Область техники, к которой относится изобретение
Настоящее изобретение относится к 2-пиридилзамещенным имидазолам, которые являются ингибиторами рецептора I типа трансформирующего фактора роста-β (TGF-β) (ALK5) и/или рецептора I типа активина (ALK4), способам их получения и их применению в медицине, в особенности при лечении и профилактике болезненного состояния, опосредованного указанными рецепторами.
Уровень техники
TGF-β обозначает семейство белков, TGF-β1, TGF-β2 и TGF-β3, которые являются плейотропными модуляторами клеточной пролиферации и дифференцировки, процесса заживления раны, продукции внеклеточного матрикса и подавления иммунного ответа. Другие представители этого надсемейства включают активины, ингибины, морфогенетические белки костей, факторы роста и дифференцировки и ингибирующее вещество Мюллера.
TGF-β1 передает сигналы через две высококонсервативные одиночные трансмембранные серин/треонинкиназы, рецепторы TGF-β I типа (ALK5) и II типа. После опосредованной лигандом олигомеризации рецептор II типа гиперфосфорилирует сериновые/треониновые остатки в области GS ALK5, что приводит к активации ALK5 путем создания связывающего сайта для белков Smad. Активированный ALK5, в свою очередь, фосфорилирует белки Smad2 и Smad3 на C-концевом SSXS-мотиве, вызывая, тем самым, их диссоциацию от рецептора и образование гетеромерного комплекса со Smad4. Комплексы Smad транслоцируются в ядро, ассоциируются со специфическими ДНК-связывающими кофакторами и комодуляторами, в конечном счете, активируя транскрипцию компонентов внеклеточного матрикса и ингибиторов разрушающих матрикс протеаз.
Активины передают сигналы таким же образом, как и TGF-β. Активины связываются с серин/треонинкиназой, рецептором II типа активина (ActRIIB), и активированный рецептор II типа гиперфосфорилирует серин/треониновые остатки в области GS ALK4. Активированный ALK4, в свою очередь, фосфорилирует Smad2 и Smad3. Последующее образование гетеро-Smad комплекса с Smad4 приводит к индуцированной активином регуляции транскрипции генов.
Многочисленные исследования на экспериментальных животных показали взаимосвязь между гломерулярной экспрессией TGF-β и фиброзом, включая Thy-1 модель пролиферативного гломерулонефрита на крысах, анти-GBM гломерулонефрит на кроликах и модель 5/6 нефрэктомии очагового сегментарного гломерулосклероза на крысах, как сообщалось недавно (например, Bitzer, M. et al., Kidney Blood Press. Res. 21: 1-12 (1998)). Нейтрализация антитела к TGF-β улучшает гистологию клубочковой зоны на Thy-1 модели нефрита (например, Border, W.A. et al., Nature 346: 371-374 (1990)).
Гипергликемические условия увеличивают синтез мРНК TGF-β и белка как в клетках проксимальных канальцев почки мыши, так и в мезангиальных клетках человека (например, Wahab, N.A. et al., Biochem. J. 316: 985-992 (1996); Rocco, M.V. et al., Kidney Int. 41: 107-114 (1992)). У больных диабетом с начальным заболеванием почек обнаруживается повышенное накопление мРНК TGF-β и белка в клубочках (например, Yoshioka, K. et al., Lab. Invest. 68: 154-163 (1993)). В почках с хроническим почечным интерстициальным фиброзом характерными признаками являются утолщение базальных мембран канальцев и увеличенный интерстициальный компартмент с интерстициальным фиброзом, характеризующимся увеличением содержания коллагенов I, III, V, VII и фибронектина (например, Eddy, A.A., J. Am. Soc. Nephrol. 7: 2495-2508 (1996)).
Экспрессия гена TGF-β и продукция белка увеличиваются в различных животных моделях пневмофиброза, включая блеомициновую, кремнеземную, асбестовую и радиационную модели (например, Phan, S.H. and Kunkel, S.L., Exp. Lung Res. 18: 29-43 (1992); Williams, A.O. et al., Am. J. Pathol. 142: 1831-1840 (1993); Rube, C.E. et al., Int. J. Radiat. Oncol. Biol. Phys. 47: 1033-1042 (2000)). Сопутствующее увеличение содержания белка TGF-β1 и экспрессии гена коллагена в срезах прилегающей ткани из идиопатического пневмофиброза наблюдалось при легочном фиброзном заболевании человека (например, Broekelmann, T.J. et al., Proc. Natl. Acad. Sci. USA 88: 6642-6646 (1991)). Повышенная продукция TGF-β описана у больных саркоидозом, пневмокониозом, асбестозом и индуцированным облучением фиброзом (например, Khalil, N. et al., Am. J. Respir. Cell. Mol. Biol. 14: 131-138 (1996); Jagirdar, J. et al., Environ. Health Perspect. 105: 1197-1203 (1997)). Антитела к TGF-β и TGF-β-растворимые рецепторы могли частично ингибировать фиброз на моделях индуцированного блеомицином легочного фиброза у грызунов (например, Giri, S.N. et al., Thorax 48: 959-966 (1993); Wang, Q. et al., Thorax 54: 805-812 (1999)). Полагают, что табачный дым является одним из наиболее важных факторов, которые могут вызывать заболевание мелких дыхательных путей с последующим развитием хронической обструктивной болезни легких (COPD) (например, Wright, J.M. et al., Am. Rev. Respir. Dis. 146: 240-262 (1992)). COPD является медленно прогрессирующим и необратимым заболеванием, характеризующимся функциональным отклонением обструкции дыхательных путей. Была выдвинута гипотеза о том, что TGF-β вовлечен в ремоделирование дыхательных путей, обнаруженное при хронических воспалительных нарушениях дыхательных путей, таких как COPD (например, Takizawa, H. Int. J. Mol. Med. 1: 367-378 (1998); Ning, W. et al., Proc. Natl. Acad. Sci. USA 101: 14895-14900 (2004)).
Звездчатые клетки печени (HSC) являются основным источником белков внеклеточного матрикса при фиброзе печени. Продукция внеклеточного матрикса активированными звездчатыми клетками печени существенно увеличивается при действии TGF-β1 (например, Friedman, S.L., Prog. Liver Dis. 14: 101-130 (1996); Pietrangelo, A., Semin. Liver Dis. 16: 13-30 (1996)). У трансгенных мышей, которые избыточно экспрессируют TGF-β1 в печени, развивается фиброз печени, наряду с такими внепеченочными патологиями, как почечный фиброз (например, Sanderson, N. et al., Proc. Natl. Acad. Sci. USA 92: 2572-2576 (1995)).
TGF-β1 и его рецепторы избыточно экспрессируются в травмированных кровеносных сосудах и в фибропролиферативных сосудистых повреждениях, приводя к избыточной продукции внеклеточного матрикса (например, Saltis, J. et al., Clin. Exp. Pharmacol. Physiol. 23: 193-200 (1996); McCaffrey, T.A. et al., J. Clin. Invest. 96: 2667-2675 (1995)).
Антитела к TGF-β снижают образование рубцов и улучшают цитоархитектуру новых слоев кожи у крыс (например, Shah, M., J. Cell. Sci. 108: 985-1002 (1995)), улучшают заживление ран роговицы у кроликов (например, Moller-Pedersen, T., Curr. Eye Res. 17: 736-747 (1998)) и усиливают заживление ран желудочных язв у крыс (например, Ernst, H., Gut 39: 172-175 (1996)).
Радиационный фиброз является частым следствием терапевтического или случайного чрезмерного воздействия облучения на нормальные ткани человека. Как сообщалось недавно, TGF-β1 играет центральную роль в инициации, развитии и стойкости радиационного фиброза (например, Martin, M. et al., Int. J. Radiat. Oncol. Biol. Phys. 47: 277-290 (2000)).
Трансплантация органов осложняется во многих случаях хроническим отторжением и для некоторых органов, таких как почка, это представляет собой основную форму гибели пересаженного органа. У пациентов-людей хроническое отторжение трансплантатов легкого и почки связано с повышенной экспрессией TGF-β в ткани (например, El-Gamel, A. et al., Eur. J. Cardiothorac. Surg. 13: 424-430 (1998); Shihab, F.S. et al., J. Am. Soc. Nephrol. 6: 286-294 (1995)).
TGF-β участвует в образовании перитонеальных спаек (например, Saed, G.M. et al., Wound Repair Regen. 7: 504-510 (1999)). Образование брюшинных и подкожных фиброзных спаек может быть предотвращено ингибиторами ALK5 и/или ALK4.
Уровни TGF-β2 в водянистой влаге глаз увеличиваются почти в половине глаз с первичной открытоугольной глаукомой (POAG) и в большинстве глаз с ювенильной глаукомой (например, Picht, G. et al., Graefes Arch. Clin. Exp. Ophthalmol. 239: 199-207 (2001)). Сообщалось, что как TGF-β1, так и TGF-β2 изоформы повышают продукцию внеклеточного матрикса в культивируемых фибробластах теноновой капсулы человека, полученных от больных псевдоэксфолиативной глаукомой и POAG (например, Kottler, U.B. et al., Exp. Eye Res. 80: 121-134 (2005)). В патентной заявке США 2007/0142376 A1 раскрыто лечение глаукомы и регулирование внутриглазного давления с использованием модулирующих средств ALK5, и ингибитор ALK5 снижает уровень фибронектина в обработанных TGF-β2 перфузируемых передних отрезках глаза человека и уровни фибронектина, ингибитора активатора плазминогена-1 (PAI-1) и C-пептида проколлагена I типа в обработанных TGF-β2 клеточных культурах трабекулярной сети.
Опухолевые клетки и стромальные клетки в опухолях на последних стадиях различных злокачественных опухолей, как правило, избыточно экспрессируют TGF-β. Это приводит к стимуляции ангиогенеза и клеточной подвижности, подавлению иммунной системы и увеличенному взаимодействию опухолевых клеток с внеклеточным матриксом (например, Hojo, M. et al., Nature 397: 530-534 (1999)). Вследствие этого, опухолевые клетки становятся более инвазивными и метастазируют в отдаленные органы (например, Maehara, Y. et al., J. Clin. Oncol. 17: 607-614 (1999); Picon, A. et al., Cancer Epidemiol. Biomarkers Prev. 7: 497-504 (1998)).
PAI-1 представляет собой основной физиологический ингибитор как активатора плазминогена тканевого типа, так и активатора плазминогена урокиназного типа. Повышенные уровни PAI-1 связаны с тромбообразованием и сосудистым заболеванием, позволяя предположить, что высокий уровень PAI-1 в плазме может вызывать состояние гиперкоагуляции путем нарушения естественного баланса между фибринолизом и коагуляцией (например, Vaughan, D.E., J. Invest. Med. 46: 370-376 (1998)). Известно, что TGF-β стимулирует экспрессию PAI-1 (например, Dennler, S. et al., EMBO J. 17: 3091-3100 (1998)). Соответственно, ингибирование продукции PAI-1 с помощью ингибитора пути передачи сигнала TGF-β может дать в результате новую фибринолитическую терапию.
Передача сигнала активина и избыточная экспрессия активина связана с патологическими нарушениями, которые включают накопление внеклеточного матрикса и фиброз (например, Matsuse, T. et al., Am. J. Respir. Cell Mol. Biol. 13: 17-24 (1995); Inoue, S. et al., Biochem. Biophys. Res. Comm. 205: 441-448 (1994); Matsuse, T. et al., Am. J. Pathol. 148: 707-713 (1996); De Bleser et al., Hepatology 26: 905-912 (1997); Pawlowski, J.E., et al., J. Clin. Invest. 100: 639-648 (1997); Sugiyama, M. et al., Gastroenterology 114: 550-558 (1998); Munz, B. et al., EMBO J. 18: 5205-5215 (1999)), воспалительные ответы (например, Rosendahl, A. et al., Am. J. Respir. Cell Mol. Biol. 25: 60-68 (2001), кахексию или истощение (Matzuk, M.M. et al., Proc. Natl. Acd. Sci. USA 91: 8817-8821 (1994); Coerver, K.A. et al., Mol. Endocrinol. 10: 534-543 (1996); Cipriano, S.C. et al., Endocrinology 141: 2319-2327 (2000)), заболевания или патологические ответы центральной нервной системы (например, Logan, A. et al., Eur. J. Neurosci. 11: 2367-2374 (1999); Logan, A. et al., Exp. Neurol. 159: 504-510 (1999); Masliah, E. et al., Neurochem. Int. 39: 393-400 (2001); De Groot, C.J.A. et al., J. Neuropathol. Exp. Neurol. 58: 174-187 (1999); John, G.R. et al., Nat. Med. 8: 1115-1121 (2002)) и гипертензию (например, Dahly, A.J. et al., Am. J. Physiol. Regul. Integr. Comp. Physiol. 283: R757-767 (2002)). Исследования показали, что TGF-β и активин могут действовать синергично, вызывая продукцию внеклеточного матрикса (например, Sugiyama, M. et al., Gastroenterology 114; 550-558 (1998)).
Следовательно, становится очевидным, что ингибирование связанного с ALK5 и/или ALK4 фосфорилирования Smad2 и Smad3 с помощью предпочтительных соединений согласно настоящему изобретению может обеспечивать лечение и профилактику нарушений, вовлекающих эти пути передачи сигнала.
В международной патентной публикации WO 00/61576 и патентной заявке США 2003/0149277 A1 раскрыты производные триарилимидазола и их применение в качестве ингибиторов ALK5. В международной патентной публикации WO 01/62756 A1 раскрыты производные пиридинилимидазола и их применение в качестве ингибиторов ALK5. В международной патентной публикации WO 02/055077 A1 раскрыто применение имидазолильных производных циклического ацеталя в качестве ингибиторов ALK5. В международной патентной публикации WO 03/087304 A2 раскрыты тризамещенные гетероарилы и их применение в качестве ингибиторов ALK5 и/или ALK4. В международной патентной публикации WO 2005/103028 A1 и патенте США 7407958 B2 раскрыты 2-пиридилзамещенные имидазолы в качестве ингибиторов ALK5 и/или ALK4. В особенности, продемонстрировано применение одного из типичных соединений, заявленных в международной патентной публикации WO 2005/103028 A1 и патенте США 7407958 B2, IN-1130, на различных животных моделях в качестве ингибиторов ALK5 и/или ALK4. IN-1130 эффективно подавляет почечный фиброз, индуцированный односторонней закупоркой мочеточника (UUO) у крыс (Moon, J.-A. et al., Kidney Int. 70: 1234-1243 (2006)), улучшает экспериментальный аутоиммунный энцефаломиелит (EAE) у SBE-luc и GFAP-luc мышей, иммунизированных MOG35-55 (Luo, J. et al., J. Clin. Invest. 117: 3306-3315 (2007)), ослабляет фиброз белочной оболочки и корректирует пенильный изгиб у крыс (Ryu, J.-K. et al., J. Sex. Med. 6: 1284-1296 (2009)), и существенно снижает объем опухоли с усиленным иммунным ответом у мышей, обработанных клеточной линией рака предстательной железы мышей Tramp C2 (Lee, G.T. et al., J. Urol. 180: 2660-2667 (2008)). А также в патентной заявке США 2008/0319012 A1 раскрыты 2-пиридилзамещенные имидазолы в качестве ингибиторов ALK5 и/или ALK4. В частности, продемонстрировано применение одного из типичных соединений, заявленных в US 2008/0319012 A1, IN-1233, на различных животных моделях в качестве ингибиторов ALK5 и/или ALK4. IN-1233 эффективно предотвращает развитие и прогрессирование легочной артериальной гипертензии на монокроталиновой модели у крыс посредством ингибирования передачи сигнала TGF-β (Long, L. et al., Circulation 119: 566-576 (2009)), а также предотвращает образование грануляционной ткани после помещения непокрытого металлического стента в уретральной модели у крыс (Kim, J. H. et al., Radiology 255: 75-82 (2010)).
Краткое описание сущности изобретения
Техническое решение
Неожиданно было обнаружено, что соединения класса 2-пиридилзамещенных имидазолов функционируют в качестве сильных и селективных ингибиторов ALK5 и/или ALK4 и, следовательно, они применимы при лечении, профилактике и ослаблении различных опосредованных ALK5 и/или ALK4 болезненных состояний, таких как гломерулонефрит, диабетическая нефропатия, волчаночный нефрит, индуцированная гипертензией нефропатия, почечный интерстициальный фиброз, почечный фиброз в результате осложнений при применении лекарственных средств, обусловленная ВИЧ нефропатия, обусловленная трансплантацией нефропатия, фиброз печени всех этиологий, связанная с различными инфекциями печеночная дисфункция, индуцированный алкоголем гепатит, нарушения желчных протоков, муковисцидоз, пневмофиброз, интерстициальное заболевание легких, острая легочная недостаточность, респираторный дистресс-синдром у взрослых, идиопатический пневмофиброз, хроническая обструктивная болезнь легких, индуцированное инфекционными или токсическими средствами заболевание легких, фиброз сердца после инфаркта миокарда, застойная сердечная недостаточность, кардиомиопатия при дилатации, миокардит, утолщение интимы, сосудистый стеноз, индуцированное гипертензией ремоделирование сосудов, легочная артериальная гипертензия, коронарный рестеноз, периферический рестеноз, каротидный рестеноз, индуцированный стентом рестеноз, атеросклероз, рубцевание хрусталика, рубцевание роговицы, пролиферативная витреоретинопатия, глаукома, внутриглазное давление, образование избыточных или гипертрофических шрамов или келоидов на коже в процессе заживления полученной при травматических или хирургических повреждениях раны, перитониальная и подкожная спайка, склеродермия, фибросклероз, прогрессирующий системный склероз, дерматомиозит, полимиозит, артрит, остеопороз, язвы, неврологическая дисфункция, эректильная дисфункция у мужчин, болезнь Пейрони, контрактура Дюпюитрена, болезнь Альцгеймера, синдром Рейно, индуцированный облучением фиброз, тромбоз, рост опухолевых метастазов, множественная миелома, меланома, глиома, глиобластомы, лейкоз, саркомы, лейомиомы, мезотелиома и карциномы легкого, молочной железы, толстого кишечника, яичника, шейки матки, печени, желчных протоков, желудочно-кишечного тракта, поджелудочной железы, предстательной железы, головы и шеи.
Описание фигур
Вышеуказанные аспекты и другие признаки настоящего изобретения будут объясняться в последующем описании во взаимосвязи с сопроводительными фигурами, где
на фиг.1 показан эффект соединений примеров 2, 60, 86, 92 и 94 на индуцированную TGF-β1 активность репортера 3TP-Luc в клетках HaCaT-3TP-Luc;
на фиг.2 показан эффект соединений примеров 2, 60, 86, 92 и 94 на индуцированную TGF-β1 активность репортера 3TP-Luc в клетках 4T1-3TP-Luc;
на фиг.3 показан эффект соединения примера 2 на индуцированную TGF-β1 Smad2/3 ядерную транслокацию в клетках MCF10A;
на фиг.4 показан эффект соединения примера 2 на индуцированную TGF-β1 клеточную миграцию в клетках MCF10A;
на фиг.5a и 5b показан эффект соединения примера 2 на индуцированную TGF-β1 клеточную инвазию в клетках 4T1. (5a). Окрашенные DAPI клетки, оставшиеся на нижней поверхности, (5b). Среднее число клеток на поле наблюдения, полученное от 5 случайных полей;
на фиг.6 показан эффект соединения примера 2 на клеточный рост клеток 4T1;
на фиг.7 показан эффект соединения примера 2 на клеточный рост клеток MCF10A;
на фиг.8a и 8b показан эффект соединения примера 3 на метастазирование опухоли молочной железы в легкое у мышей BALB/c с ксенотрансплантатом клеток 4T1. Соединение примера 3 (13,6 или 27,3 мг/кг), растворенное в воде (носителе), давали мышам перорально BID пять дней подряд в неделю в течение четырех недель. (8a). Белые пятна на поверхности легкого указывают на метастатические узлы (белые стрелки). (8b). Число метастатических узлов на всей поверхности легкого;
на фиг.9a и 9b показан эффект соединения примера 2 на метастазирование опухоли молочной железы в легкое у мышей BALB/c с ксенотрансплантатом клеток 4T1. Соединение примера 2 (5, 10, 20 или 40 мг/кг), растворенное в составе искусственного желудочного сока (носителе), давали мышам перорально пять дней подряд в неделю в течение трех недель. (9a). Белые пятна на поверхности легкого указывают на метастатические узлы. (9b). Число метастатических узлов на поверхности левой доли легкого;
на фиг.10a, 10b и 10c показан эффект соединения примера 2 на метастазирование опухоли молочной железы в легкое у мышей BALB/c с ксенотрансплантатом клеток 4T1. Соединение примера 2 (5, 10, 20 или 40 мг/кг), растворенное в составе искусственного желудочного сока (носителе), давали мышам перорально через день (три раза в неделю) в течение 24 дней. (10a). Белые пятна на поверхности легкого указывают на метастатические узлы. (10b). Число метастатических узлов на поверхности левой доли легкого. (10c). Эффект на индуцированное TGF-β1 фосфорилирование Smad2 в опухолевой ткани;
на фиг.11a, 11b и 11c показан эффект соединения примера 61 на метастазирование опухоли молочной железы в легкое у мышей BALB/c с ксенотрансплантатом клеток 4T1. Соединение примера 61 (43,6 мг/кг), растворенное в солевом растворе (носителе), давали мышам интраперитонеально через день (три раза в неделю) в течение 2,5 недель. (11a). Белые пятна на поверхности легкого указывают на метастатические узлы. (11b). Число метастатических узлов на поверхности левой доли легкого. (11c). Объем первичной опухоли;
на фиг.12a, 12b, 12c и 12d показан эффект соединения примера 61 на метастазирование опухоли молочной железы в легкое у MMTV/c-Neu мышей. Мыши, являющиеся носителями опухоли MMTV/c-Neu, получали интраперитонеально лечение соединением примера 61 (43,6 мг/кг) через день в течение трех недель. (12a). Окрашивание опухоли молочной железы и тканей легкого гематоксилином и эозином (H&E). (12b). Число гистологически обнаруживаемых метастатических поражений в легком. (12c). Объем опухоли молочной железы. (12d). Уровень мРНК β-казеина;
на фиг.13a и 13b показан эффект соединения примера 3 на метастазирование опухоли молочной железы в легкое у MMTV/c-Neu мышей. Мыши, являющиеся носителями опухоли MMTV/c-Neu, получали интраперитонеально лечение соединением примера 3 (43,6 мг/кг) через день в течение десяти недель. (13a). Уровень мРНК β-казеина. (13b). Активность MMP-9 и MMP-2 в первичной опухоли молочной железы;
на фиг.14a, 14b, 14c и 14d показан эффект соединения примера 3 на фиброз печени с перевязкой желчного протока у крыс. Соединение примера 3 (21,8 или 43,6 мг/кг), растворенное в солевом растворе (носителе), давали крысам перорально три раза в неделю в течение четырех недель, начиная после операции BDL (перевязка желчных протоков). (14a). Активность сывороточной аланинаминотрансферазы (ALT) и аспартатаминотрансферазы (AST). (14b). Уровень белка pSmad3 в печени. (14c). Уровень белков α-SMA, фибронектина и виментина в печени. (14d). Окрашивание тканей печени гематоксилином и эозином (H&E);
на фиг.15a, 15b и 15c показан эффект соединения примера 2 на фиброз печени с перевязкой желчного протока у крыс. Соединение примера 2 (5, 10 или 20 мг/кг), растворенное в составе искусственного желудочного сока (носитель), давали крысам перорально три раза в неделю в течение четырех недель, начиная после операции BDL. (15a). Активность ALT и AST. (15b). Уровень белков α-SMA и фибронектина в печени. (15c). Окрашивание тканей печени гематоксилином и эозином (H&E);
на фиг.16a и 16b показан эффект соединения примера 2 на индуцированный блеомицином фиброз легкого у мышей. Соединение примера 2 (5, 10 или 20 мг/кг), растворенное в составе искусственного желудочного сока (носителе), давали мышам перорально пять раз в неделю в течение двух недель, начиная с дня 7. (16a). Уровень белков α-SMA и фибронектина в легком. (16b). Окрашивание тканей легкого гематоксилином и эозином (H&E).
В таблице 1 показаны структуры и спектральные данные 1H-ЯМР и масс-спектров соединений примеров 1-139;
в таблице 2 показаны структуры и данные 1H-ЯМР и масс-спектров соединений примеров 140-153;
в таблице 3 показаны значения IC50 соединений выбранных примеров для фосфорилирования киназы ALK5;
в таблице 4 показаны или значения IC50, или ингибирование фосфорилирования различных киназ (в %) соединением примера 2;
в таблице 5 показано воздействие соединения примера 3 на изменение массы тела и органов у крыс с BDL; и
в таблице 6 показано воздействие соединения примера 2 на изменение массы тела и органов у крыс с BDL.
Наилучший способ осуществления
В одном варианте осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли:
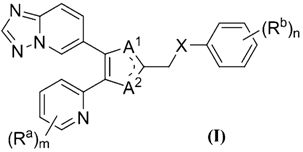
где каждый Ra независимо представляет собой H, галоген, C1-6алкил, C1-6галогеналкил, C3-6циклоалкил, OH, -O-C1-6алкил, -O-C1-6галогеналкил, -O-C3-6циклоалкил, NH2, -NH-C1-6алкил, -NH-C1-6галогеналкил, -NH-C3-6циклоалкил, -S-C1-6алкил, -S-C1-6галогеналкил, -S-C3-6циклоалкил, CN или NO2;
m равно 0, 1, 2, 3 или 4;
один из A1 и A2 представляет собой N, и другой представляет собой NR1, где R1 представляет собой H, OH, C1-6алкил, C1-6галогеналкил или C3-6циклоалкил;
X представляет собой связь, -(CH2)p-, -NR2-, -O- или -S-, где p равно 0 или 1, и R2 представляет собой H или C1-3алкил;
каждый Rb независимо представляет собой H, галоген, C1-6алкил, C1-6галогеналкил, C3-6циклоалкил, C2-6алкенил, C2-6алкинил, -(CH2)q-OR3, -(CH2)q-NR3R4, -(CH2)q-SR3, -(CH2)q-NO2, -(CH2)q-CONHOH, -(CH2)q-CN, -(CH2)q-COR3, -(CH2)q-CO2R3, -(CH2)q-CONR3R4, -(CH2)q-тетразол, -(CH2)q-CH=CH-CN, -(CH2)q-CH=CH-CO2R3, -(CH2)q-CH=CH-CONR3R4, -(CH2)q-CH=CH-тетразол, -(CH2)q-NHCOR3, -(CH2)q-NHCO2R3, -(CH2)q-CONHSO2R3, -(CH2)q-NHSO2R3, -(CH2)q-C≡C-CN, -(CH2)q-C≡C-CO2R3, -(CH2)q-C≡C-CONR3R4, -(CH2)q-C≡C-тетразол, -(CH2)q-SOR5, -(CH2)q-SO2R5 или -(CH2)r-(OR3)2, где R3 и R4 независимо представляют собой H, C1-6алкил, C1-6галогеналкил или C3-6циклоалкил; или взятые вместе с атомом азота, к которому они присоединены, образуют моноциклическое кольцо, такое как имидазол, пирролидин, пиперидин, морфолин, пиперазин и гомопиперазин; R5 представляет собой C1-6алкил, C1-6галогеналкил или C3-6циклоалкил; q равно 0, 1, 2, 3 или 4; и r равно 1, 2, 3 или 4;
n равно 0, 1, 2, 3, 4 или 5.
Используемая в настоящем описании двойная связь, обозначенная в формуле (I) пунктирными линиями, представляет собой возможные таутомерные циклические формы соединений, которые входят в объем настоящего изобретения, причем двойная связь образована с незамещенным азотом.
Предпочтительно, Ra представляет собой C1-3алкил или галоген.
Предпочтительно, m равно 1 или 2.
Предпочтительно, один из A1 и A2 представляет собой N, и другой представляет собой NR1, где R1 представляет собой H.
Предпочтительно, X представляет собой -(CH2)p- или -NR2-, где p равно 0, и R2 представляет собой H.
Предпочтительно, Rb представляет собой галоген, C1-3алкил, C1-3галогеналкил, C3-4циклоалкил, C2-4алкенил, C2-4алкинил, -(CH2)q-OR3, -(CH2)q-NR3R4, -(CH2)q-SR3, -(CH2)q-CN, -(CH2)q-COR3, -(CH2)q-CO2R3, -(CH2)q-CONR3R4, -(CH2)q-NHCOR3, -(CH2)q-NHSO2R3, -(CH2)q-SOR5 или -(CH2)q-SO2R5, где R3 и R4 независимо представляют собой H, C1-3алкил, C1-3галогеналкил или C3-4циклоалкил; или взятые вместе с атомом азота, к которому они присоединены, образуют моноциклическое кольцо, такое как имидазол, пирролидин, пиперидин, морфолин, пиперазин и гомопиперазин; R5 представляет собой метил; и q равно 0, 1 или 2.
Предпочтительно, n равно 1, 2 или 3.
Конкретные соединения по настоящему изобретению, которые могут быть приведены, включают следующие соединения и их фармацевтически приемлемые соли:
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фторанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-фторанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-фторанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2,3-дифторанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,4-дифторанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,5-дифторанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-хлоранилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-хлоранилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-хлоранилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2,3-дихлоранилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,4-дихлоранилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,5-дихлоранилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-броманилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-броманилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-броманилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-метиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-метиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-метиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2,3-диметиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,4-диметиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,5-диметиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-этиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-этиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-изопропиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-изопропиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-изопропиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-виниланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-виниланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-виниланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-этиниланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-метоксианилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-метоксианилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-метоксианилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2,3-диметоксианилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,4-диметоксианилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,5-диметоксианилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(метоксиметил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(метоксиметил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-(метоксиметил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(трифторметокси)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(трифторметокси)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-(трифторметокси)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(метилтио)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(метилтио)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-(метилтио)анилин;
2-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензонитрил;
4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фталонитрил;
2-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензамид;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензамид;
4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензамид;
2-(3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетонитрил;
2-(4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетонитрил;
1-(3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)этанон;
1-(4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)этанон;
метил-3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензоат;
метил-4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензоат;
N-(2-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетамид;
N-(3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетамид;
N-(4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетамид;
N-(2-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)метансульфонамид;
N-(3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)метансульфонамид;
N-(4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)метансульфонамид;
N1-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-N2,N2-диметилбензол-1,2-диамин;
N1-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-N3,N3-диметилбензол-1,3-диамин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(пирролидин-1-ил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-морфолиноанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-морфолиноанилин;
N3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-фтор-N1,N1-диметилбензол-1,3-диамин;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-5-(диметиламино)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-4-(диметиламино)бензонитрил;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-((диметиламино)метил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-((диметиламино)метил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(пирролидин-1-илметил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(пирролидин-1-илметил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(морфолинометил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(морфолинометил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-5-((диметиламино)метил)-2-фторанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-((диметиламино)метил)-2-фторанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фтор-3-(пирролидин-1-илметил)анилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фтор-3-(морфолинометил)анилин;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-4-((диметиламино)метил)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-2-((диметиламино)метил)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-5-((диметиламино)метил)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-4-(пирролидин-1-илметил)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-2-(пирролидин-1-илметил)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-5-(пирролидин-1-илметил)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-4-(морфолинометил)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-2-(морфолинометил)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-5-(морфолинометил)бензонитрил;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(2-(диметиламино)этиланилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(2-(диметиламино)этиланилин;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-этилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензонитрил;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-этилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фторанилин;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фтор-N-метиланилин;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)(метил)амино)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)(метил)амино)бензамид;
6-(2-бензил-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридин;
6-(2-(2-фторбензил)-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридин;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)бензамид;
6-(5-(6-метилпиридин-2-ил)-2-(феноксиметил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридин;
6-(2-((2-фторфенокси)метил)-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридин;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метокси)бензонитрил;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метокси)бензамид;
6-(5-(6-метилпиридин-2-ил)-2-(фенилтиометил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридин;
6-(2-((2-фторфенилтио)метил)-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридин.
Соединения настоящего изобретения обычно представляют собой малые органические молекулы (непептидные малые молекулы), как правило, размером приблизительно менее 1000 дальтон. Предпочтительные непептидные малые молекулы характеризуются молекулярными массами приблизительно менее 750 дальтон, более предпочтительно, приблизительно менее 500 дальтон.
Соединения формулы (I) также могут быть предоставлены в форме «пролекарства», которое разработано для высвобождения соединения формулы (I) при введении субъекту. Разработанные формы пролекарств хорошо известны в данной области техники и зависят от заместителей, содержащихся в соединении формулы (I). Например, гидроксилсодержащий заместитель может быть соединен с носителем, который делает соединение биологически неактивным до тех пор, пока он не будет удален под действием эндогенных ферментов или, например, под действием ферментов, нацеленных на конкретный рецептор или конкретную локализацию у субъекта.
Соединение формулы (I), которое является кислым по природе (например, содержащее карбоксильную или фенольную гидроксильную группу), может образовывать фармацевтически приемлемую соль, например, соль натрия, калия, кальция или золота. В объем изобретения входят также соли, образованные с такими фармацевтически приемлемыми аминами, как аммиак, алкиламины, гидроксиалкиламины и N-метилглюкамин. Соединение формулы (I) может быть обработано кислотой с образованием кислотно-аддитивных солей. Примеры таких кислот включают хлористоводородную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, серную кислоту, метансульфоновую кислоту, фосфорную кислоту, пара-бромфенилсульфоновую кислоту, угольную кислоту, янтарную кислоту, лимонную кислоту, бензойную кислоту, щавелевую кислоту, малоновую кислоту, салициловую кислоту, яблочную кислоту, фумаровую кислоту, аскорбиновую кислоту, малеиновую кислоту, уксусную кислоту и другие неорганические и органические кислоты, хорошо известные специалистам в данной области техники. Кислотно-аддитивные соли могут быть получены путем обработки соединения формулы (I) в его свободной основной форме достаточным количеством кислоты (например, хлористоводородной кислотой), с получением кислотно-аддитивной соли (например, гидрохлорида). Кислотно-аддитивная соль может быть преобразована обратно в свою свободную основную форму путем обработки соли подходящим разбавленным водным раствором основания (например, гидроксидом натрия, бикарбонатом натрия, карбонатом калия или аммиаком).
Некоторые соединения настоящего изобретения могут быть кристаллизованы или перекристаллизованы из растворителей, таких как водные и органические растворители. В таких случаях могут образовываться сольваты. В объем настоящего изобретения входят стехиометрические сольваты, включая гидраты, а также соединения, содержащие переменное количество воды, которые могут быть получены способами, такими как лиофилизация.
Соединения формулы (I) могут содержать один или более центров асимметрии, и потому могут существовать в виде энантиомеров или диастереомеров. Следует понимать, что настоящее изобретение включает как смеси, так и отдельные конкретные изомеры соединений формулы (I). Кроме того, определенные соединения формулы (I), которые содержат алкенильные группы, могут существовать в виде цис- или транс-изомеров. В каждом случае, изобретение включает как смеси, так и отдельные конкретные изомеры.
Соединения формулы (I) также могут существовать в таутомерных формах, и настоящее изобретение включает как смеси, так и отдельные конкретные таутомеры.
В настоящее изобретение также включены меченные радиоактивным изотопом производные соединений формулы (I), которые подходят для биологических исследований.
Используемый в настоящем описании термин «алкильная» группа относится к насыщенной алифатической углеводородной группе, содержащей 1-6 атомов углерода. Алкильная группа может быть неразветвленной или разветвленной. Примеры алкильной группы включают, но без ограничения, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, н-пентил и н-гексил. Алкильная группа может быть необязательно замещена одним или более заместителями, такими как алкокси, циклоалкокси, амино, нитро, карбокси, циано, галоген, гидроксил, сульфо или меркаптогруппа.
Используемый в настоящем описании термин «циклоалкильная» группа относится к алифатическому карбоциклическому кольцу, состоящему из 3-6 атомов углерода. Примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил и циклогексил.
Используемый в настоящем описании термин «галогеналкильная» группа относится к алкильной группе, содержащей один или более атомов галогена. Примеры галогеналкильных групп включают фторметил, хлорметил, бромметил и трифторметил.
Используемый в настоящем описании термин «галогеновая» группа относится к фтору, хлору, брому или йоду.
Используемый в настоящем описании термин «алкенильная» группа относится к алифатической углеродной группе, содержащей 2-6 атомов углерода и, по меньшей мере, одну двойную связь. Подобно алкильной группе алкенильная группа может быть неразветвленной или разветвленной. Примеры алкенильной группы включают, но без ограничения, винил, аллил, изопренил, 2-бутенил и 2-гексенил. Алкенильная группа может быть необязательно замещена одним или более заместителями, такими как алкокси, циклоалкокси, амино, нитро, карбокси, циано, галоген, гидроксил, сульфо или меркаптогруппа.
Используемый в настоящем описании термин «алкинильная» группа относится к алифатической углеродной группе, содержащей 2-6 атомов углерода и, по меньшей мере, одну тройную связь. Алкинильная группа может быть неразветвленной или разветвленной. Примеры алкинильной группы включают, но без ограничения, этинил, пропаргил и бутинил. Алкинильная группа может быть необязательно замещена одним или более заместителями, такими как алкокси, циклоалкокси, амино, нитро, карбокси, циано, галоген, гидроксил, сульфо или меркаптогруппа.
Используемый в настоящем описании термин «ингибитор ALK5 и/или ALK4» относится к соединению, отличному от ингибиторных Smad, например Smad6 и Smad7, которое селективно ингибирует предпочтительно рецепторы ALK5 и/или ALK4 по сравнению с рецепторами p38 или рецепторами II типа.
Используемый в настоящем описании термин «опосредованное ALK5 и/или ALK4 болезненное состояние» относится к любому болезненному состоянию, которое опосредуется (или модулируется) ALK5 и/или ALK4, например, к заболеванию, которое модулируется путем ингибирования фосфорилирования Smad2 и Smad3 в путях передачи сигнала TGF-β и/или активина.
Используемый в настоящем описании термин «язвы» применяют для включения, но без ограничения, диабетических язв, хронических язв, язв желудка и язв двенадцатиперстной кишки.
Соединения формулы (I) могут быть получены рядом известных способов из коммерчески доступных или известных исходных веществ. Если исходные вещества недоступны из коммерческого источника, то они могут быть получены способами, известными в данной области техники.
Схема 1
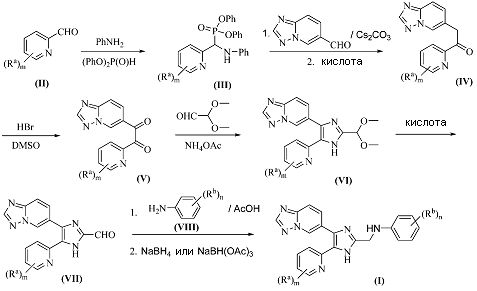
Согласно одному способу, соединения формулы (I), где A1 представляет собой N, и A2 представляет собой NH, или A1 представляет собой NH, и A2 представляет собой N, и X представляет собой -NH-, получают, как представлено на схеме 1. В частности, Ra-замещенный пиридин-2-карбальдегид (II) подвергают взаимодействию с анилином и дифенилфосфитом, с получением N,P-ацеталя (III), который может быть затем подвергнут сочетанию с [1,2,4]триазоло[1,5-a]пиридин-6-карбальдегидом, с последующим гидролизом в кислых условиях, с получением монокетона (IV). Монокетон (IV) может быть окислен до дикетона (V) при помощи HBr в ДМСО. Полученный дикетон (V) может быть конденсирован с 2,2-диметоксиацетальдегидом в присутствии ацетата аммония, с получением ацеталь-защищенного имидазола (VI), который может быть гидролизован в кислых условиях, с получением имидазол-2-карбальдегида (VII). Имидазол-2-карбальдегид (VII) может быть подвергнут сочетанию с Rb-замещенным анилином (VIII) в присутствии кислоты, такой как уксусная кислота, с получением имина, который может быть затем восстановлен восстановителем, например боргидридом натрия или триацетоксиборгидридом натрия, с получением соединения формулы (I). Значения Ra, Rb, m и n являются такими, как определено выше.
Схема 2
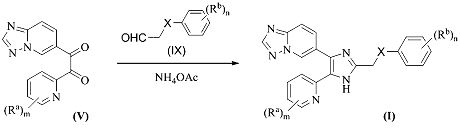
Согласно другому способу, соединения формулы (I), где A1 представляет собой N, и A2 представляет собой NH, или A1 представляет собой NH, и A2 представляет собой N, и X представляет собой -(CH2)p-, -NR2-, -O- или -S-, где p равно 0 или 1, и R2 представляет собой C1-3алкил, получают, как представлено на схеме 2. Дикетон (V) может быть конденсирован с соответствующим Rb-замещенным альдегидом (IX) в присутствии ацетата аммония, с получением соединения формулы (I). Значения Ra, Rb, m и n являются такими, как определено выше.
Схема 3
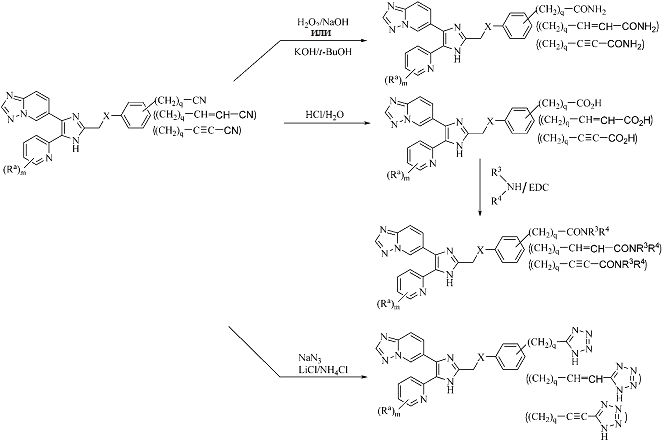
Альтернативно, если в соединениях формулы (I) Rb представляет собой -(CH2)q-CN, -(CH2)q-CH=CH-CN или -(CH2)q-C≡C-CN, то он может быть затем функционализирован с образованием соединения формулы (I), как представлено на схеме 3. Значения Ra, R3, R4, X, m и q являются такими, как определено выше.
Полученные соединения настоящего изобретения, представленные формулами (I)-(IX), могут быть разделены и очищены подходящими традиционными способами, такими как колоночная хроматография и перекристаллизация.
Соединения настоящего изобретения могут быть введены любым подходящим путем, например, с использованием перорального, буккального, сублингвального, ректального, вагинального, назального, местного или парентерального (включая внутривенное, внутримышечное, подкожное и интракоронарное) введения.
Местные составы по настоящему изобретению могут быть представлены, например, в виде мазей, кремов или лосьонов, глазных мазей и глазных или ушных капель, пропитанных повязок и аэрозолей, и могут содержать приемлемые общепринятые добавки, такие как консерванты, растворители для содействия проникновению лекарственного средства и смягчители для мазей и кремов.
Составы также могут содержать совместимые общепринятые носители, такие как основы для мазей и кремов и этанол или олеиловый спирт для лосьонов. Такие носители могут присутствовать в количестве приблизительно от 1% приблизительно до 98% состава. Чаще всего они будут составлять приблизительно до 80% состава.
Для введения человеку при терапевтическом или профилактическом лечении приведенных выше нарушений пероральные, буккальные или сублингвальные дозировки соединения формулы (I), как правило, будут находиться в диапазоне 50-5000 мг в сутки для среднего взрослого пациента (70 кг). Таким образом, для обычного взрослого пациента отдельные таблетки или капсулы содержат 25-500 мг активного соединения, в подходящем фармацевтически приемлемом растворителе или носителе, для введения в виде однократной или множественных доз, один или несколько раз в сутки. Дозировки для парентерального введения, как правило, будут находиться в диапазоне 25-250 мг на одну дозу, по необходимости. На практике лечащий врач будет определять конкретную схему дозировки, которая будет наиболее подходящей для отдельного пациента, и она будет изменяться в зависимости от возраста, массы и реакции конкретного пациента. Вышеперечисленные дозировки являются иллюстративными для среднего случая, но могут иметь место отдельные случаи, в которых могут быть использованы более высокие или более низкие диапазоны дозирования, и такие диапазоны входят в объем настоящего изобретения.
Для применения человеку соединение формулы (I) может быть введено отдельно, но, как правило, будет вводиться в смеси с фармацевтическим носителем, выбранным с учетом заданного пути введения и стандартной фармацевтической практики. Например, соединение может быть введено перорально, буккально или сублингвально в форме таблеток, содержащих такие эксципиенты, как крахмал или лактоза, или в форме капсул или суппозиториев, или отдельно, или в смеси с эксципиентами, или в форме эликсиров или суспензий, содержащих ароматизаторы или красители. Такие жидкие препараты могут быть приготовлены с такими фармацевтически приемлемыми добавками, как суспендирующее средство (например, метилцеллюлоза, полусинтетический глицерид, такой как витепсол, или смесь глицеридов, таких как смесь масла абрикосовых косточек и сложных эфиров PEG-6 или смесь PEG-8 и каприловых/каприновых глицеридов). Соединение также может быть введено путем парентеральной инъекции, например внутривенно, внутримышечно, подкожно или интракоронарно. Для парентерального введения соединение лучше всего использовать в форме стерильного водного раствора, который может содержать другие вещества, например соли или моносахариды, такие как маннит или глюкоза, чтобы сделать раствор изотоническим по отношению к крови.
Таким образом, в следующем аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль или сольват, вместе с фармацевтически приемлемым разбавителем или носителем.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли или сольвату, или содержащей их фармацевтической композиции по настоящему изобретению для применения в терапии.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или его фармацевтически приемлемой соли или сольвата, или содержащей их фармацевтической композиции по настоящему изобретению для производства лекарственного средства для лечения заболевания, опосредованного рецепторами ALK5 и/или ALK4, у млекопитающих.
Опосредованные ALK5 и/или ALK4 болезненные состояния включают, но без ограничения, гломерулонефрит, диабетическую нефропатию, нефрит при волчанке, индуцированную гипертензией нефропатию, почечный интерстициальный фиброз, почечный фиброз в результате осложнений при применении лекарственных средств, обусловленную ВИЧ нефропатию, обусловленную трансплантацией нефропатию, фиброз печени всех этиологий, связанную с различными инфекциями печеночную дисфункцию, индуцированный алкоголем гепатит, нарушения желчных протоков, муковисцидоз, пневмофиброз, интерстициальное заболевание легких, острую легочную недостаточность, респираторный дистресс-синдром у взрослых, идиопатический пневмофиброз, хроническую обструктивную болезнь легких, индуцированное инфекционными или токсическими средствами заболевание легких, фиброз сердца после инфаркта миокарда, застойную сердечную недостаточность, кардиомиопатию при дилатации, миокардит, утолщение интимы, сосудистый стеноз, индуцированное гипертензией ремоделирование сосудов, легочную артериальную гипертензию, коронарный рестеноз, периферический рестеноз, каротидный рестеноз, индуцированный стентом рестеноз, атеросклероз, рубцевание хрусталика, рубцевание роговицы, пролиферативную витреоретинопатию, глаукому, внутриглазное давление, образование избыточных или гипертрофических шрамов или келоидов на коже в процессе заживления полученной при травматических или хирургических повреждениях раны, перитониальную и подкожную спайку, склеродермию, фибросклероз, прогрессирующий системный склероз, дерматомиозит, полимиозит, артрит, остеопороз, язвы, неврологическую дисфункцию, эректильную дисфункцию у мужчин, болезнь Пейрони, контрактуру Дюпюитрена, болезнь Альцгеймера, синдром Рейно, индуцированный облучением фиброз, тромбоз, рост опухолевых метастазов, множественную миелому, меланому, глиому, глиобластомы, лейкоз, саркомы, лейомиомы, мезотелиому и карциномы легкого, молочной железы, толстого кишечника, яичника, шейки матки, печени, желчных протоков, желудочно-кишечного тракта, поджелудочной железы, предстательной железы, головы и шеи.
Настоящее изобретение дополнительно относится к способу ингибирования путей передачи сигнала TGF-β и/или активина у человека, например ингибирования фосфорилирования Smad2 или Smad3 ALK5 и/или ALK4.
Настоящее изобретение дополнительно относится к способу ослабления накопления избыточного внеклеточного матрикса у человека путем ингибирования путей передачи сигнала TGF-β и/или активина, например, ингибирования фосфорилирования Smad2 или Smad3 ALK5 и/или ALK4.
Настоящее изобретение дополнительно относится к способу лечения, профилактики или ослабления метастазирования опухолевых клеток у человека путем ингибирования TGF-β пути передачи сигнала.
Настоящее изобретение дополнительно относится к способу лечения, профилактики или ослабления карцином, опосредованных избыточной экспрессией TGF-β, у человека путем ингибирования пути передачи сигнала TGF-β.
Настоящее изобретение дополнительно относится к способу лечения, профилактики или ослабления сосудистых повреждений у человека путем ингибирования пути передачи сигнала TGF-β.
Настоящее изобретение дополнительно проиллюстрировано в последующих примерах, которые не должны приниматься как ограничивающие объем настоящего изобретения, раскрытый в формуле изобретения. В примерах масс-спектры с ионизацией электрораспылением (ESI-МС) получали на Q-TOF2 масс-спектрометре (Micromass, Manchester, UK).
ПРИМЕРЫ
Препаративный пример 1
Получение дифенил(6-метилпиридин-2-ил)(фениламино)метилфосфоната (соединение формулы (III), где Ra=CH3)
Смесь 6-метилпиридин-2-карбоксальдегида (2,12 г, 17,50 ммоль), анилина (1,63 г, 17,50 ммоль), дифенилфосфита (4,92 г, 21,00 ммоль) и октагидрата цирконилхлорида (0,56 г, 1,75 ммоль) перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь экстрагировали CH2Cl2 (3×50 мл), и CH2Cl2 раствор промывали водой (2×20 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением указанного в заголовке соединения (6,96 г, 92%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3): δ 7,51 (т, 1H, J=7,8 Гц), 7,38 (дд, 1H, J=7,6, 2,0 Гц), 7,27-7,22 (м, 4H), 7,19-7,15 (м, 2H), 7,14-7,07 (м, 4H), 7,05-7,02 (м, 3H), 6,80-6,74 (м, 3H), 5,53 (псевдо т, 1H, J=7,4 Гц), 5,36 (дд, 1H, J=21,0, 8,2 Гц), 2,54 (с, 3H).
Препаративный пример 2
Получение дифенил(6-этилпиридин-2-ил)(фениламино)метилфосфоната (соединение формулы (III), где Ra=CH2CH3)
Указанное в заголовке соединение получали, как описано в препаративном примере 1, при использовании 6-этилпиридин-2-карбоксальдегида вместо 6-метилпиридин-2-карбоксальдегида. Выход: 81%;
1Н ЯМР (400 МГц, CDCl3): δ 7,55 (т, 1H, J=7,6 Гц), 7,38 (дд, 1H, J=7,6, 2,0 Гц), 7,26-7,09 (м, 8H), 7,07-7,00 (м, 5H), 5,59 (псевдо т, 1H, J=7,0 Гц), 5,34 (дд, 1H, J=20,8, 8,0 Гц), 2,82 (кв, 2H, J=7,6 Гц), 1,28 (т, 3H, J=7,6 Гц).
Препаративный пример 3
Получение 2-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-1-(6-метилпиридин-2-ил)этанона (соединение формулы (IV), где Ra=CH3)
К перемешиваемому раствору [1,2,4]триазоло[1,5-a]пиридин-6-карбальдегида (2,50 г, 17,01 ммоль) (полученного согласно способу, описанному в WO 03/087304 A2) и дифенил(6-метилпиридин-2-ил)(фениламино)метилфосфоната (7,32 г, 17,01 ммоль) в смеси ТГФ (40 мл) и изо-PrOH (10 мл) добавляли Cs2CO3 (7,20 г, 22,11 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. В реакционную смесь по каплям добавляли раствор 3 н. HCl (25 мл) и перемешивали смесь в течение 1 ч. Затем смесь разбавляли трет-бутилметиловым эфиром (40 мл) и экстрагировали 1 н. HCl (2×35 мл). Водные экстракты нейтрализовали добавлением 50% KOH до достижения pH 7-8. Осажденные вещества собирали фильтрованием, промывали водой и сушили над P2O5 в вакууме, с получением указанного в заголовке соединения (3,41 г, 80%) в виде не совсем белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3): δ 8,61 (д, 1H, J=0,8 Гц), 8,31 (с, 1H), 7,88 (дд, 1H, J=7,6, 1,6 Гц), 7,73 (т, 1H, перекрыв., J=7,6 Гц), 7,71 (дд, 1H, перекрыв., J=9,2, 0,8 Гц), 7,54 (дд, 1H, J=9,2, 1,6 Гц), 7,37 (дд, 1H, J=7,6, 1,6 Гц), 4,62 (с, 2H), 2,67 (с, 3H).
Препаративный пример 4
Получение 2-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-1-(6-этилпиридин-2-ил)этанона (соединение формулы (IV), где Ra=CH2CH3)
Указанное в заголовке соединение получали, как описано в препаративном примере 3, при использовании дифенил(6-этилпиридин-2-ил)(фениламино)метилфосфоната вместо дифенил(6-метилпиридин-2-ил)(фениламино)метилфосфоната. Выход: 78%;
1Н ЯМР (400 МГц, CDCl3): δ 8,61 (дд, 1H, J=1,6, 0,8 Гц), 8,29 (с, 1H), 7,88 (ушир.д, 1H, J=7,6 Гц), 7,74 (т, 1H, J=7,6 Гц), 7,70 (дд, 1H, J=9,2, 0,8 Гц), 7,54 (дд, 1H, J=9,2, 1,6 Гц), 7,37 (дд, 1H, J=7,6, 0,8 Гц), 4,62 (с, 2H), 2,93 (кв, 2H, J=7,6 Гц), 1,39 (т, 3H, J=7,6 Гц).
Препаративный пример 5
Получение 1-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-2-(6-метилпиридин-2-ил)этан-1,2-диона (соединение формулы (V), где Ra=CH3)
К перемешиваемой суспензии 2-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-1-(6-метилпиридин-2-ил)этанона (6,20 г, 24,57 ммоль) в ДМСО (48 мл) по каплям при 0°C добавляли HBr (48% масс. в воде, 5,96 г, 12,4 мл) и нагревали смесь при 60-70°C. Спустя 2 ч, реакционную смесь охлаждали до 0°C, вливали в воду со льдом (20 мл) и подщелачивали до pH 10 добавлением твердого K2CO3. Смесь экстрагировали CHCl3 (2×250 мл), органическую фазу промывали водой (2×100 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси MeOH и CH2Cl2 в качестве элюента, с получением указанного в заголовке соединения (6,02 г, 92%) в виде светло-желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3): δ 9,11 (дд, 1H, J=1,6, 1,2 Гц), 8,47 (с, 1H), 8,14 (дд, 1Н, J=9,2, 1,6 Гц), 8,04 (ушир.д, 1H, J=7,6 Гц), 7,88 (дд, 1Н, J=9,2, 1,2 Гц), 7,84 (т, 1H, J=7,8 Гц), 7,42 (ушир.д, 1H, J=8,0 Гц), 2,49 (с, 3H).
Препаративный пример 6
Получение 1-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-2-(6-этилпиридин-2-ил)этан-1,2-диона (соединение формулы (V), где Ra=CH2CH3)
Указанное в заголовке соединение получали, как описано в препаративном примере 5, при использовании 2-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-1-(6-этилпиридин-2-ил)этанона вместо 2-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-1-(6-метилпиридин-2-ил)этанона. Выход: 79%;
1Н ЯМР (400 МГц, CDCl3): δ 9,11 (дд, 1H, J=1,6, 0,8 Гц), 8,42 (с, 1H), 8,08 (дд, 1H, J=9,2, 1,6 Гц), 7,98 (ушир.д, 1H, J=7,6 Гц), 7,83 (дд, 1H, перекрыв., J=9,2, 0,8 Гц), 7,82 (т, 1H, перекрыв., J=7,6 Гц), 7,38 (ушир.д, 1H, J=7,6 Гц), 2,71 (кв, 2H, J=7,6 Гц), 1,08 (т, 3H, J=7,6 Гц).
Препаративный пример 7
Получение 6-(2-(диметоксиметил)-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридина (соединение формулы (VI), где Ra=CH3)
Перемешиваемый раствор 1-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-2-(6-метилпиридин-2-ил)этан-1,2-диона (6,00 г, 22,49 ммоль) в терт-бутилметиловом эфире (120 мл) обрабатывали диметилацетальглиоксалем (60% масс. раствор в воде, 7,8 мл, 44,98 ммоль). Добавляли NH4OAc (4,33 г, 56,2 ммоль) в MeOH (60 мл) и перемешивали полученную смесь при комнатной температуре в течение 3 ч. pH реакционной смеси доводили до 8 добавлением насыщенного водного раствора NaHCO3. Реакционную смесь экстрагировали CHCl3 (2×150 мл), CHCl3 раствор промывали водой (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси MeOH и CH2Cl2 в качестве элюента, с получением указанного в заголовке соединения (6,13 г, 78%) в виде светло-желтой пены.
1Н ЯМР (400 МГц, CDCl3): δ 10,54 (ушир.с, 1H), 8,96 (с, 1H), 8,36 (с, 1H), 7,82 (дд, 1H, J=9,2, 1,6 Гц), 7,77 (дд, 1H, J=9,2, 0,8 Гц), 7,47 (т, 1H, J=7,8 Гц), 7,23 (д, 1H, J=7,6 Гц), 7,04 (д, 1H, J=8,0 Гц), 5,57 (с, 1H), 3,48 (с, 6H), 2,58 (с, 3H).
Препаративный пример 8
Получение 6-(2-(диметоксиметил)-5-(6-этилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридина (соединение формулы (VI), где Ra=CH2CH3)
Указанное в заголовке соединение получали, как описано в препаративном примере 7, при использовании 1-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-2-(6-этилпиридин-2-ил)этан-1,2-диона вместо 1-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-2-(6-метилпиридин-2-ил)этан-1,2-диона. Выход: 68%;
1Н ЯМР (400 МГц, CDCl3): δ 10,67 (ушир.с, 1H), 8,97 (ушир.с, 1H), 8,35 (с, 1H), 7,83 (дд, 1H, J=9,2, 1,6 Гц), 7,76 (дд, 1H, J=9,2, 0,8 Гц), 7,50 (т, 1H, J=7,8 Гц), 7,25 (ушир.д, 1H, J=7,6 Гц), 7,05 (д, 1H, J=8,0 Гц), 5,56 (с, 1H), 3,46 (с, 6H), 2,83 (кв, 2H, J=7,6 Гц), 1,31 (т, 3H, J=7,6 Гц).
Препаративный пример 9
Получение 4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-карбальдегида (соединение формулы (VII), где Ra=CH3)
6-(2-(Диметоксиметил)-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридин (6,00 г, 17,12 ммоль) растворяли в 1 н. HCl (120 мл) и нагревали смесь при 70°C в течение 3 ч. Реакционную смесь оставляли охлаждаться до 0°C и затем нейтрализовали добавлением насыщенного водного раствора NaHCO3. Смесь экстрагировали 10% MeOH в CHCl3 (3×200 мл), органическую фазу сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении, с получением указанного в заголовке соединения (4,69 г, 90%) в виде светло-желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3): δ 9,82 (с, 1H), 9,01 (ушир.с, 1H), 8,41 (с, 1H), 7,85 (дд, 1H, J=9,2, 0,8 Гц), 7,82 (дд, 1Н, J=9,2, 1,6 Гц), 7,55 (т, 1H, J=7,8 Гц), 7,33 (ушир.с, 1H), 7,16 (д, 1H, J=8,0 Гц), 2,60 (с, 3H).
Препаративный пример 10
Получение 4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-этилпиридин-2-ил)-1H-имидазол-2-карбальдегида (соединение формулы (VII), где Ra=CH2CH3)
Указанное в заголовке соединение получали, как описано в препаративном примере 9, при использовании 6-(2-(диметоксиметил)-5-(6-этилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридина вместо 6-(2-(диметоксиметил)-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридина. Выход: 99%;
1H ЯМР (400 МГц, ДМСО-d6): δ 9,86 (т, 1H, J=1,2 Гц), 9,59 (с, 1H), 8,43 (с, 1H), 8,21 (дд, 1H, J=9,2, 1,6 Гц), 7,82 (ушир.д, 1H, J=8,0 Гц), 7,73 (дд, 1H, J=9,2, 0,8 Гц), 7,69 (т, 1H, J=7,8 Гц), 7,08 (ушир.д, 1H, J=7,6 Гц), 2,71 (кв, 2H, J=7,6 Гц), 1,16 (т, 3H, J=7,6 Гц).
Препаративный пример 11
Получение 3-амино-5-(диметиламино)бензонитрила (соединение формулы (VIII), где Rb=3-циано-5-диметиламино)
Указанное соединение получали в 2 последовательные стадии, как описано ниже.
3-Бром-N,N-диметил-5-нитроанилин (1,73 г, 7,06 ммоль) (полученный согласно способу, описанному в J. Org. Chem. 60: 5091-5103 (2003)), пиридин (24 мл) и CuCN (1,26 г, 2,14 ммоль) добавляли в сухую герметичную пробирку. Смесь нагревали при 220°C при перемешивании в течение 3,5 ч. Реакционную смесь оставляли охлаждаться до 100°C, вливали в колбу, содержащую смесь водного аммиака (100 мл) и воды (100 мл) и экстрагировали EtOAc (2×100 мл). EtOAc раствор последовательно промывали разбавленным раствором аммиака (100 мл), водой (100 мл) и насыщенным раствором соли (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 3-(диметиламино)-5-нитробензонитрила (0,44 г, 33%) в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 7,74 (дд, 1H, J=2,0, 1,2 Гц), 7,65 (т, 1H, J=2,2 Гц), 7,11 (дд, 1H, J=2,4, 1,2 Гц), 3,10 (с, 6H).
Полученный выше 3-(диметиламино)-5-нитробензонитрил (0,42 г, 2,22 ммоль) в метаноле (80 мл) гидрировали в присутствии 10% Pd/C (0,04 г) в атмосфере водорода в течение ночи. Реакционную смесь фильтровали через слой целита и упаривали фильтрат досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением указанного в заголовке соединения (0,29 г, 80%) в виде коричневой вязкой жидкости.
1H ЯМР (400 МГц, CDCl3): δ 6,35 (дд, 1H, J=2,4, 1,6 Гц), 6,28 (дд, 1H, J=2,0, 1,6 Гц), 6,14 (т, 1H, J=2,2 Гц), 3,76 (ушир.с, 2H), 2,92 (с, 6H).
Препаративный пример 12
Получение 3-((диметиламино)метил)-2-фторанилина (соединение формулы (VIII), где Rb=3-(диметиламино)метил-2-фтор)
Указанное соединение получали в 3 последовательные стадии, как описано ниже, используя в качестве исходного вещества коммерчески доступный 2-фтор-1-метил-3-нитробензол.
Перемешиваемый раствор 2-фтор-1-метил-3-нитробензола (15,80 г, 101,94 ммоль) и N-бромсукцинимида (18,14 г, 101,94 ммоль) в CCl4 (400 мл) обрабатывали бензоилпероксидом (0,37 г, 1,52 ммоль). Смесь нагревали при температуре кипения с обратным холодильником в течение ночи и затем охлаждали до комнатной температуры. Реакционную смесь фильтровали и упаривали фильтрат досуха при пониженном давлении. Остаток растворяли в CH2Cl2 (100 мл) и снова упаривали. Фильтрат упаривали досуха при пониженном давлении и очищали остаток MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 1-(бромметил)-2-фтор-3-нитробензола (8,11 г, 34%) в виде не совсем белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3): δ 8,02 (м, 1H), 7,71 (м, 1H), 7,30 (тд, 1H, J=8,4, 1,6 Гц), 4,55 (д, 2H, J=1,6 Гц).
К перемешиваемой смеси 1-(бромметил)-2-фтор-3-нитробензола (0,70 г, 2,99 ммоль) и гидрохлорида диметиламина (0,48 г, 5,98 ммоль) в CH2Cl2 (10 мл) по каплям добавляли триэтиламин (0,91 г, 8,97 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч и упаривали досуха при пониженном давлении. Остаток разбавляли водой (10 мл) и экстрагировали EtOAc (2×25 мл). EtOAc раствора промывали водой (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 1-(2-фтор-3-нитрофенил)-N,N-диметилметанамина (0,45 г, 76%) в виде желтой вязкой жидкости.
1Н ЯМР (400 МГц, CD3OD): δ 8,04-7,99 (м, 1H), 7,78-7,73 (м, 1H), 7,37 (тд, 1H, J=8,0, 1,0 Гц), 3,64 (д, 2H, J=2,0 Гц), 2,29 (д, 6H, J=0,8 Гц).
Смесь полученного выше 1-(2-фтор-3-нитрофенил)-N,N-диметилметанамина (0,45 г, 1,93 ммоль), железного порошка (1,35 г, 2,41 ммоль), 2 н. HCl (1 мл) и этанола (5 мл) нагревали при температуре кипения с обратным холодильником при перемешивании в течение 2 ч. После охлаждения до комнатной температуры смесь фильтровали через слой целита. Фильтрат упаривали досуха при пониженном давлении, остаток разбавляли водой (10 мл) и подщелачивали до pH 10 добавлением твердого K2CO3. Водную смесь экстрагировали EtOAc (2×25 мл), EtOAc раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением указанного в заголовке соединения (0,35 г, 91%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 6,88 (тд, 1H, J=7,8, 1,0 Гц), 6,72-6,67 (м, 2H), 3,71 (ушир.с, 2H), 3,47 (д, 2H, J=1,6 Гц), 2,27 (с, 6H).
Препаративный пример 13
Получение 2-фтор-3-(пирролидин-1-илметил)анилина (соединение формулы (VIII), где Rb=2-фтор-3-(пирролидин-1-илметил))
К перемешиваемому раствору 1-(бромметил)-2-фтор-3-нитробензола (2,00 г, 8,54 ммоль) и пирролидина (0,91 г, 12,82 ммоль) в CH2Cl2 (15 мл) по каплям при 0°C добавляли триэтиламин (1,72 г, 17,08 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и затем упаривали досуха при пониженном давлении. Остаток разбавляли водой (15 мл) и экстрагировали EtOAc (2×30 мл). EtOAc раствор промывали насыщенным раствором соли (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 1-(2-фтор-3-нитробензил)пирролидина (1,20 г, 63%) в виде вязкого масла.
1H ЯМР (400 МГц, CD3OD): δ 8,02-7,97 (м, 1H), 7,81-7,76 (м, 1H), 7,36 (тд, 1H, J=8,0, 1,2 Гц), 3,81 (д, 1H, J=2,0 Гц), 2,62-2,58 (м, 4H), 1,84-1,80 (м, 4H).
Указанное в заголовке соединение получали, как описано в препаративном примере 12, при использовании 1-(2-фтор-3-нитробензил)пирролидина вместо 1-(2-фтор-3-нитрофенил)-N,N-диметилметанамина. Выход: 80%;
1H ЯМР (400 МГц, CD3OD): δ 6,87 (тд, 1H, J=8,0, 0,8 Гц), 6,77 (тд, 1H, J=8,0, 2,0 Гц), 6,68-6,64 (м, 1H), 3,67 (д, 2H, J=1,6 Гц), 2,60-2,57 (м, 4H), 1,82-1,78 (м, 4H).
Препаративный пример 14
Получение 2-фтор-3-(морфолинометил)анилина (соединение формулы (VIII), где Rb=2-фтор-3-(морфолинометил))
Перемешиваемый раствор 1-(бромметил)-2-фтор-3-нитробензола (2,50 г, 10,6 ммоль) и морфолина (2,78 г, 32,0 ммоль) в толуоле (24 мл) нагревали при температуре кипения с обратным холодильником в течение 2,5 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем промывали 1 н. NaOH (2×20 мл). Водный слой экстрагировали EtOAc (2×25 мл), объединенный раствор в толуоле и EtOAc экстракты сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 4-(2-фтор-3-нитробензил)морфолина (1,95 г, 89%) в виде светло-желтого твердого вещества.
1Н ЯМР (400 МГц, CD3OD): δ 8,02-7,97 (м, 1H), 7,83-7,78 (м, 1H), 7,36 (тд, 1H, J=8,0, 1,0 Гц), 3,70-3,67 (м, 6H), 2,51 (ушир.т, 4H, J=4,6 Гц).
Указанное в заголовке соединение получали, как описано в препаративном примере 12, при использовании 4-(2-фтор-3-нитробензил)морфолина вместо 1-(2-фтор-3-нитрофенил)-N,N-диметилметанамина. Выход: 90%;
1H ЯМР (400 МГц, CD3OD): δ 6,87 (тд, 1H, J=8,0, 0,8 Гц), 6,77 (тд, 1H, J=8,0, 2,0 Гц), 6,68-6,64 (м, 1H), 3,68 (ушир.т, 4H, J=4,8 Гц), 3,55 (д, 2H, J=1,6 Гц), 2,49 (ушир.т, 4H, J=4,8 Гц).
Препаративный пример 15
Получение 3-амино-4-((диметиламино)метил)бензонитрила (соединение формулы (VIII), где Rb=5-циано-2-(диметиламино)метил)
К перемешиваемой смеси 4-(бромметил)-3-нитробензонитрила (5,00 г, 20,74 ммоль) (полученного согласно способу, описанному в WO 07/024945 A1) и гидрохлорида диметиламина (2,03 г, 24,89 ммоль) в CH2Cl2 (70 мл) по каплям при 0°C добавляли триэтиламин (6,30 г, 62,23 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Реакционную смесь упаривали досуха при пониженном давлении, остаток разбавляли водой (20 мл) и экстрагировали CH2Cl2 (3×100 мл). CH2Cl2 раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 4-((диметиламино)метил)-3-нитробензола (3,58 г, 84%) в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 8,15 (ушир.с, 1H), 7,92 (ушир.с, 1H), 7,85 (ушир.д, 1H, J=7,2 Гц), 3,80 (с, 2H), 2,27 (с, 6H).
Указанное в заголовке соединение получали, как описано в препаративном примере 11, при использовании 4-((диметиламино)метил)-3-нитробензонитрила вместо 3-(диметиламино)-5-нитробензонитрила. Выход: 91%;
1H ЯМР (400 МГц, CDCl3): δ 7,02 (дд, 1Н, J=7,6, 0,4 Гц), 6,91 (дд, 1Н, J=7,6, 1,6 Гц), 6,84 (д, 1H, J=1,6 Гц), 5,05 (ушир.с, 2H), 3,43 (с, 2H), 2,18 (с, 6H).
Препаративный пример 16
Получение 3-амино-2-((диметиламино)метил)бензонитрила (соединение формулы (VIII), где Rb=3-циано-2-(диметиламино)метил)
К перемешиваемой смеси 2-(бромметил)-3-нитробензонитрила (1,10 г, 4,56 ммоль) (полученного согласно способу, описанному в Tetrahedron 40: 1863-1868 (1984)) и гидрохлорида диметиламина (0,74 г, 9,13 ммоль) в CH2Cl2 (15 мл) по каплям при 0°C добавляли триэтиламин (1,85 г, 18,25 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 ч и упаривали досуха при пониженном давлении. Остаток разбавляли водой (10 мл) и экстрагировали EtOAc (3×50 мл). EtOAc раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 2-((диметиламино)метил)-3-нитробензонитрила (0,75 г, 80%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 7,93 (ушир.д, 1H, J=8,0 Гц), 7,85 (дд, 1H, J=8,0, 1,4 Гц), 7,55 (т, 1H, J=8,0 Гц), 3,94 (с, 2H), 2,24 (с, 6H).
Указанное в заголовке соединение получали, как описано в препаративном примере 11, при использовании 2-((диметиламино)метил)-3-нитробензонитрила вместо 3-(диметиламино)-5-нитробензонитрила. Выход: 93%;
1Н ЯМР (400 МГц, CDCl3): δ 7,15 (т, 1H, J=7,6 Гц), 7,00 (дд, 1H, J=7,6, 0,8 Гц), 6,83 (д, 1H, J=7,6 Гц), 5,06 (ушир.с, 2H), 3,73 (с, 2H), 2,29 (с, 6H).
Препаративный пример 17
Получение 3-амино-5-((диметиламино)метил)бензонитрила (соединение формулы (VIII), где Rb=3-циано-5-(диметиламино)метил)
К перемешиваемой смеси 3-(бромметил)-5-нитробензонитрила (1,50 г, 6,22 ммоль) (полученного согласно способу, описанному в J. Org. Chem. 55: 1040-1043 (1990)) и гидрохлорида диметиламина (1,01 г, 12,44 ммоль) в CH2Cl2 (15 мл) по каплям при 0°C добавляли триэтиламин (1,88 г, 18,66 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 ч и упаривали досуха при пониженном давлении. Остаток разбавляли водой (15 мл) и экстрагировали EtOAc (3×50 мл). EtOAc раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 3-((диметиламино)метил)-5-нитробензонитрила (1,10 г, 87%) в виде вязкой жидкости.
1H ЯМР (400 МГц, CDCl3): δ 8,45 (с, 1H), 8,40 (д, 1H, J=1,6 Гц), 8,01 (с, 1H), 3,59 (с, 2H), 2,30 (с, 6H).
Указанное в заголовке соединение получали, как описано в препаративном примере 11, при использовании 3-((диметиламино)метил)-5-нитробензонитрила вместо 3-(диметиламино)-5-нитробензонитрила. Выход: 98%;
1H ЯМР (400 МГц, CDCl3): δ 6,96 (м, 1H), 6,88 (ушир.д, 1H, J=0,8 Гц), 6,80 (дд, 1Н, J=2,4, 1,6 Гц), 3,86 (ушир.с, 2H), 3,34 (с, 2H), 2,24 (с, 6H).
Препаративный пример 18
Получение 3-амино-4-(пирролидин-1-илметил)бензонитрила (соединение формулы (VIII), где Rb=5-циано-2-(пирролидин-1-илметил))
К перемешиваемому раствору 4-(бромметил)-3-нитробензонитрила (5,12 г, 21,57 ммоль) и пирролидина (1,84 г, 25,88 ммоль) в CH2Cl2 (72 мл) по каплям при 0°C добавляли триэтиламин (6,54 г, 64,71 ммоль). Смесь перемешивали при комнатной температуре в течение 1,5 ч и упаривали досуха при пониженном давлении. Остаток разбавляли водой (30 мл) и экстрагировали CH2Cl2 (3×100 мл). CH2Cl2 раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 3-нитро-4-(пирролидин-1-илметил)бензонитрила (2,24 г, 45%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3): δ 8,16 (д, 1H, J=1,6 Гц), 7,94 (ушир.д, 1H, J=8,0 Гц), 7,83 (дд, 1H, J=8,0, 1,6 Гц), 3,99 (с, 2H), 2,54 (ушир.с, 4H), 1,79 (м, 4H).
Указанное в заголовке соединение получали, как описано в препаративном примере 11, при использовании 3-нитро-4-(пирролидин-1-илметил)бензонитрила вместо 3-(диметиламино)-5-нитробензонитрила. Выход: 91%;
1Н ЯМР (400 МГц, CDCl3): δ 7,06 (д, 1H, J=7,6 Гц), 6,91 (дд, 1H, J=7,6, 1,6 Гц), 6,84 (д, 1H, J=1,6 Гц), 5,08 (ушир.с, 2H), 3,64 (с, 2H), 2,47 (ушир.с, 4H), 1,78 (ушир.с, 4H).
Препаративный пример 19
Получение 3-амино-2-(пирролидин-1-илметил)бензонитрила (соединение формулы (VIII), где Rb=3-циано-2-(пирролидин-1-илметил))
К перемешиваемому раствору 2-(бромметил)-3-нитробензонитрила (1,10 г, 4,56 ммоль) и пирролидина (0,65 г, 9,13 ммоль) в CH2Cl2 (15 мл) по каплям при 0°C добавляли триэтиламин (1,85 г, 18,25 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч и упаривали досуха при пониженном давлении. Остаток разбавляли водой (10 мл) и экстрагировали CH2Cl2 (3×50 мл). CH2Cl2 раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 3-нитро-2-(пирролидин-1-илметил)бензонитрила (0,96 г, 91%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3): δ 7,90 (д, 1H, J=7,6 Гц), 7,83 (дд, 1H, J=7,6, 0,8 Гц), 7,52 (т, 1H, J=7,6 Гц), 4,14 (с, 2H), 2,52 (ушир.с, 4H), 1,72 (ушир.с, 4H).
Указанное в заголовке соединение получали, как описано в препаративном примере 11, при использовании 3-нитро-2-(пирролидин-1-илметил)бензонитрила вместо 3-(диметиламино)-5-нитробензонитрила. Выход: 93%;
1Н ЯМР (400 МГц, CDCl3): δ 7,13 (т, 1H, J=7,8 Гц), 6,99 (дд, 1H, J=7,8, 1,2 Гц), 6,82 (д, 1H, J=8,0 Гц), 5,11 (ушир.с, 2H), 3,91 (с, 2H), 2,58 (ушир.с, 4H), 1,81 (ушир.с, 4H).
Препаративный пример 20
Получение 3-амино-5-(пирролидин-1-илметил)бензонитрила (соединение формулы (VIII), где Rb=3-циано-5-(пирролидин-1-илметил))
К перемешиваемому раствору 3-(бромметил)-5-нитробензонитрила (1,50 г, 6,22 ммоль) и пирролидина (0,53 г, 7,46 ммоль) в CH2Cl2 (15 мл) по каплям при 0°C добавляли триэтиламин (1,88 г, 18,68 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Реакционную смесь упаривали досуха при пониженном давлении, остаток разбавляли водой (15 мл) и экстрагировали EtOAc (3×50 мл). EtOAc раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 3-нитро-5-(пирролидин-1-илметил)бензонитрила (1,30 г, 90%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 8,45 (ушир.с, 1H), 8,39 (ушир.т, 1H, J=1,6 Гц), 8,02 (ушир.с, 1H), 3,78 (с, 2H), 2,56 (ушир.с, 4H), 1,84 (ушир.с, 4H).
Указанное в заголовке соединение получали, как описано в препаративном примере 11, при использовании 3-нитро-5-(пирролидин-1-илметил)бензонитрила вместо 3-(диметиламино)-5-нитробензонитрила. Выход: 85%;
1Н ЯМР (400 МГц, CDCl3): δ 6,99 (псевдо т, 1H, J=1,6 Гц), 6,94 (псевдо т, 1H, J=1,6 Гц), 6,79 (дд, 1H, J=2,4, 1,6 Гц), 3,87 (ушир.с, 2H), 3,56 (с, 2H), 2,54 (м, 4H), 1,81 (м, 4H).
Препаративный пример 21
Получение 3-амино-4-(морфолинометил)бензонитрила (соединение формулы (VIII), где Rb=5-циано-2-(морфолинометил))
К перемешиваемому раствору 4-(бромметил)-3-нитробензонитрила (7,12 г, 29,55 ммоль) и морфолина (3,09 г, 35,45 ммоль) в CH2Cl2 (98 мл) по каплям при 0°C добавляли триэтиламин (8,97 г, 88,64 ммоль). Смесь перемешивали при комнатной температуре в течение 1,5 ч и упаривали досуха при пониженном давлении. Остаток разбавляли водой (40 мл) и экстрагировали CH2Cl2 (3×100 мл). CH2Cl2 раствор промывали водой (50 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 4-(морфолинометил)-3-нитробензонитрила (5,84 г, 80%) в виде желтого твердого вещества.
1H ЯМР(400 МГц, CDCl3): δ 8,12 (с, 1H), 7,83 (ушир.с, 2H), 3,84 (с, 2H), 3,67 (т, 4H, J=4,6 Гц), 2,45 (т, 4H, J=4,6 Гц).
Указанное в заголовке соединение получали, как описано в препаративном примере 11, при использовании 4-(морфолинометил)-3-нитробензонитрила вместо 3-(диметиламино)-5-нитробензонитрила. Выход: 78%;
1Н ЯМР (400 МГц, CDCl3): δ 7,05 (д, 1H, J=8,0 Гц), 6,92 (дд, 1H, J=8,0, 1,6 Гц), 6,86 (д, 1H, J=1,6 Гц), 5,02 (ушир.с, 2H), 3,68 (ушир.т, 4H, J=4,0 Гц), 3,53 (с, 2H), 2,41 (ушир.с, 4H).
Препаративный пример 22
Получение 3-амино-2-(морфолинометил)бензонитрила (соединение формулы (VIII), где Rb=3-циано-2-(морфолинометил))
К перемешиваемому раствору 2-(бромметил)-3-нитробензонитрила (1,10 г, 4,56 ммоль) и морфолина (0,80 г, 9,13 ммоль) в CH2Cl2 (15 мл) по каплям при 0°C добавляли триэтиламин (1,85 г, 18,25 ммоль). Смесь перемешивали при комнатной температуре в течение 1,5 ч и упаривали досуха при пониженном давлении. Остаток разбавляли водой (10 мл) и экстрагировали CH2Cl2 (3×50 мл). CH2Cl2 раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 2-(морфолинометил)-3-нитробензонитрила (1,02 г, 90%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3): δ 7,90 (ушир.д, 1H, J=8,0 Гц), 7,84 (дд, 1H, J=8,0, 1,2 Гц), 7,56 (т, 1H, J=8,0 Гц), 3,99 (с, 2H), 3,60 (ушир.т, 4H, J=4,4 Гц), 2,46 (ушир.с, 4H).
Указанное в заголовке соединение получали, как описано в препаративном примере 11, при использовании 2-(морфолинометил)-3-нитробензонитрила вместо 3-(диметиламино)-5-нитробензонитрила. Выход: 82%;
1Н ЯМР (400 МГц, CDCl3): δ 7,15 (т, 1H, J=7,6 Гц), 7,01 (д, 1H, J=7,6 Гц), 6,83 (д, 1H, J=7,6 Гц), 5,00 (ушир.с, 2H), 3,78 (с, 2H), 3,69 (ушир.с, 4H), 2,50 (ушир.с, 4H).
Препаративный пример 23
Получение 3-амино-5-(морфолинометил)бензонитрила (соединение формулы (VIII), где Rb=3-циано-5-(морфолинометил))
К перемешиваемому раствору 3-(бромметил)-5-нитробензонитрила (1,50 г, 6,22 ммоль) и морфолина (0,65 г, 7,46 ммоль) в CH2Cl2 (15 мл) по каплям при 0°C добавляли триэтиламин (1,88 г, 18,66 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и затем упаривали досуха при пониженном давлении. Остаток разбавляли водой (15 мл) и экстрагировали EtOAc (3×50 мл). EtOAc раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 3-(морфолинометил)-5-нитробензонитрила (0,87 г, 85%) в виде не совсем белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3): δ 8,45 (ушир.с, 1H), 8,41 (ушир.с, 1H), 8,01 (ушир.с, 1H), 3,74 (ушир.т, 4H, J=4,4 Гц), 3,64 (с, 2H), 2,48 (ушир.т, 4H, J=4,4 Гц).
Указанное в заголовке соединение получали, как описано в препаративном примере 11, при использовании 3-(морфолинометил)-5-нитробензонитрила вместо 3-(диметиламино)-5-нитробензонитрила. Выход: 85%;
1H ЯМР (400 МГц, CDCl3): δ 7,00 (ушир.т, 1Н, J=1,6 Гц), 6,93 (ушир.с, 1H), 6,81 (дд, 1Н, J=2,4, 1,6 Гц), 3,88 (ушир.с, 2H), 3,74 (ушир.т, 4H, J=4,6 Гц), 3,44 (с, 2H), 2,47 (ушир.с, 4H).
Препаративный пример 24
Получение 2-(2-фторфенокси)ацетальдегида (соединение формулы (IX), где Rb=2-фтор, X=O). Указанное соединение получали в 2 последовательные стадии.
Перемешиваемую смесь 2-фторфенола (1,00 г, 8,92 ммоль), 2-бром-1,1-диэтоксиэтана (1,75 г, 8,92 ммоль) и K2CO3 (1,47 г, 10,7 ммоль) в безводном ДМФА (10 мл) нагревали при 110°C в течение ночи. Реакционную смесь вливали в ледяную воду (15 мл) и экстрагировали EtOAc (2×100 мл). EtOAc раствор промывали водой (25 мл) и насыщенным раствором соли (25 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 1-(2,2-диэтоксиэтокси)-2-фторбензола (1,65 г, 81%) в виде вязкой жидкости.
1Н ЯМР (400 МГц, CDCl3): δ 7,09-6,96 (м, 3H), 6,93-6,87 (м, 1H), 4,85 (т, 1H, J=5,2 Гц), 4,07 (д, 2H, J=5,2 Гц), 3,82-3,74 (м, 2H), 3,69-3,61 (м, 2H), 1,24 (т, 6H, J=7,0 Гц).
К перемешиваемому раствору 1-(2,2-диэтоксиэтокси)-2-фторбензола (1,65 г, 7,23 ммоль) в смеси 1,4-диоксана (50 мл) и воды (40 мл) при 0°C добавляли конц. HCl (17,6 мл) и перемешивали смесь при комнатной температуре в течение ночи. Реакционную смесь охлаждали до 0°C, нейтрализовали добавлением насыщенного раствора NaHCO3 и экстрагировали EtOAc (2×200 мл). EtOAc раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением указанного в заголовке соединения (0,78 г, 71%) в виде вязкой жидкости.
1Н ЯМР (400 МГц, CDCl3): δ 9,87 (с, 1H), 7,12 (ддд, 1H, J=11,4, 8,2, 1,6 Гц), 7,08-7,04 (м, 1H), 7,01-6,95 (м, 1H), 6,91 (тд, 1H, J=8,2, 1,6 Гц), 4,63 (с, 2H).
Препаративный пример 25
Получение 2-(2-фторфенилтио)ацетальдегида (соединение формулы (IX), где Rb=2-фтор, X=S)
Указанное соединение получали в 2 последовательные стадии.
Смесь 2-фтортиофенола (1,00 г, 7,80 ммоль), диэтилацеталя бромацетальдегида (1,41 мл, 9,36 ммоль) и Cs2CO3 (3,05 г, 9,36 ммоль) в безводном ДМФА (20 мл) перемешивали в атмосфере N2 при комнатной температуре в течение ночи. Реакционную смесь фильтровали через металлокерамическую воронку и разбавляли фильтрат водой (20 мл). Водную смесь экстрагировали Et2O (3×100 мл), органическую фазу сушили над безводным MgSO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением (2,2-диэтоксиэтил)(2-фторфенил)сульфана (1,81 г, 95%) в виде вязкой жидкости.
1Н ЯМР (400 МГц, CDCl3): δ 7,46-7,41 (м, 1H), 7,24-7,18 (м, 1H), 7,09-7,02 (м, 1H), 4,64 (т, 1H, J=5,6 Гц), 3,69-3,61 (м, 2H), 3,56-3,49 (м, 2H), 3,10 (д, 2H, J=5,6 Гц), 1,17 (т, 6H, J=7,2 Гц).
К перемешиваемому раствору (2,2-диэтоксиэтил)(2-фторфенил)сульфана (1,00 г, 4,09 ммоль) в смеси 1,4-диоксана (30 мл) и воды (25 мл) при 0°C добавляли конц. HCl (9 мл) и перемешивали смесь при комнатной температуре в течение 2 ч. Реакционную смесь охлаждали до 0°C, нейтрализовали добавлением насыщенного раствора NaHCO3 и экстрагировали CH2Cl2 (3×50 мл). Органическую фазу промывали водой (50 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении, с получением указанного в заголовке соединения (0,59 г, 85%) в виде вязкой жидкости, которую сразу использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3): δ 9,56 (тд, 1H, J=3,2, 1,2 Гц), 7,42-7,38 (м, 1H), 7,31-7,25 (м, 1H), 7,12-7,06 (м, 2H), 3,58 (д, 2H, J=3,2 Гц).
Препаративный пример 26
Получение 3-(метил(2-оксоэтил)амино)бензонитрила (соединение формулы (IX), где Rb=3-циано, X=NMe)
Указанное соединение получали в 3 стадии, используя в качестве исходного вещества коммерчески доступный 3-аминобензонитрил.
К перемешиваемому раствору 3-аминобензонитрила (2,50 г, 21,10 ммоль) в безводном ДМСО (30 мл) при 0°C порциями добавляли NaH (0,61 г, 25,39 ммоль), перемешивали смесь при комнатной температуре в течение 20 мин и затем обрабатывали диэтилацетальбромацетальдегидом (4,20 г, 21,10 ммоль). Спустя 2 ч, при 0°C медленно добавляли насыщенный водный NH4Cl (20 мл) и экстрагировали реакционную смесь EtOAc (2×50 мл). EtOAc раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием EtOAc и гексана в качестве элюента, с получением 3-(2,2-диэтоксиэтиламино)бензонитрила (0,86 г, 17%) в виде светло-оранжевого масла.
1H ЯМР (400 МГц, CDCl3): δ 7,22 (тд, 1H, J=7,8, 0,6 Гц), 6,97 (м, 1H), 6,84-6,81 (м, 2H), 4,67 (т, 1H, J=5,4 Гц), 3,77-3,69 (м, 2H), 3,61-3,53 (м, 2H), 3,24 (д, 2H, J=5,6 Гц), 1,24 (т, 6H, J=7,0 Гц).
К перемешиваемому раствору 3-(2,2-диэтоксиэтиламино)бензонитрила (0,84 г, 3,59 ммоль) в безводном ДМФА (5 мл) при 0°C порциями добавляли NaH (0,10 г, 4,30 ммоль). Спустя 20 мин, добавляли MeI (0,61 г, 4,30 ммоль) и перемешивали смесь при комнатной температуре в течение 6 ч. Реакционную смесь охлаждали до 0°C и по каплям добавляли водный раствор NH4Cl (10 мл). Водную смесь экстрагировали CHCl3 (2×30 мл), органическую фазу сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением 3-((2,2-диэтоксиэтил)(метил)амино)бензонитрила (0,65 г, 73%) в виде вязкой жидкости.
1H ЯМР (400 МГц, CDCl3): δ 7,28-7,24 (м, 1H), 6,96-6,93 (м, 3H), 4,60 (т, 1H, J=5,2 Гц), 3,76-3,69 (м, 2H), 3,55-3,47 (м, 2H), 3,46 (д, 2H, J=5,2 Гц), 3,02 (с, 3H), 1,20 (т, 6H, J=7,0 Гц).
К перемешиваемому раствору 3-((2,2-диэтоксиэтил)(метил)амино)бензонитрила (0,65 г, 2,61 ммоль) в безводном диоксане (6 мл) при 0°C по каплям добавляли 1 н. HCl (4,30 ммоль) и перемешивали смесь при комнатной температуре в течение 1 ч. Реакционную смесь охлаждали до 0°C, нейтрализовали добавлением водного раствора NaHCO3 и экстрагировали CHCl3 (2×30 мл). CHCl3 раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении, с получением указанного в заголовке соединения (0,24 г, 52%) в виде вязкой жидкости, которую использовали на следующей стадии без дополнительной очистки.
1Н ЯМР (400 МГц, CDCl3): δ 9,74 (т, 1H, J=0,8 Гц), 7,29 (ддд, 1H, J=8,8, 7,4, 0,8 Гц), 7,02 (ддд, 1H, J=7,4, 0,8, 1,2 Гц), 6,86 (дд, 1H, J=2,4, 1,2 Гц), 6,82 (ддд, 1H, J=8,8, 2,4, 1,2 Гц) 4,14 (д, 2H, J=0,8 Гц), 3,10 (с, 3H).
Препаративный пример 27
Получение 2-((2-фторфенил)(метил)амино)ацетальдегида (соединение формулы (IX), где Rb=2-фтор, X=NMe)
Указанное соединение получали в 3 стадии, используя в качестве исходного вещества коммерчески доступный 2-фторанилин.
Перемешиваемую смесь 2-фторанилина (2,00 г, 17,90 ммоль), диметилацетальбромацетальдегида (3,25 мл, 21,40 ммоль) и Cs2CO3 (11,70 г, 35,80 ммоль) в безводном ДМФА (10 мл) нагревали до 120°C в течение ночи. Реакционную смесь упаривали досуха при пониженном давлении и экстрагировали остаток Et2O (2×150 мл). Et2O раствор промывали водой (4×50 мл) и насыщенным раствором соли (2×50 мл), сушили над безводным MgSO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси EtOAc и гексана в качестве элюента, с получением N-(2,2-диэтоксиэтил)-2-фторанилина (2,00 г, 49%) в виде бесцветного масла.
1Н ЯМР (400МГц, CDCl3): δ 7,03-6,95 (м, 2H), 6,82-6,77 (м, 1H), 6,71-6,65 (м, 1H), 4,72 (т, 1H, J=5,6 Гц), 3,78-3,71 (м, 2H), 3,62-3,54 (м, 2H), 3,29 (д, 2H, J=5,6 Гц), 1,26-1,22 (м, 6H)/
К перемешиваемому раствору N-(2,2-диэтоксиэтил)-2-фторанилина (1,00 г, 4,40 ммоль) в безводном ДМФА (10 мл) при 0°C порциями добавляли NaH (0,16 г, 6,60 ммоль). Спустя 30 мин, добавляли MeI (0,5 мл, 8,80 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Реакционную смесь экстрагировали EtOAc (2×100 мл), EtOAc раствор промывали водой (50 мл) и насыщенным раствором соли (50 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении, с получением N-(2,2-диэтоксиэтил)-2-фтор-N-метиланилина (0,41 г, 38%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3): δ 7,05-6,96 (м, 3H), 6,83-6,81 (м, 1H), 4,69 (т, 1H, J=5,2 Гц), 3,72-3,64 (м, 2H), 3,55-3,49 (м, 2H), 3,33 (дд, 2H, J=5,2, 1,2 Гц), 2,97 (с, 3H), 1,19-1,53 (м, 6H).
К перемешиваемому раствору N-(2,2-диэтоксиэтил)-2-фтор-N-метиланилина (0,40 г, 1,60 ммоль) в 1,4-диоксане (5 мл) при 0°C по каплям добавляли 2,5н HCl (5 мл) и перемешивали смесь при комнатной температуре в течение ночи. Реакционную смесь охлаждали до 0°C, нейтрализовали добавлением насыщенного раствора NaHCO3 и экстрагировали CHCl3 (2×50 мл). CHCl3 раствор промывали водой (20 мл) и насыщенным раствором соли (20 мл), сушили над Na2SO4, фильтровали и упаривали досуха при пониженном давлении, с получением указанного в заголовке соединения (0,27 г, 98%) в виде бесцветного масла, которое использовали на следующей стадии без дополнительной очистки.
1Н ЯМР (400 МГц, CDCl3): δ 9,80 (дд, 1H, J=2,4, 1,2 Гц), 7,07-6,89 (м, 4H), 3,91 (дд, 2H, J=1,8, 0,8 Гц), 2,97 (с, 3H).
Пример получения 1
Получение N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-виниланилина (пример 37)
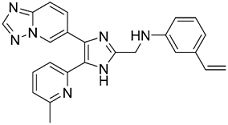
К перемешиваемому раствору 4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-карбальдегида (4,00 г, 13,14 ммоль) в 1,2-дихлорэтане (240 мл) добавляли 3-виниланилин (2,36 г, 19,71 ммоль) и AcOH (0,79 г, 13,14 ммоль) и нагревали смесь при 80°C в течение 2 ч. Реакционную смесь охлаждали до 0°C и добавляли NaBH(OAc)3 (5,56 г, 26,20 ммоль). Смесь перемешивали при 40°C в течение ночи и затем доводили pH реакционной смеси до 7-8 добавлением при 0°C 10% раствора K2CO3. Реакционную смесь экстрагировали 5% MeOH в CHCl3 (2×200 мл), органическую фазу сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси MeOH и CH2Cl2 (1/19 (об./об.)) в качестве элюента, с получением указанного в заголовке соединения (2,89 г, 63%) в виде твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 10,59 (ушир.с, 1H), 8,94 (с, 1H), 8,37 (с, 1H), 7,81 (д, 1H, J=9,2 Гц), 7,77 (д, 1H, J=9,2 Гц), 7,45 (т, 1H, J=7,8 Гц), 7,20 (ушир.д, 1H, перекрыв., J=7,6 Гц), 7,15 (т, 1H, перекрыв., J=7,8 Гц), 7,00 (д, 1H, J=8,0 Гц), 6,86 (д, 1H, J=7,6 Гц), 6,75 (т, 1H, J=2,0 Гц), 6,63 (дд, 1H, перекрыв., J=17,6, 10,8 Гц), 6,61 (дд, 1H, перекрыв., J=8,0, 2,0 Гц), 5,69 (дд, 1H, J=17,6, 0,8 Гц), 5,21 (дд, 1H, J=10,8, 0,8 Гц), 4,55 (с, 2H), 4,39 (ушир.с, 1H), 2,51 (с, 3H); МС (ESI) m/z 408,21.
Пример получения 2
Получение гидрохлорида N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-виниланилина (пример 38)
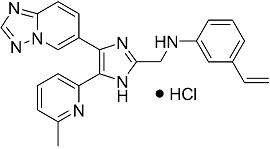
Перемешиваемую суспензию N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-виниланилина (1,00 г, 2,45 ммоль) в безводном CHCl3 (12 мл) нагревали при 50°C, с получением прозрачного раствора. CHCl3 раствор охлаждали до 0°C, добавляли 1,0M HCl в Et2O (7,36 мл, 7,36 ммоль). Спустя 5 мин, осадок фильтровали в атмосфере N2 и тщательно сушили над P2O5 в вакууме, с получением указанного в заголовке соединения (1,07 г, 98%) в виде желтого порошка.
1Н ЯМР (400 МГц, ДМСО-d6): δ 9,49 (дд, 1H, J=1,6, 0,8 Гц), 8,65 (с, 1H), 7,97 (дд, 1H, J=9,2, 0,8 Гц), 7,86 (дд, 1H, перекрыв., J=9,2, 1,6 Гц), 7,85 (т, 1H, перекрыв., J=7,8 Гц), 7,65 (д, 1H, J=8,0 Гц), 7,38 (д, 1H, J=7,6 Гц), 7,12 (т, 1H, J=7,8 Гц), 6,91 (т, 1H, J=1,6 Гц), 6,79 (д, 1H, J=7,6 Гц), 6,71 (дд, 1H, J=8,0, 1,6 Гц), 6,64 (дд, 1Н, J=17,6, 11,2 Гц), 5,80 (дд, 1Н, J=17,6, 0,8 Гц), 5,20 (дд, 1H, J=11,2, 0,8 Гц), 4,79 (с, 2H), 2,51 (с, 3H).
Пример получения 3
Получение сульфата N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-виниланилина (пример 39)
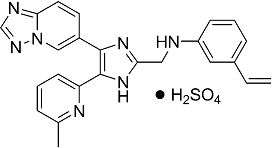
К перемешиваемой суспензии N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-виниланилина (100 мг, 0,25 ммоль) в безводном EtOH (2 мл) при 0°C добавляли 10% H2SO4 в безводном EtOH (0,20 мл, 0,37 ммоль). Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 10 мин. Реакционную смесь разбавляли безводным Et2O (8 мл) и дополнительно перемешивали в течение 10 мин. Осажденные вещества фильтровали в атмосфере N2, промывали безводным Et2O (4×4 мл) и затем тщательно сушили над P2O5 в вакууме, с получением указанного в заголовке соединения (79 мг, 64%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6): δ 9,40 (дд, 1H, J=1,6, 0,8 Гц), 8,63 (с, 1H), 7,98 (дд, 1H, J=9,2, 0,8 Гц), 7,84 (т, 1H, J=8,0 Гц), 7,76 (дд, 1H, J=9,2, 1,6 Гц), 7,43 (д, 1H, J=7,6 Гц), 7,40 (д, 1H, J=8,0 Гц), 7,13 (т, 1H, J=7,8 Гц), 6,80 (ушир.с, 1H), 6,79 (д, 1H, перекрыв., J=7,6 Гц), 6,64 (дд, 1H, перекрыв., J=17,6, 11,2 Гц), 6,63 (дд, 1H, перекрыв., J=7,6, 2,0 Гц), 5,74 (дд, 1H, J=17,6, 0,8 Гц), 5,21 (дд, 1H, J=11,2, 0,8 Гц), 4,68 (с, 2H), 2,58 (с, 3H).
Пример получения 4
Получение 3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-4-((диметиламино)метил)бензонитрила (пример 116)
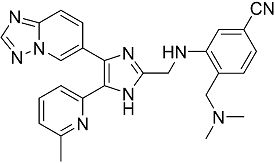
К перемешиваемому раствору 4-((1,2,4)триазоло(1,5-a)пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-карбальдегида (0,50 г, 1,64 ммоль) в 1,2-дихлорэтане (30 мл) добавляли 3-амино-4-((диметиламино)метил)бензонитрил (0,43 г, 2,46 ммоль) и AcOH (0,20 г, 3,29 ммоль) и смесь нагревали при 80°C в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в безводном MeOH (30 мл). К метанольному раствору при 0°C добавляли NaBH4 (0,25 г, 6,57 ммоль) и затем смесь оставляли нагреваться до комнатной температуры и дополнительно перемешивали в течение 3 ч. pH реакционной смеси доводили до 7-8 добавлением при 0°C 1 н. HCl и затем удаляли MeOH при пониженном давлении. Водную смесь экстрагировали CH2Cl2 (2×50 мл), CH2Cl2 раствор сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси MeOH и CH2Cl2 (1/19 (об./об.)) в качестве элюента, с получением указанного в заголовке соединения (0,60 г, 79%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 8,99 (с, 1H), 8,36 (с, 1H), 7,86 (дд, 1H, J=9,2, 1,6 Гц), 7,78 (дд, 1H, J=9,2, 0,8 Гц), 7,47 (т, 1H, J=7,6 Гц), 7,23 (ушир.д, 1H, J=7,6 Гц), 7,09 (д, 1H, J=7,6 Гц), 7,01 (д, 1H, J=7,6 Гц), 6,98 (дд, 1H, J=7,6, 1,6 Гц), 6,93 (ушир.с, 1H), 4,59 (с, 2H), 3,63 (с, 2H), 2,53 (с, 3H), 2,33 (с, 6H); МС (ESI) m/z 464,23 (MH+).
Соединения, перечисленные в последующей таблице 1, получали способом, аналогично описанному выше в примерах получения 1-4. В таблицу 1 включены данные масс-спектрометрии для указанных соединений.
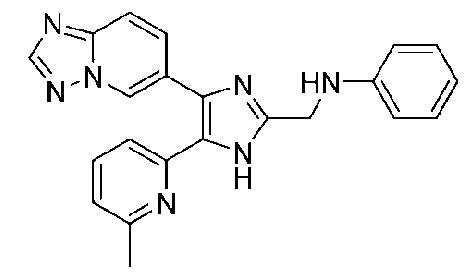
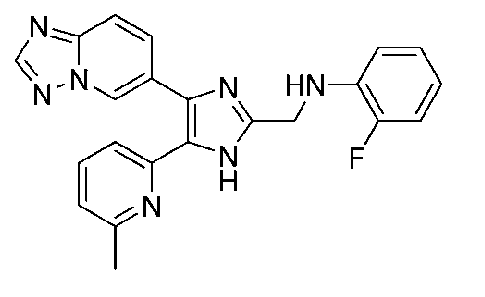
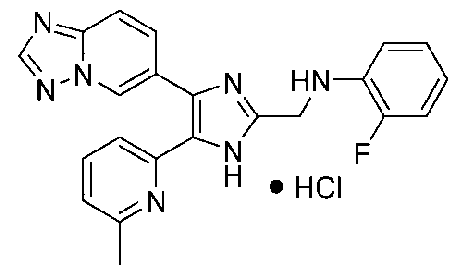
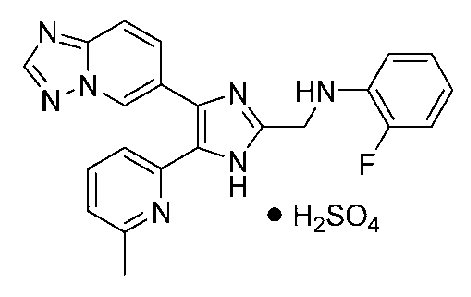
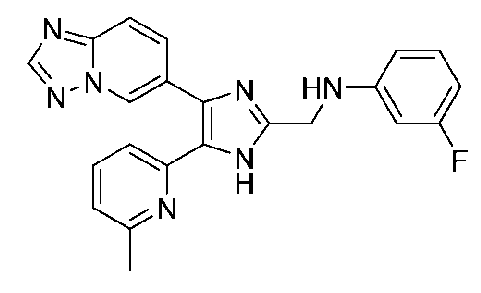
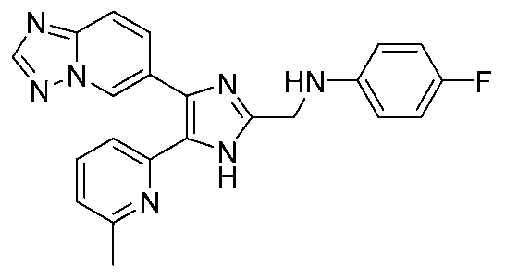
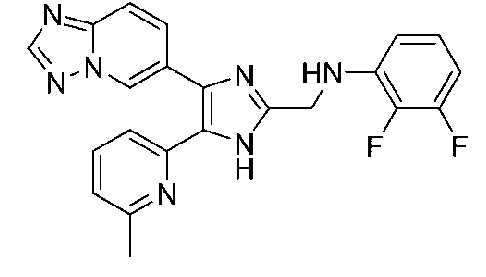
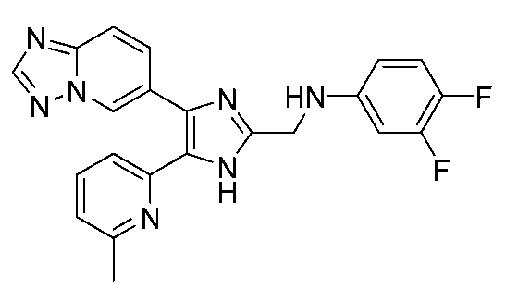
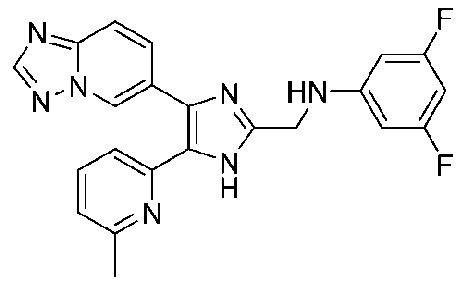
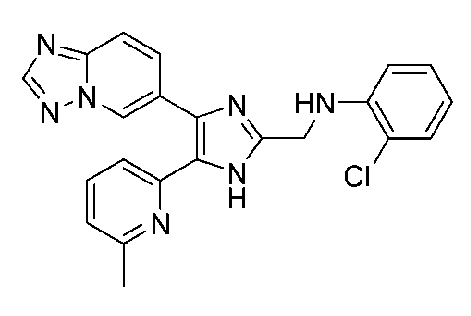
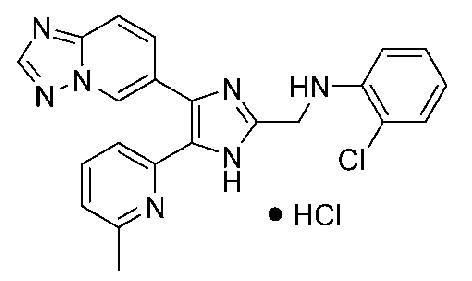
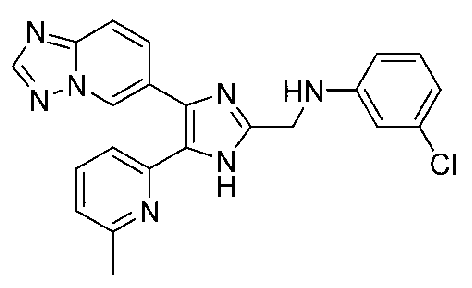
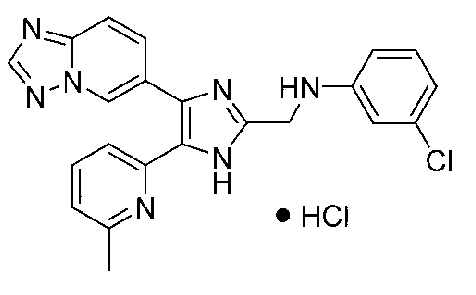
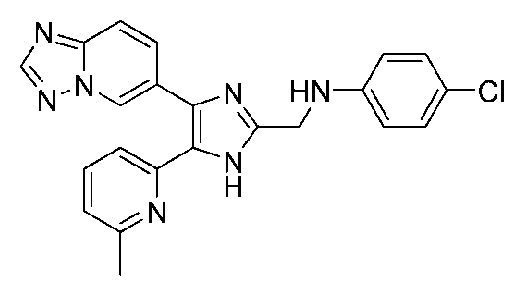
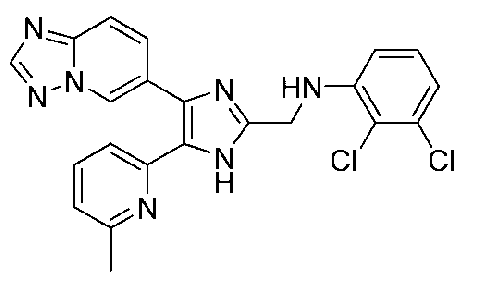
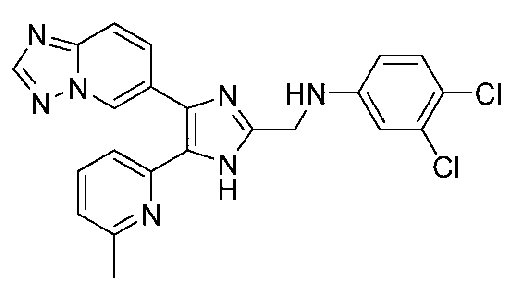
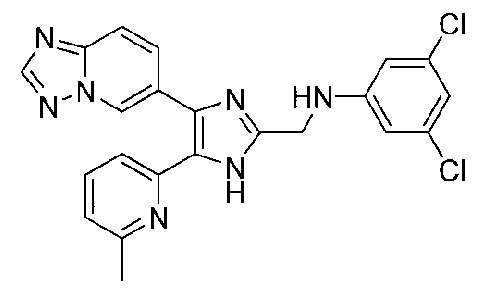
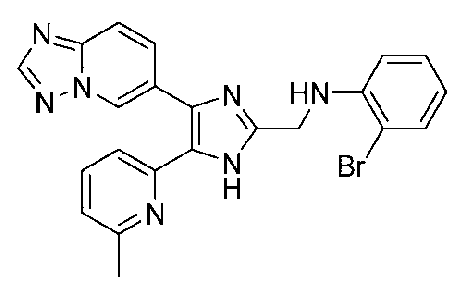
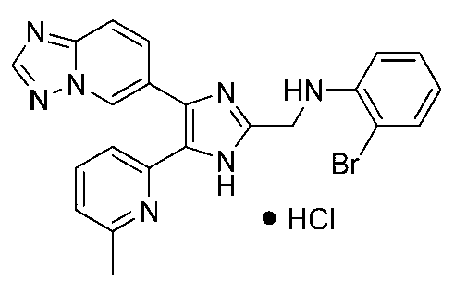
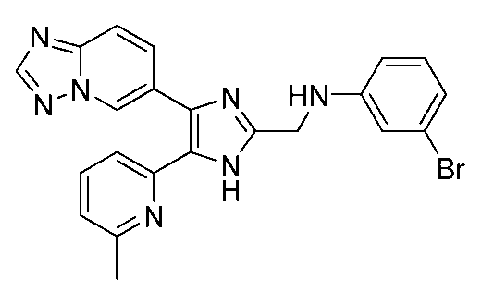
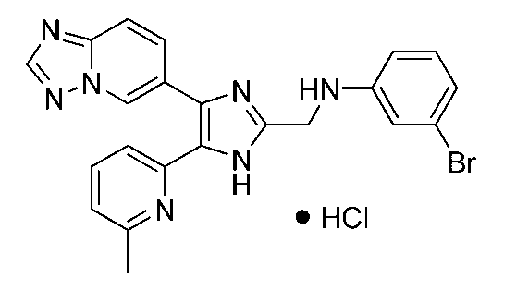
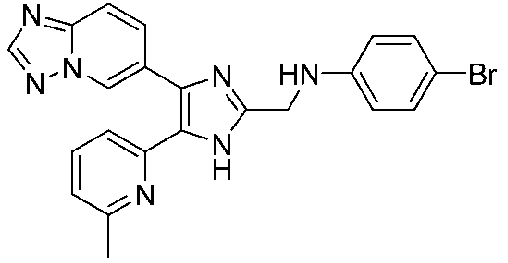
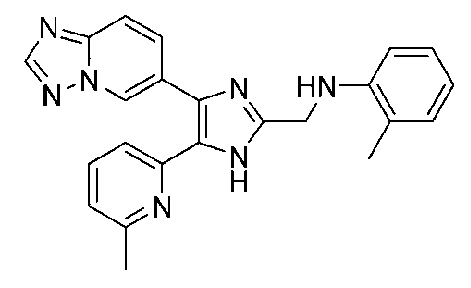
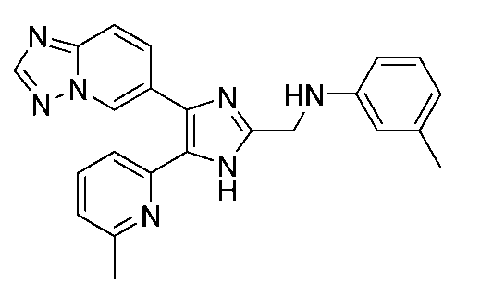
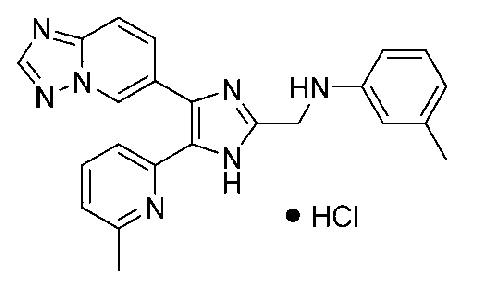
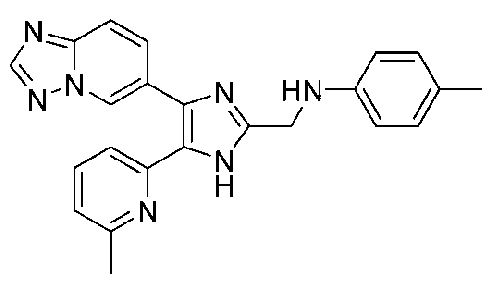
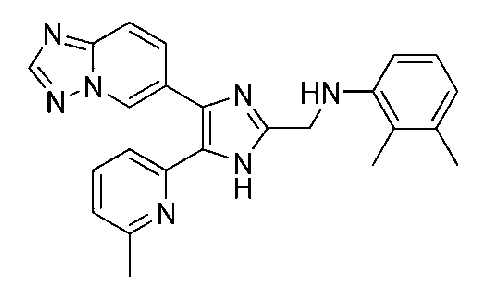
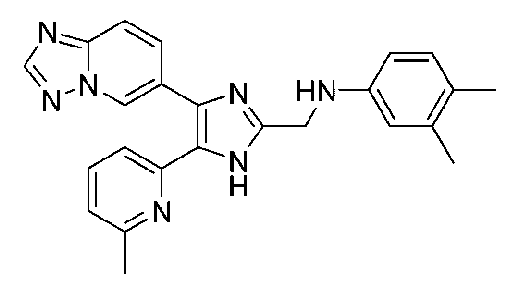
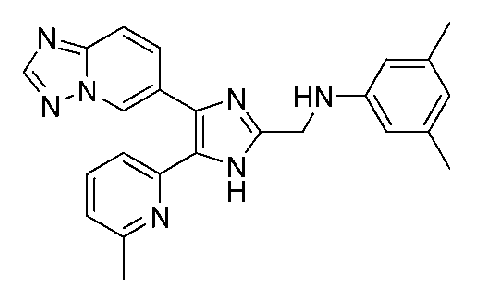
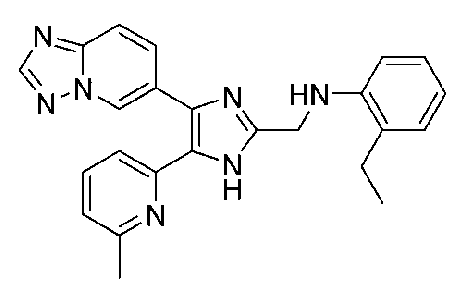
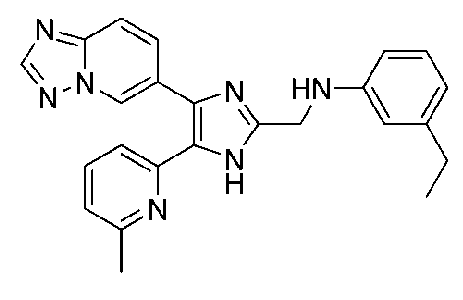
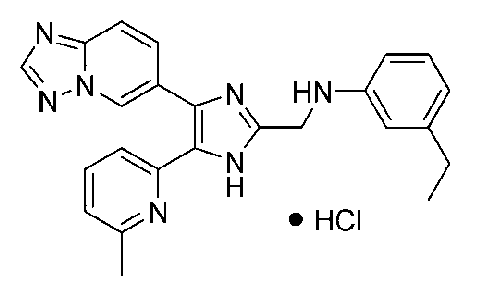
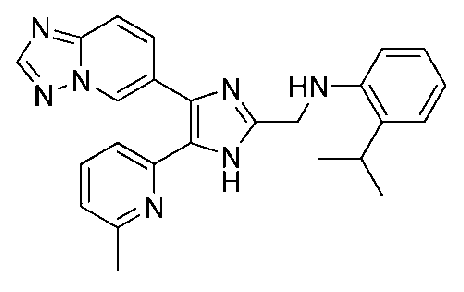
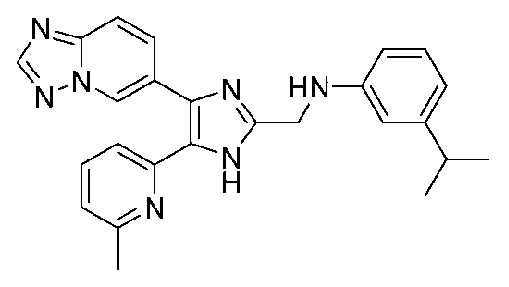
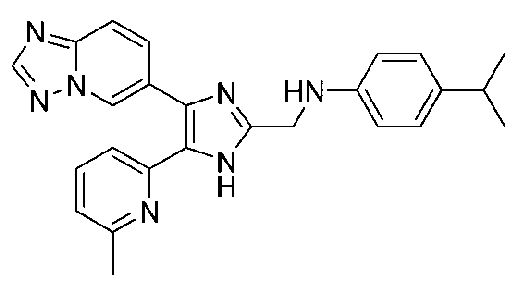
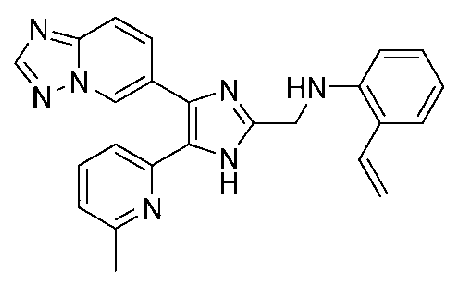
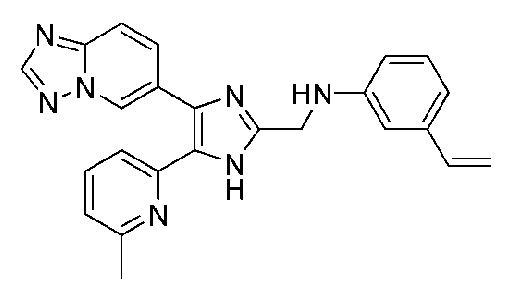
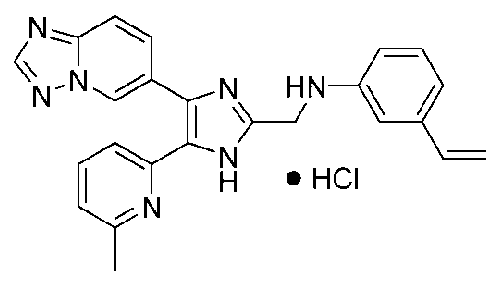
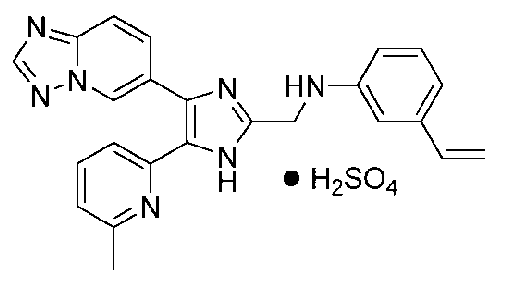
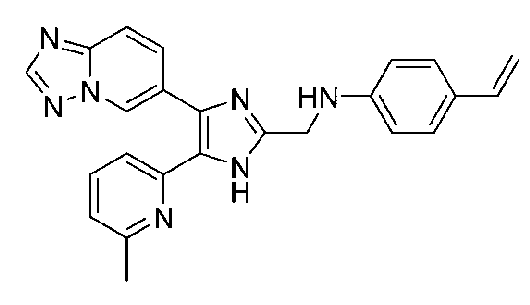
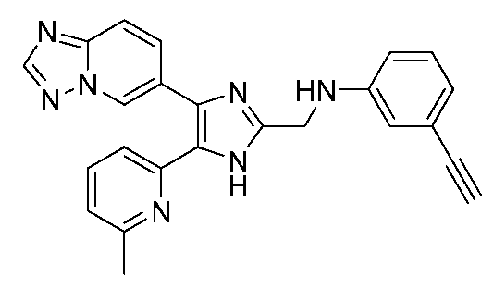
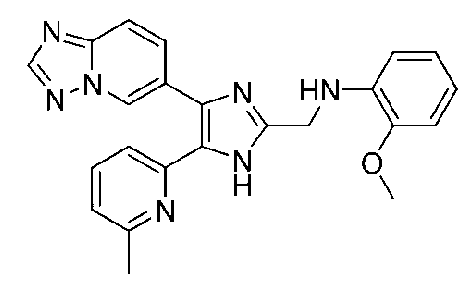
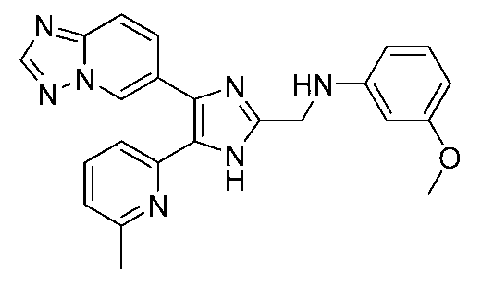
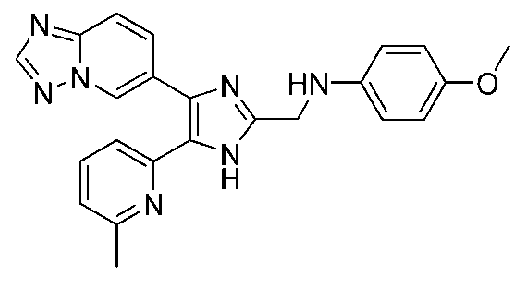
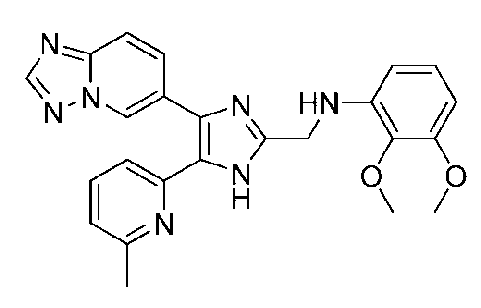
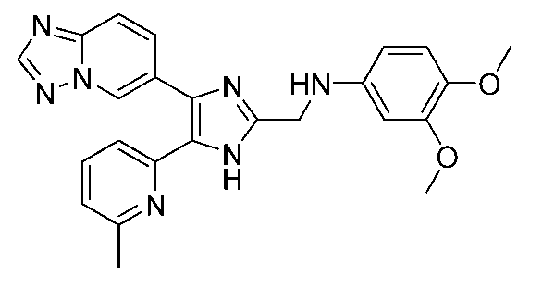
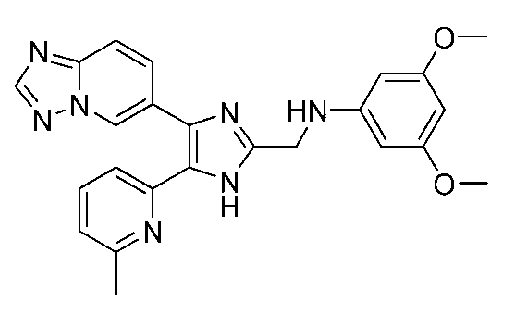
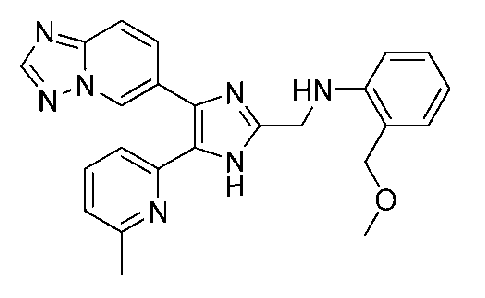
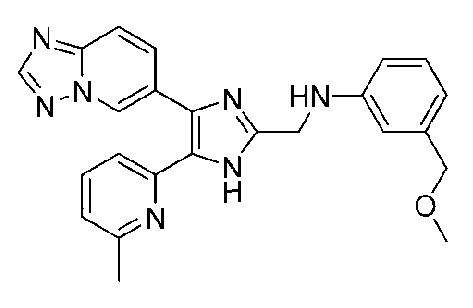
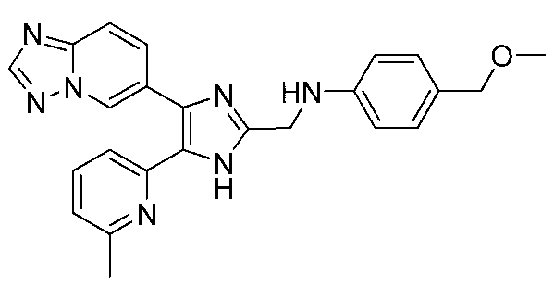
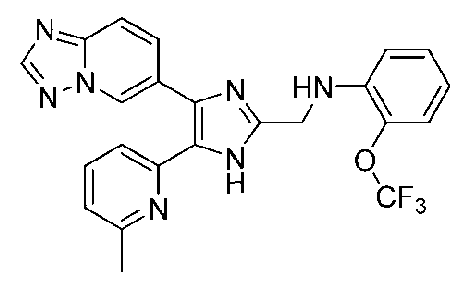
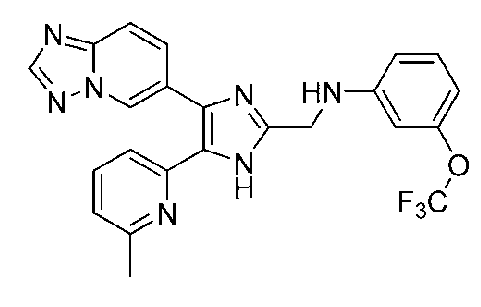
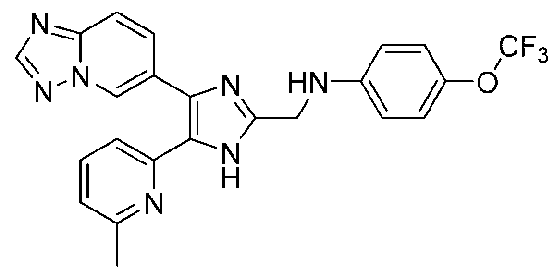
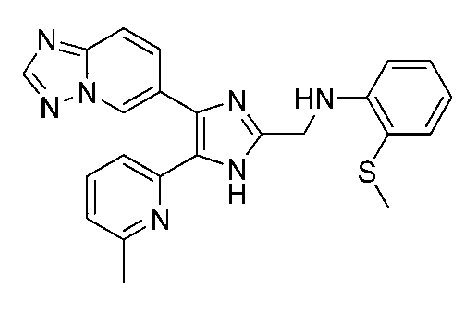
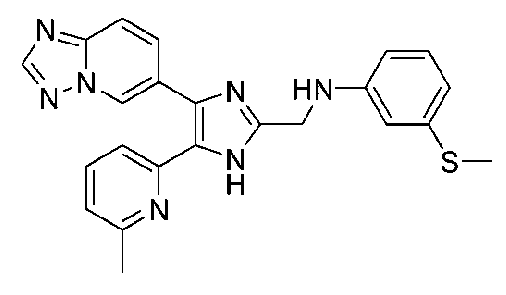
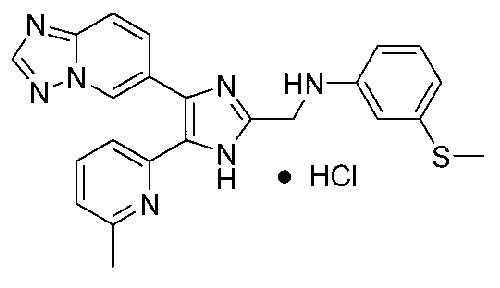
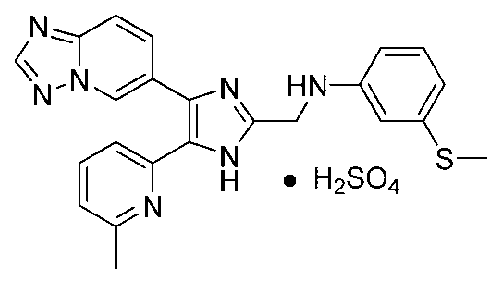
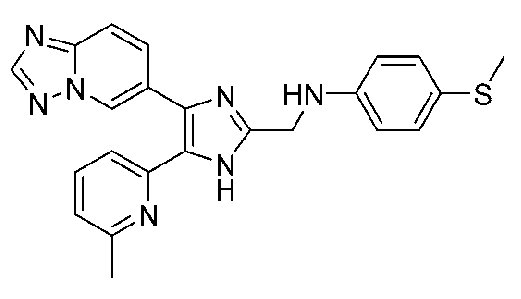
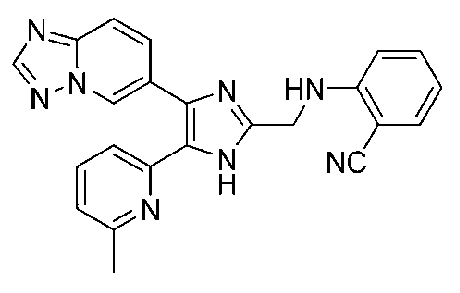
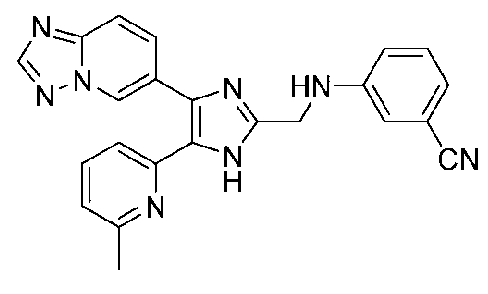
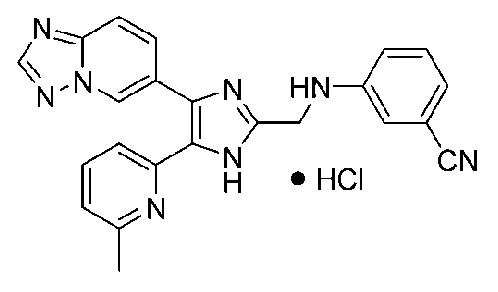
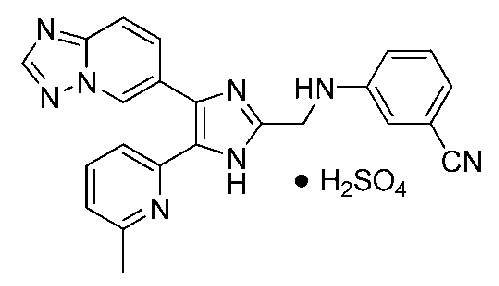
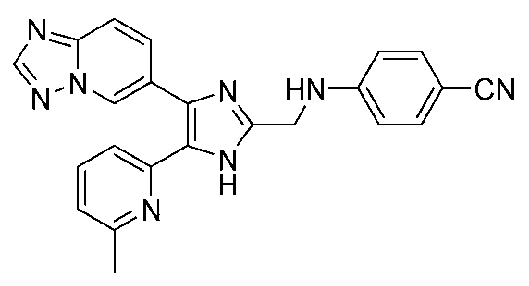
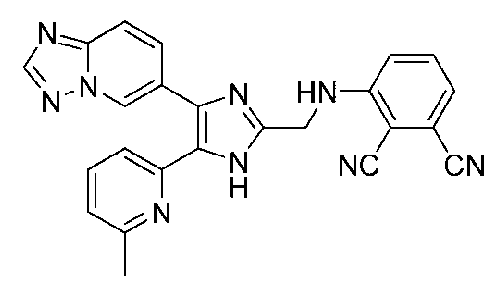
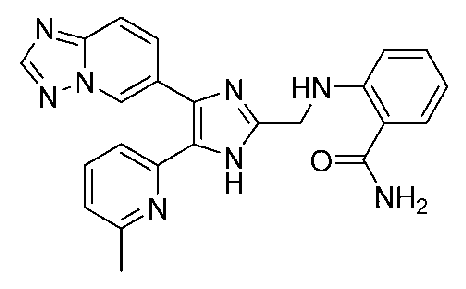
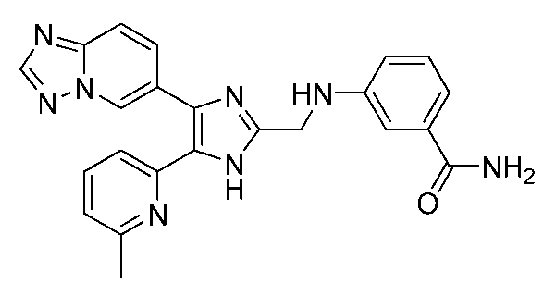
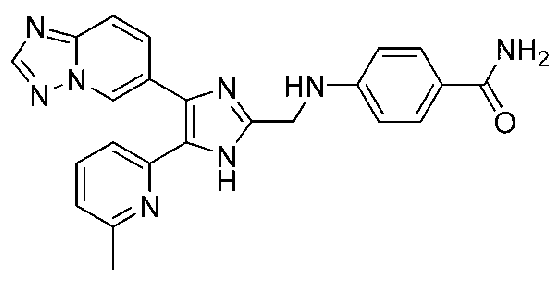
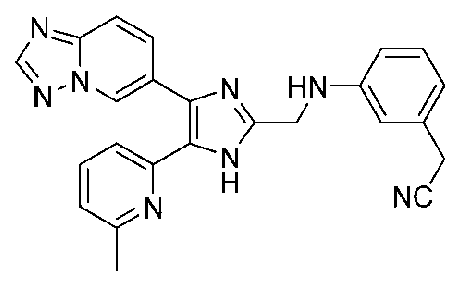
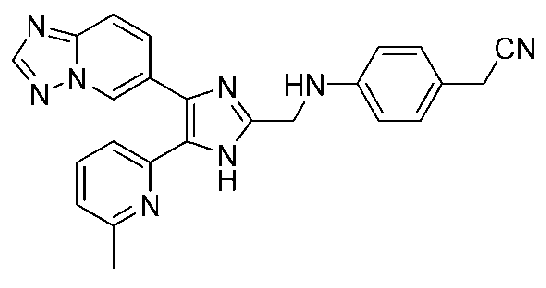
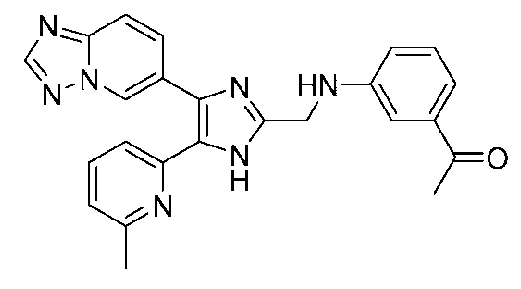
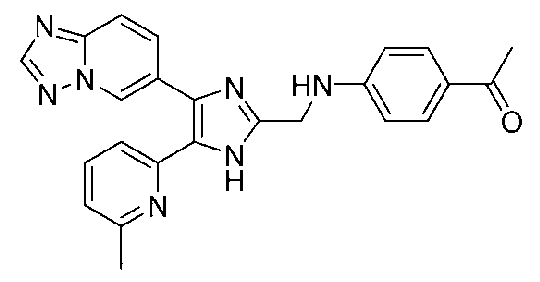
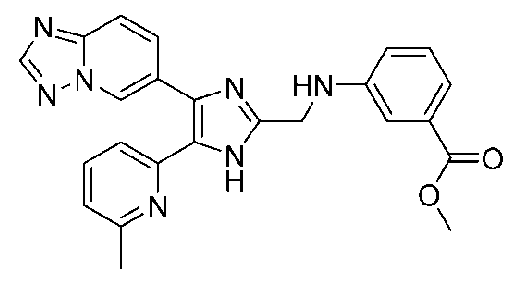
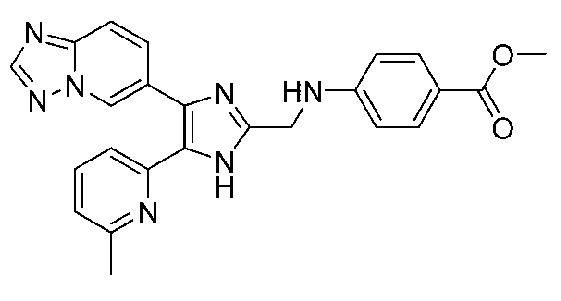
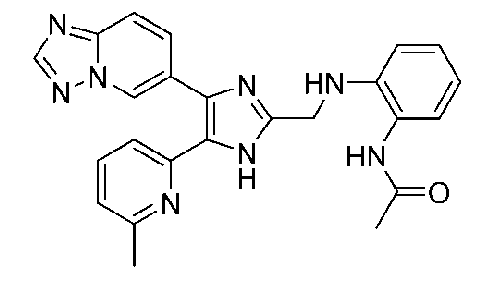
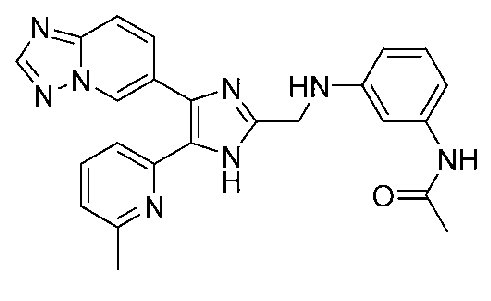
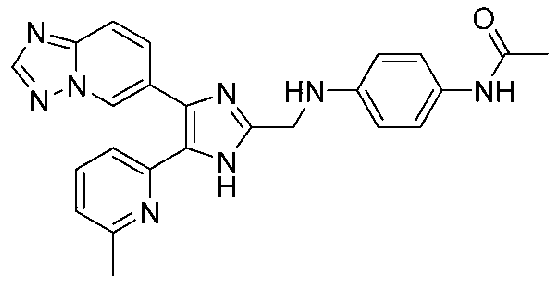
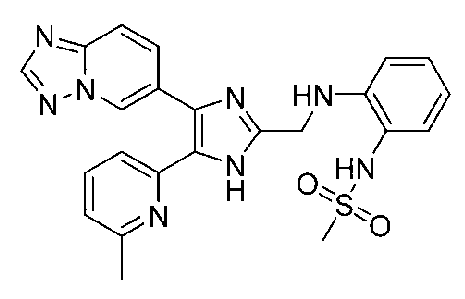
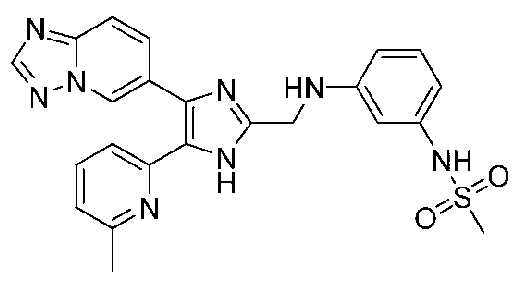
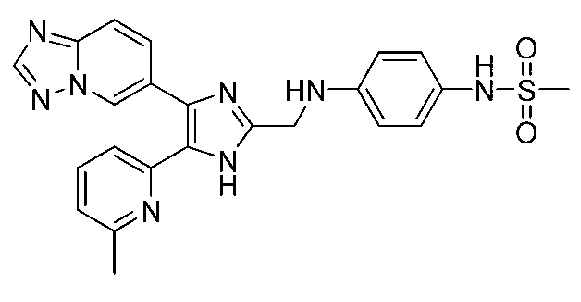
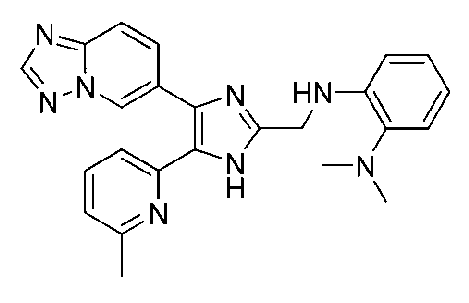
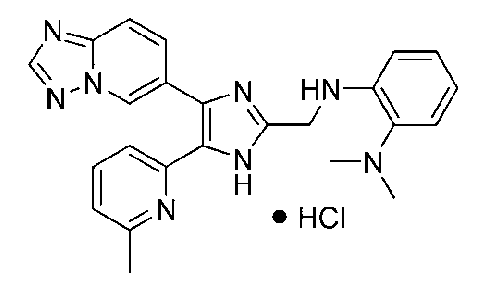
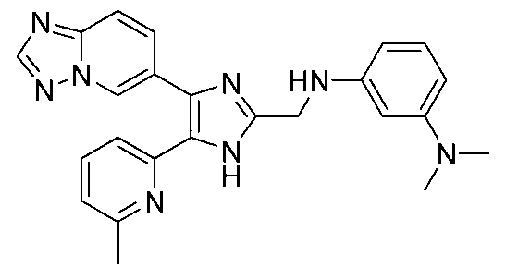
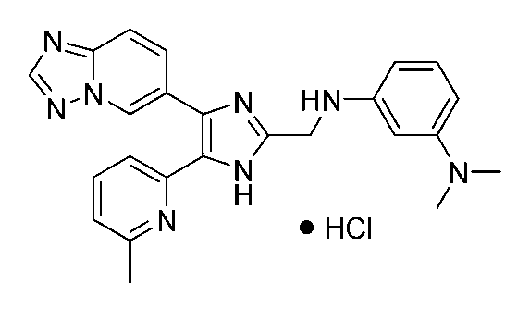
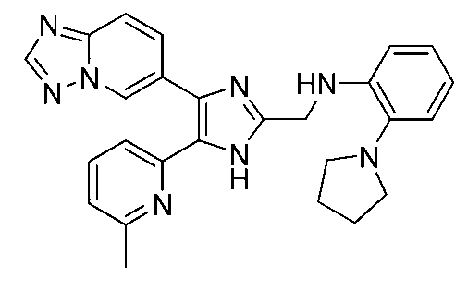
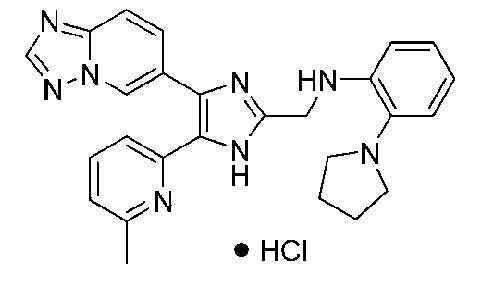
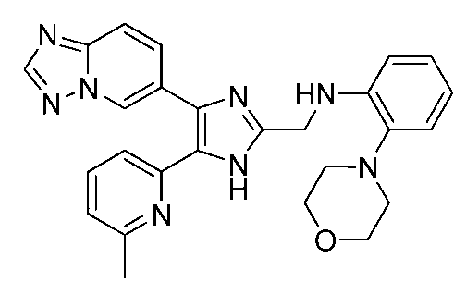
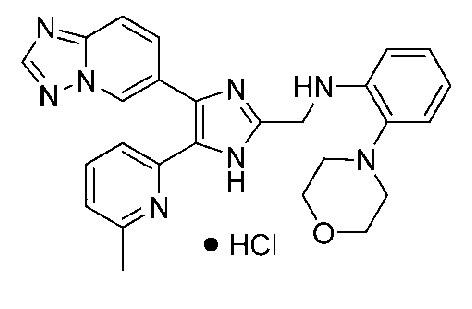
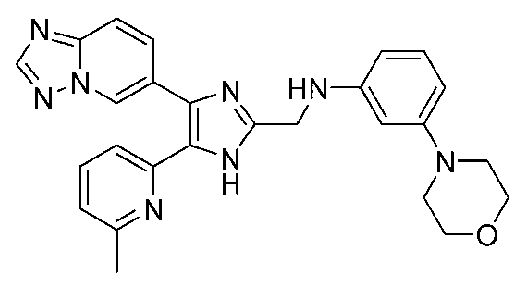
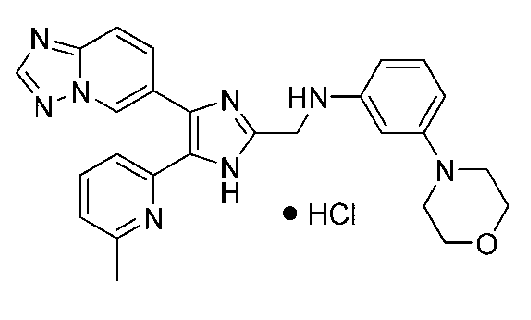
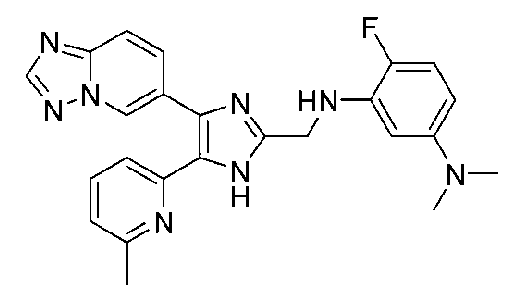
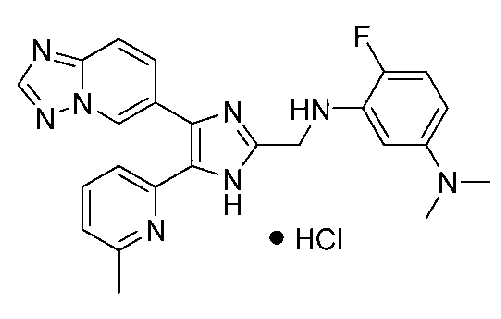
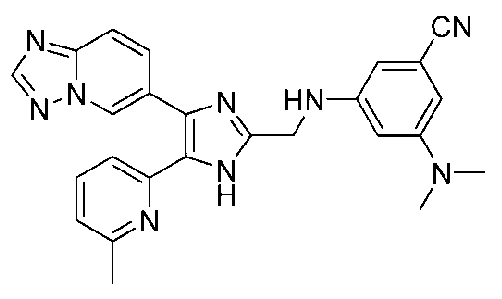
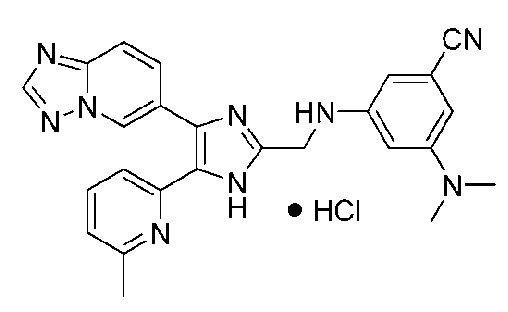
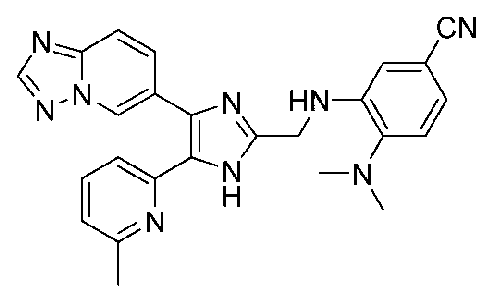
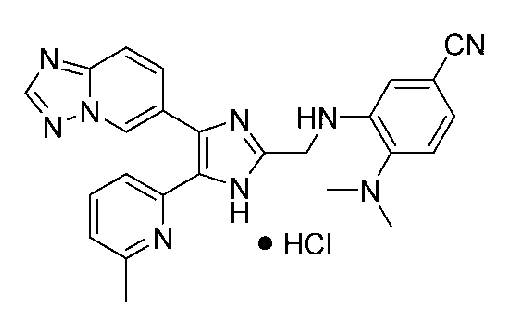
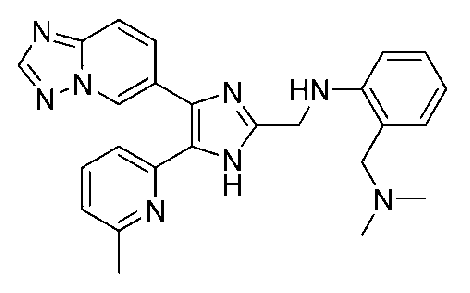
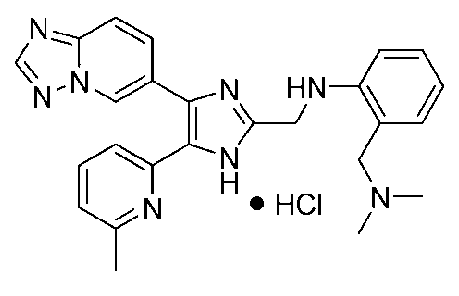
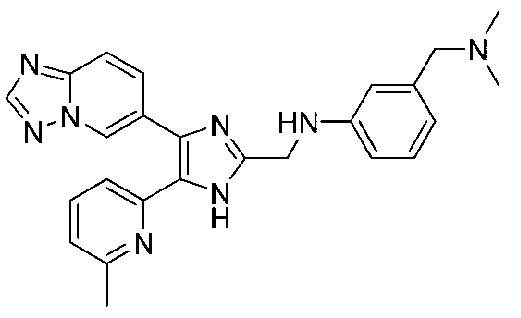
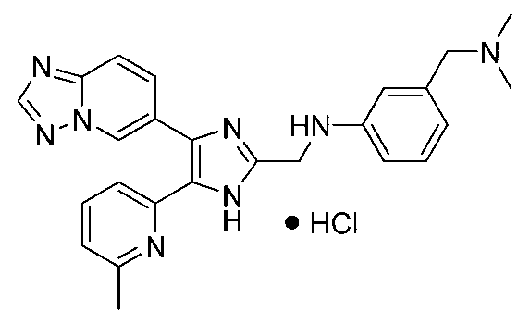
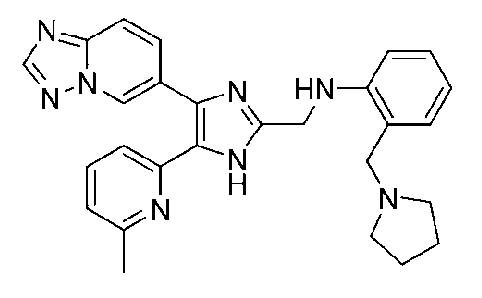
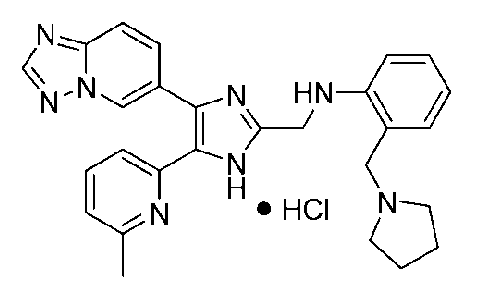
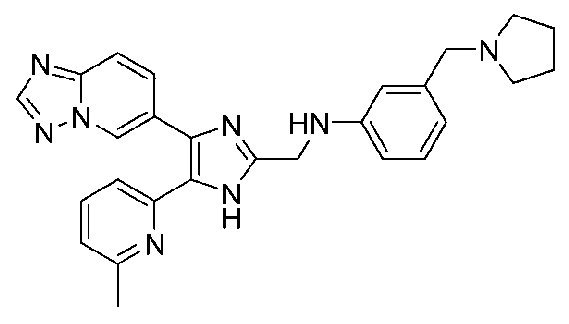
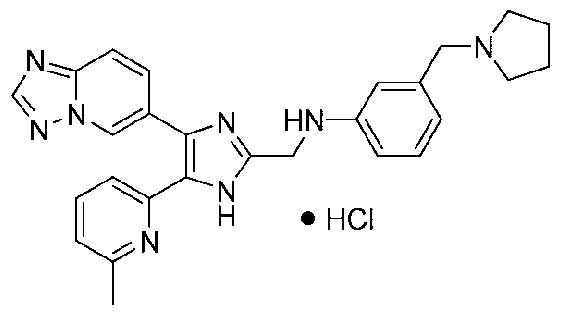
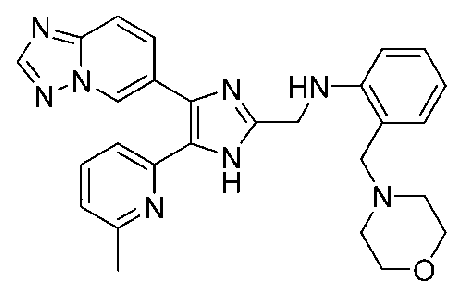
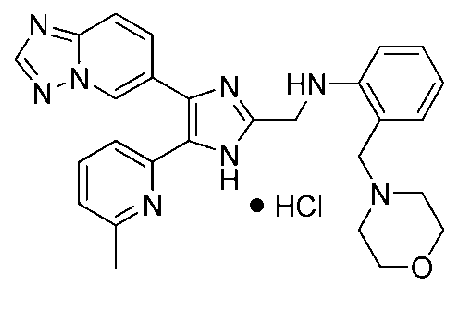
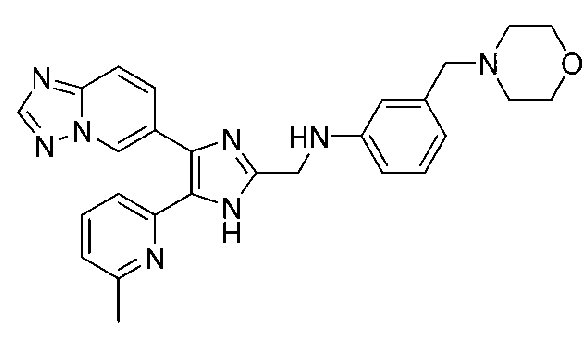
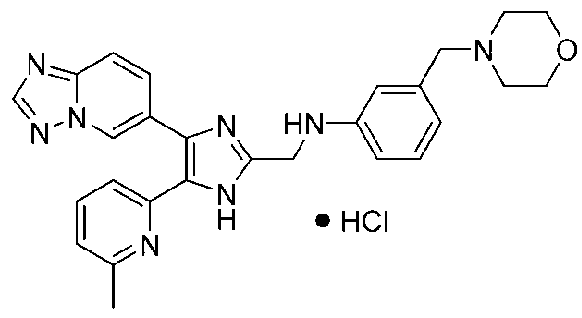
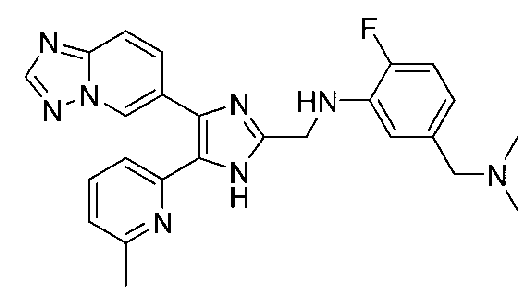
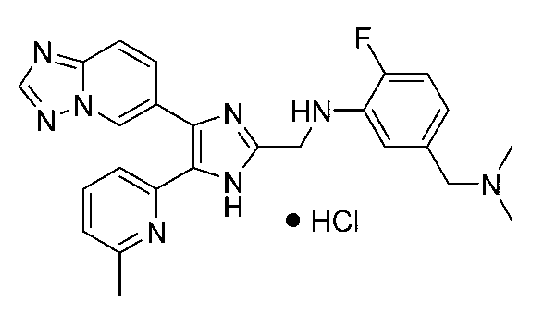
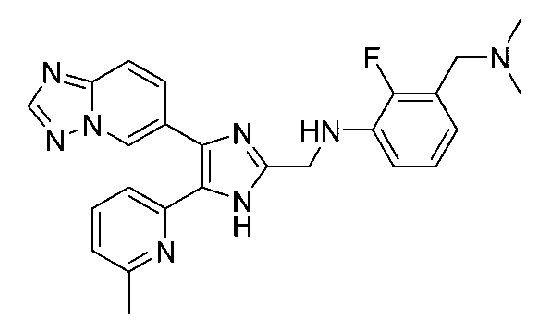
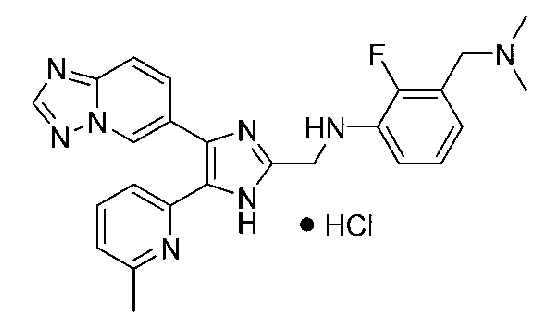
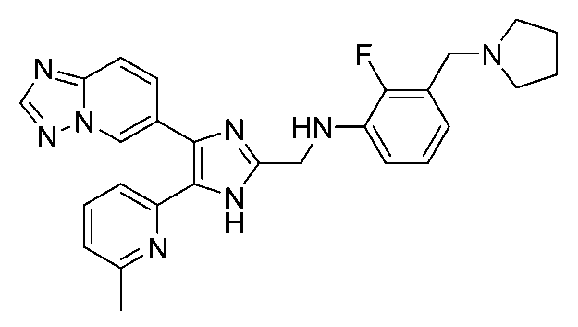
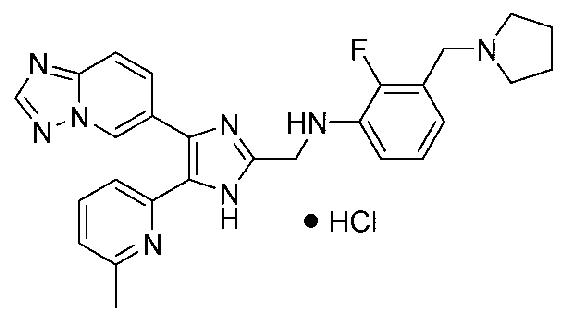
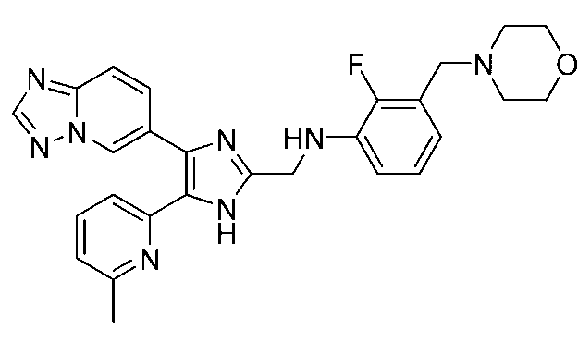
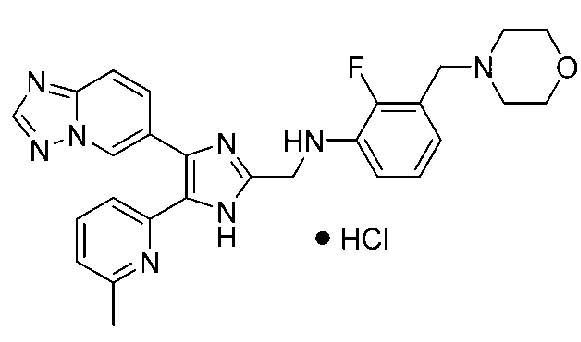
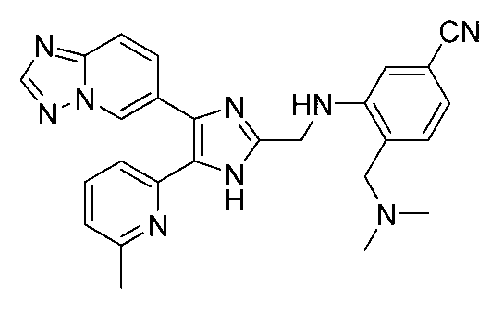
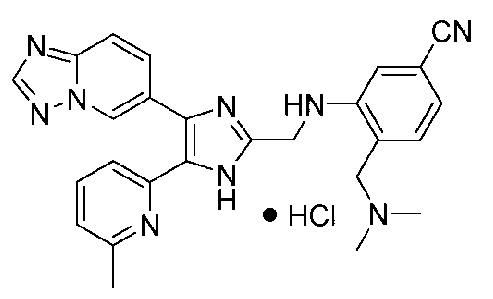
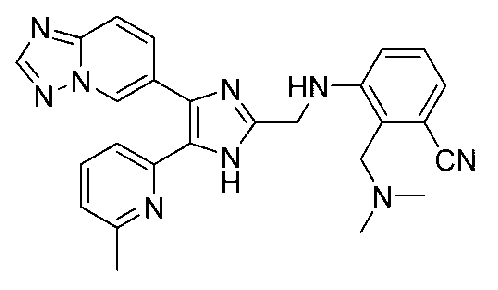
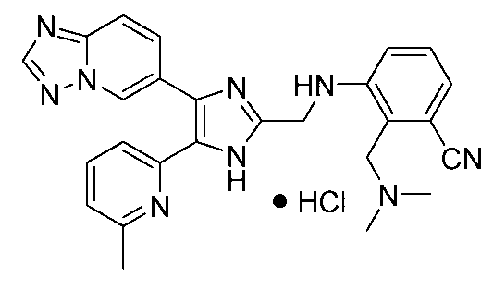
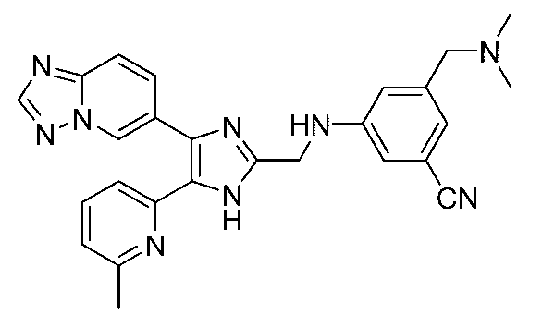
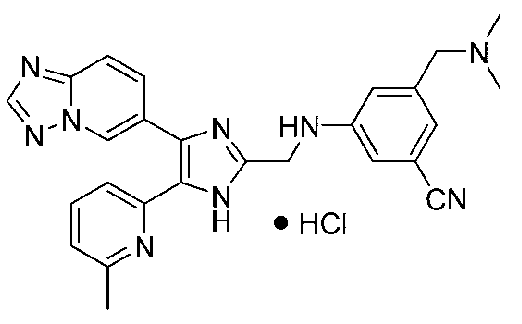
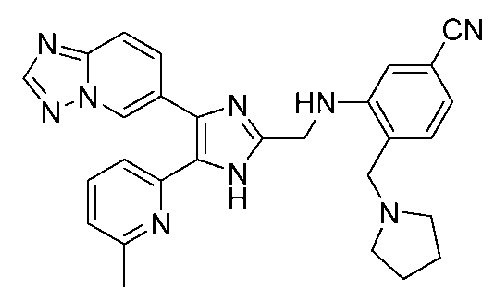
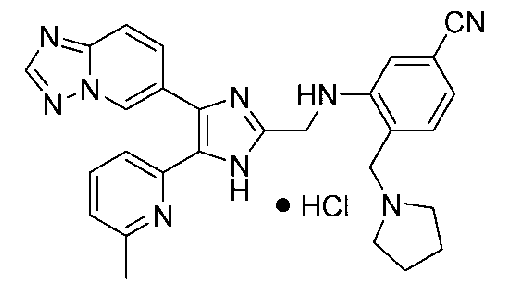
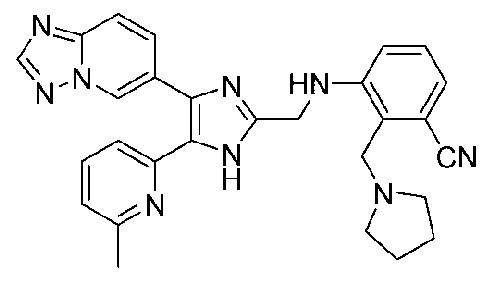
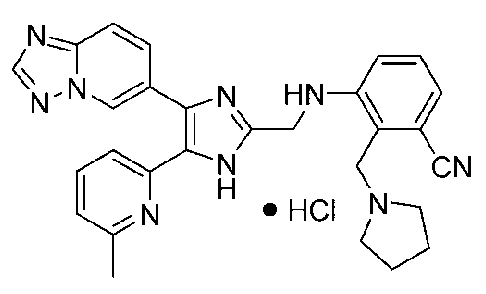
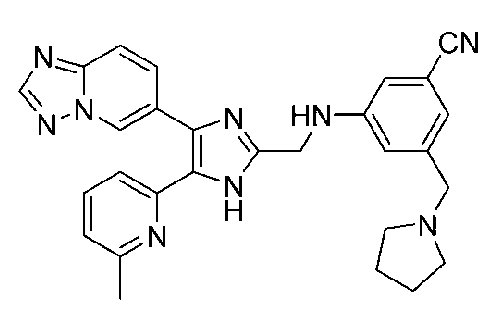
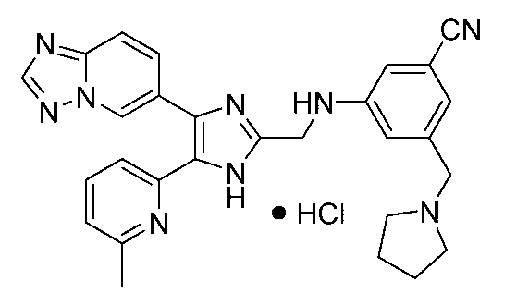
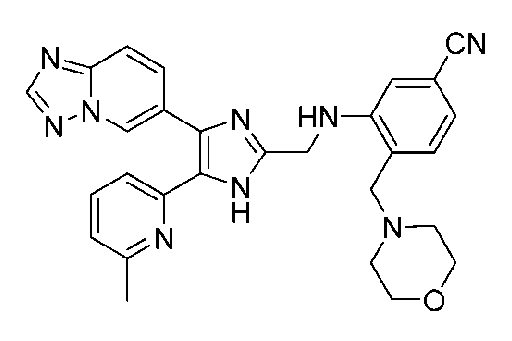
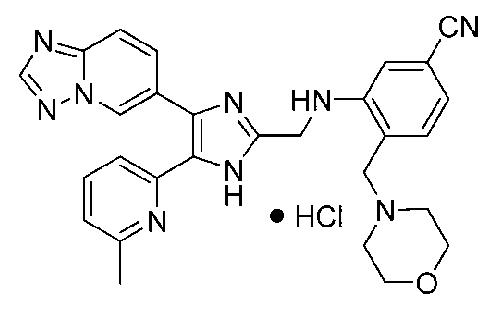
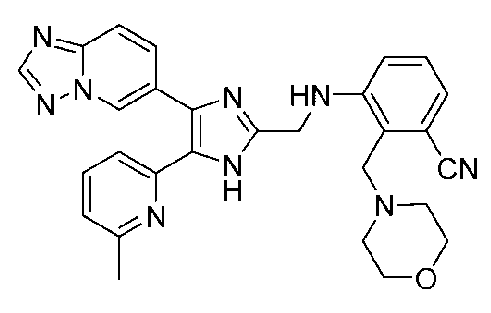
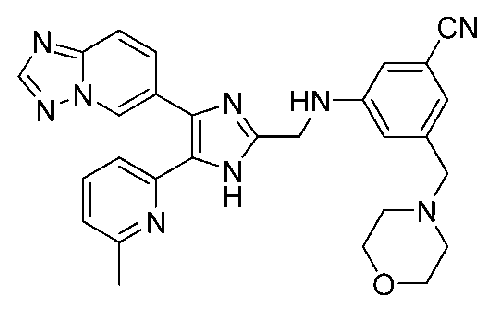
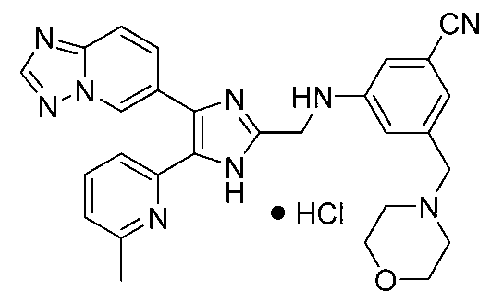
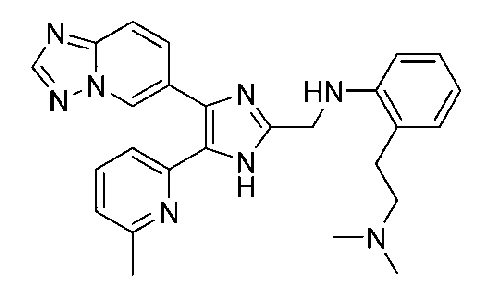
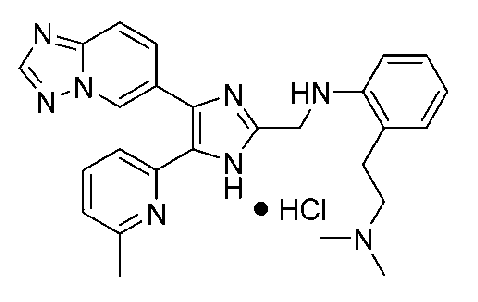
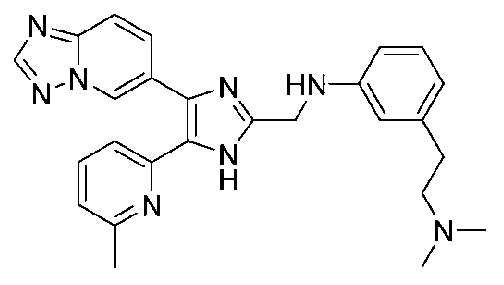
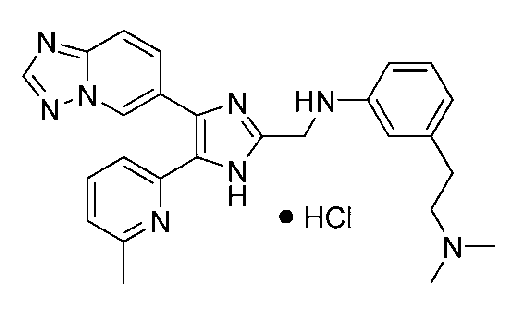
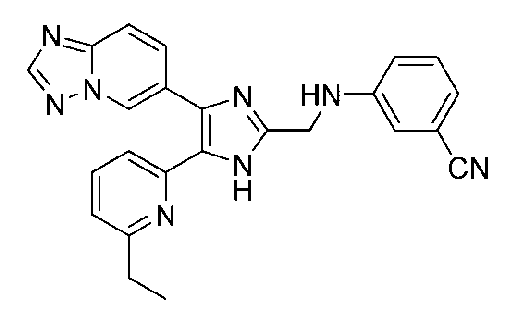
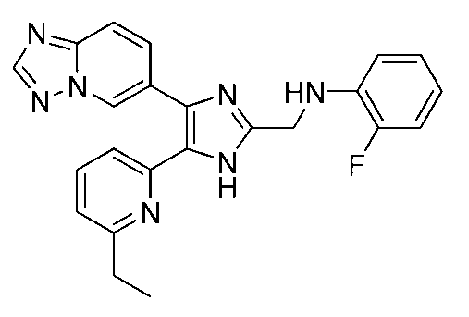
Пример получения 5
Получение N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фтор-N-метиланилина (пример 140)
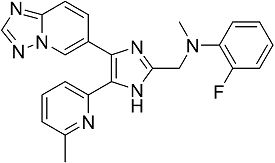
К перемешиваемому раствору 1-((1,2,4)триазоло(1,5-a)пиридин-6-ил)-2-(6-метилпиридин-2-ил)этан-1,2-диона (0,20 г, 0,75 ммоль) в смеси трет-бутилметилового эфира (10 мл) и MeOH (8 мл) добавляли 2-((2-фторфенил)(метил)амино)ацетальдегид (190 мг, 1,13 ммоль) и NH4OAc (0,15 г, 1,88 ммоль) и перемешивали смесь при комнатной температуре в течение 2 ч. pH смеси доводили до 8 добавлением насыщенного водного раствора NaHCO3. После удаления растворителя реакционную смесь экстрагировали CHCl3 (2×100 мл), CHCl3 раствор промывали водой (20 мл) и насыщенным раствором соли (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси MeOH и CH2Cl2 (1/19 (об./об.)) в качестве элюента, с получением указанного в заголовке соединения (90 мг, 32%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,97 (ушир.с, 1H), 8,37 (с, 1H), 7,82 (дд, 1H, J=9,2, 1,6 Гц), 7,78 (дд, 1H, J=9,2, 1,2 Гц), 7,49 (т, 1H, J=7,8 Гц), 7,25 (ушир.д, 1H, J=7,6 Гц), 7,14-7,06 (м, 3H), 7,04 (д, 1H, J=7,6 Гц), 7,00-6,94 (м, 1H), 4,44 (с, 2H), 2,91 (с, 3H), 2,58 (с, 3H); МС (ESI) m/z 414,20 (MH+).
Пример получения 6
Получение 3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)бензонитрила (пример 145)
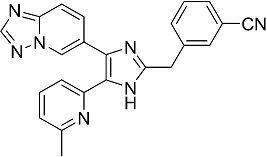
К перемешиваемому раствору 1-((1,2,4)триазоло(1,5-a)пиридин-6-ил)-2-(6-метилпиридин-2-ил)этан-1,2-диона (4,00 г, 15,02 ммоль) в смеси трет-бутилметилового эфира (30 мл) и MeOH (30 мл) добавляли 3-(формилметил)бензонитрил (полученный согласно способу, описанному в WO 02/096875 A1) (6,54 г, 45,07 ммоль) и NH4OAc (11,58 г, 150,24 ммоль) и перемешивали смесь при комнатной температуре в течение 90 мин. pH смеси доводили до 8 добавлением насыщенного водного раствора NaHCO3. После удаления растворителя смесь экстрагировали EtOAc (2×150 мл), EtOAc раствор промывали водой (50 мл) и насыщенным раствором соли (50 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси MeOH и CH2Cl2 (1/19 (об./об.)) в качестве элюента, с получением указанного в заголовке соединения (1,92 г, 33%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, ДМСО-d6) δ 12,70 (ушир.с, 1H), 9,53 (ушир.с, 1H), 8,49 (с, 1H), 7,96 (дд, 1H, J=9,2, 1,8 Гц), 7,84 (д, 1H, J=2,0 Гц), 7,82 (д, 1H, J=9,2 Гц), 7,74-7,71 (м, 2H), 7,69 (т, 1H, перекрыв., J=7,6 Гц), 7,56 (т, 1H, J=7,8 Гц), 7,47 (ушир.с, 1H), 7,15 (д, 1H, J=7,6 Гц), 4,18 (с, 2H), 2,47 (с, 3H); МС (ESI) m/z 392,18 (MH+).
Пример получения 7
Получение 3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)бензамида (пример 147)
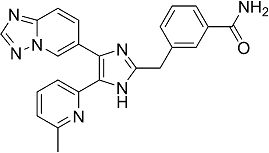
К перемешиваемому раствору 3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)бензонитрила (41 мг, 0,10 ммоль) в EtOH (2 мл) при комнатной температуре добавляли 28% H2O2 (13,9 мл, 0,11 ммоль) и 1 н. NaOH (0,39 мл, 0,39 ммоль). Смесь нагревали до 60°C в течение 1 ч, и затем при 0°C добавляли 1 н. HCl для доведения pH до 7-8. После удаления растворителя остаток экстрагировали CH2Cl2 (2×15 мл). CH2Cl2 раствор промывали водой (5 мл) и насыщенным раствором соли (5 мл), сушили над безводным Na2SO4, фильтровали и упаривали досуха при пониженном давлении. Остаток очищали MPLC на силикагеле с использованием смеси MeOH и CH2Cl2 (1/9 (об./об.)) в качестве элюента, с получением указанного в заголовке соединения (15 мг, 35%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 8,93 (ушир.с, 1H), 8,32 (с, 1H), 7,77 (с, 1H), 7,76 (дд, 1H, перекрыв., J=9,2, 1,6 Гц), 7,69 (д, 1H, J=9,2 Гц), 7,56 (д, 1H, J=8,0 Гц), 7,44 (т, 1H, J=7,6 Гц), 7,39 (д, 1H, J=7,6 Гц), 7,26 (т, 1H, J=8,0 Гц), 7,21 (д, 1H, J=7,6 Гц), 6,98 (д, 1H, J=7,6 Гц), 6,60 (ушир.с, 1H), 6,27 (ушир.с, 1H), 4,15 (с, 2H), 2,44 (с, 3H); МС (ESI) m/z 410,19.
Соединения, перечисленные в последующей таблице 2, получали способом, аналогично описанному выше в примерах получения 5-7. В таблицу 2 включены данные масс-спектрометрии для указанных соединений.
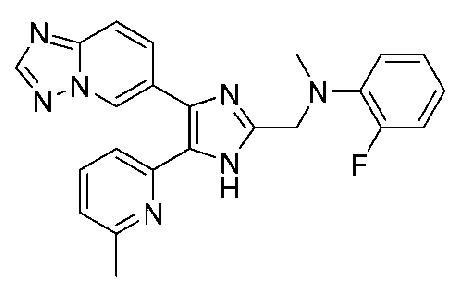
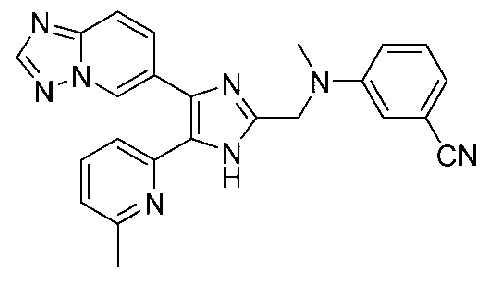
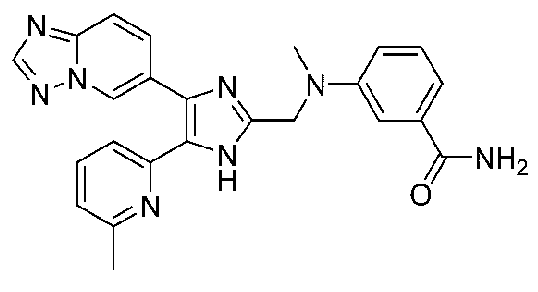
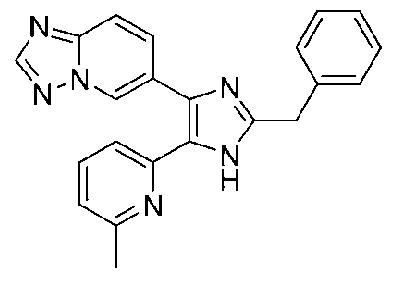
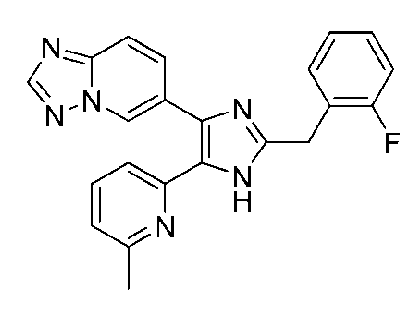
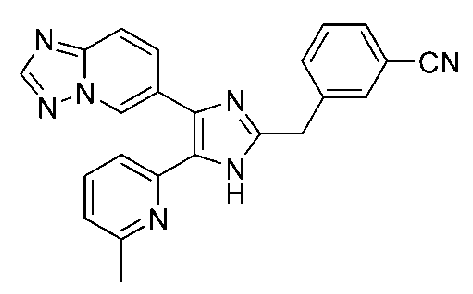
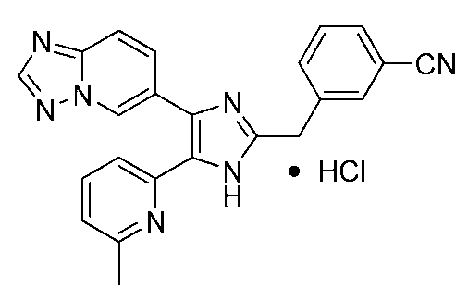
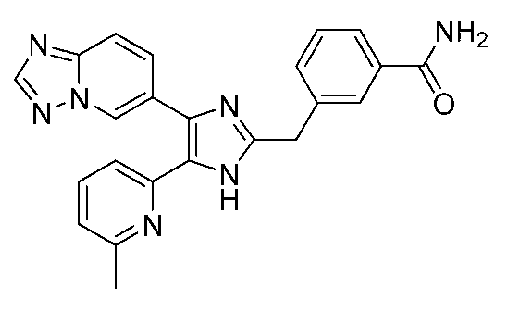
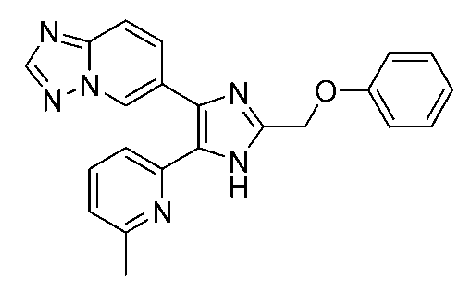
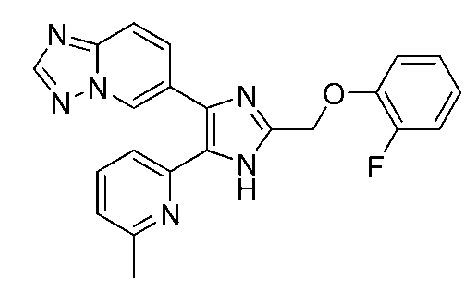
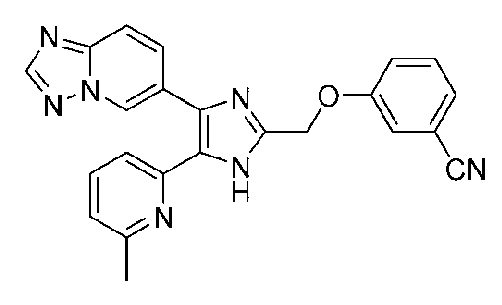
Биологические данные
Биологическая активность соединений настоящего изобретения может быть оценена с использованием следующих методов анализа.
Бесклеточный метод анализа для оценки ингибирования фосфорилирования киназы ALK5
Белок ALK5 экспрессировали в Sf9 клетках насекомых в качестве рекомбинантного GST-слитого белка человека с использованием бакуловирусной системы экспрессии. Экспрессированный белок очищали аффинной хроматографией с использованием GSH-агарозы (Sigma-Aldrich). Анализ киназы проводили в 96-луночных планшетах FlashPlatesTM от Perkin Elmer (Boston, MA, USA) в 50 мкл объеме реакции. Реакционную смесь вносили пипеткой в четыре этапа в следующем порядке: 20 мкл буфера для исследования (стандартного буфера), 5 мкл раствора ATP в H2O, 5 мкл каждого из исследуемых соединений формулы (I) в 10% ДМСО, 10 мкл GSK3 (14-27) (200 нг)/10 мкл ALK5 раствора (1 нг) (предварительно перемешанный). Реакционная смесь содержала 60 мМ HEPES-NaOH, pH 7,5, 3 мМ MgCl2, 3 мМ MnCl2, 3 мкМ Na3VO4, 1,2 мМ DTT, 50 мкг/мл PEG20000, 1 мкМ [γ-33P]-ATP (приблизительно 2,5×105 имп./мин на лунку), 200 нг/10 мкл GSK3 (14-27) и 1 нг/10 мкл ALK5. Реакционную смесь инкубировали при 30°C в течение 60 мин. Реакцию останавливали добавлением 50 мкл 2% (об./об.) H3PO4, планшеты аспирировали и дважды промывали 200 мкл 0,9% (масс./об.) NaCl. Анализ проводили с помощью роботизированной системы BeckmanCoulter Biomek 2000. Включение 33Pi (подсчет имп./мин) определяли с помощью микропланшетного сцинтилляционного счетчика (Microbeta, Wallac).
Соединения формулы (I), как правило, характеризовались значениями IC50 менее 1 мкМ; некоторые характеризовались значениями IC50 менее 0,1 мкМ и некоторые характеризовались значениями IC50 даже менее 10 нМ, как показано в таблице 3.
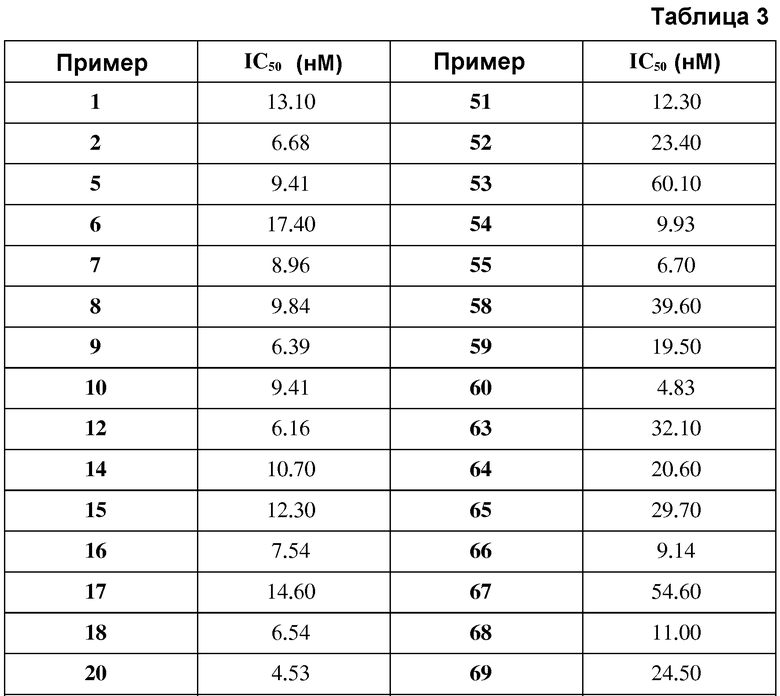
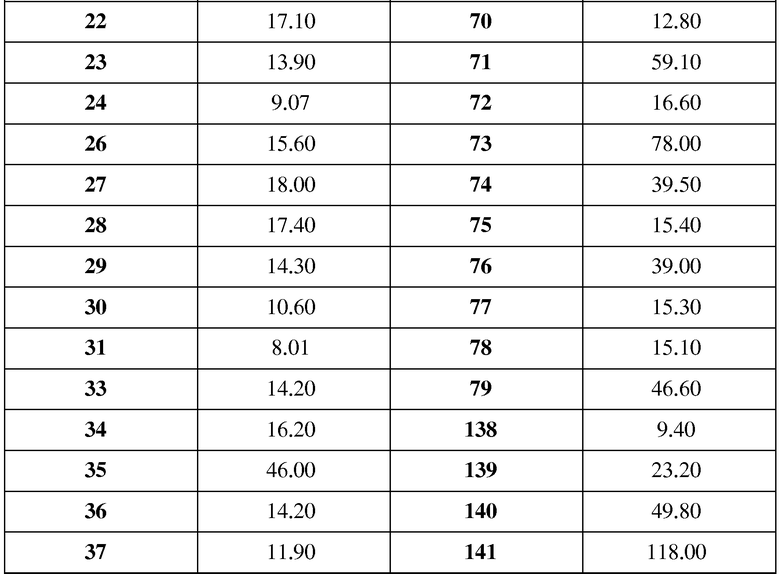
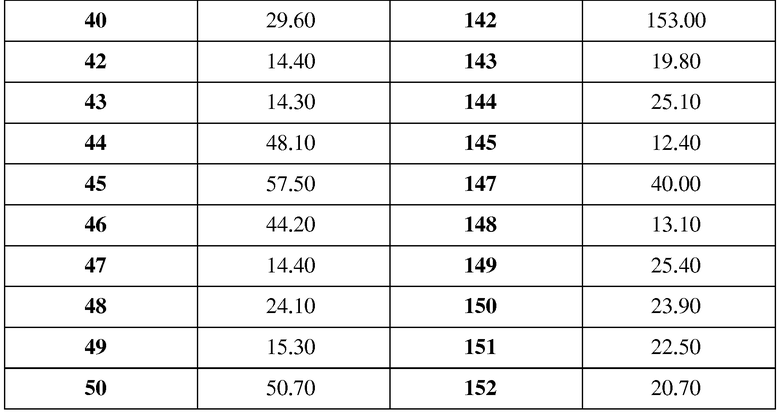
Бесклеточный метод анализа для оценки ингибирования фосфорилирования киназы ALK4
Ингибирование фосфорилирования киназы ALK4 исследуемыми соединениями формулы (I) может быть определено аналогично описанному выше для ингибирования ALK5, за исключением того, что GST-меченую ALK4 (Invitrogen Corporation) и RBER-CHKtide использовали вместо GST-меченой ALK5 и GSK3 (14-27).
Соединения формулы (I), как правило, характеризовались значениями IC50 менее 1 мкМ; некоторые характеризовались значениями IC50 менее 0,1 мкМ и некоторые характеризовались значениями IC50 даже менее 10 нМ.
Определение профиля селективности киназы
Анализ киназ проводили в 96-луночных планшетах FlashPlatesTM производства Perkin Elmer в 50 мкл объеме реакции. Реакционную смесь вносили пипеткой в четыре этапа в следующем порядке: 15 мкл раствора ATP в H2O, 20 мкл буфера для исследования (стандартного буфера), 5 мкл соединения примера 2 в 10% ДМСО, 10 мкл смеси фермент/субстрат в H2O. Реакционная смесь содержала 70 мМ HEPES-NaOH, pH 7,5, 3 мМ MnCl2, 3 мкМ Na3VO4, 1,2 мМ DTT, 1 мкМ [γ-33P]-ATP (приблизительно 6×105 имп/мин на лунку), протеинкиназу (различные количества) и субстрат (различные количества). Реакционные смеси инкубировали при 30°C в течение 60 мин. Реакцию останавливали добавлением 50 мкл 2% (об./об.) H3PO4, планшеты аспирировали и дважды промывали 200 мкл 0,9% (масс./об.) NaCl. Все анализы проводили с помощью роботизированной системы BeckmanCoulter Biomek 2000/SL. Включение 33Pi (подсчет имп./мин) определяли с помощью микропланшетного сцинтилляционного счетчика.

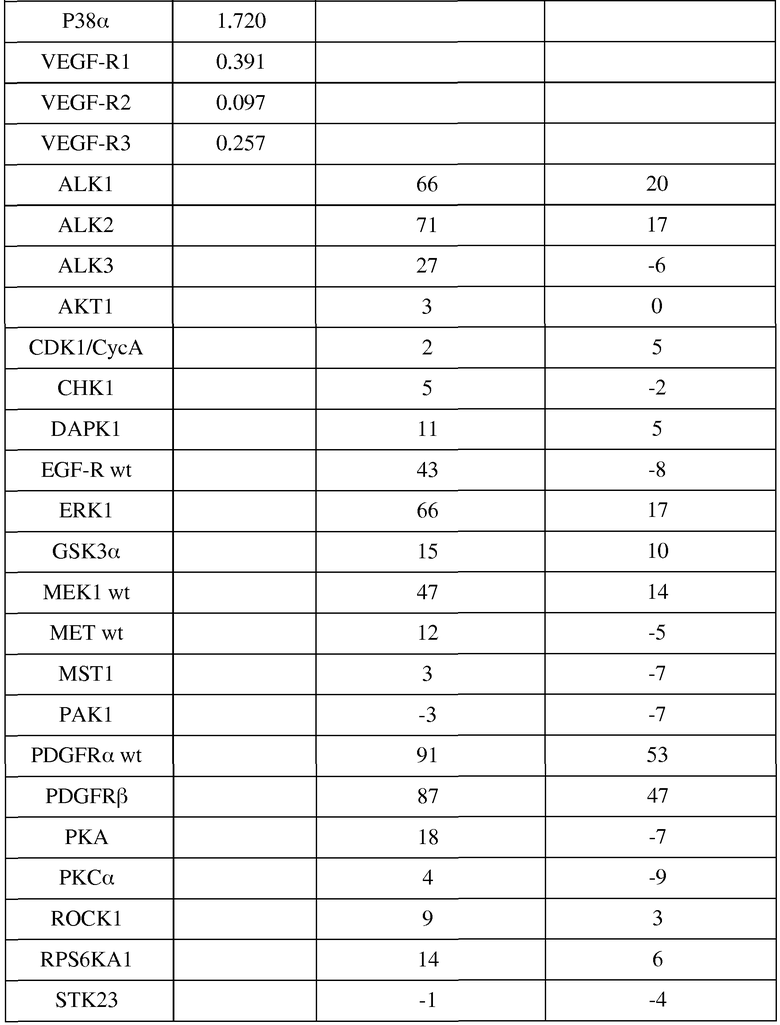
Анализ для оценки клеточного ингибирования передачи сигнала TGF-β
Стабильные клетки HaCaT-3TP-Luc или стабильные клетки 4T1-3TP-Luc, содержащие плазмиду экспрессии p3TP-Luc (neo), высевали в 96-луночный планшет с плотностью 2,5×104 клеток на лунку или 5×105 клеток на лунку, соответственно. Клетки параллельно обрабатывали TGF-β1 (2 нг/мл) в 0,2% FBS в присутствии или при отсутствии каждого из исследуемых соединений формулы (I) приблизительно при 60-70% конфлюэнтности в течение 24 часов при 37°C в 5% CO2. Клеточные лизаты получали с использованием люциферазной аналитической системы (Promega) согласно инструкции производителя и люминесценцию измеряли с помощью люминометра Micro Lumat Plus (Berthold, Germany).
Соединения формулы (I), как правило, характеризовались значениями IC50 менее 1 мкМ; некоторые характеризовались значениями IC50 менее 0,1 мкМ и некоторые характеризовались значениями IC50 даже менее 10 нМ.
Иммунофлуоресцентный анализ
Клетки MCF10A помещали на покровное стекло в 6-луночный планшет с плотностью 2×105 клеток на лунку. Спустя 12 часов, когда клетки прикреплялись, среду с 10% FBS заменяли средой с 0,5% FBS. Спустя 24 часа клетки обрабатывали TGF-β1 (2 нг/мл) с соединением примера 2 (1 мкМ) или без него в течение 2 часов. Затем клетки фиксировали 4% раствором формальдегида в течение 30 мин при комнатной температуре и гасили гасящим раствором (50 мМ NH4Cl в PBS) в течение 15 мин. После трехкратной промывки PBS клетки инкубировали с блокирующим раствором/раствором для пермеабилизации (1% BSA и 0,1% Triton X-100 в PBS) в течение 1 часа при комнатной температуре и инкубировали с антителом к Smad2/3 (BD Biosciences, Franklin Lakes, NJ, USA) в течение ночи при 4°C. Флуоресценцию визуализировали с помощью Cy3-конъюгированного козьего антитела к IgG мыши (Jackson ImmunoResearch Laboratories, Bar Harbor, ME, USA). Ядра этих же клеток окрашивали раствором DAPI. Клетки анализировали с использованием лазерного конфокального микроскопа LSM 510 META (Carl Zeiss, Germany).
Соединение примера 2 подавляло индуцированную TGF-β1 ядерную транслокацию Smad2/3 в клетках MCF10A.
Исследование заживления ран
Клетки MCF10A высевали в 6-луночный планшет с плотностью 2×105 клеток на лунку. Когда свыше 80% площади в каждой лунке было занято клетками, 10% FBS заменяли 0,2% FBS. Спустя 24 часа с помощью пластикового наконечника пипетки создавали рану и затем обрабатывали клетки TGF-β1 (2 нг/мл) в присутствии соединения примера 2 или без него (1 мкМ) в течение 16 часов. Изменение площади раны с 0 до 16 часов рассчитывали на основании программы Image J (National Institutes of Health, MD, USA) на основании фазово-контрастных изображений клеток, полученных с помощью микроскопа.
Соединение примера 2 подавляло индуцированную TGF-β1 клеточную миграцию в клетках MCF10A.
Исследование инвазии в матригель
Верхнюю поверхность камер системы Transwells (диаметр 6,5 мм, размер поры 8 мкм; Corning, Lowell, MA, USA) покрывали 20 мкл разведенного матригеля (BD Biosciences). Клетки 4T1 высеивали с плотностью 4×104 клеток в лунку на верхнюю камеру системы Transwells в бессывороточную среду с TGF-β1 или без него (2 нг/мл) в присутствии или отсутствие соединения примера 2. Нижнюю камеру заполняли 10% FBS с TGF-β1 (2 нг/мл) в присутствии или отсутствие соединения примера 2. После инкубации в течение 20 ч при 37°C в 5% CO2 клетки, оставшиеся на верхней поверхности мембраны, удаляли ватной палочкой и наблюдали за окрашенными DAPI клетками, оставшимися на нижней поверхности, с использованием флуоресцентной микроскопии. Среднее количество клеток в поле зрения получали по данным из 5 случайных полей.
Соединение примера 2 подавляло индуцированную TGF-β1 клеточную инвазию при анализе инвазии в матригель.
Изучение клеточного роста
Либо клетки 4T1, либо клетки MCF10A высевали в 96-луночный планшет с плотностью 5×103 клеток на лунку. После того как клетки прикреплялись, их обрабатывали соединением примера 2, растворенным в ДМСО, в 0,2% сывороточной среды. После инкубации в течение четырех суток жизнеспособность клеток определяли с помощью SRB анализа.
Соединение примера 2 не демонстрировало эффекта на рост клеток 4T1 и слегка увеличивало рост клеток MCF10A без статистической значимости, таким образом, давая возможность предположить, что антиметастатический эффект соединения примера 2 не был обусловлен ингибированием роста первичной опухоли.
Антиметастатический эффект на модели мышей BALB/c с ксенотрансплантатом клеток 4T1
Самок мышей BALB/c приобретали у Orient Bio Inc. (Seoul, Korea). Животных содержали в комнате с контролируемой температурой (22°C) и снабжали пищей и водой ad libitum. Клетки 4T1 (1,2×104 клеток) суспендировали в PBS и имплантировали в жировую подушку 4-ой левой молочной железы самок мышей BALB/c в возрасте 5-6 месяцев (сутки 0). В эксперименте 1 лечение начинали после имплантации опухоли (сутки 0). Соединение примера 3 (13,6 или 27,3 мг/кг), растворенное в воде, давали мышам перорально BID пять суток подряд в неделю в течение четырех недель. В эксперименте 2 лечение начинали на сутки 4. Соединение примера 2 (5, 10, 20 или 40 мг/кг), растворенное в составе искусственного желудочного сока, давали мышам перорально пять суток подряд в неделю в течение трех недель. В эксперименте 3 лечение начинали на сутки 4. Соединение примера 2 (5, 10, 20 или 40 мг/кг), растворенное в составе искусственного желудочного сока, давали мышам перорально через сутки (три раза в неделю) в течение 24 суток. В эксперименте 4 клетки 4T1 (1×104 клеток) суспендировали в PBS и имплантировали в жировую подушку 4-ой левой молочной железы самок мышей BALB/c в возрасте 10 месяцев (сутки 0). Лечение начинали на сутки 10. Соединение примера 61 (43,6 мг/кг), растворенное в солевом растворе, давали мышам интраперитонеально через сутки в течение 2,5 недель. Во всех экспериментах мышей умерщвляли спустя 24-72 ч после введения последней дозы, и немедленно инъецировали в трахею 15% раствор индийских чернил (Hardy Diagnostics) в PBS. Окрашенные индийскими чернилами легкие выделяли и удаляли краситель с помощью раствора Фекета (60% этанола, 3% формальдегида и 4% уксусной кислоты в PBS) в течение, по меньшей мере, 20 мин. На поверхности левой доли легкого подсчитывали число метастатических узлов и фотографировали легкое при помощи цифрового фотоаппарата. Размер опухоли измеряли с использованием штангенциркулей и рассчитывали объем опухоли с использованием следующего уравнения:
Объем опухоли=(0,5236)×(толщина)2×(длина).
Соединения примеров 2, 3 и 61 существенно снижали число метастатических узлов в легком.
В эксперименте 3 проводили вестерн-блоттинг для исследования эффекта соединения примера 2 на фосфорилирование Smad2 в опухолевой ткани. Либо буфер-растворитель (4 мМ HCl, 1 мг/мл BSA), либо TGF-β1 (50 нг/мышь) в буфере-растворителе вводили мышам внутривенно за 2 ч до того, как мышей умерщвляли. Полученные из мышей ткани опухоли лизировали в буфере RIPA [50 мМ Tris, pH 7,5, 150 мМ NaCl, 0,1% додецилсульфата натрия, 0,5% дезоксихолата натрия, 1% NP-40, 1 мМ NaF, 1 мМ Na3VO4, 1 мМ PMSF, смесь ингибиторов протеаз (1 таблетка смеси ингибиторов протеаз Roche Diagnostics GmbH/10 мл) (Roche)] в течение 20 мин на льду. Лизаты очищали центрифугированием при 13000 об/мин при 4°C в течение 20 мин. Содержание белка в супернатанте определяли с использованием набора для анализа белка Micro-BCA (бицинхониновая кислота) (Thermo Scientific). Лизаты, содержащие 20-50 мкг общего белка, отделяли электрофорезом на полиакриламидном геле и затем электрофоретически переносили на поливинилидендифторидные блоттинг-мембраны (Millipore, Billerica, MA, USA). Мембраны блокировали 5% BSA (Sigma-Aldrich) в PBS, содержащем 0,5% Tween-20 (PBST), в течение 1 ч и инкубировали в течение ночи при 4°C с одним из следующих антител: антитела к фосфо-Smad2 (Millipore), антитела к Smad2/3 (BD Transduction Laboratories, NJ, USA) или антитела к β-актину (Sigma-Aldrich) в PBST, содержащем 1% BSA. Мембраны трижды промывали PBST и инкубировали либо с козьим антителом против иммуноглобулина мыши, конъюгированным с пероксидазой хрена, либо с HRP-конъюгированным козьим антителом против иммуноглобулина кролика (SantaCruz Biotechnology, Santa Cruz, CA, USA) при комнатной температуре в течение 1 ч. Связанные антитела обнаруживали с использованием реагента Люминол для вестерн-блоттинга (SantaCruz Biotechnology). Интенсивность полос анализировали с использованием устройства формирования изображения с денситометром LAS-3000 (FUJIFILM, Tokyo, Japan).
Соединение примера 2 подавляло индуцированное TGF-β1 фосфорилирование Smad2 в опухолевой ткани
Антиметастатический эффект на модели рака молочной железы у мышей MMTV/c-Neu
Трансгенных самок мышей MMTV/c-Neu приобретали у Jackson Laboratory (Bar Harbor, ME, USA). Животных содержали в комнате с контролируемой температурой с SPF (22°C) и снабжали пищей и водой ad libitum. В эксперименте 1 соединение примера 61 (43,6 мг/кг), растворенное в солевом растворе, давали мышам MMTV/c-Neu в возрасте 32 недель интраперитонеально через сутки в течение трех недель. В эксперименте 2 соединение примера 3 (43,6 мг/кг), растворенное в солевом растворе, давали мышам MMTV/c-Neu в возрасте 32 недель интраперитонеально через сутки в течение десяти недель. Мышей умерщвляли через 24 ч после введения последней дозы и анализировали ткани опухоли молочной железы и легкого путем окрашивания гематоксилином и эозином (H&E). Для анализа уровня мРНК β-казеина в тканях опухоли молочной железы и легкого из этих тканей выделяли общие РНК с использованием реагента TRIzol (Invitrogen Corporation) и набора RNeasy Mini (Qiagen) согласно инструкции производителя. Из 2 мкг общих РНК синтезировали кДНК с использованием случайного праймера (Invitrogen Corporation) с помощью MMLV RTase (Invitrogen Corporation) в течение 1 ч при 37°C и проводили ПЦР амплификацию с использованием полимеразы Taq (Promega) и следующих генспецифических праймеров: мышиный GAPDH (прямой) 5'-ATG TGT CCG TCG TGG ATC TGA-3' и (обратный) 5'-TTG AAG TCG CAG GAG ACA ACC-3', мышиный β-казеин (прямой) 5'-TCC CAC AAA ACA TCC AGC C-3' и (обратный) 5'-ACG GAA TGT TGT GGA GTG G-3'. Амплифицированную ДНК анализировали электрофорезом на агарозном геле.
Соединение примера 61 существенно снижало число метастатических поражений в легком. В легком мышей MMTV/c-Neu обнаруживали значительное содержание мРНК β-казеина (маркера дифференцировки молочной железы). Соединения примеров 3 и 61 существенно ингибировали уровень экспрессии мРНК β-казеина в легком, демонстрируя их антиметастатический эффект.
Активность MMP-9 и MMP-2 в первичной опухоли молочной железы измеряли с помощью желатиновой зимографии. Ткани опухоли из мышей (30 мг) лизировали в 500 мкл буфера RIPA (50 мМ Tris, 150 мМ NaCl, 0,1% додецилсульфата натрия, 0,5% дезоксихолата натрия, 1% NP-40, ингибитор протеазы без EDTA) в течение 10-20 мин на льду. Лизаты очищали центрифугированием при 13000 об/мин при 4°C в течение 10 мин. Содержание белка супернатантов определяли с использованием набора для анализа белка Micro-BCA (Thermo Scientific). Загружаемые образцы получали путем добавления загрузочного буфера (0,5М Tris, pH 6,8, 50% глицерина, 10% SDS и 1% раствора бромфенолового синего) в лизаты, содержащие 15 мкг общего белка. Загружаемые образцы нагревали при 60°C в течение 5 мин и отделяли электрофорезом на 10% полиакриламидном геле, содержащем 0,2% желатина. Гель дважды промывали отмывочным буфером [2,5% Triton-X100, 0,05M Tris-HCl, pH 7,5 и 0,1М NaCl] в течение 30 мин при комнатной температуре. Затем гель инкубировали в инкубационном буфере [0,05М Tris-HCl, pH 7,5, 0,15М NaCl, 0,01М CaCl2, 0,02% NaN3 и 1 мкМ ZnCl2] при 37°C в течение 16-18 ч при встряхивании. Гель окрашивали 0,5% раствором кумасси голубого R250, содержащим 5% метанола и 10% уксусной кислоты, в течение 2-4 ч при комнатной температуре и дважды удаляли краситель с помощью раствора для удаления красителя (5% метанола и 10% уксусной кислоты) в течение 30 мин при комнатной температуре. Изображение геля получали с использованием устройства формирования изображения с денситометром LAS-3000 (FUJIFILM) в режиме cybergreen.
Соединение примера 3 существенно ингибировало активность MMP-9 и MMP-2 в первичной опухоли молочной железы.
Антифибротический эффект на модели фиброза печени с перевязкой желчного протока
Самцов крыс Спраг-Доули (SD) в возрасте 6 месяцев приобретали у Orient Bio Inc. В эксперименте 1 SD крыс массой 180-200 г случайным образом распределяли на пять экспериментальных групп: ложнооперированные крысы (n=5), ложнооперированные крысы, получавшие лечение соединением примера 3 (43,6 мг/кг, n=5), крысы с перевязкой желчного протока (BDL) (n=10), крысы с BDL, получавшие лечение или 21,8, или 43,6 мг/кг соединения примера 3 (n=10). В эксперименте 2 SD крыс массой 180-200 г случайным образом распределяли на пять экспериментальных групп: ложнооперированные крысы (n=5), крысы с BDL (n=10), крысы с BDL, получавшие лечение или 5, или 10, или 20 мг/кг соединения примера 2 (n=10). Для BDL животным проводили анестезию золетилом (20 мг/кг) и ксилазином (10 мг/кг), вытягивали общий желчный проток и проводили двойную перевязку с использованием шелковой нити 3-0. Первую лигатуру накладывали ниже соединения печеночного протока и вторую накладывали выше входа поджелудочного протока. Затем общий желчный проток разрезали между двойными лигатурами. У ложнооперированных крыс делали разрез в брюшине и затем зашивали без какого-либо лечения. Лечение начинали в пределах 2 ч после хирургической процедуры. Либо соединение примера 3, растворенное в солевом растворе (эксперимент 1), либо соединение примера 2, растворенное в составе искусственного желудочного сока (эксперимент 2), давали крысам перорально три раза в неделю в течение четырех недель, начиная после операции BDL. Животных содержали в комнате с контролируемой температурой (при 21°C) и снабжали их автоклавируемой пищей и водой. Спустя 48 ч после введения последней дозы животных умерщвляли и извлекали сыворотку, селезенки и печени. Печени сагиттально разрезали на несколько частей, быстро замораживали в жидком азоте и хранили при -70°C. Часть печени погружали в 10% нейтральный забуференный формалин для гистопатологических исследований. Все экспериментальные процедуры проводили согласно своим внутриорганизационным нормативам. Активность сывороточных аланинаминотрансферазы (ALT) и аспартатаминотрансферазы (AST) определяли с использованием набора для спектрофотометрического ферментного анализа (Asan Pharm. Co., Ltd., Hwaseong-si, Korea) согласно инструкции производителя. Также использовали автоматизированный прибор для исследования общей биохимии сыворотки. Образцы печени фиксировали в 10% нейтральном забуференном формалине перед рутинной обработкой в залитых парафином образцах. Делали срезы (5 мкм толщиной), окрашивали их с использованием гематоксилина и эозина (H&E) и исследовали методом световой микроскопии. Ткани печени лизировали в буфере RIPA [50 мМ Tris, pH 7,5, 150 мМ NaCl, 1 мМ EDTA, 0,1% додецилсульфата натрия, 0,5% дезоксихолата натрия, 1% NP-40, 50 мМ NaF, 1 мМ Na3VO4, 1 мМ PMSF, смесь ингибиторов протеаз (1 таблетка смеси ингибиторов протеаз Roche Diagnostics GmbH/10 мл) (Roche)] в течение 20 мин на льду. Лизаты очищали центрифугированием при 13000 об/мин при 4°C в течение 20 мин. Содержание белка в супернатанте определяли с использованием набора для анализа белка Micro-BCA (Thermo Scientific). Лизаты, содержащие 20-60 мкг общего белка, отделяли электрофорезом на 6-10% полиакриламидном геле с додецилсульфатом натрия и затем переносили на нитроцеллюлозные (Whatman®, Germany) или поливинилидендифторидные мембраны (Millipore). Мембраны блокировали 5% BSA (Sigma-Aldrich) или 5% раствором нежирного сухого молока с течение 1 ч и инкубировали в течение ночи при 4°C с одним из следующих антител: антитело кролика к фосфо-Smad3 (Cell Signaling Technology, Beverly, MA, USA), антитело кролика к α-SMA (Millipore), антитело мыши к фибронектину, антитело мыши к виментину (BD Biosciences) или антитело мыши к β-актину (Sigma-Aldrich). Мембраны трижды промывали солевым раствором с Tris-буфером и инкубировали или с HRP-конъюгированным козьим антителом против иммуноглобулина кролика, или с HRP-конъюгированным козьим антителом против иммуноглобулина мыши (SantaCruz Biotechnology) при комнатной температуре в течение 1 ч. Связанные антитела обнаруживали с использованием набора ECL (GE Healthcare, Princeton, NJ, USA). Интенсивность полос анализировали с использованием устройства формирования изображения с денситометром LAS-3000 (FUJIFILM).
Крысы с BDL демонстрировали потерю массы тела и увеличение массы органов (печени и селезенки) по сравнению с ложнооперированными контрольными крысами. Соединения примеров 2 и 3 восстанавливали потерю массы тела и снижали массу органов (печени и селезенки) у крыс с BDL. У крыс с BDL наблюдалось существенное увеличение показателей сывороточных ALT и AST по сравнению с ложнооперированными животными. Соединения примеров 2 и 3 улучшали показатели сывороточных ALT и AST у крыс с BDL. Соединения примера 3 ингибировали передачу сигнала Smad и подавляли α-SMA, фибронектин и виментин в печень крыс с BDL. Соединения примера 2 подавляли α-SMA и фибронектин в печень крыс с BDL. Печени крыс с BDL показали типичные гистологические изменения, характеризующиеся разрушением центральной±центральной архитектуры и формированием мостикового фиброза по сравнению с таковым в печени нормальных крыс. Соединения примеров 2 и 3 в большой степени отменяли индуцированное BDL гистологическое изменение.
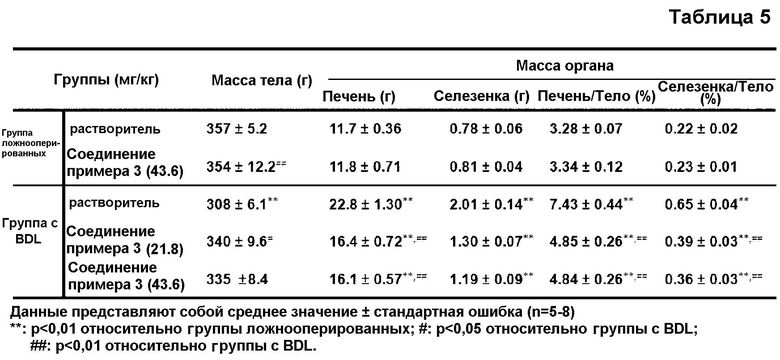
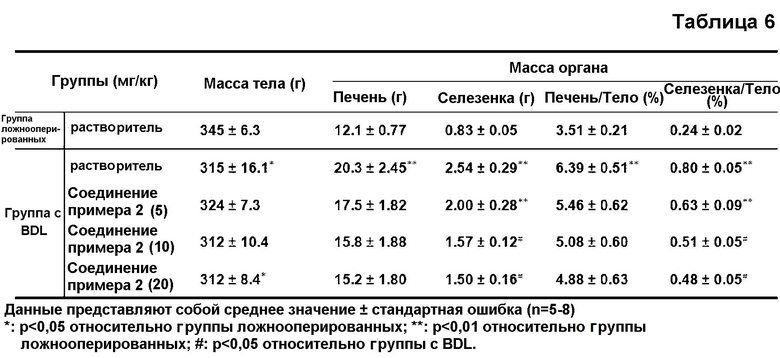
Антифибротический эффект на модели индуцированного блеомицином фиброза легкого
Самцов мышей ICR в возрасте 6 месяцев приобретали в Orient Bio Inc. Мышей массой 31-35 г случайным образом распределяли на пять экспериментальных групп: ложнооперированые контрольные мыши (солевой раствор, n=6), получавшие блеомицин (BLM) мыши (n=10), и получавшие BLM мыши, получавшие лечение либо 5, либо 10, либо 20 мг/кг соединения примера 2 (n=10). Для индукции фиброза легкого мышам проводили анестезию золетилом (10 мг/кг) и ксилазином (5 мг/кг) и давали им BLM (который давали в виде BLM сульфата, 1 мг/кг) (MBcell, Los Angeles, CA, USA), растворенный в 60 мкл солевого раствора, один раз в сутки 0 посредством интратрахеальной инстилляции. Соединение примера 2, растворенное в составе искусственного желудочного сока, давали мышам перорально пять раз в неделю в течение двух недель, начиная с суток 7. Животных содержали в комнате с контролируемой температурой (при 21°C) и снабжали автоклавированной пищей и водой. На третьей неделе после операции животных умерщвляли и удаляли легкие. Легкие сагиттально разрезали на несколько частей, быстро замораживали в жидком азоте и хранили при -70°C. Часть легких погружали в 10% нейтральный забуференный формалин для гистопатологических исследований. Все экспериментальные процедуры проводили согласно своим внутриорганизационным нормам. Образцы легкого фиксировали в 10% нейтральном забуференном формалине перед рутинной обработкой в залитых парафином образцах. Делали срезы (5 мкм толщиной), окрашивали с использованием гематоксилина и эозина (H&E) и исследовали методом световой микроскопии. Ткани легкого лизировали в буфере RIPA [50 мМ Tris, pH 7,5, 150 мМ NaCl, 1 мМ EDTA, 0,1% додецилсульфата натрия, 0,5% дезоксихолата натрия, 1% NP-40, 50 мМ NaF, 1 мМ Na3VO4, 1 мМ PMSF, смесь ингибиторов протеаз (1 таблетка смеси ингибиторов протеаз Roche Diagnostics GmbH/10 мл) (Roche)] в течение 20 мин на льду. Лизаты очищали центрифугированием при 13000 об/мин при 4°C в течение 20 мин. Содержание белка в супернатанте определяли с использованием набора для анализа белка Micro-BCA (Thermo Scientific). Лизаты, содержащие 20―50 мкг общего белка, отделяли электрофорезом на 6―10% полиакриламидном геле с додецилсульфатом натрия и затем переносили на нитроцеллюлозную мембрану (Whatman®). Мембраны блокировали 5% раствором нежирного сухого молока в течение 1 ч и инкубировали в течение ночи при 4°C или с антителом кролика к α-SMA (Millipore), или с антителом мыши к фибронектину (BD Biosciences). Мембраны трижды промывали солевым раствором с Tris-буфером и инкубировали или с HRP-конъюгированным козьим антителом против иммуноглобулина кролика, или с HRP-конъюгированным козьим антителом против иммуноглобулина мыши (SantaCruz Biotechnology) при комнатной температуре в течение 1 ч. Связанные антитела обнаруживали с использованием набора ECL (GE Healthcare). Интенсивность полос анализировали с использованием устройства формирования изображения с денситометром LAS-3000 (FUJIFILM).
В индуцированных BLM фиброзных легких было обнаружено повышенное содержание α-SMA и фибронектина по сравнению с ложнооперированными животными. Соединение примера 2 снижало содержание α-SMA и фибронектина в индуцированном BLM фиброзном легком. В тканях легкого из получавших BLM мышей обнаруживали типичную гистологию, характеризующуюся утолщением легочных межальвеолярных перегородок и их инфильтрацию воспалительными клетками с обнаружением отложений коллагена в интерстициальной ткани. Соединение примера 2 значительно снижало индуцированные BLM гистологические изменения при всех тестируемых дозах.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗОТСОДЕРЖАЩИЕ КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОДУКЦИИ БЕТА-АМИЛОИДА | 2010 |
|
RU2515976C2 |
| ИНГИБИТОРЫ ЦИТОКИНОВ | 2008 |
|
RU2485113C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ | 2009 |
|
RU2513723C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2-ПИРИДИНКАРБОКСАМИДА | 2004 |
|
RU2338744C2 |
| СОЕДИНЕНИЯ, ЯВЛЯЮЩИЕСЯ АНТАГОНИСТАМИ А3-АДЕНОЗИНОВОГО РЕЦЕПТОРА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2737157C2 |
| 6-5 КОНДЕНСИРОВАННЫЕ КОЛЬЦА КАК ИНГИБИТОРЫ С5а | 2018 |
|
RU2780338C2 |
| КОНДЕНСИРОВАННОЕ 4-ОКСОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2005 |
|
RU2358969C2 |
| ПРОИЗВОДНЫЕ 2-ПИРИДОНА | 2010 |
|
RU2551847C2 |
| НОВОЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ БЕНЗИМИДАЗОЛА | 2004 |
|
RU2329261C2 |
| ИНГИБИТОРЫ КИНАЗ, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И ДРУГИХ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2482112C2 |
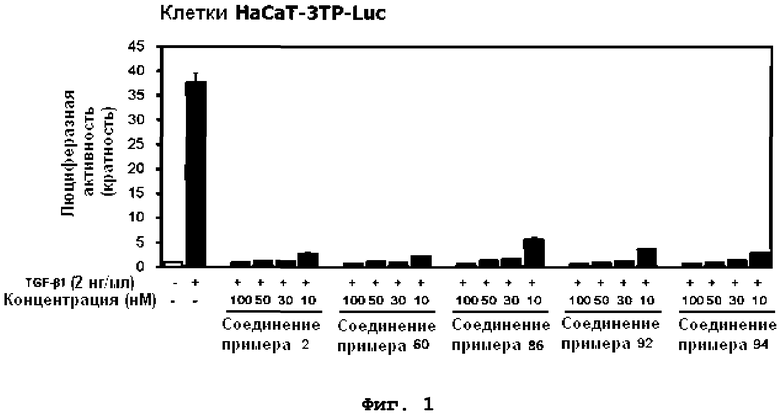
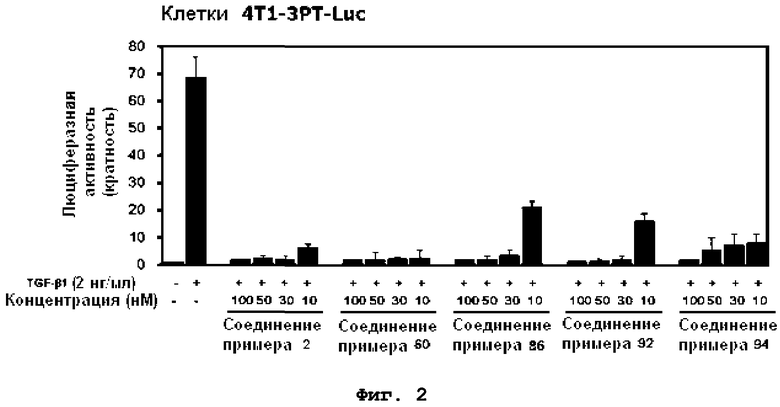
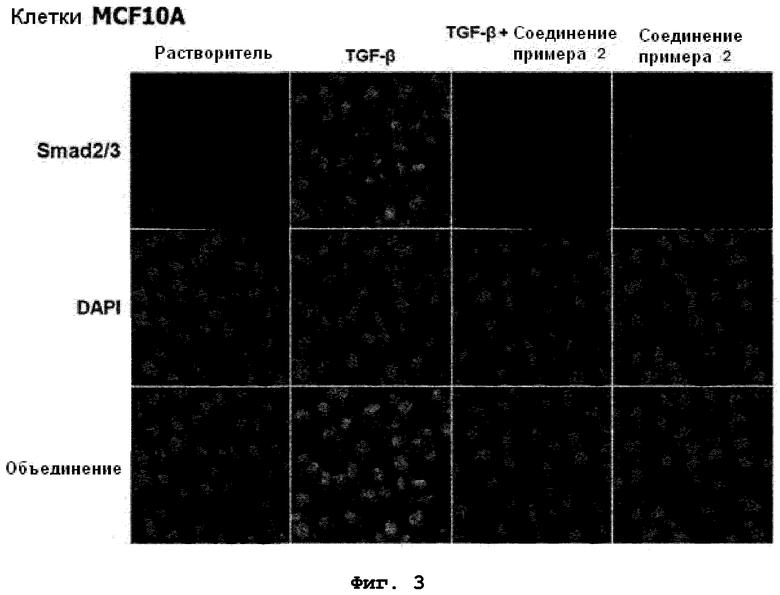
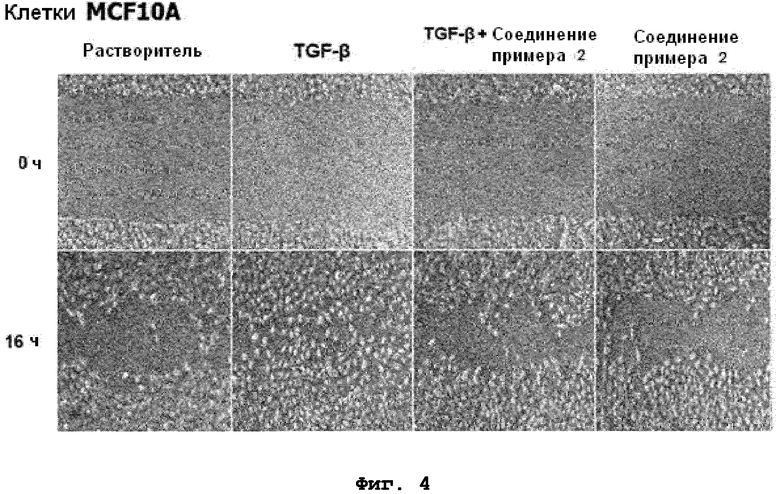
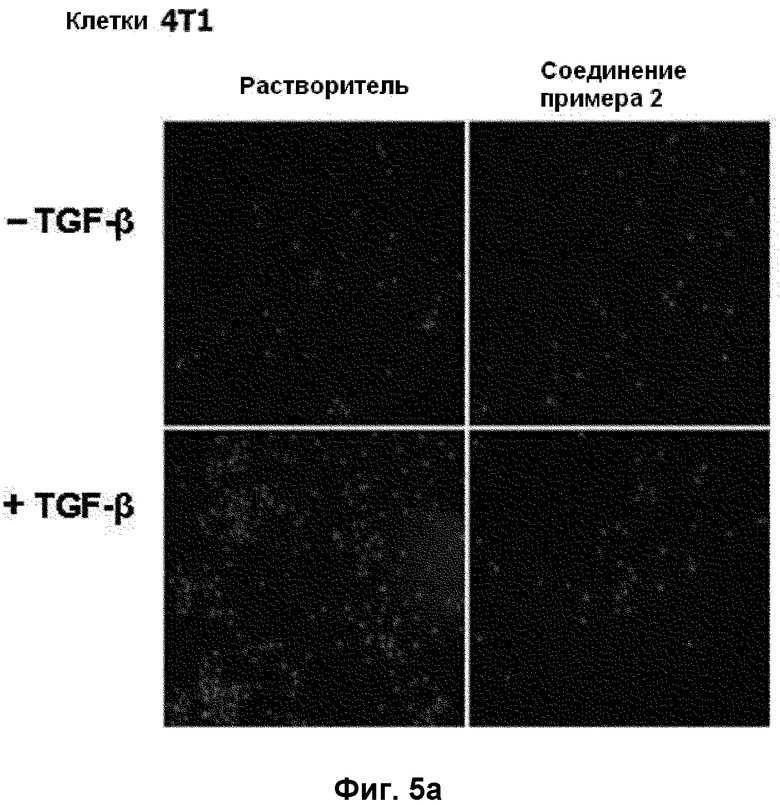
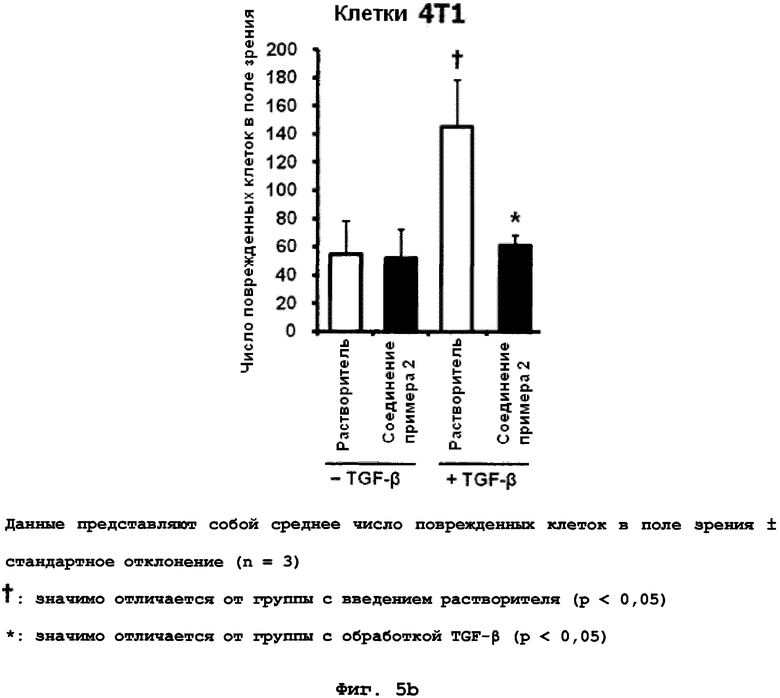
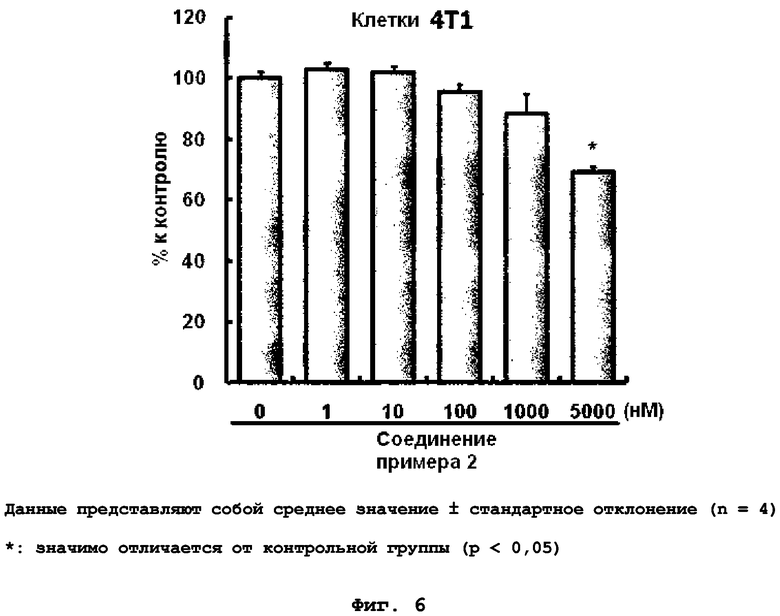
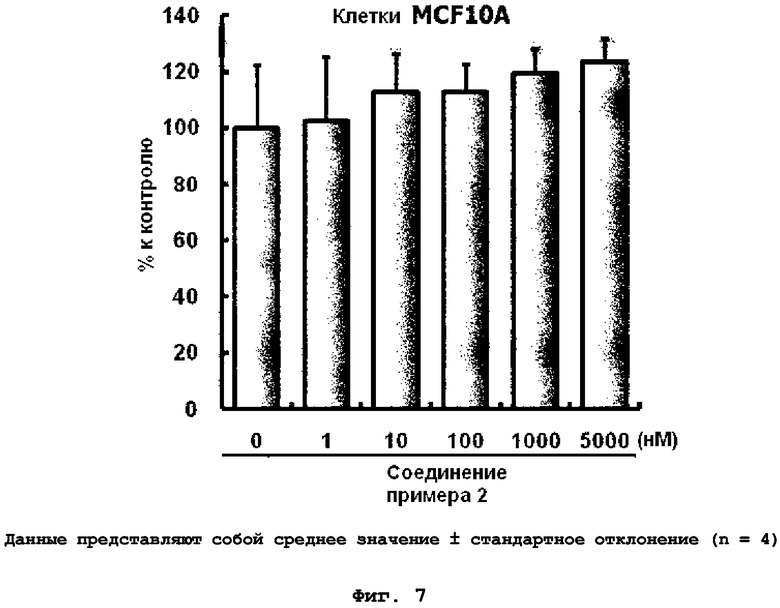
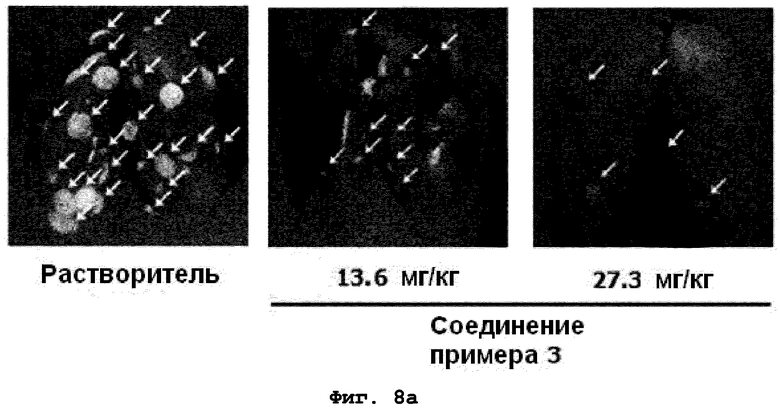
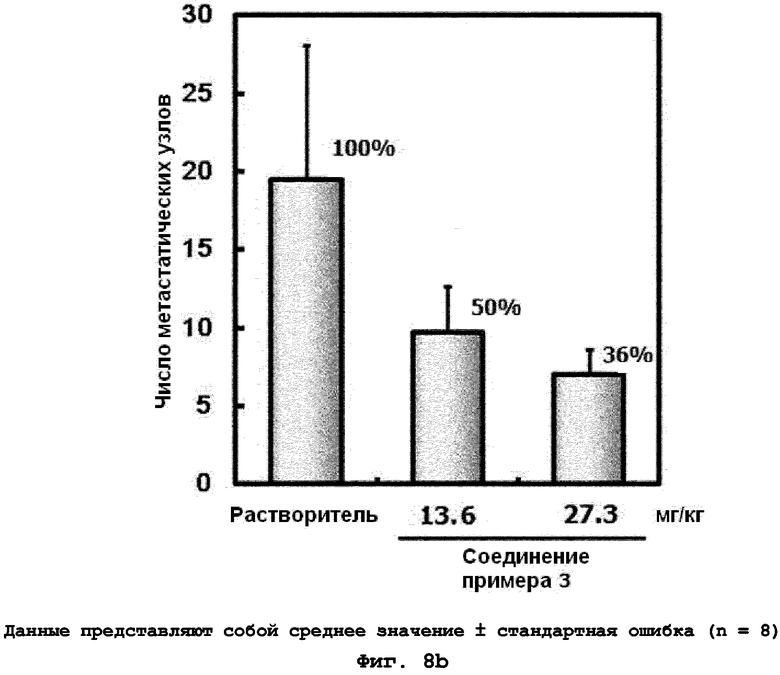
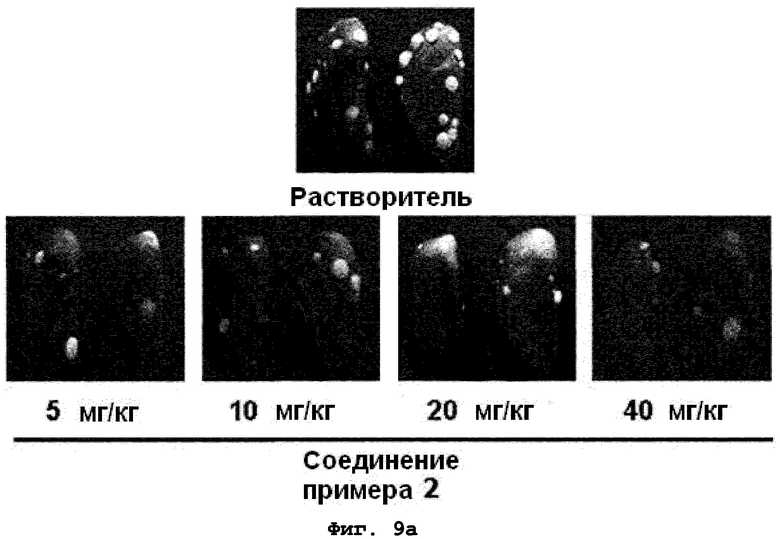
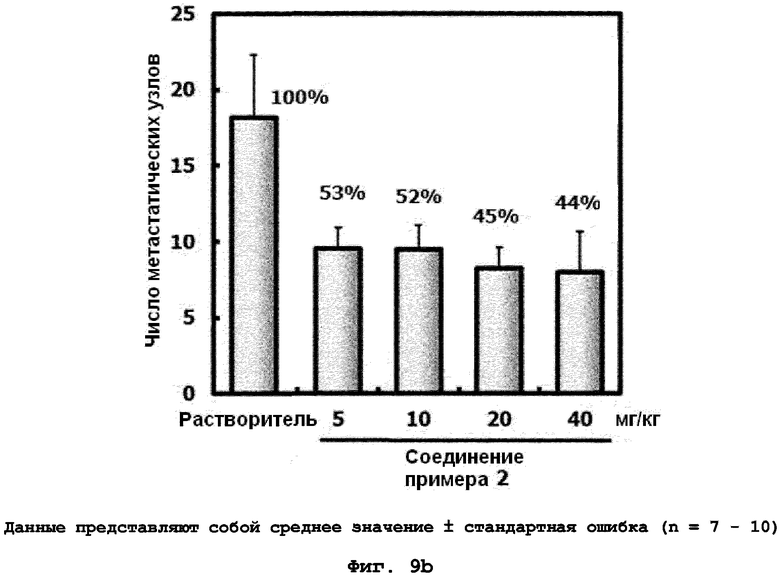
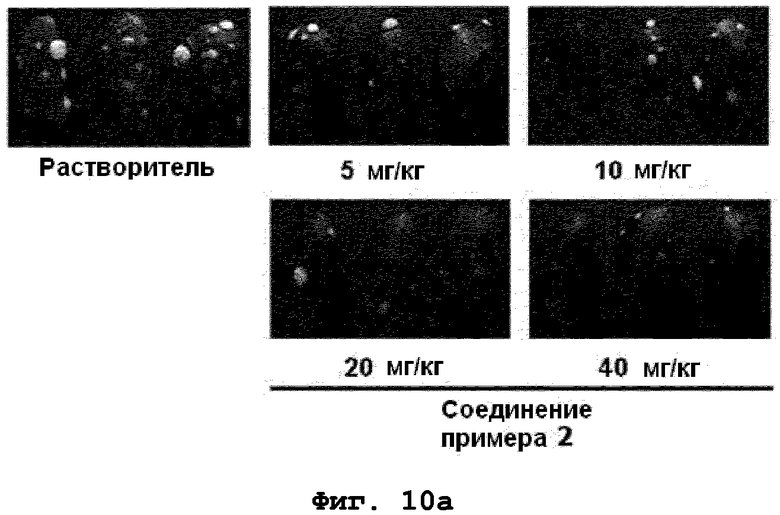
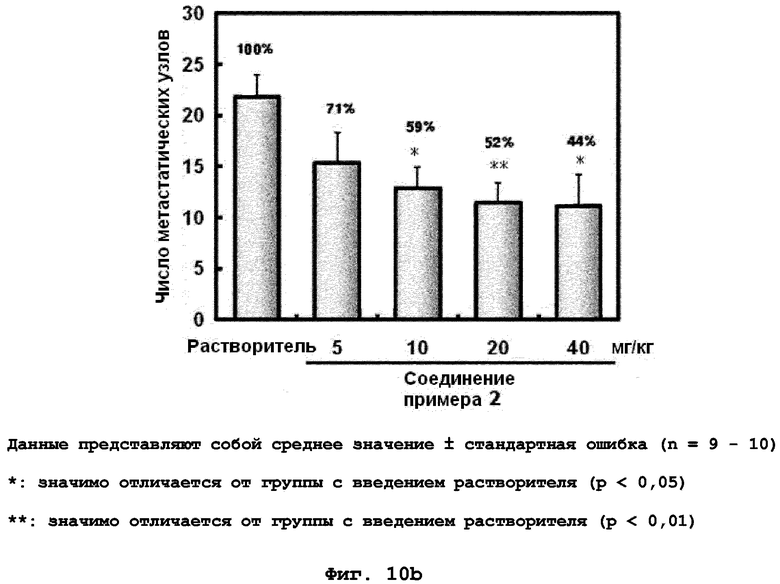
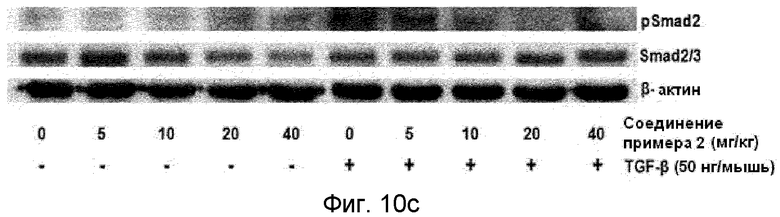

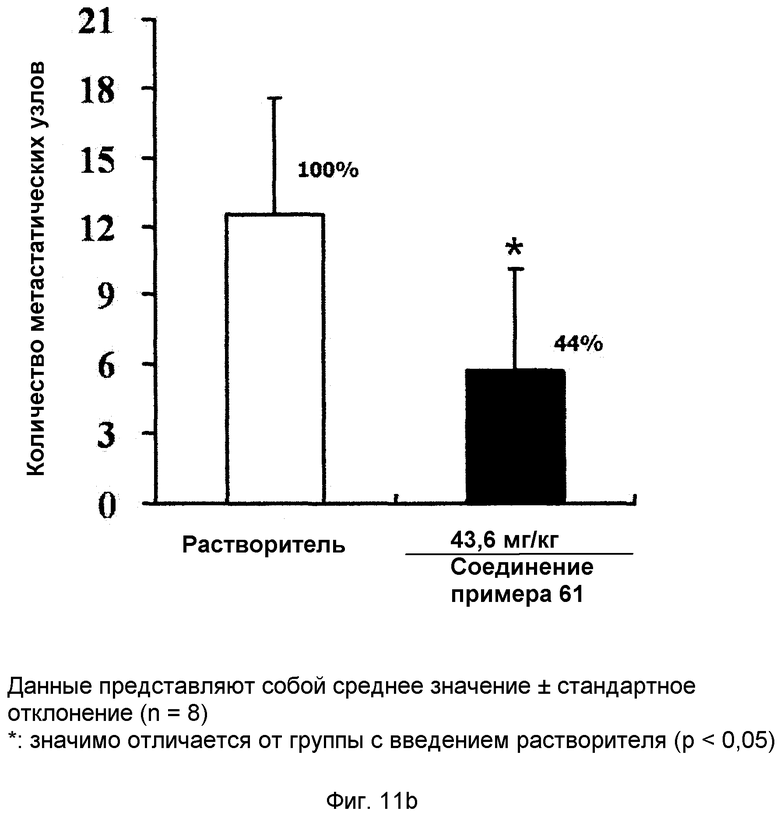
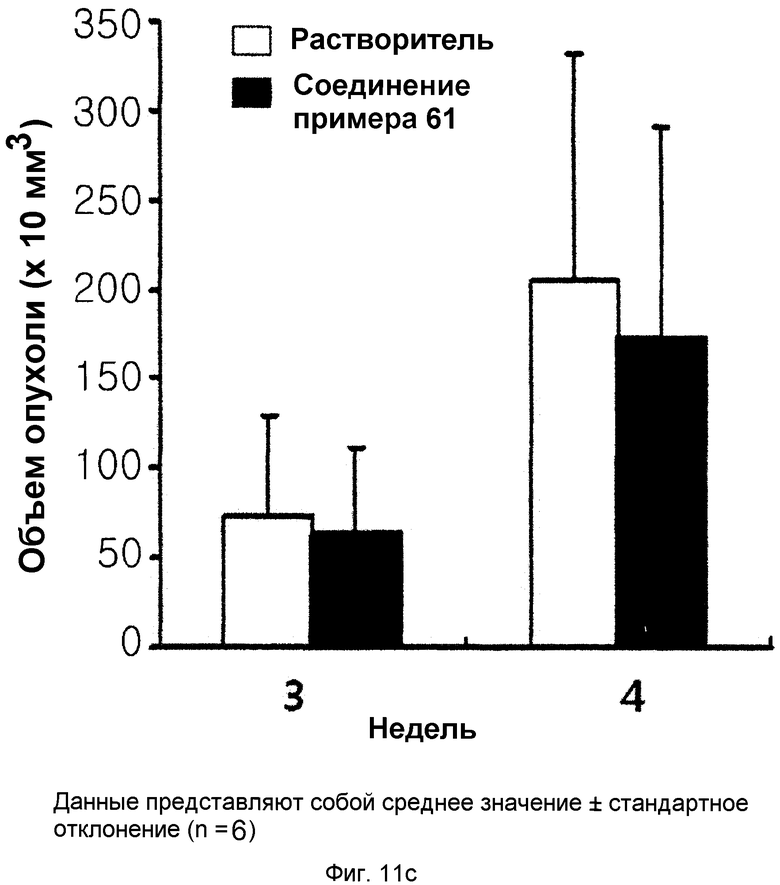
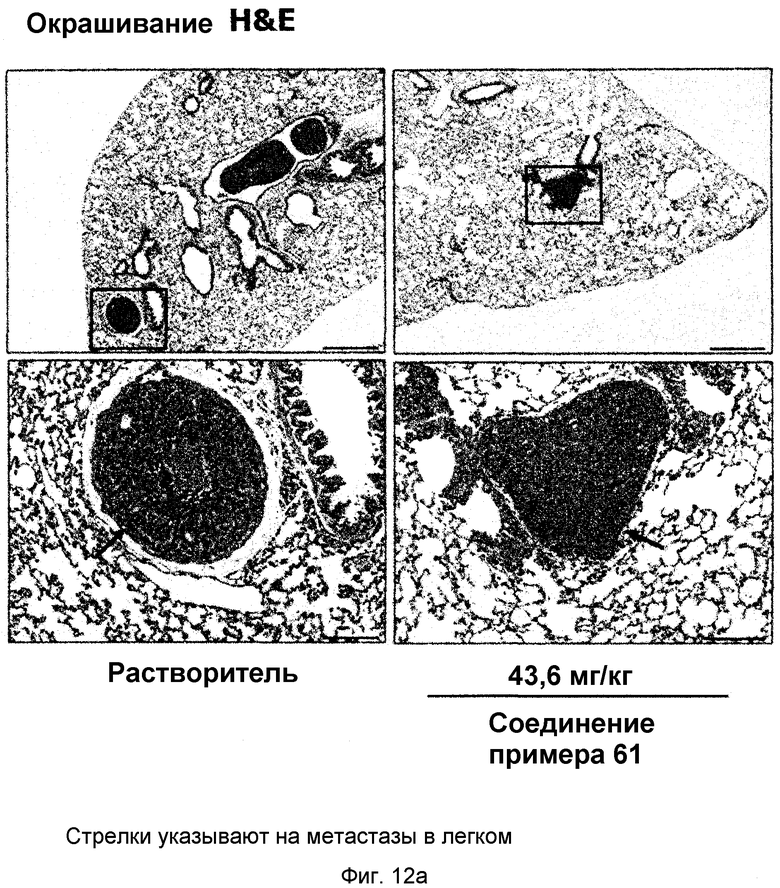
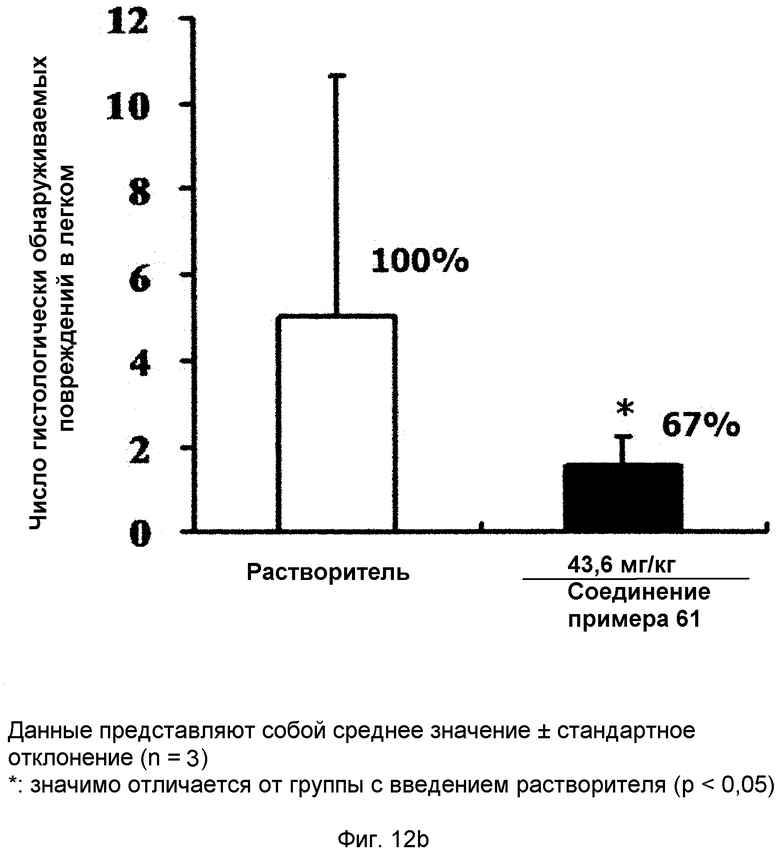
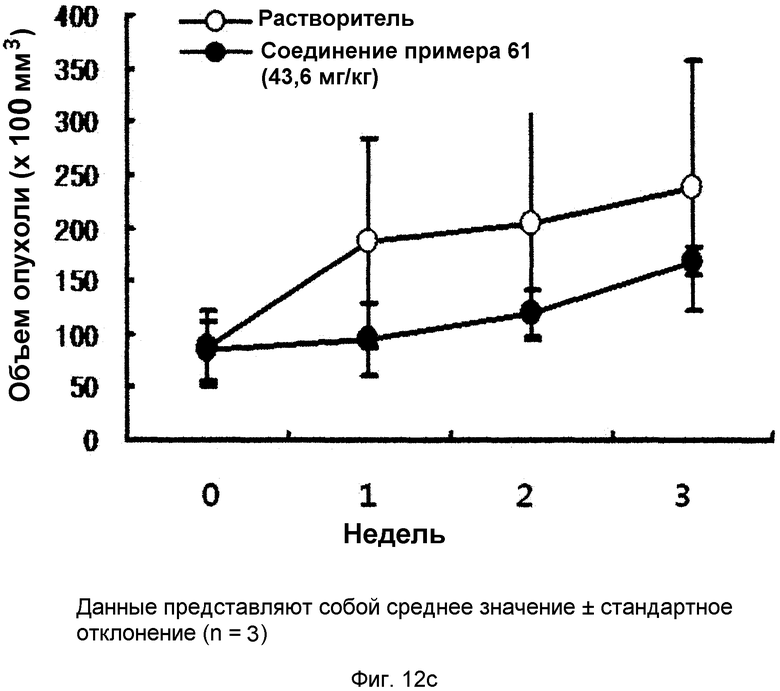
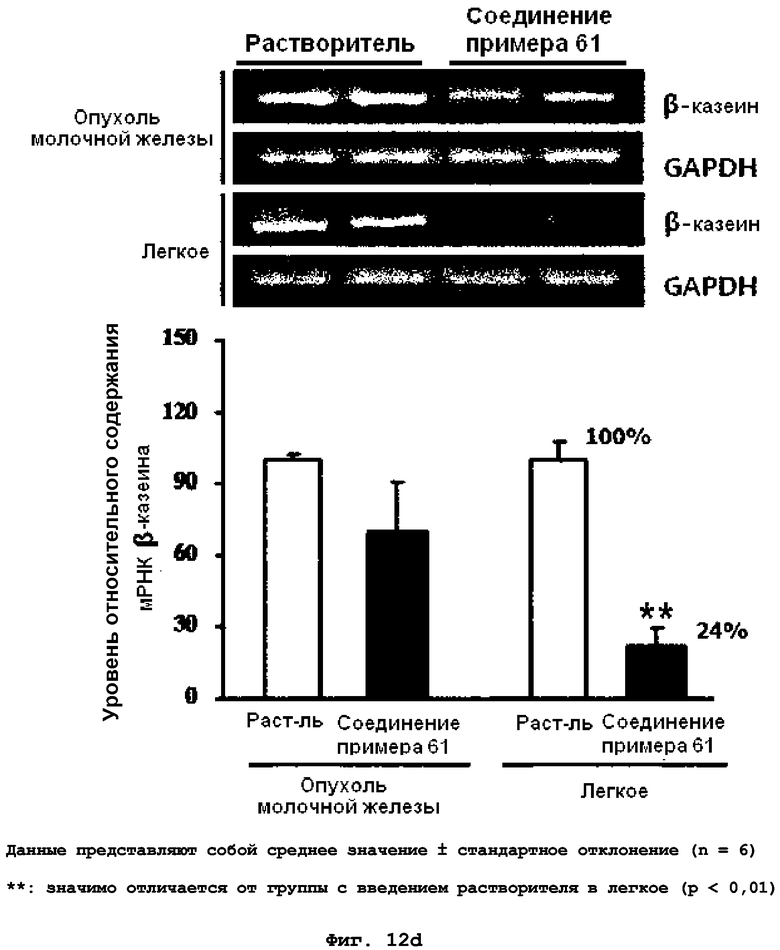
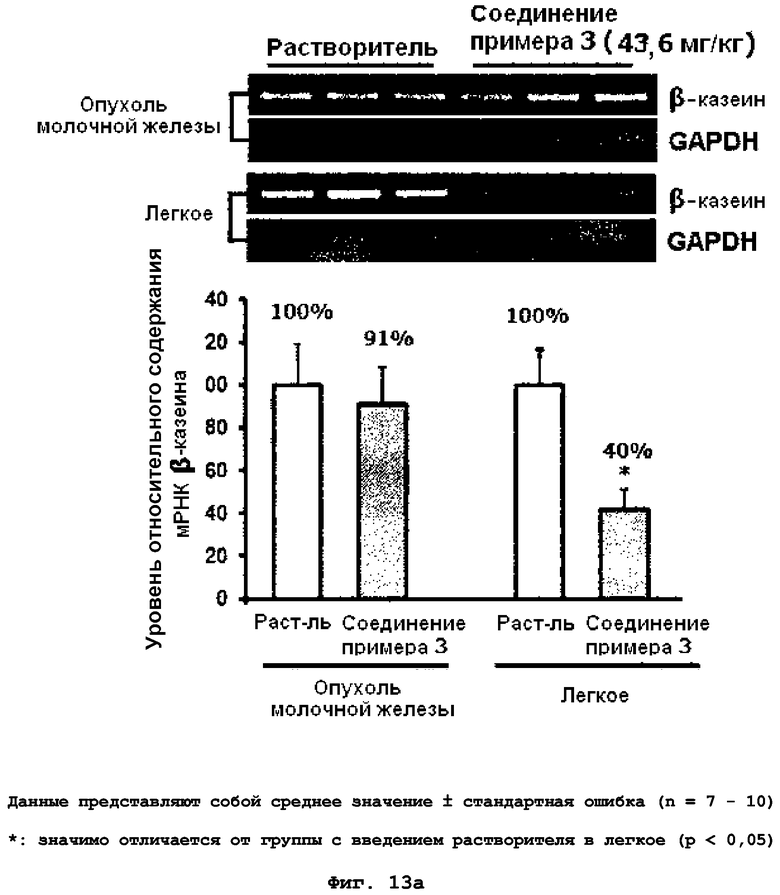
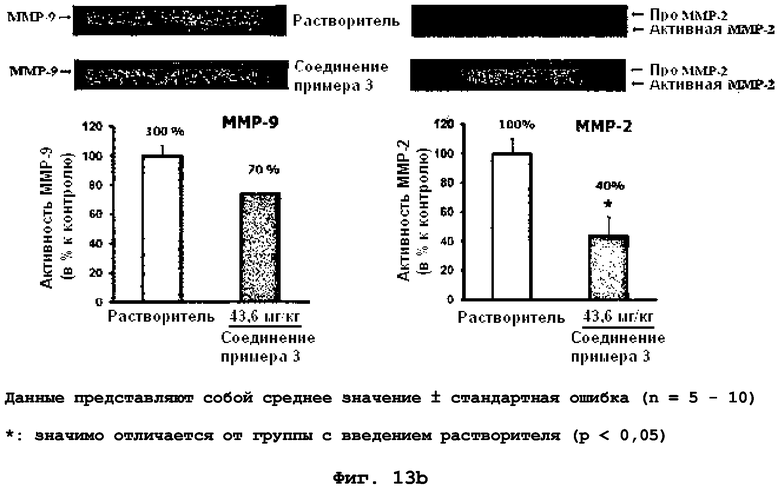
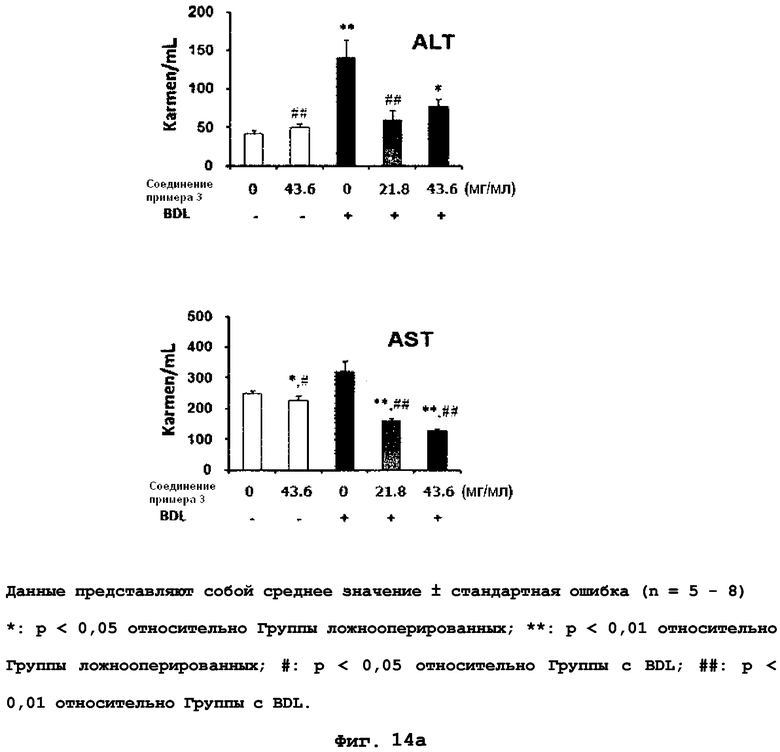
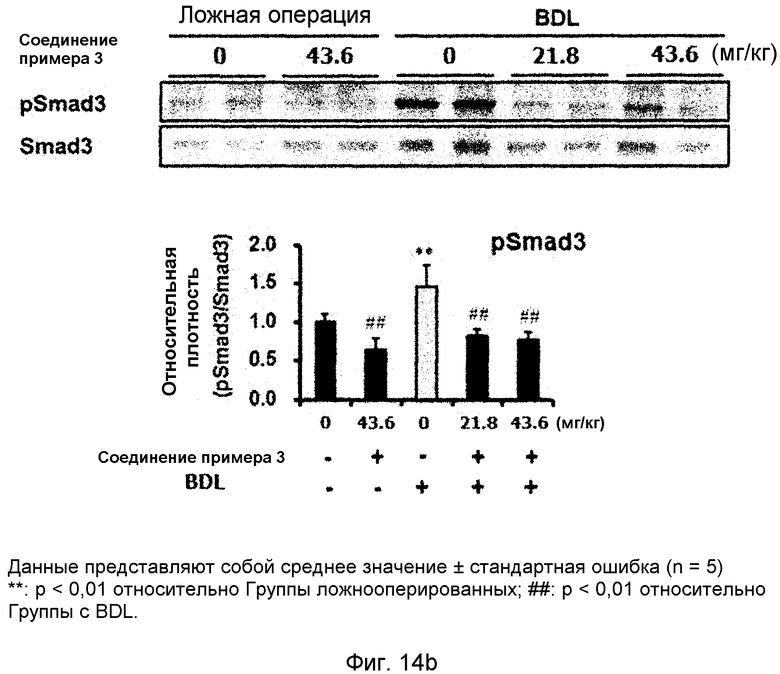
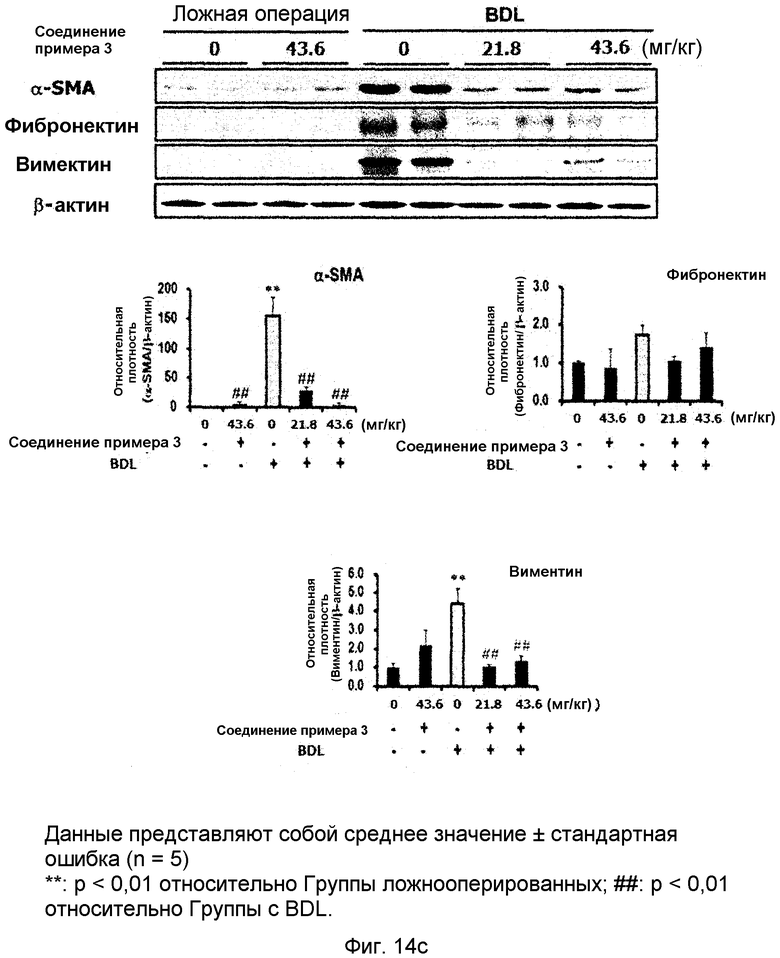
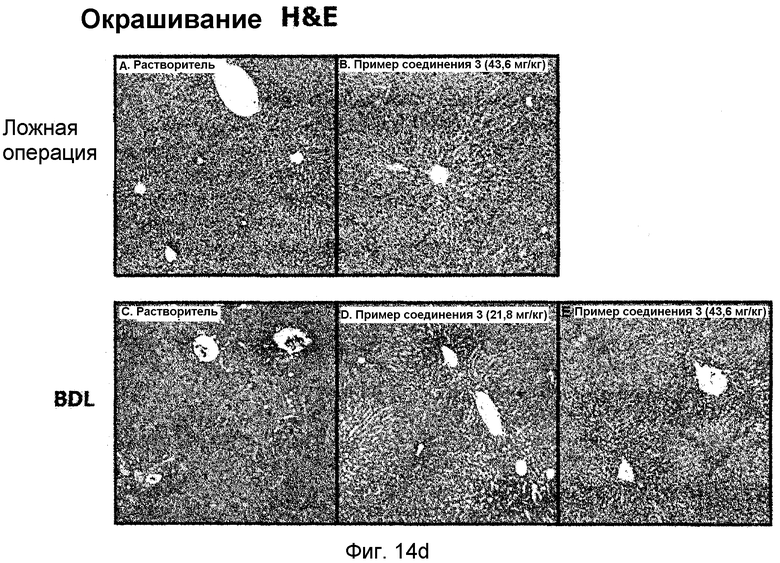
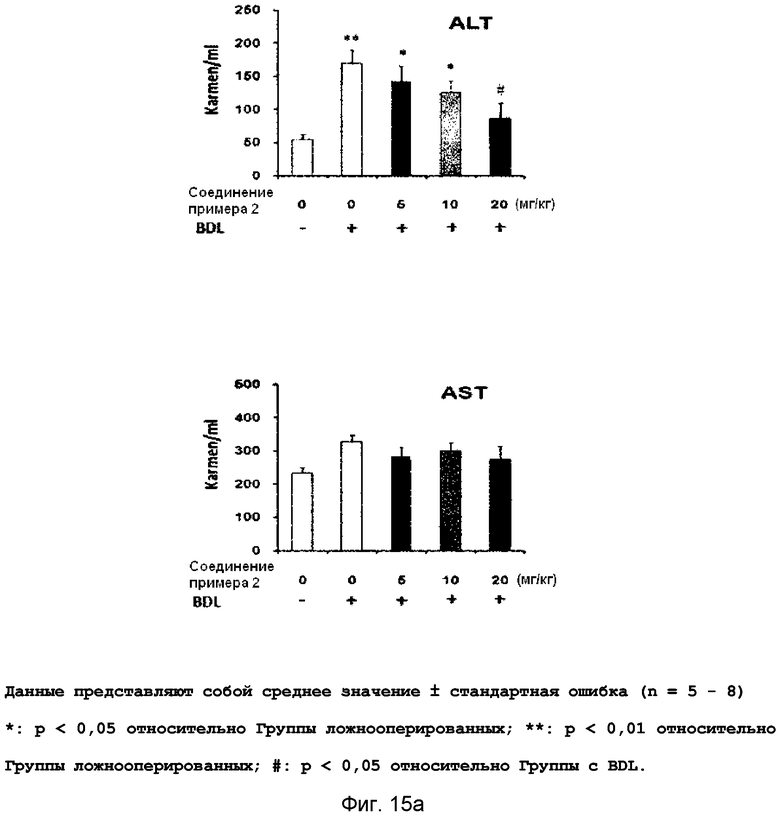
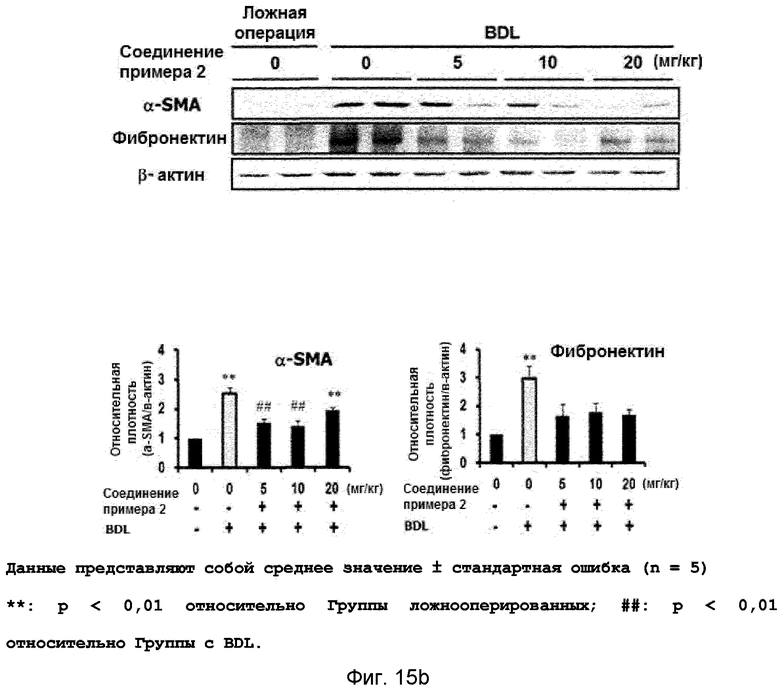
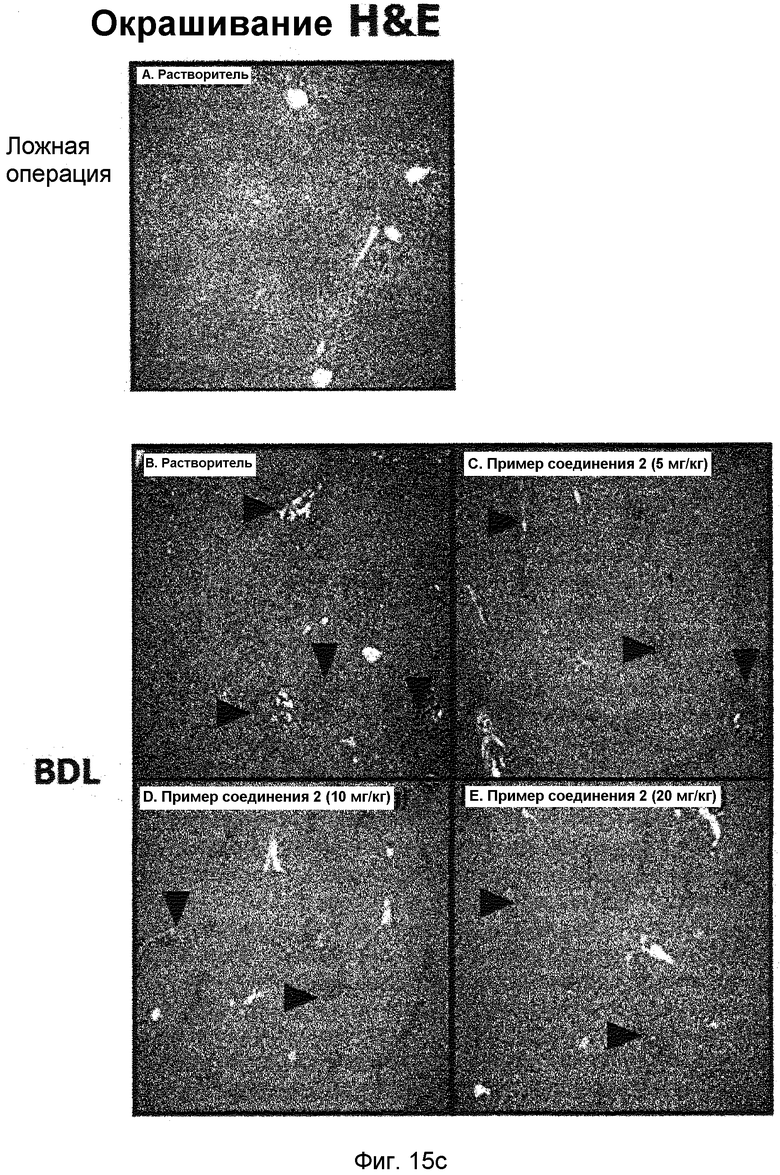
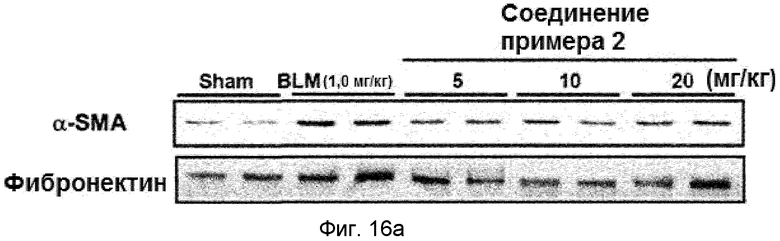
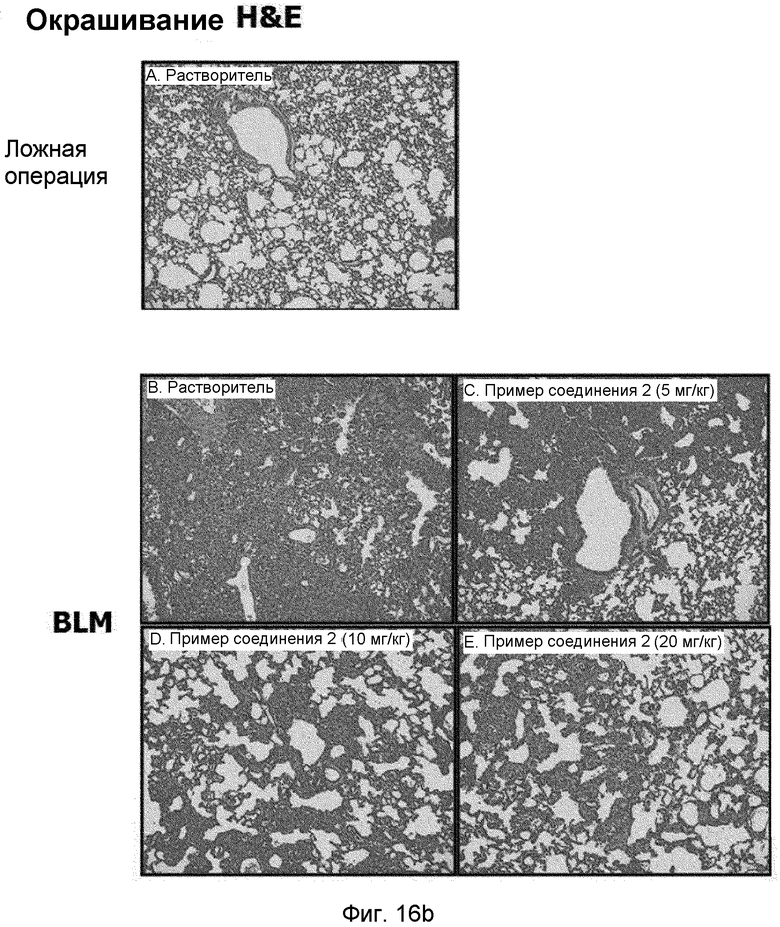
Описываются новые 2-пиридилзамещенные имидазолы общей формулы (I)
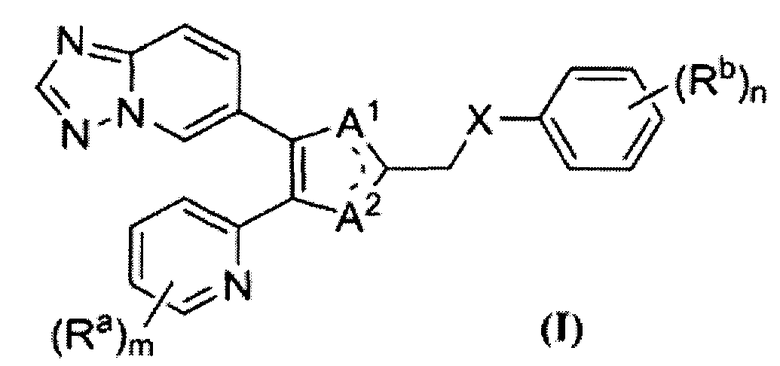
где Ra - C1-6алкил; m равно 1;
A1 = N; A2 = NR1, где R1 - водород;
X - связь, -NR2-, -O- или -S-, где R2 - водород или C1-3алкил;
Rb независимо - H, галоген, C1-6алкил, C2-6алкенил, C2-6алкинил,
-(CH2)q-OR3, где R3 - C1-6алкил или C1-6галогеналкил, и
q=0,1,
-(CH2)q-NR3R4, где R3 и R4 независимо - C1-6алкил или совместно с атомом азота - пирролидинил или морфолинил, и q=0-2;
-SR3, где R3 - C1-6алкил,
-(CH2)q-CN, где q=0 или 1,
-COR3 или -CO2R3, где R3 - C1-6алкил,
-CONR3R4, где R3 и R4 - водород,
-NHCOR3 или -NHSO2R3, где R3 означает C1-6алкил;
n равно 0, 1, 2, 3, 4 или 5;
или их фармацевтически приемлемые соли, или гидраты и фармацевтически приемлемые композиции для лечения или ослабления метастазирования опухолевых клеток, карцином, фиброза путем ингибирования активности путей передачи сигнала TGF-β или активина, или обоих. 5 н. и 1 з.п. ф-лы, 6 табл., 7 пр., 33 ил.
1. Соединение формулы (I)
где
Ra представляет собой C1-6алкил;
m равно 1;
A1 представляет собой N;
A2 представляет собой NR1, где R1 означает водород;
X представляет собой связь, -NR2-, -O- или -S-, где R2 означает водород или C1-3алкил;
каждый Rb независимо представляет собой H, галоген, C1-6алкил, C2-6алкенил, C2-6алкинил,
-(CH2)q-OR3, где R3 означает C1-6алкил или C1-6галогеналкил и
q=0 или 1,
- (CH2)q-NR3R4, где R3 и R4 независимо означают C1-6алкил или совместно с атомом азота, к которому они присоединены, образуют моноциклическое кольцо, такое как пирролидинил или морфолинил, и q=0, 1 или 2;
-SR3, где R3 означает C1-6алкил,
-(CH2)q-CN, где q=0 или 1,
-COR3, где R3 означает C1-6алкил,
-CO2R3, где R3 означает C1-6алкил,
-CONR3R4, где R3 и R4 означают водород,
-NHCOR3, где R3 означает C1-6алкил,
-NHSO2R3, где R3 означает C1-6алкил;
n равно 0, 1, 2, 3, 4 или 5;
или его фармацевтически приемлемая соль или гидрат.
2. Соединение по п.1, которое выбирают из группы, состоящей из:
N-((4-([1,2,4]триазоло [1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фторанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-фторанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-фторанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2,3-дифторанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,4-дифторанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,5-дифторанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-хлоранилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-хлоранилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-хлоранилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2,3-дихлоранилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,4-дихлоранилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,5-дихлоранилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-броманилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-броманилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-броманилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-метиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-метиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-метиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2,3-диметиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,4-диметиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,5-диметиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-этиланилина;
N-((4-([l,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-этиланилина;
N-((4-([l,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-изопропиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-изопропиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-изопропиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-виниланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-виниланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-виниланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-этиниланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-метоксианилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-метоксианилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-метоксианилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2,3-диметоксианилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,4-диметоксианилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3,5-диметоксианилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(метоксиметил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(метоксиметил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-(метоксиметил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(трифторметокси)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(трифторметокси)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-(трифторметокси)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(метилтио)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(метилтио)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-(метилтио)анилина;
2-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензонитрила;
4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фталонитрила;
2-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензамида;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензамида;
4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензамида;
2-(3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетонитрила;
2-(4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетонитрила;
1-(3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)этанона;
1-(4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)этанона;
метил-3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензоата;
метил-4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензоата;
N-(2-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетамида;
N-(3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетамида;
N-(4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)ацетамида;
N-(2-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)метансульфонамида;
N-(3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)метансульфонамида;
N-(4-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)фенил)метансульфонамида;
N1-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-N2,N2-диметилбензол-1,2-диамина;
Nl-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-N3,N3-диметилбензол-1,3-диамина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(пирролидин-1-ил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-морфолиноанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-морфолиноанилина;
N3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-4-фтор-N1,N1-диметилбензол-1,3-диамина;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламинo)-5-(диметиламино)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-4-(диметиламино)бензонитрила;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-((диметиламино)метил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-((диметиламино)метил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(пирролидин-1-илметил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(пирролидин-1-илметил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(морфолинометил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(морфолинометил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-5-((диметиламино)метил)-2-фторанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-((диметиламино)метил)-2-фторанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фтор-3-(пирролидин-1-илметил)анилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фтор-3-(морфолинометил)анилина;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-4-(диметиламино)метил)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-2-(диметиламино)метил)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-5-(диметиламино)метил)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-4-(пирролидин-1-илметил)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-2-(пирролидин-1-илметил)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-5-(пирролидин-1-илметил)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-4-(морфолинометил)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-2-(морфолинометил)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)-5-(морфолинометил)бензонитрила;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-(2-(диметиламино)этиланилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-3-(2-(диметиламино)этиланилина;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-этилпиридин-2-ил)-1H-имидазол-2-ил)метиламино)бензонитрила;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-этилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фторанилина;
N-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)-2-фтор-N-метиланилина;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)(метил)амино)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)(метил)амино)бензамида;
6-(2-бензил-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридина;
6-(2-(2-фторбензил)-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридина;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метил)бензамида;
6-(5-(6-метилпиридин-2-ил)-2-(феноксиметил)-1H-имидазол-4-ил)- [1,2,4]триазоло[1,5-a]пиридина;
6-(2-((2-фторфенокси)метил)-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридина;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1H-имидазол-2-ил)метокси)бензонитрила;
3-((4-([1,2,4]триазоло[1,5-a]пиридин-6-ил)-5-(6-метилпиридин-2-ил)-1Н-имидазол-2-ил)метокси)бензамида;
6-(5-(6-метилпиридин-2-ил)-2-(фенилтиометил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридина;
6-(2-((2-фторфенилтио)метил)-5-(6-метилпиридин-2-ил)-1H-имидазол-4-ил)-[1,2,4]триазоло[1,5-a]пиридина;
и их фармацевтически приемлемых солей и гидратов.
3. Фармацевтическая композиция, проявляющая ингибирующую активность путей передачи сигнала TGF-β или активина, или обоих, содержащая терапевтически эффективное количество одного или более соединений по п.1 или их фармацевтически приемлемой соли, или гидрата и фармацевтически приемлемый разбавитель или носитель.
4. Фармацевтическая композиция для лечения, профилактики или ослабления метастазирования опухолевых клеток, содержащая терапевтически эффективное количество одного или более соединений по п.1 или их фармацевтически приемлемой соли или гидрата.
5. Фармацевтическая композиция для лечения, профилактики или ослабления карцином, опосредованных избыточной экспрессией TGF-β, путем ингибирования пути передачи сигнала TGF-β, содержащая терапевтически эффективное количество одного или более соединений по п.1 или их фармацевтически приемлемой соли или гидрата.
6. Фармацевтическая композиция для лечения, профилактики или ослабления фиброза, содержащая терапевтически эффективное количество одного или более соединений по п.1 или их фармацевтически приемлемой соли или гидрата.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| US 4775762 A 04.10.1988 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| . | |||

