочтительно в интервале от 0,05:1 до 0,11:1. Отношение молей произведенного монооксида углерода к молям поданного способного к окислению ароматического соединения предпочтительно находится в интервале приблизительно от 0,005:1 до 0,09:1, более предпочтительно в интервале приблизительно от 0,01:1 до 0,07:1, еще более предпочтительно в интервале приблизительно от 0,015:1 до 0,05:1 и наиболее предпочтительно в интервале от 0,02:1 до 0,4.
Содержание диоксида углерода в сухом выходящем газе реактора окисления предпочтительно находится в интервале приблизительно от 0,1 до 1,5% мол., более предпочтительно в интервале приблизительно от 0,20 до 1,2% мол., еще более предпочтительно в интервале приблизительно от 0,25 до 0,9% мол. и наиболее предпочтительно в интервале от 0,30 до 0,8% мол. Содержание монооксида углерода в сухом выходящем газе реактора окисления предпочтительно находится в интервале приблизительно от 0,05 до 0,6% мол., более предпочтительно в интервале приблизительно от 0,10 до 0,5% мол., еще более предпочтительно в интервале приблизительно от 0,15 до 0,35% мол. и наиболее предпочтительно в интервале от 0,18 до 0,28% мол.
Заявители установили, что значимым фактором для уменьшения производства оксидов углерода до предпочтительных интервалов является улучшение чистоты рециркулированного растворителя и загрузки способного к окислению соединения, чтобы понизить концентрацию ароматических соединений, не имеющих неароматических углеводородных групп, в соответствии с установками настоящего изобретения - это одновременно снижает образование оксидов углерода и вредных примесей. Другим фактором является улучшение распределения пара-ксилола и окислителя в пределах реакционной емкости в соответствии с установками настоящего изобретения. Другие факторы, обеспечивающие приведенные выше предпочтительные уровни оксидов углерода, состоят в работе при градиентах в реакционной среде, как это описано в данном изобретении, для давления, температуры, концентрации способного к окислению соединения в жидкой фазе и для окислителя в газовой фазе. Другие факторы, обеспечивающие приведенные выше предпочтительные уровни оксидов углерода, состоят в работе в рамках открытий в данной заявке для предпочтительной объемной скорости реакции, давления, температуры, композиции растворителя, композиции катализатора и механической геометрии реакционной емкости.
Одно из возможных преимуществ работы в пределах предпочтительных интервалов образования оксида углерода состоит в том, что применение молекулярного кислорода может быть уменьшено, хотя и не до стехиометрических значений. Несмотря на хорошее ступенчатое изменение окислителя и способного к окислению соединения в соответствии с настоящим изобретением, избыток кислорода должен поддерживаться выше стехиометрического значения, которое рассчитано для загрузки способного к окислению соединения отдельно, чтобы допустить некоторые потери оксидов углерода и создать избыток молекулярного кислорода для контроля образования вредных примесей. Конкретно для случая, когда загрузка способного к окислению соединения представляет собой ксилол, отношение подачи массы молекулярного кислорода к массе ксилола предпочтительно находится в интервале приблизительно от 0,9:1 до 5:1, более предпочтительно в интервале приблизительно от 0,95:1 до 1,3:1 и наиболее предпочтительно в интервале от 1:1 до 1,15:1. Конкретно для загрузки ксилола усредненное по времени содержание молекулярного кислорода в сухом выходящем газе реактора окисления предпочтительно находится в интервале приблизительно от 0,1 до 6% мол., более предпочтительно в интервале приблизительно от 1 до 2% мол. и наиболее предпочтительно в интервале от 1,5 до 3% мол.
Другое возможное преимущество работы в пределах предпочтительных интервалов образования оксида углерода состоит в том, что меньшее количество ароматического соединения превращается в оксиды углерода и другие, менее полезные формы. Такое преимущество оценивается с использованием суммы молей всех ароматических соединений, выходящих из реакционной среды, поделенной на сумму молей всех ароматических соединений, поступающих в реакционную среду, в течение непрерывного периода времени, предпочтительно в течение одного часа, более предпочтительно в течение одного дня и наиболее предпочтительно в течение 30 последовательных дней. Такое отношение далее в настоящем изобретении называется «мольным отношение выживания» для ароматических соединений при прохождении через реакционную среду и выражается в числовых процентах. Если все поступающие ароматические соединения выходят из реакционной среды в виде ароматических соединений, которые все преимущественно находятся в окисленной форме поступающих ароматических соединений, то мольное отношение выживания имеет максимальное значение 100%. Если только 1 из каждых 100 поступающих ароматических молекул превращается в оксид углерода и/или другие неароматические молекулы (например, в уксусную кислоту), пока они проходят через реакционную среду, то мольное отношение выживания составляет 99%. Конкретно для случая, когда основная загрузка способного к окислению соединения состоит из ксилола, мольное отношение выживания для ароматического соединения при прохождении через реакционную среду предпочтительно находится в интервале приблизительно от 98 до 99,9%, более предпочтительно в интервале приблизительно от 98,5 до 99,8% и наиболее предпочтительно в интервале от 99,0 до 99,7%.
Другой аспект рассматриваемого изобретения включает выработку метилацетата в реакционной среде, содержащей уксусную кислоту и одно или несколько способных к окислению ароматических соединений. Такой метилацетат является относительно летучим по сравнению с водой и уксусной кислотой и, следовательно, имеет тенденцию следовать за выходящим газом, если дополнительное охлаждение или другие рабочие узлы не используются для его выделения и/или для его разрушения перед высвобождением выходящего газа обратно в окружающую среду. Образование метилацетата, таким образом, означает эксплуатационные расходы, а также капитальные затраты. По-видимому, метилацетат образуется вначале за счет объединения метильного радикала, возможно, образовавшегося вследствие разложения уксусной кислоты, с кислородом, с получением метилгидроксипероксида, за счет последующего разложения с образованием метанола и, наконец, за счет взаимодействия полученного метанола с оставшейся уксусной кислотой с образованием метилацетата. Вне зависимости от химического пути образования, как установили заявители, когда скорость выработки метилацетата является слишком низкой, образование оксидов углерода также является слишком низким, а образование вредных ароматических примесей слишком высоким. Если метилацетат образуется со слишком высокой скоростью, образование оксидов углерода также является излишне высоким, что приводит к потерям выхода по растворителю, способному к окислению, соединению и окислителю. При использовании предпочтительных вариантов осуществления изобретения отношение количества молей произведенного метилацетата к количеству молей поданного способного к окислению ароматического соединения предпочтительно находится в интервале приблизительно от 0,005:1 до 0,09:1, более предпочтительно в интервале приблизительно от 0,01:1 до 0,07:1 и наиболее предпочтительно в интервале от 0,02:1 до 0,04:1.
Когда образование диоксида углерода, монооксида углерода, их образование в сумме и/или образование метилацетата находятся ниже предпочтительных интервалов, раскрытых в настоящем изобретении, или когда мольное отношение выживания ароматических соединений находится выше предпочтительных интервалов, раскрытых в настоящем изобретении, должна быть повышена реакционная активность или должна быть понижена СОС. Один усилитель активности повышает температуру в пределах предпочтительных интервалов, описанных в настоящем изобретении. Другой усилитель активности повышает каталитическую активность, которая обеспечивается смесью каталитических химикатов и растворителя. Обычно повышение концентраций кобальта и/или брома будет усиливать реакционную активность, если они используются в рамках предпочтительных интервалов. Регулирование концентрации в пределах реакционной среды других каталитических компонентов и воды также может быть использовано для усиления реакционной активности. СОС понижают за счет снижения скорости подачи способного к окислению соединения и/или за счет повышения объема реакционной среды.
Когда образование диоксида углерода, монооксида углерода, их образование в сумме и/или образование метилацетата составляют больше, чем описанные предпочтительные интервалы, и/или когда мольное отношение выживания ароматических соединений находится выше предпочтительных интервалов, раскрытых в настоящем изобретении, предпочтительные контролирующие действия являются противоположными приведенным выше действиям, также в предпочтительных интервалах, описанных в настоящем изобретении. Отмечено, что особенно полезно повышать СОС насколько это возможно в описанных интервалах, сохраняя при этом хорошее качество окисления, которое оценивается по вредным примесям в СТК и в реакционной среде. Также отмечено, что трудно сохранить такое качество окисления при такой высокой СОС и что очень большая осторожность необходима по следующим показателям: распределение сырья, поступающего в реакционную среду; качество аэрации по всей реакционной среде; деаэрация на выходе из реакционной среды; СОС кислорода и растворенный кислород по всей реакционной среде; избыток окислителя, выходящего из реакционной среды; желаемый пространственный градиент СОС кислорода; желаемый пространственный градиент концентрации способного к окислению соединения; желаемый пространственный градиент концентрации окислителя; головное давление; желаемый пространственный градиент давления и предпочтительная температура на середине высоты реакционной смеси, и как это все обсуждено в данном описании. Дополнительно и с целью достижения более низких количеств диоксида углерода, монооксида углерода и/или их суммы и/или чтобы повысить мольное отношение выживания для ароматических соединений, заявители установили, что полезно подавлять в пределах реакционной зоны концентрацию растворимых ароматических соединений, не имеющих неароматических углеводородных групп (например, изофталевой кислоты, фталевой кислоты и бензойной кислоты); такое подавление может быть реализовано за счет использования более чистого сырья способного к окислению соединения и/или более чистого растворителя, особенно в пределах предпочтительных интервалов, описанных в настоящем изобретении для каждого компонента.
В реакционной среде, где непрерывно окисляется пара-ксилол до терефталевой кислоты при повышенной СОС, описанной в настоящем изобретении, предпочтительно, чтобы количество пара-толуиловой кислоты в жидкой фазе реакционной среды, поддерживалось в интервале приблизительно от 200 до 10000 масс.ч./млн, более предпочтительно приблизительно от 800 до 8000 масс.ч./млн и наиболее предпочтительно от 1600 до 6000 масс.ч./млн. Кроме того, конверсия пара-ксилола до терефталевой кислоты в реакционной среде предпочтительно поддерживается приблизительно выше 50% мол., более предпочтительно приблизительно выше 90% мол., еще более предпочтительно приблизительно выше 95% мол. и наиболее предпочтительно свыше 97% мол.
В одном из вариантов настоящего изобретения предпочтительно, чтобы один или несколько рабочих параметров, описанных в изобретении (в том числе количественно определенных численными значениями рабочих параметров), удерживались в течение значительного с промышленной точки зрения периода времени. Предпочтительно работа в соответствии с одним или несколькими описанными выше рабочими параметрами поддерживается, по меньшей мере, в течение приблизительно 1 часа, более предпочтительно, по меньшей мере, приблизительно 12 часов, еще более предпочтительно, по меньшей мере, приблизительно 36 часов и наиболее предпочтительно, по меньшей мере, 96 часов. Следовательно, если не указано особо, рабочие параметры, описанные в данном случае, как подразумевается, применимы к работе в стационарном, оптимальном/промышленном режиме - в отсутствие операций запуска, остановки или частичной оптимизации.
Следует отметить, что для всех численных интервалов, представленных в настоящем изобретении, верхняя и нижняя границы интервалов могут быть независимыми друг от друга. Например, численный интервал от 10 до 100 означает больше чем 10 и/или меньше чем 100. Следовательно, интервал от 10 до 100 обеспечивает поддержку для заявленного ограничения больше чем 10 (без верхней границы), для заявленного ограничения меньше чем 100 (без нижней границы), а также для всего интервала от 10 до 100 (как с верхней границей, так и с нижней границей). Кроме того, когда определение «приблизительно» используется для модификации численного значения, следует понимать, что в одном из вариантов осуществления изобретения численное значение является точным численным значением.
Изобретение описано подробно с помощью конкретных ссылок на предпочтительные варианты его осуществления, но необходимо понимать, что могут быть осуществлены изменения и модификации в рамках сути и объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИСТЕМА ПОЛУЧЕНИЯ ПОЛИКАРБОНОВОЙ КИСЛОТЫ, ИСПОЛЬЗУЮЩАЯ ОХЛАЖДЕННЫЙ МАТОЧНЫЙ РАСТВОР ИЗ ОКИСЛИТЕЛЬНОГО СЖИГАНИЯ В КАЧЕСТВЕ ЗАГРУЗКИ СИСТЕМЫ ОЧИСТКИ ОТ ЗАГРЯЗНЕНИЙ | 2007 |
|
RU2458907C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2384563C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388743C2 |
| СИСТЕМА ОКИСЛЕНИЯ, ИСПОЛЬЗУЮЩАЯ ВНУТРЕННЮЮ КОНСТРУКЦИЮ ДЛЯ УЛУЧШЕНИЯ ГИДРОДИНАМИКИ | 2006 |
|
RU2418629C2 |
| СИСТЕМА ОКИСЛЕНИЯ С ВНУТРЕННИМ ВТОРИЧНЫМ РЕАКТОРОМ | 2006 |
|
RU2448766C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2382758C2 |
| СОСТАВ СЫРОЙ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2388744C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2006 |
|
RU2435753C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388745C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2381212C2 |
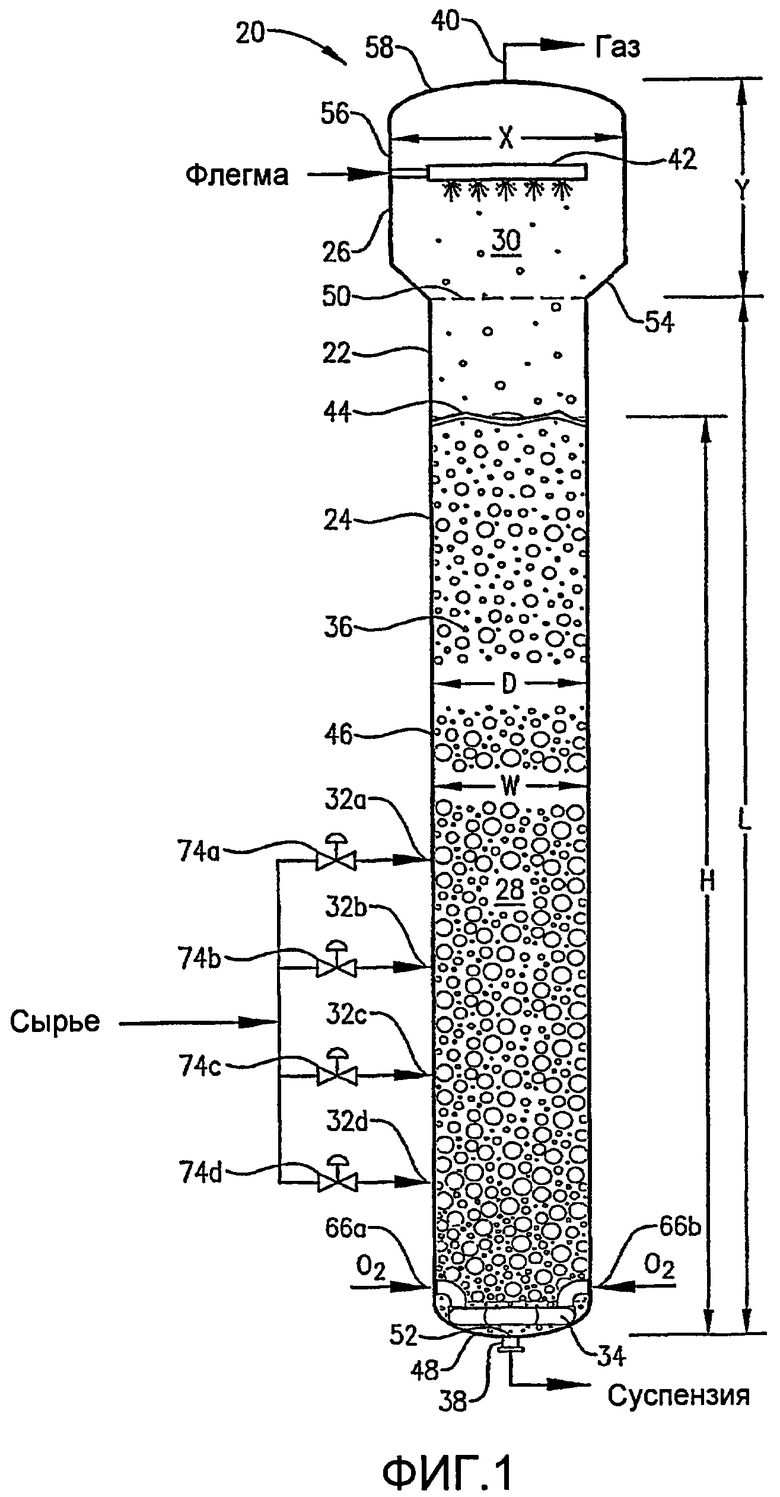
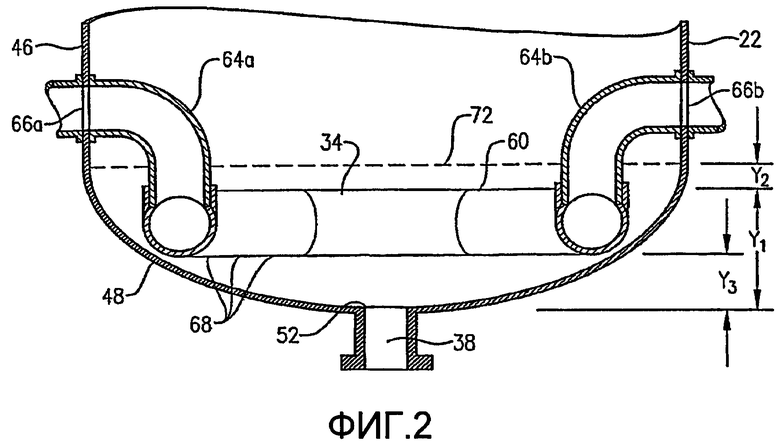
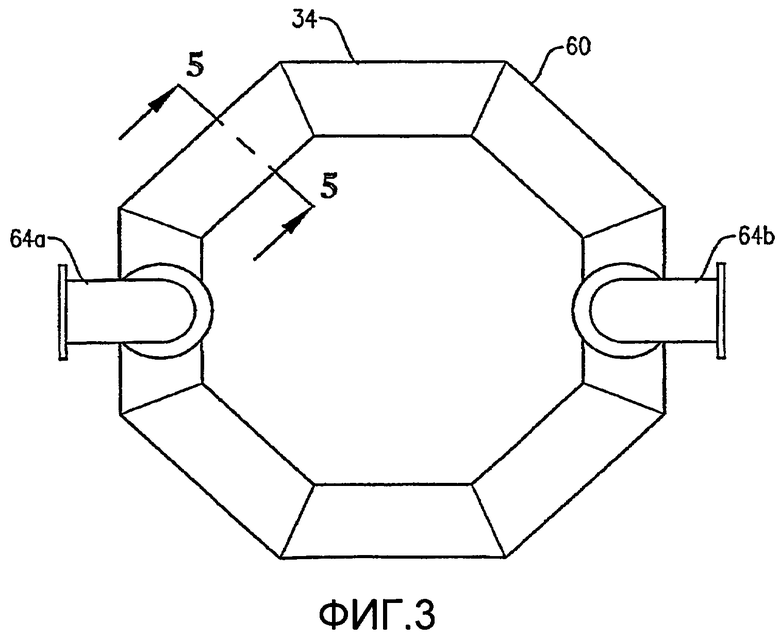
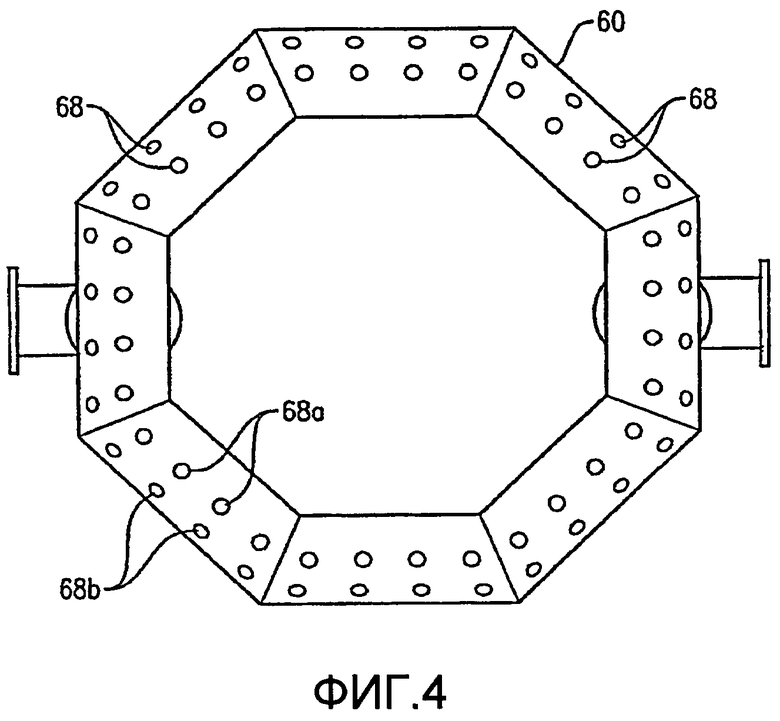
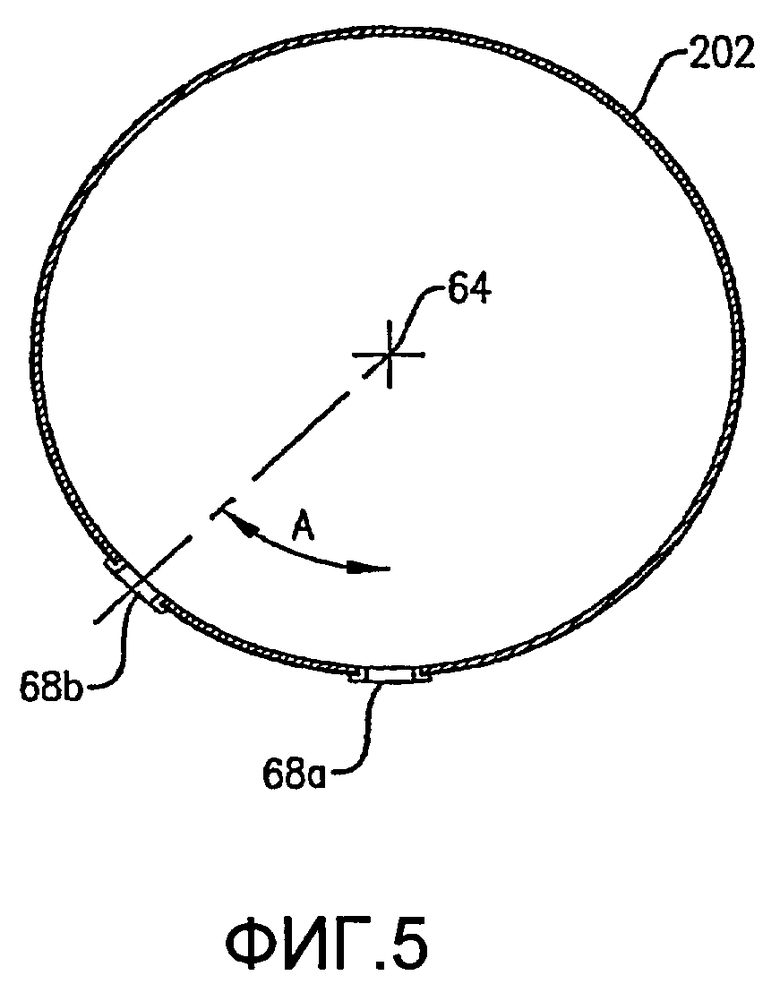
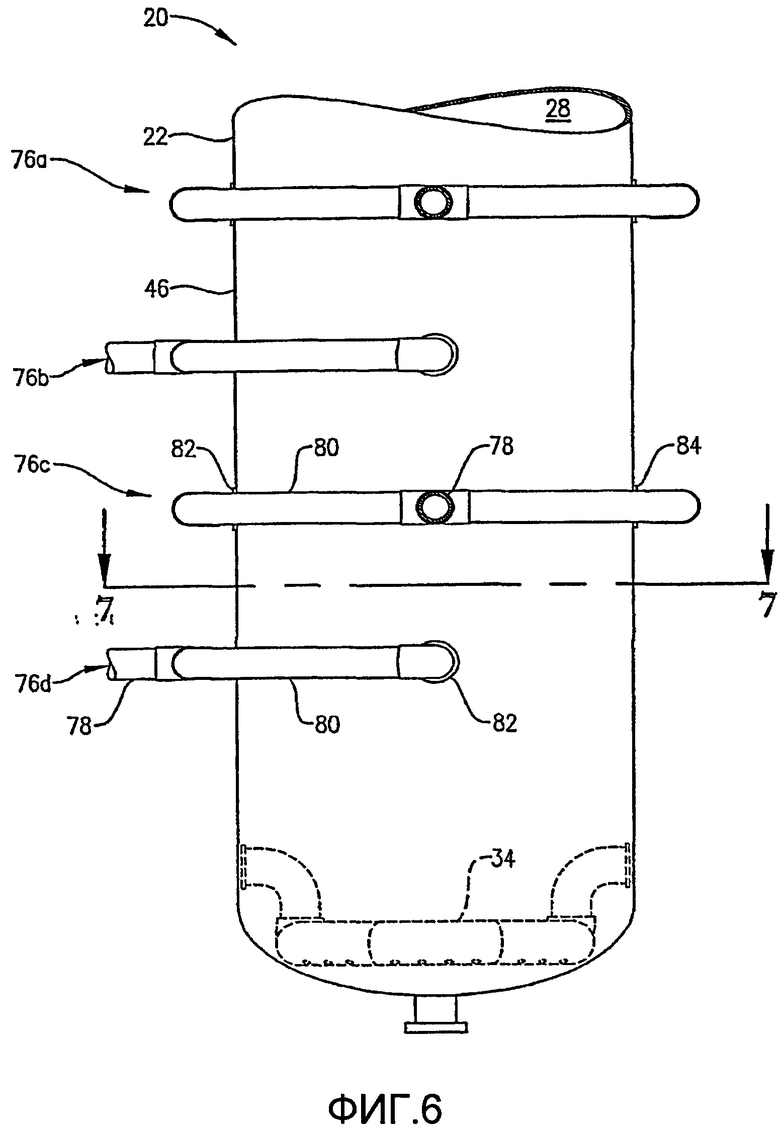
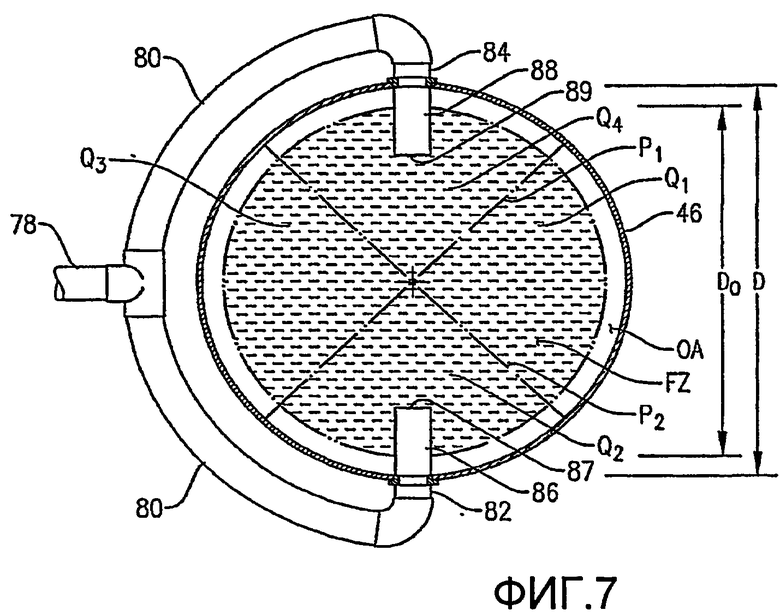
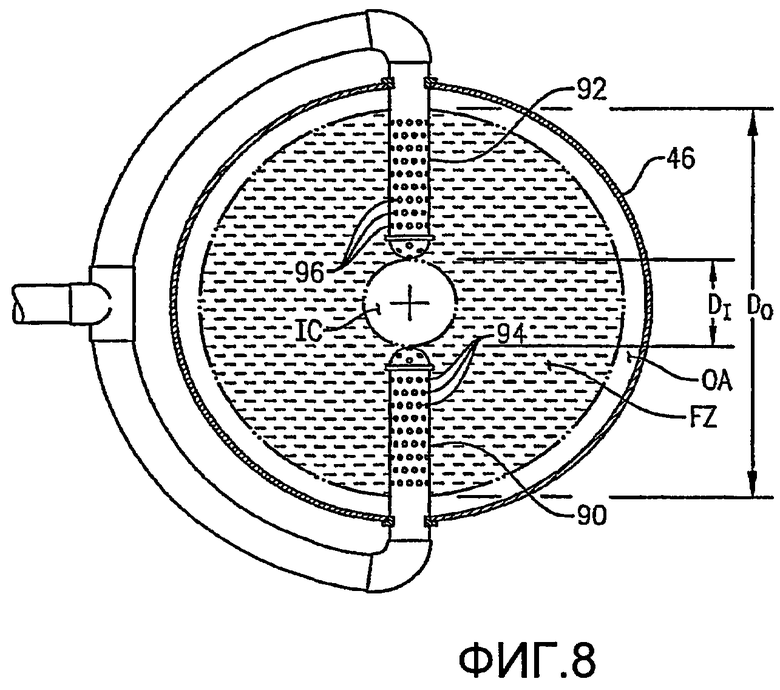
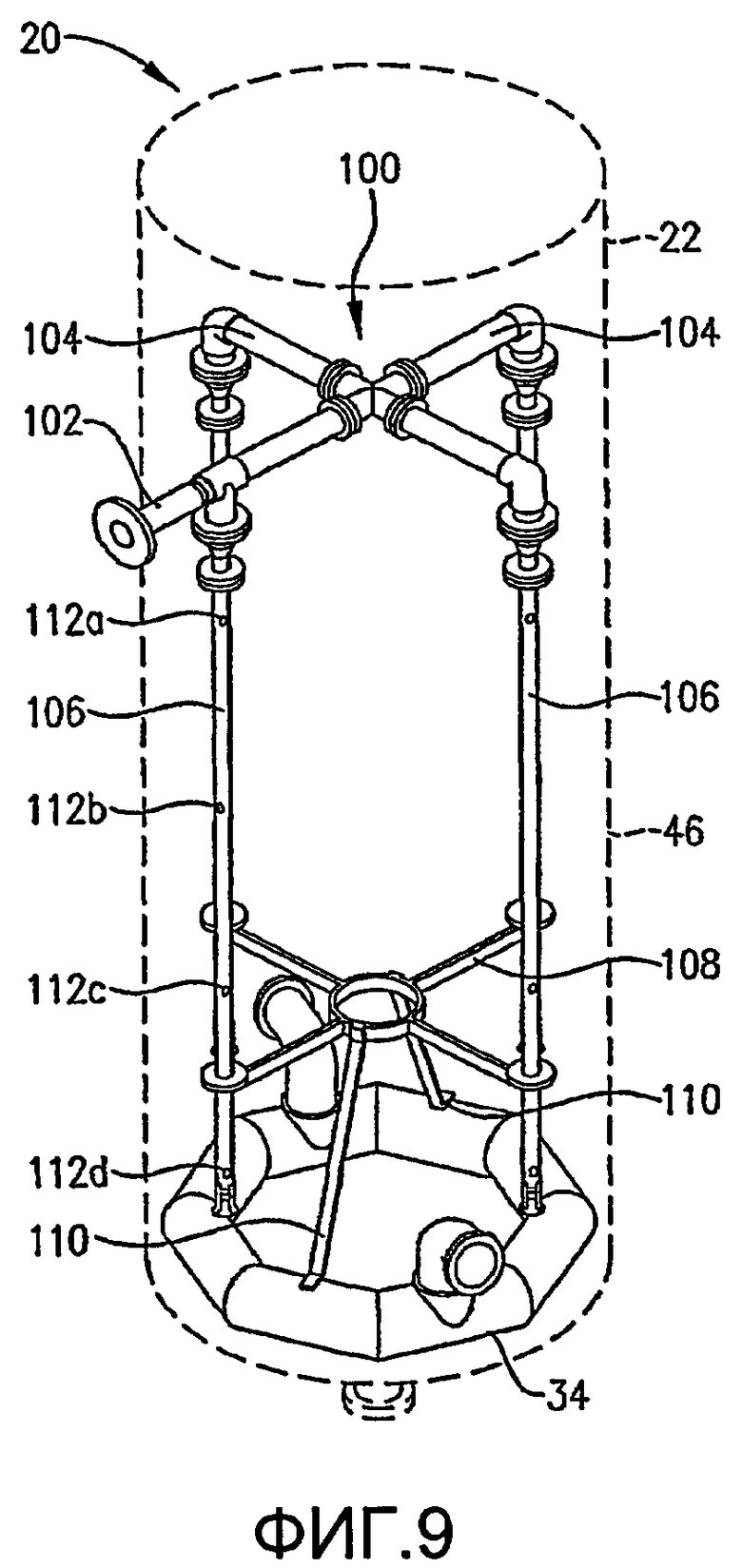
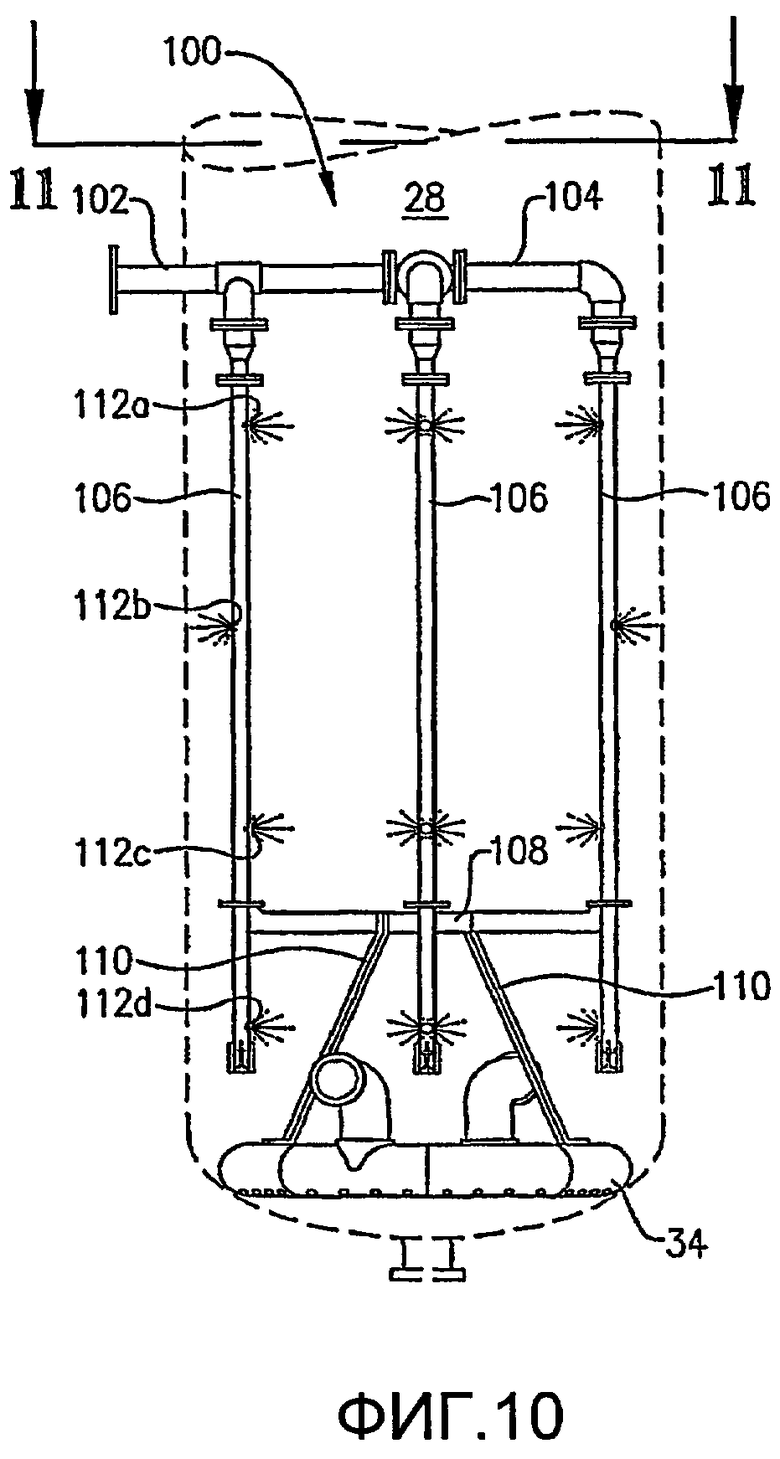
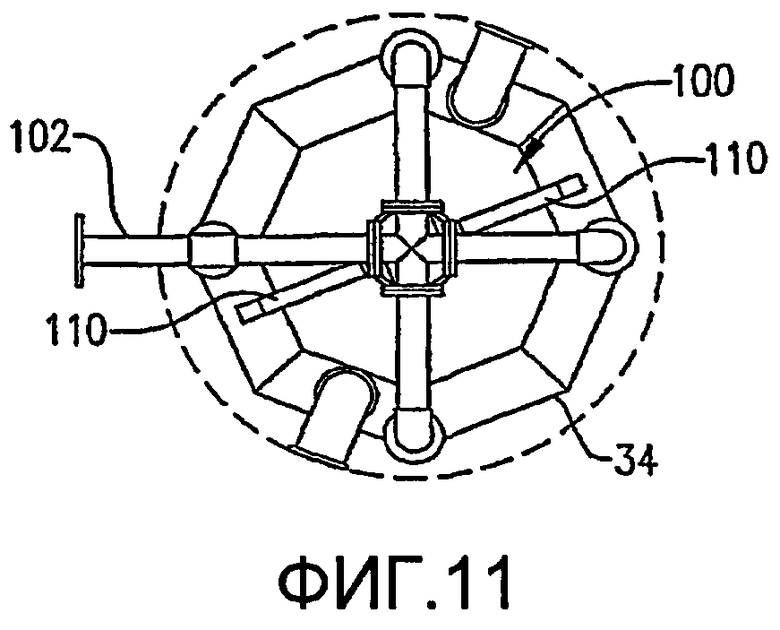
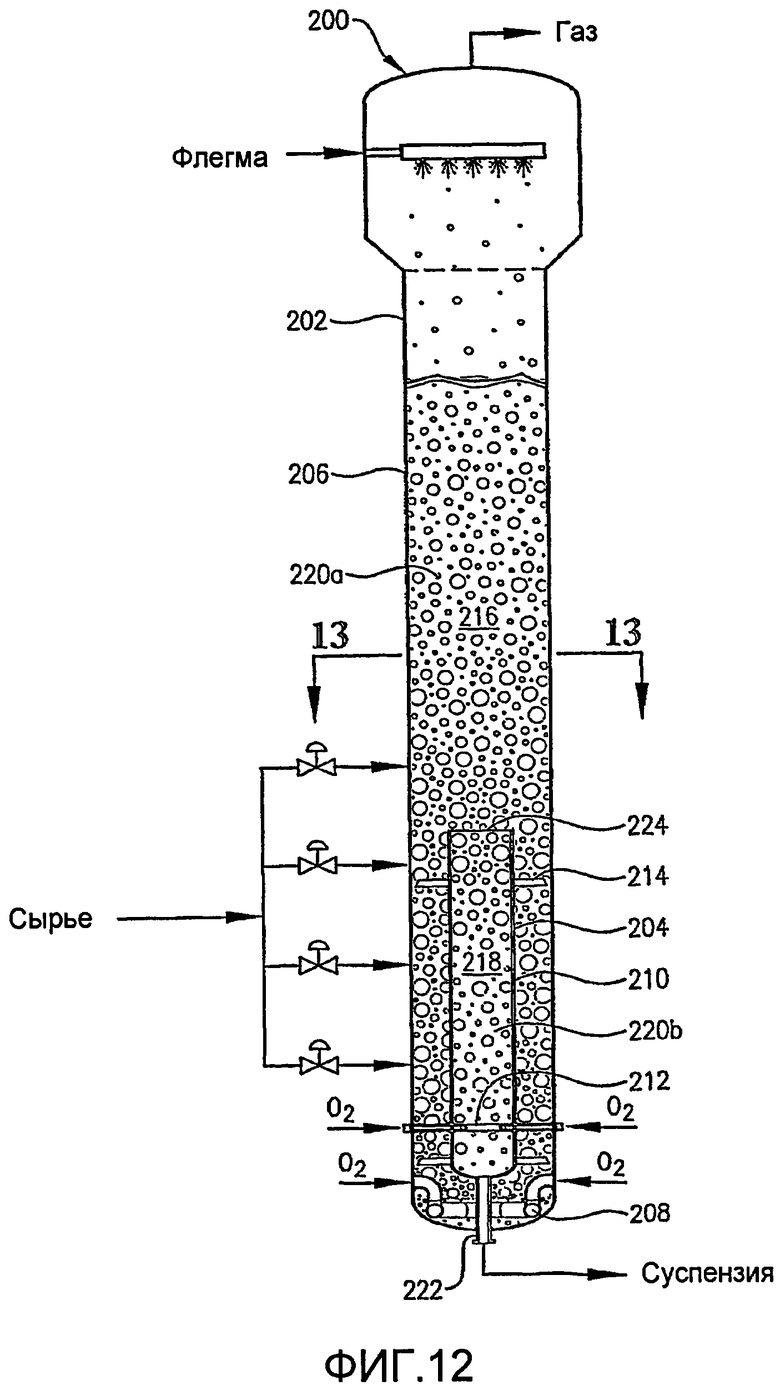
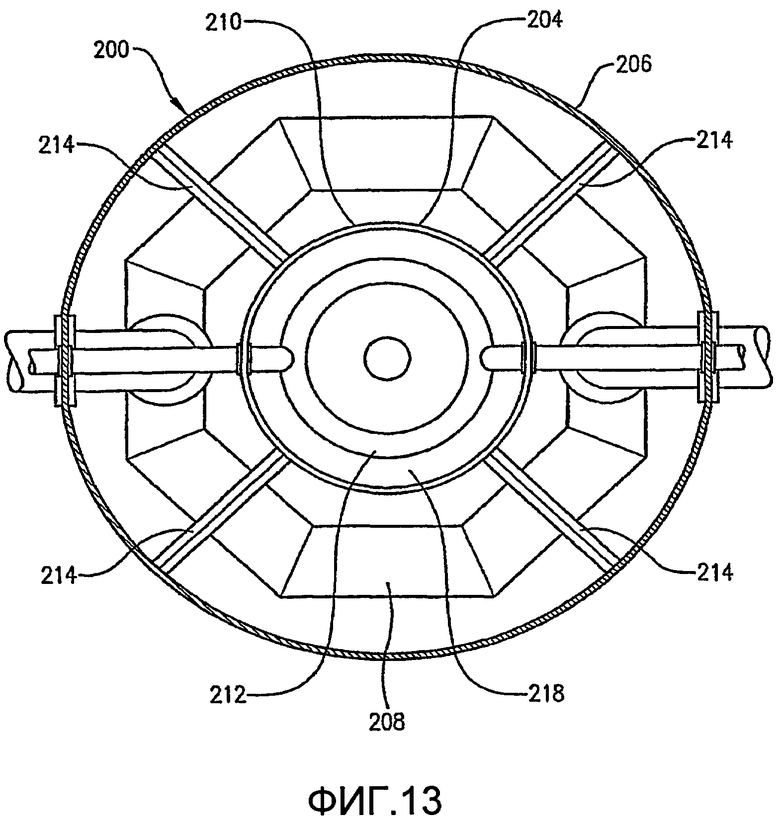
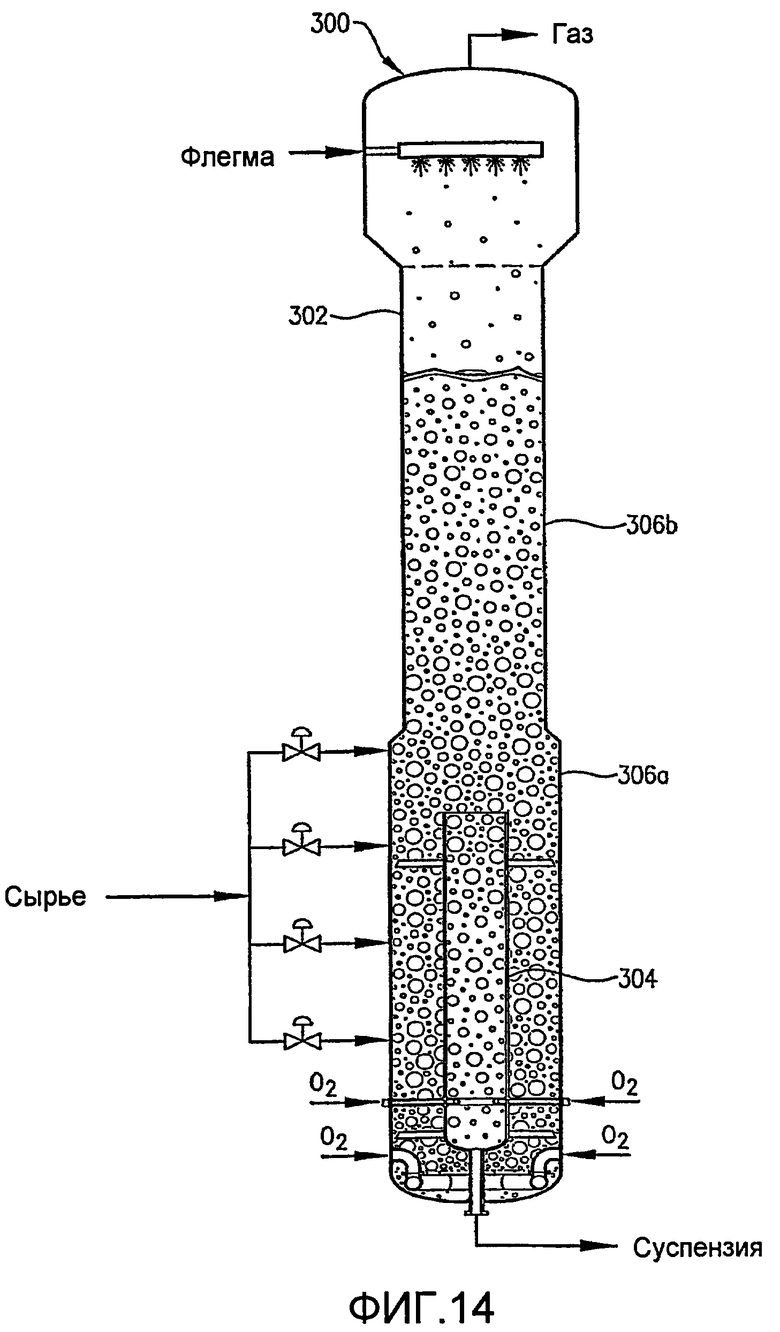
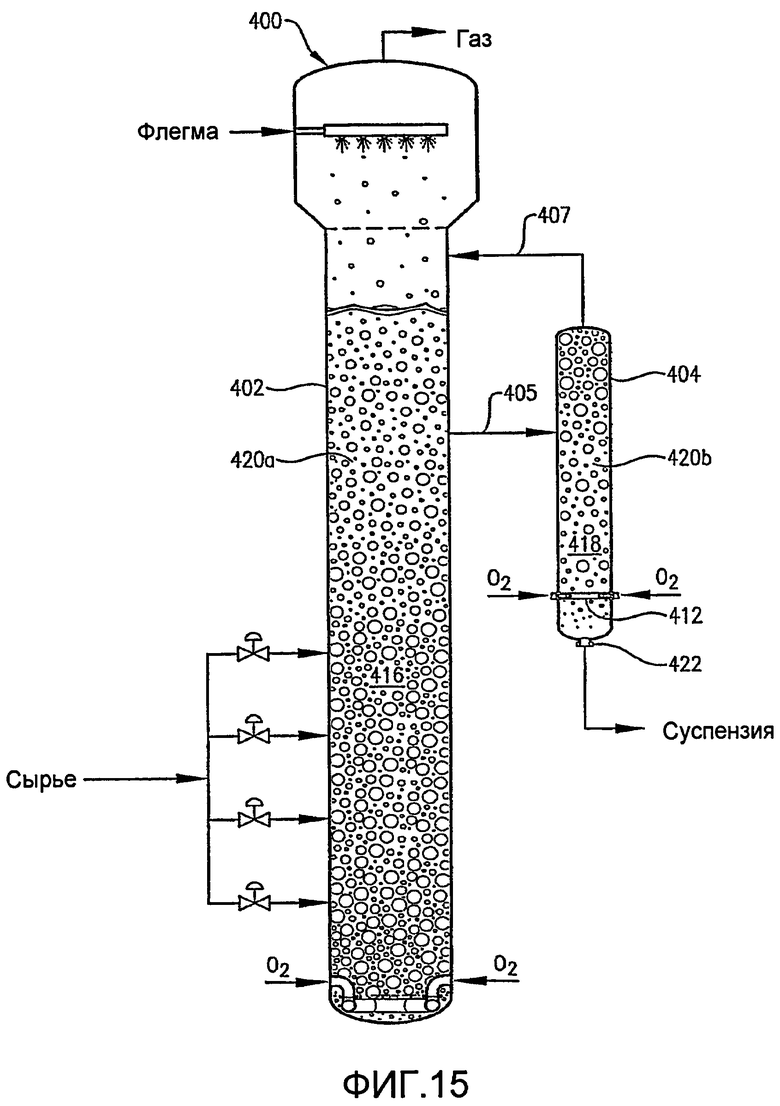
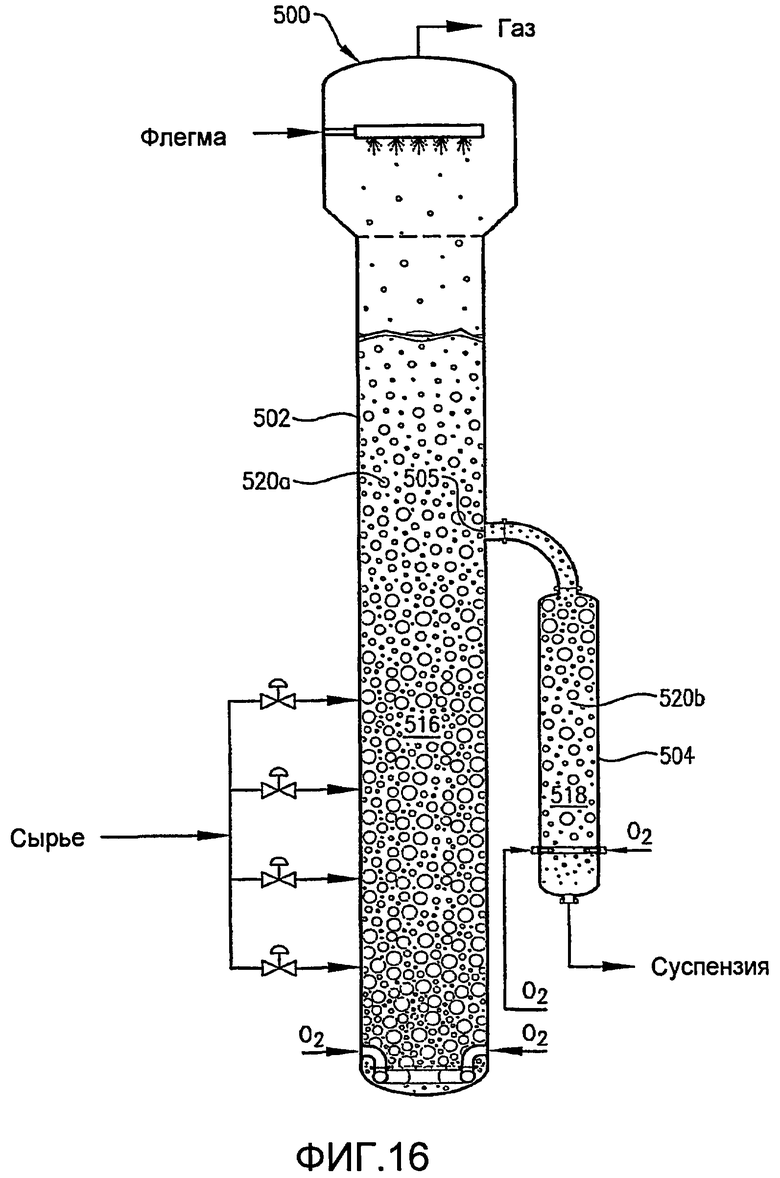
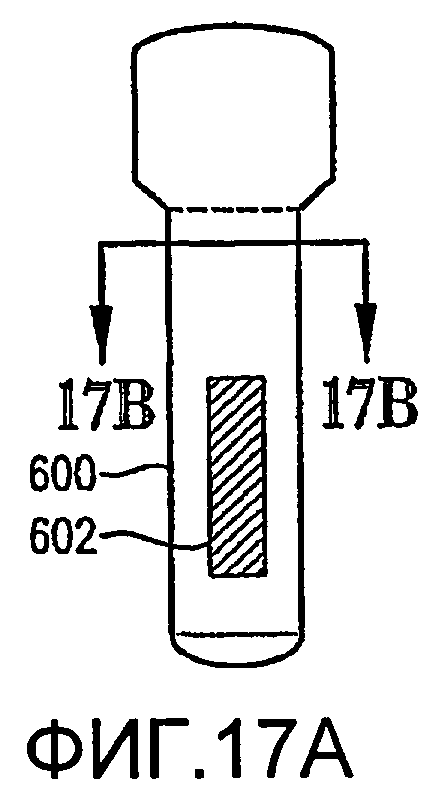



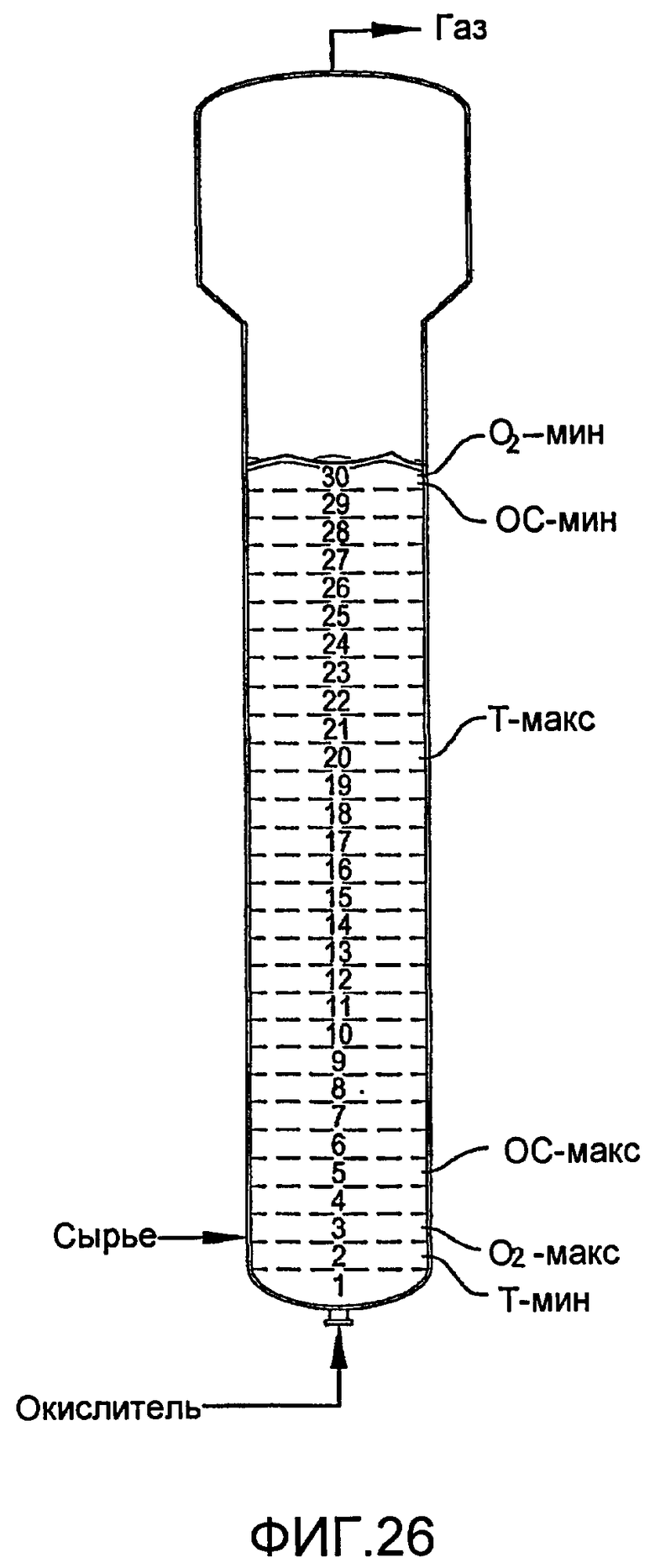
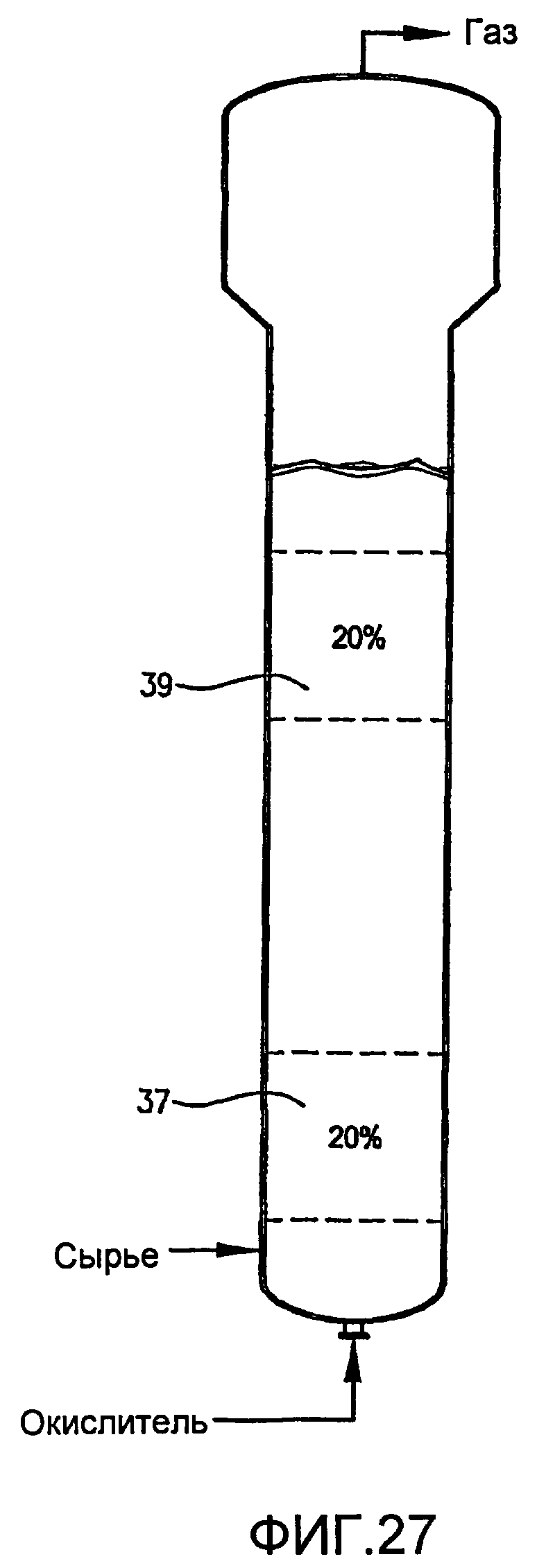





Изобретение относится к усовершенствованному способу получения композиции ароматической дикарбоновой кислоты, включающему (а) проведение окисления многофазной реакционной среды в реакторе первичного окисления с получением в результате первой суспензии; (b) проведение дополнительного окисления, по меньшей мере, части указанной первой суспензии в реакторе вторичного окисления, где указанный реактор вторичного окисления представляет собой реактор по типу барботажной колонны, причем способ дополнительно включает введение ароматического соединения в указанный реактор первичного окисления, где, по меньшей мере, приблизительно 80% мас. указанного ароматического соединения, введенного в указанный реактор первичного окисления, окисляется в указанном реакторе первичного окисления, причем головные газы перемещают из верха реактора вторичного окисления в реактор первичного окисления. Раскрыты оптимизированный процесс и оборудование для более эффективного и экономичного проведения жидкофазного окисления. Такое жидкофазное окисление проводят в реакторе по типу барботажной колонны, которая обеспечивает высокоэффективную реакцию при относительно низких температурах. Когда окисленное соединение представляет собой пара-ксилол и продуктом реакции окисления является сырая терефталевая кислота (СТК), такой продукт, СТК, может быть очищен и выделен с помощью более экономичных методик, чем когда СТК получена с помощью обычного процесса высокотемпературного окисления. 2 н. и 28 з.п. ф-лы, 4 табл., 31 ил.
1. Способ получения композиции ароматической дикарбоновой кислоты, включающий
(a) проведение окисления многофазной реакционной среды в реакторе первичного окисления с получением в результате первой суспензии;
(b) проведение дополнительного окисления, по меньшей мере, части указанной первой суспензии в реакторе вторичного окисления, где указанный реактор вторичного окисления представляет собой реактор по типу барботажной колонны,
причем способ дополнительно включает введение ароматического соединения в указанный реактор первичного окисления, где, по меньшей мере, приблизительно 80 мас.% указанного ароматического соединения, введенного в указанный реактор первичного окисления, окисляется в указанном реакторе первичного окисления,
причем головные газы перемещают из верха реактора вторичного окисления в реактор первичного окисления.
2. Способ по п.1, где указанное ароматическое соединение представляет собой пара-ксилол.
3. Способ по любому из пп.1-2, где стадия (b) включает окисление пара-толуиловой кислоты, присутствующей в указанной первой суспензии.
4. Способ по п.3, дополнительно включающий выведение второй суспензии из указанного реактора вторичного окисления, где усредненная по времени концентрация пара-толуиловой кислоты в жидкой фазе указанной второй суспензии составляет менее чем приблизительно 50 мас.% от усредненной по времени концентрации пара-толуиловой кислоты в жидкой фазе указанной первой суспензии.
5. Способ по п.4, где усредненная по времени концентрация пара-толуиловой кислоты в жидкой фазе указанной первой суспензии составляет, по меньшей мере, приблизительно 500 мас. ч/млн, и где усредненная по времени концентрация пара-толуиловой кислоты в жидкой фазе указанной второй суспензии составляет менее чем приблизительно 250 мас. ч/млн.
6. Способ по п.1, где указанный реактор первичного окисления представляет собой реактор по типу барботажной колонны.
7. Способ по п.1, где указанный реактор вторичного окисления расположен снаружи указанного реактора первичного окисления.
8. Способ по п.7, где, по меньшей мере, часть указанного реактора вторичного окисления расположена вдоль боковой стороны указанного реактора первичного окисления.
9. Способ по п.1, где указанный реактор вторичного окисления не является реактором вытеснения.
10. Способ по п.1, дополнительно включающий отведение указанной первой суспензии из указанного реактора первичного окисления через выпускное отверстие для суспензии, расположенное между нижним и верхним концами указанного реактора первичного окисления.
11. Способ по п.10, где указанный реактор первичного окисления ограничивает в нем первичную реакционную зону, имеющую максимальную высоту (Нр), где указанное выпускное отверстие для суспензии находится на расстоянии, по меньшей мере, приблизительно 0,1Нр от нижнего и верхнего концов указанной первичной реакционной зоны.
12. Способ по п.11, где указанное выпускное отверстие для суспензии находится на расстоянии, по меньшей мере, приблизительно 0,25Hi от нижнего и верхнего концов указанной первичной реакционной зоны.
13. Способ по п.1, где указанный реактор первичного окисления ограничивает в нем первичную реакционную зону, где указанный реактор вторичного окисления ограничивает в нем вторичную реакционную зону, где отношение объема указанной первичной реакционной зоны к объему указанной вторичной реакционной зоны находится в интервале приблизительно от 4:1 до 50:1.
14. Способ по п.13, где указанная первичная реакционная зона имеет отношение максимальной вертикальной высоты к максимальному горизонтальному диаметру в интервале приблизительно от 3:1 до 30:1, где указанная вторичная реакционная зона имеет отношение максимальной вертикальной высоты к максимальному горизонтальному диаметру в интервале приблизительно от 1:1 до 50:1.
15. Способ по любому из пп.13 и 14, где отношение максимального горизонтального диаметра указанной первичной реакционной зоны к максимальному горизонтальному диаметру указанной вторичной реакционной зоны находится в интервале приблизительно от 0,1:1 до 0,6:1, где отношение максимальной вертикальной высоты указанной первичной реакционной зоны к максимальной вертикальной высоте указанной вторичной реакционной зоны находится в интервале приблизительно от 0,1:1 до 0,9:1.
16. Способ по п.13, где указанная первичная реакционная зона имеет максимальный диаметр (Dp), где объемный центр тяжести указанной вторичной реакционной зоны находится на расстоянии по горизонтали, по меньшей мере, приблизительно 0,5Dp от объемного центра тяжести указанной первичной реакционной зоны.
17. Способ по п.13, где указанная первичная реакционная зона имеет максимальную высоту (Нр), где объемный центр тяжести указанной вторичной реакционной зоны находится на расстоянии по вертикали менее чем 0,5Нр от объемного центра тяжести указанной первичной реакционной зоны.
18. Реакторная система для проведения способа по п.1, включающая
- реактор первичного окисления, ограничивающий первое впускное отверстие и первое выпускное отверстие; и
- реактор вторичного окисления, ограничивающий второе впускное отверстие и второе выпускное отверстие,
где указанное первое выпускное отверстие связано передачей потока жидкости с указанным вторым впускным отверстием, где указанный реактор вторичного окисления представляет собой реактор по типу барботажной колонны.
19. Реакторная система по п.18, где указанный реактор первичного окисления представляет собой реактор по типу барботажной колонны.
20. Реакторная система по любому из пп.18 и 19, где указанный реактор вторичного окисления расположен снаружи указанного реактора первичного окисления.
21. Реакторная система по п.20, где, по меньшей мере, часть указанного реактора вторичного окисления расположена вдоль боковой стенки указанного реактора первичного окисления.
22. Реакторная система по п.18, где указанный реактор вторичного окисления не является реактором вытеснения.
23. Реакторная система по п.18, где указанный реактор первичного окисления определяет выпускное отверстие для суспензии, соединенное передачей потока жидкости с указанным реактором вторичного окисления, где указанное выпускное отверстие расположено между нижним и верхним концами указанного реактора первичного окисления.
24. Реакторная система по п.23, где указанный реактор первичного окисления ограничивает в нем первичную реакционную зону, имеющую максимальную высоту (Нр), где указанное выпускное отверстие для суспензии находится на расстоянии, по меньшей мере, приблизительно 0,1Нр от нижнего и верхнего концов указанной первичной реакционной зоны.
25. Реакторная система по п.24, где указанное выпускное отверстие для суспензии находится на расстоянии, по меньшей мере, приблизительно 0,25Нр от нижнего и верхнего концов указанной первичной реакционной зоны.
26. Реакторная система по п.18, где указанный реактор первичного окисления ограничивает в нем первичную реакционную зону, где указанный реактор вторичного окисления ограничивает в нем вторичную реакционную зону, где отношение объема указанной первичной реакционной зоны к объему указанной вторичной реакционной зоны находится в интервале приблизительно от 4:1 до 50:1.
27. Реакторная система по п.26, где указанная первичная реакционная зона имеет отношение максимальной вертикальной высоты к максимальному горизонтальному диаметру в интервале приблизительно от 3:1 до 30:1, где указанная вторичная реакционная зона имеет отношение максимальной вертикальной высоты к максимальному горизонтальному диаметру в интервале приблизительно от 1:1 до 50:1.
28. Реакторная система по любому из пп.26 и 27, где отношение максимального горизонтального диаметра указанной зоны первичного окисления к максимальному горизонтальному диаметру указанной зоны вторичного окисления находится в интервале приблизительно от 0,1:1 до 0,6:1, где отношение максимальной вертикальной высоты указанной зоны первичного окисления к максимальной вертикальной высоте указанной зоны вторичного окисления находится в интервале приблизительно от 0,1:1 до 0,9:1.
29. Реакторная система по п.26, где указанная первичная реакционная зона имеет максимальный диаметр (Dp), где объемный центр тяжести указанной вторичной реакционной зоны находится на расстоянии по горизонтали, по меньшей мере, приблизительно 0,5Dp от объемного центра тяжести указанной первичной реакционной зоны.
30. Реакторная система по п.26, где указанная первичная реакционная зона имеет максимальную высоту (Нр), где объемный центр тяжести указанной вторичной реакционной зоны находится на расстоянии по вертикали менее чем 0,5Нр от объемного центра тяжести указанной первичной реакционной зоны.
| СПОСОБ, СИСТЕМА И ДАТЧИК ПОДКЛЮЧЕНИЯ ДЛЯ ИДЕНТИФИКАЦИИ ПОРТА КОММУТАЦИОННОЙ ПАНЕЛИ | 2006 |
|
RU2310210C9 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОЙ КАРБОНОВОЙ КИСЛОТЫ | 2001 |
|
RU2259346C2 |
| US 5821270 A, 13.10.1998 | |||
| US 6800664 B1, 05.10.2004. | |||

