ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке согласно § 119(e) 35 U.S.C. испрашивается приоритет предварительной заявки на патент США порядковый № 60/700 673, поданной 19 июля 2005, содержание которой во всей его полноте включено в настоящую заявку в качестве ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к 2-(азетидин-2-он-1-ил)-замещенным алкановым кислотам, являющимся аналогами аминокислот. В частности, изобретение относится к таким алкановым кислотам, которые являются аналогами фенилаланина, цистеина, гомоцистеина и гомосерина, а также их аналогам и производным. Кроме того, настоящее изобретение относится к способам лечения млекопитающих при необходимости избавления от болезненных состояний, связанных с антагонизмом рецепторам вазопрессина V1a, V1b и V2 и чувствительных к этому антагонизму.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Аргинин вазопрессин (AVP) является нейрогипофизарным нейропептидом, вырабатываемым в гипоталамусе, причем он вовлечен в большое число биологических процессов в системе кровообращения, в периферической нервной системе (PNS) и центральной нервной системе (CNS, ЦНС). В частности, AVP действует в качестве нейротрасмиттера в мозге. Были выявлены несколько фармакологически значимых подтипов рецептора вазопрессина, включая рецепторы вазопрессина V1a, V1b и V2. Указанные рецепторы вазопрессина вовлечены в некоторые психиатрические, психологические и поведенческие болезненные состояния, включая депрессию, тревожность, аффективные расстройства и стресс, а также неопиоидное опосредование переносимости боли. Кроме того, рецепторы вазопрессина вовлечены в ряд метаболических процессов, включая гомеостаз метаболизма воды, деятельность почек, опосредование функций сердечно-сосудистой системы, а также регулирование температуры тела млекопитающих.
Например, AVP играет важную роль в возникновении депрессии, т.е. одного из наиболее распространенных серьезных расстройств ЦНС. В число потенциальных мишеней для лечения депрессии входит гипоталамно-гипофизарно-надпочечниковая ось (HPA ось), которая нарушена у многих пациентов с депрессией, а также с вызванными стрессом аффективными расстройствами (смотрите Scott and Dinam, 1998; Serradiel-Le Gal et al., 2002, причем содержание указанных источников включено в настоящую заявку в качестве ссылки). Нормализация деятельности HPA оси, по-видимому, является предварительным условием для устойчивой ремиссии симптомов депрессии при применении лекарственных средств (смотрите Steckler, et al., 1999, причем содержание указанного источника включено в настоящую заявку в качестве ссылки).
Одним из признаков большой депрессии является повышенный уровень кортизола и ACTH, связанный с нарушением регулирования оси HPA (смотрите Owens and Nemeroff, 1993; Plotsky et al.1998, причем содержание указанных работ включено в настоящую заявку в качестве ссылки). Кортиколиберин (CRH) и аргинин вазопрессин (AVP) представляют собой два основных стимулятора секреции ACTH, и современные предклинические и клинические исследования показали, что AVP важен для опосредования высвобождения ACTH во время хронического психологического стресса (смотрите Scott and Dinan, 1997, 1998, причем содержание работ включено в заявку в качестве ссылки). AVP образуется в нейронах, локализованных на паравентрикулярном ядре гипоталамуса, и активация этих нейронов вызывает выделение AVP в кровоток в системе воротной вены медианного возвышения. Однако выделение кортизола в ответ на психологический стресс у здоровых добровольцев, находящихся в тревожном состоянии, по-видимому, регулируется AVP, а не CRH (смотрите Boudarene et al., 1999, причем содержание работы включено в настоящую заявку в качестве ссылки). Хронический психологический стресс, сопровождаемый нарушением регулирования оси HPA, может вносить вклад в этиологию аффективных расстройств. Было обнаружено, что многие пациенты с большой депрессией демонстрировали повышенные уровни AVP, которые понижались по мере ослабления психического расстройства (смотрите van Londen et al., 1997&2000, причем содержание работ включено в настоящую заявку в качестве ссылки).
Кроме того, AVP транспортируется к передней доле гипофиза, где он может стимулировать высвобождение ACTH путем взаимодействия с рецептором V1b на клеточных мембранах кортикотрофов. Например, крысы, селективно выведенные для демонстрации поведения, связанного с высокой тревожностью, демонстрируют нарушение регуляции HPA оси. Воздействие антагониста рецептора V1b может ликвидировать CRH-стимулированную секрецию ACTH, демонстрируя переключение в регуляции ACTH с CRH на AVP (смотрите Keck et al., 1999, причем содержание работы включено в настоящую заявку в качестве ссылки). Также было продемонстрировано наличие рецепторов V1b в нескольких отделах ЦНС крыс и ЦНС мышей. Следовательно, считается, что антагонисты V1b, которые проникают в ЦНС, могут обладать большим терапевтическим потенциалом при аффективных расстройствах, связанных со стрессом. В настоящее время не существует антагонистов вазопрессина, которые способны преодолевать барьер кровь-мозг (Serradeil-Le Gal et al., 2002). Кроме того, существуют предклинические и клинические доказательства, что вазопрессин, действуя через рецептор V1b, способствует развитию подтипа большой депрессии, связанного с хроническим стрессом и нарушением регуляции HPA оси (смотрите Boudarene et al., 1999; Griebel et al., 2002; Scott and Dinan, 1997, 1998, причем содержание указанных работ включено в настоящую заявку в качестве ссылки).
Сообщалось, что сердечно-сосудистые расстройства являются причиной большинства случаев госпитализации людей в возрасте 65 лет и старше. Было показано, что AVP содействует патофизиологии и развитию сердечных заболеваний, включая застойной сердечной недостаточности (смотрите Schrier & Abraham "Hormones and hemodynamics in heart failure," N. Engl. J. Med. 341:577-585 (1999); Thibonnier "Vasopressin receptor antagonists in heart failure," Curr. Op. Pharmacology 3:683-687 (2003); Lee et al., "Vasopressin: A new target for the treatment of heart failure," Am. Heart J. 146:9-18 (2003), причем содержание всех перечисленных работ включено в настоящую заявку в качестве ссылки). Кроме того, координированная физиология почечной/сердечно-сосудистой систем содействует нормальной работоспособности и гомеостазу сердца. Таким образом, AVP играет также важную роль в водном и электролитическом равновесии, регулировании объема крови, тонусе гладкой мускулатуры сосудов, а также способности сердца к сокращениям и сердечном метаболизме. Каждый из перечисленных факторов входит в число основных факторов, влияющих на производительность работы сердца и его способность удовлетворять потребности организма. AVP влияет на все перечисленные факторы, в частности, за счет активации рецепторов V1a и V2. Рецепторы вазопрессина V1a локализованы в гладкой мускулатуре сосудов и кардиомиоцитах, соответственно содействуя сужению сосудов, а также синтезу белка и росту сердечных клеток. Рецепторы вазопрессина V2 локализованы в собирающих протоках почечных нефронов, содействуя обратному всасыванию свободной воды. Небольшие изменения осмолярности плазмы ощущаются рецепторами в гипоталамусе, которые регулируют нейросекреторное выделение AVP из гипофиза. При осмотической стимуляции содержание AVP в плазме может подняться от исходного уровня 3-4 пг/мл до 9-10 пг/мл. Эти умеренные изменения уровня нейрогормона AVP совместно с ренин-ангиотензин-альдостероновой системой изо дня в день регулируют равновесие воды и электролитов у здоровых субъектов.
Однако сообщалось, что роль AVP в физиологии сердечно-сосудистой системы здоровых субъектов является минимальной, и для этих людей необходимы сверхфизиологические дозы нейрогормонов, чтобы оказывать влияние на кровяное давление, способность сердца к сокращению и коронарный кровоток. В противоположность этому, AVP играет основную роль у пациентов с сердечной недостаточностью. Например, было замечено, что основные уровни AVP в плазме повышены у пациентов с сердечной недостаточностью по сравнению со здоровыми субъектами (контроль), в частности у тех, у которых имеется гипонатриемия (смотрите Goldsmith "Congestive heart failure: potential role of arginine vasopressin antagonists in the therapy of heart failure," Congest. Heart Fail. 8:251-6 (2002); Schrier and Ecder, (2001), причем содержание данных работ включено в настоящую заявку в качестве ссылки). Далее, с AVP связан пониженный диурез воды у пациентов с застойной сердечной недостаточностью (CHF), ведущий к увеличению объема крови, гипонатриемии, отеку и увеличению веса. При сердечной недостаточности повышение содержания AVP в плазме ведет к увеличению сопротивления периферических сосудов и заклиненного давления в легочных капиллярах при уменьшении минутного сердечного выброса и ударного объема сердца. Кроме того, дополнительные данные наводят на мысль, что AVP вносит вклад в гипертрофические характеристики миокарда при сердечной недостаточности (смотрите Nakamura et al., "Hypertrophic growth of cultured neonatal rat heart cells mediated by vasopressin Via receptor," Eur J Pharmacol 391:39-48 (2000); Bird et al., "Significant reduction in cardiac fibrosis and hypertrophy in spontaneously hypertensive rats (SHR) treated with a V1a receptor antagonist," (реферат) Circulation 104:186 (2001), причем содержание указанных работ включено в настоящую заявку в качестве ссылки), и клеточные/молекулярные исследования показали, что помимо этого AVP инициирует сигнальный каскад, который содействует фиброзу миокарда, как правило, наблюдаемому при развитии заболевания.
Структурные модификации вазопрессина привели к получению ряда агонистов вазопрессина (смотрите Sawyer, Pharmacol.Reviews, 13:255(1961)). Кроме того, было раскрыто несколько мощных и селективных пептидных антагонистов вазопрессина (смотрите Lazslo et al., Pharmacological Reviews, 43:73-108 (1991); Mah and Hofbauer, Drugs of the Future, 12:1055-1070 (1987); Manning and Sawyer, Trends in Neuroscience, 7:8-9 (1984)). Далее были раскрыты новые структурные классы непептидных антагонистов вазопрессина (смотрите Yamamura et al., Science, 275:572-574 (1991); Serradiel-Le Gal et al., Journal of Clinical Investigation, 92:224-231 (1993); Serradiel-Le Gal et al., Biochemical Pharmacology, 47(4):633-641 (1994)). Наконец, общий структурный класс 2-(азетидин-2-он-1-ил)-замещенных эфиров и амидов уксусной кислоты известен в качестве промежуточных продуктов синтеза при получении β-лактамовых антибиотиков (смотрите патент США № 4751299).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Было обнаружено, что некоторые соединения, входящие в общий класс 2-(азетидин-2-он-1-ил)-замещенных алкановых кислот, а также их производные являются антагонистами рецепторов вазопрессина, включая рецепторы вазопрессина V1a, V1b и V2. В настоящей заявке описаны 2-(азетидин-2-он-1-ил)-замещенные алкановые кислоты, являющиеся аналогами фенилаланина, цистеина, гомоцистеина и гомосерина, а также их аналоги, гомологи и производные. Кроме того, в заявке описаны фармацевтические композиции, которые включают терапевтически эффективные количества указанных алкановых кислот, предназначенные для лечения заболеваний и расстройств, которые зависимы от антагонизма к одному или нескольким рецепторам вазопрессина, например рецепторам V1a, V1b и V2. Дополнительно описаны способы лечения заболеваний и болезненных состояний у млекопитающих, которые связаны с нарушением функций вазопрессина и которые зависимы от антагонизма к рецепторам вазопрессина, например рецепторам V1a, V1b и V2 или их комбинации. Помимо этого описаны способы получения 2-(азетидин-2-он-1-ил)-замещенных алкановых кислот, являющихся аналогами фенилаланина, цистеина, гомоцистеина и гомосерина, а также их различных аналогов и производных.
В одном из иллюстративных вариантов осуществления настоящего изобретения описаны соединения формулы (I):
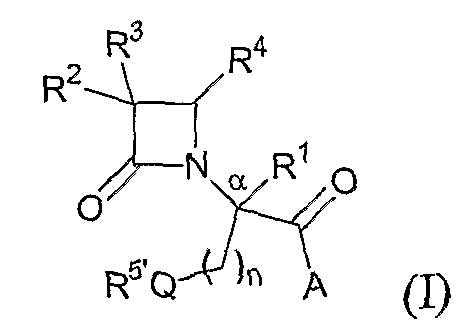
где Q представляет собой кислород, серу или окисленную серу, включая группы -S(O)- и -SO2-;
n представляет собой 1 или 2;
A представляет собой R5O, монозамещенную аминогруппу, дизамещенную аминогруппу или необязательно замещенный азотсодержащий гетероцикл, присоединенный по атому азота;
R1 представляет собой водород или C1-C6алкил;
R2 представляет собой водород, алкил, включая C1-C6алкил, алкенил, включая C2-C6алкенил, как, например, винил, аллил и т.п., алкинил, включая C2-C6алкинил, как, например, этинил, пропинил и т.п., алкокси, включая C1-C4алкокси, алкилтио, включая C1-C4алкилтио, галоген, галогеналкил, как, например, трифторметил, трифторхлорэтил и т.п., циано, формил, алкилкарбонил, включая C1-C3алкилкарбонил, алкоксикарбонил или заместитель, выбранный из группы, состоящей из -CO2R8, -CONR8R8' и -NR8(COR9);
R3 представляет собой необязательно замещенные амино, амидо, ациламидо или уреидо группу; или же R3 представляет собой азотсодержащую гетероциклическую группу, присоединенную по атому азота;
R4 представляет собой алкил, включая C1-C6алкил, алкенил, включая C2-C6алкенил, алкинил, включая C2-C6алкинил, циклоалкил, включая C3-C8циклоалкил, циклоалкенил, включая C3-C9циклоалкенил, как, например, лимонеил, пинеил и т.п., алкилкарбонил, включая C1-C3алкилкарбонил, необязательно замещенный арил, необязательно замещенный арилалкил, включая арил(C1-C4алкил), необязательно замещенный арилгалогеналкил, необязательно замещенный арилалкоксиалкил, необязательно замещенный арилалкенил, включая арил(C2-C4алкенил), необязательно замещенный арилгалогеналкенил или необязательно замещенный арилалкинил, включая арил(C2-C4алкинил);
R5 выбран из водорода, алкила, включая C1-C6алкила, циклоалкила, включая C3-C8циклоалкила, алкоксиалкила, включая (C1-C4алкокси)-(C1-C4алкила), необязательно замещенного арилалкила, включая арил(C1-C4алкила), гетероциклила, гетероциклил(C1-C4алкила) и R6R7N-(C2-C4алкила), где каждый из гетероциклилов независимо выбран из тетрагидрофурила, морфолинила, пирролидинила, пиперидинила, пиперазинила, гомопиперазинила или хинуклидинила; где указанные морфолинил, пирролидинил, пиперидинил, пиперазинил, гомопиперазинил или хинуклидинил необязательно замещены по атому азота C1-C4алкилом или необязательно замещенным арил(C1-C4алкилом);
R5' выбран из группы, состоящей из -SR15, -S(O)R15, -SO2R15, C1-C6алкила, C3-C8циклоалкила, (C1-C4алкокси)-(C1-C4алкила), необязательно замещенного арилалкила, включая арил(C1-C4алкила), гетероциклила, гетероциклил(C1-C4алкила) и R6'R7'N-(C2-C4алкила), где каждый из гетероциклилов независимо выбран из тетрагидрофурила, морфолинила, пирролидинила, пиперидинила, пиперазинила, гомопиперазинила или хинуклидинила; где указанные морфолинил, пирролидинил, пиперидинил, пиперазинил, гомопиперазинил или хинуклидинил необязательно замещены по атому азота C1-C4алкилом или необязательно замещенным арил(C1-C4алкилом);
R6 представляет собой водород или алкил, включая C1-C6алкил, и R7 представляет собой алкил, включая C1-C6алкил, циклоалкил, включая C3-C8циклоалкил, необязательно замещенный арил или необязательно замещенный арилалкил, включая арил(C1-C4алкил); или же R6 и R7 совместно с присоединенным атомом азота образуют гетероцикл, как, например, пирролидинил, пиперидинил, морфолинил, пиперазинил и гомопиперазинил; где указанные пиперазинил или гомопиперазинил необязательно замещены по атому азота заместителем R13;
R6' представляет собой водород или алкил, включая C1-C6алкил, и R7' представляет собой алкил, включая C1-C6алкил, циклоалкил, включая C3-C8циклоалкил, необязательно замещенный арил или необязательно замещенный арилалкил, включая арил(C1-C4алкил); или же R6' и R7' совместно с присоединенным атомом азота образуют гетероцикл, как, например, пирролидинил, пиперидинил, морфолинил, пиперазинил и гомопиперазинил; где указанные пиперазинил или гомопиперазинил необязательно замещены по атому азота заместителем R13';
каждый из заместителей R8 и R8' в каждом из случаев независимо выбран из водорода, алкила, включая C1-C6алкил; циклоалкила, включая C3-C8циклоалкил; необязательно замещенного арила или необязательно замещенного арилалкила, включая арил(C1-C4алкил); или же R8 и R8' совместно с присоединенным атомом азота образуют гетероцикл, как, например, необязательно замещенный пирролидинил, пиперидинил, морфолинил, пиперазинил и гомопиперазинил;
заместитель R9 выбран из водорода, алкила, включая C1-C6алкил; циклоалкила, включая C3-C8циклоалкил; алкоксиалкила, включая (C1-C4алкокси)-(C1-C4алкил); необязательно замещенного арила, необязательно замещенного арилалкила, включая арил(C1-C4алкил); необязательно замещенного гетероарила, необязательно замещенного гетероарилалкила, включая гетероарил(C1-C4алкил); и R8R8'N-(C1-C4алкила);
R13 и R13' независимо выбран из водорода, алкила, включая C1-C6алкил; циклоалкила, включая C3-C8циклоалкил; алкоксикарбонила, включая C1-C4алкоксикарбонил; необязательно замещенного арилоксикарбонила, необязательно замещенного арилалкила, включая арил(C1-C4алкил); и необязательно замещенного арилоила;
R15 выбран из группы, состоящей из C1-C6алкила, C3-C8циклоалкила, (C1-C4алкокси)-(C1-C4алкила), необязательно замещенного арил(C1-C4алкила), Y'-, Y'-(C1-C4алкила) и R6'R7'N-(C2-C4алкила); а также
гидраты, сольваты и фармацевтически приемлемые соли указанных соединений;
при условии, что, если Q представляет собой кислород, n равно 2 и R5' не является -SR15, -S(O)R15 или -SO2R15.
Другой иллюстративный вариант осуществления настоящего изобретения относится к соединениям формулы (II):
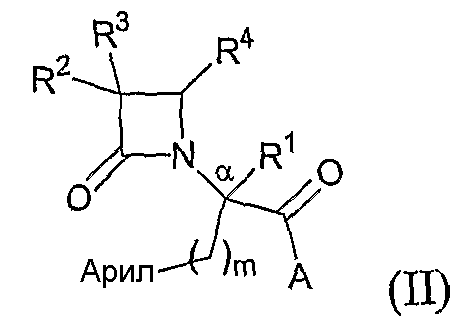
где арил представляет собой необязательно замещенную моноциклическую или полициклическую ароматическую группу;
m означает 1,2,3 или 4; и
A, R1, R2, R3 и R4 соответствуют определению, данному при описании формулы (I); а также гидратам, сольватам и фармацевтически приемлемым солям указанных соединений.
Другой иллюстративный вариант осуществления настоящего изобретения относится к соединениям формулы (III):
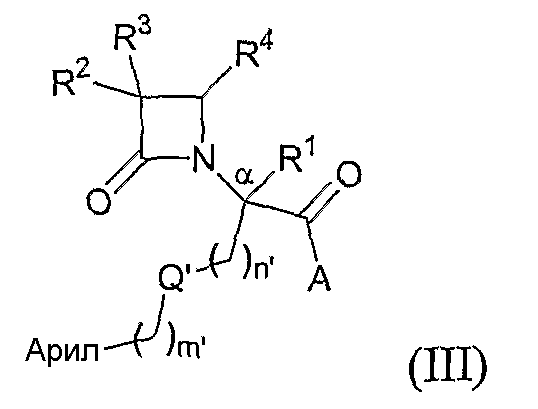
где арил представляет собой необязательно замещенную моноциклическую или полициклическую ароматическую группу;
Q' представляет собой кислород, серу или -CH2-;
n' означает 0, 1 или 2;
m' означает 0, 1 или 2; и
A, R1, R2, R3 и R4 соответствуют определению, данному при описании формулы (I); а также
гидратам, сольватам и фармацевтически приемлемым солям указанных соединений;
при условии, что, если Q' представляет собой кислород, n' означает 2; и если Q' представляет собой серу, n' означает 1 или 2.
В одном из аспектов изобретение относится к соединениям формулы (I), в которых Q представляет собой кислород, и n означает 2. В другом аспекте в изобретении описаны соединения формулы (I), в которых Q представляет собой серу и n означает 1 или 2. В другом аспекте изобретение относится к соединениям формулы (I), в которых Q представляет собой серу, n равно 1 и R5' является алкилом или необязательно замещенным арилалкилом. В другом аспекте в изобретении описаны соединения формулы (I), в которых Q является серой, n означает 2 и R5' является алкилом или необязательно замещенным арилалкилом.
В одном из аспектов в соединениях формул (II) и (III), арил представляет собой необязательно замещенный фенил, включая фенил, алкилфенил, гидроксифенил, алкоксифенил, галогенфенил, цианофенил и т.п.; необязательно замещенный пиридинил, включая 2-, 3- и 4-пиридинил, алкил 2-, 3- и 4-пиридинил, галоген 2-, 3- и 4-пиридинил и т.п.; и необязательно замещенный нафтил, включая 2- и 3- нафтил, алкилнафтил, гидроксинафтил, алкоксинафтил, галогеннафтил и т.п.
Следует понимать, что различные аспекты формул, описанных в настоящей заявке, могут быть выбраны в многочисленных сочетаниях. Иллюстративно, для любого из соединений формул (I), (II) или (III), выбирают соединения, в которых R2 представляет собой водород, R4 является арилалкенилом и A представляет собой либо моно- или дизамещенную аминогруппу либо необязательно замещенный азотсодержащий гетероцикл. В различных вариантах выбирают соединения, в которых R2 представляет собой водород или метил, R4 является арилалкенилом и A представляет собой либо моно- или дизамещенную аминогруппу, либо необязательно замещенный азотсодержащий гетероцикл. В другой иллюстративной комбинации для соединений формул (I) и (III) R2 представляет собой водород, R4 является арилалкенилом и Q и Q' представляют собой атомы серы. В различных вариантах A представляет собой либо моно- или дизамещенную аминогруппу, либо необязательно замещенный азотсодержащий гетероцикл, и n или n' означает 1. В других вариантах R1 представляет собой водород и в ряде других вариантов R4 более конкретно является необязательно замещенным фенилэтилом. Кроме того, следует понимать, что такие варианты можно комбинировать далее для определения подмножеств, выбранных из соединений, описанных в настоящем изобретении.
В другом варианте осуществления в настоящей заявке описаны фармацевтические композиции, причем указанные композиции включают одно или несколько соединений, описанных в настоящей заявке, в том числе, но не ограничиваясь этим, соединения формул (I), (II) или (III) и/или 2-(азетидин-2-он-1-ил)-замещенные аналоги фенилаланина, цистеина, гомоцистеина и гомосерина, а также производные и аналоги описанных в настоящей заявке соединений и их комбинации. 2-(азетидин-2-он-1-ил)-замещенные аналоги фенилаланина, цистеина, гомоцистеина и гомосерина и их производные и аналоги включают соединения формул (I), (II) или (III). Описанные в настоящей заявке фармацевтические композиции также включают один или несколько фармацевтически приемлемых носителей, разбавителей и/или наполнителей. В одном из иллюстративных аспектов описаны фармацевтические композиции, которые проявляют пероральную активность и/или пероральную биодоступность. В другом иллюстративном аспекте описаны фармацевтические композиции, которые дают возможность 2-(азетидин-2-он-1-ил)-замещенным аналогам фенилаланина, цистеина, гомоцистеина и гомосерина, а также их производным и аналогам проникать сквозь барьер кровь-мозг.
В другом варианте осуществления описаны способы лечения болезненных состояний у млекопитающих, чувствительных к антагонизму к рецепторам вазопрессина V1a, V1b и/или V2, в случае необходимости такого лечения. Эти способы включают стадию введения млекопитающему фармацевтически эффективного количества одного или нескольких соединений, раскрытых в настоящей заявке, включая, но не ограничиваясь этим, соединения формул (I), (II) или (III) и/или 2-(азетидин-2-он-1-ил)-замещенные аналоги фенилаланина, цистеина, гомоцистеина и гомосерина, а также их производных и аналогов, описанных в настоящей заявке, а также их комбинаций. В другом варианте осуществления способы включают стадию введения млекопитающему композиции, содержащей фармацевтически эффективное количество одного или нескольких 2-(азетидин-2-он-1-ил)-замещенных аналогов фенилаланина, цистеина, гомоцистеина и гомосерина, а также их производных и аналогов, описанных в настоящей заявке, и фармацевтически приемлемый носитель, разбавитель или наполнитель.
Иллюстративные болезненные состояния, которые чувствительны к антагонизму рецепторам вазопрессина V1a, V1b и/или V2 и поддаются лечению описанными в настоящей заявке способами, включают различные психические заболевания, связанные со стрессом, депрессию, тревожность, аффективные расстройства, обсессивно-компульсивные заболевания, импульсивность, агрессивные расстройства и т.п.; заболевания, влияющие на гомеостаз воды, почечную функцию, ингибирование метаболизма фосфатидил-инозита, регуляцию температуры и т.п.; заболевания, связанные с тошнотой, рвотой и болью; а также различные сердечно-сосудистые заболевания, включая застойную сердечную недостаточность, расстройства или состояния, связанные с агрегацией тромбоцитов и т.п. Дополнительно в настоящей заявке описаны способы лечения других болезненных состояний, которые могут подвергаться лечению, например, антагонистами рецептора окситоцина, антагонистами рецептора тахикинина, антагонистами рецептора нейрокинина 1, антагонистами рецептора нейрокинина 2 и т.п., причем указанные способы включают стадию введения пациенту, при необходимости избавления от такого заболевания или болезненного состояния, эффективного количества одного или нескольких 2-(азетидин-2-он-1-ил)-замещенных аналогов фенилаланина, цистеина, гомоцистеина и гомосерина, а также их производных и аналогов, описанных в настоящей заявке, включая соединения формул (I), (II) или (III); или же способы включают стадию введения пациенту, при необходимости избавления от такого заболевания или болезненного состояния, описанной в настоящей заявке композиции, причем композиция включает эффективное количество одного или нескольких 2-(азетидин-2-он-1-ил)-замещенных аналогов фенилаланина, цистеина, гомоцистеина и гомосерина, а также их производных и аналогов, описанных в настоящей заявке, включая соединений формул (I), (II) или (III), и фармацевтически приемлемый носитель, разбавитель и/или наполнитель.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 показано сродство к связыванию с человеческим V1b (Ki=0,07 нМ) соединения примера 9B, определенное в результате исследования конкурентного связывания, осуществленного для клеток CHO, трансфецированных человеческим рецептором V1a.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Общие химические термины, использованные в описании формул в настоящей заявке, имеют свои обычные традиционные значения. Например, термин «алкил» относится к линейному или необязательно разветвленному насыщенному углеводороду, включая, но не ограничиваясь перечисленным, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, 2-пентил, 3-пентил, неопентил, гексил, гептил, октил и т.п.
Термин «циклоалкил» относится к неразветвленному или необязательно разветвленному насыщенному углеводороду, хотя бы часть которого образует цикл, включая, но не ограничиваясь перечисленным, циклопропил, циклобутил, циклопентил, метилциклопентил, циклогексил, циклогептил, циклооктил и т.п.
Термин «алкенил» относится к линейному или необязательно разветвленному углеводороду, который содержит хотя бы одну двойную связь, включая, но не ограничиваясь перечисленным, винил или этенил, аллил или пропенил, изопропенил, 2-бутенил, 2-метил-2-пропенил, бутадиенил и т.п.
Термин «алкинил» относится к линейным или необязательно разветвленным углеводородам, которые содержат, как минимум, одну тройную связь, включая, но не ограничиваясь перечисленным, этинил, пропинил, 1-бутинил, гекс-4-ен-2-инил и т.п.
Термин «арил» относится к ароматическому циклу или гетероароматическому циклу и включает такие группы, как фурил, пирролил, тиенил, пиридинил, тиазолил, оксазолил, изоксазолил, изотиазолил, имидазолил, пиразолил, фенил, пиридазинил, пиримидинил, пиразинил, тиадиазолил, оксадиазолил, нафтил, инданил, флуоренил, хинолинил, изохинолинил, бензодиоксанил, бензофуранил, бензотиенил и т.п.
Термин «необязательно замещенный» относится к замещению одного или нескольких, например, от одного до примерно трех, атомов водорода одним или несколькими заместителями. В число заместителей входят, не ограничиваясь перечисленным, такие группы, как C1-C4алкил, C1-C4алкокси, C1-C4алкилтио, гидрокси, нитро, галоген, карбокси, циано, C1-C4галогеналкил, C1-C4галогеналкокси, амино, карбамоил, карбоксамидо, амино, алкиламино, диалкиламино, алкилалкиламино, C1-C4алкилсульфониламино и т.п.
Термин «гетероцикл» относится к неароматической циклической структуре, в которую входят один или несколько гетероатомов, например, азот, кислород, сера и т.п., причем указанный термин охватывает такие группы, как тетрагидрофурил, морфолинил, пирролидинил, пиперидинил, пиперазинил, гомопиперазинил, хинуклидинил и т.п.
Термин «алкокси» относится к алкильному или циклоалкильному заместителю, присоединенному к остальной части молекулы через атом кислорода, и указанный термин охватывает такие группы, как метоки, этокси, пропокси, изопропокси, бутокси, трет-бутокси и т.п.
Термин «ацил» включает такие термины, как «алканоил» и «ароил», и относится к алкилу, алкенилу, алкинилу, арилу и т.п., присоединенному к остальной части молекулы через карбонильную группу. В качестве иллюстрации, в число ацилов входят формил, ацетил, пропаноил, бутаноил, пентаноил, циклогексаноил, необязательно замещенный бензоил и т.п.
Термин «галоген» относится к фтору, хлору, брому и иоду.
Термин «алканоилокси» охватывает такие группы, как формилокси, ацетокси, н-пропионокси, н-бутирокси, пивалоилокси и подобные низшие алканоилокси группы.
Термины «необязательно замещенный C1-C4алкил», «необязательно замещенный C3-C8циклоалкил» и «необязательно замещенный C2-C4алкенил» относятся соответственно к алкилу, циклоалкилу или алкенилу, необязательно замещенному заместителем, описанным в настоящей заявке, включая, но не ограничиваясь перечисленным, гидроксигруппу, защищенную гидроксигруппу, алкил, защищенный карбоксил, карбамоил, бензилтио, алкилтио и т.п.
Термин «(C1-C4алкил)», использованный, например, в названиях «арил(C1-C4алкил)», «(C1-C4алкокси)-(C1-C4алкил)» и т.п., относится к насыщенной линейной или разветвленной насыщенной алкильной двухвалентной цепи, образованной одним-четырьмя атомами углерода, включающей, например, арил, C1-C4алкокси и т.п. в качестве заместителя, и этот термин относится к таким группам, как, например, бензил, фенэтил, фенпропил, α-метилбензил, метоксиметил, этоксиэтил и т.п.
Термин «необязательно замещенный фенил» подразумевает фенильный радикал, необязательно замещенный одним или несколькими заместителями, каждый из которых независимо выбран из таких групп, как C1-C4алкил, C1-C4алкокси, гидрокси, галоген, нитро, трифторметил, сульфонамидо, циано, карбамоил, амино, моно(C1-C4алкил)амино, ди(C1-C4алкил)амино, C1-C4алкилсульфониламино и инол-2-ил.
Термин «защищенная аминогруппа» относится к аминогруппе, защищенной защитной группой, которая может применяться для защиты атома азота, как, например, атома азота в β-лактамовом цикле, во время синтеза или последующих реакций. Примерами таких групп являются бензил, 4-метоксибензил, 4-метоксифенил, триалкилсилил, например триметилсилил и т.п.
Термин «защищенная карбоксигруппа» относится к карбоксигруппе, защищенной или блокированной стандартными защитными группами, обычно используемыми для временного блокирования кислотной карбоксигруппы. Примеры таких групп включают низшие алкилы, например трет-бутил, галогензамещенные низшие алкилы, например 2-иодэтил и 2,2,2-трихлорэтил, бензил и замещенный бензил, например 4-метоксибензил и 4-нитробензил, дифенилметил, алкенил, например аллил, триалкилсилил, например триметилсилил и трет-бутилдиэтилсилил, и подобные карбоксизащитные группы.
Следует понимать, что в вариантах осуществления изобретения, описанных в настоящей заявке, иллюстративным вариантом алкила является C1-C6алкил, например метил, этил, пропил, проп-2-ил и т.п.; иллюстративным вариантом алкенила является C2-C6алкенил, например винил, аллил и т.п.; иллюстративным вариантом алкинила является C2-C6алкинил, как, например, этинил, пропинил и т.п.; иллюстративным вариантом алкокси является C1-C4алкокси, например метокси, пент-3-окси и т.п.; иллюстративным вариантом алкилтио является C1-C4алкилтио, например этилтио, 3-метилбут-2-илтио и т.п.; иллюстративным вариантом алкилкарбонила является C1-C3алкилкарбонил, например ацетил, пропаноил и т.п.; иллюстративным вариантом циклоалкила является C3-C8циклоалкил; иллюстративным вариантом циклоалкенила является C3-C9циклоалкенил, например лимоненил, пиненил и т.п.; иллюстративным вариантом необязательно замещенного арилалкила является необязательно замещенный арил(C1-C4алкил); иллюстративным вариантом необязательно замещенного арилалкенила является необязательно замещенный арил(C2-C4алкенил); иллюстративным вариантом необязательно замещенного арилалкинила является необязательно замещенный арил(C2-C4алкинил); иллюстративным вариантом алкоксиалкила является (C1-C4алкокси)-(C1-C4алкил); иллюстративным вариантом необязательно замещенного гетероарилалкила является необязательно замещенный гетероарил(C1-C4алкил); и иллюстративным вариантом алкоксикарбонила является C1-C4алкоксикарбонил.
Термин «антагонист» в настоящем описании относится к полному или частичному антагонисту. Хотя может применяться частичный антагонист любой естественной активности, частичные антагонисты в качестве иллюстрации демонстрируют не менее чем примерно 50% действия антагониста или не менее чем примерно 80% действия антагониста. Указанный термин также охватывает соединения, которые являются полными антагонистами рецептора вазопрессина V1b. Имеется в виду, что описанные в настоящей заявке иллюстративные способы требуют терапевтически эффективных количеств антагонистов рецептора вазопрессина V1b; следовательно, соединения, проявляющие частичный антагонизм к рецептору вазопрессина V1b, могут вводиться в более высоких дозах, чтобы проявлять достаточную антагонистическую активность для ингибирования действий вазопрессина или агониста вазопрессина.
В одном из аспектов соединений формулы (I) A представляет собой монозамещенную аминогруппу, дизамещенную аминогруппу или необязательно замещенный азотсодержащий гетероцикл, присоединенный по атому азота.
В другом аспекте в изобретении описаны соединения формулы (I), в которых Q представляет собой кислород и n равно 2. В другом аспекте в изобретении описаны соединения формулы (I), в которых Q представляет собой серу и n равно 1 или 2. В другом аспекте в изобретении описаны соединения формулы (I), в которых Q представляет собой серу, n равно 2 и R5' представляет собой алкил или необязательно замещенный арилалкил. В другом аспекте в изобретении описаны соединения формулы (I), в которых Q представляет собой серу, n равно 2 и R5' представляет собой алкилтио или необязательно замещенный арилалкилтио.
В одном из аспектов соединений формул (II) и (III) арил представляет собой необязательно замещенный фенил, включая фенил, алкилфенил, гидроксифенил, алкоксифенил, галогенфенил, цианофенил и т.п.; необязательно замещенный пиридинил, включая 2-, 3- и 4-пиридинил, алкил 2-, 3- и 4-пиридинил, галоген 2-, 3- и 4-пиридинил и т.п.; и необязательно замещенный нафтил, включая 2- и 3-нафтил, алкилнафтил, гидроксинафтил, алкоксинафтил, галогеннафтил и т.п.
В другом аспекте в изобретении описаны соединения формулы (II), в которых арил представляет собой необязательно замещенный фенил, включая фенил, алкилфенил, гидроксифенил, алкоксифенил, галогенфенил, цианофенил и т.п.; необязательно замещенный пиридинил, включая 2-, 3- и 4-пиридинил, алкил 2-, 3- и 4-пиридинил, галоген 2-, 3- и 4-пиридинил и т.п.; и необязательно замещенный нафтил, включая 2- и 3-нафтил, алкилнафтил, гидроксинафтил, алкоксинафтил, галогеннафтил и т.п.
В другом аспекте в изобретении описаны соединения формулы (II), в которых R5' представляет собой необязательно замещенный алкил, включая необязательно замещенный C1-C6алкил, C1-C4алкил и C1-C2алкил. В другом аспекте в изобретении описаны соединения формулы (II), в которых R5' представляет собой необязательно замещенный арил(C1-C4алкил), включая фенил(C1-C4алкил) или необязательно замещенный арил(C1-C2алкил).
В другом аспекте в изобретении описаны соединения формулы (III), в которых каждый из символов n' и m' обозначает целое число 1.
В другом аспекте в изобретении описаны соединения формул (II) и (III), в которых арил представляет собой необязательно замещенный фенил. В другом аспекте в изобретении описаны соединения формул (II) и (III), в которых каждый из символов m и m' обозначает целое число 1.
В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой монозамещенную аминогруппу. В другом аспекте в изобретении описаны соединения формулы (I), в которых A представляет собой дизамещенную аминогруппу. В другом аспекте в изобретении описаны соединения формулы (I), в которых A представляет собой необязательно замещенный азотсодержащий гетероцикл, присоединенный по атому азота.
В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой аминогруппу формулы R14XN-; где R14 выбран из группы, состоящей из водорода, гидрокси, алкила, включая C1-C6алкил; алкоксикарбонила, включая C1-C4алкоксикарбонил; и бензила; и в которых X выбран их группы, состоящей из алкила, включая C1-C6алкил; циклоалкила, включая C3-C8циклоалкил; алкоксиалкила, включая (C1-C4алкокси)-(C1-C4алкил); необязательно замещенного арила, необязательно замещенного арилалкила, включая необязательно замещенный арил(C1-C4алкил); а также группы Y, Y-(C1-C4алкила), R6R7N- и R6R7N-(C2-C4алкила), где Y представляет собой гетероцикл. В одном из вариантов соединений формул (I), (II) и (III), R14 представляет собой водород.
В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой гетероциклил, имеющий формулу R14XN-, где R14 и X, совместно с присоединенным атомом азота, образуют гетероцикл, например гетероцикл, выбранный из группы, состоящей из пирролидинила, пиперидинила, пиперазинила и гомопиперазинила; причем указанный гетероцикл необязательно замещен R10, R12, R6R7N- или R6R7N-(C1-C4алкилом), как определено выше.
В одном из вариантов в изобретении описаны соединения формул (I), (II) и (III), в которых R14 и X, совместно с присоединенным атомом азота, образуют пиперидинил, необязательно замещенный по 4 положению гидрокси, алкилом, включая C1-C6алкил; циклоалкилом, включая C3-C8циклоалкил; алкокси, включая C1-C4алкокси; алкоксикарбонилом, включая (C1-C4алкокси)карбонил; гидроксиалкилоксиалкилом, включая (гидрокси(C2-C4алкилокси))-(C2-C4алкил); R6R7N-, R6R7N-алкилом, включая R6R7N-(C1-C4алкил); дифенилметилом, необязательно замещенным арилом, необязательно замещенным арил(C1-C4алкилом) или пиперидин-1-ил(C1-C4алкилом).
В другом варианте в изобретении описаны соединения формул (I), (II) и (III), в которых R14 и X, совместно с присоединенным атомом азота, образуют пиперазинил, необязательно замещенный по 4 положению алкилом, включая C1-C6алкил; циклоалкилом, включая C3-C8циклоалкил; необязательно замещенным арилом, необязательно замещенным арилалкилом, включая необязательно замещенный арил(C1-C4алкил); α-метилбензилом, и т.п., N-алкил ацетамид-2-илом, включая N-(C1-C5алкил)ацетамид-2-ил; N-(циклоалкил)ацетамид-2-илом, включая N-(C3-C8циклоалкил)ацетамид-2-ил; R6R7N-, R6'R7'N- или алкоксикарбонилом, включая (C1-C4алкоксикарбонил).
В другом варианте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой дизамещенную аминогруппу, имеющую формулу R14XN-, где R14 и X совместно с присоединенным атомом азота образуют пиперидинил, необязательно замещенный по 4 положению алкилом, включая C1-C4алкил; или гетероциклил(C1-C4алкилом).
В другом варианте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой дизамещенную аминогруппу, имеющую формулу R14XN-, где R14 и X совместно с присоединенным атомом азота образуют пиперидинил, необязательно замещенный по 4 положению пиперидинил(C1-C4алкилом), пиперазинил(C1-C4алкилом) или пирролидинил(C1-C4алкилом).
В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой монозамещенную аминогруппу. В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой дизамещенную аминогруппу. В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой необязательно замещенный азотсодержащий гетероцикл, присоединенный по атому азота.
В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой монозамещенную аминогруппу формулы XNH-; где X выбран из группы, состоящей из алкила, включая C1-C6алкил; циклоалкила, включая C3-C8циклоалкил; алкоксиалкила, включая (C1-C4алкокси)-(C1-C4алкил); необязательно замещенного арила, необязательно замещенного арилалкила, включая необязательно замещенный арил(C1-C4алкил); а также группы Y, Y-(C1-C4алкила), R6R7N- и R6R7N-(C2-C4алкила), где Y представляет собой гетероцикл.
В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой дизамещенную аминогруппу формулы R14XN-; где R14 выбран из группы, состоящей из гидрокси, алкила, включая C1-C6алкил; алкоксикарбонила, включая C1-C4алкоксикарбонил; и бензила; и в которых X выбран из группы, состоящей из алкила, включая C1-C6алкил; циклоалкила, включая C3-C8циклоалкил; алкоксиалкила, включая (C1-C4алкокси)-(C1-C4алкил); необязательно замещенного арила, необязательно замещенного арилалкила, включая необязательно замещенный арил(C1-C4алкил); а также группы Y, Y-(C1-C4алкила), R6R7N- и R6R7N-(C2-C4алкила), где Y представляет собой гетероцикл.
В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых A представляет собой необязательно замещенный гетероциклил, имеющий формулу R14XN-, где R14 и X, совместно с присоединенным атомом азота, образуют гетероцикл, например гетероцикл, выбранный из группы, состоящей из пирролидинила, пиперидинила, пиперазинила и гомопиперазинила; причем указанный гетероцикл необязательно замещен R10, R12, R6R7N- или R6R7N-(C1-C4алкилом), как определено выше.
В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых R14 и X, совместно с присоединенным атомом азота, образуют пиперидинил, необязательно замещенный по 4 положению гидрокси, алкилом, включая C1-C6алкил; циклоалкилом, включая C3-C8циклоалкил; алкокси, включая C1-C4алкокси; алкоксикарбонилом, включая (C1-C4алкокси)карбонил; гидроксиалкилоксиалкилом, включая(гидрокси(C2-C4алкилокси))-(C2-C4алкил); R6R7N-, R6R7N-алкилом, включая R6R7N-(C1-C4алкил); дифенилметилом, необязательно замещенным арилом, необязательно замещенным арил(C1-C4алкилом) или пиперидин-1-ил(C1-C4алкилом).
В другом аспекте в изобретении описаны соединения формул (I), (II) и (III), в которых R14 и X, совместно с присоединенным атомом азота, образуют пиперазинил, необязательно замещенный по 4 положению алкилом, включая C1-C6алкил; циклоалкилом, включая C3-C8циклоалкил; необязательно замещенным арилом, необязательно замещенным арилалкилом, включая необязательно замещенный арил(C1-C4алкил); α-метилбензилом, и т.п., N-алкил ацетамид-2-илом, включая N-(C1-C5алкил)ацетамид-2-ил; N-(циклоалкил)ацетамид-2-илом, включая N-(C3-C8циклоалкил)ацетамид-2-ил; R6R7N-, R6'R7'N- или алкоксикарбонилом, включая (C1-C4алкокси)карбонил).
Описаны иллюстративные соединения формул (I), (II) и (III), в которых A представляет собой дизамещенную аминогруппу, имеющую формулу R14XN-, где R14 и X совместно с присоединенным атомом азота образуют пиперидинил, необязательно замещенный по 4 положению алкилом, включая C1-C4алкил; или гетероциклил(C1-C4алкилом).
Описаны иллюстративные соединения формул (I), (II) и (III), в которых A представляет собой дизамещенную аминогруппу, имеющую формулу R14XN-, где R14 и X совместно с присоединенным атомом азота образуют пиперидинил, необязательно замещенный по 4 положению пиперидинил(C1-C4алкилом), пиперазинил(C1-C4алкилом) или пирролидинил(C1-C4алкилом).
Описаны иллюстративные соединения формул (I), (II) и (III), в которых R14 и X совместно с присоединенным атомом азота образуют гомопиперазинил, необязательно замещенный по 4 положению алкилом, включая C1-C4алкил; арилом или арил(C1-C4алкилом).
Описаны иллюстративные соединения формул (I), (II) и (III), в которых A представляет собой дизамещенную аминогруппу, имеющую формулу R14XN-, где R14 и X совместно с присоединенным атомом азота образуют гетероцикл, выбранный из группы, состоящей из пирролидинонила, пиперидинонила, 2-(пирролидин-1-илметил)пирролидин-1-ила и 1,2,3,4-тетрагидроизохинолин-2-ила.
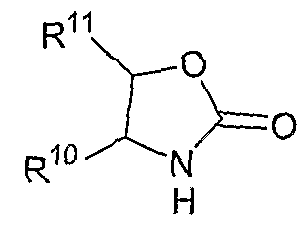
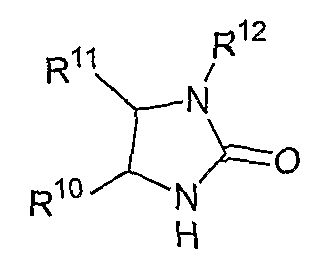
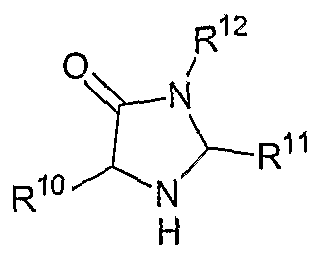
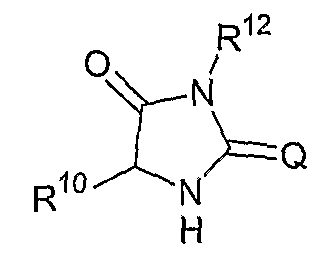
В другом аспекте соединений формул (I), (II) или (III) R3 представляет собой структуру, выбранную из группы, состоящей из:
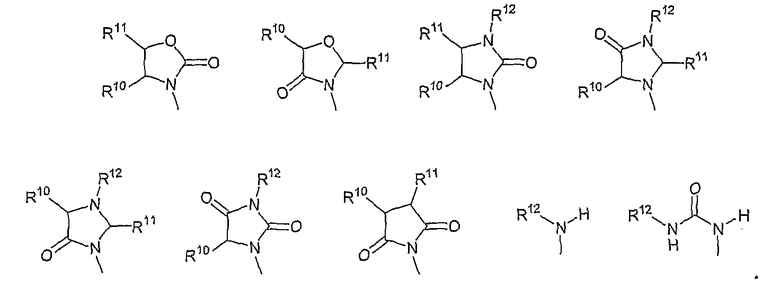
где каждый из заместителей R10 и R11 независимо выбран из водорода, необязательно замещенного алкила, включая C1-C6алкил; необязательно замещенного циклоалкила, включая C3-C8циклоалкил; алкоксиалкила, включая C1-C4алкоксиалкил; алкилкарбонилокси, включая C1-C5алкилкарбонилокси, необязательно замещенного арила, необязательно замещенного арилалкила, включая арил(C1-C4алкил); необязательно замещенного арилалкилокси, включая арил(C1-C4алкилокси); необязательно замещенного арилалкилкарбонилокси, включая арил(C1-C4алкилкарбонилокси); дифенилметокси и трифенилметокси; и
R12 выбран из водорода, алкила, включая C1-C6алкил; циклоалкила, включая C3-C8циклоалкил; алкоксикарбонила, включая C1-C4алкоксикарбонил; необязательно замещенного арилоксикарбонила, необязательно замещенного арилалкила, включая арил(C1-C4алкил); и необязательно замещенного арилоила.
В другом аспекте в изобретении описаны соединения формул (I), (II) или (III), в которых R3 представляет собой структуру, выбранную из группы, состоящей из
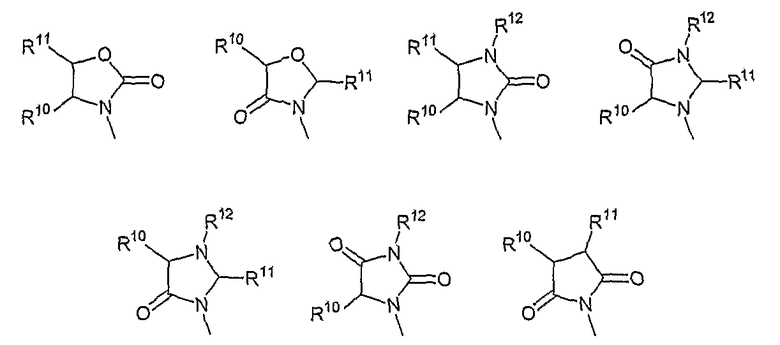
где R10, R11 и R12 соответствуют определениям, данным в настоящей заявке.
В другом аспекте в изобретении описаны соединения формул (I), (II) или (III), в которых R3 представляет собой структуру, выбранную из группы, состоящей из
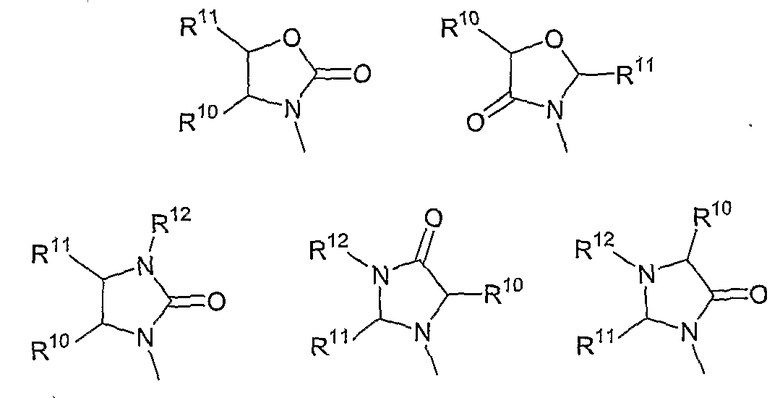
где R10, R11 и R12 соответствуют определениям, данным в настоящей заявке.
В другом аспекте в изобретении описаны соединения формул (I), (II) или (III), в которых R3 представляет собой структуру, выбранную из группы, состоящей из
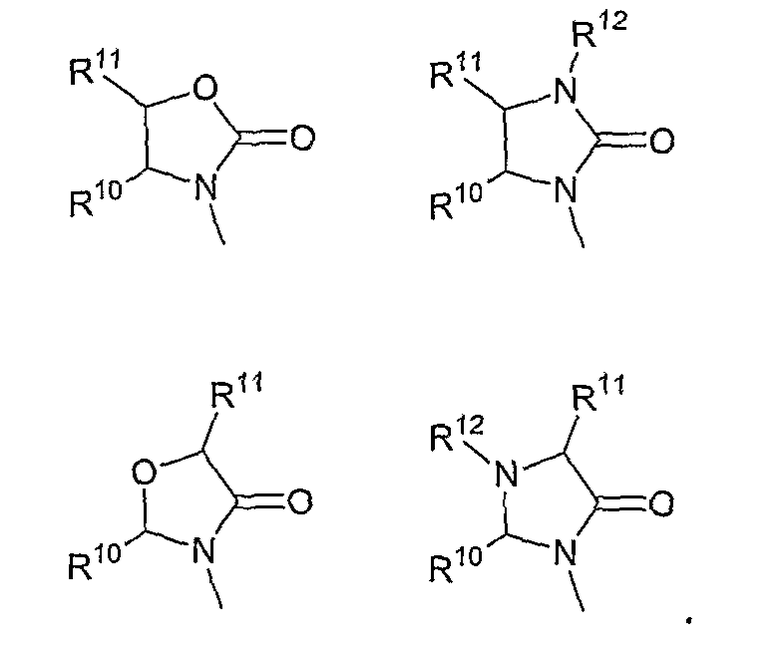
где R10,R11 и R12 соответствуют определениям, данным в настоящей заявке.
Следует понимать, что описанные выше в настоящей заявке варианты осуществления, аспекты и разновидности по настоящему изобретению могут объединяться друг с другом всеми возможными путями с получением дополнительных вариантов осуществления, аспектов и разновидностей. Например, в другом аспекте изобретение относится к соединениям формул (I), (II) и (III), в которых A представляет собой дизамещенную аминогруппу, имеющую формулу R14XN-, где R14 и X совместно с присоединенным атомом азота образуют пиперидинил, необязательно замещенный по 4 положению алкилом, включая C1-C4алкил; или гетероциклил(C1-C4алкилом); и R3 представляет собой структуру
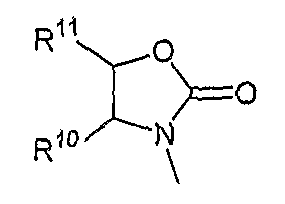
где R10 и R11 соответствуют определениям, данным в настоящей заявке.
Соединения, описанные в настоящем изобретении, включают структуру ядра азетидинона, которая содержит асимметрические атомы углерода C(3) и C(4), образуя четыре стереоизомерные конфигурации, как показано следующими формулами:
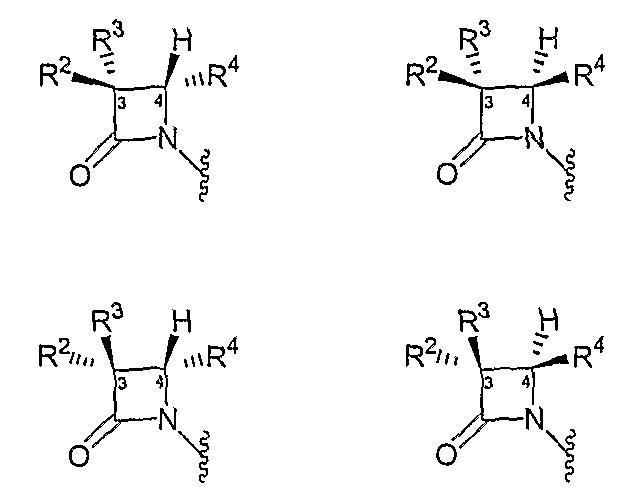
Следовательно, описанные в настоящей заявке соединения могут существовать в виде одного диастереомера, в виде рацемической смеси или в виде смеси различных диастереомеров. Предполагается, что для некоторых приложений могут применяться определенные стереоизомеры или смеси стереоизомеров, тогда как в других приложениях могут применяться другие стереоизомеры или смеси стереоизомеров. В некоторых вариантах осуществления описан один стереоизомер, например, со структурой ядра азетидинона, имеющей диастереомерную конфигурацию (3S,4R).
Кроме того, предполагается, что α-атом углерода, несущий заместитель R1, также является хиральным. Кроме того, группы, выбранные в качестве заместителей R1, R2, R3, R4 и A, также могут включать хиральные центры. Например, если R3 представляет собой 4-замещенный оксазолидин-2-он-3-ил, то положение 4 этого цикла является асимметрическим. Далее, если R3 представляет собой 2,5-дизамещенный оксазолидин-4-он-3-ил или 1,2,5-тризамещенный имидазолидин-4-он-3-ил, то каждый из атомов углерода в положениях 2 и 5 этих циклов является асимметрическим. Наконец, если R3 представляет собой сукцинимидо, и один из заместителей R14 и R15 является водородом, углерод, имеющий неводородный заместитель, также является асимметрическим. Следовательно, дополнительные стереоизомеры в своей совокупности представлены формулами (I), (II) или (III). Хотя в настоящем описании рассмотрены соединения, включающие все комбинации стереохимически чистых изомеров, принимается во внимание, что во многих случаях в описанных соединениях хотя бы один из описанных выше хиральных центров может присутствовать в одной абсолютной конфигурации. В одном иллюстративном аспекте описанные в заявке соединения имеют абсолютную конфигурацию (αR,3S,4R) или абсолютную конфигурацию (αS,3S,4R).
Иллюстративные варианты соединений, описанных в настоящем изобретении, включают классы соединений формул (I), (II) или (III), в которых:
A представляет собой R5O-;
A представляет собой R5O-, и R5 представляет собой C1-C6алкил;
A представляет собой R5O-, и R5 представляет необязательно замещенный арил(C1-C4алкил);
A представляет собой монозамещенную аминогруппу формулы XNH-;
A представляет собой дизамещенную аминогруппу, имеющую формулу R14XN-;
A представляет собой XNH- или R14XN, и X представляет собой необязательно замещенный арил(C1-C4алкил);
A представляет собой XNH- или R14XN, и X представляет собой R6R7N-(C1-C4алкил);
A представляет собой XNH- или R14XN, X представляет собой R6R7N-(C1-C4алкил) и R6 и R7 совместно с присоединенным атомом азота образуют гетероцикл;
A представляет собой R14XN, и R14 и X совместно с присоединенным атомом азота образуют гетероцикл;
A представляет собой R14XN, R14 и X совместно с присоединенным атомом азота образуют гетероцикл, и гетероцикл необязательно замещен необязательно замещенным гетероциклил(C1-C4алкилом);
A представляет собой R14XN, R14 и X совместно с присоединенным атомом азота образуют пиперидинил, и пиперидинил необязательно замещен по 4 положению гетероциклил(C1-C4алкилом), включая пиперидинил(C1-C4алкил), пиперазинил(C1-C4алкил) и пирролидинил(C1-C4алкил);
A представляет собой XNH- или R14XN-, и X представляет собой необязательно замещенный арил(C1-C4алкил);
A представляет собой XNH- или R14XN-, X представляет собой необязательно замещенный арил(C1-C4алкил), и арил представляет собой необязательно замещенный фенил;
R1 представляет собой водород;
R1 представляет собой C1-C6алкил;
R1 представляет собой C1-C2алкил;
R2 представляет собой водород;
R2 представляет собой C1-C2алкил;
R2 представляет собой метил;
R2 представляет собой метилтио;
R2 представляет собой циано;
R3 представляет собой 4-замещенный оксазолидин-2-он-3-ил;
R3 представляет собой 4,5-дизамещенный оксазолидин-2-он-3-ил;
R3 представляет собой 2-замещенный оксазолидин-4-он-3-ил;
R3 представляет собой 2-замещенный имидазолидин-4-он-3-ил;
R3 представляет собой 1,2-дизамещенный имидазолидин-4-он-3-ил;
R3 представляет собой 5-замещенный имидазолидин-2-он-1-ил;
R3 представляет собой 4,5-дизамещенный имидазолидин-4-он-1-ил;
R4 представляет собой необязательно замещенный 2-арилэт-1-ил;
R4 представляет собой необязательно замещенный 2-арилэтен-1-ил;
R5' представляет собой C1-C6алкил;
R5' представляет собой необязательно замещенный арил(C1-C4алкил).
Другие иллюстративные варианты соединений, описанных в настоящем изобретении, включают классы соединений формулы (II), в которых A, R5, X, R14, R1, R2, R3 и R4 соответствуют данным выше определениям; и в которых арил представляет собой фенил, замещенный фенил или 4-замещенный фенил.
Имеется в виду, что описанные выше классы соединений могут быть скомбинированы для получения дополнительных иллюстративных классов. Другие комбинации описанных выше классов соединений рассматриваются в настоящем изобретении.
Другие иллюстративные классы соединений описываются следующей формулой:
где Ar представляет собой необязательно замещенный фенил, необязательно замещенный пиридинил, необязательно замещенный фурил или необязательно замещенный тиенил; A представляет собой азотсодержащий гетероцикл, присоединенный по атому азота, который необязательно замещен гетероциклил(С1-С4алкилом); и R5' является необязательно замещенным арилалкилом, включая арил(С1-С4алкил).
Другие иллюстративные классы соединений описываются следующими формулами:
где Ar представляет собой необязательно замещенный фенил, необязательно замещенный пиридинил, необязательно замещенный фурил или необязательно замещенный тиенил; A представляет собой азотсодержащий гетероцикл, присоединенный по атому азота, который необязательно замещен гетероциклил(С1-С4алкилом); и R5' является необязательно замещенным арилалкилом, включая арил(С1-С4алкил).
Другие иллюстративные классы соединений описываются следующей формулой:
где Ar представляет собой необязательно замещенный фенил, необязательно замещенный пиридинил, необязательно замещенный фурил или необязательно замещенный тиенил; A представляет собой азотсодержащий гетероцикл, присоединенный по атому азота, который необязательно замещен гетероциклил(С1-С4алкилом); n означает 1, 2 или 3; и арил представляет собой необязательно замещенный фенил или необязательно замещенный нафтил.
В другом варианте осуществления описанные в настоящей заявке соединения включают основную аминогруппу. Такие амины способны образовывать соли со многими неорганическими и органическими кислотами с получением фармацевтически приемлемых кислотно-аддитивных солей. Предполагается, что в тех случаях, когда соединения описанных в настоящей заявке формул являются маслами, а не твердыми веществами, способность соединений образовывать твердые аддитивные соли будет облегчать обработку и введение описанных в настоящей заявке соединений. Кислотами, обычно применяемыми для образования таких солей, являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, фосфорная кислота и т.п., а также органические кислоты, такие как п-толуолсульфоновая кислота, метансульфоновая кислота, щавелевая кислота, п-бромфенилсульфоновая кислота, угольная кислота, янтарная кислота, лимонная кислота, бензойная кислота, уксусная кислота и т.п. Следовательно, примерами таких фармацевтически приемлемых солей являются сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, иодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацинаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, β-гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты, манделаты и т.п. Предпочтительными фармацевтически приемлемыми солями являются соли, образованные соляной кислотой, трифторуксусной кислотой, малеиновой кислотой или фумаровой кислотой.
Описанные в настоящей заявке соединения применимы в способах, направленных на обеспечение антагонизма к рецепторам вазопрессина V1a,V1b и V2. Этот антагонизм применим при лечении ряда расстройств и заболеваний, которые связаны с указанными рецепторами у млекопитающих. В качестве иллюстрации, млекопитающим, подвергаемым лечению путем введения соединений, описанных в настоящей заявке, является человек.
В другом варианте осуществления в настоящей заявке также описаны соединения, которые проникают сквозь барьер кровь-мозг. Имеется в виду, что соединения, которые преодолевают барьер кровь-мозг, могут иметь более широкое применение при лечении различных болезненных состояний, которые чувствительны к антагонистам вазопрессина. Например, необходимо понимать, что таковыми в настоящее время признаются различные подтипы депрессивных заболеваний.
В другом варианте осуществления изобретение относится к способам получения соединений формул (I), (II) или (III). В одном из аспектов описан способ получения соединений формулы:
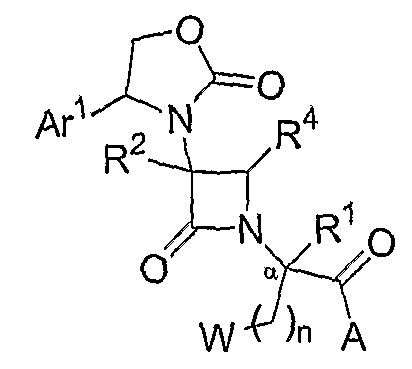
где W представляет собой QR5' или арил, согласно определениям, данным для различных вариантов осуществления в настоящей заявке; Ar1 представляет собой необязательно замещенный арил или необязательно замещенный гетероарил; и R1, R2, R4, n и A соответствуют определениям, данным для различных вариантов осуществления в настоящей заявке. Способы получения включают стадию взаимодействия соединения формулы:
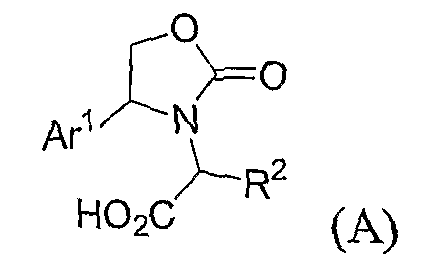
с соединением формулы
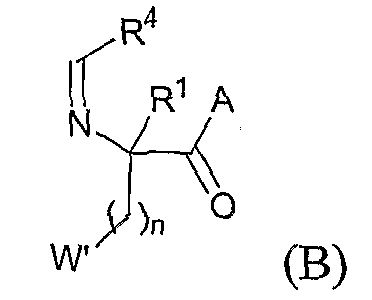
где W' представляет собой -QR5' или арил, согласно определениям, данным для различных вариантов осуществления в настоящей заявке, или W' представляет собой защищенную форму QR5' или арила, которая может быть лишена защиты или превращена в -QR5' или арил. В одном из аспектов описываемого способа, если Q представляет собой кислород, n равно 2. В одном из вариантов, описан способ получения соединений приведенной выше формулы, в которых R4 представляет собой необязательно замещенный арилэтенил. Этот способ включает стадию взаимодействия соединения формулы (A) с соединением формулы:
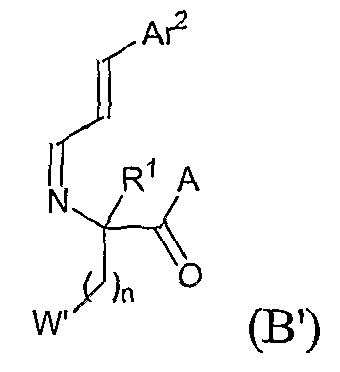
где W' представляет собой -QR5' или арил, согласно определениям, данным для различных вариантов осуществления в настоящей заявке, или W' представляет собой защищенную форму QR5' или арила, которая может быть лишена защиты или превращена в -QR5' или арил. В одном из аспектов описываемого способа, если Q представляет собой кислород, n равно 2.
Эфиры и амиды 2-(азетидинон-1-ил)уксусной кислоты, а также их аналоги и производные, описанные в настоящей заявке, в основном могут быть получены известными в технике способами синтеза, а также с применением различных методик, описанных в настоящей заявке. Как показано для соединений формул (I), (II) и (III), описанные в настоящей заявке эфиры 2-(азетединон-1-ил)алкандикарбоновой кислоты могут быть получены 2+2 циклоприсоединением подходящим образом замещенных производных уксусной кислоты (i) и иминоэфиров (ii) при обработке основанием в подходящем растворителе, как показано на схеме синтеза I, где Z представляет собой гидроксил или уходящую группу, и целое число n, а также фрагменты A, R1, R2, R3 и R4 соответствуют данным выше описаниям. Термин «уходящая группа», используемый далее по тексту заявки, относится к заместителям, например галогенам, ацилокси, бензоилокси и т.п., связанным с активированным атомом углерода, которые могут быть замещены нуклеофилом. Химические реакции, показанные на схеме синтеза I, применимы к иминам (ii), которые содержат фрагменты сложных эфиров, тиоэфиров или амидов.
Схема синтеза I
Получение соответствующих иминов (ii), получение типовых примеров требуемых ацетилгалогенидов или ангидридов (i) и методика циклоприсоединения в основном описаны в патентах США №№ 4665171 и 4751299, содержание которых включено в настоящую заявку в качестве ссылки. Имеется в виду, что в случае, если Q в соединениях (ii-a) представляет собой серу или ее окисленную форму, например сульфоксид или сульфон, это может быть несовместимо с условиями проведения некоторых реакций. В этих случаях для блокирования нежелательных реакций атома серы могут применяться правильно подобранные защитные группы. Иллюстративные защитные группы для атома серы описаны в книге Greene & Wuts “Protective Groups in Organic Synthesis”, 2d Ed., John Wiley & Sons, New York, 1991, содержание которой включено в настоящую заявку в качестве ссылки.
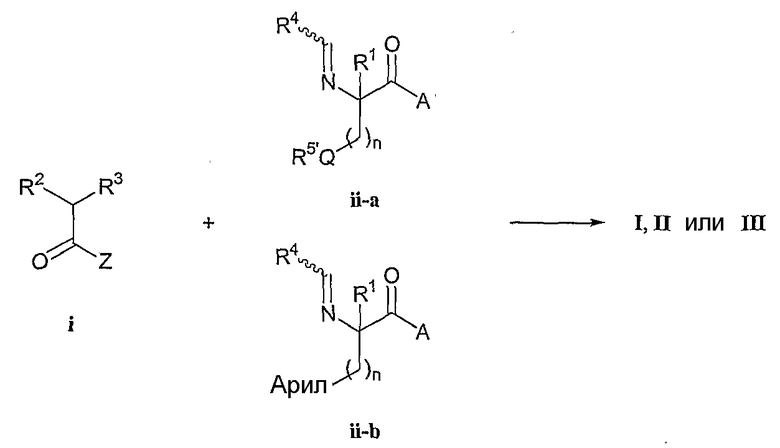
В одном иллюстративном варианте R3 представляет собой 4-замещенный оксазолидин-2-он-3-ил или 1,4,5-тризамещенный имидазолидин-2-он-3-ил. Эти соединения формул (I), (II) и (III) для которых требуется, чтобы R3 представлял собой 4-замещенный оксазолидин-2-он-3-ил или 1,4,5-тризамещенный имидазолидин-2-он-3-ил, получают из соответствующих (4-замещенных оксазолидин-2-он-3-ил) или (1,4,5-тризамещенных имидазолидин-2-он-3-ил)ацетилгалогенидов или ангидридов. Галогенангидрид или ангидрид кислоты может быть получен из соответствующим образом замещенного глицина. Этот глицин на первой стадии превращают в карбамат и затем восстанавливают с получением соответствующего спирта. Затем спирт циклизуют в 4-замещенный оксазолидин-2-он, который затем вводят в реакцию N-алкилирования с эфиром галогенуксусной кислоты. Эфир гидролизуют и полученную кислоту превращают в ацетилгалогенид или ангидрид (i). Иллюстративные примеры оксазолидинонов, которые используются в данном способе синтеза и последующих способах синтеза, описанных в настоящей заявке, включают следующие коммерчески доступные соединения.
Иллюстративные примеры имидазолидинонов и имидазолидиндионов, которые используются в данном способе синтеза и последующих способах синтеза, описанных в настоящей заявке, включают следующие коммерчески доступные соединения.
В другом иллюстративном варианте R3 представляет собой 2,5-дизамещенный оксазолидин-4-он-3-ил или 1,2,5-тризамещенный имидазолидин-4-он-3-ил. Соединения формул (I), (II) и (III), для которых требуется, чтобы R3 представлял собой 2,5-дизамещенный оксазолидин-4-он-3-ил или 1,2,5-тризамещенный имидазолидин-4-он-3-ил, получают из соответствующих (2,5-дизамещенного оксазолидин-4-он-3-ил) или (1,2,5-тризамещенного имидазолидин-4-он-3-ил)ацетилхлоридов или ангидридов соответственно. Условия реакции, применимые для получения указанных соединений, описаны в патенте США № 4772694, включенном в настоящую заявку в качестве ссылки. Вкратце, требуемый оксазолидинон или имидазолидинон получают соответственно из α-гидроксикислоты или α-аминокислоты. Имидазолоны получают, превращая α-аминокислоту, т.е. (R11)-CH(NH2)CO2H, в амид с защищенной аминогруппой, и затем конденсируя этот амид с альдегидом, т.е. (R10)-CHO, в присутствии кислоты с получением защищенного по положению 3 имидазолидин-4-она, где R10 и R11 соответствуют данным выше определениям. В положение 1 с помощью подходящего реагента может быть введена функциональная группа R12, где R12 соответствует данному выше определению, и с положения 3 может быть снята защита. Затем цикл имидазолидин-4-она алкилируют эфиром галогенуксусной кислоты, эфир расщепляют и полученную замещенную уксусную кислоту превращают в соответствующий галогенангидрид или ангидрид (i). Требуемые оксазолидиноны получают аналогичным способом из соответствующих α-гидроксикислот, т.е. (R11)-CH(OH)CO2H.
В другом иллюстративном варианте R3 представляет собой сукцинимидогруппу. Те соединения формул (I), (II) и (III), для которых требуется, чтобы R3 представлял собой сукцинимидо, получают из соответствующих 2-(сукцинимидо)ацетилгалогенидов или ангидридов. Способы получения этих реагентов описаны в патенте США № 4734498, включенном в настоящую заявку в качестве ссылки. Вкратце, эти соединения получают из винной кислоты или, если один из заместителей R10 и R11 является водородом, из яблочной кислоты. Винную кислоту ацилируют или O-алкилируют, соответствующую диацил или ди-O-алкилвинную кислоту обрабатывают ангидридом кислоты для получения янтарного ангидрида и реакцией этого янтарного ангидрида со сложным эфиром глицина получают вначале нециклический полуамидоэфир, который затем циклизуют с получением 3,4-дизамещенного эфира сукцинимидоуксусной кислоты. Расщепляют эфирную группу и превращают полученную кислоту в соответствующий галогенангидрид или ангидрид кислоты (i). Монозамещенный сукцинимидоацетил галогенангидрид или ангидрид получают из яблочной кислоты через образование янтарного ангидрида с последующим образованием сукцинимида, как описано выше.
В другом иллюстративном варианте R3 представляет собой N-замещенный амин или N'-замещенную мочевину. Те соединения формул (I), (II) и (III), для которых требуется, чтобы R3 представлял N-замещенный амин или N'-замещенную мочевину, могут быть получены из соответствующих 3-амино аналогов, защищенных фталимидом. Фталимидная защитная группа может быть удалена с использованием стандартных методик, как, например, обработкой гидразином и т.п. После снятия защиты аминогруппа может быть алкилирована одним из большого числа алкил и циклоалкил галогенидов и сульфатов, например метилиодидом, изопропилбромидом, диэтилсульфатом, циклопропилметилбромидом, циклопентилиодидом и т.п. Указанные аминогруппы также могут быть ацилированы галогенангидридами кислот, ангидридами кислот, изоцианатами, изотиоцианатами, такими как ацетилхлорид, пропионовый ангидрид, метилизоцианат, 3-трифторметилфенилизотиоцианат и т.п.
Основания, которые необходимо применять в схеме синтеза I, включают, в числе прочих, третичные алифатические амины, например триметиламин и триэтиламин, циклические третичные амины, например N-метилпиперидин и N-метилморфолин, ароматические амины, например пиридин и лутидин, а также другие органические основания, например 1,8-диазабицикло[5,4,0]ундец-7-ен (DBU).
Растворители, применимые для проведения реакций, описанных в схеме синтеза I, включают, в том числе, диоксан, тетрагидрофуран, диэтиловый эфир, этилацетат, дихлорметан, хлороформ, четыреххлористый углерод, бензол, толуол, ацетонитрил, диметилсульфоксид и N,N-диметилформамид. Предполагается, что с применением описанных в настоящей заявке способов может быть получена любая желаемая стереохимическая конфигурация целевых соединений путем выбора желаемой конфигурации у каждого хирального центра из числа отмеченных выше. Такой выбор может быть осуществлен путем применения оптически чистых исходных соединений или путем разделения смесей оптических изомеров на подходящей стадии синтеза соединений двух указанных выше формул с применением стандартных методик.
Кроме того, может быть получен азетидиноновый цикл, не содержащий всех необходимых заместителей R2, R3, R4 или R1-замещенной N-алкандиовой кислоты, или фрагмента алкоксиалкановой кислоты, но обладающий заместителями, способными в ходе дальнейших химических превращений преобразовываться в те группы, которые описаны для соединений формул (I), (II) и (III). В основном азетидиноны могут быть получены путем N-C(4) циклизации, как, например, циклизации ацилгидроксаматов (iv), в азетидиноновые промежуточные соединения (v), как показано на схеме II и продемонстрировано для соединений формулы (I), где R1, R2, R3, R4 и A соответствуют данным выше определениям, согласно методике Mattingly et al., в J. Am. Chem.Soc. (1979), 101, 3983 и Accts. Chem. Res.(1986), 19, 49, причем содержание указанных источников включено в настоящее описание в качестве ссылки. Имеется в виду, что другие гидроксаматы, например алкилгидроксаматы, арилгидроксаматы и т.п., подходят для проведения данной циклизации.
Схема синтеза II
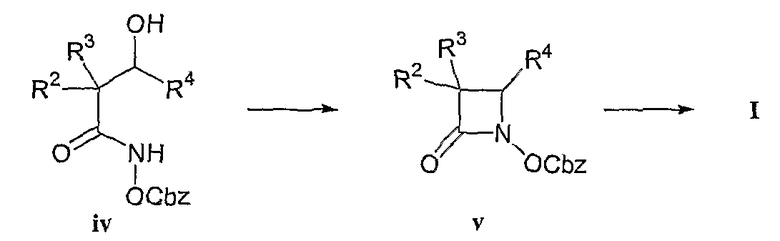
Последующие химические превращения ацилоксиазетидинона (V), предназначенные для введения, например, R1-замещенного имина аминокислоты с применением стандартных методик, иллюстративно приведут к получению соединений формул (I), (II) и (III).
Альтернативная циклизации для получения промежуточных азетидинонов, которые затем могут быть преобразованы в соединения формул (I), (II) и (III), может проводиться путем окислительной циклизации ацилгидроксаматов (vi) с получением промежуточных азетидинонов (vii), как показано на схеме синтеза III и продемонстрировано для соединений формулы (I), где R2 и R3 соответствуют данным выше определениям, и L представляет собой уходящую группу, например галогенид, согласно методике Rajendra и Miller в J. Org. Chem. (1987), 52, 4471 и Tetrahedron Lett. (1985), 26, 5385, причем содержание указанных работ включено в настоящую заявку в качестве ссылки. Группа R на схеме III представляет собой алкильный или арильный фрагмент, выбранный таким образом, чтобы при последующих превращениях обеспечить включение в молекулу заместителя R4, соответствующего данным выше определениям. Например, R может являться группой ArCH2-, где Ar представляет собой необязательно замещенную арильную группу, как в формуле (vii-a), так чтобы окислительное элиминирование HBr приводило к желаемому заместителю R4, например стирильной группе, как в формуле (vii-b). Имеется в виду, что превращение R в R4 необязательно осуществляется сразу после циклизации, и эту реакцию удобно проводить после других стадий синтеза соединений формул (I), (II) и (III). Кроме того, имеется в виду, что для осуществления циклизации подходят альтернативы показанным ацилгидроксаматам, например алкилгидроксаматы, арилгидроксаматы и т.п.
Схема синтеза III
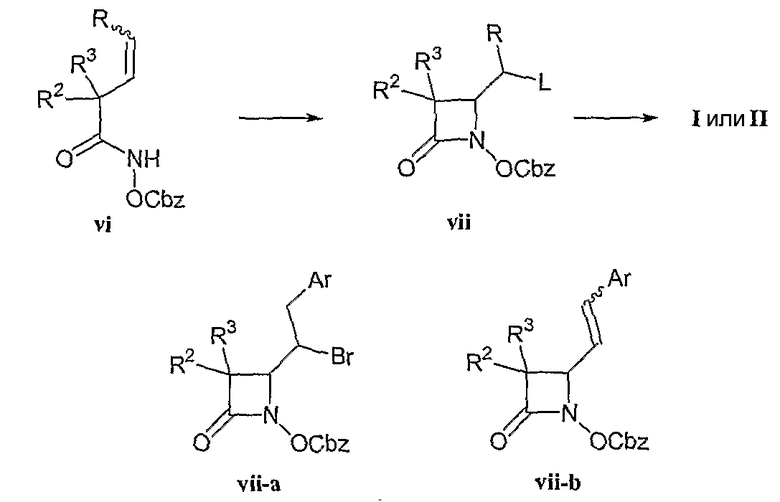
Другие применимые промежуточные соединения, такие как производные азетидинонилуксусной кислоты (x), могут быть превращены в соединения формул (I), (II) и (III), как показано для случая синтеза соединений формулы (I) на схеме синтеза IV и продемонстрировано для соединений формулы (I), где R1, R2, R3, R4, A и n соответствуют данным выше определениям. Введение фрагмента R1 и производного карбоновой кислоты R5'-Q-(CH2)n- в соединения формулы (I) может быть выполнено алкилированием аниона (x).
Схема синтеза IV
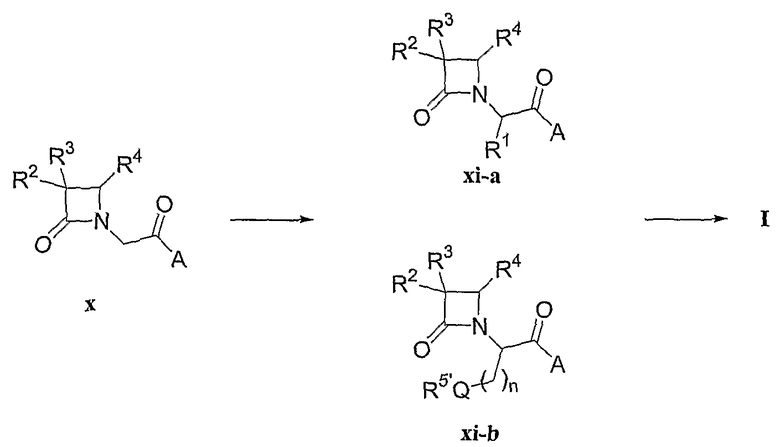
Производное уксусной кислоты (x) депротонируют и затем алкилируют алкилгалогенидом формулы R1-Z, где Z является уходящей группой, получая промежуточное соединение (xi-a). В качестве иллюстрации анион соединения (xi-a) может быть алкилирован соединением Z'-(CH2)nQR5', где Z' представляет собой уходящую группу, с получением соединения формулы (I).
Раствор производного 2-(3,4-дизамещенной азетидин-2-он-1-ил)уксусной кислоты (x) или (xi) в подходящем растворителе, например тетрагидрофуране, диоксане или диэтиловом эфире, обрабатывают ненуклеофильным основанием для получения анионов соединений (x) или (xi) соответственно. Подходящие для этой реакции основания включают диизопропиламид лития, 2,2,6,6-тетраметилпиперидинамид лития или бис(триметилсилил)амид лития. Затем для получения желаемых соединений образовавшийся анион вводят во взаимодействие с подходящим электрофилом. Применение иллюстративных электрофилов, представленных формулой арил-(CH2)n-Z, приводит к получению соответствующих соединений.
Предшествующие синтетические методики могут применяться в большинстве случаев для получения описанных в настоящей заявке соединений, включая, но не ограничиваясь перечисленным, аналоги серина, гомосерина, цистеина, гомоцистеина, фенилаланина, гомофенилаланина и других гомологов указанных соединений. Кроме того, те же способы синтеза могут применяться для получения аналогов и производных указанных соединений, как, например, аналогов тирозина, аналогов, включающих нафтил и замещенный нафтил, окисленных вариантов серусодержащих соединений, дисульфидных вариантов серусодержащих соединений, окисленных дисульфидных вариантов серусодержащих соединений и т.п.
С другой стороны, варианты соединений с дисульфидным фрагментом могут быть получены из аналогов серина и гомосерина путем превращения концевой гидроксильной группы в уходящую группу, например галоген, алкил или арилсульфонил, ацилокси и т.п., для получения соединений формулы (I) или (III), как показано на схеме V и продемонстрировано для соединений формулы (I).
Схема синтеза V
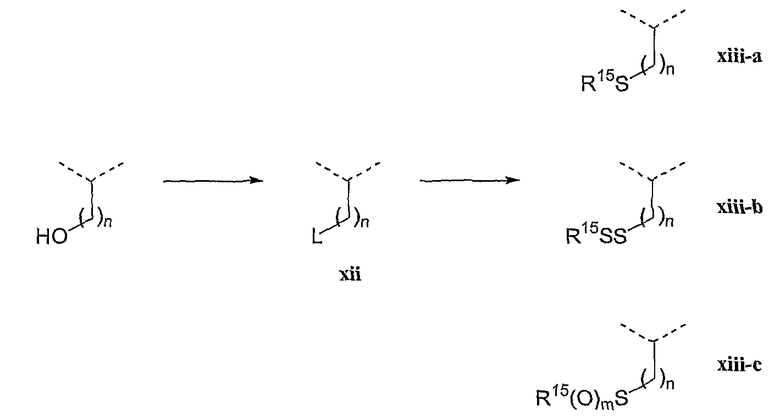
Аналоги серина и гомосерина можно превратить в соединения формулы (xii), где L является уходящей группой, применяя стандартные способы. Затем соединения (xii) могут быть превращены в соединения (xiii) обработкой сульфид-анионом, дисульфид-анионом, сульфоксид-анионом или сульфонил-анионами, где R15 соответствует данным в настоящей заявке определениям и m равно 1 или 2. Считается, что другие нуклеофилы, включая сульфонилтио, также могут применяться для замещения уходящей группы L при получении соединений (xiii).
С другой стороны, соединения с окисленными атомами серы могут быть синтезированы путем обработки полученных в результате нуклеофильного замещения тиоэфиров или дисульфидных соединений окислителем, например окислителем на основе пероксидов и т.п. В число типовых окислителей входят пероксид водорода, другие пероксиды, пероксикислоты и т.п. В случае окисления дисульфидов считается, что окислению может подвергаться только один из двух атомов серы. Кроме того, считается, что в таких условиях может быть селективно окислен атом серы, находящийся рядом с группой, которая является более сильным донором электронной плотности.
С другой стороны, соединения с окисленными атомами серы могут быть синтезированы обычной обработкой тиоэфирных или дисульфидных соединений, описанных в настоящей заявке, окислителем, например окислителем на основе пероксидов и т.п. В число типовых окислителей входят пероксид водорода, другие пероксиды, пероксикислоты и т.п. В случае окисления дисульфидов считается, что окислению может подвергаться только один из двух атомов серы. Кроме того, считается, что в таких условиях может быть селективно окислен атом серы, находящийся рядом с группой, которая является более сильным донором электронной плотности.
Соединения, полученные согласно схемам синтеза I-V, могут быть чистыми диастереомерами, смесями диастереомеров или рацематами. Фактический стереохимический состав соединения будет определяться конкретными условиями реакции, комбинацией заместителей, а также стереохимией или оптической активностью примененных реагентов. Считается, что диастереомерные смеси могут быть разделены хроматографией или фракционной кристаллизацией с получением, если это желательно, отдельных диастереомеров, с применением стандартных методик. В частности, реакции, описанные в схемах синтеза II, III и IV, создают новый хиральный центр на атоме углерода, несущем заместитель R1.
Кроме того, были описаны альтернативные способы синтеза, включая синтез нескольких представителей структурного класса эфиров и амидов замещенной 2-(азетидин-2-он-1-ил)уксусной кислоты, с целью получения β-лактамовых антибиотиков. Смотрите, например, патент США № 4751299.
Следующие далее синтезы и примеры дают дополнительную информацию о соединениях, которые являются показательными для описанного в настоящей заявке изобретения, включая способы синтеза этих соединений, но в отношении указанных синтезов и примеров не подразумевается и не следует считать, что они каким бы то ни было образом ограничивают объем настоящего изобретения. Если не указано иное, все реакции проводили при комнатной температуре, и упаривание реакционных смесей осуществляли в вакууме. Все описанные ниже соединения характеризовали стандартными аналитическими способами, включая спектроскопию ядерного магнитного резонанса (ЯМР) и масс-спектральный анализ (МС).
ПРИМЕРЫ
В каждом из приведенных ниже примеров спектр 1H ЯМР соответствовал предполагаемой структуре. Кроме того, проводили масс-спектральный анализ с использованием FAB+ для наблюдения соответствующего исходного иона (M+H)+.
ПРИМЕР 1A
(4(S)-фенилоксазолидин-2-он-3-ил)ацетилхлорид
Раствор 1,0 эквивалента (4(S)-фенилоксазолидин-2-он-3-ил)уксусной кислоты (Evans, патент США № 4665171) и 1,3 эквивалента оксалилхлорида в 200 мл дихлорметана обрабатывали каталитическим количеством безводного диметилформамида (85 мкл/миллиэквивалент производного уксусной кислоты), что приводило к энергичному выделению газа. Через 45 минут выделение газа полностью прекращалось, и реакционную смесь концентрировали при пониженном давлении, получая указанное в заглавии соединение в виде не совсем белого твердого вещества, после высушивания в вакууме в течение 2 ч.
ПРИМЕР 1B
4(R)-фенилоксазолидин-2-он-3-ил)ацетилхлорид
Соединение получали, следуя методике примера 1A, за исключением того, что использовали (4(R)-фенилоксазолидин-2-он-3-ил)уксусную кислоту вместо (4(S)-фенилоксазолидин-2-он-3-ил)уксусной кислоты (смотрите Evans & Sjogren, Tetrahedron Lett. 26:3783(1985)).
ПРИМЕР 1C
2-(4(S)-фенилоксазолидин-2-он-3-ил)пропаноилхлорид
Раствор 1 эквивалента соединения примера 3A и 1,3 эквивалентов оксалилхлорида в 200 мл CH2Cl2 (150 мл/г производного пропионовой кислоты) обрабатывали каталитическим количеством безводного ДМФА (85 мкл/ммоль производного пропионовой кислоты), что приводило к интенсивному выделению газа. Через 45 мин выделение газа полностью прекращалось, и реакционную смесь концентрировали при пониженном давлении, получая указанное в заглавии соединение в виде не совсем белого твердого вещества после высушивания в вакууме в течение 2 ч.
ПРИМЕР 2A
Метил(4(S)-фенилоксазолидин-2-он-3-ил)ацетат
Раствор (4(S)-фенилоксазолидин-2-он-3-ил)уксусной кислоты (1 г, 4,52 ммоль) (Evans в патенте США № 4665171) в 20 мл безводного метанола в течение каждого часа обрабатывали 5 эквивалентами ацетилхлорида, используя в общей сложности 20 эквивалентов. Полученный раствор перемешивали в течение ночи. Остаток, полученный после выпаривания метанола, повторно растворяли в 30 мл CH2Cl2 и обрабатывали 50 мл насыщенного водного раствора Na2CO3. Органический слой упаривали и высушивали (MgSO4), получая указанное в заглавии соединение в виде бесцветного масла (1,001 г, 94%); 1H ЯМР (CDDl3): δ 3,37 (д, J=18,0 Гц, 1H), 3,69 (c, 3H), 4,13 (т, J=8,3 Гц, 1H), 4,28 (д, J=18,0 Гц, 1H), 4,69 (т, J=8,8 Гц, 1H), 5,04 (т, J=8,4 Гц, 1H), 7,26-7,29 (м, 2H), 7,36-7,42 (м, 3H).
ПРИМЕР 2B
Метил 2-(4(S)-фенилоксазолидин-2-он-3-ил)пропаноат
Раствор соединения примера 2A (1г, 4,25 ммоль) в 10 мл безводного ТГФ при -78°C обрабатывали 4,68 мл (4,68 ммоль) 1М раствора бис(триметилсилил)амида лития в ТГФ. Реакционную смесь перемешивали в течение 1 ч при примерно -70°C и затем добавляли MeI (1,59 мл, 25,51 ммоль). После полного превращения азетидинона реакционную смесь гасили насыщенным водным раствором NH4Cl и распределяли между EtOAc и водой. Органический слой последовательно промывали насыщенным водным раствором бисульфита натрия и насыщенным водным раствором NaCl. Образовавшийся органический слой высушивали (MgSO4) и упаривали, получая указанное в заглавии соединение (смесь диастереомеров) в виде белого твердого вещества (1,06 г, 93%); 1H ЯМР (CDCl3): δ 1,07/1,53 (д/д, J=7,5 Гц, 3H), 3,59/3,74 (c/c, 3H), 3,85/4,48 (кв/кв, J-7,5 Гц, 1H), 4,10-4,14 (м, 1H), 4,60-4,64/4,65-4,69 (м/м, 1H), 4,88-4,92/4,98-5,02 (м/м, 1H), 7,24-7,40 (м, 5H).
ПРИМЕР 3A
2-(4(S)-фенилоксазолидин-2-он-3-ил)пропановая кислота
К раствору соединения примера 2B (1 г, 4,01 ммоль) в 35 мл MeOH добавляли при 0°C 14,3 мл (12,04 ммоль) 0,84М раствора LiOH в воде. Затем реакционную смесь перемешивали в течение 3 ч при комнатной температуре. По завершении гидролиза азетидинона MeOH удаляли выпариванием, неочищенный остаток растворяли в CH2Cl2 и обрабатывали насыщенным водным раствором NaCl. Полученный органический слой высушивали (MgSO4) и упаривали, получая указанное в заглавии соединение (рацемическую смесь) в виде белого твердого вещества (0,906 г, 96%). 1H ЯМР (CDCl3): δ 1,13/1,57 (д/д, J=7,5 Гц, 3H), 3,75/4,50 (кв/кв, J=7,5 Гц, 1H), 4,10-4,16 (м, 1H), 4,62-4,72 (м, 1H), 4,92-5,03 (м, 1H), 7,32-7,43 (м, 5H).
ПРИМЕР 4
Общая методика получения амидов из активированных эфирных производных
β-трет-Бутиловый эфир α-(3-трифторметил)бензиламида N-бензоксикарбонил-L-аспарагиновой кислоты. Раствор β-трет-бутилового эфира α-N-гидроксисукцинимдэфира N-бензоксикарбонил-L-аспарагиновой кислоты (1,95 г, 4,64 ммоль, Advanced ChemTech) в 20 мл сухого тетрагидрофурана обрабатывали 0,68 мл (4,74 ммоль) 3-(трифторметил)бензиламина. После завершения реакции (ТСХ, 60:40 гексан/этилацетат) смесь упаривали и полученное масло распределяли между дихлорметаном и насыщенным водным раствором бикарбоната натрия. Органический слой упаривали, получая 2,23 г (количественный выход) указанного в заглавии соединения в виде белого твердого вещества. 1H ЯМР (CDCl3): δ 1,39 (c, 9H), 2,61 (дд, J=6,5 Гц, J=17,2 Гц, 1H), 2,98 (дд, J=3,7 Гц, J=17,0 Гц, 1H), 4,41 (дд, J=5,9 Гц, J=15,3 Гц, 1H), 4,50-4,57 (м, 2H), 5,15 (c, 2H), 5,96-5,99 (м, 1H), 6,95 (c, 1H), 7,29-7,34 (м, 5H), 7,39-7,43 (м, 2H), 7,48-7,52 (м, 2H).
ПРИМЕР 5
Общая методика гидролиза трет-бутиловых сложных эфиров
Раствор трет бутилового сложного эфира в муравьиной кислоте, как правило, 1 г в 10 мл, перемешивали при комнатной температуре до тех пор, пока эфир не переставал обнаруживаться с помощью тонкослойной хроматографии (дихлорметан 95%/метанол 5%), причем типовое время реакции составляло около 3 часов. Муравьиную кислоту выпаривали при пониженном давлении, полученный твердый остаток распределяли между дихлорметаном и насыщенным водным раствором бикарбоната. Органический слой упаривали, получая не совсем белое твердое вещество, которое можно было непосредственно использовать для проведения дальнейших реакций или при желании перекристаллизовать из системы растворителей.
ПРИМЕР 6
Общая методика получения амидов из карбоновых кислот
Показана для случая получения β-трет-бутилового эфира α-(3-трифторметил)бензиламида N-бензоксикарбонил-D-аспарагиновой кислоты. Раствор 1 г (2,93 ммоль) моногидрата β-трет-бутилового эфира N-бензоксикарбонил-D-аспарагиновой кислоты (Novabiochem) в 3-4 мл дихлорметана обрабатывали последовательным добавлением 0,46 мл (3,21 ммоль) 3-(трифторметил)бензиламина, 0,44 г (3,23 ммоль) 1-гидрокси-7-бензотриазола и 0,62 г (3,23 ммоль) гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида. После как минимум 12-часового выдерживания при комнатной температуре или после завершения реакции по данным тонкослойной хроматографии (элюент 95:5 дихлорметан/метанол) реакционную смесь последовательно промывали насыщенным водным раствором бикарбоната натрия и дистиллированной водой. Органический слой упаривали, получая 1,41 г (количественный выход) указанного в заглавии соединения в виде не совсем белого твердого вещества. 1H ЯМР (CDCl3): δ 1,39 (c, 9H); 2,61 (дд, J=6,5 Гц, J=17,2 Гц, 1H); 2,98 (дд, J=4,2 Гц, J=17,2 Гц, 1H); 4,41 (дд, J=5,9 Гц, J=15,3 Гц, 1H); 4,50-4,57 (м, 2H); 5,10 (c, 2H); 5,96-6,01 (м, 1H); 6,91-7,00 (м, 1H); 7,30-7,36 (м, 5H); 7,39-7,43 (м, 2H); 7,48-7,52 (м, 2H).
ПРИМЕР 6A
N-трет-бутоксикарбонил-(S)-бензил-D-цистеин-[4-(2-(1-пиперидил)этил)]пиперидинамид
N-трет-бутилоксикарбонил-(S)-бензил-D-цистеин (0,289 г, 0,93 ммоль) и 4-[2-(1-пиперидил)этил]пиперидин (0,192 г, 0,98 ммоль) смешивали в дихлорметане (20 мл) согласно методике примера 6, получая 0,454 г (количественный выход) не совсем белого твердого вещества. 1H ЯМР (CDCl3): δ 0,89-1,15 (м, 2H); 1,39-1,44 (м, 16H); 1,54-1,61 (м, 4H); 1,62-1,71 (м, 1H); 2,21-2,35 (м, 5H); 2,49-2,58 (м, 2H); 2,66-2,74 (м, 1H); 2,79-2,97 (м, 1H); 3,67-3,76 (м, 3H); 4,48-4,51 (м, 1H); 4,72-4,75 (м, 1H); 5,41-5,44 (м, 1H); 7,19-7,34 (м, 5H).
ПРИМЕР 7А
трет-Бутиловый эфир N-[(9H-флуорен-9-ил)метоксикарбонил]-O-бензил-D-серина
N-[(9H-Флуорен-9-ил)метоксикарбонил]-O-бензил-D-серин (0,710 г, 1,70 ммоль) в дихлорметане (8 мл) обрабатывали трет-бутилацетатом (3 мл) и концентрированной серной кислотой (40 мкл) в плотно закрытой колбе при 0°C. После завершения взаимодействия (ТСХ), реакционную смесь гасили дихлорметаном (10 мл) и насыщенным водным раствором бикарбоната калия (15 мл). Органический слой промывали дистиллированной водой и упаривали. Полученный остаток очищали колоночной флэш-хроматографией (98:2 дихлорметан/метанол), получая 0,292 г (77%) бесцветного масла. 1H ЯМР (CDCl3): δ 1,44 (c, 9H); 3,68 (дд, J=2,9 Гц, J=9,3 Гц, 1H); 3,87 (дд, J=2,9 Гц, J=9,3 Гц, 1H); 4,22 (т, J=7,1 Гц, 1H); 4,30-4,60 (м, 5H); 5,64-5,67 (м, 1H); 7,25-7,39 (м, 9H); 7,58-7,61 (м, 2H); 7,73-7,76 (м, 2H).
ПРИМЕР 8A
трет-Бутиловый эфир O-бензил-D-серина
Соединение примера 7A (0,620 г, 1,31 ммоль) в дихлорметане (5 мл) обрабатывали трис(2-аминоэтил)амином (2,75 мл) в течение 5 ч. Полученную смесь дважды промывали фосфатным буфером (pH=5,5), один раз насыщенным водным раствором бикарбоната калия и упаривали, получая 0,329 г (количественный выход) указанного в заглавии соединения в виде не совсем белого твердого вещества. 1H ЯМР (CD3OD): δ 1,44 (c, 9H); 3,48 (дд, J=J'=4,2 Гц, 1H); 3,61 (дд, J=4,0 Гц, J=9,2 Гц, 1H); 3,72 (дд, J=4,6 Гц, J=9,2 Гц, 1H); 4,47 (д, J=12,0 Гц, 1H); 4,55 (д, J=12,0 Гц, 1H); 7,26-7,33 (м, 5H).
ПРИМЕР 9
Общая методика получения 2-азетидинона из имина и ацетилхлорида
Стадия 1: Общая методика получения имина из производного аминокислоты. Раствор 1 эквивалента эфира или амида α-аминокислоты в дихлорметане последовательно обрабатывали 1 эквивалентом подходящего альдегида и водоотнимающим реагентом, например, сульфатом магния или силикагелем в количестве примерно 2 г водоотнимающего реагента на грамм исходного эфира или амида α-аминокислоты. Реакционную смесь перемешивали при комнатной температуре до тех пор, пока исходные соединения полностью не вступали в реакцию, что определяли с помощью тонкослойной хроматографии. Как правило, реакции завершались в течение часа. Затем реакционную смесь фильтровали, осадок на фильтре промывали дихлорметаном, фильтрат концентрировали при пониженном давлении и получали желаемый имин, который использовали в последующей стадии без дальнейшей очистки.
Стадия 2: общая методика 2+2 циклоприсоединения имина и ацетилхлорида. Раствор имина в дихлорметане (10 мл дихлорметана/1 г имина) охлаждали до 0°C. К этому охлажденному раствору добавляли 1,5 эквивалента подходящего амина, как правило, триэтиламина, и затем по каплям добавляли раствор 1,1 эквивалента подходящего ацетилхлорида, например, описанного в примере 1А, в дихлорметане (10 мл дихлорметана/1 г подходящего ацетилхлорида). Реакционной смеси давали нагреться до комнатной температуры в течение 1 ч и затем гасили добавлением насыщенного водного раствора хлорида аммония. Полученную смесь распределяли между водой и дихлорметаном. Слои разделяли и органический слой последовательно промывали 1 н. хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Органический слой высушивали над сульфатом магния и концентрировали при пониженном давлении. Остаток можно непосредственно применять в дальнейших реакциях или, если желательно, можно очистить хроматографией или перекристаллизацией из подходящей системы растворителей.
ПРИМЕР 9А
трет-Бутил (2R)-(бензилоксиметил)-2-[3(S)-(4(S)-фенилоксазолидин-2-он-3-ил)-4(R)-(2-стирил)азетидин-2-он-1-ил]ацетат
Имин, полученный из 0,329 г (1,31 ммоль) трет-бутилового эфира O-бензил-D-серина (пример 8A) и коричного альдегида, смешивали с 2-(4(S)-фенилоксазолидин-2-он-3-ил)ацетилхлоридом (пример 1A) согласно методике примера 9 и получали 0,543 г (73%) продукта после очистки колоночной хроматографией (90:10 гексан/этилацетат); 1H ЯМР (CDCl3): δ 1,39 (c, 9H); 3,56 (дд, J=2,7 Гц, J=9,5 Гц, 1H); 3,82 (дд, J-4,8 Гц, J=9,5 Гц, 1H); 4,11 (т, J=8,3 Гц, 1H); 4,21-4,29 (м, 2H); 4,50-4,58 (м, 3H); 4,71-4,78 (м, 2H); 6,19 (дд, J=9,1 Гц, J=16,0 Гц, 1H); 6,49 (д, J=16,0 Гц, 1H); 7,07-7,11 (м, 1H); 7,19-7,40 (м, 14H).
ПРИМЕР 9B
N-[4-[2-(пиперид-1-ил)этил]пиперидин-1-ил]амид (2S)-(бензилтиометил)-2-[3(S)-(4(S)-фенилоксазолидин-2-он-3-ил)-4(R)-(2-стирил)азетидин-2-он-1-ил]уксусной кислоты
Имин, полученный из дигидрохлорида (S)-(бензил)-D-цистеин-[4-(2-(1-пиперидил)этил)]пиперидинамида (пример 11А, 0,417 г, 0,90 ммоль) и коричного альдегида в присутствии триэтиламина (0,26 мл, 1,87 ммоль), вводили в реакцию с 2-(4(S)-фенилоксазолидин-2-он-3-ил)ацетилхлоридом (пример 1A) согласно методике примера 9, получая 0,484 г (76%) продукта в виде не совсем белого твердого вещества после перекристаллизации из смеси дихлорметан/гексан. 1H ЯМР (CDCl3): δ 0,89-1,06 (м, 2H); 1,40-1,44 (м, 5H); 1,57-1,67 (м, 6H); 2,25-2,43 (м, 6H); 2,45-2,59 (м, 2H); 2,71-2,88 (м, 2H); 3,55-3,70 (м, 3H); 4,11-4,17 (м, 1H); 4,37-4,47 (м, 2H); 4,54-4,61 (м, 1H); 4,64-4,69 (м, 1H); 4,76-4,84 (м, 2H); 6,05-6,19 (м, 1H); 6,66-6,71 (м, 1H); 7,12-7,40 (м, 15H).
Соединения ПРИМЕРОВ 9C-9AD, показанные в следующей таблице, также могут быть получены с применением описанных в настоящей заявке методик, путем замены описанных выше производных серина или цистеина производными, соответствующими показанным ниже соединениям.
ПРИМЕР 10А
(2R)-(Бензоксиметил)-2-[3(S)-(4(S)-фенилоксазолидин-2-он-3-ил)-4(R)-(2-стирил)азетидин-2-он-1-ил]уксусная кислота
Соединение примера 9A (0,16 г, 0,28 ммоль) гидролизовали согласно методике, использованной в примере 5, получая 0,144 г (количественный выход) не совсем белого твердого вещества; 1H ЯМР (CDCl3): δ 3,65 (дд, J=4,0 Гц, J=9,5 Гц, 1H); 3,82 (дд, J=5,5 Гц, J=9,5 Гц, 1H); 4,11 (дд, J=7,8 Гц, J=8,8 Гц, 1H); 4,33 (c, 2H); 4,50 (д, J=5,0 Гц, 1H); 4,57 (т, J=9,0 Гц, 1H); 4,67 (дд, J=4,0 Гц, J=5,0 Гц, 1H); 4,69 (дд, J=5,0 Гц, J=9,5 Гц, 1H); 4,75 (т, J=8,0 Гц, 1H); 6,17 (дд, J=9,3 Гц, J=15,8 Гц, 1H); 6,55 (д, J=16,0 Гц, 1H); 7,09-7,12 (м, 2H); 7,19-7,42 (м, 13H).
Соединение примера 10А применяли для получения других производных амидов и эфиров, например амидов и эфиров, представленных группой А в числе соединений формул (I), (II) и (III).
ПРИМЕР 11А
Дигидрохлорид (S)-(бензил)-D-цистеин-[4-(2-(1-пиперидил)этил)]пиперидинамида
В течение ночи проводили взаимодействие N-трет-бутоксикарбонил-(S)-(бензил)-D-цистеин-[4-(2-(1-пиперидил)этил)]пиперидинамида (0,453 г, 0,93 ммоль) с ацетилхлоридом (0,78 мл, 13,80 ммоль) в безводном метаноле (15 мл). Указанное в заглавии соединение получали в виде не совсем белого твердого вещества путем упаривания реакционной смеси досуха (0,417 г, 97%). 1H ЯМР (CD3OD): δ 0,94-1,29 (м, 2H); 1,49-1,57 (м, 1H); 1,62-1,95 (м, 10H); 2,65-2,80 (м, 2H); 2,81-2,97 (м, 4H); 3,01-3,14 (м, 2H); 3,50-3,60 (м, 3H); 3,81-3,92 (м, 2H); 4,41-4,47 (м, 2H); 7,25-7,44 (м, 5H).
ПРИМЕР 12А
трет-Бутил [3(S)-(4(S)-фенилоксазолидин-2-он-3-ил)-4(R)-(2-стирил)азетидин-2-он-1-ил]ацетат
Имин, полученный из 4,53 г (34,5 ммоль) трет-бутилового эфира глицина и коричного альдегида, вводили в реакцию с 2-(4(S)-фенилоксазолидин-2-он-3-ил)ацетилхлоридом (пример 1A) по методике примера 9, получая 5,5 г (30%) соединения, указанного в заголовке, в виде бесцветных кристаллов (после перекристаллизации из н-хлорбутана); т.пл 194-195°C.
ПРИМЕР 13
Общая методика алкилирования и/или ацилирования (азетидин-2-он-1-ил)ацетата
Раствор (азетидин-2-он-1-ил)ацетата в тетрагидрофуране (0,22 М по азетидинону), как в примере 12А, охлаждали до -78°C и обрабатывали бис(триметилсилил)амидом лития (2,2 эквивалента). Полученный анион обрабатывали подходящим алкил или ацилгалогенидом (1,1 эквивалента). После того как весь азетидинон вступал в реакцию, реакционную смесь гасили насыщенным водным раствором хлорида аммония и распределяли между этилацетатом и водой. Органическую фазу последовательно промывали 1 н. хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Полученный органический слой высушивали (сульфат магния) и упаривали. Остаток очищали хроматографией на силикагеле, применяя подходящий элюент, например смесь 3:2 гексан/этилацетат.
Эту методику применяли для получения соединений формул (I), (II) и (III) альтернативным способом синтеза из общего промежуточного соединения, например трет-бутил[3(S)-(4(S)фенилоксазолидин-2-он-3-ил)-4(R)-(2-стирил)азетидин-2-он-1-ил]ацетата и родственных соединений. Кроме того, эта методика применялась для получения алкилированных и ацилированных аналогов описанных в настоящей заявке соединений, например соединений формул (I), (II) и (III), в которых заместитель R1 отличается от водорода. Далее, считается, что эта методика может быть модифицирована для введения дополнительных групп в азетидиноновый цикл с целью получения описанных в настоящей заявке соединений, в которых заместитель R2 отличается от водорода.
Считается, что эпимеры этих соединений по атому углерода, находящемуся в альфа-положении относительно азетидинонового цикла, также могут быть получены в соответствии с описанными выше методиками за счет выбора подходящих исходных соединений. Кроме того, все остальные соединения, входящие в число соединений формул (I), (II) и (III), также могут быть получены в основном по методикам предшествующих примеров.
В другом варианте осуществления соединения, описанные в настоящем изобретении, применимы в качестве антагонистов рецепторов вазопрессина V1a, V1b и V2 в способах лечения пациентов, страдающих от заболеваний и болезненных состояний, которые чувствительны к рецепторам вазопрессина V1a, V1b и V2. В качестве иллюстрации способы, описанные в настоящем изобретении, включают стадию введения субъекту или пациенту, при необходимости такого лечения, эффективного количества соединения, описываемого формулами, приведенными в настоящей заявке. Антагонизм по отношению к различным подтипам рецепторов вазопрессина сопровождается многочисленными физиологическими и терапевтическими полезными эффектами. Эти эффекты могут появляться из-за антагонизма в отношении рецепторов вазопрессина как периферической, так и центральной нервной системы. Применения, относящиеся к периферической нервной системе, включают введение антагонистов рецепторов вазопрессина V1a и/или V2 в качестве вспомогательных средств при сердечной недостаточности или в качестве антитромботических средств. Действие на центральную нервную систему включает введение антагонистов рецепторов вазопрессина V1a и/или V1b, т.е. соединений, описанных в настоящей заявке, для лечения обсессивно-компульсивных расстройств, агрессивных расстройств, депрессии, тревожности и других психологических и неврологических расстройств.
Иллюстративные болезненные состояния, которые чувствительны к антагонистам рецептора вазопрессина V2 и могут подвергаться лечению описанными в настоящей заявке способами, включают различные сердечно-сосудистые расстройства, включая расстройства или состояния, связанные с агрегацией тромбоцитов и т.п. Кроме того, в настоящем изобретении описаны способы лечения других заболеваний и болезненных состояний, которые могут подвергаться лечению, например, антагонистами рецептора окситоцина, антагонистами рецептора тахикинина, антагонистами рецептора нейрокинина 1, антагонистами рецептора нейрокинина 2 и т.п., где указанный способ включает стадию введения пациенту, при необходимости облегчения такого заболевания или болезненного состояния, эффективного количества одного или нескольких соединений из числа замещенных 2-(азетидин-2-он-1-ил)алкандикарбоновых кислот, замещенных 2-(азетидин-2-он-1-ил)гидроксиалкилалкановых кислот, замещенных 2-(азетидин-2-он-1-ил)алкилалкановых кислот, а также их аналогов и производных, описанных в настоящей заявке.
ПРИМЕР МЕТОДИКИ 1
Клетки, экспрессирующие человеческий рецептор вазопрессина V 1b
кДНК человеческого рецептора вазопрессина 1B (HV1B) (смотрите Lolait et al., "Extrapituitary expression of the rat Vlb vasopressin receptor gene" Proc. Natl. Acad. Sci. USA. 92:6783-7 (1995); de Keyzer et al., "Cloning and characterization of the human V3(Vlb) pituitary vasopressin receptor" FEBS Lett. 356:215-20 (1994); Sugimoto et al., "Molecular cloning and functional expression of a cDNA encoding the human Vlb vasopressin receptor" J. Biol. Chem. 269:27088-92 (1994)) встраивали в вектор экспрессии клеток млекопитающих PCI-neo (Promega) по сайту EcoR1. Рекомбинантную плазмиду, несущую кДНК HV1B, выделяли из трансформированных клонов E.Coli и использовали для трансфекции клеток яичника китайского хомячка (CHO-K1, ATCC). Два микрограмма ДНК рецептора HV1B вводили в 105 клеток CHO, культивированных в 6-луночном планшете, используя методику трансфекции, опосредованную Fugene-6 (Boehringer Mannheim). Через двадцать четыре часа после трансфекции клетки культивировали в условиях отбора по G-418 (0,25 мг/мл), добавленного в культуральную среду. Спустя три дня проводили ограниченное разбавление для получения изолированных клонов клеток в 96-луночных планшетах. После 2-недельного периода роста моноклоны распределяли по двум группам 12-луночных планшетов. По достижении конфлюентности одну группу лунок исследовали на способность связывать меченный тритием аргинин-вазопрессин (NEN). Вначале были выявлены девять положительных клонов из числа 60 исследованных, и клоны, показавшие наиболее высокое связывание AVP, сохраняли в качестве перманентных линий клеток для отбора соединений Serenix по сродству к HV1B.
ПРИМЕР МЕТОДИКИ 2
Клеточный анализ связывания человеческих или крысиных рецепторов V 1a , V 1b и/или V 2
Клеточные линии V1a, V1b и/или V2 (клетки, экспрессирующие человеческие или крысиные рецепторы V1a, V1b и/или V2) выращивали в среде альфа-MEM с добавкой 10% телячьей сыворотки и 250 мкг/мл G418 (Gibco, Grand Island, NY) в колбе объемом 75 см3. Для конкурентного анализа связывания клетки HV1b отделяли друг от друга с помощью не содержащего ферментов раствора для разделения клеток на основе PSB (Speciality Media, Phillipursburg, NJ), следуя инструкции производителя. Клетки помещали в 12-луночные культуральные планшеты в количестве одна колба на 18 планшетов, (количество следует регулировать в зависимости от степени конфлюентности клеток) и сохраняли в культуре в течение 2-3 дней. Затем культуральную среду удаляли, клетки один раз промывали 2 мл связывающего буфера (25 мМ Hepes, 0,25% BSA, 1xDMEM, pH=7,0) при комнатной температуре. В каждую лунку добавляли 990 мкл связывающего буфера, содержащего 1 нМ 3H-AVP и затем 10 мкл последовательно разбавленных тестируемых соединений или не меченого AVP (все растворы в ДМСО). Каждый анализ проводили в трех экземплярах и получали кривые зависимости ингибирования от дозировки, состоящие из точек, соответствующих общему связыванию (только ДМСО) и 5 концентрациям (0,1, 1,0, 10, 1000 и 1000 нм) тестируемого соединения или не меченого AVP, охватывающие IC50. Клетки инкубировали в течение 30 минут при 37°C в увлажненном инкубаторе. Затем аналитическую смесь удаляли и каждую лунку три раза промывали PBS (pH=7,4). После промывания добавляли 1 мл 2% SDS на лунку и содержимому планшетов давали осесть в течение 15 мин при комнатной температуре. Осторожно постукивали по планшетам, чтобы убедиться в отслоении разрушенных клеток. Содержимое лунок полностью переносили во флакон для подсчета сцинтилляции. Затем каждую лунку промывали 0,5 мл PBS и добавляли в соответствующий флакон. Затем добавляли сцинтилляционную жидкость (Ecoscint, National Diagnostics, Atlanta, Georgia) в количестве 3 мл на сосуд. Проводили подсчет образцов на счетчике сцинтилляции жидкостей (Beckman LS801). Рассчитывали значения IC50 и Ki, используя программу Prism Curve для построения кривых по точкам.
В описанном анализе с использованием клеток, экспрессирующих человеческие рецепторы V1a или V1b, проводили испытание соединений отдельных примеров. Значения сродства к связыванию (IC50) для иллюстративных соединений приведены в следующей таблице. Кроме того, в таблице приведены константы ингибирования (Ki) иллюстративных соединений.
сродство к связыванию (IC50 (нМ))
сродство к связыванию (Ki (нМ))
сродство к связыванию (IC50 (мкМ))
сродство к связыванию (Ki (мкМ))
ПРИМЕР МЕТОДИКИ 3
Ингибирование метаболизма фосфатидилинозита, опосредованного рецептором вазопрессина V 1b - функциональный анализ активности антагонистов
Физиологические действия вазопрессина опосредуются конкретными рецепторами, связанными с G-белком. Рецепторы вазопрессина V1a, V1b и/или V2 связаны с G-белком, который связан с cAMP. Агонистический или антагонистический характер описанных в настоящей заявке соединений может быть определен по их способности ингибировать опосредованный вазопрессином метаболизм фосфатидилинозита с применением стандартных методик, включая методики, описанной в следующих параграфах.
Клетки, экспрессирующие человеческие или крысиные рецепторы V1a, V1b и/или V2, выращивали на альфа-модифицировнной минимальной поддерживающей среде, содержавшей 10% телячью сыворотку и 0,25 мг/мл G418. За три дня до проведения анализа близкие к конфлюэнтности культуры разделяли и высевали в 6-луночные планшеты для культур тканей, причем примерно 100 лунок засевали из каждой 75 см3 колбы (разделение в эквивалентном соотношении 12:1). Каждая лунка содержала 1 мл ростовой среды с 2 мкКюри [3H] миоинозита (American Radiolabeled Chemicals, St. Louis, MO).
Все анализы проводили в трех экземплярах, за исключением исходного и 10 нМ AVP (для обоих n=6). Аргинин вазопрессин (AVP) растворяли в 0,1 н. уксусной кислоте. Испытуемые препараты растворяли в ДМСО в день эксперимента и разбавляли ДМСО до 200 раз, т.е. конечной исследованной концентрации. Потенциальные лекарственные средства и AVP (или соответствующие объемы ДМСО) по отдельности добавляли в 5 мкл ДМСО в стеклянные пробирки 12х75 мм, содержавшие 1 мл аналитического буфера (сбалансированный солевой раствор Tyrode, содержавший 50 мМ глюкозы, 10 мМ LiCl, 15 мМ HEPES, pH 7,4, 10 мкМ фосфорамидон и 100 мкМ бактирацин). Инкубирование проводили в случайном порядке. Инкубирование начинали с удаления среды, содержавшей метку, один раз промывая монослой 1 мл 0,9% NaCl и добавляя содержимое в пробирки для анализа. Планшеты инкубировали в течение 1 ч при 37°C. Инкубирование прерывали удалением инкубационной среды, добавлением 500 мкл ледяной 5% трифторуксусной кислоты и оставлением полученной смеси на 15 минут.
Продукты инкубирования разделяли на колонках BioRad Poly-Prep Econo-Columns, наполненных 0,3 мл формиатной смолы AG 1 X-8100-200. Смолу смешивали с водой в соотношении 1:1 и в каждую колонку добавляли 0,6 мл полученной смеси. Затем колонки промывали 10 мл воды. Под каждую колонку помещали сцинтилляционный сосуд (20 мл). Содержимое каждой инкубационной лунки переносили в микроколонку, после чего лунку промывали 0,5 мл дистиллированной воды, которую также добавляли в микроколонку. Затем колонки дважды промывали 5 мл 5 мМ миоинозита для элюирования свободного инозита. 1 мл аликвоты полученной жидкости переносили в новые 20 мл сцинтилляционные сосуды, добавляли 10 мл Beckman Ready Protein Plus и производили подсчет. После окончания промывки миоинозитом, пустые сцинтилляционные сосуды помещали под колонки и элюировали [3H]-содержащие инозилфосфаты троекратным добавлением 1 мл 0,5 М формиата аммония, содержавшего 0,1 н. муравьиную кислоту. Условия элюирования оптимизировали для получения моно-, бис- и трисфосфатов инозита, без элюирования более метаболически инертных тетракис-, пентакис- и гексакис фосфатов. Образцы подсчитывали на многоцелевом сцинтилляционном счетчике Beckman LS 6500 после добавления 10 мл сцинтилляционной жидкости Tru-Count High Salt Capacity.
Содержание инозит липидов измеряли путем добавления 1 мл додецилсульфата натрия (SDS) в каждую лунку, после чего давали содержимому лунки осесть в течение по крайней мере 30 мин. Растворенное содержимое каждой лунки переносили в 20 мл сцинтилляционный флакон. Добавляли 10 мл сцинтилляционной жидкости Beckman Ready Protein Plus и производили подсчет радиоактивности.
Кривые зависимости концентрация-отклик для AVP и кривые концентрация-ингибирование для тестируемых соединений по сравнению с 10 нМ AVP исследовали нелинейным методом наименьших квадратов - подгонки кривых относительно 4-параметрической логистической функции. Варьировали параметры исходного и максимального уровней инозитфосфатов, EC50 или IC50, а также коэффициент Хилла для достижения наилучшего соответствия. В процессе подгонки кривых весовые коэффициенты выбирали в соответствии с предположением, что среднеквадратичное отклонение пропорционально числу радиоактивных распадов в минуту. В каждом эксперименте строили полные кривые концентрация-отклик для AVP и значения IC50 преобразовывали в значения Ki с применением уравнения Cheng-Prusoff, основываясь на значении EC50 для AVP в том же эксперименте. Содержание инозитфосфатов выражали как число распадов в минуту на 106 распадов в минуту общего количества инозита.
Эксперименты по определению конкурентоспособности тестируемых соединений заключались в построении кривых концентрация-отклик для AVP в отсутствии и в присутствии тестируемого соединения в двух или нескольких концентрациях. Данные согласовывали со следующим логистическим уравнением, описывающим конкурентное связывание:
где Y означает число распадов в минуту для инозитфосфатов, B означает концентрацию исходных инозитфосфатов, M означает максимальное увеличение концентрации инозитфосфатов, А означает концентрацию агониста (AVP), E представляет собой EC50 для агониста, D означает концентрацию агониста, K представляет собой Ki агониста и Q означает кооперативность (коэффициент Хилла).
Эксперименты с целью определения конкуренции тестируемых соединений заключались в построении кривых концентрация-отклик для AVP в отсутствии и присутствии тестируемого соединения как минимум в пяти концентрациях. Значения Ki, которые отражали активность антагонистов AVP при образовании сигнальной молекулы IP3, рассчитывали с помощью программы PRISM на основе уравнения Cheng и Prusoff.
ПРИМЕР МЕТОДИКИ 4
Обнаружение семян золотистыми хомячками
Считается, что в определенных условиях способность хомячков обнаруживать семена может отражать уровень их тревожности. Описываемая методика исследования способности к обнаружению семян у хомячков, подвергшихся воздействию описанных в настоящей заявке соединений, представляет собой животную модель тревожности.
Мужских особей сирийских золотистых хомячков (Mesocricetus auratus) (120-130 г), полученных из лаборатории Harlan Sprague-Dawley Laboratories (Indianapolis, IN), помещали по одному в клетки из плексигласа (24 см × 24 см × 20 см), содержали в режиме обратного цикла свет:темнота (14:10; свет включали в 19:00) и снабжали пищей и водой без ограничения. Все тесты проводили во время темновой фазы суточного цикла в условиях слабого красного освещения. Перед тестированием все животные голодали в течение 20-24 часов. Через 90 минут после интраперитонеальной (IP) инъекции SRX262 (n=10) или солевого носителя (n=10) животных извлекали из их постоянной клетки и помещали во временную клетку на 2 минуты. Во время их отсутствия под подстилку в углу их постоянной клетки закладывали шесть семян подсолнечника. Животных помещали в их постоянную клетку, случайно располагая их головой по направлению в один из пустых углов клетки и фиксируя время задержки обнаружения семян во время пятиминутного периода наблюдения. Задержка в обнаружении семян уменьшалась после введения соединений, описанных в настоящей заявке, причем задержка была сравнима по величине с задержкой при введении флуоксетина, буспирона и хлордиазепоксида.
ПРИМЕР МЕТОДИКИ 5
Социальное подчинение у хомячков, исследование биохимического маркера
Имеется целая группа литературных источников, описывающих нейроэндокринные и поведенческие последствия повторяющегося социального подчинения у взрослых самцов золотистых хомячков. Поражения в драках с другими особями и переход на более низкий социальный статус вызывают у взрослых животных сильный стресс, приводя к изменению уровней адренальных и гонадальных стероидов, а также к изменениям в социальном поведении (Rose et al., 1975; Eberhart et al., 1980, 1983). Исследования взрослых самцов хомячков показали снижение уровней тестостерона и повышение уровня глюкокортикоидов после неоднократных поражений от доминантных особей своего вида (Huhman et al., 1991).
Самцов хомячков размещали и содержали, как описано выше. В течение 30 минут каждый день в течение 14 последовательных дней животных подвергали угрозе и нападению со стороны более крупных представителей вида (n=14). После этих ежедневных эпизодов травмирующего стресса животных оставляли в покое в их постоянных клетках в течение 10 дней. Во время этого восстановительного периода животным вводили соединения по настоящему изобретению (1 мг/кг/день) (n=7) или солевой раствор (n=7). В конце периода введения препаратов животных умерщвляли отсечением головы и собирали кровь из туловища для проведения радиоимунного анализа тестостерона и кортизола. Уровни тестостерона хомячков, находившихся в постоянном подчинении, были очень низкими, тогда как базовые уровни кортизона были высокими. Этот нейроэндокринный профиль изменялся под действием соединений, описанных в настоящей заявке. Собранные данные показали, что блокирование рецепторов V1b может улучшить восстановление после травмирующего стресса, например социального подчинения.
ПРИМЕР МЕТОДИКИ 6
Социальное подчинение у хомячков, исследование поведения, отбор по антидепрессивной активности
На примере хомячков использовалась модель социального подчинения в схеме «резидент-интрудер». Модель агрессии резидент/интрудер основывается на мотивации резидентного животного преследовать и бороться с интрудерами, попадающими на их территорию (Miczek 1974). Более мелкие животные, помещенные в постоянную клетку в качестве резидентов, при постоянных столкновениях с противником будут терпеть поражения и становиться социально подчиненными. Социальное подчинение является существенной природной причиной стресса в царстве животных. Животные, потерпевшие поражение и подчиненные в ходе установления доминантной иерархии или территориальных стычек, могут быть в высшей степени склонны к подчинению в будущих агонистических взаимодействиях.
Например, побежденная мышь демонстрирует меньшую агрессию и более склонное к подчинению поведение (Frishknecht et al., 1982; Williams and Lierle 1988). Крысы, постоянно терпящие поражения от более агрессивных особей, демонстрируют угнетенное поведение, характеризующееся меньшей социальной инициативой и наступательной агрессией, а также усиление защитного поведения (Van de Poll et al., 1982). Неоднократно побежденные самцы хомячков демонстрируют подчинительную реакцию при столкновениях с неагрессивными интрудерами (Potegal et al., 1993), кроме того, их нормальное репродуктивное поведение подавлено, как показывает время задержки перед спариванием с готовой к оплодотворению самкой. Кроме того, после неоднократных поражений от доминантной особи резидентная особь хомячка демонстрирует защитное поведение и страх по отношению к неагрессивным интрудерам меньшего размера (Potegal et al., 1993). Распространение подчинительного поведения на новых, не представляющих угрозы животных является примером «обусловленного поражения» (Potegal et al., 1993). Обусловленное поражение не является постоянным у взрослых хомячков, т.к. склонность к бегству и защитному поведению исчезает через несколько недель. Животных, демонстрирующих обусловленное поражение, подвергали действию соединений по настоящему изобретению и наблюдали за возвращением к нормальному агрессивному и репродуктивному поведению.
Кроме того, социальное подчинение находится под явно выраженным влиянием нейроэндокринологии животного. У взрослых животных, проигрывающих в столкновениях и испытавших снижение социального статуса, изменяются уровни адренальных и гонадальных стероидов (Rose at al., 1975; Eberhart et al., 1908, 1983). После повторяющихся поражений от доминантных сородичей взрослые самцы хомячков демонстрируют пониженные уровни тестостерона и повышенные уровни глюкокортикоидов (Huhman et al., 1991). Оценивали восстановление нормальных уровней тестостерона и кортизола у животных, которым вводили описанные в настоящей заявке соединения.
Мужских особей сирийских золотистых хомячков (Mesocricetus auratus) (120-130 г), полученных из лаборатории Harlan Sprague-Dawley Laboratories (Indianapolis, IN), помещали по одному в клетки из плексигласа (24 см × 24 см × 20 см), содержали в режиме обратного цикла свет:темнота (14:10; свет включали в 19:00) и снабжали пищей и водой без ограничения. Все тесты проводили во время темновой фазы суточного цикла в условиях слабого красного освещения. Каждое соединение тестировали в трех дозировках (100 мкг, 1 мг и 10 мг/кг) плюс солевой носитель. Было исследовано двадцать четыре особи (по шесть в группе). Животных подвергали социальному подчинению, помещая их в постоянную клетку более крупного хомячка каждый день на 30 минут в течение 14 дней подряд. Животных каждый день помещали к различным резидентным особям, так что угрозы и нападения происходили постоянно. После прекращения социального подчинения животным давали восстановиться в спокойном состоянии в их постоянных клетках в течение следующих двух недель. В это время им в течение одной недели вводили соединения, описанные в настоящей заявке, или носитель. В конце этой недели животных тестировали на агрессию по отношению к более мелким интрудерам, помещаемым в их постоянную клетку. Для животных фиксировали время задержки перед укусом, число укусов и время сжатия челюстей. На следующий день в постоянную клетку животного помещали готовую к оплодотворению самку и определяли время до начала спаривания. В конце двухнедельного периода животных умерщвляли и анализировали кровь из туловища на тестостерон и кортизол. Всех животных умерщвляли в первые два часа темновой фазы цикла свет:темнота для минимизации суточных изменений уровня кортизола. Данные для разных групп сравнивали с помощью односторонней модели ANOVA с последующими post hoc тестами Бонферрони.
ПРИМЕР МЕТОДИКИ 7
Приподнятый крестообразный лабиринт
Приподнятый крестообразный лабиринт был разработан для отбора лекарственных средств по анксиолитическому и анксиогенному действию на грызунов. Эта методика была подтверждена поведенчески, физиологически и фармакологически. Крестообразный лабиринт состоит из двух открытых ответвлений и двух закрытых ответвлений. Крысы и мыши инстинктивно стремятся осуществлять меньше заходов в открытые ответвления, чем в закрытые ответвления, и будут проводить значительно меньше времени в открытых ответвлениях. Принудительное пребывание в открытых ответвлениях связано со значительно более тревожным поведением и более высокими уровнями гормона стресса, чем принудительное пребывание в закрытых ответвлениях. Клинически эффективные анксиолитики, например, хлордиазепоксид или диазепам значительно увеличивают процент времени пребывания в открытых ответвлениях и число заходов в открытые ответвления. Напротив, анксиогенные соединения, например, иохимбин или амфетамины уменьшают число заходов в открытые ответвления и проводимое в них время.
Группу самцов мышей содержали в условиях нормального цикла свет:темнота 12:12, причем свет включали в 08:00 и снабжали пищей и водой без ограничений. Крестообразный лабиринт состоял из двух открытых ответвлений длиной 40 см, шириной 6 см без стенок. Два закрытых ответвления имели те же размеры и стенки высотой 25 см. Каждая пара ответвлений располагалась напротив другой, образуя крестообразный лабиринт. Лабиринт поднимали на высоту 50 см. Каждое соединение испытывали в трех дозировках (100 мкг, 1 мг и 10 мг/кг) плюс солевой носитель. Тестировали двадцать четыре особи (по шесть в группе) в крестообразном лабиринте через 90 минут после IP инъекции препаратов в объеме приблизительно 0,1 мл. В начале эксперимента животных помещали в конец одного из открытых ответвлений. В течение периода наблюдений продолжительностью 5 минут определяли время задержки захода животного в закрытое ответвление, время, проведенное в закрытых ответвлениях, и число заходов в открытые ответвления после первого попадания в закрытое ответвление. Данные для разных групп сравнивали с помощью односторонней модели ANOVA с последующими post hoc тестами Бонферрони.
ПРИМЕР МЕТОДИКИ 8
Импульсивность/неуместная агрессия
Импульсивность и/или неуместная агрессия могут быть определены с применением стандартного анализа поведения животных, включая модели резидент-интрудер, модели агрессии, вызванной изоляцией, и модели агрессии между самками и/или агрессии между самцами. Эти исследования можно проводить на примере мышей, крыс и/или хомячков. Аргинин вазопрессин (AVP) вовлечен в агрессивное поведение ряда видов, включая людей (смотрите Coccaro et al., “Cerebrospinal fluid vasopressin levels: correlates with aggression and serotonin function in personality-disordered subjects” Arch. Gen. Psychiatry 55:708-14(1998)). Было показано, что инфузия антагонистов рецептора вазопрессина уменьшает агрессию (смотрите Ferris & Potengal “Vasopressin receptor blockade in the anterior hypothalamus suppresses aggression in hamsters” Physiol. Behav. 44:235-39 (1988)). Исследование вазопрессин V1b-нокаутированных мышей показало уменьшение агрессивного поведения у этих животных (смотрите Wersinger et al., “Vasopressin V1b receptor knockout reduces aggressive behavior in male mice” Mol. Psychiatry 7:975-84 (2002)).
В эксперименте использовали взрослых самцов сирийских хомячков (Mesocricetus auratus, Charles River Laboratories). Хомячков, которых предполагалось использовать в качестве резидентов, поселяли по одному по крайней мере за 2 недели до начала эксперимента. Субпопуляцию самцов более мелкого размера использовали в качестве интрудеров, причем их поселяли группами (три особи/клетка) для минимизации уровня агрессии. Пара резидент-интрудер должна была иметь разницу в массе как минимум 10 г. Например диапазон масс для резидентов находился между 105 и 150 г, а диапазон масс интрудеров - между 95-140 г, хотя их абсолютные массы могли изменяться. Животных помещали в клетки из плексигласа (46,0×24,0×21,0 см) с подстилкой из стержней кукурузных початков в комнате с регулируемой температурой (например, 69°F) и влажностью, с неограниченным доступом к пище и воде, причем животных содержали при световом цикле 14:10 свет-темнота с выключением света в 12:00 пополудни. Тесты проводили при освещении красным светом в первые три часа темновой фазы цикла свет-темнота. За всеми животными осуществляли ежедневный уход в течение 10 дней до начала исследования.
С каждым отдельно живущим хомячком проводили однократный тест без применения лекарств для отбора с целью определения исходных уровней агрессии животных. В тестировании препаратов использовали только резидентных самцов, которые осуществляли как минимум один укус за тестовую сессию. Тесты с соединениями, описанными в настоящей заявке, проводили через 48 ч после отбора. Через 25 минут после введения препаратов резидентов переносили в комнату для тестирования. Интрудеров помещали в клетки резидентов через 5 минут для 10-минутного теста. Каждый резидент сталкивался не с тем интрудером, которого использовали во время фазы отбора. Следует понимать, что протоколы, использованные в этом эксперименте, согласуются с соответствующими правилами штата и федеральными нормами. Оценки поведения включали время до нападения, время до укуса и число укусов. Данные анализировали с помощью односторонней модели ANOVA, необязательно с последующими post hoc тестами Ньюмана-Кеулса. Дополнительные подробности этого исследования можно найти в Blanchard et al., “AVP V1b selective antagonist SSR 149415 blocks aggressive behaviors in hamsters” Pharmacol., Biochem.Behav.80:189-94 (2005).
ПРИМЕР МЕТОДИКИ 9
Связывание и функциональный анализ человеческого окситоцина
Окситоцин известен благодаря тому, что он играет роль гормона во время родов и лактации. Агонисты окситоцина применяются в клинической практике, чтобы вызывать лактацию; вызывать или усиливать акт родов; регулировать послеродовую атонию матки и маточное кровотечение; вызывать сжатие матки после кесарева сечения или во время других видов маточной хирургии; а также для того, чтобы вызывать терапевтическое прекращение беременности. Действуя в качестве нейротрансмиттера в центральной нервной системе, окситоцин также играет важную роль в проявлении функций центральной нервной системы, например в материнском поведении, половом поведении (включая эрекцию полового члена, лордоз и копулятивное поведение), зевоте, механизмах терпимости и подчинения, кормлении, груминге, регулировании сердечно-сосудистой системы и терморегуляции (Argiolas and Gessa, Neuroscience and Biobehavioral Reviews, 15:217-231 (1991)). Антагонисты окситоцина находят терапевтическое применение в качестве средств для отсрочки или предотвращения преждевременных родов; или для замедления или остановки родов на короткий период для проведения других терапевтических мероприятий.
Предполагается, что соединения, описанные в настоящей заявке, также являются средствами, влияющими на окситоцин. Препараты окситоцина и ряд агонистов окситоцина имеются в продаже для терапевтического применения. В последние годы были разработаны антагонисты окситоцина, ослабляющие сокращения матки, и оценена их потенциальная применимость при лечении преждевременных родов и дисменореи (Pavo et al., J. Med. Chem., 37:255-259 (1994); Akerlund et al., Br. J. Obstet. Gynaecol., 94:1040-1044 (1987); Akerlund et al., Br. J.Obstet. Gynaecol., 86:484-487 (1979)). Антагонист окситоцина атосибан был изучен клинически и показал более значительное подавление преждевременных родовых схваток по сравнению с плацебо (Goodwin et al., Am. J. Obstet. Gynecol., 170:474 (1994)).
Был клонирован и экспрессирован человеческий рецептор окситоцина (Kimura et al., Nature, 356: 526-529 (1992)), причем его можно идентифицировать по номеру доступа X64878. Чтобы продемонстрировать сродство соединений, описанных в настоящей заявке, к человеческому рецептору окситоцина, исследовали их связывание, используя линию клеток, экспрессирующих человеческий рецептор окситоцина в клетках 293 (далее по тексту называемых клеточной линией OTR), в основном следуя методике, описанной Morel et al. (Nature, 356:523-526 (1992)). Линия клеток 293 является перманентной линией первичных почечных эмбриональных человеческих клеток, трансформированных с помощью ДНК лишенного белковой оболочки человеческого аденовируса 5. Идентификационный номер ATCC CRL-1533.
Линию клеток OTR выращивали в среде DMEM (Модифицированная Далбеко эссенциальная среда, Sigma, St.Louis, MO, USA) с добавкой 10% сыворотки телят, 2мМ L-глутамина, 200 мкг гигромицина (Sigma, St.Louis, MO, USA) и 250 мкг/мл G418 (Gibco, Grand Island, NY, USA). Для получения мембран клетки OTR выращивали до конфлюэнтности в 20 вращающихся флаконах. Клетки разделяли в не содержащей ферментов среде для разделения клеток (Specialty Media, Lavalette, NJ, USA) и центрифугировали при 3200 об/мин в течение 15 минут. Осадок повторно суспендировали в 40 мл буфера Tris-HCl (гидрохлорид трис[гидроксиметил] аминометана) (50 мМ, pH 7,4) и гомогенизировали в течение 1 минуты с помощью Tekmar Tissumizer (Cincinnatti, OH USA). Суспензию центрифугировали при 40000g в течение 10 минут. Осадок повторно суспендировали и центрифугировали, как описано выше. Полученный в результате осадок суспендировали в 80 мл Tris-буфера pH 7,4 и хранили в виде 4 мл аликвот при -80°C. Для анализа аликвоты ресуспендировали в аналитическом буфере и разбавляли до концентрации 375 мкг белка на мл. Концентрацию белка определяли анализом BCA (Pierce, Rockford, IL, USA).
Аналитический буфер включал 50 мМ Tris-HCl (гидрохлорид трис[гидроксиметил]аминометана), 5 мМ MgCl2 и 0,1% альбумин бычьей сыворотки при pH 7,4. Радиоактивным лигандом для исследования связывания являлся [3H]окситоцин ([тирозил-2,6-3H]окситоцин, 48,5 Кюри/ммоль, DuPont NEN, Boston, MA, USA). Компоненты добавляли в следующем порядке: 195 мкл аналитического буфера, 200 мкл мембран OTR (75 мкг белка) в аналитическом буфере, 5 мкл тестируемого соединения в диметилсульфоксиде (ДМСО) или чистый ДМСО и 100 мкл [3H]окситоцина в аналитическом буфере (конечная концентрация 1,0 нМ). Инкубирование проводили в течение одного часа при комнатной температуре. Связанный радиоактивный лиганд отделяли от несвязанного фильтрованием на харвестере с ячейками Brandel (Gaithersburg, MD, USA) через фильтры из стекловолокна Whatman GF/B, которые замачивали в течение 2 часов в 0,3% полиэтиленимине. Фильтры промывали ледяным 50 мМ Tris-HCl (pH 7,7 при 25°C) и кружки фильтров помещали в сцинтилляционные флаконы, в которые затем добавляли 5 мл сцинтилляционной жидкости Ready Protein PlusTM и подсчитывали на сцинтилляционном счетчике для жидкостей. Каждое инкубирование осуществляли в трех экземплярах и получали кривые зависимости доза-ингибирование, которые включали точки, соответствующие общему связыванию, неспецифическому связыванию (100 мкМ окситоцин, Sigma, St. Louis, MO, USA) и 6 или 7 концентрациям тестируемого соединения, охватывающим IC50. Общее связывание, как правило, составляло примерно 1000 имп./мин и неспецифическое связывание - примерно 200 имп./мин. Значения IC50 рассчитывали по нелинейному методу наименьших квадратов подгонкой кривых к 4-параметрической логистической модели. Некоторые соединения формулы (I) продемонстрировали сродство к рецептору окситоцина.
Существует несколько биоанализов для определения агонистического или антагонистического характера соединений, демонстрирующих сродство к рецептору окситоцина. Один из этих анализов описан в патенте США № 5373089, включенном в настоящую заявку в качестве ссылки. Указанный биоанализ основан на методиках, описанных в статье Sawyer et al., (Endocrinology, 106:81 (1980)), которые, в свою очередь, основаны на сообщении Holton (Brit. J. Pharmacol., 3:328 (1948)). Способы анализа для оценки pA2 описаны Schild (Brit. J. Pharmacol., 2:189 (1947)).
ПРИМЕР МЕТОДИКИ 10
Исследование функциональной активности в отношении окситоцина
1. Животный материал: для анализа использовали 1,5 см кусок матки неоплодотворенной крысы (Holtzman) в период естественной течки.
2. Буфер/кювета для анализа: Использовали буфер Munsiks. Этот буфер содержал 0,5 мМ Mg2+. Через буферный раствор непрерывно продували смесь 95% кислорода/5% диоксида углерода, что давало значение pH 7,4. Температура аналитического раствора 37°C. Использовали 10 мл кювету для анализа, снабженную водяной рубашкой для поддержания температуры, а также входным и выходным отводами для добавления и удаления буфера.
3. Самописец/датчик: Используемый для анализа кусок ткани матки прикрепляют за один конец и соединяют с датчиком Statham Strain Gauge Force Transducer, другой конец которого, в свою очередь, присоединен к самописцу Grass Polygraph Model 79 для наблюдения за сжатием ткани.
4. Протокол анализа:
(a) Ткань приводили в равновесие с аналитическим раствором, в течение 1 часа, путем промывания новой порцией буфера каждые 15 минут. В течение всего этого времени поддерживали натяжение ткани с усилием один грамм.
(b) Ткань вначале стимулировали окситоцином в концентрации 10 нМ для привыкания ткани к среде и 4 мМ хлоридом калия (KCl) для определения максимальной реакции сжатия.
(c) Затем строили общую кривую доза-отклик для окситоцина и использовали концентрацию окситоцина, эквивалентную примерно 80% максимального отклика для оценки pA2 антагониста.
(d) Ткань подвергали действию окситоцина (Calbiochemical, San Diego, CA) в течение одной минуты и удаляли его промыванием. Перед добавлением следующей дозы агониста или антагониста оставляли трехминутную паузу. Если тестировали антагонист, перед добавлением агониста оставляли пятиминутную паузу. Агониста добавляли на одну минуту. Все отклики ткани интегрировали с применением 7P10 Grass Integrator. Для тестирования антагонистов использовали одну концентрацию окситоцина, эквивалентную 80% максимального отклика. Использовали три различные концентрации антагониста, причем две из них должны были снижать отклик на действие агониста менее чем на 50% и одна должна снижать отклик более чем на 50% (в идеальном случае эти соотношения должны были бы быть 25%, 50% и 75%). Это повторяли по три раза для каждой дозы антагониста для трех исследованных точек.
(e) Расчеты pA2 - для антагонистов рассчитывали соотношения доза-отклик (DR) и строили график Шилда, нанося точки в координатах Log(DR-1) - Log концентрации антагониста. Построенную линию обсчитывали с помощью регрессионного анализа по методу наименьших квадратов. Значение pA2 представляет собой концентрацию антагониста в точке, где регрессионная кривая пересекает нулевую точку ординаты Log(DR-1). pA2 представляет собой отрицательный логарифм концентрации антагониста, которая уменьшает отклик на действие агониста до одной второй.
ПРИМЕР МЕТОДИКИ 11
Анализ связывания рецептора тахикинина
Предполагается, что описанные в настоящей заявке соединения являются средствами, оказывающими влияние на тахикинин. Тахикинины представляют собой семейство пептидов, которые имеют общую амидированную карбокси-концевую последовательность. Субстанция P была первым пептидом из этого семейства, который удалось выделить, хотя его очистка и определение его первичной последовательности не были проведены до начала 1970-х. Между 1983 и 1984 несколько групп сообщили о выделении двух новых тахикининов млекопитающих, называемых в настоящее время нейрокинином A (известным также под названием субстанция K, нейромедин 1 и нейрокинин α), и нейрокинином B (известным также как нейромедин K и нейрокинин β). Для ознакомления с обзором этих открытий смотрите J. E. Maggio, Peptides, 6 (Supplement 3): 237-243 (1985).
Антагонисты рецептора тахикинина полезны при лечении широкого круга клинических состояний, которые характеризуются наличием избытка тахикинина. Эти клинические состояния могут включать расстройства центральной нервной системы, такие как тревожность, депрессия, психоз и шизофрения; нейродегенеративные расстройства, такие как деменция, включая старческую деменцию типа Альцгеймера, болезнь Альцгеймера, СПИД-ассоциированная деменция и синдром Дауна; демиелинизирующие заболевания, такие как рассеянный склероз и боковой амиотрофический склероз, а также другие невропатологические расстройства, такие как периферическая невропатия, например диабетическая невропатия и невропатия, вызванная химиотерапией, а также постгерпетические и другие невралгии; острые и хронические обструктивные заболевания дыхательных путей, такие как синдром дыхательной недостаточности взрослых, бронхопневмония, бронхоспазм, хронический бронхит, кашель водителей и астма; воспалительные заболевания, такие как воспалительные заболевания кишечника, псориаз, фиброз, остеоартрит и ревматоидный артрит; расстройства скелетно-мышечной системы, такие как остеопороз; аллергии, такие как экзема и ринит; расстройства, связанные с гиперчувствительностью, как, например, дерматит при контакте с сумахом ядовитым; глазные заболевания, такие как конъюнктивит, весенний конъюнктивит и т.п.; кожные заболевания, такие как контактный дерматит, диффузный нейродермит, аллергическая сыпь и другие экземоподобные дерматиты; пагубные пристрастия, такие как алкоголизм; соматические расстройства, связанные со стрессом; симпатическую рефлекторную дистрофию, такую как плечевой синдром; дистимические расстройства; неблагоприятные иммунологические реакции, такие как отторжение трансплантированных тканей и расстройства, связанные с усилением или подавлением иммунитета, такие как системная красная волчанка; желудочно-кишечные расстройства или заболевания, связанные с нейронным управлением органами брюшной полости, такие как язвенный колит, болезнь Крона, рвота и синдром раздраженного кишечника; расстройства деятельности мочевого пузыря, такие как гиперрефлексия детрузора и недержание; атеросклероз; фиброзирующие и коллагеновые заболевания, такие как склеродермия и эозинофильный фасциолез; раздражающие симптомы доброкачественной гипертрофии простаты; расстройства кровообращения, вызванные заболеваниями, расширяющими и сужающими сосуды, такими как стенокардия, мигрень и болезнь Рейно; а также боль и ноцицепцию, например, относящуюся к или связанную с любым из упомянутых ранее состояний, особенно распространением боли при мигрени.
Тахикинины широко распространены как в центральной, так и в периферической нервной системе. Высвобождаясь из нервов, они влияют на ряд биологических процессов, которые, в большинстве случаев, зависят от активации конкретных рецепторов, экспрессируемых на мембране целевых клеток. Кроме того, тахикинины вырабатываются рядом тканей, не относящихся к нервной системе. Тахикинины млекопитающих, т.е. субстанция P, нейрокинин A и нейрокинин B, действуют через три основных подтипа рецепторов, именуемых NK-1, NK-2 и NK-3 соответственно. Эти рецепторы присутствуют в различных органах.
Считают, что субстанция P, в том числе, вовлечена в нейропередачу ощущений боли, включая боли, связанной с мигренью, головной болью и артритом. Эти пептиды также вовлечены в желудочно-кишечные расстройства и заболевания желудочно-кишечного тракта, такие как воспалительные заболевания кишечника. Кроме того, тахикинины играют определенную роль во многих других заболеваниях, что обсуждается ниже.
В силу значительного числа клинических расстройств, связанных с избытком тахикининов, разработка антагонистов рецепторов тахикинина должна послужить усилению контроля за этими клиническими состояниями. Самыми первыми из обнаруженных антагонистов рецепторов тахикинина были пептидные производные. Оказалось, что эти антагонисты обладают ограниченной фармацевтической применимостью из-за их метаболической неустойчивости. В современных публикациях описаны новые классы непептидных антагонистов рецепторов тахикинина, которые в основном обладают более высокой биодоступностью при пероральном приеме и метаболической стабильностью по сравнению с ранее открытыми классами антагонистов рецепторов тахикинина. Примеры таких новых непептидных антагонистов рецепторов тахикинина можно найти в европейской патентной публикации 591 040 A1, опубликованной 6 апреля 1994; публикации согласно договору о патентной кооперации WO 94/01402, опубликованной 20 января 1994; публикации согласно договору о патентной кооперации WO 94/04494, опубликованной 3 марта 1994; публикации согласно договору о патентной кооперации WO 93/011609, опубликованной 21 января 1993; публикации согласно договору о патентной кооперации WO 94/26735, опубликованной 24 ноября 1994. Анализы, применимые для оценки антагонистов рецепторов тахикинина, хорошо известны в технике. Смотрите, например, J. Jukic et al., Life Sciences, 49:1463-1469 (1991); N. Kucharczyk et al., Journal of Medicinal Chemistry, 36:1654-1661 (1993); N. Rouissi et al., Biochemical and Biophysical Research Communications, 176:894-901 (1991).
ПРИМЕР МЕТОДИКИ 12
Анализ связывания рецептора NK-1
Антагонисты NK-1 применимы при лечении боли, особенно хронической боли, например невропатической боли, послеоперационной боли, мигреней, боли, связанной с артритом, боли, связанной с раком, хронической боли нижнего отдела спины, гистаминовых головных болей, невралгии, связанной с герпесом, боли в фантомной конечности, центральной боли, зубной боли, невропатической боли, боли, устойчивой к действию опиоидов, боли внутренних органов, хирургической боли, боли, вызванной травмой костей, боли во время схваток и родов, боли вследствие ожога, включая солнечный ожог, послеродовой боли, боли при стенокардии и болей, связанных с мочеполовым трактом, включая цистит.
Помимо лечения боли, антагонисты NK-1 особенно применимы при лечении и профилактике недержания мочи; раздражающих симптомов доброкачественной гипертрофии простаты; расстройств моторики желудочно-кишечного тракта, таких как синдром раздраженного кишечника; острых и хронических обструктивных заболеваний дыхательных путей, таких как бронхоспазм, бронхопневмония, астма и синдром дыхательной недостаточности взрослых; атеросклероза; воспалительных состояний, таких как воспалительные заболевания кишечника, язвенный колит, болезнь Крона, ревматоидный артрит, остеоартрит, нейрогенное воспаление, аллергии, ринит, кашель, дерматит, аллергическая сыпь, псориаз, конъюнктивит, рвота, вызванное раздражением сужение зрачков; отторжении трансплантированных тканей; просачивании плазмы, вследствие цитокиновой химиотерапии и т.п.; травме спинного мозга; инсульте; мозговом инсульте (ишемии); болезни Альцгеймера; болезни Паркинсона; рассеянном склерозе; боковом амиотрофическом склерозе; шизофрении; тревожности и депрессии.
Исследование связывания с радиоактивно меченным рецептором проводили с использованием варианта ранее опубликованного протокола. D.G. Payan et al., Journal of Immunology, 133:3260-3265 (1984). В этом анализе аликвоту клеток IM9 (1х106 клеток/пробирка в среде RPMI 1604 с добавкой 10% фетальной телячьей сыворотки) инкубировали с 20 пМ 125I-меченой субстанцией P, в присутствии возрастающих концентраций конкурирующего соединения в течение 45 минут при 4°C.
Клеточная линия IM9 является хорошо изученной клеточной линией которая легко доступна для общего использования. Смотрите, например, Annals of the New York Academy of Science, 190:221-234 (1972); Nature (London), 251:443-444 (1974); Proceedings of the National Academy of Sciences (USA), 71:84-88 (1974). Эти клетки культивировали по стандартной методике в RPMI 1640 с добавками 50 мкг/мл гентамицин сульфата и 10% фетальной телячьей сыворотки.
Реакцию прекращали фильтрованием через собирающую систему на основе фильтра из стекловолокна, используя фильтры, предварительно замоченные в течение 20 минут в 0,1% полиэтиленамине. Специфическое связывание меченой субстанции P определяли в присутствии 20 мМ немеченого лиганда.
ПРИМЕР МЕТОДИКИ 13
Анализ связывания рецептора NK-2
Антагонисты NK-2 применимы при лечении недержания мочи, бронхоспазма, астмы, синдрома дыхательной недостаточности взрослых, расстройств моторики желудочно-кишечного тракта, например синдрома раздраженного кишечника, а также боли.
Клетки CHO-hNK-2R, представляющие клеточную линию, полученную из клеток CHO, трансформированных для выработки человеческих рецепторов NK-2, и экспрессирующие примерно 400000 молекул этих рецепторов на клетку, выращивали в колбах объемом 75 см3 или вращающихся флаконах в минимальной поддерживающей среде (альфа модификация) с добавкой 10% фетальной бычьей сыворотки. Генетическая последовательность человеческого рецептора NK-2 приведена в работе N. P. Gerard et al., Journal of Biological Chemistry, 265:20455-20462 (1990).
Для получения мембран конфлюэнтные культуры из 30 вращающихся флаконов разделяли путем промывания каждого вращающегося флакона 10 мл солевого раствора с фосфатным буфером Далбеко (PBS) без кальция и магния с последующим добавлением 10 мл не содержащего ферментов раствора для разделения клеток (на основе PBS, от Specialty Media, Inc.). По прошествии еще 15 минут разделенные клетки собирали и центрифугировали при 1000 об/мин в течение 10 минут на клинической центрифуге. Мембраны получали гомогенизацией осадка клеток в 300 мл 50 мМ Tris-буфера, pH 7,4 с помощью гомогенизатора TEKMAR® в течение 10-15 секунд с последующим центрифугированием при 12000 об/мин (20000g) в течение 30 минут с применением ротора BECKMAN JA-14®. Осадок один раз промывали по описанной выше методике и полученный после этого осадок ресуспендировали в 100-120 мл 50 мМ буфера TRIS, pH 7,4 и 4 мл аликвоты полученной суспензии хранили замороженными при -70°C. Концентрация белка в этих препаратах составляла 2 мг/мл.
Для анализа связывания рецептора, одну 4-мл аликвоту препарата мембран CHO-hNK-2R суспендировали в 40 мл аналитического буфера, содержащего 50 мМ Tris, pH 7,4, 3 мМ хлорид магния, 0,02% альбумин бычьей сыворотки (BSA) и химостатин 4 мкг/мл. Для одного образца использовали объем гомогената 200 мкл (40 мкг белка). Радиоактивным лигандом являлся [125I]иодгистидил-нейрокинин A (New England Nuclear, NEX-252), 2200 Кюри/ммоль. Готовили препарат лиганда в аналитическом буфере с концентрацией 20 нКюри на 100 мкл; конечная концентрация во время анализа составляла 20 пМ. Неспецифическое связывание определяли с использованием 1 мкМ эледоизина. Для построения стандартной кривой концентрация-отклик использовали десять концентраций эледоизина от 0,1 до 1000 нМ.
Все образцы и стандарты добавляли в смесь для инкубирования в 10 мкл диметилсульфокисда (ДМСО) при отборе (одна доза) или в 5 мкл ДМСО для определения значений IC50. Порядок добавления компонентов в смесь для инкубирования был следующим: 190 или 195 мкл аналитического буфера, 200 мкл гомогената, 10 или 5 мкл образца в ДМСО, 100 мкл радиоактивного лиганда. Образцы инкубировали в течение 1 ч при комнатной температуре и затем фильтровали на харвестере с ячейками через фильтры, которые были предварительно замочены в течение двух часов в 50 мМ буфере Tris, pH 7,7, содержащем 0,5% BSA. Фильтр три раза промывали приблизительно 3 мл холодного 50 мМ буфера Tris, pH 7,7. Затем из фильтра выбивали кружочки, помещали их в пробирки из полистирола 12×75 мм и подсчитывали на счетчике гамма-излучения.
ПРИМЕР МЕТОДИКИ 14
Лечение рвоты
Помимо перечисленных выше показаний к лечению, описанные в настоящей заявке соединения могут применяться при лечении рвоты, включая сильную, замедленную и заранее ожидаемую рвоту, такую как рвота, вызванная химиотерапией, радиацией, токсинами, беременностью, вестибулярными расстройствами, движением, хирургическим вмешательством, мигренью и изменениями внутричерепного давления. В частности, соединения приведенных в настоящей заявке формул могут найти применение при лечении рвоты, вызванной антинеопластическими (цитотоксическими) средствами, включая теми средствами, которые обычно используются при химиотерапии рака.
Примеры таких химиотерапевтических средств включают алкилирующие средства, например азотистые иприты, этилениминовые соединения, алкилсульфонаты, а также другие соединения с алкилирующим действием, такие как нитрозомочевины, цисплатин и дакарбазин; антиметаболиты, например фолиевую кислоту, антагонисты пурина или пиримидина; ингибиторы митоза, например алкалоиды барвинка и производные подофиллотоксина; а также цитотоксические антибиотики.
Конкретные примеры химиотерапевтических средств описаны, например, в D. J. Stewart in Nausea and Vomiting: Recent Research and Clinical Advances (под ред. J.Kucharczyk et al. 1991) на стр. 177-203. Обычно применяемые химиотерапевтические средства включают цисплатин, дакарбазин (DTIC), дактиномицин, меклоретамин (азотистый иприт), стрептозоцин, циклофосфамид, кармустин (BCNU), ломустин (CCNU), доксорубицин, даунорубицин, прокарбазин, митомицин, цитарабин, этопозид, метотрексат, 5-фторурацил, винбластин, винкристин, блеомицин и хлорамбуцил. R. J. Gralla et al., Cancer Treatment Reports, 68:163-172 (1984).
Соединения описанных в настоящей заявке формул также могут найти применение в лечении рвоты, вызванной радиацией, включая радиационной терапией, например, при лечении рака, или лучевой болезнью; а также при лечении послеоперационной тошноты и рвоты.
ПРИМЕР МЕТОДИКИ 15
Ингибирование агрегации тромбоцитов
Помимо перечисленного выше известно, что рецепторы вазопрессина V2 опосредуют агрегацию тромбоцитов. Агонисты рецептора вазопрессина вызывают агрегацию тромбоцитов, тогда как антагонисты рецептора вазопрессина V2 ингибируют агрегацию тромбоцитов, осажденных под действием вазопрессина или антагонистов вазопрессина. Степень антагонистической активности соединений, описанных в настоящей заявке, может быть определена при использовании обычных способов, включая анализ, описанный в следующих параграфах.
У здоровых добровольцев отбирали кровь через прокол в вене и смешивали с гепарином (60 мл крови добавляли к 0,4 мл солевого раствора с гепарином (4 мг гепарина на мл солевого раствора)). Получали обогащенную тромбоцитами плазму (PRP), центрифугируя цельную кровь (150g) и к PRP добавляли индометацин (3 мкМ) для блокирования опосредованной тромбоцитами реакции высвобождения. PRP непрерывно перемешивали при 37°C, причем после добавления аргинин вазопрессина (AVP) (30 нМ) для инициирования агрегации, следовало изменение оптической плотности. Соединения растворяли в 50% диметилсульфоксиде (ДМСО) и добавляли к плазме (10 мкл/415 мкл PRP) перед добавлением AVP. Измеряли ингибирование в процентах AVP-индуцированной агрегации и рассчитывали IC50.
В исследованиях, использующих промытые тромбоциты, 50 мл цельной крови смешивали с 10 мл раствора цитраты/гепарин (85 мМ цитрат натрия, 64 мМ лимонная кислота, 111 мМ глюкоза, 5 ед/мл гепарин) и отделяли PRP, как описано выше. Затем PRP центрифугировали (150g) и тромбоциты ресуспендировали в физиологическом буферном растворе (10 мМ HEPES, 135 мМ хлорид натрия, 5 мМ хлорид калия и 1 мМ хлорид магния), содержащем 10 мМ индометацин. Перед началом агрегации под действием AVP (30 нМ), которая была описана выше, к перемешиваемым тромбоцитам добавляли человеческий фибриноген (0,2 мг/мл) и хлорид кальция (1 мМ).
ПРИМЕР МЕТОДИКИ 16
Поведение золотистых хомячков, связанное с мечением предметов боковой поверхностью тела
Обсессивно-компульсивные заболевания проявляются в большом разнообразии качеств и симптомов, в основном связанных с неконтролируемым стремлением больного совершать ненужные условные действия. Внешними характеристиками данного заболевания являются действия, связанные с покупками, наведением порядка, чисткой и т.п., находящиеся за пределами любой разумной потребности или разумного обоснования. Субъект, пораженный болезнью в тяжелой степени, может быть неспособен делать что-либо иное, кроме выполнения условных действий, диктуемых заболеванием. Обсессивно-компульсивные заболевания во всех своих разновидностях являются предпочтительной мишенью для лечения способами и композициями по настоящему изобретению в качестве дополнительной терапии. Применимость соединений формулы (I) при лечении обсессивно-компульсивного расстройства была продемонстрирована в следующем исследовании.
Особенно стереотипный вид поведения золотистых хомячков, а именно мечение предметов боковой поверхностью тела, может быть вызван микроинъекциями вазопрессина (10-100 нл, 1-100 мкМ) в передний отдел гипоталамуса (Ferris et al., Science, 224, 521-523 (1984); Albers and Ferris, Regulatory Peptides, 12, 257-260 (1985), Ferris et al., European Journal of Pharmacology, 154, 153-159 (1988)). Вслед за разрешающим раздражителем описываемое поведение начинается с груминга, облизывания и расчесывания больших сальных желез на дорсолатеральной части боковой поверхности тела. Приступы груминга боковых желез могут быть такими интенсивными, что шерсть на боку становится спутанной и пропитывается слюной. После груминга хомячки проявляют поведение, связанное с мечением предметов боковой поверхностью тела, которое является разновидностью запахового мечения, связанного с обонятельной передачей информации (Johnston, Physio. Behav., 51, 437-448 (1985); Ferris et al., Physio. Behav., 40, 661-664 (1987)), проявляющееся в выгибании спины и энергичном трении боковыми железами о любую вертикальную поверхность. Вызванное вазопрессином мечение боковой поверхностью тела обычно начинается в течение минуты после микроинъекции (Ferris et al., Science, 224, 521-523 (1984)). Такое поведение является специфичным для вазопрессина, тогда как микроинъекции других нейропептидов, возбуждающих аминокислот и катехоламинов не вызывают описанного мечения (Ferris et al., Science, 224, 521-523 (1984); Albers and Ferris, Regulatory Peptides, 12, 257-260 (1985)). Более того, мечение боковой поверхностью тела специфично для рецептора вазопрессина V1, т.к. это поведение селективно подавляется антагонистами рецептора V1 и активируется агонистами рецептора V1 (Ferris et al., Neuroscience Letters, 55, 239-243 (1985); Albers et al., Journal of Neuroscience, 6, 2085-2089 (1986); Ferris et al., European Journal of Pharmacology, 154, 153-159 (1988)).
Все животные, использованные в этом исследовании, являлись взрослыми самцами золотистых хомячков (Mesocricetus auratus) весом примерно 160 г. Перед поведенческими тестами животным проводили стереотаксическую операцию и давали восстановиться. Хомячков содержали при обращенном световом цикле (14 ч света, 10 ч темноты, включение света в 19:00) в клетках из плексигласа и давали пищу и воду без ограничений.
Стереотаксическую операцию проводили при обезболивании пентобарбиталом. Стереотаксические координаты были следующими: 1,1 мм впереди темени, 1,8 мм вбок от серединного сагиттального шва под углом 8° от вертикальной линии и 4,5 мм под твердой мозговой оболочкой. Носок помещали на уровне линии, соединяющей уши. Одноходовой направляющий катетер № 26 опускали до нужного участка и прикрепляли к черепу зубным цементом. Направляющий катетер закрывали обтюратором № 33, выходившим за пределы направляющего катетера на 1 мм. Внутренний катетер, использовавшийся для микроинъекций, выходил за пределы направляющего на 3 мм для достижения переднего отдела гипоталамуса.
Хомячкам проводили микроинъекцию 1 мкМ вазопрессина в объеме 150 нл. Вазопрессин вводили в виде коктейля с 200 мМ, 20 мМ, 2 мМ тестируемого соединения или сам по себе, в носителе, т.е. диметилсульфоксиде. Как вазопрессин, так и тестируемые соединения растворяли в 100% диметилсульфоксиде. Все инъекции проводили в передний отдел гипоталамуса. Подсчитывали число мечений стенок боковой поверхностью тела животных в течение 10 минут в чистой клетке.
ПРИМЕР МЕТОДИКИ 17
Применение в сочетании с ингибиторами обратного захвата серотонина
Другим аспектом настоящего изобретения является применение соединений формулы (I) в сочетании с ингибиторами обратного захвата серотонина с целью лечения обсессивно-компульсивных заболеваний, агрессивного поведения или депрессии. Соединения, применимые в качестве ингибиторов обратного захвата серотонина включают, не ограничиваясь перечисленными:
Флуоксетин, т.е. N-метил-3-(п-трифторметилфенокси)-3-фенилпропиламин, поступает на рынок в форме гидрохлорида и в виде рацемической смеси двух энантиомеров. Впервые это соединение упоминалось в патенте США № 4314081. Робертсон и соавторы J. Med. Chem., 31, 1412 (1988) описали разделение R и S энантиомеров флуоксетина и показали, что по активности в качестве ингибиторов обратного захвата серотонина энантиомеры сходны друг с другом. В настоящей заявке слово «флуоксетин» будет использоваться для обозначения любой кислотно-аддитивной соли или свободного основания и будет включать либо рацемическую смесь, либо любой из R и S энантиомеров;
Дулоксетин, т.е. N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин, обычно вводится в виде гидрохлорида и (+)энантиомера. Впервые описан в патенте США № 4956388, где показана его высокая эффективность. Слово «дулоксетин» будет использоваться в настоящей заявке для обозначения любой кислотно-аддитивной соли или молекулы свободного основания;
Венлафаксин известен в литературе, причем способ его синтеза и активность в качестве ингибитора захвата серотонина и норэпинефрина описаны в патенте США № 4761501. Венлафаксин в этом патенте определен, как «соединение A»;
Милнаципран (N,N-диэтил-2-аминометил-1-фенилциклопропанкарбоксамид) описан в патенте США № 4478836, где милнаципран получен в примере 4. Полученные в данном патенте соединения описаны в качестве антидепрессантов. Moret и соавторы в Neuropharmacology, 24, 1211-19 (1985) описывают его фармакологическую активность как ингибитора обратного захвата серотонина и норэпинефрина;
Циталопрам, т.е. 1-[3-(диметиламино)пропил]-1-(4-фторфенил)-1,3-дигидро-5-изобензофуранкарбонитрил, раскрыт в патенте США № 4136193 в качестве ингибитора обратного захвата серотонина. Его фармакология была раскрыта Christensen и соавторами Eur. J. Pharmacol., 41, 153 (1977), и сообщения о его клинической эффективности при депрессии можно найти у Dufour et. al., Int. Clin. Psychopharmacol., 2, 225 (1987), и Timmerman et. al., смотрите выше, 239;
Флувоксамин, т.е. 5-метокси-1-[4-(трифторметил)фенил]-1-пентанон-O-(2-аминоэтил)оксим описан в патенте США № 4085225. Научные статьи об этом лекарственном препарате были опубликованы Claassen et. al., Brit. J. Pharmacol., 60, 505 (1977); De Wilde et. al. J. Affective Disord., 4, 249 (1982); и Benfield и et. al. Drugs, 32, 313 (1986);
Пароксетин, т.е. транс-(-)-3-[(1,3-бензодиоксол-5-илокси)метил]-4-(4-фторфенил)пиперидин может быть обнаружен в патентах США №№ 3912743 и 4007196. Сообщения о лекарственной активности соединения: Lassen, Eur. J. Pharmacol., 47, 351 (1978); Hassan et al., Brit. J. Clin.Pharmacol., 19, 705 (1985); Laursen et al., Acta Psychiat. Scand., 71, 249 (1985); и Battegay et al., Neuropsychobiology, 13, 31 (1985); и
Сертралин, т.е. гидрохлорид (1S-цис)-4-(3,4-дихлорфенил)-1,2,3,4-тетрагидро-N-метил-1-нафтиламина, представляющий собой ингибитор обратного захвата серотонина, раскрытый в патенте США № 4536518, продается на рынке в качестве антидепрессанта.
Все упомянутые выше патенты включены в настоящую заявку в качестве ссылки.
Дополнительную терапию по данному аспекту настоящего изобретения проводят путем введения антагонистов рецептора вазопрессина V1a, описанных в настоящей заявке, совместно с ингибитором обратного захвата серотонина, любым способом, который обеспечивает эффективные уровни соединений в организме в одно и то же время. Все упомянутые соединения доступны в виде пероральных лекарственных форм и обычно вводятся перорально, и поэтому пероральное введение дополнительной комбинации является предпочтительным. Соединения могут вводиться совместно, в одной лекарственной форме или раздельно.
Данный аспект настоящего изобретения обеспечивает усиление снижения наблюдаемой концентрации вазопрессина как результата введения антагониста рецептора вазопрессина V1b, за счет введения ингибитора обратного захвата серотонина. Описываемый аспект настоящего изобретения особенно подходит для применения при лечении депрессии и обсессивно-компульсивного расстройства. Такие расстройства часто могут не поддаваться лечению ингибиторами обратного захвата серотонина, применяемыми отдельно.
Хотя имеется возможность введения соединений, применяемых в способах, описанных в настоящей заявке, непосредственно, без использования каких-либо фармацевтических составов, соединения обычно вводят в форме фармацевтических композиций, включающих фармацевтически приемлемый носитель и как минимум один действующий ингредиент. Эти композиции можно вводит различными путями, включая пероральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и интраназальный. Многие из соединений, применяемых в способах, описанных в настоящей заявке, эффективны как в виде композиций для инъекций, так и для перорального приема. Такие композиции получают способами, хорошо известными в фармацевтической технике, причем они включают как минимум одно действующее соединение. Смотрите, например, Remington's Pharmaceutical Sciences (16th ed. 1980).
При получении фармацевтических композиций, применяемых в способах, описанных в настоящем изобретении, действующий ингредиент обычно смешивают с наполнителем, разбавляют разбавителем или помещают в такой носитель, который может принимать форму капсулы, саше, бумаги или другого контейнера. Если наполнитель служит разбавителем, он может быть твердым, полутвердым или жидким веществом, которое действует в качестве носителя или среды для активного ингредиента. Так, например, композиции могут принимать форму таблеток, пилюль, порошков, ромбовидных таблеток, саше, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (твердых или в жидкой среде), мазей, содержащих, например, до 10% по массе активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильно упакованных порошков.
При приготовлении составов может потребоваться измельчить действующее соединение для обеспечения подходящего размера частиц перед смешиванием с другими ингредиентами. Если действующее соединение в основном нерастворимо, его обычно измельчают до размера частиц менее 200 меш (отв./дюйм). Если действующее соединение в основном растворимо в воде, размер частиц обычно выравнивают путем измельчения с целью обеспечения в основном однородного распределения в составе, например около 40 меш.
Некоторые примеры подходящих наполнителей включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, гуммиарабик, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу. Составы могут дополнительно включать: смазывающие средства, например, тальк, стеарат магния и минеральное масло; средства, способствующие смачиванию; эмульгирующие и суспендирующие средства; консерванты, например, метил - и пропилгидроксибензоаты; подсластители и вкусоароматические средства. Композиции, описанные в настоящей заявке, могут быть составлены таким образом, чтобы обеспечить быстрое, длительное или отсроченное высвобождение действующего ингредиента после введения пациенту, путем применения известных в технике способов.
Описываемые композиции предпочтительно получают в виде дозированных лекарственных форм, причем каждая доза содержит от примерно 0,05 до примерно 100 мг, чаще от примерно 1,0 до примерно 30 мг действующего ингредиента. Термин «дозированная лекарственная форма» относится к физически отдельным единицам, удобным в качестве разовых доз для людей и других млекопитающих, причем каждая единица содержит заранее определенное количество действующего вещества, рассчитанное на получение желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим наполнителем.
Действующие соединения в основном эффективны в широком диапазоне дозировок. Например, дневные дозы, как правило, попадают в диапазон от примерно 0,01 до примерно 30 мг/кг массы тела. В иллюстративном варианте дневные дозы могут находится в диапазоне от примерно 0,02 до примерно 10 мг/кг массы тела, в диапазоне от примерно 0,02 до примерно 1 мг/кг массы тела или в диапазоне от примерно 0,02 до примерно 0,1 мг/кг массы тела. Такие диапазоны дозировок применимы для лечения любого пациента или млекопитающего. Дополнительно, в случае лечения взрослых людей, иллюстративные дозы входят в диапазон от примерно 0,02 до примерно 15 мг/кг массы тела, или в диапазон от примерно 0,1 до примерно 10 мг/кг/день, в виде одной или нескольких доз. Однако следует понимать, что фактически вводимое количество соединения должно определяться врачом, исходя из существенных обстоятельств, включая подвергаемое лечению состояние, выбранный путь введения, конкретное вводимое соединение или соединения, возраст, вес и реакцию конкретного пациента и тяжесть его симптомов, и, следовательно, указанные выше диапазоны дозировок предназначены служить иллюстрациями и их не следует истолковывать и понимать как ограничивающие настоящее изобретение каким-либо образом. В некоторых случаях более чем уместными могут оказаться дозировки, находящиеся ниже нижней границы указанных выше диапазонов, тогда как в других случаях могут применяться еще большие дозировки без появления каких-либо вредных побочных эффектов. Считается, что такие большие дозы могут в первое время быть разделены на несколько меньших доз для введения на протяжении дня.
Тип состава, примененного для введения соединения по способам, описанным в настоящей заявке, может определяться конкретными примененными соединениями, типом фармакокинетического профиля, желательного для пути введения и соединением (соединениями), а также состоянием пациента.
ПРИМЕР СОСТАВА 1
Получали твердые желатиновые капсулы, содержащие следующие ингредиенты:
Перечисленные ингредиенты смешивают и заполняют смесью в количестве 340 мг твердые желатиновые капсулы.
ПРИМЕР СОСТАВА 2
Получали таблетки, применяя перечисленные ниже ингредиенты:
Компоненты смешивали и прессовали с получением таблеток массой 240 мг каждая.
ПРИМЕР СОСТАВА 3
Получали сухой порошок для введения с помощью ингалятора, содержащий следующие компоненты:
Действующее соединение смешивали с лактозой и смесь добавляли в аппарат для ингалирования сухих порошков.
ПРИМЕР СОСТАВА 4
Получали таблетки следующего состава, каждая из которых содержала 30 мг действующего ингредиента:
Действующий ингредиент, крахмал и целлюлозу просеивали через сито № 20 меш U.S. и тщательно смешивали. Раствор поливинилпирролидона смешивали с полученным порошком и затем просеивали через сито 16 меш U.S. Полученные таким образом гранулы высушивали при 50-60°C и просеивали через сито 16 меш U.S. Затем к гранулам добавляли натрий карбоксиметил крахмал, стеарат магния и тальк, предварительно пропущенные через сито № 30 меш U.S., и полученную смесь прессовали на агрегате для получения таблеток, получая таблетки массой 120 мг каждая.
ПРИМЕР СОСТАВА 5
Капсулы, каждая из которых содержала 40 мг лекарственного препарата, получали следующим способом:
Действующий ингредиент, целлюлозу, крахмал и стеарат магния смешивали, просеивали через сито № 20 меш U.S. и наполняли твердую желатиновую капсулу полученной смесью в количестве 150 мг.
ПРИМЕР СОСТАВА 6
Суппозитории, каждый из которых содержал 25 мг действующего ингредиента получали следующим способом:
Действующий ингредиент просеивали через сито № 60 меш U.S. и суспендировали в глицеридах насыщенных жирных кислот, предварительно расплавленных при минимально возможной температуре. Затем смесь выливали в форму для получения суппозиториев нормальной емкости 2,0 г и давали застыть.
ПРИМЕР СОСТАВА 7
Суспензии, содержавшие 50 мг лекарственного препарата на одну дозу 5,0 мл, получали следующим способом:
Микрокристаллическая целлюлоза (89%)
Лекарственный препарат, сахарозу и ксантановую смолу смешивали, пропускали через сито № 10 меш U.S. и затем смешивали с предварительно полученным раствором микрокристаллической целлюлозы и натрий карбоксиметил целлюлозы в воде. Бензоат натрия, вкусоароматическую добавку и краситель разбавляли в небольшом количестве воды и добавляли при перемешивании. Затем добавляли достаточное количество воды для получения требуемого объема.
ПРИМЕР СОСТАВА 8
Капсулы, содержащие 15 мг лекарственного средства получали следующим способом:
Действующий ингредиент, целлюлозу, крахмал и стеарат магния смешивали, просеивали через сито № 20 меш U.S. и заполняли полученным составом в количестве 425 мг твердые желатиновые капсулы.
ПРИМЕР СОСТАВА 9
Состав для внутривенного применения может быть получен следующим способом:
ПРИМЕР СОСТАВА 10
Состав для местного применения может быть получен следующим способом:
Мягкий белый парафин нагревали до плавления. Вносили жидкий парафин и эмульгирующий воск и перемешивали до растворения. Добавляли действующий ингредиент и продолжали перемешивание до его диспергирования. Затем смесь охлаждали до твердого состояния.
ПРИМЕР СОСТАВА 11
Сублингвальные или буккальные таблетки, содержащие 10 мг действующего ингредиента, могут быть получены следующим способом:
Смешивали глицерин, воду, цитрат натрия, поливиниловый спирт и поливинилпирролидон при непрерывном перемешивании, поддерживая температуру около 90°C. Затем в раствор вводили полимеры, полученный раствор охлаждали до примерно 50-55°C и медленно добавляли лекарственный препарат. Гомогенную смесь выливали в формы, изготовленные из инертного материала для получения диффузной матрицы, содержащей лекарственное средство и имеющей толщину примерно 2-4 мм. Затем эту диффузную матрицу разрезали, получая отдельные таблетки, имеющие подходящий размер.
ПРИМЕР СОСТАВА 12
В способах, описанных в настоящей заявке, другой иллюстративный состав применяется в средствах трансдермальной доставки («пластырях»). Такие трансдермальные пластыри могут применяться для обеспечения непрерывной или прерывистой инфузии описанных в настоящей заявке соединений в регулируемых количествах. Устройство и применение трансдермальных пластырей, предназначенных для доставки фармацевтических средств, хорошо известно в технике. Смотрите, например, патент США № 5023252, выданный 11 июня 1991, включенный в настоящую заявку в качестве ссылки. Можно разработать подобные пластыри, обеспечивающие непрерывную, пульсирующую доставку лекарственных средств или доставку по необходимости.
ПРИМЕР СОСТАВА 13
Часто бывает желательно или необходимо прямо или косвенно вводить фармацевтическую композицию в мозг. Методики прямого введения включают размещение катетера для введения лекарственного средства в вентрикулярную систему пациента, чтобы обойти барьер кровь-мозг. Одна из таких имплантируемых систем доставки, применяемая для переноса биологических агентов в конкретные анатомические области организма, описана в патенте США № 5011472, который включен в настоящую заявку в качестве ссылки.
ПРИМЕР СОСТАВА 14
Косвенные способы, которые в основном являются предпочтительными, как правило, включают создание композиций, в которых обеспечен перевод лекарственного соединения в скрытую форму путем превращения гидрофильных лекарственных средств в жирорастворимые лекарства или пролекарства. Перевод в скрытую форму достигается путем блокирования гидрокси, карбонильных, сульфатных и первичных аминогрупп, имеющихся в молекуле лекарственного средства, чтобы сделать лекарство более жирорастворимым и способным преодолевать барьер кровь-мозг. С другой стороны, доставка гидрофильных лекарственных средств может быть улучшена внутриартериальной инфузией гипертонических растворов, которые могут кратковременно открывать барьер кровь-мозг.
Хотя в предшествующем описании изобретение было иллюстрировано и подробно описано, эти иллюстрации и описания следует истолковывать как примерные и типовые по своему характеру, причем понимается, что были продемонстрированы и описаны только иллюстративные варианты осуществления и что защищены все изменения и модификации, которые соответствуют сути описанного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2015 |
|
RU2742773C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПОСТТРАВМАТИЧЕСКОГО СТРЕССОВОГО РАССТРОЙСТВА | 2011 |
|
RU2775783C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ ПОВРЕЖДЕНИЯ ГОЛОВНОГО МОЗГА | 2018 |
|
RU2797548C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПОСТТРАВМАТИЧЕСКОГО СТРЕССОВОГО РАССТРОЙСТВА | 2011 |
|
RU2623209C9 |
| 5-ГАЛОГЕНЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ОКСИНДОЛА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ВАЗОПРЕССИНЗАВИСИМЫХ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2484090C2 |
| ПРОИЗВОДНЫЕ ИНДОЛ-3-ИЛ-КАРБОНИЛ-СПИРО-ПИПЕРИДИНА | 2006 |
|
RU2420529C2 |
| Агонисты V1а-рецепторов | 2013 |
|
RU2634617C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ОКСИДОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛИГАНДОВ РЕЦЕПТОРА ВАЗОПРЕССИНА | 2007 |
|
RU2461556C2 |
| ИНДОЛ-3-ИЛ-КАРБОНИЛ-АЗАСПИРОПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ВАЗОПРЕССИНА | 2006 |
|
RU2418801C2 |
| ПРОИЗВОДНЫЕ ИНДОЛ-3-ИЛ-КАРБОНИЛ-ПИПЕРИДИН-БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ V1А | 2006 |
|
RU2415139C2 |
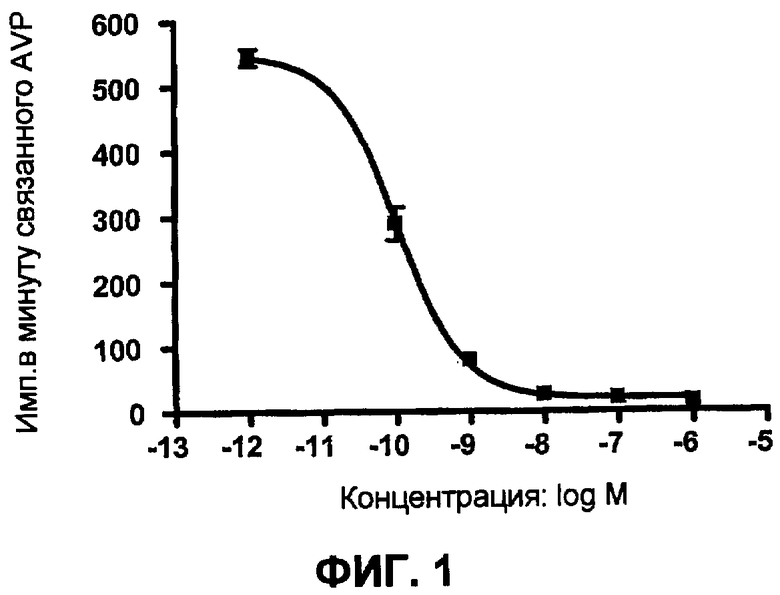
Настоящее изобретение относится к соединению формулы
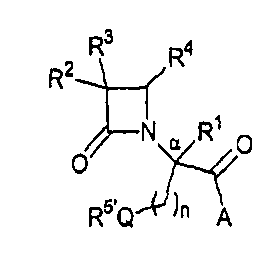
где A, Q, R1, R2, R3, R4, R5' и n представлены в п.1 формулы, а также к его гидратам, сольватам и фармацевтически приемлемым солям. Также описаны применение указанного выше соединения и фармацевтическая композиция, включающая такое соединение, для лечения болезненного состояния у млекопитающих, которое чувствительно к действию антагонистов рецепторов вазопрессина V1a, V1b или V2. 3 н. и 17 з.п. ф-лы, 13 пр., 1 ил.
1. Соединение формулы
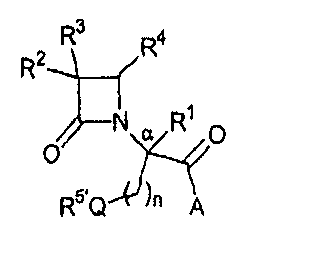
где Q представляет собой серу;
n представляет собой 1 или 2;
А представляет собой необязательно замещенный азотсодержащий гетероцикл, присоединенный по атому азота, где азотсодержащий гетероцикл представляет собой морфолинил, пирролидинил, пиперидинил, пиперазинил, гомопиперазинил или хинуклидинил, каждый из которых необязательно замещен группами R10, R12, R6R7N-, или R6R7N-(C1-C4 алкил); или азотсодержащий гетероцикл представляет собой 2-(пирролидин-1-илметил)пирролидин-1-ил или 1,2,3,4-тетрагидроизохинолин-2-ил;
R1 представляет собой водород или С1-С6алкил;
R2 представляет собой водород;
R3 выбран из группы, состоящей из:
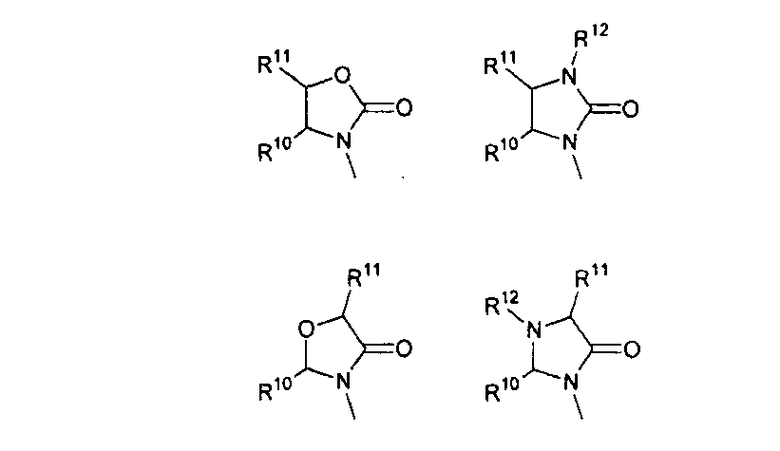
где каждый из заместителей R10 и R11 независимо выбран из водорода, необязательно замещенного С1-С6алкила, необязательно замещенного С3-С8циклоалкила; С1-С4алкоксиС1-С4алкила; С1-С6алкилкарбонилокси; необязательно замещенного арила, необязательно замещенного арил(С1-С4алкила); необязательно замещенного арил(С1- С4алкилокси); необязательно замещенного арил(С1-С4алкилкарбонилокси); дифенилметокси и трифенилметокси; и
R12 выбран из водорода, С1-С6алкила; С3-С8циклоалкила; С1-С4алкоксикарбонила; необязательно замещенного арилоксикарбонила, необязательно замещенного арил (С1-С4алкила); и необязательно замещенного арилоила;
R4 представляет собой необязательно замещенный стирил;
R5' представляет собой необязательно замещенный фенил(С1-С4алкил);
R6 представляет собой водород или С1-С6алкил, и R7 представляет собой С1-С6алкил, С3-С8циклоалкил, необязательно замещенный арил или необязательно замещенный арил(С1-С4алкил); или же R6 и R7 совместно с присоединенным атомом азота образуют гетероцикл, выбранный из группы, состоящей из пирролидинила, пиперидинила, морфолинила, пиперазинила и гомопиперазинила; где указанные пиперазинил или гомопиперазинил необязательно замещены по атому азота заместителем R13;
R13 выбран из группы, состоящей из водорода, С1-С6алкила, С3-С8циклоалкила, С1-С4алкоксикарбонила, необязательно замещенного арилоксикарбонила, необязательно замещенного арил(С1-С4алкила) и необязательно замещенного арилоила; а также гидраты, сольваты и фармацевтически приемлемые соли указанных соединений;
где каждая из указанных выше необязательно замещенных групп в каждом случае означает группу, несущую от одного до трех заместителей, независимо выбранных из группы, состоящей из С1-С4алкила, С1-С4алкокси, С1-С4алкилтио, гидрокси, нитро, галогена, карбокси, циано, С1-С4галогеналкила, С1-С4галогеналкокси, амино, карбамоила, карбоксамидо, амино, алкиламино, диалкиламино, алкилалкиламино и С1-С4алкилсульфониламино; и
где в каждой из указанных выше групп арил независимо выбран из группы, состоящей из фурила, пирролила, тиенила, пиридинила, тиазолила, оксазолила, изоксазолила, изотиазолила, имидазолила, пиразолила, фенила, пиридазинила, пиримидинила, пиразинила, тиадиазолила, оксадиазолила, нафтила, инданила, флуоренила, хинолинила, изохинолинила, бензодиоксанила, бензофуранила и бензотиенила.
2. Соединение по п.1, в котором Q представляет собой серу и n равно 1.
3. Соединение по п.1, в котором Q представляет собой серу и n равно 2.
4. Соединение по любому из пп.1-3, в котором R5' представляет собой необязательно замещенный фенил(С1-С2алкил).
5. Соединение по любому из пп.1-3, в котором R5' представляет собой бензил.
6. Соединение по любому из пп.1-3, где А представляет собой замещенный гетероцикл, где гетероцикл выбран из группы, состоящей из пирролидинила, пиперидинила и пиперазинила.
7. Соединение по любому из пп.1-3, в котором фрагмент А представляет собой пиперидинил или пиперазинил и где фрагмент А замещен по 4 положению пирролидинилом, пиперидинилом, пиперазинилом, гомопиперазинилом, пирролидинил (С1-С4алкилом), пиперидинил (С1-С4алкилом), пиперазинил (С1-С4алкилом) или гомопиперазинил (С1-С4алкилом).
8. Соединение по любому из пп.1-3, где R5' представляет собой фенил (С1-С2)алкил.
9. Соединение по любому из пп.1-3, где R3 представляет собой
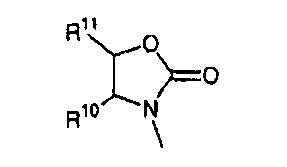
10. Соединение по п.9, где R3 представляет собой
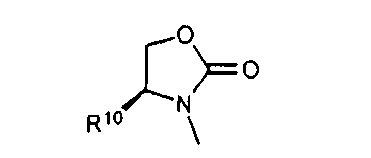
11. Соединение по любому из пп.1-3, где R5' представляет собой необязательно замещенный бензил.
12. Соединение по п.1 формулы
или его фармацевтически приемлемая соль.
13. Фармацевтическая композиция, включающая соединение по любому из пп.1-3, а также фармацевтически приемлемый носитель, разбавитель, наполнитель или их комбинацию; где соединение присутствует в количестве, эффективном для лечения болезненного состояния у млекопитающих, которое чувствительно к действию антагонистов рецепторов вазопрессина V1a, V1b или V2, при необходимости такого лечения.
14. Фармацевтическая композиция по п.13, где болезненное состояние представляет собой психическое заболевание, связанное со стрессом, депрессию, тревожность, аффективные расстройства, обсессивно-компульсивные заболевания, импульсивность, агрессивные расстройства, заболевания, влияющие на гомеостаз воды, почечную функцию, ингибирование метаболизма фосфатидил-инозита или регуляцию температуры, заболевания, связанные с тошнотой, рвотой и болью; или сердечно-сосудистое заболевание, застойную сердечную недостаточность или расстройство, связанное с агрегацией тромбоцитов, или их сочетание.
15. Применение соединения по любому из пп.1-3 для приготовления лекарственного средства, включающего эффективное количество указанного соединения, для лечения болезненного состояния у млекопитающих, которое чувствительно к действию антагонистов рецепторов вазопрессина V1a, V1b или V2, при необходимости такого лечения.
16. Применение по п.15, где композиция дополнительно включает фармацевтически приемлемый носитель, разбавитель, наполнитель или их комбинацию.
17. Применение по п.15, где болезненное состояние представляет собой психическое заболевание, связанное со стрессом, депрессию, тревожность, аффективные расстройства, обсессивно-компульсивное заболевание, импульсивность, агрессивное расстройство, заболевание, влияющее на гомеостаз воды, почечную функцию, ингибирование метаболизма фосфатидил-инозита или регуляцию температуры, заболевание, связанное с тошнотой, рвотой и болью; или сердечно-сосудистое заболевание, застойную сердечную недостаточность или расстройство, связанное с агрегацией тромбоцитов, или их сочетание.
18. Применение по п.17, где болезненное состояние представляет собой обсессивно-компульсивное заболевание, тревожность, депрессию и их сочетания.
19. Применение по п.17, где заболевание представляет собой тревожность.
20. Применение по п.17, где заболевание представляет собой депрессию.
| WO 03031407 А, 17.04.2003 | |||
| Способ совместного получения алкилкарбонатов и формиатов щелочных металлов | 1961 |
|
SU144840A1 |
| JP 56125361 А, 01.10.1981 | |||
| Ojima Iwao et al., Journal of The Chemical Society, Chemical Communications, 1987, (8), 625-626 | |||
| US 6610680 B1, 26.08.2003 | |||
| US 5759865 A, 02.06.1998 | |||
| WO 9635122 A1, 07.11.1996 | |||
| СПОСОБ И ПРИСПОСОБЛЕНИЕ ДЛЯ ПОЛУЧЕНИЯ В ВАГРАНКЕ ОБЕЗУГЛЕРОЖЕННЫХ ЖЕЛЕЗО-УГЛЕРОДИСТЫХ СПЛАВОВ, ГЛАВНЫМ ОБРАЗОМ ЧУГУНА | 1925 |
|
SU4628A1 |

