Изобретение относится к хроматографическому определению различных химических соединений и может быть использовано в медицине, биологии, экологии и допинговом контроле.
Известен способ анализа жидких препаратов на основе растительного сырья методом хроматографии, который включает извлечение летучих веществ отгонкой исходной пробы паром, концентрирование летучих веществ экстракцией низкокипящим растворителем с последующей отгонкой растворителя и наконец собственно определение компонентов методами газовой или жидкостной хроматографии [1].
Недостатком указанного технического решения является невысокая информативность получаемых данных, в частности хроматографических спектров, что препятствует однозначной идентификации химических соединений и их фрагментов в произвольных комбинациях.
Известен также способ хроматографической идентификации компонентов сложных смесей органических соединений, включающий пропускание вещества через систему последовательно соединенных колонок, заполненных сорбентом различной полярности, отбор пробы после каждой колонки, детектирование на детекторах различных типов и идентификацию определяемого вещества расчетом коэффициента чувствительности и относительного удерживания по данным двух детекторов [2].
К недостаткам указанного способа следует отнести сложность процедуры анализа, а также вероятность получения недостоверных результатов при расчетах значений удерживания разных колонок.
Известен также способ идентификации неизвестных веществ методом газовой хроматографии в сочетании с масс-спектрометрией. По указанному способу регистрируют хроматограмму как функцию времени удерживания и регистрируют масс-спектр в период времени, соответствующий выходу вещества из колонки, и сравнивают с масс-спектрами известных веществ, находящимися в базе данных, далее определяют индекс удерживания и сравнивают его с таковыми из базы данных и идентифицируют вещество по двум параметрам - масс-спектру и индексу удерживания [3].
Недостатком указанного способа, несмотря на привлекательность использования индекса удерживания, является высокая вероятность получения недостоверных результатов анализа, поскольку индексы удерживания изначально привязаны к конкретной колонке с определенными параметрами и зачастую либо не воспроизводятся, либо на другом, хотя и аналогичном оборудовании, воспроизводятся с искажением, что может привести к недостоверной интерпретации результатов анализа.
Известен также способ идентификации неизвестных веществ методом жидкостной хроматографии в сочетании с тандемной масс-спектрометрией. По указанному способу также регистрируют хроматограмму как функцию времени удерживания и регистрируют масс-спектр в период времени, соответствующий выходу вещества из колонки, и сравнивают с масс-спектрами известных веществ, находящимися в базе данных, далее определяют индекс удерживания и сравнивают его с таковыми из базы данных и идентифицируют вещество по двум параметрам - масс-спектру и индексу удерживания [4].
Недостатком указанного способа являются высокие значения граничных концентраций определяемых веществ и существенная вероятность недостоверности определения близких по строению веществ из-за идентичной фрагментации в ходе столкновительной диссоциации.
В то же время используемое в прототипе аппаратурное оформление процесса тем не менее не позволяет достичь поставленного технического результата. Авторы полагают наиболее близкими к заявляемому устройству по техническим характеристикам и достигаемому техническому результату устройства, описанные в [5 и 6].
Указанное устройство, например [6], включает смонтированный в термостате узел ввода пробы, инжектор, хроматографическую колонку (ХГК), ионно-орбитальную ловушку с детектором, соединенные с общим для системы блоком автоматического управления ходом анализа.
В указанном устройстве пробу исследуемого образца с подвижной жидкой фазой разделяют на компоненты пропусканием через хроматографическую колонку, ионизируют разделенные компоненты и детектируют ионы сепарацией через орбитально-ионную ловушку (ОИЛ) с последующим определением принадлежности каждого иона конкретному веществу на основании его аналитических характеристик.
Техническим результатом, на достижение которого направлено создание данного изобретения, является обеспечение возможности однозначной идентификации химических соединений в произвольных комбинациях за счет повышения точности определения характеристик исследуемых веществ при одновременном повышении порога чувствительности и оперативности определения.
Поставленный технический результат достигается тем, что исследуемые вещества пропускают через монолитную хроматографическую колонку и при этом скорость потока поддерживают в пределах по меньшей мере 500-1100 мкл/мин, ионизацию компонентов проводят в атмосфере азота в присутствии носителя метанол/вода, причем соотношение компонентов носителя в ходе анализа изменяют линейно от 20/80 до 90/10 в течение 5 минут, и из ионно-орбитальной ловушки на детектирование последовательно отбирают и подают ионизированные компоненты с разностью масс по меньшей мере 10-4 а.е.м.
Авторы предлагают неожиданное решение - заменить в известном устройстве обычную ХГК на монолитную. Последняя позволяет в известной степени снять ограничения по скорости подачи подвижной фазы (в нашем случае это смесь метанол/вода) в ХГК со всеми вытекающими из этого преимуществами:
- увеличить скорость подачи исследуемого вещества в ХГК до необходимых пределов,
- соответственно за единицу времени увеличить выход протонов из подвижной фазы, что позволяет ионизировать пропорционально большее количестве исследуемого вещества,
- существенно сократить продолжительность проведения анализа
- возможность эффективного использования химической ионизации компонентов исследуемого вещества.
Известно, что как атомная масса, так и молекулярная масса веществ выражается не целыми, а дробными числами и, зная по химической формуле органического соединения точную молекулярную массу его, может быть вычислен и элементный состав (программа Molecular Fragment Calculator). С другой стороны, если известна измеренная масса протонированного иона (m/z), то по этой массе можно установить брутто-формулу вещества. При этом следует подчеркнуть, что при определении массы с точностью до третьего знака (например 345,253) брутто-формула дает список из трех веществ, одно из которых соответствует станозололу, а при определении массы с точностью до четвертого знака (345,2533) брутто-формула дает только одно вещество - станозолол. Из вышеизложенного вытекает, что, чем точнее в процессе проведения анализа определена масса протонированного иона, тем выше точность определения искомого вещества.
Следует особо отметить, что с учетом вышеизложенного, заявляемый способ определения ксенобиотиков может реализоваться как с привлечением эталонных образцов исследуемых веществ, так и без него.
Что касается технологических параметров осуществления заявляемого технического решения, то следует отметить, что в тандемной масс-спектрометрии (ВЭЖХ-МС/МС) исследуемую пробу подвергают электрораспылительной ионизации в режиме регистрации положительных (или отрицательных) ионов. Характерной особенностью такого процесса ионизации является парадоксальное явление - чем меньше скорость потока элюэнта, тем продуктивнее процесс ионизации. В то же время не следует упускать из виду то обстоятельство, что ВЭЖХ-МС/МС сочетает два процесса: хроматографию и масс-спектрометрию. А эти процессы зачастую противоречивы по параметрам, поскольку низкая скорость потока элюента, хотя и способствует более эффективной ионизации, отрицательно сказывается на селективности.
В то же время, как указывалось выше, использование монолитной ХГК позволяет (за счет повышения скорости подвижной фазы) за единицу времени увеличить выход протонов из подвижной фазы, что позволяет ионизировать пропорционально большее количестве исследуемого вещества, а это приводит к более эффективному использованию химической ионизации компонентов исследуемого вещества и, соответственно, к повышению чувствительности системы. Однако и в этом случае повышение скорости подвижной фазы выше определенного уровня снижает эффективность работы монолитной ХГК, и соответственно, снижается чувствительность системы. Поэтому в заявляемом способе определения ксенобиотиков параметры проведения процесса ограничены такими пределами, которые оптимально совмещают осуществление хроматографического разделения исследуемой пробы и спектрального анализа ее.
Выход за пределы заявленных параметров проведения процесса приводит к ухудшению селективности (подача в детектор ионизированных компонентов с разностью масс 10-3), снижению чувствительности (неизменный состав подвижной фазы, снижение скорости подвижной фазы или повышение ее за заявленные пределы), резкому снижению предела чувствительности (с 3 пг/мл до 30-80 пг/мл при замене монолитной ХГК на обычную) и т.д.
Таким образом, с учетом вышеизложенного, устройство для осуществления заявляемого способа включает смонтированный в термостате узел ввода пробы, инжектор, ХГК, ионно-орбитальную ловушку (ОИЛ) с детектором, соединенные с общим для системы блоком автоматического управления ходом анализа, и дополнительно включает блок химической ионизации со встроенным генератором азота, размещенный между выходом анализируемого вещества из хроматографической колонки и входом в орбитально-ионную ловушку, и в качестве хроматографической колонки устройство включает монолитную хроматографическую колонку.
На Фиг.1 представлена блок-схема заявляемого устройства.
Устройство включает термостат 1, содержащий узел ввода пробы 2, инжектор 3, соединенный с монолитной ХГК 4, выходом 5 соединенной с блоком химической ионизации 6 с присоединенным к нему генератором азота 7. Блок химической ионизации 6 последовательно скоммутирован с ОИЛ 8 и детектором 09. Вся система объединена с блоком управления ходом анализа 10 соответствующими коммуникациями 11.
Работает устройство следующим образом.
Устанавливают рабочую температуру термостата 1 и через блок автоматического управления ходом анализа 10 задают необходимые параметры хромато-масс-спектрометрического определения анализируемой пробы. Анализируемую пробу вводят в узел ввода пробы 2, далее через инжектор 3 проба поступает в монолитную ХГК 4, где происходит разделение на компоненты, которые по выходу 5 из колонки поступают в блок химической ионизации 6, в который одновременно подают азот из генератора 7. В блоке химической ионизации 6 компоненты пробы ионизируются (коронный разряд, атмосфера азота, носитель смесь метанол-вода) и ионы (положительные) поступают в ОИЛ 8, в которой осуществляют отбор и подачу в детектор 9 ионизированных компонентов пробы, и результаты детектирования регистрируют в блоке автоматического управления хода анализа 10.
В качестве аналитической системы могут быть использованы, например, хромато-масс-спектрометр Surveyor HPLC, совмещенный с ОИЛ-масс-спектрометром (Bremen, Германия) и оснащенный ионным источником для химической ионизации с генератором азота NM30LA (Peak Scientific, США).
В качестве монолитной ХГК могут быть использованы, например, PLPR-S 300 (Polymer Labs), Chromolith (Merck), Onyx Monolith C18 (Phenomenex, Германия) и другие подобные колонки.
В качестве вспомогательного оборудования могут быть использованы:
- автоматический шейкер фирмы Glas-Col®, США - для ЖЖЭ;
- центрифуга марки Rotina 46R фирмы Hettich, (ФРГ) для получения контрастной поверхности раздела между органической и водной фазами;
- вакуумный концентратор фирмы Barnstead Inc. (США) для упаривания органического экстракта.
Для приготовления подвижных фаз и для растворения образцов сравнения могут быть использованы, например, метанол для хроматографии (Merck, Германия) и сверхчистая вода (ρ=18,2 Мом/см, полученная на установке Direct Q, Millipore).
Для жидкостно-жидкостной экстракции может быть использован диэтиловый эфир марки ч.д.а. (Медхимпром, Россия).
Ионизацию осуществляют воздействием коронного разряда в атмосфере азота при нормальном давлении.
Детектирование определяемых веществ проводят в режиме регистрации полного сканирования.
Изобретение может быть осуществлено следующим образом.
К исследуемому образцу биологической жидкости добавляют фосфатный буфер и раствор β-глюкуронидазы (Roche, Германия) и подвергают образец ферментативному гидролизу, гидролизат охлаждают, добавляют смесь карбоната калия и гидрокарбоната натрия до pH 9-10, далее образец экстрагируют диэтиловым эфиром с добавлением сульфата натрия. Органический экстракт затем центрифугируют, эфирный слой упаривают досуха, сухой остаток растворяют в метаноле. Приготовленный таким образом для анализа экстракт далее направляют на хромато-масс-спектрометрическое определение, параметры которого будут представлены в примерах конкретного осуществления заявленного технического решения.
Для лучшего понимания изобретение может быть проиллюстрировано, но не исчерпано следующими примерами его конкретного осуществления.
Пример 1.
Определение ксенобиотиков.
Анализ проводят на хромато-масс-спектрометрической системе Surveuor HPLC, совмещенной с ОИЛ-масс-спектрометром (Bremen, Германия) и оснащенной ионным источником для химической ионизации с генератором азота NM30LA (Peak Scientific, США). В качестве ХГК используют Onyx Monolith C18.
Рабочие растворы (список использованных веществ представлен ниже в Таблице 1, получены от фирмы LGC Promochem, Wesel, Германия) готовят растворением ксенобиотиков в заведомо не содержащей последних биологической жидкости (моча), и при этом содержание каждого вещества в биологической жидкости зависит от его класса (например, анаболики 10 нг/мл; бета-блокаторы и бета-агонисты 50; диуретики 200; стимуляторы 100 и т.д.). К образцу мочи (5 мл) с растворенным ксенобиотиком добавляют 1 мл фосфатного буферного раствора (pH 7,0) и 50 мкл раствора β-глюкуронидазы и подвергают образец ферментативному гидролизу (57°C, 3 часа), гидролизат охлаждают до комнатной температуры, добавляют в него 100 мг смеси карбоната калия и гидрокарбоната натрия (2:1) до pH 9-10, далее образец экстрагируют 5 мл диэтилового эфира с добавлением 500 мг сульфата натрия. Органический экстракт затем центрифугируют в течение 5 минут при 2500 об/мин, эфирный слой упаривают досуха, сухой остаток растворяют в 50 мкл метанола. Приготовленный таким образом для анализа экстракт далее направляют на хромато-масс-спектрометрическое определение при следующих параметрах: температура фокусирующего капилляра - 280°C, ток через иглу 5 мкА, скорость потока осушающего газа (азот) 20 л/час; скорость сканирования 1 скан/сек; диапазон сканирования 100-500 а.е.м.; точность определения масс ≤2 млн-1; разрешение 60000 (на половине высоты); объем вводимой пробы 10 мкл; подвижная фаза А: метанол-вода 80:20, В: метанол - вода 90:10; градиентное элюирование 0-8 мин: 50% В; 8-12 мин: 100% В; скорость потока 800 мкл/мин. Снимают и регистрируют хроматографические и масс-спектрометрические характеристики пробы: определяют время удержания, молекулярную массу, нижний предел обнаружения. Нижний предел обнаружения устанавливают следующим образом: готовят раствор ксенобиотика пяти разных концентраций в каждой из 10 разных проб биологической жидкости и анализируют 50 полученных образцов. Селективность определяют сравнением результатов анализа чистой биологической жидкости (матрица) и с добавлением соответствующего ксенобиотика. Результаты анализа представлены в Таблице 1.
ния (нг)
Пример 2. Анализ биологической жидкости, содержащей смесь ксенобиотиков.
Процедуру пробоподготовки и анализ пробы проводят как в Примере 1, за исключением того, что скорость потока поддерживают в пределах 1000 мкл/мин и анализу подвергают биологическую жидкость, содержащую болденон, тренболон и дигидроэкземестан. Результаты анализа представлены в Таблице 2.
Пример 3 (сравнительный по патенту [4])
Получение масс-спектров, определение характеристических ионов, времени удержания и пределов детектирования кортизола и кортизона
Готовят стандартные растворы аналитов (1 мг/мл): в метаноле, далее получают рабочие растворы разбавлением стандартных растворов до содержания 1,0 мкг/мл. Приготовленные таким образом рабочие растворы вводят в систему ВЭЖХ-МС/МС (хромато-масс-спектрометр с тройным квадрупольным анализатором TSQ Quantum фирмы Thermo Finnigan (США), соединенным с высокоэффективным жидкостным хроматографом модели Surveyor, оснащенным автосамплером, насосом высокого давления и дегазатором фирмы Thermo Finnigan (США). Анализ ведут при скорости потока подвижной фазы 0.2 мл/мин. Определяемые вещества разделяют градиентным элюированием при условиях: 0 мин - 15% В; 10 мин - 60% В; 15 мин - 75% В, 25 мин. - 85%, общее время анализа с учетом стабилизации системы перед вводом следующего образца составляет 35 мин. Ионизацию при атмосферном давлении осуществляют электрораспылением в режиме регистрации отрицательных ионов. Напряжение на капилляре - 3.8 кВ; температура капилляра 245°C; скорость потока осушающего газа (азот) - 0.45 л/мин; скорость потока газа (аргон) в камере соударения - 0.075 л/мин; температура в камере ионизации 200°C; давление на распылителе - 2 атм. Детектирование определяемых веществ проводят в режиме регистрации селективных реакций (SRM). Ширина пика для прекурсор-ионов и соответствующих характерных ионов на первом квадруполе (Q1) и третьем квадруполе (Q3) составляет 0.5 а.е.м. (на половине высоты), время задержки - 5 мс. Обработку полученных данных (масс-спектры, характеристические ионы, время удержания и пределы детектирования исследуемых ксенобиотиков) проводят с применением программного обеспечения Xcalibur версии 1.3 фирмы Thermo Finnigan, США (см. Таблицу 3).
Из сравнения результатов анализа по описанию и Таблиц 1 и 3 со всей очевидностью вытекает, что предлагаемый способ обнаружения ксенобиотиков при допинговом контроле и устройство для его осуществления существенно превосходят известные по достигаемому техническому результату и обеспечивают возможность однозначной идентификации химических соединений и их фрагментов в произвольных комбинациях при одновременном повышении точности и оперативности определения.
Источники информации
1. RU 2093822 C1, М.кл. G01N 30/04, публ. 1997 г.
2. RU 2069363 C1, М.кл. G01N 30/02, публ. 1996 г.
3. WO 2004/104571, М.кл. G01N 30/00, публ. 2004 г.
4. RU 2384846 C1, М.кл. G01N 33/493, публ. 2010 г.
5. Anal. Chem. 2000. V.72. V.6. P.1156 - прототип.
6. http://www.textronica.com/lcline/ltq_sens.html - прототип устройства
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ РЕТРОСПЕКТИВНОГО ОБНАРУЖЕНИЯ КСЕНОБИОТИКОВ ПРИ ДОПИНГОВОМ КОНТРОЛЕ СПОРТСМЕНОВ | 2011 |
|
RU2478207C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КСЕНОБИОТИКОВ В МОЧЕ ЧЕЛОВЕКА ПРИ ДОПИНГОВОМ КОНТРОЛЕ | 2008 |
|
RU2390773C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ДОПИНГА У ЛОШАДЕЙ | 2011 |
|
RU2489719C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПРИЕМА КОРТИКОТРОПИНОВ ПРИ ДОПИНГОВОМ КОНТРОЛЕ СПОРТСМЕНОВ | 2008 |
|
RU2384846C1 |
| СПОСОБ ВЫЯВЛЕНИЯ НЕИЗВЕСТНЫХ ВЕЩЕСТВ В БИОЛОГИЧЕСКИХ ЖИДКОСТЯХ ПАЦИЕНТОВ, ПРИНИМАВШИХ НАРКОТИЧЕСКИЕ ИЛИ ПСИХОАКТИВНЫЕ ВЕЩЕСТВА | 2009 |
|
RU2419788C2 |
| Способ определения мельдония в моче человека | 2017 |
|
RU2639475C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПРИЕМА ЭРИТРОПОЭТИНА ПРИ ДОПИНГОВОМ КОНТРОЛЕ СПОРТСМЕНОВ | 2008 |
|
RU2390779C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ НАРКОТИЧЕСКИХ И ПСИХОАКТИВНЫХ ВЕЩЕСТВ В БИОЛОГИЧЕСКИХ ЖИДКОСТЯХ | 2009 |
|
RU2390771C1 |
| СПОСОБ ОБНАРУЖЕНИЯ ЭКЗОГЕННЫХ СТЕРОИДОВ В БИОЛОГИЧЕСКОЙ ЖИДКОСТИ ЧЕЛОВЕКА | 2012 |
|
RU2483309C1 |
| СПОСОБ РАСПОЗНАВАНИЯ И КЛАССИФИКАЦИИ 3-ОКСОСТЕРОИДОВ И ИХ МЕТАБОЛИТОВ ПРИ ДОПИНГОВОМ КОНТРОЛЕ СПОРТСМЕНОВ | 2010 |
|
RU2452967C2 |
Изобретение относится к хроматографическому анализу различных химических соединений и может быть использовано в медицине, биологии, экологии и в особенности при допинговом контроле. Предложен способ обнаружения комплекса ксенобиотиков в биологических жидкостях при допинговом контроле, при котором пробу исследуемого образца разделяют на компоненты пропусканием через монолитную хроматографическую колонку, при этом скорость потока поддерживают в пределах по меньшей мере 500-1100 мкл/мин, ионизацию компонентов проводят азотом в присутствии носителя метанол/вода в соотношении 20/80 и отбор и из ионно-орбитальной ловушки на детектирование последовательно отбирают и подают ионизированные компоненты с разностью масс по меньшей мере 10-4 а.е.м. Устройство для обнаружения комплекса ксенобиотиков в биологической жидкости включает смонтированный в термостате узел ввода пробы, инжектор, монолитную хроматографическую колонку, ионно-орбитальную ловушку с детектором, блок химической ионизации со встроенным генератором азота, размещенный между выходом анализируемого вещества из хроматографической колонки и входом в орбитально-ионную ловушку, и общий для системы блок автоматического управления ходом анализа. Техническим результатом изобретения является обеспечение возможности однозначного выявления и идентификации химических соединений и их фрагментов в произвольных комбинациях при одновременной высокой достоверности, точности и высокой скорости проведения анализа. 2 н.п. ф-лы, 3 табл., 1 ил.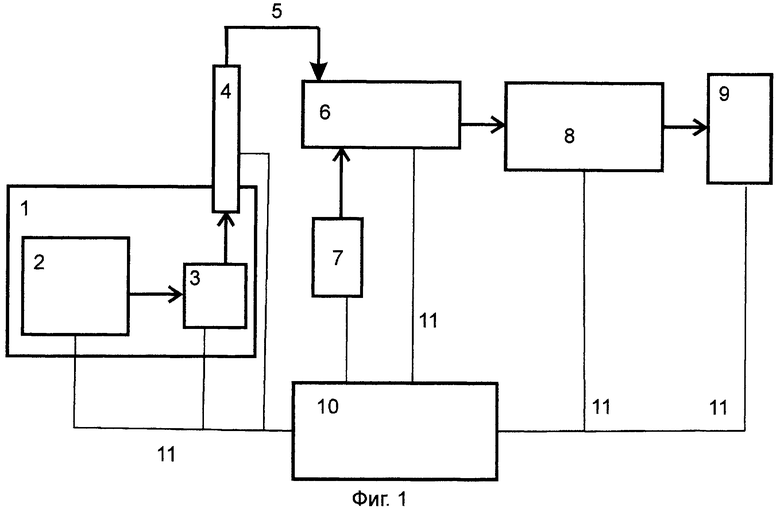
1. Способ обнаружения комплекса ксенобиотиков в биологических жидкостях при допинговом контроле, при котором готовят пробу исследуемого образца, разделяют ее на компоненты пропусканием через хроматографическую колонку с подвижной жидкой фазой, ионизируют разделенные компоненты и детектируют ионы сепарацией через орбитально-ионную ловушку с последующим определением принадлежности каждого иона конкретному веществу на основании его аналитических характеристик, отличающийся тем, что разделенные компоненты исследуемого вещества пропускают через монолитную хроматографическую колонку и при этом скорость потока поддерживают в пределах по меньшей мере 500-1100 мкл/мин, ионизацию компонентов проводят в атмосфере азота в присутствии носителя метанол/вода, причем соотношение компонентов носителя в ходе анализа изменяют линейно от 20/80 до 90/10 в течение 5 мин, и из ионно-орбитальной ловушки на детектирование последовательно отбирают и подают ионизированные компоненты с разностью масс по меньшей мере 10-4 а.е.м.
2. Устройство для обнаружения комплекса ксенобиотиков в биологической жидкости при допинговом контроле по п.1, включающее смонтированный в термостате узел ввода пробы, инжектор, хроматографическую колонку, ионно-орбитальную ловушку с детектором, соединенные с общим для системы блоком автоматического управления ходом анализа, отличающееся тем, что оно дополнительно включает блок химической ионизации со встроенным генератором азота, размещенный между выходом анализируемого вещества из хроматографической колонки и входом в орбитально-ионную ловушку, и в качестве хроматографической колонки устройство включает монолитную хроматографическую колонку.
| Anal | |||
| Chem | |||
| Термосно-паровая кухня | 1921 |
|
SU72A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Аэропланная стойка с изменяющимся сечением | 1924 |
|
SU1156A1 |
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| http://www.textronica.com/lcline/ltg_sens.html | |||
| US 7820966 B2, 26.10.2010 | |||
| СПОСОБ ОПРЕДЕЛЕНИЯ КСЕНОБИОТИКОВ В МОЧЕ ЧЕЛОВЕКА ПРИ ДОПИНГОВОМ КОНТРОЛЕ | 2008 |
|
RU2390773C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ВЕРОЯТНОЙ ТОКСИЧНОСТИ КСЕНОБИОТИКОВ ПРИМЕНИТЕЛЬНО К ЧЕЛОВЕКУ | 2002 |
|
RU2223493C2 |

