Изобретение относится к области биоорганической химии, конкретнее к синтезу псевдопептидов N-ациальных производных биогенных аминов, которые могут быть использованы в биологии и медицине в качестве модуляторов процесса перекисного окисления липидов (ПОЛ).
Антиоксиданты (АО) важный класс соединений, представляющий интерес для фармакологии и медицины. Представителями этого класса веществ являются эндогенные антиоксиданты, например супероксиддисмутаза /1/, дипептид карнозин (H- β -Ala-His-OH) /2/, декарбоксилированный аналог карнозина (N-ациальное производное гистамина b -Ala-HA) псевдопептид карцинин /3/. Кроме того, известны экзогенные АО: природный АО токоферола ацетат и синтетические - дибунол, эмоксипин, мексидол, убинон /1/.
Наиболее близким к заявляемым псевдопептидам по структуре и свойствам является антиоксидант пептидной природы карнозин.
Недостатком карнозина являются высокие действующие дозы и относительно невысокий уровень антиоксидантной активности, что в значительной мере ограничивает использование карнозина в качестве терапевтического препарата /4/.
Целью изобретения является синтез новых более эффективных антиоксидантов N-ациальных производных гистамина аналогов карнозина, а также N-ациальных производных биогенных аминов серотонина и триптамина, обладающих более низкими действующими концентрациями и, кроме того, повышенной липофильностью
аффинностью к мембранам.
Поставленная цель достигается рядом N-ацильных производных биогенных аминов общей формулы X-CO-NH-Y, где X-CO-остаток g -аминомасляной (GABA), L-пироглутаминовой (pGlu), L- и D-глутаминовой (Glu), по g -карбоксилу, кислот: b -аланина, а также лауриновой (Lau) кислоты; -NH-Y-остаток гистамина (НА), серотонина (5-НТ), триптамина (ТА); и способом их получения, заключающимся в ацилировании биогенного амина активированными по карбоксильной группе производными жирных или N-замещенных аминокислот.
Синтез предлагаемых N-аминоациальных производных биогенных аминов осуществляют классическими методами пептидной химии с применением активированных пентафторфениловых эфиров как наиболее активных из известных.
a -Аминогруппу аминокислот замещают трет-бутилоксикарбонильной (Boc) или бензилоксикарбонильной (Z) защитами a -карбоксильную функцию глутаминовой кислоты защищают бензильной (Bzl) группой. Пироглутаминовую кислоту используют в синтезе без защиты иминофункции.
Синтез осуществляют по следующей схеме 1: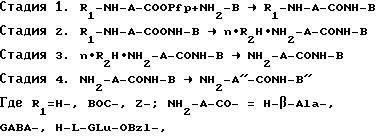
H-D-Glu-OBzl-, H-L-pGlu;
NH2-B HA, TA, 5-OBzl-TA;
NH2-A''-CO- L-Glu-; -D-Glu -NH-B'' -HA, -TA, 5-HT.
R2H HHal(HCl, HBr); CF3COOH; CF3SO3H; n 1, 2, 3.
Стадия 2 исключается в случае R1 Z. Стадия 4 осуществляется при наличии в промежуточном соединении R1-A-CONH- BNα -Z- и Bzl-групп, которые отщепляют каталитическим гидрогенолизом. Стадию 1 проводят в среде безводного апротонного растворителя, предпочтительно диметилформамида (ДМФА), 0,5 1,0 час при комнатной температуре и 24 48 часов при 0 4oC. Вос-защиту отщепляют действием хлористого водорода в органическом растворителе, предпочтительно метаноле или трифторуксусной кислоте (стадия 2).
Преимуществом данного способа синтеза является осуществление очистки и выделения (стадия 3) целевого продукта NH2-A-CONH-B, не содержащего карбоксильных групп, в виде устойчивых кристаллических свободных оснований в одну стадию с высоким выходом путем пропускания соответствующей соли через ионнообменник с четвертичными аммониевыми группами, например, АРА-10п (или 8п) производства НПО "Биолар" (Латвия) с элюцией водным спиртом, преимущественно 60% метанолом или 70% этанолом.
Так, по данному способу с применением Boc-β-Ala-OPfp и ионнообменника АРА-8п в OH- -форме получают карцинин H-b-Ala-HA в виде устойчивого кристаллического основания с высоким выходом и высокой степенью чистоты. Физико-химические характеристики опубликованы в /3/ без раскрытия способа синтеза. Преимущества данного способа синтеза карцинина видны при сравнении с известным способом его получения, заключающимся в конденсации гистамина с Nps-b-Ala-OH N,N''-дициклогексилкарбодиимидным (ДЦГК) методом.
Недостатками данного метода синтеза являются неустойчивость Nps- b -Ala-OH в свободном состоянии и необходимость его использования в виде ДЦГА соли. Кроме того, трудность удаления Nps-хлорида и его производных из реакционной смеси после отщепления Nps-защиты от Nps- b -Ala-HA, а также возможные побочные реакции по имидазолу гистамина при действии ДЦГК, обусловливают, по видимому, необходимость многократной очистки перекристаллизацией дихлоргидрата карцинина, что сказывается на величине выхода и чистоте целевого продукта /5/. Помимо перечисленных недостатков предложенного авторами /5/ метода, в работе отсутствуют характеристики конечного вещества и величина его выхода.
Другим преимуществом предлагаемого способа синтеза псевдопептидов формулы NH2-A-CONH-B, содержащих карбоксильную группу (глутамилсодержащих соединений) является одностадийный способ очистки и выделения целевого продукта в виде свободного основания путем изоэлектрического осаждения из органического растворителя, преимущественно этанола.
Синтез жирнокислотных производных биогенных аминов осуществляют путем ацилирования по аминогруппе биогенного амина хлорангидридом жирной кислоты, например, лауроилхлоридом.
Синтез осуществляют по схеме 2:
X-CO-Cl+NH2B _→ XCONHB,
где X-CO-Cl лауроилхлорид,
NH2-B гистамин,
реакцию проводят в среде апротонного органического растворителя, преимущественно ДМФА, в присутствии 1,1 экв. основания (триэтиламина), при комнатной температуре, в течение 0,5 часа. Преимуществом предлагаемого способа синтеза является простой способ очистки целевого продукта X-CONH-B путем повторной экстракции, используя различие в растворимости целевого продукта, исходных соединений и примесей в воде и органических растворителях.
Предлагаемые псевдопептиды N-ацильные производные биогенных аминов, получены с высокими выходами и высокой степенью чистоты и охарактеризованы ИК-, ЯМР-спектроскопией, элементным анализом, ТСХ и ВЭЖХ.
Предлагаемый способ получения псевдопептидов N-ацильных производных биогенных аминов иллюстрируется следующими примерами.
Индивидуальность полученных соединений проверяют методом ТСХ на пластинках Alufol (нейтр. ФРГ) в метаноле (1), на пластинах Silufol UV-254 (Чехословакия) в системах: метанол (2), изопропанол-вода-25% аммиак 6:1:3 (3), хлораформ-метанол-25% аммиак 5:3:1 (4), бутанол-уксусная кислота -вода 8:3:29 (5), хлороформ-метанол 8:2 (6), хлороформ-метанол 19:1 (7), хлороформ-метанол 9:1 (8), хлороформ-метанол 95:5 (9).
Хроматограммы проявляют хлортолидиновым реактивом, реактивом Паули, реактивом Эрлиха, нингидрином и по свечению в УФ-свете.
Температуру плавления веществ определяют на приборе Boetius (ФРГ).
ИК-спектры снимают в вазелине на приборе Shimadzu YR-35.
Углы удельного вращения измеряют на спектрополяриметре Perkin-Elmer 241-MC (Швеция) и спектрополяриметре Y-20 (Япония).
Спектры ЯМР снимают на приборе Brucker WM-250 (ФРГ) и Varian XL-400 (Япония) в дейтерированных растворителях.
Аналитическую обращенно-фазовую ВЭЖХ проводят в условиях (1): колонка Separon C-18 (3,0 • 250 мм), элюируют 0,1 M Na2HPO4 (pH 2,7, довед. H3PO4), скорость элюирования 0,2 мл/мин; в условиях (2): колонка Separon C-18 (3,0 x 250 мм), элюируют 0,1 M Na2HPO4 (pH 2,4, довед. H3PO4), скорость элюирования 0,2 мл/мин; в условиях (3): колонка Silasorb C-18 (4,0 x 125 мм), элюируют 70% ацетонитрилом в 0,05% растворе гептафтормасляной кислоты в воде; в условиях (4): колонка Hipersil ODS (250 x 3 мм), элюируют градиентом от 0% до 30% ацетонитрила в воде за 30 мин, скорость элюции 1,0 мл/мин. Препаративную обращенно-фазовую ВЭЖХ осуществляют в условиях (5): колонка Silasorb C-18 (250 x 10 мм), элюируют градиентом ацетонитрила в воде от 0% до 99% за 30 мин; скорость элюции 5,0 мин.
Пример 1. β -Аланилгистамин ( b -Ala-HA).
1.1. Na -трет. Бутилоксикарбонил β -аланилгистамин (Boc- b -Ala-HA).
К раствору 1,75 г (4,9 ммоль) Boc- b Ala OPfp в 4 мл безводного ДМФА прибавляют 0,60 г (5,0 ммоль) гистамина и перемешивают 0,5 ч при 20oC и 24 ч при 0oC. Растворитель удаляют в глубоком вакууме. Остаток сушат в вакууме над P2O5. Получают желтое аморфное вещество. Выход 1,38 г (86%), т. пл. 130 139oC, Rf 0,78 (2). ИК-спектр, см-1:1650 (амид I), 1560 (амид II).
1.2. b Аланилгистамин дихлоргидрат (2HCl- b -Ala-HA).
К 0,96 г (2,13 ммоль) Boc- b -Ala-HA добавляют 5,0 мл 3н раствора HCl в метаноле. Перемешивают 1 ч при комнатной температуре. Растворитель удаляют в вакууме, остаток промывают безводным диэтиловым эфиром, сушат в вакууме над P2O5. Выход 0,77 г (89,5%) бурого маслообразного продукта. Очищают на колонке с анионитом АРА-8п (Cl--форма), элюируя 60% раствором метанола в воде. Объединяют фракции с Rf 0,3 (3). Растворитель удаляют в вакууме. Маслообразный остаток лиофилизуют. Сушат в вакууме над P2O5. Получают аморфное стекловидное вещество желтого цвета. Выход 0,47 г (61%), т. пл. 181 183oC, Rf 0,3 (3), 0,56 (1). ИК-спектр, см-1: 1660 (амид I), 1560 (амид II). Найдено, C 37,71; H 6,33; N 21,16; Cl 26,39. C8H16N4C12O. Вычислено,C 37,76; H 6,32; N 21,26; Cl 27,79.
1.3. b -Аланилгистамин ( b -Ala-HA).
На колонку с анионитом АРА-8п (OH--форма) наносят 0,47 г b -Ala-HA•2HCl и элюируют 60% водным раствором метанола. Объединяют фракции с Rf 0,34 (3). Растворитель удаляют в вакууме, остаток лиофилизуют. Сушат в вакууме над P2O5. Получают прозрачное кристаллическое вещество. Выход 0,14 г ( 43,3% ), т. пл. 115 117oC, Rf 0,34 (3), 0,11 (1). ИК-спктр, см-1: 1650 (амид I), 1570 (амид II), 1086 (-C-N-). Спектр H1-ЯМР спектр (CD3OD), dм.д. 2,34 (т, 2H, b -CH2); 2,87 (т, 2H, a -CH2-HA); 3,30 (т, 2H, b -CH2-HA); 3,45 (т, 2H, a -CH2); 6,85 (c, 1H, CH-4-Im); 7,60 (c, 1H, CH-2-Im). Найдено, C 51,26; H 7,70; N 28,31. C8H14N4O•0,3H2O. Вычислено, C 51,22; H 7,70; N 29,05. ВЭЖХ в условиях (3): время выхода индивидуального пика 14,9 мин.
Пример 2. g -Аминобутирилгистамин.
2. 1. Na -трет-Бутилоксикарбонил- γ -аминобутирилгистамин.
К 1,80 г (4,65 ммоль) Boc-GABA-OPfp прибавляют раствор 0,52 г (4,65 ммоль) гистамина в 20 мл безводного диметилформамида. Перемешивают 1 ч при 20oC. Оставляют на 24 ч при 0oC. Растворитель удаляют в глубоком вакууме. Маслообразный остаток кристаллизуют из смеси пентана и эфира (10:12 мл). Выдерживают 24 ч при 0oC. Растворители отделяют декантированием. Остаток сушат в вакууме над P2O5. Получают прозрачное кристаллическое вещество. Выход 1,35 г ( 92,47% ), т. пл. 125 127oC, Rf 0,5 (5). H1-ЯМР спектр (CB3OD), d м. д. 1,45 (с, 9H, CH3-Bu); 1,78 (м, 2H, b -CH2); 2,22 (т, 2H, a -CH2); 2,82 (т, 2H, a -CH2-HA); 3,12 (т, 2H, b -CH2-HA); 3,52 (м, 2H, g -CH2); 6,85 (с, 1H, CH-4-Im); 7,55 (с, 1H, CH-2-Im). Найдено, C 55,01; H 7,84; N 17,87. C14H22N4O3•0,5H2O. Вычислено, C 55,72; H 7,66; N 17,80.
2.2. g -Аминобутирилгистамин.
К 1,05 г (3,33 ммоль) Boc-GABA-HA прибавляют 4,0 мл 4н. раствора HCl в безводном метаноле. Оставляют на 1 ч при 20oC. Растворитель удаляют в вакууме. Полученное желтое маслообразное вещество очищают на колонке с анионитом АРА-10п (OH--форма), элюируя 60% водным метанолом. Фракции с Rf 0,45 (4) объединяют и упаривают в вакууме при 40oC. Остаток лиофилизуют. Сушат в вакууме над P2O5. Получают аморфное стекловидное гигроскопическое вещество. Выход 0,61 г (90,77%), т.пл. 132 134oC, Rf 0,45 (4). ИК-спектр, см-1: 1590 (амид I); 1465 (амид II); H1-ЯМР спектр (CD3OD), d м.д. 1,90 (м, 2H, b -CH2); 2,35 (м, 2H, a -CH2), 2,95 (т, 2H, a -CH2-HA); 3, 30 (т, 2H, b -CH2-HA); 3,50 (т, 2H, g -CH2); 7,30 (c, 1H, CH-4-Im); 8,65 (с, 1H, CH-4-Im); 8,65 (c, 1H, CH-2-Im). Найдено, C 55,69; H 8,07; N 28,32. C9H16N4O. Вычислено, C 55,08; H 8,22; N 28,55. ВЭЖХ в условиях (2): время выхода индивидуального пика 6,41 мин.
Пример 3. L-Пироглутамилгистамин (L-pGlu-HA).
К раствору 2,00 г (15,4 ммоль) L-пироглутаминовой кислоты и 2,85 г (15,49 ммоль) пентафторфенола в 20 мл безводного диметилформамида при -10oC и интенсивном перемешивании прибавляют 3,29 г (15,9 ммоль) N,N''-дициклогексилкарбодиимида. Перемешивают 2,5 ч, при -10oC. Оставляют на 48 ч при 0oC. Осадок дициклогексилмочевины отделяют. К раствору прибавляют 1,72 г (15,9 ммоль) гистамина. Перемешивают 1 ч при 20oC и оставляют на 24 при 0oC. Растворитель удаляют в глубоком вакууме. Маслообразный остаток кристаллизуют из 7 мл сухого этанола. Оставляют на 20 ч при 0oC. Осадок отделяют фильтрованием и прибавляют 25 мл взвеси анионообменника АРА-10п (OH- форма) в 70% этаноле. Перемешивают 0,6 ч, осуществляя непрерывный хроматографический контроль в условиях (1). Раствор, содержащий вещество с Rf 0,67 (1), отделяют от ионообменника фильтрованием. Растворитель удаляют в вакууме, остаток лиофилизуют. Прозрачное аморфное вещество сушат над P2O5 в вакууме. Получают бесцветное кристаллическое вещество. Выход 1,59 г ( 79,5% ), т. пл. 206 209oC, Rf 0,67 (1), 0,44 (2), [a]D-6,0 (Cl, метанол). ИК-спектр, см-1: 1664 (амид I), 1550 (амид II), 1264 (-C-O-). H1-ЯМР спектр (D2O), dм.д. 2,0 (м, 2H, b -CH2; 2,45 (м. 2H, a - CH2); 2,95 (т, 2H, a -CH2-HA); 3,50 (т, 2H, b -CH2-HA); 4,30 (м, 1H, a -CH); 7,00 (с, 1H, CH-4-Im); 7.75 (с, 1H, CH-2-Im). Найдено, C 53,76; H 6,48; N 24,59. C10H14N4O2. Вычислено, C 54, 05; H 6,35; N 25,22. ВЭЖХ в условиях (1): время выхода индивидуального пика 10,5 мин.
Пример 4. g -D-глутамилгистамин (g-D-Glu-HA).
4.1. Na -Бензилоксикарбонил-α-бензил- g -D-глутамилгистамин (Z-D-Glu(HA)-OBzl.
К 580 мг (1 ммоль) Z-D-Glu(OPfp)-OBzl, растворенному в 1 мл ДМФА, прибавляют 110 мг (1 ммоль) гистамина при интенсивном перемешивании. После растворения прибавляют 0,2 мл (2 ммоль) N-метилморфолина. Реакционную смесь оставляют при 18 22oC на 48 часов. Растворитель удаляют в вакууме. Маслообразный остаток очищают колоночной хроматографией на силикагеле 40/100, элюируют смесью хлороформ-метанол (8:2). Фракции, содержащие целевой продукт с Rf 0,46 (6), объединяют, растворитель удаляют в вакууме. Получают прозрачное желтоватое масло. Выход 0,232 г ( 50% ). Rf 0,46 (6).
4.2. g-D-глутамилгистамин (g-D-Glu-HA).
К 190 мг (0,41 ммоль) Z-D-Glu(HA)-OBzl, растворенному в 3,0 мл метанола, прибавляют 2)) мг 10% Pd/C при интенсивном перемешивании. Гидрируют в токе водорода в течение 3 часов. Реакционную смесь фильтруют. Растворитель из фильтрата удаляют в вакууме. Получают прозрачное кристаллическое вещество. Выход 80 мг (81% ). Rf 0,26 (4), т. пл. 184 190oC. H1-ЯМР спектр (D2O), d м.д. 1,77 (к, 2H, b -CH2); 2,07 (т, 2H, a -CH2-HA); 2,52 (т, 2H, g -CH2); 3,17 (т, 2H, b -CH2-HA); 3,40 (т, 1H, a -CH); 6,67 (с, 1H, CH-4-Im); 7,47 (с, 1H, CH-2-Im). Масс-спектр m/z: [M+1H]+241,3. ВЭЖХ в условиях (1): время выхода индивидуального пика 7,4 мин.
Пример 5. g -L-глутамилтриптамин.
5.1. Na -трет-Бутилоксикарбонил- α -бензил- g -L-глутамилтрипатмин (Boc-L-Glu(TA)OBzl).
К 503 мг (1 ммоль) Boc-L-Glu(OPfp)OBzl, растворенному в 1 мл ДМФА, прибавляют 160 мг (1 ммоль) триптамина при интенсивном перемешивании. После полного растворения прибавляют 0,3 мл (3 ммоль) N-метилморфолина. Реакционную смесь оставляют при 18 22oC в темноте на 48 часов. Растворитель удаляют в вакууме. Маслообразный остаток очищают колоночной хроматографией на силикагеле 40/100, элюируют хлороформом. Фракции, содержащие целевой продукт с Rf 0,51 (7), объединяют, растворитель удаляют в вакууме. Получают прозрачное желтоватое масло. Выход 0,24 г ( 50% ). Rf 0,51 (7).
5.2. a -Бензил- g -глутамилтриптамин (H-L-Glu(TA)OBzl).
К 125 мг (0,45 ммоль) Boc-L-Glu(TA)OBzl прибавляют 1,0 мл 4н. HCl/диоксан, интенсивно перемешивают, продувают азотом. Через 30 мин к реакционной смеси прибавляют 5 мл диэтилового эфира. Выпавший маслообразный осадок отделяют от раствора декантацией и промывают 5 мл диэтилового эфира. К осадку 0,1 мл триэтиламина и 5 мл этилацетата, интенсивно перемешивают, затем реакционную смесь фильтруют. Растворитель из фильтрата удаляют в вакууме. Маслообразный остаток очищают колоночной хроматографией на силикагеле 40/100. Элюируют хлороформом, смесью хлороформ-метанол (23:2). Фракции, содержащие целевой продукт с Rf 0,27 (8), объединяют, растворитель удаляют в вакууме. Получают прозрачное масло. Выход 50 мг ( 31% ). Rf 0,27 (8).
5.3. g -Глутамилтриптамин (H-L-Glu(TA)-OH).
К 40 мг (0,1 ммоль) H-L-Glu(TA)OBzl прибавляют 2,0 мл метанола и 40 мг 10% палладия на активированном угле. Гидрируют в токе водорода в течение 1,5 часов. Pd/C отделяют фильтрованием, растворитель из фильтрата удаляют в вакууме. Получают прозрачные розовые кристаллы. Выход 22 мг ( 75% ), т. пл. 82 90oC, Rf 0,39 (4). H1-ЯМР-спектр (CD3OD), d м.д. 3,85 (т, 1H, a -CH), 2,12 (м, 2H, b -CH2), 2,43 (т, 2H, g -CH2), 2,96 (т, 2H, a -CH2-TA), 3,48 (т, 2H, b -CH2-TA), 6,99 (т, 1H, 5-CH-In), 7,05 (с, 1H, 2-CH-In), 7,06 (т, 1H, 6-CH-In), 7,55 (д, 1H, 4-CH-In). Масс-спектр m/z [M+1H]+290,1. ВЭЖХ в условиях (4): время выхода индивидуального пика 22,0 мин.
Пример 6. g -L-глутамилсеротонин (H-L-Glu(5-HT)-OH).
6.1. Na -трет.-Бутилоксикарбонил- α -бензил- g -L-глутамил(5-бензилокситриптамин) (Boc-L-Glu(5-OBzl-TA)-OBzl).
Соединение Boc-L-Glu(5-OBzl-TA)-OBzl синтезируют аналогично соединению Boc-L-Glu(TA)-OBzl (прим. 5.1. ), исходя из 503 мг (1 ммоль) Boc-L-Glu(OPfp)-OBzl и 250 мг (1 ммоль) 5-OBzlTA. Получают прозрачное желтоватое масло. Выход 350 мг ( 60% ). Rf 0,63 (7).
6.2. a -Бензил- g -глутамил(5-бензилокситриптамин)(H-Glu(5-OBzl-TA)-OBzl).
Соединение H-L-Glu(5-OBzl-TA)-OBzl синтезируют аналогично соединению H-L-Glu(TA)-OBzl (прим. 6.2.), исходя из 330 мг (0,6 ммоль) Boc-L-Glu(5-OBzl-TA)-OBzl. Получают прозрачное желтое масло. Выход 177 мг ( 61% ). Rf 0,48 (8).
6.3. g -L-глутамилсеротонин (H-L-Glu(5-HT)-OH.
Соединение H-L-Glu(5-HT)-OH синтезируют аналогично соединению H-L-Glu(TA)-OH (прим. 6.3.), исходя из 155 мг (0,32 ммоль) H-L-Glu(5-OBzl-TA)-OBzl. Получают прозрачное розовое масло, которое очищают препаративной хроматографией в условиях (5). Получают прозрачные бесцветные кристаллы. Выход 76 мг ( 80% ), Т. пл. 105 110oC, Rf 0,43 (4). H1-ЯМР спектр (CD3OD), d м. д. 3,62 (т,1H, a -CH2); 2,12 (м, 2Н,-b-CH2); 2,45 (т, 2H, g -CH2); 2,91 (т, 2H, a -CH2-TA); 3,51 (т, 2H, b -CH2-TA); 6,70 (д.д, 1H, 6-CH-In); 6,97 (д, 1H, 4-CH-In); 7,20 (д, 1H, 7-CH-In). Масс-спектр m/z [M]+304,9. ВЭЖХ в условиях (4): время выхода индивидуального пика 12,2 мин.
7. N-Лауроилгистамин.
К раствору 0,250 г (2,25 ммоль) гистамина в 6 мл сухого ДМФА при интенсивном перемешивании прибавляют 0,31 мл (2,25 ммоль) триэтиламина, затем по каплям добавляют 0,54 мл (2,48 ммоль) хлорангидрида лауриновой кислоты. Через 30 минут прибавляют 0,15 мл триэтиламина до pH 8. Растворитель удаляют в вакууме. Осадок промывают от избытка лауриновой кислоты на пористом фильтре 2 x 10 мл ацетоном и 2 x 15 мл водой. Водные промывки собирают и лиофилизуют. Получают белое кристаллическое вещество. Выход: 0,250 г ( 66,1% ), т. пл. 121 125oC, Rf 0,37 (9), ИК-спектр, см-1: 1569 (амид I); 1460 (амид II), H1-ЯМР спектр (CD3OD), d м. д. 0,91 (т, 3H, w -CH3); 1,30 (ш.с, 16H, 8-CH2); 1,56 (т, 2H, b -CY2); 2,17 (т, 2H, a -CY2); 2,92 (т, 2H, a -CH2-HA); 3,50 (т, 2H, b -CH2-HA); 7,30 (с, 1H, CH-4-Im); 8,80 (с, 1H, CH-2-Im). Найдено, C 69,67; H 10,57; N 14,29. C17H31N3O. Вычислено, C 69,52; H 10,56; N 14,31.
Влияние предлагаемого ряда псевдопептидов на процесс ПОЛ показано на модели окисления жирной (линолевой) кислоты в Fe2+-аскорбатзависимой системе по накоплению ТБК-реактивных продуктов (малонового диальдегида) с использованием методики, описанной в (6). Ингибирование аскорбатзависимого свободнорадикального окисления липидов в данной системе предлагаемым рядом псевдопептидов доходило до 60 70% при действующих концентрациях 10-2 10-6 М. Таким образом, антиоксиданты из предлагаемого ряда псевдопептидов действуют при существенно более низких концентрациях, чем карнозин, причем достигается более высокий процент ингибирования.
Данные свойства предлагаемого ряда соединений могут иметь широкое применение, в том числе в онкологии (противоопухолевое действие), иммунологии (модуляции иммунных реакций, радиопротекторное действие), в кардиологии (противоишемическое и противогипоксическое действие), в офтальмологии (лечение катаракт), для коррекции нарушений деятельности ЦНС, а также в качестве антигипоксанта при использовании гипербарической оксигенации.
Таким образом, предложенный ряд псевдопептидов N-ацильных производных биологически активных аминов модуляторов процесса перекисного окисления липидов, обладающих, по сравнению с известным аналогом карнозином, более низкими действующими концентрациями (в 102 103 раз) в аналогичных условиях и более высоким уровнем антиоксидантной активности (до 60 -67% ингибирования), с повышенной липофильностью-аффинностью к мембранам. Предложены простые и эффективные способы получения всех предлагаемых соединений.
N-ациальные производные биогенных аминов - модуляторы процесса перекисного окисления липидов и способ их получения. Использование: изобретение относится к области биоорганической химии, а также к области биологии и медицины. Сущность изобретения: N-ациальные производные биогенных аминов общей формулы X-CO-NH-Y, где X-CO-остаток амино- или жирной кислоты, а -NH-Y-остаток серотонина, триптамина, гистамина, модулирующие процесс перекисного окисления липидов. Способ получения соединений формулы XCONHY путем анилирования аминогруппы биогенного амина активированными пентафторфениловыми эфирами N-защищенных аминокислот или хлорангидридами жирных кислот в органическом растворителе с последующей очисткой ионообменной хроматографией, изоэлектрическим осаждением или повторной экстракцией из воды и органических растворителей. 2 с.п. ф-лы.
X-CO-NH-Y,
где X-CO- остаток амино или жирной кислоты, выбранной из группы: γ-аминомасляная, пироглутаминованная, β- аланин, L- или D-глутаминовая по γ-карбоксилу, лауриновая; NH-Y- остаток, выбранный из группы: серотонин, триптамин, гистамин-модуляторы перекисного окисления липидов.
X-CO-NH-Y,
где X-CO- остаток амино или жирной кислоты, выбранной из группы: γ-аминомасляная, пироглутаминовая, β- аланин, L- или D-глутаминовая по γ-карбоксилу, лауриновая; NH-Y- остаток, выбранный из группы: серотонин, триптамин, гистамин,
отличающийся тем, что аминогруппу гистамина, или триптамина, или О-бензилсеротонина ацилируют активированными пентафторфениловыми эфирами Nα- Вос- или Z-, ω-Bzl-замещенных аминокислот или хлорангидридом жирной кислоты в диметилфомамиде при 15 30oС с отщеплением от полученных продуктов Nα, ω-защитных групп в кислой среде или гидрогенолизом, с последующей очисткой и выделением целевых продуктов в виде свободных оснований путем колоночной хроматографии на анионнобменнике в ОН--форме, или изоэлектрическим осаждением, или повторной экстракцией из воды и органического растворителя.

