Область техники
Изобретение относится к области фармацевтической химии, а именно к новым органическим соединениям, влияющим на систему гемостаза, проявляющим антиагрегантные, антикоагулянтные и вазодилаторные свойства, и способу их получения.
В частности, изобретение относится к N,N'-замещенным пиперазинам, обладающим сочетанным антиагрегантным, антикоагулянтным и вазодилаторным воздействием на организм, общей формулы (I) и (II):

где R1, R2: линейный или разветвленный алкокси (C1÷C4), CH3C(=O)O;
n=1÷5; m=0÷3; Z: C=O, SO2;
X: C(=NH)NH2, C(=NH)NHC(=NH)NH2,
G - низкомолекулярная органическая или минеральная кислота, катионы натрия, калия, аммония или вода,
и

где R1, R2: линейный или разветвленный алкил (C1÷C4), линейный или разветвленный алкокси (C1÷C4);
n=1÷5; m=0÷3; Z: C=O, SO2;
X: СН2(СНОН)CH2SO3H;
G - низкомолекулярная органическая или минеральная кислота, катионы натрия, калия, аммония или вода
Предшествующий уровень техники
Заболевания системы гемостаза, в частности тромбозы, вазоконстрикция, являются ключевым звеном в патогенезе нарушений коронарного и мозгового кровообращения, что делает лечение указанных заболеваний достаточно актуальным. Успехи в лечении и профилактике нарушений гемостаза во многом связаны с применением лекарственных средств на основе физиологически активных соединений различной химической природы и механизма фармакологического действия.
Назначение антитромботических лекарственных средств снижает суммарный риск развития сердечно-сосудистых событий на четверть, нефатального инфаркта миокарда - на треть, нефатального инсульта - на четверть, сосудистой смерти - на одну шестую [McConnel H. // Br. Med. J. 2002. V.324. Р.71-86]. При этом основными направлениями антитромботической терапии являются: ингибирование агрегации тромбоцитов, целенаправленное воздействие на систему гемокоагуляции, уменьшение тромбоцитной активности эндотелия. Несмотря на большое количество препаратов, обладающих способностью подавлять активность, и агрегационную способность тромбоцитов, их клиническая эффективность при заболеваниях сердечно-сосудистой системы, требующих антиагрегантной терапии, доказана только в отношении трех групп средств - ацетилсалициловой кислоты (АСК), тиенопиридинов (тиклопидин, клопидогрель, прасугрел) и блокаторов гликопротеиновых рецепторов тромбоцитов.
Аспирин (ацетилсалициловая кислота - АСК) рассматривается как практически единственный препарат, применяющийся в целях первичной профилактики сердечно-сосудистых заболеваний [ANN Intern. Med. 2002. V.136. P.161-172], эффективность и безопасность которого подтверждена результатами многочисленных исследований [Circulation. 2004. V.110. Р.2361-2367; 30th International Stroke Conference. 2005, Abstr. P87].
К недостаткам АСК следует отнести подавление синтеза простациклинов, риск кровотечений, АСК-индуцированную гастропатию, ухудшающую переносимость и снижающую приверженность больных лечению, резистенстность больных к препарату [J. Thromb. Haemost. 2003. N1. Р.1710-1713; BMJ 2004. V.328. P.477-479; Brit. J. Clin. Pharmacol. 2008. V.66. N2. P.222-232].
Известно применение ингибитора фосфодиэстеразы - Дипиридамола для снижения частоты развития ишемических транзиторных атак, инсультов и летальности при цереброваскулярной патологии. Дипиридамол близок по терапевтическому эффекту к АСК; при совместном их применении результативность лечения повышается [Future Medicine. 2005. V.1. N1. P.19-26]. Однако применение Дипиридамола может вызывать нежелательные побочные эффекты. Так, при стенозирующем атеросклерозе артерий и наличии значительного количества коллатералей препарат может вызвать развитие синдрома обкрадывания. Поэтому назначение Дипиридамола при остром коронарном синдроме и инфаркте миокарда противопоказано [Int. Med. J. 2008. V.1. N1. P.8-14].
Представителями группы тиенопиридинов в ряду антиагрегантов являются Тиклопидин, Клопидогрель и Прасугрель. Препараты являются пролекарствами, то есть терапевтический эффект достигается за счет фармакологического действия их активных метаболитов. Достоинствами препарата Тиклопидин является снижение на 20% вероятности инсульта, на 10% - его неблагоприятных исходов, ишемии мозга или сосудистой смерти [Ann. Intern. Med. 1998. V.129. N5. Р.394-405]. Недостатками этого препарата являются низкая переносимость и часто возникающие кожные (4-15%) и желудочно-кишечные (до 20%) реакции, которые являются причинами прекращения лечения тиклопидином. Кроме того, известны случаи тромбоцитопенической пурпуры со смертельным исходом [Bennett C., Weinberg P., Rozenberg-Ben-Dror K., et al. Thrombocytopenic purpura associated with ticlopidine. Ann Intern Med 1998; 128: 541-44].
Препарат Клопидогрель, по данным CAPRIE [Lancet. 1996. V.348. Р.1329-39] во многих случаях эффективнее аспирина при длительном применении у больных с высоким риском ишемических событий. К достоинствам могут быть отнесены: лучшая, чем у тиклопидина, переносимость, включая меньшую частоту гематологических осложнений, более быстрое наступление терапевтического эффекта при использовании нагрузочных доз (300 и 600 мг), совместимость с большинством лекарственных средств, используемых в кардиологии. Недостатками препарата Клопидогрель являются резистентность (до 14% популяции) у пациентов с полиморфизмом CYP450, в особенности 2С19 [JACC. 2007. V.49. P.1505; FDA Drug Safety Communication. March 12, 2010]; существенное снижение эффективности при совместном приеме с ингибиторами протонной помпы - Омепразолом, Рабепразолом [FDA Public-health advisory: Updated safety information about a drug interaction between clopidogrel bisulfate (marketed as Plavix) and omeprazole (marketed as Prilosec and Prilosec OTC). November 17, 2009], субоптимальная реакция на препарат при наличии острого коронарного синдрома, у больных диабетом, а также у пациентов с метаболическим синдромом [JACC. 2007. V.49. Р.1505].
Препарат Тикагрелор характеризуется относительно быстрым наступлением терапевтического эффекта, выраженным ингибированием агрегационной активности тромбоцитов. Лечение тикагрелором по сравнению с Клопидогрелом снижает частоту смерти от сосудистых патологий, инфаркта миокарда, инсульта без увеличения общей частоты крупных кровотечений, но с увеличением частоты кровотечений, не связанных с инвазивными процедурами, что является существенным недостатком данного препарата, ограничивающим его применения [N. Engl. J. Med. 2009. V.361. N11. P.1045-57].
В лечебной практике применяются также прямые ингибиторы тромбина - гатраны [Am. Heart J. 2009. V.157. Р.805-810]. Так, Дабигатран демонстрирует снижение риска инсульта, в т.ч. геморрагического, кровотечений, в т.ч. жизнеугрожающих и внутричерепных, а также снижение смерти от сосудистых событий. Терапия препаратом не требует мониторинга [N. Engl. J. Med. 2009. V.361. N12. P.1139-1151]. Существенными недостатками представителей данной группы являются гепатотоксичность и внутрижелудочные кровотечения [NHS. 2009. N8], а также отсутствие сочетанного антиагрегантного, антикоагулянтного и вазодилаторного воздействия на организм.
Наиболее близкими по достигаемому эффекту к заявляемому изобретению являются препараты - блокаторы тромбоксана - Ридогрел, Озагрел, Пирмагрел, которые обладают антигипертензивными и вазодилатирующими свойствами. Препараты безопасны и эффективны при инфаркте миокарда [Cardiovascular Drug Reviews. 2008. V18. N3. Р.222-231], способны снимать бронхоспазм [Life Scinece. 1997. V.60. N18. P.1583-88].
Недостатками данной группы препаратов являются невысокая эффективность при пероральном применении.
Задачей, решаемой авторами, являлось создание более эффективных и более безопасных препаратов, обладающих сочетанным антиагрегантным, антикоагулянтным и вазодилаторным воздействием на организм.
Сущность изобретения
Технический результат был достигнут путем применения в качестве средства, обладающего сочетанной антиагрегантной, антитромболитической и вазодилаторной активностью, N,N'-замещенных пиперазинов общей формулы (I) и (II):
где R1, R2: линейный или разветвленный алкокси (C1÷C4), CH3C(=O)O;
n=1÷5; m=0÷3; Z: C=O, SO2;
Х: C(=NH)NH2, C(=NH)NHC(=NH)NH2,
G - низкомолекулярная органическая или минеральная кислота, катионы натрия, калия, аммония или вода,
и
где R1, R2: линейный или разветвленный алкил (C1÷C4), линейный или разветвленный алкокси (C1÷C4);
n=1÷5; m=0÷3; Z: C=O, SO2;
X: CH2(CHOH)CH2SO3H;
G - низкомолекулярная органическая или минеральная кислота, катионы натрия, калия, аммония иди вода.
В качестве низкомолекулярной органической кислоты используют, как правило, органические кислоты с длиной углеродной цепи C2-C4, такие как уксусная, янтарная, фумаровая и т.п. Природа органической кислоты на основные фармакологические свойства соединения (1) существенного значения не оказывает.
Указанные соединения могут использоваться как в чистом виде, так и в виде сольватов или солей - солей низкомолекулярных (C2-C4) органических или минеральных кислот, натрия, калия, аммония.
В настоящее время замещенные азотсодержащие гетероциклические соединения известны в качестве препаратов, перспективных для антитромбоцитарной терапии.
Так, в патенте США №4,370,330 предложены для стимуляции циркуляции крови новые N-триметокси-бензилпиперазины и их фармацевтически приемлемые соли общей формулы:
 , где
, где 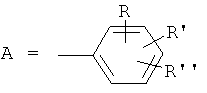
R: CF3, OH, NO2, галоген, алкил- или алкокси-; R': Н, CF3, галоген, алкил- или алкокси-заместитель; R'': Н или алкокси-заместители.
В патенте США №4,574,156 защищены в качестве средств, улучшающих циркуляцию крови, производные полиметоксибензилпиперазина общей формулы:

где R: H; OMe; n=2÷5.
В патенте США №4,368,199 предлагается использовать для лечения сосудистой и сердечной недостаточности производные 3,4,5-триметоксицинна-моилпиперазина общей формулы:
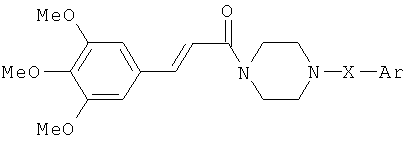
где X: -(СН2)n, n=1÷3 или -CH2-CO-, n=1, 2; Ar: замещенный фенил- или фенилдиоксоаланил - радикалы и др.
В европейском патенте №284359 описаны 1,4-дизамещенные пиперазины общей формулы:
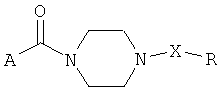
где А: пенталенил-, инденил-, инданил-, нафтил-, азуленил- и др. радикалы; R: фенил-радикал, замещенный на 1÷5 алкокси-группы; X: CH2, CO или тиокарбонил; m=2, 3 и их соли. Главным образом описаны замещенные 3,4,5-триметоксибензил- и 3,4,5-триметоксибензоилпиперазины и метод их получения, заключающийся во взаимодействии промежуточных соединений с кислотой общей формулы A-COOH или галогенангидридом общей формулы A-COW, где А: как указано выше, W: галоген. Соединения или их соли в составе фармацевтических композиций могут использоваться для ингибирования фактора активации тромбоцитов, как антиагреганты.
N,N'-замещенные пиперазины общей формулы:
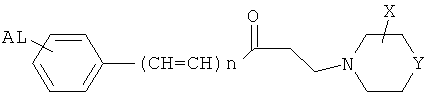
где AL: H, OH, галоген, CN, алколкси и др.; n=0÷2; Y: C, N, O; X: H, алкил, COOR и др.;
для лечения или профилактики сосудистых воспалений и тромбозов (Европейский патент №1783115).
Замещенные N-бензилпиперидинамиды, их фармацевтически приемлемые соли или гидраты используются для регуляции сердечного ритма (Европейский патент №416581, патент США №5,210,090):
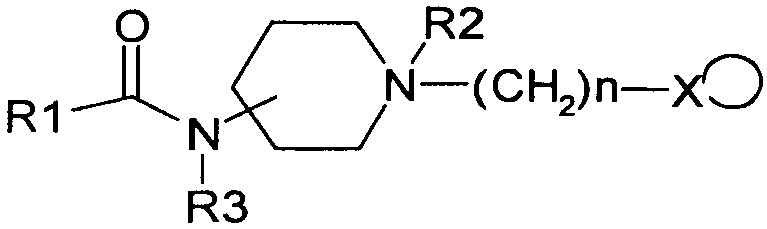
где R1: бензофуранил-радикал; n=0÷10; R2: H, алкил, О; R3: H, карбоксиалкил: X: H, пиридинил, фенил (моно- и полизамещенный на алкил, галоген, алкокси-заместитель).
Известны соединения (M.Protiva et all. Collection Czechoslov. Chem. Commun. Vol.41, p.1035-1041 (1976); CS 151752. 15.01.1974), соединения общей формулы
 , где R - метил, и
, где R - метил, и
 где R1, R2, R3-Н, OCH3, SCH3, C6H5, галоген, фенилтио-, причем как минимум один из указанных радикалов - Н в качестве веществ с гипотензивным действием.
где R1, R2, R3-Н, OCH3, SCH3, C6H5, галоген, фенилтио-, причем как минимум один из указанных радикалов - Н в качестве веществ с гипотензивным действием.
Последнее из соединений получают прибавлением сульфат S - метилизотиомочевины к кипящему раствору соединения
 , где X, R1, R2, R3 же, что и в целевом соединении.
, где X, R1, R2, R3 же, что и в целевом соединении.
Общим недостатков вышеописанных соединений является ограниченный спектр воздействия на организм, т.к. в литературе отсутствуют сведения, что подобные соединения обладают сочетанными антиагрегантными, антикоагулянтными и вазодилаторными свойствами.
Наиболее близким по структуре к заявляемым соединениям является (M.Protiva et all. Collection Czechoslov. Chem. Commun. Vol.40, p.3904-3923(1975) соединение общей формулы:
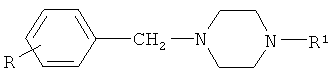 , где R, R1=H, CH3, F, NO2, CI, OCH3, SCH3
, где R, R1=H, CH3, F, NO2, CI, OCH3, SCH3
Однако при существенном отличии в структуре с заявляемыми соединениями для данного вещества отмечен узкий спектр возможного применения - в качестве вещества с гипотензивным действием.
Приведенный анализ литературы показал наличие широкого спектра биологической активности, присущего N,N'-замещенным пиперазинам, а также то, что соединения, оказывающие влияние на систему гемостаза, изучены недостаточно, в связи с чем более углубленное исследование производных N,N'-замещенных пиперазинов является актуальным.
В этой связи, а также с целью разработки эффективных и безопасных препаратов представляет интерес синтез новых производных N,N'-замещенных пиперазинов, обладающих сочетанной антиагрегантной, вазодилаторной и антитромботической активностью, высокой безопасностью, перспективных для терапии заболеваний системы гемостаза.
Варианты осуществления изобретения
Для решения данной задачи были синтезированы новые N,N'- замещенные пиперазины, биологические испытания которых продемонстрировали их высокую эффективность.
Полученные соединения подразделяются по своей структуре на две группы веществ общей формулы (II) и (III).
К первому варианту относятся N,N'-замещенные пиперазины общей формулы (II):
где R1, R2: линейный или разветвленный алкокси (C1÷C4), CH3C(=O)O;
n=1÷5; m=0÷3; Z: C=O, SO2;
X: C(=NH)NH2, C(=NH)NHC(=NH)NH2,
G - низкомолекулярная органическая или минеральная кислота.
Указанные соединения могут использоваться как в чистом виде, так и в виде сольватов или солей низкомолекулярных (C2-C4) органических или минеральных кислот.
Соединения общей формулы (II) получают путем взаимодействия N-замещенных пиперазинов общей формулы (IV)

где R1, R2: линейный или разветвленный алкокси (C1÷C4), CH3C(=O)O;
n=1÷5; m=0÷3; Z: C=O, SO2;
с карбоксамидирующими агентами или их солями в органических растворителях или воде в присутствии оснований. Процесс проводят при комнатной или повышенной температуре. В качестве карбоксамидирующих агентов может быть использовано вещество из группы: 1H-пиразол-1-карбоксамидин или дициандиамид, например 1Н-бензотриазол-1-карбоксамидин, 1H-пиразол-1-карбоксамидин, 3,5-диметил-1Н-пиразол-1-карбоксамидин, дициандиамид или вещества, образующие эти соединения при гидролизе, например соли дициандиамида, соли 1H-пиразол-1-карбоксамидина. В качестве органических растворителей могут быть использованы низшие алифатические спирты, ацетонитрил, тетрагидрофуран, диметилформамид, диметилсульфоксид, дихлорметан или их смеси, в качестве оснований - натрия или калия гидроксид, их карбонаты или гидрокарбонаты. Реакцию целесообразно проводить при pH не выше 9.0±0.5, для достижения оптимального выхода N,N'-замещенных пиперазинов формулы II;
Ко второй группе соединений (вариант 2) относятся N,N'- замещенные пиперазины общей формулы (III):
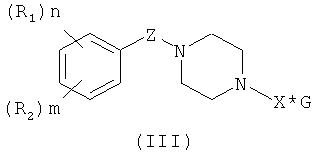
где R1, R2: линейный или разветвленный алкил (C2÷C4), линейный или разветвленный алкокси (C1÷C4);
n=1÷5; m=0÷3; Z: C=O, SO2;
X: CH2(CHOH)CH2SO3H;
G - катионы натрия, калия, аммония или вода.
Вместо указанных соединений для тех же целей могут использоваться их гидраты или фармацевтически приемлемые соли.
Способ их получения соединений общей формулы (III) заключается во взаимодействии N-замещенных пиперазинов общей формулы (V), а также их гидратов или солей:

где R1, R2: линейный или разветвленный алкил (C1÷C4), линейный или разветвленный алкокси (C1÷C4);
n=1÷5; m=0÷3; Z: C=O, SO2;
с 2-гидрокси-3-хлорпропансульфокислотой или ее солями в органических растворителях или воде в присутствии оснований.
Процесс проводят при комнатной или повышенной температуре. В качестве органических растворителей могут быть использованы низшие алифатические спирты, ацетонитрил, тетрагидрофуран, диметилформамид, диметилсульфоксид, дихлорметан или их смеси, в качестве оснований - натрия или калия гидроксид, их карбонаты или гидрокарбонаты, триэтиламин. Реакцию целесообразно проводить при pH не выше 9.0±0,5, для достижения оптимального выхода N,N'-замещенных пиперазинов формулы III.
После окончания реакции выделение конечного продукта из реакционной смеси осуществляется с помощью традиционных методов органического синтеза, выбираемых в зависимости от состава смеси - отгонкой растворителя, перекристаллизацией, переосаждением, хроматографией и т.п. Чистота полученных соединений по данным ОФ ВЭЖХ была не менее 98%.
Контроль за ходом реакции, а также оценка чистоты конечных продуктов проведены методом ОФ ВЭЖХ на хроматографе Alliance (Waters), колонка Zorbax Eclipse C18, 3.5 мкм, 3*100 мм (Agilent Technologies), подвижная фаза - смесь буферного раствора, содержащего 0.01 М натрия октансульфоната и 0.02 М натрия дигидрофосфата (pH 3.0) с ацетонитрилом, детектирование - при 230 нм.
Структура полученных соединений подтверждена данными ЯМР спектроскопии 1H, 13C, масс-спектрометрии, элементного анализа. Элементный анализ выполнен на С, Н, N, S-анализаторе Leco-932 (Leco Corporation).
Определение молекулярной массы проведено масс-спектрометрическим методом на времяпролетном масс-рефлектроне МХ-5303 с источником ионов типа «Электроспрей».
В ходе фармакобиологических исследований было показано, что синтезированные N,N'-замещенные пиперазины обладают хорошей растворимостью в воде по сравнению с известными аналогами - производными азот-содержащих гетероциклических соединений и аспирином, более широким терапевтическим интервалом и высокой безопасностью, что позволяет широко использовать их в качестве антиагрегантов и препаратов, влияющих на систему гемостаза.
В частности, при терапии патологий системы гемостаза, как показали проведенные исследования, введение N,N'-замещенных пиперазинов в организм в дозе 0.005 мМ/кг и более приводит к уменьшению патологической агрегации тромбоцитов, положительно воздействует на систему фибринолиза, обеспечивает вазодилатацию сосудов.
N,N'-замещенные пиперазины могут вводиться в организм в составе композиций, содержащих смесь физиологически-активного (действующего) вещества со вспомогательными веществами. В качестве вспомогательных веществ используются разрешенные Фармакопеей вещества, улучшающие условия получения, хранения или применения лекарственного средства, например растворители, наполнители, ароматизаторы, вкусовые добавки, стабилизаторы и т.п.
Антиагрегантные, антитромботические свойства N,N'-замещенных пиперазинов подтверждены ниже изложенными примерами.
Промышленная применимость
Пример 1
Синтез 4-(3,4,5-триметоксибензоил)пиперазин-1-карбоксимидамид фумарата гемигидрата.
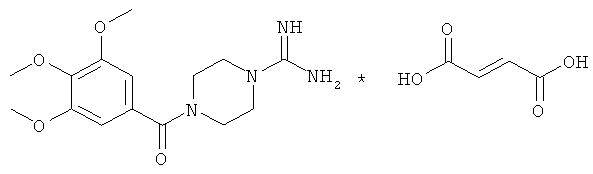
1) Синтез 3,4,5-триметоксибензоил хлорида.
В одногорлую колбу вместимостью 100 мл, снабженную обратным холодильником с газоотводящей трубкой, погруженной в раствор NaOH, помещают 20 г (94 ммоль) 3,4,5-триметоксибензойной кислоты и 45 мл абсолютного бензола. Затем прибавляют 0.5 мл ДМФА и 10.8 мл (150 ммоль) тионил хлорида. После прекращения выделения газа смесь кипятят с обратным холодильником в течение 2 ч. Раствор охлаждают до температуры 20°C, выливают в стакан с 50 мл гексана. Выпавшие белые кристаллы продукта отфильтровывают, фильтрат упаривают до 1/3 объема в вакууме при температуре 50°C. При охлаждении выделяется вторая порция продукта, которую отфильтровывают и объединяют с первой. Продукт сушат в вакууме при температуре 50÷60°C в течение 1 ч.
Выход 3,4,5-триметоксибензоил хлорида - 19.6 г (90%), т.пл. 77°C.
2) Синтез 1-(3,4,5-триметоксибензоил)пиперазин гидрохлорида.
В трехгорлую колбу вместимостью 500 мл, снабженную механической мешалкой, капельной воронкой и термометром, помещают 20.0 г (232 ммоль) пиперазина, 50 мл уксусной кислоты и 60 мл воды. К смеси прикапывают раствор 13 г (56 ммоль) 3,4,5-триметоксибензоил хлорида в 15 мл ТГФ в течение 20 мин, поддерживая температуру реакционной смеси в диапазоне 10÷15°C, затем перемешивают в течение 1 ч и оставляют на ночь. Раствор фильтруют, упаривают досуха, в вакууме при температуре 50÷60°C. К остатку прибавляют 100 мл абс. этанола, нагревают до кипения при перемешивании. Смесь охлаждают до температуры 20°C, отфильтровывают. Маточник упаривают досуха в вакууме ~20 мм рт.ст. и температуре 50÷60°C, прибавляют 100 мл ацетона и перемешивают на магнитной мешалке 30 мин при температуре 40÷50°C. Осадок отфильтровывают, промывают дважды по 50 мл ацетона и сушат в течение 12 ч при температуре 40°C.
Выход 1-(3,4,5-триметоксибензоил)пиперазин гидрохлорида - 7.8 г (44%).
ПМР-спектр 1-(3,4,5-триметоксибензоил)пиперазин гидрохлорида (Brucker 400MHz; раствор в DMSO-d6): 3.35 уш.с. (4Н; пиперазиновое кольцо); 3.58-3.78 уш. (4Н; пиперазиновое кольцо); 3.69 с. (3Н; CH3O); 3.81 с. (6H; две CH3O); 6.75 с. (2Н; ароматич.); 9.57 уш.с. (2H; NH2+).
3а) Синтез 4-(3,4,5-триметоксибензоил)пиперазин-1-карбоксимидамида фумарата при pH реакционной смеси 9±0.5.
К 4.5 г (14.2 ммоль) 1-(3,4,5-триметоксибензоил)пиперазин гидрохлорида прибавляют 1.7 г (16 ммоль) Na2CO3, 40 мл воды, 2.1 г (14.3 ммоль) 1H-пиразол-1-карбоксамидин моногидрохлорида, pH реакционной смеси составляет 9±0.5. Смесь перемешивают при температуре 20°C в течение 24 ч. Раствор упаривают в вакууме при нагревании на водяной бане и температуре 50÷80°C. К остатку прибавляют 60 мл абс. спирта и кипятят с обратным холодильником в течение 20 мин. Раствор фильтруют, фильтрат упаривают в вакууме при температуре 50÷80°C. Остаток растирают в ступке с 50 мл диэтилового эфира, затем с 50 мл ацетона, затем с 50 мл дихлорметана, растворяют в 50 мл 1 М NaOH, экстрагируют дважды по 50 мл дихлорметана. Объединенные органические вытяжки сушат над сульфатом натрия, упаривают в вакууме при комнатной температуре, растворяют в 100 мл этанола, прибавляют 1.65 г (14.2 ммоль) фумаровой кислоты, кипятят в течение 2-х ч, горячий раствор фильтруют, упаривают до 1/4 объема, выдерживают при температуре 4°C в течение 3 ч, продукт отделяют фильтрацией, высушивают в вакууме при температуре 40°C до постоянной массы, перекристаллизовывают из изопропанола и высушивают в вакууме при температуре 40°C до постоянной массы.
Выход C15H22N4O4*C4H4O4*0.5H2O, 4-(3,4,5-триметоксибензоил)-пиперазин-1-карбоксимидамида фумарата гемигидрата - 2.3 г (35%). Элементный анализ C15H22N4O4*C4H4O4*0.5H2O. Вычислено, %: С 51.00; Н 6.08; N 12.52. Найдено, %: С 51.08; Н 6.18; N 12.44. Масс-спектр, основание, найдено: m/z 322.32. Вычислено: М 322.36.
3б) Синтез 4-(3,4,5-триметоксибензоил)пиперазин-1-карбоксимидамида фумарата при pH реакционной смеси 12±0.5
К 4.5 г (14.2 ммоль) 1-(3,4,5-триметоксибензоил)пиперазин гидрохлорида прибавляют 2.07 г (37 ммоль) KOH, 40 мл воды, 2.1 г (14.3 ммоль) 1H-пиразол-1-карбоксамидин моногидрохлорида, pH реакционной смеси составляет 12±0.5. Смесь перемешивают при температуре 20°C в течение 24 ч. Раствор упаривают в вакууме при нагревании на водяной бане и температуре 50÷80°C. К остатку прибавляют 60 мл абс. спирта и кипятят с обратным холодильником в течение 20 мин. Раствор фильтруют, фильтрат упаривают в вакууме при температуре 50÷80°C. Остаток растирают в ступке с 50 мл диэтилового эфира, затем с 50 мл ацетона, затем с 50 мл дихлорметана, растворяют в 50 мл 1 М NaOH, экстрагируют дважды по 50 мл дихлорметана. Объединенные органические вытяжки сушат над сульфатом натрия, упаривают в вакууме при комнатной температуре, растворяют в 100 мл этанола, прибавляют 1.65 г (14.2 ммоль) фумаровой кислоты, кипятят в течение 2-х ч, горячий раствор фильтруют, упаривают до 1/4 объема, выдерживают при температуре 4°C в течение 3 ч, продукт отделяют фильтрацией, высушивают в вакууме при температуре 40°C до постоянной массы, перекристаллизовывают из изопропанола и высушивают в вакууме при температуре 40°C до постоянной массы.
Выход C15H22N4O4*C4H4O4*0.5H2O, 4-(3,4,5-триметоксибензоил)-пиперазин-1-карбоксимидамида фумарата гемигидрата - 1.4 г (21%). Масс-спектр, основание, найдено: m/z 322.32. Вычислено: М 322.36.
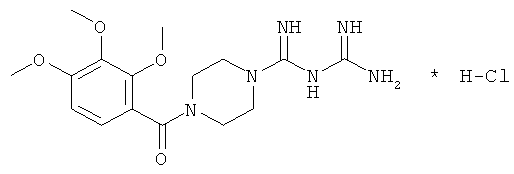
Пример 2
Синтез N-карбамидоил-4-(2,3,4-триметоксибензоил)пиперазин-1-карбоксимидамид гидрохлорида
1) Синтез 2,3,4-триметоксибензоил хлорида.
В одногорлую колбу вместимостью 100 мл, снабженную обратным холодильником с газоотводящей трубкой, погруженной в раствор NaOH, помещают 25 г (118 ммоль) 2,3,4-триметоксибензойной кислоты и 50 мл абсолютного бензола, прибавляют 0.5 мл ДМФА и 10.8 мл (150 ммоль) тионил хлорида. После прекращения выделения газа смесь кипятят с обратным холодильником в течение 2 ч. Раствор охлаждают до температуры 20°C, выливают в стакан с 50 мл гексана. Осадок отфильтровывают, фильтрат упаривают до 1/3 объема в вакууме при температуре 50÷80°С. При охлаждении выделяется вторая порция продукта, которую отфильтровывают и объединяют с первой. Продукт, белый порошок, сушат в вакууме при температуре 50÷60°C в течение 1 ч.
Выход 2,3,4-триметоксибензоил хлорида - 23.1 г (85%), т.пл. 42°C.
2) Синтез 1-(2,3,4-триметоксибензоил)пиперазин гидрохлорида.
В трехгорлую колбу вместимостью 500 мл, снабженную механической мешалкой, капельной воронкой и термометром, помещают 30.0 г (348 ммоль) пиперазина, 75 мл уксусной кислоты и 90 мл воды. К смеси прикапывают раствор 19.5 г (85 ммоль) 2,3,4-триметоксибензоил хлорида в 25 мл ТГФ в течение 30 мин, поддерживая температуру реакционной массы в диапазоне 10÷15°C. Далее перемешивают 1 ч и оставляют на ночь. Светло-желтый раствор упаривают досуха в вакууме при температуре 50÷60°C. Остаток обрабатывают 450 мл 2.5 н. HCl (1.125 моль) при охлаждении льдом и перемешивают 15 мин. Раствор фильтруют. Фильтрат упаривают досуха в вакууме -20 мм рт.ст. и температуре 50÷60°C. Остаток обрабатывают 150 мл абс. этанола и нагревают до кипения при перемешивании. Смесь охлаждают до температуры 20°C и отфильтровывают осадок пиперазина дигидрохлорида. Маточник упаривают досуха в вакууме ~20 мм рт.ст. и температуре 50÷60°C. К остатку прибавляют 150 мл ацетона и перемешивают на магнитной мешалке 30 мин при температуре 40÷50°С. Осадок отфильтровывают, дважды промывают ацетоном по 50 мл, сушат в течение 12 ч при температуре 40°C.
Выход 1-(2,3,4-триметоксибензоил)пиперазин гидрохлорида - 12.1 г (45%).
3) Синтез N-карбамимидоил-4-(2,3,4-триметоксибензоил)пиперазин-1-карбо-ксимидамида гидрохлорида.
В одногорлую колбу вместимостью 100 мл помещают 6.3 г (20 ммоль) 1-(2,3,4-триметоксибензоил)пиперазин гидрохлорида, 1.77 г (21 ммоль) дициандиамида и 50 мл 1-бутанола. Смесь кипятят с обратным холодильником в течение 10 ч, упаривают в вакууме при температуре 50÷80°C. К остатку прибавляют 100 мл этанола и кипятят с обратным холодильником в течение 0.5 ч. Горячую смесь фильтруют, осадок сушат при температуре 45°C в течение 2 сут.
Выход C16H24N6O4*HCl, N-карбамимидоил-4-(2,3,4-триметоксибензоил)пиперазин-1-карбоксимидамида гидрохлорида - 4.3 г (53%). Элементный анализ C16H24N6O4*HCl. Вычислено, %: С 47.94; Н 6.29; N 20.96. Найдено, %: С 48.63; Н 6.47; N 21.03. Масс-спектр, основание, найдено: m/z 364.38. Вычислено: М 364.40.
ПМР-спектр N-карбамимидоил-4-(2,3,4-триметоксибензоил)пиперазин-1-карбоксимидамида гидрохлорида (Brucker 400MHz; раствор в DMSO-d6): 3.17-3.70 уш. синглеты (8Н; пиперазиновое кольцо); 3.75 с. (3Н; CH3O); 3.76 с. (3H; CH3O); 3.80 с. (3H; CH3O); 6.83 с. и 6.91 с. (1Н и 1Н; ароматич.); 6.98 уш.с. и 7.38 уш.с. (4Н и 2Н; 2NH, NH2(+), NH2).
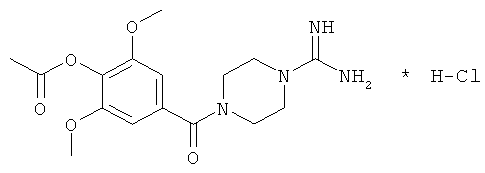
Пример 3
Синтез 4-(4-ацетилокси-3,5-диметоксибензоил)пиперазин-1-карбоксимидамид гидрохлорида
1) Синтез 1-(4-ацетилокси-3,5-диметоксибензоил)пиперазин гидрохлорида.
В трехгорлую колбу вместимостью 500 мл, снабженную механической мешалкой, капельной воронкой и термометром, помещают 20.0 г (232 ммоль) пиперазина, 50 мл уксусной кислоты и 60 мл воды, прикапывают раствор 13 г (50 ммоль) 4- ацетилокси-3,5-диметоксибензоил хлорида в 15 мл ТГФ в течение 20 мин, поддерживая температуру реакционной среды в диапазоне 10÷15°C, перемешивают в течение 1 ч, оставляют на ночь. Раствор упаривают досуха в вакууме при температуре 50÷60°C. Остаток обрабатывают 300 мл (750 ммоль) 2.5 н. HCl при охлаждении льдом, перемешивают 15 мин. Раствор фильтруют, маточник упаривают досуха в вакууме при ~20 мм рт.ст. и температуре 50÷60°C. Остаток обрабатывают 100 мл абсолютного этанола и нагревают до кипения при перемешивании. Смесь охлаждают до температуры 20°C и отфильтровывают. Маточник упаривают досуха в вакууме ~20 мм рт.ст. и температуре 50÷60°C. К остатку прибавляют 100 мл ацетона и перемешивают на магнитной мешалке 30 мин при температуре 40÷50°C. Белый осадок отфильтровывают, промывают дважды по 50 мл ацетона, сушат в течение 12 ч при температуре 40°C.
Выход 1-(4-ацетилокси-3,5-диметоксибензоил)пиперазин гидрохлорида - 7.6 г (44%).
2) Синтез 4-(4-ацетилокси-3,5-диметоксибензоил)пиперазин-1-карбоксимидамид гидрохлорида.
К 4.52 г (13.1 ммоль) 1-(4-ацетилокси-3,5-диметоксибензоил)пиперазин гидрохлорида прибавляют 1.6 г (14.8 ммоль) Na2CO3 и 40 мл воды, 1.92 г (13.1 ммоль) 1H-пиразол-1-карбоксамидин гидрохлорида. Смесь перемешивают при температуре 30°C в течение 24 ч. Раствор упаривают в вакууме при температуре 50÷80°C. К остатку прибавляют 60 мл изопропанола и кипятят с обратным холодильником в течение 20 мин. Раствор фильтруют, маточник упаривают в вакууме при температуре 50÷80°C. Остаток растирают в ступке с 50 мл диэтилового эфира, затем - с 50 мл ацетона, затем растирают с 50 мл дихлорметана и высушивают. Полученный продукт - белый порошок - перекристаллизовывают из изопропанола, сушат в течение 12 ч при температуре 40°C.
Выход C16H22N4O5*HCl, 4-(4-ацетокси-3,5-диметоксибензоил)пиперазин-1-карбоксимидамида гидрохлорида - 2.0 г (35%). Элементный анализ C16H22N4O5*HCl. Вычислено, %: С 49.68; Н 5.99; N 14.48. Найдено, %: С 49.79; Н 5.86; N 14.43. Масс-спектр, основание, найдено: m/z 350.42. Вычислено: М 350.36.
Пример 4
Синтез 3-(1-(2,3,4-триметоксибензоил)пиперазин-4-ил)-2-гидрокси-1-пропансульфоновой кислоты натриевой соли

В одногорлую колбу вместимостью 100 мл помещают 6.3 г (20 ммоль) 1-(3,4,5-триметоксибензоил)пиперазина гидрохлорида и 30 мл воды, прибавляют порциями 5.5 г (65 ммоль) NaHCO3. После прекращения выделения газа прибавляют раствор 4.10 г (20 ммоль) 2-гидрокси-3-хлорпропансульфокислоты в 30 мл воды и 0.1 г йодистого калия, pH реакционной смеси составляет 9±0.5.
Смесь кипятят с обратным холодильником в течение 5 ч. Раствор упаривают досуха в вакууме при ~20 мм рт.ст. и температуре 50÷60°C. Остаток обрабатывают 100 мл абс. этанола и кипятят с обратным холодильником в течение 0.5 ч при перемешивании. Горячий раствор отфильтровывают от неорганических солей и выдерживают в течение суток. Белый осадок отфильтровывают и сушат при температуре 45°C в течение 2 сут.
Выход C17H27N2NaO7S, натриевой соли 3-(1-(2,3,4-триметоксибензоил)-пиперазин-4-ил)-2-гидрокси-1-пропансульфоновой кислоты 4.1 г (46%).
Элементный анализ C17H25N2NaO8S. Вычислено, %: С 46.36; Н 5.72; N 6.36. Найдено, %: С 46.33; Н 5.74; N 6.41
Пример 5
Синтез 4-(2,3,4,5-тетраметоксибензоил)пиперазин-1-ил)-карбоксимидамид ацетат

1) Получение 2-бром-3,4,5-триметоксибензойной кислоты
В 1 литровой плоскодонной колбе с обратным холодильником с ловушкой для бромистого водорода, капельной воронкой, на магнитной мешалке, к раствору 24.2 г (106 ммоль) 3,4,5-триметоксибензойной кислоты в 200 мл хлороформа прибавляют 2,5 мл воды и реакционную массу доводят до кипения и прикапывают 5.9 мл (18.4 г, 115 ммоль) брома в 40 мл хлороформа за 30 минут и кипятят до обесцвечивания раствора ~10 часов. После этого реакционную смесь переносят в делительную воронку, промывают 2 раза по 100 мл воды, органическую фазу сушат 5 г безводного сульфата натрия, фильтруют от осушителя, хлороформ упаривают на роторном испарителе, получают 30.3 г (86%) светло-желтого осадка.
ПМР-спектр 2-бром-3,4,5-триметоксибензойной кислоты (Brucker 300 MHz; раствор в CDCl3): 3.91 с (3H; CH3O); 3.93 с (3H; CH3O); 3.99 с (3H; CH3O); 7.44 с (1Н, CH-Ar), 11.4 уш. синглет (1Н, CO2H)
2) Получение 2,3,4,5-тетраметоксибензойной кислоты:
В 1 литровой плоскодонной колбе на магнитной мешалке с обратным холодильником, снабженным хлоркальциевой трубкой, готовят раствор метилата натрия, растворением 7.4 г (32 ммоль) натрия в 480 мл абсолютного метанола, затем прибавляют 30.4 г (105 ммоль) 2-бром-3,4,5-триметоксибензойной кислоты, после ее растворения прибавляют 10 г (52 ммоль) йодистой меди и реакционную смесь кипятят 10 часов, отбирают пробу, определяют полноту прохождения реакции по ПМР, если в пробе нет исходной кислоты, то реакцию выделяют, если нет - продолжают нагрев, до ее окончания.
После окончания реакции из реакционной смеси (отгоняют метанол, остаток растворяют в 200 мл воды, отфильтровывают выпавшие соли меди, остаток подкисляют концентрированной соляной кислотой до pH 1-2, экстрагируют хлористым метиленом 3х100 мл, органическую фазу промывают водой 2×50 мл, сушат 5 г безводного сульфата натрия, фильтруют от осушителя, хлористый метилен упаривают на роторном испарителе, получают m=23.5 г светло-желтого масла. Масло перекристаллизовают из 150 мл смеси гексан/этилацетат (5/1), после охлаждения до +4°C осадок фильтровали, сушили в вакууме 10 Торр, 40°C, м=21,5 г (84%).
ПМР-спектр 2,3,4,5-тетраметоксибензойной кислоты (Brucker 300 MHz; раствор в CDCl3): 3.87 с (3H; CH3O); 3.92 с (3H; CH3O); 3.98 с (3H; CH3O); 4.07 с (3H; CH3O); 7.45 с (1Н, CH-Ar), 11.5 уш. синглет (1Н, CO2H).
3) Получение хлорангидрида 2,3,4,5-тетрометоксибензойной кислоты
В круглодонной колбе на 250 мл с обратным холодильником с ловушкой для хлористого водорода на магнитной мешалке, к раствору 8.6 г (35.5 ммоль) 2,3,4,5-тетрометоксибензойной кислоты в 20 мл хлористого метилена прибавляют 3.6 мл (5.95 г, 50 ммоль) хлористого тионила и три капли диметилформамида и кипятят до прекращения выделения хлористого водорода ~3 часа, затем хлористый метилен упаривают в вакууме остаток, масло растворяют в 30 мл гептана при комнатной температуре, фильтруют от осадка, маточник упаривают получают 8.5 г (92%) желтого масла, которое используют в следующей стадии без очистки.
4) Получение трет-бутил 4-(2,3,4,5-тетраметоксибензоил)пиперазин-1-карбоксилат
В трехгорлой круглодонной колбе на 250 мл с обратным холодильником, капельной воронкой и термометром, на магнитной мешалке на охлаждающей бане, к раствору 5.8 г (31 ммоль) бок-пиперазина, 4,1 г (40 моль) триэтиламина в 70 мл хлористого метилена и к охлажденному до -5°C, прикапывают раствор 7,8 г (0.03 моль) хлорангидрида 2,3,4,5-тетрометоксибензойной кислоты в 50 мл хлористого метилена с такой скоростью, чтоб температура не поднималась выше +5°C, после этого охлаждение снимают и реакционную смесь перемешивают при комнатной температуре 1 час. Далее реакционную смесь переносят в делительную воронку и промывают 2×30 водой, 2×30 насыщенным раствором бикарбоната натрия, 2×30 насыщенным раствором хлористого аммония и 2×30 водой, хлористый метилен упаривают, а остаток жетое масло растворяют в 40 мл этанола и высаживают продукт 200 мл воды. Осадок отфильтровывают, сушат в вакууме белый осадок 10 г (81%).
ПМР-спектр 2,3,4,5-тетраметоксибензоной кислоты (Brucker 300 MHz; раствор в CDCl3): 1.4 с (9Н, трет-бутил); 3.0-3.7 м (8Н, пиперазиновое кольцо); 3.8 с (6Н, 2 CH3O); 3.9 с (3H, CH3O); 3.95 с (3H, CH3O); 7.3 с (1Н, CH-Ar).
5) Получение 4-(2,3,4,5-тетрамстоксибензоил)-пиперазин-1-карбоксимидамид ацетета
В плоскодонной колбе на 100 мл с обратным холодильником на магнитной мешалке кипятят 4.1 г (10 ммоль) трет-бутил 4-(2,3,4,5-тетраметоксибензоил)пиперазин-1-карбоксилата в 20 мл хлористого метилена и 2.3 г (20 ммоль) трифторуксусной кислоты, ~5 часов (контроль по ТСХ хлористый метилен/этилацетат 6/4), далее растворители и избыток трифторуксусной кислоты упаривают в вакууме на роторном испарителе.
Остаток без анализа используют в следующей стадии.
Соединение с предыдущей стадии растворяют в 40 мл воды и порциями прибавляют 2.12 г (20 моль) карбоната натрия, после того как перестанет выделяться углекислый газ, прибавляют 1.61 г (11 ммоль) 1H-пиразол-1-карбоксамидин гидрохлорида и реакционную смесь перемешивают при комнатной температуре 24 часа. Затем воду упаривают в вакууме на роторном испарителе при температуре бани не выше 5°C, остаток защелкивают 20 мл 20% раствора гидроокиси натрия и экстрагируют 4×40 мл хлористого метилена, органическую фазу промывают 10 мл воды, сушат сульфатом магния, после фильтрования от осушителя, упаривают хлористый метилен, остаток растворяют в 20 мл воды и пропускают через анионообменную колонку со 100 мл сорбента Dowex 1×8, 200 в основной форме, собирая ~100 мл элюата, который закисляют 10 мл уксусной кислоты. После упаривания остаток кипятят с 20 мл ацетона, осадок фильтруют, промывают 10 мл ацетона и 10 мл диэтилового эфира, получают 3.3 г (66%) 4-(2,3,4,5-тетраметоксибензоил)-пиперазин-1-карбоксимидамид ацетета.
ПМР спектр 4-(2,3,4,5-тетраметоксибензоил)-пиперазин-1-карбоксимидамид ацетета (Brucker 300 MHz; раствор в CDCl3): 1.6 с (3H, CH3COOH), 3.25 м (2Н, пиперазиновое кольцо); 3.37 м (2Н, пиперазиновое (кольцо); 3.50 м (2Н, пиперазиновое кольцо); 3.67 м (2Н, пиперазиновое кольцо); 3.7 с (3H; CH3O); 3.75 с (3H; CH3O); 3.79 с (3H; CH3O); 3.85 с (3H; CH3O), 6.55 с (1Н, CH-Ar); 9.0 уш. синглет (4Н, гуанидиновая группа).
Элементный анализ C18H28N4O6. Вычислено, %: С 54.53; Н 7.12; N 14.13. Найдено, %: С 54.48; Н 7.18; N 14.21.
Пример 6
Синтез 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1 ил-карбоксимидамид гидрохлорида

1) Синтез 3,4,5-триметоксибензолсульфохлорида
В стакан на 500 мл с термометром, на магнитной мешалке с охлаждающей баней (вода/лед) помещают раствор 19.6 г диоксида серы (306 ммоль) в 60 мл уксусной кислоты и CuCl 4 г (40 ммоль), перемешивают 15 минут. К этому раствору добавляют порциями в течение 5 мин при 0°C раствор соли диазония (полученной стандартным диазотированием 8,2 г (44.8 ммоль) 3,4,5-триметоксианилина в смеси 15 мл концентрированной соляной и 40 мл уксусной кислот с помощью раствора 3,36 г (48.7) нитрита натрия в 5 мл воды при +5°С), наблюдается пенообразование и выделение азота. После прибавления всего раствора соли диазония реакционную смесь перемешивают 1 час при комнатной температуре, затем выливают в 200 мл воды и льда и экстрагируют 3×100 мл хлористого метилена, промывают 2×100 мл воды, сушат безводным сульфатом натрия, фильтруют от осушителя и упаривают хлористый метилен. Остаток 10 г хроматографируют на колонке, заполненной силикагелем (90 г, 40-60µ), элюируя хлористым метиленом, собирая 1 фракцию (по ТСХ элюент хлористый метилен, Rf=0.48), после упаривания кристаллизующееся светло-желтое масло м=7.4 г. По данным ПМР-спектра содержит 25% примеси - 3,4,5-триметокси-1-хлорбензола. Выход 3,4,5-триметоксибензолсульфохлорида - 46%. Используют без дополнительной очистки в следующей стадии.
2) Получение трет-бутил 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1-карбоксилат
В круглодонной колбе на 500 мл с капельной воронкой на магнитной мешалке с охлаждающей баней (лед/вода) к раствору 9.3 г (50 ммоль) бок-пиперазина в 200 мл дихлорметана и 10.1 г (100 ммоль) триэтиламина прикапывают раствор 9.75 г (48.7 ммоль) 3,4,5-триметоксибензолсульфохлорида в 50 мл дихлорметана, так чтобы температура реакционной смеси не превышала +5°С, затем снимают охлаждение и перемешивают реакционную смесь при комнатной температуре 2 часа, промывают 3·100 мл водой, сушат Na2SO4, упаривают в вакууме, получают белый порошок. Его растворяют в 60 мл кипящего бензола, добавляют 60 мл гексана. После охлаждения до 20°C через 1 час фильтруют белый осадок, сушат на воздухе. Получают 12.4 г (81%) трет-бутил 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1-карбоксилата.
ПМР спектр трет-бутил 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1-карбоксилат (Brucker 300 MHz; раствор в CDCl3): 1.45 с (9Н, трет-бутил), 3.05 м (4Н, пиперазиновое кольцо); 3.62 м (4Н, пиперазиновое кольцо); 3.92 С (9Н, CH3O); 6.95 с (2Н, CH-Ar).
3) Получение 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1 гидрохлорида
В круглодонной колбе на 500 мл с обратным холодильником, на магнитной мешалке, смешивают 12.4 г (29.7 ммоль) трет-бутил 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1-карбоксилата, 150 мл этанола и 10 концентрированной соляной кислоты и кипятят 1 час (контроль по ТСХ). Реакционную смесь упаривают в вакууме, получают 10.5 г (100%) белого легкого порошка - 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1 гидрохлорида.
4) Получение 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1-карбоксимидамид гидрохлорида
4-(3,4,5-триметоксифенилсульфонил)пиперазин-1 получают в виде основания путем обработки) 7.0 г (19.8 ммоль) 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1 гидрохлорида 20%-ным водным раствором NaOH и экстракцией 2·100 мл CH2Cl2. Экстракт сушили, упарили. Остаток растворили в 50 мл DMF, добавили 2.93 г (20.0 ммоль) 1H-пиразол-1-карбоксамидин гидрохлорид и перемешивали при 20°C 20 часов. Затем растворитель упарили в вакууме, остаток обработали 2·40 мл теплым ацетоном. Получившийся белый порошок сушили при 40°С. Получили 5.5 г (70%) 4-(3,4,5-триметоксифенилсульфонил)пиперазин-1-карбоксимидамид гидрохлорида.
ПМР-спектр (Brucker 400 MHz; раствор в DMSO-d6): 3.02 м (4Н, пиперазиновое кольцо); 3.39 м (4Н, пиперазиновое кольцо); 3.75 с (6Н, CH3O); 3.87 с (6Н, CH3O); 6.96 с (1Н, CH-Ar); 7.71 с (4Н, гуанидиновая группа).
Элементный анализ C14H23ClN4O5S. Вычислено, %: С 42.58; Н 5.87; N 14.19. Найдено, %: С 42.61; Н 5.91; N 14.17.
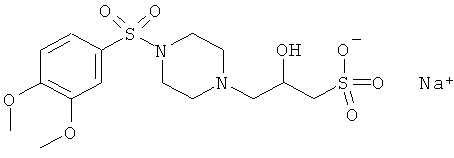
Пример 7
3-(4-(3,4-диметоксифенилсульфонил)пиперазин-1-ил)-2-гидрокси-1-пропансульфоновой кислоты натриевой соли
1) Получение трет-бутил 4-(2,3,4,5-тетраметоксифенилсульфонил)пиперазин-1-карбоксилата
В круглодонной колбе на 100 мл с капельной воронкой на магнитной мешалке с охлаждающей баней (лед/вода) к раствору 2.6 г (14 ммоль) бок-пиперазина в 40 мл дихлорметана и 2.5 г (25 ммоль) триэтиламина прикапывают раствор 3.3 г (14 ммоль) 3,4-диметоксибензолсульфохлорида в 20 мл дихлорметана, так чтобы температура реакционной смеси не превышала +5°С, затем снимают охлаждение и перемешивают реакционную смесь при комнатной температуре 2 часа, промывают 3·100 мл водой, сушат Na2SO4, упаривают в вакууме, получают белый порошок. Его растворяют в 60 мл кипящего бензола, добавляют 60 мл гексана. После охлаждения до 20°С через 1 час фильтруют белый осадок, сушат на воздухе. Получают 4.4 г (81%) трет-бутил 4-(3,4-диметоксифенилсульфонил)пиперазин-1-карбоксилата.
ПМР-спектр трет-бутил 4-(2,3,4,5-тетраметоксифенилсульфонил)-пиперазин-1-карбоксилата (Brucker 300 MHz; раствор в CDCl3): 1.45 с (9Н, трет-бутил); 2.85 м (4Н, пиперазиновое кольцо); 3.42 м (4Н, пиперазиновое кольцо); 3.98 с (6Н, CH3O); 7.12 д (1Н, CH-Ar); 7.23 с (1Н, CH-Ar); 7.35 д (1Н, CH-Ar).
2) Получение 1-(3,4-диметоксифенилсульфонил)пиперазин гидрохлорида
В круглодонной колбе на 50 мл с обратным холодильником, на магнитной мешалке, смешивают 4.4 г (11.3 ммоль) трет-бутил 4-(3,4-диметоксифенилсульфонил)пиперазин-1-карбоксилата, 20 мл этанола и 10 концентрированной соляной кислоты и кипятят 1 час (контроль по ТСХ). Реакционную смесь упаривают в вакууме, получают 3.6 г (100%) белого порошка - 4-(3,4-диметоксифенилсульфонил)пиперазин-1-карбоксимидамид гидрохлорида.
3) Получение 3 -(4-(3,4-диметоксифенилсульфонил)пиперазин-1-ил)-2-гидрокси-1-пропансульфоновой кислоты натриевой соли
В одногорлую колбу вместимостью 100 мл помещают 3.0 г (9.4 ммоль) 1-(3,4-триметоксифенилсульфонил)пиперазина гидрохлорида и 30 мл воды, прибавляют порциями 2.8 г (33 ммоль) NaHCO3. После прекращения выделения газа прибавляют раствор 1.88 г (9.1 ммоль) 2-гидрокси-3-хлорпропансульфокислоты в 10 мл воды и 0.1 г йодистого калия, pH реакционной смеси составляет 9±0.5.
Смесь кипятят с обратным холодильником в течение 5 ч. Раствор упаривают досуха в вакууме при ~20 мм рт.ст. и температуре 50÷60°C. Остаток обрабатывают 60 мл абс. этанола и кипятят с обратным холодильником в течение 0.5 ч при перемешивании. Горячий раствор отфильтровывают от неорганических солей и выдерживают в течение суток. Белый осадок отфильтровывают и сушат при температуре 45°C в течение 2 сут.
Выход C15H24N2NaO8S2, натриевой соли 3-(4-(3,4-диметоксифенилсульфонил)пиперазин-1-ил)-2-гидрокси-1-пропансульфоновой кислоты натриевой соли 2.4 г (57%).
ПМР-спектр 3-(4-(3,4-диметоксифенилсульфонил)пиперазин-1-ил)-2-гидрокси-1-пропансульфоновой кислоты натриевой соли (Brucker 300 MHz; раствор в D2O): 2.4-2.6 м (6Н, 3CH2, пиперазиновое кольцо и пропансульфоновая кислота); 2.8-3.1 м (6Н, 3CH2, пиперазиновое кольцо и пропансульфоновая кислота); 3.82 с (6Н, CH3O); 4.25 м (1Н, СН от пропансульфоновой кислоты); 7.05 д (1Н, CH-Ar); 7.18 с (1Н, CH-Ar); 7.34 д (1Н, CH-Ar).
Элементный анализ C15H23N2NaO8S2. Вычислено, %: С 40.35; Н 5.19; N 6.27. Найдено, %: С 40.44; Н 5.21; N 6.25.
Пример 8
Синтез 4-(3,4-триметоксифенилсульфонил)пиперазин-1-ил)карбоксимидамид гидрохлорида

В одногорлую колбу вместимостью 100 мл помещают 3.75 г (9.4 ммоль) 1-(3,4-триметоксифенилсульфонил)пиперазина гидрохлорида растворяют в 40 мл воды, и порциями прибавляют 1.8 г (17 ммоль) карбоната натрия, после того как перестанет выделяться углекислый газ, прибавляют 1.52 г (10.4 ммоль) 1H-пиразол-1-карбоксамидин гидрохлорид и реакционную смесь перемешивают при комнатной температуре 24 часа. Затем воду упаривают в вакууме на роторном испарителе при температуре бани не выше 50°С, остаток растворяют при нагревании в 50 мл абсолютного этанола, отфильтровывают от нерастворившихся неорганических солей, закисляют 2 мл концентрированной соляной кислоты и упаривают. Остаток после упаривания кипятят в 50 мл ацетона в течение 20 минут, фильтруют, осадок на фильтре промывают 20 мл ацетона. Сушат в вакууме при 50°С.
Получают 1.72 г (66%) 4-(3,4,5-тетраметоксифенилсульфонил)-пиперазин-1-карбоксимидамид гидрохлорида.
ПМР спектр 4-(3,4-триметоксифенилсульфонил)пиперазин-1-ил)карбоксимидамид гидрохлорида (Brucker 300 MHz; раствор в DMSO-d6): 2.96 м (4Н, 3CH2, пиперазиновое кольцо); 3.5 м (4Н, CH3, пиперазиновое кольцо); 3.95 с (6Н, CH3O); 7.1-7.2 м (2Н, CH-Ar); 7.18 с (1Н, CH-Ar); 7.34 д (1Н, CH-Ar).
Элементный анализ C13H21ClN4O4S. Вычислено, %: С 42.80; Н 5.80; N 15.36. Найдено, %: С 42.82; Н 5.83; N 15.33.
Пример 9
Синтез 4-(2,3,4-триэтоксибензоил)пиперазин-1-карбоксимидамид фумарата

1) Синтез 2,3,4-триэтоксибензоил хлорида.
В одногорлую колбу вместимостью 100 мл, снабженную обратным холодильником с газоотводящей трубкой, погруженной в раствор NaOH, помещают 30 г (118 ммоль) 2,3,4-триэтоксибензойной кислоты и 50 мл абсолютного бензола, прибавляют 0.5 мл ДМФА и 11 мл (150 ммоль) тионил хлорида. После прекращения выделения газа смесь кипятят с обратным холодильником в течение 2 ч. Раствор охлаждают до температуры 20°C, выливают в стакан с 50 мл гексана. Осадок отфильтровывают, фильтрат упаривают до 1/3 объема в вакууме при температуре 50÷80°C. При охлаждении выделяется вторая порция продукта, которую отфильтровывают и объединяют с первой. Продукт, белый порошок, сушат в вакууме при температуре 50÷60°C в течение 1 ч.
Выход 2,3,4-триэтоксибензоил хлорида - 25 г (90.5%), т.пл. 48°C.
2) Синтез 1-(2,3,4-триэтоксибензоил)пиперазин гидрохлорида.
В трехгорлую колбу вместимостью 500 мл, снабженную механической мешалкой, капельной воронкой и термометром, помещают 30.0 г (350 ммоль) пиперазина, 75 мл уксусной кислоты и 90 мл роды. К смеси прикапывают раствор 24 г (85 ммоль) 2,3,4-триэтоксибензоил хлорида в 25 мл ТГФ в течение 30 мин, поддерживая температуру реакционной массы в диапазоне 10÷15°C. Далее перемешивают 1 ч и оставляют на ночь. Светло-желтый раствор упаривают досуха в вакууме при температуре 50÷60°C. Остаток обрабатывают 450 мл 2.5 н. HCl (1.125 моль) при охлаждении льдом и перемешивают 15 мин. Раствор фильтруют. Фильтрат упаривают досуха в вакууме ~20 мм рт.ст. и температуре 50÷60°C. Остаток обрабатывают 150 мл абс. этанола и нагревают до кипения при перемешивании. Смесь охлаждают до температуры 20°C и отфильтровывают осадок пиперазина дигидрохлорида. Маточник упаривают досуха в вакууме ~20 мм рт.ст. и температуре 50÷60°C. К остатку прибавляют 150 мл ацетона и перемешивают на магнитной мешалке 30 мин при температуре 40÷50°C. Осадок отфильтровывают, дважды промывают ацетоном по 50 мл, сушат в течение 12 ч при температуре 40°C.
Выход 1-(2,3,4-триэтоксибензоил)пиперазин гидрохлорида - 15 г (50%).
3) Синтез 4-(2,3,4-триэтоксибензоил)пиперазин-1-карбоксимидамида фумарата.
В одногорлую колбу вместимостью 100 мл помещают 7.2 г (20 ммоль) 1-(2,3,4-триэтоксибензоил)пиперазин гидрохлорида, 1.8 г (21 ммоль) дициандиамида и 50 мл 1-бутанола. Смесь кипятят с обратным холодильником в течение 10 ч, упаривают в вакууме при температуре 50÷80°C. К остатку прибавляют 100 мл этанола и кипятят с обратным холодильником в течение 0.5 ч. Горячую смесь фильтруют, осадок сушат при температуре 45°C в течение 2 сут.
Выход C16H24N6O4*HCl, 4-(2,3,4-триэтоксибензоил)пиперазин-1-карбоксимидамида гидрохлорида - 5.5 г (70%).
5.9 г (15 ммоль) 4-(2,3,4-триэтоксибензил)пиперазин-1-карбоксимидамид гидрохлорида растворяют в 50 мл 1 М NaOH, экстрагируют дважды по 50 мл дихлорметана. Объединенные органические вытяжки сушат над сульфатом натрия, упаривают в вакууме при комнатной температуре, растворяют в 100 мл этанола, прибавляют 1.7 г (15 ммоль) фумаровой кислоты, кипятят в течение 2-х часов, горячий раствор фильтруют, упаривают до 1/4 объема, выдерживают при температуре 4°C в течение 3 ч. Продукт отделяют фильтрацией, высушивают в вакууме при температуре 40°C до постоянной массы, перекристаллизовывают из изопропанола и высушивают в вакууме при температуре 40°C до постоянной массы.
Выход C18H28N4O4*C4H4O4, 4-(2,3,4-триэтоксибензил)пиперазин-1-карбоксимидамид фумарата - 3 г (42%). Элементный анализ C18H28N4O4*C4H4O4. Вычислено, %: С 54.99; Н 6.71; N 11.66. Найдено, %: С 54.92; Н 6.75; N 11.63. Масс-спектр, основание, найдено: m/z 364.43. Вычислено: М 364.45.
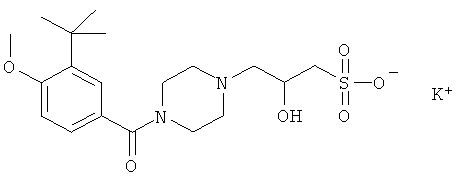
Пример 10
Синтез калиевой соли 3-(1-(3-трет-бутил-4-метоксибензоил)пиперазин-4-ил)-2-гидрокси-1-пропансульфоновой кислоты
1) Синтез N-(трет-бутилоксикарбонил)-N'-(3-трет-бутил-4-метоксибензил)-пиперазина.
В трехгорлой круглодонной колбе на 250 мл с обратным холодильником, капельной воронкой и термометром, на магнитной мешалке на охлаждающей бане, к раствору 2 г (10.7 ммоль) N-бoc-пиперазина и триэтиламина в 20 мл дихлорэтана прикапывают раствор 2.06 г (10.7 ммоль) хлоран-гидрид 3-трет-бутил-4-метоксибензойной кислоты в 20 мл сухого дихлорметана с такой скоростью, чтобы температура не поднималась выше +5°С, после этого охлаждение снимают и реакционную смесь перемешивают при комнатной температуре 1 час. Далее реакционную смесь переносят в делительную воронку и промывают 2×30 водой, 2×30 насыщенным раствором бикарбоната натрия, 2×30 насыщенным раствором хлористого аммония и 2×30 водой, хлористый метилен упаривают, а остаток желтое масло 3.5 г.
Осадок - 3.5 г используют на следующей стадии.
2) Синтез (3-трет-бутил-4-метоксибензоил)пиперазина дигидрохлорида.
К 3.5 г N-(трет-бутилокси-карбонил)-N'-(3-трет-бутил-4-метокси-бензоил)пиперазина прибавляют 30 мл 10%-ной HCl и 5 мл этанола, смесь перемешивают в течение 5 ч при температуре 20°C, упаривают досуха в вакууме при температуре 50÷80°C. К остатку прибавляют 20 мл сухого ацетона, кипятят при перемешивании в течение 20 мин, охлаждают до температуры 10°C. Через 1 ч продукт отфильтровывает, промывают 10 мл ацетона и сушат на воздухе. Выход (3-третбутил-4-метоксибензоил)пиперазин гидрохлорида 2.1 г (68%).
3) Синтез калиевой соли 3-(1-(3-трет-бутил-4-метоксибензоил)пиперазин-4-ил)-2-гидрокси-1-пропансульфоновой кислоты
В одногорлую колбу вместимостью 100 мл помещают 2.2 г (6.6 ммоль) (3-трет-бутил-4-метоксибензоил)пиперазин гидрохлорида и 30 мл воды, прибавляют порциями 5 г KHCO3. После прекращения выделения газа, прибавляют раствор 1.4 г (6.6 ммоль) натриевой соли 2-гидрокси-3-хлорпропансульфокислоты в 30 мл воды и 0.1 г йодистого калия. Смесь кипятят с обратным холодильником в течение 5 ч. Раствор упаривают досуха в вакууме при ~20 мм рт.ст. и температуре 50÷60°C. Остаток обрабатывают 50 мл абс. этанола и кипятят с обратным холодильником в течение 1 ч при перемешивании. Горячий раствор отфильтровывают от неорганических солей и выдерживают в течение суток. Белый осадок отфильтровывают и сушат при температуре 45°C в течение 2 сут.
Выход C19H29KN2O6S, калиевой соли (3-трет-бутил-4-метоксибензил)-пиперазин-4-ил)-2-гидрокси-1-пропансульфоновой кислоты - 1.3 г (45%).
Элементный анализ C19H29KN2O6S. Вычислено, %: С 50.42; Н 6.46; N 6.19. Найдено, %: С 50.61; Н 6.52; N 6.21. Масс-спектр, найдено: m/z 452.14. Вычислено: М 453.14.
Пример 11
Исследование антиагрегантных и антикоагулянтных свойств N,N'-замещенных пиперазинов на примере донорской крови человека
Кровь получали путем пункции локтевой вены донора сухой острой иглой без шприца. За 7÷10 дней до исследования отменяли прием препаратов, влияющих на функцию тромбоцитов.
В пластиковую мерную центрифужную пробирку помещали 1 мл 3.8% раствора трехзамещенного основного цитрата натрия с pH 7.4. Пунктировали локтевую вену, собирали свободно вытекающую кровь до метки 10 мл, смесь немедленно перемешивали, не допуская образования воздушных пузырей. Полученную стабилизированную кровь разливали в 2 пробирки, по 5 мл в каждую.
Для получения богатой тромбоцитами плазмы стабилизированную кровь центрифугировали 8 мин при комнатной температуре и 1000 об/мин (150 g). Повторным центрифугированием крови в течение 20 мин при 3200 об/мин (2300 g) и 22°C получали бедную тромбоцитами плазму.
Исследование антикоагулянтной активности проводилось в течение 2 ч после получения богатой тромбоцитами плазмы. Плазму стандартизовали до получения концентрации тромбоцитов 200-250×109/л, добавляя бестромбоцитарную плазму. Концентрацию тромбоцитов определяли на анализаторе СОЛАР АР 2110.
Антиагрегантные свойства N,N'-замещенных пиперазинов оценивали по их влиянию на агрегацию тромбоцитов в тестах с АДФ-, коллаген- и ристоцетин-индуцированной агрегации. Для растворения N,N'-замещенных пиперазинов и разведения реагентов использовали Трис-HCl-буфер, pH 7.4.
В качестве основной характеристики агрегационной активности тромбоцитов принята максимальная амплитуда агрегации (МА) - максимальное значение коэффициента пропускания пробы после внесения агрегирующего агента, в % к пропусканию бестромбоцитарной плазмы. Реакция фиксировалась на прямолинейном участке подъема кривой агрегации в течение 16 с.
Измерения проводили на 4-х канальном анализаторе CHRONO-LOG 490-4D (CHRONO-LOG, США). Температура в ячейке - 37°C, скорость вращения магнитной мешалки - 1200 об/мин.
0.5 мл раствора N,N'-замещенного пиперазина вносили в кювету, добавляли стандартизованную тромбоцитарную плазму (0.4 мл), перемешивали, инкубировали при 37°C в течение 5 мин. Кювету помещали в агрегометр, добавляли 0.05 мл раствора индуктора агрегации (АДФ, или коллаген, или ристоцетин) и регистрировали агрегацию тромбоцитов в течение 3-х минут.
В тесте АДФ-индуцированной обратимой агрегации тромбоцитов использовался реактив CHRONO-PAR ADP REAGENT (CHRONO-LOG, США) с концентрацией 10 мкМ. Первичную (обратимую) агрегацию тромбоцитов оценивали по реакции на добавление к плазме пороговой дозы АДФ (концентрация АДФ - 1 мкМ).
В тесте коллаген-индуцированной агрегации тромбоцитов реактив CHRONO-PAR COLLAGEN (CHRONO-LOG, США) разводили буфером до концентрации коллагена 10 мкг/мл.
Для исследования ристоцетин-индуцированной агрегации тромбоцитов реактив CHRONO-PAR RISTOCETIN (CHRONO-LOG, США) разводили буфером до концентрации ристоцетина 12 мг/мл. Измеряли амплитуду агрегации тромбоцитов через 60 с после начала реакции.
Изучение антиагрегантных свойства N,N'-замещенных пиперазинов с индуктором арахидоновой кислотой (CHRONO-LOG, США): к 450 мкл тромбоцитарной плазмы добавляли 50 мкл раствора N,N'-замещенного пиперазина, инкубировали в течение 15 мин при 36°C в кювете агрегометра CHRONO-LOG. Концентрация арахидоновой кислоты в пробе - 0.2 мМ, N,N'-замещенных пиперазинов, препарата сравнения аспирина - 4÷5 мМ/л.
Было показано, что N,N'-замещенные пиперазины имеют выраженную аспирино-подобную активность, ингибируя агрегацию тромбоцитов, индуцируемую арахидоновой кислотой (таблица 5).
Для исследования антикоагулянтных свойств N,N'-замещенных пиперазинов определяли активированное парциальное тромбопластиновое время (АПТВ) - способность N,N'-замещенных пиперазинов удлинять время свертывания бедной тромбоцитами плазмы по сравнению с контролем. Использовали набор реагентов для определения АПТВ (РосНИИГТ МЗ РФ по ТУ 9398-214-01966456-99) и коагулометр COAG-A-MATE ® ХМ (ORGANON TEKNIKA, США).
Растворы N,N'-замещенных пиперазинов смешивали с бедной тромбоцитами плазмой в соотношении 1:1, прибавляли 0.1 мл реактива АПТВ, смесь инкубировали в кювете при 37°C 5 мин, добавляли 0.1 мл 0.277% CaCl2. Определяли время свертывания, в секундах.
Результаты исследования свойств N,N'-замещенных пиперазинов представлены в таблицах 1-5 (приложение). Как следует из этих данных, N,N'-замещенные пиперазины - это соединения с селективным влиянием на факторы коагуляционного каскада и высокой антиагрегантной и антикоагулянтной активностью. Антиагрегантная и антикоагулянтная активность соединений по настоящему изобретению превышает активность препаратов
Пример 12. Исследование антиагрегантных и антикоагулянтных свойств N,N'-замещенных пиперазинов in vivo
Исследования N,N'-замещенных пиперазинов in vivo выполнялись на крысах-самцах линии Вистар массой 300±30 г (возраст 15-25 недель). Животные содержались на неограниченном потреблении корма и воды.
Изучалось влияние N,N'-замещенных пиперазинов на показатели АДФ-индуцированной агрегации тромбоцитов и фибринолитическую активность крови.
Внутривенное введение (в/в): 1 мл раствора N,N'-замещенного пиперазина или 1 мл раствора препарата сравнения озагрела вводили болюсно в хвостовую вену ненаркотизированных крыс.
Пероральное введение (в/ж): 0.2 г композиции, содержащей N,N'-замещенный пиперазин или препарат сравнения озагрел (Таблица 6), смешивали с 1.5 мл воды, суспензию вводили в/ж жестким металлическим зондом.
Дозу действующего вещества рассчитывали в мМ на 1 кг массы крысы. В контроле аналогично вводили физиологический раствор (1.5 мл).
Сразу после введения препаратов животных помещали в отдельные стандартные клетки для наблюдения.
Забор крови, стабилизированной гепарином (50 ед/мл), осуществляли в течение 40÷60 с из бедреной вены под наркозом (тиопентал-натрий, 50 мг/кг внутрибрюшинно, в 1 мл физиологического раствора). Соотношение крови и стабилизатора составляло 9:1. Кровь помещали в силиконизированные пробирки и осторожно перемешивали.
Исследование агрегационной активности тромбоцитов производили в цельной крови через 30 мин от момента забора крови.
Определение проводилось на импедансном агрегометре АИ-300 (НПО им. Коминтерна совместно с НИИ кардиологии МЗ РФ им. В.А.Алмазова) проводили при температуре 37°C и постоянной скорости перемешивания (1100 об/мин) в интактной среде (Иванов В.И. и др.; Авторское свидетельство SU 1504591 А1, 1989, бюл. N32). Электроды датчиков при соприкосновении с перемешиваемой кровью покрываются сначала тромбоцитами в один слой. При внесении индуктора развивается агрегация тромбоцитов, в результате количество тромбоцитов на электродах увеличивается. Утолщение покрывающего электроды слоя при агрегации тромбоцитов приводит к увеличению импеданса между электродами.
В кювету с магнитной мешалкой помещали 0.55 мл цельной крови крыс и проводили предынкубацию при 37°C в течение 2-3 мин в термостате. Затем в кювету помещали датчик, переносили в специальную камеру прибора и после включения перемешивания вносили индуктор агрегации - раствор динатриевой соли АДФ с концентрацией 0.25 мМ. Соотношение объемов индуктора агрегации и пробы цельной крови составляло 1:12. Определение интенсивности агрегации производили импедансным методом через 5 мин от момента ввода индуктора (Таблица 7).
Исследование влияния N,N'-замещенных пиперазинов на фибринолитическую активность крови крыс оценивали по времени спонтанного лизиса сгустка, получаемого из эуглобулиновой фракции плазмы. Метод является одним из интегральных методов оценки состояния фибринолитической системы.
Отбор стабилизированной цитратом натрия (3.2%) крови осуществляли под наркозом (тиопентал-натрий 50 мг/кг, внутрибрюшинно в 1 мл физиологического раствора), в течение 60 с из бедренной вены.
Соотношение крови и стабилизатора составляло 9:1. Стабилизированную кровь центрифугировали в течение 10 мин при скорости 1200 g. Исследовали бедную тромбоцитами плазму с реагентом для исследования спонтанного эуглобулинового фибринолиза фирмы ООО «Технология-стандарт» (Россия, Барнаул). Перед определением реагенты разводили дистиллированной водой до концентрации реагентов: кальция хлорид - 0.277%, уксусная кислота - 1%.
Для получения эуглобулиновой фракции плазмы в пробирке последовательно смешивали 8 мл воды д/и, 0.18 мл 1% уксусной кислоты и 0.5 мл плазмы. Смесь инкубировали при температуре 4÷8°C в течение 30 мин, центрифугировали при 600 g в течение 5 мин. Надосадочную жидкость сливали, пробирку опрокидывали на фильтровальную бумагу на 1 мин. Оставшийся на дне пробирки осадок эуглобулинов разводили в 0.5 мл рабочего буфера.
К 0.5 мл раствора эуглобулинов в пробирке добавляли 5 мл 0.277% раствора CaCl2, осторожно перемешивали, избегая встряхивания, инкубировали на водяной бане при температуре 37°C.
Регистрировали время (мин) от момента добавления раствора CaCl2 до полного растворения сгустка. В норме у здоровых крыс время спонтанного лизиса эуглобулинов составляет 90-180 минут. Укорочение времени лизиса свидетельствует об активации, а удлинение - об угнетении фибринолиза (Таблица 8).
Пример 13. Исследование влияния N,N'-замещенных пиперазинов на систему гемостаза. Модель экспериментального тромбоза
Исследования выполнялись на крысах-самцах линии Вистар массой 230±30 г (питомник лабораторных животных «Рапполово»,). Животные содержались на неограниченном потреблении корма (стандартный рацион для лабораторных крыс К-120 фирмы «Информ-корм», Россия) и воды.
Препарат вводили болюсно в хвостовую вену ненаркотизированным животным. В контроле вводили физраствор (5 мл/кг). Сразу после введения животных помещали в отдельные стандартные клетки для наблюдения.
Моделирование тромбоза: через 50 мин после введения препарата в левую бедренную вену вводили 1 мл раствора фотосенсибилизатора Бенгальский розовый A (Acros Organic, США) в дозе 17 мг/кг [Boselli 2007, Петрищев 2009], производили разрез длиной около 2 см на внутренней поверхности правого бедра животного. Участок бедренной артерии длиной около 5 мм освобождали от окружающих тканей, выделяли из сосудисто-нервного пучка. Под данный участок артерии подводили полоску непрозрачного черного пластика шириной 3 мм, изолируя тем самым вену и окружающие ткани от облучения. Облучение проводили при помощи диодного лазера «DPSS-лазер» (Diode Pumpd Solid State Laser, Южная Корея). Длина волны 532 нм, мощность 60 мВт, площадь облучения - 1 мм2, продолжительность облучения - 40 минут. Кровоток в бедренной артерии измеряли при помощи высокочастотной ультразвуковой допплерографии (прибор «Минимакс-Допплер-К»), частота датчика 20 мГц). Измеряли изменение скорости кровотока в процессе облучения артерии (Таблица 9).
Пример 14. Исследование вазодилаторных свойств N,N'-замещенных пиперазинов
Вазодилаторные свойства N,N'-замещенных пиперазинов оценилились по их влиянию на реактивность сосудов микроциркуляторного русла.
Исследования выполнялись на крысах-самцах линии Вистар массой 300±30 г (питомник лабораторных животных «Рапполово» РАМН). Животные содержались на неограниченном потреблении корма (стандартный рацион для лабораторных крыс К-120 фирмы «Информ-корм», Россия) и воды.
Растворы N,N'-замещенных пиперазинов и физраствор (контроль) вводили болюсно, в течение 1 мин в хвостовую вену ненаркотизированных животных из расчета 5,0 мл на 1 кг. Сразу после введения раствора животных помещали в отдельные стандартные клетки для наблюдения.
Через 30 мин после введения животных наркотизировали (тиопентал-натрий, 60 мг/кг подкожно). Через нижнесрединный доступ извлекали петлю тонкой кишки, примыкающей к мезоаппендиксу для исследования микроциркуляции в венулах и артериолах брыжейки в проходящем свете и помещали крысу на термостатируемый предметный столик микроскопа. Аппликацию раствора норадреналина (Агетан, 2 мг/мл, Laboratoire AGUETTANT, Франция) производили непосредственно на брыжейку (Fumess J.B., Marshall J.M., 1974; Янтарева Л.И., 2004).
Изучения сосудов микроциркуляторного русла проводили на базе микроскопа ЛЮМАМ И1 (ЛОМО, Россия). С помощью видеокамеры (Optics and Electronics ISTA Ltd) производили видеозапись данных на персональный Компьютер, обработку результатов осуществляли по программе Video-Test 4.0 (ISTA Ltd., Россия). Определяли: диаметр сосудистой стенки (D, мкм) до и после аппликации норадреналина, время начала замедления кровотока в сосудах под действием норадреналина (наблюдение производилось в течение 1 мин после аппликации).
Проведенные фармако-биологические испытания показали, что N,N'-замещенные пиперазины обладают антиагрегантными, антикоагулянтными и вазодилатирующими свойствами. Композиции на основе N,N'-замещенных пиперазинов положительно влияют на восстановление системы гемостаза.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВЕЩЕСТВО, ОБЛАДАЮЩЕЕ СОЧЕТАННОЙ АНТИАГРЕГАНТНОЙ, АНТИКОАГУЛЯНТНОЙ И ВАЗОДИЛАТОРНОЙ АКТИВНОСТЬЮ, И СПОСОБ ПОЛУЧЕНИЯ N, N'-ЗАМЕЩЕННЫХ ПИПЕРАЗИНОВ | 2014 |
|
RU2577039C2 |
| Способ получения производных этиленбензоила | 1972 |
|
SU439983A1 |
| НОВЫЕ КАРБОКСИЗАМЕЩЕННЫЕ ЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ КАРБОКСАМИДА | 1997 |
|
RU2199535C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ N-АКРИЛОИЛПИПЕРАЗИНА | 1990 |
|
RU2024513C1 |
| ПРОИЗВОДНЫЕ СПИРОПИПЕРИДИНА, ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ | 1999 |
|
RU2184735C2 |
| ДИПЕПТИДНИТРИЛЬНЫЕ ИНГИБИТОРЫ КАТЕПСИНА К | 2001 |
|
RU2293732C2 |
| ДИПЕПТИДНИТРИЛЬНЫЕ ИНГИБИТОРЫ КАТЕПСИНА К | 2001 |
|
RU2265601C2 |
| ПРОИЗВОДНЫЕ АЛЬФА-(N-СУЛЬФОНАМИДО)АЦЕТАМИДА КАК ИНГИБИТОРЫ БЕТА-АМИЛОИДА | 2002 |
|
RU2300518C2 |
| Способ получения производных гетразепина | 1989 |
|
SU1738089A3 |
| N-ГИДРОКСИФОРМАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2004 |
|
RU2351595C2 |
Изобретение относится к области медицины, а именно к новым органическим соединениям, а именно к N,N'-замещенным пиперазинам общей формулы (I):
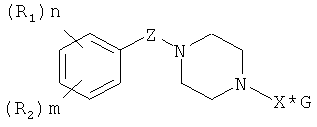
где R1, R2: линейный или разветвленный алкокси (С1÷С4), CH3C(=O)O; n=1-5; m=0-3; Z: C=O, SO2; X:C(=NH)NH2, C(=NH)NHC(-NH)NH2, G - низкомолекулярная органическая или минеральная кислота, катионы натрия, калия, аммония или вода, влияющим на систему гемостаза, проявляющим антиагрегантные, антикоагулянтные и вазодилаторные свойства, и к способу получения N,N'-замещенных пиперазинов формулы 1, путем взаимодействия N-замещенных пиперазинов общей формулы

где R1, R2: линейный или разветвленный алкокси (C1÷C4), CH3C(=O)O; n=1-5; m=0-3; Z: C=O, SO2; с 1H-пиразол-1-карбоксамидином, дициандиамидом и их солями в органических растворителях или воде при температуре 10-50°C в присутствии оснований. Новые вещества перспективны для профилактики и лечения заболеваний системы гемостаза. 4 н. и 8 з.п. ф-лы, 10 табл., 14 пр.
1. N,N'-замещенные пиперазины общей формулы (I):

где R1, R2 - инейный или разветвленный алкокси (C1÷C4), CH3C(=O)O;
n=1-5; m=0-3;
Z - C=O, SO2;
X - C(=NH)NH2, C(=NH)NHC(-NH)NH2;
G - низкомолекулярная органическая или минеральная кислота, катионы натрия, калия, аммония или вода.
2. Способ получения N,N'-замещенных пиперазинов по п.1, путем взаимодействия N-замещенных пиперазинов общей формулы
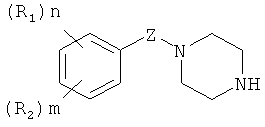
где R1, R2 - линейный или разветвленный алкокси (C1÷C4), CH3C(=O)O;
n=1-5; m=0-3; Z - C=O, SO2;
с 1H-пиразол-1-карбоксамидином, дициандиамидом и их солями в органических растворителях или воде при температуре 10-50°C в присутствии оснований.
3. Способ получения N,N'-замещенных пиперазинов по п.2, отличающийся тем, что в качестве N-замещенных пиперазинов используют их гидраты.
4. Способ получения N,N'-замещенных пиперазинов по п.2, отличающийся тем, что в качестве N-замещенных пиперазинов используют их соли.
5. Способ получения N,N'-замещенных пиперазинов по п.2, отличающийся тем, что в качестве оснований используют гидроксиды или карбонаты щелочных металлов, а также их органические основания.
6. Способ получения N,N'-замещенных пиперазинов по п.2 отличающийся тем, что в качестве органических растворителей используют низшие алифатические спирты, ацетонитрил, тетрагидрофуран, диметилформамид, диметилсульфоксид, дихлорметан или их смеси.
7. N,N'-Замещенные пиперазины общей формулы (II)

где R1, R2 - линейный или разветвленный алкил (C1÷C4), линейный или разветвленный алкокси (C1-C4);
n=1-5; m=0-3; Z - C=O, SO2;
X - CH2(CHOH)CH2SO3H,
G - низкомолекулярная органическая или минеральная кислота, катионы натрия, калия, аммония или вода.
8. Способ получения N,N'-замещенных пиперазинов по п.7 путем взаимодействия N-замещенных пиперазинов общей формулы
где R1, R2 - линейный или разветвленный алкил (C1÷C4), линейный или разветвленный алкокси (C1-C4);
n=1-5; m=0-3; Z - C=O, SO2;
с 2-гидрокси-3-хлорпропансульфокислотой или ее солями в органических растворителях или воде при температуре 10-100°C в присутствии оснований.
9. Способ получения N,N'-замещенных пиперазинов по п.8, отличающийся тем, что в качестве N-замещенных пиперазинов используют их гидраты.
10. Способ получения N,N'-замещенных пиперазинов по п.8, отличающийся тем, что в качестве N-замещенных пиперазинов используют их соли.
11. Способ получения N,N'-замещенных пиперазинов по п.8, отличающийся тем, что в качестве органических растворителей используют низшие алифатические спирты, ацетонитрил, тетрагидрофуран, диметилформамид, диметилсульфоксид, дихлорметан, хлороформ или их смеси.
12. Способ получения N,N'-замещенных пиперазинов по п.8, отличающийся тем, что в качестве оснований используют аммиак, натрия гидроксид, или калия гидроксид, или карбонат натрия, или гидрокарбонат натрия, или карбонат калия, или гидрокарбонат калия.
| Collection of Czechoslovak Chemical Communications, 41 (4), 1035-41, 1976 | |||
| Collection of Czechoslovak Chemical Communications, 40 (12), 3904-23, 1975 | |||
| Способ получения антиокислительной присадки | 1961 |
|
SU151752A1 |
| JP 44001981 B4, 28.01.1969 | |||
| 1,4,5,6-ТЕТРАГИДРО-6-ОКСО-5-(2-ПИПЕРАЗИНОЭТИЛ)-4-ФЕНИЛ-3-(4-ХЛОРФЕНИЛ)ПИРРОЛО[3, 4-С]ПИРАЗОЛА ДИГИДРОХЛОРИД, ПРОЯВЛЯЮЩИЙ ГИПОТЕНЗИВНУЮ И АНТИКОАГУЛЯНТНУЮ АКТИВНОСТИ | 2006 |
|
RU2320661C2 |

