Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к твердым формам, например, кристаллическим и аморфным формам (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида, его фармацевтическим композициям, а также к способам.
Предпосылки создания настоящего изобретения
CFTR представляет собой цАМФ/АТФ-опосредованный анионный канал, который экспрессируется в ряде типов клеток, включая абсорбирующие и секреторные эпителиальные клетки, где он регулирует поток анионов через мембрану, а также активность ионных каналов и белков. В эпителиальных клетках нормальное функционирование CFTR является важным для поддержания транспорта электролитов по всему организму, включая респираторную и пищеварительную ткань. CFTR состоит приблизительно из 1480 аминокислот, которые кодируют белок, составленный из тандемного повтора трансмембранных доменов, причем каждый из них содержит шесть трансмембранных спиралей и нуклеотид-связывающий домен. Два трансмембранных домена соединены большим, полярным, регуляторным (R)-доменом с большим количеством мест фосфорилирования, которые регулируют активность канала и клеточный транспорт.
Обнаружен и секвенирован ген, кодирующий CFTR (см. Gregory, R.J. et al. (1990) Nature 347:382-386; Rich, D.P. et al. (1990) Nature 347:358-362), (Riordan, J.R. et al. (1989) Science 245: 1066-1073). Дефект в данном гене вызывает мутации в CFTR, приводя в результате к кистозному фиброзу ("CF"), самому распространенному смертельному генетическому заболеванию у людей. Кистозным фиброзом страдает приблизительно один из 2500 младенцев в США. В общей популяции США, вплоть до 10 миллионов людей содержат единичную копию дефектного гена без явных болезнетворных эффектов. В противоположность этому, индивиды с двумя копиями CF-ассоциированного гена страдают ослабляющими и смертельными эффектами CF, включая хроническое заболевание легких.
У пациентов с кистозным фиброзом, мутации в CFTR, эндогенно экспрессируемом в респираторном эпителии, приводят к снижению апикальной секреции анионов, вызывая дисбаланс в ионном и флюидном транспорте. Возникающее в результате ослабление анионного транспорта способствует усилению накопления слизи в легком и сопровождающих ее микробных инфекций, которые, в конце концов, приводят к смерти CF пациентов. В добавление к респираторному заболеванию, CF пациенты обычно страдают желудочно-кишечным заболеванием и недостаточностью поджелудочной железы, которые, если их не лечить, приводят в результате к смерти. Кроме того, большинство мужчин с кистозным фиброзом являются бесплодными, и у женщин с кистозным фиброзом репродуктивная функция снижается. В противоположность тяжелым эффектам двух копий CF-ассоциированного гена, индивиды с одной копией CF-ассоциированного гена проявляет повышенную устойчивость к холере и обезвоживанию, являющимся результатом диареи, что объясняется относительно высокой частотой CF гена в популяции.
Сиквенс-анализ CFTR гена CF хромосом выявил ряд вызывающих заболевание мутаций (Cutting, G.R. et al. (1990) Nature 346:366-369; Dean, M. et al. (1990) Cell 61:863:870; и Kerem, B-S. et al. (1989) Science 245: 1073-1080; Kerem, B-S et al. (1990) Proc. Natl. Acad. Sci. USA 87:8447-8451). В настоящее время обнаружено более чем 1000 вызывающих заболевание мутаций в CF гене, как сообщается в научной и медицинской литературе. Самой распространенной мутацией является делеция фенилаланина в положении 508 CFTR аминокислотной последовательности, и ее обычно называют ΔF508-CFTR. Данная мутация наблюдается по существу в 70 процентах случаев кистозного фиброза, и она связана с тяжелым заболеванием. Другие мутации включают R117H и G551D.
Делеция остатка 508 в ΔF508-CFTR препятствует корректному фолдингу образующегося белка. Это приводит к неспособности мутантного белка покидать ER и перемещаться к плазматической мембране. Как результат, число каналов, присутствующих в мембране, является гораздо меньшим, чем наблюдается в клетках, экспрессирующих CFTR дикого типа. В добавление к нарушению транспорта, мутация приводит в результате к нарушению открытия ионных каналов. Вместе, снижение количества каналов в мембране и нарушение открытия ионных каналов, приводят к ослаблению транспорта анионов через эпителий, приводя в результате к нарушению ионного и флюидного транспорта. (Quinton, P.M. (1990), FASEB J. 4: 2709-2727). Однако исследования показали, что сниженное количество ΔF508-CFTR в мембране является активным, хотя и в меньшей степени, чем CFTR дикого типа. (Dalemans et al. (1991), Nature Lond. 354: 526-528; Denning et al, supra; Pasyk and Foskett (1995), J. Cell. Biochem. 270: 12347-50). В добавление к ΔF508-CFTR, другие мутации, вызывающие заболевания, в CFTR, которые приводят в результате к нарушению транспорта, синтеза и/или открытия каналов, можно подавлять или активировать для изменения секреции анионов и изменения развития и/или тяжести заболевания.
Хотя CFTR переносит ряд молекул в добавление к анионам, ясно, что его роль (транспорт анионов) представляет собой один из элементов важного механизма транспорта ионов и воды через эпителий. Другие элементы включают эпителиальный Na+ канал, ENaC, Na+/2Cl-/K+ котранспортер, Na+-K+-АТФазный насос и базолатеральные мембранные K+ каналы, которые отвечают за поглощение клеткой хлорида.
Данные элементы работают вместе для достижения направленного транспорта через эпителий за счет их селективной экспрессии и локализации в клетке. Поглощение хлорида протекает за счет координированной активности ENaC и CFTR, присутствующих в апикальной мембране, и Na-K-АТФазного насоса и Cl- каналов, экспрессирующихся на базелатеральной поверхности клетки. Вторичный активный транспорт хлорида из люминальной области приводит в результате к накоплению внутриклеточного хлорида, который затем может пассивно покидать клетку через Cl- каналы, приводя в результате к векторному транспорту. Расположение Na+/2Cl-/K+ котранспортера, Na+-K+-АТФазного насоса и базолатеральных мембранных K+ каналов на базолатеральной поверхности и CFTR в люминальной области координирует выделение хлорида через CFTR в люминальной области. Поскольку сама вода вероятно никогда не переносится активным транспортом, ее поток через эпителий зависит от небольшого трансмембранного осмотического градиента, генерируемого объемным потоком натрия и хлорида.
Как обсуждалось выше, считают, что делеция остатка 508 в ΔF508-CFTR препятствует корректному фолдингу образующегося белка, приводя в результате к неспособности данного мутантного белка покидать ER и перемещаться к плазматической мембране. Как результат, недостаточное количество зрелого белка присутствует в плазматической мембране, и перенос хлорида в эпителиальных тканях значительно ослаблен. Действительно, показано, что данное клеточное явление нарушенного процессинга в эндоплазматическом ритикулуме (ER) АТФ-связывающих кассетных транспортеров ER аппаратом является причиной не только CF заболевания, но также широкого спектра других отдельных и наследственных заболеваний. Два способа, по которым ER аппарат может неправильно работать, заключаются или в потери связывания для ER экспорта белков, приводящей к разрушению, или ER накоплении данных дефектных/неправильно свернутых белков [Aridor M, et al., Nature Med., 5(7), pp 745-751 (1999); Shastry, B.S., et al., Neurochem. International, 43, pp 1-7 (2003); Rutishauser, J., et al, Swiss Med Wkly, 132, pp 211-222 (2002); Morello, JP et al, TIPS, 21, pp. 466-469 (2000); Brass P., et al, Human Mut., 14, pp. 186-198 (1999)].
(R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид описан в опубликованной патентной заявке США US20090131492 (причем указанная публикация включена в настоящее описание во всей своей полноте посредством ссылки) в качестве модулятора CFTR активности и, таким образом, он является полезным для лечения CFTR-опосредованных заболеваний, таких как кистозный фиброз. Однако существует необходимость в стабильных твердых формах указанного соединения, которые можно легко применять в фармацевтических композициях, пригодных для применения в качестве терапевтических агентов.
Сущность настоящего изобретения
Настоящее изобретение относится к твердым формам (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида (далее "соединение 1"), который имеет структуру ниже:
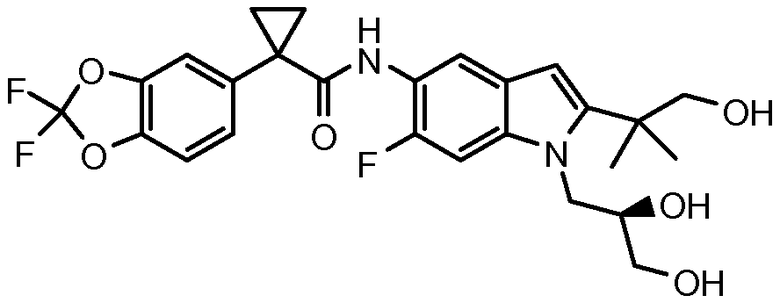
Соединение 1
Соединение 1 и его фармацевтически приемлемые композиции являются пригодными для лечения или уменьшения тяжести CFTR-опосредованных заболеваний, таких как, например, кистозный фиброз. В одном аспекте соединение 1 имеет в основном кристаллическую и бессолевую форму, называемую формой A, как описано и охарактеризовано в настоящем описании. В другом аспекте соединение 1 имеет аморфную форму, как описано и охарактеризовано в настоящем описании. Свойства твердого вещества, относящиеся к его эффективности в качестве лекарственного средства, могут зависеть от формы твердого вещества. Например, в лекарственном веществе изменение твердой формы может приводить к различиям в свойствах, таких как температура плавления, скорость растворения, пероральная абсорбция, биодоступность, в токсикологических результатах и даже в результатах клинических испытаний.
Способы, описанные в настоящем описании, можно применять для получения композиций настоящего изобретения, содержащих форму A или аморфную форму соединения 1, или обе. Количества и свойства компонентов, используемых в способах, будут описаны в настоящем описании.
Краткое описание фигур
На фиг.1 представлена порошковая рентгеновская дифрактограмма соединения 1.
На фиг.2 представлена кривая дифференциальной сканирующей калориметрии (ДСК) соединения 1.
На фиг.3 представлен график термогравиметрического анализа (ТГА) соединения 1.
На фиг.4 представлена порошковая рентгеновская дифрактограмма, рассчитанная для монокристалла формы A соединения 1.
На фиг.5 представлена реальная порошковая рентгеновская дифрактограмма формы A соединения 1, полученной методикой суспендирования (2 недели) с DCM в качестве растворителя.
На фиг.6 представлена кривая дифференциальной сканирующей калориметрии (ДСК) формы A соединения 1.
На фиг.7 представлена реальная порошковая рентгеновская дифрактограмма формы A соединения 1, полученной способом быстрого испарения ацетонитрила.
На фиг.8 представлена реальная порошковая рентгеновская дифрактограмма формы A соединения 1, полученной способом с использованием антирастворителя, такого как EtOAc и гептан.
На фиг.9 представлено конформационное изображение формы A соединения 1 на основе монокристаллического рентгеновского анализа.
На фиг.10 представлено конформационное изображение, показывающее порядок укладки формы A соединения 1.
На фиг.11 представлен твердофазный 13C-ЯМР спектр (15,0 кГц вращение) формы A соединения 1.
На фиг.12 представлен твердофазный 19F-ЯМР спектр (12,5 кГц вращение) формы A соединения 1.
На фиг.13 представлена порошковая рентгеновская дифрактограмма аморфной формы соединения 1 из способа быстрого упаривания на роторном испарителе.
На фиг.14 представлена кривая модулированной дифференциальной сканирующей калориметрии (МДСК) аморфной формы соединения 1, полученной способом быстрого упаривания на роторном испарителе.
На фиг.15 представлен график термогравиметрического анализа аморфной формы соединения 1, полученной способом быстрого упаривания на роторном испарителе.
На фиг.16 представлена порошковая рентгеновская дифрактограмма аморфной формы соединения 1, полученной способом сушки распылением.
На фиг.17 представлена кривая модулированной дифференциальной сканирующей калориметрии (МДСК) аморфной формы соединения 1, полученной способом сушки распылением.
На фиг.18 представлен твердофазный 13C-ЯМР спектр (15,0 кГц вращение) аморфной формы соединения 1.
На фиг.19 представлен твердофазный 19F-ЯМР спектр (12,5 кГц вращение) аморфной формы соединения 1.
Подробное описание настоящего изобретения
Определения
Как использовано в настоящем изобретении, следующие определения следует применять, если не указано иное.
Термин "CFTR", как применяют в настоящем описании, означает муковисцидозный трансмембранный регулятор проводимости или его мутацию, обладающий регуляторной активностью, включая, но не ограничиваясь ими, ΔF508CFTR и G551DCFTR (см., например, http://www.genet.sickkids.on.ca/cftr/, для CFTR мутаций).
Как применяют в настоящем описании, термин "аморфная" относится к твердой форме, которая состоит из неупорядоченно расположенных молекул и не обладает отличимой кристаллической решеткой.
Как применяют в настоящем описании, "кристаллическая" относится к соединениям или композициям, где структурные элементы расположены в фиксированных геометрических формах или решетках, так что кристаллические вещества имеют жесткий дальний порядок. Структурные элементы, которые образуют кристаллическую структуру, могут представлять собой атомы, молекулы или ионы. Кристаллические вещества имеют определенную температуру плавления.
Термин "модулирование", как применяют в настоящем описании, означает увеличение или уменьшение, например, активности, на измеряемую величину.
Термин "химически стабильная", как применяют в настоящем описании, означает то, что твердая форма соединения 1 не разлагается до одного или более различных химических соединений при воздействии указанных условий, например, 40°C/75% относительной влажности, в течение указанного периода времени, например, 1 дня, 2 дней, 3 дней, 1 недели, 2 недель или дольше. В некоторых вариантах осуществления менее чем 25% твердой формы соединения 1 разлагается, в некоторых вариантах осуществления менее чем приблизительно 20%, менее чем приблизительно 15%, менее чем приблизительно 10%, менее чем приблизительно 5%, менее чем приблизительно 3%, менее чем приблизительно 1%, менее чем приблизительно 0,5% формы соединения 1 разлагается в указанных условиях. В некоторых вариантах осуществления разлагается необнаруживаемое количество твердой формы соединения 1.
Термин "физически стабильная", как применяют в настоящем описании, означает то, что твердая форма соединения 1 не превращается в одну или более различных физических форм соединения 1 (например, различные твердые формы, как измерено XRPD, ДСК и т.д.) при воздействии указанных условий, например, 40°C/75% относительная влажность, в течение указанного периода времени, например 1 дня, 2 дней, 3 дней, 1 недели, 2 недель или дольше. В некоторых вариантах осуществления менее чем 25% твердой формы соединения 1 превращается в одну или более различных физических форм при воздействии указанных условий. В некоторых вариантах осуществления менее чем приблизительно 20%, менее чем приблизительно 15%, менее чем приблизительно 10%, менее чем приблизительно 5%, менее чем приблизительно 3%, менее чем приблизительно 1%, менее чем приблизительно 0,5% твердой формы соединения 1 превращается в одну или более различных физических форм при воздействии указанных условий. В некоторых вариантах осуществления неизмеримое количество твердой формы соединения 1 превращается в одну или более различных физических форм соединения 1.
Как применяют в настоящем описании, фраза "по существу аморфное соединение 1" используется взаимозаменяемо с фразами "аморфное соединение 1," "аморфное соединение 1, по существу не содержащее кристаллического соединения 1" и "по существу аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид". В некоторых вариантах осуществления по существу аморфное соединение 1 содержит менее чем приблизительно 30% кристаллического соединения 1, например, менее чем приблизительно 30% кристаллического соединения 1, например, менее чем приблизительно 25% кристаллического соединения 1, менее чем приблизительно 20% кристаллического соединения 1, менее чем приблизительно 15% кристаллического соединения 1, менее чем приблизительно 10% кристаллического соединения 1, менее чем приблизительно 5% кристаллического соединения 1, менее чем приблизительно 2% кристаллического соединения 1.
Как применяют в настоящем описании, фраза "по существу кристаллическая форма A соединения 1" используется взаимозаменяемо с фразами "форма A соединения 1" и "кристаллическая форма A соединения 1, по существу не содержащая аморфное соединение 1". В некоторых вариантах осуществления по существу кристаллическая форма A соединения 1 содержит менее чем приблизительно 30% аморфного соединения 1 или других твердых форм, например, менее чем приблизительно 30% аморфного соединения 1 или других твердых форм, например, менее чем приблизительно 25% аморфного соединения 1 или других твердых форм, менее чем приблизительно 20% аморфного соединения 1 или других твердых форм, менее чем приблизительно 15% аморфного соединения 1 или других твердых форм, менее чем приблизительно 10% аморфного соединения 1 или других твердых форм, менее чем приблизительно 5% аморфного соединения 1 или других твердых форм, менее чем приблизительно 2% аморфного соединения 1 или других твердых форм. В некоторых вариантах осуществления по существу кристаллическая форма соединения 1 содержит менее чем приблизительно 1% аморфного соединения 1 или других твердых форм.
Термин "по существу не содержащая" (как во фразе "по существу не содержащая формы X") при ссылке на указанную твердую форму соединения 1 (например, аморфную или кристаллическую форму, описанную в настоящем описании) означает то, что присутствует менее чем 20% (по массе) указанной формы (форм) или коформы (коформ) (например, кристаллической или аморфной формы соединения 1), более предпочтительно, присутствует менее чем 10% (по массе) указанной формы (форм), более предпочтительно, присутствует менее чем 5% (по массе) указанной формы (форм), и наиболее предпочтительно, присутствует менее чем 1% (по массе) указанной формы (форм).
Термин "по существу чистое" при ссылки на указанную твердую форму соединения 1 (например, аморфную или кристаллическую форму вещества, описанную в настоящем описании) означает то, что указанная твердая форма содержит менее чем 20% (по массе) остаточных компонентов, таких как альтернирующая полиморфная или изоморфная форма (формы) или коформа (коформы) соединения 1. Предпочтительно, по существу чистая твердая форма соединения 1 содержит менее чем 10% (по массе) альтернирующих полиморфных или изоморфных кристаллических форм соединения 1, более предпочтительно менее чем 5% (по массе) альтернирующих полиморфных или изоморфных кристаллических форм соединения 1, и самое предпочтительное менее чем 1% (по массе) альтернирующих полиморфных или изоморфных кристаллических форм соединения 1.
Как применяют в настоящем описании, "дисперсия" относится к дисперсной системе, где одно вещество, диспергированная фаза, распределено в виде дискретных единиц по всему второму веществу (непрерывная фаза или среда). Размер диспергированной фазы может изменяться в значительной степени (например, от коллоидных частиц нанометрового размера до размера в несколько микрон). Как правило, диспергированные фазы могут представлять собой твердые вещества, жидкости или газы. В случае твердой дисперсии, диспергированная и непрерывная фаза являются твердыми. При фармацевтическом применении, твердая дисперсия может содержать кристаллическое лекарственное средство (диспергированная фаза) в аморфном полимере (непрерывная фаза), или, альтернативно, аморфное лекарственное средство (диспергированная фаза) в аморфном полимере (непрерывная фаза). В некоторых вариантах осуществления аморфная твердая дисперсия содержит полимер, образующий диспергированную фазу, и лекарственное средство образует непрерывную фазу. В некоторых вариантах осуществления дисперсия содержит аморфное соединение 1 или по существу аморфное соединение 1.
Термин "твердая аморфная дисперсия" обычно относится к твердой дисперсии двух или более компонентов, обычно лекарственного средства и полимера, но может также содержать другие компоненты, такие как поверхностно-активные вещества или другие фармацевтические эксципиенты, когда соединение 1 является аморфным или по существу аморфным (например, по существу не содержащим кристаллического соединения 1), и физическая стабильность и/или растворение, и/или растворимость аморфного лекарственного средства увеличивается другими компонентами.
Как применяют в настоящем описании, термин "около" или "приблизительно", при использовании в связи с дозами, количествами или массовыми процентами ингредиентов композиции или дозированной формы, означает дозу, количество или массовые проценты, которые, как известно специалисту в данной области техники, обеспечивают фармакологический эффект, эквивалентный эффекту, полученному с помощью указанной дозы, количества или массовых процентов. Конкретно, термин "около" или "приблизительно" означает приемлемую ошибку для конкретной величины, как определено специалистом в данной области техники, которая частично зависит от того, какую величину измеряют или определяют. В определенных вариантах осуществления термин "около" или "приблизительно" обозначает в пределах 1, 2, 3 или 4 стандартных отклонений. В определенных вариантах осуществления термин "около" или "приблизительно" обозначает в пределах 30%, 25%, 20%, 15%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5%, 0,1% или 0,05% указанной величины или диапазона.
Сокращения "MTBE" и "DCM" обозначает метил-трет-бутиловый эфир и дихлорметан, соответственно.
Сокращение "XRPD" обозначает порошковую рентгеновскую дифракцию.
Сокращение "ДСК" обозначает дифференциальную сканирующую калориметрию.
Сокращение "ТГА" обозначает термогравиметрический анализ.
Если не указано иное, также предполагается, что структуры, показанные в настоящем описании, включают все изомерные (например, энантиомерные, диастереомерные и геометрические (или конформационные)) формы структуры; например, R и S конфигурации для каждого асимметрического центра, (Z) и (E) изомеры двойной связи, и (Z) и (E) конформационные изомеры. Следовательно, отдельные стереохимические изомеры, а также энантиомерные, диастереомерные и геометрические (или конформационные) смеси соединений настоящего изобретения включены в объем настоящего изобретения. Все таутомерные формы соединения 1 включены в настоящее изобретение. Например, соединение 1 может существовать в виде таутомеров, каждый из которых может быть включен в настоящее изобретение:
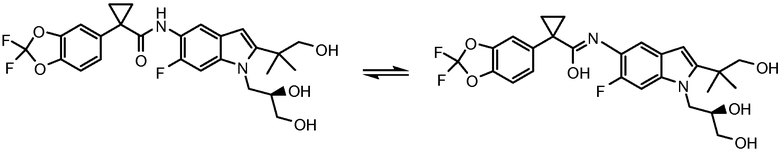
Кроме того, если не указано иное, также предполагается, что структуры, показанные в настоящем описании, включают соединения, которые отличаются наличием только одного или более изотопно обогащенных атомов. Например, соединение 1, где один или более атомов водорода заменены дейтерием или тритием, или один или более атомов углерода заменены 13C- или 14C-обогащенным углеродом, включено в объем настоящего изобретения. Данные соединения являются, например, пригодными в качестве аналитических инструментов, зондов в биологических исследованиях или соединений с улучшенным терапевтическим профилем.
В одном аспекте настоящее изобретение относится к (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамиду, характеризующемуся как кристаллическая форма A.
В другом варианте осуществления форма A характеризуется одним или более пиками при 19,3-19,7 градусах, 21,5-21,9 градусах и 16,9-17,3 градусах в порошковой рентгеновской дифракции, полученной с использованием Cu K альфа излучения. В другом варианте осуществления форма A характеризуется одним или более пиками приблизительно при 19,5, 21,7 и 17,1 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком при 20,2-20,6 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком приблизительно при 20,4 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком при 18,6-19,0 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком приблизительно при 18,8 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком при 24,5-24,9 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком приблизительно при 24,7 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком при 9,8-10,2 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком приблизительно при 10,0 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком при 4,8-5,2 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком приблизительно при 5,0 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком при 24,0-24,4 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком приблизительно при 24,2 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком при 18,3-18,7 градусах. В другом варианте осуществления форма A дополнительно характеризуется пиком приблизительно при 18,5 градусах.
В другом варианте осуществления форма A характеризуется дифракционной картиной, по существу аналогичной дифракционной картине фиг.4. В другом варианте осуществления форма A характеризуется дифракционной картиной, по существу аналогичной дифракционной картине фиг.5.
В другом аспекте настоящее изобретение относится к кристаллической форме (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида, имеющей моноклинную кристаллическую систему, C2 пространственную группу и следующие размеры монокристалла: a=21,0952(16) Å, α=90°, b=6,6287(5) Å, β=95,867(6)°, c=17,7917(15) Å и γ=90°.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей форму A и фармацевтически приемлемый носитель. В другом варианте осуществления фармацевтическая композиция дополнительно содержит дополнительный терапевтический агент. В другом варианте осуществления дополнительный терапевтический агент выбирают из муколитического средства, бронхолитического средства, антибиотика, противоинфекционного средства, противовоспалительного средства, CFTR потенциатора или питательного вещества.
В другом аспекте настоящее изобретение относится к способу получения формы A, включающему суспендирование (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида в растворителе в течение эффективного промежутка времени. В другом варианте осуществления растворитель представляет собой этилацетат, дихлорметан, MTBE, изопропилацетат, вода/этанол, вода/ацетонитрил, вода/метанол или вода/изопропиловый спирт. В другом варианте осуществления эффективная продолжительность времени составляет 24 часа до 2 недель. В другом варианте осуществления эффективная продолжительность времени составляет 24 часа до 1 недели. В другом варианте осуществления эффективная продолжительность времени составляет 24-72 часа.
В другом аспекте настоящее изобретение относится к способу получения формы A, включающему растворение (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида в растворителе и упаривание растворителя. В другом варианте осуществления растворитель представляет собой ацетон, ацетонитрил, метанол или изопропиловый спирт.
В другом аспекте настоящее изобретение относится к способу получения формы A, включающему растворение (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида в первом растворителе и добавление второго растворителя, в котором (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид не растворяется. В другом варианте осуществления первый растворитель представляет собой этилацетат, этанол, изопропиловый спирт или ацетон. В другом варианте осуществления второй растворитель представляет собой гептан или воду. В другом варианте осуществления добавление второго растворителя осуществляют при перемешивании раствора первого растворителя и (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида.
В другом аспекте настоящее изобретение относится к твердому по существу аморфному (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамиду. В другом варианте осуществления аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид содержит менее чем приблизительно 5% кристаллического (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид и фармацевтически приемлемый носитель. В другом варианте осуществления фармацевтическая композиция дополнительно содержит дополнительный терапевтический агент. В другом варианте осуществления дополнительный терапевтический агент выбирают из муколитического средства, бронхолитического средства, антибиотика, противоинфекционного средства, противовоспалительного средства, CFTR потенциатора или питательного вещества.
В другом аспекте настоящее изобретение относится к способу получения аморфного (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида, включающему растворение (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида в подходящем растворителе и удаление растворителя упариванием на роторном испарителе. В другом варианте осуществления растворитель представляет собой метанол.
В другом аспекте настоящее изобретение относится к твердой дисперсии, содержащей аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид и полимер. В другом варианте осуществления полимер представляет собой гидроксипропилметилцеллюлозу (HPMC). В другом варианте осуществления полимер представляет собой ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS).
В другом варианте осуществления полимер присутствует в количестве от 10% масс. до 80% масс. В другом варианте осуществления полимер присутствует в количестве от 30% масс. до 60% масс. В другом варианте осуществления полимер присутствует в количестве приблизительно 49,5% масс.
В другом варианте осуществления (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид присутствует в количестве от 10% масс. до 80% масс. В другом варианте осуществления (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид присутствует в количестве от 30% масс. до 60% масс. В другом варианте осуществления (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид присутствует в количестве приблизительно 50% масс.
В другом варианте осуществления твердая дисперсия дополнительно содержит поверхностно-активное вещество. В другом варианте осуществления поверхностно-активное вещество представляет собой лаурилсульфат натрия. В другом варианте осуществления поверхностно-активное вещество присутствует в количестве от 0,1% масс. до 5% масс. В другом варианте осуществления поверхностно-активное вещество присутствует в количестве приблизительно 0,5% масс.
В другом варианте осуществления полимер представляет собой ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS) в количестве 49,5% масс., поверхностно-активное вещество представляет собой лаурилсульфат натрия в количестве 0,5% масс., и (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид присутствует в количестве 50% масс.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей твердую дисперсию и фармацевтически приемлемый носитель. В другом варианте осуществления фармацевтическая композиция дополнительно содержит дополнительный терапевтический агент. В другом варианте осуществления дополнительный терапевтический агент выбирают из муколитического средства, бронхолитического средства, антибиотика, противоинфекционного средства, противовоспалительного средства, CFTR потенциатора или питательного вещества.
В другом аспекте настоящее изобретение относится к способу получения аморфного (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида, включающему сушку распылением (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида.
В другом варианте осуществления способ включает смешение (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида и подходящего растворителя, и затем сушку распылением смеси для получения аморфного (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида. В другом варианте осуществления растворитель представляет собой спирт. В другом варианте осуществления растворитель представляет собой метанол.
В другом варианте осуществления способ включает: a) получение смеси, содержащей (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид, полимер и растворитель; и b) сушку распылением смеси до формы твердой дисперсии.
В другом варианте осуществления полимер представляет собой ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS). В другом варианте осуществления полимер присутствует в количестве от 10% масс. до 80% масс. от твердой дисперсии. В другом варианте осуществления полимер присутствует в количестве приблизительно 49,5% масс. от твердой дисперсии. В другом варианте осуществления растворитель представляет собой метанол. В другом варианте осуществления смесь дополнительно содержит поверхностно-активное вещество. В другом варианте осуществления поверхностно-активное вещество представляет собой лаурилсульфат натрия (SLS). В другом варианте осуществления поверхностно-активное вещество присутствует в количестве от 0,1% масс. до 5% масс. от твердой дисперсии. В другом варианте осуществления поверхностно-активное вещество присутствует в количестве приблизительно 0,5% масс. от твердой дисперсии.
В другом варианте осуществления полимер представляет собой ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS) в количестве приблизительно 49,5% масс. от твердой дисперсии, растворитель представляет собой метанол, и смесь дополнительно содержит лаурилсульфат натрия в количестве приблизительно 0,5% масс. от твердой дисперсии.
В другом аспекте настоящее изобретение относится к способу лечения CFTR-опосредованного заболевания у субъекта, включающему введение субъекту эффективного количества формы A, аморфного (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида или твердой дисперсии аморфного (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида.
В другом варианте осуществления CFTR-опосредованное заболевание выбрано из кистозного фиброза, астмы, ХОБЛ, вызванной курением, хронического бронхита, синусита, запора, панкреатита, недостаточности поджелудочной железы, мужского бесплодия, вызванного врожденной билатеральной аплазией семявыносящих протоков (CBAVD), легкой болезни легких, идиопатического панкреатита, аллергического бронхолегочного аспергиллеза (АБЛА), заболевания печени, наследственной эмфиземы, наследственного гемохроматоза, нарушений коагуляции-фибринолиза, дефицита белка С, наследственного ангионевротического отека типа 1, нарушения обработки липидов, семейной гиперхолестеринемии, хиломикронемии 1 типа, абеталипопротеинемии, болезней лизосомальных накоплений, I-клеточной анемии/болезни Дери, мукополисахаридоза, болезни Сандгоффа/Тея-Сакса, болезни Криглера-Наджара II типа, полиэндокринопатии/гиперинсулинемии, сахарного диабета, карликовости Ларона, миелопероксидазной недостаточности, первичного гипопаратиреоза, меланомы, синдрома дефицита синтеза гликопротеинов CDG типа 1, врожденного гипертиреоза, незавершенного остеогенеза, наследственной гипофибриногенемии, ACT дефицита, несахарного диабета (DI), нейрофизеального DI, нейрогенного DI, синдрома Шарко-Мари-Туса, заболевания Пелицеуса-Мерцбахера, нейродегенеративных заболеваний, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, прогрессирующего надядерного паралича, болезни Пика, нескольких полиглутаминовых неврологических расстройств, болезни Хантингтона, спиноцеребеллярной атаксии типа I, спинной и бульбарной мышечной атрофии, дентаторубального паллидолюизиана, миотонической дистрофии, губчатой энцефалопатии, наследственной болезни Крейтцфельда-Якоба (в связи с нарушением процессинга прионных белков), болезни Фабри, синдрома Штрауслера-Шейнкера, ХОБЛ, сухих глаз, болезни Шегрена, остеопороза, остеопении, синдрома Горема, каналопатии хлоридных каналов, врожденной миотонии (формы Томпсона и Беккера), синдрома Бартера типа III, болезни Дента, гиперэкплексии, эпилепсии, лизосомальной болезни накопления, синдрома Ангельмана, первичной цилиарной дискинезии (PCD), наследственного нарушения структуры и/или функции ресничек, PCD с транспозицией органов (также известного как синдром Картагенера), PCD без транспозиции органов или цилиарной аплазии. В другом варианте осуществления CFTR-опосредованным заболеванием является кистозный фиброз. В другом варианте осуществления субъект имеет муковисцидозный трансмембранный рецептор (CFTR) с ΔF508 мутацией. В другом варианте осуществления субъект имеет муковисцидозный трансмембранный рецептор (CFTR) с R117H мутацией. В другом варианте осуществления субъект имеет муковисцидозный трансмембранный рецептор (CFTR) с G551D мутацией.
В другом варианте осуществления способ включает введение дополнительного терапевтического агента. В другом варианте осуществления терапевтический агент выбран из муколитического средства, бронхолитического средства, антибиотика, противоинфекционного средства, противовоспалительного средства, CFTR потенциатора или питательного вещества.
В другом аспекте настоящее изобретение относится к набору, содержащему форму A, аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид или твердую дисперсию, содержащую аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамид, и инструкции для их применения.
Способы получения формы A и аморфной формы соединения 1
Соединение 1 представляет собой исходное вещество, и в одном варианте осуществления его можно получить конденсацией хлорангидрида кислоты с амином согласно схемам 1-4.
Схема 1. Получение хлорангидрида кислоты
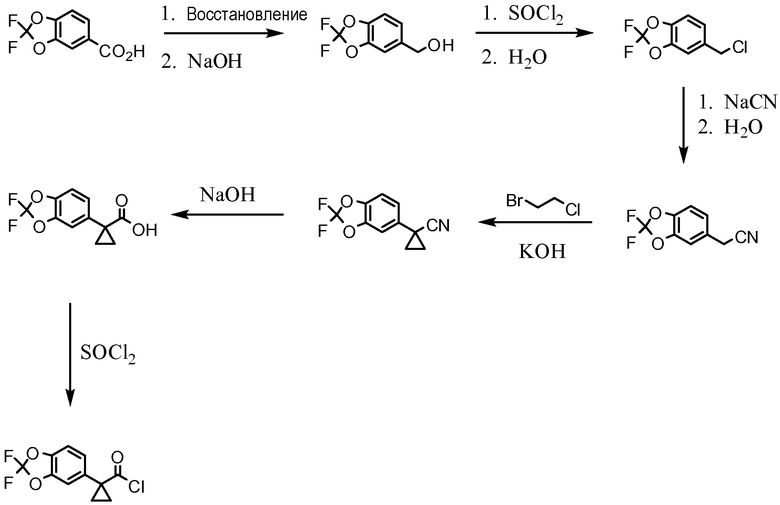
Схема 2. Альтернативное получение хлорангидрида кислоты
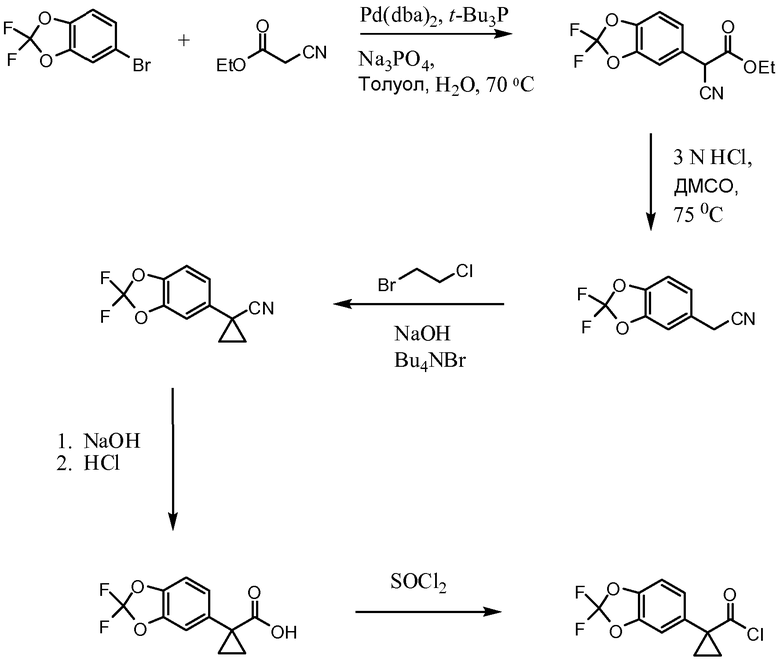
Схема 3. Получение амина
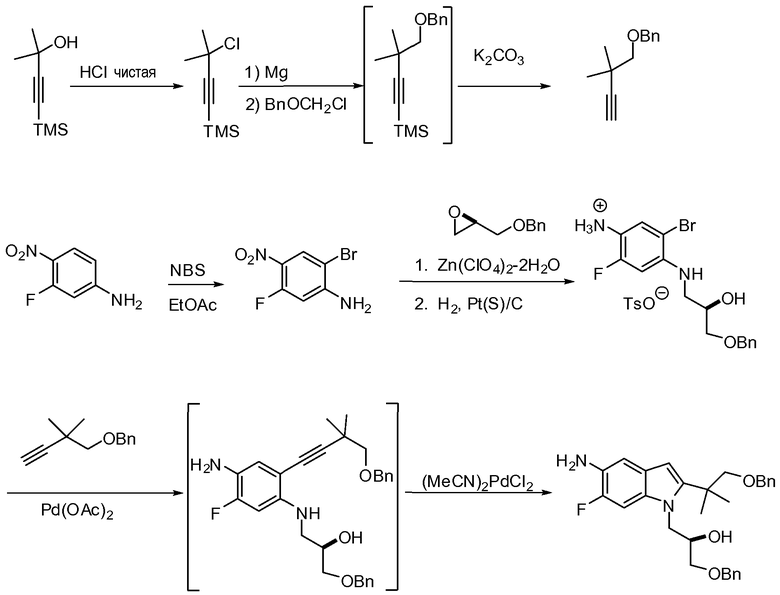
Схема 4. Получение соединения 1
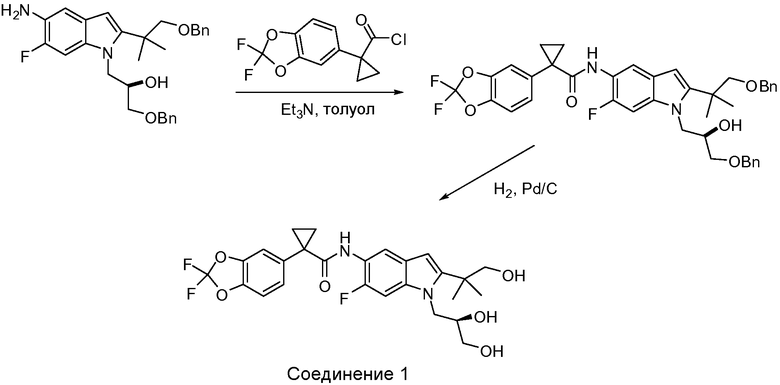
Способы получения формы A соединения 1
В одном варианте осуществления форму A получают суспендированием соединения 1 в подходящем растворителе в течение эффективного периода времени. В другом варианте осуществления подходящий растворитель представляет собой этилацетат, дихлорметан, MTBE, изопропилацетат, различные соотношения растворов вода/этанол, различные соотношения растворов вода/ацетонитрил, различные соотношения растворов вода/метанол или различные соотношения растворов вода/изопропиловый спирт. Например, различные соотношения растворов вода/этанол включают вода/этанол 1:9 (об./об.), вода/этанол 1:1 (об./об.) и вода/этанол 9:1 (об./об.). Различные соотношения растворов вода/ацетонитрил включают вода/ацетонитрил 1:9 (об./об.), вода/ацетонитрил 1:1 (об./об.) и вода/ацетонитрил 9:1 (об./об.). Различные соотношения растворов вода/метанол включают вода/метанол 1:9 (об./об.), вода/метанол 1:1 (об./об.) и вода/метанол 9:1 (об./об.). Различные соотношения растворов вода/изопропиловый спирт включают вода/изопропиловый спирт 1:9 (об./об.), вода/изопропиловый спирт 1:1 (об./об.) и вода/изопропиловый спирт 9:1 (об./об.).
Обычно, приблизительно 40 мг соединения 1 суспендируют в приблизительно 1,5 мл подходящего растворителя (целевая концентрация 26,7 мг/мл) при комнатной температуре в течение эффективного количества времени. В некоторых вариантах осуществления эффективное количество времени представляет собой приблизительно от 24 часов до приблизительно 2 недель. В некоторых вариантах осуществления эффективное количество времени представляет собой приблизительно от 24 часов до приблизительно 1 недели. В некоторых вариантах осуществления эффективное количество времени представляет собой приблизительно от 24 часов до приблизительно 72 часов. Затем, твердые вещества собирают.
В другом варианте осуществления форму A получают растворением соединения 1 в подходящем растворителе и затем упариванием растворителя. В одном варианте осуществления подходящий растворитель представляет собой растворитель, где соединение 1 имеет растворимость, больше, чем 20 мг/мл. Например, данные растворители включают ацетонитрил, метанол, этанол, изопропиловый спирт, ацетон и подобные.
Обычно, соединение 1 растворяют в подходящем растворителе, фильтруют и затем оставляют или для медленного испарения, или для быстрого испарения. Пример медленного испарения включает емкость, такую как пробирка, содержащую раствор соединения 1, с парафильмом с одним отверстием. Пример быстрого испарения включает оставление контейнера, такого как пробирка, содержащая раствор соединения 1, незакрытым. Затем твердые вещества собирают.
В другом аспекте настоящее изобретение относится к способу получения формы A, включающему растворение соединения 1 в первом растворителе и добавление второго растворителя, где соединение 1 плохо растворяется (растворимость <1 мг/мл). Например, первый растворитель может представлять собой растворитель, где соединение 1 имеет растворимость, большую, чем 20 мг/мл, например этилацетат, этанол, изопропиловый спирт или ацетон. Второй растворитель может представлять собой, например, гептан или воду.
Обычно, соединение 1 растворяют в первом растворителе и фильтруют для удаления любых зародышей кристаллов. Второй растворитель медленно добавляют при перемешивании. Твердые вещества осаждаются, и их собирают фильтрованием.
Способы получения аморфного соединения 1
Исходя из соединения 1 или формы A соединения 1, аморфную форму соединения 1 можно получить способами упаривания на роторном испарителе или способами сушки распылением.
Растворение соединения 1 в подходящем растворителе, подобном метанолу, и упаривание на роторном испарителе метанола до пены дает аморфную форму соединения 1. В некоторых вариантах осуществления баню с теплой водой используют для ускорения упаривания.
Аморфную форму соединения 1 можно также получить из формы A соединения 1, применяя способы сушки распылением. Сушка распылением представляет собой способ, который превращает жидкий поток в высушенную дисперсную форму. Необязательно, второй способ сушки, такой как сушка в псевдоожиженном слое или вакуумная сушка, можно применять для уменьшения количества остаточных растворителей до фармацевтически приемлемого уровня. Обычно, сушка распылением включает контакт высокодиспергированной жидкой суспензии или раствора и достаточного объема горячего воздуха для осуществления упаривания и сушки жидких капель. Состав, который будут сушить распылением, может представлять собой любой раствор, грубодисперсную суспензию, суспензию, коллоидную дисперсию или пасту, которые можно распылять, используя выбранное устройство для сушки распылением. В стандартном способе состав распыляют в токе теплого отфильтрованного воздуха, который упаривает растворитель и переносит высушенный продукт в сборник (например, циклон). Затем, отработанный воздух очищают от растворителя или, альтернативно, отработанный воздух направляют в конденсатор для получения и возможного повторного использования растворителя. Коммерчески доступные типы устройств можно использовать для проведения сушки распылением. Например, коммерчески доступные распылительные сушилки получают от Buchi Ltd. и Niro (например, PSD линия распылительных сушилок, полученных от Niro) (см., US 2004/0105820; US 2003/0144257).
В сушке распылением обычно используют твердую загрузку вещества приблизительно от 3% до приблизительно 30% масс., (т.е. лекарственное средство и эксципиенты), например, приблизительно от 4% до приблизительно 20% масс., предпочтительно, по меньшей мере, приблизительно 10%. В общем, верхний предел твердой загрузки определяется вязкостью (например, способностью перекачиваться) полученного в результате раствора и растворимостью компонентов в растворе. Как правило, вязкость раствора может определять размер частиц в полученном в результате порошкообразном продукте.
Методики и способы сушки распылением можно найти в Perry's Chemical Engineering Handbook, 6th Ed., R.H. Perry, D.W. Green & J.O. Maloney, eds.), McGraw-Hill book co. (1984); и Marshall "Atomization and Spray-Drying" 50, Chem. Eng. Prog. Monogr. Series 2 (1954). Как правило, сушку распылением проводят при температуре на входе приблизительно от 60°C до приблизительно 200°C, например, приблизительно от 95°C до приблизительно 185°C, приблизительно от 110°C до приблизительно 182°C, приблизительно от 96°C до приблизительно 180°C, например, приблизительно 145ºC. Сушку распылением обычно проводят при температуре на выходе приблизительно от 30°C до приблизительно 90°C, например, приблизительно от 40°C до приблизительно 80°C, приблизительно от 45°C до приблизительно 80°C например, приблизительно 75°C. Скорость потока распыления обычно составляет приблизительно от 4 кг/ч до приблизительно 12 кг/ч, например, приблизительно от 4,3 кг/ч до приблизительно 10,5 кг/ч, например, приблизительно 6 кг/ч или приблизительно 10,5 кг/ч. Скорость потока подачи обычно составляет приблизительно от 3 кг/ч до приблизительно 10 кг/ч, например, приблизительно от 3,5 кг/ч до приблизительно 9,0 кг/ч, например, приблизительно 8 кг/ч или приблизительно 7,1 кг/ч. Степень распыления обычно составляет приблизительно от 0,3 до 1,7, например, приблизительно от 0,5 до 1,5, например, приблизительно 0,8 или приблизительно 1,5.
Удаление растворителя может требовать последующую стадию сушки, такую как лотковая сушка, сушка в псевдоожиженном слое (например, приблизительно от комнатной температуры до приблизительно 100°C), вакуумная сушка, микроволновая сушка, сушка с вращающимся барабаном или вакуумная сушка в биконической сушилке (например, приблизительно от комнатной температуры до приблизительно 200°C).
В одном варианте осуществления твердую дисперсию сушат в псевдоожиженном слое.
В одном из способов растворитель содержит летучий растворитель, например растворитель, имеющий температуру плавления, менее чем приблизительно 100°C. В некоторых вариантах осуществления растворитель содержит смесь растворителей, например, смесь летучих растворителей или смесь летучего и нелетучего растворителя. При использовании смеси растворителей, смесь может содержать один или более нелетучих растворителей, например, когда нелетучий растворитель присутствует в смеси в количестве приблизительно 15%, например, менее чем приблизительно 12%, менее чем приблизительно 10%, менее чем приблизительно 8%, менее чем приблизительно 5%, менее чем приблизительно 3% или менее чем приблизительно 2%.
Предпочтительными растворителями являются такие растворители, где соединение 1 имеет растворимость, по меньшей мере, приблизительно 10 мг/мл, (например, по меньшей мере, приблизительно 15 мг/мл, 20 мг/мл, 25 мг/мл, 30 мг/мл, 35 мг/мл, 40 мг/мл, 45 мг/мл, 50 мг/мл или более). Более предпочтительные растворители включают растворители, где соединение 1 имеет растворимость, по меньшей мере, приблизительно 20 мг/мл.
Примеры растворителей, которые можно испытывать, включают ацетон, циклогексан, дихлорметан, Ν,Ν-диметилацетамид (DMA), Ν,Ν-диметилформамид (DMF), 1,3-диметил-2-имидазолидинон (DMI), диметилсульфоксид (ДМСО), диоксан, этилацетат, этиловый эфир, ледяную уксусную кислоту (HAc), метилэтилкетон (MEK), N-метил-2-пирролидинон (NMP), метил-трет-бутиловый эфир (MTBE), тетрагидрофуран (ТГФ), пентан, ацетонитрил, метанол, этанол, изопропиловый спирт, изопропилацетат и толуол. Примеры сорастворителей включают ацетон/ДМСО, ацетон/DMF, ацетон/вода, MEK/вода, ТГФ/вода, диоксан/вода. В системе двух растворителей растворители могут присутствовать в количестве приблизительно от 0,1% до приблизительно 99,9%. В некоторых предпочтительных вариантах осуществления вода представляет собой сорастворитель с ацетоном, когда вода присутствует приблизительно от 0,1% до приблизительно 15%, например, приблизительно от 9% до приблизительно 11%, например, приблизительно 10%. В некоторых предпочтительных вариантах осуществления вода представляет собой сорастворитель с MEK, когда вода присутствует приблизительно от 0,1% до приблизительно 15%, например, приблизительно от 9% до приблизительно 11%, например, приблизительно 10%. В некоторых вариантах осуществления раствор растворителей содержит три растворителя. Например, ацетон и воду можно смешивать с третьим растворителем, таким как DMA, DMF, DMI, ДМСО или HAc. В случаях, когда аморфное соединение 1 представляет собой компонент твердой аморфной дисперсии, предпочтительные растворители растворяют как соединение 1, так и полимер. Подходящие растворители включают растворители, описанные выше, например, MEK, ацетон, воду, метанол, и их смеси.
Размер частиц и диапазон температур сушки можно изменять для получения оптимальной твердой дисперсии. Как будет очевидно специалистам в данной области техники, небольшой размер частиц будет приводить к улучшенному удалению растворителя. Однако авторы обнаружили, что меньшие размеры могут приводить к рыхлым частицам, которые, при определенных обстоятельствах, не дают оптимальных твердых дисперсий для последующей обработки, такой как изготовление таблеток. При более высоких температурах может наблюдаться кристаллизация или химическое разложение соединения 1. При более низких температурах может не удаляться требуемое количество растворителя. Способы настоящего изобретения обеспечивают оптимальный размер частиц и оптимальную температуру сушки.
Как правило, размер частиц является таким, что D10 (мкм) составляет менее чем приблизительно 5, например, менее чем приблизительно 4,5, менее чем приблизительно 4,0 или менее чем приблизительно 3,5, D50 (мкм) обычно составляет менее чем приблизительно 17, например, менее чем приблизительно 16, менее чем приблизительно 15, менее чем приблизительно 14, менее чем приблизительно 13, и D90 (мкм) обычно составляет менее чем приблизительно 175, например, менее чем приблизительно 170, менее чем приблизительно 170, менее чем приблизительно 150, менее чем приблизительно 125, менее чем приблизительно 100, менее чем приблизительно 90, менее чем приблизительно 80, менее чем приблизительно 70, менее чем приблизительно 60 или менее чем приблизительно 50. Как правило, объемная плотность высушенных распылением частиц составляет приблизительно от 0,08 г/см3 до приблизительно 0,20 г/см3, например, приблизительно от 0,10 до приблизительно 0,15 г/см3, например, приблизительно 0,11 г/см3 или приблизительно 0,14 г/см3. Насыпная плотность высушенных распылением частиц обычно находится в диапазоне приблизительно от 0,08 г/см3 до приблизительно 0,20 г/см3, например, приблизительно от 0,10 до приблизительно 0,15 г/см3, например, приблизительно 0,11 г/см3 или приблизительно 0,14 г/см3, для 10 встряхиваний; от 0,10 г/см3 до приблизительно 0,25 г/см3, например, приблизительно от 0,11 до приблизительно 0,21 г/см3, например, приблизительно 0,15 г/см3, приблизительно 0,19 г/см3 или приблизительно 0,21 г/см3 для 500 встряхиваний; от 0,15 г/см3 до приблизительно 0,27 г/см3, например, приблизительно от 0,18 до приблизительно 0,24 г/см3, например, приблизительно 0,18 г/см3, приблизительно 0,19 г/см3, приблизительно 0,20 г/см3 или приблизительно 0,24 г/см3 для 1250 встряхиваний; и от 0,15 г/см3 до приблизительно 0,27 г/см3, например, приблизительно от 0,18 до приблизительно 0,24 г/см3, например, приблизительно 0,18 г/см3, приблизительно 0,21 г/см3, приблизительно 0,23 г/см3 или приблизительно 0,24 г/см3 для 2500 встряхиваний.
Полимеры
Твердые дисперсии, содержащие аморфное соединение 1 и полимер (или носитель в твердом состоянии) также включены в объем настоящего изобретения. Например, соединение 1 присутствует в виде аморфного соединения в качестве компонента твердой аморфной дисперсии. Твердая аморфная дисперсия обычно содержит соединение 1 и полимер. Примеры полимеров включают целлюлозные полимеры, такие как HPMC или HPMCAS, и полимеры, содержащие пирролидон, такие как PVP/VA. В некоторых вариантах осуществления твердая аморфная дисперсия содержит один или более дополнительных эксципиентов, таких как поверхностно-активное вещество.
В одном варианте осуществления полимер способен растворяться в водной среде. Растворимость полимеров может быть pH-независимой или pH-зависимой. Полимеры с pH-зависимой растворимостью включают один или более кишечнорастворимых полимеров. Термин "кишечнорастворимый полимер" относится к полимеру, который преимущественно растворим в менее кислом окружении кишечного тракта, по сравнению с более кислым окружением желудка, например, полимер, который является нерастворимым в кислой водной среде, но является растворимым, когда pH составляет более 5-6. Подходящий полимер должен быть химически и биологически инертным. Для того чтобы увеличить физическую стабильность твердых дисперсий, температура стеклования (Tg) полимера должна быть такой высокой, как это возможно. Например, предпочтительные полимеры имеют температуру стеклования, по меньшей мере, равную или большую, чем температура стеклования лекарственного средства (т.е. соединения 1). Другие предпочтительные полимеры имеют температуру стеклования, которая находится в пределах приблизительно от 10 до приблизительно 15ºC лекарственного средства (т.е. соединения 1). Примеры подходящих температур стеклования полимеров включают, по меньшей мере, приблизительно 90°C, по меньшей мере, приблизительно 95°C, по меньшей мере, приблизительно 100°C, по меньшей мере, приблизительно 105°C, по меньшей мере, приблизительно 110°C, по меньшей мере, приблизительно 115°C, по меньшей мере, приблизительно 120°C, по меньшей мере, приблизительно 125°C, по меньшей мере, приблизительно 130°C, по меньшей мере, приблизительно 135°C, по меньшей мере, приблизительно 140°C, по меньшей мере, приблизительно 145°C, по меньшей мере, приблизительно 150°C, по меньшей мере, приблизительно 155°C, по меньшей мере, приблизительно 160°C, по меньшей мере, приблизительно 165°C, по меньшей мере, приблизительно 170°C или, по меньшей мере, приблизительно 175°C (как измерено в сухих условиях). Не желая быть связанными теорией, считают, что лежащий в основе механизм заключается в том, что полимер с большей Tg обычно имеет меньшую молекулярную подвижность при комнатной температуре, которая может быть определяющим фактором для стабилизации физической стабильности аморфной твердой дисперсии.
Кроме того, гигроскопичность полимеров должна быть низкой, например, менее чем приблизительно 10%. С целью сравнения в данном описании, гигроскопичность полимера или композиции характеризуется приблизительно при 60% относительной влажности. В некоторых предпочтительных вариантах осуществления полимер имеет менее чем приблизительно 10% гигроскопичность, например менее чем приблизительно 9%, менее чем приблизительно 8%, менее чем приблизительно 7%, менее чем приблизительно 6%, менее чем приблизительно 5%, менее чем приблизительно 4%, менее чем приблизительно 3% или менее чем приблизительно 2% гигроскопичность. Гигроскопичность может также влиять на физическую стабильность твердых дисперсий. Обычно, влага, поглощенная полимерами, может сильно снижать Tg полимеров, а также полученных в результате твердых дисперсий, что будет дополнительно снижать физическую стабильность твердых дисперсий, как описано выше.
В одном из вариантов осуществления полимер представляет собой один или более водорастворимых полимеров или частично водорастворимых полимеров. Водорастворимые или частично водорастворимые полимеры включают, но не ограничиваются ими, целлюлозные производные (например, гидроксипропилметилцеллюлозу (HPMC), гидроксипропилцеллюлозу (HPC)) или этилцеллюлозу; поливинилпирролидоны (PVP); полиэтиленгликоли (PEG); поливиниловые спирты (PVA); акрилаты, такие как полиметакрилат (например, Eudragit® E); циклодекстрины (например, β-циклодекстрин) и сополимеры и их производные, включая, например, PVP-VA (поливинилпирролидон-винилацетат).
В некоторых вариантах осуществления полимер представляет собой гидроксипропилметилцеллюлозу (HPMC), такую как HPMCE50, HPMCE15 или HPMC60SH50).
Как обсуждается в настоящем описании, полимер может представлять собой pH-зависимый кишечнорастворимый полимер. Данный pH-зависимый кишечнорастворимый полимер включает, но не ограничивается ими, целлюлозные производные (например, ацетатфталат целлюлозы (CAP)), фталаты гидроксипропилметилцеллюлозы (HPMCP), ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS), карбоксиметилцеллюлозу (CMC) или ее соль (например, натриевую соль, такую как (CMC-Na)); ацетаттримеллитат целлюлозы (CAT), ацетатфталат гидроксипропилцеллюлозы (HPCAP), ацетатфталат гидроксипропилметилцеллюлозы (HPMCAP) и ацетатфталат метилцеллюлозы (MCAP), или полиметакрилаты (например, Eudragit® S). В некоторых вариантах осуществления полимер представляет собой ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS). В некоторых вариантах осуществления полимер представляет собой ацетатсукцинат гидроксипропилметилцеллюлозы HG качества (HPMCAS-HG).
В еще другом варианте осуществления полимер представляет собой сополимер поливинилпирролидона, например, сополимер винилпирролидона/винилацетата (PVP/VA).
В вариантах осуществления, где соединение 1 образует твердую дисперсию с полимером, например, с HPMC, HPMCAS или PVP/VA полимером, количество полимера относительно суммарной массы твердой дисперсии находится в диапазоне приблизительно от 0,1% до 99% масс. Если не указано особо, проценты лекарственного средства, полимера и других эксципиентов, как описано для дисперсии, даны в массовых процентах. Количество полимера обычно составляет, по меньшей мере, приблизительно 20%, и предпочтительно, по меньшей мере, приблизительно 30%, например, по меньшей мере, приблизительно 35%, по меньшей мере, приблизительно 40%, по меньшей мере, приблизительно 45% или приблизительно 50% (например, 49,5%). Данное количество обычно составляет приблизительно 99% или менее, и предпочтительно приблизительно 80% или менее, например, приблизительно 75% или менее, приблизительно 70% или менее, приблизительно 65% или менее, приблизительно 60% или менее, или приблизительно 55% или менее. В одном варианте осуществления полимер присутствует в количестве вплоть до приблизительно 50% суммарной массы дисперсии (и даже более конкретно, приблизительно от 40% до 50%, такой как приблизительно 49%, приблизительно 49,5% или приблизительно 50%). HPMC и HPMCAS являются доступными с различным качеством у ShinEtsu, например, HPMCAS является доступным в ряде вариантов, включая AS-LF, AS-MF, AS-HF, AS-LG, AS-MG, AS-HG. Каждый из данных типов качеств изменяется в зависимости от процентного замещения ацетата и сукцината.
В некоторых вариантах осуществления соединение 1 и полимер присутствуют приблизительно в равных количествах, например, каждый из полимера и лекарственного средства образует приблизительно половину процентной массы дисперсии. Например, полимер присутствует в количестве приблизительно 49,5%, и лекарственное средство присутствует в количестве приблизительно 50%.
В некоторых вариантах осуществления смесь соединения 1 и полимера представляет собой 1-20% масс./масс. суммарного содержания твердых компонентов нетвердой дисперсии перед сушкой распылением. В некоторых вариантах осуществления смесь соединения 1 и полимера представляет собой 5-15% масс./масс. суммарного содержания твердых компонентов нетвердой дисперсии перед сушкой распылением. В некоторых вариантах осуществления смесь соединения 1 и полимера представляет собой приблизительно 11% масс./масс. суммарного содержания твердых компонентов нетвердой дисперсии перед сушкой распылением.
В некоторых вариантах осуществления дисперсия дополнительно содержит другие вспомогательные ингредиенты, такие как поверхностно-активное вещество (например, SLS). В некоторых вариантах осуществления поверхностно-активное вещество составляет менее чем приблизительно 10% дисперсии, например, менее чем приблизительно 9%, менее чем приблизительно 8%, менее чем приблизительно 7%, менее чем приблизительно 6%, менее чем приблизительно 5%, менее чем приблизительно 4%, менее чем приблизительно 3%, менее чем приблизительно 2%, приблизительно 1%, или приблизительно 0,5%.
В вариантах осуществления включающих полимер, полимер должен присутствовать в количестве, эффективном для стабилизации твердой дисперсии. Стабилизация включает ингибирование или предотвращение кристаллизации соединения 1. Данная стабилизация будет ингибировать преобразование аморфной формы соединения 1 в кристаллическую форму. Например, полимер будет предотвращать, по меньшей мере, часть (например, приблизительно 5%, приблизительно 10%, приблизительно 15%, приблизительно 20%, приблизительно 25%, приблизительно 30%, приблизительно 35%, приблизительно 40%, приблизительно 45%, приблизительно 50%, приблизительно 55%, приблизительно 60%, приблизительно 65%, приблизительно 70%, приблизительно 75% или более) соединения 1 от превращения из аморфной в кристаллическую форму. Стабилизацию можно измерить, например, измерением температуры стеклования твердой дисперсии, измеряя скорость релаксации аморфного вещества, или измерением растворимости или биодоступности соединения 1.
Подходящие полимеры для применения в комбинации с соединением 1, например, для твердой дисперсии форма A, такой как аморфная твердая дисперсия, должны иметь одно или более из следующих свойств:
Температура стеклования полимера должна быть не менее чем приблизительно на 10-15ºC ниже температуры стеклования соединения 1. Предпочтительно, температура стеклования полимера является большей, чем температура стеклования соединения 1, и, как правило, по меньшей мере, на 50°C выше требуемой температуры хранения лекарственного продукта. Например, по меньшей мере, приблизительно 100ºC, по меньшей мере, приблизительно 105°C, по меньшей мере, приблизительно 105°C, по меньшей мере, приблизительно 110°C, по меньшей мере, приблизительно 120°C, по меньшей мере, приблизительно 130°C, по меньшей мере, приблизительно 140°C, по меньшей мере, приблизительно 150°C, по меньшей мере, приблизительно 160°C, по меньшей мере, приблизительно 160°C или более.
Полимер должен быть относительно негигроскопичным. Например, полимер должен при хранении в стандартных условиях поглощать менее чем приблизительно 10% воды, например, менее чем приблизительно 9%, менее чем приблизительно 8%, менее чем приблизительно 7%, менее чем приблизительно 6%, или менее чем приблизительно 5%, менее чем приблизительно 4% или менее чем приблизительно 3% воды. Предпочтительно полимер будет при хранении в стандартных условиях по существу не содержать поглощенную воду.
Полимер должен иметь аналогичную или лучшую растворимость в растворителях, подходящих для способов сушки распылением, по сравнению с растворимостью соединения 1. В предпочтительных вариантах осуществления полимер будет растворяться в одном или более одинаковых растворителях или системах растворителей как соединение 1. Предпочтительно, полимер является растворимым, по меньшей мере, в одном растворителе, не содержащем гидроксильных групп, таком как метиленхлорид, ацетон или их комбинации.
Полимер в комбинации с соединением 1, например, в твердой дисперсии или в жидкой суспензии, должен увеличивать растворимость соединения 1 в водной и физиологически родственной среде, и относительно растворимости соединения 1 в отсутствии полимера или относительно растворимости соединения 1 в комбинации с эталонным полимером. Например, полимер может увеличивать растворимость аморфного соединения 1 снижением количества аморфного соединения 1, которое преобразуется в кристаллическое соединение 1 или из твердой аморфной дисперсии, или из жидкой суспензии.
Полимер должен увеличивать скорость релаксации аморфного вещества.
Полимер должен увеличивать физическую и/или химическую стабильность соединения 1.
Полимер должен улучшать технологичность соединения 1.
Полимер должен улучшать одно или более свойств обработки, введения или хранения соединения 1.
Полимер не должен неблагоприятно взаимодействовать с другими фармацевтическими компонентами, например, эксципиентами.
Пригодность возможного полимера (или другого компонента) можно проверить, применяя способы сушки распылением (или другие способы), описанные в настоящем описании, для получения аморфной композиции. Возможную композицию может быть сравнена с учетом стабильности, устойчивости к образованию кристаллов или других свойств, и сравнена с эталонным составом, например, составом чистого аморфного соединения 1 или кристаллического соединения 1. Например, возможная композиция может быть протестирована для определения, задерживает ли она возникновение кристаллизации, опосредованной растворителем, или подавляет ли она процентное преобразование в указанный момент времени в контролируемых условиях, по меньшей мере, на 50%, 75%, 100% или 110%, а также эталонный состав или возможная композиция может быть протестирована для определения, обладает ли она улучшенной биодоступностью или растворимостью, относительно кристаллического соединения 1.
Поверхностно-активные вещества
Твердая дисперсия или другая композиция может содержать поверхностно-активное вещество. Поверхностно-активное вещество или смесь поверхностно-активных веществ будет обычно снижать межфазовое поверхностное натяжение между твердой дисперсией и водной средой. Подходящее поверхностно-активное вещество или смесь поверхностно-активных веществ может также увеличивать растворимость в воде и биодоступность соединения 1 из твердой дисперсии. Поверхностно-активные вещества для применения в связи с настоящим изобретением включают, но не ограничиваются ими, эфиры жирных кислот и сорбитана (например, Spans®), эфиры жирных кислот и полиоксиэтиленсорбитана (например, Tweens®), лаурилсульфат натрия (SLS), додецилбензолсульфонат натрия (SDBS) сульфосукцинатдиоктил натрия (Docusate), натриевую соль дезоксихолевой кислоты (DOSS), сорбитанмоностеарат, сорбитантристеарат, гексадецилтриметиламмонийбромид (HTAB), N-лауроилсаркозин натрия, олеат натрия, миристат натрия, стеарат натрия, пальмитат натрия, гелуцир 44/14, этилендиаминтетрауксусную кислоту (EDTA), витамин E d-альфа токоферилполиэтиленгликоль 1000 сукцинат (TPGS), лецитин, MW 677-692, моногидрат мононатрия глютаминовой кислоты, лабразоль, PEG 8 каприлик/каприковые глицериды, транскутол, диэтиленгликоль моноэтиловый эфир, солютол HS-15, полиэтиленгликоль/гидроксистеарат, таурохолевую кислоту, плюроник F68, плюроник F108 и плюроник F127 (или любые другие сополимеры полиоксиэтилена-полиоксипропилена (Pluronics®) или насыщенные полигликозилированные глицериды (Gelucirs®)). Конкретный пример поверхностно-активного вещества, которое можно использовать в связи с настоящим изобретением, включает, но не ограничивается ими, Span 65, Span 25, Tween 20, Capryol 90, плюроник F108, лаурилсульфат натрия (SLS), витамин E TPGS, плюроники и сополимеры. SLS обычно является предпочтительным.
Количество поверхностно-активного вещества (например, SLS) относительно суммарной массы твердой дисперсии может составлять от 0,1 до 15%. Предпочтительно, оно составляет приблизительно от 0,5% до приблизительно 10%, более предпочтительно приблизительно от 0,5% до приблизительно 5%, например, приблизительно 0,5-4%, приблизительно 0,5-3%, приблизительно 0,5-2%, приблизительно 0,5-1% или приблизительно 0,5%.
В определенных вариантах осуществления количество поверхностно-активного вещества относительно суммарной массы твердой дисперсии составляет, по меньшей мере, приблизительно 0,1%, предпочтительно приблизительно 0,5%. В данных вариантах осуществления поверхностно-активное вещество будет присутствовать в количестве не более чем приблизительно 15%, и предпочтительно не более чем приблизительно 12%, приблизительно 11%, приблизительно 10%, приблизительно 9%, приблизительно 8%, приблизительно 7%, приблизительно 6%, приблизительно 5%, приблизительно 4%, приблизительно 3%, приблизительно 2% или приблизительно 1%. Вариант осуществления, где поверхностно-активное вещество присутствует в количестве приблизительно 0,5% масс., является предпочтительным.
Возможные поверхностно-активные вещества (или другие компоненты) могут быть протестироваваны на пригодность для применения в настоящем изобретении способом, аналогично описанному для тестирования полимеров.
Применение, составление в композицию и введение
Фармацевтически приемлемые композиции
В другом аспекте настоящего изобретения обеспечиваются фармацевтически приемлемые композиции, где данные композиции содержат форму A соединения 1 или аморфное соединение 1, как описано в настоящем описании, и необязательно содержат фармацевтически приемлемый носитель, вспомогательное вещество или наполнитель. В определенных вариантах осуществления данные композиции необязательно дополнительно содержат один или более дополнительных терапевтических агентов.
Как описано выше, фармацевтически приемлемые композиции настоящего изобретения, кроме того, содержат фармацевтически приемлемый носитель, вспомогательное вещество или наполнитель, которые, как применяют в настоящем описании, включают любые и все растворители, разбавители или другие жидкие среды, эксципиенты для дисперсии или суспензии, поверхностно-активные агенты, изотонические агенты, загустители или эмульгаторы, консерванты, твердые связующие вещества, лубриканты и подобные, как подходит для конкретной требуемой дозированной формы. В Remington's Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) описаны различные носители, применяемые для формулирования фармацевтически приемлемых композиций и известные способы их получения. Кроме случаев, когда любая общепринятая среда-носитель является несовместимой с соединениями настоящего изобретения, таких как оказание неблагоприятного биологического воздействия или иное, взаимодействие среды вредным способом с любым другим компонентом (компонентами) фармацевтически приемлемой композиции, ее применение рассматривают включенным в объем настоящего изобретения. Некоторые примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничиваются этим, иониты, глинозем, стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота или сорбат калия, частичные глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, полиакрилаты, воски, блок-полимеры полиэтилена-полиоксипропилена, шерстяной жир, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод, желатин, тальк, эксципиенты, такие как какао-масло и воск для суппозитория, масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль или полиэтиленгликоль; эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; апирогенную воду; изотонический солевой раствор, раствор Рингера, этиловый спирт и забуференный фосфатом солевой раствор, а также другие нетоксичные совместимые лубриканты, такие как лаурилсульфат натрия и стеарат магния, а также красители, антиадгезивы, средства для покрытия, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты также могут присутствовать в композиции, по решению составителя.
Применение соединений и фармацевтически приемлемых композиций
В еще другом аспекте настоящее изобретение относится к способу лечения состояния, заболевания или расстройства, непосредственно связанного с CFTR. В определенных вариантах осуществления настоящее изобретение обеспечивает способы лечения состояния, заболевания или расстройства, непосредственно связанного с дефицитом CFTR активности, причем способы включают введение композиции, содержащей твердую форму A соединения 1 или аморфное соединение 1, описанные в настоящем описании, нуждающемуся в лечении субъекту, предпочтительно млекопитающему.
"CFTR-опосредованное заболевание", как применяют в настоящем описании, представляет собой заболевание, выбранное из кистозного фиброза, астмы, ХОБЛ, вызванной курением, хронического бронхита, синусита, запора, панкреатита, недостаточности поджелудочной железы, мужского бесплодия, вызванного врожденной билатеральной аплазией семявыносящих протоков (CBAVD), легкой болезни легких, идиопатического панкреатита, аллергического бронхолегочного аспергиллеза (АБЛА), заболевания печени, наследственной эмфиземы, наследственного гемохроматоза, нарушений коагуляции-фибринолиза, дефицита белка С, наследственного ангионевротического отека типа 1, нарушения обработки липидов, семейной гиперхолестеринемии, хиломикронемии 1 типа, абеталипопротеинемии, болезней лизосомальных накоплений, I-клеточной анемии/болезни Дери, мукополисахаридоза, болезни Сандгоффа/Тея-Сакса, болезни Криглера-Наджара II типа, полиэндокринопатии/гиперинсулинемии, сахарного диабета, карликовости Ларона, миелопероксидазной недостаточности, первичного гипопаратиреоза, меланомы, синдрома дефицита синтеза гликопротеинов CDG типа 1, врожденного гипертиреоза, незавершенного остеогенеза, наследственной гипофибриногенемии, ACT дефицита, несахарного диабета (DI), нейрофизеального DI, нейрогенного DI, синдрома Шарко-Мари-Туса, заболевания Пелицеуса-Мерцбахера, нейродегенеративных заболеваний, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, прогрессирующего надядерного паралича, болезни Пика, нескольких полиглутаминовых неврологических расстройств, болезни Хантингтона, спиноцеребеллярной атаксии типа I, спинной и бульбарной мышечной атрофии, дентаторубального паллидолюизиана, миотонической дистрофии, губчатой энцефалопатии, наследственной болезни Крейтцфельда-Якоба (в связи с нарушением процессинга прионных белков), болезни Фабри, синдрома Штрауслера-Шейнкера, ХОБЛ, сухих глаз, болезни Шегрена, остеопороза, остеопении, синдрома Горема, каналопатии хлоридных каналов, врожденной миотонии (формы Томпсона и Беккера), синдрома Бартера типа III, болезни Дента, гиперэкплексии, эпилепсии, лизосомальной болезни накопления, синдрома Ангельмана, первичной цилиарной дискинезии (PCD), наследственного нарушения структуры и/или функции ресничек, PCD с транспозицией органов (также известного как синдром Картагенера), PCD без транспозиции органов или цилиарной аплазии.
В определенных вариантах осуществления настоящее изобретение относится к способу лечения CFTR-опосредованного заболевания у человека, включающему стадию введения указанному человеку эффективного количества композиции, содержащей форму A соединения 1 или аморфное соединение 1, описанные в настоящем описании.
Согласно альтернативному предпочтительному варианту осуществления настоящее изобретение относится к способу лечения кистозного фиброза у человека, включающему стадию введения указанному человеку композиции, содержащей форму A соединения 1 или аморфное соединение 1, описанные в настоящем описании.
Согласно настоящему изобретению "эффективное количество" формы A соединения 1 или аморфного соединения 1 или их фармацевтически приемлемой композиции представляет собой количество, эффективное для лечения или снижения тяжести любого из заболеваний, указанных выше.
Форму A соединения 1 или аморфное соединение 1 или их фармацевтически приемлемую композицию можно вводить, применяя любое количество и любой способ введения, эффективный для лечения или снижения тяжести одного или более заболеваний, указанных выше.
В определенных вариантах осуществления форма A соединения 1 или аморфное соединение 1, описанные в настоящем описании, или их фармацевтически приемлемая композиция являются пригодными для лечения или снижения тяжести кистозного фиброза у пациентов, которые проявляют остаточную CFTR активность в апикальной мембране респираторного и нереспираторного эпителия. Наличие остаточной CFTR активности в области поверхности эпителия можно легко определить, применяя способы, известные в данной области техники, например, стандартные электрофизиологические, биохимические или гистохимические способы. Данные способы определяют CFTR активность с применением in vivo или ex vivo электрофизиологических способов, измерения Cl- концентрации в поте или слюне, или ex vivo биохимических или гистохимических способов для контроля плотности клеточной поверхности. Применяя данные способы, остаточную CFTR активность можно легко определить у гетерозиготных и гомозиготных пациентов для ряда различных мутаций, включая гетерозиготных и гомозиготных пациентов для самой распространенной мутации, ΔF508, а также других мутаций, таких как G551D мутация или R117H мутация.
В одном варианте осуществления форма A соединения 1 или аморфное соединение 1, описанные в настоящем описании, или их фармацевтически приемлемая композиция являются пригодными для лечения или снижения тяжести кистозного фиброза у пациентов с определенными генотипами, проявляющими остаточную CFTR активность, например, мутацией III класса (нарушение регуляции или открытия каналов), мутацией IV класса (измененная проводимость) или мутацией V класса (сниженный синтез) (Lee R. Choo-Kang, Pamela L., Zeitlin, Type I, II, III, IV, and V cystic fibrosis Tansmembrane Conductance Regulator Defects and Opportunities of Therapy; Current Opinion in Pulmonary Medicine 6:521-529, 2000). Другие генотипы пациентов, которые проявляют остаточную CFTR активность, включают пациентов, гомозиготных для одного или более классов или гетерозиготных с другим классом мутаций, включая мутации I класса, мутации II класса или неклассифицированную мутацию.
В одном варианте осуществления форма A соединения 1 или аморфное соединение 1, описанные в настоящем описании, или их фармацевтически приемлемая композиция являются пригодными для лечения или снижения тяжести кистозного фиброза у пациентов с определенными клиническими фенотипами, например, у человека с умеренным клиническим фенотипом, который обычно коррелирует с количеством остаточной CFTR активности в апикальной мембране эпителия. Данные фенотипы включают пациентов, страдающих недостаточностью поджелудочной железы, или пациентов, которым поставлен диагноз идиопатического панкреатита и врожденного двустороннего отсутствия семявыносящих протоков, или мягкой болезни легких.
Точное требуемое количество будет изменяться от субъекта к субъекту, в зависимости от вида, возраста и общего состояния субъекта, тяжести инфекции, конкретного агента, способа введения и подобных. Соединения настоящего изобретения предпочтительно формулируют в виде единичной дозированной формы для простоты введения и однородности дозирования. Выражение "единичная дозированная форма", как применяют в настоящем описании, относится к физически дискретной единице агента, подходящего для лечения пациента. Однако следует понимать, что суммарное дневное применение соединений и композиции настоящего изобретения будет определяться лечащим врачом в рамках обоснованного медицинского заключения. Конкретные значения эффективных доз для любого конкретного пациента или организма будут зависеть от ряда факторов, включая заболевание, которое подвергается лечению, и тяжести заболевания; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и рацион пациента; время введения, путь введения и скорость выведения конкретного применяемого соединения; продолжительность лечения; лекарственные средства, применяемые в комбинации или совместно с конкретным применяемым соединением, и подобных факторов, хорошо известных в медицинской области техники. Термин "пациент" или "субъект", как применяют в настоящем описании, означает животное, предпочтительно млекопитающее, и наиболее предпочтительно человека.
Фармацевтически приемлемые композиции настоящего изобретения можно вводить людям и другим животным, перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей или каплей), буккально, в виде перорального или назального спрея, или подобных, в зависимости от тяжести заболевания, которое будут лечить. В определенных вариантах осуществления соединения настоящего изобретения можно вводить перорально или парентерально в дозах приблизительно от 0,01 мг/кг до приблизительно 50 мг/кг и предпочтительно приблизительно от 1 мг/кг до приблизительно 25 мг/кг массы тела человека в день, один или более раз в день, для получения требуемого терапевтического эффекта.
В определенных вариантах осуществления количество дозы формы A соединения 1 или аморфного соединения 1 в единичной дозированной форме составляет от 100 мг до 1000 мг. В другом варианте осуществления количество дозы формы A соединения 1 или аморфного соединения 1 составляет от 200 мг до 900 мг. В другом варианте осуществления количество дозы формы A соединения 1 или аморфного соединения 1 составляет от 300 мг до 800 мг. В другом варианте осуществления количество дозы формы A соединения 1 или аморфного соединения 1 составляет от 400 мг до 700 мг. В другом варианте осуществления количество дозы формы A соединения 1 или аморфного соединения 1 составляет от 500 мг до 600 мг.
Инъецируемые препараты, например, стерильные инъецируемые водные или масляные суспензии можно формулировать согласно известному уровню техники, используя подходящие диспергирующие или смачивающие агенты и суспендирующие агенты. Стерильный инъецируемый препарат может также представлять собой стерильный инъецируемый раствор, суспензию или эмульсию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Приемлемые среды и растворители, которые можно применять, включают воду, раствор Рингера, U.S.P. и изотонический раствор хлорида натрия. Кроме того, стерильные, жирные масла общепринято применяют в качестве растворителя или суспендирующей среды. Для данной цели можно использовать любое мягкое жирное масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, используют при получении инъецируемых лекарственных средств.
Инъецируемые составы можно стерилизовать, например, фильтрованием через удерживающий бактерии фильтр или введением стерилизующего агента в форме стерильной твердой композиции, которая может быть растворена или диспергирована в стерильной воде или другой стерильной инъецируемой среде перед применением.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые можно получить смешением соединений настоящего изобретения с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозитория, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела и, следовательно, плавятся в полости прямой кишки или влагалища и высвобождают активное соединение.
Твердые дозированные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В данных твердых дозированных формах активное соединение смешивают, по меньшей мере, с одним инертным, фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или дикальций фосфат и/или a) наполнителями или разбавителями, такими как крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующими, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и камедь, с) увлажнителями, такими как глицерин, d) дезинтегрантами, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, е) агентами, замедляющими растворение, такими как парафин, f) ускорителями абсорбции, такими как соединения четвертичного аммония, g) увлажняющими агентами, такими как, например, цетиловый спирт и глицеринмоностеарат, h) абсорбентами, такими как каолин и бентонитовая глина, и i) лубрикантами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смесей. В случае капсул, таблеток и пилюль, дозированная форма может также содержать буферирующие агенты.
Твердые композиции аналогичного типа можно также применять в качестве наполнителей в мягких и твердых желатиновых капсулах, применяя такие эксципиенты, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и подобные. Твердые дозированные формы таблеток, драже, капсул, пилюль и гранул можно получить с покрытием или оболочкой, такими как кишечнорастворимое покрытие, и другими покрытиями, хорошо известными в области фармацевтического формулирования. Они могут необязательно содержать затемняющий компонент и могут также представлять собой композиции, которые высвобождают активный ингредиент (ингредиенты) только или предпочтительно в определенной части желудочно-кишечного тракта, необязательно замедленным способом. Примеры заливочных композиций, которые можно применять, включают полимерные вещества и воски. Твердые композиции аналогичного типа можно также применять в качестве наполнителей мягких и твердых желатиновых капсул, используя такие эксципиенты, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и подобные.
Активные соединения также могут быть в инкапсулированной форме с одним или более эксципиентами, как указано выше. Твердые дозированные формы в виде таблеток, драже, капсул, пилюль и гранул можно получить с покрытием и оболочкой, такой как кишечнорастворимые покрытия, покрытия, контролирующие высвобождение, и другими покрытиями, хорошо известными в области фармацевтического формулирования. В данных твердых дозированных формах активное соединение можно смешивать, по меньшей мере, с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Данные дозированные формы могут также содержать, как в общепринятой практике, дополнительные вещества, отличные от инертных разбавителей, например, лубриканты для получения таблеток и другие эксципиенты для получения таблеток, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль, дозированные формы могут также содержать буферирующие агенты. Они могут необязательно содержать затемняющие вещества и могут также представлять собой композиции, которые высвобождают только активный ингредиент (ингредиенты), или предпочтительно, в определенной части желудочно-кишечного тракта, необязательно замедленным способом. Примеры заливочных композиций, которые можно применять, включают полимерные вещества и воски.
Также следует понимать, что форму A соединения 1 или аморфное соединение 1, описанные в настоящем описании, или их фармацевтически приемлемую композицию можно применять в комбинированной терапии, т.е. форму A соединения 1 или аморфное соединение 1 можно вводить одновременно с, перед или после одного или более других требуемых терапевтических средств или медицинских процедур. Конкретная комбинация терапий (терапевтических средств или процедур) для применения в комбинированном режиме будет принимать во внимание совместимость требуемых терапевтических средств и/или процедур и желаемый терапевтический эффект, которого требуется достигнуть. Также следует понимать, что применяемые терапии могут достигать требуемого эффекта для того же заболевания (например, соединение настоящего изобретения можно вводить одновременно с другим агентом, применяемым для лечения того же заболевания), или они могут достигать различных эффектов (например, контроля любых побочных эффектов). Как применяют в настоящем описании, дополнительные терапевтические агенты, которые обычно вводят для лечения или предотвращения конкретного заболевания или состояния, являются известными в качестве "подходящих для заболевания или состояния, которое подвергают лечению".
В одном варианте осуществления дополнительный агент выбирают из муколитического средства, бронхолитического средства, антибиотика, противоинфекционного средства, противовоспалительного средства, CFTR потенциатора или питательного вещества.
В одном варианте осуществления дополнительный агент представляет собой 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту. В другом варианте осуществления дополнительный агент представляет собой N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид. В другом варианте осуществления дополнительный агент выбирают из таблицы 1:

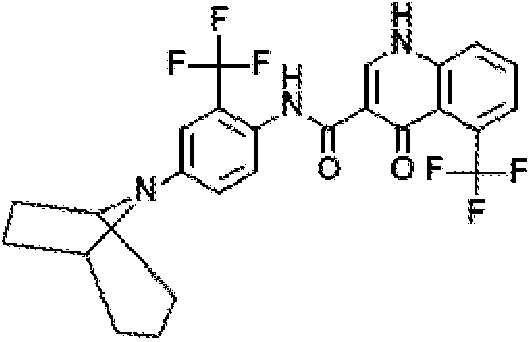
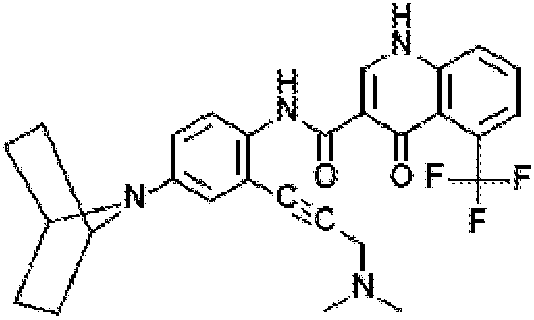
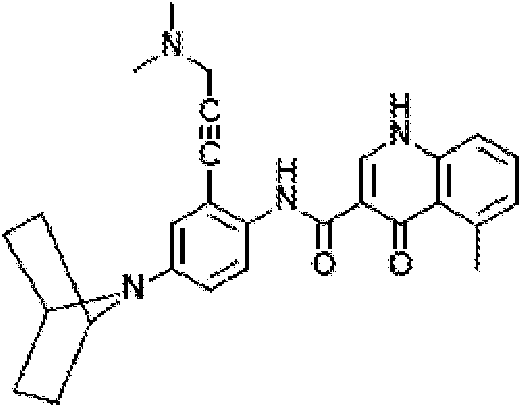
В другом варианте осуществления дополнительный агент представляет собой любую комбинацию вышеуказанных агентов. Например, композиция может содержать соединение 1, 3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойную кислоту и N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид. В другом примере, композиция может содержать соединение 1, N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид и любое одно из соединений из таблицы 1, т.е. соединения 1-14 таблицы 1 или любую их комбинацию.
В одном варианте осуществления дополнительный терапевтический агент представляет собой антибиотик. Примеры антибиотиков, пригодных в настоящем изобретении, включают тобрамицин, включая порошок тобрамицина для ингаляции (TIP), азитромицин, азтреонам, включая аэрозольную форму азтреонама, амикацин, включая его липосомальные составы, ципрофлоксацин, включая его составы, пригодные для введения путем ингаляции, левофлаксацин, включая его аэрозольные составы, и комбинации двух антибиотиков, например, фосфомицина и тобрамицина.
В другом варианте осуществления дополнительный агент представляет собой муколитическое средство. Примеры муколитических средств, пригодных в настоящем изобретении, включают Pulmozyme®.
В другом варианте осуществления дополнительный агент представляет собой бронхолитическое средство.
Примеры бронхолитических средств включают альбутерол, метапротенеролсульфат, пирбутерола ацетат, сальметерол или тетрабулинсульфат.
В другом варианте осуществления дополнительный агент является эффективным для восстановления жидкости поверхности дыхательных путей легких. Данные агенты улучшают движение соли в и из клеток, позволяя слизистой легочных путей быть более увлажненной и, следовательно, более легко очищаться. Примеры данных агентов включают гипертонический раствор NaCl для внутривенного введения, денуфозол тетранатрия ([[(3S,5R)-5-(4-амино-2-оксопиримидин-1-ил)-3-гидроксиоксолан-2-ил]метоксигидроксифосфорил][[[(2R,3S,4R,5R)-5-(2,4-диоксопиримидин-1-ил)-3,4-дигидроксиоксолан-2-ил]метоксигидроксифосфорил]оксигидроксифосфорил]гидрофосфат), или бронхитол (ингаляционный состав маннита).
В другом варианте осуществления дополнительный агент представляет собой противовоспалительный агент, т.е. агент, который уменьшает воспаление в легких. Примеры данных агентов, пригодных в настоящем изобретении, включают ибупрофен, докозагексаеновую кислоту (DHA), силденафил, ингаляционный глутатион, пиоглитазон, гидроксихлорохин или симавастатин.
В другом варианте осуществления дополнительный агент представляет собой CFTR модулятор, отличный от формы I соединения 1, т.е. агент, который обладает эффектом модулирования CFTR активности.
Примеры данных агентов включают аталурен ("PTC124®"; 3-[5-(2-фторфенил)-1,2,4-оксадиазол-3-ил]бензойная кислота), синапультид, ланковутид, депелестат (ингибитор человеческой рекомбинантной нейтрофил-эластазы), кобипростон (7-{(2R,4aR,5R,7aR)-2-[(3S)-1,1-дифтор-3-метилпентил]-2-гидрокси-6-оксаоктогидроциклопента[b]пиран-5-ил}гептановая кислота) и N-(5-гидрокси-2,4-ди-трет-бутилфенил)-4-оксо-1H-хинолин-3-карбоксамид.
В другом варианте осуществления дополнительный агент представляет собой питательное вещество.
Примеры питательных веществ включают панкрелипазу (заменитель фермента панкреатина), включая Pancrease®, Pancreacarb®, Ultrase® или Creon®, Liprotomase® (ранее Trizytek®), Aquadeks® или глютатионовую ингаляцию. В одном варианте осуществления дополнительное питательное вещество представляет собой панкрелипазу.
В другом варианте осуществления дополнительный агент представляет собой соединение, выбранное из гентамицина, куркумина, циклофосфамида, 4-фенилбутирата, миглустата, фелодипина, нимодипина, филоксина B, генистейна, апигенина, цАМФ/цГМФ модуляторов, таких как ролипрамом, силденафил, милринон, тадалафил, амринон, изопротеренол, альбутерол и альметерол, дезоксиспергуалина, HSP 90 ингибиторов, ингибиторов HSP-70, ингибиторов протеосомы, таких как эпоксомицин, лактацистин и т.д.
В другом варианте осуществления дополнительный агент представляет собой соединение, описанное в WO 2004028480, WO 2004110352, WO 2005094374, WO 2005120497 или WO 2006101740.
В другом варианте осуществления дополнительный агент представляет собой производное бензо(c)хинолизиния, которое ингибирует CFTR модулирующую активность, или бензопирановое производное, которое ингибирует CFTR модулирующую активность.
В другом варианте осуществления дополнительный агент представляет собой соединение, описанное в US7202262, US6992096, US20060148864, US20060148863, US20060035943, US20050164973, WO2006110483, WO2006044456, WO2006044682, WO2006044505, WO2006044503, WO2006044502 или WO2004091502.
В другом варианте осуществления дополнительный агент представляет собой соединение, описанное в WO2004080972, WO2004111014, WO2005035514, WO2005049018, WO2006099256, WO2006127588 или WO2007044560.
Данные комбинации являются пригодными для лечения заболеваний, описанных в настоящем описании, включая кистозный фиброз. Данные комбинации являются также пригодными в наборах, описанных в настоящем описании.
Количество дополнительного терапевтического агента, присутствующего в композициях настоящего изобретения, будет не более чем количество, которое будут обычно вводить в композиции, содержащей данный терапевтический агент в виде единственного активного агента. Предпочтительно количество дополнительного терапевтического агента в описанных в настоящем описании композициях будет находиться в диапазоне приблизительно от 50% до 100% количества, обычно присутствующего в композиции, содержащей данный агент в качестве единственного терапевтически активного агента.
Форму A соединения 1 и аморфную форму, описанные в настоящем описании, или их фармацевтически приемлемую композицию можно также вводить в композиции для покрытия имплантируемых медицинских устройств, таких как протезы, искусственные клапаны, сосудистые трансплантаты, стенты и катетеры. Соответственно, настоящее изобретение в другом аспекте включает композицию для покрытия имплантируемого устройства, содержащую форму A соединения 1 и/или аморфную форму, описанные в настоящем описании, или их фармацевтически приемлемую композицию, и в классах и подклассах в настоящем описании, и носитель, подходящий для покрытия указанного имплантируемого устройства. В еще другом аспекте настоящее изобретение включает имплантируемое устройство, покрытое композицией, содержащей форму A соединения 1 и/или аморфную форму, описанные в настоящем изобретении, или их фармацевтически приемлемую композицию, и носитель, подходящий для покрытия указанного имплантируемого устройства. Подходящие покрытия и общее получение покрытых имплантируемых устройств описано в патентах США 6099562; 5886026 и 5304121. Покрытия обычно представляют собой биосовместимые полимерные вещества, такие как гидрогельный полимер, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочная кислота, этиленвинил ацетат и их смеси. Покрытия можно необязательно дополнительно покрывать подходящим верхним покрытием фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидов или их комбинацией для придания композиции свойств контролируемого высвобождения.
Для того чтобы настоящее изобретение, описанное в данном описании, было более понятно, представлены следующие примеры. Следует понимать, что данные примеры приведены для иллюстративных целей и не интерпретируются как ограничивающие настоящее изобретение каким-либо образом.
ПРИМЕРЫ
СПОСОБЫ И МАТЕРИАЛЫ
Модуляционная дифференциальная сканирующая калориметрия (МДСК) и дифференциальная сканирующая калориметрия (ДСК)
Модуляционную дифференциальную сканирующую калориметрию (МДСК) использовали для исследования температуры стеклования аморфной формы и высушенной распылением дисперсии соединения. Дифференциальную сканирующую калориметрию (ДСК) использовали для определения температуры плавления кристаллических веществ и для распознания различных полиморфов. Данные получали, используя TA DSC Q2000 дифференциальный сканирующий калориметр (TA Instruments, New Castle, DE). Прибор калибровали индием. Образцы приблизительно 1-5 мг взвешивали в алюминиевых герметичных тиглях, которые обжимали, используя крышки с одним отверстием. Для МДСК образцы сканировали при температуре от -20°C до 220°C при скорости нагревания 2°C/минута с +/-1°C модуляцией каждые 60 секунд. Для ДСК образцы сканировали от 25°C до 220°C при скорости нагревания 10°C/мин. Данные собирали Thermal Advantage Q Series™ программным обеспечением (версия: 2.7.0.380) и анализировали Universal Analysis программным обеспечением (версия: 4.4A, сборка: 4.4.0.5) (TA Instruments, New Castle, DE).
XRPD (рентгеновская порошковая дифрактометрия)
Рентгеновскую порошковую дифрактометрию использовали для характеризации физической формы партий, полученных к настоящему времени, и характеризации различных обнаруженных полиморфов. XRPD данные соединения получали на PANalytical X'pert Pro порошковом рентгеновском дифрактометре (Almelo, the Netherlands). XRPD образец регистрировали при комнатной температуре с медным излучением (1,54060 Å). Рентгеновский луч генерировали, используя Cu запаянную лампу при 45 кВ, 40 мА с никелевым Κβ подавляющим фильтром. Оптика падающего луча состояла из переменной дивергентной щели для обеспечения постоянной иллюминированной длины на образец и на стороне отраженного луча; быстрый линейный твердофазный детектор использовали с активной длинной 2,12 градуса 2 тета, измеренные в сканирующем режиме. Порошкообразный образец упаковывали в вогнутую область кремниевого держателя на нулевом уровне, и вращение осуществляли для получения лучших статистических данных. Симметричный скан измеряли от 4-40 градусов 2 тета с размером шага 0,017 градусов и временем шага скана 15,5 секунд. Программным обеспечением для сбора данных являлся X'pert Data Collector (версия 2.2e). Программным обеспечением для анализа данных являлся или X'pert Data Viewer (версия 1.2d) или X'pert Highscore (версия: 2.2c).
Термогравиметрический анализ (ТГА)
ТГА использовали для исследования наличия остаточного растворителя в охарактеризованных партиях и определения температуры, при которой наблюдается разложение образца. ТГА данные получали на TA Q500 термогравиметрическом анализаторе (TA Instruments, New Castle, DE). Образец с массой приблизительно 2-5 мг сканировали от 25°C до 300°C при скорости нагревания 10°C/мин. Данные получали Thermal Advantage Q Series™ программным обеспечением (версия 2.5.0.255) и анализировали Universal Analysis программным обеспечением (версия 4.4A, сборка 4.4.0.5) (TA Instruments, New Castle, DE).
Определение монокристаллической структуры формы A соединения 1
Дифракционные данные получали на Bruker Apex II дифрактометре, снабженном Cu Kα источником в запаянной лампе и Apex II CCD детектором. Структуру определяли и уточняли, применяя SHELX программу (Sheldrick,G., Acta Cryst., (2008) A64, 112-122). На основе статистики интенсивностей и нулевой систематичности определяли и уточняли структуру в C2 пространственной группе. Абсолютную конфигурацию определяли, применяя аномальную дифракцию. Параметр Флэка уточнялся до 0,00 (18), показывая, что модель представляет правильный энантиомер [(R)].
Твердофазный ЯМР
Твердофазный ЯМР проводили на Bruker-Biospin 400 МГц спектрометре с широким отверстием, снабженном Bruker-Biospin 4 мм HFX зондом. Образцы помещали в 4 мм ZrO2 роторы и вращали в условиях вращения под магическим углом (MAS) со скоростью вращения 12,5 кГц. Сначала измеряли время протонной релаксации, применяя 1H MAS Ti измерение времени релаксации способом насыщения-восстановления, устанавливая правильную задержку перед запуском нового цикла 13C кросс-поляризационного (CP) MAS эксперимента. CP время контакта углеродного CPMAS эксперимента устанавливали на 2 мс. Применяли CP импульс протона с линейным изменением (от 50% до 100%). Согласование Хартмана-Хана оптимизировали на внешнем контрольном образце (глицин). Фторидный MAS спектр регистрировали с развязкой от протонов. TPPM15 последовательность развязки использовали с силой поля приблизительно 100 кГц и для 13C и для 19F накоплении.
Vitride® (бис(2-метоксиэтокси)алюмогидрид натрия [или NaAlH2(OCH2CH2OCH3)2], 65% масс. раствор в толуоле) покупали у Aldrich Chemicals.
2,2-дифтор-1,3-бензодиоксол-5-карбоновую кислоту покупали у Saltigo (партнер Lanxess Corporation).
Где-либо в настоящем описании, когда название соединения может некорректно описывать структуру соединения, структура заменяет название и руководит.
Получение соединения 1
Кислотный фрагмент
Получение (2,2-дифтор-1,3-бензодиоксол-5-ил)метанола
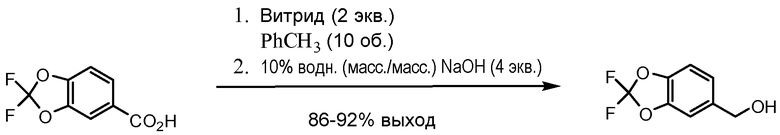
Коммерчески доступную 2,2-дифтор-1,3-бензодиоксол-5-карбоновую кислоту (1,0 экв.) суспендировали в толуоле (10 об.). Vitride® (2 экв.) добавляли черед капельную воронку при такой скорости, чтобы поддерживать температуру 15-25°C. В конце добавления температура повышалась до 40°C в течение 2 часов, затем осторожно добавляли через капельную воронку 10% (масс./масс.) водн. NaOH (4,0 экв.), поддерживая температуру 40-50°C. После перемешивания в течение дополнительных 30 минут, слои разделяли при 40°C. Органическую фазу охлаждали до 20°C, затем промывали водой (2×1,5 об.), сушили (Na2SO4), фильтровали и концентрировали, с получением неочищенного (2,2-дифтор-1,3-бензодиоксол-5-ил)метанола, который использовали непосредственно на следующей стадии.
Получение 5-хлорметил-2,2-дифтор-1,3-бензодиоксола
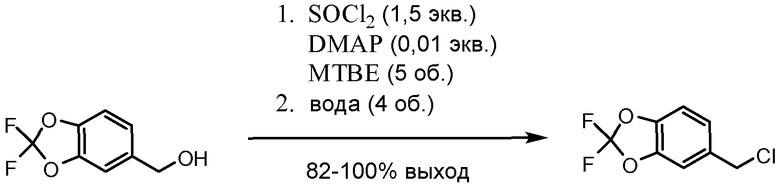
(2,2-дифтор-1,3-бензодиоксол-5-ил)метанол (1,0 экв.) растворяли в MTBE (5 об.). Добавляли каталитическое количество DMAP (1 моль%), и добавляли через капельную воронку SOCl2 (1,2 экв.). SOCl2 добавляли при такой скорости, чтобы поддерживать температуру в реакторе 15-25°C. Температура повышалась до 30°C в течение 1 часа, затем смесь охлаждали до 20°C, затем добавляли через капельную воронку воду (4 об.), поддерживая температуру менее чем 30°C. После перемешивания в течение дополнительных 30 минут, слои разделяли. Органический слой перемешивали и добавляли 10% (масс./об.) водн. NaOH (4,4 об.). После перемешивания в течение 15-20 минут, слои разделяли. Затем органическую фазу сушили (Na2SO4), фильтровали и концентрировали, с получением неочищенного 5-хлорметил-2,2-дифтор-1,3-бензодиоксола, который использовали непосредственно на следующей стадии.
Получение (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрила
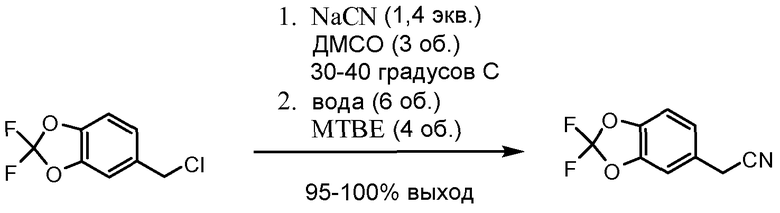
Раствор 5-хлорметил-2,2-дифтор-1,3-бензодиоксола (1 экв.) в ДМСО (1,25 об.) добавляли к суспензии NaCN (1,4 экв.) в ДМСО (3 об.), поддерживая температуру между 30-40°C. Смесь перемешивали в течение 1 часа, затем добавляли воду (6 об.), с последующим добавлением MTBE (4 об.). После перемешивания в течение 30 минут, слои разделяли. Водный слой экстрагировали MTBE (1,8 об.). Объединенные органические слои промывали водой (1,8 об.), сушили (Na2SO4), фильтровали и концентрировали, с получением неочищенного (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрила (95%), который использовали непосредственно на следующей стадии.
1Н-ЯМР (500 МГц, ДМСО) δ 7,44 (уш.с, 1H), 7.43 (д, J=8,4 Гц, 1H), 7,22 (дд, J=8,2, 1,8 Гц, 1H), 4,07 (с, 2H).
Получение (2,2-дифтор-1,3-бензодиоксол-5-ил)-1-этилацетатацетонитрила.
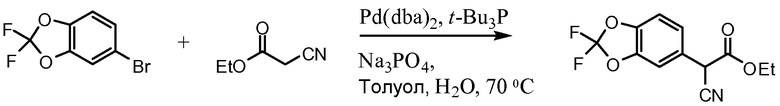
Реактор продували азотом и заполняли 900 мл толуола. Растворитель дегазировали барботированием азота в течение не менее 16 часов. Затем в реактор помещали Na3PO4 (155,7 г, 949,5 ммоль), с последующим добавлением бис(дибензилиденацетон)палладия (0) (7,28 г, 12,66 ммоль). Добавляли 10% масс./масс. раствор трет-бутилфосфина в гексане (51,23 г, 25,32 ммоль) в течение 10 минут при 23°C из капельной воронки, продутой азотом. Смесь перемешивали в течение 50 минут, затем добавляли в течение 1 минуты 5-бром-2,2-дифтор-1,3-бензодиоксол (75 г, 316,5 ммоль). После перемешивания в течение дополнительных 50 минут, к смеси добавляли этилцианоацетат (71,6 г, 633,0 ммоль) в течение 5 минут, с последующим добавлением одной порцией воды (4,5 мл). Смесь нагревали при 70°C в течение 40 минут и анализировали ВЭЖХ каждые 1-2 часа на процентную конверсию реагента в продукт. После того как наблюдали полное преобразование (обычно 100% преобразование через 5-8 часов), смесь охлаждали до 20-25°C и фильтровали через слой целита. Слой целита промывали толуолом (2×450 мл), и объединенные органические фракции концентрировали до 300 мл в вакууме при 60-65°C. Концентрат помещали в 225 мл ДМСО и концентрировали в вакууме при 70-80°C до прекращения активной отгонки растворителя. Раствор охлаждали до 20-25°C и разбавляли до 900 мл ДМСО для использования на стадии 2.
1H-ЯМР (500 МГц CDCl3) δ 7,16-7,10 (м, 2H), 7,03 (д, J=8,2 Гц, 1H), 4,63 (с, 1H), 4,19 (м, 2H), 1,23 (т, J=7,1 Гц, 3H).
Получение (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрила
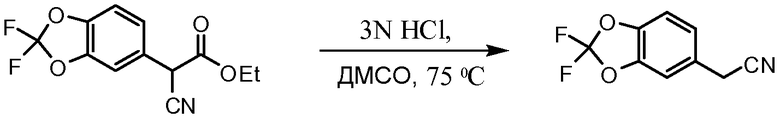
К ДМСО раствору (2,2-дифтор-1,3-бензодиоксол-5-ил)-1-этилацетатацетонитрила со стадии выше добавляли 3н. HCl (617,3 мл, 1,85 моль) в течение 20 минут, поддерживая внутреннюю температуру <40°C. Затем смесь нагревали при 75°C в течение 1 часа и анализировали ВЭЖХ каждые 1-2 час на % конверсии. Когда наблюдали конверсию >99% (обычно через 5-6 часов), реакцию охлаждали до 20-25°C и экстрагировали MTBE (2×525 мл), в течение достаточного времени для обеспечения полного разделения фаз в процессе экстракции. Объединенные органические экстракты промывали 5% NaCl (2×375 мл). Затем раствор переносили в устройство, подходящее для 1,5-2,5 мм рт. ст. вакуумной перегонки, снабженное охлажденной колбой-приемником. Раствор концентрировали в вакууме при <60°C для удаления растворителей. Затем (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрил отгоняли из полученного в результате масла при 125-130°C (температура печи) и 1,5-2,0 мм рт. ст. (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрил выделяли в виде прозрачного масла с выходом 66% из 5-бром-2,2-дифтор-1,3-бензодиоксола (2 стадии) и с ВЭЖХ чистотой 91,5% AUC (соответствует масс./масс. анализу 95%).
1Н-ЯМР (500 МГц, ДМСО) δ 7,44 (уш.с, 1H), 7,43 (д, J=8,4 Гц, 1H), 7,22 (дд, J=8,2, 1,8 Гц, 1H), 4,07 (с, 2H).
Получение (2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбонитрила
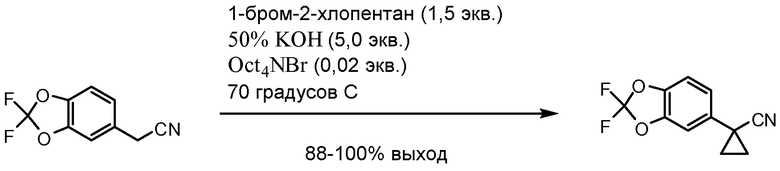
Смесь (2,2-дифтор-1,3-бензодиоксол-5-ил)ацетонитрила (1,0 экв.), 50% масс. водного KOH (5,0 экв.) 1-бром-2-хлопентана (1,5 экв.) и Oct4NBr (0,02 экв.) нагревали при 70°C в течение 1 часа. Реакционную смесь охлаждали, затем обрабатывали MTBE и водой. Органическую фазу промывали водой и насыщенным раствором соли, затем растворитель удаляли, с получением (2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбонитрила.
1H-ЯМР (500 МГц, ДМСО) δ 7,43 (д, J=8,4 Гц, 1H), 7,40 (д, J=1,9 Гц, 1H), 7,30 (дд, J=8,4, 1,9 Гц, 1H), 1,75 (м, 2H), 1,53 (м, 2H).
Получение 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбоновой кислоты

(2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбонитрил гидролизовали, используя 6M NaOH (8 экв.) в этаноле (5 об.) при 80°C в течение ночи. Смесь охлаждали до комнатной температуры, и этанол упаривали в вакууме. Остаток растворяли в воде и MTBE, добавляли 1М HCl, и слои разделяли. Затем MTBE слой обрабатывали дициклогексиламином (0,97 экв.). Суспензию охлаждали до 0°C, фильтровали и промывали гептаном, с получением соответствующей DCHA соли. Соль растворяли в MTBE и 10% лимонной кислоте и перемешивали до растворения твердых веществ. Слои разделяли, и MTBE слой промывали водой и насыщенным раствором соли. Растворитель заменяли гептаном, с последующим фильтрованием, с получением 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбоновой кислоты после сушки в вакуумной печи при 50°C в течение ночи. ESI-МС m/z рассчитано 242,04, найдено 241,58 (M+1)+;
1H-ЯМР (500 МГц, ДМСО) δ 12,40 (с, 1H), 7,40 (д, J=1,6 Гц, 1H), 7,30 (д, J=8,3 Гц, 1H), 7,17 (дд, J=8,3, 1,7 Гц, 1H), 1,46 (м, 2H), 1,17 (м, 2H).
Фрагмент амина
Получение 2-бром-5-фтор-4-нитроанилина.

В колбу помещали 3-фтор-4-нитроанилин (1,0 экв.), с последующим добавлением этилацетата (10 об.) и перемешивали, для растворения всех твердых веществ. Добавляли порциями N-бромсукцинимид (1,0 экв.), поддерживая внутреннюю температуру 22°C. В конце реакции, реакционную смесь концентрировали в вакууме на роторном испарители. Остаток суспендировали в дистиллированной воде (5 об.), для растворения и удаления сукцинимида. (Сукцинимид можно также удалить способом обработки водой). Воду декантировали, и твердое вещество суспендировали в 2-пропаноле (5 об.) в течение ночи. Полученную в результате суспензию фильтровали, и влажный осадок на фильтре промывали 2-пропанолом, сушили в вакуумной печи при 50°C в течение ночи с продувкой N2 до достижения постоянной массы. Выделяли желтовато-коричневый твердый остаток (50% выход, 97,5% AUC). Другими примесями были бром-региоизомер (1,4% AUC) и дибромовый продукт присоединения (1,1% AUC).
1H-ЯМР (500 МГц, ДМСО) δ 8,19 (1H, д, J=8,1 Гц), 7,06 (уш.с, 2H), 6.64 (д, 1H, J=14,3 Гц).
Получение тозилатной соли бензилгликолят-4-аммоний-2-бром-5-фторанилина
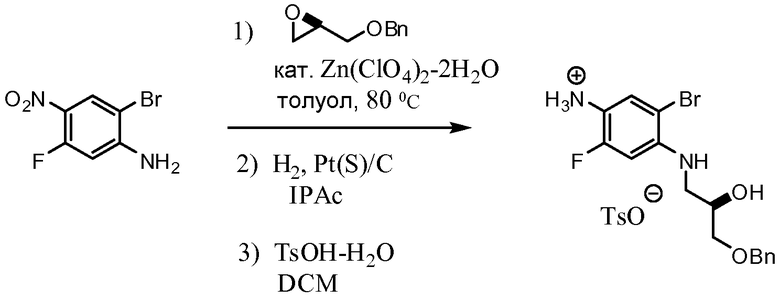
В тщательно высушенную колбу в атмосфере N2 помещали следующие реагенты: активированные порошкообразные 4Å молекулярные сита (50% масс. относительно 2-бром-5-фтор-4-нитроанилина), 2-бром-5-фтор-4-нитроанилин (1,0 экв.), дигидрат перхлората цинка (20 моль%) и толуол (8 об.). Смесь перемешивали при комнатной температуре для NMT 30 минут. Наконец, добавляли в стационарном потоке (R)-бензилглицидиловый эфир (2,0 экв.) в толуоле (2 об.). Реакционную смесь нагревали до 80°C (внутренняя температура) и перемешивали в течение приблизительно 7 часов или до того как 2-бром-5-фтор-4-нитроанилин составлял <5% AUC.
Реакцию охлаждали до комнатной температуры и добавляли целит (50% масс.), с последующим добавлением этилацетата (10 об.). Полученную в результате смесь фильтровали для удаления целита и сит и промывали этилацетатом (2 об.). Фильтрат промывали раствором хлорида аммония (4 об., 20% масс./об.). Органический слой промывали раствором бикарбоната натрия (4 об.×2,5% масс./об.). Органический слой концентрировали в вакууме на роторном испарителе. Полученную в результате суспензию растворяли в изопропилацетате (10 об.), и полученный раствор переносили в гидрогенизатор Буши.
В гидрогенизатор помещали 5% масс. Pt(S)/C (1,5 моль%), и смесь перемешивали в атмосфере N2 при 30°C (внутренняя температура). Реакцию продували N2, с последующей продувкой водородом. Давление гидрогенизатора устанавливали на 1 бар водорода, и смесь быстро перемешивали (>1200 об/мин). В конце реакции, катализатор фильтровали через слой целита и промывали дихлорметаном (10 об.). Фильтрат концентрировали в вакууме. Оставшийся изопропилацетат удаляли с дихлорметаном (2 об.), и концентрировали на роторном испарителе досуха.
Полученный в результате остаток растворяли в дихлорметане (10 об.), добавляли моногидрат п-толуолсульфокислоты (1,2 экв.) и перемешивали в течение ночи. Продукт фильтровали и промывали дихлорметаном (2 об.) и сушили в вакууме. Влажный осадок на фильтре переносили на сушильный лоток и в вакуумную печь и сушили при 45°C продувкой N2 до достижения постоянной массы. Тозилатную соль бензилгликолят-4-аммоний-2-бром-5-фторанилина выделяли в виде не совсем белого твердого остатка.
Определяли, что хиральная чистота составляла >97% э.и.
Получение (3-хлор-3-метилбут-1-инил)триметилсилана
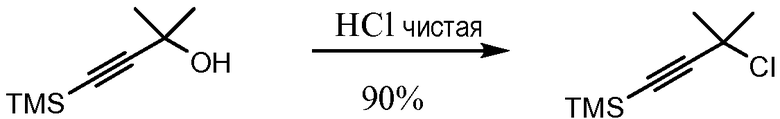
Пропаргиловый спирт (1,0 экв.) помещали в колбу. Добавляли водную хлористоводородную кислоту (37%, 3,75 об.) и начинали перемешивание. В процессе растворения твердого спирта, наблюдали небольшое поглощение тепла (5-6°C). Полученную в результате смесь перемешивали в течение ночи (16 часов), которая медленно становилась темно-красной. В 30 л реактор с рубашкой загружали воду (5 об.), которую, затем, охлаждали до 10°C. Реакционную смесь переносили медленно в воду за счет вакуума, поддерживая внутреннюю температуру смеси ниже 25°C. Добавляли гексан (3 об.), и полученную в результате смесь перемешивали в течение 0,5 часа. Фазы разделяли, и водную фазу (pH<1) сливали и отбрасывали. Органическую фазу концентрировали в вакууме, используя роторный испаритель, с получением продукта в виде красного масла.
Получение (4-(бензилокси)-3,3-диметилбут-1-инил)триметилсилана
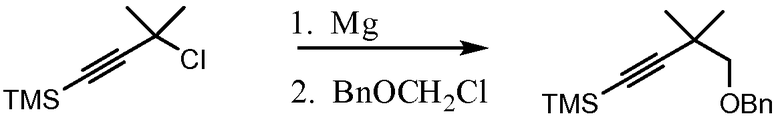
Способ A
Все дескрипторы эквивалентов и объемов в данной части представлены относительно 250 г реакции. Магниевую стружку (69,5 г, 2,86 моль, 2,0 экв.) помещали в 3 л 4-горлую колбу и перемешивали с магнитной мешалкой в атмосфере азота в течение 0,5 часа. Реактор погружали на баню со льдом и водой. Медленно добавляли в колбу при перемешивании раствор пропаргилхлорида (250 г, 1,43 моль, 1,0 экв.) в ТГФ (1,8 л, 7,2 об.) до наблюдения первоначальной экзотермической реакции (приблизительно 10°C). Образование реагента Гриньяра подтверждали IPC, применяя 1H-ЯМР спектроскопию. После завершения выделения тепла, медленно добавляли остаток раствора, поддерживая температуру смеси <15°C. Добавление требовало приблизительно 3,5 часа. Полученную в результате темно-зеленую смесь декантировали в 2 л колбу с пробкой.
Все дескрипторы эквивалентов и объемов в данной части представлены относительно 500 г реакции. В 22 л реактор помещали раствор бензилхлорметилового эфира (95%, 375 г, 2,31 моль, 0,8 экв.) в ТГФ (1,5 л, 3 об.). Ректор охлаждали на бане со льдом и водой. Две из четырех порций реагента Гриньяра, полученного выше, объединяли и затем медленно добавляли к бензилхлорметиловому эфиру через капельную воронку, поддерживая температуру смеси ниже 25°C. Добавление требовало 1,5 часов. Реакционную смесь перемешивали в течение ночи (16 часов).
Все дескрипторы эквивалентов и объемов в данной части представлены относительно 1 кг реакции. Раствор 15% хлорида аммония получали в 30 л реакторе с рубашкой (1,5 кг в 8,5 кг воды, 10 об.). Раствор охлаждали до 5°C. Две реакционные смеси с реактивом Гриньяра выше объединяли и затем переносили в раствор хлорида аммония через сосуд-коллектор. При данном гашении наблюдали экзотермическую реакцию, которое осуществляли при такой скорости, чтобы поддерживать внутреннюю температуру ниже 25°C. После завершения переноса, температуру рубашки реактора устанавливали равной 25°C. Добавляли гексан (8 л, 8 об.), и смесь перемешивали в течение 0,5 часа. После разделения фаз, водную фазу (pH 9) отделяли и отбрасывали. Оставшуюся органическую фазу промывали водой (2 л, 2 об.). Органическую фазу концентрировали в вакууме, используя 22 л роторный испаритель, с получением неочищенного продукта в виде оранжевого масла.
Способ B
Магниевую стружку (106 г, 4,35 моль, 1,0 экв.) помещали в 22 л реактор и затем суспендировали в ТГФ (760 мл, 1 об.). Реактор охлаждали на бане со льдом и водой так, чтобы температура смеси достигала 2°C. Добавляли медленно в реактор раствор пропаргилхлорида (760 г, 4,35 моль, 1,0 экв.) в ТГФ (4,5 л, 6 об.). После добавления 100 мл, добавление прекращали, и смесь перемешивали до повышения температуры до 13°C, что указывало на образование реагента Гриньяра. После завершения экзотермической реакции, медленно добавляли другие 500 мл раствора пропаргилхлорида, поддерживая температуру смеси <20°C. Образование реагента Гриньяра подтверждали IPC, применяя 1H-ЯМР спектроскопию. Медленно добавляли остаток раствора пропаргилхлорида, поддерживая температуру смеси <20°C. Добавление требовало приблизительно 1,5 часа. Полученный в результате темно-зеленый раствор перемешивали в течение 0,5 часа. Образование реагента Гриньяра подтверждали IPC, применяя 1H-ЯМР спектроскопию. Чистый бензилхлорметиловый эфир помещали в капельную воронку реактора и затем добавляли по каплям в реактор, поддерживая температуру смеси ниже 25°C. Добавление требовало 1,0 часа. Реакционную смесь перемешивали в течение ночи. Водная обработка и концентрирование осуществляли, применяя ту же методику и относительные количества веществ, как в способе A, с получением продукта в виде оранжевого масла.
Получение 4-бензилокси-3,3-диметилбут-1-ина

В 30 л реактор с рубашкой помещали метанол (6 об.), который затем охлаждали до 5°C. Добавляли в реактор гидроксид калия (85%, 1,3 экв.). Наблюдали повышение температуры на 15-20°C по мере растворения гидроксида калия. Температуру рубашки устанавливали на 25°C. Добавляли раствор 4-бензилокси-3,3-диметил-1-триметилсилилбут-1-ина (1,0 экв.) в метаноле (2 об.), и полученную в результате смесь перемешивали до завершения реакции, которую контролировали ВЭЖХ. Обычно время реакции при 25°C составляло 3-4 часа. Реакционную смесь разбавляли водой (8 об.) и затем перемешивали в течение 0,5 часа. Добавляли гексан (6 об.), и полученную в результате смесь перемешивали в течение 0,5 часа. Фазы разделяли, и затем водную фазу (pH 10-11) отделяли и отбрасывали. Органическую фазу промывали раствором KOH (85%, 0,4 экв.) в воде (8 об.), с последующей промывкой водой (8 об.). Затем органическую фазу концентрировали, используя роторный испаритель, с получением указанного в заголовке соединения в виде желто-оранжевого масла. Обычно чистота данного вещества составляла около 80% главным образом с одной присутствующей примесью.
1Н-ЯМР (400 МГц, C6D6) δ 7,28 (д, 2H, J=7,4 Гц), 7,18 (т, 2H, J=7,2 Гц), 7,10 (д, 1H, J=12 Гц), 4,35 (с, 2H), 3,24 (с, 2H), 1,91 (с, 1H), 1,25 (с, 6H).
Получение бензилгликолята 4-амино-2-(4-бензилокси-3,3-диметилбут-1-инил)-5-фторанилина
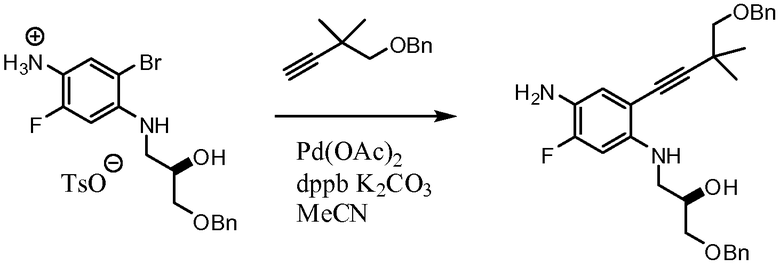
Тозилатную соль бензилгликолята 4-аммоний-2-бром-5-фторанилина преобразовывали в свободное основание перемешиванием твердого вещества в EtOAc (5 об.) и насыщенном NaHCO3 растворе (5 об.) до получения прозрачного органического слоя. Полученные в результате слои разделяли, и органический слой промывали насыщенным NaHCO3 раствором (5 об.), с последующей промывкой насыщенным раствором соли, и концентрировали в вакууме, с получением тозилатной соли бензилгликолята 4-аммоний-2-бром-5-фторанилина в виде масла.
Затем в колбу помещали тозилатную соль бензилгликолят 4-аммоний-2-бром-5-фторанилина (свободное основание, 1,0 экв.), Pd(OAc) (4,0 моль%), dppb (6,0 моль%) и порошкообразный K2CO3 (3,0 экв.) и перемешивали с ацетонитрилом (6 об.) при комнатной температуре. Полученную в результате реакционную смесь дегазировали в течение приблизительно 30 минут барбатированием N2 через трубку. Затем 4-бензилокси-3,3-диметилбут-1-ин (1,1 экв.), растворенный в ацетонитриле (2 об.), быстро добавляли и нагревали до 80°C и перемешивали до достижения полного исчезновения тозилатной соли 4-аммоний-2-бром-5-фторанилина. Реакционную суспензию охлаждали до комнатной температуры, фильтровали через слой целита и промывали ацетонитрилом (2 об.). Фильтрат концентрировали в вакууме, и остаток повторно растворяли в EtOAc (6 об.). Органический слой промывали дважды NH4Cl раствором (20% масс./об., 4 об.) и насыщенным раствором соли (6 об.). Полученный в результате органический слой концентрировали, с получением коричневого масла, и использовали таковой на следующей стадии.
Получение N-бензилгликолят-5-амино-2-(2-бензилокси-1,1-диметилэтил)-6-фториндола
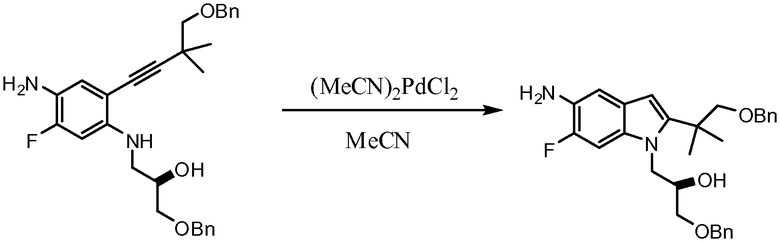
Неочищенное масло бензилгликолята 4-амино-2-(4-бензилокси-3,3-диметилбут-1-инил)-5-фторанилина растворяли в ацетонитриле (6 об.) и добавляли (MeCN)2PdCl2 (15 моль%) при комнатной температуре. Полученную в результате смесь дегазировали, используя N2 через трубку в течение приблизительно 30 минут. Затем реакционную смесь перемешивали при 80°C в атмосфере N2 в течение ночи. Реакционную смесь охлаждали до комнатной температуры, фильтровали через слой целита и промывали остаток ацетонитрилом (1 об.). Полученный в результате фильтрат концентрировали в вакууме и повторно растворяли в EtOAc (5 об.). Добавляли делоксан-II THP (5% масс. относительно теоретического выхода N-бензилгликолята 5-амино-2-(2-бензилокси-1,1-диметилэтил)-6-фториндола) и перемешивали при комнатной температуре в течение ночи. Затем смесь фильтровали через слой силикагеля (2,5 дюйма глубина, 6 дюймов диаметр фильтра) и промывали EtOAc (4 об.). Фильтрат концентрировали до темно-коричневого остатка и использовали как таковой на следующей реакции.
Повторная очистка неочищенного N-бензилгликолята 5-амино-2-(2-бензилокси-1,1-диметилэтил)-6-фториндола:
Неочищенный N-бензилгликолят-5-амино-2-(2-бензилокси-1,1-диметилэтил)-6-фториндол растворяли в дихлорметане (приблизительно 1,5 об.) и фильтровали через слой силикагеля, используя смесь 30% EtOAc/гептан, при этом удалялись примеси. Затем слой силикагеля промывали смесью 50% EtOAc/гептан для выделения N-бензилгликолята 5-амино-2-(2-бензилокси-1,1-диметилэтил)-6-фториндола до того, как в фильтрате наблюдали бледный цвет. Полученный фильтрат концентрировали в вакууме, с получением коричневого масла, которое кристаллизовалось при стоянии при комнатной температуре.
1H-ЯМР (400 МГц, ДМСО) δ 7,38-7,34 (м, 4H), 7,32-7,23 (м, 6H), 7,21 (д, 1H, J=12,8 Гц), 6,77 (д, 1H, J=9,0 Гц), 6,06 (с, 1H), 5,13 (д, 1H, J=4,9 Гц), 4,54 (с, 2H), 4,46 (уш.с, 2H), 4,45 (с, 2H), 4,33 (д, 1H, J=12,4 Гц), 4,09-4,04 (м, 2H), 3,63 (д, 1H, J=9,2 Гц), 3,56 (д, 1H, J=9,2 Гц), 3,49 (дд, 1H, J=9,8, 4,4 Гц), 3,43 (дд, 1H, J=9,8, 5,7 Гц), 1,40 (с, 6H).
Получение соединения 1
Получение бензил-защищенного соединения 1
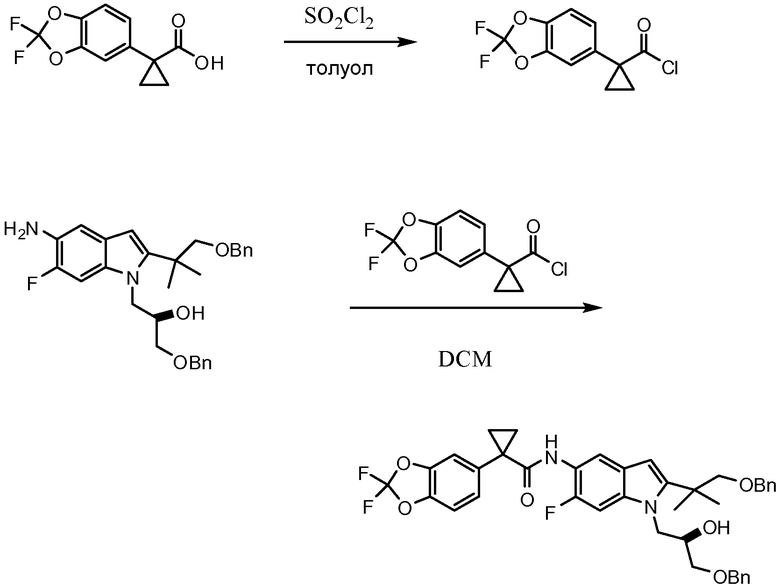
1-(2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбоновую кислоту (1,3 экв.) суспендировали в толуоле (2,5 об. относительно 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбоновой кислоты), и смесь нагревали до 60°C. Добавляли через капельную воронку SOCl2 (1,7 экв.). Полученную в результате смесь перемешивали в течение 2 часов. Толуол и избыток SOCl2 отгоняли, используя роторный испаритель. Добавляли дополнительное количество толуола (2,5 об. относительно 1-(2,2-дифтор-1,3-бензодиоксол-5-ил)циклопропанкарбоновой кислоты) и снова отгоняли. Неочищенный хлорангидрид кислоты растворяли в дихлорметане (2 об.) и добавляли через капельную воронку к смеси N-бензилгликолят 5-амино-2-(2-бензилокси-1,1-диметилэтил)-6-фториндола (1,0 экв.) и триэтиламин (2,0 экв.) в дихлорметане (7 об.), поддерживая температуру 0-3°C (внутренняя температура). Полученную в результате смесь перемешивали при 0°C в течение 4 часов и затем нагревали до комнатной температуры в течение ночи. Добавляли к реакционной смеси дистиллированную воду (5 об.) и перемешивали в течение NLT 30 минут, и слои разделяли. Органическую фазу промывали 20% масс. K2CO3 (4 об.×2), с последующей промывкой насыщенным раствором соли (4 об.) и концентрировали, с получением неочищенного бензил-защищенного соединения 1 в виде густого коричневого масла, которое дополнительно очищали фильтрованием через слой силикагеля.
Фильтрование через слой силикагеля: Неочищенное бензил-защищенное соединение 1 растворяли в этилацетате (3 об.) в присутствии активированного угля Darco-G (10% масс. относительно теоретического выхода бензил-защищенного соединения 1) и перемешивали при комнатной температуре в течение ночи. К полученной смеси добавляли гептан (3 об.) и фильтровали через слой силикагеля (2× масс. неочищенного бензил-защищенного соединения 1). Слой силикагеля промывали смесью этилацетат/гептан (1:1, 6 об.) или по существу до полного исчезновения цвета в фильтрате. Фильтрат концентрировали в вакууме, с получением бензил-защищенного соединения 1 в виде вязкого красновато-коричневого масла, и его использовали непосредственно на следующей стадии.
Повторная очистка: Бензил-защищенное соединение 1 повторно растворяли в дихлорметане (1 об. относительно теоретического выхода бензил-защищенного соединения 1) и наносили на слой силикагеля (2× масс. неочищенного бензил-защищенного соединения 1). Слой силикагеля промывали дихлорметаном (2 об. относительно теоретического выхода бензил-защищенного соединения 1), и фильтрат отбрасывали. Слой силикагеля промывали смесью 30% этилацетат/гептан (5 об.), и фильтрат концентрировали в вакууме, с получением бензил-защищенного соединения 1 в виде вязкого красновато-оранжевого масла, и его использовали непосредственно на следующей стадии.
Получение соединения 1
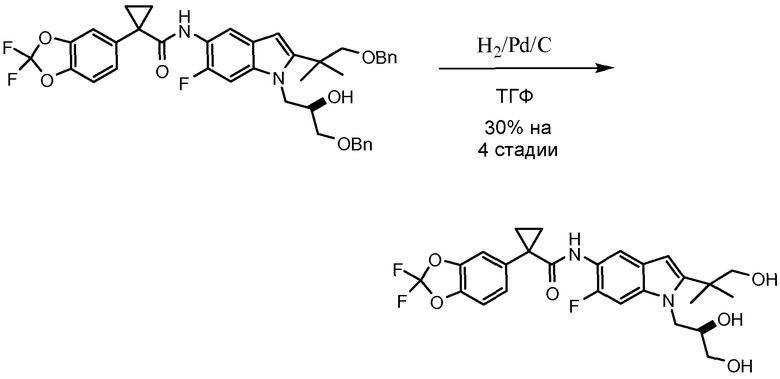
Способ A
20 л химический реактор продували три раза газообразным азотом, и затем помещали в него палладий на углероде (Evonik E 101 N/W, 5% Pd, 60% влажность, 200 г, 0,075 моль, 0,04 экв.). Затем химический реактор продували азотом три раза. Добавляли в химический реактор с помощью всасывания раствор неочищенного бензил-защищенного соединения 1 (1,3 кг, приблизительно 1,9 моль) в ТГФ (8 л, 6 об.). Реактор закрывали и затем продували три раза газообразным азотом. При легком перемешивании реактор продували три раза газообразным водородом, удаляли воздух в атмосферу разбавлением азотом. В химическом реакторе создавали давление 3 бар с помощью водорода, и скорость перемешивания увеличивали до 800 об/мин. Наблюдали быстрое поглощение водорода (растворение). После завершения поглощения, реактор нагревали до 50°C.
В целях безопасности, термостат выключали в конце каждого рабочего дня. В реакторе создавали давление 4 бар с помощью водорода и затем отключали от баллона с водородом.
После 2 полных дней реакции, добавляли к смеси дополнительное количество Pd/C (60 г, 0,023 моль, 0,01 экв.). Это осуществляли продувкой три раза газообразным азотом и затем добавлением катализатора через отверстие для добавления твердых веществ. Продолжительность реакции отслеживали как выше. После 4 полных дней, реакцию считали закончившейся, как определено с помощью ВЭЖХ, которая показала исчезновение не только исходного вещества, но также пика, соответствующего моно-бензилированного промежуточного соединения.
Реакционную смесь фильтровали через слой целита. Реактор и осадок на фильтре промывали ТГФ (2 л, 1,5 об.). Затем слой целита смачивали водой, и осадок на фильтре отбрасывали соответствующим образом. Объединенные фильтраты и ТГФ промывку концентрировали, используя роторный испаритель, с получением продукта в виде черного масла, 1 кг.
Эквиваленты и объемы следующей очистки даны относительно 1 кг неочищенного вещества. Неочищенное черное масло растворяли в смеси 1:1 этилацетат-гептан. Смесь помещали на слой силикагеля (1,5 кг, 1,5 по массе экв.) в пористую воронку, который был насыщен смесью 1:1 этилацетат-гептан. Слой силикагеля промывали смесью 1:1 этилацетат-гептан (6 л, 6 об.) и затем чистым этилацетатом (14 л, 14 об.). Элюент собирали в виде 4 фракций, которые анализировали ВЭЖХ.
Эквиваленты и объемы следующей очистки даны относительно 0,6 кг неочищенного вещества. Фракцию 3 концентрировали на роторном испарителе, с получением коричневой пены (600 г), и затем повторно растворяли в MTBE (1,8 л, 3 об.). Черно-коричневый раствор перемешивали в течение ночи при температуре окружающей среды, в процессе чего наблюдали кристаллизацию. Добавляли гептан (55 мл, 0,1 об.), и смесь перемешивали в течение ночи. Смесь фильтровали, используя воронку Бюхнера, и осадок на фильтре промывали смесью 3:1 MTBE-гептан (900 мл, 1,5 об.). Осадок на фильтре сушили на воздухе в течение 1 часа и затем сушили в вакууме при температуре окружающей среды в течение 16 часов, с получением 253 г VXc-661 в виде не совсем белого твердого остатка.
Эквиваленты и объемы следующей очистки даны относительно 1,4 кг неочищенного вещества. Фракции 2 и 3 после фильтрования на силикагеле выше, а также вещество из предыдущей реакции смешивали и концентрировали, с получением 1,4 кг черного масла. Смесь повторно фильтровали через силикагель (1,5 кг силикагеля, элюируя 3,5 л, 2,3 об. 1:1 этилацетат-гептан, затем 9 л, 6 об. чистого этилацетата), как описано выше, которая после концентрирования давала коричневое пенообразное твердое вещество (390 г).
Эквиваленты и объемы следующей очистки даны относительно 390 г неочищенного вещества. Коричневый остаток был нерастворим в MTBE, поэтому его растворяли в метаноле (1,2 л, 3 об.). Используя 4 л реактор Мортона, снабженный длинным дефлегматором, смесь отгоняли до 2 об. Добавляли MTBE (1,2 л, 3 об.), и смесь отгоняли до 2 об. Добавляли вторую порцию MTBE (1,6 л, 4 об.), и смесь отгоняли до 2 об. Добавляли третью порцию MTBE (1,2 л, 3 об.), и смесь отгоняли до 3 об. Анализ дистиллята GC показал, что он состоит приблизительно из 6% метанола. Термостат устанавливали на 48°C (ниже температуры кипения азеотропа MTBE-метанол, которая равна 52°C). Смесь охлаждали до 20°C в течение 2 часов, в течение которых наблюдали относительно быструю кристаллизацию. После перемешивания смеси в течение 2 часов добавляли гептан (20 мл, 0,05 об.), и смесь перемешивали в течение ночи (16 часов). Смесь фильтровали, используя воронку Бюхнера, и осадок на фильтре промывали смесью 3:1 MTBE-гептан (800 мл, 2 об.). Осадок на фильтре сушили на воздухе в течение 1 часа и затем сушили в вакууме при температуре окружающей среды в течение 16 часов, с получением 130 г соединения 1 в виде не совсем белого твердого остатка.
Способ B
Бензил-защищенное соединение 1 растворяли и промывали ТГФ (3 об.) для удаления любых оставшихся растворителей. Бензил-защищенное соединение 1 растворяли в ТГФ (4 об.) и добавляли в гидрогенизатор, содержащий 5% масс. Pd/C (2,55% моль, 60% влажность, Degussa E5 E101 NN/W). Внутреннюю температуру реакции устанавливали на 50°C, и реактор продували N2 (×5), с последующей продувкой водородом (×3). Давление в гидрогенизаторе устанавливали 3 бар водорода, и смесь быстро перемешивали (>1100 об/мин). В конце реакции, катализатор отфильтровывали через слой целита и промывали ТГФ (1 об.). Фильтрат концентрировали в вакууме, с получением коричневого пенообразного твердого вещества. Полученный в результате остаток растворяли в MTBE (5 об.) и добавляли 0,5н. HCl раствор (2 об.) и дистиллированную воду (1 об.). Смесь перемешивали в течение NLT 30 минут, и полученные в результате слои разделяли. Органическую фазу промывали 10% масс. K2CO3 раствором (2 об.×2), с последующей промывкой насыщенным раствором соли. Органический слой добавляли в колбу, содержащую силикагель (25% масс.), Deloxan-THP II (5% масс., 75% влажность) и Na2SO4, и перемешивали в течение ночи. Полученную в результате смесь фильтровали через слой целита и промывали 10% ТГФ/MTBE (3 об.). Фильтрат концентрировали в вакууме, с получением неочищенного соединения 1 в виде бледно-коричневой пены.
Выделение соединения 1 из маточника: вариант A
Фильтрация через слой силикагеля: Маточник концентрировали в вакууме, с получением коричневой пены, растворяли в дихлорметане (2 об.) и фильтровали через слой силикагеля (3× масс. неочищенного соединения 1). Слой силикагеля промывали смесью этилацетат/гептан (1:1, 13 об.), и фильтрат отбрасывали. Слой силикагеля промывали смесью 10% ТГФ/этилацетат (10 об.), и фильтрат концентрировали в вакууме, с получением соединения 1 в виде бледно-коричневой пены. Следовали указанному выше способу кристаллизации для выделения соединения 1.
Выделение соединения 1 из маточника: вариант B
Колоночная хроматография на силикагеле: После хроматографии на силикагеле (50% этилацетат/гексан-100% этилацетат), желаемое соединение выделяли в виде бледно-коричневой пены. Следовали вышеуказанному способу кристаллизации для выделения оставшегося соединения 1.
На фиг.1 показана порошковая рентгеновская дифрактограмма соединения 1. ДСК кривая соединения 1 показана на фиг.2. ДСК кривая на фиг.2 показывает, что соединение 1 не представляет собой чистую твердую фазу. Существует дополнительный пик при 119°C, по сравнению с формой A соединения 1 (см. фиг.6). ТГА кривая соединения 1 показана на фиг.3.
Соединение 1 можно также получить одним из нескольких синтетических способов, описанных в опубликованной патентной заявке США US20090131492, включенной в настоящее описание посредством ссылки.
Получение формы A соединения 1
Способ суспензионного типа
Для EtOAc, MTBE, изопропилацетата или DCM, приблизительно 40 мг соединения 1 добавляли в пробирку вместе с 1-2 мл любого из вышеуказанных растворителей. Суспензию перемешивали при комнатной температуре в течение от 24 часов до 2 недель, и форму A соединения 1 собирали центрифугированием суспензии (с помощью фильтра). На фиг.5 показана XRPD дифрактограмма формы A соединения 1, полученной данным способом с DCM в качестве растворителя.
Для EtOH/вода растворов, приблизительно 40 мг соединения 1 добавляли в три отдельные пробирки. В первую пробирку добавляли 1,35 мл EtOH и 0,15 мл воды. Во вторую пробирку добавляли 0,75 мл EtOH и 0,75 мл воды. В третью пробирку добавляли 0,15 мл EtOH и 1,35 мл воды. Все три пробирки перемешивали при комнатной температуре в течение 24 часов. Затем каждую суспензию центрифугировали отдельно (с фильтром), с получением формы A соединения 1.
Для растворов изопропиловый спирт/вода, приблизительно 40 мг соединения 1 добавляли в три отдельные пробирки. В первую пробирку добавляли 1,35 мл изопропилового спирта и 0,15 мл воды. Во вторую пробирку добавляли 0,75 мл изопропилового спирта и 0,75 мл воды. В третью пробирку добавляли 0,15 мл изопропилового спирта и 1,35 мл воды. Все три пробирки перемешивали при комнатной температуре в течение 24 часов. Затем каждую суспензию центрифугировали отдельно (с фильтром), с получением формы A соединения 1.
Для растворов метанол/вода, приблизительно 40 мг соединения 1 добавляли в пробирку. Добавляли 0,5 мл метанола и 1 мл воды, и суспензию перемешивали при комнатной температуре в течение 24 часов. Суспензию центрифугировали (с фильтром), с получением формы A соединения 1.
Для ацетонитрила, приблизительно 50 мг соединения 1 добавляли в пробирку вместе с 2,0 мл ацетонитрила. Суспензию перемешивали при комнатной температуре в течение 24 часов, и форму A соединения 1 собирали с помощью центрифуги (с фильтром).
Для растворов ацетонитрил/вода, приблизительно 50 мг соединения 1 растворяли в 2,5 мл ацетонитрила, с получением чистого раствора после обработки ультразвуком. Раствор фильтровали и 1 мл отбирали в пробирку. Добавляли 2,25 мл воды, с получением мутной суспензии. Суспензию перемешивали при комнатной температуре в течение 24 часов (с фильтром).
Способ медленного испарения
Приблизительно 55 мг соединения 1 растворяли в 0,5 мл ацетона, с получением чистого раствора после обработки ультразвуком. Раствор фильтровали, и 0,2 мл отбирали в пробирку. Пробирку накрывали парафильмом с одним проткнутым в нем отверстием и оставляли для выстаивания. Перекристаллизованную форму A соединения 1 собирали фильтрованием.
Способ быстрого испарения
Для изопропилового спирта, приблизительно 43 мг соединения 1 растворяли в 2,1 мл изопропилового спирта, с получением чистого раствора после обработки ультразвуком. Раствор фильтровали в пробирку и оставляли ее открытой. Перекристаллизованную форму A соединения 1 собирали фильтрованием.
Для метанола, приблизительно 58 мг соединения 1 растворяли в 0,5 мл метанола, с получением чистого раствора после обработки ультразвуком. Раствор фильтровали, и 0,2 мл отбирали в незакрытую пробирку и оставляли для выстаивания. Перекристаллизованную форму A соединения 1 собирали фильтрованием.
Для ацетонитрила, приблизительно 51 мг соединения 1 растворяли в 2,5 мл ацетонитрила, с получением чистого раствора после обработки ультразвуком. Раствор фильтровали, и половину раствора отбирали в открытую пробирку и оставляли для выстаивания. Перекристаллизованную форму A соединения 1 собирали фильтрованием. На фиг.7 показана XRPD дифрактограмма формы A соединения 1, полученной данным способом.
Способ с применением антирастворителя
Для смеси EtOAc/гептан, приблизительно 30 мг соединения 1 растворяли в 1,5 мл EtOAc, с получением чистого раствора после обработки ультразвуком. Раствор фильтровали, и 2,0 мл гептана добавляли в отфильтрованный раствор при медленном перемешивании. Раствор перемешивали в течение дополнительных 10 минут и оставляли для выстаивания. Перекристаллизованную форму A соединения 1 собирали фильтрованием. На фиг.8 показана XRPD дифрактограмма формы A соединения 1, полученной данным способом.
Для смеси изопропиловый спирт/вода, приблизительно 21 мг соединения 1 растворяли в 1,0 мл изопропилового спирта, с получением чистого раствора после обработки ультразвуком. Раствор фильтровали, с получением 0,8 мл раствора. Добавляли 1,8 мл воды при медленном перемешивании. Добавляли дополнительные 0,2 мл воды, с получением мутной суспензии. Перемешивание прекращали на 5 минут, с получением чистого раствора. Раствор перемешивали в течение дополнительных 2 минут и оставляли для выстаивания. Перекристаллизованную форму A соединения 1 собирали фильтрованием.
Для смеси этанол/вода, приблизительно 40 мг соединения 1 растворяли в 1,0 мл этанола, с получением чистого раствора после обработки ультразвуком. Раствор фильтровали и добавляли 1,0 мл воды. Раствор перемешивали в течение 1 дня при комнатной температуре. Перекристаллизованную форму A соединения 1 собирали фильтрованием.
Для смеси ацетон/вода, приблизительно 55 мг соединения 1 растворяли в 0,5 мл ацетона, с получением чистого раствора после обработки ультразвуком. Раствор фильтровали, и 0,2 мл отбирали в пробирку. Добавляли 1,5 мл воды, и затем дополнительные 0,5 мл воды, с получением мутной суспензии. Суспензию перемешивали в течение 1 дня при комнатной температуре. Форму A соединения 1 собирали фильтрованием.
В таблице 2 ниже обобщены различные способы получения формы A соединения 1.
Рентгеновская дифракционная картина, рассчитанная для монокристаллической структуры формы A соединения 1, показана на фиг.4. В таблице 3 перечислены рассчитанные пики для фиг.1.
Фактическая порошковая рентгеновская дифрактограмма формы A соединения 1 показана на фиг.5. В таблице 4 перечислены фактические пики для фиг.5.
ДСК кривая формы A соединения 1 показана на фиг.6. Температура плавания для формы A соединения 1 наблюдается приблизительно при 172-178°C.
Данные для монокристалла получены для формы A соединения 1, давая дополнительные подробности о кристаллической структуре, включая размеры решетки и упаковку.
Получение кристаллов
Кристаллы формы A соединения 1 получали медленным испарением из концентрированного раствора метанола (10 мг/мл). Отбирали бесцветные кристаллы формы A соединения 1 с размерами 0,20×0,05×0,05 мм, очищали, используя минеральное масло, устанавливали на MicroMount и центрировали на Bruker APEXil дифрактометре. Три серии 40 матриц, разделенных в обратном пространстве, получали для обеспечения матрицей ориентации и первоначальных параметров ячейки. Конечные параметры ячейки получали и корректировали на основе всего набора данных.
Эксперимент
Набор дифракционных данных в обратном пространстве получали до разрешения 0,83 Å, используя 0,5° шаг с 30 с воздействием для каждой матрицы. Данные получали при комнатной температуре [295 (2) K]. Интегрирование интенсивностей и уточнение параметров ячейки осуществляли, применяя APEXII программное обеспечение. Наблюдение кристалла после получения данных показало отсутствие следов разложения.
Данные кристалла формы A соединения 1
Геометрия: Все оцененные стандартные отклонения (за исключением оцененного стандартного отклонения в двухгранном угле между двумя среднеквадратичными плоскостями) оценивали, используя полную ковариационную матрицу. Все оцененные стандартные отклонения ячейки учитывались отдельно в оценке оцененных стандартных отклонений сторон, углов и торсионных углов; корреляции между оцененными стандартными отклонениями в параметрах ячейки применяли только тогда, когда они определялись кристаллической симметрией. Приближенный (изотропный) расчет оцененных стандартных отклонений ячейки применяли для оценки оцененных стандартных отклонений, включающих среднеквадратичные плоскости.
Параметры совокупности данных для кристалла формы A соединения 1
Сбор данных: Apex II; уточнение параметров ячейки: Apex II; обработка данных: Apex II; программа (программы), применяемая для разрешения структуры: SHELXS97 (Sheldrick, 1990); программа (программы), применяемая для уточнения структуры: SHELXL97 (Sheldrick, 1997); молекулярные графики: Mercury; программное обеспечение, применяемое для получения материалов для публикации: publCIF.
Параметры после уточнения для кристалла формы A соединения 1
Уточнение: уточнение F2 относительно всех отражений. Взвешенный R-фактор wR и точность приближения S основываются на F2, общепринятые R-факторы R основываются на F, с F, установленным на ноль, для отрицательного F2. Предельное выражение F2>2sigma(F2) применяют только для расчета R-факторов (gt) и т.д., и оно не соответствует выбору отражений для уточнения. R-факторы, основанные на F2, являются приблизительно в два раза больше, чем факторы на основе F, и R-факторы, основанные на всех данных, будут даже более большими.
Конформационные картины формы A соединения 1 на основе монокристаллического рентгеновского анализа показаны на фиг.9 и 10. Концевые группы -OH соединены через сеть водородных связей, образуя тетрамерный кластер с четырьмя соседними молекулами (фиг.10). Другая гидроксильная группа действует в качестве донора водородной связи для образования водородной связи с карбонильной группой из соседней молекулы. Кристаллическая структура показывает плотную упаковку молекул. Форма A соединения 1 является моноклинной, C2 пространственная группа, со следующими размерами элементарной ячейки: a=21,0952(16) Å, b=6,6287(5) Å, c=17,7917(15) Å, β=95,867(6)°, γ=90°.
Твердофазный 13C-ЯМР спектр формы A соединения 1 показан на фиг.11. В таблице 8 приведены химические сдвиги соответствующих пиков.
Твердофазный 19F-ЯМР спектр формы A соединения 1 показан на фиг.12. Пики со звездочкой обозначают вращение боковых цепей. В таблице 9 приведены химические сдвиги соответствующих пиков.
Получение аморфной формы соединения 1
Способ упаривания на роторном испарителе
Аморфную форму соединения 1 также получали с помощью упаривания на роторном испарителе.
Соединение 1 (приблизительно 10 г) растворяли в 180 мл MeOH и упаривали на роторном испарителе на 50°C бане до пены. ДСК (фиг.14) и XRPD (фиг.13) подтвердили аморфную форму соединения 1. На фиг.15 показана ТГА кривая аморфной формы соединения 1, полученной данным способом.
Способ сушки с распылением
9,95 г ацетатсукцината гидроксипропилметилцеллюлозы HG качества (HPMCAS-HG) взвешивали в 500 мл стакане вместе с 50 мг лаурилсульфата натрия (SLS). MeOH (200 мл) смешивали с твердым веществом. Вещество перемешивали в течение 4 часов. Для обеспечения максимального растворения, после 2 часов перемешивания раствор обрабатывали ультразвуком в течение 5 минут, затем перемешивание продолжали в течение оставшихся 2 часов. Мелкодисперсная суспензия HPMCAS оставалась в растворе. Однако визуальное наблюдение определило, что смолы не осталось на стенках реактора или не прилипло ко дну после наклонения реактора.
Форму A соединения 1 (10 г) выливали в 500 мл стакан, и систему продолжали перемешивать. Раствор сушили распылением, применяя следующие параметры:
Выделяли приблизительно 16 г аморфной формы соединения 1 (80% выход). Аморфную форму соединения 1 подтверждали XRPD (фиг.16) и ДСК (фиг.17).
Твердофазный 13C-ЯМР спектр аморфной формы соединения 1 показан на фиг.18. В таблице 10 приведены химические сдвиги соответствующих пиков.
Твердофазный 19F-ЯМР спектр аморфной формы соединения 1 показан на фиг.19. Пики со звездочкой обозначают вращение боковых цепей. В таблице 11 приведены химические сдвиги соответствующих пиков.
В таблице 12 ниже перечислены дополнительные аналитические данные для соединения 1.
АНАЛИЗЫ
Анализы на обнаружение и измерение ΔF508-CFTR корректирующих свойств соединений
Оптические способы измерения трансмембранного потенциала для анализа ΔF508-CFTR модулирующих свойств соединений
В оптическом анализе трансмембранного потенциала используют потенциалчувствительные FRET сенсоры, описанные Gonzalez и Tsien (See, Gonzalez, J.E. и R.Y. Tsien (1995) "Voltage sensing by fluorescence resonance energy transfer in single cells" Biophys J 69(4): 1272-80, и Gonzalez, J.E. и R.Y. Tsien (1997) "Improved indicators of cell membrane potential that use fluorescence resonance energy transfer" Chem Biol 4(4): 269-77) в комбинации с приборами для измерения изменения флуоресценции, такими как Voltage/Ion Probe Reader (VIPR) (см., Gonzalez, J. E., K. Oades, et al. (1999) "Cell-based assays and instrumentation for screening ion-channel targets" Drug Discov Today 4(9): 431-439).
Данные потенциалчувствительные анализы основаны на резонансном переносе энергии флуоресценции (FRET) между растворенным в мембране, потенциалчувствительным красителем, DiSBAC2(3), и флуоресцентным фосфолипидом, CC2-DMPE, который соединен с внешней стороной плазматической мембраны и действует в качестве FRET донора. Изменения потенциала мембраны (Vm) приводят к тому, что отрицательно заряженный DiSBAC2(3) перераспределяется поперек плазматической мембраны и, соответственно, изменяется количество переноса энергии из CC2-DMPE. Изменение испускания флуоресценции отслеживают, используя VIPR™ II, который представляет собой встроенное устройство для подачи жидкости и флуоресцентный детектор, предназначенный для проведения получения изображения на основе клеток в 96- или 384-луночных титрационных микропланшетах.
1. Обнаружение корректирующих соединений
Для обнаружения малых молекул, которые корректируют транспортный дефект, связанный с ΔF508-CFTR, был разработан формат анализа с одним добавлением HTS. Клетки выдерживали в бессывороточной среде в течение 16 часов при 37°C в присутствии или отсутствии (отрицательный контроль) тестируемого соединения. В качестве положительного контроля, клетки, помещенные в 384-луночные планшеты, выдерживали в течение 16 часов при 27° для "температурной коррекции" ΔF508-CFTR. Затем, клетки промывали 3× раствором Кребса-Рингера и помещали в потенциалчувствительные красители. Для активации ΔF508-CFTR, 10 мкМ форсколина и CFTR потенциатора, генистеина (20 мкМ), добавляли вместе со средой, не содержащей Cl-, в каждую лунку. Добавление среды, не содержащей Cl-, способствовало Cl- истечению в ответ на ΔF508-CFTR активацию, и полученную в результате деполяризацию мембраны оптически отслеживали, используя индикаторные для напряжения красители на основе FRET.
2. Обнаружение потенциирующих соединений
Для обнаружения потенциаторов ΔF508-CFTR, был разработан формат анализа с двойным добавлением HTS. В течение первого добавления, среду, не содержащую Cl-, с или без тестируемого соединения добавляли в каждую лунку. Через 22 секунды добавляли вторую среду, не содержащую Cl- и содержащую 2-10 мкМ форсколина, для активации ΔF508-CFTR. Внеклеточная Cl- концентрация после обоих добавлений составляла 28 мМ, которая способствовала Cl- истечению в ответ на ΔF508-CFTR активацию, и полученную в результате деполяризацию мембраны оптически отслеживали, используя индикаторные для напряжения красители на основе FRET.
3. Растворы
4. Культура клеток
NIH3T3 мышиные фибробласты, стабильно экспрессирующие ΔF508-CFTR, использовали для оптических измерений мембранного потенциала. Клетки выдерживали при 37°C в 5% CO2 и 90% влажности в модифицированной по способу Дульбекко среде Игла, дополненной 2 мМ глютамином, 10% фетальной телячьей сывороткой, 1X NEAA, β-ΜΕ, 1X pen/strep и 25 мМ HEPES в 175 см2 колбах для культур. Для всех оптических анализов, клетки высевали при 30000/лунка в 384-луночные планшеты, покрытые матригелем, и выращивали в течение 2 часов при 37°C перед выращиванием при 27°C в течение 24 часов для анализа на потенцирование. Для анализов на коррекцию, клетки выращивали при 27°C или 37°C с и без соединений в течение 16-24 часов.
Электрофизиологические анализы для определения ΔF508-CFTR модулирующих свойств соединений
1. Анализ с использованием камеры Уссинга
Эксперименты с использованием камеры Уссинга проводили на поляризованных эпителиальных клетках, экспрессирующих ΔF508-CFTR, для получения дополнительных характеристик ΔF508-CFTR модуляторов, обнаруженных в оптических анализах. FRTΔF508-CFTR эпителиальные клетки, выращенные на Costar Snapwell вставках клеточных культур, помещали в камеру Уссинга (Physiologic Instruments, Inc., San Diego, CA), и монослои непрерывно коротко замыкали, применяя систему фиксации потенциала (Department of Bioengineering, University of Iowa, IA, and, Physiologic Instruments, Inc., San Diego, CA). Трансэпителиальное сопротивление измеряли, применяя 2 мВ импульс. В данных условиях, FRT эпителий показывал сопротивление 4 ΚΩ/см2 или более. Растворы выдерживали при 27°C и барбатировали воздухом. Потенциал смещения электрода и сопротивление жидкости корректировали, применяя вставку без клеток. В данных условиях, ток отражает поток Cl- через ΔF508-CFTR, экспрессированный в апикальной мембране. ISC получали в цифровой форме, применяя MP100A-CE интерфейс и AcqKnowledge программное обеспечение (v3.2.6; BIOPAC Systems, Santa Barbara, CA).
2. Обнаружение корректирующих соединений
В стандартном протоколе применяют градиент концентрации Cl- от базолатеральной к апикальной мембране. Для создания данного градиента, нормальный раствор Рингера использовали на базолатеральной мембране, тогда как апикальный NaCl заменяли эквимолярным количеством глюконата натрия (оттитрованный до pH 7,4 NaOH) для получения большого градиента концентрации Cl- поперек эпителия. Все эксперименты проводили с интактным монослоем. Для полной активации ΔF508-CFTR, использовали форсколин (10 мкМ) и PDE ингибитор, IB MX (100 мкМ), с последующим добавлением CFTR потенциатора, генистеина (50 мкМ).
Как наблюдалось для других типов клеток, ингибирование при низких температурах FRT клеток, стабильно экспрессирующих ΔF508-CFTR, увеличивало функциональную плотность CFTR в плазматической мембране. Для определения активности корректирующих соединений, клетки выдерживали с 10 мкМ тестируемого соединения в течение 24 часов при 37°C, и затем промывали 3× перед регистрацией данных. цАМФ- и генистеин-опосредованный Isc в клетках, обработанных соединением, приводили к 27°C и 37°C контролям и выражали в виде процента активности. Предварительное выдерживание клеток с корректирующим соединением значительно снижало цАМФ- и генистеин-опосредованный Isc по сравнению с 37°C контролями.
3. Обнаружение потенциирующих соединений
В стандартном протоколе применяют градиент концентрации Cl- от базолатеральной к апикальной мембране. Для создания данного градиента, нормальный раствор Рингера использовали на базолатеральной мембране и пермеабилизировали нистатином (360 мкг/мл), тогда как апикальный NaCl заменяли эквимолярным количеством глюконата натрия (оттитрованный до pH 7,4 NaOH) для получения большого градиента концентрации Cl- поперек эпителия. Все эксперименты проводили через 30 минут после пермеабилизации нистатином. Форсколин (10 мкМ) и все тестируемые соединения добавляли к обеим сторонам вставок для культур клеток. Эффективность предполагаемых ΔF508-CFTR потенциаторов сравнивали с эффективностью известного потенциатора, генистеина.
4. Растворы
5. Культура клеток
Эпителиальные клетки крыс Фишера (FRT), экспрессирующие ΔF508-CFTR (FRTΔF508-CFTR), использовали для экспериментов с камерой Уссинга для предполагаемых ΔF508-CFTR модуляторов, обнаруженных в оптических анализах авторов. Клетки выращивали на вставках для клеточных культур Costar Snapwell и выращивали в течение 5 дней при 37°С и 5% CO2 в среде Хэма F-12, модифицированной Куном, снабженной 5% фетальной телячьей сывороткой, 100 мкг/мл пенициллина и 100 мкг/мл стрептомицина. Перед применением для получения характеристик по потенцирующей активности соединений, клетки выдерживали при 27°C в течение 16-48 часов для коррекции для ΔF508-CFTR. Для определения активности корректирующих соединений, клетки выдерживали при 27°C или 37°C с и без соединений в течение 24 часов.
6. Снятие показаний на цельной клетке
Макроскопический ΔF508-CFTR ток (IΔF508) в NIH3T3 клетках, скорректированных по температуре и тестируемому соединению, стабильно экспрессирующих ΔF508-CFTR, отслеживали, применяя перфорированный пэтч-кламп, снятие показаний для цельной клетки. Кратко, снятие показаний при фиксации потенциала IΔF508 осуществляли при комнатной температуре, используя Axopatch 200B пэтч-кламп усилитель (Axon Instruments Inc., Foster City, CA). Все показания получали при частоте отбора проб 10 кГц и низкочастотно фильтровали при 1 кГц. Пипетки имели сопротивление 5-6 ΜΩ при заполнении их внутриклеточным раствором. В данных условиях получения показаний рассчитанный потенциал реверсии для Cl- (ECl) при комнатной температуре составлял -28 мВ. Все показания имели сопротивление изоляции >20 GΩ и добавочное сопротивление <15 ΜΩ. Генерирование импульса, получение данных и анализ осуществляли, применяя ПК, снабженный Digidata 1320 A/D интерфейсом в сочетании с Clampex 8 (Axon Instruments Inc.). Баня содержала <250 мкл соляного раствора, и непрерывно перфузировалась при скорости 2 мл/минута, используя гравитационную перфузионную систему.
7. Обнаружение корректирующих соединений
Для определения активности корректирующих соединений, увеличивающей плотность активного ΔF508-CFTR в плазматической мембране, применяли описанный выше способы получения показаний с помощью перфорированного пэтч-клампа для измерения плотности тока после 24-часовой обработки корректирующими соединениями. Для полной активации ΔF508-CFTR, добавляли к клеткам 10 мкМ форсколина и 20 мкМ генистеина. В данных условиях получения показаний, плотность тока после 24-часового выдерживания при 27°C была больше, чем плотность тока, наблюдаемая после 24-часового выдерживания при 37°C. Данные результаты согласуются с известными эффектами низкотемпературного выдерживания на плотность ΔF508-CFTR в плазматической мембране. Для определения эффектов корректирующих соединений на CFTR плотности тока, клетки выдерживали с 10 мкМ тестируемого соединения в течение 24 часов при 37°C, и плотность тока сравнивали с 27°C и 37°C контролями (% активности). Перед получением показаний, клетки промывали 3× внеклеточной средой для получения показаний для удаления оставшегося тестируемого соединения. Предварительное выдерживание с 10 мкМ корректирующих соединений значительно повышало цАМФ- и генистеин-зависимый ток, по сравнению с 37°C контролями.
8. Обнаружение потенциирующих соединений
Способность ΔF508-CFTR потенциаторов повышать макроскопический ΔF508-CFTR Cl- ток (IΔF508) в NIH3T3 клетках, стабильно экспрессирующих ΔF508-CFTR, также исследовали, применяя способы получения показаний с помощью перфорированного пэтч-клампа. Потенциаторы, обнаруженные в оптических анализах, вызывали зависящее от дозы увеличение IΔF508 с аналогичной силой и эффективностью, наблюдаемыми в оптических анализах. Во всех исследуемых клетках потенциал реверсии перед и в процессе применения потенциатора составлял приблизительно -30 мВ, который представлял собой рассчитанный ECl (-28 мВ).
9. Растворы
10. Культура клеток
NIH3T3 мышиные фибробласты, стабильно экспрессирующие ΔF508-CFTR, использовали для получения показаний на цельной клетке. Клетки выдерживали при 37°C в 5% CO2 и 90% влажности в модифицированной по способу Дульбекко среде Игла, дополненной 2 мМ глютамином, 10% фетальной телячьей сывороткой, 1X NEAA, β-ΜΕ, 1X pen/strep и 25 мМ HEPES в 175 см2 колбах для культур. Для получения показаний на цельной клетке, 2500-5000 клеток высевали на покрытые поли-L-лизином покровные стекла и выращивали в течение 24-48 часов при 27°C перед применением для исследования активности потенциаторов, и выдерживали с или без корректирующего соединения при 37°C для измерения активности корректоров.
11. Получение показаний для одиночного канала
Активность одиночного канала скорректированного по температуре ΔF508-CFTR, стабильно экспрессирующегося в NIH3T3 клетках, и активности потенцирующих соединений наблюдали, применяя пэтч-кламп с конфигурацией «наружная сторона внутри». Кратко, получение показаний при фиксации потенциала активности одиночного канала проводили при комнатной температуре с Axopatch 200B пэтч-кламп усилителем (Axon Instruments Inc.). Все показания получали при частоте отбора проб 10 кГц и низкочастотно фильтровали при 400 Гц. Пэтч-пипетки были изготовлены из Corning Kovar Sealing #7052 стекла (World Precision Instruments, Inc., Sarasota, FL) и имели сопротивление 5-8 ΜΩ при заполнении внеклеточным раствором. ΔF508-CFTR активировался после отсечения добавлением 1 мМ Mg-АТФ и 75 нМ цАМФ-зависимой протеинкиназы, каталитической субъединицы (PKA; Promega Corp. Madison, WI). После стабилизации активности канала, пэтч перфузировали, используя гравитационную микроперфузионную систему. Приток помещали рядом с пэтч, что приводило в результате к полному обмену раствора в пределах 1-2 секунд. Для поддержания ΔF508-CFTR активности в процессе быстрой перфузии, добавляли к отмывающему раствору неспецифический фосфатазный ингибитор F- (10 мМ NaF). В данных условиях получения показаний, канальная активность оставалась постоянной в течение всего времени получения показаний с помощью пэтч-клампа (вплоть 60 минут). Токи, создаваемые положительным зарядом, перемещающимся из внутриклеточных растворов (анионы, перемещающиеся в противоположном направлении), показаны в качестве положительных токов. Потенциал пипетки (Vp) поддерживали равным 80 мВ.
Канальную активность анализировали по мембранным пэтчам, содержащим ≤2 активных каналов. Максимальное количество одновременных открытий каналов определяло количество активных каналов в течение эксперимента. Для определения амплитуды тока одиночного канала, данные, полученные после 120 секунд, ΔF508-CFTR активности "автономно" фильтровали при 100 Гц, и затем применяли для построения гистограммы амплитуды по всем точкам, которую согласовывали с мультигауссовыми функциями, применяя Bio-Patch Analysis программное обеспечение (Bio-Logic Comp. France). Суммарный микроскопический ток и вероятность открытого состояния канала (PO) определяли из 120 секундной канальной активности. PO определяли, применяя Bio-Patch программное обеспечение или из соотношения PO=I/i(N), где I = средний ток, i = амплитуда тока одиночного канала и N = число активных каналов в пэтче.
12. Растворы
13. Клеточная культура
NIH3T3 мышиные фибробласты, стабильно экспрессирующие ΔF508-CFTR, использовали для получения показаний пэтч-клампа в конфигурации с оторванным кусочком мембраны. Клетки выдерживали при 37°C в 5% CO2 и 90% влажности в модифицированной по способу Дульбекко среде Игла, дополненной 2 мМ глютамином, 10% фетальной телячьей сывороткой, 1X NEAA, β-ΜΕ, 1X pen/strep и 25 мМ HEPES в 175 см2 колбах для культур. Для получения показаний для одиночного канала, 2500-3000 клеток высевали на покрытых поли-L-лизином покровных стеклах и выращивали в течение 24-48 часов при 27°C перед применением.
Применяя способы, описанные выше, измеряли активность, т.е. EC50, соединения 1, и она показана в таблице 13.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2, 2-ДИФТОРБЕНЗО[d][1, 3]ДИОКСОЛ-5-ИЛ)-N-(1-(2, 3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2711481C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ МУКОВИСЦИДОЗНЫМ ТРАНСМЕМБРАННЫМ РЕГУЛЯТОРОМ ПРОВОДИМОСТИ | 2015 |
|
RU2744460C2 |
| ТВЕРДЫЕ ФОРМЫ 3-(2, 2-ДИФТОРБЕНЗО[D][1, 3] ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ | 2011 |
|
RU2579370C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2012 |
|
RU2640420C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ КАССЕТНЫХ ТРАНСПОРТЕРОВ | 2008 |
|
RU2512682C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ CFTR ЗАБОЛЕВАНИЙ | 2014 |
|
RU2718044C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ ТРАНСМЕМБРАННЫМ РЕГУЛЯТОРОМ КИСТОЗНОГО ФИБРОЗА CFTR | 2013 |
|
RU2827711C2 |
| ПРЕПАРАТЫ 3-(6-(1-(2, 2-ДИФТОРБЕНЗО[D][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ | 2013 |
|
RU2644723C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ 3-(2, 2-ДИФТОРБЕНЗО[d][1, 3]ДИОКСОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДО)-3-МЕТИЛПИРИДИН-2-ИЛ)БЕНЗОЙНУЮ КИСЛОТУ, И ИХ ВВЕДЕНИЕ | 2011 |
|
RU2592368C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ ТРАНСМЕМБРАННЫМ РЕГУЛЯТОРОМ КИСТОЗНОГО ФИБРОЗА (CFTR) | 2013 |
|
RU2692676C2 |
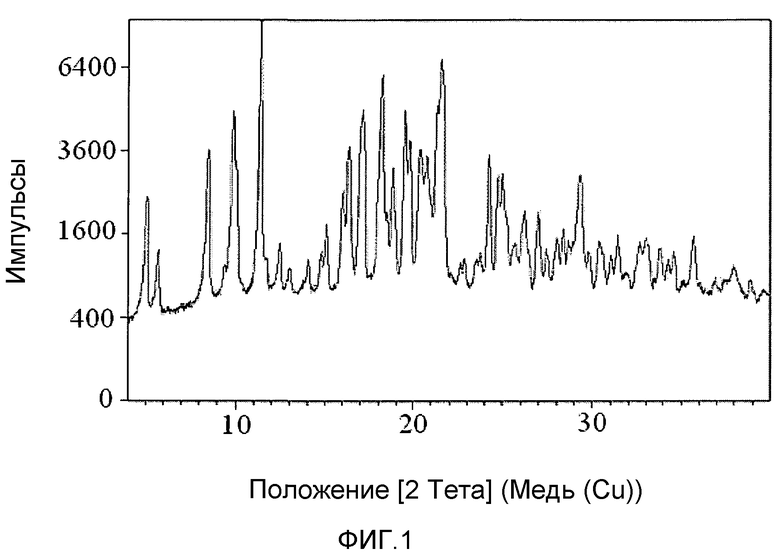
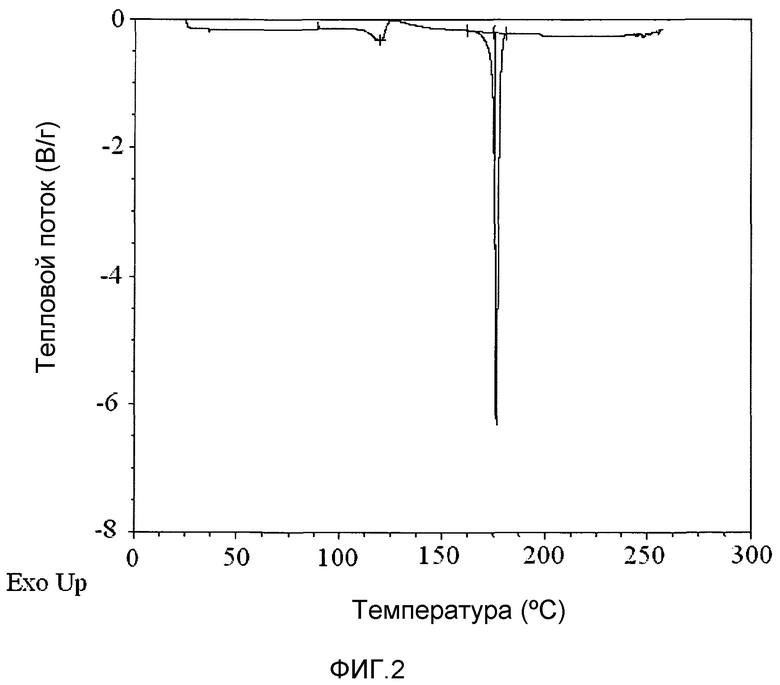
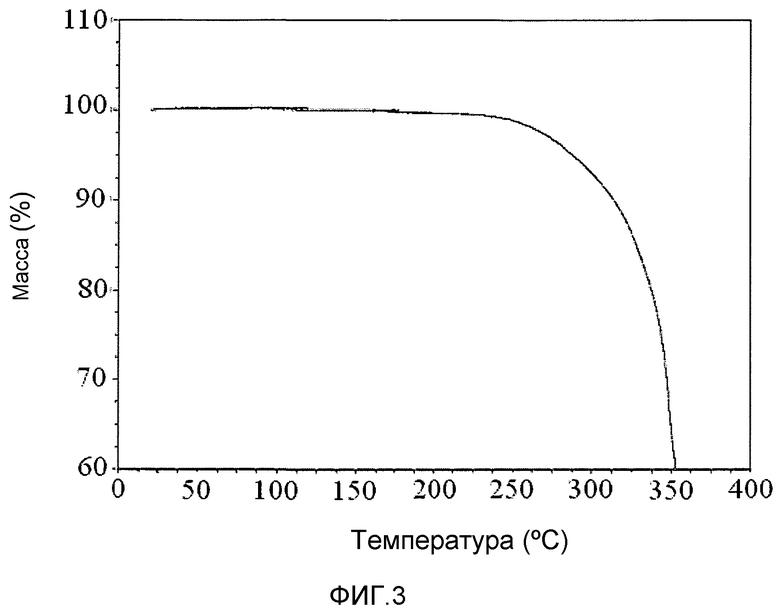
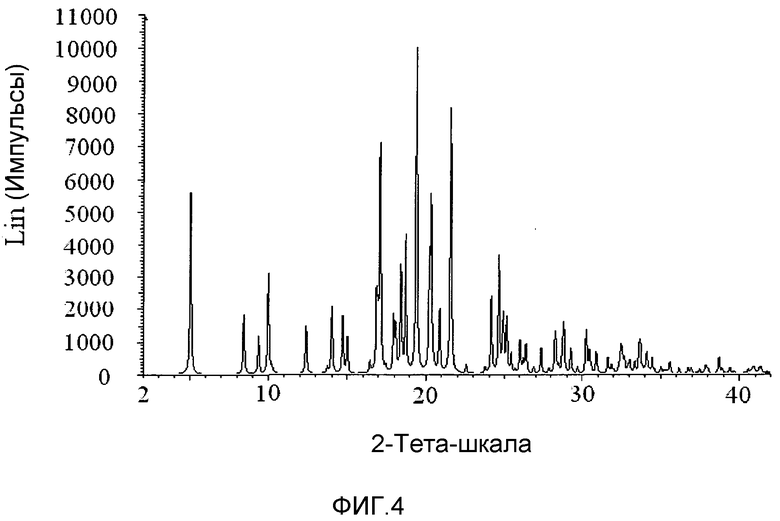
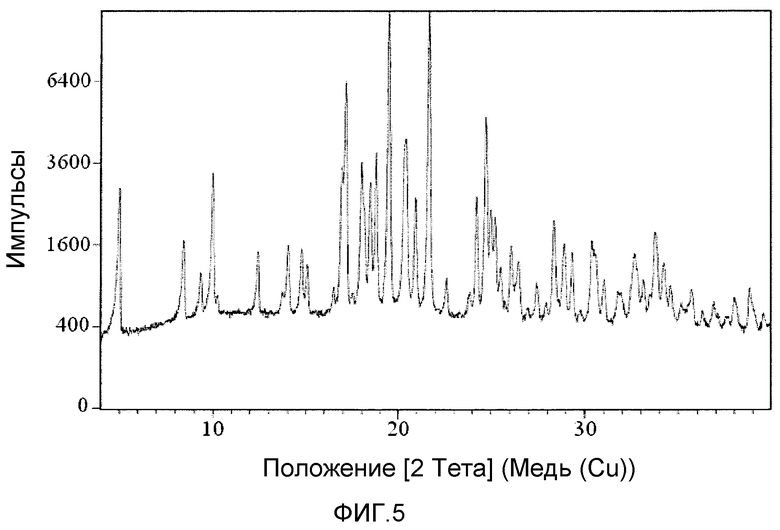
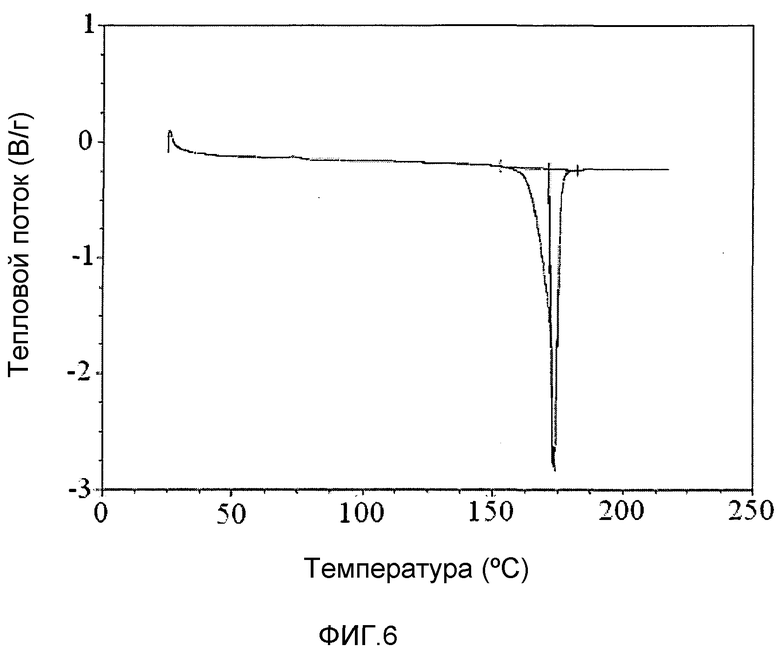
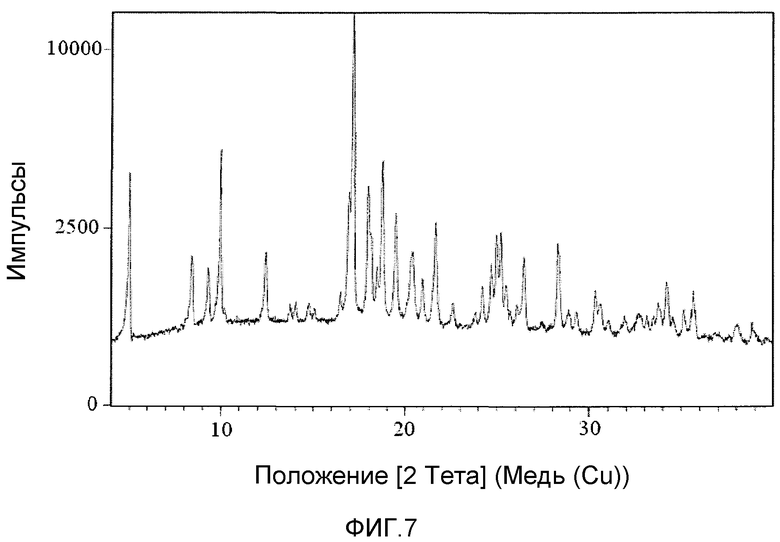
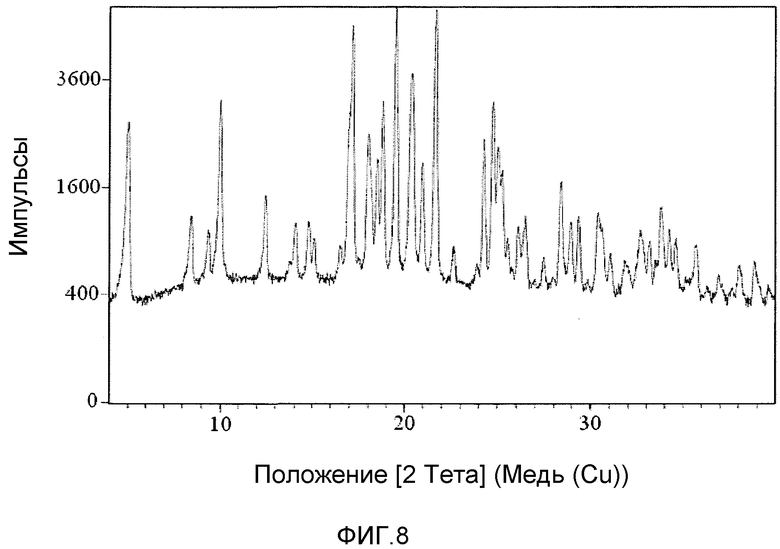
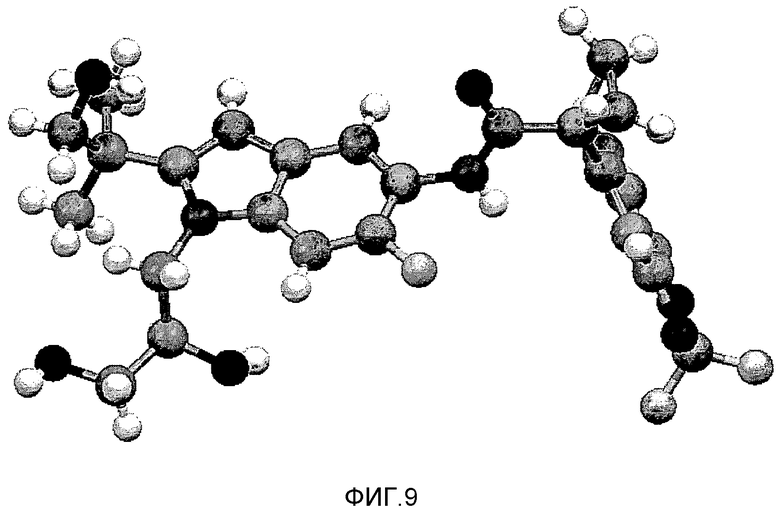
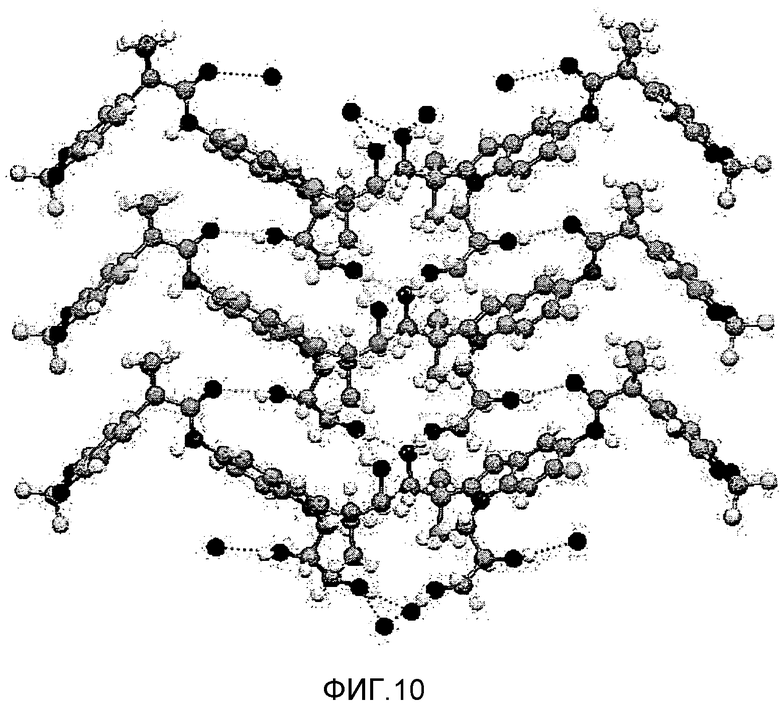
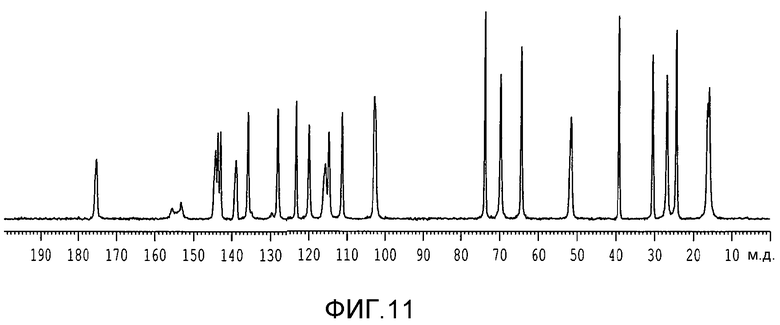
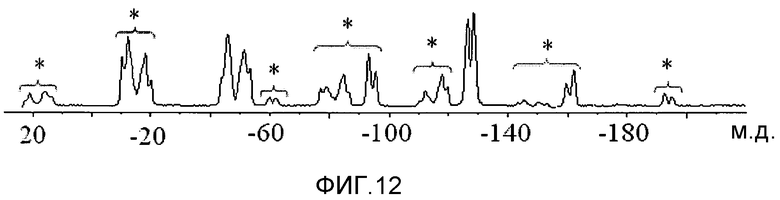
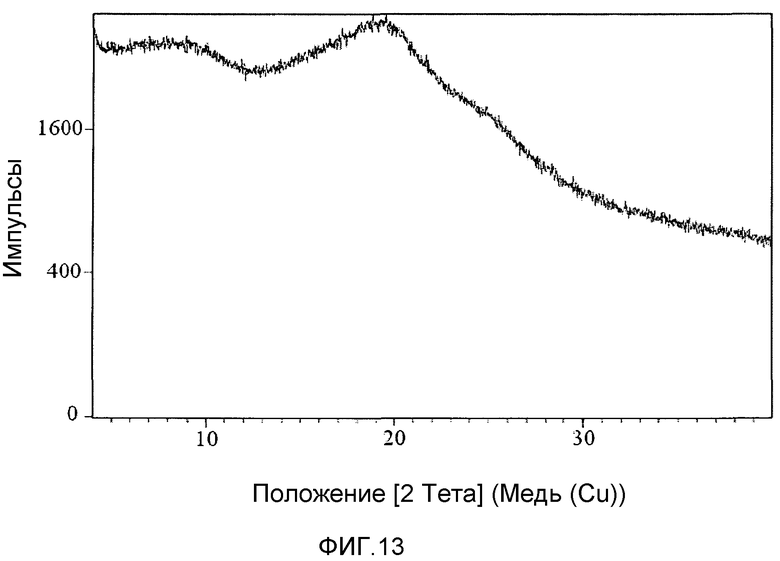
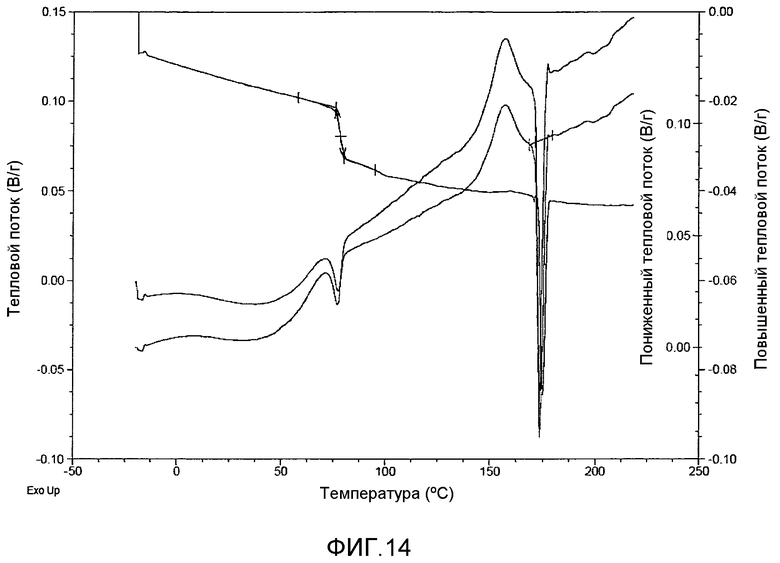
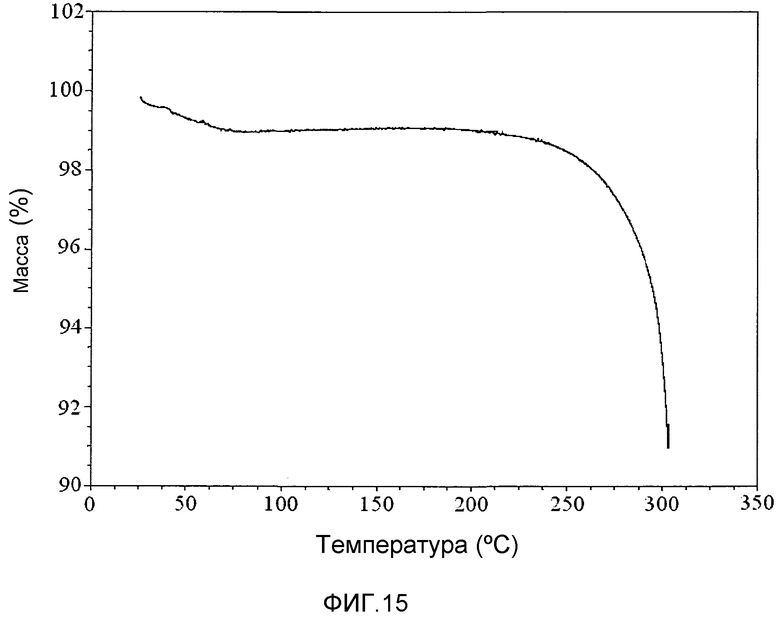
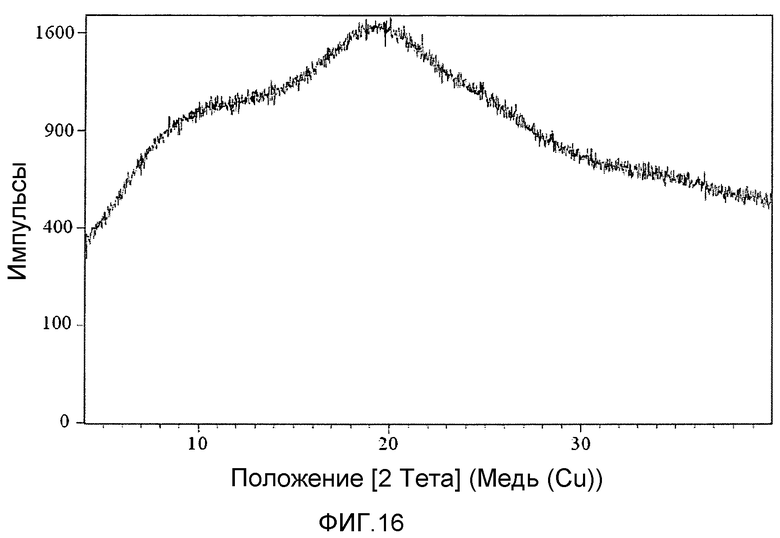
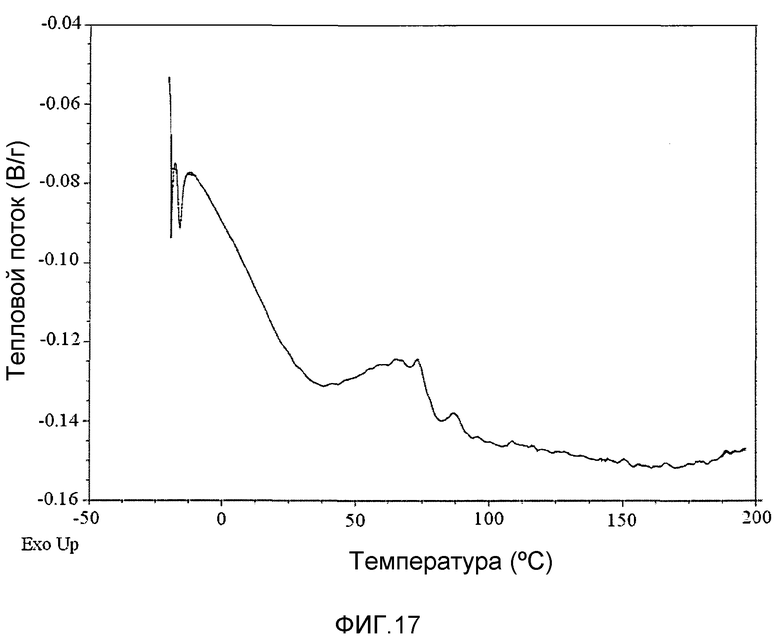
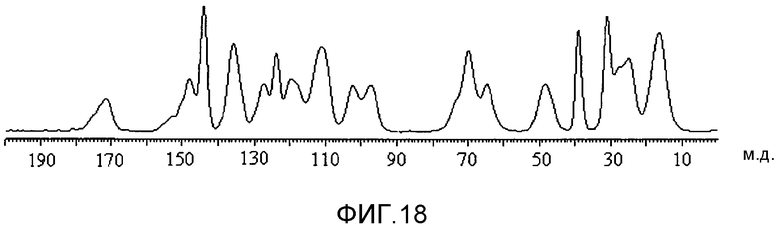
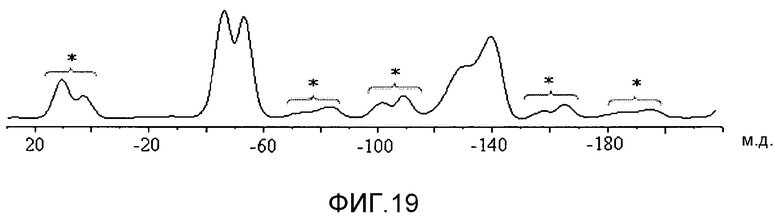
Настоящее изобретение относится к твердым формам (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1H-индол-5-ил)циклопропанкарбоксамида (соединение 1) по существу в кристаллической форме (форма A), где форма А характеризуется одним или более пиками при 19,3-19,7 градусах, 21,5-21,9 градусах и 16,9-17,3 градусах в рентгеновской порошковой дифрактометрии, полученной с применением Сu К альфа излучения, или аморфной форме, которая находится в форме высушенной распыленной дисперсии, их фармацевтическим композициям, а также к способам лечения. 14 н. и 68 з.п. ф-лы, 19 ил., 13 табл.
1. (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид, характеризующийся как кристаллическая форма А, где форма А характеризуется одним или более пиками при 19,3-19,7 градусах, 21,5-21,9 градусах и 16,9-17,3 градусах в рентгеновской порошковой дифрактометрии, полученной с применением Сu К альфа излучения.
2. Форма А по п. 1, где форма А характеризуется одним или более пиками приблизительно при 19,5, 21,7 и 17,1 градусах.
3. Форма А по п. 1, где форма А дополнительно характеризуется пиками при 20,2-20,6 градусах.
4. Форма А по п. 1, где форма А дополнительно характеризуется пиком приблизительно при 20,4 градусах.
5. Форма А по п. 1, где форма А дополнительно характеризуется пиком при 18,6-19,0 градусах.
6. Форма А по п. 1, где форма А дополнительно характеризуется пиком приблизительно при 18,8 градусах.
7. Форма А по п. 1, где форма А дополнительно характеризуется пиком при 24,5-24,9 градусах.
8. Форма А по п. 1, где форма А дополнительно характеризуется пиком приблизительно при 24,7 градусах.
9. Форма А по п. 1, где форма А дополнительно характеризуется пиком при 9,8-10,2 градусах.
10. Форма А по п. 1, где форма А дополнительно характеризуется пиком приблизительно при 10,0 градусах.
11. Форма А по п. 1, где форма А дополнительно
характеризуется пиком при 4,8-5,2 градусах.
12. Форма А по п. 1, где форма А дополнительно характеризуется пиком приблизительно при 5,0 градусах.
13. Форма А по п. 1, где форма А дополнительно характеризуется пиком при 24,0-24,4 градусах.
14. Форма А по п. 1, где форма А дополнительно характеризуется пиком приблизительно при 24,2 градусах.
15. Форма А по п. 1, где форма А дополнительно характеризуется пиком при 18,3-18,7 градусах.
16. Форма А по п. 1, где форма А дополнительно характеризуется пиком приблизительно при 18,5 градусах.
17. Форма А по п. 1, где форма А характеризуется дифракционной картиной, по существу аналогичной дифракционной картине фиг. 4.
18. Форма А по п. 1, где форма А характеризуется дифракционной картиной, по существу аналогичной дифракционной картине фиг. 5.
19. Кристаллическая форма (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида, имеющая моноклинную кристаллическую систему, С2 пространственную группу и следующие размеры элементарной ячейки:
а=21,0952(16) Å α=90°
b=6,6287 (5) Å β=95,867 (6)°
с=17,7917(15) Å γ=90°.
20. Фармацевтическая композиция, обладающая активностью модулятора CFTR, содержащая форму А по п. 1 и фармацевтически приемлемый носитель.
21. Фармацевтическая композиция по п. 20, дополнительно содержащая дополнительный терапевтический агент.
22. Фармацевтическая композиция по п. 21, где дополнительный терапевтический агент выбран из муколитического средства, бронхолитического средства, антибиотика, противоинфекционного средства, противовоспалительного средства, CFTR потенциатора или питательного вещества.
23. Способ получения формы А по п. 1, включающий суспендирование (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида в растворителе в течение эффективного количества времени.
24. Способ по п. 23, где растворитель представляет собой этилацетат, дихлорметан, МТВЕ, изопропилацетат, вода/этанол, вода/ацетонитрил, вода/метанол или вода/изопропиловый спирт.
25. Способ по п. 23, где эффективное количество времени составляет от 24 часов до 2 недель.
26. Способ получения формы А по п. 1, включающий растворение (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида в растворителе и упаривание растворителя.
27. Способ по п. 26, где растворитель представляет собой ацетон, ацетонитрил, метанол или изопропиловый спирт.
28. Способ получения формы А по п. 1, включающий растворение (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида в первом растворителе и добавление второго растворителя, в котором (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид является нерастворимым.
29. Способ по п. 28, где первый растворитель представляет собой этилацетат, этанол, изопропиловый спирт или ацетон.
30. Способ по п. 28, где второй растворитель представляет собой гептан или воду.
31. Способ по п. 28, где добавление второго растворителя осуществляют при перемешивании раствора первого растворителя и (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида.
32. Твердый по существу аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид, где твердый по существу аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид находится в форме высушенной распылением дисперсии.
33. Аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-
ил)-1Н-индол-5-ил)циклопропанкарбоксамид по п. 32, содержащий менее чем приблизительно 5% кристаллического (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида.
34. Фармацевтическая композиция, обладающая активностью модулятора CFTR, содержащая аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид по п. 32 и фармацевтически приемлемый носитель, где аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид находится в формк высушенной распылением дисперсии.
35. Фармацевтическая композиция по п. 34, дополнительно содержащая дополнительный терапевтический агент.
36. Фармацевтическая композиция по п. 35, где дополнительный терапевтический агент выбран из муколитического средства, бронхолитического средства, антибиотика, противоинфекционного средства, противовоспалительного средства, CFTR потенциатора или питательного вещества.
37. Способ получения аморфного (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида по п. 32, включающий растворение (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-
индол-5-ил)циклопропанкарбоксамида в подходящем растворителе и удаление растворителя упариванием на роторном испарителе, где аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид находится в форме высушенной распылением дисперсии.
38. Способ по п. 37, где растворитель представляет собой метанол.
39. Твердая дисперсия, обладающая активностью модулятора CFTR, содержащая аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид по п. 32 и полимер.
40. Твердая дисперсия по п. 39, где полимер представляет собой гидроксипропилметилцеллюлозу (НРМС).
41. Твердая дисперсия по п. 39, где полимер представляет собой ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS).
42. Твердая дисперсия по п. 39, где полимер присутствует в количестве от 10% масс, до 80% масс.
43. Твердая дисперсия по п. 30, где полимер присутствует в количестве от 30% масс, до 60% масс.
44. Твердая дисперсия по п. 39, где полимер присутствует в количестве приблизительно 49,5% масс.
45. Твердая дисперсия по п. 39, где (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид присутствует в количестве от 10% масс.
до 80% масс.
46. Твердая дисперсия по п. 39, где (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид присутствует в количестве от 30% масс, до 60% масс.
47. Твердая дисперсия по п. 39, где (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид присутствует в количестве приблизительно 50% масс.
48. Твердая дисперсия по п. 39, дополнительно содержащая поверхностно-активное вещество.
49. Твердая дисперсия по п. 48, где поверхностно-активное вещество представляет собой лаурилсульфат натрия.
50. Твердая дисперсия по п. 48, где поверхностно-активное вещество присутствует в количестве от 0,1% масс, до 5% масс.
51. Твердая дисперсия по п. 48, где поверхностно-активное вещество присутствует в количестве приблизительно 0,5% масс.
52. Твердая дисперсия по п. 48, где полимер представляет собой ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS) в количестве 49,5% масс., поверхностно-активное вещество представляет собой лаурилсульфат натрия в количестве 0,5% масс., и (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид присутствует в количестве 50% масс.
53. Фармацевтическая композиция, обладающая активностью модулятора CFTR, содержащая твердую дисперсию по п. 39 и фармацевтически приемлемый носитель.
54. Фармацевтическая композиция по п. 53, дополнительно содержащая дополнительный терапевтический агент.
55. Фармацевтическая композиция по п. 54, где дополнительный терапевтический агент выбран из муколитического средства, бронхолитического средства, антибиотика, противоинфекционного средства, противовоспалительного средства, CFTR потенциатора или питательного вещества.
56. Способ получения аморфного (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида, включающий сушку распылением формы А (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида.
57. Способ по п. 56, включающий смешение формы A (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида и подходящего растворителя, и затем сушку распылением смеси для получения аморфного (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида.
58. Способ по п. 57, где растворитель представляет собой спирт.
59. Способ по п. 57, где растворитель представляет собой метанол.
60. Способ по п. 56, включающий:
a) получение смеси, содержащей форму A (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида, полимер и растворитель; и
b) сушку распылением смеси для получения твердой дисперсии.
61. Способ по п. 60, где полимер представляет собой ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS).
62. Способ по п. 60, где полимер содержится в количестве от 10% масс, до 80% масс, твердой дисперсии.
63. Способ по п. 60, где полимер содержится в количестве приблизительно 49,5% масс, твердой дисперсии.
64. Способ по п. 60, где растворитель представляет собой метанол.
65. Способ по п. 60, где смесь дополнительно содержит поверхностно-активное вещество.
66. Способ по п. 65, где поверхностно-активное вещество представляет собой лаурилсульфат натрия (SLS).
67. Способ по п. 65, где поверхностно-активное вещество содержится в количестве от 0,1% масс. до 5% масс. твердой дисперсии.
68. Способ по п. 65, где поверхностно-активное вещество содержится в количестве приблизительно 0,5% масс. твердой дисперсии.
69. Способ по п. 60, где полимер представляет собой
ацетатсукцинат гидроксйпропилметилцеллюлозы (HPMCAS) в количестве приблизительно 49,5% масс.твердой дисперсии, растворитель представляет собой метанол, и смесь дополнительно содержит лаурилсульфат натрия в количестве приблизительно 0,5% масс, твердой дисперсии.
70. Способ лечения CFTR-опосредованного заболевания у субъекта, включающий введение субъекту эффективного количества формы А по п. 1, аморфного (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамида по п. 32 или твердой дисперсии по п. 39.
71. Способ по п. 70, где CFTR-опосредованное заболевание выбрано из кистозного фиброза, астмы, ХОБЛ, вызванной курением, хронического бронхита, синусита, запора, панкреатита, недостаточности поджелудочной железы, мужского бесплодия, вызванного врожденной билатеральной аплазией семявыносящих протоков (CBAVD), легкой болезни легких, идиоматического панкреатита, аллергического бронхолегочного аспергиллеза (АБЛА), заболевания печени, наследственной эмфиземы, наследственного гемохроматоза, нарушений коагуляции-фибринолиза, дефицита белка С, наследственного ангионевротического отека типа 1, нарушения переработки липидов, семейной гиперхолестеринемии, хиломикронемии 1 типа, абеталипопротеинемии, болезней лизосомальных накоплений, I-клеточной анемии/болезни Дери, мукополисахаридоза, болезни Сандгоффа/Тея-Сакса, болезни Криглера-Наджара II типа, полиэндокринопатии/гиперинсулинемии, сахарного диабета, карликовости Ларона, миелопероксидазной
недостаточности, первичного гипопаратиреоза, меланомы, синдрома дефицита синтеза гликопротеинов CDG типа 1, врожденного гипертиреоза, незавершенного остеогенеза, наследственной гипофибриногенемии, ACT дефицита, несахарного диабета (DI), нейрофизеального DI, нейрогенного DI, синдрома Шарко-Мари-Туса, заболевания Пелицеуса-Мерцбахера, нейродегенеративных заболеваний, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, прогрессирующего надъядерного паралича, болезни Пика, нескольких полиглутаминовых неврологических расстройств, болезни Хантингтона, спиноцеребеллярной атаксии типа I, спинной и бульбарной мышечной атрофии, дентаторубального паллидолюизиана, миотонической дистрофии, губчатой энцефалопатии, наследственной болезни Крейтцфельда-Якоба (в связи с нарушением процессинга прионных белков), болезни Фабри, синдрома Штрауслера-Шейнкера, ХОБЛ, сухих глаз, болезни Шегрена, остеопороза, остеопении, синдрома Горема, каналопатии хлоридных каналов, врожденной миотонии (формы Томпсона и Беккера), синдрома Бартера типа III, болезни Дента, гиперэкплексии, эпилепсии, лизосомальной болезни накопления, синдрома Ангельмана, первичной цилиарной дискинезии (PCD), наследственного нарушения структуры и/или функции ресничек, PCD с транспозицией органов (также известного как синдром Картагенера), PCD без транспозиции органов или цилиарной аплазии.
72. Способ по п. 70, где CFTR-опосредованное заболевание выбрано из кистозного фиброза, ХОБЛ, остеопороза или эмфиземы.
73. Способ по п. 70, где CFTR-опосредованное заболевание
представляет собой кистозный фиброз.
74. Способ по п. 70, где субъект имеет муковисцидозный трансмембранный рецептор (CFTR) с AF508 мутацией.
75. Способ по п. 70, где субъект имеет муковисцидозный трансмембранный рецептор (CFTR) с R117H мутацией.
76. Способ по п. 70, где субъект имеет муковисцидозный трансмембранный рецептор (CFTR) с G551D мутацией.
77. Способ по п. 70, где способ включает введение дополнительного терапевтического агента.
78. Способ по п. 77, где терапевтический агент выбран из муколитического средства, бронхолитического средства, антибиотика, противоинфекционного средства, противовоспалительного средства, CFTR потенциатора или питательного вещества.
79. Набор для модулирования активности CFTR, содержащий форму А по п. 1, аморфный (R)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-N-(1-(2,3-дигидроксипропил)-6-фтор-2-(1-гидрокси-2-метилпропан-2-ил)-1Н-индол-5-ил)циклопропанкарбоксамид по п. 32, или твердую дисперсию по п. 39, и инструкцию для их применения.
80. Фармацевтическая композиция по п. 22, где дополнительный терапевтический агент представляет собой N-(5-гидрокси-2,4-ди-трет-ьутилфенил)-4-оксо-1Н-хинолин-3-карбоксамид.
81. Фармацевтическая композиция по п. 36, где дополнительный терапевтический агент представляет собой N-(5-гидрокси-2,4-ди-трет-ьутилфенил)-4-оксо-1Н-хинолин-3-карбоксамид.
82. Фармацевтическая композиция по п. 55, где дополнительный терапевтический агент представляет собой N-(5-гидрокси-2,4-ди-
трет-ьутилфенил)-4-оксо-1Н-хинолин-3-карбоксамид.
| US 20090131492 A1 21.05.2009 | |||
| Caira M.R.,"Crystalline Polymorfphism of Organic Compounds", Topics in Current Chemistry,Springer,Berlin, v.198, 1998 | |||
| RU 2002123882 A 20.03.2004. |

