ПРЕДШЕСТВУЮЩИЙ ИЗОБРЕТЕНИЮ УРОВЕНЬ ТЕХНИКИ
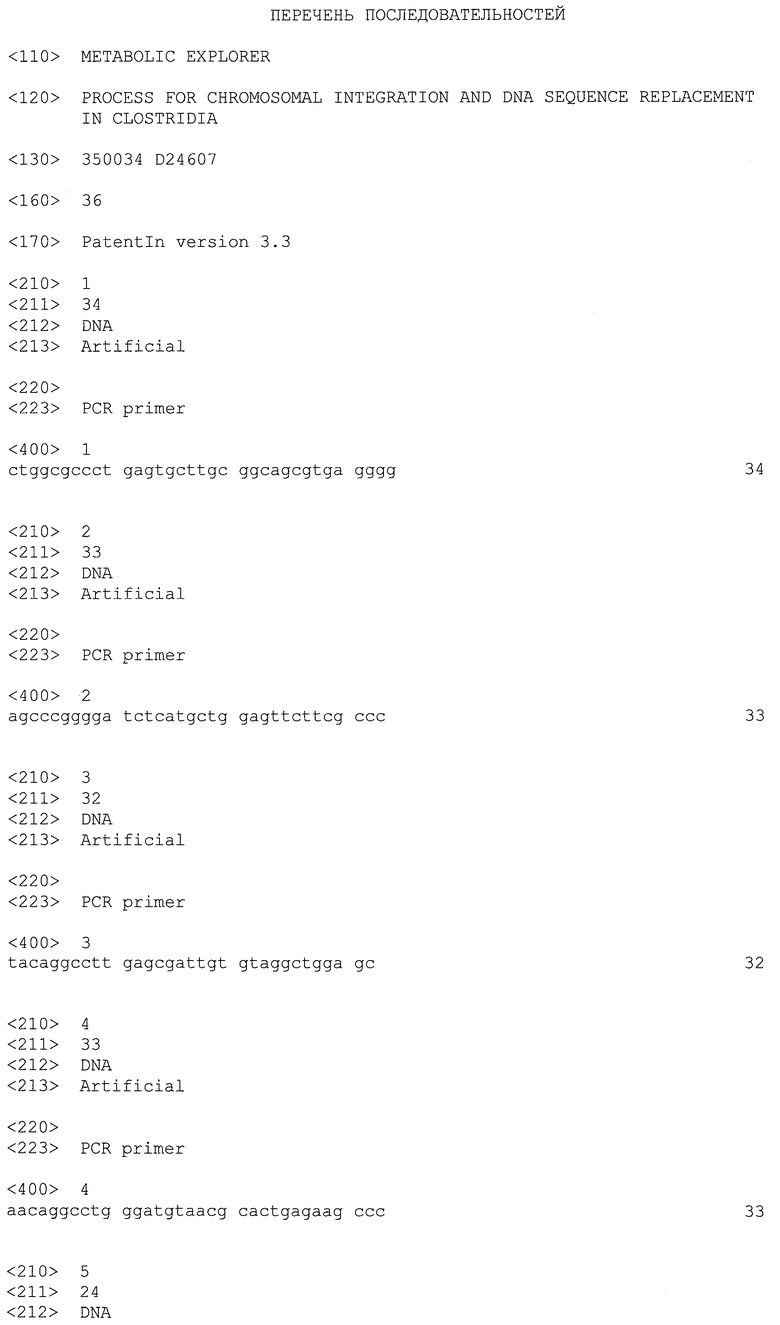
Клостридии (Clostridia) представляют собой грамположительные, анаэробные бактерии с низким содержанием GC, широко используемые в промышленности благодаря своей способности продуцировать растворители, в частности бутанол, этанол и ацетон, а также диолы типа 1,3-пропандиола, органические кислоты типа уксусной, масляной или молочной кислоты, и в вакцинах.
Конструирование рекомбинантных Clostridia составляет важную часть разработок в данной области техники. С целью улучшения возможностей их использования в промышленности штаммы Clostridium генетически модифицируют.
Для проведения этих модификаций наиболее часто используемой методикой для всех видов микроорганизмов является гомологичная рекомбинация. Трансформация и гомологичная рекомбинация в некоторых микроорганизмах всесторонне описана в данной области техники. См., например (Datsenko and Wanner; PNAS, 2000) и (Fabret et al., Molecular Microbiology, 2002).
Clostridia по своей природе не способны к трансформации, и существующие в настоящее время способы их трансформации являются неэффективными и не позволяют вносить множественные мутации. Это препятствует промышленным разработкам в данной области.
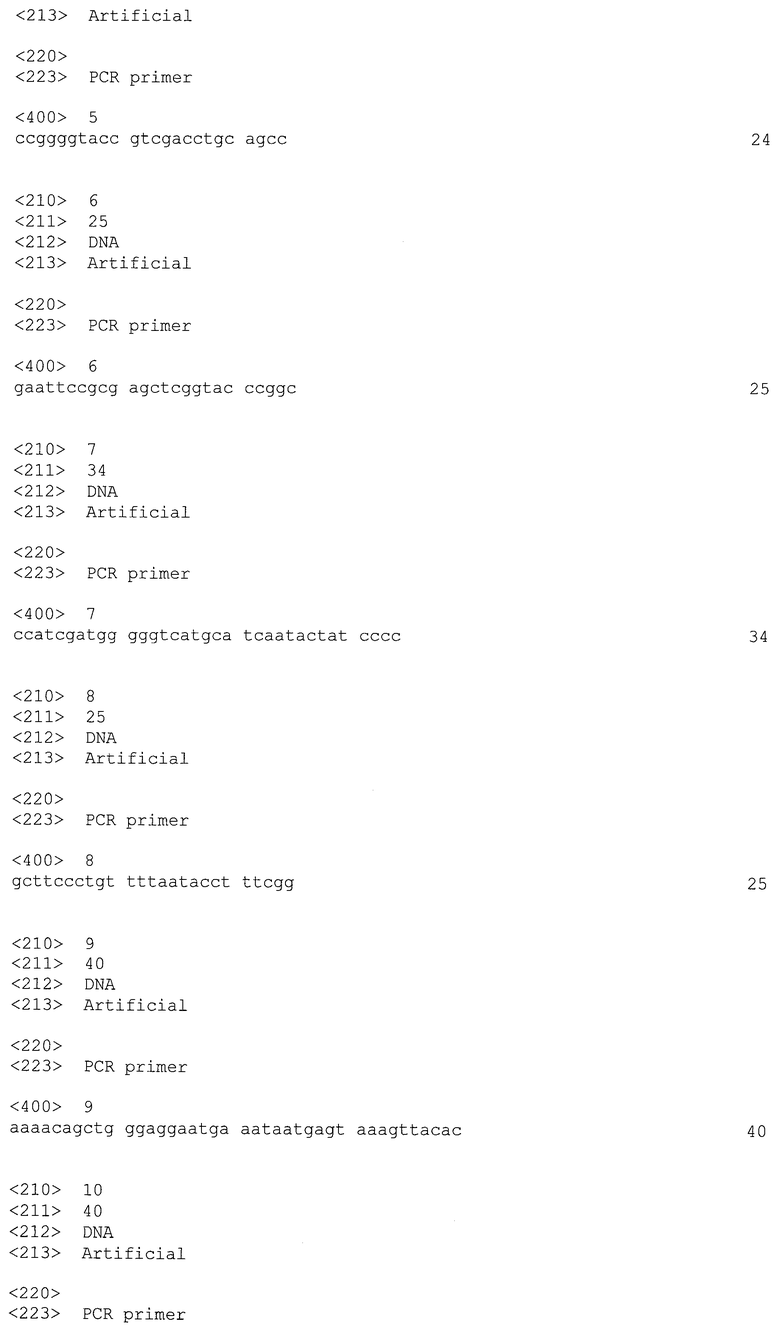
Обычно Clostridia продуцируют внеклеточные ДНКазы и рестрикционные ферменты, которые разрушают чужеродную ДНК до и после ее введения в клетки в целях проведения трансформации. Классические методы, основанные на введении ПЦР-фрагментов, которые хорошо работают во многих микроорганизмах, таких как E.coli или дрожжи, не выполняются в этих микроорганизмах, поскольку период полужизни вне клетки и внутри клетки такой ДНК-конструкции, которая подвергается рекомбинации, слишком мал и эффективность рекомбинации, как правило, низка. В других микроорганизмах эти трудности обойдены путем использования векторов, которые реплицируются в хозяине, увеличивая таким образом вероятность рекомбинационного события. Тем не менее, после завершения рекомбинационного события вектор, теперь несущий интактную последовательность ДНК-мишени, должен быть элиминирован. Эта проблема была разрешена в Lactococcus lactis (Biswas et al., J. Bacteriol., 1993) путем использования чувствительных к температуре репликонов, которые могут быть элиминированы при непермиссивной температуре. Ни один из векторов с такими характеристиками в настоящее время недоступен для Clostridia. Поэтому, конструирование мутантов в Clostridia до настоящего времени было очень трудоемким и зачастую безуспешным.
Об инактивации генов Clostridia сообщалось в следующих статьях (см. таблицу 1).
До настоящего времени инактивацию генов выполняли в Clostridia посредством трансформации с использованием кольцевой ДНК, которая не могла реплицироваться в штаммах-мишенях. Поскольку ДНКазы и эндонуклеазы рестрикции ДНК, присутствующие в Clostridia, быстро разрушают введенную ДНК и, как правило, частота рекомбинации в этом роду не очень высока, получение мутантов было очень трудоемким.
К тому же, все описанные до настоящего времени рекомбинантные штаммы (см. выше) устойчивы к MLS (макролид-линкозамид-стрептограмин) или хлорамфениколу, и соответствующие маркерные гены не могут быть удалены после осуществления рекомбинационного события. Это ограничивает количество возможных рекомбинаций количеством доступных маркеров устойчивости в этих бактериях максимум до 3. Кроме того, для промышленного применения этих бактерий, могло бы быть полезно иметь безмаркерные штаммы для того, чтобы избежать высвобождения генов устойчивости к антибиотикам в ферментационные среды.
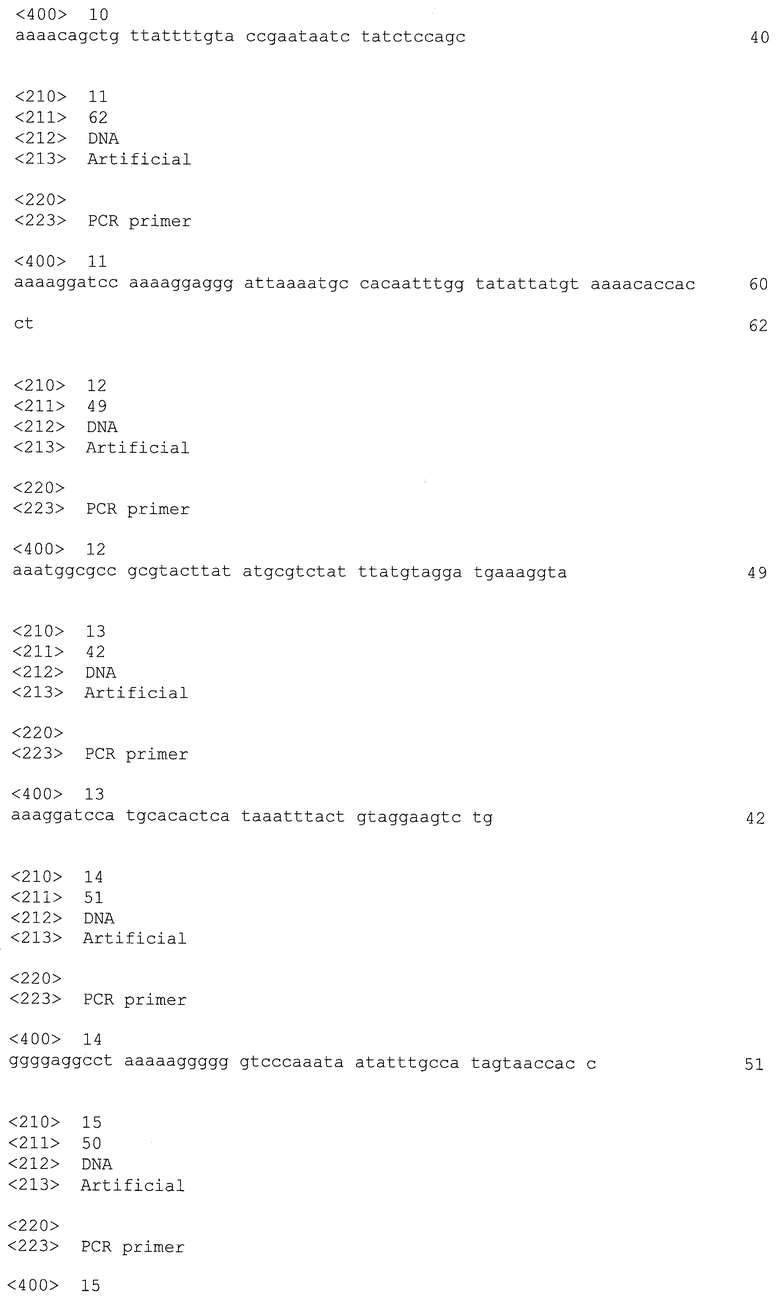
Более того, некоторые из этих штаммов, полученные в результате единичных рекомбинационных событий, имеют тот недостаток, что они не будут стабильны, если культивируются без какого-либо селекционного давления.
Следовательно, состояние данной области техники таково, что все еще существует необходимость в способе трансформации Clostridia с высокой эффективностью, с легкой в выполнении стадией селекции рекомбинантных штаммов, который позволяет осуществлять последующие замены последовательностей ДНК в одном и том же штамме, что приводит к получению рекомбинантных Clostridia, являющихся генетически стабильными и безмаркерными.
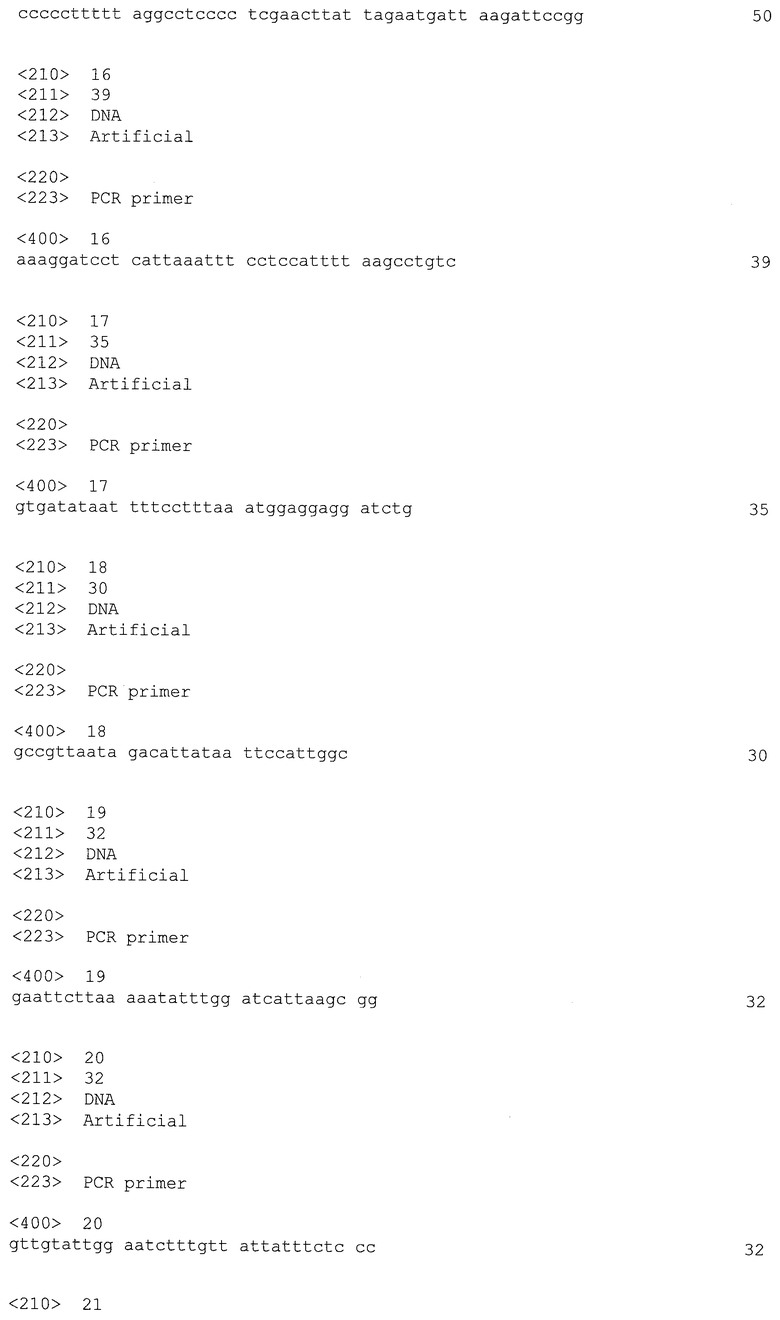
Настоящее изобретение относится к новому способу замены или делетирования последовательности ДНК в Clostridia, легкому в выполнении и применимому в промышленном масштабе. Этот способ полезен для рутинной модификации нескольких генных локусов в Clostridia.
Этот способ основан на использовании репликативного вектора, полезного для трансформации Clostridia с высокой эффективностью.
С использованием этого нового способа в геном может быть введено неограниченное количество мутаций путем элиминирования кассет устойчивости из генома и повторного использования их в успешных раундах замены последовательности ДНК.
Эффективное введение множественных мутаций в Clostridia должно позволить промышленности улучшить существующие промышленные штаммы и разработать новые способы.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно изобретению предложен способ замены последовательности ДНК-мишени посредством гомологичной рекомбинации в Clostridia, включающий:
- трансформацию указанного штамма вектором, содержащим:
- ориджин репликации, позволяющий осуществлять его репликацию в Clostridia, и
- замещающую кассету, содержащую первый маркерный ген, окруженный двумя последовательностями, гомологичными выбранным участкам вокруг последовательности ДНК-мишени, позволяющими осуществлять рекомбинацию данной кассеты, и
- второй маркерный ген,
- селекцию штаммов с интегрированной в их геном указанной кассетой, которые экспрессируют первый маркерный ген,
- селекцию штаммов с элиминированным указанным вектором, которые не экспрессируют второй маркерный ген.
Все молекулярно-биологические методики, используемые для реализации изобретения, полно описаны в Sambrook, Fritsch and Maniatis, "Molecular cloning: a laboratory manual", 2nd edition, Cold Spring Harbor Laboratory Press, 1989.
Используемый в контексте настоящего изобретения термин "замена" последовательности ДНК-мишени означает, что в локус последовательности ДНК-мишени вводят иную, нежели исходная, последовательность.
Согласно изобретению последовательность ДНК определяется как генная или межгенная последовательность. Обе они могут содержать промоторные или регуляторные последовательности.
Выражение "последовательность ДНК-мишени" обозначает любую интересуемую структуру, выбранную специалистом в данной области техники из гена, межгенного участка, промоторной или регуляторной последовательности. Так обозначаются, в частности, гены, кодирующие представляемые интерес белки, например ферменты, вовлеченные в клеточный метаболизм.
Замещающая/вставляемая последовательность ДНК может быть кодирующей или нет. Она может представлять собой мутированную последовательность гена-мишени, промоторную или регуляторную последовательность и/или маркер, как например ген устойчивости к антибиотику или дающий окраску фермент. Она может быть длиннее или короче замененной последовательности в зависимости от расстояния, разделяющего эти два гомологичных участка.
Благодаря наличию вставки экспрессия гена-мишени обычно нарушается, частично или полностью подавляется или усиливается. Замена последовательности ДНК-мишени на последовательность, близкую первоначальной, но содержащую мутации, приводит к реальной экспрессии мутированного белка, промоторной или регуляторной последовательности.
Если в результате замены последовательности ДНК-мишени происходит полное элиминирование указанной последовательности ДНК, ген квалифицируется как "делетированный".
Выражение "гомологичная рекомбинация" относится к событию замены сегмента ДНК другим таким сегментом, который имеет идентичные участки (гомологичные) или близкие к гомологичному. Это событие также называют кроссинговером ДНК.
Термин "трансформация" относится к инкорпорированию экзогенной нуклеиновой кислоты в клетку, при этом такое "приобретение" новых генов является временным (если вектор, несущий гены, "вылечивается") или постоянным (в том случае, если экзогенная ДНК интегрируется в хромосому).
Термин "вектор" относится к внехромосомному элементу, несущему гены или кассеты, который обычно существует в форме кольцевых двухцепочечных молекул ДНК, но также может представлять собой одноцепочечную молекулу ДНК. Оба термина "вектор" и "плазмида" используются независимо.
Вектором по изобретению является репликативный вектор. Он содержит по меньшей мере один ориджин репликации и предпочтительно несколько сайтов инициации репликации, что позволяет ему быть функционально активным в разных видах.
Особо предпочтительный вектор может содержать два сайта инициации репликации:
- Ori, функционально активный в Е.coli,
- RepL из pIM13, происходящего из В.subtilis, функционально активный в Clostridia (Mermelstein et al., Biotechnology, 1992).
Выражение "последовательности, гомологичные последовательности ДНК-мишени" относится к последовательностям, имеющим большую схожесть последовательности с выбранными участками последовательности-мишени.
Термин "маркерный ген" относится к последовательности, кодирующей маркерный белок под контролем регуляторных элементов, функционально активных в Clostridia. Такие белки общеизвестны в данной области техники. Например, специалист в данной области техники может использовать ген устойчивости к антибиотику, флуоресцентный или окрашивающий маркер или маркер ауксотрофности. Примеры полезных маркерных генов будут приведены ниже.
Теперь, после осуществления рекомбинационного события, вектор несет интактную последовательность ДНК-мишени и поэтому должен быть элиминирован. Элиминирование репликативного вектора обычно происходит в последующих культурах клонов в результате негативной или позитивной селекции клонов, из которых этот вектор элиминирован. Элиминирование вектора также может представлять собой активную стадию этого способа с использованием эндонуклеаз, которые специфично расщепляют последовательности ДНК, присутствующие в векторе. Как только вектор "вылечивается", штаммы более не экспрессируют второй маркерный ген и могут быть отобраны по этой характеристике.
В конкретном воплощении изобретения второй маркерный ген представляет собой ген устойчивости к антибиотику. Среди полезных генов устойчивости к антибиотику специалисту в данной области техники будет известен тот, который является наиболее соответствующим. Например, можно использовать следующие гены: ген CatP, дающий устойчивость к хлорамфениколу и тиамфениколу, или ген MLSR, дающий устойчивость к эритромицину.
В предпочтительном воплощении изобретения второй маркерный ген представляет собой встречно-селектируемый маркерный (counter-selective marker) ген.
Встречно-селектируемым маркером является ген, присутствие которого летально для микроорганизма-хозяина в некоторых случаях, как например присутствие родственного ему субстрата. Встречно-селектируемые маркеры могут быть использованы в качестве позитивной селекции на предмет утраты плазмиды.
Предпочтительно, чтобы встречно-селектируемым маркерным геном являлся ген, который восстанавливает активность отсутствующего или делетированного несущественного эндогенного гена.
Наиболее часто используемыми встречно-селектируемыми маркерами являются гены, которые придают чувствительность к сахарозе, стрептомицину или фузаровой кислоте. Они использованы для конструирования мутантов или вакцинных штаммов в нескольких бактериальных штаммах. Для подробного рассмотрения см. обзор Reyrat et at., 1998, Infection and Immunity. Встречно-селектируемые маркеры, которые можно использовать в Clostridia, включают гены, дающие чувствительность к 5-фтор-урацилу (5-FU), гамма-глутамилгидразиду (GBS) или 8-аза-2,6-диаминопурину (8ADP).
В предпочтительном воплощении встречно-селектируемый маркер представляет собой ген upp, кодирующий урацил-фосфорибозилтрансферазу, которая способствует превращению 5-фторурацила (5-FU) в токсичный продукт. Клетки, обладающие Upp-активностью, не могут расти на 6-FU-содержащей среде.
Использование этого встречно-селектируемого маркера, в частности, полезно, когда трансформированные Clostridia представляют собой Δupp и поэтому способны расти на содержащей 5-FU среде перед трансформацией и после элиминирования вектора. Штаммы с элиминированным вектором могут быть позитивно отселектированы.
Супрессорные мутации, которые могут появиться в гене upp в присутствии 5-FU, иногда могут приводить к ошибочным предположениям касательно утраты плазмиды. В предпочтительном воплощении изобретения вектор дополнительно содержит третий маркер, предпочтительно ген устойчивости к антибиотику, который дает возможность проведения второй селекции штаммов, чувствительных к антибиотику. Такая негативная селекция может быть использована в дополнение к позитивной селекции, основанной на гене upp.
В предпочтительном воплощении изобретения вектор элиминируется в результате переваривания эндонуклеазами после осуществления рекомбинационного события. Предпочтительно, вектор несет последовательности ДНК, которые распознаются рестрикционными эндонуклеазами и которые вместе с тем отсутствуют в геноме используемого вида Clostridium. Следовательно, вектор специфически разрушается без потери целостности генома Clostridium.
Рестрикционные эндонуклеазы представляют собой ферменты, которые расщепляют молекулы ДНК по положению конкретных последовательностей оснований, рестрикционному сайту эндонуклеазы. Специалист в данной области способен определить, сайт какой рестрикционной эндонуклеазы отсутствует в геноме представляющего интерес штамма Clostridium. Возможными рестрикционными эндонуклеазами, которые могут быть использованы для С.acetobutylicum, являются AscI, FseI, NotI, SfiI, SrfI. В другом воплощении могут быть использованы мегануклеазы, которые распознают большие (12-45 п.о.) сайты ДНК-мишеней, такие как I-SceI, НО или I-CreI.
В предпочтительном воплощении изобретения штамм Clostridium, который должен быть трансформирован, несет в своем геноме по меньшей мере один ген, кодирующий эндонуклеазу, которая распознает рестрикционной сайт эндонуклеазы, присутствующий в векторе. Возможно, что экспрессия рестрикционной эндонуклеазы находится под контролем индуцибельного промотора.
Индуцибельный промотор представляет собой элемент ДНК, который позволяет протекать экспрессии последовательности-мишени, обусловленной добавлением соответствующего индуктора. Например, система индуцибельного промотора в Clostridium, известная специалисту в данной области, описана в Girbal et al., 2003, Appl. Env. Microbiol. 69: 4985-8.
После осуществления рекомбинационного события и перед скринингом штаммов, в которых должен быть элиминирован вектор, можно индуцировать экспрессию рестрикционной эндонуклеазы. Рестрикционная эндонуклеаза будет расщеплять вектор, присутствующий в Clostridia, что приведет к его элиминированию.
Возможно, что ген, кодирующий рестрикционную эндонуклеазу, может быть вставлен в геном перед введением вектора в этот штамм.
В другом воплощении изобретения ген, кодирующий рестрикционную эндонуклеазу, также может увеличивать частоту рекомбинации перед элиминированием плазмиды путем увеличения количества линейной ДНК в клетках, которая, как известно, подвергается рекомбинации лучше, чем кольцевая ДНК.
В конкретном воплощении изобретения первым маркерным геном является ген устойчивости к антибиотику, введенный в середину замещающей кассеты.
В специфическом воплощении изобретения этот первый маркерный ген может быть удален из генома трансформированных штаммов Clostridium. В частности, первый маркерный ген может быть окружен двумя рекомбиназными сайтами-мишенями и затем элиминирован под действием рекомбиназы после того, как произошел акт гомологичной рекомбинации.
В предпочтительном воплощении изобретения рекомбиназа экспрессируется вторым вектором, несущим соответствующий ген, при этом указанный вектор введен в Clostridia в результате трансформации.
Предпочтительно, чтобы рекомбиназными сайтами-мишенями были FRT (Flippase Recognition Target)-последовательности (распознаваемые флиппазой последовательности-мишени). Рекомбиназа FLP (флиппаза) из Saccharomyces cerevisiae является активной для конкретной, состоящей из 34 пар оснований последовательности ДНК, обозначаемой как FRT-последовательность (мишень для рекомбиназы FLP). Если присутствуют два из этих FRT-сайтов, то фермент FLP производит двухцепочечные разрывы в цепях ДНК, обменивает концы первой FRT с концами второй последовательности-мишени и затем вновь соединяет помененные цепи. Этот процесс приводит к делетированию ДНК, которая лежит между этими двумя сайтами.
В конкретном способе реализации изобретения, последовательности, гомологичные выбранным участкам около последовательности ДНК-мишени, могут содержать мутации, затрагивающие вплоть до 10% пар оснований, включая фрагмент ДНК, используемый для рекомбинационного события.
В предпочтительном воплощении изобретения штаммы Clostridium, которые должны быть трансформированы, имеют делеции в генах, кодирующих рестрикционные эндонуклеазы. Эти штаммы выгодны тем, что их легко трансформировать без какого-либо предварительного метилирования плазмиды in vivo.
В другом предпочтительном воплощении изобретения штаммы Clostridium, которые должны быть трансформированы, имеют делеции в генах, кодирующих внеклеточные ДНКазы.
В другом воплощении изобретения штаммы Clostridium, которые должны быть трансформированы, имеют делеции в гене upp.
В другом предпочтительном воплощении изобретения штаммы Clostridium, которые должны быть трансформированы, выбраны среди Clostridium acetobutylicum, Clostridium bejeirinckii, Clostridium saccharoperbutylacetonicum, Clostridium butylicum, Clostridium butyricum, Clostridium perfringens, Clostridium tetani, Clostridium sporogenes, Clostridium thermocellum, Clostridium saccharolyticum (теперь Thermoanaerobacter saccharolyticum), Clostridium thermosulfurogenes (теперь Thermoanaerobacter thermosulfurigenes), Clostridium thermohydrosulfuricum (теперь Thermoanaerobacter ethanolicus).
Предпочтительным штаммом Clostridium является Clostridium acetobutylicum.
В предпочтительном воплощении изобретения штаммом Clostridium acetobutylicum, который должен быть трансформирован, является штамм Δ Сас15, имеющий делецию в гене, кодирующем рестрикционную эндонуклеазу Сас 824I. Этот штамм выгоден тем, что его легко трансформировать без какого-либо предварительного метилирования плазмиды in vivo.
В другом предпочтительном воплощении изобретения штаммом Clostridium acetobutylicum, который должен быть трансформирован, является штамм, имеющий делецию в гене upp, обозначаемый как Δ upp. В результате, трансформация этого штамма вектором, несущим ген upp, позволяет осуществлять селекцию штаммов, чувствительных к 5-ри-содержащей среде, и после этого позитивную селекцию штаммов, утративших плазмиду, которые более нечувствительны к S-FU-содержащей среде.
Штаммом Clostridium acetobutylicum, который должен быть трансформирован, также может быть Δ Сас15 Δ upp.
Этот способ предпочтительно используют для последующей замены двух или более генов-мишеней посредством гомологичной рекомбинации в одном и том же штамме Clostridium.
Настоящее изобретение также относится к рекомбинантному штамму Clostridium, допускающему его получение в соответствии со способом по изобретению. Предпочтительно, что способ по изобретению может быть использован для получения рекомбинантных штаммов Clostridium, где ген сас15 был делетирован (штамм Δсас15), где ген upp был делетирован (штамм Δupp) и где оба гена сас15 и upp были делетированы (штамм ΔСас15 Δupp). Настоящее изобретение также относится к вектору для трансформации данного штамма, как например описано выше.
ИЗЛОЖЕНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
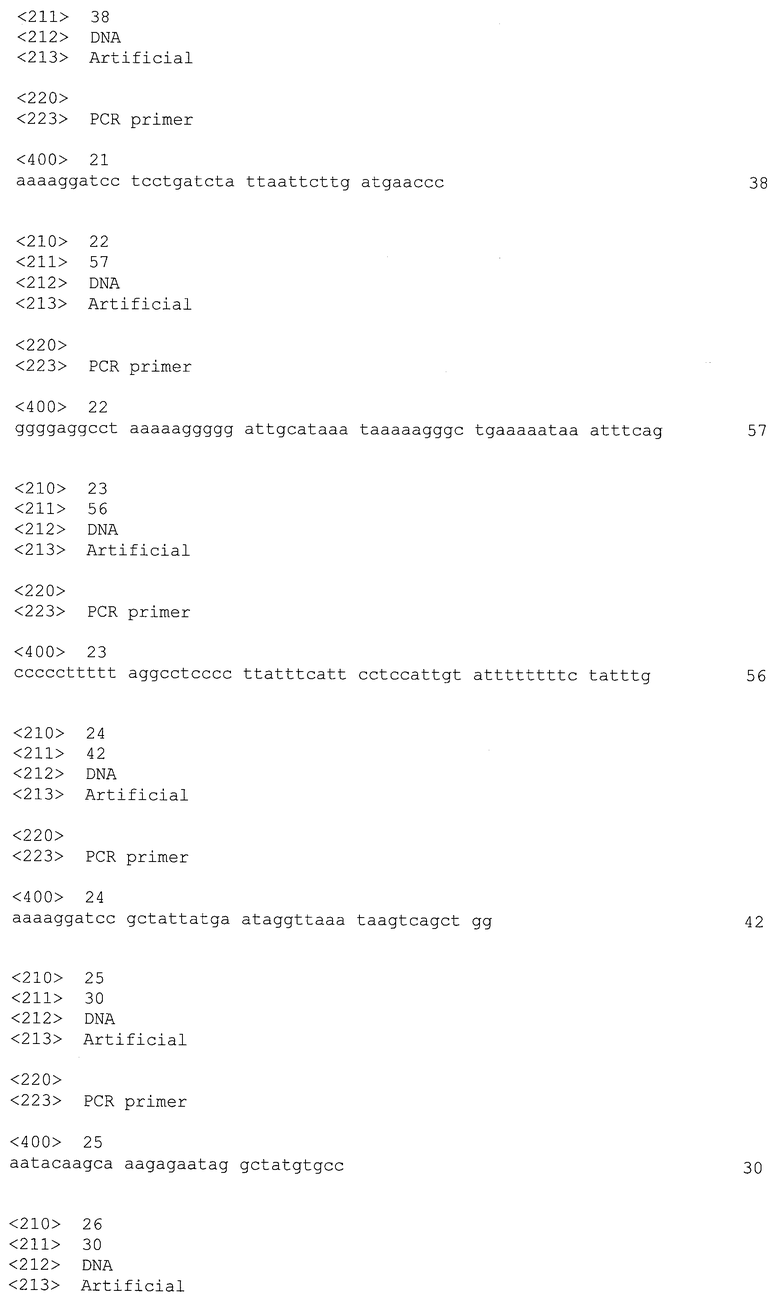
Фиг.1. Карта вектора pUC18-FRT-MLS2.
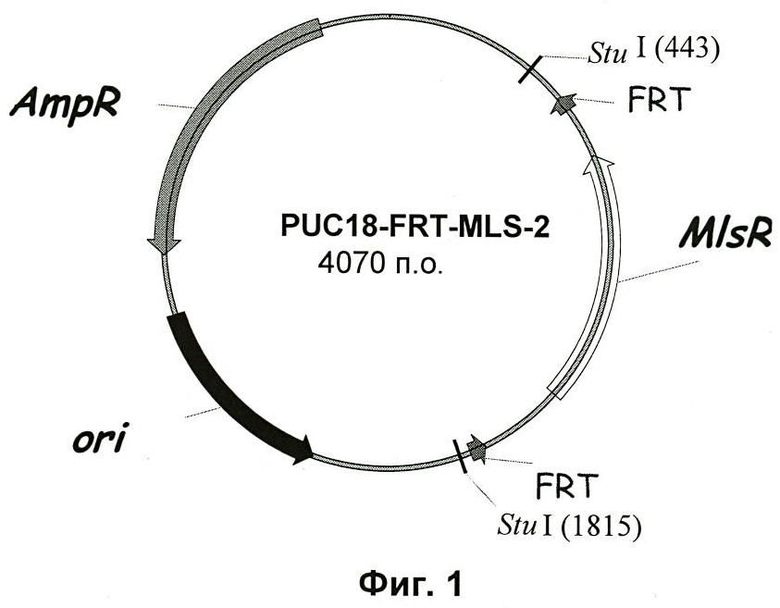
Фиг.2. Карта вектора pCons2-1.
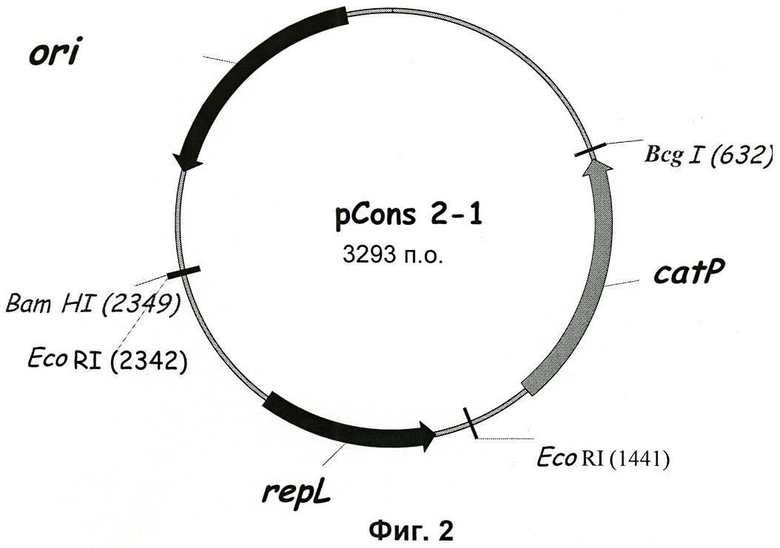
Фиг.3. Карта вектора pCIP2-1.
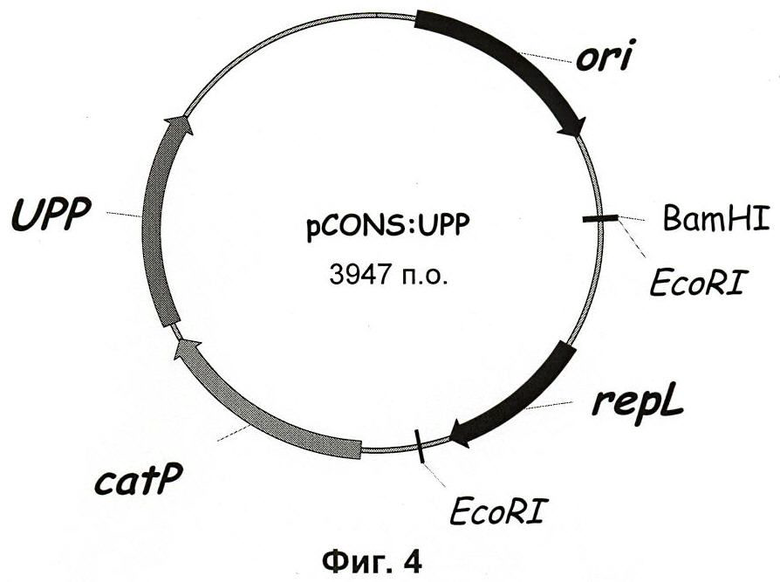
Фиг.4. Карта вектора pCons::UPP.
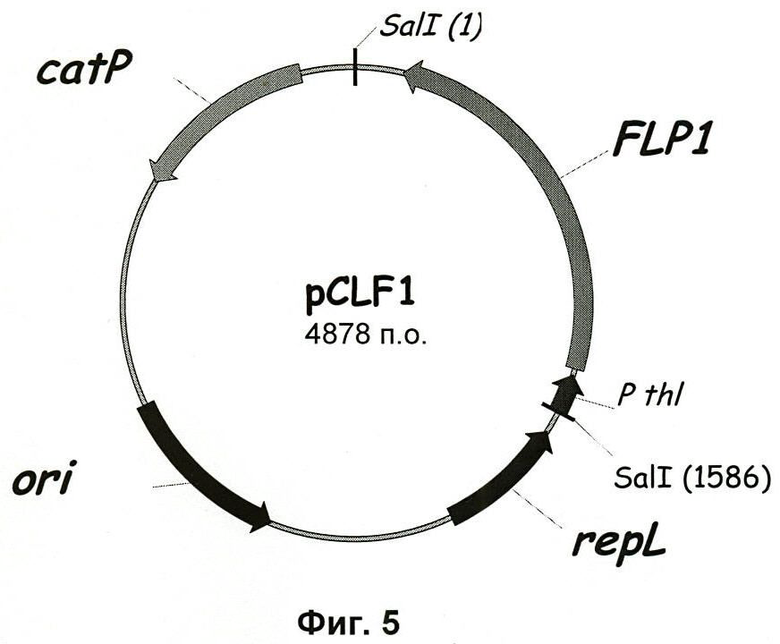
Фиг.5. Карта вектора pCLF1.
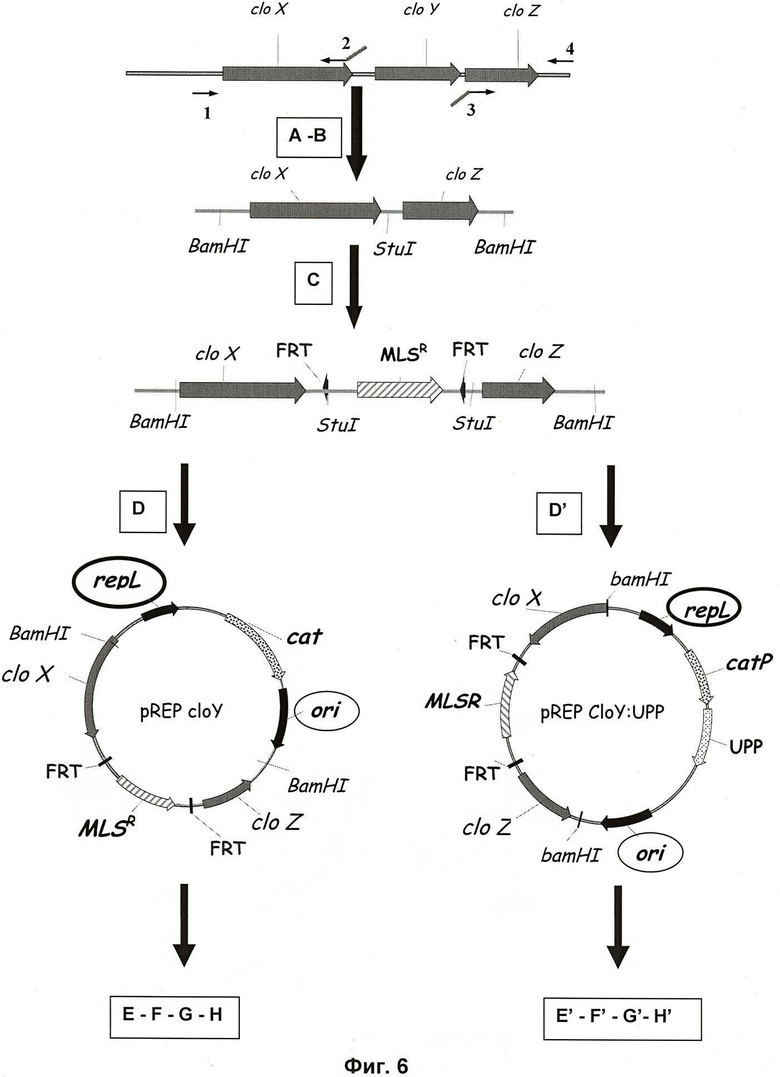
Фиг.6. Схематическое представление метода внесения делеций для Clostridia.
Следующие далее стадии выполняются последовательно:
А - амплификация двух выбранных участков около последовательности ДНК-мишени; 1, 2, 3, 4 представляют собой ПЦР-праймеры;
В - клонирование полученных ПЦР-фрагментов в клонирующий вектор рТОРО;
С - вставка маркера в сайт рестрикции StuI, присутствующий в ПЦР-праймерах;
D - клонирование замещающей кассеты в BamHI-сайт pCONS 2.1: конструирование вектора pREP cloY;
Е - трансформация Clostridium вектором pREP cloY;
F - хромосомная интеграция замещающей кассеты посредством двойного кроссинговера в процессе субкультивирования;
G - скрининг клонов с фенотипами EryR и ThiamS;
Н - ПЦР-анализ с целью контроля на предмет замены гена и утраты плазмиды;
или
D' - клонирование замещающей кассеты в BamHI-сайт pCONS::UPP: конструирование вектора pREP cloY:UPP;
Е' - трансформация Clostridium Δupp вектором pREP cloY: UPP;
F' - хромосомная интеграция замещающей кассеты посредством двойного кроссинговера в процессе субкультивирования;
G' -скрининг клонов с фенотипами EryR и 5-FUR (ThiamS);
Н' - ПЦР-анализ с целью контроля на предмет замены гена и утраты плазмиды.
ПРИМЕРЫ
Пример 1. Конструирование векторов
1.1. Конструирование pUC18-FRT-MLS2
Эта плазмида содержит ген MLSr, функционально активный в Clostridia и фланкированный двумя FRT-сайтами и двумя StuI-сайтами и полезный для конструирования замещающих кассет. Обращенную полимеразную цепную реакцию (ОПЦР) проводили, используя ДНК-полимеразу Pwo с pKD4 в качестве матричной плазмиды (Datsenko и Wanner, 2000) и олигонуклеотиды PKD4.1 и PKD4.2 в качестве праймеров для амплификации участка плазмиды с FRT-сайтами, но без маркера Kmr. Этот дефосфорилированный на конце фрагмент позже лигировали с геном MLSr, полученным после переваривания с помощью HindIII плазмиды pETSPO (Harris et al., 2002, J. Bacteriol.) и обработки фрагментом Кленова. Затем использовали соответствующую плазмиду (pKD4-Ery1) в качестве матрицы для амплификации гена MLSr, фланкированного двумя FRT-сайтами и двумя StuI-сайтами, в ПЦР-реакции, используя олигонуклеотиды FRT-MLSR-F и FRT-MLSR-R в качестве праймеров и ДНК-полимеразу Pwo. Это фрагмент клонировали непосредственно в переваренную с помощью SmaI (плазмиду) pUC18, получая плазмиду pUC18-FRT-MLS2 (Фиг.1).
ПЦР-праймеры:
1.2. Конструирование pCons 2.1
Эта плазмида содержит ориджин репликации pIM13, функционально активный в Clostridia (репликация по механизму "катящегося кольца"), ген catP, дающий устойчивость к тиамфениколу, и уникальный BamHI-сайт для клонирования замещающей кассеты. Эту плазмиду конструировали с использованием двухстадийной процедуры из плазмиды pETSPO (Harris et al., 2002, J. Bacteriol.) с удалением части полилинкера и, среди прочих, BamHI- и EcoRI-сайта. ОПЦР проводили с использованием ДНК-полимеразы Pwo с pETSPO в качестве матричной плазмиды и с олигонуклеотидами PCONSAccI и PCONSEcoRI в качестве праймеров, и ПЦР-продукт фосфорилировали и лигировали. После трансформации E.coli получали плазмиду pCons0. Затем эту плазмиду переваривали с использованием BamHI для удаления кассеты spo0A, нужный фрагмент ДНК очищали и лигировали, получая плазмиду pCons2-1. Карта pCons2-1 приведена на Фиг.2.
ПЦР-праймеры:
1.3. Конструирование вектора pCIP2-1
В этом конструировании ориджин репликации р1М13 из pCons2-1 заменяли на ориджин репликации плазмиды pSOL1, плазмиды с θ-механизмом репликации. Для этого ориджин репликации pSOL1 подвергали ПЦР-амплификации с использованием ДНК-полимеразы Pwo, применяя суммарную ДНК С.acetobutylicum в качестве матрицы и олигонуклеотиды ori-3-D и ori-4-R в качестве праймеров. Этот ПЦР-продукт клонировали в pCR-Bluntll-TOPO, полученную плазмиду переваривали, используя EcoRI, и очищали фрагмент размером 2,2 т.п.о. Аналогично, плазмиду pCons2.1 переваривали, используя EcoRI, очищали фрагмент размером 2,4 т.п.о. и лигировали его с 2,2 т.п.о. EcoRI фрагментом, содержащим ориджин репликации от pSOL1, получая плазмиду pCIP2-1 (Фиг.3).
ПЦР-праймеры:
1.4. Конструирование вектора pCons::upp
Ген upp, имеющий собственный сайт связывания с рибосомой (rbs; ribosome binding site), клонировали в pCons2.1 по BcgI-сайту, расположенному сразу вниз по течению от гена CatP, с целью конструирования искусственного оперона с upp, экспрессируемым под контролем промотора CatP. Таким образом, не было введено никаких гомологичных участков, которые бы сделали возможной хромосомную интеграцию гена upp в штамм Δсас15Δupp в последующих экспериментах по внесению делеций, в которых ген upp используют в качестве встречно-селектируемого маркера на предмет утраты плазмиды.
Ген upp вместе с его rbs подвергали ПЦР(Pfu)-амплификации из геномной ДНК С.acetobutylicum, используя олигонуклеотиды REP-UPP F и REP-UPP R в качестве праймеров. ПЦР-продукт в 664 п.о. переваривали, используя PvuII, клонировали в pCons2.1, переваренный с помощью Bcgl, и обрабатывали ДНК-полимеразой Т4 с получением дефосфорилированных концов. Таким образом, получали репликативный вектор pCons::UPP (см. Фиг.4).
ПЦР-праймеры:
REP-UPP F (SEQ ID №9): 5'-aaaacagctgggaggaatgaaataatgagtaaagttacac-3'
REP-UPP R (SEQ ID №10); 5'-aaaacagctgttattttgtaccgaataatctatctccagc-3'
1.5. Конструирование вектора pCLF1
Ген FLP1 S.cerevisiae, кодирующий рекомбиназу FLP, клонировали в вектор pCons2.1 под контролем промотора и RBS из гена тиолазы (thI) из С.acetobutylicum, что обуславливает высокий уровень экспрессии в этом микроорганизме.
Ген FLP1 подвергали ПЦР(Pfu)-амплификации, используя олигонуклеотиды FLP1-D и FLP1-R в качестве праймеров и плазмиду рСР20 (Datsenko и Wanner, 2000) в качестве матрицы.
ПЦР-праймеры:
-FLP1-D (SEQ ID №11): 5'-aaaaggatccaaaaggagggattaaaatgccacaatttggtatattatgtaaaacaccacct-3'
-FLP1-R (SEQ ID №12): 5'-aaatggcgccgcgtacttatatgcgtctatttatgtaggatgaaaggta-3'
FLP1-D имеет удлиняющий сегмент на 5'-конце, включающий BamIII-сайт и RBS-последовательность от thI.
С использованием FLP1-R вводили SfoI-сайт в 3'-конец ПЦР-продукта.
ПЦР-продукт переваривали, используя BamHI и SfoI, и напрямую клонировали в экспрессирующий вектор pSOS95, который был переварен теми же ферментами, получая плазмиду pEX-FLP1 (6281 п.о.).
Фрагмент SalI (1585 п.о.) pEX-FLP1, содержащий FLPI-экспрессирующую касету, клонировали по SalI-сайту pCONS2.1 для получения плазмиды pCLF1 (4878 п.о.) (Фиг.5).
Пример 2. Делетирование гена сас1502, кодирующего рестрикционный фермент сас824I в Clostridium acetobutylicum
Для схематического представления данного способа см. Фиг.6.
Два фрагмента ДНК, окружающие ген, кодирующий Сас824I, (САС1502) подвергали ПЦР-амплификации с использованием ДНК-полимеразы Pwo, применяя суммарную ДНК из С.acetobutylicum в качестве матрицы и две специфические пары олигонуклеотидов в качестве праймеров (см. Таблицу 2). Используя пары праймеров САС 1В-САС 2 и САС 3-САС 4В, получали ДНК-фрагменты 1493 п.о. и 999 п.о., соответственно. С использованием обоих праймеров САС 1В и САС 4В вводят SamHI-сайт, тогда как праймеры САС 2 и САС 3 имеют комплементарные удлиненные с 5'-конца последовательности, с помощью которых вводят StuI-сайт. ДНК-фрагменты САС 1 В-САС 2 и САС 3-САС 4 В соединяли в эксперименте по ПЦР-слиянию, используя праймеры САС 1В и САС 4В, и полученный фрагмент клонировали в вектор pCR4-TOPO-Blunt, получая рТОРО:сас15. По уникальному StuI-сайту рТОРО:сас15 вводили StuI-фрагмент размером 1372 п.о. от pUC18-FRT-MLS2, несущий ген устойчивости к антибиотикам MLSr с FRT-последовательностями по обеим сторонам. Замещающую кассету сас1502, полученную после переваривания полученной плазмиды с помощью BamHI, клонировали по BamHI-сайту в pCons2-1, получая плазмиду pREPCAC15, и в pCIP2.1, получая pCIPCAC15.
Плазмиды pREPCAC15 и pCIPCAC15 метилировали in vivo и использовали для трансформации С.acetobutylicum путем электропорации. После селекции на чашках Петри на предмет клонов, устойчивых к эритромицину (40 мкг/мл), по одной колонии от каждого из трансформантов культивировали в течение 24 часов в жидкой синтетической среде с эритромицином в концентрации 40 мкг/мл и затем субкультивировали в жидкой среде 2YTG без антибиотика. Соответствующие разведения наносили на обогащенный агар для клостридий (RCA) с эритромицином в концентрации 40 мкг/мл. Для селекции интегрантов, утративших векторы pREPCAC15 или pCIPCAC15, устойчивые к эритромицину клоны наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл. В то время как с использованием трансформантов pREPCAC15 было получено несколько колоний с желаемым фенотипом, никаких таких колоний при использовании трансформантов pCIPCAC15 получено не было. Этот факт демонстрирует, что θ-механизм репликации pCIPCAC15 в меньшей степени подходит для того, чтобы способствовать двойному кроссинговеру в С.acetobutylicum, нежели механизм "катящегося кольца". Генотип клонов, устойчивых к эритромицину и чувствительных к тиамфениколу, проверяли ПЦР-анализом (используя праймеры САС 0 и САС 5, локализованные вне замещающей кассеты САС15, и праймеры САС D и САС R, локализованные внутри сас1502). Штаммы Δcac15::mlsR, утратившие pREPCAC15, выделяли.
Штамм Δcac15::mlsR трансформировали вектором pCLF1, экспрессирующим ген FLP1, кодирующий рекомбиназу Flp из S.cerevisiae. После трансформации и селекции на предмет устойчивости к тиамфениколу (50 мкг/мл) на чашках Петри одну колонию культивировали на синтетической жидкой среде с тиамфениколом в концентрации 50 мкг/мл и соответствующие разведения наносили на RCA с тиамфениколом в концентрации 50 мкг/мл. Тиамфеникол-устойчивые клоны наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл. Генотип клонов с чувствительностью к эритромицину и устойчивостью к тиамфениколу проверяли ПЦР-анализом, используя праймеры САС 0 и САС 5В.
Проводили два последовательных 24-часовых культивирования штамма Δсас15 с чувствительностью к эритромицину и устойчивостью к тиамфениколу с целью удаления pCLF1. Штамм Δсас15, утративший pCLF1, выделяли в соответствии с его чувствительностью как к эритромицину, так и к тиамфениколу. Штамм обозначали как MGCΔcac15 С.acetobutylicum.

Пример 3. Делетирование гена upp, кодирующего урацил-фосфорибозилтрансферазу в Clostridium acetobutylicum Δсас15
Два фрагмента ДНК, расположенные "вверх по течению" и "вниз по течению" от upp (САС2879), подвергали ПЦР-амплификации с использованием ДНК-полимеразы Pwo, применяя суммарную ДНК из С.acetobutylicum в качестве матрицы и две специфические пары олигонуклеотидов в качестве праймеров (см. Таблицу 3). Используя пары праймеров UPP 1-UPP 2 и UPP 3-UPP 4, получали ДНК-фрагменты 1103 п.о. и 1105 п.о., соответственно. С использованием обоих праймеров UPP 1 и UPP 4 вводят BamHI-сайт, тогда как праймеры UPP 2 и UPP 3 имеют удлиненные с 5'-конца последовательности, с помощью которых вводят StuI-сайт. ДНК-фрагменты UPP 1-UPP 2 и UPP 3-UPP 4 соединяли в эксперименте по ПЦР-слиянию, используя праймеры UPP 1 и UPP 4, и полученный фрагмент клонировали в pCR4-TOPO-Blunt, получая рТОРО:upp. По уникальному StuI-сайту рТОРО:upp вводили StuI-фрагмент размером 1372 п.о. от pUC18-FRT-MLS2, несущий ген устойчивости к антибиотикам MLSr с FRT-последовательностями по обеим сторонам. Замещающую кассету upp, полученную после переваривания полученной плазмиды с помощью SamHI, клонировали в pCons2-1 по BamН1-сайту, получая плазмиду pREPUPP.
Плазмиду pREPUPP использовали для трансформации штамма С.acetobutylicuin MGCΔcac15 путем электропорации без проведения предварительного in vivo метилирования. После селекции на чашках Петри на предмет клонов, устойчивых к эритромицину (40 мкг/мл), одну колонию культивировали в течение 24 часов в жидкой синтетической среде с эритромицином в концентрации 40 мкг/мл и затем субкультивировали в жидкой среде 2YTG без антибиотика. Соответствующие разведения наносили на RCA с эритромицином в концентрации 40 мкг/мл. Для селекции интегрантов, утративших вектор pREPUPP, устойчивые к эритромицину клоны наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл. Генотип клонов, устойчивых к эритромицину и чувствительных к тиамфениколу, проверяли ПЦР-анализом (используя праймеры UPP 0 и UPP 5, локализованные вне замещающей кассеты UPP, и праймеры UPP D и UPP R, локализованные внутри UPP). Штамм Δcac15Δupp::mlsR, утративший pREPUPP, выделяли. Предыдущее делетирование сас1502 подтверждали, как описано ранее в первом примере. Штамм Δсас15Δupp::mlsR является устойчивым к 5-FU в концентрации 400 мкМ по сравнению с 50 мкМ для штамма Δсас15.
Штамм Δсас15Δupp::mlsR трансформировали вектором pCLF1, экспрессирующим ген FLP1, кодирующий рекомбиназу Flp из S.cerevisiae. После трансформации и селекции на устойчивость к тиамфениколу (50 мкг/мл) на чашках Петри одну колонию культивировали в синтетической жидкой среде с тиамфениколом в концентрации 50 мкг/мл и соответствующие разведения наносили на RCA с тиамфениколом в концентрации 50 мкг/мл. Тиамфеникол-устойчивые клоны наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл. Генотип клонов с чувствительностью к эритромицину и устойчивостью к тиамфениколу проверяли ПЦР-анализом, используя праймеры UPP 0 и UPP 5. Проводили два последовательных 24-часовых культивирования штамма Δсас15Δupp с чувствительностью к эритромицину и устойчивостью к тиамфениколу с целью удаления pCLF1. Штамм Δсас15Δupp, утративший pCLF1, выделяли в соответствии с его чувствительностью как к эритромицину, так и к тиамфениколу.

Пример 4. Делетирование гена сас3535 с использованием upp и 5-фторурацила в качестве позитивной селекции на предмет утраты плазмиды
Два фрагмента ДНК, расположенные "вверх по течению" и "вниз по течению" от гена сас3535, кодирующего вторую систему рестрикции-модификации С.acetobutylicum, подвергали ПЦР-амплификации с использованием ДНК-полимеразы Pwo, применяя суммарную ДНК из С.acetobutylicum в качестве матрицы и две специфические пары олигонуклеотидов (см. Таблицу 4). Используя пары праймеров RM 1-RM 2 и RM 3-RM 4, получали ДНК-фрагменты 1 т.п.о. и 0,9 т.п.о., соответственно. С использованием обоих праймеров RM 1 и RM 4 вводят BamHI-сайт, тогда как праймеры RM 2 и RM 3 имеют комплементарные удлиненные с 5'-конца последовательности, с помощью которых вводят StuI-сайт. ДНК-фрагменты RM 1-RM 2 и RM 3-RM 4 соединяли в эксперименте по ПЦР-слиянию, используя праймеры RM 1 и RM 4, и полученный фрагмент клонировали в вектор pCR4-TOPO-Blunt, получая плазмиду рТОРО:сас3535. По уникальному StuI рТОРО:сас3535 вводили Sful-фрагмент размером 1372 п.о. от pUC18-FRT-MLS2, несущий ген устойчивости к антибиотикам MLSr с FRT-последовательностями по обеим сторонам. Замещающую кассету САС3535, полученную после переваривания полученной плазмиды с помощью BamHI, клонировали в pCons::upp по BamHI-сайту, получая плазмиду pREPCAC3535::upp.
Плазмиду pREPCAC3535::upp использовали для трансформации штамма С.acetobutylicum MGCΔcac15Δupp путем электропорации. После селекции на чашках Петри на предмет клонов, устойчивых к эритромицину (40 мкг/мл), одну колонию культивировали в течение 24 часов в жидкой синтетической среде с эритромицином в концентрации 40 мкг/мл и 100 мкл неразбавленной культуры наносили на RCA с эритромицином в концентрации 40 мкг/мл и 5-FU в концентрации 400 мкМ. Колонии, устойчивые как к эритромицину, так и к 5-FU, наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл с целью проверки того, что устойчивость к 5-FU ассоциирована с чувствительностью к тиамфениколу. Генотип клонов, устойчивых к эритромицину и чувствительных к тиамфениколу, проверяли ПЦР-анализом (используя праймеры RM 0 и RM 5, локализованные вне замещающей кассеты САС3535, и праймеры RM D и RM R, локализованные внутри гена сас3535). Таким образом, выделяли штамм Δсас15ΔuppΔсас35::mR, утративший pREPCAC3535::upp.



| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ БИОЛОГИЧЕСКОГО ПРОИЗВОДСТВА н-БУТАНОЛА | 2007 |
|
RU2461627C2 |
| КОНСТРУКЦИИ И СПОСОБЫ ДЛЯ ПРОДУЦИРОВАНИЯ И СЕКРЕЦИИ ФЕРМЕНТОВ, РАСЩЕПЛЯЮЩИХ КЛЕТОЧНУЮ СТЕНКУ | 2009 |
|
RU2525194C2 |
| ФЕРМЕНТАТИВНОЕ ПОЛУЧЕНИЕ 1-БУТАНОЛА | 2006 |
|
RU2429295C2 |
| РЕКОМБИНАНТНЫЕ АТТЕНУИРОВАННЫЕ МИКРООРГАНИЗМЫ CLOSTRIDIUM И ВАКЦИНА | 2007 |
|
RU2445364C2 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК, СОДЕРЖАЩАЯ ГЕНЫ СИНТЕЗА БУТАНОЛА ИЗ Clostridium acetobutylicum (ВАРИАНТЫ), РЕКОМБИНАНТНЫЙ ШТАММ Lactobacillus brevis - ПРОДУЦЕНТ Н-БУТАНОЛА (ВАРИАНТЫ) И СПОСОБ МИКРОБИОЛОГИЧЕСКОГО СИНТЕЗА Н-БУТАНОЛА | 2008 |
|
RU2375451C1 |
| ФЕРМЕНТАТИВНОЕ ПОЛУЧЕНИЕ ЧЕТЫРЕХУГЛЕРОДНЫХ СПИРТОВ | 2006 |
|
RU2394913C2 |
| ПО СУЩЕСТВУ НЕТОКСИЧНЫЙ МУТЕИН АЛЬФА-ТОКСИНА Clostridium perfringens И КОДИРУЮЩАЯ ЕГО МОЛЕКУЛА НУКЛЕИНОВОЙ КИСЛОТЫ | 2007 |
|
RU2593945C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4-ДИГИДРОКСИБУТИРАТА | 2013 |
|
RU2645260C2 |
| НОВАЯ СИСТЕМА ИЗМЕНЕНИЯ ГЕНОМА ДЛЯ МИКРООРГАНИЗМОВ | 2014 |
|
RU2694316C1 |
| РЕКОМБИНАНТНЫЙ МИКРООРГАНИЗМ ДЛЯ ПОЛУЧЕНИЯ ПОЛЕЗНЫХ МЕТАБОЛИТОВ | 2012 |
|
RU2658770C2 |


Изобретение относится к области биотехнологии, а именно к способу замены последовательности ДНК-мишени посредством гомологичной рекомбинации в Clostridium acetobutylicum, способу замены двух или более генов-мишеней посредством гомологичной рекомбинации в одной и той же клетке С.acetobutylicum, трансформированной клетке Clostridium acetobutylicum, трансформирующему вектору. Способ включает трансформацию клетки Clostridium acetobutylicum вектором, содержащим: ориджин репликации, позволяющий осуществлять его репликацию в С.acetobutylicum; замещающую кассету, содержащую первый маркерный ген, окруженный двумя последовательностями, гомологичными выбранным участкам около последовательности ДНК-мишени, позволяющими осуществлять рекомбинацию данной кассеты; второй маркерный ген, представляющий собой upp встречно-селектируемый маркер. Осуществляют селекцию клеток с интегрированной в их геном указанной кассетой, которые экспрессируют первый маркерный ген. Проводят селекцию клеток с элиминированным указанным вектором, которые не экспрессируют второй маркерный ген. Предложенное изобретение позволяет получать трансформированную клетку Clostridium acetobutylicum, которая является генетически стабильной и безмаркерной. 4 н. и 27 з.п. ф-лы, 6 ил., 4 табл., 4 пр.
1. Способ замены последовательности ДНК-мишени посредством гомологичной рекомбинации в Clostridium acetobutylicum, включающий:
- трансформацию клетки Clostridium acetobutylicum вектором, содержащим:
- ориджин репликации, позволяющий осуществлять его репликацию в С. acetobutylicum, и
- замещающую кассету, содержащую первый маркерный ген, окруженный двумя последовательностями, гомологичными выбранным участкам около последовательности ДНК-мишени, позволяющими осуществлять рекомбинацию данной кассеты, и
- второй маркерный ген,
- селекцию клеток с интегрированной в их геном указанной кассетой, которые экспрессируют первый маркерный ген,
- селекцию клеток с элиминированным указанным вектором, которые не экспрессируют второй маркерный ген,
где второй маркерный ген представляет собой ген upp, встречно-селектируемый маркер.
2. Способ по п.1, где встречно-селектируемый маркер представляет собой ген, который восстанавливает активность несущественного отсутствующего или делегированного гена.
3. Способ по п.1, где вектор содержит третий маркер, который дает возможность проведения негативной селекции клеток с элиминированным указанным вектором.
4. Способ по п.1, где вектор элиминируется в результате переваривания эндонуклеазами.
5. Способ по п.4, где вектор несет последовательности ДНК, которые распознаются рестрикционными эндонуклеазами и которые вместе с тем отсутствуют в геноме используемых клеток С. acetobutylicum.
6. Способ по п.4 или 5, где используемые клетки С. acetobutylicum несут в своем геноме по меньшей мере одну эндонуклеазу, специфичную к рестрикционным сайтам, присутствующим в векторе, возможно экспрессируемую под контролем индуцибельного промотора.
7. Способ по п.1, где первый маркерный ген представляет собой ген устойчивости к антибиотику.
8. Способ по п.1, где первый маркерный ген окружен двумя рекомбиназными сайтами-мишенями.
9. Способ по п.8, где после того, как произошел акт гомологичной рекомбинации и замещающая кассета интегрировалась, первый маркерный ген, находящийся в этой кассете, элиминируется под действием рекомбиназы.
10. Способ по любому из пп.8 или 9, где указанная рекомбиназа экспрессируется геном, доставляемым вторым вектором, введенным в эти клетки.
11. Способ по любому из пп.8 или 9, где указанные рекомбиназные сайты-мишени представляют собой FRT(Flippase Recognition Target)-последовательности (распознаваемые флиппазой последовательности-мишени), а указанная рекомбиназа представляет собой рекомбиназу FLP (флиппаза).
12. Способ по п.1, где указанные последовательности, гомологичные участкам около последовательности ДНК-мишени, содержат мутации, затрагивающие включительно до 10% пар оснований, используемых для рекомбинационного события.
13. Способ по п.1, где С. acetobutylicum, которые должны быть трансформированы, имеют делеции генов, кодирующих рестрикционные эндонуклеазы.
14. Способ по п.1, где С. acetobutylicum, которые должны быть трансформированы, имеют делеции генов, кодирующих ДНКазу.
15. Способ по п.1, где С. acetobutylicum, которые должны быть трансформированы, имеют делеции гена upp.
16. Способ по п.1, где клетки Clostridium acetobutylicum, которые должны быть трансформированы, являются Δ сас15.
17. Способ по п.1, где клетки Clostridium acetobutylicum, которые должны быть трансформированы, являются Δ сас15 Δ upp.
18. Способ замены двух или более генов-мишений посредством гомологичной рекомбинации в одной и той же клетке С. acetobutylicum, причем указанный способ по любому из пп.1-17 осуществляют последовательно два или более раз.
19. Трансформированная клетка Clostridium acetobutylicum, полученная в соответствии со способом по любому из пп.1-18.
20. Трансформированная клетка С. acetobutylicum по п.19, где был делетирован ген сас15.
21. Трансформированная клетка С. acetobutylicum по п.19, где был делетирован ген upp.
22. Трансформированная клетка С. acetobutylicum по п.19, где были делетированы оба гена сас15 и upp.
23. Трансформирующий вектор, содержащий:
- ориджин репликации, позволяющий осуществлять его репликацию в С. acetobutylicum, и
- замещающую кассету, содержащую первый маркерный ген, окруженный двумя последовательностями, гомологичными выбранным участкам около последовательности ДНК-мишени, позволяющими осуществлять рекомбинацию данной кассеты, и
- ген upp второй встречно-селектируемый маркерный ген.
24. Вектор по п.23, где встречно-селектируемый маркер представляет собой ген, который восстанавливает активность несущественного отсутствующего или делетированного гена.
25. Вектор по п.23, содержащий третий маркер, который дает возможность проведения негативной селекции клеток с элиминированным указанным вектором.
26. Вектор по п.23, который элиминируется в результате переваривания эндонуклеазами.
27. Вектор по п.26, несущий последовательности ДНК, которые распознаются рестрикционными эндонуклеазами и которые вместе с тем отсутствуют в геноме используемых клеток С. acetobutylicum.
28. Вектор по п.23, где первый маркерный ген представляет собой ген устойчивости к антибиотику.
29. Вектор по п.23, где первый маркерный ген окружен двумя рекомбиназными сайтами-мишенями.
30. Вектор по п.29, где указанные рекомбиназные сайты-мишени представляют собой FRT(Flippase Recognition Target)-последовательности (распознаваемые флиппазой последовательности-мишени), а указанная рекомбиназа представляет собой рекомбиназу FLP (флиппаза).
31. Вектор по п.23, где указанные последовательности, гомологичные участкам около последовательности ДНК-мишени, содержат мутации, затрагивающие включительно до 10% пар оснований, используемых для рекомбинационного события.
| HARRIS L.M | |||
| ET AL., Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824, J Bacteriol, 2002, v.184, no.13, pp.3586-3597 | |||
| FABRET C | |||
| ET AL., A new mutation delivery system for genome-scale approaches in Bacillus subtilis, Mol Microbiol., 2002, v.46, no.1, pp.25-36 | |||
| WO 03064623 A2, 07.08.2003. |

