Настоящее изобретение относится к хинолиновым производным, способам их получения, фармацевтическим композициям, содержащим их, способу получения фармацевтических композиций и их применению в терапии.
Р2Х7 рецептор (ранее известный как P2Z рецептор), который представляет собой управляемый лигандами ионный канал, присутствует на множестве типов клеток, главным образом на тех, про которые известно, что они вовлечены в воспалительный/иммунный процесс, более конкретно, макрофагах, тучных клетках и лимфоцитах (Т и В). Активация Р2Х7 рецептора внеклеточными нуклеотидами, в частности аденозинтрифосфатом, приводит к высвобождению интерлейкина-1β (IL-1β) и образованию гигантских клеток (макрофаги/клетки микроглии), дегрануляции (тучные клетки) и пролиферации (Т-клетки), апоптозу и шеддингу L-селектина (лимфоциты). Также Р2Х7 рецепторы расположены на антиген-представляющих клетках (АРС), кератиноцитах, ацинарных клетках слюнных желез (клетках околоушной железы), гепатоцитах и мезангиальных клетках. Было бы желательным создание соединений, эффективных в качестве антагонистов Р2Х7 рецепторов, для применения в лечении воспалительных, иммунных или сердечно-сосудистых заболеваний, в этиологии которых может играть роль Р2Х7 рецептор.
Важным свойством лекарственного средства, действующего в качестве антагониста Р2Х7 рецептора, является его высокая эффективность. Кроме того, для таких лекарственных средств желательно, чтобы они обладали хорошей селективностью и фармакокинетическими свойствами для дополнительного усиления эффективности такого лекарственного средства. Например, было бы желательно, чтобы такие лекарственные средства проявляли низкую активность в отношении калиевого канала, кодируемого человеческим геном hERG (ether-a-go-go-related gene). В этом отношении низкая активность против hERG связывания in vitro является свидетельством низкой активности in vivo.
Р2Х7 антагонисты, содержащие хинолинильные группы, известны из WO 2003/080579, WO 2004/106305, WO 2005/009968 и WO 2006/059945. В настоящее время неожиданно было обнаружено, что узкий класс соединений, в общем описанных в WO 2004/106305, проявляет благоприятные фармацевтические свойства. Например, в дополнение к высокой эффективности соединения по настоящему изобретению проявляют очень низкую активность против hERG связывания, что повышает их пригодность для использования в качестве фармацевтических средств.
В соответствии с настоящим изобретением предложено соединение формулы (I) или его фармацевтически приемлемая соль
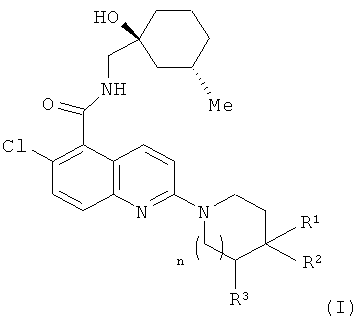 ,
,
где n равно 0 или 1;
когда n равно 0, тогда R1 представляет собой водород или метил, R2 представляет собой гидроксил, и R3 представляет собой водород; и
когда n равно 1, тогда R1 представляет собой водород, и один из R2 и R3 представляет собой гидроксил, а другой из R2 и R3 представляет собой водород.
Следует понимать, что некоторые соединения по настоящему изобретению могут существовать в сольватированной, например гидратированной, а также несольватированной формах. Следует понимать, что настоящее изобретение охватывает все такие сольватированные формы.
Соединения по настоящему изобретению демонстрируют очень высокую Р2Х7 антагонистическую активность. Кроме того, они обладают чрезвычайно низкой аффинностью в отношении калиевого канала, кодируемого человеческим геном hERG (ether-a-go-go-related gene), и поэтому являются перспективными в плане соображений безопасности.
Фармацевтически приемлемые соли соединения формулы (I) включают, но без ограничения ими, соли присоединения кислот, такие как гидрохлорид, гидробромид, фосфат, ацетат, фумарат, малеат, тартрат, цитрат, оксалат, метансульфонат или пара-толуолсульфонат. В одном воплощении изобретения фармацевтически приемлемые соли соединения формулы (I) выбраны из гидрохлорида, гидробромида, фосфата, тартрата, цитрата, оксалата, метансульфоната или пара-толуолсульфоната.
Соединения по настоящему изобретению содержат два хиральных центра, локализованных на циклогексильном кольце в формуле (I). Один из хиральных центров локализован на том атоме циклогексильного кольца, к которому непосредственно присоединен гидроксильный заместитель (положение 1), а другой локализован на том атоме циклогексильного кольца, к которому непосредственно присоединен метильный заместитель (положение 3). В настоящем изобретении стереохимическая конфигурация по обоим хиральным центрам является S-конфигурацией ((1S, 3S) стереоизомеры), как обозначается системой Кана-Ингольда-Прелога (Cahn-lngold-Prelog) и как представлено в структуре формулы (I) ниже.
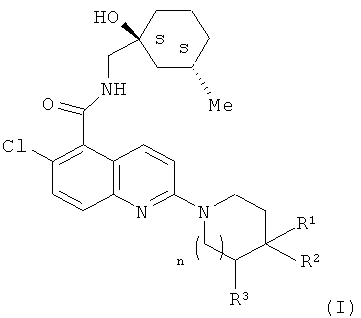
В воплощениях изобретения, где n равно 0, соединения формулы (I) содержат дополнительный хиральный центр по тому атому углерода, к которому непосредственно присоединены как R1, так и R2. Настоящее изобретение охватывает соединения всех стереохимических конфигураций по этому положению, включая их смеси.
В одном воплощении изобретения n равно 0, R1 представляет собой водород или метил, R2 представляет собой гидроксил, и R3 представляет собой водород.
В одном воплощении изобретения n равно 0, R1 представляет собой водород, R2 представляет собой гидроксил, и R3 представляет собой водород. В одном аспекте этого воплощения хиральный центр по атому углерода, к которому непосредственно присоединены R1 и R2, имеет S конфигурацию. В другом аспекте этого воплощения хиральный центр по атому углерода, к которому непосредственно присоединены R1 и R2, имеет R конфигурацию.
В одном воплощении изобретения n равно 0, R1 представляет собой метил, R2 представляет собой гидроксил, и R3 представляет собой водород. В одном аспекте этого воплощения хиральный центр по атому углерода, к которому непосредственно присоединены R1 и R2, имеет S конфигурацию. В другом аспекте этого воплощения хиральный центр по атому углерода, к которому непосредственно присоединены R1 и R2, имеет R конфигурацию.
В одном воплощении изобретения n равно 1, R1 представляет собой водород, и один из R2 и R3 представляет собой гидроксил, а другой из R2 и R3 представляет собой водород.
В одном воплощении изобретения n равно 1, R1 представляет собой водород, R2 представляет собой гидроксил, и R3 представляет собой водород.
В одном воплощении изобретения n равно 1, R1 представляет собой водород, R2 представляет собой водород, и R3 представляет собой гидроксил. Соединения согласно этому воплощению содержат дополнительный хиральный центр по атому углерода, к которому непосредственно присоединен R3. Настоящее изобретение охватывает все стереохимические конфигурации по этому положению, включая их смеси. В одном аспекте этого воплощения хиральный центр по атому углерода, к которому непосредственно присоединен R3, имеет S конфигурацию. В другом аспекте этого воплощения хиральный центр по атому углерода, к которому непосредственно присоединен R3, имеет R конфигурацию.
Соединения по настоящему изобретению содержат два хиральных центра, локализованных на циклогексильном кольце в формуле (I). Стереохимическая конфигурация по обоим этим хиральным центрам является S конфигурацией, то есть они являются (1S, 3S) стереоизомерами. Во избежание сомнений (1S, 3S) стереоизомеры по настоящему изобретению могут быть представлены в виде смеси с одним или более других возможных стереоизомеров по этим хиральным центрам, то есть (1R, 3R), (1R, 3S) и (1S, 3R) стереоизомеров. Например, (1S, 3S) стереоизомер может быть представлен в 1:1 смеси с (1R, 3R) стереоизомером.
В одном воплощении настоящего изобретения предложено соединение формулы (I), которое является оптически чистым по (1S, 3S) хиральным центрам. В еще одном воплощении настоящего изобретения предложено соединение формулы (I), которое является оптически чистым по всем его хиральным центрам.
В контексте настоящего описания термин «оптически чистый» определен в терминах энантиомерного избытка (е.е.) и диастереомерного избытка (d.e.), которые рассчитывают из соотношения разности между количествами соответствующих присутствующих энантиомеров/диастереомеров и суммой этих количеств, выраженного в процентах. Например, композиция, содержащая 95% одного энантиомера и 5% другого энантиомера, имеет энантиомерный избыток (е.е.) 90% [то есть (95-5)/(95+5)×100]. Диастереомерный избыток определяют аналогично энантиомерному избытку. Оптически чистые соединения по настоящему изобретению имеют е.е. по меньшей мере 90%. В одном воплощении изобретения оптически чистые соединения имеют е.е. по меньшей мере 95%. В другом воплощении изобретения оптически чистые соединения имеют е.е. по меньшей мере 98%. Когда соединение имеет диастереоизомеры, оптически чистые соединения имеют е.е. по меньшей мере 90% и диастереомерный избыток (d.e.) по меньшей мере 90%. В одном воплощении изобретения оптически чистые соединения имеют е.е. по меньшей мере 95% и d.e. по меньшей мере 95%. В другом воплощении изобретения оптически чистые соединения имеют е.е. по меньшей мере 98% и d.e. по меньшей мере 98%.
В одном воплощении изобретения соединение формулы (I) выбрано из:
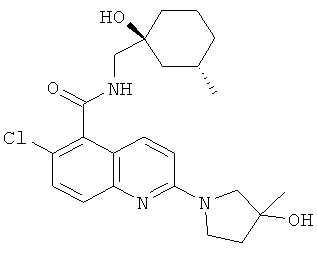
6-хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-[(3S)-3-гидроксипирролидин-1-ил]хинолин-5-карбоксамида,
6-хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-[(3S)-3-гидроксипирролидин-1-ил]хинолин-5-карбоксамида,
6-хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-(4-гидроксипиперидин-1-ил)хинолин-5-карбоксамида и
6-хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-(3-гидрокси-3-метилпирролидин-1-ил)хинолин-5-карбоксамида
или их фармацевтически приемлемой соли.
В настоящем изобретении также предложен способ получения соединения формулы (I), как оно определено выше, или его фармацевтически приемлемой соли, включающий:
(а) взаимодействие соединения формулы
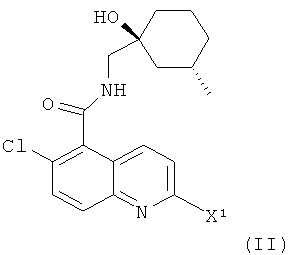
где X1 представляет собой подходящую уходящую группу (например, галоген, пара-толуолсульфонат, метансульфонат или трифторметансульфонат), с соединением формулы (III)
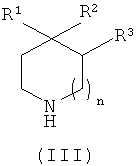 ,
,
где R1, R2, R3 и n являются такими, как определено в формуле (I), и возможно образование фармацевтически приемлемой соли этого соединения.
Взаимодействие (II) и (III) может быть осуществлено в органическом растворителе, таком как метанол, ацетонитрил, N,N-диметилформамид, диметилсульфоксид или 1-метил-2-пирролидинон, и в присутствии подходящего основания, такого как гидрид натрия, триэтиламин, диизопропилэтиламин или карбонат калия, при температуре в диапазоне от 50°С до 150°С, в частности от 80°С до 120°С, либо в микроволновом реакторе, либо в обычном тепловом режиме.
Соединения формулы (III) в виде либо свободного основания, либо соли (приемлемые соли соединения формулы (III) включают, без ограничения ими, соли присоединения кислот, такие как соль гидрохлорид, гидробромид, фосфат, ацетат, фумарат, малеат, тартрат, цитрат, оксалат, метансульфонат или п-толуолсульфонат) либо имеются в продаже, либо известны из литературы, либо могут быть получены с использованием известных методик специалистами в данной области.
Соединения формулы (II) могут быть получены посредством взаимодействия соединения формулы (IV)
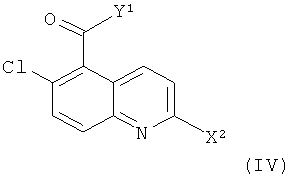 ,
,
где X2 представляет собой уходящую группу (например, галоген, пара-толуолсульфонат, метансульфонат или трифторметансульфонат) и Y1 представляет собой подходящую уходящую группу (например, гидроксил или хлор), с (1S,3S)-1-(аминометил)-3-метилциклогесанолом (соединение (V)).
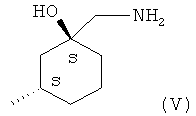
При взаимодействии (IV) и (V), где Y1 представляет собой радикал хлора, реакцию можно удобным образом осуществлять в органическом растворителе, таком как ацетон, дихлорметан, N,N-диметилформамид или 1-метил-2-пирролидинон, с подходящим основанием, таким как карбонат калия, диизопропилэтиламин или триэтиламин. Когда Y1 представляет собой гидроксильную группу, тогда может оказаться необходимым или желательным использовать агент сочетания, такой как бром-трис-пирролидино-фосфоний гексафторфосфат (PyBroP), 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDCI) или O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU). Когда Y1 представляет собой радикал хлора, такие соединения могут удобным образом быть получены посредством обработки соответствующего производного карбоновой кислоты в стандартных условиях (например, тионилхлорид или оксалилхлорид в дихлорметане).
Соединение формулы (V) является новым соединением и образует еще один аспект настоящего изобретения. Соответственно в еще одном аспекте настоящего изобретения предложено соединение, которое представляет собой (1S,3S)-1-(аминометил)-3-метилциклогексанол или его соль. В одном воплощении этого аспекта соединение (1S,3S)-1-(аминометил)-3-метилциклогексанол является оптически чистым (то есть оптически чистым, как определено для оптически чистых соединений формулы (I)). Соли (1S,3S)-1-(аминометил)-3-метилциклогексанола включают соли присоединения кислот, такие как гидрохлорид или гидробромид.
(1S,3S)-1-(Аминометил)-3-метилциклогексанол (V) может быть получен путем взаимодействия соединения формулы (VI) с подходящим образом защищенным эквивалентом аммиака, таким как фталимид (с последующей обработкой гидразином), ди-трет-бутилимидокарбонат (с последующей обработкой кислотой, например хлористым водородом), бензиламины, например 4-метоксибензиламин (с последующей обработкой 2,3-дихлор-5,6-дициано-1,4-бензохиноном (DDQ)) или, альтернативно бензиламину, N-бензил-1-метанамин или 1,1-дифенилметанамин (с последующим снятием защиты с помощью водорода в присутствии подходящего металлического катализатора).
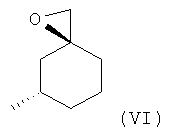
Взаимодействие между соединением формулы (VI) и бензиламином может удобным образом быть осуществлено в протонных растворителях, таких как метанол или этанол (возможно, в виде смешанной системы растворителей с толуолом), или в апротонных растворителях, таких как ацетонитрил, тетрагидрофуран или N,N-диметилформамид, при температуре в диапазоне от 25°С до 140°С, в частности от 65°С до 100°С, либо в микроволновом реакторе, либо в обычном температурном режиме. Последующее удаление бензильной защитной группы может удобным образом быть осуществлено в условиях гидрогенолиза в протонном растворителе, таком как метанол, этанол или уксусная кислота, или в апротонных растворителях, таких как этилацетат, при температуре в диапазоне от 25°С до 100°С, предпочтительно при 25°С, в атмосфере водорода при 1-5 бар (100-500 кПа), предпочтительно при 4 бар (400 кПа), в присутствии катализатора, такого как палладий на углероде, оксид платины или родия на углероде, предпочтительно палладия на углероде. Соединение (VI) известно из литературы (Alexakis A. et al., Synlett 2001, No.9, 1375). Подробный пример получения (1S,3S)-1-(аминометил)-3-метилциклогексанола приведен здесь далее в разделе «Примеры».
Соединение формулы (IV), где X2 представляет собой уходящую группу (например, галоген, паратолуолсульфонат, метансульфонат или трифторметансульфонат) и Y1 представляет собой гидроксил, может быть получено из соединения формулы (VII)
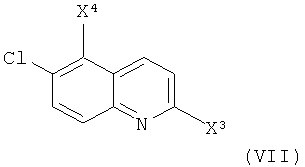 ,
,
где X3 представляет собой уходящую группу (например, галоген, паратолуолсульфонат, метансульфонат или трифторметансульфонат) и X4 представляет собой радикал иода или брома.
При превращении (VII) в (IV) взаимодействие может быть удобным образом осуществлено посредством обмена металл/галоген с последующим электрофильным гашением диоксидом углерода. Взаимодействие может быть осуществлено в органическом растворителе, таком как тетрагидрофуран, диэтиловый эфир, диглим или гексан, с металлоорганическим реагентом, таким как бутиллитий, втор-бутиллитий, трет-бутиллитий или хлорид изопропилмагния, при температуре в диапазоне от -78°С до 25°С (например, при 25°С для обмена металл/галоген и 0°С для взаимодействия с диоксидом углерода).
Соединение (VII) может быть превращено в (IV) посредством взаимодействия в условиях карбонилирования в воде в качестве растворителя при температуре от 25°С до 120°С, в атмосфере монооксида углерода от 1 до 8 бар (100-800 кПа) в присутствии металлического катализатора (например, 1,1'-бис(дифенилфосфино)ферроцендихлорпалладия(II) или 1,1'-бис(ди-трет-бутилфосфино)ферроцендихлорпалладия(II) (Pd-118)), в присутствии аминного основания (например, триэтиламина или диизопропилэтиламина).
Соединения формулы (II), где X1 представляет собой подходящую уходящую группу (например, галоген, паратолуолсульфонат, метансульфонат или трифторметансульфонат), может быть получено из соединений формулы (VII), где X3 представляет собой уходящую группу (например, галоген, паратолуолсульфонат, метансульфонат или трифторметансульфонат) и X4 представляет собой радикал иода или брома.
При превращении (VII) в (II) взаимодействие может быть удобным образом осуществлено в органическом растворителе, таком как N-метилпирролидин, N,N-диметилформамид, N,N-диметилацетамид, тетрагидрофуран или ацетонитрил, в присутствии амина (V) при температуре от 25°С до 120°С, в атмосфере монооксида углерода от 1 до 8 бар (100-800 кПа), в присутствии металлического катализатора (например, 1,1'-бис(дифенилфосфино)-ферроцендихлорпалладия(II) или 1,1'-бис(ди-трет-бутилфосфино)-ферроцендихлор палладия(II) (Pd-118)) и в присутствии аминного основания (например, триэтиламина или диизопропилэтиламина).
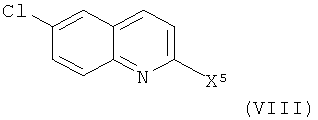
Соединения формулы (VII), где X3 представляет собой уходящую группу (например, галоген, паратолуолсульфонат, метансульфонат или трифторметансульфонат) и X4 представляет собой радикал иода или брома, могут быть получены из соединения формулы (VIII)
 ,
,
где X5 представляет собой уходящую группу (например, галоген, паратолуолсульфонат, метансульфонат или трифторметансульфонат).
В превращении (VIII) в (VII), где X3 представляет собой радикал иода, взаимодействие может быть удобным образом осуществлено в кислоте, такой как парящая серная кислота или трифликовая кислота, в присутствии источника иода, такого как иод (I2), N-иодсукцинимид (NIS) или монохлорид иода (ICI), в присутствии или в отсутствие соли металла (например, трифторметансульфоната серебра или сульфата серебра).
Соединения формулы (VIII) либо имеются в продаже, либо известны из литературы, либо могут быть получены с помощью известных методик специалистом в данной области. Например, соединение формулы (VIII), где X5 представляет собой радикал хлора, известно из литературы (Inglis S.R., et al., J. Med. Chem., 2004, 47, 5405).
Соединение формулы (VII), где X3 представляет собой радикал хлора и X4 представляет собой радикал иода, является новым соединением и образует еще один аспект настоящего изобретения. Соответственно в еще одном аспекте настоящего изобретения предложено соединение, которое представляет собой 2,6-дихлор-5-иодхинолин.
Соединения формулы (II) являются новыми соединениями и образуют еще один аспект настоящего изобретения. В одном воплощении изобретения предложены соединения формулы (II), где X1 выбран из галогена, паратолуолсульфоната, метансульфоната и трифторметансульфоната.
В другом воплощении изобретения предложено соединение формулы (II), где X1 представляет собой радикал хлора. Соответственно в еще одном аспекте настоящего изобретения предложено соединение, которое представляет собой 2,6-дихлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]-метил}хинолин-5-карбоксамид.
Специалисту будет очевидно, что в способах по настоящему изобретению некоторые функциональные группы, такие как гидрокси, карбоксильные или аминогруппы, в исходных реагентах или промежуточных соединениях могут нуждаться в защите посредством защитных групп. Так, получение соединения формулы (I) может включать на определенных стадиях введение защиты посредством одной или более защитных групп или удаление одной или более защитных групп. Введение и удаление защиты функциональных групп описаны в 'Protective Groups in Organic Synthesis', 2nd edition, T.W.Greene и P.G.M.Wuts, Wiley-lnterscience (1991) и 'Protecting Groups', P.J.Kocienski, Georg Thieme Verlag (1994). Соединения формулы (I) выше могут быть превращены в фармацевтически приемлемую соль с использованием традиционных методов.
Соединения по настоящему изобретению обладают благоприятными эффективностью, селективностью и/или фармакокинетическими свойствами. Например, соединения по настоящему изобретению обладают низкой аффинностью в отношении калиевого канала, кодируемого человеческим геном hERG (ether-a-go-go-related gene). В этом отношении взаимодействие лекарственного средства с hERG-кодируемым калиевым каналом и вследствие этого восстановление отрицательного клеточного потенциала истечением K+ могут вызывать удлинение QT-интервала, ведущее к приобретенному синдрому удлиненного QT-интервала (LQT) [М.С.Sanguinetti, С. Jiang, М.Е. Curran, M.Т.Keating, Cell 1995, 81, 299-307; и K.Finlayson et al., Eur. J. Pharm. 2004, 500, 129-142]. Это, в свою очередь, может индуцировать потенциально фатальную аритмию, известную как «torsade de points» (TdP) [W.Haferkamp et al., Eur. Heart J. 2000, 21, 1216-1331]. Новые химические соединения, если они не предназначены для сердечно-сосудистого использования, у которых отсутствуют эффекты в отношении сердечных каналов и hERG-каналов, в частности, будут поэтому демонстрировать улучшенный профиль безопасности и тем самым обеспечивать терапевтические и регуляторные преимущества по сравнению с лекарственными средствами, обладающими QT-удлиняющими эффектами. Kiss et al. (Assay Drug Dev. Technol. 2003, 1, 127-135) описывают способ оценки соединений на их способность ингибировать активность ионных каналов, таких как hERG. Springthorpe и Strandlund (WO 2005037052) описывают способ оценки соединений на их способность связываться с IKr калием (hERG).
Соединения согласно настоящему изобретению также обладают хорошей биодоступностью согласно определению посредством фармакокинетических параметров. Например, соединения согласно настоящему изобретению могут обладать низким связыванием с белками плазмы. Соединения согласно настоящему изобретению могут также обладать низкой активностью в скрининге in vitro фосфолипидоза.
Соединение по изобретению или его фармацевтически приемлемая соль могут обеспечивать преимущества в лечении:
1. заболеваний респираторного тракта: обструктивные заболевания дыхательных путей, включая астму, в том числе бронхиальную, аллергическую, наследственную, приобретенную, индуцированную физической нагрузкой, индуцированную приемом лекарственных средств (включая индуцированную приемом аспирина и NSAID (нестероидные противовоспалительные лекарственные средства)) и индуцированную пылью астму, как интермиттирующую, так и персистирующую, и всех степеней тяжести, и другие случаи гиперреактивности дыхательных путей; хроническая обструктивная болезнь легких (COPD); бронхит, в том числе инфекционный и эозинофильный бронхит; эмфизема; бронхоэктаз; цистический фиброз; саркоидоз; экзогенный аллергический альвеолит и родственные заболевания; гиперчувствительный пневмонит; пневмофиброз, в том числе криптогенный фиброзный альвеолит, идиопатическая интерстициальная пневмония, фиброз, являющийся осложнением противоопухолевой терапии и хронической инфекции, включая туберкулез и аспергиллез и другие грибковые инфекции; осложнения легочной трансплантации; васкулитоподобные и тромботические расстройства легочной сосудистой сети и легочная гипертензия; противокашлевая активность, в том числе лечение хронического кашля, ассоциированного с воспалительными и секреторными состояниями дыхательных путей, и ятрогенного кашля; острый и хронический ринит, в том числе медикаментозный ринит и вазомоторный ринит; круглогодичный и сезонный аллергический ринит, в том числе нервный ринит (сенная лихорадка); назальный полипоз; острая вирусная инфекция, в том числе простуда, и инфекция, вызванная респираторно-синцитиальным вирусом, вирусом гриппа, коронавирусом (включая атипичную пневмонию (SARS)) и аденовирусом;
2. кости и суставы: артритиды, ассоциированные с остеоартритом/остеоартрозом или включающие остеоартрит/остеоартроз, как первичный, так и вторичный по отношению, например, к врожденной дисплазии тазобедренного сустава; цервикальный и поясничный спондилит и боль в нижней области спины и шеи; ревматоидный артрит и болезнь Стилла; серонегативные спондилоартропатии, в том числе анкилозирующий спондилит, псориатический артрит, реактивный артрит и недифференцируемая спондилоартропатия; септический артрит и другие ассоциированные с инфекцией артропатии и заболевания кости, такие как туберкулез, в том числе болезнь Потта и полиартрит Понсе; острый и хронический индуцируемый отложением кристаллов синовит, в том числе уратная подагра, заболевание вследствие отложения пирофосфата кальция и воспаление сухожилий, синовиальной сумки и синовиальной жидкости, ассоциированное с апатитом кальция; болезнь Бехчета; первичный и вторичный синдром Шегрена; системный склероз и ограниченная склеродермия; системная красная волчанка, заболевание соединительной ткани смешанного типа и недифференцируемое заболевание соединительной ткани; воспалительные миопатии, в том числе дерматомиозит и полимиозит; ревматическая полимиалгия; юношеский артрит, в том числе идиопатические воспалительные артритиды суставов любой локализации и ассоциированные синдромы, ревматическая лихорадка и ее системные осложнения; васкулитиды, в том числе гигантоклеточный артериит, синдром Такаясу, синдром Чурга-Штраусса (Churg-Strauss), полиартериит нодозный, микроскопический полиартериит и васкулитиды, ассоциированные с вирусной инфекцией, реакциями гиперчувствительности, криоглобулинами и парапротеинами; боль в нижней части спины; семейная средиземноморская лихорадка, синдром Мукла-Уэльса (Muckle-Wells), семейная ирландская лихорадка, болезнь Кикучи (Kikuchi); индуцированные приемом лекарственных средств артралгии, поражения кожи при тендините и миопатии;
3. боль и перестройка соединительной ткани при скелетно-мышечных нарушениях из-за повреждения (например, спортивной травмы) или следующих заболеваний: артритиды (например, ревматоидный артрит, остеоартрит, подагра или вызванная кристаллами артропатия), другое заболевание суставов (такое как дегенерация межпозвоночного диска или дегенерация височно-челюстного сустава), заболевание с перестройкой кости (такое как остеопороз, болезнь Педжета или остеонекроз), полихондриты, склеродермия, смешанное нарушение соединительной ткани, спондилоартропатии или заболевание периодонта (такое как периодонтит);
4. кожа: псориаз, атопический дерматит, контактный дерматит или другие экзематозные поражения кожи и реакции гиперчувствительности замедленного типа; фито- и фотодерматит; себорейная экзема, герпетиформная экзема, красный плоский лишай, склеротический и атрофический лишай, гангренозная пиодермия, кожный саркоид, дискоидная красная волчанка, пузырчатка, пемфигоид, врожденный буллезный эпидермолиз, крапивница, ангионевротические отеки, васкулитиды, токсические эритемы, кожные эозинофилии, гнездная алопеция, облысение по мужскому типу, синдром Свита (Sweet), болезнь Вебера-Крисчена, множественная эритема; целлюлит, как инфекционный, так и неинфекционный; панникулит; кожные лимфомы, немеланомный рак кожи и другие диспластические поражения; индуцированные приемом лекарственных средств расстройства, в том числе стойкая лекарственная сыпь;
5. глаза: блефарит; конъюнктивит, в том числе круглогодичный и весенний аллергический конъюнктивит; иритит; передний и задний увеит; хореоидит; аутоиммунная реакция; дегенеративные или воспалительные расстройства, влияющие на сетчатку; офтальмия, в том числе симпатическая офтальмия; саркоидоз; инфекции, в том числе вирусная, грибковая и бактериальная;
6. желудочно-кишечный тракт: глоссит, гингивит, периодонтит; эзофагит, в том числе рефлюкс-эзофагит; эозинофильный гастроэнтерит, мастоцитоз, болезнь Крона, колит, в том числе неспецифический язвенный колит, проктит, зуд заднего прохода; глютеновая болезнь, синдром раздраженного кишечника, диарея невоспалительного характера и пищевые аллергии, которые проявляют действие, не связанное с кишечником (например, мигрень, ринит и экзема);
7. брюшная полость: гепатит, включая аутоиммунный, алкогольный и вирусный; фиброз и цирроз печени; холецистит; панкреатит, как острый, так и хронический;
8. мочеполовая сфера: нефрит, включая интерстициальный и гломерулонефрит; нефротический синдром; цистит, включая острый и хронический (интерстициальный) цистит и язву Хуннера (Hunner's); острый и хронический уретрит, простатит, эпидидимит, оофорит и сальпингит; вульво-вагинит; болезнь Пейрони (Peyronie's); эректильная дисфункция (мужская и женская);
9. отторжение аллотрансплантатов: острое и хроническое после, например, трансплантации почки, сердца, печени, легкого, костного мозга, кожи или роговицы, либо после переливания крови; либо хроническая болезнь «трансплантат против хозяина»;
10. ЦНС (центральная нервная система): болезнь Альцгеймера и другие деменции, включая CJD (болезнь Крейтцфельдта-Якоба) и nvCJD («новый вариант» болезни Крейтцфельдта-Якоба); амилоидоз; рассеянный склероз и другие демиелинизирующие синдромы; церебральный атеросклероз и васкулит; темпоральный артериит; тяжелая псевдопаралитическая миастения; острая и хроническая боль (острая, перемежающаяся или постоянная боль центрального или периферического происхождения), включая висцеральную боль, головную боль, мигрень, невралгию тройничного нерва, атипичную фасциальную боль, боль в суставах и костях, боль, являющуюся следствием рака и опухолевой инвазии, синдромы невропатической боли, включая диабетическую, послегерпетическую и ассоциированную с ВИЧ (вирус иммунодефицита человека) невропатии; нейросаркоидоз; осложнения центральной и периферической нервной системы вследствие злокачественных, инфекционных или аутоиммунных процессов;
11. другие аутоиммунные и аллергические расстройства, включая тиреоидит Хашимото (Hashimoto's), болезнь Гравса (Graves), болезнь Аддисона (Аddison), сахарный диабет, идиопатическую тромбоцитопеническую пурпуру, эозинофильный фасциит, синдром гипер-IgE, антифосфолипидный синдром;
12. другие нарушения с воспалительной или иммунологической компонентой; включая синдром приобретенного иммунодефицита (AIDS), проказу, синдром Сезари (Sezary) и паранеопластические синдромы;
13. сердечно-сосудистые поражения: атеросклероз, поражающий коронарную и периферическую сосудистую систему кровообращения; перикардит; миокардит, воспалительные и аутоиммунные кардиомиопатии, включая сердечный саркоид; ишемические реперфузионные повреждения; эндокардит, васкулит и аортит, включая инфекционный (например, сифилитический); васкулитиды; поражения проксимальных и периферических вен, включая флебит и тромбоз, в том числе тромбоз глубоких вен и варикозные поражения вен;
14. онкология: лечение общих злокачественных новообразований, включая рак простаты, молочной железы, легкого, яичника, поджелудочной железы, кишечника и толстой кишки, желудка, опухоли кожи и мозга, и злокачественные состояния, поражающие костный мозг (включая лейкемии) и лимфопролиферативную систему, такие как лимфома Ходжкина и не-Ходжкина; включая предупреждение и лечение метастазирования и рецидивирующих опухолей, и паранеопластические синдромы; и,
15. желудочно-кишечный тракт: желудочная болезнь (Coeliac disease), проктит, эозинофильный гастроэнтерит, мастоцитоз, болезнь Крона (Crohn), неспецифический язвенный колит, микроскопический колит, индетерминантный колит, раздражение кишечника, синдром раздраженного кишечника, невоспалительная диарея, пищевые аллергии с проявлениями, далекими ото рта, например мигрень, ринит и экзема.
Таким образом, в настоящем изобретении дополнительно предложено соединение формулы (I) или его фармацевтически приемлемая соль, как здесь определено ранее, для применения в терапии.
В еще одном аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее, в изготовлении лекарственного средства для использования в терапии.
В контексте настоящего описания термин "терапия" также включает "профилактику", если нет конкретного указания на обратное. Термины "терапевтический" и "терапевтически" следует понимать соответственно.
В еще одном аспекте изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено ранее, для лечения ревматоидного артрита.
В другом аспекте изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено ранее, для лечения воспалительного кишечного заболевания.
В другом аспекте изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено ранее, для лечения болезни Крона.
В еще одном аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее, в изготовлении лекарственного средства для использования в лечении ревматоидного артрита.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее, в изготовлении лекарственного средства для использования в лечении астмы или хронического обструктивного заболевания легких.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее, в изготовлении лекарственного средства для использования в лечении воспалительного кишечного заболевания.
В другом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее, в изготовлении лекарственного средства для использования в лечении болезни Крона.
В изобретении также предложен способ лечения ревматоидного артрита, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее, нуждающемуся в этом пациенту.
В изобретении также предложен способ лечения воспалительного кишечного заболевания, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее, нуждающемуся в этом пациенту.
В изобретении также предложен способ лечения болезни Крона, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее, нуждающемуся в этом пациенту.
В изобретении также предложен способ лечения обструктивного заболевания дыхательных путей (например, астмы или COPD), включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее, нуждающемуся в этом пациенту.
Для применения соединения по изобретению или его фармацевтически приемлемой соли для терапевтического лечения теплокровного животного, такого как человек, указанный ингредиент обычно готовят в соответствии со стандартной фармацевтической практикой в виде фармацевтической композиции.
Поэтому в другом аспекте настоящему изобретению также предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль (активный ингредиент), совместно с фармацевтически приемлемым адъювантом, разбавителем или носителем.
В еще одном аспекте настоящего изобретения также предложен способ изготовления указанной композиции, включающий смешивание активного ингредиента с фармацевтически приемлемым адъювантом, разбавителем или носителем. В зависимости от способа введения фармацевтическая композиция будет, например, содержать от 0,05 до 99 масс.% (массовых процента), в частности от 0,05 до 80 масс.%, например от 0,10 до 70 масс.%, в частности от 0,10 до 50 масс.%, активного ингредиента, причем все массовые проценты рассчитаны исходя из общей массы композиции.
Фармацевтические композиции по данному изобретению могут быть введены стандартным образом для болезненного состояния, которое желательно лечить, например посредством местного (например, в легкое и/или дыхательные пути или на кожу), перорального, ректального или парентерального введения. Для этих целей соединения по данному изобретению могут быть приготовлены в виде препарата средствами, известными в уровне техники, в форме, например, аэрозолей, сухих порошковых препаратов, таблеток, капсул, сиропов, порошков, гранул, водных или масляных растворов или суспензий, (липидных) эмульсий, диспергируемых порошков, суппозиториев, мазей, кремов, капель и стерильных водных или масляных растворов или суспензий.
Подходящей фармацевтической композицией по данному изобретению является композиция, пригодная для перорального введения в стандартной лекарственной форме, например в виде таблетки или капсулы, содержащей между 0,1 мг и 1 г активного ингредиента.
В другом аспекте фармацевтическая композиция по изобретению представляет собой композицию, пригодную для внутривенной, подкожной или внутримышечной инъекции. Каждый пациент может получать, например, внутривенную, подкожную или внутримышечную дозу от 0,01 мгкг-1 до 100 мгкг-1 соединения по данному изобретению, например в диапазоне от 0,1 мгкг-1 до 20 мгкг-1, при введении композиции от 1 до 4 раз в день. Внутривенную, подкожную или внутримышечную дозу можно вводить посредством болюсной инъекции. Альтернативно внутривенную дозу можно вводить посредством непрерывной инфузии в течение определенного периода времени. Альтернативно каждый пациент будет получать суточную пероральную дозу, которая приблизительно эквивалента суточной парентеральной дозе, при введении композиции от 1 до 4 раз в день.
Данное изобретение дополнительно относится к комбинированным терапиям, где соединение по изобретению или его фармацевтически приемлемую соль, или фармацевтическую композицию либо препарат, содержащие соединение по изобретению, вводят одновременно или последовательно либо в виде комбинированного препарата с другим терапевтическим агентом или агентами для лечения одного или более чем одного из перечисленных состояний.
В частности, для лечения воспалительных заболеваний, таких как (но без ограничения ими) ревматоидный артрит, остеоартрит, астма, аллергический ринит, хроническое обструктивное заболевание легких (COPD), псориаз и воспалительное заболевание кишечника, соединения по изобретению можно объединять с агентами, перечисленными ниже.
Нестероидные противовоспалительные агенты (далее NSAID), в том числе неселективные ингибиторы циклооксигеназ СОХ-1/СОХ-2, применяемые как местно, так и системно (такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флурбипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, азапропазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин); селективные ингибиторы СОХ-2 (такие как мелоксикам, целекоксиб, рофекоксиб, валдекоксиб, лумарококсиб, парекоксиб и эторикоксиб); ингибирующие циклооксигеназу доноры оксида азота (CINOD); глюкокортикостероиды (вводимые как местным, так и пероральным, внутримышечным, внутривенным или внутрисуставным путем); метотрексат; лефлуномид; гидроксихлороквин; d-пеницилламин; ауранофин или другие парентеральные или пероральные препараты золота; анальгетики; диацереин; внутрисуставные лекарственные средства, такие как производные гиалуроновой кислоты; и пищевые добавки, такие как глюкозамин.
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с цитокином или агонистом либо антагонистом функции цитокинов (включая агенты, которые действуют на цитокиновые сигнальные пути, такие как модуляторы системы супрессоров цитокиновых сигнальных путей, SOCS), включая альфа-, бета- и гамма-интерфероны; инсулиноподобный фактор роста типа I (IGF-1); интерлейкины (IL), в том числе IL1-17, и антагонисты или ингибиторы интерлейкинов, такие как анакинра; ингибиторы фактора некроза опухолей альфа (TNF-α), такие как моноклональные антитела против TNF (например, инфликсимаб, адалимумаб и CDP-870) и антагонисты рецепторов TNF, в том числе иммуноглобулиновые молекулы (такие как этанерцепт) и низкомолекулярные агенты, такие как пентоксифиллин. В дополнение, это изобретение относится к комбинации соединения по изобретению с моноклональным антителом, направленным на В-лимфоциты (таким как CD20 (ритуксимаб), MRA-aILl6R) и Т-лимфоциты (CTLA4-Ig, HuMax Il-15).
Настоящее изобретение также дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с модулятором функции рецепторов хемокинов, таким как антагонист CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для семейства С-С); CXCR1, CXCR2, CXCR3, CXCR4 и CXCR5 (для семейства С-Х-С) и CX3CR1 для семейства С-Х3-С.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором матриксных металлопротеаз (ММР), то есть стромелизинов, коллагеназ и желатиназ, а также аггреканазы; особенно коллагеназы-1 (ММР-1), коллагеназы-2 (ММР-8), коллагеназы-3 (ММР-13), стромелизина-1 (ММР-3), стромелизина-2 (ММР-10) и стромелизина-3 (ММР-11) и ММР-9 и ММР-12, в том числе такими агентами, как доксициклин.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором биосинтеза лейкотриенов, ингибитором 5-липоксигеназы (5-LO) или антагонистом белка, активирующего 5-липоксигеназу (FLAP), таким как: зилейтон; АВТ-761; фенлейтон, тепоксалин; Abbott-79175; Abbott-85761; N-(5-замещенный)-тиофен-2-алкилсульфонамид; 2,6-ди-трет-бутилфенол-гидразоны; метокситетрагидропираны, такие как Zeneca ZD-2138; соединение SB-210661; соединение, представляющее собой пиридинил-замещенный 2-цианонафталин, такое как L-739010; соединение, представляющее собой 2-цианохинолин, такое как L-746530; или индольное или хинолиновое соединение, такое как MK-591, MK-886 и BAY×1005.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антагонистом рецепторов лейкотриенов (LT) B4, LTC4, LTD4 и LTE4, выбранным из группы, состоящей из фенотиазинов-3-1, таких как L-651392; амидиновых соединений, таких как CGS-25019c; бензоксаламинов, таких как онтазоласт; бензокарбоксиимидаминов, таких как BIIL 284/260; и таких соединений, как зафирлукаст, аблукаст, монтелукаст, пранлукаст, верлукаст (MK-679), RG-12525, Ro-245913, иралукаст (CGP 45715А) и BAY×7195.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором фосфодиэстеразы (PDE), таким как метилксантанин, включая теофиллин и аминофиллин; селективным ингибитором изофермента PDE, включая ингибитор PDE4, ингибитор изоформы PDE4D или ингибитор PDE5.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антагонистом гистаминовых рецепторов типа 1, таким как цетиризин, лоратадин, дезлоратадин, фексофенадин, акривастин, терфенадин, астемизол, азеластин, левокабастин, хлорфенирамин, прометазин, циклизин или мизоластин, применяемым перорально, местно или парентерально.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с ингибитором протонного насоса (таким как омепразол) или гастропротективным антагонистом гистаминовых рецепторов типа 2.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антагонистом гистаминовых рецепторов типа 4.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с сосудосуживающим симпатомиметиком, представляющим собой агонист адренорецепторов альфа-1/альфа-2, таким как пропилгекседрин, фенилэфрин, фенилпропаноламин, эфедрин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид, трамазолина гидрохлорид или этилнорэпинефрина гидрохлорид.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с антихолинергическими агентами, в том числе антагонистом мускаринового рецептора (М1, М2 и М3), таким как атропин, хиосцин, гликопирролат, ипратропия бромид, тиотропия бромид, окситропия бромид, пирензепин или телензепин.
Настоящее изобретение также относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли и агониста бета-адренорецептора (включая бета-рецепторы подтипов 1-4), такого как изопреналин, сальбутамол, формотерол, сальметерол, тербуталин, орципреналин, битолтерола мезилат или пирбутерол либо их хиральный энантиомер.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с хромоном, таким как хромогликат натрия или недохромил натрия.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с глюкокортикоидом, таким как флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат, циклезонид или мометазона фуроат.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с агентом, который модулирует ядерные рецепторы гормона, такие как рецепторы, активируемые пролифератором пероксисом (PPAR).
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с иммуноглобулином (Ig) или препаратом Ig либо антагонистом или антителом, модулирующим функционирование Ig, таким как анти-IgE (например, омализумаб).
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с другим системным или местно наносимым противовоспалительным агентом, таким как талидомид или его производное, ретиноид, дитранол или кальципотриол.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с комбинациями аминосалицилатов и сульфапиридина, такого как сульфасалазин, месалазин, бальсалазид и олсалазин; и иммуномодуляторными агентами, такими как тиопурины, и кортикостероидами, такими как будесонид.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с антибактериальным агентом, таким как производное пенициллина, тетрациклин, макролид, бета-лактам, фторхинолон, метронидазол, ингалируемый аминогликозид; противовирусным агентом, включая ацикловир, фамцикловир, валацикловир, ганцикловир, цидофовир, амантадин, римантадин, рибавирин, занамавир и оселтамавир; ингибитором протеазы, таким как индинавир, нелфинавир, ритонавир и саквинавир; нуклеозидным ингибитором обратной транскриптазы, таким как диданозин, ламивудин, ставудин, залцитабин или зидовудин; или ненуклеозидным ингибитором обратной транскриптазы, таким как невирапин или эфавиренц.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с сердечно-сосудистым агентом, таким как блокатор кальциевых каналов, блокатор бета-адренорецепторов, ингибитор ангиотензин-конвертирующего фермента (АСЕ), антагонист рецепторов ангиотензина-2; агентом снижения липидов, таким как статин или фибрат; модулятором морфологии клеток крови, таким как пентоксифиллин; тромболитиком или антикоагулянтом, таким как ингибитор агрегации тромбоцитов.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с агентом, действующим на ЦНС, таким как антидепрессант (такой как сертралин), лекарственное средство против болезни Паркинсона (такое как депренил, L-ДОФА, ропинирол, прамипексол, ингибитор моноаминооксидазы В (МАОВ), такой как селегин и расагилин, ингибитор соединения Р (comP), такой как тасмар, ингибитор А-2, ингибитор обратного захвата дофамина, антагонист N-метил-D-аспартата (NMDA), никотиновый агонист, дофаминовый агонист или ингибитор нейрональной синтазы оксида азота) или лекарственное средство против болезни Альцгеймера, такое как донепезил, ривастигмин, такрин, ингибитор СОХ-2, пропентофиллин или метрифонат.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли с агентом для лечения острой или хронической боли, таким как действующий центрально или периферически анальгетик (например, опиоид или его производное), карбамазепин, фенитоин, вальпроат натрия, амитриптилин или другие антидепрессанты, парацетамол или нестероидный противовоспалительный агент.
Настоящее изобретение дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с парентерально или местно применяемым (в том числе ингалируемым) локальным обезболивающим средством, таким как лигнокаин или его производное. Соединение по настоящему изобретению или его фармацевтически приемлемую соль можно также использовать в комбинации с агентом против остеопороза, включая гормональный агент, такой как ралоксифен или бифосфонат, такой как алендронат.
Настоящее изобретение еще дополнительно относится к комбинации соединения по изобретению или его фармацевтически приемлемой соли вместе с: (1) ингибитором триптазы; (2) антагонистом фактора активации тромбоцитов (PAF); (3) ингибитором интерлейкин-превращающего фермента (ICE); (4) ингибитором инозинмонофосфатдегидрогеназы IMPDH; (5) ингибитором молекул адгезии, в том числе антагонистами VLA-4 (очень поздний антиген); (6) катепсином; (7) ингибитором киназы, таким как ингибитор тирозинкиназы (такой как Btk, Itk, Jak3 или MAP, например гефитиниб или мезилат иматиниба), серин/треонинкиназы (таким как ингибитор митоген-активируемой протеинкиназы (МАР-киназы), такой как р38, JNK, протеинкиназа А, В или С, или киназа ингибиторной киназы (IKK)), или киназы, участвующей в регуляции клеточного цикла (такой как циклин-зависимая киназа); (8) ингибитором глюкозо-6-фосфат-дегидрогеназы; (9) антагонистом рецепторов кинин-B1 либо -В2; (10) средством против подагры, например колхицином; (11) ингибитором ксантин-оксидазы, например аллопуринолом; (12) средством, способствующим выведению мочевой кислоты, например пробенецидом, сульфинпиразоном или бензбромароном; (13) стимулятором секреции гормона роста; (14) трансформирующим фактором роста (TGFβ); (15) тромбоцитарным фактором роста (PDGF); (16) фактором роста фибробластов, например основным фактором роста фибробластов (bFGF); (17) капсаициновым кремом; (18) антагонистом рецепторов тахикинина NK1 или NK3, выбранным из группы, состоящей из NKP-608C, SB-233412 (талнетант) или D-4418; (19) ингибитором эластазы, выбранным из группы, состоящей из UT-77 или ZD-0892; (20) ингибитором индуцибельной синтазы оксида азота (iNOS); (21) рецептор-гомологичной молекулой для хемоаттрактанта, экспрессированной на ТН2-клетках (такой как антагонист CRTH2); (22) ингибитором Р38; (23) агентом модулирования функционирования Toll-подобных рецепторов (TLR); (24) агентом модулирования активности другого пуринергического рецептора или (25) ингибитором фактора активации транскрипции, такого как NF-κВ, API или STATS.
Соединение по изобретению или его фармацевтически приемлемая соль могут также быть использованы в комбинации с существующим терапевтическим агентом для лечения рака, причем подходящие агенты включают, например:
(1) антипролиферативное/антинеопластическое лекарственное средство или их комбинацию, используемые в медицинской онкологии, такое как алкилирующий агент (например, цисплатин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан или нитрозомочевина); антиметаболит (например, антифолат, такой как фторпиримидин, например 5-фторурацил или тегафур, ралтитрексед, метотрексат, цитозина арабинозид, гидроксимочевина, гемцитабин или паклитаксел); противоопухолевый антибиотик (например, антрациклин, такой как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин или митрамицин); антимитотический агент (например, алкалоид барвинка, такой как винкристин, винбластин, виндезин или винорелбин, или таксоид, такой как таксол или таксотере); или ингибитор топоизомеразы (например, эпиподофиллотоксин, такой как этопозид, тенипозид, амсакрин, топотекан или камптотецин);
(2) цитостатический агент, такой как антиэстроген (например, тамоксифен, торемифен, ралоксифен, дролоксифен или иодоксифен), обратный регулятор эстрогенового рецептора (например, фулвестрант), антиандроген (например, бикалутамид, флутамид, нилутамид или ципротерона ацетат), антагонист LHRH (лютеинизирующий гормон роста) или агонист LHRH (например, гозерелин, леупрорелин или бузерелин), прогестоген (например, мерестрола ацетат), ингибитор ароматазы (например, анастрозол, летрозол, воразол или экземестан) или ингибитор 5α-редуктазы, такой как финастерид;
(3) агент, который ингибирует инвазию раковых клеток (например, ингибитор металлпротеиназ, такой как маримастат, или ингибитор рецепторного функционирования урокиназного активатора плазминогена);
(4) ингибитор функционирования ростовых факторов, например: антитело к ростовому фактору (например, анти-erbb2 антитело трастузумаб или анти-erbb2 антитело цетуксимаб [С225]), ингибитор фарнезилтрансферазы, ингибитор тирозинкиназ или ингибитор серин/треонинкиназ, ингибитор семейства эпидермальных факторов роста (например, ингибитор EGFR семейства тирозинкиназ, такой как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, AZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) или 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)-хиназолин-4-амин (CI 1033)), ингибитор семейства факторов роста тромбоцитарного происхождения или ингибитор семейства факторов роста гепатоцитарного происхождения;
(5) антиангиогенный агент, такой как агент, который ингибирует эффекты васкулярно-эндотелиального фактора роста (например, антитело против васкулярно-эндотелиального клеточного фактора роста бевацизумаб, соединение, описанное в WO 97/22596, WO 97/30035, WO 97/32856 или WO 98/13354), или соединение, которое действует по другому механизму (например, линомид, ингибитор функционирования интегрина αvβ3 или ангиостатин);
(6) агент, поражающий сосуды, такой как комбретастатин А4, или соединение, описанное в WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 или WO 02/08213;
(7) агент, используемый в антисмысловой терапии, например агент, нацеленный на одну из мишеней, указанных выше, такой как ISIS 2503, анти-rаs антисмысловой агент;
(8) агент, используемый в методологии генной терапии, например в подходах, заключающихся в замене аберрантных генов, таких как подходы в отношении аберрантного р53 или аберрантного BRCA1 или BRCA2, GDEPT (нацеленная на гены ферментная пролекарственная терапия), такие как подходы с использованием цитозиндезаминазы, тимидинкиназы или бактериальной нитроредуктазы, и подходы, состоящие в увеличении устойчивости пациента к химиотерапии или радиотерапии, такие как генная терапия множественной лекарственной устойчивости; или
(9) агент, используемый в иммунотерапевтическом подходе, например ex-vivo и in-vivo методы для увеличения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или гранулоцитарно-макрофагальный колониестимулирующий фактор, методы снижения вялости Т-клеток, методы с использованием трансфицированных иммунных клеток, таких как цитокин-трансфицированные дендритные клетки, методы с использованием цитокин-трансфицированных опухолевых клеточных линий и методы с использованием антиидиотипических антител.
Настоящее изобретение будет дополнительно проиллюстрировано путем приведения следующих иллюстративных примеров. В этих примерах ЯМР спектры снимали на спектрометре Varian Unity с частотой протонов 300 или 400 МГц. MS (масс-спектрометрия) спектры снимали на спектрометре Agilent 1100 MSD G1946D или на спектрометре Hewlett Packard HP1100 MSD G1946A. Разделения посредством препаративной HPLC (высокоэффективная жидкостная хроматография) осуществляли с помощью колонки Waters Symmetry® или Xterra®, используя смесь 0,1% водная трифторуксусная кислота:ацетонитрил, 0,1% водный аммиак:ацетонитрил или 0,1% ацетат аммония:ацетонитрил в качестве элюента. Взаимодействия в условиях микроволнового излучения осуществляли в СЕМ Discover одномодовом микроволновом реакторе. Соединения и промежуточные соединения называли с помощью пакета для составления названий IUPAC от ACD Labs, Toronto, Canada.
Пример 1
6-Хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-[(3S)-3-гидроксипирролидин-1-ил]хинолин-5-карбоксамид
a) (3S,5S)-5-Метил-1-оксаспиро[2.5]октан
Указанное в подзаголовке соединение получали в соответствии с описанной в литературе процедурой (Weijers, C.A.G.M. et al., JOC. 2005, 70, 6639-6646) посредством взаимодействия раствора трет-бутоксида калия (7,84 г) в диметилсульфоксиде (200 мл) со смесью (3S)-3-метилциклогексанона (4,0 г, более 98% е.е.) (Alexakis A. et al., Synlett 2001, No.9, 1375 и Hiemstra H. and Wynberg H., Tetrahedron Lett., 1977, 2183) и иодида триметилсульфоксония (15,4 г) в диметилсульфоксиде (100 мл), что давало указанное в подзаголовке соединение (3,5 г).
1H ЯМР δ(CDCl3) 2.62 (2Н, m), 1.86-1.56 (5H, m), 1.26 (2H, m), 0.99 (1H, m), 0.92 (3H, d), 0.86 (1H, m).
b) (1S,3S)-1-[(Бензиламино)метил]-3-метилциклогексанол
Метанольный (1 мл) раствор бензиламина (5,9 г) и (3S,5S)-5-метил-1-оксаспиро[2.5]октана (3,5 г) нагревали в микроволновом реакторе (100 Вт) в течение 30 минут при 100°С, затем концентрировали в вакууме и очищали посредством колоночной флеш-хроматографии (SiO2, 20% этилацетат/изогексан в качестве элюента) с получением указанного в подзаголовке соединения в виде бесцветного масла (4,5 г).
m/z 234 (М+Н, 100%).
c) (1S,3S)-1-(Аминометил)-3-метилциклогексанол. HCl
Смесь (1S,3S)-1-[(бензиламино)метил]-3-метилциклогексанола (4,5 г) и 5% палладия на углероде (500 мг) в метаноле (40 мл) перемешивали в атмосфере водорода при 4 бар (400 кПа) в течение 72 часов. Реакционную смесь фильтровали через целит, промывали метанолом (2х) и концентрировали в вакууме с получением (1S,3S)-1-(аминометил)-3-метилциклогексанола в виде бесцветного масла (2,5 г).
1H ЯМР δ(CDCl3) 2.53 (2H, d), 1.50-1.90 (6H, cm), 1.06 (2H, m), 0.87 (3H, d) и 0.81 (2H, m).
(1S,3S)-1-(Аминометил)-3-метилциклогексанол легко превращается в указанное в подзаголовке соединение посредством обработки его в виде раствора в диэтиловом эфире 1-молярным эквивалентом 4 M HCl в 1,4-диоксане.
d) 2,6-Дихлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-хинолин-5-карбоксамид
К перемешиваемой суспензии 2,6-дихлор-хинолин-5-карбоновой кислоты (2,40 г) (WO 2004/106305, Пример 76, стадия (b)) в дихлорметане (100 мл) добавляли оксалилхлорид (3,15 г, 2,16 мл) и каплю N,N-диметилформамида. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего летучие вещества удаляли в вакууме и остаток разводили в дихлорметане (100 мл). К этому раствору добавляли (1S,3S)-1-(аминометил)-3-метилциклогексанол. HCl (1,78 г) и диизопропилэтиламин (6,70 мл) и реакционную смесь перемешивали в течение 20 часов, после чего промывали водой. Органический слой сушили (MgSO4), фильтровали и летучие вещества выпаривали с получением сырого твердого вещества. Это вещество перекристаллизовывали из толуола с получением указанного в подзаголовке соединения в виде бежевого твердого вещества (2,5 г).
1H ЯМР δ(CDCl3) 8.24 (1Н, d), 7.99 (1Н, d), 7.71 (1Н, t), 7.46 (1Н, d), 6.34 (1H, s), 3.56 (2H, d), 1.82-1.52 (7H, m), 1.35 (1Н, td), 1.07 (1Н, t), 0.93 (3H, d), 0.88 (1Н, dd).
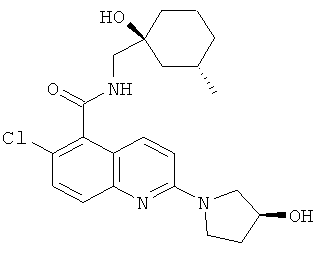
e) 6-Хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-[(3S)-3-гидрокси-пирролидин-1-ил]хинолин-5-карбоксамид
(S)-3-Гидроксипирролидин (80 мг) добавляли к суспензии продукта со стадии (d) (0,2 г) и диизопропилэтиламина (300 мкл) в ацетонитриле (3 мл). Реакционную смесь нагревали в микроволновом реакторе (100 Вт) в течение 30 минут при 120°С, после чего концентрировали в вакууме. Добавляли воду (15 мл) и суспензию обрабатывали ультразвуком в течение 10 минут. Твердое вещество отфильтровывали и сушили в вакууме в течение ночи с получением указанного в заголовке соединения в виде бежевого твердого вещества (180 мг). Т. пл. 222°С (ацетонитрил).
m/z 418 (M+H, 100%), 416 (М-Н, 100%).
1H ЯМР δ(DMSO) 8.49 (1H, t), 7.81 (1H, d), 7.54 (1H, d), 7.49 (1H, d), 6.95 (1H, d), 4.99 (1H, d), 4.42 (1H, s), 4.15 (1H, s), 3.59 (3H, m), 3.47 (1H, s), 3.28 (2H, d), 2.04 (1H, m), 1.92 (1H, m), 1.73 (1H, m), 1.64-1.41 (5H, m), 1.29 (1H, m), 1.04 (1H, t), 0.84 (3H,d), 0.75 (1H, m).
Пример 2
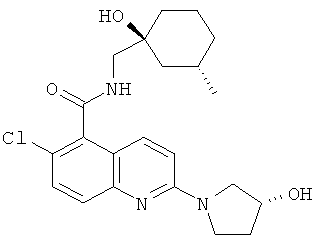
6-Хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-[(3R)-3-гидроксипирролидин-1-ил]хинолин-5-карбоксамид
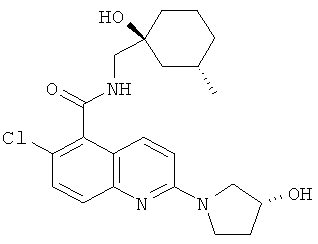
Указанное в заголовке соединение получали способом из Примера 1, стадия (е), посредством взаимодействия (R)-3-гидроксипирролидина (80 мг) (вместо (S)-3-гидроксипирролидина) с продуктом Примера 1, стадия (d) (0,2 г), и диизопропилэтиламином (300 мкл) в ацетонитриле (3 мл) с получением указанного в заголовке соединения в виде кремового твердого вещества (190 мг). Т. пл. 222-223°С (ацетонитрил).
m/z 418 (M+H, 100%).
1H ЯМР δ(CD3OD) 7.92 (1H, d), 7.65 (1H, d), 7.49 (1H, d), 6.94 (1H, d), 4.54 (1H, d), 3.69 (3H, m), 3.62 (1H, m), 3.41 (2H, s), 2.15 (1H, m), 2.10 (1H, m), 1.86-1.53 (6H, br. m), 1.40 (1H, m), 1.12 (1H, t), 0.89 (3H, d), 0.85 (1H, q).
Пример 3
6-Хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-(4-гидроксипиперидин-1-ил)хинолин-5-карбоксамид
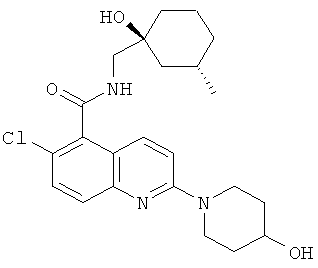
Указанное в заголовке соединение получали способом из Примера 1, стадия (е), посредством взаимодействия пиперидин-4-ола (28 мг) (вместо (S)-3-гидроксипирролидина) с продуктом Примера 1, стадия (d) (0,10 г), и диизопропилэтиламином (0,11 г) в ацетонитриле (2 мл) с получением указанного в заголовке соединения в виде белого твердого вещества (99 мг). Т. пл. 120°С с разл.
m/z 432 (М+Н, 100%), 430 (М-Н, 100%).
1H ЯМР δ(DMSO) 8.46 (1H, t), 7.79 (1H, d), 7.52 (1H, d), 7.49 (1H, d), 7.32 (1H, d), 4.71 (1H, d), 4.18 (2H, m), 4.13 (1H, s), 3.73 (1H, m), 3.37-3.20 (4H, m), 1.84-1.21 (11H, m), 1.02 (1H, t), 0.82 (3H, d), 0.73 (1H, m).
Пример 4
6-Хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-(3-гидрокси-3-метилпирролидин-1-ил)хинолин-5-карбоксамид
a) 1-Бензил-3-метилпирролидин-3-ол
Бромид метилмагния (4 мл 3 М раствора в диэтиловом эфире) по каплям добавляли к раствору 1-бензилпирролидин-3-она (1,75 г) в тетрагидрофуране (50 мл) при 0°С. Смесь перемешивали при 0°С в течение 1 часа, после чего добавляли воду и диэтиловый эфир и слои разделяли. Органический слой промывали рассолом, сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырое вещество очищали посредством колоночной флеш-хроматографии (SiO2, смесь 5% метанол/дихлорметан в качестве элюента) с получением указанного в подзаголовке соединения в виде бледно-коричневого масла (0,8 г).
1H ЯМР δ(CDCl3) 7.34-7.20 (5Н, m), 3.63 (2H, s), 3.00-2.92 (1H, m), 2.71 (1H, d), 2.37-2.28 (1H, m), 2.22 (1H, d), 1.92-1.84 (2H, m), 1.33 (3H, s).
b) 3-Метилпирролидин-3-ол
Суспензию 5% палладия на углероде в этаноле (1 мл) добавляли к раствору 1-бензил-3-метилпирролидин-3-ола (0,8 г) в метаноле (10 мл) и реакционную смесь перемешивали в атмосфере водорода при давлении 5 бар (500 кПа) в течение 4 дней, после чего фильтровали через целит и промывали этанолом (100 мл). Летучие примеси удаляли в вакууме с получением указанного в подзаголовке продукта в виде желтого масла (0,42 г).
1H ЯМР δ(CDCl3) 3.18 (1Н, m), 2.96 (1Н, m), 2.90 (1H, d), 2.68 (1H, d), 1.82 (2H, m), 1,41 (3H, s).
с) 6-Хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-(3-гидрокси-3-метилпирролидин-1-ил)хинолин-5-карбоксамид
Указанное в заголовке соединение получали способом из Примера 1, стадия (е), посредством взаимодействия 3-метилпирролидин-3-ола (0,42 г) (вместо (S)-3-гидроксипирролидина) с (1-гидрокси-3-метил-циклогексилметил)-амидом 2,6-дихлор-хинолин-5-карбоновой кислоты (0,10 г) и триэтиламином (0,38 мл) (вместо диизопропилэтиламина) в ацетонитриле (1 мл) с получением указанного в заголовке сырого соединения и в виде смеси диастереомеров 1:1. Реакционную смесь концентрировали в вакууме и продукты выделяли посредством колоночной флеш-хроматографии (SiO2, смесь 4% метанол/дихлорметан в качестве элюента) в виде бесцветного твердого вещества (70 мг). Диастереомеры разделяли посредством сверхкритичной жидкостной хроматографии (SFC) на колонке OJ Daicel, используя смесь 25% этанол/диоксид углерода в качестве элюента, с получением изомера 1 в виде бесцветного твердого вещества (18 мг), т. пл. 195-200°С с разл.,
m/z 432 (М+Н, 100%),
1H ЯМР δ(DMSO) 8.48 (1H, t), 7.80 (1H; d), 7.55-7.46 (2H, m), 6.92 (1H, d), 4.82 (1H, s), 4.14 (1H, s), 3.70-3.49 (2H, m), 3.38 (1H, d), 3.27 (1H, d), 2.54-2.46 (2H, m), 1.99-1.42 (8H, m), 1.37 (3H, s), 1.34-1.22 (1H, m), 1.04 (1H, t), 0.84 (3H, d), 0.81-0.68 (1H, m),
и изомера 2 в виде бесцветного твердого вещества (17 мг), т. пл. 200-202°С,
m/z 432 (М+Н, 100%),
1H ЯМР δ(DMSO) 8.48 (1H, t), 7.80 (1H, d), 7.54-7.48 (2H, m), 6.83 (1H, d), 4.82 (1H, s), 4.14 (1H, s), 3.70-3.48 (2H, m), 3.38 (1H, d), 3.28 (1H, d), 2.53-2.48 (2H, m), 2.02-1.42 (8H, m), 1.37 (3H, s), 1.33-1.21 (1H, m), 1.04 (1H, t), 0.84 (3H, d), 0.80-0.68 (1H, m).
Пример 5
6-Хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-[(3R)-3-гидроксипирролидин-1-ил]хинолин-5-карбоксамид (получение, альтернативное Примеру 2)
Общие условия Примера 5: ЯМР спектр снимали на спектрометре Bruker Avance 360 МГц, Bruker Avance 400 МГц или Bruker DPX250 250 МГц. Аналитические стереохимические HPLC определения выполняли применяя колонки АСЕ 3 Phenyl, 150×3 мм, и Chirapak AD-H 150×4,6 мм, используя в качестве градиента элюции 0,1% водный ацетат аммония:ацетонитрил и изократическую элюцию смесью 19,9:80:0,1 изопропанол:изогексан:триэтиламин соответственно.
а) 2,6-Дихлорхинолин
Оксихлорид фосфора (16,72 кг) загружали в сосуд, содержащий 6-хлорхинолин-2(1Н)-он (12,50 кг) (получен согласно способу Johnston K.M. et at., J. Chem. Soc. Perkin Trans. 1, 1972, 1648, и ссылки), хлорид бензилтриметиламмония (1,575 кг) и 1,2-диметоксиэтан (87,8 кг), при 70°С. В виде промывки линии загружали 1,2-диметоксиэтан (22,5 кг). Реакционную смесь перемешивали при температуре от 70°С до 75°С в течение приблизит. 6 часов, после чего партию концентрировали до приблизит. 44 л посредством вакуумной перегонки (ниже 40°С). Концентрат разбавляли дихлорметаном (253,1 кг), доводили до температуры от 38°С до 45°С и гасили посредством добавления воды (37,5 кг), поддерживая температуру в диапазоне от 38°С до 45°С. Через 70 минут партию охлаждали до температуры в диапазоне от 25°С до 30°С и обрабатывали целитом (1,30 кг) в течение 40 минут. Суспензию фильтровали под давлением через фильтровальную мембрану 1 мкм и фильтраты разбавляли дихлорметаном (87,5 кг). Фазы разделяли и водную фазу дважды экстрагировали дихлорметаном (82 кг). Объединенные органические экстракты промывали последовательно 5%-ным (масс./масс.) раствором гидрокарбоната натрия (37 л), водой (37 кг) и затем концентрировали до приблизит. 75 л при температуре в диапазоне от 25°С до 40°С. Загружали изопропанол (96,5 кг) и затем партию концентрировали до приблизит. 75 л при температуре в диапазоне от 25°С до 40°С. Загружали изопропанол (95,4 кг) и затем партию концентрировали до приблизит. 75 л при температуре в диапазоне от 25°С до 40°С. Полученную суспензию перемешивали при температуре в диапазоне от 16°С до 18°С в течение 2 часов и затем фильтровали. Осадок на фильтре промывали изопропанолом (19,7 кг) при приблизит. 20°С и затем сушили при 50°С в вакууме с получением указанного в подзаголовке соединения в виде грязно-белого твердого вещества (12,04 кг).
1H ЯМР δ(DMSO) 8.45 (1Н, d), 8.22 (1Н, d), 7.99 (1H, d), 7.86 (1H, dd), 7.68 (1H, d).
b) 2,6-Дихлор-5-иодхинолин
2,6-Дихлорхинолин (12,04 кг) загружали в трифторметансульфоновую кислоту (80,6 кг) десятью приблизительно равными порциями так, чтобы температура поддерживалась в диапазоне от 15°С до 25°С. Затем загружали N-иодсукцинимид (13,74 кг) пятью приблизительно равными порциями так, чтобы температура поддерживалась в диапазоне от 15°С до 25°С. Реакционную смесь перемешивали при температуре от 20°С до 25°С в течение приблизит. 36 часов. Температуру доводили до 15°С-20°С, смесь разбавляли дихлорметаном (159,4 кг), доводили температуру до 5°С-10°С и гасили посредством добавления воды (96,5 кг), поддерживая температуру в диапазоне от 5°С до 23°С. Суспензию осветляли с помощью фильтровальной мембраны 1 мкм и осуществляли промывку линии дихлорметаном (16,1 кг). Фазы разделяли и водную фазу экстрагировали дихлорметаном (48,2 кг). Объединенные органические экстракты промывали 5%-ным (масс./масс.) раствором гидрокарбоната натрия (48 л). Фазу гидрокарбоната натрия снова экстрагировали дихлорметаном (15,4 кг). Объединенные органические экстракты промывали 20%-ным (масс./масс.) раствором тиосульфата натрия (48 л). Фазу тиосульфата натрия снова экстрагировали дихлорметаном (16,3 кг). Объединенные органические экстракты промывали водой (47 л). Водную фазу снова экстрагировали дихлорметаном (16,4 кг). Объединенные органические экстракты снова загружали в сосуд, осуществляли промывку линии дихлорметаном (31,3 кг) и концентрировали до приблизит. 48 л при атмосферном давлении. Загружали дихлорметан (63 кг) и партию концентрировали до приблизит. 48 л при атмосферном давлении. Загружали дихлорметан (66 кг) и партию концентрировали до приблизит. 48 л при атмосферном давлении. Загружали дихлорметан (63,6 кг) и партию концентрировали до приблизит. 48 л при атмосферном давлении. Загружали дихлорметан (63,8 кг) и партию концентрировали до приблизит. 48 л при атмосферном давлении. Загружали дихлорметан (77,8 кг) и партию концентрировали до приблизит. 48 л при атмосферном давлении. Загружали ацетонитрил (47,7 кг) и партию концентрировали до приблизит. 96 л при атмосферном давлении. Загружали ацетонитрил (46,4 кг) и партию концентрировали до приблизит. 96 л при атмосферном давлении. Партию охлаждали до температуры в диапазоне от 18°С до 23°С, перемешивали в течение 2,5 часов и затем фильтровали. Осадок на фильтре дважды промывали ацетонитрилом (19,6 кг) при приблизит. 20°С и затем сушили при температуре вплоть до 55°С в вакууме с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (16,74 кг).
1H ЯМР δ(DMSO) 8.51 (1Н, d), 8.01-7.94 (2Н, m), 7.72 (1H, d).
с) 2,6-Дихлор-хинолин-5-карбоновая кислота
2,09 M хлорид изопропилмагния (27,0 л) загружали в сосуд, содержащий 2,6-дихлор-5-иодхинолин (15,0 кг) и дегазированный тетрагидрофуран (103,4 кг) при температуре от 18°С до 25°С. Загружали тетрагидрофуран (13,9 кг) в виде промывки линии. Реакционную смесь перемешивали при температуре от 18°С до 25°С в течение 15 минут, охлаждали до температуры от 10°С до 15°С и барботировали газообразным диоксидом углерода в течение приблизит. 6 часов при поддержании температуры в диапазоне от 5°С до 25°С. В сосуд при температуре от 15°С до 20°С загружали метанол (12,3 кг), перемешивали в течение приблизит. 30 минут и затем дополнительно разбавляли водой (134,0 кг). Партию концентрировали до приблизит. 135 л посредством вакуумной перегонки (ниже 40°С). Концентрат разбавляли водой (121 кг), этилацетатом (40,7 кг) и температуру доводили до 20°С-25°С. Фазы разделяли и водную фазу промывали этилацетатом (3×40,8 кг). Величину рН водной фазы доводили до рН 5,09 с помощью 1 М соляной кислоты (3,97 л) и дважды промывали трет-бутилметиловым эфиром (23,4 кг). Величину рН водной фазы доводили до рН 1,35 с помощью 2 М соляной кислоты (25,5 л), перемешивали в течение приблизит. 1 часа при 22°С и фильтровали. Осадок на фильтре промывали 4 раза водой (приблизит. 60 л) при приблизит. 20°С и затем сушили при температуре вплоть до 55°С в вакууме с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (8,74 кг).
1H ЯМР δ(DMSO) 14.41 (1Н, br s), 8.32 (1H, d), 8.10 (1H, dd), 7.97 (1H, d), 7.77 (1H, d).
d) O,O'-(S)-(1,1'-Динафтил-2,2'-диил)-N,N'-ди-(R,R)-1-фенилэтил-фосфорамидит
(R(R,R))-(+)-Бис(альфа-метилбензил)амин (0,35 кг)загружали в сосуд, содержащий трихлорид фосфора (0,1365 кг), толуол (3,15 л) и триэтиламин (0,714 л), поддерживая температуру в диапазоне от 18°С до 25°С. В виде промывки линии загружали толуол (0,35 л). Через 3,5 часа загружали раствор (S)-(-)-1,1'-би(2-нафтола) (0,445 кг) в виде раствора в тетрагидрофуране (0,70 л), поддерживая температуру в диапазоне от 20°С до 26°С. В виде промывки линии загружали тетрагидрофуран (0,35 л). Реакционную смесь перемешивали в течение18 часов при температуре окружающей среды и затем фильтровали через диоксид кремния [9 см (h) × 19 см (w)]. Продукт элюировали толуолом (10×1,75 л). Объединенные фильтраты концентрировали в вакууме до приблизит. 1 л при температуре ниже 35°С и затем разбавляли ацетонитрилом (5,6 л). Полученную суспензию охлаждали до температуры от 0°С до 5°С, перемешивали в течение приблизит. 1 часа и затем фильтровали. Осадок на фильтре промывали ацетонитрилом (2×0,70 л) и сушили под азотом на фильтре с получением указанного в подзаголовке соединения в виде бесцветного твердого вещества (0,65 кг).
1H ЯМР δ(CDCl3) 7.95-7.87 (4Н, m), 7.59 (1H, s), 7.47-7.36 (4H, m), 7.29-7.20 (3H, m), 7.18-7.03 (10Н, m), 4.58-4.44 (2H, m), 1.72 (6H, d).
e) (S)-3-Метилциклогексанон
Толуол (38,1 кг) загружали в сосуд, содержащий O,O'-(S)-(1,1'-динафтил-2,2'-диил)-N,N'-ди-(R,R)-1-фенилэтилфосфорамидит (0,448 кг) и трифторметансульфонат меди(II) (0,132 кг), и перемешивали при 21°С в течение приблизит. 90 минут. Сосуд продували аргоном, охлаждали до температуры от -25°С до -30°С и добавляли циклогекс-2-ен-1-он (14,0 кг), поддерживая температуру в диапазоне от -25°С до -30°С. Загружали толуол (12,1 кг) в виде промывки линии/сосуда. Добавляли 2 М диметилцинк в толуоле (приблизит. 84,5 л), поддерживая температуру в диапазоне от -20°С до -30°С в течение приблизит. 4,5 часов. Добавляли толуол (12,5 кг) в виде промывки линии/сосуда. Реакционную смесь перемешивали при температуре от -20°С до -30°С в течение приблизит. 10 часов, после чего партию быстро охлаждали охлажденным метанолом (54,5 кг) в течение приблизит. 45 минут, поддерживая температуру ниже 10°С. Партию разбавляли толуолом (5,5 кг), перемешивали при 7°С в течение 1 часа, нагревали до 18°С и затем перемешивали в течение еще 6 часов. Реакционную смесь фильтровали, используя фильтровальную мембрану 100 мкм и 1 мкм, и осадок на фильтре дважды промывали толуолом (13 кг) при приблизит. 20°С. Реакционную смесь концентрировали до 70 л при атмосферном давлении и затем очищали посредством пленочной дистилляции с получением указанного в подзаголовке соединения (13,13 кг) в виде раствора в толуоле.
1H ЯМР δ(CDCl3) 2.45-2.15 (3Н, m), 2.10-1.80 (4Н, m), 1.75-1.55 (1H, m), 1.40-1.25 (1H, m), 1.05 (3H, d).
f) (3S,5S)-5-Метил-1-окса-спиро[2.5]октан
Раствор трет-бутоксида калия (11,6 кг) в диметилсульфоксиде (36 кг) добавляли к смеси триметилсульфоксония иодида (22,68 кг) в диметилсульфоксиде (36,2 кг), поддерживая температуру в диапазоне от 15°С до 25°С. Загружали диметилсульфоксид (11,5 кг) в виде промывки линии/сосуда и партию перемешивали при температуре от 15°С до 25°С в течение 90 минут. К реакционной смеси добавляли толуольный раствор (61,02 кг), содержащий 3-метил-циклогексанон (10,50 кг), поддерживая температуру в диапазоне от 15°С до 25°С. Загружали толуол (9,9 кг) в виде промывки линии/сосуда. Партию перемешивали в течение 2 часов, гасили путем добавления воды (94,5 кг), поддерживая температуру в диапазоне от 15°С до 25°С, и затем фильтровали. Загружали воду (11,0 кг) в виде промывки линии/сосуда. Фазы разделяли и органическую фазу оставляли в виде раствора указанного в подзаголовке соединения (11,1 кг) в толуоле.
1H ЯМР δ(CDCl3) 2.61 (2H, dd), 2.00-1.36 (6H, m), 1.32-1.16 (2H, m), 1.05-0.80 (4H, m).
g) (1S,3S)-1-[(Бензиламино)метил]-3-метил-циклогексанол. HCl
Бензиламин (20,4 кг) загружали в толуольный раствор (72,3 кг) (3S,5S)-5-метил-1-окса-спиро[2.5]октана (9,69 кг) и изопропанола (22,2 кг), поддерживая температуру в диапазоне от 15°С до 25°С. Загружали изопропанол (15,6 кг) в виде промывки линии/сосуда и партию нагревали до температуры от 68°С до 72°С. Через приблизит. 5,5 часов партию охлаждали до 6°С и в нее загружали соляную кислоту в изопропаноле [полученную с ацетилхлоридом (24,4 кг) и изопропанолом (26,7 кг)] так, чтобы температура поддерживалась в диапазоне от 0°С до 20°С. Загружали охлажденный трет-бутилметиловый эфир (35,9 кг), поддерживая температуру в диапазоне от 0°С до 20°С, и затем перемешивали при приблизит. 10°С в течение 90 минут. Полученную суспензию фильтровали, осадок на фильтре дважды промывали трет-бутилметиловым эфиром (приблизит. 35 кг) при приблизит. 10°С и затем сушили на фильтре под вакуумом в течение приблизит. 10 часов с получением указанного в подзаголовке соединения в виде бесцветного твердого вещества (20,29 кг).
1H ЯМР δ(CDCl3) 9.40 (1Н, br s), 7.63-7.60 (2H, m), 7.44-7.36 (3H, m), 4.31 (1H, s), 4.23 (2H, s), 2.74 (2H, s), 2.10-1.45 (7H, m), 1.11 (1Н, dt), 0.83 (3H, d), 0.87-0.68 (1H, m).
h) (1S,3S)-1-Аминометил-3-метил-циклогексанол. HCl
20%-ный (масс./об.) гидроксид натрия (50 л) загружали в сосуд, содержащий (1S,3S)-1-[(бензиламино)метил]-3-метилциклогексанол. HCl (25 кг, содержание 13,93 кг) и трет-бутилметиловый эфир (88,6 кг), поддерживая температуру в диапазоне от 18°С до 25°С. Фазы разделяли и водную фазу дважды экстрагировали трет-бутилметиловым эфиром (37,2 кг). Объединенные органические экстракты снова загружали в сосуд, осуществляли промывку линии трет-бутилметиловым эфиром (20,2 кг) и концентрировали до приблизит. 75 л при атмосферном давлении. Добавляли трет-бутилметиловый эфир (52,6 кг) и партию концентрировали до приблизит. 75 л при атмосферном давлении. Загружали абсолютный этанол (118,6 кг) и партию концентрировали до приблизит. 75 л при температуре от 30°С до 45°С. Температуру доводили до приблизит. 20°С и затем загружали в продутый азотом сосуд, содержащий 5% палладий на углероде (5,00 кг). Загружали абсолютный этанол (63,0 кг) в виде промывки линии/сосуда. Сосуд помещали в атмосферу водорода, температуру доводили до 58°С-62°С и перемешивали в течение 12 часов. Реакционную смесь охлаждали до температуры от 18°С до 25°С, продували азотом и фильтровали. Осуществляли рециркуляцию абсолютного этанола (20,8 кг) через/вокруг сосуд(а)/линий(и) и осадок на фильтре для выделения продукта. Осуществляли рециркуляцию абсолютного этанола (19,7 кг) через/вокруг сосуд(а)/линии(й) и осадок на фильтре для выделения продукта. Объединенные фильтраты концентрировали до приблизит. 75 л при температуре от 30°С до 45°С и затем разбавляли диизопропиловым эфиром (132,3 кг). Реакционную смесь нагревали до флегмообразования и концентрировали до приблизит. 75 л при атмосферном давлении, загружали диизопропиловый эфир (124,6 кг) и партию концентрировали до приблизит. 75 л при атмосферном давлении. Загружали диизопропиловый эфир (123 кг) и партию концентрировали до приблизит. 75 л при атмосферном давлении. Смесь охлаждали до температуры от 5°С до 10°С и в нее загружали HCl в этаноле [получена с ацетилхлоридом (7,68 кг) и этанолом (49,9 кг)] так, чтобы температура поддерживалась в диапазоне от 5°С до 20°С. Загружали охлажденный диизопропиловый эфир (73,2 кг), поддерживая температуру в диапазоне от 5°С до 20°С и затем перемешивали при приблизит. 7°С в течение 1 часа. Полученную суспензию фильтровали, осадок на фильтре дважды промывали диизопропиловым эфиром (приблизит. 38 кг) при приблизит. 20°С и затем сушили на фильтре под вакуумом в течение приблизит. 10 часов с получением указанного в подзаголовке соединения в виде бесцветного твердого вещества (8,02 кг).
1H ЯМР δ(D2O) 3.06 (2Н, s), 1.84-1.60 (7Н, m), 1.44-1.30 (1Н, m), 1.10 (1H, t), 1.02-0.87 (4H, m).
i) 2,6-Дихлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-хинолин-5-карбоксамид
Тионилхлорид (7,2 кг) загружали в сосуд, содержащий 2,6-дихлор-хинолин-5-карбоновую кислоту (5,00 кг) и толуол (43,0 кг). В виде промывки линии загружали толуол (13,1 кг). Температуру реакционной смеси доводили до 82°С-84°С и перемешивали в течение приблизит. 7 часов. Партию охлаждали до температуры ниже 40°С и концентрировали до приблизит. 25 л посредством вакуумной перегонки при температуре от 30°С до 40°С. Загружали толуол (43,5 кг) и партию концентрировали до приблизит. 25 л при температуре от 30°С до 40°С. Загружали толуол (22,0 кг) и температуру доводили до 20°С-25°С. Этот толуольный раствор затем загружали в охлажденную смесь (1S,3S)-1-(аминометил)-3-метилциклогексанола гидрохлорида (3,70 кг), тетрагидрофурана (43,5 кг) и триэтиламина (6,26 кг) так, чтобы температура поддерживалась в диапазоне от 5°С до 10°С. Загружали толуол (4,6 кг) в виде промывки линии. Через 4 часа температуру доводили до 20°С-25°С и гасили добавлением воды (25 кг), поддерживая температуру в диапазоне от 20°С до 25°С. Температуру доводили до 40°С-45°С в течение 20 минут, после чего фазы разделяли. Органическую фазу промывали водой (25 л) при температуре в диапазоне от 40°С до 45°С и затем концентрировали до 45 л при температуре от 25°С до 40°С. Загружали толуол (42,7 кг) и партию концентрировали до приблизит. 45 л при температуре от 25°С до 40°С. Загружали толуол (43,2 кг) и партию концентрировали до приблизит. 45 л при температуре от 25°С до 40°С. Реакционную смесь затем нагревали до температуры флегмообразования для достижения полного растворения, охлаждали до температуры от 20°С до 25°С в течение 2,5 часов и затем перемешивали в течение 2,5 часов при температуре от 20°С до 25°С. Полученную суспензию фильтровали и осадок на фильтре дважды промывали толуолом (приблизит. 9 кг) при приблизит. 20°С и затем сушили при температуре вплоть до 55°С в вакууме с получением указанного в подзаголовке соединения в виде бесцветного твердого вещества (5,88 кг).
1H ЯМР δ(DMSO) 8.66 (1Н, t), 8.25 (1Н, d), 8.04 (1H, d), 7.91 (1H, d), 7.76 (1H, d), 4.24 (1H, s), 3.35-3.25 (ожидался 2Н, d, однако сигнал скрыт сигналом воды), 1.85-1.45 (6Н, m), 1.32 (1H, dt), 1.05 (1H, t), 0.92-0.71 (4H, m).
j) 6-Хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-[(3R)-3-гидроксипирролидин-1-ил]хинолин-5-карбоксамид
Метанол (36,9 кг) загружали в сосуд, содержащий 2,6-дихлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-хинолин-5-карбоксамид (5,85 кг), (R)-3-гидроксипирролидин гидрохлорид (4,15 кг), ацетонитрил (32,5 кг) и триэтиламин (9,8 кг). Сосуд нагревали до температуры флегмообразования. Через приблизит. 70 часов партию охлаждали до температуры от 40 до 45°С и осветляли пропусканием через фильтровальную мембрану 1 мкм. Метанол (4,7 л) загружали в сосуд в две промывки сосуда/линии и партию концентрировали до приблизит. 29 л при атмосферном давлении. Загружали ацетонитрил (46,8 кг) и партию концентрировали до приблизит. 29 л при атмосферном давлении. Загружали ацетонитрил (46,8 кг) и партию концентрировали до приблизит. 29 л при атмосферном давлении. Партию охлаждали до температуры от 18°С до 25°С, разбавляли водой (56,5 кг) и перемешивали в течение 1 часа. Полученную суспензию фильтровали, осадок на фильтре дважды промывали водой (приблизит. 30 кг) и затем сушили при температуре вплоть до 45°С в вакууме с получением указанного в заголовке соединения в виде бесцветного твердого вещества [5,6 кг, 100% е.е., 98,5% d.e. (избыток относительно всех возможных стереоизомеров, как определено посредством аналитической стереохимической HPLC)].
1H ЯМР δ(DMSO) 8.53 (1Н, t), 7.84 (1Н, d), 7.59-7.51 (2H, 2×d), 6.99 (1H, d), 5.03 (1Н, d), 4.45 (1Н, br s), 4.19 (1Н, s), 3.79-3.41 (4H, m), 3.31 (2H, d), 2.13-1.29 (9H, m), 1.08 (1H, t), 0.94-0.73 (4H, m).
Фармакологические анализы
Р2Х7 анализ
Известно, что некоторые соединения, такие как бензоилбензоиладенозинтрифосфат (bbATP), являются агонистами Р2Х7 рецептора, воздействуя на образование пор в плазменной мембране (Drug Development Research (1996), 37(3), p.126). Соответственно, когда рецептор активируется с помощью bbATP в присутствии этидийбромида (флуоресцентного ДНК зонда), наблюдают увеличение флуоресценции внутриклеточного ДНК-связанного этидийбромида. Увеличение флуоресценции может быть использовано в качестве меры активации Р2Х7-рецептора и тем самым для количественной оценки воздействия соединения на Р2Х7 рецептор.
Таким образом, каждое из указанных в заголовках Примеров соединений тестировали на антагонистическую активность в отношении Р2Х7 рецептора. Тест проводили в 96-луночных круглодонных микротитрационных планшетах, лунки заполняли 250 мкл тестируемого раствора, содержавшего 200 мкл суспензии клеток ТНР-1 (2,5×106 клеток/мл), содержавшей 10-4 М этидийбромида, 25 мкл высококалиевого буферного раствора, содержавшего 10-5 М bbATP, и 25 мкл высококалиевого буферного раствора, содержавшего концентрации тестируемого соединения обычно 30 мкМ - 0,001 мкМ. Планшет покрывали пластиковой крышкой и инкубировали при 37°С в течение одного часа. Затем планшет считывали во флуоресцентном планшетном ридере Perkin-Elmer, возбуждение 520 нм, эмиссия 595 нм, ширина щели: Ех 15 нм, Em 20 нм. Для целей сравнения в тесте в качестве контролей по раздельности использовали bbATP (агонист Р2Х7 рецептора) и пиридоксаль-5-фосфат (антагонист Р2Х7 рецептора). На основании полученных результатов считывания рассчитывали величину pIC50 для каждого тестируемого соединения, где эта величина представляет собой отрицательный логарифм концентрации тестируемого соединения, необходимой для снижения bbАТР агонистической активности на 50%.
Протокол связывания hERG
Анализ hERG проводили согласно процедуре, описанной в WO 2005/037052. Аффинность (pIC50) соединений в отношении субъединицы ионного канала, кодируемого человеческим hERG геном (ether-a-go-go-related gene), определяли посредством конкурентного связывания радиолиганда 3,7-бис[2-(4-нитро[3,5-3H]фенил)этил]-3,7-диазабицикло[3.3.1]нонана с мембранами НЕK (почка эмбриона человека) клеток, экспрессирующими hERG, в формате промывки на фильтре.
Мембраны инкубировали в течение 3 часов при комнатной температуре с серийными разведениями тестируемых соединений, радиолигандом 3,7-бис[2-(4-нитро[3,5-3H]фенил)этил]-3,7-диазабицикло[3.3.1]нонаном в конечной концентрации 1 нМ и буфером для анализа (10 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), 130 мМ NaCl, 5 мМ KCl, 1 мМ EGTA (этиленгликольтетрауксусная кислота), 0,8 мМ MgCl2, pH 7,4). Анализ проводили в конечном объеме 200 мкл, в присутствии 1% (об./об.) диметилсульфоксида. Неспецифическое связывание определяли путем измерения связывания 3,7-бис[2-(4-нитро[3,5-3H]фенил)этил]-3,7-диазабицикло[3.3.1]нонана в присутствии 10 мкМ астемизола. В ходе инкубации GF/B фильтровальные планшеты иммерсировали в покрывающем растворе 0,3% (об./об.) полиэтиленимина и 0,2% (масс./об.) BSA (бычий сывороточный альбумин). После инкубирования планшеты для анализа собирали на предварительно покрытые GF/B фильтровальные планшеты, используя харвестер Tomtec.
Определяли величину pIC50, определяемую как отрицательный логарифм концентрации соединения, необходимой для 50%-ного снижения связывания 3,7-бис[2-(4-нитро[3,5-3H]фенил)этил]-3,7-диазабицикло[3.3.1]нонана. Величина 'менее чем' указывает на менее чем 50%-ное ингибирование в указанной концентрации, которая является наибольшей тестированной концентрацией.
Результаты
Каждое из соединений из Примеров демонстрировало очень высокую антагонистическую активность в отношении Р2Х7, показывая величину pIC50 не менее 8,0. Более того, каждое из соединений проявляло особенно низкую активность в отношении hERG, с менее чем 50%-ным ингибированием в самой высокой тестировавшейся концентрации. В таблице 1 представлены величины Р2Х7 pIC50 и hERG pIC50 для соединений из Примеров 1-4 (изомер 2) и соединений сравнения, описанных в примерах в WO 2004/106305 (Примеры 29, 36, 44 и 50).
Соединения согласно настоящему изобретению демонстрировали значение Р2Х7 IC50 в концентрациях 10 нМ или ниже. Далее, они не проявляли достаточной активности для регистрации IC50 в отношении hERG в концентрации 100 мкМ. Соответственно, соединения по настоящему изобретению имеют соотношение P2X7:hERG аффинности более 10000. Соединения сравнения из Примеров 29, 36, 44 и 50 из WO 2004/106305 соответственно демонстрировали hERG IC50 в концентрации 32 мкМ, 8 мкМ, 13 мкМ и 13 мкМ и требовали концентрации 63 нМ, 6 нМ, 13 нМ и 32 нМ для регистрации Р2Х7 IC50. Соответственно, их соотношения аффинностей P2X7:hERG составляют 502, 1258, 1000 и 398 соответственно.
Биодоступность - крысиная PK
Для описания метаболизма соединения в организме в DMPK (фармакокинетика метаболизма лекарственных средств) используют фармакокинетические параметры и понятия. Распределение и экскреция соединения отражаются в зависимости концентрация в плазме - время. Посредством соответствующего дозирования, взятия образцов и анализа могут быть определены ключевые параметры (клиренс, объем, период полувыведения, биодоступность и так далее).
Тестируемые соединения обычно вводили внутривенно (в.в.) в правую латеральную хвостовую вену самцов крыс Sprague Dawley в дозовом уровне 3 мг/кг (1 мл/кг) в смеси DMA (N,N-диметилацетамид):вода (40:60 об./об.). Крысам осуществляли пероральное (п.о.) дозирование в дозовом уровне 5 мг/кг (2 мл/кг) в смеси 0,5% гидроксипропилметилцеллюлоза (НРМС, масс./об.)/0,1% Твин 80 (об./об.) в воде). После внутривенного введения отбирали последовательные образцы крови (200 мкл) из левой латеральной хвостовой вены через 2, 4, 8, 15, 30, 60, 120, 180, 300, 420, 720 и 1440 мин и через 0, 20, 40, 60, 120, 180, 300, 420, 720 и 1440 мин после перорального введения. Плазму получали посредством центрифугирования.
Для определения плазменных уровней тестируемого соединения по 50 мкл метанола добавляли к 50 мкл каждого из тестируемых образцов и по 40 мкл метанола добавляли аликвотам по 50 мкл контрольной плазмы, содержавшей 10 мкл добавленного аутентичного стандарта, используемого для создания калибровочной кривой и QC (контроль качества). И наконец, по 100 мкл метанола, содержавшего химически сходный внутренний стандарт, добавляли к каждому образцу, стандарту и QC, что давало конечный объем 200 мкл. Все образцы плазмы затем тщательно перемешивали и помещали при температуре -20°С по меньшей мере на один час, после чего центрифугировали. Полученные супернатанты анализировали посредством HPLC-MSMS (сочетание высокоэффективной жидкостной хроматографии и масс-спектрометрии) после того, как был создан подходящий селективный и чувствительный метод путем оптимизации как конусного напряжения, так и энергии столкновения.
Фармакокинетические параметры получали из кривой концентрация-время, используя некомпартментный анализ в WinNonLin®. Биодоступность рассчитывали используя следующее уравнение
F=AUCOп.о.·Доза в.в./АUСв.в.·Доза п.о.
П.о. биодоступность у крыс для соединения из примера 2=59%.
Протокол in vitro фосфолипидоза
Определение способности соединений индуцировать фосфолипидоз проводили посредством in vitro флуоресцентного анализа, который дает оценку накопления фосфолипидов в первичных гепатоцитах крыс. Гепатоциты выделяли от крыс Han Wistar с помощью 2-стадийного метода обработки коллагеназой. Затем гепатоциты помещали в покрытые коллагеном 96-луночные планшеты в среду Вильямса (William's). Клеткам давали возможность прилипать в течение 1 часа, затем среду заменяли раствором 250 мкг·мл-1 коллагена в клеточной культуральной среде Hepatozyme.
Клетки затем культивировали в течение 48 часов со сменой среды через 24 часа. Через 48 часов после выделения среду заменяли на Hepatozyme, дополненную флуоресцентным фосфолипидом N-(6-тетраметилродамин-тиокарбамоил)-1,2-дигексадеканоил-sn-глицеро-3-фосфоэтаноламином (DHPE-TRITC) (5 мкг·мл-1). В это время к гепатоцитам добавляли тестируемые соединения в диапазоне концентраций в серийном разведении с конечной концентрацией 0,4% диметилсульфоксида (используемого в качестве растворителя для тестируемых соединений).
Клетки инкубировали в течение еще 24 часов и затем фиксировали добавлением забуференного фосфатом физиологического раствора (PBS), содержавшего ядерный краситель Hoechst 33342 (конечная концентрация 2 мкМ), и раствора параформальдегида (конечная концентрация 4%). Планшеты выдерживали при комнатной температуре в течение 30 минут, затем промывали три раза в растворе PBS.
Изображения гепатоцитов затем получали с помощью автоматизированной микроскопической платформы (GE In Cell Analyser 3000). Затем использовали алгоритмы анализа изображений для оценки жизнеспособности клеток и накопления метки DHPE-TRITC в жизнеспособных гепатоцитах. Дискретное накопление, наблюдавшееся для тестируемых соединений, затем нормировали в категорию 0, представляя накопление, наблюдавшееся в клетках, подвергнутых воздействию только носителя, и 1, представляя клетки, подвергнутые воздействию 10 мкМ амиодарона. Определяют максимальное накопление на кривой дозового ответа тестируемого соединения, где жизнеспособность клеток составляет более 50%, как являющееся дозой, при которой наблюдали этот максимум. Также определяли наименьшую дозу, которая вызывала более 50% клеточной токсичности. Токсичность в отдельных клетках идентифицируют как изменение ядерного мечения до конденсированной точечной морфологии.
Известно, что доза, при которой наблюдают фосфолипидоз, обратно коррелирует с in vivo наличием фосфолипидоза (David K. Monteith, Ryan E. Morgan & Bartley Halstead (2006) "In vitro assays и biomarkers for drug-induced phospholipidosis". Expert Opinion on Drug Metabolism & Toxicology, vol.2 (5), pp. 687-696).
Для соединения из Примера 2 согласно настоящему изобретению не регистрировали измеримого накопления даже при максимальной тестируемой концентрации 250 мкМ. Кроме того, оно демонстрировало минимальную токсичную концентрацию более 250 мкМ.
Измерение связывания с белками плазмы
Степень связывания с белками плазмы определяли посредством равновесного диализа соединения между человеческой плазмой и водным буфером при 37°С и определения концентраций соединения в плазме и буфере посредством HPLC-MS/MS.
Ячейки для диализа (отсечение по молекулярной массе 5000) готовили посредством промывания водой с последующим вымачиванием в буфере для диализа в течение минимум 1 часа. Буфер для диализа представлял собой изотонический забуференный физиологический раствор с рН 7,4. Готовили исходные растворы соединения в диметилсульфоксиде с концентрацией 0,5 мМ.
Исходный DMSO (диметилсульфоксид) раствор соединения добавляли к плазме в соотношении 10 мкл DMSO на каждый мл плазмы. Это давало 1% DMSO в плазменном растворе с каждым соединением в концентрации 5 мкМ.
Затем готовили ячейки для диализа и одну половину ячейки заполняли 750 мкл буфера для диализа, а другую половину заполняли 750 мкл плазменного раствора соединения. Приготовленные ячейки запаивали и помещали в инкубационный бокс при 37°С. Эти ячейки затем переворачивали в течение минимум 4 часов для уравновешивания.
После уравновешивания 500 мкл буферных образцов удаляли и добавляли в сосуды для HPLC вместе со 100 мкл плазмы (образец в 6-кратно разведенной плазме) и 100 мкл плазменных образцов удаляли и добавляли в сосуды для HPLC вместе с 500 мкл буфера для диализа (образец в 6-кратно разведенной плазме).
Затем образцы анализировали посредством HPLC-MS/MS. Получали четырехточечную калибровочную кривую путем разведении исходных растворов 6-кратно разведенной плазмой в концентрациях 0,013 мкМ, 0,05 мкМ, 0,25 мкМ и 1,25 мкМ, которые инъецировали в этом порядке с последующим буферным образцом и затем плазменным образцом.
Вычисления
Концентрацию соединения в образцах определяли с помощью программного обеспечения MassLynx версия 4.1 (производится Waters/Micromass), которое автоматически рассчитывало калибровочную кривую и интерполировало концентрацию соединения в аналитах. Связывание с белками плазмы определяли из измеренной концентрации как процент соединения, связанного в плазме (% связанного) с помощью следующего уравнения:
Связывание с белками плазмы человека (% связанного) для соединения из Примера 2=88%.
| название | год | авторы | номер документа |
|---|---|---|---|
| АГОНИСТЫ РЕЦЕПТОРА МЕЛАНОКОРТИНА-4 | 2020 |
|
RU2786864C1 |
| АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2758686C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| Новые пролекарства на основе катехоламина для применения в лечении болезни Паркинсона | 2018 |
|
RU2806075C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТАГОНИСТ Р2Х РЕЦЕПТОРА И ФАКТОР НЕКРОЗА ОПУХОЛИ α | 2004 |
|
RU2350354C2 |
| ПРОЛЕКАРСТВО ФТОРСОДЕРЖАЩЕЙ АМИНОКИСЛОТЫ | 2013 |
|
RU2639868C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2747318C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2022 |
|
RU2803084C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТАГОНИСТ P2X-РЕЦЕПТОРА И НЕСТЕРОИДНОЕ ПРОТИВОВОСПАЛИТЕЛЬНОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2004 |
|
RU2338556C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2777979C2 |
Настоящее изобретение относится к новым производным хинолина формулы (I) или к их фармацевтически приемлемым солям, где n равен 0 или 1; когда n равен 0, тогда R1 представляет собой водород или метил, R2 представляет собой гидроксил, и R3 представляет собой водород; и когда n равен 1, R1 представляет собой водород, и один из R2 и R3 представляет собой гидроксил, а другой из R2 и R3 представляет собой водород. Также изобретение относится к конкретным соединениям формулы (I) и к способу получения соединений формулы (I). Технический результат: получены новые производные хинолина, обладающие высокой Р2Х7 антагонистической активностью. 3 н. и 6 з.п. ф-лы, 1 табл., 5 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
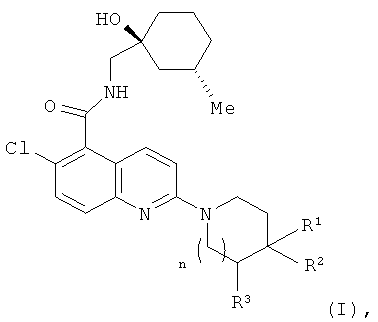
где n равен 0 или 1;
когда n равен 0, тогда R1 представляет собой водород или метил, R2 представляет собой гидроксил, и R3 представляет собой водород; и
когда n равен 1, R1 представляет собой водород, и один из R2 и R3 представляет собой гидроксил, а другой из R2 и R3 представляет собой водород.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где n равен 0, R1 представляет собой водород или метил, R2 представляет собой гидроксил, и R3 представляет собой водород.
3. Соединение по п.2 или его фармацевтически приемлемая соль, где R1 представляет собой водород.
4. Соединение по п.2 или его фармацевтически приемлемая соль, где R1 представляет собой метил.
5. Соединение по п.1 или его фармацевтически приемлемая соль, где n равен 1, R1 представляет собой водород, и один из R2 и R3 представляет собой гидроксил, а другой из R2 и R3 представляет собой водород.
6. Соединение по п.5 или его фармацевтически приемлемая соль, где R2 представляет собой гидроксил, и R3 представляет собой водород.
7. Соединение по п.5 или его фармацевтически приемлемая соль, где R2 представляет собой водород, и R3 представляет собой гидроксил.
8. Соединение формулы (I), выбранное из:
6-хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-[(3S)-3-гидроксипирролидин-1-ил]хинолин-5-карбоксамида,
6-хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-[(3R)-3-гидроксипирролидин-1-ил]хинолин-5-карбоксамида,
6-хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-(4-гидроксипиперидин-1-ил)хинолин-5-карбоксамида и
6-хлор-N-{[(1S,3S)-1-гидрокси-3-метилциклогексил]метил}-2-(3-гидрокси-3-метилпирролидин-1-ил)хинолин-5-карбоксамида,
или его фармацевтически приемлемая соль.
9. Способ получения соединения формулы (I) по любому из пп.1-8, включающий:
(а) взаимодействие соединения формулы (II)
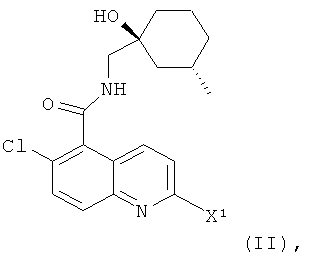
где Х1 представляет собой подходящую уходящую группу, с соединением формулы (III)
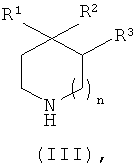 ,
,
где R1, R2, R3 и n являются такими, как определено в формуле (I), и возможно образование фармацевтически приемлемой соли соединения.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Biggs D.F | |||
| et al | |||
| Synthesis and pharmacological evaluation of some beta-beta-disudstituted analogs of acetylcholine | |||
| - JOURNAL OF MEDICINAL CHEMISTRY, vol.15, no.6, 1972, pp.642-646 | |||
| Способ получения амидов, их диастериомеров, рацематов, энантиомеров или их аддитивных солей | 1987 |
|
SU1614759A3 |
| Горячеканальная литьевая форма для изготовления полимерных изделий | 1985 |
|
SU1242396A1 |

