ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к областям молекулярной биологии и генетической инженерии микроорганизмов, в частности, дрожжей.
ВВЕДЕНИЕ
Гомологичная рекомбинация у микроорганизмов, таких как дрожжи, основана на механизме репарации двунитевых разрывов, при которой связываются фрагменты ДНК. При выявлении двунитевого разрыва ДНК экзонуклеаза разрушает оба 5ʹ-конца, после чего происходит инвазия гомологичной матрицы. Механизм синтеза ДНК репарирует обе нити, и лигирование ДНК завершает процесс без каких-либо делеций [Storici et al., (2003). PNAS USA 100: 14994-9; Haber, (2000). Trends Genet 16: 259-264]. Хотя репарация на основе гомологичной рекомбинации может происходить даже при гомологии 30 п.о., намного более эффективно она происходит в случае гомологии 200-400 п.о. [Sugawara et al., (2000). Mol. Cell. Biol. 20: 5300-5309].
Альтернативный способ репарации двунитевого разрыва основан на связывании негомологичных концов, при котором гетеродимер так называемых Ku-белков охватывает разрушенные концы хромосомы, что стимулирует связывание дополнительных белков. Такие дополнительные белки процессируют концы ДНК и лигируют их, что обычно создает делецию нескольких нуклеотидов [Storici et al., (2003), PNAS USA 100: 14994-9].
Способ репарации на основе гомологичной рекомбинации успешно применяли для конструирования плазмиды из двух используемых для совместной трансформации фрагментов ДНК, которые содержали гомологичные области [Ma et al., (1987). Gene, 58: 201-16-26]. Микроорганизмы, и в частности вид S. cerevisiae, являются легко поддающимися обработке организмами для разработки новых способов [работа Kumar and Snyder, (2001), Nat. Rev. Genet. 2: 302-312], в которых осуществляют генетическое изменение либо с использованием двунитевой ДНК, либо с использованием однонитевой ДНК [Orr-Weaver et al., (1981). PNAS USA 78: 6354-6358; Moerschell et al., (1988). PNAS USA 85: 524-528]. S. cerevisiae может принимать и собирать, по меньшей мере, 38 перекрывающихся однонитевых олигонуклеотидов и линейный двунитевой вектор в результате одного события трансформации с перекрываниями между олигонуклеотидами всего лишь 20 пар оснований и длиной 200 нуклеотидов [Gibson, (2009). Nucleic Acids Res. 37: 6984-6990].
Одним из наиболее эффективных средств функциональной характеристики неизвестных генных продуктов является полная делеция генов из хромосомы. Было разработано целенаправленное воздействие на гены с использованием ПЦР-фрагментов, фланкированных короткими гомологичными последовательностями длиной всего 35-40 п.н., которые обеспечивают возможность прямой трансформации вследствие высокой эффективности гомологичной рекомбинации у S. cerevisiae [Baudin et al., (1993). Nucleic Acids Res. 21: 3329-3330; Klinner and Schafer, (2004). FEMS Microbiol. Rev. 28: 201-223]. Однако у лагерных дрожжей S. pastorianus эффективность гомологичной рекомбинации низкая из-за сложной генетики. Поэтому лагерные штаммы требуют более длинных гомологичных перекрывающих фланкирующих участков (>400 п.н.), для того, чтобы имела место эффективная репарация двунитевых разрывов или инсерция делеционной кассеты в геномную ДНК.
Для нокаута множественных генов также необходима рециркуляция маркера. Предыдущие системы для делеции генов и последующего вырезания маркера содержали маркер, фланкированный либо прямыми повторами бактериальной последовательности hisG [Akada et al., (2002). Yeast 19: 393-402], либо двумя сайтами-мишенями сайт-специфичной рекомбиназы [McNabb et al., (1997). Biotechniques, 22: 1134-1139; Storici et al., (1999). Yeast 15: 271-283; Gueldener et al., (2002). Nucleic Acids Res. 30: e23; Iwaki and Takegawa, (2004). Biosci. Biotechnol. Biochem. 68: 545-550]. При использовании такого способа одна копия повторов сохраняется в хромосоме-мишени. Однако когда в геноме присутствует множество остаточных последовательностей, процент правильной интеграции при последующих трансформациях поддающимися негативной селекции кассетами сильно снижается [Davidson and Schiestl, (2000). Curr. Genet 38: 188-190]. Множественные сайты-мишени могут вызывать хромосомные перестройки. Например, когда в геноме одновременно располагались четыре набора повторов loxP, хромосомные перестройки происходили с частотой 50% при экспрессии рекомбиназы CRE. Это означает, что при последующем целенаправленном воздействии на тот же микроорганизм требуется дополнительный скрининг при каждом раунде, чтобы идентифицировать правильный нокаут [Delneri et al., (2000). Gene 252: 127-135].
Для преодоления указанной проблемы использовали систему для бесшовной делеции генов, в которой амплифицированную в ПЦP кассету, содержащую маркер URA3, связанный с дуплицированной последовательностью длиной 40 пар оснований, полученной из локуса-мишени, использовали для нарушения HIS3 и рециркуляции маркера без какого-либо рубцевания генома [Akada et al., (2006). Yeast 23: 399-405]. Колонии подвергали обратной селекции с использованием 5-фтороротовой кислоты для идентификации колоний, в которых утрачен URA3 вследствие рекомбинации между дуплицированными последовательностями длиной 40 пар оснований. Это приводило к делеции HIS3 без остаточных посторонних последовательностей [Akada et al., (2006). Yeast 23: 399-405]. Также было показано, что длинный участок 966 пар оснований необходим для правильного таргетинга гена. Замена такого участка короткой гомологичной последовательностью длиной 40 п.о. приводила к тому, что трансформанты не образовывались [Akada et al., (2006). Yeast 23: 399-405].
Эффективность таргетинга, отчасти, зависит от присутствия длинной гомологичной последовательности в таргетирующей конструкции [Davidson and Schiestl, (2000). Curr. Genet. 38: 188-190]. Однако гомологичную рекомбинацию у некоторых микроорганизмов, таких как, например, большинство штаммов лагерных дрожжей Saccharomyces pastorianus, трудно достичь даже в присутствии длинных гомологичных последовательностей в таргетирующей конструкции (Murakami et al., 2012. Yeast, 29: 155-165).
Когда гомологичная рекомбинация менее эффективна, шанс получения ложноположительных результатов возрастает. Ложноположительные результаты являются результатами случайных одиночных кроссоверных событий.
Настоящее изобретение преодолевает проблему, связанную с эффективным таргетингом, за счет получения набора таргетирующих конструкций, в которых правильная экспрессия селектируемого маркера зависит от события рекомбинации между таргетирующими конструкциями. Было обнаружено, что частота событий рекомбинации между таргетирующими конструкциями заметно возрастает после интеграции таргетирующих конструкций в правильный локус таргетинга. Таким образом, система таргетинга согласно настоящему изобретению, содержащая набор таргетирующих конструкций, в значительной степени повышает процент правильно интегрированных конструкций у микроорганизмов, которые экспрессируют селектируемый маркер, по сравнению с содержащей один вектор системой таргетинга. Расщепление маркера на две части ограничивает получение ложноположительных результатов из-за одиночных событий кроссинговера. Способ на основе расщепленного маркера улучшает отношение истинно положительных результатов к ложноположительным результатам (Nielsen et al., 2006. Fungal Gen. Biol. 43: 54-64).
В изобретении предлагается набор таргетирующих конструкций, содержащий первую конструкцию, содержащую первую область гомологии с геномом-мишенью микроорганизма, сайт узнавания для эндонуклеазы и первую часть селектируемого маркера, и вторую конструкцию, содержащую вторую часть селектируемого маркера, копию сайта узнавания эндонуклеазой и вторую область гомологии с геномом-мишенью микроорганизма, при этом фрагмент первой части селектируемого маркера перекрывается с фрагментом, который присутствует во второй части селектируемого маркера, обеспечивая возможность рекомбинации между первой и второй частями селектируемого маркера; при этом кодирующая последовательность, которая кодирует эндонуклеазу и которая связана с индуцируемым промотором, присутствует в первой или во второй конструкции; и при этом часть первой области гомологии с геномом-мишенью в первой конструкции дуплицирована между копией сайта узнавания эндонуклеазой и второй областью гомологии с геномом-мишенью во второй конструкции; или часть второй области гомологии с геномом-мишенью во второй конструкции дуплицирована между первой областью гомологии с геномом-мишенью и сайтом узнавания эндонуклеазой в первой конструкции.
Каждая из указанных первой и второй областей гомологии с геномом-мишенью содержит, по меньшей мере, 20 пар оснований (п.о.). В принципе, не существует верхнего предела длины указанных первой и второй областей гомологии. Однако, по практическим причинам, таким как простота и эффективность создания первой и второй конструкций, указанные первая и вторая области гомологии предпочтительно содержат от 20 п.о. до 100 т.п.о., более предпочтительно от 40 п.о. до 10 т.п.о., более предпочтительно от 50 п.о. до 5 т.п.о., более предпочтительно от 100 п.о. до 1 т.п.о..
Указанная дуплицированная область гомологии с геномом-мишенью в первой и второй таргетирующих конструкциях предпочтительно имеет длину от 20 до 500 п.о., предпочтительно от 20 до 200 п.о., предпочтительно от 40 до 100 п.о., предпочтительно примерно 80 п.о.. Указанная дуплицированная область гомологии с геномом-мишенью в первой и второй таргетирующих конструкциях позволяет бесшовное удаление маркера из генома-мишени посредством гомологичной рекомбинации.
Первая конструкция предпочтительно содержит в указанном порядке первую область гомологии с геном-мишенью микроорганизма, сайт узнавания для эндонуклеазы и первую часть селектируемого маркера. Вторая конструкция предпочтительно содержит в указанном порядке вторую часть селектируемого маркер, кодирующую последовательность, которая кодирует эндонуклеазу и которая связана с индуцируемым промотором, копию сайта узнавания эндонуклеазой, копию части первой области гомологии с геном-мишенью, которая присутствует в первой конструкции, и вторую область гомологии с геном-мишенью микроорганизма. Такая конфигурация изображена на фигуре 1.
Термин «конструкция» в используемом в настоящем описании смысле относится к искусственно сконструированному участку нуклеиновой кислоты. Предпочтительной конструкцией является вектор, предпочтительно вектор, который содержит бактериальные гены резистентности для роста в бактериях. Наиболее предпочтительной конструкцией является плазмида, линейная или кольцевая двунитевая ДНК, которая способна реплицироваться в бактериях независимо от хромосомной ДНК.
Геном-мишенью может быть любой ген микроорганизма, предпочтительно дрожжей, геномную последовательность которого необходимо изменить. Термин «ген» в используемом в настоящем описании смысле относится к части генома микроорганизма, которая содержит интронные и экзонные части гена, область промотора указанного гена и геномные последовательности, которые опосредуют экспрессию указанного гена, такие как, например, энхансерные последовательности.
Специалисту будет понятно, что таргетирующие конструкции предпочтительно можно применять для изменения гена микроорганизма. Поэтому изобретение также относится к набору таргетирующих конструкций, который содержит первую конструкцию, содержащую первую область гомологии с геном-мишенью микроорганизма, сайт узнавания для эндонуклеазы и первую часть селектируемого маркера, и вторую конструкцию, содержащую вторую часть селектируемого маркера, копию сайта узнавания эндонуклеазой и вторую область гомологии с геном-мишенью микроорганизма, при этом фрагмент первой части селектируемого маркера перекрывается с фрагментом, который присутствует во второй части селектируемого маркера, обеспечивая возможность рекомбинации между первой и второй частями селектируемого маркера; при этом кодирующая последовательность, которая кодирует эндонуклеазу и которая связана с индуцируемым промотором, присутствует в первой или во второй конструкции; и при этом часть первой области гомологии с геном-мишенью в первой конструкции дуплицирована между копией сайта узнавания эндонуклеазой и второй областью гомологии с геном-мишенью во второй конструкции; или часть второй области гомологии с геном-мишенью во второй конструкции дуплицирована между первой областью гомологии с геном-мишенью и сайтом узнавания эндонуклеазой в первой конструкции. Указанная дуплицированная область гомологии с геном-мишенью в первой и второй таргетирующих конструкциях предпочтительно имеет длину от 20 до 200 п.о., предпочтительно от 40 до 100 п.о., предпочтительно примерно 80 п.о..
Термин «изменение» геномной последовательности включает замену одного или нескольких нуклеотидов, инсерцию одного или нескольких нуклеотидов и/или делецию одного или нескольких нуклеотидов в любом месте генома, предпочтительно в гене.
Например, если первая и вторая области гомологии с геном-мишенью содержат соседние геномные последовательности гена, замена одного или нескольких нуклеотидов в первой области гомологии и/или во второй области гомологии будет приводить к изменению гена после гомологичного таргетинга с использованием набора таргетирующих конструкций согласно изобретению. Указанную замену одного или нескольких нуклеотидов предпочтительно осуществляют в области гомологии с геном-мишенью, которая присутствует в первой и во второй конструкциях.
Указанным изменением геномной последовательности предпочтительно является делеция одного или нескольких нуклеотидов, предпочтительно в любом месте внутри гена. Например, если первая и вторая области гомологии с геном-мишенью содержат геномные последовательности гена, которые разделены в геноме организма, изменение гена после гомологичного таргетинга с использованием набора таргетирующих конструкций согласно изобретению будет приводить к делеции области, которая была локализована между первой и второй областями гомологии в исходной хромосоме.
Указанным микроорганизмом предпочтительно является анеуплоидный микроорганизм, предпочтительно анеуплоидные дрожжи. Термин «анеуплоидия» в используемом в настоящем описании смысле относится к наличию аномального количества хромосом в клетке или в организме, которое отличается от нормального количества хромосом для такого организма. Анеуплоидный микроорганизм может иметь одну или несколько дополнительных хромосом или может утрачивать одну или несколько хромосом. Термин «анеуплоидный микроорганизм» включает полиплоидный микроорганизм. Известно, что у грибов анеуплоидия придает резистентность к противогрибковым лекарственным средствам и обеспечивает быструю адаптивную эволюцию [Calo et al., (2013). PLoS Pathog. 9(3): e1003181].
Указанным микроорганизмом, предпочтительно анеуплоидным микроорганизмом, предпочтительно является организм вида Ascomycota, предпочтительно Saccharomycotina, предпочтительно Saccharomyces sensu stricto (Saccharomyces paradoxus, S. mikatae, S. bayanus, S. eubayanus, S. kudriavzevii, S. paradoxus, S. arboricolus), Kazachstania, Naumovozyma, Nakaseomyces, Vanderwaltozyma, Zygosaccharomyces, Lachancea, Kluyveromyces, Eremothecium, Torulaspora, Ogataea, Debaryomyces, Clavispora, Candida, Komagataella, и/или Yarrowia. Предпочтительным организмом являются лагерные дрожжи Saccharomyces pastorianus.
S. pastorianus предположительно является гибридом S. cerevisiae и S. eubayanus. Размер генома S. pastorianus до 60% больше, чем размер генома S. cerevisiae, и содержит крупные части двух геномов. S. cerevisiae содержит гаплоидный набор из 16 хромосом размером в диапазоне от 200 до 2200 т.п.о.. Размер генома S. pastorianus составляет 24-50 миллионов оснований. Дополнительно сообщалось, что анеуплоидными видами Saccharomyces являются S. monacensis и S. uvarum. Обзор полиплоидных грибов приведен в публикации Albertin and Marullo, (2012). Proc. R Soc. B 279: 2497-2509.
Указанным селектируемым маркером предпочтительно является ауксотрофный селектируемый маркер или доминантный селектируемый маркер, которые известны специалисту. Предпочтительные ауксотрофные маркеры включают URA3, KlURA3, CaURA3, HIS3, his5, LEU2, KlLEU2, LYS2, TRP1, ADE1, ADE2 и MET15. Предпочтительные доминантные маркеры включают KanMX, Sh ble, hph, CUP1, SFA1, dehH1, PDR3-9, AUR1-C, nat, pat, ARO4-OFP, SMR1, FZF1-4 и DsdA. Сводные данные о предпочтительных маркерах, которые обычно применяют для организмов дрожжей, приведены в таблице 1.
Указанная первая конструкция предпочтительно содержит первую часть, предпочтительно первые две трети или первую половину, области, которая кодирует селектируемый маркер. Например, URA3, также называемый YEL021W, кодирует фермент оротидин-5'-фосфат (OMP)-декарбоксилазу, которая катализирует шестую ферментативную стадию биосинтеза пиримидинов de novo, превращая OMP в уридинмонофосфат (UMP). Кодируемый белок содержит 267 аминокислот, которые кодируются последовательностью нуклеиновой кислоты из 801 пар оснований (п.о.). Указанная первая конструкция предпочтительно содержит от 200 до 600 п.о. кодирующей области URA3, более предпочтительные от 300 до 500 п.о.. Вторая конструкция предпочтительно содержит от 200 до 600 п.о. кодирующей области URA3, более предпочтительные от 300 до 500 п.о..
Область перекрывания между первой и второй частями селектируемого маркера предпочтительно составляет примерно от 20 п.о. до 800 п.о., предпочтительно примерно от 50 п.о. до примерно 600 п.о., предпочтительно около 200 п.о..
Предпочтительным селектируемым маркером является URA3. URA3 кодирует оротидин-5-фосфатдекарбоксилазу (ODC-азу), которая представляет собой фермент, который катализирует реакцию, которая заключается в синтезе пиримидиновых рибонуклеотидов для РНК дрожжей. Утрата активности ODC-азы приводит к отсутствию клеточного роста в случае, когда урацил или уридин не добавлены в среду. Когда присутствует функциональный ген URA3, ауксотрофные микроорганизмы могут расти в отсутствие урацила и/или уридина. Напротив, добавление 5-фтороротовой кислоты в присутствии функционального гена URA3 приводит к образованию токсичного соединения, вызывающего гибель микроорганизмов. Поэтому URA3 позволяет осуществлять и положительных и отрицательную селекцию.
Следующий предпочтительный селектируемый маркер обеспечивается присутствием нуклеотидной последовательности, кодирующей либо агматинуреогидролазу (агматиназу) (EC.3.5.3.11), либо гидролазу гуанидинокислоты (гуанидинобутиразу; EC.3.5.3.7). Микроорганизмы, предпочтительно из семейства Saccharomytacea, включая штаммы S. cerevisiae, не способны расти на гуанидинобутирате и/или агматине в качестве единственного источника азота. Оба фермента, гидролаза гуанидинокислоты и агматиназа, катализируют образование мочевины, источника азота, обычно усваиваемого такими микроорганизмами, как S. cerevisiae. Таким образом, агматиназа и гуанидинобутираза являются важными характеристиками потенциального доминантного селектируемого маркера «приобретения функции» у таких микроорганизмов, как S. cerevisiae, при выращивании на гуанидинобутирате и/или агматине в качестве единственного источника азота. Предпочтительный ген гуанидинобутиразы кодирует белок, содержащий аминокислотную последовательность GenBank XP_456325.1 или ее ферментативно активную часть. Предпочтительный ген агматинуреогидролазы кодирует белок, содержащий аминокислотную последовательность GenBank AAC75974.1 или ее ферментативно активную часть.
Указанный селектируемый маркер связан с промотором, который управляет экспрессией селектируемого маркера в микроорганизме, и терминатором, который опосредует эффективное образование 3ʹ-концов мРНК. Указанным промотором предпочтительно является промотор дрожжей, предпочтительно промотор дрожжей, выбранный из промоторов гликолитических генов PGI1, PFK1, PFK2, FBA1, TPI1, TDH1, TDH3, PGK1, GPM1, ENO1, ENO2 и из промоторов ACT1, TEF1, AgTEF2, PMA1. Указанный промотор также можно использовать для экспрессии доминантного селектируемого маркера. Терминаторы ряда генов известны специалисту и были использованы, например, в экспрессирующих векторах, включая терминаторы генов CYC1, TRP1, ADH1, MFl, FLP и D (Romanos et al., 1992. Yeast 8: 423-488).
Первая или вторая таргетирующая конструкция содержит кодирующую последовательность, которая кодирует эндонуклеазу и которая связана с индуцируемым промотором. Эндонуклеаза предпочтительно представляет собой редкощепящую эндонуклеазу, такую как, например, PacI (последовательность узнавания мишени 5'-TTAATTAA); AscI (последовательность узнавания мишени 5ʹ-GGCGCGCC), и AsiSI (последовательность узнавания мишени 5'-GCGATCGC). PacI, AscI и AsiSI доступны из New England Biolabs. Эндонуклеаза более предпочтительно представляет собой хоминг-эндонуклеазу. Термин «хоминг-эндонуклеаза» относится к эндонуклеазам, которые кодируются либо отдельными генами с интронами, либо в виде слияния с белком хозяина, либо в виде подвергающегося самосплайсингу интеина. Предпочтительный список хоминг-эндонуклеаз приведен в таблице 2. Дополнительными примерами хоминг-нуклеаз являются I-DirI, I-NjaI, I-NanI, I-NitI, F-TevI, F-TevII, F-CphI, PI-MgaI, I-CsmI, которые все известны специалисту. Дополнительные примеры хоминг-нуклеаз приведены Benjamin K. (заявка на выдачу патента US2012/052582), которая включена в настоящее описание в виде ссылки.
Предпочтительной хоминг-нуклеазой является PI-PspI (New England Biolabs; последовательность узнавания 5ʹ-TGGCAAACAGCTATTATGGGTATTATGGGT)) или PI-SceI (New England Biolabs; последовательность узнавания 5ʹ-ATCTATGTCGGGTGCGGAGAAAGAGGTAAT). Кодирующие последовательности большинства хоминг-эндонуклеаз известны. Например, кодирующие последовательности PI-SceI и PI-PspI доступны из баз данных общего пользования (GenBank, номер доступна Z74233.1, и Genbank, номер доступна U00707.1, соответственно). Специалисту будет понятно, что последовательность, которая отличается от общедоступной последовательности для нуклеазы, все еще может кодировать нуклеазу. Например, термин «кодирующая область PI-PspI» включает последовательность, которая имеет отклонения от общедоступной последовательности, например, вследствие оптимизации кодонов, но которая все еще экспрессирует активную эндонуклеазу, которая узнает и расщепляет указанную последовательность узнавания мишени.
Указанная эндонуклеаза находится под контролем индуцируемого промотора. Термин «индуцируемый промотор» в используемом в настоящем описании смысле относится к промотору, экспрессию которого можно регулировать. Индуцируемые промоторы известны специалисту. Примерами индуцируемых промоторов, которые были использованы у дрожжей, являются промотор GAL1 и промотор GAL10, которые индуцируются галактозой, промотор SUC2, который индуцируется сахарозой, промотор MAL12, который индуцируется мальтозой; промотор CUP1, который индуцируется медью, и промоторы tetO7 и tetO2, которые индуцируются тетрациклином [Gari et al., (1997). Yeast 13: 837-48; Yen et al., (2003). Yeast 20: 1255-62]. Предпочтительным индуцируемым промотором является промотор GAL1.
Один сайт узнавания, содержащий последовательность узнавания мишени для эндонуклеазы, располагают вблизи (позади) первой области гомологии с геном-мишенью микроорганизма в первой конструкции. Копию такого сайта узнавания располагают вблизи (перед) второй областью гомологии с геном-мишенью микроорганизма во второй конструкции. Специалисту будет понятно, что когда часть первой области гомологии с геном-мишенью в первой конструкции дуплицируют между копией сайта узнавания эндонуклеазой и второй областью гомологии с геном-мишенью во второй конструкции, указанную копию сайта узнавания располагают вблизи (перед) дупликации первой области гомологии с геном-мишенью во второй конструкции. Альтернативно, сайт узнавания располагают вблизи (позади) дуплицированной части второй области гомологии с геном-мишенью в первой конструкции, когда часть второй область гомологии с геном-мишенью во второй конструкции дуплицируют в первой конструкции. Селектируемый маркер, включая последовательности промотора и терминатора, и кодирующую область эндонуклеазы, включая индуцируемый промотор, располагают между сайтом узнавания в первой конструкции и копией такого сайта узнавания во второй конструкции.
Изобретение, кроме того, относится к способу изменения генома, предпочтительно гена-мишени, у микроорганизма, включающему обеспечение поступления набора таргетирующих конструкций согласно изобретению к указанному микроорганизму и отбор микроорганизма, в котором был изменен геном. Указанный отбор микроорганизма, в котором был изменен геном, предпочтительно осуществляют в результате отбора микроорганизма, который функционально экспрессирует рекомбинированный селектируемый маркер.
Как указано в настоящем описании выше, частота событий рекомбинации между таргетирующими конструкциями заметно повышается после интеграции таргетирующих конструкций в правильный локус таргетинга. Поэтому наличие функционального рекомбинируемого селектируемого маркера является четким показателем присутствия правильно интегрированных таргетирующих конструкций в геном-мишень и, следовательно, показателем измененного генома у микроорганизма.
Как указано в настоящем описании выше, термины «процесс изменения», «изменение» и «измененный» относятся к замене одного или нескольких нуклеотидов, инсерции одного или нескольких нуклеотидов и/или делеции одного или нескольких нуклеотидов в любом месте в гене-мишени.
Замена одного или нескольких нуклеотидов может быть осуществлена в результате изменения одного или нескольких нуклеотидов в первой область гомологии и/или во второй области гомологии. Когда первая область гомологии и вторая область гомологии охватывают соседние области генома, предпочтительно ген-мишень, интеграция таргетирующих векторов будет приводить к изменению генома. При наличии указанная замена одного или нескольких нуклеотидов предпочтительно осуществляется за счет изменения одного или нескольких нуклеотидов в перекрывающейся области гомологии в геноме, которая присутствует в первая и во второй конструкции.
Указанным изменением геномной последовательности предпочтительно является делеция одного или нескольких нуклеотидов в любом месте в геноме, предпочтительно в гене. Например, если первая и вторая области гомологии с геном-мишенью содержат геномные последовательности, которые разделены в геноме организма, то изменение генома после гомологичного таргетинга с использованием набора таргетирующих конструкций согласно изобретению будет приводить к делеции области, которая локализована между первой и второй областями гомологии в исходной хромосоме.
Изобретение, кроме того, относится к способу получения микроорганизма, содержащего измененный геном, предпочтительно измененный ген, при этом способ включает обеспечение поступления набора таргетирующих конструкций согласно изобретению к указанному микроорганизму и отбор микроорганизма, в котором был изменен геном и который функционально экспрессирует рекомбинированный селектируемый маркер.
Способ получения микроорганизма, содержащего измененный геном, предпочтительно включает индукцию индуцируемого промотора для экспрессии эндонуклеазы с удалением при этом селектируемого маркера и кодирующей области эндонуклеазы, включая индуцируемый промотор, из генома-мишени.
Изобретение, кроме того, относится к микроорганизму, содержащему геномное изменение, который получен способами согласно изобретению. В случае наличия, дуплицированные области гомологии с геномом-мишенью в первой и второй таргетирующих конструкциях обеспечивают бесшовное удаление маркера из генома-мишени в результате гомологичной рекомбинации. Получаемый в результате микроорганизм содержит только изменение или изменения, которые имелись в первой и/или второй таргетирующей конструкции или которые были индуцированы рекомбинацией таргетирующих конструкций с подвергаемым таргетингу геномом, такие как инсерция в подвергаемый таргетингу геном или делеция из подвергаемого таргетингу генома.
Изобретение, кроме того, относится к микроорганизму, содержащему геномное изменение, предпочтительно изменение гена-мишени, при этом изменение включает инсерцию функционально рекомбинируемого селектируемого маркера и кодирующей последовательности для эндонуклеазы, которая связана с индуцируемым промотором, при этом геном-мишень содержит одну копию последовательность узнавания для эндонуклеазы в обоих сайтах инсерции.
Изобретение, кроме того, относится к способу получения микроорганизма, содержащего измененный геном, при этом способ включает получение микроорганизма, содержащего изменение генома, предпочтительно гена-мишени, при этом изменение включает инсерцию функционально рекомбинируемого селектируемого маркера и кодирующей последовательности для эндонуклеазы, которая связана с индуцируемым промотором, при этом геном-мишень содержит одну копию последовательности узнавания для эндонуклеазы в обоих сайтах инсерции, и индукцию индуцируемого промотора для удаления последовательностей нуклеиновой кислоты между последовательностями узнавания эндонуклеазой. И снова, в случае наличия, дуплицированные области гомологии с геном-мишенью в первой и второй таргетирующих конструкциях обеспечивают бесшовное удаление маркера из генома-мишени в результате гомологичной рекомбинации посредством обеспечения геномной ДНК небольшим гомологичным участком для эффективного повторного связывания разрушенных нитей ДНК. Получаемый в результате микроорганизм содержит только изменение или изменения, которые имелись в первой и/или второй таргетирующей конструкции или которые были индуцированы рекомбинацией таргетирующих конструкций с подвергаемым таргетингу геномом, такие как инсерция в подвергаемый таргетингу геном или делеция из генома, предпочтительно инсерция в подвергаемый таргетингу ген или делеция из подвергаемого таргетингу гена или делеция в пределах подвергаемого таргетингу гена.
Langlerouault F & Jacobs E (1995) A method for performing precise alterations in the yeast genome using a recyclable selectable marker. Nucleic Acids Res 23: 3079-3081.
Swinkels B, Selten G, Bakhuis J, Bovenberg R & Vollebregt A (1997) The use of homologous amdS genes as selectable markers.
Patent application WO97/06261.
3ʹ AACTCCTCCAAAGAGACATTTATT 5ʹ
3'---AACTCCTCC AAAGAGACATTTATT--- 5'
3' ATTGATATTGCCAGGATTCCATCGCT 5ʹ
3'---ATTGATATTGCCAG GATTCCATCGCT--- 5'
3' CTTCCAAACCGTGGAGCTACAGCCGAGTAG 5ʹ
3'---CTTCCAAACCGTG GAGCTACAGCCGAGTAG---5'
3' GCTAGGATTCCATCGCTTTAAGT 5ʹ
3'---GCTAGGATTCCATC GCTTTAAGT---5'
3' GGGCCGATTGAGACACGGTC 5ʹ
3' GACCCAAGTTTTGCAGCACTCTGTCAAACC 5ʹ
3'---GACCCAAGTTTTGCAG CACTCTGTCAAACC---5'
3'---TACGGAACGGCC CATTCAAGGCCGCGCGTA---5'
3' GTTTTGCAGCATTCAAGGCCGCGC 5'
3' TCATTACTCGGATTGCGAGTCGTT 5ʹ
3'---TCATTACTCGGATTGC GAGTCGTT---5'
3' TCATTACTCGGATTGCGAGTTGTT 5ʹ
3' GTGTAGGTATTGGTATAGTAAAAA 5ʹ
3' GTCATGATGCCAATG5ʹ
3' CGCTCGGGCATTCCCACACATGCCC
3' ATTGATACTGAGAGAATTCCATCGGTTTA
3' ACCGTTTGTCGATAATACCCATAATACCCA
3' ACAGTGTAACTCCACGTGATCAATAATG
3'---ACAGTGTAACTCCAC GTGATCAATAATG---5'
3' TCAATGCGATCCCTATTGTCCCATTATATC
3' AAAACTAAGAAACCAGTGGGACTTCATAT
3' TAACCTCCAAAACCATTGATAAATAATGG
3' AGAAAAGAGAACTAATCGGGATTAGATGC
3' TTATTAAAAGAAGAATCATTACGG
3' CAATAAATTACAAAATCATCAACC
3' ACAGTGTAACTCCACGTGATCAATAATG
3' CAGCCCGAGTATTGGGCTT
3'---CAGCCCGAGTA TTGGGCTT---5'
3' TCACCATAGT TGCGAGTCATCTAC
3' CGAATACTCATACTTCACTTGTGCAATAAG
3'---AT ACATAGAAAACGCACATGGAAATTGAAG---5'
3' ATRCGNCTRTGNCTGCCTAARA
3'---ATRCGNCTRTGNC TGCCTAARA---5'
3' TTTAACGAACGTTTGTCGATAATGCCGATA
3' GAAGTCATACGGGGCTTTG
3'---GAAGT CATACGGGGCTTTG---5'
3' GGACTGAGAGAATTCCATCGGTTT
Таблица 2. Сводные данные о хоминг-эндонуклеазах и их последовательностях-мишенях.
Сокращения: SF (структурное семейство): HI: семейство LAGLIDADG; HII: семейство GIY-YIG; HIII: семейство H-N-H; HIV: семейство His-Cys box.
D: Биологический домен источника: A: археи; B: бактерии; E: эукариоты.
SCL: Субклеточная локализация: хлороп: хлоропласт; Хр: хромосомный; митох: митохондриальный; ядерн: внехромосомный ядерный; фаг: бактериофаг
Надписи к фигурам
Фигура 1. Вектор 1 и 2 со всеми необходимыми частями для стандартной кассеты для делеции. Перекрывание 400 оснований в селектируемом маркере amDs (указано скрещенными линиями) сконструировано для рекомбинации за счет гомологии.
Фигура 2. Ненаправленный клонирующий вектор TOPO Blunt и pUC19, используемые для конструирования плазмиды с векторами 1 и 2.
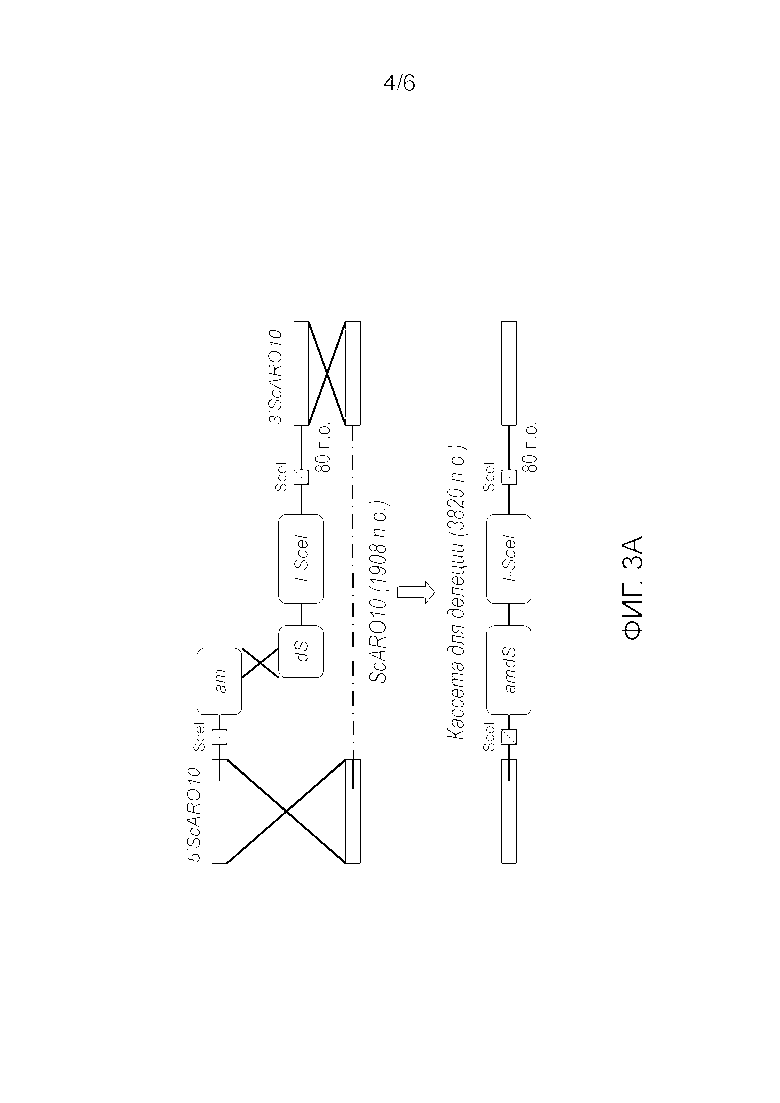
Фигура 3A: Целенаправленная делеция в гене с использованием кассеты amdS-I-SceI с гомологичными фланкирующими участками длиной 500 п.о. для нокаута ScARO10.
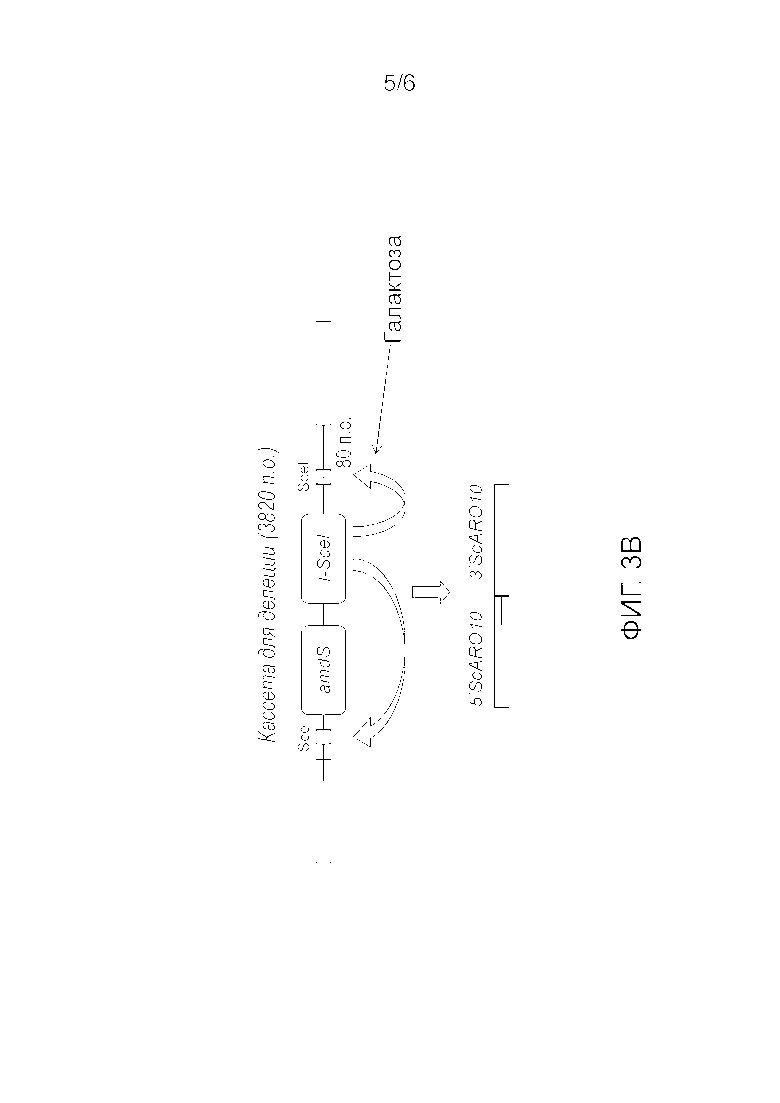
3B: Бесшовное удаление маркера с использованием GAL1p I-SceI для получения активного белка эндонуклеазы I-SceI, которая расщепляет геномную ДНК в сайтах узнавания SceI, введенный в кассету.
Фигура 4. Векторы 1 и 2, содержащие перекрывающиеся фрагменты селектируемого маркера KlGBU1, кодирующего гуанидинобутиразу.
ПРИМЕРЫ
Пример 1
Материалы и способы
Штаммы и условия культивирования
Все штаммы Saccharomyces, используемые в настоящем исследовании, перечислены в таблице I. В случае выращивания в жидкой среде встряхиваемые флаконы помещали в шейкер-инкубатор при 200 об./мин и 30°C. В случае выращивания на чашках все штаммы, за исключением CBS1483, выращивали при 30°C. CBS1483 выращивали при 20°C. Штаммы расти в разных средах: в неселективных условиях дрожжи выращивали в сложной среде с дрожжевым экстрактом, пептоном и декстрозой (YPD), содержащей 10 г/л дрожжевого экстракта, 20 г/л пептона, 22 г/л моногидрата глюкозы, pH 6), использовали синтетическую среду (SM), содержащую 5,0 г/л (NH4)2SO4, 3,0 г/л KH2PO4, 0,5 г/л MgSO4.7H2O, 1 мл/л раствора микроэлементов и 1 мл/л раствора витаминов (Verduyn et al., 1992) [Verduyn et al., (1990). J General Microbiology 136: 395-403].
В случае использования amdSYM (AgTEF2-amdS-AgTEF2ter) в качестве маркера (NH4)2SO4 заменяли на 0,6 г/л ацетамида в качестве источника азота и 6,6 г/л K2SO4, чтобы компенсировать поступление сульфата (SM-Ac). Клетки без рециркулируемого маркера отбирали на SM, содержащей 2,3 г/л фторацетамида (SM-Fac). SM, SM-Ac и SM-Fac при необходимости дополняли 20 мг/л аденина и 15 мг/л сульфата L-канавалина. Во всех экспериментах использовали 20 г/л глюкозы в качестве источника углерода. pH во всех средах доводили до значения 6,0, используя KOH. Твердые среды готовили, добавляя 2% агар к описанной выше среде.
Для индукции GAL1p источник углерода глюкозы заменяли альтернативным источником углерода галактозой в концентрации 20 г/л. При использовании жидкой среды с галактозой рост стимулировали добавлением 0,0125% глюкозы. В целях получения культур для хранения дрожжи, которые имели правильно собранные используемые при трансформации фрагменты в геномной ДНК, выращивали в жидкой культуре YPD или MM. Дрожжи с правильно собранной плазмидой выращивали в селективных условиях в MM-культуре. После достижения достаточного значения OD660 добавляли глицерин, чтобы получить концентрацию 30%, и клетки хранили при -80°C.
ДНК-методики
Полимеразная цепная реакция (ПЦР)
Осуществляли два разных способа ПЦР. В целях секвенирования клонирования применяли высокоточную ДНК-полимеразу Phusion (Finnzymes, Vantaa, Finland), чтобы иметь более точное считывание, поскольку она обладает корректирующей 3ʹ-5ʹ-экзонуклеазной активностью. В случае осуществления селекции (ПЦР дрожжевых колоний) использовали основную смесь для ПЦР DreamTaq (Fermentas GmbH, St. Leon-Rot, Germany), которая не имеет корректирующей 3ʹ-5ʹ-экзонуклеазной активностью.
Виды Saccharomyces, используемые в примере 1.
Entian & Kotter (2007)
Libkind et al. (2011)
a K.U. Leuven, Centre for Malting and Brewing Collection, Centre for Malting and Brewing, Louvain, Belgium.
Ссылки: van Dijken et al., (2000). Enzyme Microb. Technol. 26: 706-714; Entian and Kotter, (2007). 25 Yeast genetic strain and plasmid collections. Academic Press, Amsterdam, the Netherlands. In: Methods in Microbiology (Stansfield I and Stark J, eds) 36: 629-666 The Centraalbureau voor Schimmelcultures (CBS) Fungal Biodiversity Centre, (http://www.cbs.knaw.nl/index.php), Libkind et al., (2011). PNAS USA 108: 14539-14544.
Чтобы создать правильную ДНК-матрицу при осуществлении ПЦР дрожжевых колоний, небольшую часть одной колонии ресуспендировали в 15 мкл 0,02N NaOH и суспензию колонии дрожжей кипятили в течение 10 мин при 100°C. После этого 2 мкл такой клеточной суспензии использовали в качестве матрицы для ПЦР, которую осуществляли с применением основной смеси для ПЦР DreamTaq (Fermentas GmbH, St. Leon-Rot, Germany), следуя рекомендациям производителя.
Высокоточную ПЦР колоний использовали для подтверждения инсерции кассеты для делеции гена в геномную ДНК. Клетки из жидкой дрожжевой культуры (200 мкл) осаждали центрифугированием, ресуспендировали в 100 мкл 0,2 М LiAc с 1% раствором SDS и инкубировали в течение 5 минут при 70°C. После добавления 96% этанола и встряхивания осуществляли осаждение центрифугированием в течение 3 минут при 15000 g. Осадок промывали 70% этанолом и снова центрифугировали. Этанол удаляли и осадок сушили при максимальной температуре 30°C, затем добавляли 100 мкл буфера TE, чтобы растворить осадок. Остатки клеток осаждали центрифугированием в течение 30 секунд при 15000 g и использовали 5 мкл перенесенного надосадка для высокоточной ПЦР колоний.
Все праймеры, используемые в ПЦР, описаны в таблице II.
Последовательности используемых праймеров
Рестрикция
Ферменты рестрикции, используемые в настоящем проекте, были получены от поставщица Fermentas. Смесь для расщепления готовили в общем объеме либо 20 мкл, либо в 30 мкл, в зависимости от требуемого конечного количества. Общая смесь содержала 1 мкл каждого фермента рестрикции (1 единица FastDigest/мкл), 2 или 3 мкл 10-кратного буфера FastDigest и не содержащую нуклеаз деминерализованную воду. Использовали примерно 50-200 нг ДНК в общей смеси объемом 20 мкл и 100 нг - 2 мкг в общей смеси объемом 30 мкл. Смесь для расщепления инкубировали при температуре 37°C.
Гель-электрофорез
Чтобы проанализировать различные ДНК-методики, молекулы нуклеиновых кислот загружали на 1% агарозный гель (окрашенный заранее 0,001% SybrSafe) в 1x-буфере TAE (40 мМ трис-ацетат и 1 мМ EDTA). Все молекулы ДНК разделяли, прикладывая электрическое поле 100 В на 30 минут. Картины геля получали посредством экспозиции в УФ-свете.
Извлечение из геля
Чтобы очистить ДНК из геля использовали набор для экстракции из геля (Sigma-Aldrich, Zwijndrecht, The Netherlands). Представляющий интерес фрагмент ДНК вырезали из агарозного геля лезвием. ДНК экстрагировали, следуя рекомендациям производителя.
Лигирование
Липкие или тупые концы фрагментов ДНК соединяли, используя ДНК-лигазу T4 (Life Technologies Europe BV, Bleiswijk, The Netherlands). Смесь для лигирования готовили согласно стандартному протоколу производителя и инкубировали при 16°C в течение 20 часов. Плазмиды, созданные после лигирования были готовы для трансформации E. coli.
Трансформация E. coli
Небольшой объем плазмид (5 мкл) добавляли к 50 мкл химически компетентных клеток One Shot TOP10 (Life Technologies) и следовали протоколу химической трансформации E. coli, предоставленному поставщиком. Трансформированные клетки инкубировали при 37°C в течение одного часа со встряхиванием, давая возможность клеткам восстановиться и выработать резистентность к используемому антибиотику. Суспензию клеток высевали на предварительно согретые селективные чашки и инкубировали в течение ночи при 37°C. Клетки с плазмидами, получаемые в результате ненаправленного клонирования Blunt End TOPO (Life Technologies), высевали на чашки с LB-агаром (10 г триптона, 5 г дрожжевого экстракта, 10 г NaCl, 15 г/л агара), содержащие 50 мкг/мл канамицина (125 мкл) или 100 мкг/мл ампициллина (125 мкл). Клетки с плазмидами, которые получены в результате разрезании и лигирования в pUC19, высевали штрихом на LB с 100 мкг/мл ампициллина (250 мкл). После инкубации в течение ночи отдельные колонии повторно высевали штрихом на свежие предварительно нагретые селективные чашки, снова выращивая в течение ночи. После повторного посева штрихом был возможен отбор отдельных колоний E. coli без фона. Отдельные колонии инокулировали в 5 мл среды LB с соответствующим антибиотиком для выращивания в течение ночи. Перед выделением плазмид добавляли 200 мкл раствора глицерина к 800 мкл культуры, после этого образец хранили в морозильной камере при -80°C.
Выделение плазмид
Чтобы выделить плазмиды, культуру E. coli с требуемой плазмидой выращивали в течение ночи в среде LB. После осаждения образца объемом 1-5 мл следовали протоколу, прилагаемому к набору Genelute Plasmid Miniprept (Sigma-Aldrich). На последней стали использовали 50 мкл вместо 100 мкл не содержащей нуклеаз деминерализованной воды для элюирования очищенной плазмиды с колонки.
Трансформация дрожжей
Для трансформации дрожжей оптическую плотность (OD) определяли по поглощению, измеряемому с использованием спектрофотометра при длине волны 660 нм. Используя OD660 0,6-0,8 (2×107 клеток/мл для 10 трансформаций) или скорректированное на количество клеток, необходимое для отдельной трансформации, осуществляли трансформацию согласно способу с использованием ацетета лития, однонитевой ДНК-носителя и полиэтиленгликоля [Gietz and Woods, (2002). Methods in Enzymology 350: 87-96]. Дрожжевые клетки высевали штрихом на соответствующе селективные чашки.
Конструирование плазмид
Центральная часть полной кассеты для делеции гена, которая была названа amdS-ISceI, состояла из оптимизированного по кодонам гена amdS и I-SCEI под контролем промотора галактозы 1 (GAL1p), фланкированного сайтами узнавания SceI. Кассета для делеции amdS-ISceI была разделена на два фрагмента, которые содержали перекрывание 400 пар оснований в гене amdS, чтобы осуществить стадию гомологичной рекомбинации существенной части в сборке кассеты in vivo. Последовательности праймеров приведены в таблице II.
Первый фрагмент, названный вектором 1, конструировали с использованием обратного праймера Rv V1 SceI SacI, который связывается с началом кассеты amdSYM в промоторе AgTEF2 и который включал сайт узнавания SceI и сайт рестрикции SacI, и с использованием прямого праймера Fw V1 NdeI, который связывался с концом предполагаемого перекрывания 400 оснований в amdS и включал сайт рестрикции NdeI на 5ʹ-конце (фигура 1). pUGamdS использовали в качестве матрицы для конструирования вектора 1.
Для конструирования плазмиды ПЦР-фрагмент вектор 1, описанный выше, клонировали в pCR4Blunt-TOPO, получая плазмиду pUD266.
Последнюю часть amdS амплифицировали с pUGamdS [Solis-Escalante, D. et al. (2013). FEMS Yeast Research, 13: 126-39], используя прямой праймер Fw V2 NdeI, который связывался с началом предполагаемого перекрывания 400 пар оснований и включал сайт рестрикции NdeI в ПЦР-продукт, и обратный праймер Rv V2-overlap-ISceI, чтобы создать перекрывание 60 пар оснований в промоторе GAL1, который контролировал экспрессию I-SCEI. Такой фрагмент был назван вектором 2A. Кассету экспрессии GAL1p-I-SCEI амплифицировали с pUDC073 (Kuijpers et al., 2013. FEMS Yeast Research. 13: 769-81) с использованием прямого праймера Fw V2 ISceI и обратного праймера Rv V2 SceI EcoRI, которые включали сайт SceI с другой стороны кассеты и сайт EcoRI. Такой фрагмент был назван вектором 2B. ПЦР-продукты, векторы 2A и 2B, сливали посредством ПЦР с использованием Fw V2 NdeI и Rv V2 SceI EcoRI. ПЦР осуществляли, смешивая равные молярные количества обоих фрагментов с получением слитого фрагмента, названного вектором 2 (фигура 1). ПЦР-фрагмент вектор 2 клонировали в pCR4Blunt-TOPO, получая pUD267. Плазмиды проверяли в рестрикционном анализе, чтобы подтвердить ориентацию клонированного фрагмента.
Кассета для делеции гена
Для конструирования конечных фрагментов для делеции гена последовательности длиной 500 пар оснований из 5ʹ- и 3ʹ-концов ScARO10, гена, который кодирует фенилпируватдекарбоксилазу, вовлеченную в путь Эрлиха [Vuralhan. et al., (2005). Appl. Environ. Microbiol. 71: 3276-3284] амплифицировали с геномной ДНК CBS1483.
Стандартные плазмиды для конструирования векторов
В плазмидах pUD268 и pUD269 два полных фрагмента для делеции гена были расположены между прямым и обратным праймерами M13, которые использовали для амплификации кассеты для делеции amdS-ISceI.
При трансформации разных штаммов в смеси для трансформации дрожжей использовали молярные эквиваленты двух фрагментов, при этом общее количество ДНК составляло 837 нг: 337 нг вектора 1 с 5ʹ-ScARO10 и 500 нг вектора 2 с 3ʹ-ScARO10. В случае S. pastorianus CBS1483 трансформацию повторяли, используя 3,37 мкг и 5 мкг обоих указанных частей, соответственно.
Результаты
Рекомбинация для сборки in vivo pUDC114
Хотя имеется огромное количество данных, которые можно найти, об эффективности трансформации в CEN.PK113-7D, нет сравнимых данных о гомологичной рекомбинации с использованием 60 пар оснований, доступных для штаммов, используемых в данном исследовании. Поскольку полиплоидия и хромосомные перестройки могут влиять на способность клеток осуществлять гомологичную рекомбинацию, три разновидности S. cerevisiae с разной плоидностью трансформировали перекрывающимися фрагментами, перечисленными в таблице II, чтобы создать плазмиду pUDC114: гаплоидный CEN.PK113-7D, образующий IMC067, диплоидный CEN.PK122, создающий IMC076, и полиплоидный PRG410, создающий IMC077. Кроме того, полиплоидные штаммы S. pastorianus CMBS33 и CBS1483 и недавно открытый диплоидный S. eubayanus CBS12357 [Libkind et al., (2011). PNAS USA 108: 14539-14544] исследовали в отношении способности продуцировать плазмиду pUDC114 с образованием IMC064 в случае CBS12357 и IMC066 в случае CMBS33. В случае CBS1483 ожидали, что сборка не произойдет из-за короткой длины гомологичных последовательностей.
Штаммы IMC067, IMC076, IMC077, IMC066 и IMC064 были способны к рекомбинации плазмиды pUDC114 с получением гена amdS и ростом на ацетамиде в качестве единственного источника азота. Как и предполагали, CBS1483 не был способен рекомбинировать перекрывающиеся последовательности длиной 60 пар оснований с образованием плазмиды pUDC114. Вычисляли количество трансформантов на мкг ДНК. Результаты показаны в таблице VI.
Таблица VI: Средние эффективности трансформации, измеренные в виде количества трансформантов на мкг ДНК со стандартной ошибкой среднего (SEM). Трансформацию PRG410 для создания IMC077 осуществляли несколько раз, но только одна чашка содержала позитивные оранжевые колонии *
Более 2*104 трансформантов на мкг ДНК было образовано в случае гаплоидного IMC067, приблизительно 6*103 трансформантов на мкг ДНК было получено в случае диплоидного IMC076 и в случае IMC077 только 19 трансформантов на мкг ДНК было создано из полиплоидного штамма верхнего брожения PRG410. Таким образом, с увеличением плоидности разных штаммов S. cerevisiae количество трансформантов на мкг ДНК значимо уменьшается в соответствии с данными, представленными в таблице VI. В случае диплоидного S. eubayanus CBS12357 дикого типа количество трансформантов на мкг ДНК также было более чем в 10 раз меньше, чем в случае диплоидного лабораторного штамма CEN.PK122 (таблица VI). Штамм лагерного брожения S. pastorianus CMBS33 был не очень эффективным по сравнению с диплоидным лабораторным штаммом S. cerevisiae и только немного менее эффективным, чем другой исходный штамм S. eubayanus CBS12357.
Конструирование стандартных векторов.
Чтобы создать состоящий из двух частей селектируемый маркер, маркер amdS разделяли на две части, которые имели перекрывание 400 пар оснований в ORF amdS. Первую часть получали амплификацией промотора TEF2 Ashbya gossipii и первых 1138 нуклеотидов открытой рамки считывания amdS из pUGamdSY. Полученный в результате фрагмент из 1569 пар оснований содержал на своем 5ʹ-конце сайт рестрикции эндонуклеазой SceI. Клонирование такого фрагмента в плазмиде pCR4Blunt-TOPO давало pUD266. Вторую часть состоящей из двух частей маркерной системы конструировали в две стадии: 1) во вторую часть маркера включали последние 908 пар оснований селектируемого маркера amdS и терминатор AgTEF2. Такая кассета была фланкирована на 3ʹ-конце удлинением 60 п.о., комплементарным кассете SCEI. 2) Кассету эндонуклеазы SCEI, которая несла ген SCEI под контролем промотора GAL1, амплифицировали с pUDC073. На своем 3ʹ-конце такой фрагмент содержал дополнительный сайт рестрикции эндонуклеазой SceI. Затем оба фрагмента соединяли вместе посредством ПЦР со слиянием и полученный слитый фрагмент клонировали в векторе pCR4Blunt-TOPO, получая pUDC267.
Чтобы контролировать направление интеграции, осуществляли рестрикционный анализ на pUDC266 с двойным расщеплением NotI и BamH1, при этом Not1 разрезала внутри плазмиды pCR4Blunt-TOPO, а BamHI разрезала внутри вектора 1, что приводило к появлению характерной картины полос 438 и 5087 пар оснований. Для контроля направления интеграции в pUD267 осуществляли рестрикционный анализ с использованием расщепления NotI и HindIII, приводящего к характерной картине полос 815 и 5732 пар оснований.
Состоящий из двух частей фрагмент, находящийся в pUDC266, секвенировали и не выявили мутации.
Рестрикционные анализы и анализы последовательностей подтвердили корректность плазмид и то, что не произошли мутации в сконструированной последовательности. Однако анализ pUDC267 и полученной консенсусной последовательности показал наличие нескольких однонуклеотидных полиморфизмов (SNP). Один SNP действительно приводил к изменению первого нуклеотида кодона из amdS, изменяя аминокислоту триптофан на аргинин. Даже несмотря на то, что это означает замену гидрофобной аминокислоты на положительно заряженную (следовательно гидрофильную) аминокислоту, не было обнаружено изменения эффективности обусловленного ацетамидазой роста. Другие SNP и даже добавление 7 нуклеотидов были вне последовательностей amdS и I-SceI.
Получение кассеты для делеции.
Хотя центральная часть кассеты для делеции гена amdS-ISceI лигировали в стандартные векторы pUD266 и pUD267, последовательности для гомологичной рекомбинации не были интегрированы в кассету для делеции. Для получения фрагментов для гомологичной рекомбинации геномную ДНК CBS1483 амплифицировали с использованием Fw 5ʹScARO10 NotI и Rv 5ʹScARO10 SacI в случае расположенной выше части ScARO10 и с использованием Fw 3ʹScARO10 BamHI 80 п.о. и Rv 3ʹScARO10 PstI в случае расположенной ниже части. pUDC266 и pUD267 расщепляли NotI/SacI и PstI/PmeI и лигировали с 5ʹ-фрагментом (NotI/SacI) и 3ʹ-фрагментом (полученным с использованием PstI), соответственно. Такие лигирования создавали два новых вектора pUD268, несущих первый элемент селектируемой кассеты, направленный к 5ʹ-стороне локуса ScARO10, и pUD269, несущий второй элемент селектируемой кассеты, направленный к 3ʹ-стороне локуса ScARO10.
Делеция аллеля ScARO10.
Для доказательства принципа делеции гена и удаления маркера с применением нового способа с использованием кассеты amdS-ISceI с гомологичными фланкирующими участками длиной 500 пар оснований, первая стадии заключается в успешной делеции генов в нескольких штаммов. Целенаправленная делеция гена с использованием двух предварительно созданных кассет состояла из трех важных стадий рекомбинации, основанной на гомологии (фигура 3A).
Нефункциональные части amdS в обоих стандартных векторах требовалось рекомбинировать и создать функциональный селектируемый ген amdS для роста на ацетамиде. Другие две стали заключались в рекомбинация между 5ʹ-ScARO10- и 3ʹ-ScARO10-фрагментами на кассете и такими сайтами в геномной ДНК.
Чтобы оценить, можно ли применять такой новый способ делеции гена и удаления маркера по отношению к S. pastorianus, тестировали несколько штаммов. В качестве контроля трансформировали три штамма Saccharomyces cerevisiae, лабораторный гаплоидный штамм MATa CEN.PK113-7D, лабораторный диплоидный штамм MATa/MATa CEN.PK122 и промышленный штамм для верхнего брожения PRG410. Наряду с указанными штаммами дрожжей также трансформированы штаммы Saccharomyces pastorianus CBS1483 и CMBS33.
Делеция аллеля ScARO10 в штаммах семейства CEN.PK с использованием основанного на использовании двух частей подхода давала более 200 трансформантов на мкг ДНК. Напротив, трансформация промышленного штамма PRG410 давала намного меньшее количество трансформантов (0,4 трансформанта на мкг ДНК).
Тогда как делеция с использованием кассеты для делеции с созданными в ПЦР короткими фланкирующими участками была неуспешной в случае S. pastorianus CBS1483, применение способа на основе использования двух частей делало возможным проведение успешной идентификации мутантов с правильными делециями. Однако количество трансформантов оставалось низким (0,5 трансформанта на мкг ДНК) (таблица VII). Типичная трансформация CBS1483 с использованием 8,37 мкг состоящего из двух частей маркера amdS, направленного на локус ScARO10, приводила к росту 2-9 трансформантов на чашках со средой с 30 мМ ацетамида.
В случае трансформации, приводившей в результате к девяти трансформантам CBS1483, две позитивные колонии повторно высевали штрихом, чтобы получить истинные одиночные колонии, а другие 7 колоний проверяли, используя высокоточную ПЦР на колониях с наружными-внутренними праймерами Fw 5ʹScARO10/amdS и KanB r, создавая фрагмент длиной 2851 пар оснований. Все они были позитивными в отношении гомологичной рекомбинации между 5ʹ-частью ScARO10 и перекрытием 400 пар оснований в amdS, что свидетельствует о том, что все кассеты для делеции были успешно интегрированы. Был получен отдельный изолят колонии и переименован в IMK490 (CBS1483 с одним делетированным локусом Scaro10Δ::amdS).
Подобным образом получили отдельные изоляты колоний из CEN.PK113-7D, CEN.PK122, PRG410, CMBS33 и переименовали в IMK386, IMK487, IMK488 и IMK489 соответственно.
Средние эффективности трансформации, измеренные в виде количества трансформантов на мкг ДНК со стандартной ошибкой среднего (SEM)
Чтобы абсолютно гарантировать, что кассета для делеции была успешно интегрирована в целевую геномную ДНК, гомологичную рекомбинацию области перекрывания в amdS также контролировали с использованием праймеров amdS KanA f и KanB r (2164 п.о.) и гомологичную рекомбинацию 3ʹ-ScARO10 с использованием внутренних-наружных праймеров FK072 и Rv 3ʹScARO10/amdS check (1081 п.о.). Поскольку каждый штамм содержит более одной копии ScARO10, за исключением CEN.PK113-7D, присутствие других копий ScARO10 контролировали с использованием праймеров внутреннего ScARO10-Fw и внутреннего ScARO10-Rv (959 п.о.).
Результаты показали, что все штаммы, используемые в данном проекте, были способны к нокауту, по меньшей мере, одной копии ScARO10. CEN.PK113-7D scaro10Δ::amdS, другие штаммы, включая обе колонии, проверенные в случае IMK490, все еще имели, по меньшей мере, одну копию ScARO10 в своем геноме. Все колонии IMK486-IMK490 хранили и образец каждой из них повторно инокулировали в YPD, чтобы вырастить для второй стадии, удаления маркера.
Бесшовное удаление маркера.
Чтобы подтвердить, что интегрированная состоящая из двух частей конструкция может быть легко удалена из локуса-мишени с использованием индуцируемой эндонуклеазы SCEI, штамм IMK490 (CBS1483 с одним делетированным локусом Scaro10Δ::amdS) выращивали на среде с галактозой, чтобы индуцировать промотор GAL1, который является промотором, контролирующим экспрессию SCEI (фигура 3B). Штамм IMK490 выращивали на синтетической среде с галактозой в течение 48 часов. После индукции эндонуклеаза создает разрез в сайтах SceI, которые фланкируют кассету для делеции, и в то же время удаляет ее кодирующую область из хромосомы, таким образом обеспечивая рециклинг редактирующей геном конструкции.
Штаммы IMK386, IMK487, IMK488 и IMK489 обрабатывали сходным образом.
Селекцию штаммов с использованием подвергаемого обратной селекции маркера осуществляли, используя два разных способа:
1-Культивирование на галактозе
Выращенные на галактозе клетки IMK490 высевали штрихом на чашки, содержащие галактозу в качестве источника углерода и инкубировали при 30°C в течение 3 суток. Отдельные изоляты колоний ресуспендировали в 100 мкл стерильной воды и 5 мкл переносили на чашки с синтетической средой, содержащей либо аммоний, либо ацетамид в качестве единственного источника азота. Клетки на таких чашках выращивали в течение 2 суток при 30°C. Подобным образом единичные изолированные колонии IMK386, IMK487, IMK488 и IMK489 помещали на чашки с синтетической средой, содержащей либо аммоний, либо ацетамид в качестве единственного источника азота.
В случае IMK486 из 5 собранных колоний ни в одна не имела рециклинга маркера amdS, в случае IMK487 только 1 из 5 клонов имел рециклинг маркера amdS, и в случае IMK489 все пять собранных клона утрачивали маркер amdS маркер. В случае штамма IMK490 проверяли 11 колоний и 10 имели вырезанный селектируемый маркер amdS. Чтобы определить удалялся ли маркер из хромосомы, осуществляли ПЦР колоний на геномной ДНК типичного клона с использованием внешних-внешних праймеров Fw 5ʹScARO10/amdS check и Rv 3ʹScARO10/amdS check, которые давали фрагмент длиной 1426 пар оснований. При этом подтверждали, что кассета amdS-ISceI была удалена из генома.
2-Инокуляция в жидкую среду и обратная селекция на фторацетамиде.
Второй способ, применяемый для удаления маркера, заключался в инокуляции 1 мл культуры из каждого штамма, содержащего кассету amdS-ISceI, в жидкую среду с 2% галактозы в качестве основного источника углерода и 0,05% глюкозы для усиления роста разных штаммов дрожжей. После выращивания в течение 4 часов в такой жидкой среде отбирали образцы и разбавляли в 200 раз в стерильной воде, и 100 мкл высевали на чашки с синтетической средой с галактозой и фторацетамидом. Колонии, которые экспрессировали ген amdS, могли поэтому гидролизовать фторацетамид на аммоний и фторацетат, который является токсичным. Могли расти только клетки, не имеющие селектируемого маркера amdS.
В данном проекте исследовали гомологичную рекомбинацию по сборке in vivo плазмиды pUDC114 из четырех двунитевых фрагментов ДНК, которые имели перекрывания 60 пар оснований. pUDC114 содержала гены каротина crtYBEI, что приводило к получению позитивных колоний оранжевого цвета в качестве средства для быстрой идентификации. Результаты трансформации показали, что с увеличением плоидности разных штаммов S. cerevisiae количество трансформантов на мкг ДНК значимо снижалось с 2*104 трансформантов на мкг ДНК в гаплоидном CEN.PK113-7D до 6*103 трансформантов на мкг ДНК в диплоидном CEN.PK122 и, наконец, до 19 трансформантов на мкг ДНК в полиплоидном штамме верхнего брожения PRG410. В недавно открытом диплоидном S. eubayanus CBS12357 дикого типа количество трансформантов на мкг ДНК также было более чем в 10 раз меньше, чем в диплоидном лабораторном штамме CEN.PK122.
Эффективность гомологичной рекомбинации в сложных лагерных штаммах ранее не исследовали. S. pastorianus CMBS33 был в 20 раз менее эффективным, чем диплоидный CEN.PK122, но только немного менее эффективным, чем другой исходный штамм S. eubayanus CBS12357. Эффективность трансформации и гомологичной рекомбинации фрагментов pUDC114 в штамме лагерного брожения CMBS33 составляла ±300 трансформантов на мкг ДНК, но ноль в случае CBS1483. Полученный результат подтверждает, что не все штаммы лагерного брожения идентичны, при этом были отобраны генетические различия, определяемые особенностями процесса брожения.
Система для делеции гена с удалением маркера amdS эндонуклеазой I-SceI является новым способом изменения и делеции генов у штаммов лагерного брожения и лабораторных штаммов S. cerevisiae. Возможность изменения и/или делеции генов и последующего удаления маркера у S. pastorianus CBS1483 с использованием кассеты amdS-ISceI вносит существенный вклад в копилку исследователей в области пивоваренной промышленности.
Пример 2
Геномную ДНК Kluyveromyces lactis штамма ATCC 8585 получали, как описано (Burke et al., 2000. Cold Spring Harbor Laboratory. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual). ORF KLLA0F27995g, кодирующую KlGBU1, амплифицировали с геномной ДНК, используя полимеразу Phusion Hot-Start (Finnzymes) и праймеры: прямой праймер GBU1 (5ʹ-CATCCGAACATAAACAACCATGAAGGTTGCAGGATTTATATTG) и обратный праймер GBU1 (5ʹ-CAAGAATCTTTTTATTGTCAGTACTGATCAGGCTTGCAAAACAAATTGTTC). Получали кодирующую последовательность гена GBU1 K. lactis.
Набор таргетирующих конструкций, содержащих селектируемый маркер KlGBU1 со всеми необходимыми частями для стандартной кассеты для делеции представлен на фигуре 4. Перекрывание 400 оснований в селектируемом маркере KlGBU1 (указано скрещенными линиями) сконструировано для рекомбинации за счет гомологии.
| название | год | авторы | номер документа |
|---|---|---|---|
| УРЕОГИДРОЛАЗЫ В КАЧЕСТВЕ ДОМИНАНТНЫХ СЕЛЕКТИВНЫХ МАРКЕРОВ У ДРОЖЖЕЙ | 2015 |
|
RU2702766C2 |
| СПОСОБ ХРОМОСОМНОЙ ИНТЕГРАЦИИ И ЗАМЕНЫ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК В CLOSTRIDIA | 2006 |
|
RU2464317C2 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pPBS-St9, КОДИРУЮЩАЯ ПОЛИПЕПТИД СОМАТОТРОПИНА, И ШТАММ ДРОЖЖЕЙ SACCHAROMYCES CEREVISIAE ДЛЯ ПРОДУКЦИИ РЕКОМБИНАНТНОГО СОМАТОТРОПИНА | 2011 |
|
RU2465315C1 |
| СПОСОБЫ И СРЕДСТВО МОДИФИКАЦИИ РАСТИТЕЛЬНОГО ГЕНОМА В НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ, ШИРОКО ИСПОЛЬЗУЕМОЙ В ГЕНОМНОЙ ИНЖЕНЕРИИ РАСТЕНИЙ | 2011 |
|
RU2639512C2 |
| СПОСОБ ПОЛУЧЕНИЯ САЙТ-НАПРАВЛЕННЫХ ТРАНСЛОКАЦИЙ В КЛЕТКАХ SACCHAROMYCES CEREVISIAE | 2018 |
|
RU2692552C1 |
| СПОСОБ УПРАВЛЕНИЯ МЕТАБОЛИЗМОМ КЛЕТКИ | 2019 |
|
RU2731897C1 |
| РЕКОМБИНАНТНЫЙ ГЕРПЕСВИРУС КОИ (KHV) И ВАКЦИНА ДЛЯ ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЯ, ВЫЗЫВАЕМОГО KHV | 2012 |
|
RU2662768C2 |
| РЕКОМБИНАНТНЫЙ ШТАММ ESCHERICHIA COLI-ПРОДУЦЕНТ L-ТРЕОНИНА | 2013 |
|
RU2546237C1 |
| ТЕЛОМЕРАЗНАЯ ОБРАТНАЯ ТРАНСКРИПТАЗА ПТИЦ | 2007 |
|
RU2415867C2 |
| Способ получения штамма-продуцента фосфолипазы А2 Komagataella phaffii (Pichia pastoris) YIB Δleu2_PLA2S | 2020 |
|
RU2746817C1 |




Изобретение относится к биотехнологии. Предложен набор таргетирующих конструкций, включающий первую конструкции, содержащую первую область гомологии с геном-мишенью микроорганизма, сайт узнавания эндонуклеазой и первую часть селектируемого маркера, и вторую конструкцию, содержащую вторую часть селектируемого маркера, сайт узнавания эндонуклеазой и вторую область гомологии с геном-мишенью микроорганизма, причем фрагмент первой части селектируемого маркера перекрывается с фрагментом второй части селектируемого маркера, обеспечивая возможность рекомбинации между первой и второй частями селектируемого маркера. Изобретение, кроме того, относится к способу изменения генома микроорганизма и к способу получения микроорганизма. Данное изобретение обеспечивает эффективное нацеливание и изменение гена-мишени микроорганизма. 3 н. и 9 з.п. ф-лы, 4 ил., 7 табл., 2 пр.
1. Набор таргетирующих конструкций для изменения целевого генома микроорганизма, включающий первую конструкцию, содержащую в указанном порядке первую область гомологии с геномом-мишенью микроорганизма, сайт узнавания эндонуклеазой и первую часть селектируемого маркера, и вторую конструкцию, содержащую в указанном порядке вторую часть селектируемого маркера, копию сайта узнавания эндонуклеазой и вторую область гомологии с геномом-мишенью микроорганизма, при этом
каждая из первой и второй областей гомологии с геномом-мишенью содержит, по меньшей мере, 100 пар оснований (п.о.);
фрагмент первой части селектируемого маркера перекрывается с фрагментом, который присутствует во второй части селектируемого маркера, обеспечивая возможность рекомбинации между первой и второй частями селектируемого маркера, при этом указанный фрагмент предпочтительно содержит от 200 до 600 п.о.;
кодирующая последовательность, которая кодирует эндонуклеазу и которая связана с индуцируемым промотором, присутствует в первой или второй конструкции; и
часть первой области гомологии с геномом-мишенью в первой конструкции дуплицирована между копией сайта узнавания эндонуклеазой и второй областью гомологии с геномом-мишенью во второй конструкции; или часть второй области гомологии с геномом-мишенью во второй конструкции дуплицирована между первой областью гомологии с геномом-мишенью и сайтом узнавания эндонуклеазой в первой конструкции, при этом указанная дуплицированная область предпочтительно содержит от 40 до 200 п.о.
2. Набор таргетирующих конструкций по п.1, в котором перекрывающийся фрагмент селектируемого маркера имеет длину примерно 400 пар оснований (п.о.).
3. Набор таргетирующих конструкций по п.1 или 2, в котором дуплицированная область гомологии с геномом-мишенью в первой и второй таргетирующих конструкциях предпочтительно имеет длину примерно 80 п.о.
4. Набор таргетирующих конструкций по п.1 или 2, в котором эндонуклеаза является хоминг-эндонуклеазой.
5. Набор таргетирующих конструкций по п. 1 или 2, в котором селектируемым маркером является маркер ауксотрофности и/или доминантный маркер.
6. Набор таргетирующих конструкций по п.1 или 2, в котором индуцируемый промотор выбран из промотора GAL1, промотора GAL10, промотора SUC2, промотора MAL12, CUP1 и регулируемого тетрациклином промотора.
7. Набор таргетирующих конструкций по п.1 или 2, в котором микроорганизм является анеуплоидным микроорганизмом.
8. Набор таргетирующих конструкций по п.1 или 2, в котором микроорганизмом является Ascomycota, предпочтительно Saccharomycotina.
9. Набор таргетирующих конструкций по п.1 или 2, в котором микроорганизмом является Saccharomyces pastorianus.
10. Способ изменения генома микроорганизма, включающий предоставление набора таргетирующих конструкций по любому из пп.1-9 указанному микроорганизму и отбор микроорганизма, в котором был изменен геном.
11. Способ получения микроорганизма, включающего изменение генома, при этом способ включает предоставление набора таргетирующих конструкций по любому из пп.1-9 указанному микроорганизму и отбор микроорганизма, в котором был изменен геном и который функционально экспрессирует рекомбинированный селектируемый маркер.
12. Способ по п.11, дополнительно включающий индукцию индуцируемого промотора для экспрессии эндонуклеазы.
| KUIJPERS N | |||
| G | |||
| A | |||
| et al | |||
| "One-step assembly and targeted integration of multigene constructs assisted by the I-SceI meganuclease in Saccharomyces cerevisiae." FEMS yeast research, 2013 (published online 07.10.2013), 13(8):769-781 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| US 5612205 A, 18.03.1997 | |||
| QIAN W | |||
| et al | |||
| "Improved gene disruption method and Cre-loxP mutant system for multiple gene disruptions in Hansenula polymorpha." Journal of microbiological methods, 2009, 79(3): 253-259 | |||
| AKADA R | |||
| et al | |||
| "PCR-mediated seamless gene deletion and marker recycling in Saccharomyces cerevisiae." Yeast, 2006, 23(5): 399-405 | |||
| BLASCO L | |||
| et al | |||
| "A new disruption vector (pDHO) to obtain heterothallic strains from both Saccharomyces cerevisiae and Saccharomyces pastorianus." International Microbiology, 2011, 14: 201-206 | |||
| RU 2009136452 А, 10.04.2011. | |||

