ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[1] Настоящее изобретение относится к соединению, проявляющему превосходную агонистическую активность в отношении рецепторов меланокортина. Более конкретно, настоящее изобретение относится к соединению следующей формулы 1, фармацевтической композиции, содержащей соединение в качестве активного ингредиента, и его применению, и соединение настоящего изобретения проявляет превосходную агонистическую активность в отношении рецепторов мелакортина-4 и может быть особенно полезным для предотвращения или лечения ожирения, диабета, воспалений и эректильной дисфункции:
[2] [Формула 1]
[3]
[4]
[5] где R1 представляет собой C2-C5 алкил.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[6] Белок лептина представляет собой гормон, секретируемый жировыми клетками тела (адипоцитами), и количество его секреции увеличивается по мере увеличения содержания жира в организме. Белок лептина регулирует функции различных нейропептидов, продуцируемых в гипоталамусе, тем самым регулируя аппетит, содержание жира в организме и различные функции in vivo, включая энергетический метаболизм (Schwartz, et al., Nature 404, 661-671 (2000)). Сигнальная трансдукция аппетита и контроля веса с помощью лептинового белка осуществляется посредством регуляции многих нижестоящих факторов, наиболее характерными из которых являются меланокортин, агути-родственный пептид (AgRP) и гормон нейропептид Y (NPY).
[7] Когда концентрация лептина в крови увеличивается в результате чрезмерного потребления калорий in vivo, секреция белкового гормона проопиомеланокортина (POMC) в гипофизе увеличивается, а продукция AgRP и NPY снижается. Альфа-MSH (гормон, стимулирующий меланоциты), который представляет собой небольшой пептидный гормон, вырабатывается нейронами POMC, и этот гормон является агонистом рецептора меланокортина-4 (MC4R) вторичных нейронов и в конечном итоге вызывает снижение аппетита. С другой стороны, когда концентрация лептина снижается из-за дефицита калорий, увеличивается экспрессия AgRP, который является антагонистом MC4R, а также увеличивается экспрессия NPY, что в конечном итоге усиливает аппетит. То есть, в зависимости от изменений лептина, гормон альфа-MSH и гормон AgRP участвуют в регуляции аппетита, являясь агонистом и антагонистом MC4R.
[8] Гормон альфа-MSH вызывает различные физиологические реакции, связываясь с 3 подтипами MCR в дополнение к MC4R. К настоящему времени идентифицировано пять подтипов MCR. Среди подтипов известно, что MC1R в основном экспрессируется в клетках кожи и участвует в пигментации меланина, MC2R в основном экспрессируется в надпочечниках и участвует в производстве глюкокортикоидного гормона, и только ACTH (адренокортикотропный гормон), полученный из POMC, является его лигандом. MC3R и MC4R, которые в основном экспрессируются в центральной нервной системе, участвуют в регуляции аппетита, энергетического метаболизма и эффективности накопления жира в организме, а MC5R, который экспрессируется в различных тканях, как известно, регулирует экзокринную функцию (Wikberg, et al., Pharm Res 42 (5) 393-420 (2000)). В частности, активация рецептора MC4R имеет эффект эффективного снижения массы тела, вызывая снижение аппетита и повышение энергетического метаболизма, и таким образом доказано, что она является основным направлением действия при разработке лекарств от ожирения (Обзор: Wikberg, Eur. J. Pharmacol 375, 295-310 (1999)); Wikberg, et al., Pharm Res 42 (5) 393-420 (2000); Douglas et al., Eur J Pharm 450, 93-109 (2002); и OʼRahilly et al., Nature Med 10, 351-352 (2004)).
[9] Роль MC4R в контроле аппетита и веса была в первую очередь продемонстрирована в эксперименте на животной модели аномальной экспрессии белка агути (мышь агути). В случае мышей агути обнаружено, что белок агути экспрессируется в высоких концентрациях в центральной нервной системе и действует как антагонист MC4R в гипоталамусе из-за генетических мутаций, вызывая, таким образом, ожирение (Yen, TT et al., FASEB J. 8, 479-488 (1994); и Lu D., et al. Nature 371, 799-802 (1994)). В последующих результатах исследований было замечено, что AgRP (родственный агути пептид), подобный фактическому белку агути, экспрессировался в гипоталамическом нерве, и также известно, что AgRP участвует в регуляции аппетита в качестве антагонистов против MC4R (Shutter, et al., Genes Dev., 11, 593-602 (1997); и Ollman, et al. Science 278, 135-138 (1997)).
[10] Церебральное введение животному альфа-MSH, который является агонистом MC4R in vivo, демонстрирует эффект снижения аппетита, и если вводят SHU9119 (пептид) или HS014 (пептид), которые являются антагонистами MC4R, то снова наблюдается эффект повышения аппетита (Kask et al., Biochem. Biophys. Res. Comm. 245, 90-93 (1998)). Кроме того, в тесте на животных с использованием меланотана II (MTII, Ac-Nle-c[Asp-His-DPhe-Arg-Trp-Lys]-NH2) и HP228, который является агонистом, аналогичным ему, после церебрального, внутрибрюшинного или подкожного введения была подтверждена эффективность подавления аппетита, потери веса и повышения энергетического метаболизма и тому подобное. (Thiele T. E., et al. Am J Physiol 274 (1 Pt 2), R248-54 (1998); Lee M. D., et al. FASEB J 12, A552 (1998); Murphy B., et al. J Appl Physiol 89, 273-82 (2000)). Напротив, когда животному вводится репрезентативный SHU9119, наблюдается значительное и устойчивое потребление корма и увеличение веса, что дает фармакологические доказательства того, что агонисты MCR могут использоваться для лечения ожирения. Эффект снижения аппетита, который, очевидно, проявляется при введении MTII, не проявляется у мышей MC4R KO (нокаутные), и этот экспериментальный результат еще раз доказывает, что эффект снижения аппетита в основном достигается за счет активации MC4R (Marsh, et al., Nat Genet 21, 119-122 (1999)).
[11] В лечении ожирения, разработанном до настоящего времени, преобладают подавители аппетита, которые действуют на центральную нервную систему, и большинство подавителей являются лекарствами, которые модулируют действие нейромедиаторов. Их примеры включают норадреналиновые агенты (фентермин и мазиндол) и флуоксетин и сибутрамин, которые являются серотонинергическими агентами. Однако модулятор нейротрансмиттера оказывает широкий спектр эффектов на различные физиологические действия помимо подавления аппетита многочисленными рецепторами подтипа. Следовательно, модуляторы не обладают селективностью для каждого подтипа и имеют большой недостаток, заключающийся в том, что при длительном применении возникают различные побочные эффекты.
[12] С другой стороны, меланокортины являются нейропептидами, а не нейротрансмиттерами, и, учитывая, что все другие функции, кроме энергетического метаболизма, нормальны у мышей KO с геном MC4R, преимущество агонистов меланокортина в качестве точки действия состоит в том, что можно вызвать только потерю веса за счет подавления аппетита, не влияя на другие физиологические функции. В частности, рецептор представляет собой рецептор, связанный с G-белком (GPCR), который относится к наиболее успешной категории среди новых точек действия лекарственных средств, разработанных к настоящему времени, и в значительной степени отличается от точек действия в родственной области техники тем, что относительно легко можно обеспечить селективность по рецепторам подтипа.
[13] В международных публикациях №№ WO 2008/007930 и WO 2010/056022 описаны соединения-агонисты рецептора меланокортина как примеры использования рецептора меланокортина в качестве точки действия.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
[14] Целью настоящего изобретения является разработка нового соединения, представленного формулой 1, которое обладает превосходной селективной агонистической активностью в отношении рецепторов меланокортина, в частности, рецепторов меланокортина-4 (MC4R), или его фармацевтически приемлемой соли или изомера.
[15] Другой целью настоящего изобретения является разработка способа получения соединения, представленного формулой 1.
[16] Еще одной целью настоящего изобретения является разработка агонистической фармацевтической композиции рецептора меланокортина, содержащей соединение, представленное формулой 1, или его фармацевтически приемлемую соль или изомер в качестве активного ингредиента.
[17] Еще одной целью настоящего изобретения является предложение применения соединения, представленного формулой 1, или его фармацевтически приемлемой соли или изомера для профилактики или лечения ожирения, диабета, воспаления и эректильной дисфункции.
[18] Еще одним объектом настоящего изобретения является способ предотвращения или лечения ожирения, диабета, воспаления и эректильной дисфункции, включающий введение соединения, представленного формулой 1, или его фармацевтически приемлемой соли или изомера нуждающемуся субъекту. из них.
[19]
РЕШЕНИЕ ЗАДАЧИ
[20] Для достижения вышеуказанной цели в настоящем изобретении предложено соединение следующей формулы 1 или его фармацевтически приемлемая соль или изомер:
[21] [Формула 1]
[22]
[23]
[24] где R1 представляет собой C2-C5 алкил.
[25]
[26] Соединение формулы 1 по настоящему изобретению может образовывать фармацевтически приемлемую соль.
[27] Кроме того, поскольку соединения в соответствии с настоящим изобретением могут иметь асимметричный углеродный центр и асимметричную ось или асимметричную плоскость, соединения могут существовать в виде цис- или транс-изомеров, R- или S-изомеров, рацематов, смесей диастереомеров и отдельных диастереомеров, и все эти изомеры и смеси включены в объем настоящего изобретения.
[28] В настоящем описании, если не указано иное, соединение формулы 1 используется в смысле включения всего соединения формулы 1, его фармацевтически приемлемых солей и изомеров.
[29]
[30] В одном варианте осуществления в соответствии с настоящим изобретением, R1 в формуле 1 представляет собой C2-C4 алкил. В другом варианте осуществления в соответствии с настоящим изобретением R1 в формуле 1 представляет собой линейный или разветвленный C2-C4 алкил, например, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил.
[31] В другом варианте осуществления в соответствии с настоящим изобретением R1 в формуле 1 представляет собой C2 или C3 алкил. В другом варианте осуществления в соответствии с настоящим изобретением R1 в формуле 1 представляет собой линейный или разветвленный C2 или C3 алкил, например этил, н-пропил или изопропил.
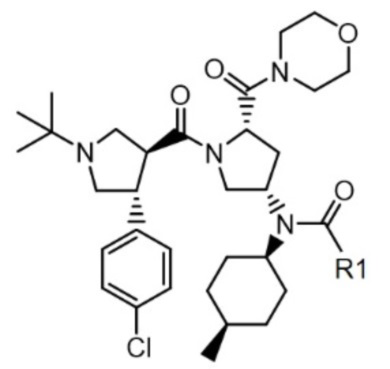
[32] В другом варианте осуществления в соответствии с настоящим изобретением соединение формулы 1 представляет собой N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)изобутирамид следующей формулы 2:
[33] [Формула 2]
[34]
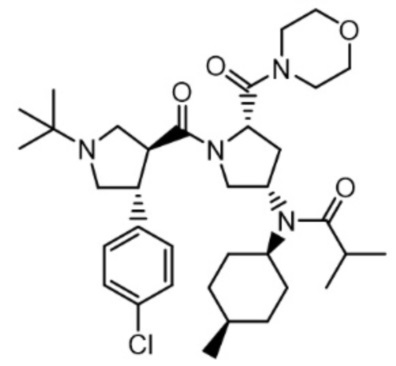
[35] В другом варианте осуществления настоящего изобретения соединение формулы 1 представляет собой N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пропионамид следующей формулы 3:
[36] [Формула 3]
[37]
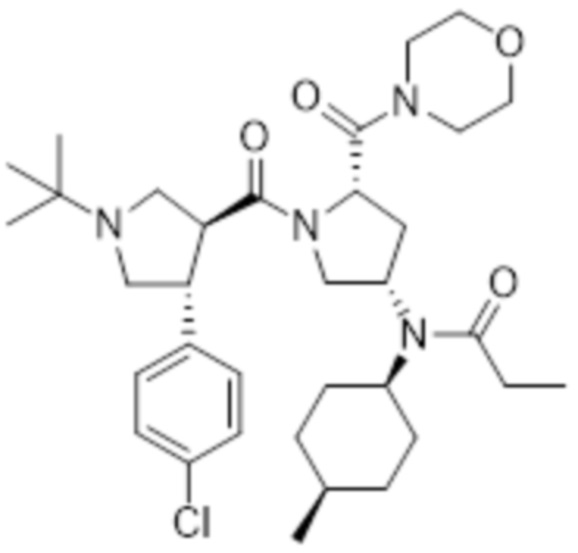
[38] В другом варианте осуществления настоящего изобретения, соединение формулы 1 представляет собой N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пиваламид следующей формулы 4:
[39] [Формула 4]
[40]
[41] 
[42] В другом варианте осуществления в соответствии с настоящим изобретением примеры фармацевтически приемлемой соли включают кислотно-аддитивные соли, образованные неорганической кислотой, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота и йодистоводородная кислота; органической карбоновой кислотой, такой как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота и малеиновая кислота; сульфоновой кислотой, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и нафталинсульфоновая кислота, но ими не ограничиваются.
[43] В другом варианте осуществления изобретения соединение представляет собой фармацевтическую соль соединения формулы 1, где R1 представляет собой этил и соль является солью неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота и йодистоводородная кислота; органической карбоновой кислоты, такой как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота и малеиновая кислота; и сульфоновой кислоты, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и нафталинсульфоновая кислота.
[44] В другом варианте осуществления изобретения соединение представляет собой фармацевтическую соль соединения формулы 1, где R1 представляет собой н-пропил и соль является солью неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота и йодистоводородная кислота; органической карбоновой кислоты, такой как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота и малеиновая кислота; и сульфоновой кислоты, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и нафталинсульфоновая кислота.
[45] В другом варианте осуществления изобретения соединение представляет собой фармацевтическую соль соединения формулы 1, где R1 представляет собой изопропил и соль является солью неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота и йодистоводородная кислота; органической карбоновой кислоты, такой как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота и малеиновая кислота; и сульфоновой кислоты, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и нафталинсульфоновая кислота.
[46] В другом варианте осуществления изобретения соединение представляет собой фармацевтическую соль соединения формулы 1, где R1 представляет собой н-бутил и соль является солью неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота и йодистоводородная кислота; органической карбоновой кислоты, такой как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота и малеиновая кислота; и сульфоновой кислоты, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и нафталинсульфоновая кислота.
[47] В другом варианте осуществления изобретения соединение представляет собой фармацевтическую соль соединения формулы 1, где R1 представляет собой изобутил и соль является солью неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота и йодистоводородная кислота; органической карбоновой кислоты, такой как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота и малеиновая кислота; и сульфоновой кислоты, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и нафталинсульфоновая кислота.
[48] В другом варианте осуществления изобретения соединение представляет собой фармацевтическую соль соединения формулы 1, где R1 представляет собой втор-бутил и соль является солью неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота и йодистоводородная кислота; органической карбоновой кислоты, такой как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота и малеиновая кислота; и сульфоновой кислоты, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и нафталинсульфоновая кислота.
[49] В другом варианте осуществления изобретения соединение представляет собой фармацевтическую соль соединения формулы 1, где R1 представляет собой трет-бутил и соль является солью неорганической кислоты, такой как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота и йодистоводородная кислота; органической карбоновой кислоты, такой как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота и малеиновая кислота; и сульфоновой кислоты, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и нафталинсульфоновая кислота.
[50] В другом варианте осуществления в соответствии с настоящим изобретением фармацевтически приемлемая соль представляет собой гидрохлорид.
[51] В другом варианте осуществления в соответствии с настоящим изобретением соединение формулы 1 представляет собой гидрохлорид N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)изобутирамида следующей формулы 5:
[52] [Формула 5]
[53]
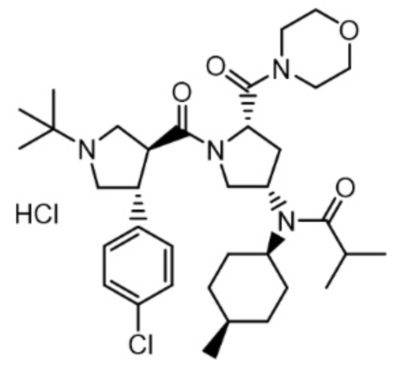
[54] В другом варианте осуществления в соответствии с настоящим изобретением соединение формулы 1 представляет собой гидрохлорид N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пропионамида следующей формулы 6:
[55] [Формула 6]
[56]
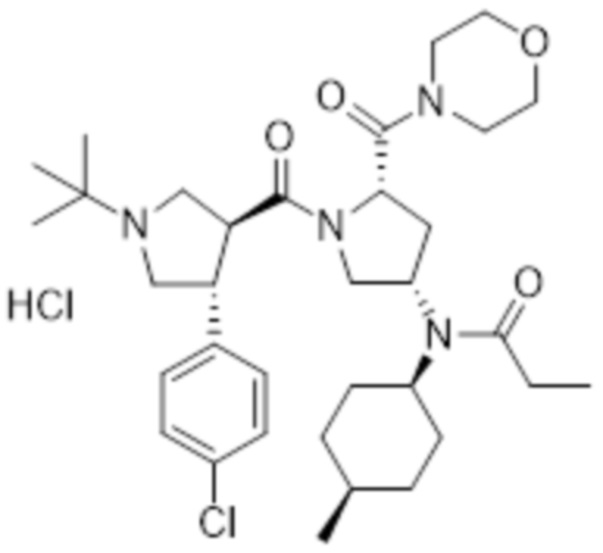
[57] В другом варианте осуществления в соответствии с настоящим изобретением соединение формулы 1 представляет собой гидрохлорид N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пиваламида следующей формулы 7:
[58] [Формула 7]
[59]
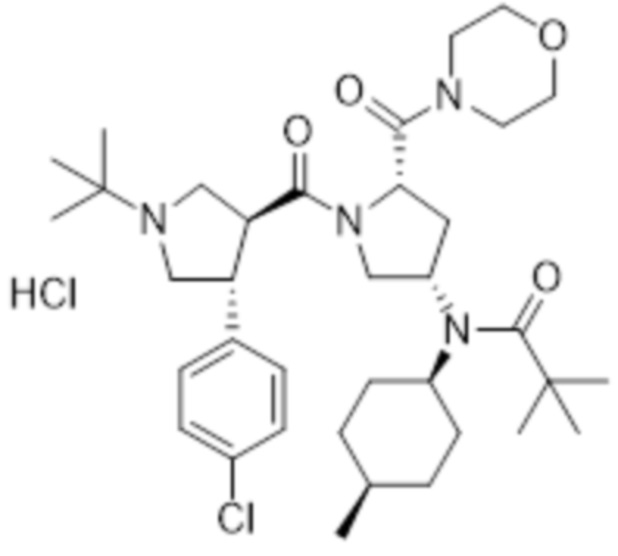
[60] В другом варианте осуществления в соответствии с настоящим изобретением гидрохлорид N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)изобутирамида вышеуказанной формулы 5, гидрохлорид N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пропионамида вышеуказанной формулы 6 и гидрохлорид N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пиваламида вышеуказанной формулы 7 может быть получен в соответствии со следующей реакционной схемой 1.
[61] [Реакционная схема 1]
[62]
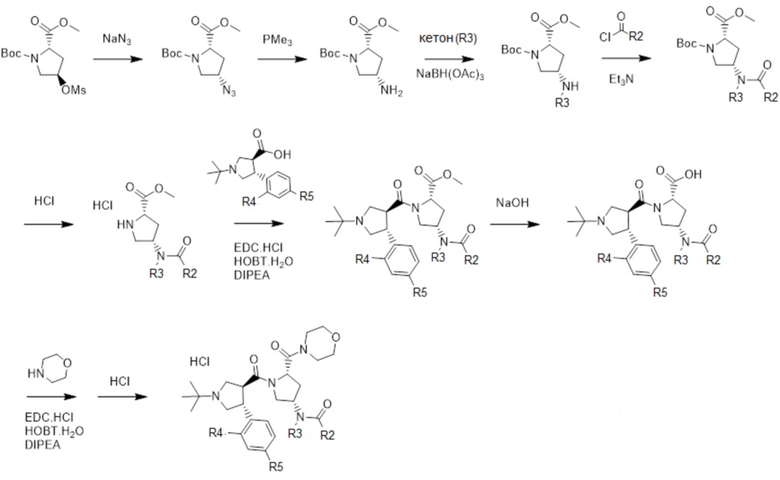
[63]
[64] На реакционной схеме 1
[65] R2 представляет собой C1-C5 алкил;
[66] R3 представляет собой C3-C8 циклоалкил, незамещенный или замещенный 1 или 2 C1-C5 алкилами; и
[67] R4 и R5, каждый, независимо, представляют собой водород или галоген.
[68]
[69] Соединение формулы 1 по настоящему изобретению проявляет превосходную агонистическую активность в отношении рецепторов меланокортина, в частности, рецепторов меланокортина-4 (MC4R), и, таким образом, в настоящем изобретении предложена также фармацевтическая композиция агонистов рецепторов меланокортина, содержащая соединение формулы 1 или его фармацевтически приемлемую соль или изомер в качестве активного ингредиента вместе с фармацевтически приемлемым носителем. В частности, композиция по настоящему изобретению проявляет превосходный эффект в предотвращении или лечении ожирения, диабета, воспаления и эректильной дисфункции, но этим не ограничивается.
[70] В настоящем описании термин «носитель» относится к соединению, которое облегчает внедрение соединения в клетки или ткани.
[71] При введении соединения по настоящему изобретению в клинических целях общая суточная доза, которую следует вводить хозяину в виде разовой дозы или отдельными дозами, предпочтительно, находится в диапазоне от 0,01 до 10 мг на 1 кг веса тела, но конкретный уровень дозы для отдельных пациентов может варьироваться в зависимости от конкретного соединения, которое будет использоваться, веса, пола, состояния здоровья и питания пациента, времени введения, метода введения и скорости выведения лекарственного средства, лекарственной смеси, а от также тяжести заболевания.
[72] Соединение по настоящему изобретению можно вводить любым путем в зависимости от цели. Например, соединение по настоящему изобретению можно вводить путем инъекции или перорально.
[73] Составы для инъекций могут быть получены с использованием подходящего диспергирующего агента, смачивающего агента или суспендирующего агента в соответствии с известными методами.
[74] Примеры твердых лекарственных форм для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы, и твердые лекарственные формы могут быть получены путем смешивания активного соединения формулы 1 по настоящему изобретению с одним или несколькими носителями, такими как инертные разбавители, лубриканты, разрыхлители и связующие.
[75]
ПОЛОЖИТЕЛЬНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[76] Соединение формулы 1 по настоящему изобретению проявляет превосходную агонистическую активность в отношении рецепторов меланокортина, в частности, рецепторов меланокортина-4 (MC4R), и, таким образом, может быть эффективно использовано для профилактики или лечения ожирения, диабета, воспалений и эректильной дисфункции.
[77] Соединение формулы 1 по настоящему изобретению оказывает целевое действие на рецептор меланокортина-4, не влияет на тревогу и депрессию, проявляя при этом эффекты потери веса и снижения питания, и его можно вводить без побочных эффектов на ингибирование гена специфических калиевых каналов сердца человека (hERG), или на проблемы безопасности, такие как мутагенез. Кроме того, соединение формулы 1 по настоящему изобретению можно вводить безопасно, поскольку отсутствует цитотоксичность и токсичность для печени.
[78]
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
[79] Далее настоящее изобретение описано более подробно с помощью препаративных примеров и примеров. Однако эти примеры являются только иллюстративными, и объем настоящего изобретения ими не ограничивается.
[80]
[81] Препаративный пример 1: Получение гидрохлорида метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)изобутирамидо)-пирролидин-2-карбоксилата
[82]
[83] 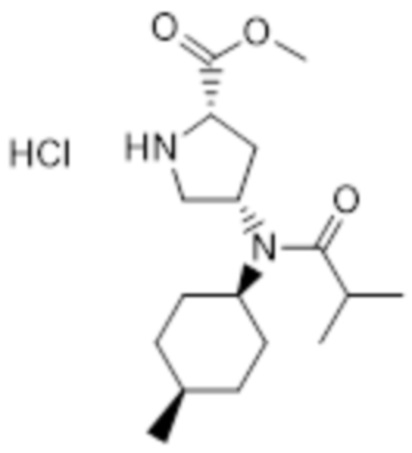
[84] Указанное в заголовке соединение получали посредством следующих стадий A, B, C, D и E.
[85]
[86] Стадия A: Получение 1-(трет-бутил) 2-метил (2S,4S)-4-азидопирролидин-1,2-дикарбоксилата
[87] 1-(трет-Бутил) 2-метил (2S,4R)-4-((метилсульфонил)окси)пирролидин-1,2-дикарбоксилат (48,5 г, 150 ммоль) растворяли в N, Nʼ-диметилформамиде (250 мл) в атмосфере азота и добавляли азид натрия (19,5 г, 300 мл). После перемешивания при температуре 80°C в течение 16 часов растворитель реакции концентрировали при пониженном давлении, добавляли воду и два раза экстрагировали этилацетатом. Органический слой промывали водным раствором хлорида натрия и водой, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого продукта (39,59 г, 98%), который использовали на следующей стадии без очистки.
[88] MS [M+H]=271 (M+1)
[89] 1H ЯМР (400 МГц, CD3OD) δ 4,43-4,37 (м, 1H), 4,35-4,27 (шир., 1H), 3,77 (с, 1,8H), 3,76 (с, 1,2H), 3,73-3,66 (м, 1H), 3,44-3,38 (м, 1H), 2,63-2,49 (м, 1H), 2,19-2,11 (м, 1H), 1,50 (с, 4,5H), 1,44 (с, 4,5H)
[90]
[91] Стадия B: Получение 1-(трет-бутил) 2-метил (2S,4S)-4-аминопирролидин-1,2-дикарбоксилата
[92] 1-(трет-Бутил) 2-метил (2S,4S)-4-азидопирролидин-1,2-дикарбоксилат (24,59 г, 91,0 ммоль), полученный на стадии A, растворяли в тетрагидрофуране (180 мл) и затем при температуре 0°C медленно добавляли 1M раствор триметилфосфин тетрагидро (109,2 мл, 109,2 ммоль). После перемешивания при той же температуре в течение одного часа, смесь перемешивали при комнатной температуре в течение трех часов. После концентрирования растворителя реакции при пониженном давлении добавляли дихлорметан (100 мл) и воду (150 мл) и перемешивали в течение около 30 минут. После разделения слоев и экстрагирования один раз дихлорметаном органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого продукта (20,62 г, 93%), который использовали на следующей стадии без очистки.
[3] MS [M+H]=245 (M+1)
[94] 1H ЯМР (400 МГц, CD3OD) δ 4,27 (м, 1H), 3,77 (с, 1,8H), 3,76 (с, 1,2H), 3,75-3,67 (м, 1H), 3,50-3,42 (м, 1H), 3,22-3,17 (м, 1H), 2,58-2,47 (м, 1H), 1,82-1,71 (м, 1H), 1,48 (с, 4,5H), 1,42 (с, 4,5H)
[95]
[96] Стадия C: Получение 1-(трет-бутил) 2-метил (2S,4S)-4-(((1S,4R)-4-метилциклогексил)амино)пирролидин-1,2-дикарбоксилата
[97] 1-(трет-Бутил) 2-метил (2S,4S)-4-аминопирролидин-1,2-дикарбоксилат (20,62 г, 84,4 ммоль), полученный на стадии B, растворяли в дихлорэтане (150 мл) и добавляли 4-метилциклогексанон (9,5 мл, 101,3 ммоль). При температуре 0°C добавляли триацетоксиборгидрид натрия (26,8 г, 126,6 ммоль) и перемешивали при комнатной температуре в течение 16 часов. Растворитель реакции концентрировали при пониженном давлении, добавляли воду и два раза экстрагировали этилацетатом. Органический слой промывали водным раствором хлорида натрия, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали колоночной хроматографией с получением указанного в заголовке соединения (22,9 г, 80%).
[98] MS [M+H]=341 (M+1)
[99] 1H ЯМР (400 МГц, CD3OD) δ 4,26 (м, 1H), 3,76 (с, 1,8H), 3,75 (с, 1,2H), 3,78-3,71 (м, 1H), 3,49-3,40 (м, 1H), 3,22-3,16 (м, 1H), 2,69-2,60 (шир., 1H), 2,58-2,46 (м, 1H), 1,87-1,77 (м, 1H), 1,73-1,63 (м, 1H), 1,62-1,35 (м, 8H), 1,48 (с, 4,5H), 1,42 (с, 4,5H), 0,96 (д, 3H)
[100]
[101] Стадия D: Получение 1-(трет-бутил) 2-метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)изобутирамидо)пирролидин-1,2-дикарбоксилата
[102] 1-(трет-Бутил) 2-метил (2S,4S)-4-(((1S,4R)-4-метилциклогексил)амино)пирролидин-1,2-дикарбоксилат (37,29 г, 109,5 ммоль), полученный на стадии C, растворяли в дихлорметане (500 мл), при температуре 0°C добавляли триэтиламин (61,1 мл, 438,1 ммоль) и медленно добавляли изобутирилхлорид (11,7 мл, 219 ммоль). После перемешивания при комнатной температуре в течение 16 часов растворитель реакции концентрировали при пониженном давлении, добавляли водный раствор гидрокарбоната натрия и два раза экстрагировали этилацетатом. Органический слой промывали водным раствором хлорида натрия и водой, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали колоночной хроматографией с получением указанного в заголовке соединения (38,79 г, 86%).
[103] MS [M+H]=411 (M+1)
[104] 1H ЯМР (400 МГц, CD3OD) δ 4,27 (м, 1H), 3,76 (с, 1,8H), 3,75 (с, 1,2H), 3,78-3,72 (м, 1H), 3,50-3,41 (м, 1H), 3,33-3,14 (м, 1H), 2,69-2,60 (м, 2H), 2,57-2,43 (м, 1H), 1,87-1,79 (м, 1H), 1,70-1,61 (м, 1H), 1,60-1,32 (м, 8H), 1,47 (с, 4,5H), 1,41 (с, 4,5H), 1,10 (дд, 6H), 0,99 (д, 3H)
[105]
[106] Стадия E: Получение гидрохлорида метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)изобутирамидо)пирролидин-2-карбоксилата
[107] 1-(трет-Бутил) 2-метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)изобутирамидо)пирролидин-1,2-дикарбоксилат (34,0 г, 82,8 ммоль), полученный на стадии D, растворяли в дихлорметане (200 мл) и при температуре 0°C добавляли 4н раствор хлористоводородной кислоты в 1,4-диоксане (82,8 мл, 331,3 ммоль). После перемешивания при комнатной температуре в течение 6 часов растворитель реакции концентрировали при пониженном давлении с получением сырого продукта (28,7 г, 99%), который использовали на следующей стадии без очистки.
[108] MS [M+H]=311 (M+1)
[109]
[110] Препаративный пример 2: Получение (3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбоновой кислоты
[111]
[112] 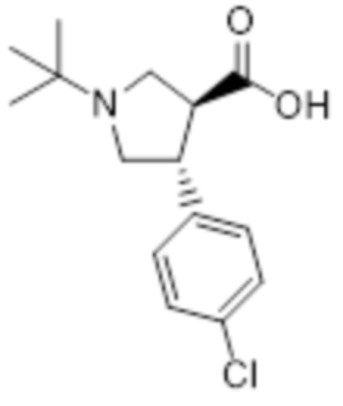
[113] Указанное в заголовке соединение получали способом, описанным в международной публикации № WO 2004/092126.
[114] MS [M+H]=282 (M+1)
[115] 1H ЯМР (400 МГц, CD3OD) δ 7,43-7,33 (м, 4H), 3,90-3,69 (м, 3H), 3,59 (дд, J=11,2, 10,0 Гц, 1H), 3,29 (дд, J=11,2, 11,2 Гц, 1H), 3,18-3,09 (м, 1H), 1,44 (с, 9H)
[116]
[117] Препаративный пример 3: Получение гидрохлорида метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пропионамидо)-пирролидин-2-карбоксилата
[118]
[119] 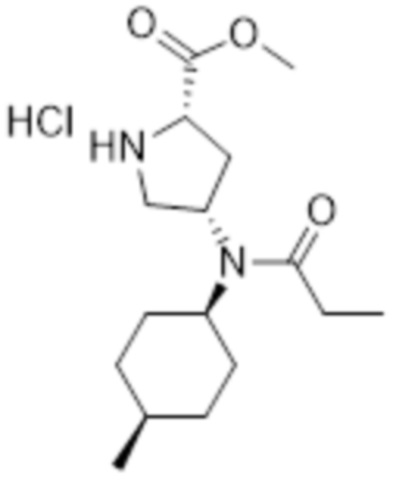
[120] Указанное в заголовке соединение получали посредством стадий A и B.
[121] Стадия A: Получение 1-(трет-бутил) 2-метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пропионамидо)пирролидин-1,2-дикарбоксилата
[123]
[124] 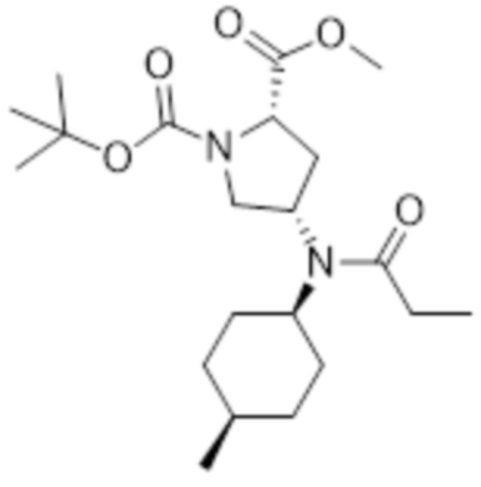
[125] Указанное в заголовке соединение (0,98 г, 84%) получали таким же образом, как на стадии D в препаративном примере 1, с использованием 1-(трет-бутил) 2-метил (2S,4S)-4-(((1S,4R)-4-метилциклогексил)амино)пирролидин-1,2-дикарбоксилата (1,0 г, 2,9 ммоль), полученного на стадии C препаративного примера 1, и пропионилхлорида (0,33 г, 3,5 ммоль).
[126] MS [M+Na]=419,5 (M+23)
[127] 1H ЯМР (400 МГц, CD3OD) δ 4,33 (м, 1H), 4,00-3,80 (м, 2H), 3,75 (м, 3H), 3,58 (м, 1H), 3,47 (м, 1H), 2,85-2,68 (м, 1H), 2,38 (кв, 2H), 2,31 (м, 1H), 1,93 (м, 1H), 1,80 (м, 2H), 1,72-1,55 (м, 4H), 1,45 (м, 2H), 1,45-1,41 (м, 9H), 1,07 (м, 6H)
[128]
[129] Стадия B: Получение гидрохлорида метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пропионамидо)пирролидин-2-карбоксилата
[130]
[131]
[132] Указанное в заголовке соединение (0,76 г, 93%) получали таким же образом, как на стадии E препаративного примера 1, с использованием 1-(трет-бутил) 2-метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пропионамидо)пирролидин-1,2-дикарбоксилата (0,98 г, 2,4 ммоль), полученного на стадии A.
[133] MS [M+H]=297,4 (M+1)
[134] 1H ЯМР (400 МГц, ДМСО-d6) δ 9,95 (шир. с, 1H), 8,63 (шир. с, 1H), 4,38 (м, 1H), 4,21 (м, 1H), 3,77 (с, 3H), 3,53 (м, 1H), 3,40 (м, 2H), 2,53 (м, 1H), 2,37 (кв, 2H), 2,24 (м, 1H), 1,88 (м, 1H), 1,68-1,55 (м, 4H), 1,52 (м, 2H), 1,40 (м, 2H), 0,97 (м, 6H)
[135]
[136] Препаративный пример 4: Получение гидрохлорида метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пиваламидо)пирролидин-2-карбоксилата
[137]
[138] 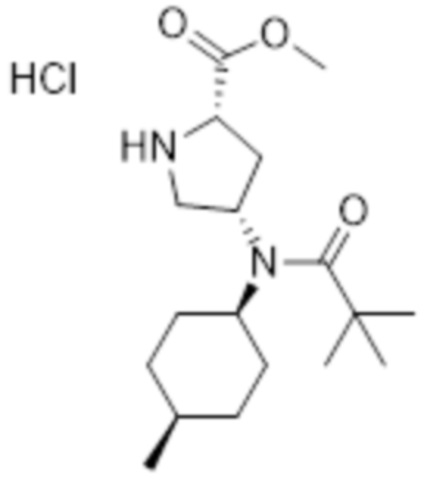
[139] Указанное в заголовке соединение получали посредством стадий A и B.
[140]
[141] Стадия A: Получение 1-(трет-бутил) 2-метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пиваламидо)пирролидин-1,2-дикарбоксилата
[142]
[143] 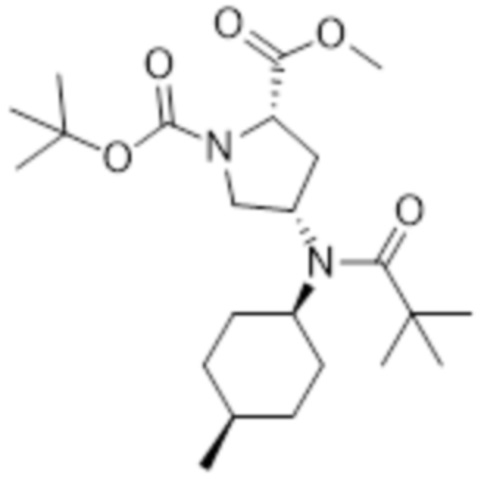
[144] Указанное в заголовке соединение получали способом, описанным в международной публикации № WO 2008/007930.
[145] MS [M+Na]=447,5 (M+23)
[146] 1H ЯМР (400 МГц, CD3OD) δ 4,34 (м, 1H), 3,90-3,75 (м, 2H), 3,73 (м, 3H), 3,45 (м, 2H), 2,75-2,60 (м, 1H), 2,30 (м, 1H), 1,95 (м, 1H), 1,85 (м, 2H), 1,66 (м, 4H), 1,50 (м, 2H), 1,45-1,41 (м, 9H), 1,25-1,20 (м, 9H), 1,05 (д, 3H)
[147]
[148] Стадия B: Получение гидрохлорида метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пиваламидо)пирролидин-2-карбоксилата
[149]
[150]
[151] Указанное в заголовке соединение (0,68 г, 99%) получали таким же образом, как на стадии E препаративного примера 1, с использованием 1-(трет-бутил) 2-метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пиваламидо)пирролидин-1,2-дикарбоксилат а(0,80 г, 1,88 ммоль), полученного на стадии A.
[152] MS [M+H]=325,4 (M+1)
[153] 1H ЯМР (400 МГц, ДМСО-d6) δ 10,24 (шир. с, 1H), 8,60 (шир. с, 1H), 4,41 (м, 1H), 4,22 (м, 1H), 3,77 (м, 3H), 3,40-3,28 (м, 3H), 2,55 (м, 1H), 2,20 (м, 1H), 1,87 (м, 1H), 1,70-1,50 (м, 6H), 1,40 (м, 2H), 1,21-1,10 (м, 9H), 1,00 (м, 3H)
[154]
[155] Пример 1: Получение гидрохлорида N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)изобутирамида
[156]
[157] 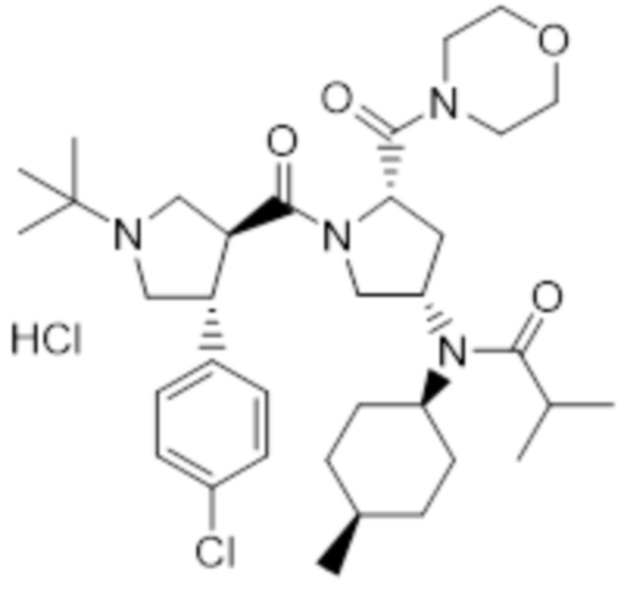
[158] Указанное в заголовке соединение получали посредством стадий A, B, C и D.
[159]
[160] Стадия A: Получение метил (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)изобутирамидо)пирролидин-2-карбоксилата
[161] Гидрохлорид метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)изобутирамидо)пирролидин-2-карбоксилата (28,7 г, 82,73 ммоль), полученный в препаративном примере 1, (3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбоновую кислоту (24,5 г, 86,87 ммоль), полученную в препаративном примере 2, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (22,2 г, 115,83 ммоль) и гидрат 1-гидроксибензотриазола (15,7 г, 115,83 ммоль) растворяли в N, Nʼ-диметилформамиде (400 мл), и медленно добавляли N, Nʼ-диизопропилэтиламин (72,0 мл, 413,66 ммоль). После перемешивания при комнатной температуре в течение 16 часов растворитель реакции концентрировали при пониженном давлении, затем добавляли 0,5н водный раствор гидроксида натрия и два раза экстрагировали этилацетатом. Органический слой промывали два раза водным раствором хлорида натрия и водой, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали колоночной хроматографией с получением указанного в заголовке соединения (41,19 г, 87%).
[162] MS [M+H]=575 (M+1)
[163]
[164] Стадия B: Получение (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)изобутирамидо)пирролидин-2-карбоновой кислоты
[165] Метил (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)изобутирамидо)пирролидин-2-карбоксилат (39,4 г, 68,62 ммоль), полученный на стадии A, растворяли в метаноле (450 мл) и добавляли 6н водный раствор гидроксида натрия (57,2 мл, 343,09 ммоль). После перемешивания при комнатной температуре в течение 16 часов и доведения pH до примерно 5 с помощью 6н водного раствора соляной кислоты реакционный раствор концентрировали при пониженном давлении. Концентрат растворяли в дихлорметане, а затем нерастворимое твердое вещество фильтровали через бумажный фильтр. Фильтрат концентрировали при пониженном давлении с получением сырого продукта (38,4 г, 99%), который использовали на следующей стадии без очистки.
[166] MS [M+H]=561 (M+1)
[167]
[168] Стадия C: Получение N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)изобутирамида
[169] (2S,4S)-1-((3S,4R)-1-(трет-Бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)изобутирамидо)пирролидин-2-карбоновую кислоту (38,4 г, 68,60 ммоль), полученную на стадии B, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (18,4 г, 96,04 ммоль) и гидрат 1-гидроксибензотриазола (13,0 г, 96,04 ммоль) растворяли в N, Nʼ-диметилформамиде (200 мл) и медленно и последовательно добавляли морфолин (5,9 мл, 68,80 ммоль) и N, Nʼ-диизопропилэтиламин (59,7 мл, 343,02 ммоль). После перемешивания при комнатной температуре в течение 16 часов реакционный раствор концентрировали при пониженном давлении, добавляли 0,5н водный раствор гидроксида натрия и два раза экстрагировали этилацетатом. Органический слой промывали два раза водным раствором хлорида натрия и водой, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали колоночной хроматографией с получением указанного в заголовке соединения (37,05 г, 86%).
[170] MS [M+H]=630 (M+1)
[171]
[172] Стадия D: Получение гидрохлорида N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)изобутирамида
[173] N-((3S,5S)-1-((3S,4R)-1-(трет-Бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)изобутирамид (5,0 г, 7,95 ммоль), полученный на стадии C, растворяли в этилацетате (50 мл) и медленно добавляли 2н раствор хлористоводородной кислоты в этилацетате (3,97 мл, 15,89 ммоль). После перемешивания при комнатной температуре в течение 30 минут растворитель реакции концентрировали при пониженном давлении. Образовавшийся сырой твердый продукт очищали растиранием, используя гексан и диэтиловый эфир, с получением указанного в заголовке соединения (5,23 г, 99%).
[174] MS [M+H]=630 (M+1)
[175] 1H ЯМР (500 МГц, CD3OD) δ 7,49-7,44 (м, 4H), 4,83 (м, 1H), 4,23-4,20 (м, 1H), 3,95-3,91 (м, 2H), 3,79-3,47 (м, 14H), 3,03-3,00 (м, 1H), 2,86-2,82 (м, 1H), 2,73-2,67 (м, 1H), 2,20-2,14 (м, 1H), 1,97 (м, 1H), 1,80-1,62 (м, 5H), 1,50 (с, 9H), 1,44-1,27 (м, 3H), 1,06-1,04 (м, 9H)
[176]
[177] Пример 2: Получение гидрохлорида N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пропионамида
[178]
[179]
[180] Указанное в заголовке соединение получали посредством стадий A, B, C и D.
[181]
[182] Стадия A: Получение метил (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)пропионамидо)пирролидин-2-карбоксилата
[183]
[184] 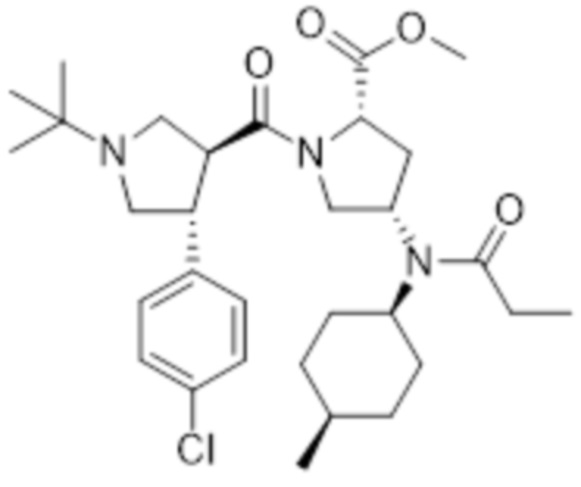
[185] Указанное в заголовке соединение (0,45 г, 35%) получали таким же образом, как на стадии A примера 1, с использованием метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пропионамидо)пирролидин-2-карбоксилата гидрохлорида (0,76 г, 2,28 ммоль), полученного в препаративном примере 3, и (3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбоновой кислоты (0,64 г, 2,28 ммоль), полученной в препаративном примере 2.
[186] MS [M+H]=560,4 (M+1)
[187] 1H ЯМР (400 МГц, CD3OD) δ 7,39-7,30 (м, 4H), 4,45 (м, 1H), 4,04 (м, 1H), 3,71 (с, 3H), 3,65-3,35 (м, 6H), 3,13 (м, 2H), 2,99 (м, 1H), 2,71 (м, 1H), 2,34 (кв, 2H), 2,20 (м, 1H), 1,92 (м, 1H), 1,75-1,55 (м, 6H), 1,42 (м, 2H), 1,22 (м, 9H), 1,03 (м, 6H)
[188]
[189] Стадия B: Получение (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)пропионамидо)пирролидин-2-карбоновой кислоты
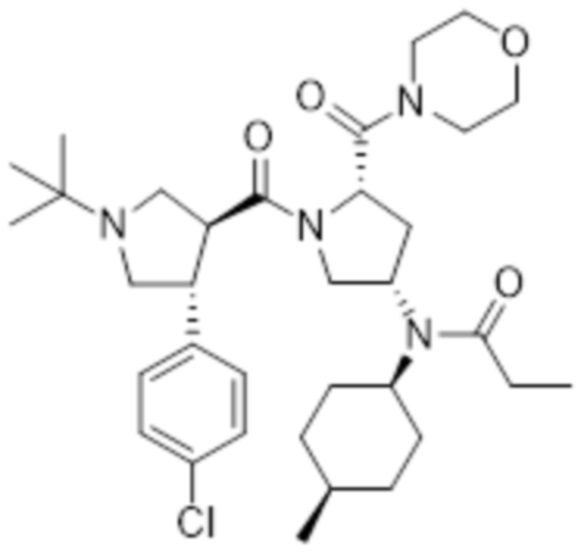
[190]
[191] 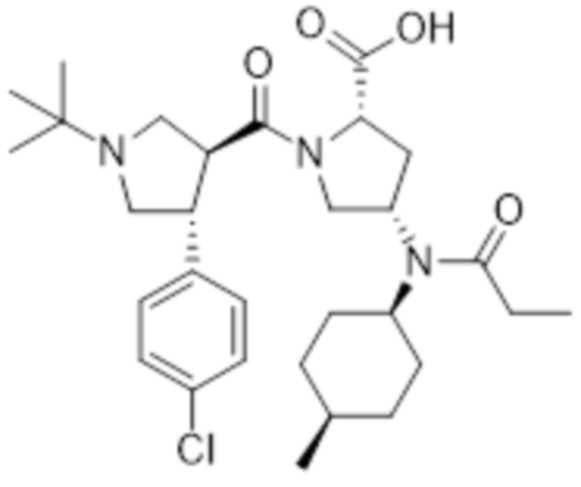
[192] Указанное в заголовке соединение (0,44 г, 99%) получали таким же образом, как на стадии B примера 1, с использованием метил (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)пропионамидо)пирролидин-2-карбоксилат (0,45 г, 0,80 ммоль), полученного на стадии A.
[193] MS [M+H]=546,4 (M+1)
[194]
[195] Стадия C: Получение N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пропионамида
[196]
[197] Указанное в заголовке соединение (0,28 г, 53%) получали таким же образом, как на стадии C примера 1, с использованием (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)пропионамидо)пирролидин-2-карбоновой кислоты (0,44 г, 0,80 ммоль), полученной на стадии B.
[198] MS [M+H]=615,5 (M+1)
[199] 1H ЯМР (400 МГц, CD3OD) δ 7,36 (м, 4H), 4,79 (м, 1H), 4,18 (м, 1H), 3,80-3,40 (м, 15H), 3,20 (м, 1H), 3,03 (м, 1H), 2,70 (м, 1H), 2,33 (кв, 2H), 2,15 (м, 1H), 1,93 (м, 1H), 1,71-1,56 (м, 6H), 1,40-1,20 (м, 11H), 1,00 (м, 6H)
[200]
[201] Стадия D: Получение гидрохлорида N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пропионамида
[2021]
[203] 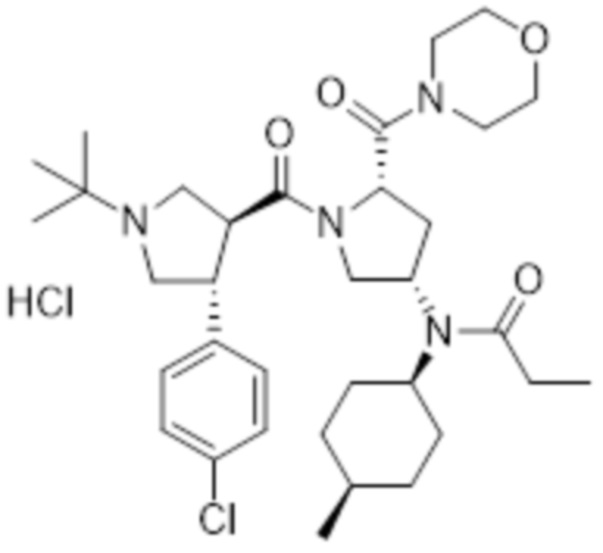
[204] Указанное в заголовке соединение (0,08 г, 94%) получали таким же образом, как на стадии D примера 1, с использованием N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пропионамида (0,08 г, 0,13 ммоль), полученного на стадии C.
[205] MS [M+H]=615,5 (M+1)
[206] 1H ЯМР (400 МГц, CD3OD) δ 7,43 (м, 4H), 4,82 (t, 1H), 4,20 (м, 1H), 4,06-3,40 (м, 15H), 2,97 (м, 1H), 2,69 (м, 1H), 2,33 (м, 2H), 2,15 (м, 1H), 1,93 (м, 1H), 1,80-1,53 (м, 5H), 1,47 (с, 9H), 1,50-1,25 (м, 4H), 1,01 (м, 6H)
[207]
[208] Пример 3: Получение гидрохлорида N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пиваламида
[209]
[210] 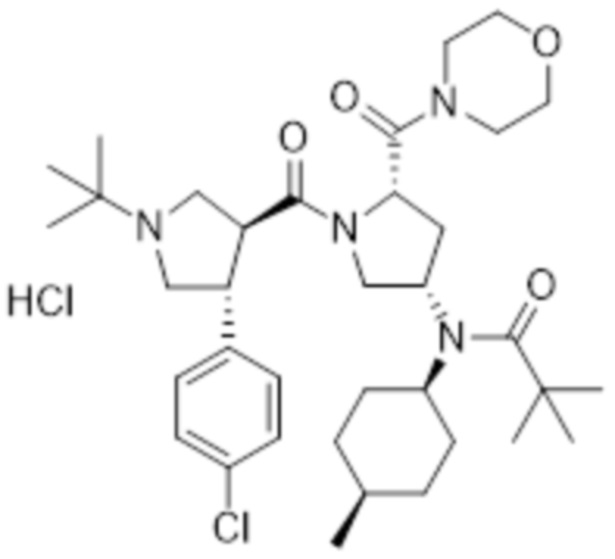
[211] Указанное в заголовке соединение получали посредством стадий A, B, C и D.
[212]
[213] Стадия A: Получение метил (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)пиваламидо)пирролидин-2-карбоксилата
[214]
[215] 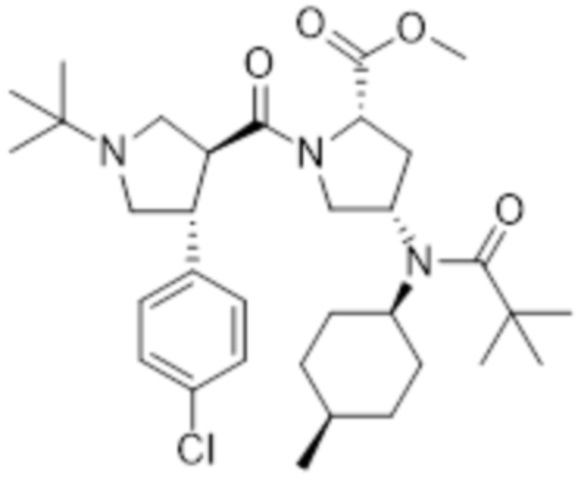
[216] Указанное в заголовке соединение (0,70 г, 66%) получали таким же образом, как на стадии A примера 1, с использованием гидрохлорида метил (2S,4S)-4-(N-((1S,4R)-4-метилциклогексил)пиваламидо)пирролидин-2-карбоксилата (0,65 г, 1,8 ммоль), полученного в препаративном примере 4, и (3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбоновой кислоты (0,50 г, 1,8 ммоль), полученной в препаративном примере 2.
[217] MS [M+H]=588,5 (M+1)
[218] 1H ЯМР (400 МГц, CD3OD) δ 7,40-7,30 (м, 4H), 4,49 (м, 1H), 4,00-3,50 (м, 4H), 3,71 (с, 3H), 3,40 (м, 3H), 3,20-3,05 (м, 2H), 3,00 (м, 1H), 2,70 (м, 1H), 2,27 (м, 1H), 1,90 (м, 1H), 1,73-1,60 (м, 6H), 1,60-1,35 (м, 2H), 1,25-1,17 (м, 18H), 1,01 (м, 3H)
[219]
[220] Стадия B: Получение (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)пиваламидо)пирролидин-2-карбоновой кислоты
[221]
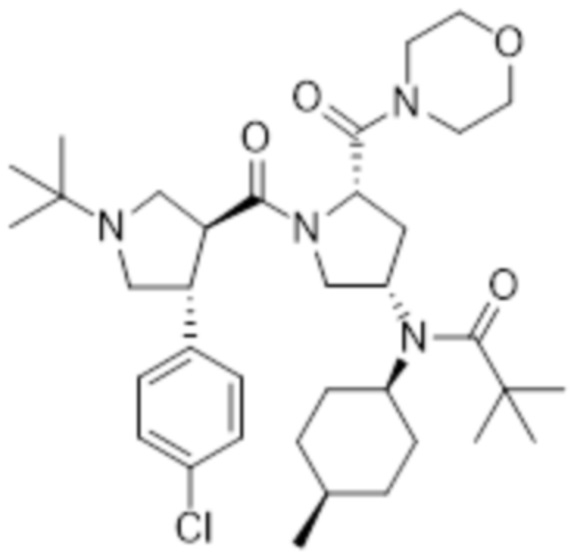
[222] 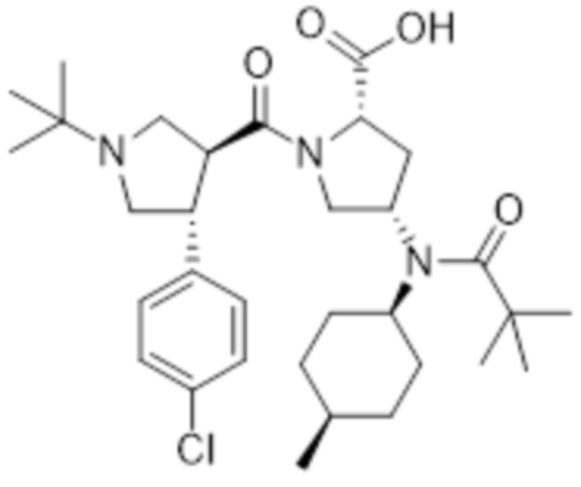
[223] Указанное в заголовке соединение (0,10 г, 99%) получали таким же образом, как на стадии B примера 1, с использованием метил (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)пиваламидо)пирролидин-2-карбоксилата (0,10 г, 0,18 ммоль), полученного на стадии A.
[224] MS [M+H]=574,4 (M+1)
[225]
[226] Стадия C: Получение N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пиваламида
[227]
[228] Указанное в заголовке соединение (0,020 г, 17%) получали таким же образом, как на стадии C примера 1, с использованием (2S,4S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-4-(N-((1S,4R)-4-метилциклогексил)пиваламидо)пирролидин-2-карбоновой кислоты (0,10 г, 0,18 ммоль), полученной на стадии B.
[229] MS [M+H]=643,5 (M+1)
[230] 1H ЯМР (400 МГц, CD3OD) δ 7,40-7,30 (м, 4H), 4,79 (м, 1H), 4,17 (м, 1H), 3,80-3,40 (м, 15H), 3,10 (м, 1H), 2,96 (м, 1H), 2,71 (м, 1H), 2,15 (м, 1H), 1,90 (м, 1H), 1,80-1,35 (м, 8H), 1,21-1,15 (м, 18H), 1,02 (м, 3H)
[231]
[232] Стадия D: Получение гидрохлорида N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пиваламида
[233]
[234]
[235] Указанное в заголовке соединение (0,29 г, 83%) получали таким же образом, как на стадии D примера 1, с использованием N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пиваламида (0,33 г, 0,51 ммоль), полученного на стадии C.
[236] MS [M+H]=643,5 (M+1)
[237] 1H ЯМР (400 МГц, CD3OD) δ 7,41 (м, 4H), 4,80 (м, 1H), 4,13 (м, 1H), 3,90 (м, 2H), 3,80-3,40 (м, 13H), 2,94 (м, 1H), 2,63 (м, 1H), 2,11 (м, 1H), 1,93 (м, 1H), 1,75 (м, 2H), 1,60 (м, 4H), 1,46 (с, 9H), 1,15 (с, 9H), 1,45-1,30 (м, 3H), 1,01 (м, 3H)
[238]
[239] Сравнительный пример 1: Получение гидрохлорида N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(2,4-дифторфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-(4,4-диметилциклогексил)ацетамида (A95)
[240]
[241] 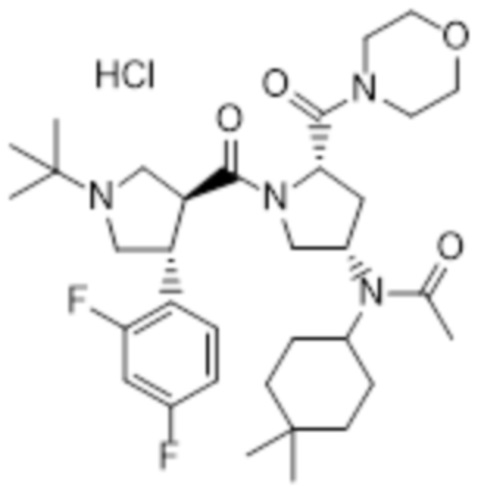
[242] Соединение A95, описанное в международной публикации № WO 2008/007930, было получено тем же способом, который там описан.
[243]
[244] Сравнительный пример 2: Получение гидрохлорида N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-(4,4-диметилциклогексил)ацетамида (A96)
[2450]
[246] 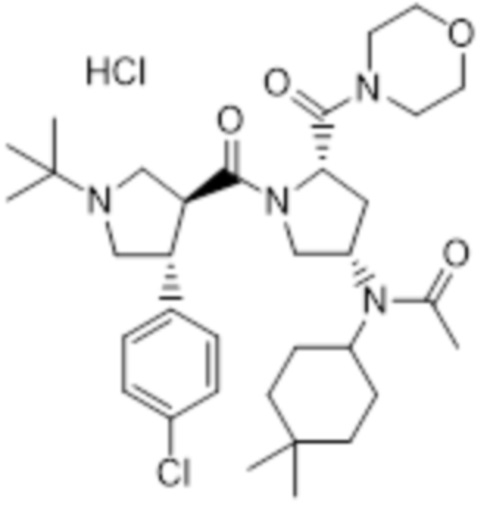
[247] Соединение A96, описанное в международной публикации № WO 2008/007930, было получено тем же способом, который там описан.
[248]
[249] Экспериментальный пример 1: Анализ люциферазы
[250] Для оценки агонистической способности против MC4R (рецептор меланокортина-4) была создана линия клеток, которая постоянно экспрессирует ген люциферазы (CRE-LUC) под контролем MC4R и CRE (элемент ответа цАМФ). После получения вектора экспрессии клеток млекопитающих (pCDNA3 (Neo)) (Invitrogen), содержащего ген MC4R, клеточные линии эмбриональной почки человека (HEK) трансформировали с использованием липофектамина 2000 (Invitrogen) вместе с вектором (pCRE-Luc) (Stratagen), экспрессирующим ген люциферазы (CRE-LUC) под контролем элемента ответа цАМФ (CRE). Трансформированные клеточные линии (HEK MC4R-Luc) инкубировали в инкубаторе при температуре 37°C в присутствии 5% CO2 в течение 24 часов с использованием модифицированной Дульбекко среды Игла (DMEM), содержащей 10% инактивированной нагреванием фетальной бычьей сыворотки (GIBCO/BRL). Клеточные линии инкубировали в течение четырех дней в присутствии модифицированной по Дульбекко среды Игла (DMEM), содержащей 10 мл селективной среды (10% инактивированной нагреванием фетальной бычьей сыворотки (GIBCO/BRL), 100 единиц мл пенициллина (GIBCO/BRL), 100 ед/мл стрептомицина (GIBCO/BRL), 800 мкг/мл генетицина (G418) (GIBCO/BRL)). Процесс удаления клеток, убитых селективной средой, путем замены среды на 10 мл новой селективной среды повторяли трижды, каждые 4 дня. Отдельные колонии, образованные окончательно отобранными и размноженными клонами, переносили под микроскопом в 24-луночный планшет для культивирования клеток, содержащий 1 мл селективной среды на лунку, и инкубировали в течение 4 дней. Форсколин (SIGMA) обрабатывали до конечной концентрации 10 мкМ, а затем инкубировали в течение пяти часов в инкубаторе при температуре 37°C в присутствии 5% CO2. Каждую лунку обрабатывали 50 мкл люциферазного реагента Bright-Glo (Promega) и оставляли при комнатной температуре на 15 минут, а затем измеряли люминесценцию каждой лунки с помощью люминометра (Victor). Клоны, демонстрирующие люминесценцию в 100 или более раз превышающую исходное значение при обработке форсколином, отбирали и использовали для измерения агонистической способности MC4R каждого соединения.
[251] Клетки HEK MC4R-Luc добавляли в каждую лунку 96-луночного люминометрического планшета для культивирования клеток (Costar) до размера 2,5×104 клеток в 100 мкл культуральной среды, а затем инкубировали в инкубаторе при температуре 37°C в присутствии 6% CO2 в течение 18 часов. Агонист MCR, разбавленный на каждой стадии концентрирования с использованием вышеуказанной культуральной среды, обрабатывали так, чтобы конечная концентрация ДМСО не превышала 1%, а затем инкубировали в течение пяти часов в инкубаторе при температуре 37°C в присутствии 6% CO2. Каждую лунку обрабатывали 50 мкл люциферазного реагента Bright-Glo (Promega) и оставляли при комнатной температуре на пять минут, а затем измеряли люминесценцию каждой лунки с помощью люминометра (Victor). Количество люминесценции, индуцированной агонистом, разбавленным на каждой стадии концентрирования, преобразовывали в относительное значение в % по отношению к количеству, проявляемому при обработке 10 мкМ NDP-α-MSH. EC0,5 MSH выражается как концентрация, которая вызывает 50% максимального количества люминесценции, которое может быть индуцировано NDP-α-MSH, а EC50 выражается как концентрация, которая вызывает 50% максимального количества люминесценции, которое может быть индуцировано каждым агонистом. Измерения проводились с использованием статистического программного обеспечения (Prizm).
[252] В таблице 1 показаны результаты измерения агонистической способности MC4R каждого соединения, полученные в вышеуказанных экспериментах, в единицах EC50 (нМ).
[253] [Таблица 1]
[254]
[255]
[256] Как показано в таблице 1, было подтверждено, что среди хорошо известных рецепторов меланокортина in vivo в отношении рецептора меланокортина-4 (MC4R), участвующего в энергетическом метаболизме и контроле веса in vivo, соединения примеров обладают более высокой агонистической способностью к MC4R, чем соединения сравнительных примеров (A95 и A96).
[257]
[258] Экспериментальный пример 2: Анализ цАМФ
[259] Рецептор меланокортина представляет собой тип рецептора, сопряженного с G-белком (GPCR), и основная роль G-белка заключается в активации вторичных преобразователей для регулирования клеточных ответов на многие физиологические стимулы посредством трансдукции сигнала. MC4R представляет собой Gs-связанный рецептор, и известно, что, если MC4R взаимодействует с агонистом, аденилатциклаза (AC) активируется, увеличивая концентрацию циклического AMP (cAMP), который является одним из вторичных преобразователей в клетках. Следовательно, можно оценить активность рецепторов меланокортина путем измерения генерации сигналов цАМФ.
[260] После того, как были установлены линии клеток, связанных с цАМФ Hunter Gs (линия клеток CHO-K1), в которых каждый из MC1R, MC3R, MC4R и MC5R был сверхэкспрессирован, чтобы можно было измерить повышение уровня цАМФ в клетках из-за реакции агонистов, клетки инокулировали в каждую лунку планшета для культивирования лейкоцитов и инкубировали в течение 24 часов в инкубаторе при температуре 37°C в присутствии 5% CO2. После инкубации среду удаляли и добавляли 15 мкл реагента 2:1 HBBS/10 мМ HEPES:cAMP XS+Ab. После добавления 5 мкл образца, разбавленного в 4 раза буфером, концентрацию носителя устанавливали равной 1% и добавляли соединение-агонист MC4R, разбавленное на каждой стадии концентрации, с последующим взаимодействием при температуре 37°C в течение 30 минут. Активность (%) каждого соединения-агониста выражается как 100%×(среднее значение RLU образца-среднее значение RLU контроля носителя)/(среднее значение RLU максимального контроля-среднее значение RLU контроля носителя), и значение было проанализировано с помощью пакета анализа данных (ChemInnovation, CA).
[261] В таблице 2 показаны результаты измерения агонистической способности рецепторов меланокортина соединений, полученных в вышеуказанных экспериментах, в значениях EC50 (нМ).
[262] [Таблица 2]
[263]
[264] Как показано в Таблице 2, было подтверждено, что среди хорошо известных рецепторов меланокортина in vivo в отношении рецептора меланокортина-4 (MC4R), участвующего в энергетическом метаболизме и контроле веса in vivo, соединения примеров обладают более высокой способностью к агонистам рецепторов, чем соединения сравнительных примеров (A95 и A96).
[265]
[266] Экспериментальный пример 3: Анализ β-аррестина
[267] Рецептор меланокортина представляет собой тип рецептора, сопряженного с G-белком (GPCR), и регулирует различные физиологические реакции путем трансдукции сигналов от многих нейротрансмиттеров. Когда GPCR фосфорилируется, β-аррестин связывается с фосфорилированной частью рецептора и играет важную роль в активации различных сигнальных путей в клетках посредством взаимодействия с другими белками. Известно, что когда рецептор меланокортина взаимодействует с агонистом, β-аррестин мобилизуется и участвует в опосредованном β-аррестином сигнальном пути. Следовательно, активность рецептора меланокортина можно оценить путем измерения β-аррестина.
[268] Была создана клеточная линия β-аррестина Pathhunter eXpress (линия клеток U2OS), в которой вместе экспрессировались β-аррестин, меченный Prolink (PK), MC1R, MC3R, MC4R, MC5R и ферментный акцептор (EA). Когда часть MCR-PK этой клеточной линии активируется, β-аррестин-EA мобилизуется, и акцептор фермента (EA) и Prolink (PK), которые являются фрагментами фермента β-галактозидазы, взаимодействуют. Активированный фермент гидролизует субстрат за счет активности β-галактозидазы, чтобы произвести хемилюминесцентный сигнал, так что активность может быть измерена. После инкубации линии клеток-аррестина Pathhunter eXpress (линия клеток U2OS) клетки инокулировали в каждую лунку планшета для культивирования клеток и инкубировали в течение 48 часов в инкубаторе при температуре 37°C в присутствии 5% CO2. После инкубации добавляли 5 мкл образца, разведенного в 5 раз буфером, концентрацию носителя устанавливали равной 1% и добавляли соединение-агонист MC4R, разбавленное на каждой стадии концентрации, с последующим взаимодействием при температуре 37°C в течение 90 минут. Активность (%) каждого соединения-агониста выражается как 100%×(среднее значение RLU образца-среднее значение RLU контроля носителя)/(среднее максимальное значение контрольного лиганда-среднее значение RLU контроля носителя), и значение было проанализированы пакетом анализа данных CBIS (ChemInnovation, CA).-
[269] В таблице 3 показаны результаты измерения активности рецептора меланокортина каждого соединения, полученные в вышеуказанных экспериментах, в значениях EC50 (нМ).
[270] [Таблица 3]
[271]
[272]
[273] Как показано в таблице 3, было подтверждено, что среди хорошо известных рецепторов меланокортина in vivo в отношении рецептора меланокортина-4 (MC4R), участвующего в энергетическом метаболизме и контроле веса in vivo, соединения примеров обладают более высокой способностью к агонистам рецепторов, чем соединения сравнительных примеров (A95 и A96).
[274]
[275] Экспериментальный пример 4: Сродство связывания
[276] Известно пять подтипов рецептора меланокортина (MCR) in vivo, и известно, что MC4R, который относится к подтипу 4, участвует в энергетическом метаболизме и контроле веса. Поскольку другие подтипы MCR участвуют в регуляции различных функций in vivo, таких как пигментация кожи, энергетический гомеостаз и экзокринные функции, обеспечение селективности в отношении MC4R соединений-агонистов MC4R очень важно для предотвращения возможных побочных эффектов в будущем. Поэтому были измерены рецепторные способности агонистов MC4R для каждого подтипа MCR.
[277] После того, как были созданы клеточная линия CHO-K1, экспрессирующая человеческий рекомбинантный MC1R, и клеточные линии HEK-293, экспрессирующие MC3R, MC4R и MC5R, из каждой клеточной линии собирали мембраны. В 96-луночном планшете для культивирования клеток 3 мкг MC1R-мембраны и 0,04 нМ 125I-NDP-α-MSH на лунку взаимодействовали при температуре 37°C в течение двух часов. 3 мкг мембраны MC3R и MC5R и 0,035 нМ 125I-NDP-α-MSH взаимодействовали при температуре 37°C в течение двух часов. 3 мкг мембраны MC3R и MC5R и 0,035 нМ 125I-NDP-α-MSH подвергали взаимодействию при температуре 37°C в течение одного часа, и 3,12 мкг мембраны MC4R и 0,02 нМ 125I-NDP-α-MSH подвергали взаимодействию при температуре 37°C в течение двух часов. В это время в каждую лунку добавляли 25 мМ адсорбционного буфера HEPES-KOH (pH 7,0), содержащего агонист MCR, разведенный на каждой стадии концентрации, и подвергали взаимодействию. Прореагировавший раствор переносили на фильтр и промывали адсорбционным буфером, а затем измеряли радиоактивность. Значение, исключая количество неспецифического связывания в присутствии 1 мкМ (MC1R) и 3 мкМ (MC3R, MC4R, MC5R) NDP-α-MSH от каждого общего количества связывания, использовали в качестве количеств специфического связывания 125I-NDP-α-МСГ. Измеряли степень, в которой специфическое связывание 125I-NDP-α-MSH ингибировалось агонистом, разбавленным при каждой ступенчатой концентрации. IC50 выражали как концентрацию каждого агониста, которая ингибировала специфическое связывание 50% 125I-NDP-α-MSH.
[278] В таблице 4 представлены результаты измерения связывания рецепторов меланокортина соединений, полученных в вышеуказанном эксперименте, в значениях Ki (нМ).
[279] [Таблица 4]
[280]
[281]
[282] Как показано в Таблице 4, было подтверждено, что среди хорошо известных рецепторов меланокортина in vivo в отношении рецептора меланокортина-4 (MC4R), участвующего в энергетическом метаболизме и контроле веса in vivo, соединения примеров обладают более высокой способностью связываться с рецепторами, чем соединения сравнительных примеров (A95 и A96).
[283]
[284] Экспериментальный пример 5: Фармакокинетика и метаболизм лекарственного средства
[285] Экспериментальный пример 5-1: Фармакокинетический профиль
[286] Для исследования фармакокинетических (PK) свойств соединения примера 1 и соединений сравнительных примеров был проведен следующий эксперимент.
[287] Для проведения PK-теста для соединения примера 1 и соединений сравнительных примеров (A95 и A96) были приготовлены мыши C57BL6 в возрасте около 7 недель, по 12 особей были распределены по каждому введенному веществу, разделены на группы и голодали для перорального приема. В день введения раствор лекарственного средства готовили с концентрацией 1 мг/мл с использованием дистиллированной воды (DW) в качестве носителя и вводили перорально из расчета 1 мл на кг массы тела каждой особи, а конечная доза составляла 10 мг/кг. Через 1, 3, 8 и 24 часа после введения у каждого из 3 особей в каждой группе собирали цельную кровь путем забора крови из сердца, а затем помещали в пробирку с гепарином для предотвращения коагуляции. После этого головной мозг каждой особи собирали и помещали в пробирку для EP, измеряли массу ткани и хранили при заморозке при температуре -20°C.
[288] В день анализа в каждую тканевую пробирку добавляли DDW в 4 раза превышающий объем массы ткани и гомогенизировали, и сохраненную плазму размораживали при комнатной температуре. Как и в случае с плазмой брали 50 мкл гомогената ткани и переносили в отдельную пробирку, и затем в каждую пробирку с плазменным и тканевым гомогенатом добавляли 200 мкл ацетонитрила (AN), что в 4 раза больше общего объема образца для проведения депротеинизации. В этом случае AN включил внутренний стандарт. Для построения калибровочной кривой был приготовлен раствор AN (с внутренним стандартом) с известными концентрациями 0,1, 0,5, 5, 50 и 500 нг/мл, и была проведена депротеинизация в 4-кратном объеме, как указано выше, в пустой плазме каждой из плазмы и мозга. Таким образом, были подготовлены окончательные калибровочные кривые от 0,4 до 2000 нг/мл для плазмы и 1, 5, 50, 500 и 5000 нг/мл для мозга. После того, как 0,5 мкл супернатанта, полученного после депротеинизации, было введено в ЖХ-МС/МС, площади пиков соединений примера и сравнительных примеров (A95 и A96) были скорректированы с площадями пиков IS, чтобы получить пиковую реакцию на каждом точку отбора пробы и выполнить преобразование концентрации по калибровочной кривой.
[289] Фармакокинетические параметры (Cmax, AUCinf, t1/2 и тому подобное) рассчитывали методом некомпартментного анализа с использованием WinNonlin 8.1 для значений концентрации в крови во времени для каждой группы введения.
[290] Фармакокинетические характеристики каждого соединения сравнивали путем сравнения воздействия, изменений периода полужизни и тому подобное для каждой группы введения лекарственного средства, и полученные значения показаны в таблицах 5 и 6. Кроме того, значения результатов в соответствии с соотношением воздействия на мозг и воздействия крови показано в таблице 7.
[291] [Таблица 5]
[292]
[293] [Таблица 6]
[294]
[295] [Таблица 7]
[296]
[297]
[298] На основании приведенных выше результатов, воздействие каждого соединения было подтверждено во всем организме и в головном мозге в порядке следования соединения примера 1, соединения сравнительного примера 2 (A96) и соединения сравнительного примера 1 (A95). Время полужизни потери в головном мозге в течение периода наблюдения выше, чем значение, наблюдаемое в крови, и предполагается, что это означает эффективное сохранение ряда веществ в месте проявления эффективности. Кроме того, было подтверждено, что, когда каждое соединение вводилось в одной и той же дозе, соединение примера имело наилучшее абсолютное воздействие и стабильность в головном мозге. Кроме того, было подтверждено, что соотношение воздействия на мозг и воздействия на кровь соединения примера 1 также было самым превосходным по сравнению с соединениями сравнительных примеров (A95 и A96).
[299] В дополнение к этим результатам, когда in vitro активность каждого соединения и стойкость лекарственного средства были всесторонне рассмотрены, наилучшую эффективность можно было ожидать от соединения из примера 1 по сравнению с соединениями из сравнительных примеров, когда была введена та же доза. Чтобы достичь эффективности определенного уровня, можно вводить более низкую дозу, и, соответственно, ожидается, что это сведет к минимуму побочные эффекты, возникающие в результате системного воздействия.
[300]
[301] Экспериментальный пример 5-2: Ингибирование CY (%)
[302] Чтобы подтвердить лекарственное взаимодействие с изоферментом CYP (цитохром P450), был проведен следующий эксперимент.
[303] Для измерения и сравнения ингибирующей способности соединения примера 1 и соединения сравнительного примера 2 (A96) были приготовлены рекомбинантные ферменты CYP1A2, 2C9, 2C19, 2D6 и 3A4. Следующая таблица 8 относится к использованию зондового субстрата, положительного контроля, изофермента и условий измерения в отношении измерения ингибирующей способности каждого вещества.
[304] [Таблица 8]
[305]
ДМСО
метаноле
[306]
[307] Инкубирование проводили в 96-луночном планшете (Costar, 3792-черный, с круглым дном), буферная система, используемая для метаболизма, представляла собой 50 мМ калий-фосфатный буфер и pH 7,4, а конечный объем реакции составлял 250 мкл. Конечные концентрации соединения примера 1, соединения сравнительного примера 2 (A96) и положительного контроля в общем буфере составляли 10 мкМ (2% метанол (об./об.)), и каждая включала отрицательный контроль (только метанол, 2% (об./об.)). Буфер, содержащий экспериментальное соединение, смешивали с таким же объемом раствора для приготовления изофермента CYP так, чтобы конечная концентрация составляла 10 мкМ, с последующим предварительным нагреванием в течение 10 минут в флюоресцентном считывающем устройстве для планшетов при температуре 37°C. В каждую лунку добавляли раствор NRS (система регенерации NADPH: 0,22 мМ β-NADP, 2,8 мМ глюкозо-6-фосфат и 0,6 единиц/мл глюкозо-6-фосфатдегидрогеназы) с последующей предварительной инкубацией в течение 30 минут. После этого реакцию инициировали добавлением субстрата в лунку, подвергнутую предварительной инкубации, и контролировали с интервалом в одну минуту при длине волны измерения для каждого субстрата в течение 30 минут. Измеренные значения, полученные для положительного контроля и соединения примера 1, сравнивали с интенсивностью флуоресценции отрицательного контроля (без ингибирования или соединения) для подтверждения ингибирующей способности каждого соединения против изофермента. Ингибирующая способность (%) против каждого изофермента, когда каждое соединение обрабатывали в концентрации 10 мкМ, показана в таблице 9. Ингибирующая активность (%) была выражена на основании следующих критериев: A=<50%, B=>50%.
[308] Обрабатывающая концентрация соединения 10 мкМ была очень высокой, и представляет собой консервативный критерий для оценки ингибирующей способности на основе такой обрабатывающей концентрации и выбора лекарства со степенью ингибирования ниже 50% в качестве кандидата. Можно считать, что это наименее вероятно, что выбранный препарат-кандидат в этом случае ингибирует изофермент CYP.
[309] [Таблица 9]
[310]
[311]
[312] Из приведенных выше результатов при обработке в той же концентрации было подтверждено, что ингибирующая способность соединения примера 1 против основного изофермента CYP была ниже или аналогична таковой у соединения (A96) сравнительного примера 2. Существует опасение, что соединение (A96) сравнительного примера 2 взаимодействует с лекарственными средствами из-за очень высокой ингибирующей способности против CYP2C9. Известно, что CYP2C9 участвует в метаболизме примерно 10% всех коммерчески доступных лекарств и играет важную роль в потере эффективности лекарств с узким терапевтическим окном. Кроме того, CYP2C9 очень важен как в исследовательских, так и в клинических аспектах, поскольку сообщалось о полиморфизме, зависящем от каждой особи. Об этом также можно узнать из того факта, что CYP2C9 представлен в списке основных изоферментов, ингибирующий эффект которых следует проверять во время разработки лекарственного средства в руководстве FDA по взаимодействию лекарств с лекарственными средствами (DDI).
[313] На основании этого было определено, что при введении соединения примера 1 лекарственное взаимодействие, вызванное ингибированием CYP, ниже, чем у соединения сравнения (A96).
[314]
[315] Экспериментальный пример 6: Фармакология
[316] Фармакологический эффект агониста рецептора меланокортина-4 соединения по настоящему изобретению оценивали на следующей модели ожирения.
[317]
[318] Экспериментальный пример 6-1: Модель ожирения у мышей, индуцированного диетой с высоким содержанием жиров
[319] Эффект агонистов рецептора меланокортина-4 на ожирение оценивали с использованием модели ожирения у мышей, индуцированного диетой с высоким содержанием жиров.
[320] Пятинедельным самцам мышей C57BL/6Ntaconic давали диету с содержанием жира 60 ккал % (D12492, Research Diet.) в течение 15 недель, чтобы вызвать ожирение. Соединение примера 1, соединения сравнительных примеров (A95 и A96) и сибутрамин в качестве положительного контроля готовили в дистиллированной воде и вводили перорально 19-недельным мышам с моделями ожирения, индуцированного диетой с высоким содержанием жиров, один раз в день с 1 по 16 день. С 1 по 16 день измеряли массу тела один раз в день, потребление пищи измеряли пять раз в неделю и потребление питьевой воды измеряли два раза в неделю. Уровень глюкозы в крови и гликированный гемоглобин измеряли на 15-й день, и всех животных умерщвляли на 17-й день. Кровь собирали через брюшную полую вену, удаляли и взвешивали печень и эпидидимальную жировую ткань. Собранную кровь помещали в пробирку с гепарином и центрифугировали для отделения плазмы, а затем выполняли биохимический анализ плазмы.
[321] В таблице 10 показана разница в скорости изменения веса носителя, измеренную на 12-й день для каждой дозы каждого соединения.
[322] [Таблица 10]
[323]
[324] (Различие в скорости изменения веса по сравнению с носителем)
[325] Значимая различие в массе тела между контрольной группой носителя и группой, получавшей соединение: *p<0,05, **p<0,01 (среднее±SEM, t-критерий Стьюдента, непарный, двусторонний)
[326]
[327] На этих моделях ожирения у мышей, в частности, когда вводили только соединение примера 1, была показана значительная потеря веса. Когда соединение примера 1 вводили в дозах 10 мг/кг и 30 мг/кг, было вызвано -9,4% и -15,1% ингибирования увеличения веса по сравнению с контрольным носителем, когда вводили растворитель, соответственно, и был продемонстрирован статистически значимый эффект потери веса по сравнению с соединениями сравнительных примеров (A95 и A96).
[328] Это вызвано превосходной агонистической способностью MC4R in vitro, превосходным абсолютным воздействием на мозг и стабильностью, а также превосходным коэффициентом воздействия на мозг по сравнению с воздействием на кровь соединения из примера 1 по сравнению с соединениями из сравнительных примеров, и считается, что значительная разница может проявляться даже при низких дозах.
[329] Кроме того, соединение примера 1 показало превосходные результаты, равные или более высокие с точки зрения эффективности, по сравнению с сибутрамином (Reductil), который является средством для лечения ожирения в родственной области, и, таким образом, ожидается, что оно будет проявлять значительную лекарственную эффективность. в реальном клиническом применении.
| название | год | авторы | номер документа |
|---|---|---|---|
| АГОНИСТЫ РЕЦЕПТОРА МЕЛАНОКОРТИНА | 2007 |
|
RU2411240C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛФЕНИЛЦИКЛОГЕКСАНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2505521C2 |
| ПИПЕРАЗИНОВОЕ ПРОИЗВОДНОЕ | 2016 |
|
RU2731913C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| ПРОЛЕКАРСТВО ФТОРСОДЕРЖАЩЕЙ АМИНОКИСЛОТЫ | 2013 |
|
RU2639868C1 |
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ ДЛЯ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК | 2008 |
|
RU2434010C2 |
| КОНЪЮГАТ ЛИГАНДА С ЦИТОТОКСИЧЕСКИМ ЛЕКАРСТВЕННЫМ СРЕДСТВОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2708461C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2018 |
|
RU2765152C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2021 |
|
RU2826628C1 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2014 |
|
RU2704112C2 |
Изобретение относится к области органической химии, а именно к соединению формулы 1 или его фармацевтически приемлемой соли, где R1 представляет собой C2-C5 алкил. Также изобретение относится к фармацевтической композиции на основе соединения формулы 1, ее применению для получения лекарственного средства и к способу лечения или профилактики указанных заболеваний. Технический результат: превосходная агонистическая активность в отношении рецепторов меланокортина, проявляемая соединением формулы 1, что может быть использовано для предотвращения или лечения ожирения, диабета, воспалений и эректильной дисфункции. 10 н. и 9 з.п. ф-лы, 10 табл., 16 пр.
Формула 1
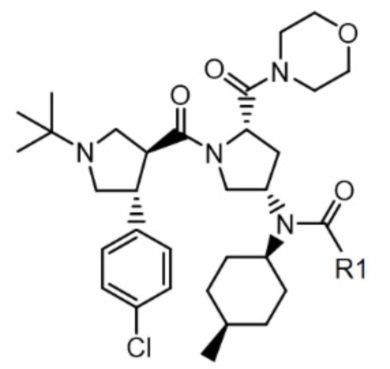
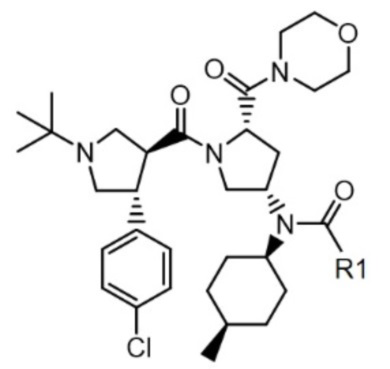
1. Соединение следующей формулы 1 или его фармацевтически приемлемая соль или стереоизомер:
[Формула 1]
 ,
,
где R1 представляет собой C2-C5 алкил.
2. Соединение или его фармацевтически приемлемая соль или стереоизомер по п.1, где R1 представляет собой C2-C4 алкил.
3. Соединение или его фармацевтически приемлемая соль или стереоизомер по п.2, где R1 представляет собой этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил.
4. Соединение или его фармацевтически приемлемая соль или стереоизомер по п.2, где соединение выбрано из следующей группы:
N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)изобутирамид,
N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пропионамид и
N-((3S,5S)-1-((3S,4R)-1-(трет-бутил)-4-(4-хлорфенил)пирролидин-3-карбонил)-5-(морфолин-4-карбонил)пирролидин-3-ил)-N-((1S,4R)-4-метилциклогексил)пиваламид.
5. Соединение или его фармацевтически приемлемая соль или стереоизомер по п.1, где фармацевтически приемлемая соль представляет собой кислотно-аддитивную соль, образованную неорганической кислотой, выбранной из группы, включающей хлористоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту, бромистоводородную кислоту и йодистоводородную кислоту.
6. Соединение или его фармацевтически приемлемая соль или стереоизомер по п.5, где фармацевтически приемлемая соль представляет собой гидрохлорид.
7. Фармацевтическая композиция агониста рецептора меланокортина, содержащая терапевтически эффективное количество соединения формулы 1 или его фармацевтически приемлемой соли или стереоизомера, определенные в любом из пп.1-6, в качестве активного ингредиента вместе с фармацевтически приемлемым носителем.
8. Фармацевтическая композиция агониста рецептора меланокортина по п.7, которая предназначена для профилактики или лечения ожирения.
9. Фармацевтическая композиция агониста рецептора меланокортина по п.7, которая предназначена для профилактики или лечения диабета.
10. Фармацевтическая композиция агониста рецептора меланокортина по п.7, которая предназначена для профилактики или лечения воспаления.
11. Фармацевтическая композиция агониста рецептора меланокортина по п.7, которая предназначена для профилактики или лечения эректильной дисфункции.
12. Применение фармацевтической композиции агониста рецептора меланокортина по п.7 при получении лекарственного средства для профилактики или лечения ожирения.
13. Применение фармацевтической композиции агониста рецептора меланокортина по п.7 при получении лекарственного средства для профилактики или лечения диабета.
14. Применение фармацевтической композиции агониста рецептора меланокортина по п.7 при получении лекарственного средства для профилактики или лечения воспаления.
15. Применение фармацевтической композиции агониста рецептора меланокортина по п.7 при получении лекарственного средства для профилактики или лечения эректильной дисфункции.
16. Способ профилактики или лечения ожирения у нуждающегося в этом субъекта, включающий введение субъекту фармацевтической композиции агониста рецептора меланокортина по п.7.
17. Способ профилактики или лечения диабета у нуждающегося в этом субъекта, включающий введение субъекту фармацевтической композиции агониста рецептора меланокортина по п.7.
18. Способ профилактики или лечения воспаления у нуждающегося в этом субъекта, включающий введение субъекту фармацевтической композиции агониста рецептора меланокортина по п.7.
19. Способ профилактики или лечения эректильной дисфункции у нуждающегося в этом субъекта, включающий введение субъекту фармацевтической композиции агониста рецептора меланокортина по п.7.
| WO 2005047251 A1, 26.05.2005 | |||
| US 20040019094 A1, 29.01.2004 | |||
| WO 2001070708 A1, 27.09.2001 | |||
| АГОНИСТЫ РЕЦЕПТОРА МЕЛАНОКОРТИНА | 2007 |
|
RU2411240C2 |
| НОВОЕ ПИРРОЛИДИНОВОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ АГОНИСТА РЕЦЕПТОРА МЕЛАНОКОРТИНА | 2015 |
|
RU2669938C2 |

