Данное изобретение относится к производным 6-(1Н-имидазо-1-ил)-2-арил- и 2-гетероарилхиназолина и хинолина, действующим как ингибиторы моноаминоксидазы (МАО) и лиганды имидазолинового рецептора, к способу их получения и к применению таких соединений, их фармацевтически приемлемых солей и сольватов и соответствующих фармацевтических композиций для фармакологического лечения депрессии и родственных расстройств, болезни Паркинсона, токсикомании, толерантности к морфину и зависимости от морфина.
Уровень техники
Депрессия представляет собой общее и опасное расстройство настроения, которое воздействует на эмоцию, познавательную способность и поведение; вместо четко определенного заболевания депрессия охватывает широкий спектр расстройств, включающих от ощущения несчастья до более тяжелых расстройств, приводящих к нетрудоспособности, таких как клиническая депрессия (также называемая большим депрессивным расстройством или униполярной депрессией), дистимическое расстройство, биполярное расстройство, атипическая депрессия, психотическая депрессия, послеродовая депрессия и сезонное аффективное расстройство (A. Doris et al. Depressive illness, Lancet, 1999, 354, 9187, 1369). Согласно Всемирной организации здравоохранения (ВОЗ/WHO), депрессия характеризуется пониженным настроением, потерей интереса или удовольствия, ощущениями виновности или низкого самоуважения, нарушениями сна и/или аппетита, слабой сконцентрированностью. Большое депрессивное расстройство, также известное как большая депрессия, представляет собой самый обычный тип депрессии с приблизительно 10-25% риском в течение жизни для населения промышленных стран. Оно характеризуется комбинацией симптомов и состояний с потерей трудоспособности, которые серьезно мешают работе и семейной жизни, традиционным особенностям (привычкам) ко сну и приему пищи и всему здоровью пациента. Дистимическое расстройство, также называемое дистимией, характеризуется длительными менее тяжелыми симптомами, которые могут не лишать человека трудоспособности, но могут мешать хорошему самочувствию человека, таким образом влияя на социальную жизнь. Биполярное расстройство, также называемое маниакально-депрессивным заболеванием, характеризуется чередующимися изменениями настроения от экстремальных высоких уровней (например, мания) до экстремально низких уровней (например, депрессия). Атипическая депрессия представляет собой подтип дистимии и большой депрессии, характеризующийся реактивностью настроения и вегетативными симптомами, подобными перееданию и чрезмерному сну. Психотическая депрессия имеет место, когда тяжелое депрессивное заболевание сопровождается некоторой формой психоза, галлюцинациями и бредом. Послеродовая депрессия, которой подвержены 10-15% женщин, диагностируется, если большой депрессивный эпизод происходит в пределах одного месяца после рождения ребенка, данное заболевание имеет симптомы, аналогичные клинической депрессии. Сезонное аффективное расстройство характеризуется началом депрессивного заболевания в течение зимних месяцев. Депрессивные и тревожные симптомы часто перекрываются. Тревожные расстройства включают посттравматическое стрессовое расстройство, паническое расстройство, агорафобию, социальную фобию, обсессивно-компульсивное расстройство. Паническое расстройство классифицируется как тревожное расстройство, так как тревога является преобладающим симптомом, панические атаки представляют собой последовательность дискретных эпизодов панического расстройства. Развитие тяжелых фобических симптомов соответствует эскалации по частоте и интенсивности панических атак, ведущих к тяжелому с потерей трудоспособности расстройству, которое влияет на профессиональную, социальную и семейную жизнь пациента. Депрессия может представлять собой основное состояние или может сосуществовать с другими серьезными медицинскими заболеваниями, такими как заболевание сердца, удар, рак, диабет и болезнь Паркинсона. Клинические исследования показали, что люди, которые имеют депрессию кроме другого серьезного медицинского заболевания, имеют тенденцию к проявлению более тяжелых симптомов как депрессии, так и медицинского заболевания, более тяжелой адаптации к их медицинскому состоянию и более высоким медицинским затратам, чем затраты без сопутствующей депрессии. Исследования подтвердили, что лечение депрессии может также помогать в улучшении результата лечения сопутствующего заболевания. Злоупотребление алкоголем, табаком и лекарственными средствами также может сосуществовать с депрессией. Фактически, статистическое изучение показало, что сосуществование расстройств настроения распространено среди людей, вовлеченных в злоупотребление алкоголем, табаком и лекарственными средствами. Депрессивные расстройства являются экстремально общими, воздействующими ежегодно на примерно 120 миллионов человек в мире. Согласно ВОЗ депрессия является ведущей причиной нетрудоспособности, и она представляет собой четвертый наиболее важный вклад в глобальное бремя болезни. Заболеваемость и смертность среди пациентов с депрессией выше, чем среди обычных пациентов. Согласно Национальному институту психического здоровья (National Institute of Mental Health (NIMH)) недавние исследования показали, как лица с большой депрессией с вероятностью более чем в четыре раза страдали бы от сердечного приступа по сравнению с контролями без депрессии. Согласно NIMH прямые и непрямые социальные затраты на депрессию доходили в течение 1990 года до примерно 30 миллиардов долларов США, при этом непрямые затраты выражаются сниженной производительностью рабочих и нарушением персональных, профессиональных и семейных взаимоотношений. Аналогичная оценка в Европе в течение 2004 года показала социальные затраты в 118 миллиардов евро, причем депрессия выделяется в качестве самого дорогостоящего расстройства головного мозга в Европе.
Согласно моноаминовой гипотезе, депрессия вызвана дисбалансом данных нейротрансмиттеров в головном мозге. Одна фармакологическая стратегия, нацеленная на преодоление данного дисбаланса, состоит в ингибировании фермента моноаминоксидазы (МАО; ЕС 1.4.3.4). Моноаминовые нейротрансмиттеры: серотонин (5-НТ), норэпинефрин (NE) и допамин широко распределены в мозге и включены в регуляцию настроения, познавательной способности, сна, страха и социального поведения. Дисфункции в механизмах контролирования данных нейротрансмиттеров часто связаны с самыми главными психиатрическими расстройствами, и лекарственные средства, нацеленные на моноаминовые нейротрансмиттеры, были исследованы и широко исследованы для лечения депрессии. МАО представляет собой FAD-зависимый фермент (флавопротеин), расположенный в основном во внешних митохондриальных мембранах нейронов и глиальных клетках, а также в других клетках периферии (т.е. эпатоциты), где она катализирует окислительное деаминирование нейротрансмиттера, ксенобиотических и эндогенных аминов. Антидепрессантный подход для ингибиторов МАО основан на факте, что ингибированием активности фермента предотвращена деактивация данных эндогенных нейротрансмиттеров, таким образом повышается как их синаптическая концентрация, так и длительность действия. Существуют две изоформы МАО: МАО-А, которая предпочтительно деаминирует серотонин, норэпинефрин и эпинефрин, а также амины, присутствующие в пище, подобные тирамину, и МАО-В, которая предпочтительно деаминирует допамины, фенилэтиламины и бензиламины (B.H. Moussa, British J. Pharmacology, 2006, 147, S287-296). Первое поколение ингибиторов МАО, неселективно и необратимо блокировавших обе изоформы МАО, приводило к побочным эффектам, таким как гипертонический криз (также называемый “сырный синдром”), особенно из-за ингибирования МАО-А, что блокирует каскад запуска метаболизма тирамина, в котором избыточные количества норэпинефрина могут вести к гипертоническому кризу. Второе поколение обратимых ингибиторов МАО-А, таких как моклобемид и брофаромин, проявляло в клинических опытах потенциальную антидепрессантную активность, но незначительную предрасположенность вызывать после проглатывания тирамина гипертонический криз в терапевтической дозировке (Bonnet U., CNS Drug Review, 2003, 9, 1, 97-140). Это потому, что обратимость допускает конкуренцию и таким образом проглоченный тирамин способен вытеснять ингибитор от фермента. Селективные обратимые ингибиторы МАО-В не вызывают гипертонический криз. Недавние исследования подтверждают, что тревожные расстройства также могут быть связаны с дисфункцией нейротрансмиссии серотонина и дисбалансами в метаболизме катехоламина. Эффективность ингибиторов МАО в лечении тревожных расстройств показана несколькими клиническими опытами и отдельными сообщениями (J. Clin. Psychiatry, 2006, 67, S12:20-26). Ингибиторы МАО-В пролонгируют активность как эндогенно, так и экзогенно полученного допамина, что делает их вариантом либо для монотерапии на ранней стадии болезни Паркинсона (PD), либо для дополнительной терапии у пациентов, лечившихся леводопой. Эффективность данного подхода к МАО-В для лечения PD клинически доказана опытами, включающими два утвержденных в США ингибитора МАО-В, расагилин и селегилин, а также использование сафинамида при присутствии в фазе III. Все данные лекарственные средства давали симптоматическое ослабление боли при использовании в качестве монотерапии или вспомогательной терапии, причем еще проявляют потенциал в качестве агентов, модифицирующих заболевание.
Имидазолиновые рецепторы, семейство неадренергических рецепторов, впервые идентифицированные Bousquet в 1984, широко распределены как в центральной, так и в периферической системе. Были идентифицированы три основных подкласса имидазолиновых связывающих сайтов (IBS): I1-IBS, который предпочтительно связывает клонидин, расположен на мембране нейронов и включен в центральное регулирование давления крови, I2-IBS, который предпочтительно связывает идазоксан, расположен преимущественно во внешней мембране митохондрии, и I3-IBS, который был идентифицирован в поджелудочной железе. Исследования по выделению белка показали, что МАО-А и МАО-В, обе, представляют собой I2-связывающие белки. Дополнительные фармакологические исследования показывали, как агонисты в I2-IBS способны ингибировать активность МАО, таким образом предоставляя альтернативный подход к ингибиторам МАО для контролирования активности как МАО-А, так и МАО-В. На нескольких моделях животных было показано, как лиганды I2-IBS способны модулировать центральные моноаминовые уровни, а также недавно было показано, как изменения в плотности I2-IBS могут быть повышены у пациентов с депрессией. Агмантин представляет собой эндогенный амин, образуемый при декарбоксилировании аргинина, который недавно предложен в качестве нейротрансмиттера в ЦНС/CNS. Недавно для агмантина и других селективных агонистов I2-IBS, таких как 2-BFI (2-бензофуранилимидазолин) и норгарман (β-карболин), были опубликованы антидепрессантные свойства на некоторых моделях животных, подтверждающие таким образом in vivo, что I2-имидазолиновый рецептор представляет собой новую фармакологическую мишень для лечения депрессии и родственных расстройств (MP Zeadan, Eur. J. Pharmacology, 2007, 565, 1-3, 125-31).
Синдром отмены наркотиков и алкоголя часто сопровождается атипической депрессией, которая вызывает возобновление потребления алкоголя и наркотиков, в соответствии с этим лечение антидепрессантами, включающее лечение ингибиторами МАО, в целом можно рассматривать как фармакологический подход для лечения злоупотребления наркотиками и алкоголем. Однако в некоторых случаях ингибиторы МАО, как было показано доклиническими и клиническими опытами, даже лучше чем другие лекарственные антидепрессанты по нескольким причинам.
Никотин вызывает толерантность и привыкание действием на центральные допаминергические пути метаболизма, таким образом, только 50% снижение в потреблениях никотина может запускать абстинентные симптомы, такие как тревожность, депрессивные симптомы, расстройства познавательной способности, расстройства сна. Применение ингибиторов МАО в качестве новой фармакотерапии для лечения зависимости от курения основано как на компенсационном действии данных лекарственных средств на допаминергический путь, так и на антидепрессивных эффектах, что должно исключать эпизоды ремиссии (T.P. George et al., Clin. Pharmacol. Ther., 2008, 83, 4, 619-21).
Злоупотребление кокаином представляет собой серьезную проблему для здоровья во многих частях мира, до настоящего времени нет утвержденных фармакологических терапий, чтобы преодолеть кокаиновую зависимость. Доклинические изучения наводят на мысль, что кокаиновая зависимость может быть обусловлена ингибированием транспортера допамина, проявляемым кокаином, который вызывает допаминовый усиленный эффект. Ингибиторы МАО и особенно ингибиторы МАО-В, как показано предварительными опытами с ингибитором МАО-В селегилином, повышая уровни моноаминов, могут помогать в преодолении кокаиновой зависимости путем противодействия снижению уровня допамина вследствие синдрома отмены наркотика (E.J. Houtsmeller, Psychopharmacology, Berl., 2004, 172, 1, 31-40).
Доклинические модели главное внимание уделяли тому, как лиганд I2-IBS усиливает анальгезирующее действие морфина и ингибирует толерантность и зависимость от опиоидов (A. Mirales et al., Eur. J. Pharmacology, 2005, 22, 518, 2-3, 234-242). Агмантин и 2-BFI наряду с другими агонистами I2-IBS были показаны для потенцирования аналгезии, вызываемой опиоидами, и ослабления развития толерантности и зависимости, в то время как антагонист I2-IBS, такой как идазоксан, полностью обращал данные эффекты. Интересно, что аналогичные эффекты потенцирования морфиновой аналгезии и предотвращения толерантности и зависимости наблюдались также в моделях на животных с ингибиторами МАО (A. Wasik et al., J. Physiol. Pharmacol., 2007, 58, 2, 235-52; K.Grasing et al., Behav. Pharmacol., 2005, 16, 1, 1-13), и подтверждены клинически для моклобемида, обратимого ингибитора МАО-А (G. Vaiva, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2002, 26, 3, 609-11).
Описание изобретения
В своей предыдущей патентной заявке WO2008/014822 авторы описали производные 2-арил- и 2-гетероарил-6-(1Н-имидазо-1-ил)хиназолина и хинолина для лечения боли и воспалительных нарушений. Позже авторы открыли, что производные 2-арил- и 2-гетероарил-6-(1Н-имидазо-1-ил)хиназолина и хинолина формулы (I) удивительным образом наделены отличными свойствами по ингибированию МАО и являются потенциальными агонистами I2-IBS. В соответствии с этим, данное изобретение относится к применению соединений формулы (I), их фармацевтически приемлемых солей и/или сольватов и соответствующих фармацевтических композиций для фармакологического лечения депрессии, включающей большое депрессивное расстройство, дистимическое расстройство, биполярное расстройство типа II, маниакальную депрессию, тревожные расстройства, включающие посттравматическое стрессовое расстройство, паническое расстройство.
Согласно целесообразности, сообщенной в уровне техники, данное изобретение еще относится к применению соединений формулы (I), их фармацевтически приемлемых солей и/или сольватов и соответствующих фармацевтических композиций для фармакологического лечения болезни Паркинсона. В другом варианте осуществления данное изобретение относится к применению соединений формулы (I), их фармацевтически приемлемых солей и/или сольватов и соответствующих фармацевтических композиций из них для фармакологического лечения синдромов отмены и чтобы избежать эпизодов ремиссии по алкоголизму, курению и наркомании, включая злоупотребление кокаином. В другом варианте осуществления данное изобретение относится к применению соединений формулы (I), их фармацевтически приемлемых солей и/или сольватов и соответствующих фармацевтических композиций, где соединения формулы (I) использованы индивидуально или в комбинации с морфином или другими опиоидными лекарственными средствами для усиления опиоидного фармакологического действия и/или для снижения дозировки опиоидного лекарственного средства. В другом варианте осуществления данное изобретение относится к применению соединений формулы (I), их фармацевтически приемлемых солей и/или сольватов и соответствующих фармацевтических композиций из них для лечения толерантности и зависимости из-за применения опиоидных лекарственных средств.
Соединения формулы (I):
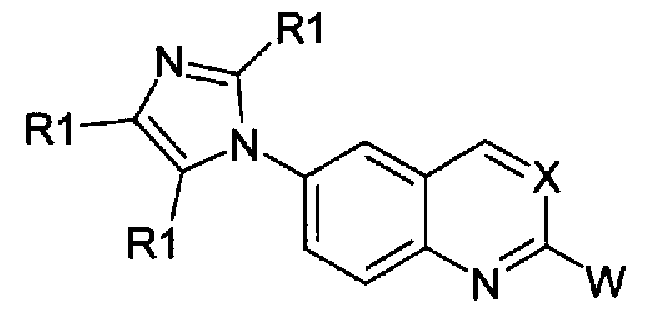
в которой
- Х независимо выбран из -СН группы или атома азота (-N);
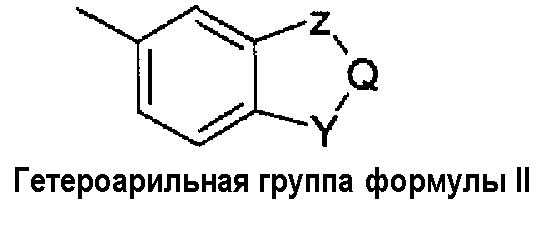
- W независимо выбран из арильной группы, гетероарильной группы или гетероарильной группы формулы II:
- когда W представляет собой арильную группу, он является незамещенным или замещенным фенилом с одним или более заместителями, независимо выбираемыми из галогена (-F, -Cl, -Br), трифторметила (-CF3), алкила (-R2), гидроксила (-OH), алкокси (-OR2), трифторметокси (-OCF3), циано (-CN), карбоксамидо (-CONHR3, или -NHCOR3, или -CONR2R3, или -NR2COR3), карбонила (-CO-R3), алкилтио или тиола (-SR3), сульфинила (-SOR3) и сульфонила (-SO2R3), причем R2 и R3 принимают значения, определенные ниже;
- когда W представляет собой гетероарильную группу, он независимо выбран из следующих гетероциклов с пятью или шестью атомами: 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, пиррол-2-ил, пиррол-3-ил, пиридин-4-ил, пиридин-3-ил, пиримидин-4-ил. Гетероциклическое кольцо может быть замещено одним или двумя заместителями, независимо выбираемыми из R1, алкокси (-OR2) или гидрокси (-OH), причем R1 и R2 принимают значения, определенные ниже;
- когда W представляет собой гетероарильную группу формулы (II), он является бензоконденсированным 5- или 6-членным гетероциклом, в котором
- Z и Y независимо выбраны из атома кислорода (-O-), атома серы (-S-) или групп -CHR3-, -CR3=, -NH-, -N=;
- Q независимо выбран из групп -CHR3, -CH=, -CR3=, -CHR3-CH2-;
при условии, что комбинация Y, Z, Q групп дает 1,3-бензодиоксол, бензофуран, 2,3-дигидробензофуран, бензотиофен, 2,3-дигидробензотиофен, индол, 2,3-дигидроиндол, бензимидазол, бензоксазол, бензотиазол, 2Н-3,4-дигидробензопиран, [1,4]-бензодиоксин, 2,3-дигидро-[1,4]-бензодиоксин (1,4-бензодиоксан);
- R1 независимо выбран из водорода (-Н), С1-С4 алкила, гидроксиметила (-CH2OH), аминометила (-CH2NH2), алкиламинометила [CH2NH(R2)] или диалкиламинометила [CH2N(R2)2], трифторметила (-CF3). C1-C4 алкильная группа представляет собой нормальную или разветвленную насыщенную или ненасыщенную C1-C4 углеводородную цепь. При условии, что в соединениях формулы (I) не более чем две R1 группы, замещающие имидазольное кольцо, представляют собой одновременно C1-C4 алкил или трифторметил (-CF3) и только одна R1 группа представляет собой гидроксиметил (-CH2OH), аминометил (-CH2NH2), алкиламинометил [CH2NH(R2)] или диалкиламинометил [CH2N(R2)2];
- R2 представляет собой С1-С6 алкильную цепь, где С1-С6 алкильная цепь имеет те же значения, как определено для С1-С4 цепи, но необязательно замещенной арилом, причем арил принимает значения, определенные выше;
- R3 независимо выбран из водорода, С1-С4 алкила, определенного выше для R1.
Соединения формулы (I), определенные выше, имеют таутомеры, объем данного изобретения включает все возможные таутомеры соединений формулы (I).
Определенные выше соединения формулы (I), когда W представляет собой арил или гетероарил формулы (I), включены в объем соединений формулы (I) в предыдущей заявке данных авторов WO 2008/014822, однако некоторые из них являются новыми соединениями, ранее не описанными в примерах патентной заявки данных авторов WO 2008/014822.
В следующем варианте осуществления данное изобретение относится к этим новым соединениям, их фармацевтически приемлемым солям и/или сольватам и соответствующим фармацевтическим композициям и их применению для фармакологического лечения тех заболеваний, которые детализированы для соединений формулы (I).
Данные новые соединения представляют собой:
[6-(2-метил-1Н-имидазол-1-ил)-2-фенил]хиназолин
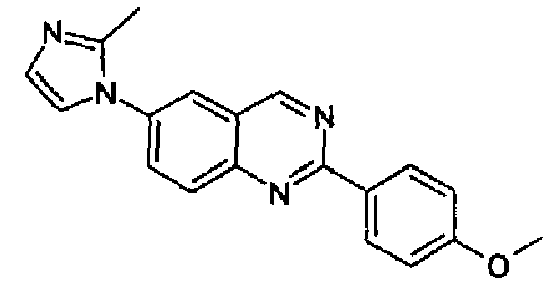
[6-(2-метил-1Н-имидазол-1-ил)-2-(4-метоксифенил)]хиназолин
[6-(4-метил-1Н-имидазол-1-ил)-2-фенил]хиназолин
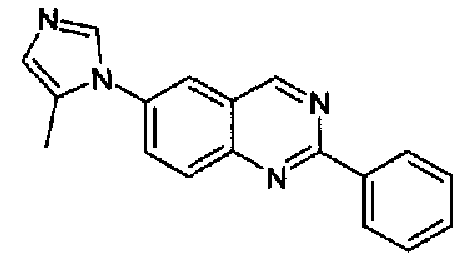
[6-(5-метил-1Н-имидазол-1-ил)-2-фенил]хиназолин
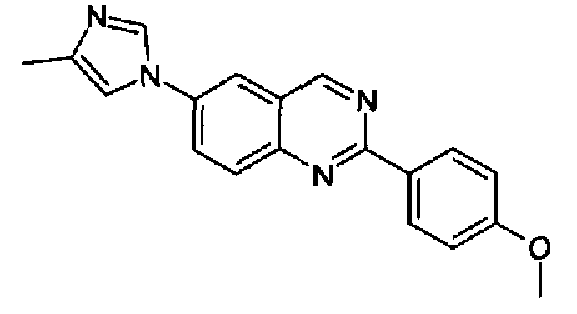
[6-(4-метил-1Н-имидазол-1-ил)-2-(4-метоксифенил)]хиназолин
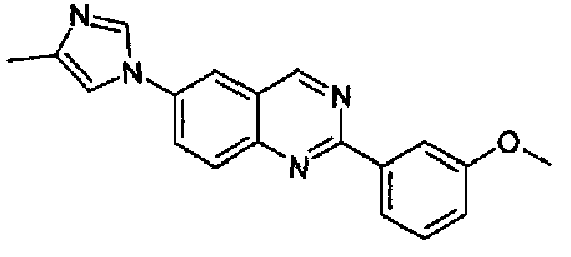
[6-(4-метил-1Н-имидазол-1-ил)-2-(3-метоксифенил)]хиназолин
[6-(4-метил-1Н-имидазол-1-ил)-2-(2-метоксифенил)]хиназолин
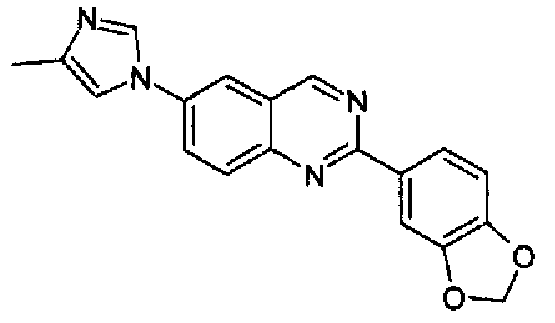
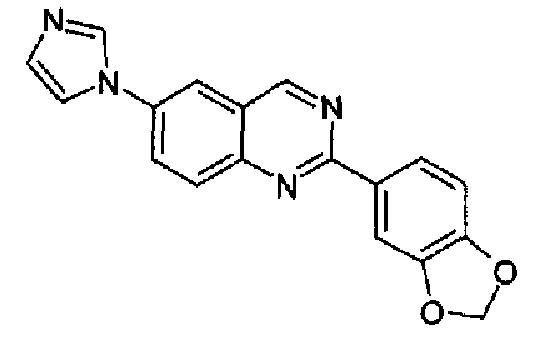
[6-(4-1Н-имидазол-1-ил)-2-(1,3-бензодиоксол-5-ил)]хиназолин
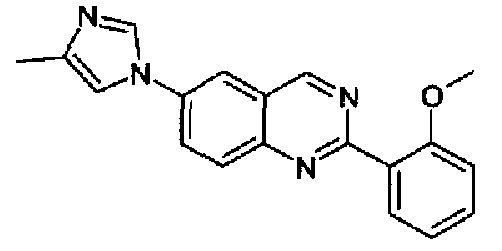
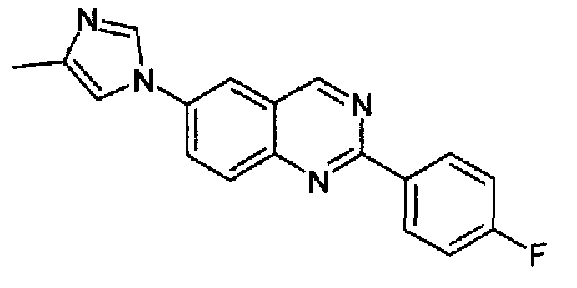
[6-(4-метил-1Н-имидазол-1-ил)-2-(4-фторфенил)]хиназолин
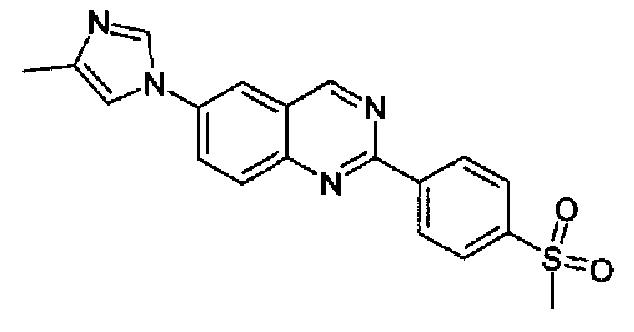
[6-(4-метил-1Н-имидазол-1-ил)-2-(4-метансульфонилфенил)]хиназолин
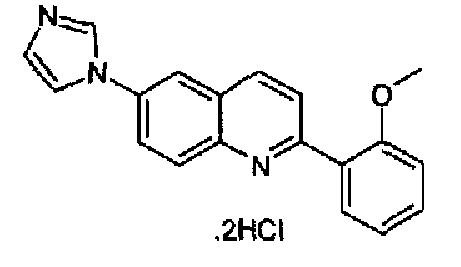
[6-(1Н-имидазол-1-ил)-2-(4-метоксифенил)]хинолин
[6-(1Н-имидазол-1-ил)-2-(2-метоксифенил)]хинолин
[6-(1Н-имидазол-1-ил)-2-(1,3-бензодиоксол-5-ил)]хинолин
[6-(1Н-имидазол-1-ил)-2-(4-фторфенил)]хинолин
[6-(1Н-имидазол-1-ил)-2-(4-диметиламинофенил)]хинолин
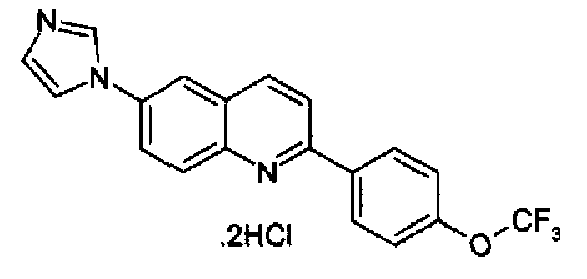
[6-(1Н-имидазол-1-ил)-2-(4-трифторметоксифенил)]хинолин
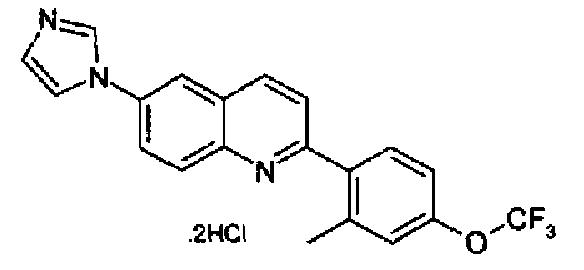
[6-(1Н-имидазол-1-ил)-2-(2-метил-4-трифторметоксифенил)]хинолин
[6-(1Н-имидазол-1-ил)-2-(4-диметиламинофенил)]хинолин
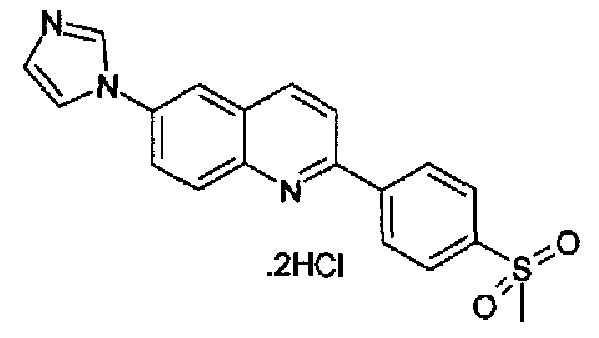
[6-(1Н-имидазол-1-ил)-2-(4-метансульфонилфенил)]хинолин
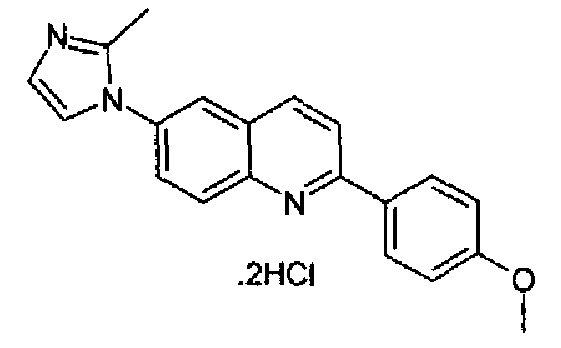
[6-(2-метил-1Н-имидазол-1-ил)-2-(4-метоксифенил)]хинолин
[6-(2-метил-1Н-имидазол-1-ил)-2-(2-метоксифенил)]хинолин
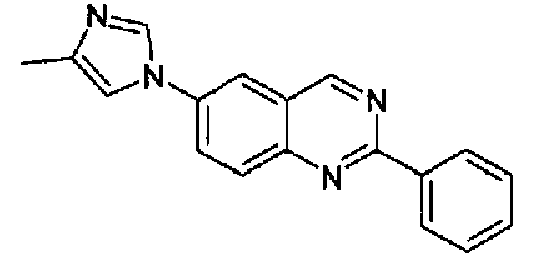
[6-(4-метил-1Н-имидазол-1-ил)-2-фенил]хинолин
[6-(4-метил-1Н-имидазол-1-ил)-2-(4-метоксифенил)]хинолин
[6-(4-метил-1Н-имидазол-1-ил)-2-(4-фторфенил)]хинолин
[6-(4-метил-1Н-имидазол-1-ил)-2-(4-метилтиофенил)]хинолин.
Названные выше соединения формулы (I), когда W представляет собой гетероарил, определенный выше, не охватываются структурной формулой соединений формулы (I) в предыдущей заявке данных авторов WO 2008/014822.
В следующем варианте осуществления данное изобретение относится к этим новым соединениям формулы (I), где W представляет собой гетероарил, определенный выше, их фармацевтически приемлемым солям и сольватам, соответствующей фармацевтической композиции и их применению для фармакологического лечения тех заболеваний, которые детализированы для соединений формулы (I).
Согласно данному изобретению соединения формулы (I) могут быть использованы в виде свободного основания, в виде фармацевтически приемлемой соли или в виде сольватной или гидратной формы. Соли соединений формулы (I) представляют собой фармацевтически приемлемые аддитивные соли с неорганическими и органическими кислотами. Репрезентативные, но не ограничивающие примеры неорганических солей соединений формулы (I) представляют собой гидрохлорид, гидросульфат, сульфат, гидрофосфат и фосфат. Соответствующие репрезентативные, но не ограничивающие примеры органических солей представляют собой метансульфонат, малеат, сукцинат, фумарат, тартрат, малонат и оксалат.
Способы получения соединений формулы (I) широко описаны в предыдущей заявке данных авторов WO 2008/014822, однако для тех соединений формулы (I), где имидазолильная группа замещена (R1 не является водородом), часто получены очень низкие выходы и сложные реакционные смеси, когда использованы способы получения соединений формулы (I), опубликованные в WO 2008/014822. В другом варианте осуществления данное изобретение относится к новым, более практичным и полезным способам для получения соединений формулы (I), отличающимся более высокими средними выходами и более простыми процедурами для выделения и очистки продукта.
В другом варианте осуществления данное изобретение относится к фармацевтическим композициям для соединений формулы (I), применимым для фармакологического лечения тех заболеваний, которые детализированы выше. В объеме данного изобретения термин «фармацевтическая композиция (лекарственный продукт)» относится к любой пероральной или парентеральной лекарственной форме, подходящей для лечения вышеприведенных патологий, которая содержит эффективное количество по меньшей мере одного из активных ингредиентов (лекарственных веществ), соединений формулы (I), их солей или сольватов и фармацевтически приемлемый носитель, эксципиенты или разбавители, определенные ниже, для перорального или парентерального введения.
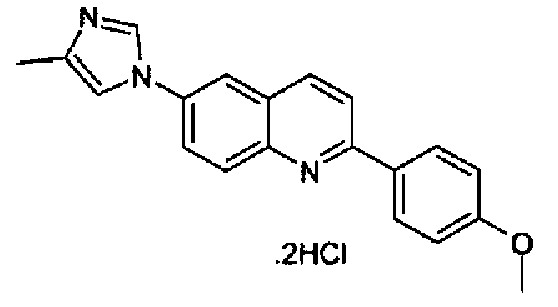
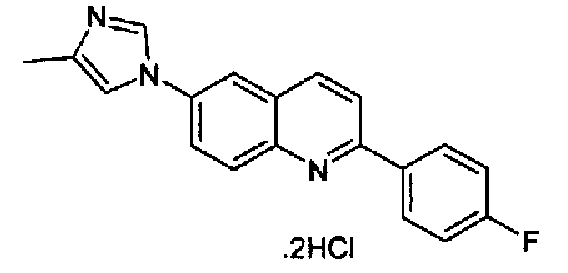
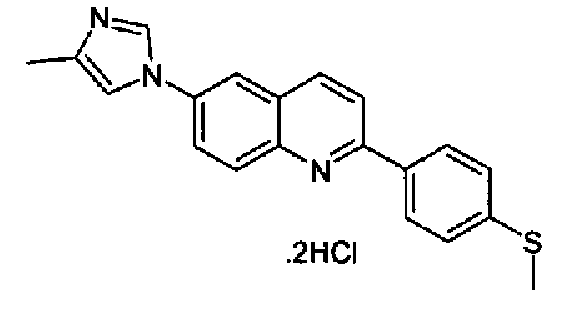
Репрезентативные, но не ограничивающие примеры соединений формулы (I) представлены в таблице 1.
Получение соединений изобретения
Соединения формулы (I) могут быть получены, как описано в WO 2008/014822, взаимодействием соединения формулы (III) с производным имидазола формулы (IV), как указано на схеме 1, в которой X, W и R1 имеют те же значения, которые определены выше для соединений формулы (I), и Hal представляет собой атом галогена, такой как фтор, хлор, бром и йод, обычно фтор или бром.
Схема 1:
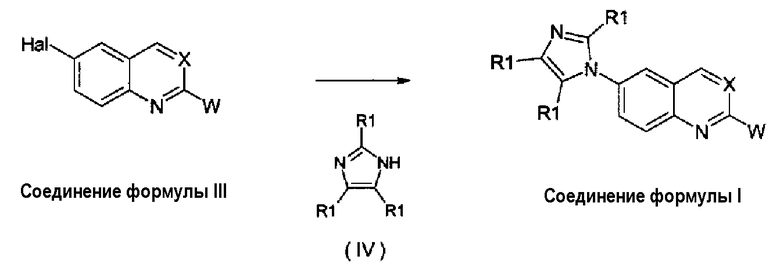
Взаимодействие соединения формулы (III) может быть осуществлено использованием производного имидазола формулы (IV) либо в виде свободного основания, либо в виде его соли со щелочным металлом (соль натрия, лития или калия) согласно общим условиям реакции, описанным в WO 2008/014822, или, точнее, использованием CuI или Cu2O в качестве катализатора, диметилэтилендиамина или 4,7-диметокси-1,10-фенантролина в качестве лигандов и диглима в качестве растворителя, карбоната цезия в качестве основания при температуре примерно 150°С в течение 20-50 часов.
Когда Х представляет собой атом азота, соединения формулы (III) могут быть получены из известных диаминов формулы (V), как показано на схеме 2.
Схема 2:
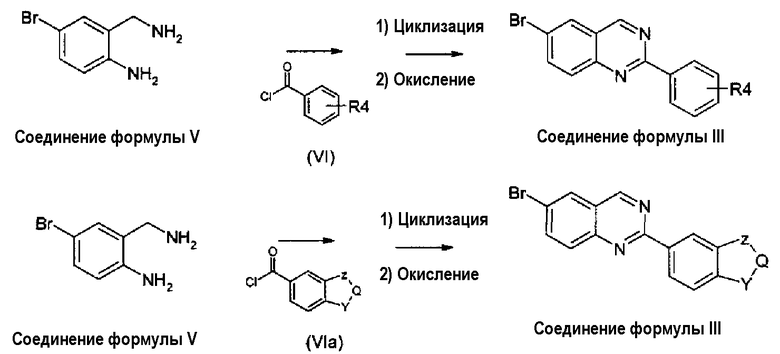
Там, где Y, Q и Z имеют те же значения, как для соединений формулы (I), R4 представляет собой любой из заместителей, данных выше в качестве заместителей для арильной группы в соединениях формулы (I). Соединения формулы (V) получены согласно известным способам, соединения формулы (VI) и (VIa) представляют собой известные соединения или получены согласно известным способам. Для стадий циклизации и окисления могут быть использованы условия реакций, описанные ранее в WO 2008/014822, однако более высокие выходы могут быть получены для большинства случаев при применении условий реакции, сообщенных в примере 1. Данная улучшенная синтетическая процедура также состоит из более простых операций, таким образом приводящих к более практическому синтетическому способу.
В альтернативном случае, соединение формулы (III), где X представляет собой атом азота (-N), может быть получено циклизацией диамина формулы (V) с ортоэфиром формулы (VII) или (VIIa), как описано на схеме 3.
Схема 3:
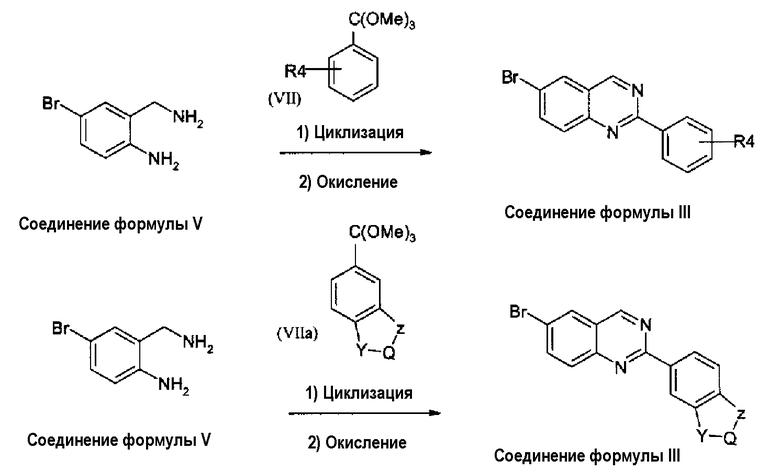
где R4, Y, Q и Z принимают значения, которые описаны выше. Реакцию циклизации ортоэфиров формулы (VII) и (VIIa) с бисаминами формулы (V) выполняют в толуоле или другом инертном органическом растворителе, используя кислотный катализатор, обычно п-толуолсульфоновую кислоту при температуре кипения с обратным холодильником в течение примерно 50 часов. Стадия окисления может быть выполнена при использовании MnO2 в дихлорметане.
В альтернативном случае, соединения формулы (I), где X представляет собой атом азота (-N), могут быть получены циклизацией диамина формулы (VIII) с солями Пиннера формулы (IX) или (IXa), как описано на схеме 4.
Схема 4:
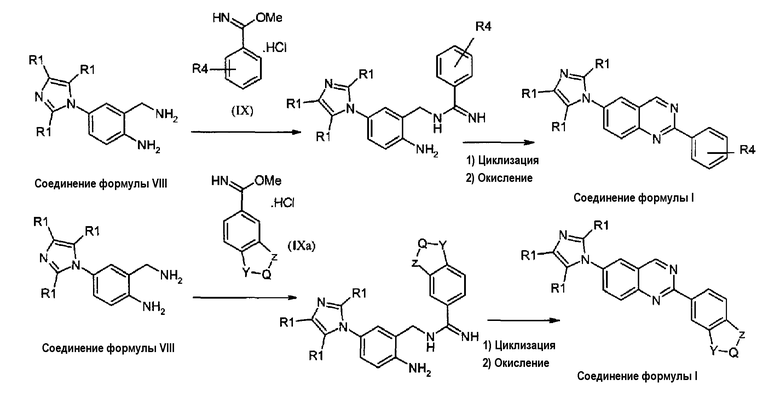
Реакция конденсации и циклизации бисаминов формулы (VIII) с солями Пиннера формулы (IX) или формулы (IXa), где R4, Y, Q и Z принимают значения, указанные выше, может быть осуществлена нагреванием реакционной смеси в спиртовом растворителе, таком как метанол, этанол или пропанол, при температуре кипения с обратным холодильником в течение примерно 1 часа. Образовавшийся промежуточный амидин затем циклизуется в соответствующий дигидрохиназолин нагреванием в уксусной кислоте. Окисление дигидрохиназолинового промежуточного продукта до соответствующего соединения формулы (I) достигается использованием MnO2 в инертном органическом растворителе, таком как дихлорметан.
Соли Пиннера формулы (IX) и (IXa) получают известными процедурами, обычно барботированием безводной хлористоводородной кислоты в спиртовой раствор соответствующего нитрила при температуре от -20°С до 0°С. Образовавшуюся соль Пиннера кристаллизуют из простого эфира, обычно трет-бутилметилового эфира.
Соединения формулы (VIII) получают согласно схеме 5, восстановлением нитрила формулы (X), который, в свою очередь, получают восстановлением соответствующего нитропроизводного формулы (XI), где R1 принимает значения как для соединений формулы (I). Соединения формулы (XI) получают нуклеофильным замещением на 5-фтор-2-цианонитробензоле с помощью имидазолилпроизводного формулы (IV).
Схема 5:
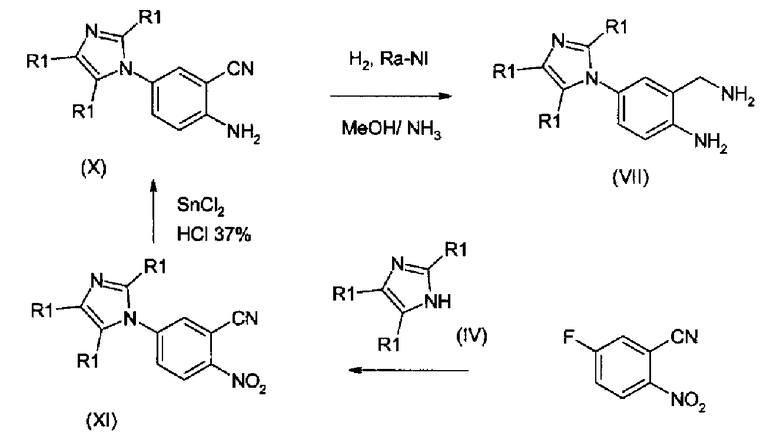
Каталитическое восстановление соединения формулы (X) для получения соединения формулы (VII) может быть выполнено с использованием в качестве катализатора никеля Ренея при давлении водорода примерно 60 бар/0,6 МПа, в метаноле или этаноле, содержащем примерно 10% аммиака (газ), при температуре 30-60°С. Превращение цианопроизводного формулы (XI) в соединение формулы (X) может быть осуществлено при использовании SnCl2 в концентрированной HCl при температуре, изменяющейся от -10°С до 0°С. Производные формулы (XI) получают взаимодействием 5-фтор-2-цианонитробензола с имидазолилпроизводными формулы (IV), в органическом растворителе, обычно ацетонитриле, при температуре 50-90°С. Когда заместитель R1 в соединении формулы (IV) находится в положении -4 и заместители R1 в других положениях представляют собой атомы водорода, могли быть получены региоизомеры соединений формулы (XI). Данные региоизомеры могут быть разделены колоночной хроматографией и/или кристаллизацией.
Соединения формулы (I), где X представляет собой -CH группу, могут быть получены из соединения формулы (XII) взаимодействием с боронатом формулы (XIII) или (XIIIa), как дано на схеме 6.
Схема 6:
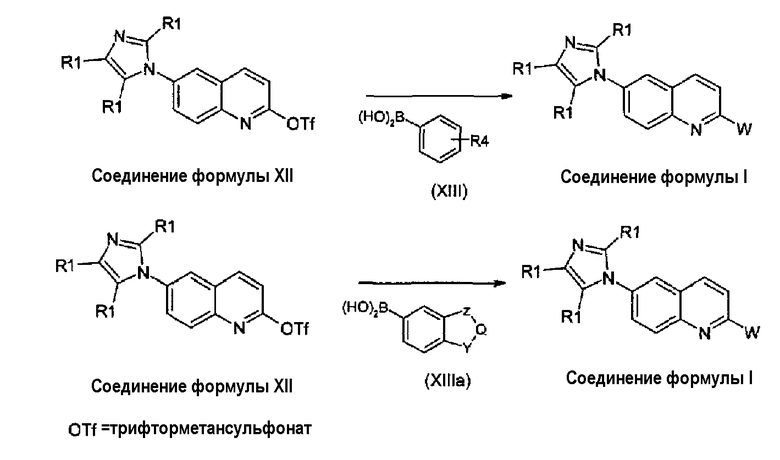
где R1, R4, W, Y, Z и Q принимают значения, которые описаны выше. Аналогичный подход, который использует конденсацию Сузуки для получения соединений формулы (I), определенной в настоящем описании, но исходя из производных 2-хлор-6-имидазолилхинолина, ранее был опубликован в патентной заявке данных авторов WO 2008/014822. Однако использование трифлатной группы вместо атома хлора, как опубликовано ранее, заметно повышает выходы реакции конденсации, а также имеются более высокие выходы по приготовлению соединений формулы (XII) в сравнении с соответствующими 2-хлорпроизводными. Взаимодействие соединения формулы (XII) с боронатом формулы (XIII) или (XIIIa) проводят в инертном органическом растворителе, таком как толуол, диметоксиэтан или тетрагидрофуран, в присутствии основания, такого как карбонат калия или карбонат цезия, с палладиевым катализатором. Тетракистрифенилфосфинпалладий или соль палладия и соответствующий лиганд могут быть использованы в качестве катализатора. Соединения формулы (XIII) или (XIIIa) являются коммерчески доступными соединениями или могут быть получены согласно способам, хорошо известным в данной области.
В альтернативном случае, соединения формулы (I) могут быть получены взаимодействием соединений формулы (XII) с арилгалогенидами, обычно арилбромидами, производными формулы (XIIIb) и (XIIIc) согласно реакции Штилле (Tetrahedron Letters, 36, 50, 9085, 1995), как показано на схеме 6а.
Схема 6а:
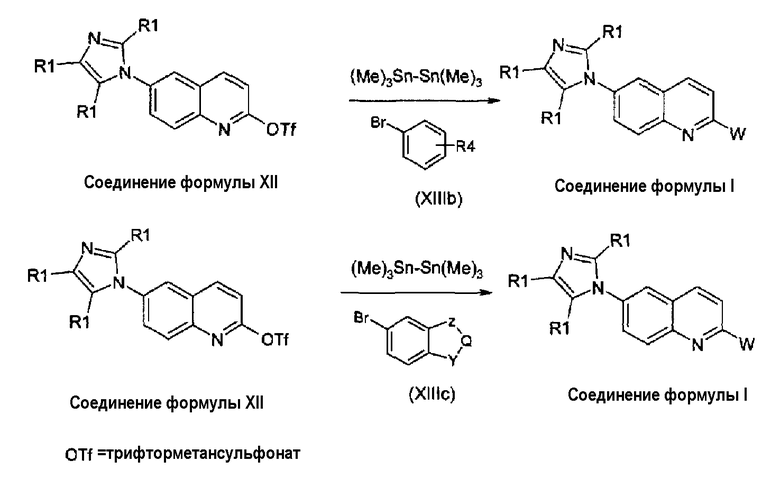
Реакция может быть выполнена или с бис(триметил)оловом или с бис(трибутил)оловом при использовании в качестве катализатора: тетракис(трифенилфосфин)палладия или трис(дибензилиденацетон)дипалладия или дихлорбис(трифенилфосфин)палладия, в присутствии хлорида лития или фторида калия, в растворителе, таком как диоксан, тетрагидрофуран, диметоксиэтан или толуол. Арилбромиды формулы (XIIIb) и (XIIIc) коммерчески доступны или могут быть получены согласно известным способам.
Соединения формулы (XII), где R1, R4, W, Y, Z и Q принимают значения, как описано выше, получены, как обозначено на схеме 7, из 2-хинолинонов формулы (XIV). 2-хинолиноны формулы (XIV) получены из соответствующих производных 2-метоксихинолина формулы (XV), которые, в свою очередь, получены из 6-бромпроизводных формулы (XVI) взаимодействием с производными имидазола формулы (IV). 2-Метокси-6-бромхинолин, соединение формулы (XVI), является известным соединением (RN: 99455-07-7).
Схема 7:
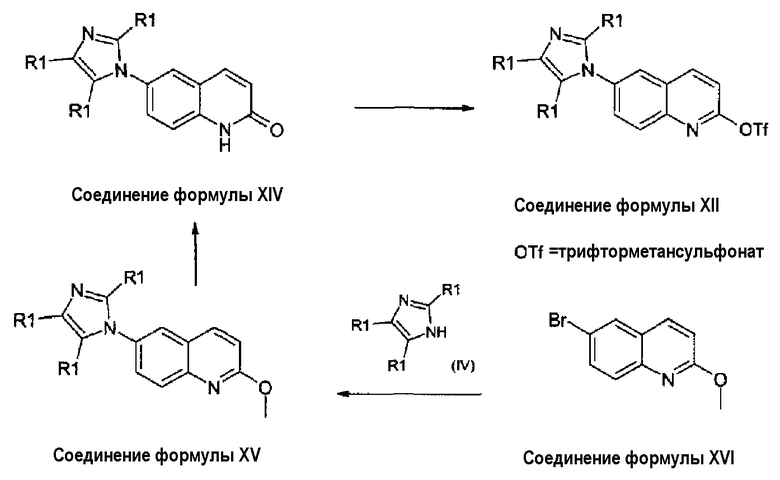
Получение соединения формулы (XII) из соединения формулы (XIV) может быть осуществлено в пиридине с применением ангидрида трифторметансульфоновой кислоты или трифторметансульфонилхлорида при 0°С/-10°С, или в дихлорметане при использовании органического основания, такого как триэтиламин или диизопропилэтиламин. В альтернативном случае, может быть использован бис-трифторметиланилид в диметилформамиде с применением гидрида натрия (NaH) в качестве основания. Превращение соединения формулы (XV) в соединение формулы (XIV) достигнуто применением хлористоводородной или бромистоводородной кислоты при температурах, изменяющихся от 25°С до температуры кипения с обратным холодильником. В альтернативном случае может быть использован BBr3 в дихлорметане. Соединения формулы (XV) получены взаимодействием 6-бром-2-метоксихинолина, формула (XVI), с имидазолом или замещенными имидазолами формулы (IV). Реакция может быть проведена с использованием соединений формулы (IV) в виде свободного основания или соответствующей соли щелочного металла, в присутствии подходящего катализатора, в растворителе, таком как диметилформамид (ДМФА/DMF), диметилсульфоксид (ДМСО/DMSO), ацетонитрил, N-метилпирролидон (NMP), диметоксиэтан, тетрагидрофуран (ТГФ/THF), толуол или ксилол, при температуре, изменяющейся от 50°С до температуры кипения с обратным холодильником. В качестве катализатора может быть использован медный катализатор, такой как CuI, смесь Cu/CuO или комплекс Cu(OTf)2·бензол, необязательно в присутствии лигандов, таких как 8-гидроксихинолин, 1,10-фенантролин, диметилэтилендиамин, дибензилиденацетон. Обычно используют основание, такое как карбонат калия, карбонат цезия, карбонат триэтиламмония. В качестве катализатора может быть также применен палладий, обычно методология Бучвальда-Хартвига (Buchwald-Hartwig) для добавления имидазола к арилбромидам, в ДМФА/DMF как растворителе, с применением либо бинапа [2,2'-бис(дифенилфосфино)-1,1'-бинафтил], либо Dppf [1,3-бис(дифенилфосфинопропан)]палладия как растворимых катализаторов, и трет-бутилата калия в качестве основания при микроволновом нагревании может быть использована для получения соединений формулы (XV).
Когда производное имидазола формулы (IV) замещено (например, R1: метил, трифторметил, гидроксиметил), соединения формулы (XIV) могут быть получены с высоким выходом циклизацией соединения формулы (XVII), как показано на схеме 8. Соединения формулы (XVII) получены из анилинов формулы (XVIII), которые, в свою очередь, получены восстановлением соединений формулы (XIX). Соединения формулы (XIX) получены взаимодействием коммерчески доступного 4-фторнитробензола с имидазолами формулы (IV).
Схема 8:
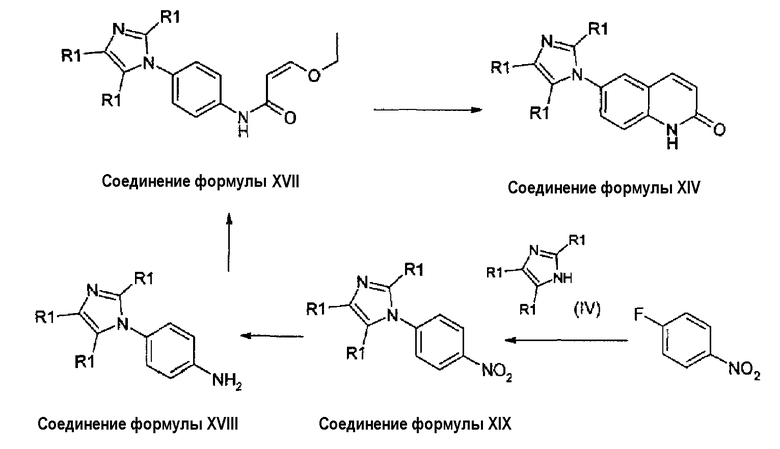
Циклизация соединения формулы (XVII) в соединение формулы (XIV) может быть осуществлена перемешиванием енольного эфира в минеральной кислоте (хлористоводородная или серная кислота), при этом температура изменяется от -10°С до +25°С. В альтернативном случае циклизация может быть проведена в инертном органическом растворителе, таком как дихлорметан, диметоксиэтан или толуол, с использованием кислоты Льюиса как катализатора. Соединения формулы (XVII) могут быть получены взаимодействием соединений формулы (XVIII) с 3-этоксиакрилоилхлоридом в пиридине или в дихлорметане в присутствии триэтиламина. Восстановление соединений формулы (XIX) может быть осуществлено при использовании SnCl2 в спирте (этанол или метанол) или каталитически с применением водорода и Pd/C или PtO2 в качестве катализатора. Соединения формулы (XIX) получены из 4-фторнитробензола и имидазолильных производных формулы (IV) согласно способам, описанным выше.
В альтернативном случае соединение формулы (I), где X представляет собой -CH группу или атом азота (-N), могут быть получены из соединения формулы (XX) взаимодействием с глиоксалем или дикарбонильным производным формулы (XXI) в присутствии формальдегида или альдегида формулы R1CHO и хлорида аммония, как дано на схеме 9.
Схема 9:
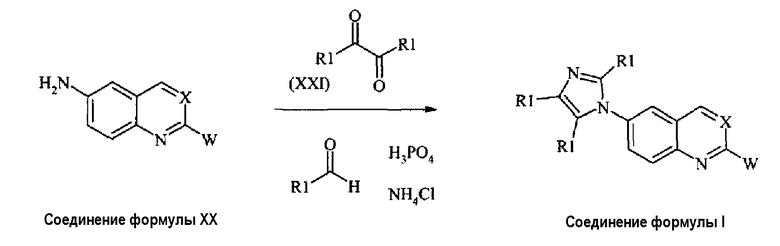
где Х, W и R1 имеют те же значения, как обсуждено выше для соединений формулы (I).
Соединения формулы (I), где все R1 представляют собой атомы водорода, могут быть получены обработкой соединений формулы (ХХ) глиоксалем, в метаноле, обычно при комнатной температуре, затем добавлением NH4Cl и формальдегида и нагреванием при кипении с обратным холодильником, в конце добавляют фосфорную кислоту. Соединения формулы (I), где имидазол замещен, могут быть получены использованием подобной процедуры, но с применением дикарбонильного соединения формулы (XXI) (где по меньшей мере один R1 представляет собой не водород) вместо глиоксаля, альдегид формулы R1CHO также может быть использован (Synthesis, 2003, 2661-2666).
Неограничивающие репрезентативные примеры для приготовлений соединений формулы (I) представлены ниже.
Пример 1: [6-(1H-имидазол-1-ил)-2-фенил]хиназолин
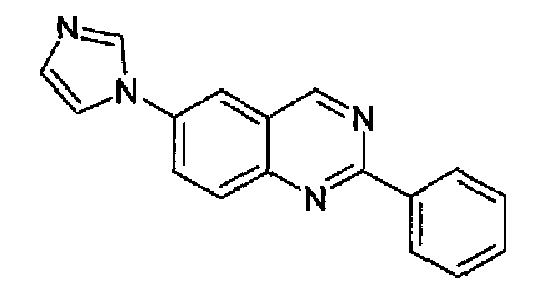
CuI (6,6 г, 0,034 моль) и диметилэтилендиамин (8,67 мл, 0,07 моль) в инертной атмосфере добавляли к 700 мл диглима при комнатной температуре (к.т.). После перемешивания в течение нескольких минут получали суспензию, к данной суспензии добавляли 6-бром-2-фенилхиназолин (62,5 г, 0,228 моль) и имидазол (31,2 г, 0,456 моль, 2 экв.), затем Cs2CO3 (74,7 г, 0,023 моль). Полученную реакционную смесь нагревали при 150°С при перемешивании в течение 46 часов. После охлаждения реакционную смесь доводили до к.т. и разбавляли водным насыщенным раствором NH4Cl (3,5 л). Добавляли этилацетат (AcOEt), отделяли органическую фазу и водную фазу экстрагировали с помощью AcOEt, собранные органические фазы промывали водой, фильтровали, сушили и концентрировали. Остаток, растворенный в смеси AcOEt/метанол (МеОН) (95:5), фильтровали через силикагель, концентрировали и кристаллизовали из смеси МеОН/гексан с получением указанного в заголовке продукта (48,7 г, выход 78%). C17H12N4; М.м./мол.масса/MW: 272,31; т.пл. 153,8-158,7°С; 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 7,23 (с, 1Н), 7,58-7,62 (м, 3Н), 8,00 (с, 1Н), 8,23 (д, 1Н), 8,39-8,63 (м, 5Н), 9,72 (с, 1Н). ИК (KBr): 1556, 1506, 1379.
6-Бром-2-фенилхиназолин
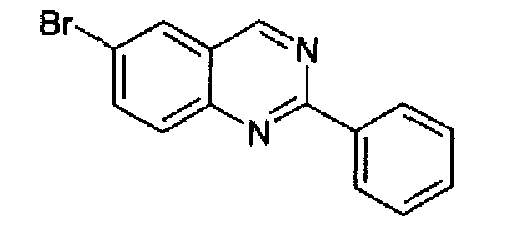
К дихлорметану (DCM) (3,5 л) добавляли 5-бром-2-аминобензиламин (137 г, 0,5 моль) и триэтиламин (ТЕА) (250 мл, 1,75 моль) при 0°С при перемешивании. Затем при перемешивании добавляли бензоилхлорид (55 мл, 0,45 моль) в DCM (500 мл) с такой скоростью, чтобы поддерживать температуру при 0-5°С. Смесь перемешивали в течение 3 часов при к.т. Добавляли воду (1 л) и органическую фазу отделяли, промывали водой и сушили. Растворитель выпаривали и к остатку (147,5 г), суспендированному в толуоле (1,5 л), добавляли SOCl2 (100 мл). Полученную суспензию нагревали при кипении с обратным холодильником в течение 72 часов. При охлаждении получался осадок, его фильтровали, промывали толуолом и суспендировали в водном аммиаке, данную суспензию экстрагировали с помощью AcOEt. Объединенные органические фазы промывали водой, сушили и концентрировали с получением дигидрохиназолинового производного в виде светло-коричневого твердого вещества (93,8 г, выход 64%). Данный дигидрохиназолин растворяли в DCM (2 л) и добавляли с перемешиванием MnO2 (56,28 г). Образовавшуюся суспензию перемешивали при к.т. в течение 18 часов. Суспензию фильтровали через целит, осадок промывали с помощью DCM и объединенный фильтрат и промывочные порции концентрировали с получением указанного в заголовке продукта в виде аморфного твердого вещества, 85,54 г (общий выход 60%; выход по стадии окисления 95%). C14H9BrN2; М.м.: 285,15; МС m/z: 286 (M+1); 1Н-ЯМР (300 МГц, d6-ДМСО) м.д.: 7,58-7,61 (м, 3Н), 8,02 (д, 1Н), 8,17 (дд, 1Н), 8,49-8,56 (м, 3Н), 9,70 (с, 1Н).
6-Бром-2-фенилхиназолин (циклизация с применением сложного ортоэфира триметилбензойной кислоты)
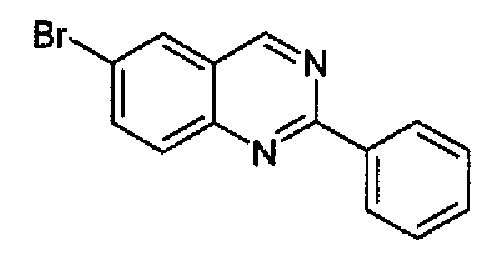
К толуолу (200 мл) добавляли 5-бром-2-аминобензиламин (9,5 г, 47,2 ммоль) и сложный ортоэфир триметилбензойной кислоты (8,2 г, 47,2 ммоль), затем п-толуолсульфоновую кислоту (1,35 г, 7,1 ммоль). Полученную суспензию перемешивали при кипении с обратным холодильником в течение 50 часов. Реакционную смесь охлаждали при к.т., разбавляли с помощью AcOEt (150 мл), промывали насыщенным бикарбонатом натрия, затем водой. Органический слой сушили и концентрировали с получением промежуточного дигидрохиназолина в виде светло-коричневого твердого вещества (8,5 г; 63%). Данный промежуточный продукт растворяли в DCM (20 мл) при к.т., затем добавляли MnO2 (5,1 г). Образовавшуюся смесь перемешивали при к.т. в течение 48 часов, затем фильтровали через целит. Фильтрат концентрировали с получением указанного в заголовке продукта в виде белого твердого вещества (8,1 г, 95%). C14H9BrN2; М.м.: 285,15; МС m/z: 286 (M+1). 1Н-ЯМР (300 МГц, d6-ДМСО) м.д.: 7,58-7,61 (м, 3Н), 8,02 (д, 1Н), 8,17 (дд, 1Н), 8,49-8,56 (м, 3Н), 9,70 (с, 1Н).
5-Бром-2-аминобензиламин
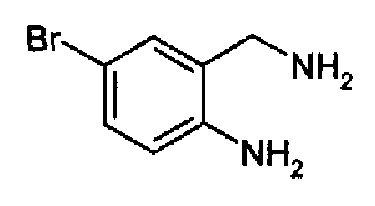
Раствор борана в ТГФ (1М, 400 мл) добавляли при 0°С к суспензии 5-бромантранилонитрила (60 г, 0,304 моль, полученный, как описано в S.M. Mackenzie et al., J. Chem. Soc. C, 1970, 17, 2298-2308) в ТГФ (450 л) в атмосфере N2. Смесь перемешивали в течение 72 часов при к.т. После охлаждения при 0°С добавляли абсолютный EtOH, затем через раствор барботировали HCl. Смесь концентрировали и остаток суспендировали в изопропиловом простом эфире. Полученное твердое вещество сушили с получением дигидрохлорида указанного в заголовке продукта (76,6 г, выход 91,4%). C7H9BrN2·2HCl, М.м.: 273,9; 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 4,13 (c, 2Н), 5,82 (c, 4Н), 7,24 (д, 1Н), 7,55 (дд, 1Н), 7,73 (с, 1Н), 8,57 (с, 2Н). Поскольку свободное основание используют на стадии циклизации, данный гидрохлорид суспендировали в водном аммиаке, перемешивали в течение нескольких минут, после чего осаждается свободное основание. Твердое вещество фильтровали и сушили (выход количественный).
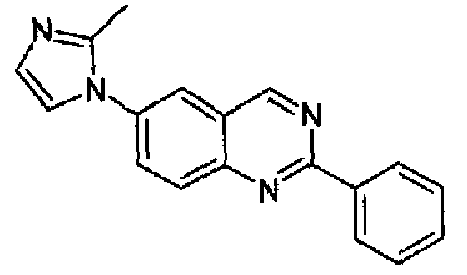
Пример 2: [6-(2-метил-1H-имидазол-1-ил)-2-фенил]хиназолин
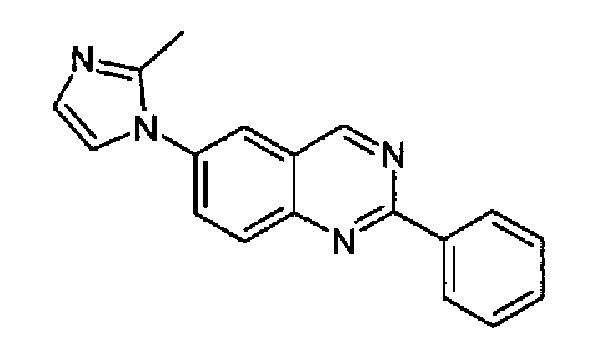
6-Бром-2-фенилхиназолин (1,43 г, 5,0 ммоль) и 2-метилимидазол (0,50 г, 6 ммоль) смешивали с PEG 400 (d:1,126, 1,0 г, 885 мкл) и 4,7-диметокси-1,10-фенантролином (186 мг, 0,75 ммоль), к данной смеси добавляли Cu2O (38,5 мг, 0,25 ммоль) и Cs2CO3 (2,29 г, 7,0 ммоль). Полученную реакционную смесь нагревали при 110°С в атмосфере аргона в течение 24 часов. После охлаждения при к.т. смесь разбавляли с помощью DCM (50 мл) и фильтровали через целит, осадок промывали с помощью DCM и объединенный фильтрат и промывочные порции упаривали досуха. Остаток очищали хроматографически (SiO2, EtOAc/MeOH 95:5). Очищенное указанное в заголовке соединение выделяли в виде бледно-желтого твердого вещества, 1,02 г (выход: 71%), т.пл.: 198,3-200,3°С. C18H14N4, М.м.: 286,34; МС: m/z 287 (M+Н); 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 2,40 (с, 1Н), 7,0 (с, 1Н), 7,40 (с, 1Н), 7,60 (м, 3Н), 8,10-8,30 (м, 3Н), 8,60 (м, 2Н), 9,80 (с, 1Н).
В альтернативном случае, 6-(2-метил-1H-имидазол-1-ил)-2-фенилхиназолин может быть получен из 4-(2-метил-1Н-имидазол-1-ил)-2-аминометиланилина:
Пример 2 (В): 6-(2-метил-1H-имидазол-1-ил)-2-фенилхиназолин (альтернативный способ)
4-(2-метил-1Н-имидазол-1-ил)-2-аминометиланилин (2,0 г, 10 ммоль) и гидрохлорид метилбензимидата (3,5 г, 20 ммоль; RN: 5873-90-5, Aldrich) растворяли в метаноле (50 мл), полученную смесь нагревали при кипении с обратным холодильником в течение 2 часов, за данное время аминометилпроизводное превращалось в соответствующий бензамидин. После этого метанол выпаривали и остаток поглощали ледяной уксусной кислотой (50 мл), реакционную смесь нагревали при кипении с обратным холодильником в течение 1,5 часов. После охлаждения при к.т. реакционную смесь разбавляли толуолом (50 мл) и упаривали. Остаток поглощали с помощью AcOEt (400 мл), промывали водным аммиаком, водой, затем сушили и концентрировали. Полученный маслянистый остаток растворяли в DCM (400 мл) и добавляли MnO2 (6,0 г, 70 ммоль) при к.т., тремя порциями, в продолжение 2 часов. Полученную суспензию перемешивали при к.т. в течение 24 часов, затем фильтровали через целит и осадок промывали с помощью DCM. Объединенный фильтрат и промывочные порции концентрировали и остаток хроматографировали на силикагеле (DCM/MEOH/NH3, 85:25:2), соответствующие объединенные фракции упаривали и остаток поглощали простым этиловым эфиром, нагревали при кипении с обратным холодильником в течение 5 минут, затем охлаждали при 25°С для кристаллизации указанного в заголовке продукта в виде бледно-коричневого порошка (2,0 г; выход: 74%). C18H14N4, М.м.: 286,34; МС: m/z 287 (M+H); 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 2,40 (с, 1Н), 7,0 (с, 1Н), 7,40 (с, 1Н), 7,60 (м, 3Н), 8,10-8,30 (м, 3Н), 8,60 (м, 2Н), 9,80 (с, 1Н).
Пример 3: [6-(2-метил-1H-имидазол-1-ил)-2-(4-метоксифенил)]хиназолин
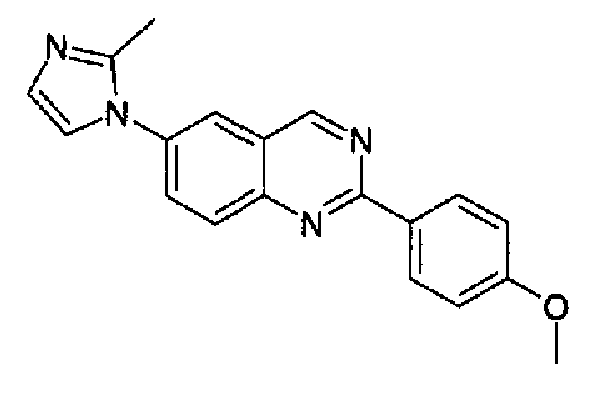
Получен аналогично с выходом 69%, исходя из 4-(2-метил-1Н-имидазол-1-ил)-2-аминометиланилина (2,0 г, 10 ммоль) и гидрохлорида метил(4-метокси)бензимидата. Бледно-серый порошок, т.пл.: 198,3-200,3°С. C19H16N4O, М.м.: 316,37. МС: m/z 317 (M+1). 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 2,42 (с, 3Н), 3,33 (с, 3Н), 7,01 (с, 1Н), 7,50 (с, 1Н), 7,59-7,63 (м, 2Н), 8,11-8,32 (м, 3Н), 8,58-8,63 (м, 2Н), 9,79 (с, 1Н). FT-IR (ATR) см-1: 1624, 1588, 1557, 1496, 1414, 1300, 1271, 1165, 843, 761.
Дигидрохлорид 4-(2-метил-1Н-имидазол-1-ил)-2-аминометиланилина
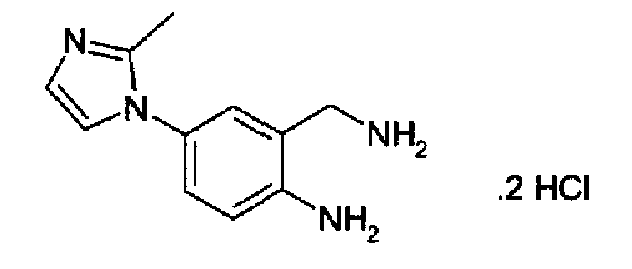
К 4-(2-метил-1Н-имидазол-1-ил)-2-цианоанилину (7,8 г; 39 ммоль), растворенному в 10% NH3/метаноле (70 мл), добавляли никель Ренея (2 г), полученную смесь гидрировали при 60°С при давлении водорода 60 бар/6 МПа в течение 12 часов. Реакционную смесь после продувания азотом фильтровали на целите, осадок промывали метанолом и объединенный фильтрат и промывочные порции упаривали, остаток растворяли в метаноле, фильтровали и барботировали HCl при 0°С с получением указанного в заголовке продукта в виде желто-оранжевого твердого вещества (6,1 г, 60%). C10H12N4·2HCl, М.м.: 263,23. МС: m/z 202 (M+1). 1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,20 (с, 3Н), 3,64 (с, 2Н), 5,35 (с, 2Н), 6,68 (д, 1Н), 6,82 (д, 1Н), 6,94 (дд, 1Н), 7,05-7,07 (м, 2Н). Свободное основание на вышеприведенной стадии получали суспендированием дигидрохлорида в концентрированном аммиаке, перемешиванием суспензии 5 мин, затем фильтрованием осадка, который промывали водой и сушили.
4-(2-метил-1Н-имидазол-1-ил)-2-цианоанилин
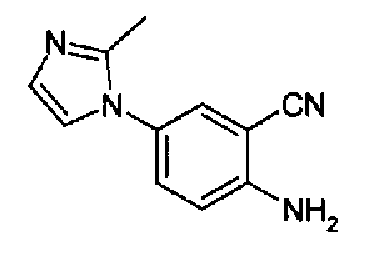
SnCl2·2H2O (60,0 г, 0,26 моль) растворяли в 37% HCl (100 мл), к данному раствору, охлажденному до -10°С, добавляли 4-(2-метил-1Н-имидазол-1-ил)-2-цианонитробензол (12,0 г, 50 ммоль) в две порции в течение 30 мин. По окончании добавлений перемешиваемой реакционной смеси позволяли принимать к.т., и после дополнительных 45 мин перемешивания ее выливали в лед/воду (250 г) и 3н КОН (500 мл). Полученную суспензию фильтровали и осадок промывали водой. Осадок суспендировали в 2М NH3/EtOH (250 мл), перемешивали в течение нескольких минут и фильтровали, фильтрат концентрировали с получением указанного в заголовке соединения в виде коричневого твердого вещества (8 г, 78%). C11H10N4, М.м.: 198,23. МС: m/z 199 (M+1). 1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,21 (с, 3Н), 4,70 (с, 2Н), 6,79 (д, 1Н), 6,97 (д, 1Н), 7,22 (дд, 1Н), 7,29 (д, 1Н).
4-(2-метил-1Н-имидазол-1-ил)-2-цианонитробензол
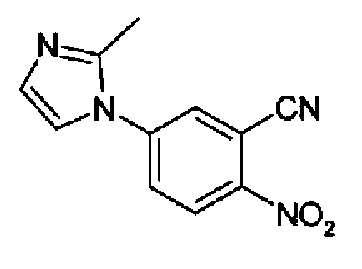
2-Циано-4-фторнитробензол (9,8 г, 59 ммоль) и 2-метилимидазол (1,45 г, 177 ммоль) растворяли в сухом ацетонитриле (300 мл), затем реакционную смесь нагревали при 90°С в течение 5 часов.
Раствор охлаждали при к.т. и выпаривали растворитель, остаток распределяли между AcOEt/0,5н HCl (5/1), отделенную органическую фазу промывали водой, насыщенным солевым раствором и затем упаривали. Оранжевый остаток кристаллизовали из смеси ацетон/гексан с получением 12,8 г (95%) указанного в заголовке соединения. Когда используют не сухой ацетонитрил, получают некоторое количество амида в качестве побочного продукта.
1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,47 (с, 3Н), 7,70 (д, 1Н), 7,12 (д, 1Н), 7,73 (дд, 1Н), 7,83 (д, 1Н), 8,46 (д, 1Н).
Пример 4: [6-(4-метил-1H-имидазол-1-ил)-2-фенил]хиназолин
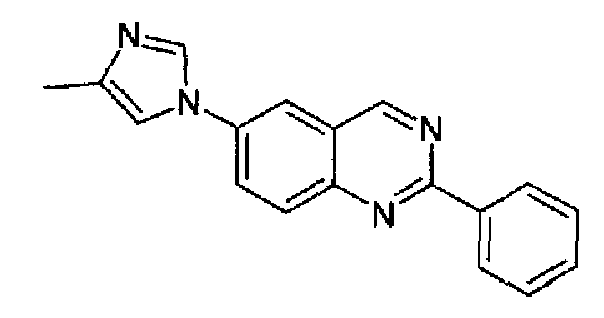
4-(4-метил-1Н-имидазол-1-ил)-2-аминометиланилин (2,0 г, 10 ммоль) и гидрохлорид метилбензимидата (1,73 г, 10 ммоль; RN: 5873-90-5, Aldrich) растворяли в метаноле (15 мл), полученную смесь нагревали при кипении с обратным холодильником в течение 2 часов, за данное время аминометилпроизводное превращалось в соответствующий бензамидин. После этого метанол выпаривали и остаток поглощали ледяной уксусной кислотой (15 мл), реакционную смесь нагревали при кипении с обратным холодильником в течение 2 часов. После охлаждения при к.т. реакционную смесь разбавляли толуолом (50 мл) и упаривали. Остаток поглощали с помощью AcOEt (200 мл), промывали водным аммиаком, затем водой, сушили и концентрировали. Маслянистый остаток растворяли в DCM (200 мл) и добавляли MnO2 (6,0 г, 70 ммоль) при к.т., тремя порциями, в продолжение 2 часов. Полученную суспензию перемешивали при к.т. в течение 22 часов, затем фильтровали через целит и осадок промывали с помощью DCM. Объединенный фильтрат и промывочные порции концентрировали и остаток поглощали простым этиловым эфиром, нагревали при кипении с обратным холодильником в течение 5 минут, затем охлаждали при 25°С для кристаллизации указанного в заголовке продукта в виде не совсем белого порошка (2,3 г; выход: 85%), плавящегося при 201,9-202,8°С. C18H14N4, М.м.: 286,34; МС: m/z 287 (M+Н); 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,21 (с, 3Н), 7,57-7,60 (м, 3Н), 7,65 (с, 1Н), 8,17 (д, 1Н), 8,33-8,38 (м, 3Н), 8,54-8,58 (м, 2Н), 9,68 (с, 1Н). FT-IR (ATR) см-1: 1626, 1585, 1555, 1503, 1442, 1390, 1253, 1060, 838, 711.
Пример 5: [6-(5-метил-1H-имидазол-1-ил)-2-фенил]хиназолин
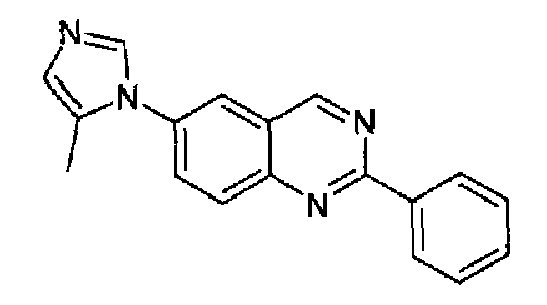
Получен аналогично с выходом 74%, исходя из 4-(5-метил-1Н-имидазол-1-ил)-2-аминометиланилина (2,0 г, 10 ммоль) и гидрохлорида метилбензимидата (1,73 г, 10 ммоль).
Бледно-коричневый порошок, т.пл.: 138,5-139,1°С. C18H14N4, М.м.: 286,34. МС: m/z 287 (M+Н); 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,27 (с, 3Н), 6,91 (с, 1Н), 7,58-7,60 (м, 3Н), 8,11 (с, 1Н), 8,21 (с, 1Н), 8,25 (м, 1Н), 8,29 (с, 1Н), 8,58 (м, 2Н), 9,78 (с, 1Н). FT-IR (ATR) см-1: 1588, 1554, 1490, 1437, 1382, 1232, 1167, 919, 812, 763, 709.
Пример 6: [6-(4-метил-1H-имидазол-1-ил)-2-(4-метоксифенил)]хиназолин
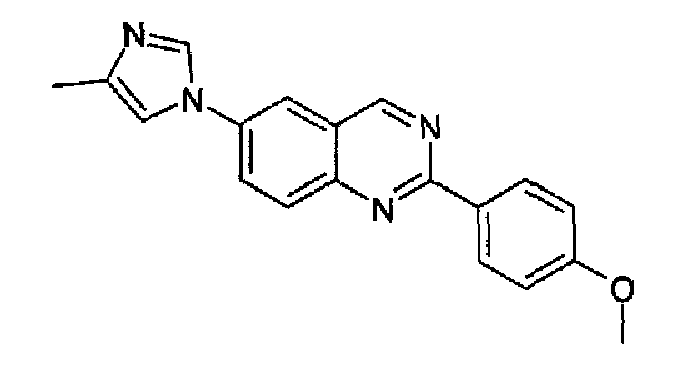
Получен аналогично с выходом 76% из 4-(4-метил-1Н-имидазол-1-ил)-2-аминометиланилина и гидрохлорида метил 4-метоксибензимидата (RN: 39739-49-6). Бесцветные кристаллы, т.пл.: 201,0-202,0°С. C19H16N4O, М.м.: 316,37; МС: m/z 317 (M+Н); 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,21 (с, 3Н), 3,85 (с, 3Н), 7,10 (д, 2Н), 7,62 (с, 1Н), 8,11 (д, 1Н), 8,28-8,33 (м, 3Н), 8,49 (д, 2Н), 9,61 (с, 1Н). FT-IR (ATR) см-1: 1627, 1580, 1515, 1388, 1377, 1252, 1167, 1017, 836.
Пример 7: [6-(4-метил-1H-имидазол-1-ил)-2-(2-метоксифенил)]хиназолин
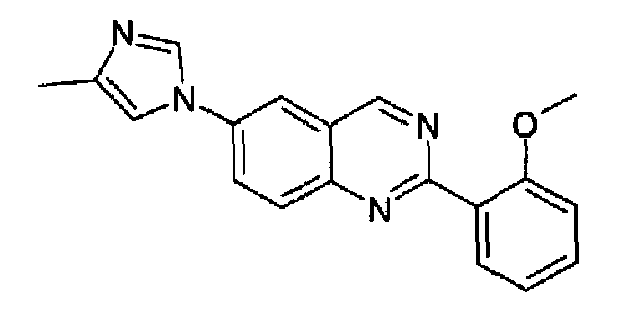
Получен аналогично с выходом 65% из 4-(4-метил-1Н-имидазол-1-ил)-2-аминометиланилина и гидрохлорида метил 2-метоксибензимидата. Бесцветные кристаллы, т.пл.: 160,6-162,0°С. C19H16N4O, М.м.: 316,37; МС: m/z 317 (M+Н); 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,21 (с, 3Н), 3,79 (с, 3Н), 6,98 (д, 1Н), 7,08 (т, 1Н), 7,11 (д, 1Н), 7,50 (т, 1Н), 7,63-7,67 (м, 2Н), 8,14 (д, 1Н), 8,27 (д, 1Н), 8,33-8,40 (м, 1Н), 9,64 (с, 1Н). FT-IR (ATR) см-1: 1560, 1507, 1398, 1243, 1060, 1023, 847, 761.
Пример 8: [6-(4-метил-1H-имидазол-1-ил)-2-(3-метоксифенил)]хиназолин
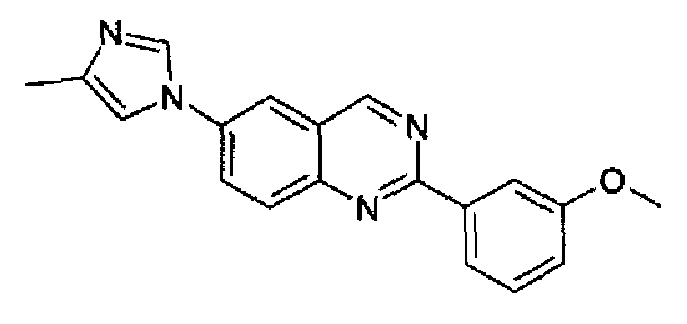
Получен аналогично с выходом 68% из 4-(4-метил-1Н-имидазол-1-ил)-2-аминометиланилина и гидрохлорида метил 3-метоксибензимидата. Бледно-желтый порошок, т.пл.: 294-296°С. C19H16N4O, М.м.: 316,37; МС: m/z 317 (M+Н); 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,22 (с, 3Н), 4,09 (с, 3Н), 7,16 (дд, 1Н), 7,50 (т, 1Н), 7,64 (с, 1Н), 8,11 (с, 1Н), 8,18 (т, 2Н), 8,35-8,40 (м, 3Н), 9,69 (с, 1Н). FT-IR (ATR) см-1: 1627, 1556, 1487, 1451, 1384, 1269, 1211, 1036, 836, 774, 719.
Пример 9: [6-(4-метил-1H-имидазол-1-ил)-2-(1,3-бензодиоксол-5-ил)]хиназолин
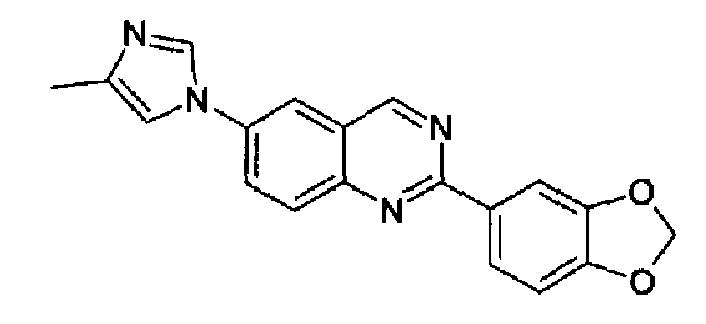
Получен аналогично с выходом 54% из 4-(4-метил-1Н-имидазол-1-ил)-2-аминометиланилина и гидрохлорида метил 1,3-метилендиоксибензимидата. Бесцветные кристаллы, т.пл.: 215,3-218,7°С. C19H14N4O2, М.м.: 330,35; МС: m/z 331 (M+Н); 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,36 (с, 3Н), 6,15 (с, 2Н), 6,98 (д, 1Н), 7,14 (с, 1Н), 7,84 (с, 1Н), 7,94 (м, 2Н), 8,13 (с, 1Н), 8,15 (д, 1Н), 8,27 (д, 1Н), 9,45 (с, 1Н). FT-IR (ATR) см-1: 1557, 1503, 1444, 1380, 1248, 1036, 826.
Пример 10: [6-(4-метил-1H-имидазол-1-ил)-2-(4-фторфенил)]хиназолин
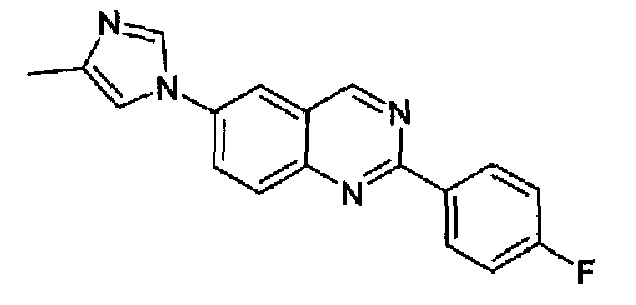
Получен аналогично с выходом 58% из 4-(4-метил-1Н-имидазол-1-ил)-2-аминометиланилина и гидрохлорида метил 4-фторбензимидата. C18H13N4F, М.м.: 304,33; МС: m/z 305 (M+Н); 1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,38 (с, 3Н), 7,18-7,38 (м, 4Н), 7,82-7,95 (м, 3Н), 8,6-8,7 (м, 2Н), 9,50 (с, 1Н). FT-IR (ATR) см-1: 1627, 1602, 1556, 1579, 1512, 1504, 1446, 1390, 1374, 1217, 1159, 1065, 836, 825, 735, 714.
Пример 11: [6-(4-метил-1H-имидазол-1-ил)-2-(4-метансульфонилфенил)]хиназолин
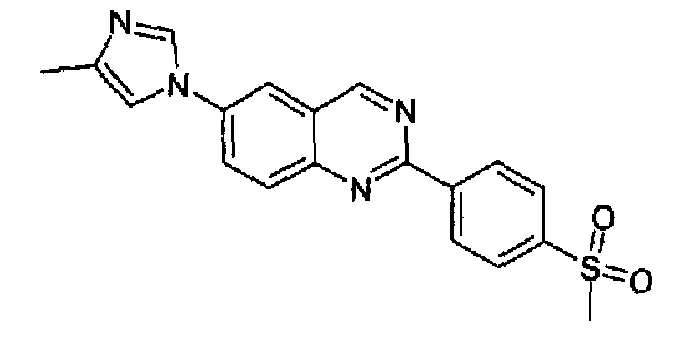
Получен аналогично с выходом 38% из 4-(4-метил-1Н-имидазол-1-ил)-2-аминометиланилина и гидрохлорида метил 4-метансульфонилбензимидата, т.пл.: 276,4-281,7°С. C19H16N4O2S, М.м.: 364,43; МС: m/z 365 (M+H); 1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,39 (с, 3Н), 3,10 (с, 3Н), 7,20 (c, 1Н), 7,90-8,10 (м, 4Н), 8,20 (дд, 4Н), 8,85 (д, 2Н), 9,60 (с, 1Н). 1Н-ЯМР (400 МГц, CDCl3) м.д.: 2,38 (с, 3Н), 3,15 (с, 3Н), 7,21 (c, 1Н), 7,93 (с, 1Н), 7,97 (с, 1Н), 7,98 (дд, 1Н), 8,14 (д, 1Н), 8,26 (д, 1Н), 8,87 (д, 1Н), 9,57 (с, 1Н).
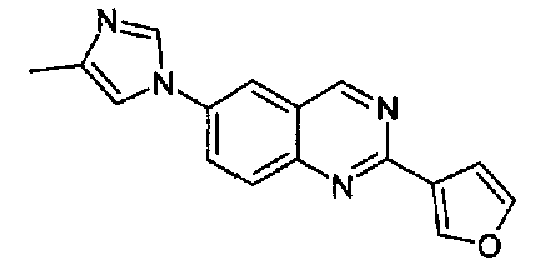
Пример 12: [6-(4-метил-1H-имидазол-1-ил)-2-(3-фурил)]хиназолин
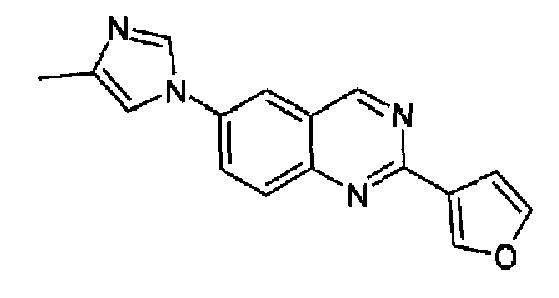
Получен аналогично с выходом 33% из 4-(4-метил-1Н-имидазол-1-ил)-2-аминометиланилина и гидрохлорида метилового сложного эфира 3-фуранкарбоксимидокислоты, т.пл.: 160,5-163,2°С. C16H12N4O, М.м.: 276,30; МС: m/z 277 (M+H); 1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,35 (с, 3Н), 7,15 (д, 2Н), 7,51 (с, 1Н), 7,80 (с, 1Н), 7,90-7,95 (м, 2Н), 8,10 (д, 1Н), 8,40 (с, 1Н), 9,40 (с, 1Н). 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,21 (с, 3Н), 7,17 (c, 1Н), 7,63 (с, 1Н), 7,87 (м, 1Н), 8,08 (д, 1Н), 8,31 (дд, 1Н), 8,35 (с, 1Н), 8,56 (с, 1Н), 9,57 (с, 1Н). FT-IR (ATR) см-1: 1629, 1588, 1576, 1558, 1501, 1379, 1148, 1059, 1007, 862, 815, 723.
4-(4-Метил-1Н-имидазол-1-ил)-2-аминометиланилин
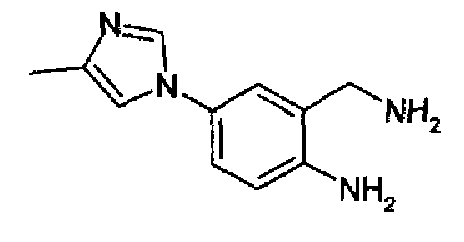
К 4-(4-метил-1Н-имидазол-1-ил)-2-цианоанилину (15,5 г; 78,2 ммоль), растворенному в 10% NH3/метаноле, добавляли никель Ренея (5 г), полученную смесь гидрировали при 60°С при давлении водорода 60 бар/0,6 МПа в течение 24 часов. Реакционную смесь, продуваемую азотом, фильтровали на целите, осадок промывали метанолом и объединенный фильтрат и промывочные порции упаривали. Остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH/2M NH3, 85:10:5), упаривали объединенные соответствующие фракции с получением очищенного указанного в заголовке продукта в виде желто-оранжевого твердого вещества (12,9 г, 82%). C11H14N4, М.м.: 202,26; 1Н-ЯМР (300 МГц, d6-ДМСО) м.д.: 2,13 (с, 3Н), 5,24 (с, 2Н), 6,67 (д, 1Н), 7,08 (дд, 1Н), 7,17 (т, 1Н), 7,23 (д, 1Н), 7,81 (д, 1Н).
4-(5-Метил-1Н-имидазол-1-ил)-2-аминометиланилин
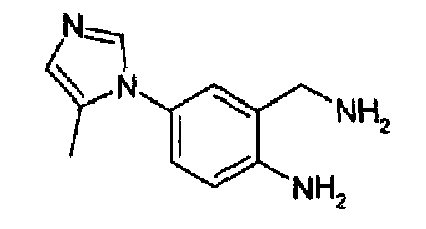
Получен с выходом 66%, как описано выше, исходя из 4-(5-метил-1Н-имидазол-1-ил)-2-цианоанилина. C11H14N4, М.м.: 202,26; 1Н-ЯМР (300 МГц, d6-ДМСО) м.д.: 2,07 (с, 3Н), 5,38 (с, 2Н), 6,89 (д, 1Н), 6,73 (т, 1Н), 6,94 (дд, 1Н), 7,06 (д, 1Н), 7,53 (д, 1Н).
4-(4-Метил-1Н-имидазол-1-ил)-2-цианоанилин
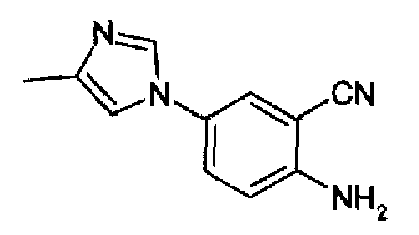
SnCl2·2H2O (119,0 г, 0,526 моль) растворяли в 37% HCl (240 мл), к данному раствору, охлажденному до -10°С, добавляли 4-(4-метил-1Н-имидазол-1-ил)-2-цианонитробензол (24,0 г, 105,0 ммоль) в пять порций в течение 20 мин. По окончании добавлений перемешиваемой реакционной смеси позволяли принимать к.т., и после дополнительных 45 мин перемешивания ее выливали в лед/воду (500 г) и 3н КОН (1,0 л). Полученную суспензию фильтровали и осадок промывали водой, остаток суспендировали в 2М NH3/EtOH (250 мл), перемешивали в течение нескольких минут и фильтровали, фильтрат концентрировали с получением указанного в заголовке соединения в виде коричневого твердого вещества (15,9 г, 76%). C11H10N4, М.м.: 198,23. 1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,2 (с, 3Н), 6,79 (д, 1Н), 6,81 (д, 1Н), 6,84 (т, 1Н), 7,75 (дд, 1Н), 7,28 (д, 1Н), 7,30-7,34 (м, 1Н), 7,57 (д, 1Н).
4-(5-Метил-1Н-имидазол-1-ил)-2-цианоанилин
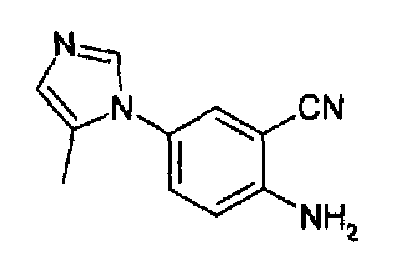
Получен с выходом 64%, как описано выше, исходя из 4-(5-метил-1Н-имидазол-1-ил)-2-цианонитробензола. C11H10N4, М.м.: 198,23; 1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,09 (с, 3Н), 6,81-6,84 (м, 2Н), 7,20 (дд, 1Н), 7,26 (д, 1Н), 7,44 (д, 1Н).
4-(4-Метил-1Н-имидазол-1-ил)-2-цианонитробензол
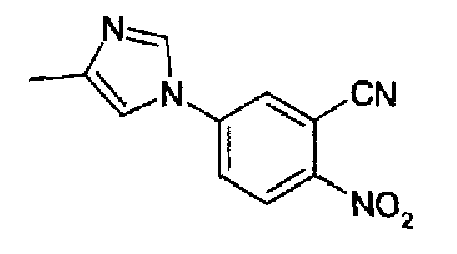
2-Циано-4-фторнитробензол (29,5 г, 177,6 ммоль) и 4-метилимидазол (29,1 г, 354,4 ммоль) растворяли в ацетонитриле (300 мл), затем реакционную смесь нагревали при 90°С в течение 3 часов.
Раствор охлаждали при к.т. и выпаривали растворитель, остаток (состоящий из примерно 85:15 региоизомерной смеси 4/5 метильных изомеров) распределяли между AcOEt/H2O (5/2), отделенную органическую фазу промывали водой, насыщенным солевым раствором и затем упаривали. Оранжевый остаток кристаллизовали из смеси ацетон/гептан. Это давало первую порцию указанного в заголовке продукта 25,0 г (62%), маточный раствор концентрировали и остаток хроматографировали на силикагеле (ацетон/гептан от 1:3 до 1:1, до 3:1) с получением еще 7,0 г (17,2%) очищенного указанного в заголовке соединения.
ТСХ: (SiO2, 245 нм) ацетон/гептан (3:1) Rf: 0,50; C11H10N4O2, М.м.: 230,23; 1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,3 (с, 3Н), 7,10 (т, 1Н), 7,63 (д, 1Н), 7,75 (дд, 1Н), 7,86 (д, 1Н), 7,91 (д, 1Н), 8,44 (д, 1Н).
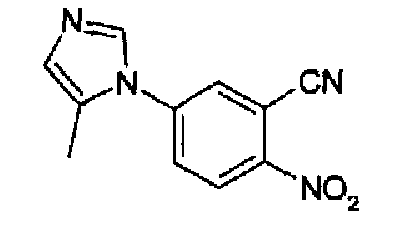
4-(5-Метил-1Н-имидазол-1-ил)-2-цианонитробензол
Получен от колоночной хроматографии, описанной выше, в виде желто-оранжевого твердого вещества (7,2 г; 17,8%).
ТСХ: (SiO2, 245 нм) ацетон/гептан (3:1) Rf: 0,30; C11H10N4O2, М.м.: 230,23; 1Н-ЯМР (300 МГц, CDCl3) м.д.: 2,3 (с, 3Н), 7,10 (т, 1Н), 7,63 (д, 1Н), 7,74 (дд, 1Н), 7,84 (д, 1Н), 8,49 (д, 1Н).
Общая процедура для получения гидрохлоридов сложных иминоэфиров
Гидрохлориды сложных иминоэфиров, используемые в данном изобретении в качестве реагентов, могут быть получены согласно процедурам, хорошо известным в литературе, например: J. Org. Chem. 69(20), 6572-6589; 2004, J. Med. Chem., 38(8), 1287-94; 1995, две представленные ниже процедуры даны в настоящем описании как примеры.
Гидрохлорид метил 4-метоксибензимидата
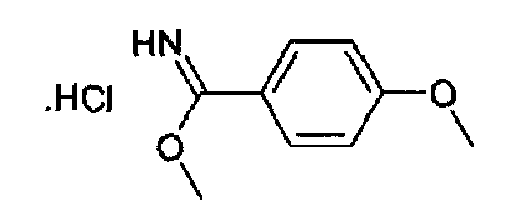
4-Метоксибензонитрил (12,5 г, 91,1 ммоль) растворяли в метаноле (140 мл), через данный охлаждаемый раствор (-5°С) барботировали газообразный HCl в течение примерно 3 часов. Реакционную смесь затем перемешивали в закрытой колбе при к.т. в течение 24 часов. Затем избыток HCl вытесняли барботированием азота и полученный раствор концентрировали, остаток поглощали посредством TBME (100 мл) и перемешивали в течение 30 мин, затем фильтровали и сушили с получением 19,0 г (выход количественный) указанного в заголовке продукта в виде бесцветного порошка. C9H11NO2·HCl, М.м.: 201,69; 1Н-ЯМР (300 МГц, D2O) м.д.: 4,20 (с, 3Н), 6,9 (м, 1Н), 7,65 (м, 1Н), 8,40 (м, 1Н).
Гидрохлорид метилового эфира 3-фуранкарбоксимидокислоты
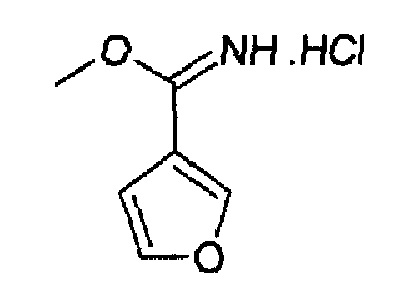
3-Фуронитрил (1,0 г; 10,8 ммоль) растворяли в сухом МеОН (12 мл), раствор охлаждали при -5°С и барботировали газообразный HCl в течение 30 мин, затем реакционный сосуд закрывали, температуре позволяли принимать к.т. и реакционную смесь перемешивали в течение ночи. Избыток HCl удаляли барботированием азота, затем растворитель выпаривали и остаток суспендировали в TBME (30 мл), фильтровали и сушили с получением 1,09 г (63%) указанного в заголовке продукта. C6H7NO2·HCl, М.м.: 161,59; 1Н-ЯМР (300 МГц, D2O) м.д.: 3,80 (с, 3Н), 4,20 (с, 3Н), 7,0 (д, 2Н), 7,90 (д, 2Н).
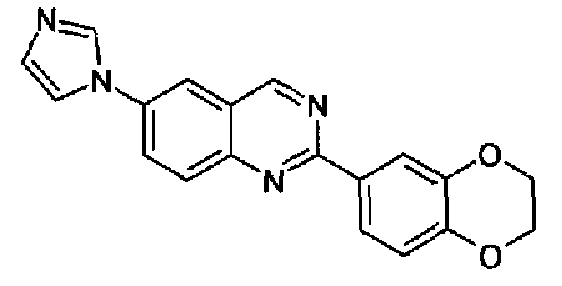
Пример 13: [6-(1H-имидазол-1-ил)-2-(1,3-бензодиоксол-5-ил)]хиназолин
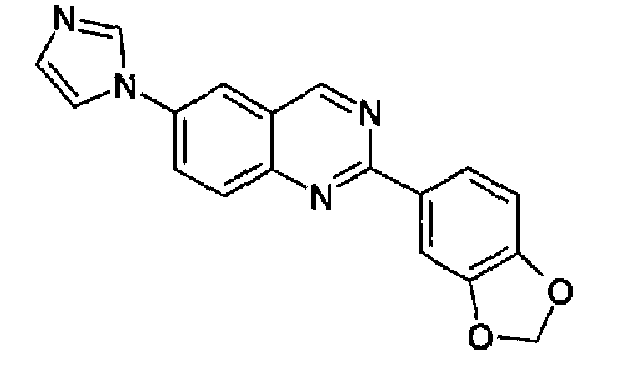
Суспензию 6-амино-2-(1,3-бензодиоксол-5-ил)хиназолина (2,5 г, 9,4 ммоль) (WO 2008/014822) и 40% водного глиоксаля (1,1 мл, 9,4 ммоль) в метаноле (20 мл) перемешивали при к.т. в течение 18 ч. Добавляли NH4Cl (1,0 г, 0,019 моль), 37% водный формальдегид (1,4 мл, 19 ммоль) и метанол (200 мл) и смесь нагревали при кипении с обратным холодильником в течение 1 ч. Добавляли 85% Н3РО4 (1,4 мл) и смесь нагревали при кипении с обратным холодильником в течение еще 4 ч. Растворитель удаляли и остаток выливали в воду и делали щелочным с помощью водного NaOH. Осадок фильтровали, промывали водой и растворяли в DCM. Продукт экстрагировали разбавленной водной HCl. К собранным водным слоям добавляли Na2CO3 и полученную смесь экстрагировали хлороформом, который промывали водой и сушили, концентрировали и полученное твердое вещество суспендировали в изопропиловом простом эфире. Твердое вещество фильтровали и сушили с получением указанного в заголовке продукта (2,0 г, выход 29%). C18H12N4O2, М.м.: 316,32; т.пл. 217-218°С. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 6,16 (с, 2Н), 7,12 (д, 1Н), 7,21 (с, 1Н), 8,00 (д, 2Н), 8,14-8,22 (м, 2Н), 8,36-8,50 (м, 3Н), 9,65 (с, 1Н). FT-IR (KBr) 1504, 1446, 1251.
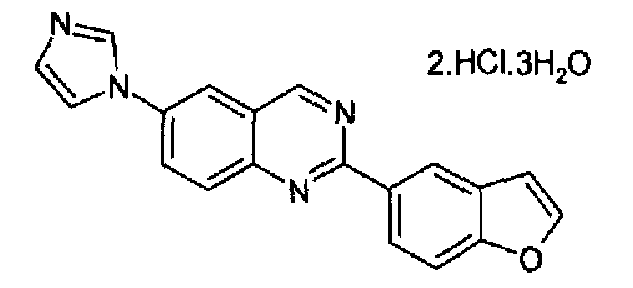
Пример 14: дигидрохлорид тригидрат [6-(1H-имидазол-1-ил)-2-(бензофуран-5-ил)]хиназолина
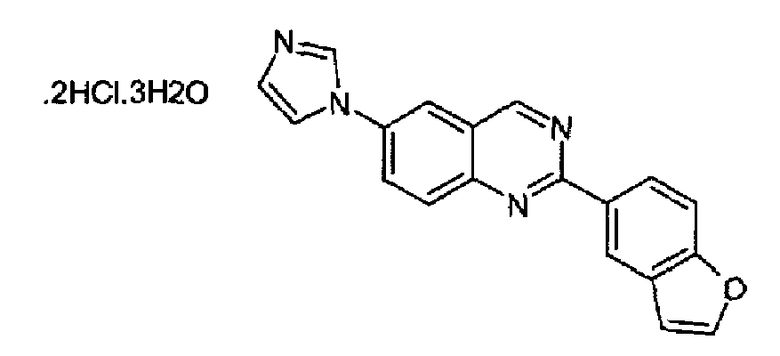
Получен аналогично с выходом 20%, исходя из 6-амино-2-(бензофуран-5-ил)хиназолина (WO 2008/014822). C19H12N4O·2HCl·3H2O; М.м.: 349,30; т.пл. 284,7-285,1°С. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 7,15 (с, 1Н), 7,78 (д, 1Н), 8,02 (с, 1Н), 8,10 (д, 1Н), 8,29 (д, 1Н), 8,47 (м, 2Н), 8,58 (д, 1Н), 8,71 (д, 1Н), 8,90 (с, 1Н), 9,78 (с, 1Н), 10,00 (с, 1Н). FT-IR (KBr): 3399, 3097, 1614.
Пример 15: [6-(1H-имидазол-1-ил)-2-(2,3-дигидро-1,4-бензодиоксин-6-ил)]хиназолин
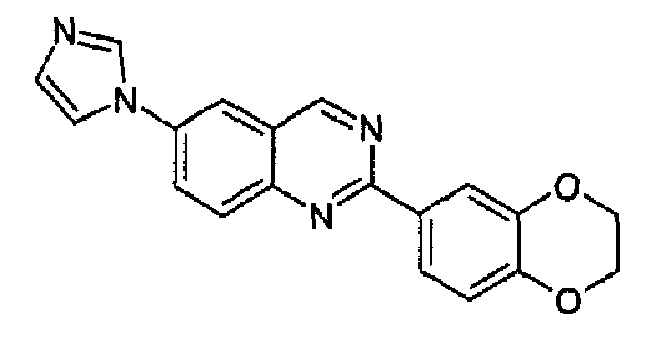
Получен аналогично с выходом 25%, исходя из 6-амино-2-(2,3-дигидро-1,4-бензодиоксин-6-ил)хиназолина (WO 2008/014822). C19H14N4O2, М.м.: 330,35; т.пл. 131,5-131,9°С; 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 4,34 (с, 4Н), 7,04 (д, 1Н), 7,21 (с, 1Н), 7,97 (д, 1Н), 8,03-8,13 (м, 2Н), 8,18 (с, 1Н), 8,32-8,43 (м, 2Н), 8,49 (с, 1Н), 9,64 (с, 1Н). FT-IR (KBr): 1555, 1507, 1286.
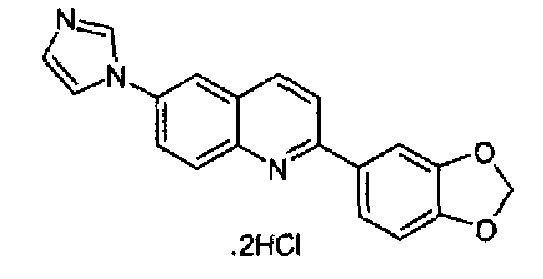
Пример 16: дигидрохлорид [6-(1H-имидазол-1-ил)-2-(1,3-бензодиоксол-5-ил)]хинолина
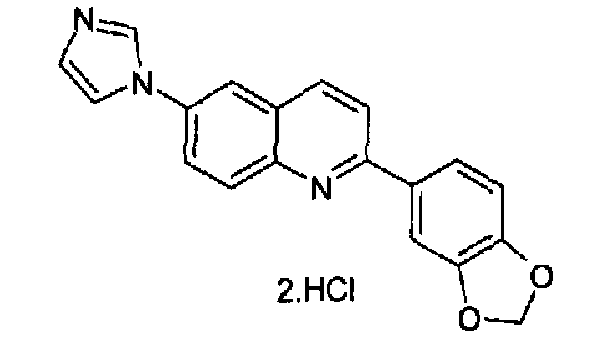
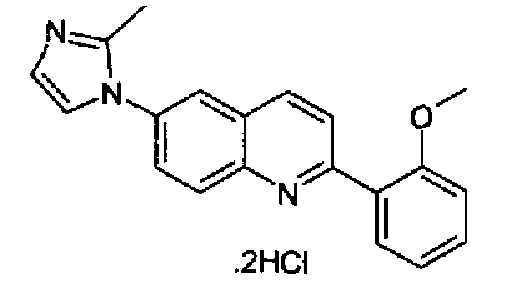
6-(1Н-имидазол-1-ил)-2-(трифторметансульфонокси)хинолин (3,0 г; 8,6 ммоль), K2CO3 (1,73; 10,4 ммоль), 3,4-метилендиоксифенилбороновую кислоту и тетракистрифенилфосфин палладий (0,8 г; 0,8 ммоль) смешивали при перемешивании в сухом толуоле (100 мл), в атмосфере аргона, при к.т. Полученную реакционную смесь нагревали при кипении с обратным холодильником в течение 15 часов, затем охлаждали при к.т. и выливали в воду (250 мл). Полученный осадок фильтровали, промывали водой, сушили и растворяли в DCM/MeOH (9:1, 10 мл), через данный раствор барботировали газообразный HCl до образования осадка, который фильтровали и сушили. Указанный в заголовке продукт получали в виде дигидрохлорида (2,57 г; выход 88%), плавящегося при 314,0-315°С. C19H13N3O2·2HCl, М.м.: 351,79. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 6,05 (с, 2Н), 7,07 (д, 1Н), 7,70-7,72 (м, 3Н), 8,01-8,29 (м, 5Н), 8,51 (д, 1Н), 9,46 (с, 1Н). FT-IR (ATR) см-1: 1602, 1495, 1443, 1265, 1254, 1110, 1029, 812.
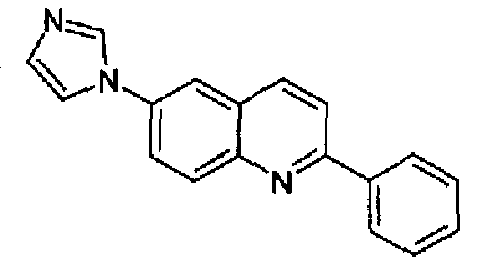
Пример 17: [6-(1H-имидазол-1-ил)-2-(фенил)]хинолин
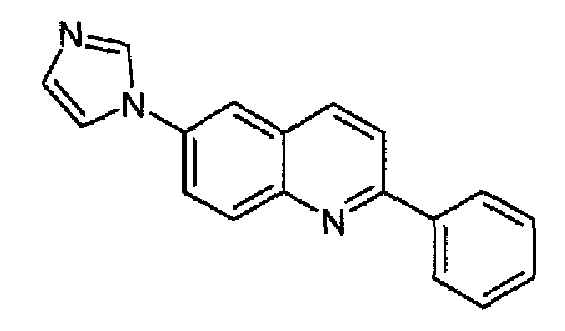
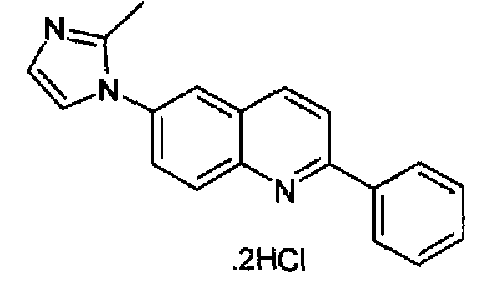
Получен аналогично в виде бесцветного твердого вещества (2,7 г; выход 89%) при использовании фенилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/метанол в виде дигидрохлорида, свободное основание получали суспендированием дигидрохлорида в концентрированном водном аммиаке, осадок фильтровали, промывали водой и сушили. Бесцветное твердое вещество (2,3 г), плавящееся при 130,6-131,4°С. C18H13N3, М.м.: 271,32. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 7,20 (м, 1Н), 7,75-7,63 (м, 3Н), 7,94 (м, 1Н), 8,11-8,32 (м, 5Н), 8,46-8,50 (м, 2Н). FT-IR (ATR) см-1: 1625, 1598, 1500, 1325, 1244, 1054, 826, 758, 655.
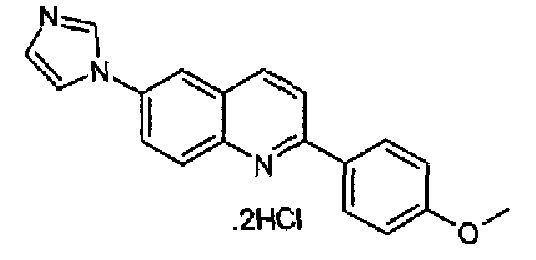
Пример 18: дигидрохлорид [6-(1H-имидазол-1-ил)-2-(4-метоксифенил)]хинолина
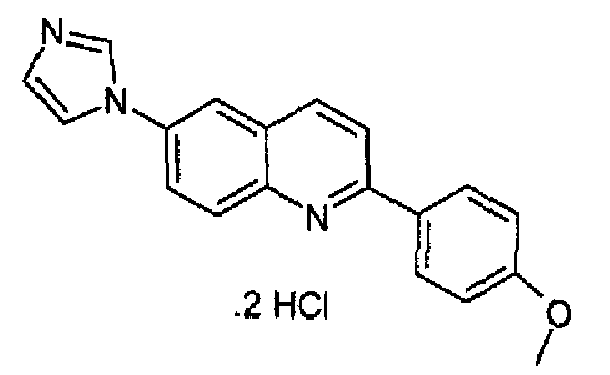
Получен аналогично с выходом 57% при использовании 4-метоксифенилбороновой кислоты в конденсации Сузуки, перекристаллизован из смеси DCM/метанол, бесцветное твердое вещество, т.пл.: 266,0-267,0°С. C19H15N3O·2HCl, М.м.: 374,27. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 3,84 (c, 3Н), 7,14 (д, 2Н), 7,87 (c, 1Н), 8,11-8,27 (м, 6Н), 8,41 (c, 1H), 8,58 (д, 1Н), 9,70 (c, 1H). FT-IR (ATR) см-1: 1597, 1510, 1272, 1184, 1014, 825, 804.
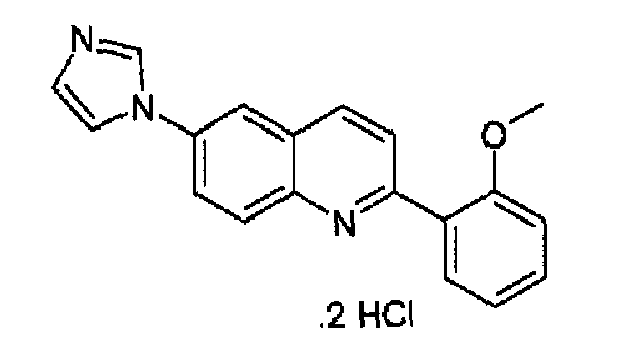
Пример 19: дигидрохлорид [6-(1H-имидазол-1-ил)-2-(2-метоксифенил)]хинолина
Получен аналогично с выходом 48% при использовании 2-метоксифенилбороновой кислоты в конденсации Сузуки, перекристаллизован из смеси DCM/МеОН, бесцветное твердое вещество, т.пл.: 251,5-252,0°С. C19H15N3O·2HCl, М.м.: 374,27. 1Н-ЯМР (200 МГц, D2O) м.д.: 3,87 (c, 3Н), 7,10-7,18 (м, 2Н), 7,40-7,68 (м, 4Н), 7,70-7,90 (м, 5Н), 8,94 (c, 1H). FT-IR (ATR) см-1: 1607, 1577, 1497, 1317, 1252, 1016, 828, 764, 745.
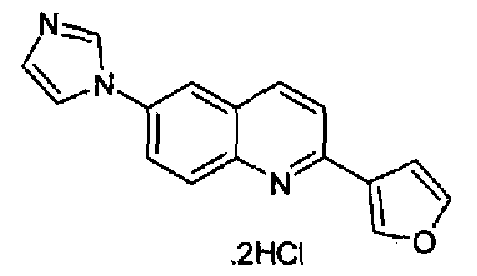
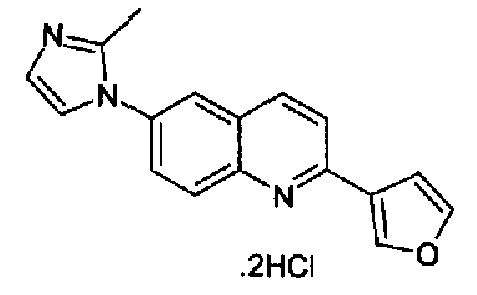
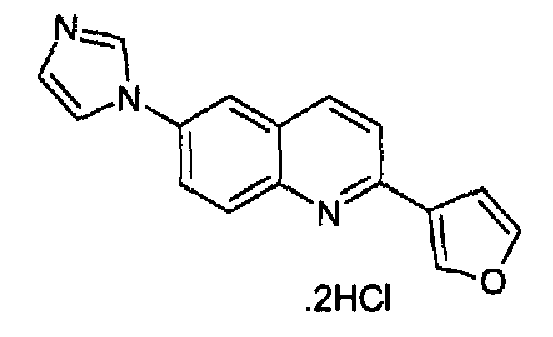
Пример 20: дигидрохлорид [6-(1H-имидазол-1-ил)-2-(3-фурил)]хинолина
Получен аналогично с выходом 81,5% при использовании 3-фурилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/МеОН как бесцветное твердое вещество, т.пл.: 293,1-295,6°С. C16H11N3O·2HCl, М.м.: 334,20. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 7,30 (c, 1Н), 7,86-7,90 (м, 2Н), 8,1 (д, 1Н), 8,15 (дд, 1Н), 8,32 (д, 2Н), 8,45 (д, 1H), 8,58 (д, 1Н), 8,68 (с, 1Н), 9,76 (c, 1H). FT-IR (ATR) см-1: 1651, 1624, 1547, 1328, 1159, 822.
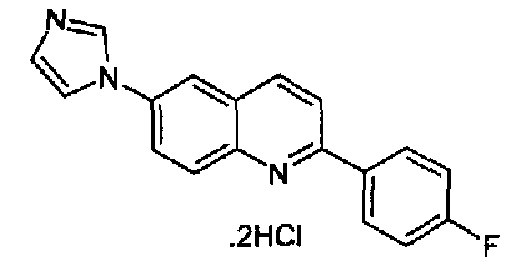
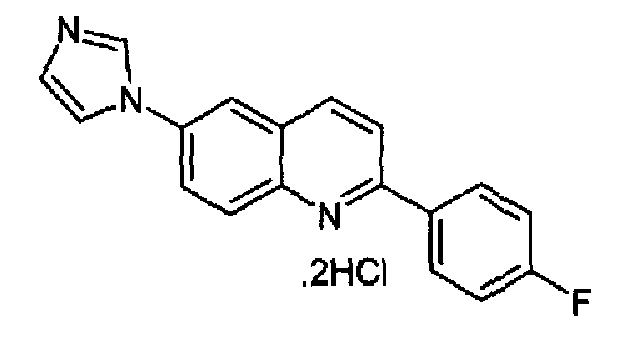
Пример 21: дигидрохлорид [6-(1H-имидазол-1-ил)-2-(4-фторфенил)]хинолина
Получен аналогично с выходом 64,5% при использовании 4-фторфенилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/МеОН, бесцветное твердое вещество, т.пл.: 280,7-282,0°С. C18H12FN3·2HCl, М.м.: 362,31. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 7,42 (т, 2Н), 8,03 (с, 1Н), 8,22-8,61 (м, 8Н), 9,94 (с, 1Н). FT-IR (ATR) см-1: 1644, 1599, 1509, 1327, 1248, 1161, 833.
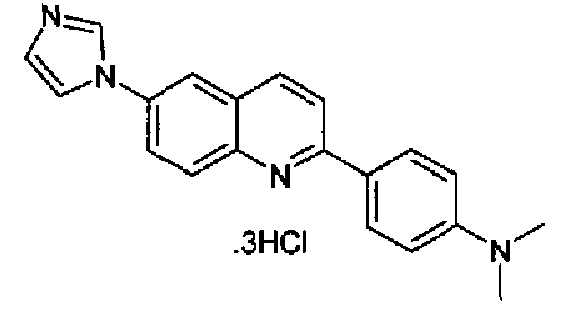
Пример 22: тригидрохлорид [6-(1H-имидазол-1-ил)-2-(4-диметиламинофенил)]хинолина
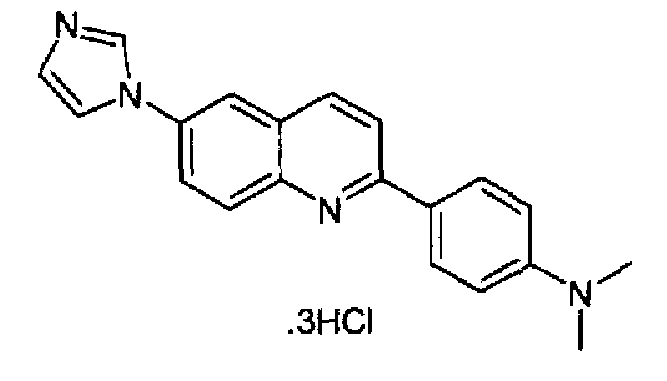
Получен аналогично в виде бесцветного твердого вещества (выход 84,5%) при использовании 4-диметиламинофенилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/МеОН, бледно-красное твердое вещество, т.пл.: 284-286°С. C20H18N4·3HCl, М.м.: 423,77. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 3,0 (с, 6Н), 6,98 (д, 2Н), 7,83 (с, 1Н), 8,14 (д, 2Н), 8,23-8,39 (м, 4Н), 8,74 (д, 2Н), 9,65 (с, 1Н). FT-IR (ATR) см-1: 1640, 1591, 1546, 1387, 1339, 1202, 1133, 812.
Пример 23: дигидрохлорид [6-(1H-имидазол-1-ил)-2-(4-трифторметоксифенил)]хинолина
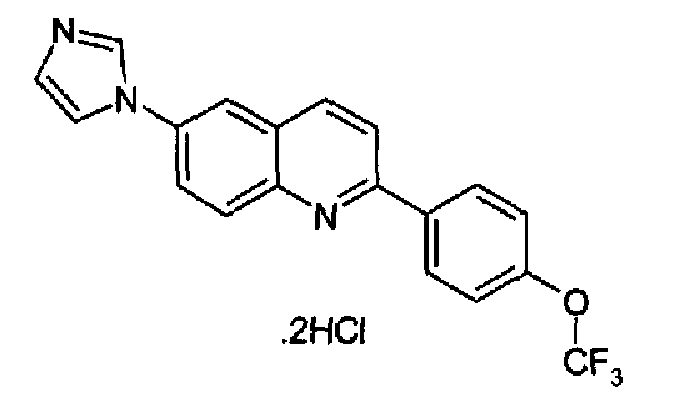
Получен аналогично в виде бесцветного твердого вещества (1,62 г; выход 78%) при использовании 4-трифторметоксифенилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/метанол, бесцветное твердое вещество, т.пл.: 260-262°С. C19H12F3N3O·2HCl, М.м.: 428,24. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 7,59 (д, 2Н), 8,03 (c, 1Н), 8,23-8,63 (м, 7Н), 9,92 (с, 1Н). FT-IR (ATR) см-1: 1619, 1326, 1251, 1184, 1149, 849, 830.
Пример 24: дигидрохлорид [6-(1H-имидазол-1-ил)-2-(2-метил-4-трифторметоксифенил)]хинолина
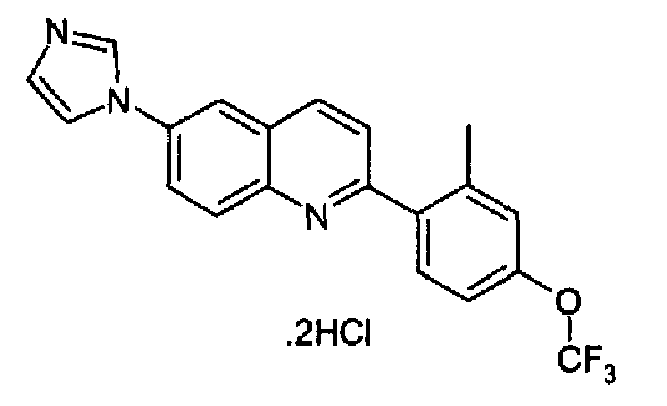
Получен аналогично с выходом 78% при использовании (2-метил-4-трифторметоксифенил)бороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/МеОН, светло-серый порошок, плавящийся при 269,7-274,5°С.
C20H14F3N3O·2HCl, М.м.: 442,27. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 2,83 (с, 3Н), 7,40 (д, 1Н), 7,77 (с, 1Н), 8,80-8,57 (м, 8Н), 9,60 (с, 1Н). FT-IR (ATR) см-1: 1638, 1616, 1270, 1224, 1149, 900, 885.
Пример 25: дигидрохлорид [6-(1H-имидазол-1-ил)-2-(4-метансульфонилфенил)]хинолина
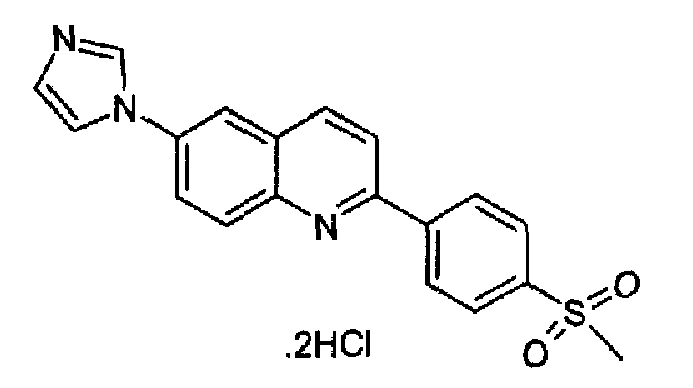
6-(1H-имидазол-1-ил)-2-(трифторметансульфонокси)хинолин (1,06 г; 2,88 ммоль) растворяли в сухом диоксане (35 мл), затем в атмосфере аргона добавляли LiCl (1,0 г, 2,88 ммоль) и гексаметилдиолово (1 г, 2,88 ммоль) и тетракистрифенилфосфинпалладий (25 мг, 0,02 ммоль). К перемешиваемой суспензии при к.т. добавляли 4-бромметансульфонилбензол (0,7 г, 3,0 ммоль), растворенный в сухом диоксане (3 мл). Полученную смесь затем нагревали при кипении с обратным холодильником в течение 48 часов, затем охлаждали при к.т. и выливали в воду (100 мл). Полученную суспензию насыщали с помощью NaHCO3 и осадок экстрагировали ACOEt. Органический слой промывали водой, затем сушили и концентрировали с получением коричневого твердого вещества. Данный продукт растворяли в смеси DCM/MeOH (9:1) и гидрохлорид осаждали барботированием газообразного HCl. После кристаллизации из воды получали указанный в заголовке продукт (600 мг, выход: 48%) в виде светло-желтого твердого вещества, плавящегося при 267,4-268,1°С. C19H15N3O2S·2HCl, М.м.: 422,33. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 3,32 (с, 3Н), 7,83 (c, 1Н), 8,14 (д, 2Н), 8,23-8,66 (м, 7Н), 9,58 (с, 1Н). FT-IR (ATR) см-1: 1600, 1508, 1298, 1140, 1090, 963, 820, 774.
6-(1H-имидазол-1-ил)-2-(трифторметансульфонокси)хинолин
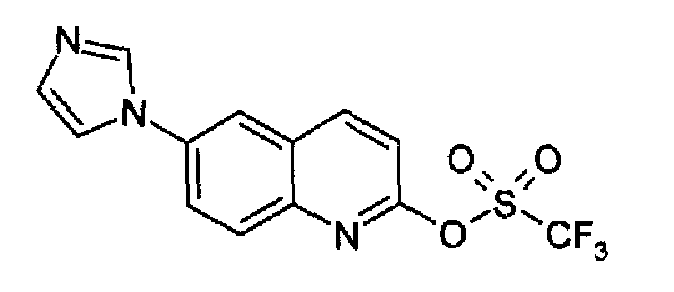
Гидрид натрия (60% суспензия в минеральном масле, 8,7 г; 219,6 ммоль) добавляли порциями при перемешивании при -3°С в атмосфере аргона к раствору гидрохлорида 6-(1Н-имидазол-1-ил)-2-хинолинона (22 г; 87,8 ммоль) в сухом ДМФА/DMF (250 мл). После завершения добавлений реакционную смесь охлаждали при -15°С и добавляли по каплям бис(трифторметилсульфонил)фениламин (37,25 г, 104,25 ммоль; RN: 37595-74-7, Aldrich), растворенный в сухом ДМФА (100 мл), с такой скоростью, чтобы поддерживать температуру реакционной смеси ниже -10°С. В конце добавления температуре реакционной смеси позволяли подниматься до к.т., и реакционную смесь перемешивали в течение еще 2 часов. Затем реакцию гасили водой (2,2 л), осадок фильтровали, промывали водой и затем гексаном. Продукт растворяли в смеси DCM/метанол (9:1; 800 мл) и сушили над Na2SO4. Концентрирование раствора вызывало кристаллизацию указанного в заголовке продукта в виде белого твердого вещества, которое фильтровали и сушили (23,6 г; 77,8%). C13H8F3N3O3S, М.м.: 343,3; MS(ESI) m/z: 344 (M+1).
1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 7,21 (с, 1Н), 7,76 (д, 1Н), 7,96 (c, 1Н), 8,17 (д, 1H), 8,32 (дд, 1Н), 8,49 (с, 2Н), 8,76 (д, 1Н).
Гидрохлорид 6-(1H-имидазол-1-ил)-2-гидроксихинолина
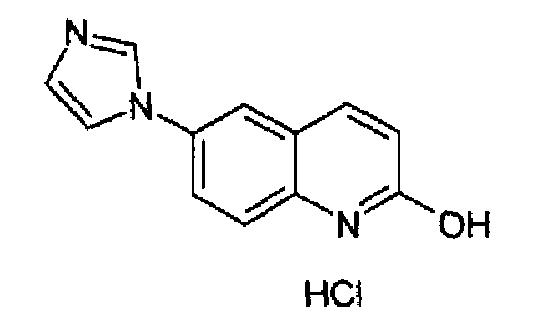
2-Метокси-6-(1H-имидазол-1-ил)хинолин (26,4 г; 115,75 ммоль) суспендировали в 3н водной HCl (170 мл), полученную реакционную смесь нагревали при кипении с обратным холодильником в течение 15 часов. Затем раствор охлаждали до 0°С, выделившийся гидрохлорид указанного в заголовке продукта фильтровали и промывали изопропанолом, затем сушили с получением 22,0 г (75,8%) продукта в виде бесцветных кристаллов, т.пл. 348,7-352,5°С. C12H9N3O·HCl, М.м.: 247,73. 1Н-ЯМР (200 МГц, D2O) м.д.: 6,40 (д, 1Н), 7,30 (д, 1Н), 7,40 (дд, 2Н), 7,48 (дд, 1Н), 7,65 (д, 1Н), 7,74 (д, 1Н), 9,60 (c, 1H).
6-(1H-имидазол-1-ил)-2-метоксихинолин
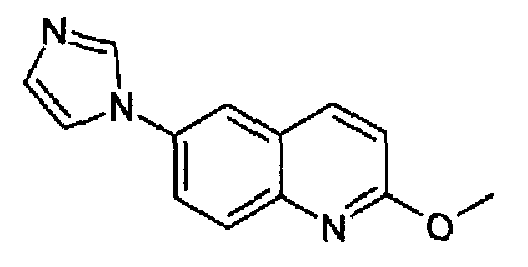
6-Бром-2-метоксихинолин (19 г; 79,8 ммоль; RN: 99455-05-7) растворяли в сухом ДМФА/DMF (100 мл) и к данному раствору добавляли имидазол (5,7 г; 84 ммоль), K2CO3 (11,6 г, 84 ммоль) и CuI (1,1 г, 4,2 ммоль) при к.т. при перемешивании и в атмосфере аргона. Полученную смесь нагревали при 150°С в течение 48 часов. Реакционную смесь охлаждали при к.т. и выливали в 2% (масс./масс.) водный раствор EDTA (600 мл), образовавшийся осадок фильтровали и промывали водой, сушили и затем суспендировали в смеси гексан/AcOEt. Полученную суспензию перемешивали в течение 10 мин, фильтровали и собранный указанный в заголовке продукт сушили, получали 14 г (выход 78%) белых кристаллов. C13H11N3O, М.м.: 225,25. MS(ESI) m/z: 226 (M+1). 1Н-ЯМР (200 МГц, CDCl3) м.д.: 3,6 (с, 3Н), 6,97 (с, 1Н), 7,51 (c, 1Н), 7,82 (д, 1H), 8,10 (дд, 1Н), 8,13 (д, 1Н), 8,27 (с, 1Н), 8,84 (д, 1Н).
6-Бром-2-метоксихинолин
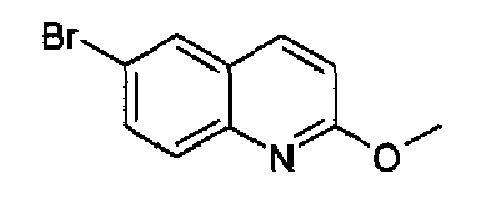
2-Хлор-6-бромхинолин (142,5 г, 0,6 моль; European Journal of Medicinal Chemistry, 35(10), 931-940; 2000; бесцветные кристаллы, т.пл.: 99,8-101,4°С) растворяли в метаноле (700 мл), затем добавляли метоксид натрия (43,9 г; 0,8 ммоль) и полученную реакционную смесь нагревали при кипении с обратным холодильником в течение 16 часов. Реакционную смесь охлаждали при к.т. и выливали в смесь лед-вода (1,8 л), указанный в заголовке продукт осаждался в виде твердого вещества кремового цвета (133 г, 95%), плавящегося при 157,9-161,1°С. C10H8BrNO2, М.м.: 238,09. MS(ESI) m/z: 239 (M+1). 1Н-ЯМР (200 МГц, CDCl3) м.д.: 4,06 (с, 3Н), 6,91 (д, 1Н), 7,64-7,75 (м, 2Н), 7,88 (д, 2H).
Пример 26: дигидрохлорид [6-(2-метил-1H-имидазол-1-ил)-2-(4-метоксифенил)]хинолина
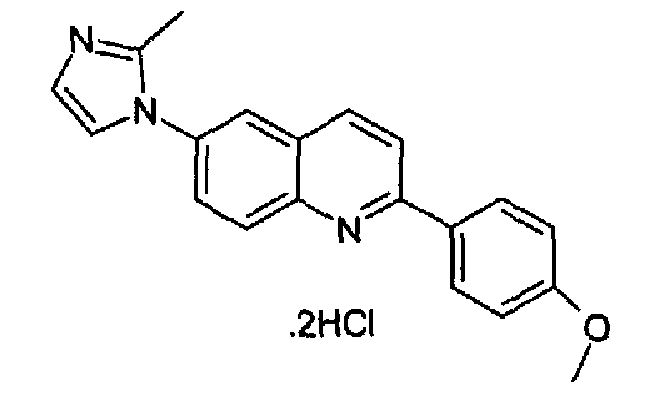
6-(1Н-2-метилимидазол-1-ил)-2-(трифторметансульфонокси)хинолин (3,77 г; 10,6 ммоль) растворяли в толуоле (100 мл), затем добавляли K2CO3 (4,40 г, 31,8 ммоль), тетракистрифенилфосфинпалладий (0,733 г, 0,6 ммоль) и 4-метоксифенилбороновую кислоту (1,74 г; 11,5 ммоль) при к.т., при перемешивании и в атмосфере аргона. Полученную реакционную смесь нагревали при кипении с обратным холодильником в течение 2 ч, затем охлаждали при к.т. и выливали в воду. Органический слой отделяли, водную фазу экстрагировали с помощью DCM и объединенные органические слои сушили, фильтровали и концентрировали. Полученное твердое вещество суспендировали в изопропиловом простом эфире, перемешивали в течение 5 мин, затем фильтровали и сушили. Твердое вещество затем растворяли в смеси DCM/метанол (9:1, 30 мл) и барботировали газообразный HCl до тех пор, пока не завершалось осаждение гидрохлорида. Данный гидрохлорид кристаллизовали из смеси изопропанол/вода с получением указанного в заголовке продукта в виде бесцветного твердого вещества (720 мг; выход 22%). C20H17N3O·2HCl, М.м.: 388,38, т.пл.: 230°С (разл.). MS (ESI) m/z: 316 (M+1). 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 2,65 (c, 3H), 3,80 (с, 3Н), 7,15 (д, 2Н), 7,90-8,10 (м, 2Н), 8,02-8,05 (м, 2Н), 8,20-8,40 (м, 4Н), 8,60 (д, 1Н). FT-IR (ATR) см-1: 1598, 1510, 1269, 1170, 1013, 835.
Пример 27: дигидрохлорид [6-(2-метил-1H-имидазол-1-ил)-2-(2-метоксифенил)]хинолина
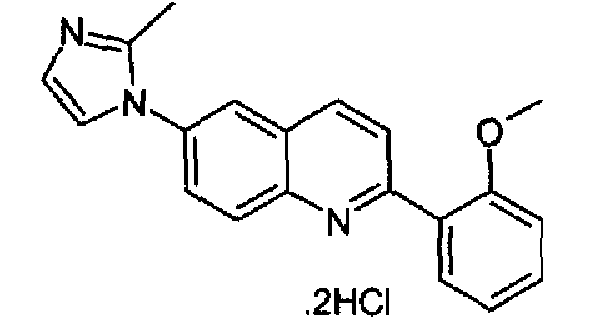
Получен аналогично с выходом 35% при использовании 2-метоксифенилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/метанол. C20H17N3O·2HCl, М.м.: 388,38, т.пл.: 235°С (разл.). 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,65 (c, 3H), 3,88 (с, 3Н), 7,17 (т, 1Н), 7,26 (д, 1Н), 7,54 (д, 1Н), 7,83-7,86 (м, 2Н), 8,02-8,05 (м, 2Н), 8,15 (д, 1Н), 8,37 (д, 1Н), 8,41 (с, 1Н), 8,61 (д, 1Н). FT-IR (ATR) см-1: 1641, 1599, 1491, 1429, 1256, 1171, 1013, 914, 761.
Пример 28: дигидрохлорид [6-(2-метил-1H-имидазол-1-ил)-2-(3-фурил)]хинолина
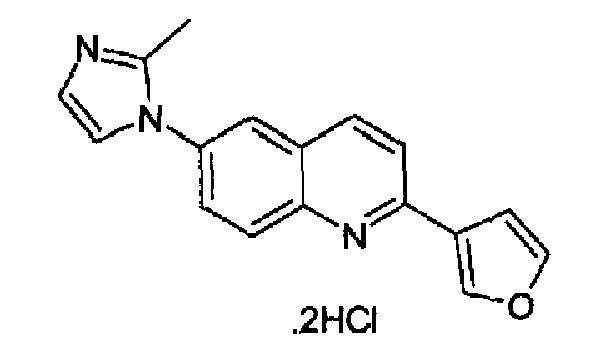
Получен аналогично с выходом 69% при использовании 3-фурилбороновой кислоты в конденсации Сузуки. Перекристаллизован из толуола, т.пл.: 240,1-243,2°С. C17H13N3O·2HCl, М.м.: 348,31. 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,64 (c, 3H), 7,17 (т, 1Н), 7,31 (с, 1Н), 7,85 (д, 1Н), 7,90 (м, 1Н), 8,0 (дд, 1Н), 8,03 (д, 1Н), 8,12 (д, 1Н), 8,28-8,32 (м, 2Н), 8,57 (д, 1Н), 8,73 (с, 1Н). FT-IR (ATR) см-1: 1647, 1620, 1595, 1499, 1368, 1280, 1170, 1152, 917, 860, 766.
Пример 29: дигидрохлорид [6-(2-метил-1H-имидазол-1-ил)-2-(фенил)]хинолина
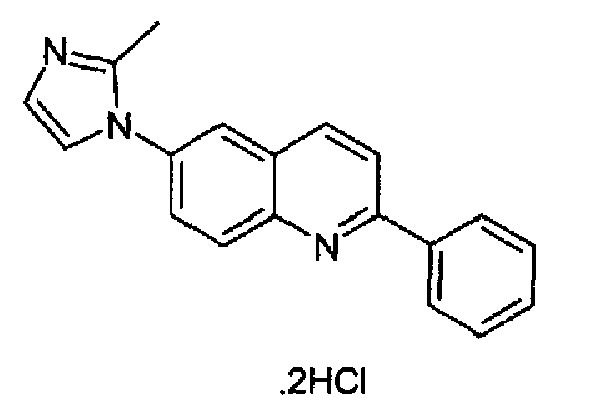
Получен аналогично с выходом 79,5% при использовании фенилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/метанол, бесцветное твердое вещество, т.пл.: 296-297°С. C19H15N3·2HCl, М.м.: 358,35. 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,64 (c, 3H), 7,60 (м, 3Н), 7,86 (д, 1Н), 8,01 (дд, 1Н), 8,03 (д, 1Н), 8,33-8,38 (м, 5Н), 8,63 (д, 1Н). FT-IR (ATR) см-1: 1642, 1615, 1591, 1522, 1504, 1433, 1323, 1273, 1168, 921, 774, 756.
6-(2-метил-1H-имидазол-1-ил)-2-(трифторметансульфонокси)хинолин
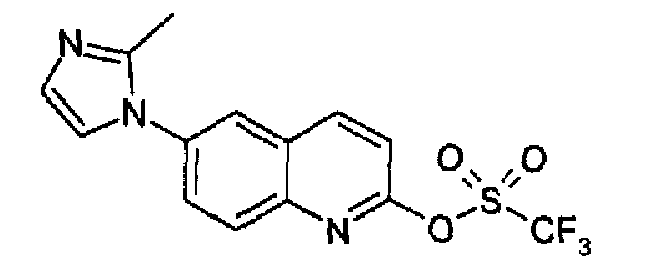
6-(2-Метил-1Н-имидазол-1-ил)-2-хинолинон (5,14 г; 17,3 ммоль) растворяли в ДМФА/DMF (40 мл), затем добавляли NaH (1,70 г, 60% дисперсия в минеральном масле, 43 ммоль) порциями при -10°С в токе аргона. Полученную смесь перемешивали при 0°С в течение 15 мин, затем охлаждали при -15°С и добавляли по каплям бис(трифторметилсульфонил)фениламин (7,22 г, 20,2 ммоль), растворенный в сухом ДМФА (25 мл). Полученную смесь перемешивали при -15°С в течение 30 мин, затем смеси позволяли принимать к.т. и перемешивали при этой температуре в течение 1,5 часов. Реакционную смесь затем выливали в воду (150 мл), образовавшийся осадок фильтровали и сушили соиспарением с толуолом. Затем продукт перемешивали с гексаном несколько минут, фильтровали и сушили с получением указанного в заголовке продукта (5,3 г; выход 88%). C14H10F3N3O3S, М.м.: 357,3; MS (ESI) m/z: 358 (M+1). 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,4 (c, 3H), 7,0 (c, 1Н), 7,52 (c, 1H), 7,79 (д, 1Н), 8,0 (дд, 1Н), 8,15 (д, 1Н), 8,31 (с, 1Н), 8,82 (д, 1Н). FT-IR (ATR) см-1: 1666, 1511, 1414, 1207, 1130, 912, 862.
Гидрохлорид 6-(2-метил-1H-имидазол-1-ил)-2-гидроксихинолина
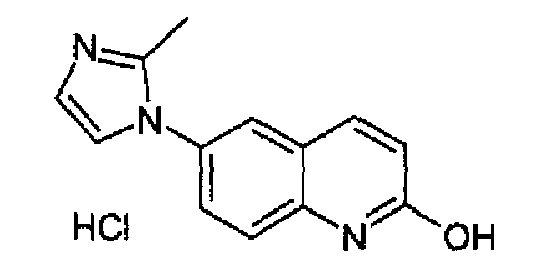
3-Этокси-N-[4-(2-метил-1Н-имидазол-1-ил)фенил]акриламид (30 г; 110 ммоль) добавляли при -5/-10°С к концентрированной серной кислоте (120 мл) и полученную смесь перемешивали при к.т. в продолжение ночи. Реакционную смесь гасили в смеси лед/вода (400 г), рН доводили до рН=8 добавлением K2CO3, осадок фильтровали и затем суспендировали в AcOEt/MeOH (9:1; 400 мл). Полученную суспензию перемешивали в течение 5 мин, неорганические соли отфильтровывали, промывали с помощью AcOEt и объединенный фильтрат и промывочные порции сушили и концентрировали. Колоночная хроматография остатка на силикагеле (AcOEt/MeOH 9:1) давала 15,3 г (62%) аморфного серого твердого вещества. Данное твердое вещество растворяли при 60°С в 3н HCl (150 мл), при охлаждении гидрохлорид кристаллизовался в виде бледно-желтого твердого вещества, которое фильтровали, промывали изопропанолом и сушили с получением 13,8 г (48%) очищенного указанного в заголовке продукта. C13H11N3O·HCl, М.м.: 261,75. 1Н-ЯМР (200 МГц, D2O) м.д.: 2,44 (с, 3Н), 6,40 (д, 1Н), 7,30 (д, 1Н), 7,40 (дд, 2Н), 7,48 (дд, 1Н), 7,65 (д, 1Н), 7,74 (д, 1H).
3-Этокси-N-[4-(2-метил-1Н-имидазол-1-ил)фенил]акриламид
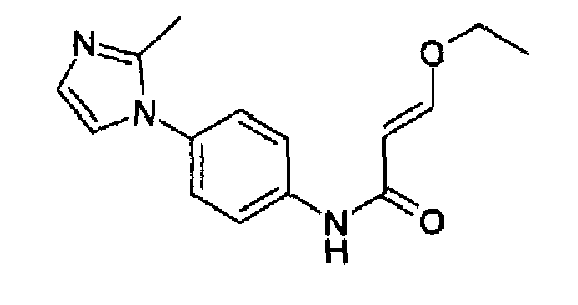
4-(2-Метил-1Н-имидазол-1-ил)анилин (40,7 г, 232 ммоль; RN: 74652-81-6, Maybridge, J. Med. Chem., 48(6), 1729-1744; 2005) растворяли в сухом пиридине (290 мл), затем к данному раствору добавляли по каплям 3-этоксиакрилоилхлорид (36,1 г, 268 ммоль) при 0°С/-10°С. Полученную смесь перемешивали при 0°С в течение 2 часов и при к.т. в продолжение ночи. Реакционную смесь гасили с помощью 100 мл воды, и пиридин отгоняли i.v., остаток поглощали водой и рН доводили до рН=10 добавлением К2СО3, полученную суспензию экстрагировали с помощью AcOEt и концентрировали. Образовавшееся твердое вещество перемешивали с гексаном и фильтровали с получением указанного в заголовке продукта (58 г; 92%). C15H17N3O2, М.м.: 271,32.
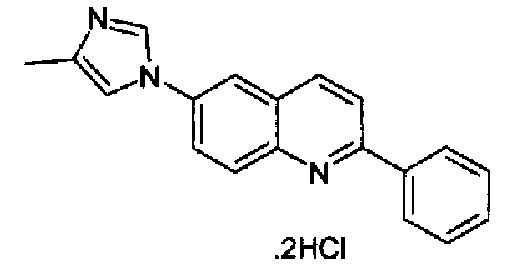
Пример 30: дигидрохлорид [6-(4-метил-1H-имидазол-1-ил)-2-(фенил)]хинолина
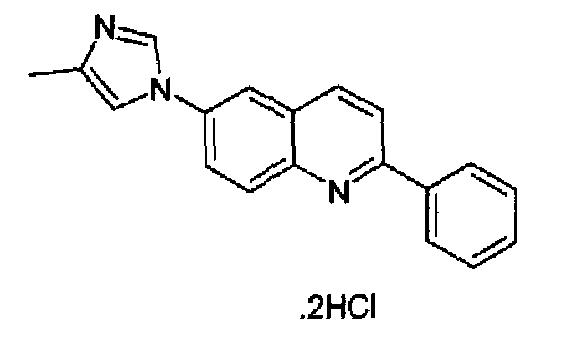
6-(4-Метил-1Н-имидазол-1-ил)-2-(трифторметансульфонокси)хинолин (1,0 г; 2,9 ммоль) растворяли в толуоле (60 мл), затем добавляли K2CO3 (1,9 г, 9,35 ммоль), тетракистрифенилфосфинпалладий (0,40 г, 0,35 ммоль) и фенилбороновую кислоту (0,42 г, 3,9 ммоль) в атмосфере аргона. Полученную смесь нагревали при кипении с обратным холодильником в течение 2 ч, затем охлаждали при к.т. и гасили водой (100 мл). Органическую фазу отделяли, водный слой экстрагировали толуолом и объединенные органические фазы концентрировали. Остаток поглощали сухим толуолом, снова концентрировали до небольшого объема, затем гидрохлорид осаждали барботированием газообразного HCl с получением указанного в заголовке продукта в виде светло-коричневого порошка (380 мг, 36%), плавящегося при 285-289°С. C19H15N3·2HCl, М.м.: 358,35. 1Н-ЯМР (200 МГц, d6-ДМСО) м.д.: 2,30 (c, 3H), 7,60 (м, 3Н), 7,86 (д, 2Н), 8,01 (д, 1Н), 8,03 (с, 1Н), 8,33-8,38 (м, 5Н), 9,62 (с, 1Н).
Пример 31: дигидрохлорид [6-(4-метил-1H-имидазол-1-ил)-2-(4-метоксифенил)]хинолина
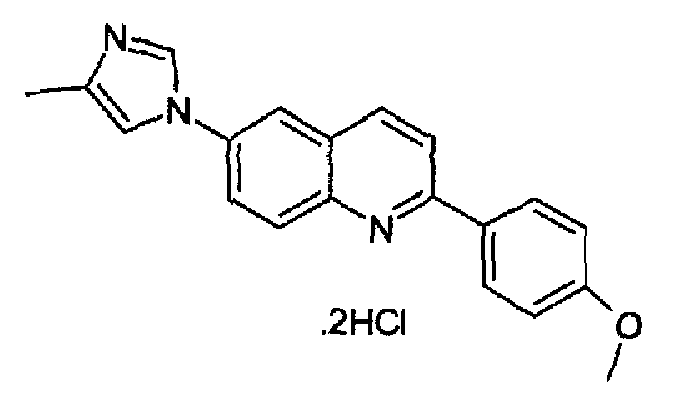
Получен аналогично с выходом 75% при использовании 4-метоксифенилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/метанол, светло-желтый порошок, т.пл.: 276-278°С разл. C20H17N3O·2HCl, М.м.: 388,38. 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,31 (c, 3H), 3,81 (с, 3Н), 7,00 (д, 2Н), 7,47 (с, 1Н), 7,77 (д, 2Н), 7,93 (т, 2Н), 8,02 (с, 1Н), 8,07 (д, 1Н), 8,55 (д, 1Н), 8,97 (с, 1Н). FT-IR (ATR) см-1: 1640, 1596, 1511, 1368, 1298, 1261, 1184, 1015, 827.
Пример 32: дигидрохлорид [6-(4-метил-1H-имидазол-1-ил)-2-(4-фторфенил)]хинолина
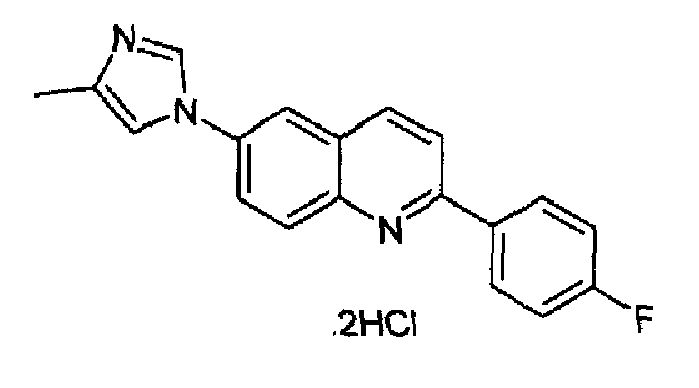
Получен аналогично с выходом 83% при использовании 4-фторфенилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/метанол, порошок кремового цвета, т.пл.: 264-268°С. C19H14FN3·2HCl, М.м.: 376,34. MS(ESI) m/z: 304 (M+1). 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,27 (c, 3H), 7,16 (т, 2Н), 7,40 (с, 1Н), 7,73-7,70 (м, 2Н), 7,84 (т, 2Н), 7,86 (с, 1Н), 7,96 (д, 1Н), 8,46 (д, 1Н), 8,90 (с, 1Н). FT-IR (ATR) см-1: 1615, 1597, 1537, 1510, 1458, 1369, 1329, 1249, 1170, 1079, 920, 840, 826.
Пример 33: дигидрохлорид [6-(4-метил-1H-имидазол-1-ил)-2-(4-метилтиофенил)]хинолина
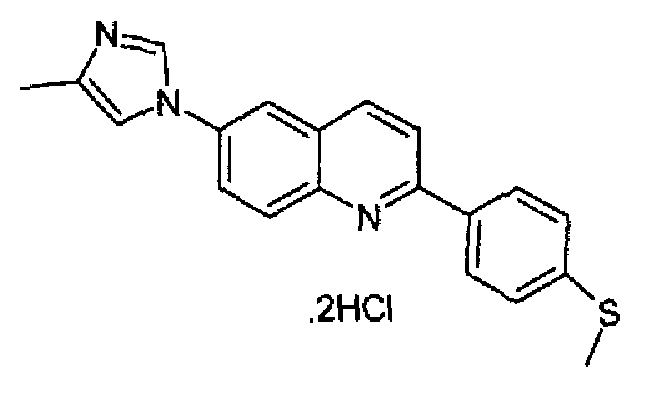
Получен аналогично с выходом 71% при использовании 4-метилтиофенилбороновой кислоты в конденсации Сузуки. Перекристаллизован из смеси DCM/метанол, светло-оранжевый порошок, т.пл.: 294-296°С. C20H17N3S·2HCl, М.м.: 404,34. MS(ESI) m/z: 332 (M+1). 1Н-ЯМР (400 МГц, d6-ДМСО) м.д.: 2,38 (c, 3H), 2,53 (c, 3Н), 7,43 (д, 2Н), 8,07-8,11 (м, 2Н), 8,22-8,28 (м, 4Н), 8,38 (с, 1Н), 8,53 (д, 1Н), 9,63 (с, 1Н). FT-IR (ATR) см-1: 1588, 1545, 1418, 1361, 1192, 1095, 1063, 976, 938, 804, 797.
6-(4-Метил-1H-имидазол-1-ил)-2-(трифторметансульфонокси)хинолин
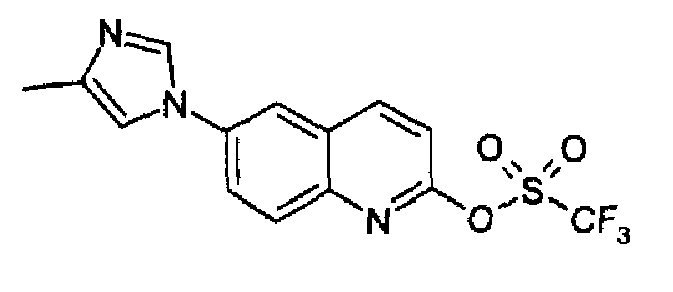
6-(4-Метил-1Н-имидазол-1-ил)-2-хинолинон (8,53 г; 28,7 ммоль) растворяли в ДМФА/DMF (80 мл), затем добавляли NaH (3,4 г, 60% дисперсия в минеральном масле, 85,4 ммоль) порциями при -7°С в токе аргона. Полученную смесь перемешивали при 0°С в течение 15 мин, затем охлаждали при -15°С и добавляли по каплям бис(трифторметилсульфонил)фениламин (13,3 г, 40,1 ммоль), растворенный в сухом ДМФА (35 мл). Полученную смесь перемешивали при -5°С в течение 20 мин, затем смеси позволяли принимать к.т. и перемешивали при этой температуре в течение 1 часа. Реакционную смесь затем выливали в воду (300 мл), образовавшийся осадок фильтровали и сушили соиспарением с DCM. Затем продукт перемешивали с гексаном несколько минут, фильтровали и сушили с получением указанного в заголовке продукта в виде коричневых кристаллов (8,0 г; выход 79%), т.пл.: 150,4-152,1°С. C14H10F3N3O3S, М.м.: 357,3; MS (ESI) m/z: 358 (M+1).
Гидрохлорид 6-(4-метил-1H-имидазол-1-ил)-2-гидроксихинолина
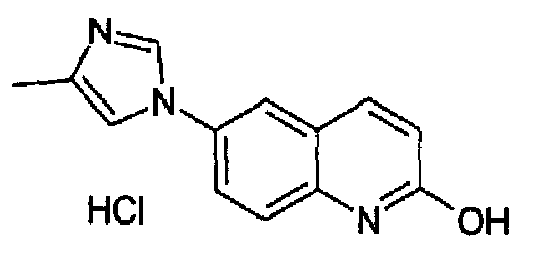
3-Этокси-N-[4-(4-метил-1Н-имидазол-1-ил)фенил]акриламид (18,0 г; 6,8 ммоль) добавляли при -5/-10°С к концентрированной серной кислоте (100 мл) и полученную смесь перемешивали при к.т. в течение 25 ч. Реакционную смесь гасили в смеси лед/вода (400 г), и перемешивали в течение еще 30 мин, затем рН доводили до рН=8 добавлением К2СО3. Осадок экстрагировали, используя AcOEt/MeOH (9:1; 4×150 мл), объединенные органические экстракты сушили и концентрировали. Остаток поглощали изопропанолом (70 мл) и добавляли при +5°С с перемешиванием водную HCl (6н, 15 мл), образовавшийся осадок фильтровали, промывали изопропанолом, затем изопропиловым простым эфиром и сушили с получением 8,53 г (44%) очищенного указанного в заголовке соединения в виде коричневого твердого вещества, т.пл.: 274-277°С.
3-Этокси-N-[4-(4-метил-1Н-имидазол-1-ил)фенил]акриламид
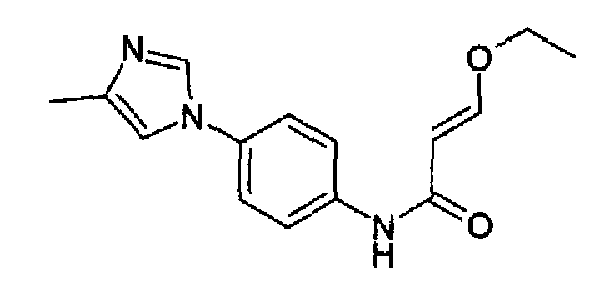
4-(4-Метил-1Н-имидазол-1-ил)анилин (15 г, 138 ммоль) растворяли в сухом пиридине (80 мл), затем свежеперегнанный 3-этоксиакрилоилхлорид (19 г, 140 ммоль) добавляли по каплям при 5°С/0°С. Полученную смесь перемешивали при 0°С в течение 1 часа и при к.т. в продолжение ночи. Реакционную смесь гасили водой (500 мл), рН доводили до рН=10 добавлением K2CO3, образовавшееся твердое вещество фильтровали, промывали водой и сушили с получением указанного в заголовке продукта в виде бледно-оранжевого порошка (18,6 г; 75%), т.пл.: 214-216°С. C15H17N3O2, M.м.: 271,32.
4-(4-Метил-1Н-имидазол-1-ил)анилин
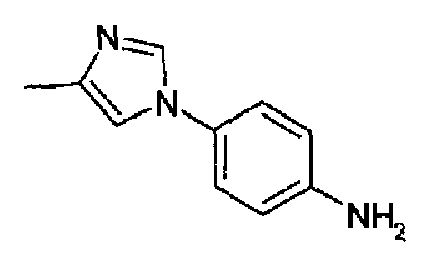
4-(4-Метил-1Н-имидазол-1-ил)нитробензол (22,5 г; 0,87 ммоль) растворяли в абсолютном этаноле (250 мл), затем порциями с охлаждением при 0°С добавляли SnCl2·2H2O (125 г; 0,55 моль). Полученную смесь перемешивали при к.т. в течение 2 часов и нагревали при кипении с обратным холодильником в течение ночи. Реакционную смесь затем охлаждали при к.т. и доводили рН до 12 добавлением 30% КОН (500 мл), затем пеллет КОН при перемешивании. Полученную суспензию фильтровали и осадок промывали этанолом, объединенный фильтрат и промывочные порции концентрировали и остаток экстрагировали с помощью DCM. Концентрирование объединенных органических экстрактов давало указанный в заголовке продукт в виде светло-коричневого твердого вещества (15,3 г; 80%), т.пл.: 122-125°С. C10H11N3, М.м. 173,22.
4-(4-Метил-1Н-имидазол-1-ил)нитробензол
4-Фторнитробензол (32,3 г; 0,229 моль) и 4-метил-1Н-имидазол (25 г; 0,3 моль) смешивали при к.т., к данной перемешиваемой смеси добавляли K2CO3 (44 г; 0,3 моль). Затем реакционную смесь нагревали при 120°С в продолжение ночи, затем охлаждали при к.т. и выливали в воду (2 л), полученную суспензию фильтровали и промывали водой. Полученное твердое вещество сушили при 60°С и кристаллизовали из этилацетата (600 мл) с получением указанного в заголовке соединения в виде желтого твердого вещества (22,5 г; 48,3%). Региоизомер, 4-(5-метил-1Н-имидазол-1-ил)нитробензол, остается в маточном растворе после кристаллизации вместе с некоторым количеством указанного в заголовке продукта (ТСХ: гексан/AcOEt 3:2). C10H9N3O2, М.м. 203,2.
Фармакологическая оценка соединений изобретения
Изучение связывания в отношении имидазолиновых рецепторов подтипа I 2 в головном мозге крысы
Опыты проводили согласно процедуре Lione L.A. et al., 1998 (Eur. J. Pharmacol., 353:123-135). Самцов крыс Wistar (240-300 г, Harlan, Италия) убивали декапитацией. Все головные мозги удаляли незамедлительно на лед и гомогенизировали в 10 объемах буферной сахарозы (0,32М в 50 мМ трис-HCl, pH 7,4 при 4°С), используя тефлон-стеклянный гомогенизатор с механическим приводом. Гомогенат центрифугировали при 1000×g в течение 10 мин при 4°С. Полученные супернатанты подвергали отстаиванию и центрифугировали при 32000×g в течение 20 мин при 4°С. Супернатанты отделяли и каждый пеллет суспендировали в 10 объемах буфера для анализа (50 мМ трис-HCl, 1 мМ MgCl2, pH 7,4 при 4°С) и вращали при 32000×g в течение 20 мин при 4°С. Пеллеты промывали дважды повторным центрифугированием при 32000×g в течение 20 мин при 4°С. Окончательные пеллеты хранили при -80°С до использования. Перед изучениями радиолигандного связывания мембранные пеллеты размораживали и промывали дополнительно 4 раза повторным получением суспензии в 10 объемах буфера для анализа (вышеприведенного) и повторным центрифугированием для удаления любых возможных эндогенных ингибиторов связывания. Содержание белка мембранных препаратов определяли, используя альбумин бычьей сыворотки в качестве стандарта (Bradford M., 1976, Anal. Biochem., 72:248-254). Для рутинных процедур (конкурентно-связывающие анализы) 250 мкл мембранной суспензии (2 мг белка/мл) инкубировали с [3H]-2BFI (2,5×10-9 M; GE Healthcare, 66 Ки/ммоль), в отсутствие или в присутствии различных концентраций тестируемых соединений. Неспецифическое связывание определяли в присутствии 10-5 М BU224 (Tocris Bioscience). Инкубирование при конечном объеме в 1 мл проводили в полистирольных 24-луночных планшетах, начинали с добавления мембранной суспензии и выполняли в течение 90 мин при 25°С. За исключением общего связывания и неспецифического связывания, точки для всех концентраций получали в двукратной повторности. Соединения тестировали в 3-5 различных концентрациях, находящихся в интервале от 10-10 М до 10-5 М концентраций. Сродство, выраженное как величина IC50 (концентрация, которая обладает 50% замещающей способностью), рассчитывали линейной регрессией (log мкМ концентрации тестируемого соединения в зависимости от % специфического остаточного связывания В/Во).
Оценка активности моноаминооксидазы (МАО)
Ингибирующую активность соединений оценивали гомогенным люминесцентным методом, методом MAO-GloTM (Promega), измеряя активность моноаминооксидаз (МАО) от рекомбинантного источника (микросомы от клеток насекомого, зараженного бакуловирусом, Sigma). Эксперименты проводили согласно процедуре Supplier's, инкубируя человеческую рекомбинантную МАО-А или МАО-В с люминогенным субстратом, производным люциферина жуков ((4S)-4,5-дигидро-2-(6-гидроксибензотиазолил)-4-тиазолкарбоновая кислота). МАО превращает данное производное люциферина в метиловый сложный эфир люциферина, и только соединения, которые препятствуют способности фермента использовать пролюминесцентный субстрат, будут вызывать изменения в образующемся люминесцентном сигнале. Анализ MAO-GloTM проводили в две стадии.
Стадия 1. Реакция МАО: субстрат МАО инкубировали с МАО-А или МАО-В (1 мкг/образец) в отсутствие (общая активность) или в присутствии (модулированная активность) тестируемых соединений. Общую активность определяли в присутствии соответствующего растворителя. Концентрации субстрата для МАО-А и МАО-В соответствуют их кажущимся значениям Кm (40 мкМ и 4 мкМ, соответственно). Реакция начиналась добавлением раствора фермента и образцы инкубировали в течение 60 мин при комнатной температуре. Для отрицательной контрольной реакции, образцы буфера (100 мМ Hepes), 5% глицерин, рН 7,5) реакции МАО фактически были включены в испытуемое соединение. Для анализа активности МАО-В, буфер для реакции МАО содержал 10% ДМСО (DMSO), чтобы повысить ферментативную активность.
Стадия 2. Определение люциферина: метиловый сложный эфир люциферина, полученный на стадии 1 действием МАО на субстрат МАО, реагирует с эстеразой и люциферазой (реагент для детекции) для генерирования света. В конце инкубации в каждую лунку добавляли реагент для определения люциферина 50 мкл, планшет инкубировали при комнатной температуре в течение 20 мин, затем люминесцентный сигнал определяли люминометром (время интеграции 0,25-1 с на лунку). Величины показаны в виде относительной световой единицы (RLU). Фактическую МАО-зависимую люминесценцию (фактическая RLU) вычисляли вычитанием средней люминесценции реакции отрицательного контроля без фермента МАО. Снижение наблюдаемого сигнала в присутствии тестируемого соединения относительно общей активности отражает его действие на активность МАО. Все соединения первоначально оценивали в 10-5 конечной концентрации, затем осуществляли кривые ингибирования для активных соединений с величинами концентраций, находящихся по меньшей мере в интервале двух порядков. Для каждой тестируемой концентрации рассчитывали процент ингибирования и линейной регрессией оценивали величину IC50.
Связывание I2 имидазолинового рецептора и анализ активности моноаминооксидазы (МАО)
Эти данные in vitro подчеркивают, как в пределах группы соединений формулы (I) можно изменением картины замещения модулировать активность I2-рецептора, ферментов МАО-А и МАО-В. Например, введение метила в кольцо имидазола позволяет сохранять активность по отношению к I2-рецептору, хотя теряется активность по ингибированию МАО (см., например, соединения в примерах 2 и 5 по сравнению с примером 1), тот же самый эффект получен с соответствующим замещением как в имидазоле, так и в фениле в положении 2 (см., например, соединения в примерах 6 и 9 по сравнению с примером 1). Соответствующее замещение в фениле в положении 2 может также модулировать активность МАО-А по сравнению с активностью МАО-В (см., например, соединения в примере 22 по сравнению с примером 17). В соответствии с этим, соединения изобретения могут быть или селективными агонистами I2-рецепторов по результатам исключительных эффективностей in vitro, или сбалансированными агонистами I2-рецепторов/МАО-А против ингибиторов МАО-В.
“Тест подвешивание за хвост” на мышах
Для оценки новых соединений-антидепрессантов разработано несколько моделей на животных. Среди них “тест подвешивание за хвост” представляет собой простую, быструю и удобную модель, в которой многие антидепрессанты снижают время иммобилизации, указывая на то, что данный параметр может быть использован как показатель антидепрессантной активности. Антидепрессантный эффект представленных примеров соединений формулы (I) оценили согласно процедуре, данной ниже. Иммобилизацию вызывали согласно процедуре Steru et al. (1985). Мышей CD1 (Harlan, Италия) индивидуально подвешивали на 75 см выше головы лейкопластырем, помещенным на 1 см от кончика хвоста. Длительность иммобилизации регистрировали в течение 5 мин. Мышей считали иммобильными, только когда они висели пассивно и полностью без движения. Соединения давали перорально, за 30 мин перед тестом в дозах, изменяющихся от 0,3 до 30 мг/кг. Собранные данные выражали как средний эффект в процентах (МРЕ), который представляет собой % ингибирования во времени иммобилизации между животными, обработанными репрезентативными соединениями формулы (I), и контролями, которые получали только наполнитель. Из данных МРЕ вычислена доза, дающая 50% снижение (ED50).
Мыши, обработанные репрезентативными соединениями формулы (I), проявляли дозозависимые активности, подобные антидепрессантной, в “тесте подвешивания за хвост” по сравнению со стандартными ссылочными лекарственными средствами.
0,3 мг/кг; IP
Как показано в таблице 4, антидепрессантный эффект примера 1 был заблокирован (ответ в зависимости от дозы сдвигался сразу) присутствием коммерчески доступного антагониста имидазолиновых (I2) рецепторов (0,3 мг/кг идазоксана). Этот эффект полностью соответствовал данным “in vitro”, показанным в таблице 2, где представленные соединения как объект данного изобретения были способны ингибировать связывание [3H]2-BFI дозой IC50 в низком микромолярном диапазоне. Это означает, что поведенческий антидепрессантный эффект репрезентативных соединений формулы (I) может быть опосредован, по меньшей мере частично, их взаимодействием с имидазолиновыми (I2) рецепторами.
CFA модель воспалительной боли на крысах: эффект репрезентативного соединения формулы (I) в усилении действия низкой дозы морфина
Эффекты соединений формулы (I) оценивали на животной модели хронической воспалительной боли. В частности, исследована их потенциальная способность повышать абсолютную анальгезирующую активность низкой дозы морфина. Недавно было показано, что использование полного адъюванта Фрейнда (CFA; Mycobacterium tuberculosis) в качестве запускающего агента для воспалительного ответа вместе с применением соответствующего протокола представляет собой подходящую модель хронической боли. Пролонгированное воспаление, вызываемое посредством CFA, широко использовано в исследованиях поведенческого болевого ответа (K. Walker, Mol. Med. Today, 1999, 5, 319-321), поскольку оно также считалось подходящим для изучения включения нейрональной пластичности в хронической боли (R. Sharif Naeini, Eur. J. Neuroscience, 2005, 22, 8, 2005-2015). Опыты проводили, как описано в литературе (C.J. Woolf, Br. J. of Pharmacology, 1997, 121, 417-424); 6 крыс использовали для каждой группы, каждый продукт тестировали в одной пероральной дозе 1,5 мг/кг в присутствии или в отсутствие фиксированной низкой дозы морфина (0,5 мг/кг; подкожно). Соединения формулы (I) вводили 24 часа спустя после интерплантарного введения, и анальгезирующую активность измеряли, начиная с 24 часов после введения. В таблице 5 результаты, полученные в модели CFA для репрезентативного соединения формулы (I) при совместном введении с низкой дозой морфина, перечислены в сравнении с той же дозой введенного одного морфина. Анальгезирующий эффект оценивали, используя тест Рендалла-Селитто (Randall-Selitto). Результаты выражали как средний эффект в процентах (МРЕ), который представляет собой разницу (%) в пороге болевой чувствительности между животными, обработанными лекарственными средствами, и контролями, которые получали только наполнитель (снижение ноцицептивного эффекта из-за нагрузки на лапу за счет увеличенной массы в сравнении с контролями, которые получали обработку посредством CFA). 100% защита означает, что животные, обработанные соединениями и CFA, могут переносить тот же раздражитель (масс.) как контрольное животное, которое не получило обработку посредством CFA.
2 ч
3 ч
4 ч
Репрезентативное соединение формулы (I) - пример 1 - введенное перорально в дозе, неспособной по своей сущности вызывать анальгезирущие эффекты, показало значительный умеренный эффект при введении в качестве добавки с низкой дозой морфина. Кроме того, повышение в эффективности вследствие обработки было присоединено к выдающемуся и удивительному повышению в длительности анальгезирующего эффекта. Абсолютный анальгезирующий эффект обработки с дополнением был в 2,29, 2,98 и 5,48 раз более эффективным, чем одним морфином спустя 2, 3 и 4 часа после введения лекарственного средства, соответственно. Эффект для 4 часов является особенно ощутимым, так как в это время животные, обработанные одним морфином, показали очень слабое снижение гипералгезии, в то время как животное с совместным введением морфина и соединения примера 1 были еще почти полностью защищены от гипералгезии.
Фармацевтические композиции
Соединения формулы I, их соли и их сольваты могут быть использованы в изготовлении подходящих лекарственных средств для терапевтического лечения депрессии и тревоги, определенных выше, для фармакологического лечения болезни Паркинсона, для фармакологического лечения синдромов отмены при злоупотреблении алкоголем, табаком и наркотиками, включая злоупотребление кокаином, и для того, чтобы избежать эпизодов ремиссии. Кроме того, соединения формулы I, их соли и их сольваты могут быть использованы сами по себе или в комбинации с морфином или другими опиоидными лекарственными средствами в изготовлении подходящих лекарственных средств для усиления опиоидного фармакологического действия и/или для снижения дозировки опиоидного лекарственного средства. Наконец, соединения формулы I, их соли и их сольваты могут быть использованы в изготовлении подходящих лекарственных средств для лечения толерантности и зависимости вследствие применения опиоидных лекарственных средств. Соединения данного изобретения могут быть введены перорально или парентерально. Используемый в настоящем описании термин «парентеральное» включает внутривенное, внутримышечное, подкожное введение. Для всех способов лечения, обсужденных в настоящем описании для соединений формулы (I), их солей или сольватов, суточная схема перорального приема лекарственных средств будет предпочтительно составлять от примерно 0,1 до примерно 20 мг/кг общей массы организма. Квалифицированным специалистом в данной области будет признано, что оптимальное количество и интервал индивидуальных дозировок соединений формулы (I) будет определен природой и степенью состояния, которое лечат. Данное изобретение также относится к композиции, подходящей для лечения вышеприведенных заболеваний, причем композиция содержит фармацевтически эффективное количество соединения формулы (I), его солей и его сольватов, и фармацевтически приемлемый носитель или разбавитель. Чтобы использовать соединение формулы (I) в терапии, его обычно следует готовить в лекарственной форме в соответствии с традиционными способами фармации и современными руководствами и релевантными хорошими лабораторными и производственными практиками. Предпочтительный путь введения для соединений изобретения является пероральным. Соединения данного изобретения могут быть приготовлены в широком разнообразии пероральных лекарственных форм, таких как капсулы, таблетки, пилюли, порошки и диспергируемые гранулы. Подходящие носители могут представлять собой одно или несколько веществ, которые могут также действовать как разбавители, вкусовые добавки, солюбилизаторы, замасливатели, суспендирующие средства, связывающие вещества. Подходящие носители включают, но без ограничения только ими, карбонат магния, стеарат магния, тальк, лактозу, пектин, декстрин, крахмал, метилцеллюлозу, натрий-карбоксиметилцеллюлозу, масло какао и тому подобное. Методики, используемые для приготовления пероральных препаратов, представляют собой обычное смешивание, гранулирование и компрессию или заполнение капсул. Другие формы, подходящие для перорального введения, включают эмульсии, сиропы и водные растворы. Эмульсии могут быть приготовлены с использованием эмульгаторов, например лецитина, пропиленгликоля или сорбитанмоноолеата. Водные растворы могут быть приготовлены растворением активного компонента в воде и добавлением подходящих окрашивающих веществ, вкусовых добавок, стабилизаторов.
Соединения данного изобретения могут быть приготовлены для парентерального введения (например, инъекцией или непрерывной инфузией) как композиция с подходящими носителями, включающими водные растворы наполнителей (т.е. физиологический раствор, декстроза) или и/или масляные эмульсии. Лекарственный продукт может быть представлен в стандартных лекарственных формах, например, в ампулах или предварительно наполненных шприцах.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ХИНОЛИНА И ХИНАЗОЛИНА СО СРОДСТВОМ К 5HT-РЕЦЕПТОРАМ | 2004 |
|
RU2402533C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2005 |
|
RU2409582C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
| Новые соединения | 2016 |
|
RU2734256C2 |
| ФЕНОКСИПИРИДИНИЛАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ PDE4-ОПОСРЕДОВАННЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2009 |
|
RU2509077C2 |
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |
| ПРОИЗВОДНЫЕ ГЛИЦИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2011 |
|
RU2585767C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И БЕНЗОМОРФОЛИНА В КАЧЕСТВЕ МОДУЛЯТОРА МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2517181C2 |
| АЛКИЛСУЛЬФОНАМИДХИНОЛИНЫ С АФФИННОСТЬЮ К РЕЦЕПТОРАМ NK-3 | 2006 |
|
RU2421447C2 |
Данное изобретение относится к области фармацевтики и медицины и касается применения соединений формулы (I), их фармацевтически приемлемых солей и сольватов для лечения депрессии и родственных расстройств, болезни Паркинсона, токсикомании, толерантности к морфину и зависимости от морфина. 6 з.п. ф-лы, 5 табл., 33 пр.
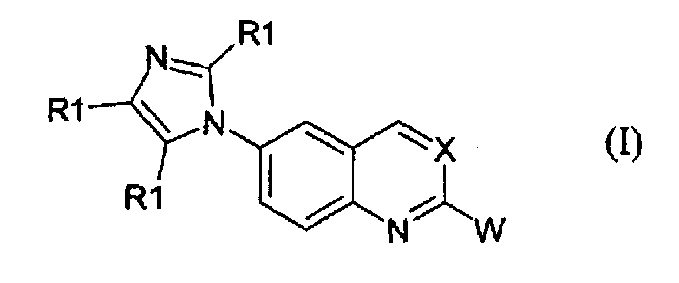
1. Применение соединений формулы (I), их фармацевтически приемлемых солей и/или сольватов для фармакологического лечения депрессии, включая большое депрессивное расстройство, дистимическое расстройство, биполярное расстройство типа II, маниакальную депрессию, тревожные расстройства и паническое расстройство,
где соединения формулы (I) представлены структурной формулой:
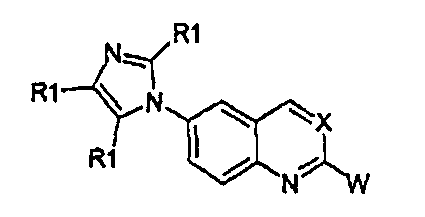
в которой X независимо выбран из -СН группы или атома азота (-N);
W независимо выбран из арильной группы или гетероарильной группы формулы II:
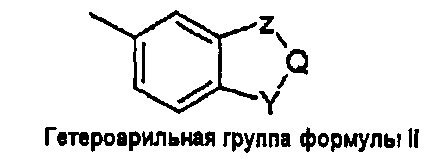
когда W представляет собой арильную группу, он является незамещенным или замещенным фенилом с одним или более заместителями, независимо выбираемыми из галогена (-F, -Cl, Вr), трифторметила (-CF3), алкила (-R2), гидроксила (-ОН), алкокси (-OR2), трифторметокси (-OCF3), циано (-CN), карбоксамидо (-CONHR3, или -NHCOR3, или -CONR2R3, или -NR2COR3), карбонила (-CO-R3), алкилтио или тиола (-SR3), сульфинила (-SOR3) и сульфонила (-SO2R3), причем R2 и R3 принимают значения, определенные ниже;
когда W представляет собой гетероарильную группу формулы (II), он является бензоконденсированным 5- или 6-членным гетероциклом, в котором
Z и Y независимо выбраны из атома кислорода (-O-), атома серы (-S-) или групп -CHR3, -CR3=-NH-, -N=;
Q независимо выбран из групп -CHR3-, -СН=, -CR3=, -CHR3- СН2-;
при условии, что комбинация Y, Z, Q групп дает 1,3-бензодиоксол, бензофуран, 2,3-дигидробензофуран, бензотиофен, 2,3-дигидробензотиофен, индол, 2,3-дигидроиндол, бензимидазол, бензоксазол, бензотиазол, 2Н-3,4-дигидробензопиран, [1,4]-бензодиоксин, 2,3-дигидро-[1,4]-бензодиоксин (1,4-бензодиоксан);
R1 независимо выбран из водорода (-Н), или С1-С4 алкила, или гидроксиметила (-СН2ОН), аминометила (-CH2NH2), алкиламинометила [-CH2NH(R2)], диалкиламинометила [-CH2N(R2)2], трифторметила (-CF3); С1-С4 алкильная группа представляет собой нормальную или разветвленную насыщенную или ненасыщенную С1-С4 углеводородную цепь; при условии, что в соединениях формулы (I) не более чем две R1 группы, замещающие имидазольное кольцо, представляют собой одновременно С1-С4 алкил или трифторметил (-CF3), и только одна R1 группа представляет собой гидроксиметил (-СН2ОН), аминометил (-CH2NH2), алкиламинометил [-CH2NH(R2)], диалкиламинометил [-CH2N(R2)2];
R2 представляет собой С1-С6 алкильную цепь; где С1-С6 алкильная цепь имеет те же значения, как определено для С1-С4 цепи, но необязательно замещенной арилом, причем арил принимает значения, определенные выше;
R3 независимо выбран из водорода, С1-C4 алкила, определенного выше для R1; включая все возможные таутомеры соединения формулы (I).
2. Применение по п.1, где W представляет собой гетероарильную группу, независимо выбираемую из следующих пента- или гексаатомных гетероциклов: 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, пиррол-2-ил, пиррол-3-ил, пиридин-4-ил, пиридин-3-ил, пиримидин-4-ил; гетероциклическое кольцо необязательно замещается одним или двумя заместителями, независимо выбираемыми из: R1, алкокси (-OR2) или гидрокси (-ОН), где R1 и R2 принимают значения, определенные в п.1.
3. Применение по п.2, их фармацевтически приемлемые соли и/или сольваты для применения в фармакологическом лечении депрессии, включая большое депрессивное расстройство, дистимическое расстройство, биполярное расстройство типа II, маниакальную депрессию, тревожные расстройства и паническое расстройство.
4. Применение соединения формулы (I) по любому из пп.1-3, их фармацевтически приемлемых солей и/или сольватов для фармакологического лечения болезни Паркинсона.
5. Применение соединения формулы (I) по любому из пп.1-3, их фармацевтически приемлемых солей и/или сольватов для фармакологического лечения симптомов отмены и устранения эпизодов ремиссии в связи со злоупотреблением алкоголем, табаком и наркотиками, включая злоупотребление кокаином.
6. Применение соединения формулы (I) по любому из пп.1-3, их фармацевтически приемлемых солей и/или сольватов самих по себе или в комбинации с морфином или другими опиоидными лекарственными средствами для усиления опиоидного фармакологического действия и/или для снижения дозировки опиоидного лекарственного средства.
7. Применение соединений формулы (I) по любому из пп.1-3, их фармацевтически приемлемых солей и/или сольватов для фармакологического лечения толерантности и зависимости, вызванной применением опиоидных лекарственных средств.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| US 7196087, B2, 27.02.2007 | |||
| RU 2005134486, A, 10.05.2006. | |||

