Область техники, к которой относится изобретение
Настоящее изобретение относится к способу селективной кристаллизации Z-формы изомера иопромида из неочищенного кристаллического иопромида, содержащего смесь E- и Z-изомеров иопромида, и к композиции, включающей Z-изомер иопромида, полученный указанным способом.
Уровень техники
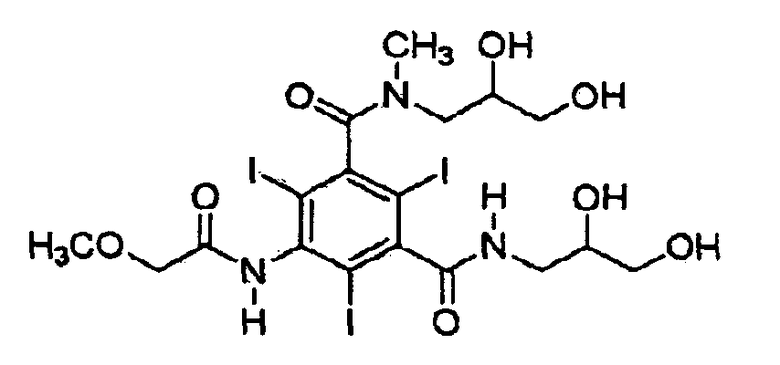
Иопромид 5-метоксиацетиламино-2,4,6-трииодизофталевой кислоты [(2,3-дигидрокси-N-метил-пропил)-(2,3-дигидроксипропил)]диамид формулы (I) является иодсодержащим рентгеноконтрастным средством, которое имеет 3 объемных атома йода в положениях 2, 4 и 6 фенильной группы, которые пространственно препятствуют свободному вращению дигидроксипропил-N-метиламиногруппы, так что имеют место два атропизомера (М. Oki, Topics in Stereochemistry, v. 14, 1983, pp. 1-81; и H. Staab et al., Tetrahedron Letters, no. 38, 1966, pp. 4593-4598).
(Формула 1)
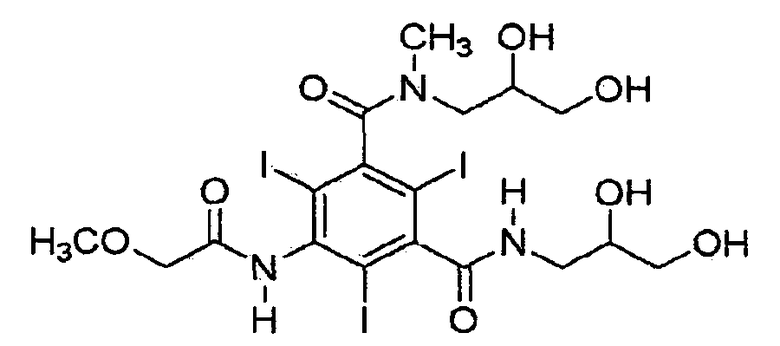
Помимо двух атропизомеров иопромида также существуют E- и Z-изомеры, образующиеся из-за пространственного затруднения свободного вращения вдоль связи между атомом углерода и атомом азота амидных связей. Соответственно, иопромид состоит из смеси четырех изомеров - Е1, E2, Z1 и Z2. В одном из атропизомеров замещенный атом азота находится выше плоскости бензольного ядра иопромида, в то время как в другом атом азота лежит ниже плоскости бензольного ядра.
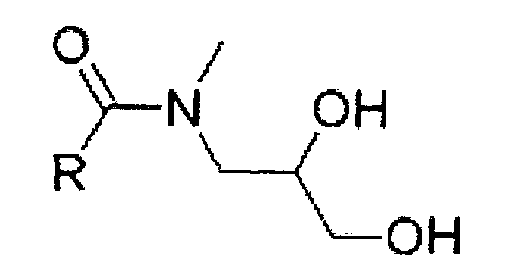
Как показано в формулах 2A и 2B, формы E и Z могут отличаться расположением заместителей вокруг связи C-N дигидрокси-N-метилпропил амидной группы.
(Формула 2А)
(Формула 2В)
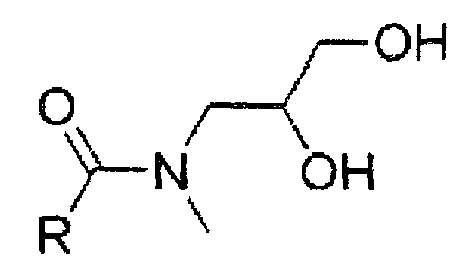
где R представляет собой фенильную группу иопромида.
Эти изомеры имеют различные физические свойства и относительное количество единственного изомера, так же как относительное содержание изомеров отрегулировано в фармацевтическом составе. Согласно фармакопее США (the Unated States Pharmacopoeia, 31st Edition, 2008, pp. 2433-2435), фармацевтический сырьевой материал должен содержать 40-50% формы 1 изомера и 49-60% формы 2 изомера, и лекарственный продукт должен содержать от 8,0 до 12,0% изомера Е1, от 9,0 до 14,0 изомера E2, от 32,0 до 40,0% изомера Z1 и от 38,0 до 46,0% изомера Z2.
С другой стороны, отсутствует эффективный способ кристаллизации иопромида в настоящее время. Патент США 4364921 раскрывает способ получения иопромида, но способ не использует заключительную стадию кристаллизации. Несмотря на то, что в европейском патенте EP 1 025 067 и английском патенте GB 2 280 436 описан способ промывки и кристаллизации иопамидола и иодиксанола, в них не раскрыта какая-либо процедура кристаллизации иопромида или изомеров иопромида.
Инъекционный состав иопромида может быть получен растворением фармацевтического сырьевого материала в воде, добавлением фармацевтически приемлемых эксципиентов к раствору, и стерилизацией его, но когда используют обычный сырьевой материал иопромида, содержащий E- и Z-изомеры, для получения фармацевтического продукта, относительное содержание изомеров в продукте часто не соответствует указанным правилам.
В международной публикации WO 2007/065534 описан способ регенерации иопромида, пригодного для фармацевтических целей, из раствора иопромида. Однако в ней просто описано выделение изомеров форм 1 и 2 и поэтому трудно выборочно получить Z-изомер иопромида.
Авторы настоящей заявки нашли способ селективного растворения и кристаллизации Z-изомера иопромида из кристаллического иопромида или концентрата иопромида, содержащего E- и Z-изомеры.
Сущность изобретения
Соответственно, целью настоящего изобретения является предоставление высокопродуктивного способа селективной кристаллизации Z-изомера иопромида из неочищенного иопромида или концентрата иопромида, содержащего смесь E- и Z-изомеров.
Другой целью настоящего изобретения является предоставление способа получения состава, включающего Z-изомер иопромида, полученного вышеупомянутым способом кристаллизации.
В соответствии с одним аспектом настоящего изобретения описан способ селективной кристаллизации Z-изомера иопромида, включающий a) растворение неочищенного иопромида или концентрата иопромида, содержащего E- и Z-изомеры в спирте; и b) нагревание образующегося спиртового раствора, чтобы селективно получить кристаллический Z-изомер иопромида.
В соответствии с другим аспектом настоящего изобретения обеспечивают способ получения состава, включающего Z-изомер иопромида, включающий i) селективную кристаллизацию Z-изомера иопромида из неочищенных кристаллов, содержащих смесь E- и Z-изомеров иопромида, или концентрата иопромида, чтобы получить кристаллический Z-изомер иопромида; и ii) растворение кристаллического Z-изомера иопромида совместно с фармацевтически приемлемым эксципиентом.
Краткое описание чертежей
Вышеупомянутые и другие цели и особенности настоящего изобретения будут становиться очевидными из следующего описания изобретения, при взятии вместе с сопровождающими рисунками, на которых, соответственно, показаны:
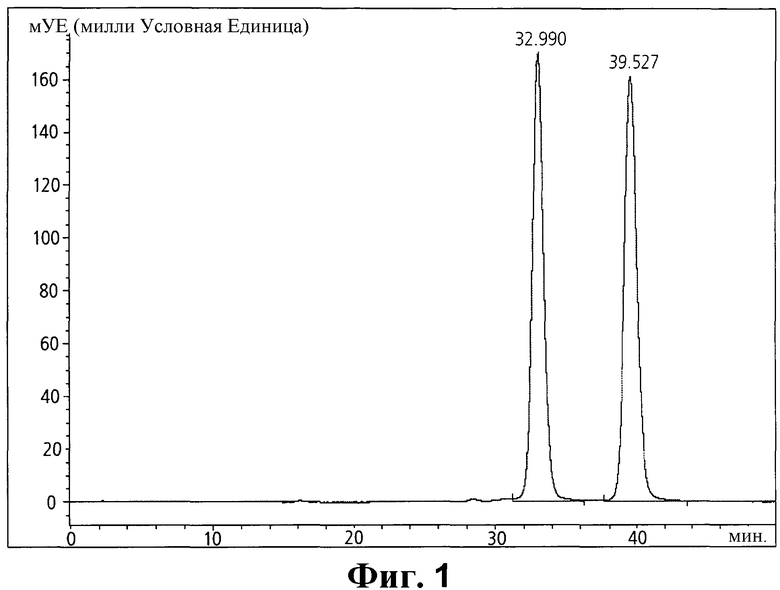
Фиг. 1 - хроматограмма кристалла иопромида по изобретению, содержащего Z-изомер, полученный в Примере 1; и
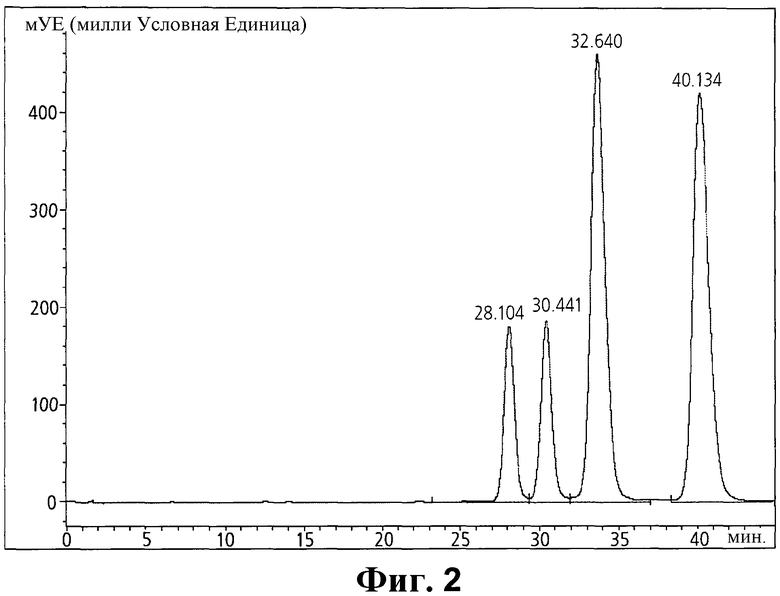
Фиг. 2 - хроматограмма кристалла неочищенного иопромида, содержащего E- и Z-изомеры.
Подробное описание изобретения
В настоящем изобретении описан способ селективной кристаллизации Z-изомера иопромида, включающий a) растворение неочищенного иопромида, содержащего смесь E- и Z-изомеров, или концентрата иопромида в спирте; и b), нагревание спиртового раствора, чтобы селективно получить кристаллический Z-изомер иопромида.
Как показано в Схеме 1, неочищенный иопромид может быть получен способом, раскрытым в патенте США 4364921:
<Схема 1>
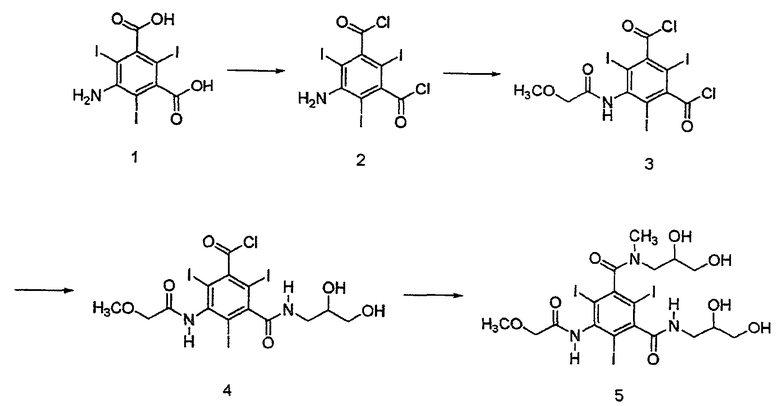
Неочищенный иопромид или концентрат иопромида могут содержать изомеры в отношении E-изомер: Z-изомер от 1:0,5 до 1:10, и от 0 до 15 вес.% влаги.
В стадии a) настоящего изобретения объем (мл) спирта, который будет использоваться для растворения неочищенных кристаллов иопромида или концентрата иопромида, является соответственно 0,1-10-кратным, предпочтительно 1-3-кратным весу (г) кристаллов неочищенного иопромида или концентрата иопромида. Предпочтительным спиртом, используемым здесь является нормальный или разветвленный алифатический С1-10 спирт, предпочтительно, метанол, этанол, изопропанол, н-бутанол, втор-бутанол, пентанол, октанол, деканол или их смесь.
В стадии b) настоящего изобретения термическая обработка может быть достигнута различными методами, известными в технологии, предпочтительно нагреванием с обратным холодильником. Z-Изомер может быть селективно кристаллизован нагреванием с обратным холодильником в течение 1-48 часов, при поддержке внешней температуры сосуда от 50 до 200°C, предпочтительно, от 80 до 180°C, более предпочтительно, от 100 до 150°C, при нормальном атмосферном давлении. Время нагревания с обратным холодильником может быть изменено в зависимости от масштаба эксперимента, то есть масса неочищенного иопромида и вышеупомянутый интервал времени предназначены иллюстрировать настоящее изобретение, не ограничивая его объем.
Содержание Z-изомера в кристаллах иопромида, полученных вышеупомянутым способом, составляет предпочтительно 85-100%, или кристаллы иопромида могут состоять из 100% Z-формы изомера или содержать 5% или меньше Е-формы изомера.
Когда кристаллизацию проводят способом по настоящему изобретению, время нагревания с обратным холодильником может быть сокращено и поэтому количество продуктов разложения из-за воздействия высокой температуры в течение долгого времени может быть снижено.
В соответствии с другим аспектом настоящего изобретения обеспечивают способ получения состава, включающего Z-изомер иопромида, включающий i) селективную кристаллизацию Z-изомера иопромида из неочищенного иопромида, включающего смесь E- и Z-изомеров иопромида или концентрата иопромида, чтобы получить кристаллический Z-изомер иопромида; и ii) растворение кристаллического Z-изомера иопромида, и фармацевтически приемлемый инертный наполнитель.
Стадия i) кристаллизации в вышеуказанном способе получения состава характеризуется включением 1) растворения неочищенного иопромида или концентрата иопромида, содержащего E- и Z-изомеры в спирте; и 2) нагревание образующегося спиртового раствора, чтобы получить кристаллический Z-изомер иопромида, и детали способа получения являются теми же самыми, что и детали вышеупомянутого способа кристаллизации Z-изомера иопромида.
Средство регуляции рН, такое как NaOH, и стабилизатор, такой как кальцийдинатриевая соль ЭДТУК (этилендиаминтетрауксусной кислоты), могут использоваться в способе получения Z-изомер-содержащего состава (смотри патент США 4634921), а водным растворителем, как используется здесь, может быть вода.
Состав, включающий Z-изомер иопромида, полученный вышеупомянутым способом, может стерилизоваться и быть составлен в форме состава для инъекций, и бесцветный и прозрачный жидкий состав предпочтителен.
С Z-изомером иопромид-содержащего состава, полученным способом по настоящему изобретению, фармацевтическое сырье делает отношения Е1, -E2, -Z1- и Z2-изомеров такими, чтобы удовлетворять стандартам, подходящим для фармацевтического состава, такого, как описан в Фармакопее Соединенных Штатов (USP). Отношение форма 1 изомера:форма 2 изомера могут рассматриваться как отношение Z1-изомер : Z2-изомер, так как отношение форма 1 изомера:форма 2 изомера обычно относится к (E1+Z1):(E2+Z2), а отношения относительных количеств Е1 к Z1 и E2 к Z2 не регулируются. В этой связи, при регулировке относительные количества изомеров иопромида в неочищенном фармацевтическом материале на основе Z1 и Z2 будут намного лучше (воспроизводимо) удовлетворять указанному стандарту.
Настоящее изобретение будет описано далее подробно в отношении следующих Примеров. Однако следует понимать, что существующее изобретение не ограничивается определенными Примерами.
Пример 1
Кристаллизация Z-изомера с применением этанола
10 г 5-метоксиацетиламино-2,4,6-трииодизофталевой кислоты [(N-метил-2,3-дигидроксипропил)-(2,3-дигидроксипропил)]диамида, содержащего 0,5 вес.% влаги (12,64 ммоль), синтезировали способом, раскрытым в патенте США 4364921. Далее, неочищенные кристаллы, полученные указанным способом, растворяли в 10 мл безводного этанола, и раствор кипятили с обратным холодильником, поддерживая внешнюю температуру сосуда при 120°C. Затем кристаллы Z-изомера иопромида, полученные кипячением с обратным холодильником в течение 6 часов и охлаждением до комнатной температуры, перемешивали в течение 1 часа и фильтровали. Выделенные твердые кристаллы промывали безводным этанолом, охлажденным до 0-5°C, и сушили при 80°C при пониженном давлении, чтобы получить 9,5 г соединения (выход 96%) в форме белого твердого вещества.
Содержание изомера в полученных кристаллах определяли жидкостной хроматографией, используя колонку 4,6 мм × 25 см (5 мкм набивка L1) согласно способу, описанному в Фармакопее Соединенных Штатов (USP), а результаты на Фиг. 2 показали, что кристаллы состояли из 46,4% Z1 и 53,6% Z2 (содержание Z-изомера 100%).
Далее, неочищенные кристаллы иопромида, полученные способом, раскрытым в патенте США 4364921, анализировали так же, как показано выше, и результаты показаны на Фиг. 1.
Пример 2
Кристаллизация Z-изомера с применением этанола
10 г 5-метоксиацетиламино-2,4,6-трииодизофталевой кислоты [(N-метил-2,3-дигидроксипропил)-(2,3-дигидроксипропил)]диамида, содержащего 3,0 вес.% влаги (12,64 ммоль), синтезировали способом, раскрытым в патенте США 4364921. Далее, неочищенные кристаллы, полученные указанным способом, растворяли в 10 мл безводного этанола и раствор кипятили с обратным холодильником, поддерживая внешнюю температуру сосуда при 120°C. Затем кристаллы Z-изомера иопромида, полученные кипячением с обратным холодильником в течение 6 часов и охлаждением до комнатной температуры, перемешивали в течение 1 часа и фильтровали. Выделенные твердые кристаллы промывали безводным этанолом, охлаждали до 0-5°C и сушили при 80°C при пониженном давлении, чтобы получить 9,5 г соединения (выход 90%) в форме белого твердого вещества.
Содержание изомеров в полученных кристаллах определяли так же, как в вышеупомянутом Примере 1.
Содержание изомеров в полученных кристаллах: 46,8% Z1, 53,2% Z2 (содержание Z-изомера: 100 %).
Пример 3
Кристаллизация Z-изомера с применением изопропанола
10 г 5-метоксиацетиламино-2,4,6-трииодизофталевой кислоты [(N-метил-2,3-дигидроксипропил)-(2,3-дигидроксипропил)]диамида, содержащего 3,0 вес.% влаги (12,64 ммоль), синтезировали способом, раскрытым в патенте США 4364921. Далее, неочищенные кристаллы, полученные указанным способом, растворяли в 10 мл изопропанола, и раствор кипятили с обратным холодильником, поддерживая внешнюю температуру сосуда при 120°C. Затем кристаллы Z-изомера иопромида, полученные кипячением с обратным холодильником в течение 6 часов и охлаждением до комнатной температуры, перемешивали в течение 1 часа и фильтровали. Выделенные твердые кристаллы промывали изопропанолом, охлаждали до 0-5°C, и сушили при 80°C при пониженном давлении, чтобы получить 9,5 г соединения (выход: 95%) в форме белого твердого вещества.
Содержание изомеров в полученных кристаллах определяли так же, как в вышеупомянутом Примере 1.
Содержание изомеров в полученных кристаллах: 45,5% Z1, 54,5% Z2 (содержание Z-изомера 100 %).
Пример 4
Кристаллизация Z-изомера с применением метанола
10 г 5-метоксиацетиламино-2,4,6-трииодизофталевой кислоты [(N-метил-2,3-дигидроксипропил)-(2,3-дигидроксипропил)]диамида, содержащего 3,0 вес.% влаги (12,64 ммоль), синтезировали способом, раскрытым в патенте США 4364921. Далее, неочищенные кристаллы, полученные указанным способом, растворяли в 5 мл изопропанола, и раствор кипятили с обратным холодильником, поддерживая внешнюю температуру сосуда при 120°C. Затем кристаллы Z-изомера иопромида, полученные кипячением с обратным холодильником в течение 12 часов и охлаждением до комнатной температуры, перемешивали в течение 1 часа и фильтровали. Выделенные твердые кристаллы промывали изопропанолом, охлаждали до 0-5°C, и сушили при 80°C при пониженном давлении, чтобы получить 8,5 г соединения (выход 85%) в форме белого твердого вещества.
Содержание изомеров в полученных кристаллах определяли так же, как в вышеупомянутом Примере 1.
Содержание изомеров в полученных кристаллах: 47,6% Z1, 52,4% Z2 (содержание Z-изомера 100 %).
Пример 5
Кристаллизация Z-изомера с применением н-бутанола
10 г 5-метоксиацетиламино-2,4,6-трииодизофталевой кислоты [(N-метил-2,3-дигидроксипропил)-(2,3-дигидроксипропил)]диамида, содержащего 3,0 вес.% влаги (12,64 ммоль), синтезировали способом, раскрытым в патенте США 4364921. Далее, неочищенные кристаллы, полученные указанным способом, растворяли в 10 мл бутанола, и раствор кипятили с обратным холодильником, поддерживая внешнюю температуру сосуда при 120°C. Затем кристаллы Z-изомера иопромида, полученные кипячением с обратным холодильником в течение 6 часов и охлаждением до комнатной температуры, перемешивали в течение 1 часа и фильтровали. Выделенные твердые кристаллы промывали н-бутанолом, охлаждали до 0-5°C, и сушили при 80°C при пониженном давлении, чтобы получить 9,5 г соединения (выход 95%) в форме белого твердого вещества.
Содержание изомеров в полученных кристаллах определяли так же, как в вышеупомянутом Примере 1.
Содержание изомеров в полученных кристаллах: 48,3% Z1, 51,7% Z2 (содержание Z-изомера 100%).
Количество свободного йодида и остаточного растворителя в каждых кристаллах иопромида, полученных в Примерах 1-5, определяли согласно методу, описанному в Фармакопее Соединенных Штатов, 31-е издание, с использованием потенциометрического титратора и газовой хроматографии, так как их сравнивали со стандартными пределами концентрата, описанными в Фармакопее Соединенных Штатов (Фармакопея) и указаниях Международной конференции по вопросам гармонизации для регистрации лекарственных препаратов для использования человеком (Конференция). Результаты показаны в Таблице 1. Стандартные пределы концентрата остаточных растворителей для метанола (MeOH), изопропанола (ИП) и н-бутанола (н-BuOH) были вычислены на основании Допустимого Ежедневного Воздействия (ДЕВ), описанного в указаниях Конференции, а стандартный предел концентрата остаточного этанола (EtOH) был вычислен на основании ДЕВ, описанного в Фармакопее.
Методы вычисления стандартных пределов концентрата остаточных растворителей с ДЕВ следующие:
1) максимальная суточная доза иопромида (Ultravist™ 370, на основании стандарта Департамента по контролю за качеством пищевых продуктов, медикаментов и косметических средств (Департамент): (769 мг/мл) предоставление 225 мл=173,025 г/день;
2) справочные пределы концентрата остаточного растворителя (частей на миллион)=(1000 предоставление ДЕВ)/суточная доза,
i) MeOH (частей на миллион)=(1000×30 мг/день)/(173,025 г/день)=173,38 частей на миллион.
ii) ИП (частей на миллион)=(1000×50 мг/день)/(173,025 г/день)=288,976 частей на миллион;
iii) н-BuOH (частей на миллион)=(1000×50 мг/день)/(173,025 г/день)=288,976 частей на миллион.
Как может быть видно из вышеуказанной Таблицы 1, кристаллы иопромида, полученные способом по настоящему изобретению, удовлетворяют стандартам, описанным в Фармакопее США и указаниях Конференции, а способ по настоящему изобретению имеет эффективность, превосходящую эффективность обычных способов с точки зрения снижения времени кипячения с обратным холодильником и снижения количества продуктов разложения.
Испытательный Пример 1
Получение фармацевтического состава и отношения изомеров в составе
62,34 г кристаллов иопромида, содержащих только Z-изомер, полученный в Примере 1 (46,4% Z1-изомера и 53,6% Z2-изомера) растворяли в 0,01 г этилендиаминтетрауксуснокислого кальция и 100 мл вторичной дистиллированной воды, и затем рН 7,2 раствора устанавливали с помощью 0,1N NaOH. Раствор выливали в емкость и стерилизовали в течение 20 минут при 120°C, чтобы получить фармацевтический состав. Как показано в Таблице 2, относительное количество изомеров иопромида в составе удовлетворяло стандартам, описанным в Фармакопее США.
Сравнительный Пример
Фармацевтический состав получали из 62,34 г кристаллов иопромида (8,76% Е1-изомера, 10,60% Е2-изомера, 39,68% Z1-изомера и 40,97% Z2-изомера) так же как в вышеуказанном Испытательном Примере 1. Затем анализировали отношения изомеров иопромида в составе. Как показано в Таблице 2, относительное количество изомеров иопромида в составе не удовлетворяло стандартам, описанным в Фармакопее США.
В то время как изобретение было описано относительно вышеупомянутых определенных вариантов, следует признать, что различные модификации и изменения могут быть сделаны в изобретении специалистами в технологии, которые также попадают в объем изобретения, как определено приложенной формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ИОПРОМИДА | 2009 |
|
RU2451667C1 |
| ПОЛУЧЕНИЕ ИОДИКСАНОЛА | 2005 |
|
RU2385316C2 |
| Способ получения фармацевтической субстанции на основе йопромида | 2017 |
|
RU2655693C1 |
| СПОСОБ ПОЛУЧЕНИЯ (R)-1-АРИЛ-2-ТЕТРАЗОЛИЛЭТИЛОВОГО ЭФИРА КАРБАМИНОВОЙ КИСЛОТЫ | 2009 |
|
RU2508290C2 |
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2,2-ДИФТОРБЕНЗО[d][1,3]ДИОКСОЛ-5-ИЛ)-N-(2,3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2573830C2 |
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2, 2-ДИФТОРБЕНЗО[d][1, 3]ДИОКСОЛ-5-ИЛ)-N-(1-(2, 3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2711481C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИГИДРОКСИЭТИЛРУТОЗИДА | 2014 |
|
RU2636939C2 |
| БИФЕНИЛЬНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2005 |
|
RU2366656C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИППУРАТА 4-(ДИ-Н-ПРОПИЛ)АМИНО-6-АМИНОКАРБОНИЛ-1,3,4,5-ТЕТРАГИДРОБЕНЗ(CD)ИНДОЛА | 1991 |
|
RU2034837C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА | 2008 |
|
RU2434009C1 |
Предложен способ селективной кристаллизации Z-изомера иодсодержащего рентгеноконтрастного средства - иопромида формулы (1), включающий а) растворение неочищенного иопромида или концентрата иопромида, содержащих Е- и Z-формы изомеров в спирте; и b) нагревание полученного спиртового раствора при 100-150°C, чтобы получить кристаллический Z-изомер иопромида. Также заявлен способ получения композиции, включающей Z-изомер иопромида формулы (1), заключающийся в i) селективной кристаллизации Z-изомера иопромида по п.1 из неочищенного иопромида, включающего смесь Е- и Z-изомеров иопромида или концентрата иопромида, чтобы получить кристаллический Z-изомер иопромида; и ii) растворении полученного кристаллического Z-изомера иопромида вместе с фармацевтически приемлемым эксципиентом. Технический результат заключается в возможности получения иопромида с высоким содержанием Z-изомера. 2 н. и 13 з.п. ф-лы, 7 пр., 2 табл., 2 ил.
1. Способ селективной кристаллизации Z-изомера иопромида, представленного формулой (1), включающий:
a) растворение неочищенного иопромида или концентрата иопромида, содержащих Е- и Z-формы изомеров в спирте; и
b) нагревание образующегося спиртового раствора при 100-150°C, чтобы получить кристаллический Z-изомер иопромида:
(Формула 1)
2. Способ по п.1, в котором содержание Z-изомера в кристаллах иопромида, полученных на стадии b), изменяется от 95 до 100%.
3. Способ по п.1, в котором спиртом является неразветвленный или разветвленный алифатический C1-9 спирт.
4. Способ по п.1, в котором объем (мл) спирта является 0,1-10-кратным весу (г) кристаллов неочищенного иопромида или концентрата иопромида.
5. Способ по п.1, в котором стадию b) нагревания проводят кипячением раствора с обратным холодильником, поддерживая внешнюю температуру сосуда от 100 до 150°C.
6. Способ по п.1, в котором отношение Е-изомера к Z-изомеру в неочищенном иопромиде или концентрате иопромида изменяется от 1:0,5 до 1:10.
7. Способ по п.1, в котором содержание влаги в неочищенном иопромиде или концентрате иопромида изменяется от 0 вес.% до 15 вес.%.
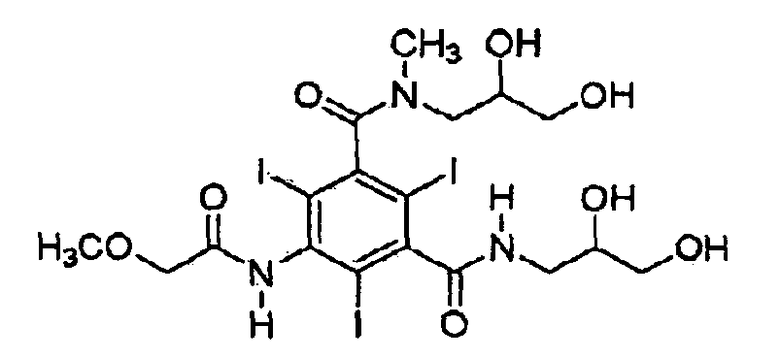
8. Способ получения композиции, включающей Z-изомер иопромида формулы (1), включающий
i) селективную кристаллизацию Z-изомера иопромида по п.1 из неочищенного иопромида, включающего смесь Е- и Z-изомеров иопромида или концентрата иопромида, чтобы получить кристаллический Z-изомер иопромида; и
ii) растворение кристаллического Z-изомера иопромида вместе с фармацевтически приемлемым эксципиентом:
(Формула 1)
9. Способ по п.8, в котором стадия i) кристаллизации включает а), растворение неочищенных кристаллов, включающих смесь Е- и Z- изомеров иопромида или концентрата иопромида в спирте; и b) нагревание образующегося спиртового раствора при 100-150°C, чтобы получить кристаллический Z-изомер иопромида.
10. Способ по п.8, в котором содержание Z-изомера в кристаллах иопромида изменяется от 95 до 100%.
11. Способ по п.9, в котором спиртом является неразветвленный или разветвленный алифатический C1-10 спирт.
12. Способ по п.9, в котором объем (мл) спирта является 0,1-10-кратным весу (г) кристаллов неочищенного иопромида или концентрата иопромида.
13. Способ по п.9, в котором стадию b) нагревания проводят кипячением раствора с обратным холодильником, поддерживая внешнюю температуру сосуда от 100 до 150°C.
14. Способ по п.9, в котором отношение Е-изомера к Z-изомеру в неочищенном иопромиде или концентрате иопромида изменяется от 1:0,5 до 1:10.
15. Способ по п.9, в котором содержание влаги в неочищенном иопромиде или концентрате иопромида изменяется от 0 вес.% до 15 вес.%.
| RU 2010148589 A, 27.05.2012 | |||
| US 7723544 B2, 25.05.2010 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |

