Область техники, к которой относится изобретение
Настоящее изобретение относится к способам идентификации групп генов и композициям для их осуществления, которые могут быть использованы для дифференцирования разных типов конкретных заболеваний, таких как рак молочной железы.
Уровень техники
Способ последовательной кластеризации. Способы биологии с большим количеством данных (НТ) находят широкое применение при исследовании заболеваний, в частности таких гетерогенных и сложных состояний, как рак молочной железы. Технологии OMIC, интерпретация и применение данных быстро прогрессируют. За последние 2-3 года в практику был внедрен целый ряд новых способов функционального анализа больших наборов данных (часто определяемых как системная биология). После многих лет применения ”радиолокационного сканирования” при выполнении «сырых» лабораторных исследований в настоящее время при исследовании заболеваний используются способы получения данных на основе анализа биологических сетей и путей. Однако несмотря на значительный прогресс научно-исследовательской работы, результаты и способы биологии с большим количеством данных еще не нашли практического применения в клиниках.
Глобальный анализ экспрессии генов в случае рака молочной железы. Рак молочной железы, злокачественное заболевание, которое занимает второе место по смертности женщин в западных странах, является сложным заболеванием (или скорее собирательным понятием, включающим целую совокупность заболеваний), охватывающим многие ткани и подтипы клеток. Полагают, что клинические проявления этого заболевания, такие как выживание, восприимчивость к лечению, возможность рецидива и риск возникновения метастазов, определяются целым рядом факторов. Рак молочной железы диагностируется путем визуализации с помощью рентгенографии, томографии и других исследований, при этом не используются диагностические ДНК и/или белковые маркеры. В настоящее время известно, что рак молочной железы не может быть классифицирован должным образом при помощи дескриптора любого одного гена/белка/фенотипа, а необходимы маркеры множества генов/белков для получения полной картины заболевания. Материально-технические средства для такого многофакторного анализа были созданы на основе технологий робастной микроматрицы и системного анализа экспрессии генов (SAGE), и в течение последних 10 лет был выполнен целый ряд крупномасштабных исследований по анализу экспрессии генов ”всего генома” в опухолях молочной железы. Двумя клинически наиболее важными вопросами, требующими решения, были подклассификация разных типов рака и обнаружение прогностических признаков образования отдаленных метастазов - главной причины смертности при раке молочной железы.
Подклассификация типов рака молочной железы.
Одним из наиболее известных исследований в области молекулярного анализа является серия работ по кластеризации типов инвазивного рака молочной железы, выполненных Sørlie et al. Указанные исследования были выполнены на основе матриц кДНК, разработанных в лаборатории Patrick Brown в Станфорде, с использованием больших наборов образцов; полученные результаты подробно описаны и сопоставимы в разных сериях исследований. Прежде всего, отдельные наборы признаков для базального и люминального эпителия, стромы и лимфоцитов были идентифицированы как характерные «черты/признаки», уникальные для опухолей. В последующих исследованиях были проанализированы 122 образца тканей (включая 115 образцов инвазивного рака молочной железы, 4 образца нормальной молочной железы) на основании двумерной неконтролируемой иерархической кластеризации ”центрального” набора из 540 генов. Набор характеристик экспрессии в опухолях соответствовали характерным признакам определенных линий клеток: два типа люминального эпителия (А и В), базальный эпителий, нормальный эпителий и ERBB+ кластеры. Люминальный эпителий типа А характеризовался высокой экспрессией рецептора эстрогена (ER) и несколькими его генами-мишенями LIV-1, HNF3A, XBP1 и GATA3. Люминальный эпителий типа В отличался более низкой экспрессией ER и высокой экспрессией GGH, LAPTMB4, NCEP1 и CCNE1. Характерный набор для базального эпителия включал кератины 5 и 17, аннексин 8, CX3CL1 и TRIM29. В базальном эпителии не был обнаружен ER и отсутствовала экспрессия генов люминального эпителия. И, наконец, кластер ERBB+ характеризовался высокой экспрессией некоторых генов группы ERBB: ERBB2, GRB7, TRAP100. В наборе признаков экспрессии нормального эпителия были обнаружены липоциты и другие неэпителиальные клетки. Указанная схема кластеризации была подтверждена другим исследованием, выполненным на такой же матрице кДНК с использованием образцов, полученных у 295 индивидов, и с привлечением независимых наборов данных, анализируемых на платформах других микроматриц. В недавно проведенном исследовании другой кластер, получивший название ”молекулярный апокрин”, характеризовался наличием рецептора андрогена (AR), отсутствием группы ER вне базального эпителия и частой амплификацией ERBB2. Необходимо отметить, что клинические фенотипы коррелировали с конкретными кластерами. Например, базальный эпителий и подтип ERBB+ были связаны с самой короткой продолжительностью жизни индивидов. Однако несмотря на надежность и пригодность для использования в исследовательской работе и в клинических условиях, указанная ”классическая” схема подклассификации является неполной и имеет недостатки. Во-первых, примерно 1/3 образцов нельзя отнести ни к одной из 5 категорий как в наборе из 122 образцов, так и в наборе из 295 образцов. Такая высокая степень неклассифицируемых индивидов ограничивает применение в клинике кластеризации на основе микроматрицы. Во-вторых, кластеры остаются гетерогенными. Указанные недостатки стали причиной создания совершенно другой классификации, описанной в настоящей заявке на патент.
Функциональный анализ глобальной экспрессии генов и других данных OMIC. Используя тысячи точек данных и представление всего генома, наборы данных OMIC подвергают строгим статистическим анализам, таким как определение значимости точек данных, контролируемая и неконтролируемая кластеризация, дисперсионный анализ ANOVA, анализ основных компонентов (РСА), и другим анализам. Однако статистический анализ является недостаточным для интерпретации ”внутренней биологии”, определяющей профили экспрессии. Функциональный анализ экспрессии генов обычно ограничивается запросом полученных статистическими способами списков генов на фоне онтологии клеточных процессов, таких как GO (http://www.geneontology.org). Подобный подход является информативным для получения ”общей картины”, например, для соотнесения характерных признаков метастазов с ”гибелью клеток, клеточным циклом, пролиферацией и репарацией ДНК”. Однако полученных данных недостаточно для ”точного картирования” набора данных на уровне взаимодействия отдельных белков и выявления комбинаций биологических событий, являющихся причиной возникновения данного состояния. Авторы настоящего изобретения рассматривают главные инструменты для анализа данных как пути, клеточные процессы и сети (сигнальные и метаболические). Пути представляют собой линейные цепи экспериментально подтвержденных последовательных биохимических трансформаций или взаимодействий сигнальных белков. Функциональные процессы представляют собой группы или категории белков, необходимые для осуществления различных клеточных функций, таких как апоптоз или окисление жирных кислот. В отличие от путей белки, участвующие в процессах, не связаны функциональными связями. Категории путей и процессов являются статическими, то есть экспериментальные данные могут быть картированы (путем связывания идентификатором точек данных) в путях и процессах, но не могут изменять их. Биологические сети имеют существенные отличия. Они представляют собой комбинации объектов - узлы (белки, метаболиты, гены и т.д.), взаимосвязанные связями (двоичные взаимодействия физических белков, одностадийные реакции обмена веществ, функциональные корреляции) и собранные ”в движении” из таблиц взаимодействия. Биологические сети являются динамичными, так как они созданы de novo из стандартных блоков (двоичные взаимодействия) и являются специфичными для каждого набора данных. Комбинация сетей, ”интерактома”, вероятно, является ближайшим представлением ”клеточного механизма” активных белков, выполняющих клеточные функции. Полный набор взаимодействий определяет потенциал клеточного механизма, миллиарды физически возможных многостадийных комбинаций. Очевидно, что лишь часть всех возможных взаимодействий активируется в любом данном состоянии, так как не все гены экспрессированы в определенный период времени в ткани и активной является только часть пула клеточных белков. Субпопуляция генов и белков с повышенной экспрессией (или пониженной экспрессией) является уникальной для данного эксперимента и улавливается ”моментальными снимками” данных, такими как микроматрицы. Созданные сети могут быть интерпретированы с учетом процессов более высокого уровня, при этом может быть выявлен механизм воздействия. Такой результат может быть достигнут путем связывания объектов сети с онтологией генов (GO) и онтологиями других процессов, а также с картами обмена веществ и передачи сигналов. Процесс анализа данных состоит из сужения списка из потенциально возможных многих тысяч точек данных до небольших наборов генов, взаимосвязанных функционально и в наибольшей степени пораженных в данном заболевании. Указанный результат достигается при помощи способов оценки пересечений функциональных категорий и вычисления релевантности к рассматриваемому набору данных (относительное насыщение путей и сетей данными).
Непредвиденная токсичность новых соединений для человека является одной из главных причин выведения лекарственного средства с рынка и из клинических испытаний. Недавно выполненный анализ лекарственных средств, поступивших на предклиническое исследование, показал, что в период между 2000 г. и 2002 г. только одна из 13 молекул, проходивших клинические исследования, достигла рынка. Вышеуказанное и другие исследования показывают, что затраты на разработку лекарственных средств, включая реализацию, возрастают и могут превышать 1 миллиард долларов. Из-за невозможности прогнозировать токсичность лекарственного средства фармацевтическая промышленность понесла убытки в размере 8 миллиардов долларов в 2003 г. Критическое значение проблемы непригодности лекарственных средств создало постоянно растущий рынок новых прогностических технологий, таких как токсикогеномика (оценка токсичности соединений для человека на основе предклинического молекулярного анализа) и фармакогеномика (оценка индивидуальной непереносимости лекарственного средства и побочных эффектов с учетом генотипа). Большинство токсикогеномных исследований представляют собой эксперименты по оценке зависимости лекарственное средство - эффект при помощи животных моделей с использованием крыс и мышей и обычно включают анализ экспрессии генов всего генома с использованием платформы микроматрицы. К современным аналитическим программным обеспечениям и справочным базам данных эмпирической экспрессии генов относятся DrugMatrix компании Iconix и ToxExpress компании GeneLogic. Указанное программное обеспечение обладает относительно низкой прогностической способностью. Однако указанную ситуацию можно кардинально изменить, благодаря созданию инструментов следующего поколения для функционального анализа и системной биологии.
Микроматрицы обеспечивают моментальный снимок экспрессии тысячи генов в данный момент времени и в определенном состоянии, при этом одной из главных задач является идентификация генов или групп генов, связанных с разными заболеваниями. Большинство способов предназначено для идентификации значимых изменений в уровнях экспрессии отдельных генов, однако изменение экспрессии может происходить на уровне путей или функционально-связанных сетевых модулей. Гораздо меньше внимания уделено одновременному использованию информации об экспрессии и взаимодействии генов. Посвященные этой теме исследования включают идентификацию биологически активных подсетей при помощи оценочных функций подсетей, и другие подходы сфокусированы на изменениях экспрессии генов в заранее определенном наборе генов, которые, как известно, являются биологически родственными. Подход, разработанный авторами настоящего изобретения, подобен по своей природе и сфокусирован на идентификации из заранее определенных сетевых модулей тех модулей, которые позволяют дифференцировать два любых состояния, то есть характеризуются характером экспрессии, который изменяется синхронно с другими состояниями. Обычные способы на основе изменения экспрессии отдельных генов не способны идентифицировать такие относительно мелкие, но одновременно происходящие изменения характера экспрессии.
Подходы на основе OMIC в механистической токсикологии. Контрольная оценка потенциально опасных токсических веществ является обязательной для пищевых и лекарственных соединений (FDA), а также для промышленных химических веществ и коммерческих соединений, выбрасываемых в окружающую среду (ЕРА). Для вышеуказанных широких категорий оценка риска токсичности от воздействия химического вещества или лекарственного средства заключается в двух связанных процессах: исследовании опасности и оценке воздействия. Общепризнанным фактом является то, что для всесторонней оценки безопасности лекарственных средств и химических веществ необходимо механистическое понимание взаимодействия химических веществ и живых систем. Большинство исследований механизмов химической токсичности (токсикогеномика) на основе OMIC сфокусировано на анализе транскрипционной экспрессии генов, и в настоящее время разрабатываются способы протеомного и метаболомного анализов для получения всестороннего представления о реакции на токсические вещества. Методы молекулярного анализа все чаще используются при оценке безопасности лекарственных средств на предклинической стадии и в клинических исследованиях заболеваний, обусловленных токсическими веществами. Токсикогеномика основана на предположении, что химические вещества с одинаковыми механизмами токсичности должны вызывать одинаковый характер экспрессии генов в пораженной ткани-мишени. Поэтому потенциальную токсичность соединения можно определить, сравнивая ее характер экспрессии генов с библиотекой стандартизированных характеристик экспрессии для ксенобиотиков с известной токсичностью. Печень и почки являются основными местами метаболизма ксенобиотиков и, следовательно, местами проявления гепатотоксичности и нефротоксичности. Поэтому справочные библиотеки характера экспрессии относятся, главным образом, к печени крыс, так как этот орган является наиболее часто используемой системой для анализа токсичности. Характер экспрессии генов может быть представлен разными способами в зависимости от используемых статистических способов (от списка генов с наибольшей амплитудой экспрессии до нервных сетей). Реакции на экспрессию лекарственных средств представлены в основном в форме специфичных для соединения или класса ”характерных генов”, то есть списков генов (биомаркеров экспрессии), характерных для определенного типа токсичности. Характерные гены обычно охватывают 50-100 сильных генов и служат для диагностики подверженности воздействию определенного химического вещества часто с прогностической точностью >90%. Характерные гены получают разными статистическими методами, такими как алгоритмы соответствия характера неконтролируемой и контролируемой кластеризации. Краткие справочные базы данных характерных признаков токсических веществ первоначально были созданы для дрожжей, а затем для грызунов.
Федеральные контролирующие агентства признают, что молекулярный анализ воздействия токсических веществ должен быть интегрирован с данными патологического и клинического анализа заболевания. Для подтверждения такой интеграции при оценке безопасности лекарственных средств FDA выпустило ”Руководство для промышленности по предоставлению фармакогеномных данных”. Аналогичным образом Совет по научной политике подготовил публикацию ”Временная политика в области геномики” и создал собственную внутреннюю ”базу для компьютерной программы токсикологического исследования в ORD”. Справочные стандарты для экспериментальной разработки и предоставления данных токсикогеномных исследований были приняты в качестве стандарта MIAME-Tox. В настоящее время отсутствует общепринятая стандартизированная экспериментальная платформа или общая схема создания базы данных для токсикогеномных исследований. В настоящее время существует несколько общедоступных, академических и коммерческих баз данных в качестве специальных репозиториев для токсикогеномных данных (таблица 1). Базы токсикогеномных и хемогеномных данных служили инструментом для поиска дополнительных специфичных для данного класса генных признаков и прогнозирования гепатотоксичности, вызываемой лекарственным средством и опосредованной окислительным стрессом, а также вызываемой лекарственным средством дегенерации почечных канальцев и нефротоксичности.
Общедоступные и коммерческие базы токсикогеномных данных, протоколы и источники
Таким образом, в фармакогенетике существует потребность в эффективных и продуктивных методах анализа баз данных путей для прогнозирования возможных новых лекарственных средств или способов лечения. Такой метод можно было бы использовать для оценки новых лекарственных средств или способов лечения на основе баз данных путей для прогнозирования предполагаемых вредных и побочных эффектов до начала дорогостоящих клинических испытаний.
Сущность изобретения
Настоящее изобретение относится к двум новым комплементарным способам, а именно к способам последовательной кластеризации и характерных признаков сетей, используемых для выполнения функционального анализа большого количества экспериментальных данных, главным образом, характера экспрессии генов всего генома. Способ последовательной кластеризации можно использовать, главным образом, для подклассификации больших наборов данных характеристик экспрессии генов для образцов генетически гетерогенной среды; характерным набором данных является набор данных большого числа индивидов для генетически сложных заболеваний человека, таких как рак, диабет, ожирение, сердечно-сосудистые заболевания и т.д. Указанный способ позволяет разделить образцы на более мелкие кластеры с четко выраженными характеристиками экспрессии для групп выбранных генов (дескрипторные группы). Каждый кластер связан с определенным различимым генотипом, клиническим результатом и определенным лечением индивида, у которого характер экспрессии относится к определенному кластеру. Способ последовательной кластеризации может быть использован в клинических испытаниях разрабатываемого лекарственного средства и для лечения индивида в клинике. В обоих случаях благодаря включению характера экспрессии генов индивида в определенную дескрипторную группу может быть определено соответствующее лечение индивида на основании конкретной дескрипторной группы.
Способ характерных признаков позволяет идентифицировать характерные модули взаимодействия белков и генов (признаки сети), экспрессия которых синхронно повышается или понижается в определенных условиях, таких как реакция клетки на лечение лекарственным средством. Указанный способ можно использовать, главным образом, для генетически гомогенных наборов данных экспрессии генов, например чистым линиям лабораторных животных, таких как мыши и крысы, линиям клеток человека и животных или генетически сходным людям. Основной областью использования способа характерных признаков сетей являются исследования реакции на лекарственное средство (токсикогеномика, исследования типа воздействия) и использование в качестве дескрипторов для кластеров экспрессии генов, идентифицированных способом последовательной кластеризации. Указанный способ можно использовать на стадиях предклинической и клинической разработки лекарственного средства или определения токсичности лекарственного средства и в качестве дескриптора для кластеров индивида.
Функции и преимущества вышеуказанных и других вариантов осуществления настоящего изобретения будут более понятны из приведенных ниже примеров. Приведенные примеры предназначены для иллюстрации преимуществ настоящего изобретения и не ограничивают объем настоящего изобретения.
Краткое описание чертежей
Прилагаемые чертежи не предназначены для масштабирования. На указанных чертежах все идентичные или почти идентичные элементы, представленные на разных фигурах, обозначены одним числом. Для простоты описания не все элементы могут быть отмечены на каждом чертеже.
На фиг.1 показана подклассификация разных типов инвазивного рака молочной железы на основании глобального анализа экспрессии.
На фиг.2 показана последовательность операций функционального анализа белков и других данных OMIC.
На фиг.3 показана последовательность операций кластеризации, описанная в настоящем изобретении.
На фиг.4 показаны бимодальные гены.
На фиг.5 показано преобразование сигналов экспрессии в соответствии с бимодальностью.
На фиг.6 показана идентификация близких групп генов в 122 образцах.
На фиг.7 показана последовательная кластеризация наборов данных 122 индивидов в семь дескрипторных групп.
На фиг.8 показана сеть для STAT1+, P1AU+ и FN1+ групп в виде входного списка.
На фиг.9 показано картирование кластеров на картах путей.
На фиг.10 показана реконструкция условных подсетей из путей.
На фиг.11 показано вычисление расстояний между образцами в пространстве экспрессии генов пути.
На фиг.12 показано исследование перекрывания групп идентифицированных генов, определяющих различные условия лечения.
На фиг.13 показаны образованные путями сети, по-разному реагирующие на лечение.
На фиг.14 показана бикластеризация генов, картированных в соответствии с путями.
На фиг.15 показана реконструкция условных подсетей из путей.
На фиг.16 показано ”прямое взаимодействие” (DI) сетей, созданных из генов, экстрагированных из большинства путей, между профилями тамоксифена и фенобарбитала.
На фиг.17 показана последовательность операций по созданию характерных признаков сетей на основании токсикогеномных данных.
На фиг.18 показаны окрестности статистически значимой сети (ориентированный граф). (А) Узел Х представляет расположенные в прямом направлении соседние элементы первого уровня с пониженной экспрессией (синий цвет) и расположенные в обратном направлении соседние элементы с повышенной экспрессией. Статистические оценки такого распределения могут быть вычислены при помощи гипергеометрического распределения. (В) Пример действительной сети. Статистически значимое распределение генов с пониженной экспрессией из индуцированного аллиловым спиртом профиля экспрессии в окрестностях киназы 1 Р70 S6. Расположенные в обратном направлении гены в основном характеризуются пониженной экспрессией (синий цвет). Указанная сеть создана при помощи программы MetaCore™ 3.0.
На фиг.19 показано сравнение характерных признаков сетей при лечении лекарственными средствами.
На фиг.20 показан управляющий интерфейс исходных данных.
На фиг.21 показана блок-схема универсальной компьютерной системы, при помощи которой могут быть выполнены разные варианты осуществления настоящего изобретения.
На фиг.22 показана блок-схема компьютерной системы хранения данных, при помощи которой могут быть выполнены разные варианты осуществления настоящего изобретения.
Подробное описание изобретения
Настоящей задачей в области фармакогенетики является создание эффективных и продуктивных способов анализа баз данных путей для прогнозирования потенциально новых лекарственных средств или способов лечения. Альтернативно базы данных могут быть использованы для оценки предлагаемых новых лекарственных средств или способов лечения по сравнению с базами данных путей с целью прогнозирования нежелательных и побочных эффектов, которые могут быть выявлены до начала дорогостоящих клинических испытаний.
Контрольная оценка возможного вреда токсических веществ является обязательной для пищевых и лекарственных соединений (FDA), а также для промышленных химических веществ и коммерческих соединений, выбрасываемых в окружающую среду (ЕРА). Для обеих указанных широких категорий оценка риска токсичности в результате воздействия химических веществ или лекарственных средств состоит из двух взаимосвязанных процессов: исследования вреда и оценки воздействия. Общепризнанным фактом является необходимость механистического понимания взаимодействия химических веществ и живых систем для полной оценки безопасности лекарственных средств и химических веществ. Большинство исследований механизмов химической токсичности (токсикогеномика) на основе OMIC сфокусировано на анализе транскрипционной экспрессии генов и анализе белкового обмена и метаболизма для всестороннего изучения реакции на токсическое вещество.
В основе токсикогеномики лежит предположение, что химические вещества с одинаковыми механизмами токсичности должны вызывать одинаковый характер экспрессии генов в пораженной ткани-мишени. Поэтому можно определить возможную токсичность соединения путем сравнения ее характера экспрессии генов с библиотекой стандартизированных характеров экспрессии ксенобиотиков, обладающих известной токсичностью. Печень и почки являются основными местами обмена ксенобиотиков и, следовательно, проявлений гепатотоксичности и нефротоксичности. Поэтому справочные библиотеки характера экспрессии относятся, главным образом, к печени крыс, так как указанная система является наиболее широко используемой системой анализа токсичности. Характер экспрессии генов может быть представлен разными способами в зависимости от используемых статистических способов (от списка генов с наиболее пораженной амплитудой экспрессии до нервных сетей). Реакции экспрессии генов на лекарственное средство представлены в основном в форме характерных генных признаков, специфичных для соединения или класса, то есть в виде списков генов (биомаркеры экспрессии), характерных для определенного типа токсичности. Характерные гены обычно включают 50-100 генов и часто позволяют диагностировать воздействие конкретного химического вещества с прогностической точностью >90%. Характерные генные признаки создают при помощи разных статистических способов, таких как алгоритмы неконтролируемой кластеризации и контролируемого соответствия характеров.
Настоящее изобретение относится к двум новым комплементарным способам, а именно к способам последовательной кластеризации и характерных признаков сетей, используемым для выполнения функционального анализа большого количества экспериментальных данных, главным образом, профилей экспрессии генов всего генома.
Способ последовательной кластеризации можно использовать главным образом для подклассификации больших наборов данных характера экспрессии генов для образцов генетически гетерогенной среды; типичным набором данных является набор данных большого числа индивидов для генетически сложных заболеваний человека, таких как рак, диабет, ожирение, сердечно-сосудистые заболевания и т.д. Указанный способ позволяет разделить образцы на более мелкие кластеры с четко выраженным характером экспрессии для групп отобранных генов (дескрипторные группы). Каждый кластер связан с определенным различимым генотипом, клиническим результатом и определенным лечением индивида, у которых характер экспрессии относится к определенному кластеру. Способ последовательной кластеризации может быть использован в клинических испытаниях разрабатываемого лекарственного средства и для лечения индивида в клинике. В обоих случаях благодаря включению характера экспрессии генов индивида в определенную дескрипторную группу может быть определено соответствующее лечение индивида на основании конкретной дескрипторной группы.
Для создания и анализа биологических сетей используются способы современной графовой теории, представленной в исследованиях шведских и немецких математиков 18 и 19 веков. Из теории произвольной сети ”по умолчанию” следует, что пары узлов соединены с одинаковой вероятностью, и степени вероятности соответствуют распределению Пуассона. Считается маловероятным наличие у любого узла большего числа краев, чем среднее значение. Анализ интерактомы дрожжей показал, что сети не являются произвольными и распределение краев является гетерогенным при наличии нескольких хорошо соединенных узлов (концентраторы) и большинства узлов с малым числом краев. Такая топология определяется как свободное масштабирование, из чего следует, что соединяемость узлов соответствует экспоненциальному ряду: Р (k)~k-γ. P (k) означает долю узлов в сети с числом связей k. Интересно отметить, что концентраторы соединены главным образом с узлами низкого уровня, признак, который сообщает биологическим сетям свойство робастности. Удаление даже значительной части узлов все же не нарушает соединения сети. Непроизвольная топология согласуется с биологическими свойствами узлов и краев. Хорошо соединенные концентраторы (определяемые в настоящем описании изобретения как верхний квартиль всех узлов с учетом числа краев) представлены главным образом эволюционно консервативными белками, так как взаимодействие налагает определенные структурные ограничения на эволюцию последовательностей.
Анализ сетей широко используется при разработке лекарственных средств и конструировании трубопроводов, как в биологии, так и химии. Данные любого типа, которые могут быть связаны с геном, белком или соединением, могут быть узнаны входными анализаторами, визуализированы и проанализированы в сетях. Поэтому почти любой эксперимент на предклинической стадии с большим количеством данных (НТ), а также исследования ДНК или обмена веществ индивида во время клинических испытаний могут быть включены в анализы сети. Наиболее важным является то, что все указанные разные наборы данных могут быть обработаны в одной и той же сети. Поэтому сети представляют собой универсальную платформу для интеграции и анализа данных, которые всегда были главным объектом биоинформатики. Сетевой анализ сложных заболеваний человека является относительно новой областью.
Способ характерных признаков сетей позволяет идентифицировать характерные модули взаимодействия белков и генов (характерные признаки сети), которые синхронно активируются или подавляются в определенных условиях, таких как реакция клетки на лечение лекарственным средством. Указанный способ можно использовать, главным образом, для генетически гомогенных наборов данных экспрессии генов, таких как чистые линии лабораторных животных, например мыши и крысы, линии клеток человека и животных или генетически сходные люди. Основными областями применения способа характерных признаков сетей являются исследования реакции на лекарственное средство (токсикогеномика, исследования типа воздействия) и использование в качестве дескрипторов для кластеров экспрессии генов, идентифицированных способом последовательной кластеризации. Указанный способ может также использоваться для предклинической и клинической разработки лекарственного средства или определения токсичности лекарственного средства и в качестве дескриптора для кластеров индивида.
Авторами настоящего изобретения ранее был разработан новый способ анализа токсикогеномных данных на основании средств анализа функциональных данных и биологических сетей. Изобретения, принадлежащие всем соавторам, описаны в прилагаемых заявках на патент, включающих заявку ”Methods for Identification of Novel Protein Drug Targets and Biomarkers Utilitizing Functional Networks”, поданную 4 августа 2006 г., и заявку на патент США 11/378928 ”System and Method For Prediction of Drug Metabolism, Toxicity, Mode of Action, and Side Effects of Novel Small Molecule Compounds”, поданную 17 марта 2006 г., описание которых полностью включено в настоящее описание в качестве ссылки.
Рассматриваемые в настоящем изобретении два способа последовательной кластеризации и характерных признаков сетей также используются для идентификации маркеров рака молочной железы и определения наиболее эффективного лечения на основании указанных маркеров.
Рак молочной железы является злокачественным заболеванием с наибольшей смертностью среди женщин в западных странах и представляет собой сложное заболевание (или скорее собирательное понятие, включающее целую совокупность заболеваний), охватывающее многие ткани и подтипы клеток. Указанный рак возникает в результате неблагоприятной серии молекулярных событий, причинно-следственные связи которых еще плохо изучены, и включает гаметические и соматические мутации, неустойчивость хромосомы, эпигенетические изменения в характере метилирования ДНК, фундаментальные изменения характера экспрессии генов и белков. Помимо анатомического характера опухоли, узлов, системы определения стадии метастазирования (TNM) для солидных опухолей, разные типы рака молочной железы делятся на пять групп с учетом характеров экспрессии генов; считается, что указанные опухоли являются высокогетерогенными в генетической среде, при этом менее 10% опухолей четко соответствуют одному из генетических биомаркеров, BRCA I и BCRA II, и подвержены влиянию экзогенных факторов, таких как рацион питания.
С учетом такой сложной этиологии считается, что подклассификация и фенотип данного заболевания, включая прогноз, ответ на лечение, возможность рецидива и риск возникновения метастазов, определяются целым рядом факторов. Неудивительно, что диагностика и прогноз на основании установленных маркеров ДНК и сывороточного белка являются достаточно неточными. Например, прогностические маркеры для 10-15% больных раком молочной железы указывают на образование метастазов в течение трех лет с момента первоначального диагноза, но во многих случаях метастазы возникают через 10 лет. Гистологическое типирование является лишь слабым прогностическим маркером метастазов, а также взаимосвязи между метастазами и опухолеположительными подмышечными лимфатическими узлами, молекулярными прогностическими маркерами отдельных генов, такими как сверхэкспрессия ERBB2, состояние рецептора эстрогена (ER), уровень BRCA и белка uPA/PAI1.
Установлено, что такая высокая степень гетерогенности не может быть должным образом описана и классифицирована при помощи одного или даже комбинации дескрипторов отдельных генов/белков/фенотипов, и необходимо создать маркеры множества генов/белков, чтобы охватить всю сложность данного заболевания. Материально-технические средства для такого многофакторного анализа были созданы на основе технологий робастной микроматрицы и системного анализа экспрессии генов (SAGE), и за последние десять лет был выполнен целый ряд крупномасштабных исследований по анализу разных аспектов экспрессии генов всего генома в опухолях молочной железы. Двумя наиболее важными в клиническом отношении целями, достигнутыми в результате выполнения указанных исследований, были подклассификация разных типов рака в зависимости от характера экспрессии и обнаружение прогностических признаков для метастазов - наиболее важной причины смертности от данной болезни.
Серия работ по кластеризации разных типов инвазивного рака молочной железы, выполненных группой исследователей из норвежского госпиталя Radium и Станфордского университета под руководством Therese Sørlie, является одним из наиболее известных исследований по анализу экспрессии генов при помощи микроматрицы. Указанное исследование было выполнено с использованием нескольких матриц кДНК на основе первоначальной стеклянной точечной матрицы, созданной 10 лет тому назад в лаборатории Patrick Brown's в Станфорде. Указанные исследования были выполнены с использованием больших наборов образцов на платформе такой же базовой матрицы при помощи таких же способов. Все это делает результаты рассмотренных исследований воспроизводимыми и сравнимыми. Важно отметить, что полученные результаты хорошо описаны, и все исходные данные представлены в Станфордской базе данных микроматриц, что позволяет использовать указанные данные для ”метаанализа” другими группами ученых - достаточно редкий случай в исследовании экспрессии генов на основе микроматриц.
В первом исследовании для идентификации кластеров коэкспрессированных генов был выбран набор из 65 хирургических образцов, полученных у 42 индивидов, и 17 культивируемых линий клеток. Характер экспрессии линий клеток был использован в качестве внутреннего маркера для корреляции кластеров коэкспрессированных генов со специфическими типами клеток в опухолях. Таким образом, был идентифицирован характер для базального и люминального эпителия, стромы и лимфоцитов. Кластеры синхронной экспрессии были связаны с такими клеточными процессами, как пролиферация и активация специфических путей передачи сигналов. Важно отметить, что исследователи использовали пары опухолей, полученные до и после химиотерапии, проводимой в течение 15 недель, при этом характер экспрессии оставался сопоставимым в парах. Поэтому указанный характер можно считать характерным признаком, уникальным для опухолей.
В последующих исследованиях было проанализировано до 122 образцов ткани (115 образцов инвазивных опухолей молочной железы, 4 образца нормальной молочной железы, 7 образцов незлокачественных опухолей). Во-первых, из 8000 экспрессированных на матрице генов был выбран центральный набор из 540 наиболее связанных генов. Затем использовали двумерную неконтролируемую иерархическую кластеризацию для разделения набора образцов на 5 отдельных кластеров с характеристическими генами, известными в качестве биомаркеров возникновения разных типов рака молочной железы. Характер экспрессии в опухолях соответствовал признакам определенных линий клеток: два типа люминального эпителия (А и В, соответственно фиг.1А и 1В); базальный эпителий (фиг.1D); нормальный эпителий (фиг.1Е) и ERBB+ кластеры (фиг.1С). Люминальный эпителий типа А характеризовался высокой экспрессией ER, LIV-1, HNF3A, XBP1 и GATA3. Люминальный эпителий типа В отличался более низкой экспрессией ER и высокой экспрессией GGH, LAPTMB4, NCEP1 и CCNE1. Набор характеристик для базального эпителия включал кератины 5 и 17, аннексин 8, CX3CL1 и TRIM29. В базальном эпителии не был обнаружен ER и отсутствовала экспрессия генов люминального эпителия типа А. И, наконец, ERBB+ кластер характеризовался высокой экспрессией некоторых генов из группы ERBB: ERBB2, GRB7, TRAP100. В наборе характеристик экспрессии нормального эпителия были обнаружены липоциты и другие неэпителиальные клетки. Со времени первой публикации в 2001 г. правильность указанной схемы кластеризации была испытана и подтверждена в другом исследовании на такой же матрице кДНК с использованием образцов, полученных у 295 индивидов, и с привлечением независимых наборов данных, полученных другими группами ученых на платформах других микроматриц.
Идентификация наборов генов, прогнозирующих образования метастазов, является второй главной целью анализа экспрессии рака молочной железы. Причиной такого интереса является низкоэффективная традиционная диагностика метастазов, которые являются главной причиной смерти при любом раке. Так как вероятность образования метастазов нельзя прогнозировать во время постановки диагноза, то 80% индивидов назначают химиотерапию. И только у 40% женщин с указанным диагнозом возникают метастазы, вызывающие смерть, поэтому более половины индивидов проходят курс ненужного лечения токсичной химиотерапией. В настоящее время считается, что экспрессия одного гена-маркера или белка-маркера не может быть достаточной для такой сложной группы заболеваний, как рак молочной железы, и глобальная экспрессия генов является, по-видимому, наиболее приемлемым способом выявления маркеров метастазов, состоящих из множества генов. Традиционные способы на основе изменений экспрессии отдельных генов не позволяют идентифицировать относительно небольшие, но одновременные изменения характера экспрессии.
Процесс анализа данных с использованием последовательной кластеризации и характерных признаков сетей был исследован в отношении подклассификации разных типов инвазивного рака молочной железы на более фундаментальной основе по сравнению с ранее применявшейся неконтролируемой и контролируемой статистической кластеризацией. Во-первых, для оценки опубликованных наборов данных, полученных у 122 и 295 больных инвазивным раком, были использованы стандартные средства анализа данных (пути, сети, клеточные процессы), но гетерогенность среди индивидов в кластерах была недопустимо высокой для выполнения прямого функционального анализа. Поэтому индивидов распределяли по группам при помощи нового метода последовательной кластеризации с последующим выполнением функционального анализа новых кластеров. При помощи комбинационного вычисления бимодальности и нового метода определения близости были идентифицированы два небольших набора генов, характерных для определенных групп индивидов. При помощи указанного способа обе группы индивидов делили на 18 биологически значимых кластеров при отсутствии неклассифицированных индивидов. Идентификация набора “специфичных для рака молочной железы” бимодальных генов основана на сравнении профилей экспрессии нормальной ткани молочной железы и опухолей, но ни в одном из опубликованных исследований не приведено достаточное число норм.
В вышеописанном исследовании под руководством Therese Sørlie, несмотря на очевидную робастность и пригодность для научно-исследовательского и клинического применения, традиционная подклассификация была неполной и страдала серьезными недостатками. Во-первых, наборы данных примерно одной трети индивидов нельзя было отнести ни к одной из пяти категорий как в наборе из 122 образцов, так и в наборе из 295 образцов; на фиг.1 показана подклассификация разных типов инвазивного рака молочной железы на основании анализа глобальной экспрессии люминального эпителия А (фиг.1А), люминального эпителия В (фиг.1В), ERBB+ (фиг.1С), базального эпителия (фиг.1D) и нормального эпителия (фиг.1Е). Очевидно, что доля неклассифицированных образцов выше, чем при использовании разных платформ микроматриц. Такая высокая степень неклассифицированных индивидов ограничивает применение указанной схемы в клиниках. Во-вторых, кластеры остаются гетерогенными с учетом экспрессии многих важных генов из характерного набора, что можно видеть при выполнении анализа функциональных сетей и путей (фиг.2). На фиг.2 показана последовательность операций функционального анализа белков и других данных OMIC. Большие наборы данных картированы в отношении канонических биологических путей, классифицированы по онтологии клеточных процессов и использованы для создания сетей, специфичных для данного заболевания. Процессы, пути и сети оценивают в зависимости от относительного насыщения точками экспериментальных данных и определяют приоритет относительно друг друга. Для сравнения сетей, созданных для разных наборов данных, используют разные статистические методы.
Один вариант осуществления изобретения относится к функциональному анализу опубликованных характеристик условных генных признаков в отношении фенотипов, имеющих важное значение в случае рака молочной железы, таких как метастазы, коэффициент выживаемости, реакция на лечение и заживление ран. Другой вариант осуществления изобретения относится к анализу опубликованных обработанных наборов генов для выявления взаимосвязи между указанными наборами, а также базовых функциональных категорий, объединяющих гены в наборе (общее регулирование транскрипции, сигнальные модули).
Один вариант осуществления изобретения относится к новому модулю или пакету программного обеспечения, который может быть создан для токсикогеномного исследования. Модуль или пакет программного обеспечения может включать последовательность операций анализа данных и предлагаемую систему составления отчетов для добровольного представления токсикогеномных данных. Модуль или пакет программного обеспечения может быть интегрирован в основные общедоступные токсикогеномных базы данных (CEBS, EDGE, Tox-MIAME express и другие), а также дополнительные пакеты программного обеспечения и базы данных.
Модуль или пакет программного обеспечения может включать аннотированные общие базы данных, относящиеся, например, к взаимодействиям белков, лекарственным средствам, заболеваниям, каноническим путям и т.д.; специфическую информацию о раке молочной железы (например, все опубликованные наборы данных большого объема, более 1000 аннотированных генов, относящихся к возникновению рака молочной железы, и карты путей рака молочной железы); инструменты для визуализации и анализа данных, такие как сети, пути, фильтры; статистические методы и средства анализа данных (например, метод формализованной подклассификации, идентификация бимодальных генов и средства сравнения на основе путей).
Процесс анализа новых данных может быть начат с анализа токсикогеномных данных и выявления специфичных для лечения биологических сетей. Во-первых, данные о взаимодействии белков из аннотированных общих баз данных, например, MetaCore™ (GeneGo, Inc.), могут быть использованы для создания примерно 15000 модулей путей, представляющих ”пространство” потенциальной функциональности. На следующей стадии данные микроматрицы, полученные для крыс, подвергнутых трем разным воздействиям (например, фенобарбитал, местранол и тамоксифен), могут быть картированы в указанных модулях. Используя корреляционное расстояние Пирсона и разные статистические методы и методы кластеризации, можно идентифицировать совокупности путей, характеризующихся ”синхронным” характером экспрессии при многочисленных повторах одного и того же лечения при одновременном наличии сильной антикорреляции с другими методами лечения.
Принимая во внимание вышеуказанные исследования рака молочной железы, было идентифицировано значительное число путей, отличающих лечение фенобарбиталом от лечения местранолом и тамоксифеном, при этом число путей, различающих лечение тамоксифеном и местранолом, является более ограниченным. Не обнаружено путей, различающих лечения тамоксифеном в двух разных концентрациях. В отличие от традиционного статистического анализа и анализа методом кластеризации экспрессии на уровне отдельных генов новый способ позволяет реконструировать объединенные сетевые модули, определяющие различия между лечением разными химическими препаратами, и выявить механизмы таких различий.
Робастная классификация разных типов рака в отдельные группы, связанные с разной реакцией на лечение, имеет важное значение для правильного выбора существующих способов лечения и создания избирательно действующих новых лекарственных средств для отдельных групп, таких как иматиниб (Glivec), гефитиниб (Iressa) и цетуксимаб (Erbutux), которые являются эффективными только в том случае, если их мишень сверхэкспрессирована или мутирована. В случае метастазов генные признаки полезны главным образом в качестве лучших прогностических факторов по сравнению с существующими маркерами для групп индивидов с плохим прогнозом, которым должны быть назначены дополнительные лечебные мероприятия.
Главными средствами для анализа данных являются пути, клеточные процессы и сети (сигнальные и метаболические). Пути представляют собой последовательные этапы реакции, которые являются биохимическими трансформациями или последовательностями событий передачи сигналов, такими как трансдукция сигналов. Функциональные процессы обычно определяются как группы или категории белков, необходимых для выполнения разных клеточных функций, таких как апоптоз или протеолиз. В отличие от путей белки в таких категориях не связаны функциональными связями. Как пути, так и категории процессов являются статическими, как было установлено в предшествующих исследованиях, то есть экспериментальные данные могут быть картированы (путем идентификации точек, связывающих данные) в путях и процессах, не изменяя их.
Биологические сети являются совершенно другими. Они представляют собой комбинации объектов (белков, метаболитов, генов и т.д.), взаимосвязанных связями (двоичные физические взаимодействия белков, одностадийные метаболические реакции, функциональные корреляции и т.д.), и могут быть скомпонованы из таблиц взаимодействия. Биологические сети являются динамичными, так как они созданы de novo из стандартных блоков в результате двоичного взаимодействия и являются специфичными для каждого набора данных. Поэтому процесс анализа данных состоит из сужения списка потенциально многих тысяч точек данных до более интерпретируемого числа. Такого результата можно достичь, используя р-значения статистического анализа, разные методы оценки для пересечения категорий и вычисления релевантности результата для данного набора данных (при относительном насыщении путей и сетей данными) (фиг.2).
Сети являются наиболее важным инструментом в функциональном анализе, так как они обеспечивают самое высокое разрешение (физически соединенные отдельные белки), однозначно описывают наборы данных и являются исчерпывающими, так как число соединяемых белков в сетях значительно превышает содержание белков во всех базах данных путей и объединенных онтологий.
Сети были использованы для оценки токсичности и метаболизма ацетаминофена (АРАР) у человека. Структура была обработана в программе MetaDrug™ (GeneGo, Inc., St. Joseph, M.I.) при помощи правил и моделей метаболического расщепления, и полученные метаболиты были отображены в сетях, соединенных с метаболизирующими ферментами. В той же сети были отображены данные экспрессии генов на микроматрице, полученные при исследовании печени крыс, интоксицированных высокой дозой АРАР. На фиг.4А показано распределение сигналов в наборе данных для бимодальных генов, специфичных для рака молочной железы. Четыре нормы указаны в окружностях. На фиг.4В показано распределение экспрессии GRB7 у 122 индивидов, при этом на оси “х” представлены первоначальные сигналы экспрессии и на оси “y“ указано число индивидов с такими сигналами. На фиг.4С показано распределение GRB7, при этом на оси “х” указаны индивиды и на оси “y” - сигналы экспрессии. Красной линией отмечен порог, который отделяет среднее значение сигналов ниже указанного порога (относительно 0) Ti=2,05. Черной линией отмечено значение µ(LGRB7)≈0,45; зеленой линией отмечено значение µ(UGRB7)≈3,65.
Дополнительная функциональность, добавляемая в анализаторы больших количеств данных (то есть MetaCore™ и MetaDrug™), включают следующие элементы.
- Полная компоновка критической массы взаимодействия белков человека в трех областях: взаимодействия лиганд-рецептор; передача сигналов между клетками и ядерный эффектор (эндогенные метаболические реакции, структурные белковые комплексы, взаимодействия на мембране). Взаимодействия из других областей интегрируются ”бесшовно”, что делает возможным системную реконструкцию состояния (такого как заболевание, реакция на лекарственное средство) на основе крупномасштабных экспериментальных наборов данных.
- Окончание точного картирования обмена веществ человека, то есть фактически все опубликованные экспериментально подтвержденные метаболические реакции соотнесены с отдельными ферментами и кодирующими ферменты генами. Точное метаболическое картирование позволяет связать передачу сигналов с обменом веществ в возникших сетях.
- Расширение анализаторов данных с включением данных обмена веществ (значения, символы эмоций, брутто-формулы Химической справочной службы (CAS)) и большое количество данных исследования соединений.
- Создание двух новых добавочных модулей для импорта и интеграции данных особого взаимодействия (например, ассоциативность коэкспрессии в ответ на воздействие соединения, дрожжевой гибрид 2 (Y2H), ChiP/CHIP или раскрывающиеся данные иммунопреципитации). Особые взаимодействия могут быть визуализированы, использованы в качестве входных данных для создания сетей, добавлены в карты путей или введены на постоянной основе в базу данных МС/MD.
- Ручная аннотация взаимодействий между генами мыши и крысы без соотнесения с человеком. Такая аннотация необходима для обнаружения различий между профилями экспрессии лекарственного средства в животных моделях и токсичностью для человека.
- Интеграция на уровне обмена массивов данных с несколькими дополнительными сторонними пакетами программного обеспечения, включая Resolver (Rosetta Inpharmatics, Seattle, W.A.), GeneSpring (Agilent Technologies, Santa Clara, C.A.) - программы для управления и статистического анализа данных экспрессии на микроматрице; Phylosopher (GeneData, Waltham, M.A.) - комплексная программа по биоинформатике; программа Biocrates/ABI для исследования обмена веществ; и Xenobase (University of Michigan Ann Arvor, M.I.) - медицинское приложение преобразования.
Один вариант осуществления изобретения относится к модулю или программному обеспечению для анализа данных, определяющему тип воздействия, побочные эффекты и токсичность. Методика состоит из оценки данных экспрессии в ответ на воздействие соединения в многочисленных функциональных дескрипторах и классификацию соединения с включением его в одну или несколько категорий токсичности на основании такой оценки. Данный процесс может быть выполнен в алгоритмах и в полуавтоматическом режиме.
Другой вариант осуществления изобретения относится к созданию библиотеки характерных признаков сетей и других функциональных дескрипторов для анализа молекулярных профилей реакции на лекарственное средство. Такие дескрипторы могут быть созданы на основе функциональных категорий. Прикладное значение такой библиотеки частично зависит от качества и объема токсикогеномных данных, используемых для анализа, которые ограничены базой данных “Химическое воздействие на биологические системы” (CEBS) (текущий и последующие выпуски, другие общедоступные базы данных и экспертные публикации). Поэтому библиотека может быть также создана в виде автоматического модуля оперативной помощи.
Другой вариант осуществления изобретения относится к процессам автоматизации и составления отчетов для функционального анализа токсикогеномных данных, которые должны оказать серьезное влияние на добровольное предоставление токсикогеномных данных компаниями-спонсорами и их рассмотрение федеральными агентствами. Предоставление токсикогеномных данных не регулируется, и вследствие этого данные предоставляются в разных форматах с использованием часто несовместимых экспериментальных условий и аналитических способов. Это в значительной степени ограничивает использование эмпирических справочных баз данных, таких как Drug Matrix™ компании Iconix (муниципалитет), предназначенных для обозревателей FDA, и увеличивает вероятность неправильной интерпретации предоставленных данных, что, в свою очередь, повышает риск направления потенциально токсичных соединений на клинические испытания и лекарственных средств на рынок сбыта.
В соответствии с настоящим изобретением сбор и анализ данных был выполнен с использованием двух наборов данных, опубликованных в вышеуказанной научной работе Sørlie. Образцы в наборе данных 122 индивидов представляли собой профили экспрессии отдельных опухолей из локально запущенных агрессивных форм рака молочной железы: 115 инвазивных опухолей, четыре нормальных образца и три незлокачественные опухоли. Набор данных 295 индивидов не включал нормальных образцов. В первоначальном исследовании 84 образца были распределены в пять неравных групп для неконтролируемой иерархической кластеризации центрального набора из 540 генов. Пять кластеров получили названия: люминальный типа А и В, базальный, ERBB2+, нормальный; каждый кластер отличался характерными наборами сверхэкспрессированных генов. 38 образцов невозможно было классифицировать с включением в любую из пяти групп. В исследовании было использовано десять подобных матриц кДНК, характеризующихся несколько разным числом генов, при наличии >8000 признаков в каждой.
В результате осуществления способа по настоящему изобретению было установлено, что 5797 уникальных генов на матрицах (определенных в соответствии с идентификацией Locus Link) были общими для всех 122 образцов. Гетерогенность образцов была поразительной. Ни один ген не был значительно сверхэкспрессирован на статистически значимом уровне у всех индивидов, только три гена были общими в четырех нормальных образцах при расхождении в 2,5 раза. При таком незначительном перекрывании образцов прямое картирование отдельных файлов в сетях было неэффективным: каждый образец характеризовался уникальным распределением путей и плохо сравнимыми сетями. Разработанный ранее способ сравнения характерных признаков сетей эффективен для относительно небольшого числа образцов и подобной генетической среды и не применим для набора из 122 файлов, содержащих полный геном человека, с неизвестной генетической средой.
Способ по настоящему изобретению также позволяет определить функциональную связь кластеров Sørlie в контексте сетей и путей, так как кластеризация была выполнена статистическими методами и функциональный анализ выходил за пределы объема выполненного исследования. Характеры экспрессии в кластерах образцов были более синхронны, чем у произвольно отобранных образцов, но вариабельность внутри кластеров была по-прежнему высокой (фиг.7 и 8). На фиг.7 показана последовательная кластеризация наборов данных 122 индивидов в семь дескрипторных групп. На фиг.7А показано распределение всех 122 образцов в 18 кластеров, и на фиг.7В изображена схема последовательной кластеризации. На фиг.8 показана сеть для STAT1+, P1AU+ и FN1+ групп в виде входного списка. На фиг.8А показано картирование сети образцов из базального кластера Sørlie. Необходимо отметить большое число зонированных генов, таких как IP10 (обведены красной линией).
Таким образом, был использован способ кластеризации, занимающий центральное место в современной теории биологических сетей и предполагающий, что биологические процессы являются дискретными и выполняются относительно небольшими модулями физически взаимосвязанных белков. Для правильного выполнения своей функции белки из одного модуля, по-видимому, синхронно экспрессированы, что можно проследить в профилях микроматрицы. В случае болезни такие модули могут быть экспрессированы не так, как в нормальном состоянии. Такие группы синхронно экспрессированных групп могут быть небольшими из-за элементного состава и ограничений топологии сетей. Указанные небольшие группы функционально связанных генов, которые необязательно высокоэкспрессированы, разбросаны по большим матрицам и, вероятно, не могут быть обнаружены при помощи радиолокационного сканирования способов кластеризации. При идентификации таких синхронно экспрессированных модулей могут быть созданы характерные признаки сети, которые должны быть типичными для кластеров образцов и, по-видимому, коррелировать с биологическими и клиническими фенотипами. Общая последовательность операций кластеризации показана на фиг.2.
В первоначальном исследовании было использовано десять сходных, но все же отличающихся матриц, и число генов, общих для всех образцов, зависело от матрицы. Для каждого общего гена вычисляли коэффициент корреляции для определения зависимости от матрицы, который определяет разницу между средними значениями для каждой матрицы и разброс между матрицами с поправкой на величину группы (матрицы), и выбирали максимальное значение для каждой группы. i - число генов на матрице; r; k1 и k2 - тип матрицы/пула генов; jk - число образцов на матрице типа k; # jk - число образцов на матрицах типа k; si jk - сигнал гена i в образце jk; µ(si jk) - среднее значение сигналов гена i на матрицах типа k; σ(si jk) - стандартное отклонение для сигналов гена i в образцах на матрицах типа k.
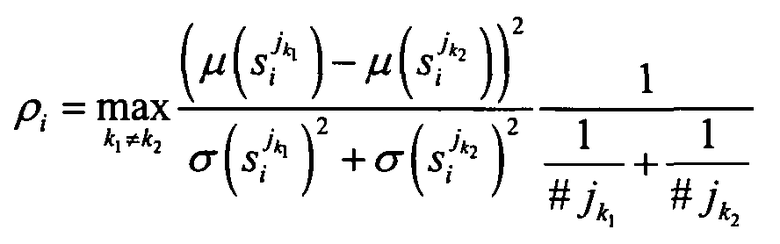
При увеличении pi корреляция с матрицей возрастает. Гены с pi>15 сильно зависят от матрицы; таким образом, в первоначальном анализе было исключено 869 зависящих от матрицы генов.
На следующем этапе значения распределения экспрессии сравнивали во всем наборе из 122 образцов для каждого гена, при этом было отмечено, что экспрессия некоторых генов имела тенденцию к кластеризации на двух уровнях или модах. Другими словами, функция экспрессии, по-видимому, является скорее дискретной при наличии двух пиков, чем непрерывной. Такая бимодальность была вычислена для каждого гена в наборах данных для 122 и 295 индивидов. Ниже приведены параметры, использованные при выполнении анализа.
- Для каждого гена было выбрано относительное пороговое значение экспрессии, и сигналы всех образцов были распределены в группы Li (ниже пороговых значений) и Ui (выше пороговых значений).
- Для каждого гена было вычислено среднее значение для группы L
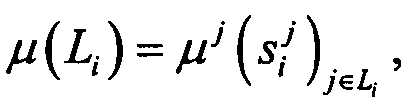
среднее значение для группы U
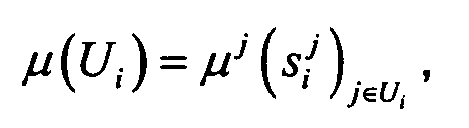
и сумма квадратов разностей между сигналами отдельных генов и средние значения в обеих группах
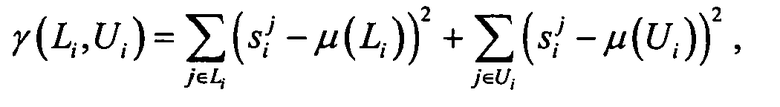
где i - число генов; j ∈ Li - число образцов, в которых сигналы для гена i помещены в группу Li; j ∈ Ui - число образцов, в которых сигналы для гена i помещены в группу Ui; si j - сигнал гена i в образце j.
- Пороговое значение для каждого гена было выбрано на основании минимизации γ(Li,Ui).
- Полученное распределение представляло собой пороговое значение, равное половине суммы средних значений для групп L и U.
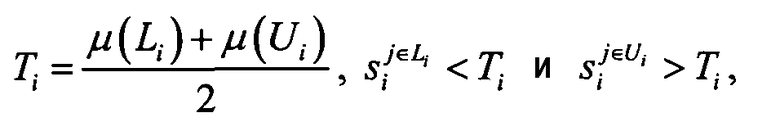
- Затем для каждого гена был вычислен коэффициент бимодальности.
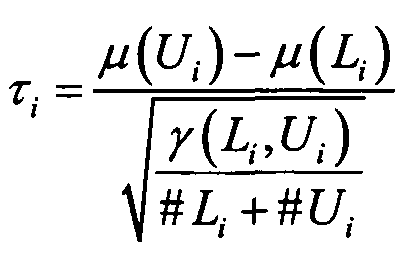
где #Li - число сигналов в группе Li, #Ui - число сигналов в группе Ui. Ген считается бимодальным, если τi>2,596. Было обнаружено 1976 таких генов; примерно 40% не зависящих от матрицы генов. Типичный бимодальный ген, GRB7, имеет τGRB7=4,762 и приведенное ниже распределение в образцах (фиг.4).
Значения экспрессии в четырех нормальных образцах были выделены в кластере с одним пиком (модой), близко расположенным для всех образцов, - совершенно неслучайное событие (фиг.4А). В таких случаях бимодальность является условной (связанной с раком молочной железы), характер экспрессии гена в случае болезни существенно отличается от нормального. Бимодальные специфичные для болезни гены могут быть весьма важны для понимания механизма болезни, а также в качестве источника биомаркеров и мишеней для лекарственного средства. Пул бимодальных генов, специфичных для рака молочной железы, был выделен следующим образом:
- Вычисляли среднее значение для нормальных индивидов
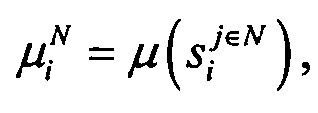
где N = число образцов, полученных у нормальных индивидов
- В случае
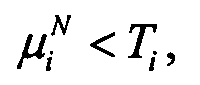
значения экспрессии генов преобразовывали в соответствии с уравнением
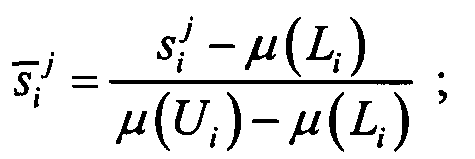
в противном случае
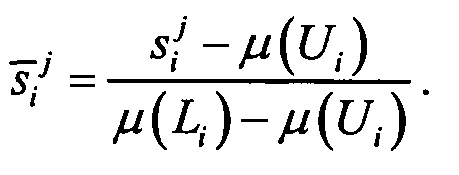
Тогда
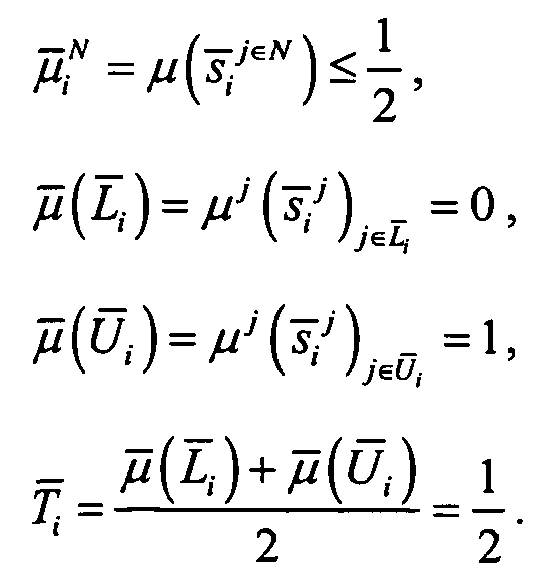
- Для каждого гена вычисляли стандартное отклонение для преобразованных значений в нормальных образцах
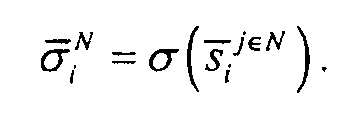
Ген считался зависящим от рака молочной железы, бимодальным геном, если его бимодальность определялась коэффициентом бимодальности τi>2,596, и если
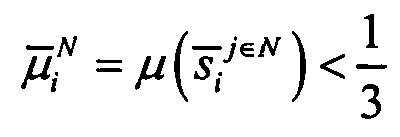
и
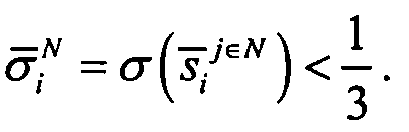
В общей сложности было идентифицировано 949 бимодальных генов, зависящих от рака молочной железы.
Затем сигналы нормализовали путем преобразования бимодальности для четкого отделения образцов индивидов с сигналами, соответствующими пику нормального состояния, от образцов индивидов с сигналами, соответствующими пику болезни. Пик, соответствующий норме, был обозначен как 0, и другой пик был обозначен как 1. Указанная нормализация необходима для минимизации расстояния между генами, разделяющими индивидов одинаковым образом. Указанная минимизация имеет важное значение. Рассмотрим гипотетический случай с тремя бимодальными генами (фиг.4А и 4В), в котором эксперименты представлены на ОХ, исходные сигналы на OY. Расстояния между генами А и С гораздо меньше, чем между А и В. Расстояние можно определить как сумму различий между сигналами у всех индивидов. Расстояние А-С равно 40; расстояние А-В >200. Однако в случае преобразования сигналов гены А и В позволяют разделить индивидов аналогичным образом, но ген С ведет себя совершенно по-другому. После нормализации исходных сигналов, как показано на фиг.5А, сигналы выглядят, как показано на фиг.5В. В принципе такое преобразование применимо к распределению экспрессии любого гена. 122 образца представлены с указанием исходных и преобразованных сигналов соответственно на фиг.5С и 5D.
Как было отмечено выше, в научной литературе описано примерно 1000 генов, имеющих отношение к раку молочной железы (с особыми SNP, мутациями, реаранжировками генома, амплификациями генов и метилированием промоторов; большим числом РНК и белков, связанных с определенными причинами возникновения болезни). Из вышеуказанных генов 56 генов были признаны генетическими маркерами рака молочной железы, 35 из указанных генов были отмечены на матрице. Пятнадцать из указанных 35 генов оказались бимодальными генами, зависящими от рака молочной железы. Одним из пятнадцати генов был ген ERBB2 из известного множества, связанного с раком молочной железы.
На следующем этапе выполнения анализа вычисляли близкие соседние группы для каждого из указанных 35 генов; близкие соседние группы определяются как гены с наиболее сходным поведением экспрессии. Указанные группы могут характеризоваться двумя параметрами: 1) гены в группе расположены близко друг от друга на векторе распределения сигналов у 122 (или 295) индивидов и 2) гены в группе расположены близко друг от друга в распределении (фиг.6). Для каждого такого гена были вычислены расстояния между его вектором и векторами всех других генов у 122 индивидов;
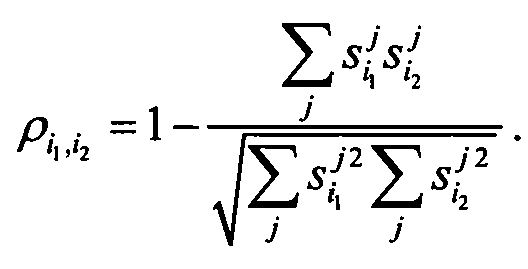
io = индекс гена у 122 индивидов,
j = номер образца,
si j = сигнал гена i в образце j после преобразования.
Расстояния между генами изменялись от 0 до 2. Гены считались соседними после преобразования, если значение р было небольшим (0,0-0,4) и было мало генов с одинаковыми значениями р относительно главного распределения, как в случае группы GRB7 (фиг.6А). Два других случая показаны на фиг.6В и 6С. В случае HMGA1 нельзя идентифицировать ни одной близкой группы. Самый близкий ген к HMGA1 находится на расстоянии 0,3, и главное распределение находится рядом с указанным геном. Расстояние от ближайшего гена до рассматриваемого гена является небольшим (<0,05), двенадцать других генов расположены на расстоянии 0,2, и ближайшая группа не определена. На фиг.6 показано, что ОХ означает относительные расстояния от рассматриваемого гена до всех 4928 генов, не зависящих от матрицы, и OY означает число генов. На фиг.6А показана четко выраженная близкая группа около ERBB2/GRB7 (в окружности); на фиг.6В отсутствует близкая группа для HMGA1 в качестве рассматриваемого гена; и на фиг.6С показана неопределенная группа для JAG2 (в окружности).
Девять генов из 35 генов соответствовали группам синхронно экспрессированных генов. Весьма удивительным является то, что все девять генов относились к набору из пятнадцати бимодальных генов, специфичных для рака молочной железы, и не к одному из 21 гена, не являющегося бимодальным геном. Четыре из девяти генов, по-видимому, входили в одни и те же группы. 29 генов, включая девять рассматриваемых генов, были идентифицированы и разделены на 7 групп (таблица 2). Важно отметить, что восемнадцать из 29 генов были бимодальными генами, зависящими от рака молочной железы.
Синхронная экспрессия в группах показана на фиг.5. Указанная экспрессия является неслучайным событием. Вероятность того, что случайный набор из 29 генов будет содержать восемнадцать бимодальных генов, составляет 4,5е-7. Гены в группах были тесно связаны в функциональном отношении и могли быть собраны в небольшие сети.
Каждая из семи групп близко расположенных соседних генов позволяет разделить 122 образца на + и - кластеры в зависимости от близости преобразованных значений экспрессии к 0 или 1 при пороговом значении, равном 0,5. Указанная процедура может быть выполнена последовательно, начиная с группы, имеющей наибольшее значение τ(L,U). В наборах данных 122 индивидов группы ERBB2 и ESR1 имеют наибольшее значение τ(L,U), и кластеризация может быть начата с любой группы. Группа ESR позволяет распределить наборы данных на кластеры ESR+ и ESR-; они делятся на четыре кластера ERBB2: ERBB+/ESR+; ERBB-/ESR+; ERBB-/ESR+; ERBB-/ESR- и так далее. На каждом этапе повторно вычисляют параметры бимодальности для каждого кластера. Деление прекращают, когда в группе остается менее 5 индивидов или когда в кластере не остается бимодальных генов.
В конечном счете, весь набор данных будет разбит на кластеры, характеризующиеся уникальным статусом каждой из семи групп дескрипторов (фиг.6). Например, любой образец в кластере 8 может быть представлен в виде цепочки: ERBB2+/ESR1+/KRT5+/СЕАСАМ-/STAT1-/PLAU-. Полную двумерную карту можно рассматривать в качестве матрицы из восемнадцати кластеров, умноженных на семь групп.
Не каждая группа позволяет окончательно выделить каждый кластер на каждом этапе. Например, статус экспрессии группы 5 (PLAU) не позволяет разделить кластеры 12 и 13, но является ключевым при разделении кластеров 8 и 9. Поэтому цепочка последовательного двустороннего анализа позволяет однозначно определить каждый кластер и отличить его от других кластеров. Два отличительных признака такой последовательной кластеризации имеют важное значение для применения указанного способа для диагностики. Во-первых, фактически все образцы были распределены в один или другой кластер по сравнению с одной третью неклассифицированных образцов при кластеризации методом Sørlie. Во-вторых, образцы в кластерах образуют конкретные сети и отображают соответствие с путями благодаря синхронной экспрессии в группах (фиг.7). Последовательная кластеризация может быть выполнена в другом порядке, например, начиная с ESR1. В таком случае могут быть получены пятнадцать кластеров, которые тесно связаны с вышеуказанными восемнадцатью кластерами. Для оценки данного способа применительно к независимому набору образцов вышеуказанные семь групп были использованы в отношении набора данных 295 индивидов, представленного на сходной, но отличающейся матрице кДНК. Все 295 образцов были разделены на 18 кластеров при помощи такой же схемы последовательной кластеризации. Из набора данных 295 индивидов были выделены две дополнительные группы (64 гена) (как и ожидалось для более крупного набора образцов).
Кластеры, выявленные в данном исследовании, являются биологически значимыми, как можно видеть по результатам картирования данных кластеров, относящихся к путям и сетям, например, набора данных 295 индивидов из исследования Sørlie. Во-первых, исходные кластеры Sørlie преобразованы в сигналы и применены к девяти группам, определенным в исследовании для картирования. Выбранные группы индивидов, относящиеся к базальному кластеру, были использованы в качестве входного списка для создания сетей. Большинство образцов базального типа характеризовалось сверхэкспрессией групп STAT1, PLAU и FN1. Однако для некоторых образцов в кластере указанные группы отличались пониженной экспрессией. В случае схемы, предложенной авторами настоящего изобретения, кластеры 1-6 также характеризуются наличием групп PLAU-FN-STAT1, но деление является более ясным, например, было произведено четкое разделение на FN1+ (кластеры 1-4) и FN1- (кластеры 5, 6).
Другой пример представлен на фиг.9. Полный образец кластера 9 из исследуемого набора данных 295 индивидов был отображен на картах путей MetaCore™, как показано на фиг.9А. Экспрессия GRB7, ERBB4 и ERBB2 сопоставимо понижена во всех образцах данного кластера. Все члены кластера 9 соответствуют кластеру люминального типа В, выявленному Chang et al. в том же наборе данных 295 индивидов. Все образцы кластера люминального типа В отображены на таких же картах путей (фиг.9А). В данном случае профили экспрессии ERBB2, GRB7 и ERBB4 не сопоставимы для всех образцов: одни гены характеризуются пониженной экспрессией и другие гены характеризуются повышенной экспрессией.
Также была произведена оценка среднего времени выживания 72 индивидов, страдающих локально запущенным раком; были использованы индивиды, обследованные Sørlie в первоначальном исследовании. Были включены только кластеры, содержащие >5 образцов индивидов. Исследование показывает, что среднее время выживания значительно колеблется от 40 месяцев для кластера 9 до 80 месяцев для кластера 14 (фиг.9В).
Как было описано выше, способ создания признаков является новым способом реконструкции специфичных для состояния сетей генетически гомогенных наборов данных, таких как токсикогеномные данные экспрессии генов, полученные при использовании животных моделей и культур клеток. Данный подход состоит из вычисления и сравнения подграфов (модулей), на которые наиболее дифференцированно влияет лекарственное средство, с использованием комбинации биологических и топологических критериев. Условные молекулярные данные (экспрессия специфичных для лекарственного средства генов) затем отображают в указанных подграфах и производят статистическую оценку (фиг.15).
Способ характерных признаков сетей был использован в отношении данных, полученных в результате выполнения нескольких токсикогеномных экспериментов, которые представляли собой реакцию на лечение лекарственными средствами крыс, в сочетании с FDA. Указанный набор данных содержит микроматрицу экспрессии генов в печени крыс, которых подвергали воздействию трех лекарственных средств: фенобарбитал, местранол и тамоксифен. Указанные данные были получены при помощи специальных двухканальных матриц кДНК. Каждое лечение включало пять биологических повторов, при этом анализировали набор из пяти образцов, полученных у животных, не подвергавшихся лечению. Биологическая модульность предполагает, что клеточные функции выполняются группами белков, временно действующих вместе. Пространство взаимодействия белков в модуле является статистически более насыщенным по сравнению с взаимодействиями между модулями, поэтому модули могут быть вычислены и визуализированы в виде подграфов в более крупных сетях.
Был получен предварительно вычисленный полный набор перекрывающихся канонических путей, выделенных из 450 карт сигнальных и метаболических путей, созданных вручную с использованием общей базы данных MetaCore™. Указанные пути затем были использованы в качестве основы для картирования экспериментальных наборов данных, а также для вычисления сетевых модулей, наиболее релевантных к конкретным путям (фиг.15). На картах указано примерно 3000 белков и более 2000 перекрывающихся путей человека. На указанных картах идентифицированы типичные входные сигналы (обозначены прямоугольными стрелками). Сетевые модули были созданы в виде цепей взаимодействий/реакций, инициируемых одним из входных сигналов и достигающих последнего белка, идентифицированного в каноническом пути; завершениями цепи часто являются факторы транскрипции и их мишени. Указанные пути представляют собой линейные модули передачи сигналов в клетках, обычно часто сигнальные каскады между вторичными мессенджерами и факторами транскрипции. Благодаря ветвлению (вариабельности перекрывающихся путей) во многих сигнальных каскадах общее число таких цепей является довольно большим - около 12000. Указанное число было ограничено выбором путей, присутствующих по меньшей мере на двух разных картах. 2200 консервативных фрагментов путей были предварительно собраны и хранились в файле следующего формата:
Идентификация пути: идентификация сети 1 → идентификация сети 2 → идентификация сети 3 →…
Идентификации сетей были распределены по спискам соответствующих идентификаций генов и файлы были введены в общую базу данных в виде списков генов. Затем были вычислены относительные расстояния между образцами в пространстве экспрессии генов отдельных путей. Подобный подход проиллюстрирован на фиг.11 и 12. В качестве примера использован путь с тремя генами (представление большего пути потребует проецирования многомерного пространства). Образцы представлены точками (векторами) в указанном пространстве в соответствии со значениями экспрессии. После импорта всех данных была использована метрическая система для вычисления расстояний между образцами в каждом пути. Например, при наличии пути, определяемого как:
Путь 1: [ген 1, ген 2, ген 3, ген 4]
и данных для каждого гена, полученных из набора данных экспрессии генов,
вычисляли расстояния для всех комбинаций образцов  , где k=2 и n = общее число образцов. Из вышеизложенного следует, что порядок расположения пар образцов не имеет значения (то есть образец 1/образец 4 имеет такое же значение, что и образец 4/образец 1). Возможные комбинации путей: образец 1/образец 2; образец 1/образец 3; образец 1/образец 4; образец 2/образец 3; образец 2/образец 4; образец 3/образец 4. Рассматриваются только расстояния между однозначными образцами, так как функция расстояния является симметричной: расстояние между образцом 1 и образом 3 аналогично расстоянию между образцом 3 и образцом 1. Для вычисления расстояний между образцами были использованы функции эвклидова расстояния и расстояния Пирсона.
, где k=2 и n = общее число образцов. Из вышеизложенного следует, что порядок расположения пар образцов не имеет значения (то есть образец 1/образец 4 имеет такое же значение, что и образец 4/образец 1). Возможные комбинации путей: образец 1/образец 2; образец 1/образец 3; образец 1/образец 4; образец 2/образец 3; образец 2/образец 4; образец 3/образец 4. Рассматриваются только расстояния между однозначными образцами, так как функция расстояния является симметричной: расстояние между образцом 1 и образом 3 аналогично расстоянию между образцом 3 и образцом 1. Для вычисления расстояний между образцами были использованы функции эвклидова расстояния и расстояния Пирсона.
Евклидово расстояние:
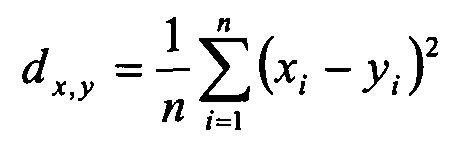
dx,y - расстояние между образцом х и образцом y,
n - число генов в пути,
i - ген,
xi - величина экспрессии гена i в образце х,
yi - величина экспрессии гена i в образце y.
Расстояние Пирсона:
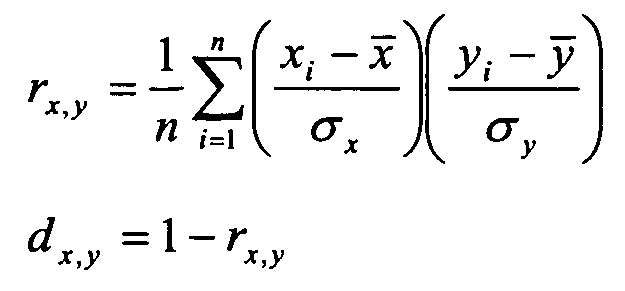
dx,y - расстояние между образцом х и образцом y,
rx,y - коэффициент корреляции для образца х/образца y,
n - число генов в пути,
i - ген,
xi - величина экспрессии гена i в образце х,
yi - величина экспрессии гена i в образце y,
х - среднее значение экспрессии для образца х,
y - среднее значение экспресии для образца y,
σх - стандартное отклонение значений экспрессии в образце х,
σy - стандартное отклонение значений экспрессии в образце y.
Для каждой функции расстояния были вычислены расстояния между образцами для всех путей:
Путь 1
…
Путь n
На фиг.11А показано вычисление расстояний между образцами в пространстве экспрессии генов пути. Показан гипотетический путь, состоящий из трех белков А, В и С. Образцы представлены в виде точек в трехмерном пространстве экспрессии генов. Следует отметить группирование образцов. Пути, соответствующие экспрессии генов (кратное значение изменений по сравнению с контрольным образцом), показаны стрелками. На фиг.11В показана кластеризация образцов в пространстве экспрессии генов пути. Для пути 1 расстояние между образцами, соответствующее одинаковому лечению, в статистическом отношении меньше расстояния, соответствующего разным способам лечения. Для пути 2 такое отличие не установлено.
На фиг.12 показано исследование перекрывания групп генов, определяющих различия в условиях лечения. На фиг.12(А) показано перекрывание генов, определяющих различия между лечением фенобарбиталом-местранолом и фенобарбиталом-тамоксифеном. Примерно 66% от общего числа генов, определяющих различия между фенобарбиталом-местранолом, позволяют также дифференцировать фенобарбитал-тамоксифен, и примерно 18% генов позволяют дифференцировать фенобарбитал и более высокие концентрации тамоксифена, но не дифференцируют фенобарбитал и более низкие концентрации тамоксифена. На фиг.12(В) показано перекрывание групп генов, определяющих различия между всеми вышеуказанными лечениями двумя лекарственными средствами.
В результате выполнения указанного способа получают матрицу расстояний между образцами в пространстве характера экспрессии генов отдельных модулей путей. Расстояния могут быть сгруппированы в соответствии с произвольно определенными типами образцов. Расстояния сравнивали у повторяющихся образцов, полученных у индивидов, которым назначали одинаковое лечение, и образцов, полученных у индивидов, проходивших разные курсы лечения. Для каждого пути была исследована статистическая гипотеза, выражающаяся в том, что среднее расстояние у повторяющихся образцов, полученных в результате выполнения аналогичного лечения, существенно отличается от среднего расстояния образцов, полученных в результате выполнения другого исследования. Если данная гипотеза находила подтверждение для пути, выполняли вычисления с целью определения двух конкретных лечений, при которых происходит кластеризация повторяющихся образцов, полученных у индивидов, проходивших одинаковый курс лечения, при этом расстояния, соответствующие разным способам лечения, имеют большее значение (фиг.11). Критерием успеха была идентификация характерных путей или групп путей, определяющих различия между реакциями на лечение, например, отличающими местранол от фенобарбитала. Выбранные пути группировали в кластеры на основании статистического t-критерия. На следующем этапе отбирали кластеры, которые позволяли установить четкое различие между двумя способами лечения. Например, пути, позволяющие установить различие между лечением местранолом и тамоксифеном, должны были удовлетворять условию, в соответствии с которым оба расстояния между повторным лечением местранолом и расстояния между повторным лечением фенобарбиталом должны быть меньше в статистическом отношении, чем расстояние, вычисленное при сравнении местранола с фенобарбиталом. В процессе отбора групп гены путей, определяющих различия между двумя конкретными способами лечения, были импортированы в общую базу данных MetaCore™ для последующего анализа.
На основании вышеописанного анализа было установлено:
- фенобарбитал по сравнению с тамоксифеном: 163 пути, содержащих в общей сложности 131 однозначный ген;
- местранол по сравнению с тамоксифеном: 3 пути, содержащих в общей сложности 15 генов; и
- местранол по сравнению с фенобарбиталом: 54 пути, содержащих в общей сложности 151 ген.
Не было обнаружено путей, определяющих различия между реакциями на разные концентрации тамоксифена. Другим важным наблюдением является то, что число путей, отличающих местранол от тамоксифена, значительно меньше, чем число путей, отличающих фенобарбитал от лечения местранолом и тамоксифеном. Не было обнаружено путей, определяющих различия между реакциями на разные концентрации тамоксифена. Местранол и тамоксифен имеют сходные структуры и биологические мишени (оба являются лигандами рецептора эстрогена); фенобарбитал отличается от вышеуказанных двух лекарственных средств в структурном отношении, а также типом воздействия (см. таблицу 3 для ознакомления с кратким обзором результатов статистического анализа).
t-Критерий по сравнению с анализом кластеризации
Списки генов из созданных групп (объединения генов в отличающихся путях) затем импортировали в текущую версию общей базы данных для исследования связности сетей. Применяли алгоритм создания сетей с ”прямым взаимодействием”. Указанный алгоритм позволяет вести поиск всех взаимодействий, соединяющих представляющие интерес гены друг с другом (фиг.16А). На сети налагали данные экспрессии, которые были превращены в логарифмические соотношения уровней экспрессии генов у животных, подвергаемых лечению и не подвергаемых лечению. Гены с повышенной экспрессией отмечены красными кружками и гены с пониженной экспрессией отмечены синими кружками. Сети содержали значительное число генов с отрицательной корреляцией разных способов лечения. Интересно отметить, что кратные значения изменения для большинства указанных генов являются относительно низкими, то есть находятся в пределах 10%-30%.
Однако указанные гены группируются во взаимосвязанную сеть с синхронной дифференцированной реакцией на разные способы лечения. Сети реакций на лекарственные средства, созданные способом по настоящему изобретению, могут характеризоваться разными параметрами. Например, содержание генов/белков можно проанализировать в таких функциональных категориях как канонические карты путей, способы GO, предварительно заданные сети (например, способы GeneGO St. Joseph, M.I.), а также категории для заболеваний и токсичностей. Подграфы характерных признаков сетей меньшего размера для генов, относящихся к отдельным способам (например, подграф митоза, фиг.16В), также могут быть представлены в качестве функционального дескриптора состояний. Распределение р-значений функциональных категорий можно рассматривать в качестве параметров сети, и функциональное расстояние между сетями может быть определено на основании числа совпадающих функциональных категорий и менее очевидных производных параметров. В соответствии с указанными распределениями сети являются по существу разными, хотя один из исходных наборов данных, использованных для создания сетей, не содержит отличий (профиль экспрессии фенобарбитала).
Следует отметить два важных преимущества способа сравнения сетей по сравнению со стандартными статистическими способами. Указанные способы проиллюстрированы на фиг.16С, на которой показана карта путей, получившая наивысшую оценку, путь CREB, для определения содержания генов в сети тамоксифена/фенобарбитала. Данные экспрессии фенобарбитала и тамоксифена были картированы и представлены гистограммами красного цвета (повышенная экспрессия на основании кратного изменения) и гистограммами синего цвета (пониженная экспрессия на основании кратного изменения). Для значений краткого изменения и Р-значений не было установлено пороговое значение. Картирование показывает удивительное соответствие точек данных в отдельных наборах данных путей. Почти все гены, реагирующие на фенобарбитал, отличались повышенной экспрессией, и гены тамоксифена отличались пониженной экспрессией. Большое число указанных генов необходимо исключить из анализа при помощи любой комбинации статистических способов, используемых в настоящее время для анализа микроматрицы. Например, слабое ограничение, включающее 1,4-кратное изменение и Р<0,1, оставляет только 5 из 32 экспрессированных генов на указанной карте (отмечены красными и синими квадратами на фиг.16С), что по существу исключает пути, подвергаемые разному воздействию, при выполнении двух способов лечения, описанных в анализе.
Для сравнения были идентифицированы 477 генов, подвергшихся наибольшему воздействию, путем выполнения стандартных испытаний по способу ANOVA и t-критерия (сравнение подвергнутых и не подвергнутых лечению животных) в двух одинаковых наборах данных. Указанные гены были отображены на картах путей в виде общих данных, таких как MetaCore™, после чего были отобраны статистически значимые карты. Карты, получившие наивысшую оценку, были сгруппированными метаболическими картами; на указанных картах были отображены определенные генетические токсические воздействия, такие как подавление цитохромов при метаболизме лекарственных средств, но при этом не был обнаружен по существу отличный характер экспрессии.
На фиг.13 показаны сети, образованные путями, по-разному реагирующими на лечение. На фиг.13А показана сеть с прямым взаимодействием, собранная из генов путей, различающих лечение местранолом и фенобарбиталом. Было картировано относительное изменение экспрессии у подвергнутых и не подвергнутых лечению крыс (логарифмические соотношения, в среднем для 5 повторов). Синими кружками отмечены гены с пониженной экспрессией, и красными кружками отмечены гены с повышенной экспрессией. На фиг.13В показаны сети, созданные из генов, выделенных из путей, различающих фенобарбитал и местранол. Следует отметить, что оба сравнения содержат значительное число отрицательно коррелированных генов.
На фиг.14 показана бикластеризация генов, которые были картированы в отношении путей. Столбцы (красные - фенобарбитал по сравнению с местранолом, зеленые - фенобарбитал по сравнению с тамоксифеном (250 нМ), синие - фенобарбитал по сравнению с тамоксифеном (500 нМ), черные - местранол по сравнению с тамоксифеном (250 нМ) и розовые - местранол по сравнению с тамоксифеном (500 нМ)) служат для обозначения генов, идентифицированных способом по настоящему изобретению в качестве генов, различающих лечение разными лекарственными средствами.
На фиг.16А показаны сети с ”прямым взаимодействием” (DI), собранные из генов, выделенных из большинства путей между профилями тамоксифена и фенобарбитала. Синими кружками обозначены гены с пониженной экспрессией; красными кружками обозначены гены с повышенной экспрессией. На фиг.16В представлен подграф из сети с прямым взаимодействием, соединяющей гены, относящиеся к способу GO исследования митоза. На фиг.16С показана получившая наивысшую оценку карта пути CREB для генов в сети А. Экспрессия генов была картирована без пороговых значений. Квадратными блоками отмечены гены в признаке, выраженном при кратном изменении >1,4 и Р<0,1. Красным квадратом обозначен фенобарбитал и синим квадратом обозначен тамоксифен в количестве 250 мг/кг. Следует отметить, что большинство генов в обоих наборах характеризуется высоким соответствием знака экспрессии, относительным уровнем в пути и уровнями предварительно установленного порогового значения (FC<1,4; Р>0,1).
На фиг.20 показан управляющий интерфейс исходных данных для модуля или пакета программного обеспечения для анализа токсичности. Пользователи могут получить стандартный набор загруженных и обработанных общедоступных данных, полученных из CEBS, GEO и других источников, и могут импортировать внутренние данные и коммерческие базы данных. Файлы организованы в виде иерархического дерева. Новые файлы загружаются при помощи многоформатного анализатора. Общий анализатор импортирует микроматрицу, данные экспрессии гена SAGE, протеомики и siРНК. Анализатор метаболизма импортирует большое количество экспериментальных данных по обмену веществ (значения CAS, символы эмоций, брутто-формулы). Анализатор взаимодействий импортирует специальные данные взаимодействия (дрожжевой гибрид 2, раскрывающийся список, текстовые ассоциации и т.д.).
Последовательность операций по созданию характерных признаков сетей можно описать следующим образом. Во-первых, исходные данные экспрессии генов предварительно обрабатывают при помощи программного обеспечения анализа микроматрицы, такого как GeneSpring™, Resolver™ или ArrayTrack™, которые интегрированы с общей базой данных, такой как MetaCore™. Предварительная обработка данных статистическими способами включает нормализацию данных, оценку качества сигнала (например, отношение сигнал/шум для каждого гена), вычисление соотношения между подвергнутыми воздействию и не подвергнутыми воздействию тканями и выполнение анализа ANOVA между повторами для идентификации генов, которые в значительной степени характеризовались повышенной/пониженной экспрессией. Таблицы с полученными данными вводят в общую базу данных, где одновременно анализируют профили экспрессии, полученные в результате применения разных способов лечения. Гены фильтруют в отношении их значимости (р-значение) и кратного изменения. Полученные наборы генов, которые позволяют установить различие между подвергнутыми воздействию и не подвергнутыми воздействию образцами, могут быть картированы при помощи одного или нескольких из шести главных инструментов функционального анализа: канонические карты путей передачи сигналов и обмена веществ в клетках; клеточные процессы, определяемые функцией GO, функциональные процессы, определяемые онтологией GeneGo, категории болезней, категории токсичности и предварительно заданные метаболические сети. После картирования данных карты, сети и категории располагаются в определенном порядке с учетом их статистической значимости для наборов данных, которую вычисляют в виде р-значений с использованием гипергеометрического распределения. Карты или процессы, являющиеся общими для большинства способов лечения в категории токсичности отбирают для создания набора характерных путей/признаков для данного класса токсических веществ.
Конечной целью такого крупномасштабного анализа является идентификация наборов по-разному экспрессированных генов с учетом трех свойств:
- Гены должны быть функционально хорошо определены в отношении клеточных процессов, путей или релевантности к болезням или токсическим веществам.
- Гены должны образовывать различимые подсети (модули) в сетях.
- Гены должны быть общими для нескольких способов лечения. Указанные группы могут соответствовать или не соответствовать категориям токсичности.
Такие модули затем могут быть использованы в качестве характерных признаков сетей для данного типа токсичности, и на их основе могут быть определены профили экспрессии для новых соединений. Важное значение имеет требование образования указанными генами хорошо соединенных сетевых модулей, так как при помощи способов анализа сети на основе топологии могут быть обнаружены сходные характеры даже тогда, когда профили экспрессии кажутся различными. Как показано на фиг.17В, два набора генов А и В перекрываются только двумя генами. Однако указанные гены характеризуются значительным числом взаимодействий в одном сетевом модуле. Таким образом, два указанных набора обладают высокой степенью функционального/механистического сходства, определяемого как совокупность перекрывающихся ”значимых” взаимодействий. Взаимодействие считается значимым при наличии по меньшей мере одного значимого гена в конце цепи. Кроме того, указанные взаимодействия образуют когерентный сетевой модуль, который далее подтверждает функциональную связь двух указанных наборов. Такое сходство совершенно не обнаруживается при сравнении списков генов (например, генных признаков) для двух способов лечения. Указанные гены могут принадлежать или не принадлежать к одной и той же функциональной категории.
На фиг.17А показана последовательность операций для создания характерных признаков сетей из токсикогеномных данных. Профили экспрессии нескольких соединений с подобными токсикологическими профилями анализируют при помощи разных функциональных фильтров, отбирают общие функциональные категории и создают сети. На фиг.17В показано функциональное сходство между наборами генов, которые характеризуются небольшим прямым перекрыванием или отсутствием такого перекрывания. Наборы генов А (красный) и В (зеленый) содержат два общих гена (подчеркнуты), но все восемь краев сети обладают сходством.
Один вариант осуществления изобретения относится к созданию новых критериев для последовательности операций реконструкции сети и разработки статистических способов квантования профилей реакций на соединения на основе топологии сети и содержания генов. Такой подход позволяет анализировать сотни профилей экспрессии в пакетном режиме и выполнять сравнительные вычисления для всего объема токсикогеномных баз данных любого размера, включая CEBS, Tox Express™, DrugMatrix™ и внутренние справочные базы данных в компаниях, производящих лекарственные средства. Интерфейс модуля также содержит несколько вариантов параметров для создания специализированных библиотек характерных признаков сетей.
Созданные сети в значительной степени перекрываются и коррелируют с фенотипами реакции на лекарственные средства и могут соответствовать или не соответствовать категориям токсичности, существующим в настоящее время (активаторы макрофагов, пролифераторы пероксисомы, генотоксические соединения и т.д.). Сравнение сетей может быть произведено на трех главных уровнях с использованием следующих параметров:
- Сравнение содержимого - самый простой тип, который показывает процентное количество генов или белков, являющихся общими для совокупности сетей.
- Сравнение графов - данный инструмент позволяет обнаружить общие подсети (связанные графы) в совокупности сетей. Данный тип несколько отличается от сравнения содержимого, так как некоторые общие узлы могут не быть частью такого связанного графа.
- Глобальное статистическое сравнение - данный инструмент позволяет вычислить главные статистические свойства всего сетевого модуля и сравнить сети с учетом указанных свойств. Такие свойства включают среднее расстояние между узлами, диаметр сети, концентраторы сети, распределение узлов по степеням и т.д.
- Локальное статистическое сравнение. Еще одним типом статистического анализа, который может быть интересен для вычисления и сравнения способов лечения, является распределение по-разному экспрессированных генов с учетом окрестностей других узлов в сети. Данный инструмент представляет собой новую концепцию сравнения сетей и более подробно рассмотрен ниже.
Концепция локальных статистических данных сетей в качестве параметров сравнения показана на фиг.18. Для каждого узла в глобальной сети могут быть определены окрестности первой, второй, третьей и т.д. степеней путем прямого обращения к базе данных. Окрестности взаимодействия узла могут быть далее разделены на разные параметры, такие как взаимодействия в обратном и прямом направлениях; механизмы взаимодействия (фосфорилирование, связывание, ковалентная модификация и т.д.); мишени фактора транскрипции; мембранные рецепторы; пары киназа-фосфатаза и т.д. Соседние сети должны быть дифференциально обогащены экспрессированными генами для разных способов лечения, и степень относительного обогащения может быть определена количественным способом.
Статистическая значимость и ее соответствие функции является ключевым вопросом; данный параметр может быть оценен при помощи способов, используемых в настоящее время в MetaCore™ при вычислении р-значений для карт, процессов, заболеваний и токсичности. По существу все окрестности могут быть представлены в виде поднабора генов, и р-значения могут быть вычислены в виде вероятности пересечения произвольного набора такого же размера с набором дифференциально экспрессированных генов для определенного способа лечения. Кроме того, р-значения могут быть вычислены для компонентов, расположенных в обратном и прямом направлениях, для выявления предполагаемых источников и приемников для активированных процессов. Например, в сети, показанной на фиг.18А, узлы, расположенные в обратном направлении от узла Х, в основном характеризуются пониженной экспрессией, в то время как узлы, расположенные в прямом направлении, в основном характеризуются повышенной экспрессией. Подробный анализ позволяет выявить количественную меру статистической значимости такого сверхпредставления. Таким образом, каждый узел в сети будет характеризоваться определенным числом локальных статистических оценок (сетевых дескрипторов), показывающих потенциальное отклонение распределения дифференциально экспрессированных генов в его окружении. При картировании профилей экспрессии для разных способов лечения в сетях может быть произведено сравнение статистических оценок, что помогает оценить их сходство.
На фиг.18 показаны окрестности статистически значимой сети (ориентированный граф). На фиг.18А узел Х характеризуется наличием в обратном направлении первичного окружения с пониженной экспрессией (синий цвет) и в прямом направлении окружения с повышенной экспрессией. Статистические оценки для такого распределения могут быть вычислены при помощи гипергеометрического распределения. На фиг.18В показана действительная сеть. Статистически значимое распределение генов с пониженной экспрессией из индуцированного аллиловым спиртом профиля экспрессии находится в окрестностях киназы 1 Р70 S6. Гены, расположенные в обратном направлении, характеризуются главным образом пониженной экспрессией (синий цвет).
Сети с наибольшим сходством в отношении токсичности при использовании разных способов лечения могут быть сгруппированы в характерный набор для данного типа. Статистические инструменты для оценки наборов заранее определенных путей и сетей могут быть подвергнуты прямому сравнению для нескольких способов лечения. Это означает, что каждую сеть, созданную отдельным профилем экспрессии, необходимо сравнить со всеми другими сетями в отношении всех характеров экспрессии. На основании вышеописанных вычислений расстояния между сетями могут быть созданы таблицы сходства сетей с последующим выполнением иерархической кластеризации сетей. На фиг.19 показано сравнение характерных признаков сетей для лечения лекарственными средствами. На фиг.19А показано вычисление оценок сходства для всех сетей в отношении нескольких способов лечения; на фиг.19В показана иерархическая кластеризации для отбора групп сетей, обладающих наибольшим сходством при использовании способов лечения в данном классе; и на фиг.19С показана форма отчетов о токсичности соединения.
Необходимо выбрать кластеры более низкого уровня наиболее сходных сетей и проанализировать их состав. Кластеры со значительным разнообразием (70%-80% всех способов лечения в одном классе) выбирают в качестве входных данных для создания специфичных для лечения характерных признаков сетей. Например, сети А, В и Е выбраны в качестве наиболее сходных сетей на фиг.19А. Необходимо исследовать показатели разных типов с учетом их вклада в вышеуказанные параметры сходства. Затем специфичные в отношении лечения сети объединяют в характерные признаки сети для данного класса токсичности.
В соответствии с одним объектом настоящего изобретения модуль или пакет программного обеспечения, предназначенный для прогнозирования токсичности соединений, может контролировать профили экспрессии обязательных генов относительно предварительно заданной совокупности характерных признаков сетей. Наборы значимых карт, сетей, процессов и других функциональных категорий могут быть вычислены на основании профиля экспрессии лекарственного средства пользователя, который можно сравнить с библиотеками характерных признаков сетей при помощи инструментальных средств оценки сходства сетей. Профиль потенциальной токсичности соединения может быть представлен в табличной форме сходства для каждой сети. В интерфейсе пользователя сходство может быть выражено как в цифровой форме, так и визуально (фиг.19С). Набор интерактивных опций позволяет пользователю создавать табличную форму по типу токсичности.
В соответствии с одним вариантом осуществления изобретения модуль или пакет программного обеспечения, предназначенный для прогнозирования токсичности соединений, может определять стандартизированную последовательность операций анализа токсикогеномных данных, создавать отчеты и включать исходные данные реакции на лекарственное средство из общедоступных баз данных, предварительно созданных карт сетей и путей для процессов, относящихся к токсикологии, а также характерные признаки сети и модули, характерные для разных типов токсичности. Исходные токсикогеномные данные экспрессии генов могут быть использованы для создания характерных признаков сетей и в качестве справочных данных. Обработанные данные (пути, категории и характерные признаки сети) могут быть использованы в качестве библиотеки функциональных дескрипторов и в качестве инструмента для картирования экспериментальных профилей токсичности для новых соединений.
В соответствии с другим вариантом осуществления изобретения модуль или пакет программного обеспечения, предназначенный для прогнозирования токсичности соединений, может включать любые или все доступные наборы токсикогеномных данных (микроматрицы, SAGE, протеомику, метаболомику), импортированные в его базу данных при помощи существующих анализаторов данных, после чего данные предварительно обрабатывают (нормализация экспериментальных данных, определение соотношения лечения и контроля, вычисление р-значений для определения согласованности образцов), аннотируют, копируют в виде дерева в администратор данных и используют в качестве справочных данных. Интуитивно-понятный модуль оперативной помощи позволяет потребителям загружать и обрабатывать оригинальные базы данных и перекрестно анализировать общедоступные, внутренние и запатентованные базы данных. При помощи универсального анализатора могут быть импортированы комплиментарные молекулярные данные почти любого типа (протеомика, метаболомика, анализы siРНК, скрининг содержимого большого объема и т.д.) и специализированные данные взаимодействия белков (анализы дрожжевых гибридов 2, эксперименты по иммунопреципитации, коэкспрессия и т.д.).
При загрузке экспериментальных наборов данных содержание генов может быть согласовано со специализированными наборами генов, уже загруженными в общую базу данных, и р-значения могут быть вычислены в виде вероятности соответствия такого же числа генов из произвольного набора для сопоставления идентификации генов в данной категории. Такое распределение р-значений для набора данных по функциональным категориям может служить в качестве количественных дескрипторов для набора данных, и профили реакций на разные лекарственные средства могут быть сравнены и кластеризованы на основании таких функциональных дескрипторов. Последовательность операций для исходного уровня функционального анализа показана на фиг.20А и 20В для трех профилей экспрессии генов крыс на микроматрице под воздействием цисплатина, этопозида и цисплатина (Johnson & Johnson (J&J), New Brunswick, N.J., данные в CEBS). Данные экспрессии могут быть также отображены в биологических сетях, предварительно созданных для процессов передачи сигналов и обмена веществ с использованием содержания генов в папке категорий в качестве входного списка. Соответствие идентификаций генов из файла представления реакций на воздействие лекарственного средства с идентификацией генов, визуализированных в сетях, обозначается цветом, и для каждой сети вычисляется р-значение (фиг.20С). Распределение генов с повышенной и пониженной экспрессией в предварительно заданных сетях может быть определено количественно в виде топологии сети и может быть также использовано в качестве функционального дескриптора.
Может быть создан целый ряд сетей аналогично сети для воспаления, показанной на фиг.20. После компоновки категорий токсичности могут быть созданы сети токсичности и интегрированы в модуль или пакет программного обеспечения для прогнозирования токсичности соединений (фиг.20А). Каждый файл соединений может быть отображен в сетях токсичности, созданных для категорий токсичности, и сети могут быть сравнены при помощи р-значений и инструментов на основе топологии для характерных признаков сетей. Поэтому все профили соединений могут быть каталогизированы с несколькими функциональными дескрипторами, после чего данные хранят в качестве справочных данных для оценки профилей экспрессии новых соединений.
В результате выполнения данного комплексного исследования каждое соединение может быть охарактеризовано рядом количественных функциональных параметров и для каждого соединения может быть вычислена общая оценка токсичности (таблица 5). Чтобы определить для профиля соединения определенную потенциальную токсичность, целый ряд функциональных индикаторов должен находиться в пределах токсичности данного типа. Пределы могут быть определены эмпирически в процессе выполнения анализа наборов данных методом J&J (New Brunswick, NJ) и с помощью других общедоступных источников. На основании окончательных значений может быть вычислена общая оценка. Вместо или помимо общей оценки может быть определен характер функциональных дескрипторов для классификации токсичности.
В соответствии с одним вариантом осуществления изобретения модуль или пакет программного обеспечения, предназначенный для прогнозирования токсичности соединений, может быть создан в виде аналитической машины для функционального анализа данных, относящихся к токсичности соединений. Модуль или пакет программного обеспечения не может быть ни универсальной платформой для биоинформатики, ни пакетом программ для анализа статистических данных, ни системой организации исходных лабораторных данных, но все указанные инструменты могут быть необходимы для современных крупномасштабных экспериментальных исследований и могут быть затребованы и использованы пользователями модуля или пакета программного обеспечения. Такие инструменты могут включать: программное обеспечение для анализа микроматриц; информационные технологии обработки медицинских/лабораторных данных; протеомику и метаболомику.
В соответствии с одним вариантом осуществления изобретения модуль или пакет программного обеспечения, предназначенный для прогнозирования токсичности соединений, может представлять собой универсальную платформу для токсикогеномных исследований и, таким образом, может производить обмен данными с разными современными информационными средами. Тремя основными стандартами для обмена биологическими данными являются SBML, PSI-MI и BioPax.
В соответствии с другим вариантом осуществления изобретения модуль или пакет программного обеспечения, предназначенный для прогнозирования токсичности соединений, может производить обмен данными со сторонними инструментальными средствами и информационными системами, используя API (интерфейс прикладных программ) для обмена информацией.
Система, предназначенная для определения кластеризации генов и характерных признаков сетей, и ее компоненты, такие как базы данных и инструментальные программные средства, могут действовать в результате использования программного обеспечения (например, С, С#, C++, Java или их комбинации), аппаратного обеспечения (например, одной или нескольких специализированных интегральных схем), программно-аппаратных средств (например, электронно-программируемой памяти) или любой комбинации указанных средств. Один или несколько компонентов системы разработки лекарственных средств могут находиться в одной компьютерной системе (например, подсистеме сбора данных), либо один или несколько компонентов могут находиться в отдельных компьютерных системах. Кроме того, каждый компонент может быть распределен по нескольким компьютерным системам и одна или несколько компьютерных систем могут быть взаимосвязаны.
Кроме того, в одной или нескольких компьютерных системах, включающих один или несколько компонентов системы разработки лекарственных средств, каждый из указанных компонентов может находиться в одной или нескольких ячейках памяти компьютерной системы. Например, разные части компонентов системы разработки лекарственных средств могут находиться в разных областях памяти (например, RAM, ROM, диск и т.д.) компьютерной системы. Одна или несколько компьютерных систем могут включать наряду с другими компонентами несколько известных компонентов, таких как один или несколько процессоров, система памяти, система запоминающих устройств для дисков, один или несколько сетевых интерфейсов и одна или несколько шин или других внутренних каналов связи, обеспечивающих соединение разных компонентов.
Система, предназначенная для определения кластеризации генов и характерных признаков сетей, может быть использована в компьютерной системе, описанной ниже со ссылкой на фиг.21 и 22.
Описанная выше система является иллюстративным вариантом системы, предназначенной для определения кластеризации генов и характерных признаков сетей. Такой иллюстративный вариант не ограничивает объем изобретения, так как любые другие многочисленные применения системы, предназначенной для определения кластеризации генов и характерных признаков сетей, например варианты содержащихся в них баз данных, также входят в объем настоящего изобретения. Ни один из пунктов приведенной ниже формулы изобретения не ограничивается каким-либо конкретным применением системы, предназначенной для определения кластеризации генов и характерных признаков сетей, за исключением тех случаев, когда такой пункт формулы изобретения включает ограничение, явно указывающее на конкретное применение.
Разные варианты осуществления изобретения могут быть выполнены в одной или нескольких компьютерных системах. Указанные компьютерные системы могут быть, например, универсальными компьютерами, в частности, на основе процессора Intel PENTIUM, процессоров Motorola PowerPC, Sun UltraSPARC, Hewlett-Packard PA-RISC или процессора любого другого типа. Следует отметить, что для определения кластеризации генов и характерных признаков сетей может быть использована одна или несколько компьютерных систем любого типа в соответствии с разными вариантами осуществления изобретения. Кроме того, система разработки программного обеспечения может быть расположена в одном компьютере или может быть распределена по нескольким компьютерам, присоединенным к коммуникационной сети.
Универсальная компьютерная система в соответствии с одним вариантом осуществления изобретения должна иметь конфигурацию, необходимую для функционирования любой описанной системы, предназначенной для определения кластеризации генов и характерных признаков сетей. Следует отметить, что указанная система может выполнять другие функции, включая связь в сети, и настоящее изобретение не ограничивается какой-либо определенной функцией или набором функций.
Например, разные объекты настоящего изобретения могут быть выполнены при помощи специализированного программного обеспечения, выполняемого в универсальной компьютерной системе 400, показанной на фиг.21. Компьютерная система 400 может включать процессор 403, присоединенный к одному или нескольким устройствам памяти 404, таким как дисковое запоминающее устройство, память или другое устройство для хранения данных. Память 404 обычно используют для хранения программ и данных во время работы компьютерной системы 400. Компоненты компьютерной системы 400 могут быть соединены при помощи соединительного механизма 405, который может включать одну или несколько шин (например, между компонентами, встроенными в тот же компьютер) и/или сеть (например, между компонентами, находящимися в отдельных компьютерах). Соединительный механизм 405 обеспечивает связь (например, передачу данных, команд) между компонентами системы 400. Компьютерная система 400 может также включать одно или несколько устройств ввода 402, например клавиатуру, мышь, трекбол, микрофон, сенсорный монитор, и одно или несколько устройств вывода 401, например принтер, дисплей, громкоговоритель. Кроме того, компьютерная система 400 может содержать один или несколько интерфейсов (не показаны), соединяющих компьютерную систему 400 с коммуникационной сетью (дополнительно или в качестве альтернативы соединительному механизму 405).
Система хранения данных 406, детально изображенная на фиг.22, обычно включает доступную для чтения и перезаписывающую энергонезависимую записывающую среду 501, в которой хранятся сигналы, определяющие выполнение программы процессором или обработку программой информации, хранящейся в среде 501. Такой средой может быть, например, память на дисках или флэш-память. Обычно в процессе работы процессор вызывает перемещение считываемых данных из энергонезависимой регистрирующей среды 501 в другую память 502, которая обеспечивает более быстрый доступ процессора к информации, чем память 501. Указанная память 502 обычно является энергозависимой оперативной памятью, такой как динамическая оперативная память (DRAM) или статическая память (SRAM). Указанная память может находиться в системе хранения данных 406 (не показана) или в системе памяти 404 (не показана). Процессор 403 обычно обрабатывает данные в памяти на интегральной схеме 404, 502 и после окончания обработки копирует данные в среду 501. Известны разные механизмы, которые управляют движением данных между средой 501 и памятью на интегральной схеме 404, 502, и настоящее изобретение не ограничено указанными механизмами. Настоящее изобретение не ограничено конкретной системой памяти 404 или системой хранения данных 406.
Компьютерная система может включать специально запрограммированные специализированные аппаратные средства, например специализированную интегральную схему (ASIC). Объекты настоящего изобретения могут быть выполнены в результате использования программного обеспечения, аппаратных средств, программно-аппаратных средств или любых их комбинаций. Кроме того, такие способы действия, системы, элементы систем и их компоненты могут быть использованы в качестве части вышеописанной компьютерной системы или независимого компонента.
Хотя компьютерная система 400 показана в качестве примера как один тип компьютерной системы, в которой могут быть выполнены разные объекты настоящего изобретения, следует отметить, что объекты настоящего изобретения не ограничены выполнением в компьютерной системе, показанной на фиг.21. Разные объекты настоящего изобретения могут быть выполнены в одном или нескольких компьютерах, имеющих другую архитектуру или компоненты, отличные от показанных на фиг.21.
Компьютерная система 400 может быть универсальной компьютерной системой, программируемой при помощи компьютерного языка программирования высокого уровня. Компьютерная система 400 может быть также использована вместе со специально запрограммированными специализированными аппаратными средствами. В компьютерной системе 400 процессор 403 обычно является стандартным процессором, таким как хорошо известный процессор класса Pentium компании Intel Corporation. Существует много других процессоров. Такой процессор обычно служит для выполнения операционной системы, которая может быть, например, операционной системой Windows 95, Windows 98, Windows NT, Windows 2000 (Windows ME) или Windows XP компании Microsoft Corporation, MAC OS System X компании Apple Computer, операционной системой Solaris компании Sun Microsystems или UNIX, доступной из разных источников. Могут быть использованы многие другие операционные системы.
Процессор и операционная система вместе определяют компьютерную платформу, для которой написаны прикладные программы на языках программирования высокого уровня. Следует отметить, что настоящее изобретение не ограничивается конкретной платформой компьютерной системы, процессором, операционной системой или сетью. Кроме того, специалистам в данной области должно быть очевидно, что настоящее изобретение не ограничивается конкретным языком программирования или компьютерной системой. Кроме того, следует отметить, что могут быть также использованы другие приемлемые языки программирования и другие приемлемые компьютерные системы.
Одна или несколько частей компьютерной системы могут быть распределены в одной или нескольких компьютерных системах (не показаны), соединенных с коммуникационной сетью. Указанные компьютерные системы могут быть также универсальными компьютерными системами. Например, разные объекты настоящего изобретения могут быть распределены в одной или нескольких компьютерных системах, конфигурация которых обеспечивает обслуживание (например, серверы) одного или нескольких компьютеров клиента или выполнение общей задачи в виде части распределенной системы. Например, разные объекты настоящего изобретения могут быть выполнены в серверной системе клиента, которая включает компоненты, распределенные в одной или нескольких серверных системах, выполняющих разные функции в соответствии с разными вариантами осуществления изобретения. Указанные компоненты могут представлять собой выполняемую программу, промежуточную программу (например, IL) или интерпретируемую программу (например, Java), которая взаимодействует по коммуникационной сети (например, Internet) при помощи протокола связи (например, TCP/IP).
Следует отметить, что настоящее изобретение не ограничено выполнением в любой конкретной системе или группе систем. Кроме того, следует отметить, что настоящее изобретение не ограничено какой-либо конкретной архитектурой, сетью или протоколом связи.
Разные варианты настоящего изобретения могут быть запрограммированы при помощи объектно-ориентированного языка программирования, такого как SmallTalk, Java, C++, Ada или C# (C-Sharp). Могут быть также использованы другие объектно-ориентированные языки программирования. Альтернативно может быть использован функциональный язык, язык сценариев и/или логический язык программирования. Разные объекты настоящего изобретения могут быть выполнены в непрограммируемой среде (например, документы, созданные в формате HTML, XML или другом формате, который при воспроизведении в окне броузера, визуализирует объекты графического интерфейса пользователя (GUI) или выполняет другие функции). Разные объекты настоящего изобретения могут быть выполнены в виде запрограммированных или незапрограммированных элементов или любой их комбинации.
После ознакомления с описанием некоторых иллюстративных вариантов осуществления настоящего изобретения специалистам в данной области должно быть очевидно, что приведенная выше информация носит иллюстративный характер и не ограничивает объем изобретения, будучи представленной только в качестве примера. Специалисту в данной области должны быть очевидны различные модификации и другие иллюстративные варианты осуществления изобретения, которые входят в объем настоящего изобретения. В частности, несмотря на то что многие примеры, приведенные в настоящем описании изобретения, включают конкретные комбинации выполнения способов или элементов системы, должно быть понятно, что указанные действия и элементы могут быть объединены любым образом с достижением аналогичных целей. Не исключена возможность выполнения действий, наличия элементов и признаков, рассмотренных в одном варианте осуществления изобретения, в других вариантах осуществления изобретения. Кроме того, в случае одного или нескольких ограничений средств и функций, указанных в приведенной ниже формуле изобретения, такие средства не ограничиваются средствами, рассмотренными в настоящем описании изобретения для выполнения указанной функции, но охватывают любые средства, известные в настоящее время или которые будут созданы в будущем, предназначенные для выполнения указанной функции.
В значении, использованном в описании изобретения или формуле изобретения, термины ”включающий”, “включая”, “содержащий”, “характеризующийся” и тому подобные носят открытый характер, то есть они использованы в значении ”включают, но не ограничиваются”. Только переходные фразы ”состоящий из” и “в основной состоящий из” являются закрытыми или полузакрытыми переходными фразами, как указано применительно к формуле изобретения в руководстве патентного ведомства США по порядку проведения экспертизы патентов (восьмое издание, второе переработанное издание, май 2004 г.), раздел 2111.03.
Термины, обозначающие порядковые числительные, такие как ”первый”, “второй”, “третий” и т.д., которые использованы в формуле изобретения для обозначения признаков формулы изобретения, сами по себе не означают приоритет или превосходство одного признака над другим или временной порядок выполнения способа, а служат только для отличия одного признака, имеющего определенное название, от другого признака с таким же названием (кроме использования порядкового числительного).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ДИАГНОСТИКИ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2013 |
|
RU2547583C2 |
| РЕГИСТРАЦИЯ РАХ2 ДЛЯ ДИАГНОСТИКИ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2009 |
|
RU2522485C2 |
| СПОСОБ ПРЕДСКАЗАНИЯ РЕЦИДИВА РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ ПРИ ЭНДОКРИННОМ ЛЕЧЕНИИ | 2011 |
|
RU2654587C2 |
| СПОСОБ ДИАГНОСТИКИ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ И РАКА ЯИЧНИКОВ ПО УРОВНЮ мРНК ММР-9 В ПЛАЗМЕ КРОВИ | 2020 |
|
RU2745424C1 |
| СПОСОБ ПРИМЕНЕНИЯ AXL В КАЧЕСТВЕ МАРКЕРА ЭПИТЕЛИАЛЬНО-МЕЗЕНХИМАЛЬНОГО ПЕРЕХОДА | 2010 |
|
RU2586493C2 |
| ПРИМЕНЕНИЕ АНТИТЕЛА ПРОТИВ PD-1 В ИЗГОТОВЛЕНИИ ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ СОЛИДНЫХ ОПУХОЛЕЙ | 2020 |
|
RU2825845C2 |
| СПОСОБ ДИАГНОСТИКИ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ ПО УРОВНЮ мРНК TGEβ и TNFα В ПЛАЗМЕ КРОВИ | 2019 |
|
RU2742209C1 |
| ДЕЗОКСИПРОИЗВОДНЫЕ ЦИТИДИНА ИЛИ УРИДИНА ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ РАКА | 2020 |
|
RU2797489C2 |
| ОПРЕДЕЛЕНИЕ МИКРОРНК В ПЛАЗМЕ ДЛЯ ОБНАРУЖЕНИЯ РАННИХ СТАДИЙ КОЛОРЕКТАЛЬНОГО РАКА | 2012 |
|
RU2611189C2 |
| ОПРЕДЕЛЕНИЕ МИКРОРНК В ПЛАЗМЕ ДЛЯ ОБНАРУЖЕНИЯ РАННИХ СТАДИЙ КОЛОРЕКТАЛЬНОГО РАКА | 2012 |
|
RU2662975C1 |


















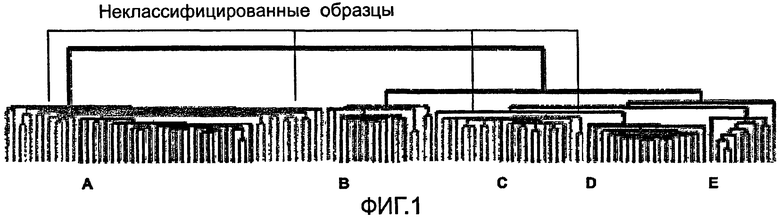
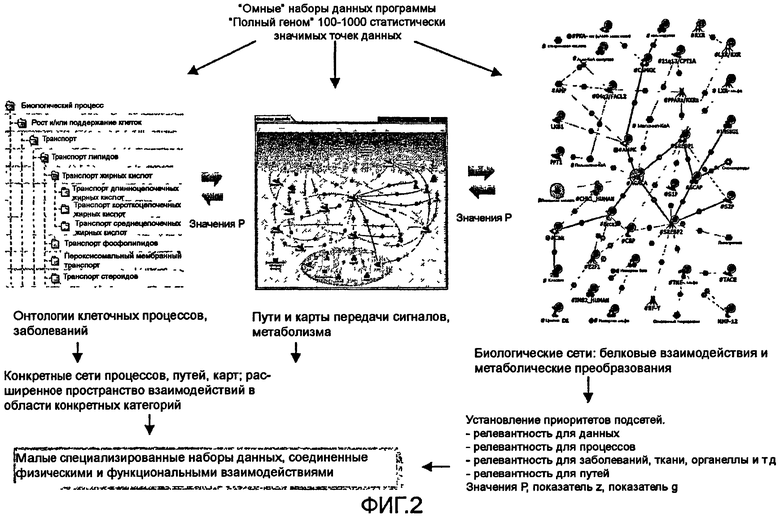
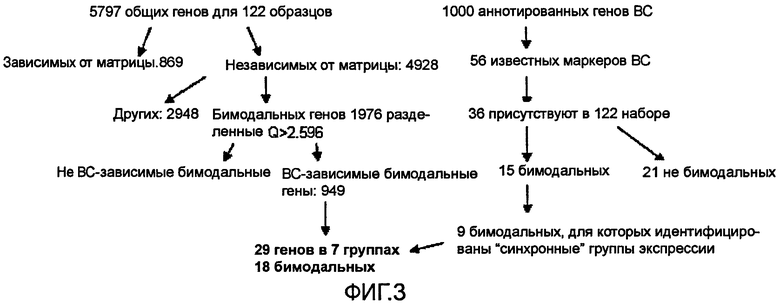
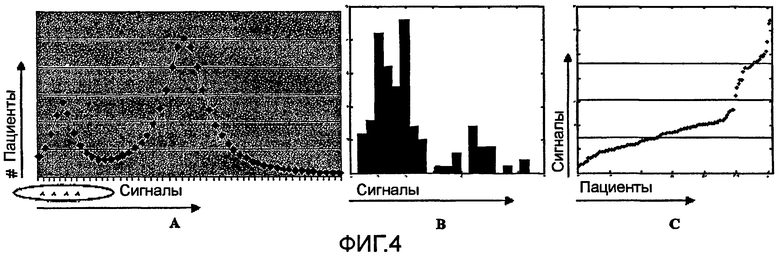
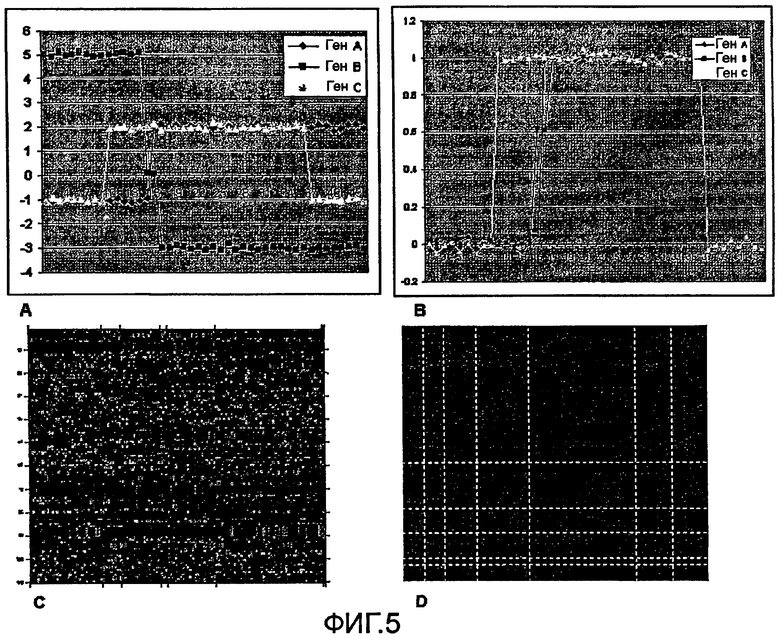
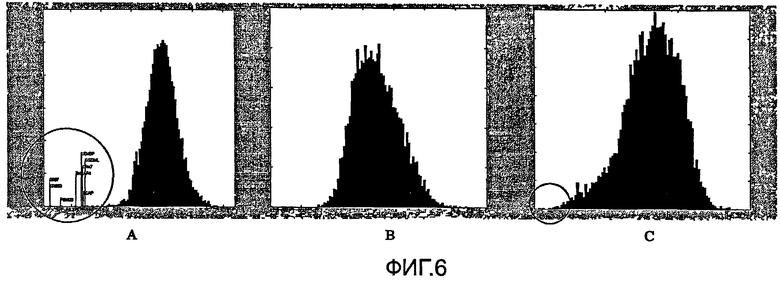
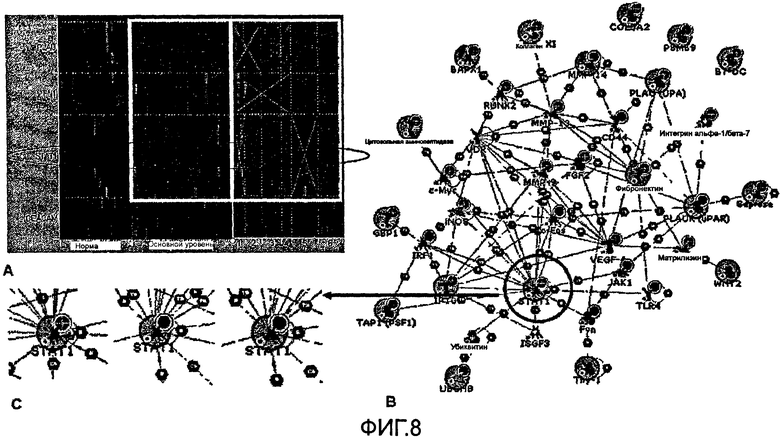
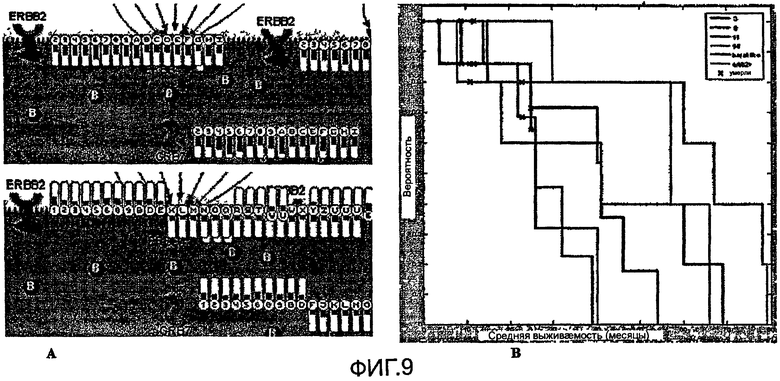
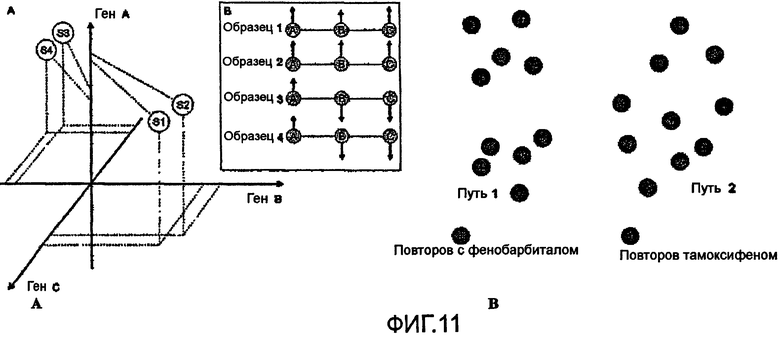
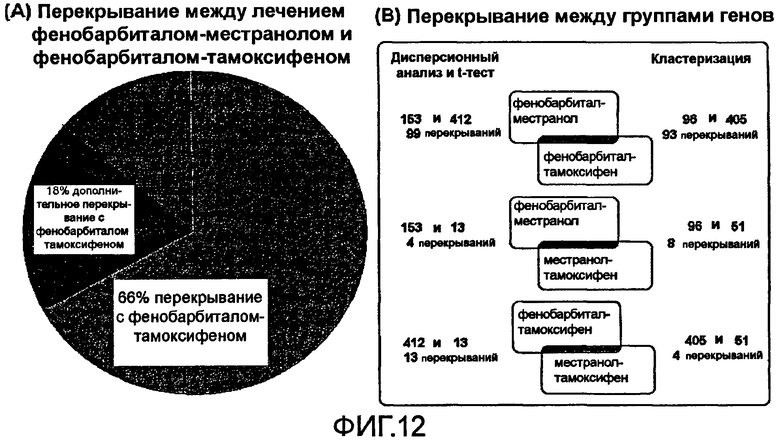
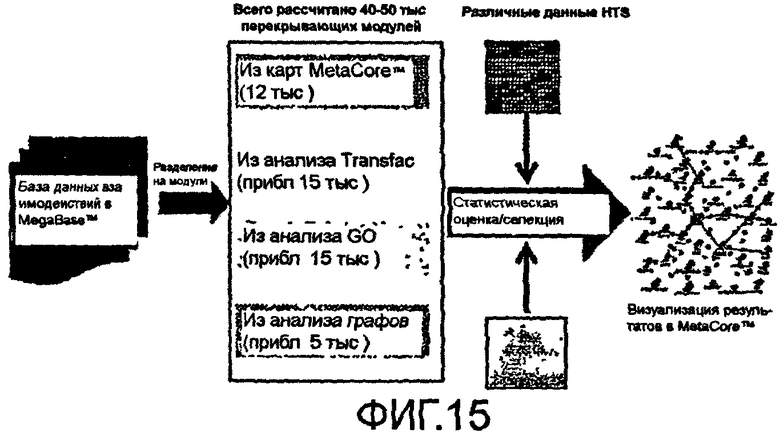
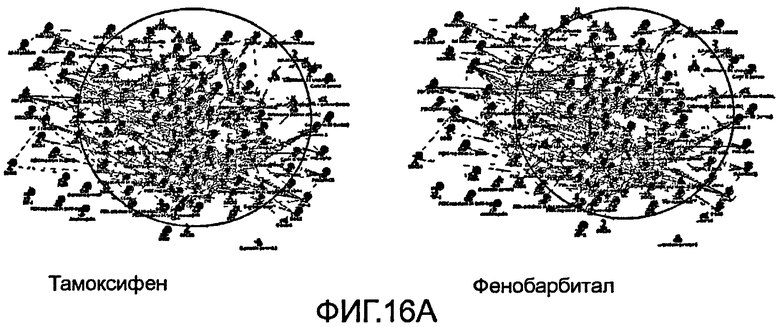
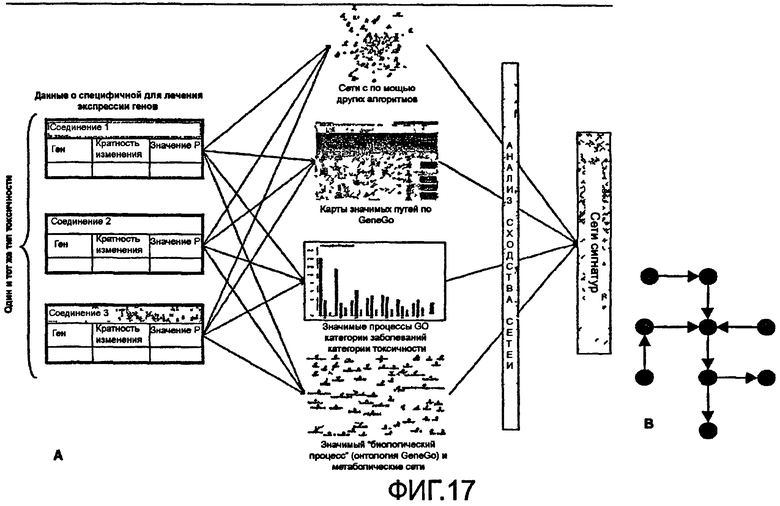
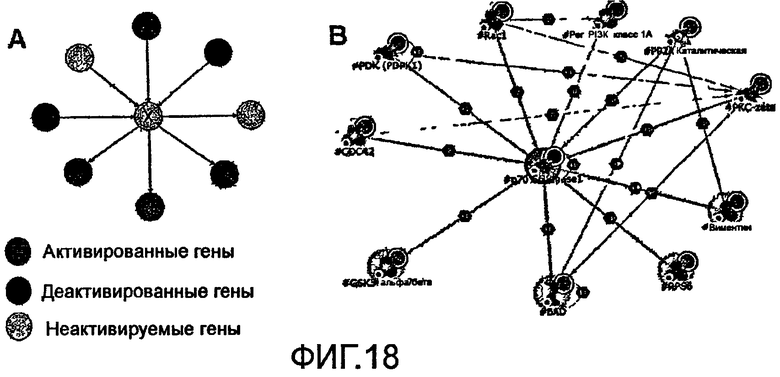
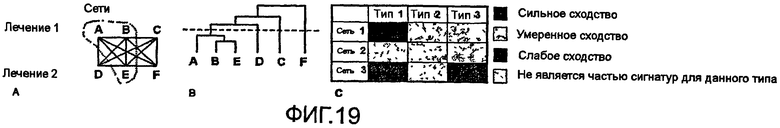
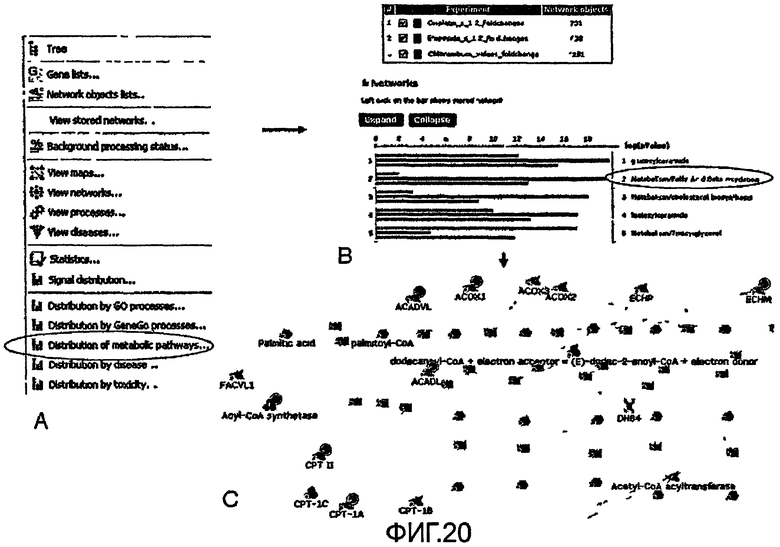
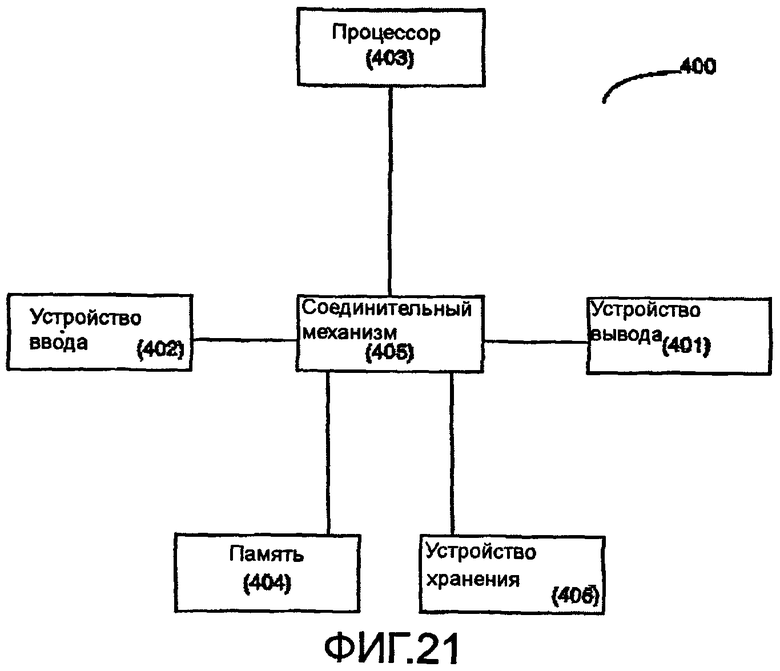
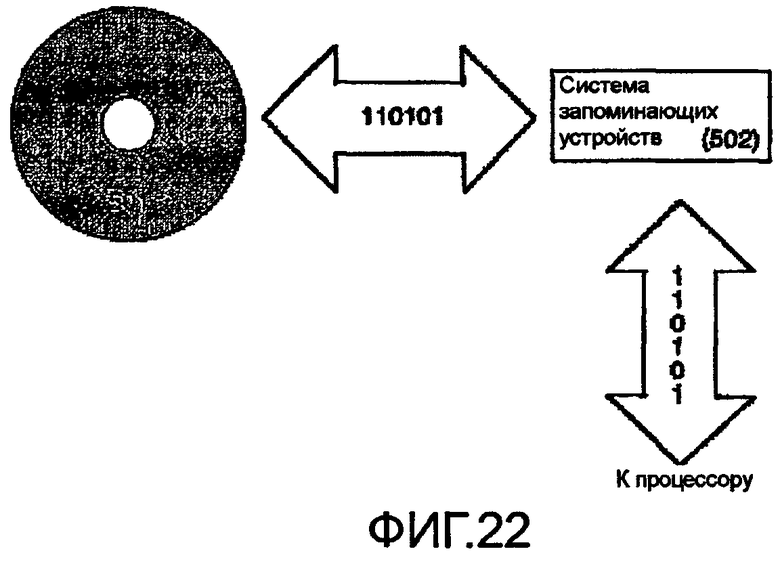
Изобретение относится к области биотехнологии, конкретно к средствам и методам диагностики и дифференцирования разных типов рака молочной железы, и может быть использовано в медицине. Изобретение основано на измерении экспрессии генов в образце рака молочной железы в следующих дескрипторных группах: (1) ERBB2, STAED3, GRB7, THRAP4, PPARBP; (2) ESR1, GATA3, ХВР1, TFF3, ACADSB; (3) KRT5, KRT17, TRIM29, GABRP; (4) KRT18, KRT8, PPP2CA, KRT8L2; (5) PLAU, COL5A2, FAP, THY1; (6) STAT1, UBE2L6, TAP1, LAP3 и (7) SEACAM5, SEACAM6, SEACAM7. При этом сверхэкспрессия большинства генов в дескрипторной группе по сравнению с образцом нормальной ткани молочной железы указывает на то, что образец ткани является положительным для данной группы, а отсутствие сверхэкспрессии большинства генов в дескрипторной группе по сравнению с образцом нормальной ткани молочной железы указывает на то, что образец ткани является отрицательным для данной группы. Комбинация положительных и/или отрицательных статусов дескрипторных групп образует набор характерных генов для классификации образца ткани рака молочной железы. Изобретение позволяет четко классифицировать тип рака молочной железы для проведения оценки клинического прогноза и токсичности лекарственного средства у пациента с раком молочной железы. 4 н. и 8 з.п. ф-лы, 22 ил., 4 табл.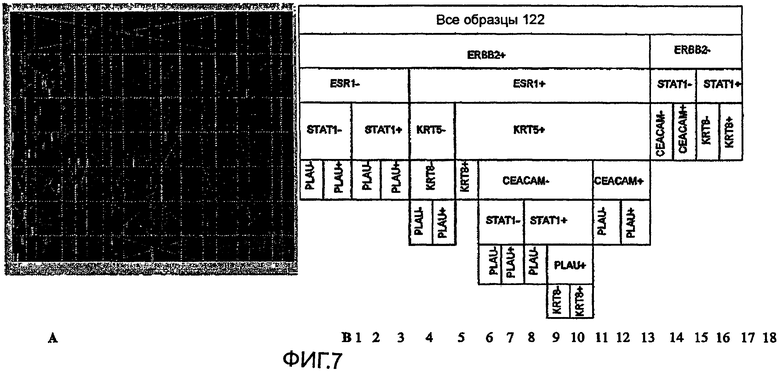
1. Изделие для применения для классификации образца ткани рака молочной железы, которое включает средство для измерения экспрессии генов в следующих дескрипторных группах:
(1) ERBB2, STAED3, GRB7, THRAP4, PPARBP;
(2) ESR1, GATA3, ХВР1, TFF3, ACADSB;
(3) KRT5, KRT17, TRIM29, GABRP;
(4) KRT18, KRT8, PPP2CA, KRT8L2;
(5) PLAU, COL5A2, FAP, THY1;
(6) STAT1, UBE2L6, TAP1, LAP3 и
(7) SEACAM5, SEACAM6, SEACAM7,
где сверхэкспрессия большинства генов в дескрипторной группе по сравнению с образцом нормальной ткани молочной груди указывает на то, что образец ткани является положительным для данной группы, а отсутствие сверхэкспрессии большинства генов в дескрипторной группе по сравнению с образцом нормальной ткани молочной груди указывает на то, что образец ткани является отрицательным для данной группы; и комбинация положительных и/или отрицательных статусов дескрипторных групп образует набор характерных генов для классификации образца ткани рака молочной железы.
2. Изделие по п.1, отличающееся тем, что средство для измерения экспрессии генов включает комплементарные олигонуклеотидные зонды или затравки.
3. Изделие по п.2, отличающееся тем, что комплементарные олигонуклеотидные зонды иммобилизованы на твердом носителе.
4. Изделие по п.3, отличающиеся тем, что иммобилизованные комплементарные олигонуклеотидные зонды образуют олигонуклеотидную матрицу.
5. Изделие по п.2, отличающееся тем, что комплементарные олигонуклеотидные праймеры подходят для выполнения полимеразной цепной реакции с обратной транскрипцией (RT-PCR).
6. Способ классификации образца ткани рака молочной железы, который включает:
(a) измерение экспрессии генов в образце ткани в следующих дескрипторных группах:
(1) ERBB2, STAED3, GRB7, THRAP4, PPARBP;
(2) ESR1, GATA3, ХВР1, TFF3, ACADSB;
(3) KRT5, KRT17, TRIM29, GABRP;
(4) KRT18, KRT8, PPP2CA, KRT8L2;
(5) PLAU, COL5A2, FAP, THY1;
(6) STAT1, UBE2L6, TAP1, LAP3 и
(7) SEACAM5, SEACAM6, SEACAM7;
(b) присвоение положительного статуса дескрипторной группе, если большинство генов в этой группе сверхэкспрессированы по сравнению с образцом нормальной ткани молочной груди;
(c) присвоение отрицательного статуса дескрипторной группе, если большинство генов в этой группе не сверхэкспрессированы по сравнению с образцом нормальной ткани молочной груди; и
(d) использование комбинации положительных и/или отрицательных статусов дескрипторных групп для образования характерных генов для классификации образца ткани рака молочной железы.
7. Способ по п.6, отличающийся тем, что стадия а) включает реакцию гибридизации или амплификации нуклеотидов.
8. Способ по п.7, отличающийся тем, что реакцию гибридизации нуклеотидов проводят на твердом носителе.
9. Способ по п.8, отличающийся тем, что реакцию гибридизации нуклеотидов проводят на олигонуклеотидной матрице.
10. Способ по п.7, отличающийся тем, что реакция амплификации нуклеотидов является полимеразной цепной реакцией с обратной транскрипцией (RT-PCR).
11. Способ оценки клинического прогноза для индивида, страдающего раком молочной железы, который включает:
a) классификацию образца ткани молочной железы, взятого у пациента с раком молочной железы, используя способ по п.6;
b) оценку клинического прогноза для индивида, страдающего раком молочной железы, по результатам, полученным на стадии а).
12. Способ оценки токсичности лекарственного средства против рака у пациента с раком молочной железы, который включает:
a) классификацию образца рака молочной железы, взятого у пациента с раком молочной железы, используя способ по п.6;
b) оценку токсичности лекарственного средства против рака у пациента с раком молочной железы по результатам, полученным на стадии а).
| CHRISTOS SOTIRIOU et al., Breast cancer classification and prognosis based on gene expression profiles from a population-based study, Proc | |||
| Natl | |||
| Acad | |||
| Sci | |||
| USA, 2003, v.100, n.18, p.10393”10398 | |||
| WO 2006103442 A, 05.10.2006 | |||
| KUN YU et al., A Molecular Signature of the Nottingham Prognostic Index in Breast Cancer, Cancer Res., 2004, v.64, n.9, |

