Область техники
Настоящее изобретение относится к новому производному фенантридиния (phenanthridinium), обладающему противоопухолевой активностью и, как ожидается, являющемуся эффективным лекарственным средством, а также к его фармацевтическому применению.
Предпосылки изобретения
В настоящее время в химиотерапии для лечения пациентов от рака применяют алкилирующие агенты, метаболические антагонисты, антибиотики, растительные алкалоиды и т.д.
Известно, что хлорид или йодид 2,3-(метилендиокси)-5-метил-7-гидрокси-8-метокси-бензо[с] фенантридиния, описанные в Chem. Pharm. Bull., 33, 1963, обладает противоопухолевой активностью (Японские патенты KOKAI 2-243628 и 3-184916). Производное бензо[с]фенантридиния и его противоопухолевая активность также описаны в Японском патенте KOKAI 5-208959.
Злокачественная опухоль имеет множество характеристик. Более того, применение этих противоопухолевых агентов вызывает резистентность к ним. Таким образом, существует потребность в разработке нового противоопухолевого агента.
Описание изобретения
Авторы данного изобретения обнаружили новое производное фенантридиния, имеющее структуру, образованную путем связывания атома азота в позиции 5 с атомом углерода в позиции 6 через алифатические углеводородные цепи, прилежащие к ним, а также обнаружили, что новое производное фенантридиния проявляет противоопухолевую активность и резистентно к химическому восстановлению и биологическим метаболическим реакциям. Эти свойства производного фенантридиния, как найдено, являются исключительно благоприятными для применения в медицине, поэтому они являются основой для настоящего изобретения.
Таким образом, настоящее изобретение относится к новому производному фенантридиния, представленному общей формулой (А):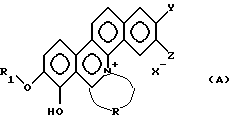
где R1 - замещенная или незамещенная низшая алифатическая углеводородная группа;
R - алифатическая углеводородная цепь, имеющая 2-6 атомов углерода, которая может быть необязательно замещена заместителем, выбранным из группы, включающей низшую алкильную группу, галоген и гидроксигруппу;
каждый из Y и Z независимо представляет собой водород, гидрокси- или низшую алкоксигруппу; или Y и Z объединяются вместе с образованием метилендиокси или фенильного кольца;
Х- - остаток кислоты или остаток водородной кислоты.
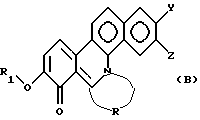
Настоящее изобретение далее относится к новому производному фенантридиния, представленному общей формулой (В):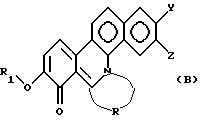
где R1 - замещенная или незамещенная низшая алифатическая углеводородная группа;
R - низшая алифатическая углеводородная цепь, имеющая 2-6 атомов углерода, которая может быть необязательно замещена заместителем, выбранным из группы, включающей низшую алкильную группу, галоген и гидроксигруппу;
каждый из Y и Z независимо представляет собой водород, гидрокси- или низшую алкоксигруппу; или Y и Z объединяются вместе с образованием метилендиокси или фенильного кольца.
Настоящее изобретение далее относится к фармацевтической композиции, включающей, в виде активного ингредиента, соединение, представленное общей формулой (А) или (В), вместе с фармакологически приемлемым носителем.
Настоящее изобретение далее относится к противоопухолевому агенту, включающему, в виде активного ингредиента, соединение, представленное общей формулой (А) или (В), вместе с фармакологически приемлемым носителем.
Настоящее изобретение далее относится к соединению, представленному общей формулой (А) или (В) для применения в фармацевтической композиции в виде активного ингредиента.
Настоящее изобретение далее относится к применению соединения, представленного общей формулой (А) или (В) при получении фармацевтической композиции для лечения или профилактики опухолей.
Настоящее изобретение далее относится к способу лечения или профилактики опухолей, включающему введение человеку соединения, представленного общей формулой (А) или (В) в эффективной дозе.
Лучший вариант осуществления изобретения
В общих формулах (А) и (В) в соответствии с настоящим изобретением, низшая алифатическая углеводородная группа включает, например, алкильную группу, имеющую 1-5 атомов углерода, и алкенилметильную группу, имеющую 3-5 атомов углерода. Примерами такой алкильной группы, имеющей 1-5 атомов углерода, являются метил, этил, пропил, изопропил, н-бутил, трет-бутил и т.д. Примерами алкенилметильной группы, имеющей 3-5 атомов углерода, являются аллил, 2-бутенил, 3-метил-2-бутенил и т.д. Эти низшие алифатические углеводородные группы могут быть необязательно замещены, и примерами таких заместителей являются гидроксигруппа, C1-C5 алкоксигруппа, C1-C5 алкоксикарбонильная группа, ацетильная группа, галоген, карбамоильная группа или фенильная группа, необязательно замещенная метоксигруппой.
Конкретными примерами алифатической углеводородной группы, которая может быть замещена или не замещена, являются метил, этил, пропил, изопропил, 2-гидроксиэтил, 2-метоксиэтил, 2-ацетоксиэтил, 2-гидроксипропил, аллил, 2-бутенил, 3-метил-2-бутенил, метоксикарбонилметил, изопропоксикарбонилметил, карбамоилметил, бензил, 4-метоксифенилметил, фторметил, трифторметил и т.д. Особенно предпочтительными являются метил, этил, аллил, 2-гидроксиэтил, 2-метоксиэтил, 2-ацетоксиэтил, карбамоилметил и трифторметил.
Алифатическая углеводородная цепь, имеющая 2-6 атомов углерода в общих формулах (А) и (В) в соответствии с настоящим изобретением, которая может быть замещена заместителем, выбранным из группы, включающей низшую алкильную группу, галоген и гидроксигруппу, относится, например, к замещенной или незамещенной полиметиленовой группе, имеющей 2-6 атомов углерода. Низшая алкильная группа, как заместитель алифатической углеводородной цепи, представляет собой алкильную группу, имеющую 1-5 атомов углерода, например, метил, этил, пропил, 1-пропил, бутил, трет-бутил, пентил и т.д. Предпочтительными примерами заместителя являются метил и этил. Примерами атома галогена являются фтор, хлор, бром и йод.
Конкретно алифатическая углеводородная цепь, имеющая 2-6 атомов углерода, которая может быть замещена или не замещена, относится к -СН2СН2-, -CH2CH2CH2-, -СН2СН(СН3)СН2-, -СН2С(СН3)2СН2-, -СН2СН(ОН)СН2-,
-CH2CHFCH2-, -CH2CF2CH2-, -СН2СНС1СН2-, -СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2-,
-СН2СН2СН2СН2СН2СН2- и т.д. Особенно предпочтительной является незамещенная полиметиленовая цепь, имеющая 3-4 атомов углерода, такая как -СН2СH2СН2- или -СН2СН2СН2СН2-.
В общих формулах (А) и (В) в соответствии с настоящим изобретением примером низшей алкоксигруппы является алкоксигруппа, имеющая 1-5 атомов углерода, такая как метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, трет-бутокси, пентокси и т.д., предпочтительно, алкоксигруппа, имеющая 1-3 атомов углерода, такая как метокси, этокси, н-пропокси или изо-пропокси.
Остаток кислоты X- в общей формуле (А) в соответствии с настоящим изобретением означает остаток кислоты, образующей нормальную соль, например, X- представляет собой ион галогена, такой как ион хлорида, бромида, иодида или фторида, либо ион сульфата, нитрата или п-толуолсульфоната. Остаток водородной кислоты означает остаток кислоты, образующей соль водородной кислоты. Остаток водородной кислоты содержит 1 или 2 атома водорода, например, ион гидросульфата, ион ангидрофосфата и т.д. Среди них ион хлорида и ион гидросульфата являются особенно предпочтительными.
В данном изобретении предпочтительными примерами соединений являются такие соединения общих формул (А) и (В), в которых R1 представляет собой метил, этил, аллил, 2-гидроксиэтил, 2-метоксиэтил, 2-ацетоксиэтил, карбамоилметил или трифторметил, R представляет собой незамещенную полиметиленовую цепь, имеющую 3-4 атома углерода, a Y и Z объединяются вместе с образованием метилендиокси или фенильного кольца.
Соединения, представленные общей формулой (А), могут быть получены в результате следующих процессов, которые подразделяются на два вида. Каждый из процессов поясняется ниже.
1. Синтез соединения, представленного общей формулой (А), где R1 - метил (в дальнейшем называемый как синтез типа 1 в общей формуле (А)):
(а) Соединение общей формулы (А), где R1 - метил, может быть синтезировано в соответствии со следующей реакционной схемой 1.
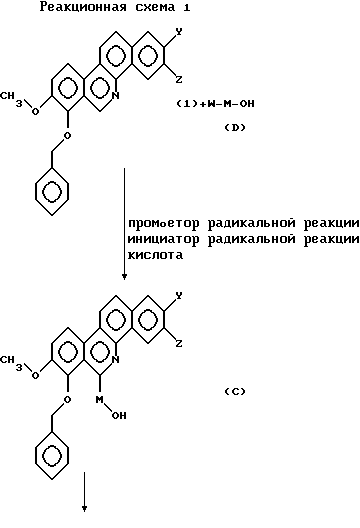
В исходных соединениях общей формулы (1), Y и Z имеют такое же значение, как и в общих формулах (А) и (В). Исходное соединение общей формулы (1), где каждый из Y и Z независимо представляет собой водород, гидрокси- или низшую алкоксигруппу, или Y и Z объединены вместе с образованием метилендиокси, т. е. производных 7-бензилокси-8-метокси-бензо[с] фенантридина, может быть синтезировано с применением способа, описанного в Японском патенте KOKAI 5-208959.
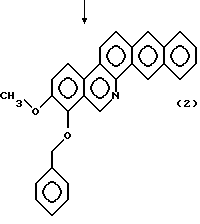
Исходное соединение общей формулы (1), где Y и Z объединены вместе с образованием фенильного кольца, т.е. производного 8-бензилокси-9-метокси-нафто[2,3-с]фенантридина, могут быть синтезированы в соответствии с реакционной схемой 3, описываемой ниже.
В общей формуле (D) М соответствует R в общих формулах (А) и (В) и представляет собой алифатическую углеводороную цепь, имеющую 2-6 атомов углерода, которая может быть необязательно замещена заместителем, выбранным из группы, включающей низшую алкильную группу, галоген и гидроксигруппу; a W представляет собой иод или бром.
Галоидалкильное соединение общей формулы (D) может быть синтезировано обычным способом.
Соединение, представленное формулой (С), может быть получено путем нагревания соединения общей формулы (1) и галоидалкильного соединения общей формулы (D) в растворителе, таком как ацетонитрил и т.д., в присутствии промотора радикальной реакции, такого как органотин гидрид, органосилан гидрид и т. д. ; инициатора радикальной реакции, такого как 2,2'-азобис(изобутиронитрил) и т.д.; и кислоты, такой как трифторуксусная кислота, при перемешивании.
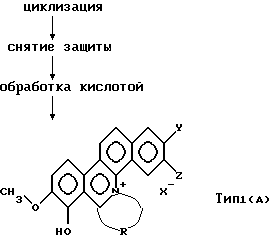
Соединение общей формулы (С) подвергают реакции с хлорангидридом кислоты, таким как метансульфонилхлорид, р-толуолсульфонилхлорид и т.д., или ангидридом кислоты, таким как трифторуксусный ангидрид и т.д., в органическом растворителе в присутствии основания, такого как триэтиламин и т.д., при охлаждении льдом до комнатной температуры с последующей обработкой реакционной смеси при температуре от комнатной до 110oС для циклизации.
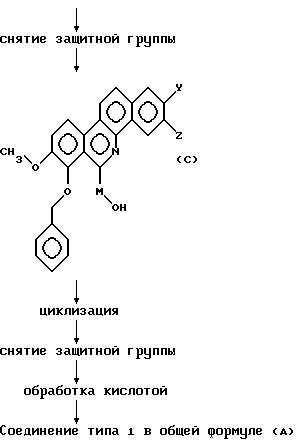
Затем осуществляют снятие защитной группы реакционного продукта, предпочтительно, без выделения и очищения продукта. Снятие защиты в данном изобретении означает удаление бензильной группы в позиции 7 в общей формуле (С).
Удаление 7-бензильной группы может быть осуществлено путем обработки при комнатной температуре до 100oС в кислых условиях, используя концентрированную соляную кислоту и т. д,
Кислотную обработку осуществляют растворением в растворителе соединения, полученного в результате снятия защиты, и добавления к раствору кислоты, например, соляной, серной, п-толуолсульфокислоты и т.д. В целом, количество кислоты составляет приблизительно 1-3 молей на моль соединения.
В соответствии с вышеописанными процедурами может быть получено соединение типа 1, представленное общей формулой (А).
(b) Соединение типа 1 в общей формуле (А) также может быть синтезировано в соответствии со следующей реакционной схемой 2.
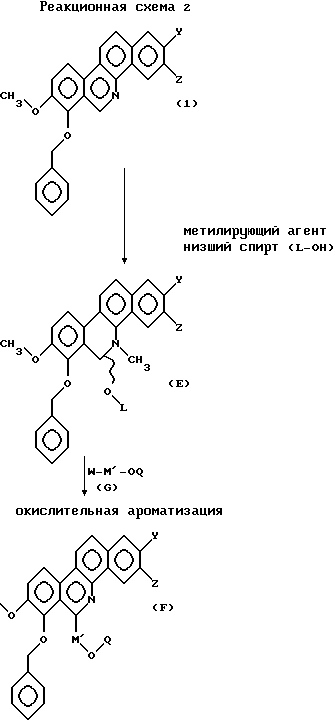
В вышеприведенной реакционной схеме W - органическая или неорганическая соль металла; L - низшая алкильная группа; М' - алифатическая углеводородная цепь, имеющая 2-6 атомов углерода, которая может быть необязательно замещена заместителем, выбранным из группы, включающей низшую алкильную группу, галоген и гидроксигруппу; a Q - защитная группа.
Реакцию соединения общей формулы (1) с метилирующим агентом проводят при нагревании и отсутствии растворителя, или путем их растворения в C6-C10 углеводородном растворителе, таком как толуол, ксилол и т.д. Реакцию обычно проводят при температуре, 50-180oС, предпочтительно, 100-150oС, обычно в течение 1-24 часов, предпочтительно, 2-10 часов.
Может быть использован любой метилирующий агент, обычно используемый для N-метилирования пиридинового кольца. Предпочтительным является метилсульфонат, используемый для метилирования, например, метилзамещенного бензолсульфоната или метилтригалогенометансульфоната. Конкретные примеры включают метил п-толуолсульфонат, метил 2-нитробензолсульфонат и метилтрифторметансульфонат.
N-метилированное таким образом соединение смешивают с низшим спиртом (L-OH), таким как этанол и т.д., предпочтительно, метанол, этанол или н-пропанол, обычно при температуре от 0oС до комнатной температуры, в присутствии основания для получения соединения общей формулы (Е).
Соединение, представленное общей формулой (Е), растворяют в апротонном растворителе, например, углеводородном растворителе, таком как бензол, толуол, ксилол и т.д.; эфирном растворителе, таком как диэтиловый эфир, диизопропиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран и т.д.; растворителе галоидного типа, таком как метиленхлорид, 1,2-дихлорэтан и т.д. Затем к раствору соединения (Е) добавляют металлоорганическое соединение общей формулы (G) в количестве 1-10 эквимолей, предпочтительно, 1-3 эквимолей, и, при необходимости, к смеси добавляют ускоритель реакции, такой как комплекс трехфтористый бор-эфир. Реакцию проводят, перемешивая смесь при температуре (-)78-(+)50oС, предпочтительно, при температуре от -20oС до комнатной температуры, в течение периода времени от 5 минут до 24 часов, предпочтительно, от 10 минут до 10 часов.
Металлоорганическое соединение общей формулы (G) может представлять собой любое металлоорганическое соединение, используемое в обычной реакции нуклеофильного замещения. В качестве металлоорганического соединения применяют, например, соединения органического лития, магния, цинка, алюминия или меди, при этом предпочтение отдается магниевоорганическому соединению. Конкретные примеры металлоорганического соединения включают 3-(трет-бутилдиметилсилокси)пропилмагний бромид, 2-метил-3-(трет-бутилдиметилсилокси)пропилмагний бромид и 4-(трет-бутилдиметилсилокси)бутилмагний бромид.
Реакционный продукт, получаемый в результате вышеописанной реакции нуклеофильного замещения, ароматизируют с окислением окисляющим агентом, получая соединение общей формулы (F). В реакции могут быть использованы различные окисляющие агенты, такие как диоксид марганца, тетраацетат свинца и дихлордицианобензохинон (DDQ), предпочтительно, диоксид марганца. Реакцию проводят при температуре 0-120oС, предпочтительно, при температуре от комнатной до 100oС, в течение 1-120 минут, предпочтительно, 5-60 минут.
Снятие защитной группы в соединении общей формулы (F) приводит к получению соединения общей формулы (С). В качестве защитной группы обычно применяют заместители, используемые для защиты гидроксигруппы, например, замещенной метильной группы, такой как метоксиметил, бензилоксиметил, тетрагидрофурил, трет -бутил, п-метоксибензил, трифенилметил и т.д.; триалкилсилильная группа, такая как трет-бутилдиметилсилил, триметилсилил и т.д.; ацильная группа, такая как ацетил, хлорацетил, бензоил, изобутирил и т.д. Для удаления защитной группы применяют подходящую процедуру для каждой защитной группы. Например, удаление защитной группы Q и защитной группы типа триалкилсилила, используемых для защиты гидроксигруппы в М', осуществляют добавлением к реакционной смеси фторида, такого как фторид тетрабутиламмония, фторид калия, фторид цезия и т.д., в растворителе, таком как тетрагидрофуран, ацетонитрил и т.д., и выдерживанием при температуре, составляющей 0-80oС, предпочтительно, 0oС до комнатной температуры. Затем соединение общей формулы (С) подвергают циклизации таким же образом, как и в реакционной схеме 1. Удалением защитной группы, а затем кислотной обработкой может быть синтезировано целевое соединение типа 1, представленное общей формулой (А).
(с) Из соединений общей формулы (1), используемых в качестве исходного соединения в реакционных схемах 1 и 2, в соответствии с реакционной схемой 3 может быть синтезировано производное 8-бензилокси-9-метокси-нафто[2,3-с] фенантридина, в котором Y и Z объединены вместе, образуя фенильное кольцо.
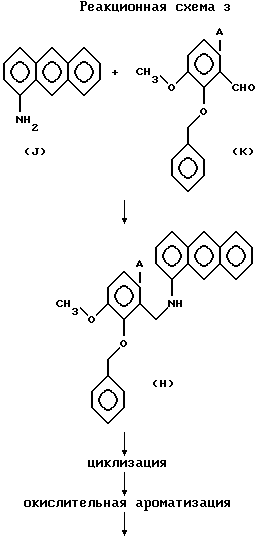
1-Аминоантрацен формулы (J) и 2-бензилокси-3-метокси-6-галогенобензальдегид формулы (К), который может быть получен способом, описанным, например, J. C. S. Perkin I., 1221 (1976) и J. Org. Chem., 53, 1708(1988), нагревают при температуре 80-110oС в толуоле или бензоле в течение 1-3 часов. Реакционную смесь концентрируют, а воду, побочный продукт, удаляют азеотропной перегонкой с толуолом или бензолом. Предпочтительно, свежий толуол или бензол добавляют в концентрат, а вышеуказанную процедуру нагревания с последующей концентрацией повторяют 2-4 раза для получения дегидратированного продукта конденсации (Шиффово основание) почти количественно. Конденсированную позицию дегиратированного продукта конденсации восстанавливают для получения соединения общей формулы (Н).
Может быть использован любой восстанавливающий агент, способный восстанавливать двойную связь углерод-азот. Восстановление предпочтительно проводят при комнатной температуре, составляющей -10-40oС, используя цианоборгидрид натрия или диметиламиноборон.
В формулах (К) и (Н) А представляет атом галогена.
Соединение общей формулы (Н) подвергают циклизации (реакция конденсации через удаление галоидводорода) в органическом растворителе, используя оловоорганическое гидридное соединение, предпочтительно, триуглеводородное (1-8 атомов углерода) соединение гидрида олова, такое как гидрид трибутилолова или гидрид триоктилолова, или диуглеводородное (1-8 атомов углерода) соединение гидрида олова, например, гидрид дифенилолова. В качестве оловоорганического гидридного соединения в этой реакции обычно предпочтительно используют гидрид трибутилолова. Реакция может быть осуществлена путем растворения соединения общей формулы (Н) и 1-6 эквимолярного количества, предпочтительно, 2-3 эквимолярного количества, оловоорганического гидридного соединения в органическом растворителе, предпочтительно, углеводородном растворителе с 6-10 атомами углерода, например, толуоле, ксилоле, бензоле и т.д., и нагревания раствора при 5-150oС, предпочтительно, 80-130oС в течение периода времени от 2 минут до 4 часов, предпочтительно, 5-60 минут, предпочтительно, в присутствии инициатора радикалов, такого как 2,2'-азобис(изобутиронитрил), 2,2'-азобис(2-метилбутиронитрил) или перекись бензоила и т.д., тем самым завершая циклизацию.
Затем реакционную смесь, предпочтительно, без выделения из нее конденсированного продукта, подвергают окислительной ароматизации по положению (месту) циклизации с окисляющим агентом. Реакцию проводят при температуре 0-120oС, предпочтительно, при температуре от комнатной до 100oС в течение 1-120 минут, предпочтительно, 5-60 минут, получая производное 8-бензилокси-4-метокси-нафто[2,3-с]фенантридина, представленного общей формулой (2). В этой реакции могут быть использованы различные окисляющие агенты. Примерами окисляющих агентов являются диоксид марганца, тетраацетат свинца и дихлордициано-бензохинон (DDQ), при этом особенно предпочтительным является диоксид марганца.
2. Синтез соединения, представленного общей формулой (А), где R1 - замещенная или незамещенная низшая алифатическая углеводородная группа, отличная от метила (в дальнейшем называемый как синтез типа 2 в общей формуле (А)):
Соединение типа 2 в общей формуле (А), где R1 - замещенная или незамещенная алифатическая углеводородная группа, отличная от метила, представлено следующей формулой: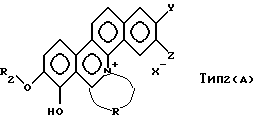
где R2 - замещенная или незамещенная алифатическая углеводородная группа, отличная от метила; a R, Y, Z и Х- имеют значения, указанные в общей формуле (А). Соединение типа 2 может быть синтезировано в соответствии со следующими процедурами. То есть, соединение общей формулы (3):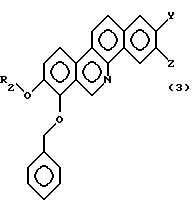
где R2 - замещенная или незамещенная алифатическая углеводородная группа, отличная от метила; a Y и Z имеют значения, указанные в общей формуле (А), используют как исходное соединение. Реакцию проводят способом, подобным реакционной схеме 1 или 2, т.е. соединение типа 2 может быть синтезировано в соответствии с реакционной схемой 1 или 2, за исключением того, что вместо соединения общей формулы (1) используют соединение общей формулы (3). Для получения соединения, содержащего, например, гидроксигруппу в качестве заместителя на алифатической углеводородной группе, представленной R2 в соединении типа 2, представленном общей формулой (А), используют соединение с гидроксигруппой, защищенной защитной группой, как R2 в соединении общей формулы (3). Защитная группа может быть легко снята, как в реакционной схеме 1 или 2.
При получении соединения общей формулы (3) соединение, в котором Y и Z каждый независимо представляет собой водород, гидрокси- или низшую алкоксигруппу, или Y и Z объединены вместе, образуя метилендиокси, может быть получено способом, описанным в Японском патенте KOKAI 5-208959, с использованием в качестве исходного материала соединения общей формулы (4)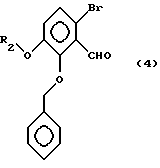
где R2 - замещенная или незамещенная алифатическая углеводородная группа, отличная от метила, способом, подобным способу получения производного 7-бензилокси-8-метокси-бензо[с] фенантридина общей формулы (1), представленной на реакционных схемах 1 и 2.
Исходное соединение общей формулы (4) может быть получено известным способом путем бромирования 2-бензилокси-3-гидроксибензальдегида, получаемым способом, описанным, например, в J. C.S. Perkin I, 1221 (1976) и J. Org. Chem. , 53, 1708 (1988), а затем взаимодействия реакционной смеси с соединением общей формулы (5):
R2-A1, (5)
где R2 имеет такое же значение, как и в формуле (4), a A1 - отщепляемая группа, такая как атом галогена, алкилсульфонильная группа и т.д., в органическом растворителе в присутствии или отсутствие основания.
В общей формуле (3) соединение, в котором Y и Z соединяются с образованием фенильного кольца, а именно, соединения общей формулы (6):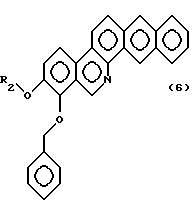
где R2 - замещенная или незамещенная алифатическая углеводородная группа, отличная от метила, может быть получено способом, подобным способу получения производного 8-бензилокси-9-метокси-нафто[2,3-с]фенантридина в вышеописанной реакционной схеме 3, за исключением того, что в качестве исходного соединения используют соединение общей формулы (4).
Полученное таким образом производное фенантридиния настоящего изобретения, представленное общей формулой (А), при реакции с основанием легко высвобождает из своей молекулы один эквивалент кислоты. Таким образом, может быть получена следующая новая структура производного фенантридиния, представленная общей формулой (В):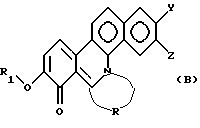
где R1, R, Y и Z имеют значения, указанные в общей формуле (А). Альтернативно, соединение общей формулы (В) легко превращают в соединение общей формулы (А) в результате реакции с кислотой.
Соединение общей формулы (В) лучше растворимо в липидах, чем соединение общей формулы (А). Однако считается, что соединение общей формулы (А) оказывает свое фармакологическое действие в виде соединения общей формулы (В) in vivo для проявления противоопухолевой активности. Более того, соединение общей формулы (В) играет роль посредника в синтезе соединения общей формулы (А). Таким образом, в реакционных схемах 1 и 2 соединение общей формулы (В) получают после снятия защиты.
В результате кислотной обработки соединения общей формулы (В), получаемого после удаления защитной группы, получают соединение общей формулы (А) в реакционных схемах 1 и 2. Также при синтезе соединения типа 2, представленного общей формулой (А), соответствующее соединение общей формулы (В) получают подобным образом после удаления защитной группы. Таким же образом в результате кислотной обработки соединения общей формулы (В) получают соединение общей формулы (А).
Соединение в соответствии с настоящим изобретением характеризуется описываемыми ниже химическими и биологическими свойствами.
Производные фенантридиния, представленные общими формулами (А) и (В), содержат замещенную или незамещенную алифатическую углеводородную цепь, представленную R в виде частичной структуры. Было обнаружено, что циклическая структура служит для пространственной защиты сайта, который, как считается, обладает многими химическими и биологическими реакционными способностями в известных производных бензо[с]фенантридиния, например, гидридросульфат 2,3-(метилендиокси)-5-метил-7-гидроксиокси-8-метокси-бензо[с]фенантридиния (Японский патент KOKAI 5-208959), и поэтому такая особенность способствует улучшенной устойчивости к химическому восстановлению и биологическим метаболическим реакциям.
Например, известный гидридросульфат 2,2-(метилендиокси) -5-метил-7-гидрокси-8-метокси-бензо[с] фенантридиния быстро восстанавливается в водном растворе в присутствии цианоборгидрида натрия как восстанавливающего агента, что приводит к исчезновению фенантридиния за несколько минут. Кроме того, при проведении in vitro метаболического исследования с использованием гомогената печени (S9), полученного из печени человека, с применением способа, описанного в Arch. Biochem. Biophys., 282, 183 (1990), наблюдается получение метаболита. Этот продукт совпадает с восстановленным продуктом, полученным в результате вышеописанного химического восстановления.
Хлорид 2,3-(метилендиокси)-7-гидрокси-8-метокси-5,6-пропано-бензо[с]фенатридиния, который является одним из соединений в соответствии с настоящим изобретением, обрабатывают цианоборгидридом натрия в водном растворе при комнатной температуре. При сравнении с известным вышеописанным гидридросульфатом 2,3-(метилендиокси)-5-метил-7-гидрокси-8-метокси-бензо[с] фенантридиния соединение в соответствии с настоящим изобретением исчезает явно постепенно. Более того, при метаболическом исследовании in vitro с использованием гомогената печени (S9), полученного из печени человека, метаболит не образуется, показывая тем самым, что соединение в соответствии с настоящим изобретением устойчиво к восстановительной метаболической реакции. Поэтому соединение в соответствии с настоящим изобретением обладает повышенной устойчивостью и, следовательно, чрезвычайно полезно как лекарственное средство.
Далее характерные примеры производного фенантридиния, представленного общей формулой (А), приведены в табл. 1. Однако подразумевается, что соединения в соответствии с настоящим изобретением не ограничиваются указанными примерами.
При использовании производного фенантридиния в соответствии с настоящим изобретением общей формулы (А) или (В) в качестве лекарственного средства, к фармацевтическим препаратам и способу их введения применимы различные известные способы. Это означает, что производное фенантридиния в соответствии с настоящим изобретением может быть введено парентерально, перорально, интраректально и т. д. Производное фенантридиния может иметь любую форму, подходящую для фармацевтических препаратов, включая инъекции, порошки, гранулы, таблетки, суппозитории и т.д. При получении фармацевтических композиций из производного фенантридиния, при необходимости, могут быть использованы различные вспомогательные вещества, применяемые при получении лекарственных средств, а именно, носители и другие добавки, например стабилизатор, консервант, успокаивающее средство, эмульгатор и т.д., если только они не оказывают вредного воздействия на активный ингредиент.
В фармацевтических препаратах содержание производного фенантридиния, представляемого общей формулой (А) или (В), может варьироваться в широком интервале в зависимости от препаративной формы, но обычно в интервале 0,01-100 мас. %, предпочтительно 0,1-50 мас.%. Остальную часть составляют носители и другие добавки, обычно используемые в лекарственных препаратах.
Доза производного фенантридиния, представленного общей формулой (А) или (В), может изменяться в зависимости от состояния пациента и т.д., однако она приблизительно составляет 50-500 мг в сутки для взрослого пациента.
Как указано выше, производные фенантридиния в соответствии с настоящим изобретением, представленные общей формулой (А) и (В), обладают противоопухолевой активностью как in vitro, так и in vivo, таким образом, ожидается, что они окажутся эффективными при лечении рака.
Далее способы получения производных фенантридиния в соответствии с настоящим изобретением и их фармакологическое действие, а также их фармацевтические композиции описаны более подробно при помощи примеров и примеров испытаний фармакологического действия. Однако предполагается, что настоящее изобретение не ограничивается только этими примерами.
Пример 1
Синтез 6-(3-гидроксипропил)-7-бензилокси-8-метокси-бензо [с] фенантридина
7-Бензилокси-8-метокси-бензо[с]фенантридин (полученный способом, описанным в Японском патенте KOKAI 5-208959, 284 мг, 0,78 ммол.) суспендируют в ацетонитриле (10 мл), и к полученной суспензии добавляют трифторуксусную кислоту (60 мкл, 0,78 ммол.), 3-бром-1-пропанол (71 мкл, 0,79 ммол.) и трис(триметилсилил)силан (481 мкл, 1,56 ммол.). Смесь перемешивают при 80oС на масляной бане. После растворения суспензии к раствору добавляют азобис(изобутиронитрил) (256 мг, 1,56 ммол.) с последующим нагреванием с дефлегмацией. Час спустя реакционной смеси дают возможность остыть до комнатной температуры, а затем к ней добавляют насыщенный водный раствор кислого углекислого натрия (60 мл) с последующей экстракцией метиленхлоридом. После промывания органического слоя насыщенным водным раствором хлорида натрия и высушивания над безводным сульфатом натрия, растворитель отгоняют в вакууме. Остаток пропускают через колонку с силикагелем (элюируя смесью 1% метанол-метиленхлорид). Основные фракции собирают и концентрируют в вакууме, получая 6-(3-гидроксипропил)-7-бензилокси-8-метокси-бензо[с] фенантридина в виде сырого продукта (чистота 50%, 142 мг, выход 21%) в желто-коричневой сухой массе.
FAB-MS (положительн.) m/z:
424 ([M+H]+), 366 ([М+Н-СН3СН2СН2ОН2]+),
333 ([М+Н-бензил]+);
1H-ЯMP (200MHz, СDСl3) δ:
9,31 (1Н, m), 8,51 (1H, d, J=9,3 Hz), 8,46 (1H, d, J=9,3 Hz), 7,95 (1Н, d, J= 9,5 Hz), 7,94 (1H, m), 7,77 (1H, m), 7,66 (1Н, m), 7,65 (1Н, d, J=9,3 Hz), 7,62-7,36 (5Н, m), 5,23 (2H, s),
4,07(3H, s), 3,82(2H, t, J=6,9Hz), 3,64(2H, t, J=6,lHz), 2,24(2H, tt, J= 6,9 и 6,lHz).
Пример 2
Синтез хлорида 7-гидрокси-8-метокси-5,6-пропано-бензо[с] фенантридиния (соединение А-1) (X-=Сl-)
Сырой продукт, 6-(3-гидроксипропил)-7-бензилокси-8-метокси-бензо[с]фенантридин (чистота 50%, 120 мг, 0,14 ммол.), синтезированный, как описано в примере 1, растворяют в метиленхлориде (4 мл). К раствору добавляют метансульфонилхлорид (22 мкл, 0,28 ммол.) и N,N-диизопропилэтиламин (50 мкл, 0,28 ммол.). Смесь перемешивают при комнатной температуре в течение 20 минут. После добавления к реакционной смеси метанола (1 мл), раствор концентрируют в вакууме. Остаток пропускают через колонку с силикагелем (элюируя смесью 8% метанол-метиленхлорид). Основные фракции собирают, а растворитель отгоняют в вакууме. Для растворения к остатку добавляют уксусную кислоту (1,6 мл) и концентрированную соляную кислоту (0,8 мл). Раствор перемешивают при 60oС на масляной бане в течение 20 минут. После охлаждения реакционной смеси до комнатной температуры, к ней добавляют насыщенный водный раствор кислого углекислого натрия (160 мл), который затем экстрагируют метиленхлоридом. Органический слой промывают водой и высушивают над безводным сульфатом натрия (содержащим соединение общей формулы (В)). Органический слой фильтруют для удаления сульфата натрия. 4М раствор хлористого водорода в диоксане добавляют к фильтрату до тех пор, пока красно-фиолетовый раствор полностью не превратится в золотисто-желтый. Раствор концентрируют в вакууме. Остаток очищают колоночной хроматографией на силикагеле (элюируя смесью 8-12% метанол-метиленхлорид), получая соединение А-1 (Х-=Сl-) (29 мг, выход 59%) в виде золотисто-желтого порошка.
FAB-MS (положительн.) m/z:
316 (М+),
1H-ЯMP (200 MHz, DMSO-d6) δ:
11,44 (1Н, brs), 8,93 (1Н, d, J=9,4 Hz), 8,93 (1Н, m), 8,61 (1Н, d, J= 9,2 Hz), 8,39 (1Н, d, J= 9,2 Hz), 8,29 (1H, m), 8,15 (1Н, d, J=9,1 Hz), 7,92-7,82 (2Н, m), 5,57 (2Н, brt, J=7,2 Hz), 4,23 (2H, brt, J=7,6 Hz), 4,10 (3H, s), 2,49 (2Н, m).
Пример 3
Синтез 2, 3-(метилендиокси) -6- (3-гидроксипропил) -7- бензилокси -8-метокси-бензо[с]фенантридина
2,3-(Метилендиокси)-7-бензилокси-8-метокси-бензо[с] фенантридина (полученного способом, описанным в Японском патенте KOKAI 5-208959) (1,228 г, 3,0 ммол.) суспендируют в ацетонитриле (48 мл), а затем к суспензии добавляют трифторуксусную кислоту (231 мкл, 3,00 ммол.), 3-бром-1-пропанол (271 мкл, 3,00 ммол.) и трис (триметилсилил) силан (1,85 мл, 6,00 ммол.). Смесь перемешивают при 80oС на масляной бане. После растворения суспензии к раствору добавляют азобис (изобутиронитрил) (0,985 г, 6,00 ммол.) с последующим нагреванием при дефлегмации. 90 минут спустя, реакционную смесь охлаждают до комнатной температуры. Отделяя осажденные кристаллы фильтрацией, исходный материал, 2,3-(метилендиокси)-7-бензилокси-8-метокси-бензо[с]фенантридин, получают обратно в виде трифторацетата (0,680 г). Фильтрат концентрируют в вакууме и к остатку добавляют насыщенный водный раствор кислого углекислого натрия (40 мл) с последующей экстракцией метиленхлоридом (50 мл). После промывания органического слоя водой (50 мл) и сушки над безводным сульфатом натрия, растворитель отгоняют в вакууме. Остаток очищают колоночной хроматографией на силикагеле (элюируя смесью 10% этилацетат-толуол), получая 2,3-(метилендиокси)-6-(3-гидроксипропил)-7-бензилокси-8-метокси -бензо[с]фенантридина (0,210 г, выход 15%) в виде светло-коричневого порошка.
FAB-MS (положительн.) m/z:
468 ([М+Н]+), 377 ([M+H-benzil]+),
1H-ЯMP (200 MHz, СDСl3) δ:
8,65 (1Н, s), 8,45 (1H, d, J=9,3 Hz), 8,31 (1H, d, J=9,1 Hz), 7,78 (1Н, d, J=9,1 Hz), 7,60 (1H, d, J=9,3 Hz), 7,57 (2H, dd, J=7,9 и 1,7 Hz), 7,49-7,37 (3H, m), 7,24 (1H, s), 6,11 (2Н, s), 5,21 (2Н, s), 4,06 (3H, s); 3,76 (2H, t, J=7,0 Hz), 3,61 (2H, t, J=6,1 Hz), 2,22 (2H, tt, J=7,0 и 6,1 Hz).
Пример 4
Синтез хлорида 2,3-(метилендиокси)-7-гидрокси-8-метокси-5,6-пропано-бензо[с]фенантридиния (соединение А-4) (X-=Сl-)
2,3-(Метилендиокси)-6-(3-оксипропил)-7-бензилокси-8-метокси-бензо[с] фенантридин (192 мг, 0,41 ммол. ) растворяют в метиленхлориде (8 мл). К раствору добавляют метансульфонилхлорид (41 мкл, 0,53 ммол.) и N,N-диизопропилэтиламин (95 мкл, 0,53 ммол.). Смесь перемешивают при комнатной температуре в течение 30 минут. К реакционной смеси добавляют метанол (1 мл) с последующим концентрированием в вакууме, получая желто-коричневый сироп (530 мг). К остатку для растворения добавляют уксусную кислоту (4 мл) и концентрированную соляную кислоту (2 мл). Раствор перемешивают при 60oС на масляной бане в течение 15 минут. Реакционную смесь охлаждают до комнатной температуры, а затем концентрируют в вакууме. Остаток растворяют в растворе 5% метанол-метиленхлорид (100 мл) и к раствору добавляют насыщенный водный раствор гидрокарбоната натрия (80 мл). Смесь энергично перемешивают до тех пор, пока оранжевый органический слой полностью не превратится в красно-фиолетовый. Органический слой отделяют и промывают водой, а затем сушат над безводным сульфатом натрия (содержащий соединение общей формулы (В)). Органический слой фильтруют для удаления сульфата натрия. 4М Раствор хлористого водорода в диоксане добавляют к фильтрату до тех пор, пока раствор не станет полностью оранжевым. Раствор концентрируют в вакууме. Раствор очищают колоночной хроматографией на силикагеле (элюируя смесью 8-12% метанол-метиленхлорид), получая соединение А-4 (Х-=Сl-) (112 мг, выход 69%) в виде золотисто-желтого порошка.
FAB-MS (положительн.) m/z:
360 (М+), UVλmaxnm:
(в 1М HCl) 438, 342, 320, 271, (в рН9) 488, 347, 335, 278;
1Н-ЯMP (200 MHz, DMSO-d6) δ:
8,72 (1H, d, J=9,2 Hz), 8,43 (1H, d, J=9,2 Hz), 8,24 (1H, s), 8,19 (1H, d, J=9,2 Hz), 8,03 (1H, d, J=9,2 Hz), 7,71 (1H, s), 6,33 (2H, s), 5,47 (2H, brt, J=7,1 Hz), 4,16 (2H, brt, J=7,7 Hz), 4,06 (3H, s), 2,44 (2H, m).
Пример 5
Синтез 2,3-(метилендиокси)-5-метил-6-этокси-7-бензилокси -8-метокси-5,6-дигидробензо[с]фенантридина
2,3-(Метилендиокси)-7-бензилокси-8-метокси-бензо[с] фенантридин (полученный способом, описанным в Японском патенте KOKAI 5-208959) (4,487 г, 10,96 ммол.) и метил 2-нитробензолсульфонат (4,30 г, 19,79 ммол.) растворяют в толуоле (90 мл). Раствор перемешивают в течение 24 часов с нагреванием при 110oС. После охлаждения реакционной смеси до комнатной температуры осажденные кристаллы отделяют фильтрацией, а затем промывают толуолом. Кристаллы суспендируют в N, N-диметилформамиде (62 мл) и к суспензии добавляют пиридин (0,62 мл). Смесь перемешивают при 60oС в течение 2 часов. После охлаждения смеси до комнатной температуры кристаллы отфильтровывают и последовательно промывают толуолом и гексаном. Полученные таким образом золотисто-желтые кристаллы суспендируют в этаноле (124 мл) и к суспензии добавляют 0,1 N водный раствор гидроксида натрия (93 мл). Смесь перемешивают до полного исчезновения золотисто-желтой окраски. Полученные светло-коричневые кристаллы отфильтровывают и промывают 50% водным этанолом, получая 2,3-(метилендиокси)-5-метил-6-этокси-7-бензилокси-8-метокси-5,6-дигидробензо[с] фенантридина (3,016 г, выход 59%) в виде светло-коричневого порошка.
FAB-MS(положительн.)m/z:
424 ([М-СН3СН2О]+);
1H-ЯMP (200 MHz, СDСl3) δ:
7,78 (1Н, d, J=8,6 Hz), 7,64 (1Н, d, J=8,6 Hz), 7,63 (1H, s), 7,46 (1H, d, J=8,6 Hz), 7,58-7,33 (5H, m), 7,11 (1H, s), 7,06 (1H, d, J=8,6 Hz), 6,04 (2H, s), 5,64 (1H, s), 5,21 (1H, d, J=10,9 Hz), 5,09 (1H, d, J=10,9 Hz) 3,94 (3H, s), 3,90 (1H, dq, J=9,6 и 7,1 Hz), 3,60 (1H, dq, J=9,6 и 7,1 Hz), 2,61 (3H, s), 1,05 (3Н, t, J=7,1 Hz).
Пример 6
Синтез 2,3-(метилендиокси)-5-метил-6-[3-(трет-бутилдиметил -силокси)пропил]-7-бензилокси-8-метокси-5,6-дигидробензо [с] фенантридина
В сухую колбу помещают магний (0,468 г, 19,3 ммол.) и к нему добавляют тетрагидрофуран (15 мл). Раствор 3-(трет-бутилдиметилсилокси)-пропилбромида (2,36 г, 9,63 ммол.) в тетрагидрофуране (15 мл), йода (несколько кристаллов) и 1,2-дибромэтана (несколько капель) порциями добавляют к смеси. Полученную смесь перемешивают при комнатной температуре в течение часа. К полученному раствору добавляют раствор 2,3-(метилендиокси)-5-метил-6-этокси-7-бензилокси-8-метокси-5,6-дигидробензо[с]фенантридина (1,01 г, 2,3 ммол.) в тетрагидрофуране с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной смеси добавляют насыщенный водный раствор хлорида аммония с последующим экстрагированием этилацетатом. Органический слой отделяют, промывают насыщенным водным раствором хлорида натрия, а затем сушат над безводным сульфатом натрия. После удаления сульфата натрия фильтрацией растворитель отгоняют в вакууме. Полученный остаток очищают колоночной хроматографией на силикагеле (элюируя смесью 33-66% метиленхлоридгексан), получая 2,3-(метилендиокси)-5-метил-6-[3-(трет-бутилдиметилсилокси)пропил] -7-бензилокси-8-метокси-5,6-дигидробензо[с]фенантридин (1,12 г, выход 96%) в виде бесцветного аморфного твердого вещества.
1H-ЯMP (200 MHz, СDСlз) δ:
7,69 (1Н, d, J=8,6 Hz), 7,64 (1Н, s), 7,57-7,35 (7Н, m), 7,09 (1Н, s), 6,96 (1Н, d, J= 8,6 Hz), 6,05-6,02 (2Н, m), 5,17 (1Н, d, J=11,4 Hz), 5,09 (1H, J= 11,4 Hz), 4,29 (1H, dd, J=9,5 и 5,1 Hz), 3,96 (3H, s), 3,45 (2H, t, J=6,8 Hz), 2,41 (3H, s), 1,80-1,53 (2H, m), 1,40-1,20 (2H, m), 0,77 (9H, s), 0,07 (3H, s), 0,09 (3H, s).
Пример 7
Синтез 2,3-(метилендиокси)-6-[3-(трет-бутилдиметилсилокси)-пропил] -7-бензилокси-8-метокси-бензо [с] фенантридина
После растворения 2,3-(метилендиокси)-5-метил-6-3(трет-бутилдиметилсилокси) пропил] -7-бензилокси-8-метокси-5,6-дигидробензо[с]фенантридина (871 мг, 1,46 ммол.) в толуоле (30 мл) к раствору добавляют активированную двуокись марганца (4,36 г). Смесь перемешивают при 100oС в течение 2 часов. Двуокись марганца удаляют фильтрацией, после этого фильтрат концентрируют в вакууме. Полученный остаток очищают колоночной хроматографией на силикагеле (элюируя смесью 33-66% метиленхлоридгексан), получая 2,3-(метилендиокси)-6-[3-(трет-бутилдиметилсилокси) пропил] -7-бензилокси-8-метокси-бензо[с] фенантридина (457 мг, выход 54%) в виде бесцветного порошка.
FAB-MS (положительн.) m/z:
582 ([М+Н]+),
1H-ЯMP (200 MHz, СDСl3) δ:
8,73 (1Н, s), 8,40 (1Н, d, J=9,4 Hz), 8,28 (1H, d, J=9,2 Hz), 7,75 (1Н, d, J=8,8 Hz), 7,60-7,50 (3Н, m), 7,47-7,35 (3Н, m), 7,22 (1H, s), 6,11 (2H, s), 5,16 (2H, s), 4,02 (3H, s), 3,75-3,63 (4H, m), 2,32-2,18 (2Н, m), 0,77 (9H, s), 0,02 (6H, s).
Пример 8
Синтез 2, 3- (метилендиокси) -6- (3-гидроксипропил) -7- бензилокси -8-метокси-бензо[с]фенантридина
После растворения 2,3-(метилендиокси)-6-[3-(трет-бутил-диметилсилокси)пропил] -7-бензилокси-8-метокси-бензо[с] фенантридина (374,4 мг, 0,644 ммол.) в тетрагидрофуране (3,2 мл) к раствору добавляют фторид тетрабутиламмония (1М раствор тетрагидрофурана, 1,9 мл). Смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют воду с последующим экстрагированием метиленхлоридом. Органическую фазу сушат над безводным сульфатом натрия, а затем концентрируют в вакууме. Полученный остаток очищают колоночной хроматографией на силикагеле (элюированной метиленхлоридом), получая 2,3-(метилендиокси)-6-(3-гидроксипропил)-7-бензилокси-8-метокси-бензо[с]фенантридина (263,7 мг, выход 88%) в виде бесцветного порошка.
Данные, полученные в результате исследования этого продукта аналитическими методами, совпадают с данными, полученными в результате исследования продукта, синтезированного в соответствии с примером 3.
Пример 9
Синтез 2,3-(метилендиокси)-5-метил-6-[4-(трет-бутилдиметил -силокси) бутил] -7-бензилокси-8-метокси-5, 6-дигидробензо [с] фенантридина
2, 3-(Метилендиокси)-5-метил-6-[4-(трет-бутилдиметилсилокси) -бутил] -7-бензилокси-8-метокси-5, 6-дигидробензо [с] фенантридина получают (выход 80%) в виде бесцветного аморфного твердого вещества способом, подобным способу, описанному в примере 8, за исключением того, что вместо 3-(трет-бутилдиметилсилокси) пропилбромида в примере 6 используют 4-(трет-бутилдиметилсилокси) бутилхлорид.
1H-ЯMP (200 MHz, СDСl3) δ:
7,70 (1Н, d, J=8,7 Hz), 7,63 (1H, s), 7,54 (1H, d, J=8,5 Hz), 7,54-7,32 (6Н, m), 7,09 (1H, s), 6,96 (1H, d, J=8,6 Hz), 6,04-6,02 (2H, m), 5,17 (1Н, d, J=11,3 Hz), 5,09 (1H, d, J=11,3 Hz), 4,29 (1H, dd, J=7,3 и 6,5 Hz), 3,96 (3H, s), 3,48 (2H, dd, J=5,9 и 5,4 Hz), 2,41 (3H, s), 1,56-1,26 (2Н, m), 0,87-0,83 (9Н, m), 0,04-0,04 (6H, m).
Пример 10
Синтез 2,3-(метилендиокси)-6-[4-(трет-бутилдиметилсилокси) -бутил] -7-бензилокси-8-метокси-бензо [с] фенантридина
После растворения 2,3-(метилендиокси)-5-метил-6-[4-(трет-бутилдиметилсилокси)бутил]-7-бензилокси-8-метокси-5,6-дигидро -бензо[с]фенантридина (300 мг, 0,49 ммол.) в толуоле (8 мл) к раствору добавляют активированную двуокись марганца (1,5 г). Смесь нагревают до дефлегмирования в течение часа. После охлаждения до комнатной температуры реакционную смесь разбавляют раствором 10% метанол-метиленхлорид (12 мл) с последующей фильтрацией. Фильтрат концентрируют в вакууме. Полученный остаток очищают колоночной хроматографией на силикагеле (элюированном метиленхлоридом), получая 2,3-(метилендиокси)-6-[4-(трет-бутилдиметилсилокси)бутил] -7-бензилокси-8-метокси-бензо[с]фенантридина (205 мг, выход 70%) в виде светло-коричневых кристаллов.
FAB-MS (положительн.) m/z:
596 ([М+Н]+),
1H-ЯMP (200 MHz, СDСl3) δ:
8,74 (1Н, s), 8,44 (1Н, d, J=9,4 Hz), 8,31 (1Н, d, J=9,1 Hz), 7,77 (1Н, d, J=8,8 Hz), 7,58 (1H, d, J=9,0 Hz), 7,60-7,55 (2Н, m), 7,47-7,35 (3Н, m), 7,24 (1H, s), 6,11 (2Н, s), 5,17 (2Н, s), 4,04 (3Н, s), 3,64 (4Н, m), 2,01 (2Н, m), 1,58 (2Н, m), 0,88 (9Н, m), 0,04 (6Н, m).
Пример 11
Синтез 2, 3- (метилендиокси) -6- (4-гидроксибутил) -7- бензилокси-8-метокси-бензо[с]фенантридина
После растворения 2,3-(метилендиокси)-6-[4-(трет-бутилдиметилсилокси) бутил] -7-бензилокси-8-метокси-бензо[с] фенантридина (532 мг, 0,89 ммол.) в тетрагидрофуране (6 мл) к раствору добавляют уксусную кислоту (102 мкл, 1,78 ммол. ) и тетрабутил-аммоний фторид (1М раствор тетрагидрофурана, 1,78 мл). Смесь перемешивают при комнатной температуре. Реакционный раствор концентрируют в вакууме. Остаток разбавляют метиленхлоридом (30 мл), который затем промывают водой. После высушивания органической фазы над безводным сульфатом натрия, раствор отгоняют в вакууме. Остаток очищают колоночной хроматографией на силикагеле (элюируя смесью 1% метанол-метиленхлорид), получая 2,3- (метилендиокси)-6-(4-гидроксибутил) -7-бензилокси-8-метокси-бензо[с] фенантридина (372 мг, выход 87%) в виде светло-желтых иголок.
FAB-MS (положительн.) m/z:
482 ([M+H]+),
1H-ЯMP (200 MHz, СDСl3) δ:
8,71 (1Н, s), 8,45 (1Н, d, J=9,3 Hz), 8,31 (1Н, d, J=9,0 Hz), 7,77 (1Н, d, J=9,0 Hz), 7,59 (1H, d, J=9,3 Hz), 7,61-7,55 (2Н, m), 7,49-7,37 (3Н, m), 7,24 (1Н, s), 6,11 (2Н, s), 5,17 (2Н, s), 4,06 (3Н, s), 3,65 (2H, dd, J=7,8 и 7,2 Hz), 3,59 (2H, t, J=6,5 Hz), 2,02 (2H, m), 1,64-1,50 (2Н, m).
Пример 12
Синтез 2,3-(метилендиокси)-7-гидрокси-8-метокси-5,6-бутано -бензо[с]фенантридиния хлорида (соединение А-7) (X-=Сl-)
2,3-(Метилендиокси)-6-(3-гидроксибутил)-7-бензилокси-8-метокси- бензо[с] фенантридин (252 мг, 0,52 ммол.) растворяют в метиленхлориде (10 мл). Метансульфонилхлорид (80 мкл, 1,03 ммол.) и N,N-диизопропилэтиламин (186 мкл, 1,04 ммол.) добавляют к раствору. Смесь перемешивают при комнатной температуре в течение 20 минут. После добавления к реакционной смеси воды (30 мл), смесь экстрагируют метиленхлоридом (30 мл). Органическую фазу последовательно промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, а затем сушат над безводным сульфатом натрия. Растворитель отгоняют в вакууме. Остаток очищают колоночной хроматографией на силикагеле (элюируя смесью 0-0,5% метанол-метиленхлорид), получая метансульфонат (210 мг, выход 72%).
Метансульфонат (186 мг, 0,33 ммол.) растворяют в толуоле (12 мл). Раствор нагревают до температуры кипения с обратным холодильником в течение двух дней. Реакционную смесь концентрируют в вакууме. К остатку добавляют уксусную кислоту (3 мл) и концентрированную соляную кислоту (1,5 мл). Смесь перемешивают при 60oС в течение 30 минут. Реакционную смесь охлаждают до комнатной температуры, а затем концентрируют в вакууме. Остаток растворяют в растворе метиленхлорида (50 мл) и к раствору добавляют насыщенный водный раствор гидрокарбоната натрия (50 мл). Смесь энергично перемешивают до тех пор, пока оранжевый органический слой не превратится полностью в красно-фиолетовый. Органический слой (содержащий соединение общей формулы (В)) отделяют и промывают водой. К органической фазе добавляют метанол (10 мл) и 1М водный раствор соляной кислоты (1 мл). Полученный раствор перемешивают (раствор вновь становится оранжевым). Раствор концентрируют в вакууме. Остаток очищают колоночной хроматографией на силикагеле (элюируя смесью 8% метанол-метиленхлорид), получая соединение А-7 (Х-=Сl-) (86 мг, выход 64%) в виде оранжевого порошка.
FAB-MS (положительн.) m/z:
374 (М+), UVλmaxnm:
(в 1М HCl) 446, 345, 325, 275,
(в рН9) 495, 353, 335, 283;
1H-ЯMP (200 MHz, СDСl3) δ:
11,41 (1H, brs), 8,66 (1H, d, J=9,2 Hz), 8,47 (1H, d, J=9,2 Hz), 8,19 (1H, d, J=8,8 Hz), 8,05 (1H, d, J=9,0 Hz), 7,90 (1H, s), 7,70 (1H, s), 6,31 (2H, s), 5,11 (2H, m), 4,26 (2H, m), 4,08 (3H, s), 2,05-1,87 (2H, m), 1,85-1,67 (2H, m).
Пример 13
Синтез N-((2'-бензилокси)-3'-метокси-6'-бромбензил)-1-антриламина
2-Бензилокси-3-метокси-6- бромбензальдегид (8,32 г, 23,2 ммол.), полученный способом, описанным в J.C.S. Perkin I, 1221 (1976) и J. Org. Chem., 53, 1708 (1988), и 1-аминоантрацен (выпускаемый "ALDRICH", 90%, 5,00 г, 23,3 ммол. ) растворяют в толуоле (230 мл). При энергичном перемешивании раствор нагревают до кипения с обратным холодильником при 120oС в течение 2 часов. Продолжая нагревание при 120oС, к реакционной смеси больше трех часов постепенно добавляют толуол (180 мл) и одновременно отгоняют растворитель. После охлаждения до комнатной температуры к смеси добавляют толуол (70 мл). При охлаждении водой к полученной смеси последовательно добавляют комплекс диметиламинборан (1,03 г, 17,5 ммол.) и уксусную кислоту (30 мл). После перемешивания при комнатной температуре в течение 75 минут к смеси при охлаждении водой добавляют 1М водный раствор соляной кислоты (130 мл). Реакционную смесь фильтруют. Фильтрат разделяют на органическую фазу и водную фазу. Водную фазу экстрагируют толуолом (200 мл • 2) и эту фракцию соединяют с ранее полученной органической фазой. Объединенную смесь сушат над безводным сульфатом натрия. Растворитель отгоняют в вакууме. Остаток очищают колоночной хроматографией на силикагеле (элюируя смесью 10% этилацетат-гексан), получая N-((2'-бензилокси)-3'-метокси-6'-бромбензил)-1-антриламин (12,77 г, количественный выход) в виде желтого порошка.
1H-ЯMP (200 MHz, СDСl3) δ:
8,86 (1Н, s), 8,38 (1H, s), 7,98 (2Н, m), 7, 50-7,07 (10Н, m), 6,57 (1Н, m), 6,20 (1Н, t, J=4,5 Hz), 5,05 (2Н, s), 4,47 (1Н, s), 4,44 (1Н, s), 3,90 (3Н, s).
Пример 14
Синтез 8-бензилокси-9-метокси-нафто[2,3-с]фенантридина
После растворения N-((2'-бензилокси)-3'-метокси-6'-бромбензил)-1-антриламина (10,81 г, 21,70 ммол.) в толуоле (1 л) к раствору добавляют гидрид триоктилолова (19,95 г, 43,43 ммол.) и температуру поднимают до 105oС. Затем к смеси добавляют 2,2'-азобис(2-метилбутиронитрил) (8,36 г, 43,46 ммол.). Раствор нагревают до кипения с обратным холодильником в течение 2 часов при 120oС. После охлаждения до комнатной температуры к полученной смеси добавляют активированную окись марганца (10,81 г). Смесь перемешивают в течение 30 минут. После добавления к ней этанола (200 мл) реакционный раствор фильтруют для удаления двуокиси марганца. Фильтрат концентрируют в вакууме. Полученный остаток кристаллизуют из смеси раствора гексан-метиленхлорид, получая 8-бензилокси-9-метокси-нафто [2,3-с] фенантридина (4,04 г, выход 45%) в виде светло-желтого порошка.
FAB-MS (положительн.) m/z:
416 ([М+Н]+),
1H-ЯMP (200 MHz, СDСl3) δ:
9,82 (1H, s), 9,65 (1H, s), 8,67 (3H, m), 8,37-8,15 (3Н, m), 7,94 (1H, d, J=9,3 Hz), 7,60 (4H, m), 7,40 (3Н, m), 5,35 (2Н, s), 4,10 (3Н, s).
Пример 15
Синтез хлорида 8-гидрокси-9-метокси-6,7-пропано-нафто [2,3-с]фенантридиния (соединение А-9) (X-=Сl-)
8-бензилокси-9-метокси-нафто[2,3-с] фенантридин (231 мг, 0,56 ммол.) суспендируют в ацетонитриле (80 мл) и к суспензии добавляют трифторуксусную кислоту (43 мкл, 0,56 ммол.), 3-бромо-1-пропанол (51 мкл, 0,56 ммол.) и трис (триметилсилил) силан (346 мкл, 1,12 ммол.). Смесь перемешивают при 80oС на масляной бане. После растворения суспендированного вещества к раствору добавляют азобис(изобутиронитрил) (184 мг, 1,12 ммол.) с последующим нагреванием с дефлегмацией. Час спустя реакционную смесь охлаждают до комнатной температуры. К остатку добавляют насыщенный водный раствор кислого углекислого натрия (50 мл), а затем экстрагируют метиленхлоридом. После промывания органического слоя водой и высушивания над безводным сульфатом натрия, растворитель отгоняют в вакууме. Остаток пропускают через колонку с силикагелем (элюируя смесью 1% метанол-метиленхлорид). Основные фракции собирают и растворитель отгоняют в вакууме. Остаток растворяют в толуоле (3 мл) и к раствору добавляют активированную двуокись марганца (100 мг). Смесь перемешивают при комнатной температуре в течение 90 минут. Двуокись марганца отфильтровывают и фильтрат концентрируют в вакууме, получая сырой 7-(3-гидроксипропил)-8-бензилокси-9-метокси-нафто[2,3-с]фенантридин (83 мг) в виде коричневой сухой массы.
Сырой продукт растворяют в метиленхлориде (3 мл). К раствору добавляют метансульфонилхлорид (13 мкл, 0,17 ммол.) и N,N-диизопропилэтиламин (30 мкл, 0,17 ммол. ). Смесь перемешивают при комнатной температуре в течение 45 минут. После добавления к реакционной смеси метанола (1 мл), смесь концентрируют в вакууме, получая сухую желто-коричневую сиропооб-разную массу. К этой массе для растворения добавляют уксусную кислоту (1,2 мл) и концентрированную соляную кислоту (0,6 мл). Раствор перемешивают при 60oС в течение 25 минут на масляной бане. Затем реакционный раствор охлаждают до комнатной температуры и к нему добавляют насыщенный водный раствор кислого углекислого натрия (100 мл). Затем смесь экстрагируют метиленхлоридом. Органическую фазу отделяют и промывают водой, а затем сушат над безводным сульфатом натрия (содержащую соединение общей формулы (В)). Сульфат натрия отфильтровывают и к раствору добавляют 4М раствор хлористого водорода в диоксане до тех пор, пока раствор не станет полностью оранжевым. Раствор концентрируют в вакууме. Остаток очищают колоночной хроматографией на силикагеле (элюируя смесью 8-12% метанол-метиленхлорид), а затем гель-фильтрационной хроматографией (элюируя водным раствором смеси 20% метанол -5 мМ соляная кислота Sephadex LH-20), получая соединение А-9 (Х-=Сl-) (10 мг, выход 4%) в виде золотисто-желтого порошка.
FAB-MS (положительн.) m/z:
366 ([М+Н]+),
1H-ЯMP (200 MHz, DMSO-d6) δ:
11,43 (1H, brs), 9,56 (1H, s), 8,88 (1H, s), 8,81 (1H, d, J=9,5 Hz), 8,61 (1H, d, J=9,2 Hz), 8,47 (1H, d, J=9,2 Hz), 8,46 (1H, m), 8,24 (1H, m), 8,17 (1H, d, J=9,2 Hz), 1, 80-7,72 (2Н, m), 5,77 (2Н, brt, J=7,1 Hz), 4 28 (2Н, brt, J=7,7 Hz), 4,11 (3H, s), 2,51 (2H, m).
Примеры фармакологических испытаний
Соединения в соответствии с настоящим изобретением подвергались испытаниям на противоопухолевую активность, а полученные результаты представлены ниже. Как показано ниже, производные фенантридиния, представленные общей формулой (А), предупреждают рост опухолевых клеток.
1. Профилактика роста раковых клеток
Раковые клетки HeLa S3, полученные из матки человека, инкубируют при 37oС в течение 24 часов в 5% СО2. Исследуемое соединение затем вводят в соприкосновение с клетками на протяжении 72 часов. После этого клетки окрашивают 0,05% метиленовой синью. Из окрашенных клеток экстрагируют пигмент. Ингибирование роста клеток определяют на основании поглощения при 660 нм, подсчитывая концентрацию 50% ингибирования роста (IC50). Результаты представлены в табл. 2.
2. Противоопухолевое действие на раковые клетки in vivo
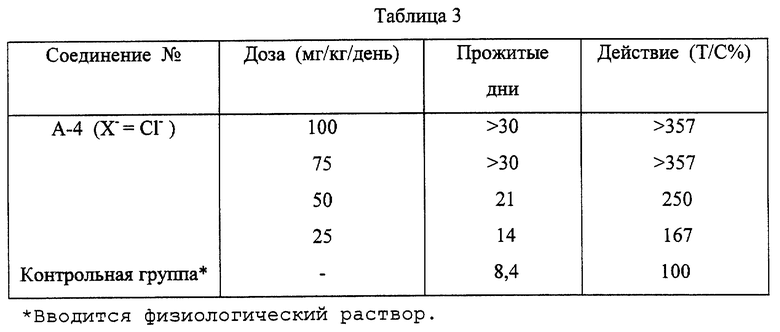
Мышиные клетки лейкемии Р388 вводят внутривенно шестинедельным самкам мышей CDF1 в дозе 105 клеток на мышь. На следующий день после пересадки опухоли им вводят разовую внутривенную инъекцию 5% глюкозного водного раствора хлорида 2,3-(метилендиокси)-7-гидрокси-8-метокси-5,6-пропано-бензо[с] фенантридиния (соединение А-4 в соответствии с настоящим изобретением (Х-= Сl-)). Противоопухолевое действие определяют, сравнивая отношение (Т/С%) дней выживания к средней величине дней выживания в контрольной группе (5 мышей). Результаты представлены в табл. 3.
3. Острая токсичность
Острую токсичность определяют путем внутривенного введения хлорида 2,3-(метилендиокси)-7-гидрокси-8-метокси-5,6-пропано-бензо[с] фенантридиния (соединение А-4 в соответствии с настоящим изобретением (Х-=Сl-)) шестинедельным самкам мышей CDF1. Животные выживают даже при дозе 100 мг/кг, оставаясь в живых.
Пример 16
Фармацевтические композиции
После взвешивания 1 г 2, 3-(метилендиокси)-7-гидрокси-8-метокси-5,6-пропано-бензо[с] фенантридиния хлорида (соединение 4-А в соответствии с настоящим изобретением, Х-=Сl-), 1 г полисорбата и 1 г Macrogol 400, эти соединения диспергируют и растворяют в 100 г стерильной воды для инъекций. Раствор фильтруют через мембранный фильтр. Фильтрат распределяют по ампулам и лиофилизуют обычным способом, получая препарат для инъекций, содержащий 50 мг на ампулу соединения А-4 (Х-=Сl-).
Пример 17
Химическое восстановление
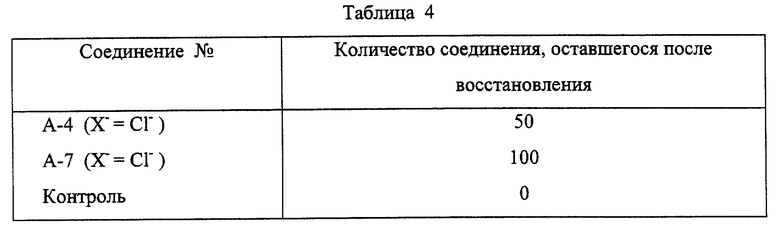
Соединение в соответствии с настоящим изобретением (0,1 мг/мл водного раствора, 0,1 мл) разбавляют метанолом (0,1 мл). К раствору добавляют водный раствор цианоборгидрида натрия (4 мг/мл, 0,02 мл). Смеси дают возможность отстояться при комнатной температуре. Реакцию прекращают, добавляя к ней 1% водную фосфорную кислоту (1 мл). Остаточное количество соединения определяют жидкостной хроматографией высокого разрешения. В качестве контроля такой же реакции подвергают кислый сернокислый 2,3-(метилендиокси)-5-метил-7-окси-8-метокси-бензо [с]фенантридиний. Результаты представлены в таблице 4.
Промышленная применимость
Производное фенантридиния в соответствии с настоящим изобретением проявляет противоопухолевую активность, устойчиво к химическому восстановлению и биологическим метаболическим реакциям и, таким образом, чрезвычайно эффективно в качестве лекарственного средства.
Как отмечалось выше, материалы данной заявки содержат данные экспериментов "in vitro" и/или "in vivo" для соединений А-4 и А-7.
Кроме того, для подтверждения реализации указанного назначения другими заявленными соединениями ниже Заявитель представляет результаты экспериментов in vitro для соединений А-1 и А-9 на раковых клетках Hela S3.
Соединения А-1 и А-9 представляют собой хлорид 7-гидрокси-8-метокси-5,6-пропанобензо[с] фенантридиния и хлорид 8-гидрокси-9-метокси-6,7-пропанонафто[2,3-с]фенантридиния, соответственно, как описано в примерах 2 и 15.
Соединение - IC50 (мкм) - Концентрация 50% ингибирования
Соединение А-1 (Х-=Сl-) - 0,15
Соединение А-9 (Х-=Сl-) - 0,06
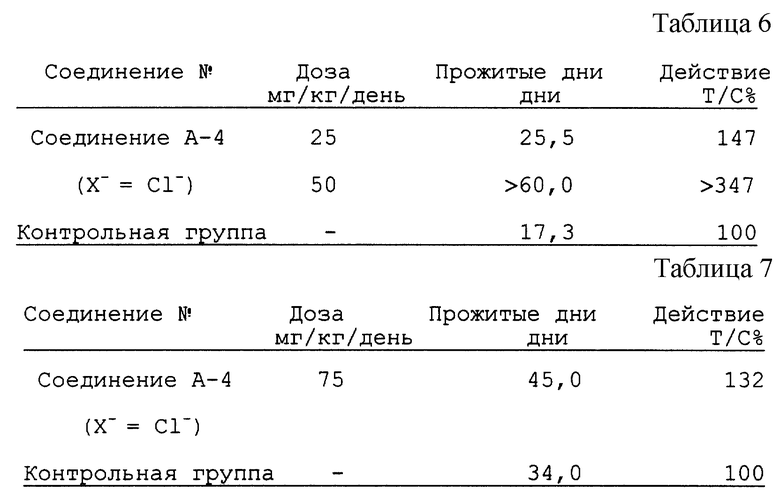
Кроме того, ниже Заявитель представляет результаты экспериментов in vivo для соединений А-1 и А-9 на прививаемой мыши лейкемии Р388 (см. табл. 5).
Из вышеизложенного становится очевидным, что заявленные соединения, отличные от А-4, также проявляют противоопухолевую активность.
Кроме того, для подтверждения реализации указанного назначения заявленных соединений в отношении других опухолей, в настоящее время Заявитель может представить результаты эксперимента in vivo для соединения А-4 на аденокарциноме ободочной кишки Colon 26 мыши и мышиной злокачественной опухоли В16, прививаемой мышам.
i) Аденокарцинома Colon 26 мыши (см. табл. 6).
ii) Злокачественная опухоль В16 мыши (см. табл. 7).
Ввиду вышеизложенного становится ясным, что заявленные соединения проявляют противоопухолевую активность на различных опухолях, таких как аденокарцинома Colon 26 мыши и мышиная злокачественная опухоль В16, прививаемая мышам, отличных от клеток лейкемии.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ АЦЕТАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И ИНГИБИТОРЫ ПРОТЕАЗ НА ИХ ОСНОВЕ | 1997 |
|
RU2181360C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ ЭФИРОВ ЭРИТРО-3-АМИНО-2-ОКСИМАСЛЯНЫХ КИСЛОТ И СООТВЕТСТВУЮЩИХ ИСХОДНЫХ КИСЛОТ | 1997 |
|
RU2169138C2 |
| ПРОИЗВОДНЫЕ БЕНЗО-/С/-ФЕНАНТРИДИНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2073674C1 |
| АНАЛОГ 3-ФЕНИЛЦИННОЛИНА И ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО НА ЕГО ОСНОВЕ | 2003 |
|
RU2324683C2 |
| ПРОИЗВОДНЫЕ [1,2,4]ТРИАЗОЛО[1,5-a]ПИРИМИДИН-2-ИЛМОЧЕВИНЫ И ЕГО ПРИМЕНЕНИЕ | 2004 |
|
RU2348636C2 |
| НОВОЕ ЦИКЛОГЕКСАНОВОЕ ПРОИЗВОДНОЕ, ЕГО ПРОЛЕКАРСТВО И ЕГО СОЛЬ И СОДЕРЖАЩЕЕ ИХ ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ОТ ДИАБЕТА | 2005 |
|
RU2394015C2 |
| СОЕДИНЕНИЯ С ГИДРОКСИКАРБОНИЛЬНЫМИ-ГАЛОГЕНАЛКИЛЬНЫМИ БОКОВЫМИ ЦЕПЯМИ | 2000 |
|
RU2247106C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ТРИТЕРПЕНОВЫЕ ПРОИЗВОДНЫЕ | 1997 |
|
RU2168517C2 |
| ПРОИЗВОДНЫЕ СПИРОКЕТАЛЕЙ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА ПРОТИВ ДИАБЕТА | 2006 |
|
RU2416617C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА | 1995 |
|
RU2137770C1 |





Изобретение относится к новым производным фенантридиния общей формулы А, где R1 - замещенная или незамещенная низшая алифатическая углеводородная группа, R - алифатическая углеводородная цепь, имеющая 2-6 атомов углерода, которая может быть необязательно замещена заместителем, выбранным из группы, включающей низшую алкильную группу, галоген или гидроксигруппу, каждый из Y и Z независимо представляет собой водород, гидрокси- или низшую алкоксигруппу, или Y и Z объединяются вместе с образованием метилендиокси или фенильного кольца, и X- - остаток кислоты или остаток водородной кислоты, а также к производным фенантридиния общей формулы В, где значения R1, R, Y, Z определены выше для формулы А. Кроме того, раскрыты соль 2,3-(метилендиокси)-7-гидрокси-8-метокси-5,6-пропано-бензо[с] фенантридиния, фармацевтическая композиция, обладающая противоопухолевой активностью, и способ ингибирования роста раковых клеток на основе представленных соединений. Изобретение может быть использовано в медицине как противоопухолевое лекарственное средство. 5 с. и 3 з.п. ф-лы, 7 табл.
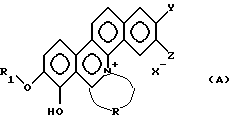
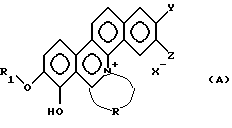
где R1 - замещенная или незамещенная низшая алифатическая углеводородная группа;
R - алифатическая углеводородная цепь, имеющая 2-6 атомов углерода, которая может быть необязательно замещена заместителем, выбранным из группы, включающей низшую алкильную группу, галоген и гидроксигруппу;
каждый из Y и Z независимо представляет собой водород, гидрокси- или низшую алкоксигруппу; или Y и Z объединяются вместе с образованием метилендиокси или фенильного кольца; и
X- - остаток кислоты или остаток водородной кислоты.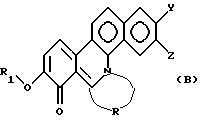
где R1 - замещенная или незамещенная низшая алифатическая углеводородная группа;
R - низшая алифатическая углеводородная цепь, имеющая 2-6 атомов углерода, которая может быть необязательно замещена заместителем, выбранным из группы, включающей низшую алкильную группу, галоген и гидроксигруппу;
каждый из Y и Z независимо представляет собой водород, гидрокси- или низшую алкоксигруппу; или Y и Z объединяются вместе с образованием метилендиокси или фенильного кольца.
| ПРОИЗВОДНЫЕ БЕНЗО-/С/-ФЕНАНТРИДИНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2073674C1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ПЕРЕВОДЧИКОВА Н.И | |||
| Противоопухолевая химиотерапия | |||
| - М.: Медицина, 1993, с.58-65. | |||

