Настоящее изобретение относится к способу получения этилена путем дегидратации этанола в паровой фазе с применением гетерополикислотного катализатора.
Этилен является важным химикатом общего назначения и мономером, который обычно производят в промышленности с помощью парового или каталитического крекинга углеводородов, полученных из сырой нефти. Однако по мере сокращения запасов сырой нефти и увеличения цен на нее возникает растущая необходимость в нахождении альтернативных экономически выгодных способов получения указанного продукта. По причине доступности этанола, получаемого ферментацией биомассы и по технологиям на основе синтез-газа, этанол становится все более важным потенциальным сырьем, из которого в будущем можно будет получать этилен.
Получение этилена химической дегидратацией этанола в паровой фазе осуществляется по хорошо известной химической реакции, которую применяют в промышленности уже много лет (см., например, Kirk Othmer Encyclopaedia of Chemical Technology (третье издание), том 9, с.411-413). Обычно эту реакцию осуществляют при повышенной температуре и пониженном давлении в присутствии кислотного катализатора, например активированного оксида алюминия или нанесенной фосфорной кислоты.
В последние годы возросло внимание к получению новых альтернативных катализаторов, обладающих улучшенными показателями. В нашей совместно поданной европейской патентной заявке №06255980.2 описан такой подход, осуществляемый с помощью способа, включающего применение нанесенного катализатора на основе гетерополикислоты (ГПК), удовлетворяющего следующему уравнению:
ОП>0,6-0,3×(загрузка ГПК/удельная поверхность катализатора), где
ОП представляет собой объем пор сухого нанесенного катализатора на основе гетерополикислоты (измеренную в мл/г катализатора); загрузка ГПК представляет собой количество гетерополикислоты, присутствующей в сухом нанесенном катализаторе на основе гетерополикислоты (измеренное в мкмоль/г катализатора), а удельная поверхность катализатора представляет собой удельную поверхность сухого нанесенного катализатора на основе гетерополикислоты (измеренную в м2/г катализатора). Способ, описанный в указанной патентной заявке, осуществляют при повышенной температуре, которая, как правило, составляет от 180 до 250°С. Описанные катализаторы обладают более высокой производительностью в отношении этилена и пониженным выходом этана, по сравнению с катализаторами, ранее описанными в данной области техники. Данные свойства предпочтительны, поскольку, во-первых, этан является нежелательным побочным продуктом, а во-вторых, его отделение от этилена в большом масштабе сложно и требует значительных затрат энергии. В европейской патентной заявке ЕР 1792885 и международной заявке WO 2007/03899, поданных авторами настоящего описания, описаны предпочтительные способы осуществления указанного процесса.
В способах данного типа, описанных в настоящей патентной заявке, поток сырья, включающий этанол, необязательно воду и другие компоненты, непрерывно подают в реактор, содержащий слой катализатора на основе гетерополикислоты, а продукты непрерывно удаляют. В установившемся режиме работы реактора поток сырья, входящий в реактор, быстро превращается в равновесную смесь воды, этанола и этоксиэтана (продукт быстрой первой стадии дегидратации этанола) около впускного отверстия. Таким образом, катализатор работает в присутствии значительного количества воды, точное количество которой зависит от вышеописанной равновесной концентрации (которая, в свою очередь, зависит от общей температуры и давления в реакторе). В действительности, условия являются еще более сложными, поскольку по мере превращения сырья в слое катализатора образуется дополнительное количество воды. Следовательно, в зависимости от геометрии реактора концентрация воды в слое катализатора может изменяться от значительной (например, в случае применения трубчатого реактора с неподвижным слоем) до очень малой (например, в реакторе с кипящим слоем). Такое колебание концентрации воды также зависит от температуры реактора и времени контакта сырья с катализатором.
Сейчас было обнаружено, что количество воды, присутствующей в процессах типа, описанного в европейской патентной заявке 06255980.2, в значительной степени влияет на относительные количества этилена и этана, получаемые при помощи катализатора. Следовательно, регулирование содержания воды является очень важным этапом для увеличения до максимума выхода этилена и минимизации образования этана в процессах такого типа. В настоящее время точный механизм воздействия воды на выходы этих продуктов неизвестен, однако в соответствии с одной из теорий предполагают, что важную роль выполняет конденсация воды в порах катализатора. Степень, в которой происходит такая конденсация, будет зависеть от того, насколько близко состояние сырьевой смеси к «точке росы» в рабочих условиях реактора.
Следовательно, настоящее изобретение обеспечивает способ осуществления работы реактора в процессах данного типа таким образом, чтобы можно было отрегулировать реактор для работы в режиме, при котором влажность катализатора в слое катализатора поддерживается на оптимальном уровне.
В соответствии с настоящим изобретением обеспечивается способ получения этилена с помощью парофазной химической дегидратации сырья, включающего этанол, воду и этоксиэтан, в реакторе при повышенных температуре и давлении в присутствии слоя катализатора, включающего нанесенную гетерополивольфрамовую кислоту, отличающийся тем, что реактор регулируют или осуществляют его работу таким образом, что он действует в режиме, удовлетворяющем следующим параметрам:
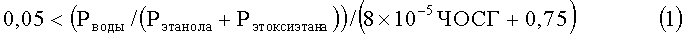
и
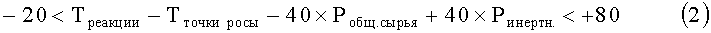 ,
,
причем
Рводы, Рэтанола и Рэтоксиэтана представляют собой парциальные давления воды, этанола и этоксиэтана соответственно в термодинамическом уравнении состава сырья при рабочей температуре и давлении процесса в МПа, ЧОСГ представляет собой часовую объемную скорость газообразного сырья, проходящего над катализатором в ч-1, Треакции представляет собой температуру реакции в °С, Тточки росы представляет собой температуру точки росы сырья при его термодинамически равновесном составе в °С при данном давлении реакции, Робщ. сырья представляет собой общее давление сырья в МПа, а Ринертн. представляет собой парциальное давление инертных веществ в сырье в МПа.
Неожиданно было найдено, что применение нанесенных гетерополикислот в режиме работы реактора, характеризуемом вышеописанными параметрами (1) и (2), позволяет увеличить до максимума выход этилена и в то же время минимизировать селективность образования этана. Дополнительное преимущество способа в соответствии с настоящим изобретением заключается в том, что образование других побочных продуктов, например этаналя, также снижается до минимума.
Можно считать, что вышеописанный параметр (2) обеспечивает меру влажности катализатора в рабочем состоянии. Полагают, что при температурах реакции, близких к точке росы конкретного применяемого сырья (при его термодинамически равновесном составе), катализатор является слишком влажным и в ходе процесса образуются неприемлемо высокие количества этана (как правило, более 600 част./млн. в составе этиленового продукта). Похожие явления также наблюдаются в случае, если температура реакции значительно превышает температуру точки росы сырья (при его термодинамически равновесном составе), когда катализатор, по существу, является слишком сухим. Только в случае, когда разница между температурой реакции и точкой росы сырья (при его термодинамически равновесном составе и с учетом количества присутствующих инертных веществ) достаточна для того, чтобы выполнялись вышеописанные параметры, этан образуется в очень низких количествах, обычно менее 600 част./млн., предпочтительно менее 250 част./млн.
Вышеописанный параметр (1) обеспечивает меру общего содержания влаги в процессе. Было найдено, что при относительно высоком содержании воды в сырье количество образующегося этана также является низким. Такое снижение количества образующегося этана дополнительно усиливается, если часовая объемная скорость газа снижается посредством увеличения степени превращения сырья в этилен. Воздействие этого явления оптимально в случае, если условия удовлетворяют параметру (1).
Что касается рабочих условий процесса, температура реакции должна составлять от 180 до 270°С, предпочтительно от 200 до 250°С, наиболее предпочтительно от 210 до 240°С, а общее давление реакции должно составлять от 1 до 3 МПа.
Для осуществления улучшений, описанных в настоящем описании, предпочтительно выбирать условия работы таким образом, чтобы количество этана в выходящем из реакторе потоке составляло, по меньшей мере, менее 1000 част./млн., предпочтительно менее 600 част./млн., наиболее предпочтительно менее 250 част./млн. Для достижения такого низкого содержания этана предпочтительно, чтобы рабочие условия были такими, чтобы для параметра (1) выполнялось следующее неравенство:
0,1<(Рводы/(Рэтанола+Рэтоксиэтана))/(8×10-5×ЧОСГ+0,75),
более предпочтительно:
0,2<(Рводы/(Рэтанола+Рэтоксиэтана))/(8×10-5×ЧОСГ+0,75),
наиболее предпочтительно:
0,3<(Рводы/(Рэтанола+Рэтоксиэтана))/(8×10-5×ЧОСГ+0,75),
а для параметра (2) выполнялось следующее неравенство:
-20<Треакции-Тточки росы-40×Робщ. сырья+40×Ринертн.<60
более предпочтительно:
0<Треакции-Тточки росы-40×Робщ. сырья+40×Ринертн.<50
наиболее предпочтительно:
20<Треакции-Тточки росы-40×Робщ. сырья+40×Ринертн.<40
Особенно ценно, что любая комбинация предпочтительных интервалов этих двух параметров способна обеспечить выгоду и входит в состав настоящего изобретения. Это дополнительно показано на Фиг.1.
Как указано выше, при работе в режиме, определенном параметрами (1) и (2), компоненты сырья в реакторе будут находиться в равновесии и сырье будет включать от 0 до 65 об.% этоксиэтана, от 0 до 15 об.% воды и от 0 до 20 об.% этилена. Часовая объемная скорость газа (ЧОСГ) будет обычно составлять от 500 до 10000 ч-1. Предпочтительно ЧОСГ составляет от 1000 до 5000 ч-1 и наиболее предпочтительно ЧОСГ составляет от 2000 до 4000 ч-1. Равновесный состав сырья можно создавать в передней части реактора, либо в предшествующем основному реакторе этерификации. Последний подход позволяет регулировать экзотермический эффект, связанный с происходящим на первой стадии процессом этерификации, таким образом, чтобы избежать появления точек локального перегрева в слое. Также предпочтительно, чтобы этанол был получен из биомассы, поскольку, в общем, в этом случае он будет содержать значительное количество воды, которую необходимо удалить перед подачей этанола в реактор. При таких условиях для получения большего преимущества можно использовать колонну реакционной дистилляции для совместной этерификации и отделения воды. В качестве альтернативы стадии этерификации и разделения можно осуществлять отдельно и без затруднений, поскольку температура кипения этоксиэтана ниже температуры кипения воды, и эти компоненты мало склонны к образованию азеотропных смесей.
В одном из предпочтительных вариантов настоящего изобретения предпочтительно применять в составе сырья инертный неспособный к конденсации разбавитель с целью регулирования степени влажности катализатора и поддержания режима работы, заданного параметром (2). Предпочтительными разбавителями являются азот, гелий, этилен, насыщенные углеводороды, находящиеся в газообразной форме при реакционных условиях, наиболее предпочтительными среди которых являются 2-метилпропан, н-бутан и этилен. Предпочтительно, чтобы содержание применяемых инертных разбавителей было таким, чтобы значение параметра (2) составляло от 25 до 30. При определенном общем давлении в реакторе применение указанных разбавителей позволяет достичь более высокой производительности в отношении этилена и более низкого выхода этанового побочного продукта. Регулирование количества разбавителя может быть важным для оптимизации продолжительности работы катализатора в реакторе. Применение разбавителя, температура кипения которого находится между температурами кипения этаналя и этилена, с одной стороны, или этоксиэтана и этилена, с другой стороны, является предпочтительным, поскольку оно облегчает отделение этилена от других указанных компонентов ниже по течению. При использовании этого подхода можно получать этилен, содержащий менее 10 част./млн. кислородсодержащих соединений. В слой катализатора, либо между слоями катализатора (если применяют несколько слоев катализатора) выгодно добавлять разбавитель с целью поддержания рабочего режима в соответствии с настоящим изобретением. Наконец, применение разбавителей облегчает применение сырья, содержащего более высокое количество воды, чем вышеуказанное сырье (обычно более 15 об.% в составе равновесной композиции, предпочтительно 20 об.% или более).
Под выражением «гетерополивольфрамовая кислота» в настоящем описании понимают как сами свободные кислоты, так и их производные. Указанные производные включают, помимо прочего, соли щелочных металлов, соли щелочноземельных металлов, аммониевые соли, свободные кислоты, объемные катионные соли и/или соли металлов (причем соли могут представлять собой полностью или частично замещенные соли) гетерополивольфрамовых кислот. Следовательно, гетерополивольфрамовые кислоты, применяемые по настоящему изобретению, представляют собой комплексные анионы с высокой молекулярной массой, включающие атомы металлов, соединенные кислородными мостиками.
Обычно каждый анион включает от 12 до 18 атомов вольфрама, соединенных кислородными мостиками. Эти атомы окружают один или более центральный атом симметричным образом. Центральные атомы предпочтительно представляют собой кремний или фосфор, но в качестве альтернативы они могут включать любой из большого разнообразия атомов групп с 1 по 8 Периодической таблицы элементов. Эти атомы могут представлять собой медь, бериллий, цинк, кобальт, никель, бор, алюминий, галлий, железо, церий, мышьяк, сурьму, висмут, хром, родий, кремний, германий, олово, титан, цирконий, ванадий, серу, теллур, марганец, никель, платину, торий, гафний, церий, ванадий, ионы сурьмы, теллур и йод. Подходящие гетерополивольфрамовые кислоты включают гетерополивольфрамовые кислоты типа Кеггина, Уэллса-Доусона и Андерсона-Эванса-Перлова. Конкретные примеры подходящих гетерополивольфрамовых кислот приведены ниже:
кроме того, данные примеры включают следующие свободные гетерополивольфрамовые кислоты или их частично замещенные соли:
Кроме того, можно применять смеси различных гетерополивольфрамовых кислот и их солей. К предпочтительным смесям для использования в способе, описанном в настоящем изобретении, относятся смеси на основе структур Кеггина или Уэллса-Доусона, более предпочтительно гетерополивольфрамовая кислота, выбираемая для применения в способе по настоящему изобретению, представляет собой либо вольфрамкремниевую кислоту, либо вольфрамфосфорную кислоту. Наиболее предпочтительно гетерополивольфрамовая кислота представляет собой 12-вольфрамкремниевую кислоту (H4[SiW12O40].×H2O).
Предпочтительно молекулярная масса гетерополивольфрамовых кислот, применяемых в соответствии с настоящим изобретением, составляет от более 700 до менее 8500, предпочтительно от более 2800 до менее 6000. Такие гетерополивольфрамовые кислоты также включают димерные комплексы.
Нанесенный катализатор на основе гетерополивольфрамовой кислоты можно удобным образом получать путем растворения выбранной гетерополивольфрамовой кислоты в подходящем растворителе, причем подходящие растворители включают полярные растворители, например воду, простые эфиры, спирты, карбоновые кислоты, кетоны и альдегиды и/или смеси вышеперечисленного; наиболее предпочтительными растворителями являются дистиллированная вода и/или этанол. Концентрация гетерополивольфрамовой кислоты в полученном кислом растворе предпочтительно составляет от 10 до 80 мас.%, более предпочтительно от 20 до 70 мас.% и наиболее предпочтительно от 30 до 60 мас.%. Указанный раствор затем добавляют к выбранному носителю (либо в качестве альтернативы носитель погружают в раствор). Фактический объем кислого раствора, добавляемого к носителю, неограничен, следовательно, его количество может быть достаточным для достижения пропитки по влагоемкости или влажной пропитки, причем влажная пропитка (то есть приготовление с применением избыточного объема кислого раствора по отношению к объему пор носителя) является предпочтительным в интересах настоящего изобретения способом.
Полученную нанесенную гетерополивольфрамовую кислоту можно модифицировать, кроме того, в водном растворе можно формировать различные соли гетерополивольфрамовой кислоты либо перед пропиткой, либо во время пропитки носителя кислым раствором, путем осуществления длительного контакта между гетерополивольфрамовой кислотой и раствором подходящей металлической соли, либо путем добавления фосфорной кислоты и/или других минеральных кислот.
При применении растворимой металлической соли для модификации носителя соль используют в желаемой концентрации в составе раствора гетерополивольфрамовой кислоты. Носитель затем оставляют смачиваться в указанном кислом растворе на необходимое время (например, на несколько часов), причем раствор периодически перемешивают или встряхивают; затем по прошествии указанного промежутка времени соль отфильтровывают подходящим способом для удаления избытка кислоты.
Если соль является нерастворимой, предпочтительно пропитывать катализатор гетерополивольфрамовой кислотой, а затем титровать предшественником соли. Этот способ может позволить улучшить дисперсию соли гетерополивольфрамовой кислоты. Также можно применять другие методики, например вакуумную пропитку.
Затем пропитанный носитель можно промыть и высушить. Это можно осуществлять с помощью любой традиционной методики разделения, включая, например, декантирование и/или фильтрование. После извлечения пропитанный носитель можно высушить, предпочтительно поместив его в печь при повышенной температуре. В качестве альтернативы, либо в дополнение к вышеописанному, можно применять эксикатор. В коммерческом масштабе данную стадию сушки, как правило, осуществляют с помощью продувания горячим инертным газом, например азотом.
Предпочтительный нанесенный катализатор на основе гетерополивольфрамовой кислоты обладает следующим свойством:
PV>0,6-0,3×[количество нанесенной ГПК/площадь поверхности катализатора], где
PV представляет собой объем пор сухого нанесенного катализатора на основе гетерополивольфрамовой кислоты (измеренный в мл/г катализатора), количество нанесенной ГПК представляет собой количество гетерополикислоты, присутствующей в сухом нанесенном катализаторе на основе гетерополивольфрамовой кислоты (измеренное в мкмоль/г катализатора), а площадь поверхности катализатора представляет собой площадь поверхности сухого нанесенного катализатора на основе гетерополивольфрамовой кислоты (измеренную в м2/г катализатора).
Количество гетерополивольфрамовой кислоты, нанесенной пропиткой на носитель, подходящим образом составляет от 10 до 80 мас.%, предпочтительно от 20 до 50 мас.% в расчете на общую массу гетерополивольфрамовой кислоты и носителя.
Массу катализатора после сушки и массу примененного носителя можно использовать для расчета массы кислоты, находящейся на носителе, путем вычитания последнего из первого, при этом получается количество нанесенной на катализатор гетерополикислоты в виде, соответствующем выражению «гетерополивольфрамовой кислоты/кг катализатора». Можно также рассчитать количество нанесенной на катализатор гетерополикислоты в «граммах гетерополивольфрамовой кислоты/литр носителя» с использованием известной или измеренной насыпной плотности носителя. Предпочтительное количество нанесенной гетерополивольфрамовой кислоты составляет от 150 до 600 г гетерополивольфрамовой кислоты/кг катализатора.
В соответствии с особенно предпочтительным вариантом настоящего изобретения среднее количество нанесенной гетерополивольфрамовой кислоты в расчете на площадь поверхности сухого нанесенного катализатора на основе гетерополивольфрамовой кислоты оставляет более чем 0,1 мкмоль/м2.
Следует отметить, что многовалентные степени окисления и степени гидратации гетерополивольфрамовых кислот, указанные выше, а также представленные типичными формулами некоторых конкретных соединений (показано выше), относятся только к «свежей» кислоте до ее нанесения пропиткой на носитель, конкретно, до того, как ее подвергают процессу дегидратации в условиях в соответствии с настоящим изобретением. Степень гидратации гетерополивольфрамовой кислоты может влиять на кислотность нанесенного катализатора и, как следствие, его активность и селективность. Таким образом, процесс пропитки или процесс дегидратации, или оба этих процесса могут приводить к изменению степени гидратации и степени окисления металлов в составе гетерополивольфрамовых кислот, то есть конкретные виды применяемых в заданных условиях процесса катализаторов могут не обеспечивать необходимые степени гидратации/окисления металлов в составе гетерополивольфрамовых кислот, использованных при пропитке носителя. Следовательно, естественно ожидать, что такие степени гидратации и окисления могут также отличаться в отработанных катализаторах после процесса дегидратации в соответствии с настоящим изобретением.
В соответствии с особенно предпочтительным вариантом настоящего изобретения количество хлорида, присутствующего в указанном нанесенном катализаторе на основе гетерополивольфрамовой кислоты или на его поверхности, составляет менее 40 част./млн., предпочтительно менее 25 част./млн., наиболее предпочтительно менее 20 част./млн.
Подходящие носители для катализаторов могут находиться в порошкообразной форме или в качестве альтернативы в гранулированной форме, или в таблетированной форме, в сферической форме, либо в виде экструдатов (включая частицы различной геометрической формы) и включают, но не ограничиваются этим, мордениты, например монтмориллонит, глины, бентонит, диатомит, оксид титана, активированный уголь, оксид алюминия, оксид кремния-оксид алюминия, совместные гели оксида кремния-оксида титана, совместные гели оксида кремния-оксида циркония, покрытый углеродом оксид алюминия, цеолиты, оксид цинка, пиролизованные в пламени оксиды. Носители могут представлять собой смешанные оксиды, нейтральные или слабоосновные оксиды. Предпочтительными являются носители на основе оксида кремния, например носители на основе силикагеля и носители, полученные гидролизом SiCl4 в пламени. Предпочтительные носители, по существу, не содержат свободных или посторонних металлов или элементов, которые могут отрицательно воздействовать на каталитическую активность системы. Таким образом, подходящие носители на основе оксида кремния обладают чистотой, составляющей, по меньшей мере, 99 мас.%. Количество примесей предпочтительно составляет менее 1 мас.%, предпочтительно менее 0,60 мас.%, наиболее предпочтительно менее 0,30 мас.%. Объем пор носителя предпочтительно составляет более 0,50 мл/г, предпочтительно более 0,8 мл/г.
Подходящие носители на основе оксида кремния включают, но не ограничиваются ими, следующие марки: Grace Davison Davicat® Grade 57, Grace Davison Davicat® 1252, Grace Davison Davicat® SI 1254, Fuji Silysia CariAct® Q15, Fuji Silysia CariAct® Q10, Degussa Aerolyst® 3045 и Degussa Aerolyst® 3043. Средний диаметр частиц носителя составляет от 2 до 10 мм, предпочтительно от 3 до 6 мм. Однако, при желании, эти частицы можно измельчать и просеивать с получением частиц меньшего размера, например от 0,5 до 2 мм.
Средний радиус пор носителя (до нанесения пропиткой гетерополивольфрамовой кислоты) составляет от 10 до 500 Å, предпочтительно от 30 до 175 Å, более предпочтительно от 50 до 150 Å и наиболее предпочтительно от 60 до 120 Å. Удельная поверхность по БЭТ предпочтительно составляет от 50 до 600 м2/г, наиболее предпочтительно от 150 до 400 м2/г. Сопротивление раздавливанию одной частицы носителя составляет по меньшей мере 1 кгс, подходящим образом не менее 2 кгс, предпочтительно не менее 6 кгс и более предпочтительно не менее 7 кгс. Насыпная плотность носителя составляет по меньшей мере 380 г/л, предпочтительно по меньшей мере, 395 г/л.
Сопротивление раздавливанию одной частицы определяли с применением измерителя сил Mecmesin, который измеряет минимальную силу, необходимую для раздавливания частицы между параллельными пластинами. Сопротивление раздавливанию основано на среднем арифметическом результате серии измерений, произведенных для, по меньшей мере, 25 частиц катализатора.
Удельную поверхность по БЭТ, объем пор, распределение пор по размерам и средний радиус пор определяли с помощью изотермы адсорбции азота при 77 К с применением статического анализатора объемной адсорбции Micromeritics TRISTAR 3000. Использованная методика является применением способов по Британским Стандартам BS4359: часть 1:1984 «рекомендации к способам газовой адсорбции (БЭТ)» и BS7591: часть 2:1992 «пористость и распределение пор материалов по размерам» - способ определения адсорбции газа. Полученные данные обрабатывали с помощью метода БЭТ (Брунауэра-Эммета-Теллера) (в диапазоне давлений от 0,05 до 0,20 Р/Р0) и метода Баррета-Джойнера-Халенды (BGH) (для диаметра пор от 20 до 1000 Å) с получением удельной площади поверхности и распределения частиц по размерам соответственно.
Подходящими ссылками на вышеприведенные методы обработки данных являются: Brunauer S., Emmett P.H., Teller E. /J. Amer. Chem. Soc., т.60, с.309, (1938) и Barrett E.P., Joyner L.G., Halenda P.P./ J. Amer. Chem. Soc., 1951, т.73, c.373-380.
Перед анализом образцы носителей и катализаторов дегазировали в течение 16 ч при 120°С вакууме, составляющем 5*10-3 Торр.
В особенно предпочтительном аспекте настоящего изобретения, дополнительно, выбранный носитель для катализатора сначала обрабатывали фторирующим агентом; авторы настоящего изобретения считают, что осуществление данного предпочтительного варианта позволит сделать катализатор более инертным и/или кислотным, таким образом удастся улучшить селективность и/или эффективность катализатора в ходе вышеупомянутого процесса дегидратации.
Чтобы убедиться в том, что реактор устойчиво работает в вышеописанном режиме, можно использовать различные подходы. Следует изначально понимать, что процесс по настоящему изобретению является, в целом, эндотермическим, следовательно, важной проблемой при поддержании условий процесса в интервале, заданном параметрами (1) и (2), приведенными выше, является подача тепла в реактор. Таким образом, в одном из предпочтительных вариантов настоящего изобретения реакцию осуществляют в серии из двух или более реакторов, работающих в адиабатическом режиме, при котором степень превращения сырья постепенно увеличивается и каждый последующий реактор работает при более высокой температуре, чем предыдущий. В качестве альтернативы, если применяют единственный реактор, можно осуществлять нагревание в нескольких точках, расположенных вдоль слоя с различными интервалами, с обеспечением возрастающего градиента температуры, таким образом, активность и селективность катализатора можно поддерживать на требуемом уровне. Зная вышеописанный режим, можно вычислить и применять оптимальные температуры и, как следствие, оптимальные требования к нагреву. В этом случае можно выгодным образом применять удаление воды и рециркуляцию потоков продукта.
Во втором предпочтительном варианте настоящего изобретения, который можно применять отдельно или в комбинации с другими предпочтительными вариантами, для регулирования времени контакта катализатора с сырьем можно изменять форму или конфигурацию реактора. Например, можно применять реактор, устроенный таким образом, что площадь его поперечного сечения увеличивается вдоль слоя катализатора. В качестве альтернативы можно применять реактор радиальной конструкции, в котором сырье поступает из центральной точки через цилиндры постепенно увеличивающегося диаметра и, следовательно, постепенно увеличивающейся активной площади поверхности.
В еще одном предпочтительном варианте время контакта гетерополикислоты и сырья можно увеличить с использованием катализаторов, степень разбавления которых увеличивается по мере глубины их расположения в слое (либо путем смешивания с разбавителем, либо с применением катализатора с постепенно увеличивающимся содержанием гетерополивольфрамовой кислоты). Такой подход позволяет облегчить регулирование работы катализатора и регулирование выделения тепла при установлении начального равновесия сырья на начальном участке слоя катализатора, как описано выше.
Полагают, что принципы, описанные выше, применимы к химической дегидратации 2-метилпропан-1-ола, 2-метилпропан-2-ола, н-пропанола и пропан-2-ола.
Далее настоящее изобретение будет проиллюстрировано с помощью ссылок на нижеприведенные примеры.
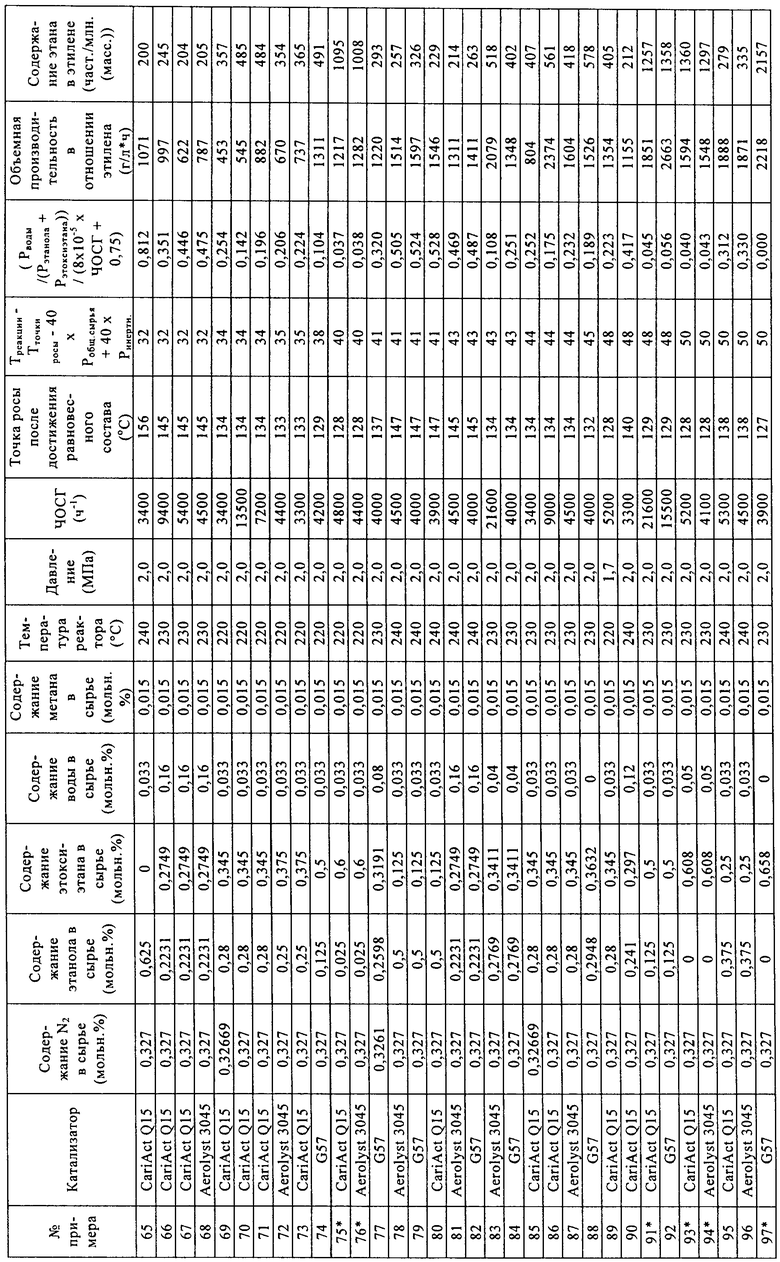
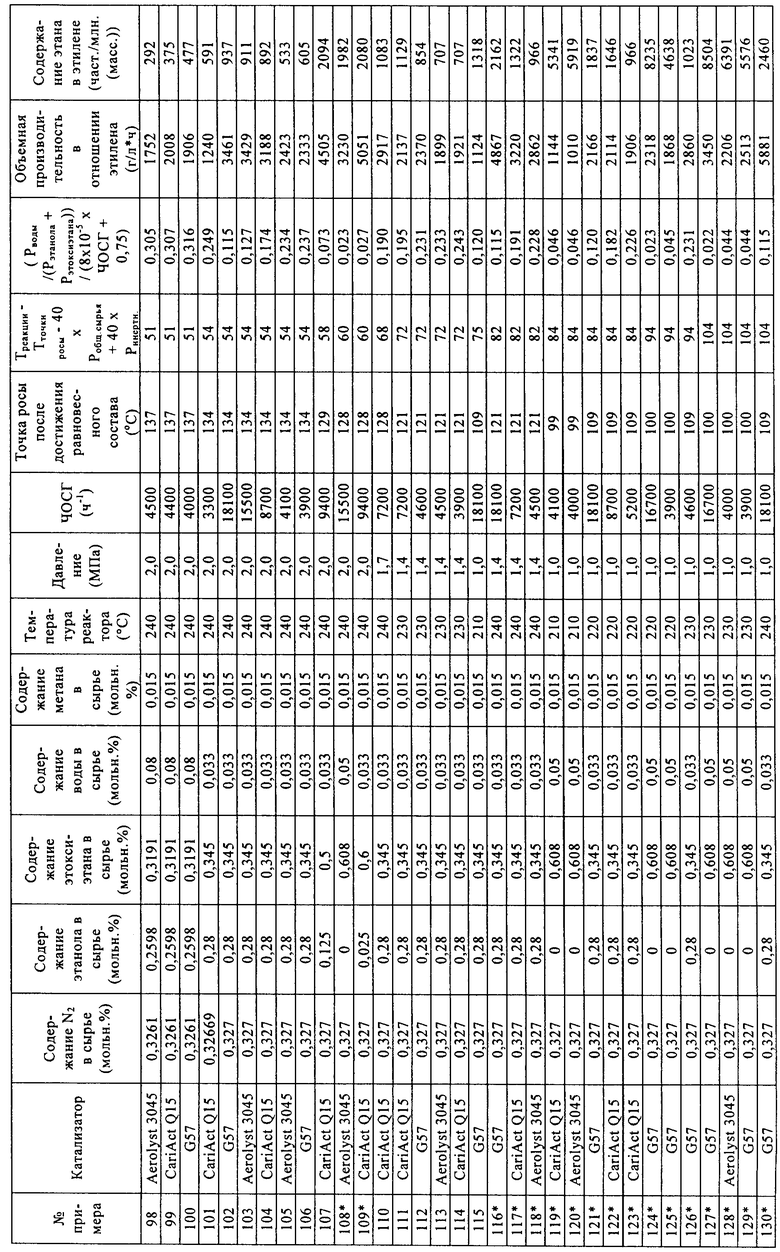
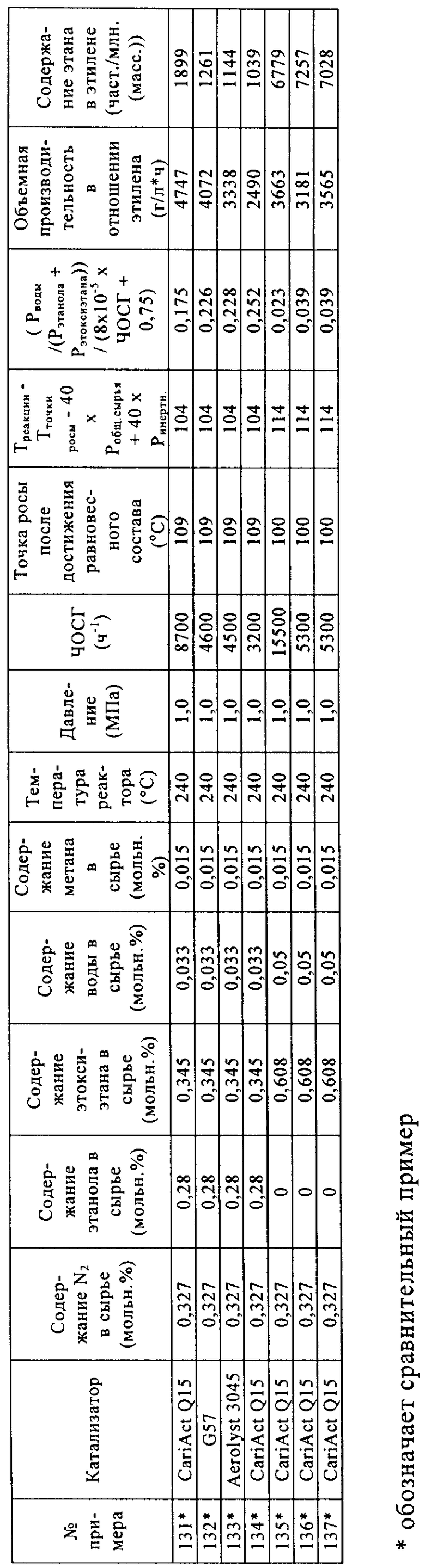
Носители, применяемые в примерах
- таблетки оксида кремния CariAct® Q15, поставлены Fuji Silysia,
- экструдаты оксида кремния Aerolyst® 3045, поставлены в виде экструдатов Degussa,
- гранулы оксида кремния Davicat® Grade 57, поставлены Grace Davison.
Свойства носителей
Материалы носителей анализировали с помощью азотной порометрии.
Приготовление катализаторов с применением воды в качестве растворителя
Кремнийвольфрамовую кислоту (H4[SiW12O40]*24H2O, М=3310,6) взвешивали, переносили в пластиковую бутылку с широким горлом и растворяли в дистиллированной воде. К этому раствору кислоты добавляли взвешенное количество материала носителя. Кислотный раствор и материал носителя оставляли смачиваться в растворе кислоты примерно на 1 ч, периодически аккуратно взбалтывая в течение этого времени с целью удаления пузырьков воздуха.
После смачивания неадсорбированный раствор кислоты удаляли путем выливания раствора кислоты с материалом носителя из пластикового контейнера на пластиковый фильтр (который содержал фильтровальную бумагу).
Катализатор оставляли на фильтре на время, составляющее примерно от 15 до 60 минут, до тех пор, пока вся жидкость не удалялась из катализатора.
После завершения удаления жидкости катализатор перемещали на керамический лоток и сушили в муфельной печи при 130°С в атмосфере азота.
Сухой твердый катализатор взвешивали и определяли количество адсорбированной на нем кремнийвольфрамовой кислоты на основании разницы в массе по отношению к начальному материалу, как указано в нижеприведенной таблице.
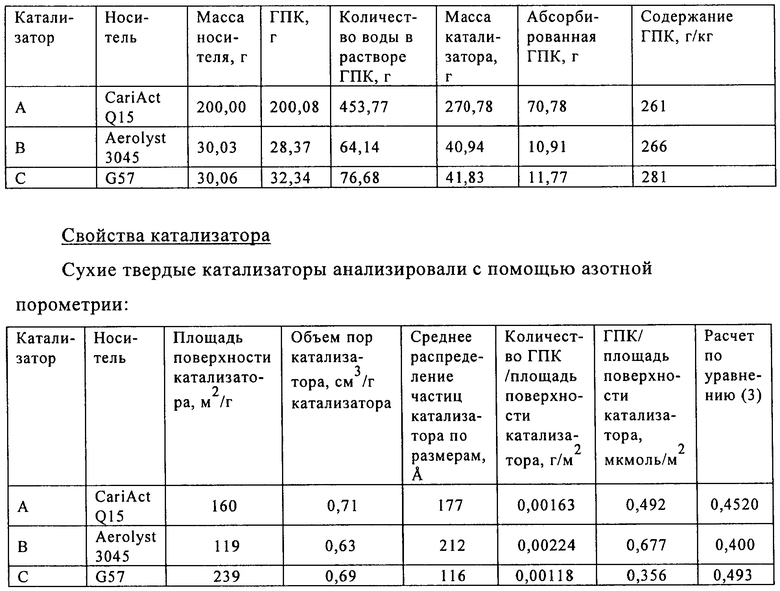
Уравнение (3)=0,6-0,3*[содержание ГПК (мкмоль/г)/площадь поверхности катализатора (м2/г)]. При расчете количества микромолей гетерополикислоты (ГПК), адсорбированной на катализаторе, полагают, что гетерополикислота является полностью гидратированной. При расчете количества микромолей адсорбированной 12-кремнийвольфрамовой кислоты (H4[SiW12O40]*24H2O) ее молярную массу (М) принимали равной 3310,6 г/моль.
Испытания катализатора
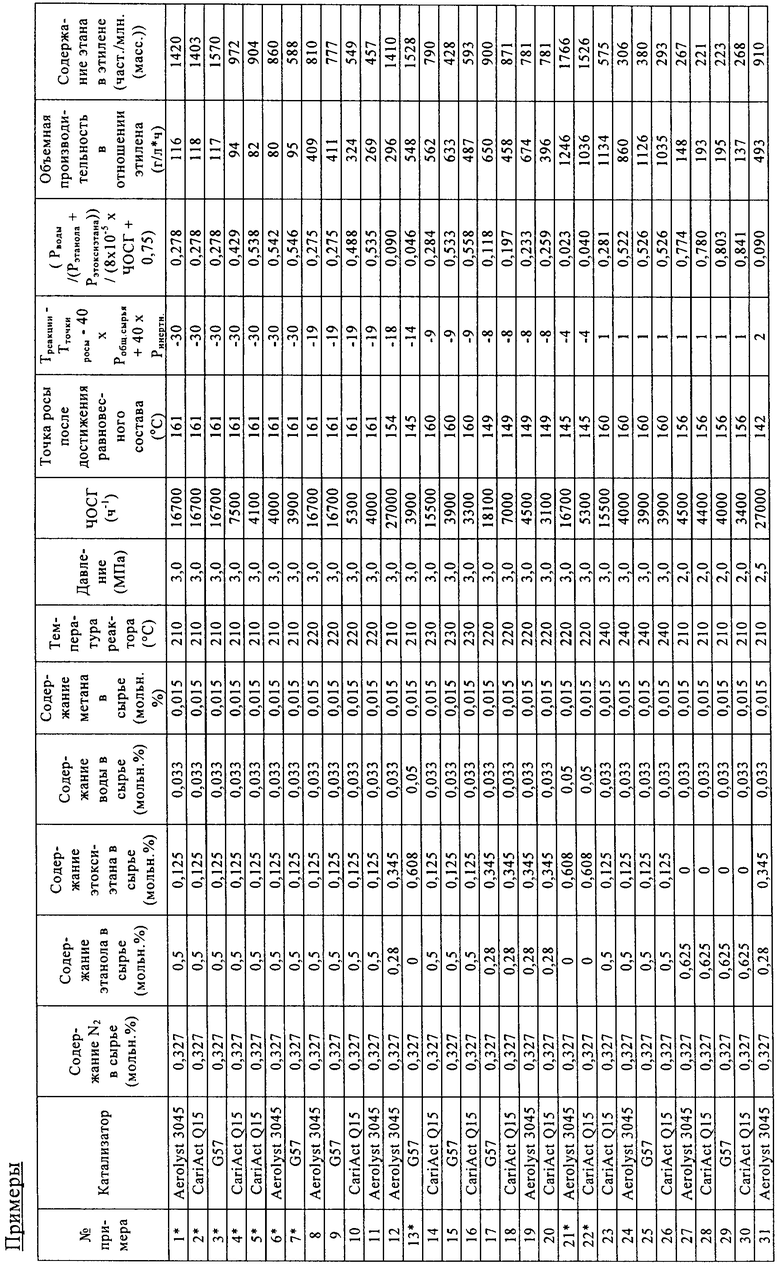
Катализаторы (диаметр частиц составлял от 125 до 180 мкм) загружали в параллельные проточные реакторы. Объем катализаторов в параллельных реакторах составлял от 0,083 до 0,97 мл. Реакторы проверяли на герметичность, затем нагревали до 220°С в токе азота. Когда температура достигала 220°С, жидкое сырье, состоящее из этанола, этоксиэтана и воды, испаряли и смешивали с азотом до подачи в реактор, содержащий катализатор. Условия реакции были следующими: давление составляло 2,0 МПа, содержание этанола составляло 28 об.%, содержание этоксиэтана составляло 34,5 об.%, содержание воды составляло 3,3 об.%, содержание азота составляло 32,7 об.%, содержание метана составляло 1,5 об.% (метан применяли в качестве внутреннего стандарта, поскольку это соединение в ходе процесса дегидратации не образуется в сколько-нибудь значительных количествах). Постоянный поток газа в каждый реактор позволял регулировать ЧОСГ в зависимости от объема загруженного катализатора. Катализаторы испытывали в таких условиях в течение 100 ч, затем изменяли состав сырья, давление и температуру. Для измерения выходов этана и этилена применяли газовую хроматографию потока продукта (в качестве внутреннего стандарта применяли азот).
Расчет точки росы
Расчет точки росы сырья при его равновесном составе осуществляли с применением реакционного блока AspenPlus RGIBBS, причем физические свойства смеси, представляющей собой текучую среду, рассчитывали с помощью модификации Соаве уравнения состояния Редлиха-Квонга.

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ИЗВЛЕЧЕНИЯ КАТАЛИЗАТОРА | 2008 |
|
RU2484900C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКЕНОВ ИЗ ОКСИГЕНАТОВ С ИСПОЛЬЗОВАНИЕМ НАНЕСЕННЫХ НА НОСИТЕЛЬ ГЕТЕРОПОЛИКИСЛОТНЫХ КАТАЛИЗАТОРОВ | 2007 |
|
RU2446011C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНА | 2006 |
|
RU2415121C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2006 |
|
RU2419596C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНА | 2006 |
|
RU2415832C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКЕНА | 2011 |
|
RU2603636C2 |
| ДЕГИДРИРОВАНИЕ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2412141C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКЕНА (ВАРИАНТЫ) | 2011 |
|
RU2593747C2 |
| РЕАКЦИОННАЯ РЕКТИФИКАЦИЯ ДЛЯ ДЕГИДРАТАЦИИ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2419595C2 |
| РЕАКЦИОННАЯ РЕКТИФИКАЦИЯ С ВОЗВРАТОМ В ПРОЦЕСС ОЛЕФИНОВ | 2006 |
|
RU2419597C2 |
Изобретения относится к способу получения этилена путем химической дегидратации в паровой фазе сырья, включающего этанол, воду и этоксиэтан, в реакторе при повышенных температуре и давлении в присутствии слоя катализатора, включающего нанесенную гетерополивольфрамовую кислоту. Способ характеризуется тем, что реактор настраивают и осуществляют его работу таким образом, что он функционирует в режиме, соответствующем следующим параметрам: 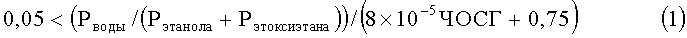
и
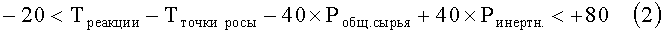
Причем Рводы, Рэтанола и Рэтоксиэтана представляют собой парциальные давления воды, этанола и этоксиэтана соответственно при термодинамически равновесном составе сырья при рабочей температуре и давлении процесса в МПа. ЧОСГ представляет собой часовую объемную скорость газообразного сырья, проходящего над катализатором, в ч-1. Треакции представляет собой температуру реакции в °С, Тточки росы представляет собой температуру точки росы сырья при его термодинамически равновесном составе в °С. Робщ.сырья представляет собой общее давление сырья в МПа, а Ринертн. представляет собой парциальное давление инертных веществ в составе сырья в МПа. Использование настоящего способа позволяет поддерживать влажность катализатора на оптимальном уровне. 12 з.п. ф-лы, 137 пр., 1 ил.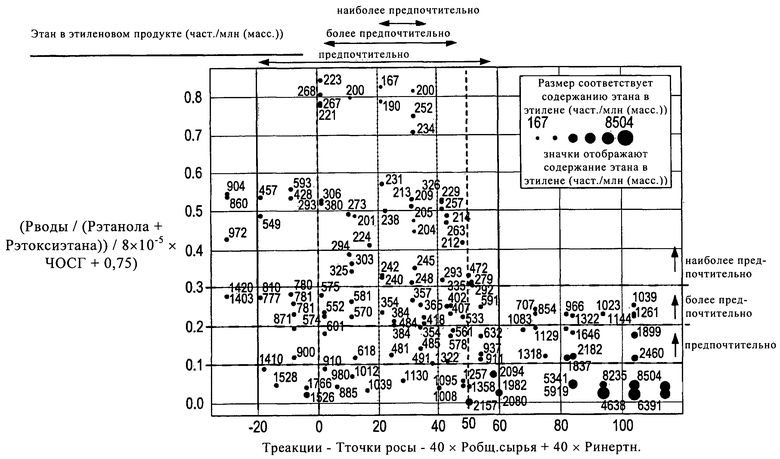
1. Способ получения этилена путем химической дегидратации в паровой фазе сырья, включающего этанол, воду и этоксиэтан, в реакторе при повышенных температуре и давлении в присутствии слоя катализатора, включающего нанесенную гетерополивольфрамовую кислоту, отличающийся тем, что реактор настраивают и осуществляют его работу таким образом, что он функционирует в режиме, соответствующем следующим параметрам:
и
,
где Рводы, Рэтанола и Рэтоксиэтана представляют собой парциальные давления воды, этанола и этоксиэтана соответственно при термодинамически равновесном составе сырья при рабочей температуре и давлении процесса, МПа; ЧОСГ представляет собой часовую объемную скорость газообразного сырья, проходящего над катализатором, ч-1; Треакции представляет собой температуру реакции, °С; Тточки росы представляет собой температуру точки росы сырья при его термодинамически равновесном составе, °С; Робщ. сырья представляет собой общее давление сырья, МПа, а Ринертн. представляет собой парциальное давление инертных веществ в составе сырья, МПа.
2. Способ по п.1, отличающийся тем, что:
0,1<(Рводы/(Рэтанола+Рэтоксиэтана))/(8×10-5×ЧОСГ+0,75).
3. Способ по п.2, отличающийся тем, что:
0,2<(Рводы/(Рэтанола+Рэтоксиэтана))/(8×10-5×ЧОСГ+0,75).
4. Способ по п.3, отличающийся тем, что:
0,3<(Рводы/(Рэтанола+Рэтоксиэтана))/(8×10-5×ЧОСГ+0,75).
5. Способ по п.1, отличающийся тем, что:
-20<Треакции-Тточки росы-40×Робщего сырья+40×Ринертн.<60.
6. Способ по п.5, отличающийся тем, что:
0<Треакции-Тточки росы-40×Робщего сырья+40×Ринертн.<50.
7. Способ по п.6, отличающийся тем, что:
20<Треакции-Тточки росы-40×Робщего сырья+40×Ринертн.<40.
8. Способ по п.1, отличающийся тем, что сырье содержит инертный не способный к конденсации разбавитель, выбранный из 2-метилпропана, н-бутана и этилена.
9. Способ по п.1, в котором химическую дегидратацию в паровой фазе осуществляют в серии из двух или более реакторов, работающих в адиабатическом режиме, причем в этих реакторах степень превращения сырья постепенно увеличивается и каждый последующий реактор работает при более высокой температуре, чем предыдущий.
10. Способ по п.8 или 9, отличающийся тем, что разбавитель вводят через множество точек в слой катализатора и/или реакторы.
11. Способ по п.1, отличающийся тем, что нанесенный катализатор на основе гетерополивольфрамовой кислоты обладает следующим свойством:
ОП>0,6-0,3× (содержание ГПК/площадь поверхности катализатора), где ОП представляет собой объем пор сухого нанесенного катализатора на основе гетерополивольфрамовой кислоты (измеренный в мл/г катализатора); содержание ГПК представляет собой количество гетерополикислоты, присутствующей в сухом нанесенном катализаторе на основе гетерополикислоты (измеренное в мкмоль/г катализатора), а площадь поверхности катализатора представляет собой удельную поверхность сухого нанесенного катализатора на основе гетерополивольфрамовой кислоты (измеренную в м2/г катализатора).
12. Способ по п.8, отличающийся тем, что равновесный состав сырья включает более 15 об.% воды.
13. Способ по п.8, отличающийся тем, что получаемый этилен содержит менее 10 млн-1 кислородсодержащих соединений.
| Контейнер для строительных камней | 1988 |
|
SU1792885A1 |
| WO 2007003899 A1, 11.01.2007 | |||
| Способ получения изобутилена | 1959 |
|
SU127252A1 |

