ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
В настоящем изобретении предусмотрены соединения, которые являются пролекарствами агониста дофамина (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола, и их применение в лечении болезни Паркинсона и/или других состояний, для которых лечение с помощью агониста дофамина является терапевтически благоприятным, таких как без ограничения синдром беспокойных ног, болезнь Хантингтона и болезнь Альцгеймера; а также нейропсихиатрических заболеваний и нарушений, таких как без ограничения шизофрения, синдром дефицита внимания с гиперактивностью и наркотическая зависимость. В настоящем изобретении также предусмотрены фармацевтические композиции, содержащие соединения по настоящему изобретению.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Болезнь Паркинсона (PD) представляет собой распространенное нейродегенеративное нарушение, которое становится все более распространенным с возрастом и, по оценкам, поражает от семи до десяти миллионов человек во всем мире. Болезнь Паркинсона является многогранным заболеванием, характеризующимся как моторными, так и немоторными симптомами. Моторные симптомы включают тремор в покое (дрожание), брадикинезию/акинезию (медлительность и скудность движений), мышечную ригидность, постуральную неустойчивость и дисфункцию походки; при этом немоторные симптомы включают нейропсихиатрические нарушения (например депрессию, психотические симптомы, тревожность, апатию, умеренное когнитивное нарушение и деменцию), а также вегетативные дисфункции и нарушения сна (Poewe et al., Nature Review, (2017) vol 3 article 17013: 1-21).
Ключевой характерной особенностью патофизиологии болезни Паркинсона является потеря пигментированных дофаминергических нейронов в компактном слое черного вещества, который обеспечивает дофаминергическую иннервацию полосатого тела и других зон головного мозга. Такая прогрессирующая нейродегенерация приводит к уменьшению уровней дофамина в полосатом теле, что в конечном итоге вызывает ряд изменений межнейронных связей в базальных ганглиях, что в конечном итоге приводит к появлению четырех основных моторных признаков болезни Паркинсона. Основной мишенью дофамина в полосатом теле являются срединные шипиковые GABA-ергические нейроны (MSN), селективно экспрессирующие рецепторы D1 или D2, продолжающиеся в виде их топографических проекций. GABA-ергический MSN, проецирующийся во внешний отдел бледного шара, также называемый стриатопаллидальным 'непрямым путем', экспрессирует рецепторы D2 (MSN-2); при этом GABA-ергический MSN, проецирующийся в ретикулярную часть черного вещества и внутренний отдел бледного шара, также называемый стриатонигральным 'прямым путем', экспрессирует рецепторы D1 (MSN-1). Уменьшение количества дофамина вследствие потери нейронов приводит к несбалансированной активности двух путей, что приводит к заметному уменьшению на выходе таламической и кортикальной активности и, в конечном итоге, моторным дисфункциям (Gerfen et al, Science (1990) 250: 1429-32; Delong, (1990) Trends in Neuroscience 13: 281-5; Alexander et Crutcher, (1990) Trends in Neuroscience 13: 266-71; и для обзора Poewe et al., Nature Review (2017) vol. 3 article 17013: 1-21).
Наиболее эффективными терапевтическими стратегиями, доступными для пациентов, страдающих болезнью Паркинсона, и направленными на контроль моторных симптомов, в основном являются непрямые и прямые агонисты дофамина. Классическая и общепринятая стандартная схема лечения включает постоянный пероральный прием L-3,4-дигидроксифенилаланина (L-DOPA), который декарбоксилируется в головном мозге с образованием дофамина. Другие подходы заключаются во введении агонистов дофаминовых рецепторов, таких как апоморфин, который действует как на подтипы рецепторов D1, так и на подтипы рецепторов D2, или прамипексол, ропинирол и другие, которые преимущественно направлены на подтипы рецепторов D2. Оптимальное облегчение в отношении моторных симптомов обеспечивается с помощью применения как L-DOPA, так и апоморфина за счет активации ими как подтипов рецепторов D1, так и подтипов рецепторов D2 и целостного уравновешивания непрямых и прямых путей (т.е. агонисты D2 при этом способствуют устранению только дисфункции непрямого пути).
L-DOPA и апоморфин, характеризующиеся структурами, изображенными ниже, в настоящее время представляют собой наиболее эффективные лекарственные средства для лечения PD в клинической практике.
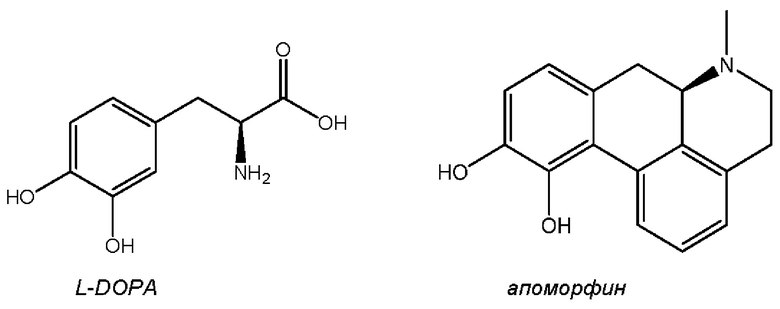
L-DOPA представляет собой пролекарство дофамина и остается наиболее эффективным лекарственным средством при лечении болезни Паркинсона с моторными симптомами. Однако после нескольких лет лечения (т.е. периода "медового месяца") возникают осложнения вследствие присущего заболеванию прогрессирования (т.е. длительной потери дофаминергических нейронов), а также плохого фармакокинетического (PK) профиля L-DOPA. Эти осложнения включают 1) дискинезию, которая представляет собой патологические непроизвольные движения, возникающие во время оптимального 'своевременного эффекта' лекарственного средства; и 2) флуктуации в фазе «выключения», что представляет собой период, в течение которого положительный эффект L-DOPA ослабевает и симптомы снова появляются или ухудшаются (Sprenger and Poewe, CNS Drugs (2013), 27: 259-272).
Прямые агонисты дофаминовых рецепторов способны активировать ауторецепторы дофамина, а также постсинаптические дофаминовые рецепторы, расположенные на срединных шипиковых нейронах MSN-1 и MSN-2. Апоморфин относится к классу агонистов дофамина с 1,2-дигидроксибензольным (катехольным) фрагментом. В сочетании с фенетиламиновым мотивом, катехоламины часто обладают низкой пероральной биодоступностью или не обладают ей, как в случае с апоморфином. Апоморфин применяют в клинической терапии PD, хотя и с использованием непероральной доставки (как правило, посредством периодического подкожного введения или непрерывной парентеральной инфузии с помощью насоса в течение дня). В случае апоморфина исследования на животных показали, что трансдермальная доставка или доставка с помощью имплантатов могут обеспечивать возможные формы введения. Однако при исследовании доставки апоморфина из имплантатов у обезьян (Bibbiani et al., Chase Experimental Neurology (2005), 192: 73-78) было установлено, что в большинстве случаев животные должны были подвергаться лечению с помощью иммунодепрессанта дексаметазона для предупреждения местного раздражения и других осложнений после операции по имплантации. Были тщательно изучены альтернативные стратегии доставки для апоморфиновой терапии при PD, такие как ингаляционные и сублингвальные составы (см., например, Grosset et al., Acta Neurol Scand. (2013), 128:166-171 and Hauser et al., Movement Disorders (2016), Vol. 32 (9): 1367-1372). Однако эти меры еще не применяются в клинической практике для лечения PD.
Альтернатива составам для неперорального применения на основе катехоламинов предусматривает применение пролекарства, обеспечивающего маскировку свободных гидроксильных групп катехола, чтобы обеспечить возможность перорального введения. Однако известной проблемой, связанной с разработкой пролекарств для клинического применения, являются трудности, связанные с прогнозированием превращения в исходное соединение в организме людей.
В литературе сообщалось о различных представляющих собой сложные эфиры пролекарствах на основе катехоламинов, таких как покрытые энтеросолюбильной оболочкой сложные эфиры N-пропилапоморфина (NPA) для дуоденальной доставки (см., например, WO 02/100377) и агонист D1-подобного рецептора адроголид, представляющий собой пролекарство, содержащее диацетил, А-86929 (Giardina and Williams; CNS Drug Reviews (2001), Vol. 7 (3): 305-316). Адроголид подвергается активному метаболизированию при первичном прохождении через печень в организме человека после перорального приема и вследствие этого обладает низкой пероральной биодоступностью (прибл. 4%). У пациентов с PD адроголид, вводимый внутривенно (IV), обладает антипаркинсонической эффективностью, сравнимой с эффективностью L-DOPA (Giardina and Williams; CNS Drug Reviews (2001), Vol. 7 (3): 305-316).
В дополнение к представляющим собой сложные эфиры пролекарствам на основе катехоламинов альтернативный подход в отношении пролекарства предусматривает маскировку двух гидроксильных групп катехола в виде соответствующего ацеталя, содержащего метилендиокси-группу (MDO), в виде ацеталя, полученного из альдегидов, за исключением формальдегида, или в виде кеталя, полученного из различных кетонов. Этот принцип получения пролекарства был описан, например, в Campbell et al., Neuropharmacology (1982); 21(10): 953-961 и в US 4543256, WO 2009/026934 и WO 2009/026935.
Еще одним предлагаемым подходом для пролекарства на основе катехоламина является образование производного енона, как предлагается, например, в WO 2001/078713 и в Liu et al., Bioorganic Med. Chem. (2008), 16: 3438-3444. Дополнительные примеры пролекарств на основе катехоламина см., например, в Sozio et al., Exp. Opin. Drug Disc. (2012); 7(5): 385-406.
Соединение (4aR,10aR)-1-н-пропил-1,2,3,4,4a,5,10,10a-октагидробензо[g]хинолин-6,7-диол, изображенное как соединение (I), указанное ниже, раскрыто в WO 2009/026934. Транс-изомер был раскрыт ранее в Liu et al., J. Med. Chem. (2006), 49: 1494-1498 и затем в Liu et al., Bioorganic Med. Chem. (2008), 16: 3438-3444, включая фармакологические данные, указывающие на то, что соединение обладает низкой пероральной биодоступностью у крыс. Рацемат впервые был раскрыт в Cannon et al., J. Heterocyclic Chem. (1980); 17: 1633-1636.
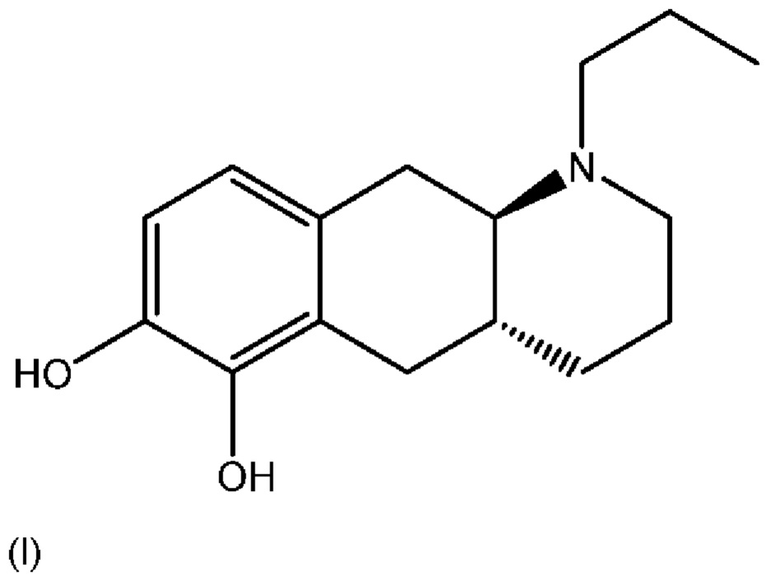
Соединение (I) является агонистом дофаминовых рецепторов со смешанной активностью в отношении D1 и D2. Три пролекарственных производных соединения (I) известны из уровня техники.
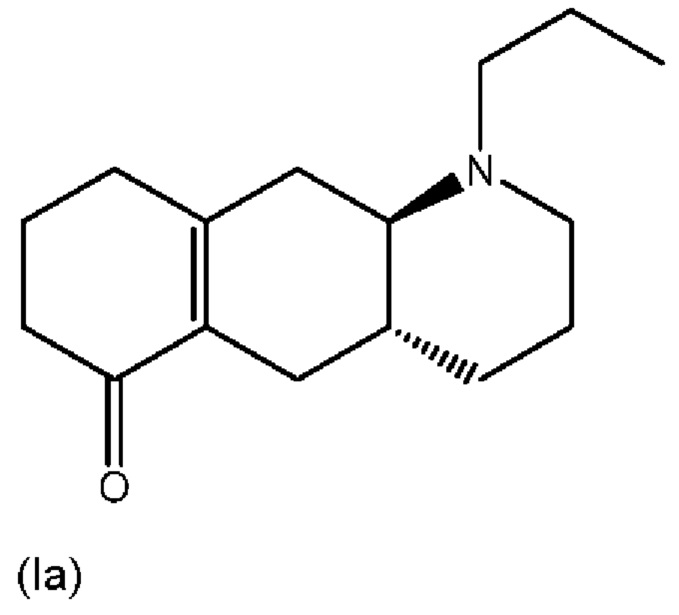
В Liu et al., J. Med. Chem. (2006), 49: 1494-1498 и Liu et al., Bioorganic Med. Chem. (2008), 16: 3438-3444 раскрыто еноновое производное формулы (Ia), изображенной ниже, которое, как было показано, превращается в активное соединение (I) в организме крыс.
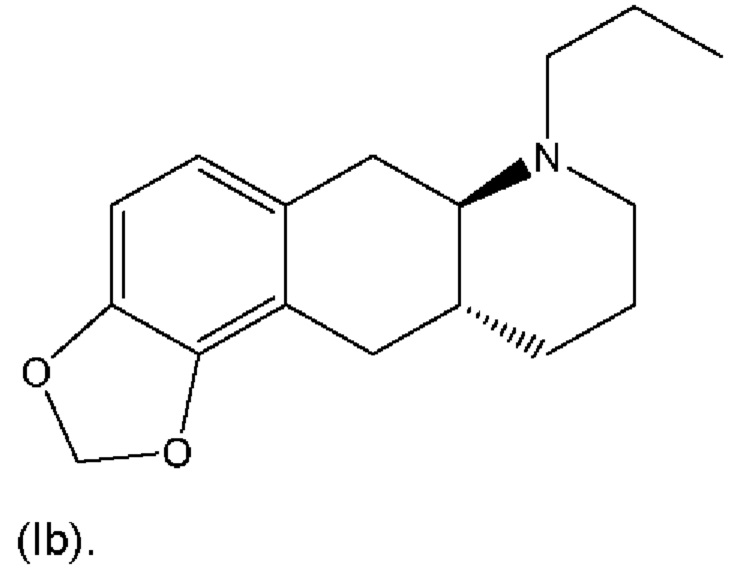
В WO 2009/026934 и WO 2009/026935 раскрыты два типа пролекарственных производных соединения (I), в том числе MDO-производное формулы (Ib) ниже:
Превращение соединения (Ib) в соединение (I) в гепатоцитах крысы и человека было продемонстрировано в WO 2010/097092. Кроме того, фармакология in vivo соединений (Ia) и (Ib), а также активного "исходного соединения" (I) была исследована на различных животных моделях, соответствующих болезни Паркинсона (WO 2010/097092). Было обнаружено, что как соединение (I), так и соединения (Ia) и (Ib) являются эффективными, указывая на то, что соединения (Ia) и (Ib) превращаются in vivo в соединение (I). Сообщалось, что все три соединения характеризовались продолжительностью действия, которая была больше, чем наблюдаемая для L-dopa и апоморфина.
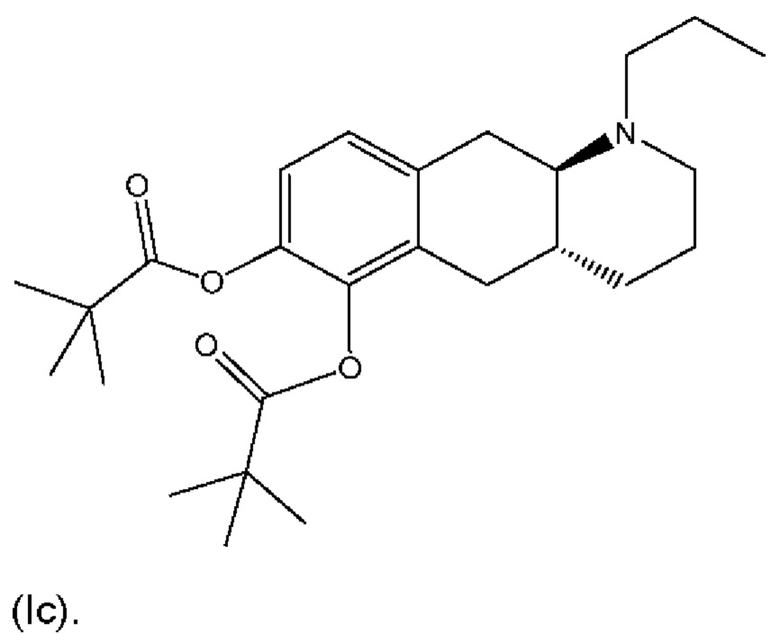
Другое пролекарство соединения (I), раскрытое в WO 2009/026934 и WO 2009/026935, является традиционным представляющим собой сложный эфир пролекарством согласно формуле (Ic):
Несмотря на долголетний интерес в данной области, очевидно, что по-прежнему существует неудовлетворенная потребность в разработке эффективных, хорошо переносимых и активных при пероральном применении лекарственных средств для лечения PD. Пролекарственное производное смешанного D1/D2-агониста, характеризующееся стабильным PK-профилем, которое может обеспечить непрерывную дофаминергическую стимуляцию, может удовлетворить такие неудовлетворенные потребности.
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям для лечения болезни Паркинсона. Более конкретно, настоящее изобретение относится к новым пролекарственным производным соединения (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола (соединения (I)). Было доказано, что соединения по настоящему изобретению являются особенно полезными для пероральной доставки соединения (I).
Соответственно, настоящее изобретение относится к соединениям формулы (Id),
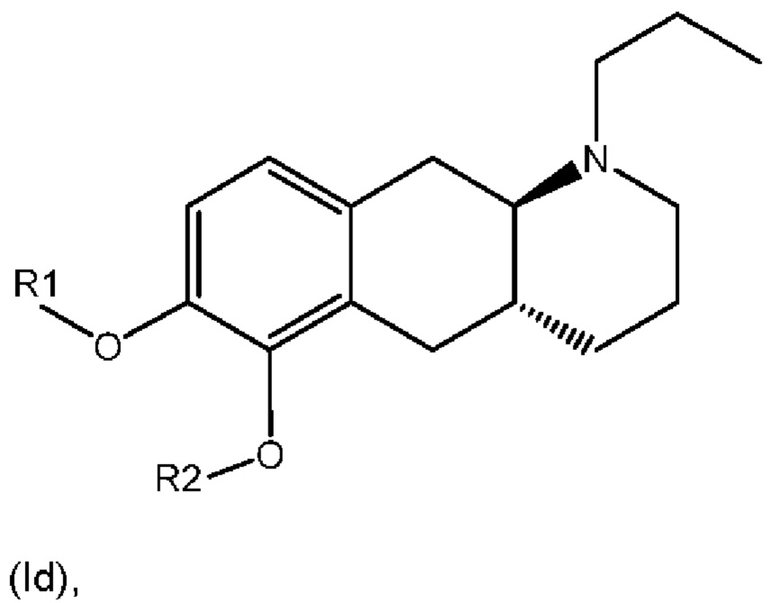
где
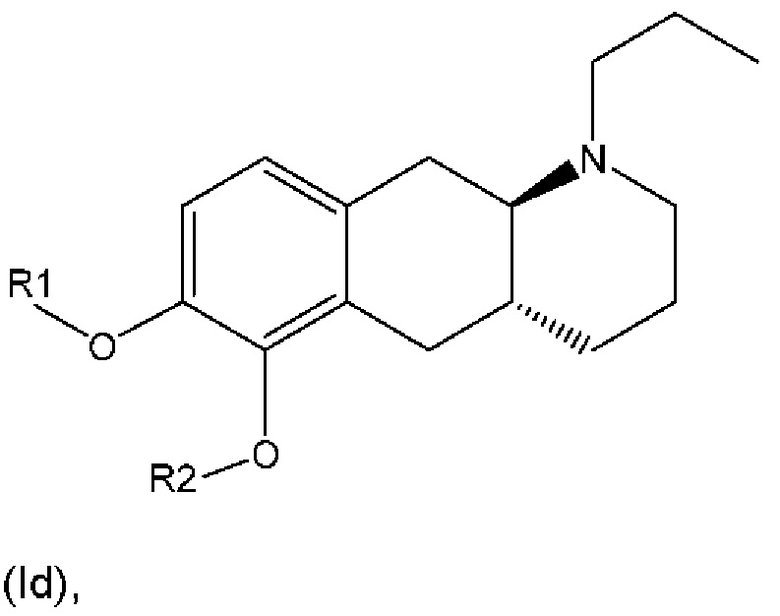
R1 представляет собой Н, и R2 выбран из одного из заместителей (i) и (ii), указанных ниже; или
R1 выбран из одного из заместителей (i) и (ii), указанных ниже, и R2 представляет собой Н; или
R1 и R2 одновременно представлены заместителем (i), указанным ниже; или
R1 и R2 одновременно представлены заместителем (ii), указанным ниже; или
R1 представляет собой заместитель (i), и R2 представляет собой заместитель (ii); или
R1 представляет собой заместитель (ii), и R2 представляет собой заместитель (i);
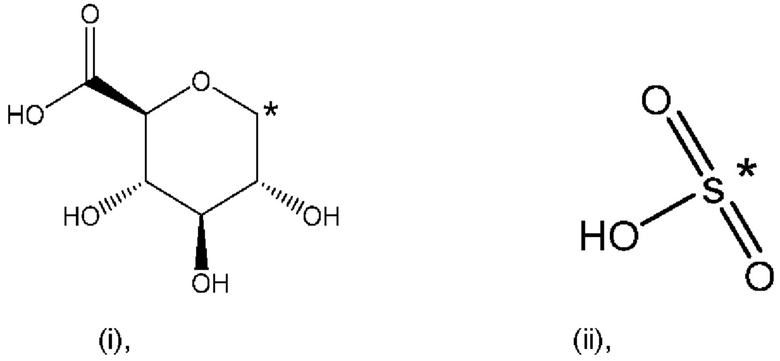
где * обозначает точку присоединения; и
при этом атом углерода в точке присоединения при заместителе (i) находится в S-конфигурации;
или их фармацевтически приемлемой соли.
В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение в соответствии с формулой (Id) или его фармацевтически приемлемую соль и одно или несколько фармацевтически приемлемых вспомогательных веществ.
В одном варианте осуществления настоящее изобретение относится к соединению формулы (Id) для применения в качестве лекарственного препарата.
В одном варианте осуществления настоящее изобретение относится к соединению формулы (Id) или его фармацевтически приемлемой соли для применения в лечении нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона, болезнь Хантингтона, синдром беспокойных ног или болезнь Альцгеймера; или нейропсихиатрического заболевания или нарушения, такого как шизофрения, синдром дефицита внимания с гиперактивностью или наркотическая зависимость.
В одном варианте осуществления настоящее изобретение относится к способу лечения нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона, болезнь Хантингтона, синдром беспокойных ног или болезнь Альцгеймера; или нейропсихиатрического заболевания или нарушения, такого как шизофрения, синдром дефицита внимания с гиперактивностью или наркотическая зависимость; при этом способ включает введение терапевтически эффективного количества соединения формулы (Id) или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
В одном варианте осуществления настоящее изобретение относится к применению соединения формулы (Id) или его фармацевтически приемлемой соли для получения лекарственного препарата, предназначенного для лечения нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона, болезнь Хантингтона, синдром беспокойных ног или болезнь Альцгеймера; или для лечения нейропсихиатрического заболевания или нарушения, такого как шизофрения, синдром дефицита внимания с гиперактивностью или наркотическая зависимость.
В контексте настоящего изобретения известно, что атом углерода в точке присоединения при заместителе (i) находится в аномерном положении (i).
ОПРЕДЕЛЕНИЯ
Соединения по настоящему изобретению
Ссылка на соединения, охватываемые настоящим изобретением, включает свободное вещество (цвиттер-ион) на основе соединений по настоящему изобретению, фармацевтически приемлемые соли соединений по настоящему изобретению, такие как соли присоединения кислоты или соли присоединения основания, и полиморфные и аморфные формы соединений по настоящему изобретению и их фармацевтически приемлемых солей. Кроме того, соединения по настоящему изобретению и их фармацевтически приемлемые соли потенциально могут существовать в несольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. Как сольватированные, так и несольватированные формы охватываются настоящим изобретением.
Фармацевтически приемлемые соли
Фармацевтически приемлемые соли в настоящем контексте предназначены для обозначения нетоксичных, то есть физиологически приемлемых солей.
Термин "фармацевтически приемлемые соли" включает фармацевтически приемлемые соли присоединения кислоты, которые представляют собой соли, образованные с неорганическими и/или органическими кислотами при атоме азота в исходной молекуле. Указанные кислоты могут быть выбраны из, например, хлористоводородной кислоты, бромистоводородной кислоты, фосфорной кислоты, азотистой кислоты, серной кислоты, бензойной кислоты, лимонной кислоты, глюконовой кислоты, молочной кислоты, малеиновой кислоты, янтарной кислоты, винной кислоты, уксусной кислоты, пропионовой кислоты, щавелевой кислоты, малоновой кислоты, фумаровой кислоты, глутаминовой кислоты, пироглутаминовой кислоты, салициловой кислоты, гентизиновой кислоты, сахарина и сульфоновых кислот, таких как метансульфоновая кислота, этансульфоновая кислота, толуолсульфоновая кислота, нафталин-2-сульфоновая кислота, 2-гидроксиэтансульфоновая кислота и бензолсульфоновая кислота.
Термин фармацевтически приемлемые соли также включает фармацевтически приемлемые соли присоединения основания, которые представляют собой соли, образованные с неорганическими и/или органическими основаниями на кислотных группах соединений формулы (Id). Указанные основания могут быть выбраны из, например, гидроксида цинка и оснований щелочных металлов, таких как гидроксид натрия гидроксид лития, гидроксид калия, и щелочноземельных оснований, таких как гидроксид кальция и гидроксид магния, и органических оснований, таких как холин, диэтиламин, триметиламин и триэтиламин.
Дополнительные примеры пригодных кислот и оснований для образования фармацевтически приемлемых солей можно найти, например, в Stahl и Wermuth (Eds) "Handbook of Pharmaceutical salts. Properties, selection, and use", Wiley-VCH, 2008.
Твердая форма
В контексте настоящего изобретения, если соединение по настоящему изобретению представлено в твердой форме, это указывает на то, что указанное соединение не растворено в какой-либо жидкости, такой как водосодержащие жидкости, органические жидкости и их смеси. Настоящее изобретение охватывает твердые формы свободного вещества (цвиттер-иона) на основе соединений по настоящему изобретению, а также твердые формы фармацевтически приемлемых солей соединений по настоящему изобретению. Термин "твердая форма" охватывает как аморфные формы соединений по настоящему изобретению и их солей, так и кристаллические формы соединений по настоящему изобретению и их солей.
Пролекарство
В контексте настоящего изобретения термины "пролекарство" или "пролекарственное производное" обозначают соединение, которое после введения живому субъекту, такому как млекопитающее, предпочтительно человек; превращается в организме в фармакологически активный фрагмент. Превращение предпочтительно происходит в организме млекопитающего, например в организме мыши, крысы, собаки, карликовой свиньи, кролика, обезьяны и/или человека. В контексте настоящего изобретения под "пролекарством соединения (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола" или "пролекарством соединения формулы (I)" или "пролекарством соединения (I)" понимают соединение, которое после введения превращается в организме в соединение (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диол. Указанное введение можно осуществлять любым традиционным путем введения фармацевтических композиций, известным из уровня техники, предпочтительно путем перорального введения.
В контексте настоящего изобретения термины "исходное соединение" и "исходная молекула" обозначают фармакологически активный фрагмент, полученный при превращении соответствующего пролекарства. Например, под "исходным соединением" одного из соединений (Ia), (Ib), (Ic) или любого из соединений по настоящему изобретению понимают соединение формулы (I).
Химическое получение
В контексте настоящего изобретения соединение, "изготовленное путем химического получения", обозначает то, что указанное соединение было получено с помощью химического способа, такого как без ограничения один из способов, описанных в экспериментальном разделе в данном документе.
Фармакокинетические определения и сокращения
Применяемый в данном документе "PK-профиль" является сокращением термина "фармакокинетический профиль". Фармакокинетические профили и фармакокинетические параметры, описанные в данном документе, основаны на данных концентрации в плазме крови в зависимости от времени, полученных для соединения формулы (I) после перорального введения дозы соединения по настоящему изобретению с применением некомпартментного моделирования. Сокращенными PK-параметрами являются: Cmax (максимальная концентрация); tmax (время до достижения Cmax); 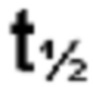 (период полувыведения); AUC0-∞ (площадь под кривой от времени введения дозы до бесконечности).
(период полувыведения); AUC0-∞ (площадь под кривой от времени введения дозы до бесконечности).
Терапевтически эффективное количество
В контексте настоящего изобретения термин "терапевтически эффективное количество" соединения означает количество, достаточное для облегчения, остановки, частичной остановки, устранения или задержки клинических проявлений данного заболевания и его осложнений при терапевтическом вмешательстве, предусматривающем введение указанного соединения. Количество, достаточное для осуществления этого, определяется как "терапевтически эффективное количество". Эффективные количества для каждой цели будут зависеть, например, от тяжести заболевания или повреждения, а также от веса и общего состояния субъекта. Следует понимать, что определения соответствующей дозировки можно достигнуть с применением общепринятых экспериментов, путем построения матрицы значений и тестирования различных точек в матрице, что находится в пределах компетенции квалифицированного врача.
В контексте настоящего изобретения "терапевтически эффективное количество" соединения по настоящему изобретению обозначает количество указанного соединения по настоящему изобретению, которое способно обеспечить такое количество соединения (I), которое является достаточным для облегчения, остановки, частичной остановки, устранения или задержки клинических проявлений определенного заболевания и его осложнений, если указанное соединение по настоящему изобретению вводят, предпочтительно пероральным путем, млекопитающему, предпочтительно человеку.
Лечение и осуществление лечения
В контексте настоящего изобретения "лечение" или "осуществление лечения" предназначено для обозначения ведения пациента и ухода за ним с целью облегчения, остановки, частичной остановки, устранения или задержки развития клинического проявления заболевания. Пациентом, подлежащим лечению, предпочтительно является млекопитающее, в частности человек.
Состояния, подлежащие лечению
Соединения по настоящему изобретению предназначены для лечения ней роде генеративных заболеваний и расстройств, как например болезнь Паркинсона, и/или других состояний, для которых лечение с помощью агониста дофамина является терапевтически благоприятным.
Терапевтические показания включают ряд расстройств центральной нервной системы, характеризующихся моторными и/или немоторными нарушениями и для которых компонентом лежащей в основе патофизиологии является дисфункция межнейронных связей, опосредованных полосатым телом. Такие функциональные нарушения можно наблюдать при нейродегенеративных заболеваниях, таких как без ограничения болезнь Паркинсона (PD), синдром беспокойных ног, болезнь Хантингтона и болезнь Альцгеймера, а также при нейропсихиатрических заболеваниях, таких как без ограничения шизофрения, синдром дефицита внимания с гиперактивностью и наркотическая зависимость.
В дополнение к нейродегенеративным заболеваниям и нарушениям существуют другие состояния, при которых увеличение дофаминергического обмена может быть благоприятным, связанные с улучшением психических функций, в том числе различных аспектов когнитивных функций. Это также может иметь положительный эффект у пациентов с депрессией, и это также можно использовать в лечении ожирения в качестве анорексигенного средства и в лечении наркотической зависимости. Это может обеспечить улучшение в случае минимальной мозговой дисфункции (MBD), нарколепсии, синдрома дефицита внимания с гиперактивностью и, возможно, негативных, позитивных, а также когнитивных симптомов шизофрении.
Синдром беспокойных ног (RLS) и синдром периодических движений конечностей (PLMD) являются альтернативными показаниями, которые клинически лечат агонистами дофамина. Кроме того, в случае импотенции, эректильной дисфункции, половой дисфункции, индуцированной SSRI, синдрома гиперстимуляции яичников (OHSS) и некоторых видов опухоли гипофиза (пролактиномы) также, вероятно, можно обеспечить улучшение путем лечения агонистами дофамина. Дофамин участвует в регуляции сердечно-сосудистой и мочевыделительной систем и, соответственно, почечную недостаточность и гипертензию можно рассматривать как альтернативные показания для применения соединений по настоящему изобретению.
Настоящее изобретение охватывает применение соединений по настоящему изобретению для лечения заболеваний и нарушений, перечисленных выше.
Комбинации
В одном варианте осуществления настоящего изобретения соединения формулы (Id) предназначены для применения в качестве используемого самостоятельно терапевтического средства в виде единственного активного соединения. В другом варианте осуществления настоящего изобретения соединения формулы (Id) можно применять в комбинации с другими средствами, применимыми в лечении нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона. Термины "комбинированное применение", "в комбинации с" и "комбинация" и т.п., применяемые в данном документе в контексте способа по настоящему изобретению, предусматривающему комбинированное введение терапевтически эффективных количеств соединения формулы (Id) и другого соединения, которое является применимым в лечении нейродегенеративного заболевания или нарушения, подразумевают введение соединения формулы (Id) одновременно или последовательно в любом порядке вместе с указанным вторым соединением.
Два соединения можно вводить одновременно или с временным промежутком между введениями двух соединений. Два соединения можно вводить либо как часть одних и тех же фармацевтических состава или композиции, либо в виде отдельных фармацевтических составов или композиций. Два соединения можно вводить в один и тот же день или в различные дни. Их можно вводить с помощью одного и того же пути введения, как например путем перорального введения, подкожной инъекции, путем трансдермального введения, путем введения препарата-депо, путем внутримышечной инъекции или внутривенной инъекции, или с помощью различных путей, где одно соединение, например, вводят перорально или размещают в виде препарата-депо, а другое соединение, например, вводят инъекцией. Два соединения можно вводить в соответствии с одними и теми же схемой или интервалом введения доз, как например один раз или два раза в сутки, один раз в неделю или один раз в месяц; или в соответствии с разными схемами введения доз, например, где одно вводят один раз в сутки, а другое вводят два раза в сутки, или один раз в неделю, или один раз в месяц.
В некоторых случаях пациент, подлежащий лечению, уже может проходить лечение с помощью одного или нескольких других соединений, применимых в лечении нейродегенеративного заболевания или нарушения, когда начинают лечение с помощью соединения формулы (Id). В других случаях пациент может уже проходить лечение с помощью соединения формулы (Id), когда начинают лечение с помощью одного или нескольких других соединений, применимых в лечении нейродегенеративного заболевания или нарушения. В других случаях лечение с помощью соединения формулы (Id) и лечение с помощью одного или нескольких других соединений, применимых в лечении нейродегенеративного заболевания или нарушения, начинают одновременно.
Соединения для комбинированного лечения
В контексте настоящего изобретения соединения, подлежащие применению в комбинации с соединением формулы (Id), могут быть выбраны из, например, L-DOPA, дроксидопы, ингибиторов МАО-В, таких как селегилин или разагилин, ингибиторов СОМТ, таких как энтакапон или толкапон, антагонистов аденозиновых 2а-рецепторов, таких как истрадефиллин, антиглутаматергических средств, таких как амантадин или мемантин, ингибиторов ацетилхолинэстеразы, таких как ривастигмин, донепезил или галантамин, и антипсихотических средств, таких как кветиапин, клозапин, рисперидон, пимавансерин, оланзапин, галоперидол, арипипразол или брекспипразол.
В дополнение к малым молекулам соединения для применения в комбинации также могут включать новые основанные на применении биологических препаратов подходы в способах лечения нейродегенеративных заболеваний или нарушений, такие как, например, применение антител, нацеленных на белок альфа-синуклеин, тау-белок или белок А-бета.
Пути введения
Фармацевтические композиции, содержащие соединение формулы (Id) либо в виде единственного активного соединения, либо в комбинации с другим активным соединением, могут быть, в частности, составлены для введения любым подходящим путем, как например пероральный, ректальный, назальный, трансбуккальный, сублингвальный, пульмональный, трансдермальный и парентеральный (например, подкожный, внутримышечный и внутривенный) пути. В контексте настоящего изобретения пероральный путь является предпочтительным путем введения.
Следует принять во внимание, что путь будет зависеть от общего состояния и возраста субъекта, подлежащего лечению, природы патологического состояния, подлежащего лечению, и активного ингредиента.
Фармацевтические составы и вспомогательные вещества
Далее термин "вспомогательное вещество" или "фармацевтически приемлемое вспомогательное вещество" относится к фармацевтическим вспомогательным веществам, в том числе без ограничения носителям, наполнителям, разбавителям, средствам против прилипания, связующим, покрытиям, красителям, разрыхлителям, ароматизаторам, веществам, улучшающим скольжение, смазывающим средствам, консервантам, сорбентам, подсластителям, растворителям, средам-носителям и вспомогательным средствам.
В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая соединение формулы (Id), такое как одно из соединений, раскрытых в экспериментальном разделе в данном документе. В настоящем изобретении также предусмотрен способ получения фармацевтической композиции, содержащей соединение формулы (Id). Фармацевтические композиции в соответствии с настоящим изобретением можно составлять с фармацевтически приемлемыми вспомогательными веществами в соответствии с традиционными методиками, такими как раскрытые в Remington, "The Science and Practice of Pharmacy", 22th edition (2012), Edited by Allen, Loyd V., Jr.
Фармацевтическая композиция, содержащая соединение по настоящему изобретению, предпочтительно представляет собой фармацевтическую композицию для перорального введения. Фармацевтические композиции для перорального введения включают твердые пероральные лекарственные формы, такие как таблетки, капсулы, порошки и гранулы; и жидкие пероральные лекарственные формы, такие как растворы, эмульсии, суспензии и сиропы, а также порошки и гранулы для растворения или суспендирования в подходящей жидкости.
Твердые пероральные лекарственные формы могут быть представлены в виде отдельных единиц (например, таблеток или твердых или мягких капсул), каждая из которых содержит предварительно определенное количество активного ингредиента и предпочтительно одно или несколько подходящих вспомогательных веществ. При необходимости твердые лекарственные формы можно покрывать оболочкой разных типов, таких как энтеросолюбильные оболочки, или их можно составлять так, чтобы обеспечивать регулируемое высвобождение активного ингредиента, такое как замедленное или пролонгированное высвобождение, в соответствии со способами, хорошо известными из уровня техники. При необходимости твердая лекарственная форма может представлять собой лекарственную форму, распадающуюся под действием слюны, такую как, например, таблетка, диспергируемая в полости рта.
Примеры вспомогательных веществ, пригодных для перорального твердого состава, включают без ограничения микрокристаллическую целлюлозу, кукурузный крахмал, лактозу, маннит, повидон, кроскармеллозу натрия, сахарозу, циклодекстрин, тальк, желатин, пектин, стеарат магния, стеариновую кислоту и низшие алкиловые простые эфиры целлюлозы. Аналогичным образом, твердый состав может включать вспомогательные вещества для составов с замедленным или пролонгированным высвобождением, известные в области техники, такие как глицерилмоностеарат и гипромеллоза. Если для перорального введения применяют твердый материал, то состав можно получать, например, путем смешивания активного ингредиента с твердыми вспомогательными веществами, а затем прессования смеси в стандартной таблетирующей машине; или состав, например, в виде порошка, гранулы или мини таблетки, может быть помещен, например, в твердую капсулу. Количество твердого вспомогательного вещества в стандартной дозе будет существенно варьировать, но, как правило, будет находиться в пределах от приблизительно 25 мг до приблизительно 1 г.
Жидкие пероральные лекарственные формы могут быть представлены в виде, например, настоек, сиропов, капель для перорального применения или наполненных жидкостью капсул. Жидкие пероральные лекарственные формы также могут быть представлены в виде порошков для растворения или суспендирования в водной или неводной жидкости. Примеры вспомогательных веществ, пригодных для перорального жидкого состава, включают без ограничения этанол, пропиленгликоль, глицерин, полиэтиленгликоли, полоксамеры, сорбит, полисорбат, моно- и диглицериды, циклодекстрины, кокосовое масло, пальмовое масло и воду. Жидкие пероральные лекарственные формы можно получать, например, растворением или суспендированием активного ингредиента в водной или неводной жидкости, или путем включения активного ингредиента в жидкую эмульсию типа "масло-в-воде" или "вода-в-масле".
В твердых и жидких пероральных составах могут использоваться дополнительные вспомогательные вещества, такие как красители, вкусовые добавки и консерванты и т.п.
Фармацевтические композиции для парентерального введения включают стерильные водные и неводные растворы, дисперсии, суспензии или эмульсии для инъекции или вливания, концентраты для инъекции или вливания, а также стерильные порошки, подлежащие ресуспендированию в стерильных растворах или дисперсиях для инъекции или вливания перед применением. Примеры вспомогательных веществ, пригодных для парентерального состава, включают без ограничения воду, кокосовое масло, пальмовое масло и растворы циклодекстринов. Водные составы должны быть подходящим образом забуферены, если необходимо, и приведены в состояние изотоничности с помощью достаточного количества соляного раствора или глюкозы.
Другие типы фармацевтических композиций включают суппозитории, ингаляторы, кремы, гели, кожные пластыри, имплантаты и составы для трансбуккального или подъязычного введения.
Необходимо, чтобы вспомогательные вещества, используемые для любого фармацевтического состава, соответствовали предполагаемому пути введения и были совместимы с активными ингредиентами.
Дозы
В одном варианте осуществления соединение по настоящему изобретению вводят в количестве от приблизительно 0,0001 мг/кг массы тела до приблизительно 5 мг/кг массы тела в день. В частности, ежедневная дозировка может находиться в диапазоне от 0,001 мг/кг массы тела до приблизительно 1 мг/кг массы тела в день. Точная дозировка будет зависеть от частоты и способа введения, пола, возраста, веса и общего состояния подлежащего лечению субъекта, природы и тяжести подлежащего лечению состояния и каких-либо сопутствующих подлежащих лечению заболеваний, предполагаемого эффекта лечения, а также других факторов, известных специалистам в данной области.
Типичная дозировка для перорального введения взрослым будет находится в диапазоне 0,01-100 мг/день соединения по настоящему изобретению, как например 0,05-50 мг/день, как например 0,1-10 мг/день или 0,1-5 мг/день. Для удобства соединения по настоящему изобретению вводят в единичной лекарственной форме, содержащей указанные соединения в количестве от приблизительно 0,01 до 50 мг, как например 0,05 мг, 0,1 мг, 0,2 мг, 0,5 мг, 1 мг, 5 мг, 10 мг, 15 мг, 20 мг или не более 50 мг, соединения по настоящему изобретению.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фигура 1: графическое изображение равновесного состояния конъюгации и деконъюгации в организме между соединением (I) и соединениями (Id-ia), (Id-ib), (Id-iia) и (Id-iib).
Фигура 2: PK-профили у крыс линии Wistar, полученные после перорального введения дозы в соответствии с примером 4. Профили получены на основе средних значений концентрации в плазме крови у 3 субъектов для каждого соединения.
Ось X: время (часы); ось Y: концентрация в плазме крови соединения (I) (пг/мл), полученная после введения дозы следующих соединений: соединения (Ia)  соединения (Ib); : соединения (Id-ia); X: соединения (Id-ib); соединения (Id-iia); +: соединения (Id-iib), X: соединения (Id-iab) и
соединения (Ib); : соединения (Id-ia); X: соединения (Id-ib); соединения (Id-iia); +: соединения (Id-iib), X: соединения (Id-iab) и  соединения (Id-iiab).
соединения (Id-iiab).
Фигуры 3 и 4: изменение локомоторной активности с течением времени (фигура 3) и общее пройденное расстояние (фигура 4) после обработки с помощью среды-носителя (Н2О, р.о.) или соединения (Id-ia) (10, 30, 100 или 300 мкг/кг, р.о.) и сравнение с видами обработки с помощью стандарта лечения (SoC): апоморфина (АРО, 3 мг/кг, s.c.), прамипексола (РРХ, 0,3 мг/кг, s.c.). Животным вводили дозу при t=60 минут после 60-мин. периода привыкания в испытательных камерах, и затем контролировали активность в течение 350 минут. Данные оценивали с применением критерия Крускала-Уоллиса с тестом множественных сравнений Данна, в результате чего получали общее значение Р<0,0001.
Фигура 3: ось X: время (мин.); ось Y: пройденное расстояние (см) ± SEM/5-минутные интервалы.
Фигура 4: ось Y: общее пройденное расстояние (см) ± SEM. Указаны уровни значимости для апостериорных сравнений (относительно группы, обработанной средой-носителем): *<0,05, **<0,01, ***<0,001, ****<0,0001.
Фигуры 5 и 6: Взаимосвязь между значениями концентрации в плазме крови соединения (Id-ia) и соединения (I) и гиперактивностью, вызванной соединением (Id-ia) (100 мкг/кг, р.о.) (фигура 5), и соответствующая взаимосвязь между значениями концентрации в плазме крови апоморфина и гиперактивностью, вызванной апоморфином (3 мг/кг, s.c.) (фигура 6).
Ось X: время (мин.); ось Y слева: пройденное расстояние (см) ± SEM/5-минутные интервалы; ось Y справа (фигура 5): концентрация в плазме крови соединения (I) (пг/мл); ось Y справа (фигура 6): концентрация в плазме апоморфина (нг/мл).
□: пройденное расстояние (см), : концентрация в плазме крови.
Фигура 7: превращение соединений (Id-ia), (Id-ib), (Id-iia), (Id-iib) и (Id-iab) в соединение (I) в гепатоцитах крысы (7а) и человека (7b).
Ось X: время (мин.); ось Y: концентрация соединения (I) (пг/мл).
: соединение (Id-ia); X: соединение (Id-ib);  соединение (Id-iia); +: соединение (Id-iib); : соединение (Id-iab).
соединение (Id-iia); +: соединение (Id-iib); : соединение (Id-iab).
Фигура 8: превращение соединений (Id-ia), (Id-ib), (Id-iia) и (Id-iib) в соединение (I) в цельной крови крысы (8а) и человека (8b).
Ось X: время (мин.); ось Y: концентрация соединения (I) (пг/мл).
: соединение (Id-ia); X: соединение (Id-ib); соединение (Id-iia); 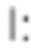 соединение (Id-iib).
соединение (Id-iib).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения идентифицировали новые соединения, которые являются пролекарствами (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола [соединения (I)], который является двойным агонистом D1/D2, характеризующиеся данными in vitro, приведенными в таблице 2.
Авторы настоящего изобретения обнаружили, что соединение (I) подвергается конъюгированию в гепатоцитах крысы и человека с образованием глюкуронидных производных (Id-ia) и (Id-ib) и сульфатных производных (Id-iia) и (Id-iib). Было показано, что конъюгаты превращаются в соединение (I) путем конъюгации и деконъюгации в организме, как показано на фиг. 1.
Известно, что глюкуронидные и сульфатные производные нестабильны в кишечнике. Производные образуются в виде высокополярных и растворимых метаболитов для облегчения элиминации соединений из организма и впоследствии легко выводятся из организма. Например, у крыс с канюлированным желчным протоком глюкуронидные и сульфатные конъюгаты часто обнаруживаются в желчи, тогда как их деконъюгат (т.е. исходное соединение) обнаруживается в фекалиях. Обратное превращение глюкуронидных и сульфатных конъюгатов в кишечнике в исходное соединение, которое затем время от времени реабсорбируется, известно как часть процесса кишечно-печеночной рециркуляции. Как упоминалось ранее, пероральное введение дозы фенэтилкатехоламинов, как например апоморфин, в целом оказалось безрезультатным из-за их низкой биодоступности. Аналогично, соединение (I) имеет недостаток в виде низкой пероральной биодоступности (Liu et al., Bioorganic Med. Chem. (2008), 16: 3438-3444). Учитывая все вышеперечисленное и принимая во внимание нестабильность глюкуронидных и сульфатных конъюгатов в желудочно-кишечном тракте, не следует ожидать, что пероральное введение дозы глюкуронидных и сульфатных конъюгатов соединения (I) можно использовать для достижения достаточной концентрации соединения в плазме крови.
Принцип применения глюкуронидных производных в качестве пролекарств для пероральной доставки был исследован для ретиноевой кислоты (Goswami et al., J. Nutritional Biochem. (2003) 14: 703-709) и для морфина (Stain-Texier et al., Drug Metab. and Disposition (1998) 26 (5): 383-387). Оба исследования показали очень низкие уровни концентрации исходных соединений после перорального введения дозы производных. Другое исследование предполагает применение буденозид-β-D-глюкуронида в качестве пролекарства для местной доставки буденозида в толстый кишечник для лечения язвенного колита исходя из плохого всасывания пролекарства самого по себе из пищеварительной системы (Nolen et al., J. Pharm Sci. (1995), 84 (6): 677-681).
Тем не менее, неожиданно авторы настоящего изобретения обнаружили, что пероральное введение дозы глюкуронидных конъюгатов (Id-ia), (Id-ib) и сульфатных конъюгатов (Id-iia) и (Id-iib), все из которых были идентифицированы как метаболиты соединения (I), у крыс и карликовых свиней обеспечивало системную концентрацию соединения (I) в плазме крови, что свидетельствует о применимости глюкуронидных и сульфатных производных соединения (I) в качестве активных при пероральном применении пролекарств соединения (I). Авторы настоящего изобретения дополнительно исследовали соединения (Id-iab) и (Id-iiab), каждое из которых замещено либо глюкуронидом, либо сульфатом при обеих гидроксильных группах катехола, и обнаружили, что эти два соединения также проявляют пролекарственную активность.
Профили концентрации в плазме крови соединения (I), полученные после перорального введения дозы соединений (Ia) и (Ib) и каждого из соединений (Id-ia), (Id-ib), (Id-iia), (Id-iib), (Id-iab) и (Id-iiab) крысам линии Wistar в соответствии с примером 4, показаны на фигуре 2. Для всех соединений дозы скорректировали по молекулярной массе до дозы, равной 300 мкг/кг соединения (Ib), соответствующей 287 мкг/кг соединения (I). Авторы настоящего изобретения обнаружили, что пероральное введение дозы соединений (Ia) и (Ib) крысам линии Wistar обеспечивает ранние и высокие пиковые концентрации соединения (I). Такие высокие пиковые концентрации у людей могут быть ассоциированы с дофаминергическими побочными эффектами, такими как, например, тошнота, рвота и головокружение. С другой стороны, введение дозы соединений (Id-ia), (Id-ib), (Id-iia), (Id-iib); (Id-iab) и (Id-iiab) обеспечивает более медленную скорость всасывания с избеганием быстрых пиковых концентраций, сопровождаемую устойчивой концентрацией соединения (I) в плазме крови. Кроме того, концентрация в плазме крови соединения (I) у крыс линии Wistar сохраняется в течение 24 часов, хотя полученное значение AUC соединения (I), как правило, ниже, чем значение AUC, полученное после введения дозы соединения (Ib). Однако, поскольку пиковые концентрации соединения (I), которые, как ожидается, будут вызывать побочные эффекты, являются более низкими, можно вводить более высокие дозы соединений (Id-ia), (Id-ib), (Id-iia), (Id-iib), (Id-iab) и (Id-iiab) для потенциального достижения более высоких общих концентраций в плазме крови соединения (I) по сравнению с достигаемыми после введения дозы соединений (Ia) и (Ib). При исследовании PK-свойств соединения (Ic) авторы настоящего изобретения обнаружили, что концентрации в плазме крови соединения (I) были чрезвычайно низкими, что делало соединение (Ic) непригодным в качестве пролекарства соединения (I) для перорального введения и подтверждало, что пероральная биодоступность соединений по настоящему изобретению является крайне непредсказуемой. PK-параметры для PK-исследований у крыс линии Wistar перечислены в таблице 3.
PK-эксперименты также были проведены с пероральным введением дозы соединений (Id-ia), (Id-ib), (Id-iia) и (Id-iib) карликовым свиньям в соответствии с примером 5. Исследование продемонстрировало, что все четыре соединения превращаются в соединение (I) в организме карликовых свиней и обеспечивают содержание в плазме крови соединения (I) после перорального введения дозы. PK параметры для данного исследования перечислены в таблице 4.
Биоконверсия соединений (Id-ia), (Id-ib), (Id-iia), (Id-iib) и (Id-iab) у человека подтверждается экспериментами из примера 1, указывающими на превращение соединений в соединение формулы (I) в гепатоцитах крысы и человека, а для (Id-ia), (Id-ib), (Id-iia), (Id-iib) в крови крысы и человека (фигуры 7 и 8).
Таким образом, в заключение, соединения по настоящему изобретению являются применимыми в качестве активных при пероральном применении пролекарств соединения (I), и наблюдалось, что у крыс они обеспечивали PK-профиль с избежанием пика Cmax, наблюдаемого в случае известных пролекарств (Ia) и (Ib), и обеспечивали значительно более высокое значение AUC соединения (I), чем соединения (Ic). Предпочтительными соединениями по настоящему изобретению являются глюкуронидные конъюгаты (Id-ia), (Id-ib) и (Id-iab).
В качестве сравнительного примера крысам линии Wistar вводили дозу одного глюкуронидного и двух сульфатных конъюгатов апоморфина ((2S,3S,4S,5R,6S)-6-[[(6aR)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ил]окси]-3,4,5-тригидрокситетрагидропиран-2-карбоновой кислоты; [(6aR)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ил]гидросульфата и [(6aR)-10-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-11-ил]гидросульфата). Введение дозы конъюгатов апоморфина перорально крысе линии Wistar при дозах до 4977 мкг/кг не обеспечивало значительной концентрации апоморфина в плазме крови (нижний предел количественного определения 500 пг/мл), за исключением 916 пг/мл в один момент времени (4 ч.) после введения дозы глюкуронидного конъюгата, что указывает на низкую пероральную биодоступность конъюгатов апоморфина или ее отсутствие. Для сравнения, пероральное введение дозы 3000 мкг/кг апоморфина самого по себе обеспечивало значение AUC в плазме крови в >100 раз ниже, чем наблюдаемое после подкожного введения 3000 мкг/кг апоморфина, что подтверждает известную плохую пероральную биодоступность апоморфина. Это дополнительно подтверждает, что пероральная доступность соединений по настоящему изобретению является крайне неожиданной (в отношении экспериментальной части см. пример 4).
Соединение (Id-ia) дополнительно исследовали в анализе локомоторной активности крыс в соответствии с примером 6. Анализ продемонстрировал дофаминергический эффект, полученный после перорального введения соединения (Id-ia), см. также фигуры 3, 4 и 5. Факт того, что соединения по настоящему изобретению, в том числе (Id-ia), не обладают дофаминергической активностью in vitro, см. также пример 2 и таблицу 1, дополнительно указывает на то, что эффект соединения (Id-ia) в анализе локомоторной активности крыс достигается путем превращения соединения (Id-ia) в соединение (I).
В заключение, важной проблемой, связанной с соединением (Ib) из уровня техники, является то, что данное соединение является агонистом рецептора 5-НТ2В. Поскольку агонисты рецептора 5-НТ2В связаны с патогенезом порока клапана сердца (VHD) после их длительного воздействия, такие соединения не подходят для применения в лечении хронических заболеваний (Rothman et al., Circulation (2000), 102: 2836-2841; and Cavero and Guillon, J. Pharmacol. Toxicol. Methods (2014), 69: 150-161). Таким образом, дополнительное преимущество соединений по настоящему изобретению заключается в том, что они не являются агонистами 5-НТ2В, см. также пример 3 и таблицу 1.
Соединения по настоящему изобретению являются пригодными в лечении нейродегенеративных заболеваний и нарушений, как например болезнь Паркинсона, и/или других состояний, для которых лечение с помощью агониста дофамина является терапевтически благоприятным. Соединения, подходящие для перорального введения, имеют потенциал в обеспечении новых стандартных способов лечения при болезни Паркинсона.
В одном варианте осуществления настоящего изобретения соединения предназначены для применения в качестве используемого самостоятельно терапевтического средства для нейродегенеративного заболевания или нарушения. В другом варианте осуществления настоящего изобретения соединения подлежат применению в комбинации с другими средствами для лечения PD, такими как соединение, выбранное из группы, состоящей из L-DOPA, ингибитора МАО-В, такого как селегилин или разагилин, ингибитора СОМТ, такого как энтакапон или толкапон, антагониста аденозиновых 2а-рецепторов, такого как истрадефиллин, антиглутаматергического средства, такого как амантадин или мемантин, ингибитора ацетилхолинэстеразы, такого как ривастигмин, донепезил или галантамин, антипсихотического средства, такого как кветиапин, клозапин, рисперидон, пимавансерин, оланзапин, галоперидол, арипипразол или брекспипразол; или в комбинации в антителом, нацеленным на белок альфа-синуклеин, тау-белок или белок А-бета.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Раскрыты следующие варианты осуществления настоящего изобретения. Первый вариант осуществления обозначен как Е1, второй вариант осуществления обозначен как Е2 и так далее.
Е1. Соединение формулы (Id),
где
R1 представляет собой Н, и R2 выбран из одного из заместителей (i) и (ii), указанных ниже; или
R1 выбран из одного из заместителей (i) и (ii), указанных ниже, и R2 представляет собой Н; или
R1 и R2 одновременно представлены заместителем (i), указанным ниже; или
R1 и R2 одновременно представлены заместителем (ii), указанным ниже; или
R1 представляет собой заместитель (i), и R2 представляет собой заместитель (ii); или
R1 представляет собой заместитель (ii), и R2 представляет собой заместитель (i);
где * обозначает точку присоединения; и
при этом атом углерода в точке присоединения при заместителе (i) находится в S-конфигурации;
или его фармацевтически приемлемая соль.
Е2. Соединение или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 1, где
R1 представляет собой Н, и R2 выбран из одного из заместителей (i) и (ii); или
R1 выбран из одного из заместителей (i) и (ii), и R2 представляет собой Н; или
R1 и R2 одновременно представлены заместителем (i); или
R1 и R2 одновременно представлены заместителем (ii).
Е3. Соединение или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 1, где R1 представляет собой Н, и R2 представляет собой заместитель (i); или
R1 представляет собой заместитель (i), и R2 представляет собой Н; или
R1 и R2 одновременно представлены заместителем (i).
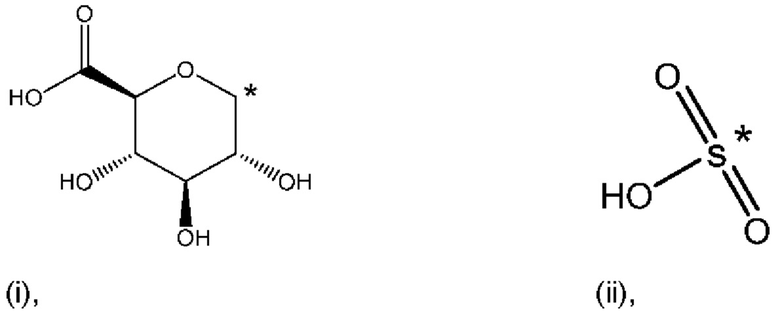
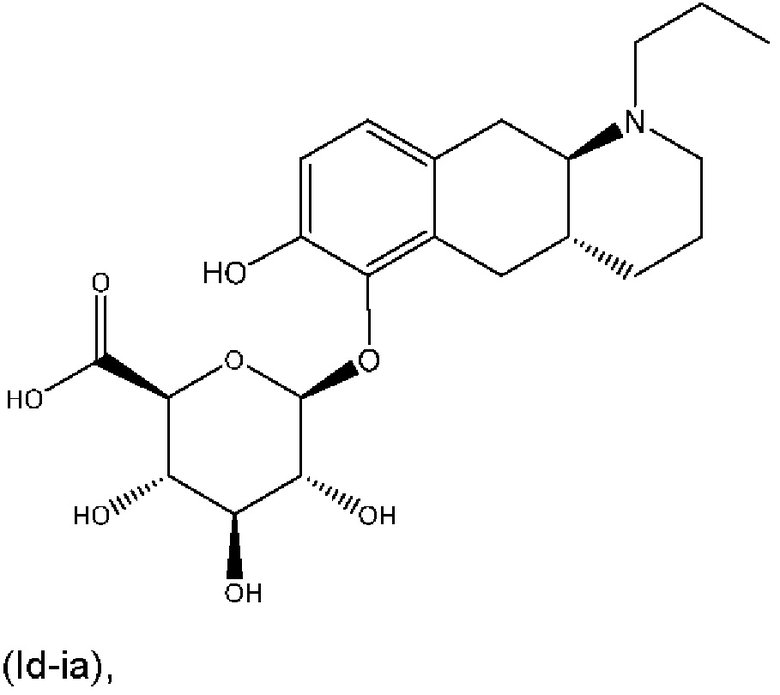
Е4. Соединение в соответствии с вариантом осуществления 1, где указанное соединение представляет собой соединение, представленное формулой (Id-ia), приведенной ниже,
или его фармацевтически приемлемая соль.
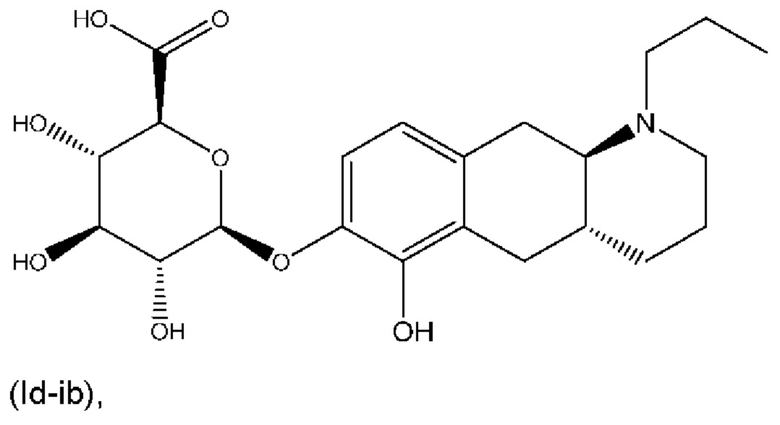
Е5. Соединение в соответствии с вариантом осуществления 1, где указанное соединение представляет собой соединение, представленное формулой (Id-ib), приведенной ниже,
или его фармацевтически приемлемая соль.
Е6. Соединение в соответствии с вариантом осуществления 1, где указанное соединение представляет собой соединение, представленное формулой (Id-iab), приведенной ниже,
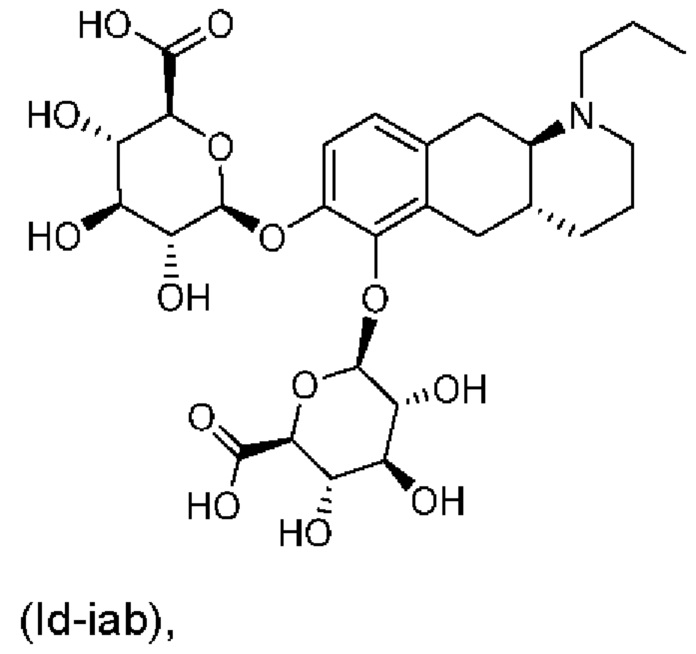
или его фармацевтически приемлемая соль.
Е7. Соединение или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 1, где R1 представляет собой Н, и R2 представляет собой заместитель (ii); или
R1 представляет собой заместитель (ii), и R2 представляет собой Н; или
R1 и R2 одновременно представлены заместителем (ii).
Е8. Соединение в соответствии с вариантом осуществления 1, где указанное соединение представляет собой соединение, представленное формулой (Id-iia), приведенной ниже,
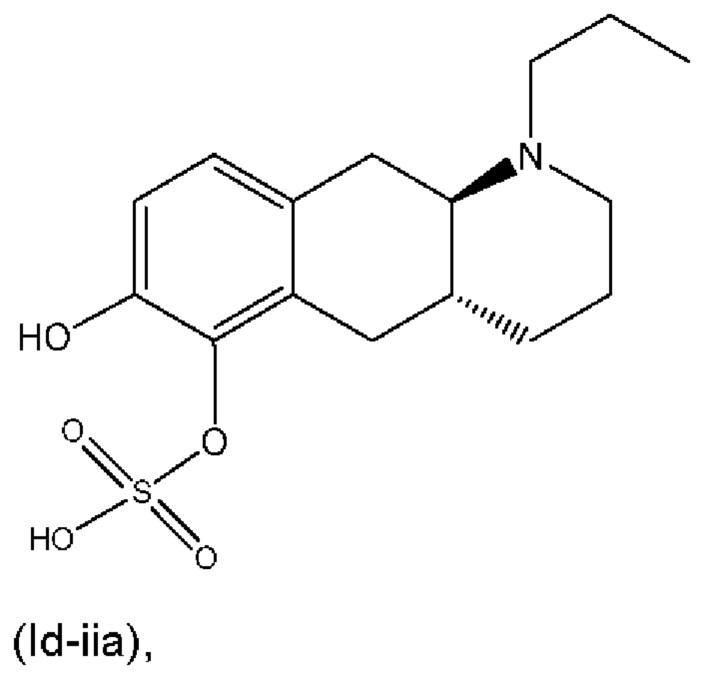
или его фармацевтически приемлемая соль.
Е9. Соединение в соответствии с вариантом осуществления 1, где указанное соединение представляет собой соединение, представленное формулой (Id-iib), приведенной ниже,
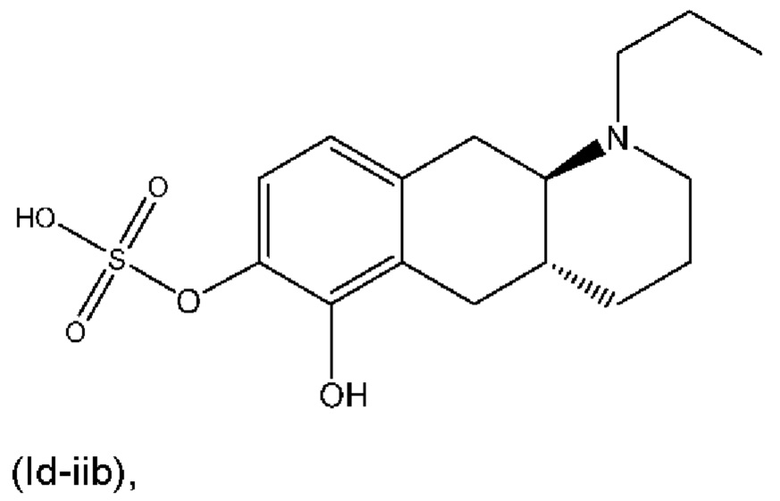
или его фармацевтически приемлемая соль.
Е10. Соединение в соответствии с вариантом осуществления 1, где указанное соединение представляет собой соединение, представленное формулой (Id-iiab), приведенной ниже,
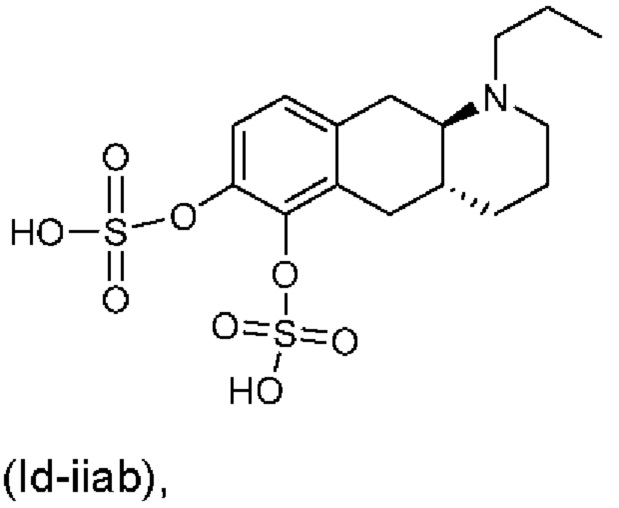
или его фармацевтически приемлемая соль.
Е11. Соединение в соответствии с вариантом осуществления 1, где соединение выбрано из группы, состоящей из:
(Id-ia): (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR,10aR)-7-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)окси)тетрагидро-2Н-пиран-2-карбоновой кислоты;
(Id-ib): (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR,10aR)-6-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)окси)тетрагидро-2Н-пиран-2-карбоновой кислоты;
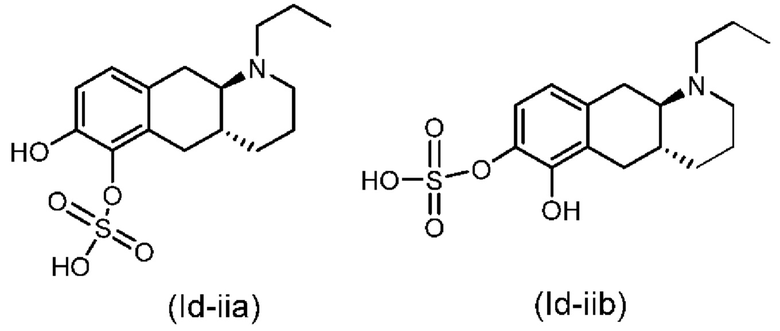
(Id-iia): (4aR,10aR)-7-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-илгидросульфата;
(Id-iib): (4aR,10aR)-6-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)гидросульфата;
(Id-iab): (2S,2'S,3S,3'S,4S,4'S,5R,5'R,6S,6'S)-6,6'-(((4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диил)бис(окси))бис(3,4,5-тригидрокситетрагидро-2Н-пиран-2-карбоновая кислота);
(Id-iiab): (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диил-бис(гидросульфат);
или фармацевтически приемлемая соль любого из этих соединений.
Е12. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-11, где указанное соединение получено вне организма млекопитающего.
Е13. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-12, где указанное соединение изготовлено путем химического получения.
Е14. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-13, где указанное соединение представлено в выделенной форме.
Е15. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-14, где указанное соединение представлено в выделенной форме, по сути не предусматривающей присутствия соединений, с которыми оно в естественных условиях находится в состоянии равновесия.
Е16. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-15, где указанное соединение представлено в выделенной форме, по сути не предусматривающей присутствия соединения формулы (I).
Е17. Соединение, которое представляет собой пролекарство соединения (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола (соединения (I)), где указанное пролекарство обеспечивает PK-профиль, где Cmax (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола составляет от 500 до 2500 пг/мл, как например от 750 до 2500 пг/мл, как например от 1000 до 2500 пг/мл, как например от 1000 до 2000 пг/мл, если указанное пролекарство вводят перорально крысе линии Wistar в дозе, соответствующей 287 мкг/кг (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола;
или фармацевтически приемлемая соль указанного соединения.
Е18. Соединение или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 17, которое представляет собой пролекарство соединения (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола (соединения (I)), где указанное пролекарство обеспечивает PK-профиль, где AUC0-∞ (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола составляет более 7000 пг*ч./мл, как например более 8000, как например более 9000, как например более 10000, как например более 11000, как например более 12000, как например более 13000, как например более 14000, как например более 15000, как например более 16000 пг*ч./мл, если указанное пролекарство вводят перорально крысе линии Wistar в дозе, соответствующей 287 мг/кг (4aR,10aR)-1-н-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола.
Е19. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 17-18, где указанный PK-профиль был получен с помощью PK-эксперимента, как описано в примере 4 в данном документе.
Е20. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-19, где указанные соединение или его фармацевтически приемлемая соль представлены в твердой форме.
Е21. Фармацевтически приемлемая соль соединения в соответствии с любым из вариантов осуществления 1-20.
Е22. Фармацевтически приемлемая соль в соответствии с вариантом осуществления 21, где указанная соль представляет собой соль присоединения кислоты соединения в соответствии с любым из вариантов осуществления 1-20.
Е23. Фармацевтически приемлемая соль в соответствии с вариантом осуществления 21, где указанная соль представляет собой соль присоединения основания соединения в соответствии с любым из вариантов осуществления 1-20.
Е24. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-23 для применения в терапии.
Е25. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-23 для применения в качестве лекарственного препарата.
Е26. Соединение или фармацевтически приемлемая соль для применения в качестве лекарственного препарата в соответствии с вариантом осуществления 25, где указанный лекарственный препарат представляет собой лекарственный препарат для перорального применения, такой как таблетка или капсула для перорального введения.
Е27. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли в соответствии с любым из вариантов осуществления 1-23 и одно или несколько фармацевтически приемлемых вспомогательных веществ.
Е28. Фармацевтическая композиция в соответствии с вариантом осуществления 27, где указанная фармацевтическая композиция предназначена для перорального введения.
Е29. Фармацевтическая композиция в соответствии с любым из вариантов осуществления 27-28, где указанная фармацевтическая композиция представляет собой фармацевтическую композицию для перорального применения.
Е30. Фармацевтическая композиция в соответствии с любым из вариантов осуществления 27-29, где указанная фармацевтическая композиция представляет собой твердую пероральную лекарственную форму.
Е31. Фармацевтическая композиция в соответствии с любым из вариантов осуществления 27-30, где указанная фармацевтическая композиция представляет собой таблетку или капсулу для перорального введения.
Е32. Фармацевтическая композиция в соответствии с любым из вариантов осуществления 27-31, где указанная фармацевтическая композиция дополнительно содержит другое средство, которое применяется в лечении нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона.
Е33. Фармацевтическая композиция в соответствии с любым из вариантов осуществления 27-31, где указанная фармацевтическая композиция дополнительно содержит соединение, выбранное из группы, состоящей из L-DOPA, ингибитора МАО-В, такого как селегилин или разагилин, ингибитора СОМТ, такого как энтакапон или толкапон, антагониста аденозиновых 2а-рецепторов, такого как истрадефиллин, антиглутаматергического средства, такого как амантадин или мемантин, ингибитора ацетилхолинэстеразы, такого как ривастигмин, донепезил или галантамин, антипсихотического средства, такого как кветиапин, клозапин, рисперидон, пимавансерин, оланзапин, галоперидол, арипипразол или брекспипразол; или антитела, нацеленного на белок альфа-синуклеин, тау-белок или белок А-бета.
Е34. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-23 для применения в лечении нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона, болезнь Хантингтона, синдром беспокойных ног или болезнь Альцгеймера; или нейропсихиатрического заболевания или нарушения, такого как шизофрения, синдром дефицита внимания с гиперактивностью или наркотическая зависимость.
Е35. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-23 для применения в лечении в соответствии с вариантом осуществления 34, где указанное нейродегенеративное заболевание или нарушение представляет собой болезнь Паркинсона.
Е36. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-23 для применения в лечении в соответствии с любым из вариантов осуществления 34-35, где указанное соединение подлежит применению в комбинации с другим средством, которое является применимым в лечении нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона.
Е37. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-23 для применения в лечении в соответствии с любым из вариантов осуществления 34-35, где указанное соединение подлежит применению в комбинации с соединением, выбранным из группы, состоящей из L-DOPA, ингибитора МАО-В, такого как селегилин или разагилин, ингибитора СОМТ, такого как энтакапон или толкапон, антагониста аденозиновых 2а-рецепторов, такого как истрадефиллин, антиглутаматергического средства, такого как амантадин или мемантин, ингибитора ацетилхолинэстеразы, такого как ривастигмин, донепезил или галантамин, антипсихотического средства, такого как кветиапин, клозапин, рисперидон, пимавансерин, оланзапин, галоперидол, арипипразол или брекспипразол; или в комбинации с антителом, нацеленным на белок альфа-синуклеин, тау-белок или белок А-бета.
Е38. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-23 для применения в лечении в соответствии с любым из вариантов осуществления 34-37, где указанное лечение проводят путем перорального введения указанного соединения.
Е39. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-23 для применения в лечении в соответствии с любым из вариантов осуществления 34-38, где указанное соединение содержится в фармацевтической композиции для перорального применения, такой как таблетка или капсула для перорального введения.
Е40. Способ лечения нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона, болезнь Хантингтона, синдром беспокойных ног или болезнь Альцгеймера; или нейропсихиатрического заболевания или нарушения, такого как шизофрения, синдром дефицита внимания с гиперактивностью или наркотическая зависимость; при этом способ включает введение терапевтически эффективного количества соединения или его фармацевтически приемлемой соли в соответствии с любым из вариантов осуществления 1-23 пациенту, нуждающемуся в этом.
Е41. Способ в соответствии с вариантом осуществления 40, где указанное нейродегенеративное заболевание или нарушение представляет собой болезнь Паркинсона.
Е42. Способ в соответствии с любым из вариантов осуществления 40-41, где указанное соединение или его фармацевтически приемлемую соль в соответствии с любым из вариантов осуществления 1-23 применяют в комбинации с другим средством, которое является применимым в лечении нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона.
43. Способ в соответствии с любым из вариантов осуществления 40-41, где указанное соединение или его фармацевтически приемлемую соль в соответствии с любым из вариантов осуществления 1-23 применяют в комбинации с соединением, выбранным из группы, состоящей из L-DOPA, ингибитора МАО-В, такого как селегилин или разагилин, ингибитора СОМТ, такого как энтакапон или толкапон, антагониста аденозиновых 2а-рецепторов, такого как истрадефиллин, антиглутаматергического средства, такого как амантадин или мемантин, ингибитора ацетилхолинэстеразы, такого как ривастигмин, донепезил или галантамин, антипсихотического средства, такого как кветиапин, клозапин, рисперидон, пимавансерин, оланзапин, галоперидол, арипипразол или брекспипразол; или в комбинации с антителом, нацеленным на белок альфа-синуклеин, тау-белок или белок А-бета.
Е44. Способ в соответствии с любым из вариантов осуществления 40-43, где указанное введение проводят пероральным путем.
Е45. Способ в соответствии с любым из вариантов осуществления 40-44, где указанное соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 1-23 содержится в фармацевтической композиции для перорального применения, такой как таблетка или капсула для перорального введения.
Е46. Применение соединения или его фармацевтически приемлемой соли в соответствии с любым из вариантов осуществления 1-23 в получении лекарственного препарата, предназначенного для лечения нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона, болезнь Хантингтона, синдром беспокойных ног или болезнь Альцгеймера; или для лечения нейропсихиатрического заболевания или нарушения, такого как шизофрения, синдром дефицита внимания с гиперактивностью или наркотическая зависимость.
Е47. Применение в соответствии с вариантом осуществления 46, где указанное нейродегенеративное заболевание или нарушение представляет собой болезнь Паркинсона.
Е48. Применение в соответствии с любым из вариантов осуществления 46-47, где указанный лекарственный препарат применяют в комбинации с другим средством, которое является применимым в лечении нейродегенеративного заболевания или нарушения, такого как болезнь Паркинсона.
Е49. Применение в соответствии с любым из вариантов осуществления 46-47, где указанный лекарственный препарат применяют в комбинации с соединением, выбранным из группы, состоящей из L-DOPA, ингибитора МАО-В, такого как селегилин или разагилин, ингибитора СОМТ, такого как энтакапон или толкапон, антагониста аденозиновых 2а-рецепторов, такого как истрадефиллин, антиглутаматергического средства, такого как амантадин или мемантин, ингибитора ацетилхолинэстеразы, такого как ривастигмин, донепезил или галантамин, антипсихотического средства, такого как кветиапин, клозапин, рисперидон, пимавансерин, оланзапин, галоперидол, арипипразол или брекспипразол; или в комбинации с антителом, нацеленным на белок альфа-синуклеин, тау-белок или белок А-бета.
Е50. Применение в соответствии с любым из вариантов осуществления 46-49, где указанный лекарственный препарат представляет собой лекарственный препарат для перорального применения, такой как таблетка или капсула для перорального введения.
В контексте настоящего изобретения известно, что атом углерода в точке присоединения при заместителе (i) (изображено в варианте осуществления 1) находится в аномерном положении (i).
Все ссылки, включая публикации, патентные заявки и патенты, цитируемые в данном документе, включены в данный документ посредством ссылки во всей своей полноте и в той же степени, как если бы было указано, что каждая ссылка индивидуально и конкретно включена посредством ссылки и приведена во всей своей полноте (в максимальной степени допускаемой законом).
Заголовки и подзаголовки применяются в данном документе исключительно для удобства, и их не следует рассматривать как ограничивающие настоящее изобретение каким-либо образом.
Описание в данном документе любого аспекта или аспектов настоящего изобретения с использованием таких терминов, как "включающий", "имеющий", "в том числе" или "содержащий", по отношению к элементу или элементам предназначено для подтверждения аналогичного аспекта или аспектов настоящего изобретения, который "состоит из", "состоит практически из" данного конкретного элемента или элементов или "по сути содержит" их, если не указано иное или это однозначно не противоречит контексту (например, композиция, описанная в данном документе как содержащая определенный элемент, должна также пониматься как описывающая композицию, состоящую из данного элемента, если не указано иное или это однозначно не противоречит контексту).
Использование всевозможных примеров или типичной фразы (в том числе "как например", "например", "к примеру" и "собственно") в данном описании предназначено исключительно для лучшего освещения настоящего изобретения и не предусматривает ограничение объема настоящего изобретения, если не указано иное.
Следует понимать, что различные аспекты, варианты осуществления, реализации и признаки настоящего изобретения, упомянутые в данном документе, могут быть заявлены по отдельности или в любом сочетании.
Настоящее изобретение включает все модификации и эквиваленты объекта, изложенного в прилагаемой к данному документу формуле изобретения согласно действующему законодательству.
СОЕДИНЕНИЯ ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ


ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
Получение соединений по настоящему изобретению
Соединения формулы (Id) можно получать с помощью способов, описанных ниже, в сочетании со способами синтеза, известными в области органической химии, или модификациями, с которыми знакомы специалисты в данной области. Исходные материалы, используемые в данном документе, являются коммерчески доступными, или их можно получить с помощью традиционных способов, известных из уровня техники, таких как способы, описанные в стандартной справочной литературе, такой как "Compendium of Organic Synthetic Methods, Vol. I-XII" (опубликованный Wiley-Interscience). Предпочтительные способы включают без ограничений способы, описанные ниже.
На схемах представлены способы, пригодные в синтезе соединений согласно настоящему изобретению. Они никоим образом не предназначены для ограничения объема настоящего изобретения.
Способы LC-MS
Аналитические данные LC-MS получали с применением способов, определенных ниже.
Способ 550. LC-MS проводили на приборе Waters Aquity UPLC-MS, состоящем из Waters Aquity, включающем управляющее устройство колонки, бинарную градиентную систему управления, органайзер для образцов, детектор PDA (работающий при 254 нМ), детектор ELS и TQ-MS, оборудованный APPI-источником, работающем в режиме положительных ионов.
Условия LC. Колонка представляла собой Acquity UPLC ВЕН С18 1,7 мкм; 2,1×50 мм, работающую при 60°С с 1,2 мл/мин. бинарного градиента, состоящего из воды + 0,05% трифторуксусной кислоты (А) и ацетонитрил/вода (95:5) + 0,05% трифторуксусной кислоты.
Градиент
Общее время прогона: 1,15 мин.
Способ 551. LC-MS проводили на приборе Waters Aquity UPLC-MS, состоящем из Waters Aquity, включающем управляющее устройство колонки, бинарную градиентную систему управления, органайзер для образцов, детектор PDA (работающий при 254 нМ), детектор ELS и TQ-MS, оборудованный APPI-источником, работающем в режиме положительных ионов.
Условия LC. Колонка представляла собой Acquity UPLC HSS Т3 1,8 мкм; 2,1×50 мм, работающую при 60°С с 1,2 мл/мин. бинарного градиента, состоящего из воды+0,05% трифторуксусной кислоты (А) и ацетонитрил/вода (95:5) + 0,05% трифторуксусной кислоты.
Градиент
Общее время прогона: 1,15 мин.
Способ 555. LC-MS проводили на приборе Waters Aquity UPLC-MS, состоящем из Waters Aquity, включающем управляющее устройство колонки, бинарную градиентную систему управления, органайзер для образцов, детектор PDA (работающий при 254 нМ), детектор ELS и TQ-MS, оборудованный APPI-источником, работающем в режиме положительных ионов.
Условия LC. Колонка представляла собой Acquity UPLC ВЕН С18 1,7 мкм; 2,1×150 мм, работающую при 60°С с 0,6 мл/мин. бинарного градиента, состоящего из воды + 0,05% трифторуксусной кислоты (А) и ацетонитрил/вода (95:5) + 0,05% трифторуксусной кислоты.
Градиент
Общее время прогона: 3,6 мин.
Способ 111. LC-MS проводили на Shimadzu LCMS-2020, состоящем из детектора PDA, работающем при 190-800 нМ, и MS, оборудованном ESI-источником, работающем в режиме положительных ионов.
Условия LC. Колонка представляла собой Phenomenex Kinetex EVO С18 2,6 мкм; 2,1×100 мм, работающую при 25°С с 0,5 мл/мин. градиента, состоящего из воды + 0,1% муравьиной кислоты (А) и ацетонитрила + 0,1% муравьиной кислоты (В).
Градиент
Общее время прогона: 18 мин.
Способ 222. LC-MS проводили на Shimadzu LCMS-2020, состоящем из детектора PDA, работающем при 190-800 нМ, и MS, оборудованном ESI-источником, работающем в режиме положительных ионов.
Условия LC. Колонка представляла собой Phenomenex Kinetex EVO С18 2,6 мкм; 2,1×100 мм, работающую при 25° с 0,5 мл/мин. градиента, состоящего из воды + 0,1% муравьиной кислоты (А) и ацетонитрила (В).
Градиент
Общее время прогона: 18 мин.
Препаративную LCMS проводили с применением способа, указанного ниже.
Система Waters AutoPurification с применением комбинированного масс/УФ-обнаружения.
Колонка: Sunfire 30×100 мм, частицы размером 5 мкм. Колонка работала при 40°С с 90 мл/мин. бинарного градиента, состоящего из воды + 0,05% трифторуксусной кислоты (А) и ацетонитрил/вода (3:5) + 0,05% трифторуксусной кислоты.
Градиент
HighRes MS проводили на Bruker Compact qTOF, оборудованном электрораспылением, работающем в режиме положительных и отрицательных ионов. Применяли прямую инфузию и выполняли калибровку с помощью формиата натрия.
Общие способы получения соединений по настоящему изобретению
Соединение (I), которое, например, можно получать, как раскрыто в WO 2009/026934, применяли в качестве промежуточного соединения в синтезе соединений по настоящему изобретению.
Вкратце, соединения (Id-ia) и (Id-ib) по настоящему изобретению можно получать из (I) путем осуществления реакции (I) с триизопропилсилилхлоридом в присутствии DIPEA (N,N-диизопропилэтиламина) в дихлорметане с получением смеси моносилилированных промежуточных соединений, представляющих собой (4aR,10aR)-1-пропил-7-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ол и (4aR,10aR)-1-пропил-6-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ол, которые затем подвергали защите с помощью трет-бутилоксикарбонильной защитной группы для обеспечения Вос-защиты с получением промежуточных соединений, представляющих собой трет-бутил((4aR,10aR)-1-пропил-7-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)карбонат [А] и трет-бутил((4aR,10aR)-1-пропил-6-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)карбонат [В При этом можно проводить последующее удаление сил ильной группы с применением TEA-3HF (триэтиламинтригидрофторида) и повторное обеспечение защиты с применением ацетилового ангидрида с получением смеси (4aR,10aR)-6-((трет-бутоксикарбонил)окси)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-илацетата и (4aR,10aR)-7-((трет-бутоксикарбонил)окси)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-илацетата. Затем можно проводить реакцию сочетания глюкуронида с применением тетра-ацетатного донора для реакции сочетания, представляющего собой ((2S,3R,4S,5S,6S)-6-(метоксикарбонил)тетрагидро-2Н-пиран-2,3,4,5-тетраилтетраацетат), в присутствии диэтилового эфирата трифторида бора (BF3-OEt2) в качестве кислотного катализатора Льюиса с получением смеси необходимых аддуктов реакции сочетания, представляющих собой (2S,3R,4S,5S,6S)-2-(((4aR,10aR)-7-ацетокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетат и (2S,3R,4S,5S,6S)-2-(((4aR,10aR)-6-ацетокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетат. Затем неочищенную смесь можно подвергать гидролизу с применением KCN во влажном метаноле с получением (Id-ia) и (Id-ib), которые можно разделить с помощью колоночной хроматографии.
Вкратце, соединения (Id-iia) и (Id-iib) по настоящему изобретению можно получать из (I) (путем осуществления реакции (I) с комплексом пиридина с триоксидом серы в пиридине с получением смеси моносульфатов (Id-iia) и (Id-iib), которые можно разделить с помощью колоночной хроматографии.
Иллюстративные соединения по настоящему изобретению
(Id-iia): (4aR,10aR)-7-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-илгидросульфат и
(Id-iib): (4aR,10aR)-6-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-илгидросульфат.
(4aR,10aR)-1-Пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола гидрохлорид (1,51 г) суспендировали в пиридине (25 мл) в атмосфере азота при комнатной температуре, добавляли комплекс пиридина с триоксидом серы (2,31 г), и суспензию перемешивали при комнатной температуре. Через 15 ч. и 23 ч. добавляли дополнительное количество комплекса пиридина с триоксидом серы (2×(2,1 г, 13,1 ммоль)) и смесь перемешивали при комнатной температуре в течение ночи.
После перемешивания в течение полных двух дней неочищенную смесь разбавляли с помощью МеОН/дихлорметан и выпаривали непосредственно на вспомогательном фильтровальном веществе. Очистка с помощью колоночной хроматографии (элюент: этилацетат/триэтиламин/МеОН, 95:5:0-70:5:25) обеспечивала соотношение двух сульфатов приблизительно 3:1. Смесь суспендировали в 10 мл МеОН, медленно добавляли 50 мл воды, и полученную суспензию перемешивали при комнатной температуре. Через 7 ч. суспензию фильтровали и осадок промывали с помощью 2×10 мл воды и высушивали в течение ночи в вакуумной печи при 40°С с получением 1,26 г неочищенного продукта в виде твердого вещества. Смесь сульфатов разделяли с применением препаративной LC-MS, и как (Id-iib), так и (Id-iia) подвергали очистке посредством растирания, путем нагревания с обратным холодильником в 50 мл МеОН и перемешивали при комнатной температуре в течение 32 ч. Суспензию фильтровали и осадок промывали с помощью 2×5 мл МеОН и высушивали в вакуумной печи при 40°С в течение ночи, затем суспендировали в 50 мл ацетонитрила и перемешивали при комнатной температуре в течение 19 ч., и осадок промывали с помощью 2×10 мл ацетонитрила и высушивали в вакуумной печи при 40°С с получением (Id-iib) (4aR,10aR)-6-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-илгидросульфата (0,52 г, 1,5 ммоль, выход 30%) в виде твердого вещества и (Id-iia) (4aR,10aR)-7-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-илгидросульфата (0,15 г, 0,45 ммоль, выход 9%) в виде твердого вещества.
(Id-iib)
LCMS (способ 555) 1,29 мин.
1Н ЯМР (600 МГц, DMSO-d6) δ 9,06 (s, 1Н), 8,89 (s, 1Н), 6,89 (d, J=8,2 Гц, 1H), 6,58 (d, J=8,3 Гц, 1H), 3,52 (d, J=12,1 Гц, 1H), 3,35-3,32 (m, 1Н), 3,31-3,22 (m, 2Н), 3,10-3,01 (m, 2Н), 2,97 (dd, J=17,4, 5,1 Гц, 1H), 2,74 (dd, J=15,6, 11,1 Гц, 1H), 2,18 (dd, J=17,4, 11,6 Гц, 1H), 1,97-1,69 (m, 5Н), 1,68-1,56 (m, 1Н), 1,35 (qd, J=13,0, 3,8 Гц, 1H), 0,96 (t, J=7,3 Гц, 3Н).
(Id-iia)
LCMS (способ 555) 1,37 мин.
1Н ЯМР (600 МГц, DMSO-d6) δ 9,07 (s, 1Н), 8,84 (s, 1Н), 6,82 (d, J=8,3 Гц, 1Н), 6,70 (d, J=8,3 Гц, 1Н), 3,51 (d, J=12,0 Гц, 1Н), 3,34-3,30 (m, 1Н), 3,26 (bs, 2Н), 3,17-2,94 (m, 3Н), 2,75-2,67 (m, 1Н), 2,35 (dd, J=17,6, 11,9 Гц, 1Н), 1,90 (t, J=13,8 Гц, 2Н), 1,85-1,69 (m, 3Н), 1,67-1,57 (m, 1Н), 1,40-1,31 (m, 1Н), 0,95 (t, J=7,3 Гц, 3Н).
(Id-iiab): (4aR,10aR)-1-пропил-1,2,3,4,4a,5,10,10а-октагидробензо[g]хинолин-6,7-диил-бис(гидросульфат) (триэтиламиновая соль)
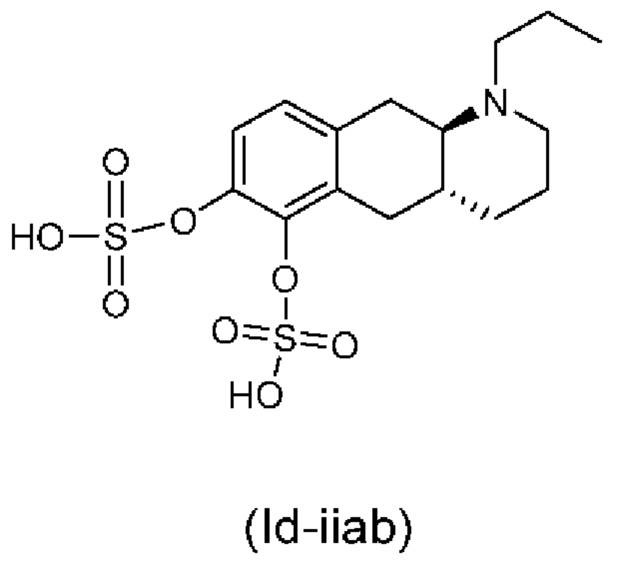
(4aR,10aR)-1-Пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола гидрохлорид (0,500 г, 1,68 ммоль) и комплекс пиридина с триоксидом серы (5,34 г, 33,6 ммоль) суспендировали в ацетонитриле (10 мл) и добавляли триэтиламин (7,02 мл, 50,4 ммоль) при комнатной температуре. Суспензию нагревали до 80°С и перемешивали в атмосфере азота в течение 16,5 ч. Обеспечивали охлаждение смеси до комнатной температуры, и выпаривали ее на вспомогательном фильтровальном веществе (10 г). Очистка с помощью колоночной хроматографии (элюент: этилацетат/триэтиламин/МеОН, 75:5:20-45:5:50) обеспечивала получение масла (1,51 г). Масло разбавляли с помощью МеОН (10 мл+3 капли DMSO), и добавляли трет-бутилметиловый эфир (МТВЕ) (2×10 мл) с помощью шприца. Маслянистое твердое вещество немедленно осаждалось. Суспензию концентрировали, и полученный осадок поглощали в МеОН (20 мл) и триэтиламине (5 мл) и фильтровали. МТВЕ (40 мл) добавляли к фильтрату в течение двух минут, и твердое вещество постепенно осаждалось. Осадок фильтровали и высушивали в вакуумной печи при 35°С в течение 15 минут с получением (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диил-бис(гидросульфат) в виде твердого вещества и в виде комплекса с триэтиламином 1:1,3 согласно анализу 1Н ЯМР (0,531 г, 0,957 ммоль, выход 57%).
LCMS (способ 555) rt=1,00 мин.
Из-за нестабильности дисульфата в кислотных условиях LCMS не обеспечивала хорошего определения чистоты.
1Н ЯМР (600 МГц, DMSO-d6) δ 8,93 (s, 1Н), 8,80 (s, 1Н), 7,40 (d, J=8,5 Гц, 1Н), 6,77 (d, J=8,6 Гц, 1Н), 3,48 (d, J=11,7 Гц, 1Н), 3,37-3,19 (m, 5Н), 3,10 (q, J=7,3 Гц, 7,8Н, триэтиламин), 3,02 (s, 1Н), 2,76-2,67 (m, 1Н), 2,46 (dd, J=17,7, 12,2 Гц, 1Н), 1,85 (d, J=11,3 Гц, 2Н), 1,81-1,56 (m, 4Н), 1,37-1,26 (m, 1Н), 1,17 (t, J=7,3 Гц, 11,7Н, триэтиламин), 0,95 (t, J=7,3 Гц, 3Н).
HRMS (ESI): рассч. масса/заряд для C16H21NO8S22- [М-2Н+] 209,5360, найденное значение 209,5360
Промежуточные соединения для получения (Id-ia), (Id-ib) и (Id-iab)
Промежуточные соединения: (4aR,10aR)-1-пропил-7-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ол и (4aR,10aR)-1-пропил-6-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ол.
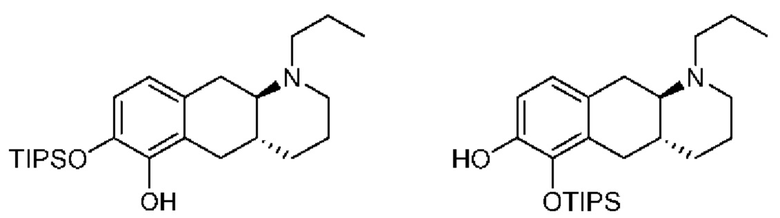
(4aR,10aR)-1-Пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола гидрохлорид (2,21 г, 7,43 ммоль) суспендировали в дихлорметане (80 мл) в атмосфере азота при комнатной температуре, добавляли N,N-диизопропилэтиламин (4,44 г, 6,0 мл, 34,4 ммоль) с последующим добавлением триизопропилсилилхлорида (2,73 г, 3,0 мл, 14,16 ммоль), и смесь перемешивали при комнатной температуре в течение 92 ч. Добавляли 10 мл МеОН, и неочищенную смесь выпаривали, дважды выпаривали совместно с дихлорметан/гептан, повторно растворяли в дихлорметане, и выпаривали непосредственно на вспомогательном фильтровальном веществе, и очищали с помощью колоночной хроматографии (элюент: н-гептан/этилацетат/триэтиламин, 100:0:0-35:60:5) с получением 3,14 г в виде смеси (4aR,10aR)-1-пропил-7-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ола (3,14 г) и (4aR,10aR)-1-пропил-6-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ола в виде масла.
Результаты ЯМР (CDCl3) демонстрировали смесь >30:1 силилированных изомеров.
Промежуточные соединения: трет-бутил((4aR,10aR)-1-пропил-7-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)карбонат [А] и трет-бутил((4aR,10aR)-1-пропил-6-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)карбонат [В].
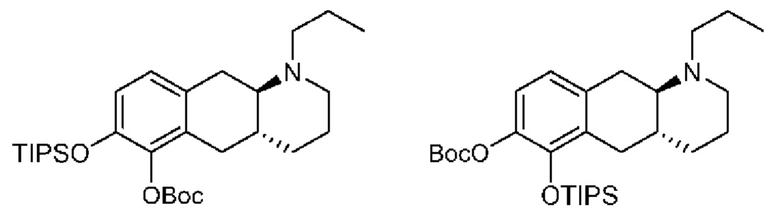
Смесь из предыдущей стадии в виде (4aR,10aR)-1-пропил-7-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ола и (4aR,10aR)-1-пропил-6-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ола (2,94 г, 7,04 ммоль) растворяли в дихлорметане (30 мл) в атмосфере азота и охлаждали до 0°С. Добавляли пиридин (6,00 мл) с последующим добавлением ди-трет-бутилдикарбоната (6,30 г), и обеспечивали нагревание реакционной смеси до комнатной температуры в течение 3-4 ч., а затем перемешивали при комнатной температуре в течение ночи. Добавляли 10 мл МеОН, и реакционную смесь выпаривали, выпаривали совместно с дихлорметан/н-гептан дважды, растворяли в дихлорметане и выпаривали на вспомогательном фильтровальном веществе.
Очистка с помощью колоночной хроматографии (элюент: н-гептан/этилацетат/триэтиламин, 100:0:0-75:20:5) обеспечивала получение смеси трет-бутил((4aR,10aR)-1-пропил-7-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)карбоната [А] и трет-бутил((4aR,10aR)-1-пропил-6-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)карбоната [В] (3,6 г) в виде масла.
Результаты ЯМР (CDCl3) после высушивания демонстрировали смесь региоизомеров.
Промежуточные соединения: (4aR,10aR)-6-((трет-бутоксикарбонил)окси)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-илацетат и (4aR,10aR)-7-((трет-бутоксикарбонил)окси)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-илацетат.
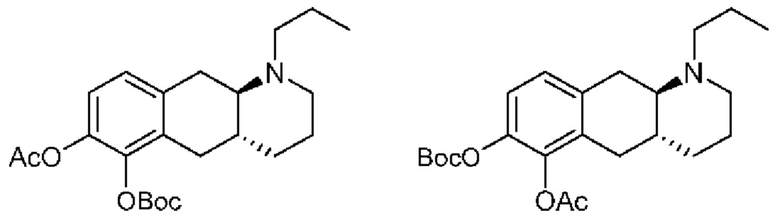
Трет-бутил((4aR,10aR)-1-пропил-7-((триизопропилсилил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)карбонат (3,600 г, 6,95 ммоль) (смесь [А]:[В] из предыдущей стадии) растворяли в THF (150 мл) в атмосфере азота при 0°С, добавляли триэтиламинтригидрофторид (2,97 г, 3,00 мл, 18,42 ммоль), и смесь перемешивали при 0°С. Через 3 ч. при 0°С добавляли пиридин (10,0 мл, 124 ммоль) и уксусный ангидрид (4,33 г, 4,00 мл, 42,4 ммоль) непосредственно в реакционную смесь при 0°С, и обеспечивали нагревание реакционной смеси до комнатной температуры. Через 16 ч. добавляли 20 мл МеОН, и реакционную смесь выпаривали, повторно растворяли в дихлорметан/гептан и выпаривали на вспомогательном фильтровальном веществе с последующей очисткой с помощью хроматографии под вакуумом на сухой колонке с получением (4aR,10aR)-6-((трет-бутоксикарбонил)окси)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-илацетата и (4aR,10aR)-7-((трет-бутоксикарбонил)окси)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-илацетата в виде масла/пены.
LCMS (способ 550) rt=0,56 мин., [М+Н]+=404 e/z.
Промежуточные соединения: (2S,3R,4S,5S,6S)-2-(((4aR,10aR)-7-ацетокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетат и (2S,3R,4S,5S,6S)-2-(((4aR,10aR)-6-ацетокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетат.
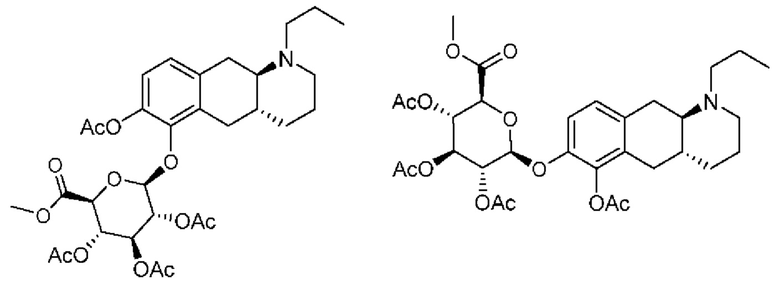
(4aR,10aR)-6-((Трет-бутоксикарбонил)окси)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-илацетат (2,489 г, 6,17 ммоль) (смесь (4aR,10aR)-6-((трет-бутоксикарбонил)окси)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-илацетата и предположительно (4aR,10aR)-7-((трет-бутоксикарбонил)окси)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-илацетата) растворяли в дихлорметане (60 мл) в атмосфере азота при комнатной температуре, добавляли (2S,3R,4S,5S,6S)-6-(метоксикарбонил)тетрагидро-2Н-пиран-2,3,4,5-тетраилтетраацетат (7,529 г, 20,01 ммоль) с последующим добавлением диэтилового эфирата трифторида бора (6,72 г, 6,0 мл, 47,3 ммоль) и смесь перемешивали при комнатной температуре в течение 5 дней. Смесь разбавляли с помощью дихлорметана и МеОН и выпаривали на вспомогательном фильтровальном веществе. Очистку проводили с помощью хроматографии под вакуумом на сухой колонке с получением смеси (2S,3R,4S,5S,6S)-2-(((4aR,10aR)-7-ацетокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетата и (2S,3R,4S,5S,6S)-2-(((4aR,10aR)-6-ацетокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетата (4,37 г) в виде пены/твердого вещества.
LC-MS (способ 555) rt=1,94 мин., [М+Н]+=620 e/z.
Промежуточное соединение: метил(2S,3S,4S,5R,6S)-6-[[(4aR,10aR)-1-пропил-6-[(2S,3R,4S,5S,6S)-3,4,5-триацетокси-6-метоксикарбонил-тетрагидропиран-2-ил]окси-3,4,4а,5,10,10а-гексагидро-2Н-бензо[g]хинолин-7-ил]окси]-3,4,5-триацетокси-тетрагидропиран-2-карбоксилат
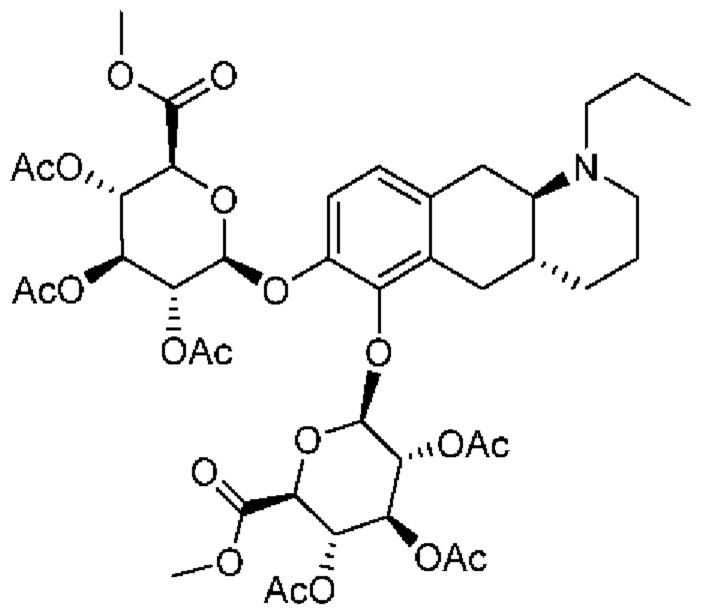
(2S,3S,4S,5R,6R)-2-(Метоксикарбонил)-6-(2,2,2-трихлор-1-иминоэтокси)тетрагидро-2H-пиран-3,4,5-триилтриацетат (1,286 г, 2,69 ммоль) и (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола гидрохлорид (0,4 г, 1,343 ммоль) растворяли в дихлорметане (5,28 г, 4,00 мл, 62,2 ммоль), затем добавляли диэтиловый эфират трифторида бора (0,381 г, 0,340 мл, 2,69 ммоль) в атмосфере азота, и смесь перемешивали в течение 3 дней в атмосфере азота в 8 мл флаконе. Добавляли дополнительное количество (2S,3S,4S,5R,6R)-2-(метоксикарбонил)-6-(2,2,2-трихлор-1-иминоэтокси)тетрагидро-2Н-пиран-3,4,5-триилтриацетата (1,286 г, 2,69 ммоль) и диэтилового эфирата трифторида бора (0,381 г, 0,340 мл, 2,69 ммоль), и смесь перемешивали в течение 4 ч., затем смесь выливали в насыщенный водный NaHCO3 (30 мл), затем экстрагировали с помощью дихлорметана (2×20 мл), и объединенные органические фазы высушивали (Na2SO4), фильтровали и выпаривали до сухого состояния под вакуумом. Неочищенную пену суспендировали в гептан/этилацетат (1:1) и перемешивали в течение ночи. Затем добавляли HCl в простом эфире (0,672 мл, 1,343 ммоль, 2 мольный) и смесь перемешивали в течение 1 ч. и выпаривали до сухого состояния под вакуумом, и добавляли МТВЕ (40 мл), и смесь нагревали с обратным холодильником и оставляли охлаждаться до комнатной температуры, затем смесь фильтровали и твердое вещество высушивали в вакуумной печи в течение 1 дня при 40°С с получением метил(2S,3S,4S,5R,6S)-6-[[(4aR,10aR)-1-пропил-6-[(2S,3R,4S,5S,6S)-3,4,5-триацетокси-6-метоксикарбонил-тетрагидропиран-2-ил]окси-3,4,4а,5,10,10а-гексагидро-2Н-бензо[g]хинолин-7-ил]окси]-3,4,5-триацетокситетрагидропиран-2-карбоксилата гидрохлорида (1,0854 г, 1,167 ммоль, выход 87%).
LC-MS, способ 550, rt=0,63 мин., [М+Н]+=895,7 e/z.
(Id-ib): (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR,10aR)-6-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)окси)тетрагидро-2Н-пиран-2-карбоновая кислота и
(Id-ia): (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR,10aR)-7-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)окси)тетрагидро-2Н-пиран-2-карбоновая кислота.
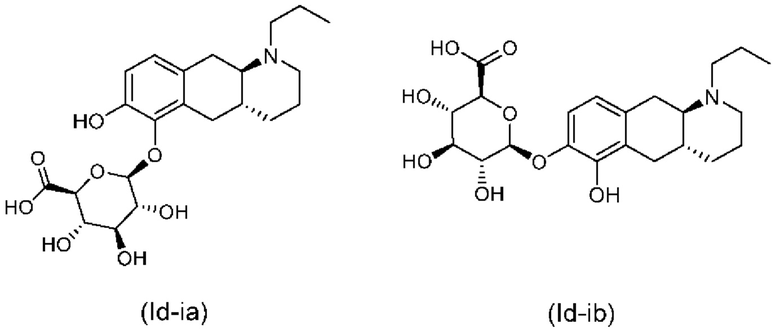
Смесь (2S,3R,4S,5S,6S)-2-(((4aR,10aR)-7-ацетокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетата и (2S,3R,4S,5S,6S)-2-(((4aR,10aR)-6-ацетокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетата (3,82 г, 6,17 ммоль) растворяли в МеОН (100 мл) и воде (20 мл), охлаждали до 0°С, добавляли цианид калия (7,295 г, 112 ммоль), и обеспечивали медленное нагревание суспензии до комнатной температуры в течение 17,5 ч. Неочищенную смесь выпаривали на вспомогательном фильтровальном веществе и высушивали. Неочищенную смесь очищали с помощью колоночной хроматографии на силикагеле (элюент: этилацетат/МеОН/вода 100:0:0-0:50:50) с получением соотношения (Id-ib) и (Id-ia), составляющего 5-6:1. Смесь разделяли с помощью препаративной LCMS.
Собранные фракции, соответствующие пику 1, содержащие (Id-ib), объединяли, выпаривали и объединяли с другой партией 186 мг (Id-ib)-TFA, которая была получена подобным образом с применением МеОН, выпаривали и высушивали с получением твердого вещества, (Id-ib) повторно суспендировали в 10 мл EtOH, и добавляли 100 мл МТВЕ, и полученную суспензию перемешивали при комнатной температуре в течение 8 ч., суспензию фильтровали, и осадок промывали с помощью 2×10 мл МТВЕ и высушивали в вакуумной печи в течение ночи с получением 1,601 г (Id-ib) в виде твердого вещества, соответствующего (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR,10aR)-6-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)окси)тетрагидро-2Н-пиран-2-карбоновой кислоте.
Собранные фракции, соответствующие пику 2, содержащие (Id-ia), объединяли, выпаривали, переносили в меньшую колбу с МеОН, выпаривали, повторно растворяли в приблизительно 12 мл МеОН, и повторно очищали с помощью препаративной LCMS, и выпаривали с получением пены/твердого вещества. Соответствующие фракции объединяли, выпаривали, переносили с МеОН в меньшую колбу и выпаривали и объединяли с другой партией 40,7 мг (Id-ia), которая была получена подобным образом. Объединенную партию растворяли в 2,5 мл EtOH, добавляли 25 мл МТВЕ, и суспензию перемешивали при комнатной температуре. Через 8 ч. суспензию фильтровали и осадок промывали с помощью 2×2,5 мл МТВЕ и высушивали в вакуумной печи в течение ночи с получением 362,2 мг (Id-ia) в виде твердого вещества, (Id-ia) суспендировали в приблизительно 10 мл EtOH, добавляли 50 мл МТВЕ, и суспензию перемешивали при комнатной температуре и фильтровали через 19 ч., и осадок промывали с помощью 2×10 мл МТВЕ и высушивали в вакуумной печи при 40°С с получением 0,279 г (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR,10aR)-7-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)окси)тетрагидро-2Н-пиран-2-карбоновой кислоты (Id-ia) в виде твердого вещества.
(Id-ib)
LCMS (способ 551) rt=0,37 мин.
1Н ЯМР (600 МГц, Метанол-d4) δ 7,02 (d, J=8,4 Гц, 1Н), 6,65 (d, J=8,4 Гц, 1Н),4,73 (d, J=7,7 Гц, 1Н), 3,89 (d, J=9,7 Гц, 1Н), 3,68-3,58 (m, 2Н), 3,54 (dd, J=9,3, 7,7 Гц, 1Н), 3,49 (t, J=9,1 Гц, 1Н), 3,47-3,36 (m, 2Н), 3,30 (dt, J=11,2, 5,6 Гц, 1Н), 3,21-3,11 (m, 3Н), 2,85 (dd, J=15,4, 11,3 Гц, 1Н),2,35 (dd, J=17,6, 11,5 Гц, 1Н), 2,12-2,02 (m, 2Н), 2,02-1,84 (m, 3Н), 1,81-1,71 (m, 1Н), 1,49 (qd, J=13,0, 3,7 Гц, 1Н), 1,09 (t, J=7,3 Гц, 3Н).
(Id-ia)
LCMS (способ 551) rt=0,39 мин.
1Н ЯМР (600 МГц, Метанол-d4) δ 6,87 (d, J=8,3 Гц, 1Н), 6,74 (d, J=8,4 Гц, 1Н), 4,62 (d, J=7,9 Гц, 1Н), 3,75 (dd, J=17,7, 4,9 Гц, 1Н), 3,66-3,62 (m, 2Н), 3,61-3,51 (m, 2Н), 3,50-3,35 (m, 3Н), 3,31-3,22 (m, 1Н), 3,14 (qd, J=12,7, 4,0 Гц, 2Н), 2,83 (dd, J=15,2, 11,3 Гц, 1Н), 2,37 (dd, J=17,7, 11,7 Гц, 1Н), 2,12 (d, J=13,4 Гц, 1Н), 2,08-2,00 (m, 1Н), 1,98-1,83 (m, 3Н), 1,81-1,71 (m, 1Н), 1,44 (qd, J=13,2,3,9 Гц, 1Н), 1,09 (t, J=7,3 Гц, 3Н).
(Id-iab): (2S,2'S,3S,3'S,4S,4'S,5R,5'R,6S,6'S)-6,6'-(((4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диил)бис(окси))бис(3,4,5-тригидрокситетрагидро-2Н-пиран-2-карбоновая кислота).
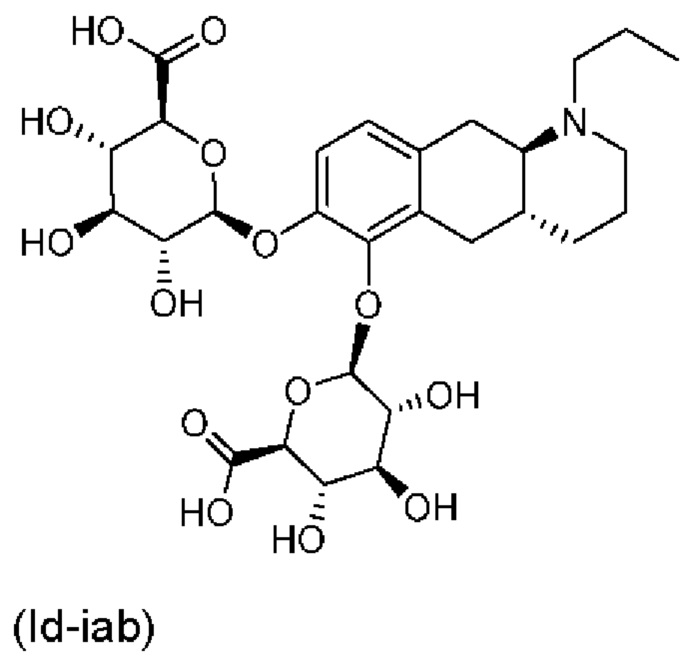
Синтез А
(1S,4aR,10aR)-1-Пропил-6,7-бис(((2S,3R,4S,5S,6S)-3,4,5-триацетокси-6-(метоксикарбонил)тетрагидро-2Н-пиран-2-ил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин (0,25 г, 0,269 ммоль) растворяли в воде (1,209 г, 1,209 мл, 67,1 ммоль), и добавляли МеОН (3,83 г, 4,84 мл, 120 ммоль) и KOH (0,393 г, 0,270 мл, 3,22 ммоль, 46%), и перемешивали в течение ночи при комнатной температуре в закрытом флаконе. В течение ночи образовывался осадок, который выделяли посредством фильтрации. Твердое вещество промывали с помощью МеОН (1,5 мл) с получением 2-калий-(2S,2'S,3S,3'S,4S,4'S,5R,5'R,6S,6'S)-6,6'-((4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диил)бис(окси))бис(3,4,5-тригидрокситетрагидро-2Н-пиран-2-карбоновая кислота) (0,096 г, 0,139 ммоль, выход 51,6%).
LC-MS способ 551 rt 0,31 мин, [М+Н]+=614,2 e/z.
Синтез В
(1S,4aR,10aR)-1-Пропил-6,7-бис(((2S,3R,4S,5S,6S)-3,4,5-триацетокси-6-(метоксикарбонил)тетрагидро-2Н-пиран-2-ил)окси)-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин (0,2585 г, 0,278 ммоль) растворяли в воде (1,250 г, 1,25 мл, 69,4 ммоль) и МеОН (3,96 г, 5 мл, 124 ммоль), и добавляли KCN (0,344 г, 5,28 ммоль), и перемешивали в течение ночи при комнатной температуре в закрытом флаконе. В течение ночи образовывался осадок, который выделяли посредством фильтрации. Твердое вещество промывали с помощью МеОН (1,5 мл) с получением 2-калий-(2S,2'S,3S,3'S,4S,4'S,5R,5'R,6S,6'S)-6,6'-(((4aR,10aR)-1-пропил-1,2,3,4,4a,5,10,10a-октагидробензо[g]хинолин-6,7-диил)бис(окси))бис(3,4,5-тригидрокситетрагидро-2Н-пиран-2-карбоновая кислота) (0,0963 г, 0,139 ммоль, выход 50,1%).
LC-MS способ 551 rt=0,34 мин, [М+Н]+=614,6 e/z.
1Н ЯМР (600 МГц, DMSO-d6) δ 7,09 (d, J=8,5 Гц, 1Н), 6,84 (d, J=8,5 Гц, 1Н), 4,91-4,79 (m, 1Н), 4,78-4,66 (m, 1Н), 3,93 (bs, 22Н (ОН/вода)), 3,42 (d, J=9,8 Гц, 1Н), 3,37-3,21 (m, 7H), 3,19 (s, 1Н), 3,11 (dd, J=16,2, 4,9 Гц, 1H), 2,90 (d, J=11,0 Гц, 1H),2,67 (ddd, J=12,9, 10,7, 5,6 Гц, 1H), 2,49 (dd, J=15,9, 10,9 Гц, 1H), 2,39-2,27 (m, 1Н), 2,15 (dt, J=17,5, 11,5 Гц, 2H), 2,05 (td, J=10,4, 4,9 Гц, 1H), 1,86 (d, J=11,7 Гц, 1H), 1,67-1,38 (m, 5Н), 1,03 (qd, J=12,3, 5,1 Гц, 1H), 0,85 (t, J=7,3 Гц, 3Н).
Дополнительные соединения, охватываемые объемом настоящего изобретения
Следующие два соединения также включены в объем настоящего изобретения:
(Id-iaiib): (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR,10aR)-1-пропил-7-сульфо-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-ил)окси)тетрагидро-2Н-пиран-2-карбоновая кислота и
(Id-iiaib): (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR,10aR)-1-пропил-6-сульфо-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-7-ил)окси)тетрагидро-2Н-пиран-2-карбоновая кислота.
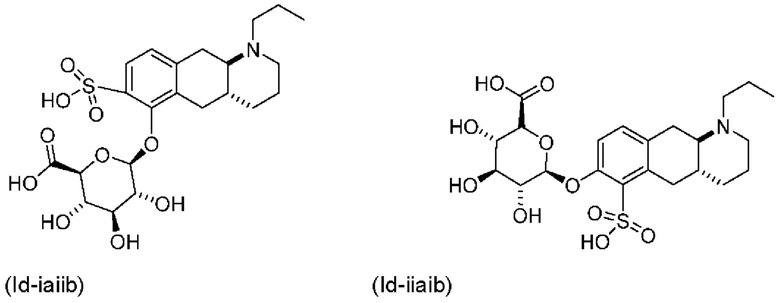
Получение апоморфиновых конъюгатов для PK-сравнения
(R)-11-Гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ила гидросульфат
1,0 г (3,19 ммоль, 1,0 экв.) полугидрата апоморфина гидрохлорида суспендировали в 3,3 мл пиридина в атмосфере аргона при комнатной температуре. 1,7 г (10,68 ммоль, 3,34 экв.). Комплекс триоксида серы с пиридином добавляли в данную суспензию, и смесь перемешивали при температуре 40°С в течение 17 часов и очищали с помощью препаративной HPLC.
Выделяли основной изомер, представляющий собой (R)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ила гидросульфат (78 мг), УФ-чистота 98,8%.
LC-MS способ 111 rt=5,18 мин, [М+Н]+=348,1 e/z.
1Н ЯМР (500 МГц, DMSO-d6) δ 9,91 (br, 1Н), 9,30 (s, 1Н), 8,27 (d, J=5,0 Гц, 1Н), 7,39 (m, 1Н), 7,20 (d, J=5,0 Гц, 1Н), 7,10 (d, J=5,0 Гц, 1Н), 6,84 (d, J=5,0 Гц, 1Н), 4,42 (bs, 1Н), 3,77 (bs, 1Н), 3,45 (d, J=10,0 Гц, 2Н), 3,25-3,21 (m, 1h), 3,20-3,10 (m, 4h), 2,71 (t, J=10,0 Гц, 1H).
(R)-10-Гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-11-ила гидросульфат
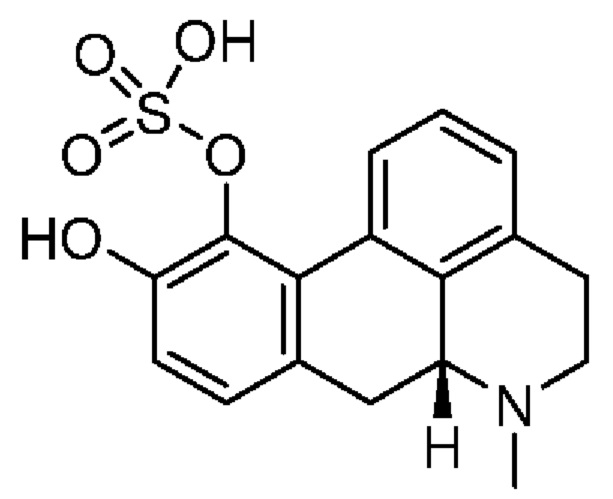
Применяли тот же способ, что и для (R)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ила гидросульфата, только небольшие изменения были сделаны для получения второстепенного компонента: продолжительность реакции уменьшили до 3 часов и тремя частями добавляли триоксид серы - пиридин. (Использовали три партии, начиная с 850 мг и дважды по 500 мг). Реакционные смеси объединяли и очищали с помощью препаративной HPLC с получением 50 мг минорного изомера, представляющего собой (R)-10-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-11-ила гидросульфат.
LC-MS способ 222 rt=4,99 мин, [М+Н]+=348,1 e/z.
1Н ЯМР (500 МГц, DMSO-d6) δ 10,00 (br, 1Н), 9,30 (s, 1Н), 8,26 (bs, 1Н), 7,23 (bs, 1Н), 7,11 (bs, 1Н), 7,01 (d, J=10,0 Гц, 1Н), 6,78 (d, J=10,0 Гц, 1Н), 3,50-2,20 (m, 7h).
Промежуточное соединение: (2S,3R,4S,5S,6S)-2-(((R)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетат
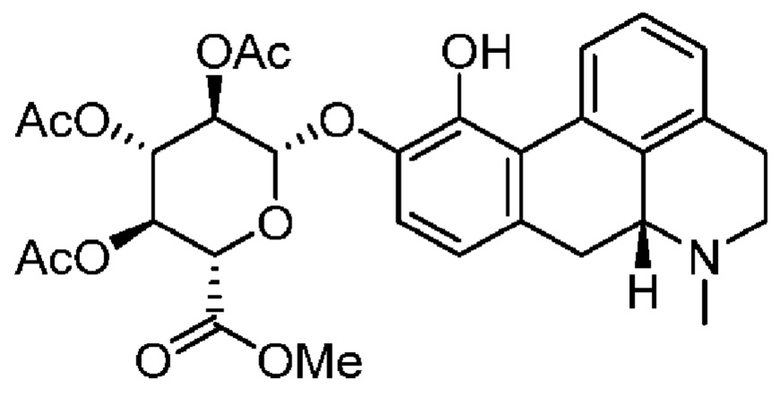
480 мг(1,796 ммоль) апоморфина (свободное основание) и 4,72 г (12,54 ммоль, 7,0 экв.) 1,2,3,4-тетра-о-ацетил-β-D-глюкопирануроната растворяли в 40 мл дихлорметана в атмосфере аргона при комнатной температуре. Исходные материалы растворялись через 10 минут с получением голубого раствора. К этому раствору добавляли 3,0 мл (3,45 г, 13,5 экв.) этилового эфирата трифторида бора в атмосфере Ar, и полученное перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь выливали в 80 мл насыщенного раствора бикарбоната натрия, перемешивали в течение 10 минут, затем разделяли. Водную фазу экстрагировали с помощью CH2Cl2 (3×40 мл). Объединенные органические фазы промывали с помощью насыщенного раствора бикарбоната натрия (1×40 мл) и солевого раствора (1×40 мл), высушивали над сульфатом натрия, фильтровали и выпаривали, получали 4,5 г твердого вещества (теоретический выход ~1,0 г). Неочищенный продукт очищали с помощью флэш-хроматографии с применением CH2Cl2:МеОН=96:4. После очистки получали 190 мг (2S,3R,4S,5S,6S)-2-(((R)-11-гидрокси-6-метил-5,6,6a,7-тетрагидро-4H-дибензо[de,g]хинолин-10-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетата.
(2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((R)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ил)окси)тетрагидро-2Н-пиран-2-карбоновая кислота
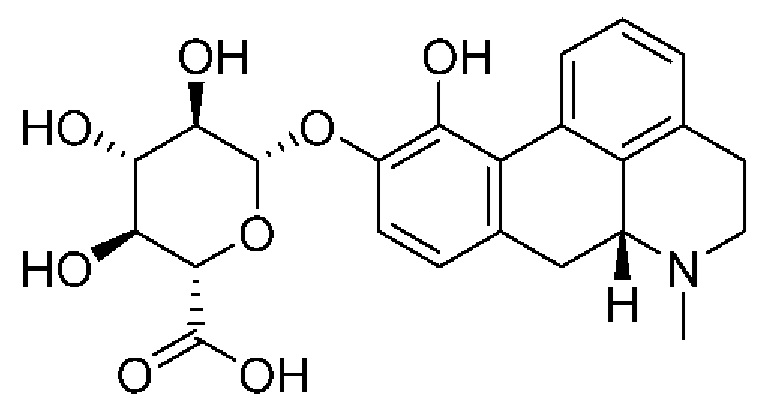
250 мг (0,432 ммоль) (2S,3R,4S,5S,6S)-2-(((R)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ил)окси)-6-(метоксикарбонил)тетрагидро-2Н-пиран-3,4,5-триилтриацетата растворяли в смеси 6,1 мл МеОН и 1,2 мл воды и охлаждали до 0°С. При этой температуре добавляли 526 мг (8,07 ммоль 18,7 экв.) KCN. Реакционную смесь перемешивали, и обеспечивали ее нагревание до комнатной температуры, и перемешивали в течение дополнительных 2 часов. Реакционную смесь фильтровали и очищали непосредственно посредством препаративной HPLC в элюенте вода-ацетонитрил-0,1% TFA. Из собранных фракций выпаривали ацетонитрил при комнатной температуре в вакууме, и водный остаток лиофилизировали с получением 133 мг TFA-соли (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((R)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ил)окси)тетрагидро-2Н-пиран-2-карбоновой кислоты в виде порошка. Ее структуру подтверждали с помощью LCMS и ЯМР.
LC-MS способ 111 rt=4,30 мин, [М+Н]+=444,2 e/z.
1Н ЯМР (500 МГц, DMSO-d6) δ 12,84 (br, 1Н), 10,00 (bs, 1Н), 8,87 (s, 1Н), 8,29 (d, J=10,0 Гц, 1Н), 7,40 (m, 1Н), 7,21 (d, J=5,0 Гц, 1Н), 7,02 (d, J=10,0 Гц, 1Н), 6,83 (d, J=5,0 Гц, 1Н), 5,77 (s, 1Н), 5,45-5,20 (m, 2Н), 4,84 (d, J=5,0 Гц, 1Н), 4,34 (bs, 1Н), 3,92 (d, J=10,0 Гц, 1Н), 3,76 (bs, 1h), 3,55-3,00 (m, 11Н, ОН/вода), 2,75-3,30 (m, 4Н).
Характеристика in vitro и in vivo соединений по настоящему изобретению
Пример 1а. Превращение соединений по настоящему изобретению в гепатоцитах крысы и человека
Соединения (Id-ia), (Id-ib), (Id-iia), (Id-iib), (Id-iab) и (Id-iiab) инкубировали по отдельности при 1 мкг/мл с гепатоцитами человека или крысы, суспендированными в DMEM (модифицированной по способу Дульбекко среде Игла) с HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислотой) при рН 7,4. Концентрация клеток при инкубации составляла 1×106 жизнеспособных клеток/мл. Инкубации проводили в стеклянных пробирках при 37°С с общим инкубируемым объемом 3,5 мл и с проведением инкубации в двух повторностях для каждого исследуемого образца. 3,5 мл суспензии гепатоцитов уравновешивали в течение 10 минут на водяной бане, установленной на 37°С, где затем инициировали инкубацию путем добавления 3,5 мкл исходного раствора исследуемого образца в DMSO (диметилсульфоксиде) и осторожного переворачивания пробирок. Конечная концентрация растворителя в инкубируемых средах составляла 0,1% DMSO. Образцы объемом 600 мкл отбирали из инкубируемых сред в заданные моменты времени, равные 0,25, 5, 15, 30 и 60 минутам, после обеспечения гомогенности суспензий гепатоцитов. Отобранный объем добавляли в криопробирки Nunc объемом 1 мл на жидкий лед, содержащие 60 мкл охлажденной льдом аскорбиновой кислоты (100 мг/мл) и 30 мкл охлажденного льдом 100 мМ 1,4-лактона сахарной кислоты в 0,5 М лимонной кислоте. Содержимое пробирок смешивали и добавляли 35 мкл раствора охлажденной льдом 20% муравьиной кислоты. Содержимое пробирок тщательно смешивали и хранили при -80°С в ожидании анализа. Способ анализа и оборудование, применяемое для анализа (I) после введения дозы (Id-ia), (Id-ib), (Id-iia) и (Id-iib), являлись такими, как описано в примерах 4 и 5 ниже в разделе "Оборудование, применяемое для анализа соединения (I) после введения дозы соединений (Ic), (Id-ia), (Id-ib), (Id-iia), (Id-iib), (Id-iab) и (Id-iiab)".
Способ анализа и оборудование, применяемое для анализа (I) после введения дозы (Id-iab) и (Id-iiab), включали смешивание равных аликвот образцов и раствора для осаждения (ацетонитрила (MeCN) с 10% метанолом (МеОН) и 1% муравьиной кислотой) с последующим центрифугированием при 4°С при 16000 g в течение 10 минут. Образцы надосадочной жидкости собирали и анализировали с помощью LC-MS/MS. Масс-спектрометр: Waters Acquity - Waters Xevo TQ-MS. Аналитическая колонка: Acquity UPLC HSS T3, 100×2,1 мм, 1,8 мкм. Подвижная фаза А: 0,2% муравьиная кислота в воде. Подвижная фаза В: 0,2% муравьиная кислота в ацетонитриле. Применяли градиент от 95/5% до 60/40 за 5 мин. Скорость потока составляла 0,3 мл/мин. Осуществляли MRM-мониторинг (I) в исследуемых образцах и в аналитических стандартах.
На фигуре 7 показано зависимое от времени превращение (Id-ia), (Id-ib), (Id-iia), (Id-iib) и (Id-iab) в соединение (I) в гепатоцитах как крысы, так и человека. В случае (Id-iiab) образование соединения (I) не поддавалось обнаружению при данных условиях исследования.
Пример 1b. Превращение соединений по настоящему изобретению в свежей крови крысы и человека
Превращение (Id-ia), (Id-ib), (Id-iia) и (Id-iib) в крови человека (среднее значение для 3 доноров) и крови крысы (среднее значение для 45 доноров) в (I) демонстрировали в свежей крови при 37°С с добавлением 1 мкг/мл (Id-ia), (Id-ib), (Id-iia) и (Id-iib) по отдельности, при этом (I) в выделенной плазме крови измеряли через 0, 5, 15, 30 и 60 минут. Способ анализа и оборудование являются такими, как описано в примерах 4 и 5 ниже в разделе "Оборудование, применяемое для анализа соединения (I) после введения дозы соединений (Ic), (Id-ia), (Id-ib), (Id-iia), (Id-iib), (Id-iab) и (Id-iiab)".
На фигуре 8 показано зависимое от времени превращение (Id-ia), (Id-ib), (Id-iia) и (Id-iib) в соединение (I) в крови как крысы, так и человека.
Пример 2. Дофаминовая агонистическая активность
Агонизм дофаминового рецептора D1
Агонизм дофаминового рецептора D1_измеряли с применением HTRF сАМР от CisBio с использованием протокола, разработанного HD Biosciences (Китай). Вкратце, анализ представляет собой анализ способом резонансного переноса энергии гомогенной флуоресценции с временным разрешением (HTRF), в котором измеряют продуцирование сАМР клетками в конкурентном иммуноанализе между нативным сАМР, продуцируемым клетками, и сАМР, меченным XL-665. Меченное криптатом антитело к сАМР визуализирует метку. Анализ проводили в соответствии с инструкциями от производителя.
Исследуемые соединения добавляли в лунки микропланшетов (384-луночный формат). Клетки HEK-293, экспрессирующие рецептор D1 человека, высевали при 1000 клеток/лунка и инкубировали 30 мин. при комнатной температуре. Метку cAMP-d2 добавляли в лунки, и затем добавляли препарат на основе меченного криптатом антитела к сАМР, и инкубировали в течение 1 ч. при комнатной температуре в темноте. Измеряли HTRF сАМР путем возбуждения донора лазером при 337 нм ("световая единица TRF") и последующим (время задержки 100 микросекунд) измерением эмиссии криптата и d2 при 615 нм и 665 нм в течение временного промежутка 200 микросекунд с временным промежутком 2000 микросекунд между повторениями/100 импульсов). Измерения HRTF проводили на планшет-ридере Envision (PerkinElmer). Сигнал HTRF рассчитывали как соотношение эмиссии при 665 нм и 615 нм. Показатель соотношения HTRF для исследуемых соединений подвергали нормализации до 0% и 100% стимуляции с применением контрольных лунок с DMSO-растворителем или 30 мкМ дофамина. Эффективность исследуемого соединения (ЕС50) оценивали с помощью нелинейной регрессии с использованием сигмоидальной кривой доза-ответ (переменный угловой коэффициент) с применением Xlfit 4 (IDBS, Гилфорд, Суррей, Великобритания, модель 205).
y=(A+((B-A)/(1+((C/x)^D)))),
где у представляет собой нормализованное значение измеренного соотношения HTRF для заданной концентрации исследуемого соединения, х представляет собой концентрацию исследуемого соединения, А представляет собой расчетную эффективность при бесконечном разбавлении соединения, и В представляет собой максимальную эффективность. С представляет собой значение ЕС50, и D представляет собой угловой коэффициент Хилла. Оценки ЕС50 получали из независимого эксперимента, и рассчитывали логарифмическое среднее значение.
Агонизм дофаминового рецептора D2
Агонизм дофаминового рецептора D2 измеряли с применением протокола анализа мобилизации кальция, разработанного HD Biosciences (Китай). Вкратце, клетки HEK293/G15, экспрессирующие рецептор D2 человека, высеивали при плотности 15000 клеток/лунка в покрытых матригелем 384-луночных планшетах с прозрачным дном и выращивали в течение 24 ч. при 37°С в присутствии 5% СО2. Клетки инкубировали с чувствительным к кальцию флуоресцентным красителем Fluo8 в течение 60-90 минут при 37°С в темноте. Исследуемые соединения получали в 3-кратно концентрированном растворе в буфере 1×HBSS с Са2+ и Mg2+. Сигнал потока кальция регистрировался непосредственно после добавления соединений из планшета для соединений в клеточный планшет для FLIPR (Molecular Devices). Данные флуоресценции подвергали нормализации с получением ответов на отсутствие стимуляции (буфер) и полную стимуляцию (1 мкМ дофамина), составляющих 0% и 100% стимуляцию соответственно. Эффективность исследуемого соединения (ЕС50) оценивали с помощью нелинейной регрессии с использованием сигмоидальной кривой доза-ответ (переменный угловой коэффициент) с применением Xlfit 4 (IDBS, Гилфорд, Суррей, Великобритания, модель 205).
y=(A+((B-A)/(1+((C/x)^D)))),
где у представляет собой нормализованное значение измеренного соотношения для заданной концентрации исследуемого соединения, х представляет собой концентрацию исследуемого соединения, А представляет собой расчетную эффективность при бесконечном разбавлении соединения, и В представляет собой максимальную эффективность. С представляет собой значение ЕС50, и D представляет собой угловой коэффициент Хилла. Оценки ЕС50 получали из независимого эксперимента, и рассчитывали логарифмическое среднее значение.
Пример 3. Анализ агонистической активности в отношении 5-НТ2В и связывания
Анализ агонистической активности в отношении 5-НТ2В
Оценку агонистической активности соединений (I), (Ia) и (Ib) в отношении рецептора 5-НТ2В человека проводили Eurofins/Cerep (Франция) с измерением влияния соединений на продуцирование инозитолмонофосфата (IP1) с применением способа обнаружения с HTRF. Вкратце, рецептор 5-НТ2В человека экспрессировался в трансфицированных клетках СНО. Клетки суспендировали в буфере, содержащем 10 мМ Hepes/NaOH (рН 7,4), 4,2 мМ KCl, 146 мМ NaCl, 1 мМ CaCl2, 0,5 мМ MgCl2, 5,5 мМ глюкозу и 50 мМ LiCl, затем распределяли в микропланшеты при плотности 4100 клеток/лунка и инкубировали в течение 30 мин. при 37°С в присутствии буфера (контроля фонового уровня), исследуемого соединения или эталонного агониста. Для контрольного измерения при стимулировании отдельные лунки для анализа содержали 1 мкМ 5-НТ. После инкубации клетки лизировали и добавляли акцептор флуоресценции (фторфен-D2-меченный IP1) и донор флуоресценции (антитело к IP1, меченное криптатом европия). Через 60 мин. при комнатной температуре измеряли перенос энергии флуоресценции при значении возбуждения lambda(Ex), равном 337 нм, и значении излучения lambda(Em), равном 620 и 665 нм, с применением планшет-ридера (Rubystar, BMG). Концентрацию IP1 определяли путем деления значения сигнала, измеренного при 665 нм, на значение сигнала, измеренного при 620 нм (соотношение). Результаты выражали в виде процента контрольного ответа на 1 мкМ 5-НТ. Стандартным эталонным агонистом являлся 5-НТ, который тестировали в каждом эксперименте при нескольких концентрациях для получения кривой концентрация-ответ, из которой рассчитывали его значение ЕС50, как описано выше для функциональных анализов в отношении дофамина.
Анализ связывания 5-НТ2В
Оценку аффинности соединений (Id-ia), (Id-ib), (Id-iia), (Id-iib) и (Id-iab) в отношении рецептора 5-НТ2В человека определяли в анализе связывания радиолиганда в Eurofins/Cerep (Франция). Гомогенаты мембран получали из клеток СНО, экспрессирующих рецептор 5НТ2В человека, инкубировали в течение 60 мин. при комнатной температуре с 0,2 нМ [125I](±)DOI (1-(4-йод-2,5-диметоксифенил)пропан-2-амин) в отсутствие или в присутствии исследуемого соединения в буфере, содержащем 50 мМ трис-HCl (рН 7,4), 5 мМ MgCl2, 10 мкМ паргилин и 0,1% аскорбиновую кислоту. Неспецифическое связывание определяют в присутствии 1 мкМ (±)DOI. После инкубации образцы быстро фильтровали в вакууме через фильтры из стекловолокна (GF/B, Packard), предварительно пропитанные 0,3% полиэтиленимином (PEI), и ополаскивали несколько раз охлажденным льдом 50 мМ трис-HCl с применением коллектора клеток на 96 образцов (Unifilter, Packard). Фильтры высушивали и измеряли радиоактивность посредством сцинтилляционного счетчика (Topcount, Packard) с применением сцинтилляционного коктейля (Microscint 0, Packard). Результаты выражены в проценте ингибирования специфического связывания контрольного радиолиганда. Стандартное эталонное соединение представляло собой (±)DOI, которое тестировали в каждом эксперименте при нескольких концентрациях с получением кривой конкуренции, из которой рассчитывали его IC50.
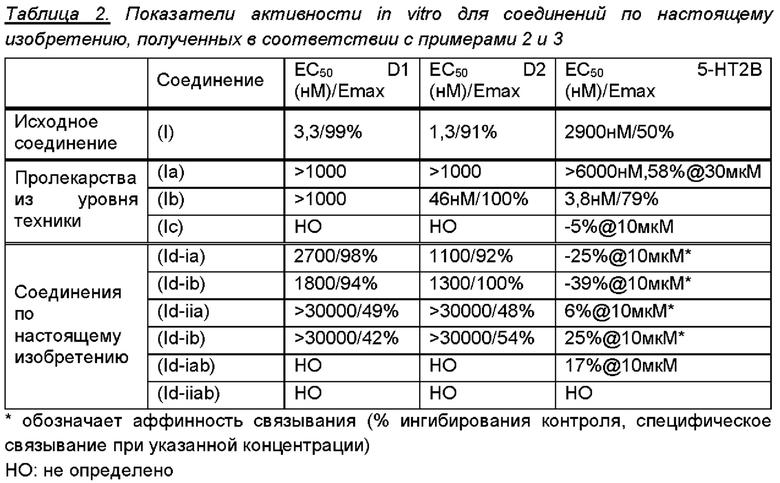
Пример 4. PK-эксперименты в отношении крыс
Для всех экспериментов образцы крови объемом примерно 0,68 мл брали из хвостовой или подъязычной вены и помещали в пробирки K3EDTA, которые были предварительно охлаждены и получены с использованием стабилизирующего раствора, состоящего из 80 мкл аскорбиновой кислоты и 40 мкл 100 мМ 1,4 лактона D-сахарной кислоты в воде. Пробирки осторожно переворачивали 6-8 раз, чтобы обеспечить тщательное перемешивание, а затем помещали в жидкий лед. Пробирку для сбора образцов помещали в жидкий лед на срок до 30 минут до центрифугирования. После удаления из жидкого льда немедленно начинали центрифугирование. Сразу после окончания центрифугирования образцы возвращали в жидкий лед. Три подобразца по 130 мкл плазмы крови переносили в каждую из трех соответствующим образом меченных криопробирок, содержащих 6,5 мкл предварительно охлажденной муравьиной кислоты (20%) (в пробирки предварительно осуществляли добавление и хранили охлажденными до применения). Крышку пробирки немедленно заменяли, и раствор плазмы крови тщательно смешивали путем осторожного переворачивания 6-8 раз. Образцы хранили замороженными при номинальной температуре -70°С в течение 60 минут после отбора образцов. Условия центрифугирования: при 3000 G в течение 10 минут при 4°С. После сбора плазму крови помещали на водный лед. Конечное хранение проводили при примерно -70°С.
Образцы плазмы крови анализировали с помощью твердофазной экстракции или непосредственного осаждения белка с последующей UPLC-MS/MS. Осуществляли MS-обнаружение с применением электрораспыления в режиме положительных ионов с мониторингом конкретного отношения массы к заряду при переходах для соединения (I) с применением внутренних стандартов для корректирования ответа. Анализировали данные зависимости концентрации от времени с применением стандартного программного обеспечения с применением подходящих некомпартментных методик с получением оценок полученных PK-параметров.
Оборудование, применяемое для анализа соединения (I) после введения дозы соединения (Ia)
Масс-спектрометр (LC-MS/MS) Waters Acquity - Sciex API 5000. Аналитическая колонка представляла собой колонку Waters ВЕН UPLC Phenyl 100×2,1 мм, размер частиц 1,7 мкм. Подвижная фаза А: 20 мМ формиат аммония (водн.) + 0,5% муравьиная кислота. Подвижная фаза В: ацетонитрил. Применяли градиент от 95/5% до 2/98 за 6,1 мин. Скорость потока составляла 0,5 мл/мин. Осуществляли MRM-мониторинг (мониторинг множественных реакций) исследуемого образца и добавленных аналитических стандартов.
Введение дозы и забор образцов крови Крысы линии Wistar Han предоставлены от Charles River Laboratories, Зульцфельд, Германия. Поддерживали искусственный автоматически контролируемый 12-часовой цикл света и темноты. Крысы получали стандартный лабораторный корм от Brogaarden (пеллеты Altromin 1324). Крысы имели неограниченный доступ к корму. Во время исследования (4-недельное исследование токсичности) крысы перорально получали один раз в день дозы (Ia) через желудочный зонд. У крыс, которым давали 300 мкг/кг (Ia), собирали образцы крови) у 3 самцов сателлитных животных в следующие моменты времени в день 29: 0,5, 1, 2, 4, 6, 8, 12 и 24 часа после введения дозы.
Оборудование, применяемое для анализа соединения (I) после введения дозы соединения (Ib)
Масс-спектрометр (LC-MS/MS) Waters Acquity - Sciex API 5000. Аналитическая колонка представляла собой колонку Waters ВЕН UPLC Phenyl 100×2,1 мм, размер частиц 1,7 мкм. Подвижная фаза А: 20 мМ формиат аммония (водн.) + 0,5% муравьиная кислота. Подвижная фаза В: ацетонитрил. Применяли градиент от 95/5% до 2/98 за 6,1 мин. Скорость потока составляла 0,5 мл/мин. Осуществляли MRM-мониторинг исследуемого образца и добавленных аналитических стандартов.
Введение дозы и забор образцов крови Крысы линии Wistar Han предоставлены от Charles River Laboratories, Великобритания. Поддерживали искусственный автоматически контролируемый 12-часовой цикл света и темноты. Крысы получали стандартный лабораторный корм (корм Teklad 2014С). Крысы имели неограниченный доступ к корму. Во время исследования (26-недельное исследование токсичности) крысы перорально получали один раз в день дозы (Ib) через желудочный зонд. У крыс, которым давали 300 мкг/кг (Ib), собирали образцы крови у 3 самцов сателлитных животных в следующие моменты времени в день 182: 0,5, 1, 2, 4, 8 и 24 часа после введения дозы.
Оборудование, применяемое для анализа соединения (I) после введения дозы соединений (Ic), (Id-ia), (Id-ib), (Id-iia), (Id-iib), (Id-iab) и (Id-iiab)
Масс-спектрометр (LC-MS/MS) Waters Acquity - Waters Xevo TQ-S. Аналитическая колонка Acquity ВЕН C18 100×2,1 мм, 1,7 мкм. Подвижная фаза А: 20 мМ NH4-формиат + 0,2% муравьиная кислота. Подвижная фаза В: ацетонитрил + 0,2% муравьиная кислота. Применяли градиент от 95/5% до 5/95% за 11,0 мин. Скорость потока составляла 0,3 мл/мин. Осуществляли MRM-мониторинг исследуемого образца и добавленных аналитических стандартов.
Введение дозы и забор образцов крови для соединений (Id-ia), (Id-ib), (Id-iia) и (Id-iib) Крысы линии Wistar Han предоставлены от Charles River Laboratories, Wiga GmbH, Германия. Поддерживали искусственный автоматически контролируемый 12-часовой цикл света и темноты. Крысы получали стандартный лабораторный корм от Brogaarden (пеллеты Altromin 1324). Крысы имели неограниченный доступ к корму. Самцам крыс линии Wistar Han перорально через желудочный зонд вводили дозу путем однократного перорального введения через желудочный зонд (Id-ia), (Id-ib), (Id-iia) и (Id-iib) соответственно. Крысам давали 633 мкг/кг (Id-ia) и (Id-ib)) или 392 мкг/кг (Id-iia) и (Id-iib)), собирали образцы крови у 3 самцов животных в следующие моменты времени в день 1: 1, 2, 4, 6, 8 и 24 часа после введения дозы.
Введение дозы и забор образцов крови для соединений (Ic), (Id-iab) и (Id-iiab) Крысы линии Wistar Han предоставлены от Envigo, Великобритания. Поддерживали искусственный автоматически контролируемый 12-часовой цикл света и темноты. Крысы получали стандартный лабораторный корм Teklad 2014С. Крысы имели неограниченный доступ к корму. Самцам крыс линии Wistar Han через желудочный зонд вводили дозу путем однократного перорального введения через желудочный зонд (Ic), (Id-iab) и (Id-iiab) соответственно. Крысам давали 793 мкг/кг (Id-iab), 703 мкг/кг (Id-iiab) и 494 мкг/кг (Ic). Собирали образцы крови у 3 самцов животных в следующие моменты времени в день 1: 1, 2, 4, 6, 8 и 24 часа после введения дозы.
Оборудование, применяемое для анализа апоморфина после введения дозы апоморфина и соответствующего глюкуронидного конъюгата, представляющего собой (((2S,3S,4S,5R,6S)-6-[[(6aR)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ил]окси]-3,4,5-тригидрокситетрагидропиран-2-карбоновую кислоту, и сульфатных конъюгатов, представляющих собой [(6aR)-11-гидрокси-6-метил-5,6,6а,7-тетрагидро-4Н-дибензо[de,g]хинолин-10-ил]гидросульфат и [(6aR)-10-гидрокси-6-метил-5,6,6а,7-тетрагидро-4H-дибензо[de,g]хинолин-11-ил]гидросульфат)
Масс-спектрометр (UPCLC-MS/MS) Waters Acquity I-Class-Waters Xevo TQ-S. Аналитическая колонка Acquity HSS Т3 C18 50×2,1 мм, 1,8 мкм. Подвижная фаза А: 10 мМ NH4-формиат, 0,2% муравьиная кислота : ацетонитрил (95:5). Подвижная фаза В: 10 мМ NH4-формиат, 0,2% муравьиная кислота : ацетонитрил (5:95). Применяли градиент от 95/5% до 5/95% за 2,40 мин. Скорость потока составляла 0,3 мл/мин. Осуществляли MRM-обнаружение исследуемых образцов и добавленных аналитических стандартов.
Введение дозы и забор образцов крови Крысы линии Wistar Han предоставлены от Charles River Laboratories, Wiga GmbH, Германия. Поддерживали искусственный автоматически контролируемый 12-часовой цикл света и темноты. Крысы получали стандартный лабораторный корм от Brogaarden (пеллеты Altromin 1324). Крысы имели неограниченный доступ к корму. Самцам крыс линии Wistar Han вводили однократную дозу апоморфина либо подкожно, либо перорально через желудочный зонд, или перорально вводили однократную дозу апоморфиновых конъюгатов через желудочный зонд. У крыс, которым вводили 3000 мкг/кг (апоморфина), или 3899 мкг/кг (сульфатного конъюгата), или 4977 мкг/кг (глюкуронидных конъюгатов) собирали образцы крови у 3 самцов животных в следующие моменты времени в день 1: 0,25, 0,5, 1, 2, 4 и 8 часов после SC-введения и 0,5, 1, 2, 4, 8 и 24 ч. после РО-введения дозы.
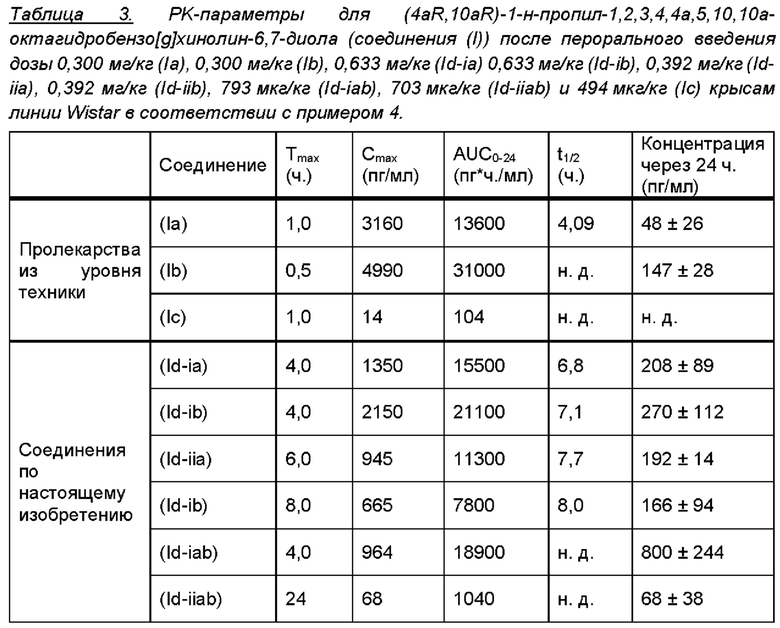
Пример 5. PK-эксперименты в отношении карликовых свиней
Образцы крови объемом примерно 0,5 мл отбирали из яремной вены с помощью шприца и помещали в предварительно охлажденные пробирки с EDTA со стабилизирующим раствором, как описано в случае крыс в примере 4. Измеряли значения концентрации соединений в плазме крови. Образцы плазмы крови анализировали с помощью твердофазной экстракции или непосредственного осаждения белка с последующей UPLC-MS/MS. Осуществляли MS-обнаружение с применением электрораспыления в режиме положительных ионов с мониторингом конкретного отношения массы к заряду при переходах для рассматриваемого соединения с применением внутренних стандартов для корректирования ответа. Анализировали данные зависимости концентрации от времени с применением стандартного программного обеспечения с применением подходящих однокомпартментных методик с получением оценок полученных PK-параметров.
Оборудование, применяемое для анализа соединения (I) после введения дозы соединений (Id-ia), (Id-ib), (Id-iia) и (Id-iib)
Масс-спектрометр (LC-MS/MS) Waters Acquity - Waters Xevo TQ-S. Аналитическая колонка Acquity ВЕН C18 100×2,1 мм, 1,7 мкм. Подвижная фаза А: 20 мМ NH4-формиат + 0,2% муравьиная кислота. Подвижная фаза В: ацетонитрил + 0,2% муравьиная кислота. Применяли градиент от 95/5% до 95/5 за 11,0 мин. Скорость потока составляла 0,3 мл/мин. Осуществляли MRM-мониторинг исследуемого образца и добавленных аналитических стандартов.
Введение дозы и забор образцов крови при фармакокинетическом исследовании однократной дозы у самки карликовой свиньи Ellegaard  предоставленной Ellegaard, Дания. Поддерживали искусственный автоматически контролируемый 12-часовой цикл света и темноты. Карликовые свиньи получали стандартный лабораторный корм от Brogaarden (пеллеты Altromin). Карликовые свиньи имели неограниченный доступ к корму. Карликовым свиньям перорально через желудочный зонд вводили соединения (Id-ia), (Id-ib), (Id-iia) и (Id-iib) соответственно. Карликовым свиньям вводили дозу 160 мкг/кг соединений (Id-ia) и (Id-ib) соответственно или 80 мкг/кг соединений (Id-iia) и (Id-iib) соответственно, собирали образцы крови у 3 самок животных в следующие моменты времени в день 1: 1, 2, 4, 6, 8, 12 и 24 часа после введения дозы.
предоставленной Ellegaard, Дания. Поддерживали искусственный автоматически контролируемый 12-часовой цикл света и темноты. Карликовые свиньи получали стандартный лабораторный корм от Brogaarden (пеллеты Altromin). Карликовые свиньи имели неограниченный доступ к корму. Карликовым свиньям перорально через желудочный зонд вводили соединения (Id-ia), (Id-ib), (Id-iia) и (Id-iib) соответственно. Карликовым свиньям вводили дозу 160 мкг/кг соединений (Id-ia) и (Id-ib) соответственно или 80 мкг/кг соединений (Id-iia) и (Id-iib) соответственно, собирали образцы крови у 3 самок животных в следующие моменты времени в день 1: 1, 2, 4, 6, 8, 12 и 24 часа после введения дозы.

Пример 6. PK/PD соединения (Id-ia)/соединения (I) у крыс в анализе гиперактивности
Животные
Всего в исследовании использовали 206 самцов крыс CD (Charles River, Германия) весом 200-250 грамм (165-190 грамм по прибытии). Животных содержали при стандартной температуре (22±1°С) и в среде с контролируемым освещением (свет включен с 7 утра до 8 вечера) с доступом к пище и воде по желанию. Эксперимент, описанный ниже, проводили в соответствии со стандартными рабочими процедурами Charles River Discovery Research Services Finland Ltd. и в соответствии с руководством национального совета по экспериментам на животных Финляндии ( ELLA) в отношении испытаний на животных.
ELLA) в отношении испытаний на животных.
Исследование локомоторной активности, тест "открытое поле"
Испытательное устройство представляет собой квадратную плексигласовую площадку (размером 40×40×40 см), на которой регистрируют траектории движения крыс с помощью монитора активности (Med. Associates Inc.). Перед началом периода испытаний крысам давали привыкнуть к их клетке для испытаний в течение 60 минут. После завершения привыкания животных обрабатывали либо соединением, либо средой-носителем и помещали обратно в установку "открытое поле". Основным измеряемым параметром теста является пройденное расстояние (регистрировали через 5-минутные отрезки времени). Общее время измерения после получения начальной обработки составляло 360 минут. Общий период наблюдения в исследовании составлял 420 минут, включая 60 минут привыкания.
Результаты
Оценивали пероральное введение соединения (Id-ia) в анализе локомоторной активности крыс и данные функциональные показатели затем коррелировали со значениями концентрации в плазме крови соединения (I). Апоморфин и прамипексол также одновременно исследовали в данном анализе в качестве препаратов сравнения (т.е. известного стандарта лечения (SoC) в области терапии болезни Паркинсона), и анализировали концентрацию в плазме крови в отношении апоморфина.
Как показано на фигуре 3, соединение (Id-ia) (от 10 до 300 мкг/кг, р.о.) увеличивает локомоторную активность с эффектом, начинающимся примерно через 2 часа после введения (приблизительно 180-минутный момент времени) и сохраняющимся до конца регистрирования (в 415-минутный момент времени). Напротив, гиперактивность, вызванная апоморфином (3 мг/кг, s.c.), является мгновенной, но кратковременной, поскольку эффект исчезает через 1,5 часа после введения (в 150-минутный момент времени). Прамипексол (0,3 мг/кг, s.c.) также вызывает увеличение активности, но его эффект проявляется через приблизительно 1 час после введения и исчезает через 2,5 часа (в 270-минутный момент времени). Общее пройденное расстояние, как видно на фигуре 4, демонстрирует значительно увеличенную активность как для соединения (Id-ia), так и для двух исследуемых препаратов сравнения, и данный эффект является эффектом, который следует ожидать от агонистов дофамина.
Параллельно с оценкой локомоторной активности отбирали образцы плазмы крови у сателлитных животных в 6 различных моментов времени (0,5, 1, 2, 3, 4 и 6 часов после введения дозы для животных, обработанных соединением (Id-ia)). Фармакокинетический анализ демонстрирует, что влияние на поведение соединения (Id-ia) (100 мкг/кг, р.о.) коррелирует со значениями концентрации в плазме крови соединения (I) (см. фигуру 5) с подтверждением того, что влияние на поведение соединения (Id-ia) обусловлено соединением (I), а не соединением (Id-ia) самим по себе. В соответствующем анализе воздействия апоморфина (через 0,25, 0,5, 1, 2, 4 и 6 часов после введения дозы) в результате получали корреляцию между значениями концентрации апоморфина в плазме крови и гиперактивным поведением (см. фигуру 6).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ТВЕРДЫЕ ФОРМЫ (2S,3S,4S,5R,6S)-3,4,5-ТРИГИДРОКСИ-6-(((4AR,10AR)-7-ГИДРОКСИ-1-ПРОПИЛ-1,2,3,4,4A,5,10,10A-ОКТАГИДРОБЕНЗО[G]ХИНОЛИН-6-ИЛ)ОКСИ)ТЕТРАГИДРО-2H-ПИРАН-2-КАРБОНОВОЙ КИСЛОТЫ | 2020 |
|
RU2820501C2 |
| СПОСОБ ИЗГОТОВЛЕНИЯ (2S,3S,4S,5R,6S)-3,4,5-ТРИГИДРОКСИ-6-(((4AR,10AR)-7-ГИДРОКСИ-1-ПРОПИЛ-1,2,3,4,4А,5,10,10А-ОКТАГИДРОБЕНЗО[G]ХИНОЛИН-6-ИЛ)ОКСИ)ТЕТРАГИДРО-2Н-ПИРАН-2-КАРБОНОВОЙ КИСЛОТЫ И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ЕЕ ПОЛУЧЕНИЯ | 2020 |
|
RU2817700C2 |
| СПОСОБ ПОЛУЧЕНИЯ (2S,3S,4S,5R,6S)-3,4,5-ТРИГИДРОКСИ-6-(((4AR,10AR)-7-ГИДРОКСИ-1-ПРОПИЛ-1,2,3,4,4A,5,10,10A-ОКТАГИДРОБЕНЗО[G]ХИНОЛИН-6-ИЛ)ОКСИ)ТЕТРАГИДРО-2H-ПИРАН-2-КАРБОНОВОЙ КИСЛОТЫ | 2020 |
|
RU2819919C2 |
| СПОСОБ ИЗГОТОВЛЕНИЯ (6AR,10AR)-7-ПРОПИЛ-6,6A,7,8,9,10,10A,11-ОКТАГИДРО-[1,3]ДИОКСОЛО[4',5':5,6]БЕНЗО[1,2-G]ХИНОЛИНА И (4AR,10AR)-1-ПРОПИЛ-1,2,3,4,4A,5,10,10A-ОКТАГИДРОБЕНЗО[G]ХИНОЛИН-6,7-ДИОЛА | 2020 |
|
RU2819924C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ | 2003 |
|
RU2345991C2 |
| ПРОИЗВОДНЫЕ БЕНЗО(G)ХИНОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2158738C2 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| БЕНЗО [F] ХИНОЛИНОН, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1995 |
|
RU2172312C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| НОВЫЕ ПРЕДШЕСТВЕННИКИ ПРОИЗВОДНЫХ ГЛУТАМАТА | 2012 |
|
RU2600981C2 |
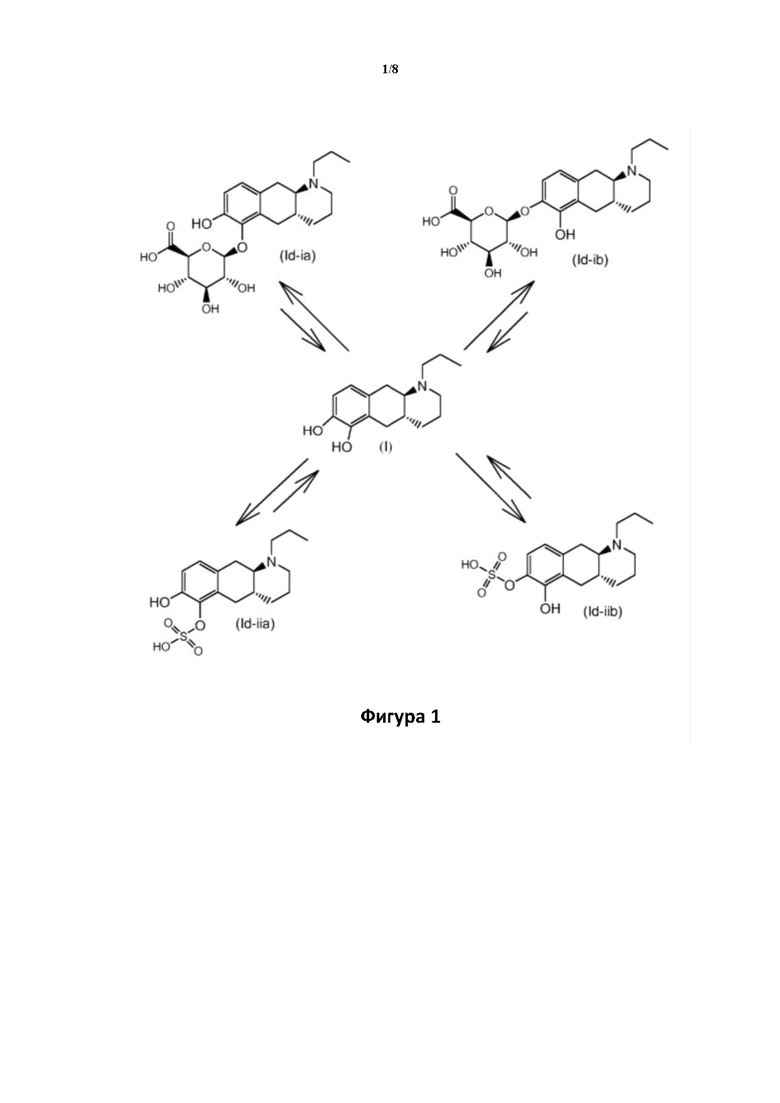
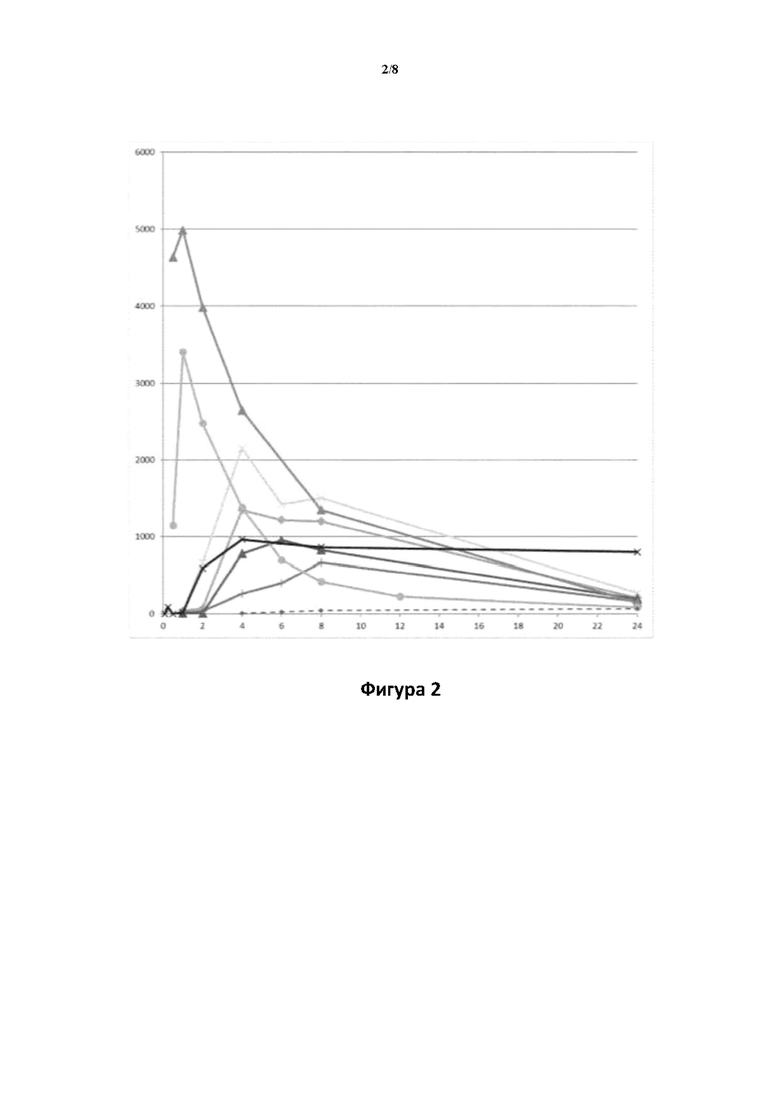

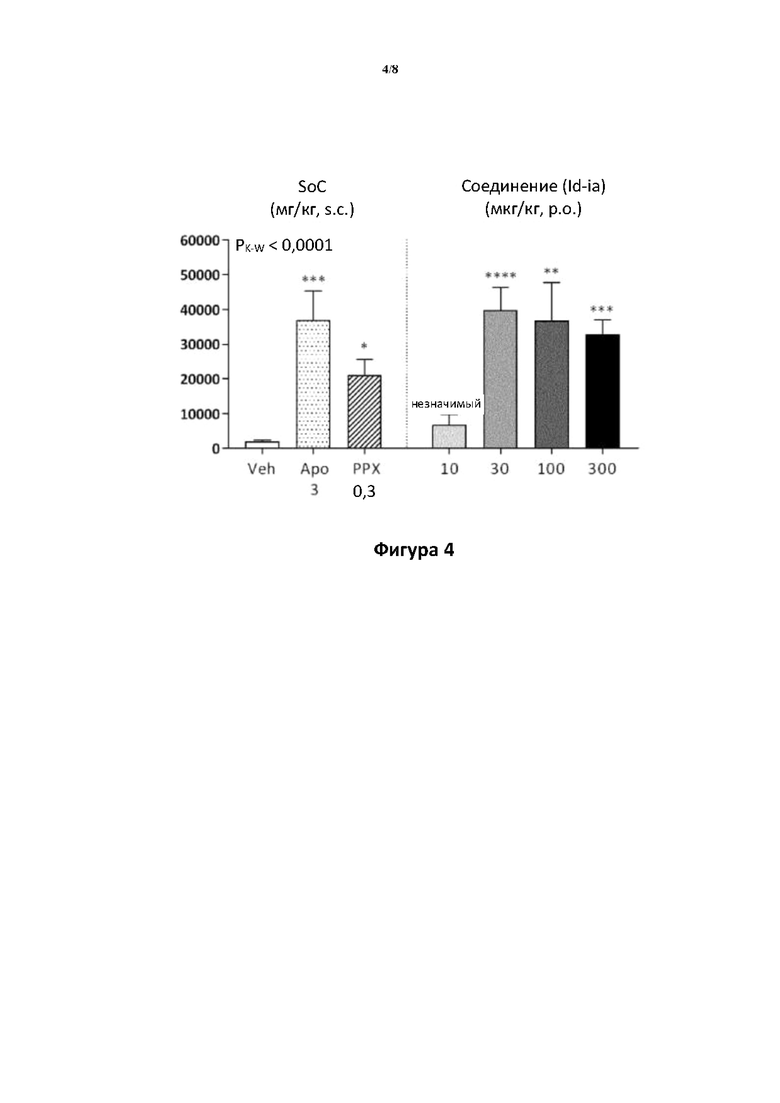
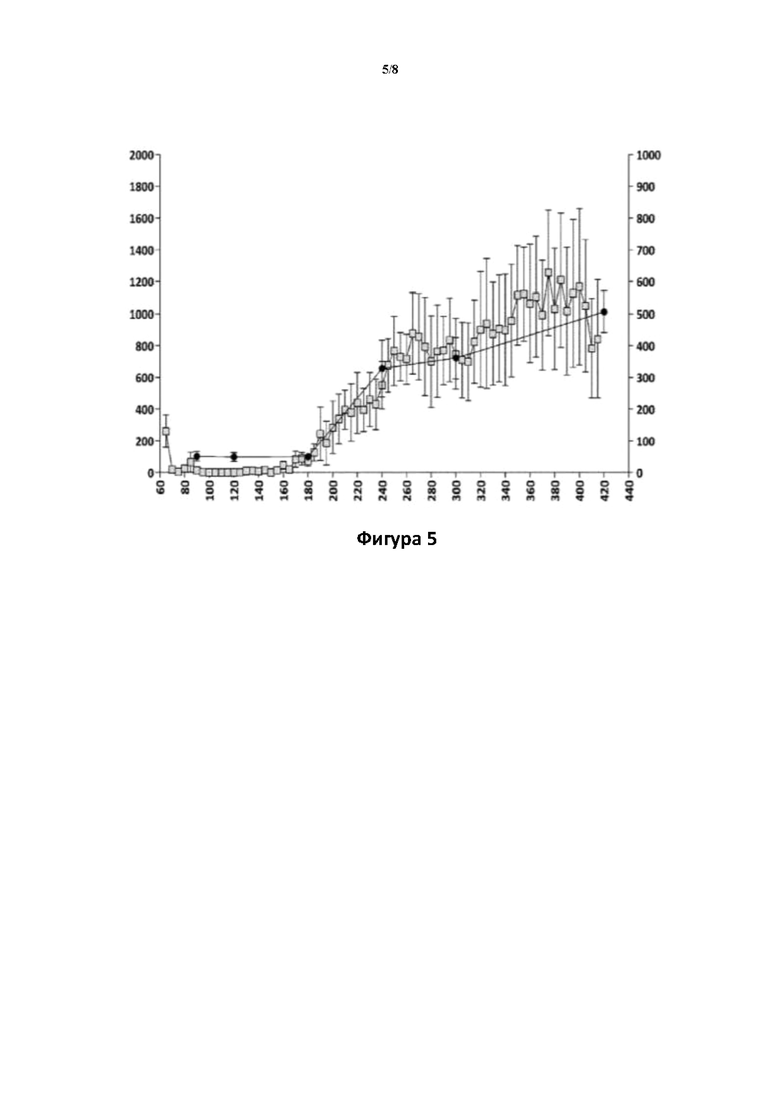
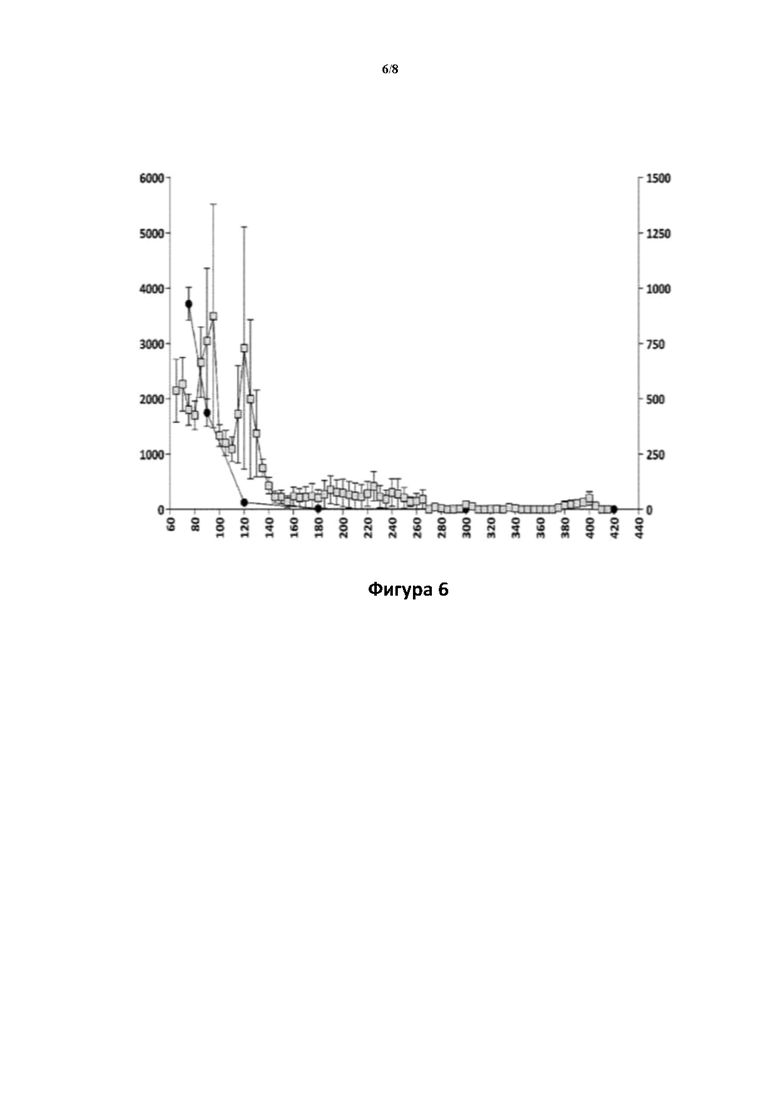
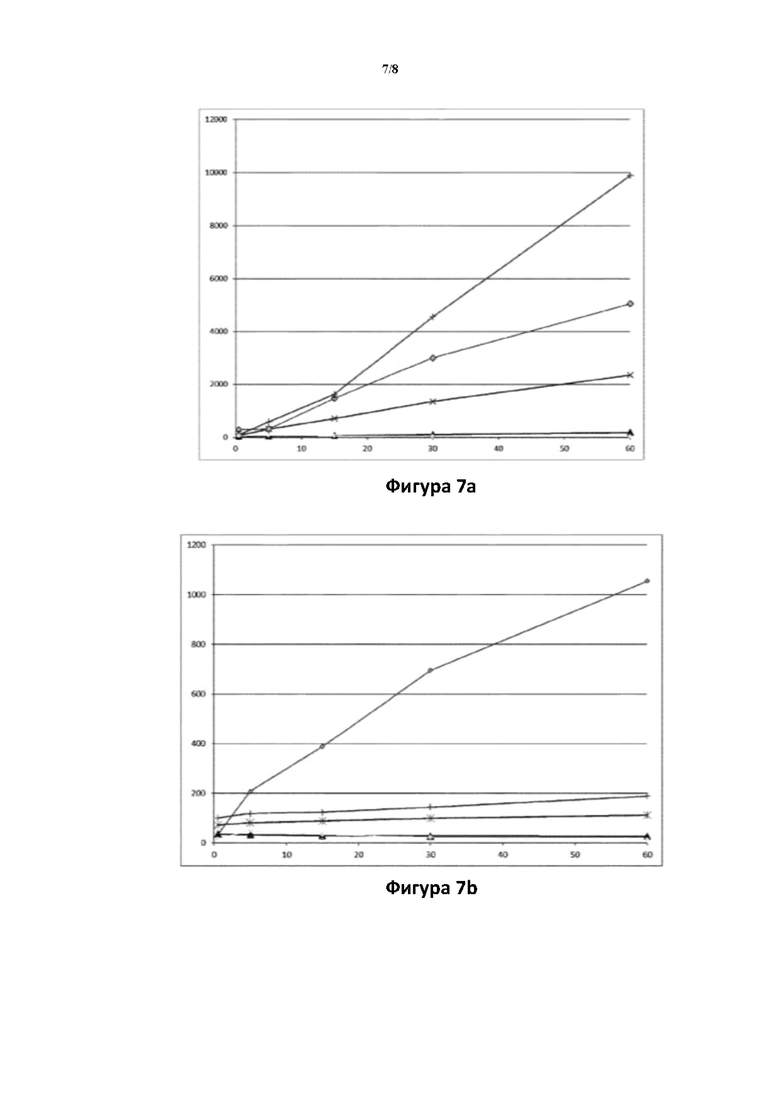
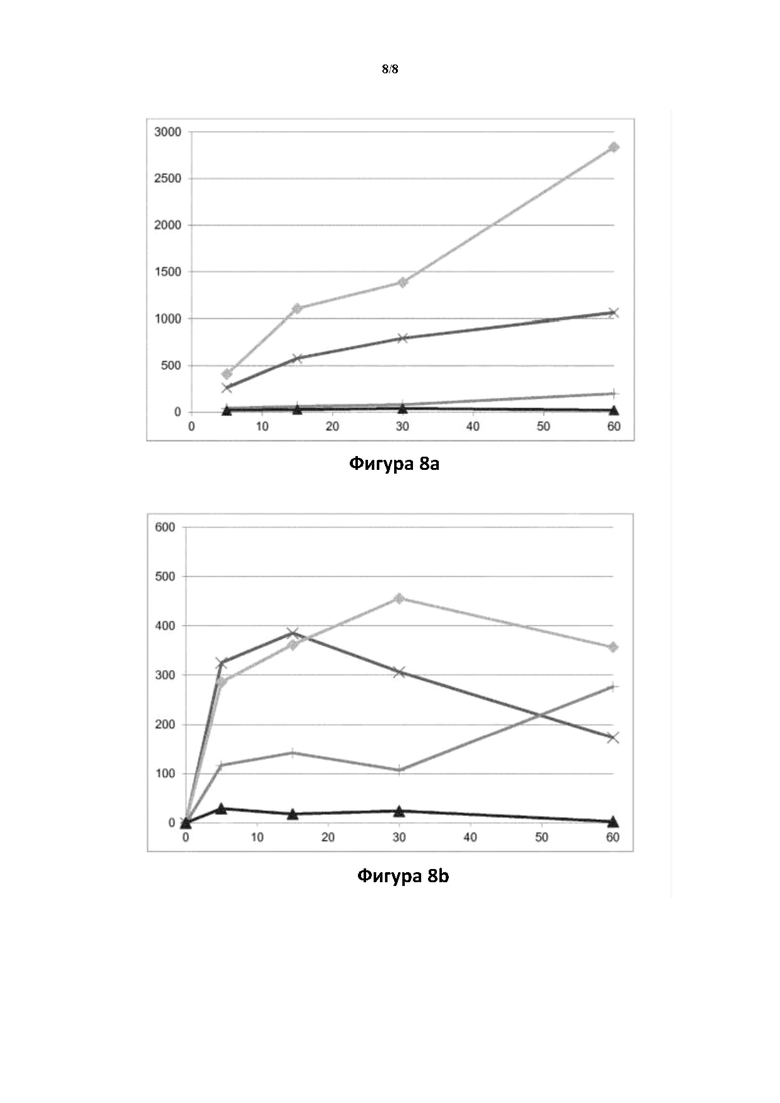
Группа изобретений относится к области органической химии и фармацевтики, а именно к пролекарствам на основе катехоламина для применения в лечении нейродегенеративных заболеваний и нарушений. Раскрыто соединение формулы (Id), где R1 представляет собой H и R2 выбран из одного из заместителей (i) и (ii); или R1 выбран из одного из заместителей (i) и (ii), указанных ниже, и R2 представляет собой H; или R1 и R2 одновременно представлены заместителем (i), указанным ниже; или R1 и R2 одновременно представлены заместителем (ii), указанным ниже; или R1 представляет собой заместитель (i) и R2 представляет собой заместитель (ii); или R1 представляет собой заместитель (ii) и R2 представляет собой заместитель (i); где * обозначает точку присоединения; и при этом атом углерода в точке присоединения при заместителе (i) находится в S-конфигурации; или его фармацевтически приемлемая соль. Кроме того, раскрыта фармацевтическая композиция, имеющая дофаминовую агонистическую активность и содержащая терапевтически эффективное количество указанного выше соединения, способы лечения нейродегенеративного и нейропсихиатрического заболевания, а кроме того, применение соединения Id для лечения заболеваний и для получения лекарственного препарата. Группа изобретений обеспечивает эффективное лечение нейродегенеративных заболеваний за счет пролекарственных соединений с оптимальными фармакокинетическимим профилями. 7 н. и 16 з.п. ф-лы, 8 ил., 4 табл., 6 пр.
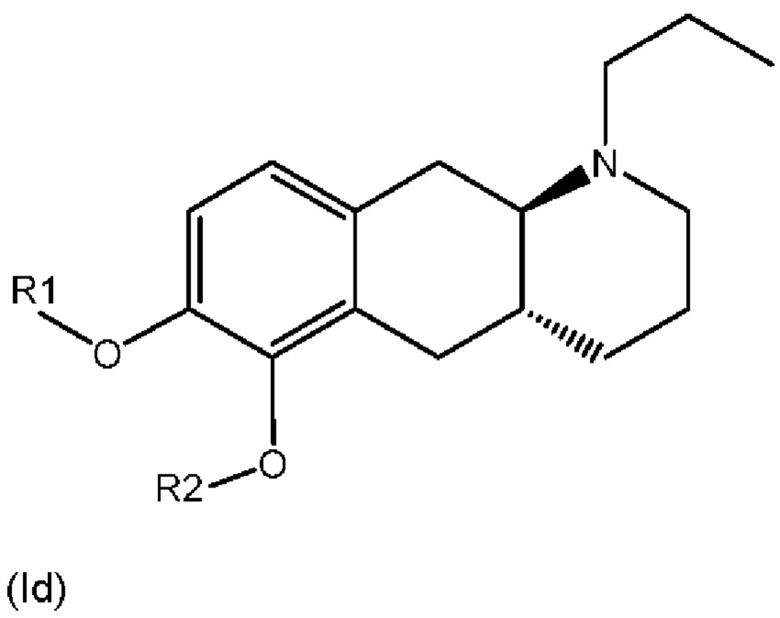
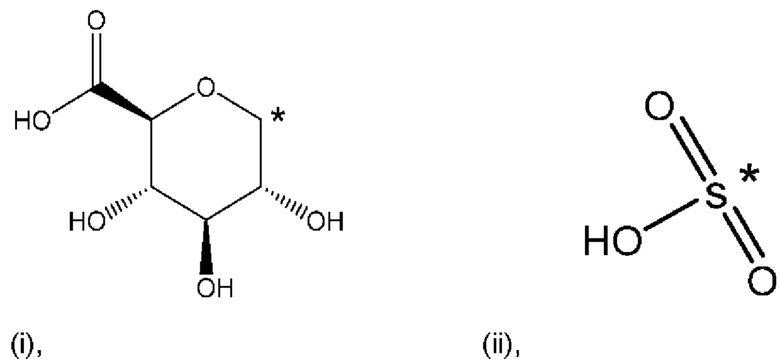
1. Соединение формулы (Id)
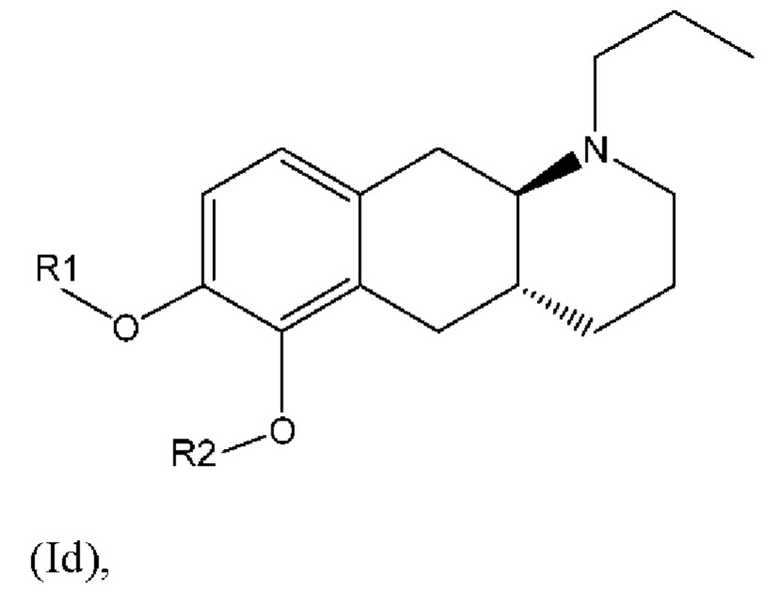
где R1 представляет собой Н и R2 выбран из одного из заместителей (i) и (ii), указанных ниже; или
R1 выбран из одного из заместителей (i) и (ii), указанных ниже, и R2 представляет собой Н; или
R1 и R2 одновременно представлены заместителем (i), указанным ниже; или
R1 и R2 одновременно представлены заместителем (ii), указанным ниже; или
R1 представляет собой заместитель (i) и R2 представляет собой заместитель (ii); или
R1 представляет собой заместитель (ii) и R2 представляет собой заместитель (i)
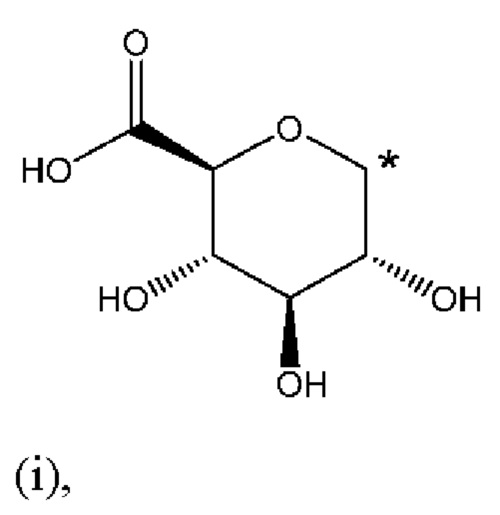
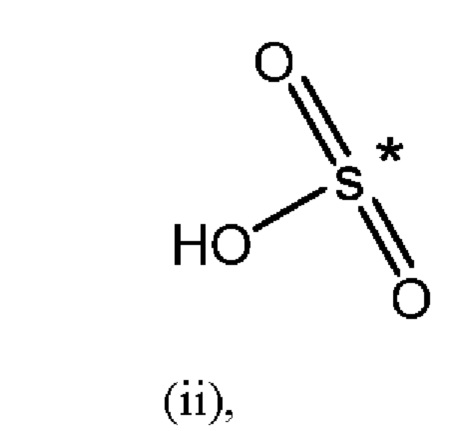
где * обозначает точку присоединения; и
при этом атом углерода в точке присоединения при заместителе (i) находится в S-конфигурации;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где соединение выбрано из группы, состоящей из
(Id-ia): (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR, 10aR)-7-гидрокси-1-пропил-1,2,3,4,4a,5,10,10a-октагидробензо[g]хинолин-6-ил)окси)тетрагидро-2H-пиран-2-карбоновой кислоты;
(Id-ib): (2S,3S,4S,5R,6S)-3,4,5-тригидрокси-6-(((4aR,10aR)-6-гидрокси-1-пропил-1,2,3,4,4a,5,10,10a-октагидробензо[g]хинолин-7-ил)окси)тетрагидро-2H-пиран-2-карбоновой кислоты;
(Id-iia): (4aR, 10aR)-7-гидрокси-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6-илгидросульфата;
(Id-iib): (4aR,10aR)-6-гидрокси-1-пропил-1,2,3,4,4a,5,10,10а-октагидробензо[g]хинолин-7-ил)гидросульфата;
(Id-iab): (2S,2'S,3S,3'S,4S,4'S,5R,5'R,6S,6'S)-6,6'-(((4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диил)бис(окси))бис(3,4,5-тригидрокситетрагидро-2Н-пиран-2-карбоновой кислоты);
(Id-iiab): (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диил-бис(гидросульфата);
или фармацевтически приемлемой соли любого из этих соединений.
3. Соединение по п. 1, где указанное соединение представляет собой соединение, представленное формулой (Id-ia), приведенной ниже,
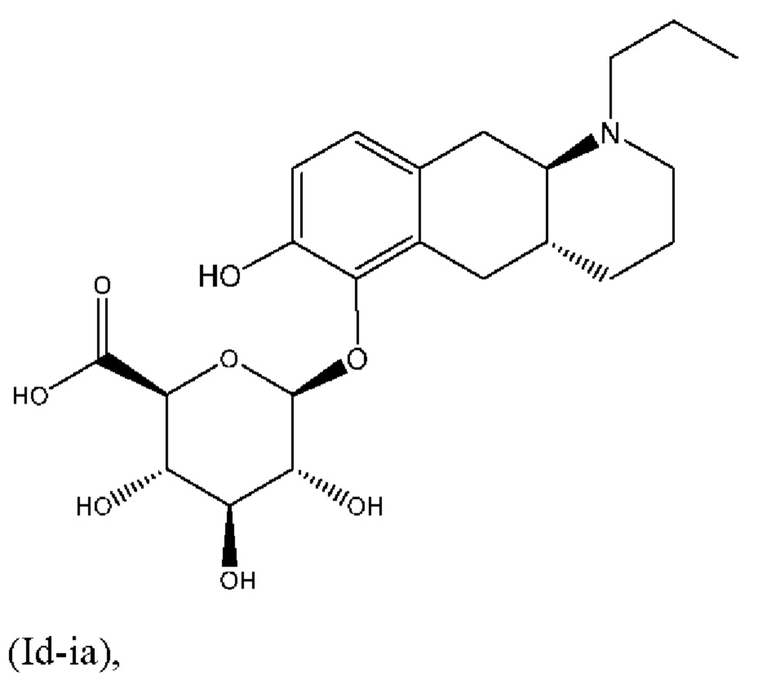
или его фармацевтически приемлемая соль.
4. Соединение по п. 1, где указанное соединение представляет собой соединение, представленное формулой (Id-ib), приведенной ниже,
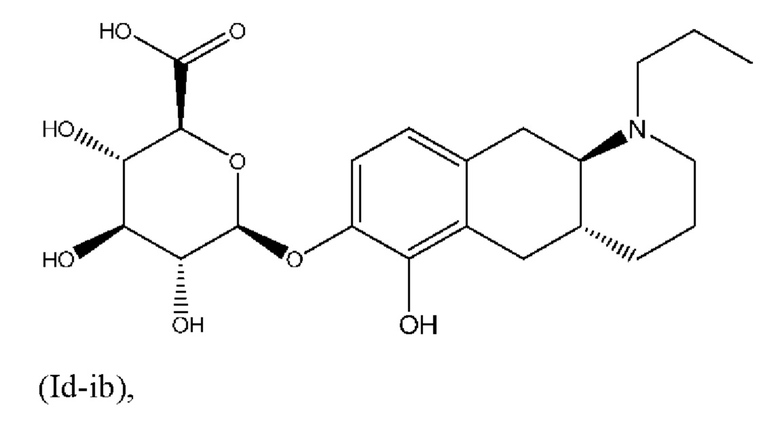
или его фармацевтически приемлемая соль.
5. Соединение по п. 1, где указанное соединение представляет собой соединение, представленное формулой (Id-iab), приведенной ниже,
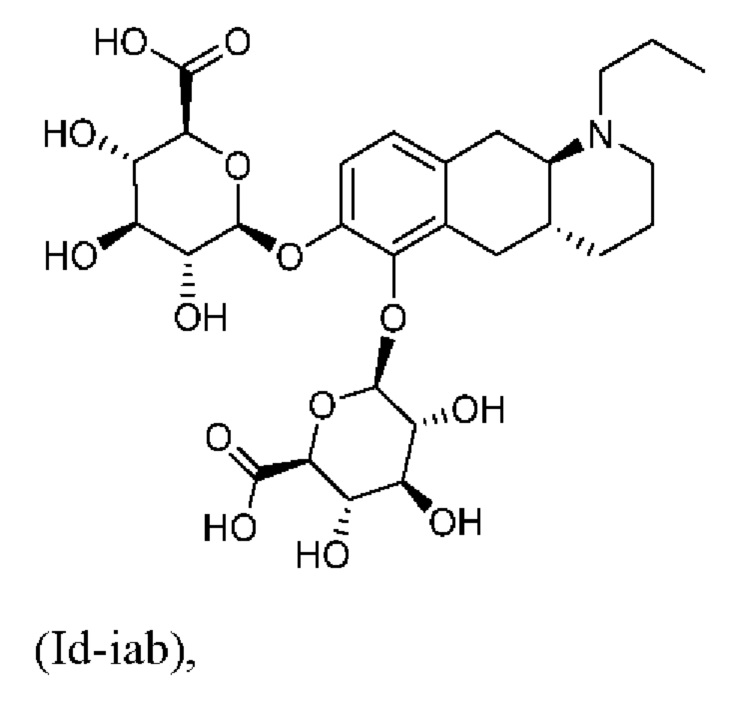
или его фармацевтически приемлемая соль.
6. Соединение по п. 1, где указанное соединение представляет собой соединение, представленное формулой (Id-iia), приведенной ниже,
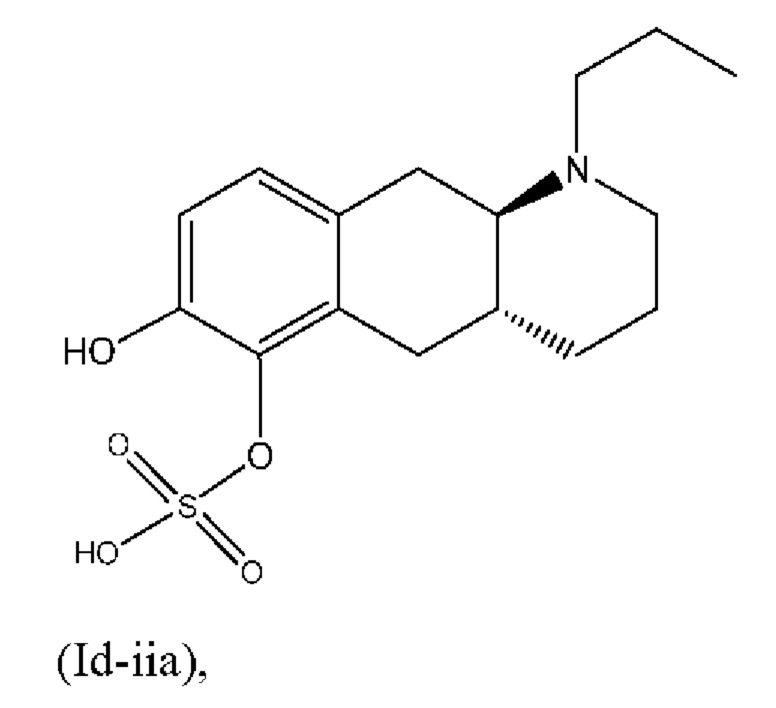
или его фармацевтически приемлемая соль.
7. Соединение по п. 1, где указанное соединение представляет собой соединение, представленное формулой (Id-iib), приведенной ниже,
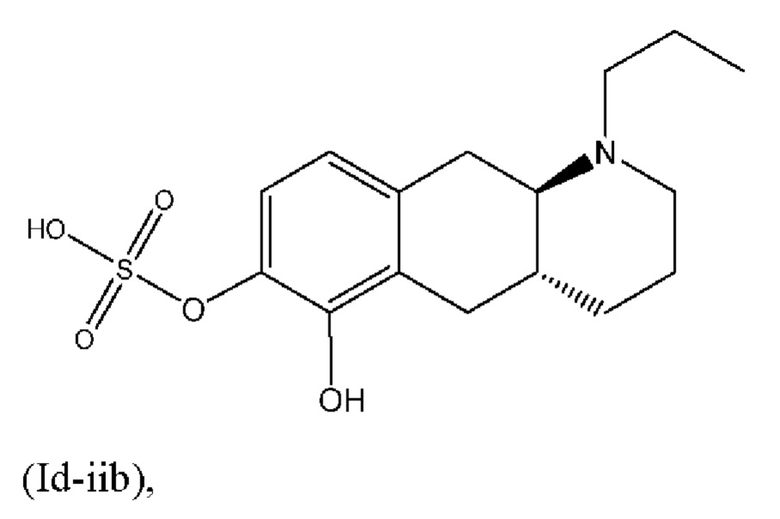
или его фармацевтически приемлемая соль.
8. Соединение по п. 1, где указанное соединение представляет собой соединение, представленное формулой (Id-iiab), приведенной ниже,
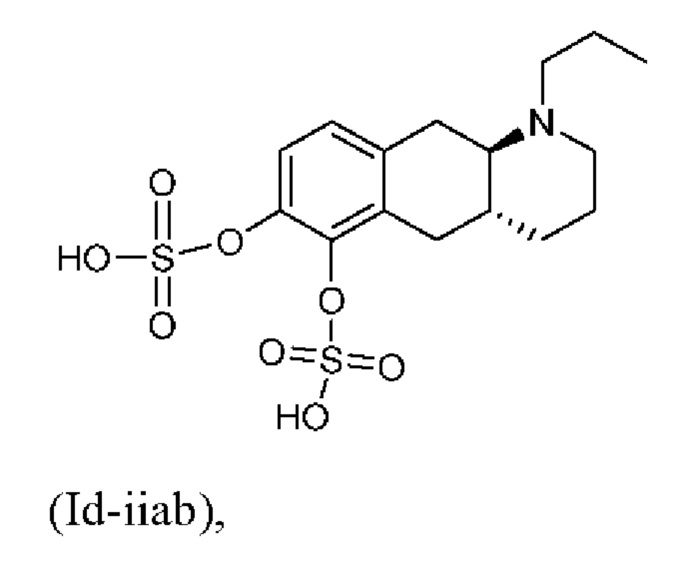
или его фармацевтически приемлемая соль.
9. Соединение по любому из пп. 1-8 или его фармацевтически приемлемая соль, где указанное соединение представлено в выделенной форме, по сути, не предусматривающей присутствия соединения формулы (I) ниже
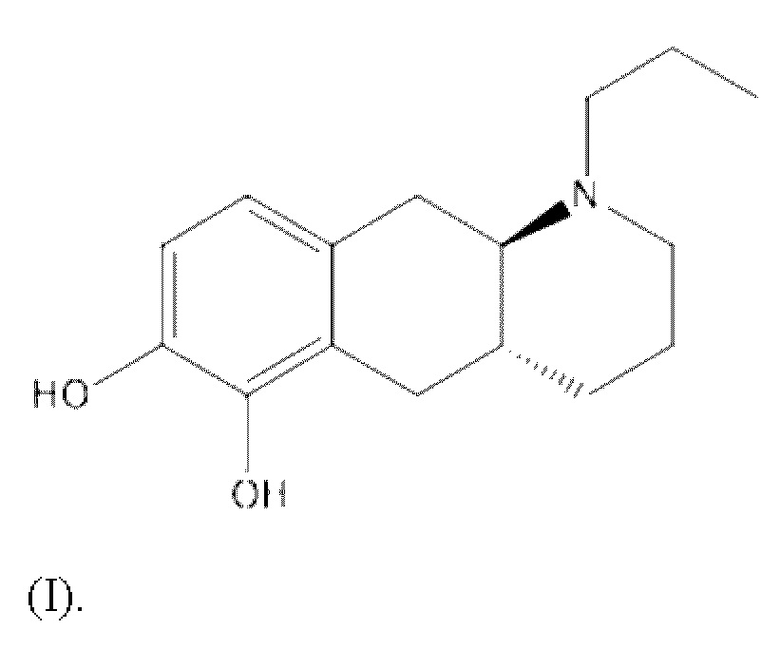
10. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-9, где указанное соединение или его фармацевтически приемлемая соль представлены в твердой форме.
11. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-10 для применения в качестве лекарственного препарата.
12. Фармацевтическая композиция, имеющая дофаминовую агонистическую активность, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-10 и одно или несколько фармацевтически приемлемых вспомогательных веществ.
13. Фармацевтическая композиция по п. 12, где указанная фармацевтическая композиция представляет собой фармацевтическую композицию для перорального применения, такую как таблетка или капсула для перорального введения.
14. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-10 для лечения заболевания, выбранного из болезни Паркинсона, болезни Хантингтона, синдрома беспокойных ног или болезни Альцгеймера.
15. Применение по п. 14, где заболевание представляет собой болезнь Паркинсона.
16. Способ лечения заболевания, выбранного из болезни Паркинсона, болезни Хантингтона, синдрома беспокойных ног или болезни Альцгеймера, при этом способ включает введение терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп. 1-10 пациенту, нуждающемуся в этом.
17. Способ по п. 16, где заболевание представляет собой болезнь Паркинсона.
18. Способ лечения нейропсихиатрического заболевания или нарушения, при этом способ включает введение терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп. 1-10 пациенту, нуждающемуся в этом.
19. Способ по п. 18, где нейропсихиатрическое заболевание или нарушение представляет собой шизофрению, синдром дефицита внимания с гиперактивностью или наркотическую зависимость.
20. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-10 для получения лекарственного препарата, предназначенного для лечения нейродегенеративного заболевания или нарушения.
21. Применение по п. 20, где нейродегенеративное заболевание или нарушение представляет собой болезнь Паркинсона, болезнь Хантингтона, синдром беспокойных ног или болезнь Альцгеймера.
22. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-10 для получения лекарственного препарата, предназначенного для лечения нейропсихиатрического заболевания или нарушения.
23. Применение по п. 22, где нейропсихиатрическое заболевание или нарушение представляет собой шизофрению, синдром дефицита внимания с гиперактивностью или наркотическую зависимость.
| WO 2010097092 А1, 02.09.2010 | |||
| Многопоршневый насос с гидравлической штангой для глубоких колодцев | 1932 |
|
SU32920A1 |
| ЛЕЧЕНИЕ ИЛИ ПРОФИЛАКТИКА СИНДРОМА ГИПЕРСТИМУЛЯЦИИ ЯИЧНИКОВ (OHSS) С ИСПОЛЬЗОВАНИЕМ АГОНИСТА ДОФАМИНА | 2006 |
|
RU2416410C2 |
| SERGE PRZEDBORSKI | |||
| Neurodegeneration: What is it and where are we? | |||
| The Journal of Clinical Investigation, 2003, Vol | |||
| Говорящий кинематограф | 1920 |
|
SU111A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

