ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к новой стабильной кристаллической форме D-изоглутамил-D-триптофана и способу ее выделения в чистой форме, свободной от неорганических солей. Данное изобретение также относится к новой стабильной аммонийной соли D-изоглутамил-D-триптофана и способу ее получения в чистой форме кристаллизацией и/или традиционной хроматографией на колонке с силикагелем.
УРОВЕНЬ ТЕХНИКИ
D-Изоглутамил-D-триптофан (также известный как H-D-iGlu-Trp-OH или тимодепрессин [Thymodepressin]) представляет собой синтетический геморегуляторный дипептид, созданный для лечения аутоиммунных заболеваний, в том числе псориаза (Sapuntsova, S. G., et al. (May 2002), Bulletin of Experimental Biology and Medicine, 133(5), 488-490). Тимодепрессин считается в России эффективным средством для лечения псориаза (патент США 5736519), где лекарственное средство на данное время присутствует на рынке как динатриевая соль в жидком препарате для инъекционного и интраназального введения. Он представляет собой иммунодепрессант и избирательно ингибирует пролиферацию клеток костного мозга, таким образом, вызывая угнетение иммунитета.
Известная твердая форма динатриевой соли D-изоглутамил-D-триптофана представляет собой аморфный порошок, который является гигроскопическим и очень сложным в обращении с технологической точки зрения. Структура динатриевой соли тимодепрессина описана в Kashirin, D. M., et al. (2000), Pharmaceutical Chemistry Journal, 34(11), 619-622. Мононатриевая соль D-изоглутамил-D-триптофана идентифицирована Службой Chemical Abstracts (CAS) и приведена в реестрах CAS REGISTRYSM, но отсутствуют публикации относительно ее получения и физических свойств. Порошковая или аморфная форма соединения, такого как D-изоглутамил-D-триптофан, предназначенного для фармацевтического применения, может создавать производственные проблемы за счет проблем с насыпной плотностью, гигроскопичностью и варьирующим содержанием воды, которое не может быть отрегулировано сушкой под вакуумом. D-Изоглутамил-D-триптофан представляет собой дипептид, и сушка аморфной формы при повышенной температуре, например 80-100°C, под вакуумом не рекомендуется.
Единственный путь синтеза H-D-iGlu-D-Trp-OH, известный из литературы, раскрыт в патенте США 5736519. В соответствии со способом, приведенным в Примере 1 патента США 5736519, изображенным в данном описании как Схема 1, Boc-D-Glu-OH (1.1) реагирует с 1,3-дициклогексилкарбодиимидом (ДЦК) с образованием циклического ангидрида (1.2). После удаления дициклогексилмочевины (ДЦМ, DCU) фильтрацией, ангидрид (1.2) реагирует с H-D-Trp-OH с образованием смеси дипептида Boc-D-iGlu-D-Trp-OH (1.3) и Boc-D-Glu-D-Trp-OH (1.4). Объединенный выход сырых Boc-D-iGlu-D-Trp-OH (1.3) и Boc-D-Glu-D-Trp-OH (1.4) составляет 70%. Однако смесь содержит только не более 35% целевого промежуточного соединения Boc-D-iGlu-D-Trp-OH (1.3). Защитную группу Boc удаляют, перемешивая растворы (1.3) и (1.4) в муравьиной кислоте как растворителе при температуре 40°C в течение 1 часа. Отношение (1.3) и (1.4) к муравьиной кислоте составляет приблизительно 1 г:8 мл (масс./об.). Продукт представляет собой смесь H-D-iGlu-D-Trp-OH (1.5) и H-D-Glu-D-Trp-OH (1.6). Поскольку пептиды (1.5) и (1.6) выделяют в равном количестве, очистка требует ионообменной хроматографии с использованием пиридинового ацетатного буфера. Выход целевого продукта H-D-iGlu-D-Trp-OH (1.5) составляет 35% от количества Boc-D-iGlu-D-Trp-OH (1.3). Таким образом, общий выход H-D-iGlu-D-Trp-OH (1.5) от количества Boc-D-Glu-OH составляет 12,25%.
Схема 1: Синтез H-D-iGlu-D-Trp-OH в соответствии с патентом США 5736519.

Способу, описанному в патенте США 5736519, свойственны несколько недостатков, как указано ниже:
1. ДЦК на стадии 1A может приводить к образованию других побочных продуктов, таких как
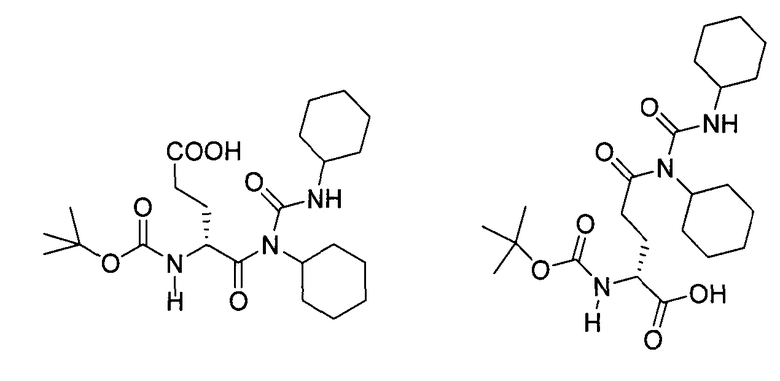
О побочных продуктах в результате соединения пептидов с ДЦК сообщалось в Marder, О., и Albericio, F. (июнь 2003), Chemical Oggi (Chemistry Today), 6-32.
2. Снятие защиты с Boc-D-iGlu-D-Trp-OH (1.3) требует повышенной температуры, и конечная очистка H-D-iGlu-D-Trp-OH требует крайне токсичного растворителя пиридина. Повышенная температура в ходе снятия защиты с (1.3) может приводить к образованию N-трет-бутилиндольного производного (1.7) как примеси (Löw, M., et. al. (1978), Hoppe-Seyler's Z. Physiol. Chem., 359(12):1643-51). Кроме того, пептид может циклизоваться с образованием глутаримида (1.8) (Pandit, U.K. (1989), Pure & Appl. Chem., Vol. 61, No. 3, pp. 423-426).
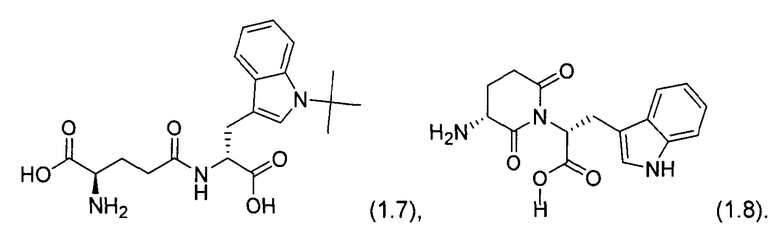
3. В ходе реакции соединения образуется только смесь Boc-D-iGlu-D-Trp-OH (1.3) и Boc-D-Glu-D-Trp-OH (1.4) в соотношении 1:1. Максимальный выход (1.3) не может превышать 50% на стадии соединения 1B. Смесь D-Glu-D-Trp-OH и D-iGlu-D-Trp-OH образуется в конце синтеза. Пептиды необходимо отделять ионообменной хроматографией и препаративной обращенно-фазовой высокоэффективной жидкостной хроматографией (ВЭЖХ). Общий выход H-D-iGlu-D-Trp-OH (1.5) составляет 12,25%, а очистка препаративной ВЭЖХ представляет собой очень длительный и неэффективный процесс. Время удерживания для двух подобных изомеров, H-D-iGlu-D-Trp-OH (1.5) и H-D-Glu-D-Trp-OH (1.6), не приведено. Повторные циклы выделения для повышения чистоты целевого изомера (1.5) являются очень неэффективными. Данный способ не может быть адаптирован к крупномасштабному производству.
4. Противоположный диастереомер L-изоглутамил-L-триптофана (также известный как H-L-iGlu-L-Trp-OH или Бестим [Bestim]) представляет собой иммуностимулятор (см. патент США 5774452). Бестим применяют в лечении язв. Он уменьшает воспалительные явления в желудке и слизистой оболочке двенадцатиперстной кишки и вызывает регресс клинических симптомов и рубцевание язвы (Tkacheva, А., et al. (2004), Eksp Klin Gastroenterol. (6):29-33, 163).
Синтез мононатриевой соли H-L-iGlu-L-Trp-OH (1:1) показан на Схеме 2 (патент США 5744452).
Схема 2: Синтез [L-iGlu-L-Trp-O-]Na+ в соответствии с патентом США 5744452.

В способе из патента США 5744452 на стадии 1 как побочный продукт образуется дициклогексилмочевина, которая должна быть удалена фильтрацией. Заявлено, что на второй стадии трифторуксусная кислота удаляет γ-o-бензиловый эфир с фрагмента глутаминовой кислоты (2.2). Бензиловый эфир (2.3) удаляется гидрированием с помощью аммония формиата в качестве донора водорода, палладиевого катализатора, натрия бикарбоната в изопропаноле при повышенной температуре с образованием мононатриевой соли H-L-iGlu-L-Trp-OH (2.4). Твердофазный синтез (2.4) также описан в указанном патенте, но триптофановый фрагмент должен быть защищен как формамид, и позже защита должна быть снята. Другие диастереомеры, L-изоглутамил-D-триптофан и D-изоглутамил-L-триптофан, также представляют собой известные соединения (патент США 5916878).
Синтез H-D-iGlu-L-Trp-OH, и H-L-iGlu-D-Trp-OH приведен на Схеме 3 и Схеме 4 соответственно (патент США 5916878).
Схема 3: Синтез H-D-изоглутамил-L-триптофана в соответствии с патентом США 5916878.
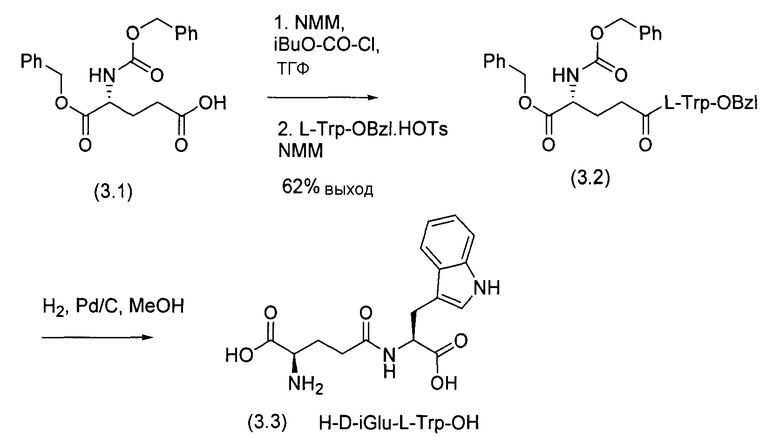
Схема 4: Синтез H-L-изоглутамил-D-триптофана в соответствии с патентом США 5916878.
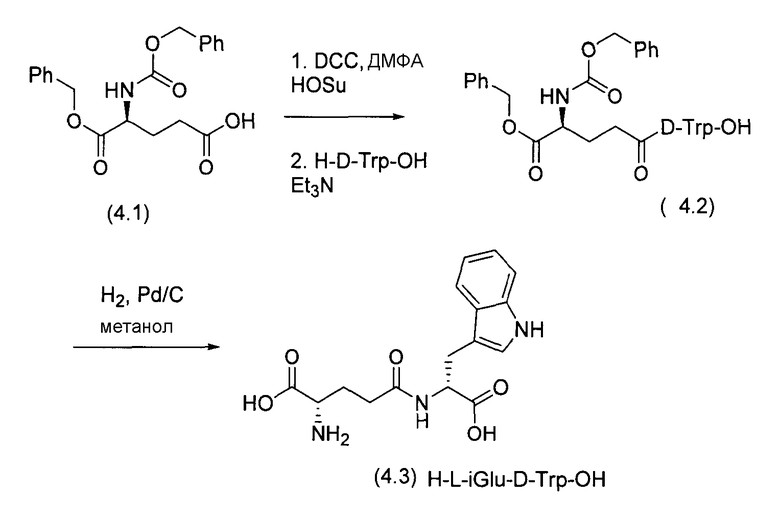
Способы, приведенные на Схемах 2, 3 и 4, могут обеспечить синтез региоспецифичного гамма-амидного продукта (2.2), (3.2) и (4.2) без образования альфа-амидного продукта, но они включают стадию гидрирования в ходе удаления бензилового эфира в соединениях (2.3), (3.2) и (4.2). Это требует использования большого количества палладиевого катализатора. Второй аспект касается частичного восстановления индольного кольца в производственном масштабе. Третий аспект касается образования глутаримида, 2-(3-амино-2,6-диоксо-пиперидин-1-ил)-3-(1H-индол-3-ил)пропионовой кислоты, в процессе гидрирования. Четвертый аспект касается стоимости. Стоимость производного CBz-Glu-OBzl, такого как (3.1) и (4.1), почти вдвое превышает цену соответствующего Boc-Glu-OBzl при производстве с помощью технологий тонкого химического синтеза. Способы на схемах 3 и 4 требуют очистки конечного продукта методом ВЭЖХ. Значение выхода составляет 33% и 35,9% соответственно. Схема 2 требует использования трифторуксусной кислоты, которая вводит другие примеси в реакцию. Кроме того, в способе на Схеме 2 используют дициклогексилкарбодиимид как пептидный конденсирующий агент. Удаление следовых количеств примесей из этого реактива представляет собой серьезную проблему в химическом производстве. Технология, таким образом, не может быть адаптирована к промышленному производству и также не может быть адаптирована для крупномасштабного производства H-D-изоглутамил-D-триптофана.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение касается новой стабильной кристаллической формы D-изоглутамил-D-триптофана и способа выделения указанного соединения в чистой форме, свободной от неорганических солей, осаждением из воды, без препаративной обращенно-фазовой высокоэффективной жидкостной хроматографии. Способ предназначен для получения чистого N-трет-бутоксикарбонил-D-изоглутамил-D-триптофана и его диэфира, свободного от N-трет-бутоксикарбонил-D-глутамил-D-триптофана, и превращения N-трет-бутоксикарбонил-D-изоглутамил-D-триптофана и его диэфира в чистый кристаллический D-изоглутамил-D-триптофан. Новый кристаллический D-изоглутамил-D-триптофан по данному изобретению легко очищать. В сравнении со способами из уровня техники, описанными выше, данное изобретение предлагает целый ряд преимуществ, как указано ниже.
Во-первых, D-изоглутамил-D-триптофан получают в кристаллической форме без применения препаративной высокоэффективной жидкостной хроматографии.
Во-вторых, ключевое промежуточное соединение Boc-D-iGlu-D-Trp-OH или соль H-D-Glu-(γ-D-Trp-OMe)-α-OBzl HCl получают с высоким выходом и высокой чистотой.
В-третьих, предложен способ превращения Boc-D-iGlu-D-Trp-OH и его диэфира или H-D-Glu-(γ-D-Trp-OMe)-α-OBzl HCl соли в D-изоглутамил-D-триптофан с высоким выходом и высокой чистотой.
В-четвертых, чистая кристаллическая форма D-изоглутамил-D-триптофана по данному изобретению неизвестна из предыдущего уровня техники. Она может применяться непосредственно в форме жидкого препарата с коррекцией pH, что устраняет потребность в использовании чрезвычайно гигроскопической и нестабильной динатриевой соли D-изоглутамил-D-триптофана.
Данное изобретение также касается новой стабильной аммонийной соли D-изоглутамил-D-триптофана и способа ее получения из N-трет-бутоксикарбонил-D-изоглутамил-D-триптофана, а также выделения такого соединения в чистой форме кристаллизацией и/или традиционной хроматографией на колонке с силикагелем.
Моноаммонийная соль D-изоглутамил-D-триптофана представляет собой стабильное твердое вещество, пригодное для дозирования с целью получения препаративных форм. Предлагается график идентификации состава для идентификации форм соли при разных значениях pH.
Новый способ получения D-изоглутамил-D-триптофана и аммонийной соли D-изоглутамил-D-триптофана (1:1) позволяет избежать вышеописанных производственных проблем и делает возможным выделение и обработку тимодепрессина и моноаммонийной соли тимодепрессина в традиционном оборудовании для химических процессов.
Объект данного изобретения - предложить пригодный способ производства D-изоглутамил-D-триптофана, который давал бы субстанцию лекарственного средства, полностью свободную от других диастереомеров, как обсуждалось выше, и способность материала сохранять стабильную форму в течение длительного периода хранения до введения в препарат.
Другой объект данного изобретения - предложить D-изоглутамил-D-триптофан, свободный от альфа-амидного изомера D-глутамил-D-триптофана.
Другой объект данного изобретения - предложить способ получения чистого D-изоглутамил-D-триптофана (H-D-iGlu-D-Trp-OH) из кислотно-аддитивной соли H-D-iGlu-D-Trp-OH, что дает продукт, по существу или полностью свободный от остатков органических растворителей и не требующий очистки обращенно-фазовой высокоэффективной жидкостной хроматографией. Твердый D-изоглутамил-D-триптофан выделяют из воды.
Другой объект данного изобретения - предложить способ получения чистого D-изоглутамил-D-триптофана (H-D-iGlu-D-Trp-OH) из основно-аддитивной соли H-D-iGlu-D-Trp-OH, который дает продукт, по существу или полностью свободный от остатков органических растворителей и не требующий очистки обращенно-фазовой высокоэффективной жидкостной хроматографией. Твердый D-изоглутамил-D-триптофан выделяют из воды.
Другой объект данного изобретения - предложить способ, в результате которого образуется D-изоглутамил-D-триптофан, по существу свободный от загрязняющих веществ в форме неорганических солей.
Другой объект данного изобретения - получение кристаллического D-изоглутамил-D-триптофана с порошковой рентгенограммой (XRPD), как показано на фиг. 1.
Другой объект данного изобретения - получение моноаммонийной соли D-изоглутамил-D-триптофана из кислотно-аддитивной соли D-изоглутамил-D-триптофана, что дает продукт, по существу или полностью свободный от остатков органических растворителей и не требующий очистки обращенно-фазовой высокоэффективной жидкостной хроматографией. Твердую аммонийную соль D-изоглутамил-D-триптофана выделяют из изопропанола и аммиака после обработки ионообменной смолой для удаления неорганических солей.
Другой объект изобретения - продуцирование моноаммонийной соли D-изоглутамил-D-триптофана с XRPD, представленной на фиг. 2.
Другой объект данного изобретения - получение аморфной моноаммонийной соли D-изоглутамил-D-триптофана, которая по существу характеризуется ИК-спектром с Фурье-преобразованием (ИК-ФП), показанным на фиг. 5.
Другой объект данного изобретения - предложить способ получения моноаммонийной соли D-изоглутамил-D-триптофана, по существу свободной от загрязняющих веществ в форме неорганических солей.
Другой объект данного изобретения - предложить способ получения кислотно-аддитивной соли, H-D-iGlu-D-Trp-OH, в частности гидрохлорида, из чистого Boc-D-iGlu-D-Trp-OH.
Другой объект данного изобретения - предложить способ получения чистого дипептида Boc-D-iGlu-D-Trp-OH без хроматографического выделения.
Другой объект данного изобретения - предложить простой способ хроматографического выделения на колонке с силикагелем для очистки D-изоглутамил-D-триптофана и его моноаммонийной соли.
Другой объект данного изобретения - предложить график идентификации состава для определения интервала pH для выделения D-изоглутамил-D-триптофана и его одновалентной моносоли. Доминирующий интервал pH для осаждения D-изоглутамил-D-триптофана из воды составляет от приблизительно 2,5 до приблизительно 3,0.
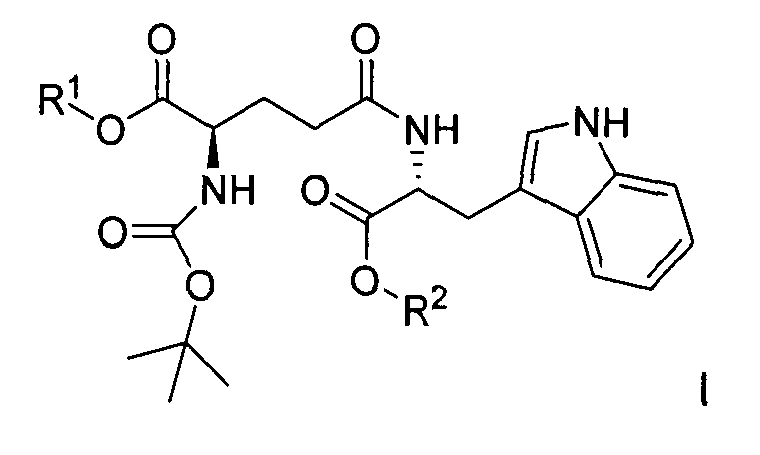
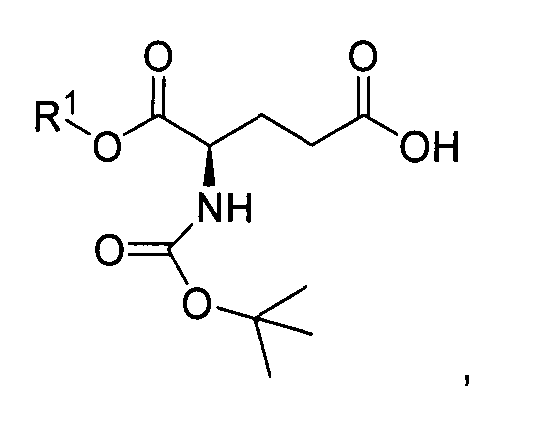
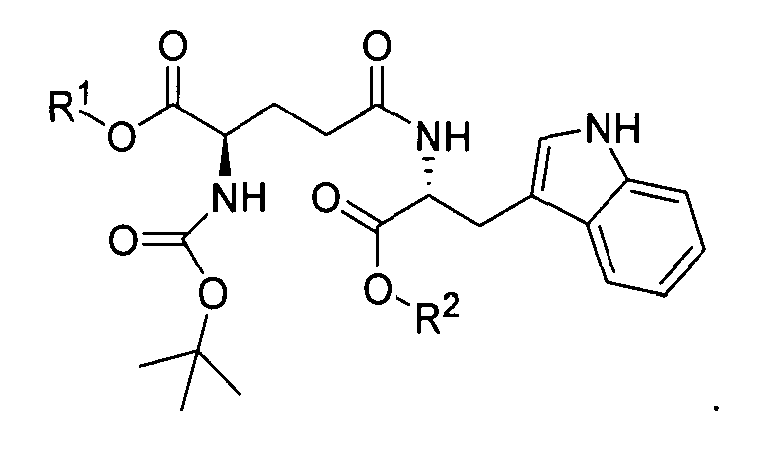
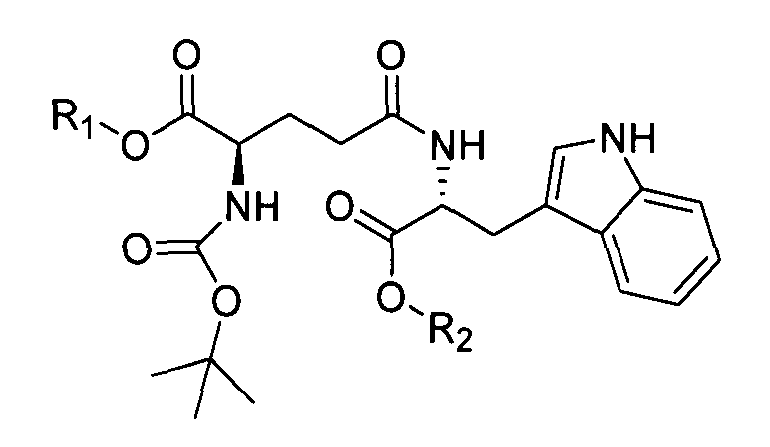
Кислотно-аддитивную соль D-изоглутамил-D-триптофана получают из дипептида Boc-D-iGlu-D-Trp-OH, образующегося в результате оснόвного гидролиза соединения формулы I:
 ,
,
где R1 выбран из группы, состоящей из C1-C4 алкила и бензила, и R2 представляет собой C1-C4 алкил, при условии что C4 алкил не является трет-бутилом,
с помощью гидроксида металла в воде и инертном растворителе в присутствии метанола с образованием Boc-D-iGlu-D-Trp-OH, свободного от других диастереомеров. Гидроксид металла выбирают из группы, состоящей из гидроксида лития, гидроксида натрия и гидроксида калия.
Соединение формулы I, в свою очередь, получают в результате пептидной конденсации Boc-D-Glu(OH)-OR1 и D-Trp-OR2, где R1 и R2 являются такими, как определено выше, с пептидными конденсирующими реагентами, такими как HOBt и ЭДК. Такой способ синтеза Boc-D-iGlu-D-Trp-OH обеспечивает значительные преимущества по сравнению с предыдущим уровнем техники, представленным в патенте США 5736519, поскольку продукт представляет собой исключительно гамма-пептидный продукт Boc-D-iGlu-D-Trp-OH, и альфа-пептидный продукт Boc-D-Glu-D-Trp-OH не может образоваться в ходе синтеза, поскольку используется Boc-D-Glu(OH)-OR1.
Снятие защиты с чистого дипептида Boc-D-iGlu-D-Trp-OH с помощью кислоты, такой как хлороводородная кислота, трифторуксусная кислота, дает кислотно-аддитивную соль. pH раствора кислотно-аддитивной соли составляет от приблизительно 2,5 до приблизительно 3,0 с получением тимодепрессина в виде твердого осадка.
Альтернативно кислотно-аддитивная соль может быть преобразована в аммонийную соль путем обработки водного раствора материала ионообменной хроматографией со смолой на основе сульфоновой кислоты. При удалении соли элюированием с использованием воды ионообменную смолу промывают смесью аммиака и изопропанола для получения сырой аммонийной соли, которую перекристаллизуют из изопропанола и воды с получением чистой моноаммонийной соли.
Раствор основно-аддитивной соли D-изоглутамил-D-триптофана получают путем снятия защиты с помощью кислоты, в частности снятия защиты с помощью HCl, из соединения формулы I, где каждый из R1 и R2 независимо выбран из группы, состоящей из C1-C4 алкила и бензила, с получением кислотно-аддитивной соли диэфира H-D-Glu-(γ-D-Trp-OR2)-α-OR1, которую далее обрабатывают гидроксидом металла в воде и инертном растворителе в присутствии метанола с образованием основно-аддитивной соли H-D-iGlu-D-Trp-OH. Гидроксид металла выбирают из группы, состоящей из гидроксида натрия, гидроксида лития и гидроксида калия. Экстракция растворителем, не смешивающимся с водой, удаляет органическую примесь в органическую фазу; водную фракцию отделяют и доводят pH до значений от приблизительно 6 до приблизительно 7 с помощью гидроксида металла. После выпаривания растворителя для уменьшения количества растворителя до соотношения растворенного вещества и растворителя менее чем приблизительно 1:8, где растворенное вещество представляет собой пептид D-изоглутамил-D-триптофан в форме основно-аддитивной соли, pH полученного раствора основно-аддитивной соли составляет от приблизительно 2,5 до приблизительно 3,0 с помощью неорганической кислоты, чтобы вызвать осаждение D-изоглутамил-D-триптофана.
Хотя не предусматривается ограничения каким-либо способом контекста и практики данного изобретения теорией или способом практики изобретения и его возможными объяснениями, считается, что при pH от приблизительно 2,5 до приблизительно 3,0 график идентификации состава на фиг. 7 и фиг. 8 показывает, что основными формами тимодепрессина являются свободный пептид (H-D-iGlu-D-Trp-OH) и одновалентная соль. Поскольку растворимость H-D-iGlu-D-Trp-OH в воде, не содержащей органических растворителей, невысока (растворимость в воде <23 мг/мл), соединение осаждается из раствора в чистой форме. XRPD материала показана на фиг. 1.
При производстве моноаммонийной соли раствор тимодепрессина при pH от приблизительно 6,0 до приблизительно 8,0 очищают ионным обменом для удаления соли. Регенерирующий раствор на основе аммиака дает чистую моноаммонийную соль после кристаллизации из изопропанола и воды. Хотя можно допускать, что моноаммонийная соль является неустойчивой и может снова превращаться в свободный дипептид, на практике соединение фактически сохраняет стабильность в течение более двух лет. Заявителем изобретена моноаммонийная соль тимодепрессина, которая представляет собой стабильный новый химический объект, который может легко кристаллизоваться из изопропанола и воды. Свойства полученного кристаллического материала показаны на фиг. 2.
Упомянутые выше способы дают чистый тимодепрессин и моноаммонийную соль без обращенно-фазовой ВЭЖХ в промышленных масштабах, но тимодепрессин и моноаммонийная соль могут быть очищены традиционной хроматографией на колонке с силикагелем с использованием указанных выше условий. Таким образом, вместо отбрасывания порции тимодепрессина в маточном растворе кристаллизации по желанию фильтрат может быть упарен и дополнительно очищен хроматографией на колонке с силикагелем.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Кристаллические соли по данному изобретению описаны в Примерах ниже.
Фиг. 1 представляет график XRPD кристаллического D-изоглутамил-D-триптофана. Параметры XRPD также могут быть представлены в значениях межплоскостных расстояний d, угла Брэгга 2θ и относительной интенсивности (выраженной как процентное содержание по отношению к наиболее интенсивной линии), как показано ниже.
Образцы порошка получены обычной техникой фронтальной упаковки и проанализированы на системе дифрактометра D8 Discovery с источником Cu-kα, действующим в режиме 45 кВ/45 мА. Система оборудована 2D-пропорциональным детектором площади (GADDS). Экспериментальные данные были получены на двух фреймах с контактом 600 сек для каждого, что охватывало интервал 3o-35o (2θ). Полученные 2D дифракционные изображения далее были интегрированы для получения стандарта, I против 2θ, характера дифракции. Данные обрабатывали с помощью разнообразного программного обеспечения для обработки данных Bruker AXS, в том числе: Eva™ 8.0 и Topas™, версия 2.1 (для анализа аппроксимации профиля и при необходимости масштабирования).
Фиг. 2 представляет собой XRPD кристаллической моноаммонийной соли D-изоглутамил-D-триптофана. Параметры XRPD могут также быть представлены в терминах межплоскостных расстояний d, угла Брэгга 2θ и относительной интенсивности (выраженной как процентное содержание по отношению к наиболее интенсивной линии), как указано ниже:
Спектры порошковой рентгенограммы для D-изоглутамил-D-триптофана и его аммонийной соли показаны на фиг. 1 и фиг. 2 ниже. Следует понимать, что значения 2 тета для графика порошковой рентгенограммы могут несколько варьировать от одного прибора к другому или от одного образца к другому, то есть приведенные значения не должны рассматриваться как абсолютные.
Фиг. 3 представляет XRPD аморфной формы D-изоглутамил-D-триптофана.
Фиг. 4 представляет характерный спектр поглощения в инфракрасной (ИК) области кристаллической моноаммонийной соли D-изоглутамил-D-триптофана.
Фиг. 5 представляет характерный спектр поглощения в инфракрасной (ИК) области аморфной моноаммонийной соли D-изоглутамил-D-триптофана.
Фиг. 6 представляет характерный спектр поглощения в инфракрасной (ИК) области кристаллического D-изоглутамил-D-триптофана.
Фиг. 7 иллюстрирует вычисление идентификации состава дипептида H-D-iGlu-D-Trp-OH и его соли с использованием значений pKa для кислотных и аминных групп. LH2 представляет собой форму двухосновной кислоты пептида H-D-iGlu-D-Trp-OH, LH представляет собой соль монокарбоновой кислоты H-D-iGlu-D-Trp-OH. Примером LH является моноаммонийная соль. L обозначает форму двухосновной соли, и ее примером является динатриевая соль пептида H-D-iGlu-D-Trp-OH.
Фиг. 8 иллюстрирует вычисление идентификации состава дипептида H-D-iGlu-D-Trp-OH и его соли с использованием экспериментально определенных значений pKa для кислотных и аминных групп. LH2 представляет собой форму двухосновной кислоты пептида H-D-iGlu-D-Trp-OH, LH представляет собой соль монокарбоновой кислоты H-D-iGlu-D-Trp-OH. Примером LH является моноаммонийная соль. L обозначает форму двухосновной соли, и ее примером является динатриевая соль пептида H-D-iGlu-D-Trp-OH.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В данном описании термин “Boc-D-Glu(OH)-OR1” обозначает структуру:
если R1 представляет собой бензил, то с химической точки зрения соединение представляет собой альфа-бензиловый эфир 2-трет-бутоксикарбониламино-D-глутаминовой кислоты.
В данном описании термин “D-Trp-OR2” обозначает структуру:
если R2 представляет собой метил, соединение представляет собой метиловый эфир D-триптофана.
В данном описании термин “Boc-D-Glu-(γ-D-Trp-OR2)-α-OR1” обозначает структуру:
В данном описании термин “H-D-Glu-(γ-D-Trp-OR2)-α-OR1” обозначает структуру:
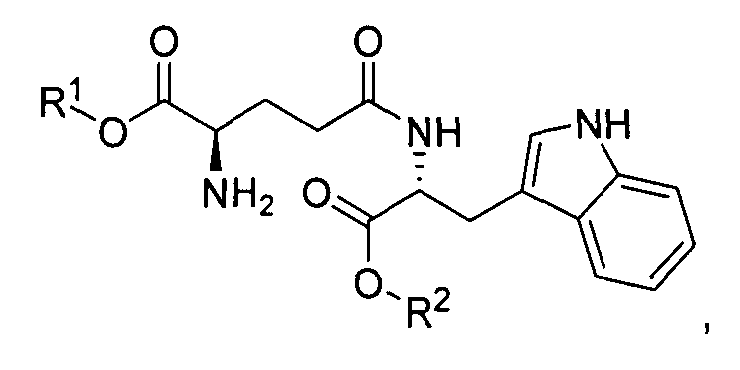
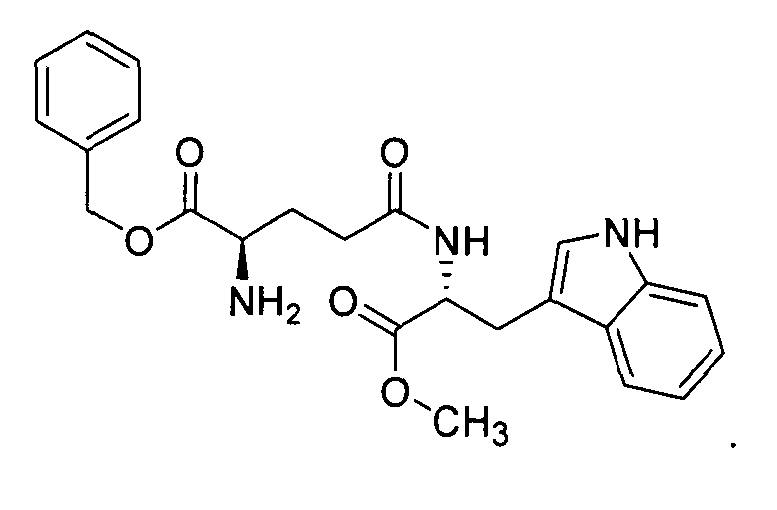
если R1 представляет собой бензил, R2 представляет собой метил, то соединение представляет собой
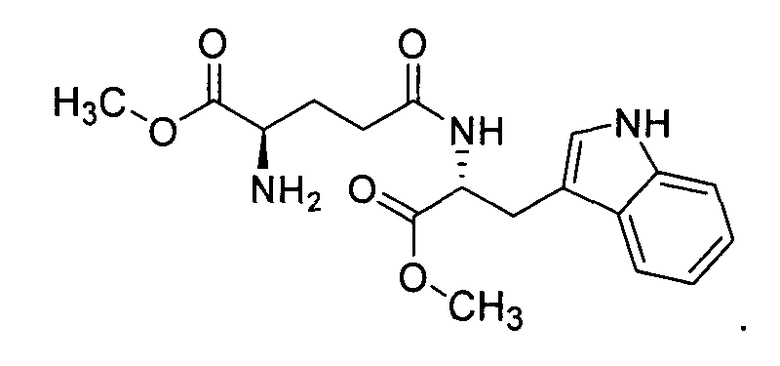
Если R1 представляет собой метил, R2 представляет собой метил, соединение представляет собой
В данном описании термин “тимодепрессин” обозначает дипептид H-D-iGlu-D-Trp-OH с химической структурой:
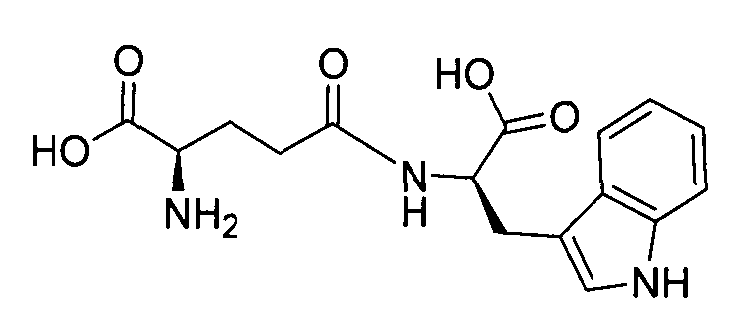 .
.
Он также может быть обозначен как H-D-Glu-(γ-D-Trp-OH)-OH.
Кислотно-аддитивная соль представляет собой соль, образованную в результате реакции амина H-D-iGlu-D-Trp-OH с неорганическими кислотами, в том числе хлороводородной кислотой, серной кислотой, бромоводородной кислотой, фосфорной кислотой и т.п., или органическими кислотами, в том числе муравьиной кислотой, уксусной кислотой, пропионовой кислотой, гликолевой кислотой, молочной кислотой, пировиноградной кислотой, щавелевой кислотой, янтарной кислотой, яблочной кислотой, винной кислотой, лимонной кислотой, трифторуксусной кислотой, бензойной кислотой, салициловой кислотой, бензолсульфоновой кислотой и толуолсульфоновой кислотой. Она может также быть образована в результате снятия защиты с производного Boc-D-iGlu-D-Trp-OH с помощью кислоты.
Основно-аддитивная соль представляет собой соль, образованную в результате реакции карбоновой кислоты H-D-iGlu-D-Trp-OH с неорганическими основаниями, в том числе натрия гидроксидом, лития гидроксидом, калия гидроксидом и т.п.
Данное изобретение направлено на способ получения H-D-iGlu-D-Trp-OH и его аммонийной соли, свободных от неорганических солей, из кислотно-аддитивных солей H-D-iGlu-D-Trp-OH, которые предпочтительно получены из дипептида Boc-D-iGlu-D-Trp-OH. Boc-D-iGlu-D-Trp-OH получают из Boc-D-Glu(OH)-OR1 и D-Trp-OR2, где R1 выбран из группы, состоящей из бензила и C1-C4 алкила, и R2 представляет собой C1-C4 алкил, при условии что C4 алкил не представляет собой трет-бутил.
Данное изобретение также направлено на способ получения H-D-iGlu-D-Trp-OH из раствора основно-аддитивной соли H-D-iGlu-D-Trp-OH, который предпочтительно получают из кислотно-аддитивной соли дипептида H-D-Glu-(γ-D-Trp-OR2)-α-OR1, где каждый из R1 и R2 независимо выбран из группы, состоящей из бензила и C1-C4 алкила.
ПРЕДПОЧТИТЕЛЬНЫЕ ВАРИАНТЫ
В пределах нескольких аспектов данного изобретения, которое сформулировано в кратком описании изобретения, последовательность стадий способа и их относительная предпочтительность описаны ниже.
В варианте данного изобретения предлагается способ получения в водной фазе свободного от неорганических солей H-D-iGlu-D-Trp-OH, который включает:
(a) получение раствора кислотно-аддитивной соли H-D-iGlu-D-Trp-OH в водной среде, в основном не содержащей органического растворителя; или получение раствора основно-аддитивной соли H-D-iGlu-D-Trp-OH в водной среде, в основном не содержащей органического растворителя;
(b) коррекцию pH до pH, соответствующего преобладающей форме, для двухосновной формы с помощью раствора гидроксида щелочного металла или неорганической кислоты, чтобы вызвать осаждение H-D-iGlu-D-Trp-OH;
(c) выделение осажденного H-D-iGlu-D-Trp-OH; и
(d) вакуумную сушку продукта, полученного на стадии (c), с образованием H-D-iGlu-D-Trp-OH.
В другом варианте данного изобретения предлагается кристаллическая форма H-D-iGlu-D-Trp-OH, представляющая собой D-изоглутамил-D-триптофан, который характеризуется XRPD, представленной в описании фигур.
В другом варианте данного изобретения предлагается кристаллический H-D-iGlu-D-Trp-OH, представляющий собой D-изоглутамил-D-триптофан, который характеризуется XRPD, проиллюстрированной на фиг. 1.
В другом варианте данного изобретения предлагается способ получения моноаммонийной соли H-D-iGlu-D-Trp-OH, свободной от неорганических солей, где способ включает следующие стадии:
(a) получение раствора кислотно-аддитивной соли H-D-iGlu-D-Trp-OH в водной среде, в основном не содержащей органического растворителя;
(b) коррекция pH до pH, соответствующего преобладающей форме, для формы одновалентной соли с помощью раствора гидроксида металла;
(c) обработка раствора со стадии (b) ионообменной смолой и элюирование водой для обмена иона металла из соли в растворе на ион водорода до тех пор, пока pH не составит от приблизительно 5,7 до приблизительно 7,0;
(d) контакт ионообменной смолы с регенерирующим раствором на основе аммиака, который осуществляет обмен ионов в растворе на целевой H-D-iGlu-D-Trp-OH, содержащийся в ионообменной смоле, с образованием, таким образом, элюата регенерирующего раствора, содержащего аммонийную соль H-D-iGlu-D-Trp-OH;
(e) выпаривание растворителя из раствора со стадии (d) с получением сырой аммонийной соли;
(f) растворение аммонийной соли со стадии (e) в воде и медленное добавление изопропанола таким образом, что образуется осадок моноаммонийной соли; и
(g) вакуумная сушка продукта со стадии (f) с образованием кристаллической формы аммонийной соли H-D-iGlu-D-Trp-OH (1:1).
Альтернативно вместо стадий (f) и (g) способ включает следующие стадии:
(h) обработка материала со стадии (d) хроматографией на силикагеле с использованием изопропанола и раствора аммиака в качестве элюента; и
(i) лиофильная сушка продукта со стадии (h) с образованием аморфной формы аммонийной соли H-D-iGlu-D-Trp-OH (1:1).
В другом варианте данного изобретения предлагается способ получения моноаммонийной соли H-D-iGlu-D-Trp-OH из кристаллического H-D-iGlu-D-Trp-OH, свободного от неорганических солей, где способ включает следующие стадии:
(a) добавление H-D-iGlu-D-Trp-OH к менее чем одному эквиваленту раствора гидроксида аммония;
(b) коррекция pH до 6-7 с помощью гидроксида аммония;
(c) выпаривание растворителя с образованием масла; добавление изопропанола при перемешивании, чтобы вызвать осаждение моноаммонийной соли;
(d) выделение осажденной аммонийной соли H-D-iGlu-D-Trp-OH и
(e) вакуумная сушка продукта, полученного на стадии (c), с получением моноаммонийной соли H-D-iGlu-D-Trp-OH.
В другом варианте данного изобретения предлагается кристаллическая аммонийная соль H-D-iGlu-D-Trp-OH (1:1), которая характеризуется XRPD, представленной в описании фигур.
В другом варианте данного изобретения предлагается кристаллическая аммонийная соль H-D-iGlu-D-Trp-OH (1:1), которая характеризуется XRPD, представленной на фиг. 2.
В другом варианте данного изобретения предлагается аморфная аммонийная соль H-D-iGlu-D-Trp-OH (1:1), которая характеризуется спектром ИК-ФП (ИК), показанным на фиг. 5.
В другом варианте данного изобретения предлагается способ получения кислотно-аддитивной соли D-изоглутамил-D-триптофана, где соль представляет собой H-D-iGlu-D-Trp-OH гидрохлорид и где способ включает:
(i) основный гидролиз соединения формулы I:
 ,
,
где R1 выбран из группы, состоящей из C1-C4 алкила и бензила, и R2 представляет собой C1-C4 алкил, при условии что C4 алкил не является трет-бутилом,
с помощью гидроксида металла в воде и инертном растворителе в присутствии метанола с образованием Boc-D-iGlu-D-Trp-OH, свободного от других диастереомеров;
(ii) снятие с помощью хлороводорода защиты с Boc-D-iGlu-D-Trp-OH со стадии (i) в инертном органическом растворителе; и выпаривание растворителя с образованием гидрохлорида H-D-iGlu-D-Trp-OH.
В другом варианте данного изобретения предлагается способ получения раствора кислотно-аддитивного гидрохлорида H-D-iGlu-D-Trp-OH, где способ включает:
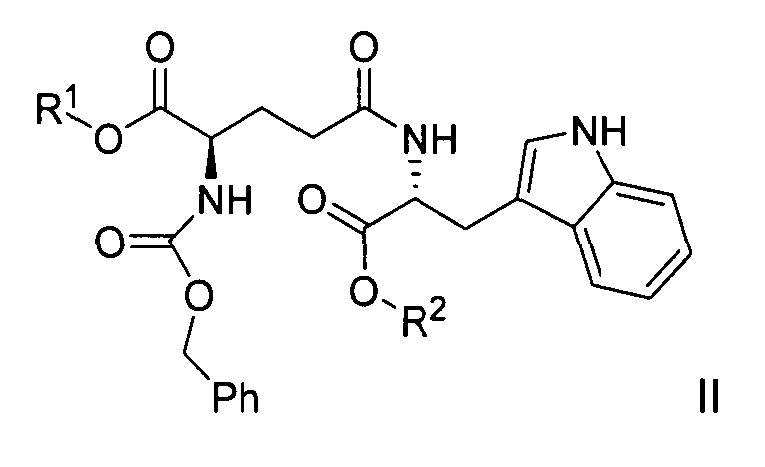
(a) гидрирование соединения формулы II:
 ,
,
где R1 представляет собой бензил, и R2 выбран из группы, состоящей из бензила и водорода,
с использованием палладия на угле в метаноле или этаноле;
(b) очистку сырого H-D-iGlu-D-Trp-OH со стадии (а) хроматографией на силикагеле с использованием изопропанола и воды в качестве элюента и
(c) обработку материала со стадии (b) хлороводородной кислотой в воде с образованием раствора гидрохлорида H-D-iGlu-D-Trp-OH в воде.
В упомянутых выше двух способах получение кислотно-аддитивной соли D-изоглутамил-D-триптофана из соединения формулы I является предпочтительным по сравнению с получением из соединения формулы II с точки зрения стоимости промежуточных химических соединений.
В другом варианте данного изобретения предлагается способ получения раствора основно-аддитивной соли H-D-iGlu-D-Trp-OH, где способ включает:
(a) снятие с помощью кислоты защиты с дипептида N-Boc-D-Glu-(γ-D-Trp-OR2)-α-OR1, где каждый из R1 и R2 независимо выбран из группы, состоящей из C1-C4 алкила и бензила;
(b) основный гидролиз продукта со стадии (a) с помощью гидроксида металла в воде и инертном растворителе в присутствии метанола, где гидроксид металла выбран из группы, состоящей из гидроксида натрия, гидроксида калия и гидроксида лития;
(c) экстракцию материала со стадии (b) растворителем, не смешивающимся с водой, и отделение водной фракции;
(d) коррекцию pH водной фракции со стадии (c) до значений в интервале от приблизительно 6 до приблизительно 7 и
(e) выпаривание растворителя из раствора со стадии (d) для образования раствора, содержащего соотношение приблизительно одной части растворенного вещества на менее чем приблизительно 8 частей воды, где растворенное вещество представляет собой основно-аддитивную соль D-изоглутамил-D-триптофана.
В другом варианте данного изобретения предлагается новое диэфирное производное H-D-Glu-(γ-D-Trp-OR2)-α-OR1 гидрохлорид, где каждый из R1 и R2 независимо выбран из группы, состоящей из бензила и C1-C4 алкила.
Соединения H-D-Glu-(γ-D-Trp-OR2)-α-OR1 неизвестны в предыдущем уровне техники. Указанные соединения могут быть использованы как промежуточные соединения для получения дипептида D-изоглутамил-D-триптофана. Альтернативно H-D-Glu-(γ-D-Trp-OR2)-α-OR1 гидрохлорид может быть использован в фармацевтическом препарате, где гидролиз эфира происходит in situ, с получением D-изоглутамил-D-триптофана при изготовлении препарата.
В варианте данного изобретения предлагается график идентификации состава, проиллюстрированный на фиг. 8, для выделения H-D-iGlu-D-Trp-OH при pH от приблизительно 2,5 до приблизительно 3,0.
СПОСОБ ПОЛУЧЕНИЯ
В данном изобретении предлагается, как изображено ниже на Схеме 5, надежная методика синтеза чистого N-(трет-бутоксикарбонил)-D-изоглутамил-D-триптофана с высоким выходом, причем указанная надежная методика отсутствует в предыдущем уровне техники (например, патент США 5736519).
Схема 5

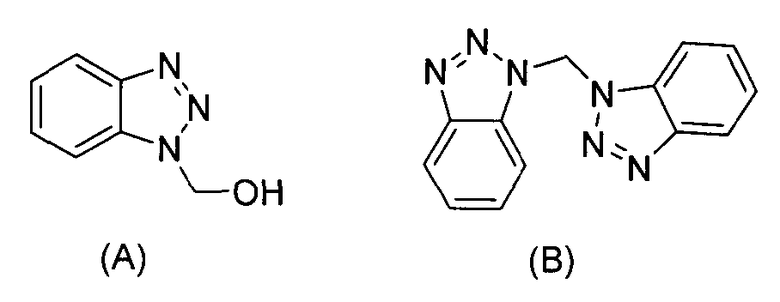
В способе по данному изобретению раствор Boc-D-Glu(OH)-OR1, где R1 представляет собой бензил, в инертном растворителе реагирует с N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлоридом (ЭДК), гидроксибензотриазолом (HOBt) и диизопропилэтиламином (ДИПЭА). Предпочтительная температура составляет от приблизительно 5 до приблизительно -5°C, и предпочтительным растворителем является дихлорметан. После перемешивания в течение от приблизительно 5 до приблизительно 30 минут, предпочтительно приблизительно 15 минут, по каплям добавляют раствор соли D-Trp-OR2 HCl, где R2 представляет собой метил, с диизопропилэтиламином (ДИПЭА). Полученный раствор перемешивают при температуре льда, предпочтительно от приблизительно -5 до приблизительно 5°C в течение приблизительно 1 часа, а затем при комнатной температуре от приблизительно 12 до приблизительно 20 часов, предпочтительно приблизительно 16 часов. Продукт Boc-D-iGlu-(D-Trp-OR2)-α-OR1 выделяют традиционными средствами. Соединение может быть легко кристаллизовано из этилацетата и гексана. Две синтетические примеси присутствуют в следовых количествах - соединения (A) и (B), которые могут быть удалены перекристаллизацией. Считается, что оба соединения образуются из реагента HOBt.
Диэфир Boc-D-Glu-(γ-D-Trp-OR2)-α-OR1 в спирте смешивают с раствором натрия гидроксида. Предпочтительное количество натрия гидроксида составляет от приблизительно 2,5 до приблизительно 5 эквивалентов на эквивалент диэфира. Более предпочтительно используют молярное соотношение от приблизительно 2 до приблизительно 3,5 моль NaOH на моль диэфирного соединения. Предпочтительное соотношение растворителя составляет приблизительно 2 мл спирта на 1 мл воды, и предпочтительное соотношение NaOH и воды составляет приблизительно 1 г на 20 мл. Такая методика выделения включает экстракцию реакционной смеси этилацетатом, и, таким образом, любая органическая примесь удаляется на этой стадии синтеза. После подкисления водную фракцию экстрагируют органическим растворителем, таким как этилацетат. Двухосновный N-(трет-бутоксикарбонил)-D-изоглутамил-D-триптофан (Boc-D-iGlu-D-Trp-OH) выделяют традиционными средствами в виде твердого вещества. Изолированный выход для двух объединенных стадий составляет 89%. Такой показатель превышает показатель методики из уровня техники (патент США 5736519). Новый способ по данному изобретению дополнительно иллюстрируется в примерах ниже.
Подробное исследование и мониторинг стадии гидролиза заявителем показали, что соединение Boc-D-iGlu-(D-Trp-OR2)-OR1, где R1 представляет собой бензил и R2 представляет собой метил, сначала взаимодействует с метанолом с образованием Boc-D-iGlu-(D-Trp-OMe)-OMe, после чего соединение гидролизуется до двухосновной кислоты. Заявителем определено, что метанол необходим для быстрого гидролиза альфа-бензилового эфира. Ввиду большого объема этилацетата, необходимого для экстракции продукта, заявитель изобрел двухфазовую методику для эффективного гидролиза Boc-D-iGlu-(D-Trp-OR2)-OR1. Перемешивают смесь Boc-D-iGlu-(D-Trp-OR2)-OR1 в трет-бутилметиловом эфире (МТБЕ) и растворе гидроксида металла, таком как раствор гидроксида лития или гидроксида натрия. Предпочтительное соотношение гидроксида металла и Boc-D-iGlu-(D-Trp-OR2)-OR1 находится в интервале от приблизительно 2,0-2,5 до 1. Добавляют метанол и смесь энергично перемешивают в течение периода от приблизительно 1 до приблизительно 6 часов, предпочтительно от приблизительно 1,5 до приблизительно 2,5 часов. Из смеси выделяют органическую фазу традиционными средствами. Данная методика устраняет необходимость в использовании большого количества этилацетата для экстракции и проиллюстрирована в примерах ниже.
В традиционном способе снимают защиту с соединения Boc-D-iGlu-D-Trp-OH с помощью органических кислот с образованием дипептида H-D-iGlu-D-Trp-OH, который требует серьезной очистки. Существуют многочисленные недостатки методик из уровня техники (патент США 5736519), в которых используют муравьиную кислоту при температуре 40°C для снятия защиты со смеси Boc-D-iGlu-D-Trp-OH и Boc-D-Glu-D-Trp-OH с образованием смеси H-D-iGlu-D-Trp-OH и H-D-Glu-D-Trp-OH. Ионообменную хроматографию и обращенно-фазовую ВЭЖХ используют для выделения продукта. Выход является низким, и методика не может быть адаптирована к крупномасштабному производству. Снятие защиты с N-трет-бутоксикарбонильной группы с помощью трифторуксусной кислоты или муравьиной кислоты образует ион трет-бутилкарбония, который может реагировать с индольным азотом с образованием N-трет-бутильного продукта (Löw, M., et. al. (1978), Hoppe-Seyler's Z. Physiol. Chem., 359(12):1643-51). Образование глутаримида (1,8) (Pandit, U.K. (1989), Pure & Appl. Chem., Vol. 61, No. 3, pp. 423-426) представляет собой еще одну проблему.
Заявителем было определено, что кислотно-аддитивная соль по данному изобретению, в частности сырой гидрохлорид, может быть легко получена с HCl в инертном растворителе, таком как этилацетат, при низкой температуре, предпочтительно от приблизительно 0°C до температуры окружающей среды. Выпаривание растворителя дает тимодепрессина гидрохлорид, который используют для получения тимодепрессина.
Для прогноза необходимого значения pH для осаждения тимодепрессина в форме двухосновной кислоты H-D-iGlu-D-Trp-OH, заявителем было проведено теоретическое вычисление по графику идентификации состава, и был сделан вывод о том, что тимодепрессин существует в двухосновной форме при значениях pH от приблизительно 2,5 до приблизительно 3,0. Данный вывод позволил изобрести способ выделения тимодепрессина без хроматографии.
Фиг. 7 иллюстрирует такой расчет. На фиг. 7 LH2 представляет собой форму дикарбоновой кислоты пептида H-D-iGlu-D-Trp-OH (то есть тимодепрессина), LH представляет собой форму соли монокарбоновой кислоты пептида H-D-iGlu-D-Trp-OH; один из таких примеров представляет собой моноаммонийную соль, L представляет собой форму соли дикарбоновой кислоты пептида H-D-iGlu-D-Trp-OH; один из таких примеров представляет собой динатриевую соль, и LH3 представляет собой кислотно-аддитивную соль тимодепрессина. Ось X показывает pH раствора. Ось Y показывает % образования по отношению к L (типичная терминология программного обеспечения) и дает молярную фракцию формы, присутствующей при конкретном значении pH. При pH от приблизительно 2,5 до приблизительно 3,0 большая часть (80%) дипептида существует, как форма дикарбоновой кислоты, и может быть осаждена из раствора, если она является нерастворимой в воде. Наше исследование показывает, что форма дикарбоновой кислоты, полученная таким способом, выявляет растворимость в воде приблизительно 23 мг/мл. При pH приблизительно 7,0 100% форм представлены формой монокарбоновой кислоты. Если противоион представляет собой натрий, то форма представляет собой тимодепрессин мононатрий.
На практике, если раствор гидрохлорида тимодепрессина растворен в воде, и значение pH доведено приблизительно до 3,0 при перемешивании, медленно образуется твердое вещество, которое фильтруют из смеси. Чистота материала по данным ЖХ превышает 97% и находится в пределах категории фармацевтической чистоты как активный фармацевтический ингредиент. Авторами определено, что данный способ превосходит предыдущий уровень техники с точки зрения производства и выделения чистого тимодепрессина (H-D-iGlu-D-Trp-OH). Отсутствует необходимость в ионообменной и обращенно-фазовой хроматографии препарата на колонке, поскольку единственным побочным продуктом является натрия хлорид, растворимый в воде. Описанная методика представляет собой прямую методику выделения и очистки тимодепрессина. Она предусматривает теоретический график идентификации состава, и способ превосходит способы из предыдущего уровня техники, которые требуют громоздкой очистки.
Значение pKa кислотных и аминных групп H-D-iGlu-D-Trp-OH определены экспериментально. График состава дипептида, построенный с использованием экспериментально определенных значений pKa, показан на фиг. 8. На фиг. 8 LH2 представляет собой тимодепрессин, LH представляет собой соль монокарбоновой кислоты, L представляет собой соль дикарбоновой кислоты, и LH3 представляет собой кислотно-аддитивную соль тимодепрессина. На оси X приведены значения pH раствора. На оси Y приведен % образования по отношению к L (типичная терминология программного обеспечения), и регистрируется молярная фракция форм, присутствующих при конкретном значении pH. Концентрацию 0,5 M используют для отображения эквивалентности 1 г тимодепрессина в 6 мл воды для целей выделения. Данная фигура показывает, что приблизительно 75% тимодепрессина (LH2) существует в форме дикарбоновой кислоты при pH 2,7. По этой причине тимодепрессин осаждается при pH 2,7 и может быть отфильтрован. Маточный раствор может быть упарен для получения второй порции тимодепрессина. График подтверждает теоретический прогноз - форма двухосновной кислоты H-D-iGlu-D-Trp-OH преобладает при pH от приблизительно 2,5 до приблизительно 3,0; указанный интервал представляет собой значение pH для выделения формы двухосновной кислоты из воды в виде осадка. Поскольку только приблизительно 80% материала будет осаждаться в чистой форме, объем маточного раствора следует уменьшить и подвергнуть второму кругу осаждения при pH от приблизительно 2,5 до приблизительно 3,0, предпочтительно при pH приблизительно 2,7.
Снятие защиты с Boc-H-D-iGlu-D-Trp-OH с помощью трифторуксусной кислоты в инертном растворителе дает соль трифторуксусной кислоты. Инертный растворитель представляет собой дихлорметан, и обычно используют смесь трифторуксусной кислоты и дихлорметана (1:1). Выпаривание растворителя дает масло, которое сушат под вакуумом для удаления остаточного растворителя. Масло диспергируют в воде. При доведении pH до приблизительно 3,0 образуется осадок твердого вещества белого цвета после перемешивания в течение периода от приблизительно 12 до приблизительно 16 часов.
При получении кислотно-аддитивной соли предпочтительно использовать HCl в инертном растворителе для образования гидрохлорида. Альтернативно соль трифторуксусной кислоты может быть образована с применением описанного выше способа. Использование соли хлороводородной кислоты в качестве кислотно-аддитивной соли является предпочтительным, поскольку снятие защиты с Boc-D-iGlu-D-Trp-OH происходит более эффективно при использовании HCl в инертном растворителе, например 3 M HCl в этилацетате. Длительность реакции существенно больше при использовании трифторуксусной кислоты. Кроме того, соль трифторуксусной кислоты D-iGlu-D-Trp-OH содержит несколько синтетических примесей, которые переходят в D-iGlu-D-Trp-OH в случае осаждения при pH от приблизительно 2,5 до приблизительно 3,0 в воде. Примеси необходимо удалять многократной перекристаллизацией.
Исходный материал Boc-D-iGlu-D-Trp-OH получают с использованием описанной раньше методологии. Кислотно-аддитивную соль необходимо сушить под вакуумом, чтобы гарантировать отсутствие органических растворителей и летучих примесей. В случае осаждения тимодепрессина из воды получают раствор кислотно-аддитивной соли в воде. Соотношение кислотно-аддитивной соли и воды находится в интервале от приблизительно 1:5 до приблизительно 1:10. Еще более предпочтительно соотношение кислотно-аддитивной соли находится в интервале от приблизительно 1:6 до приблизительно 1:8. Раствор гидроксида металла, обычно раствор гидроксида натрия, используют для осаждения продукта, но могут быть использованы гидроксид калия и растворы гидроксидов других металлов.
Сырой H-D-iGlu-D-Trp-OH также может быть получен гидрированием соединения формулы II:
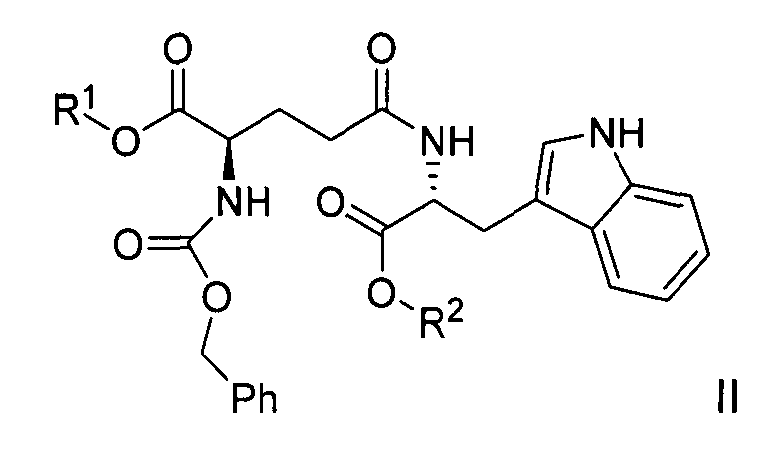 ,
,
где R1 представляет собой бензил, и R2 выбран из группы, состоящей из бензила и водорода, с использованием палладия на угле в метаноле или этаноле. После фильтрации катализатора фильтрат упаривают до состояния масла, которое дополнительно очищают хроматографией на силикагеле с использованием изопропанола и воды в качестве элюента. Полученный H-D-iGlu-D-Trp-OH может быть преобразован в соль H-D-iGlu-D-Trp-OH гидрохлорид в воде с помощью хлороводородной кислоты.
Раствор основно-аддитивной соли D-изоглутамил-D-триптофана получают снятием защиты с помощью кислоты с дипептида Boc-D-Glu-(γ-D-Trp-OR2)-α-OR1, где каждый из R1 и R2 независимо выбран из группы, состоящей из бензила и C1-C4 алкила. Например, снятие защиты с помощью HCl с Boc-D-Glu-(γ-D-Trp-OR2)-α-OR1 в инертном растворителе, таком как дихлорметан, дает соль H-D-Glu-(γ-D-Trp-OR2)-α-OR1 HCl. В случае комбинации, где R1 представляет собой бензил и R2 представляет собой метил, продукт HCl.H-D-Glu-(γ-D-Trp-OR2)-α-OR1 осаждается из дихлорметана и может быть удален фильтрацией. Обработка кислотно-аддитивной соли гидроксидом металла в инертном растворителе, таком как метанол, с целью однофазового гомогенного гидролиза или трет-бутилметиловым эфиром с целью двухфазового гидролиза дает основно-аддитивную соль H-D-iGlu-D-Trp-OH в растворе. При экстракции реакционной смеси растворителем, не смешивающимся с водой, таким как этилацетат или трет-бутилметиловый эфир, водную фракцию нейтрализуют до значений pH от приблизительно 6 до приблизительно 7 и раствор упаривают для уменьшения объема до соотношения менее чем приблизительно 1 часть растворенного вещества на 8 частей воды. Растворенное вещество, как прогнозируется вычислением идентификации состава, как показано на фиг. 8, представляет собой основно-аддитивную соль (в форме монокарбоксилата) H-D-iGlu-D-Trp-OH. Если натрия гидроксид используют как гидроксид металла, растворенное вещество будет представлять собой мононатриевую форму H-D-iGlu-D-Trp-OH в воде. Коррекция pH данного раствора до значений от приблизительно 2,5 до приблизительно 3,0 будет приводить к осаждению твердого тимодепрессина, H-D-iGlu-D-Trp-OH.
Моноаммонийная соль тимодепрессина может быть получена непосредственно из дипептида Boc-D-iGlu-D-Trp-OH. Сырую кислотно-аддитивную соль, такую как гидрохлоридная соль, полученную таким способом, как изложено выше, обрабатывают ионообменной смолой для удаления неорганической соли. Таким образом, раствор сырого тимодепрессина гидрохлорида растворяют в воде и доводят pH до значений от приблизительно 6 до приблизительно 8. Раствор обрабатывают ионообменной смолой. Преимущественно смола представляет собой смолу на базе сульфоновой кислоты. Примером является AMBERLYST® 15. Неорганическую соль удаляют, промывая водой до pH от приблизительно 5,7 до приблизительно 7. Аммиак используют как регенерирующее средство для добывания аммонийной соли тимодепрессина из смолы. Преимущественно используют концентрированный раствор аммиака и изопропанол в качестве регенерируюшего средства. Предпочтительное соотношение представляет собой концентрированный раствор аммиака и изопропанол в соотношении от приблизительно 1 до приблизительно 3-4, с конечным промывным концентрированным раствором аммиака и изопропанола в соотношении 1 часть концентрированного раствора аммиака/1 часть воды/2 части изопропанола. Раствор аммиака для промывания выпаривают при пониженном давлении до состояния масла, которое кристаллизуют с использованием изопропанола и воды с получением моноаммонийной соли в виде твердого вещества белого цвета. Предпочтительное соотношение изопропанола и воды для перекристаллизации находится в интервале от приблизительно 5:1 до приблизительно 10:1. Необходимость в колоночной хроматографии отсутствует.
В предыдущем уровне техники не существует способа очистки тимодепрессина, отличного от обращенно-фазовой жидкостной хроматографии препарата. Этот метод является чрезвычайно длительным и дорогим и не может быть адаптирован к крупномасштабному производству. Заявителем было определено, что сырой тимодепрессин может быть также очищен до фармацевтической категории чистоты флэш-хроматографией на силикагеле с использованием изопропанола и воды в качестве элюента. Предпочтительная подвижная фаза представляет собой смесь изопропанол/вода (от приблизительно 10:1 до приблизительно 5:1). Продукт выделяют традиционными средствами.
Подобным способом моноаммонийная соль также может быть очищена флэш-хроматографией на силикагеле с использованием изопропанола и концентрированного раствора аммиака в качестве элюента. Предпочтительная подвижная фаза представляет собой смесь изопропанол/раствор аммиака (от приблизительно 10:1 до приблизительно 5:1). Продукт выделяют традиционными средствами.
Моноаммонийная соль D-изоглутамил-D-триптофана, полученная в результате кристаллизации с использованием изопропанола и воды, представляет собой кристаллическое вещество. С другой стороны, если раствор моноаммонийной соли D-изоглутамил-D-триптофана сушат лиофильной сушкой, получают аморфный материал.
Обширное исследование проводилось для подтверждения того, что рацемизация хиральных центров в ходе очистки на колонке и последовательности реакций отсутствует; подробности показаны в примерах ниже.
В соответствии с данным изобретением предлагается способ синтеза Boc-D-iGlu-D-Trp-OH, свободного от альфа-изомера амида. Предлагается способ превращения Boc-D-iGlu-D-Trp-OH в кислотно-аддитивную соль тимодепрессина, в частности гидрохлорид. График идентификации предоставляет способ осаждения тимодепрессина в чистой форме при pH приблизительно 3 в воде. Кроме того, предлагается способ очистки тимодепрессина с чистотой менее чем 97% флэш-хроматографией на колонке с использованием изопропанола и воды в качестве элюента. Другой аспект данного изобретения включает традиционный способ получения моноаммонийной соли из тимодепрессина гидрохлорида. Неорганическую соль удаляют ионообменной смолой, и моноаммонийную соль добывают с использованием регенерирующего раствора на основе аммиака. Моноаммонийная соль может быть получена кристаллизацией в чистой форме. Также предлагается способ очистки моноаммонийной соли более низкой чистоты флэш-хроматографией на колонке с силикагелем с использованием изопропанола и воды в качестве элюента.
Дополнительно способ синтеза тимодепрессина, раскрытый в предыдущем уровне техники (патент США 5736519), дает сырой тимодепрессин, который должен быть очищен ионообменной хроматографией и обращенно-фазовой жидкостной хроматографией препарата. Отделение альфа-амидного продукта H-D-Glu-D-Trp-OH от гамма-амидного продукта тимодепрессина D-i-D-Glu-D-Trp-OH остается наиболее более серьезной проблемой производства. Последовательность очистки, приведенная в предыдущем уровне техники, является непригодной для крупномасштабного производства.
Применение и введение
Динатриевую соль тимодепрессина применяли для лечения псориаза. Таким образом, кристаллический тимодепрессин и моноаммонийная соль тимодепрессина по данному изобретению могут быть включены в фармацевтические композиции для введения субъекту в терапевтически активном количестве и в биологически совместимой форме, пригодной для введения in vivo, то есть в форме пептидов для введения, в которой терапевтический эффект превосходит любые токсичные эффекты.
В соответствии с графиком идентификации состава, как показано на фиг. 8, преобладающими формами при нейтральном pH являются монокарбоксилатная форма тимодепрессина, то есть мононатриевая соль дипептида D-изоглутамил-D-триптофана, если противоион представляет собой натрий. Динатриевая соль D-изоглутамил-D-триптофана является чрезвычайно гигроскопичной и очень сложной с точки зрения обработки и получения лекарственных форм. Кристаллический тимодепрессин по данному изобретению имеет XRPD, как подробно показано на фиг. 1, и его растворимость в воде составляет приблизительно 20 мг/мл. Соединение является идеальным кандидатом для замены динатриевой соли при изготовлении разнообразных препаратов. Хотя pH раствора D-изоглутамил-D-триптофана составляет приблизительно 3, может быть осуществлена коррекция значения pH с помощью раствора гидроксида натрия, карбоната натрия или бикарбоната натрия до pH от приблизительно 7 до приблизительно 7,4. Моноаммонийная соль по данному изобретению существует как в кристаллической, так и в аморфной форме. Обе формы моноаммонийной соли обладают чрезвычайно высокой растворимостью в воде. Таким образом, они также являются пригодными кандидатами для рецептур.
Введение нового кристаллического тимодепрессина и/или его моноаммонийной соли, как описано в данном описании, может быть осуществлено каким-либо из общепринятых способов введения терапевтических лекарственных средств с системной активностью. Указанные способы включают лекарственные формы для перорального, парентерального и других видов системного или местного применения или применения в форме аэрозоля.
В зависимости от предусмотренного способа введения композиции для применения могут существовать в форме твердых, полутвердых или жидких лекарственных форм, таких как таблетки, суппозитории, пилюли, капсулы, порошки, жидкости, аэрозоли, суспензии и т.п., предпочтительно в дозированных лекарственных формах, подходящих для одноразового введения точных доз. Композиции будут содержать, по меньшей мере, один традиционный фармацевтический носитель или вспомогательное вещество и кристаллический тимодепрессин или его фармацевтически приемлемую моноаммонийную соль, и дополнительно могут содержать другие лекарственные средства, фармацевтические средства, носители, адъюванты и т.п.
Для твердых композиций могут быть использованы традиционные нетоксичные твердые носители, которые включают, например, маннит, лактозу, крахмал, магния стеарат, натрия сахаринат, тальк, целлюлозу, глюкозу, сахарозу, магния карбонат и т.п., фармацевтической категории. Активное соединение, как определяется выше, может быть введено в суппозитории с использованием, например полиалкиленгликолей, например, пропиленгликоля, как носителя. Подходящие для фармацевтического введения жидкие композиции могут, например, быть получены путем растворения, диспергирования и т.п. активного соединения, как определено выше, и необязательных фармацевтических адъювантов в носителе, таком как вода, солевой раствор, водный раствор декстрозы, глицерин, этанол и т.п., с образованием таким способом раствора или суспензии. По желанию фармацевтическая композиция для введения может также содержать незначительные количества нетоксичных вспомогательных субстанций, таких как увлажняющие или эмульгирующие средства, буферные средства и т.п., например натрия ацетат, сорбитана монолаурат, натрия триэтаноламина ацетат, триэтаноламина олеат и т.п. Фактические способы получения таких лекарственных форм известны или будут очевидными для специалиста в данной отрасли; например, см. Remington: The Science and Practice of Pharmacy, David B. Troy (Ed.), Lipincott Williams & Wilkins, Philadelphia, РА, 21st Edition, 2006. Композиция или рецептура для введения в любом случае будут содержать количество активного(ых) соединения(й) в количестве, эффективном для облегчения симптомов у субъекта, подлежащего лечению.
Парентеральное введение в целом представлено инъекцией, подкожной, внутримышечной или внутривенной. Инъекционные средства могут быть изготовлены в традиционных формах, таких как жидкие растворы или суспензии, твердых формах, пригодных для изготовления раствора или суспензии в жидкости перед инъекцией, или как эмульсии. Пригодными вспомогательными веществами, например, являются вода, солевой раствор, глюкоза, глицерин, этанол и т.п. Кроме того, по желанию фармацевтические композиции для введения могут также содержать незначительные количества нетоксичных вспомогательных веществ, таких как увлажняющие или эмульгирующие средства, буферные средства и т.п., такие как натрия ацетат, сорбитана монолаурат, триэтаноламина олеат и т.п.
Для тимодепрессина или его моноаммонийной соли предпочтительно пероральное или назальное (интрабронхиальное) введение в зависимости от природы расстройства, подлежащего лечению.
Для перорального введения фармацевтически приемлемую нетоксичную композицию получают, объединяя какое-либо из обычно используемых вспомогательных веществ фармацевтической категории, такое как маннит, лактоза, крахмал, магния стеарат, натрия сахаринат, тальк, целлюлоза, глюкоза, сахароза, магния карбонат и т.п. Такие композиции приобретают форму растворов, суспензий, таблеток, пилюль, капсул, порошков, рецептур пролонгированного высвобождения и т.п. Такие композиции могут содержать от приблизительно 1% до приблизительно 95% активного ингредиента, предпочтительно от приблизительно 25% до приблизительно 70%.
Пероральное и назальное введение в легкие может также осуществляться с помощью аэрозольных лекарственных форм. Для аэрозольного введения активный ингредиент предпочтительно поставляется в тонкоизмельченной форме вместе с поверхностно-активным веществом и пропеллентом. Типичное процентное содержание активных ингредиентов составляет от приблизительно 0,01 до приблизительно 20 мас.%, предпочтительно от приблизительно 0,04% до приблизительно 1,0%.
Поверхностно-активные вещества обычно должны быть нетоксичными и предпочтительно растворимыми в пропелленте. Характерным примером таких средств являются эфиры или частичные эфиры жирных кислот, содержащие от 6 до 22 атомов углерода, такие как капроновая, октановая, лауриновая, пальмитиновая, стеариновая, линолевая, линоленовая, олестеариновая и олеиновая кислоты с многоатомным алифатическим спиртом или его циклическим ангидридом, таким как этиленгликоль, глицерин, эритритол, арабитол, маннит, сорбит, ангидрид гекситола, полученный из сорбита (эфиры сорбитана, присутствующие на рынке под названием SPAN®), и полиоксиэтиленовые и полиоксипропиленовые производные указанных эфиров. Могут использоваться смешанные эфиры, такие как смешанные или природные глицериды. Предпочтительными поверхностно-активными агентами являются олеаты или сорбитан, например, присутствующие на рынке под названиями ARLACEL®C (сорбитана сесквиолеат), SPAN®80 (сорбитана моноолеат) и SPAN®85 (сорбитана триолеат). Содержание поверхностно-активного вещества может составлять от приблизительно 0,1% до приблизительно 20 мас.% композиции, предпочтительно от приблизительно 0,25% до приблизительно 5%.
Остальная часть композиции обычно представляет собой пропеллент. Сжиженные пропелленты обычно являются газообразными в условиях окружающей среды и конденсируются под давлением. Среди пригодных сжиженных пропеллентов - низшие алканы, содержащие до пяти атомов углерода, такие как бутан и пропан, и предпочтительно фторированные или фторхлорированные алканы, такие как присутствующие на рынке под названием FREON®. Смеси вышеупомянутых компонентов также могут быть использованы.
В производстве аэрозоля емкость, оборудованную пригодным клапаном, заполняют соответствующим пропеллентом, содержащим тонкоизмельченный активный ингредиент и поверхностно-активное вещество. Таким образом, ингредиенты содержатся при повышенном давлении до выброса при срабатывании клапана.
Для местного применения такие композиции содержат эффективное количество соединения данного класса в смеси, по меньшей мере, с одним фармацевтически приемлемым нетоксичным носителем. Пригодный интервал композиции составляет от приблизительно 0,1% до приблизительно 10% активного ингредиента, и остальные представляют собой носители, предпочтительно от приблизительно 1% до приблизительно 2% активного ингредиента. Концентрация активного ингредиента в фармацевтических композициях, пригодных для местного применения, будет варьировать в зависимости от конкретной активности соединения, которую используют в зависимости от присутствующего состояния и субъекта, который подлежит лечению. Подходящие носители или растворители лекарственного средства для местного применения указанных соединений включают кремы, мази, лосьоны, эмульсии, растворы и т.п.
Например, пригодная мазь для местного применения соединений по данному изобретению содержит от приблизительно 15 до приблизительно 45% насыщенного жирного спирта, содержащего 16-24 атома углерода, такого как цетиловый спирт, стеариловый спирт, бегениловый спирт и т.п., и от приблизительно 45 до приблизительно 85 мас.% гликолевого растворителя, такого как пропиленгликоль, полиэтиленгликоль, дипропиленгликоль и смеси указанных компонентов. Мазь может также содержать от приблизительно 0 до приблизительно 15 мас.% пластификатора, такого как полиэтиленгликоль, 1,2,6-гексантриол, сорбит, глицерин и т.п.; от приблизительно 0 до приблизительно 15 мас.%, связующего средства, такого как насыщенные жирные кислоты, содержащие от 16 до 24 атомов углерода, например стеариновая кислота, пальмитиновая кислота, бегеновая кислота, амиды жирных кислот, например олеамид, пальмитамид, стеарамид, бегенамид, и эфиры жирных кислот, содержащие 16-24 атомов углерода, такие как сорбита моностеарат, полиэтиленгликоля моностеарат, полипропиленгликоль или соответствующий моноэфир других жирных кислот, таких как олеиновая кислота и пальмитиновая кислота; и от приблизительно 0 до приблизительно 20 мас.% средства, усиливающего проникновение, такого как диметилсульфоксид или диметилацетамид.
Терапевтически активное количество кристаллического тимодепрессина или его аммонийной соли может варьировать в соответствии с такими факторами, как патологическое состояние, возраст, пол и масса тела индивидуума. Схема лечения может быть изменена таким образом, чтобы обеспечить оптимальный терапевтический ответ. В целом схема ежедневного введения должна включать дозу в интервале от приблизительно 1 до приблизительно 200 мг пептида.
Далее приведены примеры характерных рецептур, которые никоим образом не ограничивают контекст приготовления разнообразных фармацевтических композиций:
Приведенные выше ингредиенты тщательным образом смешивают и прессуют в таблетки с одной риской.
Приведенные выше ингредиенты смешивают и вводят в твердую желатиновую капсулу.
Приведенные выше ингредиенты тщательно смешивают и прессуют в таблетки с одной риской.
Приведенные выше ингредиенты смешивают и вводят в твердую желатиновую капсулу.
Приведенные выше ингредиенты смешивают и вводят в твердую желатиновую капсулу.
Инъекционный препарат, буферизованный до pH приблизительно 7, получают со следующим составом:
KOH (1 н. раствор)
необходимое количество до pH 7
Инъекционный препарат, буферизованный до pH приблизительно 7, получают со следующим составом:
Суспензию для перорального применения получают со следующим составом:
Препарат для местного применения:
гидроксианизол)
Все приведенные выше ингредиенты, кроме воды, соединяют и нагревают приблизительно до 45°C при перемешивании. Затем добавляют достаточное количество воды с температурой приблизительно 45°C при энергичном перемешивании для эмульгации ингредиентов и далее добавляют необходимое количество воды до 100 г.
Ниже данное изобретение поясняется подробно со ссылкой на Примеры, но данное изобретение не ограничивается ими каким-либо образом.
Пример 1
Получение N-α-трет-бутоксикарбонил-γ-D-глутамил(α-бензиловый эфир)-D-триптофана метилового эфира или (2R)-трет-бутоксикарбониламино-(4R)-[2-(1H-индол-3-ил)-1-метоксикарбонил-этилкарбамоил]масляной кислоты бензилового эфира или N-трет-Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl.
Методика 1A:
Получение чистого справочного образца N-трет-Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl с очисткой хроматографией на силикагеле.
При перемешивании к охлажденному на льду раствору Boc-D-Glu-OBzl (6,00 г, 17,8 ммоль) в CH2Cl2 (70 мл) последовательно добавляют ЭДК (5,11 г, 26,6 ммоль), HOBt (3,60 г, 26,6 ммоль) и ДИПЭА (4,60 мл, 26,6 ммоль). Затем по каплям добавляют раствор H-D-Trp-OMe.HCl (6,77 г, 26,6 ммоль) и ДИПЭА (4,60 мл, 26,6 ммоль) в CH2Cl2 (50 мл). Полученную смесь перемешивают при температуре льда (от -3°C до 0°C) в течение 1 часа, после чего позволяют нагреться до комнатной температуры и перемешивают в течение 16 часов. Реакционную смесь упаривают до сухого состояния. Остаток распределяют между EtOAc и насыщенным раствором NaHCO3. Органическую фракцию собирают, промывают 10% раствором лимонной кислоты и затем насыщенным солевым раствором. Органическую фракцию сушат над натрия сульфатом, фильтруют и упаривают до получения густого масла. Остаток очищают колоночной хроматографией на силикагеле с использованием градиента смеси растворителей гексан/EtOAc (соотношение 8:2, 7:3 и 3:7, об./об.) в качестве элюента с получением указанного в заголовке продукта (9,40 г, 98%) в виде твердого вещества белого цвета. 1H ЯМР (ДМСО-d6) δ м.д. 10,86 (с, 1H), 8,31 (д, J=7,4 Гц, 1H), 7,49 (д, J=7,7 Гц, 1H), 7,31-7,35 (м, 7H), 7,14 (д, J=2,0 Гц, 1H), 7,06 (т, J=7,9 Гц, 1H), 6,98 (т, J=6,8 Гц, 1H), 5,12 (кв, J=5,9 Гц, 2H), 4,5 (кв, J=6,5 Гц, 1H), 3,96-4,03 (м, 1H), 3,55 (с, 3H), 2,98-3,16 (м, 2H), 2,18-2,24 (м, 2H), 1,86-1,95 (м, 1H), 1,71-1,80 (м, 1H), 1,37 (с, 9H); 13C ЯМР (ДМСО-d6) δ м.д.: 172,4 (C), 172,3 (C), 171,4 (C), 155,6 (C), 136,1 (C), 136,0 (C), 128,4 (CH), 127,9 (CH), 127,7 (CH), 127,1 (C), 123,6 (CH), 120,9 (CH), 118,4 (CH), 117,9 (CH), 111,4 (CH), 109,5 (C), 78,2 (C), 65,8 (CH2), 53,3 (CH), 53,16 (CH), 51,7 (CH3), 31,3 (CH2), 28,2 (CH3), 27,1 (CH2), 26,4 (CH2); МС (соотношение массы и заряда) 538 [M+1]+; аналитически вычислено для C29H35N3O7.0,5H2O: C, 63,72; H, 6,64; N, 7,69; найдено: C, 63,79; H, 6,06; N, 7,65.
Методика 1B:
Получение чистого образца N-трет-Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl перекристаллизацией.
Суспензию Boc-D-Glu-OBzl (60,26 г, 178,6 ммоль) в CH2Cl2 (335 мл) охлаждают приблизительно до -1oC и перемешивают в течение 15 мин. Далее последовательно добавляют ДИПЭА (46,70 мл, 268,0 ммоль), HOBt (36,20 г, 268,0 ммоль), ЭДК (51,38 г, 268,0 ммоль). Затем по каплям добавляют раствор H-D-Trp-OMe.HCl (68,25 г, 268,0 ммоль) и ДИПЭА (46,70 мл, 268,0 ммоль) в CH2Cl2 (187 мл). Полученную смесь перемешивают при температуре льда (от -1°C до -5°C) в течение 2 часов, после чего позволяют нагреться до комнатной температуры и перемешивают в течение ночи в атмосфере азота.
Реакционную смесь упаривают до сухого состояния. Остаток распределяют между EtOAc (200 мл), насыщенным раствором Na2CO3 (100 мл) и H2O (150 мл). Водную фракцию снова экстрагируют этилацетатом (200 мл). Органические фракции собирают, промывают H2O (100 мл), 10% раствором лимонной кислоты (2×200 мл) и насыщенным солевым раствором (60 мл). Органическую фракцию сушат над натрия сульфатом, фильтруют и упаривают до сухого состояния. Остаток растворяют в этилацетате (141 мл), потом добавляют гексан (106 мл). Полученную суспензию перемешивают в течение 6 часов и фильтруют. Твердое вещество тщательно промывают гексаном (100 мл), потом сушат под вакуумом в печи при 40°C, выдерживая при этой температуре в течение ночи. Получают твердое вещество практически белого цвета (81,67 г, 85%). Данные 1H ЯМР подтверждают структуру (см. Пример 1, Методика 1A).
Методика 1C:
Получение чистого образца N-трет-Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl без хроматографической очистки и определения синтетических примесей.
Boc-D-Glu-OBzl (48,0 г, 142,2 ммоль) растворяют в 270 мл дихлорметана, после чего охлаждают до 0-5°C с помощью ледяной бани. Добавляют HOBt (23,8 г, 156,4 ммоль) с последующим добавлением ДИПЭА (27,0 мл, 156,4 ммоль) и перемешивают в течение 10 мин. ЭДК (38 г, 199,1 ммоль) и предварительно смешанный раствор H-D-Trp-OMe (приготовленный из H-D-Trp-OMe.HCl [39,7 г, 156,4 ммоль] и ДИПЭА [27,0 мл, 156,4 ммоль] в 150 мл дихлорметана перемешивают при комнатной температуре в течение 20 мин) последовательно добавляют к раствору. Реакционную смесь перемешивают при 0°C, выдерживая при этой температуре в течение 2 часов и потом в течение ночи при комнатной температуре. Реакционную смесь выливают на 250 мл дистиллированной воды и экстрагируют. Органическую фракцию промывают 250 мл 10% раствора лимонной кислоты, 2×5% раствором NaHCO3 и насыщенным солевым раствором. Органическую фракцию сушат над натрия сульфатом и упаривают под вакуумом с получением пенистого твердого вещества бледно-желтого цвета.
Твердое вещество растворяют в приблизительно 250 мл этилацетата и упаривают до сухого состояния. Операцию осуществляют дважды с образованием восковидного твердого вещества. К твердому материалу добавляют 100 мл этилацетата и перемешивают при комнатной температуре. Смесь перемешивают с умеренной или большой скоростью до образования суспензии с консистенцией жидкого теста - этот процесс занимает приблизительно 45 мин (перемешивание в течение длительного периода времени может приводить к затвердению раствора в виде желатинообразной субстанции). Затем добавляют 75 мл гексана и смесь перемешивают еще в течение 10 мин. В этой точке добавляют еще 20 мл этилацетата и суспензию фильтруют немедленно с получением пушистого твердого вещества бледно-розового цвета. Твердое вещество немедленно промывают трижды по 30 мл гексана, что помогает удалить розоватое окрашивание. Фильтрат собирают и позволяют стоять неподвижно в течение 40 мин. Гранулированное твердое вещество осаждается из фильтрата. Смесь фильтруют и твердое вещество промывают трижды по 10 мл гексана.
Фильтрат собирают и упаривают до твердого состояния. Твердое вещество растворяют в 20 мл этилацетата и перемешивают до образования суспензии с консистенцией жидкого теста. Затем добавляют 40 мл гексана и смесь перемешивают в течение 5 мин. Смесь фильтруют и собранное твердое вещество промывают гексаном. Объединенные твердые вещества сушат в течение ночи в печи (35°C) под вакуумом до постоянной массы. Таким образом, получают 59,0 г (77,2%) указанного в заголовке соединения. Т.пл.: 83,1-87,5°C. Данные 1H ЯМР идентичны описанным в Примере 1, Методика 1A. Чистота по данным ВЭЖХ (% площади пика): 97,2%; время удерживания: 7,56 мин; условия ВЭЖХ: колонка Waters Symmetry C 18, 3,9×150 мм, 5 мкм; подвижная фаза: 0,035% HClO4, pH 2/CH3CN, градиент (мин-% CH3CN) 0-35, 10-90, 12-90; скорость потока: 1 мл/мин; λ: 230, 260, 280 нм.
Анализ примесей в маточном растворе.
Результаты анализа маточного раствора методом ТСХ (смесь этилацетат/гексан 50:50) показаны в таблице ниже. Оба пятна А и B флуоресцируют в УФ-свете, но дают негативный результат нингидриновых тестов. Пятна продукта и начальная линия дают положительные результаты нингидриновых тестов. Образцы А и B выделяют колоночной хроматографией на силикагеле, и их структура прояснена по данным 1H ЯМР и МС/МС. Структура А и B показана ниже.
Значение Rf в смеси этилацетат/гексан 50:50:
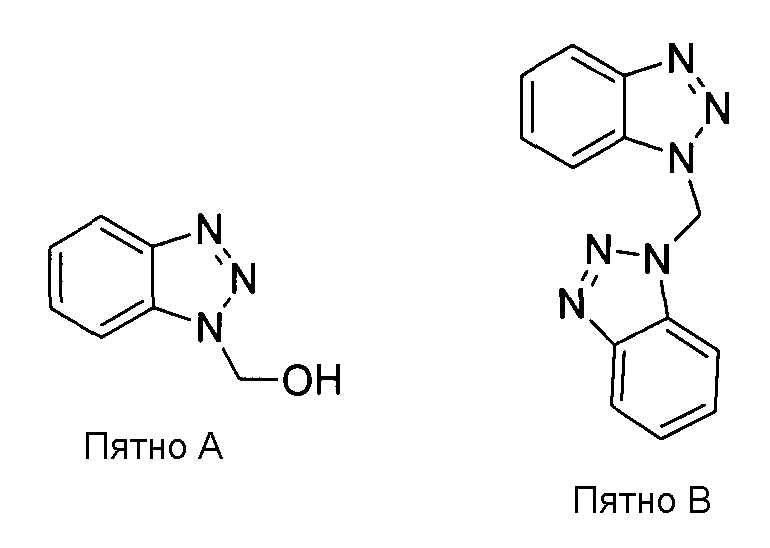
Пятна А и B отвечают примесям, связанным с HOBt. Примеси могут быть удалены перекристаллизацией. Любые следовые примеси могут быть удалены на следующей стадии гидролиза.
Пример 2
Получение N-α-трет-бутоксилкарбонил-D-изоглутамил-D-триптофана или (2R)-трет-бутоксикарбониламино-(4R)-[1-карбокси-2-(1H-индол-3-ил)-этилкарбамоил]-масляной кислоты или N-трет-α-Boc-D-iGlu-D-Trp-OH.
Методика 2A:
Однофазовый гидролиз с помощью NaOH.
При перемешивании к раствору N-трет-Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl (3,7 г, 6,9 ммоль) из Примера 1, Методика 1A, в метаноле (40 мл) добавляют раствор NaOH (1,0 г, 25 ммоль) в H2O (20 мл). Полученный раствор перемешивают при комнатной температуре в течение ночи. Реакционную смесь выливают в 1 н. раствор NaOH (100 мл) и водную смесь промывают этилацетатом (2×100 мл). Водную фракцию подкисляют 3 н. раствором HCl, после чего экстрагируют этилацетатом (2×50 мл). Органические фракции объединяют, сушат над натрия сульфатом и упаривают до сухого состояния при пониженном давлении. Получают твердое вещество белого цвета (2,7 г, 91%). Т.пл.: 148-158°C; 1H ЯМР (ДМСО-d6) δ м.д.: 12,47 (шир, 2H), 10,82 (с, 1H), 8,21 (д, J=7,8 Гц, 1H), 7,53 (д, J=7,8 Гц, 1H), 7,34 (д, J=8,1 Гц, 1H), 7,13 (д, J=2,0 Гц, 1H), 7,06 (т, J=7,5 Гц, 2H), 6,98 (т, J=7,4 Гц, 1H), 4,46 (кв, J=5,3 Гц, 1H), 3,88-3,83 (м, 1H), 3,17-2,97 (дд, J=5,2 и 8,4 Гц, 2H), 2,23-2,10 (м, 2H), 1,90-1,82 (м, 1H), 1,75-1,68 (м, 1H), 1,38 (с, 9H); 13C ЯМР (ДМСО-d6) δ м.д.: 173,9 (C), 173,4 (C), 171,5 (C), 155,6 (C), 136,1 (C), 127,2 (C), 123,5 (CH), 120,9 (CH), 118,4 (CH), 118,2 (CH), 111,4 (CH), 109,9 (C), 78,0 (C), 53,1 (CH), 52,9 (CH), 31,7 (CH2), 28,2 (CH3), 27,2 (CH2), 26,7 (CH2); ИК-ФП (KBr) ν: 3415, 3338, 2986, 1719, 1686, 1654, 1534, 1424, 1366, 1252, 1169, 1069, 744, 634, 429 см-1; МС (соотношение массы и заряда) 434 [M+1]+.
Методика 2B:
Однофазовый гидролиз с помощью LiOH.
При перемешивании к охлажденному на льду (от 0°C до 5°C) раствору Boc-D-Glu-(γ-D-Trp-OCH3)-α-OBzl (46,06 г, 85,68 ммоль) в метаноле (200 мл) добавляют раствор LiOH (10,78 г, 257,0 ммоль) в H2O (136 мл). Полученный раствор перемешивают и содержат при температуре от 0°C до 10°C, выдерживая при этой температуре в течение 3 часов. Реакционную смесь выливают в насыщенный раствор Na2CO3 (100 мл) и H2O (150 мл), водную смесь промывают этилацетатом (2×150 мл). Водную фракцию подкисляют до pH 2-3 с помощью 3 н. раствора HCl, после чего экстрагируют этилацетатом (2×200 мл). Органические фракции объединяют, сушат над натрия сульфатом и упаривают до сухого состояния при пониженном давлении. Получают твердое вещество белого цвета (36,65 г, 98,7%). Данные 1H ЯМР и МС/МС подтверждают структуру (см. Пример 2, Методика 2A).
Методика 2C:
Получение Boc-D-iGlu-D-Trp-OH без хроматографической очистки с использованием двухфазового процесса гидролиза.
Лития гидроксид (4,1 г, 97,7 ммоль) растворяют в 35 мл дистиллированной воды. Затем добавляют 65 мл метил-трет-бутилового эфира (МТБЭ) с последующим добавлением дипептида Boc-D-Glu-(γ-D-Trp-OCH3)-α-OBzl (25 г, 46,5 ммоль), полученного, как описано в Примере 1. Немедленно образуется очень густая суспензия, и 15 мл метанола и 15 МТБЕ добавляют при энергичном перемешивании. Добавляют еще 2 мл метанола и твердое вещество медленно растворяют в течение приблизительно 5 мин. Сразу после растворения материала раствор приобретает желтый/зеленый цвет, тогда как верхняя органическая фракция приобретает бледно-зеленый цвет, а водная фракция - желтый цвет. Реакционную смесь энергично перемешивают при комнатной температуре в течение 80 мин; в этой точке времени в органической фракции не остается начального материала и водная фракция содержит продукт (контроль методом ТСХ: смесь EtOAC/гексан [1:1], об./об.). Раствор выливают в делительную лейку и 2 фракции разделяют. Органическую фракцию промывают 15 мл воды. Органическая фракция приобретает розовый цвет при промывании водой. Объединенную водную фракцию промывают дважды по 30 мл этилацетата. Водную фракцию подкисляют приблизительно до pH 2 добавлением по каплям 16,6 мл 6н хлороводородной кислоты при комнатной температуре. Водную фракцию экстрагируют дважды по 50 мл этилацетата. Максимальное количество метанола добавляют в ходе второй экстракции, чтобы улучшить растворимость продукта в органической фракции. Объединенные органические фракции сушат над натрия сульфатом и упаривают в вакууме из жидкости желтого цвета с получением твердого вещества белого цвета. Твердое вещество сушат в течение ночи в печи (28°C) под вакуумом до постоянной массы.
Таким образом получают 18,6 г (выход 92%) указанного в заголовке соединения. Т.пл.: 179,0-184,6°C; Данные 1H ЯМР были идентичны описанным в Примере 2A; чистота по данным ВЭЖХ (% площади пика): 98,3%; время удерживания: 5,33 мин; условия ВЭЖХ: колонка Waters Symmetry C 18, 3,9×150 мм, 5 мкм; подвижная фаза: 0,035% HClO4, pH 2/CH3CN, градиент (T, мин-% CH3CN) 0-20, 10-90, 12-90; скорость потока: 1 мл/мин; λ:230, 260, 280 нм.
Контроль вышеуказанной реакции может быть осуществлен с помощью ВЭЖХ. В Методике 2C, выше, присутствие бензилового спирта, образующегося в результате гидролиза фрагмента бензилового эфира, можно наблюдать с помощью ТСХ (смесь EtOAC/гексан [1:1, об./об.] в качестве элюента). Бензиловый спирт образуется в результате гидролиза фрагмента бензилового эфира. Примесь, соответствующая первому пятну, является такой же, как в Примере 1, Методика 1C. Контроль с помощью ВЭЖХ и анализ методом ЖХ/МС показывают, что Boc-D-Glu-(γ-D-Trp-OCH3)-α-OBzl сначала реагирует с основой и метанолом с образованием Boc-D-Glu-(γ-D-Trp-OCH3)-α-OCH3, который затем быстро гидролизуется с получением двухосновной кислоты, Boc-D-Glu-(γ-D-Trp-OH)-OH или Boc-D-iGlu-D-Trp-OH.
Пример 3:
Получение D-изоглутамил-D-триптофана.
Методика 3A:
Получение D-изоглутамил-D-триптофана и его очистка перекристаллизацией.
Boc-D-iGlu-D-Trp-OH (20,0 г, 46,14 ммоль, из Примера 2) помещают в 3-горлую круглодонную колбу объемом 1 л, оборудованную магнитной мешалкой. Добавляют этилацетат (300 мл) и полученную суспензию охлаждают до -10°C на ледяной бане. Газообразный HCl пропускают через холодную суспензию. Интервал температуры от -4°C до -10°C поддерживают в ходе реакции и прогресс реакции контролируют с помощью ВЭЖХ. В определенной точке гетерогенная реакционная смесь превращается в прозрачный гомогенный раствор светло-розового цвета. После превращения исходного материала реакционная смесь снова превращается в суспензию. Летучие материалы затем удаляют в вакууме с получением твердого вещества светло-розового цвета. Твердое вещество растворяют в 60 мл деминерализованной воды и полученный раствор промывают дихлорметаном (2×25 мл). Затем pH водного раствора доводят приблизительно до 3,0 добавлением NaOH (10 M, приблизительно 3,6 мл) при охлаждении. Полученный раствор фильтруют для удаления любых остаточных частиц твердого вещества. Фильтрат собирают и энергично перемешивают. Твердое вещество отделяют фильтрацией. Фильтрат оставляют для последующего использования. Затем твердое вещество снова помещают в круглодонную колбу и добавляют 30 мл деминерализованной воды. Смесь энергично перемешивают и твердое вещество отделяют фильтрацией. Фильтрат оставляют для последующего использования. Твердое вещество далее промывают ледяной холодной деминерализованной водой (4×15 мл). Третий промывной водный раствор не содержит хлоридов, что подтверждается отрицательным результатом испытания с использованием AgNO3 (4% раствор). Твердое вещество сушат на воздухе, после чего помещают в вакуумную печь при 36°C, выдерживая при этой температуре в течение ночи с получением 8,5 г (чистота по данным ВЭЖХ, % площади пика: 98,3%).
Фильтраты с описанных выше стадий объединяют и осуществляют такую же процедуру перекристаллизации с получением еще 3,2 г продукта (чистота по данным ВЭЖХ, % площади пика: 98,7%). Объединенный выход для 2 порций составляет 11,7 г (75%).
Дополнительная обработка конечного фильтрата дает третью порцию (1,0 г, чистота по данным ВЭЖХ, % площади пика: 82,0%).
1H ЯМР (D2O-NaOD, pH 7,0) δ м.д.: 7,64 (д, J=7,9 Гц, 1H), 7,43 (д, J=8,1 Гц, 1H), 7,19-7,16 (м, 2H), 7,10 (т, J=7,4 Гц, 1H), 4,52-4,48 (м, 1H), 3,48 (т, J=6,1 Гц, 1H), 3,34-3,29 (м, 1H), 3,08-3,02 (м, 1H), 2,30-2,17 (м, 2H), 1,92-1,75 (м, 2H). Спектр HRPD для полученного материала показан на фиг. 1. Метод ВЭЖХ: колонка XTerra МС C18; 5 мкм, 4,6×250 мм; подвижная фаза: А = водная фракция: 4 мМ Трис, 2 мМ ЭДТА, pH 7,4; B = органическая фракция: CH3CN; программа градиента: B %: 0 мин 5%, 15 мин 55%, 30 мин 55%, 32 мин 5%, 40 мин 5%. Скорость потока = 1 мл/мин; объем инъекции = 5 мкл; λ: 222, 254, 282, 450 нм; время удерживания продукта = 6,4 мин.
Методика 3B:
Этилацетат (250 мл), предварительно охлажденный до 0°C, насыщают газообразным HCl в течение 25 мин. Добавляют Boc-D-iGlu-D-Trp-OH (15,0 г, 34,6 ммоль) с образованием суспензии. Раствор перемешивают в течение 90 мин при температуре ледяной бани. Растворитель выпаривают в вакууме с образованием твердого вещества белого цвета. Твердое вещество растворяют в 35 мл дистиллированной воды с образованием густого раствора светло-коричневого цвета. Водную фракцию промывают 30 мл дихлорметана, после чего переносят в химический стакан объемом 100 мл. С использованием pH-электрода для контроля кислотности pH доводят с 1,28 до 2,96 с использованием 3,2 мл 10 н. раствора NaOH. Раствор перемешивают в течение 1 час при комнатной температуре, медленно образуется осадок белого цвета. Твердое вещество отделяют вакуумной фильтрацией и тщательным образом промывают водой. Сырое твердое вещество суспендируют в 20 мл дистиллированной воды, перемешивая в течение 2 часов при комнатной температуре. Смесь фильтруют, твердое вещество собирают и сушат до постоянной массы в печи в вакууме в течение ночи (40°C). Таким образом, получают 8,6 г (выход 74,5%) указанного в заголовке соединения. Чистота по данным ВЭЖХ (% площади пика): 98,8%. Время удерживания: 4,21 мин; условия ВЭЖХ: колонка Waters Symmetry C 18, 3,9×150 мм, 5 мкм; подвижная фаза: 0,035% HClO4, pH 2/CH3CN, градиент (T, мин-% CH3CN) 0-10, 10-90, 12-90; скорость потока: 1 мл/мин; λ: 230, 260, 280 нм; данные 1H ЯМР отвечают структуре.
Пример 4:
Синтез моноаммонийной соли D-изоглутамил-D-триптофана (1:1) и ее выделения с помощью очистки колоночной хроматографией.
Методика 4A:
Газообразный HCl пропускают при перемешивании через охлажденный на льду (0-5°C) раствор Boc-D-Glu-(-D-Trp-OH (2,5 г, 5,8 ммоль) в этилацетате (60 мл) в течение 2,5 часов. Реакционную смесь затем упаривают до сухого состояния. Очистка остатка колоночной хроматографией на силикагеле с использованием градиента смеси растворителей изопропанола и раствора гидроксида аммония (28-30% NH4OH) (соотношение 8:2 и 7:3, об./об.) в качестве элюента дает названный в заголовке продукт (1,8 г, 84,7%) в виде твердого вещества белого цвета после выпаривания растворителя. Т.пл.: 124-128°C; 1H ЯМР (ДМСО-d6) δ м.д.: 10,98 (с, 1H), 8,25 (д, J=5,9 Гц, 1H), 7,53 (7,8 Гц, 1H), 7,30 (д, J=8,0 Гц, 1H), 7,17 (с, 1H), 7,00 (т, J=7,7 Гц, 1H), 6,92 (т, J=7,2 Гц, 1H), 4,28 (м, 1H), 3,22-3,31 (м, 2H), 2,90-2,96 (м, 1H), 2,23-2,25 (м, 2H), 1,97-1,98 (м, 1H), 1,84-1,86 (м, 1H); 13C ЯМР (ДМСО-d6) δ м.д.: 175,5 (C), 171,6 (C), 171,4 (C), 136,0 (C), 127,6 (C), 123,5 (CH), 120,5 (CH), 118,3 (CH), 117,9 (CH), 111,5 (C), 111,3 (CH), 55,3 (CH), 53,7 (CH), 32,5 (CH2), 27,8 (CH2), 27,4 (CH2); 14N ЯМР (D2O) δ м.д.: 20,4 (с); ИК-ФП (KBr) ν: 3406, 3055, 1581, 1456, 1399, 1341, 1096, 1009, 744, 535, 426 см-1; МС (соотношение массы и заряда) 334 [двухосновная кислота+1]+; аналитически вычислено для C16H22N4O5.H2O: C, 52,17; H, 6,57; N, 15,21; найдено: C, 51,95; H, 6,84; N, 14,85. Вещество представляет собой моноаммонийную соль D-изоглутамил-D-триптофана (1:1). Полученный материал аморфен, что подтверждено данными XRPD.
Методика 4B:
Газообразный HCl конденсируют в холодном этилацетате (133,4 г) при температуре -2oC (температура внешней ледяной бани) в течение 16 мин. Увеличение массы раствора составляет 21 г. Boc-D-iGlu-D-Trp-OH (3,8 г, 8,73 ммоль) растворяют в 50 мл вышеописанного раствора. Температуру полученной смеси поддерживают в интервале 0-5°C в течение 55 мин. Реакционную смесь контролируют методом ТСХ, после чего упаривают при пониженном давлении (температура роторного выпаривателя: 51-52°C) до сухого состояния. Очистка флэш-хроматографией на силикагеле с использованием градиента смеси растворителей изопропанола и раствора гидроксида аммония (28-30%) (соотношение 8:2 и 7:3, об./об.) в качестве элюента дает продукт (2,0 г, 62%) в виде твердого вещества практически белого цвета. Данные 1H ЯМР подобны данным, приведенным в Примере 4, Методика 4A. Полученный материал аморфен, что подтверждено данными XRPD.
Пример 5
Синтез моноаммонийной соли D-изоглутамил-D-триптофана (1:1).
Методика 5A:
Синтез моноаммонийной соли D-изоглутамил-D-триптофана (1:1) путем удаления неорганических солей с использованием смолы AMBERLYST®15 с последующей очисткой колоночной хроматографией.
Газообразный HCl пропускают при перемешивании через охлажденную льдом (0-5°C) суспензию полученного Boc-D-iGlu-D-Trp-OH (10,82 г, 24,96 ммоль) в этилацетате (200 мл) в течение 2 часов. Реакционную смесь далее упаривают до сухого состояния. Остаток растворяют в H2O (30 мл) и нейтрализуют до pH 6-7 с помощью 6 н. раствора NaOH. Полученный раствор загружают на хроматографическую колонку, заполненную смолой AMBERLYST®15, с последующим элюированием водой до pH 5-5,5, далее 100% изопропанолом (pH 7) и в конце смесью 25% NH4OH/ИПА (pH 10).
Фракции, которые содержат продукт, объединяют и упаривают до сухого состояния при пониженном давлении. Дополнительная очистка остатка колоночной хроматографией на силикагеле с использованием градиента смеси растворителя изопропанола и концентрированного раствора гидроксида аммония (соотношение 17:3, 4:1 и 7:5, об./об.) в качестве элюента дает названный в заголовке продукт (6,68 г, 72,7%) в виде пенистого твердого вещества светло-желтого цвета. Данные 1H ЯМР и МС/МС подтверждают структуру (см. Методику 4A). Содержание воды по данным испытания методом Карла Фишера составляет 3,7%.
Методика 5B:
Синтез моноаммонийной соли D-изоглутамил-D-триптофана (1:1) путем удаления неорганических солей с использованием смолы Amberlyst15 с последующей очисткой перекристаллизацией.
При перемешивании через охлажденную на льду (0-5°C) суспензию Boc-D-iGlu-D-Trp-OH (10,75 г, 24,80 ммоль) в этилацетате (200 мл) пропускают газообразный HCl. Реакционную смесь содержат на ледяной бане (0-5°C) в течение 2 часов. Анализ методом ТСХ (30% аммиака в изопропаноле) демонстрирует полное превращение исходного материала. Реакционную смесь выпаривают до сухого состояния в вакууме, остаток растворяют в H2O (30 мл) и нейтрализуют до pH 6-7 с помощью 10 н. NaOH. Полученный гомогенный раствор загружают на хроматографическую колонку, заполненную смолой AMBERLYST®15, с последующим элюированием водой (2450 мл) до pH 4-5,5, изопропанолом (1000 мл) и смесью 25% NH4OH/изопропанол. Фракции, которые содержат продукт, объединяют и упаривают до сухого состояния. Получают бесцветное пенистое твердое вещество, к которому добавляют изопропанол (150 мл) и H2O (30 мл). Полученную суспензию перемешивают при комнатной температуре в течение ночи. Твердое вещество отделяют вакуумной фильтрацией, тщательно промывают изопропанолом (2×60 мл), потом EtOAc (2×60 мл) и в конце сушат в вакууме в печи при 42°C, выдерживая при этой температуре в течение ночи. Получают твердое вещество практически белого цвета (6,60 г, 72,2%). Данные 1H ЯМР и МС/МС отвечают структуре (см. Пример 5). Данные XRPD полученного кристаллического материала показаны на фиг. 2. Содержание воды по данным метода Карла Фишера составляет 5,9%.
Пример 6
Синтез моноаммонийной соли D-изоглутамил-D-триптофана (1:1) из H-D-iGlu-D-Trp-OH.
Методика 6A:
Получение моноаммонийной соли D-изоглутамил-D-триптофана (1:1) с использованием CBz-D-Glu-(γ-D-Trp-OH)-γ-OBzl в качестве промежуточного соединения.
ЭДК (562 мг, 2,93 ммоль) добавляют к раствору Z-D-Glu-OBz (990 мг, 2,67 ммоль) и N-гидроксисукцинимида (337 мг, 2,93 ммоль) в ДМФА (50 мл) на ледяной бане; полученный прозрачный раствор перемешивают в течение ночи при комнатной температуре. H-D-Trp-OH (640 мг, 3,13 ммоль) и Et3N (1 мл) добавляют при комнатной температуре. Через 20 минут материал смешивают с водой и экстрагируют этилацетатом. Объединенные EtOAc фракции промывают 10% раствором лимонной кислоты и далее насыщенным солевым раствором, сушат над натрия сульфатом, фильтруют, упаривают до сухого состояния, сушат в вакууме с получением 1,49 г CBz-D-iGlu-(γ-OBzl)-D-Trp-OH. Полученный материал гидрируют с использованием 33%, мас./мас. 10% Pd/C при атмосферном давлении. Через 4 часа катализатор отфильтровывают с помощью CELITE® и фильтрат выпаривают с получением масла. Сырой продукт очищают флэш-хроматографией на колонке с использованием смеси изопропанол/NH4OH (от 80:20 до 70:30 об./об.) в качестве элюента с получением указанного в заголовке соединения (813 мг). Данные МС/МС и 1H ЯМР являются подобными соединению, полученному с использованием способа, показанного в Примере 4 выше.
Методика 6B:
Получение моноаммонийной соли D-изоглутамил-D-триптофана (1:1) из H-D-iGlu-D-Trp-OH (см. Пример 3).
H-D-iGlu-D-Trp-OH (1 г из Примера 3) смешивают с аммония гидроксидом (0,55 M, 6 мл). Смесь перемешивают и измеряют значение pH, которое равно приблизительно 4,5. Раствор гидроксида аммония (0,55 M) добавляют по каплям до pH раствора 7,0-7,5. Летучие материалы удаляют в вакууме и остаточное масло смешивают с изопропанолом. Образуется белый осадок. Через 2 часа твердое вещество (аммонийную соль) отделяют вакуумной фильтрацией. Твердое вещество сушат до постоянной массы (1 г) под глубоким вакуумом в течение 12 часов с получением аммонийной соли D-изоглутамил-D-триптофана (1:1). Содержание воды по данным метода Карла Фишера составляет 4,6%.
Пример 7
Очистка H-D-iGlu-D-Trp-OH колоночной хроматографией на силикагеле с использованием смеси изопропанола и воды.
А. Получение Cbz-D-Glu-(γ-D-Trp-OBzl)-α-OBzl.
К охлажденному на льду раствору 2,67 г Cbz-D-Glu-OBzl в 50 мл ДМФА добавляют 0,91 г N-гидроксисукцинимида (1,1 экв.) и 1,52 г ЭДК (1,1 экв.) и полученный раствор перемешивают на ледяной бане в течение 1 часа, а затем при комнатной температуре в течение ночи. К полученной реакционной смеси добавляют 2,50 г H-D-Trp-OBzl.HCl (1,05 экв.) и 3 мл Et3N при комнатной температуре. Реакция завершается через 1 час, что подтверждают данные ВЭЖХ.
Реакционную смесь гасят с помощью деминерализованной воды на ледяной бане, после чего несколько раз экстрагируют этилацетатом. Объединенные этилацетатные экстракты промывают 10% раствором лимонной кислоты и далее насыщенным солевым раствором, сушат над натрия сульфатом, фильтруют и упаривают до сухого состояния. Остаток наносят на силикагель в метаноле и смесь упаривают в вакууме. Остаток наносят на верхнюю часть колонки с влажным силикагелем и целевой продукт, Cbz-D-Glu-(γ-D-Trp-OBzl)-α-OBzl, элюируют с использованием градиента смеси растворителей (этилацетат/гексан, от 80:20 до 100:0). Целевые фракции объединяют и упаривают в вакууме с получением 4,64 г (выход 99,6%) указанного в заголовке соединения. Чистота по данным ВЭЖХ (% площади пика): 93%; условия ВЭЖХ: колонка: Symmetry C18, 3,9×150 мм, 5 мкм; подвижная фаза: 0,035% HClO4 (pH 2,5) CH3CN = градиент (мин-CH3CN %: 0-10, 10-100, 12-100, 14-50); скорость потока: 1 мл/мин; λ: 280 нм; время удерживания: 9,7 мин.
B. Очистка D-изоглутамил-D-триптофана колоночной хроматографией с использованием смеси изопропанола и воды.
К суспензии 4,0 г Cbz-D-Glu-(γ-D-Trp-OBzl)-α-Obzl, полученного, как описано в Примере 7A выше, в 150 мл смеси MeOH/H2O (95:5, об./об.) добавляют 1,5 г Pd/C (37,5% мас./мас.). Смесь гидрируют при давлении водорода 30 фунт/дюйм2. Реакция завершается через 75 мин, что подтверждают данные ВЭЖХ. Катализатор отфильтровывают на слое CELITE® и фильтрат упаривают при пониженном давлении и температуре 45°C. Далее остаток очищают флэш-хроматографией на силикагеле с использованием смеси изопропанол/H2O (соотношение 80:20 об./об.). Наиболее чистые фракции (по данным ВЭЖХ) объединяют и упаривают в вакууме. Таким образом, получают названное соединение (1,1 г, 52%) в виде порошка светло-желтого цвета. Чистота по данным ВЭЖХ (% площади пика): 99%. Данные МС/МС и 1H ЯМР отвечают целевой структуре.
Менее чистые фракции (по данным ВЭЖХ) объединяют, упаривают в вакууме с получением еще приблизительно 1,0 г (выход 48%) указанного в заголовке соединения. Чистота по данным ВЭЖХ (% площади пика): 96,7%. Данные МС/МС и 1H ЯМР соответствуют целевой структуре.
Условия ВЭЖХ являются такими же, как описано в разделе 7A выше; время удерживания H-D-iGlu-D-Trp-OH составляет 4,0 мин.
Пример 8
А. Получение H-D-Pyr-D-Trp-OH (5-оксо-D-пролил-D-триптофана) с Z-D-Glu-(γ-OEt)-OH.
Указанное в заголовке соединение представляет собой возможную синтетическую примесь H-D-iGlu-D-Trp-OH. Ее синтезируют независимо и используют как справочный образец при анализе методом ВЭЖХ продуктов, описанных в Примерах 3-7 выше.
К охлажденному на льду раствору Z-D-Glu-(γ-OEt)-OH (1 г, 3,23 ммоль) в ДМФА (60 мл) добавляют N-гидроксисукцинимид (409 мг, 3,56 ммоль), ЭДКИ (682 мг, 3,56 ммоль) и полученный раствор перемешивают на ледяной бане в течение 1 часа, после чего перемешивают при комнатной температуре в течение ночи. К полученной реакционной смеси добавляют H-D-Trp-OH (792 мг, 3,88 ммоль) и Et3N (1 мл) при комнатной температуре. Добавляют воду через 1,5 часа при температуре ледяной бани. Смесь экстрагируют несколько раз этилацетатом. Объединенные этилацетатные экстракты промывают 10% раствором лимонной кислоты, и далее солевым раствором, сушат над натрия сульфатом, фильтруют, упаривают до сухого состояния и сушат под вакуумом с получением 410 мг фракции сырого продукта А. Водную фракцию упаривают в вакууме на бане с температурой 55°C. Остаток растворяют в CH2Cl2, и органическую фракцию промывают 10% раствором лимонной кислоты (1×20 мл) и насыщенным солевым раствором, сушат над натрия сульфатом, фильтруют, упаривают до практически сухого состояния при температуре 50°C с получением фракции сырого продукта B. Обе фракции сырого продукта А и B объединяют и гидрируют над 10% Pd/C (40% мас./мас. Pd/C) при атмосферном давлении с использованием баллона с водородом при комнатной температуре в течение 2,75 часов. Реакционную смесь фильтруют через CELITE®. Фильтрат, содержащий продукт H-D-iGlu(γ-OEt)-D-Trp-OH, анализируют с помощью ВЭЖХ (такой же способ, как описано в 7A выше). Время удерживания для пика продукта составляет 4,54 мин. Фильтрат упаривают до сухого состояния при пониженном давлении на бане с температурой приблизительно 45°C. Анализ методом ВЭЖХ демонстрирует частичное превращение продукта с Rt 4,54 мин в другой продукт с пиком Rt 4,45 мин. Сырой продукт очищают флэш-хроматографией на колонке с использованием изопропанола и концентрированного раствора NH4OH (элюирование с градиентом от 90:10 до 80:20 об./об.). Анализ методом ВЭЖХ полученного материала (670 мг) демонстрирует пик со значением Rt 4,45 мин. Прояснение структуры 1H ЯМР спектроскопией показало, что продукт после обработки представляет собой циклическое соединение, показанное ниже:
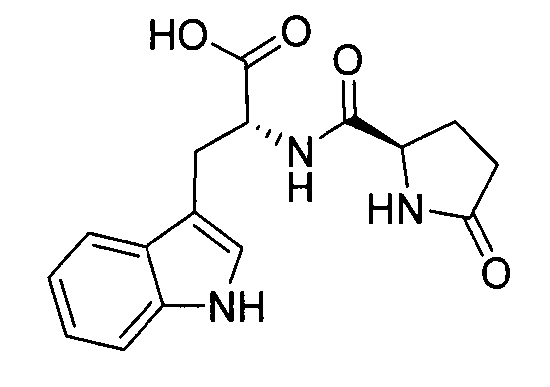
1H ЯМР (CD3OD) δ м.д.: 7,60 (д, J=7,8 Гц, 1H), 7,31 (д, J=8,0 Гц, 1H), 7,10 (с, 1H), 7,07 (т, J=7,8 Гц, 1H), 7,00 (т, J=7,4 Гц, 1H), 4,68-4,65 (м, 1H), 4,05-4,02 (м, 1H), 3,44 (дд, J=14,6 Гц, J=4,6 Гц, 1H), 3,23-3,18 (м, 1H), 2,34-2,24 (м, 1H), 2,13-2,05 (м, 1H), 2,00-1,92 (м, 1H) и 1,76-1,68 (м, 1H). МС (соотношение массы и заряда): 316 [M+1]+, 188 (100%).
13C ЯМР (CD3OD) δ м.д.: 181,6, 177,5, 174,2, 138,0, 129,3, 124,5, 122,4, 119,8, 119,5, 112,3, 111,7, 58,3, 56,3, 30,2, 28,8 и 26,5.
Метод ВЭЖХ: колонка Symmetry C18, 5 мкм, 3,9×150 мм, WAT 046980; подвижная фаза - 0,035% HClO4/CH3CN, градиент; способ: мин-CH3CN%: 0-10%, 10-100%, 12-100%, 14-50%; скорость потока: 1,0 мл/мин; детектор λ: 254 нм.
B. Независимый синтез 5-оксо-D-пролил-D-триптофана из (R)(+)-2-пирролидин-2-5-карбоновой кислоты (H-D-Pyr-D-Trp-OH).
(R)(+)-2-Пирролидин-2,5-карбоновую кислоту (2,09 г, 0,016 моль) добавляют к 250 мл дихлорметана. Добавляют диизопропилэтиламин (3,12 мл, 0,018 моль), и раствор становится прозрачным. Гидрат гидроксибензотриазола (HOBt.H2O, 2,48 г, 0,016 моль) добавляют при температуре 0°C. Добавляют ЭДКИ (4,64 г, 0,025 моль). Через 30 минут добавляют H-D-Trp-OBzl.HCl (4,76 г, 0,016 моль) с последующим добавлением диизопропилэтиламина (3,12 мл, 0,018 моль). Раствор остается прозрачным в точке времени 5 мин, и его перемешивают в течение 16 часов. Растворитель выпаривают до сухого состояния, и материал распределяют между этилацетатом и 10% хлороводородной кислотой. Этилацетатную фракцию промывают 10% раствором натрия бикарбоната, после чего насыщенным солевым раствором. Органическую фракцию сушат над натрия сульфатом и упаривают с получением пенистого твердого вещества. Полученное твердое вещество является бензил-5-оксо-D-пролил-D-триптофанатом. Без очистки образец сырого бензил-5-оксо-D-пролил-D-триптофаната (1,55 г) растворяют в метаноле (30 мл) и гидрируют над 10% Pd/C при давлении водорода 45 фунт/дюйм2 в течение 2 час. Катализатор фильтруют через целит и фильтрат выпаривают с получением пены красного цвета. Полученный материал очищают колоночной хроматографией (градиент элюирования: 10% метанола в дихлорметане и далее 50% изопропанола в дихлорметане) с получением твердого вещества (419 мг). Чистота материала может быть проверена методом ТСХ (30% водный раствор аммиака/изопропанола). Полученный материал представляет собой 5-оксо-D-пролил-D-триптофан с такими же данными ЯМР, как и для материала из раздела А.
Пример 9
Синтез моноаммонийной соли L-изоглутамил-L-триптофана (1:1).
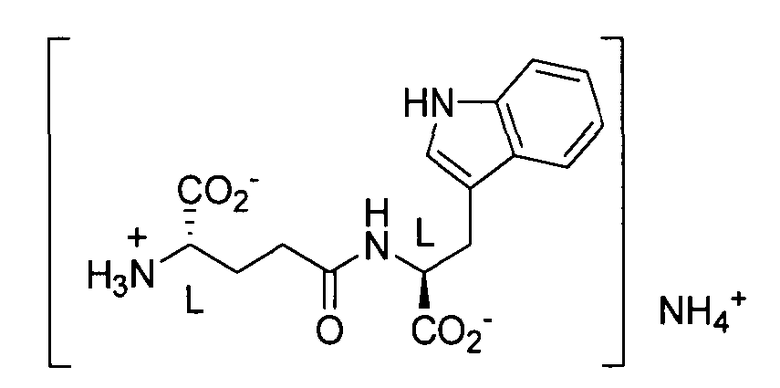
А: Синтез Boc-L-Glu-(γ-L-Trp-O-трет-Bu)-α-O-трет-Bu.
Раствор Boc-L-Glu-O-трет-Bu (1,50 г, 4,9 ммоль) в CH2Cl2 (50 мл) охлаждают до -3°C и перемешивают в течение 15 мин. Далее последовательно добавляют 1-гидроксибензотриазол (HOBt, 1,00 г, 7,4 ммоль), N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (ЭДК, 1,42 г, 7,4 ммоль) и диизопропилэтиламин (ДИПЭА, 1,30 мл, 7,4 ммоль). После этого раствор H-L-Trp-O-трет-Bu.HCl (2,20 г, 7,4 ммоль) и ДИПЭА (1,30 мл, 7,4 ммоль) в CH2Cl2 (20 мл) добавляют по каплям. Полученную смесь перемешивают при температуре льда (от -3°C до 0°C) в течение 1 часа и далее позволяют нагреться до комнатной температуры и перемешивают в течение ночи в атмосфере азота.
Реакционную смесь упаривают до сухого состояния. Остаток распределяют между EtOAc (40 мл) и насыщенным раствором NaHCO3 (100 мл). Органическую фракцию отделяют, промывают 10% раствором лимонной кислоты и далее насыщенным солевым раствором (30 мл). Органическую фракцию сушат над натрия сульфатом, фильтруют и упаривают до получения густого масла. Очистка остатка колоночной хроматографией на силикагеле с использованием градиента смеси растворителей гексан/EtOAc (соотношение 85:15, 80:20 и 60:40, об./об.) в качестве элюента дает названный в заголовке продукт (2,55 г, 95%) в виде твердого вещества белого цвета. 1H ЯМР (ДМСО-d6) δ м.д.: 10,84 (с, 1H), 8,18 (д, J=7,5 Гц, 1H), 7,52 (д, J=7,7 Гц, 1H), 7,35 (д, J=8,0 Гц, 1H), 7,12-7,15 (м, 2H), 7,06 (т, J=7,6 Гц, 1H), 6,98 (т, J=6,8 Гц, 1H), 4,41 (кв, J=6,7 Гц, 1H), 3,73-3,80 (м, 1H), 2,94-3,12 (м, 2H), 2,13-2,21 (м, 2H), 1,60-1,85 (м, 2H), 1,28-1,38 (м, 27H); 13C ЯМР (ДМСО-d6) δ м.д.: 171,5 (C), 171,4 (C), 171,1 (C), 155,5 (C), 136,1 (C), 127,2 (C), 123,5 (CH), 120,9 (CH), 118,3 (CH), 118,1 (CH), 111,3 (CH), 109,7, 80,2, 78,0, 53,9 (CH), 53,6 (CH), 31,5 (CH2), 28,2 (CH3), 27,9 (CH3), 27,6 (CH3), 27,5 (CH3), 27,2 (CH2), 26,6 (CH2); МС (соотношение массы и заряда) 546 [M+1]+; аналитически вычислено для C29H43N3O7.0,5H2O: C, 62,80; H, 8,00; N, 7,58; найдено: C, 62,69; H, 8,56; N, 7,57.
B: Синтез моноаммонийной соли L-изоглутамил-L-триптофана (1:1).
Газообразный HCl пропускают при перемешивании через охлажденный на льду (0-5°C) раствор Boc-L-Glu-(-L-Trp-O-трет-Bu)-γ-O-трет-Bu, полученный, как описано выше, (2,38 г, 4,4 ммоль) в CH2Cl2 (40 мл) в течение 5 часов. Реакционная смесь становится мутной в ходе пропускания газообразного HCl. Температуру реакции поддерживают на уровне ниже 30°C путем охлаждения на льду. Реакционную смесь далее упаривают с получением твердого вещества белого цвета (масса сырого материала = 2,80 г).
Очистка сырого образца (482 мг) с использованием обращенно-фазовой высокоэффективной флэш-хроматографии (HPFC™, Biotage) с использованием колонки C18HS M+40 и градиента смеси растворителей 15 мМ раствор NH4OAc/CH3CN в качестве элюента дает названное соединение после выпаривания растворителя и лиофильной сушки материала (150 мг). 1H ЯМР (ДМСО-d6) δ м.д.: 7,57 (д, J=7,8 Гц, 1H), 7,39 (д, J=8,1 Гц, 1H), 7,11-7,14 (м, 2H), 7,05 (т, J=7,2 Гц, 1H), 4,44-4,48 (м, 1H), 3,42 (т, J=5,7 Гц, 1H), 3,25 (дд, J=14,7, 4,7 Гц, 1H), 2,97-3,03 (м, 1H), 2,14-2,19 (м, 2H), 1,72-1,85 (м, 2H); ИК-ФП (KBr) ν: 3057, 1581, 1400, 745 см-1; МС (соотношение массы и заряда) 334 [двухосновная кислота+1]+; аналитически вычислено для C16H22N4O5.H2O: C, 52,17; H, 6,57; N, 15,21; найдено: C, 51,92; H, 6,80; N, 14,94. Вещество представляет собой моноаммонийную соль L-изоглутамил-L-триптофана (1:1).
Пример 10
Синтез моноаммонийной соли H-D-изоглутамил-L-триптофана (1:1).
А. Синтез Boc-D-Glu-(γ-L-Trp-O-трет-Bu)-α-O-трет-Bu.
Применяют методику, описанную в Примере 1A. При перемешивании к охлажденному на льду раствору Boc-D-Glu-O-трет-Bu (4,00 г, 13,2 ммоль) в CH2Cl2 (75 мл) последовательно добавляют ЭДК (3,80 г, 19,8 ммоль), HOBt (2,68 г, 19,8 ммоль) и ДИПЭА (3,50 мл, 19,8 ммоль). Полученную смесь перемешивают при температуре льда в течение 20 мин. Далее раствор H-L-Trp-O-трет-Bu.HCl (5,88 г, 19,8 ммоль) и ДИПЭА (3,50 мл, 19,8 ммоль) в CH2Cl2 (50 мл) добавляют по каплям в течение 10 мин. Полученную смесь перемешивают при температуре льда в течение 1 часа, после чего позволяют нагреться до комнатной температуры и перемешивают в течение ночи.
Реакционную смесь выпаривают до сухого состояния. Густое остаточное масло переносят в этилацетат (50 мл) и органическую фракцию последовательно промывают насыщенным раствором NaHCO3 (100 мл), 10% раствором лимонной кислоты (100 мл), насыщенным солевым раствором (100 мл) и водой (100 мл). Органическую фракцию сушат над натрия сульфатом, фильтруют и упаривают в вакууме. Очистка остатка колоночной хроматографией на силикагеле с использованием градиента смеси растворителей гексан/EtOAc (соотношение 8:2, 7:3 и 6:4 об./об.) в качестве элюента дает названный в заголовке продукт (5,88 г, 82%) в виде твердого вещества практически белого цвета. 1H ЯМР (ДМСО-d6) δ м.д.: 10,85 (с, 1H), 8,18 (д, J=7,4 Гц, 1H), 7,50 (д, J=7,7 Гц, 1H), 7,33 (д, J=8,0 Гц, 1H), 7,14-7,05 (м, 3H), 6,99 (т, J=7,1 Гц, 1H), 4,40 (кв, J=7,5 Гц, 1H), 3,81-3,75 (м, 1H), 3,12-3,07 (м, 1H), 3,01-2,95 (м, 1H), 2,18-2,15 (м, 2H), 1,86-1,83 (м, 1H), 1,73-1,65 (м, 1H), 1,39-1,29 (м, 27H); МС (соотношение массы и заряда) 568 [M+Na]+; 546 [M+1]+; аналитически вычислено для C29H43N3O7.0,75H2O: C, 62,29; H, 8,02; N, 7,51. Найдено: C, 62,43; H, 7,95; N, 7,08.
B. Синтез моноаммонийной соли H-D-Glu-(-L-Trp-OH (1:1).
Применяют методику, описанную в Примере 1B. Газообразный HCl пропускают при перемешивании через охлажденный на льду (приблизительно -5°C) раствор Boc-D-Glu-(γ-L-Trp-O-трет-Bu)-α-O-трет-Bu, полученный, как описано выше, (1,59 г, 2,91 ммоль) в этилацетате (100 мл) в течение 45 мин. Раствор из бесцветного становится мутно-желтым. Смесь перемешивают при температуре льда в течение 1 часа, после чего позволяют нагреться до комнатной температуры и перемешивают еще в течение 2 часов. По данным ВЭЖХ реакция завершается (колонка: Waters C18, 3,9×150 мм, WAT046980, подвижная фаза: градиент смеси растворителей 0,035% HClO4 [pH 2-2,5]/ацетонитрил, скорость потока: 1 мл/мин, 8: 210-270 нм).
Реакционную смесь упаривают при пониженном давлении до получения твердого вещества. Твердое вещество растворяют в ацетоне и летучие вещества удаляют при пониженном давлении. Последнюю процедуру повторяют еще дважды. Очистка остатка колоночной хроматографией с использованием градиента смеси растворителей изопропанола и раствора гидроксида аммония (28-30% NH4OH) (соотношение 85:15 и 70:30, об./об.) в качестве элюента дает названный в заголовке продукт (0,42 г, 39%) в виде пены практически белого цвета. Т.пл.: 120-130°C; 1H ЯМР (D2O) δ м.д.: 7,67 (д, J=7,9 Гц, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,18-7,22 (м, 2H), 7,13 (т, J=7,1 Гц, 1H), 4,53 (кв, J=3,8 Гц, 1H), 3,45 (т, J=5,8 Гц, 1H), 3,33 (дд, J=14,7 и 4,75 Гц, 1H), 3,07 (дд, J=14,7 и 8,8 Гц, 1H), 2,19-2,31 (м, 2H), 1,78-1,98 (м, 2H); 13C ЯМР (D2O) δ м.д.: 181,4 (C), 176,6 (C), 176,5 (C), 138,8 (C), 129,9 (C), 126,9 (CH), 124,5 (CH), 121,9 (CH), 121,4 (CH), 114,5 (CH), 113,2 (C), 58,6 (CH), 56,7 (CH), 34,2 (CH2), 30,3 (CH2), 28,9 (CH2); МС (соотношение массы и заряда) 334 [двухосновная кислота+1]+.
Пример 11
Синтез моноаммонийной соли L-изоглутамил-D-триптофана (1:1).
А. Синтез Boc-L-Glu-((-D-Trp-OMe)-α-O-трет-Bu.
Применяют методику, описанную в Примере 1A. При перемешивании к охлажденному на льду раствору Boc-L-Glu-O-трет-Bu (3,45 г, 11,4 ммоль) в CH2Cl2 (120 мл) последовательно добавляют ЭДК (3,31 г, 17,3 ммоль), HOBt (2,36 г, 17,5 ммоль) и ДИПЭА (3,0 мл, 17,1 ммоль). Полученную смесь перемешивают при температуре льда еще в течение 25 мин. Далее добавляют раствор H-D-Trp-OMe.HCl (2,20 г, 7,40 ммоль) и ДИПЭА (3,0 мл, 17,1 ммоль) в CH2Cl2 (40 мл). Полученную смесь перемешивают при температуре льда в течение 1 часа, после чего дают нагреться до комнатной температуры и перемешивают в течение ночи.
После обычной обработки, как описано в Примере 1A, очистка остатка колоночной хроматографией с использованием градиента смеси растворителей гексан/EtOAc (соотношение 8:2 и 6:4, об./об.) в качестве элюента дает названный в заголовке продукт (4,25 г, 74%) в виде пены белого цвета. 1H ЯМР (CDCl3) δ м.д.: 8,64 (с, 1H), 7,53 (д, J=7,7 Гц, 1H), 7,33 (д, J=8,0 Гц, 1H), 7,15 (т, J=7,2 Гц, 1H), 7,08 (т, J=7,5 Гц, 1H), 6,98 (с, 1H), 6,61 (д, J=7,2 Гц, 1H), 5,30 (д, J=7,8 Гц, 1H), 4,92 (кв, J=6,8 Гц, 1H), 4,16-4,17 (м, 1H), 3,67 (с, 3H), 3,28-3,34 (м, 2H), 2,16-2,27 (м, 2H), 2,05-2,14 (м, 1H), 1,79-1,89 (м, 1H), 1,42-1,43 (м, 18H); 13C ЯМР (CDCl3) δ м.д.: 172,6 (C), 172,1 (C), 171,6 (C), 171,3 (C), 155,9 (C), 136,3 (C), 127,7 (C), 123,2 (CH), 122,2 (CH), 119,6 (CH), 118,6 (CH), 111,5 (CH), 109,9 (C), 82,3 (C), 79,9 (C), 53,7 (CH), 53,3 (CH), 52,4 (CH), 32,6 (CH2), 28,9 (CH2), 28,4 (CH3), 28,1 (CH3), 27,7 (CH2); МС (соотношение массы и заряда) 504 [M+1]+; аналитически вычислено для C26H37N3O7.0,25H2O: C, 61,46; H, 7,44; N, 8,27; найдено: C, 61,36; H, 7,50; N, 7,84.
B. Синтез Boc-L-iGlu-D-Trp-OH.
При перемешивании к раствору Boc-L-Glu-((-D-Trp-OMe)-α-O-трет-Bu (3,94 г, 7,82 ммоль) в метаноле (50 мл) добавляют раствор NaOH (654 мг, 16,4 ммоль) в H2O (20 мл). Полученный раствор перемешивают при комнатной температуре в течение ночи. 1 н. Раствор NaOH (150 мл) добавляют к реакционной смеси и водный материал промывают этилацетатом (3×100 мл). Водную фракцию подкисляют 3 н. раствором HCl до pH приблизительно 2, после чего экстрагируют этилацетатом (3×100 мл). Органические фракции объединяют, сушат над натрия сульфатом и упаривают при пониженном давлении. Очистка остатка колоночной хроматографией на силикагеле с использованием градиента смеси растворителей CH2Cl2 и MeOH (соотношение 85:15, 70:30, об./об.) в качестве элюента дает названный в заголовке продукт (0,55 г, 97%) в виде пены розового цвета. 1H ЯМР (MeOD-d4) δ м.д.: 7,58 (д, J=7,7 Гц, 1H), 7,31 (д, J=8,0 Гц, 1H), 7,05-7,09 (м, 2H), 6,99 (т, J=7,3 Гц, 1H), 4,61-4,67 (м, 1H), 4,13 (шир, 1H), 3,30-3,38 (м, 2H), 3,12-3,18 (м, 1H), 2,21-2,27 (м, 2H), 1,98-2,06 (м, 1H), 1,81-1,88 (м, 1H), 1,42 (с, 9H); 13C ЯМР (MeOD-d4) δ м.д.: 158,2 (C), 138,1 (C), 129,1 (C), 124,5 (CH), 122,4 (CH), 119,9 (CH), 119,5 (CH), 112,3 (CH), 111,5 (C), 80,6 (C), 33,5 (CH2), 28,9 (CH3), 28,7 (CH2); МС (соотношение массы и заряда) 490 [M + 1]+.
C. Синтез моноаммонийной соли H-L-iGlu-D-Trp-OH (1:1).
Газообразный HCl пропускают при перемешивании через охлажденный на льду (приблизительно 0°C) раствор Boc-L-iGlu-D-Trp-OH, как описано выше, (500 мг, 1,0 ммоль) в смеси растворителей CH2Cl2 (20 мл) и EtOAc (10 мл) в течение 30 мин. Реакционную смесь перемешивают при температуре льда в течение 1 часа. По данным ВЭЖХ реакция завершается (колонка: Waters C18, 3,9×150 мм, WAT046980, подвижная фаза: градиент смеси растворителей 0,035% HClO4 [pH 2,0-2,5]/ацетонитрил, скорость потока: 1 мл/мин, 8: 210-270 нм).
Реакционную смесь выпаривают до сухого состояния с получением пены темно-пурпурного цвета. Очистка остатка колоночной хроматографией с использованием градиента смеси растворителей изопропанол/раствор гидроксида аммония (28-30% NH4OH) (соотношение 85:15 и 70:30, об./об.) в качестве элюента дает названный в заголовке продукт (333 мг, 88%) в виде густого масла оранжевого цвета. 1H ЯМР (D2O) δ м.д.: 7,64 (д, J=7,9 Гц, 1H), 7,44 (д, J=8,0 Гц, 1H), 7,16-7,19 (м, 2H), 7,10 (т, J=7,5 Гц, 1H), 4,52 (кв, J=3,7 Гц, 1H), 3,44 (т, J=6,3 Гц, 1H), 3,29-3,34 (м, 1H), 3,03-3,09 (м, 1H), 2,17-2,30 (м, 2H), 1,76-1,96 (м, 2H); 13C ЯМР (D2O) δ м.д.: 181,2 (C), 176,6 (C), 176,5 (C), 138,8 (C), 129,9 (C), 126,9 (CH), 124,5 (CH), 121,9 (CH), 121,4 (CH), 114,5 (CH), 113,1 (C), 58,5 (CH), 56,7 (CH), 51,6 (CH), 34,2 (CH2), 30,2 (CH2), 28,9 (CH2); МС (соотношение массы и заряда) 356 [двухосновная кислота+Na]+, 334 [двухосновная кислота+1]+, аналитически вычислено для C16H22N4O5.2,35H2O: C, 48,94; H, 6,85; N, 14,27; найдено: C, 48,94; H, 6,64; N, 14,28.
Пример 12
Анализ методом ВЭЖХ D-изоглутамил-D-триптофана, L-изоглутамил-L-триптофана, L-изоглутамил-D-триптофана и D-изоглутамил-L-триптофана.
Двухосновную кислоту четырех диастереомеров анализировали на хиральной колонке ВЭЖХ. D,D- и L,D-диастереомеры взяты из примеров выше, тогда как D,L-изомер получен от Bachem и L,L-изомер получен от Sigma. Анализ с использованием хиральной колонки ВЭЖХ (табл. 1) показал, что полученный D-изоглутамил-D-триптофан свободен от других диастереомеров, а именно (D,L), (L,L) и (L,D) изомеров.
Анализ H-iGlu-Trp-OH методом ВЭЖХ
В способе А образцы анализируют на хиральной колонке. Значения времени удерживания для всех четырех диастереомеров является, очевидно, разными. В способе B образцы анализируют на обычной обращенно-фазовой колонке; практически не наблюдается разницы во времени удерживания для образцов. Моноаммонийная соль D-изоглутамил-D-триптофана, раскрытая в данном изобретении, является стабильной после 2 лет хранения. Анализ методом ВЭЖХ (способ B) показал, что чистота на длине волны 254 нм составляет 99,8%.
Пример 13
А. Получение соли H-D-Glu-(γ-D-Trp-OMe)-α-OBzl HCl {(2R)-амино-(4R)-[2-(1H-индол-3-ил)-1-метоксикарбонил-этилкарбамоил]-масляной кислоты бензилового эфира гидрохлорид}.
В 3-горлую круглодонную колбу объемом 250 мл, оборудованную магнитной мешалкой, помещают Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl (20 г, 0,037 моль) и 100 мл дихлорметана с образованием прозрачного раствора при перемешивании. Раствор охлаждают на охлаждающей бане, содержащей смесь лед/NaCl, до -10°C. Газообразный HCl пропускают через холодный раствор. В ходе реакции температура находится в интервале от -4°C до -10°C. Реакция завершается приблизительно через 1 час. Твердое вещество белого цвета осаждается из раствора. Продукт в виде твердого вещества отделяют фильтрацией. Твердое вещество промывают дихлорметаном (2×40 мл), сначала сушат на воздухе, после чего сушат в вакуумной печи при температуре 42°C в течение ночи с получением 16,4 г (94%, чистота по данным ВЭЖХ 98,2%). 1H ЯМР (ДМСО-d6) δ м.д.: 10,93 (с, 1H), 8,61 (шир, 3H), 8,50 (д, J=7,4 Гц, 1H), 7,47 (д, J=7,4 Гц, 1H), 7,40-7,32 (м, 6H), 7,17 (с, 1H), 7,06 (т, J=7,4 Гц, 1H), 6,97 (т, J=7,4 Гц, 1H), 5,26-5,14 (м, 2H), 4,51-4,46 (м, 1H), 4,05-3,95 (м, 1H), 3,56 (с, 3H), 3,16-3,11 (м, 1H), 3,06-3,01 (м, 1H), 2,40-2,26 (м, 2H), 2,00-1,98 (м, 2H). Метод ВЭЖХ: колонка: XTerra МС C18 5 мкм 4,6×250 мм; подвижная фаза: А = водная фаза: 4 мМ Трис, 2 мМ ЭДТА, pH 7,4, B = органическая фаза: CH3CN. Программа градиента: B %: 0 мин 5%, 15 мин 55%, 30 мин 55%, 32 мин 5%, 40 мин 5%. Скорость потока: 1 мл/мин; объем инъекции = 5 мкл; длина волны: 222, 254, 282, 450 нм. Rt исходного материала = 25,1 мин; Rt продукта = 17,2 мин.
B. Получение (2R)-амино-(4R)-[2-(1H-индол-3-ил)-1-метоксикарбонил-этилкарбамоил]-масляной кислоты метилового эфира гидрохлорид; соль H-D-Glu-(γ-D-Trp-OMe)-α-OMe HCl.
В 3-горлую круглодонную колбу объемом 250 мл, оборудованную магнитной мешалкой, помещают Boc-D-Glu-(γ-D-Trp-OMe)-α-OMe (2,8 г, 6,06 ммоль) и метанол (30 мл) с получением прозрачного раствора при перемешивании. Раствор охлаждают на охлаждающей бане, содержащей смесь лед/NaCl, до -12°C. Газообразный HCl пропускают через холодный раствор. В ходе реакции температура находится в интервале от -12°C до +9°C. Реакция завершается приблизительно через 30 мин. Приблизительно половину реакционной смеси упаривают до сухого состояния с получением 1,3 г твердого вещества (чистота по данным ВЭЖХ 96,8%). 1H ЯМР (ДМСО-d6) δ м.д.: 10,90 (с, 1H), 8,48-8,46 (м, 4H), 7,48 (д, J=7,8 Гц, 1H), 7,34 (д, J=8,0 Гц, 1H), 7,16 (с, 1H), 7,07 (т, J=7,4 Гц, 1H), 6,98 (т, J=7,4 Гц, 1H), 4,52-4,47 (м, 1H), 4,05-3,99 (м, 1H), 3,69 (с, 3H), 3,58 (с, 3H), 3,17-3,12 (м, 1H), 3,07-3,01 (м, 1H), 2,37-2,23 (м, 2H), 1,98-1,91 (м, 2H). Метод ВЭЖХ: колонка: XTerra МС C18 5 мкм 4,6×250 мм; подвижная фаза: А = водная фаза: 4 мМ Трис, 2 мМ ЭДТА, pH 7,4; B = органическая фаза: CH3CN. Программа градиента: B %: 0 мин 5%, 15 мин 55%, 30 мин 55%, 32 мин 5%, 40 мин 5%. Скорость потока: 1 мл/мин. Объем инъекции = 5 мкл; длина волны: 222, 254, 282, 450 нм; Rt исходного материала = 18,7 мин, Rt продукта = 13,0 мин.
C. Получение соли H-D-Glu-(γ-D-Trp-OMe)-α-OBzl HCl {(2R)-амино-(4R)-[2-(1H-индол-3-ил)-1-метоксикарбонил-этилкарбамоил]-масляной кислоты бензилового эфира гидрохлорид}.
Раствор HCl в этилацетате получают пропусканием газообразного HCl, образованного в результате добавления по каплям 50 мл конц. HCl к конц. H2SO4, через охлажденный на льду (0-4°C) этилацетат (100 мл). Полученный холодный раствор кислоты помещают в капельную воронку и добавляют к тонкой суспензии Boc-D-Glu(D-Trp-OMe)-OBzl (36,45 г, 67,80 ммоль) в 150 мл этилацетата. Густая суспензия образуется в пределах 5 мин. Затем добавляют весь раствор HCl, газообразный HCl образуют, как описано выше, и пропускают непосредственно через полученную суспензию. Внутреннюю температуру поддерживают в интервале 5-10°C, охлаждая на льду. При перемешивании температуру суспензии поддерживают на уровне 5-15°C, выдерживая при этой температуре в течение 2,5 часов, после чего позволяют нагреться до комнатной температуры. Протекание реакции контролируют методом ТСХ (смесь гексан/EtOAc [1:1 об./об.] в качестве элюента). Твердое вещество собирают вакуумной фильтрацией, промывают этилацетатом (2×100 мл), после чего сушат в вакуумной печи в течение ночи. Получают твердое вещество светло-розоватого цвета (выход 27,49 г, 86%). Аналитические данные, сходные с приведенными в Примере 13A выше, за исключением некоторого количества остаточного EtOAc, обнаруженного по данным 1H ЯМР. Остаточный EtOAc может быть удален высушиванием на воздухе в течение 24-48 час перед сушкой в вакуумной печи.
D. Получение соли H-D-Glu-(γ-D-Trp-OMe)-α-OBzl HCl {(2R)-амино-(4R)-[2-(1H-индол-3-ил)-1-метоксикарбонил-этилкарбамоил]-масляной кислоты бензилового эфира гидрохлорид}
В 3-горлую круглодонную колбу помещают Boc-D-Glu-OBzl (200,0 г, 0,593 моль) и EtOAc (1,6 л). Диизопропилэтиламин (206,5 мл, 1,186 моль) добавляют к полученной суспензии белого цвета. Внутренняя температура составляет приблизительно 21°C, и наблюдается густая суспензия. Суспензия постепенно разрежается и в пределах 15 мин превращается в прозрачный раствор. Далее N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (136,4 г ЭДК, 0,711 моль) добавляют по мере образования суспензии. HCl.H-D-TrpOMe (166,1 г, 0,652 моль) добавляют порциями в течение 30 мин. Наблюдается выделение тепла, и внутренняя температура достигает 28°C. Через 2 часа внутренняя температура снижается до 25°C. Полученную суспензию энергично перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют этилацетатом (200 мл), после чего последовательно промывают 1 н. HCl (600 мл) и 5% раствором HCl (2×256 мл). Образец сырой смеси упаривают и анализируют методом ТСХ и 1H ЯМР. Содержание воды по данным метода Карла Фишера составляет 2,3%.
Содержимое колбы охлаждают на ледяной бане, и внутренняя температура составляет приблизительно 0°C. Газообразный HCl пропускают через охлажденный на льду раствор в течение 1 часа. Наблюдается выделение тепла в пределах 10 мин после пропускания газообразного HCl, и внутренняя температура достигает приблизительно 19°C. После 20 мин пропускания внутренняя температура составляет 22°C. Полученную суспензию перемешивают еще в течение часа. Твердое вещество отделяют вакуумной фильтрацией и промывают этилацетатом (2×640 мл), после чего сушат под вакуумом до постоянной массы (243,8 г). Аналитические данные сходны с приведенными в Примере 13A выше. Присутствие этилацетата (приблизительно 10% по данным протонной интеграции) подтверждается данными 1H ЯМР. Захваченный EtOAc может быть удален сушкой твердого вещества на воздухе в течение 24-48 часов перед сушкой в печи.
Пример 14
Получение раствора основно-аддитивной соли D-изоглутамил-D-триптофана, и ее превращение в тимодепрессин (D-изоглутамил-D-триптофан).
Методика 14A:
Исходный материал, соль H-D-Glu-(γ-D-Trp-OMe)-α-OBzl HCl, (4,0 г, 8,4 ммоль) помещают в 3-горлую круглодонную колбу объемом 250 мл, оборудованную магнитной мешалкой. Добавляют метанол (20 мл) с получением прозрачного раствора. Раствор охлаждают на бане, содержащей лед и соль NaCl, до -10°C. Добавляют раствор NaOH (3 н., 8,4 мл, 25,2 ммоль). ВЭЖХ применяют для контроля хода реакции. Через 2 часа анализ реакционной смеси методом ВЭЖХ показывает, что реакция еще не закончена. Добавляют раствор NaOH (3 н., 1,4 мл, 4,2 ммоль). В этой точке добавляют в целом 29,4 ммоль NaOH. Через еще 4 часа анализ реакционной смеси методом ВЭЖХ показывает, что содержание продукта в реакционной смеси составляет более 95,2%. Реакцию останавливают. Реакционную смесь подкисляют при охлаждении с помощью бани, содержащей воду со льдом, до pH 6,5 добавлением хлороводородной кислоты (6 н., ~1,3 мл, 7,8 ммоль). Полученный раствор упаривают до объема 15 мл для удаления большей части метанола. Раствор промывают этилацетатом (15 мл × 2). Раствор фильтруют и собирают фильтрат. Фильтрат дополнительно подкисляют до pH 3 добавлением хлороводородной кислоты (6 н., ~1,3 мл, 7,8 ммоль). В этой точке используют общее количество ~15,6 ммоль хлороводородной кислоты. Твердое вещество образуется в ходе перемешивания при комнатной температуре. Смесь перемешивают в течение ночи. Твердое вещество отделяют фильтрацией. Твердое вещество сушат на воздухе с получением 2,4 г сырого продукта. Далее твердое вещество снова помещают в круглодонную колбу. Добавляют деминерализованную воду (15 мл) и смесь перемешивают в течение 2 часов. Твердое вещество отделяют фильтрацией. Твердое вещество снова сушат на воздухе, после чего помещают в круглодонную колбу. Добавляют деминерализованную воду (15 мл) и смесь перемешивают в течение 1 часа. Твердое вещество отделяют фильтрацией и промывают охлажденной льдом деминерализованной водой (6 мл × 3). С помощью испытания с серебра нитратом показано, что твердое вещество не содержит хлоридов. Твердое вещество сушат на воздухе, после чего помещают в вакуумную печь при температуре 42°C, выдерживая при этой температуре в течение 19 час, с получением 1,3 г (46%, чистота по данным ВЭЖХ 98,8%). Фильтраты и водные растворы для промывания объединяют и упаривают для выделения второй порции продукта. 1H ЯМР (D2O-NaOD, pH 7,0) δ м.д.: 7,59 (д, J=7,6 Гц, 1H), 7,38 (д, J=7,6 Гц, 1H), 7,15-7,12 (м, 2H), 7,05 (т, J=7,2 Гц, 1H), 4,47-4,44 (м, 1H), 3,40 (т, J=6,1 Гц, 1H), 3,30-3,25 (м, 1H), 3,03-2,97 (м, 1H), 2,3-2,1 (м, 2H), 1,84-1,69 (м, 2H). МС (соотношение массы и заряда) 334,3 [M+1]+. Метод ВЭЖХ: колонка: XTerra МС C18 5 мкм 4,6×250 мм. Подвижная фаза: А = водная фаза: 4 мМ Трис, 2 мМ ЭДТА, pH 7,4; B = органическая фаза: CH3CN. Программа градиента: B %: 0 мин 5%, 15 мин 55%, 30 мин 55%, 32 мин 5%, 40 мин 5%. Скорость потока: 1 мл/мин. Объем инъекции = 5 мкл. Длина волны: 222, 254, 282, 450 нм. Rt продукта = 6,5 мин.
Методика 14B:
Моногидрат гидроксида лития (0,374 г, 8,9 ммоль) растворяют в 3,5 мл деминерализованной воды. Раствор помещают в одногорлую круглодонную колбу объемом 100 мл, оборудованную магнитной мешалкой. 6,5 мл Метил-трет-бутилового эфира добавляют к раствору. При комнатной температуре добавляют исходный материал, соль H-D-Glu-(γ-D-Trp-OMe)-α-OBzl HCl, (2,0 г, 4,2 ммоль) с образованием суспензии. Добавляют метанол (2 мл), большую часть твердого вещества растворяют. ВЭЖХ применяют для контроля протекания реакции. Некоторое количество исходного материала остается в реакционной смеси после перемешивания при комнатной температуре в течение ночи. Лития гидроксид моногидрат (0,190 г, 4,5 ммоль) растворяют в 2 мл воды деминерализованной и добавляют к реакционной смеси, с последующим добавлением 2 мл метанола. В этой точке добавляют общее количество 13,4 ммоль LiOH. После прохождения 4 час анализ реакционной смеси методом ВЭЖХ показывает, что реакция еще не завершилась. Моногидрат гидроксида лития (0,100 г, 2,4 ммоль) растворяют в 1 мл деминерализованной воды и добавляют к реакционной смеси. В этой точке добавляют общее количество 15,8 ммоль LiOH. Через еще 2,5 часа анализ реакционной смеси методом ВЭЖХ показывает, что содержание продукта в реакционной смеси превышает 97,5%. Реакцию останавливают. Реакционную смесь выливают в делительную воронку и 2 фракции разделяют. Водную фракцию промывают этилацетатом (15 мл × 2). Водную фракцию подкисляют при охлаждении с помощью бани, содержащей воду с льдом, до pH 6 добавлением хлороводородной кислоты (6 н., ~650 мкл, 3,9 ммоль). Водную фракцию упаривают до 5 мл и фильтруют, собирая фильтрат. Фильтрат дополнительно подкисляют до pH 3 добавлением хлороводородной кислоты (6 н., ~700 мкл, 4,2 ммоль). В этой точке используют общее количество ~8,1 ммоль хлороводородной кислоты. Твердое вещество образуется в ходе перемешивания при комнатной температуре. Твердое вещество отделяют фильтрацией. Твердое вещество сушат на воздухе, после чего снова помещают в круглодонную колбу. Добавляют деминерализованную воду (6 мл) и смесь перемешивают в течение 15 мин. Твердое вещество отделяют фильтрацией и промывают ледяной холодной деминерализованной водой (6 мл × 6). Доведено, что твердое вещество не содержит хлоридов, с помощью испытания с нитратом серебра. Твердое вещество сушат на воздухе, после чего помещают в вакуумную печь при температуре 42°C, выдерживая при этой температуре в течение 12 часов, с получением 0,44 г (31%, чистота по данным ВЭЖХ 98,5%). Фильтраты и водные растворы для промывания объединяют и упаривают для дополнительного выделения второй порции продукта. 1H ЯМР (D2O-NaOD pH 6,0) δ м.д.: 7,59 (д, J=7,7 Гц, 1H), 7,38 (д, J=7,7 Гц, 1H), 7,15-7,12 (м, 2H), 7,05 (т, J=7,2 Гц, 1H), 4,47-4,44 (м, 1H), 3,41 (т, J=6,0 Гц, 1H), 3,29-3,25 (м, 1H), 3,03-2,97 (м, 1H), 2,3-2,1 (м, 2H), 1,83-1,58 (м, 2H).
Метод ВЭЖХ: колонка: XTerra МС C18 5 мкм 4,6×250 мм. Подвижная фаза: А = водная фаза: 4 мМ Трис, 2 мМ ЭДТА, pH 7,4; B = органическая фаза: CH3CN. Программа градиента: B %: 0 мин 5%, 15 мин 55%, 30 мин 55%, 32 мин 5%, 40 мин 5%. Скорость потока 1 мл/мин. Объем инъекции = 5 мкл. Длина волны: 222, 254, 282, 450 нм. Rt продукта = 6,5 мин.
Тимодепрессин, полученный в данном изобретении, обладает растворимостью в воде от приблизительно 20 до приблизительно 23 мг/мл. Промывание водой в Методиках A и B служит целям удаления неорганических солей, таких как натрия хлорид или лития хлорид. При получении больших количеств объем воды для промывания можно контролировать, вычисляя количество присутствующей неорганической соли и с использованием растворимости для определения количества воды, необходимой для промывания продукта.
Поскольку могут быть осуществлены многочисленные изменения предпочтительных вариантов данного изобретения без отхода от контекста данного изобретения, предусматривается, что весь материал, который содержится в данном описании, должен интерпретироваться как иллюстрирующий данное изобретения, а не ограничивающий его.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ТИМОДЕПРЕССИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2536685C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОНАТРИЕВОЙ СОЛИ ИЗОГЛУТАМИЛ-ТРИПТОФАНА | 2019 |
|
RU2703991C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ МОНОНАТРИЕВОЙ СОЛИ D-ИЗОГЛУТАМИЛ-D-ТРИПТОФАНА | 2008 |
|
RU2476440C2 |
| ПЕПТИД, ОБЛАДАЮЩИЙ ИММУНОСУПРЕССИВНЫМ СВОЙСТВОМ | 1996 |
|
RU2123859C1 |
| СРЕДСТВА, УСИЛИВАЮЩИЕ СЕКРЕЦИЮ ГОРМОНА РОСТА | 2001 |
|
RU2270198C2 |
| Способ получения пептида Ac-His-Ala-Glu-Glu-NH | 2021 |
|
RU2767030C1 |
| СПОСОБ ЖИДКОФАЗНОГО СИНТЕЗА H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 2015 |
|
RU2694051C2 |
| ПРОИЗВОДНЫЕ ЛАНТИОНИНА | 2010 |
|
RU2518890C2 |
| НОВЫЕ СПОСОБЫ ПОЛУЧЕНИЯ БАРУСИБАНА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2016 |
|
RU2726414C2 |
| N-АЦИЛЬНЫЕ ПРОИЗВОДНЫЕ БИОГЕННЫХ АМИНОВ - МОДУЛЯТОРЫ ПЕРЕКИСНОГО ОКИСЛЕНИЯ ЛИПИДОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2093520C1 |
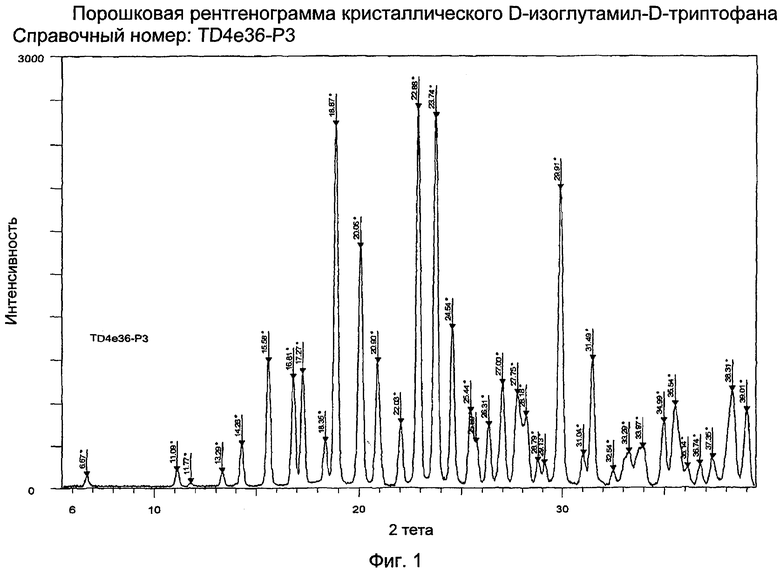
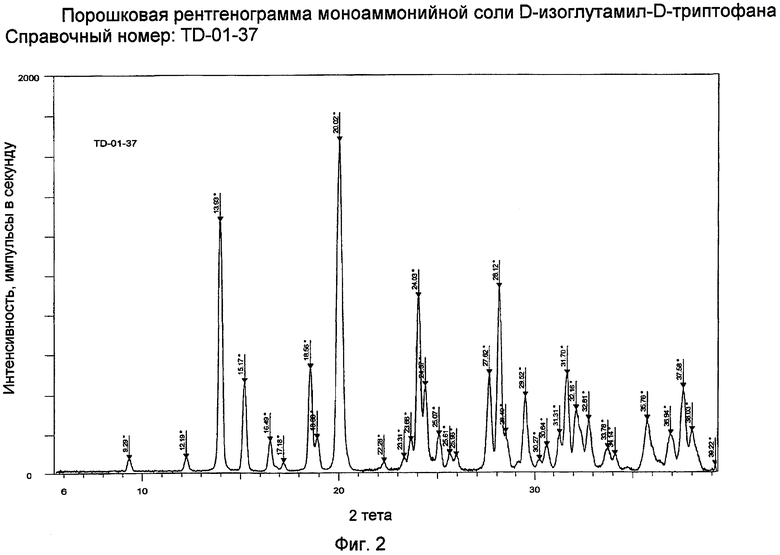
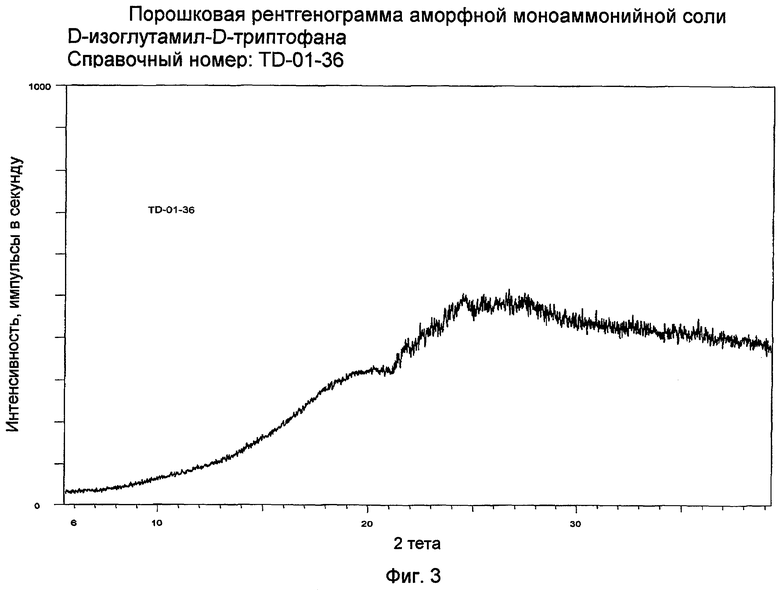

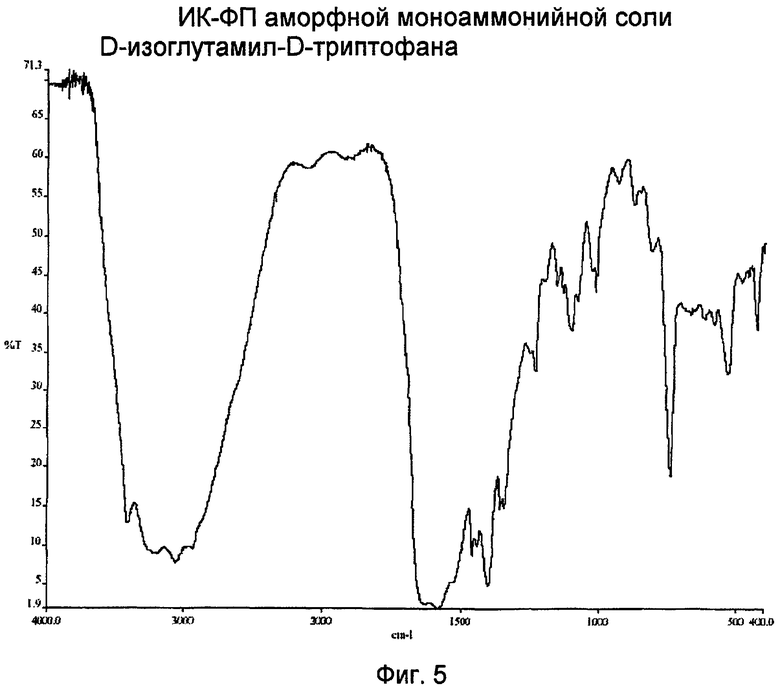
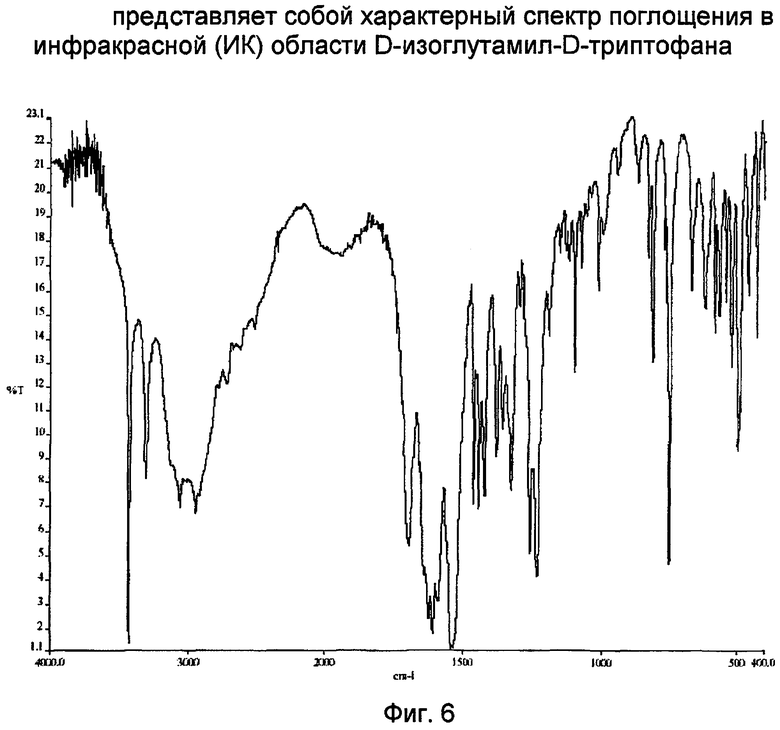
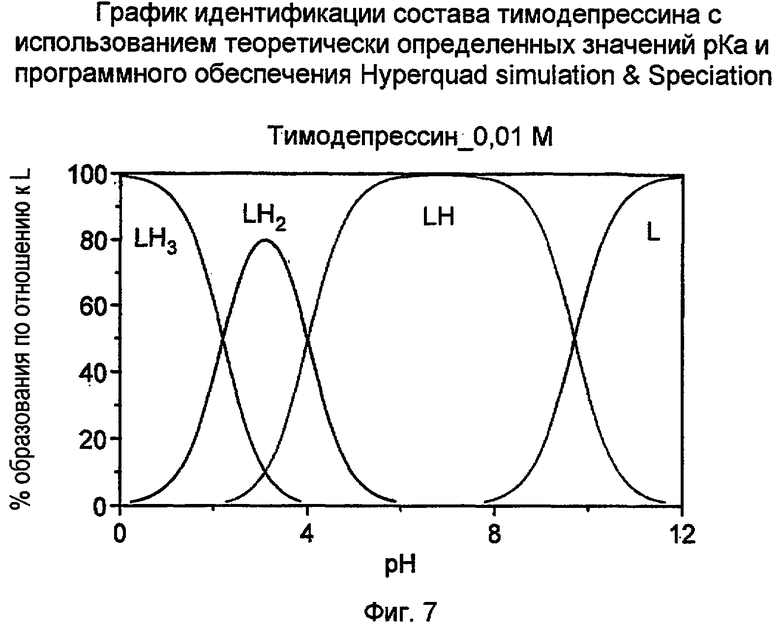
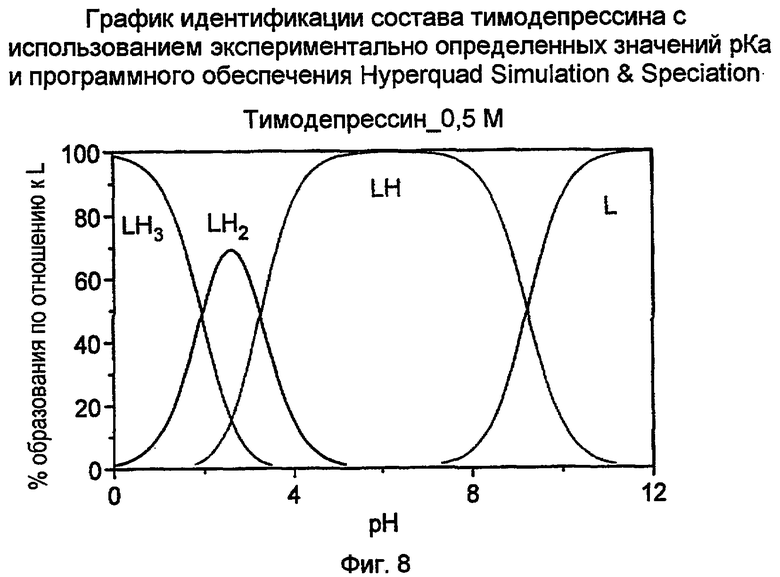
Предлагается способ получения чистого кристаллического D-изоглутамил-D-триптофана, включающий стадию снятия защиты с по существу чистого N-трет-бутоксикарбонил-D-изоглутамил-D-триптофана или его диэфира с получением по существу чистого D-изоглутамил-D-триптофана. Предлагается аморфная аммонийная соль D-изоглутамил-D-триптофана (1:1). Также предлагается способ получения чистой моноаммонийной соли D-изоглутамил-D-триптофана из по существу чистого N-трет-бутоксикарбонил-D-изоглутамил-D-триптофана. Заявлено соединение H-D-Glu-(γ-D-Trp-OR2)-α-OR1
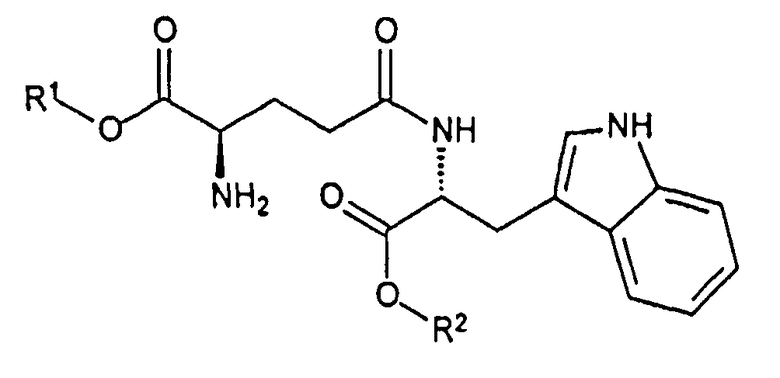
и его фармацевтически приемлемые кислотно-аддитивные соли. Также заявлена твердая фармацевтическая композиция и ее применение в качестве иммунодепрессанта или средства против псориаза, 21 н. и 30 з.п. ф-лы, 14 пр., 8 ил., 1 табл.
1. Кристаллический D-изоглутамил-D-триптофан, характеризующийся пиками на порошковой рентгенограмме, имеющими следующие значения 2θ: 18,87, 20,05, 23,74 и 29,91.
2. Кристаллический D-изоглутамил-D-триптофан, характеризующийся порошковой рентгенограммой, приведенной на фиг.1.
3. Кристаллический D-изоглутамил-D-триптофан, характеризующийся следующими параметрами порошковой рентгенограммы, представленными в значениях межплоскостных расстояний d, угла Брэгга 2θ и относительной интенсивности (выраженной как процент по отношению к наиболее интенсивной линии):
4. Способ получения D-изоглутамил-D-триптофана, свободного от неорганических солей, включающий следующие стадии:
(a) получение раствора кислотно-аддитивной соли H-D-iGlu-D-Trp-OH в водной среде, по существу, свободной от органического растворителя; или получение раствора основно-аддитивной соли H-D-iGlu-D-Trp-OH в водной среде, по существу, свободной от органического растворителя;
(b) коррекция pH до значений в интервале от приблизительно 2,0 до приблизительно 3,2 с помощью раствора гидроксида щелочного металла или неорганической кислоты, чтобы вызвать осаждение H-D-iGlu-D-Trp-OH;
(c) выделение осажденного H-D-iGlu-D-Trp-OH; и
(d) вакуумная сушка продукта, полученного на стадии (с) с получением H-D-iGlu-D-Trp-OH.
5. Способ по п.4, в котором кислотно-аддитивная соль представляет собой H-D-iGlu-D-Trp-OH гидрохлорид, полученный способом, включающим следующие стадии:
(i) основный гидролиз соединения формулы I:
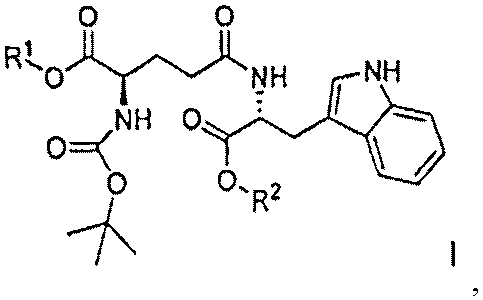
где R1 выбран из группы, состоящей из C1-C4алкила и бензила, и R2 представляет собой C1-C4алкил, при условии, что С4 алкил не является трет-бутилом,
с использованием гидроксида металла в воде и инертном растворителе, в присутствии метанола с получением Boc-D-iGlu-D-Trp-OH, свободного от других диастереомеров; и
(ii) снятие с помощью хлороводорода защиты с Boc-D-iGlu-D-Trp-OH со стадии (i) в инертном органическом растворителе; и выпаривание растворителя с получением гидрохлорида H-D-iGlu-D-Trp-OH.
6. Способ по п.4, отличающийся тем, что кислотно-аддитивная соль представляет собой H-D-iGlu-D-Trp-OH гидрохлорид, и раствор кислотно-аддитивной соли на стадии (а) получают способом, включающим:
(i) гидрирование соединения формулы II
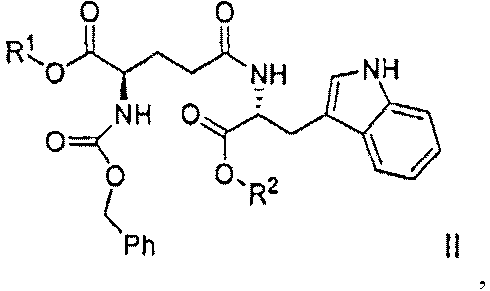
где R1 представляет собой бензил, и R2 выбран из группы, состоящей из бензила и водорода, с использованием палладия на угле в метаноле или этаноле;
(ii) очистку сырого Н-D-iGlu-D-Trp-OH со стадии (i) с помощью хроматографии на силикагеле с использованием изопропанола и воды в качестве элюента; и (iii) обработку материала со стадии (ii) хлороводородной кислотой в воде с получением раствора соли гидрохлорида H-D-iGlu-D-Trp-OH в воде.
7. Способ по п.4, в котором раствор основно-аддитивной соли H-D-iGlu-D-Trp-OH на стадии (а) получают способом, включающим:
(i) снятие с помощью кислоты защиты с дипептида Boc-D-Glu-(γ-D-Trp-OR2)-α-OR1 с получением кислотно-аддитивной соли диэфира H-D-Glu-(γ-D-Trp-OR2)-α-OR1, где каждый из R1 и R2 независимо выбран из группы, состоящей из C1-C4алкила и бензила;
(ii) основный гидролиз продукта со стадии (i) с использованием гидроксида металла в воде и инертном растворителе в присутствии метанола с получением основно-аддитивной соли H-D-iGlu-D-Trp-OH, где R1 и R2 являются такими, как определено выше, и где гидроксид металла выбран из группы, состоящей из гидроксида натрия, гидроксида калия и гидроксида лития;
(iii) экстракцию материала со стадии (ii) растворителем, не смешивающимся с водой, и отделение водного слоя;
(iv) коррекцию pH водной фазы со стадии (iii) до pH от приблизительно 6 до приблизительно 7; и
(v) выпаривание растворителя из раствора со стадии (iv) с образованием раствора, содержащего приблизительно одну часть растворенного вещества на менее чем приблизительно 8 частей воды, где растворенное вещество представляет собой основно-аддитивную соль D-изоглутамил-D-триптофана.
8. Способ получения свободного от неорганических солей H-D-iGlu-D-Trp-OH по п.4, который включает:
(a) получение раствора кислотно-аддитивной соли H-D-iGlu-D-Trp-OH в водной среде, по существу, свободной от органического растворителя;
(b) коррекцию pH до значений от приблизительно 2,0 до приблизительно 3,2 с помощью раствора гидроксида щелочного металла, чтобы вызвать осаждение H-D-iGlu-D-Trp-OH;
(c) выделение осажденного H-D-iGlu-D-Trp-OH; и
(d) вакуумную сушку продукта, полученного на стадии (с), с получением H-D-iGlu-D-Trp-OH.
9. Способ получения свободного от неорганических солей H-D-iGlu-D-Trp-OH по п.4, который включает:
(a) получение раствора основно-аддитивной соли H-D-iGlu-D-Trp-OH в водной среде, по существу, свободной от органического растворителя;
(b) коррекцию pH до значений от приблизительно 2,0 до приблизительно 3,2 с помощью раствора неорганической кислоты, чтобы вызвать осаждение H-D-iGlu-D-Trp-OH;
(c) выделение осажденного H-D-iGlu-D-Trp-OH; и
(d) вакуумную сушку продукта, полученного на стадии (с), с получением H-D-iGlu-D-Trp-OH.
10. Способ по п.8, в котором кислотно-аддитивная соль на стадии (а) представляет собой H-D-iGlu-D-Trp-OH гидрохлорид.
11. Способ по п.8 или 9, в котором pH на стадии (b) составляет от приблизительно 2,5 до приблизительно 3,0.
12. Способ по п.5, в котором R1 представляет собой бензил, и R2 представляет собой метил.
13. Способ по п.5, в котором инертный растворитель на стадии (i) выбран из группы, состоящей из метанола и метил-трет-бутилового эфира; и гидроксид металла выбран из группы, состоящей из гидроксида натрия, гидроксида лития и гидроксида калия.
14. Способ по п.9, в котором в соединении формулы Boc-D-Glu-(γ-D-Trp-OR2)-α-OR1 на стадии (a) R1 выбран из группы, состоящей из метила и бензила, и R2 выбран из группы, состоящей из метила и этила.
15. Способ по п.9, в котором инертный растворитель на стадии (b) выбран из группы, состоящей из метанола и метил-трет-бутилового эфира.
16. Кристаллическая аммонийная соль H-D-iGlu-D-Trp-OH (1:1), характеризующаяся пиками на порошковой рентгенограмме, имеющими следующие значения 2θ: 13,93, 20,02, 24,03 и 28,12.
17. Кристаллическая аммонийная соль H-D-iGlu-D-Trp-OH (1:1), характеризующаяся порошковой рентгенограммой, которая приведена на фиг.2.
18. Кристаллическая аммонийная соль H-D-iGlu-D-Trp-OH (1:1), характеризующаяся следующими параметрами порошковой рентгенограммы, представленными в значениях межплоскостных расстояний d, угла Брэгга 2θ и относительной интенсивности (выраженной как процент по отношению к наиболее интенсивной линии):
19. Аморфная аммонийная соль D-изоглутамил-D-триптофана (1:1).
20. Аморфная аммонийная соль D-изоглутамил-D-триптофана (1:1), характеризующаяся порошковой рентгенограммой, которая приведена на фиг.3.
21. Способ получения моноаммонийной соли H-D-iGlu-D-Trp-OH, свободной от неорганических солей, включающий следующие стадии:
(a) получение раствора кислотно-аддитивной соли H-D-iGlu-D-Trp-OH в водной среде, по существу, свободной от органического растворителя;
(b) коррекция pH до значений от приблизительно 3,9 до приблизительно 8,6 с помощью раствора гидроксида металла;
(c) обработка раствора со стадии (b) ионообменной смолой и элюирование водой для обмена иона металла из соли в растворе на ион водорода до тех пор, пока pH элюата не составит от приблизительно 5,7 до приблизительно 7,0;
(d) контакт ионообменной смолы с регенерирующим раствором на основе аммиака, применяемым для обмена ионов в нем на целевой H-D-iGlu-D-Trp-OH, содержащийся в ионообменной смоле, таким образом, чтобы образовался элюат регенерирующего раствора, содержащий аммонийную соль H-D-iGlu-D-Trp-OH; и
(e) выпаривание растворителя из раствора со стадии (d) с получением сырой аммонийной соли;
и дополнительно включающий следующие стадии:
(f) растворение аммонийной соли со стадии (е) в воде и медленное добавление изопропанола таким образом, что образуется осадок моноаммонийной соли; и
(g) вакуумная сушка продукта со стадии (f) с получением кристаллической формы аммонийной соли H-D-iGlu-D-Trp-OH (1:1); или
(h) обработка материала со стадии (е) хроматографией на силикагеле с применением раствора изопропанола и аммиака в качестве элюента, и удаление изопропанола путем выпаривания растворителя или растворение продукта со стадии (g) в воде с получением раствора аммонийной соли H-D-iGlu-D-Trp-OH (1:1) в воде; и
(i) лиофильная сушка продукта со стадии (h) с получением аморфной формы аммонийной соли H-D-iGlu-D-Trp-OH (1:1).
22. Способ по п.21, в котором используют по меньшей мере один раствор гидроксида металла, выбранный из раствора гидроксида натрия, раствора гидроксида лития и раствора гидроксида калия.
23. Способ по п.21 или 22, в котором кислотно-аддитивная соль представляет собой H-D-iGlu-D-Trp-OH гидрохлорид, полученный способом, включающим следующие стадии:
(i) основный гидролиз соединения формулы I:
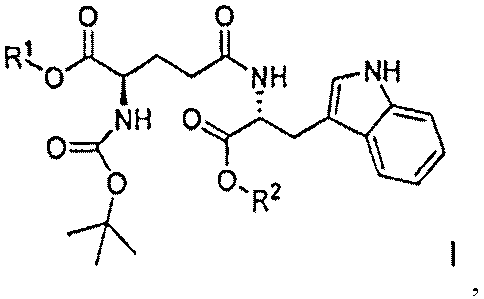
где R1 выбран из группы, состоящей из C1-C4алкила и бензила, и R2 представляет собой C1-C4алкил, при условии, что C4 алкил не является трет-бутилом,
с использованием гидроксида металла в воде и инертном растворителе, в присутствии метанола с получением Boc-D-iGlu-D-Trp-OH, свободного от других диастереомеров; и
(ii) снятие с помощью хлороводорода защиты с Boc-D-iGlu-D-Trp-OH со стадии (i) в инертном органическом растворителе; и выпаривание растворителя с получением гидрохлорида H-D-iGlu-D-Trp-OH.
24. Способ по п.21 или 22, в котором кислотно-аддитивная соль представляет собой H-D-iGlu-D-Trp-OH гидрохлорид, и раствор кислотно-аддитивной соли на стадии (а) получают способом, включающим:
(i) гидрирование соединения формулы II
где R1 представляет собой бензил, и R2 выбран из группы, состоящей из бензила и водорода, с использованием палладия на угле в метаноле или этаноле;
(ii) очистку сырого H-D-iGlu-D-Trp-OH со стадии (i) с помощью хроматографии на силикагеле с использованием изопропанола и воды в качестве элюента; и
(iii) обработку материала со стадии (ii) хлороводородной кислотой в воде с получением раствора соли гидрохлорида H-D-iGlu-D-Trp-OH в воде.
25. Способ по п.23, в котором R1 представляет собой бензил, и R2 представляет собой метил.
26. Способ по п.23, в котором инертный растворитель на стадии (i) выбран из группы, состоящей из метанола и метил-трет-бутилового эфира; и гидроксид металла выбран из группы, состоящей из гидроксида натрия, гидроксида лития и гидроксида калия.
27. Способ по п.21 или 22, в котором кислотно-аддитивная соль на стадии (а) представляет собой H-D-iGlu-D-Trp-OH гидрохлорид, значение pH, соответствующее преобладающей форме одновалентной соли на стадии (b), составляет от приблизительно 5,7 до приблизительно 7,0, и ионообменная смола на стадии (с) представляет собой AMBERLYST®15.
28. Способ получения моноаммонийной соли H-D-iGlu-D-Trp-OH из кристаллического H-D-iGlu-D-Trp-OH, свободного от неорганических солей, включающий следующие стадии:
(a) добавление кристаллического H-D-iGlu-D-Trp-OH к менее чем приблизительно одному эквиваленту раствора гидроксида аммония;
(b) коррекция pH до значений в интервале от приблизительно 6 до приблизительно 7 с помощью гидроксида аммония;
(c) выпаривание растворителя с получением масла; добавление изопропанола при перемешивании, чтобы вызвать осаждение моноаммонийной соли;
(d) выделение осажденной аммонийной соли H-D-iGlu-D-Trp-OH; и
(e) вакуумная сушка продукта, полученного на стадии (с), с получением моноаммонийной соли H-D-iGlu-D-Trp-OH.
29. Способ выделения кристаллического H-D-iGlu-D-Trp-OH из водного раствора H-D-iGlu-D-Trp-OH с использованием графического определения состава для определения процентного содержания H-D-iGlu-D-Trp-OH против pH, где процентное содержание форм H-D-iGlu-D-Trp-OH в растворе составляет более чем приблизительно 50% в растворе с pH от приблизительно 2,0 до приблизительно 3,2, что приводит к осаждению кристаллического H-D-iGlu-D-Trp-OH.
30. Соединение H-D-Glu-(γ-D-Trp-OR2)-α-OR1
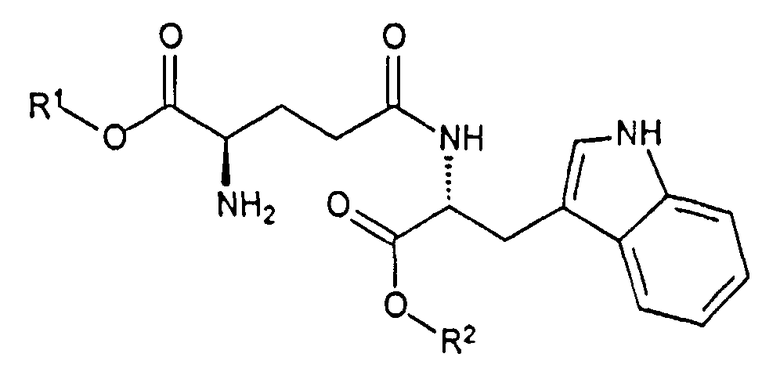
и его фармацевтически приемлемые кислотно-аддитивные соли, где каждый из R1 и R2 независимо выбран из группы, состоящей из бензила и C1-C4алкила.
31. Соединение по п.30, в котором C1-C4 алкил выбран из группы, состоящей из метила, этила, изопропила и трет-бутила.
32. Соединение по п.30 или 31, в котором R1 имеет то же значение, что и R2.
33. Соединение по п.32, в котором R1 и R2 представляют собой метил.
34. Соединение по п.32, в котором R1 и R2 представляют собой этил.
35. Соединение по п.32, в котором R1 и R2 представляют собой изопропил.
36. Соединение по п.32, в котором R1 и R2 представляют собой бензил.
37. Соединение по п.32, в котором R1 и R2 представляют собой трет-бутил.
38. Соединение по п.30 или 31, в котором R1 представляет собой этил и R2 представляет собой метил.
39. Соединение по п.30 или 31, в котором R1 представляет собой этил и R2 представляет собой изопропил.
40. Соединение по п.30 или 31, в котором R1 представляет собой трет-бутил и R2 представляет собой изопропил.
41. Соединение по п.30 или 31, в котором R1 представляет собой бензил и R2 представляет собой этил.
42. Соединение по п.30 или 31, в котором R1 представляет собой бензил или метил и R2 представляет собой этил, изопропил или трет-бутил.
43. Соединение по п.30 или 31, в котором R1 представляет собой бензил и R2 представляет собой метил.
44. Фармацевтическая композиция, являющаяся иммунодепрессантом или средством против псориаза, содержащая терапевтически эффективное количество соединения по любому из пп.30-43 по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
45. Применение терапевтически эффективного количества соединения по любому из пп.30-43 в качестве иммунодепрессанта или средства против псориаза.
46. Фармацевтическая композиция, являющаяся иммунодепрессантом или средством против псориаза, содержащая терапевтически эффективное количество аморфной моноаммонийной соли по п.19 или 20 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
47. Твердая фармацевтическая композиция, являющаяся иммунодепрессантом или средством против псориаза, содержащая терапевтически эффективное количество кристаллической соли по любому из пп.16-18 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
48. Применение аморфной моноаммонийной соли по п.19 или 20 в качестве иммунодепрессанта или средства против псориаза.
49. Применение кристаллической соли по любому из пп.16-18 в качестве иммунодепрессанта или средства против псориаза.
50. Твердая фармацевтическая композиция являющаяся иммунодепрессантом или средством против псориаза, содержащая терапевтически эффективное количество кристаллической соли соединения по любому из пп.1-3 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
51. Применение кристаллического соединения по любому из пп.1-3 в качестве иммунодепрессанта или средства против псориаза.
| US 5736519 A, 07.04.1998 | |||
| US 5744452 A, 28.04.1998 | |||
| Короткий Н.Г | |||
| и др | |||
| "Терапевтические возможности тимодепрессина у больных псориазом и механизмы его лечебного действия", Вестник дерматологии и венерологии, 2002, №4, с.58-60 | |||
| US 5916878 A, 29.06.1999 | |||
| US 5902790 A, 05.11.1999 | |||
| US 6410515 B1, 25.06.2002. |

