Родственные заявки
Настоящая заявка заявляет приоритет согласно японской патентной заявке №155062/2006, поданной 2 июня 2006 года, полное раскрытие которой включено сюда в виде ссылки.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Область техники
Настоящее изобретение относится к новым аминогликозидным антибиотикам, которые отличаются тем, что они эффективны против бактерий, которые вызывают клинически тяжелые инфекционные заболевания, в частности против устойчивых к метициллину Staphylococcus aureus (MRSA), и при этом отличаются низким уровнем нефротоксичности. Настоящее изобретение относится также к новым промежуточным соединениям, которые можно использовать для получения аминогликозидных антибиотиков.
Предпосылки изобретения
В последние годы появились бактерии, устойчивые к противомикробным агентам, которые используют для лечения инфекционных заболеваний, и лечение инфекционных заболеваний, вызываемых устойчивыми бактериями, стало основной проблемой клинической практики. В частности, MRSA известна как одна из основных устойчивых к лекарствам бактерий, которая быстро распространяется через больничную инфекцию и вызывает клинически тяжелые инфекционные заболевания, и поэтому энергично разрабатываются терапевтические агенты против инфекционных заболеваний.
Аминогликозидные антибиотики обладают широким спектром противомикробной активности от грамположительных до грамотрицательных бактерий и обладают эффективными стерилизационными характеристиками. Соответственно, ожидают, что аминогликозидные антибиотики будут действовать как многообещающие лекарственные средства, которые смогут победить различные устойчивые бактерии, включая MRSA, и исследования производных указанных соединений происходили непрерывно.
Например, в Journal of Antibiotics, Vol.24, 1971, p.485 раскрыто, что различные производные канамицина, который представляет собой аминогликозидный антибиотик, можно синтезировать и 3',4'-дезоксиканамицин B (дибекацин) можно обнаружить в производных канамицина. Дибекацин находит широкое применение в качестве химиотерапевтического агента, эффективного в отношении устойчивых бактерий с 1975 года.
В Journal of Antibiotics, Vol.26, 1973, p.412 раскрыт (S)-1-N-(4-амино-2-гидроксибутирил)дибекацин (арбекацин), полученный в результате ацилирования аминогруппы в 1-положении дибекацина аминогидроксимасляной кислотой. Далее, в японской патентной публикации №10719/1988 раскрыт способ получения арбекацина.
Арбекацин применяли в качестве терапевтического агента для лечения MRSA инфекционных заболеваний с конца 1990 года. Известно, что арбекацин обладает широким противомикробным спектром начиная с грамположительных бактерий, включая MRSA, до грамотрицательных бактерий, включая Pseudomonas aeruginosa. Прошло десять лет или более с того времени, как арбекацин стали применять в качестве терапевтического агента при MRSA инфекционных заболеваниях. Несмотря на этот факт, нет никаких сообщений о значительно повышенной устойчивости. С другой стороны, в JAPANESE SOCIETY OF CHEMOTHERAPY, Vol.50, 2002, p.494 имеется сообщение о том, что некоторые клинически выделенные MRSA обладают пониженной чувствительностью к арбекацину.
Исследования различных аналогов арбекацина непрерывно продолжались. Например, в WO 2005/070945 раскрыт тот факт, что группа соединений, отличающихся пространственной конфигурацией сайта, соответствующего 5-положению арбекацина, которые были обращены и в которые были введены различные заместители, обладает противомикробной активностью против MRSA.
С другой стороны, давно известно, что нефротоксичность является побочным эффектом действия аминогликозидных антибиотиков. Имеется также сообщение о клиническом влиянии арбекацина (Japanese Patent Laid-Open No.164696/1980) на почки (JAPANESE SOCIETY OF CHEMOTHERAPY, Vol.51, 2003, p.717).
В разделе No.F-716 44 Interscience Conference on Antimicrobal Agents and Chemotherapy (2004), авторы настоящего изобретения раскрывают относительно аналога арбекацина (соединение №TS2037: 5,4"-диэпиарбекацин), раскрытого в WO 2005/070945, результаты оценки нефротоксичности с использованием эпителиальных клеток проксимальных мочевых канальцев почек свиней и с использованием β-N-ацетил-D-глюкозаминидазы (здесь и далее обозначаемой как "NAG") как показателя. Результаты показывают, что нефротоксичность аналога арбекацина выше, чем нефротоксичность арбекацина.
Было проведено изучение различных способов для снижения нефротоксичности аминогликозидных антибиотиков. Как сообщается, совместное применение аминогликозидного антибиотика и соединения для снижения нефротоксичности является одним из таких способов. Например, известно, что фосфомицин снижает нефротоксичность некоторых аминогликозидных антибиотиков. Далее, сообщается, что в тесте на крысах совместное применение фосфомицина и арбекацина снижает нефротоксичность (The Japanese Journal of Antibiotics, Vol.47, 1994, p.664).
Недавно был изучен способ с использованием так называемого TDM (терапевтический мониторинг лекарственных средств), в котором реализуется высокая степень терапевтического действия при подавлении побочного эффекта за счет осуществления введения лекарственного средства с учетом фармакокинетики и фармакодинамики. Например, имеется сообщение, что TDM используют также для анти-MRSA лечения арбекацином (JAPANESE SOCIETY OF CHEMOTHERAPY, Vol.51, 2003, p.717).
Однако, если обычные аминогликозидные антибиотики, которые используют клинически, применяют отдельно, все еще остается необходимость в снижении нефротоксичности при сохранении широкого спектра противомикробной активности и ее высокой эффективности против бактерий, которые вызывают тяжелые инфекционные заболевания, включая MRSA. Соответственно, было желательно в области аминогликозидных антибиотиков создать новое соединение, которое обладало бы широким спектром противомикробной активности и эффективной стерилизующей способностью и в то же самое время обладало бы низким уровнем нефротоксичности. Далее, для обычных аминогликозидных антибиотиков появление бактерий, устойчивых к лекарственным средствам, стало проблемой, и все еще необходимы соединения, которые обладают превосходной противомикробной активностью также и против устойчивых к лекарственным средствам бактерий. Кроме того, если принять во внимание получение превосходных антибиотиков, критической проблемой является также изучение стабильного получения антибиотиков.
Краткое содержание изобретения
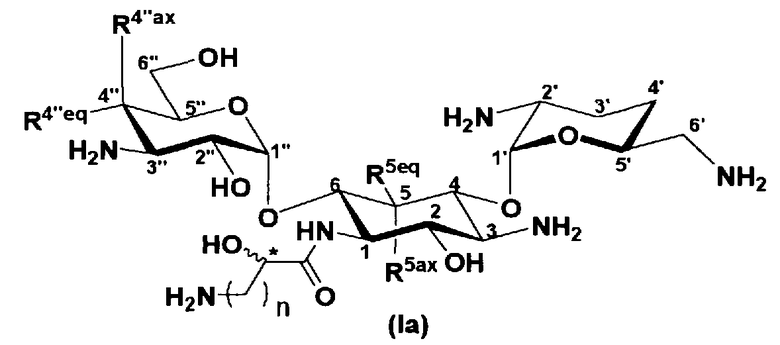
Авторам настоящего изобретения удалось получить новые соединения, представленные формулой (Ia), которые обладают широким противомикробным спектром и превосходной противомикробной активностью, так же как низким уровнем нефтротоксичности. Авторы настоящего изобретения также обнаружили, что новое соединение обладает высоким уровнем противомикробной активности против штаммов, найденных в клинических изолятах MRSA, которые обладают низким уровнем чувствительности к арбекацину.
Авторы настоящего изобретения далее обнаружили способ получения, который можно использовать для стабильного получения новых соединений, и промежуточное соединение, важное для получения указанных новых соединений.
Настоящее изобретение было создано на основании таких фактов.
Соответственно, целью настоящего изобретения является создание новых соединений, обладающих широким противомикробным спектром и превосходной противомикробной активностью, а также низким уровнем нефротоксичности, и создание синтетических промежуточных соединений, важных для получения новых соединений.
В соответствии с настоящим изобретением предложено соединение, представленное формулой (Ia), или смесь его диастереомеров относительно атома углерода, отмеченного *, или их фармакологически приемлемые соли или сольваты.
[Химическая формула 1]
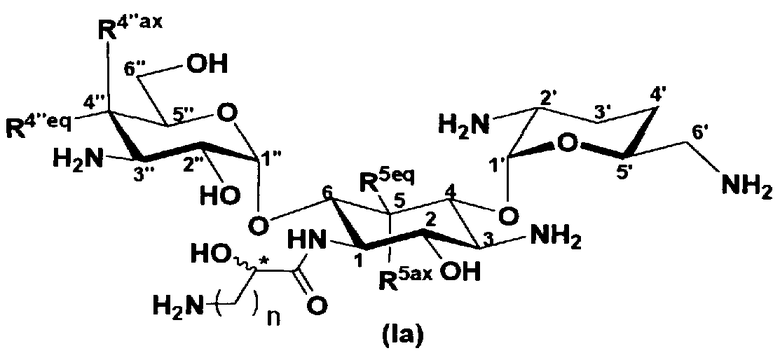
где R5ax и R5eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил,
R4"ax и R4"eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил,
n является целым числом от 1 до 4, и
конфигурация атома углерода, к которому они присоединены и обозначенному *, соответствует R или S.
Далее, в соответствии с настоящим изобретением предложено промежуточное соединение, которое можно использовать для синтеза соединения, представленного формулой (Ia).
Соединение, представленное формулой (Ia) настоящего изобретения, демонстрирует широкий противомикробный спектр и великолепную противомикробную активность и позволяет избежать тяжелой нефротоксичности. Далее, соединение, представленное формулой (Ia), может также проявлять превосходную противомикробную активность против MRSA, которые мало чувствительны к арбекацину. Тот факт, что нефротоксичность соединения, представленного формулой (Ia), ниже, чем нефротоксичность арбекацина, является преимуществом для применения в случае пациентов с инфекционными заболеваниями. Соответственно, соединение, представленное формулой (Ia), в соответствии с настоящим изобретением можно с успехом использовать для лечения инфекционных заболеваний, включая MRSA. Далее, соединение, представленное формулой (Ia), можно стабильно получать через соединения, представленные формулами (Xa), (Xb) или (XXV), которые будут раскрыты далее, и может быть с успехом использовано в качестве терапевтического агента для лечения инфекционных заболеваний.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Термин "алкил" в том смысле, как здесь использован, как группа или как часть группы, означает неразветвленную цепь, разветвленную цепь или циклический алкил, если нет других указаний. Термин "арил" означает фенил или нафтил, если нет других указаний. Термин "арилалкил" означает алкил, в котором один или более из атомов водорода заменен на арил.
Соединения, представленные формулой (Ia)
Отличительной особенностью соединений, представленных формулой (Ia) является то, что у них гидроксильная группа находится в 2-положении.
[Химическая формула 2]
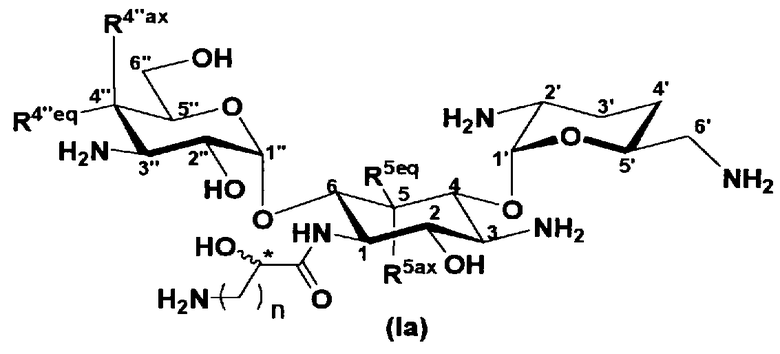
Соединение с представленной выше структурой обладает низким уровнем нефротоксичности и, в то же самое время, обладает широким спектром противомикробной активности в отношении как грамположительных бактерий, включая MRSA, до грамотрицательных бактерий, включая Pseudomonas aeruginosa, и обладает превосходной противомикробной активностью.
В соединениях, представленных формулой (Ia) в предпочтительном варианте настоящего изобретения, R5ax и R5eq отличаются друг от друга и представляют собой атом водорода или гидроксил, и R4"ax и R4"eq отличаются друг от друга и представляют собой атом водорода или гидроксил.
В соединениях, представленных формулой (Ia), n предпочтительно равно 1-3, более предпочтительно 1-2.
Далее, в соединениях, представленных формулой (Ia) в предпочтительном варианте настоящего изобретения, пространственная конфигурация гидроксильной группы в 5-положении соответствует экваториальной. Соответственно, в соединениях, представленных формулой (Ia), в представленном выше варианте R5ax представляет собой атом водорода, и R5eq представляет собой гидроксил. В соединениях, представленных формулой (Ia), в более предпочтительном варианте настоящего изобретения R4"ax и R4"eq отличаются друг от друга и представляют собой атом водорода или гидроксил.
В более предпочтительном варианте настоящего изобретения предложены соединения, представленные формулой (I), или их фармакологически приемлемые соли или сольваты.
[Химическая формула 3]
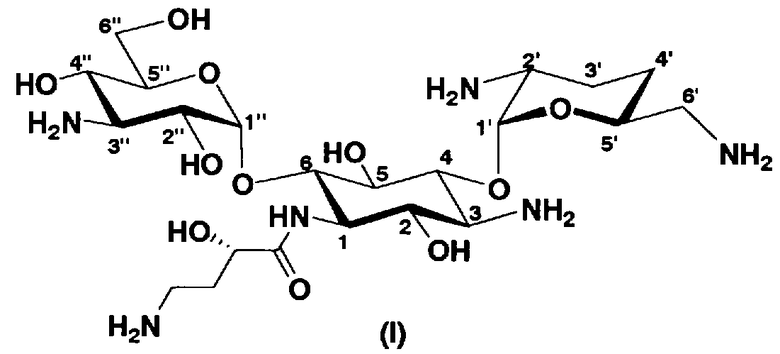
Далее, в соединениях, представленных формулой (Ia), в другом варианте настоящего изобретения, пространственная конфигурация гидроксильной группы в 5-положении соответствует аксиальной. Соответственно, в соединениях, представленных формулой (Ia), в представленном выше варианте, R5ax представляет собой гидроксил и R5eq представляет собой атом водорода. В соединениях, представленных формулой (Ia), в более предпочтительном варианте настоящего изобретения R4"ax и R4"eq отличаются друг от друга и представляют собой атом водорода или гидроксил.
Соединения, представленные формулой (Ia), могут существовать в виде солей. Примеры таких солей включают фармацевтически приемлемые нетоксичные соли. Конкретные их примеры включают гидрогалогениды, такие как гидрофториды, гидрохлориды, гидробромиды и гидроиодиды, соли неорганических кислот, такие как сульфаты, нитраты, фосфаты, перхлораты и карбонаты, соли карбоновых кислот, таких как уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, гидроксиуксусная кислота, молочная кислота, лимонная кислота, винная кислота, щавелевая кислота, бензойная кислота, миндальная кислота, масляная кислота, малеиновая кислота, пропионовая кислота, муравьиная кислота и яблочная кислота, соли аминокислот, таких как альгиновая кислота, аспарагиновая кислота и глутаминовая кислота, и соли сульфоновых кислот, таких как метансульфоновая кислота и п-толуолсульфоновая кислота. Предпочтительны соли неорганических кислот, такие как сульфаты.
Соединения, представленные формулой (Ia), или их фармакологически приемлемые соли могут существовать в виде сольватов. Предпочтительные сольваты включают гидраты или этаноляты.
Как было раскрыто выше, соединения, представленные формулой (Ia), могут быть в форме смеси диастереомеров относительно атома углерода, отмеченного *, и настоящее изобретение включает также и этот вариант.
Синтетические промежуточные соединения
Соединения, представленные формулой (Ia), можно получить следующими двумя способами. В соответствии с этими способами соединения, представленные формулой (Ia), можно с успехом получать через синтетические промежуточные соединения, которые будут раскрыты далее.
Синтетические промежуточные соединения в первом способе получения
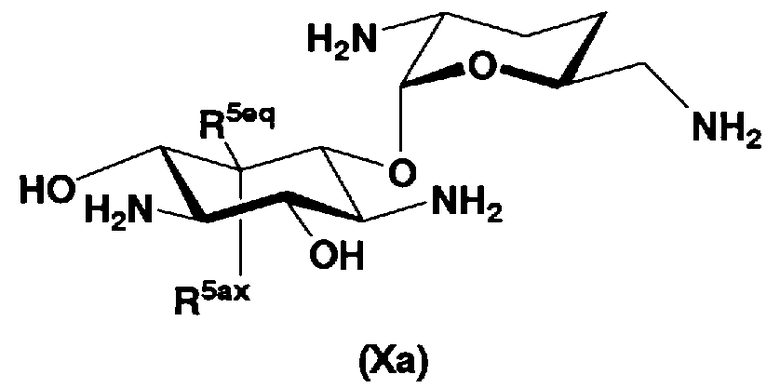
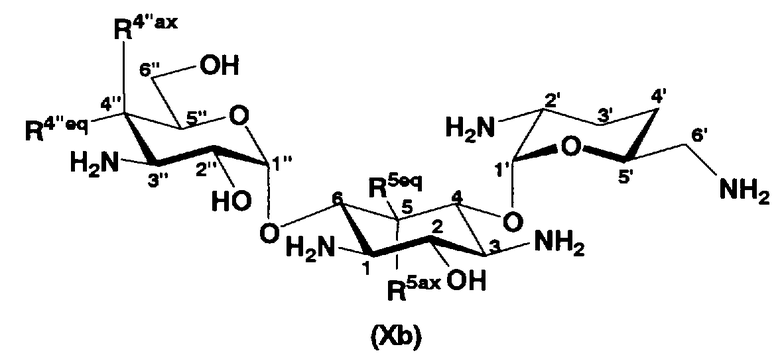
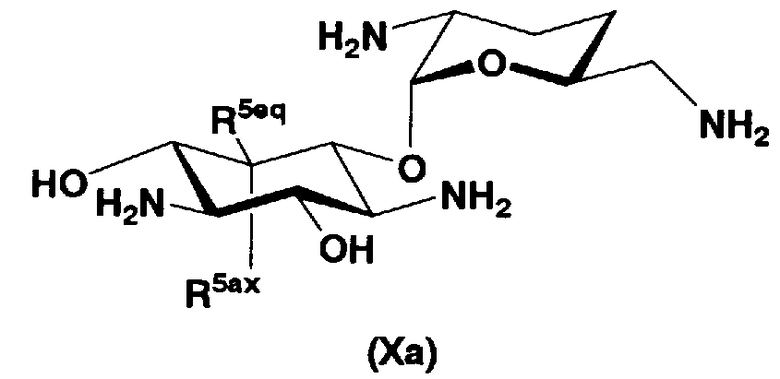
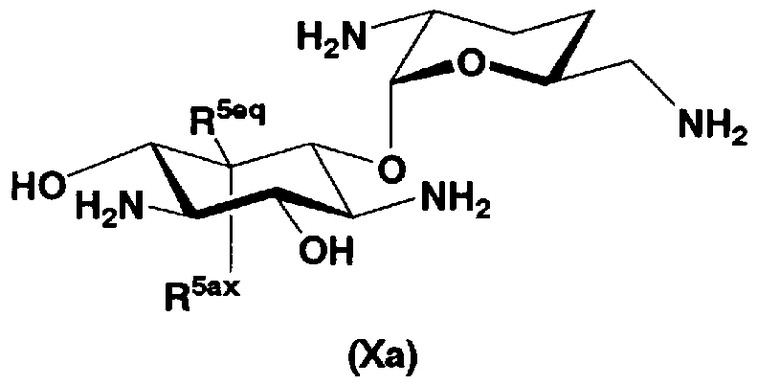
В первом способе получения в соответствии с настоящим изобретением соединения, представленные формулой (Xa) и формулой (Xb), используют как синтетические промежуточные соединения.
Соответственно, в одном варианте настоящего изобретения предложены соединения формулы (Xa)
[Химическая формула 4]
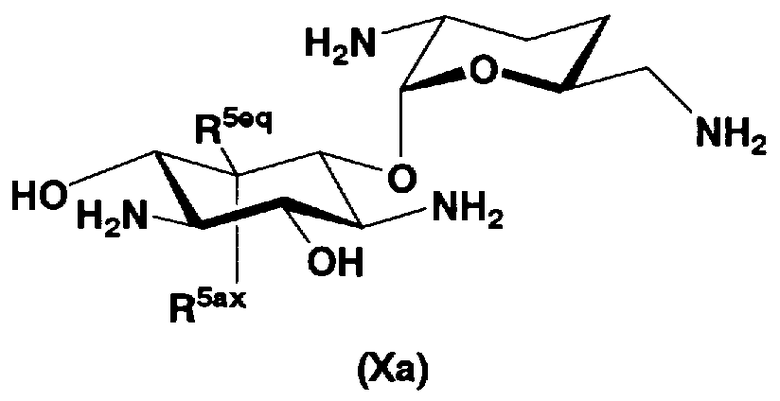
где R5ax и R5eq отличаются друг от друга и представляют собой атом водорода или гидроксил.
В другом варианте настоящего изобретения предложены соединения, представленные формулой (Xb)
[Химическая формула 5]

где R5ax и R5eq отличаются друг от друга и представляют собой атом водорода или гидроксил, и
R4"ax и R4"eq отличаются друг от друга и представляют собой атом водорода или гидроксил.
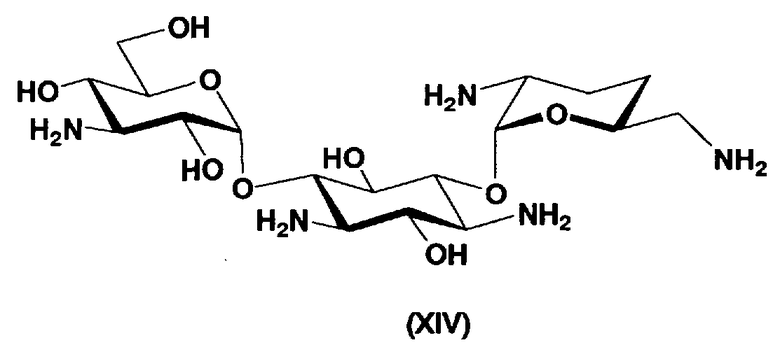
В предпочтительном варианте настоящего изобретения соединения, представленные формулой (Xb), являются соединениями, представленными формулой (XIV).
[Химическая формула 6]
Синтетические промежуточные соединения во втором способе получения настоящего изобретения
Во втором способе получения настоящего изобретения соединения, представленные формулой (XXV), или смеси их диастереомеров относительно атома углерода, отмеченного *, используют как синтетические промежуточные соединения. В соединениях, представленных формулой (XXV), в этом способе получения, пространственная конфигурация гидроксильной группы в 5-положении соответствует экваториальной. Соответственно, второй способ получения пригоден для получения соединений, которые представлены формулой (Ia) и у которых пространственная конфигурация гидроксильной группы в 5-положении соответствует экваториальной.
[Химическая формула 7]
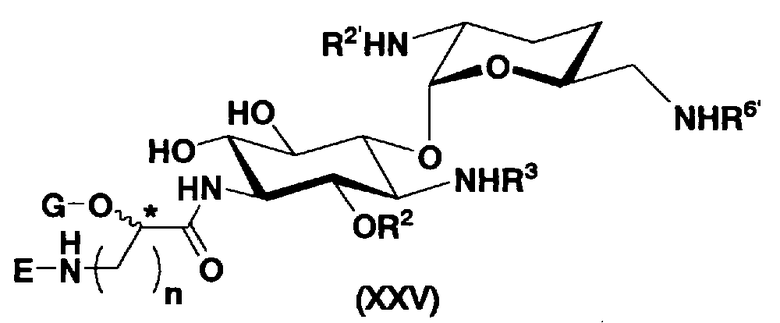
где R2 и G представляют собой защитную группу для гидроксильной группы; R3, R2', R6' и E представляют собой защитную группу для аминогруппы; n означает целое число от 1 до 4; и пространственная конфигурация атома углерода, отмеченного *, соответствует R или S.
В соединениях, представленных формулой (XXV), в более предпочтительном варианте настоящего изобретения
R2 представляет собой необязательно замещенный арилC1-3алкил,
R3, R2' и R6', которые могут быть одинаковыми или различными, представляют собой
необязательно замещенный C1-6 алкилсульфонил,
необязательно замещенный арилсульфонил или
необязательно замещенный C1-6 алкилоксикарбонил,
E представляет собой необязательно замещенный C1-6 алкилоксикарбонил и
G представляет собой атом водорода,
необязательно замещенный C1-6 алкилкарбонил или
необязательно замещенный арилкарбонил.
В соединениях, представленных формулой (XXV), арилC1-3алкильная группа, представленная R2, предпочтительно представляет собой арил C1-2 алкил, более предпочтительно бензил.
Один или более из атомов водорода в арилC1-3алкильной группе, представленной R2, необязательно заменен, например, метокси или нитро. Конкретные примеры замещенного арилC1-3алкила включают метоксибензил или нитробензил.
В соединениях, представленных формулой (XXV), C1-6 алкилсульфонильная группа, представленная R3, R2' или R6', предпочтительно представляет собой C1-3 алкилсульфонил, более предпочтительно метансульфонил.
Один или более из атомов водорода в C1-6 алкилсульфонильной группе, представленной R3, R2' или R6', необязательно заменен, например, необязательно замещенным фенилом (фенил или толил). Примеры замещенных C1-6 алкилсульфонильных групп включают бензилсульфонил или толуолсульфонил.
Один или более из атомов водорода в арилсульфонильной группе, представленной
R3, R2' или R6', необязательно заменен, например, метилом. Конкретные примеры необязательно замещенных арилсульфонильных групп включают бензилсульфонил или толуолсульфонил.
В соединениях, представленных формулой (XXV), предпочтительно, чтобы C1-6 алкилоксикарбонильная группа, представленная R3, R2' или R6', представляла собой C1-4 алкилоксикарбонил, более предпочтительно метоксикарбонил или трет-бутоксикарбонил.
Один или более из атомов водорода в C1-6 алкилоксикарбонильной группе, представленной R3, R2' или R6', необязательно заменен, например, необязательно замещенным фенилом (например фенилом, метоксифенилом или нитрофенилом). Конкретные примеры замещенных C1-6 алкилоксикарбонильных групп включают бензилоксикарбонил, трет-бутоксикарбонил, п-метоксибензилоксикарбонил или п-нитробензилоксикарбонил.
В соединениях, представленных формулой (XXV), C1-6 алкилоксикарбонильные группы, представленные E, предпочтительно представляют собой C1-3 алкилоксикарбонил, более предпочтительно метоксикарбонил или этоксикарбонил, еще более предпочтительно метоксикарбонил.
Один или более из атомов водорода в C1-6 алкилоксикарбонильной группе, представленной E, необязательно заменен, например, необязательно замещенным фенилом (например фенилом, метоксифенилом или нитрофенилом). Соответственно, конкретные примеры C1-6 алкилоксикарбонильной группы, замещенной необязательно замещенным фенилом, включают бензилоксикарбонил, п-метоксибензилоксикарбонил и п-нитробензилоксикарбонил.
В соединениях, представленных формулой (XXV), C1-6 алкилкарбонильная группа, представленная G, предпочтительно представляет собой C1-3 алкилкарбонил, более предпочтительно ацетил.
Один или более из атомов водорода в C1-3 алкилкарбонильной группе, представленной G, необязательно заменен, например, атомом галогена, таким как хлор, бром или фтор, и конкретные примеры замещенных C1-3 алкилкарбонильных групп включают трихлорацетил и трифторацетил.
В соединениях, представленных формулой (XXV), арилкарбонильная группа, представленная G, предпочтительно представляет собой бензоил.
Один или более из атомов водорода в арилкарбонильной группе, представленной G, необязательно заменен, например, фенилом, атомом галогена, таким как хлор, бром или фтор, нитро или метокси. Конкретные примеры замещенных арилкарбонильных групп включают п-фенилбензоил, п-бромбензоил, п-нитробензоил и п-метоксибензоил.
В соединениях, представленных формулой (XXV), арилC1-3алкильная группа, представленная G, предпочтительно представляет собой арилC1-2алкил, более предпочтительно бензил или трифенилметил.
Далее, один или более из атомов водорода в арилC1-3алкильной группе, представленной G, необязательно заменен, например метокси. Конкретные примеры замещенных C1-3 алкилсульфонильных групп включают п-метоксибензил.
В соединениях, представленных формулой (XXV), n обозначает 1-4, предпочтительно 1-3, более предпочтительно 1 или 2.
В соединениях, представленных формулой (XXV), в более предпочтительном варианте настоящего изобретения
R2 представляет собой арилC1-3алкил, необязательно замещенный метокси или нитро,
R3, R2' и R6', которые могут быть одинаковыми или различными, представляют собой
C1-6 алкилсульфонил, необязательно замещенный необязательно замещенным фенилом,
арилсульфонил, необязательно замещенный метилом, или
C1-6 алкилоксикарбонил, необязательно замещенный необязательно замещенным фенилом,
E представляет собой C1-6 алкилоксикарбонил, необязательно замещенный необязательно замещенным фенилом, и
G представляет собой атом водорода,
C1-6 алкилкарбонил, арилкарбонил или
арилC1-3алкил, необязательно замещенный метокси.
В соединениях, представленных формулой (XXV), в другом предпочтительном варианте настоящего изобретения,
R2 представляет собой арил C1-2 алкил, необязательно замещенный метокси или нитро,
R3, R2' и R6', которые могут быть одинаковыми или различными, представляют собой
C1-3 алкилсульфонил, необязательно замещенный необязательно замещенным фенилом,
арилсульфонил, необязательно замещенный метилом, или
C1-4 алкилоксикарбонил, необязательно замещенный необязательно замещенным фенилом,
E представляет собой C1-4 алкилоксикарбонил, необязательно замещенный необязательно замещенным фенилом, и
G представляет собой атом водорода,
C1-3 алкилкарбонил,
арилкарбонил или
арил C1-2 алкил, необязательно замещенный метокси.
В соединениях, представленных формулой (XXV), в следующем предпочтительном варианте настоящего изобретения
R2 представляет собой необязательно замещенный арилC1-3алкил,
все из R3, R2', R6' и E представляют собой необязательно замещенный C1-6 алкилоксикарбонил и
G представляет собой необязательно замещенный арилC1-3алкил.
В соединениях, представленных формулой (XXV), в следующем предпочтительном варианте настоящего изобретения
R2 представляет собой необязательно замещенный арил C1-2 алкил,
все из R3, R2', R6' и E представляют собой необязательно замещенный C1-4 алкилоксикарбонил и
G представляет собой необязательно замещенный арил C1-2 алкил.
В соединениях, представленных формулой (XXV), в следующем предпочтительном варианте настоящего изобретения
R2 представляет собой арил C1-2 алкил, необязательно замещенный метокси или нитро,
все из R3, R2', R6' и E представляют собой C1-4 алкилоксикарбонил, необязательно замещенный фенилом, необязательно замещенным метокси или нитро, и
G представляет собой арил C1-2 алкил, необязательно замещенный метокси.
В соединениях, представленных формулой (XXV), в
другом предпочтительном варианте настоящего изобретения
R2 представляет собой бензил, метоксибензил или нитробензил,
R3, R2' и R6', которые могут быть одинаковыми или различными, представляют собой метансульфонил, бензилсульфонил, п-толуолсульфонил, бензилоксикарбонил, трет-бутоксикарбонил, п-метоксибензилоксикарбонил или п-нитробензилоксикарбонил,
E представляет собой бензилоксикарбонил и
G представляет собой атом водорода, ацетил, бензоил, п-метоксибензил или трифенилметил.
В соединениях, представленных формулой (XXV), в более предпочтительном варианте настоящего изобретения
R2 представляет собой бензил, метоксибензил или нитробензил,
все из R3, R2', R6' и E представляют собой бензилоксикарбонил и
G представляет собой бензил.
Способ получения
В настоящем изобретении можно указать следующие два способа получения как способы получения соединений, представленных формулой (Ia).
Первый способ получения
В первом способе получения настоящего изобретения соединения, представленные формулой (Xa) и формулой (Xb), используют как синтетические промежуточные соединения.
В соответствии с одним из аспектов настоящего изобретения предложен способ получения соединения, представленного формулой (Ia):
[Химическая формула 8]

где R5ax и R5eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил,
R4"ax и R4"eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил, и
n является целым числом от 1 до 4,
конфигурация атома углерода, отмеченного *, соответствует R или S,
причем указанный способ включает стадии введения защитных групп в аминогруппы в соединении, представленном формулой (Xa):
[Химическая формула 9]
где R5ax и R5eq имеют указанные для формулы (Ia) значения,
осуществления взаимодействия соединения, представленного формулой (Xa), с соединением, представленным формулой (Xc):
[Химическая формула 10]
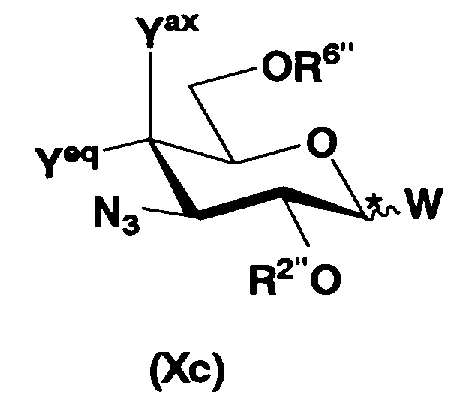
где W представляет собой отщепляемую группу; Yax и Yeq, которые могут быть одинаковыми или различными, представляют собой группу -OR4" или атом водорода; R2", R4" и R6" представляют собой защитную группу для гидроксильной группы, и конфигурация атома углерода, отмеченного *, соответствует R или S,
удаления защитных групп у полученного соединения и
превращения азидной группы в соединении в аминогруппу с получением соединения, представленного формулой (Xb):
[Химическая формула 11]
где R5ax, R5eq, R4"ax и R4"eq имеют указанные выше для формулы (Ia) значения,
необязательного введения защитных групп по функциональным группам, отличающимся от аминогруппы в 1-положении соединения, представленного формулой (Xb),
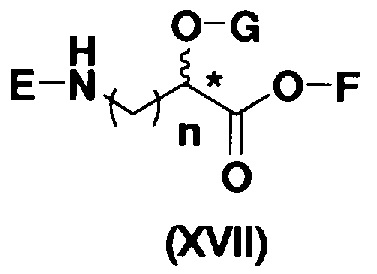
осуществления взаимодействия полученного соединения с соединением, представленным формулой (XVII):
[Химическая формула 12]
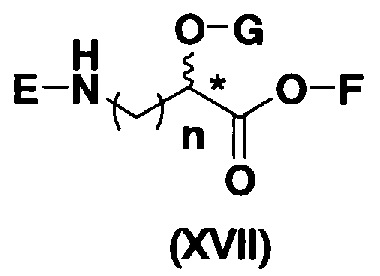
где E представляет собой защитную группу для аминогруппы; G представляет собой защитную группу для гидроксильной группы; F представляет собой атом водорода или активирующую группу для карбоновой кислоты; n является целым числом от 1 до 4; и пространственная конфигурация атома углерода, отмеченного *, соответствует R или S, и
удаления защитных групп у полученного соединения, с получением соединения, представленного формулой (Ia).
В первом способе получения настоящего изобретения соединение, представленное формулой (XVII), может быть энантиомерной смесью относительно атома углерода, отмеченного *. Соответственно, в соответствии с первым способом получения настоящего изобретения можно получить диастереомерную смесь относительно атома углерода, отмеченного *, в соединении, представленном формулой (Ia). Настоящее изобретение включает также и этот вариант.
В первом способе получения настоящего изобретения предпочтительно, чтобы гидроксил находился в 5- и 4"-положениях в соединении, представленном формулой (Ia). Соответственно, в первом способе получения настоящего изобретения в предпочтительном варианте настоящего изобретения R5ax и R5eq, которые отличаются друг от друга, представляют собой атом водорода или гидроксил; R4"ax и R4"eq, которые отличаются друг от друга, представляют собой атом водорода или гидроксил.
В соответствии с первым способом получения настоящего изобретения в соединении, представленном формулой (Ia), если гидроксил находится в 5-положении, пространственная конфигурация гидроксильной группы может быть экваториальной. Соответственно, в первом способе получения настоящего изобретения в другом предпочтительном варианте настоящего изобретения R5ax представляет собой атом водорода и R5eq представляет собой гидроксил.
В соответствии с первым способом получения настоящего изобретения в соединении, представленном формулой (Ia), если гидроксил находится в 5-положении, пространственная конфигурация гидроксильной группы может быть аксиальной. Соответственно, в первом способе получения настоящего изобретения в другом предпочтительном варианте настоящего изобретения R5ax представляет собой гидроксил и R5eq представляет собой атом водорода.
В первом способе получения настоящего изобретения предпочтительно, чтобы защитная группа, введенная в аминогруппу соединения, представленного формулой (Xa), была введена в 1-, 3-, 2'- и 6'-положение соединения, представленного формулой (Xa). Такие защитные группы включают, например, защитные группы, представленные R3, R2' и R6' в соединении, представленном формулой (XXV), или защитные группы, представленные R1, R3, R2' и R6' на схеме 2, которая будет представлена далее. Более конкретно, защитные группы, введенные в аминогруппу в 1-, 3-, 2'- и 6'-положениях соединения, представленного формулой (Xa), предпочтительно являются защитными группами, которые обычно используют в синтетической органической химии, например, они могут представлять собой необязательно замещенный алкилсульфонил, необязательно замещенный арилсульфонил или необязательно замещенный алкилоксикарбонил, более предпочтительно необязательно замещенный C1-6 алкилсульфонил, необязательно замещенный арилсульфонил или необязательно замещенный C1-6 алкилоксикарбонил, еще более предпочтительно C1-6 алкилсульфонил, необязательно замещенный необязательно замещенным фенилом, арилсульфонил, необязательно замещенный метилом, или C1-6 алкилоксикарбонил, необязательно замещенный необязательно замещенным фенилом, более предпочтителен C1-3 алкилсульфонил, необязательно замещенный необязательно замещенным фенилом, арилсульфонил, необязательно замещенный метилом, или C1-4 алкилоксикарбонил, необязательно замещенный необязательно замещенным фенилом, еще более предпочтителен метансульфонил, бензилсульфонил, п-толуолсульфонил, бензилоксикарбонил, трет-бутоксикарбонил, п-метоксибензилоксикарбонил или п-нитробензилоксикарбонил. Более предпочтителен п-толуолсульфонил или бензилоксикарбонил.
Защитная группа, введенная в функциональную группу, отличную от аминогруппы в 1-положении соединения, представленного формулой (Xb), предпочтительно вводится в аминогруппу в 2'-, 6'- и 3"-положениях соединения, представленного формулой (Xb). Защитная группа, введенная в 2'- и 6'-положения, является такой же, что и защитная группа, введенная в 2'- и 6'-положения соединения, представленного формулой (Xa). Защитная группа, введенная в аминогруппу в 3"-положении, является такой же, что и R3" на схеме 3, которая будет представлена далее. Такие защитные группы могут быть такими же как те, которые обычно используют в синтетической органической химии, и предпочтительно представляют собой необязательно замещенный алкилкарбонил, более предпочтителен C1-3 алкилкарбонил, необязательно замещенный атомом галогена, еще более предпочтителен трифторацетил.
В соединениях, представленных формулой (Xc), отщепляемая группа, представленная W, представляет собой атом галогена, такой как хлор, бром или йод, алкилтио или арилтио, более предпочтителен атом галогена, C1-3 алкилтио или арилтио, еще более предпочтителен бром или фенилтио.
Защитная группа для гидроксила, представленная R2", предпочтительно необязательно представляет собой замещенный арилалкил, более предпочтительно арил C1-2 алкил, необязательно замещенный, например, нитро, еще более предпочтительно бензил, п-метоксибензил или п-нитробензил, далее предпочтителен бензил.
Защитные группы для гидроксила, представленные R4" и R6", могут быть одинаковыми или различными, и их примеры включают защитные группы сложноэфирного типа или защитные группы типа простого эфира, предпочтительно алкилкарбонил, арилалкилкарбонил или необязательно замещенный арилалкил, более предпочтительно C1-6 алкилкарбонил, арилC1-3алкилкарбонил, или арилC1-3алкил, необязательно замещенный метокси, более предпочтительно ацетил, бензоил, бензил, п-метоксибензил или трифенилметил.
Защитная группа для гидроксильной группы, представленная R4" и R6", вместе может образовывать циклическую защитную группу. Циклическая защитная группа предпочтительно представляет собой C3-8. Их конкретные примеры включают циклические защитные группы, такие как ацетали или кетали, например циклогексилиденацеталь, изопропилиденацеталь или бензилиденацеталь.
В соединениях, представленных формулой (XVII), например, защитные группы, представленные формулой (XXV), можно указать как защитные группы для аминогруппы, представленной E. Более конкретно, защитная группа для аминогруппы, представленной E, предпочтительно представляет собой необязательно замещенный арилалкилоксикарбонил, более предпочтительно необязательно замещенный арилC1-6алкилоксикарбонил, еще более предпочтительно арилC1-6алкилоксикарбонил, необязательно замещенный необязательно замещенным фенилом, далее предпочтителен бензилоксикарбонил.
Активирующая группа для карбоновой кислоты, представленная F, является группой, которую используют в реакции для образования пептидной связи путем активирования карбоксила (метод активированных сложных эфиров), предпочтительна сукцинимидная группа, п-нитрофенил, пентафторфенил или 1-гидроксибензотриазол, более предпочтительна сукцинимидная группа.
Далее, защитной группой для гидроксильной группы, представленной G, может быть, например, защитная группа сложноэфирного типа или защитная группа типа простого эфира, и их примеры включают защитные группы, представленные формулой (XXV). Более конкретно, защитная группа для гидроксильной группы, представленной G, предпочтительно представляет собой необязательно замещенный алкилкарбонил, необязательно замещенный арилалкилкарбонил или необязательно замещенный арилалкил, более предпочтительно необязательно замещенный C1-6 алкилкарбонил, необязательно замещенный арилалкилкарбонил или необязательно замещенный арилC1-3алкил, еще более предпочтительно C1-6 алкилкарбонил; арилкарбонил; или арилC1-3алкил, необязательно замещенный, например, метокси, далее предпочтителен, например, ацетил, бензоил, бензил, п-метоксибензил или трифенилметил.
Реакционные условия для каждой стадии первого способа получения в соответствии с настоящим изобретением будут раскрыты более подробно в (1), (4) и (5), которые будут представлены далее.
Второй способ получения
Во втором способе получения настоящего изобретения в дополнение к соединениям, представленным формулой (Xa), соединения, представленные формулой (XXV), могут быть использованы в качестве синтетических промежуточных соединений. Указанный способ можно использовать для получения соединений, в которых пространственная конфигурация гидроксильной группы в 5-положении соответствует экваториальной, среди соединений, представленных формулой (Ia).
В соответствии с другим способом получения настоящего изобретения предложен способ получения соединения, представленного формулой (Ia):
[Химическая формула 13]
где
R5ax представляет собой атом водорода,
R5eq представляет собой гидроксил, и
Req, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил,
n является целым числом от 1 до 4, и
пространственная конфигурация атома углерода, отмеченного *, соответствует R или S,
причем указанный способ включает стадии
введения защитных групп в аминогруппы в 3-, 2'- и 6'-положениях, и в гидроксильную группу в 2-положении соединения, представленного формулой (Xa):
[Химическая формула 14]
где R5ax и R5eq имеют указанные для формулы (Ia) значения,
осуществления взаимодействия полученного соединения с соединением, представленным формулой (XVII):
[Химическая формула 15]
где E представляет собой защитную группу для аминогруппы;
G представляет собой защитную группу для гидроксильной группы;
F представляет собой атом водорода или активирующую группу для карбоновой кислоты;
n является целым числом от 1 до 4; и
пространственная конфигурация атома углерода, отмеченного *, соответствует R или S,
с получением соединения, представленного формулой (XXV):
[Химическая формула 16]
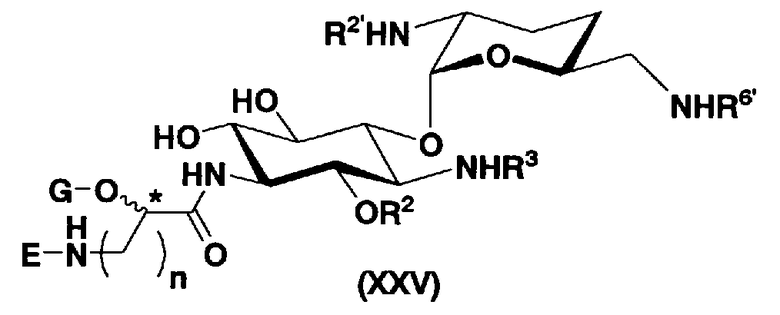
где R2 представляет собой защитную группу для гидроксильной группы; R3, R2' и R6' представляют собой защитные группы для аминогруппы, и E, G, n и пространственная конфигурация атома углерода, отмеченного *, имеют указанные для формулы (XVII) значения,
осуществления взаимодействия соединения, представленного формулой (XXV), с соединением, представленным формулой (Xc) или (Xd):
[Химическая формула 17]
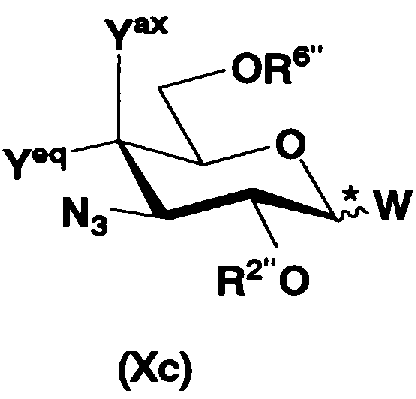
где W представляет собой отщепляемую группу; Yax и Yeq, которые могут быть одинаковыми или различными, представляют собой группу -OR4” или атом водорода; R2”, R4” и R6” представляют собой защитные группы для гидроксильной группы; и пространственная конфигурация атома углерода, отмеченного *, соответствует R или S,
[Химическая формула 18]
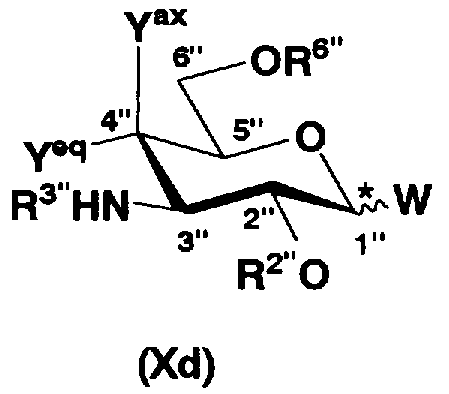
где W, Yax, Yeq, R2”, R6” и пространственная конфигурация атома углерода, отмеченного *, имеют указанные для формулы (Xc) значения, и R3" представляет собой защитную группу для аминогруппы, и
удаления защитных групп в полученном соединении и, если используют соединение, представленное формулой (Xc), превращения азидной группы в соединении в аминогруппу с получением соединения, представленного формулой (Ia).
Во втором способе получения в соответствии с настоящим изобретением соединение, представленное формулой (XVII), может быть энантиомерной смесью относительно атома углерода, отмеченного *. Соответственно, в соответствии со вторым способом получения настоящего изобретения, можно получить диастереомерную смесь относительно атома углерода, отмеченного *, в соединении, представленном формулой (Ia). Настоящее изобретение включает также и указанный вариант.
Во втором способе получения настоящего изобретения гидроксил предпочтительно находится в 4"-положении в соединении, представленном формулой (Ia). Соответственно, во втором способе получения настоящего изобретения в предпочтительном варианте настоящего изобретения R4"ax и R4"eq, которые отличаются друг от друга, представляют собой атом водорода или гидроксил.
Во втором способе получения в соответствии с настоящим изобретением конкретные воплощения защитных групп R2, R3, R2' и R6', вводимых в аминогруппу в 3-, 2'- и 6-положении, и в гидроксильную группу в 2-положении, имеют указанные выше значения. Далее, W, Yax, Yeq, R2" и R6" имеют такие же значения, как в первом способе получения.
Реакционные условия для каждой стадии во втором способе получения в соответствии с настоящим изобретением будут раскрыты подробно в (2) и (3), которые будут представлены далее.
Стадия получения синтетического промежуточного соединения (Xa)
Соединения, представленные формулой (Ia), в соответствии с настоящим изобретением можно синтезировать, используя соединение, представленное формулой (II): О-3-дезокси-4-C-метил-3-(метиламино)-β-L-арабинопиранозил-(1→6)-О-[2,6-диамино-2,3,4,6-тетрадезокси-α-D-эритроглюкопиранозил-(1→4)]-D-стрептамин (здесь и далее именуемое как "2-гидроксигентамицин C1a"), как один из исходных материалов для получения указанных соединений. Соединение, представленное формулой (II), можно с успехом использовать в качестве исходного вещества для получения соединений, представленных формулой (Xa), в представленных выше двух способах получения.
[Химическая формула 19]
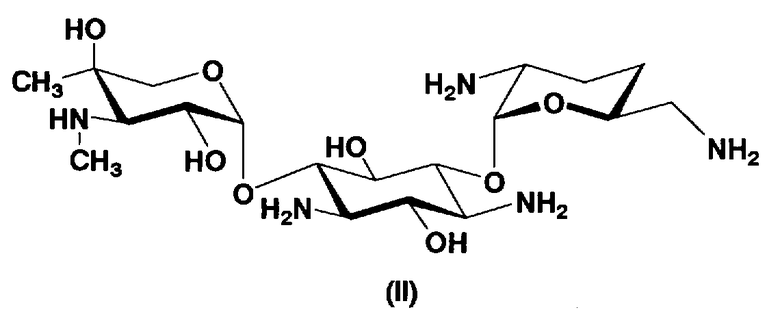
Соединение, представленное формулой (II) (2-гидроксигентамицин C1a), представляет собой соединение, полученное путем замены 2-дезоксистрептамина, который является частичным составляющим элементом в гентамицине C1a как аналога гентамицина, стрептамином. Указанное соединение можно получить обычным способом, то есть добавляя аналог дезоксистрептамина к зависимому от дезоксистрептамина продуцирующему штамму аминогликозида, культивируя продуцирующий штамм и выделяя новое аминогликозидное вещество-антибиотик, которое является веществом, полученным заменой части составляющего элемента аминогликозида, добавляемым аналогом из полученной культуры. Более конкретно, соединение, представленное формулой (II), можно получить, добавляя стрептамин в зависимый от дезоксистрептамина продуцирующий штамм гентамицина С1а и культивируя смесь. Такие продуцирующие штаммы включают, например, Micromonospora purpurea ATCC 31119. Вышеуказанный способ получения подробно раскрыт в японской открытой патентной выкладке №108041/1976, содержание которой включено сюда для ссылки. Способ получения производного аминогликозидного вещества-антибиотика и способ получения продуцирующего штамма для использования в способе получения раскрыты, например, Shier, W.T., K.L.Rinehart Jr. & D.Gottlieb et al., Proc. Nat. Acad. Sci. 63: pp.198 to 204, (1969), где неомицин является целевым соединением, и Kojima M., Sato A. et al., J. Antibiot. 26(12): pp.784-6 (1973), где целевыми соединениями являются рибостамицин и канамицин.
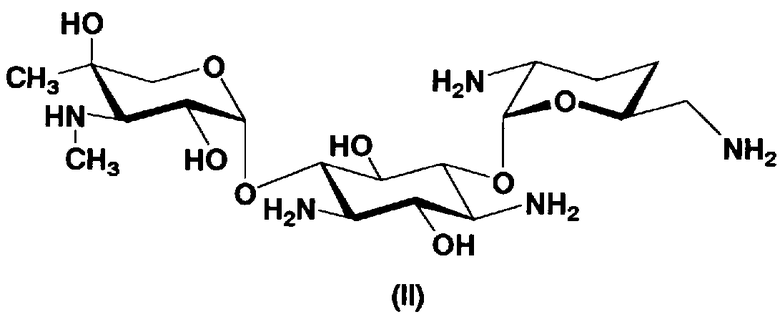
В способе получения в соответствии с настоящим изобретением, если получают соединение, представленное формулой (Ia), и в котором 5-положение гидроксильной группы соответствует экваториальному, предпочтительно, чтобы соединение, представленное формулой (II), было гидролизовано с получением соединения, представленного формулой (Xa). Условия осуществления гидролиза подробно раскрыты в первых шести стадиях на схеме 2, которая будет представлена далее.
[Химическая формула 20]
[Химическая формула 21]

где R5ax представляет собой атом водорода и R5eq представляет собой гидроксил.
В способе получения в соответствии с настоящим изобретением, если получают соединение, которое представлено формулой (Ia), и в котором пространственная конфигурация гидроксильной группы в 5-положении соответствует аксиальной, пространственная конфигурация гидроксильной группы в 5-положении соединения, представленного формулой (II), является инвертной. Так, в другом варианте настоящего изобретения способ получения соединения, представленного формулой (Ia), включает введение защитных групп в гидроксильные группы, отличные от гидроксильных групп в 4"- и 5-положениях, и в аминогруппы соединения, представленного формулой (II),
обращения пространственной конфигурации гидроксильной группы в 5-положении полученного соединения,
удаления защитных групп полученного соединения и гидролиза указанного соединения, с получением соединения, представленного формулой (Xa).
[Химическая формула 22]
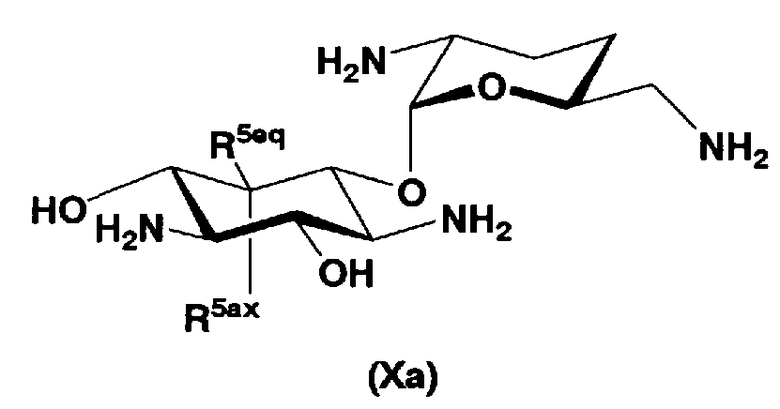
где R5ax представляет собой гидроксил и R5eq представляет собой атом водорода.
В предпочтительном варианте настоящего изобретения способ получения далее включает удаление гидроксильной группы в 4"-положении до или одновременно с обращением пространственной конфигурации гидроксильной группы в 5-положении. Указанная стадия подробно раскрыта на стадиях 5-3 до 5-5 на схеме 10, которая будет представлена далее.
Защитные группы, введенные в гидроксильные группы, отличные от гидроксильных групп в 4"- и 5-положениях, и в аминогруппы соединения, представленного формулой (II), то есть R1, R2, R3, R2', R6', R2" и R3" имеют указанные выше значения.
Способы получения в соответствии с настоящим изобретением будут классифицированы в соответствии с пространственной конфигурацией гидроксильных групп в 5- и 4"-положениях и типом способа получения и будут раскрыты более подробно.
(1) Получение соединений, в которых 5- и 4"-положения соответствуют экваториальным: первый способ получения
В первом способе получения в соответствии с настоящим изобретением, среди соединений, представленных формулой (Ia), соединения, в которых R5ax и R4"ax представляют собой атом водорода, оба, R5eq и R4"eq, представляют собой гидроксил, можно получить в соответствии со следующими тремя схемами, то есть схемой 1 (стадия 1-1 до стадии 1-5a и стадия 1-5b), схемой 2 (стадия 1-6 до стадии 1-7) и схемой 3 (стадия 1-8 до стадии 1-14).
На схеме 1 способ получения соединений, представленных формулой (Xc), использованный в способе получения в соответствии с настоящим изобретением будет раскрыт конкретно.
[Химическая формула 23]
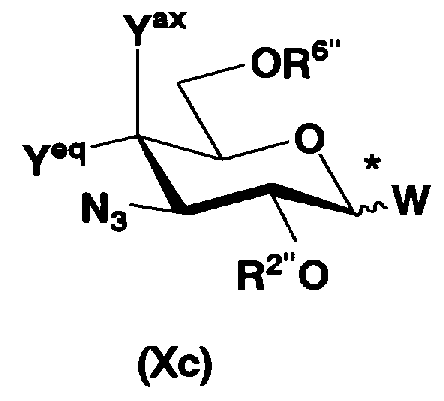
где W представляет собой отщепляемую группу; Yax представляет собой атом водорода; Yeq представляет собой группу -OR4"; R2", R4" и R6" представляют собой защитные группы для гидроксильной группы; и пространственная конфигурация атома углерода, отмеченного *, соответствует R или S.
Схема 1
Схема 1 раскрывает стадии с 1-1 до стадии 1-5a и стадию 1-5b. На схеме 1 соединения, представленные формулой (Xc), классифицированы как соединения, представленные формулой (VIII), и соединения, представленные формулой (IX) в соответствии с типом отщепляемой группы, представлены как W.
[Химическая формула 24]
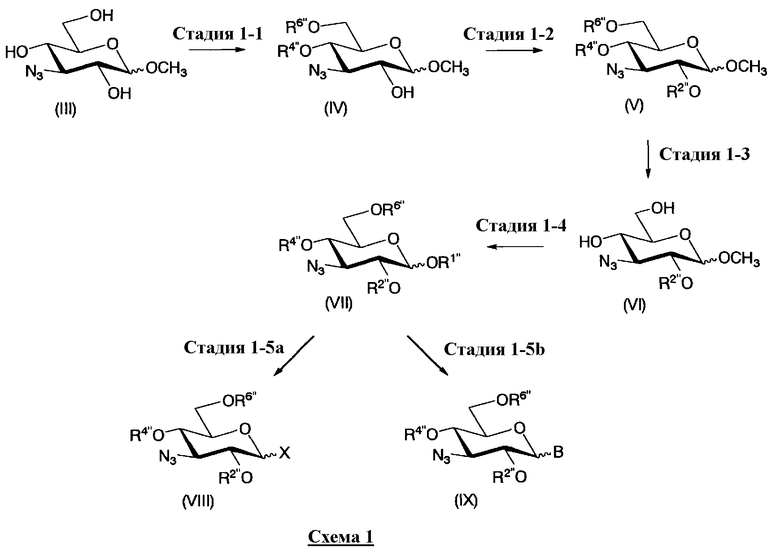
где B представляет собой отщепляемую группу, содержащую атом серы, такую как метилтио, этилтио или фенилтио, предпочтительно фенилтио; X представляет собой атом галогена, такой как хлор, бром или йод, предпочтительно атом брома; R1" представляет собой защитную группу для гидроксильной группы, предпочтительно, например, защитную группу сложноэфирного типа, такую как ацетил или бензоил, более предпочтительно ацетил; R2" представляет собой защитную группу для гидроксильной группы, предпочтительно защитную группу типа бензила, которую можно удалить способом каталитического восстановления с использованием водорода, такую как бензил, п-метоксибензил или п-нитробензил, более предпочтительно бензил; и
R4" и R6", которые могут быть одинаковыми или различными, каждый независимо представляет собой защитную группу для гидроксила, например защитную группу сложноэфирного типа, такую как ацетил или бензоил, или защитную группу типа простого эфира, такую как бензил, п-метоксибензил или трифенилметил, предпочтительно защитную группу сложноэфирного типа, такую как ацетил или бензоил, или R4" и R6" вместе представляют собой циклическую защитную группу, такую как ацеталь или кеталь, для одновременной защиты двух гидроксильных групп, например циклогексилиденацеталь, изопропилиденацеталь или бензилиденацеталь.
Стадия 1-1
Стадия 1-1 является стадией, на которой защитные группы вводят в две гидроксильные группы в 4- и 6-положениях соединения, представленного формулой (III), с получением соединения, представленного формулой (IV). Защитная группа представляет собой защитную группу типа ацеталя или кеталя, в которой R4" и R6" объединяются с образованием одной защитной группы, предпочтительно изопропилиденовой группы. Указанной стадии достигают, осуществляя взаимодействие соединения, представленного формулой (III), с кетоном, представителем которого является ацетон, или с ацеталем, представителем которого является 2,2-диметоксипропан, в присутствии кислоты.
Растворители, которые можно использовать на данной стадии, включают, например, N,N-диметилформамид, метиленхлорид, хлороформ, 1,2-дихлорэтан или этилацетат. Среди них предпочтителен N,N-диметилформамид. Кислоты, которые можно здесь использовать, включают п-толуолсульфоновую кислоту, пиридиний п-толуол сульфонат, камфорсульфоновую кислоту или хлористоводородную кислоту. Среди них предпочтительна п-толуолсульфоновая кислота.
Температура реакции находится в интервале от 20°C до температуры кипения с обратным холодильником. Время реакции составляет, например, от 1 до 24 часов.
В этой реакции, например, если используют ацеталь, представленный 2,2-диметоксипропаном, указанный способ можно адаптировать, и тогда реакцию ведут, удаляя из реакционной системы спирт, который образуется как побочный продукт, путем перегонки при пониженном давлении для ускорения реакции.
Стадия 1-2
Стадия 1-2 представляет собой стадию получения соединения, представленного формулой (V), путем введения защитной группы (R2") в гидроксильную группу в 2-положении соединения, представленного формулой (IV). Указанную стадию проводят, осуществляя взаимодействие соединения, представленного формулой (IV), с R2"X, где R2" представляет собой, например, бензил, п-метоксибензил или п-нитробензил, и X представляет собой, например, хлор, бром или йод, в присутствии основания.
Растворители, которые можно использовать на этой стадии, включают пиридин, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан или метиленхлорид. Среди них предпочтителен N,N-диметилформамид. Основания, которые можно здесь использовать, включают пиридин, лутидин, коллидин, триэтиламин, диизопропилэтиламин, 4-диметиламинопиридин, гидрид натрия и гидроксид калия. Среди них предпочтителен гидрид натрия.
Температура реакции составляет от -20°C до 50°C. Время реакции составляет от 1 до 24 часов.
Стадия 1-3
Стадия 1-3 представляет собой стадию удаления защитной группы в 4- и 6-положениях соединения, представленного формулой (V), с получением соединения, представленного формулой (VI). Указанную стадию проводят, осуществляя взаимодействие соединения, представленного формулой (V), с кислотой.
Растворители, которые можно использовать на этой стадии, включают тетрагидрофуран, диэтиловый эфир, 1,4-диоксан, метанол, метиленхлорид, хлороформ, уксусную кислоту, воду или состоящие из них смешанные растворители. Среди них предпочтителен смешанный растворитель, состоящий из уксусной кислоты и воды. Кислоты, которые можно здесь использовать, включают уксусную кислоту, трифторуксусную кислоту, хлористоводородную кислоту, серную кислоту, п-толуолсульфоновую кислоту или треххлористый бор. Среди них предпочтительна уксусная кислота.
Температура реакции находится в интервале от 0°C до температуры кипения с обратным холодильником. Время реакции составляет от 0,1 до 12 часов.
Стадия 1-4
Стадия 1-4 представляет собой стадию введения защитных групп в каждую из гидроксильных групп в 4- и 6-положениях соединения, представленного формулой (VI), и превращения метоксигруппы в 1-положении соединения, представленного формулой (VI), в ацилоксигруппу, с получением соединения, представленного формулой (VII).
Указанную стадию можно провести, например, одновременно осуществляя взаимодействие карбоновой кислоты, представленной R1"OH, такой как уксусная кислота, и ангидрида кислоты, представленного R4" 2O (или R6" 2О), такого как уксусный ангидрид, с соединением, представленным формулой (VI), в присутствии кислотного катализатора.
Растворители, которые можно использовать на вышеуказанной стадии, включают, например, метиленхлорид, хлороформ, 1,2-дихлорэтан, уксусную кислоту, уксусный ангидрид или состоящий из них смешанный растворитель. Среди них предпочтителен смешанный растворитель, состоящий из уксусной кислоты, и уксусный ангидрид. Кислоты, которые можно здесь использовать, включают хлористоводородную кислоту или серную кислоту. Предпочтительна серная кислота.
Температура реакции составляет от -20°C до 50°C. Время реакции составляет от 1 до 24 часов.
Стадию 1-4 можно осуществить в двух раздельных стадиях.
В этом случае вначале осуществляют первую стадию введения защитных групп в гидроксильную группу в 4- и 6-положениях и затем осуществляют вторую стадию превращения метоксигруппы в 1-положении в ацилоксигруппу.
Первую стадию проводят, осуществляя взаимодействие соединения, представленного формулой (VI), с ангидридом кислоты, таким как уксусный ангидрид, или галогенангидридом, таким как ацетилхлорид, в присутствии основания. Растворители, которые можно здесь использовать, включают, например, пиридин, N,N-диметилформамид, метиленхлорид, хлороформ или 1,2-дихлорэтан. Среди них предпочтителен пиридин. Основания, которые можно здесь использовать, включают триэтиламин, пиридин или 4-диметиламинопиридин. Среди них пиридин предпочтителен. Температура реакции составляет от -20°C до 50°C. Время реакции составляет от 1 до 24 часов.
На второй стадии соединение, полученное на первой стадии, подвергают взаимодействию с карбоновой кислотой, представленной R1"OH, такой как уксусная кислота, и ангидридом кислоты, представленным R1" 2О, таким как уксусный ангидрид, в присутствии кислотного катализатора.
Растворители, которые можно использовать на второй стадии, включают метиленхлорид, хлороформ, 1,2-дихлорэтан, уксусную кислоту, уксусный ангидрид или состоящие из них смешанные растворители. Среди них предпочтителен смешанный растворитель, состоящий из уксусной кислоты и уксусного ангидрида. Кислоты, которые можно здесь использовать, включают хлористый водород или серную кислоту. Предпочтительна серная кислота.
Температура реакции составляет от -20°C до 50°C. Время реакции составляет от 1 до 24 часов.
Стадия 1-5a
Стадия 1-5a представляет собой стадию превращения алкоксигруппы (OR1") в 1-положении соединения, представленного формулой (VII), в атом галогена, с получением соединения, представленного формулой (VIII). Указанную стадию проводят, осуществляя взаимодействие соединения, представленного формулой (VII), с галогенводородом, представленным HX, или галогенидом титана, представленным TiX4, где X представляет собой атом хлора или атом брома.
Растворители, которые можно использовать на этой стадии, включают метиленхлорид, хлороформ, 1,2-дихлорэтан, этилацетат или состоящие из них смешанные растворители. Среди них предпочтителен смешанный растворитель, состоящий из метиленхлорида и этилацетата.
Температура реакции составляет от -20°C до 50°C.
Время реакции составляет от 1 до 24 часов.
Стадия 1-5b
Стадия 1-5b представляет собой стадию превращения ацилоксигруппы (OR1") в 1-положении соединения, представленного формулой (VII), в тиоалкил или тиоарил в присутствии кислоты Льюиса, с получением соединения, представленного формулой (IX). Конкретно, указанную стадию проводят, осуществляя взаимодействие соединений, представленных формулой (VII), с тиолом, представленным BH, где B представляет собой, например, метилтио, этилтио или фенилтио или триметилсилилированный тиол, представленный TMS-B, где TMS представляет собой триметилсилил, и B имеет указанные выше значения, в присутствии кислоты Льюиса.
Растворители, которые можно использовать на указанной стадии, включают, например, метиленхлорид, хлороформ, 1,2-дихлорэтан, этилацетат или состоящие из них смешанные растворители. Среди них предпочтителен метиленхлорид. Кислоты Льюиса, которые можно здесь использовать, включают триметилсилилтрифлат или хлорид олова. Предпочтителен триметилсилилтрифлат. Температура реакции находится в интервале от 15-20°C до температуры кипения с обратным холодильником. Время реакции составляет от 1 до 48 часов.
Все заместители в 1-положении соединения, представленного на схеме 1, могут существовать в двух пространственных конфигурациях, то есть в аксиальной форме и в экваториальной форме. На схеме 1, указанные две пространственные конфигурации можно отделить друг от друга до использования в реакции, или альтернативно две пространственные конфигурации в смешанной форме можно использовать в реакции без разделения. Далее, соединение, представленное формулой (VIII), и соединение, представленное формулой (IX), полученное в соответствии со схемой 1, можно выделить в аксиальной форме и в экваториальной форме, которые затем используют отдельно друг от друга в соответствии со схемой 3. Альтернативно, указанные соединения можно использовать в виде смеси аксиальной формы с экваториальной формой.
Далее будет подробно раскрыт способ получения соединения, представленного формулой (Xa), включающий стадию 1-6 и стадию 1-7 в соответствии со схемой 2. На схеме 2, соединение, представленное формулой (Xa), в котором пространственная конфигурация атома водорода в 5-положении соответствует экваториальной, соответствует соединению, представленному формулой (X).
[Химическая формула 25]
где R5ax представляет собой атом водорода и R5eq представляет собой гидроксил.
[Химическая формула 26]
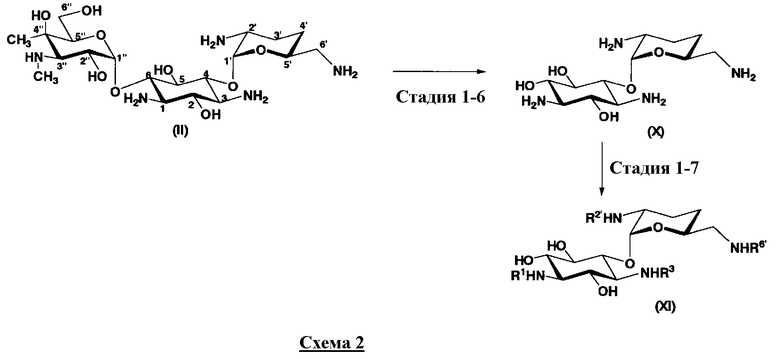
где R1, R3, R2' и R6' представляют собой защитные группы для аминогруппы, предпочтительно защитные группы, которые обычно используют в органической синтетической химии, такие как метансульфонил, бензилсульфонил, п-толуолсульфонил, бензилоксикарбонил, трет-бутоксикарбонил, п-метоксибензилоксикарбонил или п-нитробензилоксикарбонил, более предпочтительно п-толуолсульфонил или бензилоксикарбонил.
Стадия 1-6
Стадия 1-6 является стадией гидролиза соединения, представленного формулой (II) (2-гидроксигентамицин C1a), в качестве исходного соединения, с получением соединения, представленного формулой (X). Указанную стадию осуществляют, нагревая соединение, представленное формулой (II), в присутствии кислоты.
На представленной выше стадии в качестве растворителя предпочтительно используют воду. Кислоты, которые можно здесь использовать, включают хлористоводородную кислоту, серную кислоту, азотную кислоту или бромистоводородную кислоту. Среди них предпочтительна 3-5 M хлористоводородная кислота.
Температура реакции находится в интервале от 20°C до температуры кипения с обратным холодильником. Время реакции составляет от 0,5 до 24 часов.
Стадия 1-7
Стадия 1-7 является стадией введения защитных групп в четыре аминогруппы в соединении, представленном формулой (X), с получением соединения, представленного формулой (XI). Указанную стадию можно провести, осуществляя взаимодействие соединения, представленного формулой (X), с хлороформовым эфиром, таким как бензилхлороформиат, п-метоксибензилхлороформиат или п-нитробензилхлороформиат, с карбоновыми диэфирами, такими как ди-трет-бутилдикарбонат, или сульфонилирующим агентом, таким как метансульфонилхлорид, бензилсульфонилхлорид или п-толуолсульфонилхлорид в присутствии основания.
Растворители, которые можно использовать на этой стадии, включают воду, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, ацетон или состоящие из них смешанные растворители. Среди них предпочтителен смешанный растворитель, состоящий из воды и 1,4-диоксана. Основания, которые можно здесь использовать, включают гидрид натрия, карбонат калия, карбонат натрия, триэтиламин, пиридин или 4-диметиламинопиридин. Предпочтителен карбонат натрия.
Температура реакции составляет от -20°C до 50°C, и время реакции составляет от 1 до 24 часов.
Первый способ получения настоящего изобретения будет раскрыт более подробно в соответствии со схемой 3, на которой представлены стадии от 1-8 до 1-14. На схеме 3 соединение, представленное формулой (Ia), в итоге синтезируют через соединение, представленное формулой (XIV), которое представляет собой ключевое промежуточное соединение, представленное формулой (Ia).
[Химическая формула 27]
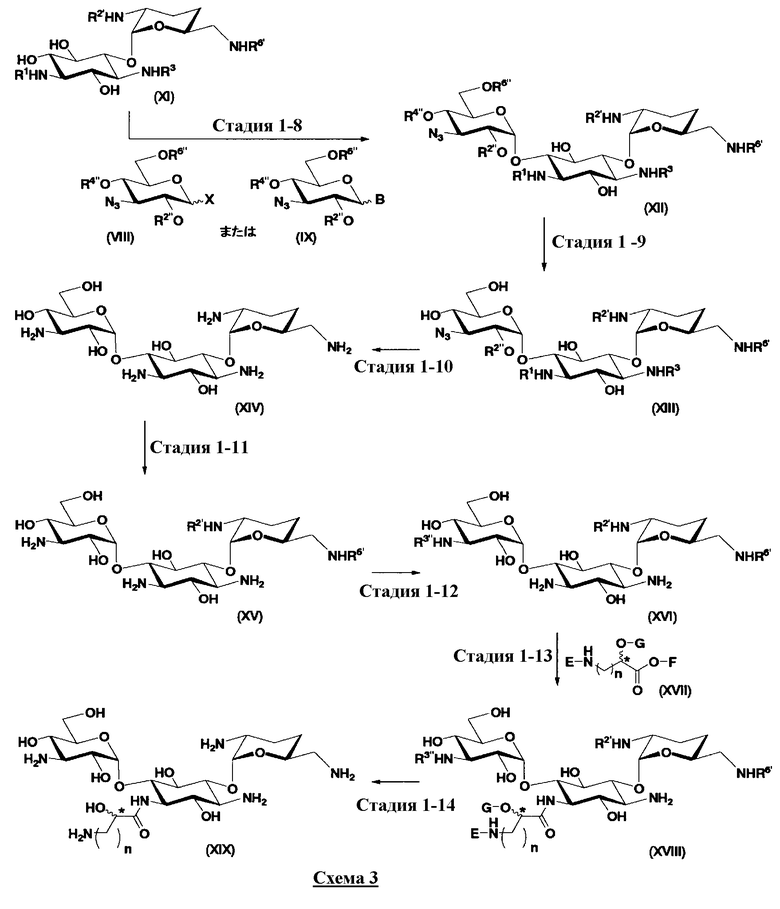
где B, X, R1, R3, R2', R6', R2", R4", R6", n и пространственная конфигурация атома углерода, отмеченного *, имеют указанные на схемах 1 и 2 значения,
E представляет собой защитную группу для амино, предпочтительно защитную группу, которую обычно используют для аминогруппы в органической синтетической химии, более предпочтительно бензилоксикарбонил,
F представляет собой атом водорода или активирующую группу для карбоновой кислоты, которую используют в реакции, которая активирует карбоксил для получения пептидной связи (метод активированных эфиров), предпочтительно, сукцинимидную группу, п-нитрофенил, пентафторфенил или 1-гидроксибензотриазол, более предпочтительна сукцинимидная группа,
G представляет собой атом водорода или защитную группу для гидроксильной группы, например защитную группу сложноэфирного типа, такую как ацетил или бензоил, или защитную группу типа простого эфира, такую как бензил, п-метоксибензил или трифенилметил, и
R3" представляет собой защитную группу для аминогруппы, предпочтительно такую, как защитные группы для аминогруппы, которые обычно используют в органической синтетической химии, более предпочтительно трифторацетил.
Стадия 1-8
Стадия 1-8 представляет собой стадию конденсации гидроксильной группы в 6-положении соединения, представленного формулой (XI), с соединением, представленным формулой (VIII), полученным на стадии 1-5a, или соединением, представленным формулой (IX), полученным на стадии 1-5b, с получением соединения, представленного формулой (XII). Указанную стадию можно провести, осуществляя взаимодействие соединения, представленного формулой (XI), с соединением, представленным формулой (VIII) или формулой (IX), в присутствии катализатора и обезвоживающего агента.
Растворители, которые можно использовать на указанной стадии, включают, например, N,N-диметилформамид, метиленхлорид, хлороформ, 1,2-дихлорэтан, диэтиловый эфир или этилацетат, предпочтительно 1,2-дихлорэтан. Катализаторы, которые можно здесь использовать, включают трифторметансульфоновую кислоту, цианид ртути, N-йодосукцинимид, трифторуксусную кислоту, бромид ртути или желтый оксид ртути. Предпочтителен цианид ртути. Обезвоживающие агенты, которые можно здесь использовать, включают, например, молекулярные сита 4Å или Drierite, предпочтительно Drierite.
Температура реакции составляет от -20°C до 60°C. Время реакции составляет от 1 до 24 часов.
Если X в формуле (VIII) соединения, полученного на стадии 1-5a, представляет собой атом брома, указанную стадию можно осуществить, используя обычную реакцию гликозилирования, используя донор бромированного сахарида, именуемую реакцией гликозилирования Кенигса-Кнорра (Chem. Ber., Vol.34, p.957 (1901)). Условия для проведения указанной реакции можно соответствующим образом определить, обратившись к обзору H.Paulsen et al. (Angew. Chem. Int. Ed. Engl., Vol.21, pp.155-173 (1982)), обзору R.R.Schmidt (Angew. Chem. Int. Ed. Engl., Vol.25, pp.212-235 (1986)) и т.п.
С другой стороны, если B в формуле (IX) соединения, полученного на стадии 1-5b, представляет собой тиофенил, указанную стадию можно осуществить, используя сообщение G.H.Veeneman et al. (Tetrahedron Letters, Vol.31, pp.1331-1334 (1990)), сообщение P.Konradsson et al. (Tetrahedron Letters, Vol.31, pp.4313-4316 (1990)) и т.п.
Стадия 1-9
Стадия 1-9 представляет собой стадию удаления защитной группы (R4") в 4"-положении и защитной группы (R6") в 6"-положении соединения, представленного формулой (XII), с получением соединения, представленного формулой (XIII). Указанную стадию можно провести, осуществляя взаимодействие соединения, представленного формулой (XII), с основанием.
Растворители, которые можно использовать на этой стадии, включают метанол, этанол, изопропиловый спирт, трет-бутиловый спирт, метиленхлорид, хлороформ, или состоящие из них смешанные растворители. Среди них предпочтителен метанол. Основания, которые можно здесь использовать, включают карбонат калия, карбонат натрия, гидроксид калия, гидроксид натрия, метоксид натрия, этоксид натрия, или трет-BuOK. Предпочтителен метоксид натрия.
Температура реакции составляет от -20°C до 60°C, и время реакции составляет от 1 до 24 часов.
Стадия 1-10
Стадия 1-10 является стадией получения соединения, представленного формулой (XIV), в качестве ключевого промежуточного соединения. Соединение, представленное формулой (XIV), составляет базовый скелет соединения, представленного формулой (Ia). Соответственно, соединение, представленное формулой (XIV), можно использовать в качестве промежуточного соединения для получения соединения, представленного формулой (Ia), и его производных, и настоящее изобретение включает также и указанный вариант.
На стадии 1-10 все защитные группы в соединении, представленном формулой (XIII), удаляют, и далее азидогруппу в 3"-положении превращают в аминогруппу, получая соединение, представленное формулой (XIV). Указанную стадию можно провести радикально, осуществляя взаимодействие соединения, представленного формулой (XIII), со щелочным металлом для удаления защитных групп для всех аминогрупп и гидроксильных групп в 2"-положении, и превращая азидогруппу в 3"-положении в аминогруппу, то есть адаптируя так называемые "условия восстановления Бирча."
Растворители, которые можно использовать на вышеуказанной стадии, включают жидкий аммиак, метиламин, этиламин, гексаметилфосфоамид, диэтиловый эфир, тетрагидрофуран или смешанный растворитель, состоящий из них, предпочтительно, жидкий аммиак. Щелочные металлы, которые можно здесь использовать, включают литий, натрий или калий, предпочтительно натрий.
Температура реакции составляет от -60°C до 20°C, и время реакции составляет от 0,5 до 24 часов.
Если защитная группа для аминогруппы в соединении, представленном формулой (XIII), представляет собой защитную группу, которую можно удалить путем каталитического восстановления с использованием водорода, например бензилоксикарбонил, п-метоксибензилоксикарбонил или п-нитробензилоксикарбонил, вышеуказанную стадию можно также провести, осуществляя взаимодействие соединения, представленного формулой (XIII), с водородом в присутствии катализатора реакции каталитического восстановления с использованием водорода. Катализаторы каталитического восстановления с использованием водорода, которые можно здесь использовать, включают палладий-на-угле, палладиевую чернь, гидроксид палладия и оксид платины. Среди них предпочтителен палладий-на-угле. В указанной реакции можно использовать любой растворитель без конкретных ограничений, если только указанный растворитель не вовлечен в реакцию. Предпочтительны метанол, этанол, тетрагидрофуран, 1,4-диоксан, состоящие из них смешанные растворители или смешанный растворитель, состоящий из вышеуказанного органического растворителя и воды.
Температура реакции составляет от 10°C до 30°C. Время реакции составляет обычно от 1 до 8 часов.
Если защитная группа для аминогруппы в соединении, представленном формулой (XIII), представляет собой трет-бутоксикарбонил, указанный способ можно также адаптировать таким образом, чтобы соединение, представленное формулой (XIII), подвергалось взаимодействию с водородом в присутствии катализатора реакции каталитического восстановления с использованием водорода для удаления защитной группы для гидроксильной группы в 2"-положении и для превращения азидогруппы в 3"-положении в аминогруппу с последующим осуществлением реакции полученного соединения с кислотой для удаления трет-бутоксикарбонила. В этом случае растворители, которые можно использовать для удаления защитной группы для аминогруппы, включают этилацетат, метиленхлорид, ацетонитрил, ацетон, анизол, воду или состоящие из них смешанные растворители. Среди них предпочтительна вода. Кислоты, которые можно здесь использовать, включают п-толуолсульфоновую кислоту, метансульфоновую кислоту, уксусную кислоту или трифторуксусную кислоту, предпочтительна трифторуксусная кислота.
Температура реакции составляет обычно от 0°C до 30°C.
Время реакции составляет от 1 до 12 часов.
Стадия 1-11
Стадия 1-11 является стадией селективного введения защитных групп (R2' и R6') в аминогруппу в 2'- и 6'-положениях соединения, представленного формулой (XIV), с получением соединения, представленного формулой (XV). Указанную стадию можно провести, осуществляя взаимодействие соединения, представленного формулой (XIV), с хлороформовым сложным эфиром, таким как бензилхлороформиат, п-метоксибензилхлороформиат или п-нитробензилхлороформиат, с карбоновым сложным эфиром, таким как ди-трет-бутилдикарбонат или N-(бензилоксикарбонилокси)сукцинимид, в присутствии соли металла.
Растворители, которые можно использовать на указанной стадии, включают, например, N,N-диметилформамид, диметилсульфоксид, метанол, этанол или изопропиловый спирт, предпочтителен метанол. Соли переходных металлов, которые можно здесь использовать, включают ацетат цинка, ацетат никеля или ацетат кобальта, предпочтительно ацетат никеля.
Температура реакции составляет от -20°C до 50°C, и время реакции составляет от 1 до 24 часов.
Стадия 1-12
Стадия 1-12 является стадией селективного введения защитных групп (R3") в аминогруппу в 3"-положении соединения, представленного формулой (XV), с получением соединения, представленного формулой (XVI). Указанную стадию можно провести, осуществляя взаимодействие соединения, представленного формулой (XV), например, с ангидридом галогенированной карбоновой кислоты, таким как трифторуксусный ангидрид или трихлоруксусный ангидрид, с галогенированным эфиром карбоновой кислоты, таким как метилтрифторацетат или этилтрифторацетат, или галогенангидридом галогенированной карбоновой кислоты.
На вышеуказанной стадии предпочтительные реагенты, которые можно здесь использовать, включают этилтрифторацетат.
Растворители, которые можно использовать на указанной стадии, включают, например, N,N-диметилформамид, диметилсульфоксид, тетрагидрофуран или 1,4-диоксан, предпочтительно N,N-диметилформамид.
Температура реакции составляет от -20°C до 50°C, и время реакции составляет от 1 до 24 часов.
Стадия 1-13
Стадия 1-13 является стадией осуществления взаимодействия аминогруппы в 1-положении соединения, представленного формулой (XVI), с производным ω-амино-α-гидроксикарбоновой кислоты, представленным формулой (XVII), с получением соединения, представленного формулой (XVIII), то есть стадией проведения реакции образования пептидной связи. Соединение, представленное формулой (XVII), представляет собой, например, производное 4-амино-2-гидроксимасляной кислоты, которое можно получить обычным способом органического синтеза, используя соответствующее исходное соединение. Альтернативно, указанное соединение можно синтезировать в соответствии со способом, представленным H. Kawaguchi et al. (Journal of Antibiotics, Vol.25, pp.695-708 (1972)). На указанной стадии, если используют соединение, представленное формулой (XVII), где F представляет собой атом водорода, используют пептидный конденсирующий агент, который обычно используют в органическом синтезе. Пептидные конденсирующие агенты включают, например, дициклогексилкарбодиимид, диизопропилкарбодиимид, N-этил-N'-диметиламинопропилкарбодиимид и его гидрохлорид, бензотриазол-1-ил-трис(диметиламино)фосфоний гексафторфосфид и дифенилфосфорилазид. Их можно использовать отдельно или альтернативно их можно использовать в комбинации с N-гидроксисукцинимидом, 1-гидроксибензотриазолом или т.п. Если используют реакцию, которая активирует карбоксильную группу для образования пептидной связи (метод активированных эфиров) в формуле (XVII), F представляет собой активирующую группу для карбоновой кислоты, выбранную из сукцинимидной группы, п-нитрофенила, пентафторфенила, 1-гидроксибензотриазола или т.п. То есть образуется соединение, называемое "активированным эфиром". В некоторых случаях указанный активированный эфир выделяют до использования.
Растворители, которые можно использовать на вышеуказанной стадии, включают N,N-диметилформамид, диметилсульфоксид, тетрагидрофуран или 1,4-диоксан. Среди них предпочтителен тетрагидрофуран.
Температура реакции составляет от -20°C до 50°C, и время реакции составляет от 1 до 48 часов.
Стадия 1-14
Стадия 1-14 представляет собой стадию удаления защитной группы в соединении, представленном формулой (XVIII), с получением соединения, представленного формулой (Ia), где как R5ax так и R4"ax представляют собой атом водорода; и оба R5eq и R4"eq представляют собой гидроксил. Указанную стадию можно провести, осуществляя взаимодействие соединения, представленного формулой (XVIII), с основанием для удаления защитной группы для аминогруппы в 3"-положении и затем осуществляя взаимодействие полученного соединения с водородом в присутствии катализатора реакции каталитического восстановления с использованием водорода для удаления оставшейся защитной группы для аминогруппы.
Растворители, которые можно использовать на вышеуказанной стадии удаления защитной группы для аминогруппы в 3"-положении, включают метанол, этанол, изопропиловый спирт, трет-бутиловый спирт, тетрагидрофуран, 1,4-диоксан, воду или состоящие из них смешанные растворители. Среди них предпочтителен смешанный растворитель, состоящий из тетрагидрофурана и воды. Основания, которые можно здесь использовать, включают водный аммиак, карбонат калия, карбонат натрия, гидроксид калия или гидроксид натрия. Среди них предпочтителен водный аммиак.
Температура реакции составляет от 0°C до 50°C, и время реакции составляет от 1 до 48 часов.
Катализаторы реакции каталитического восстановления с использованием водорода, которые можно использовать на указанной стадии удаления оставшейся защитной группы для аминогруппы, отличной от аминогруппы в 3"-положении, включают палладий-на-угле, палладиевую чернь, гидроксид палладия, никель Ренея или оксид платины. Среди них предпочтительна палладиевая чернь. Любой растворитель можно использовать без конкретных ограничений, если только указанный растворитель является инертным в отношении рассматриваемой реакции. Предпочтительные растворители включают метанол, этанол, тетрагидрофуран, 1,4-диоксан, уксусную кислоту, состоящие из них смешанные растворители или смешанный растворитель, состоящий из органического растворителя и воды. Газообразный водород можно использовать в качестве добавляемого водорода. Давление газообразного водорода может быть 1 атм, что составляет атмосферное давление. При необходимости можно также использовать сжатый газообразный водород. Что касается источников водорода, отличающихся от газообразного водорода, можно также использовать при необходимости муравьиную кислоту, соль муравьиной кислоты, циклогексен или т.п.
Температура реакции составляет от 10°C до 30°C, и время реакции составляет обычно от 1 до 8 часов.
Если защитная группа для аминогруппы в соединении, представленном формулой (XVIII), представляет собой, например, трет-бутоксикарбонил или п-метоксибензилоксикарбонил, которые можно удалить в кислотных условиях, оставшуюся защитную группу для аминогруппы, отличной от аминогруппы в 3"-положении, можно также удалить, осуществляя взаимодействие соединения, полученного в результате удаления защитной группы для аминогруппы в 3"-положении, с кислотой. В этом случае растворители, которые можно использовать на стадии удаления защитной группы для аминогруппы, включают этилацетат, метиленхлорид, ацетонитрил, ацетон, анизол, воду или состоящие из них смешанные растворители. Среди них предпочтительна вода. Кислоты, которые можно здесь использовать, включают п-толуолсульфоновую кислоту, метансульфоновую кислоту, уксусную кислоту или трифторуксусную кислоту. Среди них предпочтительна трифторуксусная кислота.
Температура реакции обычно составляет от 0°C до 30°C, и время реакции составляет от 1 до 12 часов.
(2) Получение соединений, в которых 5- и 4"-положения являются экваториальными: второй способ получения
Во втором способе получения в соответствии с настоящим изобретением среди соединений, представленных формулой (Ia), соединения, в которых оба R5ax и R4"ax представляют собой атом водорода и оба R5eq и R4"eq представляют собой гидроксил, можно получить в соответствии со следующей схемой 4 (стадия 2-1 до стадии 2-7), схемой 5 (стадия 2-8 до стадии 2-10). В этом случае исходное соединение (X) является таким же, что и соединение, синтезированное из соединения, представленного (2-гидроксигентамицином C1a), представленного формулой (II) на схеме 2.
[Химическая формула 28]
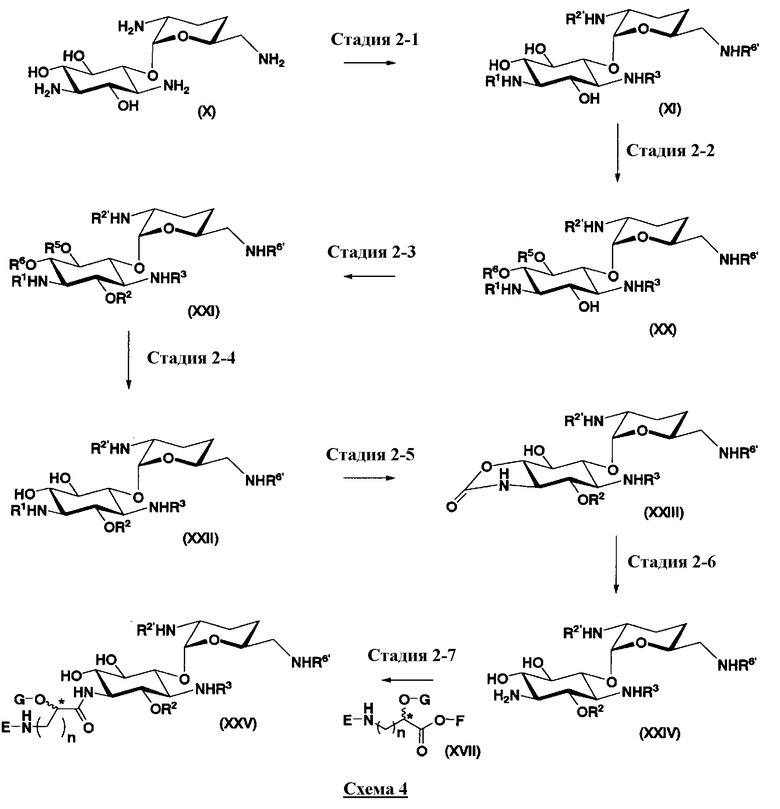
где R1, R3, R2', R6', E, F, G, n и пространственная конфигурация атома углерода, отмеченного *, имеют указанные на схемах 1 и 2 значения; R2 представляет собой защитную группу для гидроксильной группы, которую обычно используют в органическом синтезе, предпочтительно защитную группу типа бензила, которую можно удалить в реакции каталитического восстановления с использованием водорода, такую как бензил, п-метоксибензил или п-нитробензил, более предпочтительно бензил; R5 и R6 представляют собой защитные группы для гидроксильной группы, и каждая независимо представляет собой защитную группу для гидроксильной группы, или R5 и R6, взятые вместе, представляют собой циклическую защитную группу, которая одновременно защищает две гидроксильные группы, например ацеталь или кеталь, предпочтительно циклогексилиденацеталь.
На схеме 4 стадии 2-1 до стадии 2-6 представляют собой стадии введения защитной группы в аминогруппы в 3, 2' и 6'-положениях и в гидроксильную группу в 2-положении соединения, представленного формулой (X).
Стадия 2-1
Стадия 2-1 является стадией введения идентичных защитных групп в четыре аминогруппы в соединении, представленном формулой (X), синтезированном на схеме 2, с получением соединения, представленного формулой (XI). Указанную стадию можно провести, осуществляя взаимодействие соединения, представленного формулой (X), с хлороформовым эфиром, таким как бензилхлороформиат, п-метоксибензилхлороформиат или п-нитробензилхлороформиат, с диэфиром угольной кислоты, таким как ди-трет-бутилдикарбонат, или N-(бензилоксикарбонилокси)сукцинимид в присутствии основания.
Растворители, которые можно использовать на указанной стадии, включают, например, 1,2-диметоксиэтан, N,N-диметилформамид, диметилсульфоксид, метанол, этанол, изопропиловый спирт, тетрагидрофуран, 1,4-диоксан, диэтиловый эфир, метиленхлорид, хлороформ и воду. Они могут быть смешаны вместе для использования в качестве смешанного растворителя. Предпочтителен смешанный растворитель, состоящий из 1,2-диметоксиэтана или 1,4-диоксана и воды. Основания, которые можно здесь использовать, включают органические основания, такие как триэтиламин, диизопропилэтиламин, N-метилморфолин и пиридин, и неорганические основания, такие как карбонат натрия, карбонат калия и гидрокарбонат натрия. Среди них предпочтителен триэтиламин.
Температура реакции составляет от -20°C до 50°C, и время реакции составляет от 1 до 24 часов.
Стадия 2-2
Стадия 2-2 является стадией введения защитных групп (R5 и R6) в гидроксильную группу, соседнюю с 5- и 6-положениями соединения, представленного формулой (XI), с получением соединения, представленного формулой (XX). Выбираемая защитная группа для гидроксила может быть такой, что R5 и R6, каждый независимо, служит защитной группой для гидроксильной группы. Защитная группа для гидроксильной группы предпочтительно такая, что R5 и R6 вместе образуют циклическую защитную группу. Такие защитные группы включают циклогексилиденацеталь, изопропилиденацеталь и бензилиденацеталь. В этой схеме циклогексилиденацеталь предпочтителен.
Растворители, которые можно использовать на указанной стадии, включают, например, N,N-диметилформамид, метиленхлорид, хлороформ, 1,2-диметоксиэтан, 1,2-дихлорэтан, этилацетат или состоящие из них смешанные растворители. Среди них предпочтителен 1,2-диметоксиэтан. Кислоты, которые можно здесь использовать, включают п-толуолсульфоновую кислоту, пиридиний п-толуолсульфонат, камфорсульфоновую кислоту или хлористоводородную кислоту. Среди них предпочтителен пиридиний п-толуолсульфонат.
Температура реакции находится в интервале от 20°C до температуры кипения с обратным холодильником, и время реакции составляет, например, от 1 до 24 часов.
В этой реакции, если используют ацеталь, такой как 2,2-диметоксипропан или циклогексанондиметилацеталь, реакцию можно вести, удаляя спирт как побочный продукт из реакционной системы путем перегонки при пониженном давлении для ускорения реакции.
Стадия 2-3
Стадия 2-3 является стадией введения защитной группы (R2) в гидроксильную группу в 2-положении соединения, представленного формулой (XX), с получением соединения, представленного формулой (XXI). Указанную стадию можно провести, осуществляя взаимодействие соединения, представленного формулой (XX), с R2-X, где R2" представляет собой бензил, п-метоксибензил или п-нитробензил, и X представляет собой хлор, бром, йод или т.п. в присутствии основания.
Растворители, которые можно использовать на этой стадии, включают пиридин, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан или метиленхлорид. Среди них предпочтительны N,N-диметилформамид и тетрагидрофуран. Основания, которые можно здесь использовать, включают пиридин, лутидин, коллидин, триэтиламин, диизопропилэтиламин, 4-диметиламинопиридин, гидрид натрия или гидроксид калия. Среди них предпочтителен гидрид натрия.
В этой реакции можно добавлять, например, йодиды, такие как йодид натрия и тетрабутиламмонийиодид, соли серебра, такие как оксид серебра или нитрат серебра, или краун-эфир для ускорения реакции.
Температура реакции составляет от -20°C до 50°C, и время реакции составляет от 1 до 24 часов.
Стадия 2-4
Стадия 2-4 является стадией удаления защитных групп для гидроксильной группы у R5 и R6, введенных на стадии 2-3, с получением соединения, представленного формулой (XXII). Используемые реакционные условия такие же, которые обычно используют в органическом синтезе в зависимости от защитных групп, введенных в R5 и R6.
Если используют такую защитную группу для гидроксила, что R5 и R6 в формуле (XXI) вместе образуют циклическую защитную группу, указанную стадию можно провести, осуществляя взаимодействие соединения, представленного формулой (XXI), с кислотой.
Растворители, которые можно использовать на этой стадии, включают тетрагидрофуран, диэтиловый эфир, 1,4-диоксан, метанол, метиленхлорид, хлороформ, уксусную кислоту, воду или состоящие из них смешанные растворители. Среди них предпочтителен смешанный растворитель, состоящий из хлороформа и метанола, или смешанный растворитель, состоящий из уксусной кислоты и воды. Кислоты, которые можно здесь использовать, включают уксусную кислоту, трифторуксусную кислоту, хлористоводородную кислоту, серную кислоту, п-толуолсульфоновую кислоту, или трихлорид бора. Среди них предпочтительны трифторуксусная кислота или уксусная кислота.
Температура реакции находится в интервале от 0°C до температуры кипения с обратным холодильником.
Время реакции составляет от 0,1 до 12 часов.
Стадия 2-5
Стадия 2-5 является стадией удаления защитной группы (R1) для аминогруппы в 1-положении соединения, представленного формулой (XXI), и в то же самое время, образования циклического карбамата между аминогруппой и соседней гидроксильной группой в 6-положении с получением соединения, представленного формулой (XXIII).
Указанную стадию проводят, обрабатывая основанием соединение, представленное формулой (XXI). Основания, которые можно здесь использовать, включают гидрид натрия и гидроксид калия. Среди них предпочтителен гидрид натрия. Растворители, которые можно здесь использовать, включают N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан или метиленхлорид. Среди них предпочтителен N,N-диметилформамид.
Стадия 2-6
Стадия 2-6 является стадией расщепления циклического карбамата, образованного между 1- и 6-положениями соединения, представленного формулой (XXIII), с получением соединения, представленного формулой (XXIV). Указанную стадию можно осуществить, обрабатывая основанием соединение, представленное формулой (XXIII).
Основания, которые можно здесь использовать, включают неорганические основания, такие как карбонат натрия и карбонат калия, и алкоксиды металлов, такие как метоксид натрия и этоксид натрия.
Растворители, которые можно здесь использовать, включают N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, метиленхлорид, воду и состоящие из них смешанные растворители. Среди них предпочтителен смешанный растворитель, состоящий из 1,4-диоксана и воды.
Температура реакции находится в интервале от 0°C до температуры кипения с обратным холодильником, и время реакции составляет от 0,1 до 12 часов.
Стадия 2-7
Стадия 2-7 является стадией конденсации аминогруппы в 1-положении соединения, представленного формулой (XXIV), полученного на стадии 2-6, с соединением, представленным формулой (XVII), с получением соединения, представленного формулой (XXV).
Указанную стадию можно провести в тех же самых реакционных условиях, что и реакционные условия, раскрытые для стадии 1-13, или в реакционных условиях, аналогичных реакционным условиям, раскрытым для стадии 1-13 на схеме 3.
На схеме 5 представлена стадия осуществления взаимодействия соединения, представленного формулой (XXV), с соединением, представленным формулой (Xc) (стадия 2-8)
[Химическая формула 29]
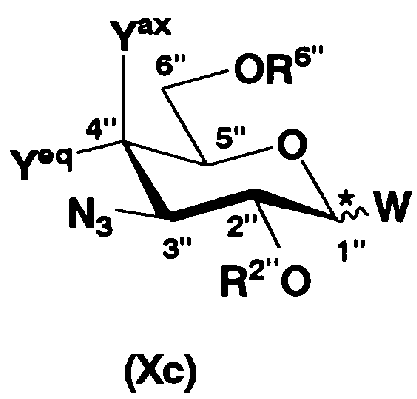
где W, Yax, Yeq, R6", R2" и пространственная конфигурация атома углерода, отмеченного *, имеют указанные выше значения.
Стадия удаления защитной группы из полученного соединения (стадия 2-9 и стадия 2-10) и стадия восстановления азидогруппы указанного соединения с получением соединения, представленного формулой (Ia) (стадия 2-10), будут раскрыты далее.
[Химическая формула 30]
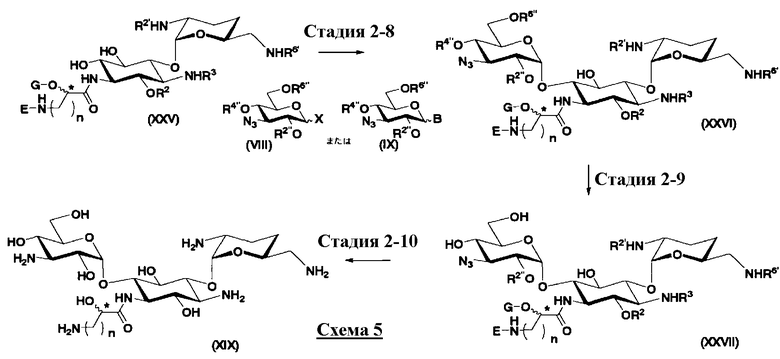
где
R2, R3, R2', R6', R2", R4", R6", B, X, E, G, n и пространственная конфигурация атома углерода, отмеченного *, имеют указанные выше значения.
Стадия 2-8
Стадия 2-8 является стадией конденсации соединения, представленного формулой (XXV), полученного в соответствии со схемой 4, с соединением, представленным формулой (Xc) (соединение, представленное формулой (VIII), или соединение, представленное формулой (IX)), с получением соединения, представленного формулой (XXVI).
Указанную стадию можно провести в тех же самых реакционных условиях, что и реакционные условия, раскрытые для стадии 1-8, или реакционные условия, аналогичные реакционным условиям, раскрытым на стадии 1-8 на схеме 3.
Стадия 2-9
Стадия 2-9 представляет собой стадию удаления двух защитных групп (R4" и R6") у гидроксильной группы в соединении, представленном формулой (XXVI), с получением соединения, представленного формулой (XXVII).
Указанную стадию можно осуществить путем гидролиза в щелочных условиях или в кислотных условиях, или обрабатывая указанное соединение нуклеофильным основанием, например метоксидом натрия или этоксидом натрия в безводных условиях.
В случае гидролиза, кроме воды и спирта в качестве растворителя в реакции можно использовать растворители, такие как метанол или этанол, и растворители, смешиваемые с водой, например тетрагидрофуран или 1,4-диоксан. Далее, можно использовать растворители, не смешиваемые с водой, например этилацетат и метиленхлорид, адаптируя реакцию двухслойного типа с водой. Растворители, которые обычно используют в органическом синтезе, можно использовать в качестве указанного растворителя в безводных условиях.
Стадия 2-10
Стадия 2-10 является стадией восстановления азидогруппы в соединении, представленном формулой (XXVII), полученном на стадии 2-9, до аминогруппы, и далее удаления трех защитных групп (R3, R2' и R6') для аминогруппы и защитной группы (R2) для гидроксильной группы в 2-положении с получением соединения, представленного формулой (XIX), которое представляет собой соединение, представленное формулой (Ia), где оба R5ax и R4"ax представляют собой атом водорода и оба R5eq и R4"eq представляют собой гидроксил.
В этой реакции реакцию восстановления азидогруппы и удаления защитной группы можно осуществить отдельно друг от друга в несколько стадий. Альтернативно, если обе реакции можно осуществить в одинаковых реакционных условиях, указанную стадию можно осуществить в одной реакции.
Реакцию с водородом в присутствии катализатора реакции каталитического восстановления с использованием водорода можно упомянуть как реакцию, в которой азидогруппу восстанавливают и превращают в аминогруппу. Катализаторы реакции каталитического восстановления с использованием водорода, которые можно здесь использовать, включают палладий-на-угле, палладиевую чернь, гидроксид палладия, никель Ренея или оксид платины. Среди них предпочтительна палладиевая чернь. Любой растворитель можно использовать без конкретных ограничений, если только указанный растворитель является инертным в указанной реакции. Предпочтительные растворители включают метанол, этанол, тетрагидрофуран, диоксан, уксусную кислоту, состоящие из них смешанные растворители или смешанный растворитель, состоящий из органического растворителя и воды. Газообразный водород можно использовать в качестве добавляемого водорода. Давление газообразного водорода может составлять 1 атм, что соответствует атмосферному давлению. При необходимости можно также использовать сжатый газообразный водород. Можно также использовать при необходимости источники водорода, которые отличаются от газообразного водорода, например муравьиную кислоту, соли муравьиной кислоты и циклогексен.
Способы восстановления азидогруппы для превращения ее в аминогруппу включают способ Staudinger et al., в котором азидосоединение подвергают взаимодействию с фосфином или фосфитом, получая иминофосфоран, который затем гидролизуют для превращения в амино (Helvetica Chemica Acta, Vol.2, p.635 (1919)). Реагенты фосфина, которые можно использовать в указанном способе, включают трифенилфосфин и триметилфосфин. Например, триметилфосфит можно упомянуть в качестве реагента фосфита. Растворители, которые можно использовать в указанной реакции, включают тетрагидрофуран, 1,4-диоксан, диэтиловый эфир, ацетонитрид, метиленхлорид, воду и состоящие из них смешанные растворители. Иминофосфоран, получаемый в качестве промежуточного соединения, можно выделить до гидролиза. Получение иминофосфорана и гидролиз можно осуществить в одну стадию, добавляя воду к реакционному растворителю.
Способ удаления трех защитных групп (R3, R2' и R6') для аминогруппы и защитной группы (R2) для гидроксильной группы в 2-положении можно выбрать, учитывая условия удаления защитных групп, которые обычно используют в органическом синтезе в зависимости от типа указанных защитных групп. Способ можно осуществить постадийно или в одну стадию.
Если защитные группы, которые можно удалить в реакции каталитического гидрирования, выбирают как для трех защитных групп (R3, R2' и R6') для аминогруппы, так и защитной группы (R2) для гидроксильной группы в 2-положении, и в этом случае, если все из R3, R2' и R6 представляют собой бензилоксикарбонил, тогда как R2 представляет собой бензил, реакцию восстановления для превращения азидогруппы в аминогруппу можно также с успехом осуществить, используя одностадийную реакцию каталитического гидрирования.
(3) Получение соединений, у которых 5- и 4"-положения являются экваториальными: второй способ получения
Во втором способе получения в соответствии с настоящим изобретением соединение, представленное формулой (Ia), можно также получить, используя соединение, представленное формулой (Xd), вместо соединения, представленного формулой (Xc).
[Химическая формула 31]
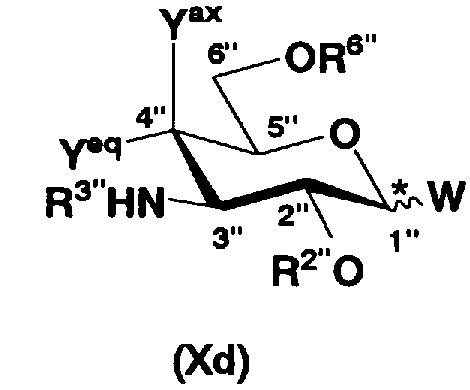
где W, Yax, Yeq, R2", R3", R6" и пространственная конфигурация атома углерода, отмеченного *, имеют указанные выше значения.
Второй способ получения будет раскрыт со ссылкой на схему 6 (стадия 3-1 до стадии 3-4) и схему 7 (стадия 3-5 до стадии 3-7).
На представленной далее схеме 6 будут раскрыты соединения, представленные формулой (Xd), где Yax представляет собой атом водорода, Yeq представляет собой гидроксил и W представляет собой отщепляемую группу (B), описываются как соединения, представленные формулой (XXXI), и будет раскрыт способ их получения.
[Химическая формула 32]
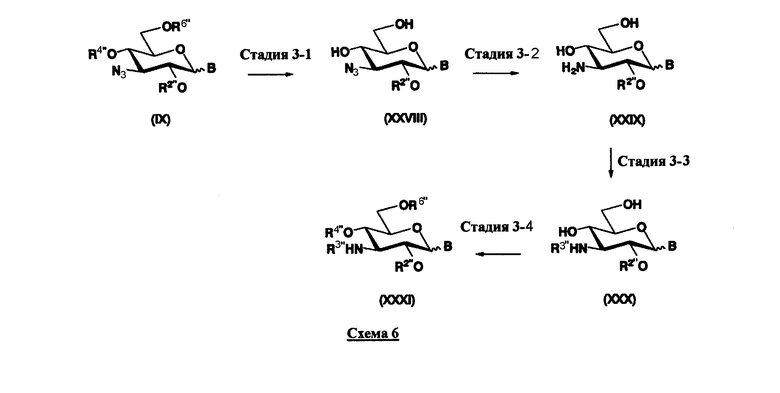
где R2", R3", R4", R6" и B имеют указанные выше значения; и
R4" и R6" предпочтительно представляют собой защитные группы сложноэфирного типа.
Стадия 3-1
Стадия 3-1 является стадией удаления двух защитных групп (R4" и R6") для гидроксильной группы в соединении, представленном формулой (IX), с получением соединения, представленного формулой (XXVIII).
Если R4" и R6" представляют собой защитные группы сложноэфирного типа, можно использовать реакцию гидролиза или реакцию сольволиза. Более предпочтительна реакция сольволиза.
Более конкретно, реакцию сольволиза можно осуществить, используя метоксид натрия, этоксид натрия или т.п. Любой растворитель можно использовать без ограничений, если только указанный растворитель является инертным в отношении указанной реакции. Предпочтительны спиртовые растворители, такие как метанол и этанол.
Температура реакции находится в интервале от -20°C до температуры кипения с обратным холодильником, и время реакции составляет от 0,1 до 12 часов.
Стадия 3-2
Стадия 3-2 является стадией восстановления азидогруппы в соединении, представленном формулой (XXVIII), в аминогрупппу, с получением соединения, представленного формулой (XXIX).
На стадии 2-10 на схеме 5 подробно раскрыта стадия восстановления азидогруппы в аминогруппу. Те же самые условия можно использовать на стадии 3-2. На схеме 6 предпочтительно используют способ Staudinger et al. (Helvetica Chemica Acta, Vol.2, p. 635 (1919)). Соединение, представленное формулой (XXIX), полученное на указанной стадии, можно выделить для использования на стадии 3-3. Альтернативно, соединение как оно есть, можно последовательно использовать в качестве исходного соединения для стадии 3-3 без выделения.
Стадия 3-3
Стадия 3-3 является стадией защиты аминогруппы соединения, представленного формулой (XXIX), с получением соединения, представленного формулой (XXX).
В качестве защитных групп для аминогруппы можно использовать обычно используемые в органическом синтезе защитные группы. Предпочтительно, защитной группой может быть такая группа, которую можно удалить в реакции каталитического гидрирования. Конкретно в качестве защитных групп можно упомянуть защитные группы типа бензила, такие как бензилоксикарбонил. Защитную группу можно ввести, например, способом, раскрытым в связи со стадией 1-7 на схеме 2, используя хлороформовый сложный эфир, раскрытый в связи со стадией 2-1 на схеме 4, или способом с использованием реагента бензилоксикарбонилирования, такого как группа N-(бензилоксикарбонилокси)сукцинимида.
Стадия 3-4
Стадия 3-4 является стадией одновременной защиты гидроксильной группы в 4-положении и гидроксильной группы в 6-положении в соединении, представленном формулой (XXX), с получением соединения, представленного формулой (XXXI). R4" и R6" на схеме 6 предпочтительно представляют собой защитные группы сложноэфирного типа, особенно предпочтителен ацетил.
Защитные группы сложноэфирного типа можно ввести способом, который обычно используют в органическом синтезе, используя ангидрид кислоты или галогенангидрид в присутствии или в отсутствие основания. Любой растворитель можно использовать без конкретных ограничений, если только указанный растворитель является инертным в отношении реакции. Педпочтителен растворитель, который может служить как в качестве растворителя, так и в качестве основания, такой как пиридин.
Температура реакции находится в интервале от -20°C до температуры кипения с обратным холодильником, и время реакции составляет от 0,1 до 72 часов.
Далее схема 7 демонстрирует стадию осуществления взаимодействия соединения, представленного формулой (XXV), с соединением, представленным формулой (XXXI) (стадия 3-5), и стадию удаления защитной группы у полученного соединения, с получением соединения, представленного формулой (XIX) (стадии 3-6 и 3-7).
[Химическая формула 33]
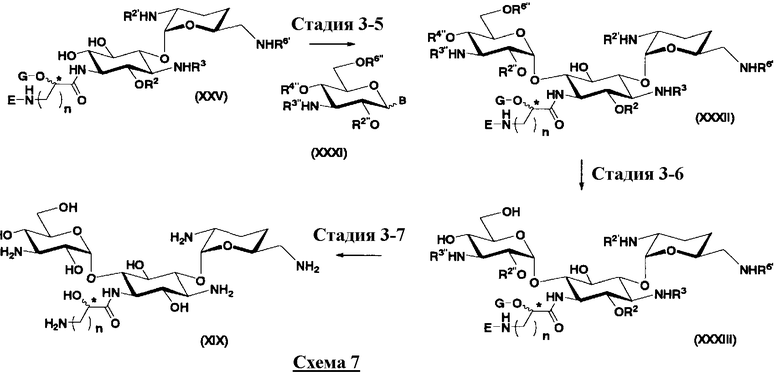
где R2, R3, R2', R6', R2", R3", R4", R6", B, E, G, n и пространственная конфигурация атома углерода, отмеченного *, имеют указанные выше значения.
Стадия 3-5
Стадия 3-5 является стадией конденсации соединения, представленного формулой (XXV), полученного на схеме 4, с соединением, представленным формулой (XXXI) на схеме 6, с получением соединения, представленного формулой (XXXII).
Конкретно реакцию конденсации можно осуществить таким же способом, который представлен на стадии 2-8 на схеме 5.
Стадия 3-6
Стадия 3-6 является стадией удаления двух защитных групп (R4" и R6") для гидроксильной группы в соединении, представленном формулой (XXXII), полученном на стадии 3-5, до превращения соединения, представленного формулой (XXXIII).
Конкретно, указанную стадию можно осуществить таким же способом, что и на стадии 2-9 на схеме 5.
Стадия 3-7
Стадия 3-7 является стадией удаления всех защитных групп (R2, R3, R2', R6', R2", R3", E и G) в соединении, представленном формулой (XXXIII), полученном на стадии 3-6, с получением соединения, представленного формулой (XIX), которое является соединением, представленным формулой (Ia), в котором оба R5ax и R4"ax представляют собой атом водорода и оба R5eq и R4"eq представляют собой гидроксил.
Все защитные группы (R2, R3, R2', R6', R2", R3", E и G) в соединении, представленном формулой (XXXIII), можно удалить постадийно или в одну стадию (если это возможно) в условиях удаления защитных групп в обычном органическом синтезе. Например, если все защитные группы в соединении, представленном формулой (XXXIII), можно удалить в реакции каталитического гидрирования, защитные группы можно удалить в одну стадию, используя те же условия реакции каталитического гидрирования, что и на стадии 1-14 на схеме 3, или условия, аналогичные условиям реакции каталитического гидрирования на стадии 1-14 на схеме 3.
(4) Получение соединения, в котором 5-положение является экваториальным и 4"-положение является аксиальным: второй способ получения
Далее, в соответствии со вторым способом получения настоящего изобретения, соединения, представленные формулой (Ia), где оба R5ax и R4"eq представляют собой атом водорода и оба R5eq и R4"ax представляют собой гидроксил, можно получить в соответствии со схемой 8 (стадия 4-1 до стадии 4-5a и стадия 4-5b) и схемой 9.
Способ получения соединений, представленных формулой (Xc), будет раскрыт подробно со ссылкой на схему 8.
[Химическая формула 34]

где W представляет собой отщепляемую группу; Yax представляет собой группу
-OR4"; Yeq представляет собой атом водорода; R2", R4" и R6" представляют собой защитные группы для гидроксильной группы; и пространственная конфигурация атома углерода, отмеченного *, соответствует R или S.
На схеме 8 соединения, представленные формулой (Xc), классифицированы в соответствии с типом отщепляемой группы, представленной W в соединениях, представленных формулой (XXXVIII), и в соединениях, представленных формулой (XXXIX).
[Химическая формула 35]
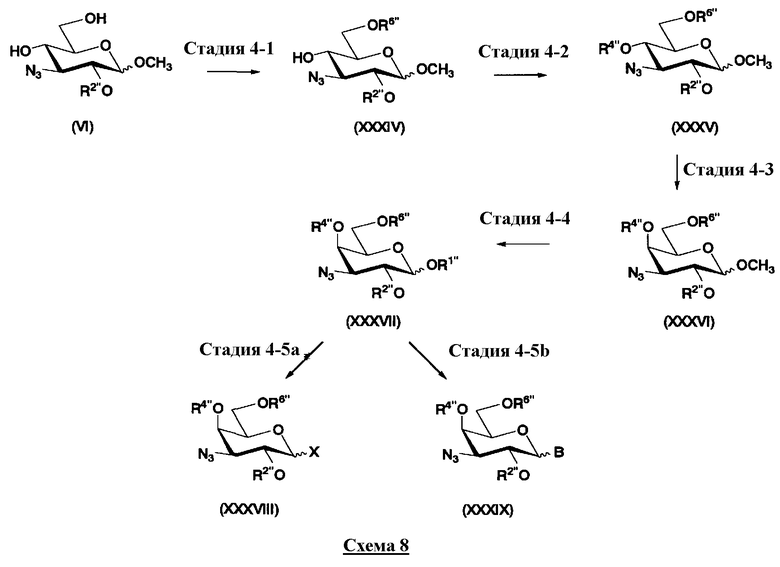
где R2" представляет собой защитную группу для гидроксильной группы, предпочтительно группу, выбранную из защитных групп типа бензильного эфира, таких как бензил и п-метоксибензил; R4" и R6", которые отличаются друг от друга, представляют собой защитную группу для гидроксильной группы, например, выбранную из защитных групп сложноэфирного типа, таких как ацетил и бензоил, и защитных групп типа сульфонила, таких как п-толуолсульфонил, метансульфонил и трифторметансульфонил; и B и X имеют указанные выше значения.
Стадия 4-1
Стадия 4-1 представляет собой стадию селективного введения защитной группы (R6") только в одну гидроксильную группу, расположенную в 6-положении из двух гидроксильных групп в соединении, представленном формулой (VI) на схеме 1, с получением соединения, представленного формулой (XXXIV).
Вводимая защитная группа предпочтительно представляет собой структурно объемную защитную группу среди защитных групп, используемых в качестве защитных групп для гидроксильной группы в органическом синтезе, конкретно предпочтительны бензоил или замещенный бензоил.
Реакцию можно осуществить таким же способом, как на стадии 3-4 на схеме 6.
Стадия 4-2
Стадия 4-2 является стадией введения отщепляемой группы в гидроксильную группу в 4-положении соединения, представленного формулой (XXXIV), с получением соединения, представленного формулой (XXXV), которое является исходным соединением на стадии 4-3 для синтеза соединения, представленного формулой (XXXVI), в котором гидроксильная группа в 4-положении была обращена.
Отщепляемая группа, обладающая более высокой способностью к отщеплению, нежели защитная группа, введенная в R6" на стадии 4-2, среди отщепляемых групп для гидроксильной группы в органическом синтезе, выбрана в качестве введенной отщепляемой группы. Конкретно выбирают отщепляемую группу сульфонильного типа. Более конкретно, предпочтительным R4" в формуле (XXXV) является, например, трифторметансульфонил.
Реакцию можно осуществить таким же способом, что и на стадии 3-4 на схеме 6.
Стадия 4-3
Стадия 4-3 является стадией утилизации отщепляемой группы в 4-положении соединения, представленного формулой (XXXV), с получением соединения, представленного формулой (XXXVI), пространственная конфигурация которого в 4-положении была обращена.
R4" в соединении, представленном формулой (XXXVI), предпочтительно является защитной группой, которая служит в качестве защитной группы для гидроксильной группы в 4-положении и может быть удалена таким же способом, что и R6" в качестве защитной группы для гидроксильной группы в 6-положении. R4" представляет собой защитную группу сложноэфирного типа, предпочтительно ацетил.
Реакцию, в которой можно получить соединение, представленное формулой (XXXVI), можно провести, осуществляя взаимодействия соли металла с карбоновой кислотой с соединением, представленным формулой (XXXV). Ацетильную группу, которая предпочтительна в качестве R4", можно с успехом ввести, используя соль уксусной кислоты, такую как ацетат цезия.
Можно использовать любой растворитель без конкретных ограничений, если только указанный растворитель является инертным в отношении рассматриваемой реакции. Указанный растворитель предпочтительно представляет собой N,N-диметилформамид.
Температура реакции находится в интервале от -20°C до температуры кипения с обратным холодильником, и время реакции составляет от 0,1 до 72 часов.
Стадия 4-4
Стадия 4-4 является стадией превращения метоксигруппы в 1-положении соединения, представленного формулой (XXXVI), в ацилоксигруппу (OR1"), с получением соединения, представленного формулой (XXXVII).
Указанную стадию можно провести таким же способом, что и на стадии 1-4 на схеме 1.
Стадия 4-5a
Стадия 4-5a является стадией превращения ацилоксигруппы (OR1") в 1-положении соединения, представленного формулой (XXXVII), в атом галогена, с получением соединения, представленного формулой (XXXVIII).
Указанную стадию можно осуществить таким же способом, что и способ на стадии 1-5a на схеме 1.
Стадия 4-5b
Стадия 4-5b является стадией превращения ацилоксигруппы (OR1") в 1-положении соединения, представленного формулой (XXXVII), в тиоалкил или тиоарил в присутствии кислоты Льюиса, с получением соединения, представленного формулой (XXXIX).
Указанную стадию можно осуществить таким же способом, что и способ на стадии 1-5b на схеме 1.
Заместитель в 1-положении соединения, представленного на схеме 8, может иметь две пространственные конфигурации, то есть может существовать в экваториальной форме и в аксиальной форме. На схеме 8 они могут быть разделены друг от друга до использования в реакции, или альтернативно, в реакции можно использовать две пространственные конфигурации в смешанной форме, как они есть. Далее, соединение, представленное формулой (XXXVIII), и соединение, представленное формулой (XXXIX), полученное на схеме 8, можно разделить на аксиальную форму и экваториальную форму, которые можно затем использовать отдельно друг от друга на схеме 9. Альтернативно, указанные соединения можно использовать в виде смеси аксиальной и экваториальной форм.
[Химическая формула 36]
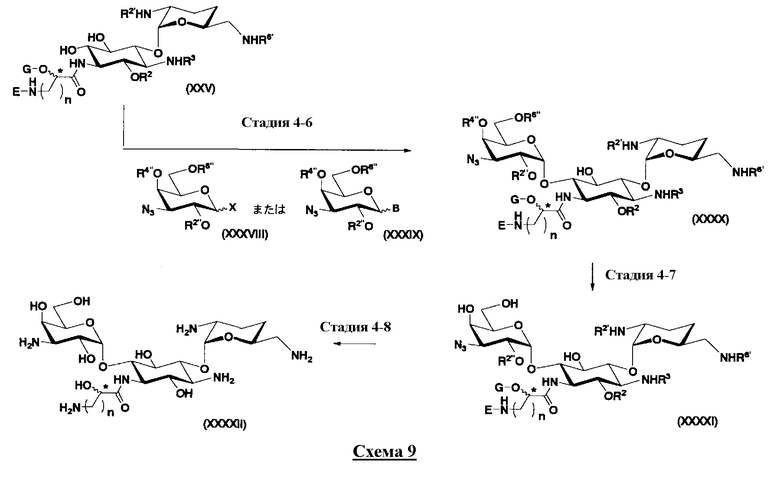
где R2, R3, R2', R6', R2", R4", R5", B, E, G, X, n и пространственная конфигурация атома углерода, отмеченного *, имеют указанные выше значения.
Стадия 4-6
Стадия 4-6 является стадией конденсации соединения, представленного формулой (XXV), полученного на схеме 4, с соединением, представленным формулой (XXXVIII), или формулой (XXXIX) на схеме 8, для получения соединения, представленного формулой (XXXX).
Реакцию конденсации можно осуществить таким же способом, что и способ на стадии 2-8 на схеме 5.
Стадия 4-7
Стадия 4-7 является стадией удаления двух защитных групп (R4" и R6") для гидроксильной группы в соединении, представленном формулой (XXXX), полученном на стадии 4-6, и превращения в соединение, представленное формулой (XXXXI).
Конкретно, указанную стадию можно осуществить таким же способом, что и способ на стадии 2-9 на схеме 5.
Стадия 4-8
Стадия 4-8 является стадией удаления всех защитных групп (R2, R3, R2', R6', R2", E и G) в соединении, представленном формулой (XXXXI), полученном на стадии 4-7, с получением соединения, представленного формулой (XXXXII), которое является соединением, представленным формулой (Ia), где R5ax и R4"eq представляют собой атом водорода и R5eq и R4"ax представляют собой гидроксил.
Конкретно, указанную стадию можно осуществить таким же способом, что и способ на стадии 2-10 на схеме 5.
(5) Получение соединения, в котором 5-положение является аксиальным и 4"-положение является экваториальным: первый способ получения
В первом способе получения в соответствии с настоящим изобретением, соединение, представленное формулой (Ia), где оба R5eq и R4"ax представляют собой атом водорода и оба R5ax и R4"eq представляют собой гидроксил, можно получить в соответствии со следующей схемой 10 (стадия 5-1 до стадии 5-6) и схемой 11 (стадия 5-7 до стадии 5-13).
Способ получения соединений, представленных формулой (Xa), в которых пространственная конфигурация гидроксильной группы в 5-положении соответствует аксиальной, будет конкретно раскрыт со ссылкой на стадии 5-1 до стадии 5-6 на схеме 10. На схеме 10 соединение, представленное формулой (Xa), в котором пространственная конфигурация гидроксильной группы в 5-положении соответствует аксиальной, соответствует соединению представленному формулой (XXXXVIII).
[Химическая формула 37]
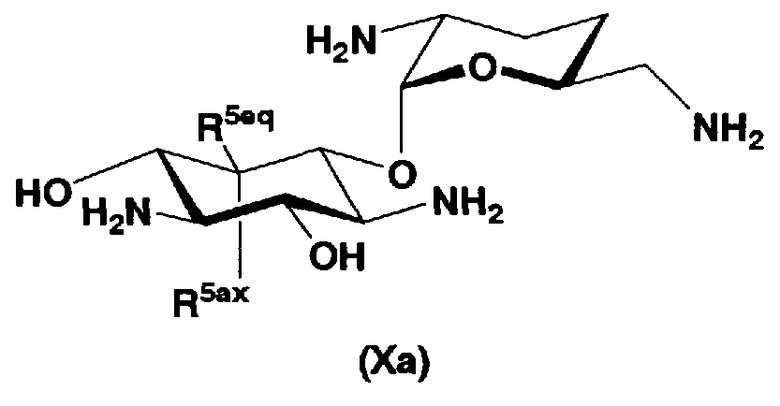
где R5ax представляет собой гидроксил и R5eq представляет собой атом водорода.
[Химическая формула 38]
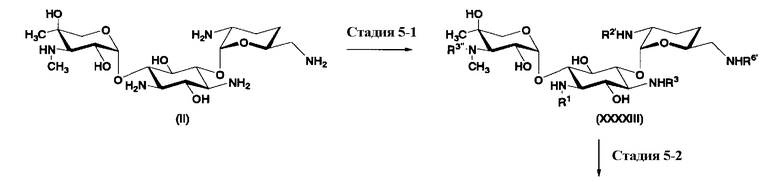
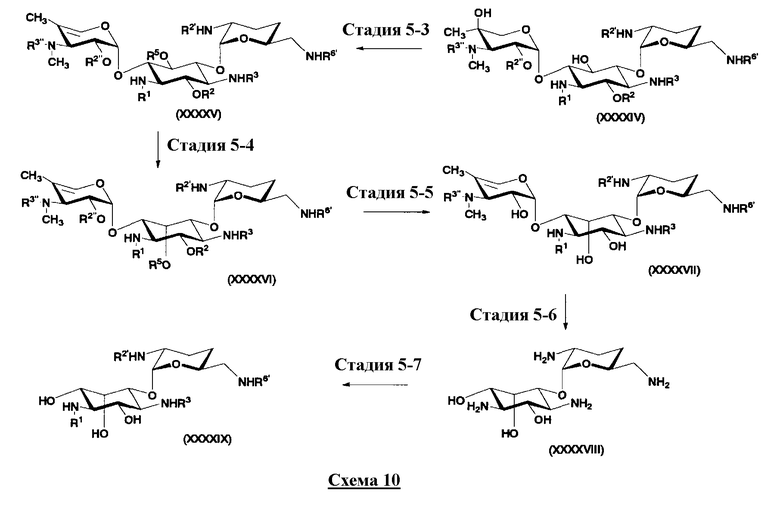
где R1, R3, R2', R6' и R3" имеют указанные выше значения; R5 представляет собой защитную группу для гидроксильной группы, и его выбирают из, например, защитных групп сложноэфирного типа, таких как ацетил или бензоил, и защитных групп типа сульфонила, таких как п-толуолсульфонил, метансульфонил или трифторметансульфонил; и R2 и R2" предпочтительно представляют собой защитные группы сложноэфирного типа.
Стадия 5-1
Стадия 5-1 является стадией введения защитных групп в пять аминогрупп в 2-гидроксигентамицине C1a, представленном формулой (II).
Введенной защитной группой является, предпочтительно, защитная группа, представленная, например, на схеме 2. На этой стадии для получения соединения, представленного формулой (XXXXIII), особенно предпочтителен трет-бутоксикарбонил. Указанную защитную группу можно ввести в условиях, подробно раскрытых в соответствии со стадией 1-7 на схеме 2.
Стадия 5-2
Стадия 5-2 является стадией введения защитных групп (R2 и R2") в две гидроксильные группы в соединении, представленном формулой (XXXXIII). Защитные группы сложноэфирного типа, такие как ацетил, предпочтительны в качестве вводимых защитных групп, и ацетил выбирают для соединения, представленного формулой (XXXXIV).
Группу ацетила можно ввести таким же способом, что и на стадии 3-4 на схеме 6.
Стадия 5-3
Стадия 5-3 является стадией введения отщепляемой группы в третичную гидроксильную группу соединения, представленного формулой (XXXXIV), в присутствии основания, чтобы вызывать отщепление и таким образом обеспечить образование ненасыщенной связи и, в то же самое время, стадией введения отщепляемой группы в гидроксильную группу в 5-положении с получением соединения, представленного формулой (XXXXV).
Подлежащую введению отщепляемую группу можно выбрать из алкилсульфонильных групп, таких как метансульфонил и трифторметансульфонил, или арилсульфонильных групп, таких как п-толуолсульфонил. Для получения соединения, представленного формулой (XXXXV), предпочтителен метансульфонил. Реакцию осуществляют, обрабатывая сульфонилхлорид и соединение, представленное формулой (XXXXIV) в присутствии основания. Основания, которые можно использовать в указанной реакции, включают органические основания, такие как триэтиламин или 4-диметиламинопиридин. Предпочтителен 4-диметиламинопиридин.
Любой растворитель можно использовать в реакции без конкретных ограничений, если только указанный растворитель является инертным в отношении реакции. Предпочтительны растворители-галогениды, такие как метиленхлорид.
Стадия 5-4
Стадия 5-4 является стадией обращения пространственной конфигурации в 5-положении соединения, представленного формулой (XXXXV), для введения защищенной гидроксильной группы и, таким образом, для синтеза соединения, представленного формулой (XXXXVI). Сложноэфирная группа предпочтительна в качестве защищенной гидроксильной группы с учетом стадии удаления защитных групп, которая будет раскрыта далее.
В соединении, представленном формулой (XXXXVI), ацетил предпочтителен.
Указанную стадию введения ацетила для обращения гидроксильной группы в 5-положении можно осуществить таким же способом, что и на стадии 4-3 на схеме 8.
Стадия 5-5
Стадия 5-5 является стадией удаления трех защитных групп (R2, R2", R5) для гидроксильных группы в соединении, представленном формулой (XXXXVI), с получением соединения, представленного формулой (XXXXVII).
В соединении, представленном формулой (XXXXVI), три защитные группы (R2, R2" и R5) для гидроксильных групп представляют собой ацетил. Их удаление можно осуществить в тех же условиях, что и на стадии 3-1 схемы 6, или в условиях, аналогичных условиям на стадии 3-1 схемы 6.
Стадия 5-6
Стадия 5-6 является стадией, на которой осуществляют реакцию кислотного гидролиза соединения, представленного формулой (XXXXVII), с получением соединения, представленного формулой (XXXXVIII).
Указанную реакцию можно осуществить со ссылкой на реакционные условия стадии 1-6 на схеме 2. Растворитель, который не ингибирует реакцию, такой как метанол, можно также дополнительно использовать для повышения растворимости соединения, представленного формулой (XXXXVII), для ускорения реакции.
Стадия 5-7
Стадия 5-7 является стадией введения защитных групп в четыре аминогруппы в соединении, представленном формулой (XXXXVIII), полученном на стадии 5-6, с получением соединения, представленного формулой (XXXXIX).
Защитная группа, которую нужно ввести, предпочтительно представляет собой защитную группу, представленную в качестве примера на схеме 2, более предпочтительно п-толуолсульфонил. Для введения защитной группы можно использовать условия, подробно раскрытые на стадии 1-7 схемы 2.
Соединение, представленное формулой (Ia), где оба R5eq и R4"ax представляют собой атом водорода и оба R5ax и R4"eq представляют собой гидроксил, можно получить в соответствии со схемой 12 (стадия 5-8 до стадии 5-14). На схеме 12 соединение представлено формулой (XXXXXVI).
Схема 11 включает стадию осуществления взаимодействия соединения, представленного формулой (XXXXIX), с соединением, представленным формулой (Xc) (стадия 5-8), стадию удаления защитных групп полученного соединения и превращения азидогруппы в соединении в аминогруппу (стадия 5-9 и стадия 5-10), стадию необязательного введения защитных групп в функциональные группы, отличные от аминогруппы в 1-положении в полученном соединении (стадия 5-11 и стадия 5-12), стадию осуществления взаимодействия полученного соединения с соединением, представленным формулой (XVII) (стадия 5-13), и стадию удаления защитных групп в полученном соединении, с получением ожидаемого соединения, представленного формулой (XXXXXVI) (стадия 5-14). На схеме 11 соединение, представленное формулой (Xc), соответствует соединению, представленному формулой (VIII) или формулой (IX).
[Химическая формула 39]
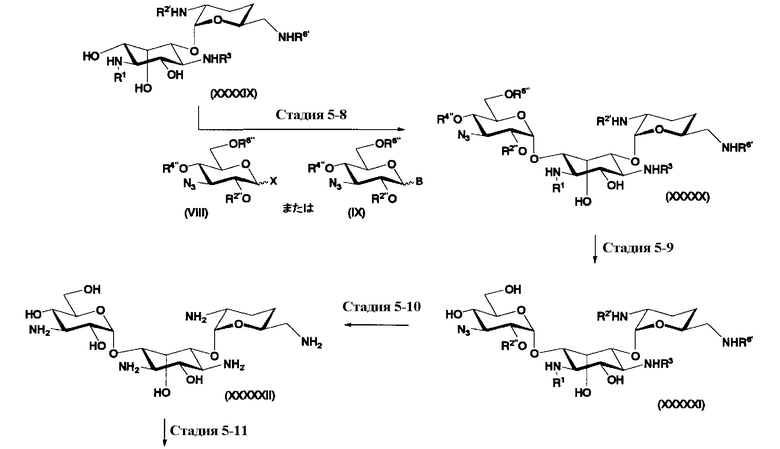
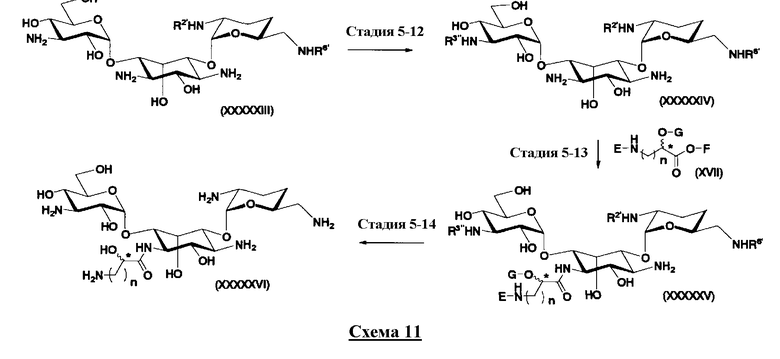
где R1, R3, R2', R6', R2", B, E, F, G, X, n и пространственная конфигурация атома углерода, отмеченного *, имеют указанные выше значения; R4" и R6" независимо представляют собой защитные группы для гидроксила, которые соответствуют защитным группам для гидроксильных групп, как определено на схеме 1, выбирают из защитных групп сложноэфирного типа, таких как ацетил и бензоил.
Стадия 5-8
Стадия 5-8 является стадией конденсации соединения, представленного формулой (XXXXIX), полученного в соответствии со схемой 10, с соединением, которое представлено формулой (VIII), полученным в соответствии со схемой 1, или соединением, представленным формулой (IX), с получением соединения, представленного формулой (XXXXX). В формуле (VIII) или формуле (IX), R4" и R6", которые являются защитными группами сложноэфирного типа для гидроксильной группы, предпочтительно представляют ацетил.
Реакцию на указанной стадии можно осуществить в таких же условиях, которые использованы на стадии 1-8 на схеме 3, или в условиях, аналогичных условиям, использованным на стадии 1-8 на схеме 3.
Стадия 5-9
Стадия 5-9 является стадией удаления двух защитных групп (R4" и R6") для гидроксильных групп в соединении, представленном формулой (XXXXX), полученном на стадии 5-8, с получением соединения, представленного формулой (XXXXXI).
Две защитные группы (R4" и R6") для гидроксильной группы в соединении, представленном формулой (XXXXX), представляют собой ацетил, и их можно удалить таким же способом, который представлен на стадии 3-1 на схеме 6.
Стадия 5-10
Стадия 5-10 является стадией восстановления азидогруппы соединения, представленного формулой (XXXXXI), полученного на стадии 5-9, до аминогруппы и далее удаления четырех защитных групп (R1, R3, R2' и R6') для аминогрупп и защитной группы (R2") для гидроксильной группы в одну стадию с получением соединения, представленного формулой (XXXXXII).
Указанная стадия является такой же, что и стадия 1-10 на схеме 3, и можно использовать условия для восстановления Бирча в условиях для стадии 1-10.
Стадия 5-11
Стадия 5-11 является стадией селективного введения защитных групп (R2' и R6') в аминогруппы в 2'- и 6'-положениях в соединении, представленном формулой (XXXXXII), полученном на стадии 5-10, с получением соединения, представленного формулой (XXXXXIII). В формуле (XXXXXIII) R2' и R6' представляют собой бензилоксикарбонил, и можно использовать условия реакции, раскрытые для стадии 1-11 на схеме 3.
Стадия 5-12
Стадия 5-12 является стадией селективного введения защитной группы (R3") в аминогруппу в 3"-положении соединения, представленного формулой (XXXXXIII), с получением соединения, представленного формулой (XXXXXIV). В формуле (XXXXXIV), предпочтительно, R3" представляет собой трифторацетил, и для проведения реакции можно использовать реакционные условия для стадии 1-12 на схеме 3.
Стадия 5-13
Стадия 5-13 является стадией осуществления взаимодействия аминогруппы в 1-положении соединения, представленного формулой (XXXXXIV), с производным ω-амино-α-гидроксикарбоновой кислоты, представленным формулой (XVII), с получением соединения, представленного формулой (XXXXXV), то есть стадию проведения реакции для образования пептидной связи. Указанную стадию можно осуществить таким же способом, что и способ стадии 1-13 на схеме 3.
Стадия 5-14
Стадия 5-14 является стадией удаления всех защитных групп в соединении, представленном формулой (XXXXXV), с получением соединения, представленного формулой (Ia), где оба R5eq и R4"ax представляют собой атом водорода и оба R5ax и R4"eq представляют собой гидроксил.
Указанную стадию можно провести таким же способом, что и способ стадии 1-14 на схеме 3.
(6) Получение соединения, в котором 5-положение соответствует аксиальному и 4"-положение соответствует аксиальному: реакция обращения пространственной конфигурации в 4"-положении
Соединение, представленное формулой (Ia), где оба R5eq и R4"eq представляют собой атом водорода и оба R5ax и R4"ax представляют собой гидроксил, можно получить в соответствии со схемой 12 (стадия 6-1 до 6-8). На схеме 12 указанное соединение представлено формулой (XXXXXXIV).
Схема 12 включает стадию введения защитных групп в функциональные группы, отличные от функциональной группы в 4"-положении соединения, представленного формулой (XXXXXVI), полученного в соответствии со схемой 11 (стадия 6-1 до стадии 6-5), стадию обращения пространственной конфигурации гидроксильной группы в 4"-положении (стадия 6-6) и стадию удаления защитных групп (стадия 6-7 и стадия 6-8).
[Химическая формула 40]
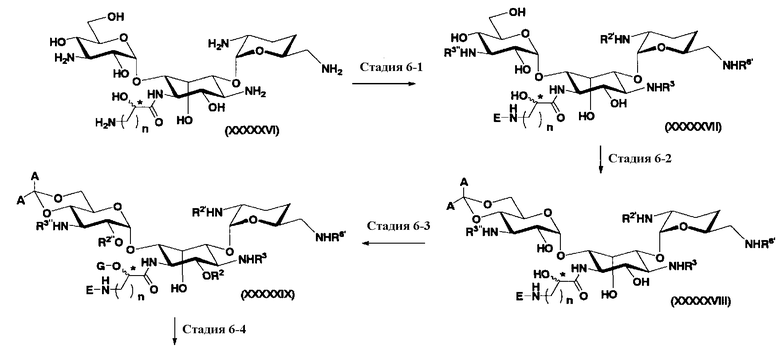
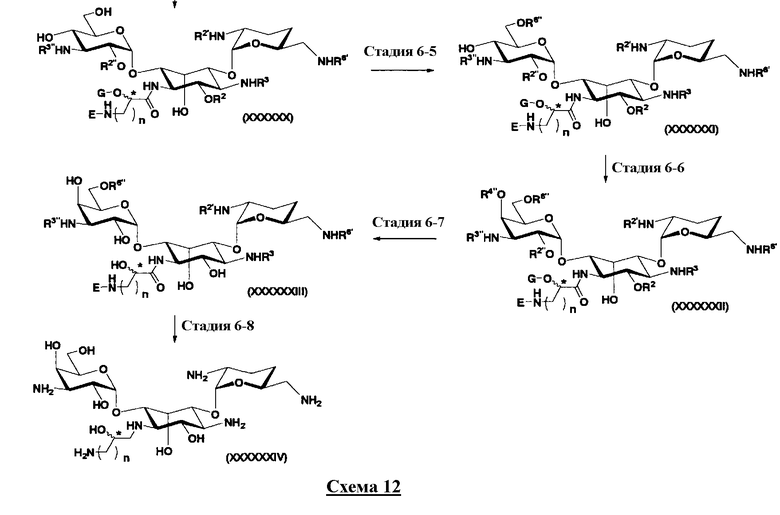
где R3, R2', R6', R3" и E представляют собой защитную группу для аминогруппы, предпочтительно трет-бутоксикарбонил; R2, R2" и G представляют собой защитную группу сложноэфирного типа, такую как ацетил и бензоил, предпочтительно ацетил; R4" представляет собой группу, выбранную из группы, состоящей из защитных групп сложноэфирного типа, таких как ацетил и бензоил и защитных групп типа сульфонила, таких как п-толуолсульфонил, метансульфонил и трифторметансульфонил; R6" представляет собой защитную группу для гидроксильной группы, которую можно удалить в кислотных условиях, такую как трифенилметил; A представляет собой C1-6 низший алкил, или две группы A вместе могут образовывать пятичленный или восьмичленный циклический алкил; и n и пространственная конфигурация атома углерода, отмеченного *, имеют указанные выше значения.
Стадия 6-1
Стадия 6-1 является стадией введения защитных групп в пять аминогрупп в соединении, представленном формулой (XXXXXVI), полученном на схеме 11, с получением соединения, представленного формулой (XXXXXVII). Указанную стадию можно осуществить способом, подробно раскрытым для стадии 1-7 на схеме 2.
Стадия 6-2
Стадия 6-2 является стадией введения циклического ацеталя, предпочтительно циклогексилиденацеталя, в качестве защитной группы в две гидроксильные группы в соединении, представленном формулой (XXXXXVII). Указанная стадия аналогична стадии 2-2 на схеме 4, и ее можно осуществить, используя реакционные условия, адаптированные на стадии 2-2 на схеме 4.
Стадия 6-3
Стадия 6-3 является стадией защиты трех гидроксильных групп в соединении, представленном формулой (XXXXXVIII), полученном на стадии 6-2, с использованием защитных групп сложноэфирного типа.
Условия для введения защитной группы сложноэфирного типа для гидроксильной группы в органическом синтезе, например условия, использованные на стадии 3-4 на схеме 6, можно использовать для осуществления указанной стадии.
Стадия 6-4
Стадия 6-4 является стадией удаления циклического ацеталя в качестве защитной группы в соединении, представленном формулой (XXXXXIX), полученном на стадии 6-3, с получением соединения, представленного формулой (XXXXXX).
Для указанной стадии можно использовать условия, адаптированные на стадии 2-4 на схеме 4.
Стадия 6-5
Стадия 6-5 представляет собой стадию введения защитной группы в первичную гидроксильную группу в соединении, представленном формулой (XXXXXX), полученном на стадии 6-4, с получением соединения, представленного формулой (XXXXXXI).
В качестве подлежащей введению защитной группы выбирают особенно объемные защитные группы, выбранные из защитных групп, используемых в качестве защитных групп для гидроксильной группы в обычном органическом синтезе, например трифенилметил. Реакцию осуществляют путем воздействия трифенилметилхлорида или трифенилметилбромида на соединение, представленное формулой (XXXXXX), в присутствии основания. Любое основание можно использовать без конкретных ограничений, если только указанное основание является инертным в отношении реакции. Предпочтительными основаниями являются, например, пиридин и 4-диметиламинопиридин. Кроме того, в указанной реакции можно использовать любой растворитель без конкретных ограничений, если только указанный растворитель является инертным в отношении реакции.
Стадия 6-6
Стадия 6-6 является стадией обращения гидроксильной группы в 4"-положении в соединении, представленном формулой (XXXXXXI) полученном на стадии 6-5, с получением соединения, представленного формулой (XXXXXXII).
Указанная стадия является стадией, аналогичной стадии 4-2 и стадии 4-3 на схеме 8, и ее можно осуществить, используя аналогичные реакционные условия. Стадию 6-6 можно также разделить на две стадии, как представлено на схеме 8.
Стадия 6-7
Стадия 6-7 является стадией удаления четырех защитных групп сложноэфирного типа (R2, R2", R4" и G) среди защитных групп для гидроксильной группы в соединении, представленном формулой (XXXXXXII), полученном на стадии 6-6, с получением соединения, представленного формулой (XXXXXXIII). Указанную стадию можно осуществить, используя стадию 2-9 на схеме 5.
Стадия 6-8
Стадия 6-8 является стадией удаления всех защитных групп в соединении, представленном формулой (XXXXXXIII), полученном на стадии 6-7, с получением соединения, представленного формулой (XXXXXXIV), которое является соединением, представленным формулой (Ia), где оба R5eq и R4"eq представляют собой атом водорода и оба R5ax и R4ax представляют собой гидроксил.
В формуле (XXXXXXIII) условия реакции для удаления защитных групп зависят от выбранной защитной группы. Например, если R3, R2', R6', R3" и E представляют собой трет-бутоксикарбонил, тогда как R6" представляет собой трифенилметил, удаление защитных групп можно осуществить в кислотных условиях, например, используя трифторуксусную кислоту.
Как раскрыто выше в соответствии со схемой реакции, представленной на схеме 12, в соединении, представленном формулой (Ia), полученном в первом или втором способе получения в соответствии с настоящим изобретением, пространственную конфигурацию гидроксила в 4"-положении можно превратить в аксиальную. Соответственно, первый или второй способ получения в соответствии с настоящим изобретением может включать введение защитных групп в функциональную группу, отличную от гидроксильной группы в 4"-положении соединения, представленного формулой (Ia), обращения пространственной конфигурации гидроксильной группы в 4"-положении и удаления защитных групп с получением соединения, представленного формулой (Ia), где пространственная конфигурация гидроксильной группы в 4"-положении была обращена. Настоящее изобретение включает также и указанный вариант.
(7) Получение соединения, в котором 5-положение соответствует аксиальному и 4"-положение соответствует аксиальному: получение синтетического промежуточного соединения, в котором 5-положение соответствует аксиальному и 4"-положение соответствует аксиальному (первый способ получения)
В первом способе получения в соответствии с настоящим изобретением соединение, представленное формулой (Ia), где оба R5eq и R4"eq представляют собой атом водорода и оба R5ax и R4"ax представляют собой гидроксил, можно получить, используя синтетическое промежуточное соединение, полученное в соответствии со схемой 13.
На схеме 13 соединение, представленное формулой (XXXXXXVII), соответствует соединению, представленному формулой (Xb), которое является синтетическим промежуточным соединением в первом способе получения в соответствии с настоящим изобретением, и в котором пространственная конфигурация гидроксильной группы в 5-положении и 4"-положении соответствует аксиальной.
[Химическая формула 41]
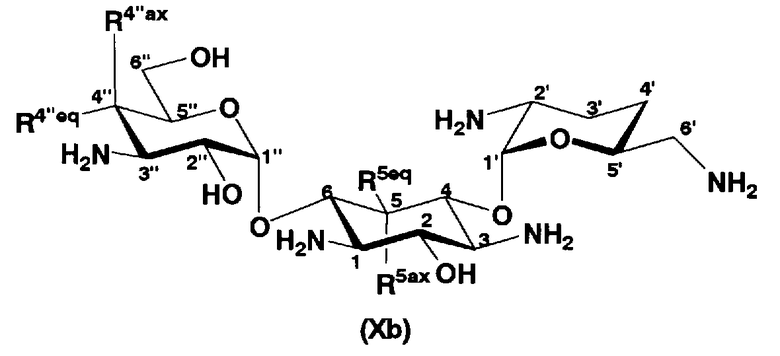
где оба R5eq и R4"eq представляют собой атом водорода; и оба R5ax и R4'ax представляют собой гидроксил.
[Химическая формула 42]
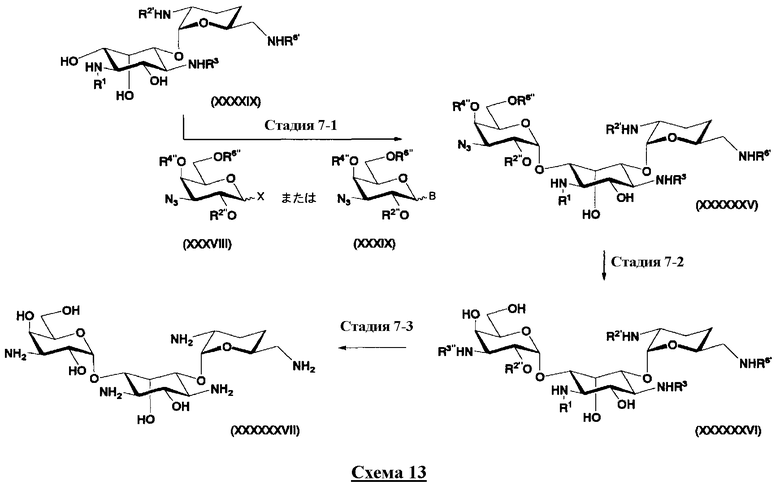
где R1, R3, R2' и R6' представляют собой защитные группы для аминогрупп, предпочтительно п-толуолсульфонил; R2" представляет собой защитную группу типа простого эфира, такую как бензил, предпочтительно бензил; R4" представляет собой защитную группу сложноэфирного типа, такую как ацетил или бензоил, предпочтительно ацетил; R6" представляет собой защитную группу сложноэфирного типа, такую как ацетил или бензоил, предпочтительно бензоил; B представляет собой отщепляемую группу, содержащую атом серы, такую как метилтио, этилтио или фенилтио, предпочтительно фенилтио; и X представляет собой атом галогена, такой как хлор, бром или йод, предпочтительно атом брома.
Стадия 7-1
Стадия 7-1 является стадией конденсации соединения, представленного формулой (XXXXIX), полученного на схеме 10, с соединением, представленным формулой (XXXVIII), или соединением, представленным формулой (XXXIX) на схеме 8, с получением соединения, представленного формулой (XXXXXXV). Указанную стадию можно осуществить способом, который был подробно раскрыт на стадии 2-8 на схеме 5.
Стадия 7-2
Стадия 7-2 является стадией удаления двух защитных групп сложноэфирного типа (R4" и R6") в соединении, представленном формулой (XXXXXXV), с получением соединения, представленного формулой (XXXXXXVI). Указанная стадия аналогична стадии 5-9 на схеме 11, и ее можно осуществить, используя реакционные условия, представленные на стадии 5-9 на схеме 11.
Стадия 7-3
Стадия 7-3 является стадией удаления всех защитных групп в соединении, представленном формулой (XXXXXXVI), полученном на стадии 7-2, восстановления азидогруппы до аминогруппы с получением соединения, представленного формулой (XXXXXXVII).
Реакционные условия для удаления защитных групп у соединения, представленного формулой (XXXXXXVII), меняется в зависимости от выбранных защитных групп. Например, если R1, R3, R2' и R6' представляют собой п-толуолсульфонил, удаление защитных групп можно осуществить, например, используя реакцию восстановления Бирча, в которой радикальную реакцию осуществляют, используя жидкий аммиак и металлический натрий.
Специалистам должно быть понятно, что после стадии 7-3 соединение, представленное формулой (Ia), можно получить, используя стадии 1-11 до стадии 1-14 на схеме 3 с получением соединения, представленного формулой (XXXXXXVII).
Соединения в соответствии с настоящим изобретением и соединения, полученные на стадиях получения, можно очистить и выделить, используя обычные способы очистки. Очистку и выделение можно осуществить, например, используя способы жидкостного разделения, перегонки, сублимации, осаждения, кристаллизации, хроматографии на нормальной колонке с силикагелем, или на колонке с силикагелем с обращенной фазой, используя хроматографическую колонку с ионообменной смолой, такой как Amberlite CG-50, Dowex 50 W × 2, или CM-Sephadex C-25, используя хроматографическую колонку с целлюлозой или т.п., используя препаративную тонкослойную хроматографию или высокоэффективную жидкостную хроматографию. Альтернативно, соединения, полученные на вышеуказанных стадиях получения, можно также соответствующим образом использовать на последующих стадиях без выделения и очистки.
Противомикробный агент
Соединения, представленные формулой (Ia) в соответствии с настоящим изобретением, или их фармакологически приемлемые соли, или их сольваты обладают превосходной противомикробной активностью против бактерий, которые вызывают инфекционные заболевания, например MRSA, staphylococcus aureus, colibacillus и Pseudomonas aeruginosa, и, предпочтительно, используют в качестве противомикробных агентов, более предпочтительно анти-MRSA агентов. Так, в соответствии с другим аспектом настоящего изобретения предложено использовать соединения в соответствии с настоящим изобретением или их фармакологически приемлемые соли или их сольваты для получения противомикробных агентов. Далее, в соответствии с еще одним предпочтительным аспектом настоящего изобретения, предложено применение соединения в соответствии с настоящим изобретением или его фармакологически приемлемой соли или его сольватов для получения анти-MRSA агента.
Фармацевтические композиции
Соединения, представленные формулой (Ia) в соответствии с настоящим изобретением, или их фармакологически приемлемые соли, или их сольваты, необязательно вместе с фармацевтически приемлемыми добавками, можно использовать в качестве фармацевтических средств. Tак, в соответствии со следующим аспектом настоящего изобретения предложена композиция, особенно фармацевтическая композиция, включающая соединение, представленное формулой (Ia), или его фармакологически приемлемую соль, или его сольват. Далее, в соответствии с другим аспектом настоящего изобретения предложено применение соединения, представленного формулой (Ia), или его фармакологически приемлемой соли, или его сольвата для получения фармацевтической композиции. Фармацевтическую композицию в соответствии с настоящим изобретением можно конкретно использовать для предотвращения и лечения инфекционных заболеваний. Предпочтительные инфекционные заболевания, для предотвращения или лечения которых очень эффективны фармацевтические композиции в соответствии с настоящим изобретением, включают внутрибольничные инфекционные заболевания и условно-патогенные инфекционные заболевания. Наиболее предпочтительными являются кожные гнойничковые заболевания, воспаление барабанной перепонки, синусит, конъюнктивит, пневмония, бронхит, сепсис, цистит, пиелонефрит, энтерит (включая пищевые отравления) и т.п.
Фармацевтическую композицию в соответствии с настоящим изобретением можно вводить пациентам парентерально или перорально, используя такие способы введения, как например, парентеральное введение (например внутривенное введение, внутримышечное введение, подкожное введение, ректальное введение или чрескожное введение), или пероральное введение в зависимости, например, от типа патогенных бактерий, характера заболевания и особенностей пациента. Далее, фармацевтическую композицию в соответствии с настоящим изобретением можно приготовить в подходящей лекарственной форме в зависимости от способа введения. Примеры таких лекарственных форм включают, например, препараты для инъекций, используемые, главным образом, например, для внутривенного введения и внутримышечного введения; препараты для наружного применения для парентерального введения, например глазные капли, ушные капли, назальные капли, глазные мази, адсорберы слизистой оболочки, дерматологические препараты, препараты для ингаляции или суппозитории; или препараты для перорального введения, например капсулы, таблетки, пилюли, тонкие порошки, гранулы, порошки, сиропы или облатки.
Вышеуказанные препараты можно получить обычным способом, используя добавки, например эксципиенты, увеличивающие объем агенты, связующие, смачивающие агенты, разрыхлители, поверхностно-активные агенты, смазывающие агенты, дисперсанты, буферные агенты, консерванты, солюбилизаторы, антисептики, корригенты, умягчители и стабилизаторы. Конкретные примеры нетоксичных добавок, которые можно здесь использовать для инъекций, глазных капель, ушных капель и назальных капель, включают растворяющие агенты или солюбилизаторы, которые могут представлять собой водные лекарственные формы или лекарственные формы, требующие растворения перед применением (например, дистиллированную воду для инъекций, физиологический солевой раствор, этанол, глицерин, пропиленгликоль, кукурузное масло и кунжутное масло), агенты, регулирующие рН (например, соли присоединения органических кислот, такие как тринатрийортофосфат и бикарбонат натрия, соли органических кислот, такие как цитрат натрия, и соли органических оснований, такие как L-лизин и L-аргинин), агенты, регулирующие тоничность растворов (например хлорид натрия, глюкоза и глицерин), буферирующие агенты (например, хлорид натрия, бензалконийхлорид и цитрат натрия), поверхностно-активные агенты (например, сорбитанмоноолеат и полисорбат 80), диспергирующие агенты (например, D-маннит), стабилизаторы (например, антиоксиданты, такие как аскорбиновая кислота, сульфит натрия и пиросульфит натрия) и хелатирующие агенты, такие как лимонная кислота и винная кислота); для глазных мазей, абсорберов слизистой оболочки, и дерматологических препаратов, составы включают, например, компоненты препаратов, пригодные в качестве мазей, кремов и пластырей (например, белый петролатум, макрогол, глицерин, жидкий парафин и хлопковую вату); для жидких препаратов для ингаляции включают, например, регуляторы pH (например, цитрат натрия и гидрид натрия), агенты, регулирующие тоничность растворов (например, хлорид натрия, бензалконийхлорид и цитрат натрия), и буферирующие агенты (например, хлорид натрия, бензалконийхлорид и цитрат натрия); для порошковых препаратов для ингаляции включают, например, лактозу в качестве носителя; и для препаратов для перорального введения и суппозиториев включают, например, эксципиенты (например, лактозу, D-маннит, кукурузный крахмал и кристаллическую целлюлозу), разрыхлители (например, карбоксиметилцеллюлозу и кальций-карбоксиметилцеллюлозу), связующие (например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и поливинилпирролидон), смазывающие агенты (например, стеарат магния и тальк), покрывающие агенты (например, шеллак, гидроксипропилметилцеллюлозу, сахарозу и оксид титана), пластификаторы (например, глицерин и полиэтиленгликоль) и субстраты (например, масло какао, полиэтиленгликоль и твердый жир).
Далее, если принять во внимание усиление терапевтического или профилактического воздействия на инфекционные заболевания, фармацевтическая композиция в соответствии с настоящим изобретением может содержать, кроме соединения в соответствии с настоящим изобретением, клинически полезный один или более из обычных противомикробных агентов (например, противомикробные агенты β-лактама (например, карбапенемы, цефалоспорины, цефамицины и пенициллины), гликопептидные противомикробные агенты, ансамициновые противомикробные агенты, аминогликозидные противомикробные агенты, хинолиновые противомикробные агенты, монобактамовые противомикробные агенты, макролидные противомикробные агенты, тетрациклиновые противомикробные агенты, хлорамфениколовые противомикробные агенты, линкомициновые противомикробные агенты, стрептограминовые противомикробные агенты, оксазолидиноновые противомикробные агенты, фосфомицины, новобиоцины, циклосерины и моеномицины). Альтернативно, фармацевтическую композицию вместе с вышеуказанным противомикробным агентом можно вводить в живой организм. Далее, если принять во внимание тот факт, что фармацевтическая композиция в соответствии с настоящим изобретением может расширить или повысить эффективность против грамотрицательных бактерий и бактерий, устойчивых против существующих противомикробных агентов, фармацевтическая композиция в соответствии с настоящим изобретением может включать, например, a drug discharge pump (efflux pump) inhibitor и ингибитор фермента (например β-лактамазы), разлагающего существующий противомикробный агент. Альтернативно, фармацевтическую композицию в соответствии с настоящим изобретением вместе с указанными ингибиторами или т.п. можно вводить в живой организм. Кроме того, если принять во внимание тот факт, что фармацевтическая композиция в соответствии с настоящим изобретением может повысить терапевтическое действие или профилактическое действие против инфекционных заболеваний, фармацевтическую композицию в соответствии с настоящим изобретением можно использовать в комбинации с соединениями, которые не обладают никакой противомикробной активностью (например, лекарственными средствами для лечения осложнений). Настоящее изобретение включает также и указанный вариант.
Как было раскрыто выше, соединение, представленное формулой (Ia) в соответствии с настоящим изобретением, или его фармакологически приемлемую соль, или их сольват можно с успехом использовать в качестве противомикробного агента или фармацевтического препарата для предотвращения или лечения инфекционных заболеваний. Так, в соответствии с другим аспектом настоящего изобретения предложен способ предотвращения или лечения инфекционных заболеваний, включающий введение соединения, представленного формулой (Ia), или его фармакологически приемлемой солью, или их сольватом, животному, включая человека. В качестве кандидатов для осуществления предотвращения или лечения используют животных, предпочтительно млекопитающих, более предпочтительно людей. Дозу соединения, представленного формулой (Ia), или его фармакологически приемлемой соли, или их сольвата могут соответствующим образом определить специалисты, например, принимая во внимание схему приема, тип патогенных бактерий и возраст, пол и вес пациентов и тяжесть заболевания. В частности, дневную дозу и число доз в день, при необходимости, можно соответствующим образом изменять.
ПРИМЕРЫ
Настоящее изобретение далее иллюстрируется следующими примерами, которые никоим образом не ограничивают изобретение.
В следующем описании все температуры приведены в градусах Цельсия.
1Н-ЯМР означает спектральный метод протонного ядерного магнитного резонанса, и 13C-ЯМР означает спектральный метод углеродного ядерного магнитного резонанса. Химические сдвиги, полученные с помощью этих методов, выражены в сдвигах (м.д.) от тетраметилсилана (ТМС) в сторону слабого магнитного поля.
МС означает масс-спектральный метод, и результаты, полученные методом ионизации рассеянием электронов (ESI), методом ионизации при атмосферном давлении (API), методом бомбардировки быстрыми атомами (FAB), и методом анализа с помощью жидкостной хромато-масс-спектрометрии (LCMS) выражают в единицах m/z (масса/заряд).
Rf значения в тонкослойной хроматографии представляют собой значения, полученные с использованием силикагельных пластин ART5715 производства Merck, и элюент, обеспечивающий получение значений Rf, указывается в скобках.
В следующих структурных формулах, Bn означает бензил, Ac - ацетил, Ph - фенил, Ts п-толуолсульфонил и Cbz -бензилоксикарбонил.
Пример 1
Получение 2-гидроксиарбекацина
[Химическая формула 43]
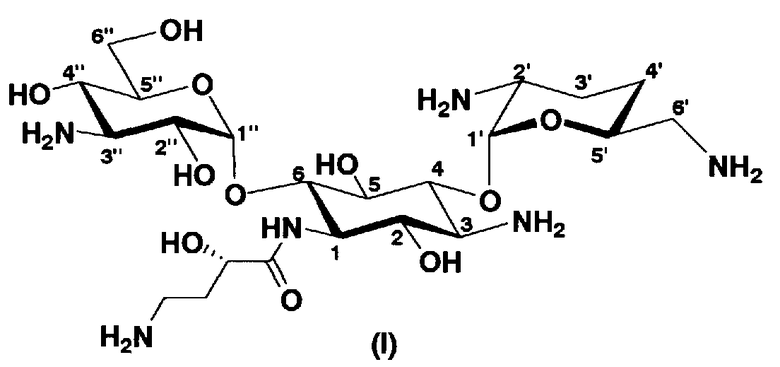
Стадия получения 1-1
Метил 3-азидо-3-дезокси-4,6-О-изопропилиден-D-глюкопиранозид
[Химическая формула 44]
Метил 3-азидо-3-дезокси-D-глюкопиранозид (32,7 г), синтезированный в соответствии со способом C.B.Barlow et al. (J. Chem. Soc., Abstracts pp.3870-3871, (June), (1965)), растворяют в 330 мл N,N-диметилформамида. К раствору добавляют 2,2-диметоксипропан (26,8 мл) и 1,92 г п-толуолсульфоновой кислоты и полученную смесь перемешивают при 50°C. Через три часа после начала перемешивания к реакционному раствору добавляют 26,8 мл 2,2-диметоксипропана; через 5,5 часа к реакционному раствору добавляют далее 2,10 г п-толуолсульфоновой кислоты; через 6,5 часа к реакционному раствору добавляют далее 17,9 мл 2,2-диметоксипропана; и через 24 часа к реакционному раствору добавляют 16,2 мл триэтиламина при охлаждении льдом, и полученную смесь концентрируют досуха, используя вакуумный насос. К остатку добавляют хлороформ (1,5 л). Полученный раствор дважды промывают 500 мл насыщенного водного раствора бикарбоната натрия и затем дважды промывают 500 мл насыщенного солевого раствора, сушат над глауберовой солью и концентрируют досуха с получением указанного в заголовке соединения (смесь α формы и β формы, 36,3 г, выход 96%) в виде сиропа коричневого цвета.
ESIMS: m/z 282 [M+Na]+
α Форма
1H-ЯМР (CDCl3): δ 4,74 (д, 1H, J=4 Гц), 3,88 (дд, 1H, J=5, 10 Гц), 3,72 (т, 1H, J=10, 10 Гц), 3,65 (дддд, 1H, J=5, 9, 10 Гц), 3,61 (т, 1H, J=10, 10 Гц), 3,55 (дт, 1H, J=4, 10, 10 Гц), 3,45 (с, 3H), 3,47 (т, 1H, J=10, 10 Гц), 2,35 (д, 1H, J=10 Гц), 1,51 (с, 3H), 1,44 (с, 3H).
β Форма
1H-ЯМР (CDCl3): δ 4,27 (д, 1H, J=8 Гц), 3,95 (дд, 1H, J=5, 10 Гц), 3,78 (т, 1H, J=10, 10 Гц), 3,64 (м, 1H), 3,56 (т, 1H, J=10, 10 Гц), 3,55 (с, 3H), 3,50 (т, 1H, J=10, 10 Гц), 3,37 (дд, 1H, J=8, 10 Гц), 2,67 (шир.с, 1H), 1,51 (с, 3H), 1, 44 (с, 3H).
Стадия получения 1-2
Метил 3-азидо-2-О-бензил-3-дезокси-4,6-О-изопропилиден-D-глюкопиранозид
[Химическая формула 45]
Указанное выше соединение (46,0 г), полученное на стадии получения 1-1, растворяют в 690 мл N,N-диметилформамида. К раствору добавляют гидрид натрия 11,4 г (60% масляная суспензия) при перемешивании при охлаждении льдом и в атмосфере азота и полученную смесь перемешивают далее при охлаждении льдом и в атмосфере азота в течение 30 минут. Бензилбромид (27,4 мл) добавляют к реакционному раствору и полученную смесь перемешивают в атмосфере азота при комнатной температуре в течение 1,5 часа. Затем реакционный раствор охлаждают льдом и доводят значение pH до 4-5, добавляя 50% водный раствор уксусной кислоты. Затем реакционную смесь концентрируют досуха и к смеси добавляют 2,0 л хлороформа. Полученный раствор дважды промывают 500 мл насыщенного водного раствора бикарбоната натрия и промывают один раз 500 мл воды. Промытый раствор сушат над глауберовой солью и концентрируют досуха с получением 73,4 г сырого продукта. Сырой продукт очищают, используя хроматографическую колонку с силикагелем (гексан:этилацетат=7:1 до 3:1), используя 350 г нейтрального силикагеля с получением указанного в заголовке соединения (смесь α формы и β формы: 55,1 г, выход 89%).
ESIMS: m/z 372 [M+Na]+.
α Форма
1H-ЯМР (CDCl3): δ 7,30-7,40 (м, 5H), 4,70-4,90 (ABкв, 2H, Jgem=12 Гц), 4,52 (д, 1H, J=4 Гц), 3,85 (дд, J=2, 10 Гц), 3,82 (т, 1H, J=10, 10 Гц), 3,66 (т, 1H, J=10, 10 Гц), 3,65 (т, 1H, J=10, 10 Гц), 3,36 (с, 3H), 3,39 (дт, 1H, J=2, 10, 10 Гц), 3,37 (дд, 1H, J=4, 10 Гц), 1,47 (с, 3H), 1,43 (с, 3H).
β Форма
1H-ЯМР (CDCl3): δ 7,30-7,40 (м, 5H), 4,71-4,88 (ABкв, 2H, Jgem=12 Гц), 4,38 (д, 1H, J=8 Гц), 3,93 (дд, J=5, 10 Гц), 3,75 (т, 1H, J=10, 10 Гц), 3,56 (с, 3H), 3,51 (т, 1H, J=10, 10 Гц), 3,45 (т, 1H, J=10, 10 Гц), 3,27 (дд, 1H, J=8, 10 Гц), 3,26 (дт, 1H, J=5, 10, 10 Гц), 1,49 (с, 3H), 1,44 (с, 3H).
Стадия получения 1-3
Метил 3-азидо-2-О-бензил-3-дезокси-D-глюкопиранозид
[Химическая формула 46]

80% водный раствор уксусной кислоты (186 мл) добавляют к 37,3 г соединения, полученного на стадии получения 1-2. Реакции предоставляют возможность протекать при 80°C и через 30 минут после начала реакции реакционный раствор охлаждают до комнатной температуры и концентрируют досуха с получением 32,9 г указанного в заголовке соединения (смесь α формы и β формы) с количественным выходом.
ESIMS: m/z 332 [M+Na]+.
α Форма
1H-ЯМР (CDCl3): δ 7,30-7,45 (м, 5H), 4,63-4,80 (ABкв, 2H, Jgem=12 Гц), 4,57 (д, 1H, J=3,5 Гц), 3,78-3,85 (м, 2H), 3,63 (дт, 1H, J=4, 4, 10 Гц), 3,44 (т, 1H, J=10, 10 Гц), 3,40 (дд, 1H, J=3,5, 10 Гц), 3,38 (т, 1H, J=10, 10 Гц), 3, 37 (с, 3H), 2,44 (д, 1H, J=3,5 Гц), 1,82 (дд, 1H, J=6, 7,5 Гц).
β Форма
1H-ЯМР (CDCl3): δ 7,30-7,45 (м, 5H), 4,71-4,92 (ABкв, 2H, Jgem=12 Гц), 4,39 (д, 1H, J=8 Гц), 3,91 (м, 1H), 3,78-3,85 (м, 2H), 3,58 (с, 3H), 3,45 (т, 1 H, J=10, 10 Гц), 3,39 (т, 1H, J=10, 10 Гц), 3,27 (дд, 1H, J=8, 10 Гц), 2,50 (д, 1H, J=2,5 Гц), 1,97 (дд, 1H, J=6, 7,5 Гц).
Стадия получения 1-4
1,4,6-три-О-ацетил-3-азидо-2-О-бензил-3-дезокси-D-глюкопираноза
[Химическая формула 47]
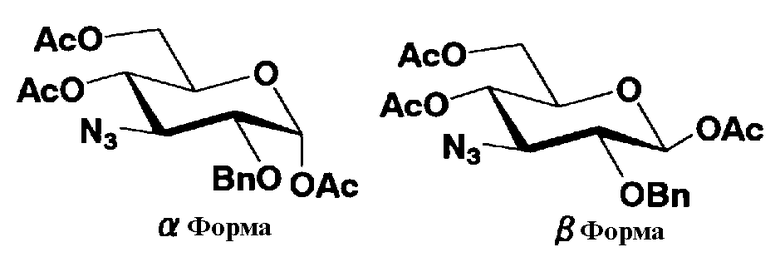
Cмесь уксусная кислота-уксусный ангидрид-концентрированная серная кислота (50:50:1) (164 мл) добавляют к и растворяют в 32,8 г соединения, полученного на стадии получения 1-3, используя ультразвуковой очиститель, и реакции предоставляют возможность протекать при комнатной температуре в течение 5 часов. Реакционный раствор добавляют по каплям при интенсивном перемешивании к 1,7 л охлажденного льдом 10% водного раствора ацетата натрия. К реакционному раствору четыре раза добавляют хлороформ (20 мл) и смесь интенсивно перемешивают при охлаждении льдом в течение 20 минут. Реакционный раствор возвращают к комнатной температуре и затем интенсивно перемешивают в течение одного часа. Реакционный раствор затем экстрагируют, используя 2,5 л хлороформа (один раз 900 мл и дважды 800 мл). Хлороформовый слой промывают один раз 500 мл насыщенного солевого раствора, дважды используя 500 мл насыщенного водного раствора бикарбоната натрия и один раз 500 мл насыщенного солевого раствора. Хлороформовый слой затем сушат над глауберовой солью и концентрируют досуха с получением 48,4 г сырого продукта. Полученный таким образом сырой продукт очищают, используя хроматографическую колонку с силикагелем (250 г) (гексан:этилацетат=5:1) с получением указанного в заголовке соединения (смесь α формы и β формы: 41,3 г, выход 92%) в виде сиропа светло-желтого цвета.
ESIMS: m/z 444 [M+Na]+.
α Форма
1H-ЯМР (CDCl3): δ 7,30-7,40 (м, 5H), 6,32 (д, 1H, J=4 Гц), 4,90 (т, 1H, J=10, 10 Гц), 4,62-4,71 (ABкв, 2H, Jgem=12 Гц), 4,22 (дд, 1H, J=5, 13 Гц), 4,01 (дд, 1H, J=2,5, 10 Гц), 3,98 (м, 1H), 3,88 (т, 1H, J=10, 10 Гц), 3,58 (дд, 1H, J=4, 10 Гц), 2,06, 2,12, 2,17 (каждый с, каждый 3H).
β Форма
1H-ЯМР (CDCl3): δ 7,30-7,40 (м, 5H), 5,62 (д, 1H, J=9 Гц), 4,91 (т, 1H, J=10, 10 Гц), 4,72-4,82 (ABкв, 2H, Jgem=12 Гц), 4,25 (дд, 1H, J=5, 13 Гц), 3,99 (дд, 1H, J=2,5, 13 H), 3,74 (ддд, 1H, J=2,5, 5, 10 Гц), 3,66 (т, 1H, J=10, 10 Гц), 3,59 (дд, 1H, J=9, 10 H), 2,07, 2,12, 2,22 (каждый с, каждый 3H).
Стадия получения 1-5a
4,6-ди-О-ацетил-3-азидо-2-О-бензил-1-бромо-3-дезокси-α-D-глюкопираноза
[Химическая формула 48]
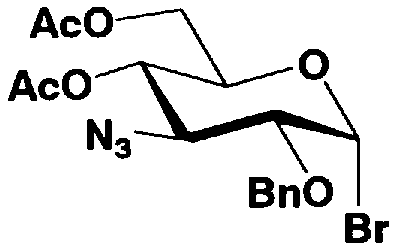
Соединение (347 мг), синтезированное на стадии получения 1-4, растворяют в смешанном растворе, состоящем из 6,2 мл метиленхлорида и 0,69 мл этилацетата. Тетрабромид титана (605 мг) добавляют к смешанному раствору при перемешивании, при охлаждении льдом и полученную смесь перемешивают при комнатной температуре в течение 14,5 часа. Реакционный раствор охлаждают льдом и к этому добавляют 30 мл охлажденного льдом метиленхлорида. Полученный раствор промывают восемь раз по 15 мл охлажденной льдом воды до тех пор, пока рН водного слоя не достигает значения pH 7. Затем полученный раствор сушат над глауберовой солью при охлаждении льдом и концентрируют досуха с получением указанного в заголовке соединения (352 мг, выход: 97%) в виде сиропа светло-желтого цвета.
1H-ЯМР (CDCl3): δ 7,20-7,45 (м, 5H), 6,32 (д, 1H, J=3,5 Гц), 4,92 (т, 1H, J=10, 10 Гц), 4,68-4,74 (ABкв, 2H, Jgem=12 Гц), 4,27 (дд, 1H, J=4,5, 12,5 Гц), 4,17 (ддд, 1H, J=2,5, 4,5, 10 Гц), 4,03 (дд, 1H, J=2,5, 12,5 Гц), 3,98 (т, 1H, J=10, 10 Гц), 3,43 (дд, 1H, J=3,5, 10 Гц), 2,13(с, 3H), 2,06 (с, 3H).
Стадия получения 1-5b
4,6-ди-О-ацетил-3-азидо-2-О-бензил-3-дезокси-1-тиофенил-α-D-глюкопираноза
[Химическая формула 49]
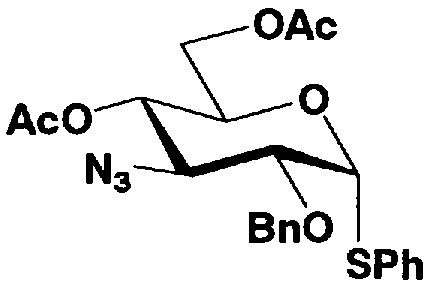
Соединение (19,9 г), полученное на стадии получения 1-4, растворяют в 200 мл метиленхлорида. К раствору добавляют триметилсилилтиофенол (26,8 мл) и 11,0 мл триметилсилилтрифлата и полученную смесь кипятят с обратным холодильником. Реакционную смесь охлаждают льдом через 41 час после начала кипячения с обратным холодильником. Затем к этому добавляют охлажденный льдом хлороформ (1,8 л). Затем реакционную смесь трижды промывают 1 л охлажденного льдом 5% водного раствора NaOH и дважды 1 л охлажденной льдом воды. Реакционную смесь далее сушат над глауберовой солью, концентрируют досуха с получением сырого продукта (22,1 г). Затем сырой продукт растворяют в 20 мл этилацетата. Затем к раствору добавляют гексан (120 мл) для кристаллизации. Полученные кристаллы промывают охлажденной льдом смесью этилацетат:гексан (1:9) с получением указанного в заголовке соединения (17,7 г, выход 79%). Затем маточный раствор и промывочную жидкость для кристаллов объединяют и концентрируют досуха с получением указанного в заголовке соединения (4,3 г).
ESIMS: m/z 494 [M+Na]+
1H-ЯМР (CDCl3): δ 7,20-7,48 (м, 10Н), 5,59 (д, 1H, J=5 Гц), 4,83 (т, 1H, J=10, 10 Гц), 4,68-4,78 (ABкв, 2H, Jgem=12 Гц), 4,46 (ддд, 1H, J=2,5, 5, 10 Гц), 4,22 (дд, 1H, J=5, 12 Гц), 3,96 (т, 1H, J=2,5, 12 Гц), 3,86 (т, 1H, J=10, 10 Гц), 3,79 (дд, 1H, J=5, 10 Гц), 2,13 (с, 3H), 1,99 (с, 3H).
Стадия получения 1-6
3',4'-дидезокси-2-гидроксинеамин
[Химическая формула 50]
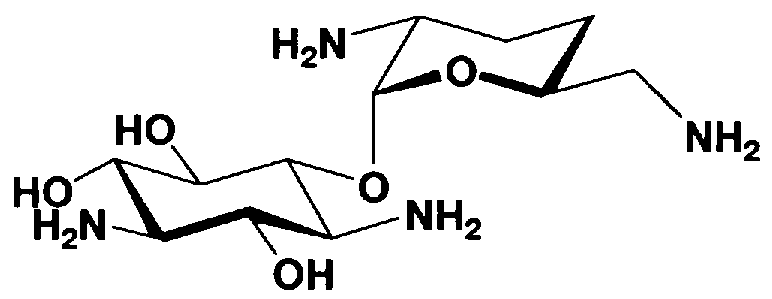
Способ A: 2-гидроксигентамицин C1a (18,0 г), представленный формулой (II), получают в соответствии с описанием японской патентной публикации №108041/1976 и очищают. Затем к полученному 2-гидроксигентамицину C1a добавляют 3 M HCl (360 мл) и полученную смесь кипятят с обратным холодильником в течение 1,5 часа. Реакционный раствор возвращают к комнатной температуре, охлаждают льдом, доводят значение pH до около 6,8, добавляя 4 M NaOH, разбавляют 1,8 л воды, и вводят в колонку с Amberlite CG-50 (уравновешенную заранее 0,005 M водным аммиаком) 500 мл. Указанную колонку промывают 1 л 0,005 M водного аммиака. Элюирование осуществляют, используя 2 л 0,1 M водного аммиака и 2 л 0,3 M водного аммиака. Фракции, содержащие указанное в заголовке соединение, объединяют и концентрируют досуха с получением 6,1 г указанного в заголовке соединения. Фракции, содержащие примеси, снова очищают, используя колонку с Amberlite CG-50 с получением 0,6 г указанного в заголовке соединения. Таким образом получают всего 6,7 указанного в заголовке соединения (монокарбонат, выход 72%).
Способ B: 2-гидроксигентамицин C1a (600 г), представленный формулой (II), получают и очищают в соответствии с описанием японской патентной публикации №108041/1976. Затем 2-гидроксигентамицин C1a (300 г, 483 ммоль в виде 2,5 карбоната) растворяют в 3 н. HCl (3 л), и полученный раствор перемешивают при 95°C в течение 70 минут. После остывания полученный раствор охлаждают льдом до 10°C, нейтрализуют и значение pH доводят до 6,88, добавляя 5 н. NaOH. Затем такую же реакцию снова осуществляют, используя 2-гидроксигентамицин C1a (300 г, 483 ммоль в виде 2,5 карбоната). Полученные две реакционные смеси объединяют и очищают, используя колонку с Amberlite CG-50 (NH4 тип; уравновешенную 0,005 M NH3, 10 л). Растворитель элюирования: 0,1 M → 0,2 M → 0,3 M NH3. Полученный продукт лиофилизируют, получая указанное в заголовке соединение (335,9 г; 781 ммоль в виде дикарбоната; выход 81%).
ESIMS: m/z 329 [M+Na]+
1H-ЯМР (26% ND3-D2O): δ 5,09 (д, 1H, J=3,5 Гц), 3,82 (м, 1H), 3,56 (т, 1H, J=10, 10 Гц), 3,30 (т, 1H, J=10, 10 Гц), 3,35 (т, 1H, J=10, 10 Гц), 3,08 (т, 1H, J=10, 10 Гц), 2,82 (дт, 1H, J=3,5, 3,5, 12 Гц), 2,78 (т, 1H, J=10, 10 Гц), 2,58-2,68 (м, 2H), 2,63 (т, 1H, J=10, 10 Гц), 1,67-1,79 (м, 2H), 1,60 (дкв, 1H, J=4, 12, 12, 12 Гц), 1,36 (дкв, 1H, J=4, 12, 12, 12 Гц).
Стадия получения 1-7
3',4'-дидезокси-2-гидроксил-1,3,2',6'-тетра-N-тозилнеамин
[Химическая формула 51]
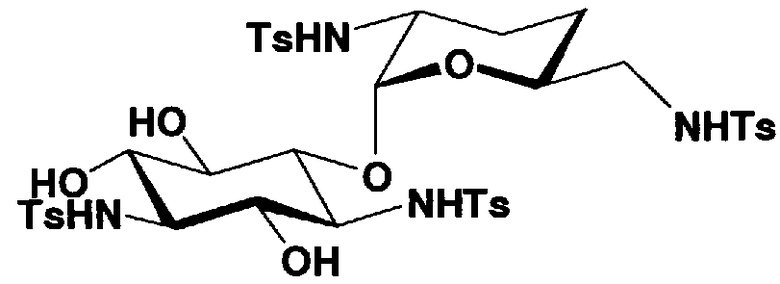
Соединение (6,7 г), полученное на стадии получения 1-6, растворяют в 67 мл воды. К раствору добавляют карбонат натрия (11,6 г) и затем добавляют 134 мл диоксана. К полученной смеси добавляют п-толуолсульфонилхлорид (20,8 г) при охлаждении льдом и полученную смесь интенсивно перемешивают в течение 15 минут при охлаждении льдом. Полученный раствор возвращают к комнатной температуре и интенсивно перемешивают в течение ночи. Затем к реакционному раствору добавляют диоксан (134 мл); через 42,5 часа 3,9 г карбоната натрия добавляют к реакционному раствору; через 88 часов к реакционному раствору добавляют 3,5 г п-толуолсульфонилхлорида; и через 112,5 часа к реакционному раствору добавляют 8,5 мл концентрированного водного аммиака, и полученную смесь перемешивают в течение 30 минут. Реакционную смесь концентрируют досуха. К остатку добавляют этилацетат (700 мл) и полученную смесь дважды промывают 300 мл воды, сушат над глауберовой солью и затем концентрируют досуха. Сырой продукт очищают, используя хроматографическую колонку с силикагелем (хлороформ:метанол=20:1), используя 250 г силикагеля, получая 3,47 г указанного в заголовке соединения. Фракцию, содержащую примеси, также обрабатывают на хроматографической колонке с силикагелем, получая 7,05 г указанного в заголовке соединения. В результате получают 10,52 г (полное количество; выход 63%) указанного в заголовке соединения.
ESIMS: m/z 945 [M+Na]+
1H-ЯМР (Пиридин-d5): δ 9,20 (д, 1H, J=7 Гц), 9,15 (д, 1H, J=8 Гц), 8,80 (д, 1H, J=8 Гц), 8,46 (т, 1H, J=6, 6 Гц), 7,90-8,18 (м, 8H), 7,00-7,15 (м, 8H), 5,97 (д, 1H, J=3 Гц), 5,09 (м, 1H), 4,14 (т, 1H, J=10, 10 Гц), 4,07 (т, 1H, J=10, 10 Гц), 3,91 (т, 1H, J=10, 10 Гц), 3,79-3,86 (м, 2H), 3,7 4 (т, 1H, J=10, 10 Гц), 3,73 (м, 1H), 3,30-3,47 (м, 2H), 2,40 (дкв, 1H, J=5, 12, 12, 12 Гц), 2,17 (с, 6H), 2,16 (с, 3H), 2,11 (с, 3H), 1,77 (м, 1H), 1,58-1,72 (м, 2H).
Стадия получения 1-8
4",6"-ди-О-ацетил-3"-азидо-2"-О-бензил-3"-дезокси-2-гидроксил-1,3,2',6'-тетра-N-тозилдибекацин
[Химическая формула 52]
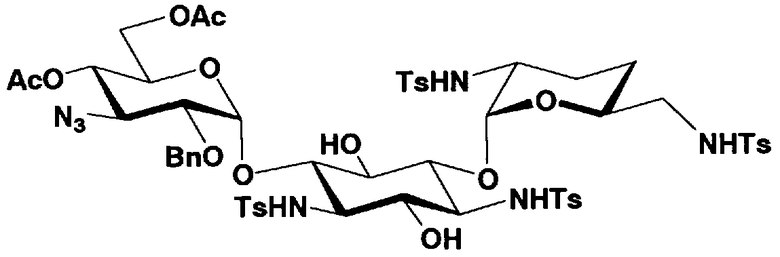
Способ A: Раствор соединения (352 мг), полученного на стадии получения 1-5a, в 7,3 мл 1,2-дихлорэтана добавляют к 363 мг соединения, полученного на стадии получения 1-7. К этому добавляют Drierite (1070 мг) и полученную смесь перемешивают при комнатной температуре в течение 30 минут. К реакционному раствору добавляют цианид ртути (397 мг) и полученную смесь перемешивают при экранировании от света при 60°C до тех пор, пока указанное соединение, полученное на стадии получения 1-5a, не исчезает, причем за ходом реакции следят, используя ТСХ. Реакционный раствор охлаждают до комнатной температуры и фильтруют через целит. Нерастворимую часть промывают 73 мл хлороформа. Полученный фильтрат и промывки объединяют, трижды промывают 36 мл насыщенного водного раствора бикарбоната натрия, трижды 36 мл 10% водного раствора иодида натрия и дважды 36 мл воды в указанном порядке, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают на хроматографической колонке с силикагелем (хлороформ:этилацетат=5:2), используя 36 г силикагеля с получением указанного в заголовке соединения (190 мг, выход 37,5%).
Способ B: Соединение (10,52 г), полученное на стадии получения 1-7, и соединение (9,67 г), полученное на стадии получения 1-5b, растворяют в 210 мл метиленхлорида. К раствору добавляют молекулярные сита 4Å (порошок 31,6 г) и полученную смесь перемешивают при комнатной температуре в течение 30 минут. Затем раствор 5,54 г N-йодсукцинимида и 0,55 мл трифторметансульфоновой кислоты в 9,45 мл метиленхлорида добавляют к реакционному раствору при перемешивании при -20°C при экранировании от света. Затем полученную смесь перемешивают при -20°C в течение 30 минут при экранировании от света. Затем к реакционному раствору добавляют 3,4 мл триэтиламина, и полученную смесь фильтруют через целит, и нерастворимую часть промывают 1,8 л хлороформа. Затем полученный фильтрат и промывки объединяют, дважды промывают 1 л насыщенного водного раствора бикарбоната натрия, дважды 1 л 10% водного раствора тиосульфата натрия и дважды 500 мл воды в указанном порядке, сушат над глауберовой солью и концентрируют досуха с получением 20,2 г сырого продукта. Затем сырой продукт очищают, используя хроматографическую колонку на 500 г силикагеля (хлороформ:этилацетат=5:2) с получением указанного в заголовке соединения (5,4 г, выход 37%).
Rf значение 0,68 (хлороформ:этилацетат=2:3).
ESIMS: m/z 1306 [M+Na]+.
1H-ЯМР (Пиридин-d5): δ 9,22 (д, 1H, J=7,5 Гц), 8,82 (д, 1H, J=8,5 Гц), 8,7 3 (т, 1H, J=6, 6 Гц), 8,25 (д, 1H, J=8,5 Гц), 7,8-8,1 (м, 10H), 7,0-7,45 (м, 11H), 6,75 (д, 1H, J=3 Гц), 6,09 (д, 1H, J=2 Гц), 5,51 (ABкв, 1H, Jgem=11 Гц), 5,23 (т, 1H, J=10, 10 Гц), 5,03 (м, 1H), 4,96 (м, 1H), 4,93 (ABкв, 1H, Jgem=11 Гц), 4,41 (т, 1H, J=10, 10 Гц), 4,38 (дд, 1H, J=4, 12 Гц), 4,23 (м, 1H), 4,07-4,21 (м, 5H), 3,83 (дд, 1H, J=3, 10 Гц), 3,78 (т, 1H, J=9, 9 Гц), 3,72 (м, 1H), 3,27 (м, 1H), 3,17 (м, 1H), 2,35 (м, 1H), 2,21 (с, 3H), 2,20 (с, 6H), 2,19 (с, 3H), 2,03 (с, 3H), 2,01 (с, 3H), 1,65 (м, 1H), 1,45-1,60 (м, 2H).
Стадия получения 1-9
3"-азидо-2"-О-бензил-3"-дезокси-2-гидроксил-1,3,2',6'-тетра-N-тозилдибекацин
[Химическая формула 53]
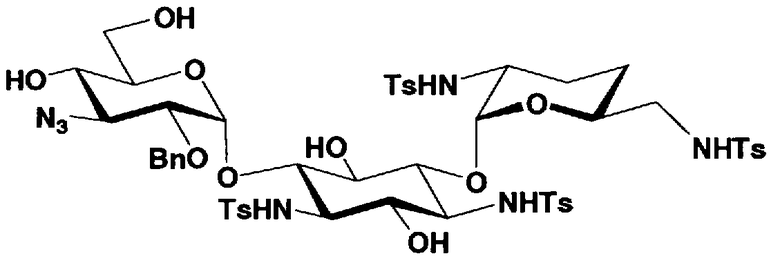
0,1% раствор (118 мл) метилата натрия в метаноле добавляют к 5,91 г соединения, полученного на стадии получения 1-8, и полученную смесь оставляют реагировать при комнатной температуре. 28% раствор (1,74 мл) метилата натрия в метаноле добавляют к реакционному раствору через 50 минут после начала реакции. Через 1,5 часа реакционный раствор нейтрализуют и доводят значение рН до 6-7, используя Dowex 50W × 2 (H+форма, 200-400 меш, замещен MeOH). Затем указанную смолу удаляют из реакционного раствора с помощью фильтрования и нерастворимую часть промывают пять раз или более метанолом. Полученный фильтрат и промывки объединяют и концентрируют досуха с получением 5,2 г сырого указанного в заголовке соединения. Указанное соединение, как оно есть, используют в следующей реакции.
Значение Rf 0,39 (хлороформ:этилацетат=2:3).
ESIMS: m/z 1222 [M+Na]+.
Стадия получения 1-10
2-гидроксидибекацин
[Химическая формула 54]

Жидкий аммиак (около 10 мл) хранят при -50°C в колбе в форме баклажана, содержащей 45,7 мг соединения, полученного на стадии получения 1-9. Затем в колбу в форме баклажана при -50°C добавляют 70 мг металлического натрия и полученную смесь интенсивно перемешивают, используя стеклянную палочку-мешалку в течение 2 часов. Метанол медленно добавляют в колбу в форме баклажана до тех пор, пока не исчезает окраска радикалов. Полученную смесь охлаждают до комнатной температуры, чтобы испарился аммиак, и в конце концентрируют досуха, используя испаритель. В колбу в форме баклажана добавляют воду (3 мл) и значение рН содержимого колбы доводят до 4-5, используя Dowex 50W×2 (H+ форма). Содержимое колбы вместе с указанной смолой вводят в колонку, набитую 2 мл той же самой смолы. Колонку промывают 20 мл воды, элюируют 1 M водным аммиаком. Позитивные в отношении нингидрина фракции объединяют и концентрируют досуха с получением 20,1 мг сырого продукта. Продукт переводят в водный раствор (10 мл), который затем вводят в колонку CM-Sephadex C-25 (уравновешенную 0,005 M водным аммиаком, 10 мл). Колонку промывают водой (10 мл). Затем осуществляют элюирование, последовательно изменяя при этом концентрацию водного аммиака с 0,05 M (50 мл) до 0,2 M (100 мл, 2 мл отсечка), и соответствующую фракцию концентрируют досуха с получением 16,4 мг указанного в заголовке соединения (моногидрат монокарбоната, выход 79%).
Rf значение 0,35 (1-бутанол:этанол:хлороформ:17% водный аммиак=4:7:2:7).
ESIMS: m/z 490 [M+Na]+.
1H-ЯМР (26% ND3-D2O): δ 5,12 (д, 1H, J=3,5 Гц), 5,01 (д, 1H, J=4 Гц), 3,88 (м, 1H), 3,83 (м, 1H), 3,75 (дд, 1H, J=2,5, 12 Гц), 3,68 (дд, 1H, J=5, 12 Гц), 3,66 (т, 1H, J=10, 10 Гц), 3,46 (дд, 1H, J=4, 10 Гц), 3,35 (т, 1H, J=10, 10 Гц), 3,28 (т, 1H, J=10, 10 Гц), 3,27 (т, 1H, J=10, 10 Гц), 3,10 (т, 1H, J=10, 10 Гц), 2,99 (т, 1H, J=10, 10 Гц), 2,83 (т, 1H, J=10, 10 Гц), 2,80 (м, 1H), 2,79 (т, 1H, J=10, 10 Гц), 2,66 (дд, 1H, J=4,5, 13 Гц), 2,62 (дд, 1H, J=7, 13 Гц), 1,68-1,77 (м, 2H), 1,60 (м, 1H), 1,37 (м, 1H).
13C-ЯМР (26% ND3-D2O): δ 102,62, 101,41, 85,70, 84,53, 75,36, 74,00, 73,67, 72,89, 71,83, 70,35, 61,35, 57,67, 56,81, 55,65, 51,35, 46,60, 28,97, 27,45.
Стадия получения 1-11
2',6'-ди-N-бензилоксикарбонил-2-гидроксидибекацин
[Химическая формула 55]
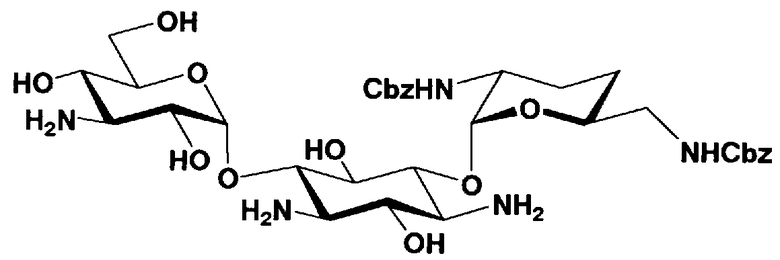
Тетрагидрат ацетата никеля (1136 мг) добавляют к соединению (625 мг; рассчитано как моногидрат монокарбоната), полученному на стадии получения 1-10. К этому добавляют метанол (25 мл) и полученную смесь обрабатывают ультразвуковым очистителем, получая гомогенный раствор. К раствору понемногу добавляют N-бензилоксикарбонилоксисукцинимид (626 мг) при охлаждении льдом в течение около 2 минут. Полученную смесь перемешивают при охлаждении льдом в течение одного часа, возвращают к комнатной температуре и далее перемешивают в течение 2,5 часа. Затем к реакционному раствору добавляют 57 мг N-бензилоксикарбонилоксисукцинимида при охлаждении льдом, возвращают к комнатной температуре и перемешивают далее в течение 2 часов. Реакционный раствор концентрируют досуха. К остатку добавляют концентрированный водный аммиак (30 мл), насыщенный хлоридом натрия, и полученную смесь трижды экстрагируют 20 мл 1-бутанола. Полученный слой бутанола концентрируют досуха. N,N-диметилформамид добавляют к 1423 мг полученного остатка и фильтруют через целит. Вещество на целите промывают N,N-диметилформамидом (20 мл×1, 10 мл×4). Полученный фильтрат и промывки концентрируют досуха с получением 1222 мг сырого продукта.
Rf значение 0,60 (1-бутанол:этанол:хлороформ:17% водный аммиак=4:7:2:7).
ESIMS: m/z 758 [M+Na]+.
1H-ЯМР (Пиридин-d5): δ 8,35 (шир.с, 1H), 7,90 (д, 1H, J=7,5 Гц), 7,15-7,55 (м, 10H), 5,62 (д, 1H, J=3 Гц), 5,41 (д, 1H, J=3 Гц), 5,33 (с, 2H), 5,30 (ABкв, 1H, Jgem=12 Гц), 5,14 (ABкв, 1H, Jgem=12 Гц), 4,64 (м, 1H), 4,44 (дд, 1H, J=2, 12 Гц), 4,42 (м, 1H), 4,28 (дд, 1H, J=5, 12 Гц), 4,24 (т, 1H, J=10, 10 Гц), 4,2 0 (дд, 1H, J=3, 10 Гц), 4,05 (м, 1H), 4,04 (т, 1H, J=10, 10 Гц), 3,90 (т, 1H, J=10, 10 Гц), 3,85 (т, 1H, J=10, 10 Гц), 3,68 (т, 1H, J=10, 10 Гц), 3,60 (т, 1H, J=10, 10 Гц), 3,52 (м, 1H), 3,39 (м, 1H), 3,29 (т, 1H, J=10, 10 Гц), 3,11 (т, 1H, J=10, 10 Гц), 2,05 (м, 1H), 1,89 (дкв, 1H, J=3,5, 13, 13, 13 Гц), 1,64 (м, 1H), 1,43 (кв, 1H, J=13, 13, 13 Гц).
Стадия получения 1-12
2',6'-ди-N-бензилоксикарбонил-2-гидрокси-3"-N-трифторацетилдибекацин
[Химическая формула 56]

Сырой продукт (1222 мг), полученный на стадии получения 1-11, растворяют в 17 мл безводного N,N-диметилформамида. К раствору добавляют этилтрифторацетат (0,15 мл) при перемешивании при охлаждении льдом. Полученную смесь возвращают к комнатной температуре и перемешивают в течение 8 часов. Реакционный раствор концентрируют досуха с получением 1416 мг продукта.
Rf значение 0,42 (часть нижнего слоя раствора хлороформ:метанол:15M водный аммиак (используют концентрированный водный аммиак)=1:1:1)
1H-ЯМР (Пиридин-d5): δ 10,65 (д, 1H, J=9 Гц), 8,35 (шир.с, 1H), 7,89 (д, 1H, J=8,5 Гц), 7,10-7,55 (м, 10H), 5,70 (д, 1H, J=3 Гц), 5,41 (д, 1H, J=3 Гц), 5,33 (с, 2H), 5,30 (ABкв, 1H, Jgem=12 Гц), 5,26 (ABкв, 1H, Jgem=12 Гц), 5,18 (м, 1H), 4,77 (м, 1H), 4,58 (т, 1H, J=10, 10 Гц), 4,50 (дд, 1H, J=3, 10 Гц), 4,31-4,46 (м, 3H), 4,18 (т, 1H, J=10, 10 Гц), 4,04 (м, 1H), 3,96 (т, 1H, J=10, 10 Гц), 3,82 (т, 1H, J=10, 10 Гц), 3,71 (т, 1H, J=10, 10 Гц), 3,51 (м, 1H), 3,40 (м, 1H), 3,31 (т, 1H, J=10, 10 Гц), 3,10 (т, 1H, J=10, 10 Гц), 2,05 (м, 1H), 1,89 (м, 1H), 1,64 (м, 1H), 1,43 (м, 1H).
Стадия получения 1-13
1-N-(4-бензилоксикарбониламино-2-(S)-гидроксибутирил)-2',6'-ди-N-бензилоксикарбонил-2-гидрокси-3"-N-трифторацетилдибекацин
[Химическая формула 57]
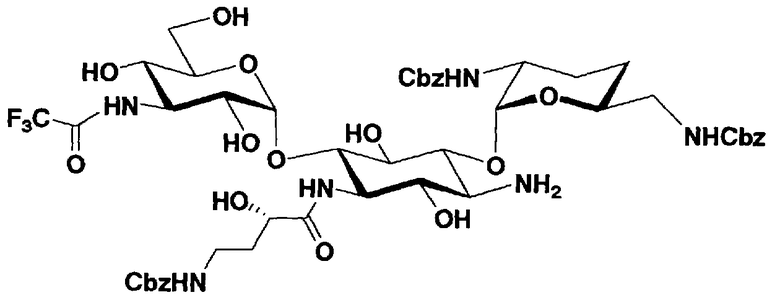
Продукт (438 мг), полученный на стадии получения 1-12, растворяют в 6,2 мл тетрагидрофурана. К полученному раствору по каплям добавляют раствор (2 мл) 149 мг сукцинимидного эфира (S)-4-N-бензилоксикарбониламино-2-гидроксимасляной кислоты, синтезированной в соответствии со способом, раскрытым Kawaguchi et al. Journal of Antibiotics, Vol.25, pp.695-708 (1972)), в тетрагидрофуране при перемешивании при охлаждении льдом за 2 минуты, и полученную смесь возвращают к комнатной температуре и перемешивают. Раствор (2 мл) 149 мг сукцинимидного эфира N-бензилоксикарбонил-4-амино-2-(S)-гидроксибутириловой кислоты в тетрагидрофуране добавляют к реакционному раствору при перемешивании при охлаждении льдом через 19 часов после начала перемешивания и полученную смесь возвращают к комнатной температуре и перемешивают. Через 20,5 часа реакционный раствор концентрируют досуха.
К остатку добавляют этилацетат (70 мл), и полученную смесь дважды промывают 14 мл насыщенного водного раствора бикарбоната натрия, дважды 14 мл воды и концентрируют досуха с получением 571 мг реакционной смеси.
Rf значение 0,59 (нижняя часть раствора хлороформ:метанол: 15 M водный аммиак (концентрированный водный аммиак)=1:1:1)
Стадия получения 1-14
2-гидроксиарбекацин
[Химическая формула 58]
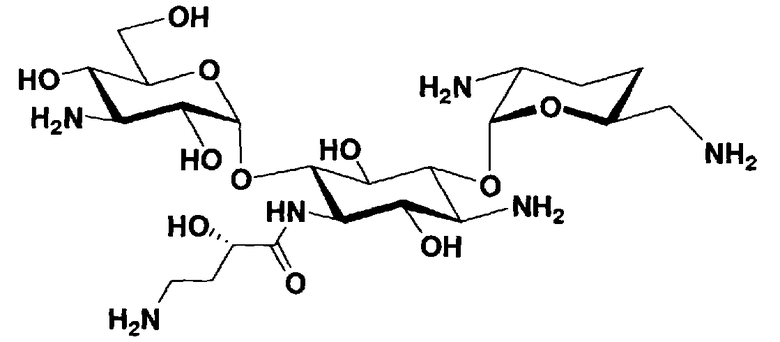
Тетрагидрофуран (22,8 мл) и 17,8 мл 3,5 M водного аммиака добавляют к 571 мг реакционной смеси, полученной на стадии получения 1-13, и смесь перемешивают при комнатной температуре в течение 20 час. Реакционный раствор концентрируют досуха. Смесь тетрагидрофуран-уксусная кислота-вода (4:1:1) (22 мл) добавляют к 624 мг полученного остатка. Затем добавляют 12 капель суспензии палладиевой черни в воде, и полученную смесь перемешивают при атмосферном давлении в течение 6 часов, сопровождая при этом барботированием в систему газообразного водорода. Затем палладиевую чернь удаляют из реакционного раствора с помощью фильтрования. Удаленную палладиевую чернь промывают водой, и полученный фильтрат и промывки объединяют и концентрируют досуха. К остатку добавляют 2 M водный аммиак (15 мл) и полученную смесь оставляют выстаиваться при комнатной температуре в течение ночи. Нерастворимую часть удаляют фильтрованием через хлопковую пробку с последующим концентрированием досуха с получением 467 мг сырого продукта. Сырой продукт растворяют в воде с получением 50 мл водного раствора и водный раствор вводят в колонку CM-Sephadex C-25 (уравновешенную 0,005 5 M водным аммиаком, 50 мл). Колонку промывают 100 мл 0,005 M водного аммиака и элюирование осуществляют, используя водный аммиак с 0,05 M (250 мл) до 0,5 M (500 мл) и затем 0,75 M (500 мл). Соответствующие фракции концентрируют, получая 39,1 мг указанного в заголовке соединения: 2-гидроксиарбекацина.
1H-ЯМР (26% ND3-D2O): δ 5,13 (д, 1H, J=3,5 Гц), 5,03 (д, 1H, J=4 Гц), 4,16 (дд, 1H, J=4, 9,5 Гц), 3,98 (м, 1H), 3,83-3,90 (м, 2H), 3,65-3,77 (м, 4H), 3,37 (т, 1H, J=10, 10 Гц), 3,34 (т, 1H, J=10, 10 Гц), 3,32 (дд, 1H, J=4, 10 Гц), 3,25 (т, 1H, J=10, 10 Гц), 2,97 (т, 1H, J=10, 10 Гц), 2,86 (т, 1H, J=10, 10 Гц), 2,82 (м, 1H), 2,70-2,78 (м, 2H), 2,65 (дд, 1H, J=5, 13 Гц), 2,62 (дд, 1H, J=7, 13 Гц), 1,86-1,96 (м, 1H), 1,69 -1,80 (м, 3H), 1,61 (дкв, 1H, J=4, 13, 13, 13 Гц), 1,37 (дкв, 1H, J=4, 13, 13, 13 Гц).
13C-ЯМР (26% ND3-D2O): δ 177,84, 102,09, 98,99, 83,65, 78,17, 75,41, 72,87, 72,23, 71,71, 71,28, 70,45, 69,81, 60,79, 56,60, 56,15, 54,88, 50,71, 46,02, 38,26, 37,23, 28,39, 26,94.
Пример 2
2,5 сульфат 2-гидроксиарбекацина (тригидрат)
[Химическая формула 59]
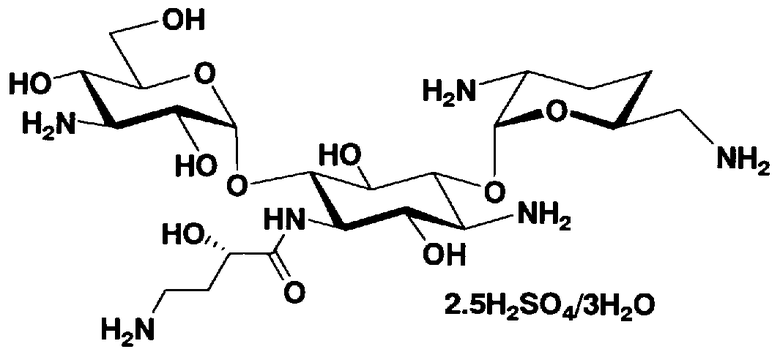
Получают водный раствор соединения (2-гидроксиарбекацин; 126,8 мг), представленного формулой (I), полученного на стадии получения 1-14 примера 1. рН водного раствора доводят до pH 7,0, добавляя 0,1 M серную кислоту, и лиофилизируют, получая 147,3 мг (тригидрата) 2,5 сульфата указанного в заголовке соединения.
Рассчитано для C22H44N6О11·2,5H2SО4·3H2О. C, 30,45; H, 6,39; N, 9,67.
Найдено: C, 30,41; H, 6,45; N, 9,46.
Пример 3:
Получение 2-гидроксиарбекацина
[Химическая формула 60]
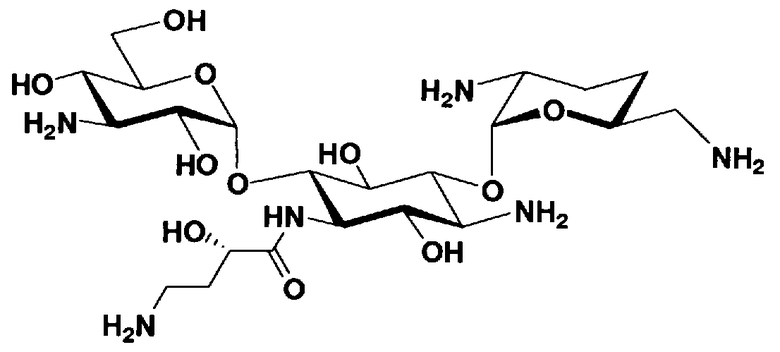
Стадия получения 3-1
1,3,2',6'-тетра-N-бензилоксикарбонил-3',4'-дидезокси-2-гидроксинеамин
[Химическая формула 61]
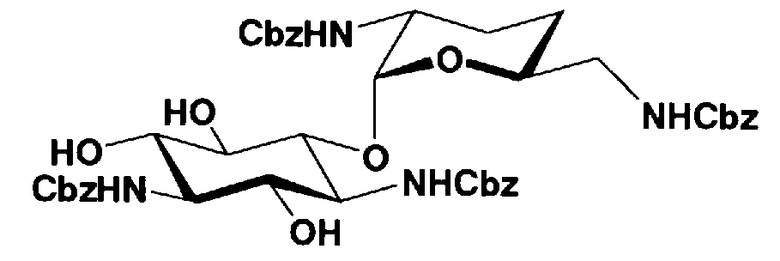
Способ A: Соединение (16,5 г, 38,3 ммоль: рассчитано как дикарбонат), полученное на стадии получения 1-6, растворяют в воде (83 мл). К раствору добавляют диметоксиэтан (165 мл), и полученную смесь перемешивают. К этому добавляют N-бензилоксикарбонилоксисукцинимид (66,8 г, 268 ммоль), затем добавляют триэтиламин (56,0 мл, 402 ммоль) и полученную смесь перемешивают в течение ночи (при этом происходит выработка тепла до 45°C). Этилацетат (490 мл) и воду (83 мл) добавляют к реакционному раствору с последующим разделением. Органический слой промывают насыщенным водным раствором бикарбоната натрия (247 мл) и 10% водным раствором тиосульфата натрия (247 мл) в указанном порядке. Органический слой сушат над безводным сульфатом магния, фильтруют и затем концентрируют при пониженном давлении. К остатку добавляют метанол (133 мл) (в виде пены) и полученную смесь перемешивают на водяной бане, нагретой до 60°C. В результате в осадок выпадают кристаллы. Полученную смесь перемешивают при комнатной температуре в течение ночи (промывки суспензии). Выпавшие в осадок кристаллы собирают с помощью фильтрования и сушат при пониженном давлении при 40°C в течение ночи с получением указанного в заголовке соединения (выход 33,8 г, количественно).
Способ B: Соединение (303 г, 704 ммоль в виде дикарбоната), полученное на стадии получения 1-6, растворяют в воде (1,5 л), и к полученному раствору добавляют 1,2-диметоксиэтан (3,0 л). К этому добавляют N-бензилоксикарбонилоксисукцинимид (1229 г, 4,93 моль, 7,0 экв), и затем добавляют триэтиламин (1013 мл, 7,40 моль, 10,5 экв), и полученную смесь перемешивают в течение ночи. К реакционному раствору добавляют этилацетат (9,1 л) и воду (1,5 л), с последующим разделением. Водный слой снова экстрагируют смесью этилацетат:1,2-диметоксиэтан=3:1 (1,8 л). Органические слои объединяют и промывают один раз 5% водным раствором бикарбоната натрия (4,6 л) и один раз 10% водным раствором тиосульфата натрия (4,6 л). Органический слой сушат над безводным сульфатом магния и затем растворитель удаляют перегонкой при пониженном давлении. К остатку добавляют метанол (2,4 л) и полученную смесь перемешивают при комнатной температуре в течение ночи. Выпавшие в осадок кристаллы отфильтровывают с получением указанного в заголовке соединения (619,1 г, 734 ммоль, выход 104%).
APIMS: m/z 843 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 7,73 (д, 1H, J=6,9 Гц), 7,12-7,56 (м, 22H), 6,82-6,95 (м, 1H), 5,67 (д, 1H, J=2,9 Гц), 5,12-5,31 (м, 8H), 4,26-4,40 (м, 1H), 3,90-4,30 (м, 7H), 3,35-3,50 (м, 2H), 1,85-2,01 (м, 2H), 1,43-1,65 (м, 2H).
Стадия получения 3-2
1,3,2',6'-тетра-N-бензилоксикарбонил-5,6-О-циклогексилиден-3',4'-дидезокси-2-гидроксинеамин
[Химическая формула 62]
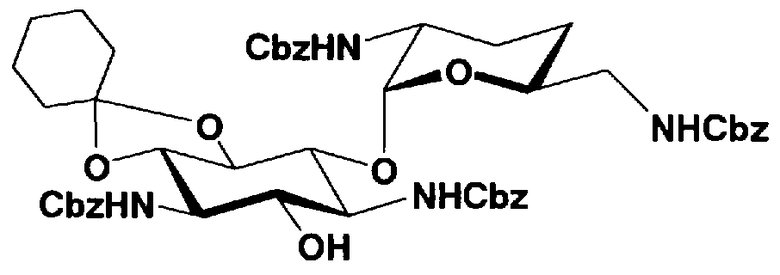
Способ A: Соединение (2,0 г, 2,4 ммоль), полученное на стадии получения 3-1, растворяют в диметоксиэтане (40 мл). К раствору добавляют 1,1-диметоксициклогексан (0,54 мл, 3,56 ммоль) и пиридиний п-толуолсульфонат (PPTS) (0,060 г, 0,24 ммоль), и полученную смесь перемешивают на масляной бане при 110°C в течение 2 часов, используя установку, включающую капельную воронку, содержащую молекулярные сита 5Å 1/16 (30 г, 40 мл) и холодильник Dimroth provided на капельной воронке (внутренняя температура 85°C). Этилацетат (40 мл) и насыщенный водный раствор бикарбоната натрия (40 мл) добавляют к реакционному раствору с последующим разделением. Органический слой промывают насыщенным водным раствором NaCl, сушат над безводным сульфатом магния и затем концентрируют при пониженном давлении. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (гексан:этилацетат=2:1 до 1:3, хлороформ:метанол=9:1) с получением указанного в заголовке соединения (1,75 г, 1,89 ммоль, выход 80%).
Способ B: Соединение (150 г, 178 ммоль), полученное на стадии получения 3-1, суспендируют в 1,2-диметоксиэтане (3,0 л). Добавляют 1,1-диметоксициклогексан (38,5 г, 267 ммоль, 1,5 экв) и пиридиний п-толуолсульфонат (4,47 г, 17,8 ммоль, 0,1 экв), и полученную смесь нагревают и кипятят с обратным холодильником, пропуская при этом через 1,5 кг молекулярных сит 5Å (1/16) в течение 4 часов. Реакционную смесь разбавляют этилацетатом (3 л) и промывают один раз 5% водным раствором бикарбоната натрия (4,6 л) и один раз 10% водным раствором тиосульфата натрия (3 л). Органический слой сушат над безводным сульфатом магния и растворитель затем удаляют перегонкой при пониженном давлении. Такую же реакцию осуществляют затем еще три раза. Полученные остатки объединяют и очищают на хроматографической колонке с силикагелем с получением указанного в заголовке соединения.
Элюент=гексан:этилацетат=2:1→1:1→1:2→1:3 (557,93 г, 604 ммоль, выход 85%).
APIMS: m/z 923 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 7,96 (д, 1H, J=6,2 Гц), 7,68 (д, 1H, J=6,8 Гц), 7,22-7,57 (м, 20H), 6,75-7,05 (м, 2H), 5,57 (с, 1H), 5,12-5,35 (м, 8H), 3,96-4,45 (м, 6H), 3,92 (т, 1H, J=10, 10 Гц), 3,79 (т, 1H, J=10, 10 Гц), 3,30-3,47 (м, 2H), 1,77-2,00 (м, 2H), 1,45-1,70 (м, 10H), 1,16-1,35 (м, 2H).
Стадия получения 3-3
2-бензилокси-1,3,2',6'-тетра-N-бензилоксикарбонил-5,6-О-циклогексилиден-3',4'-дидезоксинеамин
[Химическая формула 63]
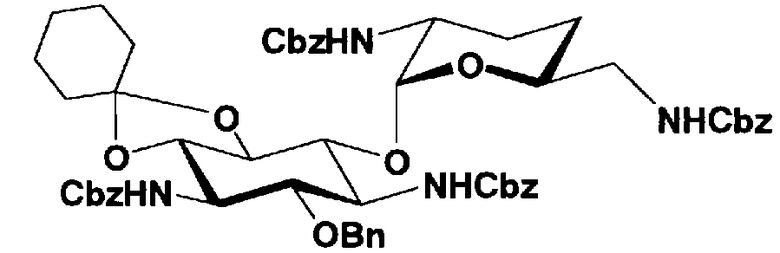
Способ A: Тетрагидрофуран (99 мл) добавляют к указанному соединению (4,970 г, 5,38 ммоль), полученному на стадии получения 3-2. 60% гидрид натрия (0,26 г, 6,50 ммоль, дисперсия в жидком парафине) и бензилбромид (1,15 мл, 9,67 ммоль) добавляют при внутренней температуре 4°C, и полученную смесь перемешивают при внутренней температуре от 7 до 8°C в течение 21 часа. К этому добавляют насыщенный водный раствор аммонийхлорида (10 мл) при внутренней температуре 4°C, и рН полученной смеси доводят до pH 7, с последующим разделением, используя 100 мл этилацетата и 50 мл воды. Органический слой сушат над сульфатом магния и растворитель удаляют перегонкой при пониженном давлении. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (толуол:этилацетат=3:1) с получением указанного в заголовке соединения (4,492 г, 4,43 ммоль, выход 83%).
Способ B: Соединение (396 г, 429 ммоль), полученное на стадии получения 3-2, растворяют в смешанном растворе, состоящем из тетрагидрофурана (8 л) и N,N-диметилформамида (158 мл), и полученный раствор охлаждают до 5°C. К раствору добавляют гидрид натрия (дисперсия в жидком парафине, 60%; 20,6 г, 515 ммоль, 1,2 экв) и бензилбромид (91,8 мл, 772 ммоль, 1,8 экв), и полученную смесь перемешивают при той же температуре в течение 6 часов. К смеси добавляют 10% водный раствор аммонийхлорида (8 л) для прекращения реакции и полученную смесь экстрагируют этилацетатом (8 л). Органический слой промывают один раз 20% солевым раствором (8 л) и один раз 10% водным раствором тиосульфата натрия (8 л) и сушат над безводным сульфатом магния. Растворитель затем удаляют перегонкой при пониженном давлении. Полученный остаток очищают на хроматографической колонке с силикагелем с получением указанного в заголовке соединения. Элюент=гексан:этилацетат=1:1→1:3 (320,24 г, 316 ммоль, выход 74%).
APIMS: m/z 1014 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 8,08-8,16 (м, 1H), 7,88-7,96 (м, 1H), 7,10-7,48 (м, 25H), 6,70-6,95 (м, 2H), 5,53 (с, 1H), 5,15-5,46 (м, 8H), 4,91 (AB, 2H, Jgem=11 Гц), 4,40-4,51 (м, 1H), 4,20-4,35 (м, 2H), 3,93-4,16 (м, 4H), 3,72 (т, 1H, J=10, 10 Гц), 3,32-3,50 (м, 2H), 1,77-2,00 (м, 2H), 1,45-1,72 (м, 10H), 1,15-1,35 (м, 2H).
Стадия получения 3-4
2-бензилокси-1,3,2',6'-тетра-N-бензилоксикарбонил-3',4'-дидезоксинеамин
[Химическая формула 64]
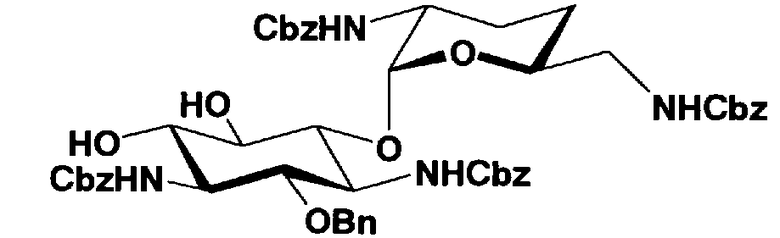
Способ A: Соединение (22,75 г, 22,5 ммоль), полученное на стадии получения 3-3, растворяют в смешанном растворе, состоящем из хлороформа (460 мл) и метанола (46 мл), и полученный раствор охлаждают до 7°C. К охлажденному раствору добавляют 90% трифторуксусную кислоту (46 мл) и полученную смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель удаляют перегонкой при пониженном давлении. К остатку добавляют метанол (460 мл) и полученную смесь подвергают суспензионной промывке в течение 2 часов, с последующим фильтрованием, получая указанное в заголовке соединение (19,47 г, 0,9 ммоль, выход 93%).
Способ B: Соединение (376 г, 371 ммоль), полученное на стадии получения 3-3, растворяют в смешанном растворе, состоящем из хлороформа (7,5 л) и метанола (750 мл), и полученную смесь охлаждают до 10°C. К охлажденному раствору добавляют 90% трифторуксусную кислоту (750 мл), и полученную смесь перемешивают при 20°C в течение 3 часов 20 минут. Растворитель удаляют перегонкой при пониженном давлении. К остатку добавляют метанол (7,5 л), суспензию перемешивают в течение ночи и фильтруют с получением указанного в заголовке соединения (313 г, 335 ммоль, выход 90%).
APIMS: m/z 933 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 7,86 (д, 1H, J=6,6 Гц), 7,62-7,70 (м, 1H), 7,10-7,55 (м, 26H), 6,80-6,95 (м, 1H), 5,64 (д, 1H, J=2,9 Гц), 5,13-5,37 (м, 8H), 4,91 (ABкв, 2H, Jgem=12 Гц), 4,29-4,37 (м, 3H), 3,90-4,17 (м, 3H), 4,02 (дд, 1H, J=9,1, 10 Гц), 3,94 (дд, 1H, J=8,8, 9,0 Гц), 3,92-3,52 (м, 2H), 1,86-2,04 (м, 2H), 1,47-1,66 (м, 2H).
Стадия получения 3-5
2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-1,6-N,О-карбонил-3',4'-дидезоксинеамин
[Химическая формула 65]
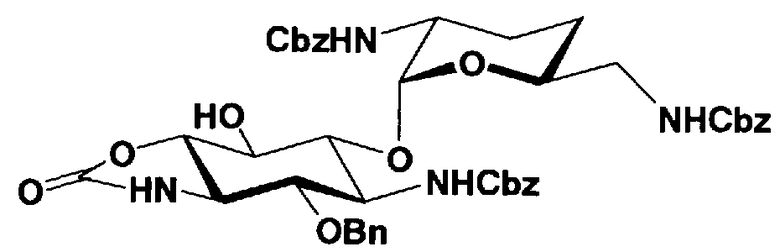
Способ A: Соединение (19,291 г, 20,7 ммоль), полученное на стадии получения 3-4, растворяют в N,N-диметилформамиде (390 мл), и полученный раствор охлаждают до 3°C. Гидрид натрия (60%, в масле; 4,968 г, 0,124 моль) добавляют к охлажденному раствору, и полученную смесь перемешивают при той же самой температуре в течение 1,5 часа. рН реакционной смеси доводят до pH около 7 при охлаждении льдом, добавляя насыщенный водный раствор аммонийхлорида (400 мл). К этому добавляют этилацетат (800 мл) и насыщенный водный раствор аммонийхлорида (400 мл), и полученную смесь перемешивают с последующим разделением. Органический слой дважды промывают 5% водным раствором аммонийхлорида (800 мл) и далее сушат над сульфатом магния. Растворитель удаляют перегонкой при пониженном давлении. К остатку добавляют метанол (400 мл). К этому добавляют затравочные кристаллы и ожидаемый продукт кристаллизуют при осторожном перемешивании. Полученные кристаллы собирают с помощью фильтрования с получением указанного в заголовке соединения. Маточный раствор концентрируют при пониженном давлении и полученный остаток подвергают суспензионной промывке смесью метанол/диизопропиловый эфир (1/1; 100 мл), получая указанное в заголовке соединение (15,70 г, 19,0 ммоль, выход 92%).
Способ B: Соединение (293 г, 314 ммоль), полученное на стадии получения 3-4, охлаждают в N,N-диметилформамиде (5,9 л) и полученный раствор охлаждают до 3°C. К охлажденному раствору добавляют гидрид натрия (дисперсия в жидком парафине, 60%; 75,4 г, 1,88 моль, 6,0 экв) и полученную смесь перемешивают при той же температуре в течение одного часа. Реакционную смесь выливают в двухслойный раствор, состоящий из 20% водного раствора аммонийхлорида (12 л) и этилацетата (5 л) при интенсивном перемешивании при охлаждении льдом. Затем к этому добавляют этилацетат (7 л) и полученную смесь перемешивают с последующим разделением. Органический слой дважды промывают 5% водным раствором аммонийхлорида (12 л) и сушат над безводным сульфатом магния. Растворитель удаляют перегонкой при пониженном давлении. К остатку добавляют метанол (5,9 л), и полученную смешанную суспензию перемешивают в течение ночи и затем фильтруют с получением указанного в заголовке соединения (248 г, 301 ммоль, выход 95%).
APIMS: m/z 825 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 8,80 (с, 1H), 8,67 (с, 1H), 8,17-8,24 (м, 1H), 7,75-7,53 (м, 1H), 7,12-7,53 (м, 20H), 6,93-7,10 (м, 1H), 5,22 (д, 1H, J=3,1 Гц), 5,11-5,39 (м, 6H), 4,88 (ABкв, 2H, Jgem=12 Гц), 3,95-4,38 (м, 7H), 3,73 (т, 1H, J=10, 10 Гц), 3,38-3,53 (м, 2H), 1,77-1,95 (м, 2H), 1,45-1,72 (м, 2H).
Стадия получения 3-6
2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезоксинеамин
[Химическая формула 66]
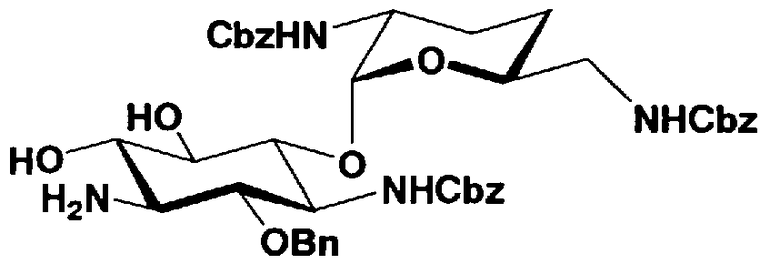
Способ A: Соединение (822 мг, 0,997 ммоль), полученное на стадии получения 3-5, растворяют в смешанной жидкости, состоящей из 1,4-диоксана (25 мл) и воды (25 мл). К раствору добавляют карбонат натрия (127 мг, 1,20 ммоль), и полученную смесь перемешивают при 80°C в течение 5 часов. Реакционную смесь охлаждают и экстрагируют один раз хлороформом (50 мл) и один раз смешанной жидкостью, состоящей из хлороформа (25 мл) и метанола (25 мл). Полученный экстракт сушат над сульфатом магния. Растворитель удаляют перегонкой при пониженном давлении. Полученный остаток растворяют в этилацетате (16 мл). К раствору добавляют затравочные кристаллы и ожидаемый продукт кристаллизуют при осторожном перемешивании, с последующим фильтрованием с получением соединения 7. Маточный раствор концентрируют при пониженном давлении и полученный остаток подвергают суспензионной промывке смесью этилацетат/диизопропиловый эфир (1/1; 4 мл), получая далее указанное в заголовке соединение (617 мг, 0,772 ммоль, выход 77%).
Способ B: Соединение (228 г, 276 ммоль), полученное на стадии получения 3-5, растворяют в смешанной жидкости, состоящей из 1,4-диоксана (6,8 л) и воды (6,8 л). Карбонат натрия (29,30 г, 276 ммоль, 1,0 экв) добавляют к полученному раствору и полученную смесь перемешивают при 80°C в течение 6 часов. Реакционную смесь охлаждают и в ней растворяют хлорид натрия (1,37 кг), и полученную смесь экстрагируют хлороформом (13,7 л). Полученный экстракт сушат над безводным сульфатом магния. Растворитель затем удаляют перегонкой при пониженном давлении с получением указанного в заголовке соединения (157,87 г, 198 ммоль, выход 71%).
FABMS: m/z 799 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 7,10-7,60 (м, 22H), 6,65-6,90 (м, 1H), 5,66 (д, 1H, J=2,9 Гц), 5,34 (с, 2H), 5,24 (ABкв, 2H, Jgem=13 Гц), 5,21 (ABкв, 2H, Jgem=13 Гц), 4,97 (ABкв, 2H, Jgem=11 Гц), 3,64-4,36 (м, 5H), 3,89 (т, 1H, J=9,0, 9,0 Гц), 3,54 (т, 1H, J=10, 10 Гц), 3,39-3,50 (м, 2H), 3,07 (т, 1H, J=10, 10 Гц), 1,87-2,02 (м, 2H), 1,45-1,65 (м, 2H).
Стадия получения 3-7
1-N-[(S)-4-бензилоксикарбониламино-2-бензилоксибутирил]-2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезоксинеамин
[Химическая формула 67]
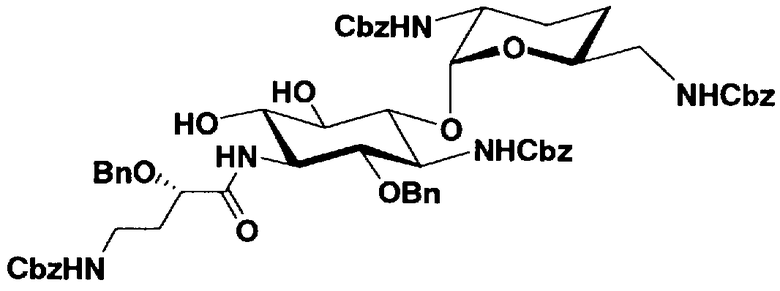
Способ A: (S)-2-бензилокси-4-бензилоксикарбониламиномасляную кислоту (9,065 г, 26,4 ммоль), синтезированную в сравнительном примере 1, растворяют в тетрагидрофуране и полученный раствор охлаждают до 2°C, к полученному раствору добавляют N-гидроксисукцинимид (3,038 г, 26,4 ммоль) и дициклогексилкарбодиимид (5,447 г, 26,4 ммоль), и полученную смесь перемешивают при комнатной температуре в течение 2 часов. Нерастворимую часть фильтруют с получением раствора активного сложного эфира. Указанное соединение (10,552 г, 13,2 ммоль), полученное на стадии получения 3-6, растворяют в тетрагидрофуране (210 мл). К раствору добавляют триэтиламин (3,7 мл, 27 ммоль) и раствор активного сложного эфира, и полученную смесь перемешивают при 50°C в течение 3,5 часа. Реакционную смесь разбавляют хлороформом (800 мл), промывают насыщенным раствором бикарбоната натрия (800 мл) и далее сушат над сульфатом магния. Растворитель удаляют перегонкой при пониженном давлении. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (хлороформ:этилацетат=1:1→хлороформ:метанол=30:1→20:1) с получением указанного в заголовке соединения (11,352 г, 10,1 ммоль, выход 76%).
Способ B: (S)-2-бензилокси-4-бензилоксикарбониламиномасляную кислоту (101,78 г, 296 ммоль, 1,5 экв), синтезированную в сравнительном примере 1, растворяют в тетрагидрофуране (2,04 л) и полученный раствор охлаждают до 3,4°C. N-гидроксисукцинимид (37,53 г, 326 ммоль, 1,65 экв) и дициклогексилкарбодиимид (67,28 г, 326 ммоль, 1,65 экв) добавляют к охлажденному раствору и полученную смесь перемешивают при 25°C в течение 3 часов. Нерастворимую часть отфильтровывают, получая раствор активного сложного эфира. Соединение (157,87 г, 198 ммоль), полученное на стадии получения 3-6, растворяют в тетрагидрофуране (3,16 л). К полученному раствору добавляют триэтиламин (41,34 мл, 296 ммоль, 1,5 экв) и раствор активного сложного эфира и полученную смесь перемешивают при 53°C в течение 4,5 часа. Перемешиваемую смесь затем разбавляют хлороформом (12,63 л) и разбавленный раствор промывают 5% водным раствором бикарбоната натрия (12,63 л) и далее сушат над безводным сульфатом магния. Растворитель удаляют перегонкой при пониженном давлении. Полученный остаток очищают, используя хроматографическую колонку с силикагелем с получением указанного в заголовке соединения. Элюент=хлороформ:ацетон=3,5:1→3:1→хлороформ:метанол=30:1→20:1. (158,45 г, 140 ммоль, выход 71%).
LCMS: m/z 1124 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 8,15 (д, 1H, J=8,3 Гц), 7,60-7,72 (м, 1H), 7,10-7,55 (м, 32H), 6,83-7,00 (м, 1H), 5,65 (д, 1H, J=2,9 Гц), 5,12-5,36 (м, 8H), 4,98 (ABкв, 2H, Jgem=11, 11 Гц), 4,60 (ABкв, 2H, Jgem=12, 12 Гц), 3,90-4,49 (м, 8H), 4,08 (т, 1H, J=9,8, 9,8 Гц), 3,38-3,70 (м, 4H), 2,20-2,36 (м, 2H), 1,85-2,01 (м, 2H), 1,45-1,65 (м, 2H).
Стадия получения 3-8
4",6"-ди-О-ацетил-3"-азидо-2",2"'-ди-О-бензил-1,3,2',6'-тетра-N-бензилоксикарбонил-3"-дезокси-2-гидроксиларбекацин
[Химическая формула 68]
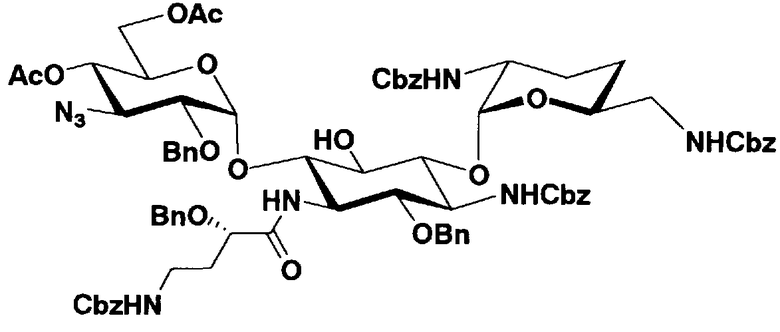
Соединение (11,249 г, 10,0 ммоль), полученное на стадии получения 3-7, и 4,6-ди-О-ацетил-3-азидо-2-О-бензил-1,3-дидезокси-1-фенилтио-α-D-глюкопиранозу (9,440 г, 20,0 ммоль), которые были высушены при пониженном давлении в течение 2 часов, растворяют в метиленхлориде (225 мл). К раствору добавляют молекулярные сита 4Å (порошок, 33,7 г), которые были высушены при пониженном давлении в течение 2 часов, и полученную смесь перемешивают при комнатной температуре в атмосфере аргона в течение одного часа. Реактор изолируют от света и охлаждают до -20°C. К этому добавляют N-йодсукцинимид (10,811 г, 48,1 ммоль) и трифторметансульфоновую кислоту (178 мл, 2,01 ммоль), и полученную смесь перемешивают при комнатной температуре в течение 2 часов. Добавляют N-йодсукцинимид (5,405 г, 24,0 ммоль) и трифторметансульфоновую кислоту (88 мл, 0,99 ммоль), и полученную смесь перемешивают при комнатной температуре в течение дополнительных двух часов. Триэтиламин (431 мл, 3,09 ммоль) добавляют при охлаждении льдом для прекращения реакции. Нерастворимую часть отфильтровывают и органический слой промывают один раз насыщенным водным раствором бикарбоната натрия (500 мл), дважды 10% водным раствором тиосульфата натрия и сушат над сульфатом магния. Растворитель удаляют перегонкой при пониженном давлении. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (хлороформ:ацетон=10:1→:/1→хлороформ:метанол=10:1), получая указанное в заголовке соединение (9,388 г, 6,32 ммоль, выход 63%). Выделяют оставшееся непрореагировавшее соединение (3,16 г, 2,81 ммоль), полученное на стадии получения 3-7.
LCMS: m/z 1485 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 8,30 (д, 1H, J=8,5 Гц), 7,75-7,83 (м, 1H), 7,20-7,67 (м, 36H), 6,92-7,05 (м, 1H), 6,75-6,90 (м, 1H), 5,92 (д, 1H, J=3,4 Гц), 5,65 (д, 1H, J=2,9 Гц), 4,94-5,35 (м, 12H), 4,62-4,76 (м, 4H), 4,35-4,50 (м, 3H), 3,92-4,35 (м, 9H), 3,71 (дд, 1H, J=3,4, 10 Гц), 3,34-3,62 (м, 4H), 2,18-2,43 (м, 2H), 2,00 (с, 3H), 1,99 (с, 3H), 1,80-1,93 (м, 2H), 1,43-1,65 (м, 2H).
Стадия получения 3-9
3"-азидо-2",2"-ди-О-бензил-1,3,2',6'-тетра-N-бензилоксикарбонил-3"-дезокси-2-гидроксиарбекацин
[Химическая формула 69]
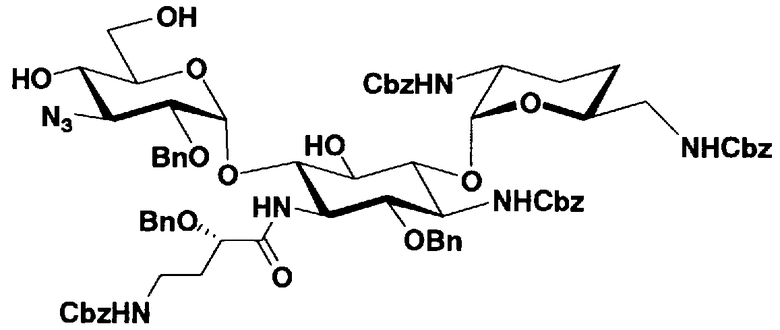
Указанное соединение (8,819 г, 5,94 ммоль), полученное на стадии получения 3-8, растворяют в смешанной жидкости, состоящей из хлороформа (180 мл) и метанола (90 мл). К раствору добавляют 0,5 M метилат натрия (метанольный раствор, 3,6 мл, 1,8 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 2 часов. К этому добавляют уксусную кислоту (0,14 мл, 2,4 ммоль) и полученную смесь промывают насыщенным водным раствором карбоната натрия (250 мл). Водный слой снова экстрагируют хлороформом (200 мл). Объединенные органические слои сушат над сульфатом магния и растворитель удаляют перегонкой при пониженном давлении. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (хлороформ:метанол=30:1) с получением указанного в заголовке соединения (7,263 г, 5,18 ммоль, выход 87%).
LCMS: m/z 1401 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 8,11 (д, 1H, J=8,3 Гц), 7,50-7,62 (м, 1H), 7,10-7,58 (м, 35H), 6,96-7,10 (м, 1H), 6,80-6,96 (м, 1H), 6,14-6,23 (м, 1H), 5,50-5,65 (м, 3H), 5,12-5,33 (м, 8H), 4,91 (ABкв, 2H, Jgem=11, 11 Гц), 4,83 (ABкв, 2H, Jgem=12, 12 Гц), 4,58 (ABкв, 2H, Jgem=12, 12 Гц), 3,85-4,51 (м, 14H), 3,37-3,64 (м, 5H), 2,20-2,42 (м, 2H), 1,80-1,95 (м, 2H), 1,45-1,65 (м, 2H).
Стадия получения 3-10
2-гидроксиарбекацин
[Химическая формула 70]
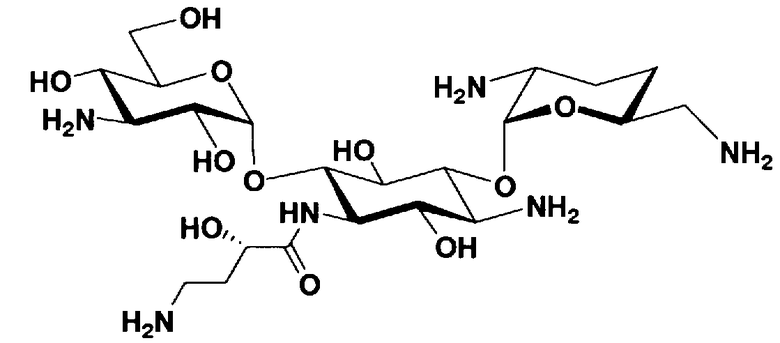
Соединение (154 мг, 0,110 ммоль), полученное на стадии получения 3-9, растворяют в 3 мл раствора смеси 1,4-диоксан:вода:1 н. хлористоводородная кислота=40:19:1. рН полученного раствора доводят до pH 1,53, к этому добавляют порошок палладиевой черни (154 мг) и полученную смесь интенсивно перемешивают в атмосфере водорода. Значение pH реакционной системы достигает 8,50 через три часа после начала интенсивного перемешивания. рН реакционной смеси доводят до 1,68, добавляя 1 н. хлористоводородную кислоту (500 мкл) и интенсивно перемешивают в атмосфере H2 в течение 15 часов. Порошок палладиевой черни удаляют с помощью фильтрования через хлопок и катализатор промывают водой. Полученный фильтрат и промывки объединяют и концентрируют досуха. Концентрат растворяют в воде, получая 10 мл раствора, который затем очищают на колонке Bio Rex 70 (уравновешенной 0,005 M водным аммиаком) с получением указанного в заголовке соединения (51,1 мг, 0,079 ммоль, выход 72%).
LCMS: m/z 569 [M+H]+.
1H-ЯМР (26% ND3-D2O): δ 5,13 (д, 1H, J=3,5 Гц), 5,03 (д, 1H, J=4 Гц), 4,16 (дд, 1H, J=4, 9,5 Гц), 3,98 (м, 1H), 3,83-3,90 (м, 2H), 3,65-3,77 (м, 4H), 3,37 (т, 1H, J=10, 10 Гц), 3,34 (т, 1H, J=10, 10 Гц), 3,32 (дд, 1H, J=4, 10 Гц), 3,25 (т, 1H, J=10, 10 Гц), 2,97 (т, 1H, J=10, 10 Гц), 2,86 (т, 1H, J=10, 10 Гц), 2,82 (м, 1H), 2,70-2,78 (м, 2H), 2,65 (дд, 1H, J=5, 13 Гц), 2,62 (дд, 1H, J=7, 13 Гц), 1,86-1,96 (м, 1H), 1,69-1,80 (м, 3H), 1,61 (дкв, 1H, J=4, 13, 13, 13 Гц), 1,37 (дкв, 1H, J=4, 13, 13, 13 Гц).
Пример 4
Получение 2-гидроксиарбекацина
[Химическая формула 71]
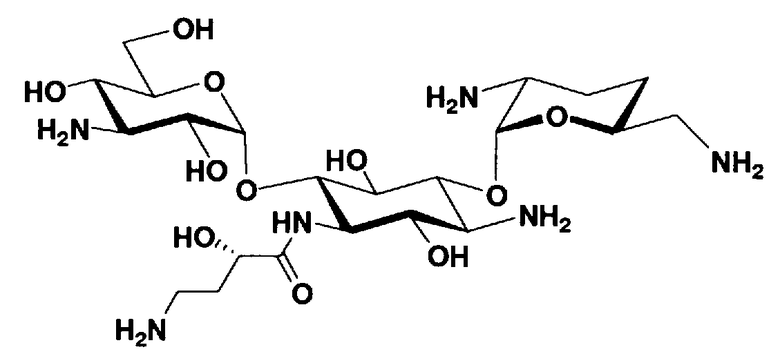
Стадия получения 4-1
3-азидо-2-О-бензил-3-дезокси-1-тиофенил-α-D-глюкопираноза
Химическая формула 72
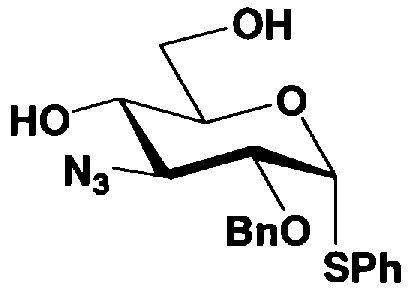
Способ A: раствор 0,13% метилат натрия/метанол (40,5 мл) и 13,5 мл хлороформа добавляют к соединению (2,67 г) со стадии получения 1-5b и полученную смесь оставляют реагировать при комнатной температуре в течение 1,5 часа. Реакционную смесь нейтрализуют (pH 6 до 7), добавляя Dowex 50 W×2 (H+ форма, замещен метанолом). Указанную смолу удаляют с помощью фильтрования с последующей промывкой метанолом (4 мл×5). Полученный фильтрат и промывки объединяют и концентрируют досуха с получением сырого продукта (2,193 г, количественно).
Способ B: Соединение (26,0 г), полученное на стадии получения 1-5b, суспендируют в 260 мл метанола и добавляют 33,3 мл раствора 0,5 M метилат натрия/метанол. Полученную смесь оставляют реагировать при комнатной температуре в течение 30 минут, и по каплям добавляют 1 мл уксусной кислоты. Реакционный раствор концентрируют досуха, получая сырой продукт, который используют, как он есть, в следующей реакции.
ESIMS: m/z 388 [M+H]+.
1H-ЯМР (CDCl3): δ 7,28-7,47 (м, 10H), 5,56 (д, 1H, J=4,6 Гц), 4,68-4,79 (ABкв, 2H, Jgem=11,7, 12,3 Гц), 4,21 (дт, 1H, J=3,9, 9,7 Гц), 3,70-3,81 (м, 4H), 3,45 (т, 1H, J=9,3, 9,5 Гц).
Стадия получения 4-2
2-О-бензил-3-бензилоксикарбониламино-1,3-дидезокси-1-фенилтио-α-D-глюкопираноза
[Химическая формула 73]
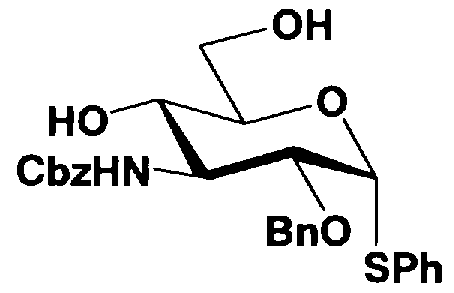
Способ A: Соединение (2,19 г), полученное способом А на стадии получения 4-1, растворяют в 44 мл (20 вес/объем) безводного тетрагидрофурана. К раствору добавляют трифенилфосфин (7,43 г, пятикратный по молям) и полученную смесь оставляют реагировать при комнатной температуре. К этому добавляют воду (0,71 мл, семикратный по молям), N-бензилоксикарбонилоксисукцинимид (1,83 г, 1,3-кратный по молям), и триэтиламин (2,37 мл, трехкратный по молям) через 18,5 часа после начала реакции, и полученную смесь перемешивают при комнатной температуре. Реакционную смесь концентрируют досуха через 2,5 часа после начала перемешивания при комнатной температуре с получением 12,6 г сырого продукта.
Способ B: Соединение (около 22,0 г), полученное способом B на стадия получения 4-1, растворяют в смешанной жидкости (550 мл, 10 вес/моль), состоящей из тетрагидрофурана/воды (1:1). Трифенилфосфин (36,1 г, 2,5-кратный по молям) к раствору добавляют и полученную смесь оставляют реагировать при комнатной температуре. N-бензилоксикарбонилоксисукцинимид (17,84 г, 1,3-кратный по молям) и 10 мл (1,3-кратный по молям) триэтиламина добавляют к этому через 2 часа после начала реакции и полученную смесь перемешивают при комнатной температуре. Реакционную смесь концентрируют досуха через 2,5 часа после начала перемешивания и промывают смесью этилацетат/гексан с получением 23,7 г сырого продукта (выход после трех стадий 87%).
APIMS: m/z 496 [M+H]+.
1Н-ЯМР (ДМСО): δ 7,52 (дт, 2H, J=1,5, 7,0 Гц), 7,23-7,36 (м, 13H), 5,78 (д, 1H, J=4,9 Гц), 5,18 (д, 1H, J=6,8 Гц), 4,97-5,12 (ABкв, 2H, Jgem=12,7, 12,9 Гц), 4,48-4,71 (ABкв, 2H, Jgem=12,2 Гц), 4,55 (т, 1H, J=5,8 Гц), 3,91 (м, 1H), 3,71 (дд, 1H, J=5,1, 10,7 Гц), 3,53 (м, 2H).
Стадия получения 4-3
4,6-ди-О-ацетил-2-О-бензил-3-бензилоксикарбониламино-1,3-дидезокси-1-фенилтио-α-D-глюкопираноза
[Химическая формула 74]
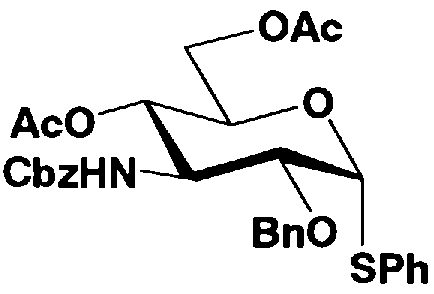
Способ A: Сырой продукт (12,61 г), полученный способом А на стадии получения 4-2, обезвоживают и растворяют в 56 мл безводного пиридина. К раствору добавляют уксусный ангидрид (5,3 мл, 10-кратный по молям) при охлаждении льдом и полученную смесь оставляют реагировать при комнатной температуре в течение 16 часов. К этому добавляют метанол (4,5 мл) (2-кратный по молям по сравнению с уксусным ангидридом), и полученную смесь оставляют выстаиваться при комнатной температуре в течение 30 минут и концентрируют досуха (азеотропная перегонка с толуолом: трижды). Затем к остатку добавляют 330 мл хлороформа и полученную смесь промывают насыщенным водным раствором бикарбоната натрия (160 мл×3), 5% водным раствором KHSО4 (160 мл×3) и дистиллированной водой (160 мл×1) в указанном порядке, сушат над глауберовой солью, и концентрируют досуха с получением 13,3 г сырого продукта. Сырой продукт очищают, используя хроматографическую колонку с силикагелем и затем кристаллизуют из смеси хлороформ/гексан, получая 3,19 г ожидаемого продукта в виде кристаллов (выход 97%).
Способ B: Часть (4,93 г) сырого продукта, полученного способом В на стадии получения 4-2, обезвоживают и растворяют в 50 мл (10 об./вес.) пиридина. К раствору добавляют уксусный ангидрид (25 мл, 5 об./вес.), и полученную смесь оставляют реагировать при комнатной температуре в течение 3 часов. К этому добавляют метанол (25 мл, 5 об./вес.) при охлаждении льдом, и полученную смесь оставляют выстаиваться при комнатной температуре в течение 30 минут. Затем добавляют 150 мл хлороформа, и полученную смесь промывают водой (150 мл×1), 2,5 н. хлористоводородной кислотой (150 мл×1), 0,5 н. хлористоводородной кислотой (150 мл×1), и насыщенным водным раствором бикарбоната натрия (150 мл×1) в указанном порядке, сушат над сульфатом магния, концентрируют досуха и кристаллизуют из смеси этилацетат/гексан с получением 5,39 г ожидаемого продукта (выход 94%).
FABMS: m/z 580 [M+H]+.
1H-ЯМР (CDCl3): δ 7,46-7,49 (м, 2H), 7,26-7,36 (м, 13H), 5,67 (д, 1H, J=5,4 Гц), 5,12 (шир.с, 2H), 4,89 (т, 1H, J=9,9, 10,3 Гц), 4,74-4,50 (ABкв, 2H, Jgem=12,2 Гц), 4,50-4,56 (м, 2H), 4,26 (дд, 1H, J=5,4, 12,2 Гц), 4,07 (дд, 1H, J=9,7, 10,3 Гц), 3,96 (дд, 1H, J=2,2, 12,2 Гц), 3,77 (шир.с, 1H), 1,98 (с, 3H), 1,94 (с, 3H).
Стадия получения 4-4
4",6"-ди-О-ацетил-2",2"'-ди-О-бензил-2-гидроксил-3,2',6',3"-тетра-N-бензилоксикарбониларбекацин
[Химическая формула 75]
Способ A: Соединение (2,401 г, 2,14 ммоль), полученное на стадии получения 3-7, и 4,6-ди-О-ацетил-2-О-бензил-3-бензилоксикарбониламино-1,3-дидезокси-1-фенилтио-α-D-глюкопиранозу (1,490 г, 2,57 ммоль), которые были высушены при пониженном давлении в течение 2 часов, растворяют в метиленхлориде (48 мл). Молекулярные сита 4Å (порошок, 7,20 г), высушенные при пониженном давлении в течение 2 часов, к раствору добавляют и полученную смесь перемешивают в атмосфере аргона при комнатной температуре в течение одного часа. Реактор экранируют от света и охлаждают до -20°C. К этому добавляют N-йодсукцинимид (1,390 г, 6,18 ммоль) и трифторметансульфоновую кислоту (38 мл, 0,43 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 2 часов. К реакционной смеси добавляют N-йодсукцинимид (0,700 г, 3,09 ммоль) и трифторметансульфоновую кислоту (19 мл, 0,22 ммоль), и полученную смесь перемешивают при комнатной температуре в течение дополнительных 2 часов. Триэтиламин (90 мл, 0,65 ммоль) добавляют при охлаждении льдом для прекращения реакции. Нерастворимую часть отфильтровывают, и органический слой промывают один раз насыщенным водным раствором бикарбоната натрия (120 мл) и дважды 10% водным раствором тиосульфата натрия, и сушат над сульфатом магния. Растворитель удаляют перегонкой при пониженном давлении и полученный остаток очищают, используя хроматографическую колонку с силикагелем (хлороформ:ацетон=10:1→7:1→хлороформ:метанол=10:1) с получением указанного в заголовке соединения (2,350 г, 1,47 ммоль, выход 69%).
Способ B: Дихлорметан (1,4 л) добавляют к соединению (70,0 г, 62,3 ммоль), полученному на стадии получения 3-7, и 4,6-ди-О-ацетил-2-О-бензил-3-бензилоксикарбониламино-1,3-дидезокси-1-фенилтио-α-D-глюкопиранозу (43,3 г, 74,7 ммоль, 1,2 экв), которая была высушена при пониженном давлении в течение ночи, и молекулярные сита 4Å (порошок, 210 г), которые были высушены при пониженном давлении в течение ночи. Полученную смесь перемешивают при комнатной температуре в атмосфере азота в течение одного часа. Реакционную смесь охлаждают до -15°C. К этому добавляют N-йодсукцинимид (67,2 г, 299 ммоль, 4,8 экв) и трифторметансульфоновую кислоту (1,1 мл, 12,5 ммоль, 0,2 экв), и полученную смесь перемешивают, экранируя от света, при -10°C в течение 50 минут. К этому добавляют N-йодсукцинимид (33,6 г, 149 ммоль, 2,4 экв) и трифторметансульфоновую кислоту (0,55 мл, 6,23 ммоль, 0,1 экв) и полученную смесь перемешивают при -10°C в течение дополнительных 50 минут. Затем к реакционной смеси добавляют триэтиламин (3,47 мл, 24,9 ммоль, 0,4 экв) для прекращения реакции. Нерастворимую часть отфильтровывают (промывают 1,4 л хлороформа), и органический слой промывают один раз 10% водным раствором тиосульфата натрия (2,8 л) и один раз 5% водным раствором бикарбоната натрия (2,8 л) и сушат над безводным сульфатом магния. Полученный остаток очищают на хроматографической колонке с силикагелем с получением указанного в заголовке соединения. Элюент=хлороформ:ацетон=7/1→5/1 (134,11 г, 84,2 ммоль, выход 68%).
LCMS: m/z 1593 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 8,22 (д, 1Н, J=3,9 Гц), 7,85 (д, 1Н, J=7,6 Гц), 7,67-7,77 (м, 1Н), 7,13-7,56 (м, 40H), 7,05-7,15 (м, 1H), 6,92-7,03 (м, 2H), 6,23-6,38 (м, 1H), 5,82 (д, 1H, J=3,2 Гц), 5,62 (с, 1H), 5,40 (т, 1H, J=10, 10 Гц), 5,12-5,35 (м, 10H), 4,87 (ABкв, 2H, J=12, 12 Гц), 4,95 (ABкв, 2H, J=11 Гц), 4,67 (кв, 1H, J=10, 10, 10 Гц), 4,60 (ABкв, 2H, J=12, 12 Гц), 3,92-4,58 (м, 13H), 3,37-3,61 (м, 4H), 2,18-2,45 (м, 2H), 2,00 (с, 3H), 1,87 (с, 3H), 1,82-1,96 (м, 2H), 1,45-1,68 (м, 2H).
Стадия получения 4-5
2",2'"-ди-О-бензил-3"-дезокси-2-гидроксил-3,2',6',3"-тетра-N-бензилоксикарбониларбекацин
[Химическая формула 76]
Соединение (0,6422 г, 0,40 ммоль), полученное на стадии получения 4-4, растворяют в смешанной жидкости, состоящей из хлороформа (12,8 мл) и метанола (6,4 мл). 0,5 M метилата натрия (метанольный раствор, 0,26 мл, 0,13 ммоль) добавляют к раствору и полученную смесь перемешивают при комнатной температуре в течение 2 часов. К этому добавляют уксусную кислоту (7,4 мкл, 0,13 ммоль) и полученную смесь промывают насыщенным водным раствором бикарбоната натрия (40 мл). Водный слой снова экстрагируют хлороформом (40 мл) и объединенные органические слои сушат над сульфатом магния. Растворитель удаляют перегонкой при пониженном давлении и остаток очищают на хроматографической колонке с силикагелем (хлороформ:метанол=30:1) с получением указанного в заголовке соединения (0,5798 г, 0,38 ммоль, выход 95%).
LCMS: m/z 1509 [M+H]+.
1H-ЯМР (Пиридин-d5): δ 8,09 (д, 1H, J=7,8 Гц), 7,60-7,70 (м, 1H), 7,57 (д, 1H, J=8,8 Гц), 7,15-7,55 (м, 40H), 6,70-7,10 (м, 2H), 6,65-6,75 (м, 1H), 6,04 (с, 1H), 5,47-5,58 (м, 3H), 5,15-5,35 (м, 10H), 4,93 (ABкв, 2H, Jgem=11, 11 Гц), 4,79 (ABкв, 2H, Jgem=12, 12 Гц), 4,61 (ABкв, 2H, Jgem=12, 12 Гц), 4,05-4,59 (м, 13H), 3,90-3,98 (м, 2H), 3,38-3,60 (м, 4H), 2,19-2,48 (м, 2H), 1,85-1,95 (м, 2H), 1,43-1,62 (м, 2H).
Стадия получения 4-6
2-гидроксиарбекацин
[Химическая формула 77]
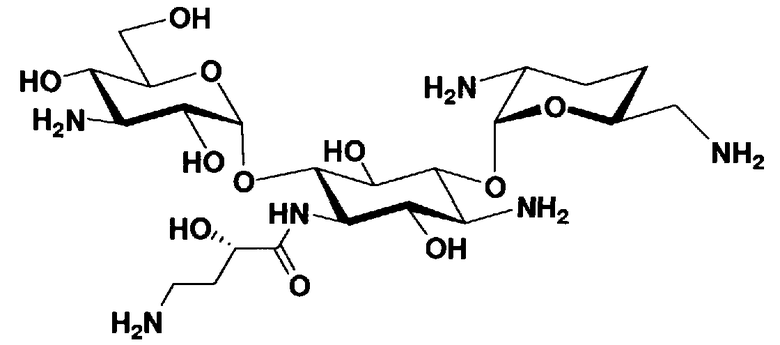
Соединение (290,0 мг, 0,192 ммоль), полученное на стадии получения 4-5, растворяют в 6 мл раствора 1,4-диоксан/вода/1 н. хлористоводородная кислота=40/19/1. рН полученного раствора доводят pH 1,54, к этому добавляют порошок палладиевой черни (145 мг), и полученную смесь интенсивно перемешивают в атмосфере водорода. Величина pH реакционной системы достигает значения 8,50 через 2 часа после начала интенсивного перемешивания. рН реакционной системы доводят до pH 1,68, добавляя 1 н. хлористоводородную кислоту (860 мкл) и интенсивно перемешивают в атмосфере водорода в течение 39 часов. Порошок палладиевой черни удаляют с помощью фильтрования через хлопок и катализатор промывают водой. Полученный фильтрат и промывки объединяют и концентрируют досуха. Полученный остаток растворяют в 10 мл воды, и полученный раствор очищают, используя колонку Amberlite CG-50 (уравновешенную 0,005 M NH4OH) с получением указанного в заголовке соединения (0,1165 г, выход 72%).
LCMS: m/z 569 [M+H]+.
1H-ЯМР (26% ND3-D2O): δ 5,13 (д, 1H, J=3,5 Гц), 5,03 (д, 1H, J=4 Гц), 4,16 (дд, 1H, J=4, 9,5 Гц), 3,98 (м, 1H), 3,83-3,90 (м, 2H), 3,65-3,77 (м, 4H), 3,37 (т, 1H, J=10, 10 Гц), 3,34 (т, 1H, J=10, 10 Гц), 3,32 (дд, 1H, J=4, 10 Гц), 3,25 (т, 1H, J=10, 10 Гц), 2,97 (т, 1H, J=10, 10 Гц), 2,86 (т, 1H, J=10, 10 Гц), 2,82 (м, 1H), 2,70-2,78 (м, 2H), 2,65 (дд, 1H, J=5, 13 Гц), 2,62 (дд, 1H, J=7, 13 Гц), 1,86-1,96 (м, 1H), 1,69-1,80 (м, 3H), 1,61 (дкв, 1H, J=4, 13, 13, 13 Гц), 1,37 (дкв, 1H, J=4, 13, 13, 13 Гц).
Пример 5
1-N-[(S)-3-амино-2-гидроксипропионил]-2-гидроксидибекацин
[Химическая формула 78]
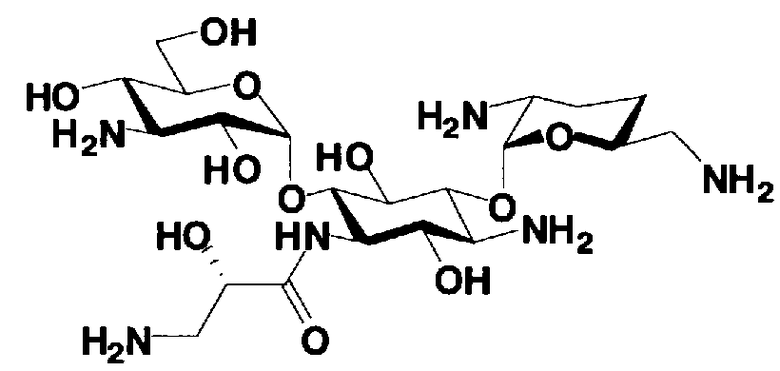
Стадия получения 5-1
1-N-[(S)-2-ацетокси-3-амино-N-бензилоксикарбонилпропионил]-2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезоксинеамин
[Химическая формула 79]
(S)-2-ацетокси-3-амино-N-бензилоксикарбонилпропионовую кислоту (21,2 мг), полученную в сравнительном примере 2, и 43,2 мг 2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезокси-2-гидроксинеамина, полученного на стадии получения 3-6 примера 3, растворяют в 0,6 мл тетрагидрофурана. К раствору добавляют 2-пропанол (1,3 мл), 0,1 мл воды и 22,4 мг 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорида (DMT-MM) и полученную смесь перемешивают при комнатной температуре в течение одного часа. Реакционный раствор концентрируют при пониженном давлении, к остатку добавляют 20 мл хлороформа, и полученную смесь промывают один раз 15 мл насыщенного водного раствора бикарбоната натрия и трижды 15 мл воды, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (3 г, хлороформ→хлороформ:метанол=→100:1→100:5→10:1) с получением указанного в заголовке соединения (43,0 мг, выход 75%).
Rf значение: 0,57 (хлороформ:метанол=10:1).
ESIMS: m/z 1084 [M+Na]+.
1H-ЯМР (Пиридин-d5): δ 9,53 (д, 1H, J=6,5 Гц), 8,08 (д, 1H, J=7,5 Гц), 7,68 (д, 1H, J=7,0 Гц), 7,49 (д, 1H, J=7,5 Гц), 7,21-7,45 (м, 26H), 5,72 (д, 1H, J=5,8 Гц), 5,44 (д, 1H, J=12,3 Гц), 5,39 (д, 1H, J=12,1 Гц), 5,22-5,37 (м, 7H), 5,17 (д, 1H, J=12,0 Гц), 5,02 (д, 1H, J=10,5 Гц), 4,59 (шир., 1H), 4,43 (дд, 1H, J=8,9, 9,3 Гц), 4,29-4,39 (м, 3H), 4,19-4,22 (м, 2H), 4,08 (дд, 1H, J=7,9, 9,3 Гц), 4,00-4,06 (м, 2H), 3,58-3,62 (м, 1H), 3,50-3,53 (м, 1H), 2,01 (шир., 2H), 1,75 (с, 3H), 1,62 (шир., 2H).
Стадия получения 5-2
1-N-[(S)-3-амино-N-бензилоксикарбонил-2-гидроксипропионил]-2,2"-ди-О-бензил-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин
[Химическая формула 80]
Соединение (225 мг), полученное на стадии получения 5-1, растворяют в 7,0 мл метиленхлорида. Соединение (246 мг), полученное на стадии получения 4-3, примера 4 и 675 мг порошка молекулярных сит 4Å, добавляют к полученному раствору, и полученную смесь перемешивают при комнатной температуре в течение одного часа. Перемешиваемую смесь охлаждают до -20°C, к ней добавляют 238 мг N-йодсукцинимида и 5,5 мкл трифторметансульфоновой кислоты и полученную смесь перемешивают, экранируя от света, при комнатной температуре в течение 3 часов. К этому добавляют триэтиламин (55 мкл) при охлаждении льдом. Реакционный раствор фильтруют через целит и нерастворимую часть промывают 30 мл хлороформа. Полученный таким образом раствор промывают один раз 20 мл насыщенного водного раствора бикарбоната натрия и дважды 15 мл 10% водного раствора тиосульфата натрия, сушат над глауберовой солью, и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (16 г, хлороформ:этилацетат=30:1→20:1→хлороформ:метанол=20:1→10:1) с получением 320 мг сырого продукта, содержащего 4",6"-ди-О-ацетил-1-N-[(S)-2-ацетокси-3-амино-N-бензилоксикарбонилпропионил]-2,2"-ди-О-бензил-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин в качестве ожидаемого продукта.
Rf значение: 0,56 (хлороформ:этилацетат=2:3):ожидаемый продукт.
ESIMS: m/z 1553 [M+Na]+: ожидаемый продукт.
Сырой продукт (320 мг) растворяют в 6,0 мл метанола. К раствору добавляют боргидрид натрия (7,1 мг) при охлаждении льдом и полученную смесь перемешивают при комнатной температуре в течение 1 часа. К этому добавляют ацетон (0,5 мл) при охлаждении льдом и полученную смесь перемешивают при комнатной температуре в течение 15 минут. Полученную смесь затем разбавляют 30 мл хлороформа, к разбавленному раствору добавляют 20 мл воды с последующим разделением. Органический слой сушат над глауберовой солью и концентрируют досуха с получением 272 мг сырого продукта (4",6"-ди-О-ацетил-1-N-[(S)-2-ацетокси-3-амино-N-бензилоксикарбонилпропионил]-2,2"-ди-О-бензил-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин).
Rf значение: 0,56 (хлороформ:этилацетат=2:3).
ESIMS: m/z 1553 [M+Na]+.
Сырой продукт (272 мг) растворяют в 5,4 мл хлороформа и 2,7 мл метанола. К раствору добавляют 28% раствор (9 мкл) метилата натрия в метаноле при охлаждении льдом и полученную смесь перемешивают при комнатной температуре в течение 2 часов. К этому добавляют уксусную кислоту (0,1 мл) при охлаждении льдом, полученную смесь разбавляют 20 мл хлороформа и разбавленный раствор промывают 10 мл насыщенного водного раствора бикарбоната натрия, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (20 г, хлороформ→хлороформ:метанол=99:1→97:3) с получением указанного в заголовке соединения (141 мг, выход 47%).
Rf значение: 0,18 (хлороформ:этилацетат=2:3).
1H-ЯМР (Пиридин-d5): δ 8,81 (д, 1H, J=6,6 Гц), 8,40 (д, 1H, J=7,5 Гц), 8,31 (д, 1H, J=7,4 Гц), 7,91 (шир., 1H), 7,65 (д, 1H, J=7,5 Гц), 7,22-7,51 (м, 36H), 5,78 (д, 1H, J=5,8 Гц), 5,73 (д, 1H, J=6,1 Гц), 5,44 (д, 1H, J=12,1 Гц), 5,39 (д, 1H, J=12,1 Гц), 5,18-5,38 (м, 10H), 5,11 (м, 1H), 5,07 (д, 1H, J=10,5 Гц), 5,01 (д, 1H, J=11,8 Гц), 4,94 (д, 1H, J=10,5 Гц), 4,78 (шир., 1H), 4,69-4,75 (м, 4H), 4,45 (шир., 1H), 4,42 (дд, 1H, J=8,5, 9,2 Гц), 4,37 (м, 1H), 4,33 (м, 1H), 4,29 (дд, 1H, J=7,7, 8,9 Гц), 4,26 (дд, 1H, J=7,9, 9,5 Гц), 4,00-4,05 (м, 2H), 3,82-3,89 (м, 2H), 3,58-3,62 (м, 1H), 3,49-3,53 (м, 1H), 1,93-2,00 (м, 2H), 1,58 (шир., 2H).
ESIMS: m/z 1427 [M+Na]+.
Стадия получения 5-3
1-N-[(S)-3-амино-2-гидроксипропионил]-2-гидроксидибекацин
[Химическая формула 81]
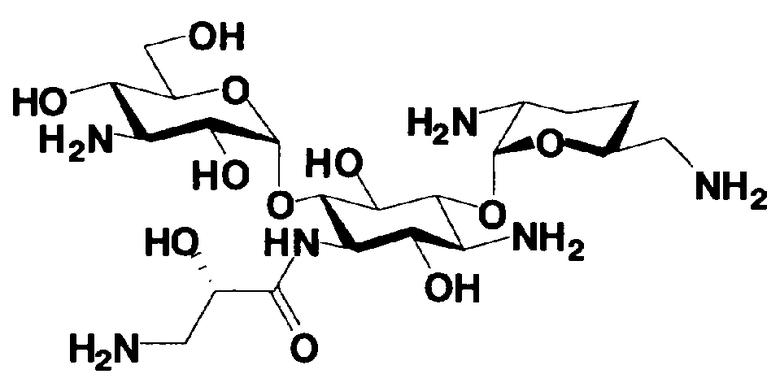
Соединение (141 мг), полученное на стадии получения 5-2, растворяют в 7 мл смеси тетрагидрофуран:вода:уксусная кислота (4:1:1). После этого добавляют семь капель смеси палладиевая чернь/вода в качестве катализатора, и полученную смесь интенсивно перемешивают при комнатной температуре в атмосфере водорода в течение 13 часов, причем в течение этого времени катализатор заменяют один раз с периодичностью 13 часов. Реакционный раствор фильтруют через хлопок и маточный раствор концентрируют при пониженном давлении. К остатку добавляют воду и полученный раствор снова концентрируют досуха при пониженном давлении. Полученный остаток растворяют в 5 мл воды. Полученный раствор нейтрализуют 0,1 M водным аммиаком и загружают в колонку Amberlite CG-50 (уравновешенную 0,005 M водным аммиаком, 5 мл), колонку промывают 0,005 M водным аммиаком (5 мл). Элюирование осуществляют, используя 0,10 M→0,25 M→0,50 M→0,75 M водный аммиак (каждого по 10 мл). Соответствующие фракции концентрируют досуха с получением указанного в заголовке соединения (1,5 карбонат 0,75 гидрат, 47 мг (71%)).
Rf значение: 0,18 (хлороформ:метанол:28% водный аммиак:этанол=4:6:7:2).
1H-ЯМР (DCl-D2O, pD ~3): δ 5,86 (д, 1H, J=3,4 Гц), 5,17 (д, 1H, J=3,8 Гц), 4,56 (дд, 1H, J=4,4, 7,8 Гц), 4,25 (дт, 1H, 4,0, 12,5), 4,16 (т, 1H, J=10,1 Гц), 4,13 (т, 1H, J=10,0 Гц), 4,05 (м, 1H), 3,97 (дд, 1H, J=1,9, 8,9 Гц), 3,94 (дд, 1H, J=1,8, 9,6 Гц), 3,88 (дд, 1H, J=3,1, 8,9 Гц), 3,77-3,84 (м, 3H), 3,72 (т, 1H, J=10,1 Гц), 3,60-3,64 (м, 1H), 3,49 (т, 1H, J=10,5 Гц), 3,37-3,45 (м, 2H), 3,33 (дд, 1H, J=2,1, 8,0 Гц), 3,27 (дд, 1H, J=1,9, 6,5 Гц), 3,16 (дд, 1H, J=6,9, 13,4 Гц), 2,07-2,13 (м, 2H), 1,92-1,98 (м, 1H), 1,60-1,66 (м, 1H).
13C-ЯМР (DCl-D2O, pD ~3): δ 174,06, 98,72, 95,19, 78,33, 74,95, 74,57, 72,41, 68,23, 68,06, 67,55, 66,38, 65,89, 60,10, 55,83, 55,50, 55,19, 49,07, 42,92, 42,22, 25,65, 20,84.
Рассчитано для C21H42N6O11 • 1,5H2CO3 • 0,75 H2O: C, 40,87; H, 7,09; N, 12,71. Найдено: C, 40,99; H, 7,07, N, 12,65.
Пример 6
1-N-[(S)-5-амино-2-гидроксипентаноил]-2-гидроксидибекацин
[Химическая формула 82]
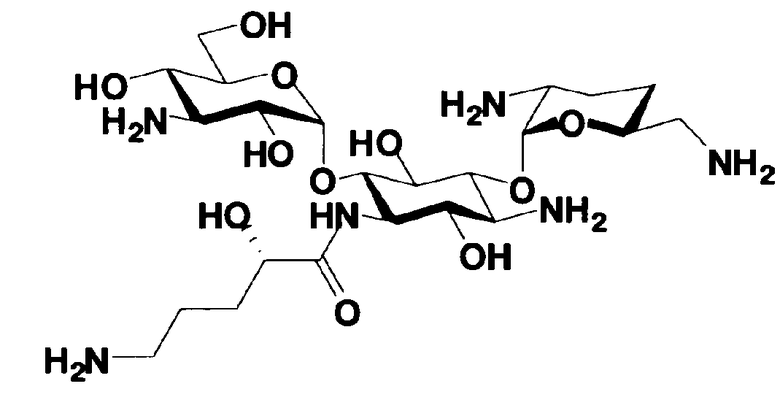
Стадия получения 6-1
1-N-[(S)-5-амино-2-бензилокси-N-бензилоксикарбонилпентаноил]-2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезоксинеамин
[Химическая формула 83]
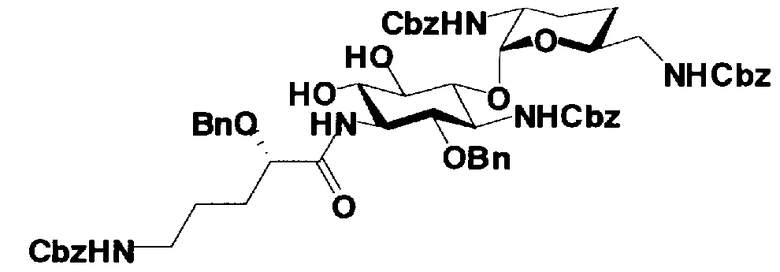
(S)-5-амино-2-бензилокси-N-бензилоксикарбонилпентановую кислоту (10 мг), полученную в сравнительном примере 3, и 22 мг 2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезокси-2-гидроксинеамина, полученного на стадии получения 3-6 примера 3, растворяют в 0,33 мл тетрагидрофурана. К раствору добавляют 2-пропанол (0,70 мл, вода 40 мкл) и 9 мг 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорида (DMT-MM), и полученную смесь перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор концентрируют при пониженном давлении. К остатку добавляют хлороформ (20 мл), и полученный раствор промывают один раз 10 мл насыщенного водного раствора бикарбоната натрия и трижды 10 мл воды, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают на хроматографической колонке с силикагелем (5 г, хлороформ→хлороформ:метанол=99:1→97:3→10:1) с получением указанного в заголовке соединения (22 мг, выход 72%).
Rf значение: 0,60 (хлороформ:метанол=10:1).
ESIMS: m/z 1160 [M+Na]+.
1H-ЯМР (Пиридин-d5): δ 8,66 (д, 1H, J=8,0 Гц), 8,43 (д, 1H, J=7,2 Гц), 8,11 (д, 1H, J=7,8 Гц), 7,87 (шир., 1H), 7,70 (шир., 1H), 7,60 (д, 2H, J=7,4 Гц), 7,39-7,51 (м, 8H), 7,21-7,31 (м, 20H), 5,79 (д, 1H, J=5,8 Гц), 5,41 (д, 1H, J=12,5 Гц), 5,38 (д, 1H, J=9,9 Гц), 5,31-5,35 (м, 2H), 5,28 (шир., 2H), 5,21 (д, 1H, J=12,5 Гц), 5,03-5,19 (м, 4H), 4,78 (д, 1H, J=11,8 Гц), 4,65 (кв, 1H, J=9,4 Гц), 4,52 (т, 1H, J=9,5 Гц), 4,49 (д, 1H, J=11,8 Гц), 4,37 (шир., 2H), 4,19-4,22 (м, 1H), 4,17 (т, 1H, J=7,3 Гц), 4,10 (т, 1H, J=7,7 Гц), 4,06 (дд, 1H, J=7,7, 9,6 Гц), 3,60-3,67 (м, 1H), 3,52-3,55 (м, 1H), 3,34 (м, 2H), 2,13 (шир., 2H), 1,99-2,09 (м, 3H), 1,90 (дт, 1H, J=5,9, 8,0 Гц), 1,62 (шир., 2H).
Стадия получения 6-2
1-N-[(S)-5-амино-2-бензилокси-N-бензилоксикарбонилпентаноил]-2,2"-ди-О-бензил-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин
[Химическая формула 84]
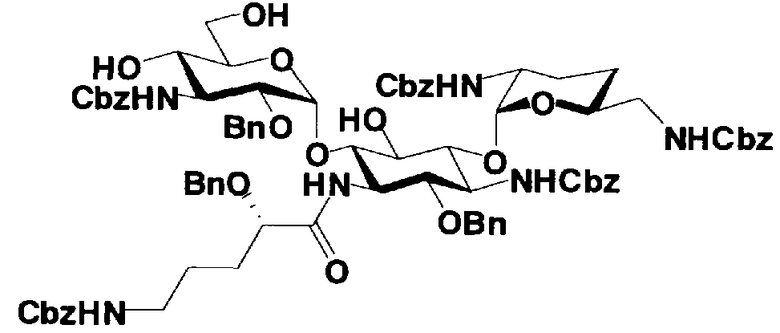
Соединение (350 мг), полученное на стадии получения 6-1, растворяют в 8,8 мл метиленхлорида. К раствору добавляют соединение (364 мг), полученное на стадии получения 4-3 примера 4, и 1000 мг порошка молекулярных сит 4Å, и полученную смесь перемешивают при комнатной температуре в течение одного часа. Реакционный раствор охлаждают до -20°C, к охлажденному раствору добавляют 350 мг N-йодсукцинимида и 8,1 мкл трифторметансульфоновой кислоты, и полученную смесь перемешивают, экранируя от света, при комнатной температуре в течение 5 часов. К этому добавляют триэтиламин (81 мкл) при охлаждении льдом и реакционную смесь фильтруют через целит. Нерастворимую часть промывают 20 мл хлороформа. Полученный таким образом раствор промывают один раз 15 мл насыщенного водного раствора бикарбоната натрия и дважды 15 мл 10% водного раствора тиосульфата натрия, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (25 г, хлороформ:этилацетат=4:1→хлороформ:метанол=20:1→10:1) с получением 467 мг сырого продукта, содержащего 4",6"-ди-О-ацетил-1-N-[(S)-5-амино-2-бензилокси-N-бензилоксикарбонилпентаноил]-2,2"-ди-О-бензил-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин в качестве ожидаемого продукта.
Rf значение: 0,51 (хлороформ:этилацетат=2:3): ожидаемый продукт.
ESIMS: m/z 1630 [M+Na]+: ожидаемый продукт.
Сырой продукт (467 мг) растворяют в 9,3 мл метанола, добавляют 20 мг боргидрида натрия при охлаждении льдом при комнатной температуре в течение 1,5 часа. К этому добавляют ацетон (1,0 мл) при охлаждении льдом, полученную смесь перемешивают при комнатной температуре в течение 15 минут и полученную смесь разбавляют 50 мл хлороформа. К этому добавляют воду (25 мл) с последующим разделением. Органический слой сушат над глауберовой солью и затем концентрируют досуха с получением 381 мг сырого продукта (4",6"-ди-О-ацетил-1-N-[(S)-5-амино-2-бензилокси-N-бензилоксикарбонилпентаноил]-2,2"-ди-О-бензил-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин).
Rf значение: 0,51 (хлороформ:этилацетат=2:3).
ESIMS: m/z 1630 [M+Na]+.
Сырой продукт (381 мг) растворяют в 7,6 мл хлороформа и 3,8 мл метанола. К раствору добавляют 28% раствор (13 мкл) метилата натрия в метаноле при охлаждении льдом, и реакционную смесь оставляют реагировать при комнатной температуре в течение 2 часов. К этому добавляют уксусную кислоту (0,1 мл) при охлаждении льдом и полученную смесь разбавляют 25 мл хлороформа. После этого разбавленный раствор промывают 15 мл насыщенного водного раствора бикарбоната натрия, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (30 г, хлороформ→хлороформ:метанол=99:1→97:3) с получением указанного в заголовке соединения 229 мг (выход 49%).
Rf значение: 0,22 (хлороформ:этилацетат=2:3).
ESIMS: m/z 1545 [M+Na]+.
1H-ЯМР (Пиридин-d5): δ 8,69 (шир., 1H), 8,38 (д, 1H, J=7,9 Гц), 7,65-7,72 (м, 2H), 7,60 (шир., 4H), 7,20-7,52 (м, 38H), 5,78 (шир., 1H), 5,72 (д, 1H, J=6,1 Гц), 5,20-5,39 (м, 13H), 5,09-5,11 (м, 1H), 5,05 (д, 1H, J=9,0 Гц), 5,01 (т, 1H, J=8,0 Гц), 4,89 (д, 1H, J=12,1 Гц), 4,68-4,80 (м, 6H), 4,48 (д, 1H, J=12,1 Гц), 4,15-4,34 (м, 5H), 4,06 (шир., 2H), 3,93 (шир., 1H), 3,88 (т, 1H, J=8,1 Гц), 3,52-3,59 (м, 1H), 3,31-3,37 (м, 1H), 1,87-2,20 (м, 5H), 1,78 (шир., 1H), 1,62 (шир., 2H).
Стадия получения 6-3
1-N-[(S)-5-амино-2-гидроксипентаноил]-2-гидроксидибекацин
[Химическая формула 85]
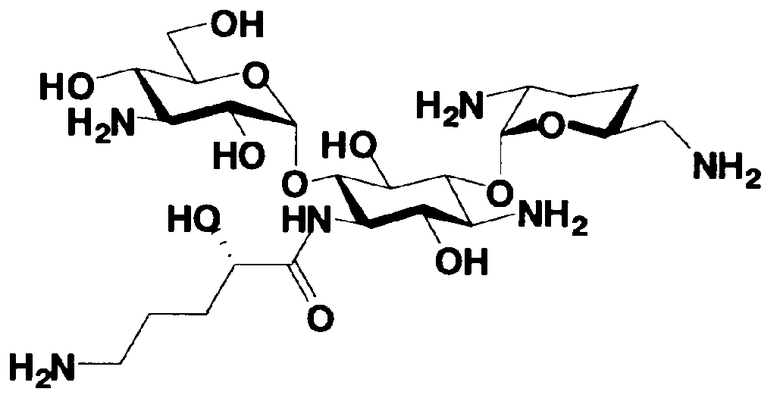
Указанное соединение (229 мг), полученное на стадии получения 6-2, растворяют в 9,2 мл смеси 1,4-диоксан:вода:1 н. хлористоводородная кислота (40:19:1), и добавляют шесть капель смеси палладиевая чернь/вода в качестве катализатора. Полученную смесь интенсивно перемешивают при комнатной температуре в атмосфере газообразного водорода в течение 14 часов. В этом случае катализатор заменяют один раз с периодичностью 14 часов. После того как реакция останавливается, реакционный раствор фильтруют через хлопок, добавляют 6 мл насыщенного водного раствора бикарбоната натрия с последующим разделением с использованием 9 мл этилацетата. Водный слой снова экстрагируют 9 мл этилацетата. Органические слои объединяют, сушат над глауберовой солью и концентрируют досуха. Полученный остаток (161 мг) растворяют в 8 мл смеси тетрагидрофуран:вода:уксусная кислота (4:1:1). В качестве катализатора добавляют смесь палладиевая чернь/вода (девять капель) и полученную смесь интенсивно перемешивают при комнатной температуре в атмосфере газа-водорода в течение 19 часов. В этом случае катализатор заменяют трижды с периодичностью 19 часов. Реакционный раствор фильтруют через хлопок и маточный раствор концентрируют при пониженном давлении. После этого снова добавляют воду и полученную смесь снова концентрируют при пониженном давлении. Полученный остаток растворяют в 5 мл воды. Полученный раствор нейтрализуют 0,1 M водным аммиаком и вводят в колонку с Amberiite CG-50 (уравновешенную 0,005 M водным аммиаком, 5 мл), и колонку промывают 0,005 M водным аммиаком (5 мл). Элюирование осуществляют, используя 0,10 M→0,25 M→0,50 M→0,75 M водный аммиак (каждый по 10 мл). Соответствующие фракции концентрируют досуха с получением 48 мг (41%) указанного в заголовке соединения (2,25 карбонат, 2,75 гидрат).
Rf значение: 0,20 (хлороформ:метанол:28% водный аммиак:этанол=4:6:7:2).
1H-ЯМР (26% ND3-D2O): δ 5,05 (д, 1H, J=3,3 Гц), 4,95 (д, 1H, J=3,7 Гц), 4,02 (дд, 1H, J=3,3, 8,0 Гц), 3,89 (дт, 1H, 2,0, 8,1), 3,81 (т, 1H, J=10,1 Гц), 3,75-3,79 (м, 1H), 3,61-3,70 (м, 4H), 3,29 (т, 1H, J=10,2 Гц), 3,24 (т, 1H, J=10,2 Гц), 3,21 (т, 1H, J=10,4 Гц), 3,17 (т, 1H, J=9,8 Гц), 2,88 (т, 1H, J=10,0 Гц), 2,78 (т, 1H, J=10,0 Гц), 2,75 (дт, 1H, J=1,7, 12,0 Гц), 2,51-2,59 (м, 4H), 1,70-1,79 (м, 1H), 1,59-1,66 (м, 2H), 1,42-1,57 (м, 4H), 1,25-1,32 (м, 1H).
13C-ЯМР (26% ND3-D2O): δ 178,16, 102,70, 99,16, 83,44, 78,36, 75,58, 72,92, 72,59, 72,26, 72,11, 71,48, 70,07, 61,08, 57,19, 56,63, 56,27, 54,97, 50,89, 45,99, 41,06, 32,03, 28,42, 26,94.
Рассчитано для C23H46N6О11·2,25 H2CО3·2,75 H2О: C, 39,30; H, 7,31; N, 10,89.
Найдено: C, 39,08; H, 7,14; N, 11,05.
Пример 7
1-N-[(S)-6-амино-2-гидроксигексаноил]-2-гидроксидибекацин
[Химическая формула 86]

Стадия получения 7-1
1-N-[(S)-6-амино-2-бензилокси-N-бензилоксикарбонилгексаноил]-2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезоксинеамин
[Химическая формула 87]
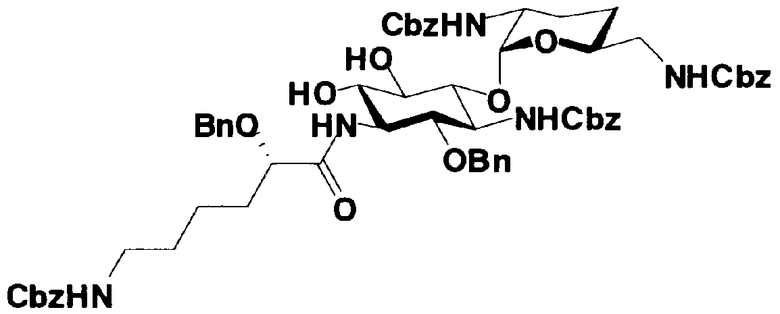
(S)-6-амино-2-бензилокси-N-бензилоксикарбонилгексановую кислоту (160 мг), полученную в сравнительном примере 4, и 344 мг 2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезокси-2-гидроксинеамин, полученного на стадии получения 3-6 примера 3, растворяют в 5,2 мл тетрагидрофурана. К раствору добавляют 2-пропанол (10,3 мл), 0,7 мл воды и 155 мг 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорида (DMT-MM) и полученную смесь перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор концентрируют при пониженном давлении. К остатку добавляют хлороформ (50 мл) и полученную смесь промывают один раз 25 мл насыщенного водного раствора бикарбоната натрия и трижды 20 мл воды, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (25 г, гексан:этилацетат=3:1) с получением указанного в заголовке соединения (355 мг, выход 72%).
Rf значение: 0,62 (хлороформ:метанол - 10:1).
ESIMS: m/z 1174 [M+Na]+ .
Стадия получения 7-2
1-N-[(S)-6-амино-2-бензилокси-N-бензилоксикарбонилгексаноил]-2,2"-О-бензил-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин
[Химическая формула 88]
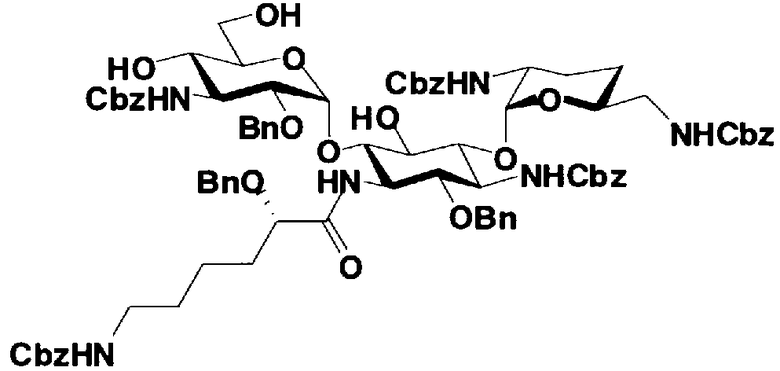
Соединение (350 мг), полученное на стадии получения 7-1, растворяют в 8,8 мл метиленхлорида. К раствору добавляют соединение (352 мг), полученное на стадии получения 4-3 примера 4, и 1000 мг порошка молекулярных сит 4Å, и полученную смесь перемешивают при комнатной температуре в течение одного часа. Реакционный раствор охлаждают до -20°C. К охлажденному раствору добавляют N-йодсукцинимид (341 мг) и 7,9 мкл трифторметансульфоновой кислоты. Полученную смесь перемешивают, экранируя от света, при комнатной температуре в течение 3 часов. К этому добавляют триэтиламин (79 мкл) при охлаждении льдом. Реакционную смесь фильтруют через целит и нерастворимую часть промывают 20 мл хлороформа. Полученный таким образом раствор промывают один раз 15 мл насыщенного водного раствора бикарбоната натрия и дважды 15 мл 10% водного раствора тиосульфата натрия, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (25 мг, хлороформ:этилацетат=4:1→хлороформ:метанол=20:1→10:1) с получением 433 мг сырого продукта содержащего 4",6"-О-ацетил-1-N-[(S)-6-амино-2-бензилокси-N-бензилоксикарбонилгексаноил]-2,2"-О-бензил-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин в качестве ожидаемого продукта.
Rf значение: 0,51 (хлороформ:этилацетат=2:3): ожидаемый продукт.
ESIMS: m/z 1643 [M+Na]+: ожидаемый продукт.
Сырой продукт (433 мг) растворяют в 8,7 мл метанола. К раствору добавляют боргидрид натрия (19 мг) при охлаждении льдом и полученную смесь перемешивают при комнатной температуре в течение 1,5 часа. К смеси добавляют ацетон (1,0 мл) при охлаждении льдом и полученную смесь перемешивают при комнатной температуре в течение 15 минут. После этого реакционную смесь разбавляют 50 мл хлороформа. Добавляют воду (25 мл) с последующим разделением. Органический слой сушат над глауберовой солью и затем концентрируют досуха, получая 357 мг сырого продукта (4",6"-О-ацетил-1-N-[(S)-6-амино-2-бензилокси-N-бензилоксикаpбонилгексаноил]-2,2"-О-бензил-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин).
ESIMS: m/z 1643 [M+Na]+.
Сырой продукт (357 мг) растворяют в 7,2 мл хлороформа и 3,6 мл метанола. К раствору добавляют 28% раствор смеси метилат натрия-метанол (13 мкл) при охлаждении льдом и полученную смесь перемешивают при комнатной температуре в течение 2 часов. К этому добавляют уксусную кислоту (0,5 мл) при охлаждении льдом и полученную смесь разбавляют 25 мл хлороформа. Разбавленный раствор промывают 15 мл насыщенного водного раствора бикарбоната натрия, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (28 г, хлороформ→хлороформ:метанол=99:1→97:3) с получением указанного в заголовке соединения (207 мг, выход 44%).
Rf значение: 0,22 (хлороформ:этилацетат=2:3).
ESIMS: m/z 1560 [M+Na]+.
1H-ЯМР (Пиридин-d5): δ 8,39 (д, 1H, J=7,9 Гц), 7,76 (д, 1H, J=8,9 Гц), 7,63 (шир., 2H), 7,21-7,55 (м, 42H), 5,81 (шир., 1H), 5,74 (шир., 1H), 5,40 (шир., 1H), 5,33-5,38 (м, 3H), 5,36 (т, 1H, J=6,4 Гц), 5,21-5,27 (м, 3H), 5,19 (д, 1H, J=11,9 Гц), 4,79-5,08 (м, 8H), 4,75 (д, 1H, J=11,9 Гц), 4,69 (д, 1H, J=11,9 Гц), 4,22-4,51 (м, 8H), 3,95-4,13 (м, 4H), 3,50-3,61 (м, 2H), 3,27 (шир., 2H), 2,02 (шир., 2H), 1,94 (шир., 2H), 1,42-1,64 (м, 6H).
Стадия получения 7-3
1-N-[(S)-6-амино-2-гидроксигексаноил]-2-гидроксидибекацин
[Химическая формула 89]

Соединение (180 мг), полученное на стадии получения 7-2, растворяют в 9,0 мл смеси тетрагидрофуран:вода:уксусная кислота (4:1:1). К раствору добавляют смесь палладиевая чернь/вода (девять капель) в качестве катализатора и полученную смесь интенсивно перемешивают в атмосфере газообразного водорода при комнатной температуре в течение 33 часов. В указанном случае катализатор заменяют шесть раз с периодичностью 33 часа. Реакционный раствор фильтруют через хлопок и маточный раствор концентрируют при пониженном давлении. После этого к остатку снова добавляют воду и полученный раствор снова концентрируют при пониженном давлении. Полученный остаток растворяют в 5 мл воды. Полученный раствор нейтрализуют, используя 0,1 M водный аммиак, и вводят в колонку с Amberlite CG-50 (уравновешенную 0,005 M водным аммиаком, 5 мл), и колонку промывают 0,005 M водным аммиаком (5 мл). Элюирование осуществляют, используя 0,10 M →0,25 M→0,50 M→0,75 M водный аммиак (каждый по 10 мл), и соответствующие фракции концентрируют досуха с получением указанного в заголовке соединения (1,5 тригидрат карбоната, 52 мг, выход 60%).
1H-ЯМР (26% ND3-D2O): δ 5,16 (д, 1H, J=3,2 Гц), 5,05 (д, 1H, J=3,3 Гц), 4,05 (дд, 1H, J=3,5, 8,8 Гц), 4,00 (д, 1H, 9,8), 3,90 (т, 1H, J=9,8 Гц), 3,82-3,87 (м, 1H), 3,69-3,79 (м, 4H), 3,40 (т, 1H, J=9,2 Гц), 3,29-3,37 (м, 2H), 3,27 (т, 1H, J=9,8 Гц), 2,98 (т, 1H, J=10,0 Гц), 2,88 (т, 1H, J=9,8 Гц), 2,85 (дт, 1H, J=4,0, 12,1 Гц), 2,61-2,70 (м, 4H), 1,71-1,83 (м, 3H), 1,59-1,69 (м, 2H), 1,33-1,56 (м, 5H).
13C-ЯМР (26% ND3-D2O): δ 178,24, 102,02, 99,07, 83,37, 78,30, 76,65, 75,43, 73,62, 72,90, 72,39, 72,07, 71,41, 70,04, 61,06, 56,65, 56,16, 54,96, 50,76, 45,95, 41,06, 34,20, 28,40, 26,98, 23,09.
Рассчитано для C24Н48N6O11·1,5 H2CO3·3H2O: C, 41,18; H, 7,72; N, 11,30. Найдено: C, 41,20; H, 7,73; N, 11,43.
Пример 8
1-N-[(R)-4-амино-2-гидроксибутирил]-2-гидроксидибекацин
[Химическая формула 90]
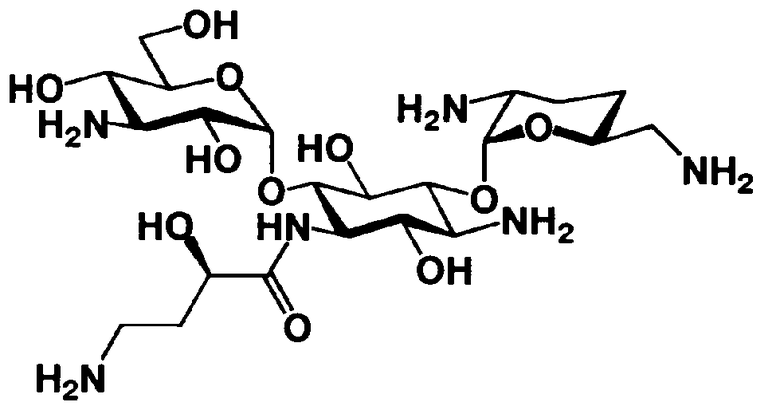
Стадия получения 8-1
2-О-бензил-1-N-[2-(R)-О-бензил-4-бензилоксикарбониламинобутирил]-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезокси-2-гидроксинеамин
[Химическая формула 91]
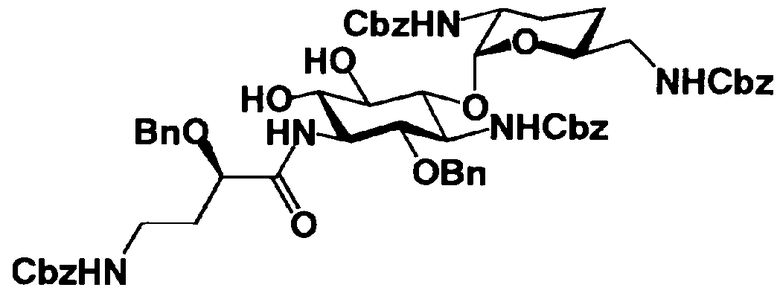
2-бензилокси-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезокси-2-гидроксинеамин (420 мг), полученный на стадии получения 3-6 примера 3, и 216 мг (R)-2-бензилокси-4-бензилоксикарбониламиномасляной кислоты, полученной в сравнительном примере 5, растворяют в 18 мл растворителя (2-пропанол:тетрагидрофуран:вода=30:10:3). К раствору добавляют 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолинийхлорид (здесь и далее обозначаемый как DMT-MM) (218 мг) и полученную смесь перемешивают при комнатной температуре. DMT-MM (146 мг) добавляют через 2 часа после начала перемешивания, полученную смесь перемешивают и через 3,5 часа реакционную смесь концентрируют досуха. К остатку добавляют хлороформ (50 мл) и полученный раствор промывают один раз 25 мл насыщенного водного раствора бикарбоната натрия и дважды водой. Промытый раствор сушат над сульфатом магния и концентрируют досуха, получая 686 мг сырого продукта. Сырой продукт очищают на хроматографической колонке с силикагелем (50 г, хлороформ→хлороформ:метанол=99:1→97:3→95:5→10:1) с получением указанного в заголовке соединения (528 мг, 85%).
Rf значение: 0,71 (хлороформ:метанол=10:1).
ESIMS: m/z 1146 [M+Na]+.
1H-ЯМР (Пиридин-d5 при 80°C): δ 8,04 (д, 1H, J=7 Гц), 7,55 (шир.д, 1H, J=6 Гц), 7,12-7,50 (31H), 7,00 (шир.с, 1H), 6,82 (шир.с, 1H), 5,64 (д, 1H, J=3 Гц), 5,29 (с, 2H), 5,10-5,27 (4H), 4,95 (с, 2H), 4,50-4,80 (ABкв, 2H, Jgem=11,5 Гц), 4,36 (т, 1H, J=8, 8 Гц), 4,28 (м, 1H), 4,20-4,25 (3H), 4,16 (т, 1H, J=9, 9 Гц), 4,08 (т, 1H, J=9, 9 Гц), 3,98 (м, 1H), 3,91 (т, 1H, J=11, 11 Гц), 3,37-3,60 (м, 4H), 2,12-2,30 (м, 2H), 1,85-2,00 (м, 2H), 1,43-1,68 (м, 2H).
Стадия получения 8-2
2,2"-ди-О-бензил-1-N-[2-(R)-О-бензил-4-бензилоксикарбониламинобутирил]-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин
[Химическая формула 92]
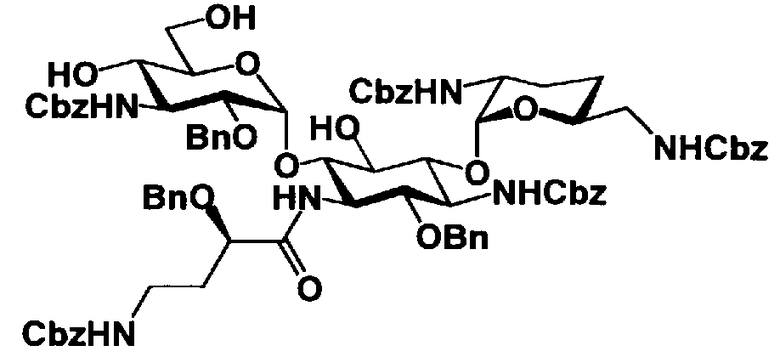
Соединение (497 мг), полученное на стадии получения 4-3 примера 4, и 482 мг соединения, полученного на стадии получения 8-1, растворяют в 20 мл метиленхлорида. К раствору добавляют порошок молекулярных сит 4Å (1,445 г) и полученную смесь перемешивают в атмосфере аргона при комнатной температуре в течение 2 часов. К перемешиваемому раствору добавляют N-йодсукцинимид (482 мг) и 7,6 мкл трифторметансульфоновой кислоты при -20°C при перемешивании. Реакционную смесь возвращают к комнатной температуре и перемешивают, экранируя от света в течение 2 часов. Соединение (248 мг), полученное на стадии получения 4-3 примера 4, 241 мг N-йодсукцинимида и 3,8 мкл трифторметансульфоновой кислоты, добавляют к реакционному раствору через 3 часа после начала перемешивания, и через 4 часа к этому добавляют 27 мкл триэтиламина при охлаждении льдом. Полученную смесь фильтруют через целит, удаляя нерастворимую часть. Нерастворимую часть промывают 40 мл хлороформа. Объединенные органические слои промывают один раз 50 мл насыщенного водного раствора бикарбоната натрия, трижды 50 мл 10% водного раствора тиосульфата натрия и дважды 50 мл воды, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку на силикагеле (130 г, хлороформ:ацетон=10:1→7:1→3:1) с получением 530 мг сырого продукта, содержащего 4",6"-ди-О-ацетил-2,2"-ди-О-бензил-1-N-[2-(R)-О-бензил-4-бензилоксикарбониламинобутирил]-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацин в качестве ожидаемого продукта.
Rf значение: 0,42 (хлороформ:этилацетат=2:3): ожидаемый продукт.
ESIMS: m/z 1615 [M+Na]+: ожидаемый продукт.
Сырой продукт (527 мг) растворяют в 10,5 мл метанола. К раствору добавляют боргидрид натрия (24,2 мг) при перемешивании и охлаждении льдом и полученную смесь перемешивают при комнатной температуре в течение 1 часа. К этому добавляют ацетон (0,38 мл) при охлаждении льдом и полученную смесь перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь концентрируют при пониженном давлении. Остаток растворяют в 50 мл хлороформа, и полученный раствор трижды промывают 25 мл воды, сушат над сульфатом магния и концентрируют досуха с получением 524 мг сырого продукта (4",6"-ди-О-ацетил-2,2"-ди-О-бензил-1-N-[2-(R)-О-бензил-4-бензилоксикарбониламинобутирил]-3,2',6',3"-тетра-N-бензилоксикарбонил-2-гидроксидибекацина).
Rf значение: 0,42 (хлороформ:этилацетат=2:3).
ESIMS: m/z 1615 [M+Na]+.
Хлороформ (10,5 мл), 5 мл метанола и раствор 28% метилат натрия - метанол (21,2 мг)/метанол (0,24 мл) добавляют к сырому продукту, и полученную смесь перемешивают при комнатной температуре. К этому добавляют уксусную кислоту (6,3 мкл) через 2,5 часа после начала перемешивания и полученную смесь промывают 30 мл насыщенного водного раствора бикарбоната натрия. Водный слой снова экстрагируют 30 мл хлороформа. Объединенные органические слои сушат над сульфатом магния и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (50 г, хлороформ:метанол=30:1) с получением указанного в заголовке соединения (337,5 мг, выход 50%).
Rf значение: 0,56 (хлороформ:метанол=10:1)
ESIMS: m/z 1531 [M+Na]+
1H-ЯМР (Пиридин-d5 при 80ºС): δ 8,01 (д, 1H, J=8 Гц), 7,57 (шир.с, 1H), 7,55 (шир.с, 1H), 7,31-7,49 (м, 16H), 7,13-7,31 (м, 24H), 6,98 (шир.с, 1H), 6,86 (шир.с, 1H), 6,80 (шир.с, 1H), 5,68 (д, 1H, J=3 Гц), 5,54 (д, 1H, J=2,5 Гц), 5,12-5,32 (м, 10H), 4,90 (с, 2H), 4,71-4,95 (ABкв, 2H, Jgem=12 Гц), 4,57-4,70 (ABкв, 2H, Jgem=12 Гц), 4,58 (т, 1H, J=10, 10 Гц), 4,47 (т, 1H, J=9, 9 Гц), 4,43-4,59 (м, 2H), 4,31 (м, 1H), 4,27 (дд, 1H, J=6, 7 Гц), 4,24-4,37 (м, 2H), 3,87-4,14 (м, 6H), 3,54 (м, 2H), 3,40 (м, 2H), 2,30 (м, 1H), 2,24 (м, 1H), 1,84-1,96 (м, 2H), 1,44-1,63 (м, 2H).
Стадия получения 8-3
1-N-[(R)-4-амино-2-гидроксибутирил]-2-гидроксидибекацин
[Химическая формула 93]
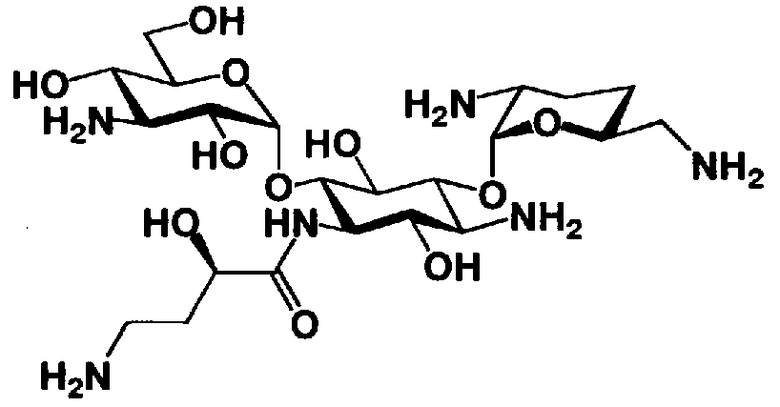
Соединение (310 мг), полученное на стадии получения 8-2, растворяют в 21,6 мл смеси тетрагидрофуран-уксусная кислота-вода (4:1:1). К раствору добавляют палладиевую чернь (20 капель), суспендированную в воде, и полученную смесь перемешивают при комнатной температуре в течение 6,5 часа, барботируя при этом в систему водород. Катализатор удаляют с помощью фильтрования, к этому добавляют палладиевую чернь (20 капель), свежесуспендированную в воде, и полученную смесь перемешивают при комнатной температуре в течение 12 часов, барботируя при этом в систему водород. Катализатор удаляют фильтрованием, к этому добавляют палладиевую чернь (десять капель), свежесуспендированную в воде, и полученную смесь перемешивают при комнатной температуре в течение 14,5 часа, барботируя при этом в систему водород. Катализатор удаляют с помощью фильтрования и промывают водой. Полученный фильтрат и промывки объединяют и концентрируют досуха. Полученный остаток растворяют в 30 мл воды. Полученный раствор вводят в колонку Amberlite CG-50 (уравновешенную 0,005 M водным аммиаком, 15 мл), и колонку промывают 30 мл 0,005 M водного аммиака. Элюирование осуществляют, используя 0,1 M (30 мл)→0,25 M (30 мл)→0,5 M (30 мл)→0,75 M (30 мл)→1,0 M водный аммиак (60 мл), и соответствующие фракции концентрируют досуха с получением указанного в заголовке соединения (112 мг, 0,5 дигидрат карбоната, 86%).
Rf значение: 0,13 (хлороформ:метанол:15 M водный аммиак (концентрированный водный аммиак):этанол=4:6:7:2).
1H-ЯМР (26% ND3-D2O): δ 5,16 (д, 1H, J=3,5 Гц), 5,01 (д, 1H, J=4 Гц), 4,21 (дд, 1H, J=3,5, 9 Гц), 3,98 (м, 1H), 3,93 (т, 1H, J=10,5, 10,5 Гц), 3,87 (м, 1H), 3,75 (т, 1H, J=10, 10 Гц), 3,72-3,80 (м, 2H), 3,69 (т, 1H, J=10, 10 Гц), 3,40 (т, 1H, J=10, 10 Гц), 3,35 (т, 1H, J=10, 10 Гц), 3,32 (дд, 1H, J=4, 10,5 Гц), 3, 28 (т, 1H, J=10, 10 Гц), 2,96 (т, 1H, J=10, 10 Гц), 2,88 (т, 1H, J=10, 10 Гц), 2,84 (дт, 1H, J=4, 4, 13 Гц), 2,76 (м, 2H), 2,66 (дд, 1H, J=5, 13,5 Гц), 2,64 (дд, 1H, J=7,5, 13,5 Гц), 1,92 (м, 1H), 1,70-1,84 (м, 3H), 1,64 (дкв, 1H, J=3,5, 13, 13, 13 Гц), 1,39 (м, 1H).
13C-ЯМР (26% ND3-D2O): δ 178,39, 102,05, 99,61, 83,52, 79,21, 75,49, 72,90, 72,38, 71,72, 71,47, 70,78, 70,02, 60,97, 56,33, 56,27, 54,97, 50,78, 45,98, 37,96, 37,08, 28,41, 26,94.
Рассчитано для C22H44N6O11• 0,5 H2CO3•2H2O. C, 42,51; H, 7,77; N, 13,22. Найдено: C, 42,51; H, 7,74; N, 13,26.
Пример 9
Получение 4"-эпи-2-гидроксиарбекацина
[Химическая формула 94]
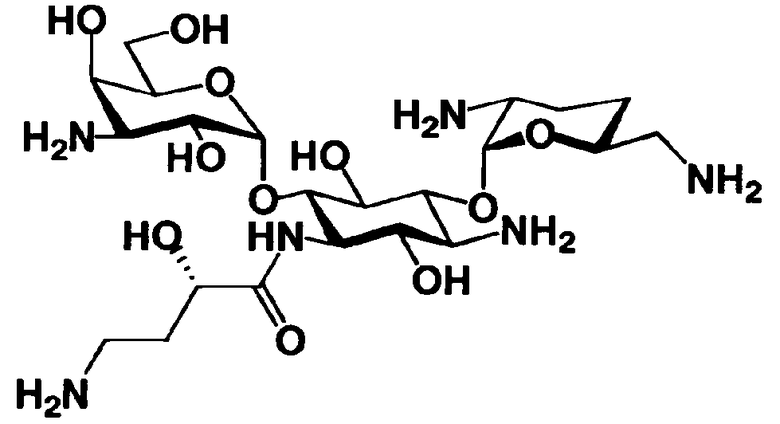
Стадия получения 9-1
3"-азидо-3,2',6',4"'-тетра-N-бензилоксикарбонил-2,2",2"'-три-О-бензил-3"-дезокси-4"-эпи-2-гидроксиарбекацин
[Химическая формула 95]

2-О-бензил-1-N-[2-(S)-О-бензил-4-бензилоксикарбониламинобутирил]-3,2',6'-три-N-бензилоксикарбонил-3',4'-дидезокси-2-гидроксинеамин (39 мг), полученный на стадии получения 3-6 примера 3, растворяют в 1,2 мл метиленхлорида. К раствору добавляют соединение (50,3 мг), полученное на стадии получения 10-5b примера 10, и 117 мг порошка молекулярных сит 4Å, и полученную смесь перемешивают при комнатной температуре в течение 30 минут. К этому добавляют N-йодсукцинимид (39 мг) и 0,91 мкл трифторметансульфоновой кислоты при -20°C при перемешивании, и полученную смесь перемешивают, экранируя от света, при -20°C в течение 3 часов. К этому добавляют триэтиламин (1,9 мкл) при -20°C, и реакционную смесь фильтруют через целит. Нерастворимую часть промывают 10 мл хлороформа. Объединенные органические слои промывают один раз 3 мл насыщенного водного раствора бикарбоната натрия и дважды 3 мл 10% водного раствора тиосульфата натрия, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (4 г, хлороформ:этилацетат=30:1→20:1, хлороформ:метанол=20:1→10:1) с получением 41 мг сырого продукта.
Rf значение: 0,65 (хлороформ:этилацетат=2:3).
Сырой продукт (41 мг) растворяют в 1,2 мл смешанной жидкости, состоящей из метиленхлорида и 0,1% раствора метилата натрия, в метаноле (метиленхлорид:раствор=2:1) при охлаждении льдом, и полученный раствор возвращают к комнатной температуре, и реакции дают протекать в течение 2 часов. К реакционной смеси добавляют 50% водный раствор уксусной кислоты (0,8 мл) при охлаждении льдом. Полученную смесь разбавляют 10 мл хлороформа и разбавленный раствор промывают 3 мл насыщенного водного раствора бикарбоната натрия, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают на хроматографической колонке с силикагелем (4 г, хлороформ→хлороформ:метанол=99:1→97:3) с получением указанного в заголовке соединения (24,1 мг, выход 51%).
Rf значение: 0,24 (хлороформ:этилацетат=2:3).
ESIMS: m/z 1423 [M+Na]+.
1H-ЯМР (Пиридин-d5): δ 8,79 (д, 1Н, J=7 Гц), 8,43 (д, 1H, J=9 Гц), 7,90 (т, 1H, J=6, 6 Гц), 7,76 (м, 1H), 7,18-7,71 (м, 35H), 7,04 (шир.с, 1H), 6,59 (шир.с, 1H), 6,07 (д, 1H, J=3,5 Гц), 5,74 (д, 1H, J=3 Гц), 5,06-5,44 (ABкв, 2H, Jgem=12 Гц), 5,24-5,41 (м, 4H), 5,04 (шир.с, 1H), 4,70-4,80 (м, 3H), 4,57 (дд, 1H, J=4, 10 Гц), 4,36-4,51 (м, 4H), 4,18-4,35 (м, 2H), 4,00-4,15 (м, 2H), 3,68 (м, 1H), 3,60 (м, 2H), 3,51 (м, 1H), 2,48 (м, 1H), 2,34 (м, 1H), 1,95 (м, 2H), 1,61 (м, 2H).
Стадия получения 9-2
4"-эпи-2-гидроксиарбекацин
[Химическая формула 96]
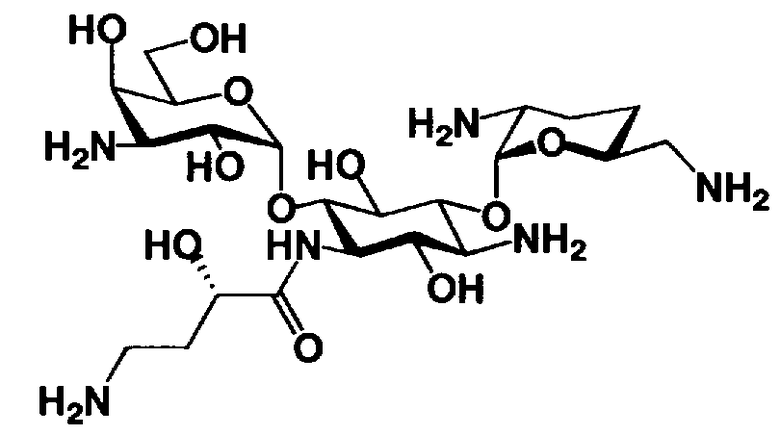
Жидкий аммиак (около 5 мл) вводят при -50°C в реактор формы типа баклажана, содержащем 24 мг соединения, полученного на стадии получения 9-1. В реактор добавляют металлический натрий (31 мг) при -50°C и полученную смесь интенсивно перемешивают стеклянной палочкой-мешалкой в течение 2 часов. Постепенно добавляют твердый аммонийхлорид до тех пор, пока окраска радикалов не исчезает. Температуру системы возвращают к комнатной температуре для испарения аммиака. И, наконец, содержимое колбы концентрируют досуха, используя испаритель. К остатку добавляют воду (2,4 мл) и pH полученного раствора доводят до рН 7, добавляя 1 M водный аммиак. Нейтрализованный раствор вводят в колонку с Amberlite CG-50 (уравновешенную 0,005 M водным аммиаком, 3 мл), и колонку промывают 0,005 M водным аммиаком (6 мл). Элюирование осуществляют, используя 0,1 M→0,2 M→0,3 M→0,5 M→0,8 M водный аммиак (каждого по 6 мл). Соответствующие фракции концентрируют досуха с получением 7,3 мг указанного в заголовке соединения (выход 53%, 2,5 карбонат 4,5 гидрат).
Rf значение: 0,16 (хлороформ:метанол:15 M водный аммиак (концентрированный водный аммиак):этанол=4:6:7:2)
1H-ЯМР (26% ND3-D2O): δ 5,08 (шир.с, 1H), 5,00 (шир.с, 1H), 4,13 (м, 2H), 3,75-3,90 (м, 3H), 3,50-3,75 (м, 4H), 3,47 (шир.д, 1H, J=10 Гц), 3,20-3,40 (м, 2H), 2,72-2,86 (м, 3H), 2,68 (м, 2H), 2,58 (шир.с, 2H), 1,83 (м, 1H), 1,48-1,75 (м, 3H), 1,31 (м, 1H).
Рассчитано для C22H44N6O11·2,5 H2CO3·4,5 H2O. C 36,57; H, 7,26; N, 10,44. Найдено: C, 36,65; H, 7,01; N, 10,57.
Пример 10
5-эпи-2-гидроксиарбекацин
[Химическая формула 97]
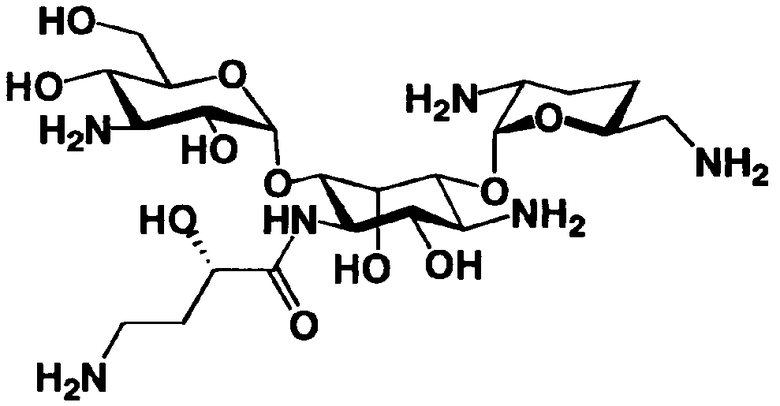
Стадия получения 10-1
Метил 3-азидо-6-О-бензоил-2-О-бензил-3-дезокси-D-глюкопиранозид
[Химическая формула 98]
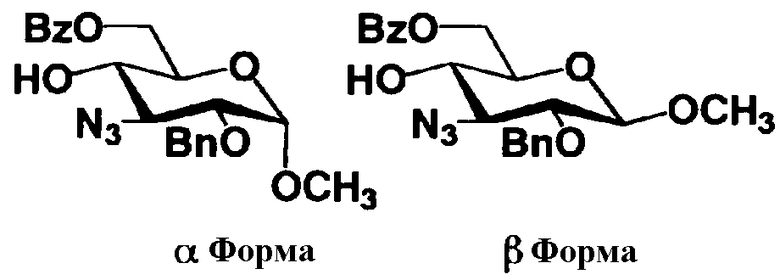
Метил 3-азидо-2-О-бензил-3-дезокси-D-глюкопиранозид (93 мг) растворяют в 0,84 мл пиридина. К раствору добавляют бензоилхлорид (45 мкл) при -20°C при перемешивании, и полученную смесь перемешивают при -20°C в течение одного часа. К раствору добавляют воду (14 мкл), и полученную смесь перемешивают при -20°C в течение 30 минут. Содержимое концентрируют досуха. Добавляют хлороформ (10 мл), и полученную смесь трижды промывают 5 мл насыщенного водного раствора бикарбоната натрия, трижды 5 мл 5% водного раствора бисульфата калия и трижды 5 мл воды, сушат над глауберовой солью и концентрируют при пониженном давлении. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (25 г, гексан:этилацетат=3:1), с получением указанного в заголовке соединения (97 мг, выход 78%).
Rf значение: 0,72 (хлороформ:метанол=20:1).
α Форма
1H-ЯМР (CDCl3): δ 7,30-8,10 (м, 10H), 4,62-4,79 (ABкв, 2H, Jgem=12 Гц), 4,73 (дд, 1H, J=4, 12 Гц), 4,60 (д, 1H, J=4 Гц), 4,45 (дд, 1H, J=3, 12 Гц), 3,87 (ддд, 1H, J=3, 4, 10 Гц), 3,85 (т, 1H, J=10, 10 Гц), 3,39 (с, 3H), 3,38 (дд, 1H, J=4, 10 Гц), 3,31 (дт, 1H, J=4, 10 Гц), 2,95 (д, J=4 Гц).
β Форма
1H-ЯМР (CDCl3): δ 7,30-8,10 (м, 10H), 4,70-4,91 (ABкв, 2H, Jgem=12 Гц), 4,74 (дд, 1H, J=4, 12 Гц), 4,53 (дд, 1H, J=4, 12 Гц), 4,38 (д, 1H, J=8 Гц), 3,59 (с, 3H), 3,57 (дт, 1H, J=4, 4, 12 Гц), 3,50 (т, 1H, J=10, 10 Гц), 3,37 (дт, 1H, J=4, 10 Гц), 3,26 (дд, 1H, J=8, 10 Гц).
Стадия получения 10-2
Метил 3-азидо-6-О-бензоил-2-О-бензил-3-дезокси-4-О-трифторметансульфонил-D-глюкопиранозид
[Химическая формула 99]
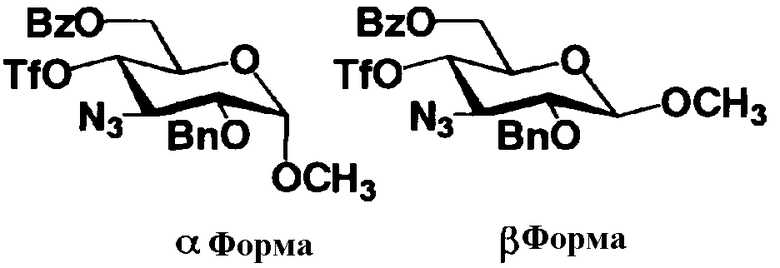
Cоединение (3,30 г), полученное на стадии получения 10-1, растворяют в 40 мл метиленхлорида. К раствору добавляют пиридин (4,8 мл), затем к этому добавляют 3,4 мл безводной трифторметансульфоновой кислоты при -20°C при перемешивании и полученную смесь перемешивают при -20°C в течение 1 часа. Добавляют метанол (0,81 мл) и полученную смесь перемешивают при -20°C в течение 20 минут. К смеси добавляют хлороформ (300 мл) и полученную смесь трижды промывают 170 мл охлажденного льдом насыщенного водного раствора бикарбоната натрия, трижды 170 мл 5% водного раствора бисульфата калия и трижды 170 мл охлажденного льдом полунасыщенного солевого раствора. Промытый раствор сушат над глауберовой солью при охлаждении льдом и концентрируют досуха при охлаждении льдом, получая указанное в заголовке соединение (3,60 г, 99%). Полученное соединение нестабильно и поэтому его сразу используют в следующей реакции.
Rf значение: 0,45 (гексан:этилацетат=3:1)
Стадия получения 10-3
Метил 3-азидо-4-O-ацетил-6-О-бензоил-2-О-бензил-3-дезокси-D-галактопиранозид
[Химическая формула 100]
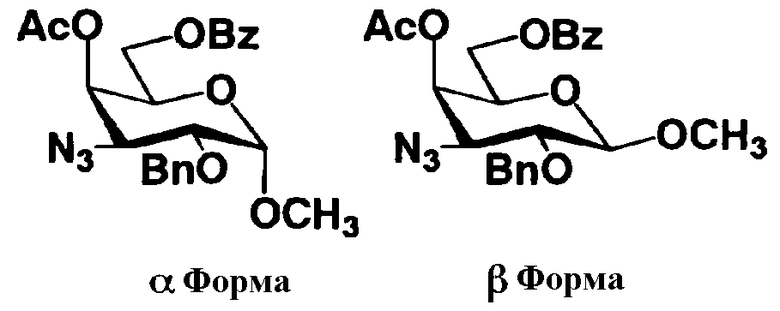
Соединение (3,60 г), полученное на стадии получения 10-2, растворяют в 33 мл N,N-диметилформамида. К раствору добавляют ацетат цезия (7,66 г) и полученную смесь оставляют реагировать при комнатной температуре в течение 1 часа. К этому добавляют этилацетат (330 мл) и полученную смесь дважды промывают 180 мл воды, дважды 180 мл насыщенного водного раствора бикарбоната натрия и дважды полунасыщенным солевым раствором, сушат над глауберовой солью и концентрируют досуха с получением указанного в заголовке соединения (3,72 г, выход 99%).
Rf значение: 0,40 (гексан:этилацетат=3:1).
α Форма
1H-ЯМР (CDCl3): δ 7,20-8,10 (м, 10H), 5,47 (д, 1H, J=4 Гц), 4,72-4,95 (ABкв, 2H, Jgem=12 Гц), 4,25 (дд, 1H, J=7, 11 Гц), 4,69 (д, 1H, J=4 Гц), 4,38 (ддд, 1H), 4,21 (дд, 1H, J=3, 7 Гц), 3,82 (дд, 1H, J=4, 11 Гц), 4,01 (дд, 1H, J=5, 11 Гц), 3,65 (с, 3H), 2,38 (с, 3H).
β Форма
1H-ЯМР (CDCl3): δ 7,20-8,10 (м, 10H), 5,45 (д, 1H, J=4 Гц), 4,62-4,82 (ABкв, 2H, Jgem=12 Гц), 4,50 (дд, 1H, J=7, 12 Гц), 4,39 (д, 1H, J=8 Гц), 4,21 (ддд, 1H), 3,94 (т, 1H, J=7, 7 Гц), 3,64 (дд, 1H, J=8, 10 Гц), 3,61 (дд, 1H, J=4, 10 Гц), 3,39 (с, 3H), 2,16 (с, 3H).
Стадия получения 10-4
3-азидо-1,4-ди-О-ацетил-6-О-бензоил-2-О-бензил-3-дезокси-D-галактопираноза
[Химическая формула 101]
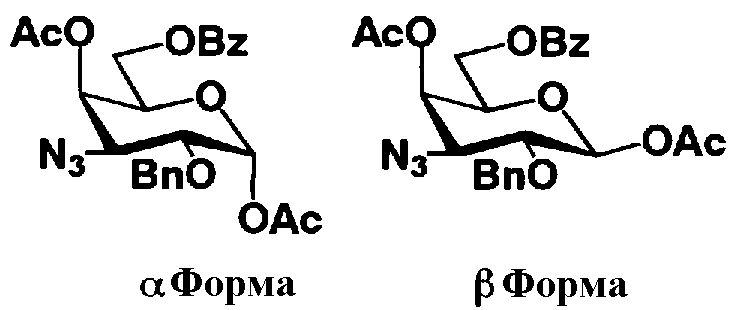
Соединение (3,72 г), полученное на стадии получения 10-3, растворяют в 74 мл смеси уксусная кислота:уксусный ангидрид:серная кислота (25:25:1), и полученный раствор перемешивают при комнатной температуре в течение 3 часов. К этому добавляют хлороформ (740 мл), и полученную смесь трижды промывают 350 мл насыщенного водного раствора бикарбоната натрия и трижды 350 мл воды, сушат над глауберовой солью и концентрируют досуха с получением указанного в заголовке соединения (3,66 г, выход 93%).
Rf значение: 0,32 (гексан:этилацетат=3:1).
α Форма
1H-ЯМР (CDCl3): δ 7,30-8,10 (м, 10Н), 6,46 (д, 1H, J=4 Гц), 5,55 (дд, 1H), 4,64-4,73 (ABкв, 2H, Jgem=12 Гц), 4,40 (дд, 1H, J=7, 11 Гц), 4,35 (м, 1H), 4,19 (дд, 1H, J=6, 11 Гц), 3,96 (м, 2H), 2,16 (с, 3H), 2,14 (с, 3H).
β Форма
1H-ЯМР (CDCl3): δ 7,30-8,10 (м, 10H), 5,69 (д, J=8 Гц), 5,51 (дд, 1H), 4,74-4,86 (ABкв, 2H, Jgem=12 Гц), 4,42 (м, 1H, J=7, 11 Гц), 4,25 (дд, 1H, J=7, 11 Гц), 4,11 (м, 1H), 3,79 (дд, 1H, J=8, 10 Гц), 3,72 (дд, 1H, J=3, 10 Гц), 2,19 (с, 3H), 2,09 (с, 3H).
Стадия получения 10-5a
3-азидо-4-О-ацетил-6-O-бензоил-2-О-бензил-1-бромо-3-дезокси- α-D-галактопираноза
[Химическая формула 102]
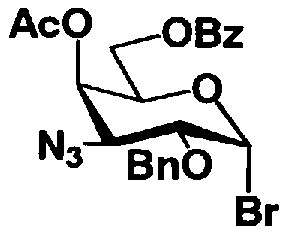
Соединение (25 мг), полученное на стадии получения 10-4, растворяют в 0,5 мл растворителя (метиленхлорид:этилацетат=9:1). Добавляют тетрабромид титана (29 мг) при охлаждении льдом при перемешивании. Полученную смесь возвращают к комнатной температуре и перемешивают в течение 1 часа. Реакционный раствор охлаждают льдом. К охлажденному раствору добавляют охлажденный льдом метиленхлорид (2 мл), и полученную смесь промывают шесть раз 2 мл охлажденной льдом воды, сушат над глауберовой солью и концентрируют досуха с получением указанного в заголовке соединения (27 мг, выход 83%). Полученное соединение нестабильно и его немедленно используют в следующей реакции.
Rf значение: 0,82 (гексан:этилацетат=3:2).
1H-ЯМР (CDCl3): δ 7,15-8,10 (т, 10Н), 6,49 (д, 1Н, J=4 Гц), 5,54 (дд, 1Н, J=2, 4 Гц), 4,45 (дд, 1H, J=7, 11 Гц), 4,39-4,43 (ABкв, 2H, Jgem=7 Гц), 4,33 (ддд, 1H), 4,27 (дд, 1H, J=6, 11 Гц), 4,06 (дд, 1H, J=4, 11 Гц), 3,74 (дд, 1H, J=4, 11 Гц), 2,16 (с, 3H).
Стадия получения 10-5b
3-азидо-4-О-ацетил-6-О-бензоил-2-О-бензил-1,3-дидезокси-1-тиофенил-α-D-галактопираноза
[Химическая формула 103]
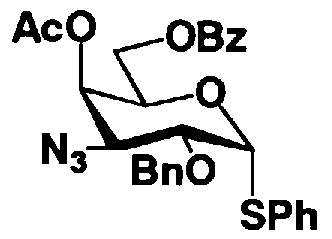
Соединение (300 мг), полученное на стадии получения 10-4, растворяют в 5 мл метиленхлорида. К раствору добавляют фенилтиотриметилсилан (353 мкл) и 135 мкл триметилсилилтрифторметансульфоната и полученную смесь кипятят с обратным холодильником при перемешивании. Реакционный раствор охлаждают льдом 6 часов после начала кипения с обратным холодильником. К охлажденному раствору добавляют охлажденный льдом метиленхлорид (24 мл) и полученную смесь дважды промывают 10 мл охлажденного льдом 5% водного раствора гидроксида натрия и дважды 10 мл охлажденной льдом воды, сушат над глауберовой солью и концентрируют досуха, с последующей перекристаллизацией из смеси этилацетат/гексан, получая указанное в заголовке соединение (204 мг, выход 62%).
Rf значение: 0,33 (гексан:этилацетат=4:1).
1H-ЯМР (CDCl3): δ 7,0-8,0 (м, 15H), 5,76 (д, 1H, J=5,5 Гц), 5,52 (д, 1H, J=3 Гц), 4,82 (шир.т, 1H, J=6,5, 6,5 Гц), 4,69-4,80 (ABкв, 2H, J=11 Гц), 4,36 (дд, 1H, J=7,5, 11,5 Гц), 4,25 (дд, 1H, J=5, 11,5 Гц), 4,17 (дд, 1H, J=5,5, 10,5 Гц), 3,92 (дд, 1H, J=3, 10,5 Гц), 2,16 (с, 3H).
Стадия получения 10-6
1,3,2',6',3"-пента-N-трет-бутоксикарбонил-2-гидроксигентамицин C1a
[Химическая формула 104]
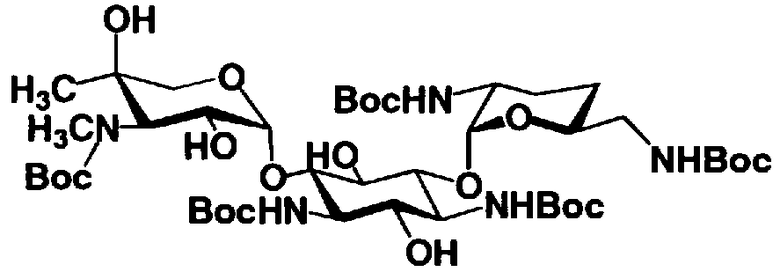
2-гидроксигентамицин C1a (10,0 г) (2,5 сульфат) растворяют в 140 мл воды. К раствору добавляют триэтиламин (30 мл). Добавляют раствор (180 мл) 28,1 г ди-трет-бутилдикарбоната в 1,4-диоксане и полученную смесь перемешивают при 60°C в течение 2 часов. К этому добавляют концентрированный водный аммиак (17 мл) и полученную смесь перемешивают при 60°C в течение 30 минут. Реакционную смесь возвращают к комнатной температуре и концентрируют досуха. К остатку добавляют воду (1 л) и полученный раствор перемешивают в течение ночи. Образовавшийся осадок собирают фильтрованием, промывают водой и сушат при пониженном давлении с получением указанного в заголовке соединения (12,4 г, выход 91%).
Rf значение: 0,73 (используют часть нижнего слоя смеси хлороформ:метанол:15 M водный аммиак (концентрированный водный аммиак)=1:1:1).
ESIMS: m/z 988 [M+Na]+.
Стадия получения 10-7
2,2"-ди-О-ацетил-1,3,2',6',3"-пента-N-трет-бутоксикарбонил-2-гидроксигентамицин C1a
[Химическая формула 105]
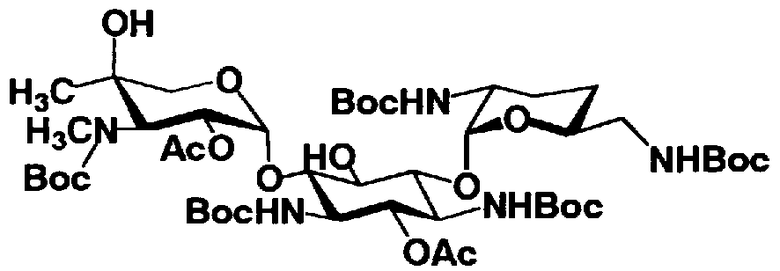
Соединение (12,4 г), полученное на стадии получения 10-6, растворяют в 250 мл пиридина. К раствору добавляют уксусный ангидрид (36,3 мл) при охлаждении льдом, и полученную смесь возвращают к комнатной температуре и оставляют реагировать. Через 88 часов после начала реакции к реакционной смеси добавляют 31,3 мл метанола при охлаждении льдом и полученную смесь перемешивают при охлаждении льдом в течение 30 минут. Реакционную смесь концентрируют досуха. К остатку добавляют хлороформ (1,2 л), и полученный раствор трижды промывают 600 мл насыщенного водного раствора бикарбоната натрия, трижды 600 мл 5% водного раствора бисульфата калия и один раз 600 мл воды, сушат над глауберовой солью и концентрируют при пониженном давлении с получением указанного в заголовке соединения (13,8 г, количественно).
Rf значение: 0,81 (хлороформ:метанол=10:1).
ESIMS: m/z 1072 [M+Na]+.
Стадия получения 10-8
2,2"-ди-О-ацетил-1,3,2',6',3"-пента-N-трет-бутоксикарбонил-4"-ено-5-О-метансульфонил-2-гидроксигентамицин C1a
[Химическая формула 106]
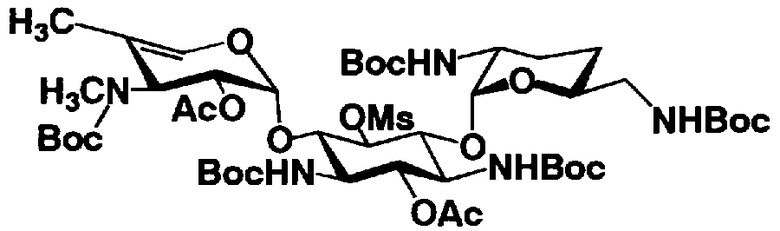
Соединение (13,5 г), полученное на стадии получения 10-7, растворяют в 270 мл метиленхлорида. К раствору добавляют 4-диметиламинопиридин (23,6 г) и 10,9 мл метансульфонилхлорида при охлаждении льдом, и полученную смесь возвращают к комнатной температуре и перемешивают в течение 45 часов. Реакционную смесь разбавляют 1 л хлороформа, и разбавленный раствор трижды промывают 600 мл насыщенного водного раствора бикарбоната натрия, трижды 600 мл 5% водного бисульфата калия и дважды 600 мл воды, сушат над глауберовой солью и затем концентрируют при пониженном давлении с получением 19,3 г сырого продукта. Сырой продукт очищают, используя хроматографическую колонку с силикагелем (200 г, хлороформ→хлороформ:метанол=50:1) с получением указанного в заголовке соединения (10,8 г, выход 76%).
Rf значение: 0,44 (хлороформ:метанол=30:1).
ESIMS: m/z 1132 [M+Na]+.
Стадия получения 10-9
2,5,2"-три-О-ацетил-1,3,2',6',3"-пента-N-трет-бутоксикарбонил-4"-ено-5-эпи-2-гидроксигентамицин C1a
[Химическая формула 107]
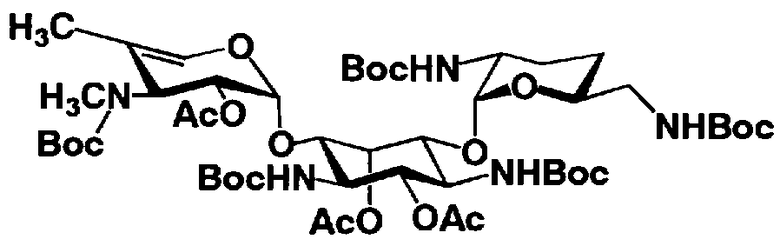
Соединение (10,5 г), полученное на стадии получения 10-8, растворяют в 105 мл N,N-диметилформамида. К раствору добавляют ацетат цезия (16,8 г, высушенного при 120°C в течение 6 часов) и полученную смесь перемешивают при 100°C в течение 16 часов. Реакционную смесь разбавляют 1,1 л этилацетата, промывают один раз 300 мл воды, дважды 300 мл насыщенного солевого раствора, сушат над глауберовой солью и концентрируют досуха с получением указанного в заголовке соединения (10,1 г, выход 99%).
Rf значение: 0,36 (хлороформ:метанол=30:1).
ESIMS: m/z 1096 [M+Na]+.
Стадия получения 10-10
1,3,2',6',3"-пента-N-трет-бутоксикарбонил-4"-ено-5-эпи-2-гидроксигентамицин C1a
[Химическая формула 108]
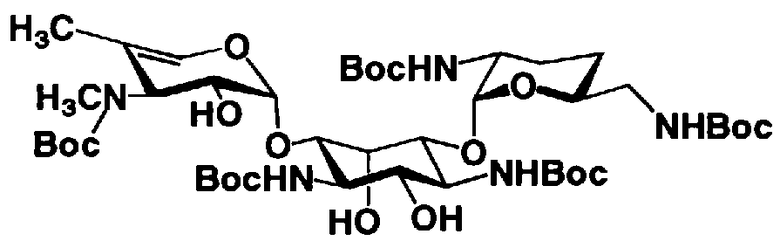
1% раствор (240 мл) метилата натрия в метаноле добавляют к соединению (10,1 г), полученному на стадии получения 10-9, и полученную смесь оставляют реагировать при комнатной температуре в течение 41 часа. Реакционную смесь нейтрализуют, добавляя Dowex 50 W×2 (H+форма, замещенная метанолом). Указанную смолу удаляют с помощью фильтрования и полученный фильтрат концентрируют досуха с получением указанного в заголовке соединения (8,2 г, выход 92%).
Rf значение: 0,24 (хлороформ:метанол=30:1).
ESIMS: m/z 970 [M+Na]+.
Стадия получения 10-11
3',4'-дидезокси-5-эпи-2-гидроксинеамин
[Химическая формула 109]
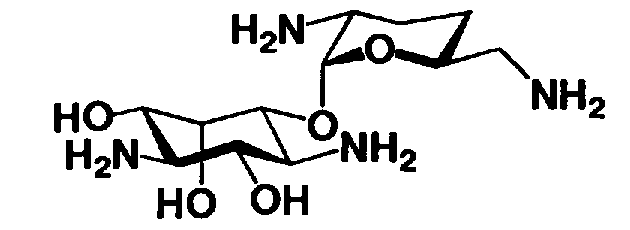
Смесь 6 M хлористоводородная кислота-метанол (1:1) (164 мл) добавляют к соединению (8,2 г), полученному на стадии получения 10-10, полученную смесь оставляют реагировать при комнатной температуре в течение 7 часов. Реакционную смесь оставляют далее реагировать при 70°C в течение 14 часов. рН реакционного раствора доводят до pH 6,6, добавляя 4 M гидроксид натрия при охлаждении льдом. Полученный раствор затем разбавляют 1,3 л воды, и разбавленный раствор вводят в колонку с Amberlite CG-50 (уравновешенную 0,005 M водным аммиаком) (900 мл), и элюирование осуществляют последовательно: 0,005 M→0,1 M→0,2 M→0,3 M→0,4 M→0,5 M водный аммиак с получением указанного в заголовке соединение (1,54 г, 41% в виде дикарбоната).
Rf значение: 0,16 (хлороформ:метанол:15 M водный аммиак (концентрированный водный аммиак):вода=1:4:1:1).
1H-ЯМР (26% ND3-D2O): δ 4,90 (д, 1Н, J=3 Гц), 4,16 (с, 1Н), 3,76 (м, 1H), 3,43 (д, 1H, J=10 Гц), 3,29 (д, 1H, J=10 Гц), 3,04 (т, 1H, J=10, 10 Гц), 3,00 (т, 1H, J=10, 10 Гц), 2,90 (т, 1H, J=10, 10 Гц), 2,76 (м, 1H), 2,61 (дд, 1H, J=4,5, 13,5 Гц), 2,57 (дд, 1H, J=7, 13,5 Гц), 1,6-1,75 (м, 3H), 1,35 (м, 1H).
13C-ЯМР (26% ND3-D2O): δ 96,51, 75,99, 75,31, 72,47, 71,09, 68,40, 54,32, 53,38, 50,32, 45,84, 28,35, 27,01.
Стадия получения 10-12
3',4'-дидезокси-5-эпи-1,3,2',6'-тетра-N-тозил-2-гидроксинеамин
[Химическая формула 110]
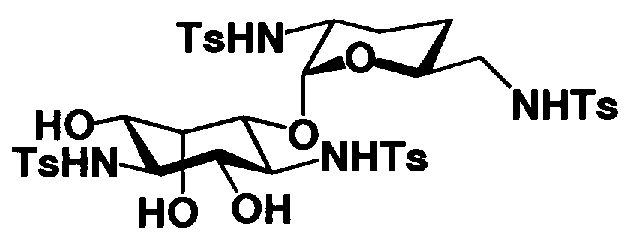
Соединение (202 мг, 0,470 ммоль, рассчитано как дикарбонат), полученное на стадии получения 10-11, растворяют в 2,0 мл воды. К раствору добавляют карбонат натрия (421 мг) при охлаждении льдом. К этому добавляют 1,4-диоксан (4,0 мл) и 541 мг тозилхлорида, и полученную смесь возвращают к комнатной температуре и оставляют реагировать. Через 2 часа после начала реакции к реакционной смеси добавляют 20 мл воды. Полученную смесь экстрагируют трижды 10 мл хлороформа, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (10 г, хлороформ:метанол=29:1) с получением указанного в заголовке соединения (383 мг, 88%).
Rf значение: 0,43 (хлороформ:метанол=10:1).
ESIMS: m/z 945 [M+Na]+.
1Н-ЯМР (Пиридин-d5): δ 9,41 (д, 1H, J=7 Гц), 8,88 (д, 1H, J=9 Гц), 8,57 (т, 1H, J=6, 6 Гц), 8,31 (шир., 1H), 7,89-8,15 (м, 8H), 6,96-7,25 (м, 8H), 5,22 (д, 1H, J=3 Гц), 4,86 (м, 1H), 4,73 (кв, 1H, J=10, 10, 10 Гц), 4,60 (шир.с, 1H), 4,43 (дт, 1H, J=7, 10, 10 Гц), 4,00 (дд, 1H, J=2, 10,5 Гц), 3,86 (т, 1H, J=10, 10 Гц), 3,77 (дд, 1H, J=2, 10,5 Гц), 3,72 (м, 1H), 3,15-3,32 (м, 2H), 2,26 (м, 1H), 2,09, 2,13, 2,19, 2,21 (каждый с, каждый 3H), 1,51-1,69 (м, 3Н).
Стадия получения 10-13
4",6"-ди-О-ацетил-3"-азидо-2"-О-бензил-3"-дезокси-5-эпи-1, 3,2',6'-тетра-N-тозил-2-гидроксидибекацин
[Химическая формула 111]
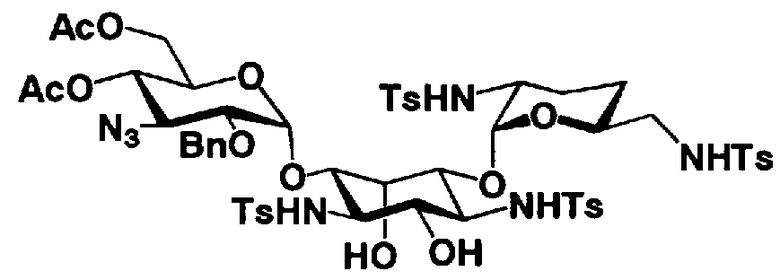
Способ A: Раствор соединения (372 мг), полученного на стадии получения 1-5a примера 1, в 21 мл метиленхлорида добавляют к указанному соединению (711 мг), полученному на стадии получения 10-12. К этому добавляют Drierite (2,16 г) и полученную смесь перемешивают при комнатной температуре в течение 3 часов. К смеси добавляют цианид ртути (795 мг) и полученную смесь перемешивают, экранируя от света, при комнатной температуре в течение 104 часов. Реакционную смесь фильтруют через целит для выделения нерастворимой части. Нерастворимую часть промывают 50 мл хлороформа. Объединенные органические слои дважды промывают 35 мл насыщенного водного раствора бикарбоната натрия, дважды 35 мл 10% водного раствора иодида натрия и один раз 35 мл воды, сушат над глауберовой солью и затем концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (10 г, хлороформ→хлороформ:этилацетат=9:1→1:1→2:3, получая указанное в заголовке соединение (426 мг, выход 44%). В этом случае выделяют 385 мг (54%) исходного соединения.
Способ B: Соединение (20,5 мг), полученное на стадии получения 10-12, и 11,4 мг соединения, полученного на стадии получения 1-5b примера 1, растворяют в 0,4 мл метиленхлорида. К раствору добавляют порошок молекулярных сит 4Å (63 мг) и полученную смесь перемешивают при комнатной температуре в течение 1 часа. К этому добавляют N-йодсукцинимид (5,9 мг) и раствор 0,6 мкл трифторметансульфоновой кислоты в 0,1 мл метиленхлорида при -20°C при перемешивании с последующим перемешиванием при -20°C при экранировании от света в течение 15 часов. Реакционную смесь фильтруют через целит для выделения нерастворимой части. Нерастворимую часть промывают 2 мл хлороформа. Объединенные органические слои дважды промывают 2 мл насыщенного водного раствора бикарбоната натрия, дважды 2 мл 10% водного раствора тиосульфата натрия и дважды 2 мл воды, сушат над глауберовой солью и затем концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (10 г, хлороформ→хлороформ:этилацетат=19:1→9:1→1:1→2:3) с получением указанного в заголовке соединения (7,7 мг, выход 27%). В этом случае выделяют 7,9 мг (38%) исходного соединения.
Rf значение: 0,19 (хлороформ:этилацетат=5:2).
ESIMS: m/z 1306 [M+Na]+.
1Н-ЯМР (Пиридин-d5): δ 8,99 (д, 1Н, J=9 Гц), 8,72 (м, 1H), 8,57 (т, 1H, J=6, 6 Гц), 8,53 (м, 1H), 7,05-8,05 (м, 21H), 5,70 (д, 1H, J=3,5 Гц), 5,49 (д, 1H, J=3 Гц), 5,29 (т, 1H, J=10, 10 Гц), 4,84-5,20 (ABкв, 2H, Jgem=12 Гц), 5,13 (шир.с, 1H), 4,86 (м, 1H), 4,73 (м, 1H), 4,65-4,77 (м, 3H), 4,55 (дд, 1H, J=4, 13, 5 Гц), 4,12-4,21 (м, 2H), 4,16 (т, 1H, J=10, 10 Гц), 3,92 (т, 1H, J=10, 10 Гц), 3,79 (дд, 1H, J=3,5, 10 Гц), 3,63 (м, 1H), 3,27 (м, 2H), 2,20 (м, 1H), 2,14, 2,17, 2,21, 2,23 (каждый с, каждый 3H), 2,00, 2,05 (каждый с, каждый 3H), 1,49-1,57 (м, 3H).
Стадия получения 10-14
3"-азидо-2"-О-бензил-3"-дезокси-5-эпи-1,3,2',6'-тетра-N-тозил-2-гидроксидибекацин
[Химическая формула 112]
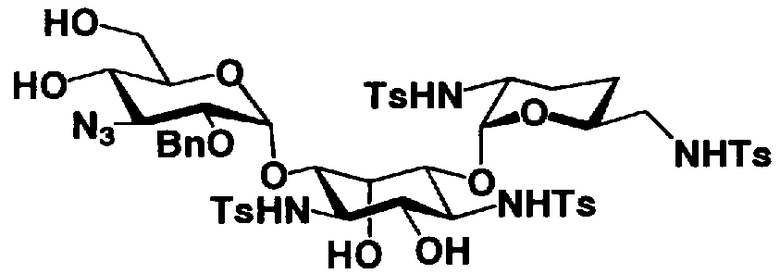
0,5% раствор (13,4 мл) метилата натрия в метаноле добавляют к соединению (673 мг), полученному на стадии получения 10-13, и полученную смесь оставляют реагировать при комнатной температуре в течение 1 часа. Реакционную смесь нейтрализуют, используя Dowex 50 W × 2 (H+форма, замещенная метанолом). Указанную смолу удаляют с помощью фильтрования и полученный фильтрат концентрируют досуха с получением указанного в заголовке соединения (593 мг, 94%).
Rf значение: 0,21 (хлороформ:этилацетат=1:1).
ESIMS: m/z 1222 [M+Na]+.
Стадия получения 10-15
5-эпигидроксидибекацин
[Химическая формула 113]
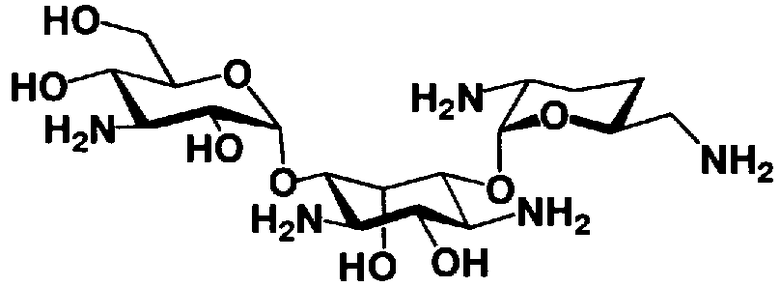
Жидкий аммиак (120 мл) помещают при -50°C в реактор в форме баклажана, содержащий 593 мг соединения, полученного на стадии получения 10-14. Металлический натрий (980 мг) добавляют при -50°C, и полученную смесь интенсивно перемешивают стеклянной палочкой-мешалкой в течение 2 часов. Метанол постепенно добавляют до тех пор, пока окраска радикалов не исчезает. Полученную смесь возвращают к комнатной температуре для испарения аммиака и в конце концентрируют досуха, используя испаритель. К остатку добавляют воду (43 мл) и рН полученного раствора доводят до pH 4-5, используя Dowex 50 W×2 (H+форма). Содержимое реактора, включая указанную смолу, как оно есть, добавляют в колонку, набитую 15 мл той же самой смолы. Колонку промывают 160 мл воды и элюирование осуществляют, используя 1 M водный аммиак (отсечка 80 мл). Позитивную в отношении нингидрина фракцию (фр.2) концентрируют досуха с получением 299 мг сырого продукта. Сырой продукт растворяют в воде (60 мл). Полученный водный раствор вводят в колонку с CM-Sephadex C-25 (уравновешенную 0,005 M водным аммиаком, 60 мл), и колонку промывают водой (120 мл). Элюирование осуществляют, используя 0,05 M (300 мл)→0,2 M водный аммиак (675 мл, отсечка 12 мл). Соответствующую фракцию (фр.38-50) концентрируют досуха с получением указанного в заголовке соединения (183 мг, выход 67,5%, в виде моногидрата монокарбоната).
Rf значение: 0,29 (1-бутанол:этанол:хлороформ:
17% водный аммиак=4:7:2:7)
1H-ЯМР (26% ND3-D2O): δ 4,99 (д, 1H, J=4 Гц), 4,92 (д, 1H, J=3 Гц), 4,47 (шир.с, 1H), 3,84 (шир.д, 1H, J=12 Гц), 3,83 (м, 1H), 3,79 (м, 1H), 3,62 (дд, 1H, J=7,5, 12,5 Гц), 3,49 (дд, 1H, J=2, 10 Гц), 3,42 (дд, 1H, J=4, 10,5 Гц), 3,37 (дд, 1H, J=2, 10 Гц), 3,17 (т, 1H, J=10, 10 Гц), 3,15 (т, 1H, J=11, 11 Гц), 3,12 (т, 1H, J=10, 10 Гц), 3,10 (т, 1H, J=10, 10 Гц), 3,06 (т, 1H, J=10, 10 Гц), 2,79 (м, 1H), 2,65 (дд, 1H, J=4,5, 13,5 Гц), 2,60 (дд, 1H, J=7,5, 13,5 Гц), 1,64-1,78 (м, 3H), 1,37 (м, 1H).
13C-ЯМР (26% ND3-D2O): δ 102,43, 96,75, 83,11, 76,41, 75,21, 74,00, 72,95, 71,40, 71,06, 68,07, 62,13, 55,39, 54,37, 53,69, 50,84, 46,49, 28,92, 27,62.
Стадия получения 10-16
2',6'-ди-N-бензилоксикарбонил-5-эпи-2-гидроксидибекацин
[Химическая формула 114]
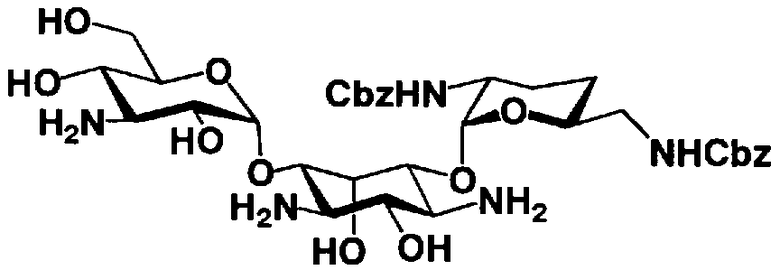
Тетрагидрат ацетата никеля (478 мг) добавляют к 224 мг (рассчитано как моногидрат монокарбоната) соединения, полученного на стадии получения 10-15. К этому добавляют метанол (9,0 мл) и полученную смесь превращают в гомогенный раствор, используя ультразвуковой очиститель (2-3 минуты, зеленый цвет). Добавляют N-бензилоксикарбонилоксисукцинимид (263 мг) порциями при охлаждении льдом за промежуток времени 2 минут. Полученную смесь перемешивают при охлаждении льдом в течение 1 часа. Полученную смесь возвращают к комнатной температуре и перемешивают далее в течение 2,5 часа. Реакционную смесь концентрируют досуха. Концентрированный водный аммиак, насыщенный хлоридом натрия (15 мл), добавляют к остатку и полученную смесь экстрагируют пять раз, используя 10 мл 1-бутанола. Полученный экстракцией слой бутанола концентрируют досуха. N,N-диметилформамид добавляют к 1594 мг полученного остатка и полученную смесь фильтруют через целит. Вещество, оставшееся на целите, промывают N,N-диметилформамидом (4 мл×6). Полученный фильтрат и промывки концентрируют досуха, получая 512 мг сырого продукта.
Rf значение: 0,41 (часть нижнего слоя смеси хлороформ:метанол:15 M водный аммиак (используют концентрированный водный аммиак)=1:1:1).
Стадия получения 10-17
2',6'-ди-N-бензилоксикарбонил-5-эпи-2-гидрокси-3"-N-трифторацетилдибекацин
[Химическая формула 115]
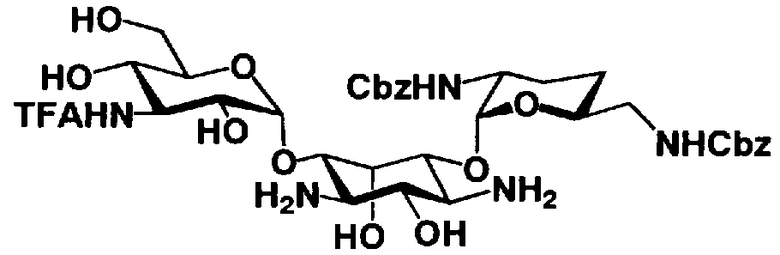
Сырой продукт (512 мг), полученный на стадии получения 10-16, растворяют в 7,1 мл безводного N,N-диметилформамида. К полученному раствору добавляют этилтрифторацетат (74 мл) при охлаждении льдом и при перемешивании. Полученную смесь возвращают к комнатной температуре и перемешивают в течение 16,5 часа. Реакционный раствор концентрируют досуха, получая 609 мг продукта.
Rf значение: 0,49 (часть нижнего слоя смеси хлороформ:метанол:15 M водный аммиак (используют концентрированный водный аммиак)=1:1:1).
Стадия получения 10-18
1-N-(4-бензилоксикарбониламин-2-(S)-гидроксибутирил)-2',6'-ди-N-бензилоксикарбонил-5-эпи-2-гидрокси-3"-N-трифторацетилдибекацин
[Химическая формула 116]
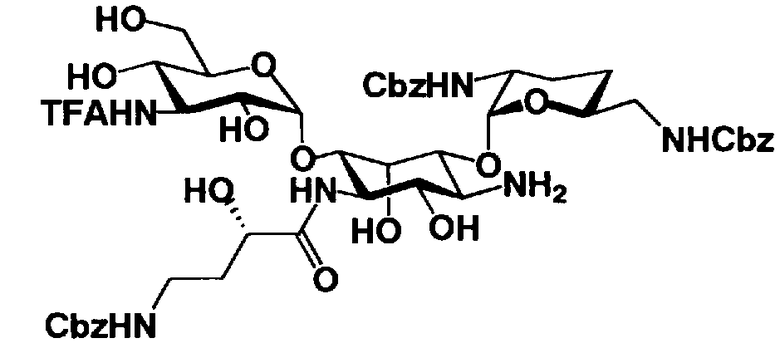
Сырой продукт (609 мг), полученный на стадии получения 10-17, растворяют в 12 мл безводного тетрагидрофурана. Затем раствор (6 мл) 219 мг сукцинимидного эфира N-бензилоксикарбонил-4-амино-2-(S)-гидроксимасляной кислоты в тетрагидрофуране, синтезированного в соответствии со способом Kawaguchi et al. (Journal of Antibiotics, Vol.25, pp.695-708 (1972)), добавляют по каплям к раствору продукта за период 3 минуты при охлаждении льдом и при перемешивании. Полученную смесь возвращают к комнатной температуре и перемешивают. Через 3,5 часа после начала перемешивания раствор (0,92 мл) 34 мг сукцинимидного эфира N-бензилоксикарбонил-4-амино-2-(S)-гидроксимасляной кислоты в тетрагидрофуране добавляют к реакционному раствору при охлаждении льдом при перемешивании, и полученную смесь возвращают к комнатной температуре и перемешивают. Еще через 18,5 часа после начала перемешивания реакционный раствор концентрируют досуха. К остатку добавляют этилацетат (150 мл), и полученную смесь дважды промывают 30 мл насыщенного водного раствора бикарбоната натрия и дважды 30 мл воды и концентрируют досуха с получением 604 мг реакционной смеси.
Rf значение: 0,67 (часть нижнего слоя смеси хлороформ:метанол:15 M водный аммиак (используют концентрированный водный аммиак)=1:1:1).
Стадия получения 10-19
5-эпи-2-гидроксиарбекацин
[Химическая формула 117]
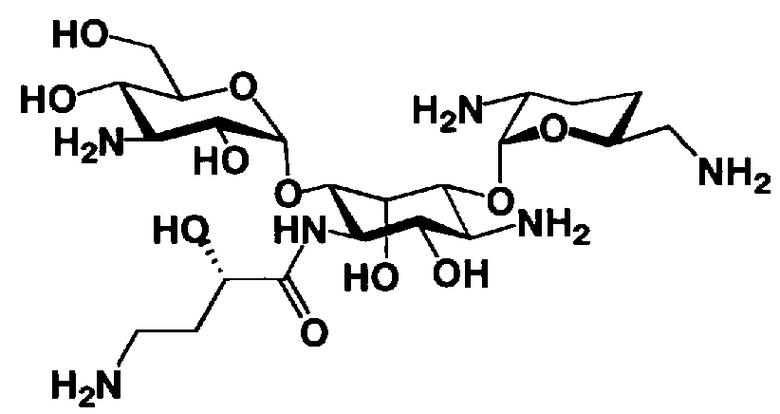
Тетрагидрофуран (20,5 мл) и 15,4 мл 3,5 M водного аммиака добавляют к реакционной смеси (604 мг), полученной на стадии получения 10-18, и полученную смесь перемешивают при комнатной температуре в течение 44 часов. Реакционный раствор концентрируют досуха. Добавляют смесь тетрагидрофуран-уксусная кислота-вода (4:1:1) (33 мл) к 656 мг полученного остатка. Затем к этому добавляют 10 капель суспензии палладиевой черни в воде и полученную смесь перемешивают при атмосферном давлении в течение 5 часов, продувая при этом систему водородом. Затем палладиевую чернь удаляют из реакционного раствора с помощью фильтрования. Палладиевую чернь промывают водой, и полученный фильтрат и промывки объединяют и концентрируют досуха. К остатку добавляют 2 M водный аммиак, и полученную смесь оставляют выстаиваться при комнатной температуре в течение ночи. Нерастворимую часть удаляют с помощью фильтрования через ватную пробку и концентрируют досуха с получением 466 мг сырого продукта. Полученный сырой продукт растворяют в 60 мл воды с получением водного раствора. Указанный водный раствор вводят в колонку с CM-Sephadex C-25 (уравновешенную 0,005 M водным аммиаком, 60 мл). Колонку промывают 120 мл 0,005 M водным аммиаком. Элюирование осуществляют, используя водный аммиак, концентрация которого меняется от 0,05 M (300 мл) до 0,5 M (600 мл), и 0,75 M (600 мл) водного аммиака. Соответствующие фракции концентрируют с получением указанного в заголовке соединения: 5-эпи-2-гидроксиарбекацин (65 мг, 2,5 тригидрат карбоната, 20% за четыре стадии).
Rf значение: 0,11 (1-бутанол:этанол:хлороформ:17% водный аммиак=4:7:2:7).
1H-ЯМР (26% ND3-D2O): δ 4,95 (д, 1H, J=4 Гц), 4,92 (д, 1H, J=3 Гц), 4,44 (т, 1H, J=2, 2 Гц), 4,17 (т, 1H, J=10, 10 Гц), 4,16 (дд, 1H, J=4, 10 Гц), 3,85 (дд, 1H, J=2, 12 Гц), 3,79 (дд, 1H, J=2, 10,5 Гц), 3,78 (м, 1H), 3,77 (м, 1H), 3,60 (дд, 1H, J=7, 12 Гц), 3,51 (дд, 1H, J=2, 10,5 Гц), 3,34 (т, 1H, J=10, 10 Гц), 3,12 (дд, 1H, J=4, 10 Гц), 3,21 (т, 1H, J=10, 10 Гц), 3,14 (т, 1H, J=10, 10 Гц), 3,02 (т, 1H, J=10, 10 Гц), 2,80 (м, 1H), 2,70-2,80 (м, 2H), 2,66 (дд, 1H, J=5, 13,5 Гц), 2,61 (дд, 1H, J=7,5, 13,5 Гц), 1,91 (м, 1H), 1,64-1,80 (м, 4H), 1,37 (м, 1H).
13C-ЯМР (26% ND3-D2O): δ 178,69, 101,16, 96,93, 78,02, 76,08, 74,06, 73,28, 72,82, 71,34, 70,92, 68,35, 62,01, 55,40, 54,18, 53,77, 50,69, 46,29, 38,64, 37,58, 28,74, 27,43.
Пример 11
5,4"-ди-эпи-2-гидроксиарбекацин
[Химическая формула 118]
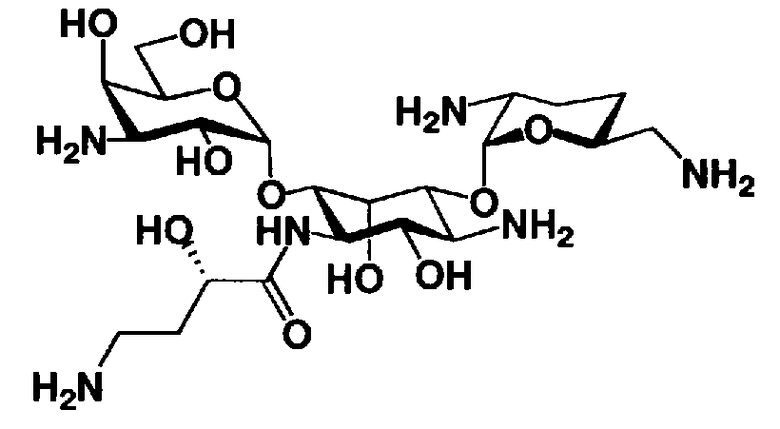
Стадия получения 11-1
3,2',6',3",4"'-пента-N-трет-бутоксикарбонил-5-эпи-2-гидроксиарбекацин
[Химическая формула 119]
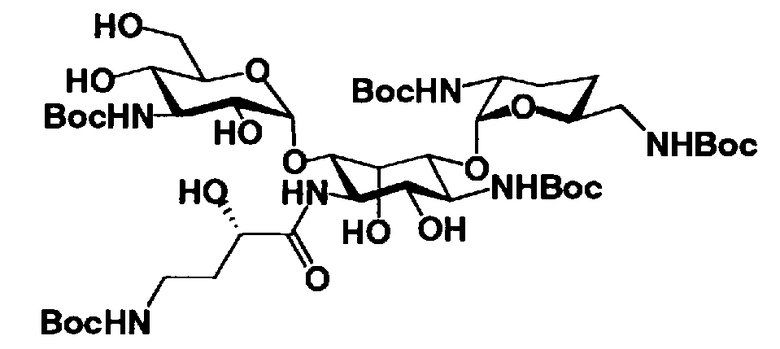
Соединение (65 мг), полученное на стадии получения 10-19 примера 10, растворяют в 0,91 мл воды. К раствору добавляют триэтиламин (0,16 мл), затем добавляют раствор 175 мг ди-трет-бутил дикарбоната в 1,4-диоксане (1,17 мл) и полученную смесь перемешивают при 60°C в течение 1,5 часа. К этому добавляют концентрированный водный аммиак (0,11 мл) и полученную смесь перемешивают при 60°C в течение 30 минут. Реакционную смесь возвращают к комнатной температуре и концентрируют досуха. Дважды осуществляют азеотропную перегонку полученного остатка, используя метанол, с получением указанного в заголовке соединения (103 мг, количественно).
Rf значение: 0,53 хлороформ:метанол:15 M водный аммиак (концентрированный водный аммиак)=5:1:0,1).
ESIMS: m/z 1091 [M+Na]+.
Стадия получения 11-2
3,2',6',3",4"'-пента-N-трет-бутоксикарбонил-4",6"-О-циклогексилиден-5-эпи-2-гидроксиарбекацин
[Химическая формула 120]
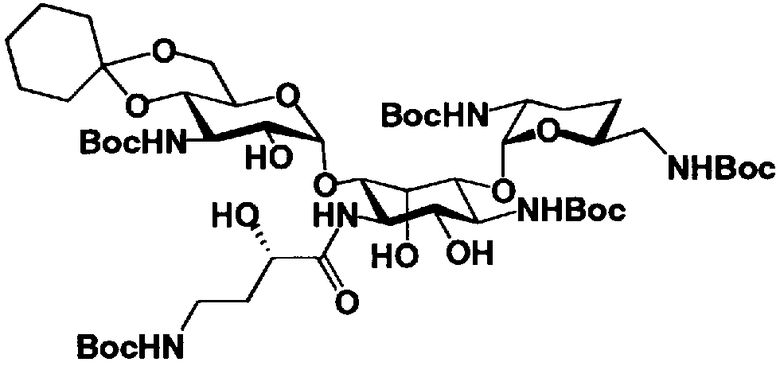
Указанное соединение (103 мг), полученное на стадии получения 11-1, растворяют в 2,0 мл N,N-диметилформамида. К раствору добавляют циклогексанон диизопропилацеталь (58 мкл) и 12,4 мг п-толуолсульфоновой кислоты, и полученную смесь оставляют реагировать при комнатной температуре в течение 1 часа. К этому добавляют насыщенный водный раствор бикарбоната натрия (20 мл), и полученный осадок собирают с помощью фильтрования, промывают водой, и сушат при пониженном давлении с получением указанного в заголовке соединения (103 мг, 88%).
Rf значение: 0,53 (хлороформ:метанол=10:1).
ESIMS: m/z 1171 [M+Na]+.
Стадия получения 11-3
2,2",2"'-три-O-ацетил-3,2',6',3",4"'-пента-N-трет-бутоксикарбонил-4",6"-O-циклогексилиден-5-эпи-2-гидроксиарбекацин
[Химическая формула 121]
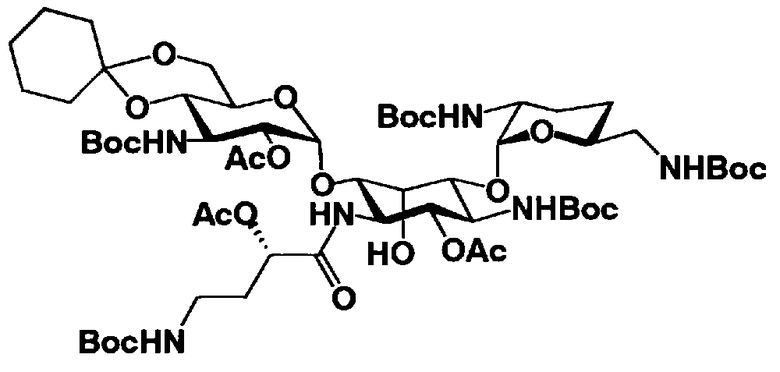
Указанное соединение (103 мг), полученное на стадии получения 11-2, растворяют в 2,1 мл пиридина. К раствору добавляют при охлаждении льдом уксусный ангидрид (0,13 мл), и полученную смесь возвращают к комнатной температуре и оставляют реагировать. Через 18,5 часа после начала реакции добавляют 0,11 мл метанола при охлаждении льдом, и полученную смесь оставляют выстаиваться при комнатной температуре в течение 30 минут, и концентрируют досуха. К остатку добавляют хлороформ (10 мл). Полученный раствор дважды промывают 3 мл насыщенного водного раствора бикарбоната натрия, дважды 3 мл 5% водного раствора бисульфата калия и дважды 3 мл воды, сушат над глауберовой солью и концентрируют досуха с получением указанного в заголовке соединения (117 мг, количественно).
Rf значение: 0,39 (хлороформ:метанол=15:1).
ESIMS: m/z 1297 [M+Na]+.
Стадия получения 11-4
2,2",2"'-три-O-ацетил-3,2',6',3",4"'-пента-N-трет-бутоксикарбонил-5-эпи-2-гидроксиарбекацин
[Химическая формула 122]
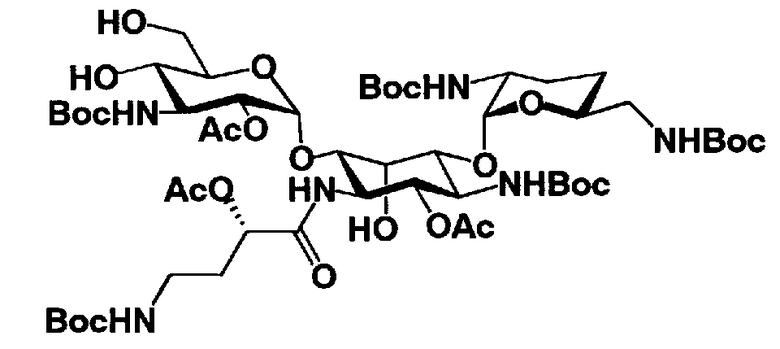
Указанное соединение (117 мг), полученное на стадии получения 11-3, растворяют в 5,17 мл смеси хлороформ:метанол (10:1). К полученному раствору добавляют 90% водный раствор трифторуксусной кислоты (0,47 мл) при охлаждении льдом и при перемешивании, и полученную смесь возвращают к комнатной температуре и перемешивают. Через 30 минут после начала перемешивания добавляют 5,3 мл хлороформа. Полученную смесь промывают один раз 3 мл воды, дважды 3 мл насыщенного водного раствора бикарбоната натрия и дважды 3 мл полунасыщенного солевого раствора, сушат над глауберовой солью и концентрируют досуха с получением указанного в заголовке соединения (107 мг, количественно).
Rf значение: 0,46 (хлороформ:метанол=10:1).
ESIMS: m/z 1217 [M+Na]+.
Стадия получения 11-5
2,2",2"'-три-О-ацетил-3,2',6',3",4"'-пента-N-трет-бутоксикарбонил-5-эпи-2-гидрокси-6"-О-тритиларбекацин
[Химическая формула 123]
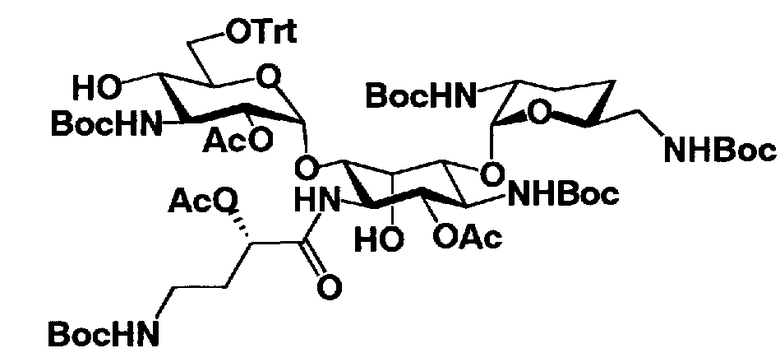
Указанное соединение (83 мг), полученное на стадии получения 11-4, растворяют в 1,7 мл пиридина. К раствору добавляют 4-диметиламинопиридин (25 мг) и 116 мг тритилхлорида и полученную смесь оставляют реагировать при 65°C. Через 17 часов после начала реакции полученную смесь возвращают к комнатной температуре. К этому добавляют метанол (0,08 мл) и полученную смесь оставляют выстаиваться в течение 1 часа. Полученную смесь затем концентрируют досуха. К остатку добавляют хлороформ (8 мл). Полученный раствор дважды промывают 3 мл насыщенного водного раствора бикарбоната натрия, трижды 3 мл 5% водного раствора бисульфата калия и трижды 3 мл воды, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (3,4 г, толуол:этилацетата:ацетон=6:1:1), с получением указанного в заголовке соединения (65 мг, 65%).
Rf значение: 0,57 (хлороформ:метанол=10:1).
ESIMS: m/z 1459 [M+Na]+.
Стадия получения 11-6
2,2",4",2"'-тетра-О-ацетил-3,2',6',3",4"'-пента-N-трет-бутоксикарбонил-5,4"-ди-эпи-2-гидрокси-6"-О-тритиларбекацин
[Химическая формула 124]
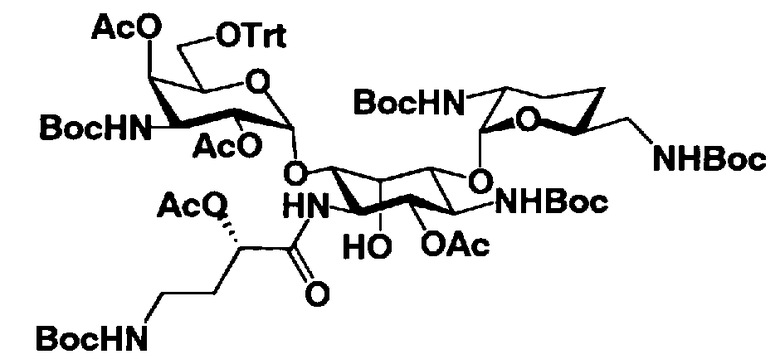
Указанное соединение (112 мг), полученное на стадии получения 11-5, растворяют в 2,3 мл метиленхлорида в атмосфере азота. К раствору добавляют пиридин (0,13 мл). Затем добавляют ангидрид трифторметансульфоновой кислоты (66 мкл) в атмосфере азота при -78°C при перемешивании. Полученную смесь перемешивают при -20°C в атмосфере азота в течение 1 часа. К смеси добавляют метанол (79 мкл) при -20°C и сразу же к этому добавляют 14 мл охлажденного льдом хлороформа. Полученную смесь дважды промывают 7 мл охлажденного льдом 10% водного раствора бисульфата калия, дважды 7 мл охлажденного льдом насыщенного водного раствора бикарбоната натрия и дважды 7 мл охлажденной льдом воды, затем сушат над глауберовой солью при охлаждении льдом в течение 10 минут. Высушенный раствор концентрируют при пониженном давлении при охлаждении льдом. Когда раствор превращается в сироп, концентрирование прекращают.
Полученный сироп растворяют в 1,1 мл N,N-диметилформамида. К раствору добавляют ацетат цезия (150 мг) (высушенный при 120°C в течение 6 часов) и полученную смесь перемешивают в атмосфере азота при комнатной температуре в течение 18 часов. Реакционный раствор разбавляют 46 мл этилацетата, и разбавленный раствор промывают один раз 11 мл воды и трижды 11 мл полунасыщенного солевого раствора, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (12 г, толуол:этилацетата:ацетон=4:1:1), с получением указанного в заголовке соединения (75 мг выход 65%).
Rf значение: 0,35 (толуол:этилацетат:ацетон=3:1:1).
ESIMS: m/z 1501 [M+Na]+.
Стадия получения 11-7
3,2',6',3",4"'-пента-N-трет-бутоксикарбонил-5,4"-ди-эпи-2-гидрокси-6"-О-тритиларбекацин
[Химическая формула 125]
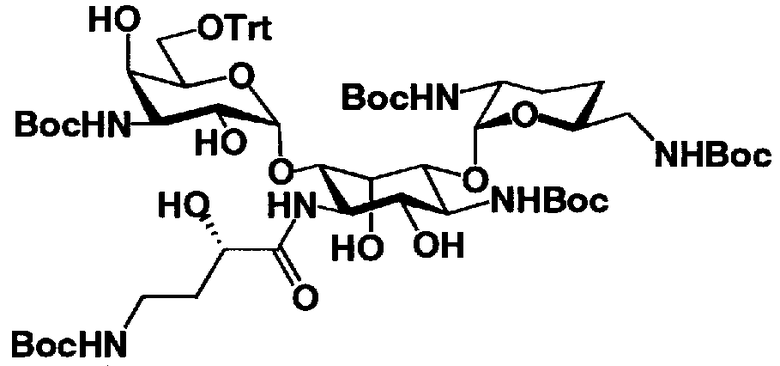
Указанное соединение (75 мг), полученное на стадии получения 11-6, растворяют в 2,3 мл 0,5% раствора метилата натрия в метаноле и полученную смесь оставляют реагировать при комнатной температуре в течение 1 часа. Реакционную смесь нейтрализуют, используя Dowex 50 W×2 (H+ форма, замещенная метанолом), и указанную смолу удаляют с помощью фильтрования. Полученный фильтрат концентрируют досуха с получением указанного в заголовке соединения (64 мг, 96%).
Rf значение: 0,60 (толуол:этилацетат:ацетон=1:1:1).
Стадия получения 11-8
5,4"-ди-эпи-2-гидроксиарбекацин
[Химическая формула 126]
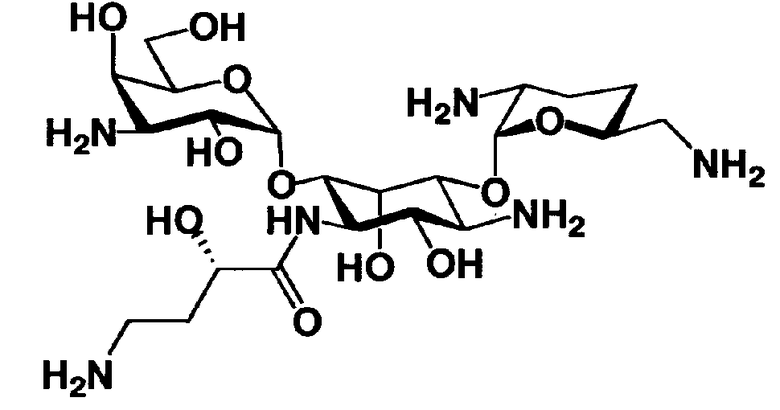
Соединение (64 мг), полученное на стадии получения 11-7, растворяют в 1,6 мл 90% водного раствора трифторуксусной кислоты при охлаждении льдом. Полученный раствор оставляют реагировать при охлаждении льдом в течение 2 часов. Реакционную смесь концентрируют досуха. К остатку добавляют воду (10 мл) и полученный раствор трижды промывают 3 мл диэтилового эфира. Водный слой концентрируют досуха. К остатку добавляют 0,005 M водного аммиака (10 мл) (pH 6-7) и полученную смесь вводят в колонку с CM-Sephadex C-25 (уравновешенную 0,005 M водным аммиаком, 10 мл). Колонку промывают 30 мл 0,005 M водного аммиака и элюирование осуществляют, используя водный аммиак в концентрациях, которые меняются от 0,2 M (50 мл) до 0,5 M (200 мл). Соответствующие фракции концентрируют с получением 28,6 мг указанного в заголовке соединения: 5-эпи-2-гидроксиарбекацина (73% в виде 2,5 тригидрата карбоната).
Rf значение: 0,09 (хлороформ:метанол:15 M водный аммиак (концентрированный водный аммиак):этанол=4:6:7:2)
1Н ЯМР (DCl-D2O, pD ~3): δ 5,44 (1H, J=3,5 Гц), 5,16 (д, 1H, J=4 Гц), 4,77 (д, 1H, J=3 Гц), 4,35 (дд, 1H, J=4,5, 9 Гц), 4,31 (т, 1H, J=11, 11 Гц), 4,17 (д, 1H, J=3 Гц), 4,14 (м, 1H), 4,13 (д, 1H, J=3, 11 Гц), 4,11 (м, 1H), 4,02 (дд, 1H, J=3, 11 Гц), 3,98 (дд, J=4, 11 Гц), 3,81 (т, 1H, J=11, 11 Гц), 3,75-3,80 (м, 2H), 3,71 (т, 1H, J=11, 11 Гц), 3,67 (дд, 1H, J=3, 11 Гц), 3,63 (м, 1H), 3,28 (дд, 1H, J=3,5, 13,5 Гц), 3,21 (т, 2H, J=7, 7 Гц), 3,11 (дд, 1H, J=7,5, 13,5 Гц), 2,23 (м, 1H), 2,05-2,13 (м, 2H), 2,00 (м, 1H), 1,94 (м, 1H), 1,64 (м, 1H).
13C-ЯМР (DCl-D2O, pD~3): δ 177,13, 100,42, 90,53, 76,63, 71,90, 70,56, 70,02, 68,33, 66,26, 66,20, 66,18, 65,57, 61,72, 53,73, 53,49, 52,59, 48,59, 42,96, 37,36, 31,34, 25,86, 21,32.
Рассчитано для C22H44N6O11·2,5H2CО3-3H2О. C, 37,84; H, 7,13; N, 10,81. Найдено: C, 37,51; H, 7,49; N, 10,96.
Пример 12
5,4"-ди-эпи-2-гидроксидибекацин
[Химическая формула 127]
Стадия получения 12-1
4"-О-ацетил-3"-азидо-6"-O-бензоил-2"-О-бензил-3"-дезокси-5,4"-ди-эпи-1,3,2',6'-тетра-N-тозил-2-гидроксидибекацин
[Химическая формула 128]
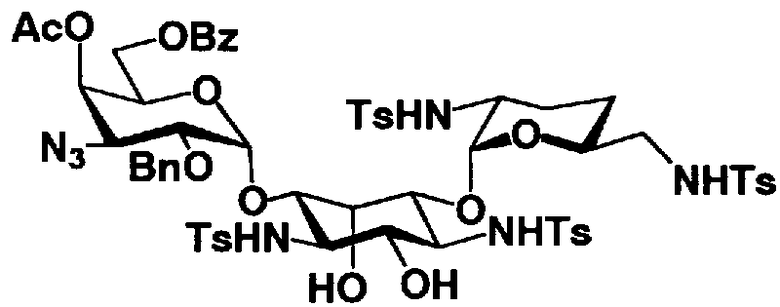
Раствор соединения (667 мг), полученного на стадии получения 10-5a, в 34 мл метиленхлорида добавляют к соединению (1,16 г), полученному на стадии получения 10-12. К этому добавляют Drierite (3,43 г) и полученную смесь перемешивают при комнатной температуре в течение 3 часов. К реакционному раствору добавляют цианид ртути (1,27 г) и полученную смесь перемешивают, экранируя от света, при комнатной температуре в течение 42 часов. Реакционный раствор фильтруют через целит для удаления нерастворимой части. Нерастворимую часть промывают 90 мл хлороформа. Объединенные органические слои дважды промывают 60 мл насыщенного водного раствора бикарбоната натрия, дважды 60 мл 10% водного раствора иодида натрия и один раз 60 мл воды, сушат над глауберовой солью и концентрируют досуха. Полученный остаток очищают, используя хроматографическую колонку с силикагелем (40 г, хлороформ→5 хлороформ:этилацетат=1:1→этилацетат) с получением указанного в заголовке соединения (529 мг, 31%). В этом случае выделяют 423 мг (36%) исходного соединения.
Rf значение: 0,20 (хлороформ:этилацетат=5:2).
ESIMS: m/z 1368 [M+Na]+.
1Н-ЯМР (Пиридин-d5): δ 9,02 (д, 1H, J=7 Гц), 8,83 (д, 1H, J=7 Гц), 8,50 (м, 1H), 8,48 (т, 1H, J=6, 6 Гц), 7,03-8,28 (м, 26H), 6,00 (д, 1H, J=3 Гц), 5,85 (д, 1H, J=2 Гц), 5,58 (шир.с, 1H), 4,81-5,26 (ABкв, 2H, Jgem=12 Гц), 5,20 (шир.с, 1H), 5,07 (м, 1H), 4,80-4,95 (м, 4H), 4,77 (т, 1H, J=10, 10 Гц), 4,72 (м, 1H), 4,68 (т, 1H, J=10, 10 Гц), 4,35 (м, 1H), 4,26 (шир.д, 1H, J=11 Гц), 4,23 (дд, 1H, J=4, 10 Гц), 3,90 (т, J=10, 10 Гц), 3,63 (м, 1H), 3,29 (м 2H), 2,26 (м, 1H), 2,00, 2,07, 2,15, 2,18, 2,22 (каждый с, каждый 3H), 1,48-1,64 (м, 3H).
Стадия получения 12-2
5,4"-ди-эпи-2-гидроксидибекацин
[Химическая формула 129]
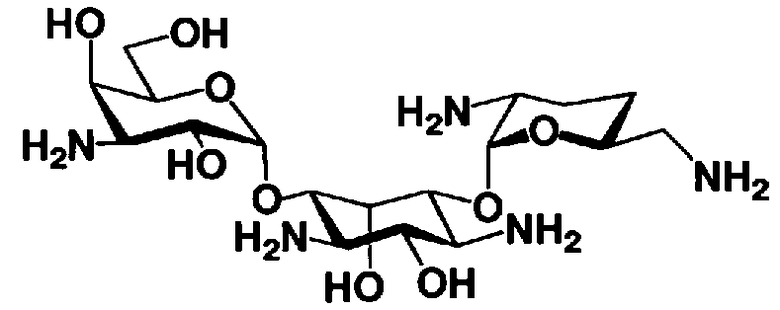
0,5 M раствор (12 мл) метилата натрия в метаноле добавляют к соединению (529 мг), полученному на стадии получения 12-1, и полученную смесь оставляют реагировать при комнатной температуре в течение 2 часов. Реакционную смесь нейтрализуют, используя Dowex 50 W × 2 (H+форма, замещенная метанолом). Указанную смолу удаляют с помощью фильтрования и полученный фильтрат концентрируют досуха с получением сырого продукта.
ESIMS: m/z 1222 [M+Na]+.
Жидкий аммиак (около 25 мл) помещают при -50°C в колбу типа баклажана, содержащую сырой продукт, и добавляют в нее 732 мг металлического натрия при -50°C. Полученную смесь интенсивно перемешивают стеклянной палочкой-мешалкой в течение 2 часов. Метанол постепенно добавляют до тех пор, пока не исчезает окраска радикалов. Полученную смесь возвращают к комнатной температуре для испарения аммиака и затем концентрируют досуха, используя испаритель. К остатку добавляют воду (32 мл), и рН полученного раствора доводят до pH 4-5, используя Dowex 50 W×2 (H+ форма), и содержимое колбы, включая указанную смолу, как оно есть, вводят в колонку, набитую 10 мл той же самой смолы. Колонку промывают 120 мл воды и элюирование осуществляют, используя 1 M водный аммиак. Позитивную в отношении нингидрина фракцию концентрируют досуха с получением сырого продукта. Сырой продукт переводят в водный раствор (40 мл). Водный раствор вводят в колонку с CM-Sephadex C-25 (уравновешенную 0,005 M водным аммиаком, 20 мл) и колонку промывают водой (40 мл). Элюирование осуществляют, используя 0,05 M→0,2 M→0,5 M водный аммиак (каждый по 40 мл), и соответствующую фракцию концентрируют досуха с получением указанного в заголовке соединения (104 мг, 43%, в виде моногидрата монокарбоната).
Rf значение: 0,15 (1-бутанол:этанол:хлороформ:17% водный аммиак=4:7:2:7).
ESIMS: m/z 468 [M+H]+, 490 [M+Na]+.
1H-ЯМР (26% ND3-D2O): δ 5,03, (д, 1H, J=4 Гц), 4,92 (д, 1H, J=3 Гц), 4,51 (т, 1H, J=2, 2 Гц), 4,08 (м, 1H), 3,85 (д, 1H, J=2 Гц), 3,80 (м, 1H), 3,68 (д, 2H, J=5,5 Гц), 3,60 (дд, 1H, J=4, 10,5 Гц), 3,49 (дд, 1H, J=2, 10 Гц), 3,36 (дд, 1H, J=2, 10 Гц), 3,14 (т, 1H, J=10, 10 Гц), 3,12 (т, 1H, J=10, 10 Гц), 3,10 (т, 1H, J=10, 10 Гц), 2,98 (дд, 1H, J=3, 10,5 Гц), 2,79 (м, 1H), 2,64 (дд, 1H, J=4, 13,5 Гц), 2,60 (дд, 1H, J=7,5, 13,5 Гц), 1,64-1,77 (м, 3H), 1,36 (м, 1H).
13C-ЯМР (26% ND3-D2O): δ 102,91, 96,82, 82,89, 76,46, 75,24, 73,81, 71,40, 71,07, 70,02, 68,09, 62,70, 54,36, 53,70, 52,49, 50,87, 46,51, 28,95, 27,61.
Сравнительный пример 1
Получение (S)-2-бензилокси-4-бензилоксикарбониламиномасляной кислоты
[Химическая формула 130]
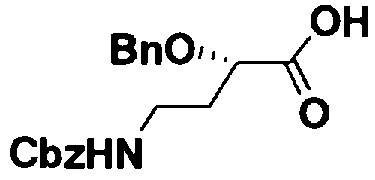
(S)-4-бензилоксикарбониламино-2-гидроксимасляную кислоту (5,25 г, 20,7 ммоль), синтезированную в соответствии со способом Kawaguchi et al. (Journal of Antibiotics, Vol.25, pp.695-708 (1972)), растворяют в 60 мл N,N-диметилформамида. При охлаждении на бане со льдом добавляют оксид бария (15,9 г, 103,7 ммоль). Через 2 минуты после добавления оксида бария к этому добавляют гидроксидоктагидрат бария (13,4 г, 42,5 ммоль) при охлаждении на бане со льдом. Через 10 минут к этому добавляют бензилбромид (7,5 мл, 63,1 ммоль) при охлаждении на бане со льдом. Через 5 минут реакционный раствор возвращают к комнатной температуре и интенсивно перемешивают. Через 1,5 часа завершение реакции подтверждают данные ТСХ (система элюента=хлороформ:этилацетат:уксусная кислота=10:5:1), и к этому добавляют воду (0,5 мл) при охлаждении на бане со льдом. Полученную смесь разбавляют хлороформом (165 мл) и к этому добавляют 4 н. HCl (70 мл). Затем добавляют воду (25 мл) с последующим разделением на слои. Водный слой экстрагируют хлороформом (50 мл). Органический слой и экстрагированный органический слой объединяют и промывают насыщенным водным раствором NaCl. Промытый раствор сушат над сульфатом магния. Растворитель удаляют перегонкой при пониженном давлении и полученный остаток очищают на хроматографической колонке с силикагелем (силикагель 60, нейтральные сферы, 160 г) (хлороформ:этилацетат=4:1→хлороформ:метанол=10:1→хлороформ:метанол:уксусная кислота=10:1:0,1 (600 мл)) с получением (S)-2-бензилокси-4-бензилоксикарбониламиномасляной кислоты (2,86 г, 8,34 ммоль).
LCMS: m/z 344 [M+H]+.
1Н-ЯМР (ДМСО-d6): δ 12,8 (с, 1H), 7,25-7,35 (м, 10H), 7,26 (д, 1H, J=4,2 Гц), 5,01 (с, 2H), 4,49 (ABкв, 2H, Jgem=12 Гц), 3,97 (дд, 1H, J=3,7, 8,8 Гц), 3,08-3,18 (м, 2H), 1,67-1,93 (м, 2H).
Сравнительный пример 2
(S)-2-ацетокси-3-амино-N-бензилоксикарбонилпропановая кислота
[Химическая формула 131]
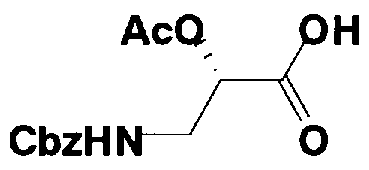
Стадия 1
Дифенилметиловый эфир (S)-3-амино-N-бензилоксикарбонил-2-гидроксипропановой кислоты
[Химическая формула 132]
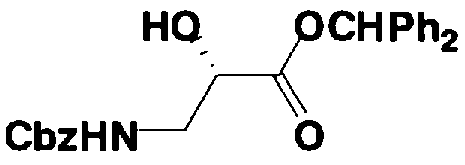
N-бензилоксикарбонилизосерин (20 мг), синтезированный в соответствии со способом R.D. Westland et al. (Carbohydr. Res., Vol.28, pp. 268-280 (1973)), растворяют в 0,6 мл тетрагидрофурана. К раствору добавляют дифенилметилазид (24 мг) и полученную смесь перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор концентрируют при пониженном давлении, и остаток очищают на хроматографической колонке с силикагелем (2 г, гексан:этилацетат=5:1→4:1→3:1), получая указанное в заголовке соединение (33 мг, количественно).
Rf значение: 0,54 (гексан:этилацетат=1:1).
1Н-ЯМР (CDCl3): δ 7,25-7,35 (м, 15H), 6,91 (с, 1H), 5,05 (шир., 2H), 4,39 (с, 1H), 3,68-3,71 (м, 1H), 3,49-3,58 (дд, 1H, J=6,1, 6,9 Гц), 3,21 (м, 1H).
Стадия 2
(S)-2-ацетокси-3-амино-N-бензилоксикарбонилпропановая кислота
[Химическая формула 133]
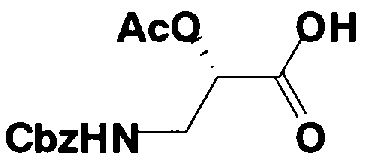
Соединение (263 мг), полученное на стадии получения 1, растворяют в 1,0 мл метиленхлорида. К раствору добавляют пиридин (70 мкл). К раствору добавляют уксусный ангидрид (20 мкл) и 0,8 мг 4-диметиламинопиридина при 0°C. Реакционную смесь возвращают к комнатной температуре и перемешивают в течение 2,5 часа. Добавляют метанол (100 мкл) и полученную смесь перемешивают при комнатной температуре в течение 20 минут для завершения реакции. К этому добавляют хлороформ (20 мл), и полученную смесь промывают 15 мл воды, сушат над глауберовой солью и концентрируют досуха, получая дифенилметиловый эфир (S)-3-амино-2-ацетокси-N-бензилоксикарбонилпропановой кислоты (28,6 мг (99%)).
Rf значение: 0,62 (гексан:этилацетат=1:1).
1Н-ЯМР (CDCl3): δ 7,25-7,37 (м, 15H), 6,87 (с, 1H), 5,26 (дд, 1H, J=4,8, 5,2 Гц), 5,06 (шир., 2H), 4,93 (с, 1H), 3,66-3,75 (м, 2H), 2,11 (с, 3H).
Вышеуказанные соединения (28,6 мг) растворяют в 0,6 мл хлороформа. К раствору добавляют трифторуксусную кислоту (0,6 мл) при 0°C и полученную смесь перемешивают при комнатной температуре в течение 2 часов. Трижды осуществляют азеотропную перегонку реакционной смеси с толуолом при пониженном давлении досуха с получением указанного в заголовке соединения (21,2 мг) в виде сырого продукта.
Rf значение: 0,44 (хлороформ:этилацетат:уксусная кислота=10:5:1).
1Н-ЯМР (CDCl3): δ 7,26-7,35 (м, 5H), 5,25 (дд, 1H, J=4,8, 5,1 Гц), 5,09 (шир., 2H), 4,86 (с, 1H), 3,62-3,70 (м, 2H), 2,12 (с, 3H).
Сравнительный пример 3
(S)-5-амино-2-бензилокси-N-бензилоксикарбонилпентановая кислота
[Химическая формула 134]
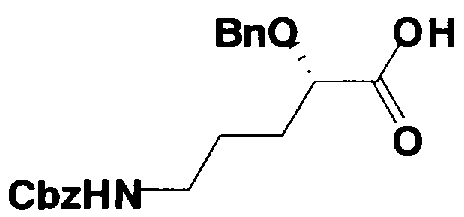
(S)-5-амино-N-бензилоксикарбонил-2-гидроксипентановую кислоту (21 мг), синтезированную в соответствии со способом R.D.Westland et al. (Carbohydr. Res., Vol.28, pp.268-280 (1973)), растворяют в 0,24 мл N,N-диметилформамида. К раствору добавляют при охлаждении льдом оксид бария (64 мг) и полученную смесь перемешивают в течение 2 минут. Затем добавляют гидроксид бария (5,4 мг) и полученную смесь перемешивают далее при той же температуре в течение 10 минут. К смеси добавляют бензилбромид (30 мкл). Через 5 минут после начала перемешивания реакционную смесь возвращают к комнатной температуре и полученную смесь интенсивно перемешивают в течение 1,5 часа. Добавляют воду (15 мл) при охлаждении льдом и полученную смесь разбавляют 30 мл хлороформа. К разбавленному раствору добавляют 4 н. HCl (0,28 мл) и полученную смесь перемешивают в течение 5 минут. Реакционный раствор разделяют, и водный слой снова экстрагируют 20 мл хлороформа. Полученный органический слой и полученный выше органический слой объединяют и сушат над сульфатом магния. Полученный таким образом раствор концентрируют при пониженном давлении и полученный остаток очищают, используя хроматографическую колонку с силикагелем (30 г, гексан:этилацетат=4:1→хлороформ:метанол=10:1) с получением указанного в заголовке соединения (11 мг, 39%).
Rf значение: 0,46 (хлороформ:этилацетат:уксусная кислота=10:5:1).
1Н-ЯМР (CDCl3): δ 7,32-7,35 (м, 10Н), 5,08 (шир., 2H), 4,70 (д, 1H, J=11,5 Гц), 4,46 (д, 1H, J=11,5 Гц), 4,00 (т, 1H, J=4,99 Гц), 3,19 (шир., 2H), 1,84 (дт, 2H, J=4,99, 7,0 Гц), 1,66 (тт, 2H, J=7,0, 7,2 Гц).
Сравнительный пример 4
(S)-6-амино-2-бензилокси-N-бензилоксикарбонилгексановая кислота
[Химическая формула 135]
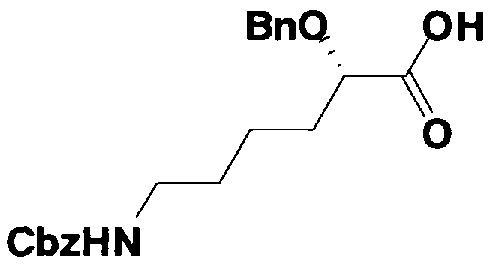
(S)-6-амино-N-бензилоксикарбонил-2-гидроксигексановую кислоту (450 мг), синтезированную в соответствии со способом R.D.Westland et al. (Carbohydr. Res., Vol.28, pp.268-280 (1973)), растворяют в 5,4 мл N,N-диметилформамида. Добавляют при охлаждении льдом оксид бария (1230 мг) и полученную смесь перемешивают в течение 2 минут. Затем к этому добавляют гидроксид бария (1061 мг) и полученную смесь перемешивают далее при той же температуре в течение 10 минут. После этого добавляют 0,57 мл бензилбромида. Через 5 часов после добавления бензилбромида полученную смесь возвращают к комнатной температуре и полученную смесь интенсивно перемешивают в течение 1,5 часа. К этому добавляют воду (15 мл) при охлаждении льдом. Полученную смесь разбавляют 30 мл хлороформа. К смеси добавляют 4 н. HCl (6,0 мл) и полученную смесь перемешивают в течение 5 минут. Реакционный раствор разделяют с помощью фильтрования. Водный слой снова экстрагируют 25 мл хлороформа. Органический слой и полученный выше слой объединяют и сушат над сульфатом магния. Полученный раствор концентрируют при пониженном давлении и полученный остаток очищают, используя хроматографическую колонку с силикагелем (32 мг, гексан:этилацетат=3:1) с получением указанного в заголовке соединения (196 мг, 33%).
Rf значение: 0,47 (хлороформ:этилацетат:уксусная кислота=10:5:1).
1Н-ЯМР (CDCl3): δ 7,29-7,34 (м, 10H), 5,09 (шир., 2H), 4,74 (д, 1H, J=11,7 Гц), 4,46 (д, 1H, J=11,7 Гц), 3,97 (т, 1H, J=5,7 Гц), 3,16 (шир., 2H), 1,77-1,83 (м, 2H), 1,41-1,50 (м, 4H).
Сравнительный пример 5
(R)-2-бензилокси-4-бензилоксикарбониламиномасляная кислота
[Химическая формула 136]
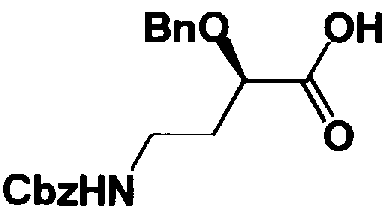
4-бензилоксикарбониламино-2-(R)-гидроксимасляную кислоту (2,245 г), синтезированную в соответствии со способом H.Naito et al. (J. Antibiot, Vol.26, pp.297-301 (1973)), растворяют в 25,6 мл N,N-диметилформамида. К раствору добавляют оксид бария (6,795 г) при охлаждении льдом при перемешивании и полученную смесь интенсивно перемешивают в течение 2 минут. К реакционной смеси добавляют октагидрат гидроксида бария (5,593 г) и полученную смесь интенсивно перемешивают при охлаждении льдом в течение 10 минут. К этому добавляют бензилбромид (3,16 мл) и полученную смесь интенсивно перемешивают при охлаждении льдом в течение 5 минут. Реакционную смесь возвращают к комнатной температуре и интенсивно перемешивают в течение 1,5 часа. Добавляют воду (0,48 мл) при охлаждении льдом и полученную смесь разбавляют 70 мл хлороформа. К разбавленному раствору добавляют 4 M хлористоводородную кислоту (31 мл). Затем к этому добавляют воду (11 мл) с последующим разделением. Водный слой экстрагируют 22 мл+10 мл хлороформа. Объединенные органические слои промывают 30 мл насыщенного солевого раствора, сушат над сульфатом магния и концентрируют досуха с получением 4,54 г сырого продукта. Полученный продукт очищают, используя хроматографическую колонку с 70 г нейтрального силикагеля (хлороформ→хлороформ:этилацетат=4:1→хлороформ:метанол=10:1→хлороформ:метанол-уксусная кислота=10:1:0,1) с получением указанного в заголовке соединения (1,107 г, 36%).
Rf значение: 0,57 (хлороформ:этилацетат:уксусная кислота=10:5:1).
ESIMS: m/z 342 [M-H]-.
1Н-ЯМР (CDCl3): δ 7,25-7,40 (10H), 5,06 (с, 2H), 4,94 (шир.с, 1H), 4,40-4,76 (АВкв, 2H, Jgem=11 Гц), 4,06 (т, 1H, J=5,5 5,5 Гц), 3,35 (м, 2H), 2,02 (м, 2H).
Тестовый пример 1
Противомикробная активность
МИК соединения, представленного формулой (I), полученного в примере 1, против MRSA штамма, специфически и клинически выделенного MRSA штамма (n=9), который обладает низкой восприимчивостью к арбекацину и против которого минимальная ингибирующая концентрация (МИК, мкг/мл) составляет от 4 до 8, определяют, используя метод разбавления на агарных планшетах в соответствии со стандартным методом Japan Society of Chemotherapy (Chemotherapy, Vol.29, pp.76 to 79, 1981).
В результате указанное соединение, представленное формулой (I), имеет значение МИК от 0,5 до 2. Указанное соединение, представленное формулой (I), обладает более высокой противомикробной активностью, чем арбекацин, против вышеуказанного MRSA штамма, обладающего низкой чувствительностью к арбекацину.
Тестовый пример 2
Противомикробная активность
МИК 2-гидроксиарбекацинов, полученных в примерах 1-4, и соединений, полученных в примерах 5, 6, 8, 9, 10 и 11, против MRSA штамма, специфически и клинически выделенного MRSA штамма, который отличается от штамма, использованного в тестовом примере 1 и который обладает высокой устойчивостью в отношении арбекацина и против которого величина МИК (мкг/мл) составляет 128, измеряют, используя способ разбавления на агарных планшетах в соответствии со стандартным способом, раскрытым в Japan Society of Chemotherapy (Chemotherapy, Vol.29, pp.76 to 79, 1981).
В результате оказалось, что указанные соединения имеют величину МИК от 2 до 32, что указывает на то, что указанные соединения, представленные формулой (I), обладают более высокой противомикробной активностью, чем арбекацин, против MRSA штамма, демонстрирующего высокую устойчивость к арбекацину.
Тестовый пример 3
Оценка воздействия на почки нормальных мышей
Систему оценки воздействия на почки создают на основании препаративного метода в модели почечного заболевания, используя гентамицин (KIDNEY и DIALYSIS, Vol.31, 1991, дополнительный тираж, "Renal Disease Model", p.423 "Drug-induced Renal Damage and its Examination Method", Kidney и Dialysis Editorial Committee, опубликованный TOKYOIGAKUSYA). Воздействие соединения, представленного формулой (I), полученного в примере 1, на почки оценивают, используя указанную оценочную систему. В качестве контрольных групп используют группу, которой вводят физиологический солевой раствор, и группу, которой вводят арбекацин. В каждой из групп по четыре мыши (Crj: CD-I(ICR) восьминедельные самки мышей).
В представленной выше системе оценок осуществляют оценку нефротоксичности, используя NAG в качестве показателя. NAG существует в лизосоме в каждой из тканей человека и животных и представляет собой фермент, который превращает мукополисахарид в гликопротеин, например превращает β-N-ацетил-D-глюкозаминид в β-N-ацетил-D-глюкозамин, и в избытке присутствует в эпителиальных клетках в проксимальном мочевом канальце почек, в котором особенно и накапливаются аминогликозидные антибиотики. Если повреждены почки и особенно их проксимальные мочевые канальцы, NAG выделяется в мочу для повышения количества NAG в моче. Соответственно считают, что повышение количества NAG в моче свидетельствует о повреждении проксимальных мочевых канальцев, и используют этот показатель в качестве одного из биохимических маркеров для оценки уровня почечной недостаточности в клинических тестах. Количество NAG определяют исследовательским методом, например способом MCP-NAG (Outline of Latest Internal Medicine, Vol.4, Clinical Laboratory - Kensa No Susumekata To Deta No Yomikata (How To Forward Examination and How To Read Data) - <Naika Soron 4 (General Remarks Of Internal Medicine 4> p.274, Imura et al. (ed.), опубликованным Nakayama Syoten Co., Ltd., 1994).
Раствор физиологического солевого раствора соединения в соответствии с настоящим изобретением с концентрацией 12 мг/мл вводят мышам в каждой группе в дозе 120 мг/кг/день (в двух раздельных дозах; утром и после полудня) в течение 4 дней путем повторного внутрибрюшинного введения. Выделенную естественным образом мочу собирают в течение около 24 часов после окончания введения утром на 4 день, и количество NAG в моче измеряют методом MCP-NAG. Точно такую же обработку осуществляют для группы, которой вводят физиологический солевой раствор, и для группы, которой вводят арбекацин.
Полученные результаты, представленные в таблице 1, демонстрируют средние значения количества NAG для каждой группы.
Как видно из таблицы 1, количество NAG в моче для группы, которой вводят указанное соединение, представленное формулой (I), меньше, чем количество NAG для группы, которой вводят арбекацин.
Сравнительный тестовый пример 1
Воздействие арбекацина и аналога арбекацина TS2037 (5,4"-диэпиарбекацин), полученного в соответствии с описанием WO 2005/070945, на почки оценивают тем же способом, что и в тестовом примере 3. В результате количество NAG в моче для группы, которой вводят TS2037, составляет 358,8 (мМЕ), тогда как количество NAG в моче для группы, которой вводят арбекацин, составляет 168,5 (мМЕ). То есть количество NAG для аналога арбекацина TS2037 оказалось больше, чем количество NAG для арбекацина.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ ИНГИБИРОВАНИЯ РОСТА ВИРУСОВ И/ИЛИ ВИРУЛИЦИДНЫЙ СПОСОБ И НОВЫЙ АНАЛОГ ПИРАЗИННУКЛЕОТИДА ИЛИ ПИРАЗИННУКЛЕОЗИДА | 2002 |
|
RU2292894C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 7Н-ПИРРОЛО[2,3-D] ПИРИМИДИНА И ЕГО ИНТЕРМЕДИАТА | 2016 |
|
RU2755618C2 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| ПРОИЗВОДНЫЕ АМИНОКИСЛОТЫ | 1994 |
|
RU2127261C1 |
| ПИРРОЛО[2,3-D]ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2712269C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2018 |
|
RU2780892C2 |
| НОВОЕ ПРОИЗВОДНОЕ АМИНА ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2668550C2 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2014 |
|
RU2678327C2 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2667060C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БЕНЗОЛА ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2276137C2 |
Раскрыт новый аминогликозидный антибиотик, который обладает превосходной противобактериальной активностью против вызывающей инфекцию бактерии, в частности MRSA, и также обладает низкой токсичностью в отношении почек. Также описан способ получения указанного антибиотика. Более конкретно, раскрыты соединение, представленное формулой (Iа), где значения радикалов указаны в описании, или его фармакологически приемлемая соль или сольват, или смесь диастереомеров соединения; противобактериальный агент, включающий соединение, фармакологически приемлемую соль, сольват или диастереомерную смесь; и способ получения указанного соединения, 10 н. и 13 з.п. ф-лы, 21 пр., 1 табл.
1. Соединение, представленное формулой (Iа) или его фармакологически приемлемая соль или сольват, или их диастереомерная смесь относительно атома углерода, отмеченного *
химическая формула 1
где
R5ах и R5eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил,
R4”ах и R4”eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил,
n является целым числом от 1 до 4, и
пространственная конфигурация атома углерода, отмеченного *, соответствует R или S.
2. Соединение по п.1, или его фармакологически приемлемая соль или сольват, или их диастереомерная смесь относительно атома углерода, отмеченного *, где
R5ax и R5eq, которые являются различными, представляют собой атом водорода или гидроксил, и
R4”ах и R4”eq, которые являются различными, представляют собой атом водорода или гидроксил.
3. Соединение по п.2, или его фармакологически приемлемая соль или сольват, или их диастереомерная смесь относительно атома углерода, отмеченного *, где
R5ax представляет собой атом водорода и
R5eq представляет собой гидроксил.
4. Соединение по п.2, или его фармакологически приемлемая соль или сольват, или их диастереомерная смесь относительно атома углерода, отмеченного *, где
R5ax представляет собой гидроксил и
R5eq представляет собой атом водорода.
5. Соединение, представленное формулой (I), или его фармакологически приемлемая соль или сольват:
химическая формула 2
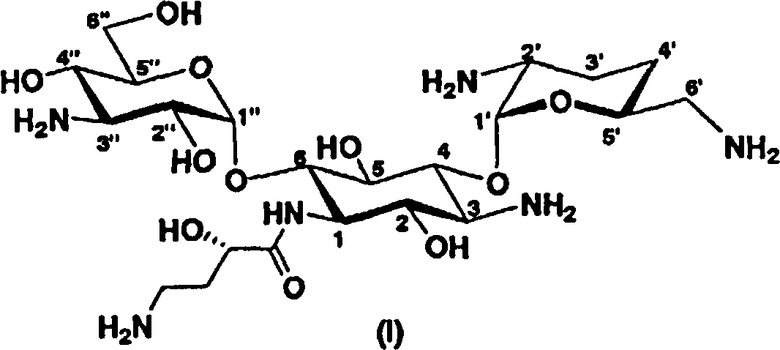
6. Соединение, представленное формулой (Ха):
химическая формула 3
где R5ax и R5eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил.
7. Соединение, представленное формулой (XXV):
химическая формула 4
где R2 представляет собой необязательно замещенный фенил-С1-3алкил,
R3, R2' и R6', которые могут быть одинаковыми или различными, представляют собой необязательно замещенный С1-6 алкилсульфонил,
необязательно замещенный фенилсульфонил или
необязательно замещенный С1-6 алкилоксикарбонил,
Е представляет собой необязательно замещенный С1-6 алкилоксикарбонил и
G представляет собой атом водорода,
необязательно замещенный С1-6 алкилкарбонил,
необязательно замещенный фенилкарбонил или
необязательно замещенный фенилС1-3алкил,
n является целым числом от 1 до 4, и
пространственная конфигурация атома углерода, отмеченного *, соответствует R или S,
или смесь его диастереомеров относительно атома углерода, отмеченного *.
8. Соединение по п.7, или смесь его диастереомеров относительно атома углерода, отмеченного *, где
R2 представляет собой фенилС1-3алкил, необязательно замещенный метокси или нитро,
R3, R2' и R6', которые могут быть одинаковыми или различными, представляют собой
С1-6 алкилсульфонил, необязательно замещенный необязательно замещенным фенилом, фенилсульфонил, необязательно замещенный метилом, или С1-6 алкилоксикарбонил, необязательно замещенный необязательно замещенным фенилом,
Е представляет собой С1-6 алкилоксикарбонил, необязательно замещенный необязательно замещенным фенилом, и
G представляет собой атом водорода, С1-6 алкилкарбонил, фенилкарбонил, фенилС1-3алкил, необязательно замещенный метокси.
9. Соединение по п.7, или смесь его диастереомеров относительно атома углерода, отмеченного *, где
R2 представляет собой бензил, метоксибензил или нитробензил,
R3, R2' и R6', которые могут быть одинаковыми или различными, представляют собой метансульфонил, бензилсульфонил, п-толуолсульфонил, бензилоксикарбонил, трет-бутоксикарбонил, п-метоксибензилоксикарбонил или п-нитробензилоксикарбонил,
Е представляет собой бензилоксикарбонил и
G представляет собой атом водорода, ацетил, бензоил, бензил, п-метоксибензил или трифенилметил.
10. Соединение, представленное формулой (Xb):
химическая формула 5
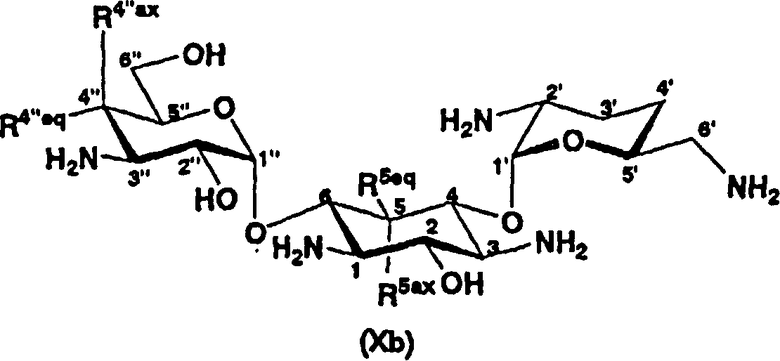
где R5ax и R5eq, которые могут являются различными и представляют собой атом водорода или гидроксил, и
R4”ах и R4”eq, которые являются различными, представляют собой атом водорода или гидроксил.
11. Соединение, представленное формулой (XIV):
химическая формула 6
12. Способ получения соединения, представленного формулой (Iа):
химическая формула 7
где R5ax и R5eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил,
R4”ах и R4”eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил,
n является целым числом от 1 до 4 и
пространственная конфигурация атома углерода, отмеченного *,
соответствует R или S,
включающий стадии
введения защитных групп в аминогруппы в соединении, представленном формулой (Ха):
химическая формула 8
где R5ax и R5eq имеют указанные для формулы (Iа) значения,
осуществления взаимодействия соединения, представленного формулой (Ха), с соединением, представленным формулой (Хс):
химическая формула 9
где W представляет собой отщепляемую группу; Yax и Yeq, которые могут быть одинаковыми или различными, представляют
собой группу -OR4” или атом водорода; R2”, R4” и R6” представляют собой защитную группу для гидроксила; и пространственная конфигурация атома углерода, отмеченного *, соответствует R или S,
удаления защитной группы у полученного соединения и превращения азидогруппы в указанном соединении в аминогруппу с получением соединения, представленного формулой (Хb):
химическая формула 10
где R5ax, R5eq, R4”ах и R4”eq имеют указанные для формулы (Iа) значения,
необязательного введения защитной группы по функциональным группам, отличающимся от аминогруппы в 1-положении соединения, представленного формулой (Xb),
осуществления взаимодействия полученного соединения с соединением, представленным формулой (XVII):
химическая формула 11
где Е представляет собой защитную группу для аминогруппы; G представляет собой защитную группу для гидроксильной группы; F представляет собой атом водорода или активирующую группу для карбоновой кислоты; n является целым числом от 1 до 4; и пространственная конфигурация атома углерода, отмеченного *, соответствует R или S, и
удаления защитной группы у полученного соединения с получением указанного соединения, представленного формулой (Iа).
13. Способ по п.12, где
R5ax и R5eq, которые являются различными, представляют собой атом водорода или гидроксил, и
R4”ах и R4”eq, которые являются различными, представляют собой атом водорода или гидроксил.
14. Способ по п.12, где
R5ax представляет собой атом водорода, и
R5eq представляет собой гидроксил.
15. Способ по п.12, где
R5ax представляет собой гидроксил, и
R5eq представляет собой атом водорода.
16. Способ получения соединения, представленного формулой (Iа):
химическая формула 12
где R5ax представляет собой атом водорода,
R5eq представляет собой гидроксил,
R4”ах и R4”eq, которые могут быть одинаковыми или различными, представляют собой атом водорода или гидроксил,
n является целым числом от 1 до 4, и
пространственная конфигурация атома углерода, отмеченного *, соответствует R или S,
включающий стадии
введения защитных групп по аминогруппам в 3-, 2'- и 6'-положениях и по гидроксильной группе в 2-положении соединения, представленного формулой (Ха):
химическая формула 13
где R5ax и R5eq имеют указанные для формулы (Iа) значения, осуществления взаимодействия полученного соединения с соединением, представленным формулой (XVII):
химическая формула 14
где Е представляет собой защитную группу для аминогруппы; G представляет собой защитную группу для гидроксильной группы; F представляет собой атом водорода или активирующую группу для карбоновой кислоты; n является целым числом от 1 до 4; и пространственная конфигурация атома углерода, отмеченного *, соответствует R или S,
с получением соединения, представленного формулой (XXV):
химическая формула 15
где R2 представляет собой защитную группу для гидроксильной группы; R3, R2' и R6' представляют собой защитную группу для аминогруппы; и Е, G, n и пространственная конфигурация атома углерода, отмеченного *, имеют указанные для формулы (XVII) значения,
осуществление взаимодействия указанного соединения, представленного формулой (XXV), с соединением, представленным формулой (Хс) или (Xd):
химическая формула 16
где W представляет собой отщепляемую группу; Yax и Yeq, которые могут быть одинаковыми или различными, представляют собой группу -OR4" или атом водорода; R2”, R4” и R6” представляют собой защитную группу для гидроксильной группы; и пространственная конфигурация атома углерода, отмеченного *, соответствует R или S,
химическая формула 17
где W, Yax и Yeq, R2”, R6” и пространственная конфигурация атома углерода, отмеченного *, имеют указанные для формулы (Хс) значения, и R3” представляет собой защитную группу для аминогруппы, и
удаления защитных групп у полученного соединения и, если используют соединение, представленное формулой (Хс), превращения азидогруппы указанного соединения в аминогруппу, с получением соединения, представленного формулой (Iа).
17. Способ по п.14 или 16, где соединение, представленное формулой (Ха), где R5ax представляет собой атом водорода, и R5eq представляет собой гидроксил, получают путем гидролиза соединения, представленного формулой (II):
химическая формула 18
18. Способ по п.15, где соединение, представленное формулой (Ха), где R5ax представляет собой гидроксил, и R5eq представляет собой атом водорода, получают путем введения защитных групп по гидроксильным группам, отличающимся от групп в 4"- и 5-положениях, и по аминогруппам в соединении, представленном формулой (II):
химическая формула 19
обращения пространственной конфигурации гидроксильной группы в 5-положении полученного соединения и
удаления защитной группы у полученного соединения, и последующего гидролиза указанного соединения с получением соединения, представленного формулой (Ха), где R5ax представляет собой гидроксил, и R5eq представляет собой атом водорода.
19. Способ по п.18, который дополнительно включает до или одновременно с обращением пространственной конфигурации гидроксильной группы в 5-положении удаление гидроксильной группы в 4”-положении.
20. Противомикробная фармацевтическая композиция, содержащая соединение по п.1 или 5, или его фармакологически приемлемую соль или сольват, или их диастереомерную смесь относительно атома углерода, отмеченного *, и фармацевтически приемлемую добавку.
21. Фармацевтическая композиция по п.20, где противомикробная фармацевтическая композиция является анти-MRSA фармацевтической композицией.
22. Применение соединения по п.1 или 5, или его фармакологически приемлемой соли или сольвата, или их диастереомерной смеси относительно атома углерода, отмеченного *, для получения противомикробной фармацевтической композиции.
23. Применение соединения по п.22, где противомикробная фармацевтическая композиция является анти-MRSA фармацевтической композицией.
| US 4107424 А, 15.08.1978 | |||
| JP 7252290 А, 03.10.1995 | |||
| US 4410516 А, 18.10.1983 | |||
| WO 2005070945 A1, 04.08.2005 | |||
| JP 56020598 A, 26.02.1981 | |||
| Способ получения 1N-( @ -окси- @ -аминоалканоил)-6N-метил-3,4-дидеоксиканамицина В | 1975 |
|
SU965359A3 |
| Способ получения 3,4дидезоксиканамицина в | 1971 |
|
SU511007A3 |
| US 3982996 A, 28.09.1976 | |||
| US 4455419 A, 19.06.1984. | |||

