Настоящее изобретение относится к макроциклическим соединениям, обладающим ингибиторной активностью в отношении репликации вируса гепатита C (HCV). Кроме того, оно относится к композициям, содержащим такие соединения в качестве активных ингредиентов, а также к способам получения таких соединений и композиций.
Во всем мире вирус гепатита C служит основной причиной хронического заболевания печени и стал центром внимания значительного количества медицинских исследований. HCV является представителем семейства вирусов Flaviviridae из рода hepacivirus и является близкородственным роду flavivirus, который включает в себя ряд вирусов, участвующих в заболеваниях человека, таких как вирус денге и вирус желтой лихорадки, и семейству pestivirus вирусов животных, которое включает в себя вирус диареи быков (BVDV). HCV представляет собой позитивно-смысловой, однонитевой РНК-содержащий вирус с геномом приблизительно из 9600 оснований. Геном содержит как 5'-, так и 3'-нетранслируемые области, которые воспроизводят вторичные структуры РНК, и центральную открытую рамку считывания, которая кодирует единый полипротеин длиной приблизительно из 3010-3030 аминокислот. Полипротеин содержит продукты, которые кодируются десятью генами и генерируются из полипротеина-предшественника в результате целой серии ко- и посттрансляционных эндопротеолитических расщеплений, опосредованных как хозяйскими, так и вирусными протеазами. Вирусные структурные протеины включают в себя ядерный нуклеокапсидный протеин и два оболочечных гликопротеина E1 и E2. Неструктурные (NS) протеины обуславливают некоторые жизненно важные вирусные ферментативные функции (геликаза, полимераза, протеаза), а также протеины с неизвестной функцией. Репликация вирусного генома опосредована РНК-зависимой РНК-полимеразой, представленной неструктурным протеином 5b (NS5B). Было доказано, что кроме полимеразы важными для репликации РНК HCV являются функции вирусной геликазы и протеазы, обе из которых представлены бифункциональным NS3-протеином. Кроме сериновой протеазы NS3, HCV кодирует также и металлопротеиназу в NS2-области.
После первоначальной острой инфекции у большинства инфицированных развивается хронической гепатит, поскольку HCV реплицируется предпочтительно в гепатоцитах, хотя не является непосредственно цитопатическим. В частности, отсутствие достаточно интенсивного ответа T-лимфоцитов и высокая предрасположенность вируса к мутации, по-видимому, способствуют высокой скорости распространения хронической инфекции. Хронический гепатит может прогрессировать до фиброза печени, приводя на конечной стадии заболевания к циррозу печени и HCC (гепатоклеточной карциноме), что является главной причиной трансплантации печени.
Существует 6 основных генотипов HCV и более 50 подтипов, которые географически распространены неодинаково. Тип 1 HCV представляет собой генотип, преобладающий в Европе и США. Обширная генетическая гетерогенность HCV имеет важные диагностические и клинические последствия, возможно объясняющие трудности в разработке вакцин и недостаточный ответ на терапию.
Передача вируса HCV может происходить через контакт с зараженной кровью или кровепродуктами, например, после переливания крови или внутривенного введения лекарственного средства. Внедрение диагностических тестов, применяемых при скрининге крови, ведет к снижению частоты случаев HCV после переливания. Однако, с учетом медленного прогресса в отношении болезни печени в конечной стадии, существующие инфекции будут сохраняться в течение десятилетий, обеспечивая серьезное медицинское и экономическое бремя.
Современные терапии HCV основаны на (пегилированном) интерфероне-альфа (IFN-α) в сочетании с рибавирином. Такая комбинированная терапия дает в результате долговременный ответ на вирус более чем у 40% пациентов, инфицированных вирусами генотипа 1, и приблизительно у 80% пациентов, инфицированных вирусами генотипов 2 и 3. Наряду с ограниченной эффективностью в отношении HCV типа 1, такая комбинированная терапия имеет значительные побочные эффекты и плохо переносится многими пациентами. Основные побочные эффекты включают в себя гриппоподобные симптомы, гематологические нарушения и психоневрологические симптомы. Следовательно, существует необходимость в более эффективных, удобных и лучше переносимых способах лечения.
Недавно в качестве клинических кандидатов внимание привлекли два пептидомиметика-ингибитора HCV-протеазы, а именно BILN-2061, описанный в заявке WO00/59929, и VX-950, описанный в заявке WO03/87092. Ряд подобных ингибиторов HCV-протеазы также описан в научной и патентной литературе. В настоящее время уже очевидно, что продолжительное введение BILN-2061 или VX-950 способствует селекции мутантов HCV, которые резистентны к соответствующему лекарственному средству, так называемых мутантов, «ускользающих» от лекарственного средства. Такие мутанты, «ускользающие» от лекарственного средства, имеют характерные мутации в геноме HCV-протеазы, а именно D168V, D168A и/или A156S. Соответственно, чтобы обеспечить потерпевших неудачу пациентов вариантами лечения, требуются дополнительные лекарственные средства с другими характеристиками резистентности, а комбинированная терапия с помощью нескольких лекарственных средств, вероятно, станет нормой в будущем, даже для терапии первого порядка.
Кроме того, опыт работы с HIV-лекарственными средствами, в частности с ингибиторами HIV-протеазы, делает акцент на том факте, что недостаточно оптимальная фармакокинетика и сложные схемы приема лекарственного средства быстро приводят к нечаянным ошибкам в соблюдении требований приема. Это в свою очередь означает, что 24-часовая минимальная концентрация (минимальная концентрация в плазме) соответствующих лекарственных средств при схеме приема против HIV часто опускается ниже пороговых значений IC90 или ED90 в течение значительных отрезков дня. Считается, что 24-часовой минимальный пороговый уровень, составляющий, по меньшей мере, IC50, и, более реалистично, IC90 или ED90, имеет существенное значение для сдерживания роста мутантов, «ускользающих» от лекарственного средства. Достижение нужной фармакокинетики и метаболизма лекарственного средства для обеспечения таких минимальных пороговых уровней предъявляет строгие требования к созданию (к дизайну) лекарственных средств. Сильная природа пептидомиметиков-ингибиторов HCV-протеазы известного уровня техники с многократными пептидными связями создает фармакокинетические затруднения для эффективных схем приема лекарственного средства.
Необходимы ингибиторы HCV, которые могут преодолеть недостатки современной HCV-терапии, такие как побочные эффекты, ограниченная эффективность, появление резистентности и ошибки в соблюдении требований приема.
Заявка WO04/094452 относится к макроциклическим изохинолиновым пептидным ингибиторам HCV. Также описаны композиции, содержащие соединения, и способы, в которых соединения применяются для ингибирования HCV.
В заявке WO05/010029 описаны азапептидные макроциклические ингибиторы сериновой протеазы гепатита C; фармацевтические композиции, содержащие вышеупомянутые соединения, предназначенные для введения субъекту, страдающему от HCV-инфекции; и способы лечения HCV-инфекции у субъекта путем введения фармацевтической композиции, содержащей упомянутые соединения по настоящему изобретению.
Настоящее изобретение относится к ингибиторам HCV, которые имеют преимущества по одному или нескольким нижеперечисленным фармакологическим взаимосвязанным свойствам, то есть активности, пониженной цитотоксичности, улучшенной фармакокинетике, улучшенному профилю резистентности, приемлемой дозировке и уменьшению количества приемов.
Кроме того, соединения по настоящему изобретению обладают относительно низкой молекулярной массой и легко синтезируются, исходя из исходных материалов, которые имеются в продаже или легко доступны благодаря процедурам синтеза, известным в данной области.
Настоящее изобретение относится к ингибиторам репликации HCV, которые можно представить формулой (I):
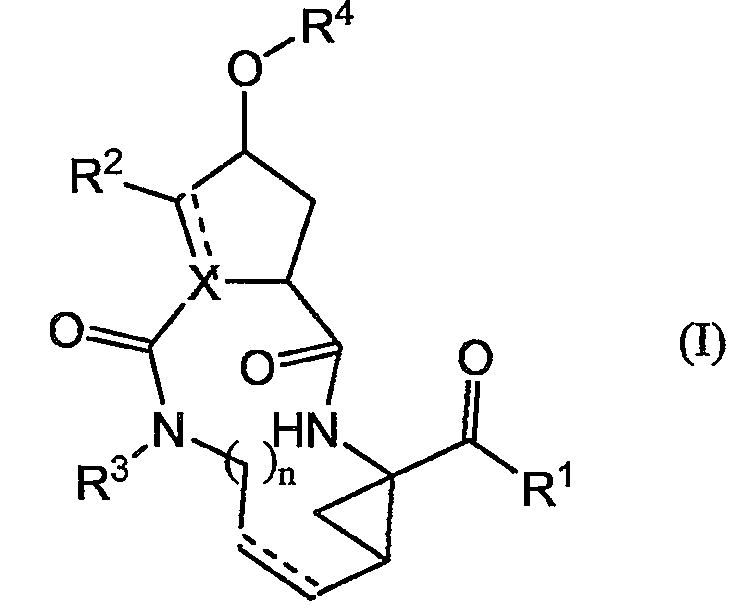
и к их N-оксидам, солям и стереоизомерам, в которых
X представляет собой N, CH и, когда X содержит двойную связь, он представляет собой C;
R 1 представляет собой -OR5, -NH-SO2R6;
R 2 представляет собой водород и, когда X представляет собой C или CH, R 2 также может представлять собой C1-6-алкил;
R 3 представляет собой водород, C1-6-алкил, C1-6-алкокси-C1-6-алкил или C3-7-циклоалкил;
R 4 представляет собой изохинолинил, необязательно замещенный одним, двумя или тремя заместителями, каждый из которых независимо выбран из C1-6-алкила, C1-6-алкокси, гидрокси, галогена, полигалоген-C1-6-алкила, полигалоген-C1-6-алкокси, амино, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонила, C1-6-алкилкарбониламино, арила и Het;
n равно 3, 4, 5 или 6;
в которой каждая пунктирная линия (представленная как ----) представляет собой необязательную двойную связь;
R 5 представляет собой водород; арил; Het; C3-7-циклоалкил, необязательно замещенный C1-6-алкилом; или C1-6-алкил, необязательно замещенный C3-7-циклоалкилом, арилом или Het;
R 6 представляет собой арил; Het; C3-7-циклоалкил, необязательно замещенный C1-6-алкилом; или C1-6-алкил, необязательно замещенный C3-7-циклоалкилом, арилом или Het;
каждый арил в виде группы или части группы представляет собой фенил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из галогена, гидрокси, нитро, циано, карбоксила, C1-6-алкила, C1-6-алкокси, C1-6-алкокси-C1-6-алкила, C1-6-алкилкарбонила, амино, моно- или ди-C1-6-алкиламино, азидо, меркапто, полигалоген-C1-6-алкила, полигалоген-C1-6-алкокси, C3-7-циклоалкила, пирролидинила, пиперидинила, пиперазинила, 4-C1-6-алкилпиперазинила, 4-C1-6-алкилкарбонилпиперазинила и морфолинила; где морфолинильные и пиперидинильные группы необязательно могут быть замещены одним или двумя C1-6-алкильными радикалами; и
каждый Het в виде группы или части группы представляет собой 5- или 6-членное насыщенное, частично ненасыщенное или полностью ненасыщенное гетероциклическое кольцо, содержащее от 1 до 4 гетероатомов, каждый из которых независимо выбран из азота, кислорода и серы, и упомянутое гетероциклическое кольцо необязательно замещено одним, двумя или тремя заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, гидрокси, нитро, циано, карбоксила, C1-6-алкила, C1-6-алкокси, C1-6-алкокси-C1-6-алкила, C1-6-алкилкарбонила, амино, моно- или ди-C1-6-алкиламино, азидо, меркапто, полигалоген-C1-6-алкила, полигалоген-C1-6-алкокси, C3-7-циклоалкила, пирролидинила, пиперидинила, пиперазинила, 4-C1-6-алкилпиперазинила, 4-C1-6-алкилкарбонилпиперазинила и морфолинила, и где морфолинильные и пиперидинильные группы необязательно могут быть замещены одним или двумя C1-6-алкильными радикалами.
Изобретение дополнительно относится к способам получения соединений формулы (I), N-оксидов, аддитивных солей, четвертичных аминов, комплексов с металлами и их стереохимически изомерных форм, их промежуточных продуктов и применению промежуточных продуктов для получения соединений формулы (I).
Изобретение относится к соединениям формулы (I), как таковым, их N-оксидам, аддитивным солям, четвертичным аминам, комплексам с металлами и их стереохимически изомерным формам для применения в качестве лекарственного средства. Изобретение дополнительно относится к фармацевтическим композициям, содержащим вышеупомянутые соединения, предназначенным для введения субъекту, страдающему от HCV-инфекции. Фармацевтические композиции могут содержать комбинации вышеупомянутых соединений с другими средствами против HCV.
Изобретение также относится к применению соединения формулы (I) или его N-оксида, аддитивной соли, четвертичного амина, комплекса с металлом или их стереохимически изомерных форм для производства лекарственного средства, предназначенного для ингибирования репликации HCV. Или изобретение относится к способу ингибирования репликации HCV у теплокровного животного упомянутым способом, включающим в себя введение эффективного количества соединения формулы (I) или его N-оксида, аддитивной соли, четвертичного амина, комплекса с металлом или их стереохимически изомерных форм.
Если не указано иначе, в качестве применяемых выше и в дальнейшем используются следующие определения.
Термин «галоген» относится к фтору, хлору, брому и йоду.
Термин «полигалоген-C1-6-алкил» в виде группы или части группы, например, в полигалоген-C1-6-алкокси, определяется как моно- или полигалогензамещенный C1-6-алкил, в частности, C1-6-алкил, замещенный одним, двумя, тремя, четырьмя, пятью, шестью или более атомами галогенов, такой как метил или этил с одним или несколькими атомами фтора, например, дифторметил, трифторметил, трифторэтил. Предпочтительным является трифторметил. Также включены перфтор-C1-6-алкильные группы, которые представляют собой C1-6-алкильные группы, в которых все атомы водорода замещены атомами фтора, например, пентафторэтил. В том случае, когда к алкильной группе присоединяется более одного атома галогена, в пределах определения полигалоген-C1-6-алкила атомы галогенов могут быть одинаковыми или разными.
Применяемый здесь термин «C1-4-алкил» в виде группы или части группы определяет насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 4 атомов углерода, такие как, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил; «C1-6-алкил» охватывает C1-4-алкильные радикалы и их высшие гомологи, содержащие 5 или 6 атомов углерода, такие как, например, 1-пентил, 2-пентил, 3-пентил, 1-гексил, 2-гексил, 2-метил-1-бутил, 2-метил-1-пентил, 2-этил-1-бутил, 3-метил-2-пентил и т.п. Среди C1-6-алкильных групп интерес представляет C1-4-алкил.
Термин «C2-6-алкенил» в виде группы или части группы определяет углеводородные радикалы с прямой и разветвленной цепью, содержащие насыщенные углерод-углеродные связи и, по меньшей мере, одну двойную связь и содержащие от 2 до 6 атомов углерода, такие как, например, этенил (или винил), 1-пропенил, 2-пропенил (или аллил), 1-бутенил, 2-бутенил, 3-бутенил, 2-метил-2-пропенил, 2-пентенил, 3-пентенил, 2-гексенил, 3-гексенил, 4-гексенил, 2-метил-2-бутенил, 2-метил-2-пентенил и т.п. Среди C2-6-алкенильных групп интерес представляет C2-4-алкенил.
Термин «C2-6-алкинил» в виде группы или части группы определяет углеводородные радикалы с прямой и разветвленной цепью, содержащие насыщенные углерод-углеродные связи и, по меньшей мере, одну тройную связь и содержащие от 2 до 6 атомов углерода, такие как, например, этинил, 1-пропинил, 2-пропинил, 1-бутинил, 2-бутинил, 3-бутинил, 2-пентинил, 3-пентинил, 2-гексинил, 3-гексинил и т.п. Среди C2-6-алкинильных групп интерес представляет C2-4-алкинил.
C3-7-циклоалкил относится к циклопропилу, циклобутилу, циклопентилу, циклогексилу и циклогептилу.
C1-6-алкандиил определяет двухвалентные насыщенные углеводородные радикалы с прямой и разветвленной цепью, содержащие от 1 до 6 атомов углерода, такие как, например, метилен, этилен, 1,3-пропандиил, 1,4-бутандиил, 1,2-пропандиил, 2,3-бутандиил, 1,5-пентандиил, 1,6-гександиил и т.п. Среди C1-6-алкандиильных групп интерес представляет C1-4-алкандиил.
C1-6-алкокси означает C1-6-алкилокси, в которой C1-6-алкил имеет указанное выше значение.
Уже применяемый здесь термин (=O) или оксо образует карбонильный фрагмент, когда присоединяется к атому углерода, сульфоксидный фрагмент, когда присоединяется к атому серы, и сульфонильный фрагмент, когда два упомянутых «оксо» присоединяются к атому серы. Всякий раз, когда кольцо или кольцевая система замещена оксо-группой, атом углерода, с которым соединяется оксо-группа, представляет собой насыщенный атом углерода.
Радикал Het представляет собой гетероцикл, который указан в данном описании и формуле изобретения. Примеры Het включают в себя, например, пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазинолил, изотиазинолил, тиазолил, изотиазолил, оксадиазолил, тиадиазолил, триазолил (включая 1,2,3-триазолил, 1,2,4-триазолил), тетразолил, фуранил, тиенил, пиридил, пиримидил, пиридазинил, пиразолил, триазинил и т.п. Среди радикалов Het представляют интерес те радикалы Het, которые являются ненасыщенными, в частности радикалы, имеющие ароматический характер. Дополнительный интерес представляют те радикалы Het, которые содержат один или два атома азота.
Каждый из радикалов Het, упомянутых в данном и следующих параграфах, необязательно может быть замещен некоторым числом и типом заместителей, упомянутых в определениях соединений формулы (I) или любых подгрупп соединений формулы (I). Некоторые из радикалов Het, упомянутых в данном и следующих параграфах, могут быть замещены одним, двумя или тремя гидрокси-заместителями. Такие гидрокси-замещенные кольца могут встречаться в виде своих таутомерных форм, содержащих кетогруппы. Например, 3-гидроксипиридазиновый фрагмент может встречаться в своей таутомерной форме 2H-пиридазин-3-она. Когда Het представляет собой пиперазинил, он предпочтительно замещен в своем положении 4 заместителем, присоединенным к 4-азоту с помощью атома углерода, например, 4-C1-6-алкилом, 4-полигалоген-C1-6-алкилом, C1-6-алкокси-C1-6-алкилом, C1-6-алкилкарбонилом, C3-7-циклоалкилом.
Представляющие интерес радикалы Het включают в себя, например, пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, оксадиазолил, тиадиазолил, триазолил (включая 1,2,3-триазолил, 1,2,4-триазолил), тетразолил, фуранил, тиенил, пиридил, пиримидил, пиридазинил, пиразолил, триазинил или любые из таких гетероциклов, конденсированных с бензольным кольцом, такие как индолил, индазолил (в частности, 1H-индазолил), индолинил, хинолинил, тетрагидрохинолинил (в частности, 1,2,3,4-тетрагидрохинолинил), изохинолинил, тетрагидроизохинолинил (в частности, 1,2,3,4-тетрагидроизохинолинил), хиназолинил, фталазинил, бензимидазолил, бензоксазолил, бензизоксазолил, бензотиазолил, бензоксадиазолил, бензотиадиазолил, бензофуранил, бензотиенил.
Радикалы Het пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, 4-замещенный пиперазинил предпочтительно присоединяются с помощью своего атома азота (то есть 1-пирролидинил, 1-пиперидинил, 4-тиоморфолинил, 4-морфолинил, 1-пиперазинил, 4-замещенный 1-пиперазинил).
Следует отметить, что положения радикалов на любом молекулярном фрагменте, указанные в определениях, могут находиться на таком фрагменте где угодно, при условии, что он химически стабилен.
Если не указано иначе, радикалы, применяемые в определениях переменных, включают в себя все возможные изомеры. Например, пиридил включает в себя 2-пиридил, 3-пиридил и 4-пиридил; пентил включает в себя 1-пентил, 2-пентил и 3-пентил.
Когда любая переменная в любом составляющем элементе встречается более одного раза, каждое определение является независимым.
Всякий раз, когда в дальнейшем применяется термин «соединения формулы (I)» или «настоящие соединения» или подобные термины, это означает, что они включают в себя соединения формулы (I), их пролекарства, N-оксиды, аддитивные соли, четвертичные амины, комплексы с металлами и стереохимически изомерные формы. Один из вариантов осуществления изобретения включает в себя соединения формулы (I) или любую указанную здесь подгруппу соединений формулы (I), а также N-оксиды, соли в виде их возможных стереоизомерных форм. Другой вариант осуществления включает в себя соединения формулы (I) или любую указанную здесь подгруппу соединений формулы (I), а также соли в виде их возможных стереоизомерных форм.
Соединения формулы (I) имеют несколько центров хиральности и существуют в виде стереохимически изомерных форм. Применяемый здесь термин «стереохимически изомерные формы» определяет все возможные соединения, состоящие из одинаковых атомов, связанных одинаковой последовательностью связей, но имеющие разные трехмерные структуры, которые не являются взаимно заменяемыми и которыми соединения формулы (I) могут обладать.
При ссылке на примеры, в которых для обозначения абсолютной конфигурации хирального атома в заместителе применяется (R) или (S), соединение рассматривается в целом и без отрыва заместителя.
Если не упомянуто или не указано иначе, химическое название соединения охватывает смесь всех возможных стереохимически изомерных форм, которыми упомянутое соединение может обладать. Упомянутая смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры упомянутого соединения. Разумеется, все стереохимически изомерные формы соединений по настоящему изобретению как в чистой форме, так и смешанные друг с другом подлежат включению в объем настоящего изобретения.
Чистые стереоизомерные формы соединений и промежуточных продуктов, которые здесь упоминаются, определяются как изомеры, по существу не содержащие других энантиомерных или диастереомерных форм той же самой основной молекулярной структуры упомянутых соединений или промежуточных продуктов. В частности, термин «стереоизомерно чистый» относится к соединениям или промежуточным продуктам, содержащим избыток стереоизомера, по меньшей мере, от 80% (то есть минимум 90% одного изомера и максимум 10% других возможных изомеров) вплоть до избытка стереоизомера 100% (то есть 100% одного изомера и отсутствие другого изомера), более конкретно, к соединениям или промежуточным продуктам, содержащим избыток стереоизомера от 90% вплоть до 100%, еще более конкретно, содержащим избыток стереоизомера от 94% вплоть до 100% и наиболее конкретно, содержащим избыток стереоизомера от 97% вплоть до 100%. Термины «энантиомерно чистый» и «диастереомерно чистый» в обсуждаемом вопросе следует понимать подобным образом, однако в таком случае они относятся соответственно к избытку энантиомера и избытку диастереомера в смеси.
Чистые стереоизомерные формы соединений и промежуточных продуктов по данному изобретению можно получать путем применения известных в данной области процедур. Например, энантиомеры можно отделять друг от друга с помощью избирательной кристаллизации их диастереомерных солей с оптически активными кислотами или основаниями. Их примерами являются винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота и камфорсульфокислота. Альтернативно энантиомеры можно разделять с помощью хроматографических способов, применяя хиральные неподвижные фазы. Упомянутые чистые стереохимически изомерные формы также можно получать из соответствующих стереохимически чистых изомерных форм подходящих исходных материалов, при условии, что происходит стереоспецифическая реакция. Если требуется определенный стереоизомер, предпочтительно упомянутое соединение синтезировать с помощью стереоспецифических способов получения. В таких способах преимущественно будут использоваться энантиомерно чистые исходные материалы.
Диастереомерные рацематы соединений формулы (I) можно получать по отдельности с помощью традиционных способов. Подходящими физическими способами разделения, которые преимущественно можно использовать, например, являются избирательная кристаллизация и хроматография, например колоночная хроматография.
Для некоторых из соединений формулы (I), их N-оксидов, солей, сольватов, четвертичных аминов или комплексов с металлами и промежуточных продуктов, применяемых для их получения, абсолютные стереохимические конфигурации экспериментально не определялись. Специалист в данной области способен определить абсолютную конфигурацию таких соединений, применяя известные в данной области способы, такие как, например, рентгенодифракционный метод.
Также разумеется, что настоящее изобретение включает в себя все изотопы атомов, встречающихся в настоящих соединениях. Изотопы включают в себя те атомы, которые имеют одинаковый атомный номер, но разные массовые числа. В качестве обычного примера и без ограничения изотопы водорода включают в себя тритий и дейтерий. Изотопы углерода включают в себя C-13 и C-14.
Применяемый на всем протяжении данного текста термин «пролекарство» означает фармакологически приемлемые производные, такие как сложные эфиры, амиды и фосфаты, такие, что полученный в результате продукт биотрансформации производного в организме (in vivo) представляет собой активное лекарственное средство, которое определено в соединениях формулы (I). Здесь включена ссылка на публикацию авторов Goodman и Gilman (The Pharmacological Basis of Therapeutics, 8-е издание, McGraw-Hill, Int. Ed. 1992, «Biotransformation of Drugs», стр. 13-15), описывающая пролекарства в целом. Пролекарства предпочтительно обладают прекрасной растворимостью в воде, повышенной биодоступностью и в процессе обмена веществ в организме (in vivo) легко превращаются в активные ингибиторы. Пролекарства соединения по настоящему изобретению можно получать путем модификации присутствующих в соединении функциональных групп таким образом, чтобы модификации расщеплялись до исходного соединения либо с помощью обычной манипуляции, либо в организме (in vivo).
Предпочтительными являются фармацевтически приемлемые, сложноэфирные пролекарства, которые гидролизуются в организме (in vivo) и являются производными от тех соединений формулы (I), которые содержат гидрокси- или карбоксильную группу. Гидролизуемый в организме (in vivo) сложный эфир представляет собой сложный эфир, который гидролизуется в организме человека или животного с образованием исходной кислоты или спирта. Подходящие, фармацевтически приемлемые сложные эфиры для карбокси-группы включают в себя сложные C1-6-алкоксиметиловые эфиры, например, метоксиметиловый, сложные C1-6-алканоилоксиметиловые эфиры, например, сложные пивалоилоксиметиловые, фталидиловые эфиры, сложные C3-8-циклоалкоксикарбонилокси-C1-6-алкиловые эфиры, например, 1-циклогексилкарбонилоксиэтиловый; сложные 1,3-диоксолен-2-онилметиловые эфиры, например, 5-метил-1,3-диоксолен-2-онилметиловый; и сложные C1-6-алкоксикарбонилоксиэтиловые эфиры, например, 1-метоксикарбонилоксиэтиловый, которые можно образовывать в соединениях по данному изобретению на любой карбокси-группе.
Гидролизуемый в организме (in vivo) сложный эфир соединения формулы (I), содержащего гидрокси-группу, включает в себя неорганические сложные эфиры, такие как сложные эфиры фосфорной кислоты, простые α-ацилоксиалкиловые эфиры и родственные соединения, которые в результате гидролиза сложного эфира в организме (in vivo) разлагаются, давая при этом исходную гидрокси-группу. Примеры простых α-ацилоксиалкиловых эфиров включают в себя ацетоксиметокси и 2,2-диметилпропионилоксиметокси. Выбор гидролизуемого в организме (in vivo) сложного эфира, образующего группы для гидрокси, включает в себя алканоил, бензоил, фенилацетил и замещенные бензоил и фенилацетил, алкоксикарбонил (для получения сложных алкилкарбонатных эфиров), диалкилкарбамоил и N-(диалкиламиноэтил)-N-алкилкарбамоил (для получения карбаматов), диалкиламиноацетил и карбоксиацетил. Примеры заместителей на бензоиле включают в себя морфолинo- и пиперазиногруппы, соединенные с кольцевым атомом азота через посредство метиленовой группы в положении 3 или 4 бензоильного кольца.
Соли соединения формулы (I) для терапевтического применения представляют собой те соли, в которых противоион является фармацевтически приемлемым. Однако также могут найти применение соли кислот и оснований, которые не являются фармацевтически приемлемыми, например, для получения или очистки фармацевтически приемлемого соединения. Все соли, являются они фармацевтически приемлемыми или нет, включены в объем настоящего изобретения.
Разумеется, что упомянутые выше фармацевтически приемлемые аддитивные соли кислот и оснований содержат терапевтически активные, нетоксичные формы аддитивных солей кислот и оснований, которые способны образовывать соединения формулы (I). Фармацевтически приемлемые аддитивные соли кислот можно удобно получать путем обработки основной формы такой подходящей кислотой. Подходящие кислоты включают в себя, например, неорганические кислоты, такие как галогенводородные кислоты, например, хлористоводородную или бромистоводородную кислоту, серную, азотную, фосфорную и тому подобные кислоты; или органические кислоты, такие как, например, уксусная, пропановая, гидроксиуксусная (гликолевая), молочная, пировиноградная, щавелевая (то есть этандиовая), малоновая, янтарная (то есть бутандиовая), малеиновая, фумаровая, яблочная (то есть гидроксибутандиовая), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и тому подобные кислоты.
С другой стороны, упомянутые формы солей путем обработки подходящим основанием можно преобразовывать в форму свободного основания.
Соединения формулы (I), содержащие кислотный протон, путем обработки подходящими органическими и неорганическими основаниями также можно преобразовывать в формы их аддитивных солей с нетоксичными металлами или аминами. Формы подходящих солей оснований включают в себя, например, соли аммония, соли щелочных и щелочноземельных металлов, например, соли лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, например, соли бензатина, N-метил-D-глюкамина, гидрабамина, и соли с аминокислотами, такими как, например, аргинин, лизин и т.п.
Применяемый выше термин аддитивная соль также включает в себя сольваты, которые соединения формулы (I), а также их соли способны образовывать. Такие сольваты представляют собой, например, гидраты, алкоголяты и т.п.
Применяемый выше термин «четвертичный амин» определяет соли четвертичного аммония, которые способны образовывать соединения формулы (I) при взаимодействии атома основного азота соединения формулы (I) и подходящего кватернизирующего агента, такого как, например, необязательно замещенный алкилгалогенид, арилгалогенид или арилалкилгалогенид, например, метилиодид или бензилиодид. Также можно применять другие реагенты с подходящими удаляемыми группами, такие как алкилтрифторметансульфонаты, алкилметансульфонаты и алкил-п-толуолсульфонаты. Четвертичный амин содержит положительно заряженный атом азота. Фармацевтически приемлемые противоионы включают в себя хлор, бром, йод, трифторацетат и ацетат. Выбранный противоион можно вводить, применяя ионообменные смолы.
Подразумевается, что N-оксидные формы настоящих соединений включают в себя соединения формулы (I), в которых один или несколько атомов азота окислены до так называемого N-оксида.
Следует учитывать, что соединения формулы (I) могут образовывать связи с металлами, иметь хелатообразующие и комплексообразующие свойства, и следовательно, могут существовать в виде комплексов с металлами или хелатов металлов. Разумеется, что такие металлированные производные соединений формулы (I) подлежат включению в объем настоящего изобретения.
Некоторые из соединений формулы (I) также могут существовать в своей таутомерной форме. Разумеется, такие формы, хотя и в неявной форме указанные в вышеупомянутой формуле, подлежат включению в объем настоящего изобретения.
Как упомянуто выше, соединения формулы (I) содержат несколько центров асимметрии. Для того, чтобы более эффективно ссылаться на каждый из таких центров асимметрии, будет применяться система нумерации, указанная в следующей структурной формуле:
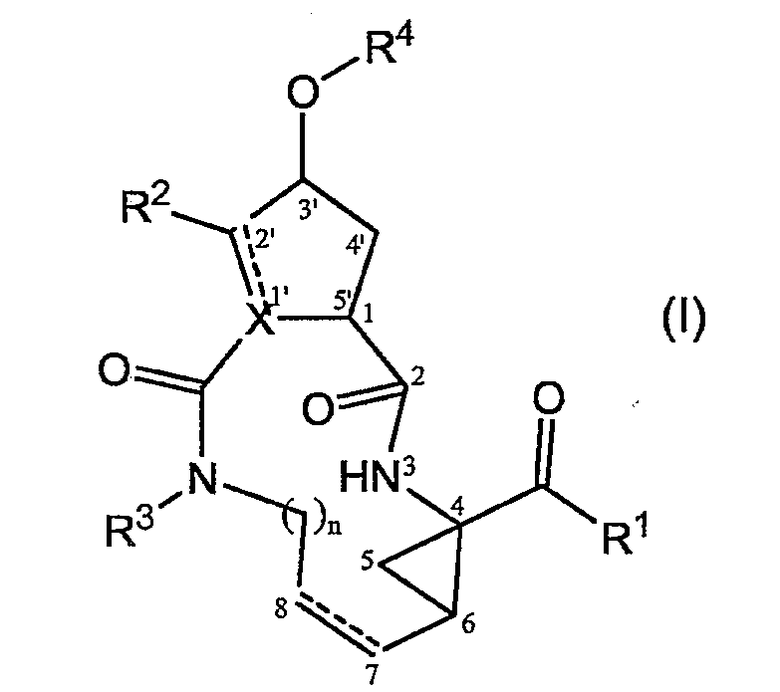
Центры асимметрии находятся в положениях 1, 4 и 6 макроцикла, а также на атоме углерода 3' в 5-членном кольце, атоме углерода 2', когда заместитель R2 представляет собой C1-6-алкил, и на атоме углерода 1', когда X представляет собой CH. Каждый из таких центров асимметрии может встречаться в своей R- или S-конфигурации.
Стереохимия в положении 1 предпочтительно соответствует стереохимии конфигурации L-аминокислоты, то есть стереохимии L-пролина.
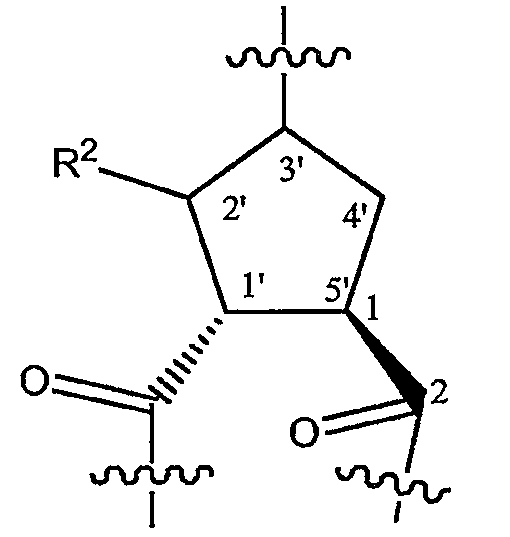
Когда X представляет собой CH, 2 карбонильные группы, замещающие циклопентановое кольцо в положениях 1' и 5', предпочтительно находятся в транс-конфигурации. Карбонильный заместитель в положении 5' предпочтительно находится в той конфигурации, которая соответствует конфигурации L-пролина. Карбонильные группы, замещающие в положениях 1' и 5', предпочтительно находятся в структуре следующей формулы, как изображено ниже:
Соединения формулы (I) включают в себя циклопропильную группу, которая представлена ниже в структурном фрагменте:
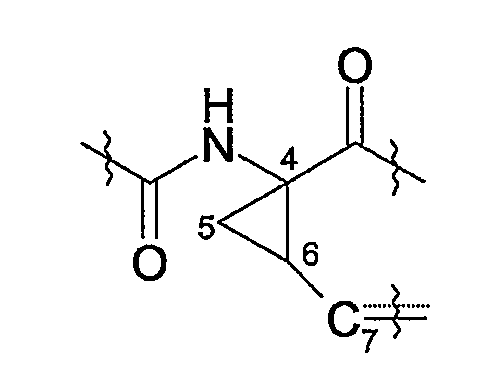
в котором C7 представляет собой атом углерода в положении 7, и атомы углерода в положении 4 и 6 являются асимметрическими атомами углерода циклопропанового кольца.
Невзирая на другие возможные центры асимметрии, находящиеся в других сегментах соединений формулы (I), присутствие двух таких центров асимметрии означает, что соединения могут существовать в виде смесей диастереомеров, таких как диастереомеры соединений формулы (I), в которых атом углерода в положении 7 имеет конфигурацию либо син- по отношению к карбонилу, либо син- по отношению к амиду, как показано ниже.
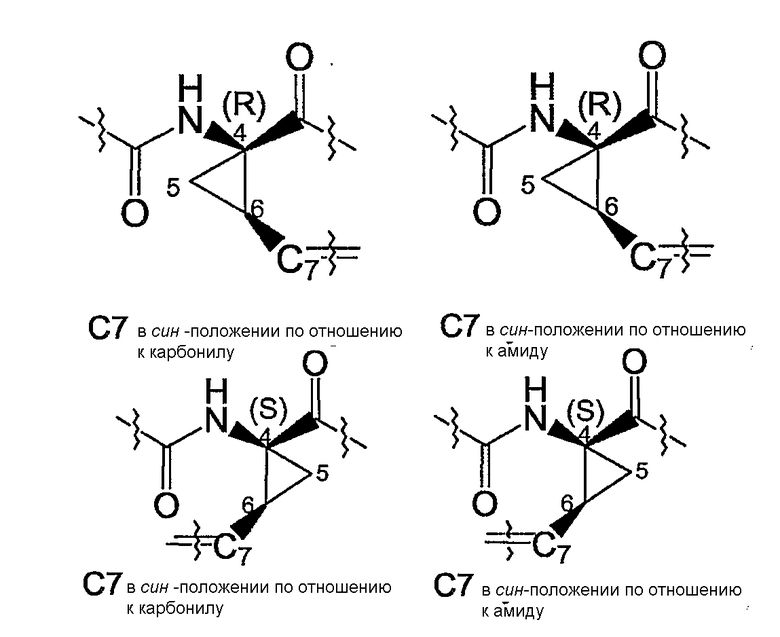
Один из вариантов осуществления изобретения относится к соединениям формулы (I), в которых атом углерода в положении 7 имеет син-конфигурацию по отношению к карбонилу. Другой вариант осуществления изобретения относится к соединениям формулы (I), в которых конфигурация на атоме углерода в положении 4 представляет собой R-конфигурацию. Специфическую подгруппу соединений формулы (I) представляют соединения, в которых атом углерода в положении 7 имеет син-конфигурацию по отношению к карбонилу и в которых конфигурация на атоме углерода в положении 4 представляет собой R-конфигурацию.
Соединения формулы (I) могут включать в себя пролиновый остаток (когда X представляет собой N) или циклопентильный или циклопентенильный остаток (когда X представляет собой CH или C). Предпочтительными являются соединения формулы (I), в которых заместитель в положении 1 (или 5') и заместитель -О-R4 (в положении 3') находятся в транс-конфигурации. Особый интерес представляют соединения формулы (I), которые в положении 1 имеют конфигурацию, соответствующую L-пролину, и заместитель -О-R4 находится в транс-конфигурации по отношению к положению 1. Предпочтительно соединения формулы (I) имеют такую стереохимию, которая указана ниже в структурных формулах (I-a) и (I-b):
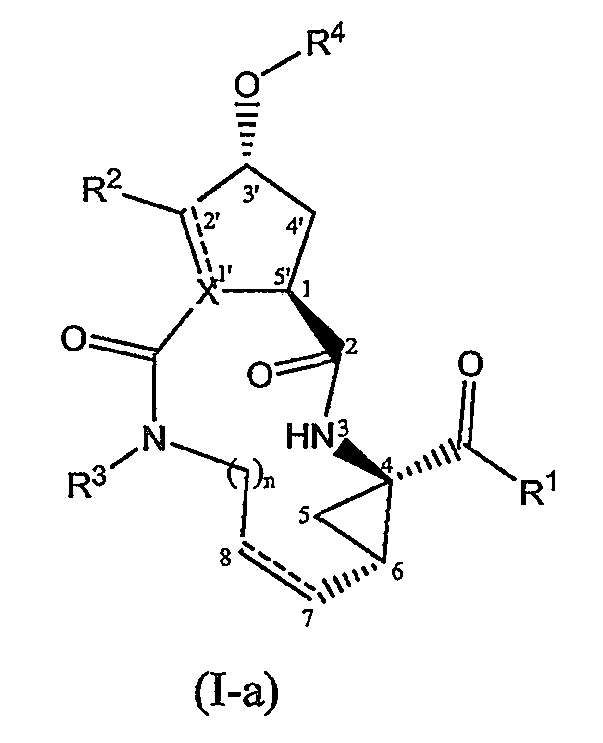
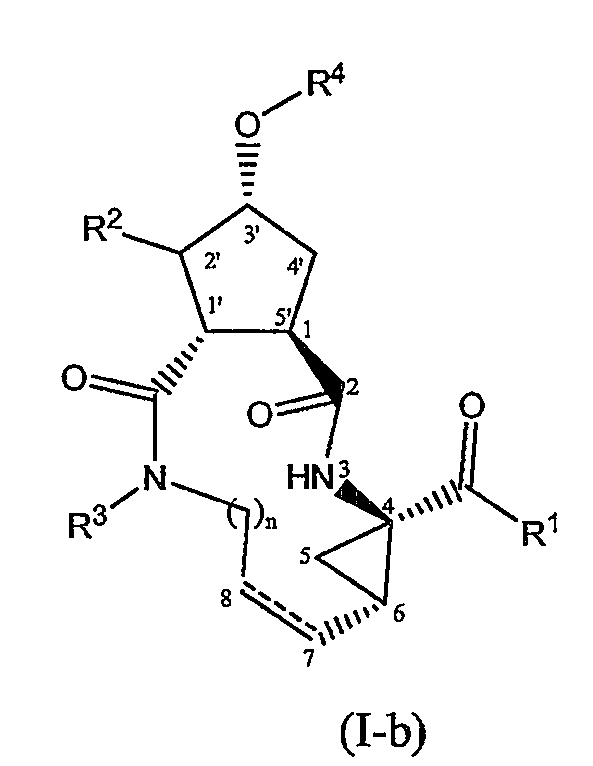
Один из вариантов осуществления настоящего изобретения относится к соединениям формулы (I) или формулы (I-a), или к любой подгруппе соединений формулы (I), в которых осуществляется одно или несколько из следующих условий:
(a) R2 представляет собой водород;
(b) X представляет собой азот;
(c) между атомами углерода 7 и 8 присутствует двойная связь.
Один из вариантов осуществления настоящего изобретения относится к соединениям формулы (I) или формул (I-a), (I-b), или к любой подгруппе соединений формулы (I), в которых осуществляется одно или несколько из следующих условий:
(a) R2 представляет собой водород;
(b) X представляет собой CH;
(c) между атомами углерода 7 и 8 присутствует двойная связь.
Особыми подгруппами соединений формулы (I) являются подгруппы, представленные следующими структурными формулами:
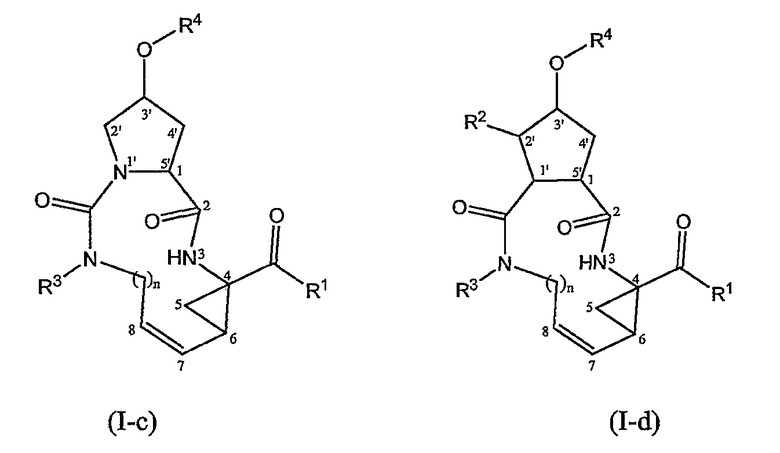
Среди соединений формулы (I-c) и (I-d) особый интерес представляют собой соединения, имеющие стереохимические конфигурации соединений формул (I-a) и (I-b) соответственно.
Двойная связь между атомами углерода 7 и 8 в соединениях формулы (I) или в любой подгруппе соединений формулы (I) может находиться в цис- или в транс-конфигурации. Предпочтительно двойная связь между атомами углерода 7 и 8 находится в цис-конфигурации, которая изображена в формулах (I-c) и (I-d).
В соединениях формулы (I) или в любой подгруппе соединений формулы (I) между атомами углерода 1' и 2' может присутствовать двойная связь, которая изображена ниже в формуле (I-e).
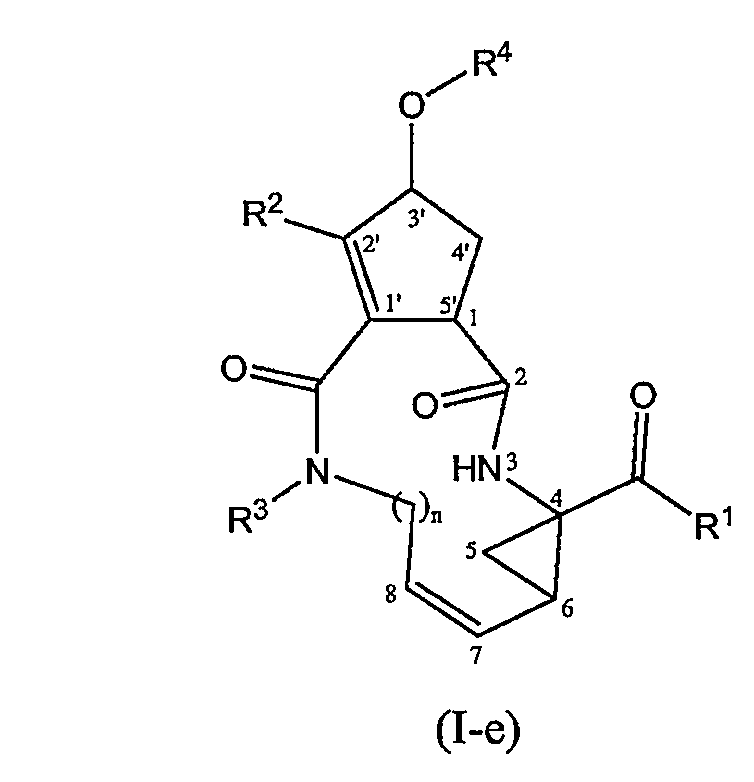
Еще одну особую подгруппу соединений формулы (I) представляют соединения, представленные следующими структурными формулами:
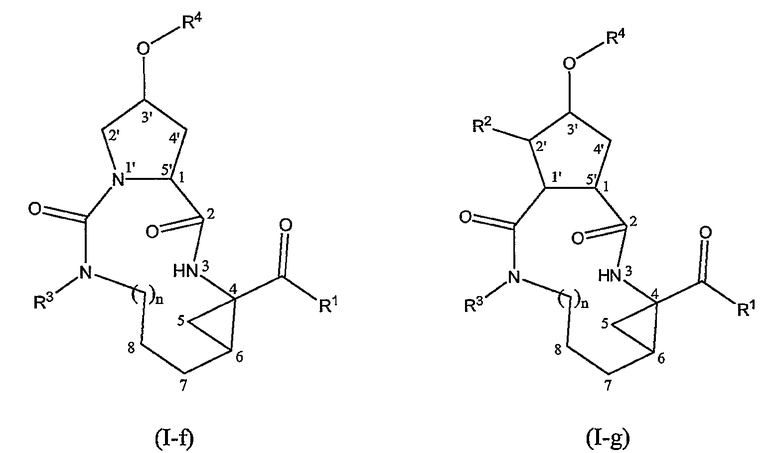
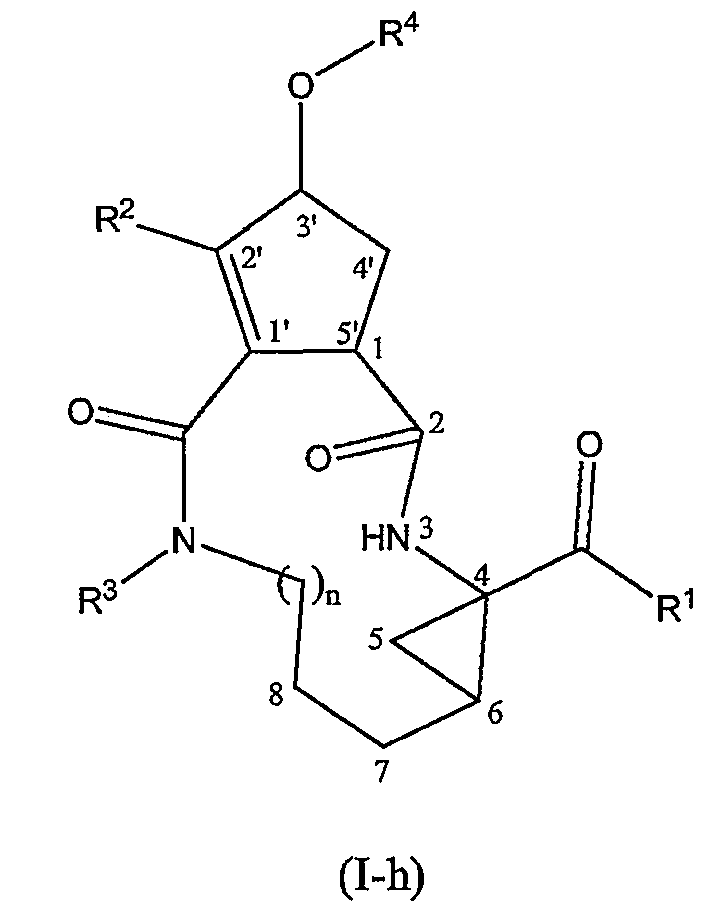
Среди соединений формул (I-f), (I-g) или (I-h) особый интерес представляют соединения, имеющие стереохимическую конфигурацию соединений формул (I-a) и (I-b).
В формулах (I-a), (I-b), (I-c), (I-d), (I-e), (I-f), (I-g) и (I-h), где это применимо, X, n, R1, R2, R3 и R4 имеют значения, указанные в определениях соединений формулы (I) или в любой из указанных здесь подгрупп соединений формулы (I).
Следует понимать, что определенные выше подгруппы соединений формул (I-a), (I-b), (I-c), (I-d), (I-e), (I-f), (I-g) или (I-h), а также любых других указанных здесь подгрупп, также включают в себя любые пролекарства, N-оксиды, аддитивные соли, четвертичные амины, комплексы с металлами и стереохимически изомерные формы таких соединений.
Когда n равно 2, заключенный в скобки фрагмент (-CH2-)«n» в соединениях формулы (I) или в любой подгруппе соединений формулы (I) соответствует этандиилу. Когда n равно 3, заключенный в скобки фрагмент (-CH2-)«n» в соединениях формулы (I) или в любой подгруппе соединений формулы (I) соответствует пропандиилу. Когда n равно 4, заключенный в скобки фрагмент (-CH2-)«n» в соединениях формулы (I) или в любой подгруппе соединений формулы (I) соответствует бутандиилу. Когда n равно 5, заключенный в скобки фрагмент (-CH2-)«n» в соединениях формулы (I) или в любой подгруппе соединений формулы (I) соответствует пентандиилу. Когда n равно 6, заключенный в скобки фрагмент (-CH2-)«n» в соединениях формулы (I) или в любой подгруппе соединений формулы (I) соответствует гександиилу. Соединения, в которых n равно 4 или 5, представляют собой особые подгруппы соединений формулы (I).
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых
(a) R1 представляет собой -OR5, в частности группу, в которой R5 представляет собой C1-6-алкил, такой как метил, этил или трет-бутил, и наиболее предпочтительно, когда R5 представляет собой водород; или
(b) R1 представляет собой -NHS(O)2R6, в частности группу, в которой R6 представляет собой C1-6-алкил, C3-C7-циклоалкил, необязательно замещенный C1-6-алкилом, или арил, например, в которых R6 представляет собой метил, циклопропил, метилциклопропил или фенил.
Дополнительные варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых
(а) R2 представляет собой водород;
(b) R2 представляет собой C1-6-алкил, предпочтительно метил.
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых
(a) X представляет собой N, C (X присоединяется с помощью двойной связи) или CH (X присоединяется с помощью одинарной связи), и R2 представляет собой водород;
(b) X представляет собой C (X присоединяется с помощью двойной связи), и R2 представляет собой C1-6-алкил, предпочтительно метил.
Дополнительные варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых
R3 представляет собой водород;
(b) R3 представляет собой C1-6-алкил;
(d) R3 представляет собой C1-6-алкокси-C1-6-алкил или C3-7-циклоалкил.
Предпочтительные варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R3 представляет собой водород или C1-6-алкил, более предпочтительно водород или метил.
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой изохинолин-1-ил, необязательно моно, ди- или тризамещенный C1-6-алкилом, C1-6-алкокси, гидрокси, галогеном, трифторметилом, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонилом, арилом, Het; где каждый арил или Het независимо и необязательно замещен галогеном, C1-6-алкилом, C1-6-алкокси, полигалоген-C1-6-алкокси, амино, моно- или ди-C1-6-алкиламино, C3-7-циклоалкилом (в частности, циклопропилом), пирролидинилом, пиперидинилом, пиперазинилом, 4-C1-6-алкилпиперазинилом (в частности, 4-метилпиперазинилом) или морфолинилом.
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой изохинолин-1-ил, необязательно моно-, ди- или тризамещенный метилом, этилом, изопропилом, трет-бутилом, метокси, этокси, трифторметилом, трифторметокси, фтором, хлором, бромом, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонилом, фенилом, метоксифенилом, цианофенилом, галогенфенилом, пиридилом, C1-4-алкилпиридилом, пиримидинилом, морфолинилом, пиперазинилом, C1-4-алкилпиперазинилом, пирролидинилом, пиразолилом, C1-4-алкилпиразолилом, тиазолилом, C1-4-алкилтиазолилом, циклопропилтиазолилом или моно- или ди-C1-4-алкиламинотиазолилом.
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой
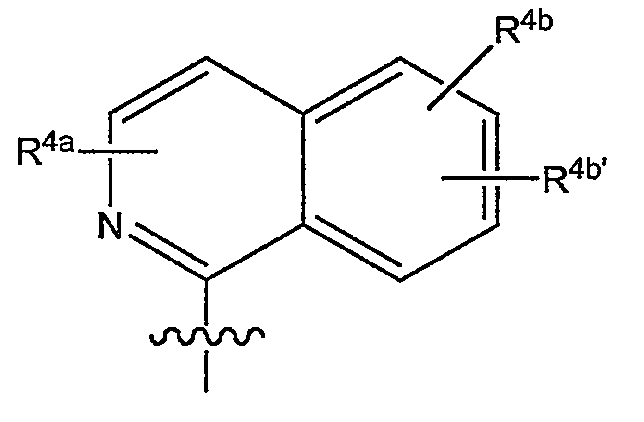
в которой в данной и следующих структурных формулах, представляющих варианты осуществления радикала R4, каждый R4a, R4b, R4b' независимо представляет собой любой из заместителей, выбранных из заместителей, упомянутых в качестве возможных заместителей моноциклических или бициклических коьцевых систем R1, которые указаны в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I).
Конкретно R4a может представлять собой водород, галоген, C1-6-алкил, C1-6-алкокси, моно- или C1-6-алкиламино, амино, арил или Het; каждый упомянутый арил или Het независимо и необязательно замещен любым из заместителей Het или арила, упомянутых в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I); или конкретно каждый упомянутый арил или Het независимо и необязательно замещен C1-6-алкилом, C1-6-алкокси, полигалоген-C1-6-алкокси, амино, моно- или ди-C1-6-алкиламино, галогеном, морфолинилом, пиперидинилом, пирролидинилом, пиперазинилом, 4-C1-6-алкилпиперазинилом (таким как 4-метилпиперазинил); и
каждый R4b и R4b' независимо представляет собой водород, C1-6-алкил, C1-6-алкокси, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонил, гидрокси, галоген, трифторметил, арил или Het; каждый упомянутый арил или Het независимо и необязательно замещен любым из заместителей Het или арила, упомянутых в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I); или конкретно каждый упомянутый арил или Het независимо и необязательно замещен C1-6-алкилом, C1-6-алкокси, полигалоген-C1-6-алкокси, амино, моно- или ди-C1-6-алкиламино; морфолинилом, пиперидинилом, пирролидинилом, пиперазинилом, 4-C1-6-алкилпиперазинилом (таким как 4-метилпиперазинил).
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4а представляет собой радикал
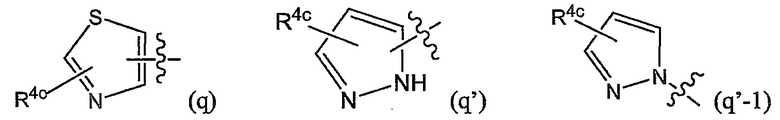
или, в частности, в которых Rla выбран из группы, состоящей из:
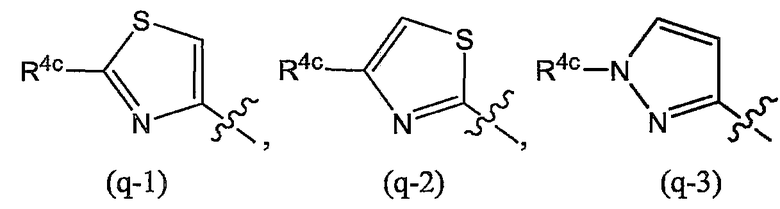 или
или 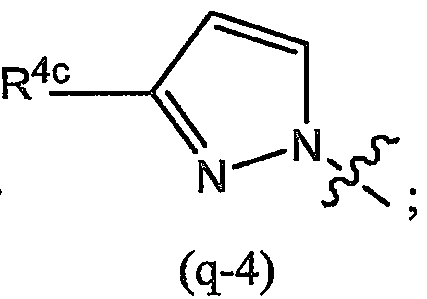
в которых, когда возможно, атом азота может содержать заместитель R4c или быть связанным с остатком молекулы;
в которых каждый R4c независимо представляет собой любой из заместителей Het, упомянутых в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I);
или, конкретно, каждый R4c независимо представляет собой водород, галоген, C1-6-алкил, амино или моно- или ди-C1-6-алкиламино, морфолинил, пиперидинил, пирролидинил, пиперазинил, 4-C1-6-алкилпиперазинил (такой как 4-метилпиперазинил); и в которых морфолинильные и пиперидинильные группы необязательно могут быть замещены одним или двумя C1-6-алкильными радикалами;
более конкретно, каждый R4c независимо представляет собой водород, галоген, C1-6-алкил, амино или моно- или ди-C1-6-алкиламино;
и когда R4c замещен (является заместителем) на атоме азота, он предпочтительно представляет собой углеродсодержащий заместитель, который связан с атомом азота через посредство атома углерода или одного из своих атомов углерода; и в случае которого R4c предпочтительно представляет собой C1-6-алкил.
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой:
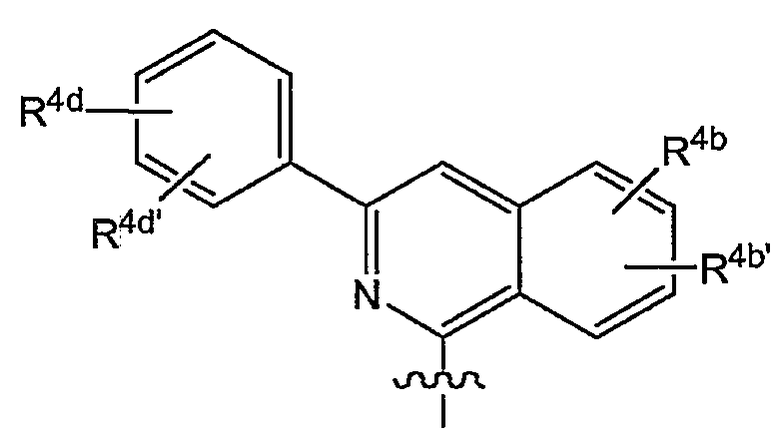
в которой каждый R4b и R4b' независимо представляет собой указанную выше группу; или конкретно каждый R4b и R4b' независимо представляет собой водород, C1-6-алкил, C1-6-алкокси, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонил, гидрокси, галоген, трифторметил, арил или Het; и
R4d и R4d' независимо представляют собой любой из заместителей арила, упомянутых в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I); или конкретно R4d или R4d' независимо представляют собой водород, C1-6-алкил, C1-6-алкокси или галоген.
Дополнительные варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой:
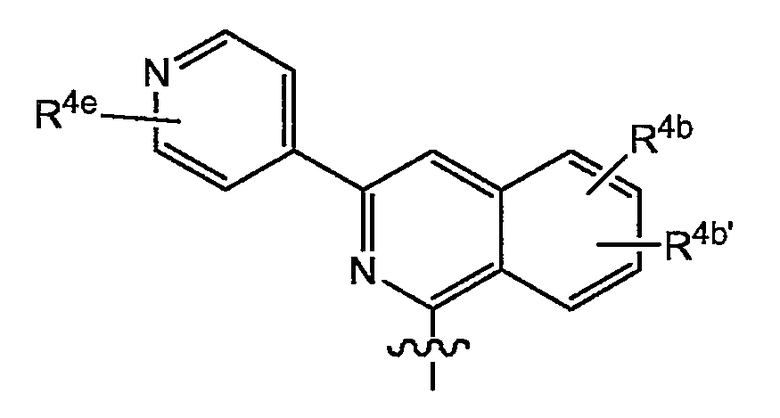
в которой каждый R4b и R4b' независимо представляет собой указанную выше группу; или, конкретно, каждый R4b и R4b' независимо представляет собой водород, C1-6-алкил, C1-6-алкокси, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонил, гидрокси, галоген, трифторметил, арил или Het; и
R4e представляет собой любой из заместителей арила, упомянутых в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I); или конкретно R4e представляет собой водород, C1-6-алкил, C1-6-алкокси или галоген.
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой:
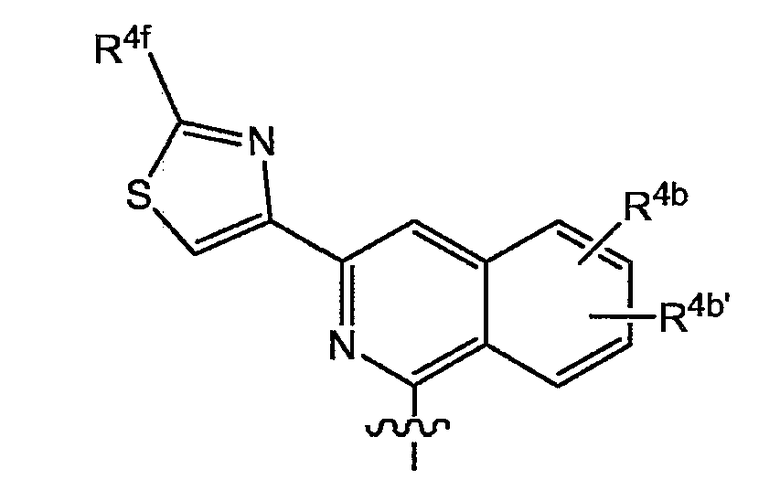
в которой каждый R4b и R4b' представляет собой указанную выше группу; или конкретно каждый R4b и R4b' независимо представляет собой водород, C1-6-алкил, C1-6-алкокси, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонил, гидрокси, галоген, трифторметил; предпочтительно R4b представляет собой C1-6-алкокси, более предпочтительно метокси; и
R4f представляет собой любой из заместителей арила, упомянутых в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I); или конкретно R4f представляет собой водород, C1-6-алкил, амино, моно- или ди-C1-6-алкиламино, пирролидинил, пиперидинил, пиперазинил, 4-C1-6-алкилпиперазинил (в частности, 4-метилпиперазинил) или морфолинил.
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой:
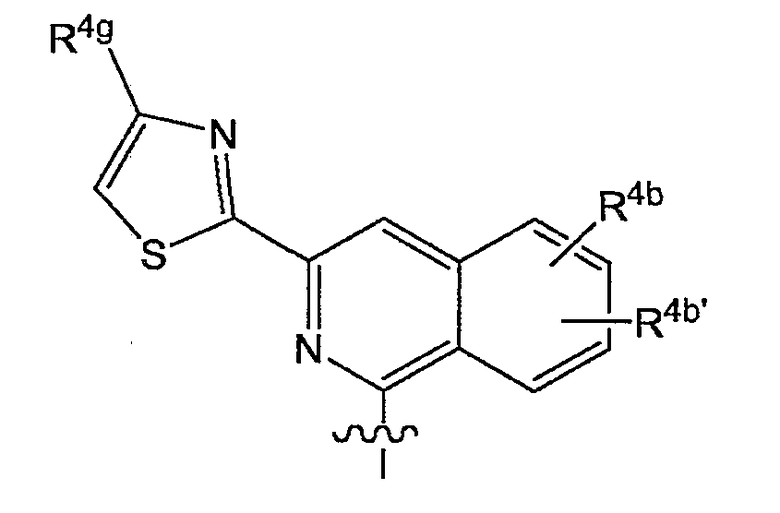
в которой каждый R4b и R4b' представляет собой указанную выше группу; или конкретно каждый R4b и R4b' независимо представляет собой водород, C1-6-алкил, C1-6-алкокси, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонил, гидрокси, галоген, трифторметил; предпочтительно R4 представляет собой C1-6-алкокси, наиболее предпочтительно метокси, галоген, или C1-3-алкил; и
R4g представляет собой любой из заместителей арила, упомянутых в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I); или конкретно R4g представляет собой водород, C1-6-алкил, амино, моно- или ди-C1-6-алкиламино, пирролидинил, пиперидинил, пиперазинил, 4-C1-6-алкилпиперазинил (в частности, 4-метилпиперазинил) или морфолинил.
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой:
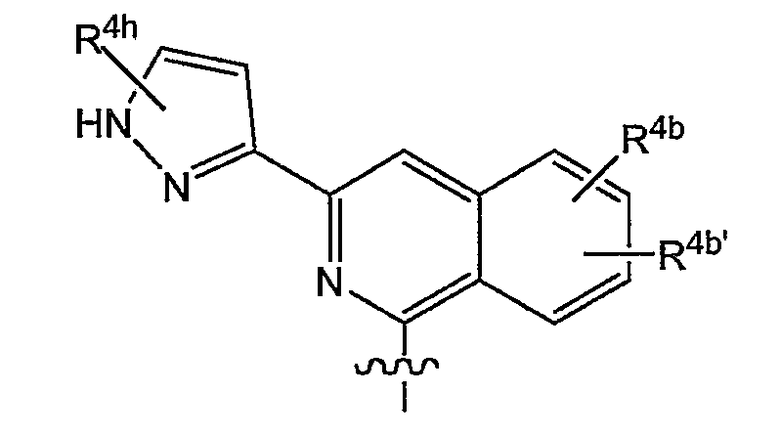
в которой каждый R4b и R4b' представляет собой указанную выше группу; или конкретно каждый R4b и R4b' независимо представляет собой водород, C1-6-алкил, C1-6-алкокси, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонил, гидрокси, галоген, трифторметил; предпочтительно R4b представляет собой C1-6-алкокси, наиболее предпочтительно метокси, галоген или C1-3-алкил; и
R4h представляет собой любой из заместителей арила, упомянутых в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I); или конкретно R4h представляет собой водород, C1-6-алкил, амино, моно- или ди-C1-6-алкиламино, пирролидинил, пиперидинил, пиперазинил, 4-C1-6-алкилпиперазинил (в частности, 4-метилпиперазинил) или морфолинил; и в которой R4h также может быть замещен (являться заместителем) на одном из атомов азота пирразольного кольца, в случае которого он предпочтительно представляет собой C1-6-алкил.
Варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой:
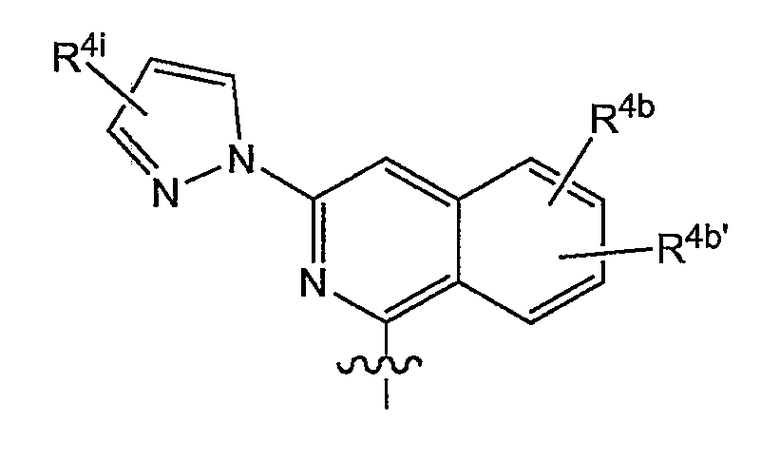
в которой каждый R4b и R4b' представляет собой указанную выше группу; или конкретно каждый R4b и R4b' независимо представляет собой водород, C1-6-алкил, C1-6-алкокси, моно- или ди-C1-6-алкиламино, моно- или ди-C1-6-алкиламинокарбонил, гидрокси, галоген, трифторметил; предпочтительно R4b представляет собой C1-6-алкокси, наиболее предпочтительно метокси, галоген, или C1-3-алкил; и
R4i представляет собой любой из заместителей арила, упомянутых в описаниях соединений формулы (I) или любой из подгрупп соединений формулы (I); или конкретно R4i представляет собой водород, C1-6-алкил, амино, моно- или ди-C1-6-алкиламино, пирролидинил, пиперидинил, пиперазинил, 4-C1-6-алкилпиперазинил (в частности, 4-метилпиперазинил) или морфолинил.
Предпочтительные варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой:
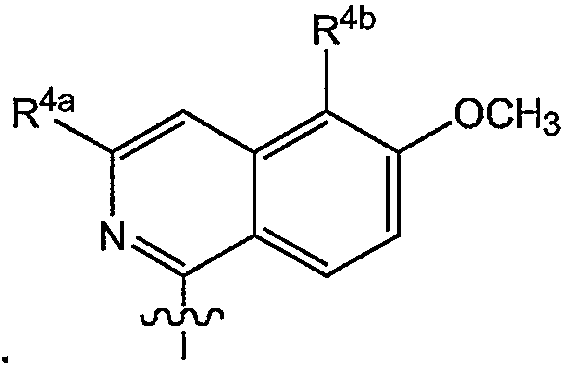
в которой R4a определен в любой из групп или подгрупп соединений формулы (I); и
R4b представляет собой водород, галоген или трифторметил.
Кроме того, предпочтительные варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой:
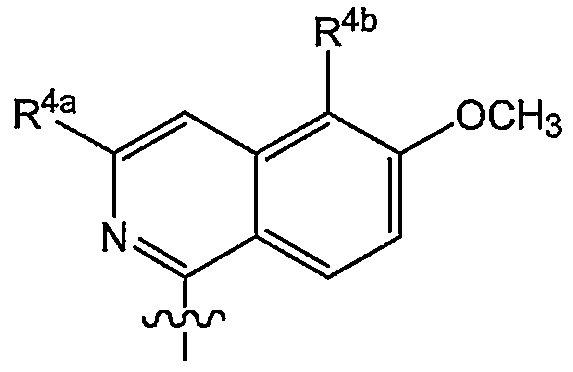
в которой R4a представляет собой метокси, этокси или пропокси; и
R4b представляет собой водород, фтор, бром, хлор, йод, метил, этил, пропил или трифторметил.
Кроме того, предпочтительные варианты осуществления изобретения относятся к соединениям формулы (I) или любой из подгрупп соединений формулы (I), в которых R4 представляет собой:
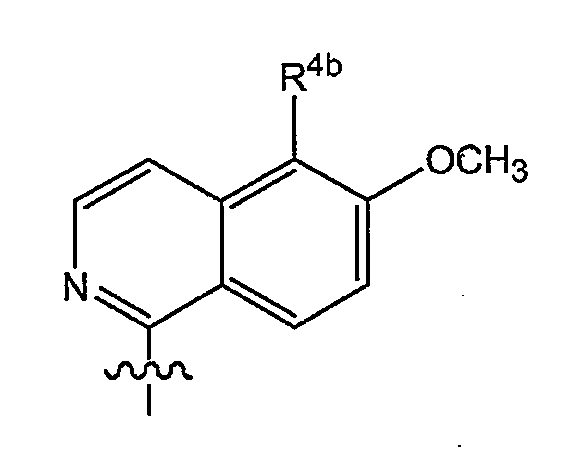
в которой R4b представляет собой водород, галоген или трифторметил.
Соединения формулы (I) состоят из трех структурных элементов P1, P2, P3. Структурный элемент P1 дополнительно содержит концевую часть P1'. Карбонильная группа, ниже в соединении (I-c) отмеченная звездочкой, может быть частью либо структурного элемента P2, либо структурного элемента P3. По химическим соображениям структурный элемент P2 соединений формулы (I), в котором X представляет собой C, включает в себя карбонильную группу, присоединенную в положении 1'.
Соединение структурных элементов P1 с P2, P2 с P3 и P1 с P1' (когда R1 представляет собой -NH-SO2R6) предусматривает образование амидной связи. Соединение элементов P1 и P3 предусматривает образование двойной связи. Соединение структурных элементов P1, P2 и P3 для получения соединений (I-i) или (I-j) может выполняться в любой заданной последовательности. Одна из стадий предусматривает циклизацию, с помощью которой образуется макроцикл.
Ниже представлены соединения (I-i), которые относятся к соединениям формулы (I), в которых атомы углерода C7 и C8 соединены двойной связью, и соединения (I-j), которые относятся к соединениям формулы (I), в которых атомы углерода C7 и C8 соединены одинарной связью. Соединения формулы (I-j) можно получать из соответствующих соединений формулы (I-i) путем восстановления двойной связи в макроцикле.
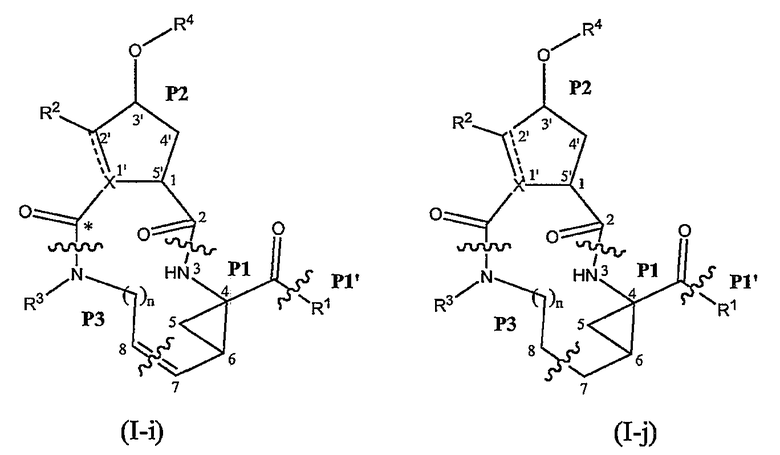
Разумеется, что описанные в дальнейшем процедуры синтеза должны быть применимы в равной степени к рацематам, стереохимически чистым промежуточным продуктам или конечным продуктам или любым смесям стереоизомеров. Рацематы или смеси стереоизомеров можно разделять на стереоизомерные формы на любой стадии процедур синтеза. В одном из вариантов осуществления изобретения промежуточные продукты и конечные продукты имеют стереохимию, указанную выше для соединений формулы (I-a) и (I-b).
В одном из вариантов осуществления изобретения соединения (I-i) получают, образуя сначала амидные связи, а затем образуя двойную связь между P3 и P1, с сопутствующей циклизацией до макроцикла.
В предпочтительном варианте осуществления изобретения соединения (I), в которых связь между атомами углерода C7 и C8 является двойной связью и которые представляют собой указанные выше соединения формулы (I-i), можно получать, как кратко изложено на следующей реакционной схеме:
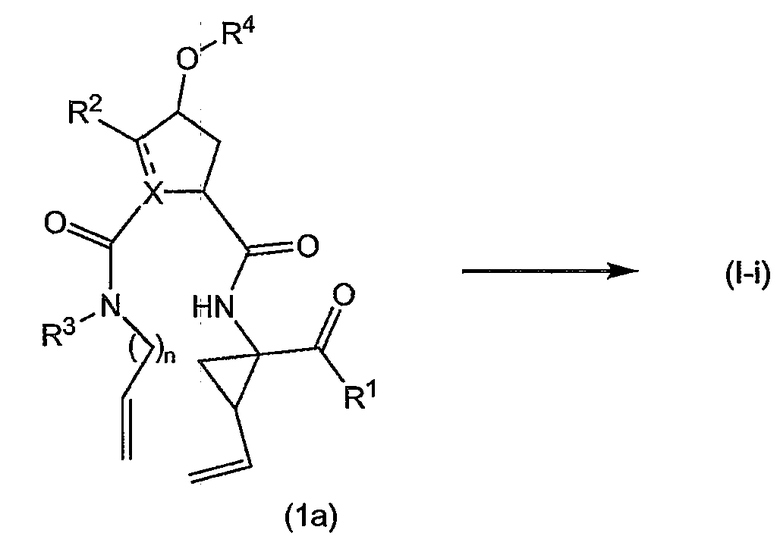
Образование макроцикла можно осуществлять с помощью реакции метатезиса олефинов в присутствии подходящего металлического катализатора, такого как, например, катализатор на основе Ru, о котором сообщается в публикациях: Miller S.J., Blackwell H.E., Grubbs R.H., J. Am. Chem. Soc. 118, (1996), 9606-9614; Kingsbury J.S., Harrity J.P.A., Bonitatebus P.J., Hoveyda A.H., J. Am. Chem. Soc. 121, (1999), 791-799; и Huang и др., J. Am. Chem. Soc. 121, (1999), 2674-2678; например, катализатор Ховейды-Граббса (Hoveyda-Grubbs).
Можно применять устойчивые на воздухе рутениевые катализаторы, такие как хлорид бис(трициклогексилфосфин)-3-фенил-1H-инден-1-илиденрутения (Neolyst M1®) или дихлорид бис(трициклогексилфосфин)-[(фенилтио)метилен]рутения (IV). Другие катализаторы, которые можно применять, представляют собой катализаторы Граббса первого и второго поколения, то есть бензилиден-бис(трициклогексилфосфин)дихлоррутений и (1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(фенилметилен)-(трициклогексилфосфин)рутений, соответственно. Особый интерес представляют катализаторы Ховейды-Граббса первого и второго поколения, которые представляют собой дихлор(o-изопропоксифенилметилен)(трициклогексилфосфин)рутений(II) и 1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(o-изопропоксифенилметилен)рутений соответственно. Также для такой реакции можно применять другие катализаторы, содержащие другие переходные металлы, такие как Mo.
Реакции метатезиса можно проводить в подходящем растворителе, например, таком как простые эфиры, например ТГФ, диоксан; галогенированные углеводороды, например, дихлорметан, CHCl3, 1,2-дихлорэтан и т.п., углеводороды, например, толуол. В предпочтительном варианте осуществления изобретения реакцию метатезиса проводят в толуоле. Такие реакции проводят при повышенных температурах в атмосфере азота.
Соединения формулы (I), в которых связь между атомами углерода C7 и C8 в макроцикле представляет собой одинарную связь, то есть соединения формулы (I-j), можно получать из соединений формулы (I-i) путем восстановления двойной связи C7-C8 в соединениях формулы (I-i). Такое восстановление можно проводить с помощью каталитического гидрирования водородом в присутствии катализатора на основе благородных металлов, например, таких как Pt, Pd, Rh, Ru или никель Ренея. Интерес представляет Rh на оксиде алюминия. Реакцию гидрирования предпочтительно проводят в растворителе, например, таком как спирт, такой как метанол, этанол, или в простом эфире, таком как ТГФ, или в их смесях. К таким растворителям или смесям растворителей также можно добавлять воду.
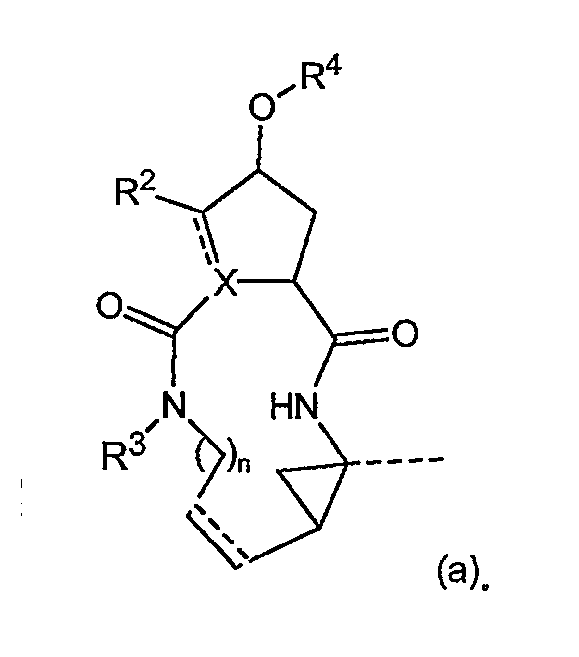
Группу R1 можно присоединять к структурному элементу P1 на любой стадии синтеза, то есть до или после циклизации, или до или после циклизации и восстановления, как описано выше. Соединения формулы (I), в которой R1 представляет собой -NHSO2R6 [упомянутые соединения представлены формулой (I-k-1)], можно получать путем присоединения группы R1 к P1 с помощью образования между обоими фрагментами амидной связи. Подобным образом соединения формулы (I), в которых R1 представляет собой -OR5, то есть соединения (I-k-2), можно получать путем присоединения группы R1 к P1 с помощью образования сложноэфирной связи. В одном из вариантов осуществления изобретения группы -OR5 вводятся на последней стадии синтеза соединений (I), как кратко изложено на следующих реакционных схемах, на которых G представляет собой группу:
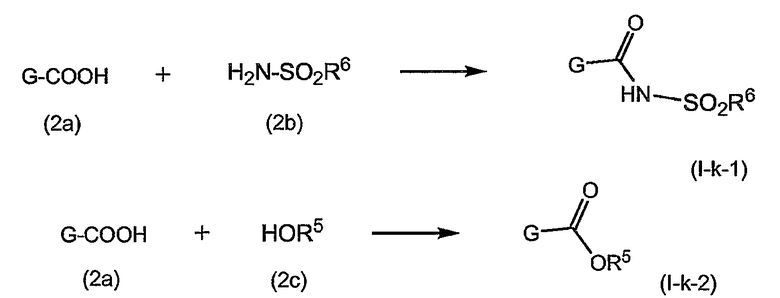
Промежуточный продукт (2a) можно связывать с амином (2b) с помощью реакции образования амида, такой как любая из описанных в дальнейшем процедур образования амидной связи. В частности, (2a) можно обрабатывать конденсирующим агентом, например, N,N' -карбонилдиимидазолом (CDI), EEDQ, IIDQ, EDCI или гексафторфосфатом бензотриазол-1-илокси-трис-пирролидинoфосфония (имеется в продаже под торговой маркой PyBOP®) в растворителе, таком как простой эфир, например, ТГФ или галогенированном углеводороде, например, дихлорметане, хлороформе, дихлорэтане, и проводить реакцию с требуемым сульфонамидом (2b), предпочтительно после взаимодействия (2a) с конденсирующим агентом. Реакции (2a) с (2b) предпочтительно осуществляют в присутствии основания, например, триалкиламина, такого как триэтиламин или диизопропилэтиламин, или в присутствии 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU). Промежуточный продукт (2a) также можно переводить в активированную форму, например в активированную форму общей формулы G-CO-Z, в которой Z представляет собой галоген или остаток активного сложного эфира, например, Z представляет собой арилокси-группу, такую как фенокси, п-нитрофенокси, пентафторфенокси, трихлорфенокси, пентахлорфенокси и т.п.; или Z может представлять собой остаток смешанного ангидрида. В одном из вариантов осуществления изобретения G-CO-Z представляет собой хлорангидрид (G-CO-Cl) или смешанный ангидрид (G-CO-O-CO-R или G-CO-O-CO-OR, в последнем случае R представляет собой, например, C1-4-алкил, такой как метил, этил, пропил, изопропил, бутил, т-бутил, изобутил или бензил). Активированную форму G-CO-Z подвергают взаимодействию с сульфонамидом (2b).
Активация карбоновой кислоты в (2a), как описано для упомянутых выше реакций, может приводить к реакции внутренней циклизации промежуточного азалактона формулы
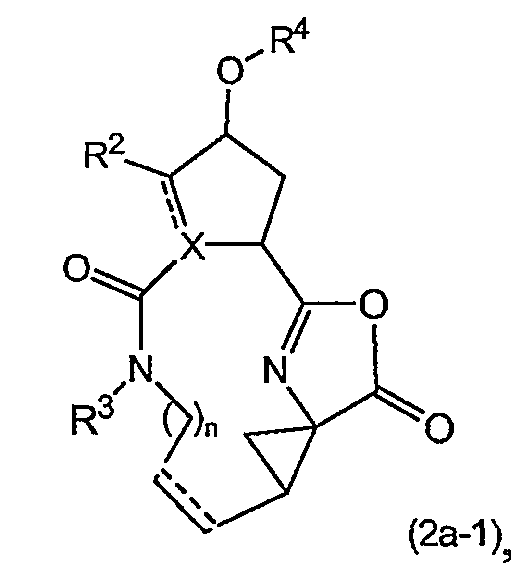
в которой значения X, R2, R3, R4, n такие, как указано выше, и в которой стереогенные центры могут иметь указанные выше стереохимические конфигурации, например, как в соединениях (I-a) или (I-b). Промежуточные продукты (2a-1) можно выделять из реакционной смеси, применяя традиционную методику, и выделенный промежуточный продукт (2a-1) затем подвергать взаимодействию с (2b), или реакционную смесь, содержащую (2a-1), можно дополнительно подвергать взаимодействию с (2b) без выделения (2a-1). В одном из вариантов осуществления изобретения, когда реакцию с конденсирующим агентом осуществляют в несмешивающимся с водой растворителе, реакционную смесь, содержащую (2a-1), можно промывать водой или слегка щелочной водой для того, чтобы удалить все водорастворимые побочные продукты. Полученный таким образом промытый раствор затем можно подвергать взаимодействию с (2b) без дополнительных стадий очистки. С другой стороны, выделение промежуточных продуктов (2a-1) может обеспечить некоторые преимущества в том смысле, что выделенный продукт после необязательной дополнительной очистки можно подвергать взаимодействию с (2b), получая при этом меньшее количество побочных продуктов и добиваясь более простого осуществления реакции.
Промежуточный продукт (2a) можно связывать со спиртом (2c) с помощью реакции образования сложного эфира. Например, (2a) и (2c) подвергают взаимодействию совместно с удалением воды либо физически, например, путем азеотропного удаления воды, либо химически, применяя дегидратирующий агент. Промежуточный продукт (2a) также можно переводить в активированную форму G-CO-Z, такую как упомянутые выше активированные формы, и затем подвергать взаимодействию со спиртом (2c). Реакции образования сложных эфиров предпочтительно проводят в присутствии основания, такого как карбонат или гидрокарбонат щелочного металла, например, гидрокарбонат натрия или калия, или третичного амина, такого как амины, упомянутые здесь в связи с реакциями образования амидов, в частности, в присутствии триалкиламина, например, триэтиламина. Растворители, которые можно применять в реакциях образования сложных эфиров, включают в себя простые эфиры, такие как ТГФ; галогенированные углеводороды, такие как дихлорметан, CH2Cl2; углеводороды, такие как толуол; полярные апротонные растворители, такие как ДМФА, DMSO, DMA; и тому подобные растворители.
Соединения формулы (I), в которых R3 представляет собой водород (упомянутые соединения представлены формулой (I-1)), также можно получать путем удаления защитной группы PG из соответствующего азот-защищенного промежуточного продукта (3a), как показано на следующей реакционной схеме. Защитная группа PG, в частности, представляет собой любою из азот-защитных групп, упомянутых в дальнейшем, и может быть удалена с помощью процедур, также упомянутых в дальнейшем:
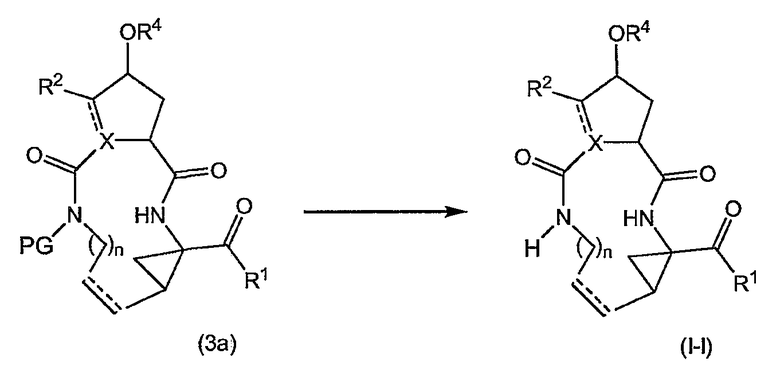
Исходные материалы (3a) для указанной выше реакции можно получать, следуя процедурам получения соединений формулы (I), но применяя промежуточные продукты, в которых группа R3 представляет собой PG.
Соединения формулы (I) также можно получать путем взаимодействия промежуточного продукта (4a) промежуточным продуктом (4b), как кратко изложено на следующей реакционной схеме, на которой различные радикалы имеют указанные выше значения:
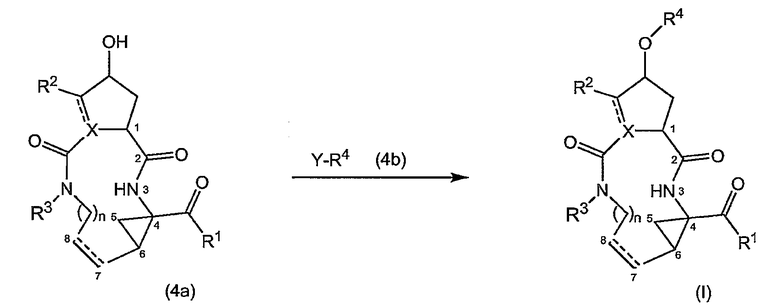
Y в (4b) представляет собой гидрокси или удаляемую группу LG, такую как галогенид, например, бромид или хлорид, или арилсульфонильную группу, например, мезилат, трифлат или тозилат и т.п.
В одном из вариантов осуществления изобретения реакция (4a) с (4b) представляет собой реакцию О-арилирования, и Y представляет собой удаляемую группу. Такую реакцию можно осуществлять, следуя процедурам, описаным в публикации E. M. Smith и др. (J. Med. Chem. (1988), 31, 875-885). В частности, такую реакцию проводят в присутствии основания, предпочтительно сильного основания, в реакционно-инертном растворителе, например в одном из растворителей, упомянутых в случае образования амидной связи.
В конкретном варианте осуществления изобретения исходный материал (4a) подвергается взаимодействию с (4b) в присутствии основания, которое является достаточно сильным, чтобы отнимать водород от гидрокси-группы, например, представляет собой щелочь на основе гидрида щелочного металла, такого как LiH или гидрид натрия, или алкоголят щелочного металла, такой как метилат или этилат натрия или калия, трет-бутилат калия, в реакционно-инертном растворителе, типа диполярного апротонного растворителя, например, DMA, ДМФА и т.п. Полученный в результате алкоголят подвергается взаимодействию с арилирующим агентом (4b), в котором, как упомянуто выше, Y представляет собой подходящую удаляемую группу. Превращение (4a) в (I) с применением такого типа реакции О-арилирования не изменяет стереохимическую конфигурацию на атоме углерода, несущем гидрокси-группу.
Альтернативно реакцию (4a) с (4b) также можно осуществлять с помощью реакции Мицунобу (Mitsunobu, 1981, Synthesis, January, 1-28; Rano и др., Tetrahedron Lett., 1995, 36, 22, 3779-3792; Krchnak и др., Tetrahedron Lett., 1995, 36, 5, 6193-6196; Richter и др., Tetrahedron Lett., 1994, 35, 27, 4705-4706). Такая реакция включает в себя обработку промежуточного продукта (4a) соединением (4b), в котором Y представляет собой гидроксил, в присутствии трифенилфосфина и активирующего агента, такого как диалкилазокарбоксилат, например, диэтилазодикарбоксилат (DEAD), диизопропилазодикарбоксилат (DIAD) или т.п. При реакции Мицунобу изменяется стереохимическая конфигурация на атоме углерода, несущем гидрокси-группу.
Альтернативно, для получения соединений формулы (I) сначала образуют амидную связь между структурными элементами P2 и P1, с последующим сочетанием структурного элемента P3 с фрагментом P1 в P1-P2, и последующим образованием карбаматной или сложноэфирной связи между P3 и фрагментом P2 в P2-P1-P3 с сопутствующим замыканием цикла.
Еще один альтернативный способ синтеза заключается в образовании амидной связи между структурными элементами P2 и P3 с последующим сочетаним структурного элемента P1 с фрагментом P3 в P3-P2, и с образованием в конце амидной связи между P1 и P2 в P1-P3-P2 с сопутствующим замыканием цикла.
Структурные элементы P1 и P3 можно присоединять к последовательности P1-P3. Если необходимо, можно восстанавливать двойную связь, соединяющую P1 и P3. Образованную таким образом последовательность P1-P3, либо восстановленную, либо нет, можно связывать со структурным элементом P2 и образованную таким образом последовательность P1-P3-P2 затем циклизировать путем образования амидной связи.
Структурные элементы P1 и P3 в любом из предыдущих подходов можно соединять с помощью образования двойной связи, например, с помощью реакции метатезиса олефинов, описанной в дальнейшем, или с помощью реакции типа реакции Виттига. Если необходимо, образованную таким образом двойную связь можно восстанавливать подобно тому, как описано выше для превращения (I-i) в (I-j). Двойную связь также можно восстанавливать на более поздней стадии, например, после добавления третьего структурного элемента или после образования макроцикла. Структурные элементы P2 и P1 соединяются с помощью образования амидной связи, и P3 и P2 соединяются с помощью образования карбамата или сложного эфира.
Концевую часть P1' можно связывать со структурным элементом P1 на любой стадии синтеза соединений формулы (I), например, до или после сочетания структурных элементов P2 и P1; до или после сочетания структурного элемента P3 с P1; или до или после замыкания цикла.
Сначала можно получать отдельные структурные элементы и затем связывать их вместе или, альтернативно, можно связывать вместе предшественников структурных элементов и изменять их на более поздней стадии до требуемого молекулярного состава.
Чтобы избежать побочных реакций, можно защищать функциональные группы в каждом из структурных элементов.
Образование амидных связей можно осуществлять, применяя стандартные процедуры, такие как процедуры, применяемые для связывания аминокислот при синтезе пептидов. Последний предусматривает дегидратирующее сочетание карбоксильной группы одного реагента с аминогруппой другого реагента с образованием соединяющей амидной связи. Образование амидной связи можно осуществлять путем взаимодействия исходных материалов в присутствии конденсирующего агента или путем преобразования карбоксильной функциональной группы в активную форму, такую как активный сложный эфир, смешанный ангидрид или хлорангидрид или бромангидрид карбоновых кислот. Общие описания таких реакций сочетания и применяемых в них реагентов можно найти в общих руководствах по химии пептидов, например, в публикации M. Bodanszky, «Peptide Chemistry», 2nd rev. ed., Springer-Verlag, Berlin, Germany, (1993).
Примеры реакций сочетания с образованием амидной связи включают в себя азидный способ синтеза, способ смешанных ангидридов угольной-карбоновых кислот (с применением изобутилхлорформиата), карбодиимидный способ (с применением дициклогексилкарбодиимида, диизопропилкарбодиимида или водорастворимого карбодиимида, такого как N-этил-N'-[(3-диметиламино)пропил]карбодиимид), способ активированных сложных эфиров (например, п-нитрофениловый, п-хлорфениловый, трихлорфениловый, пентахлорфениловый, пентафторфениловый, N-гидроксисукцинимидоэфир и тому подобные сложные эфиры), способ с применением K-реагента Вудворда, 1,1-карбонилдиимидазольный (CDI или N,N'-карбонилдиимидазольный) способ, способ с применением фосфорных реагентов или окислительно-восстановительные способы. Некоторые из таких способов можно усовершенствовать путем добавления подходящих катализаторов, например, при карбодиимидном способе путем добавления 1-гидроксибензотриазола, DBU (1,8-диазабицикло[5.4.0]ундец-7-ен) или 4-DMAP. Дополнительными конденсирующими агентами являются гексафторфосфат (бензотриазол-1-илокси)трис-(диметиламино)фосфония либо сам по себе, либо в присутствии 1-гидроксибензотриазола или 4-DMAP; или тетрафторборат 2-(1H-бензотриазол-1-ил)-N,N,N'N'-тетраметилурония, или гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония. Такие реакции сочетания можно осуществлять либо в растворе (в жидкой фазе), либо в твердой фазе.
Предпочтительно образование амидной связи осуществляют, применяя N-этилоксикарбонил-2-этилокси-1,2-дигидрохинолин (EEDQ) или N-изобутилоксикарбонил-2-изобутилокси-1,2-дигидрохинолин (IIDQ). В отличие от классического ангидридного способа EEDQ и IIDQ не требуют ни основания, ни низких температур реакции. Обычно процедура предусматривает взаимодействие эквимолярных количеств карбоксильного и аминного компонентов в органическом растворителе (можно применять большое разнообразие растворителей). Затем в избытке добавляют EEDQ или IIDQ и дают возможность смеси перемешиваться при комнатной температуре.
Реакции сочетания предпочтительно осуществляют в инертном растворителе, таком как галогенированные углеводороды, например дихлорметан, хлороформ, диполярных апротонных растворителях, таких как ацетонитрил, диметилформамид, диметилацетамид, DMSO, HMPT, простые эфиры, такие как тетрагидрофуран (ТГФ).
Во многих случаях реакции сочетания осуществляются в присутствии подходящего основания, такого как третичный амин, например триэтиламин, диизопропилэтиламин (DIPEA), N-метилморфолин, N-метилпирролидин, 4-DMAP или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Температура реакции может находиться в диапазоне от 0°C и 50°C, и время взаимодействия может находиться в диапазоне от 15 минут до 24 часов.
Функциональные группы в структурных элементах, которые соединяются вместе, можно защищать, чтобы предотвратить образование нежелательных связей. Подходящие защитные группы, которые можно применять, перечислены, например, в публикациях: Greene, «Protective Groups in Organic Chemistry», John Wiley & Sons, New York (1999) и «The Peptides: Analysis, Synthesis, Biology», т. 3, Academic Press, New York (1987).
Карбоксильные группы можно защищать в виде сложного эфира, который можно удалять, получая при этом карбоновую кислоту. Защитные группы, которые можно применять, включают в себя: 1) сложные алкиловые эфиры, такие как метиловый, триметилсилиловый и трет-бутиловый; 2) сложные арилалкиловые эфиры, такие как бензиловый и замещенный бензиловый; или 3) сложные эфиры, которые можно удалять с помощью слабого основания или слабого восстановителя, такие как сложные трихлорэтиловые и фенациловые эфиры.
Аминогруппы можно защищать с помощью различных N-защитных групп, таких как:
1) ацильные группы, такие как формил, трифторацетил, фталил и п-толуолсульфонил;
2) ароматические карбаматные группы, такие как бензилоксикарбонил (Cbz или Z) и замещенные бензилоксикарбонилы, и 9-флуоренилметилоксикарбонил (Fmoc);
3) алифатические карбаматные группы, такие как трет-бутилоксикарбонил (Boc), этоксикарбонил, диизопропилметоксикарбонил и аллилоксикарбонил;
4) циклические алкилкарбаматные группы, такие как циклопентилоксикарбонил и адамантилоксикарбонил;
5) алкильные группы, такие как трифенилметил, бензил или замещенный бензил, такой как 4-метоксибензил;
6) триалкилсилил, такой как триметилсилил или т-Bu-диметилсилил; и
7) тиолсодержащие группы, такие как фенилтиокарбонил и дитиасукциноил.
Аминозащитными группами, представляющими интерес, являются Boc и Fmoc-группы.
Предпочтительно аминозащитную группу удаляют перед следующей стадией сочетания. Удаление N-защитных групп можно осуществлять, следуя известным в данной области процедурам. Когда применяется Boc-группа, выбор способов заключается в выборе трифторуксусной кислоты, чистой или в дихлорметане, или HCl в диоксане или в этилацетате. Полученную в результате соль аммония затем нейтрализуют либо перед сочетанием, либо в процессе его проведения с помощью основных растворов, таких как водные буферы или третичные амины в дихлорметане, или ацетонитриле, или диметилформамиде. Когда применяется Fmoc-группа, выбор реагентов включает пиперидин или замещенный пиперидин в диметилформамиде, хотя можно применять любой вторичный амин. Снятие защиты осуществляют при температуре от 0°C до комнатной температуры, обычно около 15-25°C или 20-22°C.
Другие функциональные группы, которые могут вмешиваться в реакции сочетания структурных элементов, также можно защищать. Например, гидроксильные группы можно защищать в виде простых бензиловых или замещенных бензиловых эфиров, например, простого 4-метоксибензилового эфира, сложных бензоиловых или замещенных бензоиловых эфиров, например, сложного 4-нитробензоилового эфира, или с помощью триалкилсилильных групп (например, триметилсилила или трет-бутилдиметилсилила).
Дополнительные аминогруппы можно защищать с помощью защитных групп, которые можно удалять избирательно. Например, когда в качестве α-аминозащитной группы применяется Boc-группа, подходящими защитными группами для следующей боковой цепи являются: п-толуолсульфонильные (тозильные) фрагменты, которые можно применять для защиты дополнительных аминогрупп; простые бензиловые (Bn) эфиры, которые можно применять для защиты гидрокси-групп; и сложные бензиловые эфиры, которые можно применять для защиты дополнительных карбоксильных групп. Или когда в качестве α-аминозащитной группы выбирается Fmoc-группа, обычно приемлемыми являются защитные группы на основе трет-бутила. Например, Boc-группу можно применять для дополнительных аминогрупп; простые трет-бутиловые эфиры - для гидроксильных групп; и сложные трет-бутиловые эфиры - для дополнительных карбоксильных групп.
Любую из защитных групп можно удалять на любой стадии процедуры синтеза, однако предпочтительно защитные группы любой из функциональных групп, не участвующих в реакции, удалять после завершения построения макроцикла. Удаление защитных групп можно осуществлять тем или иным способом, который диктуется выбором защитных групп, и такие способы хорошо известны специалисту в данной области.
Промежуточные продукты формулы (1a), в которой X представляет собой N (упомянутые промежуточные продукты представлены формулой (1a-1)), можно получать, исходя из промежуточных продуктов (5a), которые подвергаются взаимодействию с алкенамином (5b) в присутствии агента, вводящего карбонильную группу, как кратко изложено на следующей реакционной схеме:
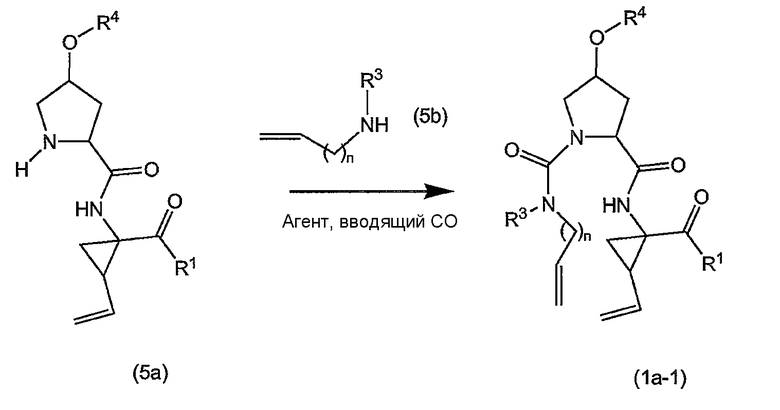
Агенты, вводящие карбонильную группу (CO), включают в себя фосген или производные фосгена, такие как карбонилдиимидазол (CDI) и т.п. В одном из вариантов осуществления изобретения соединение (5a) подвергают взаимодействию с агентом, вводящим CO, в присутствии подходящего основания и растворителя, которые могут представлять собой основания и растворители, применяемые в реакциях образования амидов, как описано выше. В конкретном варианте осуществления изобретения основание представляет собой гидрокарбонат, например NaHCO3 или третичный амин, такой как триэтиламин и т.п., и растворитель представляет собой простой эфир или галогенированный углеводород, например ТГФ, CH2Cl2, CHCl3 и т.п. Затем добавляют амин (5b), получая при этом промежуточные продукты (1a-1), как указано на вышеприведенной схеме. Альтернативный путь синтеза с применением подобных условий реакции предусматривает сначала взаимодействие агента, вводящего CO, с алкенамином (5b) и затем взаимодействие образованного при этом промежуточного продукта с (5a).
Промежуточные продукты (1a-1) альтернативно можно получать следующим образом:
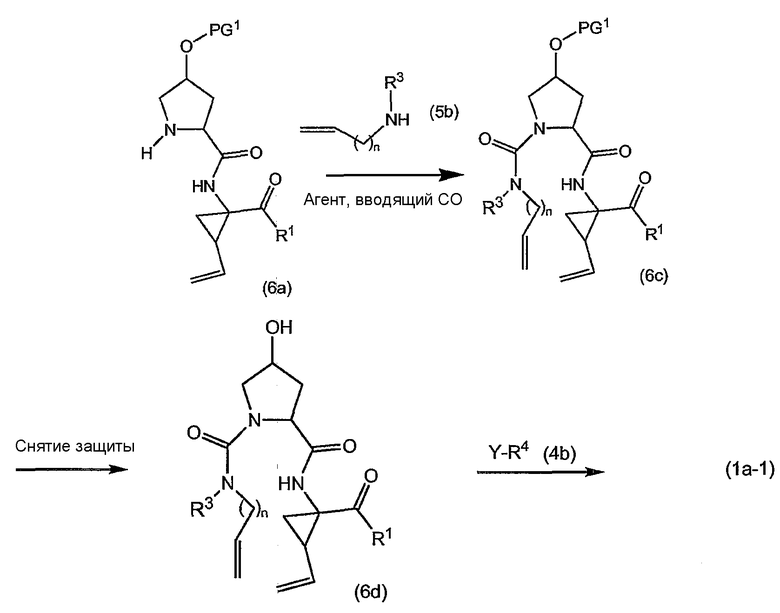
PG1 представляет собой O-защитную группу, которая может представлять собой любую из упомянутых здесь групп и, в частности, представляет собой бензоильную или замещенную бензоильную группу, такую как 4-нитробензоил. В последнем случае такую группу можно удалять с помощью реакции с гидроксидом щелочного металла (LiOH, NaOH, KOH), в частности, когда PG1 представляет собой 4-нитробензоил, с помощью LiOH в водной среде, содержащей воду и водорастворимый органический растворитель, такой как алканол (метанол, этанол) и ТГФ.
Промежуточные продукты (6a) подвергают взаимодействию с (5b) в присутствии агента, вводящего карбонильную группу, подобно описанному выше, и в результате такой реакции получают промежуточные продукты (6c). С них снимают защиту, в частности, применяя упомянутые выше условия реакции. Полученный спирт (6d) подвергают взаимодействию с промежуточными продуктами (4b), как описано выше для реакции (4а) с (4b) и такая реакция приводит к промежуточным продуктам (1а-1).
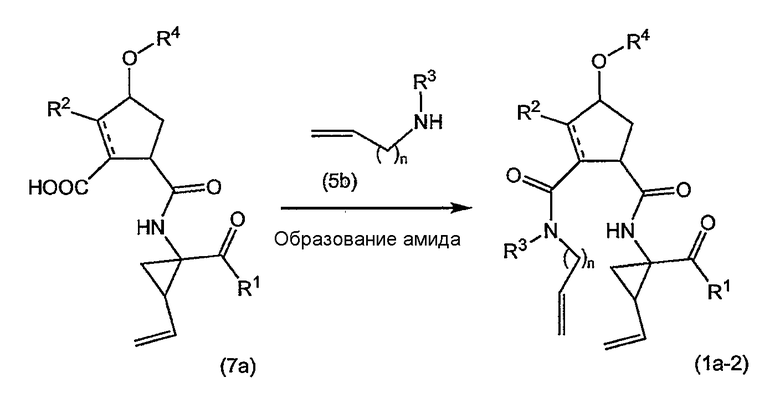
Промежуточные продукты формулы (1a), в которых X представляет собой C [упомянутые промежуточные продукты представлены формулой (1a-2)], можно получать с помощью реакции образования амидов, исходя из промежуточных продуктов (7a), которые подвергают взаимодействию с амином (5b), как показано на следующей реакционной схеме, применяя условия реакции для получения амидов, такие как описанные выше:
Промежуточные продукты (1a-1) альтернативно можно получать следующим образом:
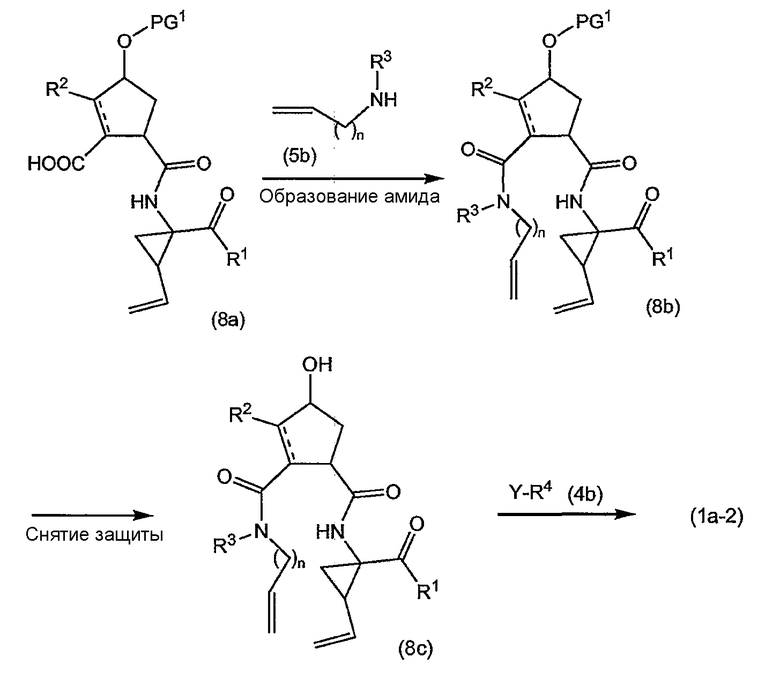
PG1 представляет собой описанную выше O-защитную группу. Можно применять такие же условия проведения реакции, как описанные выше: образование амида, как описано выше, удаление PG1, как в описании защитных групп, и введение R4, как в реакциях (4a) с реагентами (4b).
Промежуточные продукты формулы (2a) можно получать сначала циклизацией открытого амида (9a) до сложного макроциклического эфира (9b), который в свою очередь преобразуется в (2a) следующим образом:
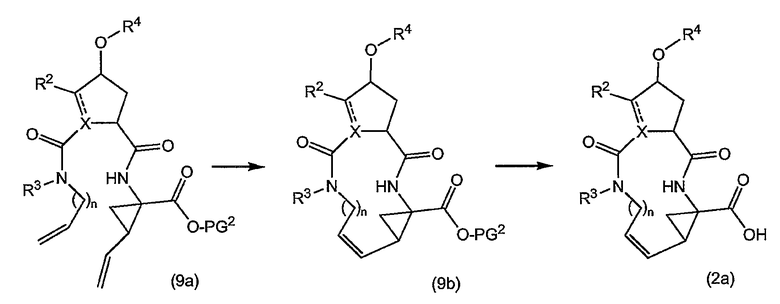
PG2 представляет собой карбоксил-защитную группу, например, одну из упомянутых выше карбоксил-защитных групп, в частности, сложный C1-4-алкиловый или бензиловый эфир, например, сложный метиловый, этиловый или т-бутиловый эфир. Реакция (9a) с образованием (9b) является реакцией метатезиса и осуществляется, как описано выше. Группу PG2 удаляют, также следуя описанным выше процедурам. Когда PG1 представляет собой сложный C1-4-алкиловый эфир, ее удаляют щелочным гидролизом, например, с NaOH или предпочтительно с LiOH, в водосодержащем растворителе, например, в смеси C1-4-алканол/вода. Бензильную группу можно удалять с помощью каталитического гидрирования.
При альтернативном синтезе промежуточные продукты (2a) можно получать следующим образом:
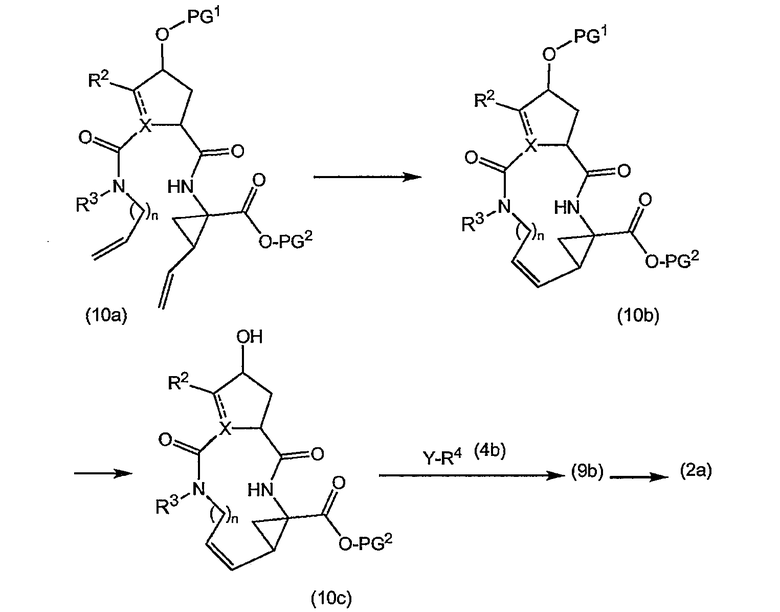
Группу PG1 выбирают так, чтобы она была способна удаляться селективно по отношению к PG2. PG2 может представлять собой, например, сложные метиловые или этиловые эфиры, которые можно удалять при обработке гидроксидом щелочного металла в водной среде, в тех случаях, когда PG1, например, представляет собой т-бутил или бензил. PG2 может представлять собой сложные т-бутиловые эфиры, удаляемые в слабокислых условиях, или PG1 может представлять собой сложные бензиловые эфиры, удаляемые с помощью сильной кислоты или путем каталитического гидрирования, в последних двух случаях PG1, например, представляет собой сложный эфир бензойной кислоты, такой как сложный эфир 4-нитробензойной кислоты.
Сначала промежуточные продукты (10a) подвергают циклизации до сложных макроциклических эфиров (10b), затем с последних снимают защиту путем удаления группы PG1 с получением соединения (10c), которое подвергают взаимодействию с промежуточными продуктами (4b) с последующим удалением карбоксил-защитной группы PG2. Циклизацию, снятие защиты PG1 и PG2 и сочетание с (4b) проводят так, как описано выше.
Группы R1 можно вводить на любой стадии синтеза либо в виде последней стадии, как описано выше, либо раньше, до образования макроцикла. На следующей схеме вводится группа R1, представляющая собой -NH-SO2R6 или -ОR5 (которые указаны выше):
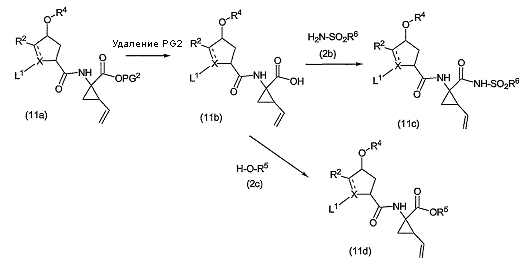
На приведенной выше схеме PG2 представляет собой группу, которая определена выше, и L1 представляет собой группу P3
в которой n и R3 определены выше, и когда X представляет собой N, L1 также может представлять собой азот-защитную группу (PG, которая определена выше), и когда X представляет собой C, L1 также может представлять собой группу -COOPG2a, в которой группа PG2a представляет собой карбоксил-защитную группу, подобную PG2, но в которой PG2a удаляется селективно по отношению к PG2. В одном из вариантов осуществления изобретения PG2a представляет собой т-бутил, а PG2 представляет собой метил или этил.
Промежуточные продукты (11c) и (11d), в которых L1 представляет собой группу (b), соответствуют промежуточным продуктам (1a) и могут дополнительно обрабатываться, как указано выше.
Сочетание структурных элементов P1 и P2
Структурные элементы P1 и P2 соединяют друг с другом, применяя реакцию образования амида, следуя описанным выше процедурам. Структурный элемент P1 может содержать карбоксил-защитную группу PG2 (как в (12b)) или может быть уже соединен с группой P1' (как в (12c)). L2 представляет собой N-защитную группу (PG) или группу (b), как указано выше. L3 представляет собой гидрокси, -OPG1 или группу -О-R4, указаную выше. Когда на любой из следующих реакционных схем L3 представляет собой гидрокси, перед каждой реакционной стадией ее можно защищать в виде группы -OPG1 и, если необходимо, затем снова снимать защиту с образованием свободной гидрокси-функции. Подобно тому, как описано выше, гидрокси-функцию можно преобразовывать в группу -О-R4.
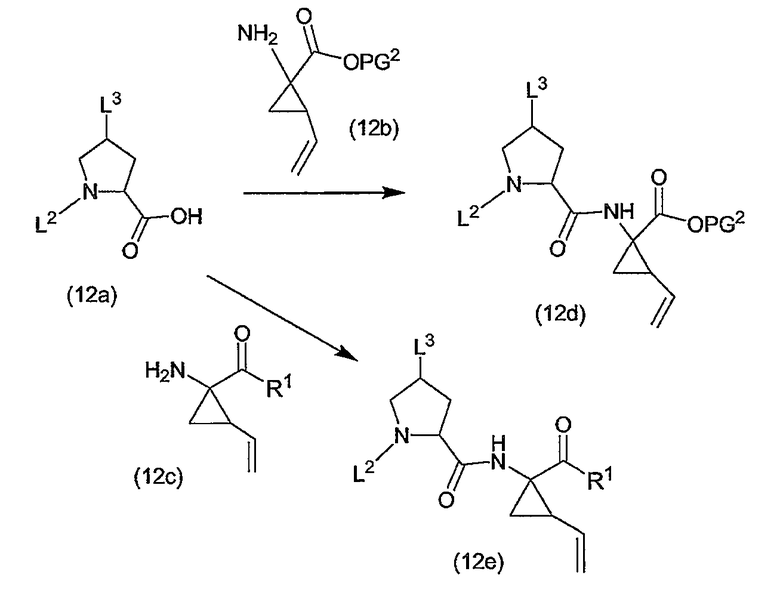
В процедуре, описанной на приведенной выше схеме, циклопропиламинокислоту (12b) или (12c) связывают с кислотной функцией (группой) структурного элемента P2 (12a) с образованием амидной связи, следуя описанным выше процедурам. Получают промежуточные продукты (12d) или (12e). Когда в последнем продукте L2 представляет собой группу (b), полученные в результате продукты представляют собой последовательности P3-P2-P1, охватывающие некоторые из промежуточных продуктов (11c) или (11d) на предыдущей реакционной схеме. Удаление из соединения (12d) кислотозащитной группы в подходящих для применяемой защитной группы условиях с последующим сочетанием с амином H2N-SO2R6 (2b) или с HOR5 (2c), как описано выше, вновь дает промежуточные продукты (12e), в которых -COR1 представляют собой амидные или сложноэфирные группы. Когда L2 представляет собой N-защитную группу, ее можно удалять, получая при этом промежуточные продукты (5a) или (6a). В одном из вариантов осуществления изобретения PG в данной реакции представляет собой BOC-группу и PG2 представляет собой метил или этил. Кроме того, когда L3 представляет собой гидрокси, исходный материал (12a) представляет собой Boc-L-гидроксипролин. В конкретном варианте осуществления изобретения PG представляет собой BOC, PG2 представляет собой метил или этил, и L3 представляет собой -О-R4.
В одном из вариантов осуществления изобретения L2 представляет собой группу (b) и указанные реакции включают в себя сочетание P1 с P2-P3, которое приводит к промежуточным продуктам (1a-1) или (1a), упомянутым выше. В другом варианте осуществления изобретения L2 представляет N-защитную группу PG, которая указана выше, и реакция сочетания приводит к промежуточным продуктам (12d-1) или (12e-1), из которых группу PG можно удалять, применяя упомянутые выше условия реакции и получая при этом промежуточные продукты (12-f) или соответственно (12g), которые охватывают промежуточные продукты (5a) и (6a), которые указаны выше:
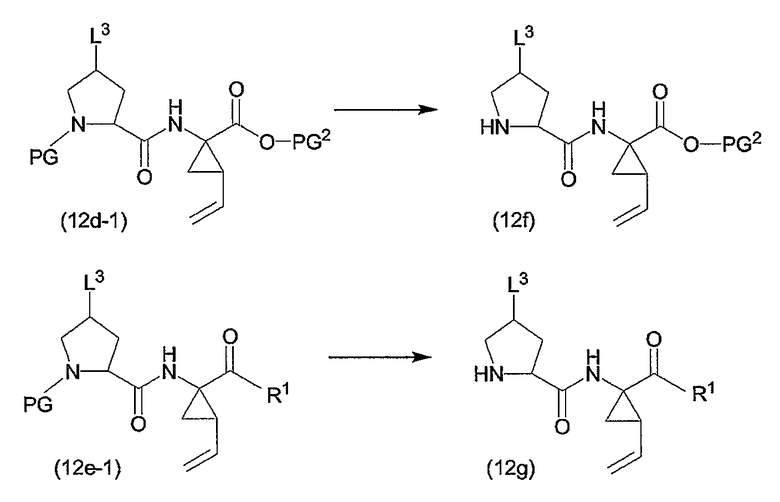
В одном из вариантов осуществления изобретения группа L3 на приведенных выше схемах представляет собой группу -O-PG1, которую можно вводить в исходный материал (12a), в котором L3 представляет собой гидрокси. В таком случае группу PG1 выбирают таким образом, чтобы ее можно было удалять селективно по отношению к группе L2, представляющей собой PG.
Подобным образом структурные элементы P2, в которых X представляет собой C, которые представляют собой циклопентановые или циклопентеновые производные, можно соединять со структурными элементами P1, как кратко изложено на следующей схеме, на которой R1, R2, L3 имеют указанные выше значения, а PG2 и PG2a представляют собой карбоксил-защитные группы. Группу PG2a обычно выбирают так, чтобы ее можно было удалять селективно по отношению к группе PG2. Удаление группы PG2a из (13c) приводит к промежуточным продуктам (7a) или (8a), которые можно подвергать взаимодействию с (5b), как описано выше.
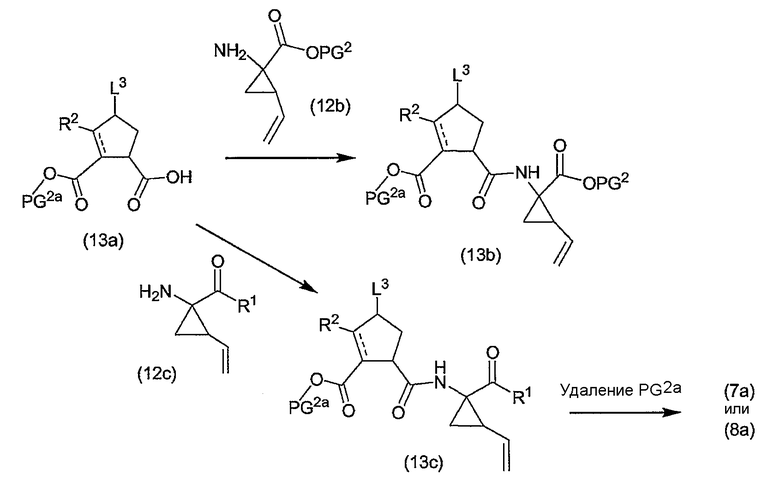
В конкретном варианте осуществления изобретения, когда X представляет собой C, R2 представляет собой H и когда X и атом углерода, несущий R2, соединены одинарной связью (P2 представляет собой циклопентановый фрагмент), PG2a и L3, взятые вместе, образуют связь, а структурный элемент P2 представлен формулой:
Бициклическую кислоту (14a) подвергают взаимодействию с (12b) или (12c) подобно тому, как описано выше, с получением (14b) и (14c) соответственно, в которых кольцо лактона раскрывается, давая промежуточные продукты (14c) и (14e). Раскрытие кольца лактонов можно осуществлять, применяя процедуры гидролиза сложных эфиров, например, применяя описанные выше условия реакции для щелочного удаления группы PG1 из (9b), в частности, применяя основные условия, такие как гидроксид щелочного металла, например, NaOH, KOH, и, в частности, LiOH.
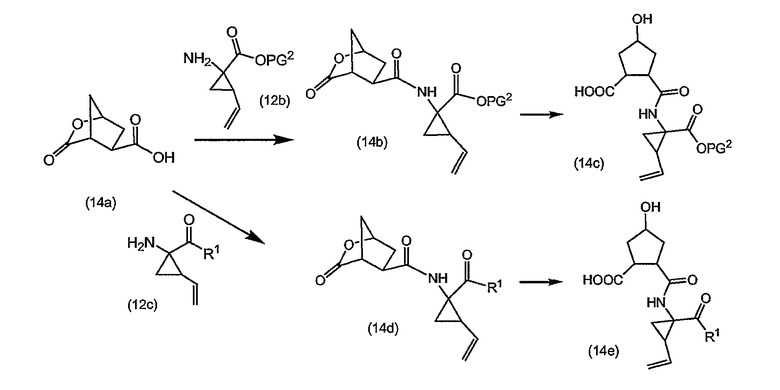
Промежуточные продукты (14c) и (14e) можно обрабатывать дополнительно, как описано в дальнейшем.
Сочетание структурных элементов P3 и P2
Что касается структурных элементов P2, которые содержат пирролидиновый фрагмент, структурные элементы P3 и P2 или P3 и P2-P1 соединяют, применяя реакцию образования карбамата и следуя процедурам, описанным выше для сочетания (5a) с (5b). Общая процедура сочетания структурных элементов P2, содержащих пирролидиновый фрагмент, представлена на следующей реакционной схеме, на которой L3 представляет собой указанную выше группу, а L4 представляет собой группу -O-PG2, группу
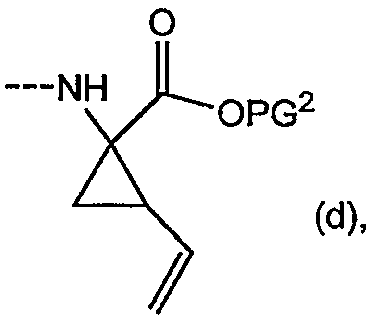 или группу
или группу 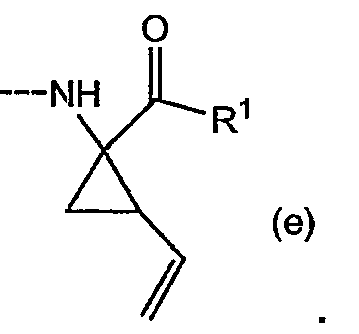
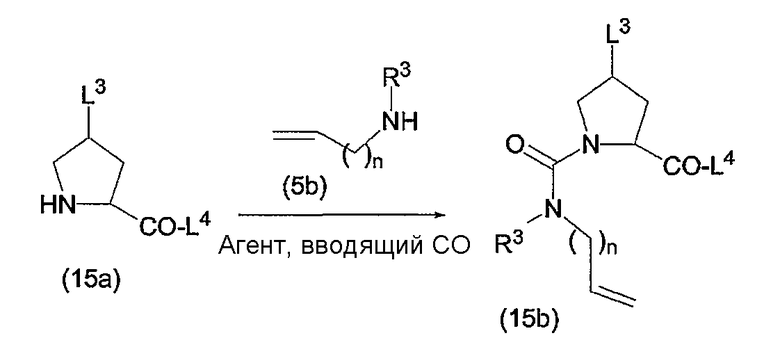
В одном из вариантов осуществления изобретения L4 в (15a) представляет собой группу -OPG2; группу PG2 можно удалять и полученную в результате кислоту связывать с циклопропиламинокислотами (12a) или (12b), получая при этом промежуточные продукты (12d) или (12e), в которых L2 представляет собой радикал (d) или (e).
Общая процедура сочетания элементов P3 с элементом P2 или с элементом P2-P1, в которых P2 представляет собой циклопентан или циклопентен, показана на следующей схеме:
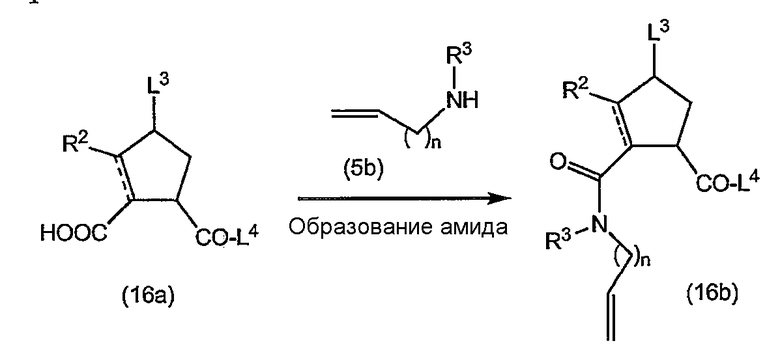
В конкретном варианте осуществления L3 и L4, взятые вместе, могут образовывать лактоновый мостик, как в соединении 14a, и сочетание элемента P3 с элементом P2 осуществляется следующим образом:
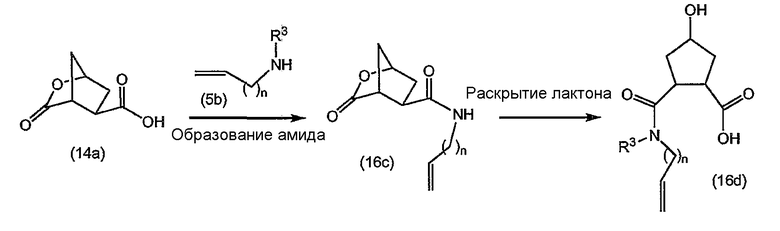
Бициклический лактон (14a) подвергают взаимодействию с соединением (5b) при осуществлении реакции образования амида с получением амида (16c), в котором лактоновый мостик раскрывается с получением соединения (16d). Условия реакции образования амида и реакции раскрытия лактона описаны выше или в дальнейшем. Промежуточный продукт (16d) в свою очередь можно связывать с группой P1 группа, как описано выше.
Реакции на приведенных выше схемах осуществляют, применяя такие же процедуры, которые описаны для реакций (5a), (7a) или (8a) с (5b) и, в частности, упомянутые выше реакции, в которых L4 представляет собой группу (d) или (e), соответстветствуют реакциям (5a), (7a) или (8a) с (5b), которые описаны выше.
Структурные элементы P1, P1', P2 и P3, применяемые при получении соединений формулы (I), можно получать, исходя из известных в данной области промежуточных продуктов. Ряд таких синтезов описан далее более подробно.
Отдельные структурные элементы можно сначала получать и затем связывать вместе или, альтернативно, можно связывать вместе предшественников структурных элементов и модифицировать их на более поздней стадии до требуемого молекулярного состава.
Функциональные группы в каждом из структурных элементов можно защищать, чтобы предотвратить побочные реакции.
Синтез структурных элементов P2
Структурные элементы P2 содержат либо пирролидиновый, циклопентановый, либо циклопентеновый фрагмент, замещенный группой -О-R4.
Структурные элементы P2, содержащие пирролидиновый фрагмент, можно получать из имеющегося в продаже гидроксипролина.
Получение структурных элементов P2, которые содержат циклопентановое кольцо, можно осуществлять, как показано на схеме ниже.
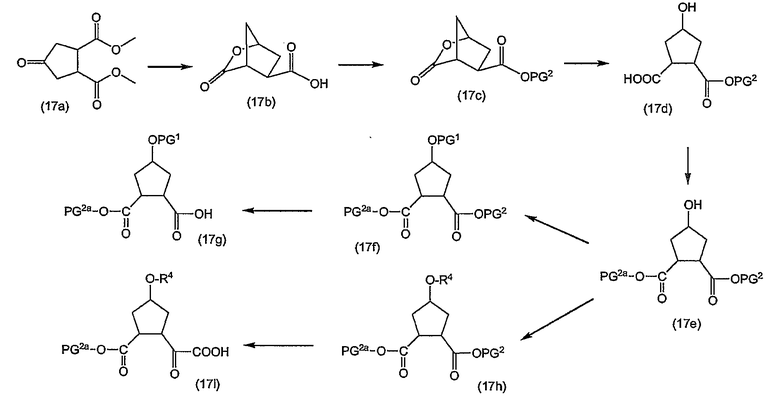
Бициклическую кислоту (17b) можно получать, например, из 3,4-бис(метоксикарбонил)циклопентанона (17a), как описано в публикации Rosenquist и др., Acta Chem. Scand. 46 (1992) 1127-1129. Первая стадия данной процедуры предусматривает восстановление кетогруппы с помощью восстановителя типа борогидрида натрия в растворителе, таком как метанол, с последующим гидролизом сложных эфиров и заключительным замыканием цикла до бициклического лактона (17b) с помощью процедур образования лактона, в частности, с применением уксусного ангидрида в присутствии слабого основания, такого как пиридин. Затем можно защитить функциональные группы карбоновой кислоты в (17b), вводя подходящую карбоксил-защитную группу, такую как группа PG2, которая представляет собой указанную выше группу, тем самым обеспечивая получение сложного бициклического эфира (17c). Группа PG2, в частности, под воздействием кислоты является лабильной группой, такой как т-бутильная группа, и вводится, например, путем обработки изобутеном в присутствии кислоты Льюиса или ди-трет-бутилдикарбонатом в присутствии основания, такого как третичный амин типа диметиламинопиридина или триэтиламина, в растворителе типа дихлорметана. Раскрытие кольца лактона (17c) с помощью описанных выше условий реакции, в частности, с помощью гидроксида лития приводит к кислоте (17d), которую можно дополнительно применять в реакциях сочетания со структурными элементами P1. Свободную кислоту (17d) также можно защищать, предпочтительно с помощью кислотозащитной группы PG2a, которая удаляется селективно по отношению к группе PG2, а гидрокси-функцию можно преобразовывать в группу -OPG1 или в группу -O-R4. Продукты, получаемые при удалении группы PG2, представляют собой промежуточные продукты (17g) и (17i), которые соответствуют промежуточным продуктам (13a) или (16a), указанным выше.
Промежуточные продукты с определенной стереохимией можно получать путем разделения промежуточных продуктов в приведенной выше последовательности реакций. Например, (17b) можно разделять, следуя известным в данной области процедурам, например, с помощью образования активной формы соли с оптически активным основанием или с помощью хиральной хроматографии, и полученные в результате стереоизомеры можно дополнительно обрабатывать, как описано выше. Группы OH и COOH в (17d) находятся в цис-положении. Транс-аналоги можно получать путем изменения стереохимии у атома углерода, несущего OH-группу (функцию), с помощью специфических реагентов в реакциях введения OPG1 или -OR4, которые меняют стереохимию, например, применяя такие реакции, как реакция Мицунобу.
В одном из вариантов осуществления изобретения промежуточные продукты (17d) связывают с элементами P1 (12b) или (12c) с помощью реакций сочетания, которые соответствуют сочетанию (13a) или (16a) с аналогичными элементами P1, применяя аналогичные условия. Последующее введение заместителя -OR4, как описано выше, с последующим удалением кислотозащитной группы PG2 приводит к промежуточным продуктам (8a-1), которые представляют собой подкласс промежуточных продуктов (7a), или части промежуточных продуктов (16a). Продукты реакции, полученные после удаления PG2, можно дополнительно связывать со структурным элементом P3. В одном из вариантов осуществления изобретения PG2 в (17d) представляет собой т-бутил, который можно удалять в кислотных условиях, например, с помощью трифторуксусной кислоты.
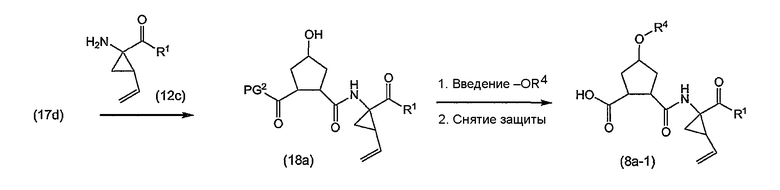
Ненасыщенный структурный элемент P2, то есть циклопентеновое кольцо, можно получать, как проиллюстрировано ниже на схеме.

С помощью реакции бромирования-элиминирования 3,4-бис(метоксикарбонил)циклопентанона (17a), которая описана в публикации Dolby и др., J. Org. Chem. 36 (1971) 1277-1285, с последующим восстановлением функциональной кетогруппы с помощью восстановителя типа борогидрида натрия, получают циклопентенол (19a). С помощью избирательного гидролиза сложного эфира, например, с помощью гидроксида лития в растворителе типа смеси диоксана и воды, получают сложный моноэфир гидрокси-замещенного циклопентенола (19b).
Ненасыщенный структурный элемент P2, в котором R2 может также отличаться от водорода, можно получать, как показано ниже на схеме.
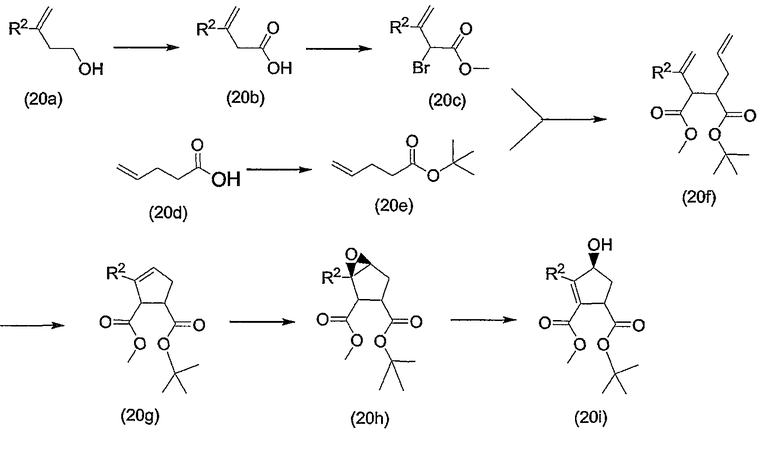
Окисление имеющегося в продаже 3-метил-3-бутен-1-ола (20a), в частности, с помощью окислителя типа хлорхромата пиридиния, приводит к (20b), который преобразуют до соответствующего сложного метилового эфира, например, путем обработки ацетилхлоридом в метаноле, с последующей реакцией бромирования бромом, получая при этом сложный α-бромэфир (20c). Последний можно затем конденсировать со сложным алкениловым эфиром (20e), полученным из (20d) с помощью реакции образования сложного эфира. Сложный эфир (20e) предпочтительно представляет собой сложный т-бутиловый эфир, который можно получать из соответствующей кислоты (20d), имеющейся в продаже, например, путем обработки ди-трет-бутилдикарбонатом в присутствии основания типа диметиламинопиридина. Промежуточный продукт (20e) обрабатывают основанием, таким как литийдиизопропиламид, в растворителе типа тетрагидрофурана, и подвергают взаимодействию с (20c), получая при этом сложный алкениловый диэфир (20f). Циклизацией (20f) с помощью реакции метатезиса олефинов, осуществляемой как описано выше, получают циклопентеновое производное (20g). Для получения эпоксида (20h) можно осуществлять стереоизбирательное эпоксидирование (20g), применяя способ асимметрического эпоксидирования по Якобсену. И наконец, реакция раскрытия эпоксида в основных условиях, например, при добавлении основания, в частности, DBN (1,5-диазабицикло[4.3.0]нон-5-ена), приводит к спирту (20i). Необязательно двойную связь в промежуточном продукте (20i) можно восстанавливать, например, с помощью каталитического гидрирования, применяя катализатор типа палладия на углероде и получая при этом соответствующее циклопентановое соединение. Сложный т-бутиловый эфир можно удалять с получением соответствующей кислоты, которую затем связывают со структурным элементом P1.
Группу -O-R4 можно вводить в пирролидиновые, циклопентановые или циклопентеновые кольца на любой удобной стадии синтеза соединений по настоящему изобретению. Один из подходов заключается в том, что сначала в упомянутые кольца вводят группу -O-R4 и затем добавляют другие требуемые структурные элементы, то есть P1 (необязательно с концевой частью P1') и P3 с последующим образованием макроцикла. Другой подход заключается в сочетании структурных элементов P2, не содержащих заместителя -O-R4, с каждым P1 и P3, и добавлении группы -O-R4 либо до, либо после образования макроцикла. В последней процедуре фрагменты P2 содержат гидрокси-группу, которую можно защищать с помощью гидрокси-защитной группы PG1.
Группы -R4 можно вводить в структурные элементы P2 путем взаимодействия гидрокси-замещенных промежуточных продуктов (21a) или (21b) с промежуточными продуктами (4b) подобно тому, как описано выше для синтеза (I), исходя из (4a). Такие реакции представлены ниже на схемах, на которых значение L2 указано выше и L5 и L5a независимо друг от друга представляют собой гидрокси или карбоксил-защитную группу -ОPG2 или -ОPG2a, или L5 может также представлять собой группу Р1, такую, как указанные выше группы (d) или (е), или L5a может также представлять собой группу Р3, такую, как указанная выше группа (b). Группы PG2 или PG2a указаны выше. Когда группы L5 и L5a представляют собой PG2 или PG2a, их выбирают так, чтобы каждую группу можно было удалять селективно по отношению к другой. Например, одна из групп L5 и L5a может представлять собой метильную или этильную группу, а другая - бензильную или т-бутильную группу.
В одном из вариантов осуществления изобретения в соединении (21a) L2 представляет собой PG, и L5 представляет собой -OPG2, или в соединении (21d) L5a представляет собой -OPG2, и L5 представляет собой -OPG2, и группы PG2 удаляют, как описано выше.
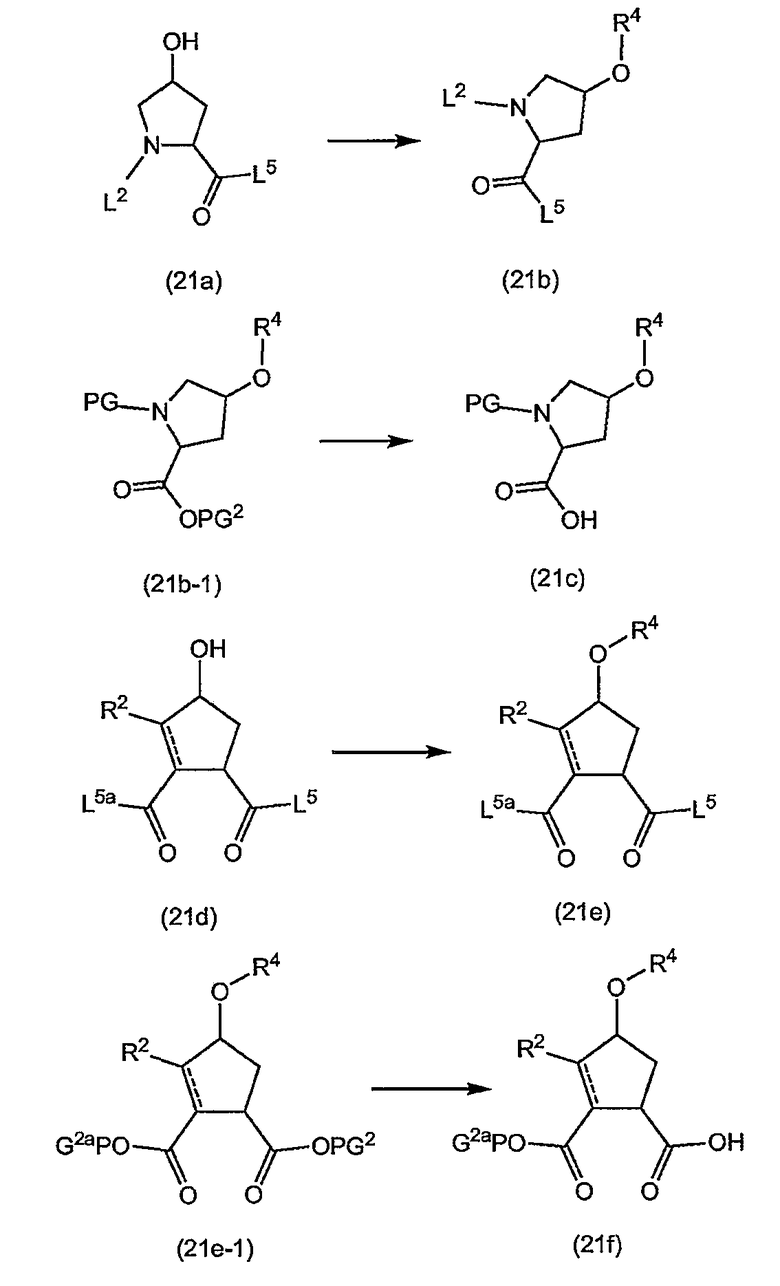
В другом варианте осуществления изобретения группа L2 представляет собой BOC, L5 представляет собой гидрокси и исходный материал (21a) представляет собой имеющийся в продаже BOC-гидроксипролин или любую другую его стереоизомерную форму, например, BOC-L-гидроксипролин, в частности, транс-изомер последнего. Когда L5 в соединении (21b) представляет собой карбоксил-защитную группу, ее можно удалять, следуя описанным выше процедурам, с получением (21с). В еще одном варианте осуществления изобретения PG в соединении (21b-1) представляет собой Boc, и PG2 представляет собой низший сложный алкиловый эфир, в частности, сложный метиловый или этиловый эфир. Гидролиз последнего сложного эфира до кислоты можно осуществлять с помощью стандартных процедур, например, проводя кислотный гидролиз с помощью хлористоводородной кислоты в метаноле или проводя гидролиз с помощью гидроксида щелочного металла, такого как NaOH, в частности, с помощью LiOH. В другом варианте осуществления изобретения гидрокси-замещенные циклопентановые или циклопентеновые аналоги (21d) преобразуют до (21е), которые, когда L5 и L5a представляют собой -ОPG2 или -ОPG2a, можно преобразовывать до соответствующих кислот (21f) путем удаления группы PG2. Удаление PG2a из (21е-1) приводит к аналогичным промежуточным продуктам.
Промежуточные продукты (4b), которые представляют собой производные изохинолина, можно получать с помощью известных в данной области процедур. Например, в патенте США 2005/0143316 описаны разнообразные способы синтеза изохинолинов в виде промежуточных продуктов R4-ОH или R4-LG. Методика синтеза таких изохинолинов описана в публикации N. Briet и др., Tetrahedron, 2002, 5761 и приведена ниже, где R4a, R4b и R4b' являются заместителями на изохинолиновом фрагменте, имеющими значения, указанные здесь для заместителей группы R4.
Производные коричной кислоты (22b) преобразовывают до 1-хлоризохинолинов по трехстадийному способу. Полученные хлоризохинолины (22e) можно затем связывать с описанными здесь производными гидроксипирролидина, гидроксициклопентана или гидроксициклопентена. На первой стадии карбоксильные группы коричных кислот (22b) активируют, например, обработкой C1-6-алкилхлорформиатом (в частности, метил- или этилхлорформиатом) в присутствии основания. Полученные смешанные ангидриды затем обрабатывают азидом натрия, получая при этом ацилазиды (22c). Для образования ацилазидов из карбоновых кислот пригодны некоторые другие способы, например, карбоновую кислоту можно обрабатывать дифенилфосфорилазидом (DPPA) в апротонном растворителе, таком как метиленхлорид, в присутствии основания. На следующей стадии ацилазиды (22c) преобразуют в соответствующие изохинолоны (22d), в частности, путем нагревания ацилазидов в растворителе с высокой температурой кипения, таком как простой дифениловый эфир. Исходные коричные кислоты (22d) имеются в продаже или их можно получать из соответствующих бензальдегидов (22a) путем непосредственной конденсации с малоновыми кислотами или их производными или используя реакцию Виттига. Промежуточные изохинолоны (22d) можно преобразовать в соответствующие 1-хлоризохинолины обработкой галогенирующим агентом, таким как оксихлорид фосфора.
Группы R4, которые представляют собой изохинолины, также можно получать, следуя процедурам, описанным в публикации K. Hirao, R. Tsuchiya, Y. Yano, H. Tsue, Heterocycles 42(1) 1996, 415-422.
Альтернативным способом синтеза изохинолиновой кольцевой системы является процедура Померанца-Фриче. Такой способ начинается с превращения производного бензальдегида (23a) в функционализированный имин (23b), который затем преобразуют в изохинолиновую кольцевую систему при обработке кислотой при повышенной температуре. Такой способ, в частности, применим для получения изохинолиновых промежуточных продуктов, которые замещены в положении C8, обозначенном звездочкой. Промежуточные изохинолины (23c) можно преобразовывать до соответствующих 1-хлорхинолинов (23e) по двухстадийному способу. Первая стадия включает в себя образование N-оксида изохинолина (23d) путем обработки изохинолина (23c) пероксидом, таким как мета-хлорнадбензойная кислота в подходящем растворителе, таком как дихлорметан. Промежуточное соединение (23d) преобразуют до соответствующего 1-хлоризохинолина при обработке галогенирующим агентом, таким как оксихлорид фосфора.
Еще один способ синтеза изохинолиновой кольцевой системы показан на схеме ниже.
В данном способе производное орто-алкилбензамида (24a) в анионной форме получают при обработке сильным основанием, таким как трет-бутиллитий в растворителе, таком как ТГФ, и затем конденсируют с производным нитрила, получая при этом изохинолин (24b). Последний можно преобразовывать в соответствующий 1-хлоризохинолин с помощью описанных выше способов. R' и R" в соединении (24a) представляют собой алкильные группы, в частности C1-4-алкильные группы, например, метил или этил.
На следующей схеме показан дополнительный способ синтеза изохинолинов:
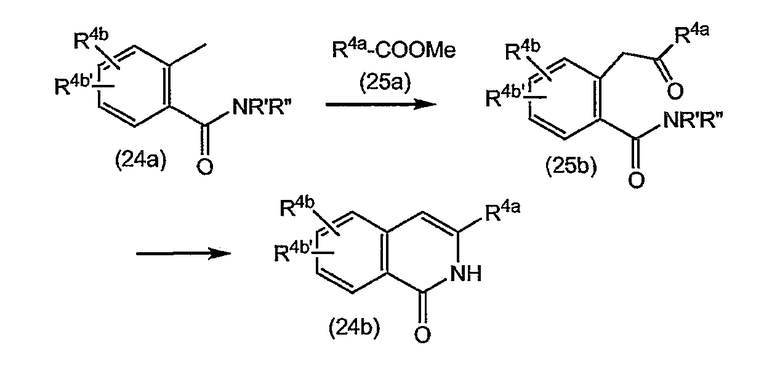
Промежуточное соединение (24a) депротонируют, применяя сильное основание, как описано выше. R' и R" имеют указанные выше значения. Полученный промежуточный анион конденсируют со сложным эфиром (25a), получая при этом промежуточный кетон (25b). В следующей реакции последний промежуточный продукт (25b) подвергают взаимодействию с аммиаком или солью аммония, например ацетатом аммония при повышенной температуре, что приводит к образованию изохинолона (24b).
В приведенном выше синтезе можно применять ряд карбоновых кислот с общей структурой (25a). Такие кислоты либо имеются в продаже, либо их можно получать с помощью процедур, известных в данной области. Пример получения производных 2-(замещенного)аминокарбоксиаминотиазола (25a -1), следуя процедуре, описанной в публикации Berdikhina и др., Chem. Heterocycl. Compd. (Engl. Transl.) (1991), 427-433, показан на следующей реакционной схеме, которая иллюстрирует получение 2-карбокси-4-изопропилтиазола (25а-1):
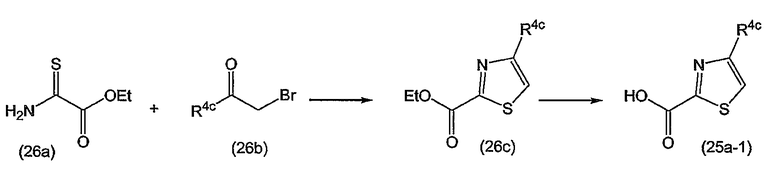
Этилтиооксамат (26a) подвергают взаимодействию с β-бромкетоном (26b) с образованием сложного эфира тиазолилкарбоновой кислоты (26c), который подвергается гидролизу до соответствующей кислоты (25a-1). Сложный этиловый эфир в таких промежуточных продуктах можно заменять другими карбоксил-защитными группами PG2, как указано выше. На приведенной выше схеме R4c имеет указанные выше значения и, в частности, R4c представляет собой C1-4-алкил, более конкретно изопропил.
Бромкетон (26b) можно получать из 3-метилбутан-2-она (MIK) с помощью силилирующего агента (такого как TMSCl) в присутствии подходящего основания (в частности, LiHMDS) и брома.
Синтез дополнительных карбоновых кислот (25a), в частности замещенных аминотиазолкарбоновых кислот (25a-2), проиллюстрирован ниже:
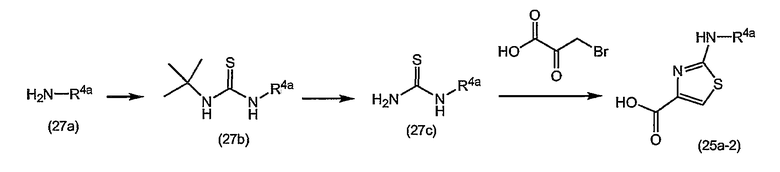
Тиомочевину (27c) с различными заместителями R4а, которые, в частности, представляют собой C1-6-алкил, можно образовывать путем взаимодействия подходящего амина (27a) с трет-бутилизотиоцианатом в присутствии основания типа диизопропилэтиламина в растворителе типа дихлорметана с последующим удалением трет-бутильной группы в кислотных условиях. При последующей конденсации производного тиомочевины (27c) с 3-бромпировиноградной кислотой получают тиазолкарбоновую кислоту (25a-2).
Еще один дополнительный способ получения изохинолинов проиллюстрирован на следующей реакционной схеме:
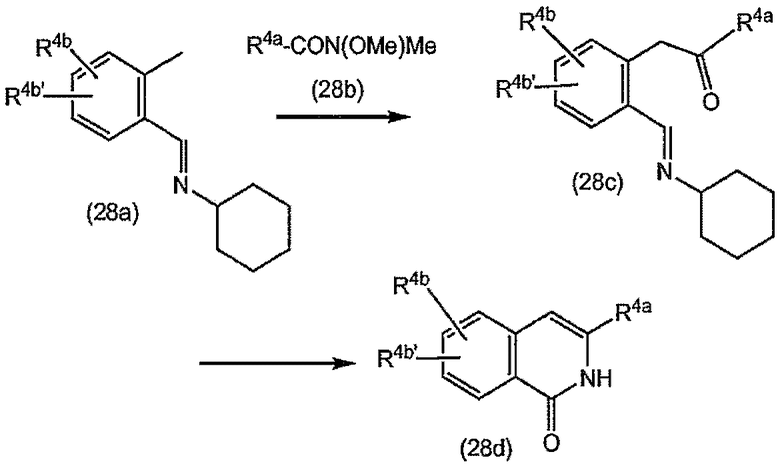
На первой стадии данного способа производное орто-алкиларилимина (28a) подвергают обработке в условиях депротонирования (например, втор-бутиллитий, ТГФ), и полученный анион конденсируют с производным активированной карбоновой кислоты, таким как амид Вайнреба (28b). Полученный кетоимин (28c) преобразуют в изохинолин (28d) путем конденсации с ацетатом аммония при повышенных температурах. Полученные при этом изохинолины можно преобразовывать до соответствующих 1-хлоризохинолинов с помощью описанных здесь способов.
Описанные здесь изохинолины либо как таковые, либо включенные в гидроксипирролидиновые, гидроксициклопентановые или гидроксициклопентановые фрагменты соединений формулы (I) или любых из упомянутых здесь промежуточных продуктов, могут быть дополнительно функционализированы. Пример такой функционализации проиллюстрирован ниже.
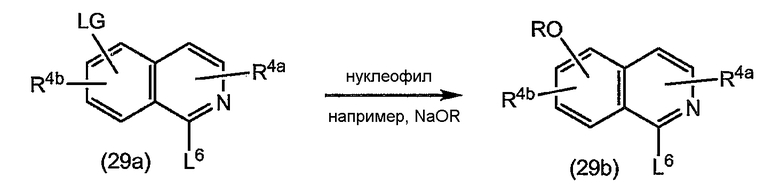
На приведенной выше схеме показано превращение 1-хлор-6-фторизохинолина в соответствующий 1-хлор-6-C1-6-алкоксиизохинолиновый фрагмент (29b) при обработке соединения (29a) алкоголятом натрия или калия в растворителе из спирта, из которого получен алкоголят. L6 на приведенной выше схеме представляет собой галоген или группу
R на приведенной выше схеме представляет собой C1-6-алкил, и LG представляет собой удаляемую группу. В одном из вариантов осуществления LG представляет собой фтор. L7 и L8 представляют собой различные заместители, которые могут быть присоединены в указанных положениях фрагмента P2, в частности, группы, такие как OL5 или L8, могут представлять собой группу P1, и L7 может представлять собой группу P3, или L7 и L8, взятые вместе, могут образовывать остаток макроциклической кольцевой системы соединений формулы (I).
На следующей схеме приведен пример модификации изохинолинов с помощью реакций Сузуки. Такие сочетания можно использовать для функционализации изохинолина в каждом из положений кольцевой системы при условии, что упомянутое кольцо подходящим образом активировано или функционализировано, например, с помощью хлора.
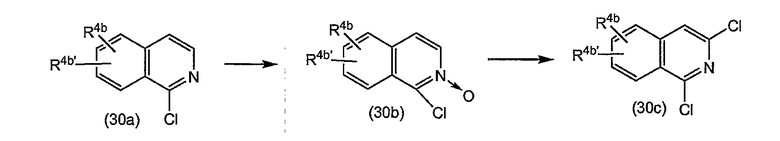
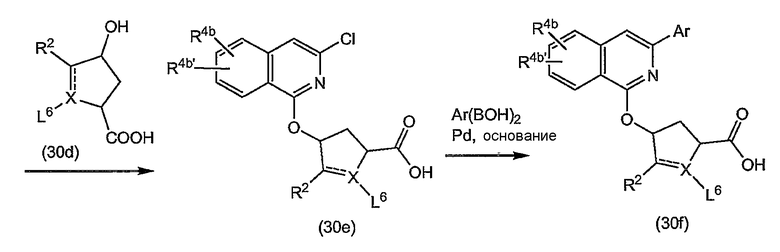
Указанная последовательность начинается с 1-хлоризохинолина (30a), который при обработке пероксидом, таким как мета-хлорнадбензойная кислота, преобразуют до соответствующего N-оксида (30b). Последний промежуточный продукт преобразуют до соответствующего 1,3-дихлоризохинолина (30c) при обработке галогенирующим агентом, например оксихлоридом фосфора. Промежуточный продукт (30c) можно связывать с промежуточным продуктом (30d), в котором L6 представляет собой группу PG, когда X представляет собой N, или L6 представляет собой группу -COOPG2, когда X представляет собой C, применяя описанные здесь способы для введения -O-R4-групп и получая при этом промежуточный продукт (30e). Промежуточный продукт (30e) дериватизируют, применяя реакцию сочетания по Сузуки с арилбороновой кислотой в присутствии палладиевого катализатора и основания в растворителе, таком как ТГФ, толуол или диполярный апротонный растворитель, такой как ДМФА, получая при этом промежуточный C3-арилизохинолин (30f). В таких способах сочетания также можно использовать гетероарилбороновые кислоты для получения C3-гетероарилизохинолинов.
Реакции сочетания по Сузуки изохинолиновых систем с арильными или гетероарильными группами также можно использовать на более поздней стадии синтеза при получении соединений формулы (I). Изохинолиновые кольцевые системы также можно функционализировать, используя другие катализируемые палладием реакции, такие как реакции сочетания Хека, Соногаширы или Стилле, как проиллюстрировано, например, в патенте США 2005/1043316.
Синтез структурных элементов P1
Циклопропанаминокислота, применяемая для получения фрагмента P1, имеется в продаже или ее можно получать, применяя известные в данной области процедуры.
В частности, сложный аминовинилциклопропилэтиловый эфир (12b) можно получать согласно процедуре, описанной в заявке WO 00/09543, или процедуре, проиллюстрированной на следующей схеме, на которой PG2 представляет собой указанную выше карбоксил-защитную группу:
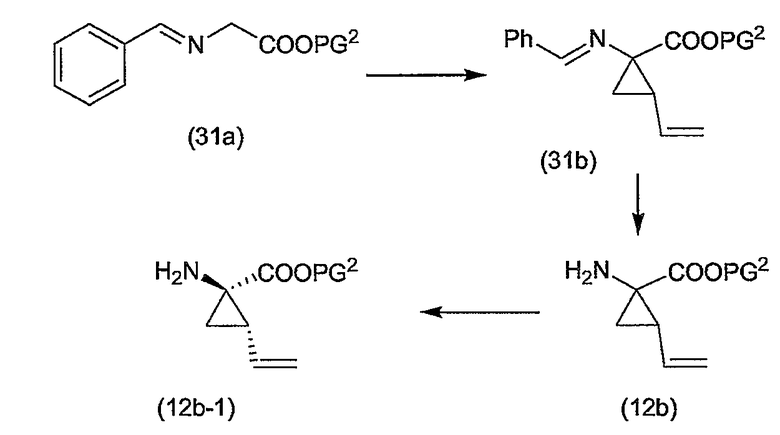
Обработкой имеющегося в продаже или легко получаемого имина (31а) 1,4-дигалогенбутеном в присутствии основания получают соединение (31b), которое после гидролиза приводит к циклопропиламинокислоте (12b), содержащей аллильный заместитель в син-положении по отношению к карбоксильной группе. Разделение смеси энантиомеров (12b) приводит к (12b-1). Разделение осуществляют с помощью известных в данной области процедур, таких как ферментативное разделение; кристаллизация с хиральной кислотой; или получение химических производных; или с помощью хиральной колоночной хроматографии. Промежуточные продукты (12b) или (12b-1) можно связывать с подходящими производными Р2, как описано выше.
Структурные элементы P1 для получения соединений общей формулы (I), в которых R1 представляет собой -OR5 или -NH-SO2R6, можно получать путем взаимодействия аминокислот (32a) с подходящим спиртом или амином соответственно в стандартных условиях, предназначенных для образования сложного эфира или амида. Циклопропиламинокислоты (32a) получают путем введения N-защитной группы PG, удаления PG2 и преобразования аминокислот (32a) до амидов (12c-1) или сложных эфиров (12c-2), которые являются подгруппами промежуточных продуктов (12c), как кратко изложено на следующей реакционной схеме, на которой PG представляет собой указанную выше группу:
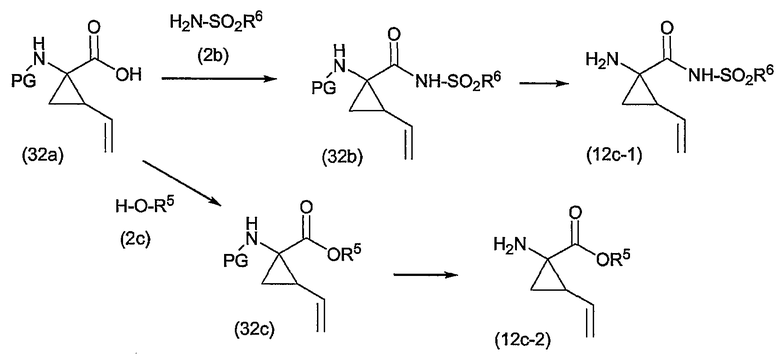
Реакция (32a) с амином (2b) является процедурой образования амида и может осуществляться согласно описанной выше процедуре. Подобная реакция с (2с) является реакцией образования сложного эфира. Обе реакции можно осуществлять, следуя описанным выше процедурам. Такая реакция приводит к промежуточным продуктам (32b) или (32с), из которых аминозащитная группа удаляется стандартными способами, такими как описанные выше способы. Такое удаление в свою очередь приводит к требуемому промежуточному продукту (12c-1). Исходные материалы (32a) можно получать из вышеупомянутых промежуточных продуктов (12b) с помощью первоначального введения N-защитной группы PG и последующего удаления группы PG2.
В одном из вариантов осуществления изобретения реакцию (32a) с (2b) осуществляют путем обработки аминокислоты конденсирующим агентом, например, N,N'-карбонилдиимидазолом (CDI) или т.п. в растворителе типа ТГФ с последующим взаимодействием с (2b) в присутствии основания, такого как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Альтернативно аминокислоту можно обрабатывать с помощью (2b) в присутствии основания типа диизопропилэтиламина с последующей обработкой конденсирующим агентом, таким как гексафторфосфат бензотриазол-1-илокси-трис-пирролидинoфосфония (имеется в продаже под маркой PyBOP®), для введения сульфонамидной группы.
Промежуточные продукты (12c-1) или (12c-2) в свою очередь можно связывать с подходящими пролиновыми, циклопентановыми или циклопентеновыми производными, как описано выше.
Синтез структурных элементов P3
Структурные элементы P3 имеются в продаже или их можно получать согласно методикам, известным специалисту в данной области. Одна из таких методик показана ниже на схеме, и в ней применяются моноацилированные амины, такие как трифторацетамид или Boc- защищенный амин.
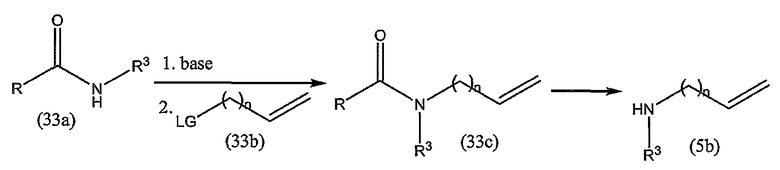
На приведенной выше схеме R вместе с группой CO образует N-защитную группу, в частности, R представляет собой т-бутокси, трифторметил; значения R3 и n определены выше и LG представляет собой удаляемую группу, в частности галоген, например хлор или бром.
Моноацилированные амины (33a) обрабатывают сильным основанием, таким как гидрид натрия, и затем подвергают взаимодействию с LG-C5-8-алкенильным реагентом (33b), в частности с галоген-C5-8-алкенилом для образования соответствующих защищенных аминов (33c). Снятие защиты с (33c) обеспечивает получение (5b), которые являются структурными элементами P3. Снятие защиты будет зависеть от функциональной группы R таким образом, что если R представляет собой т-бутокси, снятие защиты с соответствующих Boc-защищенных аминов можно осуществлять при обработке кислотой, например трифторуксусной кислотой. Альтернативно, когда R представляет собой, например, трифторметил, удаление группы R осуществляют с помощью основания, например, гидроксида натрия.
На следующей схеме иллюстрируется еще один способ получения структурного элемента P3, а именно синтез первичных C5-8-алкениламинов по Габриэлю, который можно осуществлять с помощью обработки фталимида (34a) основанием, таким как NaOH или KOH, и с помощью (33b), который указан выше, с последующим гидролизом промежуточного N-алкенилимида до образования первичного C5-8-алкениламина (5b-1).
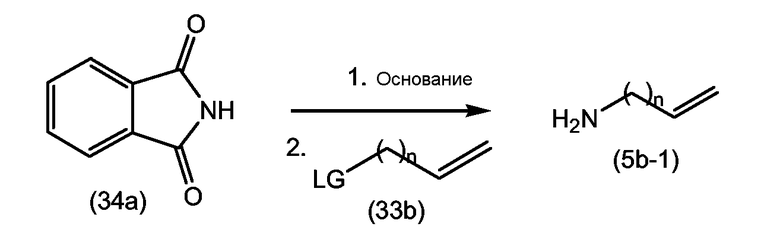
На приведенной выше схеме n имеет указанное выше значение.
Соединения формулы (I) можно преобразовывать друг в друга, следуя известным в данной области реакциям трансформации функциональных групп. Например, аминогруппы можно N-алкилировать, нитрогруппы - восстанавливать до аминогрупп, атом галогена можно заменять на атом другого галогена.
Соединения формулы (I) можно преобразовывать до соответствующих N-оксидных форм, следуя известным в данной области процедурам преобразования трехвалентного азота в его N-оксидную форму. Упомянутую реакцию N-окисления обычно можно осуществлять путем взаимодействия исходного материала формулы (I) с подходящим органическим или неорганическим пероксидом. Подходящие неорганические пероксиды включают в себя, например, пероксид водорода, пероксиды щелочных или щелочноземельных металлов, например, пероксид натрия, пероксид калия; подходящие органические пероксиды могут включать в себя пероксикислоты (надкислоты), такие как, например, бензолкарбопероксикислота или галоген-замещенная бензолкарбопероксикислота, например, 3-хлорбензолкарбопероксикислота, пероксоалкановые кислоты, например, пероксоуксусная (надуксусная) кислота, алкилгидропероксиды, например, трет-бутилгидропероксид. Подходящими растворителями являются, например, вода, низшие спирты, например, этанол и т.п., углеводороды, например, толуол, кетоны, например, 2-бутанон, галогенированные углеводороды, например, дихлорметан, и смеси таких растворителей.
Стереохимически чистые изомерные формы соединений формулы (I) можно получать, применяя известные в данной области процедуры. Диастереомеры можно разделять физическими способами, такими как избирательная кристаллизация, и хроматографическими способами, например, противоточным распределением, жидкостной хроматографией и т.п.
Соединения формулы (I) можно получать в виде рацемических смесей энантиомеров, которые можно отделять друг от друга, следуя известным в данной области процедурам разделения. Рацемические соединения формулы (I), которые являются достаточно основными или кислотными, можно преобразовывать в соответствующие диастереомерные формы солей путем взаимодействия с подходящей хиральной кислотой или хиральным основанием соответственно. Упомянутые диастереомерные формы солей затем разделяют, например, с помощью избирательной или дробной кристаллизации и выделяют из них энантиомеры с помощью щелочи или кислоты. Альтернативный способ разделения энантиомерных форм соединений формулы (I) предусматривает жидкостную хроматографию, в частности жидкостную хроматографию с применением хиральной неподвижной фазы. Упомянутые стереохимически чистые изомерные формы также можно получать из соответствующих стереохимически чистых изомерных форм подходящих исходных материалов при условии, что осуществляется стереоспецифическая реакция. Если требуется определенный стереоизомер, предпочтительно упомянутое соединение можно синтезировать с помощью стереоспецифических способов получения. В таких способах можно преимущественно использовать энантиомерно чистые исходные материалы.
При дополнительном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I), которое здесь указано, или соединения любой из подгрупп соединений формулы (I), которые здесь указаны, и фармацевтически приемлемый носитель. Терапевтически эффективное количество в данном контексте означает количество, достаточное для профилактического воздействия на вирусную инфекцию, для стабилизации или уменьшения вирусной инфекции и, в частности, вирусной HCV-инфекции у инфицированных субъектов или субъектов, подвергающихся риску быть инфицированными. При еще одном дополнительном аспекте изобретение относится к способу получения фармацевтической композиции, которая здесь указана, который включает в себя непосредственное смешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы (I), которое здесь указано, или соединения любой из подгрупп соединений формулы (I), которые здесь указаны.
Следовательно, с целью введения соединения по настоящему изобретению или любую их подгруппу можно получать в различных фармацевтических формах. В качестве подходящих композиций здесь можно сослаться на все композиции, обычно используемые для систематического введения лекарственных средств. Для получения фармацевтических композиций по данному изобретению эффективное количество конкретного соединения, необязательно в форме аддитивной соли или комплекса с металлом, в качестве активного ингредиента объединяют для получения непосредственной смеси с фармацевтически приемлемым носителем, где носитель может принимать большое разнообразие форм в зависимости от формы препарата, требуемой для введения. Такие фармацевтические композиции требуются в виде стандартной лекарственной формы, подходящей, в частности, для введения перорально, ректально, подкожно или путем парентеральной инъекции. Например, при получении композиций в лекарственной форме для перорального применения можно использовать любую из обычных фармацевтических сред, такую как, например, воду, гликоли, масла, спирты и т.п., в случае жидких препаратов для перорального применения, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазки, связующие вещества, дезинтегрирующие агенты и т.п., в случае порошков, пилюль, капсул и таблеток. Благодаря удобству их введения, таблетки и капсулы являются наиболее предпочтительными стандартными лекарственными формами для перорального применения, в случае которых безусловно используются твердые фармацевтические носители. В композициях для парентерального применения носитель обычно будет содержать стерильную воду, по меньшей мере, в значительной степени, хотя могут быть включены другие ингредиенты, например, для облегчения растворимости. Например, можно получать растворы для инъекций, в которых носитель содержит физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также можно получать суспензии для инъекций, в случае которых можно использовать подходящие жидкие носители, суспендирующие агенты и т.п. Также включены препараты в твердой форме, которые предназначены для превращения в препараты в форме жидкости незадолго до применения. В композициях, подходящих для подкожного введения, носитель необязательно содержит вещество, способствующее проникновению внутрь, и/или подходящий смачивающий реагент, необязательно объединенный с подходящими добавками любой природы в незначительных пропорциях, которые не оказывают значительного отрицательного воздействия на кожу.
Соединения по настоящему изобретению также можно вводить с помощью пероральной ингаляции или инсуффляции с помощью способов и препаратов, используемых в данной области для введения таким путем. Таким образом, в общем случае соединения по настоящему изобретению можно вводить в легкие в форме раствора, суспензии или сухого порошка, раствор является предпочтительным. Для введения настоящих соединений подходит любая система, разработанная для доставки растворов, суспензий или сухих порошков с помощью пероральной ингаляции или инсуффляции.
Таким образом, настоящее изобретение также относится к фармацевтической композиции, адаптированной для введения путем ингаляции или инсуффляции через рот и содержащей соединение формулы (I) и фармацевтически приемлемый носитель. Соединения по настоящему изобретению предпочтительно вводятся с помощью ингаляции раствора в виде распыленных или аэролизованных доз.
Особенно предпочтительно получать вышеупомянутые фармацевтические композиции в стандартной лекарственной форме для облегчения введения и равномерности дозировки. Применяемый здесь термин «стандартная лекарственная форма» относится к физически дискретным элементам, подходящим в качестве однократных дозировок; каждый элемент содержит заданное количество активного ингредиента, рассчитанное для получения требуемого терапевтического эффекта, совместно с требуемым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая таблетки с насечкой или таблетки с покрытием), капсулы, пилюли, суппозитории, пакетики с порошком, облатки, растворы или суспензии для инъекции и т.п., и их сегрегированные множества.
Соединения формулы (I) проявляют антивирусные свойства. Вирусные инфекции и ассоциированные с ними заболевания, излечиваемые с помощью соединений и способов по настоящему изобретению, включают в себя инфекции, провоцируемые HCV и другими патогенными флавивирусами, такими как вирус желтой лихорадки, вирус лихорадки Денге (типы 1-4), вирус энцефалита Сент-Луис, вирус японского энцефалита, вирус энцефалита долины Муррея, вирус лихорадки Западного Нила и вирус Кунджин. Заболевания, ассоциированные с HCV, включают в себя прогрессирующий фиброз печени, воспаление и некроз, приводящие к циррозу печени, заболеванию печени в конечной стадии и HCC; и заболевания, ассоциированные с другими патогенными флавивирусами, включают в себя желтую лихорадку, лихорадку денге, геморрагическую лихорадку и энцефалит. Кроме того, ряд соединений по настоящему изобретению проявляют активность в отношении мутировавших штаммов HCV. Кроме того, многие из соединений по настоящему изобретению обладают благоприятным фармакокинетическим профилем и свойствами, привлекательными с точки зрения биодоступности, включая приемлемый период полувыведения, AUC (площадь под кривой), пиковые значения и отсутствие неблагоприятных эффектов, таких как недостаточно быстрая реакция и ретенция в тканях.
Антивирусную активность соединений формулы (I) в отношении HCV in vitro испытывали на клеточной системе репликации HCV, основанной на публикации Lohmann и др., (1999) Science, 285:110-113, с дополнительными модификациями, описанными в публикации Krieger и др., (2001) Journal of Virology, 75: 4614-4624, которая дополнительно представлена в разделе «примеры». Данная модель, хотя и не является полной моделью HCV-инфекции, широко распространена в качестве наиболее надежной и эффективной модели автономной репликации РНК HCV среди существующих. Соединения, проявляющие антивирусную активность в отношении HCV в этой клеточной модели, рассматриваются в качестве соединений-кандидатов при дальнейшем развитии средств для лечения HCV-инфекций у млекопитающих. Следует принимать во внимание, что важно проводить различие между соединениями, которые специфически препятствуют проявлению функций HCV, и соединениями, которые вызывают цитотоксические или цитостатические эффекты в модели подавления репликации РНК репликона HCV и, как следствие, вызывают уменьшение концентрации РНК HCV или концентрации сцепленного репортерного фермента. Известны исследования в области оценки клеточной цитотоксичности, например, на основе активности митохондриальных ферментов с помощью флуорогенных редокс-красителей, таких как резазурин. Кроме того, существуют защитные клеточные экраны для оценки неселективного ингибирования активности сцепленного репортерного гена, такого как люцифераза светляка. Подходящие типы клеток могут быть наделены стабильной трансфекцией с помощью репортерного люциферазного гена, экспрессия которого зависит от конститутивно активного генного промотора, и такие клетки могут быть использованы в качестве защитных экранов для элиминации неселективных ингибиторов.
Благодаря своим антивирусным свойствам, в частности, антивирусным свойствам в отношении HCV, соединения формулы (I) или любая их подгруппа, их пролекарства, N-оксиды, аддитивные соли, четвертичные амины, комплексы с металлами и стереохимически изомерные формы применимы для лечения индивидуумов, подвергшихся действию вирусной инфекции, в частности HCV-инфекции, и для профилактики таких инфекций. В общем случае соединения по настоящему изобретению можно применять для лечения теплокровных животных, инфицированных вирусами, в частности флавивирусами, такими как HCV.
Следовательно, соединения по настоящему изобретению или любую их подгруппу можно применять в качестве лекарственных средств. Упомянутое применение в качестве лекарственного средства или способ лечения включает в себя системное введение субъектам, инфицированным вирусом, или субъектам, подверженным вирусным инфекциям, количество, эффективное для борьбы с состояниями, ассоциированными с вирусной инфекцией, в частности, с HCV-инфекцией.
Настоящее изобретение также относится к применению настоящих соединений или любой их подгруппы для производства лекарственного средства для лечения или профилактики вирусных инфекций, в частности HCV-инфекции.
Кроме того, настоящее изобретение относится к способу лечения теплокровного животного, инфицированного вирусом или подвергающегося риску инфицирования вирусом, в частности вирусом HCV; упомянутый способ включает в себя введение эффективного количества антивирусного соединения формулы (I), которое здесь указано, или соединения любой из подгрупп соединений формулы (I), которые здесь указаны.
При комбинированной терапии в качестве лекарственного средства также можно применять комбинацию ранее известного антивирусного в отношении HCV соединения, такого как, например, интерферон-α (IFN-α), пегилированный интерферон-α и/или рибавирин, и соединения формулы (I). Термин «комбинированная терапия» относится к продукту, содержащему в обязательном порядке (a) соединение формулы (I) и (b) необязательно другое антивирусное в отношении HCV соединение, в виде комбинированного препарата для одновременного, отдельного или последовательного применения при лечении HCV-инфекций, в частности, при лечении инфекций, вызванных HCV.
Антивирусные в отношении HCV соединения охватывают средства, выбранные из ингибитора полимеразы HCV, ингибитора протеазы HCV, ингибитора другого объекта в жизненном цикле HCV, и иммуномодулирующее средство, антивирусное средство и их комбинации.
Ингибиторы полимеразы HCV включают в себя, но не ограничиваются перечисленным, NM283 (валопицитабин), R803, JTK-109, JTK-003, HCV-371, HCV-086, HCV-796 и R-1479.
Ингибиторы протеаз HCV (NS2-NS3-ингибиторы и NS3-NS4A-ингибиторы) включают в себя, но не ограничиваются перечисленным, соединения по заявке WO02/18369 (см., например, строки 9-22 на стр. 273, и от строки 4 на стр. 274 до строки 11 на стр. 276,); BILN-2061, VX-950, GS-9132 (ACH-806), SCH-503034 и SCH-6. Дополнительные средства, которые можно применять, представляют собой средства, описанные в заявках WO98/17679, WO00/056331 (Vertex); WO98/22496 (Roche); WO99/07734 (Boehringer Ingelheim), WO2005/073216, WO2005073195 (Medivir) и структурно подобные средства.
Ингибиторы других объектов в жизненном цикле HCV включают в себя NS3-геликазу; ингибиторы металлопротеазы; ингибиторы антисмысловых олигонуклеотидов, такие как ISIS-14803, AVI-4065 и т.п.; малые интерферирующие РНК (миРНК), такие как SIRPLEX-140-N и т.п.; кодируемую вектором короткую шпильку РНК (РНК-шпилька); ДНК-ферменты; HCV-специфические рибозимы, такие как гептазим, RPI.13919 и т.п.; ингибиторы входа, такие как HepeX-C, HuMax-HepC и т.п.; ингибиторы α-глюкозидазы, такие как целгосивир, UT-231B и т.п.; KPE-02003002; и BIVN 401.
Иммуномодулирующие средства включают в себя, но не ограничиваются перечисленным, соединения природных и рекомбинантных изоформ интерферона, включая α-интерферон, β-интерферон, γ-интерферон, ω-интерферон и т.п., такие как Intron А®, Roferon-A®, Canferon-A300®, Advaferon®, Infergen®, Humoferon®, Sumiferon MP®, Alfaferone®, IFN-beta®, Feron® и т.п.; соединения интерферона, дериватизированные (пегилированные) полиэтиленгликолем, такие как PEG интерферон-α-2a (Pegasys®), PEG интерферон-α-2b (PEG-Intron®), пегилированный IFN-α-con1 и т.п.; препараты пролонгированного действия и производные соединений интерферона, такие как α-альбуферон - интерферон, конденсированный с альбумином, и т.п.; соединения, которые стимулируют синтез интерферона в клетках, такие как резиквимод и т.п.; интерлейкины; соединения, которые усиливают развитие ответа хелперных T-клеток 1-типа, такие как SCV-07 и т.п.; TOLL-подобные агонисты рецепторов, такие как CpG-10101 (актилон), изаторибин и т.п.; тимозин α-1; ANA-245; ANA-246; гистаминдигидрохлорид; тетрахлордекаоксид пропагермания; амплиген; IMP-321; KRN-7000; антитела, такие как сивасир, XTL-6865 и т.п.; профилактические и терапевтические вакцины, такие как InnoVac C, HCV E1E2/MF59 и т.п.
Другие антивирусные средства включают в себя, но не ограничиваются перечисленным, рибавирин, амантадин, вирамидин, нитазоксанид; телбивудин; NOV-205; тaрибавирин; ингибиторы входа во внутреннюю область рибосом; вирусные ингибиторы широкого спектра, такие как ингибиторы IMPDH (например, соединения, описанные в патентах США №№ 5807876, 6498178, 6344465, 6054472, в заявках WO97/40028, WO98/40381, WO00/56331, микофеноловая кислота и ее производные, включая, но не ограничиваясь перечисленным, VX-950, препарат Мerimepodib (VX-497), VX-148 и/или VX-944); или комбинации любых средств, из указанных выше.
Таким образом, для борьбы с HCV-инфекциями или для их лечения соединения формулы (I) можно совместно вводить в сочетании, например, с интерфероном-α (IFN-α), пегилированным интерфероном-α и/или рибавирином, а также терапевтическими средствами на основе антител, нацеленных против HCV-эпитопов, малыми интерферирующими РНК (миРНК), рибозимами, ДНК-ферментами, антисмысловой РНК, антагонистами небольших молекул, например NS3-протеазы, NS3-геликазы и NS5B-полимеразы.
Соответственно, настоящее изобретение относится к применению соединения формулы (I) или любой из его подгрупп, которые указаны выше, для производства лекарственного средства, применимого для ингибирования активности HCV у млекопитающего, инфицированного вирусами HCV, когда упомянутое лекарственное средство применяется для комбинированной терапии; упомянутая комбинированная терапия предпочтительно включает в себя соединение формулы (I) и другое соединение-ингибитор HCV, например, (пегилированный) IFN- α и/или рибавирин.
При еще одном аспекте получают комбинации соединения формулы (I), которое здесь указано, и соединения, направленного против ВИЧ. Последние предпочтительно представляют собой те ингибиторы ВИЧ, которые обладают положительным действием на метаболизм лекарственного средства и/или фармакокинетику, которая улучшает биодоступность. Примером такого ингибитора ВИЧ является ритонавир.
В связи с этим в настоящем изобретении дополнительно обеспечивается комбинация, содержащая (a) ингибитор NS3/4a-протеазы HCV формулы (I) или его фармацевтически приемлемая соль; и (b) ритонавир или его фармацевтически приемлемая соль.
Соединение ритонавир и его фармацевтически приемлемые соли и способы их получения описаны в заявке WO94/14436. О предпочтительных лекарственных формах ритонавира см. патент США № 6037157 и цитируемые в нем документы: патенты США №№ 5484801, 08/402690 и заявки WO95/07696 и WO95/09614. Ритонавир имеет следующую формулу:
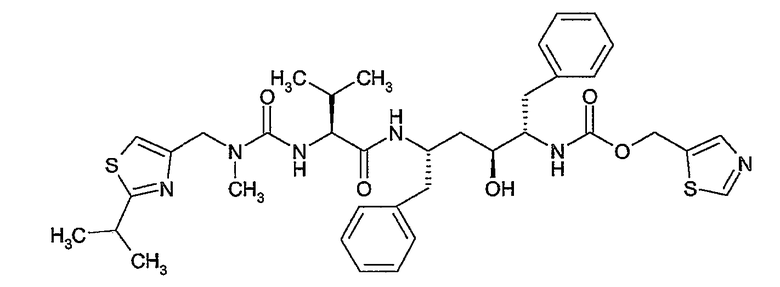
При дополнительном варианте осуществления изобретения комбинация, содержащая (a) ингибитор NS3/4a-протеазы HCV формулы (I) или его фармацевтически приемлемую соль; и (b) ритонавир или его фармацевтически приемлемую соль; дополнительно содержит дополнительное соединение против HCV, выбранное из описанных здесь соединений.
В одном из вариантов осуществления настоящего изобретения предлагается способ получения описанной здесь комбинации, включающий в себя стадию объединения ингибитора NS3/4a-протеазы HCV формулы (I) или его фармацевтически приемлемой соли и ритонавира или его фармацевтически приемлемой соли. При альтернативном варианте осуществления настоящего изобретения предлагается способ, при котором комбинация содержит один или несколько дополнительных средств, которые здесь описаны.
Комбинации по настоящему изобретению можно применять в качестве лекарственных средств. Упомянутое применение в качестве лекарственного средства или способ лечения включает в себя систематическое введение субъектам, инфицированным HCV, количества, эффективного для борьбы с состояниями, ассоциированными с HCV и другими патогенными флави- и пестивирусами. Следовательно, комбинации по настоящему изобретению можно применять для производства лекарственного средства, применимого для лечения, профилактики или борьбы с инфекцией или заболеванием, ассоциированным с HCV-инфекцией у млекопитающего, в частности, для лечения состояний, ассоциированных с HCV и другими патогенными флави- и пестивирусами.
В одном из вариантов осуществления настоящего изобретения предлагается фармацевтическая композиция, содержащая комбинацию по любому из описанных здесь вариантов осуществления и фармацевтически приемлемый наполнитель. В частности, в настоящем изобретении предлагается фармацевтическая композиция, содержащая (a) терапевтически эффективное количество ингибитора NS3/4a-протеазы HCV формулы (I) или его фармацевтически приемлемой соли, (b) терапевтически эффективное количество ритонавира или его фармацевтически приемлемой соли и (c) фармацевтически приемлемый наполнитель. Фармацевтическая композиция, кроме того, необязательно содержит дополнительное средство, выбранное из ингибитора полимеразы HCV, ингибитора протеазы HCV, ингибитора другого объекта в жизненном цикле HCV и иммуномодулирующего средства, антивирусного средства и их комбинаций.
Композиции можно получать в подходящих фармацевтических лекарственных формах, таких как описанные выше лекарственные формы. Каждый из активных ингредиентов можно получать отдельно и препараты можно вводить совместно или в виде одного препарата, содержащего оба ингредиента, и, если необходимо, можно применять дополнительные активные ингредиенты.
Подразумевается, что применяемый здесь термин "композиция" охватывает продукт, содержащий специфические ингредиенты, а также любой продукт, который получается в результате, прямо или косвенно, из комбинации специфических ингредиентов.
В одном из вариантов осуществления изобретения описанные здесь комбинации, также можно получать в виде комбинированного препарата для одновременного, отдельного или последовательного применения в ВИЧ-терапии. В таком случае соединение общей формулы (I) или любой из его подгрупп, входит в состав фармацевтической композиции, содержащей другие фармацевтически приемлемые наполнители, а ритонавир входит в состав отдельной фармацевтической композиции, содержащей другие фармацевтически приемлемые наполнители. Удобно, когда две такие отдельные фармацевтические композиции могут представлять собой часть набора для одновременного, отдельного или последовательного применения.
Таким образом, отдельные компоненты комбинации по настоящему изобретению в течение курса терапии можно вводить отдельно, в разное время или одновременно в виде разделенных на части или единых комбинированных форм. Следовательно, следует понимать, что настоящее изобретение включает в себя все такие схемы одновременного или альтернативного лечения, и термин «введение» должен интерпретироваться соответственно. При предпочтительном варианте осуществления изобретения отдельные лекарственные формы вводятся приблизительно одновременно.
В одном из вариантов осуществления изобретения комбинация по настоящему изобретению содержит количество ритонавира или его фармацевтически приемлемой соли, которое является достаточным, чтобы клинически улучшить биодоступность ингибитора NS3/4a-протеазы HCV формулы (I) по сравнению с биодоступностью в том случае, когда упомянутый ингибитор NS3/4a-протеазы HCV формулы (I) вводится в одиночку.
В другом варианте осуществления изобретения комбинация по настоящему изобретению содержит количество ритонавира или его фармацевтически приемлемой соли, которое является достаточным для увеличения, по меньшей мере, одной из фармакокинетических переменных ингибитора NS3/4a-протеазы HCV формулы (I), выбранных из t1/2, Cmin, Cmаx, Css, AUC при 12 ч или AUC при 24 ч, по сравнению с упомянутыми значениями, по меньшей мере, одной фармакокинетической переменной в случае, когда ингибитор NS3/4a-протеазы HCV формулы (I) вводится в одиночку.
Дополнительный вариант осуществления изобретения относится к способу улучшения биодоступности ингибитора NS3/4a-протеазы HCV, включающему в себя введение субъекту, нуждающемуся в таком улучшении, указанной выше комбинации, содержащей терапевтически эффективное количество каждого из компонентов упомянутой комбинации.
При дополнительном варианте осуществления изобретение относится к применению ритонавира или его фармацевтически приемлемой соли в качестве улучшающей добавки, по меньшей мере, для одной из фармакокинетических переменных ингибитора NS3/4a-протеазы HCV формулы (I), выбранных из t1/2, Cmin, Cmаx, Css, AUC при 12 ч или AUC при 24 ч, при условии, что упомянутое применение не осуществляется на практике в отношении организма человека или животного.
Применяемый здесь термин «индивидуальный» относится к животному, предпочтительно к млекопитающему, наиболее предпочтительно к человеку, который является объектом лечения, наблюдения или эксперимента.
Биодоступность определяется как доля вводимой дозы, достигающая большого круга кровообращения; t1/2 представляет собой период полувыведения или время, необходимое для того, чтобы концентрация в плазме понизилась до половины ее исходного значения. Css представляет собой концентрацию в стационарном состоянии, то есть концентрацию, при которой скорость ввода лекарственного средства равна скорости его удаления. Cmin определяется как самая низкая (минимальная) концентрация, измеренная в течение интервала между приемами лекарственного средства. Cmax представляет собой самую высокую (максимальную) концентрацию, измеренную в течение интервала между приемами лекарственного средства. AUC определяется как площадь под кривой зависимости «концентрация в плазме-время» для определенного периода времени.
Комбинации по настоящему изобретению можно вводить человеку согласно схемам приема, определенным для каждого компонента, содержащегося в упомянутых комбинациях. Компоненты, содержащиеся в упомянутых комбинациях, можно вводить вместе или отдельно. Ингибиторы NS3/4a-протеазы формулы (I) или любой их подгруппы и ритонавир, его фармацевтически приемлемую соль или его сложный эфир, могут иметь уровни дозировки в диапазоне от 0,02 до 5,0 граммов в день.
Когда ингибитор NS3/4a-протеазы HCV формулы (I) и ритонавир вводятся в комбинации, подходящее весовое отношение ингибитора NS3/4a-протеазы HCV формулы (I) к ритонавиру находится в диапазоне приблизительно от 40:1 до приблизительно 1:15, или приблизительно от 30:1 до приблизительно 1:15, или приблизительно от 15:1 до приблизительно 1:15, обычно приблизительно от 10:1 до приблизительно 1:10, и более конкретно, приблизительно от 8:1 до приблизительно 1:8. Также применимы весовые отношения ингибиторов NS3/4a-протеазы HCV формулы (I) к ритонавиру, находящиеся в диапазоне приблизительно от 6:1 до приблизительно 1:6 или приблизительно от 4:1 до приблизительно 1:4, или приблизительно от 3:1 до приблизительно 1:3, или приблизительно от 2:1 до приблизительно 1:2, или приблизительно от 1,5:1 до приблизительно 1:1,5. При одном из аспектов количество по массе ингибиторов NS3/4a-протеазы HCV формулы (I) равно или больше, чем количество по массе ритонавира, при котором подходящее весовое отношение ингибитора NS3/4a-протеазы HCV формулы (I) к ритонавиру находится в диапазоне приблизительно от 1:1 до приблизительно 15:1, обычно приблизительно от 1:1 до приблизительно 10:1, и более конкретно, приблизительно от 1:1 до приблизительно 8:1. Также применимы весовые отношения ингибитора NS3/4a-протеазы HCV формулы (I) к ритонавиру, находящиеся в диапазоне приблизительно от 1:1 до приблизительно 6:1, или приблизительно от 1:1 до приблизительно 5:1, или приблизительно от 1:1 до приблизительно 4:1, или приблизительно от 3:2 до приблизительно 3:1, или приблизительно от 1:1 до приблизительно 2:1 или приблизительно от 1:1 до приблизительно 1,5:1.
Применяемый здесь термин «терапевтически эффективное количество» означает такое количество активного соединения или компонента или фармацевтического средства, которое вызывает биологический или лечебный ответ в тканях, системе, у животного или человека, который в свете настоящего изобретения является желательным для исследователя, ветеринарного врача, лечащего врача или другого практикующего врача и который включает в себя частичное снятие симптомов заболевания, подвергаемого лечению. Поскольку настоящее изобретение относится к комбинациям, содержащим два или более средств, «терапевтически эффективное количество» представляет собой такое количество взятых вместе средств, при котором суммарный эффект вызывает требуемый биологический или лечебный ответ. Например, терапевтически эффективное количество композиции, содержащей (a) соединение формулы (I) и (b) ритонавир, может представлять собой количество взятых вместе соединения формулы (I) и ритонавира, обладающее суммарным эффектом, который терапевтически эффективен.
В общем случае предполагается, что эффективное суточное количество антивирусного средства может составлять от 0,01 мг/кг до 500 мг/кг массы тела, более предпочтительно от 0,1 мг/кг до 50 мг/кг массы тела. Возможно, что подходящим будет введение требуемой дозы в виде одной, двух, трех, четырех или более (суб)доз с подходящими интервалами на протяжении дня. Упомянутые (суб-)дозы можно получать в виде стандартных лекарственных форм, например, содержащих от 1 до 1000 мг, и, в частности, от 5 до 200 мг активного ингредиента на стандартную лекарственную форму.
Точная дозировка и частота введения зависят от конкретно применяемого соединения формулы (I), конкретного состояния, подвергаемого лечению, тяжести состояния, подвергаемого лечению, возраста, веса, пола, степени нарушения и общего физического состояния конкретного пациента, а также от другого лекарственного лечения, принимаемого индивидуально, как хорошо известно специалисту в данной области. Кроме того, очевидно, что упомянутое эффективное суточное количество можно уменьшать или повышать в зависимости от реакции подвергаемого лечению субъекта и/или в зависимости от оценки врача, прописывающего соединения по настоящему изобретению. Следовательно, упомянутые выше диапазоны эффективных суточных количеств носят только рекомендательный характер.
Согласно одному из вариантов осуществления изобретения ингибитор NS3/4a-протеазы HCV формулы (I) и ритонавир можно вводить совместно один или два раза в день, предпочтительно перорально, при этом количество соединений формулы (I) на дозу составляет приблизительно от 1 до приблизительно 2500 мг, а количество ритонавира на дозу составляет приблизительно от 1 до приблизительно 2500 мг. При другом варианте осуществления изобретения количества на дозу при совместном введении один или два раза в сутки составляют приблизительно от 50 до приблизительно 1500 мг соединения формулы (I) и приблизительно от 50 до приблизительно 1500 мг ритонавира. При еще одном варианте осуществления изобретения количества на дозу при совместном введении один или два раза в сутки составляют приблизительно от 100 до приблизительно 1000 мг соединения формулы (I) и приблизительно от 100 до приблизительно 800 мг ритонавира. При еще одном варианте осуществления изобретения количества на дозу при совместном введении один или два раза в сутки составляют приблизительно от 150 до приблизительно 800 мг соединения формулы (I) и приблизительно от 100 до приблизительно 600 мг ритонавира. При еще одном варианте осуществления изобретения количества на дозу при совместном введении один или два раза в сутки составляют приблизительно от 200 до приблизительно 600 мг соединения формулы (I) и приблизительно от 100 до приблизительно 400 мг ритонавира. При еще одном варианте осуществления изобретения количества на дозу при совместном введении один или два раза в сутки составляют приблизительно от 200 до приблизительно 600 мг соединения формулы (I) и приблизительно от 20 до приблизительно 300 мг ритонавира. При еще одном варианте осуществления изобретения количества на дозу при совместном введении один или два раза в сутки составляют приблизительно от 100 до приблизительно 400 мг соединения формулы (I) и приблизительно от 40 до приблизительно 100 мг ритонавира.
Типичные комбинации соединение формулы (I) (мг)/ритонавир (мг) при дозировке один или два раза в сутки включают в себя 50/100, 100/100, 150/100, 200/100, 250/100, 300/100, 350/100, 400/100, 450/100, 50/133, 100/133, 150/133, 200/133, 250/133, 300/133, 50/150, 100/150, 150/150, 200/150, 250/150, 50/200, 100/200, 150/200, 200/200, 250/200, 300/200, 50/300, 80/300, 150/300, 200/300, 250/300, 300/300, 200/600, 400/600, 600/600, 800/600, 1000/600, 200/666, 400/666, 600/666, 800/666, 1000/666, 1200/666, 200/800, 400/800, 600/800, 800/800, 1000/800, 1200/800, 200/1200, 400/1200, 600/1200, 800/1200, 1000/1200 и 1200/1200. Другие типичные комбинации соединение формулы (I) (мг)/ритонавир (мг) при дозировке один или два раза в сутки включают в себя 1200/400, 800/400, 600/400, 400/200, 600/200, 600/100, 500/100, 400/50, 300/50, и 200/50.
В одном из вариантов осуществления настоящего изобретения предлагается изделие промышленного производства, содержащее композицию, эффективную для лечения HCV-инфекции или для ингибирования NS3-протеазы HCV; и упаковочный материал, содержащий этикетку, на которой указано, что композицию можно применять для лечения инфекции, вызванной вирусом гепатита C; в котором композиция содержит соединение формулы (I) или любой его подгруппы, или описанную здесь комбинацию.
Другой вариант осуществления настоящего изобретения относится к набору или контейнеру, содержащему соединение формулы (I) или любой его подгруппы, или комбинацию, согласно изобретению объединяющую ингибитор NS3/4a-протеазы HCV формулы (I) или его фармацевтически приемлемую соль и ритонавир или его фармацевтически приемлемую соль в количестве, эффективном для применения в качестве стандарта или реагента в тесте или испытании по определению способности потенциальных фармацевтических препаратов ингибировать NS3/4a-протеазу HCV, рост HCV или то и другое. Такой аспект изобретения может найти применение в фармацевтических исследовательских программах.
Соединения и комбинации по настоящему изобретению можно применять в высокопроизводительных анализах целевых анализируемых образцов, таких как анализы по измерению эффективности упомянутой комбинации для лечения HCV.
Примеры
Следующие примеры предназначены для иллюстрации настоящего изобретения и при этом не ограничивают его.
Пример 1
Получение типичных промежуточных продуктов
Синтез 1-гидрокси-3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолина (6)
Стадия A
При перемешивании к раствору 3-метил-2-бутанона (27,0 г, 313 ммоль) в метаноле (150 мл) добавляли бром (50 г, 313 ммоль). Давали возможность реакции протекать (обесцвечивание) ниже 10°C. Затем перемешивание продолжали при комнатной температуре в течение 30 мин прежде, чем добавить воду (100 мл). Спустя 15 мин смесь разбавляли водой (300 мл) и четыре раза экстрагировали Et2О-простым диэтиловым эфиром. Эфирные экстракты последовательно промывали 10% раствором Na2CO3, водой, насыщенным раствором соли и сушили (Na2SO4), получая при этом 42 г (81%) требуемого продукта в виде жидкости.
Стадия В
К кипящему раствору этилтиооксамата (13,3 г, 100 г, 100 ммоль) в этаноле (100 мл) по каплям добавляли 1-бром-3-метилбутан-2-он (17,6 г, 106 ммоль) в течение 15 мин. Раствор кипятили с обратным холодильником в течение 1 час. Раствор добавляли к 250 мл холодной воды со льдом и подщелачивали концентрированным раствором аммиака. Полученную смесь дважды экстрагировали этилацетатом AcOEt. Органическую фазу промывали насыщенным раствором соли, сушили (Na2SO4) и упаривали при пониженном давлении. Сырой продукт очищали с помощью колоночной хроматографии с элюентом от дихлорметана до дихлорметана с 2% MeOH (метанола), получая при этом 13,1 г (65%) требуемого продукта: 1Н ЯМР (СDСl3): 7,20 (c, 1H); 4,49 (м, 2Н); 3,25 (м, 1Н); 1,42 (т, 3Н); 1,35 (д, 6Н).
Стадия C
К раствору N,N-диэтил-4-метокси-2-метилбензамида 4 (2,4 г, 11 ммоль) в безводном ТГФ (30 мл) при -78°C в атмосфере азота по каплям добавляли н-BuLi (8,9 мл, 2,5 M раствор в гексане). Раствор оставляли при -78°C в течение дополнительных 30 мин. Затем по каплям добавляли раствор тиазола 3 в ТГФ (5 мл). Спустя 2 ч реакционную смесь распределяли между холодной водой со льдом и AcOEt (этилацетатом). При очистке с помощью колоночной хроматографии (этилацетат AcOEt/петролейный эфир/CH2Cl2, 1:2:1) получали 1,8 г (43%) требуемого продукта 4 в виде желтоватого масла: >95% чистоты с помощью ЖХ/МС.
Стадия D
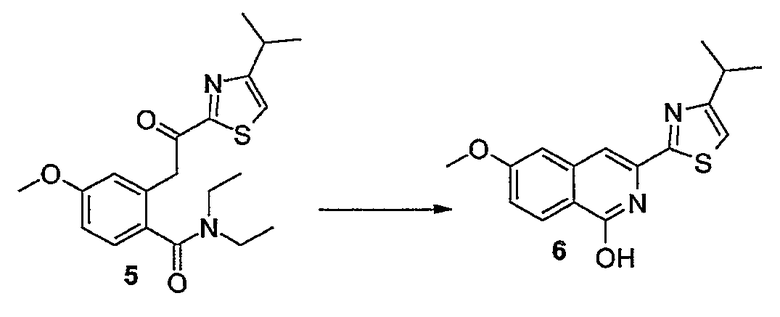
Смесь продукта 5 (1,98 г, 5,29 ммоль) и ацетата аммония (12,2 г, 159 ммоль) нагревали при 140°C в запаянной трубке в течение 1 ч, затем охлаждали до комнатной температуры. Реакционную смесь распределяли между холодной водой со льдом и CH2Cl2, сушили (Na2SO4) и фильтровали через диоксид кремния, получая при этом 1,59 г (78%) требуемого продукта 6 в виде белого порошка m/z = 301 (M+H)+.
Синтез 1-гидрокси-3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолина (7)
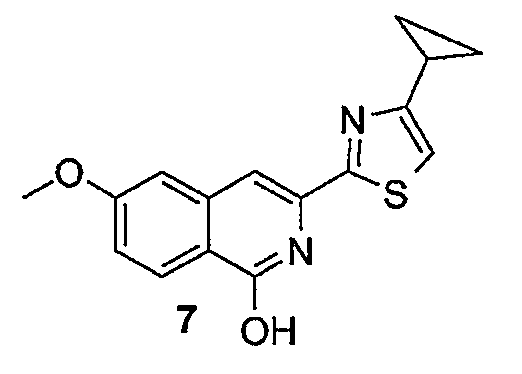
Указанный в заголовке продукт получали из метилциклопропилкетона, следуя процедурам, описанным для синтеза 1-гидрокси-3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолина 6.
Синтез 1,3-дихлор-6-метоксиизохинолина (12)
Стадия А
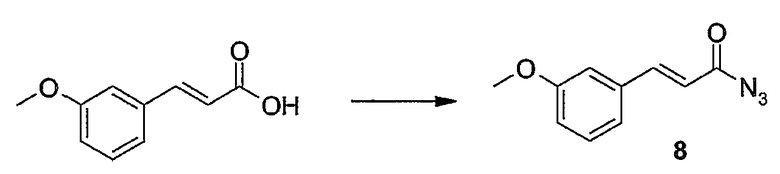
К суспензии 3-метоксикоричной кислоты (49,90 г, 280 ммоль) в ацетоне (225 мл) добавляли при 0°C в атмосфере азота триэтиламин (80,5 мл, 578 ммоль). Спустя 10 мин при 0°C добавляли по каплям этилхлорформиат (46,50 г, 429 ммоль), поддерживая при этом температуру при 0°C. Спустя 1 час при 0°C медленно добавляли раствор азида натрия (27,56 г, 424 ммоль) в воде (200 мл), затем реакционной смеси давали нагреваться до комнатной температуры. Спустя 16 ч реакционную смесь выливали в воду (500 мл) и выпаривали ацетон. Остаток экстрагировали толуолом, получая при этом раствор 8, который применяли как таковой на следующей стадии.
Стадия В
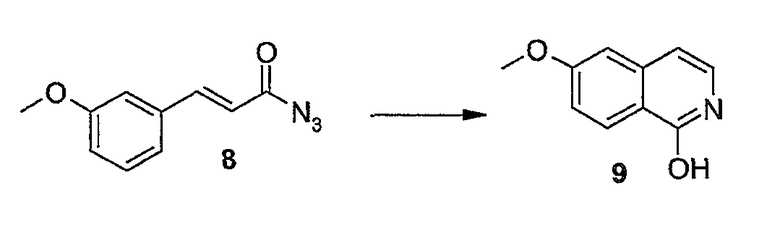
Раствор продукта 8 с предыдущей стадии в толуоле добавляли по каплям к нагретому раствору дифенилметана (340 мл) и трибутиламина (150 мл) при 190°C. Толуол сразу отгоняли с помощью аппарата Дина-Старка. После завершения добавления температуру реакционной смеси повышали до 210°C в течение 2 ч. После охлаждения выпавший в осадок продукт собирали при фильтровании и промывали гептаном, получая при этом 49,1 г (29%) требуемого продукта 9 в виде белого порошка: m/z = 176 (M+H)+; 1Н ЯМР (СDСl3): 8,33 (д, J=8,9 Гц, 1H); 7,13 (д, J=7,2 Гц, 1Н); 7,07 (дд, J=8,9 Гц, 2,5 Гц, 1Н); 6,90 (д, J=2,5 Гц, 1Н); 6,48 (д, J=7,2 Гц, 1Н), 3,98 (c, 3H).
Стадия С
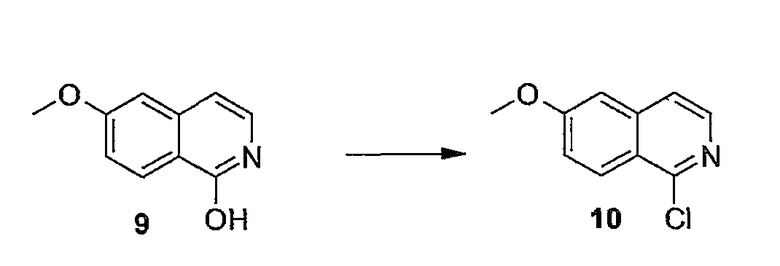
К соединению 9 (10,0 г, 57 ммоль) медленно добавляли оксихлорид фосфора (25 мл) и полученную смесь нагревали при осторожном кипячении с обратным холодильником в течение 3 ч. После завершения реакции оксихлорид фосфора выпаривали. Остаток выливали в холодную воду со льдом (40 мл) и доводили pH до 10 раствором NaOH в воде (50%). Смесь экстрагировали CHCl3, промывали насыщенным раствором соли, сушили (Na2SO4), фильтровали и упаривали. Остаток очищали с помощью колоночной хроматографии (CH2Cl2), получая при этом 8,42 г требуемого продукта 10 в виде желтого твердого вещества: m/z = 194 (M+H)+; 1Н ЯМР (СDСl3): 8,21 (д, J=9,3 Гц, 1H); 8,18 (д, J=5,7 Гц, 1Н); 7,47 (д, J=5,6 Гц, 1Н); 7,28 (дд, J=9,3 Гц, 2,5 Гц, 1Н); 7,06 (д, J=2,5 Гц, 1Н), 3,98 (c, 3H).
Стадия D
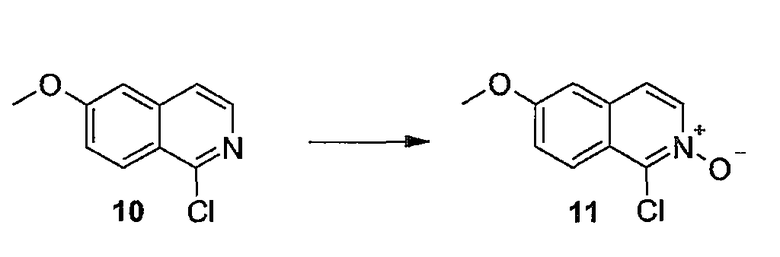
К раствору продукта 10 (2,70 г, 13,9 ммоль) в CH2Cl2 (10 мл) при 0°C небольшими порциями добавляли метахлорнадбензойную кислоту (6,41 г, 28,6 ммоль). После 30 мин при 0°C реакционную смесь нагревали до комнатной температуры в течение 12 ч. Затем реакционную смесь распределяли между 1N раствором NaOH и CH2Cl2 и последовательно промывали 1N раствором NaOH и насыщенным раствором соли. Органический слой сушили (Na2SO4), фильтровали и упаривали, получая при этом 1,89 г (64%) требуемого продукта 11 в виде оранжевого твердого вещества: m/z = 209,9 (M+H)+.
Стадия Е
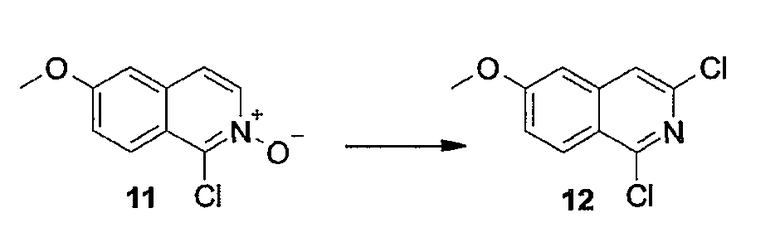
Раствор 11 (1,86 г, 8,86 ммоль) в оксихлориде фосфора (18 мл) нагревали при кипячении с обратным холодильником в течение 3 ч. Затем оксихлорид фосфора выпаривали в вакууме. Остаток выливали в холодную воду со льдом (50 мл) и pH доводили до 10 50% раствором NaOH в воде. Смесь экстрагировали CHCl3, органический слой промывали насыщенным раствором соли, сушили (Na2SO4), фильтровали и упаривали. Сырой материал очищали с помощью колоночной хроматографии (CH2Cl2), получая при этом 350 мг (17%) требуемого продукта 12 в виде желтого твердого вещества: m/z = 227,9 (M+H)+; 1Н ЯМР (СDСl3): 8,16 (д, J=9,3 Гц, 1H); 7,50 (с, 1Н); 7,25 (дд, J=9,3 Гц, 2,5 Гц, 1Н); 6,98 (д, J=2,5 Гц, 1Н); 3,98 (c, 3H).
Синтез 4-бром-1-гидрокси-6-метоксиизохинолина (13)
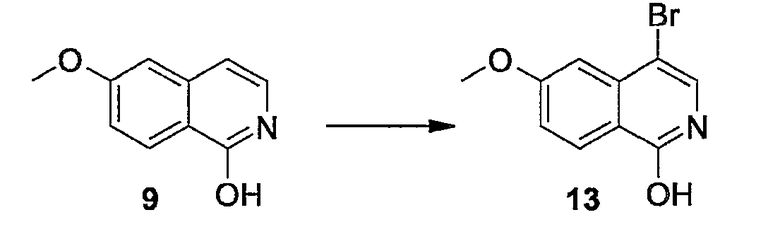
N-бромсукцинимид (2,33 г, 14,3 ммоль) добавляли к раствору продукта 9 (2,06 г, 11,8 ммоль) в ДМФА (40 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем ДМФА выпаривали и к остатку добавляли CH2Cl2. Полученную суспензию нагревали при 45°C в течение 15 мин. Белое твердое вещество отфильтровывали и промывали простым изопропиловым эфиром, получая при этом 2,07 г (69%) требуемого продукта 13: m/z = 253,7 (M+H)+; 1Н ЯМР (DMSO d6): 8,14 (д, J=8,8 Гц, 1H); 7,52 (с, 1Н); 7,17 (дд, J=8,8 Гц, 2,5 Гц, 1Н); 7,11 (д, J=2,4 Гц, 1Н); 3,83 (c, 3H).
Синтез 5-бром-1-хлор-6-метоксиизохинолина (19)
Стадия А
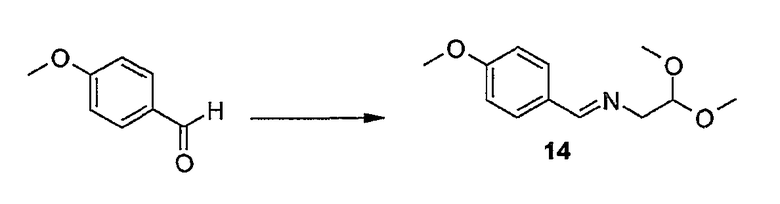
Эквимолярный раствор п-метоксибензальдегида (10 г, 73,5 ммоль) и аминоацетальдегиддиметилацеталя (7,93 г, 75,4 ммоль) в толуоле (50 мл) кипятили с обратным холодильником в течение ночи в аппарате Дина-Старка. Затем раствор упаривали в вакууме, получая при этом требуемый продукт 14, который применяли на следующей стадии без дополнительной очистки: m/z = 224 (M+H)+.
Стадия В
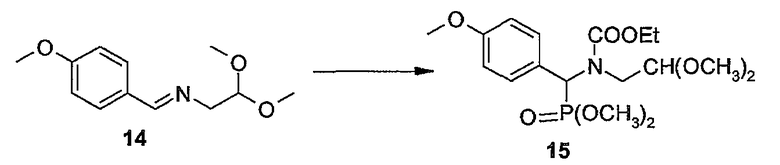
К раствору продукта 14 (73,5 ммоль) в сухом ТГФ (50 мл) при -10°C и энергичном перемешивании добавляли этилхлорформиат (8,02 г, 73,9 ммоль). После 30 мин реакционной смеси давали нагреться до комнатной температуры и добавляли триметилфосфит (10,6 г, 85,2 ммоль). Спустя 15 ч летучие вещества выпаривали в вакууме. Полученное масло 3 раза выпаривали совместно с толуолом, получая при этом требуемый продукт 15 в виде масла: m/z = 406 (M+H)+.
Стадия С
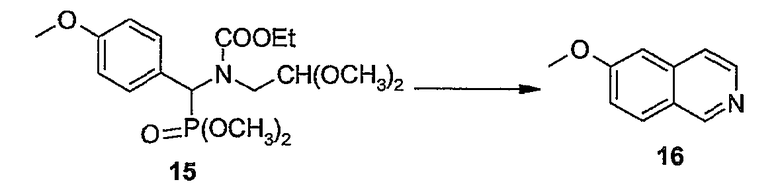
Материал (15), полученный на стадии B, растворяли в CH2Cl2 (200 мл) и охлаждали до 0°C. Затем добавляли тетрахлорид титана (86,0 г, 453 ммоль) и кипятили раствор с обратным холодильником в течение ночи. Реакционной смеси давали охладиться до комнатной температуры. Затем добавляли раствор NaOH (73 г) в воде (500 мл) и смесь перемешивали в течение 10 мин. Осадок TiO2 отфильтровывали и фильтрат экстрагировали 3N раствором HCl. pH водного слоя доводили до 10 с помощью NaOH. Продукт экстрагировали CH2Cl2, сушили (Na2SO4) и упаривали, получая при этом 5,32 г (45%) требуемого продукта 16, который применяли без дополнительной очистки на следующей стадии: m/z = 160 (M+H)+.
Стадия D
6-Метоксиизохинолин 16 (5,32 г, 33,4 ммоль) медленно добавляли при 0°C к концентрированной H2SO4 (33,5 мл). Смесь охлаждали до -25°C и добавляли NBS (7,68 г, 43,2 ммоль) с такой скоростью, чтобы температура реакции оставалась в интервале между -25°C и -22°C. Смесь перемешивали при -22°C в течение 2 ч и при -18°C в течение 3 ч, затем выливали на дробленый лед. pH доводили до 9, применяя концентрированный водный раствор NH3, и щелочную суспензию затем экстрагировали простым диэтиловый эфиром. Объединенные органические фракции промывали 1N раствором NaOH и водой, сушили (Na2SO4), фильтровали и упаривали досуха, получая при этом 5,65 г (71%) требуемого продукта 17: m/z = 237,8 (M+H)+.
Стадия Е
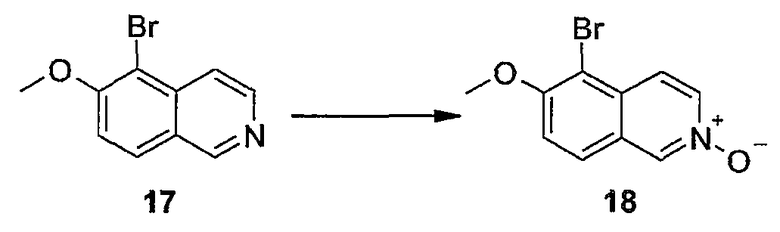
Метахлорнадбензойную кислоту (6,73 г, 30 ммоль) при 0°C добавляли к раствору продукта 17 (5,65 г, 24 ммоль) в CH2Cl2 (50 мл). После 30 мин при 0°C реакционной смеси давали нагреться до комнатной температуры в течение 3,5 ч. Затем дополнительно добавляли CH2Cl2 (300 мл) и полученную смесь последовательно промывали 1N раствором NaOH и насыщенным раствором соли. Органический слой сушили (MgSO4), фильтровали и упаривали, получая при этом 6,03 г (100%) требуемого продукта 18, который применяли как таковой на следующей стадии: m/z = 253,9 (M+H)+.
Стадия F
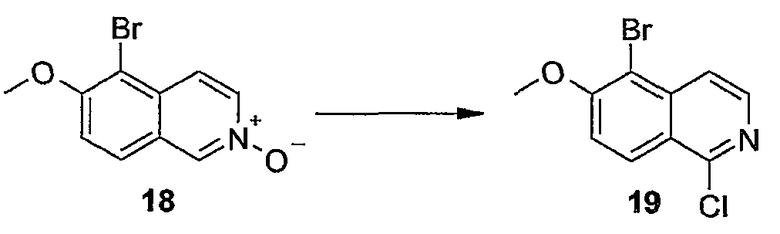
К охлажденному продукту 18 (6,03 г, 23,7 ммоль) медленно добавляли оксихлорид фосфора (60 мл) и полученную смесь затем нагревали при осторожном кипячении с обратным холодильником в течение 30 мин. После завершения реакции оксихлорид фосфора выпаривали. Остаток выливали в холодную воду со льдом (50 мл) и доводили pH до 10 с помощью NaOH. Смесь экстрагировали CHCl3, органический слой промывали насыщенным раствором соли, сушили (Na2SO4), фильтровали и упаривали. Сырой материал очищали с помощью колоночной хроматографии (CH2Cl2), получая при этом 1,15 г (18%) указанного в заголовке продукта в виде белого порошка: m/z = 271,7 (M+H)+; 1Н ЯМР (СDСl3): 8,17 (д, J=9,3 Гц, 1H); 8,28 (д, J=6,0 Гц, 1Н); 7,94 (д, J=6,0 Гц, 1Н); 7,4 (д, J=9,3 Гц, 1Н).
Синтез (гекс-5-енил)(метил)амина (21)
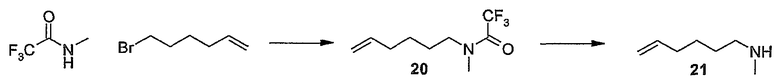
Стадия A
К раствору N-метилтрифторацетамида (25 г) в ДМФА (140 мл) при 0°C медленно добавляли гидрид натрия (1,05 экв.). Смесь перемешивали в течение 1 ч при комнатной температуре в атмосфере азота. Затем добавляли по каплям раствор бромгексена (32,1 г) в ДМФА (25 мл) и смесь нагревали до 70°C в течение 12 ч. Реакционную смесь выливали в воду (200 мл) и экстрагировали простым диэтиловым эфиром (4×50 мл), сушили (MgSO4), фильтровали и упаривали, получая при этом 35 г требуемого продукта 20 в виде желтоватого масла, которое применяли без дополнительной очистки на следующей стадии.
Стадия B
К раствору продукта 20 (35 г) в метаноле (200 мл) добавляли по каплям раствор гидроксида калия (187,7 г) в воде (130 мл). Смесь перемешивали при комнатной температуре в течение 12 ч. Затем реакционную смесь выливали в воду (100 мл) и экстрагировали простым диэтиловым эфиром (4×50 мл), сушили (MgSO4), фильтровали и отгоняли простой диэтиловый эфир при атмосферном давлении. Полученное масло очищали дистилляцией в вакууме (давление 13 мм Hg, 50°C), получая при этом 7,4 г (34%) указанного в заголовке продукта 21 в виде бесцветного масла. 1Н ЯМР (СDСl3): δ 5,8 (м, 1H); 5 (ддд, J=17,2 Гц, 3,5 Гц, 1,8 Гц, 1Н); 4,95 (м, 1Н); 2,5 (т, J=7,0 Гц, 2Н); 2,43 (с, 3H); 2,08 (кв, J=7,0 Гц, 2Н); 1,4 (м, 4Н); 1,3 (шир.с, 1Н).
Пример 2
Получение 17-[3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (29)
Стадия A
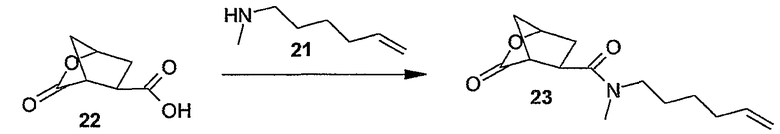
3-Оксо-2-оксабицикло[2.2.1]гептан-5-карбоновую кислоту 22 (500 мг, 3,2 ммоль) в 4 мл ДМФА добавляли при 0°C к гексафторфосфату 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (НATU) (1,34 г, 3,52 ммоль) и N-метилгекс-5-ениламину (435 мг, 3,84 ммоль) в ДМФА (3 мл) с последующим добавлением N,N-диизопропилэтиламина (DIPEA). После перемешивания в течение 40 мин при 0°C смесь перемешивали при комнатной температуре в течение 5 час. Затем растворитель выпаривали, остаток растворяли в EtOAc (этилацетате 70 мл) и промывали насыщенным раствором NaHCO3 (10 мл). Водный слой экстрагировали этилацетатом EtOAc (2×25 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl (20 мл), сушили (Na2SO4) и упаривали. При очистке флэш-хроматографией (этилацетат EtOAc/петролейный эфир, 2:1) получали 550 мг (68%) требуемого продукта 23 в виде бесцветного масла: m/z = 252 (M+H)+.
Стадия В
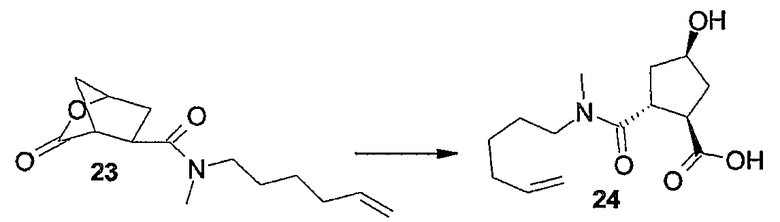
К лактонамиду 23 при 0°C добавляли раствор LiOH (105 мг в 4 мл воды). Спустя 1 час превращение завершалось (ВЭЖХ). Смесь подкисляли до pH 2-3 1N раствором HCl, экстрагировали этилацетатом (AcOEt), сушили (MgSO4), упаривали, несколько раз упаривали совмсетно с толуолом и сушили в высоком вакууме в течение ночи, получая при этом 520 мг (88%) требуемого продукта 24: m/z = 270 (M+H)+.
Стадия С
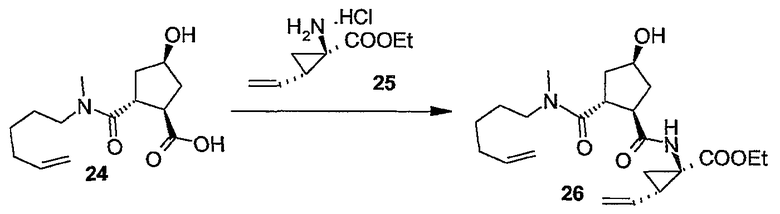
К продукту 24 (8,14 г, 30,2 ммоль) добавляли гидрохлорид сложного этилового эфира 1-(амино)-2-(винил)циклопропанкарбоновой кислоты 25 (4,92 г, 31,7 ммоль) и HATU (12,6 г, 33,2 ммоль). Смесь охлаждали на бане со льдом в атмосфере аргона и затем последовательно добавляли ДМФА (100 мл) и DIPEA (12,5 мл, 11,5 ммоль). После 30 мин при 0°C раствор перемешивали при комнатной температуре в течение дополнительных 3 ч. Затем реакционную смесь распределяли между этилацетатом EtOAc и водой, последовательно промывали 0,5 N раствором HCl (20 мл) и насыщенным раствором NaCl (2×20 мл) и сушили (Na2SO4). При очистке флэш-хроматографией (этил ацетат (AcOEt)/CH2Cl2/ петролейный эфир, 1:1:1) получали 7,41 г (60%) требуемого продукта 26 в виде бесцветного масла: m/z = 407 (M+H)+.
Стадия D
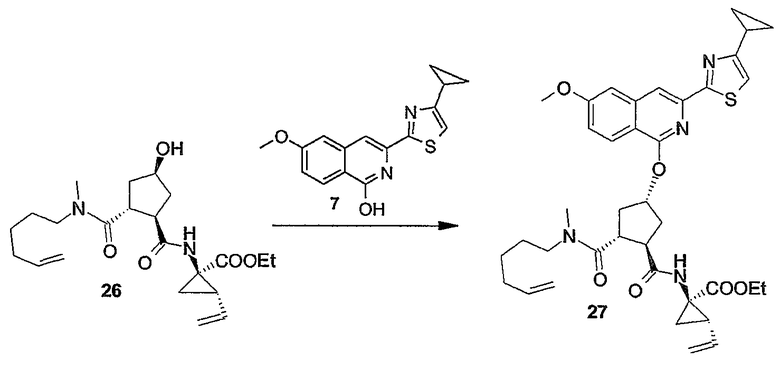
К раствору продукта 26 (300 мг, 0,738 ммоль), изохинолина 7 (308 мг, 1,03 ммоль) и трифенилфосфина (271 мг, 1,03 ммоль) в сухом ТГФ (15 мл) при -20°C в атмосфере азота добавляли DIAD (218 мкл, 1,11 ммоль). Затем реакционная смесь нагревалась до комнатной температуры. Спустя 1,5 ч растворитель выпаривали и сырой продукт очищали флэш-хроматографией на колонке (градиент смеси петролейный эфир/CH2Cl2/простой эфир от 3:1,5:0,5 до 1:1:1), получая при этом 290 мг требуемого продукта, загрязненного побочными продуктами (90% чистоты). При второй очистке (тот же самый элюент) получали 228 мг (43%) требуемого продукта 27: m/z = 687 (M+H)+. 1Н ЯМР (СDСl3): 8,11-7,98 (м, 1H); 7,98 (с, 1Н); 7,13-7,10 (м, 2Н); 6,89 (с, 1Н); 5,78-5,69 (м, 2H); 5,30-5,25 (м, 1Н); 5,11-5,09 (м, 1Н); 4,99-4,87 (м, 2Н); 4,15-4,08 (м, 2Н); 3,92 (с, 3Н); 3,71-3,58 (м, 1Н); 3,48-3,15 (м, 4Н); 3,03 (с, 3Н); 2,90-2,85 (м, 2Н); 2,60-2,25 (м, 2Н); 2,11-1,82 (м, 6H); 1,55-1,10 (м, 7Н); 0,98-0,96 (м, 4Н).
Стадия Е
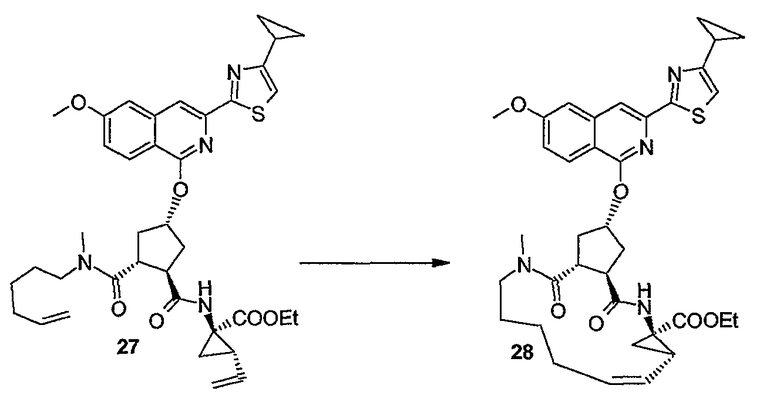
Раствор продукта 27 (220 мг, 0,32 ммоль) и катализатор Ховейды-Граббса 1-го поколения (19 мг, 0,032 ммоль) в сухом и дегазированном 1,2-дихлорэтане (400 мл) нагревали при 70°C в атмосфере азота в течение 12 ч. Затем выпаривали растворитель и остаток очищали с помощью хроматографии на силикагеле (петролейный эфир/CH2Cl2/простой диэтиловый эфир EtO2: 3:1:1), получая при этом 180 мг (85%) требуемого продукта 28: m/z = 659 (M+H)+. 1Н ЯМР (СDСl3): 8,11-8,08 (м, 1H); 7,98 (с, 1Н); 7,10-7,19 (м, 2Н); 7,09 (с, 1Н); 6,88 (с, 1H); 5,70-5,78 (м, 1Н); 5,61-5,69 (м, 1Н); 5,18-5,29 (м, 1Н); 4,63-4,69 (м, 1Н); 4,05-4,15 (м, 3Н); 3,92 (с, 3Н); 4,01-4,08 (м, 1Н); 3,28-3,26 (м, 1Н); 3,06 (с, 3Н); 2,88-3,05 (м, 2Н); 2,61-2,69 (м, 2H); 2,10-2,41 (м, 3Н); 1,90-2,02 (м, 4Н); 1,71-1,90 (м, 3Н); 0,87-1,62 (м, 9Н).
Стадия F
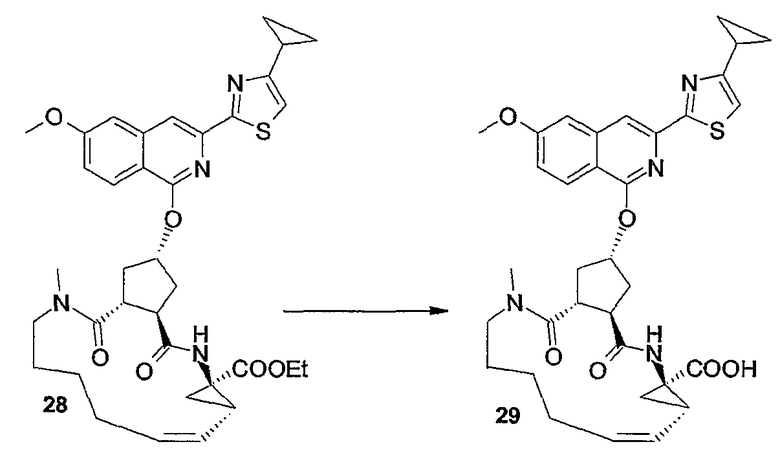
При перемешивании к раствору продукта 28 в ТГФ (15 мл) и метаноле MeOH (10 мл) добавляли раствор LiOH (327 мг) в воде (3 мл). Спустя 48 ч растворитель выпаривали и остаток распределяли между водой и простым диэтиловым эфиром. Водный слой подкисляли (pH 3) и экстрагировали этилацетатом AcOEt, сушили (MgSO4) и упаривали. Остаток кристаллизовали из простого диэтилового эфира, получая при этом 128 мг (74%) требуемого соединения 29: m/z = 631 (M+H)+. 1Н ЯМР (СDСl3): 8,00-8,03 (д, J=9,0 Гц, 1H); 7,86 (с, 1Н); 7,12 (с, 1Н); 7,10 (дд, J=9,0 Гц, 2,4 Гц, 1Н); 7,06 (д, J=2,4 Гц, 1H); 6,87 (с, 1Н); 5,64-5,71 (м, 1Н); 5,57-5,61 (м, 1Н); 5,16 (т, J=9,5 Гц, 1Н); 4,57-4,64 (м, 1Н); 3,92 (с, 3Н); 3,52-3,60 (м, 1Н); 3,25-3,37 (м, 1Н); 2,42-2,68 (м, 4Н); 2,17-2,33 (м, 3Н); 2,08-2,17 (м, 2H); 1,71-2,00 (м, 5Н); 1,33-1,62 (м, 5Н); 0,96-0,99 (м, 4Н).
Пример 3
Получение N-[17-[3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (30)
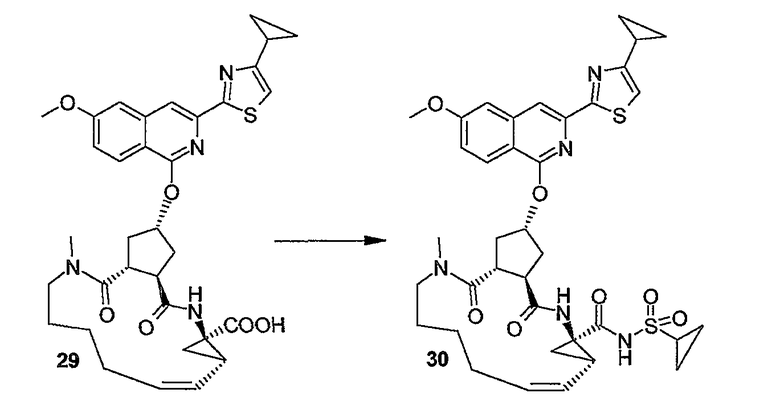
Смесь соединения 29 (91 мг, 0,14 ммоль) и 1,1'-карбонилдиимидазола (CDI) (47 мг, 0,29 ммоль) в сухом ТГФ (7 мл) нагревали при кипячении с обратным холодильником в течение 2 ч в атмосфере азота. ЖХ/МС-анализ показывал один пик промежуточного продукта (RT = 5,37). Если требуется, производное азалактона при желании можно выделить. Реакционную смесь охлаждали до комнатной температуры и добавляли циклопропилсульфонамид (52 мг, 0,43 ммоль). Затем добавляли DBU (50 мкл, 0,33 ммоль), перемешивали реакционную смесь при комнатной температуре в течение 1 ч и затем нагревали при 55°C в течение 24 ч. Растворитель выпаривали и остаток распределяли между этилацетатом AcOEt и подкисленной водой (pH 3). Сырой материал очищали с помощью колоночной хроматографии (этилацетат AcOEt/CH2Cl2/петролейный эфир, 1:1:1). Остаток кристаллизовали в простом диэтиловом эфире и фильтровали, получая при этом требуемое соединение, загрязненное циклопропилсульфонамидом. Полученный материал растирали в 3 мл воды, фильтровали, промывали водой и сушили в течение ночи с помощью высоковакуумного насоса, получая при этом 60 мг (57%) требуемого соединения 30 в виде слегка желтого порошка: m/z = 734 (M+H)+. 1Н ЯМР (СDСl3): 10,94 (с, 1H); 8,08 (д, J=8,6 Гц, 1Н); 8,00 (с, 1Н); 7,12-7,15 (м, 2Н); 6,91 (с, 1H); 6,35 (с, 1Н); 5,74-5,77 (м, 1Н); 5,63-5,69 (м, 1Н); 5,06 (т, J=10,4 Гц, 1Н); 4,60 (т, J=12,3 Гц, 1Н); 3,93 (с, 3Н); 3,35-3,42 (м, 2Н); 3,04 (с, 3H); 2,89-2,96 (м, 2Н); 2,52-2,52 (м, 2Н); 2,37-2,45 (м, 2Н); 2,10-2,32 (м, 2Н); 1,61-1,93 (м, 4Н); 1,3-1,51 (м, 4Н); 0,90-1,30 (м, 8Н).
Пример 4
Получение 17-[3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (31)
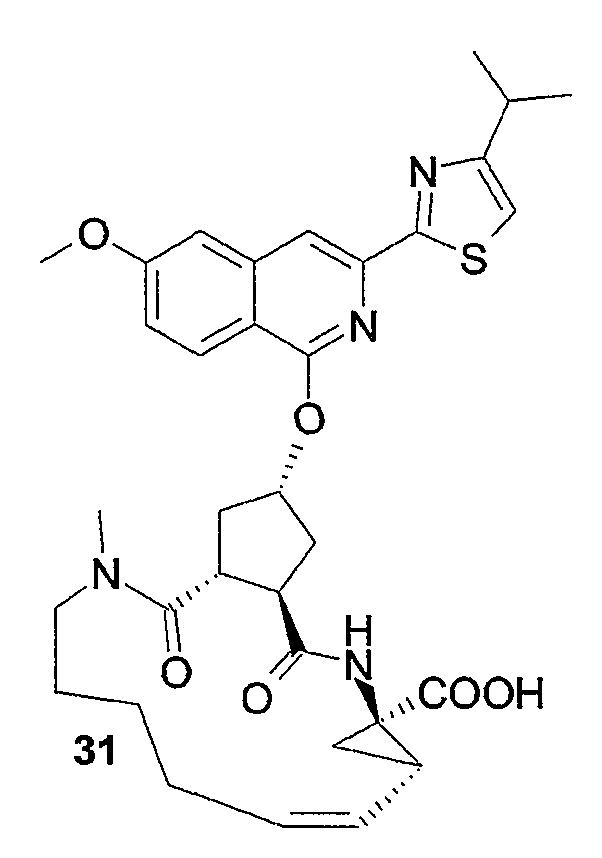
Указанный в заголовке продукт получали из 1-гидрокси-3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолина 6, следуя процедурам, описанным для 17-[3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты 29 (пример 2): m/z = 633 (M+H)+. 1Н ЯМР (СDСl3): 8,03 (д, J=8,9 Гц, 1H); 7,91 (с, 1Н); 7,20 (с, 1Н); 7,08-7,13 (м, 2Н); 6,93 (с, 1H); 5,61-5,69 (м, 2Н); 5,17 (т, J=9,5 Гц, 1Н); 4,57-4,64 (м, 1Н); 3,92 (с, 3Н); 3,55-3,63 (м, 1Н); 3,25-3,36 (м, 1Н); 3,11-3,20 (м, 1H); 3,05 (с, 3Н); 2,72-2,83 (м, 1Н); 2,53-2,66 (м, 2Н); 2,40-2,51 (м, 1Н); 2,17-2,32 (м, 2Н); 1,89-1,93 (м, 2Н); 1,71-1,83 (м, 2Н); 1,43-1,60 (м, 2Н); 1,37 (дд, J=6,9 Гц, 2,5 Гц, 6H); 1,18-1,36 (м, 1H).
Пример 5
Получение N-[17-[3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (32)
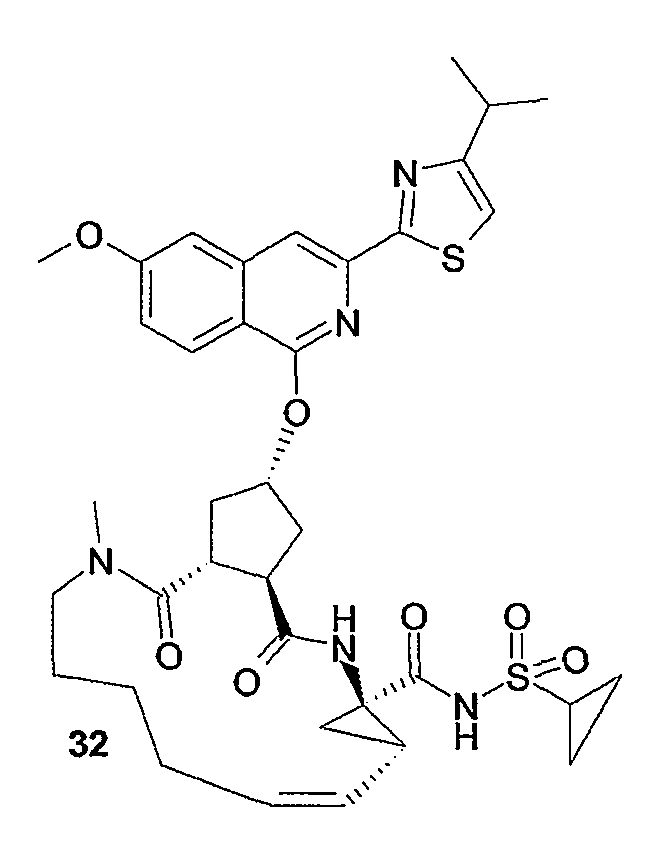
Указанный в заголовке продукт получали из 17-[3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты 31, следуя процедурам, описанным для N-[17-[3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида 30 (пример 3): m/z = 736 (M+H)+. 1Н ЯМР (СDСl3): 10,90 (с, 1H); 8,02-8,09 (м, 2Н); 7,11-7,14 (м, 2Н); 6,96 (с, 1Н); 6,29 (с, 1H); 5,78-5,83 (м, 1Н); 5,62-5,69 (м, 1Н); 5,06 (т, J=10,5 Гц, 1Н); 4,56-4,64 (м, 1Н); 3,93 (с, 3Н); 3,37-3,42 (м, 2Н); 3,15-3,21 (м, 1H); 3,04 (с, 3Н); 2,89-2,98 (м, 2Н); 2,52-2,61 (м, 2Н); 2,23-2,43 (м, 3Н); 1,64-1,93 (м, 4Н); 1,31-1,50 (м, 10Н); 1,18-1,30 (м, 2Н); 0,96-1,15 (м, 2Н).
Пример 6
Получение 17-[3-(2-изопропиламинотиазол-4-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (33)
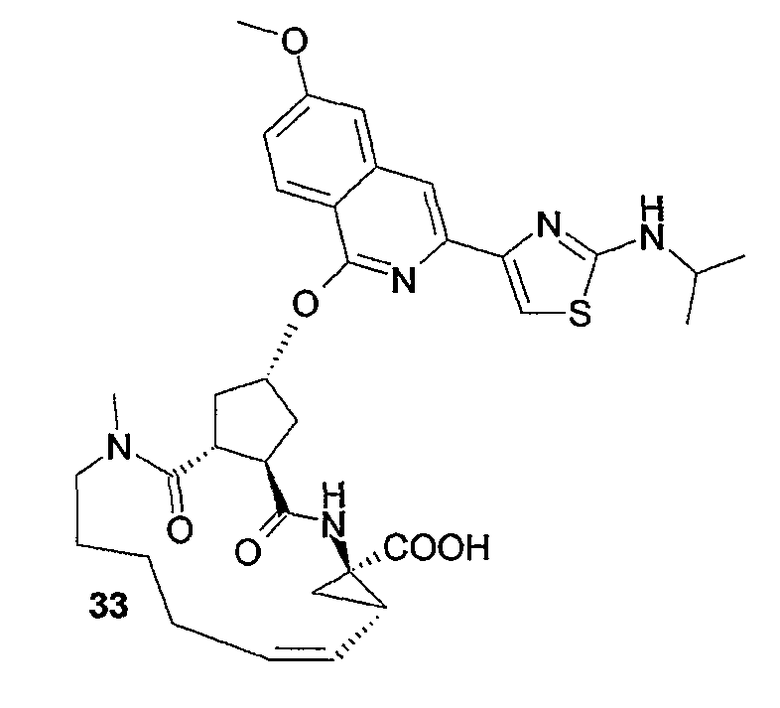
Указанный в заголовке продукт получали из 1-гидрокси-3-(2-изопропиламинотиазол-4-ил)-6-метоксиизохинолина, следуя процедурам, описанным для 17-[3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты 29 (пример 2): m/z = 648 (M+H)+. 1Н ЯМР (СDСl3): 8,03 (д, J=9,0 Гц, 1H), 7,69 (с, 1H), 7,19 (с, 1H), 7,04-7,11 (м, 3H), 5,60-5,68 (м, 2H), 5,20 (т, J=9,2 Гц, 1H), 4,54-4,61 (м, 1H), 3,93 (с, 3H), 3,54-3,70 (м, 2H), 3,12-3,20 (м, 1H), 2,83 (с, 3H), 2,35-2,60 (м, 4H), 2,11-2,30 (м, 2H), 1,80-1,93 (м, 2H), 1,69-1,79 (м, 2H), 1,40-1,51 (м, 2H), 1,30 (д, J=13,1 Гц, 6H), 1,10-1,21 (м, 2H).
Пример 7
Получение N-[17-[3-(2-изопропиламинотиазол-4-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (34)
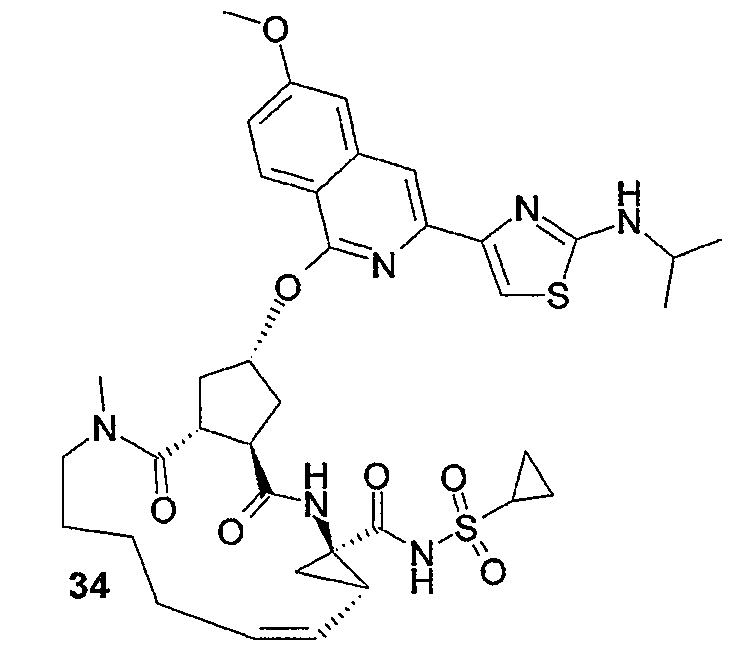
Указанный в заголовке продукт получали из 17-[3-(2-изопропиламинотиазол-4-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты 33, следуя процедурам, описанным для N-[17-[3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида 30 (пример 3): m/z = 751 (M+H)+. 1Н ЯМР (СDСl3): 10,90 (с, 1H), 8,04 (д, J= 9,3 Гц, 1H), 7,75 (с, 1H), 7,04-7,07 (м, 3H), 6,32 (с, 1H), 5,80-5,84 (м, 1H), 5,62-5,69 (м, 1H), 5,06 (т, J=10,3 Гц, 2H), 4,58-4,65 (м, 1H), 3,91 (с, 3H), 3,71-3,79 (м, 1H), 3,24-3,41 (м, 2H), 3,03 (с, 3H), 2,71-2,97 (м, 2H), 2,57-2,60 (м, 2H), 2,30-2,41 (м, 2H), 2,15-2,30 (м, 1H), 1,78-2,02 (м, 4H), 0,87-1,58 (м, 14 H).
Пример 8
Получение 17-[3-(пиразол-1-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (22)
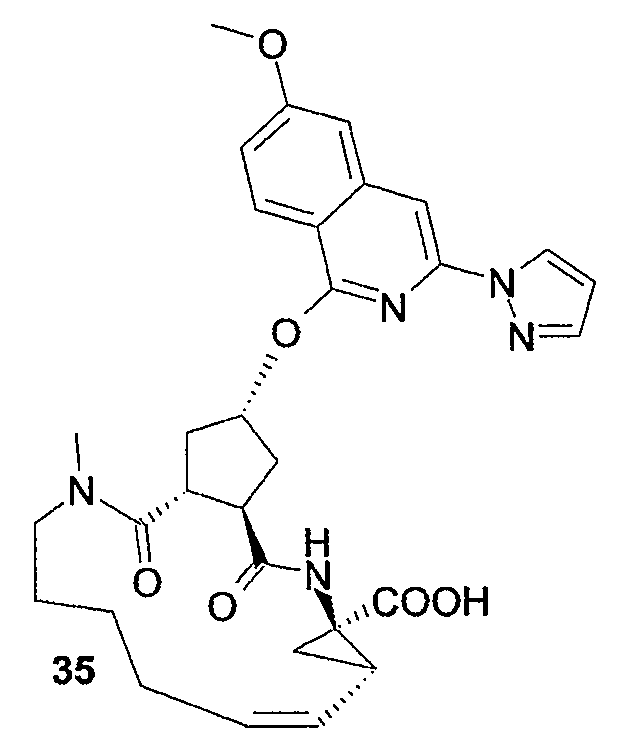
Указанный в заголовке продукт получали из 1-гидрокси-3-(пиразол-1-ил)-6-метоксиизохинолина, следуя процедурам, описанным для 17-[3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты 29 (пример 2): m/z = 574 (M+H)+. 1Н ЯМР (СDСl3): 8,45 (д, J=2,3 Гц, 1H), 8,03 (д, J=8,9 Гц, 1H), 7,71 (д, J=1,0 Гц, 1H), 7,64 (с, 1H), 7,22 (с, 1H), 7,01-7,05 (м, 2H), 6,43-6,45 (м, 1H), 5,63-5,70 (м, 2H), 5,18 (дд, J=10,3 Гц, 2,0 Гц, 1H), 4,53-4,42 (м, 1H), 3,90 (с, 3H), 3,58-3,67 (м, 1H), 3,26-3,35 (м, 1H), 3,02 (с, 3H), 2,65-2,77 (м, 1H), 2,59-2,68 (м, 1H), 2,35-2,58 (м, 2H), 2,15-2,30 (м, 2H), 1,89-2,05 (м, 2H), 1,70-1,75 (м, 2H), 1,18-1,61 (м, 4H).
Пример 9
Получение N-[17-[3-(пиразол-1-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (36)
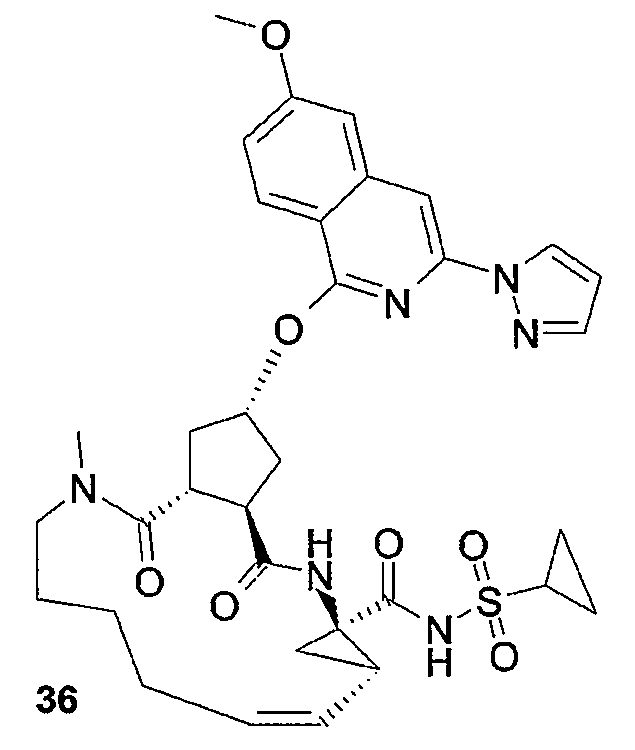
Указанный в заголовке продукт получали из 17-[3-(пиразол-1-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты 22, следуя процедурам, описанным для N-[17-[3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида 30 (пример 3): m/z = 677 (M+H)+. 1Н ЯМР (СDСl3): 8,49 (д, J=2,4 Гц, 1H), 8,06 (д, J=9,7 Гц, 1H), 7,74 (д, J=6,4 Гц, 2H), 7,04-7,08 (м, 2H), 6,46-6,48 (м, 1H), 6,37 (шир. с, 1H), 5,71-5,82 (м, 1H), 5,63-5,69 (м, 1H), 5,06 (т, J=10,5 Гц, 1H), 4,58-4,65 (м, 2H), 3,93 (с, 3H), 3,36-3,44 (м, 2H), 3,04 (с, 3H), 2,80-2,95 (м, 2H), 2,50-2,62 (м, 2H), 2,33-2,45 (м, 2H), 2,20-2,31 (м, 1H), 1,80-2,00 (м, 4H), 1,32-1,70 (м, 2H), 1,17-1,30 (м, 2H), 0,90-1,15 (м, 4H).
Пример 10
Синтез 17-(3-хлор-6-метоксиизохинолин-1-илокси)- 13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (42)
Стадия А
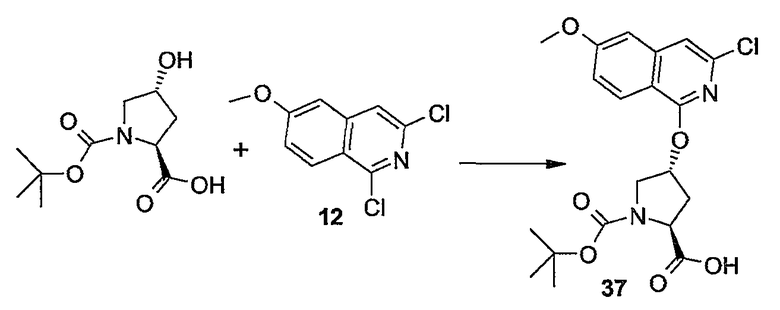
К раствору Boc-гидроксипролина (760 мг, 3,29 ммоль) в DMSO (50 мл) добавляли трет-бутилат калия (1,11 г, 9,87 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч перед добавлением 1,3-дихлор-6-метоксиизохинолина 12 (750 мг, 3,29 ммоль). Спустя 12 ч при комнатной температуре в атмосфере азота реакционную смесь гасили холодной водой со льдом, подкисляли до pH 4 раствором HCl, экстрагировали этилацетатом AcOEt, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали, получая при этом 1,39 г (90%) соединения 37 в виде белого твердого вещества: m/z = 423 (M+H)+. 1Н ЯМР (СDСl3): 8,10 (д, J=9,3 Гц, 1H), 7,15 (д, J=2,4 Гц, 1H), 7,10 (дд, J=9,3 Гц, 2,5 Гц, 1H), 6,9 (с, 1H), 5,80-5,67 (шир. с, 1H), 4,45 (т, J=7,9, 1H), 3,95 (с, 3H), 3,80-3,90 (шир. с, 1H), 3,70-3,80 (м, 1H), 2,75-2,6 (м, 1H), 2,35-2,45 (м, 1H), 1,50 (с, 9H).
Стадия В
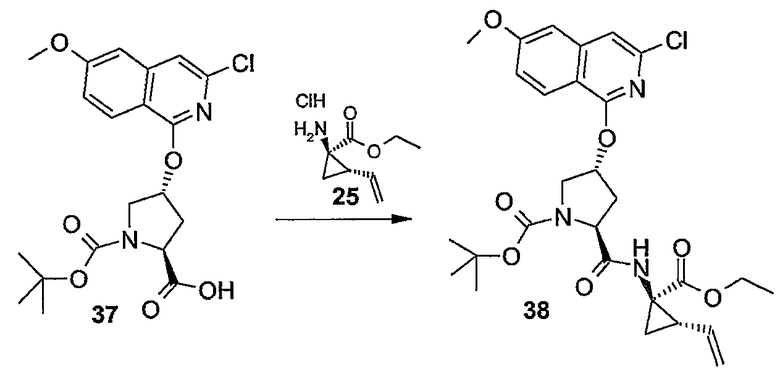
Раствор соединения 37 (1,25 г, 2,96 ммоль), гидрохлорида сложного этилового эфира 1-амино-2-винилциклопропанкарбоновой кислоты 25 (526 мг, 2,96 ммоль), HATU (1,12 г, 2,96 ммоль) и DIPEA (1,29 мл, 7,39 ммоль) в ДМФА (50 мл) перемешивали при комнатной температуре в атмосфере азота. Спустя 12 ч добавляли дихлорметан и раствор последовательно промывали водным раствором NaHCO3 и водой. Органический слой сушили (MgSO4) и упаривали. Остаток очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2/MeOH, 95:5), получая при этом 1,5 г (90%) требуемого продукта 38 в виде желтой пены: m/z = 561 (M+H)+. 1Н ЯМР (СDСl3): 8,10 (д, J=9,3 Гц, 1H), 7,50 (с, 1H), 7,25 (дд, J=9,3 Гц, 2,5 Гц, 1H), 6,98 (д, J=2,4 Гц, 1H), 5,80-5,67 (м, 1H), 5,29 (д, J=17,1 Гц, 1H), 5,12 (д, J=10,3 Гц, 1H), 4,45-4,5 (шир. с, 1H), 4,1-4,18 (м, 2H), 3,95 (с, 3H), 3,8-3,9 (шир. с, 1H), 3,7-3,8 (м, 1H), 3,25-3,35 (м, 2H), 2,35-2,45 (м, 1H), 2,1-2,2 (м, 1H), 1,5-2 (м, 6H), 1,5 (с, 9H).
Стадия С
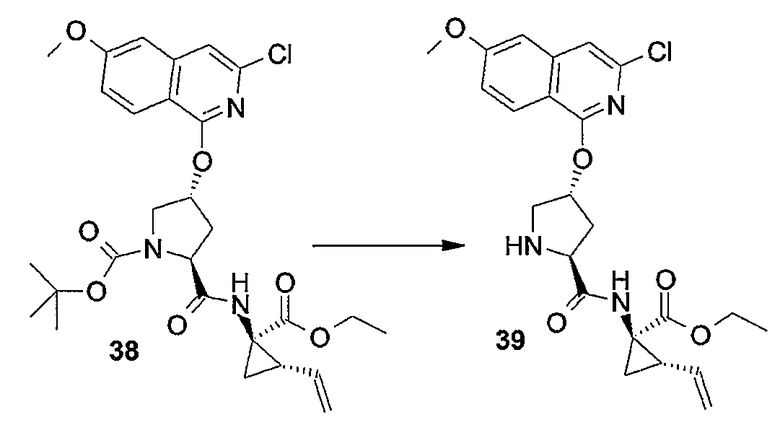
Раствор продукта 38 (3,0 г, 5,36 ммоль) в TFA-DCM 1:2 (3 мл) перемешивали при комнатной температуре в течение 60 мин. Затем добавляли толуол (3 мл) и полученную смесь упаривали досуха, получая при этом требуемый продукт 39 (степень чистоты после ВЭЖХ >95%), который применяли на следующей стадии без дополнительной очистки: m/z = 460 (M+H)+.
Стадия D
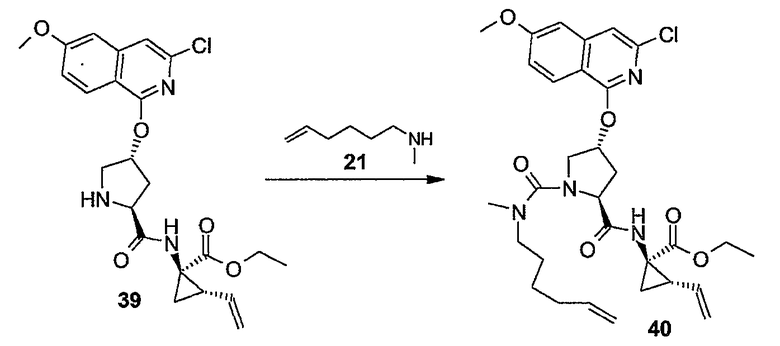
Гидрокарбонат натрия (1,83 г, 21,7 ммоль) добавляли к раствору продукта 39 (1,0 г, 2,17 ммоль) в тетрагидрофуране (25 мл). Затем добавляли фосген (1,6 мл, 1,9 M в толуоле, 4,5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем фильтровали. Растворитель упаривали и остаток растворяли в дихлорметане (25 мл). Затем добавляли гидрокарбонат натрия (1,83 г, 21,7 ммоль) с последующим добавлением (гекс-5-енил)(метил)амина 21 (1,2 г, 8,04 ммоль). После 12 ч при комнатной температуре реакционную смесь фильтровали. Фильтрат распределяли между водой и дихлорметаном. Органический слой сушили (MgSO4), фильтровали и упаривали. Остаток очищали с помощью колоночной хроматографии на диоксиде кремния (CH2Cl2/EtOAc, 95:5), получая при этом 0,80 г (69,3%) требуемого продукта 40: m/z = 600 (M+H)+. 1Н ЯМР (СDСl3): 8,10 (д, J=9,3 Гц, 1H), 7,50 (с, 1H), 7,39 (с, 1H), 7,25 (дд, J=9,3 Гц, 2,4 Гц, 1H), 6,98 (д, J=2,4 Гц, 1H), 5,81-5,62 (м, 2H), 5,56 (т, J=3,8 Гц 1H), 5,29 (дд, J=1,3 Гц, 17,2 Гц, 1H), 5,12 (дд, J=1,5 Гц, 10,4 Гц, 1H), 5,00-4,86 (м, 3H), 4,35 (т, J=7,5 Гц, 2H), 3,98 (с, 3H), 3,48-3,37 (м, 1H), 3,10-3,00 (м, 1H), 2,87 (с, 3H), 2,77-2,67 (м, 2H), 2,41-2,32 (м, 1H), 2,10 (дд, J= 8,6 Гц, 17,4 Гц, 1H), 1,98 (дд, J= 14,4 Гц, 7,1 Гц, 2H), 1,88 (дд, J=5,6 Гц, 8,1 Гц, 1H), 1,57-1,46 (м, 3H), 1,35-1,18 (м, 5H).
Стадия Е
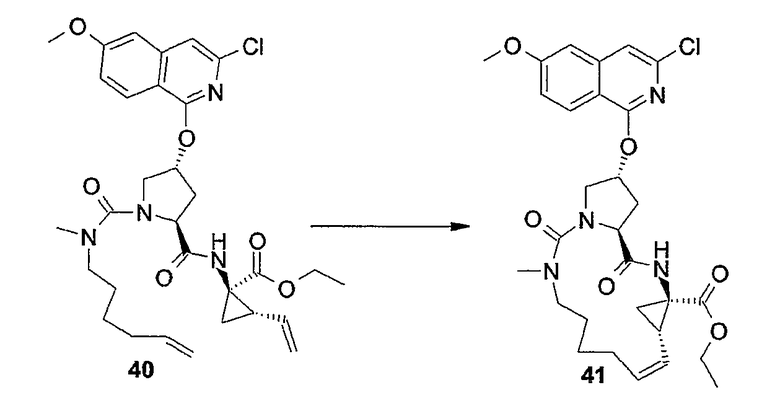
К раствору продукта 40 (1,3 г, 2,17 ммоль) в дегазированном сухом дихлорэтане (1 л) добавляли катализатор Ховейды-Граббса 1-го поколения (261 мг, 20 мол. %). Затем реакционную смесь нагревали до 70°C в течение 20 ч в атмосфере азота. Полученную смесь охлаждали до комнатной температуры и концентрировали с помощью ротационного выпаривания. Полученное масло очищали с помощью колоночной хроматографии на диоксиде кремния (CH2Cl2/EtOAc 90/10), получая при этом 720 мг (58%) указанного в заголовке продукта 41 в виде твердого вещества бежевого цвета: m/z = 572 (M+H)+. 1Н ЯМР (СDСl3): 7,95 (д, J=9,1 Гц, 1H), 7,55 (с, 1Н), 7,15 (с, 1H), 7,10 (дд, J=9,1 Гц, 2,4 Гц, 1H), 6,91 (д, J=2,4 Гц, 1H), 5,85 (шир. с, 1H), 5,65 (дд, J=18,2 Гц, 8,0 Гц, 1H), 5,15 (т, J=10,0 Гц, 1H), 4,80 (т, J=7,2 Гц, 1H), 4,19-4,28 (м, 2H), 4,05 (дд, J=3,7 Гц, J=11,3 Гц, 1H), 3,90 (с, 3H), 3,69 (д, J=11,5 Гц, 1H), 3,49-3,58 (м, 1H), 3,00-3,10 (м, 1H), 2,90 (с, 3H), 2,45-2,55 (м, 2H), 2,30-2,45 (м, 1H), 2,10-2,20 (м, 1H), 1,90-1,95 (м, 3H), 1,50-1,70 (м, 2H), 1,20-1,45 (м, 5H).
Стадия F
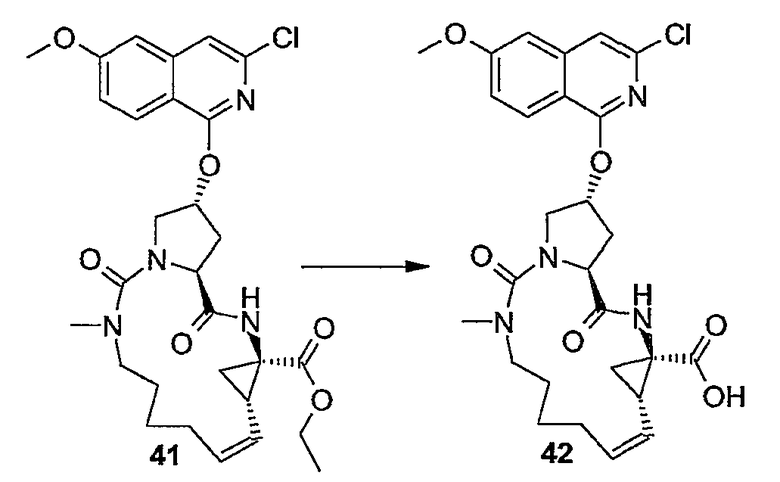
К раствору продукта 41 (100 мг, 0,18 ммоль) в тетрагидрофуране (5 мл) и метаноле (2 мл) добавляли гидроксид лития (150 мг, 3,6 ммоль) в воде (3 мл). Спустя 48 ч при комнатной температуре добавляли воду и pH полученного раствора доводили до 3 1N раствором HCl. Затем реакционную смесь экстрагировали этилацетатом, сушили (Na2SO4) и упаривали. Остаток растирали с простым диэтиловым эфиром и фильтровали, получая при этом 85 мг (89%) указанного в заголовке продукта 42 в виде белого порошка: m/z = 544 (M+H)+. 1Н ЯМР (СDСl3): 7,95 (д, J=9,1 Гц, 1H), 7,55 (с, 1H), 7,15 (с, 1H), 7,10 (дд, J=9,1 Гц, 2,4 Гц, 1H), 6,90 (д, J=2,4 Гц, 1H), 5,85 (шир. с, 1H), 5,65 (дд, J=18,2 Гц, 8,0 Гц, 1H), 5,15 (т, J=10,0 Гц, 1H), 4,80 (т, J=7,2 Гц, 1H), 4,05 (дд, J=11,3 Гц, 3,7 Гц, 1H), 3,90 (с, 3H), 3,70-3,80 (м, 1H), 3,60 (д, J=11,3 Гц, 1H), 2,85 (с, 3H), 2,80-2,85 (м, 1H), 2,25-2,50 (м, 4H), 1,95-2,00 (м, 1H), 2,90 (дд, J=8,6 Гц, 5,9 Гц, 1H), 1,55-1,60 (м, 3H), 1,30-1,50 (м, 3H).
Пример 11
Синтез N-[17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (43)
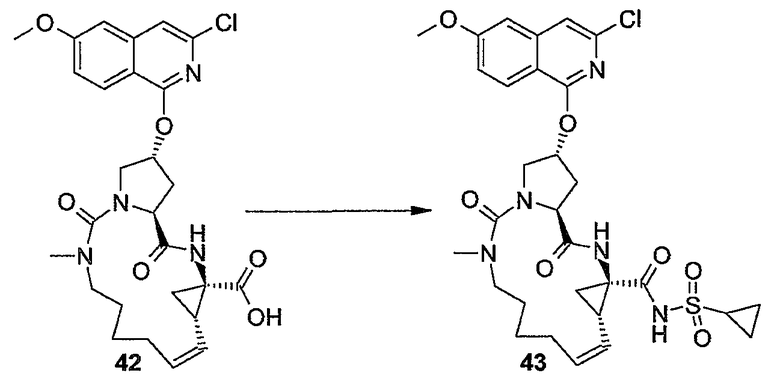
Раствор 17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (42) (80 мг, 0,147 ммоль) и карбонилдиимидазола (48 мг, 0,295 ммоль) в сухом ТГФ (25 мл) перемешивали при кипячении с обратным холодильником в атмосфере азота в течение 3 ч. Затем реакционную смесь охлаждали до комнатной температуры и добавляли циклопропилсульфонамид (54 мг, 0,442 ммоль) и DBU (52 мг, 0,34 ммоль). Полученный раствор перемешивали при 50°C в течение 48 ч. Затем реакционную смесь распределяли между этилацетатом AcOEt и водой. Органический слой сушили (MgSO4), фильтровали и упаривали. Остаток очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2/EtOAc, 95:05), получая при этом указанный в заголовке продукт, загрязненный циклопропилсульфонамидом. Полученное твердое вещество растирали в течение 10 мин с водой, фильтровали, промывали водой, сушили в высоком вакууме, снова растирали в простом диэтиловом эфире и фильтровали, получая при этом 37 мг (39%) указанного в заголовке продукта 43 в виде белого порошка: m/z = 647 (M+H)+. 1Н ЯМР (СDСl3): 10,40 (шир. с, 1H), 7,95 (д, J=9,1 Гц, 1H), 7,55 (с, 1H), 7,15 (с, 1H), 7,10 (дд, J=9,12 Гц, 2,4 Гц, 1H), 6,90 (д, J=2,4 Гц, 1H), 5,85 (шир. с, 1H), 5,65 (дд, J=18,2 Гц, 8,0 Гц, 1H), 5,15 (т, J=10,0 Гц, 1H), 4,8 (т, J=7,2 Гц, 1H), 4,10 (дд, J=11,3 Гц, 3,8 Гц, 1H), 3,9 (с, 3H), 3,60-3,70 (м, 1H), 3,6 (д, J=11,3 Гц, 1H), 3,10-3,20 (м, 1H), 2,90-3,00 (м, 1H), 2,85 (с, 3H), 2,4-2,6 (м, 3H), 1,90-2,20 (м, 3H), 1,25-1,60 (м, 7H), 0,90-1,10 (м, 2H).
Пример 12
Синтез 17-(5-бром-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (50)
Стадия A
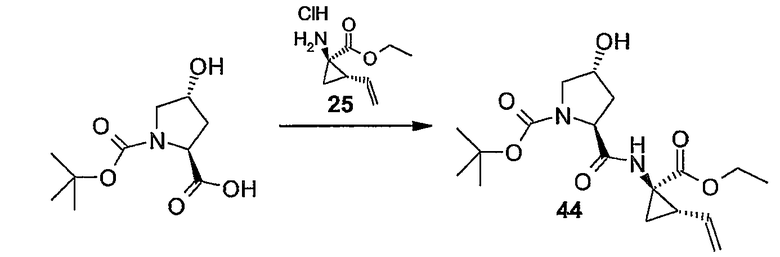
К раствору Boc-гидроксипролина (1,0 г, 4,4 ммоль), циклопропиламинокислоты 25 (825 мг, 4,3 ммоль), HATU (1,7 г, 4,48 ммоль) в ДМФА (10 мл) добавляли диизопропилэтиламин (1,9 мл, 10,9 ммоль). После 2 ч при комнатной температуре добавляли дихлорметан (200 мл). Раствор последовательно промывали насыщенным раствором NaHCO3 и воды. Органический слой сушили и концентрировали. Остаток очищали с помощью колоночной хроматографии (CH2Cl2/EtOAc, 50:50), получая при этом 1,2 г (76%) требуемого продукта 44: m/z = 369 (M+H)+. 1Н ЯМР (СDСl3): 5,80-5,67 (м, 1H), 5,32-5,24 (д, J=17,1 Гц, 1H), 5,16-5,08 (д, J=10,3 Гц, 1H), 4,61-4,45 (м, 1H), 4,45-4,29 (шир.с, 1H), 4,23-4,03 (м, 2H), 3,78-3,39 (м, 2H), 2,14-1,97 (м, 1H), 1,97-1,81 (шир. с, 1H), 1,81-1,32 (м, 12 H), 1,22 (т, J=7,1 Гц, 3H).
Стадия В
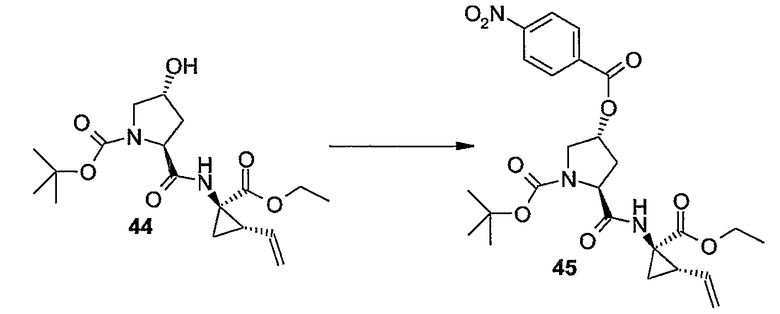
К раствору продукта 44 (5,42 г, 7,4 ммоль) в CH2Cl2 (100 мл) при 0°C добавляли триэтиламин (3,2 мл, 22 ммоль). Спустя 5 мин при 0°C добавляли по каплям раствор пара-нитробензоилхлорида (3,26 г, 18 ммоль) в CH2Cl2 (50 мл). Затем реакционной смеси давали нагреться до комнатной температуры. Спустя 20 ч раствор выливали в холодную воду со льдом, промывали насыщенным раствором соли, сушили (Na2SO4), фильтровали и упаривали. Сырой остаток очищали с помощью колоночной хроматографии (CH2Cl2/EtOAc, 90:10), получая при этом 2,15 г (56%) требуемого продукта 45: m/z = 518 (M+H)+. 1Н ЯМР (СDСl3): 8,30 (д, J=8,8 Гц, 2H), 8,16 (д, J=8,8 Гц, 2H), 5,82-5,70 (м, 1H), 5,59-5,54 (м, 1H), 5,31 (дд, J=17,2 Гц, 1,5 Гц, 1H), 5,14 (д, J=10,1 Гц, 1H), 4,56-4,40 (шир. с, 1H), 4,26-4,15 (м, 2Н), 3,80-3,67 (м, 2Н), 2,16-2,06 (м, 1Н), 1,98-1,84 (шир.с, 1H), 1,59-1,48 (шир.с, 1H), 1,48-1,38 (шир.с, 12H), 1,28-1,21 (м, 3H).
Стадия С
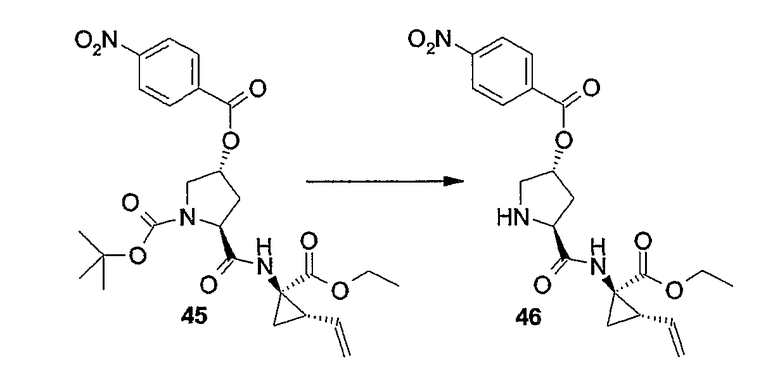
Раствор продукта 45 (2,15 г, 4,15 ммоль) в TFA-DCM 1:2 (80 мл) оставляли при комнатной температуре в течение 4 ч. Затем добавляли толуол (10 мл) и раствор упаривали досуха, получая при этом требуемое соединение 46 (степень чистоты после ВЭЖХ >95%): m/z = 418 (M+H)+.
Стадия D
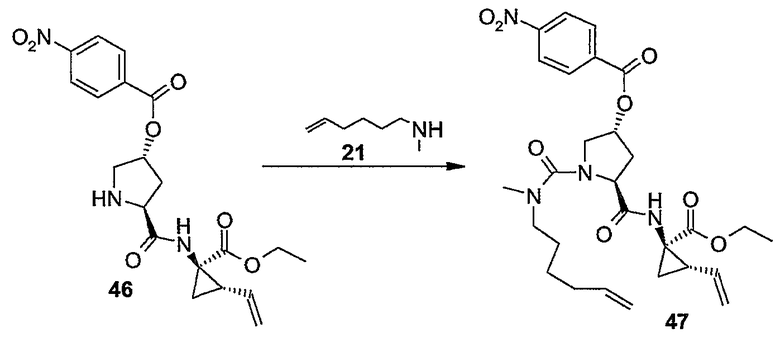
К смеси соединения 46 (1,73 г, 4,14 ммоль) и гидрокарбоната натрия (3,53 г, 42 ммоль) в ТГФ (35 мл) добавляли фосген (1,6 мл, 1,9 M в толуоле 9,28 г, 4,5 экв.). После 1,5 ч при комнатной температуре реакционную смесь фильтровали, полученный фильтрат упаривали и сырой продукт повторно растворяли в дихлорметане (35 мл). Затем добавляли гидрокарбонат натрия (3,35 г, 42 ммоль) с последующим добавлением (гекс-5-енил)(метил)амина 21 (1,1 г, 9,67 ммоль). После 12 ч при комнатной температуре реакционную смесь фильтровали. Затем добавляли воду и экстрагировали смесь дихлорметаном. Объединенные органические слои сушили (MgSO4), фильтровали и упаривали. Остаток очищали с помощью колоночной хроматографии на диоксиде кремния (CH2Cl2/EtOAc, 95:5) получая при этом 2,31 г (79%) требуемого продукта 47: m/z = 557 (M+H)+. 1Н ЯМР (СDСl3): 8,28 (д, J=8,9 Гц, 2H), 8,13 (д, J=8,9 Гц, 2H), 7,39 (с, 1Н), 5,81-5,62 (т, 2H), 5,56 (т, J=3,8 Гц, 1H), 5,29 (дд, J=17,2 Гц, 1,3 Гц, 1H), 5,12 (дд, J=10,4 Гц, 1,52 Гц, 1H), 5,00-4,86 (м, 3H), 4,20-4,06 (м, 2H), 3,79 (дд, J=12,1 Гц, 3,5 Гц, 1H), 3,57 (дд, J=12,1 Гц, 1,8 Гц, 1H), 3,48-3,37 (м, 1H), 3,10-3,00 (м, 1H), 2,87 (с, 3H), 2,77-2,67 (м, 1H), 2,41-2,32 (м, 1H), 2,10 (дд, J=8,6, 17,4 Гц, 1H), 1,98 (дд, J=14,4 Гц, 7,1 Гц, 2H), 1,88 (дд, J=8,1 Гц, 5,6 Гц, 1H), 1,57-1,46 (м, 3H), 1,35-1,18 (м, 5H).
Стадия Е
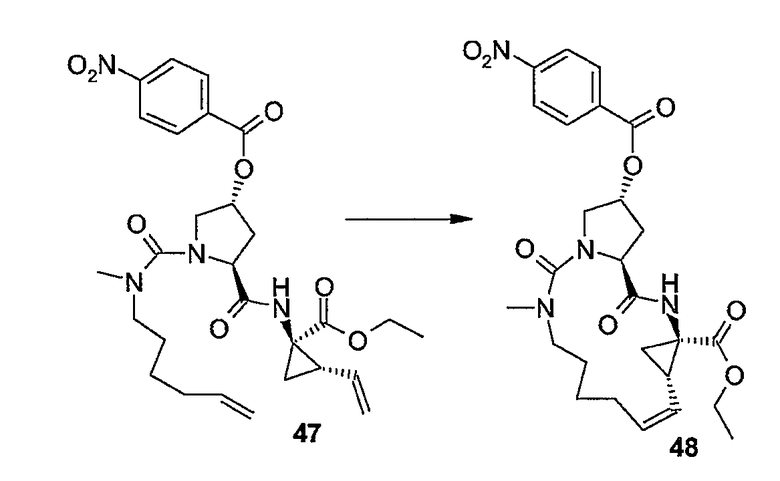
Смесь продукта 47 (1,8 г, 3,28 ммоль) и катализатора Ховейды-Граббса 1-го поколения (400 мг, 20 мол. %) в дегазированном сухом дихлорэтане (2,0 л) нагревали до 70°C в атмосфере азота в течение 20 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали с помощью ротационного выпаривания. Остаток очищали с помощью колоночной хроматографии (CH2Cl2/EtOAc, 90:10), получая при этом 888 мг (51%) требуемого соединения 48 в виде твердого вещества бежевого цвета: m/z = 529 (M+H)+. 1Н ЯМР (СDСl3): 8,28 (д, J=8,8 Гц, 2H), 8,16 (д, J=8,8 Гц, 2H), 7,47 (с, 1H), 5,76-5,67 (м, 1H), 5,62-5,57 (т, J=3,5 Гц, 1H), 5,29 (дд, J=10,5 Гц, 7,8 Гц, 1H), 4,82 (дд, J=9,8 Гц, 7,1 Гц, 1H), 4,18-4,07 (м, 2H), 4,00-3,88 (м, 2H), 3,55 (д, J=11,6 Гц, 1H), 3,07-2,97 (м, 1H), 2,91 (с, 3H), 2,64-2,54 (м, 1H), 2,48-2,29 (м, 2H), 2,16 (дд, 1H, J=17,4 Гц, 8,6 Гц, 1H), 1,96-1,83 (м, 3H), 1,80-1,61 (м, 2H), 1,45-1,25 (м, 2H), 1,22 (т, J=7,1 Гц, 3H).
Стадия F
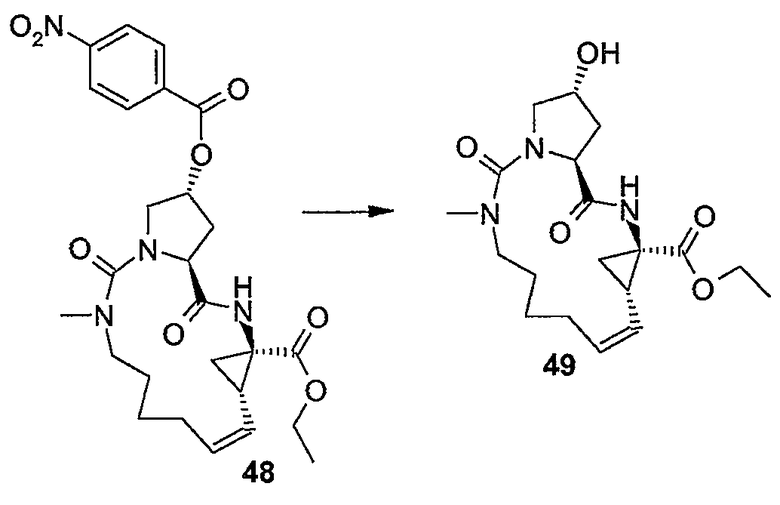
К раствору соединения 48 (451 мг, 853 ммоль) в ТГФ (25 мл) при 0°C добавляли раствор гидроксида лития (71 мг, 1,66 ммоль) в воде (5 мл). После 3 ч при 0°C реакционную смесь разбавляли водой (25 мл), затем подкисляли до pH 3 1N раствором HCl. Полученный раствор экстрагировали этилацетатом AcOEt, сушили (MgSO4), фильтровали и упаривали. Остаток очищали с помощью колоночной хроматографии (CH2Cl2/MeOH, 90:10), получая при этом 234 мг (72%) продукта 49: 1Н ЯМР (СDСl3): 8,18 (с, 1H), 7,66 (с, 1H), 5,69 (дд, J=18,0 Гц, 7,6 Гц, 1H), 5,37 (т, J=9,6 Гц, 1H), 4,68 (дд, J=9,6 Гц, 7,6 Гц, 1H), 4,78-4,11 (шир.с, 1Н)), 4,18-3,91 (м, 2H), 3,79-3,61 (м, 2H), 3,34 (д, J=11,1 Гц, 1H), 3,19-3,06 (м, 1H), 2,85 (с, 3H), 2,34-2,09 (м, 4H), 2,00-1,89 (м, 2H), 1,73 (дд, J=8,8 Гц, 5,6 Гц, 1H), 1,69-1,52 (м, 2H), 1,40-1,27 (м, 2H), 1,20 (т, J=7,1 Гц, 3H).
Стадия G
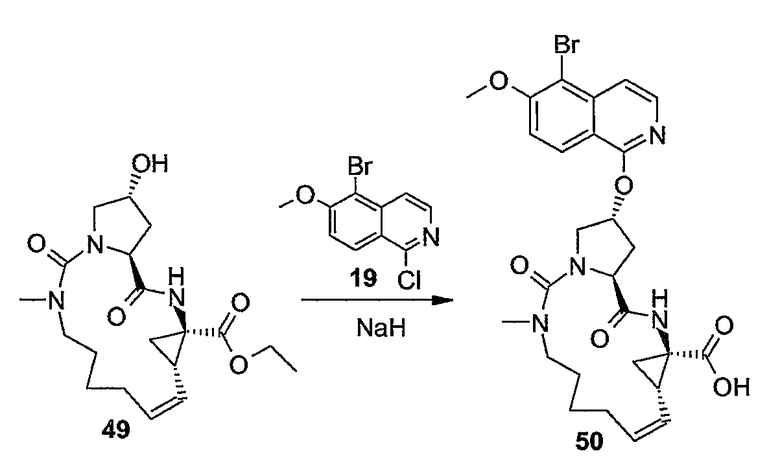
К раствору продукта 49 (260 мг, 0,683 ммоль) в ДМФА (8 мл) при 0°C добавляли небольшими порциями гидрид натрия (68,25 мг, 1,7 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч. Затем в атмосфере азота добавляли изохинолин 19 (241 мг, 0,887 ммоль) в виде одной порции. Смеси давали нагреться до комнатной температуры. Спустя 20 ч реакционную смесь выливали в холодную воду со льдом (20 мл) и экстрагировали CH2Cl2, сушили (Na2SO4), фильтровали и упаривали. При очистке с помощью колоночной хроматографии (CH2Cl2/MeOH, 96/4), с последующим гидролизом сложного эфира, как описано ранее, получали 159 мг (40%) указанного в заголовке продукта 50 в виде белого порошка: m/z = 588 (M+H)+. 1Н ЯМР (СDСl3): 8,13 (д, J=9,1 Гц, 1H), 7,97 (д, J=6,2 Гц, 1H), 7,54 (д, J=6,2 Гц, 1H), 7,39-7,30 (шир.с, 1H), 7,22 (д, J=9,2 Гц, 1H), 5,90-5,83 (шир.с, 1H), 5,71 (дд, J=17,9 Гц, 8,1 Гц, 1H), 5,18 (т, J=10,1 Гц, 1H), 4,79 (дд, J=9,1 Гц, 7,3 Гц, 1H), 4,10-3,97 (м, 4H), 3,81-3,66 (м, 1H), 3,62 (д, 1H, J=11,6 Гц, 1H), 3,19-3,05 (м, 1H), 2,85 (с, 3H), 2,59-2,22 (м, 4H), 2,01-1,90 (м, 1H), 1,89 (дд, J=8,6 Гц, 5,8 Гц, 1H), 1,70 (дд, J=9,8 Гц, 6,1 Гц, 1H), 1,67-1,58 (м, 2H), 1,43-1,28 (м, 2H).
Пример 13
Синтез N-[17-(5-бром-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (51)
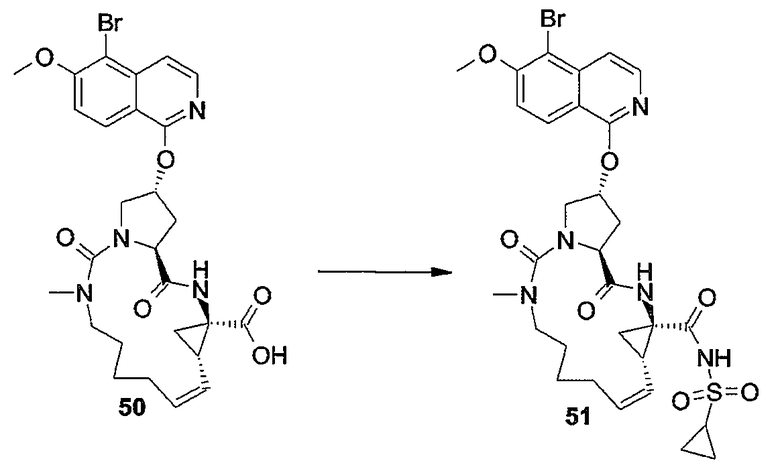
Раствор продукта 50 (151 мг, 0,257 ммоль) и карбонилдиимидазола (162 мг, 0,437 ммоль) в сухом ТГФ (10 мл) при перемешивании кипятили с обратным холодильником в атмосфере азота в течение 2 ч. Если требуется, производное азалактона при желании можно выделить. Затем реакционную смесь охлаждали до комнатной температуры и добавляли циклопропилсульфонамид (58 мг, 0,482 ммоль) и DBU (76 мг, 0,502 ммоль). Полученный раствор перемешивали при 50°C в течение 12 ч, затем охлаждали до комнатной температуры. Реакционную смесь гасили водой и экстрагировали этилацетатом AcOEt, сушили (MgSO4), фильтровали и упаривали. Сырой материал очищали с помощью колоночной хроматографии (CH2Cl2/EtOAc, 95:5). Полученное твердое вещество растирали с водой, фильтровали, сушили в высоком вакууме, растирали с простым диэтиловым эфиром и снова сушили в высоком вакууме, получая при этом 138 мг (77%) указанного в заголовке продукта 51 в виде белого порошка: m/z = 691 (M+H)+. 1Н ЯМР (СDСl3): 10,70 (шир. с, 1H), 8,12 (д, J=9,1 Гц, 1H), 7,97 (д, J=6,3 Гц, 1H), 7,54 (д, J=6,3 Гц, 1H), 7,21 (д, J=9,1 Гц, 1H), 6,70 (шир.с, 1H), 5,88 (шир.с, 1H), 5,74 (дд, J=17,3 Гц, 8,3 Гц, 1H), 5,16 (т, J=10,4 Гц, 1H), 4,74 (дд, J=9,4 Гц, 7,3 Гц, 1H), 4,11-3,98 (м, 4H), 3,69-3,55 (м, 2Н), 3,27-3,10 (м, 1Н), 3,02-2,89 (м, 1H), 2,83 (с, 3H), 2,58-2,35 (м, 3H), 2,29-2,13 (м, 1H), 2,11-1,92 (м, 2H), 1,75-0,76 (м, 9H).
Пример 14
Синтез N-[17-(5-(4-метил-3-пиридил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (52)

Раствор продукта 51 (17,3 мг, 0,025 ммоль), 6-метилпиридин-3-бороновой кислоты (5,9 мг, 0,028 ммоль), тетракис-трифенилфосфинпалладия (8,2 мг, 0,005 ммоль) и карбоната натрия (5,8 мг, 0,055 ммоль) в ДМФА (2 мл) нагревали до 90°C в течение 20 ч. Затем реакционную смесь охлаждали до комнатной температуры и выпаривали растворитель. Остаток очищали с помощью ВЭЖХ, получая при этом 3,7 мг (21%) указанного в заголовке продукта 52 в виде белого порошка, m/z = 703 (M+H)+. 1Н ЯМР (СDСl3): 10,6 (шир.с, 1H), 8,8 (с, 1H), 8,12 (д, J=9,1 Гц, 1H), 7,97 (д, J=6,3 Гц, 1H), 7,9 (д, J=9,0 Гц, 1H), 7,54 (д, J=6,3 Гц, 1H), 7,3 (д, J=9,0 Гц, 1H), 7,21 (д, J=9,1 Гц, 1H), 6,68 (шир. с, 1H), 5,87 (шир. с, 1H), 5,74 (дд, J=17,3 Гц, 8,3 Гц, 1H), 5,16 (т, J=10,4 Гц, 1H), 4,74 (дд, J=9,4 Гц, 7,3 Гц, 1H), 4,11-3,98 (м, 4H), 3,69-3,55 (м, 2H), 3,27-3,10 (м, 1H), 3,02-2,89 (м, 1H), 2,83 (с, 3H), 2,58-2,35 (м, 3H), 2,50 (с, 3H), 2,29-2,13 (м, 1H), 2,11-1,92 (м, 2H), 0,75-1,76 (м, 9H).
Пример 15
Синтез N-[17-(5-(4-метоксифенил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (53)
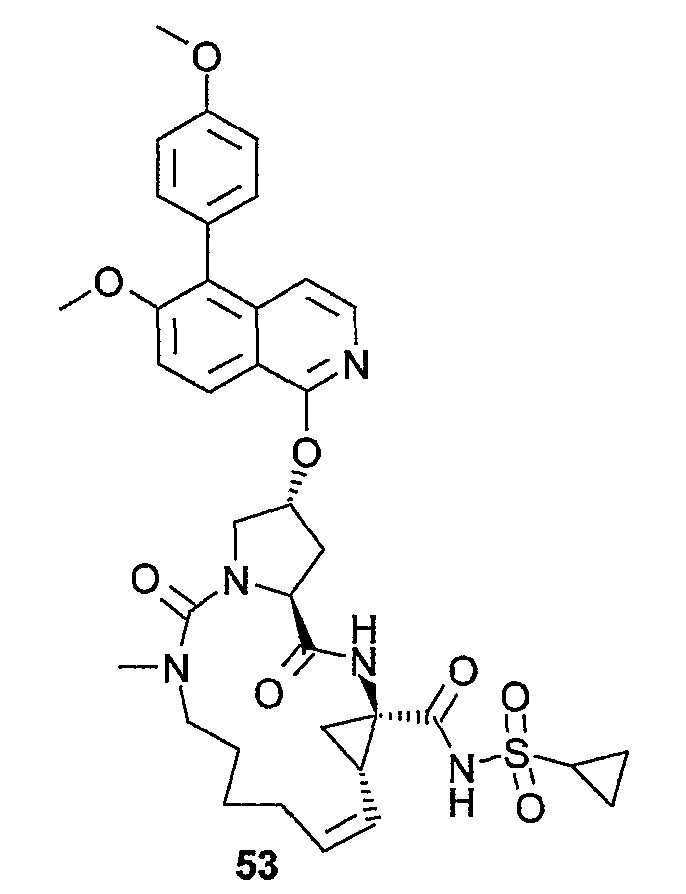
Указанный в заголовке продукт получали из N-[17-(5-бром-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (51, пример 13) и 4-метоксибензолбороновой кислоты, следуя процедуре, описанной для N-[17-(5-(4-метил-3-пиридил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (52, Пример 14); m/z = 718 (M+H)+.
Пример 16
Синтез N-[17-[5-фенил-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (54)
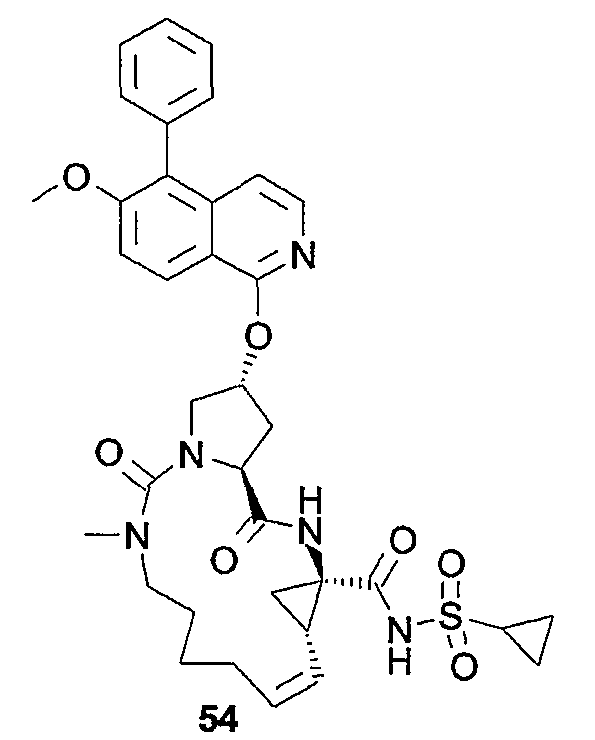
Указанный в заголовке продукт получали из N-[17-(5-бром-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (51, пример 13) и бензолбороновой кислоты, следуя процедуре, описанной для N-[17-[5-(4-метил-3-пиридил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (52, пример 14): m/z = 688 (M+H)+.
Пример 17
Синтез 17-(6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (55)
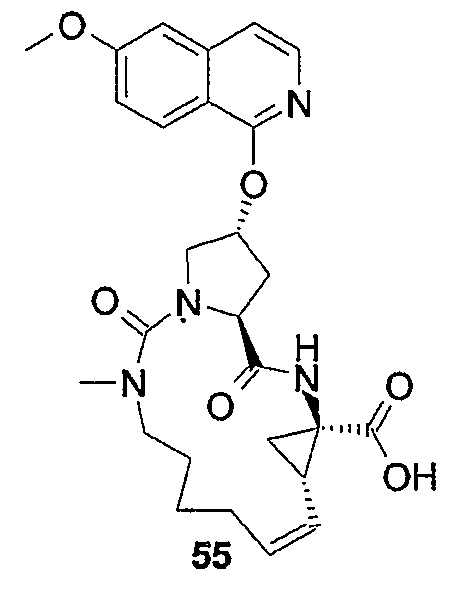
Указанный в заголовке продукт 55 получали из 1-хлор-6-метоксиизохинолина 10, следуя таким же процедурам, которые описаны для получения 17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (42, пример 10): m/z = 509 (M+H)+. 1Н ЯМР (СDСl3): 7,98 (д, J=9,2 Гц, 1H), 7,9 (д, J=6,1 Гц, 1H), 7,2 (с, 1H), 7,1 (дд, J=9,2 Гц, 2,4 Гц, 1H), 7,10 (д, J=6,1 Гц, 1Н), 6,90 (д, J=2,4 Гц, 1H), 5,85 (шир. с, 1H), 5,65 (дд, J=18,2 Гц, 8,0 Гц, 1H), 5,15 (т, J=10,0 Гц, 1H), 4,80 (т, J=7,2 Гц, 1H), 4,05 (дд, J=11,3 Гц, 3,7 Гц, 1H), 3,90 (с, 3H), 3,70-3,80 (м, IB), 3,60 (д, J=11,3 Гц, 1H), 2,85 (с, 3H), 2,80-2,85 (м, 1H), 2,25-2,50 (м, 4H), 1,95-2,00 (м, 1H), 2,90 (дд, J=8,6 Гц, 5,9 Гц, 1H), 1,55-1,60 (м, 3H), 1,30-1,50 (м, 3H).
Пример 18
Синтез N-[17-(3-фенил-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (56)
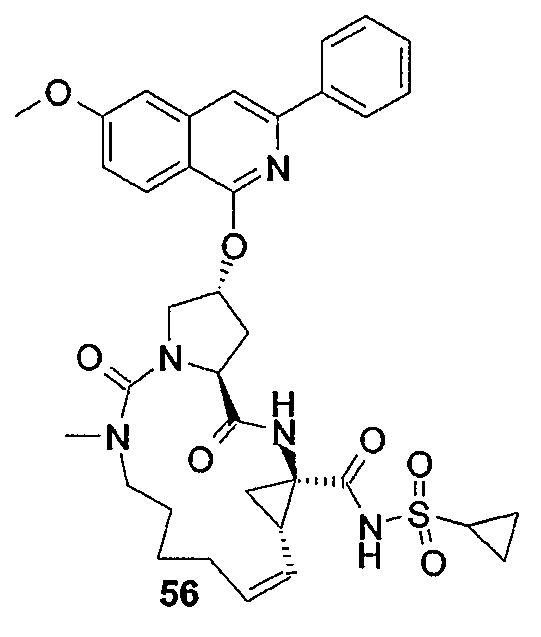
Указанный в заголовке продукт 56 получали из 17-(6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (55), следуя таким же процедурам, которые описаны для получения N-[17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамид (43, пример 11): m/z = 688.
Пример 19
Синтез 17-(3-(4-трифторметоксифенил)-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (57)
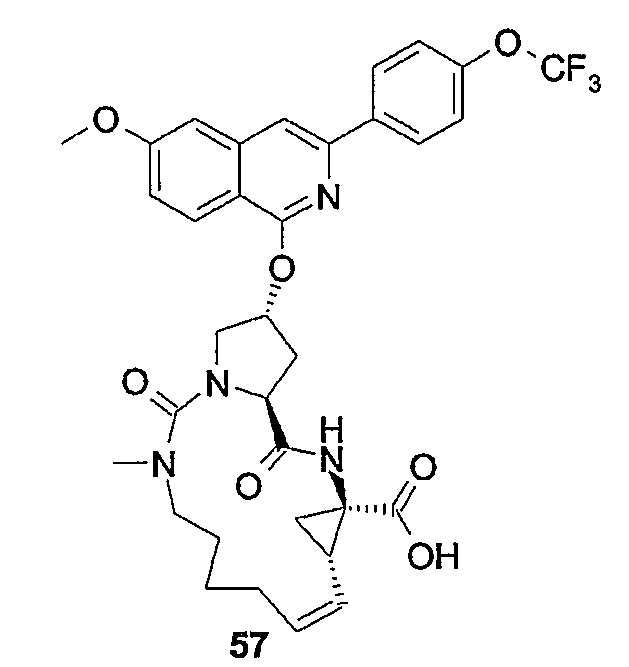
Указанный в заголовке продукт 57 получали из 1-хлор-3-[4-(трифторметил)фенил]-6-метоксиизохинолина, следуя таким же процедурам, которые описаны для получения 17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (42, пример 10): m/z = 669 (M+H)+. 1Н ЯМР (СDСl3): 8,08 (д, J=8,4 Гц, 2H), 8,02 (д, J=9,1 Гц, 1H), 7,55 (с, 1H), 7,30 (д, J=8,4 Гц, 2H), 7,11 (дд, J=9,1 Гц, 1,5, 1H), 7,05 (д, J=1,5 Гц, 1H), 6,07-5,95 (шир.с, 1H), 5,71 (дд, J=8,8 Гц, J=17,4 Гц, 1H), 5,24-5,09 (м, 1H), 4,84-4,79 (м, 1H), 4,14-4,03 (м, 1H), 3,92 (с, 3H), 3,77-3,58 (м, 3H), 3,20-3,07 (м, 1H), 2,86 (с, 3H), 2,63-2,38 (м, 3H), 2,38-2,22 (м, 1H), 2,01-1,84 (м, 2H), 1,74-1,38 (м, 5H).
Пример 20
Синтез N-[17-(3-(4-трифторметоксифенил)-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (58)
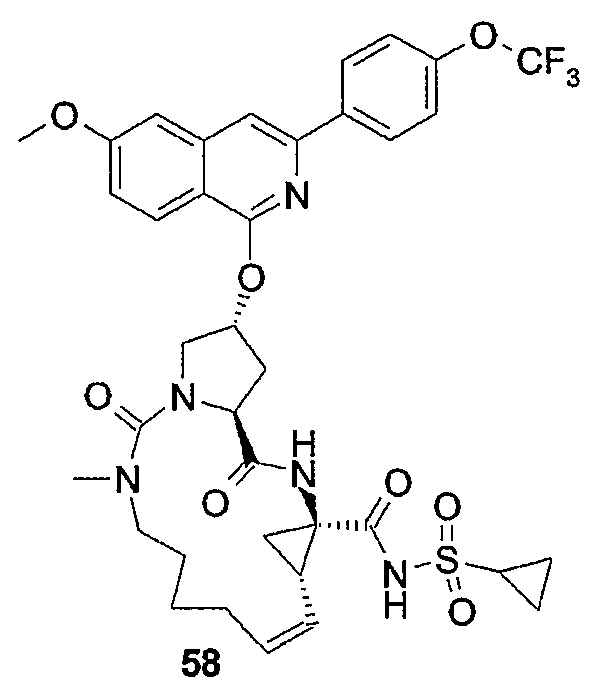
Указанный в заголовке продукт 58 получали из 17-(3-(4-трифторметоксифенил)-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (57), следуя таким же процедурам, которые описаны для получения N-[17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (43, пример 11): m/z = 772 (M+H)+. 1Н ЯМР (СDСl3): 10,63-10,57 (шир. с, 1H), 8,00 (д, J=8,5 Гц, 2H), 7,94 (д, J=9,0 Гц, 1H), 7,49 (с, 1H), 7,26 (д, J=8,5 Гц, 2H), 7,01 (дд, J=9,0 Гц, 2,4, 1H), 6,98 (д, J=2,4 Гц, 1H), 6,79-6,72 (шир.с, 1H), 5,98-5,92 (м, 1Н), 5,67 (дд, J=7,8 Гц, J=18,9 Гц, 1H), 5,09 (т, J=10,4 Гц, 1H), 4,71 (т, J=8,1 Гц, 1H), 4,03 (дд, J=11,0 Гц, 4,0, 1H), 3,85 (с, 3H), 3,64 (д, J=11,0 Гц, 1H), 3,61-3,53 (м, 1H), 3,15-3,03 (м, 1H), 2,93-2,82 (м, 1H), 2,77 (с, 3H), 2,54-2,38 (м, 3H), 2,25-2,08 (м, 1H), 2,04-1,87 (м, 2H), 1,66-0,86 (м, 9H).
Пример 21
Синтез 17-(4-бром-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (65)
Стадия А
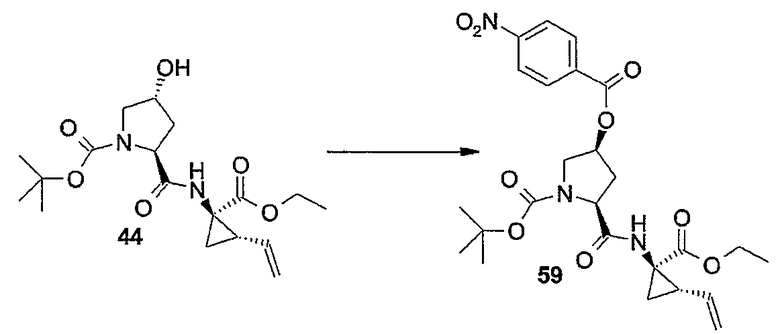
К раствору продукта 44 (10 г, 27 ммоль), 4-нитробензойной кислоты (6,8 г, 41 ммоль) и трифенилфосфина (11 г, 41 ммоль) в сухом ТГФ (200 мл) при 0°C в атмосфере азота добавляли DIAD (8,2 г, 41 ммоль). Затем реакционную смесь нагревали до комнатной температуры. Спустя 12 ч растворитель выпаривали и сырой продукт очищали с помощью флэш-хроматографии на колонке (градиент EtOAc/CH2Cl2 от 95/5 до75/25), получая при этом 8,1 г (58%) требуемого продукта, m/z = 518 (M+H)+. 1Н ЯМР (СDСl3): 8,20 (с, 4H), 5,65-5,80 (м, 1H), 5,55 (шир. с, 1H), 5,2 (дд, J=17,0 Гц, 10,2 Гц, 1H), 4,4-4,5 (м, 1H), 3,9-4,1 (м, 2H), 3,75-3,85 (м, 1H), 3,6-3,7 (м, 1H), 2,0-2,1 (м, 1H), 1,80-1,90 (м, 1H), 1,50-1,70 (м, 5), 1,50 (с, 9H), 1,10 (т, J=7,1 Гц, 3H).
Стадия В
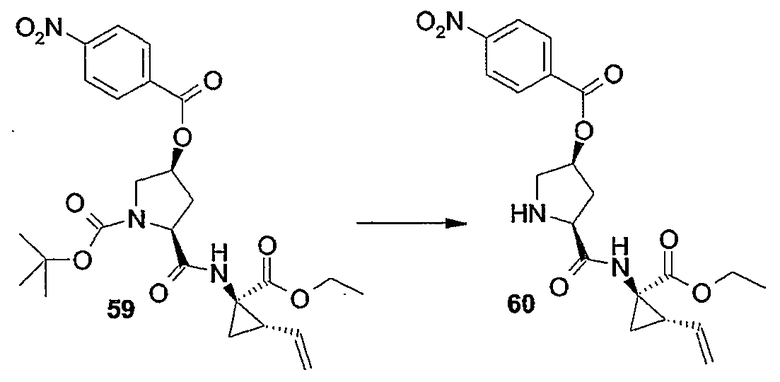
Раствор продукта 59 (6,89 г, 13,3 ммоль) в TFA-DCM 1:4 (250 мл) оставляли при комнатной температуре в течение 4 ч. Затем добавляли толуол (30 мл) и раствор упаривали досуха, получая при этом требуемое соединение 60 (степень чистоты после ВЭЖХ >97%): m/z = 418 (M+H)+.
Стадия C
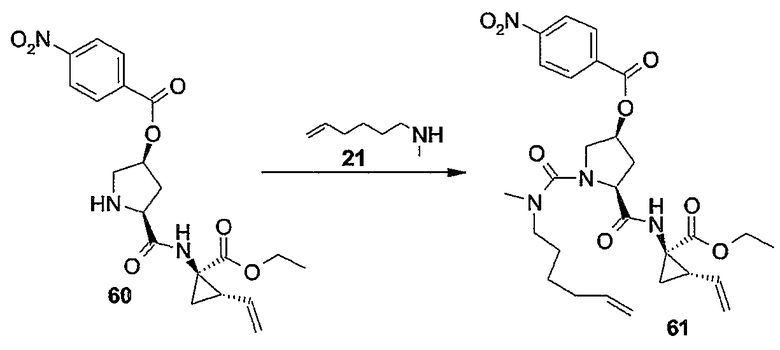
К смеси соединения 60 (5,56 г, 13,3 ммоль) и гидрокарбоната натрия (11,5 г, 137 ммоль) в ТГФ (120 мл) добавляли фосген (1,6 мл, 1,9 M в толуоле, 4,5 экв.). После 1,5 ч при комнатной температуре реакционную смесь фильтровали, полученный фильтрат упаривали и сырой остаток повторно растворяли в дихлорметане (35 мл). Затем добавляли гидрокарбонат натрия (11,55 г, 137 ммоль) с последующим добавлением (гекс-5-енил)(метил)амина 21 (2,65 г, 23,4 ммоль). После 12 ч при комнатной температуре реакционную смесь фильтровали. Затем добавляли воду и смесь экстрагировали дихлорметаном. Объединенные органические слои сушили (MgSO4), фильтровали и упаривали. Остаток очищали с помощью колоночной хроматографии на диоксиде кремния (CH2Cl2/EtOAc, 95:5), получая при этом 7,41 г (58%) требуемого продукта 61: m/z = 557 (M+H)+. 1Н ЯМР (СDСl3): 8,28 (д, J=8,9 Гц, 2H), 8,13 (д, J=8,9 Гц, 2H), 7,39 (с, 1H), 5,81-5,62 (м, 2H), 5,56 (т, J=3,8 Гц, 1H), 5,29 (дд, J=17,2 Гц, 1,3 Гц, 1H), 5,12 (дд, J=10,4 Гц, 1,52 Гц, 1H), 5,00-4,86 (м, 3H), 4,20-4,06 (м, 2H), 3,79 (дд, J =12,1 Гц, 3,5 Гц, 1H), 3,57 (дд, J=12,1 Гц, 1,8 Гц, 1H), 3,48-3,37 (м, 1H), 3,10-3,00 (м, 1H), 2,87 (с, 3H), 2,77-2,67 (м, 1H), 2,41-2,32 (м, 1H), 2,10 (дд, J=8,6, 17,4 Гц, 1H), 1,98 (дд, J=14,4 Гц, 7,1 Гц, 2H), 1,88 (дд, J=8,1 Гц, 5,6 Гц, 1H), 1,57-1,46 (м, 3H), 1,35-1,18 (м, 5H).
Стадия D
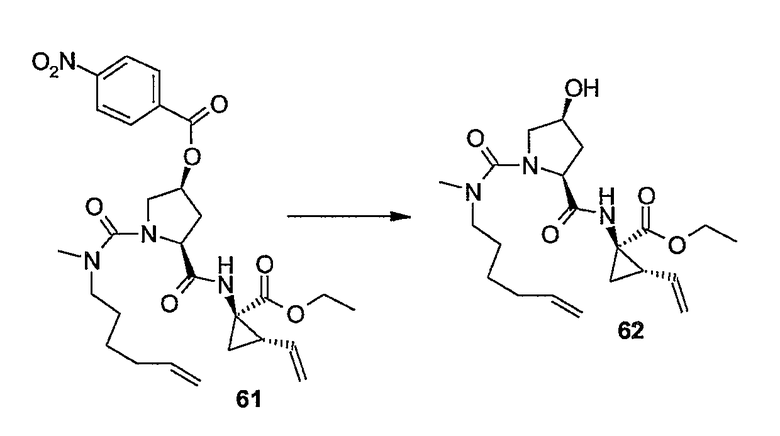
К раствору продукта 61 (4,34 г, 6,39 ммоль) в ТГФ (180 мл) при 0°C добавляли раствор гидроксида лития (632 мг, 14,8 ммоль) в воде (40 мл). После 2 ч при 0°C реакционную смесь разбавляли водой (25 мл), затем подкисляли до pH 6 1N раствором HCl. Полученный раствор экстрагировали этилацетатом AcOEt, сушили (Na2SO4), фильтровали и упаривали. Остаток очищали с помощью колоночной хроматографии (CH2Cl2/MeOH, 96:04) получая при этом 2,1 г (80%) продукта 62: m/z = 408 (M+H)+. 1Н ЯМР (СDСl3): 5,84-5,68 (м, 2H), 5,29 (дд, J=17,2 Гц, 1,3 Гц, 1H), 5,12 (дд, J=10,4 Гц, 1,52,1H), 5,05-4,93 (м, 2H), 4,78 (дд, J=9,1 Гц, 1,77, 1H), 4,60 (д, J=9,1, 1H), 4,46-4,37 (м, 1H), 4,24-4,05 (м, 2H), 3,66 (д, J=10,4 Гц, 1H), 3,43 (дд, J=10,4 Гц, 4,55, 1H), 3,37-3,26 (м, 1H), 3,17-3,07 (м, 1H), 2,88 (с, 3H), 2,29-2,02 (м, 5H), 1,87 (дд, J=8,3 Гц, 5,6, 1H), 1,67-1,52 (м, 3H), 1,49 (дд, J=9,8 Гц, 5,31, 1H), 1,44-1,38 (м, 2H), 1,22 (т, J=7,1 Гц, 3H).
Стадия Е
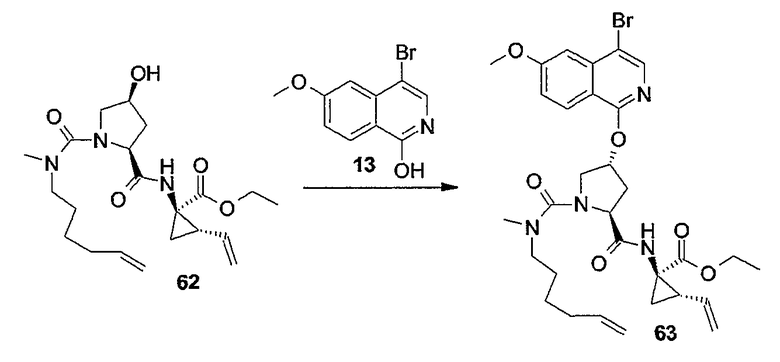
К раствору продукта 62 (900 мг, 2,208 ммоль), изохинолина 13 (673 мг, 2,65 ммоль) и трифенилфосфина (810 мг, 3,1 ммоль) в сухом ТГФ (50 мл) при -25°C в атмосфере азота добавляли DIAD (669 мг, 3,31 ммоль). Затем реакционную смесь оставляли при теспературе от -10 до -15°C в течение 3 ч. Смесь выливали в холодную воду со льдом и экстрагировали этилацетатом. Объединенные органические слои сушили (MgSO4), фильтровали и упаривали. Остаток очищали с помощью флэш-хроматографии на колонке (градиент EtOAc/CH2Cl2, 90/10), получая при этом 1 г требуемого продукта 63: m/z = 644 (M+H)+.
Стадия F

Смесь продукта 63 (1 г, 1,55 ммоль) и катализатора Ховейды-Граббса 1-го поколения (186 мг, 310 ммоль) в дегазированном сухом дихлорэтане (1,0 л) нагревали до 70°C в атмосфере азота в течение 20 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали с помощью ротационного выпаривания. Остаток очищали с помощью колоночной хроматографии (CH2Cl2/EtOAc, 90:10), получая при этом 360 мг (38%) требуемого соединения 64 в виде твердого вещества бежевого цвета: m/z = 616 (M+H)+.
Стадия G
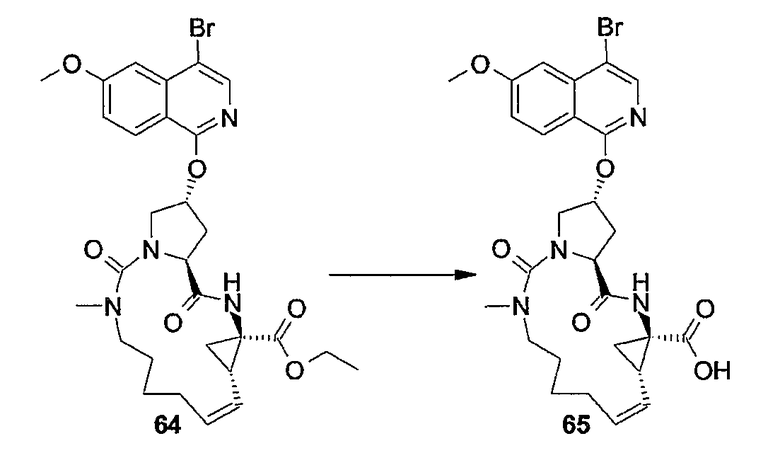
К раствору соединения 64 (360 мг, 0,585 ммоль) в тетрагидрофуране (15 мл) и метаноле (5 мл) добавляли гидроксид лития (375 мг, 8,77 ммоль) в воде (3 мл). После 48 ч при комнатной температуре добавляли воду и pH полученного раствора доводили до 3 1N раствором HCl. Затем реакционную смесь экстрагировали с этилацетатом EtOA, сушили (Na2SO4) и упаривали. Остаток растирали с простым диэтиловым эфиром и фильтровали, получая при этом 300 мг (87%) указанного в заголовке продукта 65 в виде белого порошка: m/z = 588 (M+H)+. 1Н ЯМР (СDСl3): 8,5 (с, 1H), 8,2 (д, J=9,1 Гц, 1H), 7,35 (шир. с, 1H), 7,3 (д, J=2,5 Гц, 1H), 7,17 (дд, J=9,1 Гц, 2,5 Гц, 1H), 5,8-5,85 (шир. с, 1H), 5,7 (дд, J=18,3 Гц, 7,8 Гц, 1H), 5,15 (т, J=10,0 Гц, 1H), 4,80 (дд, J=9,2 Гц, 7, 1H), 4,05 (дд, J=11,2 Гц, 4 Гц, 1H), 3,95 (с, 3H), 3,70-3,80 (м, 1H), 3,60 (д, J=11,2 Гц, 1H), 3-3,1 (м, 1H), 2,85 (с, 3H), 2,40-2,50 (м, 3H), 2,25-2,40 (м, 1H), 1,85-1,95 (м, 3H), 1,6-1,7 (м, 4H).
Пример 22
Синтез N-[17-(4-бром-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (66)
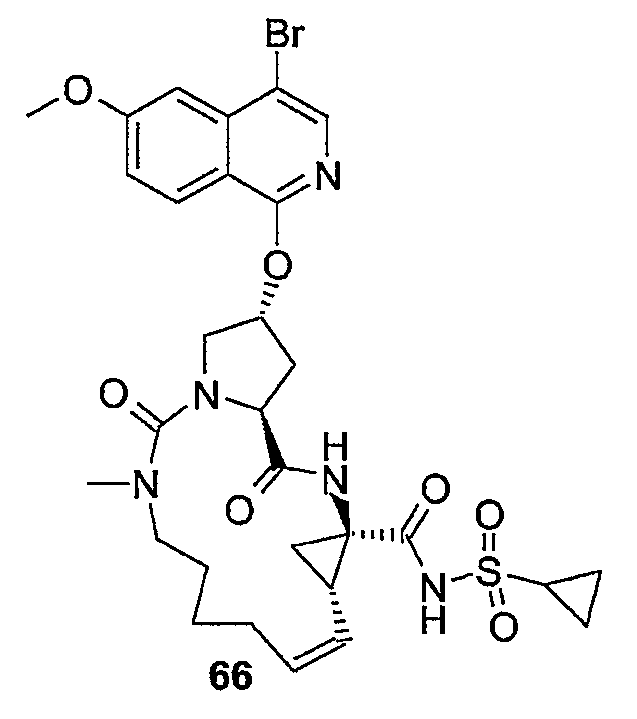
Указанный в заголовке продукт 66 получали из 17-(4-бром-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (65), следуя таким же процедурам, которые описаны для получения N-[17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (43, пример 11): m/z = 691 (M+H)+. 1Н ЯМР (СDСl3): 10,7 (шир. с, 1H), 8,09 (д, J=9,1 Гц, 1H), 7,3 (д, J=2,4 Гц, 1H), 7,25 (с, 1H), 7,15 (дд, J=9,1 Гц, 2,4, 1H), 7 (шир. с, 1H), 5,8 (шир. с, 1H), 5,74 (дд, J=18,2 Гц, 8,1H), 5,16 (т, J=10,3 Гц, 1H), 4,74 (дд, J=9,3 Гц, 7, 1H), 4,05 (дд, J=11,1 Гц, 4,1H), 3,95 (с, 3H), 3,6 (д, J=11,1 Гц, 1H), 3,1-3,2 (м, 1H), 2,9-3 (м, 1H), 2,83 (с, 3H), 2,4-2,5 (м, 3H), 2,19-2 (м, 2H), 2,5-2,7 (м, 4H), 1,4-1 (м, 3H), 1,2-1,35 (м, 2H), 1,05-1,15 (м, 1H), 0,95-1 (м, 1H).
Пример 23
Синтез 17-(3-пиразол-1-ил-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (67)
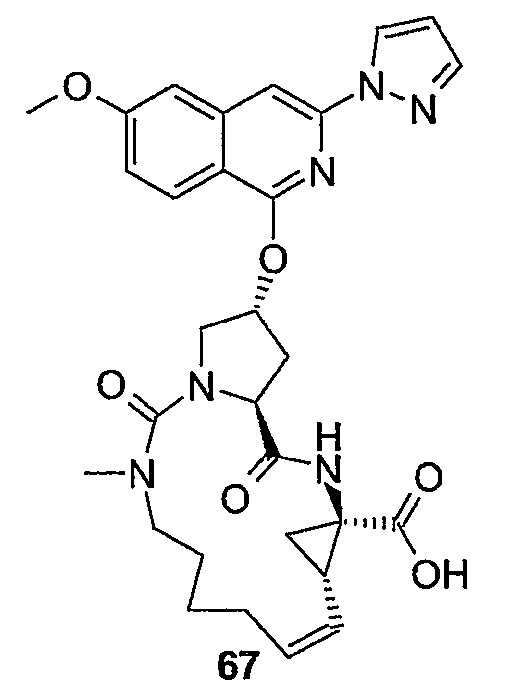
Указанный в заголовке продукт 67 получали из 1-гидрокси-6-метокси-3-(пиразол-1-ил)изохинолина, следуя таким же процедурам, которые описаны для получения 17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (42, пример 10): m/z = 575 (M+H)+.
Пример 24
Синтез N-[17-(3-пиразол-1-ил-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (68)
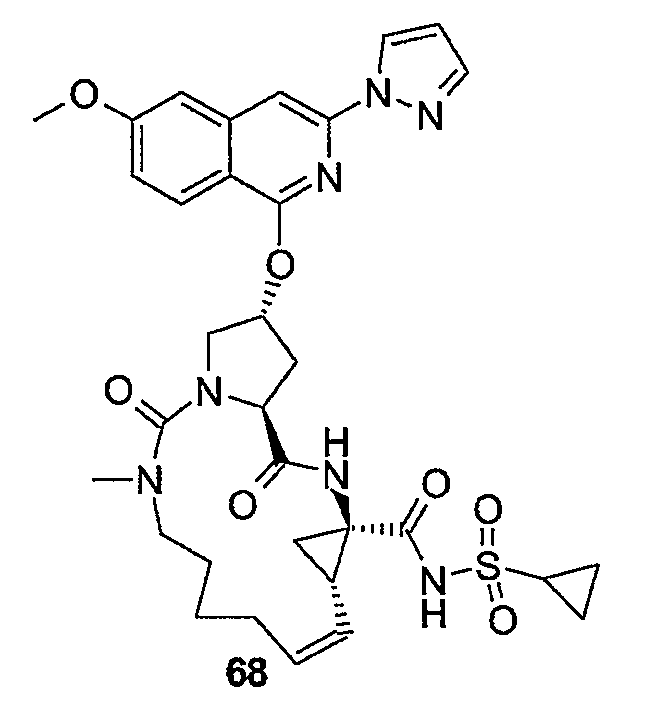
Указанный в заголовке продукт 68 получали из 17-(3-пиразол-1-ил-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (67), следуя таким же процедурам, которые описаны для получения N-[17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (43, пример 11): m/z = 678 (M+H)+. 1Н ЯМР (СDСl3): 10,5 (шир. с, 1 H), 8,4 (дд, J=2,5 Гц, 0,5, 1H), 8 (д, J=9,8 Гц, 1H), 7,75 (с, 2H), 7,00-7,10 (м, 2H), 6,55 (с, 1H), 6,45 (дд, J=2,5 Гц, 0,5, 1H), 5,95 (шир. с, 1H), 5,75 (дд, J=18,1 Гц, 8 Гц, 1H), 5,1 (т, J=10,3 Гц, 1H), 4,75 (т, J=7,0 Гц, 1H), 4,1 (дд, J=11,0 Гц, 4,3, 1H), 3,90 (с, 3H), 3,70 (д, J=11,0 Гц, 1H), 3,10-3,20 (м, 1H), 2,90-3,01 (м, 1H), 2,85 (с, 3H), 2,50-2,62 (м, 3H), 2,20-2,30 (м, 1H), 1,90-2,00 (м, 2H), 1,55-1,60 (м, 4H), 1,30-1,50 (м, 6H).
Пример 25
Синтез 17-[3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (69)

Указанный в заголовке продукт 69 получали из 1-гидрокси-3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолина (6), следуя таким же процедурам, которые описаны для получения 17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (42, пример 10): m/z = 634 (M+H)+.
Пример 26
Синтез N-[17-[3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолин-1- илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (70)
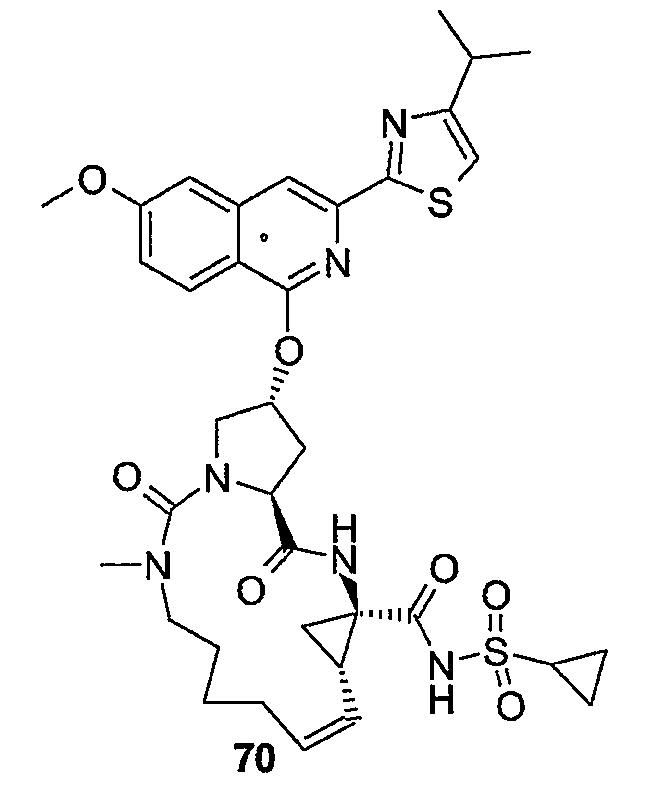
Указанный в заголовке продукт 70 получали из 17-[3-(4-изопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (69), следуя таким же процедурам, которые описаны для получения N-[17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (43, пример 11): m/z = 737 (M+H)+.
Пример 27
Синтез 17-[3-(2-изопропиламинотиазол-4-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбоновой кислоты (71)
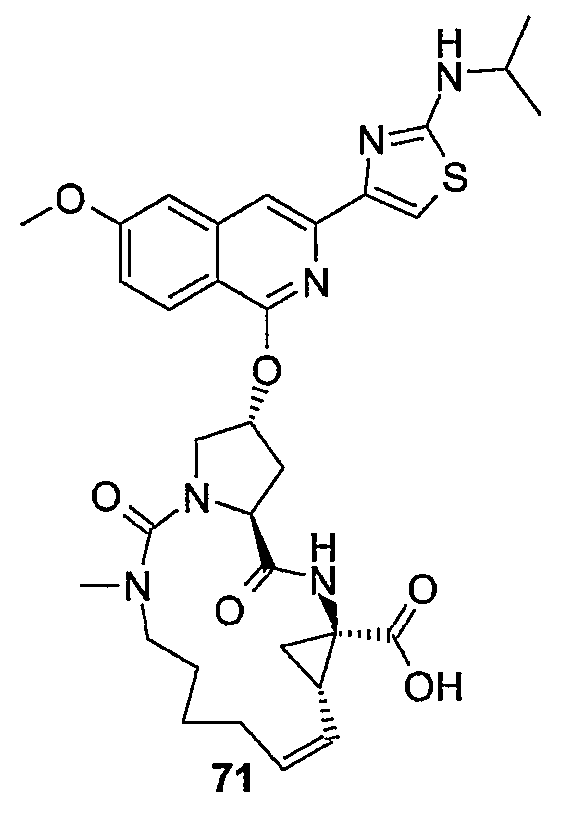
Указанный в заголовке продукт 71 получали из 1-гидрокси-3-(2-изопропиламинотиазол-4-ил)-6-метоксиизохинолина, следуя таким же процедурам, которые описаны для получения 17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (42, пример 10): m/z = 649 (M+H)+.
Пример 28
Синтез N-[17-[3-(2-изопропиламинотиазол-4-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.0 4,6 ]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (72)
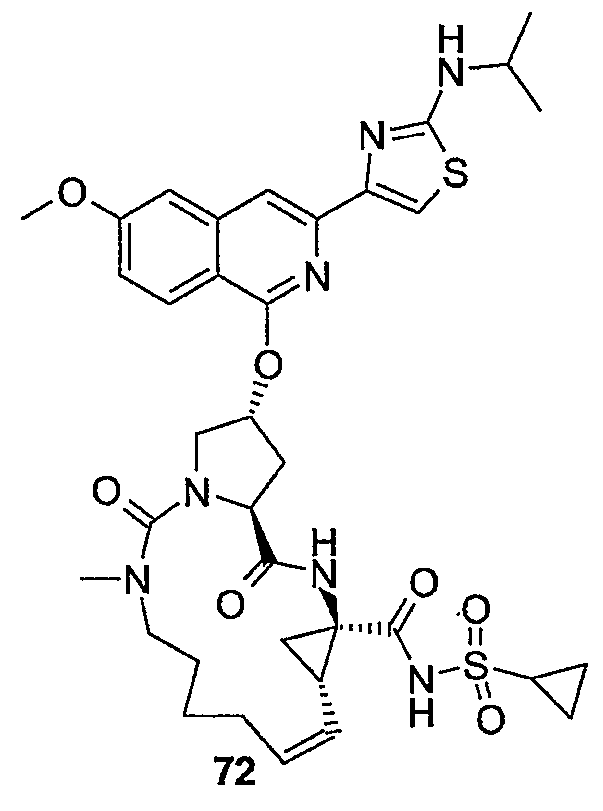
Указанный в заголовке продукт 72 получали из 17-[3-(2-изопропиламинотиазол-4-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (71), следуя таким же процедурам, которые описаны для получения N-[17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (43, пример 11): m/z = 752 (M+H)+. 1Н ЯМР (СDСl3): 0,93-1,03 (м, 1H), 1,05-1,15 (м, 1H), 1,18-1,28 (м, 3H), 1,32 (д, J=6,6 Гц, 3H), 1,34 (д, J=6,6 Гц, 3H), 1,36-1,67 (м, 4H), 1,93-2,07 (м, 2H), 2,22-2,36 (м, 1H), 2,45-2,65 (м, 3H), 2,85 (с, 3H), 2,91-3,00 (м, 1H), 3,05-3,17 (м, 1H), 3,64-3,80 (м, 3H), 3,89 (с, 3H), 4,08 (дд, J=3,8 Гц, J=10,9 Гц, 1H), 4,78 (т, J=8,1 Гц, 1H), 5,15 (т, J=10,4 Гц, 1H), 5,21-5,36 (шир. с, 1H), 5,73 (дд, J=8,1 Гц, J=18,4 Гц, 1H), 5,94-6,02 (м, 1H), 6,92-6,99 (шир. с, 1H), 7,00-7,07 (м, 2H), 7,22 (с, 1H), 7,74 (с, 1H), 7,95 (д, J=8,6 Гц, 1H), 10,54-10,99 (шир. с, 1H).
Пример 29
Синтез 18-[5-бром-6-метоксиизохинолин-1-илокси]-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0 4,6 ]нонадец-7-ен-4-карбоновой кислоты (74)
Стадия А
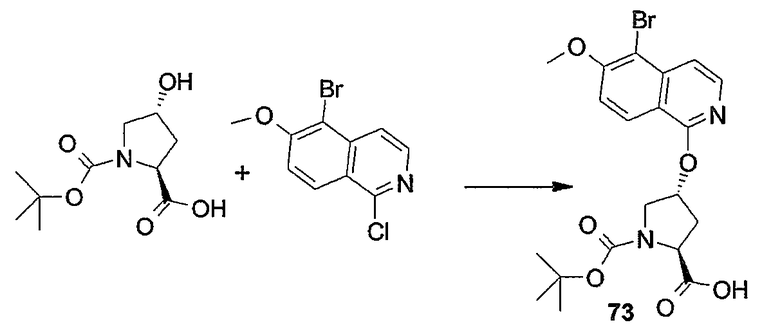
К раствору Boc-гидроксипролина (1,15 г, 4,99 ммоль) в ТГФ (50 мл) добавляли NaH (60% в минеральном масле, 500 мг, 12,5 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч перед добавлением 5-бром-6-метоксиизохинолина (1,36 г, 4,99 ммоль). После 48 ч при комнатной температуре в атмосфере азота реакционную смесь гасили холодной водой со льдом, подкисляли до pH 4 раствором HCl и экстрагировали этилацетатом, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали. Остаток очищали с помощью колоночной хроматографии (градиент EtOAc/CH2Cl2, от 5:95 до 50:50), получая при этом 751 мг (32,2%) продукта 73 в виде белого твердого вещества: m/z = 468 (M+H)+.
Синтез 18-[5-бром-6-метоксиизохинолин-1-илокси]-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0 4,6 ]нонадец-7-ен-4-карбоновой кислоты (74)
Стадия В
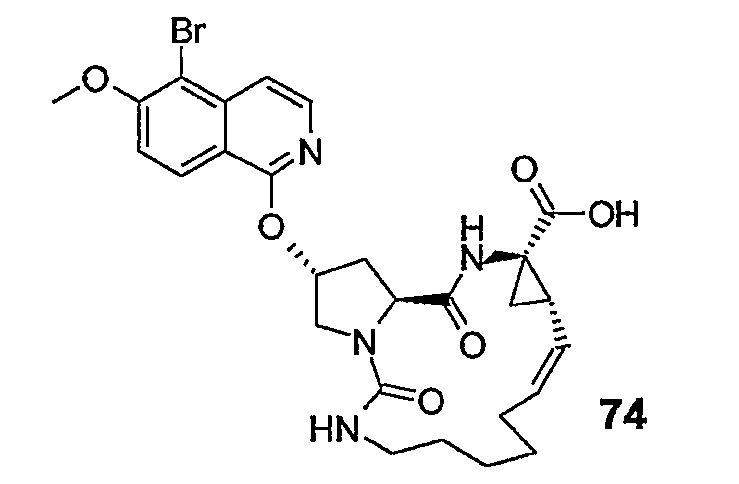
Указанное в заголовке соединение получали из промежуточного продукта 73 и гепт-8-енамина, следуя процедуре (стадии B-F), описанной для 17-(3-хлор-6-метоксиизохинолин-1-илокси)-13-метил-2,14-диоксо-3,13,15-триазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбоновой кислоты (42): m/z = 588 (M+H)+.
Пример 30
Синтез N-[18-[5-бром-6-метоксиизохинолин-1-илокси]-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.0 4,6 ]нонадец-7-ен-4-карбонил](циклопропил)сульфонамида (75)
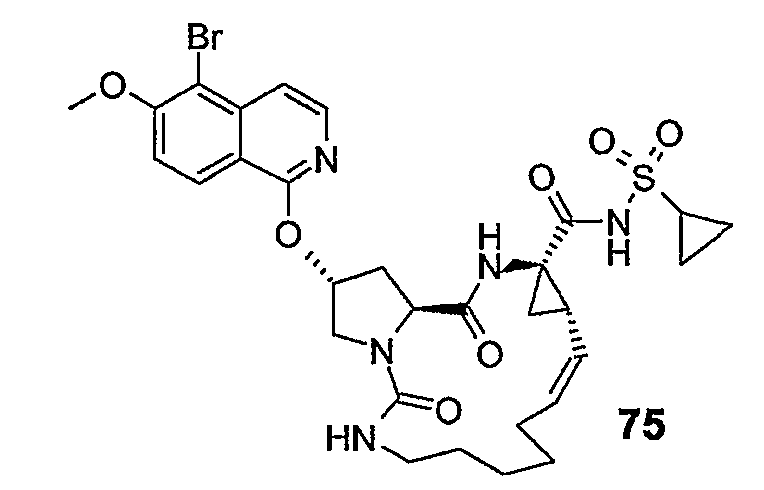
Указанное в заголовке соединение получали из 18-[5-бром-6-метоксиизохинолин-1-илокси]-2,15-диоксо-3,14,16-триазатрицикло[14.3.0.04,6]нонадец-7-ен-4-карбоновой кислоты (74), следуя процедуре, описанной для синтеза N-[17-[3-(4-циклопропилтиазол-2-ил)-6-метоксиизохинолин-1-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.04,6]октадец-7-ен-4-карбонил](циклопропил)сульфонамида (30): m/z = 691 (M+H)+.
Пример 31
Синтез кристаллического циклопентана
Синтез сложного трет-бутилового эфира 3-оксо-2-оксабицикло[2.2.1]гептан-5-карбоновой кислоты (77)
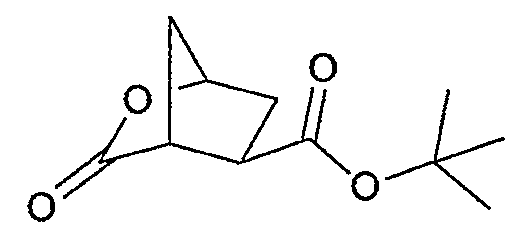
При перемешивании к раствору продукта 76 (180 мг, 1,15 ммоль) в 2 мл CH2Cl2 в инертной атмосфере аргона при 0°C добавляли DMAP (14 мг, 0,115 ммоль) и Boс2O (252 мг, 1,44 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали и сырой продукт очищали с помощью флэш-хроматографии на колонке (градиент смеси толуол/этилацетат 15:1, 9:1, 6:1, 4:1, 2:1), получая при этом указанное в заголовке соединение (124 мг, 51%) в виде белых кристаллов.
1Н-ЯМР (СDСl3): δ 1,45 (с, 9H), 1,90 (д, J=11,0 Гц, 1H), 2,10-2,19 (м, 3H), 2,76-2,83 (м, 1H), 3,10 (с, 1H), 4,99 (с, 1H); 13C-ЯМР (75,5 MГц, CD3OD): δ 27,1, 33,0, 37,7, 40,8, 46,1, 81,1, 81,6, 172,0, 177,7.
Альтернативный способ получения соединения 77
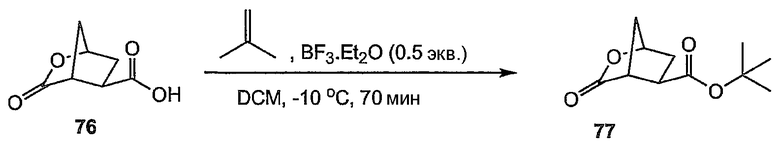
Соединение 76 (13,9 г, 89 ммоль) растворяли в дихлорметане (200 мл) и затем охлаждали приблизительно до -10°C в атмосфере азота. Затем в раствор барботировали изобутилен до тех пор, пока общий объем не увеличивался приблизительно до 250 мл, получая при этом мутный раствор. Добавляли BF3-Et2O (5,6 мл, 44,5 ммоль, 0,5 экв.) и реакционную смесь оставляли приблизительно при -10°C в атмосфере азота. Спустя 10 мин получали прозрачный раствор. Соста реакционной смеси контролировали с помощью ТСХ (смесь этилацетат-толуол 3:2, подкисленная несколькими каплями уксусной кислоты и смесь гексан-этилацетат EtOAc 4:1, окрашенная основным раствором перманганата). Через 70 мин, когда оставались только следы соединения 76, к реакционной смеси добавляли насыщенный водный раствор NaHCO3 (200 мл) и затем смесь энергично перемешивали в течение 10 мин. Органический слой промывали насыщенным раствором NaHCO3 (3 × 200 мл) и насыщенным раствором соли (1 x 150 мл), затем сушили сульфитом натрия, фильтровали и остаток упаривали до маслянистого остатка. При добавлении к остатку гексана продукт выпадал в осадок. При добавлении дополнительного количества гексана и нагревании до кипения с обратным холодильником получали прозрачный раствор, из которого кристаллизовался продукт. Кристаллы собирали при фильтровании и промывали гексаном (при комнатной температуре), затем сушили на воздухе в течение 72 ч, получая при этом бесцветные игольчатые кристаллы (12,45 г, 58,7 ммоль, 66%)
Пример 32
Активность соединений формулы (I)
Анализ репликонов
Исследовали активность соединений формулы (I) в отношении ингибирования репликации РНК HCV в клеточном анализе. Анализ показал, что соединения формулы (I) обладают активностью против репликонов HCV, функциональных в клеточной культуре. Клеточный анализ основан на бицистронной экспрессирующей конструкции, которая описана в публикации Lohmann и др., Science т.285, стр. 110-113 (1999), с модификациями, описанными в публикации Krieger и др., Journal of Virology 75: 4614-4624(2001), для стратегии многоцелевого скрининга. По существу, способ заключается в следующем.
В анализе использовали линию стабильно трансфицированных клеток Huh-7 luc/neo (далее упоминаемую как Huh-Luc). Данная линия клеток содержит РНК, кодирующую бицистронную экспрессирующую конструкцию, содержащую области NS3-NS5B дикого типа HCV типа 1b, транслируемые с сайта внутренней посадки рибосом (IRES) из вируса энцефаломиокардита (EMCV), с предшествующей репортерной группой (FfL-люцифераза) и группой селективного маркера (neoR, неомицинфосфотрансфераза). Конструкция граничит с 5' и 3'-NTR (нетранслируемыми областями) из вируса HCV типа 1b. Непрерывное культивирование репликон, содержащих клеток в присутствии G418 (neoR) зависит от репликации РНК HCV. Стабильно трансфицированные клетки, содержащие репликон, экспрессируемый РНК HCV, которая реплицируется автономно и на высоких уровнях, кодируя, наряду с прочим, люциферазу, используют для скрининга антивирусных соединений.
Репликонсодержащие клетки высевали в 384-луночные планшеты в присутствии различных концентраций испытуемых и контрольных соединений. После инкубации в течение трех дней определяли репликацию HCV, анализируя активность люциферазы (используя стандартные субстраты и реагенты для люциферазного анализа и устройство для визуализации микропланшетов Perkin Elmer ViewLuxTm ultraHTS). Репликонсодержащие клетки в контрольных культурах в отсутствие каких бы то ни было ингибиторов отличались высокой экспрессией люциферазы. Осуществляли мониторинг ингибиторной активности соединения в отношении активности люциферазы на клетках Huh-Luc, получая кривую зависимости доза-эффект для каждого испытуемого соединения. Затем рассчитывали значения EC50, которые соответствуют количеству соединения, необходимому для уменьшения уровня выявленной активности люциферазы на 50%, или, более конкретно, определяли способность РНК генетически связанного репликона HCV к репликации.
Анализ ингибирования
Цель данного анализа in vitro состояла в определении степени ингибирования протеазных комплексов NS3/4A вируса HCV соединениями согласно изобретению. Такой анализ показывает, насколько эффективными могут быть соединения согласно изобретению при ингибировании протеолитической активности NS3/4A вируса HCV.
Ингибирование полноразмерного протеолитического фермента NS3 гепатита C определяли в основном так, как описано в публикации Poliakov, Prot Expression & Purification 25 363 371 (2002). Вкратце, степень гидролиза депсипептидного субстрата Ac-DED(Edans)EEAbuψ[COO]ASK(Dabcyl)-NH2 (AnaSpec, San Jose, USA), определяли спектрофлуориметрически в присутствии кофактора пептида KKGSWTVGRIVLSGK (Ake Engström, Department of Medical Biochemistry и Microbiology, Uppsala University, Sweden). [Landro, 1997 #Biochem 36 9340-9348]. Фермент (1 нM) инкубировали в 50 мM HEPES, pH 7,5, 10 мM DTT, 40% глицерина, 0,1% н-октил-D-глюкозида, с 25 мкM кофактора NS4A и ингибитором при 30°C в течение 10 мин, после чего инициировали реакцию путем добавления 0,5 мкM субстрата. Ингибиторы растворяли в DMSO, подвергали действию ультразвука в течение 30 сек и перемешивали. Между измерениями растворы хранили при -20°C.
Конечную концентрацию DMSO в анализируемом образце доводили до 3,3%. Осуществляли коррекцию степени гидролиза с учетом эффектов внутри фильтра, согласно опубликованным в литературе процедурам [Liu, 1999 Analytical Biochemistry 267 331-335]. Значения Ki оценивали с помощью нелинейного регрессионного анализа (GraFit, Erithacus Software, Staines, MX, UK), используя модель конкурентного ингибирования и фиксированное значение для Km (0,15 мкM). Все предпринятые измерения проводили, по меньшей мере, в двух параллельных пробах.
В следующей таблице перечислены соединения, которые получены согласно любому из упомянутых выше примеров. В таблице также указаны активности протестированных соединений.
№
анализ репликонов
ферментный анализ
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2441870C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2486189C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2009 |
|
RU2518471C2 |
| МАКРОЦИКЛИЧЕСКИЕ ФЕНИЛКАРБАМАТЫ, ИНГИБИРУЮЩИЕ HCV | 2008 |
|
RU2490261C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2714197C2 |
| ПРОИЗВОДНЫЕ БЕНЗОТИАЗИНОВ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВ | 2010 |
|
RU2523791C2 |
| СОЕДИНЕННЫЕ МОСТИКОВОЙ СВЯЗЬЮ N-ЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДО-ИНГИБИТОРЫ ГАММА-СЕКРЕТАЗЫ | 2006 |
|
RU2422443C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2419619C2 |
| МАКРОЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ГЕПАТИТА C | 2000 |
|
RU2247126C2 |
| ПИРИМИДИН-ЗАМЕЩЕННЫЕ МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ HCV | 2008 |
|
RU2481340C2 |
Описываются новые макроциклические соединения формул

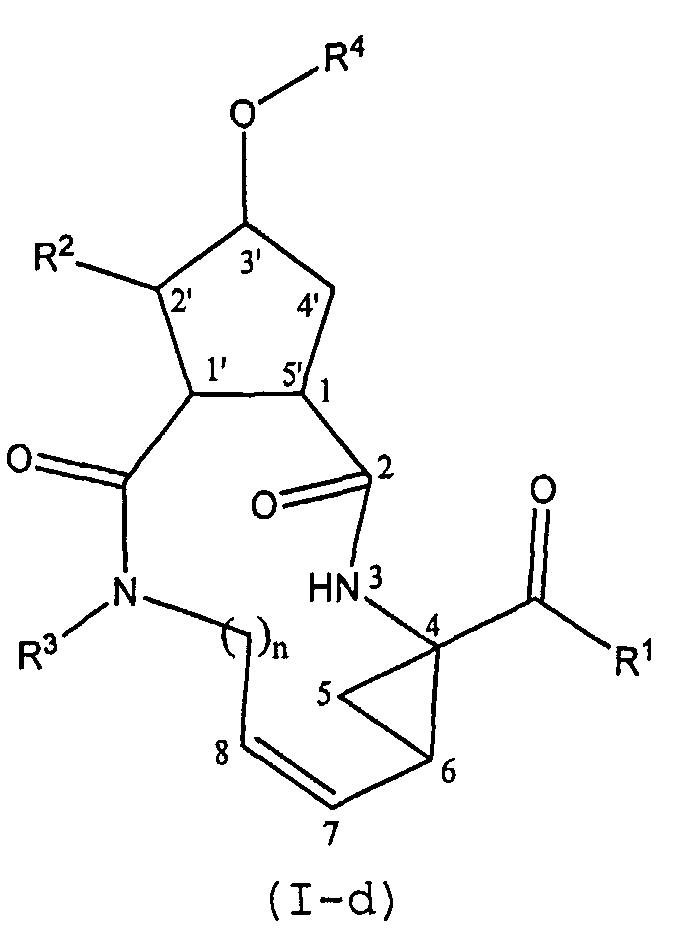
их фармацевтически приемлемые соли или стереоизомеры, где R1 = -OR5, -NH-SO2R6; R2 = водород; R3 = С1-6-алкил; R4 = изохинолинил, возможно замещенный; n равно 4 или 5; R5 = водород; R6 = С3-7-циклоалкил, и фармацевтическая композиция, их содержащая. Новые соединения обладают ингибиторной активностью в отношении репликации вируса гепатита С (HCV) и могут найти применение в медицине. 2 н. и 4 з.п. ф-лы, 1 табл.
1. Соединение формулы
его фармацевтически приемлемая соль или стереоизомер, где
R1 представляет собой -OR5, -NH-SO2R6;
R2 представляет собой водород;
R3 представляет собой C1-6-алкил;
R4 представляет собой изохинолинил, необязательно замещенный одним, двумя или тремя заместителями, каждый из которых независимо выбран из С1-6-алкокси, галогена, фенила, который в свою очередь может быть замещен С1-6-алкокси или полигалоген-С1-6-алкокси, пиридинила, замещенного С1-6-алкилом, пиразолила или тиазолила, необязательно замещенного С3-7-циклоалкилом, C1-6-алкилом или моно-C1-6-алкиламиногруппой;
n равно 4 или 5;
R5 представляет собой водород;
R6 представляет собой С3-7-циклоалкил.
2. Соединение по п.1, где R4 представляет собой
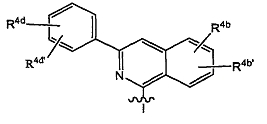
где каждый R4b и R4b' независимо представляет собой водород, C1-6-алкокси, трифторметил; и
R4d и R4d' независимо представляют собой водород, С1-6-алкил, C1-6-алкокси или галоген.
3. Соединение по любому из пп.1 и 2, где R4 представляет собой
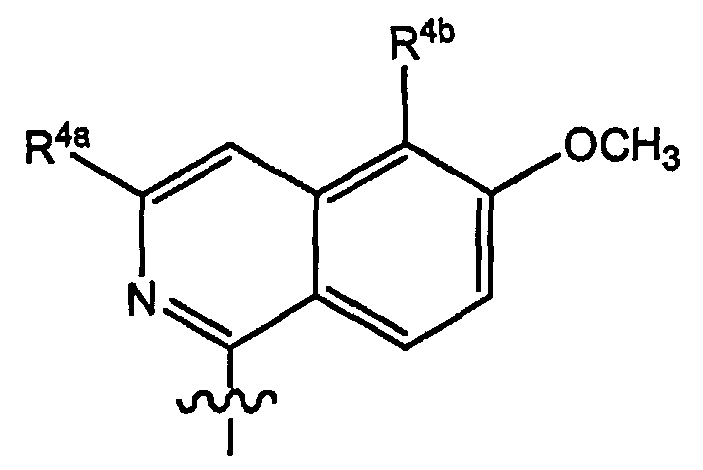
где R4a выбран из следующих фрагментов
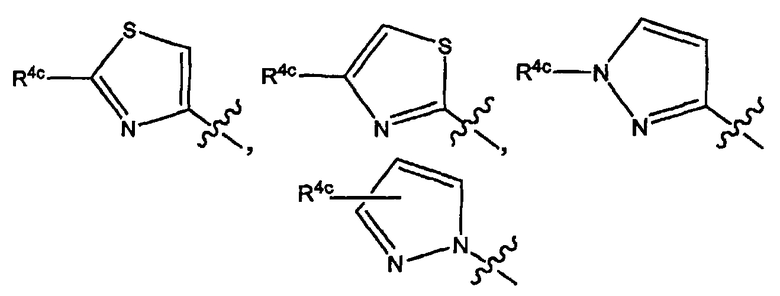
где каждый R4c независимо представляет собой водород, С1-6-алкил или моно-С1-6-алкиламино; и
R4b представляет собой водород или галоген.
4. Соединение по п.1, где
R1 представляет собой -NH-SO2R6, где R6 представляет собой циклопропил или метилциклопропил.
5. Соединение по п.1, отличающееся от фармацевтически приемлемой соли.
6. Фармацевтическая композиция, обладающая ингибиторной активностью в отношении репликации HCV, содержащая носитель и в качестве активного ингредиента эффективное против вируса количество соединения по любому из пп.1-5.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| RU 20281947 C1, 20.08.2006. | |||

