ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к производным глицина, действующим в качестве антагонистов мускариновых рецепторов, к способам их получения, к содержащим их композициям и к их терапевтическому применению.
УРОВЕНЬ ТЕХНИКИ
В настоящее время в качестве антагонистов мускариновых (M) рецепторов для индуцирования бронходилатации при лечении респираторных заболеваний используют четвертичные аммониевые соли. Примерами широко известных антагонистов мускариновых рецепторов являются, например, ипратропия бромид и тиотропия бромид.
Для лечения воспалительных или обструктивных заболеваний дыхательных путей, таких как астма и хроническое обструктивное заболевание легких (COPD), были предложены различные классы химических соединений, действующих как селективные антагонисты M3 рецептора.
Производные карбамата хинуклидина и их применение в качестве антагонистов M3 раскрыты, например, в WO 02/051841, WO 03/053966 и WO 2008/012290.
В настоящее время указанные антагонисты мускариновых рецепторов М и M3 вводят с помощью ингаляции, для того чтобы доставить лекарственное средство непосредственно на место его воздействия, ограничивая тем самым его системное действие и любой нежелательный побочный эффект в результате системной абсорбции. Но, несмотря на то, что при ингаляционном введении может быть уменьшено системное воздействие, тем не менее, соединения известного уровня техники могут, по меньшей мере потенциально, проявлять нежелательные побочные эффекты в результате системной абсорбции.
Поэтому чрезвычайно важно создать антагонисты M3 рецептора, способные оказывать местное воздействие, обладающие при этом высокой селективностью и нестабильностью в плазме крови. Указанные лекарственные средства сразу после абсорбции распадаются на неактивные соединения, которые лишены любых системных побочных эффектов, характерных для антагонистов мускариновых рецепторов.
В WO 2010/072338 описаны соединения азонийбицикло[2.2.2]октана, действующие в качестве антагонистов мускариновых рецепторов, и, кроме того, обладающие упомянутыми выше желательными с точки зрения терапии характеристиками.
Как это ни удивительно, но было обнаружено, что производные глицина, подробно описанные ниже, обладают еще более высокой селективностью и нестабильностью в плазме крови, чем эти указанные последними соединения, и, соответственно, минимальными побочными эффектами.
Таким образом, соединения настоящего изобретения представляют собой слабые наркотики, так как они способны вызывать более устойчивый бронходилатирующий эффект в легких, и более последовательно и быстро трансформироваться в неактивные метаболиты после прохождения через плазму крови человека.
Такое их свойство обеспечивает большие преимущества с точки зрения безопасности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к производным глицина общей формулы (I) или (VI), действующим в качестве антагонистов мускариновых рецепторов, к способам их получения, к содержащим их композициям, к их терапевтическому применению и к комбинациям с другими фармацевтическими активными ингредиентами, в число которых входят, например, активные ингредиенты, применяемые в настоящее время при лечении респираторных заболеваний, например, бета2-агонисты, кортикостероиды, ингибиторы P38 MAP киназы, ингибиторы IKK2, ингибиторы HNE, ингибиторы PDE4, модуляторы лейкотриена, нестероидные противовоспалительные средства и регуляторы слизи.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В частности, изобретение относится к производным глицина общей формулы (I)

где
R1 выбирают из группы, состоящей из -H, линейного или разветвленного (C1-C10)алкила, (C2-C6)алкенила, арила, (C3-C8)циклоалкила, (C5-C10)гетероциклоалкила, арил(C1-C6)алкила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -N(R5)(R6), -CN, -CON(R5)2, -NHCO(R5), -COR5, -CO2R5, (C1-C10)алкилсульфанила, (C1-C10)алкилсульфинила, (C1-C10)алкилсульфонила, (C1-C10)алкила, (C1-C10)алкилкарбоксила, (C1-C10)алкокси, арила, арилокси и гетероарила;
G выбирают из группы, состоящей из -OC(O)-, -SO2- и -C(O)-;
R2 выбирают из группы, состоящей из -H, (C1-C10)алкила и арил(C1-C6)алкила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -N(R5)2, -CN, -CON(R5)2, -NHCO(R5), -CO(R5), -CO2(R5), (C1-C10)алкилсульфанила, (C1-C10)алкилсульфинила, (C1-C10)алкилсульфонила, (C1-C10)алкила, (C1-C10)алкилкарбоксила, (C1-C10)алкокси, арила, арилокси и гетероарила;
R3 выбирают из группы, состоящей из -H, (C1-C10)алкила, арила, (C3-C8)циклоалкила, гетероарила, арил(C1-C6)алкила и гетероарил(C1-C6)алкила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -N(R5)2, -CN, -CON(R5)2, -NHCO(R5), -CO(R5), -CO2(R5), (C1-C10)алкилсульфанила, (C1-C10)алкилсульфинила, (C1-C10)алкилсульфонила, (C1-C10)алкила, (C1-C10)алкилкарбоксила, (C1-C10)алкокси, арила, арилокси, арил(C1-C10)алкиленокси и гетероарила;
R6 выбирают из группы, состоящей из фрагментов формулы (i), (ii), (iii) и (iv)

где
m=1, 2 или 3;
n=1, 2 или 3;
A- представляет собой физиологически приемлемый анион;
R4 представляет собой группу формулы (Y)
-(CH2)p-P-(CH2)q-W
(Y)
где
p представляет собой 0 или целое число от 1 до 4;
q представляет собой 0 или целое число от 1 до 4;
P отсутствует или его выбирают из группы, состоящей из -O-, -S-, -SO-, -SO2-, -C(O)-, -N(R5)- -CH=CH-, -N(R5)(SO2)-, -N(R5)(COO)-, -N(R5)(C(O))-, -S(O2)N(R5)-, -CO(O)N(R5)- и -C(O)N(R5)-;
W выбирают из группы, состоящей из H, линейного или разветвленного (C1-C10)алкила, (C2-C6)алкенила, арила, (C3-C8)циклоалкила, (C5-C10)гетероциклоалкила, арил(C1-C6)алкила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -N(R5)2, -CN, -CON(R5)2, -NH(COR5), -CO(R5), -CO2(R5), (C1-C10)алкилсульфанила, (C1-C10)алкилсульфинила, (C1-C10)алкилсульфонила, (C1-C10)алкила, (C1-C10)алкилкарбоксила, (C1-C10)алкокси, арила, арилокси и гетероарила;
R5 и R6 независимо выбирают из группы, состоящей из -H, (C1-C10)алкила, (C1-C10)алкокси, (C2-C6)алкинила, (C2-C6)алкенила, (C3-C7)циклоалкила, (C3-C7)циклоалкил-(C1-C10)алкила, гетероарила, (C1-C10)алкилгетероарила и арила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -CN, -CONH2, (C1-C10)алкилсульфанила, (C1-C10)алкилсульфинила, (C1-C10)алкилсульфонила, (C1-C10)алкила, (C1-C10)алкилкарбоксила, (C1-C10)алкокси, арила, арилокси и гетероарила, и
их фармацевтически приемлемым солям.
Настоящее изобретение также относится к соединениям общей формулы (VI):
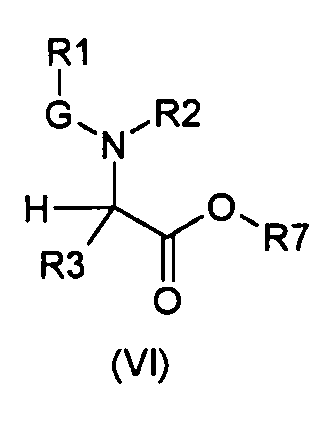
где
R1, R2, R3 и G определены выше;
R7 выбирают из группы, состоящей из фрагментов формулы (v), (vi), (vii) и (viii)
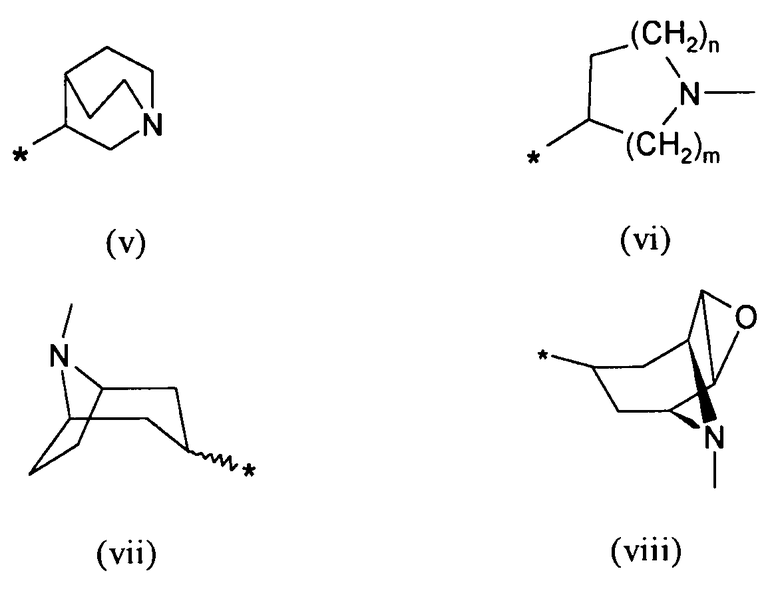
где
m и n определены выше.
Символ звездочки внутри групп формул (i)-(viii) обозначает точку присоединения к остальной части молекулы.
В настоящем изобретении, если не указано иначе, то термин "галоген" включает атомы фтора, хлора, брома и йода.
Выражение "(C1-C10)алкил" относится к алкильным группам с линейной или разветвленной цепью, в которых число углеродных атомов составляет от 1 до 10. Примерами указанных групп являются метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, гексил, гептил, октил, нонил, децил и другие подобные группы.
Выражение "(C2-C6)алкенил" относится к линейным или разветвленным углеродным цепям с одной или более двойными связями. Примерами указанных групп являются этенил, пропенил, бутенил, пентенил, гексенил и другие подобные группы.
Выражение "(C1-C10)алкокси" относится к определенным выше алкил-окси (например, алкокси) группам. Примерами указанных групп являются метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, пентокси, гексокси и другие подобные группы.
Аналогично, выражения "(C1-C10)алкилсульфанил", "(C1-C10)алкилсульфинил", "(C1-C10)алкилсульфонил" или "(C1-C10)алкилкарбоксил" относятся, соответственно, к алкил-S-, алкил-SO-, алкил-SO2- или алкил-COO группам.
Выражение "(C3-C8)циклоалкил" относится к циклическим неароматическим углеводородным группам с числом углеродных атомов от 3 до 8. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и другие подобные группы.
Выражение "(C5-C10)гетероциклоалкил" относится к (C5-C10)циклоалкилу, в котором один или более атомов водорода заменены одним или более атомами галогена, которые могут быть одинаковыми или различными.
Выражение "арил" относится к моно- или би- или трициклическим кольцевым системам, которые имеют от 6 до 20 атомов в кольце, предпочтительно, от 6 до 15, и где по меньшей мере одно кольцо является ароматическим.
Выражения "арил(C1-C6)алкил", "гетероарил(C1-C6)алкил и (C3-C8)циклоалкил(C1-C6)алкил относятся к (C1-C6)алкильным группам, дополнительно замещенным арильными, гетероарильными или циклоалкильными кольцами.
Выражение "арилокси" относится к -O-арильной группе. Примером может служить фенилокси.
Выражение "(C1-C10)алкилен" относится к цепи с числом -CH2-групп от 1 до 10. Примером может служить метилен.
Выражение "(C1-C10)алкиленокси" относится к -O(C1-C10)алкилену.
Выражение "арил(C1-C10)алкиленокси" относится к (C1-C10)алкиленокси, дополнительно замещенному арилом. Примером может служить бензилокси.
Выражение "гетероарил" относится к моно, би- или трициклическим кольцевым системам, которые имеют от 5 до 20 атомов в кольце, предпочтительно, от 5 до 15, в которых по меньшей мере одно кольцо является ароматическим, и в которых по меньшей мере один атом в кольце является гетероатомом или гетероароматической группой (например, N, NH, S или O).
Примеры подходящих арильных или гетероарильных моноциклических систем включают, например, фрагменты тиофена, бензола, пиррола, пиразола, имидазола, изоксазола, оксазола, оксадиазола, изотиазола, тиазола, пиридина, имидазолидина и фурана и другие подобные фрагменты.
Примеры подходящих арильных или гетероарильных бициклических систем включают фрагменты бензодиоксола, нафталина, бифенилена, пурина, птеридина, бензотриазола, хинолина, изохинолина, индола, изоиндола, бензотиофена, дигидробензодиоксина, дигидробензодиоксепина и бензооксазина и другие подобные фрагменты.
Предпочтительно, чтобы физиологически приемлемые анионы A- включали анионы, выбранные из хлорида, бромида, йодида, трифторацетата, формиата, сульфата, фосфата, метансульфоната, нитрата, малеата, ацетата, цитрата, фумарата, тартрата, оксалата, сукцината, бензоата и п-толуолсульфоната.
Помимо аниона A-, во всех случаях, когда в соединениях формулы (I) дополнительно присутствуют основные аминогруппы, наряду с ранее упомянутыми анионами могут присутствовать и дополнительные физиологически приемлемые анионы. Аналогично, в случае присутствия кислотных групп, таких как группы COOH, могут также присутствовать и соли соответствующего физиологически приемлемого катиона, например, включающие ионы щелочных или щелочноземельных металлов.
Первая предпочтительная группа соединений общей формулы (I) или (VI) включает соединения, в которых G выбирают из группы, состоящей из -OC(O)-, -SO2- и -C(O)-, R1 выбирают из группы, состоящей из линейного или разветвленного (C1-C10)алкила, арила, (C3-C8)циклоалкила, арил(C1-C6)алкила, (C2-C6)алкенила, (C5-C10)гетероциклоалкила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из атомов галогена, -N(R5)(R6), (C1-C10)алкила, (C1-C10)алкилкарбоксила, (C1-C10)алкокси, арилокси и гетероарила; R2 представляет собой H; и R3 и R6 определены выше.
Еще более предпочтительными внутри этого класса являются соединения общей формулы (I) или (VI), в которых R1 выбирают из группы, состоящей из метила, этила, метоксиэтоксила, трет-бутила, этенила, циклогексила, фенила, метоксифенила, хлорфенила, дифторфенила, диметилтиазола, трифторэтила, фенилэтила, циклопентила, метилэтоксила, оксофенилэтила, тиофенила, тиазолила, фторфенила, аминофенила, трет-бутоксикарбониламинофенила и метилфенила.
Другая предпочтительная группа соединений общей формулы (I) или (VI) внутри этого класса включает соединения, в которых G, R1 и R2 определены выше; R3 выбирают из группы, состоящей из (C1-C10)алкила, арила и гетероарила, необязательно замещенного одной или более группами, выбранными из атомов галогена, (C1-C10)алкила, (C1-C10)алкокси и арил(C1-C10)алкиленокси; и R6 определен выше.
Еще более предпочтительной группой соединений общей формулы (I) или (VI) являются соединения, в которых G, R1, R2 и R3 определены выше; R6 выбирают из группы, состоящей из фрагментов формулы (i), (ii) и (iii), где A- определен выше, R4 представляет собой группу формулы (Y), где p равняется 0, 1 и 3, P представляет собой -C(O)-, q равняется 0, W выбирают из группы, состоящей из (C1-C10)алкила, арила, гетероарила, (C5-C10)гетероциклоалкила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, (C1-C10)алкила, (C1-C10)алкокси, -OH и (C1-C10)алкилкарбоксила.
Еще более предпочтительными внутри этого класса являются соединения общей формулы (I) или (VI), в которых W выбирают из группы, состоящей из фенила, бензотиоксола, тиофенила и тиазолила, необязательно замещенного одним или более атомами галогена, OH, метила и метилкарбоксила.
Согласно конкретным вариантам осуществления, настоящее изобретение предлагает следующие соединения:
Соединения формулы (I) и (VI) могут иметь по меньшей мере один хиральный центр, когда R3 не является H.
Кроме того, очевидно, что в соединениях формулы (I) и (VI), в зависимости от значений R1, R2, R6 и R7, могут присутствовать дополнительные центры асимметрии. Поэтому изобретение также включает любой из оптических стереоизомеров, диастереоизомеров и их смеси в любом соотношении.
В одном из предпочтительных вариантов осуществления хиральный центр на кольцах (i), (iii), (iv), (v), (vii), (viii) и (ii), (vi), когда m и n являются различными, имеет R конфигурацию.
Так как в настоящем изобретении не всегда определена абсолютная конфигурация диастереоизомеров, их указывают в примерах как диастереоизомер 1, 2 или как их смесь.
Изобретение также предлагает фармацевтические композиции соединений формулы (I) или (VI) самих по себе или в комбинации или в смеси с одним или более фармацевтически приемлемыми носителями и/или эксципиентами.
Изобретение также предлагает фармацевтические композиции, применяемые для ингаляционного введения, такие как, например, порошки для ингаляции; содержащие пропеллент дозируемые аэрозоли или несодержащие пропеллент составы для ингаляции.
Изобретение также предлагает соединения формулы (I) или (VI) для применения в качестве лекарственного средства.
Изобретение также предлагает соединения формулы (I) или (VI) для применения при лечении бронхообструктивных или воспалительных заболеваний, предпочтительно, астмы или хронического бронхита или хронического обструктивного заболевания легких (COPD).
В дополнительном аспекте изобретение предлагает применение соединений формулы (I) или (VI) для изготовления лекарственного средства для предотвращения и/или лечения бронхообструктивных или воспалительных заболеваний, предпочтительно, астмы или хронического бронхита или хронического обструктивного заболевания легких (COPD).
Изобретение также предлагает способ предотвращения и/или лечения бронхообструктивных или воспалительных заболеваний, предпочтительно, астмы или хронического бронхита или хронического обструктивного заболевания легких (COPD), который включает введение субъекту, в случае, если это ему необходимо, терапевтически эффективного количества соединения общей формулы (I) или (VI).
Изобретение также предлагает фармацевтические композиции, применяемые для ингаляционного введения, такие как порошки для ингаляции; содержащие пропеллент дозируемые аэрозоли или несодержащие пропеллент составы для ингаляции.
Изобретение также относится к устройству, которое может представлять собой однодозовый или многодозовый ингалятор сухого порошка, ингалятор отмеренных доз и небулайзер мягких аэрозолей, включающему соединения формулы (I) или (VI).
Изобретение также относится к набору, включающему упомянутые выше фармацевтические композиции в подходящем флаконе или контейнере, и устройство, которое может представлять собой однодозовый или многодозовый ингалятор сухого порошка, ингалятор отмеренных доз и небулайзер мягких аэрозолей, в которых предусмотрена фиксация упомянутых выше флакона или контейнера.
Настоящее изобретение также относится к способу получения соединения формулы (I) или (VI), который включает:
(a) реакцию соединений общей формулы (III)

(b) с соединениями общей формулы (II)
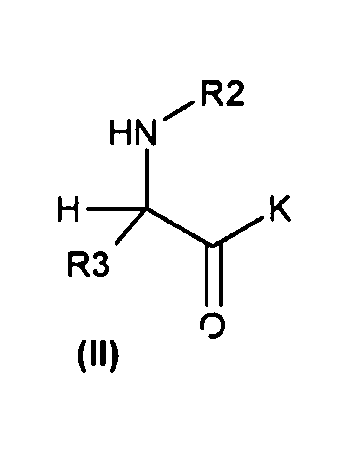
с получением соответствующих соединений общей формулы (IV)

(c) реакцию сочетания между соединениями общей формулы (IV) и (V)

С получением соединения общей формулы (VI)

(d) алкилирование соединений общей формулы (VI) алкилирующими агентами общей формулы (Y)
-(CH2)p-P-(CH2)q-W
(Y),
соединенных с группой A, которая является подходящей уходящей группой, с получением соединения общей формулы (I), где p, P, q, W, R1, R2, R3, R6 и R7 имеют упомянутые выше значения.
Настоящее изобретение также относится к способу получения соединения формулы (VI), который включает:
(a) обработку кислоты формулы (IV) одним или более эквивалентами конденсирующего агента с получением активированного промежуточного соединения;
(b) реакцию активированного промежуточного соединения со спиртом общей формулы (V).
Настоящее изобретение также относится к способу получения соединения формулы (VI), который включает:
(a) превращение соединения формулы (IV), где K=OH, в соответствующий ацилгалогенид формулы (IV), где K=галогенид
(b) реакцию ацилгалогенида формулы (IV) с соединением формулы (V).
Соединения формулы (I) или (VI) могут быть получены известными методами.
Исходные материалы для получения соединений формулы (I) или (VI), так же как и любой реагент способа, являются известными соединениями или их можно легко получить с помощью известных методик.
Условия проведения химических реакций, которые могут быть использованы в способе изобретения, описаны более подробно ниже, и они, кроме того, приводятся на следующей схеме 1.

Схема 1
Методика получения соединений формулы (VI) и (I)
Соединения, описанные в настоящем изобретении, наиболее удобно получать, исходя из соединений общей формулы (II), в которых K может являться или гидроксильной группой, или подходящей защитной группой для гидроксильной группы (например, K=(C1-C10)алкокси, такая как OMe). Соединения общей формулы (II) могут быть подвергнуты реакции с соединениями общей формулы (III), в которых z является подходящей уходящей группой, такой как галогенид (то есть, хлор, бром, фтор), или кислород, замещенный другой R1-G группой (например, когда R1 представляет собой трет-бутильную группу, G представляет собой COO группу, и z представляет собой -O-G-R1 группу, соединение (III) является ди-трет-бутилдикарбонатом или Boc ангидридом; когда R1 представляет собой метил, G представляет собой CO, и z представляет собой -O-G-R1 группу, соединение (III) является уксусным ангидридом; когда R1 представляет собой CF3, G представляет собой SO2, и z представляет собой -O-G-R1 группу, соединение (III) является трифторметансульфоновым ангидридом). Эта реакция может быть осуществлена в соответствии со стандартными методиками, описанными в литературе. В обычной методике соединения формулы (III) добавляют к раствору соединений формулы (II) в соответствующем растворителе (например, дихлорметане, этилацетате, тетрагидрофуране и воде) с получением соответствующих соединений общей формулы (IV). Реакцию удобно промотировать с помощью основания, такого как триэтиламин, пиридин, 4-диметиламинопиридин и гидроксид натрия. Эту реакцию обычно проводят в температурном интервале от 0°C до 130°C в течение времени от 30 минут до 74 часов. Реакция может быть проведена при обычном нагревании (используя масляную баню) или при микроволновом нагревании. Реакция может быть проведена в открытом сосуде или в герметизированной колбе.
Реагенты общей формулы (III) производятся промышленностью или могут быть получены в соответствии со стандартными методиками, описанными в литературе. Когда z в соединениях формулы (III) представляет собой гидроксильную группу (z=OH), она может быть или превращена в подходящую уходящую группу (такую как галогенид или кислород, замещенный другой R1-G группой с образованием ангидрида), или может быть подвергнута конденсации с соединениями формулы (II) при стандартных условиях реакции амидирования и условиях реакции сочетания пептида.
Реакция сочетания между соединениями общей формулы (IV) и (V) может быть осуществлена различными путями (обзор подходящих реакций приведен в монографии Carey, F.A. and Sundeberg, R.J. Advanced Organic Chemistry, Third Edition (1990), Plenum Press, New York and London, pg 145).
В частности, в случае, когда K представляет собой защитную группу для гидроксильной группы, защитная группа должна быть удалена перед проведением реакции сочетания с соединением (V). Например, если K=OMe, может быть проведен гидролиз эфирного фрагмента путем обработки соединения (IV), где K=OMe, соответствующим водным раствором основания, выбранного из гидроксида натрия, лития и калия, в подходящих растворителях (например, тетрагидрофуране, диоксане и других подобных растворителях). Реакция протекает при комнатной температуре в течение времени от 1 часа до 36 часов.
Первый вариант - в обычной методике, соединения (VI) могут быть получены путем реакции конденсации между соединениями формулы (V) и (IV), где K=OH, при стандартных условиях реакции амидирования и условиях реакции сочетания пептида. Например, обработка соединения (IV) одним или более эквивалентами выпускаемого промышленностью конденсирующего агента, такого как карбодиимид (например Ν,Ν'-дициклогексилкарбодиимид (DCC) и другие подобные), например, в присутствии N-гидроксибензотриазола (HOBt) и затем взаимодействие активированного промежуточного соединения со спиртом (V) приводит к образованию соединений формулы (VI). В реакционной смеси может также присутствовать органическое основание, такое как триэтиламин или 4-диметиламинопиридин. Активированное промежуточное соединение может быть или выделено, или предварительно образовано или образовано in situ. Подходящие растворители для реакции сочетания включают, но этим не ограничивая, галогенуглеводородные растворители (например, дихлорметан), тетрагидрофуран, диоксан и ацетонитрил. Реакция протекает в температурном интервале от 0°C до 170°C в течение времени в интервале от приблизительно 1 часа до 72 часов. Реакция может быть проведена при обычном нагревании (используя масляную баню) или при микроволновом нагревании. Реакция может быть проведена в открытом сосуде или в герметизированной колбе.
Второй вариант - в некоторых вариантах осуществления настоящего изобретения соединение формулы (IV) где K=OH, сначала превращают в соответствующий ацилгалогенид (IV), где K=галогенид. Эта активация может быть осуществлена в соответствии с одной из стандартных методик, описанных в литературе. Например, обработка кислоты (IV), где K=OH, одним или более эквивалентами оксалилхлорида в присутствии каталитического количества диметилформамида (DMF) в галогенуглеводородном растворителе, таком как дихлорметан, при температуре в интервале от 0°C до 35°C, дает требуемый ацилхлорид (IV), где K=Cl.
Спирт (V) затем подвергают взаимодействию с ацилгалогенидом (IV), используя известные методы. Реакция может быть промотирована основанием, таким как триэтиламин, пиридин и 4-диметиламинопиридин, в соответствующем растворителе (например, дихлорметане). Эту реакцию проводят в температурном интервале от 0°C до 130°C в течение времени от 1 часа до 74 часов. Реакция может быть проведена при обычном нагревании (используя масляную баню) или при микроволновом нагревании. Реакция может быть проведена в открытом сосуде или в герметизированной колбе.
Третий вариант - в качестве альтернативы, ацилирование спирта (V) с получением соединений общей формулы (VI) может быть осуществлено с помощью методик, в которых превращают in situ кислоту (IV), где K=OH, в соответствующие ацилгалогениды. Например, спирты (V) подвергают взаимодействию с кислотами (IV), где K=OH, в присутствии трифенилфосфина и галогенуглеводородного растворителя, такого как тетрахлорид углерода или дихлорметан, при приблизительно комнатной температуре, максимально в течение 16 часов (Lee, J.B. J.Am.Chem.Soc, 1966, 88, 3440).
Четвертый вариант - в другом способе получения соединений настоящего изобретения кислота (IV), где K=OH, может быть активирована с помощью других выпускаемых промышленностью активирующих агентов, таких как бромтрипирролидинофосфония гексафторфосфат (PyBrOP) или карбонилимидазол, в подходящем растворителе (например, дихлорметане, тетрагидрофуране и DMF), при приблизительно комнатной температуре. Последующая реакция активированного промежуточного соединения со спиртом (V) дает требуемое соединение формулы (VI). Реакция может также требовать использования органического основания, такого как диизопропилэтиламин и другие подобные основания, и обычно протекает при приблизительно комнатной температуре.
Пятый вариант - в другом способе получения соединений настоящего изобретения соединения (VI) могут быть эффективно получены путем реакции конденсации между кислотами (IV), где K=OH, и спиртом (V) при стандартных условиях реакции Мицунобу (Kumara Swamy, K.C., Chem. Rev. 2009, 109, 2551-2651). Например, кислоты (IV) и спирт (V) подвергают взаимодействию в присутствии фосфина (например, трифенилфосфина) и азадикарбоксилатного эфира (например, диэтил азодикарбоксилата или диизопропил азодикарбоксилата) в апротонном растворителе, таком как тетрагидрофуран. Реакция обычно протекает в температурном интервале от 0°C до 100°C в течение времени в интервале от 30 минут до 72 часов.
В некоторых вариантах осуществления настоящего изобретения в качестве защитной группы в соединениях общей формулы (VI) наиболее удобно использовать группу R1-G-, и она может быть удалена с получением соединений общей формулы (VIII). Эти защитные группы выбирают, используют и удаляют в соответствии со стандартными методами органического синтеза (Green T.W. and Wuts P.G.M. (1991) Protecting Groups in Organic Synthesis, John Wiley et Sons). Например, если R1 в соединениях (VI) представляет собой трет-бутил, и G представляет собой O(CO), трет-бутилоксикарбонильная (boc) защитная группа может быть расщеплена путем обработки соединений общей формулы (VI) с помощью протонной кислоты, такой как хлористоводородная кислота, трифторуксусная кислота и другие подобные кислоты. Подходящие растворители для проведения реакции снятия защиты включают, но этим не ограничивая, 1,4-диоксан, тетрагидрофуран, дихлорметан и метанол. Вода может также присутствовать в реакционной смеси. Реакция протекает в температурном интервале от 0°C до 80°C в течение времени в интервале от нескольких минут до 72 часов.
Соединения общей формулы (VIII) затем могут быть подвергнуты реакции сочетания с соединениями общей формулы (III) с получением соединений (VI), используя известные методики. Например, условия проведения реакции сочетания могут быть выбраны из условий, описанных для проведения реакции сочетания между соединением (II) и (III) на схеме 1.
Соединение общей формулы (VI) может быть получено или в виде индивидуального диастереоизомера, или в виде смеси диастереоизомеров. Например, в случае, когда R7 представляет собой группу формулы (V), спирт имеет или R, или S конфигурацию. Если используют R-энантиомер, соединение формулы (VI) может быть получено в S-R конфигурации, в R-R конфигурации или в виде смеси диастереоизомеров (R-R и S-R конфигурация).
Смесь диастереоизомеров может быть превращена в соединения формулы (I) схемы 1 или разделена с получением двух индивидуальных диастереоизомеров, которые, в свою очередь, могут быть превращены в соединения формулы (I) схемы 1. Это разделение может быть осуществлено с помощью известных методов. Эти методы включают, но этим не ограничивая, хроматографическую очистку, очистку с помощью препаративной ВЭЖХ и кристаллизацию. Например, два диастереоизомера могут быть разделены флэш-хроматографией на силикагеле, элюируя подходящими растворителями или смесью растворителей, таких как DCM и метанол, и другие подобные. В другом способе настоящего изобретения разделение диастереоизомеров может быть осуществлено, используя колонку, заполненную хиральной неподвижной фазой, например, Chiralpack® AY или Chiralcel OD®, или Chiralcel OZ®, и элюируя, например, ацетонитрилом и/или смесью ацетонитрила и спирта. В качестве варианта, разделение диастереоизомеров удобнее всего осуществлять путем кристаллизации из подходящего растворителя (например, этилового эфира и ацетона), в виде свободного основания или после образования подходящей соли (например, соли D-винной кислоты)).
Алкилирование соединений общей формулы (VI) алкилирующими агентами общей формулы (Y), связанных с подходящей уходящей группой A, выбранной из группы, состоящей из галогенида (то есть, брома, йода, хлора) и сульфонатного эфира (то есть, тозилата, трифлата, мезилата), дает соединения общей формулы (I). Этот тип реакции подробно описан в литературе при нескольких различающихся условиях, например, реакция может быть проведена без растворителя или в подходящем растворителе, выбранном из ацетонитрила, этилацетата, DMF, DMSO и тетрагидрофурана. Реакция обычно протекает при температурах от 0°C до 170°C в течение времени в интервале от нескольких минут до 72 часов. Реакция может быть проведена при обычном нагревании (используя масляную баню) или при микроволновом нагревании. Реакция может быть проведена в открытом сосуде или в герметизированной колбе.
Соединения общей формулы (I) в схеме 1 могут рассматриваться в качестве конечных продуктов или могут быть подвергнуты дальнейшему взаимодействию с получением других соединений общей формулы (I). Так, например, фрагмент R1, R2, R3 или R6 группы в общей формуле (I) может быть подвергнут реакциям окисления, восстановления или расщепления (например, для удаления необходимой защитной группы) с получением других конечных соединений общей формулы (I).
Настоящее изобретение также предлагает фармацевтические композиции соединений формулы (I) или (VI) в смеси с одним или более фармацевтически приемлемыми носителями, например, носителями, описанными в монографии Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Введение соединений изобретения может быть осуществлено, в зависимости от потребностей пациента, например, перорально, назально, парентерально (подкожно, внутривенно, внутримышечно, интрастернально и путем инфузии), ингаляционно, ректально, вагинально, местно, локально, трансдермально и окулярно.
Для введения соединений изобретения могут быть использованы различные твердые пероральные дозированные формы, включающие такие твердые формы, как таблетки, желатиновые капсулы, капсулы, каплеты, гранулы, пастилки и порошки. Соединения настоящего изобретения могут быть введены сами по себе или вместе с различными известными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмалы) и эксципиентами, включающими, но этим не ограничивая, суспендирующие средства, солюбилизаторы, буферные вещества, связующие, дезинтегранты, консерванты, окрашивающие вещества, вещества, корригирующие вкус и запах лекарственного средства, лубриканты и другие подобные эксципиенты. Предпочтительными также являются капсулы, таблетки и гелеобразные препараты с замедленным высвобождением активного ингредиента.
Для введения соединений изобретения могут быть также использованы различные жидкие пероральные дозированные формы, включающие водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие дозированные формы могут также содержать подходящие известные инертные разбавители, такие как вода, эксципиенты, такие как консерванты, смачивающие средства, подсластители, вещества, корригирующие вкус и запах лекарственного средства, а также средства для эмульгирования и/или суспендирования соединений изобретения. Соединения изобретения могут быть инъецированы, например, внутривенно, в форме изотонического стерильного раствора. Также являются возможными и другие дозированные формы.
Суппозитории для ректального введения соединений настоящего изобретения могут быть приготовлены путем смешения соединения с подходящим эксципиентом, таким как масло какао, салицилаты и полиэтиленгликоли.
Дозированные формы для вагинального введения могут представлять собой крем, гель, пасту, пену, или спрей, содержащие помимо активного ингредиента известные в области фармацевтики подходящие носители.
Фармацевтическая композиция для местного введения может находиться в форме кремов, мазей, линиментов, лосьонов, порошков, спреев и капель, подходящих для введения на коже, в глаз, ухо или нос. Местное введение может также включать трансдермальное введение с помощью таких средств, как трансдермальные пластыри.
Для лечения заболеваний дыхательных путей, соединения согласно изобретению предпочтительно вводить путем ингаляции.
Композиции для ингаляции включают порошки для ингаляции; содержащие пропеллент дозируемые аэрозоли или несодержащие пропеллент составы для ингаляции.
Для введения в виде сухого порошка могут применяться известные однодозовые и многодозовые ингаляторы. В этом случае порошок может быть засыпан в желатиновые, пластмассовые или другие капсулы, картриджи или блистерные упаковки, или в резервуар.
К соединениям изобретения в форме порошка могут быть добавлены разбавитель или носитель, обычно нетоксичные и химически инертные по отношению к соединениям изобретения, например, лактоза или любая другая добавка, применяемая для улучшения свойств вдыхаемой фракции.
Аэрозоли для ингаляции, содержащие газ-пропеллент, такой как гидрофторалканы, могут содержать соединения изобретения или в форме раствора, или в диспергированной форме. Составы с пропеллентом могут также содержать другие ингредиенты, такие как сорастворители, стабилизаторы и необязательно другие эксципиенты.
Несодержащие пропеллент составы для ингаляции, включающие соединения изобретения, могут находиться в форме растворов или суспензий в водной, спиртовой или водноспиртовой среде, и их доставка может быть осуществлена с помощью струйных или ультразвуковых небулайзеров или небулайзеров мягких аэрозолей.
Соединения изобретения могут быть введены в виде единственного активного ингредиента или в комбинации с другими фармацевтическими активными ингредиентами, включающими лекарственные средства, которые в настоящее время применяют при лечении респираторных заболеваний, например, бета2-агонисты, кортикостероиды, ингибиторы P38 MAP киназы, ингибиторы IKK2, ингибиторы HNE, ингибитор PDE4, модуляторы лейкотриена, нестероидные противовоспалительные средства и регуляторы слизи.
Изобретение также предлагает комбинации соединения формулы (I) или (VI) с β2-агонистом, выбранным из группы, состоящей из GSK-642444, индакатерола, милветерола, арформотерола, сальбутамола, левалбутерола, тербуталина, AZD-3199, BI-1744-CL, LAS-100977, бамбутерола, изопротеренола, прокатерола, кленбутерола, репротерола, фенотерола и ASF-1020.
Изобретение также предлагает комбинации соединения формулы (I) или (VI) с кортикостероидом, выбранным из группы, состоящей из пропионата, циклесонида, мометазона фуроата и будесонида.
Изобретение также предлагает комбинации соединения формулы (I) или (VI) с ингибитором P38, выбранным из группы, состоящей из семапимода, талмапимода, пирфенидона, PH-797804, GSK-725, минокина и лосмапимода.
Изобретение также предлагает комбинации соединения формулы (I) или (VI) с ингибитором IKK2.
Изобретение также предлагает комбинации соединения формулы (I) или (VI) с ингибитором HNE, выбранным из группы, состоящей из AAT, ADC-7828, Aeriva, TAPI, AE-3763, KRP-109, AX-9657, POL-6014, AER-002, AGTC-0106, respriva, AZD-9668, zemaira, AAT IV, PGX-100, элафина, SPHD-400, проластина C и ингалируемого проластина.
Изобретение также предлагает комбинации соединения формулы (I) или (VI) с ингибитором PDE4, выбранным из группы, состоящей из AN-2728, AN-2898, CBS-3595, апремиласта, ELB-353, KF-66490, K-34, LAS-37779, IBFB-211913, AWD-12-281, ципамфиллина, циломиласта, рофлумиласта, BAY19-8004 и SCH-351591, AN-6415, indus-82010, TPI-PD3, ELB-353, CC-11050, GSK-256066, оглемиласта, OX-914, тетомиласта, MEM-1414 и RPL-554.
Изобретение также предлагает комбинации соединения формулы (I) или (VI) с модулятором лекотриена, выбранным из группы, состоящей из монтелукаста, зафирлукаста и пранлукаста.
Изобретение также предлагает комбинации соединения формулы (I) или (VI) с нестероидным противовоспалительным препаратом, выбранным из группы, состоящей из ибупрофена и кетопрофена.
Изобретение также предлагает комбинации соединения формулы (I) или (VI) с регулятором слизи, выбранным из группы, состоящей из INS-37217, диквафозола, сибенадета, CS-003, талнетанта, DNK-333, MSI-1956 и гефитиниба.
Дозирование соединений настоящего изобретения зависит от ряда факторов, включающих конкретное заболевание, подвергаемое лечению, тяжесть симптомов, способ введения, частоту введения и интервал между введением лекарственного средства, конкретно используемое соединение, активность, токсилогические свойства и фармакокинетические свойства соединения.
Желательно, чтобы соединения формулы (I) или (VI) вводили, например, при дозе от 0,001 до 1000 мг/сутки, предпочтительно, от 0,1 до 500 мг/сутки.
При ингаляционном введении соединений формулы (I) или (VI), предпочтительно, чтобы их доза составляла от 0,001 до 500 мг/сутки, предпочтительно, от 0,1 до 200 мг/сутки.
Соединения формулы (I) или (VI) могут быть введены для предотвращения и/или лечения любого заболевания, при котором оказывают положительное воздействие антагонисты M3. Указанное заболевание включает: заболевания, при которых имеет место воспаление, такие как астма и хроническое обструктивное заболевание легких, острый ринит; заболевания желудочно-кишечного тракта, такие как язвенная болезнь; заболевания сердечно-сосудистой системы, такие как острый инфаркт миокарда; заболевания мочеполовых путей, такие как почечная колика; отравление антихолинэстеразным средством и грибами; применения в анестезии; применения в офтальмологии.
Они также включают неврологические и психические расстройства, такие как паркинсонизм и укачивание в транспорте.
Предпочтительно, чтобы соединения формулы (I) или (VI) вводили для предотвращения и/или лечения респираторных заболеваний, таких как умеренные и тяжелые случаи астмы и хронического обструктивного заболевания легких.
Другие респираторные заболевания включают бронхит, бронхиолит, бронхоэктаз, острый назофарингит, острый и хронический синусит, гайморит, фарингит, тонзиллит, ларингит, трахеит, эпиглоттит, круп, хроническое заболевание гланд и аденоидов, гипертрофию гланд и аденоидов, перитонзиллярный абсцесс, ринит, абсцесс или язва в носу, пневмонию, вирусную и бактериальную пневмонию, бронхопневмонию, грипп, экзогенно-аллергический альвеолит, пневмокониоз шахтеров, асбестоз, пневмокониоз, пневмопатию, респираторные состояния вследствие действия химических паров, паров и других внешних реагентов, эмфизему, плеврит, пневмоторакс, абсцесс легкого и средостения, застой крови в легких и отек легких, послевоспалительный фиброз легких, другие альвеолярные и париетоальвеолярные пневмопатии, идиопатический фиброзирующий альвеолит, синдром Хаммена-Рича, ателектаз, синдром острой дыхательной недостаточности, острую дыхательную недостаточность, медиастинит.
Далее изобретение будет описано с помощью следующих примеров.
I=промежуточные соединения
C=соединения.
ПРИМЕР 1
Получение (R)-3-((R)-2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (диастереоизомеров 1 C2)

Схема 2
Получение (R)-((R)-хинуклидин-3-ил) 2-(трет-бутоксикарбониламино)-2-фенилацетата (диастереоизомера 1 C1)
Смесь (R)-2-(трет-бутоксикарбониламино)-2-фенилуксусной кислоты (1,00 г, 3,98 ммоль), (R)-хинуклидин-3-ола (0,51 г, 3,98 ммоль), 1H-бензо[d][1,2,3]триазол-1-ола (0,64 г, 4,78 ммоль) и DCC (0,98 г, 4,78 ммоль) перемешивали при комнатной температуре в течение ночи. Затем THF удаляли под вакуумом, и остаток распределяли между EtOAc и 2M раствором K2CO3. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали досуха. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 98/2 до 95/5) с получением (R)-((R)-хинуклидин-3-ил) 2-(трет-бутоксикарбониламино)-2-фенилацетата (612 мг; 43% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,74 (д, 1Н), 7,17-7,52 (м, 5Н), 5,19 (д, 1Н), 4,58-4,82 (м, 1Н), 2,99 (ддд, 1Н), 2,55-2,69 (м, 3Н), 2,32-2,46 (м, 1Н), 2,18 (д, 1Н), 1,79-1,98 (м, 1Н), 1,44-1,71 (м, 3Н), 1,40 (с, 9Н), 1,11-1,33 (м, 1Н);
LC-MS (ESI POS): 361,4 (MH+).
Получение (R)-3-((R)-2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (диастереоизомеров 1 C2)
2-Бром-1-фенилэтанон (27,6 мг, 0,14 ммоль) добавляли к раствору (R)-((R)-хинуклидин-3-ил) 2-(трет-бутоксикарбониламино)-2-фенилацетата (диастереоизомера 1 C1) (50 мг, 0,14 ммоль) в EtOAc (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Осадок собирали вакуумной фильтрацией и промывали с помощью Et2O с получением (R)-3-((R)-2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (65 мг; 84% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,92-8,03 (м, 2Н), 7,89 (д, 1Н), 7,70-7,80 (м, 1Н), 7,56-7,68 (м, 2Н), 7,30-7,52 (м, 5Н), 5,30 (д, 1Н), 5,17-5,24 (м, 1Н), 5,15 (с, 2Н), 4,02-4,19 (м, 1Н), 3,42-3,83 (м, 5Н), 2,30-2,42 (м, 1Н), 1,75-2,12 (м, 4Н), 1,42 (с, 9Н);
LC-MS (ESI POS): 479,09 (M+).
ПРИМЕР 2
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанхлорида (C2)

Схема 3
Получение 2-(трет-бутоксикарбониламино)-2-фенилуксусной кислоты (13):
Смесь 2-амино-2-фенилуксусной кислоты (2,00 г, 13,2 ммоль) и ди-трет-бутилдикарбоната (3,47 г, 15,9 ммоль) в гидроксиде натрия (50 мл, 100 ммоль) и ацетоне (50 мл) перемешивали при комнатной температуре в течение 1 часа. Ацетон удаляли при пониженном давлении, водную фазу подкисляли до pH приблизительно 5 с помощью HCl и дважды экстрагировали с помощью EtOAc. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали досуха с получением 2-(трет-бутоксикарбониламино)-2-фенилуксусной кислоты (1,42 г; 43% выход). Соединение использовали на следующей стадии без дополнительной очистки.
Получение (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C1)
Смесь 2-(трет-бутоксикарбониламино)-2-фенилуксусной кислоты (I3) (2,00 г, 7,96 ммоль), (R)-хинуклидин-3-ола (1,21 г, 9,55 ммоль), HOBt (1,46 г, 9,55 ммоль) и DCC (1,97 г, 9,55 ммоль) в сухом THF (70 мл) перемешивали при комнатной температуре в течение ночи. Затем THF выпаривали, и неочищенное вещество обрабатывали с помощью DCM и промывали дважды с помощью 2M K2CO3 и солевого раствора. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали досуха. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=95/5) с получением (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (1,68 г; 58,5% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д.
Диастереоизомер 1: 7,73 (д, 1Н), 7,12-7,54 (м, 5Н), 5,19 (д, 1Н), 4,52-4,84 (м, 1Н), 2,99 (ддд, 1Н), 2,54-2,70 (м, 3Н), 2,31-2,47 (м, 1Н), 2,12-2,24 (м, 1Н), 1,84-1,92 (м, 1Н), 1,47-1,71 (м, 2Н), 1,40 (с, 9Н), 1,06-1,36 (м, 2Н);
Диастереоизомер 2: 7,73 (д, 1Н), 7,12-7,54 (м, 5Н), 5,19 (д, 1Н), 4,52-4,84 (м, 1Н), 2,99 (ддд, 1Н), 2,54-2,70 (м, 5Н), 1,69-1,79 (м, 1Н), 1,47-1,71 (м, 2Н), 1,40 (с, 9Н), 1,06-1,36 (м, 2Н);
LC-MS (ESI POS): 361,16 (MH+).
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанхлорида (C2)
2-Хлор-1-фенилэтанон (30,0 мг, 0,19 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C1) (70,0 мг, 0,19 ммоль) в EtOAc (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, затем добавляли Et2O (1 мл), и реакционную смесь подвергали воздействию ультразвука. Твердое вещество собирали вакуумной фильтрацией с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанхлорида (54,6 мг; 55% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,94-8,05 (м, 2Н), 7,89 (д, 1Н), 7,70-7,82 (м, 1Н), 7,54-7,70 (м, 2Н), 7,28-7,53 (м, 5Н), 5,27-5,37 (м, 1Н), 5,08-5,27 (м, 2Н), 3,95-4,28 (м, 1Н), 3,44-3,88 (м, 5Н), 2,19 и 2,37 (уш.с, 1H), 1,52-2,12 (м, 4Н), 1,41 (с, 9Н);
LC-MS (ESI POS): 479,21 (M+).
Соединения, приведенные в таблице 1, получали, как описано выше для C2, исходя из соединения C1 и соответствующих производимых промышленностью алкилирующих агентов.

1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,17-8,24 (м, 1Н), 8,05-8,15 (м, 1Н), 7,88 (д, 1Н), 7,23-7,56 (м, 6Н), 5,24-5,37 (м, 1Н), 5,13-5,23 (м, 1Н), 4,99-5,10 (м, 1Н), 3,96-4,21 (м, 1Н), 3,43-3,81 (м, 5Н), 2,12-2,23 и 2,31-2,39 (м, 1Н), 1,46-2,12 (м, 4Н), 1,41 (с, 9H)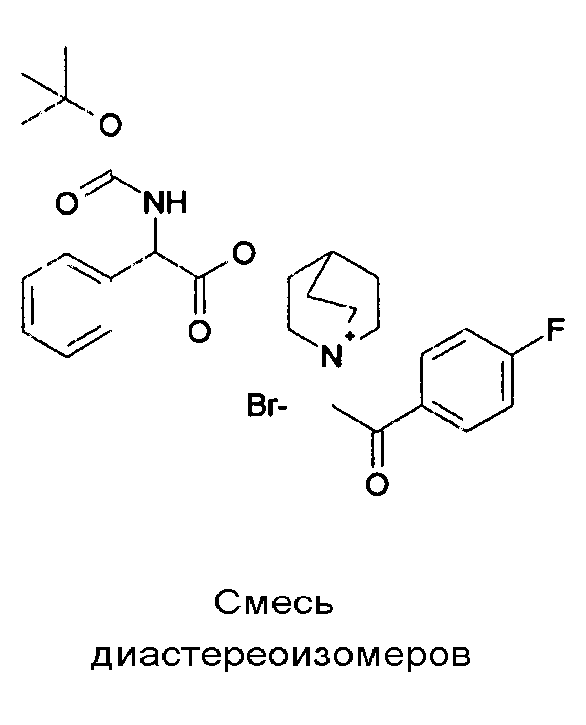
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,00-8,13 (м, 2Н), 7,88 (д, 1Н), 7,25-7,52 (м, 7Н), 5,26-5,36 (м, 1Н), 5,16-5,26 (м, 1Н), 5,02-5,16 (м, 1Н), 3,91-4,28 (м, 1Н), 3,43-3,86 (м, 5Н), 2,15-2,23 и 2,31-2,41 (м, 1Н), 1,55-2,12 (м, 4Н), 1,41 (с, 9Н)

1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,88 (д, 1Н), 7,72-7,85 (м, 2Н), 7,55-7,74 (м, 2Н), 7,26-7,52 (м, 5Н), 5,03-5,46 (м, 3Н), 3,98-4,22 (м, 1Н), 3,38-3,83 (м, 5Н), 2,15-2,25 и 2,31-2,44 (м, 1Н), 1,57-2,13 (м, 4Н), 1,41 (с, 9Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,91-8,03 (м, 1Н), 7,88 (д, 1Н), 7,70-7,85 (м, 1Н), 7,29-7,54 (м, 7Н), 5,11-5,45 (м, 2Н), 4,90-5,11 (м, 1Н), 3,96-4,28 (м, 1Н), 3,45-3,86 (м, 5Н), 2,15-2,23 и 2,31-2,42 (м, 1Н), 1,51-2,13 (м, 4Н), 1,41 (с, 9H)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,96-8,19 (м, 1Н), 7,88 (д, 1Н), 7,26-7,63 (м, 7Н), 5,11-5,44 (м, 2Н), 4,88-5,04 (м, 1Н), 3,95-4,22 (м, 1Н), 3,45-3,84 (м, 5Н), 2,14-2,23 и 2,32-2,42 (м, 1Н), 1,53-2,13 (м, 4Н), 1,42 (с, 9Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,93-8,04 (м, 2Н), 7,88 (д, 1Н), 7,66-7,76 (м, 2Н), 7,30-7,52 (м, 5Н), 5,26-5,38 (м, 1Н), 5,16-5,25 (м, 1Н), 5,04-5,16 (м, 1Н), 3,94-4,23 (м, 1Н), 3,43-3,82 (м, 5Н), 2,19 и 2,37 (уш.с, 1Н), 1,53-2,12 (м, 4Н), 1,41 (с, 9H)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,81-7,93 (м, 3Н), 7,26-7,51 (м, 7Н), 5,25-5,37 (м, 1Н), 5,14-5,25 (м, 1Н), 4,97-5,14 (м, 1Н), 3,94-4,19 (м, 1Н), 3,45-3,82 (м, 5Н), 2,42 (с, 3Н), 2,19 и 2,37 (уш.с, 1Н), 1,54-2,13 (м, 4Н), 1,41 (с, 9Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,91-8,07 (м, 2Н), 7,88 (д, 1Н), 7,26-7,54 (м, 5Н), 6,99-7,22 (м, 2Н), 5,25-5,42 (м, 1Н), 5,16-5,25 (м, 1Н), 5,03-5,16 (м, 2Н), 3,99-4,24 (м, 1Н), 3,88 (с, 3Н), 3,45-3,80 (м, 5Н), 2,15-2,22 и 2,31-2,40 (м, 1Н), 1,49-2,14 (м, 4Н), 1,41 (с, 9H)

1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 10,70 (уш.с, 1Н), 7,75-7,98 (м, 2Н), 7,25-7,56 (м, 5Н), 6,86-6,96 (м, 2Н), 5,10-5,40 (м, 2Н), 4,94-5,06 (м, 1Н), 3,97-4,21 (м, 1Н), 3,45-3,87 (м, 5Н), 2,31-2,42 (м, 1Н), 1,53-2,09 (м, 4Н), 1,41 (с, 9H)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,13-8,21 (м, 2Н), 8,04-8,13 (м, 2Н), 7,88 (д, 1Н), 7,31-7,54 (м, 5Н), 5,26-5,38 (м, 1Н), 5,08-5,26 (м, 2Н), 4,02-4,26 (м, 1Н), 3,91 (с, 3Н), 3,43-3,79 (м, 5Н), 2,19 и 2,37 (уш.с, 1Н), 1,58-2,15 (м, 4Н), 1,41 (с, 9Н)
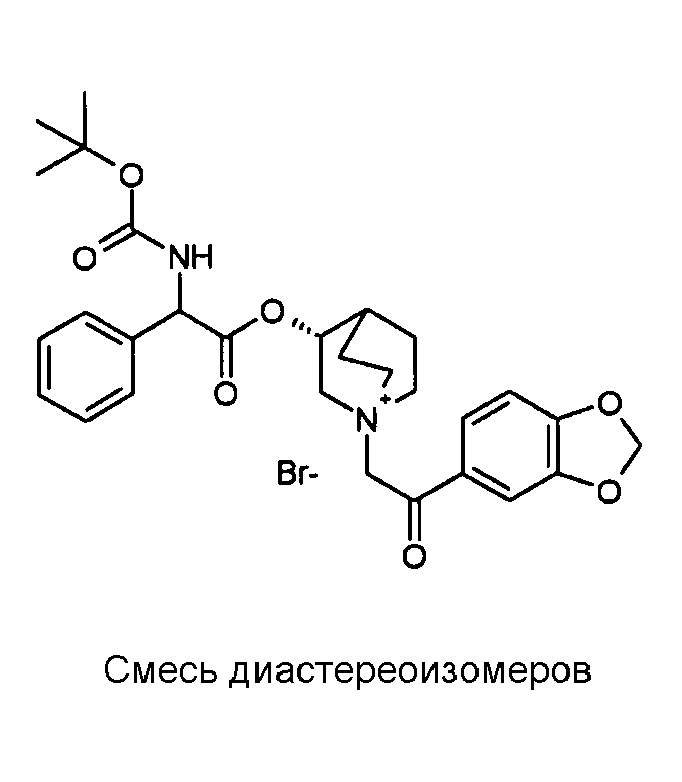
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,88 (д, 1Н), 7,57-7,68 (м, 1Н), 7,27-7,51 (м, 6Н), 7,14 (д, 1Н), 6,19 (с, 2Н), 5,12-5,41 (м, 2Н), 4,90-5,12 (м, 1Н), 3,89-4,26 (м, 1Н), 3,38-3,79 (м, 5Н), 2,15-2,23 и 2,31-2,41 (м, 1Н), 1,51-2,13 (м, 4Н), 1,41 (с, 9Н)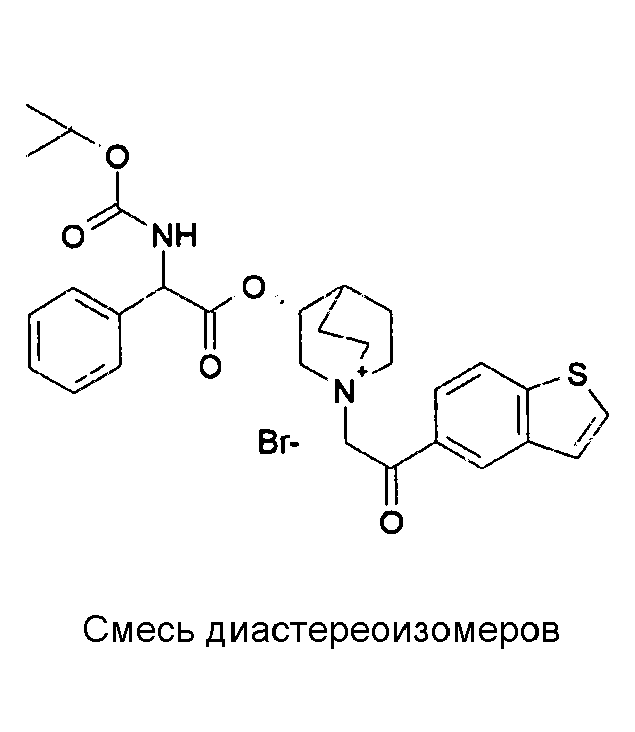
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,56 и 8,58 (д, 1Н), 8,24 и 8,25 (д, 1Н), 7,81-8,03 (м, 2Н), 7,66 (д, 1Н), 7,32-7,53 (м, 5Н), 5,04-5,40 (м, 3Н), 3,97-4,25 (м, 1Н), 3,45-3,86 (м, 5Н), 2,16-2,24 и 2,34-2,41 (м, 1Н), 1,52-2,12 (м, 4Н), 1,42 (с, 9H)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,80-8,12 (м, 2Н), 7,19-7,57 (м, 6Н), 5,09-5,38 (м, 2Н), 4,83-5,05 (м, 1Н), 3,97-4,16 (м, 1Н), 3,35-3,82 (м, 5Н), 2,13-2,23 и 2,32-2,40 (м, 1Н), 1,50-2,09 (м, 4Н), 1,41 (с, 9Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,36-8,41 (м, 1Н), 8,18-8,28 (м, 1Н), 7,88 (д, 1Н), 7,25-7,54 (м, 5Н), 5,24-5,40 (м, 1Н), 5,08-5,25 (м, 3Н), 3,97-4,25 (м, 1Н), 3,47-3,88 (м, 5Н), 2,13-2,24 и 2,32-2,42 (м, 1Н), 1,54-2,13 (м, 4Н), 1,41 (с, 9Н)
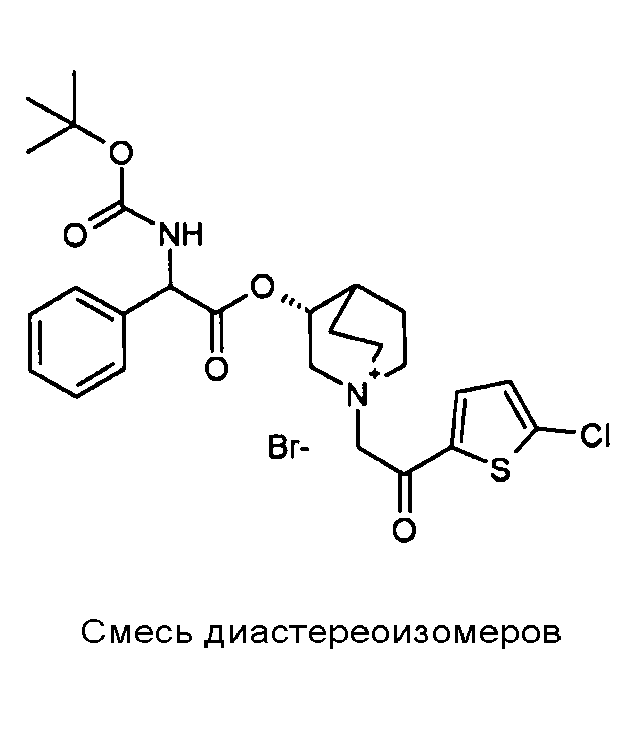
ПРИМЕР 3
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азонийбицикло[2.2.2]-октанбромида (C17)
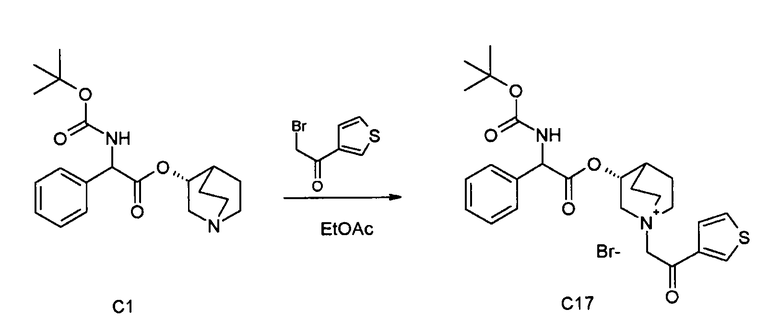
Схема 4
2-Бром-1-(тиофен-3-ил)этанон (39,8 мг, 0,19 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C1) (70,0 мг, 0,19 ммоль) в этилацетате (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли Et2O (1 мл) и твердое вещество собирали вакуумной фильтрацией. Соединение затем очищали флэш-хроматографией (DCM/MeOH=95/5) с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-(тиофен-3-ил)этил)-1-азонийбицикло[2.2.2]октанбромида (55,6 мг; 51% выход).
lH ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,60-8,65 (м, 1Н) 7,88 (д, 1Н) 7,75 и 7,73 (дд, 1Н) 7,51-7,61 (м, 1Н) 7,27-7,50 (м, 5Н) 5,10-5,39 (м, 2Н) 4,93-5,08 (м, 1Н) 4,02-4,22 (м, 1Н) 3,43-3,79 (м, 5Н) 2,30-2,41 (м, 1Н) 1,85-2,11 (м, 5Н) 1,41 (с, 9Н);
LC-MS (ESI POS): 485,06 (M+).
ПРИМЕР 4
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-метил-1-азонийбицикло[2.2.2]октанйодида (C18)

Схема 5
Метилйодид (8,6 мкл, 0,14 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C1) (50 мг, 0,14 ммоль) в этилацетате (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли Et2O (1 мл), и осадок собирали вакуумной фильтрацией и сушили под вакуумом при 40°C. Продукт затем очищали препаративной ВЭЖХ с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-метил-1-азонийбицикло[2.2.2]октанйодида (24,2 мг; 35% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,85 (д, 1Н) 7,20-7,55 (м, 5H) 5,26 и 5,31 (д, 1Н) 4,93-5,18 (м, 1Н) 3,83 (ддд, 1Н) 3,30-3,53 (м, 3Н) 3,09-3,27 (м, 1Н) 3,16 (дт, 1Н) 2,93 и 2,95 (с, 3Н) 2,04-2,14 и 2,25-2,33 (м, 1Н) 1,52-2,03 (м, 4Н) 1,40 (с, 9Н);
LC-MS (ESI POS): 375,20 (M+).
Соединение, приведенное в таблице 2, получали, как описано выше для C18, исходя из промежуточного соединения C1 и (3-бромпропокси)бензола.
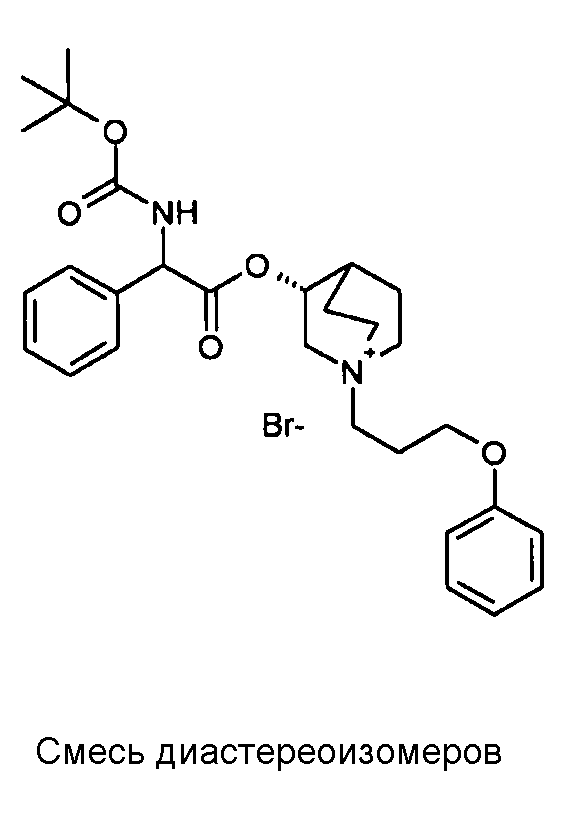
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,87 (д, 1Н) 7,18-7,58 (м, 7Н) 6,81-7,08 (м, 3Н) 5,27 и 5,32 (д, 1Н) 5,00-5,20 (м, 1Н) 4,02 и 4,04 (т, 2Н) 3,73-3,95 (м, 1Н) 3,31-3,55 (м, 6Н) 2,97-3,20 (м, 1Н) 2,29-2,39 (м, 1Н) 1,51-2,22 (м, 6Н) 1,40 (с, 9H)
ПРИМЕР 5
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-(2-метоксифенил)-2-оксоэтил)-1-азонийбицикло-[2.2.2]октанбромида (C20)

Схема 6
2-Бром-1-(2-метоксифенил)этанон (45,8 мг, 0,20 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C1) (72,0 мг, 0,20 ммоль) в ацетонитриле (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Продукт собирали вакуумной фильтрацией с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-(2-(2-метоксифенил)-2-оксоэтил)-1-азонийбицикло-[2.2.2]октанбромида (112 мг; 95% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,88 (д, 1Н), 7,76-7,84 (м, 1Н), 7,64-7,75 (м, 1Н), 7,30-7,54 (м, 5Н), 7,23-7,30 (м, 1Н), 7,03-7,19 (м, 1Н), 5,24-5,36 (м, 1Н), 5,11-5,24 (м, 1Н), 4,89 (с, 2Н), 4,03-4,23 (м, 1Н), 3,95 и 3,96 (с, 3Н), 3,45-3,84 (м, 5Н), 2,13-2,23 и 2,31-2,42 (м, 1Н), 1,51-2,12 (м, 4Н), 1,41 (с, 9Н);
LC-MS (ESI POS): 509,33 (M+).
Соединение, приведенное в таблице 3, получали, как описано выше для C20, исходя из промежуточного соединения C1 и 3-(хлорметил)-5-фенил-1,2,4-оксадиазола.

1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,09-8,28 (м, 2Н), 7,84-7,97 (м, 1Н), 7,62-7,84 (м, 3Н), 7,18-7,51 (м, 5Н), 5,25 и 5,32 (д, 1Н), 5,04-5,16 (м, 1Н), 4,87 (с, 2Н), 3,99-4,25 (м, 1Н), 3,35-3,82 (м, 5Н), 2,13-2,23 и 2,29-2,40 (м, 1Н), 1,52-2,09 (м, 4Н), 1,37 и 1,39 (с, 9Н)
ПРИМЕР 6
Получение (3R)-1-(2-трет-бутокси-2-оксоэтил)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-азонийбицикло[2.2.2]-октанбромида (C22)

Схема 7
Трет-бутил 2-бромацетат (29,5 мкл, 0,20 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C1) (72 мг, 0,20 ммоль) в ацетонитриле (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Продукт собирали вакуумной фильтрацией и затем очищали флэш-хроматографией (DCM/MeOH = от 98/2 до 95/5) с получением (3R)-1-(2-трет-бутокси-2-оксоэтил)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-азонийбицикло[2.2.2]октанбромида (97 мг; 87% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,87 (д, 1Н), 7,20-7,54 (м, 5Н), 5,23-5,44 (м, 1Н), 5,08-5,22 (м, 1Н), 4,21 и 4,23 (с, 2Н), 3,88-4,08 (м, 1Н), 3,37-3,72 (м, 5Н), 2,09-2,22 и 2,30-2,40 (м, 1Н), 1,76-2,08 (м, 4Н), 1,46 и 1,48 (с, 9Н), 1,41 (с, 9Н);
LC-MS (ESI POS): 475,33 (M+).
Соединения, приведенные в таблице 4, получали, как описано выше для C22, исходя из промежуточного соединения C1 и 2-бром-1-(2-нитрофенил)этанона или 2-бром-1-(2-гидроксифенил)этанона.

1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,30 (м, 1Н), 7,94-8,08 (м, 1Н), 7,74-7,94 (м, 3Н), 7,24-7,55 (м, 5Н), 5,26-5,36 (м, 1Н), 5,18-5,27 (м, 1Н),5,02 и 5,05 (с, 2Н), 4,04-4,25 (м, 1Н), 3,45-3,81 (м, 5Н), 2,17-2,25 и 2,30-2,43 (м, 1Н), 1,54-2,16 (м, 4Н), 1,41 (с, 9Н)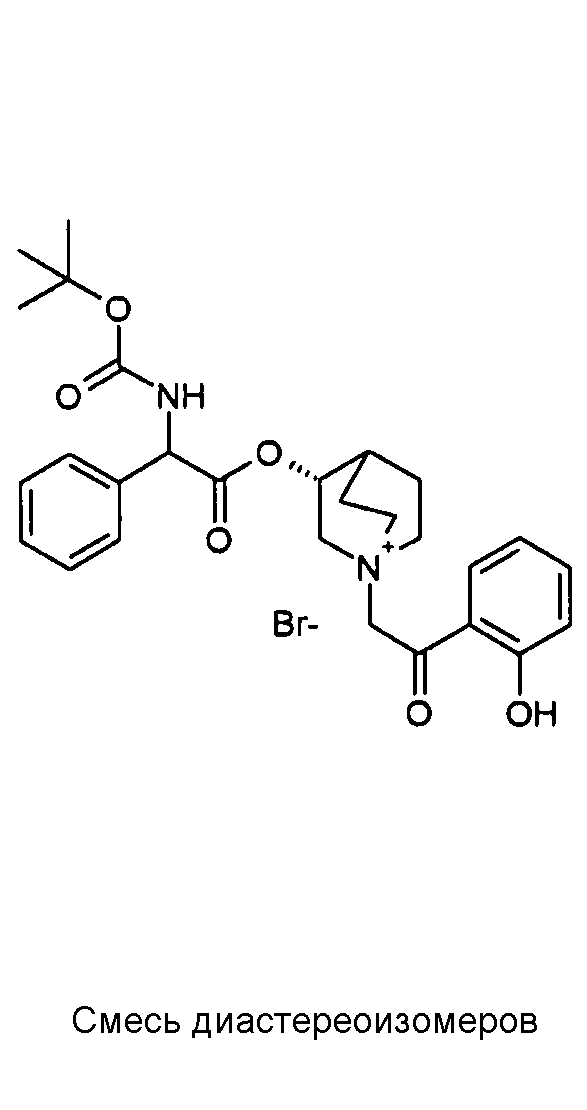
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 11,17 (уш.с, 1Н), 7,82-7,94 (м, 1Н), 7,73-7,82 (м, 1Н), 7,50-7,63 (м, 1Н), 7,43-7,50 (м, 2Н), 7,25-7,43 (м, 3Н), 7,01-7,10 (м, 1Н), 6,91-7,01 (м, 1Н), 5,24-5,44 (м, 1Н), 5,07-5,25 (м, 1Н), 4,83-5,07 (м, 2Н), 3,96-4,23 (м, 1Н), 3,46-3,83 (м, 5Н), 2,13-2,24 и 2,31-2,42 (м, 1Н), 1,47-2,12 (м, 4Н), 1,41 (с, 9Н)
ПРИМЕР 7
Получение (3R)-3-(2-(метоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (С27)

Схема 8
Получение (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25)
К раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C1) (1,25 г, 3,47 ммоль) в THF (20 мл) добавляли по каплям 37% хлористоводородную кислоту (2,00 мл, 24,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали с получением (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (1,16 г; количественный выход) в виде твердого вещества. Соединение использовали на следующей стадии без дополнительной очистки.
Получение (R)-хинуклидин-3-ил 2-(метоксикарбониламино)-2-фенилацетата (C26)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (150 мг, 0,45 ммоль) в DCM (5 мл) добавляли триэтиламин (188 мкл, 1,35 ммоль) и метил карбонохлоридата (41,7 мкл, 0,54 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и затем растворитель выпаривали. Остаток обрабатывали с помощью EtOAc и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4 и выпаривали с получением (R)-хинуклидин-3-ил 2-(метоксикарбониламино)-2-фенилацетата (58,0 мг; 40% выход). Соединение использовали на следующей стадии без дополнительной очистки.
Получение (3R)-3-(2-(метоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (C27)
К раствору (R)-хинуклидин-3-ил 2-(метоксикарбониламино)-2-фенилацетата (C26) (58,0 мг, 0,18 ммоль) в EtOAc (3 мл) и CH3CN (1 мл) добавляли 2-бром-1-фенилэтанон (39,9 мг, 0,20 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, и затем растворитель выпаривали. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 95/5 до 92/8), и полученный продукт растирали с i-Pr2O с получением (3R)-3-(2-(метоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (55,2 мг; 58,6% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,17-8,30 (м, 1Н) 7,98 (дд, 2Н) 7,71-7,84 (м, 1Н) 7,61 (тд, 2Н) 7,30-7,52 (м, 5Н) 5,36 (дд, 1Н) 5,20-5,30 (м, 1Н) 5,16 (д, 2Н) 4,01-4,23 (м, 1Н) 3,61 (с, 3Н) 3,45-3,81 (м, 5Н) 2,39 (с, 1Н) 1,90-2,11 (м, 3Н) 1,54-1,71 (м, 1Н);
LC-MS (ESI POS): 437,12 (M+).
ПРИМЕР 8
Получение (R)-хинуклидин-3-ил 2-амино-2-фенилацетата (I30)

Схема 9
Получение 2-(бензилоксикарбониламино)-2-фенилуксусной кислоты (I28)
К раствору 2-амино-2-фенилуксусной кислоты (500 мг, 3,31 ммоль) в 2 н. растворе гидроксида натрия (1,65 мл, 3,31 ммоль), перемешиваемому при 0°C, последовательно добавляли по каплям из двух разных шприцов бензил карбонохлоридат (512 мкл, 3,64 ммоль) и 2 н. раствор гидроксида натрия (1,82 мл, 3,64 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 45 минут и выпадал осадок. Добавляли воду и раствор экстрагировали с помощью Et2O. Водную фазу подкисляли с помощью 1N HCl и требуемый продукт экстрагировали снова с помощью Et2O. Объединенные органические фазы сушили над Na2SO4, фильтровали и выпаривали с получением 2-(бензилоксикарбониламино)-2-фенилуксусной кислоты (855 мг; 91% выход).
Получение (R)-хинуклидин-3-ил 2-(бензилоксикарбониламино)-2-фенилацетата (C29)
К раствору 2-(бензилоксикарбониламино)-2-фенилуксусной кислоты (I28) (855 мг, 3,00 ммоль) в THF (20 мл) добавляли (R)-хинуклидин-3-ол (457 мг, 3,60 ммоль), N,N'-метандиилиден-дициклогексанамин (742 мг, 3,60 ммоль) и 1H-бензо-[d][1,2,3]триазол-1-ол (486 мг, 3,60 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали. Остаток обрабатывали с помощью DCM, нерастворимое твердое вещество отфильтровывали и прозрачный раствор промывали дважды с помощью Na2CO3 и солевого раствора, сушили над Na2SO4 и выпаривали. Полученное неочищенное вещество очищали флэш-хроматографией на силикагеле (DCM/MeOH=9/1) с получением (R)-хинуклидин-3-ил 2-(бензилоксикарбониламино)-2-фенилацетата (925 мг; 78% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,26 (д, 1Н), 7,19-7,54 (м, 10Н), 5,28 и 5,29 (д, 1Н), 5,10 (д, 1Н), 5,05 (д, 1Н), 4,59-4,80 (м, 1Н), 2,95-3,07 (м, 1Н), 2,09-2,71 (м, 5Н), 1,70-1,80 и 1,81-1,99 (м, 1Н), 1,01-1,70 (м, 4Н).
Получение (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I30)
Раствор (R)-хинуклидин-3-ил 2-(бензилоксикарбониламино)-2-фенилацетата (C29) (100 мг, 0,25 ммоль) в MeOH (7 мл) и 37% хлористоводородной кислоте (20,8 мкл, 0,25 ммоль) перемешивали при комнатной температуре в атмосфере водорода (25 фунт/кв.дюйм) в течение 3 часов в аппарате фирмы Parr в присутствии палладия на активированном угле (10 мг, 9,40 мкмоль). Катализатор отфильтровывали и растворитель выпаривали. Полученное масло очищали с помощью картриджа с катионообменной смолой SCX, элюируя с помощью MeOH и затем MeOH/NH4OH (97/3), с получением (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (55,0 мг; 65,1% выход).
ПРИМЕР 9
Получение (3R)-3-(2-(бензилоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (C31)

Схема 10
К раствору (R)-хинуклидин-3-ил 2-(бензилоксикарбониламино)-2-фенилацетата (C29) (100 мг, 0,25 ммоль) в EtOAc (5 мл) добавляли 2-бром-1-фенилэтанон (55,5 мг, 0,28 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали и полученное неочищенное вещество очищали флэш-хроматографией на силикагеле (DCM/MeOH=93/7) с получением (3R)-3-(2-(бензилоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (127,5 мг; 85% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,22-8,52 (м, 1Н), 7,90-8,07 (м, 2Н), 7,69-7,83 (м, 1Н), 7,54-7,69 (м, 2Н), 7,19-7,54 (м, 10Н), 5,39 и 5,40 (д, 1Н), 4,99-5,29 (м, 5Н), 3,93-4,23 (м, 1Н), 3,39-3,78 (м, 5Н), 2,10-2,23 и 2,30-2,44 (м, 1Н), 1,40-2,12 (м, 4Н);
LC-MS (ESI POS): 513,25 (M+).
ПРИМЕР 10
Получение (R)-((R)-хинуклидин-3-ил) 2-(бензилоксикарбониламино)-2-фенилацетата (диастереоизомера 1 C31)

Схема 11
Получение (R)-((R)-хинуклидин-3-ил) 2-(бензилоксикарбониламино)-2-фенилацетата (диастереоизомера 1 C29)
(R)-хинуклидин-3-ол (214 мг, 1,68 ммоль), N,N'-метандиилидендициклогексанамин (347 мг, 1,68 ммоль) и 1H-бензо-[d][1,2,3]триазол-1-ол (227 мг, 1,68 ммоль) добавляли к раствору (R)-2-(бензилоксикарбониламино)-2-фенилуксусной кислоты (400 мг, 1,40 ммоль) в THF (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали. Добавляли DCM и нерастворимое твердое вещество отфильтровывали. Органическую фазу промывали дважды с помощью Na2CO3 и солевого раствора, сушили над Na2SO4 и выпаривали. Неочищенное вещество очищали флэш-хроматографией на силикагеле (DCM/MeOH=9/1) с получением (R)-((R)-хинуклидин-3-ил) 2-(бензилоксикарбониламино)-2-фенилацетата (63 мг; 11,4% выход). Также выделяют ((R)-хинуклидин-3-ил 2-(бензилоксикарбониламино)-2-фенилацетат (300 мг; 54,2% выход)).
1H ЯМР (300 МГц, ДМСО-d6) δ М.д. 8,26 (д, 1Н), 7,14-7,58 (м, 10Н), 5,28 (д, 1Н), 5,10 (д, 1Н), 5,05 (д, 1Н), 4,59-4,79 (м, 1Н), 3,01 (дд, 1Н), 2,54-2,70 (м, 3Н), 2,32-2,45 (м, 1Н), 2,22 (д, 1Н), 1,82-1,99 (м, 1H), 1,35-1,68 (м, 3Н), 1,16-1,35 (м, 1Н).
Получение (R)-3-((R)-2-(бензилоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (диастереоизомера 1 C31)
К раствору (R)-((R)-хинуклидин-3-ил) 2-(бензилоксикарбониламино)-2-фенилацетата (40 мг, 0,10 ммоль) в EtOAc (3 мл) добавляли 2-бром-1-фенилэтанон (22,2 мг, 0,11 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали. Неочищенное вещество очищали флэш-хроматографией на силикагеле (DCM/MeOH=93/7) с получением (R)-3-((R)-2-(бензилоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (45,0 мг; 74,8% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,37 (д, 1Н), 7,87-8,04 (м, 2Н), 7,69-7,82 (м, 1Н), 7,56-7,69 (м, 2Н), 7,18-7,54 (м, 10Н), 5,38 (д, 1Н), 5,17-5,26 (м, 1Н), 5,15 (с, 2Н), 5,09 (с, 2Н), 3,95-4,21 (м, 1Н), 3,42-3,75 (м, 5Н), 2,31-2,44 (м, 1Н), 1,39-2,12 (м, 4Н);
LC-MS (ESI POS): 513,17 (M+).
ПРИМЕР 11
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(винилоксикарбониламино)ацетокси)-1-азонийбицикло[2.2.2]октанбромида (C33)

Схема 12
Получение (R)-хинуклидин-3-ил 2-фенил-2-(винилоксикарбониламино)ацетата (C32)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (110 мг, 0,33 ммоль) в DCM (5 мл) добавляли триэтиламин (138 мкл, 0,99 ммоль) и винил карбонохлоридат (36,2 мкл, 0,40 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем растворитель выпаривали. Остаток обрабатывали с помощью EtOAc и промывали водой и солевым раствором, сушили над Na2SO4 и выпаривали досуха с получением (R)-хинуклидин-3-ил 2-фенил-2-(винилоксикарбониламино)ацетата (70 мг; 64% выход). Продукт использовали на следующей стадии без дополнительной очистки.
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(винилоксикарбониламино)ацетокси)-1-азонийбицикло[2.2.2]-октанбромида (C33)
К раствору (R)-хинуклидин-3-ил 2-фенил-2-(винилоксикарбониламино)ацетата (C32) (70,0 мг, 0,21 ммоль) в EtOAc (2 мл) и ацетонитриле (0,5 мл) добавляли 2-бром-1-фенилэтанон (46,4 мг, 0,23 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и затем растворитель выпаривали. Полученное твердое вещество растирали с i-Pr2O/EtOAc (1/1) с получением (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(винилокси-карбониламино)ацетокси)-1-азонийбицикло[2.2.2]октанбромида (82,4 мг; 73% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,76 и 8,78 (д, 1Н), 7,90-8,08 (м, 2Н), 7,71-7,80 (м, 1Н), 7,56-7,67 (м, 2Н), 7,30-7,54 (м, 5Н), 7,07-7,20 (м, 1Н), 5,41 и 5,42 (д, 1Н), 5,20-5,30 (м, 1Н), 5,15 и 5,17 (с, 2Н), 4,78 (дд, 1Н), 4,53 (дд, 1Н), 3,97-4,22 (м, 1Н), 3,43-3,83 (м, 5Н), 2,15-2,24 и 2,35-2,44 (м, 1Н), 1,50-2,14 (м, 4Н);
LC-MS (ESI POS): 449,27 (M+).
ПРИМЕР 12
Получение (3R)-3-(2-(этоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (C35)

Схема 13
Получение (R)-хинуклидин-3-ил 2-(этоксикарбониламино)-2-фенилацетата (C34)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (110 мг, 0,33 ммоль) в DCM (5 мл) добавляли триэтиламин (138 мкл, 0,99 ммоль) и этил карбонохлоридат (38 мкл, 0,40 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и затем растворитель выпаривали. Добавляли EtOAc и органическую фазу промывали водой и солевым раствором, сушили над Na2SO4 и выпаривали с получением (R)-хинуклидин-3-ил 2-(этоксикарбониламино)-2-фенилацетата (60 мг; 55% выход). Продукт использовали на следующей стадии без дополнительной очистки.
Получение (3R)-3-(2-(этоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (C35)
К раствору (R)-хинуклидин-3-ил 2-(этоксикарбониламино)-2-фенилацетата (C34) (60,0 мг, 0,18 ммоль) в EtOAc (2 мл) и ацетонитриле (0,5 мл) добавляли 2-бром-1-фенилэтанон (39,5 мг, 0,20 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и растворитель выпаривали. Полученное твердое вещество растирали в i-Pr2O/EtOAc (1/1) с получением (3R)-3-(2-(этоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (76 мг; 80% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,11-8,29 (м, 1Н), 7,91-8,03 (м, 2Н), 7,70-7,81 (м, 1Н), 7,56-7,67 (м, 2Н), 7,29-7,54 (м, 5Н), 5,35 и 5,36 (д, 1Н), 5,20-5,26 (м, 1Н), 5,16 и 5,18 (с, 2Н), 4,09-4,22 (м, 1Н), 4,06 (кв., 2Н), 3,43-3,84 (м, 5Н), 2,15-2,24 (м, 1Н), 1,52-2,14 и 2,32-2,42 (м, 4Н), 1,19 (т, 3Н);
LC-MS (ESI POS): 451,27 (M+).
ПРИМЕР 13
Получение (3R)-3-(2-((2-метоксиэтокси)карбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (C37)

Схема 14
Получение (R)-хинуклидин-3-ил 2-((2-метоксиэтокси)-карбониламино)-2-фенилацетата (C36)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (150 мг, 0,45 ммоль) в DCM (5 мл) добавляли триэтиламин (188 мкл, 1,35 ммоль) и 2-метоксиэтил карбонохлоридат (63 мкл, 0,54 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и затем растворитель выпаривали. Остаток обрабатывали с помощью EtOAc и органическую фазу промывали водой и солевым раствором, сушили над Na2SO4 и выпаривали досуха, получая (R)-хинуклидин-3-ил 2-((2-метоксиэтокси)карбониламино)-2-фенилацетат (73,0 мг; 45% выход).
Получение (3R)-3-(2-((2-метоксиэтокси)карбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (C37)
К раствору (R)-хинуклидин-3-ил 2-((2-метоксиэтокси)-карбониламино)-2-фенилацетата (C36) (73,0 мг, 0,20 ммоль) в EtOAc (3 мл) и ацетонитриле (1 мл) добавляли 2-бром-1-фенилэтанон (44,1 мг, 0,22 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали и полученное неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 95/5 до 93/7). Продукт растирали с i-Pr2O с получением (3R)-3-(2-((2-метоксиэтокси)карбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (61,3 мг; 54% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,23-8,44 (м, 1Н) 7,89-8,08 (м, 2Н) 7,69-7,83 (м, 1Н) 7,54-7,68 (м, 2Н) 7,29-7,52 (м, 5Н) 5,36 (дд, 1Н) 5,08-5,28 (м, 3Н) 4,05-4,21 (м, 3Н) 3,44-3,80 (м, 7Н) 3,26 (с, 3Н) 2,32-2,43 (м, 1Н) 1,88-2,12 (м, 3Н) 1,69-1,88 (м, 1Н);
LC-MS (ESI POS): 481,18 (M+).
ПРИМЕР 14
Получение (3R)-3-(2-(циклогексилоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (C39)
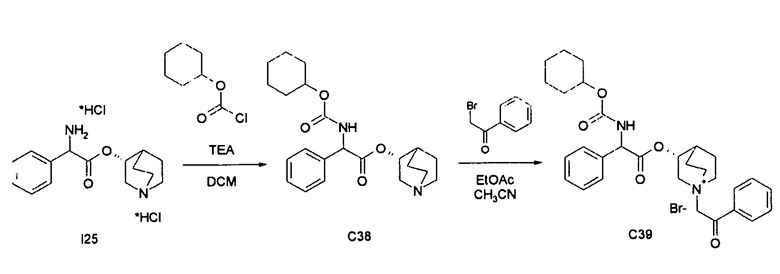
Схема 15
Получение (R)-хинуклидин-3-ил 2-(циклогексилоксикарбониламино)-2-фенилацетата (C38)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (150 мг, 0,45 ммоль) в DCM (5 мл) добавляли триэтиламин (188 мкл, 1,35 ммоль) и циклогексил карбонохлоридат (78 мкл, 0,54 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и затем растворитель выпаривали.
Остаток обрабатывали с помощью EtOAc и промывали водой и солевым раствором, сушили над Na2SO4 и выпаривали досуха с получением (R)-хинуклидин-3-ил 2-(циклогексилоксикарбониламино)-2-фенилацетата (105 мг; 60% выход).
Получение (3R)-3-(2-(циклогексилоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C39)
К раствору (R)-хинуклидин-3-ил 2-(циклогексилоксикарбониламино)-2-фенилацетата (C38) (105 мг, 0,27 ммоль) в EtOAc (3 мл) добавляли 2-бром-1-фенилэтанон (59,5 мг, 0,30 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 95/5 до 93/7) с получением (3R)-3-(2-(циклогексилоксикарбониламино)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (83,8 мг; 53% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,13 (м, 1Н) 7,90-8,04 (м, 2Н) 7,69-7,84 (м, 1Н) 7,61 (тд, 2Н) 7,25-7,53 (м, 5Н) 5,35 (дд, 1Н) 5,05-5,28 (м, 3Н) 4,41-4,67 (м, 1Н) 3,91-4,23 (м, 1Н) 3,49-3,76 (м, 5Н) 2,12-2,24 (м, 1Н) 1,89-2,12 (м, 3Н) 1,57-1,89 (м, 5Н) 1,11-1,56 (м, 6Н);
LC-MS (ESI POS): 505,19 (M+).
ПРИМЕР 15
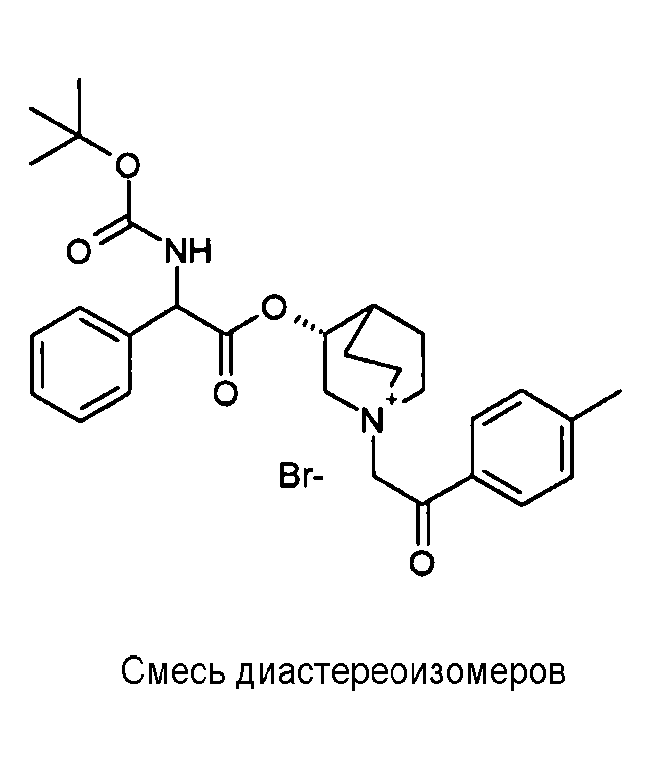
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-п-толилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C42)

Схема 16
Получение 2-(трет-бутоксикарбониламино)-2-п-толилуксусной кислоты (I40):
К суспензии 2-амино-2-п-толилуксусной кислоты (1,00 г, 6,05 ммоль) в THF (30 мл) и воде (30 мл) добавляли 2н раствор гидроксида натрия (30,3 мл, 60,5 ммоль) и ди-трет-бутилдикарбонат (2,64 г, 12,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем выпаривали THF. Оставшуюся водную фазу охлаждали и подкисляли с помощью 37% HCl до pH 1. Требуемое соединение экстрагировали с помощью EtOAc и органическую фазу промывали солевым раствором, сушили над Na2SO4 и выпаривали с получением 2-(трет-бутоксикарбониламино)-2-п-толилуксусной кислоты (1,29 г; 80% выход).
Получение (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-п-толилацетата (C41)
К раствору 2-(трет-бутоксикарбониламино)-2-п-толилуксусной кислоты (I40) (1,29 г, 4,86 ммоль) в THF (70 мл) добавляли Ν,Ν'-метандиилидендициклогексанамин (1,20 г, 5,83 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (0,79 г, 5,83 ммоль) и (R)-хинуклидин-3-ол (0,74 г, 5,83 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и затем растворитель выпаривали. Остаток обрабатывали с помощью DCM, нерастворимое твердое вещество отфильтровывали, и органический раствор промывали дважды водным раствором Na2CO3 и затем солевым раствором, сушили над Na2SO4 и выпаривали досуха. Неочищенное вещество очищали флэш-хроматографией (EtOAc/MeOH = от 8/2 до 7/3) с получением (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-п-толилацетата (1,03 г; 57% выход).
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-п-толилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C42)
К раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-п-толилацетата (C41) (100 мг, 0,27 ммоль) в EtOAc (3 мл) добавляли 2-бром-1-фенилэтанон (58,5 мг, 0,29 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 36 часов. Растворитель выпаривали, и остаток сначала растирали с i-Pr2O, а затем очищали флэш-хроматографией (DCM/MeOH = от 95/5 до 9/1) с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-п-толилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (94,4 мг; 62% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,92-8,06 (м, 2Н), 7,70-7,86 (м, 2Н), 7,54-7,68 (м, 2Н), 7,27-7,42 (м, 2Н), 7,12-7,25 (м, 2Н), 4,91-5,36 (м, 3Н), 3,92-4,21 (м, 1Н), 3,44-3,85 (м, 5Н), 2,34-2,40 (м, 1Н), 2,30 (с, 3Н), 1,53-2,22 (м, 4Н), 1,41 (с, 9Н);
LC-MS (ESI POS): 493,61 (M+).
ПРИМЕР 16
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(4-метоксифенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C45)
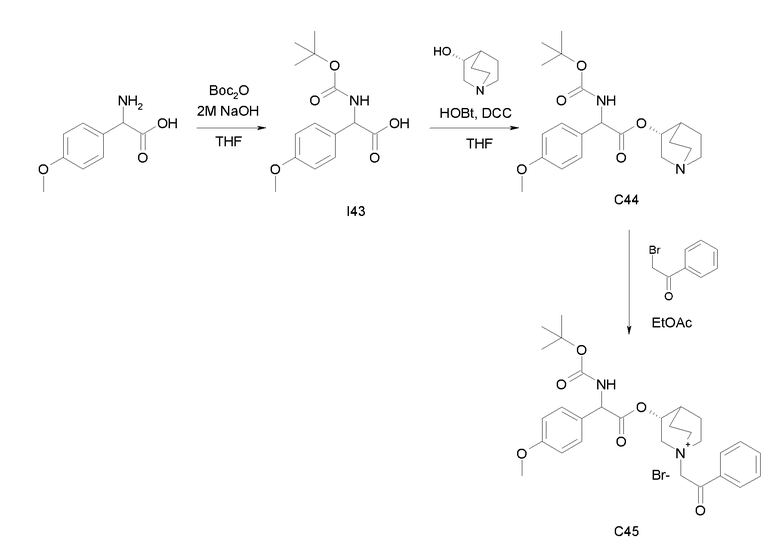
Схема 17
Получение 2-(трет-бутоксикарбониламино)-2-(4-метоксифенил)уксусной кислоты (I43)
К суспензии 2-амино-2-(4-метоксифенил)уксусной кислоты (360 мг, 1,99 ммоль) в THF (30 мл) и воде (30 мл) добавляли 2 н. раствор гидроксида натрия (20 мл, 40,0 ммоль) и ди-трет-бутилдикарбонат (867 мг, 3,97 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. THF выпаривали и оставшуюся водную фазу охлаждали и подкисляли с помощью 37% HCl до pH 1. Требуемое соединение экстрагировали с помощью EtOAc и органическую фазу промывали солевым раствором, сушили над Na2SO4 и выпаривали с получением 2-(трет-бутоксикарбониламино)-2-(4-метоксифенил)уксусной кислоты (430 мг; 77% выход).
Получение (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-метоксифенил)ацетата (C44)
К раствору 2-(трет-бутоксикарбониламино)-2-(4-метоксифенил)-уксусной кислоты (I43) (1,39 г, 4,94 ммоль) в сухом THF (60 мл) добавляли Ν,Ν'-метандиилидендициклогексанамин (1,22 г, 5,94 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (802 мг, 5,94 ммоль) и (R)-хинуклидин-3-ол (755 мг, 5,94 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и растворитель выпаривали. Остаток обрабатывали с помощью DCM, нерастворимое твердое вещество отфильтровывали, и органический раствор промывали дважды водным раствором Na2CO3 и затем солевым раствором, сушили над Na2SO4 и выпаривали. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 95/5 до 93/7) с получением (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-метоксифенил)ацетата (630 мг; 33% выход).
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(4-метокси-фенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C45)
2-Бром-1-фенилэтанон (39,3 мг, 0,20 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-метоксифенил)ацетата (C44) (70,0 мг, 0,18 ммоль) в EtOAc (2 мл) и ацетонитриле (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали и полученное бесцветное масло растирали сначала с i-Pr2O/EtOAc (10/1), а затем с i-Pr2O с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-(4-метоксифенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (60,9 мг; 58% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,88-8,05 (м, 2Н), 7,71-7,84 (м, 2Н), 7,54-7,66 (м, 2Н), 7,27-7,44 (м, 2Н), 6,80-7,02 (м, 2Н), 5,18-5,26 (м, 2Н), 5,16 (с, 2Н), 4,02-4,20 (м, 1Н), 3,76 (с, 3Н), 3,47-3,72 (м, 5Н), 2,33-2,44 (м, 1Н), 1,78-2,17 (м, 4Н), 1,41 (с, 9Н);
LC-MS (ESI POS): 509,15 (M+).
ПРИМЕР 17
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C48)
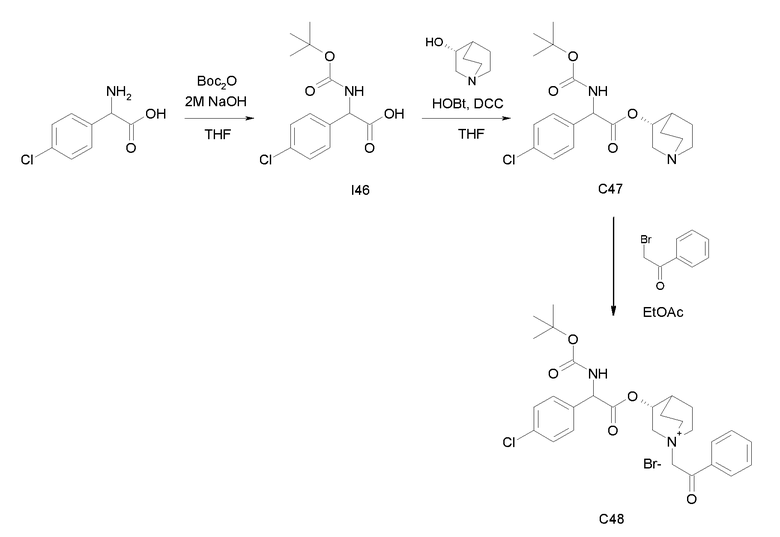
Схема 18
Получение 2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)уксусной кислоты (I46):
К суспензии 2-амино-2-(4-хлорфенил)уксусной кислоты (1,50 г, 8,08 ммоль) в THF (30 мл) и воде (30 мл) добавляли 2 н. раствор гидроксида натрия (40,4 мл, 81,0 ммоль) и ди-трет-бутилдикарбонат (3,53 г, 16,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. THF выпаривали, и оставшуюся водную фазу охлаждали и подкисляли с помощью 37% HCl до pH 1. Требуемое соединение экстрагировали с помощью EtOAc, и органическую фазу промывали солевым раствором, сушили над Na2SO4 и выпаривали с получением 2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)уксусной кислоты (2,17 г; 94% выход).
Получение (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)ацетата (C47):
К раствору 2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)-уксусной кислоты (I46) (2,17 г, 7,58 ммоль) в сухом THF (70 мл) добавляли Ν,Ν'-метандиилидендициклогексанамин (1,88 г, 9,10 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (1,23 г, 9,10 ммоль) и (R)-хинуклидин-3-ол (1,16 г, 9,10 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и растворитель выпаривали. Остаток обрабатывали с помощью DCM, нерастворимое твердое вещество отфильтровывали, и органический раствор промывали дважды водным раствором Na2CO3 и затем солевым раствором, сушили над Na2SO4 и выпаривали. Неочищенное вещество очищали флэш-хроматографией (EtOAc/MeOH = от 8/2 до 85/15) с получением (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)ацетата (1,33 г; 44% выход).
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C48):
2-Бром-1-фенилэтанон (55,4 мг, 0,28 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)ацетата (C47) (100 мг, 0,25 ммоль) в EtOAc (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 36 часов, затем добавляли вторую порцию 2-бром-1-фенилэтанона (50,4 мг, 0,25 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение еще 48 часов. Органическую фазу промывали водным раствором Na2CO3, сушили над Na2SO4 и выпаривали. Неочищенное вещество растворяли в ацетонитриле (3 мл) и добавляли 2-бром-1-фенилэтанон (60,0 мг, 0,30 ммоль). Реакционную смесь нагревали при микроволновом излучении при 100°C в течение 45 минут. Растворитель выпаривали и неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 95/5 до 9/1) с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (62,5 мг; 42% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,86-8,03 (м, 3Н), 7,69-7,82 (м, 1Н), 7,55-7,67 (м, 2Н), 7,37-7,55 (м, 4Н), 5,34 (д, 1Н), 5,16-5,25 (м, 1Н), 5,05-5,16 (м, 1Н), 3,96-4,16 (м, 1Н), 3,43-3,81 (м, 5Н), 2,32-2,42 (м, 1Н), 1,79-2,15 (м, 4Н), 1,41 (с, 9Н);
LC-MS (ESI POS): 513,16 (M+).
ПРИМЕР 18
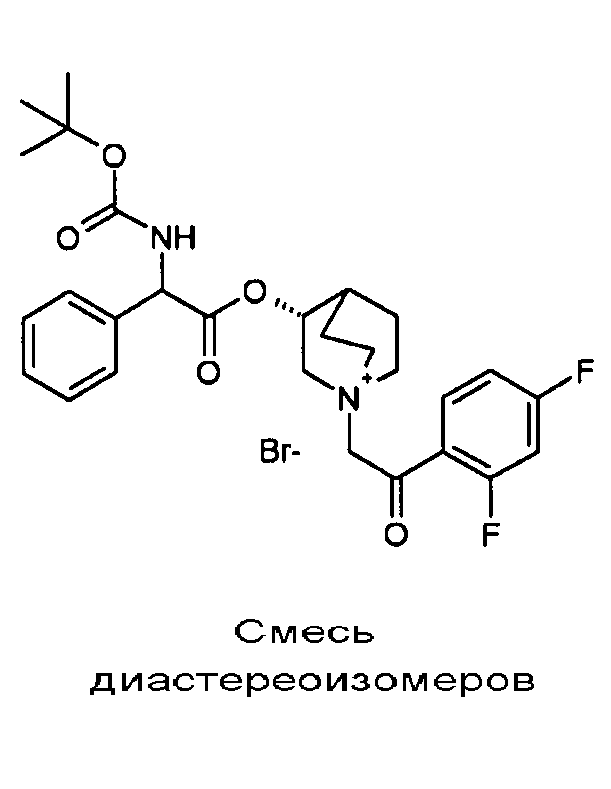
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(4-фторфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C51)
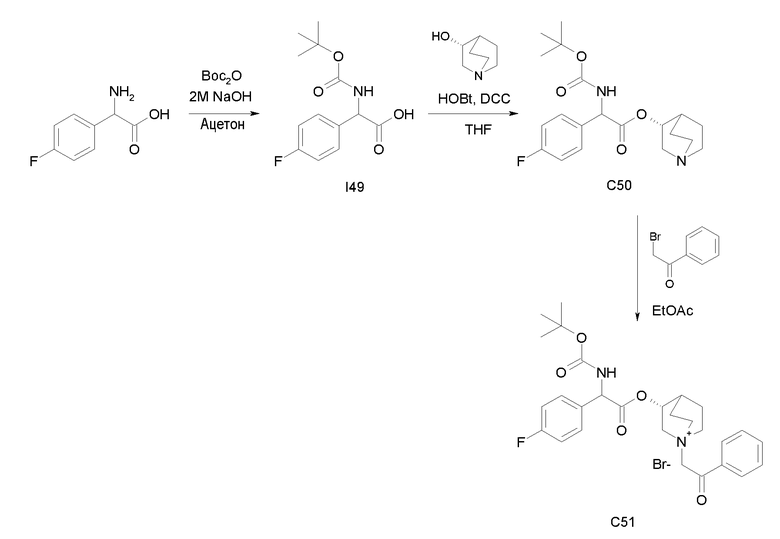
Схема 19
Получение 2-(трет-бутоксикарбониламино)-2-(4-фторфенил)-уксусной кислоты (I49)
К раствору 2-амино-2-(4-фторфенил)уксусной кислоты (1,00 г, 5,91 ммоль) в ацетоне (50 мл) и 2 н. растворе гидроксида натрия (50 мл, 100 ммоль) добавляли ди-трет-бутилдикарбонат (1,29 г, 5,91 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Ацетон выпаривали, водную фазу подкисляли с помощью 37% HCl до pH 1. Продукт экстрагировали с помощью EtOAc, и органическую фазу промывали водой и солевым раствором, сушили над Na2SO4 и выпаривали. Полученное масло растирали с петролейным эфиром с получением 2-(трет-бутоксикарбониламино)-2-(4-фторфенил)уксусной кислоты (725 мг; 45% выход).
Получение (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-фторфенил)ацетата (C50)
К раствору 2-(трет-бутоксикарбониламино)-2-(4-фторфенил)-уксусной кислоты (I49) (725 мг, 2,69 ммоль) в THF (20 мл) добавляли Ν,Ν'-метандиилидендициклогексанамин (667 мг, 3,23 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (437 мг, 3,23 ммоль) и (R)-хинуклидин-3-ол (411 мг, 3,23 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и растворитель выпаривали. Остаток обрабатывали с помощью DCM, нерастворимое твердое вещество отфильтровывали, и органический раствор промывали дважды водным раствором Na2CO3 и затем солевым раствором, сушили над Na2SO4 и выпаривали. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 95/5 до 93/7) с получением (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-фторфенил)ацетата (410 мг; 40% выход).
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(4-фторфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C51)
2-Бром-l-фенилэтанон (34,7 мг, 0,17 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-фторфенил)ацетата (C50) (60,0 мг, 0,16 ммоль) в EtOAc (2 мл) и ацетонитриле (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали. Полученный остаток растирали с i-Pr2O/EtOAc (10/1) и затем с i-Pr2O с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-(4-фторфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (46,1 мг; 50% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,93-8,02 (м, 2Н), 7,91 (д, 1Н), 7,69-7,80 (м, 1Н), 7,57-7,67 (м, 2Н), 7,47-7,57 (м, 2Н), 7,17-7,31 (м, 2Н), 5,33 (д, 1Н), 5,17-5,26 (м, 1Н), 5,15 (с, 2Н), 4,07 (ддд, 1Н), 3,44-3,79 (м, 5Н), 2,37 (уш.с, 1Н), 1,74-2,23 (м, 4Н), 1,41 (с, 9Н);
LC-MS (ESI POS): 497,14 (M+).
ПРИМЕР 19
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(3-фторфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C54)
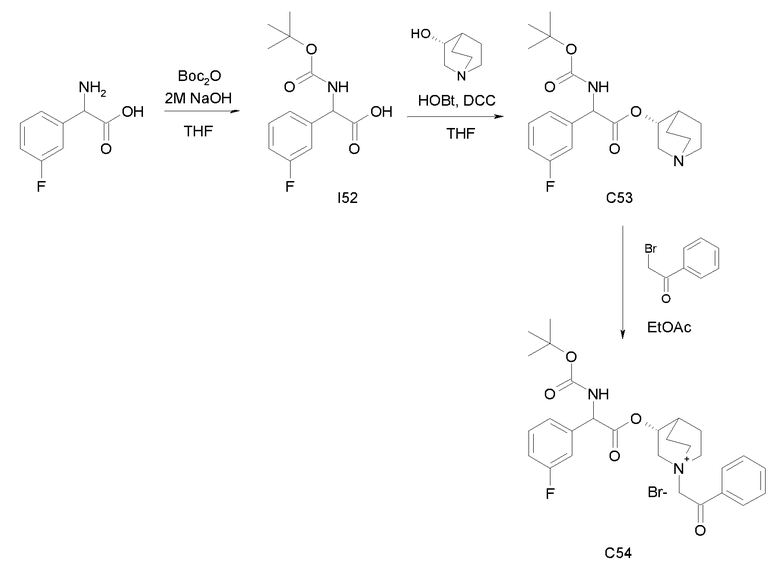
Схема 20
Получение 2-(трет-бутоксикарбониламино)-2-(3-фторфенил)-уксусной кислоты (I52):
К суспензии 2-амино-2-(3-фторфенил)уксусной кислоты (1,00 г, 5,91 ммоль) в THF (30 мл) и воде (30 мл) добавляли 2 н. раствор гидроксида натрия (29,6 мл, 59,1 ммоль) и ди-трет-бутилдикарбонат (2,58 г, 11,8 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Выпаривали THF, водную фазу охлаждали и подкисляли с помощью 37% HCl до pH 1. Требуемое соединение экстрагировали с помощью EtOAc и органическую фазу промывали солевым раствором, сушили над Na2SO4 и выпаривали с получением 2-(трет-бутоксикарбониламино)-2-(3-фторфенил)уксусной кислоты (1,10 г; 69% выход).
Получение (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(3-фторфенил)ацетата (C53):
К раствору 2-(трет-бутоксикарбониламино)-2-(3-фторфенил)-уксусной кислоты (I52) (1,10 г, 4,09 ммоль) в THF (50 мл) добавляли Ν,Ν'-метандиилидендициклогексанамин (1,01 г, 4,90 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (0,66 г, 4,90 ммоль) и (R)-хинуклидин-3-ол (0,62 г, 4,90 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и растворитель выпаривали. Остаток обрабатывали с помощью DCM, нерастворимое твердое вещество отфильтровывали, и органический раствор промывали дважды водным раствором Na2CO3 и затем солевым раствором, сушили над Na2SO4 и выпаривали с получением (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(3-фторфенил)-ацетата (1,55 г; количественный выход).
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(3-фторфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C54)
2-Бром-1-фенилэтанон (63,1 мг, 0,32 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(3-фторфенил)ацетата (C53) (100 мг, 0,26 ммоль) в EtOAc (3 мл) и ацетонитриле (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=94/6) с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-(3-фторфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (70,7 мг; 46% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,87-8,07 (м, 3Н), 7,70-7,83 (м, 1Н), 7,55-7,69 (м, 2Н), 7,38-7,50 (м, 1Н), 7,27-7,38 (м, 2Н), 7,08-7,27 (м, 1Н), 5,32-5,49 (м, 1Н), 5,19-5,27 (м, 1Н), 5,09-5,19 (м, 1Н), 3,93-4,23 (м, 1Н), 3,42-3,90 (м, 5Н), 2,15-2,26 и 2,32-2,42 (м, 1Н), 1,59-2,13 (м, 4Н), 1,41 (с, 9Н);
LC-MS (ESI POS): 497,22 (M+).
ПРИМЕР 20
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(2-фторфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C57)

Схема 21
Получение 2-(трет-бутоксикарбониламино)-2-(2-фторфенил)-уксусной кислоты (I55):
К суспензии 2-амино-2-(2-фторфенил)уксусной кислоты (1,00 г, 5,91 ммоль) в THF (30 мл) и воде (30 мл) добавляли 2 н. раствор гидроксида натрия (29,6 мл, 59,1 ммоль) и ди-трет-бутилдикарбонат (2,58 г, 11,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Выпаривали THF, водную фазу охлаждали до 0°C и подкисляли с помощью 37% HCl до pH 1. Требуемое соединение экстрагировали с помощью EtOAc и органическую фазу промывали солевым раствором, сушили над Na2SO4 и выпаривали с получением 2-(трет-бутоксикарбониламино)-2-(2-фторфенил)уксусной кислоты (1,11 г; 70% выход).
Получение (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(2-фторфенил)ацетата (C56)
К раствору 2-(трет-бутоксикарбониламино)-2-(2-фторфенил)-уксусной кислоты (I55) (1,11 г, 4,12 ммоль) в THF (50 мл) добавляли Ν,Ν'-метандиилидендициклогексанамин (1,02 г, 4,95 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (0,67 г, 4,95 ммоль) и (R)-хинуклидин-3-ол (0,63 г, 4,95 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и растворитель выпаривали. Остаток обрабатывали с помощью DCM, нерастворимое твердое вещество отфильтровывали, и органический раствор промывали дважды водным раствором Na2CO3 и затем солевым раствором, сушили над Na2SO4 и выпаривали с получением (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(2-фторфенил)ацетата (1,56 г; количественный выход).
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(2-фторфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C57)
2-Бром-1-фенилэтанон (57,9 мг, 0,29 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(2-фторфенил)ацетата (C56) (100 мг, 0,26 ммоль) в EtOAc (3 мл) и ацетонитрила (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=94/6) с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-(2-фторфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азоний-бицикло[2.2.2]октанбромида (95,1 мг; 62% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,90-8,10 (м, 3Н), 7,70-7,81 (м, 1Н), 7,61 (тд, 2Н), 7,47-7,56 (м, 1Н), 7,37-7,47 (м, 1Н), 7,15-7,30 (м, 2Н), 5,49-5,68 (м, 1Н), 5,20-5,37 (м, 1Н), 5,04-5,20 (м, 1Н), 3,99-4,28 (м, 1Н), 3,39-3,89 (м, 5Н), 2,32-2,43 (м, 1Н), 1,48-2,15 и 2,15-2,25 (м, 4Н), 1,41 (с, 9Н);
LC-MS (ESI POS): 497,18 (M+).
ПРИМЕР 21
Получение 3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-8-метил-8-(2-оксо-2-фенилэтил)-8-азонийбицикло[3.2.1]-октанбромида (C59)

Схема 22
Получение 8-метил-8-азабицикло[3.2.1]октан-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C58)
К раствору 2-(трет-бутоксикарбониламино)-2-фенилуксусной кислоты (400 мг, 1,59 ммоль) в THF (20 мл) добавляли Ν,Ν'-метандиилидендициклогексанамин (394 мг, 1,91 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (258 мг, 1,91 ммоль) и 8-метил-8-азабицикло[3.2.1]октан-3-ол (270 мг, 1,91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали. Остаток обрабатывали с помощью DCM, нерастворимое вещество отфильтровывали, и прозрачный раствор промывали дважды водным раствором Na2CO3 и солевым раствором, сушили над Na2SO4 и выпаривали с получением 8-метил-8-азабицикло-[3.2.1]октан-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (460 мг; 77% выход). Продукт использовали на следующей стадии без дополнительной очистки.
Получение 3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-8-метил-8-(2-оксо-2-фенилэтил)-8-азонийбицикло[3.2.1]-октанбромида (C59)
К раствору 8-метил-8-азабицикло[3.2.1]октан-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C58) (230 мг, 0,61 ммоль) в DMF (15 мл) и ацетонитриле (5 мл) добавляли 2-бром-1-фенилэтанон (134 мг, 0,68 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, и затем растворитель выпаривали. Неочищенное вещество сначала растирали с i-Pr2O, и затем очищали флэш-хроматографией (DCM/MeOH=94/6) с получением 3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-8-метил-8-(2-оксо-2-фенилэтил)-8-азонийбицикло[3.2.1]октанбромида (196 мг; 56% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,96-8,05 (м, 2Н) 7,88 (д, 1H) 7,67-7,79 (м, 1Н) 7,52-7,66 (м, 2Н) 7,29-7,51 (м, 5Н) 5,29 (д, 1Н) 5,09 (с, 2Н) 5,00-5,07 (м, 1Н) 4,27-4,42 (м, 1Н) 4,15-4,27 (м, 1Н) 3,26 (с, 3Н) 2,57-2,76 (м, 2Н) 2,04-2,42 (м, 3Н) 1,87-2,02 (м, 1Н) 1,48-1,80 (м, 2Н) 1,40 (с, 9Н);
LC-MS (ESI POS): 493,30 (M+).
ПРИМЕР 22
Получение 3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-8,8-диметил-8-азонийбицикло[3.2.1]октанйодида (C60)
Схема 23
К раствору 8-метил-8-азабицикло[3.2.1]октан-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C58) (230 мг, 0,61 ммоль) в DMF (15 мл) добавляли йодметан (42,1 мкл, 0,68 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и затем растворитель выпаривали. Неочищенное вещество растирали с i-Pr2O и затем очищали флэш-хроматографией (DCM/MeOH = от 95/5 до 9/1) с получением 3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-8,8-диметил-8-азонийбицикло[3.2.1]октанйодида (88,2 мг: 28% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,85 (д, 1Н) 7,22-7,62 (м, 5Н) 5,26 (д, 1Н) 4,98 (т, 1Н) 3,76-3,87 (м, 1Н) 3,65-3,76 (м, 1Н) 3,06 (с, 3Н) 2,96 (с, 3Н) 2,57-2,65 (м, 2Н) 1,95-2,23 (м, 3Н) 1,87 (д, 1Н) 1,46-1,67 (м, 2Н) 1,40 (с, 9Н);
LC-MS (ESI POS): 389,25 (M+).
ПРИМЕР 23
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-метил-1-(2-оксо-2-фенилэтил)пирролидиния бромида (C62)

Схема 24
Получение (R)-1-метилпирролидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C61)
К раствору 2-(трет-бутоксикарбониламино)-2-фенилуксусной кислоты (I3) (400 мг, 1,59 ммоль) в THF (20 мл) добавляли (R)-1-метилпирролидин-3-ол (193 мг, 1,91 ммоль), N,N'-метандиилидендициклогексанамин (394 мг, 1,91 ммоль) и 1H-бензо-[d][1,2,3]триазол-1-ол (258 мг, 1,91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем растворитель выпаривали. Остаток обрабатывали с помощью DCM, нерастворимое вещество отфильтровывали и прозрачный раствор промывали дважды водным раствором Na2CO3 и солевым раствором, сушили над Na2SO4 и выпаривали с получением (R)-1-метилпирролидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (340 мг; 64% выход).
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-метил-1-(2-оксо-2-фенилэтил)пирролидиния бромида (C62)
К раствору (R)-1-метилпирролидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C61) (170 мг, 0,51 ммоль) в DMF (5 мл) и ацетонитриле (3 мл) добавляли 2-бром-1-фенилэтанон (111 мг, 0,56 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и растворитель выпаривали. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=9/1) с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-метил-1-(2-оксо-2-фенилэтил)пирролидиния бромида (152 мг; 56% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,69-8,06 (м, 4Н), 7,54-7,69 (м, 2Н), 7,20-7,53 (м, 5Н), 5,14-5,56 (м, 4Н), 3,63-4,26 (м, 4Н), 3,11 и 3,30 (с, 3Н), 2,56-2,71 (м, 1Н), 1,93-2,40 (м, 1Н), 1,32 и 1,42 (с, 9Н);
LC-MS (ESI POS): 453,28 (M+).
ПРИМЕР 24
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-метил-1-(2-оксо-2-(тиофен-2-ил)этил)пирролидиния бромида (C63)

Схема 25
К раствору (R)-1-метилпирролидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетата (C61) (170 мг, 0,51 ммоль) в DMF (5 мл) и ацетонитриле (3 мл) добавляли 2-бром-1-(тиофен-2-ил)этанон (115 мг, 0,56 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали и неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=9/1) с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-фенилацетокси)-1-метил-1-(2-оксо-2-(тиофен-2-ил)этил)-пирролидиния бромида (132 мг; 48% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,13-8,32 (м, 1Н), 7,98-8,08 (м, 1Н), 7,83 и 7,90 (д, 1Н), 7,13-7,53 (м, 6Н), 5,37-5,56 (м, 1Н), 5,03-5,37 (м, 3Н), 3,61-4,19 (м, 4Н), 3,10 и 3,24 (с, 3Н), 2,55-2,71 (м, 1Н), 1,91-2,38 (м, 1Н), 1,34 и 1,41 (с, 9Н);
LC-MS (ESI POS): 459,23 (M+).
ПРИМЕР 25
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(фенилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]октанхлорида (C65)

Схема 26
Получение (R)-хинуклидин-3-ил 2-амино-2-фенилацетата (I30)
(R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-фенилацетат (C1) (5,17 г, 14,3 ммоль) растворяли в THF (47,8 мл) и при перемешивании при комнатной температуре добавляли по каплям 37% HCl (4,71 мл, 57,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали и остаток растворяли в DCM/MeOH (9/1; 10 мл) и добавляли приблизительно 3 г SiO2. Растворитель выпаривали, твердое вещество загружали в колонку с силикагелем и элюировали с помощью DCM/MeOH/NH4OH (9/1/0,1) с получением (R)-хинуклидин-3-ил 2-амино-2-фенилацетата (2,23 г; 60% выход).
Получение (R)-хинуклидин-3-ил 2-фенил-2-(фенилсульфонамидо)ацетата (C64)
(R)-хинуклидин-3-ил 2-амино-2-фенилацетат (I30) (100 мг, 0,38 ммоль) растворяли в DCM (4 мл) и TEA (0,11 мл, 0,77 ммоль). Добавляли бензолсульфонилхлорид (59 мкл, 0,46 ммоль) и раствор перемешивали при комнатной температуре в течение 1 часа. Летучие компоненты выпаривали и остаток очищали флэш-хроматографией (DCM/MeOH/NH4OH=95/5/0,3) с получением (R)-хинуклидин-3-ил 2-фенил-2-(фенилсульфонамидо)ацетата (101 мг; 66% выход).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(фенилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]октанхлорида (C65)
К раствору (R)-хинуклидин-3-ил 2-фенил-2-(фенилсульфонамидо)ацетата (C64) (96 мг, 0,24 ммоль) в EtOAc (1,6 мл) и ацетонитриле (0,8 мл) добавляли 2-хлор-1-фенилэтанон (40,8 мг, 0,26 ммоль). Раствор перемешивали при комнатной температуре в течение ночи. Раствор концентрировали под вакуумом и неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 95/5 до 9/1) с получением (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(фенилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]-октанхлорида (99 мг; 74% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,06 (уш.с, 1Н) 7,94-8,04 (м, 2Н) 7,71-7,83 (м, 3Н) 7,45-7,66 (м, 5Н) 7,24-7,41 (м, 5Н) 5,26 (д, 1Н) 5,22 (д, 1Н) 5,13-5,21 (м, 1Н) 4,95-5,11 (м, 1Н) 3,94-4,17 (м, 1Н) 3,47-3,79 (м, 5Н) 2,10-2,26 (м, 1Н) 1,69-2,08 (м, 4Н);
LC-MS (ESI POS): 519,27 (M+).
ПРИМЕР 26
Получение (3R)-1-(2-оксо-2-(тиазол-2-ил)этил)-3-(2-фенил-2-(фенилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (С66)

Схема 27
Получение (R)-хинуклидин-3-ил 2-фенил-2-(фенилсульфонамидо)ацетата (C64)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (0,67 г, 2,01 ммоль) в DCM (25 мл) последовательно добавляли триэтиламин (0,84 мл, 6,04 ммоль) и бензолсульфонилхлорид (0,31 мл, 2,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и затем растворитель выпаривали. Остаток обрабатывали с помощью EtOAc и промывали водой и солевым раствором, сушили над Na2SO4 и выпаривали с получением (R)-хинуклидин-3-ил 2-фенил-2-(фенилсульфонамидо)ацетата (370 мг; 46% выход).
Получение (3R)-1-(2-оксо-2-(тиазол-2-ил)этил)-3-(2-фенил-2-(фенилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C66)
К раствору (R)-хинуклидин-3-ил 2-фенил-2-(фенилсульфонамидо)ацетата (C64) (90,0 мг, 0,22 ммоль) в EtOAc (2 мл) и ацетонитриле (5 мл) добавляли 2-бром-1-(тиазол-2-ил)этанон (50,9 мг, 0,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 39 часов, затем добавляли вторую порцию 2-бром-1-(тиазол-2-ил)этанона (46,3 мг, 0,22 ммоль) и реакционную смесь перемешивали при 60°C в течение 15 часов. Растворитель удаляли при пониженном давлении и полученное твердое вещество сначала очищали флэш-хроматографией (гексан/EtOAc=9/1), а затем препаративной ВЭЖХ с получением (3R)-1-(2-оксо-2-(тиазол-2-ил)этил)-3-(2-фенил-2-(фенилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (13,6 мг; 10% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,99 (д, 1Н) 8,39 (т, 1Н) 8,24 (д, 1Н) 7,71-7,83 (м, 2Н) 7,44-7,64 (м, 3Н) 7,22-7,40 (м, 5Н) 5,10-5,22 (м, 3Н) 4,94-5,10 (м, 1Н) 3,97-4,14 (м, 1Н) 3,53-3,76 (м, 5Н) 2,12-2,23 (м, 1Н) 1,47-2,11 (м, 4Н);
LC-MS (ESI POS): 526,13 (M+).
ПРИМЕР 27
Получение (3R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-(2-фенил-2-(фенилсульфонамидо)ацетокси)-1-азонийбицикло-[2.2.2]октана 2,2,2-трифторацетата (C67)
Схема 28
К раствору (R)-хинуклидин-3-ил 2-фенил-2-(фенилсульфонамидо)ацетата (C64) (90,0 мг, 0,22 ммоль) в EtOAc (2 мл) и ацетонитриле (5 мл) добавляли 2-хлор-1-(тиофен-2-ил)этанон (39,7 мг, 0,25 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Затем снова добавляли 2-хлор-1-(тиофен-2-ил)этанон (36,1 мг, 0,22 ммоль) и реакционную смесь нагревали при 60°C в течение 15 часов. Растворитель удаляли и неочищенное вещество очищали флэш-хроматографией (гексан/EtOAc=9/1) и затем препаративной ВЭЖХ с получением (3R)-1-(2-оксо-2-(тиофен-2-ил)этил)-3-(2-фенил-2-(фенилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (11,9 мг; 8% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,99 (д, 1Н), 8,20 и 8,21 (дд, 1Н), 8,05 и 8,07 (дд, 1Н), 7,70-7,85 (м, 2Н), 7,43-7,66 (м, 3Н), 7,22-7,39 (м, 6Н), 5,12-5,21 (м, 1Н), 5,04-5,09 (м, 1Н), 4,99 и 5,02 (с, 2Н), 3,93-4,13 (м, 1Н), 3,29-3,56 (м, 5Н), 2,11-2,23 (м, 1Н), 1,44-2,10 (м, 4Н);
LC-MS (ESI POS): 525,11 (M+).
ПРИМЕР 28
Получение (3R)-3-(2-(4-метоксифенилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C69)

Схема 29
Получение (R)-хинуклидин-3-ил 2-(4-метоксифенилсульфонамидо)-2-фенилацетата гидрохлорида (C68):
(R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорид (I25) (150 мг, 0,45 ммоль) суспендировали в DCM (6 мл) и обрабатывали триэтиламином (99 мкл, 1,35 ммоль). Затем добавляли 4-метоксибензол-1-сульфонилхлорид (112 мг, 0,54 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель выпаривали, и неочищенное вещество растворяли в EtOAc и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали. Неочищенное вещество растирали с Et2O с получением (R)-хинуклидин-3-ил 2-(4-метоксифенилсульфонамидо)-2-фенилацетата гидрохлорида (105 мг; 50% выход).
Получение (3R)-3-(2-(4-метоксифенилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C69)
(R)-хинуклидин-3-ил 2-(4-метоксифенилсульфонамидо)-2-фенилацетата гидрохлорид (C68) (96 мг, 0,22 ммоль) растворяли в DCM и промывали 1M раствором K2CO3. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали. Полученное твердое вещество растворяли в EtOAc (2 мл) и добавляли 2-бром-1-фенилэтанон (49,2 мг, 0,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем промывали водным раствором K2CO3, сушили над Na2SO4, фильтровали и выпаривали с получением (3R)-3-(2-(4-метоксифенилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (75 мг; 53% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,84 (д, 1Н), 7,88-8,04 (м, 2Н), 7,66-7,82 (м, 3Н), 7,53-7,66 (м, 2Н), 7,25-7,44 (м, 5Н), 6,97-7,11 (м, 2Н), 5,12-5,19 (м, 1Н), 4,95-5,12 (м, 2Н), 3,96-4,15 (м, 1Н), 3,81 (с, 3Н), 3,38-3,76 (м, 5Н), 2,13-2,24 (м, 1Н), 1,74-2,13 (м, 4Н);
LC-MS (ESI POS): 549,19 (M+).
ПРИМЕР 29
Получение (3R)-3-(2-(4-хлорфенилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C71)

Схема 30
Получение (R)-хинуклидин-3-ил 2-(4-хлорфенилсульфонамидо)-2-фенилацетата (C70)
К раствору (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (150 мг, 0,45 ммоль) в DCM (5 мл) и триэтиламине (190 мкл, 1,35 ммоль) добавляли 4-хлорбензол-1-сульфонилхлорид (114 мг, 0,54 моль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем растворитель выпаривали. Неочищенное вещество обрабатывали с помощью EtOAc и промывали водой, солевым раствором и затем раствором K2CO3. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали досуха с получением (R)-хинуклидин-3-ил 2-(4-хлорфенилсульфонамидо)-2-фенилацетата (62 мг; 32% выход), который использовали на следующей стадии без дополнительной очистки.
Получение (3R)-3-(2-(4-хлорфенилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (C71)
К раствору (R)-хинуклидин-3-ил 2-(4-хлорфенилсульфонамидо)-2-фенилацетата (C70) (62 мг, 0,14 ммоль) в EtOAc (2,5 мл) добавляли 2-бром-1-фенилэтанон (31,2 мг, 0,16 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Затем растворитель выпаривали и неочищенное вещество растирали с Et2O с получением (3R)-3-(2-(4-хлорфенилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (38 мг; 42% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,11 (д, 1Н), 7,88-8,07 (м, 2Н), 7,69-7,85 (м, 3Н), 7,51-7,69 (м, 4Н), 7,20-7,44 (м, 5Н), 5,17-5,26 (м, 1Н), 4,92-5,17 (м, 2Н), 3,95-4,19 (м, 1Н), 3,39-3,79 (м, 5Н), 2,15-2,25 (м, 1Н), 1,76-2,15 (м, 4Н);
LC-MS (ESI POS): 553,13 (M+).
ПРИМЕР 30
Получение (3R)-3-(2-(3,4-дифторфенилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C73)

Схема 31
Получение (R)-хинуклидин-3-ил 2-(3,4-дифторфенилсульфонамидо)-2-фенилацетата (C72)
(R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорид (I25) (110 мг, 0,33 ммоль) суспендировали в DCM (4 мл) и добавляли триэтиламин (121 мкл, 1,65 ммоль), получая раствор. Добавляли 3,4-дифторбензол-1-сульфонилхлорид (53,1 мкл, 0,40 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли с помощью DCM и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали досуха. Остаток растирали с Et2O с получением (R)-хинуклидин-3-ил 2-(3,4-дифторфенилсульфонамидо)-2-фенилацетата (40 мг; 28% выход), который использовали на следующей стадии без дополнительной очистки.
Получение (3R)-3-(2-(3,4-дифторфенилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C73)
К раствору (R)-хинуклидин-3-ил 2-(3,4-дифторфенилсульфонамидо)-2-фенилацетата (C72) (40 мг, 0,09 ммоль) в EtOAc (3 мл) добавляли 2-бром-1-фенилэтанон (20,1 мг, 0,10 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем растворитель выпаривали и неочищенное вещество сначала растирали с петролейным эфиром, а затем очищали препаративной ВЭЖХ с получением (3R)-3-(2-(3,4-дифторфенилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (13 мг; 21% выход).
1H ЯМР (300 МГц, ацетон) δ м.д. 8,89 и 8,96 (д, 1Н), 8,00-8,16 (м, 2Н), 7,18-7,91 (м, 11Н), 5,52 и 5,58 (д, 1Н), 5,44 и 5,51 (д, 1Н), 5,28 и 5,35 (д, 1Н), 5,18-5,29 (м, 1Н), 4,29-4,49 (м, 2Н), 3,75-4,28 (м, 4Н), 2,39-2,53 (м, 1Н), 2,11-2,36 (м, 3Н), 1,69-2,03 (м, 1Н);
LC-MS (ESI POS): 555,12 (M+).
ПРИМЕР 31
Получение (3R)-3-(2-(2,4-диметилтиазол-5-сульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C75)
Схема 32
Получение (R)-хинуклидин-3-ил 2-(2,4-диметилтиазол-5-сульфонамидо)-2-фенилацетата (C74)
(R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорид (I25) (116 мг, 0,35 ммоль) суспендировали в DCM (6 мл) и добавляли триэтиламин (102 мкл, 1,39 ммоль), получая прозрачный раствор. Добавляли 2,4-диметилтиазол-5-сульфонилхлорид (88,0 мг, 0,42 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли с помощью DCM и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали досуха. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=85/15) с получением (R)-хинуклидин-3-ил 2-(2,4- диметилтиазол-5-сульфонамидо)-2-фенилацетата (67 мг; 44,2% выход).
Получение (3R)-3-(2-(2,4-диметилтиазол-5-сульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октана 2,2,2-трифторацетата (C75)
К раствору (R)-хинуклидин-3-ил 2-(2,4-диметилтиазол-5- сульфонамидо)-2-фенилацетата (C74) (67,0 мг, 0,15 ммоль) в EtOAc (2 мл) добавляли 2-бром-1-фенилэтанон (33,7 мг, 0,17 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель выпаривали и неочищенное вещество очищали препаративной ВЭЖХ, получая (3R)-3-(2-(2,4-диметилтиазол-5-сульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетат (18 мг; 18% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,43 и 9,44 (д, 1Н), 7,89-8,05 (м, 2Н), 7,70-7,82 (м, 1Н), 7,56-7,69 (м, 2Н), 7,24-7,46 (м, 5Н), 5,15-5,26 (м, 2Н), 5,13 (с, 2Н), 3,94-4,21 (м, 1Н), 3,43-3,62 (м, 5Н), 2,55 и 2,58 (с, 3Н), 2,39 и 2,42 (с, 3Н), 2,06-2,20 и 2,20-2,30 (м, 1Н), 1,47-2,05 (м, 4Н);
LC-MS (ESI POS): 553,13 (M+).
ПРИМЕР 32
Получение (3R)-3-(2-(метилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (C77)

Схема 33
Получение (R)-хинуклидин-3-ил 2-(метилсульфонамидо)-2-фенилацетата (C76)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (120 мг, 0,36 ммоль) в DCM (6 мл) последовательно добавляли триэтиламин (79 мкл, 1,08 ммоль) и метансульфонилхлорид (33,4 мкл, 0,43 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем летучие компоненты выпаривали. Неочищенное вещество обрабатывали с помощью EtOAc и промывали водой и солевым раствором. Органический слой сушили над Na2SO4, фильтровали и выпаривали досуха. Неочищенное вещество растирали с Et2O с получением (R)-хинуклидин-3-ил 2-(метилсульфонамидо)-2-фенилацетата (38,9 мг; 32% выход).
Получение (3R)-3-(2-(метилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (C77)
К раствору (R)-хинуклидин-3-ил 2-(метилсульфонамидо)-2-фенилацетата (C76) (38,9 мг, 0,11 ммоль) в EtOAc (1 мл) и ацетонитриле (0,5 мл) добавляли 2-бром-1-фенилэтанон (25,2 мг, 0,13 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, затем растворитель выпаривали. Неочищенное вещество растирали с Et2O с получением (3R)-3-(2-(метилсульфонамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (40 мг; 64,7% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,37 (д, 1Н), 7,90-8,05 (м, 2Н), 7,69-7,83 (м, 1Н), 7,55-7,67 (м, 2Н), 7,29-7,55 (м, 5Н), 5,27-5,34 (м, 1Н), 5,20-5,27 (м, 1Н), 5,10-5,20 (м, 2Н), 4,00-4,23 (м, 1Н), 3,46-3,81 (м, 5Н), 2,89 и 2,93 (с, 3Н), 2,22 и 2,39 (м, 1Н), 1,50-2,15 (м, 4Н);
LC-MS (ESI POS): 457,17 (M+).
ПРИМЕР 33
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(2,2,2-трифторэтилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]-октанбромида (C79)

Схема 34
Получение (R)-хинуклидин-3-ил 2-фенил-2-(2,2,2-трифтор-этилсульфонамидо)ацетата (C78)
К раствору (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (120 мг, 0,36 ммоль) в DCM (4 мл) и триэтиламине (105 мкл, 1,44 ммоль) добавляли 2,2,2-трифторэтансульфонилхлорид (47,5 мкл, 0,43 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и затем разбавляли с помощью DCM и промывали водой, солевым раствором и раствором 1M K2CO3. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=9/1) с получением (R)-хинуклидин-3-ил 2-фенил-2-(2,2,2-трифторэтилсульфонамидо)-ацетата (60 мг; 41% выход).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(2,2,2-трифторэтилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]-октанбромида (C79)
К раствору (R)-хинуклидин-3-ил 2-фенил-2-(2,2,2-трифторэтилсульфонамидо)ацетата (C78) (60 мг, 0,15 ммоль) в EtOAc (2 мл) добавляли 2-бром-1-фенилэтанон (35,3 мг, 0,18 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем растворитель выпаривали и неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=9/1) с получением (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(2,2,2-трифторэтилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]октан-бромида (5 мг; 6% выход).
1H ЯМР (300 МГц, Ацетонитрил-d3) δ м.д. 7,91-8,06 (м, 2Н), 7,71-7,83 (м, 1Н), 7,67 (д, 1Н), 7,56-7,64 (м, 2Н), 7,37-7,56 (м, 5Н), 5,36 и 5,37 (д, 1Н), 5,16-5,31 (м, 1Н), 4,91 и 4,98 (д, 1Н), 4,84 и 4,91 (д, 1Н), 3,94-4,33 (м, 3Н), 3,50-3,93 (м, 5Н), 2,32-2,42 и 2,42-2,55 (м, 1Н), 2,00-2,23 (м, 3Н), 1,79-1,92 (м, 1Н);
LC-MS (ESI POS): 525,10 (M+).
ПРИМЕР 34
Получение ((3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(фенилметилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]-октанбромида (C81)
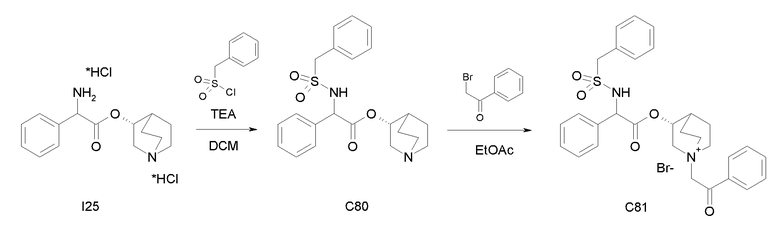
Схема 35
Получение (R)-хинуклидин-3-ил 2-фенил-2-(фенилметилсульфонамидо)ацетата (C80)
К раствору (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (100 мг, 0,30 ммоль) в DCM (4 мл) и триэтиламине (65,8 мкл, 0,90 ммоль) добавляли фенилметансульфонилхлорид (68,6 мг, 0,36 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 13 часов. Выпаривали DCM, и неочищенное вещество обрабатывали с помощью EtOAc и промывали 1M раствором Na2CO3, водой и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали с получением (R)-хинуклидин-3-ил 2-фенил-2-(фенилметилсульфонамидо)ацетата (74 мг; 59,5% выход).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(фенилметилсульфонамидо)ацетокси)-1-азонийбицикло[2.2.2]октанбромида (C81)
К раствору (R)-хинуклидин-3-ил 2-фенил-2-(фенилметилсульфонамидо)ацетата (C80) (41 мг, 0,10 ммоль) в EtOAc (2 мл) добавляли 2-бром-1-фенилэтанон (21,7 мг, 0,11 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель выпаривали, и неочищенное вещество растирали с Et2O с получением (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-(фенилметилсульфонамидо)ацетокси)-1-азонийбицикло-[2.2.2]октанбромида (50 мг; 82% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,42 (д, 1Н), 7,88-8,07 (м, 2Н), 7,69-7,81 (м, 1Н), 7,53-7,67 (м, 2Н), 7,24-7,52 (м, 10Н), 5,18-5,33 (м, 1Н), 5,08-5,14 (м, 1Н), 5,13 (д, 1Н), 4,44 (д, 1Н), 4,37 (д, 1Н), 4,02-4,21 (м, 1Н), 3,45-3,84 (м, 5Н), 2,32-2,42 (м, 1Н), 1,43-2,15 (м, 4Н);
LC-MS (ESI POS): 533,23 (M+).
ПРИМЕР 35
Получение (3R)-3-(2-(4-фторфенил)-2-(фенилсульфонамидо)-ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C84)
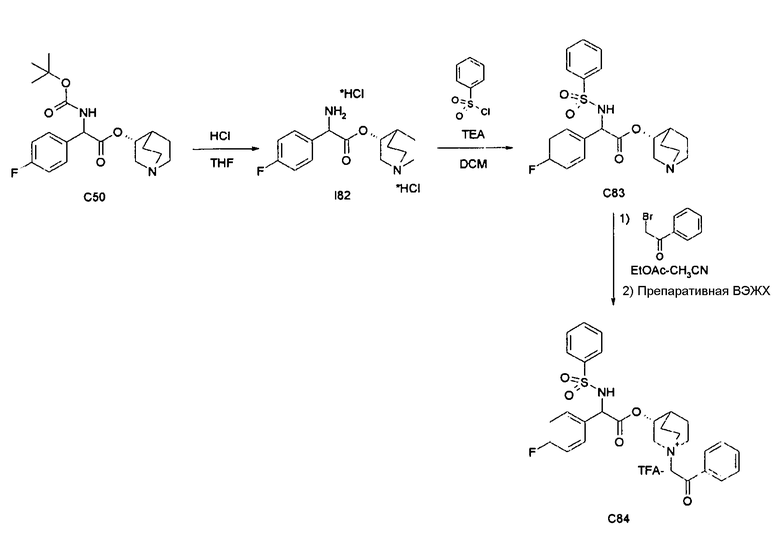
Схема 36
Получение (R)-хинуклидин-3-ил 2-амино-2-(4-фторфенил)ацетата дигидрохлорида (I82)
К раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-фторфенил)ацетата (C50) (350 мг, 0,92 ммоль) в THF (20 мл) добавляли 37% хлористоводородную кислоту (1,0 мл, 12,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Затем снова добавляли вторую порцию 37% хлористоводородной кислоты (1,0 мл, 12,2 ммоль) и реакционную смесь дополнительно перемешивали при комнатной температуре в течение 24 часов. Затем выпаривали под вакуумом легколетучие компоненты с получением (R)-хинуклидин-3-ил 2-амино-2-(4-фторфенил)ацетата дигидрохлорида (325 мг; количественный выход).
Получение (R)-хинуклидин-3-ил 2-(4-фторфенил)-2-(фенилсульфонамидо)ацетата (C83)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-(4-фторфенил)-ацетата дигидрохлорида (I82) (325 мг, 0,92 ммоль) в DCM (10 мл) добавляли триэтиламин (386 мкл, 2,78 ммоль) и бензолсульфонилхлорид (142 мкл, 1,11 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и затем растворитель выпаривали. Остаток обрабатывали с помощью EtOAc и промывали 1M раствором Na2CO3, водой и затем солевым раствором, сушили над Na2SO4 и выпаривали досуха с получением (R)-хинуклидин-3-ил 2-(4-фторфенил)-2-(фенилсульфонамидо)ацетата (150 мг; 39% выход).
Получение (3R)-3-(2-(4-фторфенил)-2- (фенилсульфонамидо)-ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C84)
К раствору (R)-хинуклидин-3-ил 2-(4-фторфенил)-2-(фенилсульфонамидо)ацетата (C83) (150 мг, 0,36 ммоль) в EtOAc (2 мл) и ацетонитриле (2 мл) добавляли 2-бром-1-фенилэтанон (78 мг, 0,39 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали и неочищенное вещество очищали препаративной ВЭЖХ с получением (3R)-3-(2-(4-фторфенил)-2-(фенилсульфонамидо)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октан-2,2,2-трифтор-ацетата (97,9 мг; 42% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,02 (д, 1Н), 7,89-8,09 (м, 2Н), 7,66-7,81 (м, 3Н), 7,32-7,68 (м, 7Н), 7,02-7,21 (м, 2Н), 5,20-5,30 (м, 1Н), 5,11 и 5,14 (с, 2Н), 4,94-5,10 (м, 1Н), 3,94-4,14 (м, 1Н), 3,38-3,66 (м, 5Н), 2,14-2,25 (м, 1Н), 1,53-2,09 (м, 4Н);
LC-MS (ESI POS): 537,18 (M+).
ПРИМЕР 36
Получение (3R)-3-(2-бензамидо-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанхлорида (C86)

Схема 37
Получение (R)-хинуклидин-3-ил 2-бензамидо-2-фенилацетата (C85)
К раствору (R)-хинуклидин-3-ил 2-амино-2-фенилацетата (I30) (100 мг, 0,38 ммоль) в DCM (4 мл) и триэтиламине (0,08 мл, 0,58 ммоль) добавляли бензоилхлорид (58,0 мкл, 0,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов и затем выпаривали летучие компоненты. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH/NH4OH=95/5/0,3) с получением (R)-хинуклидин-3-ил 2-бензамидо-2-фенилацетата (70 мг; 50% выход).
Получение (3R)-3-(2-бензамидо-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанхлорида (C86)
К раствору (R)-хинуклидин-3-ил 2-бензамидо-2-фенилацетата (C85) (70 мг, 0,19 ммоль) в EtOAc (1 мл) и ацетонитриле (1 мл) добавляли 2-хлор-1-фенилэтанон (32,7 мг, 0,21 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, затем растворители выпаривали и остаток растирали с EtOAc (8 мл) с получением (3R)-3-(2-бензамидо-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанхлорида (72 мг; 72% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,38 (д, J=7,04 Гц, 1Н), 7,88-8,11 (м, 4Н), 7,69-7,87 (м, 1Н), 7,12-7,68 (м, 10Н), 5,78 (д, J=7,04 Гц, 1Н), 5,26 (с, 2Н), 5,15-5,24 (м, 1Н), 4,05-4,27 (м, 1Н), 3,81-3,94 (м, 1Н), 3,54-3,81 (м, 4Н), 2,19-2,31 (м, 1Н), 1,91-2,14 (м, 2Н), 1,74-1,91 (м, 1Н), 1,61-1,74 (м, 1Н);
LC-MS (ESI POS): 483,02 (M+).
ПРИМЕР 37
Получение (3R)-3-(2-бензамидо-2-фенилацетокси)-1-(2-(4- фторфенил)-2-оксоэтил)-1-азонийбицикло[2.2.2]октан-2,2,2- трифторацетата (C87)

Схема 38
Получение (R)-хинуклидин-3-ил 2-бензамидо-2-фенилацетата (C85)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (0,67 г, 2,01 ммоль) в DCM (25 мл) последовательно добавляли триэтиламин (0,84 мл, 6,04 ммоль) и бензоилхлорид (0,28 мл, 2,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем растворитель выпаривали. Неочищенное вещество обрабатывали с помощью EtOAc и промывали водой и солевым раствором, сушили над Na2SO4 и выпаривали с получением (R)-хинуклидин-3-ил 2-бензамидо-2-фенилацетата (310 мг; 42% выход).
Получение (3R)-3-(2-бензамидо-2-фенилацетокси)-1-(2-(4-фторфенил)-2-оксоэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C87)
К раствору (R)-хинуклидин-3-ил 2-бензамидо-2-фенилацетата (C85) (77,5 мг, 0,21 ммоль) в EtOAc (1 мл) и ацетонитриле (5 мл) добавляли 2-хлор-1-(4-фторфенил)этанон (40,4 мг, 0,23 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов и затем растворитель выпаривали. Неочищенное вещество растирали с i-Pr2O/EtOAc (5/1) и затем очищали препаративной ВЭЖХ с получением (3R)-3-(2-бензамидо-2-фенилацетокси)-1-(2-(4-фтор-фенил)-2-оксоэтил)-1-азонийбицикло-[2.2.2]октана 2,2,2-трифторацетата (50,8 мг; 39% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,27 (д, 1Н), 7,99-8,16 (м, 2Н), 7,84-7,99 (м, 2Н), 7,28-7,67 (м, 10Н), 5,66-5,84 (м, 1Н), 5,26 (м, 1Н), 5,14 и 5,15 (с, 2Н), 3,97-4,30 (м, 1Н), 3,36-3,73 (м, 5Н), 2,18-2,26 и 2,38-2,46 (м, 1Н), 1,57-2,17 (м, 4Н);
LC-MS (ESI POS): 501,08 (M+).
Соединения, представленные в таблице 5, получали, как описано выше для C87, исходя из соединения C85 и подходящих выпускаемых промышленностью алкилирующих агентов.
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 10,72 (уш.с, 1Н), 9,25 и 9,26 (д, 1Н), 7,90-8,05 (м, 2Н), 7,80-7,90 (м, 2Н), 7,30-7,65 (м, 8Н), 6,82-6,99 (м, 2Н), 5,65-5,78 (м, 1Н), 5,17-5,33 (м, 1Н), 5,03 и 5,05 (уш.с, 2Н), 4,02-4,26 (м, 1Н), 3,60-3,86 (м, 5Н), 2,17-2,26 и 2,34-2,46 (м, 1Н), 1,51-2,15 (м, 4Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,25 и 9,27 (д, 1Н), 8,38 (д, 1Н), 8,24 (д, 1Н), 7,87-7,99 (м, 2Н), 7,33-7,60 (м, 8Н), 5,67-5,73 и 5,74-5,78 (м, 1Н), 5,22-5,32 (м, 1Н), 5,18 и 5,20 (с, 2Н), 4,03-4,29 (м, 1Н), 3,56-3,89 (м, 5Н), 2,18-2,25 и 2,37-2,47 (м, 1Н), 1,34-2,15 (м, 4Н)

1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,26 (дд, 1Н) 8,61 (ддд, 1Н) 7,86-8,03 (м, 2Н) 7,68-7,81 (м, 1Н) 7,31-7,64 (м, 9Н) 5,65-5,82 (м, 1Н) 5,18-5,33 (м, 1Н) 5,02 (д, 2Н) 4,03-4,24 (м, 1Н) 3,73-3,85 (м, 4Н) 2,18-2,46 (м, 1Н) 1,74-2,14 (м, 4Н) 1,52-1,74 (м, 1Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,37 и 9,45 (д, 1Н), 8,18-8,30 (м, 1Н), 8,12 и 8,14 (дд, 1Н), 7,89-8,02 (м, 2Н), 7,17-7,69 (м, 9Н), 5,74 и 5,80 (д, 1Н), 5,19-5,29 (м, 1Н), 5,16 и 5,17 (с, 2Н), 4,07-4,30 (м, 1Н), 3,50-4,02 (м, 5Н), 2,17-2,30 и 2,35-2,42 (м, 1Н), 1,50-2,14 (м, 4Н)
ПРИМЕР 38
Получение (3R)-3-(2-ацетамидо-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C93)

Схема 39
Получение (R)-хинуклидин-3-ил 2-ацетамидо-2-фенилацетата (C92)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (140 мг, 0,42 ммоль) в DCM (10 мл) последовательно добавляли триэтиламин (175 мкл, 1,26 ммоль) и ацетилхлорид (35,8 мкл, 0,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем растворитель выпаривали. Остаток обрабатывали небольшим количеством EtOAc и нерастворимое вещество отфильтровывали. Органическую фазу выпаривали досуха, получая (R)-хинуклидин-3-ил 2-ацетамидо-2-фенилацетат (127 мг; количественный выход).
Получение (3R)-3-(2-ацетамидо-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C93)
К раствору (R)-хинуклидин-3-ил 2-ацетамидо-2-фенилацетата (C92) (127 мг, 0,42 ммоль) в EtOAc (2 мл) и ацетонитриле (5 мл) добавляли 2-бром-1-фенилэтанон (92 мг, 0,46 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 72 часов. Растворитель выпаривали и неочищенное вещество очищали препаративной ВЭЖХ с получением (3R)-3-(2-ацетамидо-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азоний-бицикло[2.2.2]-октана 2,2,2-трифторацетата (20,5 мг; 9% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,77 и 8,79 (д, 1Н), 7,89-8,04 (м, 2Н), 7,70-7,82 (м, 1Н), 7,55-7,69 (м, 2Н), 7,29-7,54 (м, 5Н), 5,44 и 5,48 (д, 1Н), 5,18-5,27 (м, 1Н), 5,15 и 5,18 (с, 2Н), 3,98-4,21 (м, 1Н), 3,52-3,81 (м, 5Н), 1,94 (с, 3Н), 1,58-2,45 (м, 5Н);
LC-MS (ESI POS): 421,16 (M+).
ПРИМЕР 39
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-пиваламидоацетокси)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C95)

Схема 40
Получение (R)-хинуклидин-3-ил 2-фенил-2-пиваламидоацетата (C94)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (I25) (140 мг, 0,42 ммоль) в DCM (5 мл) последовательно добавляли триэтиламин (175 мкл, 1,26 ммоль) и пивалоилхлорид (62,1 мкл, 0,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем растворитель выпаривали. Остаток обрабатывали с помощью EtOAc и промывали раствором Na2CO3, водой и солевым раствором, сушили над Na2SO4, фильтровали и выпаривали с получением (R)-хинуклидин-3-ил 2-фенил-2-пиваламидоацетата (95 мг; 66% выход).
Получение (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-пиваламидоацетокси)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C95)
К раствору (R)-хинуклидин-3-ил 2-фенил-2-пиваламидоацетата (C94) (95 мг, 0,28 ммоль) в EtOAc (2 мл) и ацетонитриле (2 мл) добавляли 2-бром-1-фенилэтанон (60,4 мг, 0,30 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали и неочищенное вещество очищали препаративной ВЭЖХ с получением (3R)-1-(2-оксо-2-фенилэтил)-3-(2-фенил-2-пиваламидоацетокси)-1-азонийбицикло-[2.2.2]октана 2,2,2-трифторацетата (118,6 мг; 75% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,21 (т, 1Н) 7,91-8,05 (м, 2Н) 7,69-7,83 (м, 1Н) 7,55-7,69 (м, 2Н) 7,26-7,54 (м, 5Н) 5,40-5,54 (м, 1Н) 5,16 (д, 2Н) 5,08-5,34 (м, 1Н) 4,01-4,34 (м, 3Н) 3,43-3,79 (м, 3Н) 2,32-2,45 (м, 1Н) 1,87-2,16 (м, 3Н) 1,74 (с, 1Н) 1,17 (с, 9Н);
LC-MS (ESI POS): 463,19 (M+).
Соединения, представленные в таблице 6, получали, как описано выше для C95, обрабатывая промежуточное соединение I25 подходящими выпускаемыми промышленностью ацилгалогенидами, и затем подвергая кватернизации с помощью 2-бром-1-фенилэтанона и очистке препаративной ВЭЖХ.
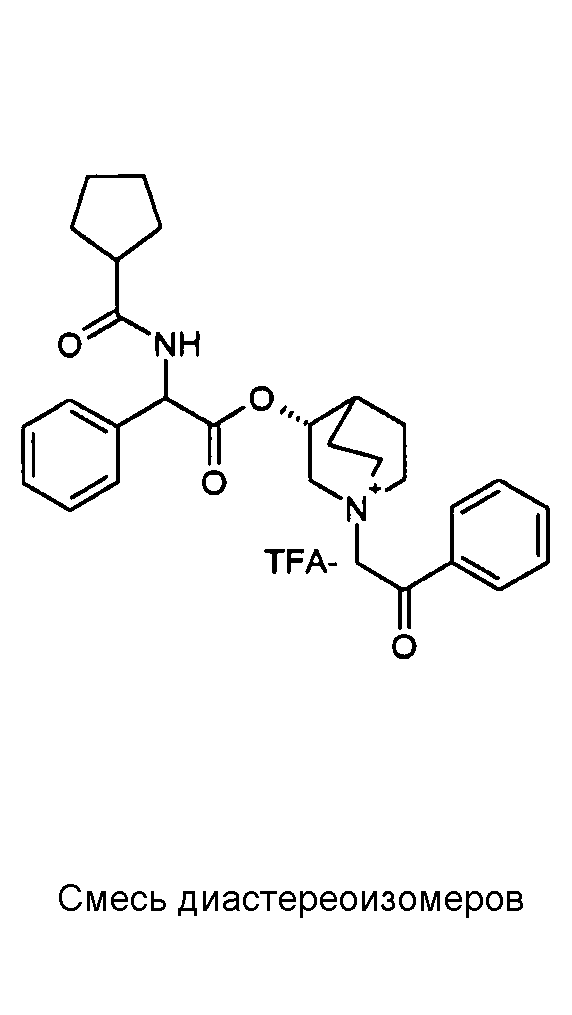
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,68 (т, 1Н) 7,88-8,10 (м, 2Н) 7,69-7,83 (м, 1Н) 7,54-7,69 (м, 2Н) 7,30-7,53 (м, 5Н) 5,37-5,54 (м, 1Н) 5,16 (д, 2Н) 5,05-5,31 (м, 1Н) 3,98-4,22 (м, 1Н) 3,61-3,85 (м, 5Н) 2,70-2,86 (м, 2Н) 2,34-2,43 (м, 1Н) 1,88-2,15 (м,3H) 1,47-1,86 (м, 8H)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,01 и 9,04 (д, 1Н), 7,90-8,08 (м, 2Н), 7,69-7,81 (м, 1Н), 7,56-7,69 (м, 2Н), 7,36-7,56 (м, 5Н), 7,11-7,35 (м, 5Н), 5,43 и 5,47 (д, 1Н), 5,17-5,28 (м, 1Н), 5,01-5,17 (м, 2Н), 3,94-4,22 (м, 1Н), 3,37-3,76 (м, 7Н), 2,09-2,19 и 2,31-2,41 (м, 1Н), 1,30-2,09 (м, 4Н)

1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,05 (дд, 1Н) 7,90-8,05 (м, 2Н) 7,70-7,83 (м, 1Н) 7,55-7,68 (м, 2Н) 7,33-7,53 (м, 5Н) 5,50 (дд, 1Н) 5,09-5,30 (м, 1Н) 5,17 (д, 2Н) 4,12-4,23 (м, 1Н) 4,08 (кв., 2Н) 3,53-3,76 (м, 5Н) 3,32-3,48 (м, 2Н) 2,36-2,44 (м, 1Н) 1,88-2,18 (м, 3Н) 1,53-1,88 (м, 1Н) 1,17 (т, 3Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,98 (дд, 1Н) 7,88-8,07 (м, 2Н) 7,70-7,84 (м, 1Н) 7,55-7,68 (м, 2Н) 7,34-7,55 (м, 5Н) 7,20-7,34 (м, 2Н) 6,87-7,06 (м, 3Н) 5,59 (т, 1Н) 5,18-5,31 (м, 1Н) 5,15 (д, 2Н) 4,65 (с, 2Н) 4,01-4,23 (м, 1Н) 3,60-3,84 (м, 4Н) 2,33-2,44 (м, 1Н) 1,58-2,14 (м, 5Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,26 и 9,28 (д, 1Н) 7,89-8,11 (м, 3Н) 7,70-7,89 (м, 2H) 7,37-7,66 (м, 7Н) 7,15-7,21 (м, 1Н) 5,68 и 5,71 (д, 1Н) 5,21-5,37 (м, 1Н) 5,16 и 5,17 (уш.с, 2Н) 4,01-4,27 (м, 1Н) 3,71-3,89 (м, 5Н) 2,17-2,25 и 2,36-2-46 (м, 1H) 1,50-2,15 (м, 4Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д.9,42 и 9,45 (д, 1Н) 8,03-8,21 (м, 2Н) 7,92-8,03 (м, 2Н) 7,70-7,84 (м, 1Н) 7,48-7,68 (м, 4Н) 7,30-7,48 (м, 3Н) 5,79 и 5,82 (д, 1Н) 5,21-5,35 (м, 1Н) 5,15 (с, 2Н) 4,04-4,23 (м, 1Н) 3,38-3,89 (м, 5Н) 2,20-2,27 и 2,34-2,44 (м, 1Н) 1,52-2,14 (м, 4Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,28 и 9,30 (д, 1Н) 7,87-8,13 м, 4Н) 7,69-7,85 (м, 1Н) 7,50-7,69 (м, 4Н) 7,37-7,50 (м, 3Н) 7,24-7,36 (м, 2Н) 5,70 и 5,74 (д, 1Н) 5,16 и 5,18 (с, 2Н) 5,05-5,37 (м, 1Н) 4,00-4,27 (м, 1Н) 3,50-3,85 (м, 5Н) 2,17-2,25 и 2,36-2,47 (м, 1Н) 1,57-2,14 (м, 4Н)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,36 и 9,38 (д, 1Н) 7,90-8,06 (м, 2Н) 7,68-7,88 (м, 3Н) 7,50-7,68 (м, 5Н) 7,34-7,50 (м, 4Н) 5,71 и 5,74 (д, 1Н) 5,21-5,44 (м, 1Н) 5,16 и 5,18 (с, 2Н) 3,99-4,25 (м, 1Н) 3,48-3,87 (м, 5Н) 2,20-2,27 и 2,39-2,46 (м, 1Н) 1,57-2,15 (м, 4Н)

1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,18 (д, 1Н) 7,96-8,01 (м, 2Н) 7,82-7,90 (м, 2Н) 7,72-7,80 (м, 1Н) 7,60-7,67 (м, 2Н) 7,52-7,60 (м, 2Н) 7,34-7,48 (м, 3Н) 7,21-7,32 (м, 2Н) 5,73 (д, 1Н) 5,21-5,32 (м, 1Н) 5,18 (с, 2Н) 4,06-4,27 (м, 1Н) 3,73-3,85 (м, 1Н) 3,51-3,73 (м, 4Н) 2,36 (с, 3Н) 2,16-2,26 (м, 1Н) 1,49-2,11 (м, 4Н)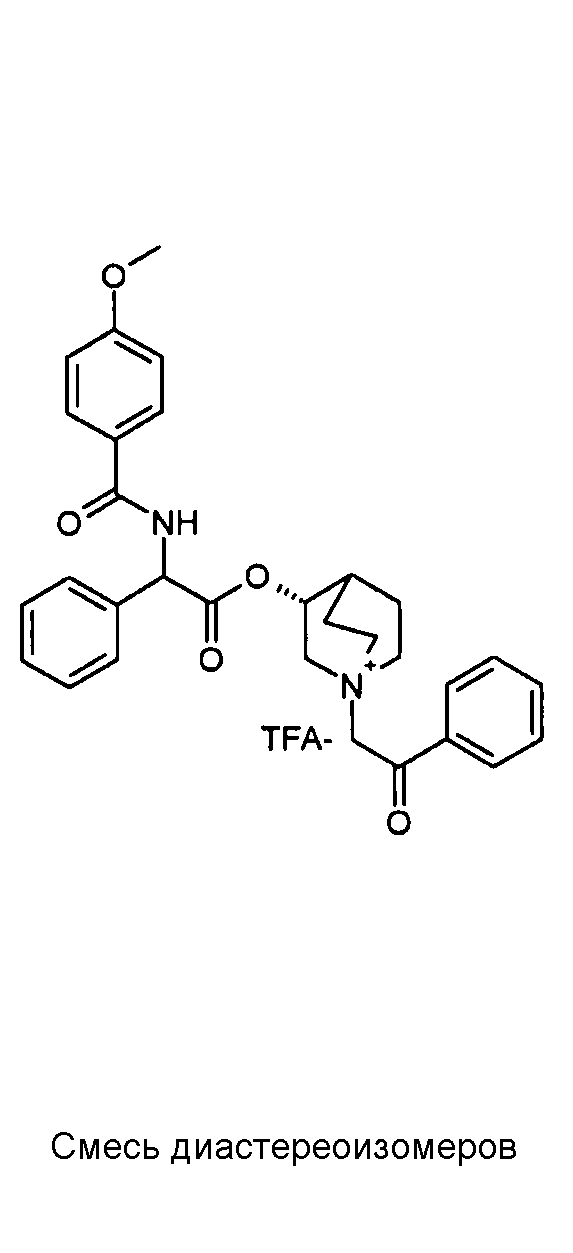
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,08 и 9,10 (д, 1Н) 7,85-8,08 (м, 4Н) 7,69-7,83 (м, 1Н) 7,50-7,68 (м, 4Н) 7,29-7,50 (м, 3Н) 6,88-7,10 (м, 2Н) 5,68 и 5,72 (д, 1Н) 5,16 и 5,17 (с, 2Н) 5,06-5,37 (м, 1Н) 4,05-4,24 (м, 1Н) 3,82 (с, 3Н) 3,45-3,77 (м, 5Н) 2,20-2,26 и 2,37-2,46 (м, 1Н) 1,58-2,14 (м, 4Н)
ПРИМЕР 40
Получение (3R)-3-(2-бензамидо-2-(4-метоксифенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C108)

Схема 41
Получение (R)-хинуклидин-3-ил 2-амино-2-(4-метоксифенил)-ацетата дигидрохлорида (I106)
К раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-метоксифенил)ацетата (C44) (560 мг, 1,43 ммоль) в THF (20 мл) добавляли 37% хлористоводородную кислоту (1,0 мл, 12,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Затем добавляли вторую порцию 37% хлористоводородной кислоты (1,0 мл, 12,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение еще 48 часов. Растворитель выпаривали с получением (R)-хинуклидин-3-ил 2-амино-2-(4-метоксифенил)ацетата дигидрохлорида (521 мг; количественный выход).
Получение (R)-хинуклидин-3-ил 2-бензамидо-2-(4-метоксифенил)ацетата (C107)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-(4-метоксифенил)ацетата дигидрохлорида (I106) (325 мг, 0,89 ммоль) в DCM (10 мл) добавляли триэтиламин (374 мкл, 2,68 ммоль) и бензоилхлорид (125 мкл, 1,07 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем растворитель выпаривали. Остаток обрабатывали с помощью EtOAc и промывали раствором Na2CO3, водой и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали с получением (R)-хинуклидин-3-ил 2-бензамидо-2-(4-метоксифенил)ацетата (145 мг; 41% выход). Продукт использовали на следующей стадии без дополнительной очистки.
Получение (3R)-3-(2-бензамидо-2-(4-метоксифенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C108)
К раствору (R)-хинуклидин-3-ил 2-бензамидо-2-(4-метоксифенил)ацетата (C107) (145 мг, 0,37 ммоль) в EtOAc (2 мл) и ацетонитриле (2 мл) добавляли 2-бром-1-фенилэтанон (80 мг, 0,40 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали и неочищенное вещество очищали препаративной ВЭЖХ с получением (3R)-3-(2-бензамидо-2-(4-метоксифенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (52,4 мг; 23% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,16 (д, 1Н), 7,87-8,07 (м, 4H), 7,69-7,82 (м, 1Н), 7,53-7,69 (м, 3Н), 7,38-7,53 (м, 4Н), 6,89-7,10 (м, 2Н), 5,50-5,69 (м, 1Н), 5,19-5,29 (м, 1Н), 5,16 (с, 2Н), 4,02-4,26 (м, 1Н), 3,78 (с, 3Н), 3,50-3,75 (м, 5Н), 2,36-2,47 (м, 1Н), 1,66-2,23 (м, 4Н);
LC-MS (ESI POS): 513,12 (M+).
ПРИМЕР 41
Получение (3R)-3-(2-бензамидо-2-п-толилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C111)

Схема 42
Получение (R)-хинуклидин-3-ил 2-амино-2-п-толилацетата дигидрохлорида (I109)
К раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-п-толилацетата (C41) (250 мг, 0,67 ммоль) в THF (50 мл) добавляли 37% хлористоводородную кислоту (1,0 мл, 12,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали с получением (R)-хинуклидин-3-ил 2-амино-2-п-толилацетат дигидрохлорида (232 мг; количественный выход).
Получение (R)-хинуклидин-3-ил 2-бензамидо-2-п-толилацетата (C110)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-п-толилацетата дигидрохлорида (I109) (116 мг, 0,33 ммоль) в DCM (5 мл) добавляли триэтиламин (139 мкл, 1,00 ммоль) и бензоилхлорид (46,5 мкл, 0,40 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем растворитель выпаривали. Остаток обрабатывали с помощью EtOAc и промывали раствором Na2CO3, водой и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали с получением (R)-хинуклидин-3-ил 2-бензамидо-2-п-толилацетата (70,0 мг; 55% выход). Продукт использовали на следующей стадии без дополнительной очистки.
Получение (3R)-3-(2-бензамидо-2-п-толилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C111)
2-Бром-1-фенилэтанон (40,5 мг, 0,20 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-бензамидо-2-п-толилацетата (C110) (70,0 мг, 0,18 ммоль) в EtOAc (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем снова добавляли 2-бром-1-фенилэтанон (36,8 мг, 0,18 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Растворитель выпаривали и неочищенное вещество очищали препаративной ВЭЖХ с получением (3R)-3-(2-бензамидо-2-п-толилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октана 2,2,2-трифторацетата (54,8 мг; 48% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,19 и 9,20 (д, 1Н), 7,86-8,05 (м, 4Н), 7,71-7,84 (м, 1Н), 7,52-7,71 (м, 3Н), 7,39-7,52 (м, 4Н), 7,17-7,33 (м, 2Н), 5,65 и 5,69 (д, 1Н), 5,21-5,31 (м, 1Н), 5,16 и 5,18 (с, 2Н), 4,00-4,26 (м, 1Н), 3,38-3,89 (м, 5Н), 2,19-2,27 и 2,38-2,46 (м, 1Н), 2,33 (с, 3Н), 1,48-2,17 (м, 4Н).
LC-MS (ESI POS): 497,12 (M+).
ПРИМЕР 42
Получение (3R)-3-(2-бензамидо-2-(4-хлорфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C114)

Схема 43
Получение (R)-хинуклидин-3-ил 2-амино-2-(4-хлорфенил)ацетата дигидрохлорида (I112)
К раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(4-хлорфенил)ацетата (C47) (250 мг, 0,63 ммоль) в THF (5 мл) добавляли 37% хлористоводородную кислоту (1,0 мл, 12,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали с получением (R)-хинуклидин-3-ил 2-амино-2-(4-хлорфенил)ацетата дигидрохлорида (233 мг; количественный выход).
Получение (R)-хинуклидин-3-ил 2-бензамидо-2-(4-хлорфенил)-ацетата (C113)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-(4-хлорфенил)-ацетата дигидрохлорида (I112) (0,12 г, 0,32 ммоль) в DCM (5 мл) добавляли триэтиламин (0,13 мл, 0,95 ммоль) и бензоилхлорид (44,0 мкл, 0,38 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Затем растворитель выпаривали, и остаток обрабатывали с помощью EtOAc и промывали раствором Na2CO3, водой и солевым раствором. Органический слой сушили над Na2SO4, фильтровали и выпаривали с получением (R)-хинуклидин-3-ил 2-бензамидо-2-(4-хлорфенил)ацетата (0,126 г; количественный выход).
Получение (3R)-3-(2-бензамидо-2-(4-хлорфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C114)
К раствору (R)-хинуклидин-3-ил 2-бензамидо-2-(4-хлорфенил)-ацетата (C113) (126 мг, 0,32 ммоль) с EtOAc (3 мл) добавляли 2-бром-1-фенилэтанон (69,2 мг, 0,35 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем добавляли вторую порцию 2-бром-1-фенилэтанона (62,9 мг, 0,32 ммоль) и реакционную смесь перемешивали в течение еще 6 часов. Растворитель выпаривали и неочищенное вещество очищали препаративной ВЭЖХ с получением (3R)-3-(2-бензамидо-2-(4-хлорфенил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октана 2,2,2-трифторацетата (46,0 мг; 23% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,29 и 9,31 (д, 1Н), 7,87-8,07 (м, 4Н), 7,71-7,82 (м, 1Н), 7,54-7,68 (м, 5Н), 7,44-7,54 (м, 4Н), 5,76 и 5,78 (д, 1Н), 5,21-5,31 (м, 1Н), 5,16 и 5,17 (уш.с, 2Н), 3,97-4,25 (м, 1Н), 3,43-3,93 (м, 5Н), 2,20-2,31 и 2,33-2,46 (м, 1H), 1,58-2,18 (м, 4Н);
LC-MS (ESI POS): 517,12 (M+).
ПРИМЕР 43
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(тиофен-2-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанхлорида (C116)

Схема 44
Получение (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(тиофен-2-ил)ацетата (C115)
Смесь 2-(трет-бутоксикарбониламино)-2-(тиофен-2-ил)уксусной кислоты (710 мг, 2,76 ммоль), (R)-хинуклидин-3-ола (421 мг, 3,31 ммоль), DCC (683 мг, 3,31 ммоль), HOBT (507 мг, 3,31 ммоль) в THF (20 мл) перемешивали при комнатной температуре в течение ночи. Выпаривали THF, неочищенное вещество распределяли между EtOAc и 1M раствором K2CO3. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали досуха. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=98/2) с получением (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(тиофен-2-ил)ацетата (509 мг, 50,3% выход).
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-2-(тиофен-2-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанхлорида (C116)
2-Хлор-l-фенилэтанон (43,0 мг, 0,28 ммоль) добавляли к раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-2-(тиофен-2-ил)ацетата (102 мг, 0,28 ммоль) в EtOAc (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Растворитель выпаривали и неочищенное вещество растирали с Et2O (2 мл) и сушили под вакуумом с получением (3R)-3-(2-(трет-бутоксикарбониламино)-2-(тиофен-2-ил)ацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанхлорида (70 мг, 48,3% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,02-8,07 (м, 1Н), 7,87-8,02 (м, 2Н), 7,68-7,84 (м, 1Н), 7,56-7,68 (м, 2Н), 7,52 (дд, 1Н), 7,12-7,26 (м, 1Н), 7,03 (дд, 1Н), 5,42-5,65 (м, 1Н), 5,17-5,33 (м, 1Н), 5,21 (с, 2Н), 4,07-4,29 (м, 1Н), 3,47-3,85 (м, 5Н), 2,21-2,33 и 2,35-2,45 (м, 1Н), 1,97-2,19 (м, 2Н), 1,66-1,97 (м, 2Н), 1,42 (с, 9Н);
LC-MS (ESI POS): 485,09 (M+).
ПРИМЕР 44
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-3-(1H-индол-3-ил)пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (C118)
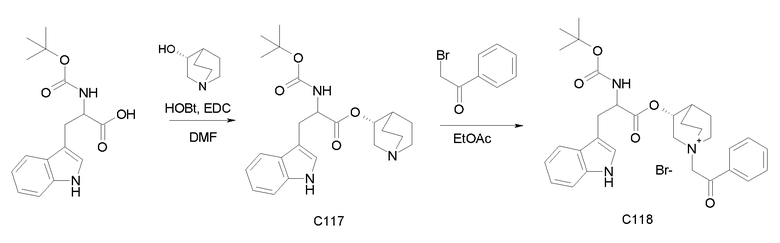
Схема 45
Получение (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-3-(1H-индол-3-ил)пропаноата (C117)
Раствор 2-(трет-бутоксикарбониламино)-3-(1H-индол-3-ил)-пропановой кислоты(100 мг, 0,33 ммоль), (R)-хинуклидин-3-ола (50,1 мг, 0,39 ммоль), HOBT (53,3 мг, 0,39 ммоль) и EDC (76 мг, 0,39 ммоль) в DMF (2 мл) нагревали при 90°C в течение 1,5 часов при микроволновом излучении. Растворитель выпаривали. Остаток обрабатывали с помощью EtOAc и промывали 1 н. раствором Na2CO3, водой и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и выпаривали с получением (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-3-(1H-индол-3-ил)пропаноата (95 мг, 69,9% выход), который использовали на следующей стадии без дополнительной очистки.
Получение (3R)-3-(2-(трет-бутоксикарбониламино)-3-(1H-индол-3-ил)пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (C118)
К раствору (R)-хинуклидин-3-ил 2-(трет-бутоксикарбониламино)-3-(1H-индол-3-ил)пропаноата (95 мг, 0,23 ммоль) в EtOAc (3 мл) добавляли 2-бром-1-фенилэтанон (54,9 мг, 0,28 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Твердое вещество собирали фильтрацией и очищали флэш-хроматографией на силикагеле (DCM/MeOH=94/6) с получением (3R)-3-(2-(трет-бутоксикарбониламино)-3-(1H-индол-3-ил)пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (31,0 мг, 22% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 10,87 (уш.с, 1Н), 7,93-8,05 (м, 2Н), 7,70-7,81 (м, 1Н), 7,58-7,69 (м, 2Н), 7,53 (д, 1Н), 7,28-7,41 (м, 2Н), 7,17-7,25 (м, 1Н), 7,04-7,14 (м, 1Н), 6,96-7,04 (м, 1Н), 5,10 и 5,16 (с, 2Н), 4,98-5,15 (м, 1Н), 4,23-4,39 (м, 1Н), 3,98-4,23 (м, 1Н), 3,46-3,85 (м, 5Н), 3,01-3,24 (м, 2Н), 1,66-2,16 (м, 5Н), 1,26 и 1,36 (с, 9H);
LC-MS (ESI POS): 532,32 (M+).
ПРИМЕР 45
Получение ((R)-3-((S)-3-(4-(бензилокси)фенил)-2-(трет-бутоксикарбониламино)пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (диастереоизомера 1 C120)

Схема 46
Bz обозначает бензильную группу.
Получение (S)-((R)-хинуклидин-3-ил) 3-(4-(бензилокси)фенил)-2-(трет-бутоксикарбониламино)пропаноата (диастереоизомера 1 C119)
Смесь (S)-3-(4-(бензилокси)фенил)-2-(трет-бутоксикарбониламино)пропановой кислоты (500 мг, 1,35 ммоль), DCC (333 мг, 1,61 ммоль) и HOBT (247 мг, 1,61 ммоль) в сухом THF (10 мл) перемешивали при комнатной температуре в атмосфере азота в течение 1 часа. Добавляли (R)-хинуклидин-3-ол (205 мг, 1,61 ммоль) и реакционную смесь перемешивали в течение 16 часов. Растворитель выпаривали и остаток распределяли между насыщенным раствором NaHCO3 и EtOAc. Органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали и выпаривали досуха. Остаток растирали с Et2O (30 мл) и нерастворимый материал отфильтровывали. Раствор выпаривали с получением (S)-((R)-хинуклидин-3-ил) 3-(4-(бензилокси)фенил)-2-(трет-бутоксикарбониламино)пропаноата (590 мг, 91% выход).
Получение (R)-3-((S)-3-(4-(бензилокси)фенил)-2-(трет-бутоксикарбониламино)пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (диастереоизомера 1 C120)
К раствору (S)-((R)-хинуклидин-3-ил) 3-(4-(бензилокси)-фенил)-2-(трет-бутоксикарбониламино)пропаноата (590 мг, 1,23 ммоль) в EtOAc (15 мл) добавляли порциями 2-бром-1-фенилэтанон (232 мг, 1,17 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали и остаток растирали с Et2O, фильтровали и сушили под вакуумом с получением (R)-3-((S)-3-(4-(бензилокси)фенил)-2-(трет-бутоксикарбониламино)пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (720 мг, 86% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,88-8,11 (м, 2Н), 7,70-7,84 (м, 1Н), 7,56-7,69 (м, 2Н), 7,26-7,50 (м, 6Н), 7,18 (м, 2Н), 6,94 (м, 2Н), 5,19 (с, 2Н), 5,15-5,26 (м, 1Н), 5,07 (с, 2Н), 4,06-4,32 (м, 2Н), 3,50-3,83 (м, 5Н), 2,79-3,09 (м, 2Н), 2,19 (уш.с, 1Н), 1,96-2,13 (м, 2Н), 1,74-1,96 (м, 2Н), 1,35 (с, 9Н);
LC-MS (ESI POS): 599,37 (M+);
[α]D= -22,52 (c=0,5, MeOH).
ПРИМЕР 46
Получение (R)-3-((S)-2-(трет-бутоксикарбониламино)-3-(4-гидроксифенил)пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (диастереоизомера 1 C122)

Схема 47
Получение (S)-((R)-хинуклидин-3-ил) 2-(трет-бутоксикарбониламино)-3-(4-гидроксифенил)пропаноата (диастереоизомера 1 C121)
Смесь (R)-3-((S)-3-(4-(бензилокси)фенил)-2-(трет-бутоксикарбониламино)пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (720 мг, 1,06 ммоль) и 10% Pd/C (100 мг, 0,09 ммоль) в MeOH (30 мл) гидрировали при 25 фунт/кв.дюйм в течение 2 часов. Катализатор отфильтровывали и растворитель выпаривали досуха. Остаток растворяли в DCM и фильтровали через картридж с сильной катионообменной смолой. Раствор выпаривали с получением (S)-((R)-хинуклидин-3-ил) 2-(трет-бутоксикарбониламино)-3-(4-гидроксифенил)пропаноата (260 мг, 62,9% выход).
Получение (R)-3-((S)-2-(трет-бутоксикарбониламино)-3-(4-гидроксифенил)пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанбромида (диастереоизомера 1 C122)
К раствору (S)-((R)-хинуклидин-3-ил) 2-(трет-бутоксикарбониламино)-3-(4-гидроксифенил)пропаноата (260 мг, 0,67 ммоль) в сухом ацетонитриле (10 мл) добавляли порциями 2-бром-1-фенилэтанон (126 мг, 0,63 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель выпаривали, и остаток очищали флэш-хроматографией (DCM/MeOH=85/15), и извлекаемый продукт растирали с i-PrOH с получением (R)-3-((S)-2-(трет-бутоксикарбониламино)-3-(4-гидроксифенил)-пропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (74 мг, 18,8% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 9,22 (с, 1Н), 7,85-8,05 (м, 2Н), 7,76 (т, 1Н), 7,62 (т, 2Н), 7,33 (д, 1Н), 7,05 (м, 2Н), 6,69 (м, 2Н), 5,20 (с, 2Н), 4,31 (д, 1Н), 4,01-4,25 (м, 2Н), 3,47-3,78 (м, 5Н), 2,75-3,02 (м, 2Н), 2,20 (уш.с, 1Н), 1,76-2,13 (м, 4Н), 1,36 (с, 9Н);
LC-MS (ESI POS): 509,33 (M+);
[α]D=-15,48 (c=0,5, MeOH).
ПРИМЕР 47
Получение (R)-3-((S)-2-(трет-бутоксикарбониламино)-3-фенилпропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октанбромида (диастереоизомера 1 C124)

Схема 48
Получение (S)-((R)-хинуклидин-3-ил) 2-(трет-бутоксикарбониламино)-3-фенилпропаноата (диастереоизомера 1 C123)
Смесь (S)-2-(трет-бутоксикарбониламино)-3-фенилпропановой кислоты (500 мг, 1,88 ммоль), DCC (467 мг, 2,26 ммоль) и HOBT (346 мг, 2,26 ммоль) в сухом THF (15 мл) перемешивали в течение 30 минут при комнатной температуре. Затем добавляли порциями (R)-хинуклидин-3-ол (288 мг, 2,26 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали, остаток растворяли в EtOAc и нерастворимое вещество отфильтровывали. Органическую фазу промывали насыщенным раствором NaHCO3 и солевым раствором, сушили над Na2SO4, фильтровали и выпаривали с получением (S)-((R)-хинуклидин-3-ил) 2-(трет-бутоксикарбониламино)-3-фенилпропаноата (600 мг, 85% выход).
Получение (R)-3-((S)-2-(трет-бутоксикарбониламино)-3-фенилпропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (диастереоизомера 1 C124)
К раствору (S)-((R)-хинуклидин-3-ил) 2-(трет-бутоксикарбониламино)-3-фенилпропаноата (600 мг, 1,60 ммоль) в EtOAc (10 мл) добавляли порциями 2-бром-1-фенилэтанон (303 мг, 1,52 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение трех дней. Осадок собирали фильтрацией и сушили под вакуумом с получением (R)-3-((S)-2-(трет-бутоксикарбониламино)-3-фенилпропаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло-[2.2.2]октанбромида (740 мг, 81% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 7,94-8,05 (м, 2Н), 7,69-7,81 (м, 1Н), 7,54-7,69 (м, 2Н), 7,41 (м, 1Н), 7,17-7,36 (м, 5Н), 5,20 (с, 2Н), 5,07-5,31 (м, 1Н), 4,21-4,38 (м, 1Н), 4,15 (дд, 1Н), 3,48-3,86 (м, 5Н), 2,77-3,13 (м, 2Н), 2,15-2,25 (м, 1Н), 1,99-2,15 (м, 2Н), 1,75-1,99 (м, 2Н), 1,35 (с, 9H);
LC-MS (ESI POS): 493,45 (M+);
[α]D= -19,72 (c=0,5, MeOH).
ПРИМЕР 48
Получение (3R)-3-(2-(2-амино-2-фенилацетамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C127)

Схема 49
Получение (R)-хинуклидин-3-ил 2-(2-(трет-бутоксикарбониламино)-2-фенилацетамидо)-2-фенилацетата (C125)
К суспензии (R)-хинуклидин-3-ил 2-амино-2-фенилацетата дигидрохлорида (500 мг, 1,50 ммоль) в THF (15 мл) и DCM (5 мл) добавляли триэтиламин (591 мкл, 4,25 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Затем добавляли DCC (309 мг, 1,50 ммоль), HOBT (203 мг, 1,50 ммоль) и 2-(трет-бутоксикарбониламино)-2-фенилуксусную кислоту (314 мг, 1,25 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали, остаток обрабатывали с помощью DCM и нерастворимое вещество отфильтровывали. Органический раствор промывали дважды 1 н. раствором Na2CO3 и солевым раствором, сушили над Na2SO4 и выпаривали. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH=95/5) с получением (R)-хинуклидин-3-ил 2-(2-(трет-бутоксикарбониламино)-2-фенилацетамидо)-2-фенилацетата (210 мг, 34% выход).
Получение (3R)-3-(2-(2-(трет-бутоксикарбониламино)-2-фенилацетамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанхлорида (C126)
К раствору (R)-хинуклидин-3-ил 2-(2-(трет-бутоксикарбониламино)-2-фенилацетамидо)-2-фенилацетата (210 мг, 0,42 ммоль) в EtOAc (3 мл) и ацетонитриле (3 мл) добавляли 2-хлор-1-фенилэтанон (65,8 мг, 0,42 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Добавляли еще 2-хлор-1-фенилэтанон (19,7 мг, 0,13 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель выпаривали и неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 94/6 до 93/7) с получением (3R)-3-(2-(2-(трет-бутоксикарбониламино)-2-фенилацетамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанхлорида (65,3 мг, 23,7% выход).
Получение (3R)-3-(2-(2-амино-2-фенилацетамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]-октана 2,2,2-трифторацетата 2,2,2-трифторуксусной кислоты (C127)
К охлажденному до 0°C раствору (3R)-3-(2-(2-(трет-бутоксикарбониламино)-2-фенилацетамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октанхлорида (50 мг, 0,08 ммоль) в диоксане (4 мл) добавляли хлористый водород (4M раствор в диоксане, 23 мкл) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Добавляли еще хлористого водорода (4M раствор в диоксане, 193 мкл) и реакционную смесь перемешивали при комнатной температуре в течение 7 часов. Затем добавляли 37% хлористоводородную кислоту (0,5 мл, 6,09 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Растворитель выпаривали и полученное неочищенное вещество очищали препаративной ВЭЖХ с получением (3R)-3-(2-(2-амино-2-фенилацетамидо)-2-фенилацетокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата 2,2,2-трифторуксусной кислоты (14,8 мг, 25,9% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,95-9,30 (м, 1Н), 7,86-8,11 (м, 2Н), 7,69-7,86 (м, 1Н), 7,52-7,69 (м, 2Н), 7,22-7,50 (м, 11Н), 5,30-5,57 (м, 2Н), 5,00-5,30 (м, 3Н), 4,03-4,17 (м, 1Н), 3,43-3,78 (м, 5Н), 2,14-2,22 и 2,32-2,45 (м, 1Н), 1,88-2,13 (м, 2Н), 1,71-1,88 (м, 1Н), 1,50-1,71 (м, 1Н), 1,36 и 1,38 (с, 9Н);
LC-MS (ESI POS): 612,53 (M+).
ПРИМЕР 49
Получение (3R)-3-(2-бензамидо-3-метилбутаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C130)

Схема 50
Получение 2-бензамидо-3-метилбутановой кислоты (C128)
Раствор 2-амино-3-метилбутановой кислоты (500 мг, 4,27 ммоль) в 2 н. растворе NaOH (2,35 мл, 4,69 ммоль) перемешивали при комнатной температуре в течение 30 минут, затем реакционную смесь охлаждали до 0°C и последовательно добавляли по каплям из двух разных шприцев бензоилхлорид (471 мкл, 4,05 ммоль) и 2н раствор NaOH (2,35 мл, 4,69 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли воду, водную фазу промывали с помощью Et2O, затем подкисляли с помощью 1M HCl и опять экстрагировали с помощью Et2O, сушили над Na2SO4 и выпаривали. Остаток растирали с i-Pr2O с получением 2-бензамидо-3-метилбутановой кислоты (780 мг, 83% выход).
Получение (R)-хинуклидин-3-ил 2-бензамидо-3-метилбутаноата (C129)
К раствору 2-бензамидо-3-метилбутановой кислоты (780 мг, 3,53 ммоль) в THF (25 мл) добавляли (R)-хинуклидин-3-ол (538 мг, 4,23 ммоль), DCC (873 мг, 4,23 ммоль) и HOBt (572 мг, 4,23 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали. Добавляли EtOAc и нерастворимое вещество отфильтровывали. Органический раствор промывали дважды 1н раствором Na2CO3 и солевым раствором, сушили над Na2SO4 и выпаривали досуха. Неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 99/1 до 8/2) с получением (R)-хинуклидин-3-ил 2-бензамидо-3-метилбутаноата (436 мг, 37,4% выход).
Получение (3R)-3-(2-бензамидо-3-метилбутаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C130)
К раствору (R)-хинуклидин-3-ил 2-бензамидо-3-метилбутаноата (218 мг, 0,66 ммоль) в EtOAc (2 мл) и ацетонитриле (5 мл) добавляли 2-бром-1-фенилэтанон (158 мг, 0,79 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали и остаток растирали с ацетонитрилом. Неочищенное вещество очищали препаративной ВЭЖХ с получением (3R)-3-(2-бензамидо-3-метилбутаноилокси)-1-(2-оксо-2-фенилэтил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (15,5 мг, 4,2% выход)
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,64 и 8,66 (д, 1Н), 7,94-8,05 (м, 2Н), 7,83-7,94 (м, 2Н), 7,69-7,81 (м, 1Н), 7,41-7,66 (м, 5Н), 5,22-5,32 (м, 1Н), 5,18 и 5,20 (с, 2Н), 4,26-4,45 (м, 1Н), 4,07-4,24 (м, 1Н) 3,53-3,85 (м, 5Н), 1,79-2,44 (м, 6Н), 1,06 (д, 3Н), 1,04 (д, 3Н);
LC-MS (ESI POS): 449,24 (M+).
ПРИМЕР 50
Получение (3R)-3-(2-бензамидо-3-метилбутаноилокси)-1-(2-оксо-2-(тиофен-2-ил)этил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (C131)
Схема 51
К раствору (R)-хинуклидин-3-ил 2-бензамидо-3-метилбутаноата (218 мг, 0,66 ммоль) в EtOAc (2 мл) и CH3CN (5 мл) добавляли 2-бром-1-(тиофен-2-ил)этанон (162 мг, 0,79 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель выпаривали и неочищенное вещество очищали флэш-хроматографией (DCM/MeOH = от 98/2 до 85/15).
Продукт растирали с i-Pr2O и затем очищали препаративной ВЭЖХ с получением (3R)-3-(2-бензамидо-3-метилбутаноилокси)-1-(2-оксо-2-(тиофен-2-ил)-этил)-1-азонийбицикло[2.2.2]октана 2,2,2-трифторацетата (30,6 мг, 8,2% выход).
1H ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,66 (д, 1Н), 8,21 (дд, 1Н), 8,07 (дд, 1Н), 7,82-7,94 (м, 2Н), 7,53-7,63 (м, 1Н), 7,43-7,53 (м, 2Н), 7,31-7,39 (м, 1Н), 5,18-5,31 (м, 1Н), 4,93-5,17 (м, 2Н), 4,32 и 4,39 (т, 1Н), 4,08-4,23 (м, 1Н), 3,58-3,86 (м, 5Н), 1,72-2,43 (м, 6Н), 0,93-1,16 (м, 6Н);
LC-MS (ESI POS): 455,26 (M+).
Исследование биологической активности
ПРИМЕР 51
Примеры исследования радиолигандного связывания клонированных мускариновых рецепторов человека и бета адренергических рецепторов человека
Клонированные клетки CHO-K1, экспрессирующие M1-, M2-, M3-рецепторы человека (Euroscreen, Swissprot P11229, P08172, P20309, Genbank: J02960, соответственно), собирали в несодержащий Ca++/Mg++ забуференный фосфатом физиологический раствор и центрифугировали при 1500 об/мин в течение 10 минут при 4°C. Осадки ресуспендировали в очень холодном буфере A (15 мМ Tris-HCl pH 7,4, 2 мМ MgCl2, 0,3 мМ EDTA, 1 мМ EGTA). Клонированные клетки, экспрессирующие M1-, M2- и M3-рецепторы, гомогенизировали с помощью лабораторного гомогенизатора PBI Polytron (режим 5 в течение 15 секунд). Неочищенную фракцию мембран собирали путем проведения двух последовательных стадий центрифугирования при 40000 g в течение 20 минут при 4°C, между которыми осуществляли стадию промывки в буфере A.
Осадки, полученные от клеток трех линий, окончательно ресуспендировали в буфере C (75 мМ Tris HCl pH 7,4, 12,5 мМ MgCl2, 0,3 мМ EDTA, 1 мМ EGTA, 250 мМ сахароза), и аликвоты хранили при -80°C.
В день проведения эксперимента замороженные мембраны M1-, M2- и M3-рецептора ресуспендировали в буфере D (50 мМ Tris-HCl pH 7,4, 2,5 мМ MgCl2, 1 мМ EDTA). Неселективный мускариновый радиолиганд [3H]-N-метилскополамин (Mol. Pharmacol. 45:899-907) использовали для метки связывающих сайтов M1, M2 и M3. Каждый из экспериментов по связыванию проводили два раза (концентрационные кривые по десяти точкам) в 96-луночных планшетах при концентрации радиолиганда 0,1-0,3 нМ. Неспецифическое связывание определяли в присутствии 10 мкΜ холодного N-метилскополамина. Образцы (конечный объем 0,75 мл) инкубировали при комнатной температуре в течение 120 минут в случае анализа связывания M1, в течение 60 минут в случае анализа связывания M2 и в течение 90 минут в случае анализа связывания M3.
Реакцию останавливали путем быстрой фильтрации через фильтрующие планшеты GF/B Unifilter и двух промывок (0,75 мл) холодным буфером, используя коллектор клеток Packard Filtermate Harvester. Радиоактивность на фильтрах измеряли микропланшетным сцинтилляционным счетчиком TopCount NXT (фирмы Canberra Packard).
В настоящих исследованиях значения Ki испытуемых соединений определяли из наблюдаемых значений IC50 известными методами. Более низкое значение Ki указывает, что испытуемое соединение имеет более высокое сродство к связыванию рецептора.
Отношение Ki M2/Ki M3 для типичных соединений общей формулы (I) на схеме 1, составляет от 30 до 157.
Взаимодействие с M3 мускариновыми рецепторами может быть оценено по результатам in vitro исследований, при которых оценивают активность испытуемых соединений и момент окончания продуцируемого ими ингибирующего действия после отмывки от антагонистов выделенной трахеи морской свинки, и по продолжительности действия in vivo в отношении бронхоспазма у морской свинки, вызванного ацетилхолином.
ПРИМЕР 52
Стабильность в плазме
Для иллюстрации распада соединений, исследовали стабильность соединения изобретения в человеческой плазме через 1 час и через 5 часов. Вкратце, 10 мкл исходного раствора 250 мкΜ соединения в ацетонитриле добавляли к 1 мл человеческой плазмы, и образцы инкубировали при 37°C. Плазму (50 мкл) отбирали через 1 и 5 часов инкубирования и добавляли к 140 мкл ацетонитрила, добавляя верапамил в качестве внутреннего стандарта (250 нг/мл). Образцы анализировали методом тандемной хроматомасс-спектрометрии (HPLC-MS/MS).
Стабильность в плазме рассчитывали как процент оставшегося соединения через 1 час и 5 часов путем деления площади пика, полученной через 1 или 5 часов, на площадь пика в момент времени 0.
Через 5 часов инкубирования значения стабильности типичных соединений общей формулы (I) в схеме 1 в плазме составляли от 0,6 до 13,1%, указывая на то, что соединения изобретения являются очень нестабильными в плазме человека.
Изобретение относится к соединениям общей формулы (I) или их фармацевтически приемлемым солям, обладающим действием антагониста мускариновых рецепторов. В формуле (I) R1 выбирают из группы, состоящей из линейного или разветвленного (C1-С10)алкила, (С2-С6)алкенила, арила, (С3-С8)циклоалкила, арил(C1-C6)алкила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -N(R5)(R6′), -NHCO(R5), -CO2R5, (С1-C10)алкила, (C1-С10)алкокси и арилокси; G выбирают из группы, состоящей из -ОС(О)-, -SO2- и -С(O)-; R2 представляет собой -Н; R3 выбирают из группы, состоящей из (C1-С10)алкила, арила, гетероарила и гетероарил(С1-С6)алкила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -ОН, (C1-С10)алкила, (C1-С10)алкокси, арилокси и арил(C1-С10)алкиленокси; R6 выбирают из группы, состоящей из фрагментов формулы (i), (ii) и (iii), где m=1, 2 или 3; n=1, 2 или 3; А- представляет собой физиологически приемлемый анион; R4 представляет собой группу формулы (Y), где p представляет собой 0 или целое число от 1 до 4; q представляет собой 0 или целое число от 1 до 4; P отсутствует или его выбирают из группы, состоящей из -О- и -С(О)-; W выбирают из группы, состоящей из Н, линейного или разветвленного (C1-С10)алкила, арила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -ОН, -NO2, -CO2(R5), (C1-С10)алкила, (C1-С10)алкокси и арила; R5 и R6′ независимо выбирают из группы, состоящей из -Н, (C1-С10)алкила и (С1-С10)алкокси. Изобретение относится также к соединениям общей формулы (VI), фармацевтической композиции, устройству, включающему эту композицию, и применению соединений формулы (I) или (VI) для изготовления лекарственного средства для предотвращения и/или лечения бронхообструктивных или воспалительных заболеваний. 5 н. и 8 з.п. ф-лы, 6 табл., 52 пр.
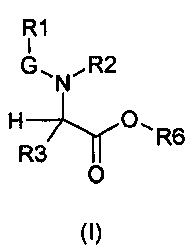
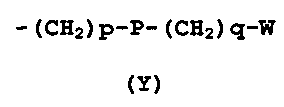
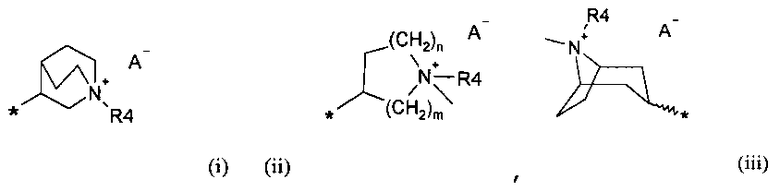
1. Соединение общей формулы (I)
где
R1 выбирают из группы, состоящей из линейного или разветвленного (C1-С10)алкила, (С2-С6)алкенила, арила, (С3-С8)циклоалкила, арил(C1-C6)алкила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -N(R5)(R6′), -NHCO(R5), -CO2R5, (С1-C10)алкила, (C1-С10)алкокси и арилокси;
G выбирают из группы, состоящей из -ОС(О)-, -SO2- и -С(O)-;
R2 представляет собой -Н;
R3 выбирают из группы, состоящей из (C1-С10)алкила, арила, гетероарила и гетероарил(С1-С6)алкила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -ОН, (C1-С10)алкила, (C1-С10)алкокси, арилокси и арил(C1-С10)алкиленокси;
R6 выбирают из группы, состоящей из фрагментов формулы (i), (ii) и (iii)
,
где
m=1, 2 или 3;
n=1, 2 или 3;
А- представляет собой физиологически приемлемый анион;
R4 представляет собой группу формулы (Y)
,
где
p представляет собой 0 или целое число от 1 до 4;
q представляет собой 0 или целое число от 1 до 4;
P отсутствует или его выбирают из группы, состоящей из -О- и -С(О)-;
W выбирают из группы, состоящей из Н, линейного или разветвленного (C1-С10)алкила, арила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, -ОН, -NO2, -CO2(R5), (C1-С10)алкила, (C1-С10)алкокси и арила;
R5 и R6′ независимо выбирают из группы, состоящей из -Н, (C1-С10)алкила и (С1-С10)алкокси;
и его фармацевтически приемлемые соли.
2. Соединение по п.1, где G выбирают из группы, состоящей из -ОС(О)-, -SO2- и -С(O)-, R1 выбирают из группы, состоящей из линейного или разветвленного (C1-С10)алкила, арила, (С3-С8)циклоалкила, (С2-С6)алкенила и гетероарила, необязательно замещенного одним или более заместителями, выбранными из атомов галогена, -N(R5)(R6′), (C1-С10)алкила, (C1-С10)алкокси и арилокси.
3. Соединение по п.1, где R1 выбирают из группы, состоящей из метила, этила, трет-бутила, этенила, циклогексила, фенила, метоксифенила, хлорфенила, дифторфенила, диметилтиазола, трифторэтила, циклопентила, тиофенила, тиазолила, фторфенила, аминофенила, трет-бутоксикарбониламинофенила и метилфенила.
4. Соединение по п.1, где R3 выбирают из группы, состоящей из (C1-С10)алкила, арила и гетероарила, необязательно замещенного одной или более группами, выбранными из атомов галогена, (C1-С10)алкила, (С1-С10)алкокси и арил(C1-С10)алкиленокси.
5. Соединение по п.1, где R6 выбирают из группы, состоящей из фрагментов формулы (i) и (ii), R4 представляет собой группу формулы (Y), где p равняется 0, 1 и 3, P представляет собой СО, q равняется 0, W выбирают из группы, состоящей из (C1-С10)алкила, арила, гетероарила, необязательно замещенного одним или более заместителями, выбранными из группы, состоящей из атомов галогена, (C1-С10)алкила, (С1-С10)алкокси и ОН.
6. Соединение по п.5, где W выбирают из группы, состоящей из фенила, бензотиоксола, тиофенила и тиазолила, необязательно замещенного одним или более атомами галогена, ОН и метилом.
7. Соединение общей формулы (VI)
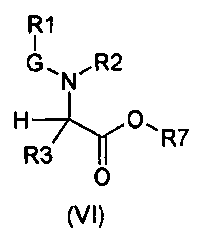 ,
,
где
R1, R2, R3 и G определены в п.1;
R7 выбирают из группы, состоящей из фрагментов формулы (v), (vi) и (vii)
 ,
,
где
m и n определены в п.1.
8. Фармацевтическая композиция, обладающая действием антагониста мускариновых рецепторов, содержащая соединение формулы (I) или (VI), определенное в любом одном из пп.1-7, в эффективном количестве с одним или более фармацевтически приемлемыми носителями и/или эксципиентами.
9. Соединение формулы (I) или (VI) по любому одному из пп.1-7, действующее в качестве антагониста мускариновых рецепторов, для применения в качестве лекарственного средства при лечении бронхообструктивных или воспалительных заболеваний.
10. Соединение формулы (I) или (VI) по любому одному из пп.1-7, действующее в качестве антагониста мускариновых рецепторов, для применения при лечении бронхообструктивных или воспалительных заболеваний, предпочтительно астмы или хронического бронхита, или хронического обструктивного заболевания легких (COPD).
11. Применение соединения формулы (I) или (VI) по любому одному из пп.1-7 для изготовления лекарственного средства для предотвращения и/или лечения бронхообструктивных или воспалительных заболеваний, предпочтительно астмы или хронического бронхита, или хронического обструктивного заболевания легких (COPD), опосредованных действием мускариновых рецепторов.
12. Фармацевтическая композиция по п.8, вводимая путем ингаляции, такая как порошки для ингаляции, содержащие пропеллент дозируемые аэрозоли или не содержащие пропеллент составы для ингаляции.
13. Устройство, включающее фармацевтическую композицию по п.12, представляющее собой однодозовый или многодозовый ингалятор сухого порошка, ингалятор отмеренных доз и небулайзер мягких аэрозолей.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| КАРБАМАТЫ ХИНУКЛИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2296762C2 |

