Область техники, к которой относится изобретение
Настоящее изобретение относится к ингибиторам PARP и ингибиторам полимеризации тубулина, и в настоящем изобретении предлагаются соединения и композиции, содержащие раскрываемые соединения. Кроме того, в настоящем изобретении предлагаются способы применения раскрываемых ингибиторов PARP и ингибиторов полимеризации тубулина, например, в качестве лекарственного средства.
Предпосылки к созданию изобретения
Нуклеарный фермент поли(АДФ-рибозо)полимераза-1 (PARP-1) является членом семейства ферментов PARP. Указанное растущее семейство состоит из ферментов PARP, таких как, например: PARP-1, PARP-2, PARP-3 и Vault-PARP; и ферментов танкираз (TANK), таких как, например: TANK-1 и TANK-2. PARP называют также поли(аденозин-5'-дифосфорибозо)полимеразой или PARS (поли(АДФ-рибозо)синтетаза).
Танкиразы (TANK) были идентифицированы в качестве компонентов теломерного комплекса человека. Было также высказано предположение, что они играют определенные роли в контролировании митотического веретена и регулировании направления миграции везикул, а также могут служить в качестве каркасов для белков, которые принимают участие в различных других клеточных процессах. Теломеры, которые играют существенную роль в обеспечении жизнедеятельности и устойчивости хромосом, поддерживаются теломеразой, специализированной обратной транскриптазой. TANK представляют собой (АДФ-рибозо)трансферазы, которые обладают некоторыми свойствами как сигнальных, так и цитоскелетных белков. Они содержат домен PARP, который катализирует поли-АДФ-рибозилирование субстратных белков, домена SAM (sterile alpha motif), который является общим для некоторых сигнальных молекул, и домена ANK, включающего от 16 до 24 анкириновых повтора, которые присутствуют также в цитоскелетном белке анкирине. Домен ANK взаимодействует со многими другим белками, включая теломерный белок фактор-1 связывания теломерного повтора (TRF-1). Поэтому указанные белки называют TRF1-взаимодействующими анкирин-связанными АДФ-рибозополимеразами (TANK).
Одной из функций TANK является АДФ-рибозилирование TRF-1. Функционирование теломеров человека контролируется комплексом связанных с теломерами белков, который включает два теломер-специфичных ДНК-связывающих белка, TRF-1 и TRF-2. TRF-2 защищает концы хромосом, а TRF-1 регулирует длину теломера. АДФ-рибозилирование подавляет способность TRF-1 связываться с теломерной ДНК. Указанное поли-АДФ-рибозилирование TRF-1 высвобождает TRF-1 из теломеров, тем самым раскрывая теломерный комплекс, и открывает доступ к теломеразе. Таким образом, TANK функционируют в качестве позитивного регулятора длины теломеров, позволяя осуществлять удлинение теломеров под действием теломеразы.
На некоторые из ролей TANK указывает идентичность белков, с которыми они взаимодействуют - чувствительная к инсулину аминопептидаза, белки Mc11 (которые являются членами семейства Bc1-2), ядерный антиген-1 Эпштейна-Барра, ядерный белок и белок митотического аппарата, а также цитоплазменный и гетерохроматиновый фактор TAB182 - и их разнообразные внутриклеточные локализации (ядерные поры, аппарат Гольджи и митотические центросомы).
Танкираза-2 (TANK-2) отличается от танкиразы-1 (TANK-1) тем, что у нее отсутствует N-концевой домен HPS (составлен гомополимерными повторами остатков His, Pro и Ser), который обнаруживается в TANK-1. Тем не менее, она, вероятно, обладает некоторыми перекрывающимися функциями с танкиразой-1, при условии, что оба белка имеют сходные субклеточные локализации, связаны друг с другом и связывают многие из одинаковых белков.
PARP-1 является основным нуклеарным белком, имеющим размер 116 кДа, который включает три домена: N-концевой ДНК-связывающий домен, который содержит две цинкосодержащие пальцеообразные области, домен аутомодифицирования и С-концевой каталитический домен. Указанный фермент синтезирует поли(АДФ-рибозу) - разветвленный полимер, который содержит более 200 единиц АДФ-рибозы. Белки-акцепторы поли(АДФ-рибозы) прямо или опосредованно участвуют в поддержании целостности ДНК. Они включают гистоны, белки HMG, топоизомеразы, ДНК- и РНК-полимеразы, ДНК-лигазы и Ca2+- и Mg2+-зависимые эндонуклеазы, факторы репарации одноцепочечных разрывов и факторы восстановления путем удаления поврежденного участка. Белок PARP экспрессируется с высоким уровнем во многих тканях и в основном в иммунной системе, сердце, мозге и клетках зародышевой линии. В обычных физиологических условиях PARP проявляет минимальную активность. Тем не менее, повреждение ДНК вызывает немедленное вплоть до 500-кратное увеличение активности PARP. В результате продукция поли(АФД-рибозы) имеет три последствия: во-первых, вызываемое повреждением ДНК поли-АФД-рибозилирование N- или C-концевых хвостов гистона H1 и H2B или селективное взаимодействие указанных белков со свободной или связанной с PARP-1 поли(АФД-рибозой) приводит к смягчению 30-нм хроматиновых волокон и увеличивает доступ к разрывам; во-вторых, она сигнализирует о возникновении и степени повреждения ДНК, так что клетка может установить адаптивный ответ в соответствии с тяжестью повреждения (репарация ДНК или самоубийство клеток); в-третьих, она опосредует быстрый рекрутмент факторов репарации одноцепочечного разрыва и факторов восстановления путем удаления поврежденного участка.
Одноцепочечные разрывы (SSB) спонтанно возникают во всех клетках. В отсутствие активности PARP-1 указанные SSB могут при репликации превратиться в двухцепочечные разрывы (DSB), которые могут привести к разрушению репликационных вилок. DSB идентифицируют по их эпигенетической отметке, фосфорилированию варианта корового гистона H2AX (γH2AX). Очень быструю локальную деконденсацию хроматина, который независимо от γH2AX возникает в DSB, можно отнести на счет продукции поли(АФД-рибозы), которую локально опосредует PARP-1.
Кроме того, эволюционные или внешние стимулы, такие как стероиды или тепловой шок, индуцируют активацию PARP-1 и зависимое от поли(АФД-рибозы) отделение гистонов от хроматина, и тем самым благоприятствуют раскрытию структуры хроматина, что в отсутствие разрывов ДНК может позволить активацию транскрипции.
Интенсивная активация PARP в клетках, страдающих от массового повреждения ДНК, приводит к значительной элиминации NAD+. Короткий полупериод существования поли(АДФ-рибозы) приводит к быстротечности функционального цикла. Как только поли(АДФ-рибоза) образовалась, она быстро разлагается под действием конститутивно активной поли(ADP-рибозо)гликогидролазы (PARG), а также фосфодиэстеразы и (АДФ-рибозо)протеинлиазы. PARP и PARG составляют цикл, в котором большое количество NAD+ превращается в АДФ-рибозу. Менее чем за час избыточное стимулирование PARP может вызвать падение уровня NAD+ и АТФ до уровня, составляющего меньше, чем 20% от нормального. Подобный сценарий особенно пагубен при ишемии, когда кислородная недостаточность уже подвергла значительному риску выработку энергии клетками. Полагают, что последующее образование свободных радикалов при реперфузии является основной причиной повреждения тканей. Часть падения концентрации АТФ, что типично для многих органов при ишемии и реперфузии, можно связать с элиминацией NAD+, вызванной функциональным циклом поли(АДФ-рибозы). Таким образом, ожидают, что ингибирование PARP или PARG сохраняет уровень клеточной энергии и тем самым обеспечивает возможность ишемизированным тканям выжить после инсульта.
Как указано выше, внутриклеточная локализация нескольких PARP указывает на физиологическую роль поли-АФД-рибозилирования при регулировании клеточного деления.
Вероятно, TANK-1 требуется для полимеризации поли(АФД-рибозы), связанной с митотическим веретеном. Активность TANK-1 по отношению к поли-АФД-рибозилированию может иметь большое значение для точного образования и поддержания биполярности веретена. Кроме того, было показано, что PARP-активность TANK-1 необходима для нормального отделения теломеров перед анафазой. Препятствия для проявления PARP-активности танкиразы приводят к аберрантному митозу, который вызывает блокировку цикла транзиторных клеток, вероятно, вследствие активации контрольной точки веретена, за которой следует смерть клетки. Поэтому ожидают, что ингибирование транкираз вызывает цитотоксическое действие на разрастающиеся опухолевые клетки.
PARP-1 и PARP-2 локализуются в центросомах, где они взаимодействуют с кинетохорными белками. Удаление гена Parp-2 у мышей приводит к значительному вызванному повреждением ДНК неправильному разделению хромосом, связанному с кинетохорными дефектами, которое указывает на то, что PARP-2 обладает важной охранной функцией для целостности перицентрического гетерохроматина. Кроме того, PARP-1 ассоциируется с центросомами, связывающими цепь контролирования повреждений ДНК с митотической контрольной точки.
Надежно установлена кардинальная роль PARP при репарации разрывов цепи ДНК, особенно в тех случаях, когда они вызваны непосредственно ионизирующим излучением или проявляются опосредованно после ферментативной репарации повреждений ДНК, вызываемых метилирующими агентами, ингибиторами топоизомеразы I типа и другими химиотерапевтическими средствами, такими как цисплатин и блеомицин. Различные исследования с использованием мышей-“нокаутов”, моделей транс-доминантного ингибирования (сверхэкспрессия ДНК-связывающего домена), антисмысловых ингибиторов и ингибиторов с небольшой молекулярной массой демонстрируют роль PARP при репарации и выживании клеток после индуцирования повреждения ДНК. Ингибирование ферментативной активности PARP должно приводить к повышенной чувствительности опухолевых клеток к обработкам, вызывающим повреждение ДНК.
Сообщалось, что ингибиторы PARP эффективны при радиосенсибилизации (гипоксической) опухолевых клеток и эффективно препятствуют восстановлению опухолевых клеток при потенциально летальном и сублетальном повреждении ДНК после радиационной терапии, в первую очередь за счет способности ингибиторов препятствовать воссоединению разрывов нити ДНК и за счет воздействия на несколько путей передачи сигнала о повреждении ДНК.
В патенте США № 5177075 рассмотрено несколько изохинолинов, которые применяли для усиления летального действия на опухолевые клетки ионизирующего излучения или химиотерапевтических средств. Weltin et al., “Effect of 6(5-Phenanthridinone), an Inhibitor of Poly(ADP-ribose) Polymerase, on Cultured Tumor Cells”, Oncol. Res., 6: 9, 399-403 (1994) обсуждают ингибирование активности PARP, уменьшение пролиферации опухолевых клеток и выраженный синергический эффект в том случае, когда опухолевые клетки подвергают совместной обработке с алкилирующим средством.
Обзоры, касающиеся современного уровня техники, опубликованы в Li and Zhang, IDrugs 2001, 4(7): 804-812, Ame et al., Bioassays 2004, 26: 882-883 и Nguewa et al., Progress in Biophysics & Molecular Biology 2005, 88: 143-172.
Утрата PARP-1 усиливает образование разрывов ДНК, которые залечиваются путем гомологической рекомбинации без непосредственного регулирования самого процесса гомологической рекомбинации. Наследственный рак груди обычно связан с наследуемыми дефектами в одном из аллелей BRAC1 или BRAC2. BRAC1 и BRAC2 важны для гомологической рекомбинации. Оставшийся функциональный аллель BRCA1 или BRCA2 может быть потерян в некоторых клетках, тем самым способствуя онкогенезу. Таким образом, возникающие опухоли дефицитны по BRCAl или BRCA2 (например, BRCA2 -/-), в то время как соматические клетки сохраняют функциональные белки BRCA (BRCA2 +/-). Подавление активности PARP в BRCA1- или BRCA2-дефектном окружении может привести к возникновению повреждений ДНК, которые обычно залечиваются путем родственного хроматидного обмена, что приводит к хроматидным отклонениям от нормы и утрате жизнеспособности. При условии повышенной чувствительности BRCA-дефективных клеток для достижения терапевтического эффекта могут потребоваться лишь относительно небольшие уровни ингибиторов PARP-1. Это еще один пример того, как ингибиторы несущественного белка репарации ДНК могут использоваться в качестве единственного средства для лечения опухолей.
Согласно обзору Horvath и Szabo (Drug News Perspect 20(3), April 2007, 171-181), самые последние исследования показывают, что ингибиторы PARP повышают смертность раковых клеток в основном вследствие того, что ингибиторы препятствуют репарации ДНК на различных уровнях. Недавние исследования также показывают, что ингибиторы PARP подавляют развитие кровеносных сосудов либо за счет ингибирования экспрессии фактора роста, либо за счет подавления индуцированных фактором роста пролиферативных клеточных ответов. Указанные наблюдения также могут быть причастны к способам противоракового воздействия, которое ингибиторы PARP оказывают в условиях in vivo.
Кроме того, исследование, проведенное Tentori et al. (Eur. J. Cancer, 2007, 43 (14) 2124-2133), показывает, что ингибиторы PARP аннулируют миграцию, вызванную действием VEGF или плацентарного фактора роста, и препятствуют образованию трубочкоподобных сетей в клеточных системах, а также уменьшают развитие кровеносных сосудов в условиях in vivo. Указанное исследование также показывает, что индуцированное фактором роста развитие кровеносных сосудов отсутствует у лишенных PARP-1 мышах-нокаутах. Результаты данного исследования подтверждают необходимость нацеливаться на PARP для оказания действия, направленного против развития кровеносных сосудов, и придают новый терапевтический смысл использованию ингибиторов PARP при лечении рака.
Ингибиторы PARP по настоящему изобретению проявляют также противораковую активность, связанную с прерыванием полимеризации тубулина.
Тубулин представляет собой гетеродимер двух родственных белков, обозначаемых как α- и β-тубулин. Тубулин полимеризуется с образованием структур, которые называют микротрубочки. Микротрубочки представляют собой высоко активные цитоскелетные элементы и играют критическую роль во многих процессах клеток эукариотов, включая митоз, подвижность клеток, форму клеток, транспорт внутриклеточных органелл и межклеточные взаимодействия.
Для протекания правильного процесса деления клеток важно, чтобы микротрубочки были способны полимеризоваться и деполимеризоваться. Микротрубочки в митотическом веретене более активны, чем микротрубочки в неделящихся клетках, и таким образом, на них могут быть нацелены агенты, которые влияют на активность микротрубочек. Изменяя полимеризацию/деполимеризацию микротрубочек, указанные агенты влияют на образование митотического веретена, останавливают делящиеся клетки в фазе G2/M клеточного цикла и, в конце концов, приводят к смерти клеток в результате апоптоза. Поскольку опухолевые клетки обладают высокими скоростями пролиферации, то на них могут быть нацелены указанные антимитотические агенты.
Были идентифицированы три основных класса тубулин-связывающих лекарств, а именно: аналоги колхицина, алкалоиды винка и таксаны, каждый из которых имеет специфический участок для связывания в молекулах β-тубулина. Паклитаксел и родственные таксаны представляют собой класс лекарств, которые стабилизируют микротрубочки, т.е. контролируют процесс, который, в конце концов, приводит к замораживанию структур микротрубочек, так что они не могут реструктурироваться. Последующая остановка митоза индуцирует механизм апоптоза, который приводит к смерти клеток. Второй класс указанных соединений, аналоги колхицина, а также несколько других соединений, связываются с одним и тем же участком в β-тубулине, что и колхицин, и нарушают процесс полимеризации и образования микротрубочек. Третий класс соединений, винбластины и другие родственные лекарства на основе винка, связываются с участком Vinca и препятствуют образованию микротрубочек, а также дестабилизируют микротрубочки.
Тубулин является также мишенью при лечении болезненных состояний, которые зависят от или являются результатом аномального образования кровеносных сосудов (неоваскуляризация), таких как злокачественные опухоли. В таких случаях цитоскелет эндотелиальных клеток сосудов разрушается за счет деполимеризации микротрубочек, что является следствием ингибирования полимеризации тубулина, приводящей к образованию микротрубочек. Длина микротрубочек зависит от скорости деполимеризации по сравнению со скоростью полимеризации. Деполимеризация микротрубочек путем ингибирования полимеризации приводит к изменению морфологии клеток эндотелия, что может вызвать блокирование или остановку кровотока. В случае раковых опухолей поток крови к болезненным тканям прекращается, тем самым опухоль лишаются кислорода и питательных веществ, что приводит к смерти клеток вследствие некроза. Неоваскулярные системы более чувствительны к действию указанных агентов, поскольку они более зависимы от цитоскелета микротрубочек, чем нормальные, здоровые эндотелиальные клетки сосудов, которые также поддерживаются цитоскелетными структурами на основе актина. Для ряда ингибиторов полимеризации тубулина, которые нацеливаются на участок связывания колхицина в тубулине, воздействия на сосуды можно добиться при более низкой концентрации in vivo, по сравнению с антипролиферативным способом воздействия. Таким образом, агенты, которые нацеливаются на участок связывания колхицина в тубулине потенциально могут обладать двойным действием, т.е. оказывать антимитотическое и антиваскулярное воздействие.
Сохраняется потребность в эффективной и действенной противораковой терапии, которая эффективна против опухолей, в настоящее время неизлечимых или трудно поддающихся лечению, которая эффективна против опухолей, обладающих множественной лекарственной устойчивостью, и которая вызывает минимальные побочные эффекты. В настоящем изобретении предлагаются соединения, композиции для ингибирования и способы ингибирования активности PARP и ингибирования связывания тубулина, с целью лечения рака. Соединения и композиции по настоящему изобретению отличаются от известных из области техники тем, что они способны оказывать двойное действие (ингибирование PARP и ингибирование связывания тубулина). Кроме того, они обладают высокой ингибиторной активностью по отношению к TANK, приводящей к усиленному противораковому действию, что делает их наиболее пригодными для терапии с использованием одного агента. Они также пригодны для повышения эффективности химиотерапии и радиационной терапии, при этом основной эффект от их воздействия заключается в запускании механизма отмирания клеток в условиях повреждения ДНК.
Уровень техники
В WO 03/101985, опубликованной 11 декабря 2003, раскрываются производные 2-оксо-1,3,4-тригидрохиназолинила для лечения расстройств, связанных с пролиферацией клеток.
В EP 1487800, опубликованном 2 октября 2005, раскрывается фенантридиноны в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В EP 1687277, опубликованном 16 июня 2005, раскрываются 6-алкенил- и 6-фенилалкилзамещенные 2-хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В EP 1709011, опубликованном 16 июня 2005, раскрываются 6-фенилалкилзамещенные 2-хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В EP 1709012, опубликованном 16 июня 2005, раскрываются 6-замещенные 2-хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В EP 1694653, опубликованном 30 июня 2005, раскрываются 6-циклогексилалкилзамещенные 2-хинолиноны и 2-хиноксалиноны в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В WO 2005/097750, опубликованной 2 октября 2005, раскрываются замещенные пиридоны в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В WO 2005/117876, опубликованной 15 декабря 2005, раскрываются низкомолекулярные ингибиторы, обладающие двойным ингибиторным действием на рак и развитие кровеносных сосудов.
В WO 2006/003146, опубликованной 12 января 2006, раскрываются производные хиназолинона в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В WO 2006/003147, опубликованной 12 января 2006, раскрываются производные фталазина в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В WO 2006/003148, опубликованной 12 января 2006, раскрываются производные хиназолиндиона в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В WO 2006/003150, опубликованной 12 января 2006, раскрываются замещенные производные 2-алкилхиназолинона в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В WO 2007/025009, опубликованной 1 марта 2007, раскрываются аналоги инденоизохинолинона в качестве ингибиторов поли(АДФ-рибозо)полимеразы.
В WO 2007/095628, опубликованной 23 августа 2007, раскрываются пиразолохинолиноны в качестве действенных ингибиторов PARP.
В WO 2008/107478, опубликованной 12 сентября 2008, раскрываются производные хинолинона в качестве ингибиторов PARP и TANK.
Публикация Tentori et al, European Journal of Cancer, vol. 43, no. 14, 2007 касается ингибирования поли(АДФ-рибозо)полимеразы (PARP) или делеции гена PARP-1, которая сдерживает развития кровеносных сосудов.
Описание изобретения
Настоящее изобретение касается соединений формулы (I)
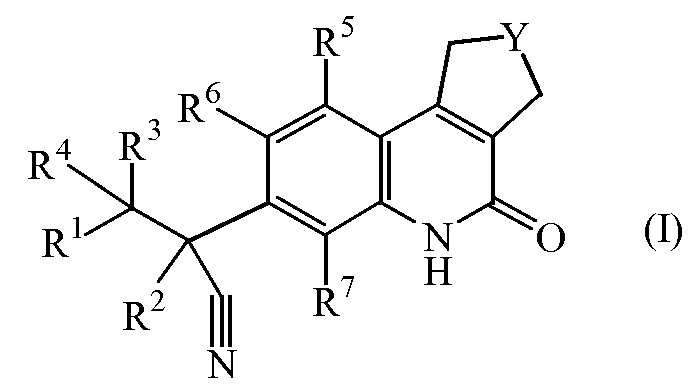
включая их стереохимически изомерные формы;
где
Y обозначает CH2 или CH2-CH2;
R1 обозначает арил или Het;
где арил обозначает фенил или нафталинил;
где Het обозначает тиенил, пирролил, пирролинил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, оксадиазолил, триазолил, тетразолил, тиадиазолил, фуранил, пиперидинил, пиридинил, пиридазинил, пиримидинил, пиперазинил, пиразинил, триазинил, индолизинил, азаиндолизинил, индолил, индолинил, бензотиенил, индазолил, бензоксазолил, бензимидазолил, бензофуранил, бензотиазолил, бензотриазолил, хроманил, пуринил, хинолинил, циннолинил, фталазинил, хиназолинил, хиноксазолинил, нафтиридинил или птеридинил;
два атома углерода в ариле или Het могут быть соединены мостиковой связью (т.е. образуют би- или трициклический фрагмент), при этом двухвалентный радикал выбран из
каждый арил, Het, имеющий мостиковую связь арил или имеющий мостиковую связь Het может быть замещен одним, двумя, тремя, четырьмя или пятью заместителями, каждый из которых независимо выбран из атома галогена, циано, нитро, гидроксикарбонила, C1-6алкила, C2-6алкенила, C2-6алкинила, C3-6циклоалкила, C3-6цикло-алкиламино, метилэтиламино, аминоC3-6циклоалкила, галогеноC1-6алкила, тригалогеноC1-6алкила, C1-6алкилкарбонила, C1-6алкилоксикарбонила, C2-6алкенилкарбонила, оксима, C1-6алкилоксима, амидоксима, -C≡C-CH2O-CH3, -C≡C-CH2N(CH3)2, -C≡C-Si(CH3)3, гидроксиC1-6алкила, гидроксиC2-6алкенила, гидроксиC2-6алкинила, цианоC1-6алкила, цианоC2-6алкенила, аминокарбонилC1-6алкила, C1-6алкилсульфонилC1-6алкила, C1-6алкилсульфонилC2-6алкенила, C1-6алкилсульфонилC2-6алкинила, -PO(OC1-6алкил)2, -B(OH)2, -S-CH3, SF5, C1-6алкилсульфонила, -NR8R9, C1-6алкилNR8R9, -OR8, -C1-6алкилOR8, -CONR8R9, пиперидинилC1-6алкила, пиперазинилC1-6алкила, C1-6алкилпиперазинилC1-6алкила, морфолинилC1-6алкила, пиперидинила, пиперазинила, C1-6алкилпиперазинила, морфолинила, фенила, тиенила, пиразолила, пирролила, пирролидинила, пиридинила, пиримидинила, оксадиазолила, имидазолила, имидазолилC2-6алкинила, C1-6алкилимидазолилC2-6алкинила, цианопиридинила, фенилC1-6алкила, фенилC2-6алкенила, морфолинилC1-6алкила, C1-6алкилоксифенила, тригалогеноC1-6алкилфенила, метилпиразолила, галогенопиримидинила или диметиламинопирролидинила; или
R1 обозначает радикал формулы
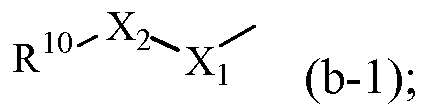
где X1 обозначает CH2, NH или N-CH3;
где X2 обозначает CH2, C=O, O, NH или N-CH3;
где R10 обозначает фенил, пиридинил, пиридазинил или пиримидинил, где каждый фенил, пиридинил, пиридазинил или пиримидинил может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, гидрокси, циано, C1-6алкила, амино, полигалгеноC1-6алкила или C1-6алкилокси; или
R1 обозначает радикал формулы
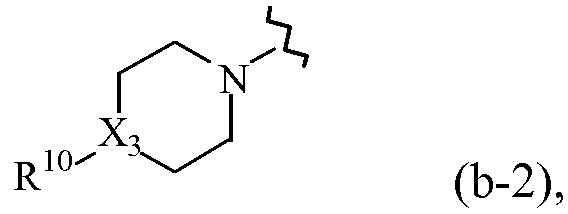
где X3 обозначает CH или N;
R2 обозначает метил, этил, пропил или C3-6циклоалкил;
каждый R3 и R4 независимо выбран из атома водорода, метила, этила, пропила, гидрокси, трифторметила, метилокси; или же R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют циклопропильный цикл или радикал формулы С(=О);
каждый R5 и R6 независимо выбран из атома водорода, атома галогена, C1-6алкилокси, циано, C1-6алкила, -OCH2CH2NR8R9, -CH2OCH2CH2NR8R9, -OCH2CH2CH2NR8R9;
R7 обозначает атом водорода, метил или атом фтора;
каждый R8 и R9 независимо выбран из атома водорода, атома галогена, C1-6алкила, C2-6алкенила, C2-6алкинила, карбонила, C1-6алкилсульфонилC1-6алкила, C1-6алкилоксиC1-6алкила, гидроксиC1-6алкила, дигидроксиC1-6алкила, цианоC1-6алкила, тригалогеноC1-6алкила, фенилC1-6алкила, (диC1-6алкил)аминоC1-6алкила, C1-6алкилсульфонила, морфолинилC1-6алкила, морфолинилкарбонила, пиперазинилC1-6алкила, C1-6алкил-пиперазинилC1-6алкила, пиперидинилC1-6алкила, тиоморфолинилC1-6алкила, C3-6циклоалкилметила, пиридинила, пиримидинила, фенила, галогенофенила, оксанилC1-6алкила, C1-6алкилсульфонилC1-6алкила или C1-6алкилкарбониламиноC1-6алкила;
их N-оксидных форм, их фармацевтически приемлемых аддитивных солей и их сольватов.
Соединения формулы (I) и промежуточные соединения по настоящему изобретению могут также существовать в их таутомерных формах. Предполагается, что подобные формы, хотя они не указаны подробно в вышеприведенной формуле, должны быть включены в объем настоящего изобретения. Следует понимать, что таутомерные формы соединений формулы (I) включают такие соединения формулы (I), в которых, например, енольная группа превращается в кето-группу (кето-енольная таутомерия).
Всякий раз, когда гетероциклические системы в R1 содержат фрагмент -CH2-, -CH= или -NH-, заместители или остаток молекулы могут быть присоединены к каждому атому углерода или атому азота, при этом предполагается, что один или оба атома водорода у одного и того же атома углерода могут быть замещены.
Ниже разъясняется ряд терминов, которые использованы в предыдущих и последующих определениях. Указанные термины используются как самостоятельно, так и в составных терминах.
В приведенных выше и далее по тексту определениях термин атом галогена является общим термином для атомов фтора, хлора, брома и иода; C1-6алкил обозначает насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода, такие как, например, метил, этил, пропил, бутил, пентил, гексил, 1-метилэтил, 2-метилпропил, 2-метилбутил, 2-метилпенил и т.п.; галогеноС1-6алкил означает C1-6алкил, содержащий один галогеновый заместитель, например, фторметил; тригалогеноС1-6алкил означает C1-6алкил, содержащий три идентичных или различных галогеновых заместителя, например, трифторметил; полигалогеноС1-6алкил в качестве группы или части группы обозначает C1-6алкил, замещенный одним или несколькими, например, 2, 3, 4 или 5 атомами галогена, в частности, обозначает метил, замещенный одним или несколькими атомами фтора, например, дифторметил или трифторметил, 1,1-дифторэтил, 1,1-дифтор-2,2,2-трифторэтил и т.п. В том случае, когда в рамках определения полигалогеноС1-6алкила к C1-6алкильной группе присоединено более одного атома галогена, они могут быть одними и теми же или разными; C2-6-алкенил обозначает углеводородные радикалы с прямой или разветвленной цепью, содержащие двойную связь, в частности, одну двойную связь, и имеющие от 2 до 6 атомов углерода, такие как, например, этенил, 2-пропенил, 3-бутенил, 2-пентенил, 3-пентенил, 3-метил-2-бутенил и т.п.; C2-6-алкинил обозначает углеводородные радикалы с прямой или разветвленной цепью, содержащие тройную связь, в частности, одну тройную связь, и имеющие от 2 до 6 атомов углерода, такие как, например, этинил, 2-пропинил, 3-бутинил, 2-бутинил, 2-пентинил, 3-пентинил, 3-гексинил и т.п.; C3-6-циклоалкил включает циклические углеводородные группы, содержащие от 3 до 6 атомов углерода, такие как циклопропил, циклобутил, циклопентил, циклогексил и т.п.
Термин “фармацевтически приемлемые аддитивные соли” означает фармацевтически приемлемые кислотно-аддитивные или основно-аддитивные соли. Следует понимать, что указанные выше или ниже фармацевтически приемлемые кислотно-аддитивные или основно-аддитивные соли включают терапевтически активные нетоксичные кислотно-аддитивные и нетоксичные основно-аддитивные солевые формы, которые способны образовывать соединения формулы (I). Соединения формулы (I), обладающие свойствами основания, могут быть превращены в их фармацевтически приемлемые кислотно-аддитивные соли обработкой указанной основной формы подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогеноводородные кислоты, в частности, хлористоводородная или бромистоводородная кислота; серная; азотная; фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропановая, гидроксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная (т.е. бутандионовая кислота), малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты.
Соединения формулы (I), обладающие кислотными свойствами, могут быть превращены в их фармацевтически приемлемые основно-аддитивные соли обработкой указанной кислотной формы соответствующим органическим или неорганическим основанием. Подходящие солевые формы включают, например, аммониевые соли, соли щелочных и щелочноземельных металлов, в частности, соли лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, в частности, соли бензатина, N-метил-D-глюкамина, гидрабамина, и соли с аминокислотами, такими как, например, аргинин, лизин и т.п.
Пригодными для терапевтического применения солями соединений формулы (I) являются такие соли, в которых противоион является фармацевтически приемлемым. Тем не менее, соли кислот и оснований, которые не являются фармацевтически приемлемыми, могут также использоваться, например, при получении или очистке фармацевтически приемлемого соединения. Все соли, независимо от того, являются они фармацевтически приемлемыми или нет, включены в объем настоящего изобретения.
Четвертичная аммониевая соль соединения формулы (I) обозначает указанное соединение, которое способно образоваться по реакции между основным атомом азота соединения формулы (I) и подходящим кватернизующим агентом, таким как, например, необязательно замещенный алкилгалогенид, арилгалогенид или арилалкилгалогенид, в частности метилиодид или бензилиодид. Могут также применяться другие реагенты с легко уходящей группой, такие как, например, алкил трифторметансульфонаты, алкил метансульфонаты и алкил п-толуолсульфонаты. Четвертичная аммониевая соль имеет, по меньшей мере, один положительно заряженный атом азота. Фармацевтически приемлемые противоионы включают ионы хлора, брома, иода, трифторацетата и ацетата. Четвертичные аммониевые соли соединений формулы (I) входят в объем настоящего изобретения.
Термин сольваты включает гидраты и формы присоединения растворителя, которые способны образовать соединения формулы (I), и их фармацевтически приемлемые аддитивные соли. Примерами подобных форм являются, в частности, гидраты, алкоголяты и т.п.
Приведенный выше и далее по тексту настоящего описания термин стереохимически изомерные формы соединений формулы (I) обозначает все возможные соединения, составленные из одних и тех же атомов, которые соединены друг с другом той же самой последовательностью связей, но имеющие различные трехмерные структуры и не являющиеся взаимосовмещаемыми, которые могут иметь соединения формулы (I). Если не отмечено или не указано иное, то химическое обозначение соединений охватывает смесь всех возможных стереохимически изомерных форм, которыми может обладать указанное соединение. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Следует понимать, что в объем настоящего изобретения входят все стереохимически изомерные формы соединений формулы (I), как в чистой форме, так и в смеси друг с другом.
Особый интерес представляют такие соединения формулы (I), которые являются стереохимически чистыми.
Чистые стереоизомерные формы соединений и промежуточных соединений в контексте настоящего описания определяются как изомеры, практически не содержащие другие энантиомерные или диастереомерные формы той же самой основной молекулярной структуры указанных соединений или промежуточных соединений. В частности, термин "стереоизомерно чистый" касается соединений или промежуточных соединений, имеющих стереоизомерный избыток, который составляет, по крайней мере, 80% (т.е. минимум 80% одного изомера и максимум 20% других возможных изомеров) вплоть до стереоизомерного избытка 100 % (т.е. 100% одного изомера и никаких других), более предпочтительно, соединения и промежуточные соединения имеют стереоизомерный избыток от 90% до 100%, еще более предпочтительно, имеют стереоизомерный избыток от 94% до 100% и, наиболее предпочтительно, имеют стереоизомерный избыток от 97% до 100%. Термины "энантиомерно чистый" и "диастереомерно чистый" следует понимать по аналогии, однако в этом случае термины имеют отношение, соответственно, к энантиомерному избытку и диастереомерному избытку в рассматриваемой смеси.
Если соединение имеет один хиральный центр, и два энантиомера указанного соединения были разделены, то звездочка "*" на чертеже указывает, что абсолютная стереохимическая конфигурация энантиомера не была определена.
Следует понимать, что N-оксидные формы соединений формулы (I) включают такие соединения формулы (I), в которых один или несколько третичных атомов азота окислены в так называемые N-оксиды, в частности, включают такие N-оксиды, где N-окислению подвергнуты один или несколько атомов азота пиперидина или пиперазина.
Соединения формулы (I) могут быть превращены в соответствующие N-оксидные формы по известным из области техники способам преобразования трехвалентного атома азот в его N-оксидную форму. Указанную реакцию N-окисления можно в общем случае осуществить по реакции исходного вещества формулы (I) с соответствующим органическим или неорганическим пероксидом. Подходящие неорганические пероксиды включают, например, пероксид водорода, пероксиды щелочного металла или щелочноземельного металла, например, пероксид натрия, пероксид калия; подходящие органические пероксиды могут включать пероксикислоты, такие как, например, надбензойная кислота, или галогензамещенная надбензойная кислота, например 3-хлорнадбензойная кислота, пероксоалкановые кислоты, например, надуксусную кислоту, алкилгидропероксиды, например, гидропероксид трет-бутила. Подходящими растворителями являются, например, вода, низшие спирты, в частности, этанол и т.п., углеводороды, в частности, толуол, кетоны, в частности, 2-бутанон, галоидированные углеводороды, в частности, дихлорметан, и смеси подобных растворителей.
Предполагается, что настоящее изобретение включает также любые изотопы атомов, присутствующих в соединениях по настоящему изобретению. Например, изотопы водорода включают тритий и дейтерий, а изотопы углерода включают C-13 и C-14.
Всякий раз, когда далее по тексту настоящего описания используется термин "соединения формулы (I)", следует понимать, что он включать также N-оксидные формы, фармацевтически приемлемые кислотно-аддитивные или основно-аддитивные соли, их сольваты и все их стереоизомерные формы.
Первую группу представляющих интерес соединений составляют такие соединения формулы (I), для которых справедливо одно или несколько из следующих ограничений:
a) Y обозначает CH2-CH2;
b) арил обозначает фенил;
c) Het обозначает пиридинил, пиримидинил, бензимидазолил или индазолил;
d) каждый арил или Het может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, циано, C1-6алкила, C1-6алкилоксикарбонила, C1-6алкилNR8R9 или -OR8;
e) X1 обозначает CH2 или N-CH3;
f) X2 обозначает CH2, C=O или O;
g) R10 обозначает фенил, который может быть замещен циано-группой;
h) R2 обозначает метил;
j) R3 и R4 обозначают атом водорода;
k) R5 и R6 обозначают атом водорода;
l) R7 обозначает атом водорода; или
m) каждый из R8 и R9 независимо выбран из атома водорода, атома галогена, C1-6алкила или тригалогеноC1-6алкила.
Вторую группу представляющих интерес соединений составляют такие соединения формулы (I), в которых R1 обозначает пиридинил или пиримидинил.
Третью группу представляющих интерес соединений составляют такие соединения формулы (I) или одна из вышеуказанных групп представляющих интерес соединений формулы (I), для которых справедливо одно или несколько из следующих ограничений:
a) Y обозначает CH2-CH2;
b) R1 обозначает фенил, пиридинил или пиримидинил;
c) каждый фенил, пиридинил или пиримидинил может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, циано или C1-6алкилокси;
e) X1 обозначает CH2;
f) X2 обозначает O;
g) R10 обозначает фенил, замещенный циано-группой;
d) R2 обозначает метил;
e) R3 и R4 обозначают атом водорода;
h) R5 и R6 обозначают атом водорода; или
i) R7 обозначает атом водорода.
Группа предпочтительных соединений состоит из таких соединений формулы (I), где Y обозначает CH2-CH2; арил обозначает фенил; Het обозначает пиридинил, пиримидинил, бензимидазолил или индазолил; каждый арил или Het может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, циано, C1-6алкила, C1-6алкилоксикарбонила, -C1-6алкилNR8R9 или -OR8; X1 обозначает CH2 или N-CH3; X2 обозначает CH2, C=O или O; R10 обозначает фенил, который может быть замещен циано-группой; R2 обозначает метил; R3 и R4 обозначают атом водорода; R5 и R6 обозначают атом водорода; R7 обозначает атом водорода; и каждый R8 и R9 независимо выбран из атома водорода, атома галогена, C1-6алкила или тригалогеноC1-6алкила.
Группа более предпочтительных соединений состоит из таких соединений формулы (I), где Y обозначает CH2-CH2; R1 обозначает фенил, пиридинил или пиримидинил; каждый фенил, пиридинил или пиримидинил может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, циано или C1-6алкилокси; X1 обозначает CH2; X2 обозначает O; R10 обозначает фенил, замещенный циано-группой; R2 обозначает метил; R3 и R4 обозначают атом водорода; R5 и R6 обозначают атом водорода; а R7 обозначает атом водорода.
Наиболее предпочтительными соединениями являются Соединение №6, Соединение №5b, Соединение №7, Соединение №4 или Соединение №17.
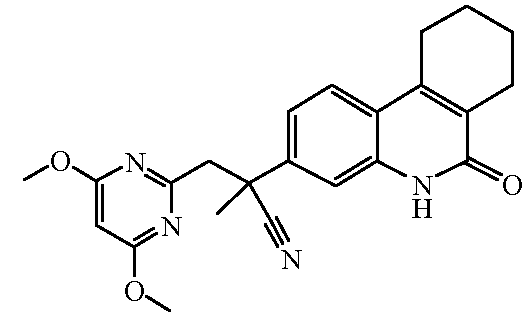
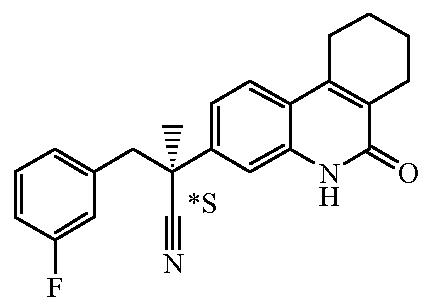
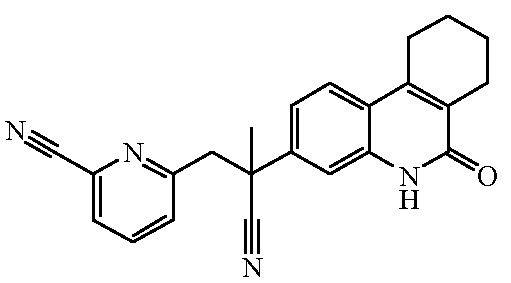
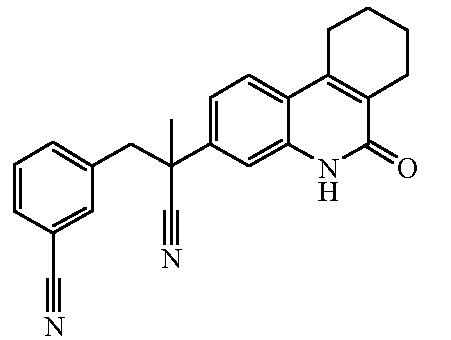
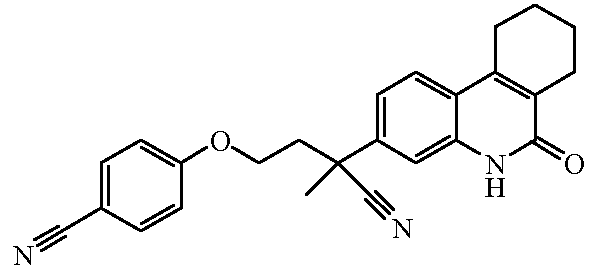
и их N-оксидные формы, их фармацевтически приемлемые аддитивные соли и их сольваты; предпочтительно, их фармацевтически приемлемые аддитивные соли и их сольваты, более предпочтительно, их фармацевтически приемлемые аддитивные соли.
Соединения формулы (I) могут быть получены по общим способам, которые приведены ниже в настоящем описании. Исходные вещества и некоторые из промежуточных соединений являются известными соединениями и коммерчески доступны или же могут быть получены в соответствии с обычными методиками проведения реакций, известными из области техники.
Некоторые из препаративных способов более подробно описаны далее. Другие способы получения конечных соединений формулы (I) приведены в примерах.
Соединения формулы (I) можно получить путем гидролиза промежуточные соединений формулы (II) согласно известным из области техники способам, подвергая промежуточные соединения формулы (II) действию подходящих реагентов, таких как хлористоводородная кислота, в присутствии инертного растворителя, такого как диоксан.
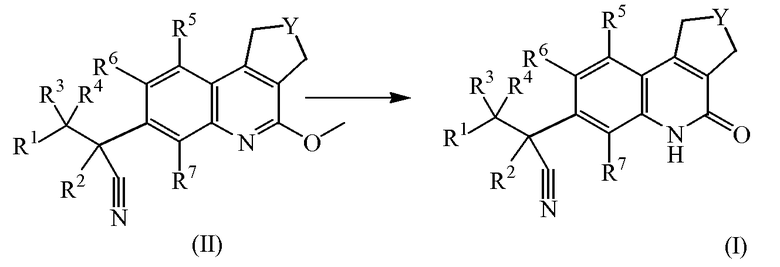
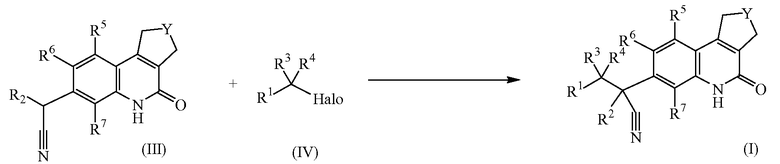
В качестве альтернативы, соединения формулы (I) можно получить, добавляя избыток основания, например, калиевой соли 2-метил-2-пропанола или диизопропиламида лития к промежуточным соединениям формулы (III) в присутствии промежуточных соединений формулы (IV), где Halo обозначает атом хлора или брома, в подходящем растворителе, таком как тетрагидрофуран, диоксан или диметилформамид.
Настоящее изобретение касается также промежуточных соединений формулы (II)
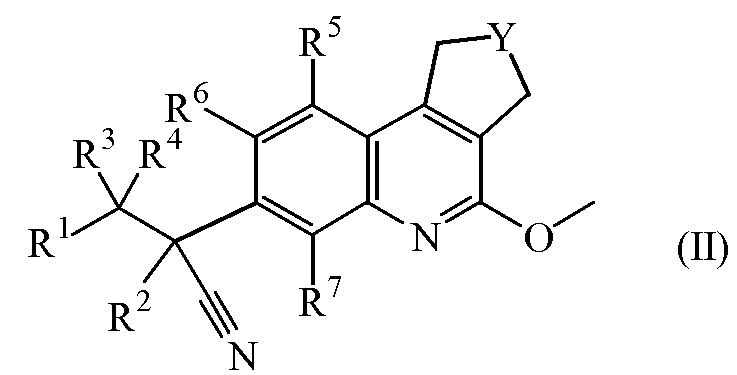
включая их стереохимически изомерные формы,
где
Y обозначает CH2 или CH2-CH2;
R1 обозначает арил или Het;
где арил обозначает фенил или нафталинил;
где Het обозначает тиенил, пирролил, пирролинил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, оксадиазолил, триазолил, тетразолил, тиадиазолил, фуранил, пиперидинил, пиридинил, пиридазинил, пиримидинил, пиперазинил, пиразинил, триазинил, индолизинил, азаиндолизинил, индолил, индолинил, бензотиенил, индазолил, бензоксазолил, бензимидазолил, бензофуранил, бензотиазолил, бензотриазолил, хроманил, пуринил, хинолинил, циннолинил, фталазинил, хиназолинил, хиноксазолинил, нафтиридинил или птеридинил;
два атома углерода в ариле или Het могут быть соединены мостиковой связью (т.е. образуют би- или трициклический фрагмент), при этом двухвалентный радикал выбран из
каждый арил, Het, имеющий мостиковую связь арил или имеющий мостиковую связь Het может быть замещен одним, двумя, тремя, четырьмя или пятью заместителями, каждый из которых независимо выбран из атома галогена, циано, нитро, гидроксикарбонила, C1-6алкила, C2-6алкенила, C2-6алкинила, C3-6циклоалкила, C3-6цикло-алкиламино, метилэтиламино, аминоC3-6циклоалкила, галогеноC1-6алкила, тригалогеноC1-6алкила, C1-6алкилкарбонила, C1-6алкилоксикарбонила, C2-6алкенилкарбонила, оксима, C1-6алкилоксима, амидоксима, -C≡C-CH2O-CH3, -C≡C-CH2N(CH3)2, -C≡C-Si(CH3)3, гидроксиC1-6алкила, гидроксиC2-6алкенила, гидроксиC2-6алкинила, цианоC1-6алкила, цианоC2-6алкенила, аминокарбонилC1-6алкила, C1-6алкилсульфонилC1-6алкила, C1-6алкилсульфонилC2-6алкенила, C1-6алкилсульфонилC2-6алкинила, -PO(OC1-6алкил)2, -B(OH)2, -S-CH3, SF5, C1-6алкилсульфонила, -NR8R9, C1-6алкилNR8R9, -OR8, -C1-6алкилOR8, -CONR8R9, пиперидинилC1-6алкила, пиперазинилC1-6алкила, C1-6алкилпиперазинилC1-6алкила, морфолинилC1-6алкила, пиперидинила, пиперазинила, C1-6алкилпиперазинила, морфолинила, фенила, тиенила, пиразолила, пирролила, пирролидинила, пиридинила, пиримидинила, оксадиазолила, имидазолила, имидазолилC2-6алкинила, C1-6алкилимидазолилC2-6алкинила, цианопиридинила, фенилC1-6алкила, фенилC2-6алкенила, морфолинилC1-6алкила, C1-6алкилоксифенила, тригалогеноC1-6алкилфенила, метилпиразолила, галогенопиримидинила или диметиламинопирролидинила; или
R1 обозначает радикал формулы
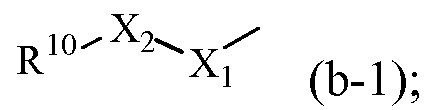
где X1 обозначает CH2, NH или N-CH3;
где X2 обозначает CH2, C=O, O, NH или N-CH3;
где R10 обозначает фенил, пиридинил, пиридазинил или пиримидинил, где каждый фенил, пиридинил, пиридазинил или пиримидинил может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, гидрокси, циано, C1-6алкила, амино, полигалгеноC1-6алкила или C1-6алкилокси; или
R1 обозначает радикал формулы
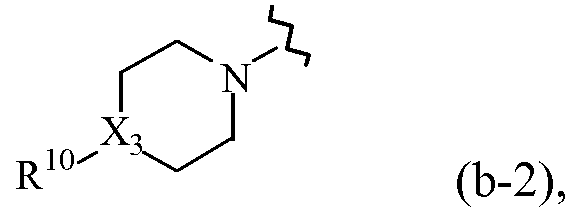
где X3 обозначает CH или N;
R2 обозначает метил, этил, пропил или C3-6циклоалкил;
каждый R3 и R4 независимо выбран из атома водорода, метила, этила, пропила, гидрокси, трифторметила, метилокси; или же R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют циклопропильный цикл или радикал формулы С(=О);
каждый R5 и R6 независимо выбран из атома водорода, атома галогена, C1-6алкилокси, циано, C1-6алкила, -OCH2CH2NR8R9, -CH2OCH2CH2NR8R9, -OCH2CH2CH2NR8R9;
R7 обозначает атом водорода, метил или атом фтора;
каждый R8 и R9 независимо выбран из атома водорода, атома галогена, C1-6алкила, C2-6алкенила, C2-6алкинила, карбонила, C1-6алкилсульфонилC1-6алкила, C1-6алкилоксиC1-6алкила, гидроксиC1-6алкила, дигидроксиC1-6алкила, цианоC1-6алкила, тригалогеноC1-6алкила, фенилC1-6алкила, (диC1-6алкил)аминоC1-6алкила, C1-6алкилсульфонила, морфолинилC1-6алкила, морфолинилкарбонила, пиперазинилC1-6алкила, C1-6алкилпиперазинилC1-6алкила, пиперидинилC1-6алкила, тиоморфолинилC1-6алкила, C3-6циклоалкилметила, пиридинила, пиримидинила, фенила, галогенофенила, оксанилC1-6алкила, C1-6алкилсульфонилC2-6алкила или C1-6алкилкарбониламиноC1-6алкила;
их N-оксидных форм, их фармацевтически приемлемых аддитивных солей и их сольватов.
Группы представляющих интерес, предпочтительных, более предпочтительных и наиболее предпочтительных соединений могут быть определены для соединений формулы (II) в соответствии с группами, которые определены для соединений формулы (I).
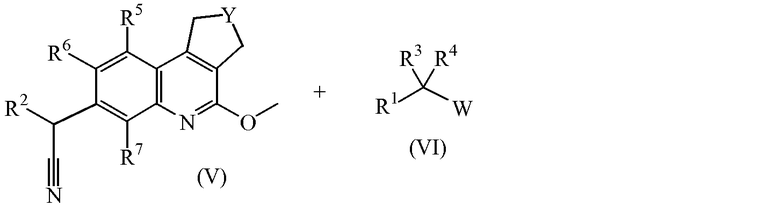
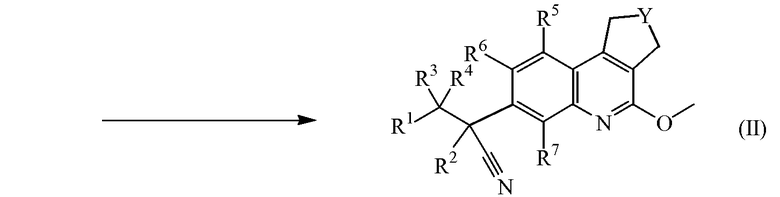
Промежуточные соединения формулы (II) можно получить, добавляя калиевую соль 2-метил-2-пропанола к промежуточным соединениям формулы (V) в присутствии промежуточных соединений формулы (VI), где W обозначает уходящую группу, такую как атом хлора, брома или мезилат, в подходящем растворителе, таком как тетрагидрофуран.
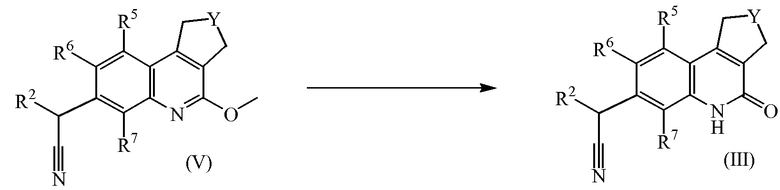
Соединения формулы (III) можно получить, подвергая промежуточные соединений формулы (V) действию подходящих реагентов, таких как хлористоводородная кислота, в присутствии инертного растворителя, в частности, диоксана.
Промежуточные соединения формулы (V) можно получить, добавляя смесь калиевой соли 2-метил-2-пропанола и тозилметилизоцианида в диметилсульфоксиде к промежуточному соединению формулы (VIII) в подходящем растворителе, таком как метанол.
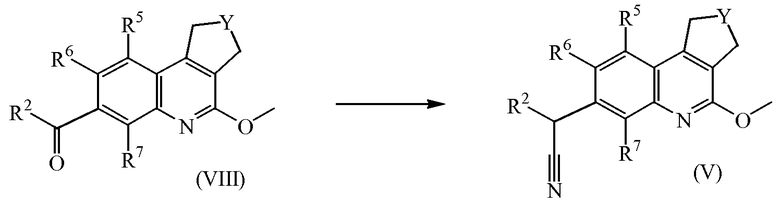
Промежуточные соединения формулы (VIII) можно получить, обработав промежуточное соединение формулы (IX) литийорганическим реагентом, таким как, например, п-бутиллитий, в инертном растворителе, в частности, в тетрагидрофуране, а затем ввести указанное промежуточное соединение в реакцию с промежуточным соединением формулы (X).
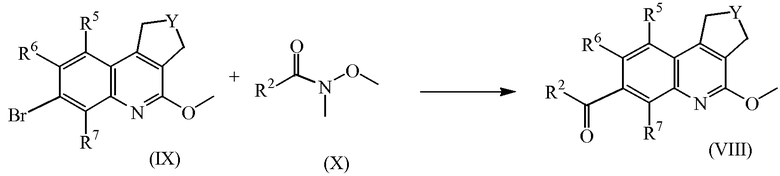
Промежуточные соединения формулы (VIII) можно также получить, преобразуя промежуточные соединения формулы (XI) в присутствии подходящего окислителя, такого как диоксид марганца в подходящем растворителе, таком как диоксан, или в присутствии тетраоксида калия и марганца и трис[2-(2-метоксиэтокси)этил]амина в подходящем растворителе, таком как дихлорметан.
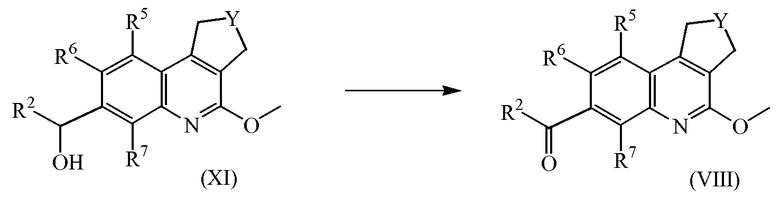
Промежуточные соединения формулы (XI) можно получить, обработав промежуточное соединение формулы (IX) литийорганическим реагентом, таким как, в частности, п-бутиллитий, в инертном растворителе, в частности, в тетрагидрофуране, а затем ввести указанное промежуточное соединение в реакцию с промежуточным соединением формулы (XII).
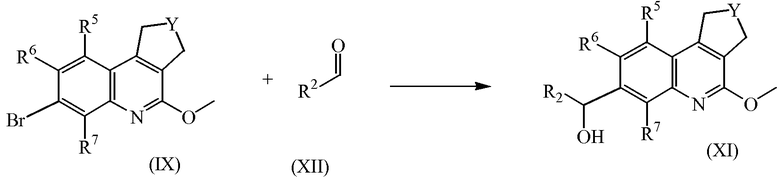
Промежуточные соединения формулы (IX) можно получить, добавив раствор натриевой соли в метаноле к промежуточным соединениям формулы (XIII), где Halo независимо означает атом хлора или брома, в подходящем растворителе, таком как метанол.
Промежуточные соединения формулы (XIII) можно получить по реакции промежуточных соединений формулы (XIV) с подходящим галоидирующим агентом, например, оксихлоридом фосфора, при соответствующей температуре, предпочтительно, при кипячении.
Промежуточные соединения формулы (XIV) можно получить, нагревая промежуточные соединения формулы (XV) в кислой среде, предпочтительно, в серной кислоте.
Соединения формулы (I) или их промежуточные соединения могут также быть превращены друг в друга посредством известных из области техники реакции или способов трансформации функциональных групп. Некоторые из подобных преобразований уже приведены выше в настоящем описании. Другими примерами являются гидролиз сложных эфиров карбоновых кислот с образованием соответствующей карбоновой кислоты или спирта; гидролиз амидов с образованием соответствующих карбоновых кислот или аминов; гидролиз нитрилов с образованием соответствующих амидов; аминогруппы в имидазоле или фениле могут быть замещены атомом водорода по известным из области техники реакциям диазотирования с последующей заменой диазо группы атомом водорода; спирты могут быть превращены в сложные эфиры и простые эфиры; первичные амины могут быть превращены во вторичные или третичные амины; двойные связи можно гидрировать в соответствующую простую связь; радикал иода в фенильной группе можно преобразовать в сложноэфирную группу введением моноксида углерода в присутствии подходящего палладиевого катализатора; радикал иода в фенильной группе может быть превращен в C2-6алкинильную группу или ее производное (например, -C≡C-Si(CH3)3 или гидроксиC2-6алкинил) по реакции с соответствующим C2-6алкинильным соединением или его производным в присутствии подходящего палладиевого катализатора; радикал -C≡C-Si(CH3)3 в фенильной группе может быть преобразован в -C≡CH в присутствии подходящего основания.
Некоторые из соединений формулы (I) и некоторые из промежуточных соединений по настоящему изобретению могут содержать асимметричный атом углерода. Чистые стереохимически изомерные формы указанных соединений и указанных промежуточные соединений могут быть получены известными из области техники способами. Например, диастереоизомеры могут быть разделены с помощью физических способов, таких как селективная кристаллизация или методы хроматографии, в частности, распределение в противотоке, жидкостная хроматография и подобные методы. Энантиомеры можно получить из рацемических смесей, преобразуя вначале указанные рацемические смеси с помощью подходящего расщепляющего агента, такого как, например, хиральные кислоты, в смеси диастереомерных солей или соединений; затем проводят физическое разделение указанных смесей диастереомерных солей или соединений, например, с помощью селективной кристаллизации, жидкостной хроматографии в сверхкритической среде или с использованием хроматографических методов, например, жидкостной хроматографии и подобных методов; и, наконец, преобразуют указанные разделенные диастереомерные соли или соединения в соответствующие энантиомеры. Чистые стереохимически изомерные формы можно также получить из чистых стереохимически изомерных форм подходящих промежуточных соединений и исходных веществ, при условии, что происходящие реакции протекают стереоспецифично.
Настоящее изобретение относится также с соединениям формулы (I), определение которых приведено выше, для использования в качестве лекарственных средств; в частности, для использования при лечении расстройства, опосредованного полимеризацией тубулина, PARP-опосредованного заболевания или TANK-опосредованного заболевания, для использования с целью подавления роста опухолей, для использования с целью ингибирования роста клеток.
Соединения по настоящему изобретению обладают способностью ингибировать PARP или ингибировать полимеризацию тубулина, что подтверждается приведенной далее экспериментальной частью.
Термин “PARP” в данном описании означает белок, способный осуществлять поли-АДФ-рибозилирование. В рамках указанного термина PARP охватывает все белки, кодируемые геном parp, их мутанты и их подвергнутые альтернативному сплайсингу белки. Кроме того, в данном описании термин “PARP” включает аналоги PARP, гомологи и ортологи у других животных.
Термин “PARP” включает, однако этим не ограничиваясь, PARP-1. В понятие указанного термина могут входить PARP-2, PARP-3, Vault-PARP (PARP-4), PARP-7 (TiPARP), PARP-8, PARP-9 (Bal), PARP-10, PARP-11, PARP-12, PARP-13, PARP-14, PARP-15, PARP-16, TANK-1, TANK-2 и TANK-3.
Термин "ингибитор PARP" используют для идентификации соединения, которое способно взаимодействовать с PARP или TANK и подавлять ее активность, в частности, ее ферментативную активность. Ингибирование ферментативной активности PARP или TANK снижает способность PARP или TANK продуцировать поли(АДФ-рибозу) или индуцировать поли-АДФ-рибозилирование субстрата. Подобное ингибирование, предпочтительно, является специфичным, т.е. ингибитор PARP снижает способность PARP продуцировать поли(АДФ-рибозу) или индуцировать поли-АДФ-рибозилирование субстрата при концентрации, которая ниже чем концентрация ингибитора, требуемая для того, чтобы оказать некоторое другое, не связанное с этим биологическое действие.
Термин “соединение, способное ингибировать полимеризацию тубулина” или “ингибитор полимеризации тубулина” используют для идентификации соединения, которое
- стабилизирует микротрубочки, ингибирует деполимеризацию микротрубочек, стабилизирует микротрубочки или замораживает структуру микротрубочек,
- препятствует полимеризации микротрубочек и препятствует образованию микротрубочек,
- дестабилизируют микротрубочки и препятствуют образованию микротрубочек.
Соединения по настоящему изобретению являются TANK-специфичными ингибиторами PARP. Термин “TANK-специфичные ингибиторы PARP” используют для идентификации соединений, которые снижают ферментативную активность членов семейства TANK (в частности, TANK-2) при концентрации, которая ниже чем концентрация ингибитора, требуемая для того, чтобы вызвать ингибирование другого фермента PARP, такого как, в частности, PARP-1.
Настоящее изобретение предусматривает также использование соединений для получения лекарственного средства для лечения любых заболеваний или расстройств у животных, в частности, у человека, которые описаны ниже.
Настоящее изобретение предусматривает также использование соединений формулы (I) для получения лекарственного средства для лечения расстройства, которое опосредуют PARP, TANK или полимеризация тубулина.
Поскольку они способны связывать PARP, то соединения по настоящему изобретению могут применяться в качестве соединений-стандартов или соединений-меток, и в этом случае один из атомов в молекуле может быть заменен, например, радиоактивным изотопом.
В настоящем изобретении рассматривается также фармацевтическая композиция, содержащая фармацевтически приемлемый носитель, а в качестве активного ингредиента - терапевтически эффективное количество соединения по настоящему изобретению.
Для приготовления фармацевтической композиции по настоящему изобретению эффективное количество конкретного соединения в форме основно-аддитивной или кислотно-аддитивной соли, в качестве активного ингредиента, тщательно смешивают с фармацевтически приемлемым носителем, при этом носитель может принимать самые разнообразные формы в зависимости от вида препарата, требуемого для введения. Указанные фармацевтические композиции, предпочтительно, готовят в виде стандартной лекарственной формы, предназначенной, преимущественно, для введения перорально, ректально, подкожно или в виде парентеральной инъекции. Например, при приготовлении композиций в пероральной дозировочной форме может использоваться любая обычно применяемая в фармацевтике среда, такая как, например, вода, гликоли, масла, спирты и т.п. в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, лубриканты, связующие, разрыхлители и т.п. в случае порошков, пилюль, капсул и таблеток. Благодаря простоте их введения, таблетки и капсулы представляют наиболее удобную пероральную форму единицы дозирования, и в этом случае, очевидно, используют твердые фармацевтические носители. Для парентеральных композиций носитель обычно, по крайней мере, в значительной степени представляет собой стерильную воду, хотя могут быть включены и другие ингредиенты, например, с целью улучшения растворимости. Могут быть приготовлены, например, растворы для инъекций, в которых носителем является физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Могут быть также приготовлены суспензии для инъекций, в которых могут использоваться подходящие жидкие носители, суспендирующие агенты и т.п. В композициях, пригодных для подкожного введения, носитель необязательно включает средство, облегчающее проникновение, и/или подходящее смачивающее средство, которое необязательно комбинируют в небольших пропорциях с любыми типами подходящих добавок, при этом добавки не оказывают значительного вредного воздействия на кожу. Указанные добавки могут облегчить нанесение на кожу и/или могут оказаться полезными для приготовления требуемых композиций. Указанные композиции могут применяться различными способами, например, в виде чрескожных пластырей, в виде средств для точечного воздействия, в виде мази. Особенно удобно, для простоты ведения и обеспечения однородности дозировки, составлять вышеуказанные фармацевтические композиции в форме стандартной лекарственной формы. Стандартная лекарственная форма в контексте настоящего описания и формулы изобретения относится к физически дискретным единицам, пригодным для однократной дозировки, при этом каждая единица содержит определенное количество активного ингредиента, которое, согласно расчету, должно произвести требуемый терапевтический эффект, вместе с нужным фармацевтическим носителем. Примерами подобных стандартных лекарственных форм являются таблетки (включая таблетки с насечкой или таблетки, покрытые оболочкой), капсулы, пилюли, пакетики с порошком, облатки, растворы или суспензии для инъекций, порции, соответствующие чайной ложке, порции, соответствующие столовой ложке, и т.п. и разделенные части указанных стандартных лекарственных форм.
Соединения по настоящему изобретению могут оказывать лечебное действие или препятствовать повреждению тканей, вызванному поражением или смертью клеток вследствие некроза или апоптоза; могут ослабить повреждение нервных или сердечно-сосудистых тканей, включая повреждение, вызванное очаговой ишемией, инфарктом миокарда и повреждением вследствие реперфузии; могут вылечивать различные болезни и условия, вызванные или усугубляемые активностью PARP; могут расширять или увеличивать время жизни или пролиферативную способность клеток; могут изменять экспрессию генов в стареющих клетках; могут усиливать чувствительность клеток к действию радиоактивных и/или химиотерапевтических препаратов. В общем случае, ингибирование активности PARP предохраняет клетки от потери энергии, предотвращая в случае нервных клеток необратимую деполяризацию нейронов и тем самым обеспечивают нейрозащиту.
По вышеуказанным причинам настоящее изобретение касается также способа введения терапевтически эффективного количества идентифицированных выше соединений, которое достаточно для ингибирования активности PARP, лечения или создания препятствий к повреждению тканей, вызванному поражением или смертью клеток вследствие некроза или апоптоза, воздействия на нейронную активность, не обусловленную токсичностью NMDA, воздействия на нейронную активность, обусловленную токсичностью NMDA, лечения повреждений нервных тканей, вызванных ишемией и поражением вследствие реперфузии, лечения неврологических расстройств и нейродегенеративных болезней; предупреждения или лечения васкулярных травм; лечения или предупреждения сердечно-сосудистых заболеваний; лечения других состояний и/или заболеваний, таких как возрастная мышечная дегенерация, СПИД и возрастные заболевания иммунной системы, воспаление, подагра, артрит, атеросклероз, кахексия, рак, дегенеративные болезни скелетных мышц, включая физиологическое репликативное старение, диабет, травма головы, воспалительные болезни кишечника (такие как колит и болезнь Крона), мышечная дистрофия, остеоартрит, остеопороз, хроническая и/или острая боль (такая как нейропатическая боль), почечная недостаточность, ишемия сетчатки, септический шок (такой как эндотоксический шок) и старение кожи; увеличения жизненного цикла и пролиферативной способности клеток; изменения экспрессии генов в стареющих клетках; повышения чувствительности (гипоксической) опухолевых клеток к действию химиотерапевтических и/или радиоактивных препаратов. Настоящее изобретение касается также способа лечения болезней и состояний у животного, в частности, человека, который включает введение указанному животному терапевтически эффективного количества идентифицированных выше соединений.
В частности, настоящее изобретение касается также способа лечения, предупреждения или задержки неврологического расстройства у животного, при этом способ включает введение указанному животному, в частности человеку, терапевтически эффективного количества идентифицированных выше соединений. Неврологическое расстройство выбрано из группы, которая включает периферическое заболевание нервной системы, вызванное физическим повреждением или болезненным состоянием, травму головного мозга, физическое повреждение спинного мозга, удар, вызванный повреждением головного мозга, очаговую ишемию, глобальную ишемию, поражение, вызванное реперфузией, демиелинизирующее заболевание и неврологическое расстройство, связанное с нейродегенерацией.
Настоящее изобретение предусматривает также применение соединений формулы (I) для ингибирования активности PARP, с целью лечения, предупреждения или задержки повреждения тканей, вызываемого поражением или отмиранием клеток вследствие некроза или апоптоза, с целью лечения, предупреждения или задержки неврологического расстройства у животного.
Термин “предупреждение нейродегенерации” включает способность предупреждать нейродегенерацию у пациентов, для которых вновь установлен диагноз развития нейродегенеративного заболевания, а также у пациентов, которые находятся в группе риска развития нового дегенеративного заболевания, а также способность препятствовать дальнейшему развитию нейродегенерации у пациентов, которые уже страдают от нейродегенеративного заболевания или имеют симптомы нейродегенеративного заболевания.
Термин “лечение” в настоящем описании включает любое воздействие на заболевание и/или состояние у животного, в частности у человека, и включает: (i) предупреждение возникновения заболевание и/или состояние у субъекта, который может быть предрасположен к данному заболеванию и/или состоянию, но у которого оно еще не диагностировано; (ii) задержку заболевания и/или состояния, т.е. подавление его развития; (iii) облегчение заболевания и/или состояния, т.е. создание предпосылок для регрессии заболевания и/или состояния. Преимущественно, термин “лечение” имеет значение, соответствующее пунктам (ii) или (iii).
Термин “радиосенсибилизирующее вещество” в контексте настоящего описания означает молекулу, преимущественно, молекулу с низкой молекулярной массой, которую назначают животным в терапевтически эффективном количестве, с целью повышения чувствительности клеток к действию ионизирующего излучения и/или для облегчения лечения заболеваний, которые могут вылечиваться с помощью ионизирующего излучения. Заболевания, которые могут вылечиваться с помощью ионизирующего излучения, включают неопластические заболевания, доброкачественные и злокачественные опухоли и раковые клетки. Предусматривается также лечение с помощью ионизирующего излучения других заболеваний, которые не указаны в настоящем описании.
Термин “вещество, повышающее чувствительность к действию химиотерапевтических препаратов” в контексте настоящего описания означает молекулу, преимущественно, молекулу с низкой молекулярной массой, которую назначают животным в терапевтически эффективном количестве, с целью повышения чувствительности клеток к химиотерапии и/или для облегчения лечения заболеваний, которые могут вылечиваться с помощью химиотерапевтических препаратов. Заболевания, которые могут вылечиваться с помощью химиотерапии, включают неопластические заболевания, доброкачественные и злокачественные опухоли и раковые клетки. Предусматривается также химиотерапевтическое лечение других заболеваний, которые не указаны в настоящем описании.
В настоящем изобретении предлагается способ подавления аномального роста клеток, включая измененные клетки, путем введения эффективного количества соединения по настоящему изобретению. Аномальный рост клеток относится к росту клеток, который не зависит от нормальных регулирующих механизмов (в частности, потеря контактного ингибирования). Он включает подавление роста опухоли как непосредственно за счет остановки роста, терминальной дифференциации и/или апоптоза раковых клеток, так и опосредованно путем ингибирования неоваскуляризации опухолей.
Соединения, композиции и способы по настоящему изобретению наиболее пригодны для лечения или предупреждения повреждения тканей, которое вызывается смертью или поражением клеток вследствие некроза или апоптоза.
Соединения по настоящему изобретению могут быть “противораковыми средствами” и этот термин охватывает также “средства против роста опухолевых клеток” и “антинеопластические средства”.
В настоящем изобретении предлагается также способ подавления роста клеток путем введения эффективного количества соединения по настоящему изобретению субъекту, в частности млекопитающему (и наиболее предпочтительно, человеку), которое нуждается в подобном лечении.
Например, способы по настоящему изобретению пригодны для лечения рака и повышения чувствительности опухолевых клеток при раке к действию химиотерапевтических и/или радиоактивных препаратов.
Примеры опухолей, включая злокачественные образования у взрослых и детей, которые могут подавляться соединениями по настоящему изобретению, включают, однако этим не ограничиваясь, рак легких, включая мелкоклеточный рак легких и не мелкоклеточной рак легких (в частности, аденокарциному), рак поджелудочной железы (в частности, карциному поджелудочной железы, такую как, например, экзокринный рак поджелудочной железы), рак толстого кишечника (в частности, колоректальную карциному, такую как, например, аденокарцинома толстого кишечника и аденома толстого кишечника), рак пищевода, сквамозную карциному полости рта, карциному языка, рак желудка, рак печени, рак носоглотки, гематопоэтические опухоли лимфоидного направления дифференцировки (в частности, острый лимфоцитарный лейкоз, лимфому В-клеток, лимфому Буркитта), неходжкинскую лимфому (в частности, лимфому из клеток зоны мантии), болезнь Ходжкина, миелоидный лейкоз (например, острый миелогенный лейкоз (AML) или хронический миелогенный лейкоз (CML)), острый лимфобластический лейкоз, хронический лимфолейкоз (CLL), фолликулярный рак щитовидной железы, миелодиспластический синдром (MDS), опухоли мезенхимальной природы, саркомы мягких тканей, липосаркомы, стромальную саркому желудочно-кишечного тракта, злокачественные опухоли периферических нервных стволов (MPNST), саркомы Эвинга, лейомиосаркомы, мезенхимальные хондросаркомы, лимфосаркомы, фибросаркомы, рабдомиосаркомы, меланомы, тератокарциномы, нейробластомы, опухоли мозга, медуллобластомы, глиомы, доброкачественную опухоль кожи (в частности, керотоакантомы), карциномы груди (в частности, рак груди в прогрессирующей стадии), рак почки, нейробластому, рак яичников, цервикальный рак, карциному эндометрия, рак мочевого пузыря, рак предстательной железы, включая развившуюся болезнь и гормонорезистентный рак предстательной железы, рак яичек, остеосаркому, рак головы и шеи, эпидермальную карциному, множественную миелому (в частности, стойкую множественную миелому), мезотелиому. Конкретные виды рака, которые можно лечить с помощью соединений по настоящему изобретению, являются рак груди, колоректальный рак, не мелкоклеточной рак легких, острый миелогенный лейкоз (AML).
В качестве другого аспекта настоящего изобретения рассматривается комбинация ингибитора PARP или соединения, обладающего способностью связывать тубулин, формулы (I) с другим противораковым средством, в частности, для использования в медицине, более конкретно - при лечении рака и родственных заболеваний.
Для лечения вышеуказанных состояний соединения по настоящему изобретению, предпочтительно, могут применяться в комбинации с одним или несколькими другими лекарственными средствами, в частности, с другим противораковыми средствами или фармацевтическими препаратами, усиливающими действие другого препарата, которые используются при лечении рака. Примеры противораковых средств или фармацевтических препаратов, усиливающих действие другого препарата (вспомогательных лекарственных средств), включают, однако этим не ограничиваясь:
координационные соединения платины, например, цисплатин, необязательно в сочетании с амифостином, карбоплатином или оксалиплатином;
таксановые соединения, например, паклитаксел, стабилизированный белком паклитаксел (Abraxane™) или доцетаксел;
ингибиторы топоизомеразы I, такие как камптотециновые соединения, например, иринотекан, SN-38, топотекан, гидрохлорид топотекана;
ингибиторы топоизомеразы II, такие как противоопухолевые эпиподофиллотоксины или производные подофиллотоксина, например, этопозид, фосфат этопозида или тенипозид;
противоопухолевые алкалоиды винка, например, винбластин, винкристин или винорелбин;
противоопухолевые нуклеозидные производные, например, 5-фторурацил, лейковорин, гемцитабин, гидрохлорид гемцитабина, капецитабин, кладрибин, флударабин, неларабин;
алкилирующие агенты, такие как азотистый иприт или нитрозомочевина, например, циклофосфамид, хлорамбуцил, кармустин, тиотепа, мефалан (мелфалан), ломустин, алтретамин, бусулфан, дакарбазин, эстрамустин, ифосфамид, необязательно в сочетании с месна, пипоброман, прокарбазин, стрептозоцин, телозоломид, урацил;
противоопухолевые производные антрациклина, например, даунорубицин, доксорубицин, необязательно в сочетании с дексразоксаном, доксил, идарубицин, митоксантрон, эпирубицин, гидрохлорид эпирубицина, валрубицин;
молекулы, которые нацеливаются на рецептор IGF-1, например, пикроподофилин;
производные тетракарцина, например, тетрокарцин A;
глюкокортикоиден, например, преднизон;
антитела, например, трастузумаб (антитело HER2), ритуксимаб (антитело CD20), гемтузумаб, гемтузумаб озогамицин, цетуксимаб, пертузумаб, бевацизумаб, алемтузумаб, экулизумаб, ибритумомаб тиуксетан, нофетумомаб, панитумумаб, тозитумомаб, CNTO 328;
антагонисты рецептора эстрогена, или селективные модуляторы рецептора эстрогена, или ингибиторы синтеза эстрогена, например, тамоксифен, фулвестрант, торемифен, дролоксифен, фаслодекс, ралоксифен или летрозол;
ингибиторы ароматазы, такие как эксеместан, анастрозол, летразол, тестолактон и ворозол;
агенты дифференцировки, такие как ретиноиды, витамин D или ретиноевая кислота и агенты, блокирующие метаболизм ретиноевой кислоты (RAMBA), например, аккутан;
ингибиторы ДНК-метилтрансферазы, например, азацитидин или децитабин;
антифолаты, например, преметрексед динатрий;
антибиотики, антиномицин D, блеомицин, митомицин C, дактиномицин, карминомицин, дауномицин, левамизол, пликамицин, митрамицин;
антиметаболиты, например, клофарабин, аминоптерин, арабинозид цитозина или метотрексат, азацитидин, цитарабин, флоксуридин, пентостатин, тиогуанин;
агенты, индуцирующие апоптоз, и антиангиогенные средства, такие как ингибиторы Bcl-2, например, YC 137, BH 312, ABT 737, госсипол, HA 14-1, TW 37 или декановая кислота;
тубулин-связывающие агенты, например, комбрестатин, колхицины или нокодазол;
ингибиторы киназы (в частности, ингибиторы EGFR (рецептор эпителиального фактора роста), MTKI (многоцелевые ингибиторы киназы), ингибиторы mTOR), например, флавоперидол, мезилат иматиниба, эрлотиниб, гефитиниб, дазатиниб, лапатиниб, дитозилат лапатиниба, сорафениб, сунитиниб, малеат сунитиниба, темсиролимус;
ингибиторы фарнезилтрансферазы, например, типифарниб;
ингибиторы гистондеацетилазы (HDAC), например, бутират натрия, субероиланилид гидроксамидной кислоты (SAHA), депсипептид (FR 901228), NVP-LAQ824, R306465, JNJ-26481585, трихостатин A, вориностат;
ингибиторы пути убиквитин-протеасома, например, PS-341, MLN.41 или бортезомиб;
препарат Yondelis;
ингибиторы теломеразы, например, теломестатин;
ингибиторы матричной металлопротеиназы, например, батимастат, маримастат, приностат или метастат;
рекомбинантные интерлейкины, например, альдеслейкин, денилейкин дифтитокс, интерферон альфа 2a, интерферон альфа 2b, пегинтерферон альфа 2b;
ингибиторы MAPK;
ретиноиды, например, алитретиноин, бексаротен, третиноин;
триоксид мышьяка;
аспарагиназы;
стероиды, например, пропионат дромостанолона, ацетат мегестрола, нандролон (деканоат, фенпропионат), дексаметазон;
агонисты или антагонисты гонадотропин-высвобождающего гормона, например, абареликс, ацетат госерелина, ацетат гистрелина, ацетат лейпролида;
талидомид, леналидомид;
меркаптопурин, митотан, памидронат, пегадемаза, пегаспаргаза, расбуриказа;
миметики BH3, например, ABT-737;
ингибиторы MEK, например, PD98059, AZD6244, CI-1040;
аналоги колониестимулирующего фактора, например, филграстим, пегфилграстим, сарграмостин; эритропоэтин и его аналоги (в частности, дарбепоэтин альфа); интерлейкин 11; опрелвекин; золедронат, золедроновая кислота; фентанил; бисфосфонат; палифермин.
Термин “координационное соединение платины” используют в настоящем описании для обозначения любого координационного соединения платины, ингибирующего рост опухолевой клетки, которое содержит платину в форме иона. Координационное соединение платины, преимущественно, назначают с дозой от 1 до 500 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 50 до 400 мг/м2, в частности, для цисплатина доза составляет приблизительно 75 мг/м2, а для карбоплатина - приблизительно 300 мг/м2 за один курс лечения.
Термин “таксановые соединения” указывает на класс соединений, которые содержат систему таксанового цикла и которые относятся к или которые могут быть выделены из отдельных видов тисовых деревьев (Taxus). Таксановые соединения, преимущественно, назначают с дозой от 50 до 400 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 75 до 250 мг/м2, в частности, для паклитаксела доза составляет приблизительно от 175 до 250 мг/м2, а для доцетаксела - приблизительно от 75 до 150 мг/м2 за один курс лечения.
Термин “ингибиторы топоизомеразы” используют для обозначения ферментов, которые способны изменять топологию ДНК в клетках эукариотов. Они играют критическую роль для важных функций клеток и пролиферации клеток. Существуют два класса топоизомераз в клетках эукариотов, а именно: типа I и типа II. Топоизомераза I представляет собой мономерный фермент с молекулярной массой приблизительно 100000. Фермент связывается с ДНК и вызывает временный однонитевой разрыв, раскручивает двойную спираль (или позволяет ей раскрутиться), а затем перед отделением от нити ДНК вновь залечивает разрыв. Топоизомераза II имеет сходный механизм действия, который включает индуцирование разрывов нити ДНК или образование свободных радикалов.
Термин “камптотециновые соединения” используют для обозначения соединений, которые связаны с родительским камптотециновым соединением или которые могут быть выделены из родительского камптотецинового соединения, представляющего собой водорастворимый алкалоид, выделяемый из китайского дерева Camptothecin acuminata и индийского дерева Nothapodytes foetida. Камптотециновое соединение, преимущественно, назначают с дозой от 0,1 до 400 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 1 до 300 мг/м2, в частности, для иринотекана доза составляет приблизительно от 100 до 350 мг/м2, а для топотекана - приблизительно от 1 до 2 мг/м2 за один курс лечения.
Термин “подофиллотоксиновые соединения” используют для обозначения соединений, которые связаны с родительским подофиллотоксином, или которые могут быть выделены из родительского подофиллотоксина, экстрагируемого из растения мандрагора. Противоопухолевое производное подофиллотоксина, преимущественно, назначают с дозой от 30 до 300 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 50 до 250 мг/м2, в частности, для этопозида доза составляет приблизительно от 35 до 100 мг/м2, а для тенпозида - приблизительно от 50 до 250 мг/м2 за один курс лечения.
Термин “противоопухолевые алкалоиды винка” используют для обозначения соединений, которые связаны с растением барвинок малый или которые могут быть получены из экстрактов растения барвинок малый (Vinca rosea). Противоопухолевый алкалоид винка, преимущественно, назначают с дозой от 2 до 30 мг на квадратный метр (мг/м2) площади поверхности тела, в частности, для винбластина доза составляет приблизительно от 3 до 12 мг/м2, для винкристина доза составляет приблизительно от 1 до 2 мг/м2, для винорелбина доза составляет от 10 до 30 мг/м2 за один курс лечения.
Противоопухолевое нуклеозидное производное, преимущественно, назначают с дозой от 200 до 2500 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 700 до 1500 мг/м2, в частности, для 5-FU доза составляет от 200 до 500 мг/м2, для гемцитабина доза составляет приблизительно от 800 до 1200 мг/м2, а для капецитабина - приблизительно от 1000 до 2500 мг/м2 за один курс лечения.
Термин "алкилирующие агенты" охватывает разнообразную группу химических реагентов, общей особенностью которых является способность вводить, в определенных физиологических условиях, алкильные группы в биологически важные макромолекулы, такие как ДНК. Для большинства наиболее важных агентов, таких как азотистые иприты и нитрозомочевины, активные алкилирующие фрагменты генерируются in vivo после протекания сложных дегенеративных реакций, некоторые из которых являются ферментативными. Наиболее важными фармакологическими действиями алкилирующих агентов являются такие, которые нарушают фундаментальные механизмы, связанные с пролиферацией клетки в синтезе конкретных ДНК и делении клетки. Способность алкилирующих агентов мешать функциям ДНК и нарушать целостностью в быстро пролиферирующих тканях оставляет основу для их применений в терапии и многих из их токсических свойств. Алкилирующие агенты, такие как азотистый иприт или нитрозомочевина, преимущественно, назначают с дозой от 100 до 500 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 120 до 200 мг/м2, в частности, для циклофосфамида доза составляет приблизительно от 100 до 500 мг/м2, для хлорамбуцила доза составляет приблизительно от 0,1 до 0,2 мг/м2, для кармустина доза составляет приблизительно от 150 до 200 мг/м2, а для ломустина доза составляет приблизительно от 100 до 150 мг/м2 за один курс лечения.
Термин “противоопухолевые производные антрациклина” включает антибиотики, получаемые из грибов Strep. peuticus var. caesius, и их производные, отличающиеся тем, что они содержат тетрациклиновую кольцевую структуру с необычным сахаром, даунозамином, присоединенным гликозидной связью. Противоопухолевые производные антрациклина, преимущественно, назначают с дозой от 10 до 75 мг на квадратный метр (мг/м2) площади поверхности тела, например, от 15 до 60 мг/м2, в частности, для доксорубицина доза составляет приблизительно от 40 до 75 мг/м2, для даунорубицина доза составляет приблизительно от 25 до 45 мг/м2, а для идарубицина доза составляет приблизительно от 10 до 15 мг/м2 за один курс лечения.
Было установлено, что амплификация белка рецептора эпидермального фактора роста 2 человека (HER2) при первичных карциномах груди коррелирует с неблагоприятным прогнозом для некоторых пациентов. Трастузумаб представляет собой получаемое из рекомбинантной ДНК тщательно очищенное гуманизированное моноклональное антитело IgG1, которое с большим сродством и высокой специфичностью связывается с внеклеточным доменом рецептора HER2.
Многие из видов рака груди имеют рецепторы эстрогена, и рост указанных опухолей может стимулироваться эстрогеном. Термин “антагонисты рецептора эстрогена” и “селективные модуляторы рецептора эстрогена” используют для обозначения конкурентных ингибиторов связывания эстрадиола с рецептором эстрогена (ER). Селективные модуляторы рецептора эстрогена, связанные с ER, индуцируют изменение трехмерной формы рецептора, модулируя его связывание с эстрогеновым ответным элементом (ERE) в молекуле ДНК.
У женщин в период менопаузы основным источником циркулирующего в организме эстрогена является превращение андрогенов надпочечников и яичников (андростендиона и тестостерона) в эстрогены (эстрон и эстрадиол) ферментом ароматазой в периферических тканях. Потеря эстрогена вследствие ингибирования или дезактивирования ароматазы, представляет собой эффективное и избирательное лечение в период менопаузы для некоторых пациенток с гормонозависимым раком груди.
Термин “агенты дифференцировки” охватывает соединения, которые различными способами могут ингибировать пролиферацию клеток и вызывать дифференцировку. Известно, что витамин D и ретиноиды играют основную роль в регулировании роста и дифференцировки самых разнообразных нормальных и злокачественных типов клеток. Агенты, блокирующие метаболизм ретиноевой кислоты (RAMBA's), повышают уровни эндогенной ретиноевой кислоты путем ингибирования опосредованного цитохромом Р450 метаболизма ретиноевых кислот.
Изменения в метилировании ДНК являются наиболее часто встречающимися аномалиями при неоплазии у человека. Гиперметилирование среди промоторов избранных генов обычно связано с дезактивацией участвующих в процессе генов. Термин “ингибиторы метилтрансферазы ДНК” используют для обозначения соединений, которые действуют путем фармакологического ингибирования метилтрансферазы ДНК и повторной активации экспрессии гена-супрессора опухолей.
Термин “ингибиторы киназы” включает действенные ингибиторы киназы, которые принимают участие в развитии клеточного цикла и программированной смерти клеток (апоптоза).
Термин “ингибиторы фарнезилтрансферазы” используют для обозначения соединений, которые предназначены для предотвращения фарнезилирования Ras и других внутриклеточных белков. Было показано, что они оказывают действие на пролиферацию и выживание злокачественных клеток.
Термин “ингибитор гистондеацетилазы” используют для обозначения соединения, которое способно взаимодействовать с гистондеацетилазой и ингибировать ее активность, в частности, ее ферментативную активность. Ингибирование ферментативной активности гистондеацетилазы означает снижение способности гистондеацетилазы удалять ацетильную группу из гистона.
Термин “другие ингибиторы пути убиквитин-протеасома“ используют для идентификации соединений, которые ингибируют нацеленную деструкцию клеточных белков в протеасоме, включая белки-регуляторы клеточного цикла.
Термин “ингибитор теломеразы” касается соединений, которые нацеливаются, снижают или ингибируют активность теломеразы, в частности, соединений, которые ингибируют рецептор теломеразы.
Термин “ингибитор матричной металлопротеиназы” включает, однако этим не ограничиваясь, пептидомиметик коллагена и непептидомиметические ингибиторы.
Соединения по настоящему изобретению могут применяться в качестве “радиосенсибилизирующего вещества” и/или “вещества, повышающего чувствительность к действию химиотерапевтических препаратов”.
Известно, что радиосенсибилизирующие вещества повышают чувствительность раковых клеток к токсическому действию ионизирующего излучения.
В литературе предложено несколько механизмов действия радиосенсибилизирующих веществ, в том числе следующие: радиосенсибилизирующие вещества гипоксических клеток (в частности, 2-нитроимидазольные соединения и производные диоксида бензотриазина) имитируют кислород или же ведут себя как биовосстанавливающие агента при гипоксии; радиосенсибилизирующие вещества отличные от гипоксических клеток (например, галогенсодержащие пиримидины) могут быть аналогами оснований ДНК и избирательно встраиваться в ДНК раковых клеток и, таким образом, способствуют вызванному излучением разрыву молекул ДНК и/или препятствуют протеканию нормальных механизмов репарации ДНК; при лечении болезни были предложены также различные другие потенциальные механизмы действия радиосенсибилизирующих веществ.
Во многих методиках лечения рака в настоящее время используют радиосенсибилизирующие вещества в сочетании с рентгеновским излучением. Примеры активизируемых рентгеновским излучением радиосенсибилизирующих веществ включают, однако этим не ограничиваясь, следующие вещества: метронидазол, мизонидазол, десметилмизонидазол, пимонидазол, этанидазол, ниморазол, митомицин C, RSU 1069, SR 4233, EO9, RB 6145, никотинамид, 5-бромдеоксиуридин (BUdR), 5-иоддеоксиуридин (IUdR), бромдеоксицитидин, фтордеоксиуридин (FudR), гидроксимочевину, цисплатин и их терапевтически эффективные аналоги и производные.
При фотодинамической терапии (PDT) в качестве активирующего излучения для сенсибилизаторов используют видимый свет. Примеры фотодинамических радиосенсибилизирующих веществ включают, однако этим не ограничиваясь, следующие вещества: производные гематопорфирина, фотофрин, производные бензопорфирина, этиопорфирин олова, феоборбид-a, бактериохлорофилл-a, нафталоцианины, фталоцианины, фталоцианин цинка и их терапевтически эффективные аналоги и производные.
Радиосенсибилизирующие вещества могут назначаться в сочетании с терапевтически эффективным количеством одного или нескольких других соединений, которые включают, однако этим не ограничиваясь, следующие вещества: соединения, которые способствуют включению радиосенсибилизирующих веществ в клетки-мишени; соединения, которые контролируют поток терапевтических агентов, питательных веществ и/или кислорода в клетки-мишени; химиотерапевтические средства, которые воздействуют на опухоль при дополнительном действии или без дополнительного действия излучения; или другие терапевтически эффективные соединения для лечения рака или другого заболевания. Примеры дополнительных терапевтических агентов, которые могут использоваться в сочетании с радиосенсибилизирующими веществами, включают, однако этим не ограничиваясь, 5-фторурацил, лейковорин, 5′-амино-5′-деокситимидин, кислород, карбоген, переливание эритроцитов, перфторуглеводороды (в частности, Fluosol 10 DA), 2,3-DPG, BW12C, блокаторы кальциевых каналов, пентоксифиллин, соединения, препятствующие развитию кровеносных сосудов, гидралазин и LBSO. Примеры химиотерапевтических средств, которые могут использоваться в сочетании с радиосенсибилизирующими веществами, включают, однако этим не ограничиваясь: адриамицин, камптотецин, карбоплатин, цисплатин, даунорубицин, доцетаксел, доксорубицин, интерферон (альфа, бета, гамма), интерлейкин 2, иринотекан, паклитаксел, топотекан и их терапевтически эффективные аналоги и производные.
Вещества, повышающего чувствительность к действию химиотерапевтических препаратов, могут назначаться в сочетании с терапевтически эффективным количеством одного или нескольких других соединений, которые включают, однако этим не ограничиваясь, следующие вещества: соединения, которые способствуют включению веществ, повышающих чувствительность к действию химиотерапевтических препаратов, в клетки-мишени; соединения, которые контролируют поток терапевтических агентов, питательных веществ и/или кислорода в клетки-мишени; химиотерапевтические средства, которые воздействуют на опухоль, или другие соединения, терапевтически эффективные для лечения рака или другого заболевания.
Соединения формулы (I) могут также использоваться для обнаружения или идентификации PARP и, в частности, рецептора PARP-1 или рецептора танкиразы. С указанной целью в соединения формулы (I) может вводиться метка. Указанную метку можно выбрать из группы, которая включает радиоизотоп, спиновую метку, метку антигена, ферментную метку, флуоресцентную группу или хемилюминесцентную группу.
Настоящее изобретение касается также комбинации по настоящему изобретению для использования при лечении, например, с целью подавления роста опухолевых клеток.
Настоящее изобретение касается также комбинации по настоящему изобретению для подавления роста опухолевых клеток.
Настоящее изобретение касается также способа подавления роста опухолевых клеток у человека, при этом способ включает введение указанному субъекту эффективного количества комбинации по настоящему изобретению.
В настоящем изобретении предлагается также способ подавления аномального роста клеток, включая измененные клетки у человека, при этом способ включает введение указанному субъекту эффективного количества комбинации по настоящему изобретению.
Могут назначаться другие медицинские средства и ингибиторы PARP, обладающие способностью связывать тубулин, как одновременно (в частности, в виде отдельных композиций или композиций для однократного приема), так и последовательно в любом порядке. В последнем случае два соединения вводят в течение времени, в количестве и способом, которые достаточны для достижения благоприятного или синергического эффекта. Следует понимать, что предпочтительный способ, порядок введения и соответствующие дозировочные количества и схемы принятия лекарств для каждого соединения в комбинации будут зависеть от конкретного назначаемого лекарственного средства и ингибитора PARP, пути введения, конкретной вылечиваемой опухоли и конкретного вылечиваемого реципиента. Оптимальный способ и путь введения, а также дозировочные количества и схемы принятия лекарств могут быть легко установлены специалистами в данной области техники с помощью обычных методов и с учетом приведенной в настоящем описании информации.
Специалисты смогут легко определить эффективное количество из результатов испытаний, которые приведены далее в настоящем описании. В общем случае предполагается, что эффективное количество составит от 0,001 мг/кг до 100 мг/кг массы тела, в частности, от 0,005 мг/кг до 10 мг/кг массы тела. Может оказаться удобным назначать требуемую дозу в виде двух, трех, четырех или большего количества субдоз через соответствующие интервалы в течение дня. Указанные субдозы могут быть составлены в виде стандартной разовой лекарственной формы, которая, например, содержит от 0,05 до 500 мг и, в частности, от 0,1 мг до 200 мг активного ингредиента в одной разовой лекарственной форме.
Точная дозировка и частота введения зависят от конкретного используемого соединения формулы (I), конкретного состояния, лечение которого проводят, тяжести состояния, лечение которого проводят, возраста, веса, пола, степени развития расстройства и общего физического состояния конкретного пациента, а так же от другого лекарственного средства, которое индивид может принимать, что хорошо известно специалистам. Кроме того, очевидно, что указанное эффективное ежедневно назначаемое количество может быть снижено или увеличено в зависимости от ответной реакции вылечиваемого субъекта и/или в зависимости от оценки врача, предписывающего составы по настоящему изобретению.
В зависимости от способа введения фармацевтическая композиция, предпочтительно, включает от 0,05 до 99% масс., более предпочтительно, от 0,1 до 70% масс., и еще более предпочтительно, от 0,1 до 50% масс. активного ингредиента и от 1 до 99,95% масс., более предпочтительно, от 30 до 99,9% масс., и еще более предпочтительно, от 50 до 99,9% масс. фармацевтически приемлемого носителя (все проценты приведены относительно общей массы композиции).
Следующие примеры поясняют настоящее изобретение.
Экспериментальная часть
Далее по тексту настоящего описания “ДМФА” обозначает N,N-диметилформамид, “DCM” обозначает дихлорметан, “DIPE” обозначает диизопропиловый эфир, “ДМСО” обозначает диметилсульфоксид, “EtOAc” обозначает этилацетат, “MeOH” обозначает метанол, “ТГФ” обозначает тетрагидрофуран.
Для некоторых соединений, содержащих 1 хиральный центр, абсолютная стереохимическая конфигурация стереогенного атома углерода в настоящем описании экспериментально не установлена. В таких случаях первую выделенную стереохимически изомерную форму обозначают как “энантиомер А”, а вторую - как “энантиомер В”, не приводя дальнейшую ссылку на действительную стереохимическую конфигурацию. Тем не менее, указанная действительная стереохимическая конфигурация “энантиомерной А” и “энантиомерной В” формы может быть однозначно определена специалистами при помощи хорошо известных из области техники методов, таких как, например, дифракционный рентгеновский анализ. Методики выделения подробно описаны ниже.
А. Получение промежуточных соединений
Пример А1
а) Получение промежуточного соединения 1  .
.
Раствор 4-циклогексен-1-илморфолина (0,0265 мол) в DCM (25 мл) по каплям при комнатной температуре добавляют к раствору 1-бром-3-изоцианатобензола (0,0252 мол) в DCM (30 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 час, а затем упаривают досуха, получая 11 г (91%) промежуточного соединения 1, которое без дальнейшей очистки используют на следующей стадии.
b) Получение промежуточного соединения 2 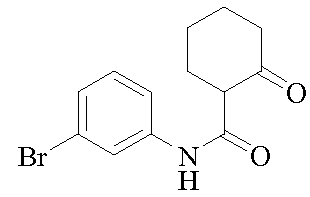 .
.
Раствор промежуточного соединения 1 (0,0252 мол) в концентрированной серной кислоте (40 мл) перемешивают при температуре 100°С в течение 30 мин, затем охлаждают до комнатной температуры, выливают в ледяную воду и вновь перемешивают при комнатной температуре в течение 1 час и 30 мин. Осадок отфильтровывают, промывают водой, диэтиловым эфиром и сушат, получая 8,4 г промежуточного соединения 2.
c) Получение промежуточного соединения 3 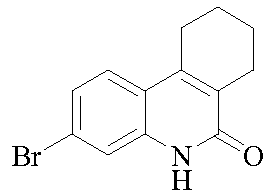 .
.
Раствор промежуточного соединения 2 (0,0017 мол) в 96%-ной серной кислоте (2 мл) перемешивают при температуре 100°С в течение 15 мин, затем осторожно выливают в ледяную воду, фильтруют, дважды промывают водой и сушат, получая 0,441 г (93%) промежуточного соединения 3, которое без дальнейшей очистки используют на следующей стадии.
d) Получение промежуточного соединения 4a 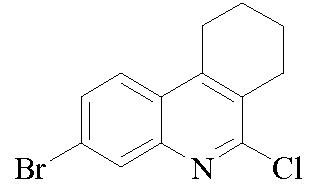 .
.
Раствор промежуточного соединения 3 (0,023 мол) в оксихлориде фосфора (100 мл) кипятят с обратным холодильником при перемешивании до полного растворения, оставляют на 10 мин, охлаждают до комнатной температуры, очень медленно (в течение одного часа) при комнатной температуре и при энергичном перемешивании выливают в воду. Твердое вещество отфильтровывают и сушат, получая 5,07 г (74%) промежуточного соединения 4a; температура плавления: 84°С.
e) Получение промежуточного соединения 4 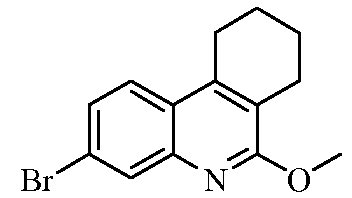 .
.
Раствор метоксида натрия (0,101 мол) добавляют к раствору промежуточного соединения 4a (0,0253 мол) в метаноле (100 мл) при комнатной температуре. Полученную смесь кипятят с обратным холодильником при перемешивании в течение 15 час, охлаждают до комнатной температуры и выливают в воду. Смесь экстрагируют с помощью DCM, органический слой отделяют, промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 6,1 г (83%) промежуточного соединения 4.
f) Получение промежуточного соединения 5 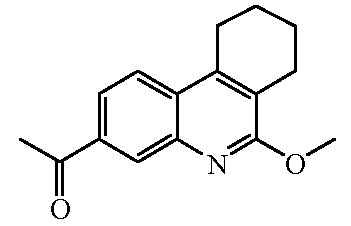 .
.
В инертной атмосфере трибутил(1-этоксивинил)олово (0,018 мол), а затем дихлорбис(трифенилфосфин)палладий(II) (0,0008 мол) добавляют к раствору промежуточного соединения 4 (0,016 мол) в сухом толуоле (29 мл). Смесь перемешивают при нагревании до 80°С в течение 18 час, охлаждают до комнатной температуры и фильтруют через целит. Органический слой промывают 2М раствором HCl, затем насыщенным раствором соли до тех пор, пока не будет достигнуто значение рН 7, сушат (над MgSO4), фильтруют и растворитель упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc от 95/5 до 80/20). Собирают чистые фракции и растворитель упаривают, получая 1,3 г (31%) промежуточного соединения 5.
g) Получение промежуточного соединения 6 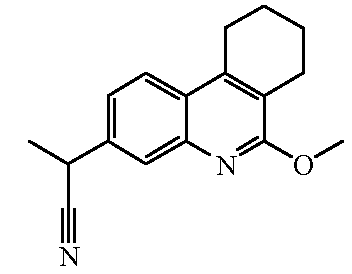 .
.
В токе азота порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,023 мол), а затем МеОН (2,4 мл) к раствору п-толуолсульфонилметилизоцианида (0,012 мол) в ДМСО (12 мл). Полученную смесь перемешивают при 10°С в течение 15 мин. Добавляют раствор промежуточного соединения 5 (0,0051 мол) в ДМСО (1,3 мл). Смесь перемешивают при 10°С в течение 1 час и 15 мин и экстрагируют с помощью DCM и EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (элюент: петролейный эфир/EtOAc от 90/10 до 50/50). Собирают чистые фракции и растворитель упаривают, получая 0,923 г (38%) промежуточного соединения 6 (в виде масла).
h) Получение промежуточного соединения 7 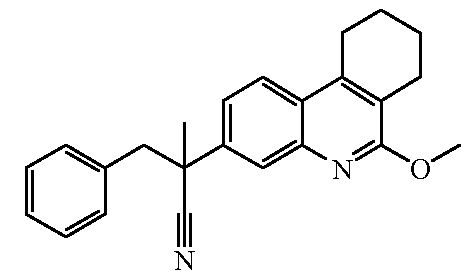 .
.
Гидрид натрия (60%-ная дисперсия в масле) (0,0005 мол) медленно добавляют при 0°С к раствору промежуточного соединения 6 (0,0003 мол) в ДМФА (1,4 мл) в токе N2. Смесь перемешивают в течение 5 мин. Добавляют бромметилбензол (0,0005 мол). Смесь перемешивают при комнатной температуре в течение 18 час. Вновь добавляют гидрид натрия (0,0005 мол). Смесь перемешивают в течение 3 час, реакцию прерывают, добавив насыщенный раствор хлорида аммония, и экстрагируют с помощью EtOAc. Органический слой промывают насыщенным раствором соли, сушат (над MgSO4), фильтруют и растворитель упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (элюент: петролейный эфир/EtOAc от 95/5 до 90/10). Собирают чистые фракции и растворитель упаривают, получая 0,159 г (99%) промежуточного соединения 7.
i) Получение промежуточного соединения 7a и 7b
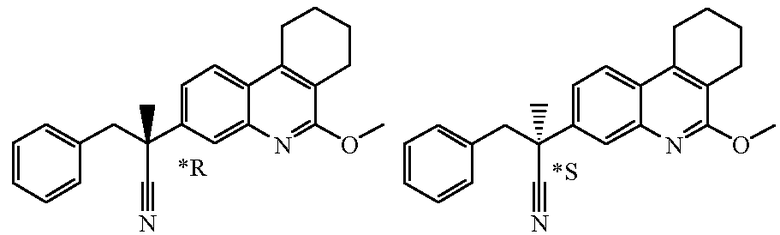 .
.
Промежуточное соединение 7 (0,28 г, 0,0008 мол) разделяют на его энантиомеры методом хроматографии в сверхкритической среде на хиральной колонке с силикагелем (колонка Chiralpak®, элюент: CO2/EtOH/iPrOH/изопропиламин: 70/15/15/0,3). Собирают чистые фракции и растворитель упаривают, получая 0,115 г (29%) промежуточного соединение 7a (энантиомер А) и 0,120 г (30%) промежуточного соединение 7b (энантиомер B).
Пример А2
Получение промежуточного соединения 8 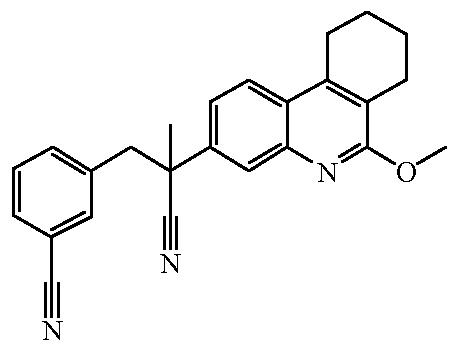 .
.
Гидрид натрия (60%-ная дисперсия в масле) (0,0006 мол) в токе N2 добавляют порциями при 0°С к раствору промежуточного соединения 6 (0,0004 мол) в ДМФА (1,7 мл). Смесь перемешивают в течение 5 мин. Добавляют 3-бромметилбензонитрил (0,0006 мол). Смесь перемешивают при комнатной температуре в течение 18 час. Вновь добавляют гидрид натрия (0,0006 мол). Смесь перемешивают еще в течение 3 час, реакцию прерывают насыщенным раствором хлорида аммония и экстрагируют с помощью EtOAc. Органический слой промывают насыщенным раствором соли, сушат (над MgSO4), фильтруют и растворитель упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 80/20). Собирают чистые фракции и растворитель упаривают, получая 0,0963 г (52%) промежуточного соединения 8 (в виде масла).
Пример А3
Получение промежуточного соединения 9 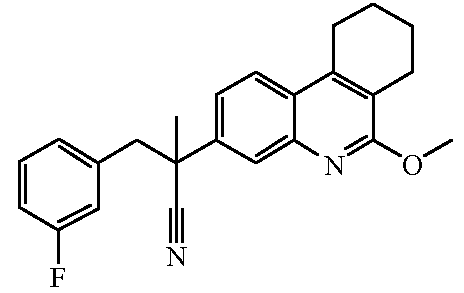 .
.
В токе N2 порциями при 5°С добавляют 2-метилпропан-2-олят калия (0,0011 мол) к раствору промежуточного соединения 6 (0,0006 мол) и 1-бромметил-3-фторбензола (0,0008 мол) в ТГФ (5 мл). Полученную смесь перемешивают при комнатной температуре в течение 5 час, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 95/5). Собирают чистые фракции и растворитель упаривают, получая 0,33 г (78%) промежуточного соединения 9.
Пример А4
Получение промежуточного соединения 10 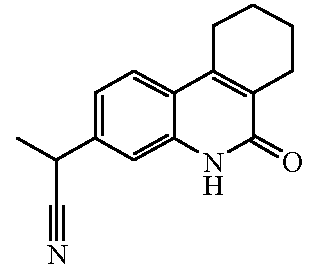 .
.
Смесь промежуточного соединения 6 (0,0009 мол) в 3н растворе HCl (2 мл) и 1,4-диоксане (2 мл) перемешивают при 80°С в течение 4 час, затем охлаждают до комнатной температуры, подщелачивают 10%-ным раствором К2СО3. Осадок отфильтровывают, промывают водой, а затем диэтиловым эфиром и сушат, получая 0,08 г (35%) промежуточного соединения 10.
Пример А5
Получение промежуточного соединения 11 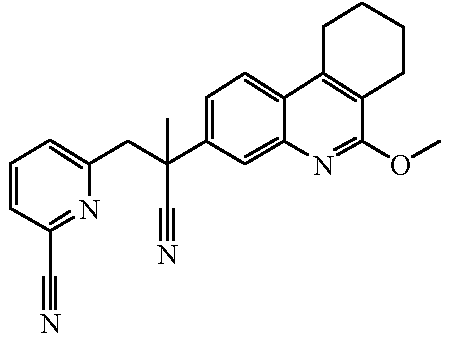 .
.
В токе N2 порциями при 5°С добавляют 2-метилпропан-2-олят калия (0,0015 мол) к раствору промежуточного соединения 6 (0,0007 мол) и 6-бромметилпиридин-2-карбонитрила (0,0011 мол) в ТГФ (5 мл). Полученную смесь перемешивают при комнатной температуре в течение 5 час, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают, получая 0,25 г промежуточного соединения 11, которое без дальнейшей очистки используют на следующей стадии реакции.
Пример А6
Получение промежуточного соединения 12 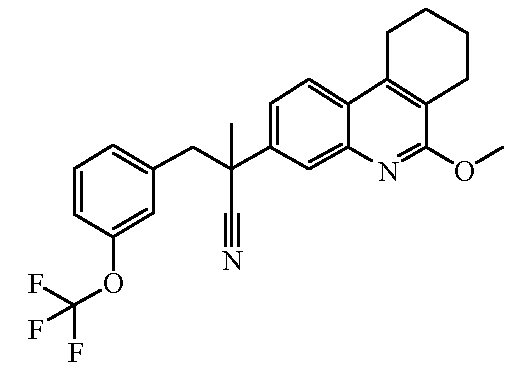 .
.
В токе N2 порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,0011 мол) к раствору промежуточного соединения 6 (0,00056 мол) и 1-бромметил-3-трифторметоксибензола (0,0008 мол) в ТГФ (10 мл). Смесь перемешивают при комнатной температуре в течение 5 час, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают, получая 0,245 г (99%) промежуточного соединения 12.
Пример А7
Получение промежуточного соединения 13 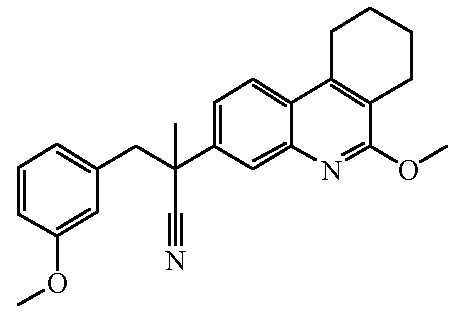 .
.
В токе N2 порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,0011 мол) к раствору промежуточного соединения 6 (0,0006 мол) и 1-бромметил-3-метоксибензола (0,0008 мол) в ТГФ (5 мл). Смесь перемешивают при комнатной температуре в течение 5 час, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают, получая 0,215 г (99%) промежуточного соединения 13.
Пример А8
Получение промежуточного соединения 14 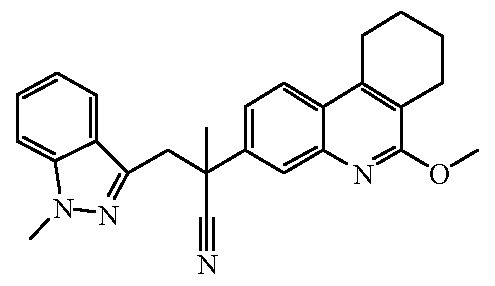 .
.
В токе N2 порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,0011 мол) к раствору промежуточного соединения 6 (0,0006 мол) и 3-хлорметил-1-метил-1Н-индазола (0,0008 мол) в ТГФ (5 мл). Смесь перемешивают при комнатной температуре в течение 5 час, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат, получая 0,15 г (65%) промежуточного соединения 14.
Пример А9
Получение промежуточного соединения 15 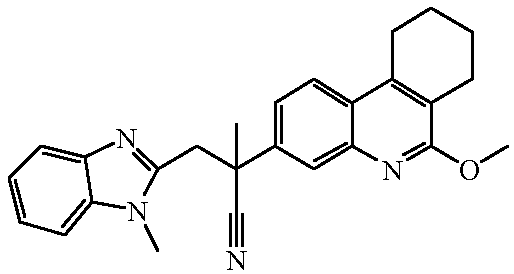 .
.
В токе N2 порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,0017 мол) к раствору промежуточного соединения 6 (0,0006 мол) и 2-хлорметил-1-метил-1Н-бензимидазола (0,0008 мол) в ТГФ (5 мл). Смесь перемешивают при комнатной температуре в течение 5 час, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток очищают колоночной хроматографией на силикагеле (элюент: DCM 100, затем DCM/MeOH/NH4OH 98/2/0,2; 3-5 мкм). Собирают чистые фракции и растворитель упаривают, получая 0,114 г (49%) промежуточного соединения 15.
Пример А10
Получение промежуточного соединения 16 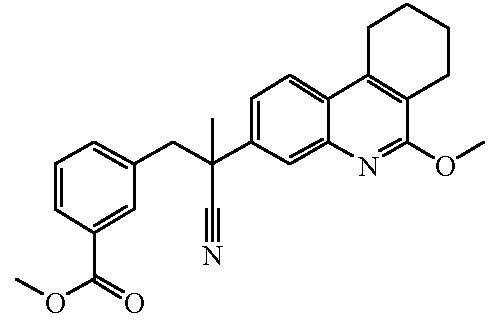 .
.
В токе N2 порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,0049 мол) к раствору промежуточного соединения 6 (0,0024 мол) и метилового эфира 3-бромметилбензойной кислоты (0,0036 мол) в ТГФ (20 мл). Смесь перемешивают при комнатной температуре в течение 5 час, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 80/20; 15-40 мкм). Собирают чистые фракции и растворитель упаривают, получая 0,392 г (39%) промежуточного соединения 16.
Пример А11
Получение промежуточного соединения 17 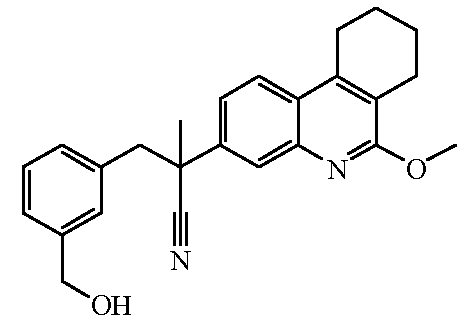 .
.
В токе N2 при 5°С добавляют раствор промежуточного соединения 16 (0,0009 мол) в сухом ТГФ (3 мл) к раствору алюмогидрида лития (0,0009 мол) в сухом ТГФ (4 мл). Смесь перемешивают при температуре 5°С в течение 1 час. Медленно добавляют EtOAc, а затем воду. Осадок отделяют фильтрованием через слой целлита. Фильтрат экстрагируют с помощью EtOAc. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 0,29 г (66%) промежуточного соединения 17.
Пример А12
a) Получение промежуточного соединения 18 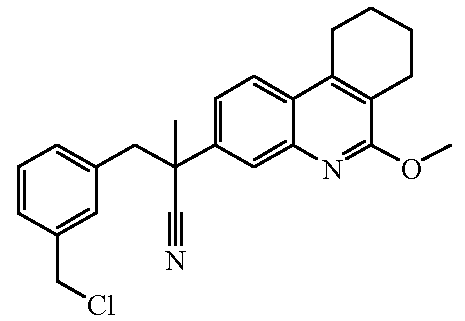 .
.
При 5°С добавляют тионилхлорид (0,09 мл) к раствору промежуточного соединения 17 (0,0006 мол) в DCM (3 мл). Смесь перемешивают при температуре 5°С в течение 1 час, затем перемешивают при комнатной температуре в течение 3 час и упаривают досуха, получая 0,22 г (95%) промежуточного соединения 18, которое без дальнейшей очистки используют на следующей стадии.
b) Получение промежуточного соединения 19
Смесь соединений
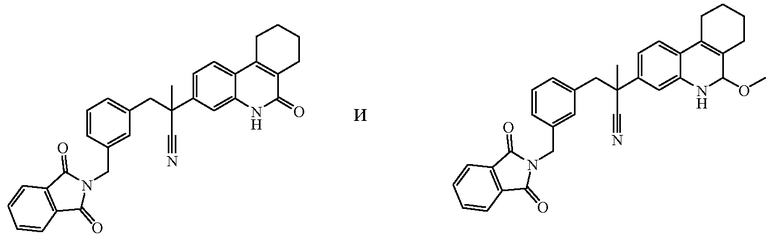 .
.
Смесь промежуточного соединения 18 (0,0005 мол), изоиндол-1,3-диона (0,0011 мол) и К2СО3 (0,0011 мол) в ДМФА (3 мл) перемешивают при температуре 100°С в течение 4 час, затем охлаждают до комнатной температуры, выливают в воду и экстрагируют с помощью EtOAc. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 0,32 г промежуточного соединения 19, которое без дальнейшей очистки используют на следующей стадии.
c) Получение промежуточного соединения 20 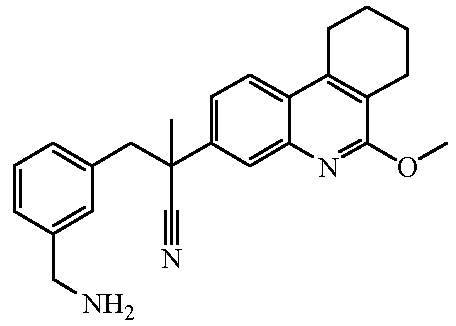 .
.
Моногидрат гидразина (0,0019 мол) добавляют по каплям к раствору промежуточного соединения 19 (0,0006 мол) в этаноле (4 мл). Смесь перемешивают при температуре 80°С в течение 4 час, выливают в холодную воду и экстрагируют с помощью EtOAc. Органический слой промывают водой и насыщенным раствором соли, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток очищают колоночной флэш-хроматографией на силикагеле (элюент: DCM/MeOH/NH4OH от 98/2/0,2 до 88/12/1,2; 3-5 мкм). Собирают чистые фракции и растворитель упаривают досуха. Остаток (0,034 г) обрабатывают с помощью DCM и диэтилового эфира, получая 0,03 г (13%) промежуточного соединения 20; температура плавления: 80°С.
Пример А13
Получение промежуточного соединения 21 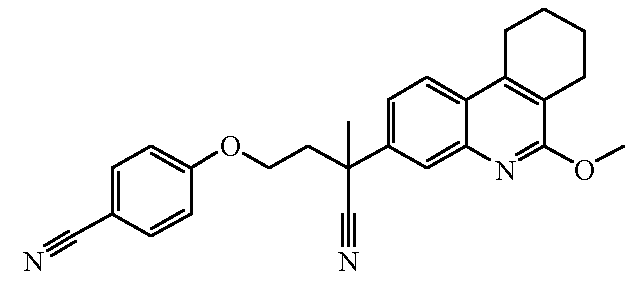 .
.
В токе N2 порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,0011 мол) к раствору промежуточного соединения 6 (0,0006 мол) и 4-(2-бромэтокси)бензонитрила (0,0008 мол) в ТГФ (5 мл). Смесь оставляют на ночь перемешиваться при комнатной температуре, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток очищают колоночной хроматографией на силикагеле (элюент: DCM/циклогексан 80/20; 3-5 мкм). Собирают чистые фракции и растворитель упаривают, получая 0,12 г (52%) промежуточного соединения 21.
Пример А14
a) Получение промежуточного соединения 22  .
.
Раствор промежуточного соединения 6 (0,0019 мол) в 1,2-диметоксиэтане (1 мл) порциями при 10°С добавляют в токе N2 к раствору 2-метилпропан-2-олята калия (0,0022 мол) в 1,2-диметоксиэтане (3 мл). Раствор перемешивают при комнатной температуре в течение 1 час. Добавляют дибромметан (0,0028 мол). Раствор перемешивают при комнатной температуре в течение 2 час, выливают в холодную воду и экстрагируют с помощью EtOAc. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 0,5 г (74%) промежуточного соединения 22, которое без дальнейшей очистки используют на следующей стадии.
b) Получение промежуточного соединения 23  .
.
Смесь промежуточного соединения 22 (0,0014 мол) и калиевого производного фталимида (0,0017 мол) в ДМФА (10 мл) оставляют перемешиваться на ночь при температуре 140°С, выливают в холодную воду и экстрагируют с помощью EtOAc. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 0,44 г (74%) промежуточного соединения 23, которое без дальнейшей очистки используют на следующей стадии.
c) Получение промежуточного соединения 24 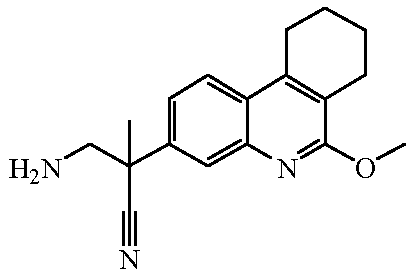 .
.
Смесь промежуточного соединения 23 (0,001 мол) и моногидрата гидразина (0,01 мол) в этаноле (10 мл) перемешивают при температуре 80°С в течение 3 час, затем охлаждают до комнатной температуры, выливают в холодную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 0,3 г (98%) промежуточного соединения 24 (в виде масла).
d) Получение промежуточного соединения 25 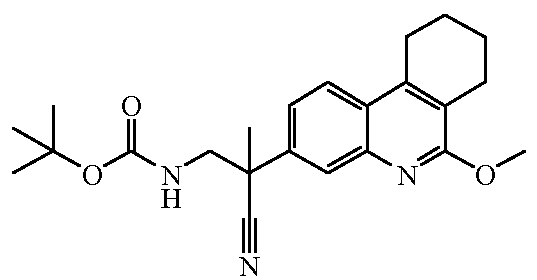 .
.
Ди-трет-бутилдикарбонат (0,0011 мол) при 10°С в токе N2 добавляют к раствору промежуточного соединения 24 (0,001 мол) в ТГФ (14 мл). Смесь перемешивают при комнатной температуре в течение 5 час, выливают в холодную воду и экстрагируют с помощью DCM. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток очищают колоночной хроматографией на силикагеле (элюент: DCM 100, затем DCM/MeOH/NH4OH 99/1/0,1; 3,4 мкм). Собирают чистые фракции и растворитель упаривают досуха, получая 0,197 г (47%) промежуточного соединения 25 (в виде масла).
e) Получение промежуточного соединения 26 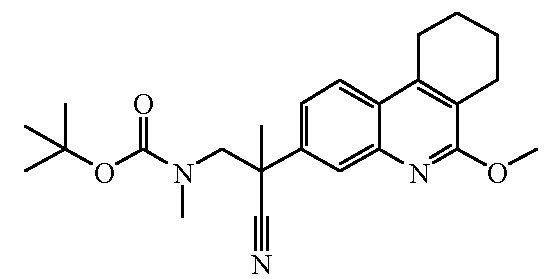 .
.
Гидрид натрия (0,0006 мол) в токе N2 добавляют при 10°С к раствору промежуточного соединения 25 (0,0005 мол) в ДМФА (3 мл). Смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют иодметан (0,0005 мол). Раствор перемешивают при комнатной температуре в течение 4 час, выливают в холодную воду и экстрагируют с помощью EtOAc. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 0,14 г (71%) промежуточного соединения 26 (в виде масла).
f) Получение промежуточного соединения 27 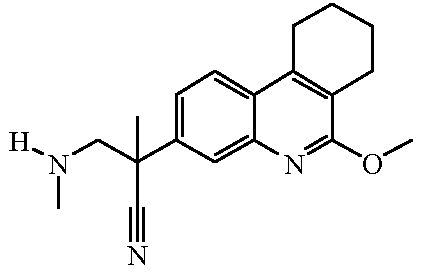 .
.
Трифторуксусную кислоту (0,0168 мол) добавляют при комнатной температуре к раствору промежуточного соединения 26 (0,0003 мол) в DCM (8 мл). Смесь перемешивают при комнатной температуре в течение 4 час, выливают в холодную воду, подщелачивают с помощью NH4OH и экстрагируют с помощью DCM. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток очищают колоночной флэш-хроматографией на силикагеле (элюент: от DCM 100 до DCM/MeOH/NH4OH 98/2/0,1). Собирают чистые фракции и растворитель упаривают досуха, получая 0,07 г (71%) промежуточного соединения 27 (в виде масла).
g) Получение промежуточного соединения 28 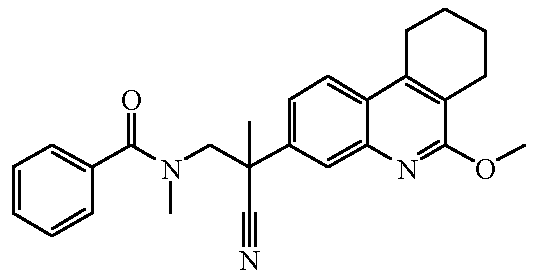 .
.
Бензоилхлорид (0,0002 мол) в токе N2 добавляют при 10°С к раствору триэтиламина (0,0002 мол) и промежуточного соединения 27 (0,0001 мол) в DCM (1,5 мл). Смесь перемешивают при температуре 10°С в течение 3 час, выливают в холодную воду и экстрагируют с помощью DCM. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 0,074 г промежуточного соединения 28.
Пример А15
Получение промежуточного соединения 29 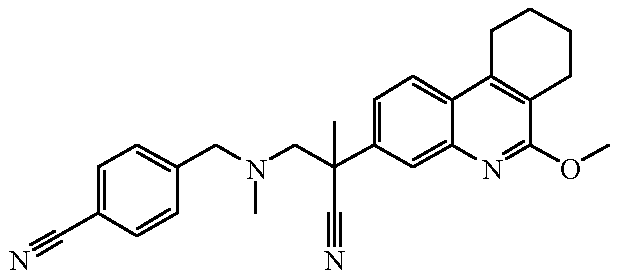 .
.
В токе N2 добавляют при 10°С 4-бромметилбензонитрил (0,0002 мол) к раствору промежуточного соединения 27 (0,0001 мол) и триэтиламина (0,0002 мол) в DCM (2 мл).
Смесь перемешивают при температуре 10°С в течение 3 час, затем охлаждают до комнатной температуры, выливают в холодную воду и экстрагируют с помощью DCM. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 0,026 г (42%) промежуточного соединения 29 (в виде масла).
Пример А16
Получение промежуточного соединения 30 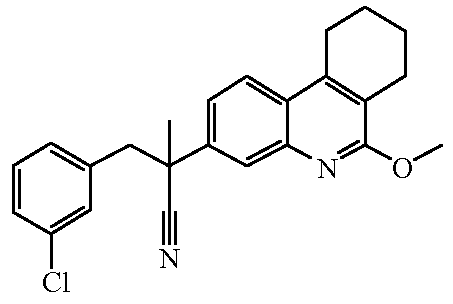 .
.
В токе N2 порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,0011 мол) к раствору промежуточного соединения 6 (0,00056 мол) и 1-бромметил-3-хлорбензола (0,0008 мол) в ТГФ (10 мл). Смесь перемешивают при комнатной температуре в течение 5 час, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха, получая 0,245 г (99%) промежуточного соединения 30.
В. Получение соединений по изобретению
Пример B1
a) Получение соединения 1 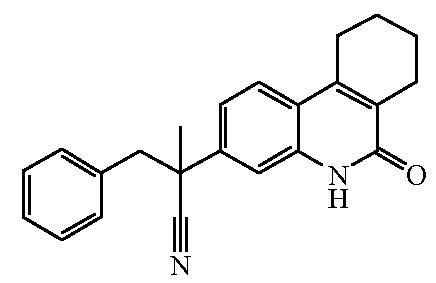 .
.
3н раствор HCl (620 мкл) добавляют к раствору промежуточного соединения 7 (0,0003 мол) в 1,4-диоксане (2,5 мл). Смесь перемешивают при температуре 80°С в течение 18 час, затем охлаждают до комнатной температуры, прерывают реакцию, добавив NaOH (0,1М), и экстрагируют с помощью EtOAc. Органический слой промывают насыщенным раствором соли, сушат (над MgSO4), фильтруют и растворитель упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (элюент: петролейный эфир/EtOAc 50/50). Собирают чистые фракции и растворитель упаривают, получая 0,0238 г (22%) соединения 1.
b) Получение соединений 2 и 3
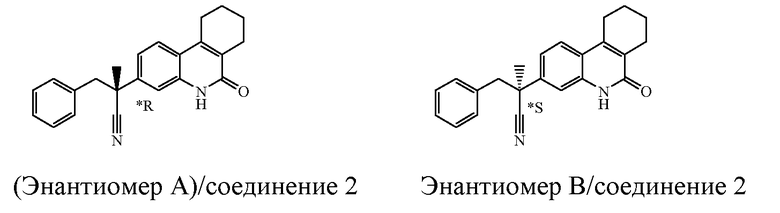 .
.
Раствор промежуточного соединения 7b (0,0004 мол) в 3н HCl (3 мл) и 1,4-диоксане (3 мл) перемешивают при температуре 80°С в течение 5 час, затем охлаждают до комнатной температуры. Осадок отфильтровывают, промывают водой, затем диэтиловым эфиром и сушат в вакууме, получая 0,09 г (78%) соединения 3; температура плавления: 250°С; [α]D 20=+89,87 (ДМФА; c=0,375).
Раствор промежуточного соединения 7a (0,0003 мол) в 3н HCl (3 мл) и 1,4-диоксане (3 мл) перемешивают при температуре 80°С в течение 5 час, затем охлаждают до комнатной температуры. Осадок отфильтровывают, промывают водой, затем диэтиловым эфиром и сушат в вакууме, получая 0,083 г (75%) соединения 2; температура плавления: 245°С; [α]D 20=-94,78 (ДМФА; c=0,35).
Пример B2
Получение соединения 4 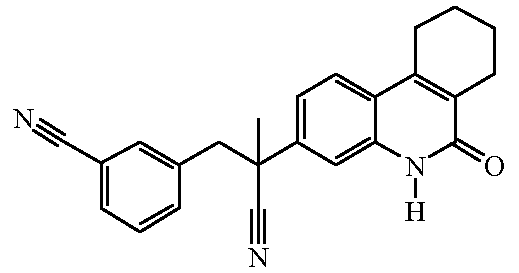 .
.
3н раствор HCl (620 мкл) добавляют к раствору промежуточного соединения 8 (0,0002 мол) в 1,4-диоксане (2,5 мл). Смесь перемешивают при температуре 80°С в течение 18 час, затем охлаждают до комнатной температуры, прерывают реакцию, добавив NaOH (0,1М), и экстрагируют с помощью EtOAc. Органический слой промывают насыщенным раствором соли, сушат (над MgSO4), фильтруют и растворитель упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 25/75). Собирают чистые фракции и растворитель упаривают, получая 0,01 г (14%) соединения 4.
Пример B3
a) Получение соединения 5  .
.
Смесь промежуточного соединения 9 (0,0006 мол) в 3н растворе HCl (2 мл) и 1,4-диоксане (4 мл) перемешивают при температуре 80°С в течение 18 час, затем охлаждают до комнатной температуры, фильтруют, промывают водой, затем диэтиловым эфиром и сушат в вакууме. Остаток и фильтрат смешивают друг с другом и переносят в EtOAc и 10%-ный раствор К2СО3. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат, получая 0,049 г (75%) соединения 5; температура плавления: 217°С.
b) Получение соединений 5a и 5b
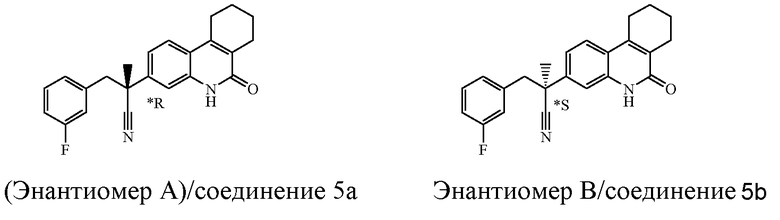 .
.
Раствор промежуточного соединения 9 (0,0009 мол) в 3н HCl (6 мл) и 1,4-диоксане (6 мл) перемешивают при температуре 80°С в течение 4 час, а затем охлаждают до комнатной температуры. Осадок отфильтровывают, промывают водой и диэтиловым эфиром и сушат. Остаток разделяют на его энантиомеры методом колоночной хроматографии (элюент: МеОН 100). Собирают две фракции и растворитель упаривают. Энантиомер А кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат, получая 0,102 г (32%) соединения 5a; температура плавления: 226°С; [α]D 20=+90,7 (ДМФА; c=0,34). Энантиомер В кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат, получая 0,098 г (31%) соединения 5b; температура плавления: 222°С; [α]D 20=-82,09 (ДМФА; c=0,335).
Пример B4
Получение соединения 6 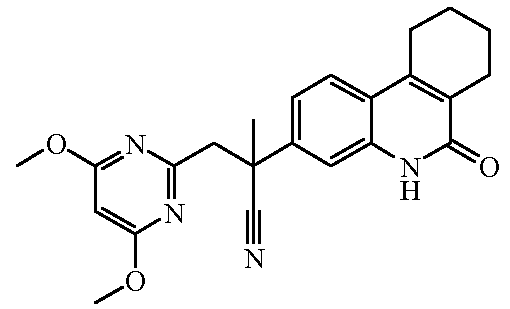 .
.
В токе N2 порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,0008 мол) к раствору промежуточного соединения 10 (0,0003 мол) и 2-(хлорметил)-4,6-диметоксипиримидина (0,0008 мол) в ТГФ (5 мл). Смесь оставляют на ночь перемешиваться при комнатной температуре, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток кристаллизуют из DIPE. Осадок отфильтровывают и сушат, получая 0,056 г (43%) соединения 6; температура плавления: 193°С.
Пример B5
Получение соединения 7 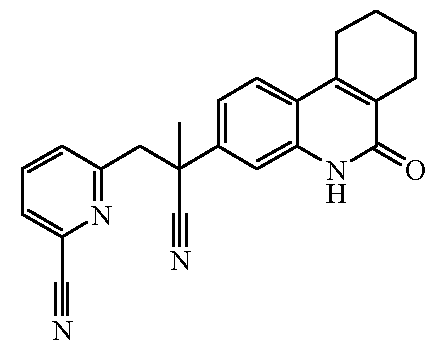 .
.
Раствор промежуточного соединения 11 (0,0009 мол) в 3н HCl (3 мл) и 1,4-диоксане (3 мл) перемешивают при температуре 80°С в течение 4 час, затем охлаждают до комнатной температуры, подщелачивают 10%-ным раствором К2СО3 и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток кристаллизуют из DIPE. Осадок отфильтровывают и сушат, получая 0,16 г (66%) соединения 7; температура плавления: 210°С.
Пример B6
Получение соединения 8 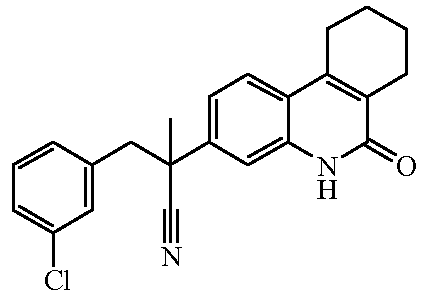 .
.
Раствор промежуточного соединения 30 (0,0005 мол) в 3н HCl (3 мл) и 1,4-диоксане (1,3 мл) перемешивают при температуре 80°С в течение 4 час, а затем охлаждают до комнатной температуры. Осадок отфильтровывают, промывают водой и диэтиловым эфиром и сушат в вакууме, получая 0,11 г (52%) соединения 8; температура плавления: 235°С.
Пример B7
Получение соединения 9 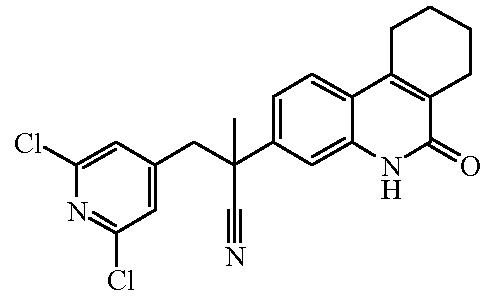 .
.
В токе N2 порциями при 10°С добавляют 2-метилпропан-2-олят калия (0,0016 мол) к раствору промежуточного соединения 10 (0,0008 мол) и 2,6-дихлор-4-хлорметилпиридина (0,001 мол) в ТГФ (15 мл). Смесь оставляют на ночь перемешиваться при комнатной температуре, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток очищают колоночной хроматографией на силикагеле (элюент: DCM/MeOH 98/2; 15-40 мкм). Собирают чистые фракции и растворитель упаривают, получая 0,07 г (21%) соединения 9; температура плавления: 212°С.
Пример B8
Получение соединения 10 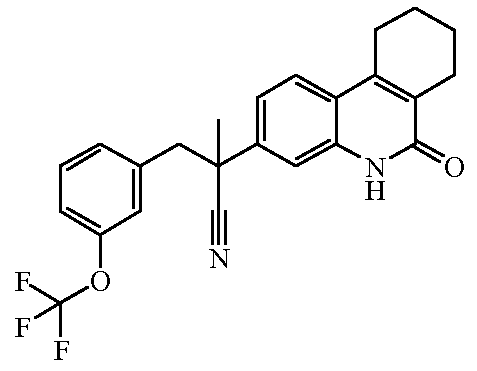 .
.
Раствор промежуточного соединения 12 (0,0005 мол) в 3н HCl (5 мл) и 1,4-диоксане (5 мл) оставляют перемешиваться на ночь при температуре 80°С, затем охлаждают до комнатной температуры, выливают в ледяную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат, получая 0,077 г (32%) соединения 10; температура плавления: 186°С.
Пример B9
Получение соединения 11 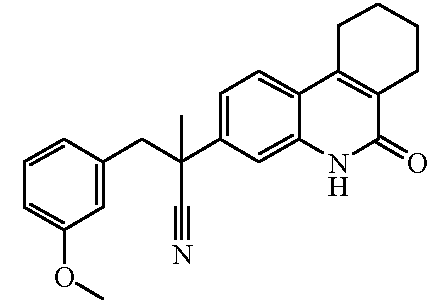 .
.
Раствор промежуточного соединения 13 (0,0005 мол) в 3н HCl (5 мл) и 1,4-диоксане (5 мл) оставляют перемешиваться на ночь при температуре 80°С, а затем охлаждают до комнатной температуры. Остаток отфильтровывают, промывают водой, затем диэтиловым эфиром и сушат в вакууме, получая 0,127 г (61%) соединения 11; температура плавления: 209°С.
Пример B10
Получение соединения 12 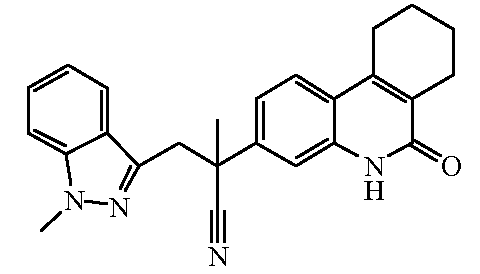 .
.
Раствор промежуточного соединения 14 (0,0003 мол) в 3н HCl (2 мл) и 1,4-диоксане (2 мл) перемешивают при температуре 80°С в течение 5 час, а затем охлаждают до комнатной температуры. Остаток отфильтровывают, промывают водой, затем диэтиловым эфиром и сушат в вакууме, получая 0,057 г (53%) соединения 12; температура плавления: 256°С.
Пример B11
Получение соединения 13 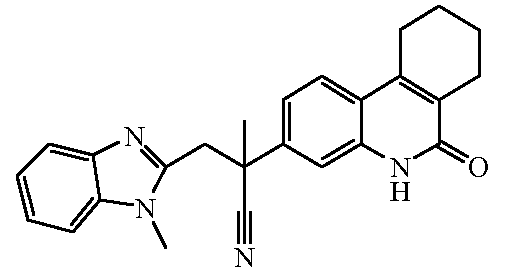 .
.
Раствор промежуточного соединения 15 (0,0003 мол) в 3н HCl (3 мл) и 1,4-диоксане (3 мл) перемешивают при температуре 80°С в течение 5 час, затем охлаждают до комнатной температуры, подщелачивают 10%-ным раствором К2СО3 и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток кристаллизуют из диэтилового эфира. Осадок отфильтровывают и сушат в вакууме, получая 0,065 г (61%) соединения 13, температура плавления: 260°С.
Пример B12
Получение соединения 14 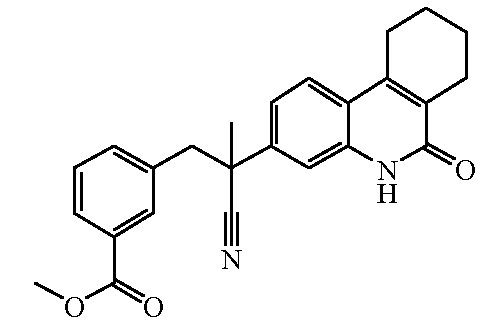 .
.
Раствор промежуточного соединения 16 (0,00007 мол) в 3н HCl (0,51 мл) и 1,4-диоксане (0,51 мл) перемешивают при температуре 80°С в течение 4 час, затем выливают в ледяную воду, подщелачивают 10%-ным раствором К2СО3 и экстрагируют с помощью EtOAc. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток переносят в DCM и диэтиловый эфир. Осадок отфильтровывают и сушат, получая 0,016 г (56%) соединения 14; температура плавления: 126°С.
Пример B13
Получение соединения 15 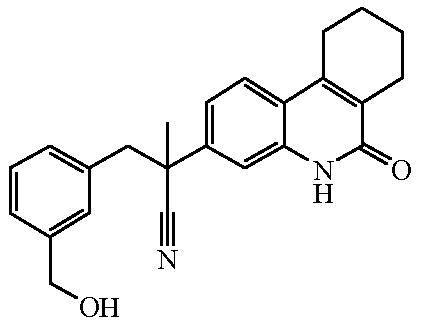 .
.
Раствор промежуточного соединения 17 (0,00007 мол) в 3н HCl (0,51 мл) и 1,4-диоксане (0,51 мл) перемешивают при температуре 80°С в течение 3 час, затем охлаждают до комнатной температуры, подщелачивают 10%-ным раствором К2СО3 и экстрагируют с помощью EtOAc. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток кристаллизуют из DIPE. Осадок отфильтровывают и сушат, получая 0,0025 г (9%) соединения 15; температура плавления: 131°С.
Пример B14
Получение соединения 16 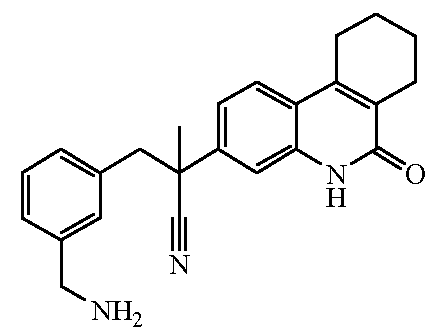 .
.
Моногидрат гидразина (0,0019 мол) добавляют по каплям к раствору промежуточного соединения 19 (0,0006 мол) в этаноле (4 мл). Смесь перемешивают при температуре 80°С в течение 4 час, выливают в холодную воду и экстрагируют с помощью EtOAc. Органический слой промывают водой и насыщенным раствором соли, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток очищают колоночной хроматографией на силикагеле (элюент: DCM/MeOH/NH4OH от 98/2/0,2 до 88/12/1,2; 3,4 мкм). Собирают чистые фракции и растворитель упаривают. Остаток переносят в DCM и диэтиловый эфир. Осадок отфильтровывают и сушат, получая 0,0173 г (7%) соединения 16; температура плавления: 80°С.
Пример B15
Получение соединения 17 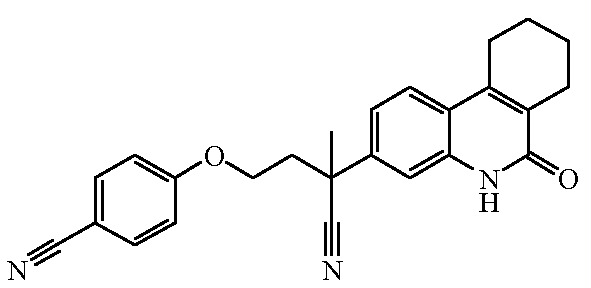 .
.
Раствор промежуточного соединения 21 (0,0003 мол) в 3н HCl (4 мл) и 1,4-диоксане (4 мл) перемешивают при температуре 80°С в течение 4 час, затем охлаждают до комнатной температуры и подщелачивают 10%-ным раствором К2СО3. Осадок отфильтровывают, промывают водой, затем диэтиловым эфиром и сушат, получая 0,069 г (59%) соединения 17; температура плавления: 169°С.
Пример B16
Получение соединения 18 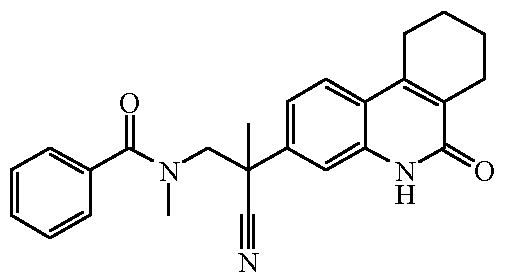 .
.
Раствор промежуточного соединения 28 (0,0002 мол) в 3н HCl (1,7 мл) и 1,4-диоксане (1,7 мл) перемешивают при температуре 80°С в течение 4 час, выливают в холодную воду и экстрагируют с помощью EtOAc. Органический слой отделяют, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток кристаллизуют из DIPE. Осадок отфильтровывают и сушат, получая 0,032 г (44%) соединения 18; температура плавления: 248°С.
Пример B17
Получение соединения 19 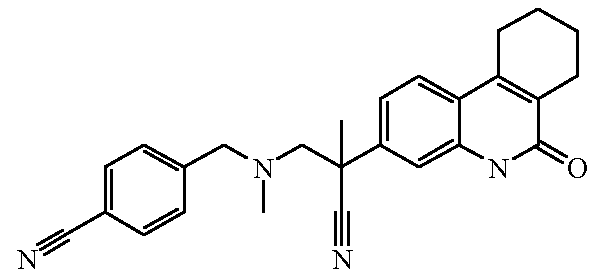 .
.
Раствор промежуточного соединения 29 (0,00007 мол) в 3н HCl (1 мл) и 1,4-диоксане (1 мл) перемешивают при температуре 80°С в течение 4 час, затем выливают в холодную воду, подщелачивают 10%-ным раствором К2СО3 и экстрагируют с помощью EtOAc. Органический слой промывают водой, сушат (над MgSO4), фильтруют и растворитель упаривают досуха. Остаток переносят в DCM и диэтиловый эфир. Осадок отфильтровывают и сушат, получая 0,025 г (87%) соединения 19; температура плавления: 254°С.
В таблице F-1 перечислены соединения, которые получают в соответствии с приведенными выше примерами.
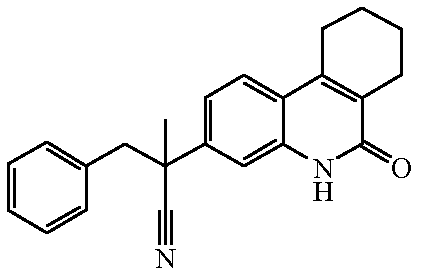
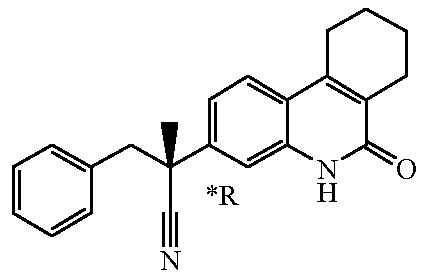
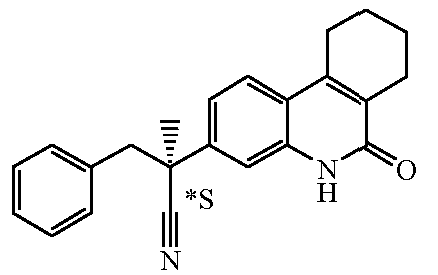
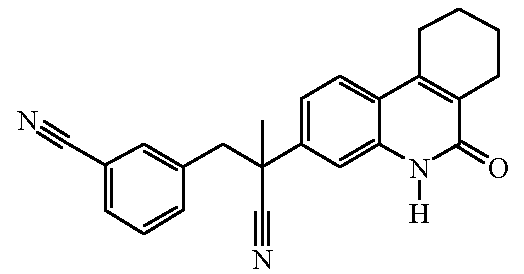
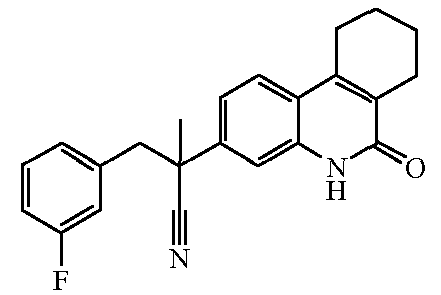
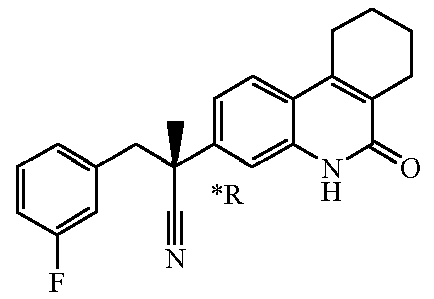
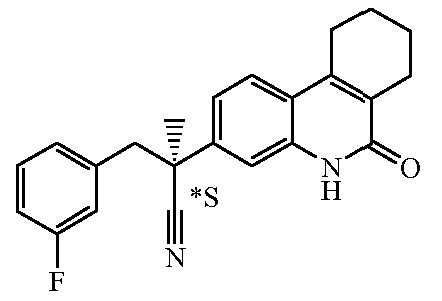
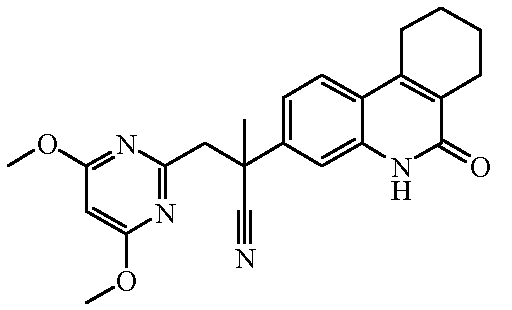
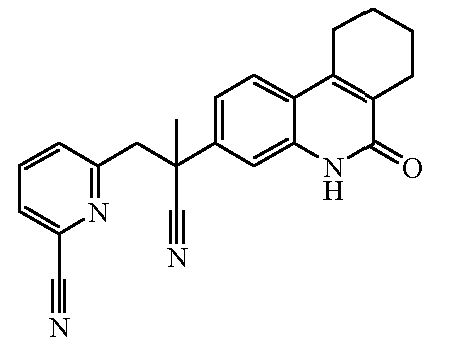
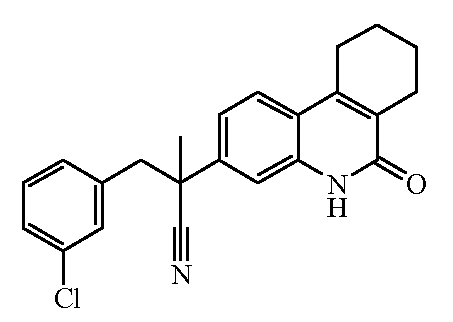
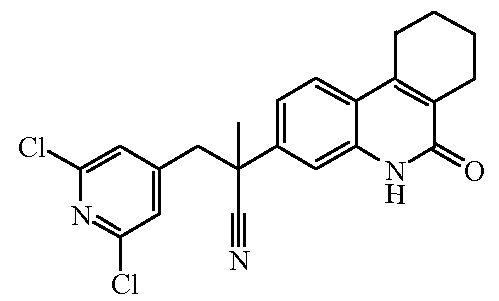
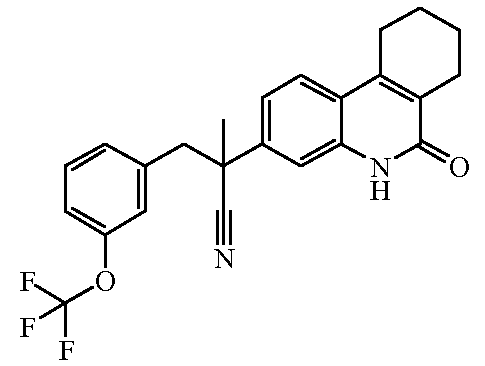
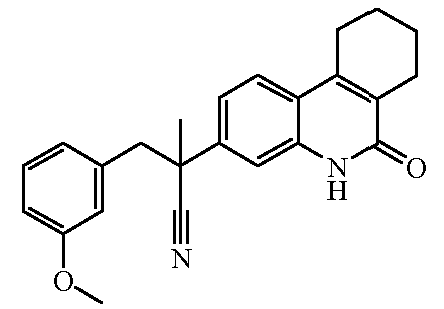
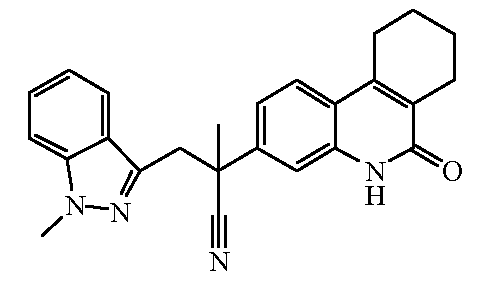
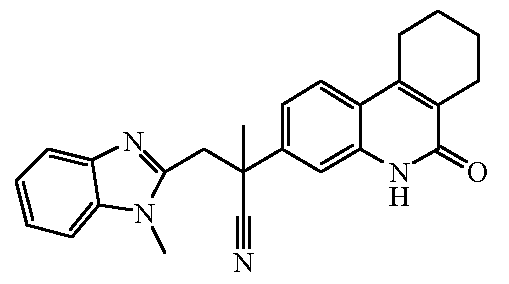
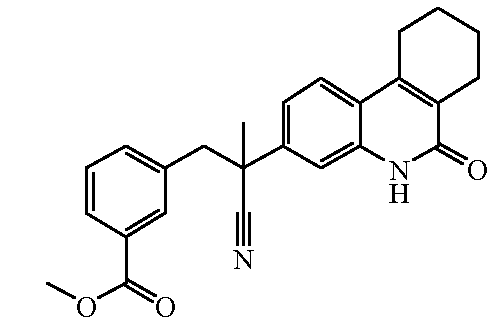
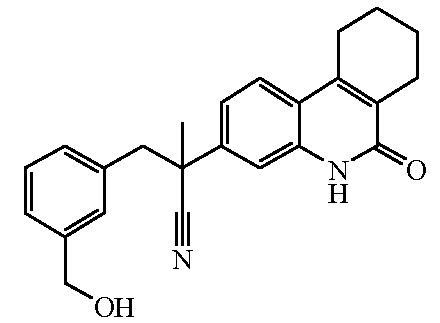
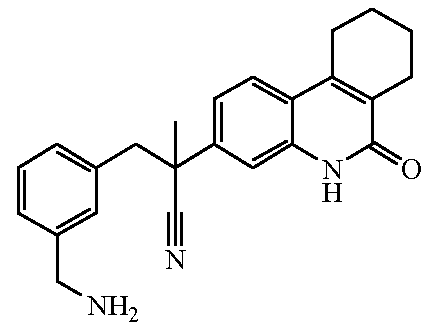
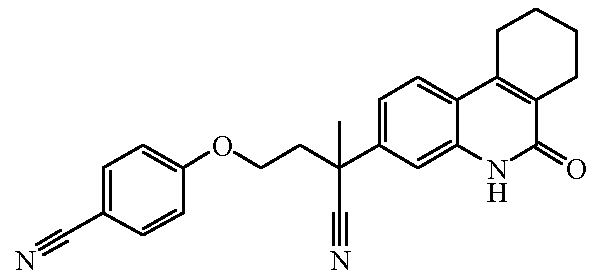
Аналитическая часть
ЖХ/МС
Общая методика проведения ЖХ/МС.
Измерения методом ВЭЖХ проводят, используя насос Waters 1512 с диодно-матричным детектором (DAD) фирмы Waters, аутосамплер Gilson 215 и колонку, которая указана в приведенных ниже способах. Часть потока из колонки направляют в масс-спектрометр. В зависимости от типа соединения применяют либо ионизацию электрораспылением, либо химическую ионизацию при атмосферном давлении (APCI).
Типичные условия при электрораспылении: напряжение на капиллярной игле 3,5 кВ, напряжение на конусе 25 В. Температуру источника поддерживают в интервале 120-150°С (точную температуру устанавливают для каждого соединения в отдельности). Типичные условия APCI: ток коронного разряда 17 мкА, напряжение на конусе 25 В, температура десольватации 350°С, а температуру источника поддерживают в интервале 140-160°С (точную температуру устанавливают для каждого соединения в отдельности).
Масс-спектры получают, проводя сканирование от 100 до 650 или 1000, если необходимо, например, в течение 1 сек, используя выдержку времени 0,1 сек. В качестве газа-распылителя применяют азот.
ЖХ/МС - Методика
В дополнение к общей методике: ВЭЖХ с обращенной фазой проводят на Waters Xterra MS с колонкой C18 5 мкм (4,6×100 мм; картридж plus guard) со скоростью потока 2 мл/мин. Две подвижные фазы (подвижная фаза А: вода с 10 мМ раствором бикарбоната аммония в ультрачистой воде; подвижная фаза В: ацетонитрил) используют для проведения процесса в градиентных условиях от 95% А до 95% В со скоростью потока 2 мл/мин в течение 3,5 мин и выдержкой в течение 2 мин. Как правило, инжектируемые объемы составляют от 2 мкл до 7 мкл включительно.
С. Фармакологические примеры
С1. In vitro сцинтилляционный проксимальный анализ (SPA) для ингибиторной активности по отношению к PARP-1
Соединения по настоящему изобретению тестируют в условиях in vitro, взяв за основу анализ по методу SPA (правом собственности обладает компании GE healthcare).
В принципе, анализ основан на хорошо отработанном методе SPA для установления поли-АДФ-рибозилирования биотинилированных белков-мишеней, т.е. гистонов. Указанное рибозирование индуцируют с помощью фермента PARP-1, активированного содержащей однонитевой разрыв ДНК, и динуклеотида [3H]-никотинамидаденин ([3H]-NAD+) в качестве донора АДФ-рибозила.
Гистоны (тип II-A, поставщик: Sigma) биотинилируют, используя набор реагентов для биотинилирования фирмы Amersham, и хранят в виде аликвот при температуре минус 20°С. Готовят основной раствор, содержащий 100 мг/мл шариков SPA-поливинилтолуол (PVT) (поставщик: Amersham) в забуференном фосфатом физиологическом растворе. Готовят основной раствор, содержащий 61,6 нМ [3H]-NAD+, добавляя [3H]-NAD+ (0,1 мКи/мл, поставщик: Perkin Elmer) в буферный раствор для инкубирования (50 мМ Tris/HCl, pH 8; 0,2 мМ DTT; 4 мМ MgCl2). Готовят 4 мМ раствор NAD+ (поставщик: Sigma). Фермент PARP-1 человека получают от компании Trevigen. Биотинилированные гистоны и шарики PVT-SPA смешивают и предварительно инкубируют в течение 30 мин при комнатной температуре. Фермент PARP-1 (концентрация зависит от партии фермента) смешивают с содержащей однонитевой разрыв ДНК и полученную смесь предварительно инкубируют при температуре 4°С в течение 30 мин. Равные части указанных раствора гистоны/шарики PVT-SPA и раствора фермент PARP-1/ДНК смешивают и 75 мкл полученной смеси вместе с 1 мкл раствора соединения по изобретению в ДМСО и 25 мкл [3H]-NAD+ на лунку помещают на 96-луночный планшет для микротитрования. Конечная концентрация в смеси для инкубирования составляет 2 мкг/мл для биотинилированных гистонов, 2 мг/мл для шариков PVT-SPA, 0,25 мкг/мл для содержащей однонитевой разрыв ДНК и в диапазоне 0,1-0,2 мкг/мл для фермента PARP-1. После инкубирования смеси в течение 20 мин при комнатной температуре реакцию прерывают, добавляя 100 мкл 4 мМ раствора NAD+ в воде (конечная концентрация 2 мМ) и планшеты смешивают. Шарики седиментируют центрифугированием (10 мин, 800 об/мин), и планшеты переносят в TopCountNXT™ (Packard) для проведения сцинтилляционных подсчетов, а значения выражают числом импульсов в минуту. При каждом эксперименте параллельно проводят манипуляции с контролями (содержащими фермент PARP-1 и ДМСО без соединения по изобретению), пустыми инкубациями (содержащими ДМСО, но не содержащими ни фермент PARP-1, ни ДНК или соединение по изобретению) и образцами (содержащими фермент PARP-1, ДНК и соединение по изобретению, растворенное в ДМСО). Все тестируемые соединения растворяют, а позднее дополнительно разбавляют в ДМСО. Строят кривую зависимости от дозы, при этом соединения по изобретению тестируют с концентрациями в диапазоне от 10-5М до 3×10-9М. При проведении каждого теста значения для пустых инкубаций вычитают как из значений для контролей, так и значений для образцов. Контроли показывают максимальную ферментативную активность PARP-1. Для каждого образца число импульсов в минуту выражают в виде процента от среднего числа импульсов в минуту для контролей. В тех случаях, когда это возможно, рассчитывают значения IC50 (концентрация лекарства, необходимая для снижения на 50% ферментативной активности PARP-1, по сравнению с контролем), используя линейную интерполяцию между экспериментальными точками, расположенными чуть выше и чуть ниже 50%-ного уровня. В настоящем описании эффект тестируемых соединений выражают в виде pIC50 (отрицательный логарифм значения IC50). В качестве соединения сравнения для оценки результатов SPA анализа используют 4-амино-1,8-нафталимид. Тестируемые соединения показывают ингибиторную активность при различных концентрациях (см. таблицу 2).
С2. In vitro сцинтилляционный проксимальный (SPA) для ингибиторной активности по отношению к TANK-2
Соединения по настоящему изобретению тестируют в условиях in vitro, взяв за основу анализа по методу SPA с использованием планшетов Ni Flash (96-луночных или 384-луночных). В принципе, анализ основан на методе SPA для установления аутополи-АДФ-рибозилирования белка TANK-2 с использованием динуклеотида [3H]-никотинамидаденин ([3H]-NAD+) в качестве донора АДФ-рибозила.
Готовят основной раствор, содержащий 100 нМ [3H]-NAD+/NAD (0,1 мКи/мл, поставщик: Perkin Elmer) и 25 мкМ (Sigma) в буферном растворе для анализа (60 мМ Tris/HCl, pH 7,4; 0,9 мМ DTT; 6 мМ MgCl2). Фермент TANK-2 получают, как описано в EP 1238063. 60 мкл буфера для анализа вместе с 1 мкл раствора соединения в ДМСО, 20 мкл [3H]-NAD+/NAD и 20 мкл фермента TANK-2 (конечная концентрация 8 мкг) добавляют в каждую лунку 96-луночного планшета с никелевым покрытием (Perkin Elmer). После инкубирования смеси в течение 120 мин при комнатной температуре реакцию прерывают, добавив 60 мкл стоп-реагента (42,6 мг NAD в 6 мл H2O). Планшеты закрывают с помощью приспособления для заклеивания планшетов и помещают в TopCountNXT™ (Packard) для проведения сцинтилляционных подсчетов. Значения выражают количеством импульсов в минуту. При каждом эксперименте параллельно проводят манипуляции с контролями (содержащими фермент TANK-2 и ДМСО без соединения), пустыми инкубациями (содержащими ДМСО, но не содержащими ни фермент TANK-2, ни соединение по изобретению) и образцами (содержащими фермент TANK-2 и соединение по изобретению, растворенное в ДМСО). Все тестируемые соединения растворяют, а позднее дополнительно разбавляют в ДМСО. Прежде всего, соединения тестируют при концентрации 10-5M. В том случае, когда соединения показывают активность при 10-5M, строят кривые зависимости от дозы, где соединения тестируют с концентрацией в диапазоне от 10-5 M до 3×10-8M. В каждом тесте значение для пустых инкубаций вычитают как из значений для контролей, так и значений для образцов. Контроли показывают максимальную ферментативную активность TANK-2. Для каждого образца число импульсов в минуту выражают в виде процента от среднего числа импульсов в минуту для контролей. В тех случаях, когда это возможно, рассчитывают значения IC50 (концентрация лекарства, необходимая для снижения на 50% ферментативной активности TANK-2, по сравнению с контролем), используя линейную интерполяцию между экспериментальными точками, расположенными чуть выше и чуть ниже 50%-ного уровня. В настоящем описании эффект тестируемых соединений выражают в виде pIC50 (отрицательный логарифм значения IC50). В качестве соединений сравнения для оценки результатов SPA анализа используют 3-аминобензамид и 4-амино-1,8-нафталимид. В настоящем описании приведены данные анализа для 96-луночных планшетов. В анализах с применением 384-луночных планшетов используют те же самые конечные концентрации, а объемы соответствующим образом корректируют. Если имеются результаты для 96-луночных планшетов, то эти результаты представлены в таблице 2, в противном случае приведены результаты для 384-луночных планшетов.
Пример С.4. Определение антипролиферативной активности.
Клетки HCT116 рака толстого кишечника человека, полученные из ATCC, выращивают в среде Маккоя 5A, дополненной с помощью 2 мМ L-глутамина, 50 мкг/мл гентамицина и 10%-ной сывороткой плода коровы, инактивированной тепловой обработкой.
Клетки PC-3 рака предстательной железы человека, полученные из ATCC, выращивают в среде Хэма F12, дополненной с помощью 1 мМ натриевой соли пировиноградной кислоты, 1,5 г/л бикарбоната натрия, 50 мкг/мл гентамицина, не являющимися незаменимыми аминокислотами и 10%-ной сывороткой плода коровы.
Реагенты, используемые при проведении анализа Alamar Blue
Резазурин приобретают у компании Aldrich (продукт № 199303). Ферроцианид калия, феррицианид калия, KH2PO4 и K2HPO4 покупают у компании Sigma (продукты №№ P9387, P8131, P5655 и P8281, соответственно).
Калиево-фосфатный буфер 0,1 M (PPB) готовят следующим образом: 2,72 г KH2PO4 и 13,86 г K2HPO4 растворяют в 500 мл воды, прошедшей очистку в элементе milli-Q, pH доводят до значения 7,4 и доводят объем до 1 л с помощью воды, прошедшей очистку в элементе milli-Q; буферный раствор стерилизуют фильтрованием и хранят при комнатной температуре. Готовят свежий маточный раствор резазурина (PPB-A), растворяя 45 мг резазурина в 15 мл забуференного фосфатом физиологического раствора. 30 мМ раствор феррицианида калия (PPB-B) готовят, растворяя 0,987 г феррицианида калия в 100 мл PPB. 30 мМ раствор ферроцианида калия (PPB-C) готовят, растворяя 1,266 г ферроцианида калия в 100 мл PPB.
Смесь PPB-A, PPB-B и PPB-C получают путем смешивания равных объемов соответствующих растворов. Рабочий раствор резазурина (далее в описании обозначают как раствор "Alamar Blue") получают путем 20-кратного (об./об.) разбавления указанной смеси в PPB и стерилизации посредством фильтрования; раствор Alamar Blue можно хранить при температуре 4°C не более 2 недель.
Методика проведения анализа Alamar Blue
Для проведения экспериментов на 384-луночных планшетах клетки высевают с плотностью 4,5×103 клеток/мл на 384-луночные планшеты Falcon (Life Technologies, Merelbeke, Belgium) черного цвета с прозрачным дном, используя 45 мкл культуральной среды. Дают клеткам в течение 24 час прикрепиться к пластику. Тестируемые соединения предварительно разбавляют (1/50 в культуральной среде) и в лунки добавляют по 5 мкл указанных предварительно разбавленных растворов. После 4 дней инкубирования в каждую лунку добавляют 10 мкл раствора Alamar Blue и клетки инкубируют еще в течение 4 час (HCT116) или 24 час (PC-3) при температуре 37°C. Интенсивность флуоресценции измеряют для каждой лунки с помощью флуоресцентного планшет-ридера (Fluorskan, Labsystems, длина волны возбуждения 540 нм, длина волны испускания 590 нм).
Антипролиферативную активность рассчитывают в виде процента оставшихся жизнеспособных клеток в подвергнутых обработке образцах, по сравнению с контролями (не подвергавшиеся обработке клетки). Полученный результат для каждого из условий проведения эксперимента является средним из трех последовательно проведенных исследований. Если необходимо, эксперименты повторяют, чтобы получить полные кривые зависимости ответной реакции от концентрации. Там, где это возможно, значения IC50 (концентрация лекарства, необходимая для снижения роста клеток на 50%, по сравнению с контролем) рассчитывают, используя пробит-анализ ранжированных данных (Finney, D. J., Probit Analyses, 2nd Ed. Chapter 10, Graded Responses, Cambridge University Press, Cambridge 1962). В настоящем описании эффект тестируемых соединений выражают в виде значений pIC50 (отрицательный логарифм величины IC50) (см. таблицу 3).
Пример С.5. Анализ полимеризации
Анализ полимеризации тубулина представляет собой адаптацию анализа, первоначально описанного Bonne, D. et al. (J. Biol. Chem., 1985, 260:2819-25). Набор реагентов для проведения анализа приобретают у компании Cytoskeleton, Inc. (номер по каталогу BK011), а анализ проводят, как описано у производителя, со следующими модификациями. Анализ проводят на 384-луночных черных планшетах Proxiplate (Perkin Elmer), а объемы соответствующим образом корректируют. Реакции проводят до конечного объема 10 мкл. Соединения добавляют к 25 мкл реакционной смеси на 96-луночных планшетах PP (Corning) на льду, а затем 10 мкл полученной смеси диспергируют в виде двух одинаковых серий на 384-луночные планшеты Proxiplates, предварительно подогретые до температуры 37°С, которые помещают в планшет-ридер Fluoroskan Ascent (Thermo Scientific). Измерения флуоресценции проводят через каждую минуту в течение одного часа. Определяют максимальный наклон для каждой лунки (линейная регрессия через 4 последовательные точки) и полимеризацию рассчитывают как процент полимеризации, которая наблюдается в отсутствие соединения. Проводят измерения для соединений вначале при концентрации 20 мкМ, а затем при 5 мкМ для тех соединений, которые показывают более чем 50%-ное ингибирование при 20 мкМ, по сравнению с полимеризацией, которая наблюдается в отсутствие соединения по изобретению. Результаты представлены в таблице F-2 в виде оценки в баллах, которую устанавливают следующим образом: соединениям, которые показывают ингибирование от 0 до 50% при 20 мкМ, присваивают 1 балл; соединениям, которые показывают ингибирование больше чем 50% при 5 мкМ, присваивают 3 балла. Соединения, которым присваивают 2 балла, определяют как соединения, показывающие более чем 50%-ное ингибирование при 20 мкМ и менее чем 50%-ное ингибирование при 5 мкМ.
Пример С.6 Анализ Eb1 Comet (разрушение микротрубочек)
Анализ Eb1 Comet основывается на детектировании белка Eb1 на положительном конце полимеризующихся микротрубочек (Mimori-Kiyosue, 2000) с использованием метода непрямой флуоресценции. Нарушение динамики микротрубочек за счет деполимеризации или стабилизации приводит к ограничению распространения Eb1 на растущих концах микротрубочек, и этот процесс можно визуально установить по исчезновению цитоплазматических очагов, содержащих Eb1.
Если коротко, то клетки PC3 рака предстательной железы человека, полученные из Американской коллекции тканевых культур, выращивают на 96-луночных планшетах (Greiner, номер по каталогу 655090) в среде Хэма F12 в соответствии с рекомендациями поставщика (ATCC). Клетки в течение 1 час при температуре 37°С подвергают обработке соединениями по изобретению, растворенными в ДМСО (конечная концентрация в ДМСО составляет 0,6%). Затем культуральную среду удаляют отсасыванием и клетки фиксируют, добавив холодный метанол (-20°C). После 15 мин инкубирования при температуре минус 20°C клетки дважды промывают средой DPBS (Gibco), содержащей 0,5% Triton X-100. К клеткам добавляют антитело Eb1 мыши (BD Transduction Laboratories, номер по каталогу 610534) (в виде 1/250 разбавления в среде DPBS, содержащей 1% бычьего сывороточного альбумина) и оставляют на ночь инкубироваться при комнатной температуре. Антитело затем удаляют и клетки дважды промывают средой DPBS, 0,5% Triton X-100. Добавляют вторичное антитело козла против мыши, конъюгированное с флуоресцентным красителем Alexa 488 (Molecular Probes) в виде 1/500 разбавления в среде DPBS, содержащей 1% бычьего сывороточного альбумина, и инкубируют при 37°C в течение 1 час. Клетки дважды промывают с помощью DPBS, 0,5% Triton X-100, а затем добавляют среду DPBS, содержащую Triton X-100, и добавляют 1/5000 Hoechst 33342 (Molecular Probes). Микроскопию, основанную на визуализации очагов Eb1, проводят с помощью клеточного анализатора IN Cell Analyser 1000 (Amersham Biosciences), используя объектив с увеличением 20×. Зависимое от соединений разрушение микротрубочек визуально наблюдают по исчезновению очагов Eb1. Наименьшую активную концентрацию (LAC) определяют как концентрацию, при которой очаги Eb1 отсутствуют, по крайней мере, у 50% подвергшихся обработке клеток. В настоящем описании эффекты тестируемых соединений выражают как pLAC (отрицательный логарифм значения LAC) (см. таблицу 3).
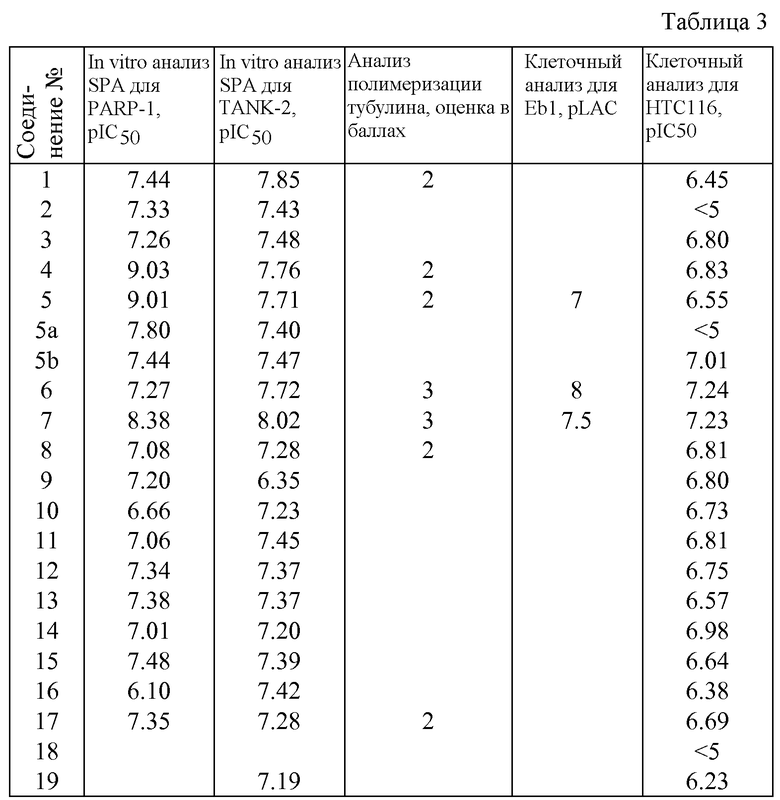
D. Пример композиции: Таблетки, покрытые оболочкой
Приготовление сердцевины таблетки
Смесь 100 г соединения формулы (I), 570 г лактозы и 200 г крахмала тщательно смешивают, а затем увлажняют раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона приблизительно в 200 мл воды. Влажную смесь порошков просеивают, сушат и вновь просеивают. Затем добавляют 100 г микрокристаллической целлюлозы и 15 г гидрированного растительного масла. Полученную смесь хорошо перемешивают и спрессовывают в таблетки, получая 10000 таблеток, каждая из которых содержит 10 мг соединения формулы (I).
Оболочка
К раствору 10 г метилцеллюлозы в 75 мл денатурированного этанола добавляют раствор 5 г этилцеллюлозы в 150 мл дихлорметана. Затем добавляют 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола, расплавляют 10 г полиэтиленгликоля и растворяют в 75 мл дихлорметана. Последний раствор добавляют к ранее полученному раствору и к смеси добавляют 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной суспензии красителя, а затем всю смесь гомогенизуют. Сердцевины таблеток покрывают полученной указанным образом смесью в устройстве для нанесения покрытий.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕЖДУ MDM2 И Р53 | 2007 |
|
RU2436784C2 |
| ПИРИДАЗИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2008 |
|
RU2490265C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОПИРАЗИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ FGFR-КИНАЗ ДЛЯ ЛЕЧЕНИЯ РАКОВЫХ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2602233C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ, АКТИВИРУЮЩЕЙ КАНАЛЫ | 2007 |
|
RU2419625C2 |
| ПРОТИВОРАКОВЫЕ БЕНЗОПИРАЗИНЫ, ДЕЙСТВУЮЩИЕ ПОСРЕДСТВОМ ИНГИБИРОВАНИЯ FGFR-КИНАЗ | 2012 |
|
RU2639863C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕЖДУ MDM2 И P53 | 2008 |
|
RU2477724C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ПОЛИ(АДФ-РИБОЗА)ПОЛИМЕРАЗ | 2005 |
|
RU2344138C2 |
| ЗАМЕЩЕННЫЕ АМИНОМАСЛЯНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ НЕПРИЛИЗИНА | 2012 |
|
RU2604522C2 |
| ПТЕРИДИНЫ В КАЧЕСТВЕ FGFR ИНГИБИТОРОВ | 2013 |
|
RU2702906C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНЫХ ТИРОЗИНКИНАЗ | 2009 |
|
RU2518089C2 |
Настоящее изобретение относится к соединениям формулы (I), их применению в качестве ингибиторов полимеризации тубулина и в качестве ингибиторов PARP, а также в составе фармацевтических композиций, содержащих указанные соединения формулы (I), где R1, R2, R3, R4, R5, R6, R7 и Y имеют значения, приведенные в формуле изобретения. Изобретение также относится к промежуточным соединениям для получения соединений формулы (I) и к способу получения промежуточных соединений. 9 н. и 7 з.п. ф-лы, 3 табл., 16 пр. получения промежуточных соединений, 17 пр. получения целевых соединений, 6 фармакологических пр., 1 пр. композиции.
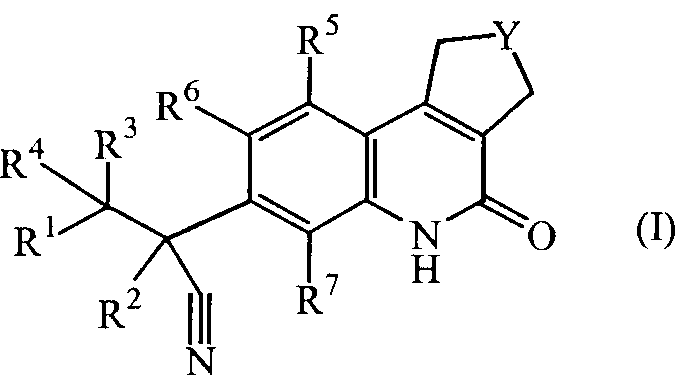
1. Соединение формулы (I),
включая его стереохимически изомерную форму;
где Y обозначает СН2-СН2;
R1 обозначает арил или Het;
где арил обозначает фенил;
где Het обозначает пиридинил, пиримидинил, индазолил, или бензимидазолил;
каждый арил или Het может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, циано, C1-6алкила, C1-6алкилоксикарбонила, гидроксиС1-6алкила, C1-6алкилNR8R9 или -OR8; или
R1 обозначает радикал формулы
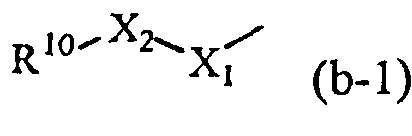 ;
;
где X1 обозначает СН2, NH или N-СН3;
где Х3 обозначает СН2, С=O или О;
где R10 обозначает фенил, необязательно замещенный одним циано; или
R2 обозначает метил, этил или пропил;
R3 и R4 представляют собой водород;
R5 и R6 представляют собой водород;
R7 обозначает атом водорода;
каждый R8 и R9 независимо выбран из атома водорода, C1-6алкила или тригалогеноС1-6алкила;
или его фармацевтически приемлемая аддитивная соль.
2. Соединение по п.1, где
каждый арил или Het может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, циано, C1-6алкила, C1-6алкилоксикарбонила, -С1-6алкилNR8R9 или -OR8; X1 обозначает СН2 или N-СН3; и R2 обозначает метил.
3. Соединение по п.1, где
R1 обозначает фенил, пиридинил или пиримидинил; каждый фенил, пиридинил или пиримидинил может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, циано или С1-6алкилокси; X1 обозначает СН2; Х2 обозначает О; R10 обозначает фенил, замещенный циано-группой; и R2 обозначает метил.
4. Соединение по п.1, где соединение выбрано из следующих соединений:
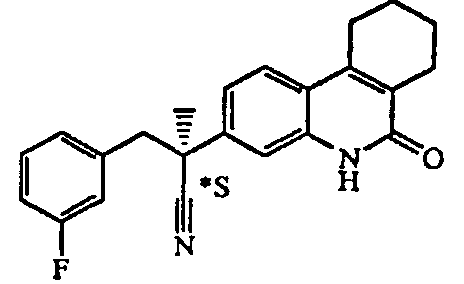
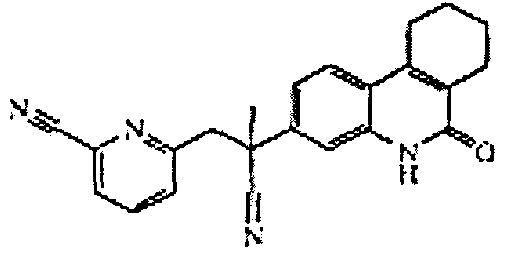
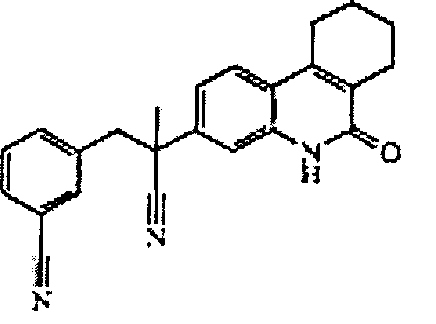
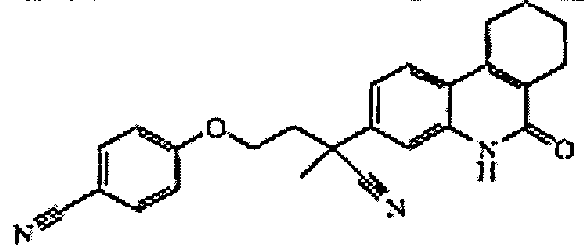
или их фармацевтически приемлемых аддитивных солей.
5. Соединение по любому из пп.1-4 для использования в качестве лекарственного средства, обладающего свойствами ингибитора PARP и ингибитора полимеризации тубулина.
6. Фармацевтическая композиция, обладающая свойствами ингибитора PARP и ингибитора полимеризации тубулина, содержащая фармацевтически приемлемый носитель, и, в качестве активного ингредиента, терапевтически эффективное количество соединения по любому из пп.1-4.
7. Способ получения фармацевтической композиции по п.6, где тщательно смешивают фармацевтически приемлемый носитель и соединение по любому из пп.1-4.
8. Применение соединений по любому из пп.1-4 для получения лекарственного средства, предназначенного для лечения расстройства, которое опосредуют PARP или полимеризация тубулина.
9. Соединение формулы (II)
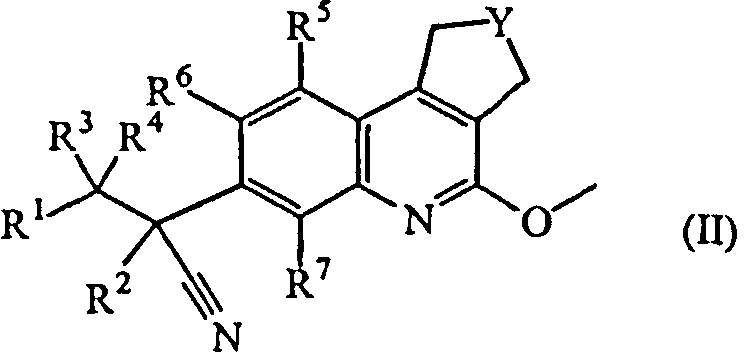
включая их стереохимически изомерные формы;
где Y обозначает СН2-СН2;
R1 обозначает арил или Het;
где арил обозначает фенил;
где Het обозначает пиридинил, пиримидинил, индазолил, или бензимидазолил;
каждый арил или Het может быть замещен одним или двумя заместителями, каждый из которых независимо выбран из атома галогена, циано, C1-6алкила, C1-6алкилоксикарбонила, гидроксиС1-6алкила, C1-6алкилNR8R9, или -OR8; или
R1 обозначает радикал формулы
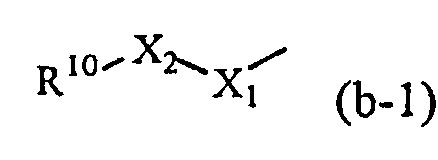 ;
;
где X1 обозначает СН2, NH или N-СН3;
где Х2 обозначает СН2, С=O или О;
где R10 обозначает фенил, необязательно замещенный одним циано; или
R2 обозначает метил, этил или пропил;
R3 и R4 представляют собой водород;
R5 и R6 представляют собой водород;
R7 обозначает атом водорода;
каждый R8 и R9 независимо выбран из атома водорода, С1-6алкила или тригалогеноС1-6алкила;
его фармацевтически приемлемая аддитивная соль.
10. Способ получения соединение по п.9, который включает добавление калиевой соли 2-метил-2-пропанола к промежуточному соединению формулы (V) в присутствии промежуточного соединения формулы (VI), где W обозначает уходящую группу, в подходящем растворителе,
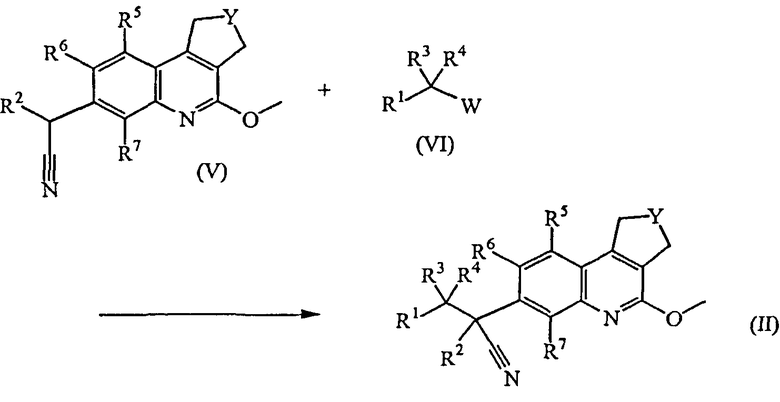
где переменные определены по п.1.
11. Соединение по любому из пп.1-4 для использования в качестве лекарственного средства для лечения рака.
12. Соединение по п.11 для использования в качестве лекарственного средства для лечения рака легких.
13. Соединение по п.11 для использования в качестве лекарственного средства для лечения рака груди.
14. Применение соединений по любому из пп.1-4 для получения лекарственного средства для лечения рака.
15. Применение соединений по любому из пп.1-4 для получения лекарственного средства для лечения рака легких.
16. Применение соединений по любому из пп.1-4 для получения лекарственного средства для лечения рака груди.
| RU 2001108579 А, 10.03.2002 | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| ПРОИЗВОДНЫЕ ФЕНАНТРИДИНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ, СПОСОБ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК | 1997 |
|
RU2183626C2 |

