Область техники, к которой относится изобретение
Настоящее изобретение относится к макроциклическим индольным производным, обладающим ингибиторной активностью в отношении репликации вируса гепатита C (HCV). Изобретение дополнительно относится к композициям, содержащим такие соединения в качестве активных ингредиентов, а также к способам получения таких соединений и композиций.
Уровень техники
Вирус гепатита C во всем мире является основной причиной хронического заболевания печени и стал центром внимания значительного числа медицинских исследований. HCV является членом семейства вирусов Flaviviridae рода hepacivirus и близко связан с родом flavivirus, который включает ряд вирусов, вовлеченных в заболевания человека, таких как вирус денге и вирус желтой лихорадки, и с семейством пестивирусов (pestivirus) животных, которое включает вирус бычьей вирусной диареи (BVDV). HCV представляет собой положительно-смысловой одноцепочечный РНК-вирус с геномом приблизительно из 9600 оснований. Геном содержит как 5', так и 3' нетранслируемые области, которые принимают вторичные структуры РНК, и центральную открытую рамку считывания, которая кодирует единственный полипротеин, состоящий приблизительно из 3010-3030 аминокислот. Полипротеин кодирует десять генных продуктов, которые генерируются из предшественника полипротеина посредством серии сочетающихся со- и пост-трансляционных эндопротеолитических расщеплений, опосредованных с помощью протеаз как хозяина, так и вируса. Структурные белки вируса включают ядерный нуклеокапсидный белок и два обрамляющих гликопротеина Е1 и Е2. Неструктурные (NS) белки кодируют некоторые неотъемлемые вирусные ферментативные функции (геликаза, полимераза, протеаза), а также белки с неизвестными функциями. Репликация вирусного генома опосредована РНК-зависимой РНК-полимеразой, кодируемой неструктурным белком 5b (NS5B). Было показано, что в дополнение к полимеразе функции как вирусной геликазы, так и протеазы, кодируемые в бифункциональном белке NS3, являются существенными для репликации РНК HCV. Помимо серинпротеазы NS3 HCV также кодирует металлопротеиназу в области NS2.
Репликация HCV предпочтительно протекает в гепатоцитах, но не является непосредственно цитопатической, приводя к персистирующей инфекции. В частности, оказалось, что недостаток сильной ответной реакции Т-лимфоцитов и высокая склонность вируса к мутации промотируют высокую скорость хронической инфекции. Существует 6 основных генотипов HCV и более 50 подтипов, которые географически распределены различным образом. HCV типа 1 является доминирующим генотипом в США и Европе. Например, HCV типа 1 насчитывает от 70 до 75 процентов всех инфекций HCV в США. Большая генетическая гетерогенность HCV имеет важные диагностические и клинические последствия, возможно, объясняя трудности в разработке вакцин и недостаточную ответную реакцию на лечение. Как оценивается, 170 миллионов человек во всем мире инфицировано вирусом гепатита С. После первоначальной острой инфекции у большинства инфицированных индивидуумов развивается хронический гепатит, который может прогрессировать в фиброз печени, приводящий к циррозу - конечной стадии заболевания печени, и HCC (гепатоклеточной карциноме) (National Institutes of Health Consensus Development Conference Statement: Management of Hepatitis C. Hepatology, 36, 5 Suppl. S3-S20, 2002). Цирроз печени, возникший вследствие инфекции HCV, является ответственным приблизительно за 10000 смертей в год только в США и является основной причиной для трансплантаций печени. Передача HCV может происходить за счет контакта с зараженной кровью и продуктами крови, например, после переливания крови или внутривенного применения лекарственных средств (наркотиков). Введение диагностических тестов, применяемых при скрининге крови, привело к нисходящей тенденции случаев HCV после переливания крови. Однако учитывая медленное развитие заболевания печени до конечной стадии, существующие инфекции будут продолжать оставаться серьезной медицинской и экономической проблемой на протяжении десятилетий (Kim W.R., Hepatology, 36, 5 Suppl. S30-S34, 2002).
Существующие в настоящее время способы терапии HCV основаны на (пегилированном) интерфероне-альфа (IFN-α) в сочетании с рибавирином. Такая комбинированная терапия приводит к устойчивому вирусологическому ответу более чем у 40% пациентов, инфицированных вирусами генотипа 1, и приблизительно у 80% пациентов, инфицированных генотипами 2 и 3. Наряду с ограниченной эффективностью в отношении вируса HCV типа 1, комбинированная терапия имеет существенные побочные эффекты и плохо переносится большинством пациентов. Например, в зарегистрированных исследованиях пегилированного интерферона и рибавирина заметные побочные эффекты приводили к приостановке лечения у приблизительно от 10 до 14 процентов пациентов. Основные побочные эффекты при комбинированной терапии включают гриппоподобные симптомы, гематологические аномалии и нейропсихиатрические симптомы. Разработка более эффективных, удобных и легче переносимых способов лечения является важной задачей общественного здравоохранения. Таким образом, лечение данного хронического заболевания является неудовлетворенной клинической потребностью, поскольку существующая в настоящее время терапия является эффективной только частично и ограничена нежелательными побочными эффектами.
Одной из областей особого внимания является поиск ингибиторов NS5b РНК-зависимой РНК-полимеразы (RdRp). В неинфицированной клетке-хозяине не существует близких структурных гомологов данной полимеразы, и обнаружение ингибиторов упомянутой полимеразы могло бы обеспечить более специфический способ воздействия. Ингибиторы, которые в настоящее время находятся на стадии исследования, можно отнести к категории либо нуклеозидных ингибиторов (NI), либо ненуклеозидных ингибиторов (NNI). Ингибиторы NI конкурируют непосредственно с нуклеотидными субстратами за связывание с высококонсервативными активными участками. Более высокой специфичности можно достичь с помощью ингибиторов NNI, которые могут взаимодействовать обычно только со структурно родственными полимеразами за пределами высококонсервативного активного участка на специфическом аллостерическом участке.
Описаны индольные производные с ингибиторной активностью в отношении HCV. В патентной заявке WO 2007/092000 описаны тетрациклические индольные производные в качестве ингибиторов NS5B HCV для лечения и/или профилактики инфицирования вирусом HCV. В заявке США 2008/0146537 описаны циклопропильные конденсированные индолобензазепиновые ингибиторы NS5B HCV. В патентной заявке WO 2008/075103 описаны макроциклические индольные производные, применимые для лечения или профилактики инфицирования вирусом гепатита C.
В настоящее время предварительные клинические исследования закончились высоким количеством неудач, тем самым привлекая внимание к необходимости заниматься поиском новых ингибиторов NS5b. Существует высокая медицинская потребность в надежном и эффективном противовирусном (HCV) лечении. Такие ингибиторы HCV могут преодолевать недостатки существующей в настоящее время терапии HCV, такие как побочные эффекты, ограниченная эффективность, возникновение резистентности и нарушения комплаентности, а также повышать устойчивый вирусологический ответ. В частности, когда терапевтические соединения обладают хорошей биодоступностью и благоприятным фармакокинетическим и метаболическим профилем.
Сущность изобретения
Было обнаружено, что некоторые макроциклические индольные производные проявляют противовирусную активность в отношении субъектов, инфицированных HCV, с полезными свойствами, относящимися к одному или нескольким из следующих параметров: противовирусная эффективность, благоприятный мутантный статус, отсутствие токсичности, благоприятный фармакокинетический и метаболический профиль, легкость получения и введения препарата. Следовательно, такие соединения применимы для лечения инфекций HCV или борьбы с ними.
Настоящее изобретение относится к ингибиторам репликации HCV, которые можно представить в виде формулы (I),

включая стереохимически изомерные формы, и солям, гидратам и сольватам,
где R1 выбран из атома водорода, атома галогена и C1-4-алкокси;
R2 выбран из C1-4-алкила и C3-6-циклоалкила;
R4 представляет собой C3-7-циклоалкил, необязательно замещенный атомом галогена; - n равно 1 или 2;
Y выбран из

 и
и

- a равно 2, 3, 4 или 5;
- каждый b независимо равен 1 или 2;
- R3 и R3' независимо выбраны из атома водорода, C1-6-алкила и C3-6-циклоалкила.
Изобретение дополнительно относится к способам получения соединений формулы (I), включая их стереохимически изомерные формы, и солей, гидратов или сольватов, их промежуточных продуктов и применению промежуточных продуктов для получения соединений формулы (I).
Изобретение относится к соединениям формулы (I) как таковым, включая их стереохимически изомерные формы, и солям, гидратам или сольватам для применения в качестве лекарственного препарата. Изобретение относится к соединениям формулы (I) как таковым, включая их стереохимически изомерные формы, и солям, гидратам или сольватам для лечения гепатита C. Изобретение дополнительно относится к фармацевтическим композициям, содержащим носитель и эффективное против вируса количество соединения формулы (I), которое здесь указано. Фармацевтические композиции могут содержать комбинации вышеупомянутых соединений с другими средствами против вируса HCV. Изобретение также относится к вышеупомянутым фармацевтическим композициям для введения субъекту, страдающему от инфекции HCV. Фармацевтические композиции могут дополнительно содержать комбинации вышеупомянутых соединений или фармацевтические композиции со средствами против ВИЧ. Поэтому изобретение также относится к вышеупомянутым фармацевтическим композициям для введения субъекту, страдающему от со-инфекции HCV/ВИЧ.
Изобретение также относится к применению соединения формулы (I), включая его стереохимически изомерные формы, или солей, гидратов или сольватов для получения лекарственного средства для ингибирования репликации HCV. Изобретение также относится к применению соединения формулы (I), включая его стереохимические изомерные формы, или солей, гидратов или сольватов для производства лекарственного препарата для профилактики или лечения состояний, ассоциированных с HCV. Изобретение также относится к способу ингибирования репликации HCV у теплокровного животного; упомянутый способ включает введение эффективного количества соединения формулы (I), включая его стереохимически изомерные формы, или солей, гидратов или сольватов. Изобретение также относится к способу профилактики или лечения состояний теплокровного животного, ассоциированных с HCV; упомянутый способ включает введение эффективного количества соединения формулы (I), включая его стереохимически изомерные формы, или солей, гидратов или сольватов. Изобретение дополнительно относится к способу профилактики или лечения со-инфекции HCV/ВИЧ у теплокровного животного; упомянутый способ включает введение эффективного количества соединения формулы (I), включая его стереохимически изомерные формы или солей, гидратов или сольватов.
Подробное описание изобретения
Теперь настоящее изобретение будет описано дополнительно. Далее более подробно описаны различные аспекты или варианты осуществления изобретения. Каждый аспект или вариант осуществления изобретения, описанный таким образом, может быть объединен с любым другим аспектом (аспектами) или вариантом (вариантами) осуществления изобретения, если ясно не указано обратное. В частности, любой признак, указанный в качестве предпочтительного или благоприятного, может быть объединен с любым другим признаком или признаками, указанными в качестве предпочтительных или благоприятных при формулировке конкретного варианта осуществления изобретения.
Если не указано иначе, употребляются следующие определения, применяемые ранее и в дальнейшем.
Для целей настоящего изобретения термины "субъект" или "инфицированный субъект" или "пациент" относятся к индивидууму, инфицированному HCV, который нуждается в лечении.
Термином "гало" или "галоген" характеризуется фтор, хлор, бром и иод.
Применяемый здесь термин "C1-4-алкил" в виде группы или части группы обозначает насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 4 атомов углерода, например, такие как метил, этил, проп-1-ил, проп-2-ил, бут-1-ил, бут-2-ил, изобутил, 2-метилпроп-1-ил; термин "C1-3-алкил" в виде группы или части группы обозначает насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 3 атомов углерода, например, такие как метил, этил, проп-1-ил, проп-2-ил. Термин "C1-6-алкил" охватывает C1-3-алкильные и C1-4-алкильные радикалы и их более высокие гомологи, содержащие 5 или 6 атомов углерода, например, такие как пент-1-ил, пент-2-ил, пент-3-ил, гекс-1-ил, гекс-2-ил, 2-метилбут-1-ил, 2-метилпент-1-ил, 2-этилбут-1-ил, 3-метилпент-2-ил и т.п. Среди C1-6-алкильных групп интерес представляет C1-4-алкил и C1-3-алкил.
Термин "C1-6-алкилен" в виде группы или части группы относится к C1-6-алкильным группам, которые являются двухвалентными, то есть с двумя одинарными связями для соединения с двумя другими группами. Неограничивающие примеры алкиленовых групп включают метилен, этилен, метилметилен, пропилен, этилэтилен, 1-метилэтилен и 1,2-диметилэтилен.
Термином "C3-7-циклоалкил" характеризуется циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Термином "C3-6-циклоалкил" обозначается циклопропил, циклобутил, циклопентил и циклогексил. Термином "C3-5-циклоалкил" обозначается циклопропил, циклобутил и циклопентил.
Термин "C1-4-алкокси" или "C1-4-алкилокси" в виде группы или части группы относится к радикалу, содержащему формулу -ORa, в которой Ra представляет собой C1-4-алкил, который определен выше. Неограничивающие примеры подходящей C1-4-алкоксигруппы включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси и трет-бутокси.
Следует отметить, что места расположения радикала на любом молекулярном фрагменте, применяемом в определениях, могут находиться на таком фрагменте где угодно, при условии, что он является химически стабильным.
Радикалы, применяемые в определениях переменных, включают все возможные изомеры, если не указано иначе. Например, пиперидинил включает пиперидин-1-ил, пиперидин-2-ил, пиперидин-3-ил и пиперидин-4-ил; пентил включает пент-1-ил, пент-2-ил и пент-3-ил. Когда любая переменная встречается в любом элементе более одного раза, каждое определение является независимым.
Во всех применяемых далее случаях термин "соединения формулы (I)" или "настоящие соединения" или подобные термины означают, что в них включены соединения формулы (I), включая их стереохимически изомерные формы, и соли, гидраты или сольваты. Один из вариантов осуществления изобретения включает соединения формулы (I) или любую указанную здесь подгруппу, включая возможные стереохимически изомерные формы, а также соли, гидраты и сольваты. Еще один вариант осуществления включает соединения формулы (I) или любые указанные здесь подгруппы, включая возможные стереохимически изомерные формы, а также их соли, гидраты и сольваты.
Во всех применяемых далее случаях термин "необязательно замещенный" означает, что в него включены незамещенные, а также замещенные, по меньшей мере, одним из указанных замещающих радикалы. В качестве примера, "C1-4-алкил, необязательно замещенный атомом хлора", означает, что включен незамещенный C1-4-алкил, а также C1-4-алкил, замещенный атомом хлора.
Соединения формулы (I) могут содержать один или несколько центров хиральности и могут существовать в виде стереохимически изомерных форм. Термин "стереохимически изомерные формы" в применяемом здесь значении означает все возможные соединения, состоящие из одинаковых атомов, связанных одинаковой последовательностью связей, но имеющих разные трехмерные структуры, которыми могут обладать соединения формулы (I).
Что касается примеров, где (R) или (S) применяется для обозначения абсолютной конфигурации хирального атома в заместителе, обозначение относится к рассмотрению всего соединения в целом, а не к заместителю в отдельности.
В одном из аспектов настоящее изобретение относится к соединениям формулы (I)

включая стереохимически изомерные формы, и их N-оксидам, солям, гидратам и сольватам, в которой Y, R1, R2, R4 и n имеют такие же значения, которые здесь указаны.
Конкретные подгруппы соединений формулы (I) представляют собой соединения формулы (II), (III), (IV), (V), (VI) и (VII)

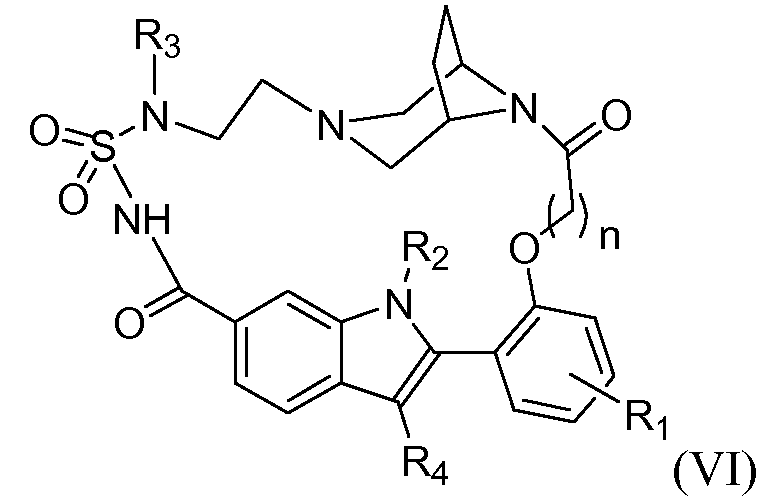 и
и
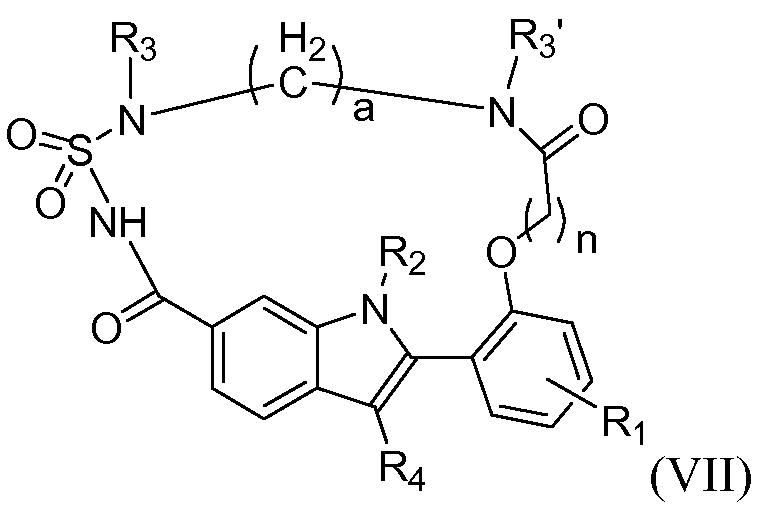
где Y, R1, R2, R3, R3', R4, a и n имеют те же самые значения, которые указаны для соединений формулы (I) или независимо для любого из вариантов их осуществления, который указан здесь.
Когда Y соответствует формуле

 или
или  ,
,
понятно, что Y может быть ориентирован в двух направлениях, то есть пиперазинильный или 3,8-диазабицикло[3.2.1]октановый фрагмент может быть соединен с сульфонамидной группой, в то время как алифатический амин соединяется с карбонильной группой, или пиперазинильный или 3,8-диазабицикло[3.2.1]октановый фрагмент соединен с карбонильной группой, а алифатический амин соединен с сульфонамидной группой.
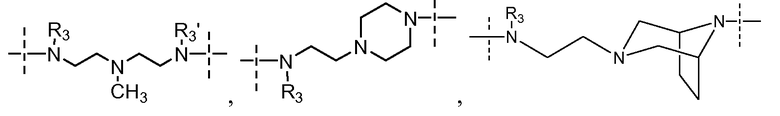
Варианты осуществления настоящего изобретения относятся к соединениям формулы (I) или любой их конкретной подгруппы, которая здесь указана, для которых применяется одно или несколько следующих ограничений:
- Y выбран из -N(R3)-(CH2)4-N(R3)-

и
- Y выбран из -N(R3)-(CH2)а-N(R3)-, где а равно 4 или 5
 и
и 
- Y выбран из -N(CH3)-(CH2)4-N(CH3)-


и
- Y выбран из
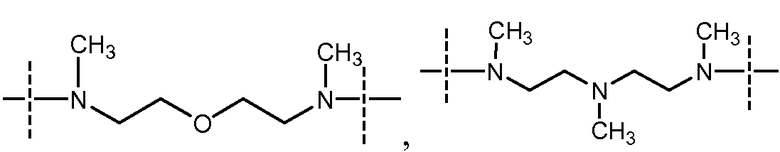 и
и
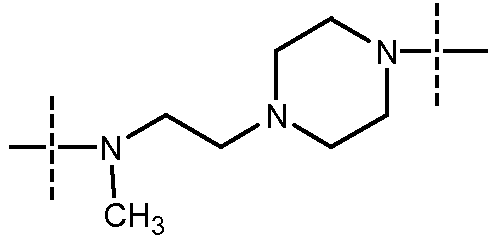
в которой пиперазиновый фрагмент соединен с карбонилом, а алифатический амин - с сульфонилом;
- Y выбран из -N(R3)-(CH2)5-N(R3)- и

- Y представляет собой
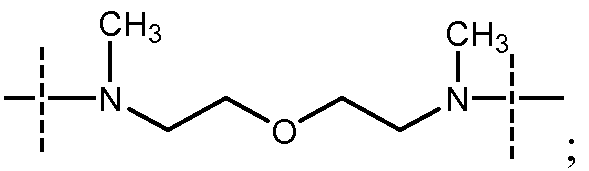
- Y представляет собой

в которой пиперазиновый фрагмент соединен с карбонилом, а алифатический амин - с сульфонилом;
- Y содержит цепь длиной в 7 атомов;
- R3 и R3' независимо выбраны из группы, состоящей из атома водорода, метила, этила, изопропила и циклопропила;
- R3 и R3' независимо выбраны из группы, состоящей из атома водорода и метила;
- R3 и R3' представляют собой метил;
- R1 выбран из группы, содержащей атом водорода, атом хлора, атом фтора или метокси;
- R1 представляет собой атом водорода или метокси или атом фтора;
- R1 представляет собой атом водорода;
- R1 расположен на бензольном кольце в мета- или пара-положении относительно связи, соединяющей бензольную группу с индольной группой;
- R1 расположен на бензольном кольце в пара-положении относительно связи, соединяющей бензольную группу с индольной группой;
- R2 выбран из метила, этила, изопропила, циклопентила и циклопропила;
- R2 представляет собой метил или изопропил;
- R2 представляет собой метил;
- R4 выбран из циклопентила, циклогексила и фторциклогексила, в частности 2-фторциклогексила;
- R4 представляет собой циклогексил;
- n равно 1;
- a равно 4 или 5;
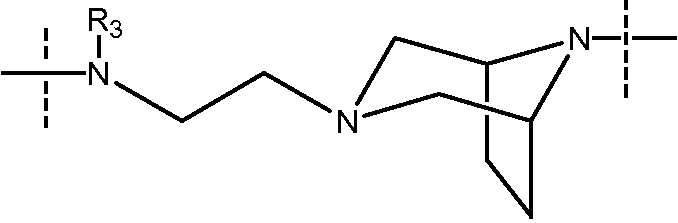
В еще одном конкретном варианте осуществления изобретения, когда Y может представлять собой

 или
или 
пиперазинильный или 3,8-диазабицикло[3.2.1]октановый фрагмент соединен с карбонильной группой, а алифатический амин соединен с сульфонамидной группой.
В одном из вариантов осуществления изобретение относится к соединениям, независимо, формул (II), (III), (IV), (V), (VI) или (VII), в которых R4 представляет собой циклогексил или 2-фторциклогексил.
В еще одном варианте осуществления изобретение относится к соединениям формул (II), (III), (IV), (V), (VI) или (VII), в которых R1 представляет собой атом водорода, метокси, атом хлора или атом фтора.
В еще одном варианте осуществления изобретение относится к соединениям формул (II), (III), (IV), (V), (VI) или (VII), в которых R2 представляет собой метил, этил или изопропил.
В еще одном варианте осуществления изобретение относится к соединениям формул (II), (III), (IV), (V), (VI) или (VII), в которых n равно 1.
В конкретном варианте осуществления изобретение относится к соединениям формулы (I), выбранным из группы, содержащей:

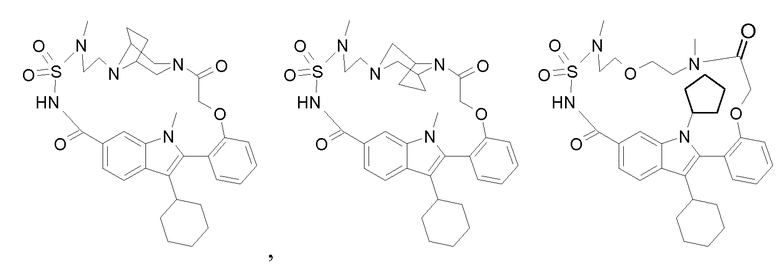 и
и
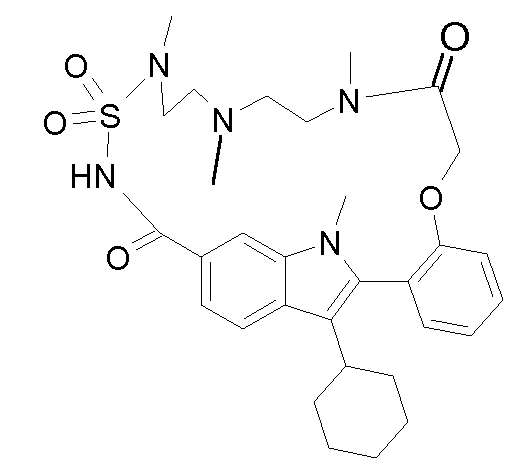
включая стереохимически изомерные формы, и солям, сольватам или гидратам.
Если не упоминается или не указано иначе, химическое обозначение соединения охватывает смесь нескольких или всех возможных стереохимически изомерных форм, которыми упомянутое соединение могло бы обладать. Упомянутая смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры упомянутого соединения. Подразумевается, что в объем настоящего изобретения включены все стереохимически изомерные формы соединений по настоящему изобретению как в чистой форме, так и в смеси друг с другом.
Чистые стереоизомерные формы соединений и промежуточных продуктов, которые упоминаются здесь, определяются как изомеры, по существу не содержащие других энантиомерных или диастереомерных форм той же основной молекулярной структуры упомянутых соединений или промежуточных продуктов. В частности, термин "стереоизомерно чистые" относится к соединениям или промежуточным продуктам, содержащим, по меньшей мере, 80%-ный избыток стереоизомера (то есть с минимальным содержанием 90% одного возможного изомера и максимальным содержанием 10% других возможных изомеров) до 100%-ного избытка (то есть 100% содержание одного изомера и отсутствие других); более предпочтительно - к соединениям или промежуточным продуктам со стереоизомерным избытком в диапазоне от 90% до 100%; еще более предпочтительно - со стереоизомерным избытком в диапазоне от 94% до 100%, и наиболее предпочтительно - со стереоизомерным избытком в диапазоне от 97% до 100%. Термины "энантиомерно чистые" и "диастереомерно чистые" следует понимать аналогичным образом, но в отношении энантиомерного избытка и диастереомерного избытка рассматриваемой смеси соответственно.
Чистые стереоизомерные формы соединений и промежуточных продуктов по данному изобретению можно получать с помощью процедур, известных в данной области техники. Например, энантиомеры могут быть отделены друг от друга с помощью селективной кристаллизации их диастереомерных солей с оптически активными кислотами или основаниями. Примерами таких кислот являются винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота и камфосульфоновая кислота. Альтернативно энантиомеры можно разделять хроматографическими способами с применением хиральных неподвижных фаз. Упомянутые стереохимически чистые изомерные формы также можно получать из соответствующих стереохимически чистых изомерных форм подходящих исходных материалов, при условии, что осуществляется стереоспецифическая реакция. В частности, если требуется конкретный стереоизомер, упомянутое соединение синтезируют с помощью стереоспецифических способов получения. При таких способах преимущественно будут использоваться энантиомерно чистые исходные материалы.
Диастереомерные рацематы соединений формулы (I) или любой их подгруппы можно получать по отдельности с помощью традиционных способов. Подходящие физические способы разделения, которые можно предпочтительно использовать, например, представляют собой селективную кристаллизацию и хроматографию, например, колоночную хроматографию.
Абсолютную стереохимическую конфигурацию некоторых соединений формулы (I), их N-оксидов, солей, гидратов или сольватов и промежуточных продуктов, применяемых при их получении, экспериментально не определяли. Специалист в данной области техники способен определить абсолютную конфигурацию таких соединений с применением способов, известных в данной области техники, например, таких как рентгеновская дифракция.
Однако те соединения формулы (I), в химической структуре которых отсутствует хиральный или стереогенный центр, могут иметь преимущество, связанное с облегчением задачи их синтеза в промышленном масштабе и/или повышением экономической эффективности синтеза.
Конкретными подгруппами соединений формулы (I) являются соединения формулы (IA) и (IB).
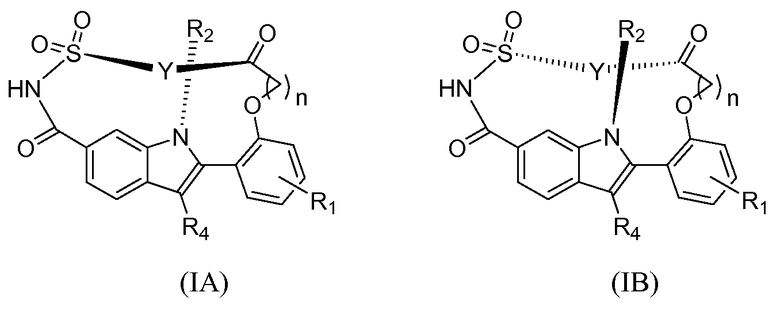
Соединения формулы (IA) и (IB) являются изомерами, и в зависимости от длины двухвалентного линкера Y и R2 (например, когда R2 представляет собой изопропил) соединения формулы (IA) и (IB) могут не находиться в равновесии, но находиться в соответствующей "заторможенной" конформации, то есть быть стабильными в своих соответствующих конформациях. Конформация соединений формулы (I), например (IA) или (IB), влияет на характеристики соединений, в том числе на их метаболическую стабильность, фармакокинетическую и биологическую активности.
Конкретными вариантами осуществления соединений формулы (IA) и (IB) являются соединения формулы (IA-I) и (IB-I).

Также подразумевается, что настоящее изобретение включает все изотопы атомов, встречающихся в настоящих соединениях. Изотопы включают такие атомы, имеющие одинаковый атомный номер, но различные масс.вес. В качестве общего примера и без ограничения изотопы водорода включают тритий и дейтерий. Изотопы углерода включают C-13 и C-14.
Соли соединений формулы (I), предназначенные для терапевтического применения, представляют собой те соли, в которых противоион является фармацевтически приемлемым. Однако соли кислот и оснований, которые являются фармацевтически неприемлемыми, также могут находить применение, например, при получении или очистке фармацевтически приемлемого соединения. Все соли, являются ли они фармацевтически приемлемыми или нет, включены в объем настоящего изобретения.
Подразумевается, что упомянутые выше фармацевтически приемлемые соли кислот и оснований содержат терапевтически активные нетоксичные формы аддитивных солей кислот и оснований, которые способны образовывать соединения формулы (I). Фармацевтически приемлемые аддитивные соли кислот можно удобно получать путем обработки основной формы такой подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористоводородную или бромистоводородную кислоту, серную, азотную, фосфорную и тому подобные кислоты; или органические кислоты, например, такие как уксусная, пропановая, гидроксиуксусная, молочная, пировиноградная, щавелевая (то есть этандионовая), малоновая, янтарная (то есть бутандионовая кислота), малеиновая, фумаровая, яблочная (то есть гидроксибутандионовая кислота), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и тому подобные кислоты.
С другой стороны, упомянутые солевые формы могут быть превращены в форму свободного основания при обработке подходящим основанием.
Соединения формулы (I) или любой их подгруппы, содержащие кислотный протон, также могут превращаться в форму нетоксичных основно-аддитивных солей металлов или форму аминных солей при обработке подходящими органическими и неорганическими основаниями. Подходящие основные солевые формы включают, например, аммониевые соли, соли щелочных и щелочноземельных металлов, например, соли лития, натрия, калия, магния, кальция и т.п.; соли с органическими основаниями, например, бензатином, N-метил-D-глюкамином, гидрабаминовые соли и соли с аминокислотами, например, такими как аргинин, лизин и т.п.
N-оксидные формы настоящих соединений представляют собой соединения формулы (I) или любой их подгруппы, в которых один или несколько атомов азота окислены до так называемого N-оксида. Обычно такие N-оксиды могут образовываться после введения соединения формулы (I) субъекту и метаболизирования в организме. Альтернативно такие N-оксиды могут быть химически синтезированы с применением способов, известных в данной области техники.
Некоторые из соединений формулы (I) или любой их подгруппы и промежуточные продукты также могут существовать в одной или нескольких таутомерных формах. Хотя в упомянутой выше формуле такие формы не указаны в явной форме, подразумевается, что они включены в объем настоящего изобретения. Соответственно, соединения и промежуточные продукты могут присутствовать в виде смеси таутомеров или в виде индивидуального таутомера.
В изобретении особенное предпочтение отдается соединениям формулы I или любой их подгруппы, которые в описанных ниже анализах ингибирования имеют значение ингибирующей концентрации менее 100 мкМ, в частности менее 50 мкМ, более точно менее 10 мкМ, в частности, менее 5 мкМ, еще более точно менее 1 мкМ, предпочтительно менее 100 нМ, и в частности, менее 50 нМ, как определено данными соответствующего анализа, такого как анализы, применяемые ниже в разделе "Примеры".
Следует понимать, что в настоящем документе подразумевается, что указанные выше подгруппы соединений формулы (I), а также любая другая указанная здесь подгруппа включает стереохимически изомерные формы и любые соли, гидраты и сольваты таких соединений.
Получение соединений формулы (I)
Общие схемы синтеза
Соединения формулы (I) могут быть получены согласно различным способам B, C, D, E, F, описанным ниже, из индольных производных A-6, синтезированных согласно описанному здесь способу A

в которой R1, R2, R4, n имеют значения, указанные для соединений формулы (I) или их подгрупп; Ra выбран из метила и трет-бутила, и Rb выбран из метила или трет-бутила. Синтез A-I описан в патентной заявке WO 2003/010140A2, публикации Journal of Medicinal Chemistry 2005, т. 48(5), стр. 1314-1317 и в патентной заявке WO 2006/029912Al.
Способ A
Схема 1

Соединения формулы A-3 можно получать с помощью реакции кросс-сочетания по Сузуки между соединениями A-I, содержащими заместитель R4 и сложноэфирную метильную или трет-бутильную группу, и производным бороновой кислоты A-2, содержащим заместитель R1 и гидроксил, защищенный подходящей защитной группой PG1, такой как бензильная группа. Кроме того, подходящие защитные группы, которые можно применять, перечислены, например, в публикациях: Greene, "Protective Groups in Organic Chemistry", John Wiley & Sons, New York (1999) и "The Peptides: Analysis, Synthesis, Biology", т. 3, Academic Press, New York (1987). Указанную реакцию можно осуществлять в присутствии палладиевого катализатора, такого как диCl-бис(трифенилфосфино)-Pd(II), и основания, такого как карбонат калия, в подходящем растворителе, таком как смесь диметоксиэтан/вода или толуол/этанол/вода, в инертной атмосфере.
Соединения A-4 можно получать алкилированием соединений A-3, с применением алкилгалогенидного производного, например метилиодида в присутствии основания, такого как гидрид натрия, карбонат калия, карбонат цезия и т.п., в присутствии подходящего растворителя, такого как ДМФА, ТГФ, ацетонитрил и т.п. Соединения A-5 можно получать путем удаления с фенольной группы защитной группы PG1 с помощью способов, известных в данной области техники. Подходящие способы снятия применяемой защиты PG1 описаны в публикациях: Greene, "Protective Groups in Organic Chemistry", John Wiley & Sons, New York (1999) и "The Peptides: Analysis, Synthesis, Biology ", т. 3, Academic Press, New York (1987). Например, бензильную защитную группу можно удалять с помощью каталитического гидрирования с применением палладиевого катализатора в подходящем растворителе, таком как метанол.
Соединения A-6 можно получать с помощью реакции алкилирования фенола с применением, например, галогенацетатного производного формулы X-CH2-CO-O-Rb, в которой X представляет собой галоген. Реакцию алкилирования можно осуществлять в присутствии основания, такого как карбонат калия, карбонат цезия и т.п., в подходящем растворителе, таком как ДМФА, ТГФ, ацетонитрил и т.п. Заместитель Rb может представлять собой метил, когда Ra представляет собой метил или трет-бутил, или трет-бутил, когда Ra представляет собой метил.
Способ B
Схематическое описание синтеза соединений формулы (I) приведено на схеме 2. Способ основывается на соединении формулы A-6.
Соединения формулы B-I можно получать региоселективным гидролизом сложного эфира, содержащего группу Rb. Для тех соединений A-6, в которых Rb представляет собой метильную группу, и Ra представляет собой трет-бутильную группу или метильную группу, региоселективный гидролиз Rb-сложного эфира можно осуществлять в основных условиях с применением гидроксида, такого как LiOH или NaOH, в полярных растворителях, таких как вода, спирт, такой как метанол или этанол, тетрагидрофуран (ТГФ) или в их смеси. Альтернативно, когда Rb представляет собой трет-бутильную группу, региоселективный гидролиз сложного эфира, содержащего группу Rb, можно осуществлять в кислотных условиях с применением, например, TFA в подходящем растворителе типа DCM.
Монозащищенный бифункциональный Y, полученный из реагента формулы PG2-Y-H, в которой Y имеет значение, указанное для формулы (I) или ее подгруппы, затем можно связывать с группой карбоновой кислоты в соединениях B-I для образования амидной связи, приводя к соединениям B-2. "PG2" в применяемом здесь значении представляет собой подходящую аминозащитную группу, выбранную из групп, известных в данной области техники. Подходящие защитные группы, которые можно применять в качестве защитной группы PG2, перечислены, например, в публикации: Greene, "Protective Groups in Organic Chemistry", John Wiley & Sons, New York (1999). В частности, PG2 представляет собой трет-бутилоксикарбонильную (Boc) защитную группу или 2-нитробензолсульфонильную (нозил) группу.
Образование амидных связей можно осуществлять с применением стандартных процедур, таких как процедуры, применяемые для связывания аминокислот в пептидном синтезе. Такое связывание подразумевает дегидратирующее связывание карбоксильной группы одного реагента с аминогруппой другого реагента с образованием соединяющей амидной связи. Образование амидной связи можно осуществлять путем взаимодействия исходных материалов в присутствии конденсирующего агента или путем преобразования карбоксильной функциональности в активную форму, такую как активный сложный эфир, смешанный ангидрид или хлорангидрид или бромангидрид карбоновой кислоты. Общие описания таких реакций сочетания и применяемых в них реагентов можно найти в общепринятых руководствах по химии пептидов, например, в публикации M. Bodanszky, "Peptide Chemistry", 2nd rev. ed., Springer-Verlag, Berlin, Germany, (1993).
Схема 2

Примеры реакций сочетания с образованием амидной связи включают азидный способ, способ смешанных ангидридов карбон-карбоновых кислот (изобутилхлорформиат), карбодиимидный способ (с использованием дициклогексилкарбодиимида (DCC), диизопропилкарбодиимида (DIC) или водорастворимого карбодиимида, такого как N-этил-N'-[3-(диметиламино)пропил]карбодиимид (EDC)), способ активированных сложных эфиров (например, п-нитрофенил, п-хлорфенил, трихлорфенил, пентахлорфенил, пентафторфенил, имидо-N-гидроксиянтарный и тому подобные сложные эфиры), способ с использованием К-реагента Вудворда, 1,1-карбонилдиимидазольный (CDI или N,N'-карбонилдиимидазол) способ, способы с использованием фосфорсодержащих реагентов или окислительно-восстановительные способы. Некоторые из указанных способов можно усовершенствовать путем добавления подходящих катализаторов, например, при карбодиимидном способе путем добавления 1-гидроксибензотриазола или 4-диметиламинопиридина (4-DMAP). Дополнительными конденсирующими агентами являются гексафторфосфат (бензотриазол-1-илокси)-трис-(диметиламино)фосфония, либо сам по себе, либо в присутствии 1-гидроксибензотриазола или 4-DMAP; или тетрафторборат 2-(1H-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония или гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония. Такие реакции сочетания можно осуществлять либо в растворе (жидкая фаза), либо на твердой фазе.
Реакции сочетания, в частности, проводят в инертном растворителе, таком как галогенированные углеводороды, например дихлорметан (DCM), хлороформ; в биполярных апротонных растворителях, таких как ацетонитрил, диметилформамид (ДМФА), диметилацетамид, DMSO, HMPT, простых эфирах, таких как тетрагидрофуран (ТГФ).
Во многих случаях реакции сочетания осуществляют в присутствии подходящего основания, такого как третичный амин, например триэтиламин, диизопропилэтиламин (DIPEA), N-метилморфолин, N-метилпирролидин, 4-DMAP или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Температура реакции может находиться в диапазоне от 0°C до 50°C, продолжительность реакции может находиться в диапазоне от 15 минут до 24 часов.
Удаление защитной группы PG2 согласно способам, известным в данной области техники, может приводить к соединениям B-3. Например, когда PG2 представляет собой Boc-защитную группу, PG2 можно удалять путем обработки соединений B-2 трифторуксусной кислотой (TFA) в подходящем растворителе, таком как DCM. Когда PG2 представляет собой нозильную группу, PG2 можно удалять путем обработки соединений B-2 тиолом типа меркаптоуксусной кислоты или тиофенола, в растворе или в твердой фазе, в присутствии основания, такого как карбонат цезия или LiOH, в подходящем растворителе, таком как ДМФА, ТГФ. Когда Ra представляет собой трет-бутильную группу, и PG2 представляет собой Boc-защитную группу, удаление PG2, как описано выше, может приводить к соединению B-3 с Ra, представляющим собой OH.
На следующей стадии введения сульфамида соединения B-3 могут подвергаться взаимодействию с сульфамидом в подходящем растворителе, например диоксане, в условиях нагревания, например, приблизительно до 100°C. Указанная реакция может протекать при микроволновом облучении и приводить к соединениям B-4. Альтернативно, сульфамидный фрагмент можно вводить путем взаимодействия соединения B-3 с аминосульфонилхлоридом в присутствии подходящего основания, такого как триэтиламин, DIPEA или пиридин, в подходящем растворителе, таком как хлорированный растворитель типа DCM, или в ДМФА, ТГФ.
Сохранившуюся функциональную группу сложного эфира в соединениях B-4, то есть -CO-O-Ra, затем можно подвергать гидролизу с применением условий, известных в данной области техники, включая омыление в основной среде, как описано выше, тем самым приводя к соединениям B-5. Для завершения указанной реакции может потребоваться нагревание. Для гидролиза функциональной группы сложного эфира в соединениях B-4 также можно применять кислотные условия, например TFA в подходящем растворителе типа DCM, когда Ra представляет собой трет-бутильную группу.
Соединения (I) можно получать макроциклизацией путем образования внутримолекулярной ацилсульфамидной связи в присутствии конденсирующих агентов, таких как CDI, которые превращают карбоксильную группу в реакционноспособный ацилимидазол при нагревании. Затем полученный ацилимидазол можно очищать перед добавлением подходящего основания, такого как DBU, для того, чтобы осуществить замыкание кольца, которое может протекать в условиях нагревания. Растворители, применяемые в указанных реакциях, могут включать ацетонитрил или ТГФ. Для того чтобы обеспечить замыкание кольца, также можно применять другие конденсирующие агенты или условия, такие как агенты и условия, известные в данной области техники или описанные выше в настоящем документе.
Способ C
Схема 3

Альтернативный способ, приводящий к соединениям B-3, который проиллюстрирован на схеме 3, может представлять собой образование амидной связи между соединениями B-1 и симметричной двухвалентной цепью Y, применяемой в избытке, по сравнению с соединениями B-1. Такую амидную связь можно синтезировать как описано выше, в частности, с применением конденсирующего агента, такого как гексафторфосфат [диметиламино-([1,2,3]триазоло[4,5-b]пиридин-3-илокси)метилен]диметиламмония (HATU), в присутствии основания, такого как DIPEA, и в подходящем растворителе типа DCM, ДМФА, или более конкретно - ТГФ. Чтобы получить соединения (I), соединения B-3 затем можно подвергать взаимодействию, как описано выше в способе B.
Способ D
Схема 4

Соединения B-4 можно получать непосредственно из соединений B-1 аналогично описанному выше синтезу соединений B-2, но с применением двухвалентной цепи Y, содержащей один сульфамидный фрагмент вместо защитной группы, то есть H2N-SO2-Y-H. Сульфамид можно вводить в H-Y-H путем нагревания реагента формулы H-Y-H, который может быть либо монозащищен подходящей защитной группой PG2 (то есть PG2-Y-H), либо нет, если он является симметричным, с сульфамидом в подходящем растворителе, таком как диоксан, при микроволновом облучении. Затем защитную группу PG2 можно удалять способами, известными в данной области техники, например, путем взаимодействия с TFA в дихлорметане, когда защитная группа представляет собой Boc-защитную группу, получая при этом моносульфамид, дериватизированный цепью Y, формулы H2N-SO2-Y-H.
Способ E
Соединения формулы B-2 или B-3 перед удалением PG2 из соединений B-2 могут подвергаться манипуляциям с функциональными группами, таким как алкилирование или восстановительное аминирование, и/или реакции, приводящей к сульфамиду B-4.
Способ F
Сложный эфир, содержащий группу Ra, в соединениях A-6 можно селективно подвергать гидролизу, как описано выше для способа B. Например, когда Ra представляет собой трет-бутильную группу, и Rb представляет собой метильную группу, Ra можно удалять в кислотных условиях с применением, например, TFA в подходящем растворителе типа DCM, получая при этом производное карбоновой кислоты F-1.
Сочетание соединения F-1 с реагентом H2N-SO2-Y-PG2 приводит к ацилсульфамидному соединению F-2. Упомянутое сочетание можно осуществлять с применением условий, описанных для последней стадии способа B. В частности, конденсирующий агент, применяемый для активации карбоксильной группы карбоновой кислоты F-1, может представлять собой CDI в подходящем растворителе типа ацетонитрила или ТГФ, в условиях нагревания. Добавление сульфамидной цепи в присутствии основания, такого как DBU, затем может приводить к соединениям F-2. PG2 представляет собой подходящую аминозащитную группу, выбранную из групп, известных в данной области техники. В частности, в способе E-F PG2 представляет собой Boc-защитную группу. Удаление защитной группы PG2 соединений F-2, согласно способам, известным в данной области техники, может приводить к соединениям F-3. Такие способы включают взаимодействие соединений F-2 с TFA в подходящем растворителе, таком как DCM, когда PG2 представляет собой Boc-защитную группу.
Схема 5
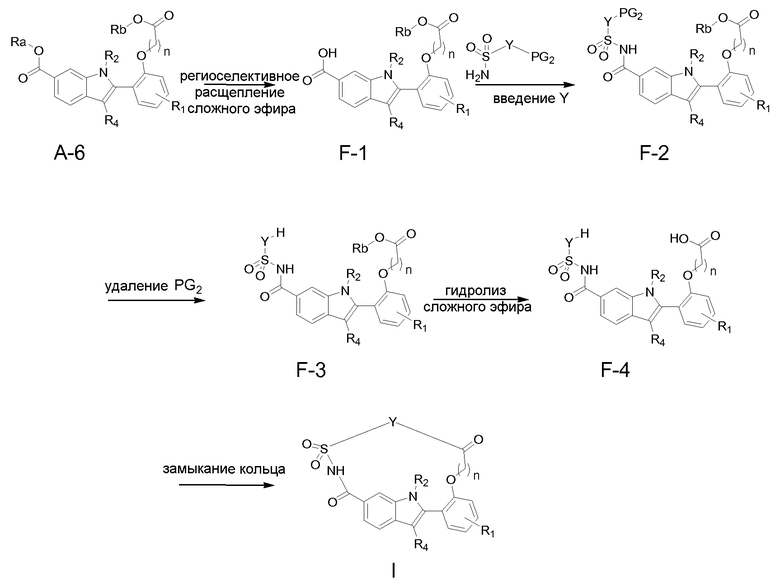
Функциональную группу сложного эфира в соединениях F-3 (в которых Rb представляет собой метильную группу) затем можно подвергать гидролизу с применением условий, известных в данной области техники, включая омыление в основной среде, как описано выше, получая при этом соединения F-4.
Альтернативно соединения F-2 можно подвергать реакции омыления в основной среде, чтобы гидролизовать сложный эфир, содержащий Rb, перед удалением аминозащитной группы PG2 с применением описанных выше условий, получая при этом соединения F-4.
Соединения (I) можно получать макроциклизацией соединений F-4 путем образования внутримолекулярной амидной связи в присутствии конденсирующих агентов, как описано в способе B. В частности, указанную стадию образования амида можно осуществлять в условиях высокого разбавления.
Стереохимически чистые изомерные формы соединений формулы (I) или любой их подгруппы можно получать путем применения процедур, известных в данной области техники. Диастереомеры можно разделять физическими способами, такими как селективная кристаллизация, и хроматографическими способами, например, при противоточном распределении, жидкостной хроматографии и т.п.
Соединения формулы (I) или любых их подгрупп можно получать в виде рацемических смесей энантиомеров, которые можно отделять друг от друга с помощью следующих известных в данной области техники процедур разделения. Рацемические соединения формулы (I) или любой их подгруппы, которые являются достаточно основными или кислотными, можно превращать в соответствующие диастереомерные солевые формы путем взаимодействия с подходящей хиральной кислотой или хиральным основанием, соответственно. Упомянутые диастереомерные солевые формы затем выделяют, например, путем селективной или дробной кристаллизации и высвобождают из них энантиомеры с помощью щелочи или кислоты. Альтернативный способ разделения энантиомерных форм соединений формулы (I) или любых их подгрупп включает жидкостную хроматографию, в частности жидкостную хроматографию с применением хиральной неподвижной фазы. Упомянутые стереохимически чистые изомерные формы также могут быть получены из соответствующих стереохимически чистых изомерных форм подходящих исходных материалов, при условии, что осуществляется стереоспецифическая реакция. В частности, если требуется конкретный стереоизомер, упомянутое соединение может быть синтезировано с помощью стереоспецифических способов получения. При таких способах могут преимущественно использоваться энантиомерно чистые исходные материалы.
При дополнительном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) или любых его подгрупп, которые здесь указаны, и фармацевтически приемлемый носитель. Терапевтически эффективное количество в данном контексте представляет собой количество, достаточное, чтобы профилактически противодействовать вирусной инфекции, стабилизировать или уменьшать вирусную инфекцию, и в частности, вирусную инфекцию HCV у инфицированных субъектов или субъектов, подвергающихся риску быть инфицированными. При еще одном дополнительном аспекте данное изобретение относится к способу получения фармацевтической композиции, которая здесь указана, который включает тщательное смешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы (I) или любых его подгрупп, которые здесь указаны.
Следовательно, согласно варианту осуществления настоящего изобретения соединения формулы (I) или любой их подгруппы можно получать в виде различных фармацевтических форм, предназначенных для введения. Понятно, что все композиции, обычно используемые для систематического введения лекарственных средств, включены в настоящее изобретение в качестве подходящих композиций. Для получения фармацевтических композиций по настоящему изобретению эффективное количество конкретного соединения в качестве активного ингредиента, необязательно в солевой форме или комплекса с металлом, при тщательном смешивании объединяют с фармацевтически приемлемым носителем, где носитель может принимать большое разнообразие форм в зависимости от формы препарата, требуемой для введения. Такие фармацевтические композиции желательно получать в стандартной лекарственной форме, подходящей, в частности, для введения перорально, ректально, подкожно или путем парентеральной инъекции. Например, при получении композиций в виде лекарственной формы для перорального введения можно использовать любую из обычных фармацевтических сред, например, такую как вода, гликоли, масла, спирты и тому подобное в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие, средства для улучшения распадаемости таблеток и тому подобное в случае порошков, драже, капсул и таблеток. Из-за легкости их введения таблетки и капсулы представляют собой наиболее предпочтительные стандартные лекарственные формы для перорального введения, в случае которых, разумеется, используются твердые фармацевтические носители. Носитель для парентеральных композиций обычно будет содержать стерильную воду, по меньшей мере, в значительной степени, хотя могут быть включены и другие ингредиенты, например, для улучшения растворимости. Например, можно получать растворы, в которых носитель содержит физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также можно получать суспензии для инъекций, в случае которых можно использовать подходящие жидкие носители, суспендирующие средства и т.п. Также включены препараты в твердой форме, в отношении которых подразумевается, что непосредственно перед применением они должны преобразовываться в препараты в жидкой форме. В композициях, подходящих для подкожного введения, носитель необязательно содержит средство, усиливающее проникновение и/или подходящее увлажняющее средство, необязательно объединенные с подходящими добавками любой природы в незначительных количествах, добавки которых не оказывают значительного вредного воздействия на кожу.
Соединения по настоящему изобретению также можно вводить посредством пероральной ингаляции или инсуффляции с помощью способов и препаратов, используемых в данной области техники для введения таким путем. Таким образом, в большинстве случаев соединения по настоящему изобретению можно вводить в легкие в форме раствора, суспензии или сухого порошка, предпочтительно в форме раствора. Для введения настоящих соединений подходит любая система, разработанная для доставки растворов, суспензий или сухих порошков посредством пероральной ингаляции или инсуффляции.
Было обнаружено, что соединения по настоящему изобретению после перорального введения демонстрируют благоприятный фармакокинетический профиль, то есть высокую концентрацию в печени и высокое концентрационное отношение печень/плазма. Предпочтительно, чтобы соединения, которые ингибируют репликацию HCV, демонстрировали высокие концентрации в печени, поскольку репликация HCV происходит в печени. Высокое концентрационное отношение печень/плазма может уменьшать побочный эффект и/или снижать минимальную дозировку.
Таким образом, настоящее изобретение также относится к фармацевтической композиции, адаптированной для введения путем ингаляции или инсуффляции через рот, содержащей соединение формулы (I) или любых его подгрупп и фармацевтически приемлемый носитель. В частности, соединения по настоящему изобретению вводят посредством ингаляции раствора в виде распыленных или аэрозольных доз.
Для облегчения введения и равномерности дозировки особенно предпочтительно получать вышеупомянутые фармацевтические композиции в стандартной лекарственной форме. Стандартная лекарственная форма в применяемом здесь значении относится к физически дискретным единицам, подходящим в качестве однократных доз, когда каждая единица содержит заданное количество активного ингредиента, рассчитанное таким образом, чтобы производить требуемый терапевтический эффект, совместно с необходимым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая таблетки с риской или с покрытием), капсулы, драже, суппозитории, пакетики с порошком, облатки, растворы или суспензии для инъекций и т.п., и их отдельные множества.
Соединения формулы (I) и любой их подгруппы проявляют противовирусные свойства. Вирусные инфекции и ассоциированные с ними заболевания, поддающиеся лечению с помощью соединений и способов согласно настоящему изобретению, включают такие инфекции, вызванные вирусом HCV и другими патогенными флавивирусами, такими как вирус желтой лихорадки, вирус лихорадки денге (1-4 типов), вирус энцефалита Сент-Луис, вирус японского энцефалита, вирус энцефалита долины Муррея, вирус лихорадки Западного Нила и вирус Kunjin. Заболевания, ассоциированные с HCV, включают прогрессирующий фиброз печени, воспаление и некроз, приводящие к циррозу - конечной стадии заболевания печени, и HCC; и заболевания, ассоциированные с другими патогенными флавивирусами, включают желтую лихорадку, лихорадку денге, геморрагическую лихорадку и энцефалит.
Однако соединения по изобретению также могут быть особенно привлекательны благодаря тому факту, что они лишены активности против других вирусов, в частности, против ВИЧ. ВИЧ-инфицированные пациенты часто страдают от со-инфекций, таких как HCV. Лечение таких пациентов ингибитором HCV, который также ингибирует ВИЧ, может приводить к возникновению резистентных штаммов ВИЧ.
Благодаря своим противовирусным свойствам, в частности своим противовирусным свойствам против HCV-инфекции соединения формулы (I) или любой их подгруппы, включая стереохимически изомерные формы и их N-оксиды, соли, гидраты и сольваты, применимы для лечения индивидуумов, подвергшихся вирусной инфекции, в частности, инфекции HCV, и для профилактики таких инфекций. В большинстве случаев соединения по настоящему изобретению могут быть применимы при лечении теплокровных животных, инфицированных вирусами, в частности флавивирусами, такими как HCV.
Следовательно, соединения по настоящему изобретению или любой их подгруппы можно применять в качестве лекарственных препаратов. Упомянутое применение в качестве лекарственного препарата или способ лечения включает систематическое введение инфицированным вирусом субъектам или субъектам, восприимчивым к вирусным инфекциям, количества, эффективного для борьбы с состояниями, ассоциированными с вирусной инфекцией, в частности с вирусной инфекцией HCV.
Настоящее изобретение также относится к применению настоящих соединений или любой их подгруппы для производства лекарственного препарата для лечения или профилактики вирусных инфекций, в частности вирусной инфекции HCV.
Кроме того, настоящее изобретение относится к способу лечения теплокровного животного, инфицированного вирусом или подвергающегося риску инфицирования вирусом, в частности вирусом HCV; упомянутому способу, включающему введение противовирусно эффективного количества соединений формулы (I) или любых соединений из их подгрупп, которые здесь указаны.
Настоящее изобретение также относится к комбинациям соединения формулы (I) или любой из его подгруппы, которая здесь указана, с другими средствами против HCV-инфекции. В одном из вариантов осуществления изобретение относится к комбинации соединения формулы (I) или любой его подгруппы, по меньшей мере, с одним средством против HCV-инфекции. В конкретном варианте осуществления изобретение относится к комбинации соединения формулы (I) или любой его подгруппы, по меньшей мере, с двумя средствами против HCV-инфекции. В конкретном варианте осуществления изобретение относится к комбинации соединения формулы (I) или любой его подгруппы, по меньшей мере, с тремя средствами против HCV-инфекции. В конкретном варианте осуществления изобретение относится к комбинации соединения формулы (I) или любой его подгруппы, по меньшей мере, с четырьмя средствами против HCV-инфекции.
В качестве лекарственного препарата в комбинированной терапии можно применять комбинацию ранее известного соединения против HCV-инфекции, такого как интерферон-α (IFN-α), пегилированный интерферон-α, рибаварин или их комбинация, и соединения формулы (I) или любой его подгруппы. В варианте осуществления термин "комбинированная терапия" относится к продукту, содержащему в обязательном порядке (a) соединение формулы (I) и (b), по меньшей мере, одно другое соединение против HCV-инфекции, в качестве объединенного препарата для одновременного, отдельного или последовательного применения в лечении HCV-инфекций, в частности в лечении инфекций с HCV.
Соединения против HCV-инфекции охватывают средства, выбранные из ингибиторов полимеразы HCV, R-7128, MK-0608, ABT-33, VCH759, PF-868554, GS9190, NM283, валопицитабина, PSI-6130, XTL-2125, NM-107, R7128 (R4048), GSK625433, R803, R-1626, BILB-1941, HCV-796, JTK-109 и JTK-003, ANA-598, IDX-184, MK-3281, MK-1220, бензимидазольных производных, бензо-1,2,4-тиадиазиновых производных, фенилаланиновых производных, A-831 и A-689; ингибиторов HCV-протеаз (NS2-NS3 и NS3-NS4A), соединений, описанных в патентной заявке WO02/18369 (см., например, стр. 273, строки 9-22 и от стр. 274, строка 4 до стр. 276, строка 11), BI-1335, TMC435350, MK7009, ITMN-191, BILN-2061, VX-950, BILN-2065, BMS-605339, VX-500, SCH 503034; ингибиторов других мишеней в жизненном цикле HCV, включая ингибиторы геликазы и металлопротеазы, ISIS-14803; иммуномодулирующих средств, таких как α-, β- и γ-интерфероны, такие как rIFN-α-2b, rIFN-α-2ba, консенсусный IFN-α (инферген), ферон, реаферон, интермакс α, rIFN-β, инферген + актиммун, IFN-омега с DUROS, альбуферон, локтерон, ребиф, пероральный IFN-α, IFN-α-2b XL, AVI-005, пегилированный инферген, соединений пегилированного дериватизированного интерферона-α, таких как пегилированный rIFN-α-2b, пегилированный rIFN-α-2a, пегилированный IFN-β, соединений, которые стимулируют синтез интерферона в клетках, интерлейкинов, агонистов toll-подобных рецепторов (TLR), соединений, которые усиливают развитие клеточного ответа Т-хелперов типа 1 и тимозина; других противовирусных средств, таких как рибаварин, аналоги рибаварина, такие как ребетол, копегус и вирамидин (тарибавирин), амантадин и тельбивудин, ингибиторов внутренней посадки рибосомы, ингибиторов α-глюкозидазы-1, таких как MX-3253 (целгосивир) и UT-231B, гепатопротекторов, таких как IDN-6556, ME-3738, LB-84451 и MitoQ, вирусных ингибиторов широкого спектра, таких как IMPDH-ингибиторы (например, соединения, описанные в патентах США №№ 5807876, 6498178, 6344465, 6054472, патентных заявках WO97/40028, WO98/40381, WO00/56331, микофеноловая кислота и ее производные, и включая, но не ограничиваясь перечисленным, VX-497, VX-148, и/или VX-944); и других лекарственных средств для лечения HCV, таких как задаксин, нитазоксанид, BIVN-401 (виростат), PYN-17 (алтирекс), KPE02003002, актилон (CPG-10101), KRN-7000, цивацир, GI-5005, ANA-975, XTL-6865, ANA-971, NOV-205, тарвацин, EHC-18, NIM811, DEBIO-025, VGX-410C, EMZ-702, AVI 4065, бавитуксимаб и оглуфанид; или комбинаций любых вышеупомянутых препаратов.
Таким образом, для борьбы с HCV-инфекциями или их лечением соединения формулы (I) или любых их подгрупп можно вводить совместно в комбинации, например, с интерфероном-α (IFN-α), пегилированным интерфероном-α, рибаварином или их комбинацией, а также с терапевтическими средствами, основанными на антителах, нацеленных против эпитопов HCV, коротких интерферирующих РНК (si-РНК), рибозимах, ДНКзимов, антисмысловой РНК, низкомолекулярных антагонистах, например, NS3-протеазы, NS3-геликазы и NS5B-полимеразы.
Комбинации по настоящему изобретению можно применять в качестве лекарственных препаратов. Соответственно, настоящее изобретение относится к применению соединения формулы (I) или любой его подгруппы, которая указана выше, для производства лекарственного препарата, применимого для ингибирования активности HCV у млекопитающего, инфицированного вирусами HCV, при котором упомянутый лекарственный препарат применяется в комбинированной терапии; упомянутая комбинированная терапия, в частности, включает соединение формулы (I) и, по меньшей мере, одно другое соединение, ингибирующее HCV, например IFN-α, пегилированный IFN-α, рибаварин или их комбинацию.
Кроме того, известно, что большой процент пациентов, инфицированных вирусом иммунодефицита человека 1 (ВИЧ), также инфицированы вирусом гепатита HCV, то есть они совместно инфицированы HCV/ВИЧ. По-видимому, ВИЧ-инфекция неблагоприятно влияет на все стадии HCV-инфекции, приводя к повышенной вирусной персистенции и ускоренному развитию связанного с HCV заболевания печени. В свою очередь, HCV-инфекция может влиять на лечение ВИЧ-инфекции, увеличивая частоту возникновения печеночной токсичности, вызванную противовирусными препаратами.
Следовательно, настоящее изобретение также относится к комбинациям соединения формулы (I) или любой его подгруппы со средствами против ВИЧ-инфекции. Также в качестве лекарственного препарата можно применять комбинацию одного или нескольких дополнительных соединений против ВИЧ-инфекции и соединение формулы (I) или любой его подгруппы. В частности, упомянутую комбинацию можно применять для ингибирования репликации HCV и ВИЧ.
Термин "комбинированная терапия" также охватывает продукт, содержащий (a) соединение формулы (I) или любой его подгруппы, (b), по меньшей мере, одно соединение против ВИЧ-инфекции и (c) необязательно, по меньшей мере, одно другое соединение против- HCV-инфекции в виде комбинированного препарата для одновременного, отдельного или последовательного применения для лечения HCV и ВИЧ-инфекций, в частности для лечения HCV и ВИЧ-инфекций или для профилактики или лечения состояний, ассоциированных с HCV и ВИЧ.
Таким образом, настоящее изобретение также относится к продукту, содержащему (a), по меньшей мере, одно соединение формулы (I) или любой его подгруппы, и (b) одно или несколько дополнительных соединений против ВИЧ-инфекции в виде комбинированного препарата для одновременного, отдельного или последовательного применения для противовирусного лечения HCV- и ВИЧ-инфекций. Различные лекарственные средства могут быть объединены в одном препарате вместе с фармацевтически приемлемыми носителями. Упомянутые соединения против ВИЧ-инфекции могут представлять собой любые известные противоретровирусные соединения, такие как сурамин, пентамидин, тимопентин, кастаноспермин, декстран (декстран-сульфат), фоскарнет-натрий (тринатрийфосфоноформиат); нуклеозидные ингибиторы обратной транскриптазы (NRTI), например зидовудин (AZT), диданозин (ddI), зальцитабин (ddC), ламивудин (3ТС), ставудин (d4T), эмтрицитабин (FTC), абакавир (АВС), амдоксовир (DAPD), элвуцитабин (АСН-126443), AVX 754 ((-)-dOTC), фозивудин тидоксил (FZT), фосфазид, HDP-990003, KP-1461, MIV-210, рацивир (PSI-5004), UC-781 и т.п.; ненуклеозидные ингибиторы обратной транскриптазы (NNRTI), такие как делавирдин (DLV), эфавиренз (EFV), невирапин (NVP), дапивирин (ТМС120), этравирин (ТМС125), рилпивирин (ТМС278), DPC-082, (+)-каланолид A, BILR-355 и т.п.; нуклеотидные ингибиторы обратной транскриптазы (NtRTI), например, тенофовир ((R)-PMPA) и фумарат тенофовирдисопроксила (TDF) и т.п.; нуклеотид-конкурирующие ингибиторы обратной транскриптазы (NcRTI), например NcRTI-1 и т.п.; ингибиторы транс-активирующих протеинов, такие как ТАТ-ингибиторы, например, RO-5-3335; BI-201 и т.п.; ингибиторы REV; ингибиторы протеазы, например ритонавир (RTV), саквинавир (SQV), лопинавир (АВТ-378 или LPV), индинавир (IDV), ампренавир (VX-478), ТМС-126, нельфинавир (AG-1343), атазанавир (BMS 232632), дарунавир (TMC114), фосампренавир (GW433908 или VX-175), бреканавир (GW-640385, VX-385), P-1946, PL-337, PL-100, типранавир (PNU-140690), AG-1859, AG-1776, Ro-0334649 и т.п.; ингибиторы входа, которые включают ингибиторы слияния (например, энфувиртид (Т-20)), ингибиторы присоединения и ингибиторы со-рецепторов; последние включают антагонистов CCR5 (например, анкривирок, CCR5mAb004, маравирок (UK-427,857), PRO-140, TAK-220, TAK-652, викривирок (SCH-D, SCH-417,690)) и антагонистов CXR4 (например, AMD-070, KRH-27315); примерами ингибиторов входа являются PRO-542, TNX-355, BMS-488043, BlockAide/CRTM, FP 21399, hNMO1, нонакин, VGV-I; ингибитором нагноения, например, является РА-457; ингибиторы вирусной интегразы, например, ралтегравир (МК-0518), элвитегравир (JTK-303, GS-9137), BMS-538158; рибозимы; иммуномодуляторы; моноклональные антитела; генотерапия; вакцины; siРНК; антисмысловые РНК; бактерициды; ингибиторы "цинковых пальцев".
Следовательно, с помощью настоящей композиции можно удобно лечить пациентов, инфицированных HCV, также страдающих от состояний, ассоциированных с ВИЧ, или даже другие патогенные ретровирусы, такие как СПИД, СПИД-ассоциированный комплекс (ARC), прогрессирующая генерализованная лимфаденопатия (PGL), а также хронические заболевания ЦНС, вызванные ретровирусами, например, такие как деменция и рассеянный склероз, опосредованные ВИЧ.
Композиции можно получать в виде препаратов в подходящих фармацевтических лекарственных формах таких, как описанные выше лекарственные формы. Каждый из активных ингредиентов может быть приготовлен по отдельности, и препараты можно вводить совместно или в виде одного препарата, содержащего оба ингредиента и, если требуется, можно обеспечить дополнительные активные ингредиенты.
Подразумевается, что в применяемом здесь значении термин "композиция" охватывает продукт, содержащий указанные ингредиенты, а также любой продукт, который прямо или косвенно получается из комбинации указанных ингредиентов.
Термин "терапевтически эффективное количество" в применяемом здесь значении означает количество активного соединения или компонента или фармацевтического средства, которое вызывает биологический или медицинский ответ в ткани, системе, у животного или человека, которого в свете настоящего изобретения добивается исследователь, ветеринар, лечащий врач или другой врач-клиницист, который включает частичное снятие симптомов заболевания, подвергаемого лечению. Поскольку настоящее изобретение также относится к комбинациям, содержащим два или более средств, "терапевтически эффективное количество" в контексте комбинаций также означает количество средств, взятых вместе для того, чтобы комбинированный эффект вызывал требуемый биологический или медицинский ответ. Например, терапевтически эффективное количество композиции, содержащей (a) соединение формулы (I), и (b) - другое средство против HCV-инфекции, могло бы означать количество соединения формулы (I) и количество другого средства против HCV-инфекции, которые, когда взяты вместе, обладают комбинированным эффектом, который является терапевтически эффективным.
Подразумевается, что в большинстве случаев дневное, эффективное противовирусное количество может составлять от 0,01 мг/кг до 500 мг/кг массы тела, в частности от 0,1 мг/кг до 50 мг/кг массы тела. Подходящим образом можно вводить необходимую дозу в виде двух, трех, четырех или более субдоз с подходящими интервалами в течение дня. Упомянутые субдозы можно готовить в виде стандартных лекарственных форм, например, содержащих от 1 до 1000 мг, и в частности, от 5 до 200 мг активного ингредиента на стандартную лекарственную форму.
Как хорошо известно специалистам в данной области техники, точная дозировка и частота введения зависят от конкретного применяемого соединения формулы (I), конкретного состояния, подвергаемого лечению, тяжести данного состояния, подвергаемого лечению, возраста, массы тела, пола, степени расстройства и общего физического состояния конкретного пациента, а также другого лекарственного лечения, которое может принимать индивидуум. Кроме того, очевидно, что упомянутое дневное эффективное количество может снижаться или повышаться в зависимости от ответной реакции субъекта, подвергаемого лечению, и/или в зависимости от оценки врача, прописывающего соединения по настоящему изобретению. Следовательно, упомянутые выше диапазоны дневного эффективного количества являются только рекомендациями.
В одном из вариантов осуществления настоящего изобретения предлагается производственное изделие, содержащее композицию, эффективную для лечения HCV-инфекции или для ингибирования NS5B-полимеразы HCV; и упаковочный материал, содержащий этикетку, на которой указано, что композиция может применяться для лечения инфекции вируса гепатита C; в котором композиция содержит соединение формулы (I) или любой его подгруппы, или комбинацию, которая здесь описана.
Еще один вариант осуществления настоящего изобретения относится к набору или контейнеру, содержащему соединение формулы (I) или любой его подгруппы в количестве, эффективном для применения в качестве стандарта или реагента для проведения испытания или анализа для определения способности потенциальных фармацевтических средств ингибировать NS5B-полимеразу HCV, рост HCV или то и другое. Такой аспект изобретения может найти свое применение в фармацевтических исследовательских программах.
Соединения и комбинации по настоящему изобретению можно применять в высокопроизводительных анализах анализируемых веществ-мишеней, таких как анализы по измерению эффективности упомянутой комбинации для лечения HCV.
ПРИМЕРЫ
Подразумевается, что следующие примеры предназначены для иллюстрации настоящего изобретения и не ограничивают его. Если не указано иначе, очистку синтезированных соединений с помощью колоночной хроматографии или флэш-хроматографии осуществляют на колонке с силикагелем.
Если не указано иначе, конечные продукты характеризовали с помощью ЖХ/МС-анализа с применением колонки SunFire C18 3,5 мк, 4,6 x 100 мм и двух подвижных фаз: подвижной фазы A (10 мМ формиата аммония (NH4OOCH)+0,1% HCOOH в H2O) и подвижной фазы B (CH3CN). Температура колонки составляла 50°C, скорость потока 2 мл/мин, и градиент подвижной фазы A и подвижной фазы B характеризовался следующим образом:
Характеристики, получаемые с помощью ЖХ/МС-анализа, представляют собой время удерживания (Rt) для ВЭЖХ и подтверждение молекулярной массы (m/z).
Пример 1
Синтез 11,11-диоксида 25-циклогексил-4,10,19-триметил-5,6,9,10-тетрагидро-2H,8H-14,18:17,20-ди(метено)-1,7,11,4,10,12,19-бензодиоксатиатетраазациклодокозин-3,13(4H,12H,19H)-диона 10

Стадия 1

Раствор метил-2-бром-3-циклогексил-1H-индол-6-карбоксилата (1, 6,14 г, 18,3 ммоль), 2-(бензилокси)фенилбороновой кислоты (5,00 г, 21,9 ммоль) и карбоната калия (5,80 г, 42 ммоль) в 450 мл смеси 1,2-диметоксиэтан/вода (4:1) тщательно продували аргоном. Затем добавляли хлорид транс-бис(трифенилфосфин)палладия(II) (0,641 г, 0,91 ммоль) и нагревали реакционную смесь при 70°C в атмосфере аргона в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом (AcOEt). Объединенные органические слои промывали насыщенным раствором NaHCO3 и насыщенным раствором соли, сушили над безводным Na2SO4 и фильтровали. Отфильтрованный раствор концентрировали в вакууме, получая при этом метил-2-[2-(бензилокси)фенил]-3-циклогексил-1H-индол-6-карбоксилат 2: m/z = 440 (M+Н)+.
Стадия 2

К раствору промежуточного продукта 2 (9,4 г, 21,4 ммоль) в сухом ДМФА добавляли NaH (810 мг, 32,1 ммоль). Затем при комнатной температуре добавляли иодметан (3,64 г, 25,7 ммоль). Спустя 12 часов реакционную смесь распределяли между водой (pH 6) и AcOEt. Органический слой сушили над безводным Na2SO4, фильтровали и затем упаривали. Остаток очищали колоночной хроматографией на диоксиде кремния (градиент: гептан/AcOEt от 1:0 до 80:20), получая при этом 7,8 г (80%) метил-2-[2-(бензилокси)фенил]-3-циклогексил-1-метил-1H-индол-6-карбоксилата 3: m/z = 454 (M+Н)+.
Стадия 3

Раствор промежуточного продукта 3 (4,00 г) в MeOH (36 мл) и AcOH (4 мл) подвергали гидрированию в присутствии гидроксида палладия в качестве катализатора. Спустя 12 часов реакционную смесь фильтровали и упаривали фильтрат, получая при этом метил-3-циклогексил-2-(2-гидроксифенил)-1-метил-1H-индол-6-карбоксилат 4: m/z = 364 (M+Н)+.
Стадия 4

Раствор промежуточного продукта 4 (4,00 г, 11,0 ммоль), трет-бутил-2-бромацетата (2,36 г, 12,1 ммоль) и карбоната калия (3,04 г, 22,0 ммоль) перемешивали при комнатной температуре. Спустя 72 часа реакционную смесь концентрировали в вакууме и остаток распределяли между CH2Cl2 и водой. Органический слой сушили, остаток растворяли в CH2Cl2 (20 мл) и добавляли TFA (20 мл). После 2 часов при комнатной температуре реакционную смесь упаривали в вакууме, получая при этом 4,2 г (90%) {2-[3-циклогексил-6-(метоксикарбонил)-1-метил-1H-индол-2-ил]фенокси}уксусной кислоты 5: m/z = 422 (M+Н)+.
Стадия 5
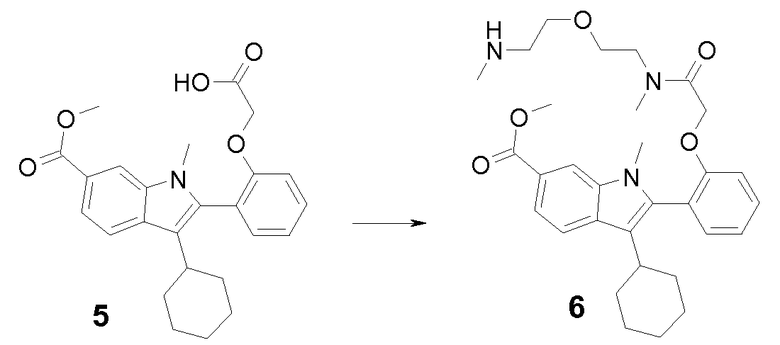
Раствор промежуточного продукта 5 (650 мг, 1,54 ммоль), N-метил-2-(метиламиноэтилокси)этиламина (1,02 г, 7,71 ммоль), диизопропилэтиламина (808 мкл, 4,63 ммоль) и HATU (880 мг, 2,31 ммоль) в сухом ТГФ (25 мл) перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали в вакууме. Остаток распределяли между AcOEt и водой. Органический слой сушили над безводным Na2SO4 и упаривали, получая при этом 750 мг (91%) метил-3-циклогексил-1-метил-2-{2-[2-(метил{2-[2-(метиламино)этокси]этил}амино)-2-оксоэтокси]фенил}-1H-индол-6-карбоксилата 6: m/z = 536 (M+Н)+.
Стадия 6

Раствор промежуточного продукта 6 (620 мг, 1,16 ммоль) и сульфамида (900 мг, 9,36 ммоль) в диоксане (10 мл) нагревали при 100°C в микроволновой печи в течение 60 минут. Реакционную смесь охлаждали до комнатной температуры и затем упаривали в вакууме. Остаток растирали с водой, фильтровали и промывали водой. Порошок растворяли в AcOEt. Раствор сушили над безводным Na2SO4, фильтровали и упаривали фильтрат, получая при этом 610 мг (86%) метил-3-циклогексил-1-метил-2-(2-{2-[метил-(2-{2-[метил(сульфамоил)амино]этокси}этил)амино]-2-оксоэтокси}фенил)-1H-индол-6-карбоксилат 7 в виде желтоватого порошка: m/z = 615 (M+Н)+.
Стадия 7
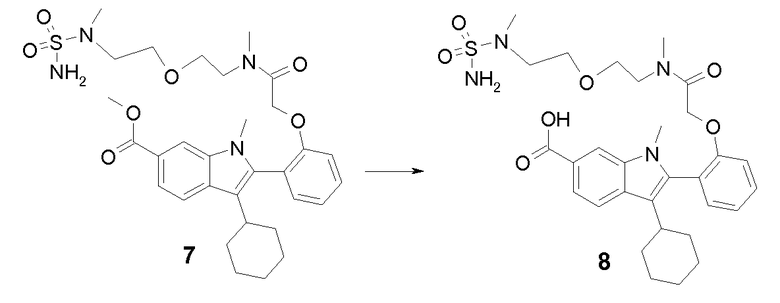
Раствор NaOH (1,00 г, 25 ммоль) в воде (5 мл) добавляли при перемешивании к раствору промежуточного продукта 7 (370 мг, 0,602 ммоль) в MeOH (30 мл) и ТГФ (10 мл). Спустя 5 часов раствор концентрировали в вакууме. Затем с помощью уксусной кислоты (AcOH) доводили pH до 5. Затем реакционную смесь экстрагировали AcOEt, органический слой сушили над безводным Na2SO4, фильтровали и упаривали фильтрат, получая при этом 300 мг (83%) 3-циклогексил-1-метил-2-(2-{2-[метил(2-{2-[метил(сульфамоил)амино]этокси}этил)амино]-2-оксоэтокси}фенил)-1H-индол-6-карбоновой кислоты 8 в виде белого порошка: m/z = 601 (M+Н)+.
Стадия 8

К раствору промежуточного продукта 8 (300 мг, 0,50 ммоль) в сухом ацетонитриле (CH3CN) (25 мл) при перемешивании добавляли карбонилдиимидазол (405 мг, 2,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, после чего наблюдалось полное превращение. Полученный раствор упаривали, остаток очищали флэш-хроматографией на диоксиде кремния (градиент: AcOEt/CH3CN от 1:0 до 0:1), получая при этом 315 мг (97%) 2-{2-[3-циклогексил-6-(1H-имидазол-1-илкарбонил)-1-метил-1H-индол-2-ил]фенокси}-N-метил-N-(2-{2-[метил(сульфамоил)амино]этокси}этил)ацетамида 9 в виде белого порошка: m/z = 651 (M+Н)+.
Стадия 9

К раствору 9 (315 мг, 0,48 ммоль) в CH3CN (5 мл) добавляли DBU (147 мг, 0,97 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем pH реакционной смеси с помощью AcOH доводили до 5. Раствор упаривали. Остаток очищали сначала колоночной хроматографией на диоксиде кремния (градиент AcOEt/CH3CN от 1:0 до 0:1) и затем препаративной ВЭЖХ, получая при этом требуемый продукт 11,11-диоксид 25-циклогексил-4,10,19-триметил-5,6,9,10-тетрагидро-2H,8H-14,18:17,20-ди(метено)-1,7,11,4,10,12,19-бензодиоксатиатетраазациклодокозин-3,13(4H, 12H,19H)-диона 10: m/z = 583 (M+Н)+, Rt = 5,13 мин.
Пример 2
Синтез 10,10-диоксида 24-циклогексил-4,9,18-триметил-4,5,6,7,8,9-гексагидро-13,17:16,19-ди(метено)-1,10,4,9, 11,18-бензоксатиатетраазациклогеникозин-3,12(2H,11H,18H)-диона 15

Стадия 1

Соединение 11 синтезировали с 88%-ным выходом из промежуточного продукта 5 и N,N'-диметилбутилендиамина согласно процедуре, о которой сообщалось в связи с синтезом метил-3-циклогексил-1-метил-2-[2-(2-{метил[4-(метиламино)бутил]амино}-2-оксоэтокси)фенил]-1H-индол-6-карбоксилата 6: m/z = 520 (M+Н)+.
Стадия 2
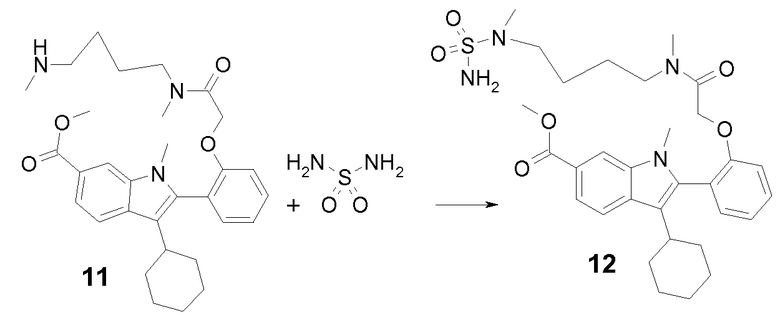
Метил-3-циклогексил-1-метил-2-{2-[2-(метил{4-[метил(сульфамоил)амино]бутил}амино)-2-оксоэтокси]фенил}-1H-индол-6-карбоксилат 12 синтезировали с 68%-ным выходом из промежуточного продукта 11 согласно процедуре, о которой сообщалось в связи с синтезом промежуточного продукта 7: m/z = 599 (M+Н)+.
Стадия 3

3-циклогексил-1-метил-2-{2-[2-(метил{4-[метил(сульфамоил)амино]бутил}амино)-2-оксоэтокси]фенил}-1H-индол-6-карбоновую кислоту 13 синтезировали с 78%-ным выходом из промежуточного продукта 12 согласно процедуре, о которой сообщалось в связи с синтезом промежуточного продукта 8: m/z = 585 (M+Н)+.
Стадия 4

2-{2-[3-циклогексил-6-(1H-имидазол-1-илкарбонил)-1-метил-1H-индол-2-ил]фенокси}-N-метил-N-{4-[метил(сульфамоил)амино]бутил}ацетамид 14 синтезировали с 92%-ным выходом из промежуточного продукта 13 согласно процедуре, о которой сообщалось в связи с синтезом промежуточного продукта 9: m/z = 635 (M+Н)+.
Стадия 5

10,10-диоксид 24-циклогексил-4,9,18-триметил-4,5,6,7,8,9-гексагидро-13,17:16,19-ди(метено)-1,10,4,9,11,18-бензоксатиатетраазациклогеникозин-3,12(2H,11H,18H)-диона 15 синтезировали с 12%-ным выходом из промежуточного продукта 14 согласно процедуре, о которой сообщалось в связи с синтезом продукта 10: m/z = 567 (M+Н)+, Rt = 5,07 мин.
Пример 3
Синтез 11,11-диоксида 25-циклогексил-10,19-диметил-5,6,7,8,9,10-гексагидро-2H-14,18:17,20-ди(метено)-1,11,4,10,12,19-бензоксатиатетраазациклодокозин-3,13(4H,12H,19H)-диона 21

Стадия 1
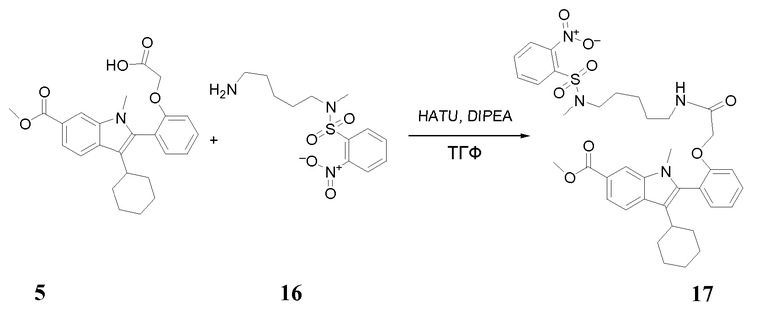
Раствор 2-(2-(3-циклогексил-6-(метоксикарбонил)-1-метил-1H-индол-2-ил)фенокси)уксусной кислоты 5 (1 г, 2,373 ммоль), N-(5-аминопентил)-N-метил-2-нитробензолсульфонамида 16 (0,715 г, 1 экв.), диизопропилэтиламина (0,92 г, 3 экв.) и HATU (1,353 г, 1,5 экв.) в сухом ТГФ (25 мл) перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь последовательно выливали в воду, экстрагировали дихлорметаном, сушили над MgSO4 и концентрировали. Полученный остаток очищали флэш-хроматографией с применением градиента метанола в DCM в качестве элюента, получая при этом 1,27 г (76%-ный выход) указанного в заголовке продукта метил-3-циклогексил-1-метил-2-(2-{2-[(5-{метил[(2-нитрофенил)сульфонил]амино}пентил)амино]-2-оксоэтокси}фенил)-1H- индол-6-карбоксилата 17; m/z = 705 (M+Н)+.
Стадия 2

К раствору метил-3-циклогексил-1-метил-2-(2-{2-[(5-{метил[(2-нитрофенил)сульфонил]амино}пентил)амино]-2-оксоэтокси}фенил)-1H-индол-6-карбоксилата 17 (1,27 г, 1,802 ммоль) в сухом ДМФА (50 мл) добавляли тиофенол (0,397 г, 2 экв.) и карбонат цезия (1,174 г, 2 экв.) при комнатной температуре (RT). Реакционную смесь перемешивали в течение 40 часов, затем последовательно выливали в воду со льдом, экстрагировали дихлорметаном, сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией с применением градиента этилацетата в DCM в качестве элюента, получая при этом 600 мг (64%) указанного в заголовке продукта метил-3-циклогексил-1-метил-2-[2-(2-{[5-(метиламино)пентил]амино}-2-оксоэтокси)фенил]-1H-индол-6-карбоксилата 18 в виде белой пены; m/z = 520 (M+Н)+.
Стадия 3
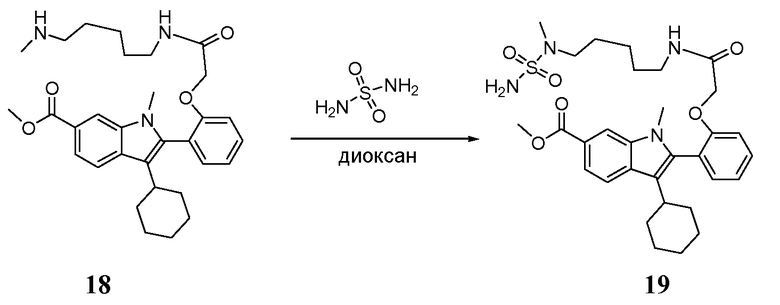
К раствору метил-3-циклогексил-1-метил-2-[2-(2-{[5- (метиламино)пентил]амино}-2-оксоэтокси)фенил]-1H-индол-6-карбоксилата 18 (0,460 г, 0,885 ммоль) в диоксане (10 мл) добавляли сульфамид (0,851 г, 8,85 ммоль). Полученную смесь перемешивали при 100°C в микроволновой печи в течение 4 часов, затем при 105°C в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры, затем концентрировали. Остаток растирали с дихлорметаном и полученный осадок избытка сульфамида отфильтровывали. Затем удаляли растворитель и остаток очищали колоночной хроматографией с применением градиента метанола в дихлорметане, получая при этом 447 мг (84%) указанного в заголовке продукта метил-3-циклогексил-1-метил-2-{2-[2-({5-[метил(сульфамоил)амино]пентил}амино)-2-оксоэтокси]фенил}-1H- индол-6-карбоксилата 19; m/z = 599 (M+Н)+.
Стадия 4
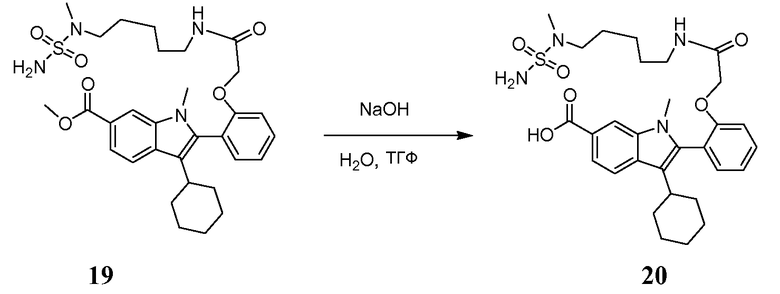
К раствору метил-3-циклогексил-1-метил-2-{2-[2-({5-[метил(сульфамоил)амино]пентил}амино)-2-оксоэтокси]фенил}-1H- индол-6-карбоксилата 19 (480 мг, 0,802 ммоль) в ТГФ (50 мл) добавляли раствор гидроксида натрия (1,283 г, 40 экв.) в воде. Полученную смесь перемешивали при комнатной температуре (RT) в течение ночи. Затем смесь последовательно выливали в воду, подкисляли с помощью HCl до pH=5, экстрагировали дихлорметаном, сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией с применением градиента метанола в DCM, получая при этом 330 мг (70%) указанного в заголовке продукта 3-циклогексил-1-метил-2-{2-[2-({5-[метил(сульфамоил)амино]пентил}амино)-2-оксоэтокси]фенил}-1H-индол-6-карбоновой кислоты 20 в виде твердого белого вещества; m/z = 585 (M+Н)+.
Стадия 5

К раствору 3-циклогексил-1-метил-2-{2-[2-({5-[метил(сульфамоил)амино]пентил}амино)-2-оксоэтокси]фенил}-1H- индол-6-карбоновой кислоты 20 (330 мг, 0,554 ммоль) в сухом ацетонитриле (CH3CN) (25 мл) при перемешивании добавляли карбонилдиимидазол (110 мг, 0,677 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после которых наблюдалось полное превращение. Затем реакционную смесь разбавляли ацетонитрилом (25 мл) и добавляли DBU (172 мг, 2 экв.). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем растворитель удаляли в вакууме, и полученный остаток последовательно растворяли в дихлорметане, два раза промывали 2M раствором HCl, затем насыщенным раствором соли. Полученный органический слой сушили над сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией с применением градиента этилацетата в дихлорметане, получая при этом 220 мг (69%) указанного в заголовке продукта 11,11-диоксида 25-циклогексил-10,19-диметил-5,6,7,8,9,10-гексагидро-2H-14,18:17,20-ди(метено)-1,11,4,10,12,19-бензоксатиатетраазациклодокозин-3,13(4H,12H,19H)-диона 21 в виде белого порошка; m/z = 567 (M+Н)+.
1H ЯМР (400 МГц, CDCl3) δ м.д. 0,85-0,9 (м, 1H), 1,00-1,40 (м, 7H), 1,4-1,5 (м, 2H), 1,75-1,85 (м, 7H), 2,2-2,25 (м, 1H), 2,5-2,6 (м, 1H), 3,00-3,1 (м, 1H), 3,2-3,3 (м, 2H), 3,25 (с, 3H), 3,5 (с, 3H), 4,3 (д, J=14 Гц, 1H), 4,6 (д, J=14 Гц, 1H), 5,5 (м, 1H), 6,9 (д, J=7,84 Гц, 1H), 7,3 (дд, J=8,1 и J=7 Гц, 1H), 7,4 (д, J=7 Гц, 1H), 7,55 (дд, J=8,1 и J=7 Гц, 1H), 7,6 (д, J=8 Гц, 1H), 7,9 (д, J=8 Гц, 1H), 8 (с, 1H).
Пример 4
Синтез 20,20-диоксида 30-циклогексил-12,21-диметил-4-окса-20-тиа-1,12,19,21,24-пентаазапентацикло[22.2.2.1 11,14 .1 13,17 .0 5,10 ]триаконта-5,7,9,11(30),13(29),14,16-гептаен-2,18-диона 22
Синтез указанного в заголовке соединения 22 осуществляли согласно 5-стадийной процедуре, о которой сообщалось в связи с синтезом соединения 21, с применением на первой стадии N-метил-2-нитро-N-(2-(пиперазин-1-ил)этил)бензолсульфонамида вместо N-(5-аминопентил)-N-метил-2-нитробензолсульфонамида 16, получая при этом 33 мг твердого белого вещества; m/z = 594 [M+H]+.
1H ЯМР (400 МГц, CDCl3) δ м.д. 0,5-0,8 (м, 1H), 1,1-1,3 (м, 8H), 1,5-1,8 (м, 7H), 2,05-2,1 (м, 1H), 2,45-2,5 (м, 2H), 2,6-2,65 (м, 1H), 2,75-3,00 (м, 2 H), 2,95 (с, 3H), 3,47 (с, 3H), 3,48-3,5 (м, 1H), 3,95-4,00 (м, 1H), 4,4 (д, J=14 Гц, 1H), 4,75 (д, J=14 Гц, 1H), 7,05 (д, J=8,3 Гц, 1H), 7,2 (дд, J=7,4 и J=7,3 Гц, 1H), 7,4 (д, J=7,3 Гц, 1H), 7,5 (дд, J=8,3 и J=7,4 Гц, 1H), 7,6 (д, J=8,4 Гц, 1H), 7,85 (д, J=8,4 Гц, 1H), 7,95 (с, 1H).
Пример 5
Синтез 10,10-диоксида 24-циклогексил-9,18-диметил-4,5,6,7,8,9-гексагидро-13,17:16,19-ди(метено)-1,10,4,9,11,18-бензоксатиатетраазациклогеникозин-3,12(2H,11H,18H)-диона 32
Стадия 1
Исходный материал для синтеза сложный эфир (2-бром-3-циклогексил-6-трет-бутилиндола) 23 описан в заявке США 2007270406A1

К раствору промежуточного продукта 23 (4 г, 10,57 ммоль) в сухом ТГФ добавляли NaH (400 мг, 15,86 ммоль). Затем при комнатной температуре добавляли иодметан (3 г, 21,15 ммоль). Спустя 12 часов реакционную смесь распределяли между водой (pH 6) и AcOEt. Органический слой сушили над безводным Na2SO4, фильтровали и затем упаривали. Остаток очищали колоночной хроматографией на диоксиде кремния (градиент гептан/EtOAc от 1:0 до 80:20), получая при этом 3,8 г (92%) трет-бутил-2-бром-3-циклогексил-1-метил-1H-индол-6-карбоксилата 24: m/z = 393 (M+Н)+.
Стадия 2

Раствор трет-бутил-2-бром-3-циклогексил-1-метил-1H-индол-6-карбоксилата (24, 500 мг, 1,27 ммоль), 2-гидроксифенилбороновой кислоты (211 мг, 1,53 ммоль) и карбоната калия (1,35 г, 12,74 ммоль) в 100 мл смеси 1,2-диметоксиэтан/вода (4:1) тщательно продували аргоном. Затем добавляли хлорид транс- бис(трифенилфосфин)палладия(II) (0,147 г, 0,127 ммоль) и в течение 12 часов нагревали реакционную смесь при 100°C в атмосфере аргона. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом (EtOAc). Объединенные органические слои промывали насыщенным раствором NaHCO3 и насыщенным раствором соли, сушили над безводным Na2SO4 и фильтровали. Отфильтрованный раствор концентрировали в вакууме, получая при этом трет-бутил-3-циклогексил-2-(2-гидроксифенил)-1-метил-1H-индол-6-карбоксилат 25: m/z = 406 (M+Н)+.
Стадия 3

Раствор промежуточного продукта 25 (0,6 г, 1,5 ммоль), метил-2-бромацетата (0,281 г, 1,83 ммоль) и карбоната калия (0,423 г, 3,06 ммоль) в 50 мл MeCN перемешивали при комнатной температуре. Спустя 12 часов реакционную смесь концентрировали в вакууме и остаток распределяли между CH2Cl2 и водой. Органический слой сушили и упаривали в вакууме, получая при этом 0,7 г (96%) трет-бутил-3-циклогексил-2-[2-(2-метокси-2-оксоэтокси)фенил]-1-метил-1H-индол-6-карбоксилата 26: m/z = 478 (M+Н)+.
Стадия 4
К раствору трет-бутил-3-циклогексил-2-(2-(2-метокси-2-оксоэтокси)фенил)-1-метил-1H-индол-6-карбоксилата 26 (0,7 г, 1,46 ммоль) в 50 мл дихлорметана добавляли трифторуксусную кислоту (4,2 г, 36,6 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Удаляли растворитель, затем остаток очищали колоночной хроматографией с применением дихлорметана/метанола, получая при этом 0,59 г (96%) 3-циклогексил-2-[2-(2-метокси-2-оксоэтокси)фенил]-1-метил-1H-индол-6-карбоновой кислоты 27: m/z = 422 (M+Н)+.
Стадия 5
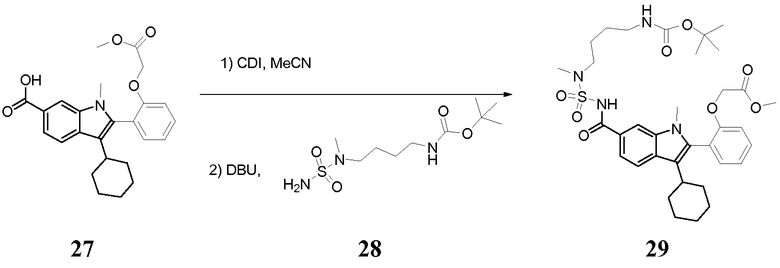
К раствору промежуточного продукта 27 (590 мг, 1,4 ммоль) в сухом ацетонитриле (CH3CN) (30 мл) при перемешивании добавляли карбонилдиимидазол (567 мг, 3,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после которых наблюдалось полное превращение. Добавляли трет-бутил-4-(метил(сульфамоил)амино)бутилкарбамат 28 (394 мг, 1,4 ммоль) и DBU (426 мг, 2,8 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем растворитель удаляли в вакууме, и полученный остаток последовательно растворяли в дихлорметане, два раза промывали раствором 2M HCl, затем насыщенным раствором соли. Полученный органический слой сушили над сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией с применением градиента этилацетата в дихлорметане, получая при этом 675 мг (70%) указанного в заголовке метил-[2-(6-{[{4-[(трет-бутоксикарбонил)амино]бутил}(метил)сульфамоил]карбамоил}-3-циклогексил-1-метил-1H-индол-2-ил)фенокси]ацетат 29 в виде белого порошка; m/z = 685 (M+Н)+.
Стадия 6

К раствору промежуточного продукта 29 (0,675 г, 1 ммоль) в 50 мл дихлорметана добавляли трифторуксусную кислоту (2,86 г, 20 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Удаляли растворитель, затем остаток очищали колоночной хроматографией с применением дихлорметана/метанола, получая при этом 0,55 г (95%) метил-[2-(6-{[(4-аминобутил)(метил)сульфамоил]карбамоил}-3-циклогексил-1-метил-1H-индол-2-ил)фенокси]ацетата 30: m/z = 585 (M+Н)+.
Стадия 7

К раствору промежуточного продукта 30 (550 мг, 0,941 ммоль) в ТГФ и метаноле (4/1) (50 мл) добавляли раствор гидроксида лития (79 мг, 1,88 ммоль) в воде. Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь последовательно выливали в воду, подкисляли до pH=5 с помощью HCl, экстрагировали дихлорметаном, сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией с применением градиента метанола в DCM, получая при этом 500 мг (93%) указанного в заголовке продукта [2-(6-{[(4-аминобутил)(метил)сульфамоил]карбамоил}-3-циклогексил-1-метил-1H-индол-2-ил)фенокси]уксусной кислоты 31 в виде твердого белого вещества; m/z = 571 (M+Н)+.
Стадия 8

К раствору 2-(2-(6-(N-(4-аминобутил)-N-метилсульфамоилкарбамоил)-3-циклогексил-1-метил-1H-индол-2-ил)фенокси)уксусной кислоты 31 (40 мг, 0,07 ммоль) в 50 мл ТГФ добавляли HATU (40 мг, 0,1 ммоль) и диизопропилэтиламин (27 мг, 0,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь последовательно выливали в воду, экстрагировали дихлорметаном, сушили над MgSO4 и концентрировали. Полученный остаток очищали флэш-хроматографией с применением градиента метанола в DCM в качестве элюента, получая при этом 5 мг (13%-ный выход) указанного в заголовке продукта 10,10-диоксида 24-циклогексил-9, 18-диметил-4,5,6,7,8,9-гексагидро-13,17:16,19-ди(метено)-1,10,4,9,11,18-бензоксатиатетраазациклогеникозин-3,12(2H,11H,18H)-диона 32; m/z = 553 (M+Н)+.
1H ЯМР (400 МГц, CDCl3) δ м.д. 0,85-0,9 (м, 1H), 1,00-1,40 (м, 7H), 1,4-1,5 (м, 2H), 1,75-1,85 (м, 5H), 2,2-2,25 (м, 1H), 2,5-2,6 (м, 1H), 3,00-3,1 (м, 1H), 3,2-3,3 (м, 2H), 3,25 (с, 3H), 3,5 (с, 3H), 4,3 (д, J=14 Гц, 1H), 4,6 (д, J=14 Гц, 1H), 5,5 (м, 1H), 6,9 (д, J=7,84 Гц, 1H), 7,3 (дд, J=8,1 и J=7 Гц, 1H), 7,4 (д, J=7 Гц, 1H), 7,55 (дд, J=8,1 и J=7 Гц, 1H), 7,6 (д, J=8 Гц, 1H), 7,9 (д, J=8 Гц, 1H), 8 (с, 1H).
Пример 6
Синтез 10,10-диоксида 24-циклогексил-18-циклопентил-4,9-диметил-4,5,6,7,8,9-гексагидро-13,17:16,19-ди(метено)-1,10,4,9,11,18-бензоксатиатетраазациклогеникозин-3, 12(2H,11H,18H)-диона 40
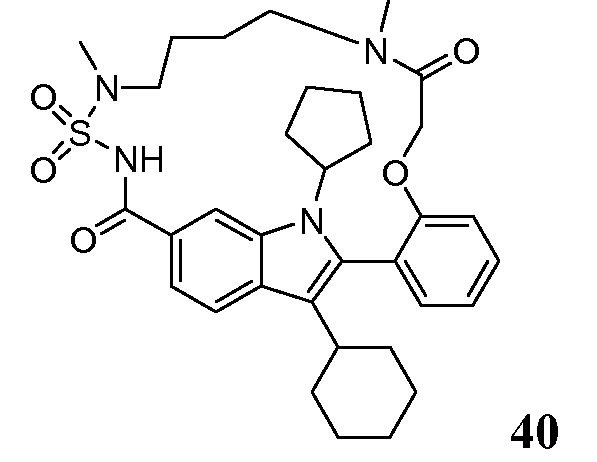
Стадия 1
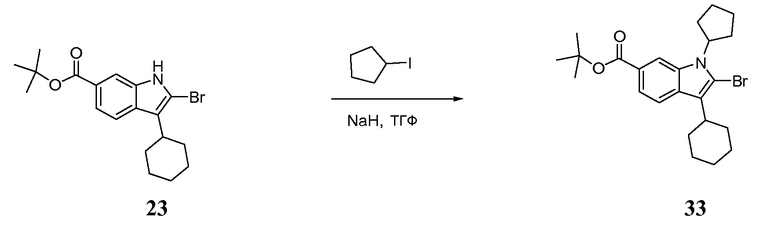
Для получения соединения 33 из соединения 23 и иодциклопентана в качестве исходного материала применяли такую же процедуру, как для получения соединения 24, получая при этом 500 мг (15%-ный выход) трет-бутил-2-бром-3-циклогексил-1-циклопентил-1H-индол-6-карбоксилата 33; m/z = 447 (M+Н)+.
Стадия 2

Для получения соединения 34 из промежуточного продукта 33 и 2-гидроксифенилбороновой кислоты в качестве исходного материала применяли такую же процедуру, как для получения соединения 25, получая при этом 400 мг (28%-ный выход) трет-бутил-3-циклогексил-1-циклопентил-2-(2-гидроксифенил)-1H-индол-6-карбоксилата 34; m/z = 460 (M+Н)+.
Стадия 3

Для получения соединения 35 из промежуточного продукта 34 и метил-2-бромацетата в качестве исходного материала применяли такую же процедуру, как для получения соединения 26, получая при этом 450 мг (97%-ный выход) трет-бутил-3-циклогексил-1-циклопентил-2-[2-(2-метокси-2-оксоэтокси)фенил]-1H-индол-6-карбоксилата 35; m/z = 532 (M+Н)+.
Стадия 4

Для получения соединения 36 из промежуточного продукта 35 в качестве исходного материала применяли такую же процедуру, как для получения соединения 31, получая при этом 410 мг (94%-ный выход) {2-[6-(трет-бутоксикарбонил)-3-циклогексил-1-циклопентил-1H-индол-2-ил]фенокси}уксусной кислоты 36; m/z = 518 (M+Н)+
Стадия 5

Для получения соединения 37 из промежуточного продукта 36 и N,N'-диметилбутилендиамина в качестве исходного материала применяли такую же процедуру, как для получения соединения 6, получая при этом 450 мг (92%-ный выход) трет-бутил-3-циклогексил-1-циклопентил-2-[2-(2-{метил[4-(метиламино)бутил]амино}-2-оксоэтокси)фенил]-1H-индол-6-карбоксилата 37; m/z = 617 (M+Н)+
Стадия 6
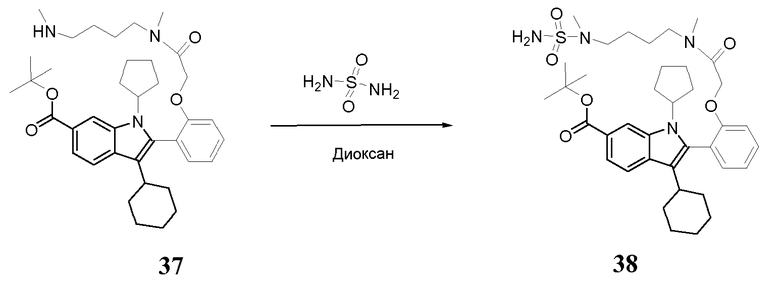
Для получения соединения 38 из промежуточного продукта 37 в качестве исходного материала применяли такую же процедуру, как для получения соединения 19, получая при этом 400 мг (79%-ный выход) трет-бутил-3-циклогексил-1-циклопентил-2-{2-[2-(метил-{4- [метил(сульфамоил)амино]бутил}амино)-2-оксоэтокси]фенил}-1H-индол-6-карбоксилата 38; m/z = 695 (M+Н)+
Стадия 7

Для получения соединения 39 из промежуточного продукта 38 в качестве исходного материала применяли такую же процедуру, как для получения соединения 27, получая при этом 150 мг (41%-ный выход) 3-циклогексил-1-циклопентил-2-{2-[2-(метил-{4-[метил(сульфамоил)амино]бутил}амино)-2-оксоэтокси]фенил}-1H-индол-6-карбоновой кислоты 39; m/z = 639 (M+Н)+.
Стадия 8
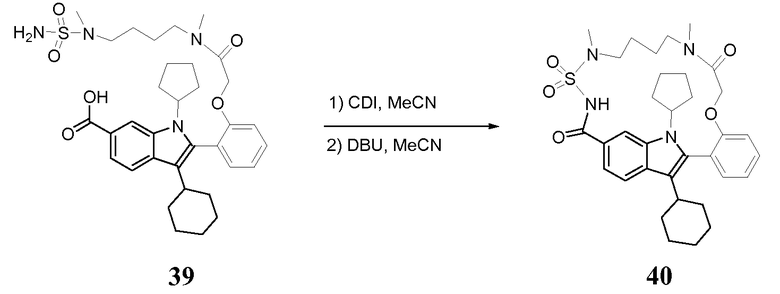
Для получения соединения 40 из промежуточного продукта 39 в качестве исходного материала применяли такую же процедуру, как для получения соединения 21, получая при этом 30 мг (21%-ный выход) 10,10-диоксида 24-циклогексил-18-циклопентил-4,9-диметил-4,5,6,7,8,9-гексагидро-13,17:16,19-ди(метено)-1,10,4,9,11,18-бензоксатиатетраазациклогеникозин-3,12(2H,11H,18H)-диона 40; m/z = 621 (M+Н)+
1H ЯМР (400 МГц, CDCl3) δ м.д. 0,85-0,9 (м, 1H), 1,00-1,40 (м, 7H), 1,5-1,6 (м, 5H), 1,75-1,85 (м, 6H), 2-2,3 (м, 6H), 2,35-2,5 (м, 2H), 2,6 (с, 3H), 2,9 (с, 3H), 3,25-3,35 (м, 1H), 3,9-3,95 (м, 1H), 4,2 (д, J=14 Гц, 1H), 4,4 (д, J=14 Гц, 1H), 7 (д, J=7,9 Гц, 1H), 7,25 (дд, J=8,1 и J=7 Гц, 1H), 7,4 (д, J=8,1 Гц, 1H), 7,5 (дд, J=7,9 и J=7 Гц, 1H), 7,55 (д, J=8,1 Гц, 1H), 7,85 (д, J=8,1 Гц, 1H), 7,9 (с, 1H).
Пример 7
Синтез 11,11-диоксида 25-циклогексил-23-фтор-10,19-диметил-5,6,7,8,9,10-гексагидро-2H-14,18: 17,20-ди(метено)-1,11,4,10,12,19-бензоксатиатетраазациклодокозин-3,13(4H,12H,19H)-диона 50

Стадия 1

Для получения соединения 41 из метил-2-бром-3-циклогексил-1H-индол-6-карбоксилата 1 и 2-(бензилокси)-4-фторфенилбороновой кислоты в качестве исходного материала применяли такую же процедуру, как для получения соединения 25, получая при этом 14,9 мг (87%-ный выход) метил 2-[2-(бензилокси)-4-фторфенил]-3-циклогексил-1H-индол-6-карбоксилата 41; m/z = 458 (M+Н)+.
Стадия 2
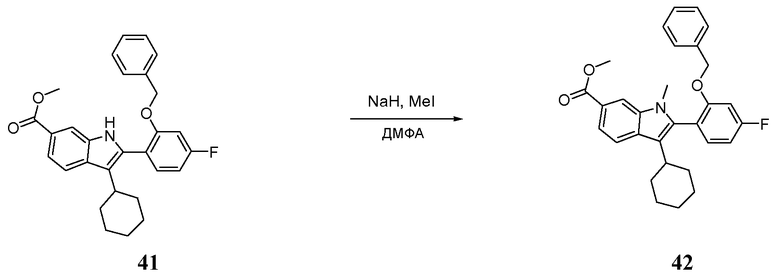
Для получения соединения 42 из промежуточного продукта 41 в качестве исходного материала применяли такую же процедуру, как для получения соединения 3, получая при этом метил-2-[2-(бензилокси)-4-фторфенил]-3-циклогексил-1-метил-1H-индол-6-карбоксилат 42 с количественным выходом: m/z = 472 (M+Н)+.
Стадия 3

Для получения соединения 43 из промежуточного продукта 42 в качестве исходного материала применяли такую же процедуру, как для получения соединения 4, получая при этом метил-3-циклогексил-2-(4-фтор-2-гидроксифенил)-1-метил-1H-индол-6-карбоксилат 43 с 96%-ным выходом; m/z = 382 (M+Н)+.
Стадия 4

Для получения соединения 44 из промежуточного продукта 43 и трет-бутил-2-бромацетата в качестве исходного материала применяли такую же процедуру, как для получения соединения 26, получая при этом метил-2-[2-(2-трет-бутокси-2-оксоэтокси)-4-фторфенил]-3-циклогексил-1-метил-1H-индол-6-карбоксилат 44 с количественным выходом; m/z = 496 (M+Н)+.
Стадия 5
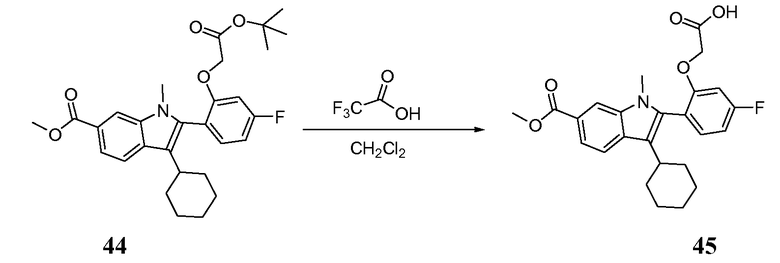
Для получения соединения 45 из промежуточного продукта 44 в качестве исходного материала применяли такую же процедуру, как для получения соединения 27, получая при этом {2-[3-циклогексил-6-(метоксикарбонил)-1-метил-1H-индол-2-ил]-5-фторфенокси}уксусную кислоту 45 с количественным выходом; m/z = 440 (M+Н)+.
Стадия 6

Для получения соединения 46 из промежуточного продукта 45 и N-(5-аминопентил)-N-метил-2-нитробензолсульфонамида 16 в качестве исходного материала применяли такую же процедуру, как для получения соединения 17, получая при этом 891 мг (68%-ный выход) метил-3-циклогексил-2-(4-фтор-2-{2-[(5-{метил[(2-нитрофенил)сульфонил]амино}пентил)амино]-2-оксоэтокси}фенил)-1-метил-1H-индол-6-карбоксилата 46; m/z = 723 (M+Н)+.
Стадия 7

Для получения соединения 47 из промежуточного продукта 46 в качестве исходного материала применяли такую же процедуру, как для получения соединения 18, получая при этом 520 мг (78%-ный выход) метил-3-циклогексил-2-[4-фтор-2-(2-{[5-(метиламино)пентил]амино}-2-оксоэтокси)фенил]-1-метил-1H-индол-6-карбоксилата 47; m/z = 538 (M+Н)+.
Стадия 8

Для получения соединения 48 из промежуточного продукта 47 в качестве исходного материала применяли такую же процедуру, как для получения соединения 19, получая при этом 590 мг (98%-ный выход) метил-3-циклогексил-2-{4-фтор-2-[2-({5-[метил(сульфамоил)амино]пентил}амино)-2-оксоэтокси]фенил}-1-метил-1H-индол-6-карбоксилата 48; m/z = 617 (M+Н)+.
Стадия 9

Для получения соединения 49 из промежуточного продукта 48 в качестве исходного материала применяли такую же процедуру, как для получения соединения 8, получая при этом 399 мг (67%-ный выход) 3-циклогексил-2-{4-фтор-2-[2-({5-[метил(сульфамоил)амино]пентил}амино)-2-оксоэтокси]фенил}-1-метил-1H-индол-6-карбоновой кислоты 49; m/z = 603 (M+Н)+.
Стадия 10
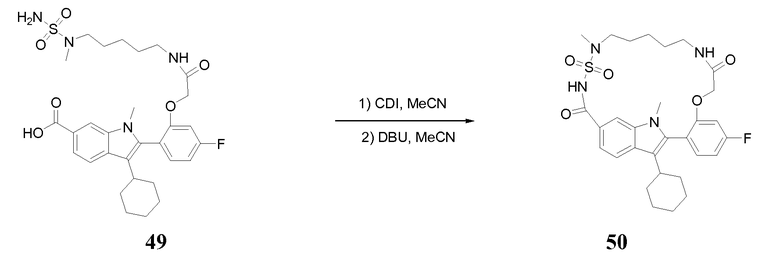
Для получения соединения 50 из промежуточного продукта 49 в качестве исходного материала применяли такую же процедуру, как для получения соединения 21, получая при этом 220 мг (57%-ный выход) 11,11-диоксида 25-циклогексил-23-фтор-10,19-диметил-5,6,7,8,9,10-гексагидро-2H-14,18:17,20-ди(метено)-1,11,4,10,12,19-бензоксатиатетраазациклодокозин-3,13(4H,12H, 19H)-диона 50; m/z = 585 (M+Н)+.
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 0,55-0,65 (м, 1H), 1,00-1,10 (м, 2H), 1,12-1,27 (м, 5H), 1,52-1,58 (м, 2H), 1,64-1,78 (м, 6H), 2,25-2,29 (м, 1H), 2,95 -3,05 (м, 5H), 3,28-3,38 (м, 2H), 3,47 (с, 3H), 4,40 (д, J=13,5 Гц, 1H), 4,49 (д, J=13,7 Гц, 1H), 5,88 (т, J=5,2 Гц, 1H), 7,03-7,08 (м, 1H), 7,18 (д, J=10,5 Гц, 1H), 7,42-7,47 (м, 1H), 7,62 (д, J=8,5 Гц, 1H), 7,84 (д, J=8,4 Гц, 1H), 8,39 (с, 1H), 11,62 (с, 1H).
Пример 8
Синтез 11,11-диоксида 25-циклогексил-22-фтор-10,19-диметил-5,6,7,8,9,10-гексагидро-2H-14,18:17,20-ди(метено)-1,11,4,10,12,19-бензоксатиатетраазациклодокозин-3,13(4H,12H,19H)-диона 51

Синтез указанного в заголовке соединения 51 осуществляли согласно 10-стадийной процедуре, о которой сообщалось в связи с синтезом соединения 50, с применением на первой стадии 2-(бензилокси)-5-фторфенилбороновой кислоты, и получая при этом 431 мг твердого белого вещества; m/z = 585 [M+Н]+.
1H ЯМР (400 МГц, CHCl3-d6) δ м.д.0,64-0,72 (м, 1H), 0,96-1,01 (м, 2H), 1,09-1,13 (м, 1H), 1,18-1,32 (м, 3H), 1,40-1,47 (м, 1H), 1,59 (ушир.с, 4H), 1,70-1,85 (м, 6H), 2,15-2,22 (м, 1H), 2,52-2,65 (м, 1H), 3,05-3,09 (м, 1H), 3,22 (с, 3H), 3,24-3,31 (м, 1H), 3,53 (с, 3H), 4,33 (д, J=12,5 Гц, 1H), 4,41 (д, J=14 Гц, 1H), 5,36-5,40 (м, 1H), 6,94 (дд, J=8,1 и J=3,4 Гц, 1H), 7,14 (д, J=7,8 Гц, 1H), 7,22 (д, J=7,6 Гц, 1H), 7,56 (д, J=8,3 Гц, 1H), 7,91 (д, J=8,3 Гц, 1H), 7,99 (s, 1H), 8,93 (ушир.с, 1H).
Пример 9
Синтез 11,11-диоксида 25-циклогексил-4,10-диметил-19-(1-метилэтил)-5,6,9,10-тетрагидро-2H,8H-14,18:17,20-ди(метено)-1,7,11,4,10,12,19-бензодиоксатиатетраазациклодокозин-3,13(4H,12H,19H)-диона 52

Синтез указанного в заголовке соединения 52 осуществляли согласно 8-стадийной процедуре, о которой сообщалось в связи с синтезом соединения 40, с применением на первой стадии 2-иодпропана и N-метил-2-(метиламиноэтилокси)этиламина на стадии 5, получая при этом 300 мг белого твердого вещества; m/z = 611 [M+Н]+.
1H ЯМР (400 МГц, CDCl3) δ м.д. 1,1-1,2 (м, 4H), 16-1,75 (м, 11H), 2,2 (с, 3H), 2,25-2,3 (м, 2H), 2,4-2,5 (м, 1H), 3,00-3,1 (м, 2H), 3,2 (с, 3H), 3,3-3,35 (м, 1H), 3,4-3,48 (м, 2H), 3,5-3,55 (м, 1H), 4,1-4,25 (м, 2H), 4,35-4,5 (м, 2H), 6,88 (д, J=8,1 Гц, 1H), 7,25 (дд, J=8,2 и J=7,5 Гц, 1H), 7,35-7,4 (м, 2H), 7,5 (дд, J=8,1 и J=7,5 Гц, 1H), 7,8 (д, J=8,3 Гц, 1H), 8,12 (с, 1H), 8,5 (ушир.с, 1H).
Пример 10
Синтез 10,10-диоксида 24-циклогексил-22-метокси-4,9,18-триметил-4,5,6,7,8,9-гексагидро-13,17:16,19-ди(метено)-1,10,4,9,11,18-бензоксатиатетраазациклогеникозин-3,12(2H,11H,18H)-диона 53

Синтез указанного в заголовке соединения 53 осуществляли согласно 9-стадийной процедуре, о которой сообщалось в связи с синтезом соединения 10, с применением на первой стадии 2-(бензилокси)-4-метоксифенилбороновой кислоты и N,N'-диметилбутилендиамина на стадии 5, получая при этом 57 мг твердого белого вещества; m/z = 597 [M+Н]+.
1H ЯМР (400 МГц, CHCl3-d6) δ м.д. 1,04-1,08 (м, 1H), 1,20-1,36 (м, 4H), 1,37-1,46 (м, 4H), 1,69 (с, 3H), 1,73-1,76 (м, 2H), 1,78-1,98 (м, 4H), 2,06-2,15 (м, 1H), 2,65-2,71 (м, 1H), 3,12 (с, 3H), 3,25-3,30 (м, 1H), 3,58 (s, 3H), 3,72 (д, J=13,1 Гц, 1H), 3,87 (с, 3H), 3,92-4,01 (м, 1H), 4,07 (д, J=13,1 Гц, 1H), 6,76 (д, J=2,1 Гц, 1H), 6,80 (дд, J=8,5 и 2,4 Гц, 1H), 7,32 (д, J=8,3 Гц, 1H), 7,58 (д, J=8,7 Гц, 1H), 7,72 (с, 1H), 7,85 (д, J=8,5 Гц, 1H), 8,36 (ушир.с, 1H).
Пример 11
Синтез 11,11-диоксида 25-циклогексил-23-метокси-4,10,19-триметил-5,6,9,10-тетрагидро-2H,8H-14,18:17,20-ди(метено)-1,7,11,4,10,12,19-бензодиоксатиатетраазациклодокозин-3,13(4H,12H,19H)-диона 54

Синтез указанного в заголовке соединения 54 осуществляли согласно 9-стадийной процедуре, о которой сообщалось в связи с синтезом соединения 10, с применением на первой стадии 2-(бензилокси)-4-метоксифенилбороновой кислоты, получая при этом 9 мг твердого белого вещества; m/z = 613 [M+Н]+.
1H ЯМР (400 МГц, CHCl3-d6) δ м.д. 1,15-1,38 (м, 4H), 1,70-1,75 (м, 2H), 1,78-1,90 (м, 4H), 2,17 (с, 3H), 2,30-2,38 (м, 1H), 2,45-2,51 (м, 1H), 2,52-2,63 (м, 1H), 3,02-3,09 (м, 1H), 3,22 (с, 3H), 3,26-3,33 (м, 1H), 3,47 (с, 3H), 3,52-3,56 (м, 2H), 3,70-3,78 (м, 1H), 3,90 (с, 3H), 4,07-4,15 (м, 1H), 4,41 (с, 2H), 6,51 (д, J=2,1 Гц, 1H), 6,72 (дд, J=8,2 и J=2,2 Гц, 1H), 7,31 (д, J =8,3 Гц, 1H), 7,45 (д, J=8,0 Гц, 1H), 7,74 (с, 1H), 7,82 (д, J=8,3 Гц, 1H), 8,75 (ушир.с, 1H).
Пример 12
Синтез N-(5-аминопентил)-N-метил-2-нитробензолсульфонамида 16
Стадия 1

К охлажденному раствору трет-бутил-(5-аминопентил)карбамата (20 г, 99 ммоль) в 250 мл дихлорметана добавляли 2-нитробензол-1-сульфонилхлорид (23 г, 104 ммоль). Полученную смесь перемешивали при 0°C, затем по каплям добавляли диизопропилэтиламин (19,7 г, 144 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в воду с лимонной кислотой, органический слой отделяли и последовательно сушили над MgSO4, фильтровали и концентрировали, получая при этом 32,6 г (85%) трет-бутил-(5-{[(2-нитрофенил)сульфонил]амино}пентил)карбамата 55 в виде белых иголочек m/z= 388 [M+H]+.
Стадия 2

К раствору трет-бутил-(5-{[(2-нитрофенил)сульфонил]амино}пентил)карбамата 55 (32,6 г, 84 ммоль) в ацетоне (300 мл) добавляли карбонат калия (23,2 г, 168 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем по каплям добавляли метилиодид (17,88 г, 126 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в воду и добавляли дихлорметан. Органический слой отделяли и последовательно сушили над MgSO4, фильтровали и концентрировали. Остаток растирали с простым диизопропиловым эфиром, получая при этом 31,6 г (93%) трет-бутил-(5-{метил[(2-нитрофенил)сульфонил]амино}пентил)карбамата 56 в виде белого порошка; m/z = 402 [M+H]+.
Стадия 3

Для получения соединения 16 из промежуточного продукта 56 в качестве исходного материала применяли такую же процедуру, как для получения соединения 27, получая при этом N-(5-аминопентил)-N-метил-2-нитробензолсульфонамид 16 с количественным выходом; m/z = 302 (M+H)+
Пример 13
Активность соединений формулы (I)
Клеточный анализ
Соединения формулы (I) исследовали на активность в отношении ингибирования репликации РНК вируса HCV в (a) клеточном анализе (анализах). Данный анализ показал, что соединения формулы (I) ингибируют функциональную клеточную линию, реплицирующую HCV, также известную как HCV-репликоны. Клеточный анализ основан на бицистронной экспрессируемой конструкции, как описано в публикации Lohmann и др. (1999) Science vol. 285 pp. 110-113 с модификациями, описанными в публикации: Krieger и др. (2001) Journal of Virology 75: 4614-4624, при многоцелевой стратегии скрининга. Здесь описаны два клеточных анализа HCV: анализ репликона (результат которого выражается в виде EC50) и анализ транзитного репликона (результат которого выражается в виде EC50-T).
Анализ репликона
В анализе использовали стабильно трансфицированную клеточную линию Huh-7 luc/neo (далее упоминаемую как Huh-Luc). В указанной клеточной линии скапливается РНК, кодирующая бицистронную экспрессирующую конструкцию, содержащую области NS3-NS5B дикого типа HCV типа 1b, транслируемые из участка внутренней посадки рибосомы (IRES) вируса энцефаломиокардита (EMCV), с предшествующей репортерной частью (FfL-люцифераза) и частью селективного маркера (neoR, неомицинфосфотрансфераза). Конструкция была ограничена 5' и 3' NTR (нетранслируемыми областями) HCV типа 1b. Непрерывная культура клеток репликона в присутствии G418 (neoR) зависит от репликации РНК HCV. Для скрининга противовирусных соединений использовали стабильно трансфицированные клетки репликона, которые экспрессируют РНК HCV, которая реплицируется автономно и до высоких уровней, кодируя, помимо прочего, люциферазу.
Клетки репликона размещали в 384-луночных планшетах в присутствии тестируемых и контрольных соединений, которые добавляли в различных концентрациях. После инкубации в течение трех дней измеряли репликацию HCV путем анализа активности люциферазы (с использованием стандартных субстратов и реагентов для анализа люциферазы и устройства для визуализации микропланшетов Perkin Elmer ViewLuxTM ultraHTS). Клетки репликона в контрольных культурах в отсутствие какого-либо ингибитора отличались высокой экспрессией люциферазы. Ингибиторную активность соединения контролировали на клетках Huh-Luc, получая кривую доза-ответ для каждого тестируемого соединения. Затем рассчитывали значения EC50, где данное значение представляло собой количество соединения, необходимое для снижения на 50% уровня детектируемой активности люциферазы, или более конкретно- способности генетически связанной РНК репликона HCV к репликации.
Анализ транзитного репликона
В анализе использовали бицистронную экспрессирующую конструкцию, содержащую области NS3-NS5B дикого типа HCV типа 1b (conlb) с адаптивными мутациями в культуре клеток ET-клона (E1202G, T 12801, K1846T) и транслируемыми из участка внутренней посадки рибосомы (IRES) вируса энцефаломиокардита (EMCV), с предшествующей репортерной частью (FfL-люцифераза), полученной на основе участка IRES вируса полиомиелита. Конструкция была ограничена 5' и 3' NTR (нетранслируемыми областями) HCV типа 1b. Из данной конструкции генерировали РНК и трансфицировали в подвергаемые обработке Huh7-клетки (Koutsoudakis G, Herrmann E, Kallis S, Bartenschlager R, Pietschmann T. The level of CD81 cell surface expression is a key determinant for productive entry of hepatitis C virus into host cells. J. Virol. 2007; 81 (2): 588-598). Трансфицированная РНК HCV реплицируется автономно, транзитно и до высоких уровней, кодируя, помимо прочего, люциферазу, и используется для тестирования противовирусных соединений.
Конструирование челночного вектора. Кассета вектора, сконструированная для челночного NS5B, была получена из плазмиды pFKi341Luc_NS3-3'-ET (20). Репликон, полученный из такой плазмиды, состоит из NS3-3'-UTR-последовательности HCV генотипа Ib штамма Conl и содержит две адаптивные мутации в клеточной культуре в NS3 (E1202G и T1280I) и одну в NS4B (K1846T). С помощью набора QuickChange для сайт-направленного мутагенеза производства Stratagene (La Jolla, CA) с использованием плазмиды pFKi341Luc_NS3-3'-ET были образованы два сайта рестрикции AflII: первый в 3'NCR непосредственно после стоп-кодона NS5B, и второй на 8 аминокислот выше участка расщепления NS5A/NS5B. Поскольку было установлено, что такой ограниченный с обеих сторон участок ScaI, необходимый для линеаризации плазмиды перед транскрипцией in vitro, должен присутствовать в полимеразной последовательности некоторых клинических изолятов, он был подвергнут мутации до XbaI. Эндогенный участок XbaI гена люциферазы светлячка удаляли путем введения молчащей мутации.
Конструирование химерных репликонов NS5B. кДНК, кодирующую C-конец NS5A (остатки 440-447) и полноразмерную форму NS5B (остатки 1-591), амплифицировали из клинических изолятов с использованием праймеров специфического подтипа с 5'-областью (16 нуклеотидов), комплементарной челночному вектору, и 3'-областью (13-19 нуклеотидов), сконструированной таким образом, чтобы она была комплементарна последовательностям клинических изолятов. Ампликоны клонировали в челночный вектор NS5B с использованием способа клонирования In-Fusion (набор для клонирования In-Fusion Dry Down PCR, Clontech). Индивидуальные клоны или пулы всех клонов, полученных после трансформации, были использованы для транскрипции in vitro и для тестирования в анализе транзитных репликонов. Данные для соединений согласно настоящему изобретению получали с использованием двух химерных репликонов от двух клинических изолятов HCV генотипа 1a, 1a_H77 и 1a_6 (номера доступа в банке генов AF011751 и EF523592, соответственно).
Анализ транзитных репликонов. Десять микрограмм транскрибированного in vitro репликона линейной РНК трансфицировали в клетки, нагруженные Huh7, и проводили количественный анализ репликации репликона после 48 часов инкубации (37°C, 5% CO2) путем измерения люминесценции после добавления субстрата люциферазы (Steady Lite Plus; Perkin Elmer). Активность соединения в отношении таких репликонов, то есть ингибиторную активность, измеряли в сериях девятиточечного разведения, получая кривую реакции доза-ответ для каждого тестируемого соединения. Затем рассчитывали значения EC50, которые представляют собой количество соединениий, необходимых для снижения на 50% уровня детектируемой люциферазной активности или, более конкретно, способности генетически связанного репликона РНК HCV к репликации. Полученные при измерении значения EC50 приведены в таблице 1. EC50-T Con1b получали с помощью репликона дикого типа генотипа 1b; EC50-T CI1, и EC50-T CI2 получали с помощью химерных репликонов клинических изолятов 1a_H77 и 1a_6, соответственно.
Анализ ингибирования действия фермента
a) Клонирование, экспрессия и очистка NS5B
Кодирующую последовательность для NS5B (генотип 1b, консенсусный штамм Con1), не содержащую 21 C-концевых остатков, амплифицировали из плазмиды pFKl389/ns3-3' (номер доступа в банке генов AJ242654), плазмиды pCV-H77C (номер доступа в банке генов AF 011751.1) и субклонировали в плазмиду pET21b, как описано ранее (Pauwels и др., J. Virol, 2007). Экспрессирующие конструкции NS5BΔC21 переносили в клетки E. coli штамма Rosetta 2 (DE3) (Novagen, Madison, WI). Осуществляли посев одной колонии в ста миллилитрах LB-среды, дополненной карбенициллином (50 мкг/мл) и хлорамфениколом (34 мкг/мл), выращивали в течение ночи и переносили в свежую LB-среду, дополненную 3% этанола, карбенициллином и хлорамфениколом в соотношении 1:200. Клетки выращивали до оптической плотности 0,6 при 600 нм, после чего температуру выращивания экспрессируемых культур смещали до 20°C после индукции изопропил-1-тио-β-D-галактопиранозидом и MgCl2 в конечной концентрации 0,4 мМ и 10 мкМ, соответственно. После 18 часов индукции клетки собирали путем центрифугирования и повторно суспендировали в буфере 20 мМ Tris-HCl, pH 7,5, 300 мМ NaCl, 10% глицерина, 0,1% NP40, 4 мМ MgCl2, 5 мМ DTT, дополненном коктейлем ингибиторов протеаз, не содержащим ЭДТА (EDTA-free Complete Protease Inhibitor, Roche, Basel, Switzerland). Клеточные суспензии разрушали ультразвуком и инкубировали с 10-15 мг/л ДНКазы I (Roche, Basel, Switzerland) в течение 30 мин. Клеточный дебрис удаляли путем ультрацентрифугирования при 30000×g в течение 1 часа, и осветленный клеточный лизат замораживали и хранили перед очисткой при -80°C.
Осветленный клеточный лизат размораживали и затем наносили на предварительно упакованную 5-миллилитровую колонку HisTrap FF, уравновешенную 25 мМ HEPES-буфером, pH 7,5, содержащим 500 мМ NaCl, 10% глицерина и 5 мМ DTT. Белки элюировали 500 мМ имидазолом при скорости потока 1 мл/мин. Фракции, содержащие представляющий интерес белок, наносили на предварительно упакованную, обессоленную колонку 26/10 HiPrep, уравновешенную 25 мМ HEPES-буфера, pH 7,5, содержащего 250 мМ NaCl, 10% глицерина и 5 мМ DTT. Затем фракцию, соответствующую вытесненному буфером пику NS5B, наносили на 6 миллилитровую колонку Resource S. Белок элюировали в условиях увеличивающегося градиента соли и собирали фракции. Степень чистоты белка определяли на предварительно залитых гелях Nu-PAGE (Invitrogen, Carlsbad, CA). Очищенные образцы NS5B концентрировали с использованием концентраторов Centri-Prep (Millipore, Billerica, MA, USA) и определяли концентрации белка методом спектрофотометрии на спектрофотометре Nanodrop (Nanodrop Technologies, Wilmington, DE, USA).
b) Анализ РНК-зависимой РНК-полимеразы
Измерение полимеразной активности NS5B HCV осуществляли путем оценки количества меченного радиоактивным изотопом ГТФ, встроенного посредством фермента во вновь синтезированную РНК с использованием гетерополимерного комплекса матрица/праймер РНК. Анализ RdRp проводили в 384-луночных планшетах. 2,5 нМ очищенного фермента NS5B предварительно инкубировали в течение 10 мин с 150 нМ 5'-биотинилированного олиго(rG13)праймера, 15 нМ матрицы поли(rC) в 18 мМ буфере Tris-HCl, pH 7,5, содержащем 5 мМ MgCl2, 20,5 мМ KCl, 17 мМ NaCl и 2,5 мМ DTT. Тестируемые соединения добавляли к преформированному комплексу полимераза-матрица и инкубировали при комнатной температуре в течение 15 мин перед добавлением 600 нМ ГТФ и 0,13 мкCi [3H]ГТФ. 30 мкл реакционной смеси инкубировали при комнатной температуре в течение 2 часов перед тем, как остановить реакцию путем добавления 30 мкл бус SPA, покрытых стрептавидином (GE Healthcare, Uppsala, Sweden), в 0,5 M ЭДТА. Реакцию (30 мкл) останавливали через 2 часа при 25°C при добавлении 30 мкл бус SPA, покрытых стрептавидином (GE Healthcare, Uppsala, Sweden) в 0,5M ЭДТА. После инкубации при 25°C в течение 30 минут планшет считывали с помощью сцинциляционного микропланшетного счетчика Packard TopCount (30 сек/лунку, задержка счета 1 мин) и рассчитывали значения IC50. Значения IC50 представляют собой концентрацию соединения, необходимую для снижения на 50% количества продуцируемой РНК, которое измеряют путем детектирования встроенного радиоактивно меченного ГТФ. Полученные значения IC50 представлены в таблице 1.
В следующей таблице 1 перечислены соединения по любому из приведенных выше примеров, активность которых была определена согласно примеру 3.
Анализ ферментативного связывания
Соединения формулы (I) исследовали на предмет кинетики их ферментативного связывания или аффинности с применением способа, основанного на поверхностном плазмонном резонансе (SPR), то есть прибора Biacore (Биакор). Считается, что медленная диссоциация ингибирующего соединения от его вирусной мишени (низкий koff, низкий Kd) потенциально уменьшает развитие лекарственной резистентности в отношении противовирусных лекарственных средств (Dierynck и др., 2007. Journal of Virology, vol.81, No. 24, 13845-13851). Все измерения осуществляли с использованием прибора Biacore T100 (GE Healthcare). Очищенные HIS6-меченные NS5BΔC21-полимеразы были иммобилизованы на поверхности сенсорного чипа NTA (GE Healthcare) с использованием нековалентного захвата в иммобилизационном буфере (20 мМ MOPS pH 7,4, 500 мМ NaCl, 0,005% Tween-P20, 1 мМ DTT, 50 мкМ EDTA).
Все исследования кинетики взаимодействия осуществляли при 25°C. Ингибиторы серийно разводили в подвижном буфере (20 мМ Tris-HCl pH 7,4, 150 мМ NaCl, 50 мкМ ЭДТА, 1 мМ DTT, 0,005% Tween-P20), содержащем 5% диметилсульфоксида (DMSO). Применяли кинетику одностадийного цикла, при которой каждую из 5 увеличивающихся концентраций соединения впрыскивали за 1 одностадийный цикл в течение 300 с и в течение 1200 с контролировали диссоциацию. Между циклами поверхность сенсора полностью восстанавливалась. Данные анализировали с применением одновременного нелинейного регрессионного анализа (общая аппроксимация данных), адаптированного для кинетики одностадийного цикла, с использованием для оценки программного обеспечения Biacore T100 BiaEval 2.0 (GE Healthcare). Индивидуальные константы скорости kon и koff и полученную константу аффинности (IQ=koff/kon) определяли путем оценки кинетических параметров сенсограмм. Кинетические модели рассчитаны для эффектов объемного и ограниченного массопереноса. Каждый анализ осуществляли, по меньшей мере, в двух независимых экспериментах. Скорость диссоциации при кинетическом взаимодействии можно пересчитать во время удерживания соединения (время полужизни в состоянии диссоциации t1/2=ln(2)/koff), представляющее собой время взаимодействия между полимеразой и ее ингибитором.
Полученные экспериментально константы скорости ассоциации (kon), константы скорости диссоциации (koff), полученная константа аффинности (IQ) и время полужизни в состоянии диссоциации (t1/2) для соединений формулы (I) или их подгрупп на ферменте NS5B дикого типа генотипов 1a, 1b и 4a, приведены в таблице 2.
Пример 14
Фармацевтические композиции на основе соединений формулы (I)
Получение препарата
Активный ингредиент, в данном случае соединение формулы (I), растворяли в органическом растворителе, таком как этанол, метанол или метиленхлорид, предпочтительно в смеси этанола и метиленхлорида. Полимеры, такие как сополимер поливинилпирролидона с винилацетатом (ПВП-ВА) или гидроксипропилметилцеллюлозой (ГПМЦ), обычно с вязкостью 5 мПз, растворяют в органических растворителях, таких как этанол, метанол, метиленхлорид. Соответственно полимер растворяют в этаноле. Растворы полимера и соединения смешивают и затем сушат с помощью распылительной сушки. Отношение соединение/полимер выбирали в диапазоне от 1/1 до 1/6. Промежуточные диапазоны составляли 1/1,5 и 1/3. Подходящее отношение составляло 1/6. Высушенным распылением порошком, представляющим собой твердую дисперсию, затем наполняли капсулы для введения. Количество лекарственного средства, загружаемое в одну капсулу, находилось в диапазоне от 50 до 100 мг в зависимости от размера применяемой капсулы.
Таблетки с покрытием в виде пленки
Получение ядра таблетки
Смесь из 100 г активного ингредиента, в данном случае соединения формулы (I), 570 г лактозы и 200 г крахмала тщательно смешивают и затем увлажняют раствором, состоящим из 5 г додецилсульфата натрия и 10 г поливинилпирролидона приблизительно в 200 мл воды. Увлажненную порошкообразную смесь пропускают через сито, сушат и снова пропускают через сито. Затем добавляют 100 г микрокристаллической целлюлозы и 15 г гидрированного растительного масла. Всю смесь тщательно смешивают и прессуют в таблетки, получая при этом 10000 таблеток, каждая из которых содержит 10 мг активного ингредиента.
Покрытие
К раствору 10 г метилцеллюлозы в 75 мл денатурированного этанола добавляют раствор 5 г этилцеллюлозы в 150 мл дихлорметана. Затем добавляют 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. 10 г полиэтиленгликоля плавят и растворяют в 75 мл дихлорметана. Полученный раствор добавляют к пленкообразователю (раствору производных целлюлозы), затем к смеси добавляют 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной суспензии красителя и полученную смесь гомогенизируют. Ядра таблеток покрывают полученной таким образом смесью в аппарате для получения покрытия.
Пример 15
Фармакокинетический анализ после однократного перорального введения
Трем крысам - самцам линии Спраг Доули (Sprague-Dawley) давали однократную пероральную дозу 10 мг/кг исследуемых соединений. Соединения вводили через желудочный зонд в виде раствора в PEG400/2% витамин E - токоферола полиэтиленгликольсукцинат (витамин E-ТПГС). Спустя 7 часов после введения животных умерщвляли и брали образцы плазма и печени. Образцы анализировали с применением подходящего исследования методом ЖХ-МС/МС для определения концентрации соединения в печени и отношения печень/плазма спустя 7 часов после введения соединения. Полученные результаты обобщены в таблице 3. Было обнаружено, что спустя 7 часов после перорального введения исследуемых соединений их концентрации в печени крыс линии Спраг Доули (Sprague-Dawley) были высокими и что исследуемые соединения показывают очень высокое отношение печень/плазма.
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2437886C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2441870C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2486189C2 |
| МАКРОЦИКЛИЧЕСКИЕ ФЕНИЛКАРБАМАТЫ, ИНГИБИРУЮЩИЕ HCV | 2008 |
|
RU2490261C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2436787C2 |
| ФОСФОРАМИДАТНЫЕ ПРОИЗВОДНЫЕ НУКЛЕОЗИДОВ | 2010 |
|
RU2553656C2 |
| ПИРИМИДИН-ЗАМЕЩЕННЫЕ МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ HCV | 2008 |
|
RU2481340C2 |
| ПРОИЗВОДНЫЕ БИС-БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2540897C2 |
| ДИАРИЛОВЫЕ ЭФИРЫ | 2010 |
|
RU2528231C2 |
| ФЕНИЛЭТИНИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2538507C2 |
Изобретение относится к соединению формулы (I)

включая его стереохимически изомерные формы, гидраты и сольваты, где R1 выбран из атома водорода, атома галогена и C1-4-алкокси; R2 выбран из C1-4-алкила и C3-6-циклоалкила; R4 представляет собой C3-7-циклоалкил; n равно 1; Y выбран из


И

a равно 4 или 5; R3 и R3' - независимо выбраны из атома водорода и C1-6-алкила. Изобретение также относится к противовирусной фармацевтической композиции на основе указанного соединения формулы (I). Технический результат: получены новые соединения и фармацевтические композиции на их основе, которые могут найти применение в медицине в терапии HCV. 3 н. и 9 з.п. ф-лы, 3 табл., 15 пр.
1. Соединение формулы (I)
включая его стереохимически изомерные формы, гидраты и сольваты, где:
- R1 выбран из атома водорода, атома галогена и C1-4-алкокси;
- R2 выбран из C1-4-алкила и C3-6-циклоалкила;
- R4 представляет собой C3-7-циклоалкил;
- n равно 1;
- Y выбран из
и
- a равно 4 или 5;
- R3 и R3' независимо выбраны из атома водорода и C1-6-алкила.
2. Соединение по п.1, где Y выбран из -N(CH3)-(CH2)4-N(CH3)-,

и
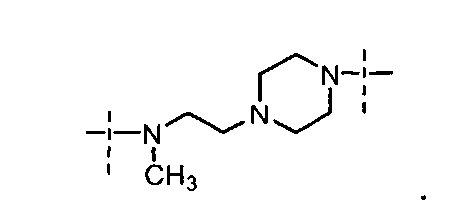
3. Соединение по п.1 или п.2, где R1 представляет собой атом водорода или метокси, или атом фтора.
4. Соединение по п.1 или п.2, где R2 выбран из метила, этила, изопропила и циклопропила.
5. Соединение по п.1 или п.2, где R4 выбран из циклогексила.
6. Соединение по п.1 или п.2, где n равно 1.
7. Соединение по п.1, выбранное из
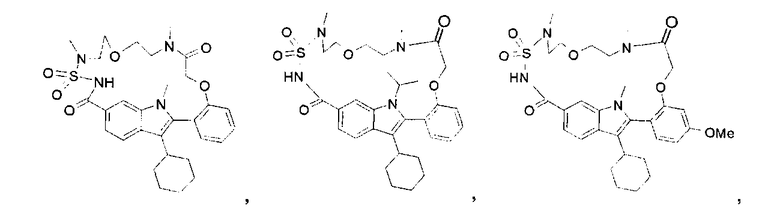


включая его стереохимически изомерные формы и соли, сольваты или гидраты.
8. Фармацевтическая композиция, содержащая носитель и эффективное противовирусное количество соединения по любому из пп.1-7 в качестве активного ингредиента.
9. Фармацевтическая композиция по п.8, дополнительно содержащая по меньшей мере одно другое соединение против вируса HCV.
10. Соединение по п.1 для ингибирования репликации HCV.
11. Применение соединения по любому из пп.1-7 для получения лекарственного средства для ингибирования репликации HCV.
12. Фармацевтическая композиция по п.8 для ингибирования репликации HCV.
| WO 2008075103 A1, 26.06.2008 | |||
| МАКРОГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2001 |
|
RU2275373C2 |

