Настоящее изобретение относится к новым производным бензимидазола и их применению в качестве потенциальных блокаторов кальциевых каналов при лечении или профилактике хронической стабильной стенокардии, гипертензии, ишемии (почечной и сердечной), сердечной аритмии, включая предсердную фибрилляцию, сердечной гипертрофии или застойной хронической недостаточности, к фармацевтическим композициям, содержащим эти производные, и к способам их получения. Бензимидазольные производные по настоящему изобретению могут быть также использованы, самостоятельно или в фармацевтических композициях, для лечения почечных заболеваний, диабета и их осложнений, гиперальдостеронизма, эпилепсии, нейропатической боли или рака у человека и других млекопитающих.
Многие кардиоваскулярные нарушения ассоциируются с «кальциевой перегрузкой», являющейся следствием нарушенного повышенного притока кальция через плазматическую мембрану гладкомышечных клеток сердца и сосудов. Существуют 3 главных пути, по которым экстрацеллюлярный кальций может входить в эти клетки: 1) рецептор-активируемые кальциевые каналы, 2) лиганд-управляемые кальциевые каналы и 3) потенциал-управляемые кальциевые каналы (VOCs).
VOCs классифицированы по шести главным категориям: L (долгоживущие), Т (транзиторные), N (нейрональные), Р (клетки Пуркенье), Q (подобные Р) и R (остаточные или стойкие).
L-тип кальциевых каналов отвечает за внутреннее движение кальция, которое инициирует сокращение в кардиальных и гладкомышечных клетках, что предполагает возможность применения блокаторов этих каналов в сердечно-сосудистой терапии. С этой точки зрения L-тип блокаторов кальциевых каналов применяется в клинике с начала 60-х годов и в настоящее время рекомендуется в качестве первой линии при лечении систолической-диастолической гипертензии и стенокардии.
Т-тип кальциевых каналов найден в различных тканях, таких как коронарная и периферическая сосудистая сеть, синусно-предсердный узел и волокна Пуркенье, головной мозг, надпочечники и почки. Благодаря такому широкому распределению можно надеяться, что блокаторы Т-типа кальциевых каналов обладают сердечно-сосудистой защитой, оказывают действие на нарушения сна, ухудшение настроения, депрессию, мигрень, гиперальдостеронемию, преждевременные роды, недержание мочи, старение мозга и нейродегенеративные нарушения, такие как болезнь Альцгеймера.
Мибефрадил (Posicor®), первый блокатор L-типа и Т-типа кальциевых каналов, продемонстрировал наилучший эффект среди блокаторов кальциевых каналов, предпочтительно действуя на L канал. Мибефрадил применялся для лечения гипертензии и стенокардии без проявления негативных побочных эффектов, часто проявляемых блокаторами L каналов, подобных инотропным препаратам, рефлекторной тахикардии, вазоконстрикторной гормональной секреции или периферического отека. Кроме того, мибефрадил показал потенциальный кардиозащитный эффект (Villame, Cardiovascular Drugs and Therapy, 15, 41-28, 2001; Ramires, J. Mol. Cell Cardiol., 30, 475-83, 1998), почечный защитный эффект (Honda, Hypertension, 19, 2031-37, 2001) и положительный эффект при лечении сердечной недостаточности (Clozel, Proceedings Association American Physicians, 111, 429-37, 1999).
Несмотря на огромные потребности в соединениях такого профиля мибефрадил, однако, был отозван с рынка в 1998 г. (через год после его выпуска) из-за неприемлемого лекарственного взаимодействия с CYP 3A4. Кроме того, были также выявлены ЭКГ нарушения (например, QT пролонгирование) и взаимодействие с опосредуемым MDR-1 оттоком дигоксина (du Souich, Clin. Pharmacol. Ther., 67, 249-57, 2000; Wandel, Drug Metab. Dispos., 28, 895-8, 2000).
Вследствие этого появилась необходимость в новых соединениях, проявляющих действие блокаторов T/L-типа кальциевых каналов, но имеющих улучшенный безопасный профиль по сравнению с мибефрадилом.
Соединения настоящего изобретения являются потенциальными блокаторами T/L каналов и поэтому применяются при заболеваниях, в которых участвуют оба Т и L каналы.
i) Первый объект изобретения включает бензимидазольные производные формулы (I)
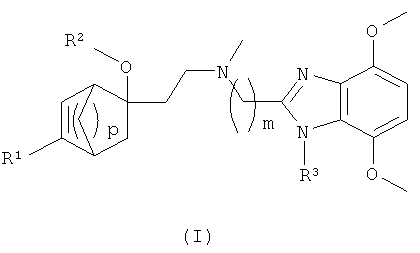
где
R1 представляет собой арил, который является незамещенным или моно-, ди- или три-замещенным, где заместители независимо друг от друга выбирают из группы, включающей С1-4алкил, С1-4алкоксигруппу, галоген и трифторметил;
R2 представляет собой водород или -CO-R21;
R21 представляет собой С1-5алкил, С1-3фторалкил или С3-6циклоалкил;
m представляет собой целое число 2 или 3;
p представляет собой целое число 2 или 3; и
R3 представляет собой водород или С1-5алкил.
В следующих параграфах представлены определения различных химических фрагментов для соединений согласно изобретению. Названные определения предназначены для единообразного применения во всем описании и в формуле изобретения, если не указано иначе и если определения не подлежат более широкому или, наоборот, более узкому толкованию.
Термин "С1-5алкил" обозначает линейную или разветвленную алкильную группу, содержащую от 1 до 5 атомов углерода, предпочтительно содержащую от 1 до 4 атомов углерода. Термин "Cx-yалкил" (x и y при этом являются целыми числами) относится к линейной или разветвленной алкильной группе, содержащей от 1 до 5 атомов углерода. Примерами С1-5алкильных групп являются метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, изобутил, н-пентил и изопентил. Предпочтительными являются метил, этил, н-пропил и изопропил. Наиболее предпочтительным является метил. Для заместителя R21 наиболее предпочтительным является изопропил.
Термин "С1-3фторалкил" обозначает линейную или разветвленную C1-3алкильную группу, которая является замещенной 1-7 атомами фтора. Примеры C1-3фторалкильных групп включают трифторметил, 2-фторэтил, 2,2-дифторэтил, 2,2,2-трифторэтил и пентафторэтил. Предпочтительными являются трифторметил, 2,2,2-трифторэтил и пентафторэтил. Наиболее предпочтительной группой является трифторметил. Для заместителя R наиболее предпочтительной является 2,2,2-трифторэтил.
Термин «С3-6циклоалкил» обозначает насыщенную циклоалкильную группу, содержащую от 3 до 6 атомов углерода. Примеры С3-6циклоалкильных групп включают циклопропил, циклобутил, циклопентил и циклогексил. Для заместителя R21 циклопропил является наиболее предпочтительной группой.
Термин «С1-5алкоксигруппа» обозначает группу формулы С1-5алкил-О-, в которой термин С1-5алкил имеет значение, приведенное ранее. Термин «Cx-yалкоксигруппа» (x и y при этом являются целыми числами) относится к линейной или разветвленной алкоксигруппе, содержащей от x до y атомов углерода. Примеры С1-5алкоксигрупп включают метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу и трет-бутоксигруппу. Предпочтительными являются метоксигруппа и этоксигруппа.
Термин «галоген» подразумевает фтор, хлор, бром или йод, предпочтительно фтор или хлор.
Термин «арил» обозначает фенильную или нафтильную группу. Предпочтительной является фенильная группа. Арильная группа может быть незамещенной или моно-, ди- или три-замещенной, где заместители независимо друг от друга выбирают из группы, включающей С1-4алкил, С1-4алкоксигруппу, галоген и трифторметил. В подвариантах арильная группа является предпочтительно незамещенной. Примеры «арильных» групп включают фенил, нафтил, 2-метилфенил, 3-метилфенил, 4-метилфенил, 3,4-диметилфенил, 2,3-диметилфенил, 2,4-диметилфенил, 2,6-диметилфенил, 3,4-диметилфенил, 3,5-диметилфенил, 2-метоксифенил, 3-метоксифенил, 4-метоксифенил, 2,3-диметоксифенил, 3,4-диметоксифенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 3,4-дифторфенил, 3-хлорфенил, 2,3-дихлорфенил, 3,4-дихлорфенил, 2-трифторметилфенил, 3-трифторметилфенил и 4-трифторметилфенил. Предпочтительным является фенил.
Приведенные далее варианты осуществления изобретения включают:
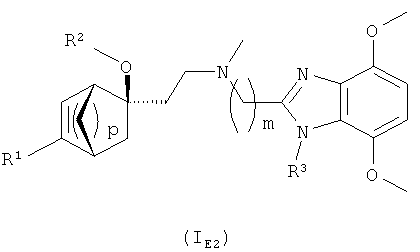
ii) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно варианту i), в которых конфигурация мостикового циклогексенового фрагмента является такой, где R2-O-заместитель и мостик -(СН2)p- циклогексенового фрагмента находятся в цис-положении (то есть абсолютная конфигурация является такой, как изображено или в формуле (IE1), или в формуле (IE2) ниже).
iii) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно варианту i), где абсолютная конфигурация является такой, как изображено в формуле (IE1)

iv) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно варианту i), где абсолютная конфигурация является такой, как изображено в формуле (IE2)
v) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно одному из вариантов i)-iv), где R1 представляет собой незамещенным фенил.
vi) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно вариантам i)-v), где p представляет собой целое число 2.
vii) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно вариантам i)-v), где p представляет собой целое число 3.
viii) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно одному из вариантов i)-vii), где R2 представляет собой -CO-R21.
ix) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно одному из вариантов i)-viii), где R21 представляет собой С1-5алкил или С3-6циклоалкил.
х) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно одному из вариантов i)-ix), где R21 представляет собой С1-5алкил (в частности, изопропил).
xi) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно одному из вариантов i)-vii), где R2 представляет собой водород.
xii) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно одному из вариантов i)-xi), где m представляет собой целое число 3.
xiii) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно одному из вариантов i)-xii), где R3 представляет собой водород.
xiv) Следующий вариант осуществления изобретения относится к соединениям формулы (I) согласно одному из вариантов i)-xii), где R3 представляет собой C1-5алкил (в частности, метил).
Соединения формулы (I) содержат стереогенные или асимметрические центры, такие как асимметрические атомы углерода. Соединения формулы (I) могут вследствие этого представлять собой смесь стереоизомеров или, предпочтительно, чистые стереоизомеры. Смеси стереоизомеров могут быть разделены с помощью методов, известных специалистам в области техники.
Предпочтительные соединения формулы (I) выбирают из группы, включающей:
(1R,2R,4R)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол;
(1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол и
(1R*,5R*,6R*)-6-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-ол.
Кроме того, следующие предпочтительные соединения формулы (I) согласно варианту i) выбирают из группы, включающей:
(1R,2R,4R)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты;
(1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты; и
(1R*,5R*,6R*)-6-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-иловый эфир изомасляной кислоты.
Относительная конфигурация стереоизомеров обозначается следующим образом: например, (1R*,5R*,6R*)-6-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-иловым эфиром изомасляной кислоты называют
(1R,5R,6R)-6-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}-этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-иловый эфир изомасляной кислоты,
(1S,5S,6S)-6-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}-этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-иловый эфир изомасляной кислоты,
или смеси этих двух энантиомеров.
Все, что используется для множественной формы соединений, солей, фармацевтических композиций, заболеваний, применимо также к единичному соединению, соли или им подобным.
Любая ссылка на соединение формулы I или ICE в этом описании может быть отнесена также к солям (и особенно к фармацевтически приемлемым солям) таких соединений в качестве соответствующей и целесообразной.
Термин «фармацевтически приемлемые соли» относится к нетоксичным, неорганическим или органическим кислотно- и/или основно-аддитивным солям. Ссылка может быть сделана на публикацию: «Salt selection for basic drugs», Int. J. Pharm., (1986), 33, 201-217.
Соединения формул (I), (IE1)) и/или (IE2) и их фармацевтически приемлемые соли могут быть применены в качестве лекарственных средств, например в виде фармацевтических композиций для энтерального или парентерального введения.
Получение фармацевтических композиций может быть осуществлено методом, известным любому специалисту в области техники (см., например: Remington, The Science and Practice of Pharmacy, 21-e изд. (2005), Часть 5, «Pharmaceutical Manufacturing» [published by Lippincott Williams & Wilkins]), путем внесения описанных соединений формулы I или их фармацевтически приемлемых солей, необязательно в комбинации с другими терапевтически приемлемыми веществами, в лекарственно принятую форму для введения вместе с приемлемым, нетоксичным, инертным, терапевтически совместимым твердым или жидким носителем и, если необходимо, стандартными фармацевтическими наполнителями.
Соединения формулы (I) или их фармацевтически приемлемые соли применимы для получения лекарственного средства для лечения или профилактики хронической стабильной стенокардии, гипертензии, ишемии (почечной и сердечной), сердечной аритмии, включая предсердную фибрилляцию, сердечную гипотонию или застойную сердечную недостаточность.
Соединения формулы (I) или их фармацевтически приемлемые соли применимы также для получения лекарственного средства для следующей группы заболеваний, отдельно или в любой комбинации:
для лечения почечных заболеваний, диабета и их осложнений, гиперальдостеронизма, эпилепсии, нейрональной боли или рака у человека и других млекопитающих;
для применения в качестве антифибриллярного агента, антиастматического агента, антиатеросклеротического агента, добавки к кардиоплегическим растворам при экстракорпоральном кровообращении, добавки к тромболитической терапии, в качестве антиагрегантного агента или в качестве агента для лечения нестабильной стенокардии;
для лечения или профилактики гипертензии, в частности портальной гипертензии, гипертензии после лечения эритропоэтином и гипертензии вследствие низкого содержания ренина;
для применения при гипоксических или ишемических заболеваниях или в качестве противоишемического агента для лечения, например, сердечной, почечной и церебральной ишемии и реперфузии (например, имеющей место после хирургического вмешательства при экстракорпоральном кровообращении), коронарного и церебрального вазоспазма и им подобных, при терапии периферических сосудистых заболеваниях (например, болезнь Рейно, перемежающаяся хромота, болезнь Такаясу), при серповидно-клеточных заболеваниях, включая инициирование и/или продолжение родовых схваток;
для лечения или профилактики нарушений, относящихся к почечной, клубочковой и мезангиальной клеточной функции, включая острую и хроническую почечную недостаточность, диабетическую нефропатию, индуцированную гипертензией нефропатию, клобучковый инсульт, почечные нарушения, связанные с возрастом или диализом, нефросклероз, нефротоксичность, связанную с визуализацией и контрастным агентом и с циклоспорином, почечную ишемию, первичный везикоуретеральный рефлюкс или клобучковый склероз;
для применения в терапии инфаркта миокарда, при лечении сердечной гипертрофии, первичной и вторичной легочной гипертензии, терапии застойной сердечной недостаточности, включая ингибирование фиброза, ингибирование расширения левого желудочка, ремоделирование и дисфункцию или рестеноз после ангиопластики или стентирования;
для лечения эндотоксемии, или эндотоксинового шока, или геморрагического шока;
для лечения сексуальной дисфункции как у мужчин (эректильная дисфункция, например, вследствие сахарного диабета, повреждения позвоночника, радикальной простатэктомии, психогенетической этиологии и других причин), так и у женщин посредством улучшения притока крови к гениталиям, в частности к кавернозным телам;
для профилактики и/или улучшения при раке или поражении концевого органа, связанном с клеточной пролиферацией;
для терапии метаболических нарушений или хронических воспалительных заболеваний, инсулинзависимого и неинсулинзависимого сахарного диабета и их осложнений (например, нейропатия, ретинопатия), гиперальдостеронизма, ремоделирования костей, псориаза, артрита, ревматоидного артрита, саркоидозного остеоартрита или экзематозного дерматита;
для лечения токсикоза печени и синдрома детской внезапной смерти, раннего и прогрессирующего заболевания и поражения печени, включающих сопутствующие осложнения (например, гепатотоксичность, фиброз, цирроз), разрушительных последствий новообразований, таких как гипертензия вследствие гемангиоперицитомы, спастических заболеваний уринарного тракта и/или мочевого пузыря, гепаторенального синдрома, иммунологических заболеваний, включая васкулит, такой как красная волчанка, рассеянный склероз, смешанная криоглобулинемия, фиброза, ассоциированного с почечной недостаточностью, и гепатотоксичности;
для применения при желудочно-кишечных заболеваниях, таких как язвенный колит, болезнь Крона, повреждение слизистых оболочек желудка, язвенное воспалительное заболевание кишечника и ишемическое заболевание кишечника, заболевания желчного пузыря или желчных протоков, таких как холангит, панкреатит, регуляция клеточного роста, начальная простатическая гипертрофия или трансплантация, или в качестве антидиаррейного агента;
для лечения нарушений, включающих сужение бронхов или хроническое или острое воспаление, такое как обструктивная болезнь легких и отек легких;
для облегчения боли, включая нейропатическую боль, периферическую боль и боль, ассоциированную с раковым заболеванием, такую как боль при раке простаты или раке костей;
для лечения васкулярных нарушений центральной нервной системы, таких как инсульт, преходящие ишемические атаки, мигрень и субарахноидальное кровотечение, поведенческие нарушения центральной нервной системы, для лечения деменции, включая болезнь Альцгеймера, старческую деменцию и сосудистую деменцию, эпилепсию или нарушение сна; или
для снижения общей заболеваемости и/или смертности в результате лечения или профилактики представленных выше заболеваний.
Изобретение также относится к способу профилактики или лечения заболеваний или нарушений, отмеченных выше, заключающемуся во введении пациенту фармацевтически активного количества соединения формулы (I).
Кроме того, соединения формулы (I) могут быть успешно использованы в комбинации с одним или более агентами, выбранными из агентов, снижающих липидный уровень, таких как статины, антикоагулянтов, таких как кумарины, антитромботических агентов, таких как клопидогель, β-блокаторов и других кардиозащитных агентов.
Кроме того, любые предпочтения, указанные для соединений формулы (I) (будь то сами соединения, их соли, композиции, содержащие соединения или их соли, применение соединений или их солей и т.п.) применимы также к соединениям формулы (IE1) и/или (IE2) и наоборот.
Получение соединений формулы (I):
Следующий аспект изобретения относится к способу получения соединений формулы (I) настоящего изобретения. Полученные соединения могут быть также превращены в их фармацевтически приемлемые соли общеизвестным способом.
В общем, все химические превращения могут быть проведены с использованием хорошо известных стандартных методов, описанных в литературе или представленных ниже на схемах 1-3. Если не указано иначе, общие группы или R1, R2, R3, р и m имеют значения, приведенные для формулы (I). Другие используемые аббревиатуры указаны в экспериментальной части. В некоторых случаях общие группы R1, R2, R3 могут не совпадать с приведенными на схемах ниже, так как требуют использования защитных групп (PG). Применение защитных групп хорошо известно из области техники (см., например, «Protective Groups in Organic Synthesis», T.W.Greene, P.G.M. Wuts, Wiley-Interscience, 1999). В рамках настоящей дискуссии введение таких защитных групп обсуждается в случае их необходимости.
Соединения формулы (I) получают с помощью следующих методов, представленных ниже на схеме 1.
Соединения формулы (I), где R2 представляет собой водород, могут быть получены посредством омыления сложного эфира (К) в стандартных щелочных условиях с помощью LiOH или NaOH в растворителях, подобных этанолу, метанолу, ТГФ или воде, при комнатной температуре или в стандартных кислых условиях с помощью водного раствора HCl или ТФК в растворителях, подобных этанолу, метанолу, ТГФ, ДХМ или воде, при комнатной температуре с получением производных кислоты (1.1). Эту кислоту вводят затем в реакцию конденсации с бензимидазольным производным (ВВ) с получением амидных производных (1.2), используя стандартные агенты конденсации, такие как ЭДК, ГОБТ или РуБОФ в присутствии основания, такого как NEt3 или ДИПЭА, и в растворителях, таких как ТГФ, ДХМ или ДМФ, предпочтительно при комнатной температуре. Амид (1.2) затем восстанавливают, получая требуемые соединения формулы (I), где R представляет собой водород, используя стандартные восстанавливающие агенты типа LiAlH4 или Red-Al в соответствующем растворителе, таком как толуол, в температурном интервале от 0°С до комнатной температуры.
Схема 1

Спирты соединений формулы (I), где R2 представляет собой В, могут быть введены в реакцию ацилирования с использованием стандартных реагентов, таких как хлорангидриды кислот, ангидриды кислот, хлорформиаты, изоцианаты или карбамоилхлориды, при необходимости в присутствии кислоты Льюиса, такой как MgBr2, или в присутствии основания, такого как NEt3, в инертных растворителях, таких как ДХМ или ТГФ, при температурах от 0°С до 65°С, с получением соединений формулы (I), где R2 представляет собой -COR21.
Ключевые промежуточные соединения (К) получают согласно схеме 2. Дикетоны (2.1) и монозащищенные кетоны (2.2) могут быть получены известными методами (Can. J. Chem., 1992, 70, 974-980, Can. J. Chem., 1968, 46, 3713-17, J. Org. Chem., 1978, 43, 4648-4650).
Схема 2

Алкилирование кетона (2.2) нуклеофилами типа реагентов Гриньяра или литирующих реагентов (получаемых из бромсоединения с помощью бутиллития в стандартных реакционных условиях), таких как фенилмагнийбромид, в соответствующем растворителе типа Et2O или ТГФ, при температурах от -78°С до комнатной температуры приводит к спиртам (2.3).
Гидролиз кеталя спиртового производного (2.3), последующее элиминирование воды в стандартных условиях проведения дегидратации и применение TsOH в соответствующих растворителях, таких как ацетон, предпочтительно при комнатной температуре, приводит к кетонам (2.4).
Альтернативно, этот процесс удаление защиты/элиминирование может быть осуществлен в две стадии. Кеталь спиртового производного (2.3) гидролизуют, как описано выше, с использованием TsOH в растворителях, подобных ацетону, при комнатной температуре, что приводит к кетону (2.5). Элиминирование воды может быть проведено в стандартных условиях при использовании Ms-Cl в присутствии основания, подобного NEt3, и в соответствующих растворителях типа ДХМ, при температурах от 0°С до комнатной температуры, или при использовании реагента Бургесса в соответствующих растворителях типа ТГФ, при температурах от 0°С до комнатной температуры, что приводит к кетоновому производному (2.4).
В другом варианте дикетон (2.1) может быть селективно моноалкилирован непосредственно до производного (2.5) с помощью соответствующего нуклеофила типа реагентов Гриньяра, в стандартных растворителях, подобных Et2O или ТГФ, при температурах около 0°С. Элиминирование воды может быть проведено в тех же стандартных условиях, которые приведены выше.
Производные кетона (2.4) превращают в требуемые ключевые промежуточные соединения (К) под действием нуклеофилов, таких как реагенты Гриньяра или литированные алкильные группы, такие как литированный трет-бутилацетат (получаемый in situ при использовании трет-бутилбромацетата, н-бутиллития и диизопропиламина, при температуре -50°С, в смеси соответствующих растворителей, таких как толуол/ТГФ или гексан/ТГФ), при температурах от -50°С до комнатной.
Синтез бензимидазольных производных (ВВ) (схема 1) представлен на схеме 3. Соответствующее замещенное дианилинпроизводное (3.1), которое синтезируют, например, из 1,4-диметокси-2,3-динитробензола (Eur. J. Org. Chem., 2006, 2786-2794), используя стандартные методы, приведенные далее в экспериментальной части, вводят в реакцию конденсации с соответственно защищенным, коммерчески доступным производным N-алкиламиноалкановой кислоты, используя стандартные реагенты и условия проведения конденсации, такие как ЭДК/ГОБТ в присутствии основания, такого как ДИПЭА, NEt3, ДМАП, в растворителях типа ТГФ, ДХМ, при комнатной температуре с получением анилиновых производных (3.2), где PG является аминозащитной группой, такой как Кбз или Бок. Нагревание производных (3.2), предпочтительно при микроволновом излучении, до 150°С, без или в соответствующих растворителях, таких как толуол или уксусная кислота, приводит к защищенным аминоалкилбензимидазольным производным (3.3). Необязательно, в случае когда R3 обозначает алкил, заместитель может быть введен с использованием стандартных реакций, таких как алкилирование соответствующими алкилгалогенидами в присутствии основания, такого как NaH или К2СО3, в растворителе типа ДМФ или ТГФ, при температуре около 0°С. Удаление защитной группы с использованием стандартных методов (гидрирование для PG = Кбз; ТФК или HCl для PG = Бок) дает требуемые аминоалкилбензимидазольные производные (ВВ).
Схема 3

Когда соединения формулы (I) получают в виде смеси энантиомеров, энантиомеры могут быть разделены с использованием методов, известных специалистам в области техники, например, путем получения и разделения энантиомерных солей, или с помощью ВЭЖХ на хиральной стационарной фазе, такой как Regis Whelk-O1 (R,R) (10 мкм) колонка, Daicel ChiralCel OD-H (5-10 мкм) колонка или Daicel ChiralPak IA (10 мкм) или AD-H (5 мкм) колонка. Типичные условия проведения ВЭЖХ включают применение изократной смеси элюента А (EtOH, в присутствии или в отсутствие амина, такого как NEt3, диэтиламин) и элюента Б (гексан), со скоростью истечения от 0,8 до 150 мл/мин.
Экспериментальная часть
Следующие примеры иллюстрируют изобретение, не ограничивая, однако, его объема.
Все температуры представлены в °С.
Соединения охарактеризованы с помощью:
1Н-ЯМР (400 МГц) или 13С-ЯМР (100 МГц) (Bruker; химические сдвиги приведены в м.д. относительно используемого растворителя; мультиплеты: s = одиночный, d = дублет, t = триплет; q = квартет, р = пентлет, hex = гексет, hept = гептет, m = мультиплет, константы сопряжения приведены в Гц);
ЖХ-МС (Finnigan Navigator с HP 1100 Binary Pump и DAD, колонка: 4,6×50 мм, Zorbax SB-AQ, 5 мкм, 120 Å, градиент: 5-95% ацетонитрила в воде, 1 мин, с 0,04% трифторуксусной кислоты, скорость истечения: 4,5 мл/мин), tR (время удерживания) приведено в мин;
ТСХ (ТСХ-пластины от фирмы Merck, силикагель 60 F254); или с помощью температур плавления.
Соединения очищают с помощью препаративной ВЭЖХ (колонка: X-terra RP18, 50×19 мм, 5 мкм, градиент: 10-95% ацетонитрила в воде, содержащей 0,5% муравьиной кислоты), или с помощью колоночной хроматографии на силикагеле.
Рацематы могут быть разделены на энантиомеры с помощью ВЭЖХ (предпочтительные условия: Daicel, ChiralCel OD 20×250 мм, 10 мкм, 4% этанола в гексане, скорость истечения 10-20 мл/мин).
Аббревиатуры (используемые в данном описании):
Ас - ацетил, безв. - безводный, Бок - трет-бутоксикабонил, БСА - бычий сывороточный альбумин, Bu - бутил, Кбз - бензилоксикарбонил, реагент Бургесса - гидроксид (метоксикарбонилсульфамоил)триэтиламмония, ДЦК - дициклогексилкарбодиимид, ДИПА - диизопропиламин, ДМАП - диметиламинопиридин, ДИРЭА - диизопропилэтиламин (основание Хюнига, этилдиизопропиламин), ДМФ - диметилформамид, ДМСО - диметилсульфоксид, DMEM - модифицированная по способу Дульбекко среда Игла, ЭА - этилацетат, ЭДК - N-(3-диметиламинопропил)-N'-этилкарбодиимид, Et - этил, EtOAc - этилацетат, EtOH - этанол, EtO2 - диэтиловый эфир, экв. - эквивалент(ы), ч - час(ы), HBSS - сбалансированный солевой раствор Хенкса, ГОБТ - 1-гидроксибензотриазол, ВЭХЖ - высокоэффективная жидкостная хроматография, ЖХ-МС - жидкостная хроматография-масс-спектрометрия, MeCN - ацетонитрил, Me - метил, МеОН - метанол, мин - минута(ы), Ms - метансульфонил, NEt3 - триэтиламин, Pd/C - палладий на угле, ОАс - ацетат, Ph - фенил, РуБОФ - гексафторфосфат бензотриазол-1-илокси-трис-пирролидинфосфония, преп.- препаративный, насыщ. - насыщенный, трет - третичный (трет-бутил = t-бутил = третичный бутил), ТФК - трифторуксусная кислота, ТГФ - тетрагидрофуран, ТСХ - тонкослойная хроматография, Red-Al - натрий-бис(2-метоксиэтокси)алюминийгидрид, tR - время удерживания, Ts - п-толуолсульфонил, TsOH - п-толуолсульфоновая кислота.
Получение промежуточных соединений
Общие методы получения ключевых промежуточных соединений (К):
Ключевые промежуточные соединения (К1А) и (К2А), которые представляют собой бицикло[2.2.2]окт-5-ен-2-ил или бицикло[3.2.2]нон-8-ен-6-ил, получают в виде смеси основного рацемата, имеющего относительную конфигурацию (R*,R*,R*) (то есть мостик -(СН2)p- циклогексенового фрагмента находится в цис-положении по отношению к группе -OR2, представляющей собой гидроксигруппу) и находящегося в меньшем количестве рацемата, имеющего относительную конфигурацию (R*,S*,R*) или (R*,R*,S*), то есть мостик-(СН2)р- (где р представляет собой 2 или 3 соответственно) циклогексенового фрагмента находится в транс-положении по отношению к группе -OR2, представляющей собой гидроксигруппу. Находящиеся в большем и меньшем количестве рацематы могут быть разделены, как описано для ключевого промежуточного соединения (К1А) в методе А1.5. Если не указано иначе, основной рацемат выделяют и используют для получения соединений примеров, приведенных ниже.
К1А: трет-Бутиловый эфир рац-(1R*,2R*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты
К1А.1 (метод А1.1): рац-(1R*,4R*)бицикло[2.2.2]октан-2,5-дион
2-(Триметилсилокси)-1,3-циклогексадиен (25 мл) и α-ацетоксиакрилонитрил (13 мл) перемешивают и нагревают при температуре 150°С в закрытом сосуде в течение 22 ч. Полученное темно-оранжевое вязкое масло растворяют в 200 мл МеОН. Затем прикалывают раствор 2,2 г метоксида натрия в 150 мл МеОН, после чего реакционную смесь перемешивают в течение 3 ч при комнатной температуре, переносят в смесь льда с водой и экстрагируют ДХМ. Органические фазы концентрируют в вакууме, а сырой остаток очищают с помощью колоночной хроматографии, используя EtOAc/гептан (в соотношении 1:2) и получая при этом 7,9 г рац-(1R*,4R*)бицикло[2.2.2]октан-2,5-диона.
ЖХ-МС: tR=0,44 мин.
К1А.2 (метод А1.2): рац-(1R*,4R*)-спиро[бицикло[2.2.2]октан-2,2'-[1,3]диоксолан]-5-он
К 4,0 г рац-(1R*,4R*)бицикло[2.2.2]октан-2,5-диона (промежуточное соединение К1А.1), растворенного в 120 мл толуола, добавляют 1,7 мл этиленгликоля и 0,27 г TsOH, после чего раствор нагревают при энергичном перемешивании с обратным холодильником в течение 3,5 ч. Реакционную смесь затем охлаждают до комнатной температуры, гасят с помощью насыщенного раствора NaHCO3, экстрагируют Et2O и органическую фазу выпаривают. Сырой продукт очищают с помощью колоночной хроматографии, используя гексан/EtOAc (в соотношении 7:3) и получая при этом 2,41 г рац-(1R*,4R*)-спиро[бицикло[2.2.2]октан-2,2'-[1,3]диоксолан]-5-она в виде желтого масла.
ЖХ-МС: tR=0,64 мин; [М+Н+CH3CN]+: 224,35.
К1А.3 (метод А1.3): Смесь рац-(7R*,8R*,10R*) и рац-(7R*,8S*,10R*)-7,10-(1,2-этилен)-8-фенил-1,4-диокса-спиро[4.5]декан-8-ола
К раствору 2,41 г рац-(1R*,4R*)-спиро[бицикло[2.2.2]октан-2,2'-[1,3]диоксолан]-5-она (промежуточное соединение К1А.2) в 80 мл Et2O прикапывают 14,5 мл фенилмагнийбромида (1-молярный раствор в Et2O) в течение 10 мин, после чего реакционную смесь перемешивают в течение 4 ч при комнатной температуре. Затем реакционную смесь осторожно гасят льдом, добавляют 8 мл 2-нормального раствора HCl и фазы разделяют. Органическую фазу выпаривают и сырой продукт очищают с помощью колоночной хроматографии, используя гептан/EtOAC (в соотношении 7:3), получая при этом 0,37 г 7,10-(1,2-этилен)-8-фенил-1,4-диокса-спиро[4.5]декан-8-ола в виде бесцветного масла. (Разделение диастереомеров с помощью колоночной хроматографии возможно, но осуществляется, если указано.)
ЖХ-МС: tR=0,84 мин; [M-H2O+H]+: 243,34.
К1А.4 (метод А1.4): рац-(1R*,4R*)-5-Фенилбицикло[2.2.2]окт-5-ен-2-он
К раствору 0,54 г 7,10-(1,2-этилен)-8-фенил-1,4-диокса-спиро[4.5]декан-8-ола (промежуточное соединение К1А.3) в 20 мл ацетона добавляют 200 мг TsOH, после чего смесь перемешивают в течение 2 дней при комнатной температуре. Реакционную смесь гасят с помощью насыщенного водного раствора NaHCO3, экстрагируют этилацетатом и органическую фазу выпаривают. Сырой продукт очищают с помощью колоночной хроматографии, используя гептан/EtOAc (в соотношении 7:3) и получая при этом 0,34 г рац-(1R*,4R*)-5-фенилбицикло[2.2.2]окт-5-ен-2-она в виде бесцветного масла.
ЖХ-МС: tR=0,93 мин; [М+Н+CH3CN]+: 240,11.
К1А.5 (метод А1.5): трет-Бутиловый эфир рац-(1R*,2R*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты и трет-бутиловый эфир рац-(1R*,2S*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты
К раствору 0,51 мл ДИПА в 0,5 мл ТГФ прикапывают 2,2 мл н-бутиллития (1,6-молярный раствор в гексане) при температуре -20°С. Через 10 мин добавляют 0,5 мл толуола и раствор перемешивают в течение 30 мин, после чего реакционную смесь охлаждают до температуры -50°С, добавляют 0,73 мл трет-бутилацетата и перемешивание продолжают в течение 1 ч при температуре -50°С. Затем добавляют 0,32 г рац-(1R*,4R*)-5-фенилбицикло[2.2.2]окт-5-ен-2-она (промежуточное соединение К1А.4), растворенного в 1 мл ТГФ, и раствор перемешивают при температуре от -50 до -20°С в течение 2,5 ч. Реакционную смесь переносят в смесь льда с водным раствором HCl, органическую фазу отделяют, промывают и выпаривают. Сырой реакционный продукт очищают с помощью колоночной хроматографии, используя смесь гептан/EtOAc (в соотношении 9:1) и получая при этом 0,30 г основного рацемата, трет-бутилового эфира рац-(1R*,2R*,4R*)-2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты, в виде белого твердого вещества, и 0,07 г второстепенного рацемата, трет-бутилового эфира рац-(1R*,2S*,4R*)-2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты, в виде бесцветного масла.
ЖХ-МС (основной рацемат): tR=1,06 мин; [M-(CH3)3-H2O+H]+: 241,11.
ЖХ-МС (второстепенный рацемат): tR=1,05 мин; [М+Н]+: 315,18.
К1А.6: трет-Бутиловый эфир (1S,2S,4S)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты и трет-бутиловый эфир (1R,2R,4R)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты
трет-Бутиловый эфир рац-(1R*,2R*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты разделяют на соответствующие энантиомеры, используя препаративную хиральную ВЭЖХ (колонка: Daicel ChiralPak AD-H, 20×250 мм, 5 мкм; гексан/EtOH в соотношении 95:5, скорость истечения 16 мл/мин), хиральную аналитическую ВЭЖХ (Daicel ChiralPak AD-H, 4,6×250 мм, 5 мкм; гексан/EtOH в соотношении 95:5, скорость истечения 0,8 мл/мин):
Энантиомер A: tR=6,70 мин;
Энантиомер Б: tR=7,93 мин.
К2А: трет-Бутиловый эфир рац-(1R*,5R*,6R*)-(6-гидрокси-8-фенилбицикло[3.2.2]нон-8-ен-6-ил)уксусной кислоты
К2А.1 (метод А1.6): Смесь рац-(1R*,5R*,8R*) и рац-(1R*,5R*,8S*)-8-гидрокси-8-фенилбицикло[3.2.2]нонан-6-она
К суспензии 1,4 г рац-(1R*,5R*)бицикло[3.2.2]нонан-6,8-диона (синтезирован согласно известным методам: Can. J. Chem., 1968, 46, 3713-3717) в 45 мл Et2O прибавляют 10,3 мл фенилмагнийбромида (1-молярный раствор в ТГФ) в течение 15 мин при температуре 0°С, а затем перемешивают в течение 2 ч при комнатной температуре, после чего реакционную смесь охлаждают до температуры 0°С, гасят ледяной водой, подкисляют водным раствором HCl и экстрагируют Et2O. Органическую фазу промывают рассолом, высушивают над MgSO4 и концентрируют в вакууме, получая сырое названное в заголовке соединение в виде желтого масла.
ЖХ-МС: tR=0,79 мин; [M+H+CH3CN)+: 272,33.
К2А.2 (метод А1.7): рац-(1R*,5R*)-8-фенилбицикло[3.2.2]нон-8-ен-6-он
Полученный выше 8-гидрокси-8-фенилбицикло[3.2.2]нонан-6-он (промежуточное соединение К2А.1) растворяют в 55 мл ацетона, добавляют 1,7 г TsOH, смесь перемешивают при комнатной температуре в течение ночи, после чего добавляют еще одну порцию (3,5 г) TsOH и перемешивание продолжают дополнительно в течение 5 ч. Реакционную смесь затем разбавляют этилацетатом, органическую фазу промывают насыщенным водным раствором NaHCO3 и выпаривают. Сырой продукт очищают с помощью колоночной хроматографии, используя смесь гептан/EtOAc (в соотношении 4:1), получая при этом 0,9 г рац-(1R*,5R*)-8-фенилбицикло[3.2.2]нон-8-ен-6-она в виде желтоватого масла.
ЖХ-МС: tR=0,99 мин; [M+H]+: 213,11.
K2A.3: трет-Бутиловый эфир рац-(1R*,5R*,6R*)-(6-гидрокси-8-фенилбицикло[3.2.2]нон-8-ен-6-ил)уксусной кислоты
Получают из рац-(1R*,5R*)-8-фенилбицикло[3.2.2]нон-8-ен-6-она (промежуточное соединение К2А.2), используя метод А1.5.
ЖХ-МС (основной рацемат): tR=1,11 мин; [М-(CH3)3-H2O+Н]+: 254,02.
ББ. [3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламин
ББ.1. 3,6-Диметоксибензол-1,2-диамин
3,6-Диметоксибензол-1,2-диамин синтезируют, растворяя 6,0 г 1,4-диметокси-2,3-динитробензола (Eur. J. Org. Chem., 2006, 2786-2794) в 220 мл EtOH, трижды продувают азотом и добавляют 600 мг 10%-ного Pd/C. Реакционную смесь перемешивают в атмосфере водорода (баллон), добавляют через 2 дня еще 300 мг 10%-ного Pd/C и смесь перемешивают дополнительно в течение 24 ч. Фильтрование через слой целлита, промывание EtOH и EtOAc и концентрированно в вакууме дает 4,3 г 3,6-диметоксибензол-1,2-диамина в виде черного твердого вещества.
ЖХ-МС: tR=0,48 мин; [M+H]+: 169,09.
ББ.2. Бензиловый эфир [3-(2-амино-3,6-диметоксифенилкарбамоил)пропил]-метилкарбаминовой кислоты
К раствору 3,1 г 4-(бензилоксикарбонилметиламино)масляной кислоты в 80 мл ДХМ добавляют 6,5 мл ДИПЭА, 1,8 г ГОБТ, 2,6 г ЭДК и 154 мг ДМАП. После перемешивания в течение 10 мин добавляют 2,1 г 3,6-диметоксибензол-1,2-диамина, растворенного в 20 мл ДХМ, и смесь перемешивают при комнатной температуре в течение ночи. Затем реакционную смесь гасят насыщенным водным раствором NaHCO3, фазы разделяют и органическую фазу промывают рассолом, высушивают над MgSO4 и концентрируют в вакууме, получая сырое названное в заголовке соединение в виде черного масла.
ЖХ-МС: tR=0,88 мин; [М+Н]+: 402,06.
ББ.3. Бензиловый эфир [3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метилкарбаминовой кислоты
К смеси полученного выше сырого бензилового эфира 3-(2-амино-3,6-диметокси-фенилкарбамоил)пропил]метилкарбаминовой кислоты в 16 мл толуола добавляют 4 мл ДМФ и 1,9 г TsOH, после чего реакцию нагревают при температуре 150°С в течение 2 ч в микроволновой печи. Затем добавляют насыщенный водный раствор NaHCO3 и фазы разделяют. Органическую фазу промывают рассолом, высушивают над MgSO4, концентрируют в вакууме, фильтруют через слой силикагеля, используя EtOAc, и снова концентрируют. Очистка с помощью колоночной хроматографии с использованием этиоацетата дает 2,7 г бензилового эфира 3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метилкарбаминовой кислоты в виде коричневой смолы.
ЖХ-МС: tR=0,85 мин; [M+H]+: 384,62.
ББ.4. [3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламин
Раствор 2,6 г бензилового эфира 3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метилкарбаминовой кислоты в 60 мл EtOH трижды продувают азотом, после чего добавляют 260 мг 10%-ного Pd/C. Реакционную смесь затем перемешивают в атмосфере водорода (баллон) в течение 5 ч при комнатной температуре. Фильтрование через слой целлита, промывание EtOH и концентрирование в вакууме дает 1,7 г 3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламина в виде коричневой пены.
ЖХ-МС: tR=0,57 мин; [М+Н]+: 250,13.
Примеры
Пример 1: рац-(1R*,2R*,4R*)-2-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол
1.1 (метод П1.1): рац-(1R*,2R*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусная кислота
К раствору 4,0 г трет-бутилового эфира рац-(1R*,2R*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты в 25 мл EtOH добавляют 2,1 г гидрата LiOH, 8 мл Н2О и 22 мл МеОН. Реакционную смесь перемешивают при комнатной температуре в течение 3 дней и затем концентрируют. Остаток распределяют между водой и Et2O. Водный слой отделяют и подкисляют 1-нормальным раствором HCl, в результате чего образуется белое твердое вещество, которое отфильтровывают, промывают 5 мл разбавленной HCl и высушивают в вакууме, получая 3,2 г рац-(1R*,2R*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты в виде белого твердого вещества.
ЖХ-МС: tR=0,86 мин; [М-Н2О+Н]+: 241,28.
1.2 (метод П1.2): рац-(1R*,2R*,4R*)-N-[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]-2-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)-N-метилацетамид
К раствору 280 мг рац-(1R*,2R*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты в 7 мл ТГФ прибавляют 0,58 мл ДИПЭА, 175 мг ГОБТ и 250 мг ЭДК при комнатной температуре. После перемешивания в течение 10 мин добавляют 270 мг 3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламина и реакционную смесь перемешивают при комнатной температуре в течение ночи. Затем реакционную смесь гасят насыщенным водным раствором NaHCO3, фазы разделяют и органическую фазу промывают водой и рассолом, высушивают над MgSO4 и концентрируют в вакууме. Очистка с помощью колоночной хроматографии с использованием смеси EtOAc/МеОН (в соотношении от 5:1 до 2:1) дает 475 мг рац-(1R*,2R*,4R*)-N-[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]-2-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)-N-метилацетамида в виде белой пены.
ЖХ-МС: tR=0,91 мин; [M+H]+: 490,06.
1.3 (метод П1.3): рац-(1R*,2R*,4R*)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол
К раствору 310 мг рац-(1R*,2R*,4R*)-N-[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]-2-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)-N-метилацетамида в 8 мл толуола добавляют по каплям 0,77 мл Red-Al (65%-ный раствор в толуоле) при температуре 0°С. После перемешивания в течение 10 мин при температуре 0°С охлаждающую баню удаляют и перемешивание продолжают в течение 3 ч при комнатной температуре. Реакционную смесь затем осторожно переносят в смесь 1-молярного ледяного раствора NaOH и перемешивают в течение 10 мин. Водную фазу экстрагируют толуолом, объединенные органические фазы промывают рассолом, высушивают над MgSO4 и концентрируют в вакууме. Очистка с помощью колоночной хроматографии с использованием смеси EtOAc/МеОН (в соотношении 2:1) дает 230 мг рац-(1R*,2R*,4R*)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ола в виде белой пены.
ЖХ-МС: tR=0,79 мин; [M+H]+: 476,13.
Пример 1А: (1R*,2R*,4R*)-2-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир рац-изомасляной кислоты
1А.1 (метод П1.4): (1R*,2R*,4R*)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир рац-изомасляной кислоты
К раствору 199 мг рац-(1R*,2R*,4R*)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ола в 4 мл ДХМ добавляют 0,2 мл NEt3 и 0,1 мл изобутирилхлорида при температуре 0°С. Реакционную смесь при перемешивании в течение ночи оставляют самопроизвольно нагреваться до комнатной температуры. Затем смесь гасят насыщенным водным раствором NaHCO3, фазы разделяют и водную фазу экстрагируют ДХМ. Объединенные органические фазы промывают рассолом, высушивают над MgSO4 и концентрируют в вакууме. Остаток растворяют в 3 мл EtOAc, добавляют силикагель и 1,5 мл МеОН и смесь энергично перемешивают в течение 7 дней, после чего смесь фильтруют, промывают смесью EtOAc/MeOH (в соотношении 2:1) и выпаривают. Очистка с помощью колоночной хроматографии с использованием смеси EtOAc/MeOH (в соотношении от 5:1 до 3:1 + 0,1% NEt3) дает 186 мг (1R*,2R*,4R*)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-илового эфира рац-изомасляной кислоты в виде бежевой пены.
ЖХ-МС: tR=0,90 мин; [M+H]+: 546,23.
1А.2 (Метод П1.5): Дихлорид (1R*,2R*,4R*)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-илового эфира рац-изомасляной кислоты
Полученный выше продукт может быть переведен в соответствующую дигидрохлоридную соль с помощью следующего метода.
К раствору 186 мг (1R*,2R*,4R*)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-илового эфира рац-изомасляной кислоты в 2 мл EtOAc добавляют 0,3 мл 3-молярного раствора HCl в EtOAc при температуре 0°С. Реакционную смесь выпаривают досуха без нагревания, получая 199 мг дихлорида (1R*,2R*,4R*)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-илового эфира рац-изомасляной кислоты.
Пример 2: (1R,2R,4R)-2-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол или (1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол
2.1: (1R,2R,4R)-(2-Гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусная кислота или (1S,2S,4S)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусная кислота
Получают согласно методу П1.1 в примере 1, используя энантиомер Б трет-бутилового эфира рац-(1R*,2R*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты (см. К1А.6).
ЖХ-МС: tR=0,91 мин; [M-H2O+H]+: 241,10.
2.2: (1R,2R,4R)-2-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол или (1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол
Получают согласно методам П1.2-П1.3 примера 1, используя полученную выше (2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусную кислоту.
ЖХ-МС: tR=0,78 мин; [М+Н]+: 476,09.
Пример 2А: (1R,2R,4R)-2-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты или (1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты
Получают согласно методу П1.4 примера 1А, используя полученный выше 2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол (соединение примера 2).
ЖХ-МС: tR=0,89 мин; [М+H]+: 546,19.
Пример 3: (1R,2R,4R)-2-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол или (1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол
3.1: (1R,2R,4R)-(2-Гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусная кислота или (1S,2S,4S)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусная кислота
Получают согласно методу П1.1 примера 1, используя энантиомер А трет-бутилового эфира рац-(1R*,2R*,4R*)-(2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусной кислоты (см. К1А.6).
ЖХ-МС: tR=0,91 мин; [M-H2O+H]+: 241,16.
3.2: (1R,2R,4R)-2-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол или (1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол
Получают согласно методам П1.2-П1.3 примера 1, используя полученную выше (2-гидрокси-5-фенилбицикло[2.2.2]окт-5-ен-2-ил)уксусную кислоту.
ЖХ-МС: tR=0,79 мин; [М+Н]+: 476,09.
Пример 3А: (1R,2R,4R)-2-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты или (1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты
Получают согласно методу П1.4 примера 1А, используя полученный выше 2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-ол (соединение примера 3).
ЖХ-МС: tR=0,89 мин; [M+H]+: 546,11.
Пример 4: рац-(1R*,5R*,6R*)-6-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-ол
4.1: рац-(1R*,5R*,6R*)-(6-Гидрокси-8-фенилбицикло[3.2.2]нон-8-ен-6-ил)уксусная кислота
Получают согласно методу П1.1 примера 1, используя трет-бутиловый эфир рац-(1R*,5R*,6R*)-6-гидрокси-8-фенилбицикло[3.2.2]нон-8-ен-6-ил)уксусной кислоты (см. К2А.3).
ЖХ-МС: tR=0,96 мин; [M+Na+H]+: 296,10.
4.2: рац-(1R*,5R*,6R*)-6-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-ол
Получают согласно методам П1.2-П1.3 примера 1, используя рац-(1R*,5R*,6R*)-(6-гидрокси-8-фенилбицикло[3.2.2]нон-8-ен-6-ил)уксусную кислоту.
ЖХ-МС: tR=0,80 мин; [M+H]+: 490,06.
Пример 4А: (1R*,5R*,6R*)-6-(2-{[3-(4,7-Диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-иловый эфир рац-изомасляной кислоты
Получают согласно методу П1.4 примера 1А, используя рац-(1R*,5R*,6R*)-6-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-ол.
ЖХ-МС: tR=0,91 мин; [M+H]+: 560,05.
Биологические тесты
Анализ L каналов в условиях in vitro
L канальная антогонистическая активность (IC50) соединений формулы (I) определяется в соответствии со следующим экспериментальным методом.
Человеческие эмбриональные почечные клетки (НЕК293), экспрессируемые человеческим Cav1.2 каналом в дополнение к вспомогательным субъединицам β-2а и α2δ-1, выращивают в культуральной среде (DMEM, содержащая 10% инактивированной нагреванием фетальной телячьей сыворотки (ФТС), 100 U/мл пенициллина, 100 мкг/мл стрептомицина, 100 мкг/мл G418, 40 мкг/мл цеосина и 100 мкг/мл гидромицина). Клетки высеивают в количестве 20.000 клеток/ячейку в 384-ячеистые черные стерильные планшеты с прозрачным дном (покрытые поли-L-лизином, фирма Becton Dickinson). Засеянные планшеты инкубируют в течение ночи при температуре 37°С в 5% СО2. KCl-раствор готовят в виде 80 мМ раствора для разведения в буфере для анализа (HBSS, содержащий 0,1% альбумина бычьей сыворотки, 20 мМ HEPES, 0,375 г/л NaHCO3, скорректированный до рН 7,4 с помощью NaOH) для использования при анализе с конечной концентрацией 20 мМ. Антагонисты получают в виде 10 мМ растворов для разведения в ДМСО, затем разводят в 384-ячеистых планшетах сначала в ДМСО, затем в буфере для анализа для получения 3-кратного разведения. На следующий день проведения анализа добавляют 25 мкл буфера для окрашивания (HBSS, содержащий 20 мМ HEPES, 0,375 г/л NaHCO3 и 3 мкМ флюоресцентного кальциевого индикатора fluo-4 AM (1 мМ раствора для разбавления в ДМСО, содержащего 10% плюроника) в каждую ячейку засеянного планшета. 384-ячеистые клеточные планшеты инкубируют в течение 60 мин при температуре 37°С в 5% CO2, после чего дважды промывают 50 мкл на ячейку, используя буфер для анализа и оставляя 50 мкл/ячейку этого буфера для достижения равновесия при этой температуре (30-60 мин). В флюоресцентный томографический планшет-ридер (FLIPR, фирма Molecular Devices) антагонисты добавляют в объеме 25 мкл/ячейку, инкубируют в течение 3 мин и, наконец, добавляют 25 мкл/ячейку KCl-раствора для клеточной деполяризации. Флюоресценцию измеряют с 2-секундным интервалом в течение 8 мин и площадь флюоресцентного пика сравнивают с площадью флюоресцентного пика, индуцированного 20 мМ KCl, с растворителем вместо антагониста. Для каждого антагониста IC50 величина (концентрация в нМ соединения, необходимого для ингибирования 50% KCl-индуцируемого флюоресцентного ответа) измерялась вплоть до 10 мкМ.
Соединения примеров 1, 2, 3, 4 не тестировались в анализе. IC50 величины соединений примеров 1А, 2А, 3А и 4А составляли от 156 до 439 нМ со средней величиной 305 нМ.
Анализ Т каналов в условиях in vitro
Т канальная антагонистическая активность (IC50 величины) соединений формулы (I) определяется согласно следующему экспериментальному методу, данные которого представлены в таблице 1.
Человеческие эмбриональные почечные клетки (НЕК293), экспрессируемые человеческим Cav3.1, Cav3.2 или Cav3.3 каналом, соответственно, выращивают в культуральной среде (DMEM, содержащая 10% инактивированной нагреванием фетальной телячьей сыворотки (ФТС), 100 U/мл пенициллина, 100 мкг/мл стрептомицина и 1 мг/мл G418). Клетки высеивают в количестве 20.000 клеток/ячейку в 384-ячеистые черные стерильные планшеты с прозрачным дном (покрытые поли-L-лизином, фирма Becton Dickinson). Засеянные планшеты инкубируют в течение ночи при температуре 37°С в 5% СО2. Са2+-раствор готовят в виде 100 мМ раствора для разведения в 100 мМ тетраэтиламмонийхлорида (ТЭА-хлорид), 50 мМ HEPES, 2,5 мМ CaCl2, 5 мМ KCl, 1 мМ MgCl2, скорректированный до рН 7,2 с помощью ТЭА-гидрохлорида, для использования при анализе с конечной концентрацией 10 мМ. Антагонисты получают в виде 10 мМ растворов для разведения в ДМСО, затем разводят в 384-ячеистых планшетах сначала в ДМСО, затем в 100 мМ ТЭА-хлорида, 50 мМ HEPES, 2,5 мМ CaCl2, 5 мМ KCl, 1 мМ MgCl2, скорректированного до рН 7,2 с помощью ТЭА-гидрохлорида, для получения 9-кратного разбавления. На следующий день проведения анализа добавляют 25 мкл буфера для окрашивания (HBSS, содержащий 20 мМ HEPES, 0,375 г/л NaHCO3 и 3 мкМ флюоресцентного кальциевого индикатора fluo-4 AM (1 мМ раствора для разбавления в ДМСО, содержащего 10% плюроника) в каждую ячейку засеянного планшета. 384-ячеистые клеточные планшеты инкубируют в течение 60 мин при температуре 37°С в 5% СО2, после чего дважды промывают 50 мкл на ячейку, используя HBSS, содержащий 0,1% БСА, 20 мМ HEPES, 0,375 г/л NaHCO3, оставляя 50 мкл/ячейку этого буфера для достижения равновесия при этой температуре (30-60 мин). В флюоресцентный томографический планшет-ридер (FLIPR, фирма Molecular Devices) антагонисты добавляют в объеме 6,25 мкл/ячейку, инкубируют в течение 3 мин и, наконец, добавляют 6,25 мкл/ячейку Са2+ раствора. Флюоресценцию измеряют с 2-секундным интервалом в течение 8 мин и площадь флюоресцентного пика сравнивают с площадью флюоресцентного пика, индуцированного 10 мМ Са2+, с растворителем вместо антагониста. Для каждого антагониста IC50 (концентрация в нМ соединения, необходимого для ингибирования 50% Са2+-индуцируемого флюоресцентного ответа) измерялась вплоть до 10 мкМ.
Воздействие на изолированные сердца по методу Лангедорффа (Lgdff)
Соединения были протестированы на их потенциальную способность восстановления кровяного давления и их действие на сокращение сердечной мышцы. ЕС50 величины на изолированных сердцах мышей определялись соответственно литературным публикациям (Doring HJ., The isolated perfused heart according to Langendorff technique-function-application, Physiol. Bohemoslov., 1990, 39(6), 481-504; Kligfield P, Horner H, Brachfeld N., A model of graded ischemia in the isolated perfused rat heart, J. Appl. Physiol., 1976, Jun, 40(6), 1004-8).
Соединение примера 1А, исследованное с помощью метода, описанного выше в эксперименте Лангендорффа, имело показатель ЕС50, составляющий 5 нМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛИ (1R*,2R*,4R*)-2-(2-{[3(4,7-ДИМЕТОКСИ-1Н-БЕНЗОИМИДАЗОЛ-2-ИЛ)ПРОПИЛ]МЕТИЛАМИНО}ЭТИЛ)-5-ФЕНИЛБИЦИКЛО[2.2.2]ОКТ-5-ЕН-2-ИЛОВОЙ ИЗОМАСЛЯНОЙ КИСЛОТЫ | 2009 |
|
RU2516247C2 |
| МОСТИКОВЫЕ ШЕСТИЧЛЕННЫЕ ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2503663C2 |
| N-ДИГИДРОКСИАЛКИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 2-ОКСОИМИДАЗОЛА | 2006 |
|
RU2414456C2 |
| ВТОРИЧНЫЕ АМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕНИНА | 2007 |
|
RU2425032C2 |
| КОНДЕНСИРОВАННОЕ ХИНОЛИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2384571C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2625309C2 |
| МОДУЛЯЦИЯ ВОСПАЛИТЕЛЬНЫХ И МЕТАСТАТИЧЕСКИХ ПРОЦЕССОВ | 2005 |
|
RU2377988C2 |
| ПРОИЗВОДНЫЕ БИЦИКЛО[2,2,1]ГЕПТ-7-ИЛАМИНА И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2442771C2 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ, ПРОТИВОВИРУСНЫЙ АКТИВНЫЙ КОМПОНЕНТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2010 |
|
RU2452735C1 |
| ЛИГАНДЫ НИКОТИНОВОГО РЕЦЕПТОРА АЛЬФА-7, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2005 |
|
RU2418797C2 |
Настоящее изобретение относится к соединениям формулы (I) или к фармацевтически приемлемым солям такого соединения, где R1 представляет собой незамещенный фенил; R2 представляет собой -CO-R21; R21 представляет собой С1-5алкил; m представляет собой целое число 3; р представляет собой целое число 2 или 3; и R3 представляет собой водород. Также изобретение относится к конкретным соединениям формулы (I), к фармацевтической композиции на основе соединения формулы (I) и к применению соединения формулы (I). Технический результат: получены новые соединения, полезные в качестве блокаторов T/L каналов. 3 н. и 3 з.п. ф-лы, 1 табл., 8 пр.
1. Соединение формулы (I)
где R1 представляет собой незамещенный фенил;
R2 представляет собой -CO-R21;
R21 представляет собой С1-5алкил;
m представляет собой целое число 3;
р представляет собой целое число 2 или 3; и
R3 представляет собой водород;
или фармацевтически приемлемая соль такого соединения.
2. Соединение формулы (I) по п.1, где конфигурация мостикового циклогексенового фрагмента является такой, что R2-О-заместитель и мостик -(СН2)р- циклогексенового фрагмента находятся в цис-положении;
или фармацевтически приемлемая соль такого соединения.
3. Соединение формулы (I) по п.1, выбранное из следующих соединений:
(1R,2R,4R)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты,
(1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты,
(1R,5R,6R)-6-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-иловый эфир изомасляной кислоты и
(1S,5S,6S)-6-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-8-фенилбицикло[3.2.2]нон-8-ен-6-иловый эфир изомасляной кислоты,
или фармацевтически приемлемая соль такого соединения.
4. Соединение формулы (I) по п.1, выбранное из следующих соединений:
(1R,2R,4R)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты,
(1S,2S,4S)-2-(2-{[3-(4,7-диметокси-1Н-бензимидазол-2-ил)пропил]метиламино}этил)-5-фенилбицикло[2.2.2]окт-5-ен-2-иловый эфир изомасляной кислоты,
или фармацевтически приемлемая соль такого соединения.
5. Фармацевтическая композиция для лечения или профилактики хронической стабильной стенокардии, гипертензии, почечной или сердечной ишемии, сердечной аритмии, включая предсердную фибрилляцию, сердечной гипертрофии или застойной сердечной недостаточности, содержащая в качестве активного компонента соединение формулы (I) по одному из пп.1-4 или его фармацевтически приемлемую соль и по крайней мере один терапевтически инертный наполнитель.
6. Применение соединения формулы (I) по одному из пп.1-4 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики хронической стабильной стенокардии, гипертензии, почечной или сердечной ишемии, сердечной аритмии, включая предсердную фибрилляцию, сердечной гипертрофии или застойной сердечной недостаточности.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| US 4808605 A, 28.02.1989 | |||
| Способ растворения смеси гафниевых и циркониевых солей фосфорной кислоты | 1925 |
|
SU3947A1 |

