Область техники, к которой относится изобретение
Настоящее изобретение относится к макроциклическим соединениям, обладающим ингибиторной активностью в отношении NS3 серинпротеазы HCV (вируса гепатита С). Оно относится также к композициям, включающим указанные соединения в качестве активных ингредиентов, а также к способам получения указанных соединений и композиций.
Уровень техники
Вирус гепатита С (hepatitis C virus - HCV) во всем мире является главной причиной хронического поражения печени и стал основным предметом глубоких медицинских исследований. HCV является представителем семейства флавивирусов (Flaviviridae) в роду гепацивирусов и тесно связан с родом флавивирусов (flavivirus), который включает ряд вирусов, вовлеченных в такие заболевания человека, как вирус лихорадки Денге и вирус желтой лихорадки, и с семейством пестивирусов животных, которое включает вирус бычьей вирусной диареи (bovine viral diarrhea virus - BVDV). Геном HCV включает как 5', так и 3' нетранслируемые области, которые воспринимают вторичные структуры РНК, и центральную открытую рамку считывания, которая кодирует единственный полипротеин. Полипротеин кодирует десять генных продуктов, которые генерируются из предшественника полипротеина с помощью организованных рядов ко- и посттрансплантационных эндопротеолитических расщеплений, опосредуемых как протеазами организма-хозяина, так и протеазами вируса. Структурные белки вируса включают нуклеокапсидный белок ядра и два охватывающих его гликопротеина Е1 и Е2. Неструктурные (non-structural - NS) белки кодируют некоторые ферменты вируса жизненно важных функций (такие как геликаза, полимераза, протеаза), а также белки неизвестных функций. Репликация генома вируса опосредуется РНК-зависимой РНК-полимеразой, кодированной неструктурным белком 5b (NS5B). Было показано, также как полимеразная функция вируса, геликазная и протеазная функции, кодируемые в бифункциональном NS3 белке, имеют большое значение для репликации HCV РНК. Помимо NS3 серинпротеазы HCV кодирует также металлопротеиназу в NS2 области.
После первичного острого инфицирования у большинства инфицированных пациентов развивается хронический гепатит С, поскольку HCV воспроизводится предпочтительно в гепатоцитах, но не является непосредственно цитопатическим. В частности, отсутствие сильного Т-лимфоцитного ответа и высокого сродства вируса к мутации, по-видимому, способствует высокой скорости развития хронического инфицирования. Хронический гепатит может развиваться до фиброза печени, приводящего к циррозу, конечной стадии поражения печени, и гепатоцеллюлярного рака (hepatocellular carcinoma - HCC), что делает его основной причиной трансплантации печени.
Существует 6 основных генотипов и свыше 50 подтипов HCV, которые распространены в разных географических зонах. 1 тип HCV является доминирующим типом в Европе и США. Обширная генетическая гетерогенность HCV имеет важные диагностические и клинические последствия, возможно объясняющие трудности в разработке вакцины и отсутствие ответной реакции на современную терапию.
Перенос HCV может осуществляться через контакт с зараженной кровью или препаратами, полученными из зараженной крови, например, в результате переливания крови или внутривенного применения лекарственных средств. Внедрение диагностических тестов, используемых при отборе крови, привело к тенденции снижения посттрансфузионной заболеваемости HCV. Однако вследствие медленного развития заболевания печени до конечной стадии развития болезни, инфекции, уже имеющие место в настоящее время, будут продолжать представлять серьезную медицинскую и экономическую проблему на протяжении десятилетий.
Применяющиеся в настоящее время терапевтические методы лечения HCV основываются на (пэгилированном) интерфероне-альфа (IFN-α) в сочетании с рибофлавином. Такая комбинированная терапия приводит к стабильному вирусологическому ответу у более чем 40% пациентов, инфицированных вирусом генотипа 1, и примерно у 80% пациентов, инфицированных генотипами 2 и 3. Помимо ограниченной эффективности в отношении HCV типа 1 такая комбинированная терапия сопровождается значительными побочными эффектами и плохо переносится многими пациентами. Основные побочные эффекты включают симптомы, подобные симптомам при заболевании гриппом, гематологические отклонения и нейропсихиатрические симптомы. Следовательно, существует потребность в более эффективных, удобных и легче переносимых способах лечения.
В научной и патентной литературе описано много ингибиторов HCV протеазы. Длительное введение ингибиторов HCV протеазы обычно приводит к появлению резистентных HCV мутантов, так называемых «мутантов, отключающих действие лекарственных средств». Они имеют характеристические мутации в протеазном геноме HCV, в частности D168V, D168Y и/или A165S. Поэтому существует потребность в дополнительных лекарственных средствах с различными характеристиками резистентности для предоставления пациентам с такими отклонениями выбора лечения. Такие лекарственные средства могут найти применение в комбинированной терапии, которая, как ожидается, станет нормой в будущем даже в качестве терапии первой линии.
Практический опыт применения лекарственных средств против ВИЧ, в частности ингибиторов ВИЧ протеазы, показывает, что суб-оптимальная фармакокинетика и сложные схемы дозировки быстро приводят к случайным нарушениям режима и схемы приема лекарственных средств. Это, в свою очередь, означает, что нижняя концентрация (минимальная концентрация в плазме) для соответствующих лекарственных средств в режиме лечения ВИЧ зачастую на длительные периоды времени в течение суток падает ниже порогового значения IC90 или ED90. Считается, что при минимальной концентрации в течение 24 часов, равной, по меньшей мере, IC50, точнее IC90 или ED90, существенно замедляется развитие мутантов, «отключающих действие лекарственных средств».
Достижение нужной фармакокинетики и метаболизма лекарственного средства для получения таких нижних уровней концентрации обуславливает жесткое требование к разработке лекарственного средства. Известные ингибиторы HCV протеазы с множеством пептидных связей предъявляют дополнительные фармакокинетические требования к эффективным режимам дозировки.
Таким образом, существует потребность в ингибиторах HCV, которые могут преодолевать такие недостатки современной терапии HCV, как побочные эффекты, ограниченная эффективность, развитие резистентности и неэффективность лечения вследствие нарушения схемы приема лекарственного средства.
Настоящее изобретение относится к ингибиторам репликации HCV, которые проявляют, по меньшей мере, одно улучшенное свойство относительно соединений предшествующего уровня. В частности, ингибиторы согласно настоящему изобретению являются наилучшими по одному или нескольким из таких связанных фармакологических свойств как эффективность, сниженная цитотоксичность, улучшенная фармакокинетика, улучшенный профиль резистентности, приемлемая дозировка и содержание действующего вещества в таблетке.
Сущность изобретения
Настоящее изобретение относится к ингибиторам репликации HCV, которые могут быть представлены формулой (I)

включая их стереоизомеры, где
A представляет собой -C(=O)OR1, -C(=O)-NH-SO2-R2, -C(=O)C(=O)NR3aR3b, -C(=O)-NH-SO2-NR3aR3b, -C(=O)NH-P(=O)(OR4a)(R4b) или -P(=O)(OR4a)(R4b),
где
R1 представляет собой водород; арил; Het; C3-7циклоалкил, необязательно замещенный C1-6алкилом; или C1-6алкил, необязательно замещенный C3-7циклоалкилом, арилом или Het;
R2 представляет собой арил; Het; C3-7циклоалкил, необязательно замещенный C1-6алкилом; или C1-6алкил, необязательно замещенный C3-7 циклоалкилом, арилом или Het;
R3a и R3b каждый независимо представляют собой водород; C1-6алкил, необязательно замещенный C1-6алкокси-, гидроксильной группой, галогеном, C3-7циклоалкилом, арилом или Het; арил; C2-6алкенил; Het; C3-7циклоалкил, необязательно замещенный C1-6алкилом; или R3a и R3b вместе с атомом азота, к которому они присоединены, образуют группу Het1; и R3a также может представлять собой C1-6алкокси;
R4a представляет собой водород, C1-6алкил, C2-6алкенил, C3-7циклоалкил, арил или C1-6алкил, необязательно замещенный C3-7циклоалкилом или арилом;
R4b представляет собой R4b', OR4b' или NHR4b';
R4b' представляет собой C1-6алкил, C2-6алкенил, C3-7циклоалкил, арил или C1-6алкил, необязательно замещенный C3-7циклоалкилом или арилом;
X представляет собой N, CH и, когда X соединен двойной связью, то представляет собой C;
E представляет собой NR5 или, когда X представляет собой N, тогда E представляет собой NR5 или CR6aR6b;
R5 представляет собой водород, C1-6алкил, C1-6алкоксиC1-6алкил или C3-7циклоалкил;
R6a и R6b независимо представляют собой водород или C1-6алкил, или R6a и R6b вместе с атомом углерода, к которому они присоединены, образуют C3-7циклоалкил;
n равно 3, 4, 5 или 6;
каждая пунктирная линия ----- независимо представляет собой необязательную двойную связь;
R7 представляет собой водород или, когда X представляет собой C или CH, R7 также может представлять собой C1-6алкил;
R8 представляет собой радикал формулы
R8a и R9a каждый независимо представляет собой водород, C1-6алкил, C2-6алкенил, C1-6алкокси, гидроксильную группу, галоген, полигалогенC1-6алкил, циано, амино, моно- или C1-6диалкиламино;
каждый R9 независимо представляет собой C1-6алкил, необязательно замещенный C1-6алкокси, гидроксильной группой или галогеном; C3-7циклоалкил; C2-6алкенил; C1-6алкокси; C3-7циклоалкилокси; арилокси; Het-O-; гидроксильную группу; циано; полигалогенC1-6алкил; моно- или C1-6диалкиламино;
каждый R10 независимо представляет собой водород, C1-6алкил, C2-6алкенил, C1-6алкокси, гидроксильную группу, галоген, полигалогенC1-6алкил, циано, амино, моно- или C1-6диалкиламино;
каждый арил независимо представляет собой фенил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из галогена, гидроксильной группы, нитро, циано, карбоксила, C1-6алкила, C1-6алкокси, C1-6алкоксиC1-6алкила, C1-6алкилкарбонила, аминогруппы, моно- или диC1-6алкиламино, азидо, меркапто, C1-6алкилтио, полигалогенC1-6алкила, полигалогенC1-6алкокси, C3-7циклоалкила и Het1;
каждый Het независимо представляет собой 5- или 6-членный насыщенный, частично ненасыщенный или полностью ненасыщенный гетероцикл, содержащий 1, 2, 3 или 4 гетероатома, каждый из которых независимо выбран из атомов азота, кислорода и серы, причем указанный гетероцикл является необязательно замещенным одним, двумя или тремя заместителями, каждый из которых независимо выбран из галогена, гидроксильной группы, нитро, циано, карбоксила, C1-6алкила, C1-6алкокси, C1-6алкоксиC1-6алкила, C1-6алкилкарбонила, аминогруппы, моно- или диC1-6алкиламино, азидо, меркапто, полигалогенC1-6алкила, полигалогенC1-6алкокси, C3-7циклоалкила, Het1;
каждый Het1 независимо представляет собой пирролидинил, пиперидинил, пиперазинил, 4-C1-6алкилпиперазинил, 4-C1-6алкилкарбонилпиперазинил и морфолинил, где морфолинильная и пиперидинильная группы могут быть необязательно замещены одним или двумя C1-6алкильными радикалами;
или их N-оксиды, фармацевтически приемлемые соли или фармацевтически приемлемые сольваты.
Изобретение относится к соединениям формулы (I) как таковым и их N-оксидам, фармацевтически приемлемым аддитивным солям и стереохимически изомерным формам для применения в качестве лекарственного средства. Изобретение относится также к фармацевтическим композициям, включающим упомянутые выше соединения, для введения субъекту, страдающему HCV инфекцией. Фармацевтические композиции могут включать комбинации упомянутых выше соединений с другими средствами, обладающими противовирусной активностью в отношении HCV.
Изобретение относится также к применению соединения формулы (I), его N-оксида, фармацевтически приемлемой аддитивной соли или стереохимически изомерной формы для производства лекарственного средства для ингибирования HCV репликации. Или изобретение относится к способу ингибирования HCV репликации в организме теплокровного животного, причем указанный способ включает введение эффективного количества соединения формулы (I), его N-оксида, фармацевтически приемлемой аддитивной соли или стереохимически изомерной формы.
Подробное описание изобретения
Используемые выше и далее термины имеют следующие определения (если не указано иного значения).
Термин «C1-4алкил», когда используется в описании изобретения для определения группы или части группы, означает насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащей от 1 до 4 атомов углерода, такие как, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил; термин «C1-6алкил» включает C1-4алкильные радикалы и их высшие гомологи, содержащие 5 или 6 атомов углерода, такие как, например, 1-пентил, 2-пентил, 3-пентил, 1-гексил, 2-гексил, 2-метил-1-бутил, 2-метил-1-пентил, 2-этил-1-бутил, 3-метил-2-пентил и т.п. В ряду C1-6алкилов интерес представляет C1-4алкил.
Термин «C2-6алкенил» в определении группы или части группы означает углеводородные радикалы с прямой или разветвленной цепью, содержащей насыщенные углерод-углеродные связи и, по меньшей мере, одну двойную связь, и включающие в себя от 2 до 6 атомов углерода, такие как, например, этенил (или винил), 1-пропенил, 2-пропенил (или аллил), 1-бутенил, 2-бутенил, 3-бутенил, 2-метил-2-пропенил, 2-пентенил, 3-пентенил, 2-гексенил, 3-гексенил, 4-гексенил, 2-метил-2-бутенил, 2-метил-2-пентенил и т.п. В ряду C2-6алкенилов интерес представляет C2-4алкенил.
Термин «C3-7циклоалкил» представляет собой общее название таких радикалов, как циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термин «C1-6алкокси» означает C1-6алкилокси, в котором «C1-6алкил» представляет собой радикал, определенный выше и присоединенный к атому кислорода, т.е. -O-C1-6алкил. В ряду C1-6алкокси интерес представляют метокси, этокси и пропокси.
Термин «галоген» представляет собой общее название таких атомов как фтор, хлор, бром и йод, в частности, фтор или хлор.
Термин «полигалогенC1-6алкил» при определении группы или части группы, например, в группе «полигалогенC1-6алкокси», означает моно- или полигалогензамещенный C1-6алкил, в частности C1-6алкил, замещенный одним, двумя, тремя, четырьмя, пятью, шестью или большим количеством атомов галогена, такой как метил или этил с одним или несколькими атомами фтора, например, дифторметил, трифторметил, трифторэтил. Предпочтительным является трифторметил. Указанный термин включает также перфторC1-6алкильные группы, которые представляют собой C1-6алкильные группы, в которых все атомы водорода замещены атомами фтора, например, пентафторэтил. В случае, когда более одного атома галогена присоединены к алкильной группе, которая определена как полигалогенC1-6алкил, атомы галогена могут быть одинаковыми или разными.
Фрагмент (=О) или оксо, который упоминался выше, образует карбонильную группу, когда присоединен к атому углерода, сульфоксидную группу, когда присоединен к атому серы, сульфонильную группу, когда два указанных фрагмента присоединены к атому серы. Атом углерода, к которому присоединена оксо-группа, является насыщенным атомом углерода независимо от того, цикл или циклическая система замещена оксо-группой.
Радикал Het представляет собой гетероцикл, который определен в данном описании и в формуле изобретения. Примеры Het включают, например, пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазинолил, изотиазинолил, тиазолил, изотиазолил, оксадиазолил, тиадиазолил, триазолил (включая 1,2,3-триазолил, 1,2,4-триазолил), тетразолил, фуранил, тиенил, пиридил, пиримидил, пиридазинил, триазинил и т.п. Среди Het радикалов особый интерес представляют радикалы, которые являются ненасыщенными, в частности, радикалы ароматической природы. Среди Het радикалов особый интерес представляют также радикалы, содержащие один или два атома азота.
Каждый из Het радикалов, упомянутых в описании, может быть необязательно замещенным различным количеством различного вида заместителями, указанными в определениях соединений формулы (I) или любой из подгрупп соединений формулы (I). Некоторые из Het радикалов, упомянутых в описании, могут быть замещены одной, двумя или тремя гидрокси-заместителями. Такие гидрокси-замещенные циклы могут существовать в таутомерных формах, содержащих кето-группы. Например, 3-гидроксипиридазин может существовать в таутомерной форме как 2H-пиридазин-3-он. Когда Het представляет собой пиперазинил, он предпочтительно является замещенным в 4-положении заместителем, присоединенным к атому азота через атом углерода, например, 4-C1-6алкилом, 4-полигалогенC1-6алкилом, C1-6алкоксиC1-6алкил, C1-6алкилкарбонилом, C3-7циклоалкилом.
Представляющие интерес Het радикалы включают, например, пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, оксадиазолил, тиадиазолил, триазолил (включая 1,2,3-триазолил, 1,2,4-триазолил), тетразолил, фуранил, тиенил, пиридил, пиримидил, пиридазинил, пиразолил, триазинил или любой из указанных гетероциклов, конденсированный с бензольным циклом, например, индолил, индазолил (в частности, 1H-индазолил), индолинил, хинолинил, тетрагидрохинолинил (в частности, 1,2,3,4-тетрагидрохинолинил), изохинолинил, тетрагидроизохинолинил (в частности, 1,2,3,4-тетрагидроизохинолинил), хиназолинил, фталазинил, бензимидазолил, бензоксазолил, бензизоксазолил, бензотиазолил, бензоксадиазолил, бензотиадиазолил, бензофуранил, бензотиенил.
Het радикалы пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, 4-замещенный пиперазинил предпочтительно присоединены через включенный в них атом азота (т.e. представляют собой 1-пирролидинил, 1-пиперидинил, 4-тиоморфолинил, 4-морфолинил, 1-пиперазинил, 4-замещенный 1-пиперазинил).
Следует отметить, что положение радикалов на любом фрагменте молекулы, упомянутом в определениях, может быть произвольным, если при этом фрагмент является химически стабильным.
Радикалы, используемые в определениях переменных, включают все возможные изомеры, если не указано иного. Например, термин «пиридил» включает 2-пиридил, 3-пиридил и 4-пиридил; термин «пентил» включает 1-пентил, 2-пентил и 3-пентил.
Когда любая переменная встречается более одного раза на любом фрагменты, каждое определение является независимым.
Предполагается, что термины «соединения формулы (I)», «данные соединения» или аналогичные термины, используемые в описании, включают соединения формулы (I), их N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы. Один вариант осуществления изобретения включает соединения формулы (I) или любую подгруппу соединений формулы (I), упомянутую в описании, а также их фармацевтически приемлемые соли и возможные стереоизомерные формы.
Соединения формулы (I) содержат несколько центров хиральности и существуют в стереохимически изомерных формах. Термин «стереохимически изомерные формы», когда используется в описании, означает все возможные соединения, составленные из одних и тех же атомов, соединенных в такой же последовательности связей, но имеющие разные трехмерные структуры, которые не могут совмещаться при наложении и которыми могут обладать соединения формулы (I).
Что касается примеров, где для обозначения абсолютной конфигурации хирального атома в заместителе используется символ (R) или (S), указанное обозначение учитывает все соединение в целом, а не заместитель в отдельности.
За исключением особо оговоренных или особо выделенных случаев, химическое определение соединения включает смесь всех возможных стереохимически изомерных форм, которыми может обладать указанное соединение. Такая смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Подразумевается, что все стереохимически изомерные формы соединений согласно настоящему изобретению, в том числе чистая форма и смесь каждой одной формы с другой, включены в область настоящего изобретения.
Термин «стереоизомерно чистые формы соединений и промежуточных продуктов», когда упоминается в описании, означает изомеры, по существу свободные от других энантиомерных или диастереомерных форм этой же основной структуры указанных соединений или промежуточных продуктов. В частности, термин «стереоизомерно чистые» относится к соединениям или промежуточным продуктам со стереохимическим избытком в интервале от, по меньшей мере, 80% (т.е. минимум 90% одного и максимум 10% другого из возможных изомеров) и до 100% (т.е. 100% одного изомера и отсутствие другого из возможных изомеров), точнее к соединениям или промежуточным продуктам со стереоизомерным избытком от 90% до 100%, более точно со стереоизомерным избытком от 94% до 100% и наиболее точно со стереомерным избытком от 97% до 100%. Термины «энантиомерно чистые» и «диастереомерно чистые» следует понимать аналогично, но в отношении энантиомерного избытка и дистереомерного избытка рассматриваемой смеси, соответственно.
Чистые стереоизомерные формы соединений и промежуточных продуктов согласно настоящему изобретению могут быть получены в соответствии с методиками, известными в данной области техники. Например, энантиомеры могут разделяться селективной кристаллизацией их диастереомерных солей с оптически активными кислотами или основаниями. Примерами таких кислот являются винная кислота, дибензоилвинная кислота, дитолуолвинная кислота и камфорсульфоновая кислота. Альтернативно, энантиомеры могут разделяться хроматографическими методами с использованием хиральных стационарных фаз. Указанные стереохимически чистые изомерные формы также могут быть получены из соответствующих стереохимически чистых изомерных форм подходящих исходных веществ при условии, что реакция протекает стереоспецифически. Предпочтительно, если необходим специфический стереоизомер, указанное соединение синтезируют стереоспецифическими методами получения. В таких методах будут преимущественно применяться энантиомерно чистые исходные вещества.
Диастереомерные рацематы соединений формулы (I) могут быть получены отдельно подходящими методами. Подходящими методами физического разделения, которые преимущественного могут применяться, являются, например, селективная кристаллизация и хроматография, например, колоночная хроматография.
Для некоторых из соединений формулы (I), их N-оксидов, фармацевтически приемлемых аддитивных солей и сольватов, а также промежуточных продуктов, используемых при их получении, абсолютная стереохимическая конфигурация экспериментально не определялась. Квалифицированный специалист данной области техники может определить абсолютную конфигурацию таких соединений с использованием методов, известных в данной области техники, таких как, например, дифракция рентгеновских лучей.
Подразумевается также, что настоящее изобретение включает все изотопы атомов, которые содержат соединения согласно настоящему изобретению. Изотопы включают атомы с одинаковыми атомными номерами, но разными массовыми числами. В качестве общего примера, но без ограничения, изотопы водорода включают тритий и дейтерий. Изотопы углерода включают С-13 и С-14.
Фармацевтически приемлемые аддитивные соли включают терапевтически активные нетоксичные кислотно- и основно-аддитивные солевые формы соединений формулы (I). Фармацевтически приемлемые кислотно-аддитивные соли могут быть традиционно получены обработкой основной формы такой подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например, хлористоводородную или бромистоводородную кислоту, серную, азотную, фосфорную и т.п. кислоты; или органические кислоты, такие как, например, уксусная, пропановая, гидроксиуксусная, молочная, пирувиновая, щавелевая (т.е. этандионовая), малоновая, янтарная (т.е. бутандионовая кислота), малеиновая, фумаровая, яблочная (т.е. гидроксибутандионовая кислота), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламиновая, салициловая, п-аминосалициловая, памоиновая кислота и т.п. кислоты. И наоборот, указанные солевые формы могут быть превращены в свободное основание обработкой подходящим основанием.
Соединения формулы (I), содержащие кислотный протон, также могут быть превращены в нетоксичные формы солей металлов или амино-аддитивные солевые формы обработкой подходящими органическими и неорганическими основаниями. Подходящие основные солевые формы включают, например, аммониевые соли, соли щелочных и щелочно-земельных металлов, например, лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, например, бензатином, N-метил-D-глюкамином, гидраминные соли и соли с аминокислотами, такими как, например, аргинин, лизин и т.п.
Подразумевается также, что термин «аддитивные соли» включает сольваты, которые способны образовывать соединения формулы (I), а также их соли. Такими сольватами являются, например, гидраты, алкоголяты, например, этаноляты, пропаноляты и т.п.
Подразумевается, что N-оксидные формы соединений согласно настоящему изобретению включают соединения формулы (I), в которых один или несколько атомов азота окислены до, так называемого, N-оксида.
Некоторые из соединений формулы (I) могут существовать в их таутомерной форме. Подразумевается, что такие формы, хотя точно и не указаны в представленной выше формуле, включены в область настоящего изобретения.
Как указано выше, соединения формулы (I) содержат несколько асимметрических центров. Для более точного определения каждого из этих асимметрических центров будет использоваться система нумерации, представленная на приведенной ниже структурной формуле.
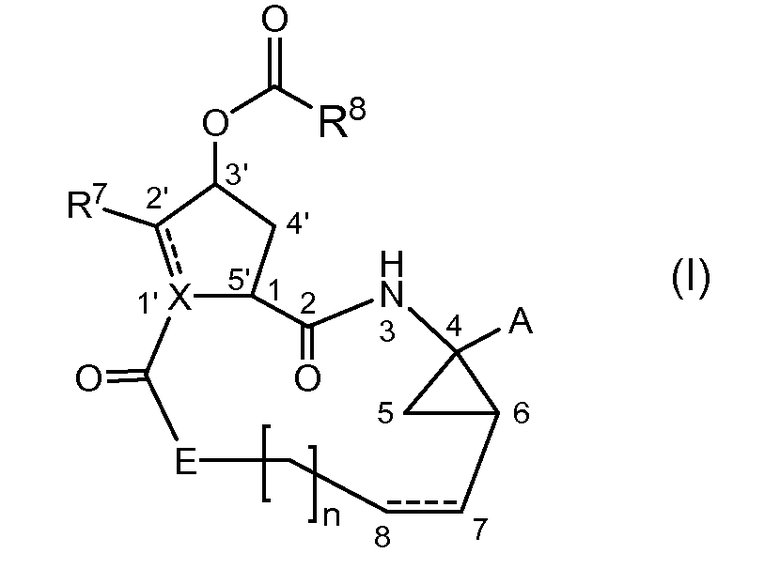
Асимметрические центры находятся в положениях 1, 4 и 6 макроцикла, а также при атоме углерода 3' в 5-членном цикле, при атоме углерода 2', когда заместитель R7 представляет собой С1-6алкил, и при атоме углерода 1', когда Х представляет собой СН. Каждый из этих асимметрических центров может существовать в R или S конфигурации.
Когда Х представляет собой N, стереохимия в положении 1 предпочтительно соответствует конфигурации L-аминокислоты, т.е. L-пролину, как показано ниже.

Когда Х представляет собой СН, 2 карбонильные группы, замещенные в положениях 1' и 5' циклопентанового цикла, предпочтительно находятся в транс-конфигурации. Карбонильный заместитель в положении 5' предпочтительно находится в конфигурации, которая соответствует конфигурации L-пролина. Карбонильные группы, замещенные в положениях 1' и 5', предпочтительно находятся в положениях, которые представлены ниже на структуре следующей формулы:

Соединения формулы (I) включают циклопропильную группу, которая представлена на структурном фрагменте ниже:

где С7 представляет собой углерод в положении 7, и атомы углерода в положении 4 и 6 представляют собой асимметрические атомы углерода в циклопропановом цикле. Наличие этих двух асимметрических центров означает, что соединения могут существовать в виде смесей диастереомеров, таких как диастереомеры соединений формулы (I), где углерод в положении 7 находится в цис-конфигурации по отношению к карбонилу или в цис-конфигурации по отношению к амиду, как показано ниже.

Один вариант осуществления изобретения относится к соединениям формулы I, в которых атом углерода в положении 7 находится в цис-конфигурации относительно карбонила. Другой вариант осуществления изобретения относится к соединениям формулы (I), в которых конфигурация при атоме углерода в положении 4 является R-конфигурацией. Конкретной подгруппой соединений формулы (I) является подгруппа соединений, в которых атом углерода в положении 7 находится в цис-конфигурации по отношению к карбонилу и конфигурация при атоме углерода в положении 4 является R-конфигурацией.
В соответствии с еще одним вариантом осуществления изобретения циклопропильная группа (С4-С5-С6) присоединена к группе А, которая представляет собой фосфонатную группу -Р(=О)(OR4a)(R4b). Согласно данному варианту, атом углерода в положении 7 находится в цис-конфигурации относительно фосфонатной группы или относительно амидной группы, как показано на структурном фрагменте ниже:

Еще один вариант осуществления изобретения относится к соединениям формулы (I), в которых атом углерода в положении 7 находится в цис-конфигурации относительно фосфонатной группы. Другой вариант осуществления изобретения относится к соединениям формулы (I), в которых конфигурация при атоме углерода в положении 4 является S-конфигурацией. Конкретной подгруппой соединений формулы (I) является подгруппа соединений, в которых атом углерода в положении 7 находится в цис-конфигурации по отношению к фосфонатной группе и конфигурация при атоме углерода в положении 4 является S-конфигурацией.
Соединения формулы (I) могут включать пролиновый остаток (т.е. Х представляет собой N), циклопентильный или циклопентенильный остаток (т.е. Х представляет собой СН или С, соответственно). В соответствии с одним вариантом осуществления данного изобретения соединения включают следующие структурные фрагменты:

Другие варианты осуществления изобретения представляют собой соединения формулы (I) или любые подгруппы соединений формулы (I), где R7 представляет собой метил, Е представляет собой NR5, X представляет собой С, соединенный через двойную связь с атомом углерода, несущим R7.
Предпочтительными являются соединения формулы (I), в которых заместитель в положении 1 (или 5') и пиримидиновый заместитель в положении 3', присоединенный через эфирную связь, находятся в транс-конфигурации. Особый интерес представляют соединения формулы (I), в которых положение 1 имеет конфигурацию, соответствующую L-пролину, и пиримидиновый заместитель в положении 3', присоединенный через эфирную связь, находится в транс-конфигурации относительно положения 1.
Предпочтительно соединения формулы (I) имеют стереохимию, которая показана в структурах формул (I-a) и (I-b) ниже:

Один вариант осуществления настоящего изобретения относится к соединениям формулы (I) или формул (I-a), (I-b), или любой подгруппы соединений формулы (I), в которых выполняется одно или несколько из следующих условий:
(a) R7 представляет собой водород;
(b) Х представляет собой азот;
(с) Е представляет собой NR5;
(d) двойная связь находится между 7 и 8 атомами углерода.
Еще один вариант осуществления настоящего изобретения относится к соединениям формулы (I), формул (I-a), (I-b) или любой подгруппы соединений формулы (I), в которых выполняется одно или несколько из следующих условий:
(a) R7 представляет собой водород;
(b) Х представляет собой азот;
(с) Е представляет собой CR6aR6b;
(d) двойная связь находится между 7 и 8 атомами углерода.
Еще один вариант осуществления настоящего изобретения относится к соединениям формулы (I) или формул (I-a), (I-b), или любой подгруппы соединений формулы (I), в которых выполняется одно или несколько из следующих условий:
(a) R7 представляет собой водород;
(b) Х представляет СН;
(с) Е представляет собой NR5, где R5 принимает значения, определенные выше, в частности R5 представляет собой водород или C1-6алкил;
(d) двойная связь находится между 7 и 8 атомами углерода.
Конкретными подгруппами соединений формулы (I) являются подгруппы соединений, представленные структурными формулами (I-c), (I-d) и (I-e) ниже:

Среди соединений формул (I-c), (I-d) и (I-e) соединения со стереохимической конфигурацией, показанной в формулах (I-a) и (I-b), соответственно, представляют особый интерес.
Двойная связь между атомами 7 и 8 в соединениях формулы (I) или в любой подгруппе соединений формулы (I) может иметь цис- или транс-конфигурацию. Предпочтительно, двойная связь между атомами углерода 7 и 8 имеет цис-конфигурацию, как показано в формулах (I-c), (I-d) и (I-е).
Другие конкретные подгруппы соединений формулы (I) включают соединения, представленные следующими структурными формулами:

Особый интерес среди соединений формул (I-f), (I-g) или (I-h) представляют соединения со стереохимической конфигурацией соединений формул (I-a) и (I-b).
В соединениях формул (I-a), (I-b), (I-c), (I-d), (I-e), (I-f), (I-g) или (I-h), когда это применимо, A, E, X, n, R5, R7, R8 и R9 принимают значения, указанные в определениях соединений формулы (I) или любой из подгрупп соединений формулы (I), определенных в описании.
Следует представлять, что описанные выше подгруппы соединений формулы (I-a), (I-b), (I-c), (I-d) или (I-e), а также любая другая подгруппа, определенная в описании, включает также любые N-оксиды, аддитивные соли и стереохимически изомерные формы таких соединений.
Когда в соединениях формулы (I) или в любой подгруппе соединений формулы (I) n равно 2, фрагмент -СН2-, заключенный в скобки с индексом “n”, соответствует этандиилу. Когда в соединениях формулы (I) или в любой подгруппе соединений формулы (I) n равно 3, фрагмент -СН2-, заключенный в скобки с индексом “n”, соответствует пропандиилу. Когда в соединениях формулы (I) или в любой подгруппе соединений формулы (I) n равно 4, фрагмент -СН2-, заключенный в скобки с индектом “n”, соответствует бутандиилу. Когда в соединениях формулы (I) или в любой подгруппе соединений формулы (I) n равно 5, фрагмент -СН2-, заключенный в скобки с индектом “n”, соответствует пентандиилу. Когда в соединениях формулы (I) или в любой подгруппе соединений формулы (I) n равно 6, фрагмент -СН2-, заключенный в скобки с индексом “n”, соответствует гександиилу. Особый интерес представляет подгруппа соединений формулы (I), в которых n равно 4 или 5.
Вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), в которых А представляет собой -С(=О)OR1, в частности, где R1 представляет собой С1-6алкил, такой как метил, этил или трет-бутил, наиболее предпочтительно, где R1 представляет собой водород.
Еще одним вариантом осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), в которых А представляет собой -С(=О)-NH-SO2-R2, в частности, где R2 представляет собой С3-7циклоалкил, фенил или группу Het, например, тиазолил или пиридил, который необязательно замещен одним или несколькими заместителями, такими как один или два заместителя, выбранные из С1-6алкила, С1-6алкокси, трифторметила и галогена, или, в частности, один или два заместителя, выбранные из метила, фтора и хлора. Например, R2 может представлять собой 1-метилциклопропил.
Еще одним вариантом осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), где А представляет собой -С(=О)С(=О)NR3aR3b, в частности, где R3a и R3b независимо выбраны из водорода, С1-6алкила, необязательно замещенного арилом, и С2-6алкенила. В одном варианте осуществления изобретения один из R3a и R3b представляет собой водород, а другой представляет собой 3-пропенил, циклопропилметил или циклопропил. В еще одном варианте осуществления изобретения R3a и R3b оба представляют собой водород.
Еще одним вариантом осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), в которых А представляет собой -С(=О)-NH-P(=О)(OR4a)(R4b), в частности, где R4a представляет собой С1-6алкил, в особенности этил или изопропил, R4b представляет собой OR4b', и R4b' представляет собой С1-6алкил, такой как этил или изопропил.
Еще одним вариантом осуществления изобретения являются соединения формулы (I), в которых А представляет собой -P(=О)(OR4a)(R4b), в частности, где R4a представляет собой С1-6алкил, в особенности этил или изопропил, R4b представляет собой OR4b' и R4b' представляет собой С1-6алкил, в особенности этил или изопропил.
Еще одним вариантом осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), где
(a) R5 представляет собой водород; С1-6алкил; С1-6алкоксиС1-6алкил; или С3-7циклоалкил;
(b) R5 представляет собой водород или С1-6алкил;
(с) R5 представляет собой водород.
Предпочтительными вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), где R5 представляет собой водород или С1-6алкил, более предпочтительно водород или метил.
Еще один вариант осуществления изобретения относится к соединениям формулы (I), (I-e) или любой подгруппе соединений формулы (I), где R6a и R6b независимо представляют собой водород или С1-6алкил, например, метил. Предпочтительно R6a представляет собой водород, и R6b представляет собой метил, или, более предпочтительно, R6a и R6b оба представляют собой водород.
Вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), где
(а) R8 представляет собой радикал формулы

(b) R8 представляет собой радикал формулы
(с) R8 представляет собой радикал формулы

Вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), где
(d) R8 представляет собой радикал формулы

(e) R8 представляет собой радикал формулы

(f) R8 представляет собой радикал формулы

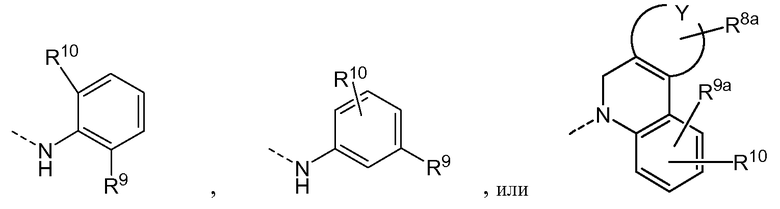
Вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), в которых R9 и R10 или один из R9 и R10 принимают(ет) следующие значения:
R9 представляет собой С1-6алкил (например, метил, этил или изопропил); С1-6алкокси (например, метокси, этокси или изопропокси); арилокси; Het-O-; циано; или R9 представляет собой С1-6алкокси (например, метокси, этокси или изопропокси) или арилокси (например, фенокси или 4-метоксифенокси);
R10 представляет собой водород; С1-6алкил (например, метил, этил или изопропил); С1-6алкокси (например, метокси, этокси или изопропокси); циано.
В абзаце выше арил и Het принимают значения, описанные выше или далее, в частности, арил представляет собой фенил, необязательно замещенный С1-6алкокси (например, метокси, этокси или изопропокси), точнее, 4-замещенный фенил; и Het, в частности, представляет собой пиридил или пиримидинил.
Предпочтительными вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), в которых один из R9 представляет собой водород, метокси или циано.
Предпочтительными вариантами изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), где R9 представляет собой циано, С1-6алкилоксикарбонил, моно- и ди(С1-6алкиламино), галоген, амино, С1-6алкокси, арилокси, С1-6алкил, Het; или где R9 представляет собой циано, С1-6алкилоксикарбонил (например, метоксикарбонил), моно(С1-6алкиламино) (например, метиламино), галоген (например, хлор), С1-6алкокси (например, метокси), фенокси, С1-6алкил (например, метил), тиазолил, необязательно замещенный С1-6алкилом (например, 2-метил-4-тиазолил).
Предпочтительными вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), в которых R10 представляет собой водород, С1-6алкокси (например, метокси) или галоген.
Предпочтительными вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), в которых один из R9 и R10 представляет собой галоген (в частности фтор) или трифторметил. Другими предпочтительными вариантами являются соединения, в которых R9 представляет собой галоген (в частности фтор) или трифторметил, и R10 представляет собой водород.
Предпочтительными вариантами изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), в которых R8 представляет собой радикал формулы

где R9 представляет собой циано или метил и R10 представляет собой водород или метокси; или где R9 представляет собой циано или метокси и R10 представляет собой водород.
Другими предпочтительными вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), где R8 представляет собой радикал формулы
 , или формулы
, или формулы  или формулы
или формулы

где выполняется любое или оба из следующих условий (а) и (b):
(a) R9 представляет собой циано, С1-6алкилоксикарбонил, моно- и ди(С1-6алкиламино), галоген, амино, С1-6алкокси, арилокси, С1-6алкил, Het; или где R9 представляет собой циано, С1-6алкилоксикарбонил (например, метоксикарбонил), моно(С1-6алкиламино) (например, метиламино), галоген (например, хлор), С1-6алкокси (например, метокси), фенокси, С1-6алкил (например, метил), тиазолил, необязательно замещенный С1-6алкилом (например, 2-метил-4-тиазолил);
(b) R10 представляет собой водород, С1-6алкокси (например, метокси) или галоген.
Вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), где группа  представляет собой группу
представляет собой группу
 , которая имеет следующую структуру:
, которая имеет следующую структуру:

Вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), где группа  принимает значения, определенные в предыдущем абзаце, и R8a, R9a и R10 или один из R8a, R9a и R10, в частности, принимают следующие значения:
принимает значения, определенные в предыдущем абзаце, и R8a, R9a и R10 или один из R8a, R9a и R10, в частности, принимают следующие значения:
R8a и R9a независимо представляют собой водород, С1-6алкил (например, метил, этил или изопропил); С1-6алкокси (например, метокси, этокси или изопропокси); арилокси; Het-O; циано; или R10 представляет собой водород, С1-6алкокси (напрмер, метокси, этокси или изопропокси) или арилокси (например, фенокси или 4-метоксифенокси);
R8a и R9a независимо представляют собой водород; С1-6алкил (например, метил, этил или изопропил); С1-6алкокси (например, метокси, этокси или изопропокси); циано; или R10 представляет собой водород, С1-6алкокси (например, метокси, этокси или изопропокси).
Вариантами осуществления изобретения являются соединения формулы (I) или любой подгруппы соединений формулы (I), в которых R8 представляет собой водород.
В предыдущих абзацах арил и Het принимают значения, определенные ранее, в частности, арил представляет собой фенил, необязательно замещенный С1-6алкокси (например, метокси, этокси или изопропокси), точнее, 4-замещенный фенил; и Het, в частности, представляет собой пиридил или пиримидинил.
Предпочтительными вариантами осуществления изобретения являются соединения формулы (I) или любой из подгрупп соединений формулы (I), в которых один из R9 представляет собой водород, метокси или циано.
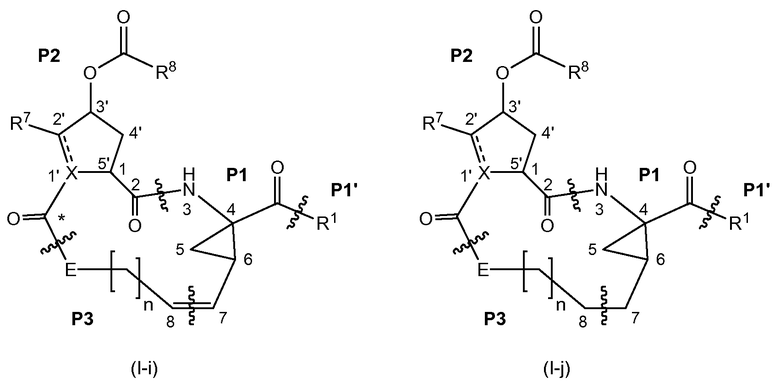
Соединения формулы (I) состоят из трех структурообразующих блоков Р1, Р2, Р3. Структурообразующий блок Р1 дополнительно содержит концевую часть («хвост») Р1'. Карбонильная группа, помеченная звездочкой в соединении (I-i), представленном ниже, может быть частью структурообразующего блока Р2 или структурообразующего блока Р3. С химической точки зрения понятно, что структурообразующий блок Р2 соединений формулы (I), где Х представляет собой С, включает карбонильную группу, присоединенную в положении 1'.
Связывание структурных блоков Р1 с Р2, Р2 с Р3 и Р1 с Р1' (когда R1 представляет собой -NH-SO2R2) происходит с образованием амидной связи. Связывание блоков Р1 и Р3 происходит с образованием двойной связи. Связывание структурообразующих блоков Р1, Р2 и Р3 для получения соединений (I-i) или (I-j) может осуществляться в любой последовательности. Одна из стадий включает циклизацию, в результате которой образуется макроцикл.
Ниже показаны соединения (I-i), представляющие собой соединения формулы (I), в которых атомы углерода С7 и С8 соединены двойной связью, и соединения (I-j), представляющие собой соединения формулы (I), в которых атомы углерода С7 и С8 соединены одинарной связью. Соединения формулы (I-j) могут быть получены из соответствующих соединений формулы (I-i) восстановлением двойной связи в макроцикле.
Подразумевается, что методики синтеза, описанные далее, применимы для рацематов, стереохимически чистых промежуточных или конечных продуктов или любых стереоизомерных смесей. Рацематы или стереохимические смеси могут разделяться на стереоизомерные формы на любой стадии методик синтеза. В одном варианте осуществления изобретения промежуточные продукты и конечные продукты имеют стереохимию, определенную выше в соединениях формулы (I-a) или (I-b).
В приведенном далее описании R11 представляет собой радикал

В одном варианте осуществления изобретения соединения (I-i) получают посредством образования амидных связей с последующим образованием мостика двойной связи между Р3 и Р1 с одновременной циклизацией до макроцикла.
В еще одном варианте осуществления изобретения соединения (I), в которых связь между С7 и С8 является двойной связи, то есть соединения формулы (I-i), определенные выше, могут быть получены в соответствии со следующей схемой синтеза:
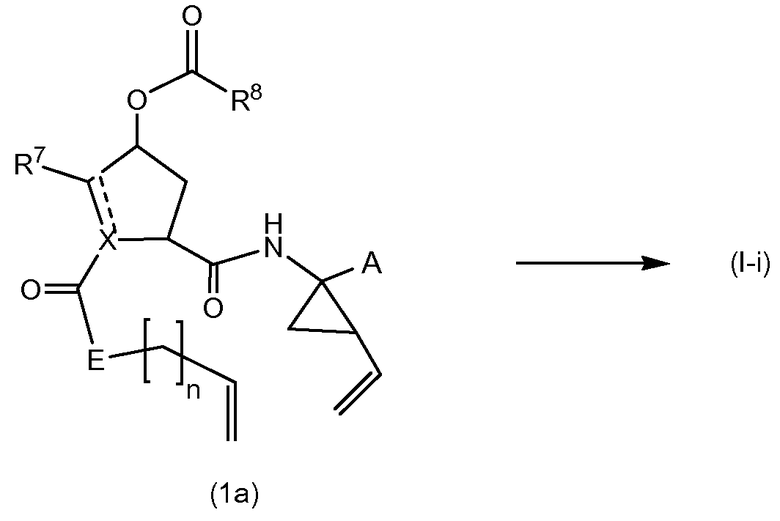
Образование макроцикла может осуществляться через реакцию обмена олефинов в присутствии подходящего металлического катализатора, такого как катализатор на основе рутения, описанный в публикациях Miller, S.J., Blackwell, H.E., Grubbs, R.H. J. Am. Chem. Soc. 118, (1996), 9606-9614; Kingsbury, J. S., Harrity, J. P. A., Bonitatebus, P. J., Hoveyda, A. H., J. Am. Chem. Soc. 121, (1999), 791-799; и Huang et al., J. Am. Chem. Soc. 121, (1999), 2674-2678; например, катализатора Говейды-Граббса (Hoveyda-Grubbs).
Могут применяться стабильные на воздухе катализаторы, такие как хлорид бис(трициклогексилфосфин)-3-фенил-1Н-инден-1-илиденрутения (Neolyst M1®) или дихлорид бис(трициклогексилфосфин)[(фенилтио)метилен]рутения (IV). Другими катализаторами, которые могут применяться, являются катализаторы Граббса первого и второго поколений, т.е. бензилиден-бис(трициклогексилфосфин)дихлоррутений и (1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(фенилметилен)(трициклогексилфосфин)рутений, соответственно. Особый интерес представляют катализаторы Говейды-Граббса первого и второго поколений, которые представляют собой дихлор(о-изопропоксифенилметилен)(трициклогексилфосфин)рутений(II) и 1,3-бис-(2,4-триметилфенил)-2-имидазолидинилиден)дихлор(о-изопропоксифенилметилен)рутений, соответственно. В этой реакции могут также применяться и другие катализаторы, содержащие другие переходные металлы, такие как Мо.
Реакции обмена могут проводиться в подходящем растворителе, таком как, например, простые эфиры, в частности, ТГФ, диоксан; галогенированные углеводороды, например, дихлорметан, CHCl3, 1,2-дихлорэтан и т.п., углеводороды, например, толуол. В предпочтительном варианте осуществления изобретения реакция обмена проводится в толуоле. Такие реакции проводятся при повышенных температурах в атмосфере азота.
Соединения формулы (I), в которых связь между С7 и С8 в макроцикле представляет собой одинарную связь, т.е. соединения формулы (I-j), могут быть получены из соединений формулы (I-i) восстановлением С7-С8 двойной связи в соединениях формулы (I-i). Такое восстановление может осуществляться посредством каталитического гидрирования водородом в присутствии катализатора благородного металла, такого как, например, Pt, Pd, Rh, Ru или никель Ренея. Особый интерес представляет Ph на алюминии. Реакция гидрирования предпочтительно проводится в растворителе, таком как, например, спирт, в частности, метанол, этанол, или простой эфир, такой как ТГФ, или в их смеси. К этим растворителям или смесям растворителей может добавляться вода.
Группа А может присоединяться к структурообразующему блоку Р1 на любой стадии синтеза, т.е. до или после циклизации либо до или после циклизации с восстановлением, как описано выше. Соединения формулы (I), в которых А представляет собой -CO-NHSO2R2, т.е. соединения формулы (I-k-1), могут быть получены присоединением группы А к Р1 посредством образования амидной связи между этими двумя фрагментами. Аналогично, соединения формулы (I), в которых R1 представляет собой -C(=O)OR1, т.е. соединения формулы (I-k-2), могут быть получены присоединением группы R1 к Р1 посредством образования сложноэфирной связи. В одном варианте осуществления изобретения группы -C(=O)OR1, вводятся на последней стадии синтеза соединений (I), как показано на представленной ниже схеме синтеза, где G представляет собой группу

Промежуточный продукт (2а) может сочетаться с сульфонамидом (2b) посредством реакции образования амида, например в соответствии с любой из методик образования амидной связи, описанных ниже. В частности, (2а) может обрабатываться агентом сочетания, например, N,N'-карбонилдиимидазолом (CDI), EEDQ, IIDO, EDCI или бензотриазол-1-илокси-трис-пирролидинофосфонием гексафторфосфатом (коммерчески доступным как PyBOP®), в растворителе, таком как простой эфир, например, ТГФ, или галогенированный углеводород, например, дихлорметан, хлороформ, дихлорэтан, и подвергаться взаимодействию с целевым сульфонамидом (2b), предпочтительно после взаимодействия (2а) с агентом сочетания. Взаимодействия (2а) с (2b) предпочтительно проводятся в присутствии основания, например, триалкиламина, такого как триэтиламин или диизопропилэтиламин, или 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU). Промежуточное соединение (2а) также может превращаться в активированную форму, например, активированную форму общей формулы G-CO-Z, где Z представляет собой галоген или остаток активного сложного эфира, например, Z представляет собой арилокси-группу, такую как фенокси, п-нитрофенокси, пентафторфенокси, трихлорфенокси, пентахлорфенокси и т.п.; или Z может представлять собой остаток смешанного ангидрида. В одном варианте осуществления изобретения G-CO-Z представляет собой хлорангидрид (G-CO-Cl) или смешанный ангидрид кислоты (G-CO-O-CO-R или G-CO-O-CO-OR, R в последней формуле представляет собой С1-4алкил, такой как метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил или бензил). Активированная форма G-CO-Z подвергается взаимодействию с сульфонамидом (2b).
Соединения формулы (I), в которых A представляет собой -C(=O)-NH-P(=O)(OR4a)(R4b), то есть соединения формулы (I-k-3), могут быть получены посредством образования амидной связи между промежуточным продуктом (2а) и фосфорамидатом (2d) в соответствии с методиками образования амидной связи, описанным далее. В частности, (2а) может подвергаться обработке агентом сочетания в подходящем растворителе с последующим взаимодействием с фосфорамидатом (2d) предпочтительно в присутствии основания, такого как гидрид натрия, предпочтительно после взаимодействия (2а) с агентом сочетания. Промежуточное соединение (2а) также может подвергаться превращению в активированную форму, например, активированную форму общей формулы G-CO-Z, где Z представляет собой галоген или остаток активного сложного эфира, например, Z представляет собой арилоксигруппу, такую как фенокси, п-нитрофенокси, пентафторфенокси, трихлорфенокси, пентахлорфенокси и т.п.; или Z может представлять собой остаток смешанного ангидрида. В одном варианте осуществления изобретения G-CO-Z представляет собой хлорангидрид (G-CO-Cl) или смешанный ангидрид кислоты (G-CO-O-CO-R или G-CO-O-CO-OR, где R представляет собой C1-4алкил, такой как метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил, или бензил). Активированная форма G-CO-Z подвергается взаимодействию с целевым соединением (2b). Агент сочетания, растворитель и основание могут быть такими, как описано далее в общем описании получения амидных связей.

Активация карбоновой кислоты в (2а), как описано в реакциях, представленных выше, может приводить к промежуточной реакции циклизации с получением промежуточного азалактона формулы

где Х, E, R7, R11 и n принимают значения, описанные выше, и где стереогенные центры могут иметь стереохимическую конфигурацию, которая описана выше, например, как в (I-a) или (I-b). Промежуточные соединения (2а-1) могут быть выделены из реакционной смеси с использованием традиционных методик, и выделенное промежуточное соединение (2а-1) затем подвергается взаимодействию с (2b), или реакционная смесь, содержащая (2а-1) может подвергаться взаимодействию с (2b) или (2d) без выделения (2а-1). В одном варианте осуществления изобретения, где взаимодействие с агентом сочетания проводится в не смешивающимся с водой растворителе, реакционная смесь, содержащая (2а-1), может промываться водой или слабо подщелоченной водой для удаления всех не растворимых в воде побочных продуктов. Полученный таким образом промывной раствор может затем подвергаться взаимодействию с (2b) или (2d) без дополнительных стадий очистки. С другой стороны, выделение промежуточных соединений (2а-1) может обеспечивать определенные преимущества в том, что выделенный продукт после необязательной дополнительной очистки может подвергаться взаимодействию с (2b) или (2d), что приводит к уменьшению образования побочных продуктов и облегчает проведение реакции.
Промежуточный продукт (2а) может сочетаться со спиртом (2с) посредством реакции образования сложного эфира. Например, (2а) и (2с) подвергаются взаимодействию с удалением воды, которое осуществляется либо физически, например, азеотропным удалением воды, либо химически с использованием дегидратирующего агента. Промежуточное соединение (2а) может также превращаться в активированную форму G-CO-Z, такую как активированные формы, упомянутые выше, и далее взаимодействовать со спиртом (2с). Реакции образования сложного эфира предпочтительно проводятся в присутствии основания, такого как карбонат или гидрокарбонат щелочного металла, например, гидрокарбонат натрия или калия, или третичный амин, такой как амины, упомянутые в описании реакций образования амида, в частности, триалкиламин, например, триэтиламин. Растворители, которые могут использоваться в реакциях образования сложного эфира, включают простые эфиры, такие как ТГФ; галогенированные углеродороды, такие как дихлорметан, CH2Cl2; углеводороды, такие как толуол; полярные апротонные растворители, такие как ДМФА, ДМСО, ДМА; и т.п. растворители.
Соединения формулы (I), в которых Е представляет собой NH, то есть соединения формулы (I-l), также могут быть получены удалением группы PG, которая является защитной группой азота, из соответствующего промежуточного соединения (3а) в соответствии с представленной ниже схемой реакции. Защитная группа PG, в частности, представляет собой любую защитную группу атома азота из упомянутых далее и может удаляться с использованием методик, также описанных ниже:
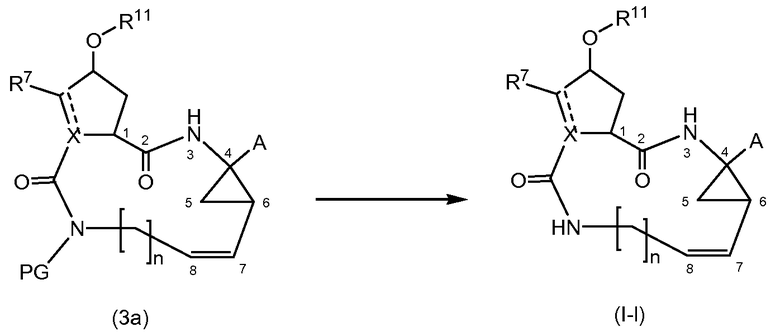
Исходные вещества (3а) в представленной выше реакции могут быть получены в соответствии с методиками получения соединений формулы (I), но с использованием промежуточных соединений, в которых группа R5 представляет собой защитную группу атома азота PG, как определено выше.
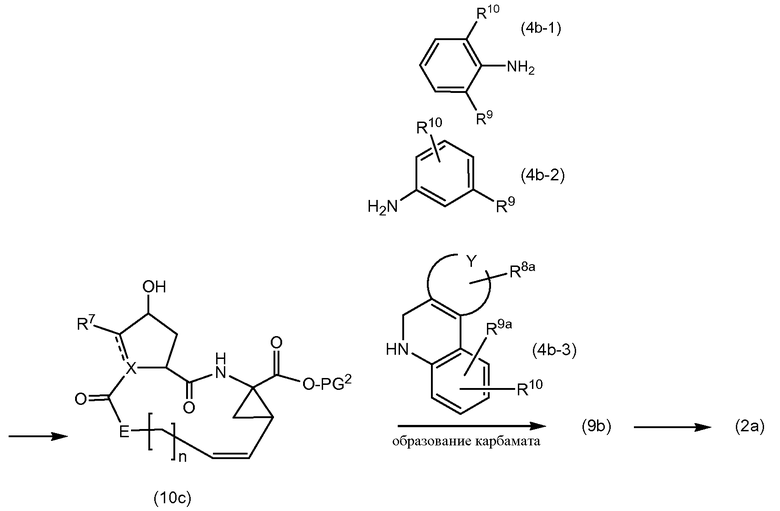
Соединения формулы (I) могут быть получены взаимодействием промежуточного соединения (4а) с амином (4b-1), (4b-2) или (4b-3) в присутствии карбамат-образующего реагента, как показано в представленной далее схеме синтеза, где различные радикалы принимают значения, определенные выше:

Взаимодействие промежуточных продуктов (4а) с карбамат-образующим реагентом проводится в таких же растворителях и основаниях, которые используются для образования амидной связи, как описано далее.
Реакции образования карбамата могут проводиться с использованием различных методов, в частности, взаимодействием аминов с алкилхлорформиатами; взаимодействием спиртов с карбамоилхлоридами или изоцианатами; посредством реакций, включающих комплексы металлов или реагенты переноса ацильной группы (см., например, Greene, T. W. и Wuts, P. G. M., "Protective Groups in Organic Synthesis"; 1999; Wiley и Sons, p. 309-348). Для синтеза карбаматов из некоторых исходных веществ, в том числе аминов, может использоваться моноксид углерода и некоторые металлические катализаторы. В качестве катализаторов могут использоваться такие металлы как палладий, иридий, уран и платина. Могут использоваться также способы синтеза карбаматов с применением диоксида углерода, описанные в литературе (см., например, Yoshida, Y., et al., Bull. Chem. Soc. Japan 1989, 62, 1534; Aresta, M., et al., Tetrahedron, 1991, 47, 9489).
Один подход к получению карбаматов включает применение промежуточных соединений

где Q представляет собой удаляемую группу, такую как галоген, в частности, хлор и бром, или группу, используемую в активных сложных эфирах для образования амидной связи, таких как упомянутые выше, например, фенокси или замещенная фенокси-группа, такая как п-хлор- или п-нитрофенокси, трихлорфенокси, пентахлорфенокси, N-гидроксисукцинимидил и т.п. Промежуточные соединения (4d) могут быть получены из спиртов (4а) и фосгена с получением, таким образом, хлорформиата или перемещением хлора из последнего промежуточного соединения в промежуточные соединения (5а), т.е. промежуточные соединения формулы (4d), в которых Q представляет собой Q1. В данной методике и методиках последующих реакций Q1 представляет собой любой из активных фрагментов сложных эфиров, таких как фрагменты, упомянутые выше. Промежуточные продукты (4d) подвергаются взаимодействию с аминами (4b-1), (4b-2) или (4b-3) с получением таким образом соединений формулы (I).
Промежуточные соединения (4е), которые представляют собой промежуточные соединения (4d), в которых Q представляет собой Q1, также могут быть получены взаимодействием спирта (4а) с карбонатами Q1-CO-Q1, такими как, например, бисфенол, бис-(замещенный фенол) или бис-N-гидроксисукцинимидилкарбонаты:

Представленные выше реакции получения промежуточных соединений (4d) могут проводиться в присутствии оснований и растворителей, упомянутых выше для синтеза амидных связей, в частности, триэтиламина в качестве основания и дихлорметана в качестве растворителя.
Альтернативно, для получения соединения формулы (I) сначала проводится реакция образования амидной связи между структурообразующими блоками Р2 и Р1 с последующим сочетанием структурообразующего блока Р3 с фрагментом Р1 в Р1-Р2 и затем реакция образования карбаматной или эфирной связи между фрагментами Р3 и Р2 в Р2-Р1-Р3 с одновременным закрытием цикла.
Еще одна альтернативная методика синтеза заключается в образовании амидной связи между структурообразующими блоками Р2 и Р3 с последующим сочетанием структурообразующего блока Р1 с фрагментом Р3 в Р3-Р2 и далее с образованием амидной связи между Р1 и Р2 в Р1-Р3-Р2 с одновременным закрытием цикла.
Структурообразующие блоки Р1 и Р3 могут связываться в последовательности Р1-Р3. Если необходимо, двойная связь, связывающая Р1 и Р3, может подвергаться восстановлению. Полученная таким образом последовательность Р1-Р3, восстановленная или нет, может сочетаться со структурообразующим блоком Р2 с образованием таким образом последовательности Р1-Р3-Р2, циклизуемой далее посредством образования амидной связи.
Структурообразующие блоки Р1 и Р3 в любом из описанных выше подходов могут сочетаться посредством образования двойной связи, например, посредством обменной реакции олефинов, описанной далее, или с помощью реакции Виттига. Если необходимо, полученная таким образом двойная связь может подвергаться восстановлению способом, аналогичным описанному выше для превращения (I-i) в (I-j). Двойная связь также может подвергаться восстановлению на более поздней стадии, т.е. после присоединения третьего структурообразующего блока или после образования макроцикла. Структурообразующие блоки Р2 и Р1 связываются посредством образования амидной связи, и Р3 и Р2 связываются посредством образования группы карбамата или сложного эфира.
Остаток Р1' может присоединяться к структурообразующему блоку Р1 на любой стадии синтеза соединений формулы (I), например, до или после соединения структурообразующих блоков Р2 и Р1; до или после присоединения структурообразующего блока Р3 к Р1; или до или после закрытия цикла.
Вначале могут быть получены отдельные структурообразующие блоки, которые затем могут сочетаться вместе, или, альтернативно, предшественники структурообразующих блоков могут сначала сочетаться вместе и затем модифицироваться на последней стадии с получением молекулы желательной структуры.
Функциональные группы в каждом из структурообразующих блоков могут защищаться для предупреждения побочных реакции.
Образование амидных связей может проводиться с использованием стандартных методик, таких как методики, используемые для сочетания аминокислот в синтезе пептидов. Последний включает дегидративное сочетание карбоксильной группы одного реагента с аминогруппой другого реагента с образованием связующей амидной связи. Образование амидной связи может проводиться посредством взаимодействия исходных веществ в присутствии агента сочетания или посредством превращения карбоксильной функциональности в активную форму, такую как активный сложный эфир, смешанный ангидрид либо хлорангидрид или бромангидрид карбоновой кислоты. Общее описание таких реакций сочетания и реагентов, используемых в них, можно найти в учебниках по химии пептидов, например, в публикации M. Bodanszky, “Peptide Chemistry”, 2nd rev. ed., Springer-Verlag, Berlin, Germany, (1993).
Примеры реакций сочетания с образованием амидной связи включают азидный способ, способ смешанного ангидрида карбоновой кислоты (изобутилхлорформиат), карбодиимидный способ (с использованием дициклогексилкарбодиимида, диизопропилкарбодиимида или растворимого в воде карбодиимида, такого как N-этил-N'-[(3-диметиламино)пропил]карбодиимид), способ активного сложного эфира (например, п-нитрофенилового, п-хлорфенилового, трихлорфенилового, пентахлорфенилового, пентафторфенилового, N-гидроксисукцинимидо и т.п. сложных эфиров), способ реагента-К Вудворда, способ 1,1-карбонилдиимидазола (CDI или N,N'-карбонилдиимидазола), способы с применением фосфор-содержащих реагентов или окисления-восстановления. Некоторые из этих способов могут усовершенствоваться добавлением подходящих катализаторов, например, карбодиимидный метод может улучшаться добавлением 1-гидроксибензотриазола, DBU (1,8-диазабицикло[5.4.0]ундец-7-ен) или 4-DMAP. Дополнительными агентами сочетания являются гексафторфосфат (бензотриазол-1-илокси)трис-(диметиламино)фосфония (сам по себе, либо в присутствии 1-гидроксибензотриазола или 4-DMAP); или тетрафторборат 2-(1Н-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония или гексаторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония. Эти реакции сочетания могут проводиться в растворе (в жидкой фазе) или в твердой фазе.
Предпочтительное образование амидной связи проводится с применением N-этилоксикарбонил-2-этилокси-1,2-дигидрохинолина (EEDQ) или N-изобутилоксикарбонил-2-изобутилокси-1,2-дигидрохинолина (IIDQ). В отличие от классической методики с применением ангидрида, при применении EEDQ и IIDQ не требуется ни основания, ни низких температур реакции. Обычно методика включает взаимодействие эквимолярных количеств карбоксильного и аминного компонентов в органическом растворителе (могут использоваться самые разные растворители). Затем в избытке добавляется EEDQ или IIDQ и смесь выдерживают при комнатной температуре с перемешиванием.
Реакции сочетания предпочтительно проводятся в инертном растворителе, таком как галогенированнные углеводороды, например, дихлорметан, хлороформ, диполярные апротонные растворители, такие как ацетонитрил, диметилформамид, диметилацетамид, ДМСО, НМРТ, простые эфиры, такие как тетрагидрофуран (ТГФ).
Во многих примерах реакции сочетания проводятся в присутствии подходящего основания, такого как третичный амин, например, триэтиламин, диизопропилэтиламин (DIPEA), N-метилморфолин, N-метилпирролидин, 4-DMAP или 1,8-диазобицикло[5.4.0]ундец-7-ен (DBU). Температура реакции может находится в интервале от 0°С до 50°С, и время реакции может составлять от 15 минут до 24 часов.
Функциональные группы в структурообразующих блоках, которые связаны вместе, могут защищаться для предупреждения образования нежелательных связей. Подходящие защитные группы, которые могут использоваться, приведены, например, в публикациях Greene, «Protective Groups in Organic Chemistry”, John Wiley & Sons, New York (1999) и «The Peptides: Analysis, Synthesis, Biology”, Vol. 3, Academic Press, New York (1987).
Карбоксильные группы могут защищаться с получением группы сложного эфира, которая может расщепляться с получением группы карбоновой кислоты. Защитные группы, которые могут применяться, включают 1) группы сложных алкиловых эфиров, таких как метиловый, триметилсилиловый или трет-бутиловый; 2) группы сложных арилалкиловых эфиров, таких как бензиловый и замещенный бензиловый; или 3) группы сложных эфиров, которые могут расщепляться под действием слабого основания или слабого восстановителя, такие как группы сложных трихлорэтилового и фенацилового эфиров.
Аминогруппы могут защищаться с помощью различных N-защищающих групп, таких как:
1) ацильные группы, такие как формильная, трифторацетильная, фталильная и п-толуолсульфонильная;
2) ароматические карбаматные группы, такие как бензилоксикарбонильная (Cbz или Z) группа, замещенные бензиоксикарбонилы и 9-флуоренилметилоксикарбонил (Fmoc);
3) алифатические карбаматные группы, такие как трет-бутилоксикарбонил (Boc), этоксикарбонил, диизопропилметоксикарбонил и аллилоксикарбонил;
4) циклические алкилкарбаматные группы, такие как циклопентилоксикарбонил и адамантилоксикарбонил;
5) алкильные группы, такие как трифенилметил, бензил или замещенный бензил, такой как 4-метоксибензил;
6) триалкилсилил, такой как триметилсилил или трет-бутилдиметилсилил; и
7) группы, содержащие тиольную группу, такие как фенилтиокарбонил и дитиасукциноил.
Защитными группами, представляющими особый интерес, являются Вос и Fmoc.
Предпочтительно защитная группа аминогруппы расщепляется перед следующей стадией сочетания. Удаление N-защищающих групп может осуществляться в соответствии с известными в данной области техники методиками. Когда используется Вос-группа, выбираемыми методами являются применение трифторуксусной кислоты (чистой или в дихлорметане) или HCl в диоксане либо в этилацетате. Полученная соль аммония затем нейтрализуется перед сочетанием или in situ с помощью основных растворов, таких как водные буферы или третичные амины в дихлорметане, ацетонитриле или диметилформамиде. Когда используется Fmoc-группа, выбираемыми реагентами являются пиперидин или замещенный пиперидин в диметилформамиде, но может использоваться и любой вторичный амин. Удаление защиты проводится при температуре в интервале от 0°С до комнатной температуры, обычно в интервале примерно 15-25°С или 20-22°С.
Другие функциональные группы, которые могут влиять на реакции сочетания структурообразующих блоков, также могут быть защищены. Например, гидроксильные группы могут защищаться с получением групп простых бензиловых или замещенных бензиловых эфиров, например, простого 4-метоксибензилового эфира, сложного бензоилового или замещенного бензоилового эфиров, например, сложного 4-нитробензоилового эфира, или с помощью триалкилсилильных групп (например, триметилсилильной или трет-бутилдиметилсилильной группы).
Дополнительные аминогруппы могут защищаться с помощью защитных групп, способных подвергаться селективному расщеплению. Например, когда в качестве α-аминозащищающей группы используются Вос, подходящими являются следующие защитные группы боковых цепей: п-толуолсульфонильные (тозильные) фрагменты могут применяться для защиты дополнительных аминогрупп; простые бензиловые (Bn) эфиры могут применяться для защиты гидроксильных групп; и сложные бензиловые эфиры могут применяться для защиты дополнительных карбоксильных групп. Когда для защиты α-аминогруппы выбрана Fmoc, походящими защитными группами обычно являются трет-бутиловые группы. Например, Вос может применяться для дополнительных аминогрупп; группы простых трет-бутиловых эфиров - для гидроксильных групп; и группы сложных трет-бутиловых эфиров - для дополнительных карбоксильных групп.
Любые из защитных групп могут удаляться на любой стадии методики синтеза, но предпочтительно защитные группы любых функциональных групп, не вовлеченных в стадии реакций синтеза, удаляются после завершения образования макроцикла.
Удаление защитных групп может осуществляться любым способом, определенным выбором защитных групп, которые хорошо известны специалисту данной области техники.
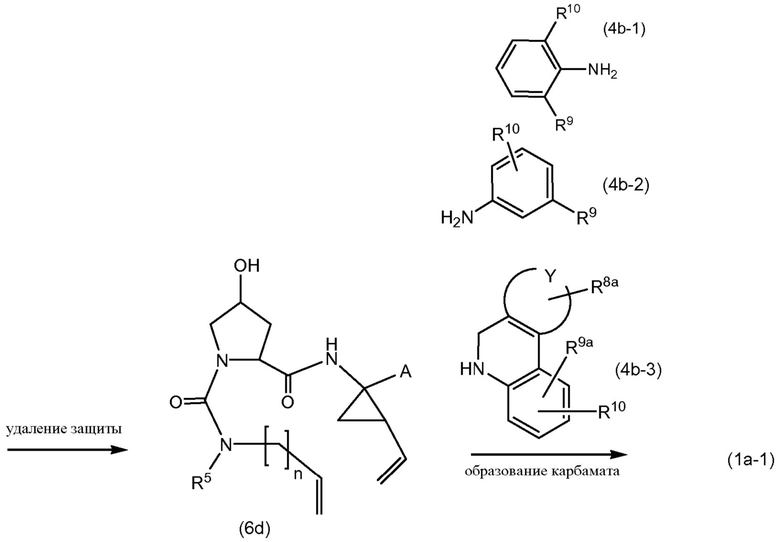
Промежуточные продукты формулы (1a), в которых Х представляет собой N, то есть промежуточные продукты формулы (1а-1), могут быть получены исходя из промежуточных соединений (5а), которые подвергаются взаимодействию с алкенамином (5b) в присутствии агента введения карбонила, как представлено на приведенной далее схеме реакции.

Агенты введения карбонильной группы (СО) включают фосген или производные фосгена, такие как карбонилдиимидазол (CDI) и т.п. В одном варианте осуществления изобретения соединение (5а) подвергается взаимодействию с агентом введения СО-группы в присутствии подходящего основания и в растворителе, которые могут представлять собой основания и растворители, используемые в реакциях образования амидной связи, описанных выше. В конкретном варианте осуществления изобретения основание представляет собой гидрокарбонат, например, NaHCO3, или третичный амин, такой как триэтиламин и т.п., и растворитель представляет собой простой эфир или галогенированный углеводород, например, ТГФ, CH2Cl2, CHCl3 и т.п. После этого добавляется амин (5b) с получением промежуточных продуктов (1а-1), как показано на схеме выше. Альтернативный способ с использованием аналогичных условий реакции включает сначала взаимодействие агента введения СО с алкенамином (5b) и последующее взаимодействие полученного таким образом промежуточного продукта с соединением (5а).
Промежуточные продукты (1а-1) могут быть альтернативно получены следующим образом:

PG1 представляет собой О-защищающую группу, которая может представлять собой любую из групп, упомянутых в описании, и в частности, представляет собой бензоильную или замещенную бензоильную группу, такую как 4-нитробензоил. В последнем примере эта группа может удаляться взаимодействием с гидроксидом щелочного металла (LiOH, NaOH, KOH), в частности, когда PG1 представляет собой 4-нитробензоил, взаимодействием с LiOH в водной среде, включающей воду и растворимый в воде органический растворитель, такой как алканол (метанол, этанол) и ТГФ.
Промежуточные соединения (6а) подвергаются взаимодействию с алкенамином (5b) в присутствии агента введения карбонильной группы, как описано выше, и указанная реакция приводит к получению промежуточных продуктов (6с). Из полученных соединений удаляются защитные группы, в частности, с использованием условий реакций, описанных выше. Полученный спирт (6d) подвергается взаимодействию с промежуточными продуктами (4b-1), (4b-2) или (4b-3), как описано выше в описании взаимодействия (4а) с (4b-1), (4b-2) или (4b-3), и указанная реакция приводит к получению промежуточных продуктов (1а-1).
Промежуточные продукты формулы (1а), где Х представляет собой С, то есть промежуточные продукты формулы (1а-2), могут быть получены с помощью реакции образования амида, исходя из промежуточных продуктов (7а), которые подвергаются взаимодействию с алкенамином (5b), как представлено на приведенной далее схеме реакции, с использованием условий реакции получения амидов, которые описаны выше.

Альтернативно, промежуточные продукты (1а-2) могут быть получены следующим образом:

PG1 представляет собой О-защищающую группу, которая описана выше. Могут использоваться условия реакции, описанные выше: образование амида, как описано выше, удаление PG1 в соответствии с описанием защитных групп и введение -OR11, как в реакциях (4а) с аминами (4b-1), (4b-2) или (4b-3).
Промежуточные продукты формулы (2а) могут быть получены циклизацией открытого амида (9а) с получением макроциклического сложного эфира (9b), который, в свою очередь, подвергается превращению в (2а) следующим образом:

PG2 представляет собой защитную группу карбоксильной группы, например, одну из защитных групп карбоксильной группы, упомянутых выше, в частности, группу сложного С1-4алкилового или бензилового эфира, например, сложного метилового, этилового или трет-бутилового эфира. Реакция превращения (9а) в (9b) является реакцией обмена и проводится как описано выше. Группа PG2 удаляется в соответствии с методиками, также описанными выше. Когда PG2 представляет собой группу С1-4алкилового эфира, она удаляется щелочным гидролизом, например, с NaOH или, предпочтительно, LiOH в водном растворителе, например, смесью С1-4алканол/вода. Бензильная группа может удаляться каталитическим гидрированием.
В альтернативном синтезе промежуточные соединения (2а) могут быть получены следующим образом:

PG1 группа выбирается таким образом, чтобы она селективно расщеплялась с образованием PG2. PG2 может представлять собой, например, группу сложного метилового или этилового эфиров, которая может удаляться обработкой гидроксидом щелочного металла в водной среде, в этом случае PG1 представляет собой, например, трет-бутил или бензил. PG2 группа может представлять собой группу сложного трет-бутилового эфира, которая может удаляться в слабых кислотных условиях, или PG1 может представлять собой группу простого бензилового эфира, которая может удаляться с помощью сильной кислоты или каталитическим гидрированием, в двух последних случаях PG1 представляет собой, например, группу сложного бензойного эфира, такую как группа 4-нитробензойного эфира.
Сначала промежуточные соединения (10а) подвергаются циклизации до сложных макроциклических эфиров (10b), удаление защиты из последних осуществляется удалением PG1 группы с получением (10с), который подвергается взаимодействию с (4b) в соответствии с реакцией образования карбамата, как описано выше, с последующим удалением группы PG2, защищающей карбоксильную группу. Циклизация, удаление защитных групп PG1 и PG2, а также реакция образования карбамата с (4b-1), (4b-2) или (4b-3) описаны выше.
Группы А могут вводиться на любой стадии синтеза, либо на последней стадии, как описано выше, либо ранее до образования макроцикла. В представленной далее схеме вводятся группы А, которые представляют собой -CO-NH-SO2R2 или -CO-OR5 (как определено выше):

В представленной выше схеме PG2 принимает значения, определенные выше, и L1 представляет собой Р3 группу 
где n и R5 принимают значения, определенные выше, и Х представляет собой N, L1 также может представлять собой группу -COOPG2a, где группа PG2a представляет собой защитную группу карбоксильной группы, аналогичную PG2, но группа PG2a способна селективно расщепляться с получением группы PG2. В одном варианте осуществления изобретения PG2a представляет собой трет-бутил, и PG2 представляет собой метил или этил.
Промежуточные продукты (11с) и (11d), где L1 представляет собой группу (b), соответствуют промежуточным продуктам (1а) и могут подвергаться технологической обработке, как определено выше.
Сочетание структурообразующих блоков Р1 и Р2
Структурообразующие блоки Р1 и Р2 связываются с использованием реакции образования амида в соответствии с методиками, описанными выше. Структурообразующий блок Р1 может содержать группу PG2, защищающую карбоксильную группу (как в (12b)), или может быть уже соединенным с Р1' группой (как в (12с)). L2 представляет собой N-защищающую группу (PG) или группу (b), как описано выше. L3 представляет собой гидроксильную группу, -OPG1 или группу -O-R11, которые определены выше. Когда в любой из представленных далее схем реакций L3 представляет собой гидроксильную группу, перед каждой реакционной стадией она может защищаться и может быть представлена как группа -OPG1 и, если нужно, затем защитная группа может удаляться с получением свободной функциональной гидроксильной группы. Аналогично, как описано выше, гидроксильная функциональная группа может превращаться в группу -O-R11.

В методике, представленной выше, схемы циклопропиламинокислота (12b) или (12с) сочетается с кислотной функциональной группой Р2 структурообразующего блока (12а) с образованием амидного мостика в соответствии с методиками, описанными выше. Таким образом получают промежуточные продукты (12d) или (12е). Когда в последних L2 представляет собой группу (b), полученные продукты представляют собой последовательности Р3-Р2-Р1, включающие некоторые из промежуточных продуктов (11с) или (11d) в предыдущей схеме реакции. Удаление защитной группы кислоты в (12d) с использованием подходящих условий для используемой защитной группы с последующим сочетанием с сульфонамидом H2N-SO2R2 (2b), фосфорамидом (2d) или HOR1 (2c), как описано выше, снова приводит к получению промежуточных продуктов (12е), где -А представляет собой амидную группу или группу сложного эфира. Когда L2 представляет собой N-защищающую группу, она может удаляться с получением промежуточных продуктов (5а) или (6а). В одном варианте осуществления изобретения PG в такой реакции представляет собой ВОС группу, и PG2 представляет собой метил или этил. Когда L3 представляет собой гидроксильную группу, исходное соединение (12а) представляет собой Вос-L-гидроксипролин. В конкретном варианте осуществления изобретения PG представляет собой ВОС группу, PG2 представляет собой метил или этил, L3 представляет собой -O-R11.
В одном варианте осуществления изобретения L2 представляет собой группу (b), и представленные реакции включают сочетание Р1 с Р2-Р3, что приводит к получению промежуточных продуктов (1а-1) или (1а), описанных выше. В другом варианте осуществления изобретения L2 представляет собой N-защищающую группу PG, которая определена выше, и реакция сочетания приводит к получению промежуточных продуктов (12d-1) или (12е-1), из которых группа PG может удаляться с использованием условий реакции, описанных выше, с получением промежуточных продуктов (12-f) или (12-g), соответственно, которые включают и промежуточные продукты (5а) и (6а), определенные выше:

В одном варианте осуществления изобретения группа L3 в приведенных выше схемах представляет собой группу -О-PG1, которая может быть введена в исходное вещество (12а), где L3 представляет собой гидроксильную группу. В этом примере PG1 выбрана таким образом, что она способна селективно расщепляться для получения группы L2, которая представляет собой PG.
В аналогичном способе структурообразующие блоки Р2, в которых Х представляет собой С и которые представляют собой производные циклопентана или циклопентена, могут связываться со структурообразующими блоками Р1 как показано на приведенной далее схеме, где А, R7, L3 принимают значения, определенные выше, и PG2 и PG2a представляют собой защитные группы карбоксильной группы. PG2a обычно выбирается таким образом, чтобы она могла селективно расщепляться с получением группы PG2. Удаление группы PG2a в (13с) приводит к получению промежуточных продуктов (7а) или (8а), которые могут подвергаться взаимодействию с (5b), как описано выше.

В конкретном варианте осуществления изобретения, где Х представляет собой С, R7 представляет собой Н и Х и углерод, к которому присоединена группа R7, соединены одинарной связью (Р2 представляет собой циклопентановый фрагмент), PG2a и L3 вместе образуют связь, и структурообразующий блок Р2 соответствует формуле

Бициклическая кислота (14а) подвергается взаимодействию с (12b) или (12с), как описано выше, с получением (14b) и (14с), соответственно, где лактон раскрывается с получением промежуточных продуктов (14с) и (14е). Лактоны могут подвергаться раскрытию с использованием методик гидролиза сложных эфиров, например, в условиях, описанных выше для щелочного удаления PG1 группы в (9b), в частности, с использованием основных условий, таких как гидроксид щелочного металла, например, NaOH, KOH, в особенности LiOH.
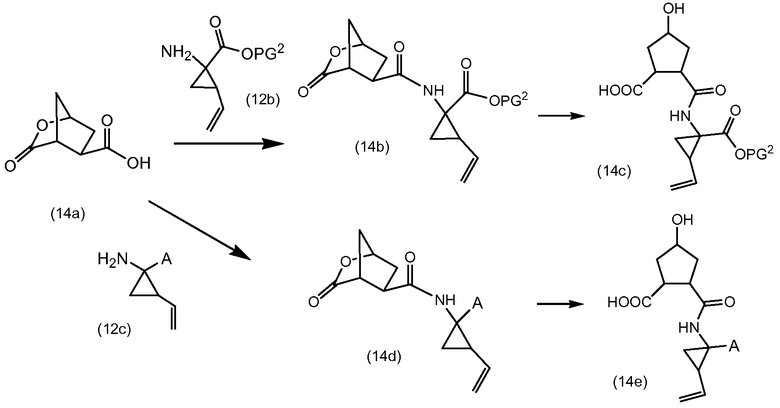
Промежуточные продукты (14с) и (14е) могут подвергаться дальнейшей обработке, как описано ниже.
Сочетание структурообразующих блоков Р3 и Р2
Когда структурообразующие блоки Р2 содержат пирролидиновый фрагмент, структурообразующие блоки Р3 и Р2 или Р3 и Р2-Р1 сочетаются с использованием реакции образования карбамата в соответствии с методиками, описанными выше для сочетания (5а) с (5b). Общая методика сочетания Р2 блоков, содержащих пирролидиновый фрагмент, представлена на приведенной далее схеме реакции, где L3 принимает значения, определенные выше, и L4 представляет собой группу -О-PG2, группу
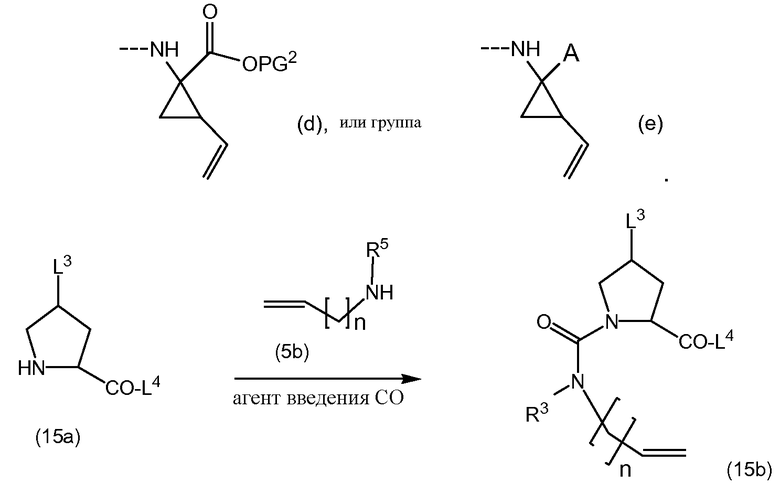
В одном варианте осуществления изобретения L4 в (15а) представляет собой группу -OPG2, где PG2 группа может удаляться и полученная кислота может сочетаться с циклопропиламинокислотами (12а) или (12b) с получением промежуточных продуктов (12d) или (12е), в которых L2 представляет собой радикал (d) или (е).
Общая методика сочетания блоков Р3 с блоком Р2 или с блоком Р2-Р1, где Р2 представляет собой циклопентан или циклопентен, представлена на приведенной далее схеме. L3 и L4 принимают значения, определенные выше.

В конкретном варианте осуществления изобретения L3 и L4 вместе образуют лактоновый мостик, как в (14а), и сочетание блока Р3 с блоком Р2 осуществляется следующим образом:

Бициклический лактон (14а) подвергается взаимодействию с (5b) в соответствии с реакцией образования амида с получением амида (16с), лактоновый мостик в котором раскрывается с получением (16d). Условия реакций образования амида и раскрытия лактона описаны выше или далее. Промежуточный продукт (16d), в свою очередь, может сочетаться с группой Р1, как описано выше.
Реакции, показанные в представленных выше схемах, проводятся с использованием методик, которые описаны выше для реакций (5а), (7а) или (8а) с (5b), в частности, описанные выше реакции, где L4 представляет собой группу (d) или (е), соответствуют реакциям (5а), (7а) или (8а) с (5b), как описано выше.
Структурообразующие блоки Р1, Р1', Р2 и Р3, используемые при получении соединений формулы (I), могут быть получены из известных в данной области техники промежуточных продуктов. Ряд таких синтезов описан далее более подробно.
Сначала могут быть получены отдельные структурообразующие блоки, которые затем могут соединяться вместе или, альтернативно, предшественники структурообразующих блоков могут соединяться вместе и модифицироваться на более поздней стадии до молекулы желательного строения.
Функциональные группы в каждом из структурообразующих блоков могут защищаться для предупреждения побочных реакций.
Синтез структурообразующих блоков Р2
Структурообразующие блоки Р2 содержат пирролидиновый, циклопентановый или циклопентеновый фрагмент, замещенный группой -O-R11.
Структурообразующие блоки Р2, содержащие пирролидиновый фрагмент, могут быть получены из коммерчески доступного гидроксипролина.
Получение структурообразующих блоков Р2, которые содержат циклопентановое кольцо, может осуществляться в соответствии со схемой, представленной ниже.

Бициклическая кислота (17b) может быть получена, например, из 3,4-бис(метоксикарбонил)циклопентанона (17а), как описано в публикации Rosenquist et al., Acta Chem. Scand. 46 (1992) 1127-1129. Первая стадия в данной методике включает восстановление кето-группы таким восстановителем, как борогидрид натрия, в растворителе, таком как метанол, с последующим гидролизом сложных эфиров и, наконец, замыканием кольца бициклического лактона (17b) в соответствии с методиками образования лактона, в частности, посредством применения уксусного ангидрида в присутствии слабого основания, такого как пиридин. Функциональная группа карбоновой кислоты в (17b) может после этого защищаться посредством введения подходящей защитной группы, такой как группа PG2, которая определена выше, с получением, таким образом, сложного бициклического эфира (17с). Группа PG2, в частности, является кислотно-лабильной, такой как трет-бутильная группа, и вводится, например, обработкой изобутеном в присутствии кислоты Льюиса или ди-трет-бутилдикарбонатом в присутствии основания, такого как третичный амин, например, диметиламинопиридин или триэтиламин, в растворителе, таком как дихлорметан. Раскрытие лактона (17с) в условиях, описанных выше, в частности, в присутствии гидроксида лития, приводит к получению кислоты (17d), которая может использоваться далее в реакциях сочетания со структурообразующими блоками Р1. Свободная кислотная группа в (17d) также может защищаться, предпочтительно защитной группой кислоты PG2a, которая селективно расщепляется до группы PG2, и гидроксильная функция может превращаться в группу -OPG1 или в группу -O-R11. Промежуточные продукты (17g) и (17i), полученные после удаления группы PG2, соответствуют промежуточным продуктам (13а) или (16а), определенным выше.
Промежуточные продукты со специфической стереохимией могут быть получены разделением промежуточных продуктов в описанной выше последовательности реакций. Например, (17b) может быть разделен в соответствии с известными методиками, например, посредством взаимодействия солевой формы с оптически активным основанием или методом хиральной хроматографии, и полученные стереоизомеры могут подвергаться дальнейшей обработке, как описано выше. Группы ОН и СООН в (17d) находятся в цис-положении. Транс-аналоги могут быть получены инвертированием стереохимии при атоме углерода, к которому присоединена функциональная группа -ОН, с применением специфических реагентов в реакциях введения -OPG1 или -O-R11, при которых происходит инвертирование стереохимии, таких как, например, реакция Митсунобу.
В одном варианте осуществления изобретения промежуточные продукты (17d) сочетаются с блоками Р1 (12b) и (12с) в результате реакция сочетания, которые соответствуют сочетанию (13а) или (16а) с этими же блоками Р1 с использованием таких же условий. Последовательное введение -O-R11 заместителя, как описано выше, и далее удаление защитной группы кислотной группы PG2 приводит к получению промежуточных продуктов (8а-1), которые представляют собой подгруппу промежуточных продуктов (7а) или часть промежуточных продуктов (16а). Продукты реакции удаления PG2 могут далее сочетаться со структурообразующим блоком Р3. В одном варианте осуществления изобретения PG2 в (17d) представляет собой трет-бутил, который может удаляться в кислотных условиях, например, при обработке трифторуксусной кислотой.

Ненасыщенный структурообразующий блок Р2, т.е. циклопентеновое кольцо, может быть получен как показано на схеме, приведенной ниже.
Реакция бромирования-отщепления 3,4-бис(метоксикарбонил)циклопентанона (17а), как описано в публикации Dolby et al., J. Org. Chem. 36 (1971) 1277-1285, с последующим восстановлением функциональной кето-группы таким восстановителем, как борогидрид натрия, приводит к получению циклопентенола (19а). Селективный гидролиз сложного эфира с использованием, например, гидроксида лития, в растворителе, таком как смесь диоксана и воды, приводит к получению гидрокси-замещенного сложного моноэфирного циклопентенола (19b).
Ненасыщенный структурообразующий блок Р2, где R7 также возможно отличен от водорода, может быть получен как показано на схеме, представленной ниже.
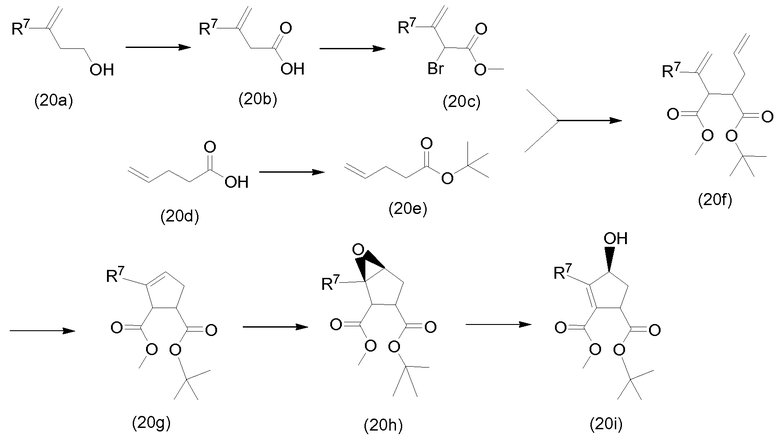
Окисление коммерчески доступного 3-метил-3-бутен-1-ола (20а), в частности таким окислителем, как хлорхромат пиридиния, приводит к получению (20b), который подвергается превращению в соответствующий сложный метиловый эфир, например, обработкой ацетилхлоридом в метаноле, с последующей реакцией бромирования бромом с получением сложного α-бромэфира (20с). Последний затем может подвергаться конденсированию со сложным алкениловым эфиром (20е), полученным из (20d) посредством реакции образования сложного эфира. Сложный эфир (20е) предпочтительно представляет собой трет-бутиловый эфир, который может быть получен из соответствующей коммерчески доступной кислоты (20d), например, обработкой ди-трет-бутилдикарбонатом в присутствии основания, такого как диметиламинопиридин. Промежуточный продукт (20е) подвергается обработке основанием, таким как диизопропиламид лития, в растворителе, таком как тетрагидрофуран, и взаимодействует с (20с) с получением сложного алкенилового диэфира (20f). Циклизация (20f) посредством обменной реакции олефинов, осуществляемой как описано выше, обеспечивает получение производного циклопентена (20g). Стереоселективное эпоксидирование (20g) может проводиться с использованием метода асимметричного эпоксидирования Джакобсена (Jacobsen) c получением эпоксида (20h). И наконец, реакция раскрытия эпоксида в основных условиях, например, посредством добавления основания, в частности DBN (1,5-диазабицикло-[4.3.0]нон-5-ена), приводит к получению спирта (20i). Двойная связь в промежуточном продукте (20i) может необязательно подвергаться восстановлению, например, каталитическим гидрированием с использованием катализатора, такого как палладий на углероде, с получением соответствующего циклопентанового соединения. Группа сложного трет-бутилового эфира может удаляться c получением соответствующей кислоты, которая последовательно сочетается со структурообразующим блоком Р1.
Группа -O-R11 может вводиться в пирролидиновый, циклопентановый или циклопентеновый циклы на любой удобной стадии синтеза соединений согласно настоящему изобретению. Одним из подходов является первичное введение -O-R11 группы в указанные циклы и последовательное добавление других нужных структурообразующих блоков, т.е. Р1 (необязательно с хвостом Р1') и Р3 и затем образование макроцикла. Другим подходом является сочетание структурообразующих блоков Р2, не содержащих -O-R11 заместителя, с каждым из блоков Р1 и Р3 и введение -O-R11 группы перед либо после образования макроцикла. В последней методике фрагменты Р2 содержат гидроксильную группу, которая может защищаться защитной группой гидроксильной группы PG1.
R11 группы могут вводиться в структурообразующие блоки Р2 посредством взаимодействия гидрокси-замещенных промежуточных продуктов (21а) или (21b) с промежуточными продуктами (4b) аналогично тому, как описано выше для синтеза (I), исходя из (4а). Эти реакции представлены на схемах ниже, где L2 принимает значения, определенные выше, и L5 и L5a независимо друг от друга представляют собой защитную группу гидроксильной или карбоксильной группы -OPG2 или -OPG2a, соответственно, или L5 может также представлять собой Р1 группу, такую как группа (d) или (е), которые определены выше, или L5a также может представлять собой Р3 группу, такую как группа (b), которая определена выше. Группы PG2 и PG2a принимают значения, определенные выше. Когда группы L5 и L5a представляют собой PG2 или PG2a, они выбираются таким образом, чтобы каждая группа могла подвергаться селективному расщеплению с получения другой. Например, одна группа из L5 и L5a может представлять собой метильную или этильную группу, а другая представляет собой бензильную или трет-бутильную группу.
В одном варианте осуществления изобретения в (21а) L2 представляет собой -OPG2, или в (21d) L5a представляет собой -OPG2 и L5 представляет собой -OPG2, и группы PG2 удаляются, как описано выше.



В другом варианте осуществления группа L2 представляет собой ВОС, L5 представляет собой гидроксильную группу, а исходное вещество (21а) представляет собой коммерчески доступный ВОС-гидроксипролин или его любую другую стереоизомерную форму, например, ВОС-L-гидроксипролин, в частности, его транс-изомер. Когда L5 в (21b) представляет собой защитную группу карбоксильной группы, он может удаляться в соответствии с методиками, описанными выше для (21с). В еще одном варианте осуществления изобретения PG в (21b-1) представляет собой Вос, и PG2 представляет собой сложный эфир низшего алкила, в частности, сложный метиловый или этиловый эфир. Гидролиз последнего сложного эфира до кислоты может проводиться в соответствии со стандартными методиками, например, кислотным гидролизом с применением соляной кислоты в метаноле или гидроксида щелочного металла, такого как NaOH или, более предпочтительно, с применением LiOH. В еще одном варианте осуществления изобретения гидрокси-замещенные циклопентановый или циклопентеновый аналоги (21d) подвергаются превращению в (21е), где L5 и L5a представляют собой -OPG2 или -OPG2a, и полученный продукт далее может подвергаться превращению в соответствующие кислоты (21f) посредством удаления группы PG2. Удаление PG2a в (21е-1) приводит к получению аналогичных промежуточных продуктов.
Синтез структурообразующих блоков Р1
Циклопропанаминокислота, используемая для получения фрагмента Р1, является коммерчески доступной или может быть получена в соответствии с методиками, известными в данной области техники.
В частности, сложный аминовинилциклопропилэтиловый эфир (12b) может быть получен в соответствии с методикой, описанной в WO 00/09543, или как представлено на приведенной далее схеме, где PG2 представляет собой защитную группу карбоксильной группы, которая определена выше:

Обработка коммерчески доступного или легко синтезируемого имина (31а) 1,4-дигалогенбутеном в присутствии основания приводит к получению (31b), который после гидролиза обеспечивает получение циклопропиламинокислоты (12b), содержащей аллильный заместитель в syn-положении относительно карбоксильной группы. Разделение энантиомерной смеси (12b) приводит к получению (12b-1). Разделение проводится в соответствии с известными методиками, такими как ферментативное разделение; кристаллизация с хиральной кислотой; химическая дериватизация; или хиральная колоночная хроматография. Промежуточные продукты (12b) или (12b-1) могут сочетаться с подходящими производными Р2, как описано выше.
Структурообразующие блоки Р1 для получения соединений согласно общей формуле (I), в которых А представляет собой -COOR1, -CO-NH-SO2R2 или -CO-NH-PO(OR4a)(OR4b), могут быть получены взаимодействием аминокислот (32а) с подходящим спиртом, сульфонамидом или фосфорамидом, соответственно, в стандартных условиях образования получения эфира или амида. Циклопропиламинокислоты (32а) получают введением N-защищающей группы PG и удалением PG2, и полученные PG-защищенные аминокислоты (32а) подвергаются превращению в амиды (12с-1) или сложные эфиры (12с-2), которые представляют собой подгруппы промежуточных продуктов (12с), как представлено на приведенной далее схеме, где PG принимает значения, определенные выше.

Взаимодействие (32а) с амином (2b) или с (2d) представляет собой методику образования амида. Аналогичная реакция с (2с) представляет собой реакцию образования сложного эфира. Оба типа реакций могут осуществляться в соответствии с методиками, описанными выше. Эта реакция приводит к получению промежуточных продуктов (32b), (32b-1) или (32с), из которых защитная группа аминогруппы удаляется стандартными методами, такими как методы, описанные выше. Это, в свою очередь, приводит к получению целевого промежуточного продукта (12с-1), (12с-1а) или (12с-2). Исходные вещества (32а) могут быть получены из упомянутых выше промежуточных продуктов (12b). Исходные вещества (32а) могут быть получены из упомянутых выше промежуточных продуктов (12b) сначала введением N-защищающей группы PG и последующим удалением группы PG2.
В одном варианте осуществления изобретения взаимодействие (32а) с (2b) или с (2d) осуществляется обработкой аминокислоты агентом сочетания, например, N,N'-карбонилдиимидазолом (CDI) или т.п., в растворителе, таком как ТГФ, с последующим взаимодействием с (2b) или с (2b-1) в присутствии основания, такого как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Альтернативно, аминокислота может подвергаться обработке (2b) или (2d) в присутствии основания, такого как диизопропилэтиламин, или гидридом натрия в случае (2d) с последующей обработкой агентом сочетания, таким как гексафторфосфат бензотриазол-1-илокси-трис-пирролидинофосфония (коммерчески доступен как PyBOP®) для введения сульфонамидной группы.
Структурообразующие блоки Р1 для получения соединений согласно общей формуле (I), где А представляет собой -C(=O)C(=O)NR3aR3b, удобно получают в соответствии с приведенной далее схемой.

PG2a в исходном веществе (32d) представляет собой алкильную группу, в частности, С1-6алкил, такой как метил или этил. (32d) может быть получен в результате реакции образования сложного эфира (32а) с подходящим алканолом или введением азот-защищающей группы PG в (12b), как описано выше. Восстановление сложноэфирной группы в производном аминокислоты (32d) до соответствующего гидроксиметиленового промежуточного продукта (32е), проводимое, например, с помощью борогидрида лития с последующим окислением полученной гидроксиметиленовой группы с использованием умеренного окислителя, такого как, например, периодинан Десса-Мартина, обеспечивает получение альдегида (32f). Взаимодействие альдегида (32f) с подходящим производным изонитрила в присутствии карбоновой кислоты, такой как трифторуксусная кислота (ТФУК), в присутствии основания, например, пиридина, в соответствии с реакцией Пассерини (которая описана, например, в Org. Lett., Vol. 2, № 18, 2000) приводит к получению сложного эфира карбоновой кислоты полученного α-гидроксиамида, например, в случае ТФУК - к получению трифторацетата. Группа сложного эфира карбоновой кислоты в полученном таким образом α-гидроксиамиде далее может удаляться с использованием стандартных методик, например, с использованием основных условий, таких как LiOH, что обеспечивает получение α-гидроксиамида (32g). Удаление группы PG приводит к получению промежуточных продуктов (32h), которые могут сочетаться с Р2 группой. Гидроксильная функциональная группа в (32g) может подвергаться окислению до соответствующего α-кетоамида, но для предотвращения побочных реакций в последующих реакциях (таких как удаление N-защищающей группы, сочетание с Р2 фрагментом и т.д.) используется α-гидроксиамид или его предшественник - сложный эфир карбоновой кислоты. Окисление α-гидроксильной группы Р1 фрагмента затем проводится на любой удобной стадии синтеза, например, после сочетания с Р2 фрагментом или на более поздних стадиях синтеза, например, на последней стадии, с использовании слабого окислителя, такого как, например, периодинан Десса-Мартина, получая таким образом соединения формулы I или промежуточные продукты, в которых А представляет собой -C(=O)C(=O)NR3aR3b.
Описанная выше методика, т.е. восстановление сложного эфира, окисление до альдегида, взаимодействие с изонитрилом, также могут проводиться на более поздних стадиях методики синтеза, например, после образования макроцикла.
Структурообразующие блоки Р1, которые могут применяться для получения соединений общей формулы (I), где А представляет собой -C(=O)NH-P(=O)(OR4a)(R4b) или -P(=O)(OR4a)(R4b)фосфонат, могут быть получены в соответствии с методиками, описанными в WO 2006/020276. В конкретных примерах соединения формулы (I), где А представляет собой -P(=O)(OR4a)(R4b), могут быть получены следующим образом:

Исходное 32i подвергается взаимодействию с основанием, в частности с CsOH, предпочтительно в присутствии катализатора фазового перехода, такого как хлорид триэтилбензиламмония, и добавляется 32j, образующих циклопроцикльное кольцо с боковой виниловой цепью, т.е. циклопропилфосфонат 32k. Защитная группа группы фенил-СН= удаляется в кислотных условиях (например, HCl в дихлорметане) с получением 32l. Последнее вещество может разделяться на стереоизомеры с помощью известных методик, например, получением соли с оптически активной кислотой, например, с дибензоил-L-винной кислотой, и удаление группы винной кислоты из соли приводит к получению 32m. Аналоги, отличные от эфилфосфонатов, могут быть получены из исходных веществ 32i, содержащих группы сложных эфиров, отличные от группы сложного этилового эфира. Исходные 32i являются известными веществами или могут быть легко получены с использованием известных методов.
Промежуточные продукты (12с-1) или (12с-2), в свою очередь, могут сочетаться с подходящими производными пролина, циклопентана или циклопентена, как описано выше.
Синтез структурообразующих блоков Р3
Структурообразующие блоки Р3 являются коммерчески доступными или могут быть получены в соответствии с методиками, известными специалисту данной области техники. Одна из таких методик представлена на схеме ниже, где используются моноацилированные амины, такие как трифторацетамид или Вос-защищенный амин.

На представленной выше схеме R вместе с группой СО образует N-защищающую группу, в частности, R представляет собой трет-бутокси, трифторметил; R5 и n принимают значения, определенные выше, и LG представляет собой удаляемую группу, в частности, галоген, например, хлор или бром.
Моноацилированные амины (33а) подвергаются обработке сильным основанием, таким как гидрид натрия, и последовательно подвергаются взаимодействию с реагентом LG-C5-8алкенилом (33b), в частности галогенС5-8алкенилом, для получения соответствующих защищенных аминов (33с). Удаление защиты из (33с) приводит к получению (5b), которые являются структурообразующими блоками Р3. Удаление защиты будет определяться функциональной группой R; таким образом, если R представляет собой трет-бутокси, удаление защиты из соответствующего Вос-защищенного амина может проводиться посредством кислотной обработки, например, обработкой трифторуксусной кислотой. Альтернативно, когда R представляет собой, например, трифторметил, удаление R группы проводится обработкой основанием, например, гидроксидом натрия.
Приведенная далее схема иллюстрирует еще один способ получения структурообразующего блока Р3, а именно - синтез первичных С5-8алкениламинов по методу Габриеля, который может проводиться обработкой фталамида (34а) основанием, таким как NaOH или КОН, и (33b), который определен выше, с последующим гидролизом промежуточного N-алкенилимида для получения первичного С5-8алкениламина (5b-1).
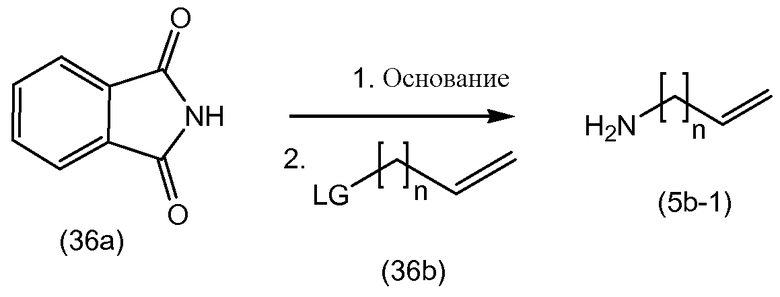
В приведенной выше схеме n принимает значения, определенные выше.
Каждое из соединений формулы (I) может подвергаться превращению в любое другое в соответствии с известными реакциями трансформации функциональных групп. Например, аминогруппы могут подвергаться N-алкилированию, нитрогруппы восстанавливаться до аминогрупп, один атом галогена может заменяться на другой атом галогена.
Промежуточные продукты, используемые для получения соединений формулы (I), являются известными соединениями или аналогами известных соединений, которые могут быть получены при модификации известных методик, которые может осуществить квалифицированный специалист.
Соединения формулы (I) могут подвергаться превращению в соответствующие N-оксидные формы в соответствии с известными методиками превращения трехвалентного азота в его N-оксидную форму. Указанная реакция N-окисления обычно может проводиться посредством взаимодействия исходного соединения формулы (I) с подходящим органическим или неорганическим пероксидом. Подходящие неорганические пероксиды включают, например, пероксид водорода, пероксиды щелочных или щелочно-земельных металлов, например, пероксид натрия, пероксид калия; подходящие органические пероксиды могут включать оксикислоты, такие как, например, бензолкарбоновая оксикислота или галогензамещенная бензолкарбоновая оксикислота, например, 3-хлорбензолкарбоновая оксикислота, пероксоалкановые кислоты, например, пероксиуксусная кислота, алкилгидропероксиды, например, трет-бутилгидропероксид. Подходящими растворителями являются, например, вода, низшие спирты, например, этанол и т.п., углеводороды, например, толуол, кетоны, например, 2-бутанон, галогенированные углеводороды, например, дихлорметан и смеси таких растворителей.
Стереохимически чистые изомерные формы соединений формулы (I) могут быть получены в соответствии с методиками, известными в данной области техники. Диастереомеры могут разделяться физическими методами, такими как селективная кристаллизация, и хроматографическими методами, например, методом распределения противотока, жидкостной хроматографии и т.п.
Соединения формулы (I) могут быть получены в виде рацемических смесей энантиомеров, которые могут разделяться с помощью известных в данной области техники методик. Рацемические соединения формулы (I), которые являются достаточно основными или кислотными, могут подвергаться превращению в соответствующие диастереомерные солевые формы взаимодействием с подходящей хиральной кислотой или подходящим хиральным основанием, соответственно. Указанные диастереомерные солевые формы затем разделяются, например, селективной или фракционной кристаллизацией, и энантиомеры высвобождаются с помощью щелочи или кислоты. Альтернативные способы разделения энантиомерных форм соединений формулы (I) включают жидкостную хроматографию, в частности, жидкостную хроматографию с применением хиральной стационарной фазы. Указанные стереохимически чистые изомерные формы также могут быть получены из соответствующих стереохимически чистых изомерных форм подходящих исходных веществ при условии, что реакция протекает стереоспецифически. Предпочтительно, если необходимо получать специфический стереоизомер, указанное соединение может синтезироваться стереоспецифическими способами получения. В этих способах могут преимущественно применяться энантиомерно чистые исходные вещества.
В соответствии с дополнительным аспектом, настоящее изобретение относится к фармацевтической композиции, включающей терапевтически эффективное количество соединения формулы (I), как определено в описании, или соединение любой из подгрупп соединений формулы (I), как определено в описании, и фармацевтически приемлемый носитель. Термин «терапевтически эффективное количество» в данном контексте означает количество, достаточное для профилактического действия против вирусной инфекции, для стабилизация или снижения вирусной инфекции, в частности HCV инфекции, у зараженных субъектов или субъектов, подверженных риску заражения. В соответствии с еще одним аспектом, настоящее изобретение относится к способу получения фармацевтической композиции, как она определена в описании, который включает тщательное смешение фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы (I), как оно определено в описании, или соединением любой из подгрупп соединений формулы (I), как оно определено в описании.
Следовательно, соединения согласно настоящему изобретению или любой из его подгрупп для удобного применения могут вводиться в различные фармацевтические формы. В качестве подходящих композиций могут быть перечислены все композиции, обычно используемые для системного введения лекарственных средств. Для приготовления фармацевтических композиций согласно настоящему изобретению эффективное количество конкретного соединения, необязательно в форме аддитивной соли металла или комплекса металла, в качестве активного ингредиента объединяется в однородной смеси с фармацевтически приемлемым носителем, форма которого может зависеть от желательной формы препарата. Такие фармацевтические композиции желательно представляют собой стандартную лекарственную форму, подходящую для введения перорально, ректально, чрескожно или парентеральной инъекцией. Например, при приготовлении композиций в лекарственной форме для перорального введения может применяться любая традиционно используемая фармацевтическая среда, такая как, например, вода, гликоли, масла, спирты и т.п., в случае жидких препаратов для перорального введения, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, дезинтегрирующие добавки и т.п., в случае порошков, пилюль, капсул и таблеток. Благодаря легкости введения, таблетки и капсулы представляют собой наиболее преимущественные лекарственные формы стандартной дозы для перорального введения, в которых применяются стандартные твердые фармацевтические носители. Носитель для приготовления композиций для парентерального введения будет обычно включать стерильную воду, по меньшей мере, в большей части, хотя могут вводиться и другие ингредиенты, например, добавки для повышения растворимости. Например, могут приготавливаться растворы для инъекций, в которых носитель включает раствор соли (физиологический раствор), раствор глюкозы или смесь раствора соли и раствора глюкозы. Могут приготавливаться суспензии для инъекций, в которых могут применяться подходящие жидкие носители, суспендирующие агенты и т.п. Композиции включают также препараты в твердой форме, предназначенные для превращения непосредственно перед применением в препараты жидкой формы. В композициях, подходящих для чрескожного введения, носитель необязательно включает добавку, повышающую проникновение, и/или подходящий смачивающий агент, необязательно объединенный с минимальными количествами подходящих добавок любой природы, которые не оказывают значительного неблагоприятного воздействия на кожу.
Соединения согласно настоящему изобретению могут также вводиться перорально ингаляцией или инсуффляцией с помощью методов и препаратов, применяемых для такого введения. Таким образом, обычно композиции согласно настоящему изобретению могут вводиться в легкие в форме раствора, суспензии или сухого порошка, причем предпочтительным является раствор. Любая система, разработанная для доставки растворов, суспензий или сухих порошков пероральной ингаляцией или инсуффляцией, является подходящей для введения соединений согласно настоящему изобретению.
Следовательно, настоящее изобретение предоставляет также фармацевтическую композицию, приспособленную для введения ингаляцией или инсуффляцией через рот и включающую соединение формулы (I) и фармацевтически приемлемый носитель. Предпочтительно, соединения согласно настоящему изобретению вводятся ингаляцией раствора в распыляемых или разбрызгиваемых лекарственных формах.
Особенно удобно приготавливать описанные выше фармацевтические композиции в виде стандартных лекарственных форм для простоты введения и унифицированной дозировки. Термин «стандартная лекарственная форма», когда используется в описании, относится к физически дискретным частицам (единицам), подходящим для применения в качестве разовых доз, причем каждая единица содержит предопределенное количество активного ингредиента, рассчитанное для получения желательного терапевтического действия, в комбинации с необходимым фармацевтическим носителем. Примерами таких лекарственных форм стандартной дозы являются таблетки (включая таблетки с насечками и таблетки, покрытые оболочкой), капсулы, пилюли, свечи, пакетированные порошки, вафли, растворы или суспензии для инъекций и т.п., а также их отдельные множества.
Соединения формулы (I) проявляют противовирусные свойства. Вирусные инфекции и связанные с ними заболевания могут лечиться с использованием соединений и способов согласно настоящему изобретению, включая инфекции, вызываемые HCV и другими патогенными флавивирусами, такими как вирус желтой лихорадки, вирус лихорадки Денге (типы 1-4), возбудитель энцефалита Св. Льюиса, возбудитель японского энцефалита, вирус энцефалита долины Муррея, вирус лихорадки Западного Нила и вирус Кунджина. Заболевания, связанные с HCV, включают прогрессирующий фиброз печени, воспаление и некроз, приводящий к циррозу, поражение печени конечной стадии и НСС; заболевания, связанные с другими патогенными флавивирусами, включают желтую лихорадку, лихорадку Денге, геморрагическую лихорадку и энцефалит. Кроме того, ряд соединений согласно настоящему изобретению являются активными в отношении мутированных линий HCV. Более того, многие из соединений согласно настоящему изобретению показывают благоприятный фармакокинетический профиль и обладают привлекательными свойствами в плане биологической доступности, включая приемлемый период полураспада, величину площади под кривой (area under curve - AUC) и амплитудные значения, а также отсутствие неблагоприятных явлений, таких как недостаточно быстрое начало действия и удерживание в тканях.
Противовирусная активность соединений формулы (I) в отношении HCV в условиях in vitro может испытываться в клеточной системе репликона HCV, как описано в публикации Lohmann et al. (1999) Science 285:110-113, c дополнительными модификациями, описанными в публикации Krieger et al. (2001) Journal of Virology 75:4614-4624 (введенной в описание в виде ссылки), типичный пример которой приведен в разделе «Примеры». Данная модель, хотя и не является моделью полной инфекции для HCV, широко используется в качестве наиболее надежной и эффективной модели автономной репликации HCV РНК, доступной в настоящее время. Соединения, проявляющие противовирусную активность в отношении HCV в данной клеточной модели, рассматриваются в качестве потенциальных лекарственных средств для лечения HCV инфекций у млекопитающих, подлежащих дальнейшему изучению. Следует представлять, что важно отличать соединения, которые специфически воздействуют на функции HCV, от соединений, которые вызывают цитотоксический или цитостатический эффекты в модели репликона HCV и как следствие вызывают снижение концентраций РНК HCV или связанных считывающих ферментов. Методы анализа, известные в области оценки клеточной цитотоксичности, основаны, например, на активности митохондриальных ферментов и включают применение флурогенных окислительно-восстановительных красителей, таких как резазурин. Кроме того, существуют фильтр подсчета клеток для оценки неселективного ингибирования связанной активности репортер-гена, такого как люцифераза светлячка. Клетки подходящих типов могут быть снабжены стабильной трансфекцией люциферазным геном-репортером, экспрессия которого зависит от конститутивно активного гена-промотера, и такие клетки могут использоваться в качестве фильтра-счетчика для исключения неселективных ингибиторов.
Благодаря противовирусным свойствам, в частности противовирусной активности в отношении HCV, соединения формулы (I) или любой их подгруппы, их N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы могут применяться для лечения пациентов, зараженных вирусом, в частности вирусом, который представляет собой HCV, и для профилактики вирусных инфекций, в частности HCV инфекций. Обычно соединения согласно настоящему изобретению могут применяться для лечения теплокровных животных, инфицированных вирусами, в частности, флавивирусами, такими как HCV.
Таким образом, соединения согласно настоящему изобретению или любой их подгруппы могут применяться в качестве лекарственного средства. Указанное применение в качестве лекарственного средства или способ лечения включает системное введение субъектам, инфицированным вирусом, или субъектам, восприимчивым к вирусным инфекциям, количества, эффективного для подавления состояний, связанных с вирусной инфекцией, в частности HCV инфекцией.
Настоящее изобретение относится также к применению соединений согласно настоящему изобретению или любой их подгруппы в производстве лекарственного средства для лечения или профилактики вирусной инфекции, в частности HCV инфекции.
Кроме того, настоящее изобретение относится к способу лечения теплокровного животного, инфицированного вирусом или подверженного риску инфицирования вирусом, в частности HCV, причем указанный способ включает введения противовирусно эффективного количества соединения формулы (I), как определено в описании, или соединения любой из подгрупп соединений формулы (I), как определено в описании.
Обычно подразумевается, что суточное противовирусно эффективное количество должно составлять от 0,01 мг/кг до 500 мг/кг массы тела, от 0,1 мг/кг до 50 мг/кг массы тела или от 0,5 мг/кг до 5 мг/кг максы тела. Может быть приемлемо вводить необходимую дозу в виде двух, трех, четырех или большего количества меньших доз через подходящие интервалы времени в течение дня. Указанные меньшие дозы могут приготавливаться в виде лекарственных форм стандартной дозы, например, содержащих от 1 до 1000 мг, в частности от 5 до 200 мг, активного ингредиента на лекарственную форму стандартной дозы.
Изобретение относится также к комбинации соединения формулы (I), включая его стереоизомерную форму, фармацевтически приемлемую соль или фармацевтически приемлемый сольват, и другого противовирусного соединения, в частности другого соединения, обладающего противовирусной активностью в отношении HCV. Термин «комбинация» может относиться к изделию, содержащему (а) соединение формулы (I), которое определено выше, и (b) необязательно другое противовирусное в отношении HCV соединение, в виде комбинированного препарата для одновременного, раздельного или последовательного применения при лечении HCV инфекций.
Соединения, обладающие противовирусной активностью в отношении HCV, которые могут применяться в таких комбинациях, включают лекарственные средства, выбранные из ингибитора HCV полимеразы, ингибитора HCV протеазы, ингибитора другой мишени в жизненном цикле HCV, и иммуномодуляторное лекарственное средство и их комбинации. Ингибиторы HCV полимеразы включают NM283 (валопицитабин), R803, JTK-109, JTK-003, HCV-371, HCV-086, HCV-796 и R-1479. Ингибиторы HCV протеаз (NS2-NS3 ингибиторы и NS3-NS4A ингибиторы) включают соединения, описанные в WO 02/18369 (см., например, страницу 273, строки 9-22 и со страницы 274 строки 4 до страницы 276 строки 11); BILN-2061, VX-950, GS-9132 (ACH-806), SCH-503034 и SCH-6. Дополнительные агенты, которые могут применяться, представляют собой лекарственные средства, описанные в WO 98/17679, WO 00/056331 (Vertex); WO 98/22496 (Roche); WO 99/07734, (Boehringer Ingelheim), WO 2005/073216, WO 2005/073195 (Medivir) и структурно схожие агенты.
Ингибиторы других мишеней в жизненном цикле HCV включают ингибиторы NS3 геликазы; ингибиторы металлопротеазы; ингибиторы антисенс-олигонуклеотидов, такие как ISIS-14803, AVI-4065 и т.п.; малые интерферирующие РНК (siRNA), такие как SIMPLEX-140-N; вектор-инкодируемые короткие РНК, образующие шпильки (shRNA); ДНКзимы; HCV специфические рибозимы, такие как гептазим, RPI.13919 и т.п.; ингибиторы проникновения, такие как HepeX-C, HuMax-HepC и т.п.; ингибиторы альфа-глюкозидазы, такие как целгосивир, UT-231B и т.п.; KPE-02003002; и BIVN 401.
Иммуномодуляторы включают изоформные соединения природного и рекомбинатного интерферона, включая α-интерферон, β-интерферон, γ-интерферон, ω-интерферон и т.п., такие как Intron A®, Roferon-A®, Canferon-A300®, Advaferon®, Infergen®, Humoferon®, Sumiferon MP®, Alfaferone®, IFN-beta®, Feron® и т.п.; полиэтиленгликолевые (пэгилированные) производные интерферона, такие как ПЭГ-интерферон-α-2а (Pegasys®), ПЭГ интерферон-α-2b (PEG-Intron®), пэгилированный IFN-α-con1 и т.п.; препараты интерферонов длительного действия и производные интерфероновых соединений, такие как интерферон, слитый с альбумином - альбуферон-α и т.д.; соединения, которые стимулируют синтез интерферона в клетках, такие как резиквимод и т.п.; интерлейкины; соединения, которые повышают выработку ответа хелпера Т клеток 1 типа, такие как SCV-07 и т.п.; агонисты TOLL-подобных рецепторов, такие как CpG-10101 (актилон), изаторибин и т.п.; тимозин α-1; ANA-245; ANA-246; гистамин-дигидрохлорид; пропагерманий; тетрахлордекаоксид; амплиген; IMP-321; KRN-7000; антитела, такие как сивацир (civacir), XTL-6865 и т.п.; и профилактические и терапевтические вакцины, такие как InnoVac C, HCV E1E2/MF59 и т.п.
Другие противовирусные средства включают рибавирин, амантадин, вирамидин, нитазоксанид; телбивудин; NOV-205; тарибавирин; ингибиторы внутреннего проникновения рибосом; ингибиторы вирусов широкого спектра действия, такие как IMPDH ингибиторы, и микофенольная кислота и ее производные, включая, но без ограничения, VX-950, меримеподиб (VX-497), VX-148 и/или VX-944); или комбинация любых из перечисленных выше противовирусных средств.
Особыми средствами, подходящими для применения в указанных комбинациях, являются интерферон-α (IFN-α), пэгилированный интерферон-α или рибавирин, а также терапевтические средства на основе антител против HCV эпитопов, малые интерферирующие РНК (siRNA), рибозимы, ДНКзимы, антисмысловые ДНК, антагонисты небольших молекул, например, NS3 протеазы, NS3 геликазы и NS5B полимеразы.
В соответствии с еще одним аспектом, предоставлена комбинация соединения формулы (I), которое определено выше, и соединения, обладающего противовирусной активностью в отношении ВИЧ. Последнее предпочтительно представляет собой ингибиторы ВИЧ, которые обладают положительным влиянием на метаболизм лекарственного средства и/или фармакокинетику, что улучшает его биологическую доступность. Примером такого ингибитора ВИЧ является ритонавир. Само по себе настоящее изобретение предоставляет также комбинацию, включающую (а) ингибитор HCV NS3/4а протеазы формулы (I) или его фармацевтически приемлемую соль; и (b) ритонавир или его фармацевтически приемлемую соль. Соединение ритонавир, его фармацевтически приемлемые соли и способы их получения описаны в WO 94/14436, в Патенте США 6037157, и ссылках, приведенных в них; в Патенте США № 5484801, Заявке на Патент США 08/402690, WO95/07696 и WO95/09614 раскрыты предпочтительные лекарственные формы ритонавира. Один вариант осуществления изобретения относится к комбинации, включающей (а) ингибитор HCV NS3/4a протеазы формулы (I) или его фармацевтически приемлемую соль; и (b) ритонавир или его фармацевтически приемлемую соль; необязательно включающей дополнительное соединение, обладающее противовирусной активностью в отношении HCV и выбранное из соединений, упомянутых выше.
Изобретение также относится к способу получения комбинации, которая определена выше, включающему стадию объединения соединения формулы (I), которое определено выше, и другого лекарственного средства, такого как противовирусное, в том числе противовирусное в отношении HCV или ВИЧ, лекарственное средство, в частности, противовирусное средство, упомянутое выше.
Указанные комбинации могут найти применение в производстве лекарственного средства для лечения HCV инфекции или инфекции другого патогенного флави- или пестивируса у млекопитающего, инфицированного им, указанная комбинация, в частности, включает соединение формулы (I), которое определено выше, и интерферон-α (IFN-α), пэгилированный интерферон-α или рибавирин. Или изобретение предоставляет способ лечения млекопитающего, в частности человека, инфицированного HCV или другим патогенным флави- или пестивирусом, способ включает введение указанному млекопитающему эффективного количества комбинации, которая определена в описании. В частности, указанный способ лечения включает системное введение указанной комбинации в таком количестве, которое является эффективным в лечении клинических состояний, связанных с HCV инфекцией.
В одном варианте осуществления изобретения упомянутые выше комбинации представлены в форме фармацевтической композиции, которая включает активные ингредиенты, описанные выше, и носитель, который описан выше. Каждый из активных ингредиентов может быть представлен в форме отдельного препарата и такие препараты могут вводиться совместно, или активные ингредиенты могут быть представлены в форме единого препарата, содержащего оба активных ингредиента и, если нужно, дополнительные активные ингредиенты. В первом примере комбинации также могут быть представлены в виде комбинированного препарата для одновременного, отдельного или последовательного применения в HCV терапии. Указанная композиция также может принимать любую из форм, описанных выше. В одном варианте осуществления изобретения оба ингредиента введены в одну лекарственную форму, такую как комбинированная лекарственная форма фиксированной дозы. В особом варианте настоящее изобретение предоставляет фармацевтическую композицию, включающую (а) терапевтически эффективное количество соединения формулы (I), его стереоизомерной формы, фармацевтически приемлемой соли или фармацевтически приемлемого сольвата, (b) терапевтически эффективное количество ритонавира или его фармацевтически приемлемой соли и (с) носитель.
Отдельные компоненты комбинаций согласно настоящему изобретению могут вводиться отдельно в разное время в течение курса терапевтического лечения, одновременно в виде нескольких доз, вводимых через определенные интервалы, или в разовой дозе. Подразумевается, что настоящее изобретение включает все такие схемы одновременного или поочередного лечения, и термин «введение» следует интерпретировать соответственно. В предпочтительном варианте осуществления изобретения раздельные лекарственные формы вводятся одновременно.
В одном варианте осуществления изобретения комбинации согласно настоящему изобретению содержат количество ритонавира или его фармацевтически приемлемой соли, достаточное для клинического улучшения биологической доступности ингибитора HCV NS3/4а протеазы формулы (I) по сравнению с биологической доступностью ингибитора HCV NS3/4а протеазы формулы (I), вводимого отдельно. Или комбинации согласно настоящему изобретению содержат количество ритонавира или его фармацевтически приемлемой соли, достаточное для повышения, по меньшей мере, одной из фармакокинетических переменных ингибитора HCV NS3/4а протеазы формулы (I), выбранных из t1/2, Cmin, Cmax, CSS, площади под кривой за период, равный 12 часам, или площади под кривой за период, равный 24 часам, относительно указанной, по меньшей мере, одной фармакокинетической переменной ингибитора HCV NS3/4а протеазы, вводимого отдельно.
Комбинации согласно настоящему изобретению могут вводиться людям в дозах, находящихся в интервалах значений, специфических для каждого компонента, содержащегося в указанных комбинациях, например, соединение формулы (I), как оно определено выше, и ритонавир или его фармацевтически приемлемая соль могут иметь уровни дозировок в интервале от 0,02 до 5,0 г/сутки.
Массовое соотношение соединения формулы (I) и ритонавира может находиться в интервале от примерно 30:1 до примерно 1:15, от примерно 15:1 до примерно 1:10, от примерно 15:1 до примерно 1:1, от примерно 10:1 до примерно 1:1, от примерно 8:1 до примерно 1:1, от примерно 1:5 до 1:1 или до примерно 5:1, от примерно 3:1 до примерно 1:1 или от примерно 2:1 до примерно 1:1. Соединение формулы (I) и ритонавир могут вводиться совместно один или два раза в сутки, предпочтительно перорально, где количество соединения формулы (I) на дозу находится в интервале от примерно 1 до примерно 2500 мг, от примерно 50 до примерно 1500 мг, от примерно 100 до примерно 1000 мг, от примерно 200 до примерно 600 мг или от примерно 100 до примерно 400 мг; и количество ритонавира на дозу находится в интервале от 1 до примерно 2500 мг, от примерно 50 до примерно 1500 мг, от примерно 100 до примерно 800 мг, от примерно 100 до примерно 400 мг или от 40 до 100 мг ритонавира.
Примеры
Приведенные далее примеры предназначены для иллюстрации настоящего изобретения, но не ограничивают его области. В некоторых примерах показано получение структурообразующих блоков, которые могут сочетаться с любым другим подходящим структурообразующим блоком, описанным в изобретении, а не только со структурообразующими блоками типичных примеров конечных продуктов формулы I.
Пример 1


Стадия a: этиловый эфир 1-[(3-оксо-2-оксабицикло[2.2.1]гептан-5-карбонил)амино]-2-винилциклопропанкарбоновой кислоты (1a)
К раствору 3-оксо-2-оксабицикло[2.2.1]гептан-5-карбоновой кислоты (857 мг, 5,5 ммоль) в ДМФА (14 мл) и ДХМ (25 мл) при комнатной температуре добавляют этиловый эфир 1-амино-2-винилциклопропанкарбоновой кислоты (1,15 г, 6,0 ммоль), HATU (2,29 г, 6,0 ммоль) и DIPEA (3,82 мл, 22 ммоль). Реакционную смесь перемешивают в атмосфере N2 при температуре окружающей среды в течение 1 часа. ЖХ/МС анализ показывает полную конверсию, после чего реакционную смесь концентрируют в вакууме. Остаток снова растворяют в ДХМ (100 мл) и 0,1 M HCl (водн.) и слои разделяют. Органическую фазу промывают NaHCO3 (водн.) и раствором соли, сушат (MgSO4) и фильтруют. Растворитель удаляют в вакууме, получая соединение, указанное в заглавии (1,6 г, 99%). ЖХ/МС (метод А): tR=2,46 мин, >95%, m/z (ESI+)=294(MH+).
Стадия b: диизопропилэтиламинная соль 2-(1-этоксикарбонил-2-винилциклопропилкарбамоил)-4-гидроксициклопентанкарбоновой кислоты (1b)
К раствору сложного эфира (1a) (800 мг, 2,73 ммоль) в воде (15 мл) в емкости микроволнового реактора объемом 20 мл добавляют DIPEA (1,2 мл, 6,8 ммоль) и магнитную мешалку. Емкость герметично закрывают и полученную несмешивающуюся взвесь энергично встряхивают перед внесением в микроволновую полость. После предварительного перемешивания в течение 1 мин. реакционную смесь облучают в течение 40 минут при установленной температуре 100°C. После охлаждения до 40°C прозрачный раствор концентрируют в вакууме и полученное масло коричневого цвета выпаривают 3 раза с ацетонитрилом для удаления остаточной воды. Полученное сырое соединение, указанное в заглавии, в форме DIPEA соли сразу используют в следующей стадии. ЖХ/МС (метод А): tR=1,29 мин, >95%, m/z (ESI+)=312(MH+).
Стадия c: этиловый эфир 1-{[2-(гекс-5-енилметилкарбамоил)-4-гидроксициклопентанкарбонил]амино}-2-винилциклопропанкарбоновой кислоты (lc)
Сырую кислоту (1b) (5,5 ммоль) растворяют в ДХМ (50 мл) и ДМФА (14 мл), после чего при комнатной температуре добавляют HATU (2,09 г, 5,5 ммоль), гекс-5-енилметиламин (678 мг, 6,0 ммоль) и DIPEA (3,08 мл, 17,5 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. ЖХ/МС анализ показывает полную конверсию, после чего реакционную смесь концентрируют в вакууме. Остаток снова растворяют в EtOAc (100 мл) и органический слой промывают 0,1 M HCl (водн.), K2CO3 (водн.) и раствором соли, сушат (MgSO4) и фильтруют. Растворитель удаляют в вакууме с получением масла, которое очищают флэш-хроматографией (диоксид кремния, EtOAc:MeOH), получая указанное в заглавии соединение (1,65 г, 74%). ТСХ (диоксид кремния): MeOH:EtOAc 5:95, Rf=0,5; ЖХ/МС (метод А): tR=3,44 мин, >95%, m/z (ESl+)=407(MH+).
Стадия d: этиловый эфир 1-{[4-этоксиметокси-2-(гекс-5-енилметил-карбамоил)циклопентанкарбонил]амино}-2-винилциклопропанкарбоновой кислоты (1d)
К раствору диена 1c (7,94 г, 0,02 моль) в ДХМ и DIPEA (9,7 мл, 6 экв.) с перемешиванием при 0°C (ледяная баня) добавляют хлорметилэтилэфир (2,79 г, 3 экв.). Реакционную смесь перемешивают при комнатной температуре в течение ночи, концентрируют на роторном испарителе и очищают колоночной хроматографией на силикагеле (элюирование с градиентом: EtOAc/петролейный эфир, 1:1->1:0), получая чистое соединение, указанное в заглавии, в виде желтоватого сиропа (6,67 г, 74%).
Стадия e: этиловый эфир 17-этоксиметокси-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-4-карбоновой кислоты (1e)
Диен (1d) (1,75 г) растворяют в сухом дихлорэтане (800 мл, 0,0046 М раствор). Через полученный раствор в течение 10 минут барботируют аргон, затем добавляют катализатор Ховейды-Граббса 1-го покол. (20 мг 3,5% (моль)) и реакционную смесь перемешивают при 95°C в течение ночи. В реакционную смесь добавляют дополнительную порцию катализатора (25 мг) (при барботировании аргона) и реакционную смесь перемешивают при 97°C. Реакционную смесь охлаждают до комнатной температуры, добавляют поглотитель (100 мг) и реакционную смесь перемешивают в течение 4 часов при комнатной температуре. Реакционную смесь фильтруют, фильтрат концентрируют на роторном испарителе и остаток очищают колоночной хроматографией на силикагеле, получая чистое соединение, указанное в заглавии (1,23 г, выход 75%).
Стадия f: 17-этоксиметокси-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-4-карбоновая кислота (1f)
Сложный эфир 1e (2,26 г) смешивают с ТГФ (20 мл), MeOH (20 мл) и раствором LiOH (1N 20 мл) и перемешивают при 55°C в течение ~17 часов. Реакционную смесь концентрируют на роторном испарителе, разбавляют водой (30-50 мл) и подкисляют 10% лимонной кислотой до pH 3-4. Мутный раствор экстрагируют этилацетатом (3x50 мл). Объединенные органические экстракты промывают водой и раствором соли и сушат над сульфатом магния. Осушитель удаляют фильтрацией и этилацетат удаляют на роторном испарителе. Остаток сушат в высоком вакууме, получая 2 г указанного в заголовке соединения в виде белой пены. Полученный продукт используют в следующей стадии без дополнительной очистки.
Стадия g: (17-этоксиметокси-13-метил-2,14-диоксо-3.13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-4-карбонил)амид циклопропансульфоновой кислоты (1g)
Кислоту 1f растворяют в сухом ДХМ (20 мл). В раствор добавляют EDC (1,2 экв.) и реакционную смесь перемешивают в течение 3 часов при комнатной температуре. ЖХ-МС показывает полную конверсию исходного вещества. Раствор разбавляют ДХМ и промывают водой (3×20 мл). Водную фазу экстрагируют ДХМ и объединенные ДХМ экстракты промывают раствором соли, сушат над сульфатом магния, фильтруют и концентрируют на роторном испарителе, получая сироп коричневатого цвета, который используют в следующей стадии без дополнительной очистки.
Сироп растворяют в сухом ДХМ (20 мл), к полученному раствору добавляют циклопропансульфонамид (1,1 экв.) и затем DBU. Раствор перемешивают при комнатной температуре в течение 17 часов. Ход реакции отслеживают ЖХ-МС. Реакционную смесь разбавляют ДХМ (70 мл), промывают 10% лимонной кислотой (2x20 мл) и раствором соли, сушат над сульфатом магния, концентрируют на роторном испарителе и очищают колоночной хроматографией на YCM диоксиде кремния (примерно 50 г, элюирование: эфир), получая указанное в заглавии соединение в виде белой пены (83%), (M+H)+ 512.
Стадия h: (17-гидрокси-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-4-карбонил)амид циклопропансульфоновой кислоты (1h)
Этоксиэфир 1g растворяют в смеси ТГФ/MeOH/вода (1:1:1, общий объем 30 мл), после чего при перемешивании добавляют 2,5 мл конц. соляной кислоты. Реакционную смесь перемешивают в течение ночи при комнатной температуре, контролируя ход реакции с помощью ЖХ-МС. Затем реакционную смесь выливают в насыщ. водный раствор NaHCO3 (50 мл) и упаривают до половины объема на роторном испарителе. Полученную смесь подкисляют 10% лимонной кислотой и экстрагируют ДХМ (3×20 мл). Объединенные органические фазы промывают раствором соли, сушат над сульфатом магния, фильтруют, концентрируют на роторном испарителе и сушат в высоком вакууме в течение ночи. Полученный продукт используют далее без дополнительной очистки.
Стадия i: 4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (2-цианофенил)карбаминовой кислоты (1i)
Спирт 1h (11 мг) растворяют в сухом дихлорметане (2 мл), к полученному раствору добавляют 2-изоцианобензонитрил (2 экв.) и затем триэтиламин (5 мкл). Реакционную смесь перемешивают в течение ночи при комнатной температуре и затем концентрируют на роторном испарителе. Очистка остатка препаративной ВЭЖХ (вода/ацетонитрил с добавлением 0,1% ТФУК, элюирование с градиентом: 30-80) приводит к получению указанного в заглавии соединения (5 мг, 27%), [M+H]+ 598.
Пример 2

(4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (2-феноксифенил)карбаминовой кислоты (2)
Спирт 1h (11 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но используя 2-изоцианофеноксибензоловый эфир вместо 2-изоцианбензонитрила, получая указанное в заглавии соединение (10 мг, 63%), [M+H]+ 665.
Пример 3

(4-циклопропансульфониламинокарбонил-13-метил-
2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (2-метоксифенил)карбаминовой кислоты (3)
Спирт 1h (11 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 2-изоцианометоксибензилового эфира вместо 2-изоцианбензонитрила, получая указанное в заголовке соединение (9 мг, 58%), [M+H]+ 603.
Пример 4

4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадец-7-ен-17-иловый эфир (3-циано-5-метоксифенил)карбаминовой кислоты (4)
Спирт 1h (15 мг) растворяют в сухом ДХЭ, к полученному раствору добавляют 20 мг бикарбоната натрия и затем 2 мл раствора фосгена в толуоле (20%). Реакционную смесь перемешивают при комнатной температуре в течение 3 часа (полная конверсия до хлоримидата в соответствии с данными ЖХ-МС), затем концентрируют на роторном испарителе и сушат от избытка фосгена в высоком вакууме (1,5 часа). Сухую реакционную смесь переносят в пробирку микроволнового реактора (2-5 мл), смешивают с сухим ДХЭ (3 мл), 3-амино-5-метоксибензонитрилом (2 экв.), карбонатом калия (9 мг, 1,5 экв.), распыленными молекулярными ситами (4Å, 5 мг) и выдерживают в микроволновой печи при 100°C в течение 45 мин. Реакционную смесь пропускают через слой диоксида кремния (элюирование: ДХМ, затем 10% метанол в ДХМ). Полученные фракции, содержащие целевое карбаматное соединение, объединяют, концентрируют на роторном испарителе и очищают колоночной хроматографией на YMC диоксиде кремния (15 г, элюирование: смесь этилацетат/петролейный эфир (1:3) для удаления избытка анилина, затем дихлорметан и далее 2% метанол в дихлорметане), получая указанное в заголовке соединение в виде порошка (15 мг, 8%), [M+H]+ 628.
Пример 5
Метиловый эфир 3-(4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-илоксикарбониламино)бензойной кислоты (5)
Спирт 1h (20 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но используя метиловый эфир 3-аминобензойной кислоты вместо 3-амино-5-метоксибензонитрила и получая указанное в заголовке соединение (10 мг, 36%), [M+H]+ 631.
Пример 6

Стадия a: (17-этоксиметокси-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-4-карбонил)амид 1-метилциклопропансульфоновой кислоты (6a)
Кислоту 1f подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия g), но с использованием метилциклопропансульфонамида вместо циклопропансульфонамида, получая указанное в заголовке соединение.
Стадия b: (17-гидрокси-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-4-карбонил)амид 1-метилциклопропансульфоновой кислоты (6b)
Этоксиэфир 3a обрабатывают в соответствии с методикой, описанной в примере 1 (стадия h), получая указанное в заголовке соединение.
Стадия c: 13-метил-4-(1-метилциклопропансульфониламинокарбонил)-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (5-циано-2-метоксифенил)карбаминовой кислоты (6c)
Спирт 3b (30 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 3-амино-4-метоксибензонитрила вместо 3-амино-5-метокси-бензонитрила, получая указанное в заголовке соединение (12 мг, 33%), [M+H]+ 642.
Пример 7

13-метил-4-(1-метил-циклопропансульфониламинокарбонил)-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (3-метилкарбамоилфенил)карбаминовой кислоты (7)
Спирт 3b (34 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 3-амино-N-метилбензамида вместо 3-амино-5-метоксибензонитрила, получая указанное в заголовке соединение (8 мг, 19%), [M+H]+ 644.
Пример 8

4-(1-метилциклопропансульфониламинокарбонил)-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (3-хлорфенил)карбаминовой кислоты (5)
Спирт 3b (35 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 3-хлоранилина вместо 3-амино-5-метоксибензонитрила, получая указанное в заголовке соединение (27 мг, 66%), [M+H]+ 622.
Пример 9

13-метил-4-(1-метил-циклопропансульфониламинокарбонил)-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (3-диметиламинофенил)карбаминовой кислоты (6)
Спирт 3b (80 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 3-диметиламиноанилина вместо 3-амино-5-метоксибензонитрила, получая указанное в заголовке соединение (50 мг, 40%), [M+H]+ 63.
Пример 10

4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (3-метоксифенил)карбаминовой кислоты (10)
Спирт 1h (6 мг) растворяют в сухом дихлорметане (2 мл) и к полученному раствору добавляют сначала 3-изоцианометоксибензол (2 экв.), затем триэтиламин (5 мкл). Реакционную смесь перемешивают в течение ночи при комнатной температуре и затем концентрируют на роторном испарителе. Очистка остатка препаративной ВЭЖХ (элюирование: вода/ацетонитрил с 0,1% ТФУК, градиент 30-80) приводит к получению указанного в заглавии соединения (2 мг, 27%), [M+H]+ 603.
Пример 11
4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (3-феноксифенил)карбаминовой кислоты (11)
Спирт 1h (10 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 3-изоцианофеноксибензола вместо 3-изоцианометоксибензола, получая указанное в заголовке соединение (11 мг, 72%), [M+H]+ 665.
Пример 12

4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (3-цианофенил)карбаминовой кислоты (12)
Спирт 1h (10 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 3-изоцианобензонитрила вместо 3-изоцианометоксибензола, получая указанное в заголовке соединение (10 мг, 75%), [M+H]+ 598.
Пример 13

13-метил-4-(1-метилциклопропансульфониламинокарбонил)-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (3-цианофенил)карбаминовой кислоты (13)
Спирт 3b (20 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 7 (стадия i), но с использованием 3-изоцианобензонитрила вместо 3-изоцианометоксибензола, получая указанное в заголовке соединение (13 мг, 51%), [M+H]+ 612.
Пример 14

13-метил-4-(1-метил-циклопропансульфониламинокарбонил)-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (3-метоксифенил)карбаминовой кислоты (14)
Спирт 3b (18 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 7 (стадия i) получая указанное в заголовке соединение (14 мг, 59%), [M+H]+ 617.
Пример 15

4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир м-толилкарбаминовой кислоты (15)
Спирт 1h (17 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 3-изоцианотолуола вместо 3-изоцианометоксибензола, получая указанное в заголовке соединение (6 мг, 26%), [M+H]+ 587.
Пример 16

4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (2-фтор-5-метилфенил)карбаминовой кислоты (16)
Спирт 1h (23 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 4-фтор-3-изоцианотолуола вместо 3-изоцианометоксибензола, получая указанное в заголовке соединение (14 мг, 46%), [M+H]+ 605.
Пример 17

4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-l7-иловый эфир [3-(2-метилтиазол-4-ил)фенил]карбаминовой кислоты (17)
Спирт 1h (30 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 4-(3-изоцианофенил)-2-метилтиазола вместо 3-изоцианометоксибензола, получая указанное в заголовке соединение (19 мг, 49%), [M+H]+ 670.
Пример 18
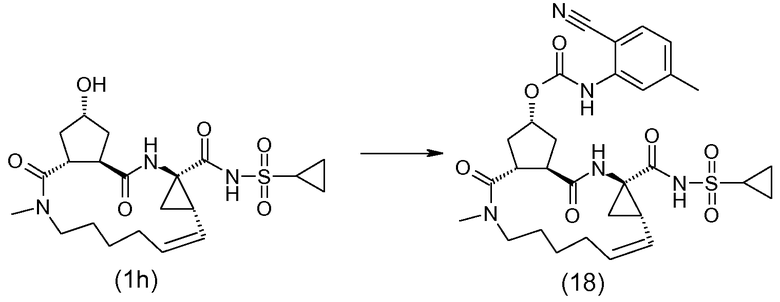
4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-17-иловый эфир (2-циано-5-метилфенил)карбаминовой кислоты (18)
Спирт 1h (30 мг) подвергают взаимодействию в соответствии с методикой, описанной в примере 1 (стадия i), но с использованием 2-амино-4-метилбензонитрила вместо 3-амино-5-метокси-бензонитрила, получая указанное в заголовке соединение (25 мг, 70%).
Пример 19

Стадия a: трет-бутиловый эфир (1-гидроксиметил-2-винилциклопропил)карбаминовой кислоты (19a)
К раствору этилового эфира 1-трет-бутоксикарбониламино-2-винилциклопропанкарбоновой кислоты (0,51 г, 2,0 ммоль) в ТГФ (10 мл) при 0°C добавляют 2M раствор борогидрида лития (4 мл, 8 ммоль). Реакционную смесь анализируют ТСХ (смесь гексан-этилацетат (7:3), подкрашенная с использованием сульфата аммониймолибдата-церия в водной 10% серной кислоте) и после перемешивания в течение ночи при комнатной температуре реакцию осторожно гасят с помощью водной 10% лимонной кислоты (25 мл, добавление по каплям при 0°C). Полученную смесь промывают дихлорметаном (3×10 мл) и объединенные органические слои сушат (Na2SO4), фильтруют и концентрируют. Флэш-хроматография остатка (элюирование: гексан-этилацетат (1:1)) с последующим концентрированием подходящих фракций и сушкой остатка в вакууме в течение ночи приводит к получению продукта в виде бесцветного сиропа (0,407 г, 1,91 ммоль, 96%).
1H ЯМР (400 МГц, CDC13): δ 0,98 (м, 1H), 1,15 (м, 1H), 1,44 (с, 9H), 1,84 (м, 1H), 3,20 (уш. с, 1H), 3,60 (дд, 1H), 3,78 (уш.м, 1H), 5,10-5,26 (м, 3H), 5,70 (м, 1H).
Стадия b: трет-бутиловый эфир (1-формил-2-винилциклопропил)карбаминовой кислоты (19b)
К раствору спирта 19a (0,152 г, 0,71 ммоль) в дихлорметане (5 мл) при комнатной температуре с перемешиванием добавляют периодинан Десса-Мартина (0,33 г, 0,78 ммоль). Ход реакции контролируют ТСХ (гексан-этилацетат (3:2), УФ-визуализация и нанесение пятен с использованием молибдата аммония-сульфата церия в водном 10% растворе соляной кислоты). Окрашивание указывает на довольно чистое взаимодействие, но УФ контроль указывает на наличие нескольких побочных продуктов. Спустя 1 час полученный желтовато-красный раствор разбавляют дихлорметаном (20 мл), затем промывают смесью (1:1) 10% водный раствор тиосульфата натрия/насыщенный водный раствор гидрокарбоната натрия (3×20 мл), затем сушат (Na2SO4), фильтруют и концентрируют. Флэш-хроматография остатка с использованием элюирования со ступенчатым градиентом (этилацетат в гексане 20-30 %) с последующим концентрированием подходящих фракций и сушкой остатка в вакууме в течение ночи приводит к получению указанного в заглавии соединения в форме бесцветного масла (0,054 г, 0,255 ммоль, 36 %).
Стадия c: трет-бутиловый эфир [1-(трет-бутилкарбамоилгидроксиметил)-2-винилциклопропил]карбаминовой кислоты (19c)
К раствору альдегида 19b (0,054 г, 0,255 ммоль) и трет-бутилизонитрила (0,043 мл, 0,38 ммоль) в дихлорметане (1 мл) и пиридине (0,083 мл, 1,02 ммоль) в атмосфере азота добавляют трифторуксусную кислоту (0,039 мл, 0,51 ммоль). Смесь выдерживают в течение 30 минут при комнатной температуре и далее перемешивают в течение 2 дней. После того, как ТСХ (7:3 гексан-этилацетат) и ЖХ-МС показывают примерно 60% конверсию, реакционную смесь разбавляют этилацетатом (10 мл). Раствор последовательно промывают водной 10% лимонной кислотой (3×5 мл) и насыщенным водным раствором гидрокарбоната натрия (3×5 мл), затем сушат (Na2SO4), фильтруют и концентрируют. Остаток обрабатывают смесью (1:1:1) водн. 1M LiOH/ТГФ/MeOH (1,5 мл) в течение 10 минут при комнатной температуре, после этого разбавляют водной 10% лимонной кислотой и переносят в этилацетат, затем сушат (Na2SO4), фильтруют и концентрируют. Колоночная хроматография остатка (элюирование: гексан-этилацетат (7:3)) с последующим концентрированием подходящих фракций и сушкой остатка в вакууме в течение ночи приводит к получению продукта в виде твердого бесцветного вещества (0,027 г, 0,086 ммоль, 34%).
1H ЯМР (400 МГц, CDCl3): δ 1,24 (м, 1H), 1,33-1,40 (м, 10 H), 1,44 (с, 9H), 1,87 (м, 1H), 3,65 (д, 1H), 5,21 (м, 3H), 5,50 (д, 1H), 5,89 (м, 1H), 7,03 (уш. с, 1H).
α-Гидроксиамид-производные соединений формулы (I) согласно настоящему изобретению получают удалением N-ВОС из указанного в заголовке соединения с последующим сочетанием полученного амина с кислотой, такой как кислота 1f, в соответствии с методикой, описанной в примере 1 (стадия g), с последующим удалением защиты гидроксильной группы и реакцией карбамоилирования в соответствии с методиками примера 1 (стадии h и I, соответственно).
Общая методика окисления α-гидроксиамидов до α-кетоамидов:
Обычно α-гидроксиамид растворяют в дихлорметане (20-30 мл/г) при комнатной температуре, затем добавляют периодинан Десса-Мартина (1,1 экв.) и состав реакционной смеси анализируют с помощью ТСХ и ЖХ-МС. После завершения реакции или близко к завершению реакции смесь разбавляют дихлорметаном и затем промывают смесью водн. 10% тиосульфат натрия/насыщ. водн. раствор гидрокарбоната натрия (1:1) (х3), затем сушат (Na2SO4), фильтруют и концентрируют. Остаток очищают колоночной хроматографией или препаративнной ЖХ.
Пример 20

Стадия a: 1-{[2-(гекс-5-енилметилкарбамоил)-4-гидроксициклопентанкарбонил]амино}-2-винилциклопропанкарбоновая кислота (20a)
Соединение 1c (493 мг, 1,21 ммоль) растворяют в ДМФА (1 мл) и полученный раствор переносят в реакционную емкость микроволнового реактора объемом 20 мл. Затем добавляют водный раствор LiOH (2 M, 10,5 мл) и магнитную мешалку (stirbar). Реакционную емкость герметично закрывают и полученную взвесь энергично встряхивают перед внесением в микроволновый реактор. Реакционную смесь обрабатывают микроволнами в течение 30 минут при температуре 130°C. Реакционную смесь охлаждают до 40°C, полученный прозрачный раствор подкисляют до pH 2 с помощью водной HCl (1 M, 24 мл) и экстрагируют EtOAc (3x20 мл). Объединенные органические слои промывают раствором соли, сушат (MgSO4) и фильтруют. Растворитель выпаривают в вакууме, получая указанное в заглавии соединение (410 мг, 90 %). ЖХ/МС >95%, м/z (ESI+)=379(MH+).
Стадия b: (1-циклопропансульфониламинокарбонил-2-винилциклопропил)амид 2-(гекс-5-енилметиламинокарбонил)-4-гидроксициклопентанкарбоновой кислоты (20b)
Сырую кислоту 20a (410 мг, 1,09 ммоль) растворяют в ДМФА (1,5 мл) и ДХМ (4,5 мл) и к полученному раствору при комнатной температуре добавляют EDAC (417 мг, 2,18 ммоль). Смесь выдерживают с перемешиванием при комнатной температуре в течение 10 минут. К смеси добавляют DMAP (133 мг, 1,09 ммоль) и снова выдерживают смесь при комнатной температуре в течение 20 минут. Затем к смеси добавляют предварительно смешанный раствор амида циклопропансульфоновой кислоты (527 мг, 4,36 ммоль) и DBU (663 мг, 4,36 ммоль) в ДМФА (2 мл) и ДХМ (2 мл) и полученную смесь выдерживают в микроволновой печи при 100°C в течение 30 минут. Полученный раствор красного цвета концентрируют в вакууме и снова растворяют в EtOAc (20 мл). Органическую фазу промывают 1 M HCl (водн.) (3×10 мл) и раствором соли (10 мл), сушат (MgSO4) и фильтруют. Растворитель выпаривают в вакууме, получая сырой сульфонамид, который далее очищают хроматографией (диоксид кремния, EtOAc/MeOH, 97,5:2,5), получая указанное в заглавии соединение (403 мг, 77%); ЖХ/МС, >95%, м/z (ESI+)=482(MH+).
Соединения формулы (I) могут быть получены из промежуточных продуктов (20b) посредством реакции карбамоилирования, проведенной любым из методов, описанных выше, например, в соответствии с методиками, описанными в примере 1 (стадия i) или в примере 10, с последующей реакцией закрытия цикла в соответствии с методикой, описанной в примере 1 (стадия е).
Пример 21


Стадия a: трет-бутиловый эфир 2-(1-этоксикарбонил-2-винилциклопропилкарбамоил)-4-гидроксипирролидин-1-карбоновой кислоты (21a)
Boc-защищенный 4-гидроксипролин (4 г, 17,3 ммоль), HATU (6,9 г, 18,2 ммоль) и этиловый эфир 1-амино-2-винилциклопропанкарбоновой кислоты, полученный в соответствии с методикой, описанной в WO03/099274, (3,5 г, 18,3 ммоль), растворяют в ДМФА (60 мл) и охлаждают до 0°C на ледяной бане. К смеси добавляют диизопропилэтиламин (DIPEA) (6 мл). Ледяную баню удаляют и смесь оставляют на ночь при температуре окружающей среды. Затем добавляют дихлорметан (~80 мл), органическую фазу промывают водным раствором гидрокарбоната натрия, лимонной кислотой, водой, раствором соли и сушат над сульфатом натрия. Очистка флэш-хроматографией (элюирование: эфир →7% метанол в эфире) приводит к получению чистого соединения, указанного в заглавии (6,13 г, 96%)
Стадия b: трет-бутиловый эфир 2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-4-(4-нитробензоилокси)пирролидин-1-карбоновой кислоты (21b)
Соединение 21a (11,8 г, 32,0 ммоль) и пиридин (27 мл, 305 ммоль) растворяют в ДХМ (200 мл) и охлаждают до 0°C, добавляют 4-нитробензоилхлорид (6,6 г, 35,6 ммоль) и полученный раствор перемешивают при комнатной температуре в течение ночи. Реакционную смесь промывают NaHCO3 (водн.), водным раствором лимонной кислоты и раствором соли, сушат над MgSO4 и упаривают на диоксиде кремния. Сырой продукт очищают колоночной хроматографией на силикагеле (EtOAc/н-гептан: 50:50), получая 11,84 г (72%) указанного в заголовке соединения.
Стадия c: 5-(1-этоксикарбонил-2-винилциклопропилкарбамоил)пирролидин-3-иловый эфир 4-нитробензойной кислоты (21c)
Из соединения 21b (11,84 г, 22,9 ммоль) удаляют защитную группу с помощью ТФУК (30 мл), растворенной в ДХМ (100 мл), и затем обрабатывают методом, известным в данной области техники, получая указанное в заголовке соединение (9,37 г, 98%).
Стадия d: 5-(1-этоксикарбонил-2-винилциклопропилкарбамоил)-1-[гепт-6-енил-(4-метоксибензил)карбамоил]пирролидин-3-иловый эфир 4-нитробензойной кислоты (21d)
Амин 21c (4,68 г, 11,2 ммоль) растворяют в ТГФ (100 мл), добавляют NaHCO3 (насыщенный раствор) (прим. 5 мл) и затем раствор фосгена (20% в толуоле, 11,6 мл, 22,5 ммоль). Реакционную смесь энергично перемешивают в течение 1 часа и затем фильтруют, упаривают и снова растворяют в ДХМ (100 мл). К смеси добавляют NaHCO3 (насыщенный раствор) (прим. 5 мл) и затем гепт-6-енил-(4-метоксибензил)амин (3,92 г, 16,8 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи, фильтруют и упаривают на диоксиде кремния. Сырой продукт очищают колоночной хроматографией на силикагеле (EtOAc/н-гептан: 25/75), получая указанное в заголовке соединение (6,9 г, 91%).
Стадия e: этиловый эфир 14-(4-метоксибензил)-18-(4-нитробензоилокси)-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0*4,6*]нонадец-7-ен-4-карбоновой кислоты (21e)
Диен 21d (406 мг, 0,6 ммоль) растворяют в ДХЭ (250 мл) и дегазируют. К раствору добавляют катализатор Говейды-Граббса 2-го поколения (26 мг, 0,042 ммоль) и раствор кипятят с обратным холодильником в течение 3 часов. После этого смесь упаривают и остаток используют непосредственно в следующей стадии.
Стадия f: этиловый эфир 18-гидрокси-14-(4-метоксибензил)-2,15-диоксо-3,14,16-триазатрицикло-[14.3.0.0*4,6*]нонадец-7-ен-4-карбоновой кислоты (21f)
Сырое соединение 21e (445 мг) растворяют в смеси ТГФ (20 мл), MeOH (10 мл) и воды (10 мл). После охлаждения смеси до 0°C добавляют 1M LiOH (2 мл). Спустя 1,5 часа гидролиз завершают, к смеси добавляют HOAc (1 мл) и раствор упаривают до объема примерно 10 мл. Добавляют воду и смесь экстрагируют ДХМ (2x30 мл). Объединенные органические экстракты промывают NaHCO3 (водн.), водой, раствором соли и сушат над MgSO4. Сырой продукт очищают колоночной хроматографией на силикагеле (элюирование с градиентом: ДХМ/MeOH от 100/0 до 80/20), получая указанное в заголовке соединение (201 мг, 67%).
Стадия g: этиловый эфир 18-этоксиметокси-14-(4-метокси-бензил)-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0*4.6*]нонадец-7-ен-4-карбоновой кислоты (21g)
К раствору спирта 21f (1,35 г, 2,70 ммоль, 75% чистота) и N-этилдиизопропиламина (1,42 мл, 8,1 ммоль) в дихлорметане (15 мл), охлажденному до 0°C, при перемешивании добавляют хлорметилэтилэфир (0,5 мл, 5,4 ммоль). Смеси дают возможность нагреться при перемешивании до комнатной температуры, после чего смесь охлаждают до 0°С, добавляют дополнительное количество N-этилдиизопропиламина (1 мл, 5,7 ммоль) и хлорметилэтилэфира (0,3 мл, 3,2 ммоль) и полученную смесь перемешивают в течение 16 часов при комнатной температуре. Далее реакционную смесь переносят в колонку с силикагелем (ступенчатое элюирование с градиентом: этилацетат в гексане 50-80%). Концентрирование подходящих фракций приводит к получению указанного в заглавии соединения в виде сиропа коричневатого цвета, который кристаллизуется на воздухе (0,8 г, 53%). LR-МС: C30H44N3O7 - вычислено: 558; найдено: 558 [M+H].
Стадия h: 18-этоксиметокси-14-(4-метоксибензил)-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0*4,6*]нонадец-7-ен-4-карбоновая кислота (21h)
Раствор сложного эфира 21g (0,775 г, 1,39 ммоль) в смеси ТГФ-метанол-водный 1M LiOH (1:1:1) (36 мл) перемешивают при комнатной температуре в течение 3,5 часов, после чего ТСХ (95:5 и 9:1 дихлорметан-метанол) и ЖХ-МС показывают полную конверсию исходного вещества в карбоновую кислоту. После этого реакционную смесь упаривают приблизительно до 1/3 объема, затем разбавляют водой (10 мл) и подкисляют до приблизительно pH 4, используя 10% лимонную кислоту (60 мл), в процессе чего образуется осадок. Смесь промывают этилацетатом (3×25 мл) и объединенные органические слои промывают раствором соли (2×50 мл), затем сушат (Na2SO4) фильтруют и упаривают. Остаток концентрируют из толуола (3×10 мл), получая сырое соединение, указанное в заголовке, в виде не совсем белой пены (0,75 г, количественный выход). LR-МС: C28H40N3O7: вычислено 530; найдено 530 [M-H].
Стадия i: соединение 21i
К раствору карбоновой кислоты 21h (примерно 1,39 ммоль) в дихлорметане (10 мл) при комнатной температуре добавляют N-этил-N'-(3-диметиламинопропил)карбодиимид × HCl (0,32 г, 1,67 ммоль), затем перемешивают в течение ночи, после чего ЖХ-МС показывает полную конверсию кислоты в продукт. После этого реакционную смесь разбавляют дихлорметаном (10 мл), промывают водой (3×10 мл), затем сушат (Na2SO4) фильтруют и упаривают с получением твердого бесцветного вещества (выход сырого продукта: 0,7 г), которое сразу используют в следующей стадии. LR-МС: C28H38N3O6: вычислено 512; найдено 512 [M+H].
Стадия j: [18-этоксиметокси-14-(4-метокси-бензил)-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0*4,6*]нонадец-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (21j)
К раствору сырого оксазолинона 21i (0,328 г, 0,64 ммоль) в дихлорметане (4 мл) при перемешивании добавляют циклопропилсульфонамид (0,117 г, 0,96 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (0,19 мл, 1,3 ммоль), затем перемешивают при комнатной температуре в течение ночи. Реакционную смесь анализируют ЖХ-МС, затем разбавляют дихлорметаном (20 мл), промывают последовательно водной 10% лимонной кислотой (3×15 мл) и раствором соли (1×15 мл), затем сушат (Na2SO4), фильтруют и концентрируют, получая не совсем белую пену. Колоночная хроматография остатка (ступенчатое элюирование с градиентом: этилацетат в толуоле 60-100%) с последующим концентрированием и сушкой подходящих фракций приводит к получению указанного в заглавии соединения в виде бесцветной пены (0,27 г, 66% для 3 стадий).
1H ЯМР: (500 МГц, ДМСО-d6): δ 0,9-1,6 (м, 14H), 1,80 (м, 1H), 1,90 (м, 1H), 2,0-2,2 (м, 3H), 2,25 (м, 1H), 2,95 (м, 1H), 3,05 (м, 1H), 3,3-3,4 (м, 2H), 3,50 (кв, 2H), 3,7-3,8 (м, 4H), 3,97 (д, 1H), 4,3-4,4 (м, 2H), 4,55 (д, 1H), 4,63 (м, 2H), 5,12 (м, 1 H), 5,70 (м, 1H), 6,88 (д, 2H), 7,19 (д, 2H), 8,12 (с, 1H). LR-МС: C31H45N4O8S: вычислено 633; найдено 633 [M+H].
Стадия k: (18-гидрокси-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0*4,6*]нонадец-7-ен-4-карбонил)амид циклопропансульфоновой кислоты (21k)
Раствор ацеталя 21j (0,038 г, 0,06 ммоль) в смеси ТГФ-метанол-2M водн. соляная кислота (1:1:1) (1,5 мл) перемешивают при комнатной температуре в течение 30 минут, затем добавляют дополнительную порцию соляной кислоты (0,1 мл) и полученную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь нейтрализуют с использованием насыщенного водного раствора гидрокарбоната натрия, затем концентрируют на диоксид кремния. Флэш-хроматография остатка (элюирование: этилацетат/метанол 9:1) приводит к получению продукта в виде бесцветной пены (0,020 г, 73%). LR-МС: C20H29N4O6S: вычислено 453; найдено 453 [M-H].
Ингибиторы согласно настоящему изобретению получают из промежуточного продукта 21k посредством реакции карбамоилирования с использованием любого из способов, описанных выше, например, в примере 1 (стадия i) или в примере 10.
Пример 22

Стадия a: [18-этоксиметокси-14-(4-метоксибензил)-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0*4,6*]нонадец-7-ен-4-карбонил]амид 1-метилциклопропансульфоновой кислоты (22a)
К раствору оксазолинона 21i (0,372 г, 0,73 ммоль) в дихлорметане (4 мл) при перемешивании добавляют циклопропилметилсульфонамид (0,147 г, 1,09 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (0,22 мл, 1,45 ммоль). Полученную смесь перемешивают при комнатной температуре в течение ночи. Обработка и хроматография реакционной смеси как описано в примере 19 (стадия j) приводит к получению целевого продукта в виде бесцветного сиропа, который кристаллизуется на воздухе (0,31 г, 65% для 3 стадий). 1H ЯМР (500 МГц, ДМСО-d6): δ 0,92 (м, 2H), 1,1-1,6 (м, 15H), 1,78 (м, 1H), 1,88 (м, 1H), 2,0-2,1 (м, 3H), 2,26 (м, 1H), 3,02 (м, 1H), 3,2-3,4 (м, 2H), 3,49 (кв, 2H), 3,7-3,8 (м, 4H), 3,95 (д, 1H), 4,3-4,4 (м, 2H), 4,54 (д, 1H), 4,6-4,7 (м, 2H), 5,06 (м, 1H), 5,69 (м, 1H), 6,88 (д, 2H), 7,19 (д, 2H), 8,22 (с, 1H), 11,23 (с, 1H). LR-МС: C32H47N4O8S: вычислено 647; найдено 647 [M+H].
Стадия b: (18-гидрокси-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0*4,6*]нонадец-7-ен-4-карбонил)амид 1-метилциклопропансульфоновой кислоты (22b)
Из ацеталя 22a (0,301 г, 0,465 ммоль) удаляют защитную группу, используя смесь дихлорметан/трифторуксусная кислота/H2O (2:1:0,1) (6,2 мл), при комнатной температуре в течение 4 часов, затем концентрируют на диоксиде кремния и очищают флэш-хроматографией (элюирование: этилацетат/метанол 9:1), получая продукт в виде бесцветной пены (0,065 г, 30%). LR-МС: C21H33N4O6S. Вычислено 469; найдено 469 [M+H].
Соединения формулы (I) получают из промежуточного продукта 22b в соответствии с реакцией карбамоилирования с использованием любого из способов, описанных выше, например, в примере 1 (стадия i) или в примере 10.
Пример 23

Стадия i: 4-циклопропансульфониламинокарбонил-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-17-иловый эфир 6H-фенантридин-5-карбоновой кислоты (1i)
5,6-дигидрофенантридин (1k) получают в соответствии со следующей методикой: в круглодонную колбу объемом 500 мл, снабженную магнитной мешалкой и обратным холодильником, загружают 6(5H)-фенантридинон (1000 мг, 5123 мкмоль), ТГФ (250 мл) (тонкая суспензия). Через содержимое колбы барботируют азот, в смесь добавляют 2M раствор BH3-диметилсульфидного комплекса в ТГФ (10 мл). Полученную смесь кипятят с обратным холодильником при перемешивании в течение 24 часов. После этого ТСХ показывает, что реакция еще не завершена (этилацетат/гептан 1/1). Растворители удаляют при пониженном давлении. К остатку добавляют воду (50 мл) и этилацетат (100 мл). Водный слой промывают этилацетатом (3×100 мл). Органические слои объединяют, сушат (сульфат натрия), твердый осадок удаляют фильтрацией и растворители удаляют при пониженном давлении. Остаток очищают колоночной хроматографией с диоксидом кремния (элюирование с градиентом: гептан до 50% этилацетат в гептане). Целевые фракции объединяют и растворители удаляют при пониженном давлении, получая продукт в виде твердого не совсем белого вещества (270 мг, выход: 29%). ЖХ-МС: 182 (M+E)+.
Спирт 1h (15 мг) растворяют в сухом ДХЭ, к полученному раствору добавляют 20 мг бикарбоната натрия и затем 2 мл раствора фосгена в толуоле (20%). Реакционную смесь перемешивают при комнатной температуре в течение 3 часа, затем концентрируют на роторном испарителе и избыток фосгена удаляют в высоком вакууме (1,5 часа). Сухую реакционную смесь переносят в реакционную емкость микроволнового реактора (2-5 мл), смешивают с сухим ДХЭ (3 мл), 5,6-дигидрофенантридином (1k) (2 экв.), карбонатом калия (9 мг, 1,5 экв.), распыленными молекулярными ситами (4Å, 5 мг) и полученную смесь выдерживают в микроволновом реакторе при 100°C в течение 45 минут. Реакционную смесь пропускают через небольшой слой диоксида кремния (элюирование: ДХМ, затем 10% метанол в ДХМ). Полученные фракции, содержащие целевое карбаматное соединение, объединяют, концентрируют на роторном испарителе и очищают колоночной хроматографией на YMC диоксиде кремния (15 г, элюирование: этилацетат/петролейный эфир (1:3) для удаления избытка 5,6-дигидрофенантридина (1k), затем дихлорметан и далее 2% метанол в дихлорметане), получая указанное в заголовке соединение в виде порошка.
Пример 24
(17-гидрокси-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4,6*]октадец-7-ен-4-карбонил)амид 1-метилциклопропансульфоновой кислоты (24)
Соединение (6b), полученное в соответствии с описанной выше методикой, подвергают взаимодействию с циклическим амином (23k), как описано в примере 23, получая метилциклопропиловый аналог соединения (23i).
Активность соединений формулы (I)
Оценка биологического действия на репликон
Соединения формулы (I) исследуют на активность ингибирования HCV РНК репликации в клеточном анализе. Данный опыт показывает, что соединения формулы (I) проявляют активность в отношении HCV репликонов, функциональных в клеточной культуре. Клеточный анализ основан на конструкции экспрессии бицистрона, как описано в публикации Lohmann et al. (1999) Science vol. 285 pp. 110-113 с модификациями, описанными в публикации Krieger et al. (2001) Journal of Virology 75: 4614-4624, в многоцелевой стратегии скрининга. По существу, метод заключается в следующем.
В опыте используют стабильно трансфектированную клеточную линию Huh-7 luc/neo (далее в описании называемую Huh-Luc). Данная клеточная линия несет в себе конструкцию РНК кодирующей экспрессии бицистрона, включающую области NS3-NS5B дикого типа HCV типа 1b, транслированные из сайта внутреннего связывания рибосомы (Internal Ribosome Entry Site - IRES) вируса энцефаломиокардита (encephalomyocarditis virus - EMCV), которой предшествует область считывания (FfL-люцифераза) и подбираемая область маркера (neoR, неомицинфосфотрансфераза). Конструкция ограничена нетранслируемыми областями 5' и 3' (non-translated regions - NTR) из HCV типа 1b. Непрерывная культура клеток репликона в присутствии G418 (neoR) зависит от репликации HCV РНК. Стабильно трансфективнованные клетки репликона, которые экспрессируют HCV РНК, воспроизводятся автономно и до высоких уровней и кодируют inter alia люциферазу, используются для скрининга противовирусных соединений.
Репликоновые клетки помещают на 384-луночные микропланшеты в присутствии опытного и контрольного соединений, которые добавляют в различных концентрациях. Микропланшеты инкубируют в течение трех дней, после чего количественно определяют репликацию HCV посредством анализа активности люциферазы (с использованием стандартных опытных субстратов и реагентов люциферазы, а также микропланшетного визуализатора Perkin Elmer ViewLuxTm ultraHTS). Репликоновые клетки в контрольных культурах имеют высокую экспрессию в отсутствии любого ингибитора. Ингибиторная активность соединения в отношении активности люциферазы отслеживают на Huh-Luc клетках, получая кривую доза-эффект для каждого опытного соединения. Затем вычисляют значения EC50, которые представляют собой количество соединения, необходимое для 50% снижения определяемой активности люциферазы, или, точнее, способность генетически связанного HCV репликона РНК к репликации.
Анализ ингибирования
Цель данного анализа in vitro состоит в количественном определении ингибирования комплексов HCV NS3/4A протеаза соединениями согласно настоящему изобретению. Данный анализ обеспечивает определение того, как эффективные соединения согласно настоящему изобретению должны ингибировать HCV NS3/4A протеолитическую активность.
Ингибирование фермента NS3 протеазы гепатита C полной длины количественно определяют по существу в соответствии с методикой, описанной в публикации Poliakov, 2002 Prot Expression & Purification 25 363 371. Кратко, гидролиз депсипептидного субстрата, Ac-DED(Edans)EEAbuψ[COO]ASK(Dabcyl)-NH2 (AnaSpec, San Jose, USA), определяют спектрофотометрически в присутствии пептидного кофактора, KKGSVVIVGRIVLSGK (Äke Engström, Department of Medical Biochemistry и Microbiology, Uppsala University, Sweden). [Landro, 1997 #Biochem 36 9340-9348]. Фермент (1 нМ) инкубируют в 50 мМ HEPES, pH 7,5, 10 мМ DTT, 40% глицерин, 0,1% н-октил-D-глюкозид в присутствии 25 мкМ NS4A кофактора и ингибитора при 30°C в течение 10 минут, после чего реакции инициируют добавлением 0,5 мкМ субстрата. Ингибиторы растворяют в ДМСО, обрабатывают ультразвуком в течение 30 секунд с вихревым перемешиванием. Между измерениями растворы хранят при -20°C.
Конечную концентрацию ДМСО в опытном образце доводят до 3,3%. Скорость гидролиза корректируют для эффектов внутреннего фильтра в соответствии с опубликованными методиками [Liu, Analytical Biochemistry, 1999, vol. 267, pp. 331-335]. Значения Ki определяют посредством нелинейного регрессионного анализа (GraFit, Erithacus Software, Staines, MX, UK), используя модель конкурентного ингибирования и фиксированное значение Km (0,15 мкМ). Для всех измерений осуществляют, по меньшей мере, две репликации.
Соединения согласно настоящему изобретению предпочтительно обладают высокой эффективностью в отношении вируса HCV немутантного и мутантного типа, в особенности в отношении вируса, включающего мутации «отключения лекарственных средств». Мутации «отключения лекарственных средств» представляют собой мутации, которые возникают у пациентов вследствие селективного антивирусного воздействия на предшествующем уровне и придают повышенную стойкость к этому противовирусному средству.
Ингибирование некоторых мутантных HCV, показанное соединениями согласно настоящему изобретению, определяют в соответствии с методикой, описанной в WO2004/039970.
A156T и D168V представляют собой особенно релевантные мутанты «отключения лекарственных средств» в контексте терапевтического лечения HCV с использованием ингибиторов NS3 протеазы, и соединения согласно настоящему изобретению предпочтительно обладают низкими значениями Ki в отношении этих мутантов.
В приведенной далее таблице 1 представлены соединения, которые были получены в соответствии с методикой любого из приведенных выше примеров. Активности испытываемых соединений также представлены в таблице 1. Условные обозначения A, B, C, D, E, и F соответствуют следующим значениям:
A соответствует EC50>10 мкМ;
B соответствует EC50 в интервале от 10 мкМ до 1 мкМ;
C соответствует EC50 в интервале от 0,99 мкМ до 200 нМ;
D соответствует EC50 в интервале от 199 нМ до 0,5 нМ.
E соответствует Ki>100 нМ;
F соответствует Ki в интервале от 100 нМ до 30 нМ;
G соответствует Ki в интервале от 29,9 нМ до 0,1 нМ;
Оценка репликации
Ферментный анализ
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИМИДИН-ЗАМЕЩЕННЫЕ МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ HCV | 2008 |
|
RU2481340C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2437886C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2441870C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2486189C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2436787C2 |
| ПТЕРИДИНЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ HCV, И СПОСОБЫ ПОЛУЧЕНИЯ ПТЕРИДИНОВ | 2006 |
|
RU2447074C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2009 |
|
RU2518471C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА, ПРИМЕНИМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ FGFR КИНАЗЫ | 2014 |
|
RU2701517C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2654857C2 |
| ПТЕРИДИНЫ В КАЧЕСТВЕ FGFR ИНГИБИТОРОВ | 2013 |
|
RU2702906C2 |
Описываются макроциклические фенилкарбаматы формулы (I)
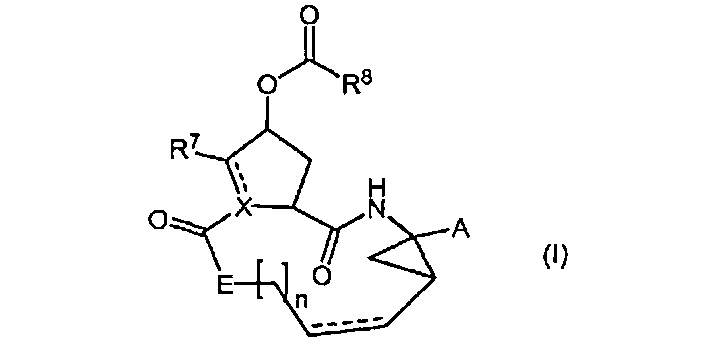
где A представляет собой -C(=O)OR1 или -C(=O)-NH-SO2-R2, где R1 представляет собой водород или C1-С6алкил; R2 представляет собой C3-7циклоалкил, фенил, тиазолил или пиридил, каждый из которых является необязательно замещенным одним или несколькими заместителями, выбранными из C1-6алкила, C1-6алкокси, трифторметила и галогена; X представляет собой N или CH; E представляет собой NR5; R5 представляет собой водород, C1-6алкил, C1-6алкоксиC1-6алкил или C3-С7Циклоалкил; n равно 4 или 5; где пунктирная линия -----, прилегающая к фрагменту -(CH2)n-, представляет собой двойную связь; и где пунктирная линия ----- в пятичленном цикле, включающем X, представляет собой простую связь и R7 представляет собой водород; R8 такой, как указано в формуле изобретения, или его N-оксид, фармацевтически приемлемая аддитивная соль или фармацевтически приемлемый сольват, обладающие противовирусной активностью, которые могут применяться в качестве HCV ингибиторов; а также фармацевтические композиции, включающие указанные соединения в качестве активного ингредиента. 4 н. и 10 з.п. ф-лы, 24 пр. 1 табл.
1. Соединение формулы (I):

включая его стереоизомеры, где
A представляет собой -C(-O)OR1 или -C(=O)-NH-SO2-R2,
где R1 представляет собой водород или C1-C6алкил;
R2 представляет собой C3-7циклоалкил, фенил, тиазолил или пиридил, каждый из которых является необязательно замещенным одним или несколькими заместителями, выбранными из C1-6алкила, C1-6алкокси, трифторметила и галогена;
X представляет собой N или CH;
E представляет собой NR5;
R5 представляет собой водород, C1-6алкил, C1-6алкоксиC1-6алкил или C3-7циклоалкил;
n равно 4 или 5;
где пунктирная линия-----, прилегающая к фрагменту -(CH2)n-, представляет собой двойную связь; и
где пунктирная линия ----- в пятичленном цикле, включающем X, представляет собой простую связь и R7 представляет собой водород;
R8 представляет собой радикал формулы

или группа  представляет собой группу:
представляет собой группу:

которая имеет следующую структуру


где R8a и R9a каждый независимо представляет собой водород, C1-6алкил, C2-6алкенил, C1-6алкокси, гидроксильную группу, галоген, полигалоген C1-6алкил, циано, амино, моно- или диC1-6 алкиламиногруппу;
каждый R9 независимо представляет собой C1-6алкил, необязательно замещенный C1-6алкокси, гидроксильной группой или галогеном; C3-7циклоалкил; C2-6алкенил; C1-6алкокси; C3-7циклоалкилокси; арилокси; Het-O-; гидроксильную группу; циано;
полигалоген C1-6алкил; моно- или ди C1-6алкиламино;
каждый R10 независимо представляет собой водород, C1-6алкил, C2-6алкенил, C1-6алкокси, гидроксильную группу, галоген, полигалогенC1-6алкил, циано, амино, моно- или ди C1-6алкиламино;
где каждый арил независимо представляет собой фенил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из галогена, гидроксильной группы, нитро, циано, карбоксила, C1-6алкила, C1-6алкокси, C1-6алкоксиC1-6алкила, C1-6алкилкарбонила, амино, моно- или ди C1-6алкиламино, азидо, меркапто, C1-6алкилтио, полигалоген C1-6алкила, полигалоген C1-6алкокси, C3-7циклоалкила и Het1;
каждый Het независимо представляет собой 5- или 6-членный насыщенный, частично ненасыщенный или полностью ненасыщенный гетероцикл, содержащий 1, 2, 3 или 4 гетероатома, каждый из которых независимо выбран из атомов азота, кислорода и серы, причем указанный гетероцикл является необязательно замещенным одним, двумя или тремя заместителями, каждый из которых независимо выбран из галогена, гидроксильной группы, нитро, циано, карбоксила, C1-6алкила, C1-6алкокси, C1-6алкоксиC1-6алкила, C1-6алкилкарбонила, амино, моно- или ди C1-6алкиламино, азидо, меркапто, полигалоген C1-6алкила, полигалоген C1-6алкокси, C3-7циклоалкила, Het1;
каждый Het1 независимо представляет собой пирролидинил, пиперидинил, пиперазинил, 4-C1-6алкилпиперазинил, 4-C1-6алкилкарбонилпиперазинил и морфолинил, где морфолинильная и пиперидинильная группы могут быть необязательно замещенными одним или двумя C1-6алкильными радикалами;
или его N-оксид, фармацевтически приемлемая аддитивная соль или фармацевтически приемлемый сольват.
2. Соединение по п.1, где X представляет собой N.
3. Соединение по п.1, где R8 представляет собой радикал формулы
 ; или
; или
R8 представляет собой радикал формулы
 ; или
; или
R8 представляет собой радикал формулы

4. Соединения по п.1, где R8 представляет собой радикал формулы

5. Соединение по п.3 или 4, где R9 представляет собой C1-6алкил (например, метил, этил или изопропил); C1-6алкокси (например, метокси, этокси или изопропокси); арилокси; Het-O-;
циано; или R9 представляет собой C1-6алкокси (например, метокси, этокси или изопропокси) или арилокси (например, фенокси или 4-метоксифенокси).
6. Соединение по п.3 или 4, где R10 представляет собой водород; C1-6алкил (например, метил, этил или изопропил); C1-6алкокси (например, метокси, этокси или изопропокси); циано.
7. Соединение по п.1, где группа

имеет структуру

8. Соединение по п.7, где R8, R8a, R9 представляют собой водород.
9. Соединение по п.1, где арил представляет собой фенил, необязательно замещенный C1-6алкокси-группой, и Het представляет собой пиридил или пиримидинил.
10. Соединение по п.1, где A представляет собой -C(=O)-NH-SO2R2, в частности, где R2 представляет собой C3-7циклоалкил, фенил или группу Het, например, тиазолил или пиридил, каждый из которых является необязательно замещенным одним или несколькими, например одним или двумя, заместителями, выбранными из C1-6алкила, C1-6алкокси, трифторметила и галогена, или в частности одним или двумя заместителями, выбранными из метила, фтора или хлора; или A представляет собой C(=O)OR1, где R1 представляет собой водород или C1-6алкил, такой как метил.
11. Фармацевтическая композиция, обладающая противовирусной активностью, включающая в качестве активного ингредиента соединение формулы (I) по любому из пп.1-10, и носитель.
12. Соединение по п.1 для применения в качестве лекарственного средства, обладающего противовирусной активностью.
13. Применение соединения по любому из пп.1-10 для получения лекарственного средства для лечения или профилактики вирусной инфекции.
14. Применение по п.13, где вирусная инфекция представляет HCV инфекцию.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| WO 00/059929 A, 12.10.2000 | |||
| Приспособление для нанесения и высверливания центров в валах и т.п. цилиндрических предметах | 1927 |
|
SU7738A1 |

