ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к новым кристаллическим и аморфным фармацевтически приемлемым солям D-изоглутамил-D-триптофана. В частности, данное изобретение касается калиевой соли D-изоглутамил-D-триптофана (1:1), литиевой соли D-изоглутамил-D-триптофана (1:1), кальциевой соли D-изоглутамил-D-триптофана (2:1), магниевой соли D-изоглутамил-D-триптофана (2:1) и органических аммонийных солей D-изоглутамил-D-триптофана (1:1), свойства которых улучшены по сравнению с аморфным D-изоглутамил-D-триптофаном, кристаллическим D-изоглутамил-D-триптофаном и динатриевой солью D-изоглутамил-D-триптофана. Данное изобретение также относится к способам получения таких новых солей D-изоглутамил-D-триптофана.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Соединение D-изоглутамил-D-триптофан (также известное как H-D-iGlu-D-Trp-OH или тимодепрессин) представляет собой синтетический геморегуляторный дипептид следующей формулы:

Тимодепрессин представляет собой свободную двухосновную кислоту, которой присвоен регистрационный номер® согласно Chemical Abstracts Service (CAS) 186087-26-3. Патент США 5736519 раскрывает H-D-iGlu-D-Trp-OH и способ его получения, в ходе которого соединение очищают ионообменной хроматографией. Соединение является иммунодепрессантом и выборочно ингибирует пролиферацию гемопоэтических клеток-прекурсоров и стимулирует апоптоз гранулоцитов и апоптоз лимфоцитов (Sapuntsova, S.G., et al. (May 2002), Bulletin of Experimental Biology and Medicine, 133(5), 488-490).
Тимодепрессин в настоящее время продается в России в виде динатриевой соли D-изоглутамил-D-триптофана в виде жидкого препарата для инъекций и в форме для интраназального применения для лечения псориаза и атопического дерматита. Твердая форма динатриевой соли D-изоглутамил-D-триптофана представляет собой аморфный порошок, который является гигроскопичным и очень сложным в обращении. Молекулярная формула динатриевой соли D-изоглутамил-D-триптофана C16H17N3Na2O5. Ее химическая структура представлена ниже:
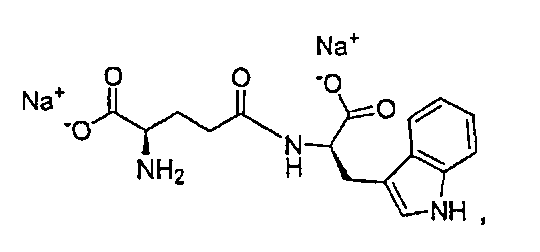
и описана в Kashirin, D.М., et al. (2000), Pharmaceutical Chemistry Journal, 34(11), 619-622.
При проведении исследований авторы изобретения установили, что лиофилизированная динатриевая соль D-изоглутамил-D-триптофана является чрезвычайно гигроскопичной, в течение нескольких минут превращается в гель на воздухе и является сложной в обращении.
Порошкообразная или аморфная форма соединения, предназначенного для фармацевтического применения, может приводить к производственным проблемам, связанным с насыпной плотностью, гигроскопичностью и переменным содержанием воды, которое не может быть отрегулировано сушкой в вакууме. D-изоглутамил-D-триптофан представляет собой дипептид, и сушка аморфной формы при повышенной температуре, например, 80-100°C, в вакууме не рекомендуется. Таким образом, существуют серьезные трудности, с которыми сталкиваются в ходе способа очистки динатриевой соли D-изоглутамил-D-триптофана и получения чистой динатриевой соли в производственных масштабах. Более того, не существует опубликованной методики ее получения.
Мононатриевая соль D-изоглутамил-D-триптофана идентифицирована Chemical Abstracts Services (CAS) и приведена в реестрах CAS REGISTRYSM под регистрационным номером CAS® 863988-88-9. Однако, нет никаких ссылок, где упоминалось бы вещество, и, следовательно, отсутствуют опубликованные данные относительно его идентичности, физических и/или химических свойств, характеристик или способа его получения. Лиофилизированный порошок мононатриевой и динатриевой соли пептидных лекарственных препаратов не может обеспечить контролируемого диапазона насыпной плотности порошка для рецептуры. Он может требовать значительных инвестиций в технологию диспергирования лиофилизированного материала.
Таким образом, существует необходимость в разработке альтернативных кристаллических фармацевтически приемлемых солей D-изоглутамил-D-триптофана. Такие кристаллические соли в целом могут быть очищены более простыми способами по сравнению с аморфной формой и могут обладать другими предпочтительными свойствами, например, с точки зрения их специфической кристаллической формы, и/или их характеристик растворимости, и/или отсутствия гигроскопичности, и/или их характеристик стабильности, в том числе свойств их термической стабильности, и/или их способности подвергаться окислительному разложению.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Задача настоящего изобретения состояла в разработке ряда новых стабильных, фармацевтически приемлемых солей тимодепрессина, пригодных для получения фармацевтических препаратов.
Не все соли D-изоглутамил-D-триптофана являются химически стабильными (например, аморфная динатриевая соль). Однако, авторы изобретения получили новые стабильные металлические и органические аммонийные соли D-изоглутамил-D-триптофана (H-D-i-Glu-D-Trp-OH), которые являются предметом настоящего изобретения.
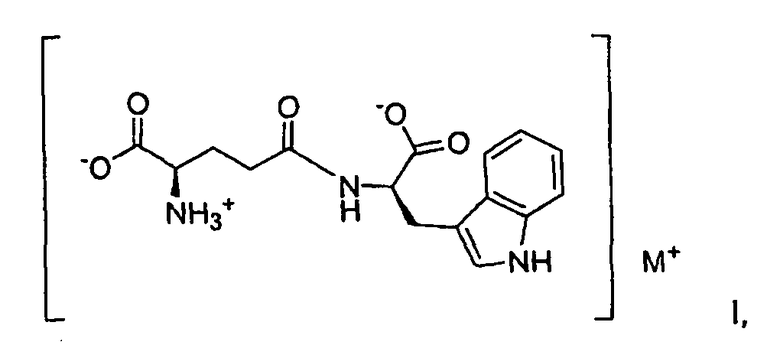
В одном аспекте по настоящему изобретению предлагаются новые соли D-изоглутамил-D-триптофана. Такие новые формы соли представляют собой соединения формулы I:

где M выбирают от группы, состоящей из лития и калия;
формулы II:
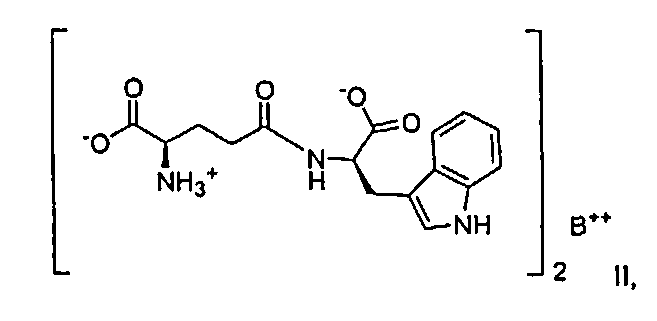
где B выбирают от группы, состоящей из магния и кальция; и формулы III:
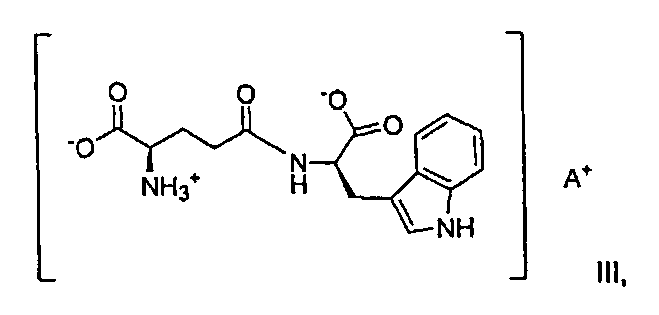
где A выбирают из группы, состоящей из трет-бутиламмония, трис(гидроксиметил)метиламмония и метил-(2,3,4,5,6-пентагидроксигексил)аммония.
Конкретно, в новых солях формулы I катион лития или калия заменяет один атом водорода в карбоксильном фрагменте соединения D-изоглутамил-D-триптофана. Авторы изобретения установили, что новые соли формулы I по настоящему изобретению, где M представляет собой калий (т.е. монокалиевая соль D-изоглутамил-D-триптофана), могут существовать в различных формах, в частности, в аморфной или в некристаллической форме, а также в кристаллической форме. Авторы изобретения также установили, что новые соли формулы I по настоящему изобретению, где M представляет собой литий (т.е. монолитиевая соль D-изоглутамил-D-триптофана), могут существовать в различных формах, в частности, в аморфной или некристаллической форме, а также в кристаллической форме. Таким образом, данное изобретение касается монолитиевых и монокалиевых солей D-изоглутамил-D-триптофана в любой из их форм.
В другом аспекте настоящего изобретения предлагаются новые литиевые соли и калиевые соли D-изоглутамил-D-триптофана, соединения формулы I.
В еще одном аспекте настоящего изобретения предлагается кристаллическая калиевая соль D-изоглутамил-D-триптофана, соединение формулы I.
В еще одном аспекте настоящего изобретения предлагается кристаллическая литиевая соль D-изоглутамил-D-триптофана, соединения формулы I.
Данное изобретение также касается кристаллической формы магниевой соли D-изоглутамил-D-триптофана и полукристаллической формы кальциевой соли D-изоглутамил-D-триптофана, где катион кальция или магния заменяет один атом водорода в карбоксильном фрагменте соединения D-изоглутамил-D-триптофана. Кальциевая или магниевая соль D-изоглутамил-D-триптофана образуется в соотношении 2:1, как иллюстрируется формулой II.
Авторы изобретения установили, что магниевая соль по настоящему изобретению (т.е. магниевая соль тимодепрессина [1:2]) существует в кристаллической форме, в то время как кальциевая соль по настоящему изобретению (т.е. кальциевая соль тимодепрессина [1:2]) является полукристаллической с процентом кристалличности, не превышающим примерно 67%.
В другом аспекте настоящего изобретения предлагается новая кристаллическая магниевая соль D-изоглутамил-D-триптофана, соединение формулы II.
В еще одном аспекте настоящего изобретения предлагается кальциевая соль D-изоглутамил-D-триптофана, соединение формулы II.
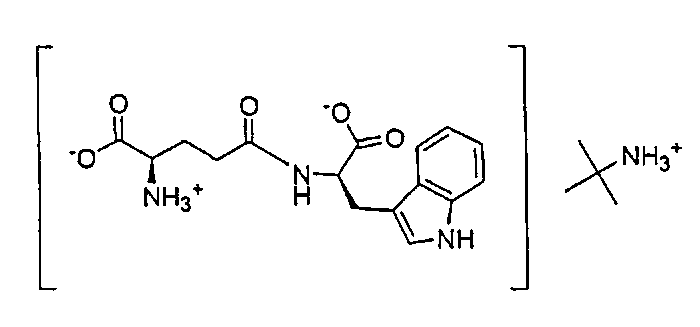
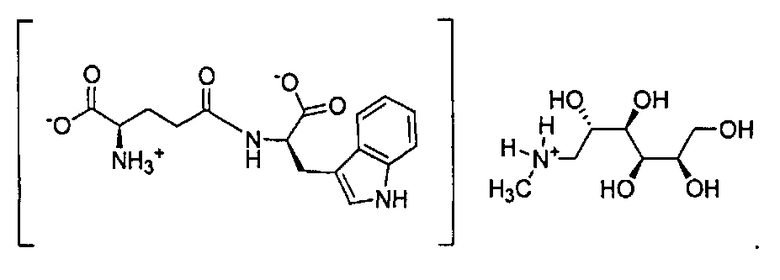
Данное изобретение также касается органических солей амина формулы III, где A выбирают из группы, состоящей из трет-бутиламмония, трис(гидроксиметил)метиламмония и метил-(2,3,4,5,6-пентагидроксигексил)-аммония. Авторы изобретения установили, что такие соли формулы III являются аморфными.
В другом аспекте настоящего изобретения предлагается новая трет-бутиламмонийная, трис(гидроксиметил)метиламмонийная, метил-(2,3,4,5,6-пентагидроксигексил)-аммонийная соль D-изоглутамил-D-триптофана, соединение формулы III.
В еще одном аспекте настоящего изобретения предлагается способ получения солей формул I, II и III из дипептида D-изоглутамил-D-триптофана.
В еще одном аспекте настоящего изобретения предлагается способ получения указанных солей D-изоглутамил-D-триптофана (соединение формулы I и II) в результате солевого обмена D-изоглутамил-D-триптофана аммонийной соли (1:1).
В более ранней патентной заявке, поданной в Канаде 28 ноября 2006 г., заявитель раскрывает способы получения D-изоглутамил-D-триптофана и его моноаммонийной соли, новой стабильной кристаллической формы D-изоглутамил-D-триптофана и его моноаммонийной соли. D-изоглутамил-D-триптофан и его моноаммонийная соль, используемая в настоящем изобретении, могут быть получены при помощи способа, описанного в вышеуказанной патентной заявке.
В другом аспекте настоящего изобретения предлагается фармацевтическая композиция, в состав которой входит любая из новых солей, описанных выше, и как минимум один фармацевтически приемлемый носитель.
Фармацевтическая композиция может быть получена путем объединения любой из новых солей, описанных выше, и как минимум одного фармацевтически приемлемого носителя. В другом аспекте настоящего изобретения предлагается способ создания фармацевтической композиции, в состав которой входит любая из новых солей, описанных выше, и как минимум один фармацевтически приемлемый носитель.
В другом аспекте настоящего изобретения предлагается применение любой из новых солей, описанных в данном изобретении, с получением лекарственного средства для лечения псориаза у субъекта, нуждающегося в таком лечении.
Другой признак кристаллических солей по настоящему изобретению состоит в том, что они могут также использоваться в качестве промежуточных соединений в получении некристаллической соли для того, чтобы обеспечить выделение некристаллической соли с уровнем чистоты и однородностью, которые подходят для препарата, соответствующего строгим фармацевтическим требованиям и спецификациям. Примерами данных солей являются литиевая соль, натриевая, калиевая и аммонийная соли. Методы перекристаллизации в общем удаляют примеси в ходе способа, в то время как очистка аморфной субстанции пептидного лекарственного средства требует препаративной обращенно-фазовой жидкостной хроматографии с высоким давлением, которая является не экономичной.
Другие и дополнительные преимущества и признаки настоящего изобретения будут очевидны специалистам в данной отрасли из следующего подробного описания, которое сопровождается соответствующими фигурами.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На приложенных фигурах:
На Фиг.1А представлена порошковая рентгенограмма (XRPD) калиевой соли D-изоглутамил-D-триптофана (1:1).
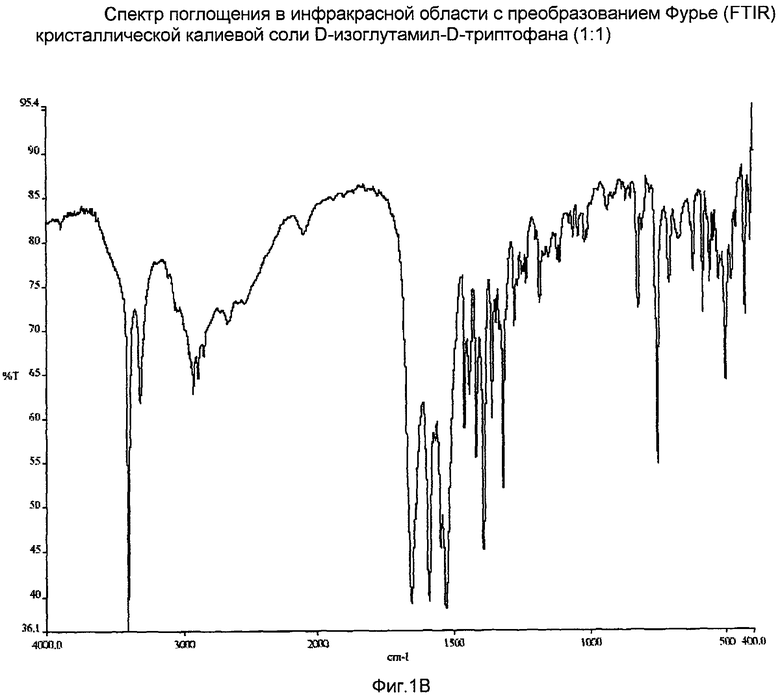
На Фиг.1В представлен инфракрасный спектр с преобразованием Фурье (FTIR) кристаллической калиевой соли D-изоглутамил-D-триптофана (1:1).
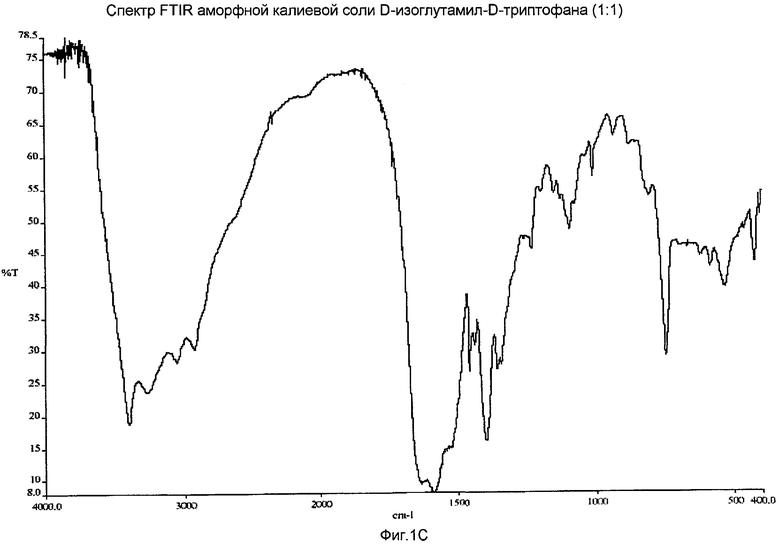
На Фиг.1C представлен спектр FTIR аморфной калиевой соли D-изоглутамил-D-триптофана (1:1).
На Фиг.2A представлена XRPD образца литиевой соли D-изоглутамил-D-триптофана (1:1).
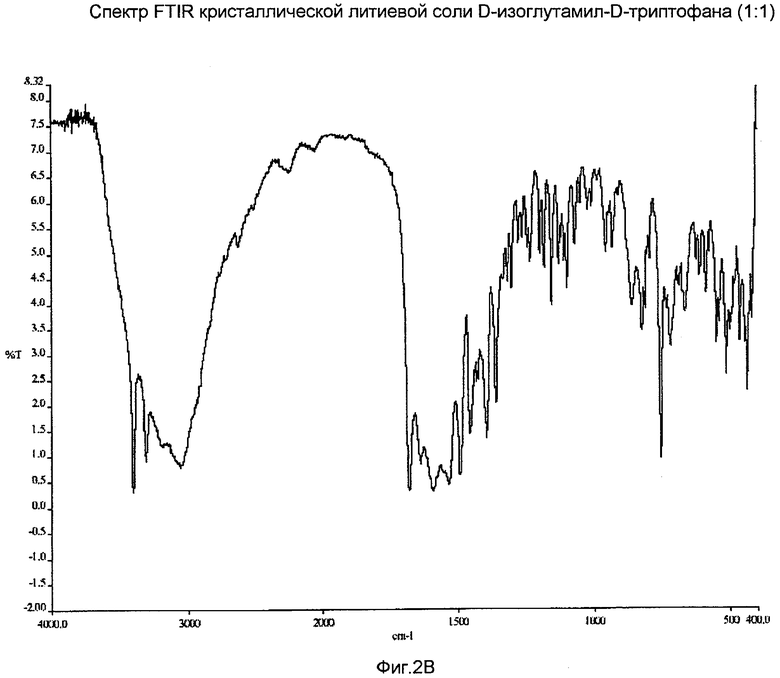
На Фиг.2B представлен спектр FTIR кристаллической литиевой соли D-изоглутамил-D-триптофана (1:1).
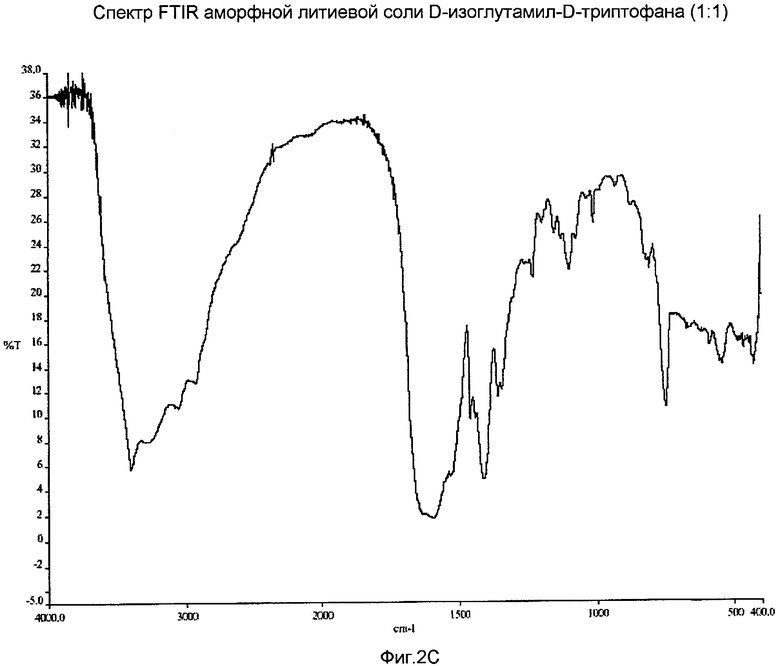
На Фиг.2C представлен спектр FTIR аморфной литиевой соли D-изоглутамил-D-триптофана (1:1).
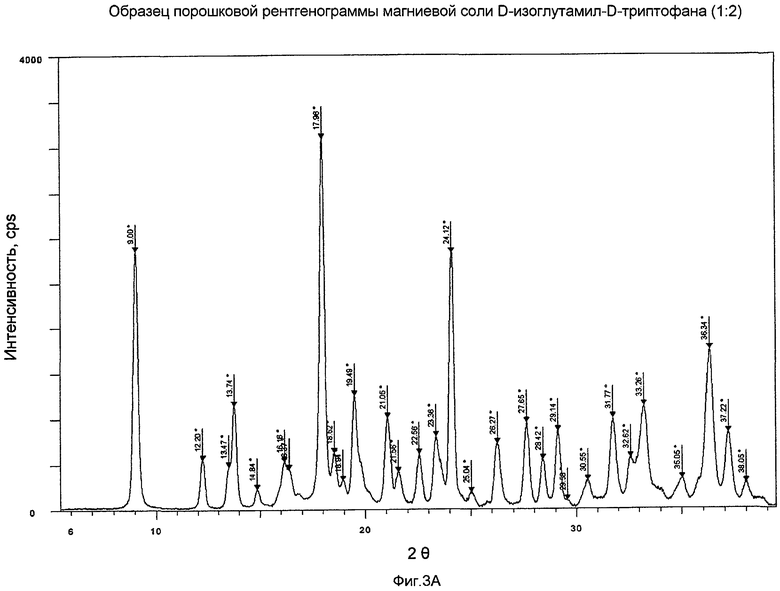
На Фиг.3A представлен XRPD магниевой соли D-изоглутамил-D-триптофана (1:2).
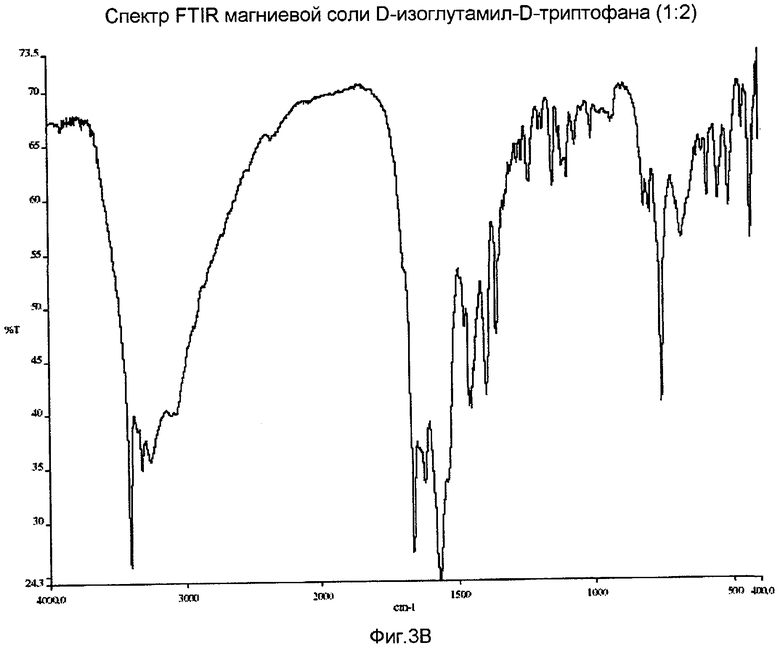
На Фиг.3B представлен спектр FTIR магниевой соли D-изоглутамил-D-триптофана (1:2).
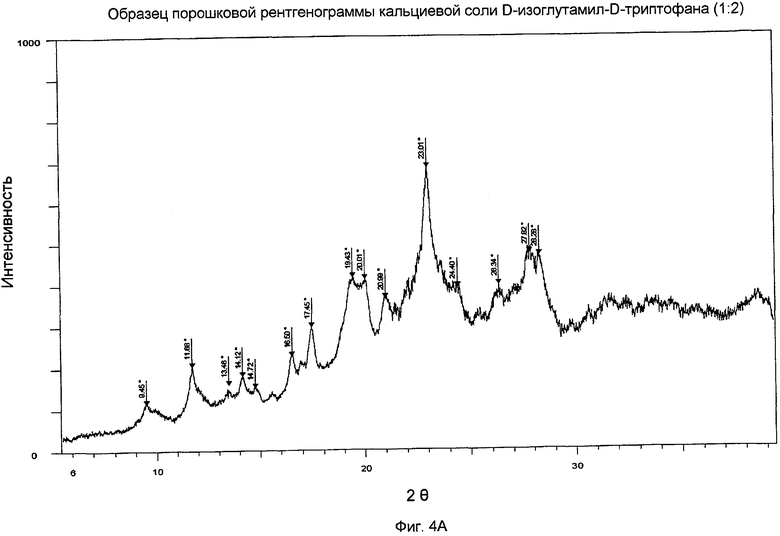
Фиг.4A представлен XRPD кальциевой соли D-изоглутамил-D-триптофана (1:2).
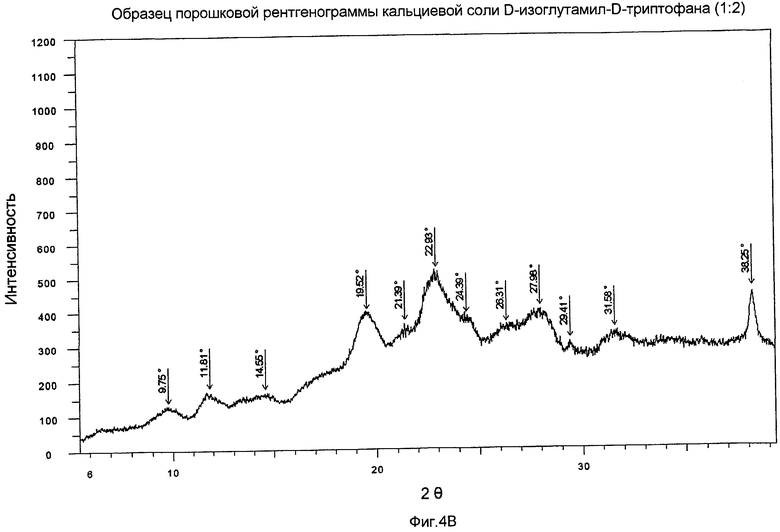
На Фиг.4В представлен XRPD кальциевой соли D-изоглутамил-D-триптофана (1:2).
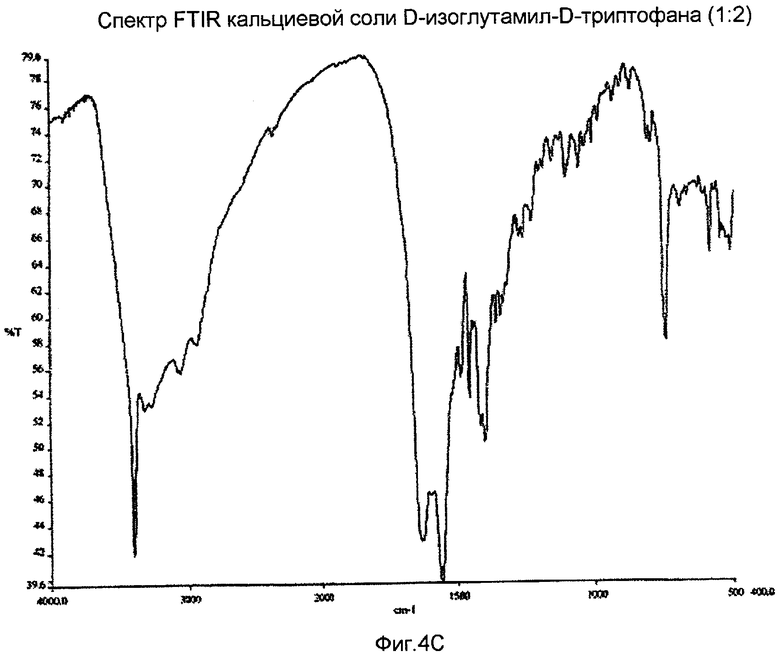
На Фиг.4C представлен спектр FTIR кальциевой соли D-изоглутамил-D-триптофана (1:2), полученный с использованием материала с фиг.4A.
На Фиг.5 показан график идентификации состава дипептида, D-изоглутамил-D-триптофана при различных показателях pH.
На Фиг.6 показан спектр FTIR аморфной соли трет-бутиламина и D-изоглутамил-D-триптофана (1:1).
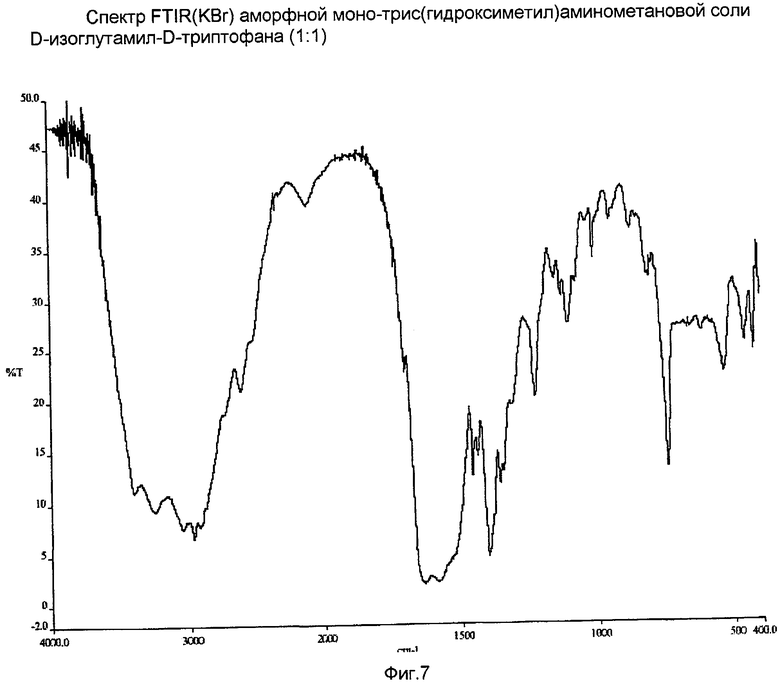
На Фиг.7 представлен спектр FTIR аморфной моно-трис(гидроксиметил)аминометановой соли D-изоглутамил-D-триптофана (1:1).
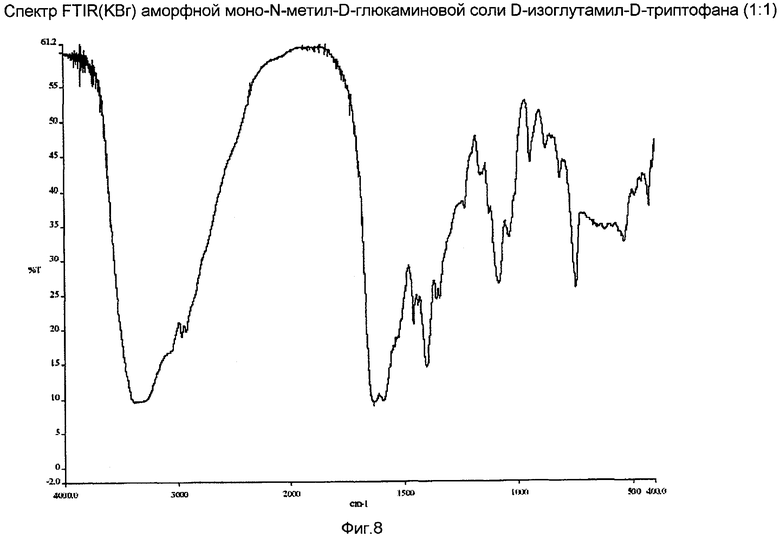
На Фиг.8 представлен спектр FTIR аморфной моно-N-метил-D-глюкаминовой соли D-изоглутамил-D-триптофана (1:1).
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ИЗОБРЕТЕНИЯ
Как было указанно выше, данное изобретение касается новых металлических солей D-изоглутамил-D-триптофана и органических солей амина D-изоглутамил-D-триптофана.
В данном изобретении D-изоглутамил-D-триптофан является свободной двухосновной кислотой
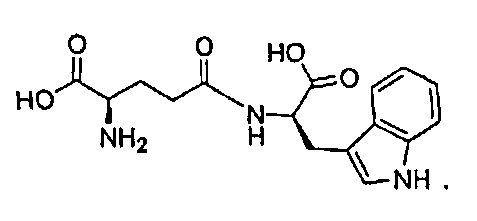
Химия аминокислот или простых дипептидов осложнена тем фактом, что группа -NH2 представляет собой основание и группа -CO2H представляет собой кислоту. Таким образом, в водном растворе ион H+ перемещается от одного конца молекулы к другому с образованием цвиттер-иона:

Цвиттер-ионы одновременно электрически заряжены и электрически нейтральны. Они содержат позитивные и негативные заряды, но суммарный заряд на молекуле равен нулю. Хотя образование соли не связано с какой-либо теорией, аминокислотный фрагмент iGlu H-D-iGlu-D-Trp-OH существует в виде цвиттер-иона, и, таким образом, присутствует только одна группа -CO2H, которая остается доступной для образования соли, если только 1 экв. одновалентного гидроксида металла, 0,5 экв. двухвалентного гидроксида металла B(OH)2 или один эквивалент органического амина используется для коррекции pH до нейтральных значений. Примерами одновалентных гидроксидов металлов являются гидроксид натрия, гидроксид лития и гидроксид калия. Примерами двухвалентных гидроксидов металлов являются гидроксид кальция и гидроксид магния.
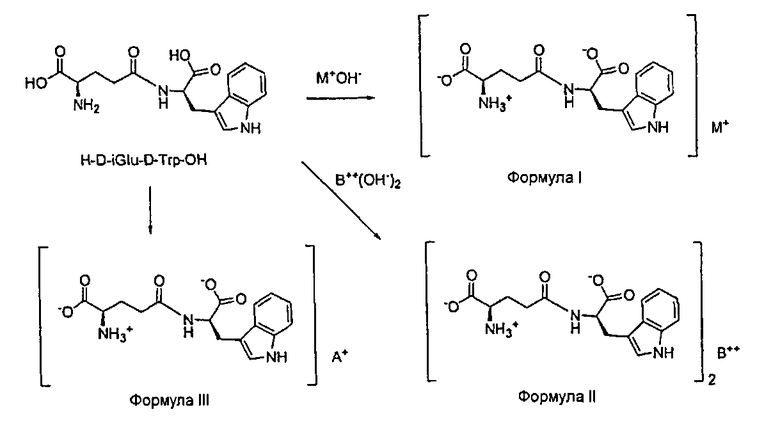
Если соль одновалентного металла и H-D-iGlu-D-Trp-OH формулы I изображается в формате, показанном выше, только одна группа CO2H может связаться с одним ионом одновалентного металла с образованием соли формулы I. Примерами таких одноосновных солей по настоящему изобретению являются калиевая и литиевая соли (1:1). Примерами двухвалентных металлических солей по настоящему изобретению являются магниевая и кальциевая соли. Примерами органических солей амина по настоящему изобретению являются соли трет-бутиламмония, трис(гидроксиметил)метиламмония и метил-(2,3,4,5,6-пентагидроксигексил)аммония.
Хотя в предшествующем уровне техники свободно используется термин «тимодепрессин» для обозначения как свободного двухосновного D-изоглутамил-D-триптофана, так и его динатриевой соли, в пределах контекста настоящего изобретения, тимодепрессин представляет собой свободную двухосновную кислоту D-изоглутамил-D-триптофана с молекулярной формулой C16H19N3O5, а динатриевая соль представляет собой соединение с молекулярной формулой C16H17N3Na2O5. Они представляют собой два различных химических вещества с различными физико-химическими свойствами.
В данном изобретении монолитиевая или монокалиевая соль образуется заменой одного атома водорода карбоксильной группы ионом металла, лития или калия, со структурой, показанной в формуле I выше. Конкретные структуры показаны ниже:
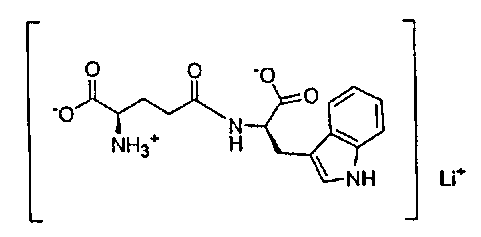
Литиевая соль формулы I
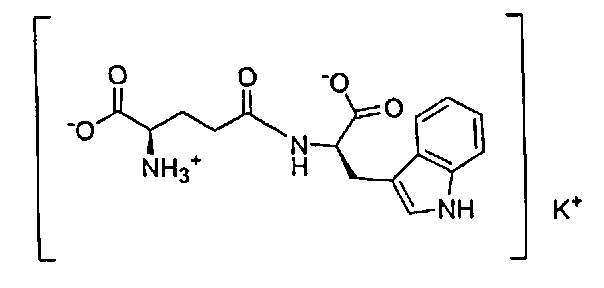
Калиевая соль формулы I
В данном изобретении магниевая или кальциевая соль образуется заменой одного атома водорода карбоксильной группы ионом металла, магния или кальция, со структурой, показанной в формуле II выше. Термин “кальциевая соль D-изоглутамил-D-триптофана (2:1)” в данном описании относится к Ca(D-изоглутамил-D-триптофан)2. Подобным образом, термин “магниевая соль D-изоглутамил-D-триптофана (2:1)” относится здесь к Mg(D-изоглутамил-D-триптофан)2. Конкретные структуры показаны ниже:
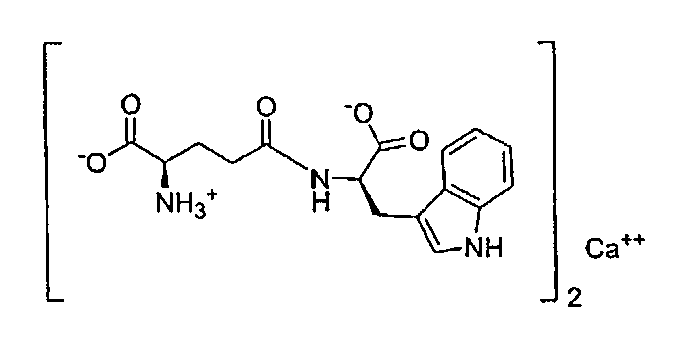
Кальциевая соль формулы II
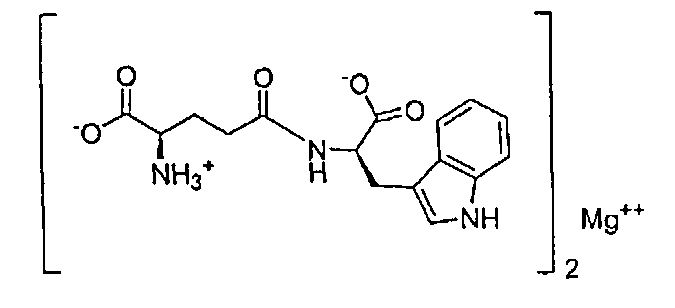
Магниевая соль формулы II
В данном изобретении органические соли амина относятся к солям пептида и органического амина. Например, трет-бутиламин, N-метил-D-глюкамин и трометамин представляют собой органические амины. Органическая соль амина по настоящему изобретению образована заменой одного атома водорода карбоксильной группы органическим амином со структурой, показанной в формуле III выше. Например, органическая соль амина, образованная из трет-бутиламина и D-изоглутамил-D-триптофана, в данном описании обозначает соль D-изоглутамил-D-триптофана трет-бутиламмония (1:1). Конкретная структура показана ниже:
Органическая соль амина по настоящему изобретению, образованная из N-метил-D-глюкамина и D-изоглутамил-D-триптофана, в данном описании обозначает соль D-изоглутамил-D-триптофана метил-(2,3,4,5,6-пентагидроксигексил)аммония (1:1). Конкретная структура показана ниже:
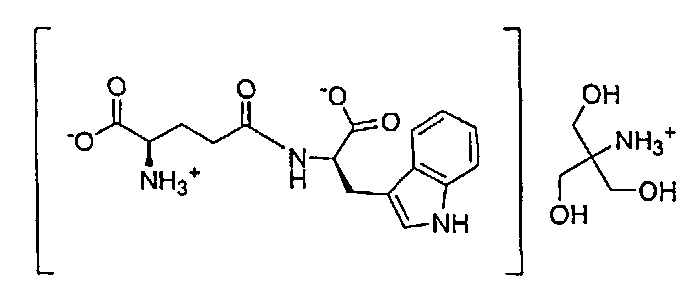
Органическая соль амина по настоящему изобретению, образованная из трометамина и D-изоглутамил-D-триптофана, в данном описании обозначает соль D-изоглутамил-D-триптофана трис(гидроксиметил)метиламмония (1:1). Конкретная структура показана ниже:
Фармацевтически приемлемая соль лекарственной субстанции является химически стабильной и может использоваться в фармацевтической композиции. В отличие от простых ароматических углеводородов, тимодепрессин представляет собой дипептид с несколькими функциональными группами. Дипептид D-изоглутамил-D-триптофан содержит альфа-аминогруппу, две карбоксильные группы и индольный азот в пределах одной молекулы. Идеальная соль должна быть такой, для которой значение pH раствора близко к 7 или находится в диапазоне слабоосновных значений pH. В ходе исследования авторы изобретения с целью разработки новой соли, свободной от недостатков динатриевой соли D-изоглутамил-D-триптофана, использовали графическое определение состава (фиг.5) для того, чтобы определить соли с идеальным pH раствора и растворимостью, приемлемые для фармацевтических препаратов. Фиг.5 представляет собой график идентификации состава, H-D-iGlu-D-Trp-ОН в интервале pH от 0 до 12 с использованием экспериментально определенных значений pKa. На фиг.5, LH2 = H-D-iGlu-D-Trp-OH, LH = соль монокарбоновой кислоты, L = соль дикарбоновой кислоты и LH3 = кислотно-аддитивная соль H-D-iGlu-D-Trp-OH. На оси X показано значение pH раствора, в то время как на оси Y показано количество форм, присутствующих при конкретном значении pH. Исходя из практического опыта авторов изобретения, они используют 6 мл воды на грамм H-D-iGlu-D-Trp-OH для целей выделения, что соответствует концентрации раствора 0,5 М.
Согласно графическому определению состава, показанному на фиг.5, pH для формы двухосновной кислоты составляет от приблизительно 2,7 до приблизительно 3 в растворе. pH раствора для металлической соли формулы I, где M представляет собой калий или литий; двухосновной соли формулы II, где В представляет собой кальций или магний, составляет приблизительно 7.
Другие соли, например, соли формулы IV
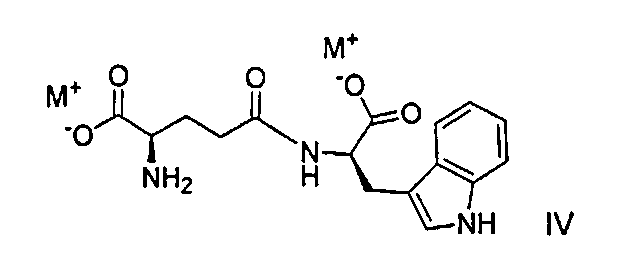
где M является таким, как определено выше, соли формулы V
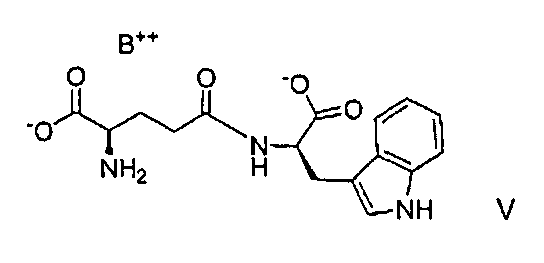
где B является таким, как определено выше, и соли формулы VI
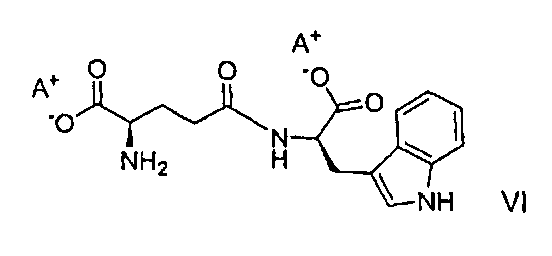
где A является таким, как определено выше, представляют собой новые соли.
Динатриевая соль (формула IV, где M = Na) является менее стабильной химической формой в качестве твердого вещества. Она чрезвычайно гигроскопична, и ее очень трудно отвешивать при исследовании рецептур. В растворе, в соответствии с графическим определением состава (фиг.5), pH превышает 9,0, и значение pH раствора должно быть доведено до значений от приблизительно 7,0 до приблизительно 7,4 при изготовлении препарата.
Как указано выше, авторы изобретения установили, что калиевая соль формулы I может существовать в аморфной или некристаллической форме, а также в кристаллической форме, в зависимости от условий, при которых она была получена, как описано более подробно ниже. Данное изобретение касается монокалиевой соли тимодепрессина в любой из ее форм.
В одном из вариантов настоящего изобретения предлагается калиевая соль формулы I в аморфной форме.
В другом варианте настоящего изобретения предлагается калиевая соль формулы I в кристаллической форме.
В другом варианте настоящего изобретения кристаллическая калиевая соль формулы I охарактеризована порошковой рентгенограммой, полученной при λ=1,542 Å с применением источника излучения Cu Kα, которая содержит пики в области следующих значений 2θ - 9,91, 14,84, 15,81, 18,97, 19,76, 24,04, 24,36, 24,82, 25,48, 27,49, 27,94, 28,42, 30,82, 31,28, 31,69, 32,17, 34,35, 35,81 и 36,96°.
В другом варианте настоящего изобретения кристаллическая калиевая соль формулы I характеризуется порошковой рентгенограммой, в существенной мере сходной с показанной на фиг.1А.
Как указано выше, авторы изобретения определили, что литиевая соль формулы I может существовать в аморфной или некристаллической форме, а также в кристаллической форме, в зависимости от условий, при которых она была получена, как описано более подробно ниже. Данное изобретение касается монолитиевой соли тимодепрессина в любой из ее форм.
В одном варианте настоящего изобретения предлагается литиевая соль формулы I в аморфной форме.
В другом варианте настоящего изобретения предлагается литиевая соль формулы I в кристаллической форме.
В другом варианте настоящего изобретения кристаллическая литиевая соль формулы I охарактеризована порошковой рентгенограммой, полученной при λ=1,542 Å с применением источника излучения Cu Kα, которая содержит пики в области следующих значений 2θ - 13,57, 15,53, 18,71, 20,11, 23,34, 24,1, 25,09, 27,31, 27,72, 28,39, 29,31, 30,19, 31,21, 32,06, 33,05, 33,62 и 37,41°.
В другом варианте настоящего изобретения кристаллическая литиевая соль формулы I характеризуется порошковой рентгенограммой, в существенной мере сходной с показанной на фиг.2А.
Как указано выше, авторы изобретения установили, что магниевая соль формулы II существует в кристаллической форме.
В другом варианте настоящего изобретения предлагается магниевая соль формулы II в кристаллической форме.
В другом варианте настоящего изобретения кристаллическая магниевая соль формулы II, которая охарактеризована порошковой рентгенограммой, полученной при λ=1,542 Å с применением источника излучения Cu Kα, которая содержит пики в области следующих значений 2θ - 12,2, 13,74, 14,84, 16,16, 17,96, 18,52, 18,94, 19,49, 21,05, 21,56, 22,56, 23,36, 24,12, 26,27, 27,65, 28,42, 29,14, 30,55, 31,77, 32,62, 33,26, 35,05, 36,34, 37,22 и 38,05°.
В другом варианте настоящего изобретения кристаллическая магниевая соль формулы II характеризуется порошковой рентгенограммой, в существенной мере сходной с показанной на фиг.3A.
Процент кристалличности тимодепрессина кальция или кальциевой соли формулы II по настоящему изобретению находится на уровне ниже приблизительно 67%, более предпочтительно - ниже приблизительно 50%, и наиболее предпочтительно - ниже приблизительно 25%.
В другом варианте настоящего изобретения предлагается кальциевая соль формулы II со степенью кристалличности ниже приблизительно 67%.
Общая кристалличность, измеренная методом порошкового рентгеноструктурного анализа, обеспечивает дополнительную полезную информацию для фармацевтических материалов, которые содержат какой-либо аморфный материал, образованный в ходе процедуры синтеза. Она также является ценным показателем с точки зрения контроля долгосрочных изменений в кристаллических материалах. Не будучи связанным с какими-либо конструктивными и композиционными особенностями, показатель "процент кристалличности" может быть подходящим индикатором стабильности конкретного материала как функции времени. Способ определения процента кристалличности соединения по настоящему изобретению описан в примере ниже. Характерные образцы XRPD кальциевой соли D-изоглутамил-D-триптофана показаны на фиг.4A и 4B.
В другом варианте настоящего изобретения предлагается соль трет-бутиламмония формулы III, как показано ниже:
В другом варианте настоящего изобретения предлагается соль трис(гидроксиметил)метиламмония формулы III, как показано ниже:
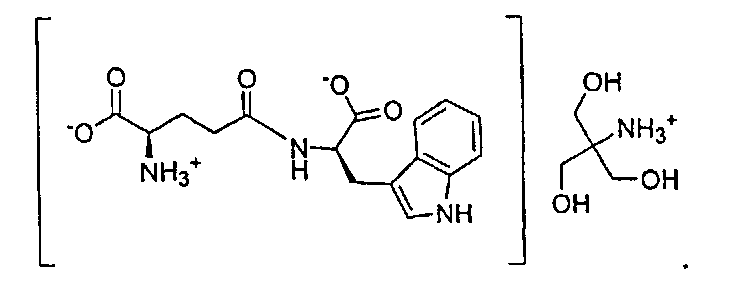
В другом варианте настоящего изобретения предлагается соль метил-(2,3,4,5,6-пентагидроксигексил)аммония формулы III, как показано ниже:
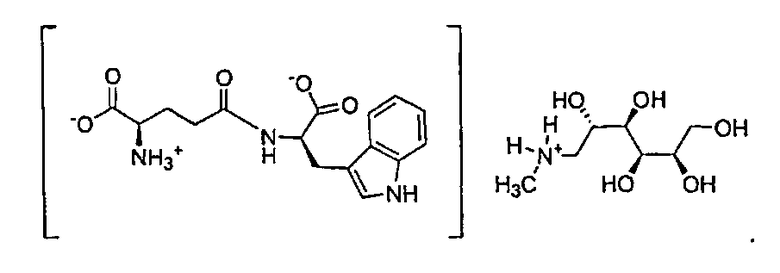
В другом варианте настоящего изобретения предлагается способ получения литиевой или калиевой соли формулы I, который включает (а) взаимодействие D-изоглутамил-D-триптофана в воде с гидроксидом лития или калия; (b) концентрирование раствора до образования масла и добавление изопропанола при перемешивании для осаждения соли; отделение полученного осадка; сушку продукта в вакууме с получением металлической соли формулы I, где М представляет собой литий или калий.
В другом варианте настоящего изобретения предлагается способ получения натриевой соли формулы IV, который включает (а) взаимодействие D-изоглутамил-D-триптофана в воде с гидроксидом натрия; (b) концентрирование раствора до образования масла и добавление изопропанола при перемешивании для осаждения соли; отделение полученного осадка; сушку продукта в вакууме с получением металлической соли формулы IV, где M представляет собой натрий.
В частности, соль карбоксилата металла формулы I образуется при взаимодействии смеси H-D-iGlu-D-Trp-OH с гидроксидом металла в количестве менее чем приблизительно 1 экв., например, гидроксидом калия или гидроксидом лития, и коррекцией pH при помощи того же гидроксида металла до приблизительно 7,0. Выпаривание растворителя дает масло, которое обрабатывают изопропанолом для осаждения твердой соли. Соль выделяют обычными средствами и сушат в вакууме с получением продукта формулы I.
В другом варианте настоящего изобретения предлагается способ получения кальциевой соли формулы II, который включает (a) взаимодействие D-изоглутамил-D-триптофана в воде с гидроксидом кальция; (b) концентрирование при перемешивании для осаждения соли; отделение полученного осадка и сушку продукта в вакууме с получением металлической соли формулы II, где В представляет собой кальций.
В частности, кальциевую соль получают смешиванием дипептида H-D-iGlu-D-Trp-OH с гидроксидом кальция, предпочтительно приблизительно 0,48-0,49 экв. гидроксида кальция на 1 экв. H-D-iGlu-D-Trp-OH, при температуре ледяной бани при перемешивании в течение нескольких часов, предпочтительно от приблизительно 2,5 до приблизительно 4 часов, с получением раствора. Предпочтительное количество воды составляет приблизительно 12,5 мл воды на 1 грамм H-D-iGlu-D-Trp-OH. pH раствора доводили до приблизительно 6 при помощи насыщенного раствора Ca(OH)2, и нерастворимые частицы отфильтровывали. Фильтрат упаривали до уровня от приблизительно 14 до приблизительно 16% от начального объема. При перемешивании в течение периода от приблизительно 14 до приблизительно 18 часов при комнатной температуре образуется твердое вещество, которое отфильтровывали. Кальциевую соль сушили в вакууме.
В другом варианте настоящего изобретения предлагается способ получения магниевой соли формулы II, который включает (a) взаимодействие D-изоглутамил-D-триптофана с этоксидом магния в изопропаноле; (b) концентрирование раствора с получением твердого вещества; смешивание раствора с водой; отфильтровывание нерастворимых частиц; разбавление фильтрата водой при перемешивании для осаждения соли; отделение полученного осадка и сушку в вакууме соли формулы II, где B представляет собой магний.
В частности, магниевую соль получают добавлением H-D-iGlu-D-Trp-OH к смеси этоксида магния в изопропаноле при температуре ледяной бани, предпочтительно используют приблизительно 0,48-0,49 экв. этоксида магния на 1 экв. H-D-iGlu-D-Trp-OH. Смесь перемешивают в течение периода от приблизительно 3 до приблизительно 10 часов, предпочтительно от приблизительно 4 до приблизительно 5 часов. pH раствора проверяли, отбирая образец и смешивая его с несколькими каплями воды. Добавляли дополнительное количество этоксида магния, предпочтительно, от приблизительно 0,1 до приблизительно 0,12 экв. этоксида магния, и перемешивали в течение периода от приблизительно 10 до приблизительно 18 часов, предпочтительно, в течение периода от приблизительно 14 до приблизительно 16 часов. pH раствора проверяли, отбирая образец и смешивая его с несколькими каплями воды, pH составлял приблизительно 7,0. Раствор упаривали при пониженном давлении с получением твердого вещества, которое растворяется в воде. Нерастворимые частицы отфильтровывали, и фильтрат упаривали с получением твердого вещества. Твердое вещество перемешивали с водой для образования суспензии, и далее перемешивали в течение периода от приблизительно 3 до приблизительно 6 часов с образованием осадка. Соль магния отфильтровывали и сушили в вакууме.
В другом варианте настоящего изобретения предлагается способ получения органической соли амина формулы III, который включает (a) взаимодействие D-изоглутамил-D-триптофана с органическим амином в воде, где органический амин представляет собой трет-бутиламин, или N-метилглюкамин, или трометамин; и (b) концентрирование раствора, совместное испарение с изопропанолом; добавление ацетона для осаждения соли; отделение полученного осадка и сушку продукта в вакууме с получением органической аммонийной соли формулы III, где A представляет собой трет-бутиламмоний или трис(гидроксиметил)метиламмоний или метил-(2,3,4,5,6-пентагидроксигексил)аммоний.
В частности, соединение формулы III, где противоион представляет собой органический амин, получали смешиванием органического амина с H-D-iGlu-D-Trp-OH в воде при температуре окружающей среды, перемешивая смесь в течение периода времени от приблизительно 12 до приблизительно 18 часов. Растворитель упаривали совместно с изопропанолом и упаривали в вакууме с получением твердого вещества, которое перемешивали с ацетоном и фильтровали. Примеры органических аминов выбраны из группы, состоящей из трис(гидроксиметил)аминометана, N-метилглюкамина и трет-бутиламина.
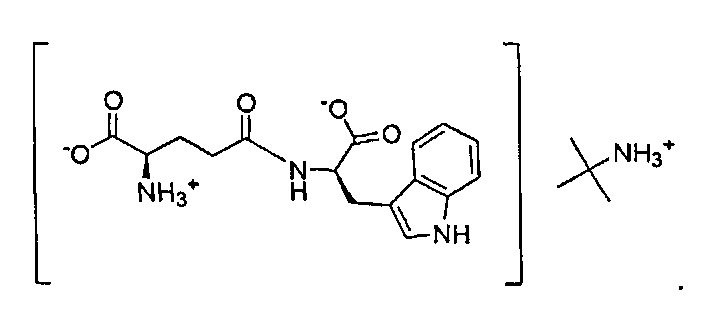
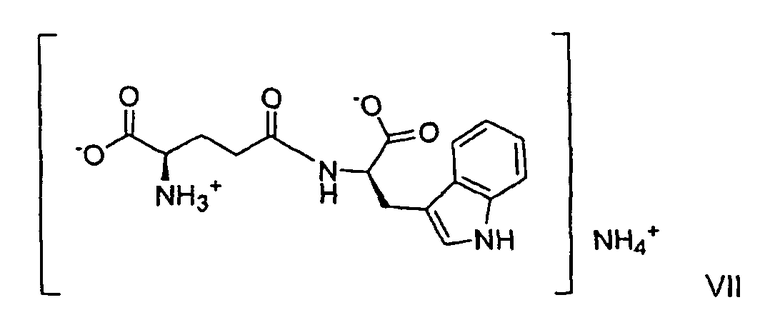
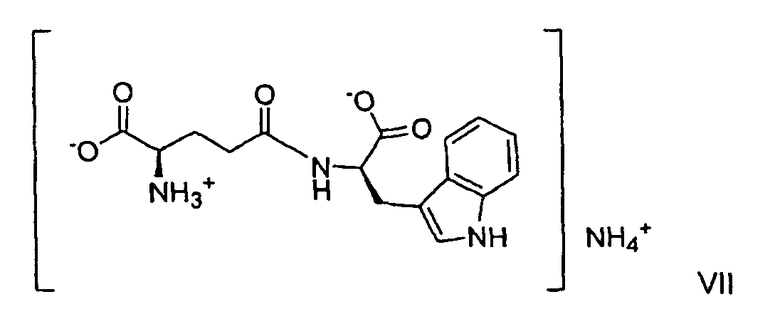
В другом варианте настоящего изобретения предлагается способ солевого обмена для получения соли D-изоглутамил-D-триптофана, который включает (a) взаимодействие аммонийной соли, представленной формулой VII,
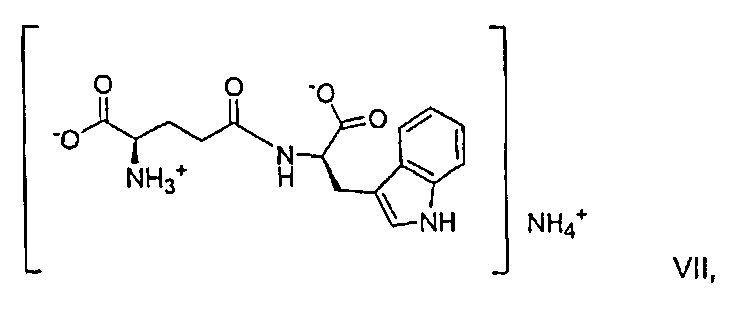
в водном растворе с приблизительно 1 экв. гидроксида металла, представленным MOH, где M представляет собой калий или литий; и (b) выпаривание растворителя с получением твердого вещества, которое перемешивают с водой и изопропанолом с получением соединения формулы I, где М представляет собой калий или литий.
В другом варианте настоящего изобретения предлагается способ солевого обмена для получения соли D-изоглутамил-D-триптофана, который включает (a) взаимодействие аммонийной соли, представленной формулой VII, в водном растворе с приблизительно 0,5 экв. гидроксида металла B(OH)2, где B представляет собой кальций или магний; (b) выпаривание растворителя с получением твердого вещества, которое перемешивают с водой и изопропанолом с получением соединения формулы II, где В представляет собой кальций или магний.
Соединение формулы I может быть получено солевым обменом. Стабильная аммонийная соль H-D-iGlu-D-Trp-OH используется в качестве исходного материала. Ниже представлен характерный способ получения соединения формулы I. Раствор H-D-iGlu-D-Trp-OH, аммонийной соли (1:1) перемешивают с гидроксидом металла в воде и перемешивают в течение периода от приблизительно 15 минут до приблизительно 2 часов, предпочтительно, в течение периода от приблизительно 15 минут до приблизительно 45 минут. Растворитель удаляют выпариванием и оставшуюся жидкость перемешивают с изопропанолом с получением осадка. Гидроксид металла выбирают из группы, состоящей из гидроксида лития и гидроксида калия. Порошковые рентгенограммы D-изоглутамил-D-триптофана калиевой соли (1:1) и D-изоглутамил-D-триптофана литиевой соли (1:1) показывают, что они представляют собой кристаллические вещества.
Если раствор H-D-iGlu-D-Trp-OH, аммонийной соли (1:1) смешивают с гидроксидом металла в воде, с последующим перемешиванием в течение периода от приблизительно 15 минут до приблизительно 2 часов, предпочтительно, в течение периода времени от приблизительно 15 минут до приблизительно 45 минут, а затем высушивают лиофильной сушкой, материал, полученный таким образом, является аморфным. Аморфные формы натриевых, калиевых или литиевых солей H-D-iGlu-D-Trp-OH могут быть получены данным способом.
Соединение формулы II может быть получено в результате обмена аммонийной соли с гидроксидом кальция или гидроксидом магния. Например, суспензию H-D-iGlu-D-Trp-OH и металлический гидроксид в воде, например, гидроксид кальция или гидроксид магния, нагревают до приблизительно 50-65°C в течение периода от приблизительно 1 до приблизительно 4 часов. Растворитель удаляют выпариванием. Оставшуюся жидкость перемешивают с изопропанолом для осаждения H-D-iGlu-D-Trp-OH металлической соли (2:1). Если металл представляет собой магний, полученная металлическая соль H-D-iGlu-D-Trp-OH магния (2:1) является кристаллической, что подтверждается порошковой рентгенограммой. Если металл представляет собой кальций, выделенный H-D-iGlu-D-Trp-OH кальций (2:1) является полукристаллическим веществом, со степенью кристалличности менее чем приблизительно 67%.
14N-ЯМР представляет собой полезную методику характеристики моноаммонийной соли тимодепрессина. Металлические соли формулы I и II, полученные указанным выше способом, в существенной мере свободны от аммонийной соли, о чем свидетельствует отсутствие сигнала NH4 + по данным 14N-ЯМР.
Авторы изобретения использовали графический метод определения состава (фиг.5) для того, чтобы вычислить диапазон pH для солевых форм дипептида H-D-iGlu-D-Trp-OH. Как показано на фиг.5, соль формулы I, или II, или III преобладает при значениях pH от приблизительно 6 до приблизительно 8 и делает их идеальными кандидатами для применения препарата или введения в фармацевтические композиции. Она является особенно подходящей для жидких препаратов, сублингвальных таблеток, назальных капель и спреев.
ПРИМЕНЕНИЕ И ВВЕДЕНИЕ
Калиевые, литиевые, кальциевые, магниевые соли D-изоглутамил-D-триптофана и органические соли амина и D-изоглутамил-D-триптофана по настоящему изобретению могут быть включены в фармацевтические композиции для введения субъекту в терапевтически активном количестве и в биологически совместимой форме, пригодной для введения in vivo, т.е. в форме пептидов для введения, в которой терапевтический эффект превосходит любые токсические эффекты.
В соответствии с методом графического определения состава, как показано на фиг.5, преобладающими формами при нейтральных значениях pH являются монокарбоксилатная форма тимодепрессина, то есть мононатриевая соль дипептида D-изоглутамил-D-триптофана, если противоион представляет собой натрий. Динатриевая соль D-изоглутамил-D-триптофана является чрезвычайно гигроскопичной, и ее очень сложно отмеривать.
Аморфная или кристаллическая форма солей по настоящему изобретению представляют собой идеальные кандидатуры для замены динатриевой соли при изготовлении различных препаратов, пригодных для лечения таких же состояний и/или заболеваний, для лечения которых применяют тимодепрессин, например, псориаза. Введение новых кристаллических и аморфных солей по настоящему изобретению, как описано в данном описании, может быть осуществлено любым из общепринятых способов введения терапевтических лекарственных средств с системной активностью. Указанные способы включают лекарственные формы для перорального, парентерального и других видов системного или местного применения или применения в форме аэрозоля.
В зависимости от предусмотренного способа введения композиции для применения могут существовать в форме твердых, полутвердых или жидких лекарственных форм, таких как таблетки, суппозитории, пилюли, капсулы, порошки, жидкости, аэрозоли, суспензии и т.п., предпочтительно в дозированных лекарственных формах, подходящих для однократного введения точных доз. Композиции будут содержать как минимум один традиционный фармацевтический носитель или вспомогательное вещество и кристаллический тимодепрессин или его фармацевтически приемлемую моноаммонийную соль, и дополнительно могут содержать другие лекарственные средства, фармацевтические средства, носители, адъюванты и т.п.
Для твердых композиций могут быть использованы традиционные нетоксичные твердые носители, которые включают, например, маннит, лактозу, крахмал, магния стеарат, натрия сахаринат, тальк, целлюлозу, глюкозу, сахарозу, магния карбонат и т.п., фармацевтической категории. Активное соединение, как определяется выше, может быть введено в суппозитории, с использованием, например, полиалкиленгликолей, например, пропиленгликоля, как носителя. Подходящие для фармацевтического введения жидкие композиции могут, например, быть получены путем растворения, диспергирования и т.п., активного соединения, как определено выше, и необязательных фармацевтических адъювантов в носителе, таком как вода, солевой раствор, водный раствор глюкозы, глицерин, этанол и т.п., с образованием таким способом раствора или суспензии. По желанию, фармацевтическая композиция для введения может также содержать незначительные количества нетоксичных вспомогательных субстанций, таких как увлажняющие или эмульгирующие средства, буферизующие средства и т.п., например, натрия ацетат, сорбитана монолаурат, натрия триэтаноламина ацетат, триэтаноламина олеат и т.п. Фактические способы получения таких лекарственных форм известны или будут очевидными для специалиста в данной отрасли; например, см. Remington: The Science and Practice of Pharmacy, 21st Edition, 2006, Part 5, Pharmaceutical Manufacturing, Chapters 37, 39, 41-47 and 50, pp.702-719, 745-775, 802-938 и 1000-1017 (ранее известна как Remington's Pharmaceutical Sciences), David B. Troy (Ed.), Lipincott Williams & Wilkins, Балтимор, Мериленд. Композиция или рецептура для введения в любом случае будут содержать количество активного(ых) соединения(й) в количестве, эффективном для облегчения симптомов у субъекта, нуждающегося в лечении.
Парентеральное введение в целом характеризуется инъекцией, подкожной, внутримышечной или внутривенной. Инъекционные средства могут быть введены в традиционные формы, такие как жидкие растворы или суспензии, твердые формы, пригодные для изготовления раствора или суспензии в жидкости перед инъекцией, или как эмульсии. Пригодными вспомогательными веществами, например, являются вода, солевой раствор, декстроза, глицерин, этанол и т.п. Кроме того, по желанию, фармацевтические композиции для введения могут также содержать незначительные количества нетоксичных вспомогательных веществ, таких как увлажняющие или эмульгирующие средства, pH буферизующие средства и т.п., такие как натрия ацетат, сорбитана монолаурат, триэтаноламина олеат и т.п.
Для солей по настоящему изобретению предпочтительно пероральное или назальное (бронхиальное) введение, в зависимости от природы расстройства, подлежащего лечению.
Для перорального введения фармацевтически приемлемую нетоксичную композицию получают, соединяя какое-либо из обычно используемых вспомогательных веществ фармацевтической категории, такое как маннит, лактоза, крахмал, магния стеарат, натрия сахаринат, тальк, целлюлоза, глюкоза, сахароза, магния карбонат и т.п. Такие композиции могут существовать в форме растворов, суспензий, таблеток, пилюль, капсул, порошков, рецептур с пролонгированным высвобождением и т.п. Такие композиции могут содержать от приблизительно 1% до приблизительно 95% активного ингредиента, предпочтительно, от приблизительно 25% до приблизительно 70%.
Пероральное и назальное введение в легкие может также осуществляться с помощью аэрозольных лекарственных форм. Для аэрозольного введения активный ингредиент предпочтительно поставляется в тонко измельченной форме вместе с поверхностно-активным веществом и пропеллентом. Типичное процентное содержание активных ингредиентов составляет от приблизительно 0,01 до приблизительно 20% по массе, предпочтительно, от приблизительно 0,04% до приблизительно 1,0%.
Поверхностно-активные вещества обычно должны быть нетоксичными, и предпочтительно растворимыми в пропелленте. Характерным примером таких средств являются эфиры или частичные эфиры жирных кислот, содержащих от 6 до 22 атомов углерода, таких как капроновая, каприловая, лауриновая, пальмитиновая, стеариновая, линолевая, линоленовая, олестеариновая и олеиновая кислоты, с многоатомным алифатическим спиртом или его циклическим ангидридом, таким как этиленгликоль, глицерин, эритритол, арабитол, маннит, сорбит, ангидрид гекситола, полученный из сорбита (эфиры сорбитана, присутствующие на рынке под названием SPANS®) и полиоксиэтиленовые и полиоксипропиленовые производные указанных эфиров. Могут использоваться смешанные эфиры, такие как смешанные или природные глицериды. Предпочтительными поверхностно-активными агентами являются олеаты или сорбитан, например, присутствующие на рынке под названиями ARLACEL® C (сорбитана сесквиолеат), SPAN® 80 (сорбитана моноолеат) и SPAN® 85 (сорбитана триолеат). Содержание поверхностно-активного вещества может составлять от приблизительно 0,1% до приблизительно 20% по массе композиции, предпочтительно, от приблизительно 0,25% до приблизительно 5%.
Остальная часть композиции обычно представляет собой обычный пропеллент. Сжиженные пропелленты обычно являются газообразными в условиях окружающей среды и конденсируются под давлением. Среди пригодных сжиженных пропеллентов - низшие алканы, содержащие до пяти атомов углерода, такие как бутан и пропан; и, предпочтительно, фторированные или фторхлорированные алканы, такие как присутствующие на рынке под названием FREON®. Смеси вышеуказанных компонентов также могут быть использованы.
При получении аэрозоля емкость, оборудованную пригодным клапаном, заполняют соответствующим пропеллентом, содержащим тонко измельченный активный ингредиент и поверхностно-активное вещество. Таким образом, ингредиенты содержатся при повышенном давлении до выброса при срабатывании клапана.
Для местного применения такие композиции содержат эффективное количество соединения данного класса в смеси как минимум с одним фармацевтически приемлемым нетоксичным носителем. Пригодный для композиции интервал составляет от приблизительно 0,1% до приблизительно 10% активного ингредиента; остальная часть композиции представляет собой носители; предпочтительно, от приблизительно 1% до приблизительно 2% активного ингредиента. Концентрация активного ингредиента в фармацевтических композициях, пригодных для местного применения, будет варьировать в зависимости от активности конкретного соединения, которое применяют в соответствии с присутствующим состоянием и субъектом, который подлежит лечению. Подходящие носители или растворители лекарственного средства для местного применения указанных соединений включают кремы, мази, лосьоны, эмульсии, растворы и т.п.
Например, пригодная мазь для местного применения соединений по данному изобретению содержит от приблизительно 15 до приблизительно 45% насыщенного жирного спирта, содержащего 16-24 атома углерода, такого как цетиловый спирт, стеариловый спирт, бегениловый спирт и т.п., и от приблизительно 45 до приблизительно 85% по массе гликолевого растворителя, такого как пропиленгликоль, полиэтиленгликоль, дипропиленгликоль и смеси указанных компонентов. Мазь может также содержать от приблизительно 0 до приблизительно 15% по массе пластификатора, такого как полиэтиленгликоль, 1,2,6-гексантриол, сорбит, глицерин и т.п.; от приблизительно 0 до приблизительно 15% по массе связующего средства, такого как насыщенные жирные кислоты, содержащие от 16 до 24 атомов углерода, например, стеариновая кислота, пальмитиновая кислота, бегеновая кислота, амиды жирных кислот, например, олеамид, пальмитамид, стеарамид, бегенамид и сложные эфиры жирных кислот, содержащие 16-24 атомов углерода, такие как сорбита моностеарат, полиэтиленгликоля моностеарат, полипропиленгликоль или соответствующий моноэфир других жирных кислот, таких как олеиновая кислота и пальмитиновая кислота; и от приблизительно 0 до приблизительно 20% по массе средства, усиливающего проникновение, такого как диметилсульфоксид или диметилацетамид.
Терапевтически активное количество солей по настоящему изобретению может варьироваться в соответствии с такими факторами, как патологическое состояние, возраст, пол и масса тела индивидуума. Схема лечения может быть изменена таким образом, чтобы обеспечить оптимальный терапевтический ответ. В целом, схема ежедневного введения должна включать дозу в интервале от приблизительно 1 до приблизительно 200 мг пептида.
Далее приведены примеры характерных рецептур, которые никоим образом не ограничивают контекст получения разнообразных фармацевтических композиций по настоящему изобретению:
Перечисленные выше ингредиенты тщательно смешивают и прессуют в таблетки с одной риской.
Перечисленные выше ингредиенты смешивают и вводят в твердую желатиновую капсулу.
Перечисленные выше ингредиенты смешивают и прессуют в таблетки с одной риской.
Перечисленные выше ингредиенты смешивают и вводят в твердую желатиновую капсулу.
Перечисленные выше ингредиенты смешивают и вводят в твердую желатиновую капсулу.
Инъекционный препарат, буферизованный до pH приблизительно 7, получают со последующим составом:
количество до pH 7
количество до 20 мл
Инъекционный препарат, буферизованный до pH приблизительно 7, получают со последующим составом:
количество до 1 мл
количество до pH 7
Суспензию для перорального применения получают со следующим составом:
Препарат для местного применения:
Все приведенные выше ингредиенты, кроме воды, объединяют и нагревают приблизительно до 45°C при перемешивании. Затем добавляют достаточное количество воды с температурой приблизительно 45°C при энергичном перемешивании для эмульгации ингредиентов и далее добавляют необходимое количество воды до 100 г.
Ниже данное изобретение поясняется подробно со ссылкой на Примеры, но данное изобретение никоим образом не ограничивается ими.
В данном изобретении предлагается фармацевтическая композиция, которая содержит литиевую или калиевую соль формулы I в любой из ее форм и одно или больше фармацевтически приемлемых вспомогательных веществ.
В данном изобретении также предлагается фармацевтическая композиция, которая содержит кальциевую или магниевую соль формулы II в любой из ее форм и одно или больше фармацевтически приемлемых вспомогательных веществ.
В данном изобретении дополнительно предлагается фармацевтическая композиция, которая содержит органическую соль амина формулы III в любой из ее форм и одно или больше фармацевтически приемлемых вспомогательных веществ.
Дальнейшие детали предпочтительных вариантов по настоящему изобретению проиллюстрированы в следующих примерах, которые следует трактовать как не ограничивающие относительно приложенной формулы изобретения.
ПРИМЕРЫ
Пример 1:
Получение калиевой соли D-изоглутамил-D-триптофана (1:1) из D-изоглутамил-D-триптофана и гидроксида калия
В круглодонную колбу объемом 100 мл, оборудованную магнитной мешалкой, помещали 5 мл раствора гидроксида калия (0,5 н). Раствор охлаждали до 0°C на ледяной бане, и к нему добавляли твердый H-D-iGlu-D-Trp-OH (1,00 г, 3 ммоль). Смесь перемешивали и одновременно pH раствора доводили до приблизительно 6,0, добавляя несколько капель раствора гидроксида калия (0,5 н). Раствор фильтровали для удаления частиц твердого вещества. Фильтрат упаривали до сухого состояния при температуре бани приблизительно 30°C с получением твердого вещества. После сушки в вакууме при комнатной температуре в течение ночи получали соль с количественным выходом и чистотой ВЭЖХ (процент области пика) 98,3%. Метод ВЭЖХ; колонка: XTerra МС C18; 5 мкм, 4,6×250 мм; подвижная фаза: A = водная фаза: 4 мМ Трис, 2 мМ ЭДТА, pH 7,4; B = органическая фаза: CH3CN; градиент: B%: 0 мин 5%, 15 мин 55%, 30 мин 55%, 32 мин 5%, 35 мин 5%; скорость потока: 1 мл/мин; объем инъекции: 5 мкл; λ: 222, 254, 282, 450 нм; время удерживания продукта: 6,41 мин. Образец XRPD данного кристаллического материала показан на фиг.1A; содержание воды по данным анализа методом Карла-Фишера составляло 0,7%; УФ (вода, c = 23,8 мкМ, λmax нм): 221 (ε 33270), 280 (ε 5417); МС (соотношение массы и заряда): 372,0 [M]+, 334,2 [C16H20N3O5]+, 187,9 (100%). Спектр FTIR (KBr) показан на фиг.1B.
Пример 2:
A. Получение монокалиевой соли D-изоглутамил-D-триптофана (1:1) из моноаммонийной соли D-изоглутамил-D-триптофана (1:1)
Раствор H-D-iGlu-D-Trp-OH, моноаммонийной соли (1:1), (1,66 г, 4,05 ммоль) и гидроксида калия (253 мг, 4,50 ммоль) в воде (20 мл) перемешивали при комнатной температуре в течение 15 мин. pH раствора составлял приблизительно 9. Реакционную смесь упаривали при пониженном давлении до объема приблизительно 1 мл. После охлаждения до комнатной температуры добавляли изопропанол до момента образования осадка твердого вещества. Полученную суспензию перемешивали при комнатной температуре в течение 15 мин, затем фильтровали. Твердое вещество промывали изопропанолом (2×20 мл) и этилацетатом (20 мл), затем сушили в вакууме в печи с температурой 42°C, выдерживая при этой температуре в течение ночи. Получали твердое вещество практически белого цвета (1,49 г, выход 99%). Содержание воды по данным анализа методом Карла-Фишера составляло 2,5%. Данные анализа (образец XRPD, FTIR и МС спектры) сходны с описанными в Примере 1.
B. Получение аморфной формы калиевой соли D-изоглутамил-D-триптофана (1:1) из моноаммонийной соли D-изоглутамил-D-триптофана (1:1).
Раствор H-D-iGlu-D-Trp-ОН, моноаммонийной соли (1:1), (517 мг, 1,40 ммоль) и гидроксида калия (82 мг, 1,46 ммоль) в воде (10 мл) перемешивали при комнатной температуре в течение 30 минут. Полученную смесь высушивали лиофильной сушкой в течение ночи. Продукт получали в виде твердого вещества практически белого цвета с количественным выходом. Образец XRPD спектра подтвердил, что данный материал является аморфным. 1H ЯМР (D2O) δ: 7,69 (д, J=7,9 Гц, 1H), 7,48 (д, J=8,2 Гц, 1H), 7,23 (т, J=7,6 Гц, 1H), 7,22 (с, 1H), 7,16 (т, J=7,4 Гц, 1H), 4,59 (дд, J=8,7, 4,8 Гц, 1H), 3,51 (дд, J=6,8, 5,8 Гц, 1H), 3,38 (дд, J=14,8, 4,8 Гц, 1H), 3,11 (дд, J=14,8, 8,8 Гц, 1H), 2,20-2,49 (м, 2H) и 1,85-1,94 (м, 2H); 13C ЯМР (D2O) δ: 181,4, 177,0, 176,6, 138,8, 129,9, 126,9, 124,5, 121,9, 121,4, 114,5, 113,2, 58,6, 57,0, 34,6 (CH2), 30,2 (CH2) и 29,3 (CH2); содержание воды по данным анализа методом Карла-Фишера составляло 5,4%; спектр FTIR (KBr) показан на фиг.1C; МС (соотношение массы и заряда): 371,7 [M]+, 334,2 [C16H20N3O5]+, 187,9 (100%); чистота ВЭЖХ (процент области пика): 99,8%, время удерживания: 5,04 мин; условия ВЭЖХ: колонка Waters Symmetry С18, 3,9×150 мм, 5 мкм; подвижная фаза: 0,035% НСlO4, pH 2/CH3CN, 85:15 изократическая, скорость потока: 1 мл/мин; λ: 220, 254, 280 нм.
Пример 3:
A. Получение литиевой соли D-изоглутамил-D-триптофана (1:1) из моноаммонийной соли D-изоглутамил-D-триптофана (1:1) и гидроксида лития моногидрата
Раствор H-D-iGlu-D-Trp-OH, моноаммонийной соли (1:1), (1,40 г, 3,80 ммоль) и гидроксида лития моногидрат (159 мг, 3,80 ммоль) в воде (20 мл) перемешивали при комнатной температуре в течение 20 мин. pH раствора составлял приблизительно 9. Реакционную смесь выпаривают при пониженном давлении до объема растворителя приблизительно 2 мл. После охлаждения до комнатной температуры добавляли изопропанол до образования осадка твердого вещества. Полученную суспензию перемешивали при комнатной температуре в течение 20 мин, затем фильтровали. Твердое вещество промывали изопропанолом (2×20 мл) и этилацетатом (20 мл), затем сушили в вакууме в печи с температурой 42°C в течение ночи. Продукт получали в виде твердого вещества практически белого цвета с количественным выходом. Образец XRPD данного кристаллического материала показан на фиг.2A. Содержание воды по данным анализа методом Карла-Фишера 10,7%. MC (соотношение массы и заряда): 340,1 [M+1]+, 334,3 [C16H20N3O5]+, 187,9 (100%). Спектр FTIR (KBr) показан на фиг.2В.
B. Раствор H-D-iGlu-D-Trp-OH, моноаммонийной соли (1:1), (480 мг, 1,30 ммоль) и гидроксида лития моногидрат (57 мг, 1,36 ммоль) в воде (10 мл) перемешивали при комнатной температуре в течение 30 мин. Полученную смесь сушили лиофильной сушкой в течение ночи. Продукт получали в виде твердого вещества практически белого цвета с количественным выходом. Образец XRPD подтвердил, что данный материал является аморфным. 1H ЯМР (D2O) δ: 7,69 (д, J=7,8 Гц, 1H), 7,50 (д, J=8,2 Гц, 1H), 7,23 (т, J=7,1 Гц, 1H), 7,22 (с, 1H), 7,16 (т, J=7,5 Гц, 1H), 4,57 (дд, J=8,7, 4,8 Гц, 1H), 3,36-3,43 (м, наложение т и дд, 2H), 3,12 (дд, J=14,7, 8,7 Гц, 1H), 2,20-2,35 (м, 2H) и 1,78-1,92 (м, 2H); 13C ЯМР (D2O) δ: 181,4, 178,1, 176,7, 138,8, 129,9, 126,9, 124,5, 121,9, 121,4, 114,5, 113,2, 58,6, 57,1, 34,7 (CH2), 30,2 (CH2) и 29,3 (CH2); спектр FTIR (KBr) показан на фиг.2С; содержание воды по данным анализа методом Карла-Фишера составляло 11,5%. Спектр МС подобен описанному в Примере 3A; чистота ВЭЖХ (процент области пика): 99,8%, время удерживания: 5,10 мин. Применяли условия ВЭЖХ, описанные в Примере 2B.
Пример 4:
Получение литиевой соли D-изоглутамил-D-триптофана (1:1) из D-изоглутамил-D-триптофана и гидроксида лития моногидрата
A. В круглодонной колбе объемом 100 мл, оборудованной магнитной мешалкой, растворяли гидроксида лития моногидрат (125,8 мг, 2,99 ммоль) в 10 мл воды. Раствор охлаждали до 0°C при помощи ледяной бани. H-D-iGlu-D-Trp-OH (1,00 г, 3 ммоль) суспендировали в растворе. Твердое вещество медленно растворяли на протяжении 2,5 часов с образованием прозрачного бледно-розового раствора. После дополнительных 30 минут перемешивания смесь нагревали до комнатной температуры. Раствор фильтровали и осторожно упаривали до объема приблизительно 4 мл. Медленно добавляли изопропанол (25 мл) до начала образования твердого вещества. Раствор фильтровали и твердое вещество разделяли на две равные части.
B. Одну часть твердого вещества, полученного по методике А, промывали изопропанолом (2×15 мл). Твердое вещество сначала сушили на воздухе, а затем сушили в вакууме в печи (35°C) в течение ночи. Содержание воды по данным анализа методом Карла-Фишера составляло 10,6%. Образец XRPD и MC и FTIR (KBr) спектры данного соединение сходны с описанными в Примере 3А.
C. Вторую часть твердого вещества промывали изопропанолом (2×15 мл), затем этилацетатом (2×10 мл). Твердое вещество сначала сушили на воздухе, а затем сушили в вакууме в печи (35°C) в течение ночи. Образец XRPD и FTIR (KBr) спектр данного соединение сходны с описанными в Примере 3A.
Объединенный выход материала, полученного по методикам B и C, составляет 0,99 г (выход 97,6%).
Пример 5:
Получение магниевой соли D-изоглутамил-D-триптофана (1:2) из D-изоглутамил-D-триптофана
В круглодонную колбу объемом 100 мл, оборудованную магнитной мешалкой, помещали этоксид магния (Aldrich, 98%, 0,206 г, 1,76 ммоль) и изопропанол (15 мл). Раствор охлаждали до 0°C на ледяной бане и добавляли твердый H-D-iGlu-D-Trp-OH (1,20 г, 3,60 ммоль). Суспензию белого цвета перемешивали при комнатной температуре в течение 4 часов. 2-3 капли реакционной смеси помещали в пробирку и добавляли несколько капель деминерализованной воды. Смесь встряхивали с образованием прозрачного раствора. pH раствора составлял 4,0-4,5. К реакционной смеси добавляли этоксид магния (Aldrich, 98%, 0,050 г, 0,43 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. 2-3 капли суспензии белого цвета помещали в пробирку и добавляли несколько капель деминерализованной воды. Смесь встряхивали с получением прозрачного раствора. pH раствора составлял приблизительно 7,0. Смесь выпаривали до сухого состояния при температуре бани 30°C с получением твердого вещества белого цвета. Остаток растворяли в 15 мл деминерализованной воды с получением раствора желтого цвета. Последний фильтровали для удаления частиц твердого вещества. Фильтрат выпаривали до сухого состояния при температуре бани 30°C с получением твердого вещества. Твердое вещество суспендировали в деминерализованной воде (20 мл) и смесь перемешивали в течение 3 часов. Твердое вещество отделяли при помощи фильтрации и промывали ледяной деминерализованной водой (2×6 мл). Твердое вещество сначала сушили на воздухе, а затем помещали в вакуумную печь при 42°C, выдерживая при этой температуре в течение ночи. Таким образом, 0,88 г (выход 72%, чистота ВЭЖХ [процент области пика]: 99,1%) продукта было получено. Применяли метод ВЭЖХ, описанный в Примере 1. Время удержания для данного продукта составляло 6,39 мин. Образец XRPD спектра данного кристаллического материала показан на фиг.3A. Содержание воды по данным анализа методом Карла-Фишера составляло 12,2%. МС (соотношение массы и заряда): 689,3 [M]+, 334,2 [C16H20N3O5]+ 187,9 (100%). УФ (вода, c = 11,7 мкМ, λmax нм): 221 (ε 57906), 280 (ε 9449). Спектр FTIR (KBr) показан на фиг.3B.
Пример 6:
Получение магниевой соли D-изоглутамил-D-триптофана (1:2) из моноаммонийной соли D-изоглутамил-D-триптофана (1:1)
Суспензию D-изоглутамил-D-триптофана, моноаммонийной соли (1:1), (1,53 г, 4,15 ммоль) и гидроксида магния в H2O (20 мл) нагревали при температуре 55-60°C, выдерживая при этой температуре в течение 3 часов. Полученную суспензию желтоватого цвета выпаривали при пониженном давлении до объема приблизительно 1-2 мл. Затем добавляли изопропанол (30 мл). Суспензию перемешивали в течение 20 мин при комнатной температуре, после чего фильтровали. Твердое вещество промывали последовательно изопропанолом (2×20 мл) и этилацетатом (20 мл), затем сушили в вакуумной печи при 42°C, выдерживая при этой температуре в течение ночи. Получали твердое вещество желтоватого цвета (1,5 г). Содержание воды по данным анализа методом Карла-Фишера составляло 8,8%. Данные анализа (образец XRPD и FTIR и МС спектры) сходны с описанными в Примере 5.
Пример 7:
Получение кальциевой соли D-изоглутамил-D-триптофана (1:2) из D-изоглутамил-D-триптофана (1:1) и гидроксида кальция
В круглодонную колбу объемом 100 мл, оборудованную магнитной мешалкой, помещали гидроксид кальция (Aldrich, 99,99%, менее 3% кальция карбоната, 0,2603 г, 3,51 ммоль) и деминерализованную воду (30 мл). Мутный раствор охлаждали до 0°C на ледяной бане, и добавляли твердый H-D-iGlu-D-Trp-OH (2,404 г, 7,2 ммоль). Смесь перемешивали в течение 2,5 часов с образованием прозрачного, слегка розоватого раствора. pH раствора доводили до 6,0, добавляя насыщенный раствор гидроксида кальция. Раствор фильтровали для удаления частиц твердого вещества. Фильтрат разделяли на два равных объема (приблизительно по 20 мл каждый): Раствор A и Раствор B.
Раствор А упаривали при помощи роторного испарителя до объема приблизительно 4-5 мл с использованием водяной бани с температурой приблизительно 30°C. Раствор остается прозрачным. Полученный концентрированный раствор интенсивно перемешивали при комнатной температуре в течение 17 часов с получением твердого вещества. Твердое вещество отделяли фильтрацией и промывали ледяной деминерализованной водой (3×6 мл). Твердое вещество сначала сушили лиофильной сушкой, после чего сушили в вакуумной печи при 40°C, выдерживая при этой температуре в течение ночи с получением твердого вещества 0,70 г (55%, чистота ВЭЖХ по данным % области пика: 97,7%). Применяли метод ВЭЖХ, описанный в Примере 1. Время удержания для данного продукта составляло 6,39 мин. Образец XRPD спектра данного материала показан на фиг.4А. Содержание воды по данным анализа методом Карла-Фишера составляло 5,4%. МС (соотношение массы и заряда): 705,6 [M+1]+, 334,2 [C16H20N3O5]+, 187,9 (100%). УФ (вода, с=10,8 мкМ, λmax нм): 221 (ε 61014), 280 (ε 9943). Спектр FTIR (KBr) показан на фиг.4C.
Раствор В испаряли до сухого состояния. Добавляли деминерализованную воду (6 мл) и смесь перемешивали в течение 16 часов. Нерастворимое твердое вещество фильтровали и сушили под высоким вакуумом при 35°C температуре в течение 48 часов (0,53 г). Образец XRPD сходен с показанным на фиг.4А.
Пример 8:
Получение кальциевой соли D-изоглутамил-D-триптофана (1:2) из моноаммонийной соли D-изоглутамил-D-триптофана (1:1) и гидроксида кальция
Суспензию моноаммонийной соли (1:1) D-изоглутамил-D-триптофана, (1,49 г, 4,06 ммоль) и гидроксида кальция (150 мг, 2,03 ммоль) в воде (20 мл) нагревали до температуры 55-60°C, выдерживая при этой температуре в течение 1 часа. Полученный раствор упаривали при пониженном давлении до объема приблизительно 1-2 мл. Добавляли изопропанол (30 мл). Суспензию перемешивали в течение 20 мин при комнатной температуре, затем фильтровали. Твердое вещество промывали последовательно изопропанолом (2×20 мл) и этилацетатом (20 мл), затем сушили в печи при температуре 42°C, выдерживая при этой температуре в течение ночи. Получали твердое вещество практически белого цвета (1,45 г). Образец XRPD данного полукристаллического материала показан на фиг.4B. Данный материал имеет более низкую степень кристалличности, чем материал из Примера 7. Содержание воды по данным анализа методом Карла-Фишера составляло 6,2%. МС (соотношение массы и заряда): 705,4 [M+1]+, 334,2 [C16H20N3O5]+, 187,9 (100%).
Пример 9:
Определение процента кристалличности кальциевой соли D-iGlu-D-Trp с помощью метода порошкового рентгеноструктурного анализа
Общая степень кристалличности, измеренная с помощью техники XRPD, обеспечивает дополнительную полезную информацию для фармацевтических материалов, которые содержат какой-либо аморфный материал, образованный в ходе синтеза. Она также представляет собой подходящий показатель с точки зрения контроля долгосрочных изменений в кристаллических материалах. Не будучи связанным с какими-либо конструктивными и композиционными свойствами, показатель "процентная кристалличность" может быть подходящим индикатором стабильности конкретного материала как функция времени.
Процент кристалличности обычно измеряют как соотношение между частью дифракции кристаллической части образца, IC, и полной дифракцией того же образца, IC+B. Значения IC могут быть получены после соответствующего вычитания рассеянной части из фона, IB.
Для такого вида анализов, дифракцию измеряют как общую область под кривой:
- цельного образца, полученного в результате синтеза (данные можно корректировать с учетом воздушного рассеивания) - ITOTAL,
- только пики (IC) после вычитания фона (коррекция с учетом воздушного рассеивания),
- только основа с коррекцией с учетом воздушного рассеивания - (IB).
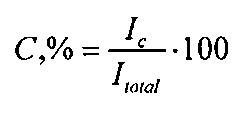 , где Ttotal=Ic+Ib.
, где Ttotal=Ic+Ib.
Следует отметить, что такое измерение не рекомендуется как стандартизированное из-за того, что очень сложно (практически невозможно) сравнить межлабораторные результаты. Каждый дифрактометр имеет свой собственный масштаб уровня фона и высоты пика, а также площади под кривой в зависимости от многих инструментальных факторов, а также факторов подготовки образца.
Описанный выше подход возможен по большей части для дифрактометров "точка-детектора", в которых детектор перемещается с синхронизированной скоростью, в 2 раза большей, чем скорость перемещения образца, для того, чтобы гарантировать постоянное соотношение θ/2 θ.
Однако, в данном исследовании, в связи с техническими проблемами системы дифракции «точка-детектор» D5000, все образцы были проанализированы на системе D8, оборудованной 2D детектором площади, и вышеприведенный подход не мог быть применен. Таким образом, была разработана другая техника измерения кристалличности образцов, проанализированных на такой системе. Выбрана только узкая часть 2D изображения дифракции с одинарным пиком в центре и соседней области, где будет измеряться фон.
Образцы, полученные в Примере 8, демонстрируют % кристалличности приблизительно 18-20%, в то время как образцы, полученные способом из Примера 7, демонстрируют диапазон % кристалличности приблизительно 25-50%. Процент кристалличности образца может быть улучшен, если образец растворить в воде. Нерастворимые частицы отфильтровывали и твердому веществу давали медленно осаждаться из раствора.
Все образцы были измерены для вычисления их кристалличности с использованием узкого диапазона 21-25° с самым сильным отражением при 13°. Фон вычитали как линейный и использовали эмпирический коэффициент корреляции 2,5.
Как было указано выше, результаты, полученные таким образом для кристалличности полукристаллических образцов, не следует рассматривать, как абсолютные. Ни один из образцов данного структурного типа не обладает кристаллическим порядком, достаточно высоким для того, чтобы рассматриваться как абсолютный эталон. В Примерах 7 и 8 получена кальциевая соль тимодепрессина (1:2) с различным процентом кристалличности.
Пример 10:
Получение моно-трет-бутиламиновой соли D-изоглутамил-D-триптофана (1:1) из D-изоглутамил-D-триптофана и трет-бутиламина
A. К суспензии D-iGlu-D-Trp (1,00 г, 3,00 ммоль) в 25 мл деминерализованной воды добавляли 0,7 мл (2,22 экв.) трет-бутиламина при комнатной температуре. Реакционная смесь оставалась прозрачной, и pH раствора составлял приблизительно 9. После перемешивания при комнатной температуре в течение 1 часа добавляли изопропанол и летучие материалы удаляли в вакууме. Оставшееся твердое вещество суспендировали в ацетоне и отделяли при помощи вакуумной фильтрации. Твердое вещество сушили в вакууме при температуре 40°C в течение ночи с получением 1,16 г (выход 95%) моносоли амина. 1H ЯМР подтвердил, что продукт представляет собой моноаддитивную соль. Образец XRPD подтвердил, что данный материал является аморфным. 1H ЯМР (D2O) δ: 7,71 (д, J=8,6 Гц, 1H), 7,39 (д, J=8,1 Гц, 1H), 7,13-7,16 (м, 2H), 7,07 (т, J=7,6 Гц, 1H), 4,48 (дд, J=8,3, 4,9 Гц, 1H), 3,44 (т, J=6,4 Гц, 1H), 3,28 (дд, J=14,8, 4,7 Гц, 1H), 3,02 (дд, J=14,7, 8,7 Гц, 1H), 2,18-2,26 (м, 2H), 1,76-1,97 (м, 2H) и 1,26 (с, 9H). 14N-ЯМР (D2O) δ (м.д.): 40,2 (ушир.) и 56,3 (с), *NH4 NO3 применяли как внешний стандарт с установкой опорного сигнала на уровне 20,689 м.д. Содержание воды по данным анализа методом Карла-Фишера составляло 4,0%. МС (соотношение массы и заряда): 407,3 [M+1]+ (слабый), 334,2 [C16H20N3O5]+, 187,9 (100%). ИК спектр показан на фиг.6. УФ (вода, c = 34,8 мкМ, λmax нм): 220 (ε 31067), 280 (ε 5112).
В. К суспензии D-iGlu-D-Trp (1,00 г, 3,00 ммоль) в 25 мл деминерализованной воды добавляли 0,31 мл (1,0 экв.) трет-бутиламина при комнатной температуре (RT). Реакционная смесь оставалась прозрачной, и pH раствора составлял приблизительно 9. После перемешивания при комнатной температуре в течение 1 часа добавляли изопропанол и летучие материалы удаляли в вакууме. Оставшееся твердое вещество суспендировали в ацетоне и твердое вещество отделяли при помощи вакуумной фильтрации. Твердое вещество сушили в вакууме при температуре 40°C в течение ночи с получением 1,16 г (выход 95%) соли амина. Данные анализа для данного соединения (XRPD, 1Н ЯМР, MC, FTIR) сходны с описанными в Примере 10A выше.
Пример 11:
Получение соли монотрис(гидроксиметил)аминометана D-изоглутамил-D-триптофана (1:1) из D-изоглутамил-D-триптофана и трис(гидроксиметил)аминометана (TRIS)
К суспензии D-iGlu-D-Trp (1,00 г, 3,00 ммоль) в 20 мл деминерализованной воды добавляли раствор 363 мг (1,0 экв.) трис(гидроксиметил)аминометана (TRIS) в 15 мл деминерализованной воды при комнатной температуре. Реакционная смесь оставалась прозрачной, и pH раствора составлял приблизительно 7. После перемешивания при комнатной температуре в течение ночи добавляли изопропанол и летучие материалы удаляли в вакууме. Попытки перекристаллизовать соединение с использованием смеси изопропанол/вода или метанол/диэтиловый эфир были неудачными. Оставшееся твердое вещество суспендировали в ацетоне и перемешивали при комнатной температуре в течение 1 часа, твердое вещество отделяли при помощи вакуумной фильтрации. Твердое вещество сушили в вакууме при температуре 40°C в течение ночи с получением 1,33 г продукта (выход 97,5%). 1Н ЯМР (D2O) δ: 7,61 (д, J=7,9 Гц, 1H), 7,41 (д, J=8,1 Гц, 1H), 7,14-7,17 (м, 2H), 7,08 (т, J=7,4 Гц, 1H), 4,48 (дд, J=8,5, 4,8 Гц, 1H), 3,64 (с, 6H), 3,46 (т, J=6,0 Гц, 1H), 3,28 (дд, J=14,8, 4,7 Гц, 1H), 3,02 (дд, J=14,7, 8,7 Гц, 1H), 2,17-2,28 (м, 2H) и 1,74-1,90 (м, 2H). Содержание воды по данным анализа методом Карла-Фишера составляло 3,3%. МС (соотношение массы и заряда): 454,9 [M+1]+ (слабый), 334,0 [C16H20N3O5]+, 187,9 (100%). ИК спектр показан на фиг.7; УФ (вода, c = 36,4 мкМ, λmax нм): 220 (ε 28373), 280 (ε 4537).
Пример 12:
Получение моно-N-метил-D-глюкаминовой соли D-изоглутамил-D-триптофана (1:1) из D-изоглутамил-D-триптофана и N-метил-D-глюкамина
К суспензии D-iGlu-D-Trp (1,00 г, 3 ммоль) в 20 мл деминерализованной воды добавляли раствор 586 мг (1,0 экв.) N-метил-D-глюкамина в 15 мл деминерализованной воды при комнатной температуре. Реакционную смесь перемешивали в течение выходных при комнатной температуре. Реакционная смесь оставалась прозрачной, и pH раствора составлял приблизительно 7. Добавляли изопропанол и летучие материалы удаляли в вакууме. Оставшееся твердое вещество суспендировали в ацетоне и твердое вещество отделяли при помощи вакуумной фильтрации. Твердое вещество сушили в вакууме при температуре 40°C в течение ночи с получением продукта с количественным выходом. Образец XRPD подтвердил, что данный материал является аморфным. 1Н ЯМР (D2O) δ: 7,61 (д, J=7,9 Гц, 1H), 7,41 (д, J=8,1 Гц, 1H), 7,15-7,18 (м, 2H), 7,08 (т, J=7,5 Гц, 1H), 4,47 (дд, J=8,6, 4,8 Гц, 1H), 3,99-4,02 (м, 1H), 3,70-3,75 (м, 2H), 3,65-3,68 (м, 1H), 3,54-3,60 (м, 2H), 3,45 (т, J=6,2 Гц, 1H), 3,27 (дд, J=14,8, 4,7 Гц, 1H), 3,02-3,13 (м, 3H), 2,68 (с, 3H), 2,19-2,26 (м, 2H) и 1,75-1,95 (м, 2H). 14N ЯМР (D2O) δ (м.д.): 29,6 и 39,2 (ушир. наложение), *NH4 NO3 применяли как внешний стандарт с установкой опорного сигнала на уровне 20,689 м.д. Содержание воды по данным анализа методом Карла-Фишера составляло 3,1%. МС (соотношение массы и заряда): 529,5 [M+1]+, 334,2 [C16H20N3O5]+, 187,9 (100%). ИК спектр показан на фиг.8. УФ (вода, c = 41,2 мкМ, λmax нм): 220 (ε 27341), 280 (ε 4419).
Пример 13:
Пример методики получения D-изоглутамил-D-триптофана, моноаммонийной соли (1:1) из H-D-iGlu-D-Trp-OH
H-D-iGlu-D-Trp-OH (1 г) смешивали с гидроксидом аммония (0,55 М, 6 мл). Смесь перемешивали; pH смеси составляет приблизительно 4,5. Раствор гидроксида аммония (0,55 М) добавляли по каплям до тех пор, пока pH раствора не достигнет 7,0-7,5. Летучие материалы удаляли в вакууме и оставшееся масло смешивали с изопропанолом. Образовывался белый осадок. Через 2 часа твердую аммонийную соль отделяли при помощи вакуумной фильтрации. Твердое вещество сушили до постоянной массы (1 г) в глубоком вакууме в течение 12 часов с получением D-изоглутамил-D-триптофана, аммонийной соли (1:1).
Хотя предпочтительные варианты изобретения описаны в данном описании, специалистам в данной области будет понято, что могут быть осуществлены вариации без отхода от духа изобретения или контекста приложенной формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЙ D-ИЗОГЛУТАМИЛ-D-ТРИПТОФАН И МОНОАММОНИЙНАЯ СОЛЬ D-ИЗОГЛУТАМИЛ-D-ТРИПТОФАНА | 2007 |
|
RU2483077C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ МОНОНАТРИЕВОЙ СОЛИ D-ИЗОГЛУТАМИЛ-D-ТРИПТОФАНА | 2008 |
|
RU2476440C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОНАТРИЕВОЙ СОЛИ ИЗОГЛУТАМИЛ-ТРИПТОФАНА | 2019 |
|
RU2703991C1 |
| ТАРТРАТ 3-((1R,3R)-1-(2,6-ДИФТОР-4-((1-(3-ФТОРПРОПИЛ)АЗЕТИДИН-3-ИЛ)АМИНО)ФЕНИЛ)-3-МЕТИЛ-1,3,4,9-ТЕТРАГИДРО-2H-ПИРИДО[3,4-b]ИНДОЛ-2-ИЛ)-2,2-ДИФТОРПРОПАН-1-ОЛА, ЕГО ТВЕРДЫЕ ФОРМЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2809220C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2007 |
|
RU2446156C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2014 |
|
RU2654855C2 |
| ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ С НЕМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ | 2003 |
|
RU2351314C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА TLR7/TLR8 | 2019 |
|
RU2792005C2 |
| Твёрдые формы цефтолозана | 2014 |
|
RU2703457C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ N-[4-(ХЛОРДИФТОРМЕТОКСИ)ФЕНИЛ]-6-[(3R)-3-ГИДРОКСИПИРРОЛИДИН-1-ИЛ]-5-(1H-ПИРАЗОЛ-5-ИЛ)ПИРИДИН-3-КАРБОКСАМИДА | 2020 |
|
RU2836337C2 |




Изобретение относится к фармацевтически приемлемым кристаллическим или аморфным солям D-изоглутамил-D-триптофана, способам их получения, фармацевтическим композициям, которые их содержат, и их применению для получения фармацевтических композиций для лечения различных состояний и/или заболеваний. В частности, данное изобретение относится к калиевой соли D-изоглутамил-D-триптофана (1:1) и магниевой соли D-изоглутамил-D-триптофана (2:1). 20 н. и 2 з.п. ф-лы, 15 ил., 13 пр.
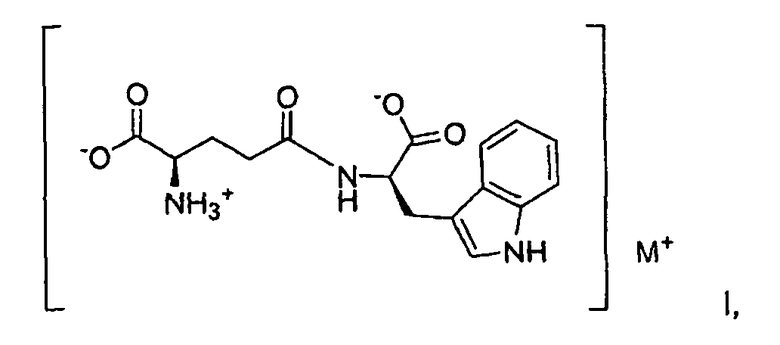
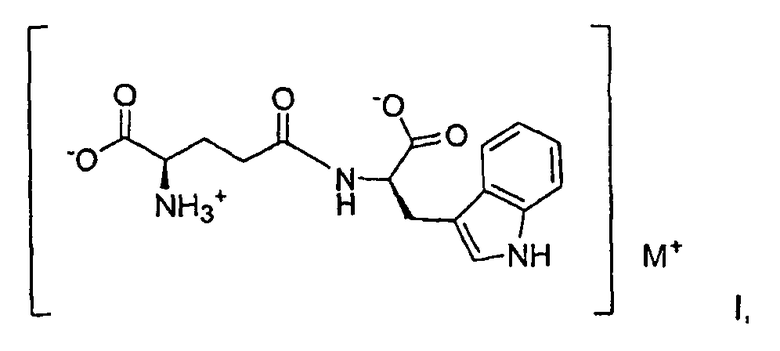
1. Фармацевтически приемлемая соль D-изоглутамил-D-триптофана формулы I:
где М представляет собой калий и где соль находится в кристаллической форме, которая характеризуется пиками на порошковой рентгенограмме в области следующих значений 2-θ: 9,91, 14,84, 15,81, 18,97, 19,76, 24,04, 24,36, 24,82, 25,48, 27,49, 27,94, 28,42, 30,82, 31,28, 31,69, 32,17, 34,35, 35,81 и 36,96°.
2. Фармацевтически приемлемая соль по п.1, которая характеризуется порошковой рентгенограммой, представленной на фиг.1А.
3. Фармацевтически приемлемая соль D-изоглутамил-D-триптофана формулы I:
где М представляет собой калий и где соль находится в аморфной форме, которая характеризуется инфракрасным спектром с преобразованием Фурье (FTIR), представленным на Фиг.1C.
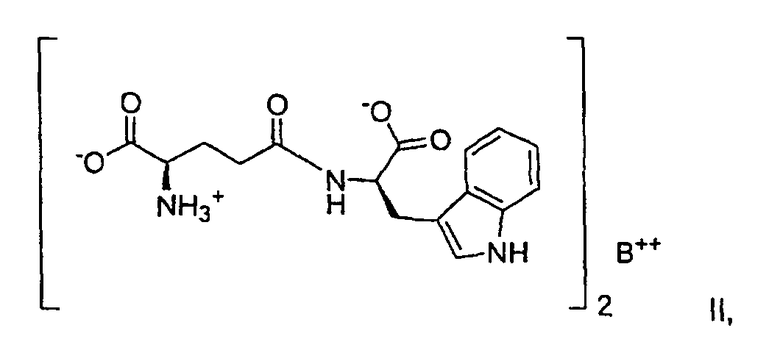
4. Фармацевтически приемлемая соль D-изоглутамил-D-триптофана формулы II:
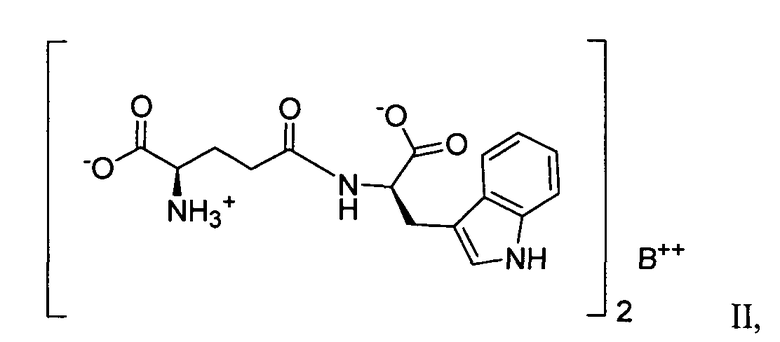
где B представляет собой магний и где соль находится в кристаллической форме, которая характеризуется пиками на порошковой рентгенограмме в области следующих значений 2-θ: 12,2, 13,74, 14,84, 16,16, 17,96, 18,52, 18,94, 19,49, 21,05, 21,56, 22,56, 23,36, 24,12, 26,27, 27,65, 28,42, 29,14, 30,55, 31,77, 32,62, 33,26, 35,05, 36,34, 37,22 и 38,05°.
5. Фармацевтически приемлемая соль по п.4, которая характеризуется порошковой рентгенограммой, представленной на фиг.3А.
6. Способ получения металлической соли D-изоглутамил-D-триптофана формулы I:
где М представляет собой калий и где соль находится в кристаллической форме, которая характеризуется пиками на порошковой рентгенограмме в области следующих значений 2-θ: 9,91, 14,84, 15,81, 18,97, 19,76, 24,04, 24,36, 24,82, 25,48, 27,49, 27,94, 28,42, 30,82, 31,28, 31,69, 32,17, 34,35, 35,81 и 36,96° или которая характеризуется порошковой рентгенограммой, представленной на фиг.1А, причем указанный способ включает
(a) взаимодействие D-изоглутамил-D-триптофана с гидроксидом калия в воде с получением раствора;
(b) концентрирование раствора до образования масла;
(c) добавление изопропанола к маслу при перемешивании для осаждения соли с получением осадка;
(d) отделение полученного осадка; и
(e) сушку продукта в вакууме с получением соли калия формулы I.
7. Способ получения металлической соли D-изоглутамил-D-триптофана формулы II:
где В представляет собой магний и где соль находится в кристаллической форме, которая характеризуется пиками на порошковой рентгенограмме в области следующих значений 2-θ:12,2, 13,74, 14,84, 16,16, 17,96, 18,52, 18,94, 19,49, 21,05, 21,56, 22,56, 23,36, 24,12, 26,27, 27,65, 28,42, 29,14, 30,55, 31,77, 32,62, 33,26, 35,05, 36,34, 37,22 и 38,05° или которая характеризуется порошковой рентгенограммой, представленной на фиг.3А, причем указанный способ включает
(а) взаимодействие D-изоглутамил-D-триптофана с этоксидом магния в изопропаноле с получением раствора;
(b) концентрирование раствора до получения твердого вещества;
(c) смешивание твердого вещества с водой;
(d) отделение нерастворимых частиц фильтрацией с получением фильтрата;
(e) разбавление фильтрата водой при перемешивании для осаждения продукта;
(f) отделение продукта; и
(g) сушку продукта в вакууме с получением металлической соли магния формулы II.
8. Способ солевого обмена для получения фармацевтически приемлемой соли D-изоглутамил-D-триптофана, представленной общей формулой I:
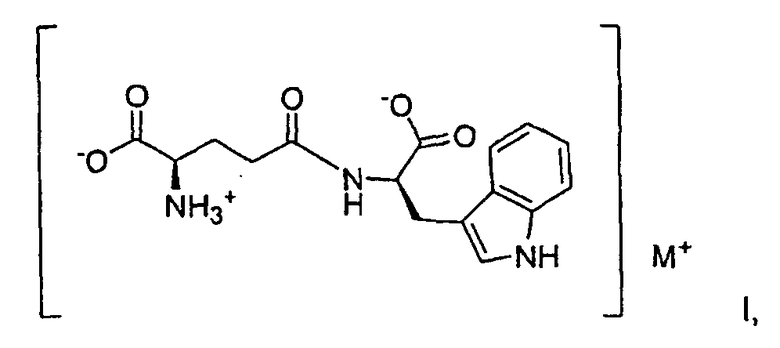
где М представляет собой калий и где соль находится в кристаллической форме, которая характеризуется пиками на порошковой рентгенограмме в области следующих значений 2-θ: 9,91, 14,84, 15,81, 18,97, 19,76, 24,04, 24,36, 24,82, 25,48, 27,49, 27,94, 28,42, 30,82, 31,28, 31,69, 32,17, 34,35, 35,81 и 36,96° или которая характеризуется порошковой рентгенограммой, представленной на фиг.1А, причем указанный способ включает
(а) взаимодействие аммонийной соли, представленной формулой VII:
в водном растворе с 1 экв. гидроксида металла, представленным МОН, где М является таким, как определено выше;
(b) выпаривание растворителя с получением твердого вещества; и
(c) перемешивание твердого вещества с водой и изопропанолом с получением фармацевтически приемлемой соли формулы I.
9. Способ солевого обмена для получения фармацевтически приемлемой соли D-изоглутамил-D-триптофана, представленной общей формулой I:
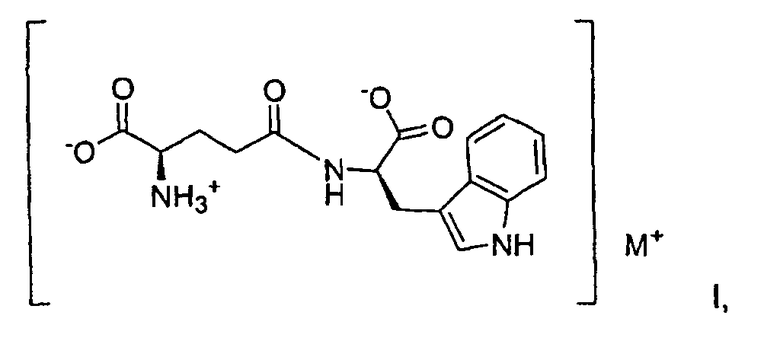
где М представляет собой калий и где соль находится в аморфной форме, которая характеризуется инфракрасным спектром с преобразованием Фурье (FTIR), представленным на Фиг.1C, причем указанный способ включает
(а) взаимодействие аммонийной соли, представленной формулой VII:
в водном растворе с 1 экв. гидроксида металла, представленным МОН, где М является таким, как определено выше;
(b) перемешивание раствора в течение 30 минут; и
(c) лиофильную сушку раствора с получением фармацевтически приемлемой соли формулы I.
10. Способ солевого обмена для получения фармацевтически приемлемой соли D-изоглутамил-D-триптофана, представленной общей формулой II:
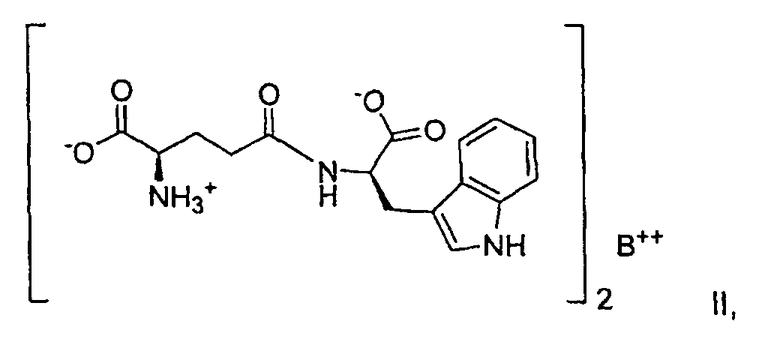
где В представляет собой магний и где соль находится в кристаллической форме, которая характеризуется пиками на порошковой рентгенограмме в области следующих значений 2-θ: 12,2, 13,74, 14,84, 16,16, 17,96, 18,52, 18,94, 19,49, 21,05, 21,56, 22,56, 23,36, 24,12, 26,27, 27,65, 28,42, 29,14, 30,55, 31,77, 32,62, 33,26, 35,05, 36,34, 37,22 и 38,05° или которая характеризуется порошковой рентгенограммой, представленной на фиг.3А, причем указанный способ включает
(а) взаимодействие аммонийной соли, представленной формулой VII:
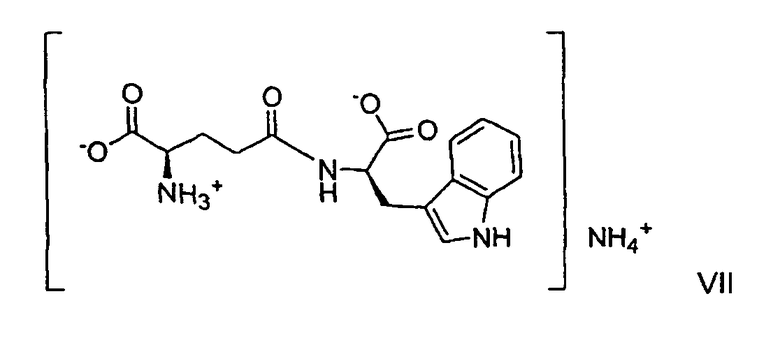
в водном растворе с приблизительно 0,5 экв. гидроксида металла В(ОН)2, где В является таким, как определено выше;
(b) выпаривание растворителя с получением твердого вещества; и
(c) перемешивание твердого вещества с водой и изопропанолом с получением фармацевтически приемлемой соли формулы II.
11. Фармацевтическая композиция, обладающая свойствами иммунодепрессанта, содержащая терапевтически эффективное количество фармацевтически приемлемой соли по любому из пп.1-5 и по меньшей мере одно вспомогательное вещество.
12. Способ получения фармацевтической композиции, включающий объединение фармацевтически приемлемой соли по любому из пп.1-5 по крайней мере с одним фармацевтически приемлемым носителем.
13. Применение эффективного количества фармацевтически приемлемой соли по любому из пп.1-5 в качестве иммунодепрессанта.
14. Применение эффективного количества фармацевтически приемлемой соли по любому из пп.1-5 в качестве средства против псориаза.
15. Применение эффективного количества фармацевтически приемлемой соли по любому из пп.1-5 в качестве средства для лечения атопического дерматита.
16. Применение эффективного количества фармацевтически приемлемой соли по любому из пп.1-5 в качестве селективного ингибитора пролиферации гемопоэтических клеток-прекурсоров.
17. Применение эффективного количества фармацевтически приемлемой соли по любому из пп.1-5 в качестве стимулятора апоптоза гранулоцитов и апоптоза лимфоцитов.
18. Применение эффективного количества фармацевтической композиции по п.11 для лечения псориаза у нуждающегося в этом пациента.
19. Применение эффективного количества фармацевтической композиции по п.11 для лечения иммунодепрессивных заболеваний у нуждающегося в этом пациента.
20. Применение эффективного количества фармацевтической композиции по п.11 для лечения атопического дерматита у нуждающегося в этом пациента.
21. Применение эффективного количества фармацевтической композиции по п.11 для селективного ингибирования пролиферации гемопоэтических клеток-прекурсоров у нуждающегося в этом пациента.
22. Применение эффективного количества фармацевтической композиции по п.11 для стимуляции апоптоза гранулоцитов и апоптоза лимфоцитов у нуждающегося в этом пациента.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| US 6911431 B1,28.06.2005 | |||
| US 5736519 A, 07.04.1998 | |||
| US 5744452 A, 28.04.1998 | |||
| ПЕПТИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2107692C1 |
| ПЕПТИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2107691C1 |
| Устройство для фиксации помольных стаканов на вибростоле измельчителя | 1979 |
|
SU869808A1 |
| КРИСТАЛЛИЧЕСКИЙ D-ИЗОГЛУТАМИЛ-D-ТРИПТОФАН И МОНОАММОНИЙНАЯ СОЛЬ D-ИЗОГЛУТАМИЛ-D-ТРИПТОФАНА | 2007 |
|
RU2483077C2 |
| US 6410515 B1, 25.06.2002 | |||
| H.IYO et al, "Sequence-Dependent Interaction of Acidic Amino Acid with Guanine Base in | |||

