Изобретение относится к биотехнологии, а именно к созданию мыши, предназначенной для моделирования состояний, при которых существует риск развития синдрома внезапной сердечной смерти, и может быть использовано для выявления групп риска развития синдрома внезапной сердечной смерти, выявления его электрофизиологических, биохимических, иммунологических предикторов и разработки новых медицинских технологий профилактики и лечения.
В клинической практике внезапную сердечную смерть определяют как циркуляторный коллапс, вызывающий летальный исход в течение часа после появления первых симптомов и обусловленный чаще всего аритмиями сердца. В общей популяции внезапная сердечная смерть - явление достаточно редкое, встречающееся с частотой 0,1-0,2%. В структуре общей смертности на долю внезапной сердечной смерти приходится около 10%.
В настоящее время моделирование синдрома внезапной сердечной смерти на мелких лабораторных животных подразумевает преимущественно получение методами генной инженерии мышей, склонных к развитию структурных и функциональных изменений сердца и электрической нестабильности миокарда, на фоне которых развиваются угрожающие жизни нарушения сердечного ритма.
Известна трансгенная мышь, у которой на фоне гиперэкспрессии в сердце индуцибельной NO-синтетазы (iNOS) достаточно часто развивается аритмия, вызывающая внезапную смерть животных (Mungrue IN, Gros R, You X, et al. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J Clin Invest. 2002, v.109, N6, 735-43).
Способ получения таких мышей предполагает интегрирование в их геном двух трансгенов: человеческой iNOS и промотора гена легких цепей альфа-миозина (αМНС) вместе с реагирующим на тетрациклин активатором транскрипции (tTA). Второй трансген (αMtTA) необходим для того, чтобы обеспечить тканеспецифичную экспрессию продуктов первого трансгена в кардиомиоцитах.
Процесс достижения поставленной цели включает несколько основных этапов:
а) встраивание основного трансгена (ген полноразмерной iNOS человека) в бактериальную плазмиду и молекулярное клонирование рекомбинантной плазмиды в клетках непатогенного штамма дрожжей;
б) введение трансгена в пронуклеусы оплодотворенной яйцеклетки мышей с помощью специального манипулятора для получения в достаточном количестве модифицированных яйцеклеток;
в) перенос жизнеспособных модифицированных яйцеклеток в репродуктивные органы самки-реципиента;
г) скрининг потомства самок-реципиентов модифицированных яйцеклеток для селекции особей, несущих в геноме трансген iNOS;
д) введение в геном iNOS+-мышей второго (дополнительного) трансгена путем их скрещивания с αMtTA+-мышами.
Первым недостатком данного способа является то, что он подразумевает проведение ряда высокотехнологичных процедур, включая манипуляции с молекулами нуклеиновых кислот in vitro и перенос созданных конструкций генов в живой организм. В число таких процедур входит получение плазмидного вектора со встроенной в него человеческой ДНК и маркерным геном β-галактозидазы, клонирование рекомбинантной ДНК в микробных клетках, введение клонированного гена с помощью микропипеток и специального манипулятора в пронуклеусы извлеченных из яйцеводов самок оплодотворенных яйцеклеток, анализ ДНК мышей методами PCR и Саузерн-блот гибридизации. Эти процедуры являются дорогостоящими, длительными, требуют соответствующего оборудования и специальных знаний и умений исследователя в области биотехнологии и генной инженерии.
Второй недостаток связан с тем, что конечная эффективность трансгенеза лимитирована сегодняшним уровнем развития технологии переноса трансгена в яйцеклетки, поскольку количество яйцеклеток, в которых интеграция трансгена в геном и его экспрессия в тканях носят стабильный характер, а также количество мышей, развивающихся из трансформированной яйцеклетки, невелико.
Третий недостаток обусловлен тем, что гиперэкспрессия у мышей человеческого трансгена iNOS ассоциирована со значительной смертностью трансгенных (iNOS+/αMtTA+) мышей в эмбриональном периоде.
Четвертый недостаток обусловлен тем, что для компенсации отмеченной выше внутриутробной гибели трансгенных мышей в водный рацион беременных самок вводят доксициклин, временно "выключая" экспрессию iNOS трансгена. Длительное потребление беременными самками мышей антибиотика может оказывать неучтенное влияние на их потомство и привносить ограничение в работу с ними, например, в области исследования иммунной системы, на которую малые дозы антибиотиков оказывают супрессорный эффект.
Известны трансгенные mdx мыши, у самок которых синдром внезапной сердечной смерти манифестирует во время беременности или вскоре после родов (Elsherif L, Huang MS, Shai SY, Yang Y, Li RY, Chun J, Mekany MA, Chu AL, Kaufman SJ, Ross RS. Combined deficiency of dystrophin and betal integrin in the cardiac myocyte causes myocardial dysfunction, fibrosis and calcification. Circ Res. 2008; v.102, N9: 1109-17).
Способ получения таких мышей предполагает дополнительную хромосомную перестройку в геноме мышей mdx, затрагивающую ген бета 1-интегрина. Способ осуществляют путем скрещивания мышей mdx c мышами, у которых вызывают делецию в гене бета1-интегрина.
Мышей с делецией в гене бета1-интегрина получают путем программируемого нокаута генов, основанного на использовании известных в генной инженерии технологий - гомологичной и сайт-специфичной рекомбинаций ДНК. Получение нокаутированых по гену бета1-интегрина мышей включает несколько основных этапов:
а) создание векторной конструкции, несущей LoxP сайты, фланкирующие подлежащую удалению последовательность гена бета1-интегрина;
б) введение сконструированного вектора методом электропорации в эмбриональные стволовые клетки мыши и отбор среди них клеток, в которых гомологичная рекомбинация чужеродной и клеточной ДНК была успешной;
в) повторную электропорацию и интегрирование в геном отобранных стволовых клеток последовательности гена Cre-рекомбиназы под контролем тканеспецифичного или индуцибельного промотора;
г) отбор линии стволовых клеток с инактивированным геном бета1-интегрина;
д) введение отобранных на предыдущем этапе стволовых клеток в бластоцисты и имплантацию химерных зародышей в матку ложно беременных самок;
е) скрининг потомков химерных мышей и получение особей, гомозиготных по мутации в гене бета1-интегрина;
ж) получение путем скрещивания нокаутированных мышей и MLC2v мышей особей с селективной инактивацией в сердце гена бета1-интегрина.
Необходимость специального дорогостоящего оборудования, культур эмбриональных стволовых клеток, дополнительных линий мышей, а также потребность в квалифицированных сотрудниках, владеющих методами генной инженерии, является недостатком, общим для данного способа и способа, описанного в первом примере.
Самки нокаутированных мышей гибнут в течение беременности или вскоре после родов на фоне ассоциированных с беременностью изменений сердечной гемодинамики. Это обстоятельство осложняет воспроизводство нокаутированных мышей, что является вторым недостатком данного способа.
Данный способ получения генетически модифицированных мышей был выбран нами в качестве прототипа.
Нами была поставлена задача разработки мышиной модели состояний, при которых существует риск развития синдрома внезапной сердечной смерти.
Технический результат осуществления предложенного способа состоит в создании модели состояний, угрожающих развитием синдрома внезапной сердечной смерти, на основе легко поддерживаемой в лабораторных условиях новой генетической разновидности страдающих дистрофин-дефицитной кардиомиопатией мышей mdx, не требует для своего получения сложных биотехнологических манипуляций и позволяет длительно мониторировать состояние здоровья модельных животных в естественных для них условиях.
Сущность изобретения. Получены трансгенные мыши, предназначенные для моделирования состояний, угрожающих развитием синдрома внезапной сердечной смерти при дистрофин-дефицитной кардиомиопатии, имеющие мутацию в экзоне 23 гена мышечного белка дистрофина, расположенного на Х хромосоме, и генетический дефект, ответственный за проявление фенотипических признаков кожно-окулярного альбинизма с отсутствием расщепления по окрасу шерсти в репродуктивной колонии.
Для получении модельных животных в геном мышей mdx необходимо включить вторую мутацию, находящуюся, как и мутация в гене дисторофина, в гомозиготном состоянии.
Для этого используют оптимальную для выведения дистрофин-дефицитных mdx-альбиносов схему скрещивания, при которой не теряется мутация в гене дистрофина, а ген/гены, ответственные за появление фенотипических признаков кожно-окулярного альбинизма, переходят в гомозиготное состояние.
В лабораторных условиях требуется наличие небольшой колонии черных mdx мышей. При скрещивании используют гомозитных черных самок mdx/mdx и самцов-альбиносов (X/Y), мутация в гене дистрофина у которых отсутствует. Потомки от этого скрещивания (F1), гетерозиготы, имеют черный окрас, несут мутацию в гене дистрофина и гене/генах, ответственных за отсутствие пигментации кожных покровов. Второе поколение (F2), получаемое при последующих скрещиваниях гомозиготных черных самок с самцами F1, имеет черный окрас и несет мутацию в гене дистрофина в гомозиготном состоянии. Ген, ответственный за отсутствие пигментации кожных покровов, находится у таких мышей в гетерозиготном состоянии. Среди потомков от скрещивания между собой самок и самцов второго поколения (F3) начинается выщепление белых особей, несущих мутацию в гене дистрофина и мутацию в "гене альбинизма" в гомозиготном состоянии.
В силу такой особенности полученных белых особей, как отсутствие расщепления по окраске, их можно использовать для создания репродуктивной колонии mdx-альбиносов.
Получение мышей, предназначенных для моделирования состояний, угрожающих развитием синдрома внезапной сердечной смерти при дистрофин-дефицитной кардиомиопатии, требует соблюдения условий, направленных на обеспечение их повышенной жизнестойкости путем использования пищевого рациона с содержанием белка не менее 24%. Для достижения указанного условия может быть использован, например, полнорационный экструдированный комбикорм марки Премиум.
Контроль генетической принадлежности mdx-альбиносов осуществляют путем регулярной ДНК-диагностики наличия мутации в экзоне 23 гена дистрофина. Объектом анализа служат образцы ДНК, выделяемой из кончика хвоста мышей. Дополнительными критериями наличия данной мутации являются два фенотипических признака: а) высокий уровень активности (от 2000 до 10000 и более ЕД) креатинкиназы сыворотки крови; б) отсутствие дистрофина в мышечных волокнах, подтверждаемое при гистохимическом анализе срезов скелетных мышц и сердца (дистрофин-дефицитные мыши).
Отличительной характеристикой мышей является длительность периода восстановления после минимальных нагрузок, связанных с проведением малотравматичных для мышей манипуляций: осмотра животного, записи ЭКГ, взятия крови путем венесекции и пр. В течение этого периода, продолжающегося от 15 до 30 и более минут, возвращенные в клетку мыши неподвижно лежат на ее дне. У части из них без видимых предвестников может наступить внезапная сердечная смерть, подтверждаемая данными посмертного исследования.
Предлагаемый способ реализован в представленном ниже протоколе исследования иммунологических предикторов синдрома внезапной сердечной смерти на модели mdx-альбиносов.
11.08.10 у шести самцов в возрасте 1,5 лет (N1, N2, N3, N4, N5, N6) из репродуктивной колонии mdx-альбиносов (клетка Т2-39) и двух контрольных самцов (N7, N8) того же возраста (клетка Т1-67) провели электрокардиографический анализ сердечной деятельности и взяли 100 мкл крови из хвостовой вены для иммунологического исследования.
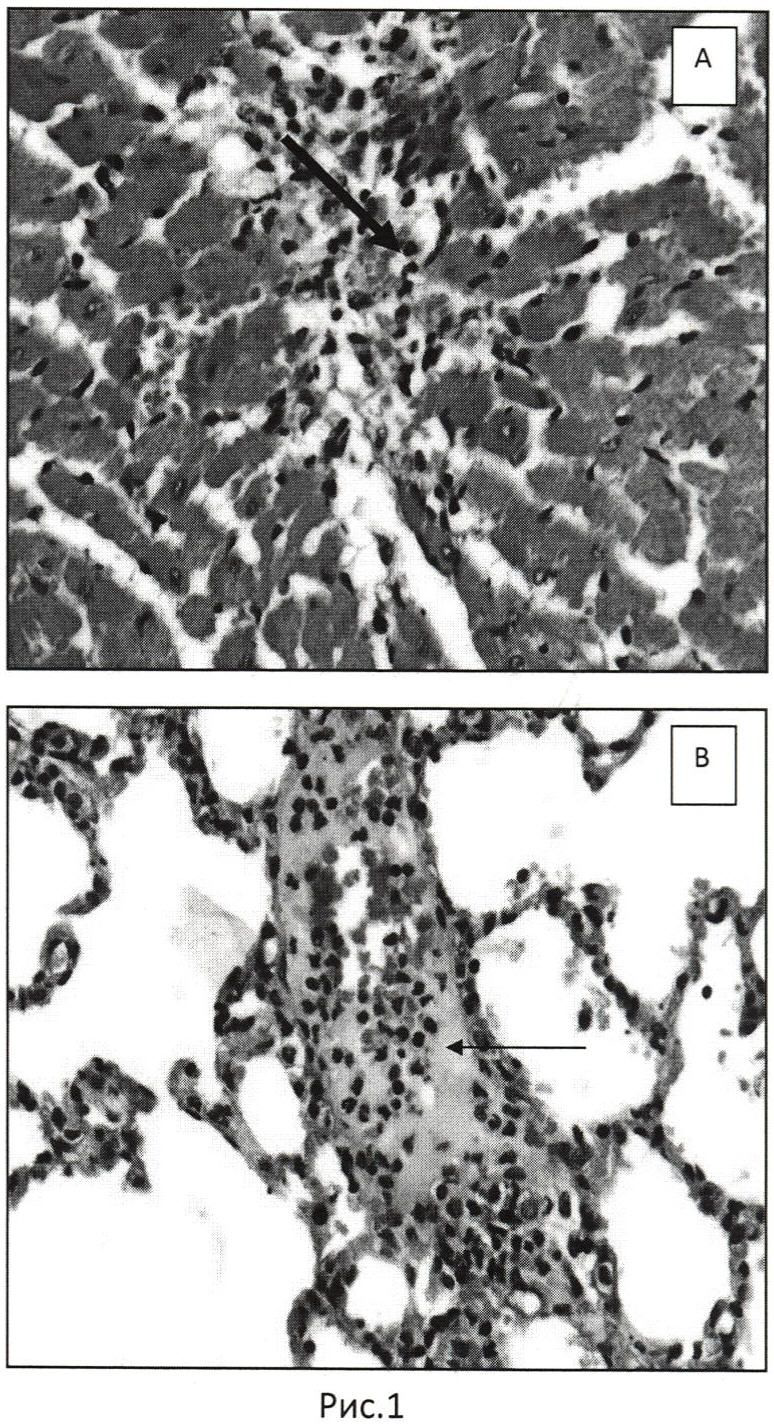
Контрольные самцы, возвращенные в клетку после проведенных с ними манипуляций, сразу же начинали активное передвижение по ней. Mdx-альбиносам для восстановления двигательной активности понадобилось примерно 20 минут, в течение которых мыши неподвижно лежали на дне клетки. Один из шести дистрофин-дефицитных самцов (N2) внезапно, без каких-либо предвестников, умер. Посмертное исследование обнаружило признаки острой сердечной недостаточности, развившейся на фоне органического поражения миокарда (кардиомиопатии) в виде многочисленных мелких очагов некроза кардиомиоцитов лейкоцитарной реакцией по периферии и очагов продуктивного воспаления (рис.1А). В гистологических препаратах легких обнаружено острое полнокровие и отек легочной ткани (рис.1В).
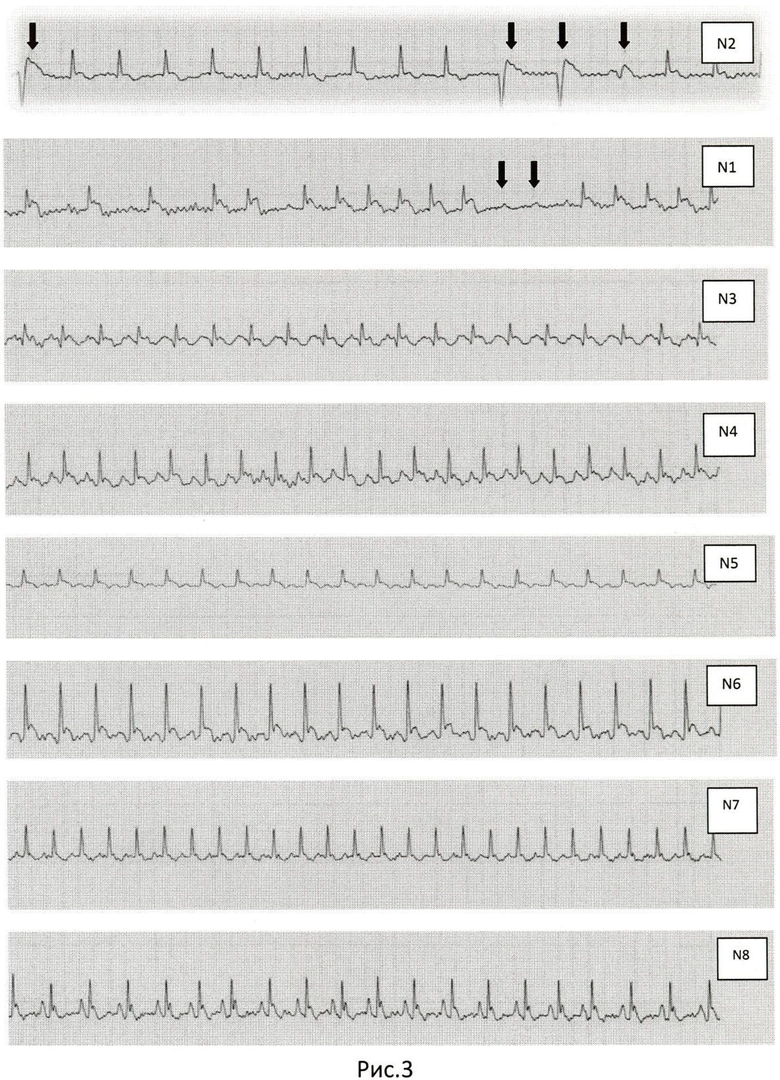
Согласно данным электрокардиографического анализа все шесть mdx-альбиносов имели риск внезапной аритмической смерти в силу наличия у них такого маркера электрической нестабильности миокарда, как удлинение QT интервала (рис.2). На момент записи ЭКГ отчетливые прогностически неблагоприятные нарушения сердечного ритма обнаружены у двух мышей.
У самца N2, у которого реализовался риск внезапной сердечной смерти, на записанной незадолго до нее электрокардиограмме (рис.3), выявлена экстрасистолия высоких градаций (по Лауну и Вольфу), угрожающая трансформацией в фибрилляцию желудочков, несущую непосредственную угрозу жизни.
У самца N1 имела место значимая для течения и прогноза кардиомиопатии атриовентрикулярная блокада 2 степени с длительными периодами асистолии (рис.3).
У двух самцов с дистрофин-дефицитной кардиомиопатией (N5 и N6 на рис.3) зарегистрирован такой предиктор внезапной сердечной смерти, как очень низкая вариабельность сердечного ритма (величина SDNN составляла 0,71 и 0,79 мс по сравнению с 6 мс и 1,4 мс в контроле).
Mdx-альбиносы, оставшиеся на данный момент в живых, образовали группу длительного наблюдения за состоянием сердечно-сосудистой и иммунной систем, в частности для выявления возможных иммунных предикторов внезапной сердечной смерти.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения мышиной модели для изучения синдрома Леша-Нихена путем внесения делеции p.Val8del в ген hprt1 | 2021 |
|
RU2768048C1 |
| Способ получения мышиной модели для изучения миодистрофии Дюшенна и вариантов ее терапии | 2023 |
|
RU2815936C1 |
| ТРАНСГЕННЫЕ МЫШИ, ЭКСПРЕССИРУЮЩИЕ ЛИПОПРОТЕИН (А) ЧЕЛОВЕКА С ОТКЛЮЧЕННЫМ ГЕНОМ ВИТАМИНА С, И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДЕЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2721254C2 |
| Способ получения линии гуманизированных мышей, содержащих инсерцию 3974insT в гене mGrin2a (mice glutamate [NMDA] receptor subunit epsilon-1), приводящую к преждевременному прекращению трансляции белка grin2a | 2021 |
|
RU2764650C1 |
| Способ получения линии гуманизированных мышей, трансгенных по hACE2 | 2020 |
|
RU2757114C1 |
| ЖИВОТНАЯ МОДЕЛЬ, ЭКСПРЕССИРУЮЩАЯ ЛЮЦИФЕРАЗУ ПОД КОНТРОЛЕМ ПРОМОТОРА ОСНОВНОГО БЕЛКА МИЕЛИНА (MBP-LUCI), И ПРИМЕНЕНИЕ МОДЕЛИ ДЛЯ ВИЗУАЛИЗАЦИИ БИОЛЮМИНЕСЦЕНЦИИ IN VIVO | 2010 |
|
RU2601128C2 |
| Линия мышей, трансгенных по альфа-цепи Т-клеточного рецептора клеток памяти, для изучения их функциональной активности | 2017 |
|
RU2691484C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРАНСГЕННОЙ МЫШИ, НЕ СОДЕРЖАЩЕЙ ФУНКЦИОНАЛЬНЫЙ РЕЦЕПТОР-1 РИЛИЗИНГ-ФАКТОРА КОРТИКОТРОПИНА, СПОСОБ ИДЕНТИФИКАЦИИ АГОНИСТА ИЛИ АНТАГОНИСТА РИЛИЗИНГ-ФАКТОРА КОРТИКОТРОПИНА, УРОКОРТИНА ИЛИ ЛИГАНДА СЕМЕЙСТВА РИЛИЗИНГ-ФАКТОРА КОРТИКОТРОПИНА И СПОСОБ СКРИНИНГА СОЕДИНЕНИЙ, КОТОРЫЕ ЯВЛЯЮТСЯ АНАЛОГАМИ ИЛИ АГОНИСТАМИ КОРТИКОСТЕРОНА ИЛИ КОРТИКОТРОПИНА, С ИСПОЛЬЗОВАНИЕМ ТАКОЙ МЫШИ | 1999 |
|
RU2236127C2 |
| Способ получения генно-модифицированных лабораторных животных с нуль-аллелем гена P2rx3 | 2022 |
|
RU2805173C1 |
| ИНДУКТОР СЧИТЫВАНИЯ И ТЕРАПЕВТИЧЕСКИЙ АГЕНТ ДЛЯ ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ МУТАЦИЯМИ ТИПА НОНСЕНС-МУТАЦИЙ | 2011 |
|
RU2571060C2 |
Изобретение относится к медицине. Сущность изобретения состоит в получении на основе мышей mdx, генетическим маркером которых является мутация в экзоне 23 гена дистрофина, репродуктивной колонии белых дистрофин-дефицитных мышей. У таких мышей склонность к развитию синдрома внезапной сердечной смерти появляется в результате введения в геном мышей mdx гена/генов, ответственных за развитие кожно-окулярного альбинизма. Изобретение позволяет модулировать заболевания человека на животных и может быть использовано для выявления групп риска внезапной сердечной смерти, выявления ее электрофизиологических, биохимических, иммунологических предикторов и разработки новых медицинских технологий профилактики и лечения. 3 ил.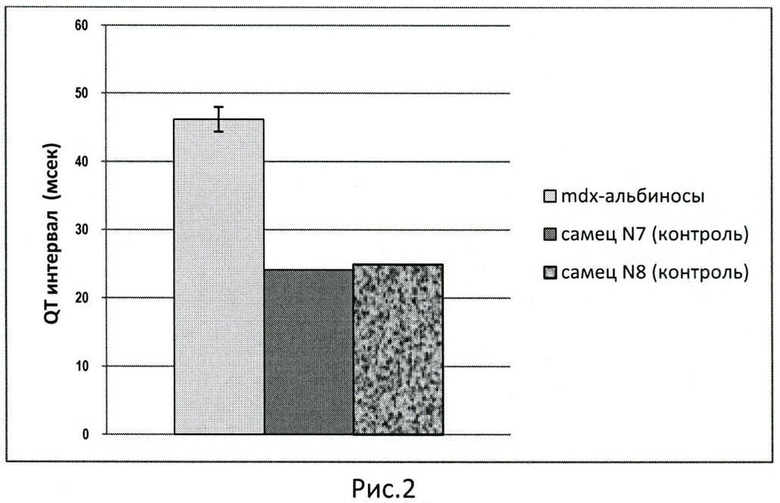
Применение трансгенной мыши, имеющей мутацию в экзоне 23 гена мышечного белка дистрофина, расположенного на X хромосоме, и генетический дефект, ответственный за проявление фенотипических признаков кожно-окулярного альбинизма с отсутствием расщепления по окрасу шерсти в репродуктивной колонии, для моделирования состояний, угрожающих развитием синдрома внезапной сердечной смерти при дистрофин-дефицитной кардиомиопатии.
| АВЕТИСОВ С.Э | |||
| и др | |||
| Экстраокулярные мышцы mdx-мышей как мишень для клеточной терапии: гистологическая характеристика с использованием компьютерного морфометрического анализа | |||
| Клеточная трансплантология и тканевая инженерия, т.3, №3, 2008, с.47-50 | |||
| КРИВОВ Л.И | |||
| и др | |||
| Новая генетическая разновидность MDX мышей: исследование особенностей |

