Область техники, к которой относится изобретение
Изобретение относится, помимо прочего, к области очистки пептидов, в частности циклических или нециклических пептидов, их аналогов или производных. Более конкретно, изобретение относится к упрощенному или оптимизированному способу очистки циклических пептидов из композиции, содержащей указанный пептид и, по меньшей мере, одну родственную примесь, хроматографическими способами, позволяющими получать желаемый конечный продукт с высокими выходами, селективностью и степенью чистоты. Усовершенствованный способ, в частности, пригоден для получения эптифибатида, эксенатида, атозибана, незиритида и родственных производных и аналогов. Получаемые полипептиды имеют высокую степень чистоты, которая составляет, по меньшей мере, 96% и, предпочтительно, составляет, по меньшей мере, около 99%.
Уровень техники
Существенным аспектом при получении рекомбинантных (генно-инженерных) пептидов, включая циклические пептиды, их производные и аналоги, предназначенные для терапевтического использования с целью лечения людей или животных, является рассматриваемый способ очистки, предназначенный для получения продукта с достаточно высокими селективностью, выходом и степенью чистоты таким образом, что желаемый продукт по существу не содержит примесей посторонних белков, которые могут образовываться в процессе синтеза.
В процессе синтеза пептидов образуются различные виды примесей, например диастереомеры, продукты гидролиза лабильных амидных связей, делеционные последовательности, образующиеся преимущественно при твердофазном пептидном синтезе, пептиды внедрения и побочные продукты, в частности полиморфные формы, образующиеся при снятии защитных групп на последней стадии синтеза. Таковыми часто являются некоторые побочные продукты, связанные с образованием циклических пептидов, содержащих дисульфидные связи [Bodansky и соавт., Principles of Peptide Synthesis; Springer-Verlag; Berlin, 1993]. В данной области техники существует необходимость в разработке хроматографических способов очистки, которые можно было бы использовать в больших масштабах при минимальном количестве стадий, требуемых для отделения желаемого пептида, содержащегося в маточном растворе, от сложной смеси родственных примесей. В сущности, большинство примесей, образующихся в процессе синтеза, нельзя удалить одним-единственным хроматографическим способом, но этого можно достичь сочетанием нескольких способов, например хроматографии с обращенной фазой и катионообменной хроматографии, что позволяет получать лекарственный препарат, лекарственное вещество в рецептурном буферном растворе, описанным в настоящем изобретении.
В заявленном изобретении используются два хроматографических способа, а именно хроматография с обращенной фазой и катионообменная хроматография.
Для получения желаемого конечного результата в отношении чистоты и выхода продукта применяется ряд хроматографических способов. Хроматография с обращенной фазой является одним из наиболее эффективных способов очистки, основной принцип разделения которой основан на гидрофобных взаимодействиях. Обращено-фазовая жидкостная хроматография (ОФ-ЖХ) и обращенно-фазовая высокоэффективная жидкостная хроматография (ОФ-ВЭЖХ) широко используются для очистки молекул, таких как пептиды и белки, получаемых либо синтетическим, либо рекомбинантным способом. Методами ОФ-ЖХ и ОФ-ВЭЖХ можно эффективно разделять близкородственные примеси, эти методы уже используются для очистки разнообразных молекул (Lee и соавт., "Preparative HPLC," 8th Biotechnology Symposium, Pt. 1, 593-610 (1988)). Кроме того, ОФ-ЖХ и ОФ-ВЭЖХ успешно используются для очистки молекул, в частности белков, в промышленных масштабах (Olsen и соавт., 1994, J. Chromatog. A, 675, 101).
Принцип ионообменной хроматографии (ИОХ) включает два различных подхода: анионный и катионный обмены в соответствии с зарядом лигандов на ионообменной смоле. Традиционный ИОХ способ очистки обычно включает несколько стадий: стадии уравновешивания, нанесения или загрузки, промывки, элюирования и регенерации (см. Remington's Pharmaceutical Sciences, Gennaro, ed., Mack Publishing Co., Easton, Pa., 1990, или Remington: The Science and Practice of Pharamacy, 19th Edition (1995)).
Каждый параметр хроматографической процедуры играет существенную роль при получении желаемого белкового продукта. Для крупномасштабной очистки пептидов на обращенно-фазовых смолах используются различные хроматографические среды (матрицы). К числу наиболее распространенных сред относятся смолы с С-4, С-8 и С-18 алкильными цепями, закрепленные на поверхности диоксида кремния. К одним из важных параметров относятся форма и размер смоляных частиц неподвижной фазы. К числу других существенных параметров относятся тип буферной системы, скорость потока, рН и т.д. Несмотря на усовершенствования способов очистки пептидов, некоторые очищенные пептиды все еще содержат неприемлемые противоионы в нежелательных количествах. В контексте данного предлагаемого принципа действия дается ссылка на альтернативный способ очистки, который позволяет преодолеть проблемы, стоящие в данной области техники.
US 2006148699 относится к способу очистки пептидов на основе ОФ-ВЭЖХ, который включает стадию промывки колонки водным раствором фармацевтически приемлемой соли, содержащей противоион, и элюирования пептида с колонки смесью органических растворителей и кислоты, содержащей фармацевтически приемлемый противоион, при этом рН водного раствора составляет, по меньшей мере, 6. В последующих зависимых пунктах формулы изобретения также более конкретно заявляются циклические, нециклические пептиды и эптифибатид.
B WO 2005100388 раскрывается ОФ-ВЭЖХ способ очистки эксендина-4 с помощью ацетонитрилводной градиентной смеси, который позволяет извлекать продукт со степенью чистоты 97,5%.
WO 2005019262 относится к использованию смолы на основе сополимера стирола и дивинилбензола для ОФ-ВЭЖХ очистки глюкагонподобных пептидов.
В данной области техники существует необходимость в эффективном хроматографическом способе эффективного разделения молекул, таких как циклические пептиды, после их твердофазного синтеза с целью получения конечных пептидных продуктов особой чистоты. Такая необходимость могла бы быть удовлетворена в том случае, если способ позволяет воспроизводить, насколько это возможно, выход, чистоту, производительность и рабочие условия хроматографического процесса, в котором элюирование осуществляется с использованием определенной смеси растворителей, в определенном рН диапазоне и с учетом других соответствующих факторов. Рабочую методику можно успешно использовать для промышленного разделения.
Раскрытие изобретения
Основная цель настоящего изобретения состоит в обеспечении способа очистки пептида от смеси, содержащей, по меньшей мере, одну родственную примесь.
Другая цель настоящего изобретения состоит в обеспечении способа очистки посредством обращенно-фазовой высокоэффективной жидкостной хроматографии и ионообменной хроматографии.
Еще одна цель настоящего изобретения состоит в обеспечении способа очистки пептидов, которые могут быть циклическими или нециклическими и выбраны из группы, состоящей из эптифибатида, эксенатида, атозибана или незиритида и родственных аналогов и производных.
Соответственно, настоящее изобретение касается способа очистки пептида из смеси, содержащей, по меньшей мере, одну родственную примесь, при этом указанный способ включает стадию контактирования смеси пептидов с матрицей колонки для обращенно-фазовой высокоэффективной жидкостной хроматографии и/или матрицей для ионообменной хроматографии с целью получения очищенного пептида; способа очистки пептида с использованием обращенно-фазовой высокоэффективной жидкостной хроматографии из смеси, содержащей, по меньшей мере, одну родственную примесь, при этом указанный способ включает стадии: заполнения колонки для обращенно-фазовой высокоэффективной жидкостной хроматографии полимерной смолой, закрепленной на диоксиде кремния, с последующим уравновешиванием колонки буферным раствором органической кислоты, содержащим 5% полярного растворителя; загрузки пептидной композиции, содержащей, по меньшей мере, одну родственную примесь, на хроматографическую колонку со скоростью потока по большей мере 100-400 см/ч; промывки колонки тем же самым буферным раствором, использовавшимся на стадии (а); и элюирования очищенного продукта с колонки линейным градиентом концентраций (от 8 до 14%) с получением очищенного пептидного продукта; а также касается способа очистки пептида с помощью ионообменной хроматографии из смеси, содержащей, по меньшей мере, одну родственную примесь, при этом указанный способ включает стадии: уравновешивания колонки для катионообменной хроматографии (катионообменной колонки) водным буферным раствором слабой кислоты; загрузки очищенного пептида на колонку для обращенно-фазовой высокоэффективной жидкостной хроматографии; промывки колонки и элюирования пептида буферным раствором, используемым на стадии (а) с получением очищенного пептидного продукта; способа очистки пептида из смеси, содержащей, по меньшей мере, одну родственную примесь, при этом указанный способ включает стадии: заполнения колонки для обращенно-фазовой высокоэффективной жидкостной хроматографии полимерной смолой, закрепленной на диоксиде кремния, с последующим уравновешиванием колонки буферным раствором органической кислоты, содержащим 5% полярного растворителя; загрузки пептидной композиции, содержащей, по меньшей мере, одну родственную примесь, на колонку со скоростью потока по большей мере 100-400 см/ч с последующей промывкой колонки буферным раствором, используемым на стадии (а); элюирования очищенного продукта с колонки линейным градиентом концентраций (от 8 до 14%), загрузки продукта, элюированного с колонки для обращенно-фазовой высокоэффективной жидкостной хроматографии, на катионообменную колонку, уравновешенную водным буферным растворов слабой кислоты, с последующей промывкой колонки и элюированием пептидного продукта элюирующим буфером с получением очищенного пептидного продукта.
Краткое описание чертежей
Фиг 1. Хроматограмма, отображающая чистоту эптифибатида.
Фиг 2. Хроматограмма, отображающая чистоту атозибана.
Фиг 3. Хроматограмма, отображающая чистоту незиритида.
Фиг 4. Хроматограмма, отображающая чистоту эксенатида.
Осуществление изобретения
Настоящее изобретение относится к способу очистки пептида из смеси, содержащей, по меньшей мере, одну родственную примесь, при этом указанный способ включает стадию контактирования смеси пептидов с матрицей для обращенно-фазовой высокоэффективной жидкостной хроматографии и/или матрицей для ионообменной хроматографии, приводящей к получению очищенного пептида.
В другом варианте осуществления настоящего изобретения указанный пептид представляет собой циклический или нециклический пептид, выбранный из группы, состоящей из эптифибатида, эксенатида, атозибана или незиритида и родственных аналогов или производных.
В еще одном варианте осуществления настоящего изобретения указанная пептидная смесь контактирует в любой последовательности с указанной матрицей, состоящей из полимерной смолы, закрепленной на диоксиде кремния.
В еще одном варианте осуществления настоящего изобретения смола может быть выбрана из группы, включающей Сефадекс, Сефадекс LH20, Сефадекс G-25, Сефадекс G-10, Сефарозу, Супердекс, метилакрилатную смолу, карбоксиметилцеллюлозу, сульфопропилцеллюлозу, карбоксиметилсефадекс, сульфопропилсефадекс, сульфопропилсефарозу и карбоксиметилсефарозу, предпочтительно полистирол или полидивинилбензол.
В еще одном варианте осуществления настоящего изобретения размер частиц и размер пор смоляных гранул варьируется в диапазоне 1-50 мкм и 100-500 Å, соответственно.
В еще одном варианте осуществления настоящего изобретения, в указанном способе очистке используется градиентная смесь с концентрацией полярного органического буферного растворителя от 2 до 30% в водной фазе, содержащей буферный раствор органической кислоты.
В еще одном варианте осуществления настоящего изобретения указанным полярным буферным растворителем является ацетонитрил.
В другом варианте осуществления настоящего изобретения указанный буферный раствор органической кислоты выбран из группы, включающей лимонную кислоту, уксусную кислоту, хлорную кислоту и муравьиную кислоту.
В другом варианте осуществления настоящего изобретения молярная концентрация буферного раствора варьируется в диапазоне 10-50 мМ.
В другом варианте осуществления настоящего изобретения очистка проводится при рН, варьирующемся в диапазоне 2-9.
В другом варианте настоящего осуществления изобретения указанный способ дополнительно включает другую необязательную стадию эксклюзионной хроматографии.
В другом варианте осуществления настоящего изобретения посредством используемого в нем способа получается очищенный пептидный продукт, степень чистоты которого составляет от 97 до 100%.
В другом варианте осуществления настоящего изобретения степень чистоты продукта составляет, по меньшей мере, 96%.
Настоящее изобретение относится к способу очистки пептида с использованием обращенно-фазовой высокоэффективной жидкостной хроматографии из смеси, содержащей, по меньшей мере, одну родственную примесь, при этом указанный способ включает стадии:
а) наполнения колонки для обращенно-фазовой высокоэффективной жидкостной хроматографии полимерной смолой, закрепленной на диоксиде кремния, с последующим уравновешиванием колонки буферным раствором органической кислоты, содержащим 5% полярного растворителя;
b) загрузки пептидной композиции, содержащей, по меньшей мере, одну родственную примесь, на хроматографическую колонку со скоростью потока по большей мере 100-400 см/ч;
c) промывки колонки тем же самым буферным раствором, что использовался на стадии (а); и
d) элюирования очищенного продукта с колонки линейным градиентом концентраций (от 8 до 14%) с получением очищенного пептидного продукта.
Настоящее изобретение относится к способу очистки пептида с использованием ионообменной хроматографии из смеси, содержащей, по меньшей мере, одну родственную примесь, при этом указанный способ включает стадии:
a) уравновешивания колонки для катионообменной хроматографии водным буферным раствором слабой кислоты;
b) загрузки колонки для катионобменной хроматографии очищенным пептидом; и
c) промывки колонки и элюирования пептида тем же самым буферным раствором, что использовался на стадии (а), с получением очищенного пептидного продукта.
Настоящее изобретение относится к способу очистки пептида из смеси, содержащей, по меньшей мере, одну родственную примесь, при этом указанный способ включает стадии:
a) наполнения колонки для обращенно-фазовой высокоэффективной жидкостной хроматографии полимерной смолой, закрепленной на диоксиде кремния, с последующим уравновешиванием колонки буферным раствором органической кислоты, содержащим 5% полярного растворителя;
b) загрузки пептидной композиции, содержащей, по меньшей мере, одну родственную примесь, на колонку со скоростью потока по большей мере 100-400 см/ч с последующей промывкой колонки тем же самым буферным раствором, что использовался на стадии (а);
c) элюирования очищенного продукта с колонки линейным градиентом концентраций (от 8 до 14%);
d) загрузки продукта, элюированного с колонки для обращенно-фазовой высокоэффективной жидкостной хроматографии, на катионообменную колонку, уравновешенную водным буферным раствором слабой кислоты; и
e) промывки колонки и элюирования пептидного продукта элюирующим буфером с получением очищенного пептидного продукта.
В другом варианте осуществления настоящего изобретения смола может быть выбрана из группы, состоящей из сефадекса, метилакрилатной смолы, карбоксиметилцеллюлозы, карбоксиметилсефадекса, сульфопропилцеллюлозы и сульфопропилсефадекса, предпочтительно из полистирола или полидивинилбензола.
В еще одном варианте осуществления настоящего изобретения размер частиц и размер пор смоляных гранул варьируется в диапазоне 1-50 мкм и 100-500 Å, соответственно.
В еще одном варианте осуществления настоящего изобретения указанный способ, дополнительно включает другую необязательную стадию эксклюзионной хроматографии.
В еще одном варианте осуществления настоящего изобретения указанный пептид представляет собой циклический или нециклический пептид, выбранный из группы, состоящей из эптифибатида, эксенатида, атозибана или незиритида и родственных аналогов или производных.
В еще одном варианте осуществления настоящего изобретения полярным буферным растворителем является ацетонитрил.
В еще одном варианте осуществлении настоящего изобретения буферный раствор органической кислоты выбран из группы, состоящей из лимонной кислоты, уксусной кислоты и муравьиной кислоты.
В еще одном варианте осуществления настоящего изобретения молярная концентрация используемого буферного раствора варьируется в диапазоне 10-50 мМ.
В еще одном варианте осуществления настоящего изобретения очистка проводится при рН, варьирующемся в диапазоне 2-9.
В еще одном варианте осуществления настоящего изобретения посредством используемых в нем способов получается очищенный пептидный продукт, степень чистоты которого составляет от 97 до 100%.
В еще одном варианте осуществления настоящего изобретения степень чистоты пептида, такого как эптифибатид, эксенатид, атозибан и незиритид, составляет, по меньшей мере, 96%.
Вышеизложенные и другие цели настоящего изобретения достигнуты в специально выделенном способе получения и очистки циклических или нециклических пептидных соединений, образующихся после твердофазного синтеза.
Способ очистки пептидов включает в себя хроматографическую очистку посредством обращенно-фазовой высокоэффективной жидкостной хроматографии с использованием полярного буферного растворителя в концентрации от 2 до 20%, предпочтительно ацетонитрила, в водной фазе, содержащей органический кислотный буфер с рН от 2 до 5.
Цель настоящего изобретения состоит в обеспечении хроматографической среды/системы растворителей, с помощью которой осуществляется обращенно-фазовая высокоэффективная жидкостная хроматография и катионообменная хроматография.
В широком смысле, настоящее изобретение относится к ОФ-ВЭЖХ хроматографическому способу очистки пептида из смеси, содержащей указанный пептид и родственные примеси, который включает стадии: разделения указанного пептида и указанных родственных примесей, содержащихся в указанной смеси, путем элюирования на колонке для ОФ-ВЭЖХ, наполненной полимерной смолой, уравновешенной буферным раствором органической кислоты с 5% содержанием полярного растворителя, загрузки раствора пептида на колонку с желаемой скоростью потока по большей мере 360 см/ч, промывки колонки 50 мМ раствором органической кислоты с пониженным содержанием (5%) полярного органического растворителя, элюирования пептидного продукта указанной комбинацией буферов с линейным градиентом концентраций от 8 до 14%.
Специалисты в данной области техники понимают, что существуют различные переменные параметры, которые можно регулировать в процессе проведения хроматографических способов настоящего изобретения. К таким параметрам относятся условия загрузки и элюирования, такие как ионная сила, состав буфера, рН, температура, добавление малого количества органического растворителя и т.д. Однако такие параметры регулируются обычными способами и специалисты в данной области техники могут легко установить оптимальные условия.
Прежде чем объяснять как минимум один вариант осуществления изобретения более подробно на примере иллюстративных чертежей, экспериментов, результатов и методик, необходимо понимать, что изобретение не ограничивается в отношении подробностей построения и расположения компонентов, изложенных в следующем описании или проиллюстрированных на чертежах, в экспериментах и/или результатах.
Возможны и другие варианты осуществления изобретения или изобретение может быть воплощено на практике или осуществлено различными способами. Подразумевается, что, как таковые, определения, используемые в данном документе, даются в самом широком смысле, а также, что варианты осуществления являются примерными, а не исчерпывающими. Также надо понимать, что формулировки и терминология, используемые в данном документе, даются с целью описания и не следует рассматривать их как ограничительные.
Данное изобретение относится к способам и методикам, используемым при очистке циклических/нециклических пептидов, обладающих биологической активностью, схожей с активностью природных пептидов, и, более конкретно, к способам, в которых используется ОФ-ВЭЖХ для отделения активного пептидного соединения от других веществ, которые не обладают такой активностью и поэтому могут рассматриваться в качестве примесей. Очищенный пептидный продукт приготавливают в рецептурном растворе с помощью катионообменной хроматографии или комбинации катионообменной и эксклюзионной хроматографии.
В одном аспекте настоящее изобретение обеспечивает способ очистки пептида, полипептида или белка, получаемого твердофазным синтезом, при этом указанный способ включает первоначальную стадию очистки пептидного образца посредством обращенно-фазовой хроматографии на колонке из полимерной смолы в хроматографических условиях, достаточных для получения указанного пептида со степенью чистоты около 99%, с последующим концентрированием элюированного пептида посредством катионообменной хроматографии в виде буферного раствора для приготовления готового лекарственного средства.
Другое важное преимущество способа разделения согласно настоящему изобретению состоит в том, что его можно постепенно масштабировать подходящим и воспроизводимым образом. Кроме того, способ настоящего изобретения дает продукты, которые превосходят продукты, получаемые другими известными до настоящего времени способами очистки и образуются с более высоким выходом.
Элюирование пептида в раствор для приготовления лекарственного средства (далее - "рецептурный раствор") имеет преимущества по сравнению с традиционными способами. Традиционные способы включают лиофилизацию очищенного пептида и повторное растворение лиофилизированного порошка до добавления вспомогательных средств.
Элюирование пептида в рецептурный раствор устраняет необходимость в использовании лиофильной сушки и последующем повторном растворении.
Таким образом, при использовании либо по отдельности, либо в сочетании со стандартными методиками экстракции и хроматографирования способы экстрагирования данного изобретения позволяют выделять циклические пептиды заявленного изобретения с высоким выходом и высокой степенью чистоты при меньшем количестве стадий по сравнению с тем, что требуется при использовании традиционных способов.
Особенностью изобретения в другом аспекте является способ очистки белка, который включает в себя стадии загрузки смеси, содержащей соединение, на колонку для ОФ-ВЭЖХ и элюирования образца, содержащего пептид, органическим растворителем, взаимодействия образца с ионообменной смолой в условиях, обеспечивающих связывание соединения со смолой, и промывки органического растворителя, оставшегося на колонке, водным буферным раствором.
Для эффективного осуществления настоящего изобретения с целью эффективной очистки требуется выбор правильной комбинации используемой хроматографической колонки, значения рН и ионной силы буферного раствора.
Каждый параметр хроматографической процедуры играет существенную роль для получения желаемого белкового продукта. Буферная система, используемая для очистки, способна улучшать разделение примесей и интересуемой молекулы за счет гидрофобности соединений. При указанном значении рН буферная система элюирует примеси раньше в процессе градиентного элюирования. Это достигается за счет того, что примеси являются менее гидрофобными по сравнению с интересующей молекулой при указанных условиях.
В настоящем изобретении применяются забуференные растворы, содержащие полярный растворитель, в качестве органических элюентов для ОФ-ВЭЖХ очистки циклических пептидов, их аналогов или производных, рН которых находится в диапазоне от 2 до 8. Настоящее изобретение обеспечивает повышенную эффективность разделения, более высокую чистоту и удобство для промышленного использования по сравнению с существующим уровнем техники в области ОФ-ВЭЖХ очистки циклических пептидов с использованием вышеупомянутых систем растворителей. Неожиданно, разделение целевых циклических пептидных соединений и родственных примесей улучшено за счет новой методологии, используемой в настоящем изобретении, которая дает более чистые циклические пептидные продукты.
Вместе с гидрофобностью соединений на разделение влияет рН буферной системы. Изменение рН, происходящее на различных стадиях хроматографического разделения, влияет на подвижность соединений на колонке.
Гранулы, используемые для разделения, представляют собой сополимер стирола и дивинилбензола с размером частиц 10 мкм и размером пор 300 Å. Загружаемый образец обладает способностью связываться с гранулами, поскольку за счет размера пор регулируется площадь поверхности, доступная для молекул. Гранулы обладают высокой рН-устойчивостью, поскольку они состоят из полимера, и за счет размера частиц можно достигнуть более лучшего разделения.
Состав рецептурного раствора
Эптифибатид
1. Для инъекций: 1 мл содержит 2 мг эптифибатида, 5,25 мг лимонной кислоты, раствор с рН 5,25, регулируемый добавлением уксусной кислоты, и воду (оставшееся количество).
2. Для инфузий: 1 мл содержит 0,75 мг эптифибатида, 5,25 мг лимонной кислоты, раствор с рН 5,25, регулируемый добавлением уксусной кислоты, и воду (оставшееся количество).
Атозибан
1. Для инъекций: 1 мл содержит 7,5 мг атозибана, 50 мг маннита, 4,5 мг лимонной кислоты, раствор с рН 4,5, регулируемый добавлением HCI, и воду (оставшееся количество).
Определения терминов
При описании и изложении формулы настоящего изобретения будет использована следующая терминология в соответствии с определениями, изложенными далее.
Термин "полипептид", "белок", "пептид" относится к полимеру аминокислот и не относится к конкретной длине продукта; таким образом, пептиды, олигопептиды и белки включены в определение полипептида. Этот термин также не относится к или исключает постэкспрессионные модификации полипептида, хотя химические или постэкспрессионные модификации этих полипептидов могут быть включены или исключены в виде особых вариантов осуществления. Поэтому, к примеру, модификации полипептидов, которые включают ковалентное связывание гликозильных, ацетильных, фосфатных, липидных групп и т.п., несомненно охватываются термином полипептид. Кроме того, полипептиды с данными модификациями могут быть определены как индивидуальные частицы, включаемые или исключаемые из настоящего изобретения. В одном варианте осуществления молекула представляет собой полипептид или его родственный аналог или производное. Предпочтительно, полипептидом является циклический пептид. Согласно другому предпочтительному варианту осуществления полипептидом является нециклический пептид. В еще одном варианте осуществления полипептид выбран из группы, включающей эптифибатид, эксенатид, атозибан или незиритид.
Термин "очистка" пептида из композиции, содержащей пептид и одну или несколько загрязняющих примесей, обозначает повышение степени очистки пептида, содержащегося в композиции, за счет снижения содержания, по меньшей мере, одной загрязняющей примеси в пептидной композиции.
"Примесь" - это вещество, которое отличается от желаемого полипептидного продукта или интересуемого белка. К различным видам примесей относятся (но перечень этим не ограничивается) диастереомеры, продукты гидролиза лабильных амидных связей, делеционные последовательности, образующиеся преимущественно при твердофазном пептидном синтезе, пептиды внедрения и побочные продукты, в частности полиморфные формы, образующиеся при снятии защитных групп на последней стадии синтеза.
Термин "хроматография" относится к процессу, посредством которого интересуемое растворенное вещество отделяется от других растворенных веществ, содержащихся в смеси, за счет различий в скоростях, с которыми индивидуальные растворенные вещества смеси двигаются по неподвижной фазе под воздействием подвижной фазы, или за счет различий процессов связывания и элюирования.
Используемый в этом документе термин "высокоэффективная жидкостная хроматография" относится к такому хроматографическому способу, в котором частицы (неподвижной фазы), используемые для заполнения колонки, имеют небольшие одинаковые размеры (от 3 до 50 микрон) с небольшим отклонением от выбранного размера. В таком способе хроматографии обычно применяется относительно высокое (порядка 3,4-24,1 МПа) давление на входе. Термины "ионный обмен" и "ионообменная хроматография" относятся к хроматографическому способу, в котором интересуемое растворенное вещество (например, белок), содержащееся в смеси, взаимодействует с заряженным соединением, связанным (например, за счет ковалентных связей) с ионообменным веществом твердой фазы, так что интересуемое растворенное вещество неспецифически взаимодействует с заряженным соединением в большей или меньшей степени по сравнению с растворенными примесями или загрязняющими веществами, содержащимися в смеси. Загрязняющие растворенные вещества, содержащиеся в смеси, смываются с колонки, содержащей ионообменное вещество, быстрее или медленнее, чем интересуемое растворенное вещество, или связываются или выводятся из смолы лучше в сравнении с интересуемым растворенным веществом. К "ионообменной хроматографии", в частности, относится катионообменная, анионообменная хроматография и хроматография смешанного типа. За стадией катионообменной хроматографии может следовать стадия ОФ-ВЭЖХ или наоборот. Предпочтительно за стадией катионообменной хроматографии следуют другие хроматографические стадии.
"Катионообменная хроматография" - это процесс, при котором положительно заряженные ионы связываются с отрицательно заряженной смолой.
"Загрузочный буфер" - это буфер, который используется для загрузки композиции, содержащей молекулы интересуемого полипептида и одну или несколько примесей, на ионообменную смолу. Загрузочный буфер имеет такую проводимость и/или такой рН, при котором молекулы интересуемого полипептида (и обычно одна или несколько примесей) связываются с ионообменной смолой или при котором интересуемый белок протекает через колонку, в то время как примеси остаются связанными со смолой.
"Полярным буферным растворителем" может являться любой растворитель, который растворяет ионные соединения или ковалентные соединения, способные к диссоциации на ионы, и используется в качестве буфера. В контексте настоящего изобретения, предпочтительным полярным растворителем является ацетонитрил.
Типичный пример способа разделения и очистки пептидов изобретения, получаемых твердофазным синтезом, включает следующие стадии:
i) наполнения колонки для ОФ-ВЭЖХ полимерной смолой, уравновешенной буферным раствором органической кислоты, содержащим 5% полярного растворителя;
ii) загрузки пептидной композиции, содержащей, по меньшей мере, одну родственную примесь, на колонку со скоростью потока менее либо равной около 100-400 см/ч;
iii) промывки колонки тем же самым буферным раствором, что использовался на стадии (i); и
iv) элюирования очищенного продукта с колонки линейным градиентом концентраций от 8 до 14%.
Элюированный очищенный раствор пептида затем загружают на катионообменную колонку, что способствует концентрированию продукта. Концентрирование указанного пептида в сочетании с элюированием его рецептурным буфером посредством катионообменной хроматографии включает следующие стадии:
а) уравновешивания катионообменной колонки водным буферным раствором слабой кислоты;
b) загрузки пептида, очищенного посредством ОФ-ВЭЖХ, на колонку;
с) промывки колонки и элюирования пептидного продукта буферным раствором, используемым на стадии (а).
Буфер А представляет собой 1-5% раствор ацетонитрила, а буфер В - 10-50 мМ буферный раствор органической кислоты; образец загружается со скоростью потока по большей мере 100-400 см/ч. Используемые градиенты концентраций варьируются в зависимости от образца пептида, подвергаемого очистке.
Первая стадия способа, описываемого в этом документе, включает очистку молекул из смесей, содержащих их, путем загрузки смесей на колонку для обращенно-фазовой жидкостной хроматографии. Колонка может представлять собой колонку низкого или высокого давления (ЖХВД), последняя наполняется средой, диаметр частиц которой составляет менее 20 мкм. Колонка, предпочтительно, наполняется средой, диаметр частиц которой составляет 5-40 мкм, более предпочтительно, 10-40 мкм и, наиболее предпочтительно, 10-15 мкм. В контексте настоящего изобретения, размер частиц смолы, наполняемой в колонку, составляет 10 мкм. Таким образом, колонка, предпочтительно, представляет собой колонку для ЖХВД, используемую, в частности, для очистки пептидов. Предпочтительно, размер пор колонки составляет 100-4000 Å, более предпочтительно 100-500 Å. В контексте настоящего изобретения, размер пор смолы, наполняемой в колонку, составляет 300 Å. Длина колонки составляет, предпочтительно, 10-50 см, более предпочтительно, 25-35 см.
рН элюирующего буфера может составлять от 2 до 9, либо от 3 до 8, от 4 до 8 или от 5 до 8, хотя значение рН или диапазон значений рН при элюировании определяется в зависимости от интересуемого пептида и типа осуществляемого хроматографического способа. Соответствующий диапазон значений рН загрузочного, промывного или элюирующего буфера легко определяется стандартными способами, при этом интересуемый белок должен извлекаться в активной форме. К примерам элюирующих буферов, используемых для данной цели, относятся цитратный или ацетатный буфер. Матрицей колонки может являться любой подходящий материал, включая матрицу на основе полимерной смолы, диоксида кремния или метакриловой смолы. Предпочтительно, матрицей является AMBERCHROM HPR10. К катионообменным смолам, которые предполагается использовать для осуществления настоящего изобретения, относятся сульфопропилсефароза, гидрогелевая полимеризованная керамическая гранула, карбоксиметилцеллюлоза, гидрофильные сферические гранулы полимера, карбоксиметилсефадекс, сульфопропилцеллюлоза, сульфопропилсефадекс и т.п. К предпочтительным в настоящее время катионообменным смолам относятся сульфопропилсефароза и гидрогелевые полимеризованные керамические гранулы, при этом сульфопропилсефароза в настоящее время является наиболее предпочтительной катионообменной смолой для использования при осуществлении настоящего изобретения благодаря ее доступности и отличным техническим характеристикам. Эксклюзионными смолами, используемыми в настоящем изобретении, являются Сефадекс LH-20, Сефадекс G-25, Сефадекс G-10. Скорость потока обычно составляет 20-400 см/ч или 4-40 объемов колонки (ОК)/ч в зависимости от того, является ли хроматография кислотной или нейтральной. Предпочтительно, пептид загружают на колонку со скоростью потока по большей мере 360 см/ч.
В случае хроматографического разделения посредством ОФ-ВЭЖХ емкость загрузки пептида на колонку обычно составляет 2-15 г/л в расчете на содержание молекул и примесей. Емкость загрузки ионообменных колонок составляет по большей мере 70 г/л на стадии концентрирования.
Эти и другие неограничительные варианты осуществления настоящего изобретения легко понятны для обычного специалиста в данной области техники при чтении раскрытия и прилагаемой формулы изобретения. Надо понимать, что данное изобретение не ограничивается отдельными описанными способами и процессами, конечно же, возможны вариации по желаемым белковым/пептидным продуктам и способам. Также надо понимать, что используемая в данном документе терминология дается только с целью описания отдельных вариантов осуществления, а не с целью ограничения.
Следует понимать, что использование приведенного способа очистки, описанного в примерах, для достижения разделения с высоким разрешением в сочетании с компонентами, используемыми для разделения, делает его особо эффективным для получения желаемого пептида простым, удобным и недорогим способом.
Технические приемы настоящей заявки дополнительно поясняются с помощью следующих примеров. Однако не следует понимать эти примеры как ограничение объема настоящего изобретения. Следующие примеры представляют собой предпочтительные варианты осуществления настоящего изобретения.
Пример 1
Трифторацетат эптифибатида со степенью чистоты 66%, полученный твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Трифторацетат эптифибатида сначала растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация эптифибатида - менее 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 50 мМ уксусной кислоте. Отфильтрованный раствор эптифибатида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5%) ацетонитрила в 50 мМ уксусной кислоте. Чистый продукт элюируют с колонки линейным градиентом концентраций (8-14%) ацетонитрила (буфера В) в 50 мМ уксусной кислоте (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 23-26 МПа (бар). Эптифибатид, полученный данным способом, имеет степень чистоты 98,7% и получается с выходом 54%. Условия проведения ВЭЖХ анализа эптифибатида представлены в таблице ниже.
Очищенный раствор эптифибатида загружают на катионообменную колонку для проведения концентрирования эптифибатида и элюирования его в рецептурный буфер, который мог бы представлять собой концентрат готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации и заполнять его во флаконы в виде готовой лекарственной формы.
Катионообменную колонку уравновешивают 27 мМ лимонной кислотой с рН 2,7. Очищенный эптифибатид загружают на катионообменную колонку после разбавления водой в соотношении 1:1 до концентрации по меньшей мере 65 г/л основы. Колонку промывают 27 мМ лимонной кислотой с рН 2,7. Элюирование проводят 27 мМ лимонной кислотой с рН 5,25. Полученный элюат с концентрацией по меньшей мере 9 г/л разбавляют до необходимой концентрации и заполняют во флаконы в виде готовой лекарственной формы. Перепад давлений вдоль колонки во время процесса составляет от 0,5 до 0,7 МПа (бар) при скорости потока 180 см/ч. Эптифибатид, полученный данным способом, имеет степень чистоты 98,6% и получается с выходом 100%.
Пример 2
Трифторацетат эптифибатида со степенью чистоты 69,5%, полученный твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Трифторацетат эптифибатида вначале растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация эптифибатида - по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают. рН ацетата натрия доводят до 3,0 добавлением уксусной кислоты.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают 10 мМ раствором ацетата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила. Отфильтрованный раствор эптифибатида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают 10 мМ раствором ацетата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (9-12%) ацетонитрила (буфера В) в 10 мМ растворе ацетата натрия (рН 3,0) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 25-29 МПа (бар). Эптифибатид, полученный данным способом, имеет степень чистоты 98,6% и получается с выходом 43,0%.
Очищенный раствор эптифибатида загружают на катионообменную колонку для проведения концентрирования эптифибатида и элюирования его в рецептурный буфер, который мог бы представлять собой концентрат готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации и заполнять его во флаконы в виде готовой лекарственной формы.
Катионообменную колонку уравновешивают 27 мМ лимонной кислотой с рН 2,7. Очищенный эптифибатид загружают на катионообменную колонку после разбавления водой в соотношении 1:1 до концентрации по меньшей мере 65 г/л основы. Колонку промывают 27 мМ лимонной кислотой с рН 2,7. Элюирование проводят 27 мМ лимонной кислотой с рН 5,25. Полученный элюат с концентрацией по меньшей мере 9 г/л разбавляют до необходимой концентрации и заполняют во флаконы в виде готовой лекарственной формы. Перепад давлений вдоль колонки во время процесса составляет от 0,5 до 0,7 МПа (бар) при скорости потока 180 см/ч. Эптифибатид, полученный данным способом, имеет степень чистоты 98,6% и получается с выходом 100%.
Пример 3
Трифторацетат эптифибатида со степенью чистоты 69,5%, полученный твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Трифторацетат эптифибатида вначале растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация эптифибатида - по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ лимонной кислоте (рН 2,5). Отфильтрованный раствор эптифибатида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ лимонной кислоте (рН 2,5). Чистый продукт элюируют с колонки линейным градиентом концентраций (9-12%) ацетонитрила (буфера В) в 10 мМ лимонной кислоте (рН 2,5) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет от 22 до 28 МПа (бар). Эптифибатид, полученный данным способом, имеет степень чистоты 98,6% и получается с выходом 57%.
Очищенный раствор эптифибатида загружают на катионообменную колонку для проведения концентрирования эптифибатида и элюирования его в рецептурный буфер, который мог бы представлять собой концентрат готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации и заполнять его во флаконы в виде готовой лекарственной формы.
Катионообменную колонку уравновешивают 27 мМ лимонной кислотой с рН 2,7. Очищенный эптифибатид загружают на катионообменную колонку после разбавления водой в соотношении 1:1 до концентрации по меньшей мере 50 г/л основы. Колонку промывают 27 мМ лимонной кислотой с рН 2,7. Элюирование проводят 27 мМ лимонной кислотой с рН 5,25. Полученный элюат с концентрацией по меньшей мере 9 г/л разбавляют до необходимой концентрации и заполняют во флаконы в виде готовой лекарственной формы. Перепад давлений вдоль колонки во время процесса составляет от 0,5 до 0,7 МПа (бар) при скорости потока 180 см/ч. Эптифибатид, полученный данным способом, имеет степень чистоты 98,6% и получается с выходом 100%.
Пример 4
Трифторацетат эптифибатида со степенью чистоты 66%, полученный твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Трифторацетат эптифибатида вначале растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация эптифибатида - по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 0,05% хлорной кислоте (рН 1,70). Отфильтрованный раствор эптифибатида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит <10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5%) ацетонитрила в 0,05% хлорной кислоте (рН 1,70). Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (8-12%) ацетонитрила (буфера В) в 0,05% хлорной кислоте (рН 1,70) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 28-32 МПа (бар). Эптифибатид, полученный данным способом, имеет степень чистоты 96,6% и получается с выходом 40%.
Очищенный раствор эптифибатида загружают на катионообменную колонку для проведения концентрирования эптифибатида и элюирования его в рецептурный буфер, который мог бы представлять собой концентрат готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации и заполнять его во флаконы в виде готовой лекарственной формы.
Катионообменную колонку уравновешивают 27 мМ лимонной кислотой с рН 2,7. Очищенный эптифибатид загружают на катионообменную колонку после разбавления водой в соотношении 1:1 до концентрации по меньшей мере 50 г/л основы. Колонку промывают 27 мМ лимонной кислотой с рН 2,7. Элюирование проводят 27 мМ лимонной кислотой с рН 5,25. Полученный элюат с концентрацией по меньшей мере 9 г/л разбавляют до необходимой концентрации и заполняют во флаконы в виде готовой лекарственной формы. Перепад давлений вдоль колонки во время процесса составляет от 0,5 до 0,7 МПа (бар) при скорости потока 180 см/ч. Эптифибатид, полученный данным способом, имеет степень чистоты 98,6% и получается с выходом 100%.
Пример 5
Трифторацетат эптифибатида со степенью чистоты 69,5%, полученный твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Трифторацетат эптифибатида вначале растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация эптифибатида - по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают 10 мМ раствором формиата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила.
Отфильтрованный раствор эптифибатида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают 10 мМ раствором формиата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (5-17%) ацетонитрила (буфера В) в 10 мМ растворе формиата натрия (рН 3,0) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 30-33 МПа (бар). Эптифибатид, полученный данным способом, имеет степень чистоты 96,8% и получается с выходом 54,0%.
Очищенный раствор эптифибатида загружают на катионообменную колонку для проведения концентрирования эптифибатида и элюирования его в рецептурный буфер, который мог бы представлять собой концентрат готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации и заполнять его во флаконы в виде готовой лекарственной формы.
Катионообменную колонку уравновешивают 27 мМ лимонной кислотой с рН 2,7. Очищенный эптифибатид загружают на катионообменную колонку после разбавления водой в соотношении 1:1 до концентрации по меньшей мере 50 г/л основы. Колонку промывают 27 мМ лимонной кислотой с рН 2,7. Элюирование проводят 27 мМ лимонной кислотой с рН 5,25. Полученный элюат с концентрацией по меньшей мере 9 г/л разбавляют до необходимой концентрации и заполняют во флаконы в виде готовой лекарственной формы. Перепад давлений вдоль колонки во время процесса составляет от 0,5 до 0,7 МПа (бар) при скорости потока 180 см/ч. Эптифибатид, полученный данным способом, имеет степень чистоты 98,6% и получается с выходом 100%.
Пример 6
Трифторацетат эптифибатида со степенью чистоты 69,5%, полученный твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Трифторацетат эптифибатида вначале растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация эптифибатида - по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ борной кислоте (рН 4,0). Отфильтрованный раствор эптифибатида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ борной кислоте (рН 4,0). Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (5-17%) ацетонитрила (буфера В) в 10 мМ борной кислоте (рН 4,0) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 30-33 МПа (бар). Эптифибатид, полученный данным способом, имеет степень чистоты 98,0% и получается с выходом 51,0%.
Очищенный раствор эптифибатида загружают на катионообменную колонку для проведения концентрирования эптифибатида и элюирования его в рецептурный буфер, который мог бы представлять собой концентрат готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации и заполнять его во флаконы в виде готовой лекарственной формы.
Катионообменную колонку уравновешивают 27 мМ лимонной кислотой с рН 2,7. Очищенный эптифибатид загружают на катионообменную колонку после разбавления водой в соотношении 1:1 до концентрации по меньшей мере 50 г/л основы. Колонку промывают 27 мМ лимонной кислотой с рН 2,7. Элюирование проводят 27 мМ лимонной кислотой с рН 5,25. Полученный элюат с концентрацией по меньшей мере 9 г/л разбавляют до необходимой концентрации и заполняют во флаконы в виде готовой лекарственной формы. Перепад давлений вдоль колонки во время процесса составляет от 0,5 до 0,7 МПа (бар) при скорости потока 180 см/ч. Эптифибатид, полученный данным способом, имеет степень чистоты 98,6% и получается с выходом 100%.
Пример 7
Атозибан
Неочищенную соль атозибана со степенью чистоты 73,5%, полученную твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Неочищенную соль атозибана вначале растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация атозибана - по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 50 мМ уксусной кислоте. Отфильтрованный раствор атозибана загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (9%) ацетонитрила в 50 мМ уксусной кислоте. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (9-12%) ацетонитрила (буфера В) в 50 мМ уксусной кислоте (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 26-32 МПа (бар). Атозибан, полученный данным способом, имеет степень чистоты 98,6% и получается с выходом 57%. Условия проведения ЖХВД анализа атозибана представлены в таблице ниже.
Пример 8
Неочищенную соль атозибана со степенью чистоты 73,5%, полученную твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Неочищенную соль атозибана вначале растворяют в 5% растворе ацетонитрила в 50 мМ уксусной кислоте, получая прозрачный раствор с концентрацией продукта по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 50 мМ уксусной кислоте. Отфильтрованный раствор атозибана загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5 и 9%) ацетонитрила в 50 мМ уксусной кислоте. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (9-13%) ацетонитрила (буфера В) в 50 мМ уксусной кислоте (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 26-32 МПа (бар). Атозибан, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 71,0%.
Очищенный раствор атозибана загружают на катионообменную колонку для проведения концентрирования. Катионообменную колонку (КО) уравновешивают 5 мМ уксусной кислотой с рН 3,3. Очищенный атозибан загружают на катионообменную колонку после разбавления водой в соотношении 1:1. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 50 г/л смолы. Колонку промывают 5 мМ уксусной кислотой с рН 3,3. Элюирование проводят 500 мМ раствором ацетата аммония с рН 7,8. Концентрация полученного элюата составляет по меньшей мере 15 г/л. Перепад давлений вдоль колонки во время процесса составляет от 0,2 до 0,3 МПа (бар) при скорости потока 60 см/ч. Атозибан, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 80%.
Элюат, смытый с катионообменной колонки, вводят на эксклюзионную колонку, заменяя буфер на уксусную кислоту, с целью получения концентрата готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации компонентами рецептуры и заполнять его во флаконы в виде готовой лекарственной формы.
Эксклюзионную колонку уравновешивают раствором уксусной кислоты с низкой концентрацией (2-5 мМ). Объем образца (элюат после КО), вводимого на колонку, составляет 30% от объема колонки. Элюат с колонки собирают так, что концентрация атозибана составляет по меньшей мере 15 г/л. Процесс проводят при скорости потока 15 см/ч и перепаде давления вдоль колонки по большей мере 3 МПа (бар). Элюат, смытый с колонки, представляет собой концентрат, который разбавляют до необходимой концентрации, добавляя компоненты рецептуры, и заполняют во флакон.
Пример 9
Неочищенную соль атозибана со степенью чистоты 84,1%, полученную твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Неочищенную соль атозибана вначале растворяют в 5% растворе ацетонитрила в 50 мМ уксусной кислоте, получая прозрачный раствор с концентрацией продукта по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают 10 мМ раствором ацетата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила. Отфильтрованный раствор атозибана загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают 10 мМ раствором ацетата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (13-20%) ацетонитрила (буфера В) в 10 мМ растворе ацетата натрия (рН 3,0) (буфер А) в количестве 20 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 19-22 МПа (бар). Атозибан, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 94,0%.
Очищенный раствор атозибана загружают на катионообменную колонку для проведения концентрирования. Катионообменную колонку (КО) уравновешивают 5 мМ уксусной кислотой с рН 3,3. Очищенный атозибан загружают на катионообменную колонку после разбавления водой в соотношении 1:1. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 50 г/л смолы. Колонку промывают 5 мМ уксусной кислотой с рН 3,3. Элюирование проводят 500 мМ раствором ацетата аммония с рН 7,8. Концентрация полученного элюата составляет по меньшей мере 15 г/л. Перепад давлений вдоль колонки во время процесса составляет от 0,2 до 0,3 МПа (бар) при скорости потока 60 см/ч. Атозибан, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 80%.
Элюат, смытый с катионообменной колонки, вводят на эксклюзионную колонку, заменяя буфер на уксусную кислоту, с целью получения концентрата готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации компонентами рецептуры и заполнять его во флаконы в виде готовой лекарственной формы.
Эксклюзионную колонку уравновешивают раствором уксусной кислоты с низкой концентрацией (2-5 мМ). Объем образца (элюат после КО), вводимого на колонку, составляет 30% от объема колонки. Элюат с колонки собирают так, что концентрация атозибана составляет по меньшей мере 15 г/л. Процесс проводят при скорости потока 15 см/ч и перепаде давления вдоль колонки по большей мере 3 МПа (бар). Элюат, смытый с колонки, представляет собой концентрат, который разбавляют до необходимой концентрации, добавляя компоненты рецептуры, и заполняют во флакон.
Пример 10
Неочищенную соль атозибана со степенью чистоты 81,5%, полученную твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Неочищенную соль атозибана вначале растворяют в 5% растворе ацетонитрила в 50 мМ уксусной кислоте, получая прозрачный раствор с концентрацией продукта по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ лимонной кислоте (рН 3,0). Отфильтрованный раствор атозибана загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ лимонной кислоте (рН 3,0). Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (12-20%) ацетонитрила (буфера В) в 10 мМ лимонной кислоте (рН 3,0) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 22-23 МПа (бар). Атозибан, полученный данным способом, имеет степень чистоты 99,8% и получается с выходом 73,0%.
Очищенный раствор атозибана загружают на катионообменную колонку для проведения концентрирования. Катионообменную колонку (КО) уравновешивают 5 мМ уксусной кислотой с рН 3,3. Очищенный атозибан загружают на катионообменную колонку после разбавления водой в соотношении 1:1. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 50 г/л смолы. Колонку промывают 5 мМ уксусной кислотой с рН 3,3. Элюирование проводят 500 мМ раствором ацетата аммония с рН 7,8. Концентрация полученного элюата составляет по меньшей мере 15 г/л. Перепад давлений вдоль колонки во время процесса составляет от 0,2 до 0,3 МПа (бар) при скорости потока 60 см/ч. Атозибан, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 80%.
Элюат, смытый с катионообменной колонки, вводят на эксклюзионную колонку, заменяя буфер на уксусную кислоту, с целью получения концентрата готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации компонентами рецептуры и заполнять его во флаконы в виде готовой лекарственной формы.
Эксклюзионную колонку уравновешивают раствором уксусной кислоты с низкой концентрацией (2-5 мМ). Объем образца (элюат после КО), вводимого на колонку, составляет 30% от объема колонки. Элюат с колонки собирают так, что концентрация атозибана составляет по меньшей мере 15 г/л. Процесс проводят при скорости потока 15 см/ч и перепаде давления вдоль колонки по большей мере 3 МПа (бар). Элюат, смытый с колонки, представляет собой концентрат, который разбавляют до необходимой концентрации, добавляя компоненты рецептуры, и заполняют во флакон.
Пример 11
Неочищенную соль атозибана со степенью чистоты 80,2%, полученную твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Неочищенную соль атозибана вначале растворяют в 5% растворе ацетонитрила в 50 мМ уксусной кислоте, получая прозрачный раствор с концентрацией продукта по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 0,05% хлорной кислоте (рН 1,70). Отфильтрованный раствор атозибана загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5%) ацетонитрила в 0,05% хлорной кислоте (рН 1,70). Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (12-20%) ацетонитрила (буфера В) в 0,05% хлорной кислоте (рН 1,70) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 23-24 МПа (бар). Атозибан, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 94,0%.
Очищенный раствор атозибана загружают на катионообменную колонку для проведения концентрирования. Катионообменную колонку (КО) уравновешивают 5 мМ уксусной кислотой с рН 3,3. Очищенный атозибан загружают на катионообменную колонку после разбавления водой в соотношении 1:1. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 50 г/л смолы. Колонку промывают 5 мМ уксусной кислотой с рН 3,3. Элюирование проводят 500 мМ раствором ацетата аммония с рН 7,8. Концентрация полученного элюата составляет по меньшей мере 15 г/л. Перепад давлений вдоль колонки во время процесса составляет от 0,2 до 0,3 МПа (бар) при скорости потока 60 см/ч. Атозибан, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 80%.
Элюат, смытый с катионообменной колонки, вводят на эксклюзионную колонку, заменяя буфер на уксусную кислоту, с целью получения концентрата готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации компонентами рецептуры и заполнять его во флаконы в виде готовой лекарственной формы.
Эксклюзионную колонку уравновешивают раствором уксусной кислоты с низкой концентрацией (2-5 мМ). Объем образца (элюат после КО), вводимого на колонку, составляет 30% от объема колонки. Элюат с колонки собирают так, что концентрация атозибана составляет по меньшей мере 15 г/л. Процесс проводят при скорости потока 15 см/ч и перепаде давления вдоль колонки по большей мере 3 МПа (бар). Элюат, смытый с колонки, представляет собой концентрат, который разбавляют до необходимой концентрации, добавляя компоненты рецептуры, и заполняют во флакон.
Пример 12
Неочищенную соль атозибана со степенью чистоты 73,5%, полученную твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Неочищенную соль атозибана вначале растворяют в 5% растворе ацетонитрила в 50 мМ уксусной кислоте, получая прозрачный раствор с концентрацией продукта по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 0,05% хлорной кислоте (рН 3,0). Отфильтрованный раствор атозибана загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5%) ацетонитрила в 0,05% хлорной кислоте (рН 3,0). Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (12-20%) ацетонитрила (буфера В) в 0,05% хлорной кислоте (рН 3,0) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 26-32 МПа (бар). Атозибан, полученный данным способом, имеет степень чистоты 99,4% и получается с выходом 71,0%.
Очищенный раствор атозибана загружают на катионообменную колонку для проведения концентрирования. Катионообменную колонку (КО) уравновешивают 5 мМ уксусной кислотой с рН 3,3. Очищенный атозибан загружают на катионообменную колонку после разбавления водой в соотношении 1:1. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 50 г/л смолы. Колонку промывают 5 мМ уксусной кислотой с рН 3,3. Элюирование проводят 500 мМ раствором ацетата аммония с рН 7,8. Концентрация полученного элюата составляет по меньшей мере 15 г/л. Перепад давлений вдоль колонки во время процесса составляет от 0,2 до 0,3 МПа (бар) при скорости потока 60 см/ч. Атозибан, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 80%.
Элюат, смытый с катионообменной колонки, вводят на эксклюзионную колонку, заменяя буфер на уксусную кислоту, с целью получения концентрата готовой лекарственной формы. Элюат можно разбавлять до необходимой концентрации компонентами рецептуры и заполнять его во флаконы в виде готовой лекарственной формы.
Эксклюзионную колонку уравновешивают раствором уксусной кислоты с низкой концентрацией (2-5 мМ). Объем образца (элюат после КО), вводимого на колонку, составляет 30% от объема колонки. Элюат с колонки собирают так, что концентрация атозибана составляет по меньшей мере 15 г/л. Процесс проводят при скорости потока 15 см/ч и перепаде давления вдоль колонки по большей мере 3 МПа (бар). Элюат, смытый с колонки, представляет собой концентрат, который разбавляют до необходимой концентрации, добавляя компоненты рецептуры, и заполняют во флакон.
Пример 13
Незиритид
Неочищенную соль незиритида со степенью чистоты 59,0%, полученную твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Неочищенный порошок незиритида вначале растворяют в 10% растворе ацетонитрила в 50 мМ уксусной кислоте, получая прозрачный раствор с концентрацией продукта по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают 10 мМ раствором ацетата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила. Отфильтрованный раствор незиритида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают 10 мМ раствором ацетата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (5-15%) ацетонитрила (буфера В) в 10 мМ растворе ацетата натрия (рН 3,0) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 28-33 МПа (бар). Незиритид, полученный данным способом, имеет степень чистоты 92,5% и получается с выходом 50,0%. Условия проведения ВЭЖХ анализа незиритида представлены в таблице ниже.
Пример 14
Неочищенную соль незиритида со степенью чистоты 59,0%, полученную твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Неочищенный порошок незиритида вначале растворяют в 10% растворе ацетонитрила в 50 мМ уксусной кислоте, получая прозрачный раствор с концентрацией продукта по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ лимонной кислоте (рН 3,0). Отфильтрованный раствор незиритида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ лимонной кислоте (рН 3,0). Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (5-15%) ацетонитрила (буфера В) в 10 мМ лимонной кислоте (рН 3,0) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 28-33 МПа (бар). Незиритид, полученный данным способом, имеет степень чистоты 97,2% и получается с выходом 33,0%.
Пример 15
Неочищенную соль незиритида со степенью чистоты 59,0%, полученную твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Неочищенный порошок незиритида вначале растворяют в 10% растворе ацетонитрила в 50 мМ уксусной кислоте, получая прозрачный раствор с концентрацией продукта по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают 10 мМ раствором формиата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила. Отфильтрованный раствор незиритида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают 10 мМ раствором формиата натрия (рН 3,0) с пониженным содержанием (5%) ацетонитрила. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (12-20%) ацетонитрила (буфера В) в 10 мМ растворе формиата натрия (рН 3,0) (буфер А) в количестве 25 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 24-28 МПа (бар). Незиритид, полученный данным способом, имеет степень чистоты 98,7% и получается с выходом 18,0%.
Пример 16
Эксенатид
Трифторацетат эксенатида со степенью чистоты 56%, полученный твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Трифторацетат эксенатида вначале растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация эксенатида - по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ лимонной кислоте (рН 5,0). Отфильтрованный раствор эксенатида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 5 г/л смолы. После загрузки колонку промывают раствором с пониженным содержанием (5%) ацетонитрила в 10 мМ лимонной кислоте (рН 5,0). Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (30-33%) ацетонитрила (буфера В) в 10 мМ лимонной кислоте (рН 5,0) (буфер А) в количестве 20 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 15-25 МПа (бар). Эксенатид, полученный данным способом, имеет степень чистоты 92,2% и получается с выходом 49%. Условия проведения ВЭЖХ анализа эксенатида представлены в таблице ниже.
Элюат, полученный по вышеуказанному способу, разбавляют три раза водой, что способствует его связыванию на колонке. Элюат эксенатида, полученный по методике, описанной в примере 1, со степенью чистоты 92,0% разбавляют три раза водой, что способствует его связыванию на колонке. Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å) от компании Rohm and Haas, уравновешивают раствором с 10% содержанием ацетонитрила в 50 мМ уксусной кислоте. Полученный образец эксенатида со степенью чистоты 92% загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 5 г/л смолы. После загрузки колонку промывают 20% раствором ацетонитрила в 50 мМ уксусной кислоте. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (25-28%) ацетонитрила (буфера В) в 50 мМ уксусной кислоте (буфер А) в количестве 20 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 20-25 МПа (бар). Эксенатид, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 50%.
Пример 17
Трифторацетат эксенатида со степенью чистоты 46,2%, полученный твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Трифторацетат эксенатида вначале растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация эксенатида - по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают 10 мМ раствором ацетата натрия (рН 4,0) с пониженным содержанием (10%) ацетонитрила. Отфильтрованный раствор эксенатида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают 10 мМ раствором ацетата натрия (рН 4,0) с пониженным содержанием (10%) ацетонитрила. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (28-33%) ацетонитрила (буфера В) в 10 мМ растворе ацетата натрия (рН 4,0) (буфер А) в количестве 20 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 18-20 МПа (бар). Эксенатид, полученный данным способом, имеет степень чистоты 94,2% и получается с выходом 57%.
Элюат эксенатида со степенью чистоты 94,2%, полученный по методике вышеуказанного примера, разбавляют три раза водой, что способствует его связыванию на колонке.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å) от компании Rohm and Haas, уравновешивают раствором с 10% содержанием ацетонитрила в 50 мМ уксусной кислоте. Полученный образец эксенатида со степенью чистоты 94,2% загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают 20% раствором ацетонитрила в 50 мМ уксусной кислоте. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (25-28%) ацетонитрила (буфера В) в 50 мМ уксусной кислоте (буфер А) в количестве 20 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 20-25 МПа (бар). Эксенатид, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 50%.
Пример 18
Трифторацетат эксенатида со степенью чистоты 46,2%, полученный твердофазным синтезом, подвергают очистке на колонке, наполненной полимерной смолой. Трифторацетат эксенатида вначале растворяют в смеси ацетонитрила и 50 мМ уксусной кислоты (1:1), получая прозрачный раствор. Полученный раствор дополнительно разбавляют 50 мМ уксусной кислотой, так что концентрация ацетонитрила составляет 5%, а концентрация эксенатида - по большей мере 2 г/л. Перед загрузкой на колонку раствор отфильтровывают.
Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å), уравновешивают 10 мМ раствором формиата натрия (рН 4,0) с пониженным содержанием (10%) ацетонитрила. Отфильтрованный раствор эксенатида загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают 10 мМ раствором формиата натрия (рН 4,0) с пониженным содержанием (10%) ацетонитрила. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (27-35%) ацетонитрила (буфера В) в 10 мМ растворе формиата натрия (рН 4,0) (буфер А) в количестве 20 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 24-29 МПа (бар). Эксенатид, полученный данным способом, имеет степень чистоты 94,9% и получается с выходом 52%.
Элюат эксенатида со степенью чистоты 94,9%, полученный по методике вышеуказанного примера, разбавляют три раза водой, что способствует его связыванию на колонке. Колонку, наполненную смолой Amberchrom HPR10 (размер частиц 10 мкм и размер пор 300 Å) от компании Rohm and Haas, уравновешивают раствором с 10% содержанием ацетонитрила в 50 мМ уксусной кислоте. Полученный образец эксенатида со степенью чистоты 94,2% загружают на колонку со скоростью потока по большей мере 360 см/ч. Загрузку пептида на колонку проводят до тех пор, пока его концентрация не составит по большей мере 10 г/л смолы. После загрузки колонку промывают 20% раствором ацетонитрила в 50 мМ уксусной кислоте. Чистый продукт элюируют с колонки осуществлением линейного градиента концентраций (25-28%) ацетонитрила (буфера В) в 50 мМ уксусной кислоте (буфер А) в количестве 20 ОК. Перепад давлений вдоль колонки в процессе очистки составляет 20-25 МПа (бар). Эксенатид, полученный данным способом, имеет степень чистоты 99,6% и получается с выходом 50%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХРОМАТОГРАФИЧЕСКИЕ СПОСОБЫ И СОЕДИНЕНИЯ, ОЧИЩЕННЫЕ ЭТИМИ СПОСОБАМИ | 2010 |
|
RU2508294C2 |
| ПРЕПАРАТИВНЫЙ ХРОМАТОГРАФИЧЕСКИЙ СПОСОБ НА ОСНОВЕ НЕЛИНЕЙНОГО ГРАДИЕНТА И ПРОДУКТЫ, ОЧИЩЕННЫЕ ЭТИМ СПОСОБОМ | 2010 |
|
RU2489441C1 |
| СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО ИНСУЛИНА ГЛАРГИНА | 2008 |
|
RU2495131C2 |
| СПОСОБ ОЧИСТКИ ГОЗЕРЕЛИНА | 2015 |
|
RU2578414C1 |
| ХРОМАТОГРАФИЧЕСКИЕ СПОСОБЫ | 2008 |
|
RU2464066C2 |
| СПОСОБ ОЧИСТКИ ФАКТОРА СВЕРТЫВАНИЯ КРОВИ VIII | 2009 |
|
RU2698392C2 |
| УЛУЧШЕНИЕ АФФИННОЙ ХРОМАТОГРАФИИ ИММУНОГЛОБУЛИНОВ ПУТЕМ ПРИМЕНЕНИЯ ФЛОКУЛЯЦИИ ДО ЗАХВАТА | 2020 |
|
RU2794431C1 |
| Способ очистки фолликулостимулирующего гормона | 2020 |
|
RU2803653C1 |
| СПОСОБ ОЧИСТКИ ФАКТОРА СВЕРТЫВАНИЯ КРОВИ VIII | 2009 |
|
RU2567811C2 |
| СПОСОБ ОЧИСТКИ РЕКОМБИНАНТНОГО ИЛ-36РА (ВАРИАНТЫ) | 2018 |
|
RU2700751C1 |
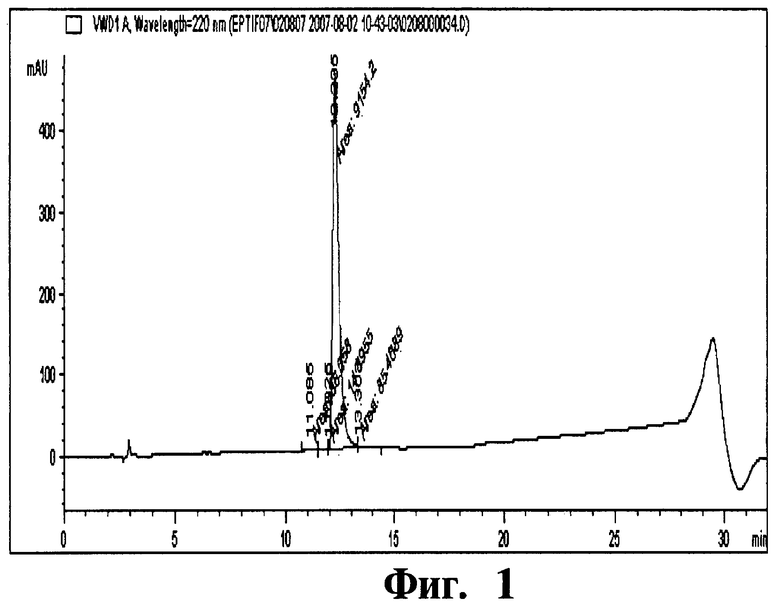
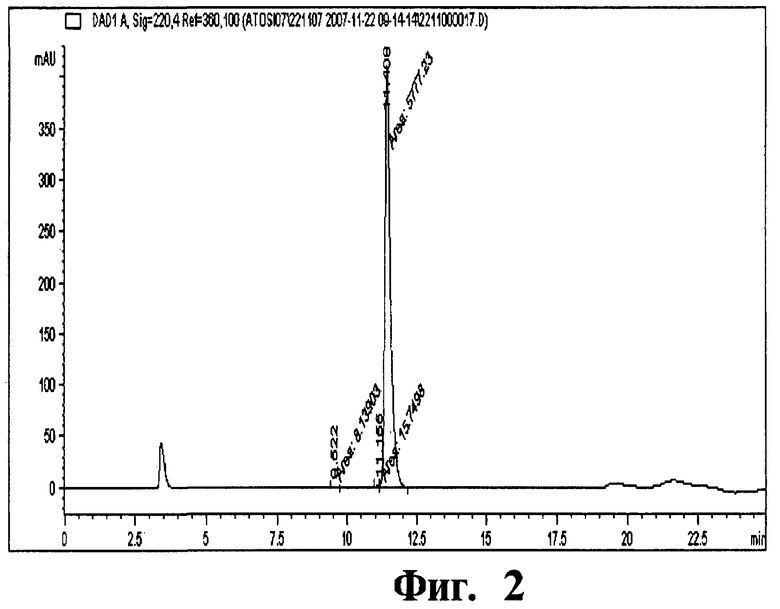
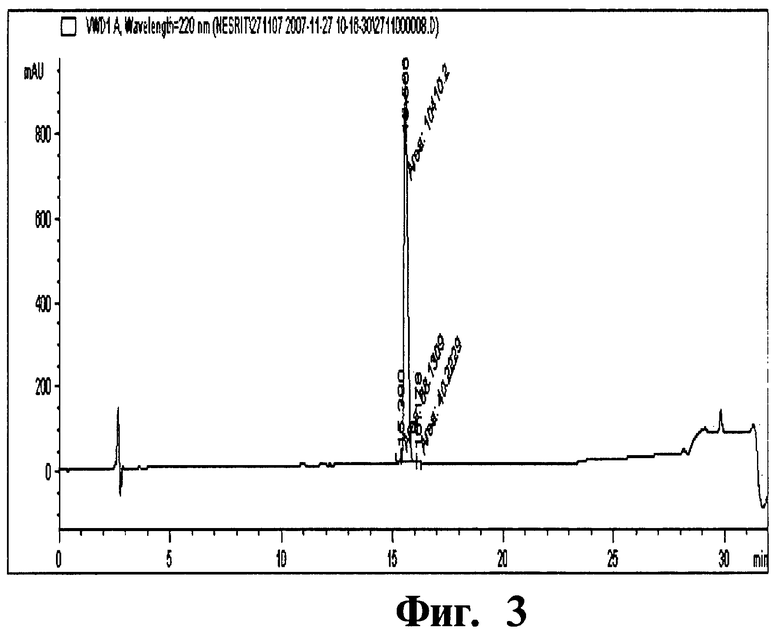
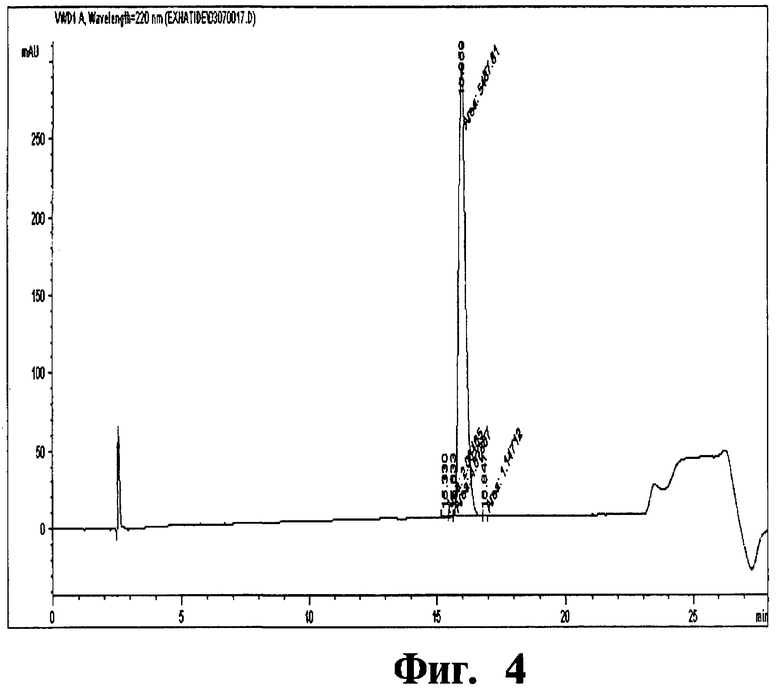
Изобретение относится к способу очистки циклического или нециклического пептида, выбранного из группы пептидов, включающей эптифибатид, эксенатид, атозибан и незиритид или их комбинацию, из смеси, содержащей по меньшей мере одну примесь, включающему контактирование указанной смеси с матрицей для ОФ-ВЭЖХ и матрицей для ионообменной хроматографии и получение очищенного пептидного продукта с чистотой по меньшей мере 96% и, предпочтительно, составляет, по меньшей мере, около 99%. 10 з.п. ф-лы, 4 табл., 4 ил., 18 пр.
1. Способ очистки циклического или нециклического пептида, выбранного из группы пептидов, включающей эптифибатид, эксенатид, атозибан и незиритид или их комбинацию, из смеси, содержащей по меньшей мере одну примесь, включающий контактирование указанной смеси с матрицей для ОФ-ВЭЖХ и матрицей для ионообменной хроматографии и получение очищенного пептидного продукта с чистотой по меньшей мере 96%, при котором наполняют колонки для ОФ-ВЭЖХ полимерной смолой, которую уравновешивают буферным раствором органической кислоты, содержащим 5% ацетонитрила, загружают пептидную смесь, содержащую, по меньшей мере, одну примесь, на колонку со скоростью потока по большей мере приблизительно 100÷400 см/ч, промывают колонки тем же самым буферным раствором, что и при использовании на стадии наполнения колонки для ОФ-ВЭЖХ, элюируют очищенный пептид с колонки линейным градиентом концентраций от 8 до 14%, уравновешивают колонку для катионообменной хроматографии водным буферным раствором слабой кислоты, загружают пептид, очищенный посредством ОФ-ВЭЖХ, на колонку для катионообменной хроматографии, промывают колонки и элюируют пептидный продукт буферным раствором, используемым на уравновешивания колонки для катионообменной хроматографии, и получают очищенный пептид с чистотой по меньшей мере 96%.
2. Способ по п.1, в котором указанная полимерная смола выбрана из группы, включающей Сефадекс, метилакрилатную смолу, карбоксиметилцеллюлозу, карбоксиметилсефадекс, сульфопропилцеллюлозу, сульфопропилсефадекс и полимерную смолу, закрепленную на диоксиде кремния.
3. Способ по п.1, в котором указанной полимерной смолой является полистирол или полидивинилбензол.
4. Способ по п.1, в котором размер частиц гранул указанной полимерной смолы составляет 1÷50 мкм.
5. Способ по п.1, в котором размер пор гранул указанной полимерной смолы составляет 100÷500 Å.
6. Способ по п.1, в котором дополнительно проводят эксклюзионную хроматографию.
7. Способ по п.1, в котором указанная органическая кислота выбрана из группы, включающей лимонную кислоту, уксусную кислоту, хлорную кислоту и муравьиную кислоту.
8. Способ по п.1, в котором молярная концентрация указанного буферного раствора органической кислоты составляет 10÷50 мМ.
9. Способ по п.1, в котором указанная слабая кислота выбрана из группы, включающей лимонную кислоту и уксусную кислоту.
10. Способ по п.1, в котором молярная концентрация указанного буферного раствора слабой кислоты составляет 5 мМ.
11. Способ по п.1, в котором очистку проводят при рН от 2 до 9.
| Способ отделения протеинов | 1976 |
|
SU688124A3 |
| WO 2005019262 А1, 03.03.2005 | |||
| WO 2007071767 А1, 28.06.2007 | |||
| US 6492327 В2, 10.12.2003 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| WO 2005100388 A1, 27.10.2005 | |||
| JP 6256399 A, 13.09.1994 | |||
| TERANISHI H et al: "Isolation and characterization of four VIP-related peptides from red-bellied newt, Cynops pyrrhogaster", REGULATORY PEPTIDES, 2004, vol | |||
| Устройство для разметки подлежащих сортированию и резанию лесных материалов | 1922 |
|
SU123A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

