ОБЛАСТЬ ТЕХНИКИ
Настоящее раскрытие иллюстрирует полезность использования агентов для образования ионных пар при препаративной очистке. В частности, раскрытие относится к использованию агентов для образования ионных пар в обращенно-фазовой препаративной хроматографии, позволяя получать высокую степень чистоты целевого конечного продукта. Раскрытие показывает, что агенты для образования ионных пар оказывают существенное влияние на требуемую чистоту полипептидов.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Для получения желаемого конечного результата в плане чистоты и выхода используют ряд различных хроматографических методик. Обращенно-фазовая хроматография является одним из наиболее эффективных используемых способов очистки. Обращенно-фазовая жидкостная хроматография ("ОФ-ЖХ") и обращенно-фазовая высокоэффективная жидкостная хроматография ("ОФ-ВЭЖХ") широко используются для очистки молекул, таких как пептиды и белки, получаемых синтетическими или рекомбинантными методами. Методы ОФ-ЖХ и ОФ-ВЭЖХ позволяют эффективно разделять близкородственные примеси и применяются для очистки многих разноплановых молекул (Lee et al. "Preparative HPLC (Препаративная ВЭЖХ)", 8th Biotechnology Symposium, Pt. 1, 593-610 (1988)). Кроме того, ОФ-ЖХ и ОФ-ВЭЖХ с успехом используются для очистки молекул, в частности, белков, в промышленном масштабе (Olsen et al., 1994, J. Chromatog. A, 675, 101).
Использование агентов для образования ионных пар, таких как триэтиламин (ТЭА), трифторуксусная кислота (ТФК), гексансульфоновая кислота (ГСК) и так далее, в аналитических методах исследования и их влияние на разрешение белковых пиков является хорошо известным фактом, Shayne Сох Gad; Handbook of Pharmaceutical Biotechnology (Справочник по фармацевтической биотехнологии). Агент для образования ионных пар адсорбируется на неподвижной фазе за счет ее углеводородной цепи, образующей двойной электрический слой, сообщающий поверхности неподвижной фазы электрический потенциал. Благодаря этому молекулы белков/пептидов участвуют как в ионном обмене с ионами электролита подвижной фазы, так и в адсорбционном эффекте с обращенной фазой неподвижной фазы (Frederick F. Cantwell et al., 1984, Journal of Pharmaceutical and Biomedical Analysis, Volume 2, Pages 153-164).
Текущее раскрытие относится к использованию агентов для образования ионных пар при препаративной очистке белков и пептидов.
В частности, данное раскрытие относится к использованию ГСК и ТЭА в ОФ-ВЭЖХ препаративной линейной хроматографии.
Агенты для образования ионных пар, используемые в ОФ-ВЭЖХ, по существу содержат длинную углеводородную цепь с ионизируемой группой. Такие молекулы в подвижной фазе образуют ионную пару с поверхностными зарядами белка/пептида. За счет этого образования ионных пар увеличивается гидрофобность пептида/белка, приписываемая углеводородной цепи агента для образования ионных пар. Такое взаимодействие зависит от поверхностных зарядов. Разные белки смеси имеют разные поверхностные заряды и, следовательно, по-разному связываются с неподвижной фазой, что улучшает разрешение между пиками белков. Адсорбированные агенты для образования ионных пар сообщают поверхности неподвижной фазы заряд, характерный для их функциональной группы. Это отталкивает одноименные заряды, тем самым изменяя селективность различных белков смеси.
В традиционной хроматографии наблюдается тот факт, что при увеличении нагрузки на колонку разрешение между примесями и целевым белком ухудшается. Полосы белка при элюировании имеют тенденцию к слиянию, что сказывается на качестве хроматографии в плане чистоты препарата и выхода. Текущее раскрытие устраняет эту проблему, являющуюся ключом к возможности производства белков. Более высокая нагрузка на колонку, большая, чем в случае белка, подаваемого в колонку для аналитического определения, является значимой для усовершенствования процесса и обуславливает стоимость и выход данного процесса. Текущее раскрытие показывает, что агенты для образования ионных пар оказывают существенное влияние на чистоту белков даже при увеличении нагрузки белка. Текущее раскрытие демонстрирует использование образования ионных пар для улучшения выхода на хроматографической стадии.
ЗАДАЧИ ИЗОБРЕТЕНИЯ
Основной задачей настоящего изобретения является разработка хроматографического способа очистки (выделения) полипептидов из смеси.
Другой основной задачей настоящего изобретения является получение аналога инсулина, такого как аспарт, гларгин и лизпро.
Еще одной основной задачей настоящего изобретения является получение очищенных полипептидов, таких как атозибан и эптифибатид.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Таким образом, настоящее изобретение относится к хроматографическому способу очистки полипептида из смеси, содержащей по меньшей мере одну родственную примесь, при этом указанный способ включает в себя стадию использования ОФ-ВЭЖХ с агентом для образования ионных пар, имеющим концентрацию в диапазоне приблизительно от 0,01% до 2%, в сочетании с органическим модификатором, имеющим концентрацию в диапазоне приблизительно от 5% до 85%; к хроматографическому способу очистки полипептида из смеси, содержащей по меньшей мере одну родственную примесь, при этом указанный способ включает в себя стадии: а) заполнения ОФ-ВЭЖХ колонки силикагелевой смолой (С4-С18), уравновешенной с помощью приблизительно от 5% до 85% органического модификатора, b) нанесения на колонку полипептидной смеси со скоростью приблизительно от 180 см/ч до 360 см/ч, с) промывки колонки агентом для образования ионных пар, имеющим концентрацию в диапазоне приблизительно от 0,05% до 1%, в сочетании с органическим модификатором, имеющим концентрацию в диапазоне приблизительно от 5% до 85%, и d) обеспечения линейного градиента приблизительно от 10% до 70% для элюирования очищенного полипептида с колонки; к аналогу инсулина, полученному с помощью способа, как указано выше, с чистотой в диапазоне приблизительно от 90% до 100%; очищенному аспарту, очищенному до степени чистоты по меньшей мере 98%; очищенному гларгину, очищенному до степени чистоты по меньшей мере 99%; очищенному лизпро, очищенному до степени чистоты по меньшей мере 97%; полипептиду, полученному с помощью способа, как указано выше, с чистотой в диапазоне приблизительно от 90% до 100%; очищенному атозибану, очищенному до степени чистоты по меньшей мере 99,14%; и очищенному эптифибатиду, очищенному до степени чистоты по меньшей мере 94%.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к хроматографическому способу очистки полипептида из смеси, содержащей по меньшей мере одну родственную примесь, при этом указанный способ включает в себя стадию использования ОФ-ВЭЖХ с агентом для образования ионных пар, имеющим концентрацию в диапазоне приблизительно от 0,01% до 2%, в сочетании с органическим модификатором, имеющим концентрацию в диапазоне приблизительно от 5% до 85%.
Согласно одному из вариантов осуществления настоящего изобретения наиболее предпочтительно, чтобы агент для образования ионных пар и органический модификатор имели концентрацию в диапазоне приблизительно от 0,05% до 1% и приблизительно от 8% до 65% соответственно.
Согласно другому варианту осуществления настоящего изобретения агент для образования ионных пар выбирают из группы, включающей в себя гексансульфоновую кислоту, трифторуксусную кислоту, пентафторпропановую кислоту, триэтиламин и гептафтормасляную кислоту.
Согласно еще одному варианту осуществления настоящего изобретения органический модификатор выбирают из группы, включающей в себя ацетонитрил, этанол, метанол и изопропанол.
Согласно еще одному варианту осуществления настоящего изобретения полипептид выбирают из группы, включающей в себя аналог инсулина, эптифибатид и атозибан.
Согласно еще одному варианту осуществления настоящего изобретения инсулиновыми аналогами являются аспарт, лизпро и гларгин.
Настоящее изобретение также относится к хроматографическому способу очистки полипептида из смеси, содержащей по меньшей мере одну родственную примесь, при этом указанный способ включает в себя стадии: а) заполнения ОФ-ВЭЖХ колонки силикагелевой смолой (C4-C18), уравновешенной с помощью приблизительно от 5% до 85% органического модификатора, b) нанесения на колонку полипептидной смеси со скоростью приблизительно от 180 см/ч до 360 см/ч, с) промывки колонки агентом для образования ионных пар, имеющим концентрацию в диапазоне приблизительно от 0,05% до 1%, в сочетании с органическим модификатором, имеющим концентрацию в диапазоне приблизительно от 5% до 85%, и d) обеспечения линейного градиента приблизительно от 10% до 70% для элюирования очищенного полипептида с колонки.
Согласно одному из вариантов осуществления настоящего изобретения силикагелевая смола предпочтительно представляет собой С8.
Согласно другому варианту осуществления настоящего изобретения хроматографическую очистку проводят при величине рН в диапазоне приблизительно от 2,5 до 8,5.
Согласно еще одному варианту осуществления настоящего изобретения смола имеет размер частиц в диапазоне приблизительно от 5 мкм до 40 мкм, предпочтительно приблизительно от 7 мкм до 20 мкм и наиболее предпочтительно приблизительно от 10 мкм до 13 мкм.
Согласно еще одному варианту осуществления настоящего изобретения зерно смолы имеет размер пор в диапазоне приблизительно от 50Å до 2000Å, предпочтительно приблизительно от 100Å до 500Å и, наиболее предпочтительно 120Å.
Согласно еще одному варианту осуществления настоящего изобретения чистота полипептида лежит в диапазоне приблизительно от 90% до 100%, предпочтительно составляет по меньшей мере 99%.
Настоящее изобретение также относится к аналогу инсулина, полученному с помощью способа, как указано выше, с чистотой в диапазоне приблизительно от 90% до 100%.
Настоящее изобретение также относится к очищенному аспарту, очищенному до степени чистоты по меньшей мере 98%.
Настоящее изобретение также относится к очищенному гларгину, очищенному до степени чистоты по меньшей мере 99%.
Настоящее изобретение также относится к очищенному лизпро, очищенному до степени чистоты по меньшей мере 97%.
Настоящее изобретение также относится к очищенному полипептиду, полученному с помощью способа, как указано выше, с чистотой в диапазоне приблизительно от 90% до 100%.
Настоящее изобретение также относится к очищенному атозибану, очищенному до степени чистоты по меньшей мере 99,14%.
Настоящее изобретение также относится к очищенному эптифибатиду, очищенному до степени чистоты по меньшей мере 94%.
Изобретение относится к использованию агентов для образования ионных пар, таких как триэтиламин, трифторуксусная кислота, гексансульфоновая кислота, пентафторпропановая кислота и тому подобные, в ОФ-ВЭЖХ препаративной линейной хроматографии полипептидов. В частности, изобретение относится к использованию агентов для образования ионных пар при посредстве обращенно-фазовой препаративной линейной хроматографии аналогов инсулина и пептидов.
Еще одной целью и преимуществом настоящего изобретения является повышенная чистота целевого белка даже при увеличении нагрузки белка.
Определение терминов
Если в данном контексте не указано иное, научные и технические термины, используемые применительно к настоящему раскрытию, должны иметь значения, хорошо понятные специалистам в данной области техники. Более того, если иное не требуется контекстом, единичные термины должны включать в себя множественные, а множественные термины должны включать единичные. Методы и технологии настоящего изобретения обычно выполняют в соответствии со стандартными методами, известными в данной области техники.
Термин «полипептид», «белок», «пептид» относится к полимеру, состоящему из аминокислот, и не относится к определенной длине продукта; тем самым пептиды, олигопептиды и белки включены в определение полипептид. Этот термин также не относится или исключает постэкспрессионные модификации полипептида, хотя химические или постэкспрессионные модификации этих полипептидов могут быть включены или исключены в виде конкретных вариантов осуществления. Вследствие этого, например, модификации полипептидов, включающие ковалентное присоединение гликозильных групп, ацетильных групп, фосфатных групп, липидных групп и тому подобного, несомненно, включены в термин полипептид. Более того, полипептиды с такими модификациями могут быть определены как отдельные виды, включаемые или исключаемые из настоящего изобретения. Согласно одному из вариантов осуществления молекула представляет собой полипептид или его родственные аналоги либо их производные. Согласно предпочтительному варианту осуществления полипептид выбирают из аналогов инсулина, таких как аспарт, лизпро и гларгин. Предпочтительно, чтобы полипептид был циклическим пептидом. Согласно другому предпочтительному варианту осуществления полипептид является нециклическим пептидом. Согласно еще одному предпочтительному варианту осуществления полипептид выбирают из группы, включающей в себя эптифибатид и атозибан.
Термин "аналог инсулина" предназначен для охвата любой формы "инсулина", как определено выше, где одна или несколько аминокислот в полипептидной цепи замещена (замещены) альтернативной аминокислотой, и/или где одна или несколько аминокислот исключены, или где одна или несколько дополнительных аминокислот добавлены в полипептидную цепь. Аналог инсулина выбирают из группы, включающей в себя аспарт, лизпро и гларгин.
Инсулин аспарт является аналогом человеческого инсулина, представляющим собой быстродействующий парэнтеральный агент для снижения сахара в крови. Аспарт является гомологом регулярного человеческого инсулина за исключением единственной замены аминокислоты пролина в положении В28 аспартамовой кислотой, его получают с помощью метода рекомбинантных ДНК.
Инсулин гларгин отличается от человеческого инсулина тем, что аминокислота аспарагин в положении 21 на А-цепи инсулина замещена глицином и два аргинина добавлены в С-конец В-цепи. Инсулин гларгин также является аналогом человеческого инсулина, представляющим собой быстродействующий парэнтеральный агент для снижения сахара в крови.
Инсулин лизпро является аналогом человеческого инсулина, представляющим собой быстродействующий парэнтеральный агент для снижения сахара в крови. Химически это Lys (В28), Pro (В29)-аналог человеческого инсулина, полученный с обратной последовательностью аминокислот в положениях 28 и 29 на В-цепи инсулина.
Атозибан - синтетический пептид, является ингибитором гормонов окситоцина и вазопрессина. Это антагонист рецептора окситоцина, эффективен при лечении острых приступов при преждевременных родах. Он является конкурентным антагонистом окситоцина на маточных рецепторах окситоцина и разработан в качестве новой токолитической терапии при преждевременной родовой деятельности. Атозибан еще называют ADH (антидиуретическим гормоном).
Эптифибатид - антитромбоцитарный препарат класса ингибиторов гликопротеина IIb/IIIa. Его циклическое гептапептидное белковое производное обнаружено в яде юго-восточного карликового гремучника. Он принадлежит к классу так называемых аргинин-глицин-аспартат-миметиков и обратимо связывается с тромбоцитами.
Термин "хроматография" относится к процессу, с помощью которого целевое анализируемое вещество, входящее в состав смеси, отделяют от других растворенных веществ смеси в результате разницы в скоростях прохождения отдельными растворенными веществами смеси через неподвижную фазу под действием подвижной фазы либо в процессе связывания или элюирования.
Термин "высокоэффективная жидкостная хроматография" при использовании в данном контексте относится к такой хроматографической процедуре, при которой частицы (неподвижная фаза), используемые для заполнения колонки, имеют небольшой размер (от 3 до 50 микрон) и регулярную структуру с незначительными отклонениями от выбранного размера. При такой хроматографии, как правило, используют относительно высокие (порядка 500-3500 пси) давления.
Термин "примесь" относится к любому продукту, не имеющему общей природы с целевым белком. Примеси, как правило, являются родственными продуктами. Примесь, являющаяся целевой при очистке технического инсулина аспарт с помощью использования агентов для образования ионных пар, представляет собой безлидерный предшественник дез-В-аргинин-инсулина аспарт. Он отличается от инсулина аспарт тем, что имеет удлинение из 5 аминокислот на С-конце В-цепи, этими 5 аминокислотами являются Arg-Asp-Ala-Asp-Asp, что показывает, что при кислых рН он будет иметь избыточный положительный заряд по отношению к аспарту.
"Агенты для образования ионных пар" обычно представляют собой ионные соединения, образующие ионные пары с противоположно заряженными ионами. Как правило, агенты для образования ионных пар, используемые в ОФ-ВЭЖХ, содержат углеводородную цепь, придающую определенную гидрофобность, вследствие чего агенты для образования ионных пар могут удерживаться на колонке для обращенно-фазовой хроматографии. Типичные агенты для образования ионных пар, способные образовывать ионные пары с белками, могут использоваться согласно настоящему раскрытию и могут включать в себя трифторуксусную кислоту (ТФК), гексансульфоновую кислоту (ГСК), пентафторпропановую кислоту (ПФПК), триэтиламин (TEA), гептафтормасляную кислоту (ГФМК) и тому подобное.
В особенности данное изобретение относится к использованию ГСК и ТЭА в ОФ-ВЭЖХ препаративной линейной хроматографии, примеры чего представлены в предпочтительных вариантах осуществления. ГСК имеет структуру, состоящую из 6С длинной углеводородной цепи с сернокислотной 
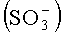
Триэтиламин (ТЭА) в качестве хорошо известного агента для образования ионных пар элюирует белок в форме близких полос, и это свойство в настоящее время используется при более высокой нагрузке (препаративной нагрузке) для улучшения выходов и чистоты в случае ОФ-хроматографии.
"Органические модификаторы" представляют собой неполярные растворители, используемые при ОФ-хроматографии, которые при низких концентрациях способствуют связыванию анализируемых молекул с неподвижной фазой и при более высоких концентрациях элюируют их. В текущем раскрытии используют различные органические модификаторы, такие как ацетонитрил, изопропанол, этанол, метанол, диметилформамид и т.д.
"Дезокта" представляет родственную продукту примесь, образующуюся во время стадии трипсинизации процесса. Она характеризуется отсутствием 8 аминокислот в направлении С-конца В-цепи инсулина и его аналогов.
0,85RRT, 0,9RRT, 1,05RRT и 1,07RRT представляют собой родственные продукту примеси, образующиеся во время реакций некоторых промежуточных продуктов, которые образуются во время процесса. Эти примеси являются следствием большого числа реакционных стадий, вовлеченных в процесс получения атозибана.
0,86RRT, 1,22RRT, 1,8RRT и 2,1RRT представляют собой примеси, образующиеся вследствие большого числа реакционных стадий, вовлеченных в процесс получения эптифибатида. Они являются, как правило, промежуточными продуктами реакций, и некоторые образуются во время процесса. Они могут быть промежуточными продуктами, содержащими защищенные группы, что предохраняет их от превращения в продукт.
Настоящее изобретение относится к использованию агентов для образования ионных пар для получения по существу чистых аналогов инсулина и пептида с чистотой в диапазоне 90%-100%. Инсулиновые аналоги могут быть выбраны из группы, состоящей из аспарта, лизпро и гларгина. Белок или пептид представляет собой атозибан и эптифибатид.
Целью настоящего изобретения является использование по меньшей мере от 0,05% до 1% (мас./об.) агента для образования ионных пар, при этом способ основан на ОФ-ВЭЖХ. Наиболее предпочтительно использовать агент для образования ионных пар в диапазоне 0,05%-1% (мас./об.).
В широком аспекте настоящее изобретение относится к использованию образования ионных пар, применяемому для очистки целевого белка, включающей следующие стадии:
равновесное насыщение неподвижной фазы для подготовки ее к связыванию с целевым анализируемым веществом с последующей нагрузкой, где сырой или неочищенный материал, содержащий анализируемое вещество, проходит через неподвижную фазу. Образец наносят со скоростью приблизительно 180-360 см/ч. Используемые градиенты варьируют в зависимости от образца пептида, подлежащего очистке.
Первая стадия процесса включает в себя очистку молекул из смесей, содержащих их, путем нанесения смесей на колонку для обращенно-фазовой жидкостной хроматографии. Предпочтительно, чтобы колонка была заполнена средой, имеющей диаметр частиц приблизительно 5-40 мкм, более предпочтительно приблизительно 10-40 мкм и наиболее предпочтительно приблизительно 10-13 мкм. Предпочтительно чтобы колонка имела размер пор приблизительно 100-2000 ангстрем, более предпочтительно приблизительно 100-500 ангстрем. В контексте настоящего изобретения размер пор смолы, заполняющей колонку, составляет 120 ангстрем.
Далее следует промывка для удаления несвязанных молекул, элюирование связанного анализируемого вещества с неподвижной фазы и регенерация для извлечения всех крепко связанных молекул, которые не элюируются в данных условиях элюирования. Для уравновешивания и промывки используют 5%-65% концентрации различных органических модификаторов, таких как ацетонитрил, изопропанол, этанол, метанол, диметилформамид и т.д. Очистку проводят при величине рН в диапазоне 2,5-8,5.
Средой в колонке может быть любой подходящий материал, включая смолу на полимерной основе, среду на основе силикагеля или метакрилатную среду.
Один из аспектов настоящего изобретения заключается в использовании агентов для образования ионных пар в ОФ-ВЭЖХ препаративной линейной хроматографии для получения по существу чистых белков и пептидов. Согласно настоящему раскрытию могут использоваться типичные агенты для образования ионных пар, способные образовывать ионные пары с белками, включающие в себя трифторуксусную кислоту (ТФК), гексансульфоновую кислоуа (ГСК), пентафторпропановую кислоту (ПФПК), триэтиламин (TEA), гептафтормасляную кислоту (ГФМК), гептафтормасляную кислоту (ГФМК) и т.д.
В особенности данное изобретение относится к использованию ГСК и ТЭА в ОФ-препаративной линейной хроматографии, примерами чего являются предпочтительные варианты осуществления. ГСК имеет структуру, состоящую из 6С длинной углеводородной цепи с сернокислотной

Триэтиламин (ТЭА) также работает аналогично ГСК, при этом ТЭА образует ионные пары с отрицательно заряженными фрагментами на поверхности белков и увеличивает гидрофобность белков.
Еще одна цель настоящего изобретения относится к использованию агента для образования ионных пар, способствующего изменению селективности примеси, близкородственной целевому белку и создающей основную проблему при очистке.
Примеси в основном являются родственными продукту. Примесь, являющаяся целевой при очистке технического инсулина аспарт с помощью использования агентов для образования ионных пар, представляет собой безлидерный предшественник дез-В-аргинин-инсулина аспарт.Он отличается от инсулина аспарт тем, что имеет удлинение из 5 аминокислот на С-конце В-цепи, этими 5 аминокислотами являются Arg-Asp-Ala-Asp-Asp, что показывает, что при кислых рН он будет иметь избыточный положительный заряд по отношению к аспарту, являющемуся целевым для ГСК. В инсулине гларгин ГСК способствует отделению предшественника инсулина гларгин, поскольку тот обладает избыточными положительными зарядами по сравнению с инсулином гларгин.
Еще одним аспектом настоящего изобретения является повышенная чистота целевого белка даже в случае увеличения нагрузки белка.
Эффективное осуществление настоящего изобретения требует использования индивидуализации правильной комбинации хроматографической матрицы, величины рН и ионной силы буферного раствора для эффективной очистки.
Величина рН буферной системы влияет на разделение в сочетании с гидрофобностью соединений. Изменение величины рН, описанное во время различных стадий хроматографического разделения, оказывает влияние на подвижность соединений на колонке.
Приведенные выше описания конкретных вариантов осуществления настоящего изобретения представлены в целях иллюстрации и описания. Они не являются всеобъемлющими и не ограничивают изобретение определенными раскрытыми формами. На основании вышеизложенных идей возможны различные модификации и изменения. Кроме того, может быть внесено множество изменений для адаптирования частной ситуации, материала, химического соединения, процесса, стадии или стадий процесса к целям, объему и сущности настоящего изобретения. Все такие модификации подпадают под объем прилагаемой формулы изобретения.
Технология текущей заявки на патент дополнительно проработана с помощью следующих примеров. Однако примеры не следует рассматривать как ограничивающие объем изобретения.
Следующие примеры предназначены для дополнительной иллюстрации вариантов осуществления настоящего изобретения, но не предназначены для ограничения объема изобретения. Хотя они являются типичными для того, чтобы их можно было использовать, в качестве альтернативы могут использоваться и другие способы, методики или технологии, известные специалистам в данной области техники.
Более полное понимание может быть получено с помощью следующих конкретных примеров, приведенных исключительно для иллюстративных целей и не предназначенных для ограничения объема изобретения.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Контрольный пример 1
Технический инсулин аспарт с чистотой ~75% 10-кратно разбавляли очищенной водой и добавляли ИПС (изопропиловый спирт) до получения конечной концентрации 5%. Техническую смесь очищали на колонке KromasilTM (100Å - 13 мкм - С8). Подвижной фазой А был 250 мМ раствор ацетата натрия, рН 4,0, а подвижной фазой В был изопропанол. Градиентное элюирование от 15% до 18% подвижной фазы В в подвижной фазе А осуществляли с помощью 20 объемов колонки. Наблюдалось отделение дезоктаинсулина аспарт, однако отделения безлидерного предшественника дез-В-аргинин-инсулина аспарт достигнуто не было. Общая чистота доходила до ~90%.
Пример 1
Технический инсулин аспарт с чистотой ~75% 10-кратно разбавляли очищенной водой и добавляли ИПС до получения конечной концентрации 5%. Техническую смесь очищали на колонке KromasilTM (100Å- 13 мкм - С8). Подвижной фазой А была 1% гексансульфоновая кислота (ГСК) (мас./об.) в 250 мМ растворе ацетата натрия, рН 4,0, а подвижной фазой В был изопропанол. Градиентное элюирование 18%-22% подвижной фазы В в подвижной фазе А проводили с помощью 20 объемов колонки. При добавлении ГСК наблюдалось эффективное извлечение дезокта-инсулина аспарт, а также снижение содержания безлидерного предшественника дез-В-аргинин-инсулина аспарт от уровня ~2,77% до менее чем 0,27%, при этом общая достигнутая чистота составляла ~97,85%.
Контрольный пример 2
Технический инсулин аспарт с чистотой ~75% 10-кратно разбавляли очищенной водой и добавляли этиловый спирт до конечной концентрации 10%. Техническую смесь очищали на колонке KromasilTM (100Å - 13 мкм - С8). Подвижной фазой А был 25 мМ раствор сульфата аммония в 250 мМ растворе ацетата натрия, рН 4,0, а подвижной фазой В был этиловый спирт. Градиентное элюирование 26%-32% подвижной фазы В в подвижной фазе А проводили с помощью 20 объемов колонки. Содержание безлидерного предшественника дез-В-аргинин-инсулина аспарт было снижено от уровня ~2% до 1%. Примесь дезокта-инсулина аспарт удалялась полностью, а содержание моногликозилированного инсулина аспарт было уменьшено с ~3% до 1,3%. При этом общая достигнутая чистота составляла ~94%.
Пример 2
Технический инсулин аспарт с чистотой ~75% 10-кратно разбавляли очищенной водой и добавляли этиловый спирт до конечной концентрации 10%. Техническую смесь очищали на колонке KromasilTM (100Å - 13 мкм - С8). Подвижной фазой А была 1% гексансульфоновая кислота (ГСК) (мас./об.) в 25 мМ растворе сульфата аммония в 250 мМ растворе ацетата натрия, рН 4,0, а подвижной фазой В был этиловый спирт. Градиентное элюирование 25%-35% подвижной фазы В в подвижной фазе А проводили с помощью 20 объемов колонки. Добавление ГСК снижало содержание безлидерного предшественника дез-В-аргинин-инсулина аспарт от уровня ~3% до менее чем 0,3%. Дезокта-инсулин аспарт удалялся полностью, а уровни моногликозилированного инсулина аспарт снижались с ~2% до 0,3%. При этом общая достигнутая чистота составляла ~98%.
Контрольный пример 3
Технический инсулин аспарт с чистотой ~67% 10-кратно разбавляли очищенной водой и добавляли метиловый спирт до конечной концентрации 10%. Техническую смесь очищали на колонке KromasilTM (100Å - 13 мкм - С8). Подвижной фазой А был 25 мМ раствор сульфата аммония в 250 мМ растворе ацетата аммония, рН 4,0, а подвижной фазой В был метиловый спирт. Градиентное элюирование 45%-55% подвижной фазы В в подвижной фазе А проводили с помощью 20 объемов колонки. Содержание безлидерного предшественника дез-В-аргинин-инсулина аспарт было снижено от уровня ~2% до менее чем 1%. Примесь моногликозилированного инсулина аспарт не показала какого-либо существенного снижения. Снижение составило от 2% до 1,5%. Содержание дезокта-инсулина аспарт было уменьшено от ~10% до менее чем ~2%. При этом общая достигнутая чистота составляла ~89%.
Пример 3
Технический инсулин аспарт с чистотой ~67% 10-кратно разбавляли очищенной водой и добавляли метиловый спирт до конечной концентрации 10%. Техническую смесь очищали на колонке KromasilTM (100Å - 13 мкм - С8). Подвижной фазой А была 1% гексансульфоновая кислота (ГСК) (мас./об.) в 25 мМ растворе сульфата аммония в 250 мМ растворе ацетата аммония, рН 4,0, а подвижной фазой В был метиловый спирт. Градиентное элюирование 55%-65% подвижной фазы В в подвижной фазе А проводили с помощью 20 объемов колонки. Добавление ГСК очищало безлидерный предшественник дез-В-аргинин-инсулина аспарт от уровня ~2% до менее чем 1%. Примесь моногликозилированного инсулина аспарт снижалась от 3% до менее чем 0,5%. Содержание дезокта-инсулина аспарт было уменьшено от ~10% до менее чем ~2%. При этом общая достигнутая чистота составляла ~92%.
Пример 4
Технический инсулин аспарт с чистотой ~67% 10-кратно разбавляли очищенной водой и добавляли метиловый спирт до конечной концентрации 10%. Техническую смесь очищали на колонке KromasilTM (100Å - 13 мкм - С8). Подвижной фазой А была 1% гексансульфоновая кислота (ГСК) (мас./об.) в 25 мМ растворе сульфата аммония в 250 мМ растворе ацетата аммония, рН 4,0, а подвижной фазой В был метиловый спирт. Проводили градиентное элюирование 20%-50% подвижной фазы В в подвижной фазе А с помощью 5 объемов колонки с последующим элюированием 55%-65% подвижной фазы В в подвижной фазе А с помощью 20 объемов колонки. Добавление ГСК очищало безлидерный предшественник дез-В-аргинин-инсулина аспарт от уровня ~2% до менее чем 1% и моногликозилированную примесь - от ~3% до 0,6%. Содержание дезокта-инсулина аспарт было снижено от ~10% до менее чем ~2%. При этом общая достигнутая чистота составляла ~92%.
Пример 5
Технический инсулин аспарт с чистотой ~67% 10-кратно разбавляли очищенной водой и добавляли метиловый спирт до конечной концентрации 10%. Техническую смесь очищали на колонке KromasilTM (100Å - 13 мкм - С8). Подвижной фазой А была 1% гексансульфоновая кислота (ГСК) (мас./об.) в 25 мМ растворе сульфата аммония в 250 мМ растворе ацетата аммония, рН 3,5, а подвижной фазой был метиловый спирт. Градиентное элюирование 55%-60% подвижной фазы В в подвижной фазе А проводили с помощью 20 объемов колонки. Добавление ГСК очищало моногликозилированный инсулин аспарт от уровня ~3% до менее чем 0,4%. Содержание дезокта-инсулина аспарт было снижено от ~10% до менее чем ~1%. При этом общая достигнутая чистота составляла ~90%.
Контрольный пример 6
Частично очищенный инсулин гларгин с чистотой ~94% 3-кратно разбавляли очищенной водой и добавляли метиловый спирт до конечной концентрации 10%. Техническую смесь очищали на колонке KromasilTM(100Å - 13 мкм - С8). Подвижной фазой А был 100 мМ раствор сульфата аммония, рН 4,0, а подвижной фазой В был метиловый спирт. Градиентное элюирование 51%-59% подвижной фазы В в подвижной фазе А проводили с помощью 20 объемов колонки. Уровни предшественника инсулина гларгин были снижены с ~2,0% до 1,2%, а уровни DesВ32-R-инсулина гларгин не снижались. При этом общая достигнутая чистота составляла ~97,5%.
Пример 6
Частично очищенный инсулин гларгин с чистотой ~94% 3-кратно разбавляли очищенной водой и добавляли метиловый спирт до конечной концентрации 10%. Техническую смесь очищали на колонке KromasilTM (100Å - 13 мкм - С8). Подвижной фазой А была 0,5% гексансульфоновая кислота (ГСК) (масс/об.) в 100 мМ растворе сульфата аммония, рН 4,0, а подвижной фазой В был метиловый спирт. Градиентное элюирование 51%-59% подвижной фазы В в подвижной фазе А проводили с помощью 20 объемов колонки. Добавление ГСК очищало предшественник инсулина гларгин от уровня ~2,5% до менее чем 0,8%, a DesB32-R - от 0,7% до менее чем 0,2%. При этом общая достигнутая чистота составляла ~99,2%.
Пример 7
Частично очищенный инсулин лизпро с чистотой ~77% 3-кратно разбавляли очищенной водой и добавляли ацетонитрил до конечной концентрации 10%. Техническую смесь очищали на колонке KromasilTM (100Å - 13 мкм - С8). Подвижной фазой А был 1% триэтиламин (TEA) (об./об.) в 20 мМ растворе хлорида магния и 100 мМ растворе Tris, рН 8,5, а подвижной фазой В был ацетонитрил. Градиентное элюирование 24%-30% подвижной фазы В в подвижной фазе А проводили с помощью 25 объемов колонки. Добавление ТЭА снижало уровень безлидерного предшественника дез-В-аргинин-инсулина лизпро от 15% до менее чем 1%. При этом общая достигнутая чистота составляла ~97,5%.
Контрольный пример 8
Технический атозибан после одной стадии очистки имеет чистоту ~95%. Пул элюата после первой стадии разбавляли таким образом, чтобы довести концентрацию растворителя до 5%. Нагрузку затем очищали на колонке Daiso (100Å - 10 мкм - С8). Подвижной фазой А была 50 мМ уксусная кислота, а подвижной фазой В - ацетонитрил. Проводили градиентное элюирование от 9% до 12% подвижной фазы В в подвижной фазе А с помощью 15 объемов колонки с последующим элюированием от 12 до 17% подвижной фазы В в подвижной фазе А с помощью 10 объемов колонки. Достигнутая степень чистоты составляла ~96%, свидетельствуя о полном удалении только 1,05RRT примесей. Примеси 0,85RRT и 0,9RRT не показали какого-либо уменьшения.
Пример 8
Технический атозибан после одной стадии очистки имеет чистоту ~95%. Пул элюата после первой стадии разбавляли таким образом, чтобы довести концентрацию растворителя до 5%. Нагрузку затем очищали на колонке Daiso (120Å - 10 мкм - С8). Подвижной фазой А была 0,05% гексансульфоновая кислота (ГСК) (масс/об.) в 50 мМ растворе уксусной кислоты, а подвижной фазой В был ацетонитрил. Проводили градиентное элюирование от 10% до 17% подвижной фазы В в подвижной фазе А с помощью 25 объемов колонки. Добавление ГСК способствовало повышению чистоты до 98,6%, свидетельствуя о полном удалении примесей 0,85RRT, 0,90RRT, 1,05RRT и 1,07RRT.
Контрольный пример 9
Технический атозибан после одной стадии очистки имеет чистоту ~95%. Пул элюата после первой стадии разбавляли таким образом, чтобы довести концентрацию растворителя до 5%. Нагрузку затем очищали на колонке Daiso (120Å - 10 мкм - С8). Подвижной фазой А был 50 мМ фосфатный буфер с величиной рН 8,0, а подвижной фазой В - ацетонитрил. Проводили градиентное элюирование от 18% до 24% подвижной фазы В в подвижной фазе А с помощью 25 объемов колонки. Достигнутая степень чистоты составляла 98,5%. Содержание 0.97RRT было снижено от ~1,7% до -0,6%. Примесь 0.95RRT не удалялась, а содержание примесей 1,05RRT и 1,07RRT было уменьшено с -0,8% до ~0,3%.
Пример 9
Технический атозибан после одной стадии очистки имеет чистоту ~95%. Пул элюата после первой стадии разбавляли таким образом, чтобы довести концентрацию растворителя до 5%. Нагрузку затем очищали на колонке Daiso (120Å - 10 мкм - С8). Подвижной фазой А был 0,2% триэтиламин (TEA) (об./об.) в 50 мМ фосфатном буфере с величиной рН 8,0, а подвижной фазой В был ацетонитрил. Проводили градиентное элюирование от 20% до 24% подвижной фазы В в подвижной фазе А с помощью 25 объемов колонки. Добавление ТЭА способствовало повышению чистоты до 99,14%, свидетельствуя о полном удалении примесей 0,95RRT, 0,97RRT и 50% уменьшении содержания примесей 1,05RRT и 1,07RRT.
Пример 10
Технический эптифибатид имеет чистоту ~58%. Нагрузку готовили растворением технического вещества в 50 мМ растворе уксусной кислоты и 5% ацетонитрила. Нагрузку затем очищали на колонке Daiso (120Å - 10 мкм - С8). Подвижной фазой А была 0,1% трифторуксусная кислота (ТФК) (об./об.), а подвижной фазой В был ацетонитрил. Проводили градиентное элюирование от 8% до 14% подвижной фазы В в подвижной фазе А с помощью 25 объемов колонки. Добавление ТФК способствовало повышению чистоты до 94%, свидетельствуя о полном удалении примесей 0,86RRT, 1,22RRT, 1,8RRT и 2,1RRT.
Раскрыты предпочтительные варианты осуществления настоящего изобретения. Однако специалисту в данной области техники будет понятно, что под идеи данного изобретения могут подпадать определенные модификации. Вследствие этого для определения истинного объема и содержания изобретения следует ознакомиться со следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРЕПАРАТИВНЫЙ ХРОМАТОГРАФИЧЕСКИЙ СПОСОБ НА ОСНОВЕ НЕЛИНЕЙНОГО ГРАДИЕНТА И ПРОДУКТЫ, ОЧИЩЕННЫЕ ЭТИМ СПОСОБОМ | 2010 |
|
RU2489441C1 |
| СПОСОБ ОЧИСТКИ ЦИКЛИЧЕСКОГО ИЛИ НЕЦИКЛИЧЕСКОГО ПЕПТИДА | 2008 |
|
RU2461564C2 |
| СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО ИНСУЛИНА ГЛАРГИНА | 2008 |
|
RU2495131C2 |
| ОЧИСТКА АНАЛОГОВ ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-1 | 2020 |
|
RU2816581C2 |
| Водная композиция инсулина аспарта и инъецируемый инсулиновый препарат | 2022 |
|
RU2832344C2 |
| ХРОМАТОГРАФИЧЕСКИЕ СПОСОБЫ | 2008 |
|
RU2464066C2 |
| СПОСОБ ПОЛУЧЕНИЯ АНАЛОГОВ ИНСУЛИНА ИЗ ИХ СООТВЕТСТВУЮЩИХ ПРЕДШЕСТВЕННИКОВ (ВАРИАНТЫ) | 2008 |
|
RU2458989C1 |
| СТАБИЛИЗИРОВАННЫЕ ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ АНАЛОГОВ ИНСУЛИНА И/ИЛИ ПРОИЗВОДНЫЕ ИНСУЛИНА | 2014 |
|
RU2702345C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОГО ИНСУЛИНА | 1992 |
|
RU2087151C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИНСУЛИНОВЫХ СОЕДИНЕНИЙ | 2002 |
|
RU2376379C2 |
Изобретение относится к хроматографическому способу очистки аналога инсулина, выбранного из аспартата, лизпро и гларгина, атозибана или эптифибатида из смеси, содержащей по меньшей мере одну родственную примесь. В способе используют агенты для образования ионных пар в ОФ-препаративной линейной хроматографии, позволяя получать высокую степень чистоты целевого конечного продукта. 6 з.п. ф-лы, 10 пр.
1. Хроматографический способ очистки аналога инсулина, выбранного из аспарта, лизпро и гларгина, атозибана или эптифибатида из смеси, содержащей по меньшей мере одну родственную примесь, при величине рН в диапазоне приблизительно от 2,5 до 8,5, включающий стадии:
a) заполнения ОФ-ВЭЖХ колонки силикагелевой смолой (С4-С18), уравновешенной приблизительно от 5% до 85% органического модификатора;
b) нанесения смеси, содержащей аналог инсулина, атозибан или эптифибатид, на колонку со скоростью приблизительно 180 см/ч до 360 см/ч;
c) промывки колонки агентом для образования ионных пар, имеющим концентрацию в диапазоне приблизительно от 0,05% до 1%, в сочетании с органическим модификатором, имеющим концентрацию в диапазоне приблизительно от 5% до 85%, при величине рН в диапазоне приблизительно от 2,5 до 8,5; и
d) элюирования очищенного аналога инсулина, атозибана или эптифибатида с колонки с использованием линейного градиента приблизительно от 10% до 70%.
2. Способ по п.1, отличающийся тем, что силикагелевая смола предпочтительно представляет собой С8.
3. Способ по п.1, отличающийся тем, что смола имеет размер частиц в диапазоне приблизительно от 5 мкм до 40 мкм, предпочтительно приблизительно от 7 мкм до 20 мкм и наиболее предпочтительно приблизительно от 10 мкм до 13 мкм.
4. Способ по п.1, отличающийся тем, что частицы смолы имеют размер пор в диапазоне приблизительно от 50Å до 2000Å, предпочтительно приблизительно от 100Å до 500Å и наиболее предпочтительно 120Å.
5. Способ по п.1, отличающийся тем, что агент для образования ионных пар выбирают из группы, включающей в себя гексансульфоновую кислоту, трифторуксусную кислоту, пентафторпропановую кислоту, триэтиламин и гептафтормасляную кислоту.
6. Способ по п.1, отличающийся тем, что органический модификатор выбирают из группы, включающей в себя ацетонитрил, этанол, метанол и изопропанол.
7. Способ по любому из пп.1-7, отличающийся тем, что чистота аналога инсулина, атозибана или эптифибатида составляет приблизительно от 90% до 100%, предпочтительно по меньшей мере 99%.
| RU 2004118503 A, 15.11.2002 | |||
| СПОСОБ ОЧИСТКИ ЦИКЛИЧЕСКОГО ИЛИ НЕЦИКЛИЧЕСКОГО ПЕПТИДА | 2008 |
|
RU2461564C2 |
| US 5290920 A, 01.03.1994 | |||
| Автономный инвертор | 1973 |
|
SU515228A1 |
| WO 2007095661 A1, 30.08.2007 | |||
| А.Б | |||
| Романчиков и др | |||
| "Генно-инженерный инсулин человека | |||
| VII | |||
| Повышение эффективности хроматографического разделения при использовании принципа бифункциональности", Биоорганическая химия, 1997, т.23, стр.98-103 | |||

