Область техники, к которой относится изобретение
Изобретение относится к макроциклическим соединениям, имеющим ингибиторную активность по отношению к серин-протеазе NS3 из HCV. Кроме того, он относится к композициям, содержащим эти соединения в качестве активных ингредиентов, а также к способам получения этих соединений и композиций.
Уровень техники
Вирус гепатита С (HCV) представляет собой основную причину хронического заболевания печени по всему миру и стал центром сосредоточения значительных медицинских исследований. HCV является членом семейства вирусов Flaviviridae рода hepacivirus, и является близким роду flavivirus, который включает в себя ряд вирусов, вовлеченных в заболевания человека, такие как вирус лихорадки денге и вирус желтой лихорадки, и к животному семейству pestivirus, которые включают в себя вирус бычьей вирусной диареи (BVDV). Геном HCV содержит как 5', так и 3' нетранслируемые области, которые адаптируют вторичные структуры РНК, и центральную открытую рамку считывания, которая кодирует единственный полипротеин. Полипротеин кодирует десять генных продуктов, которые генерируются из полипротеина предшественника с помощью оркестрированной серии со- и посттрансляционных эндопротеолитических расщеплений, медиируемых протеазами как хозяина, так и вируса. Вирусные структурные белки включают в себя сердцевинный белок нуклеокапсида и два оболочных гликопротеина Е1 и Е2. Неструктурные (NS) белки кодируют некоторые основные ферментативные функции вируса (геликазы, полимеразы, протеазы), а также белки с неизвестными функциями. Репликация вирусного генома опосредуется с помощью РНК-зависимой РНК полимеразы, кодируемой с помощью неструктурного белка 5b (NS5B). В дополнение к полимеразе, функции как геликазы, так и протеазы вируса, которые кодируются в бифункциональном белке NS3, как показано, являются основными для репликации РНК из HCV. В дополнение к серин-протеазе NS3, HCV кодирует также металлопротеиназу в области NS2.
После начальной острой инфекции, у большинства инфицированных индивидуумов развивается хронический гепатит, поскольку HCV реплицируется предпочтительно в гепатоцитах, но не является непосредственно цитопатическим. В частности, отсутствие энергичной реакции Т-лимфоцитов и высокая склонность вируса к мутации, видимо, способствуют высокой доле хронической инфекции. Хронический гепатит может развиваться в фиброз печени, приводящий к циррозу, заболеванию печени конечной стадии, и к НСС (гепатоцеллюлярная карцинома), что делает его ведущей причиной трансплантации печени.
Имеется 6 главных генотипов HCV и более чем 50 субтипов, которые по-разному распределяются географически. HCV типа 1 является доминирующим генотипом в Европе и США. Широкая генетическая гетерогенность HCV имеет важные диагностические и клинические последствия, вероятно, объясняя сложности при разработке вакцины и отсутствие реакции на современную терапию.
Передача HCV может осуществляться через контакт с зараженной кровью или продуктами крови, например, после переливания крови или внутривенного приема лекарственных средств. Введение диагностических тестов, используемых при скрининге крови, привело к понижающемуся тренду для частоты возникновения HCV после переливания. Однако благодаря медленному развитию заболевания печени конечной стадии, существующие инфекции будут продолжать представлять собой серьезную медицинскую и экономическую проблему в течение десятилетий.
Современная терапия HCV основывается на (пегилированном) интерфероне-альфа (IFN-α) в сочетании с рибавирином. Эта комбинированная терапия дает задержанную вирологическую реакцию более чем у 40% пациентов, инфицированных вирусом генотипа 1, и примерно у 80% тех, которые инфицированы генотипами 2 и 3. Кроме ограниченной эффективности по отношению HCV типа 1, эта комбинированная терапия имеет значительные побочные воздействия и плохо переносится многими пациентами. Главные побочные воздействия включают в себя симптомы, сходные с гриппом, гематологические аномалии и нейропсихиатрические симптомы. Следовательно, имеется необходимость в более эффективных, удобных и лучше переносимых видах лечения.
Ряд сходных ингибиторов протеазы HCV описан в академической и патентной литературе. Так WO 2005/073195 раскрывает пептидомиметики, которые ингибируют NS3 протеазу вируса гепатита С (HCV). Данный документ описывает соединения общей формулы VI, несущие необязательно замещенную пиримидильную группу, предназначенные для лечения гепатита С.
Также известны макролитические соединения общей формулы (I), которые включают 4-оксо-пиримидинильную группу, которые могут использоваться для лечения гепатита С (WO 2007/014919).
WO 2007/014925 раскрывает ингибиторы HCV репликации. Данные соединения представляют собой макролитические соединения, также несущие 4-оксо-пиримидинильную группу, которые могут использоваться для лечения гепатита С.
Длительное введение ингибиторов протеазы HCV обычно приводит к селекции резистентных мутантов HCV, так называемых ускользнувших мутантов. Они имеют характерные мутации в геноме протеазы HCV, а именно D168V, D168Y и/или A165S. Соответственно, имеется необходимость в дополнительных лекарственных средствах для различных паттернов резистентности для пациентов, не поддающихся лечению. Такие лекарственные средства могут найти применение в комбинированной терапии, которая, как ожидается, станет нормой в будущем, даже для первичного лечения.
Эксперимент с лекарственными средствами против ВИЧ, в частности с ингибиторами протеазы ВИЧ, говорит, что субоптимальная фармакокинетика и сложные режимы дозирования быстро приводят к необратимым неудачам, связанным с совместимостью. Это в свою очередь означает, что средняя 24-часовая концентрация (минимальная концентрация в плазме) для соответствующих лекарственных средств в режиме ВИЧ часто падает ниже порога IC90 или ED90 в течение большей части дня. Считается, что 24-часовой уровень, равный, по меньшей мере, IC50, а более реалистически, IC90 или ED90, является главным условием для замедления развития ускользнувших мутантов.
Достижение необходимой фармакокинетики и метаболизма лекарственных средств, чтобы сделать возможным установление таких средних уровней, создает жесткий вызов при конструировании лекарственных средств. Известные ингибиторы протеазы HCV с множеством пептидных связей накладывают дополнительные фармакокинетические ограничения на эффективные режимы дозирования.
Имеется необходимость в ингибиторах HCV, которые могут преодолеть недостатки современной терапии HCV, такие как побочные воздействия, ограниченная эффективность, возникновение резистентности и отказы по совместимости.
Настоящее изобретение относится к ингибиторам репликации HCV, которые демонстрируют, по меньшей мере, одно улучшенное свойство по сравнению с теми соединениями, которые известны из литературы. В частности, ингибиторы по настоящему изобретению являются превосходными по одному или нескольким из следующих свойств, связанных с фармакологией, то есть по сильнодействию, пониженной цитотоксичности, улучшенной фармакокинетике, улучшенному профилю резистентности, приемлемым проблемам с дозированием и таблетированием. Ингибиторы HCV по настоящему изобретению являются особенно привлекательными благодаря их хорошей активности против мутантных штаммов HCV.
Краткое описание изобретения
Настоящее изобретение относится к ингибиторам репликации HCV, которые могут быть представлены формулой (I):
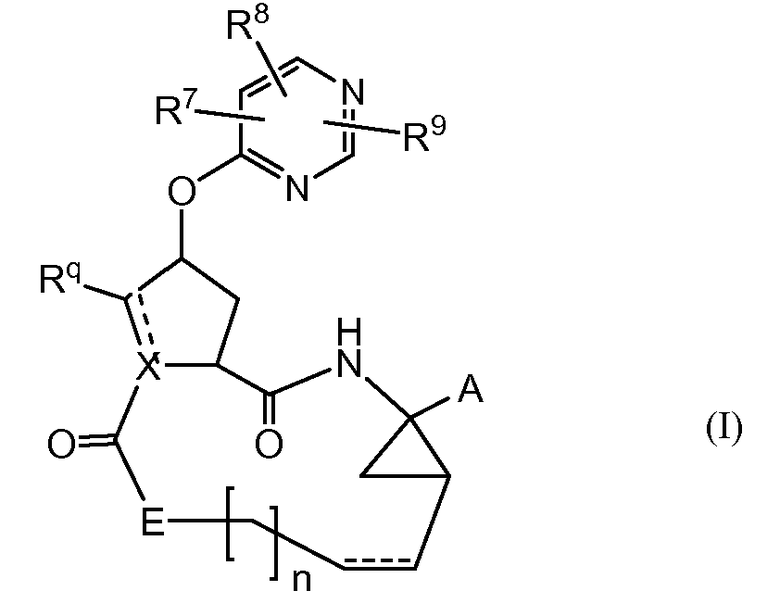
где
A представляет собой -C(=O)OR1, -C(=O)-NH-SO2-R2, -C(=O)C(=O)NR3aR3b, -C(=O)-NH-SO2-NR3aR3b, -C(=O)NH-P(=O)(OR4a)(R4b), или -P(=O)(OR4a)(R4b), где;
R1 представляет собой водород; арил; Het; C3-7циклоалкил, необязательно замещенный C1-6алкилом; или C1-6алкил, необязательно замещенный C3-7циклоалкилом, арилом или Het;
R2 представляет собой арил; Het; C3-7циклоалкил, необязательно замещенный C1-6алкилом; или C1-6алкил, необязательно замещенный C3-7циклоалкилом, арилом или Het;
каждый из R3a и R3b независимо представляют собой водород; C1-6алкил, необязательно замещенный C1-6алкокси, гидрокси, галогеном, C3-7циклоалкилом, арилом или Het; арил; C2-6алкенил; Het; C3-7циклоалкил, необязательно замещенный C1-6алкилом; или R3a и R3b вместе с атомом азота, к которому они присоединены, образуют группу Het1; и R3a может также представлять собой C1-6алкокси;
R4a представляет собой водород, C1-6алкил, C2-6алкенил, C3-7циклоалкил, арил или C1-6алкил, необязательно замещенный C3-7циклоалкилом или арилом;
R4b представляет собой R4b', OR4b' или NHR4b';
R4b' представляет собой C1-6алкил, C2-6алкенил, C3-7циклоалкил, арил или C1-6алкил, необязательно замещенный C3-7циклоалкилом или арилом;
X представляет собой N, CH, и когда X несет двойную связь, он представляет собой C;
Rq представляет собой водород, или когда X представляет собой C или CH, Rq может также представлять собой C1-6алкил;
E представляет собой NR5, или когда X представляет собой N, тогда E представляет собой NR5 или CR6aR6b;
R5 представляет собой водород, C1-6алкил, C1-6алкоксиС1-6алкил, или C3-7циклоалкил;
R6a и R6b независимо представляют собой водород или C1-6алкил, или R6a и R6b вместе с атомом углерода, к которому они присоединены, образуют C3-7циклоалкил;
n равен 3, 4, 5 или 6;
каждая пунктирная линия ----- независимо представляет собой необязательную двойную связь;
R7 и R8 независимо представляют собой C1-6алкил, необязательно замещенный C1-6алкокси, -NRaRb, гидрокси, галогеном, C3-7циклоалкилом или арилом; C3-7циклоалкил; арил; Het; C2-6алкенил; C1-6алкокси; C3-7циклоалкилокси; арилокси; Het-O-; гидрокси; циано; галоген; полигалоген-C1-6алкил; -NRaRb; и R7 может также представлять собой водород;
R9 представляет собой водород или C1-6алкил;
Ra представляет собой H, C1-6алкил, C1-6алкокси;
Rb представляет собой H; C3-7циклоалкил; C1-6алкил, необязательно замещенный C3-7циклоалкилом или арилом; или Ra и Rb вместе с атомом азота, к которому они присоединены, образуют Het1;
каждый из арилов независимо представляет собой фенил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из галогена, гидрокси, нитро, циано, карбоксила, C1-6алкила, C1-6алкокси, C1-6алкоксиС1-6алкила, C1-6алкилкарбонила, амино, моно- или диC1-6алкиламино, азидо, меркапто, C1-6алкилтио, полигалогенC1-6алкила, полигалогенC1-6алкокси, C3-7циклоалкила и Het1;
каждый из Het независимо представляет собой 5- или 6-членное насыщенное, частично ненасыщенное или полностью ненасыщенное гетероциклическое кольцо, содержащее 1, 2, 3 или 4 гетероатома, каждый из которых независимо выбирается из азота, кислорода и серы, указанное гетероциклическое кольцо является необязательно замещенным одним, двумя или тремя заместителями, каждый из них независимо выбирается из галогена, гидрокси, нитро, циано, карбоксила, C1-6алкила, C1-6алкокси, C1-6алкоксиС1-6алкила, C1-6алкилкарбонила, амино, моно- или диC1-6алкиламино, азидо, меркапто, полигалогенC1-6алкила, полигалогенC1-6алкокси, C3-7циклоалкила, Het1;
каждый из Het1 независимо представляет собой пирролидинил, пиперидинил, пиперазинил, 4-C1-6алкил-пиперазинил, 4-C1-6алкилкарбонилпиперазинил и морфолинил, и при этом морфолинильные и пиперидинильные группы могут быть необязательно замещенными одним или двумя C1-6алкильными радикалами;
или их N-оксиды, фармацевтически приемлемые аддитивные соли, или их стереоизомеры.
Настоящее изобретение, кроме того, относится к способам получения соединений формулы (I), а также к использованию промежуточных соединений при получении соединений формулы (I).
Настоящее изобретение относится к соединениям формулы (I) самим по себе и к их N-оксидам, фармацевтически приемлемым аддитивным солям и их стереохимически изомерным формам, для применения в качестве лекарственного средства. Настоящее изобретение, кроме того, относится к фармацевтическим композициям, содержащим указанные соединения, для введения субъекту, страдающему от инфекции HCV. Фармацевтические композиции могут содержать комбинации указанных выше соединений с другими агентами против HCV.
Настоящее изобретение также относится к применению соединения формулы (I), его N-оксида, фармацевтически приемлемой аддитивной соли или стереохимически изомерной формы, для получения лекарственного средства для ингибирования репликации HCV. Или настоящее изобретение относится к способу ингибирования репликации HCV у теплокровного животного, указанный способ включает в себя введение эффективного количества соединения формулы (I), его N-оксида, фармацевтически приемлемой аддитивной соли или стереохимически изомерной формы.
Подробное описание изобретения
Как используется до и после этого, применяются следующие определения, если не отмечается иного.
Как используется в описании "C1-4алкил" как группа или часть группы определяет насыщенные углеводородные радикалы с прямой или разветвленной цепью, имеющие от 1 до 4 атомов углерода, такие, например, как метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил; "C1-6алкил" охватывает C1-4алкильные радикалы и их высшие гомологи, имеющие 5 или 6 атомов углерода, такие, например, как 1-пентил, 2-пентил, 3-пентил, 1-гексил, 2-гексил, 2-метил-1-бутил, 2-метил-1-пентил, 2-этил-1-бутил, 3-метил-2-пентил и тому подобное. Среди C1-6алкилов интерес представляет C1-4алкил.
Термин "C2-6алкенил" как группа или часть группы определяет углеводородные радикалы с прямой и разветвленной цепью, имеющие насыщенные связи углерод-углерод и, по меньшей мере, одну двойную связь, и имеющие от 2 до 6 атомов углерода, такие, например, как этенил (или винил), 1-пропенил, 2-пропенил (или аллил), 1-бутенил, 2-бутенил, 3-бутенил, 2-метил-2-пропенил, 2-пентенил, 3-пентенил, 2-гексенил, 3-гексенил, 4-гексенил, 2-метил-2-бутенил, 2-метил-2-пентенил и тому подобное. Среди C2-6алкенилов интерес представляет собой C2-4алкенил.
C3-7циклоалкил является общим наименованием для циклопропила, циклобутила, циклопентила, циклогексила и циклогептила.
C1-6алкокси означает C1-6алкилокси, где C1-6алкил является таким, как определено выше, и связан с атомом кислорода, то есть представляет собой -O-C1-6алкил. Среди C1-6алкокси интерес представляют собой метокси, этокси и пропокси.
Термин галоген является общим наименованием для фтора, хлора, брома и йода, в частности, фтора или хлора.
Термин "полигалогенC1-6алкил" как группа или часть группы, например, в полигалогенC1-6алкокси, определяется как C1-6алкил, замещенный одним или несколькими атомами галогенов, в частности, C1-6алкил, замещенный одним, двумя, тремя, четырьмя, пятью, шестью или более атомами галогена, такой как метил или этил с одним или несколькими атомами фтора, например, дифторметил, трифторметил, трифторэтил. Предпочтительным является трифторметил. Также включенными являются перфторC1-6алкильные группы, которые представляют собой C1-6алкильные группы, где все атомы водорода замещены атомами фтора, например, пентафторэтил. В случае, когда в пределах определения полигалогенC1-6алкила более одного атома галогена присоединяется к алкильной группе, атомы галогена могут быть одинаковыми или различными.
Как использовалось в описании ранее, термин (=O) или оксо образует карбонильный остаток, когда присоединяется к атому углерода, сульфоксидный остаток, когда присоединяется к атому серы, и сульфонильный остаток, когда два указанных термина присоединяются к атому серы. Когда кольцо или кольцевая система является замещенной оксо группой, атом углерода, с которым связана оксо, представляет собой насыщенный углерод.
Радикал Het представляет собой гетероцикл, как определено в этом описании и в формуле изобретения. Примеры Het включают в себя, например, пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, пирролил, пиразонил, имидазолил, оксазолил, изоксазолил, тиазинолил, изотиазинолил, тиазолил, изотиазолил, оксадиазолил, тиадиазолил, триазолил (включая 1,2,3-триазолил, 1,2,4-триазолил), тетразолил, фуранил, тиенил, пиридил, пиримидил, пиридазинил, триазинил и тому подобное. Среди радикалов Het интерес представляют те, которые являются ненасыщенными, в частности, имеющими ароматический характер. Кроме того, интерес представляют те радикалы Het, которые имеют один или два атома азота.
Каждый из радикалов Het, рассмотренных в этом и в следующем абзаце, может быть необязательно замещенным некоторым количеством и видом заместителей, рассмотренных в определениях соединений формулы (I) или любой из подгрупп соединений формулы (1). Некоторые из радикалов Het, рассмотренных в этом и в следующем абзаце, могут быть замещенными одним, двумя или тремя гидрокси заместителями. Такие гидрокси-замещенные кольца могут существовать как их таутомерные формы, несущие кето-группы. Например, 3-гидроксипиридазиновый остаток может существовать в своей таутомерной форме 2H-пиридазин-3-она. Когда Het представляет собой пиперазинил, он предпочтительно является замещенным в своем 4-положении заместителем, связанным с 4-азотом с помощью атома углерода, например, 4-C1-6алкилом, 4-полигалогенC1-6алкилом, C1-6алкоксиС1-6алкилом, C1-6алкилкарбонилом, C3-7циклоалкилом.
Представляющие интерес радикалы Het включают в себя, например, пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, пирролил, пиразонил, имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, оксадиазолил, тиадиазолил, триазолил (включая 1,2,3-триазолил, 1,2,4-триазолил), тетразолил, фуранил, тиенил, пиридил, пиримидил, пиридазинил, пиразонил, триазинил или любой из таких гетероциклов, конденсированный с бензольным кольцом, такой как индолил, индазолил (в частности, 1H-индазолил), индолинил, хинолинил, тетрагидрохинолинил (в частности, 1,2,3,4-тетрагидрохинолинил), изохинолинил, тетрагидроизохинолинил (в частности, 1,2,3,4-тетрагидроизохинолинил), хиназолинил, фталазинил, бензимидазолил, бензоксазолил, бензизоксазолил, бензотиазолил, бензоксадиазолил, бензотиадиазолил, бензофуранил, бензотиенил.
Радикалы Het пирролидинил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, 4-замещенный пиперазинил предпочтительно связываются через их атом азота, (то есть как 1-пирролидинил, 1-пиперидинил, 4-тиоморфолинил, 4-морфолинил, 1-пиперазинил, 4-замещенный 1-пиперазинил).
Необходимо отметить, что положения радикалов в любом молекулярном остатке, используемом в определении, может находиться на таком остатке где угодно, постольку, поскольку он является химически стабильным.
Радикалы, используемые в определениях переменных, включают в себя все возможные изомеры, если не указано иного. Например, пиридил включает в себя 2-пиридил, 3-пиридил и 4-пиридил; пентил включает в себя 1-пентил, 2-пентил и 3-пентил.
Когда любая переменная встречается в любом составляющем более одного раза, каждое определение является независимым.
Когда он используется далее, термин "соединения формулы (I)", или "настоящие соединения" или сходные термины, как подразумевается, включают в себя соединения формулы (I), их N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы. Один из вариантов осуществления включает в себя соединения формулы (I) или любую подгруппу соединений формулы (I), определенную среди них, и их фармацевтически приемлемые аддитивные соли и возможные стереоизомерные формы.
Соединения формулы (I) имеют несколько центров хиральности и существуют как стереохимически изомерные формы. Термин "стереохимически изомерные формы", как используется в описании, определяет все возможные соединения, состоящие из одинаковых атомов, связанных с помощью одной и той же последовательности связей, но имеющие различные трехмерные структуры, которые не являются взаимозаменяемыми, которыми могут обладать соединения формулы (I).
При упоминании случаев, где (R) или (S) используют для обозначения абсолютной конфигурации хирального атома в заместителе, обозначение осуществляют, принимая во внимание соединение в целом, а не заместитель отдельно.
Если не указывается или не рассматривается иного, химическое обозначение соединения охватывает смесь всех возможных стереохимически изомерных форм, которыми может обладать указанное соединение. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Все стереохимически изомерные формы соединений по настоящему изобретению, как в чистой форме, так и смешанные друг с другом, считаются охваченными рамками настоящего изобретения.
Чистые стереоизомерные формы соединений и промежуточных соединений, как рассмотрено в описании, определяются как изомеры, по существу не содержащие других энантиомерных или диастереомерных форм этой же основной молекулярной структуры указанных соединений или промежуточных соединений. В частности, термин "стереоизомерно чистый" относится к соединениям или промежуточным соединениям, имеющим стереоизомерный избыток, по меньшей мере, от 80% (то есть минимум 90% одного изомера и максимум 10% других возможных изомеров) до стереоизомерного избытка 100% (то есть 100% одного изомера и отсутствие другого), более конкретно, соединения или промежуточные соединения, имеющие стереоизомерный избыток от 90% до 100%, еще более конкретно, имеющие стереоизомерный избыток от 94% до 100% и наиболее конкретно, имеющие стереоизомерный избыток от 97% до 100%. Термины "энантиомерно чистый" и "диастереомерно чистый" должны пониматься сходным образом, но тогда принимаются во внимание энантиомерный избыток и диастереомерный избыток, соответственно, для рассматриваемой смеси.
Чистые стереоизомерные формы соединений и промежуточных соединений по настоящему изобретению могут быть получены посредством применения процедур, известных из литературы. Например, энантиомеры могут отделяться друг от друга посредством селективной кристаллизации их диастереомерных солей вместе с оптически активными кислотами или основаниями. Их примеры представляют собой винную кислоту, дибензоилвинную кислоту, дитолуоилвинную кислоту и камфорсульфоновую кислоту. Альтернативно, энантиомеры могут разделяться с помощью хроматографических методик с использованием хиральных неподвижных фаз. Указанные чистые стереохимически изомерные формы могут также быть получены из соответствующих чистых стереохимически изомерных форм соответствующих исходных материалов, при условии, что реакция осуществляется стереоспецифично. Предпочтительно, если желательным является конкретный стереоизомер, указанное соединение синтезируют с помощью стереоспецифичных способов получения. Эти способы преимущественно используют энантиомерно чистые исходные материалы.
Диастереомерные рацематы соединений формулы (I) могут быть получены отдельно с помощью обычных способов. Соответствующие физические способы разделения, которые могут преимущественно использоваться, представляют собой, например, селективную кристаллизацию и хроматографию, например колоночную хроматографию.
Для некоторых соединений формулы (I), их N-оксидов, фармацевтически приемлемых аддитивных солей и их сольватов, и промежуточных соединений, используемых при их получении, абсолютная стереохимическая конфигурация экспериментально не определена. Специалист в данной области способен определить абсолютную конфигурацию таких соединений с использованием известных из литературы способов, таких, например, как дифракция рентгеновского излучения.
Настоящее изобретение, так же как предполагается, включает в себя все изотопы атомов, имеющихся в настоящих соединениях. Изотопы включают в себя эти атомы, имеющие одинаковое атомное число, но различные массовые числа. В качестве общего примера и без ограничения, изотопы водорода включают в себя тритий и дейтерий. Изотопы углерода включают в себя C-13 и C-14.
Фармацевтически приемлемые аддитивные соли включают в себя терапевтически активные нетоксичные формы солей присоединения кислот и оснований для соединений формулы (I). Фармацевтически приемлемые кислотно-аддитивные соли обычно могут быть получены посредством обработки формы основания такой соответствующей кислотой. Соответствующие кислоты включают в себя, например, неорганические кислоты, такие как галогенводородные кислоты, например, хлористоводородную или бромистоводородную кислоту, серную, азотную, фосфорную и тому подобные кислоты; или органические кислоты, такие, например, как уксусная, пропановая, гидроксиуксусная, молочная, пировиноградная, щавелевая (то есть этандионовая), малоновая, янтарная (то есть бутандионовая кислота), малеиновая, фумаровая, яблочная (то есть гидроксилбутандионовая кислота), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламиновая, салициловая, п-аминосалициловая, памовая и тому подобные кислоты. Наоборот, указанные солевые формы могут преобразовываться посредством обработки соответствующим основанием в форму свободного основания.
Соединения формулы (I), содержащие кислотный протон, могут также преобразовываться в их нетоксичные формы солей присоединения металлов или аминов посредством обработки соответствующими органическими и неорганическими основаниями. Соответствующие формы основных солей включают в себя, например, соли аммония, соли щелочных и щелочноземельных металлов, например, соли лития, натрия, калия, магния, кальция и тому подобное, соли с органическими основаниями, например, соли с бензатином, N-метил-D-глюкамином, гидрабамином, и соли с аминокислотами, такими, например, как аргинин, лизин и тому подобное.
Термин аддитивные соли, как подразумевается, также включает в себя сольваты, которые соединения формулы (I), а также их соли, могут образовывать. Такие сольваты представляют собой, например, гидраты, алкоголяты, например, этаноляты, пропаноляты и тому подобное.
N-оксидные формы настоящих соединений, как подразумевается, включают в себя соединения формулы (I), где один или несколько атомов азота окисляются до так называемого N-оксида.
Некоторые соединения формулы (I) могут также существовать в их таутомерной форме. Такие формы, хотя не указаны в явном виде в формуле выше, как предполагается, включаются в рамки настоящего изобретения.
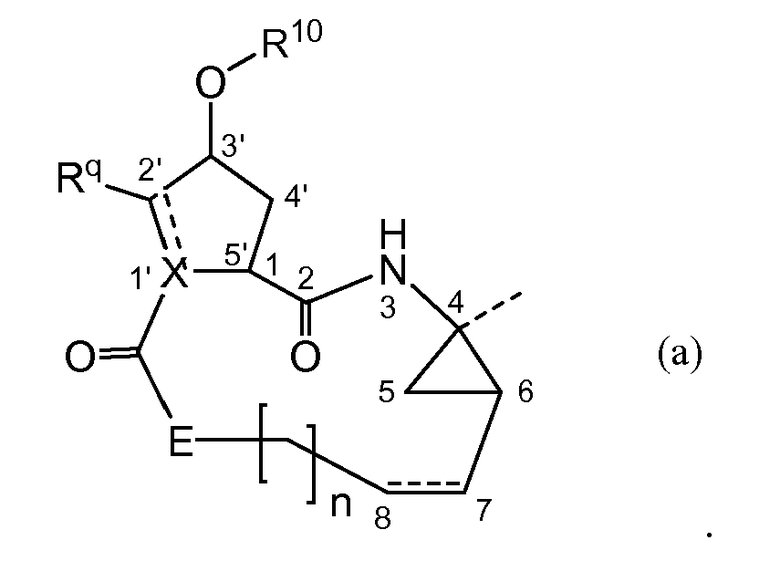
Как рассмотрено выше, соединения формулы (I) имеют несколько асимметричных центров. Чтобы более эффективно упоминать каждый из этих асимметричных центров, будет использоваться система нумерации, как показано на следующей структурной формуле.

Асимметричные центры присутствуют в положениях 1, 4 и 6 макроцикла, а также на атомах углерода 3' в 5-членном кольце, на атоме углерода 2', когда заместитель Rq представляет собой C1-6алкил, и на атоме углерода 1', когда X представляет собой CH. Каждый из этих асимметричных центров может существовать в их R или S конфигурации.
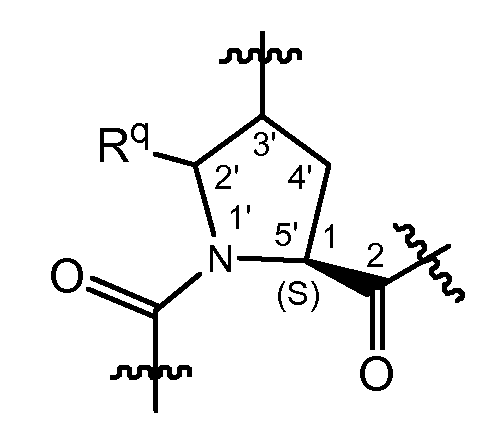
Когда X представляет собой N, стереохимия в положении 1 предпочтительно соответствует стереохимии конфигурации L-амино кислоты, то есть для L-пролина, как показано ниже.
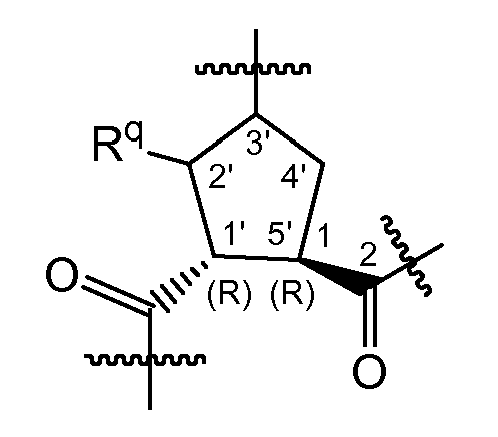
Когда X представляет собой CH, 2 карбонильные группы, замещенные в положениях 1' и 5' циклопентанового кольца, предпочтительно находятся в транс-конфигурации. Карбонильный заместитель в положении 5' предпочтительно находится в такой конфигурации, которая соответствует конфигурации L-пролина. Карбонильные группы, замещенные в положениях 1' и 5', предпочтительно являются такими, как изображено ниже в структуре следующей формулы:
Соединения формулы (I) включают в себя циклопропильную группу, как представлено в структурном фрагменте ниже:
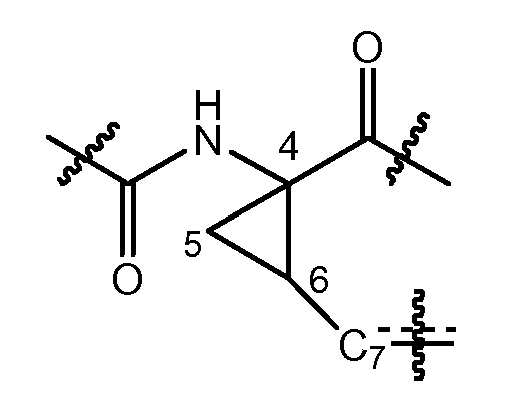
где C7 представляет собой атом углерода в положении 7 и атомы углерода в положении 4 и 6 представляют собой асимметричные атомы углерода циклопропанового кольца. Присутствие этих двух асимметричных центров означает, что соединения могут существовать в виде смесей диастереомеров, таких как диастереомеры соединений формулы (I), где атом углерода в положении 7 находится либо в цис-положении по отношению к карбонилу, либо в цис-положении по отношению к амиду, как показано ниже.
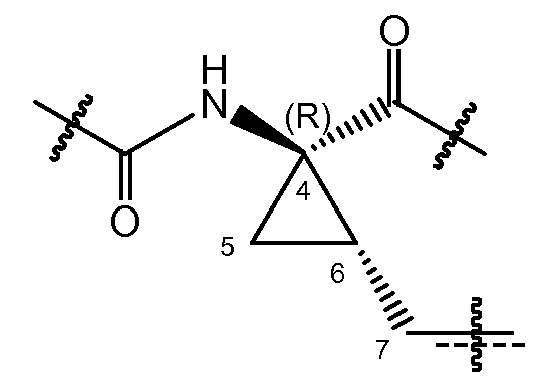
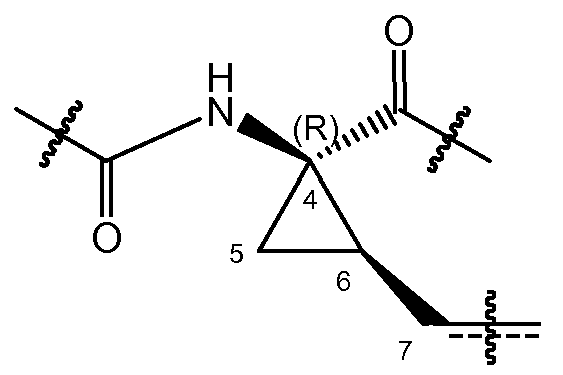
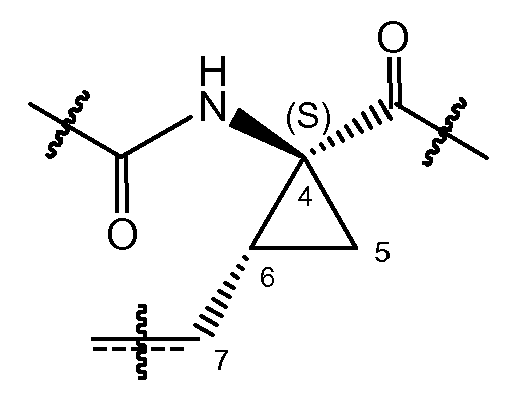
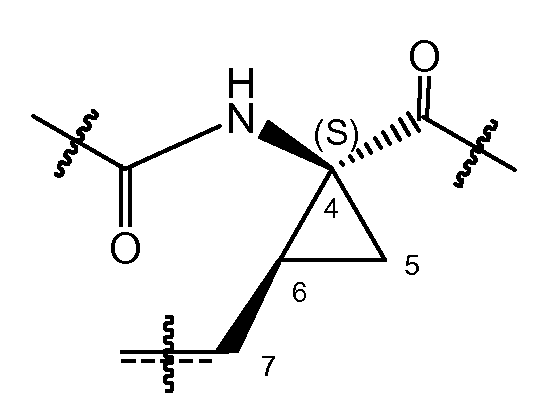
Один из вариантов осуществления относится к соединениям формулы I, где атом углерод в положении 7 находится в цис-положении по отношению к карбонилу. Другой вариант осуществления относится к соединениям формулы (I), где конфигурация на атоме углерода в положении 4 представляет собой R. Конкретная подгруппа соединений формулы (I) представляет собой такую, где атом углерода в положении 7 находится в цис-положении по отношению к карбонилу и где конфигурация на атоме углерода в положении 4 представляет собой R.
В соответствии с одним из вариантов осуществления циклопропильная группа (C4-C5-C6) связывается с группой A, которая представляет собой фосфонатную группу -P(=O)(OR4a)(R4b). В соответствии с этим вариантом осуществления, атом углерода в положении 7 находится в цис-положении либо к фосфонату, либо к амиду, как представлено в структурном фрагменте ниже:
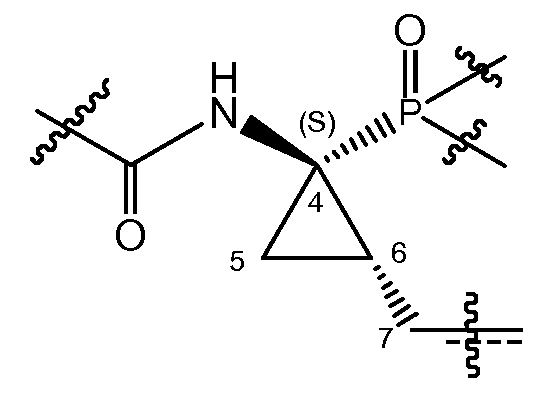
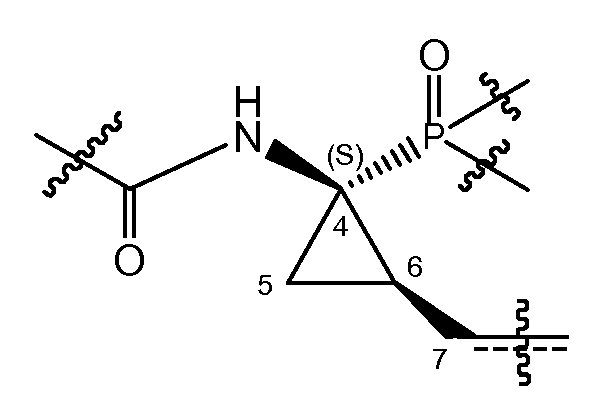
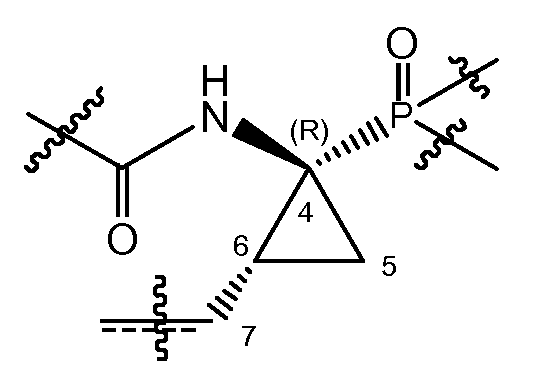
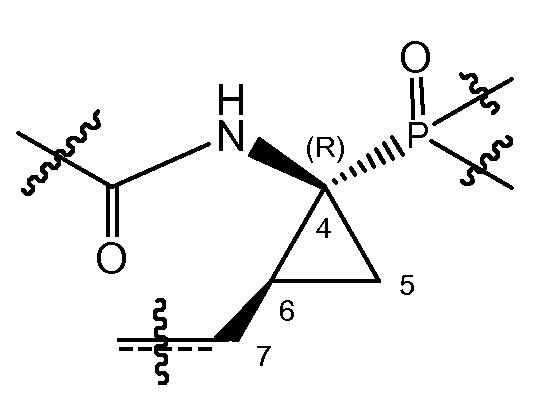
Один из вариантов осуществления относится к соединениям формулы (I), где атом углерода в положении 7 находится в цис-положении по отношению к фосфонату. Другой вариант осуществления относится к соединениям формулы (I), где конфигурация на атоме углерода в положении 4 представляет собой S. Конкретная подгруппа соединений формулы (I) представляет собой такую, где атом углерода в положении 7 находится в цис-положении по отношению к фосфонату и где конфигурация на атоме углерода в положении 4 представляет собой S.
Соединения формулы (I) могут содержать пролиновый остаток, то есть X представляет собой N, или циклопентильный или циклопентенильный остаток, то есть X представляет собой CH или C, соответственно. В соответствии с одним из вариантов осуществления настоящего изобретения соединения содержат частичные структуры:
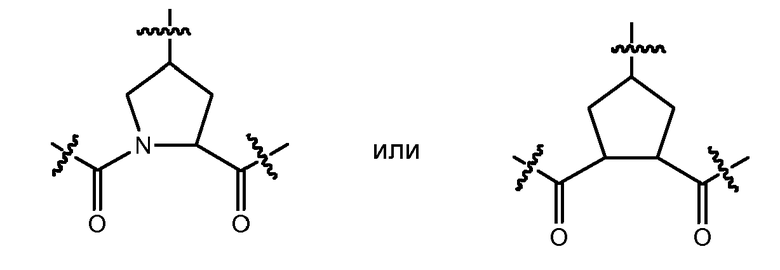
Дополнительные варианты осуществления настоящего изобретения представляют собой соединения формулы (I) или любую из подгрупп соединений формулы (I), где Rq представляет собой метил, E представляет собой NR5, X представляет собой CRz и Rz образует двойную связь с атомом углерода, несущим Rq.
Предпочтительными являются соединения формулы (I), где заместитель в 1 (или 5') положении и связанный с простым эфиром пиримидиновый заместитель в положении 3' находится в транс-конфигурации. Особенный интерес представляют собой соединения формулы (I), где положение 1 имеет конфигурацию, соответствующую L-пролину, и связанный с простым эфиром пиримидиновый заместитель в положении 3' находится в транс-конфигурации по отношению к положению 1.
Предпочтительно, соединения формулы (I) имеют стереохимию, как показано на структурах формул (I-a) и (I-b), ниже:
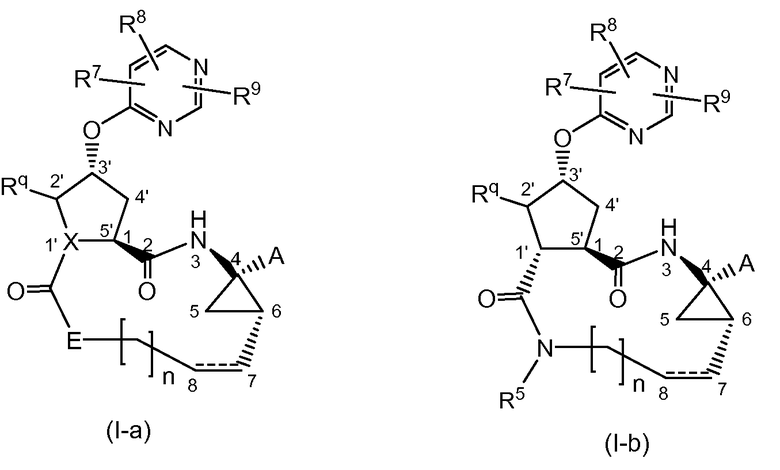
Один из вариантов осуществления настоящего изобретения относится к соединениям формулы (I) или формул (I-a), (I-b) или к любой подгруппе соединений формулы (I), где выполняется одно или несколько из следующих условий:
(a) Rq представляет собой водород;
(b) X представляет собой азот;
(c) E представляет собой NR5;
(d) присутствует двойная связь между атомами углерода 7 и 8.
Другой вариант осуществления настоящего изобретения относится к соединениям формулы (I) или формул (I-a), (I-b), или к любой подгруппе соединений формулы (I), где выполняется одно или несколько из следующих условий:
(a) Rq представляет собой водород;
(b) X представляет собой азот;
(c) E представляет собой CR6aR6b;
(d) присутствует двойная связь между атомами углерода 7 и 8.
Другой вариант осуществления настоящего изобретения относится к соединениям формулы (I) или формул (I-a), (I-b), или к любой подгруппе соединений формулы (I), где выполняется одно или несколько из следующих условий:
(a) Rq представляет собой водород;
(b) X представляет собой CH;
(c) E представляет собой NR5, где R5 является таким, как определено выше, в частности, R5 представляет собой водород или C1-6алкил;
(d) присутствует двойная связь между атомами углерода 7 и 8.
Конкретные подгруппы соединений формулы (I) представляют собой те, которые представлены ниже структурными формулами (I-c), (I-d) и (I-e):
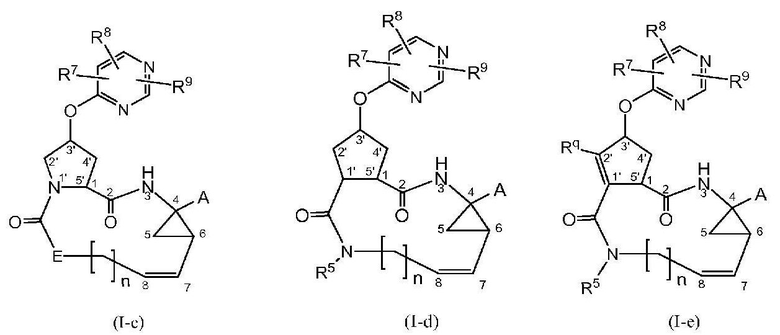
Среди соединений формулы (I-c), (I-d) и (I-e) те, которые имеют стереохимическую конфигурацию, показанную в формулах (I-a) и (I-b), соответственно, представляют собой особенный интерес.
Двойная связь между атомами углерода 7 и 8 в соединениях формулы (I) или в любой подгруппе соединений формулы (I) может находиться в цис- или в транс-конфигурации. Предпочтительно, двойная связь между атомами углерода 7 и 8 находится в цис-конфигурации, как показано в формулах (I-c), (I-d) и (I-e).
Другие конкретные подгруппы соединений формулы (I) представляют собой те, которые представлены следующими структурными формулами:
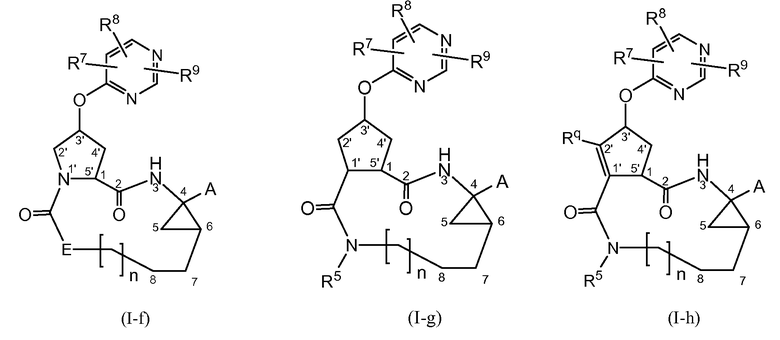
Особенный интерес среди соединений формул (I-f), (I-g) или (I-h) представляют собой те, которые имеют стереохимическую конфигурацию соединений формул (I-a) и (I-b).
В (I-a), (I-b), (I-c), (I-d), (I-e), (I-f), (I-g) или (I-h), где это применимо, A, E, X, n, Rq, R5, R7, R8 и R9 являются такими, как указано в определениях соединений формулы (I) или в любой из подгрупп соединений формулы (I), определенных в описании.
Необходимо понять, что определенные выше подгруппы соединений формул (I-a), (I-b), (I-c), (I-d) или (I-e), а также любая другая подгруппа, определенная в описании, как подразумевается, включают в себя также любые N-оксиды, аддитивные соли и стереохимически изомерные формы таких соединений.
Когда N равно 2, остаток -CH2- в скобках перед "n" соответствует этандиилу в соединениях формулы (I) или в любой подгруппе соединений формулы (I). Когда n равно 3, остаток -CH2- в скобках перед "n" соответствует пропандиилу в соединениях формулы (I) или в любой подгруппе соединений формулы (I). Когда n равно 4, остаток -CH2- в скобках перед "n" соответствует бутандиилу в соединениях формулы (I) или в любой подгруппе соединений формулы (I). Когда n равно 5, остаток -CH2- в скобках перед "n" соответствует пентандиилу в соединениях формулы (I) или в любой подгруппе соединений формулы (I). Когда n равно 6, остаток -CH2- в скобках перед "n" соответствует гександиилу в соединениях формулы (I) или в любой подгруппе соединений формулы (I). Конкретные подгруппы соединений формулы (I) представляют собой такие соединения, где n равно 4 или 5.
Варианты осуществления настоящего изобретения представляют собой соединения формулы (I) или любые подгруппы соединений формулы (I), где A представляет собой -C(=O)OR1, в частности, где R1 представляет собой C1-6алкил, такой как метил, этил или трет-бутил, а наиболее предпочтительно, где R1 представляет собой водород.
Другой вариант осуществления настоящего изобретения представляют собой соединения формулы (I) или любые подгруппы соединений формулы (I), где A представляет собой -C(=O)-NH-SO2R2, в частности, где R2 представляет собой C3-7циклоалкил, фенил или группу Het, например, тиазолил или пиридил, любой из них является необязательно замещенным одним или более, например одним или двумя заместителями, выбранными из C1-6алкила, C1-6алкокси, трифторметила и галогена, или в частности, одним или двумя заместителями, выбранными из метила, фтора и хлора. Например, R2 может представлять собой 1-метилциклопропил.
Другой вариант осуществления настоящего изобретения представляют собой соединения формулы (I) или любые подгруппы соединений формулы (I), где A представляет собой -C(=O)C(=O)NR3aR3b, в частности, где R3a и R3b независимо выбираются из водорода, C1-6алкила, необязательно замещенного арилом, и C2-6алкенила. В одном из вариантов осуществления, один из R3a и R3b представляет собой водород, а другой представляет собой 3-пропенил, циклопропилметил или циклопропил. В дополнительном варианте осуществления, как R3a, так и R3b представляют собой водород.
Другой вариант осуществления настоящего изобретения представляют собой соединения формулы (I) или любые подгруппы соединений формулы (I), где A представляет собой -C(=O)-NH-P(=O)(OR4a)(R4b), в частности, где R4a представляет собой C1-6алкил, в частности этил или изопропил, и R4b представляет собой OR4b, и R4b' представляет собой C1-6алкил, такой как этил или изопропил.
Другой вариант осуществления настоящего изобретения представляют собой соединения формулы (I) или любой из подгрупп соединений формулы (I), где A представляет собой -P(=O)(OR4a)(R4b), в частности, где R4a представляет собой C1-6алкил, в частности, этил или изопропил, и R4b представляет собой OR4b', и R4b' представляет собой C1-6алкил, в частности этил или изопропил.
Другие варианты осуществления настоящего изобретения представляют собой соединения формулы (I) или любые подгруппы соединений формулы (I), где
(a) R5 представляет собой водород; C1-6алкил; C1-6алкоксиС1-6алкил или C3-7циклоалкил;
(b) R5 представляет собой водород или C1-6алкил;
(c) R5 представляет собой водород.
Предпочтительные варианты осуществления настоящего изобретения представляют собой соединения формулы (I) или любые подгруппы соединений формулы (I), где R5 представляет собой водород или C1-6алкил, более предпочтительно, водород или метил.
Еще один вариант осуществления относится к соединениям формул (I), (I-e) или к любой подгруппе соединений формулы (I), где R6a и R6b независимо представляют собой водород или C1-6алкил, например, метил. Предпочтительно, R6a представляет собой водород и R6b представляет собой метил, или более предпочтительно, как R6a, так и R6b представляют собой водород.
Еще один вариант осуществления относится к соединениям формулы (I) или к любой подгруппе соединений формулы (I), где R7 и R8, каждый, независимо выбираются из C1-6алкила, необязательно замещенного C1-6алкокси, гидрокси, галогеном или арилом; арила; Het; C2-6алкенила; C1-6алкокси; арилокси; Het-O-; гидрокси; циано; галогена; и -NRaRb; и R7 может представлять собой водород; или где R7 и R8, каждый, независимо выбираются из C1-6алкила, необязательно, замещенного C1-6алкокси или арилом; арила; Het; C2-6алкенила; C1-6алкокси; арилокси; Het-O-; гидрокси; циано; галогена; -NRaR; и R7 может представлять собой водород; или, где R7 и R8, каждый, независимо выбираются из C1-6алкила, необязательно замещенного C1-6алкокси или арилом; арила; пиридила; C1-6алкокси; арилокси; пиридил-O- или -NRaRb; где Ra и Rb, каждый, независимо представляют собой водород или C1-6алкил, или группа -NRaRb представляет собой Het1; и R7 может представлять собой водород; или где R7 и R8, каждый, независимо выбираются из C1-6алкила, C1-6алкокси; арила; и -NRaRb; где Ra и Rb, каждый, независимо представляют собой водород или C1-6алкил, или группа -NRaRb представляет собой морфолинил; и R7 может представлять собой водород. Конкретные подгруппы соединений этого варианта осуществления представляют собой такие, где R9 представляет собой водород.
Конкретный вариант осуществления настоящего изобретения относится к соединениям формулы (I) или к любой их подгруппе, где связанный с простым эфиром пиримидинильный остаток имеет следующую структуру:

где R7, R8 и R9 являются такими, как определено выше.
В этом варианте осуществления представляют интерес следующие дополнительные подгруппы соединений (I) или любых их подгрупп: то есть, это такие, где применимы одно или несколько из следующих определений:
R9 представляет собой водород или C1-6алкил, в частности, водород или метил; или R9 представляет собой водород;
R7 и R8 независимо представляют собой C1-6алкил, галоген, C1-6алкокси, амино, моно- или диC1-6алкил-амино, арил, Het, или Het1; в частности, где арил, Het или Het1 представляет собой пиридил, тиазолил, оксазолил, пиразонил, фенил, пиперидинил, или морфолинил, где указанный пиридил, тиазолил, оксазолил, пиразонил, фенил может быть необязательно замещенным одним или двумя заместителями, выбранными из C1-6алкила, C1-6алкокси, галогена, амино и C1-6алкокси-C1-6алкила; или одним или двумя, или одним, заместителем (заместителями), выбранными из C1-6алкила, галогена и C1-6алкокси или выбранными из метокси, хлора или фтора; и где указанный пиперидинил или морфолинил может быть необязательно замещенным одним или двумя C1-6алкилами; и где
R7 может также представлять собой водород.
Конкретные подгруппы в этом варианте осуществления представляют собой такие, где применимы в группах (a) или (b) одно или несколько из следующих определений:
R7 и R8 независимо представляют собой галоген (например, хлор), C1-6алкил, C1-6алкокси (например, метокси или изопропокси), морфолинил, пиперидинил, амино, моно- или диC1-6алкиламино; или R7 представляет собой водород, C1-6алкил, C1-6алкокси (например, метокси или изопропокси), морфолинил, моно- или диC1-6алкиламино (например, метиламино или изопропиламино); и где
R7 может также представлять собой водород;
R9 представляет собой водород или C1-6алкил, в частности, водород или метил; или R9 представляет собой водород.
Конкретные подгруппы в этом варианте осуществления представляют собой такие, где в группах (a) или (b) применимы одно или несколько из следующих определений:
R7 представляет собой водород; C1-6алкил; C1-6алкокси; морфолинил; пиперидинил; фенил, необязательно замещенный галогеном, C1-6алкилом или C1-6алкокси; амино; моно- или диC1-6алкиламино; или R7 представляет собой водород; C1-6алкокси (например, метокси или изопропокси); морфолинил; фенил, необязательно замещенный C1-6алкокси; моно- или диC1-6алкиламино (например, метиламино или изопропиламино);
R8 представляет собой C1-6алкил, фенил, морфолинил, моно- или диC1-6алкиламино; или R8 представляет собой C1-6алкил (например, трет-бутил), фенил, морфолинил, моно- или диC1-6алкиламино (например, метиламино или изопропиламино).
Другой вариант осуществления относится к соединениям формулы (I) или к любым их подгруппам, где заместители R7 и R8 в группах (a) или (b) независимо выбираются из арила, Het, -O-арила, -O-Het, C1-6алкила, необязательно замещенного арила или Het. Арил и Het в предыдущих определениях, в частности, могут представлять собой необязательно замещенный фенил, пиридил, пиперидинил, морфолинил, пиразонил и тиазолил, каждый, необязательно замещаются, как определено выше, в частности, для R7 и R8, представляющих собой фенил, пиридил, пиразонил или тиазолил, метокси, хлором или фтором.
Другой вариант осуществления относится к соединениям формулы (I) или к любым их подгруппам, где заместители R7 и R8 независимо представляют собой C1-6алкил, например, трет-бутил, или C1-6алкокси, например, метокси, или где R8 представляет собой фенил или пиридил, необязательно замещенный.
Другой вариант осуществления относится к таким соединениям формулы (I) или к любым их подгруппам, которые представляют собой такие, где пиримидинильный радикал (a) или (b) представляет собой:
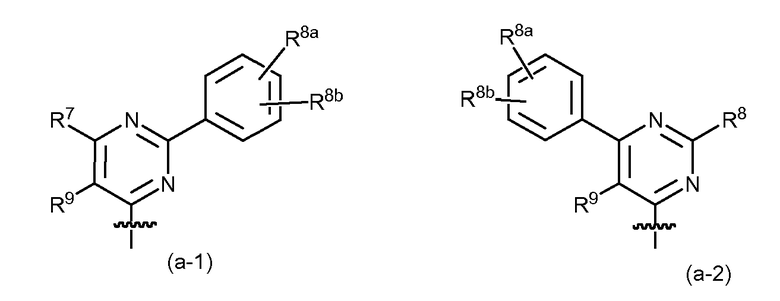
где R7 и R8 независимо представляют собой фенил или C1-6алкокси, например, метокси; R9 представляет собой водород или C1-6алкил; в частности, R9 представляет собой водород или метил; или R9 представляет собой водород; R8a или R8b представляет собой необязательный заместитель арила, как определено выше, в частности, R8a или R8b представляют собой водород, C1-6алкокси, например, метокси, или галоген, например, фтор или хлор. Когда один из R8a или R8b является иным, чем водород, он может, в частности, быть замещенным в пара положении фенильного кольца.
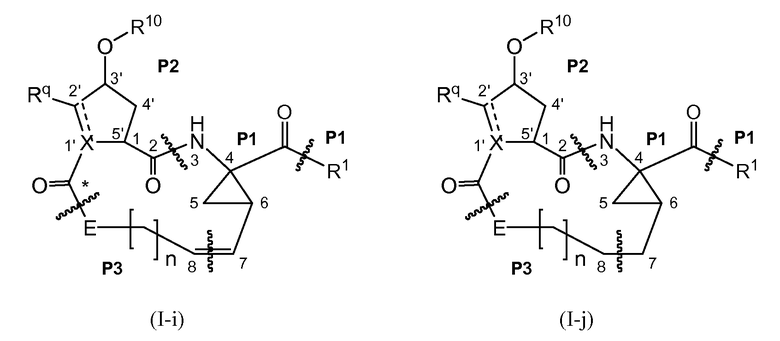
Соединения формулы (I) состоят из трех структурных элементов P1, P2, P3. Структурный элемент P1 дополнительно содержит хвост P1'. Карбонильная группа, отмеченная ниже звездочкой в соединении (I-c), может представлять собой часть либо структурного элемента P2, либо структурного элемента P3. По чисто химическим причинам, структурный элемент P2 соединений формулы (I), где X представляет собой C, включает в себя карбонильную группу, присоединенную в положении 1'.
Связывание структурных элементов P1 с P2, P2 с P3 и P1 с P1' (когда R1 представляет собой -NH-SO2R2) включает в себя образование амидной связи. Связывание структурных элементов P1 и P3 включает в себя формирование двойной связи. Связывание структурных элементов P1, P2 и P3 для получения соединения (I-i) или (I-j) может осуществляться в любой заданной последовательности. Одна из стадий включает в себя циклизацию, при этом образуется макроцикл.
В следующем далее описании, представлении соединений и схемах реакций R10 представляет собой пиримидинильную группу:
Ниже представлены соединения (I-i), которые представляют собой соединения формулы (I), где атомы углерода C7 и C8 соединяются двойной связью, и соединения (I-j), которые представляют собой соединения формулы (I), где атомы углерода C7 и C8 соединяются одинарной связью. Соединения формулы (I-j) могут быть получены из соответствующих соединений формулы (I-I) посредством восстановления двойной связи в макроцикле.
Процедуры синтеза, описанные далее, как подразумевается, могут применяться также для рацематов, стереохимически чистых промежуточных соединений или конечных продуктов, или для любых стереоизомерных смесей. Рацематы или стереохимические смеси могут разделяться на стереоизомерные формы на любой ступени процедур синтеза. В одном из вариантов осуществления, промежуточные соединения и конечные продукты имеют стереохимию, определенную выше в соединениях формулы (I-a) и (I-b).
В одном из вариантов осуществления, соединения (I-i) получают посредством формирования сначала амидных связей и последующего образования двойной связи между P3 и P1 с одновременной циклизацией макроцикла.
В одном из вариантов осуществления, соединения (I), где связь между C7 и C8 представляет собой двойную связь, которые являются соединениями формулы (I-i), как определено выше, могут быть получены, как показано на следующей схеме реакции:
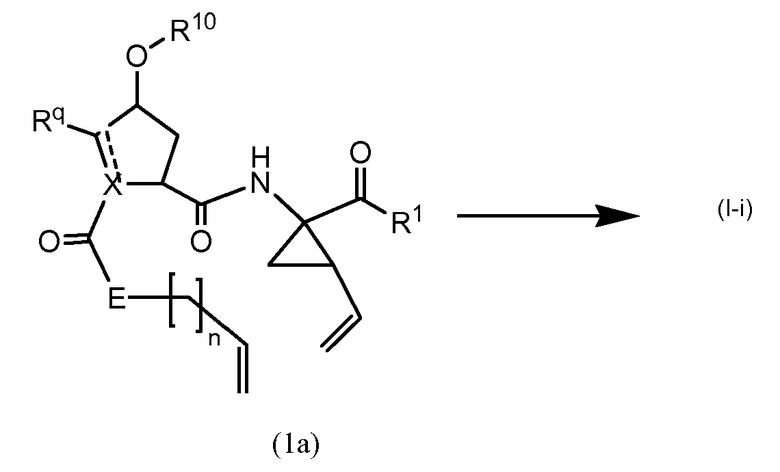
Образование макроцикла может осуществляться посредством реакции метатезиса олефинов в присутствии соответствующего металлического катализатора, такого например, как катализатор на основе Ru, о котором сообщается в Miller, S.J., Blackwell, H.E., Grubbs, R.H. J. Am. Chem. Soc. 118, (1996), 9606-9614; Kingsbury, J. S., Harrity, J. P. A., Bonitatcbus, P. J., Hovejda, A. H., J. Am. Chem. Soc. 121, (1999), 791-799; и Huang et al., J. Am. Chem. Soc. 121, (1999), 2674-2678; например, катализатора Ховейды-Граббса.
Могут использоваться стабильные на воздухе рутениевые катализаторы, такие как бис(трициклогексилфосфин)-3-фенил-1H-инден-1-илиденрутений хлорид (Neolyst М1®) или бис(трициклогексилфосфин)-[(фенилтио)метилен]рутений (IV) дихлорид. Другие катализаторы, которые могут использоваться, представляют собой катализаторы Граббса первого и второго поколения, например, бензилиден-бис(трициклогексилфосфин)дихлоррутений и (1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(фенилметилен)-(трициклогексилфосфин)рутений, соответственно. Особенный интерес представляют катализаторы Ховейды-Граббса первого и второго поколения, которые представляют собой дихлор(o-изопропоксифенилметилен)(трициклогексилфосфин)рутений(II) и 1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор-(o-изопропоксифенилметилен)рутений, соответственно. Также, для этой реакции могут использоваться другие катализаторы, содержащие другие переходные металлы, такие как Mo.
Реакции метатезиса могут осуществляться в соответствующем растворителе, таком, например, как простые эфиры, например, ТГФ, диоксан; галогенированные углеводороды, например, дихлорметан, CHCl3, 1,2-дихлорэтан и тому подобное, углеводороды, например, толуол. В предпочтительном варианте осуществления, реакцию метатезис осуществляют в толуоле. Эти реакции осуществляют при повышенных температурах в атмосфере азота.
Соединения формулы (I), где связь между C7 и C8 в макроцикле представляет собой одинарную связь, то есть соединения формулы (I-j) могут быть получены из соединений формулы (I-i) посредством восстановления двойной связи C7-C8 в соединениях формулы (I-i). Это восстановление может осуществляться посредством каталитического гидрирования водородом в присутствии катализатора на основе благородного металла, такого, например, как Pt, Pd, Rh, Ru или никель Ренея. Интерес представляет Rh на окиси алюминия. Реакцию гидрирования предпочтительно осуществляют в растворителе, таком, например, как спирт, такой как метанол, этанол, или в простом эфире, таком как ТГФ, или в их смесях. Вода также может добавляться к этим растворителям или смесям растворителей.
Группа A может соединяться со структурным элементом P1 на любой ступени синтеза, то есть до или после циклизации, или до или после циклизации и восстановления, как рассмотрено в описании выше. Соединения формулы (I), где A представляет собой -CO-NHSO2R2, указанные соединения представлены формулой (I-k-1), могут быть получены посредством связывания группы A с P1 посредством формирования амидной связи между обоими остатками. Подобным же образом, соединения формулы (I), где R1 представляет собой -C(=O)OR', то есть соединения (I-k-2) могут быть получены посредством связывания группы R1 с P1 посредством формирования сложноэфирной связи. В одном из вариантов осуществления, группы -C(=O)OR1 вводят на последней стадии синтеза соединений (I), как показано на следующих далее схемах реакций, где G представляет собой группу:
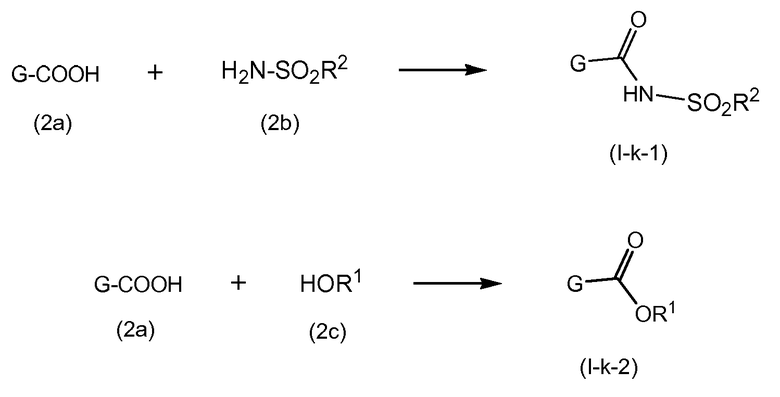
Промежуточное соединение (2a) может связываться с сульфонамидом (2b) посредством реакции образования амида, такой как любая из процедур формирования амидной связи, описанных далее. В частности, (2a) может обрабатываться агентом для связывания, например, N,N'-карбонилдиимидазолом (CDI), EEDQ, IIDQ, EDCI или бензотриазол-1-ил-окси-трис-пирролидинофосфоний гексафторфосфатом (коммерчески доступным как PyBOP®), в растворителе, таком как простой эфир, например, ТГФ, или галогенированный углеводород, например, дихлорметан, хлороформ, дихлорэтан, и взаимодействовать с желаемым сульфонамидом (2b), предпочтительно, после взаимодействия (2a) с агентом для связывания. Реакции (2a) с (2b) предпочтительно осуществляют в присутствии основания, например, триалкиламина, такого как триэтиламин или диизопропилэтиламин, или 1,8-диазабицикло[5.4.0]ундек-7-ен (DBU). Промежуточное соединение (2a) может также преобразовываться в активированную форму, например, в активированную форму общей формулы G-CO-Z, где Z представляет собой галоген или остаток активного сложного эфира, например, Z представляет собой арилокси группу, такую как фенокси, п-нитрофенокси, пентафторфенокси, трихлорфенокси, пентахлорфенокси и тому подобное; или Z может представлять собой остаток смешанного ангидрида. В одном из вариантов осуществления, G-CO-Z представляет собой хлорангидрид (G-CO-Cl) или смешанный ангидрид кислоты (G-CO-O-CO-R или G-CO-O-CO-OR, R в последнем представляет собой, например, C1-4алкил, такой как метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил, или бензил). Активированная форма G-CO-Z взаимодействует с сульфонамидом (2b).
Соединения формулы (I), где A представляет собой -C(=O)-NH-P(=O)(OR6a)(R6b), указанные соединения представлены формулой (I-k-3), могут быть получены посредством образования амидной связи между промежуточным соединением (2a) и фосфорамидатом (2d), следуя процедурам образования амидной связи, описанным далее. В частности, (2a) может обрабатываться агентом для связывания в соответствующем растворителе, предпочтительно, в присутствии основания, с последующим взаимодействием с фосфорамидатом (2d), предпочтительно, после взаимодействия (2a) с агентом для связывания. Промежуточное соединение (2a) может также преобразовываться в активированную форму, например, в активированную форму общей формулы G-CO-Z, где Z представляет собой галоген или остаток активного сложного эфира, например, Z представляет собой арилокси группу, такую как фенокси, п-нитрофенокси, пентафторфенокси, трихлорфенокси, пентахлорфенокси и тому подобное; или Z может представлять собой остаток смешанного ангидрида. В одном из вариантов осуществления, G-CO-Z представляет собой хлорангидрид (G-CO-Cl) или смешанный ангидрид кислоты (G-CO-O-CO-R или G-CO-O-CO-OR, R в последнем представляет собой, например, C1-4алкил, такой как метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил, или бензил). Активированная форма G-CO-Z взаимодействует с желаемым (2d). Агент для связывания, растворитель и основание могут быть такими, как описано далее в общем описании получения амидных связей.
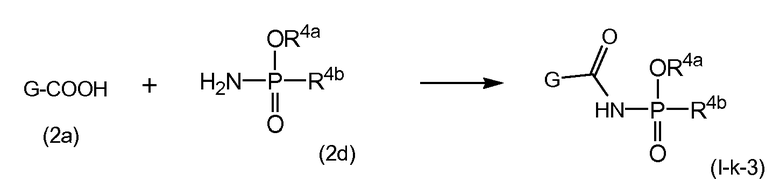
Активирование карбоновой кислоты в (2a), как описано в указанных выше реакциях, может приводить к реакции внутренней циклизации, к азалактоновому промежуточному соединению формулы
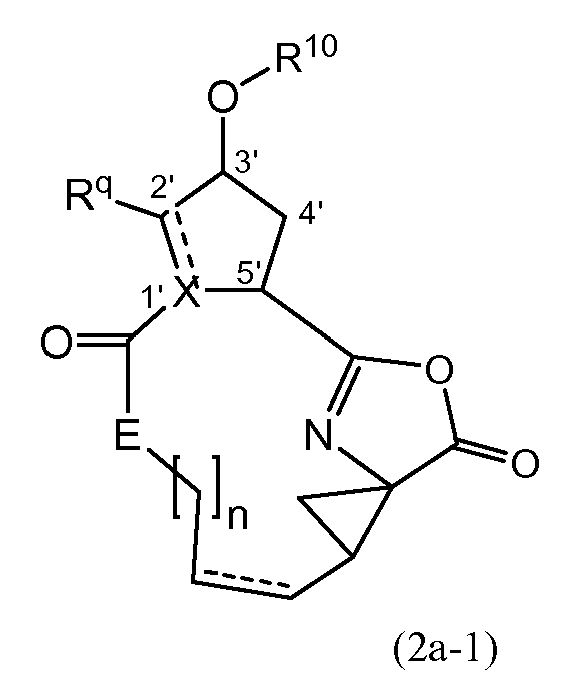
где X, E, Rq, R10 и n являются такими, как определено выше, и где стереогенные центры могут иметь стереохимическую конфигурацию, как определено выше, например, как в (I-a) или (I-b). Промежуточные соединения (2a-1) могут быть выделены из реакционной смеси с использованием обычной методологии, и выделенное промежуточное соединение (2a-1) затем взаимодействует с (2b) или (2d), или реакционная смесь, содержащая (2a-1), может взаимодействовать дополнительно с (2b) или (2d) без выделения (2a-1). В одном из вариантов осуществления, где реакция с агентом для связывания осуществляется в растворителе, не смешиваемом с водой, реакционную смесь, содержащую (2a-1), могут промывать просто водой или чуть основной водой для удаления всех водорастворимых побочных продуктов. Полученный таким образом промытый раствор может затем взаимодействовать с (2b) или (2d) без дополнительных стадий очистки. Выделение промежуточных соединений (2a-1), с другой стороны, может обеспечить определенные преимущества в том, что выделенный продукт после необязательной дополнительной очистки может взаимодействовать с (2b) или (2d), что приводит к получению меньшего количества побочных продуктов и к более легкому извлечению реакционной смеси.
Промежуточное соединение (2a) может связываться со спиртом (2c) с помощью реакции образования сложного эфира. Например, (2a) и (2c) взаимодействуют вместе, с удалением воды либо физически, например, посредством азеотропного удаления воды, либо химически, посредством использования дегидратирующего агента. Промежуточное соединение (2a) может также преобразовываться в активированную форму G-CO-Z, такую как активированные формы, рассмотренные выше, а впоследствии взаимодействовать со спиртом (2c). Реакция образования сложных эфиров предпочтительно осуществляют в присутствии основания, такого как карбонат или гидрокарбонат щелочного металла, например, бикарбонат натрия или калия, или третичный амин, такой как амины, рассмотренные в описании в связи с реакциями образования амидов, в частности, триалкиламин, например, триэтиламин. Растворители, которые могут использоваться в реакциях образования сложных эфиров, включают в себя простые эфиры, такие как ТГФ; галогенированные углеводороды, такие как дихлорметан, CH2Cl2; углеводороды, такие как толуол; полярные апротонные растворители, такие как ДМФ, ДМСО, DMA; и тому подобные растворители.
Соединения формулы (I), где E представляет собой NH, указанные соединения представлены как (I-1), могут также быть получены посредством удаления защитной группы PG с соответствующего азот-защищенного промежуточного соединения (3a), как на следующей далее схеме реакции. Защитная группа PG, в частности, представляет собой любую из азот-защитных групп, рассмотренных далее, и может удаляться с использованием процедур, также рассмотренных далее:
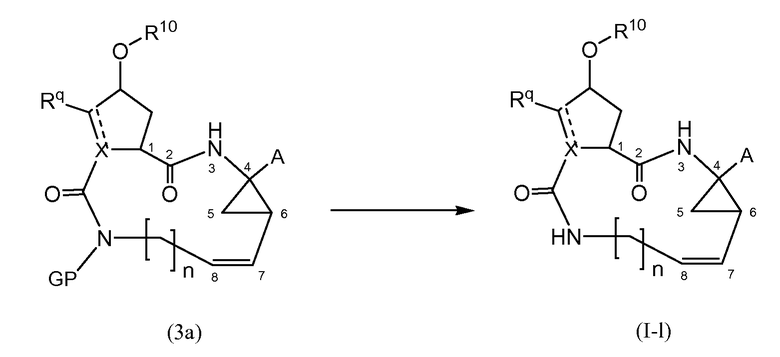
Исходные материалы (3a) в указанной выше реакции могут быть получены, следуя процедурам для получения соединений формулы (1), но с использованием промежуточных соединений, где группа R5 представляет собой PG.
Соединения формулы (I) могут также быть получены посредством взаимодействия промежуточного соединения (4a) с промежуточным соединением (4b), как показано на следующей схеме реакции, где различные радикалы имеют обозначения, определенные выше:
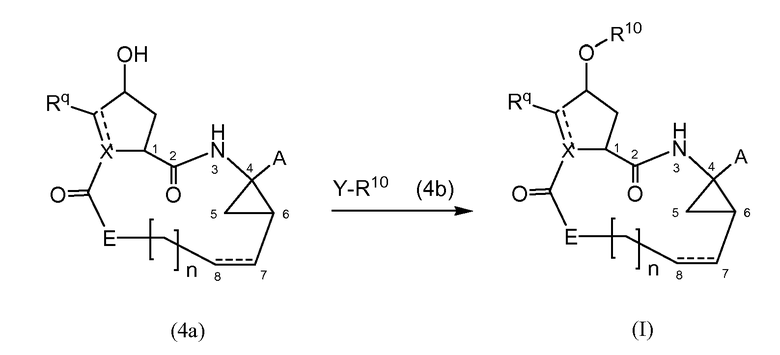
Y в (4b) представляет собой гидрокси или уходящую группу LG, такую как галогенид, например, бромид или хлорид, или арилсульфонильную группу, например, мезилат, трифлат или тозилат и тому подобное.
В одном из вариантов осуществления, реакция (4a) с (4b) представляет собой реакцию O-арилирования и Y представляет собой уходящую группу. Эта реакция может осуществляться, следуя процедурам, описанным E. M. Smith et al. (J. Med. Chem. (1988), 31, 875-885). В частности, эту реакцию осуществляют в присутствии основания, предпочтительно, сильного основания, в реакционно инертном растворителе, например, в одном из растворителей, рассмотренных для образования амидной связи.
В конкретном варианте осуществления, исходный материал (4a) взаимодействует с (4b) в присутствии основания, которое является достаточно сильным, чтобы отделить водород от гидрокси группы, например, щелочи из гидрида щелочного металла, такого как LiH или гидрид натрия, или алкоксида щелочного металла, такого как метоксид или этоксид натрия или калия, трет-бутоксид калия, в реакционно инертном растворителе, подобном диполярному апротонному растворителю, например, DMA, ДМФ, и тому подобное. Полученный алкоголят взаимодействует с арилирующим агентом (4b), где Y представляет собой соответствующую уходящую группу, как рассмотрено выше. Преобразование (4a) в (I) с использованием этого типа реакции O-арилирования не изменяет стереохимическую конфигурацию на атоме углерода, несущем гидрокси группу.
Альтернативно, реакция (4a) с (4b) может также осуществляться с помощью реакции Мицунобу (Mitsunobu, 1981, Synthesis, January, 1-28; Rano et al., Tetrahedron Lett., 1995, 36, 22, 3779-3792; Krchnak et al., Tetrahedron Lett., 1995, 36, 5, 6193-6196; Richter et al., Tetrahedron Lett., 1994, 35, 27, 4705-4706). Эта реакция включает в себя обработку промежуточного соединения (4a) с помощью (4b), где Y представляет собой гидрокси, в присутствии трифенилфосфина и активирующего агента, такого как диалкилазокарбоксилат, например, диэтилазодикарбоксилата (DEAD), диизопропилазодикарбоксилата (DIAD) или чего-либо подобного. Реакция Мицунобу изменяет стереохимическую конфигурацию на атоме углерода, несущем гидрокси группу.
Альтернативно, для получения соединений формулы (I), сначала формируется амидная связь между структурными элементами P2 и P1, с последующим присоединением структурного элемента P3 к остатку P1 в P1-P2, и с последующим образованием карбаматной или сложноэфирной связи между P3 и остатком P2 в P2-P1-P3 с одновременным замыканием кольца.
Еще одна альтернативная методология синтеза представляет собой образование амидной связи между структурными элементами P2 и P3, с последующим связыванием структурного элемента P1 с остатком P3 в P3-P2 и с образованием последней амидной связи между P1 и P2 в P1-P3-P2 с одновременным замыканием кольца.
Структурные элементы P1 и P3 могут связываться с последовательностью P1-P3. Если это желательно, двойная связь, связывающая P1 и P3, может восстанавливаться. Полученная таким образом последовательность P1-P3, либо восстановленная, либо нет, может связываться со структурным элементом P2, и сформированная таким образом последовательность P1-P3-P2 впоследствии циклизируется посредством образования амидной связи.
Структурные элементы P1 и P3 в любом из предыдущих подходов могут связываться с помощью формирования двойной связи, например, посредством реакции метатезиса олефинов, описанной далее, или реакции типа Виттига. Если это желательно, сформированная таким образом двойная связь может восстанавливаться, подобно тому, как описано выше для преобразования (I-i) в (I-j). Двойная связь может также восстанавливаться на более поздней ступени, например, после добавления третьего структурного элемента или после образования макроцикла. Структурные элементы P2 и P1 связываются посредством образования амидной связи, и P3 и P2 связываются посредством образования карбамата или сложного эфира.
Хвост P1' может соединяться со структурным элементом P1 на любой ступени синтеза соединений формулы (I), например, до или после связывания структурных элементов P2 и P1; до или после связывания P3 структурного элемента с P1 или до или после замыкания кольца.
Индивидуальные структурные элементы могут сначала получаться, впоследствии связываться вместе, или, альтернативно, предшественники структурных элементов могут связываться вместе и модифицироваться на более поздней ступени до желаемой молекулярной композиции.
Функциональные группы каждого из структурных элементов могут защищаться для предотвращения побочных реакций.
Образование амидных связей может осуществляться с использованием стандартных процедур, таких как те, которые используются для связывания аминокислот в пептидном синтезе. Последний включает в себя дегидративное связывание карбоксильной группы одного реагента с амино-группой другого реагента с образованием связывающей амидной связи. Образование амидной связи может осуществляться посредством взаимодействия исходных материалов в присутствии агента для связывания или посредством преобразования карбоксильной функциональной группы в активную форму, такую как активный сложный эфир, смешанный ангидрид или хлорангидрид или бромангидрид карбоновой кислоты. Общие описания таких реакций связывания и реагентов, используемых в них, можно найти в общих справочниках по пептидной химии, например, M. Bodanszky, "Peptide Chemistry", 2nd rev. ed., Springer-Verlag, Berlin, Germany, (1993).
Примеры реакций связывания с образованием амидной связи включают в себя азидный способ, способ со смешанным ангидридом угольной-карбоновой кислоты (изобутилхлорформиатом), способ с карбодиимидом (дициклогексилкарбодиимидом, диизопропилкарбодиимидом или водорастворимым карбодиимидом, таким как N-этил-N'-[(3-диметиламино)пропил]карбодиимид), способ с активным сложным эфиром (например, п-нитрофенильным, п-хлорфенильным, трихлорфенильным, пентахлорфенильным, пентафторфенильным, имидо N-гидроксисукциновым и подобными сложными эфирами), K-способ с реагентом Вудворда, способ с 1,1-карбонилдиимидазолом (CDI или N,N'-карбонилдиимидазолом), способы с реагентами на основе фосфора или окисления-восстановления. Некоторые из этих способов могут быть улучшены посредством добавления соответствующих катализаторов, например, для способа с карбодиимидом, посредством добавления 1-гидроксибензотриазола, DBU (1,8-диазабицикло-[5.4.0]ундек-7-ена) или 4-DMAP. Другие агенты для связывания представляют собой (бензотриазол-1-илокси)трис-(диметиламино)фосфоний гексафторфосфат, либо сам по себе, либо в присутствии 1-гидроксибензотриазола, или 4-DMAP; или 2-(1H-бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат или O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат. Эти реакции связывания могут осуществляться либо в растворе (жидкая фаза), либо в твердой фазе.
Предпочтительное образование амидной связи осуществляют с использованием N-этилоксикарбонил-2-этилокси-l,2-дигидрохинолина (EEDQ) или N-изобутилокси-карбонил-2-изобутилокси-1,2-дигидрохинолина (IIDQ). В отличие от классической процедуры с ангидридом, EEDQ и IIDQ не требуют присутствия основания или низких температур реакции. Как правило, процедура включает в себя взаимодействие эквимолярных количеств карбоксильного и аминового компонентов в органическом растворителе (могут использоваться самые разнообразные растворители). Затем добавляют избыток EEDQ или IIDQ и смеси позволяют перемешиваться при комнатной температуре.
Реакции связывания предпочтительно осуществляют в инертном растворителе, таком как галогенированные углеводороды, например, дихлорметан, хлороформ, диполярные апротонные растворители, такие как ацетонитрил, диметилформамид, диметилацетамид, ДМСО, HMPT, простые эфиры, такие как тетрагидрофуран (ТГФ).
Во многих случаях реакции связывания осуществляют в присутствии соответствующего основания, такого как третичный амин, например, триэтиламин, диизопропилэтиламин (DIPEA), N-метилморфолин, N-метилпирролидин, 4-DMAP или l,8-диазабицикло[5.4.0]ундек-7-ен (DBU). Температура реакции может находиться в пределах между 0°C и 50°C, и время реакции может находиться в пределах между 15 мин и 24 час.
Функциональные группы в структурных элементах, которые связываются вместе, могут защищаться для предотвращения образования нежелательных связей. Соответствующие защитные группы, которые могут использоваться, перечисляются, например, в Greene, "Protective Groups in Organic Chemistry", John Wiley & Sons, New York (1999), и в "The Peptides: Analysis, Synthesis, Biology", Vol. 3, Academic Press, New York (1987).
Карбоксильные группы могут защищаться как сложный эфир, который может расщепляться с получением карбоновой кислоты. Защитные группы, которые могут использоваться, включают в себя 1) сложные алкиловые эфиры, такие как метиловый, триметилсилиловый и трет-бутиловый; 2) сложные арилалкиловые эфиры, такие как бензиловый и замещенный бензиловый; или 3) сложные эфиры, которые могут расщепляться с помощью слабого основания или слабых восстановительных средств, такие как сложные трихлорэтиловый и фенациловый эфиры.
Аминогруппы могут защищаться с помощью разнообразных N-защитных групп, таких как:
1) ацильные группы, такие как формил, трифторацетил, фталил и п-толуолсульфонил;
2) ароматические карбаматные группы, такие как бензилоксикарбонил (Cbz или Z) и замещенные бензилоксикарбонилы, и 9-флуоренилметилоксикарбонил (Fmoc);
3) алифатические карбаматные группы, такие как трет-бутилоксикарбонил (Boc), этоксикарбонил, диизопропилметоксикарбонил и аллилоксикарбонил;
4) циклические алкильные карбаматные группы, такие как циклопентилоксикарбонил и адамантилоксикарбонил;
5) алкильные группы, такие как трифенилметил, бензил или замещенный бензил, такой как 4-метоксибензил;
6) триалкилсилил, такой как триметилсилил или t-Bu диметилсилил; и
7) тиол-содержащие группы, такие как фенилтиокарбонил и дитиасукциноил.
Интересные амино-защитные группы представляют собой Boc и Fmoc.
Предпочтительно амино-защитная группа отщепляется перед следующей стадией связывания. Удаление N-защитных групп может осуществляться после процедур, известных в данной области. Когда используют группу Boc, выбранные способы представляют собой трифторуксусную кислоту, чистую или в дихлорметане, или HCl в диоксане или в этилацетате. Полученную соль аммония затем нейтрализуют либо перед связыванием, либо in situ с помощью основных растворов, таких как водные буферы или третичные амины в дихлорметане или в ацетонитриле или диметилформамиде. Когда используют группу Fmoc, выбранные реагенты представляют собой пиперидин или замещенный пиперидин в диметилформамиде, но можно использовать любой вторичный амин. Снятие защиты осуществляют при температуре в пределах между 0°C и комнатной температурой, обычно, примерно при 15-25°C или 20-22°C.
Другие функциональные группы, которые могут вмешиваться в реакции связывания структурных элементов, также могут защищаться. Например, гидроксильные группы могут защищаться как бензиловые или замещенные простые бензиловые эфиры, например, простой 4-метоксибензиловый эфир, сложные бензоиловые или замещенные бензоиловые эфиры, например, сложный 4-нитробензоиловый эфир, или с помощью триалкилсилильных групп (например, триметилсилила или трет-бутилдиметилсилила).
Другие аминогруппы могут защищаться с помощью защитных групп, которые могут отщепляться селективно. Например, когда используют Boc как α-амино-защитную группу, следующие группы для защиты боковых цепей являются пригодными для использования: п-толуолсульфонильные (тозильные) остатки могут использоваться для защиты других аминогрупп; простые бензиловые (Bn) эфиры могут использоваться для защиты гидроксигрупп; и сложные бензиловые эфиры могут использоваться для защиты других карбоксильных групп. Или когда Fmoc выбирают для защиты α-амино, как правило, приемлемыми являются защитные группы на основе трет-бутила. Например, Boc может использоваться для других аминогрупп; простые трет-бутиловые эфиры для гидроксильных групп и сложные трет-бутиловые эфиры для других карбоксильных групп.
Любая защитная группа может удаляться на любой ступени процедур синтеза, но предпочтительно, защитные группы любых функциональных групп, не вовлеченные в стадии реакции, удаляют после завершения создания макроцикла. Удаление защитных групп может осуществляться любым способом, который диктуется выбором защитных групп, параметры которых хорошо известны специалистам в данной области.
Промежуточные соединения формулы (1a), где X представляет собой N, указанные промежуточные соединения представлены формулой (1a-1), могут быть получены, начиная с промежуточных соединений (5a), которые взаимодействуют с алкенамином (5b) в присутствии агента для введения карбонила, как показано на следующей схеме реакции.
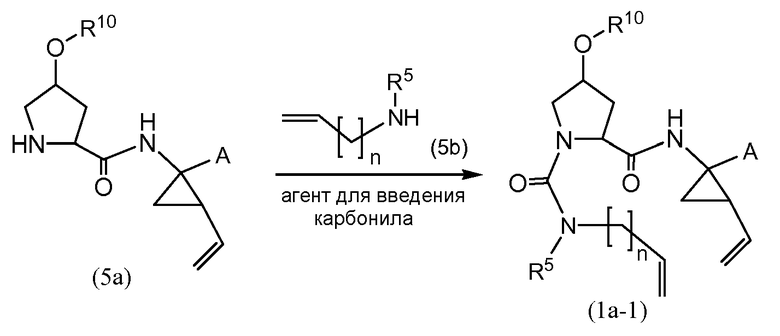
Агенты для введения карбонила (CO) включают в себя фосген, или производные фосгена, такие как карбонилдиимидазол (CDI), и тому подобное. В одном из вариантов осуществления (5a) взаимодействует с агентом для введения карбонила CO в присутствии соответствующего основания и растворителя, который может представлять собой основания и растворители, используемые в реакциях образования амида, как описано выше. В конкретном варианте осуществления, основание представляет собой бикарбонат, например, NaHCO3, или третичный амин, такой как триэтиламин и тому подобное, а растворитель представляет собой простой эфир или галогенированный углеводород, например, ТГФ, CH2Cl2, CHCl3, и тому подобное. После этого добавляют амин (5b), получая при этом промежуточные соединения (1a-1), как на указанной выше схеме. Альтернативный путь с использованием сходных условий реакции включает в себя сначала взаимодействие агента для введения CO с алкенамином (5b), а затем взаимодействие полученного таким образом промежуточного соединения с (5 a).
Промежуточные соединения (1a-1) могут альтернативно быть получены следующим образом:
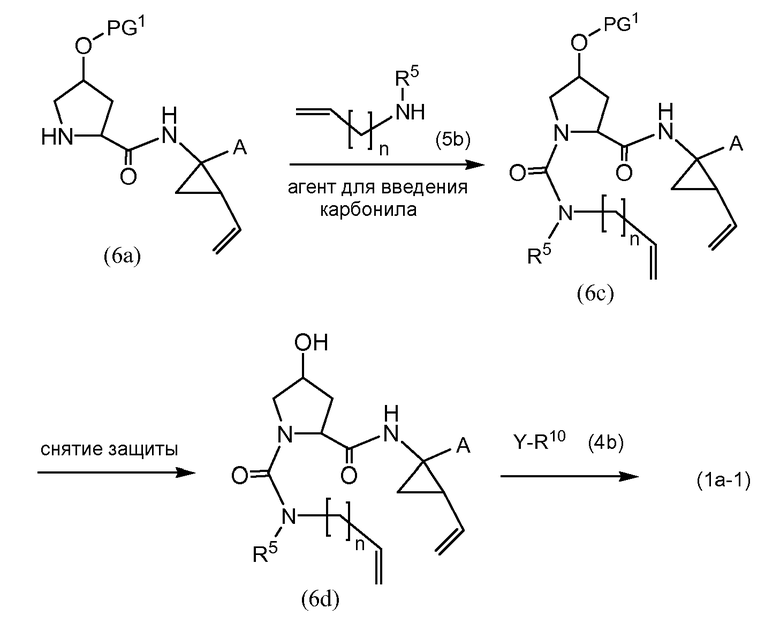
PG1 представляет собой O-защитную группу, которая может представлять собой любую из групп, рассмотренных в описании, и в частности, представляет собой бензоильную или замещенную бензоильную группу, такую как 4-нитробензоил. В последнем случае эта группа может удаляться посредством реакции с гидроксидом щелочного металла (LiOH, NaOH, KOH), в частности, когда PG1 представляет собой 4-нитробензоил, с LiOH, в водной среде, содержащей воду и водорастворимый органический растворитель, такой как алканол (метанол, этанол), и ТГФ.
Промежуточные соединения (6a) взаимодействуют с (5b) в присутствии агента для введения карбонила, подобно тому, как описано выше, и эта реакция дает промежуточные соединения (6c). С них снимают защиту, в частности с использованием условий реакции, рассмотренных выше. Полученный спирт (6d) взаимодействует с промежуточными соединениями (4b), как описано выше для взаимодействия (4a) с (4b), и эта реакция дает промежуточные соединения (1a-1).
Промежуточные соединения формулы (1a), где X представляет собой C, указанные промежуточные соединения представлены формулой (1a-2), могут быть получены посредством реакции образования амида, начиная с промежуточных соединений (7a), которые взаимодействуют с амином (5b), как показано на следующей далее схеме реакции, с использованием условий реакции для получения амидов, таких как те, которые описаны выше.
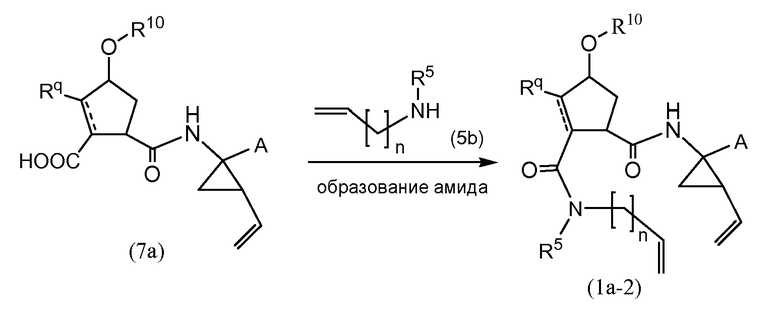
Промежуточные соединения (1a-2) могут альтернативно быть получены следующим образом:
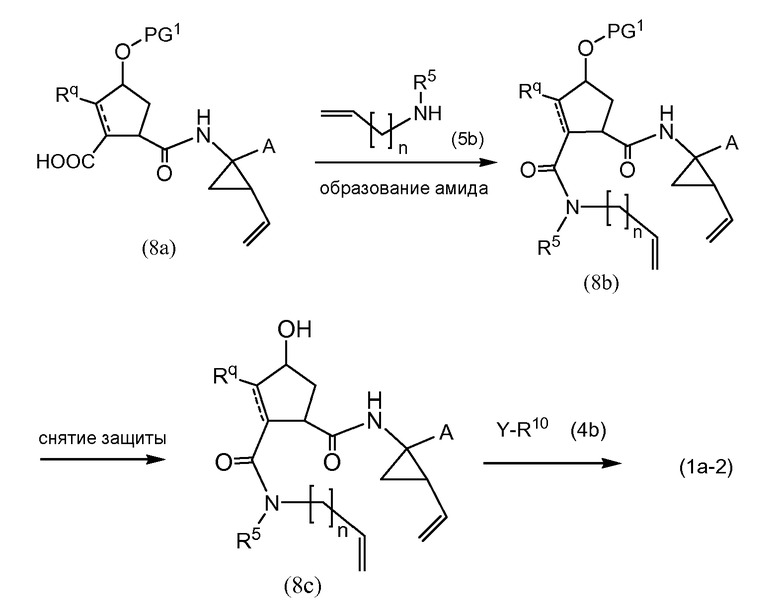
PG1 представляет собой O-защитную группу, как описано выше. Могут использоваться такие же условия реакции, как описано выше: образование амида, как описано выше, удаление PG1, как в описании защитных групп, и введение R10, как в реакциях (4a) с реагентами (4b).
Промежуточные соединения формулы (2a) могут быть получены посредством циклизации сначала открытого амида (9a) до сложного макроциклического эфира (9b), который, в свою очередь, преобразуется в (2a) следующим образом:
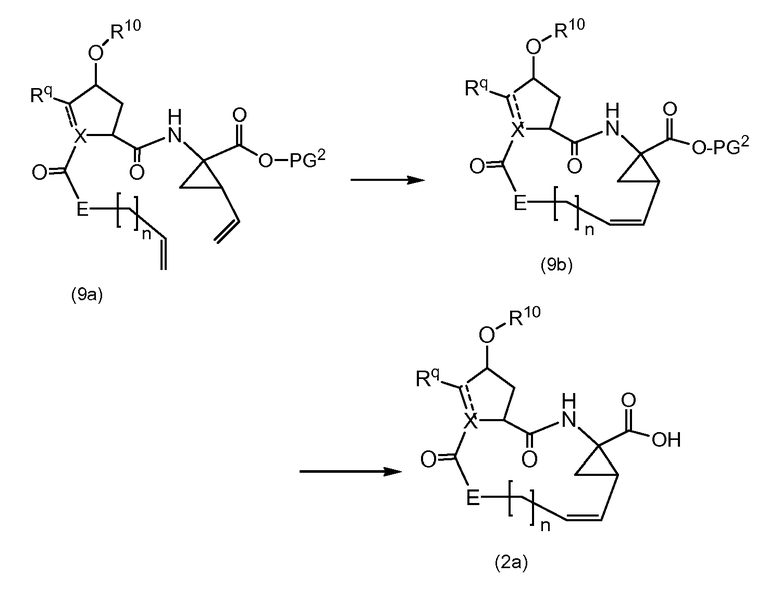
PG2 представляет собой карбоксил-защитную группу, например, одну из карбоксил-защитных групп, рассмотренных выше, в частности, сложный C1-4алкиловый или бензиловый эфир, например, сложный метиловый, этиловый или трет-бутиловый эфир. Реакция от (9a) до (9b) представляет собой реакцию метатезиса и осуществляется, как описано выше. Группу PG2 удаляют, следуя процедурам, также описанным выше. Когда PG2 представляет собой сложный C1-4алкиловый эфир, ее удаляют с помощью щелочного гидролиза, например, с помощью NaOH или, предпочтительно, LiOH, в водном растворителе, например, в смеси C1-4алканол/вода. Бензильная группа может удаляться с помощью каталитического гидрирования.
В альтернативном синтезе, промежуточные соединения (2a) могут быть получены следующим образом:
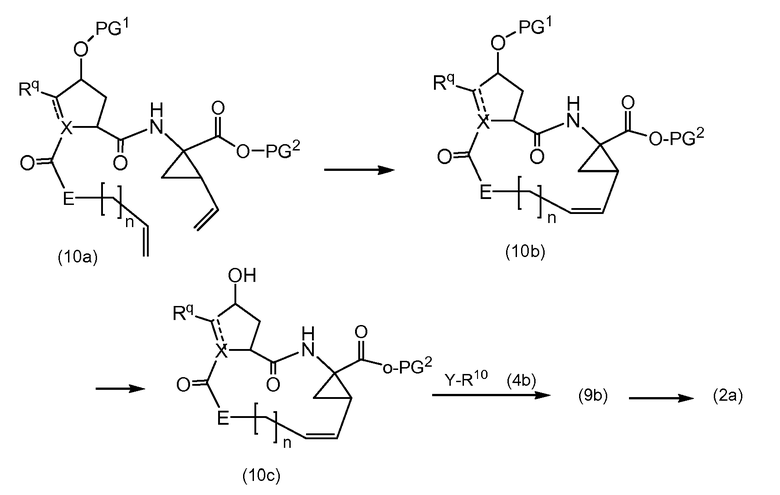
Группу PG1 выбирают так, что она может селективно расщепляться с получением PG2. PG2 может представлять собой, например, сложные метиловые или этиловые эфиры, которые могут удаляться с помощью обработки гидроксидом щелочного металла в водной среде, в этом случае PG1 представляет собой, например, трет-бутил или бензил. PG2 может представлять собой сложные трет-бутиловые эфиры, которые могут удаляться при слабо кислотных условиях, или PG1 может представлять собой простые бензиловые эфиры, которые могут удаляться с помощью сильной кислоты или с помощью каталитического гидрирования, в последних двух случаях PG1 представляет собой, например, сложный эфир бензойной кислоты, такой как сложный эфир 4-нитробензойной кислоты.
Сначала, промежуточные соединения (10a) циклизируются до сложных макроциклических эфиров (10b), с последних снимают защиту посредством удаления группы PG1 до (10c), которая взаимодействует с промежуточными соединениями (4b), с последующим удалением карбоксил-защитной группы PG2. Циклизация, снятие защиты PG1 и PG2 и связывание с (4b) являются такими, как описано выше.
Группы A могут вводиться на любой ступени синтеза, либо как последняя стадия, как описано выше, либо раньше, перед образованием макроцикла. На следующей далее схеме, вводятся группы A, представляющие собой -CO-NH-SO2R2 или -CO-OR5 (которые являются такими, как определено выше):
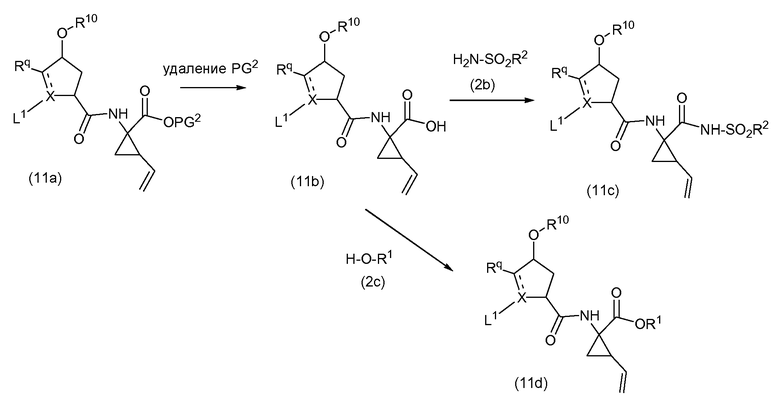
На приведенной выше схеме PG2 является таким, как определено выше, и L1 представляет собой группу P3

где n и R5 являются такими, как определено выше и, где X представляет собой N, L1 может также представлять собой азот-защитную группу (PG, как определено выше) и, где X представляет собой C, L1 может также представлять собой группу -COOPG2a, где группа PG2a представляет собой карбоксил-защитную группу, сходную с PG2, но где PG2a может селективно расщепляться с получением PG2. В одном из вариантов осуществления PG2a представляет собой трет-бутил, и PG2 представляет собой метил или этил.
Промежуточные соединения (11c) и (11d), где L1 представляет собой группу (b), соответствуют промежуточным соединениям (1a) и могут обрабатываться в дальнейшем, как определено выше.
Связывание структурных элементов P1 и P2
Структурные элементы P1 и P2 связываются с использованием реакции образования амида, следуя процедурам, описанным выше. Структурный элемент P1 может иметь карбоксил-защитную группу PG2 (как в (12b)) или может уже быть связанным с группой P1' (как в (12c)). L2 представляет собой N-защитную группу (PG) или группу (b), как определено выше. L3 представляет собой гидрокси, -OPG1 или группу -O-R10, как определено выше. Когда на любой из следующих схем реакций L3 представляет собой гидрокси, перед каждой стадией реакции, она может защищаться как группа -OPG1 и, если это желательно, затем может освобождаться от защиты с получением свободной гидрокси функциональной группы. Подобным же образом, как описано выше, гидрокси функциональная группа может преобразовываться в группу -O-R10.
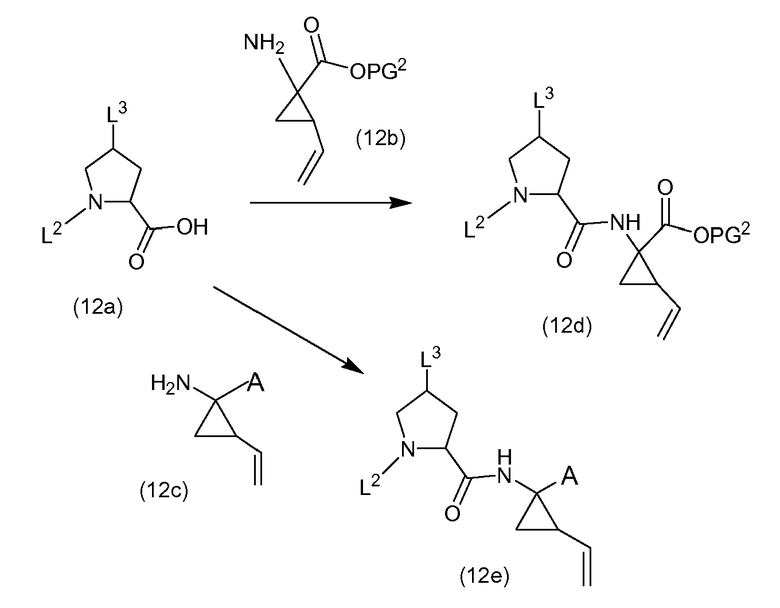
В процедуре на указанной выше схеме, циклопропиламинокислота (12b) или (12c) связывается с кислотной функциональной группой структурного элемента P2 (12a) с образованием амидной связи, следуя процедурам, описанным выше. Получают промежуточные соединения (12d) или (12e). Когда в последнем L2 представляет собой группу (b), полученные в результате продукты представляют собой последовательности P3-P2-P1, охватывающие некоторые промежуточные соединения (11c) или (11d) в предыдущей схеме реакции. Удаление кислотной защитной группы в (12d), с использованием соответствующих условий для используемой защитной группы, с последующим связыванием с амином H2N-SO2R2 (2b), с HOR1 (2c) или фосфорамидатом (4d), как описано выше, опять дает промежуточные соединения (12e), где -A представляют собой амидные или сложноэфирные группы. Когда L2 представляет собой N-защитную группу, она может удаляться с получением промежуточных соединений (5a) или (6a). В одном из вариантов осуществления, PG в этой реакции представляет собой группу BOC и PG2 представляет собой метил или этил. Когда дополнительная L3 представляет собой гидрокси, исходный материал (12a) представляет собой Boc-L-гидроксипролин. В конкретном варианте осуществления, PG представляет собой BOC, PG2 представляет собой метил или этил и L3 представляет собой -O-R10.
В одном из вариантов осуществления, L2 представляет собой группу (b) и эти реакции включают в себя связывание P1 с P2-P3, которое приводит к получению промежуточных соединений (1a-1) или (1a), рассмотренных выше. В другом варианте осуществления L2 представляет собой N-защитную группу PG, которая является такой, как определено выше, и реакция связывания приводит к получению промежуточных соединений (12d-l) или (12e-1), с которых может удаляться группа PG, с использованием условий реакций, рассмотренных выше, с получением промежуточных соединений (12-f) или, соответственно, (12g), которые охватывают промежуточные соединения (5a) и (6a), как определено выше:
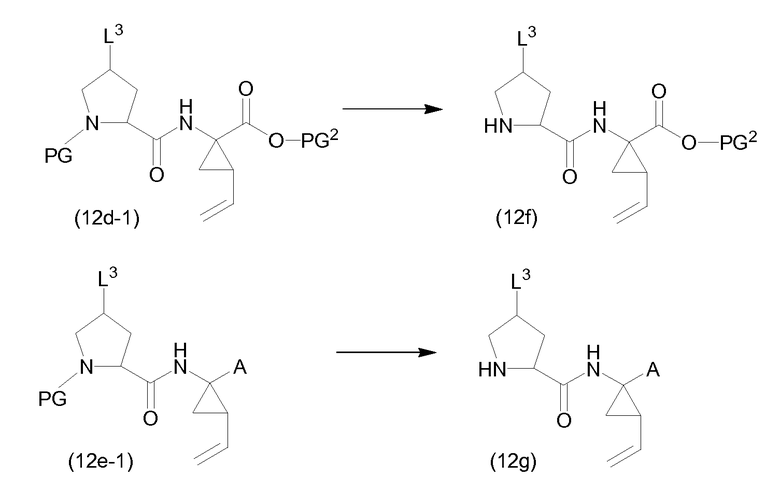
В одном из вариантов осуществления, группа L3 в указанных выше схемах представляет собой группу -O-PG1, которая может вводиться на исходном материале (12a), где L3 представляет собой гидрокси. В этом случае PG1 выбирают так, что она может селективно расщепляться с получением группы L2, представляющей собой PG.
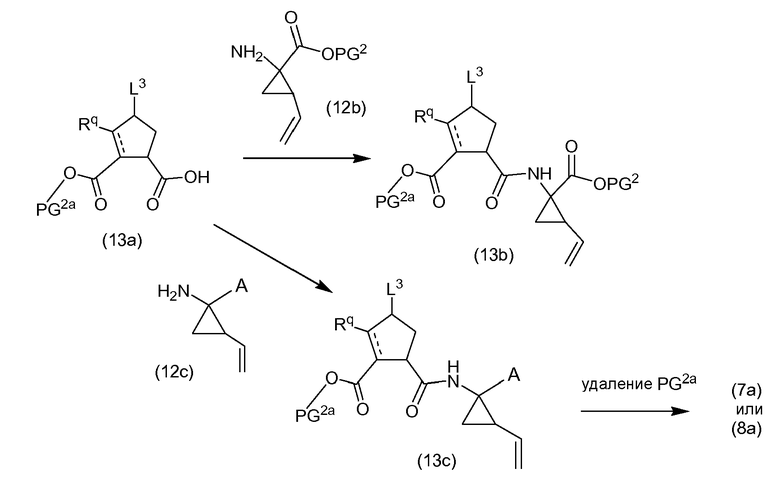
Подобным же образом, структурные элементы P2, где X представляет собой C, которые представляют собой производные циклопентана или циклопентена, могут связываться со структурными элементами P1, как показано на следующей далее схеме, где A, Rq, L3 являются такими, как определено выше, и PG2 и PG2a представляют собой карбоксил-защитные группы. Как правило, PG2a выбирают так, что она может селективно расщепляться с получением группы PG2. Удаление группы PG2 в (13c) дает промежуточные соединения (7a) или (8a), которые могут взаимодействовать с (5b), как описано выше.
В конкретном варианте осуществления, где X представляет собой C, Rq представляет собой H, и когда X и несущая атом углерода Rq связываются одинарной связью (P2 представляет собой циклопентановый остаток), PG2a и L3, взятые вместе, образуют связь, и структурный элемент P2 представлен формулой:
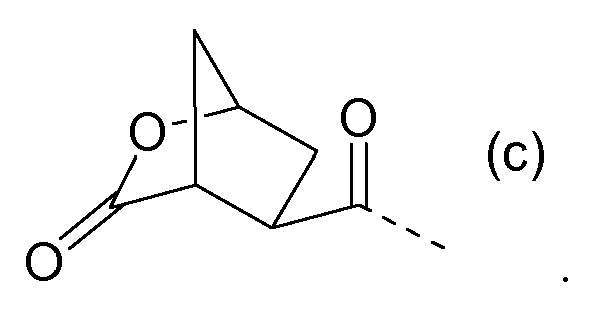
Бициклическая кислота (14a) взаимодействует с (12b) или (12c), подобно тому, как описано выше, до получения (14b) и (14c), соответственно, где лактон открывается с получением промежуточных соединений (14c) и (14e). Лактоны могут открываться с использованием процедур сложноэфирного гидролиза, например, с использованием условий реакций, описанных выше для щелочного удаления группы PG1 в (9b), в частности, с использованием основных условий, таких как гидроксид щелочного металла, например, NaOH, KOH, в частности, LiOH.
Промежуточные соединения (14c) и (14e) могут дополнительно обрабатываться, как описано далее.
Связывание структурных элементов P3 и P2
Для структурных элементов P2, которые имеют пирролидиновый остаток, структурные элементы P3 и P2 или P3 и P2-P1 связываются с использованием реакции образования карбамата, следуя процедурам, описанным выше для связывания (5a) с (5b). Общая процедура для связывания структурных элементов P2, имеющих пирролидиновый остаток, представлена на следующей схеме реакции, где L3 является таким, как определено выше, и L4 представляет собой группу -O-PG2, группу

В одном из вариантов осуществления L4 в (15a) представляет собой группу -OPG2, группа PG2 может удаляться и полученная кислота связывается с циклопропиламинокислотами (12a) или (12b) с получением промежуточных соединений (12d) или (12e), где L2 представляет собой радикал (d) или (e).
Общая процедура связывания структурных элементов P3 со структурным элементом P2 или со структурным элементом P2-P1, где P2 представляет собой циклопентан или циклопентен, показана на следующей далее схеме. L3 и L4 являются такими, как определено выше.
В конкретном варианте осуществления L3 и L4, взятые вместе, могут образовывать лактоновый мостик, как в (14a), и связывать структурный элемент P3 со структурным элементом P2 следующим образом:
Бициклический лактон (14a) взаимодействует с (5b) в реакции образования амида с получением амида (16c), в котором лактоновый мостик раскрывается до (16d). Условия реакции для реакций образования амида и открывания лактона являются такими, как описано выше или далее. Промежуточное соединение (16d), в свою очередь, может связываться с группой P1, как описано выше.
Реакции на указанных выше схемах осуществляют с использованием таких же процедур, как описано выше для реакций (5a), (7a) или (8a) с (5b) и, в частности, указанные выше реакции, где L4 представляет собой группу (d) или (e), соответствуют реакциям (5a), (7a) или (8a) с (5b), как описано выше.
Структурные элементы P1, P1', P2 и P3, используемые для получения соединений формулы (I), могут быть получены, начиная с промежуточных соединений, известных в данной области. Ряд таких путей синтеза описываются далее более подробно.
Индивидуальные структурные элементы могут сначала получаться, а затем связываться вместе, или альтернативно, предшественники структурных элементов могут связываться вместе и модифицироваться на более поздней ступени до желаемой молекулярной композиции.
Функциональные группы каждого из структурных элементов могут защищаться для предотвращения побочных реакций.
Синтез структурных элементов P2
Структурные элементы P2 содержат либо пирролидиновый, циклопентановый, либо циклопентеновый остаток, замещенный группой -O-R10.
Структурные элементы P2, содержащие пирролидиновый остаток, могут быть получены из коммерчески доступного гидроксипролина.
Получение структурных элементов P2, которые содержат циклопентановое кольцо, может осуществляться, как показано на схеме ниже.
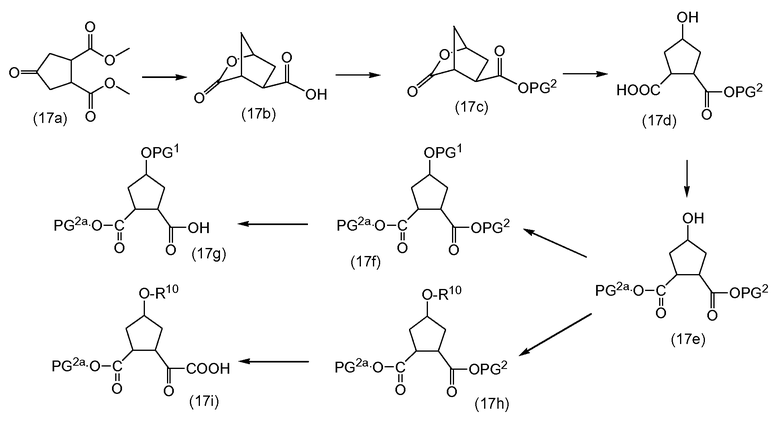
Бициклическая кислота (17b) может быть получена, например, из 3,4-бис(метоксикарбонил)циклопентанона (17a), как описано в Rosenquist et al., Acta Chem. Scand. 46 (1992) 1127-1129. Первая стадия этой процедуры включает в себя восстановление кето-группы с помощью восстанавливающего агента, подобного боргидриду натрия, в растворителе, таком как метанол, с последующим гидролизом сложных эфиров и конечного замыкания кольца до бициклического лактона (17b) с использованием процедур образования лактона, в частности, посредством использования уксусного ангидрида в присутствии слабого основания, такого как пиридин. Функциональная группа карбоновой кислоты в (17b) может затем защищаться с помощью введения соответствующей карбоксил-защитной группы, такой как группа PG2, которая является такой, как определено выше, с получением, таким образом, сложного бициклического эфира (17c). Группа PG2, в частности, представляет собой кислотно-лабильную, например, трет-бутильную группу и вводится, например, с помощью обработки изобутеном в присутствии кислоты Льюиса или с помощью ди-трет-бутилдикарбоната в присутствии основания, такого как третичный амин, подобный диметиламинопиридину или триэтиламину, в растворителе, подобном дихлорметану. Открывание лактона (17c) с использованием условий реакции, описанных выше, в частности, с гидроксидом лития, дает кислоту (17d), которая может использоваться далее в реакциях связывания со структурными элементами P1. Свободная кислота в (17d) может также защищаться, предпочтительно, с помощью кислотной защитной группы PG2a, которая может селективно расщепляться с получением PG2, и гидроксифункциональная группа может преобразовываться в группу -OPG1 или в группу -O-R10. Продукты, полученные при удалении группы PG2, представляют собой промежуточные соединения (17g) и (17i), которые соответствуют промежуточным соединениям (13a) или (16a), определенным выше.
Промежуточные соединения с конкретной стереохимией могут быть получены посредством разрешения промежуточных соединений в указанной выше последовательности реакций. Например, (17b) может быть разрешено, следуя процедурам, известным в данной области, например, под воздействием на солевые формы с помощью оптически активного основания или с помощью хиральной хроматографии, и полученные стереоизомеры могут дополнительно обрабатываться, как описано выше. Группы OH и COOH в (17d) находятся в цис-положении. Транс аналоги могут быть получены посредством инверсии стереохимии на атоме углерода, несущем функциональную группу OH, посредством использования конкретных реагентов в реакциях, для введения OPG1 или O-R10, которые инвертируют стереохимию, например, посредством применения реакции Мицунобу.
В одном из вариантов осуществления, промежуточные соединения (17d) связываются со структурными элементами P1 (12b) или (12c) с помощью реакций связывания, соответствующих связыванию (13a) или (16a), с такими же структурными элементами P1, с использованием таких же условий. Последующее введение заместителя -O-R10-, как описано выше, с последующим удалением кислотной защитной группы PG2 дает промежуточные соединения (8a-1), которые представляют собой подкласс промежуточных соединений (7a) или часть промежуточных соединений (16a). Продукты реакции удаления PG2 могут дополнительно связываться со структурным элементом P3. В одном из вариантов осуществления PG2 в (17d) представляет собой трет-бутил, который может удаляться при кислотных условиях, например, с помощью трифторуксусной кислоты.
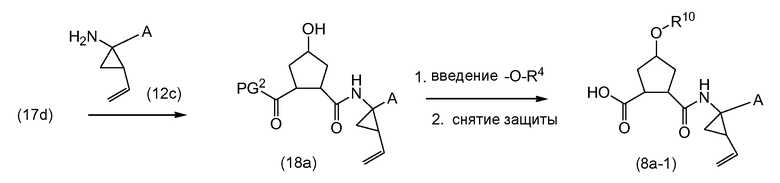
Ненасыщенный структурный элемент P2, то есть циклопентеновое кольцо, может быть получен, как иллюстрируется на схеме ниже.
Реакция бромирования-элиминирования 3,4-бис(метоксикарбонил)циклопентанона (17a), как описано в Dolby et al., J.Org. Chem. 36 (1971) 1277-1285, с последующим восстановлением функциональной кето-группы с помощью восстанавливающего агента, подобного боргидриду натрия, дает циклопентенол (19a). Селективный сложноэфирный гидролиз с использованием, например, гидроксида лития в растворителе, подобном смеси диоксана и воды, дает гидрокси-замещенный сложный циклопентеноловый моноэфир (19b).
Ненасыщенный структурный элемент P2, где Rq может также быть иным, чем водород, может быть получен, как показано на схеме ниже.
Окисление коммерчески доступного 3-метил-3-бутен-1-ола (20a), в частности, с помощью окислительного агента, подобного пиридиний хлорхромату, дает (20b), которое преобразуется в соответствующий сложный метиловый эфир, например, посредством обработки ацетилхлоридом в метаноле, с последующей реакцией бромирования с бромом, с получением сложного α-бромо эфира (20c). Последний может затем конденсироваться вместе со сложным алкениловым эфиром (20e), полученным из (20d), с помощью реакции образования сложного эфира. Сложный эфир в (20e) предпочтительно представляет собой сложный трет-бутиловый эфир, который может быть получен из соответствующей коммерчески доступной кислоты (20d), например, посредством обработки ди-трет-бутилдикарбонатом в присутствии основания, подобного диметиламинопиридину. Промежуточное соединение (20e) обрабатывают основанием, таким как литий диизопропиламид, в растворителе, подобном тетрагидрофурану, и оно взаимодействует с (20c) с получением сложного алкенилового диэфира (20f). Циклизация (20f) с помощью реакции метатезиса олефина, осуществляемая, как описано выше, дает производное циклопентена (20g). Стереоселективное эпоксидирование (20g) может осуществляться с использованием способа асимметричного эпоксидирования Якобсена с получением эпоксида (20h). Наконец, реакция открывания эпоксида при основных условиях, например, посредством добавления основания, в частности, DBN (1,5-диазабицикло[4.3.0]нон-5-ена), дает спирт (20i). Необязательно, двойная связь в промежуточном соединении (20i) может восстанавливаться, например, с помощью каталитического гидрирования с использованием катализатора, подобного палладию на угле, с получением соответствующего циклопентанового соединения. Сложный трет-бутиловый эфир может удаляться с получением соответствующей кислоты, которая впоследствии связывается со структурным элементом P1.
Группа -O-R10 может вводиться на пирролидиновом, циклопентановом или циклопентеновом кольцах на любой удобной ступени синтеза соединений в соответствии с настоящим изобретением. Один из подходов заключается в том, чтобы сначала ввести группу -O-R10 в указанные кольца, а впоследствии добавить другие желаемые структурные элементы, то есть P1 (необязательно с хвостом P1') и P3, с последующим образованием макроцикла. Другой подход заключается в связывании структурных элементов P2, не несущих заместителях -O-R10, с каждым из P1 и P3, и в добавлении группы -O-R10 либо до, либо после образования макроцикла. В последней процедуре, остатки P2 имеют гидрокси группу, которая может защищаться с помощью гидрокси-защитной группы PG1.
Группы R10 могут вводиться на структурных элементах P2 посредством взаимодействия гидрокси-замещенных промежуточных соединений (21a) или (21b) с промежуточными соединениями (4b), подобно тому, как описано выше, для синтеза (I), начиная с (4a). Эти реакции представлены на схемах, ниже, где L2 является таким, как определено выше, и L5 и L5a независимо друг от друга представляют собой гидрокси, карбоксил-защитную группу -OPG2 или -OPG2a, или L5 может также представлять собой группу P1, такую как группа (d) или (e), как определено выше, или L5a может также представлять собой группу P3, такую как группа (b), как определено выше. Группы PG2 и PG2a являются такими, как определено выше. Когда группы L5 и L5a представляют собой PG2 или PG2a, они выбираются так, что каждая группа может селективно расщепляться с получением другой группы. Например, одна из L5 и L5a может представлять собой метильную или этильную группу, а другая - бензильную или трет-бутильную группу.
В одном из вариантов осуществления в (21a), L2 представляет собой PG и L5 представляет собой -OPG2, или в (21d), L5a представляет собой -OPG2 и L5 представляет собой -OPG2, и группы PG2 удаляют, как описано выше.
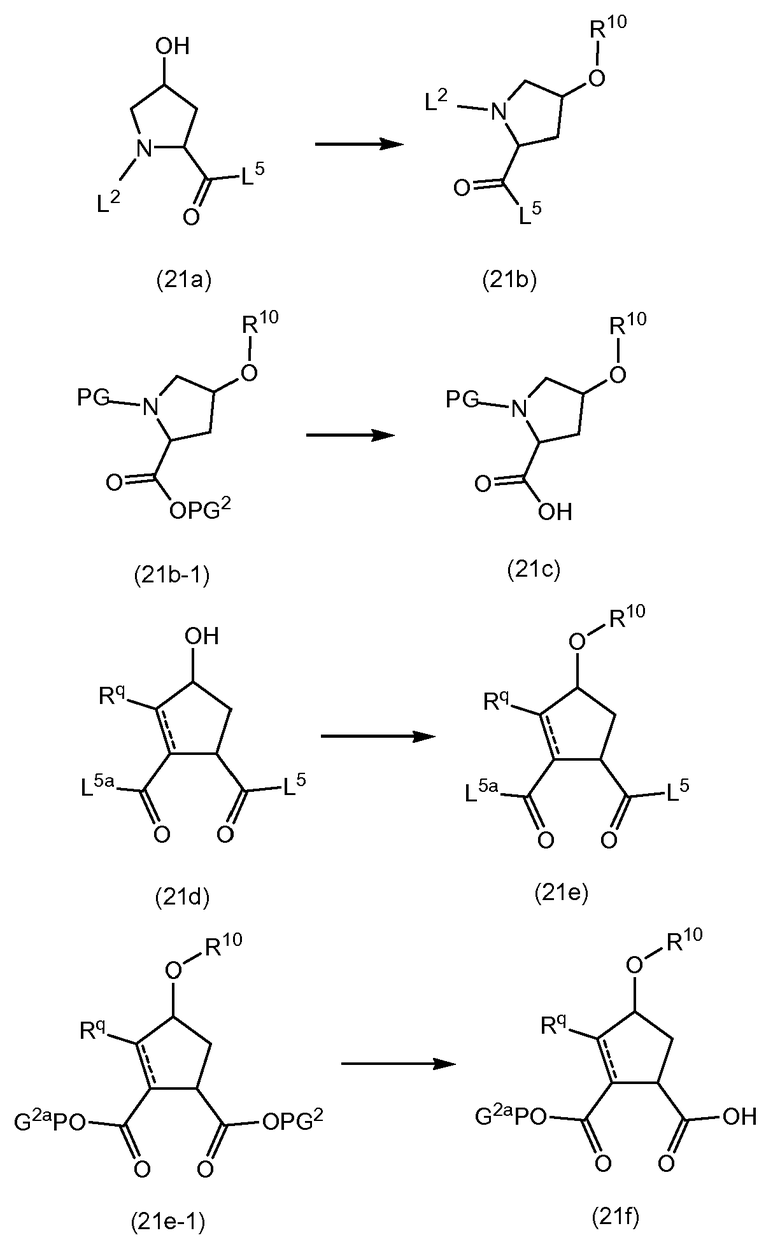
В другом варианте осуществления группа L2 представляет собой BOC, L5 представляет собой гидрокси и исходный материал (21a) представляет собой коммерчески доступный BQC-гидроксипролин, или любую другую его стереоизомерную форму, например, BOC-L-гидроксипролин, в частности, транс-изомер последнего. Когда L5 в (21b) представляет собой карбоксил-защитную группу, она может удаляться, следуя процедурам, описанным выше, до получения (21c). Еще в одном варианте осуществления PG в (21b-1) представляет собой Boc и PG2 представляет собой сложный низший алкиловый эфир, в частности, сложный метиловый или этиловый эфир. Гидролиз последнего сложного эфира до кислоты может осуществляться с помощью стандартных процедур, например, кислотного гидролиза с помощью хлористоводородной кислоты в метаноле или с помощью гидроксида щелочного металла, такого как NaOH, в частности, с помощью LiOH. В другом варианте осуществления, гидрокси-замещенные аналоги циклопентана или циклопентена (21d) преобразуются в (21e), которое, когда L5 и L5a представляют собой -OPG2 или -OPG2a, могут преобразовываться в соответствующие кислоты (21f) посредством удаления группы PG2. Удаление PG2a в (21e-1) приводит к получению подобных промежуточных соединений.
Синтез структурных элементов P1
Циклопропановая аминокислота, используемая при получении фрагмента P1, является коммерчески доступной или может быть получена с использованием процедур, известных в данной области.
В частности, сложный аминовинилциклопропилэтиловый эфир (12b) может быть получен в соответствии с процедурой, описанной в заявке на Международный патент WO 00/09543, или как иллюстрируется на следующей далее схеме, где PG2 представляет собой карбоксил-защитную группу, как определено выше:
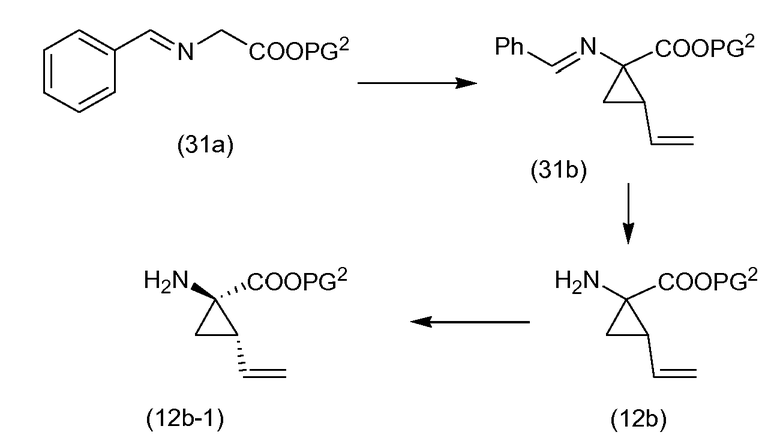
Обработка коммерчески доступного или легко получаемого имина (31a) 1,4-дигалоген-бутеном в присутствии основания дает (31b), которое после гидролиза дает циклопропиловую аминокислоту (12b), имеющую аллильный заместитель syn по отношению к карбоксильной группе. Разрешение энантиомерной смеси (12b) приводит к получению (12b-1). Разрешение осуществляют с использованием процедур, известных в данной области, таких как ферментативное разделение; кристаллизация с хиральной кислотой или химическая дериватизация; или с помощью хроматографии на хиральной колонке. Промежуточные соединения (12b) или (12b-1) могут связываться с соответствующими производными P2, как описано выше.
Структурные элементы P1 для получения соединений в соответствии с общей формулой (I), где A представляет собой -COOR1, -CO-NH-SO2R2 или -CO-NH-PO(OR4a)(OR4b), могут быть получены посредством взаимодействия аминокислот (32a) с соответствующим спиртом или амином, соответственно, при условиях, стандартных для образования сложного эфира или амида. Циклопропиловые аминокислоты (32a) получают посредством введения N-защитной группы PG и удаления PG2, и получаемые PG защищенные аминокислоты (32a) преобразуют в амиды (12c-1) или сложные эфиры (12c-2), которые представляют собой подгруппу промежуточных соединений (12c), как показано на следующей далее схеме реакций, где PG является таким, как определено выше.
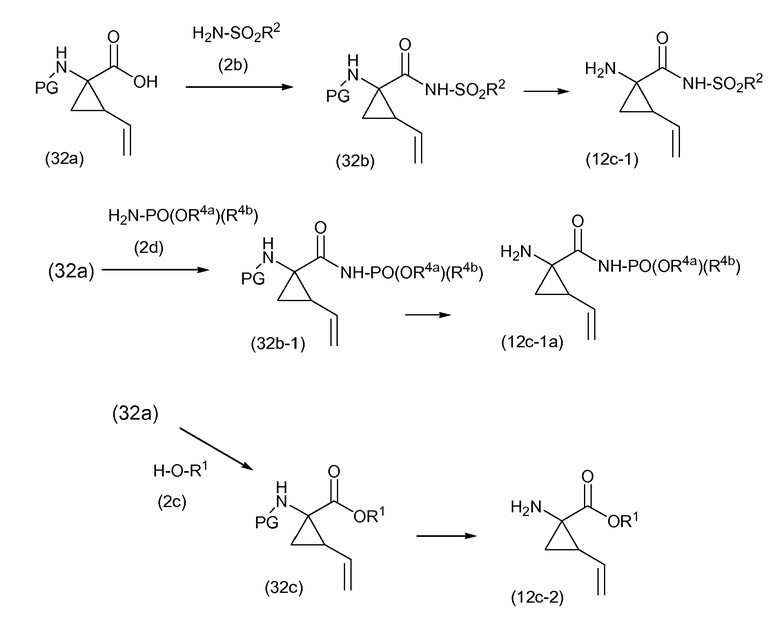
Реакция (32a) с сульфонамидамином (2b) или с фосфорамидатом (2d) представляет собой процедуру образования амида. Реакция, сходная с (2c), представляет собой реакцию образования сложного эфира. Оба типа реакций могут осуществляться, следуя процедурам, описанным выше. Это реакция дает промежуточные соединения (32b), (32b-1) или (32c), из которых амино-защитную группу удаляют с помощью стандартных способов, таких как те, которые описаны выше. Это, в свою очередь, приводит к получению желаемого промежуточного соединения (12c-1), (12c-1a), или (12c-2). Исходные материалы (32a) могут быть получены из рассмотренных выше промежуточных соединений (12b) посредством введения сначала N-защитной группы PG, а затем удаления группы PG2.
В одном из вариантов осуществления взаимодействие (32a) с (2b) или с (2d) осуществляется посредством обработки аминокислоты агентом для связывания, например, N,N'-карбонилдиимидазолом (CDI) или чем-либо подобным, в растворителе, сходным с ТГФ, с последующим взаимодействием с (2b) или с (2d) в присутствии основания, такого как 1,8-диазабицикло[5.4.0]ундек-7-ен (DBU). Альтернативно, аминокислота может обрабатываться (2b) или (2d) в присутствии основания, подобного диизопропилэтиламину, с последующей обработкой агентом для связывания, таким как бензотриазол-1-ил-окси-трис-пирролидино-фосфоний гексафторфосфат (коммерчески доступный как PyBOP®), для осуществления введения сульфонамидной группы.
Структурные элементы P1 для получения соединений в соответствии с общей формулой (I), где A представляет собой -C(=O)C(=O)NR3aR3b, обычно получают как, показано на следующей схеме.
PG2a в исходном материале (32d) представляет собой алкильную группу, в частности, C1-6алкил, такой как метил или этил. (32d) может быть получено посредством реакции образования сложного эфира (32a) с соответствующим алканолом или посредством введения азот-защитной группы PG на (12b), как описано выше. Восстановление сложноэфирной группы в производном аминокислоты (32d) до соответствующего промежуточного соединения гидроксиметилена (32e), осуществляемое, например, посредством обработки боргидридом лития, с последующим оксилением полученной гидроксиметиленовой группы с использованием слабого окислителя, такого, например, как периодинан Десса-Мартина, дает альдегид (32f). Реакция последнего альдегида (32f) с соответствующим производным изонитрила и в присутствии карбоновой кислоты, такой как трифторуксусная кислота (TFA), в присутствии основания, например, пиридина, в реакции Пассерини (как описано, например, в Org. Lett., Vol. 2, No 18, 2000), дает сложный эфир карбоновой кислоты и полученного α-гидрокси амида, например, в случае TFA, трифторацетата. Сложный эфир карбоновой кислоты в полученном таким образом α-гидрокси амиде может затем удаляться с использованием стандартных процедур, например, с использованием основных условий, таких как LiOH, с получением таким образом α-гидрокси амида (32g). Удаление группы PG приводит к получению промежуточных соединений (32h), которые могут связываться с группой P2. Гидрокси функциональная группа в (32g) может окисляться до соответствующего α-кето амида, но для предотвращения побочных реакций α-гидрокси амид или его предшественник сложный эфир карбоновой кислоты используют в дальнейших реакциях (таких как удаление N-защитной группы, связывание с остатком P2, и тому подобное). Окисление α-гидрокси группы остатка P1 затем осуществляют на любой удобной ступени синтеза, например, после его связывания с остатком P2 или на более поздних ступенях синтеза, например, на последней стадии, с использованием слабого окислителя, такого, например, как периодинан Десса-Мартина, с получением таким образом соединения формулы I или промежуточных соединений, где A представляет собой -C(=O)C(=O)NR3aR3b.
Указанная выше процедура, то есть восстановление сложного эфира, окисление до альдегида, реакция с изонитрилом, также может осуществляться на более поздних ступенях процедуры синтеза, например, после получения макроцикла.
Структурные элементы P1, пригодные для получения соединений в соответствии с общей формулой (I), где A представляет собой -C(=O)NH-P(=O)(OR4a)(R4b) или -P(=O)(OR4a)(R4b), фосфонат, могут быть получены, следуя процедурам, описанным в заявке на Международный патент WO 2006/020276. В частности, конкретные соединения формулы (I), где A представляет собой -P(=O)(OR4a)(R4b), могут быть получены следующим образом:
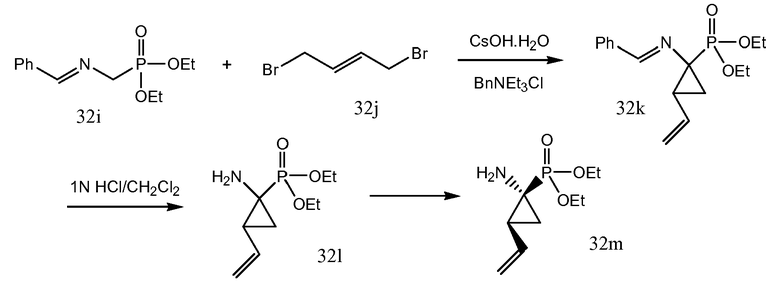
Исходный материал 32i взаимодействует с основанием, в частности, с CsOH, предпочтительно, в присутствии катализатора с переносом фазы, такого как триэтилбензиламмоний хлорид, и добавляют 32j, с образованием циклопропильного кольца с винильной боковой цепью, то есть циклопропилфосфоната 32k. Фенил-CH=-защитную группу удаляют при кислотных условиях (например, HCl в дихлорметане), с получением 32l. Последнее может разрешаться на его стереоизомеры с использованием методики, известной в данной области, например, посредством формирования соли с оптически активной кислотой, например, с дибензоил-L-винной кислотой, которая после удаления производного винной кислоты дает 32m. Аналоги иные, чем этилфосфонаты, могут быть получены из исходных материалов 32i, имеющих сложноэфирные группы, иные, чем этильные. Исходные материалы 32i представляют собой известные материалы или могут легко быть получены с использованием способов, известных в данной области.
Промежуточные соединения (12c-1) или (12c-2) в свою очередь могут связываться с соответствующими пролиновыми, циклопентановыми или циклопентеновыми производными, как описано выше.
Пиримидиновая группа (то есть радикал R10) может вводиться при получении структурных элементов P2, как описано выше, или на последней ступени синтеза, даже как последняя стадия.
Исходные материалы для введения группы R10 (например, R10-OH и аналогов) могут быть получены, как показано в следующих схемах реакций.
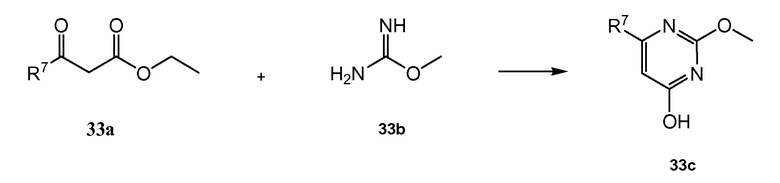
Соответствующим образом замещенный сложный β-кето эфир (33a), несущий группу R7 в β-положении, взаимодействует с 2-метилизомочевиной (33b) в присутствии основания, такого как метоксид натрия, в растворителе, подобном метанолу, с получением дизамещенного пиримидинола (33c).
Альтернативный способ для различных замещенных пиримидинов использует общее промежуточное соединение, которое впоследствии может преобразовываться в пиримидиновые производные с различными структурами замещения. Способ для этого промежуточного соединения (34d) является следующим.
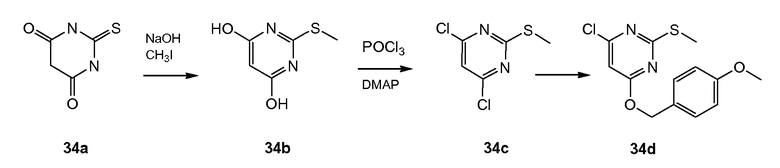
Селективное алкилирование атома серы в тиобарбитуровой кислоте (34a) посредством обработки метилйодидом или любым другим сходным алкилирующим агентом в присутствии основания, такого как гидроксид натрия или что-либо подобное, дает производное простого тиоэфира (34b). Замещение гидрокси-группы хлором с помощью галогенирующего агента, например, посредством обработки трихлоридом фосфора, в присутствии основания, такого как диметиламинопиридин или что-либо подобное, дает (34c), при последующем взаимодействии с соответствующим производным бензилового спирта, например, с п-метоксибензиловым спиртом, в присутствии основания, такого как NaH, дает метоксибензил-защищенный пиримидинол (34d).
В другом аспекте, настоящее изобретение относится к промежуточному соединению (34d), новому соединению, которое служит в качестве удобного промежуточного соединения для получения соединения формулы (I). Промежуточное соединение (34d) обеспечивает большую гибкость для дальнейшего синтеза, поскольку оно может селективно взаимодействовать в различных положениях пиримидинового кольца в любом желаемом порядке, либо на данной ступени, либо на более поздней ступени, например, в конце синтеза. Например, функциональная группа простого тиоэфира может окисляться до соответствующего сульфона, с последующей реакцией нуклеофильного замещения с различными нуклеофилами, такими как реагенты Гриньяра, алкоголяты или амины, с получением пиримидинов, несущих C-, O- или N-связанный заместитель, соответственно, в 2-положении. Альтернативно, заместитель хлор может быть заменен соответствующим алкоголятом, таким как метоксид или что-либо подобное, или могут использоваться условия связыванием Штилле или Сузуки для введения C-связанного заместителя, например, арильной или гетероарильной группы.
Следующая далее схема иллюстрирует способ введения O-связанных и C-связанных заместителей в положенииях 4 и 2 пиримидинола (3d), соответственно.
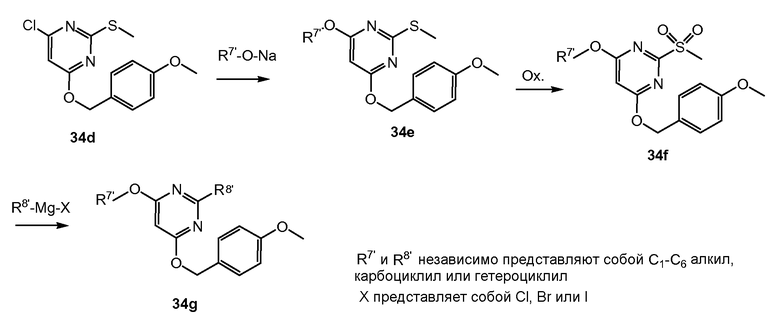
Замещение заместителя хлора в (34d) с помощью алкоксилирующего агента, такого как алкоксид щелочного металла, дает производное простого эфира (34e). Окисление серы с использованием соответствующего окислителя, например, хлорфенила mCPBA, до (34f), с последующим замещением сульфоновой группы в (34f) посредством использования соответствующего реагента Гриньяра дает алкилированный пиримидинол (34g).
C-связанный заместитель может вводиться в положении 4 промежуточного соединения производного пиримидинола 3d, например, посредством связывания Сузуки или Штилле, как иллюстрируется на следующей схеме.
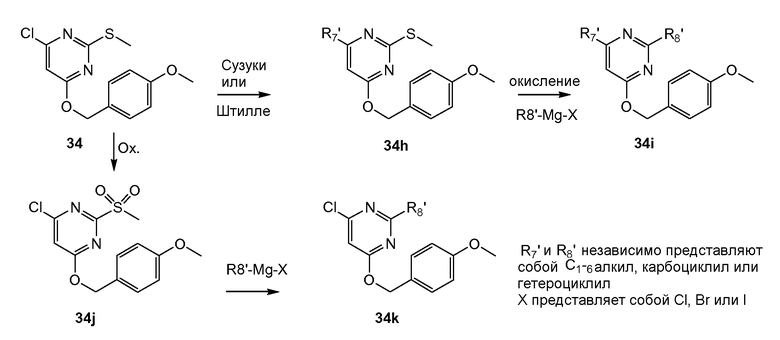
Воздействие на соединение хлора (34d) условий связывания Сузуки или Штилле дает алкилированное соединение (34h), которое впоследствии может окисляться до сульфона, а затем взаимодействовать с желаемым нуклеофилом, например, с реагентом Гриньяра, как описано выше, с получением диалкилированного пиримидинола (34i). Альтернативно, стадия окисления-замещения промежуточного соединения (34d) может осуществляться с получением сначала (34j), и заместитель хлор замещают соответствующим нуклеофилом, либо непосредственно после получения (34k), либо на более поздней ступени синтеза, например, как последнюю стадию, когда пиримидиновое производное связывается с остатком P2.
Синтез структурных элементов P3
Структурные элементы P3 являются доступными коммерчески или могут быть получены в соответствии с методиками, известными специалистам в данной области. Одна из этих методик показана на схеме ниже и использует моноацилированные амины, такие как трифторацетамид или Boc-защищенный амин.
На указанной выше схеме R вместе с группой CO образует N-защитную группу, в частности, R представляет собой трет-бутокси, трифторметил; R5 и n являются такими, как определено выше, и LG представляет собой уходящую группу, в частности, галоген, например, хлор или бром.
Моноацилированные амины (33a) обрабатывают сильным основанием, таким как гидрид натрия, а впоследствии они взаимодействуют с реагентом LG-C5-8алкенилом (33b), в частности, с галогенC5-8алкенилом, с образованием соответствующих защищенных аминов (33c). Снятие защиты с (33c) дает (5b), которые представляют собой структурные элементы P3. Снятие защиты будет зависеть от функциональной группы R, таким образом, если R представляет собой трет-бутокси, снятие защиты с соответствующего Boc-защищенного амина может осуществляться с помощью кислотной обработки, например, трифторуксусной кислотой. Альтернативно, когда R представляет собой, например, трифторметил, удаление группы R осуществляют с помощью основания, например, гидроксида натрия.
Следующая далее схема иллюстрирует еще один способ получения структурного элемента P3, а именно синтез Габриеля первичных C5-8алкениламинов, который может осуществляться посредством обработки фталимида (34a) основанием, таким как NaOH или KOH, и (33b), которое является таким, как определено выше, с последующим гидролизом промежуточного соединения N-алкенилимида для генерирования первичного C5-8алкениламина (5b-1).
На указанной выше схеме n является таким, как определено выше.
Соединения формулы (I) могут преобразовываться друг в друга, следуя реакциям преобразования функциональных групп, известным в данной области. Например, амино-группы могут N-алкилироваться, нитро группы восстанавливаться до амино-группы, атом галогена может заменяться другим галогеном.
Ряд промежуточных соединений, используемых для получения соединений формулы (I), представляют собой известные соединения или являются аналогами известных соединений, которые могут быть получены, следуя модификациям методик, известных в данной области, легко доступных для специалистов.
Соединения формулы (I) могут преобразовываться в соответствующие формы N-оксидов, следуя процедурам, известным в данной области для преобразования трехвалентного азота в его N-оксидную форму. Указанная реакция N-окисления может, как правило, осуществляться посредством взаимодействия исходного материала формулы (I) с соответствующим органическим или неорганическим пероксидом. Соответствующие неорганические пероксиды включают в себя, например, перекись водорода, пероксиды щелочных или щелочноземельных металлов, например, пероксид натрия, пероксид калия; соответствующие органические пероксиды могут включать в себя перокси кислоты, такие, например, как бензолкарбопероксовая кислота или галоген-замещенные бензолкарбопероксовые кислоты, например, 3-хлорбензолкарбопероксовую кислоту, пероксоалкановые кислоты, например, пероксоуксусную кислоту, алкилгидропероксиды, например, трет-бутил гидропероксид. Соответствующие растворители представляют собой, например, воду, низшие спирты, например, этанол и тому подобное, углеводороды, например, толуол, кетоны, например, 2-бутанон, галогенированные углеводороды, например, дихлорметан, и смеси таких растворителей.
Чистые стереохимически изомерные формы соединений формулы (I) могут быть получены посредством применения процедур, известных в данной области. Диастереомеры могут разделяться с помощью физических способов, таких как селективная кристаллизация и хроматографические методики, например, противоточное распределение, жидкостная хроматография и тому подобное.
Соединения формулы (I) могут быть получены в виде рацемических смесей энантиомеров, которые могут отделяться друг от друга, следуя процедурам разрешения, известных в данной области. Рацемические соединения формулы (1), которые являются достаточно основными или кислотными, могут преобразовываться в соответствующие диастереомерные солевые формы посредством взаимодействия с соответствующей хиральной кислотой, хиральным основанием, соответственно. Указанные диастереомерные солевые формы впоследствии разделяются, например, посредством селективной или фракционной кристаллизации, и энантиомеры высвобождаются после нее с помощью щелочи или кислоты. Альтернативный способ выделения энантиомерных форм соединений формулы (I) включает в себя жидкостную хроматографию, в частности, жидкостную хроматографию с использованием хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы могут также быть получены из соответствующих чистых стереохимически изомерных форм соответствующих исходных материалов, при условии, что реакция осуществляется стереоспецифично. Предпочтительно, если желаемым является конкретный стереоизомер, указанное соединение может синтезироваться посредством стереоспецифичных способов получения. Эти способы могут преимущественно использовать энантиомерно чистые исходные материалы.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I), как определено в описании, или соединения любой из подгрупп соединений формулы (I), как определено в описании, и фармацевтически приемлемый носитель. Терапевтически эффективное количество в этом контексте представляет собой количество, достаточное, чтобы действовать профилактическим образом против вирусной инфекции, стабилизируя или уменьшая ее, и в частности, вирусную инфекцию HCV, у инфицированных субъектов или у субъектов, имеющих риск инфицирования. Еще в одном дополнительном аспекте, настоящее изобретение относится к способу получения фармацевтической композиции, как определено в описании, который включает в себя тщательное перемешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы (I), как определено в описании, или соединения из любой из подгрупп соединений формулы (I), как определено в описании.
По этой причине, соединения по настоящему изобретению или любая их подгруппа могут приготавливаться в виде различных фармацевтических форм для целей введения. В качестве соответствующих композиций могут быть упомянуты все композиции, используемые обычно для системного введения лекарственных средств. Для получения фармацевтических композиций по настоящему изобретению, эффективное количество конкретного соединения, необязательно в форме аддитивной соли или комплекса с металлом, в качестве активного ингредиента объединяется в тесной смеси с фармацевтически приемлемым носителем, этот носитель может принимать разнообразные формы, в зависимости от формы препарата желательной для введения. Эти фармацевтические композиции являются желательными в соответствующей стандартной дозированной форме, в частности, для перорального, ректального, подкожного введения, или с помощью парентеральной инъекции. Например, при получении композиций в пероральной дозированной форме, может использоваться любая из обычных фармацевтических сред, такие, например, как вода, гликоли, масла, спирты и тому подобное, в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, лубриканты, связующие вещества, разрыхляющие агенты и тому подобное, в случае порошков, пилюль, капсул и таблеток. Благодаря простоте их введения, таблетки и капсулы представляют собой наиболее преимущественные пероральные стандартные дозированные формы, в этом случае, очевидно, используют твердые фармацевтические носители. Для парентеральных композиций, носитель обычно будет содержать стерильную воду, по меньшей мере, в основном, хотя могут включаться и другие ингредиенты, например, для облегчения растворимости. Например, могут быть получены растворы для инъекций, в которых носитель включает в себя солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Суспензии для инъекций также могут приготавливаться, в этом случае могут использоваться соответствующие жидкие носители, суспендирующие агенты и тому подобное. Также включаются препараты твердых форм, предназначенные для преобразования, незадолго до использования, в препараты в жидкой форме. В композициях, пригодных для подкожного введения, носитель необязательно содержит агент для улучшения проницаемости и/или соответствующий смачивающий агент, необязательно объединенный с соответствующими добавками любой природы в малых пропорциях, эти добавки не вносят значительного вредного воздействия на кожу.
Соединения по настоящему изобретению могут также вводиться посредством пероральной ингаляции или инсуфляции посредством способов и препаратов, используемых в данной области для введения этим путем. Таким образом, в целом соединения по настоящему изобретению, могут вводиться в легкие в форме раствора, суспензии или сухого порошка, раствор является предпочтительным. Любая система, разработанная для доставки растворов, суспензий или сухих порошков посредством пероральной ингаляции или инсуфляции, являются пригодной для введения настоящих соединений.
Таким образом, настоящее изобретение также предусматривает фармацевтическую композицию, адаптированную для введения с помощью ингаляции или инсуфляции через рот, содержащую соединение формулы (I) и фармацевтически приемлемый носитель. Предпочтительно, соединения по настоящему изобретению вводятся посредством ингаляции раствора в распыленных или аэрозольных дозах.
Является особенно преимущественным приготовление указанных фармацевтических композиций в стандартной дозированной форме для простоты введения и однородности дозирования. Стандартная дозированная форма, как используется в описании, относится к физически отдельным единицам, пригодным в качестве стандартных доз, каждая доза содержит заданное количество активного ингредиента, вычисленное для получения желаемого терапевтического воздействия, в сочетании с необходимым фармацевтическим носителем. Примеры таких стандартных дозированных форм представляют собой таблетки (включая разделяемые таблетки или таблетки с покрытиями), капсулы, пилюли, суппозитории, пакеты с порошками, вафли, растворы или суспензии для инъекций и тому подобное, и их разделенные наборы.
Соединения формулы (I) показывают противовирусные свойства. Вирусные инфекции и связанные с ними заболевания, которые могут лечиться с использованием соединений и способов по настоящему изобретению, включают в себя инфекции, переносимые HCV и другими патогенными флавивирусами, такими как желтая лихорадка, лихорадка Денге (типы 1-4), энцефалит Сент-Луиса, японский энцефалит, энцефалит долины Мюррэя, вирус западного Нила и вирус Кунжин. Заболевания, связанные с HCV, включают в себя прогрессирующий фиброз печени, воспаление и некроз, приводящие к циррозу, заболевание печени последней стадии и HCC (гепатоцеллюлярную карциному); а для других патогенных флавивирусов, заболевания включают в себя желтую лихорадку, лихорадку Денге, геморрагическую лихорадку и энцефалит. Кроме того, ряд соединений по настоящему изобретению являются активными против мутантных штаммов HCV. В дополнение к этому, многие соединения по настоящему изобретению показывают благоприятный фармакокинетический профиль и имеют привлекательные свойства с точки зрения биологической доступности, включая приемлемые значения половинного времени жизни, AUC (площади под кривой) и пиковые значения, и отсутствие неблагоприятных явлений, таких как недостаточно быстрое появление и удерживание в тканях.
Противовирусная активность in vitro соединений формулы (I) против HCV может исследоваться в клеточной системе с HCV репликонами на основе Lohmann et al. (1999) Science 285:110-113, с дополнительными модификациями, описанными Krieger et al. (2001) Journal of Virology 75: 4614-4624 (включается сюда в качестве ссылки), которая дополнительно иллюстрируется в разделе Примеры. Эта модель, хотя и не представляет собой полную модель инфекции HCV, широко принята как наиболее устойчивая и эффективная модель автономной репликации РНК HCV, доступная в настоящее время. Соединения, демонстрирующие активность против HCV в клеточной модели, рассматриваются как кандидаты для дальнейшей разработки при лечении инфекций HCV у млекопитающих. Будет понятно, что важно отличать соединения, которые конкретно вмешиваются в функционирование HCV, от тех, которые оказывают цитотоксические или цитостатические воздействия в модели HCV репликона, и как следствие, вызывают уменьшение концентрации РНК в HCV или связанного репортерного фермента. Анализы известны в области оценок клеточной цитотоксичности, например, на основе активности митохондриальных ферментов с использованием флюорогенных окислительно-востановительных красителей, таких как ресазурин. Кроме того, существуют скрининговые анализы с подсчетом клеток для оценки неселективного ингибирования активности связывания репортерного гена, такие как люциферазой светлячков. Соответствующие типы клеток могут снабжаться стабильной трансфекцией репортерного гена люциферазы, экспрессия которого зависит от промотора конститутивно активного гена, и такие клетки могут использоваться в качестве сравнительного скринингового анализа для устранения неселективных ингибиторов.
Благодаря их противовирусным свойствам, в частности, их анти-HCV свойствам, соединения формулы (I) или любая их подгруппа, их N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы являются пригодными для лечения индивидуумов, инфицированных вирусом, в частности, вирусом, который представляет собой HCV, и для профилактики вирусных инфекций, в частности, инфекций HCV. Как правило, соединения по настоящему изобретению могут быть пригодными для лечения теплокровных животных, инфицированных вирусами, в частности флавивирусами, такими как HCV.
По этой причине, соединения по настоящему изобретению или любая их подгруппа могут использоваться в качестве лекарственного средства. Указанное использование в качестве лекарственного средства или способа лечения включает в себя системное введение инфицированным вирусом субъектам или субъектам, склонным к вирусным инфекциям, количества, эффективного для борьбы с состояниями, связанными с вирусными инфекциями, в частности, с инфекцией HCV.
Настоящее изобретение также относится к использованию настоящих соединений или любой их подгруппы при получении лекарственного средства для лечения или предотвращения вирусной инфекции, в частности, инфекции HCV.
Кроме того, настоящее изобретение относится к способу лечения теплокровных животных, инфицированных вирусом или имеющих риск инфицирования вирусом, в частности, HCV, указанный способ включает в себя введение антивирусно эффективного количества соединения формулы (I), как определено в описании, или соединения из любой из подгрупп соединений формулы (I), как определено в описании.
Как правило, предполагается, что противовирусно эффективное ежедневное количество должно составлять от 0,01 мг/кг до 500 мг/кг массы тела или от 0,1 мг/кг до 50 мг/кг массы тела, или от 0,5 мг/кг до 5 мг/кг массы тела. Может быть правильным введение необходимой дозы как двух, трех, четырех или более субдоз через соответствующие интервалы в течение дня. Указанные субдозы могут приготавливаться как стандартные дозированные формы, например, содержащие от 1 до 1000 мг, и в частности, от 5 до 200 мг активного ингредиента на стандартную дозированную форму.
Точная дозировка и частота введения зависят от конкретного используемого соединения формулы (I), от конкретного состояния, которое лечится, от тяжести состояния, которое лечится, от возраста, массы, пола, степени расстройства и общего физического состояния конкретного пациента, а также от другого лекарственного средства, которое может принимать индивидуум, как хорошо известно специалистам в данной области. Кроме того, ясно, что указанное эффективное ежедневное количество может понижаться или увеличиваться в зависимости от реакции субъекта, который лечится, и/или в зависимости от оценки врача, предписывающего соединения по настоящему изобретению. Пределы эффективных ежедневных количеств, рассмотренных выше, являются, следовательно, только примерными инструкциями.
Настоящее изобретение также относится к сочетанию соединения формулы (I), включая его стереоизомерную форму, фармацевтически приемлемую соль или их фармацевтически приемлемый сольват, и другого противовирусного соединения, в частности, другого анти-HCV соединения. Термин "сочетание" может относиться к продукту, содержащему (a) соединение формулы (1), как определено выше, и (b) необязательно, другое анти-HCV соединение, в качестве объединенного препарата для одновременного, раздельного или последовательного использования при лечении инфекций HCV.
Анти-HCV соединения, которые могут использоваться в таких комбинациях, включают в себя агенты, выбранные из ингибитора полимеразы HCV, ингибитора протеазы HCV, ингибитора другой мишени в жизненном цикле HCV и иммуномодуляторного агента, и их сочетаний. Ингибиторы полимеразы HCV включают в себя NM283 (валопицитабин), R803, JTK-109, JTK-003, HCV-371, HCV-086, HCV-796 и R-1479. Ингибиторы протеаз HCV (ингибиторы NS2-NS3 и ингибиторы NS3-NS4A) включают в себя соединения из заявки на Международный патент WO 02/18369 (смотри, например, страницу 273, строки 9-22, и от страницы 274, строка 4 до страницы 276, строка 11); BILN-2061, VX-950, GS-9132 (ACH-806), SCH-503034 и SCH-6. Дополнительные агенты, которые могут использоваться, представляют собой те, которые описаны в заявках на Международный патент WO 98/17679, WO 00/056331 (Vertex); WO 98/22496 (Roche); WO 99/07734, (Boehringer Tungelheim), WO 2005/073216, WO 2005/073195 (Medivir), и структурно родственные агенты.
Ингибиторы других мишеней в жизненном цикле HCV включают в себя геликазу NS3; ингибиторы металлопротеазы; ингибиторы из антисмысловых олигонуклеотидов, такие как ISIS-14803, AVI-4065 и тому подобное; siРНК, такие как SIRPLEX-140-N, и тому подобное; кодируемые в вектор РНК с короткими шпилечными петлями (shРНК); ферменты ДНК; HCV-специфичные рибозимы, такие как гептазим, RPI.13919 и тому подобное; ингибиторы входа, такие как HepeX-C, HuMax-HepC, и тому подобное; ингибиторы альфа-глюкозидазы, такие как целгозивир, UT-231B, и тому подобное; KPE-02003002 и BIVN 401.
Иммуномодуляторные агенты включают в себя соединения природных и рекомбинантных изоформ интерферона, включая α-интерферон, β-интерферон, γ-интерферон, ω-интерферон и тому подобное, такие как Intron A®, Roferon-A®, Canferon-A300®, Advaferon®, Infergen®, Humoferon®, Sumiferon MP®, Alfaferone®, IFN-beta®, Feron®, и тому подобное; дериватизированные полиэтиленгликолем (пегилированные) соединения интерферона, такие как PEG интерферон-α-2a (Pegasys®), PEG интерферон-α-2b (PEG-Intron®), пегилированный IFN-α-conl, и тому подобное; препараты длительного действия и производные интерферона, такие как конденсированный с альбумином интерферон альбуферон-α, и тому подобное; соединения, которые стимулируют синтез интерферона в клетках, такие как резиквимод, и тому подобное; интерлейкины; соединения, которые усиливают развитие реакции хелперных T лимфоцитов типа 1, такие как SCV-07, и тому подобное; агонисты рецепторов типа TOLL, такие как CpG-10101 (актилон), изаторибин, и тому подобное; тимозин α-1; ANA-245; ANA-246; гистамин дигидрохлорид; пропагерманий; тетрахлордекаоксид; амплиген; IMP-321; KRN-7000; антитела, такие как цивацир, XTL-6865, и тому подобное; и профилактические и терапевтические вакцины, такие как InnoVac C, HCV E1E2/MF59, и тому подобное.
Другие противовирусные агенты включают в себя рибавирин, амантадин, вирамидин, нитазоксанид; телбивудин; NOV-205; тарибавирин; ингибиторы внутреннего входа в рибосому; вирусные ингибиторы широкого спектра, такие как ингибиторы IMPDH, и микофенольную кислоту и ее производные, и включая, но, не ограничиваясь этим VX-950, меримеподиб (VX-497), VX-148 и/или VX-944); или комбинации любых препаратов, указанных выше.
Конкретные агенты для использования в таких комбинациях включают в себя интерферон-α (IFN-α), пегилированный интерферон-α или рибавирин, а также терапевтические средства на основе антител, нацеленных против эпитопов HCV, малые интерферирующие РНК (siРНК), рибозимы, ДНК ферменты, антисмысловую РНК, низкомолекулярные анатагонисты, например, протеазы NS3, NS3 геликазы и полимеразы NS5B.
В другом аспекте предусматриваются комбинации соединения формулы (I), как определено в описании, и соединения анти-ВИЧ. Последние предпочтительно представляют собой такие ингибиторы ВИЧ, которые имеют положительное воздействие на метаболизм лекарственных средств и/или фармакокинетику, которая улучшает биологическую доступность. Пример такого ингибитора ВИЧ представляет собой ритонавир. Как таковое, настоящее изобретение, кроме того, предусматривает комбинацию, содержащую (a) ингибитор протеазы NS3/4a из HCV формулы (I) или его фармацевтически приемлемую соль; и (b) ритонавир или его фармацевтически приемлемую соль. Соединение ритонавир, его фармацевтически приемлемые соли и способы его получения описаны в заявке на Международный патент WO 94/14436, в патенте США №6037157 и в ссылках, цитируемых в описании: патент США №5484801, заявка на патент США №08/402690, заявка на Международный патент WO WO95/07696 и заявка на Международный патент WO WO95/09614, описывают предпочтительные дозированные формы ритонавира. Один из вариантов осуществления относится к комбинации, содержащей (a) ингибитор протеазы NS3/4a из HCV формулы (I) или его фармацевтически приемлемую соль; и (b) ритонавир или его фармацевтически приемлемую соль; она необязательно содержит дополнительное анти-HCV соединение, выбранное из соединений, рассмотренных выше.
Настоящее изобретение также относится к способу получения комбинации, как раскрыто в описании, включающему в себя стадию объединения соединения формулы (I), как определено выше, и другого агента, такого как противовирусный, включая агент анти-HCV или анти-ВИЧ, в частности, те, которые рассмотрены выше.
Указанные комбинации могут найти применение при получении лекарственного средства для лечения инфекции HCV или другого патогенного флави- или пестивируса у млекопитающего, инфицированного ими, указанная комбинация, в частности, содержит соединение формулы (I), как определено выше, и интерферона-α (IFN-α), пегилированного интерферона-α, или рибавирина. Или настоящее изобретение предусматривает способ лечения млекопитающего, в частности, человека, инфицированного HCV или другим патогенным флави- или пестивирусом, включающий в себя введение указанному млекопитающему эффективного количества комбинации, как раскрыто в описании. В частности, указанное лечение включает в себя системное введение указанной комбинации, и эффективное количество представляет собой такое количество, которое является эффективным при лечении клинических состояний, связанных с инфекцией HCV.
В одном из вариантов осуществления, комбинации, рассмотренные выше, приготавливаются в форме фармацевтической композиции, которая содержит активные ингредиенты, описанные выше, и носитель, как описано выше. Каждый из активных ингредиентов может приготавливаться отдельно, и препараты могут вводиться совместно, или может предусматриваться один препарат, содержащий их оба и, по желанию, дополнительные активные ингредиенты. В первом случае, комбинации могут также приготавливаться как объединенный препарат для одновременного, раздельного или последовательного использования при терапии HCV. Указанная композиция может принимать любую из форм, описанных выше. В одном из вариантов осуществления, оба ингредиента приготавливаются в одной дозированной форме, такой как фиксированная комбинированная доза. В конкретном варианте осуществления, настоящее изобретение предусматривает фармацевтическую композицию, содержащую (a) терапевтически эффективное количество соединения формулы (I), включая его стереоизомерную форму или его фармацевтически приемлемую соль, или его фармацевтически приемлемый сольват, и (b) терапевтически эффективное количество ритонавира или его фармацевтически приемлемой соли, и (c) носитель.
Индивидуальные компоненты комбинаций по настоящему изобретению могут вводиться по-отдельности, в различные моменты времени, в течение курса терапии или одновременно в разделенных или единых комбинировканных формах. Настоящее изобретение, как подразумевается, охватывает все такие режимы одновременного или чередующегося лечения и термин "введение" должен интерпретироваться соответствующим образом. В предпочтительном варианте осуществления, раздельные дозированные формы вводятся одновременно.
В одном из вариантов осуществления, комбинации по настоящему изобретению содержат количества ритонавира или его фармацевтически приемлемой соли, которые достаточны для клинического улучшения биологической доступности ингибиторов протеазы NS3/4a из HCV формулы (I) по сравнению с биологической доступностью, когда указанный ингибитор протеазы NS3/4a из HCV формулы (I) вводится сам по себе. Или комбинации по настоящему изобретению содержат количество ритонавира или его фармацевтически приемлемой соли, которое является достаточным для увеличения, по меньшей мере, одного из фармакокинетических переменных ингибитора протеазы NS3/4a из HCV формулы (I), выбранного из t1/2, Cmin, Cmax, Css, AUC через 12 часов или AUC через 24 часа, по отношению к указанному, по меньшей мере, одному фармакокинетическому переменному, когда ингибитор протеазы NS3/4a из HCV формулы (I) вводится сам по себе.
Комбинации по настоящему изобретению могут вводиться людям в конкретных пределах дозировки для каждого компонента, содержащегося в указанных комбинациях, например, соединение формулы (I), как определено выше, и ритонавир или его фармацевтически приемлемая соль могут иметь уровни дозировки в пределах от 0,02 до 5,0 г/день.
Массовое отношение соединения формулы (I) к ритонавиру может находиться в пределах примерно от 30:1 примерно до 1:15 или примерно от 15:1 примерно до 1:10, или примерно от 15:1 примерно до 1:1, или примерно от 10:1 примерно до 1:1, или примерно от 8:1 примерно до 1:1, или примерно от 1:5 до 1:1, примерно до 5:1, или примерно от 3:1 примерно до 1:1, или примерно 2:1 до 1:1. Соединение формулы (I) и ритонавир могут вводиться совместно один или два раза в день, предпочтительно, перорально, где количество соединения формулы (I) на одну дозу составляет примерно от 1 примерно до 2500 мг или примерно от 50 примерно до 1500 мг, или примерно от 100 примерно до 1000 мг, или примерно от 200 примерно до 600 мг, или примерно от 100 примерно до 400 мг; и количество ритонавира на дозу составляет от 1 примерно до 2500 мг или примерно от 50 примерно до 1500 мг, или примерно от 100 примерно до 800 мг, или примерно от 100 примерно до 400 мг, или от 40 примерно до 100 мг ритонавира.
ПРИМЕРЫ
Следующие далее примеры предназначены для иллюстрации настоящего изобретения, но не для ограничения его. Некоторые примеры показывают получение структурных элементов, которые могут связываться с любым другим соответствующим структурным элементом, рассмотренным в описании, а не только со структурными элементами иллюстрируемых конечных продуктов формулы I.
Пример 1
Стадия a: Сложный трет-бутиловый эфир 2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-4-гидрокси-пирролидин-1-карбоновой кислоты (1a)
Boc-защищенный 4-гидроксипролин (4 г, 17,3 ммоль), HATU (6,9 г, 18,2 ммоль) и сложный этиловый эфир 1-амино-2-винил-циклопропанкарбоновой кислоты, полученный, как описано в заявке на Международный патент WO03/099274 (3,5 г, 18,3 ммоль), растворяют в диметилформамиде (ДМФ) (60 мл) и охлаждают до 0°С на ледяной бане. Добавляют диизопропилэтиламин (DIPEA) (6 мл). Ледяную баню удаляют, и смесь оставляют при температуре окружающей среды в течение ночи. Затем добавляют дихлорметан (DCM) (около 80 мл) и органическую фазу промывают водным раствором гидрокарбоната натрия, лимонной кислотой, водой, насыщенным раствором соли и сушат над сульфатом натрия. Очистка с помощью флэш-хроматографии (простой эфир → 7% метанола в простом эфире) дает чистое указанное в заголовке соединение (6,13 г, 96%).
Стадия b: Сложный трет-бутиловый эфир 2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-4-(4-нитро-бензоилокси)-пирролидин-1-карбоновой кислоты (1b)
Соединение со стадии a (6,13 г, 16,6 ммоль), 4-нитробензойную кислоту (4,17 г, 25 ммоль) и PPh3 (6,55 г, 25 ммоль) растворяют в тетрагидрофуране (ТГФ) (130 мл). Раствор охлаждают до ~0°C и медленно добавляют диизопропилазидокарбоксилат (5,1 г, 25 ммоль). Затем охлаждение убирают, и смесь оставляют в течение ночи при условиях окружающей среды. Добавляют водный раствор гидрокарбоната натрия (60 мл), и смесь экстрагируют дихлорметаном. Очистка с помощью флэш-хроматографии (пентан-простой эфир, 2:1 → пентан-простой эфир, 1:2 → 2% метанола в простом эфире) дает чистое указанное в заголовке соединение (6,2 г, 72%).
Стадия c: Сложный 5-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)пирролидин-3-иловый эфир 4-нитро-бензойной кислоты (1c)
Соединение со стадии b (6,2 г, 12 ммоль) растворяют в охлажденной на льду смеси трифторметансульфоновой кислоты 33% в дихлорметане. Затем ледяную баню удаляют, и смесь оставляют при комнатной температуре в течение ~1,5 час. Растворитель выпаривают и добавляют 0,25 M карбонат натрия, и смесь экстрагируют дихлорметаном. Выпаривание дает указанное в заголовке соединение (4,8 г, 95%) в виде желтоватого порошка.
Стадия d: Сложный 5-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-1-(гекс-5-енил-метил-карбамоил)-пирролидин-3-иловый эфир 4-нитро-бензойной кислоты (Id)
Амин 1c (4,5 г, 10,8 ммоль) растворяют в ТГФ (160 мл). Добавляют столовую ложку гидрокарбоната натрия, а затем фосген (11,3 мл, 20% в толуоле). Смесь энергично перемешивают в течение 1 часа. Смесь фильтруют и повторно растворяют в дихлорметане (160 мл). Добавляют гидрокарбонат натрия (~ столовую ложку), а затем гидрохлоридамин (2,9 г, 21,6 ммоль). Реакционную смесь оставляют при комнатной температуре в течение ночи. Очистка с помощью флэш-хроматографии (простой эфир → 3% метанола в простом эфире) дает чистое указанное в заголовке соединение (5,48 г, 91%),
Стадия e: Сложный этиловый эфир 13-метил-l7-(4-нитро-бензоилокси)-2,14-диоксо-3,l3,15-триазатрицикло-[13.3.0.0*4.6*]октадек-7-ен-4-карбоновой кислоты (1e)
Диен 1d (850 мг, 1,53 ммоль) растворяют в 1,5 л дегазированного и сухого 1,2-дихлорэтана и нагревают с обратным холодильником в атмосфере аргона в течение ночи. Добавляют поглотитель кислорода (MP-TMT, P/N 800470 от Argonaut technologies, ~1/2 чайной ложки), и смесь перемешивают в течение 2 час, фильтруют и концентрируют при пониженном давлении. Сырой продукт кристаллизуют из дихлорметана/н-гексана, с получением указанного в заголовке соединения (600 мг, 74%).
Стадия f: Сложный этиловый эфир 17-гидрокси-13-метил-2,14-диоксо-3,13,15-триаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновой кислоты (1f)
Соединение 1e (200 мг, 0,38 ммоль) растворяют в смеси метанола/ТГФ/воды, 1:2:1, (20 мл) и охлаждают на ледяной бане. Медленно добавляют гидроксид лития (1,9 мл, 1M). Смесь перемешивают в течение 4 час при 0°C, затем нейтрализуют водным раствором уксусной кислоты (20 мл) и экстрагируют дихлорметаном. Органическую фазу промывают бикарбонатом, водой, насыщенным раствором соли и сушат над сульфатом магния. Очистка с помощью хроматографии (2% метанола в дихлорметане → 4%) дает указанное в заголовке соединение в виде сероватого порошка (80%).
Стадия g: 2-Метокси-6-фенилпиримидин-4-ол (1g)
Натрий (4,14 г, 180 ммоль) растворяют в сухом метаноле (120 мл), и раствор охлаждают до 0-5°C. Добавляют сульфат O-метилизомочевины (10,3 г, 60 ммоль) и этилбензоилацетат (11,5 г, 60 ммоль), и смесь перемешивают в течение двух часов при комнатной температуре, а затем нагревают с обратным холодильником в течение двенадцати часов. Смесь выпаривают, подкисляют раствором 2M HCl и экстрагируют три раза этилацетатом и три раза DCM. Органическую фазу сушат и выпаривают, и остаток суспендируют в простом диэтиловом эфире, твердый продукт отфильтровывают, промывают и сушат, что дает указанное в заголовке соединение, (1,0 г) MS+1=203.
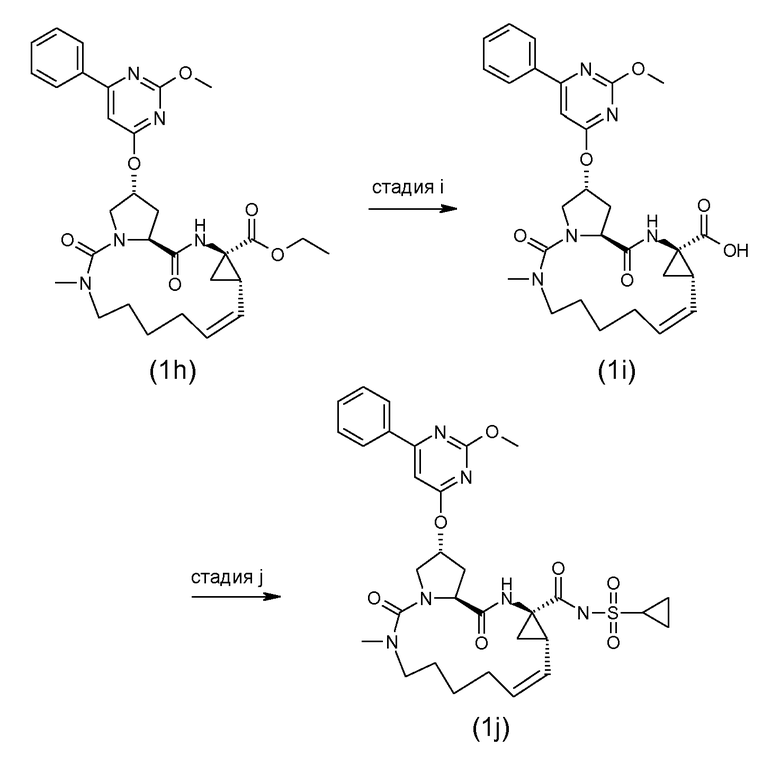
Стадия h: Сложный этиловый эфир 17-(2-Метокси-6-фенил-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13,15-триаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновой кислоты (1h)
Соединение 1f (250 мг, 0,659 ммоль) и соединение 1g (160 мг, 0,791 ммоль) и PPb, (432 мг, 1,648 ммоль) суспендируют в ТГФ (30 мл) и ДМФ (2 мл) при 0°C. Добавляют DIAD (0,32 мл, 1,648 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют простой эфир, и некоторую часть PPtt3O отфильтровывают. Реакционную смесь концентрируют, и остаток очищают с помощью колоночной флэш-хроматографии (DCM/ MeOH 95/5), что дает указанное в заголовке соединение (193 мг, 52%), MS (M+H)+564.
Стадия i: 17-(2-Метокси-6-фенил-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13,15-триаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновая кислота (1i)
Соединение 1h (95 мг, 0,169 ммоль) растворяют в смеси ТГФ:MeOH:H2O 2:1:1 (24 мл). Добавляют LiOH (1M, 1,7 мл) и смеси позволяют перемешиваться при комнатной температуре в течение ночи. Добавляют 5% лимонную кислоту, а затем DCM, и после экстрагирования органический слой сушат (MgSO4), фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии (DCM/MeOH 98/2 → 4/6), что дает указанное в заголовке соединение (78 мг, 86%), MS (M+H)+536.
Стадия j: [17-(2-Метокси-6-фенил-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13,15-триаза-трицикло[13.3.0.0*4,6*]октадек-7-ен-4-карбонил-амид циклопропансульфоновой кислоты (1i)
Соединение 1i (78 мг, 0,146 ммоль) и EDAC (34 мг, 0,175 ммоль) растворяют в DCM (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа (ЖХ-МС показывает присутствие промежуточного соединения). Добавляют амид циклопропансульфоновой кислоты (20 мг, 0,161 ммоль) и DBU (46 мкл, 0,307 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 3,5 час. Добавляют лимонную кислоту (5%), и органический слой отделяют и промывают насыщенным раствором соли, сушат (MgSO4), фильтруют и выпаривают. Остаток очищают с помощью препаративной ВЭЖХ, что дает чистое указанное в заголовке соединение (50 мг, 54%), (M+H)+639).
Пример 2
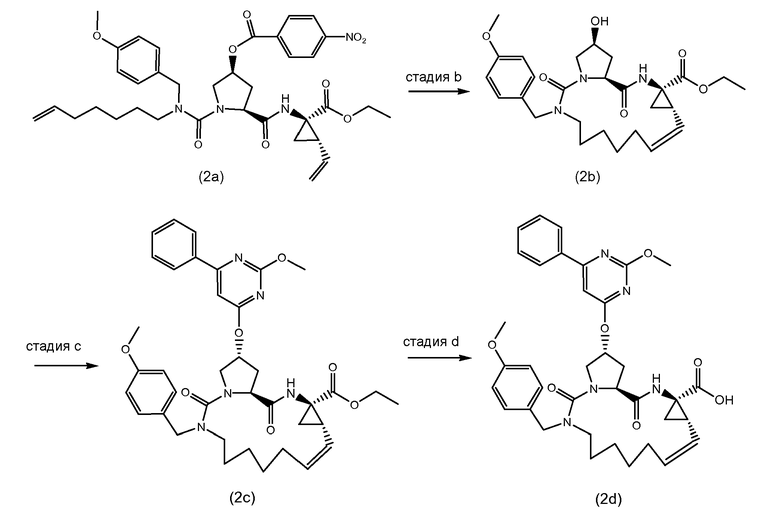
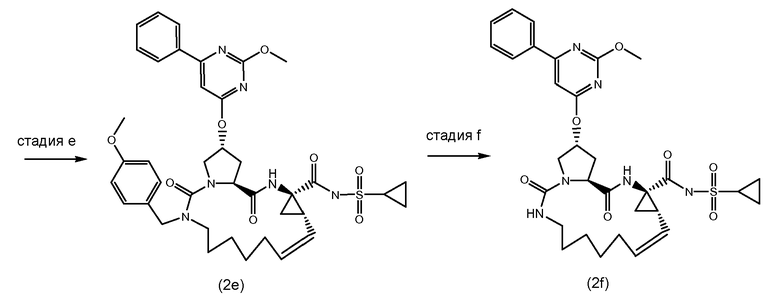
Стадия a: Сложный 5-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-1-[гепт-6-енил-(4-метокси-бензил)-карбамоил]-пирролидин-3-иловый эфир 4-нитро-бензойной кислоты (2a)
К раствору соединения 1c (4,5 г, 10,8 ммоль) в ТГФ (160 мл) добавляют NaHCO3 (1 столовую ложку) и фосген в толуоле (1,93 M, 11,5 мл, 22 ммоль). Смесь энергично перемешивают в течение 1 час при комнатной температуре, а затем фильтруют и выпаривают. Остаток растворяют в CH2Cl2 (160 мл), и добавляют NaHCO3 (1 столовую ложку) и гепт-5-енил-(p-метоксибензил)амин (4,3 г, 18,5 ммоль). После перемешивания в течение ночи при комнатной температуре реакционную смесь фильтруют и выпаривают досуха. Колоночная флэш-хроматография на силикагеле (EtOAc:толуол 25:75 → 40:60) дает указанное в заголовке соединение (6,59 г, 90%) в виде светло-коричневого сиропа.
Стадия b: Сложный этиловый эфир 18-гидрокси-14-(4-метокси-бензил)-2,15-диоксо-3,14,16-триаза-трицикло-[14.3.0.0*4,6*]нонадек-7-ен-4-карбоновой кислоты (2b)
Соединение 2a (1 г, 1,48 ммоль) растворяют в 1,2-дихлорэтане (2 л). Смесь дегазируют в течение 15 мин с использованием потока аргона. Добавляют катализатор Ховейды-Граббса (II) (50 мг, 5% моль), и смесь нагревают с обратным холодильником в течение 4 час. Растворитель выпаривают, и сырой сложный эфир растворяют в ТГФ (100 мл), метаноле (50 мл) и воде (50 мл). Смесь охлаждают до 0°C на ледяной бане. Добавляют водный раствор гидроксида лития (20 мл, 1M), и смесь перемешивают при 0°C в течение 4 час. Затем объем удваивают посредством добавления воды и смесь подкисляют уксусной кислотой. Экстрагирование (дихлорметан) с последующей колоночной флэш-хроматографией (метанол 1 → 5% в простом эфире) дает чистое указанное в заголовке соединение (450 мг, 61%).
MS (M+H)+ 500.
Стадия c: Сложный этиловый эфир 14-(4-метокси-бензил)-18-(2-метокси-6-фенил-пиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло[14.3.0.0*4.6*]нонадек-7-ен-4-карбоновой кислоты (2c)
Спирт 2b (500 мг, 1 ммоль), пиримидинол (1 г) (243 мг, 1,2 ммоль), PPh3 (656 мг, 2,5 ммоль) и DIAD (0,49 мл, 2,5 ммоль) растворяют в ТГФ (40 мл) и ДМФ (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель выпаривают, добавляют простой эфир, и PPh3O отфильтровывают. Очистка с помощью колоночной хроматографии, толуол/EtOAc, 9:1, дает указанное в заголовке соединение (680, 99%) MS (M+H)+ 684.
Стадия d: 14-(4-Метоксибензил)-18-(2-метокси-6-фенилпиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло[14.3.0.0*4.6*]нонадек-7-ен-4-карбоновая кислота (2d)
Соединение 2c (0,680 мг, 0,996 ммоль) растворяют в смеси ТГФ:MeOH:H2O 2:1:1 (144 мл). Добавляют LiOH (1 M, 10 мл) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют 5% лимонную кислоту, а затем DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают. Остаток очищают с помощью колоночной хроматографии, DCM:MeOH 9:1, что дает указанное в заголовке соединение (380 мг, 58%), MS (M+H)+ 656.
Стадия e: [14-(4-метоксибензил)-18-(2-метокси-6-фенил-пиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло[14.3.0.0*4.6*]нонадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (2e)
Соединение 2d (200 мг, 0,305 ммоль) растворяют в DCM (10 мл). Добавляют EDAC (70 мг, 0,366 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют амид циклопропансульфоновой кислоты (41 мг, 0,336 ммоль) и DBU (96 (мкл, 0,641 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 72 час. Добавляют 5% лимонную кислоту, и органический слой отделяют и промывают насыщенным раствором соли, сушат, фильтруют и выпаривают. Этот материал используют непосредственно на следующей стадии без дополнительной очистки.
Стадия f: [18-(2-Метокси-6-фенилпиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло[14.3.0.0*4.6*]нонадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (2f)
Соединение 2e растворяют в смеси TFA:DCM, 1:2, (12 мл) и перемешивают при комнатной температуре в течение 1 часа. Добавляют NaHCO3, и органический слой отделяют, сушат, фильтруют и выпаривают. Остаток очищают с помощью препаративной ВЭЖХ, что дает чистое указанное в заголовке соединение (32 мг, 16% на двух стадиях). MS (M+H)+ 639.
Пример 3
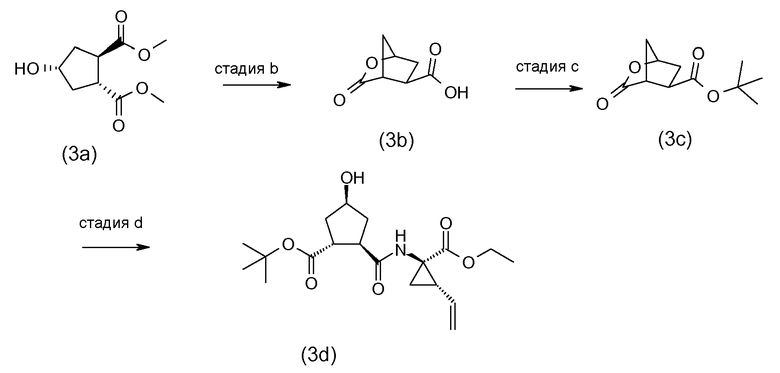
Стадия a: Сложный диметиловый эфир 4-гидрокси-циклопентан-1,2-дикарбоновой кислоты (3a)
Боргидрид натрия (1,11 г, 0,029 моль) добавляют к перемешиваемому раствору сложного диметилового эфира (1R,2S)-4-оксо-циклопентан 1,2-дикарбоновой кислоты (4,88 г, 0,0244 моль) в метаноле (300 мл) при 0°C. Через 1 час реакцию гасят 90 мл насыщенного солевого раствора, концентрируют и экстрагируют этилацетатом. Органические фазы объединяют, сушат, фильтруют и концентрируют. Сырой продукт очищают с помощью колоночной флэш-хроматографии (толуол/этилацетат 1:1), что дает указанное в заголовке соединение (3,73 г, 76%) в виде желтого масла.
Стадия b: 3-Оксо-2-окса-бицикло[2.2.1]гептан-5-карбоновая кислота (3b)
Гидроксид натрия (1M, 74 мл, 0,074 моль) добавляют к перемешиваемому раствору соединения 3a (3,73 г, 0,018 моль) в метаноле (105 мл) при комнатной температуре. Через 4 час реакционную смесь нейтрализуют с помощью 3M HCl, выпаривают и выпаривают совместно с толуолом несколько раз. Добавляют пиридин (75 мл) и Ac2O (53 мл), и реакционной смеси дают возможность для встряхивания в течение ночи при комнатной температуре. Затем смесь совместно выпаривают с толуолом и очищают с помощью колоночной флэш-хроматографии (этилацетат + 1% уксусная кислота), что дает указанное в заголовке соединение (2,51 г, 88%) в виде желтого масла.
Стадия c: Сложный трет-бутиловый эфир 3-оксо-2-окса-бицикло[2.2.1]гептан-5-карбоновой кислоты (3c)
Соединение 3b (13,9 г, 89 ммоль) растворяют в дихлорметане (200 мл), а затем охлаждают приблизительно до -10°C в атмосфере азота. Изобутилен барботируют в растворе до тех пор, пока общий объем не увеличится приблизительно до 250 мл, что дает "мутный раствор". Добавляют BF3 × Et2O (5,6 мл, 44,5 ммоль, 0,5 экв.), и реакционную смесь поддерживают приблизительно при -10°C в атмосфере азота. Через 10 мин получают прозрачный раствор. Реакцию отслеживают с помощью ТСХ (EtOAc-толуол 3:2 подкисляют с помощью нескольких капель уксусной кислоты и гексан-EtOAc, 4:1, окрашивают основным раствором перманганата). Через 70 мин остаются только следы соединения 13, и к реакционной смеси добавляют водный насыщенный раствор NaHCO3 (200 мл), затем ее энергично перемешивают в течение 10 мин. Органический слой промывают насыщенным раствором NaHCO3 (3×200 мл) и насыщенным раствором соли (1×150 мл), затем сушат над сульфитом натрия, фильтруют и концентрируют в масле, содержащем малые капли. При добавлении гексана к остатку продукт измельчают. Добавление дополнительного гексана и нагрев с обратным холодильником дают прозрачный раствор, из которого продукт кристаллизуют. Кристаллы собирают с помощью фильтрования и промывают гексаном (комнатная температура), затем сушат на воздухе в течение 72 час, что дает указанное в заголовке соединение в виде бесцветных иголок (12,45 г, 58,7 ммоль, 66% от первого сбора).
1H-ЯМР (300 МГц, CD3OD) δ 1,45 (с, 9H), 1,90 (д, J=11,0 Гц, 1H), 2,10-2,19 (м, 3H), 2,76-2,83 (м, 1H), 3,10 (с, 1H), 4,99 (с, 1H); 13C-ЯМР (75,5 МГц, CD3OD) δ 27,1, 33,0, 37,7, 40,8, 46,1, 81,1, 81,6, 172,0, 177,7.
Стадия d: Сложный трет-бутиловый эфир (1R,2R,4S)-2-((1R,2S)-1-этоксикарбонил-2-винил-циклопропилкарбамоил)-4-гидрокси-циклопентанкарбоновой кислоты (3d)
Соединение 3c (56 мг, 0,264 ммоль) растворяют в диоксане/воде 1:1 (5 мл), и смесь охлаждают до 0°C. Добавляют 1 M гидроксида лития (0,52 мл, 0,520 ммоль), и смесь перемешивают при 0°C в течение 45 минут, после этого смесь нейтрализуют с помощью 1M хлористоводородной кислоты, выпаривают и выпаривают совместно с толуолом. Кристаллический остаток растворяют в ДМФ (5 мл) и добавляют гидрохлорид сложного этилового эфира (1R,2S)-1-амино-2-винилцикло-пропанкарбоновой кислоты (60 мг, 0,313 ммоль) и диизопропилэтиламин (DIEA) (138 мкл, 0,792 ммоль), и раствор охлаждают до 0°C. Добавляют HATU (120 мг, 0,316 ммоль), и смесь перемешивают в течение 0,5 час при 0°C, а затем в течение дополнительных 2 час при комнатной температуре. Затем смесь выпаривают и экстрагируют EtOAc, промывают насыщенным раствором соли, сушат, фильтруют и концентрируют. Очистка с помощью колоночной флэш-хроматографии (толуол/EtOAc 1:1) дает указанное в заголовке соединение (86 мг, 89%) в виде бесцветного масла. Полученное масло кристаллизуют из этилацетата-гексана.
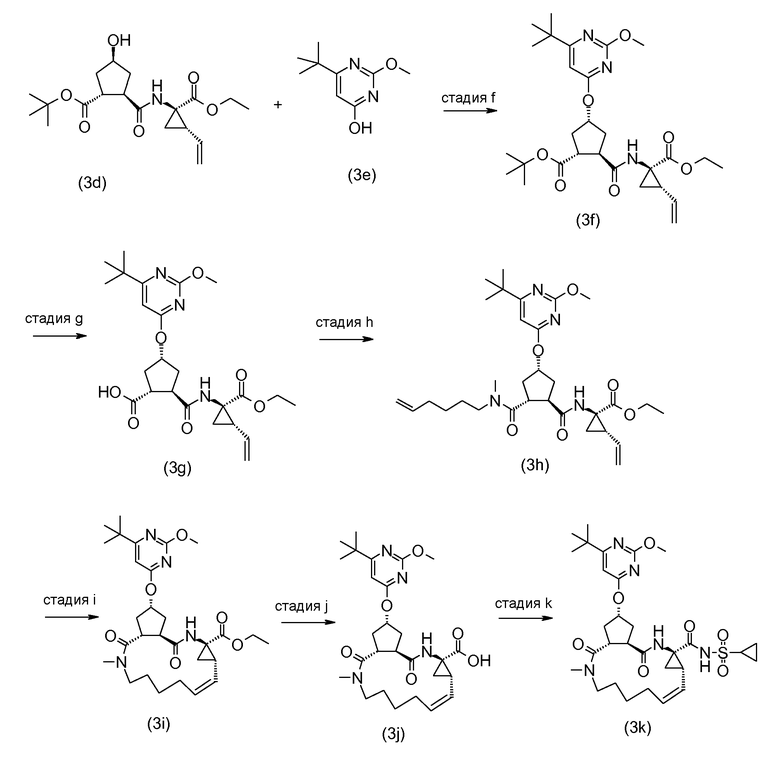
Стадия e: 6-трет-бутил-2-метокси-пиримидин-4-ол (3e)
К метоксиду натрия (5,67 г, 0,105 моль) в MeOH (60 мл) (0°C) добавляют гидрохлорид O-метилизомочевины (6,1 г, 0,055 моль) и этилпивалоилацетат (8,6 г, 0,05 моль). Смесь нагревают при 60°C в течение 20 минут, а затем перемешивают при комнатной температуре в течение дополнительных 30 минут. После выпаривания растворителя остаток растворяют в 25 мл H2O. Добавляют HCl для получения pH 6. Продукт отфильтровывают, что дает указанное в заголовке соединение в виде белого твердого продукта. (4,4 г, 88%), MS (M+H)+ 183.
Стадия f: Сложный трет-бутиловый эфир 4-(6-трет-бутил-2-метоксипиримидин-4-илокси)-2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)циклопентанкарбоновой кислоты (3f)
Соединение 3d (0,55 г, 1,5 ммоль), 6-трет-бутил-2-метокси-пиримидин-4-ол (0,33 г, 1,8 ммоль) и PPh3 (0,99 г, 3,75 ммоль) суспендируют в ТГФ (40 мл) при 0°C. Добавляют DIAD (0,74 мл, 3,75 ммоль) и реакционной смеси дают возможность перемешиваться при комнатной температуре в течение ночи. Растворитель выпаривают, и остаток очищают с помощью колоночной хроматографии (DCM/MeOH 95/5), что дает указанное в заголовке соединение (677 мг, 85%), MS (M+H)+ 532.
Стадия g: 4-(6-трет-Бутил-2-метоксипиримидин-4-илокси)-2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)циклопентанкарбоновая кислота (3g)
Соединение 3f растворяют в DCM (20 мл). Добавляют триэтилсилан (0,6 мл, 3,75 ммоль) и TFA (20 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1,5 час. Растворитель совместно выпаривают с толуолом, и сырой остаток очищают с помощью колоночной хроматографии (DCM → DCM/MeOH (90/10)), что дает указанное в заголовке соединение, (583 мг, 96%), MS (M+H)+ 476.
Стадия h: Сложный этиловый эфир 1-{[4-(6-трет-бутил-2-метоксипиримидин-4-илокси)-2-(гекс-5-енил-метилкарбамоил)-циклопентанкарбонил]амино}-2-винил-циклопропанкарбоновой кислоты (3h)
Соединение 3g (583 мг, 1,227 ммоль) растворяют в 15 мл сухого ДМФ. Добавляют DIEA (0,96 мл, 5,522 ммоль), N-метилгексенамин × HCl (270 мг, 1,779 ммоль) и HATU (676 мг, 1,779 ммоль) при 0°C, и реакционную смесь перемешивают при комнатной температуре в течение ночи. ДМФ выпаривают, и остаток растворяют в EtOAc и промывают насыщенным раствором NaHCO3 (водн.), H2O и насыщенным раствором соли. Органический слой сушат (MgSO4), фильтруют и выпаривают. Очистка с помощью колоночной хроматографии (гептан/EtOAc) дает чистое указанное в заголовке соединение (550 мг, 78%), MS (M+H)+ 573.
Стадия i: Сложный этиловый эфир 17-(6-трет-бутил-2-метоксипиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновой кислоты (3i)
Соединение 3h (500 мг, 0,87 ммоль) и катализатор Ховейды-Граббса, 2-ое поколение (50 мг), растворяют в дегазированном и сухом DCE (500 мл). Смесь нагревают с обратным холодильником в течение ночи в атмосфере N2. Материал перемешивают с окисью кремния, растворитель выпаривают, и остаток очищают с помощью колоночной хроматографии, EtOAc/гептан, 30:70 → 50:50, что дает указанное в заголовке соединение (350 мг, 74%), MS (M+H)+ 543.
Стадия j: 17-(6-трет-Бутил-2-метоксипиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновая кислота (3j)
Соединение 3i (350 мг, 0,645 ммоль) растворяют в смеси ТГФ:MeOH:H2O, 2:1:1 (100 мл). Добавляют LiOH (1 M, 6,5 мл), и реакционную смесь перемешивают при 60°C в течение ночи. Смесь подкисляют посредством добавления лимонной кислоты, а затем экстрагируют три раза EtOAc. Объединенные органические слои сушат (MgSO4), фильтруют и выпаривают. Полученный остаток очищают с помощью колоночной хроматографии (DCM/MeOH, 98/2 → 94/6), что дает указанное в заголовке соединение (320 мг, 97%), MS (M+H)+ 515.
Стадия k: [17-(6-трет-Бутил-2-метоксипиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (3k)
Соединение 3j (120 мг, 0,233 ммоль) растворяют в DCM (10 мл). Добавляют EDAC (54 мг, 0,28 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 2,5 час. Добавляют амид циклопропансульфоновой кислоты (31 мг, 0,256 ммоль) и DBU (73 мкл, 0,489 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют 5% лимонную кислоту, и органический слой промывают насыщенным раствором соли, сушат, фильтруют и выпаривают. Очистка с помощью препаративной ВЭЖХ дает чистое указанное в заголовке соединение (65 мг, 45%), MS (M+H)+ 618.
Пример 4
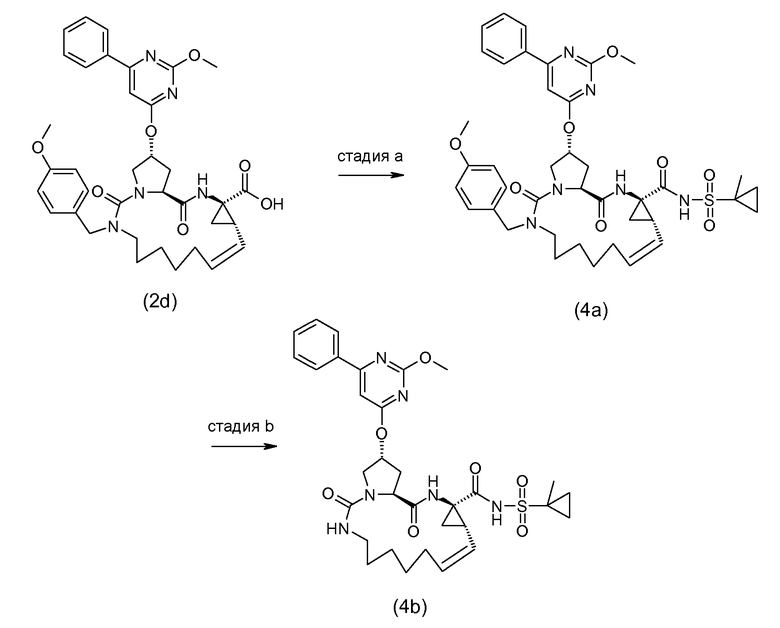
Стадия a: [14-(4-Метоксибензил)-18-(2-метокси-6-фенил-пиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло-[14.3.0.0*4.6*]нонадек-7-ен-4-карбонил]амид 1-метил-циклопропансульфоновой кислоты (4a)
Соединение 2d (130 мг, 0,198 ммоль) растворяют в DCM (10 мл). Добавляют EDAC (46 мг, 0,238 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 2,5 час. Добавляют амид метилциклопропансульфоновой кислоты (30 мг, 0,3218 ммоль) и DBU (63 мкл, 0,416 ммоль), и смесь перемешивают при комнатной температуре в течение ночи. Добавляют 5% лимонную кислоту, и органический слой промывают насыщенным раствором соли, сушат, фильтруют и выпаривают. Материал используют непосредственно на следующей стадии без дополнительной очистки.
Стадия b: [18-(2-Метокси-6-фенилпиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло[14.3.0.0*4.6*]нонадек-7-ен-4-карбонил]амид 1-метил-циклопропансульфоновой кислоты (4b)
Соединение 4a растворяют в смеси TFA:DCM, 1:2 (12 мл), и перемешивают при комнатной температуре в течение 1 часа. Добавляют NaHCO3, и органический слой сушат, фильтруют и выпаривают. Полученный остаток очищают с помощью препаративной ВЭЖХ, что дает чистое указанное в заголовке соединение (36 мг, 16% после двух стадий), MS (M+H)+ 639.
Пример 5
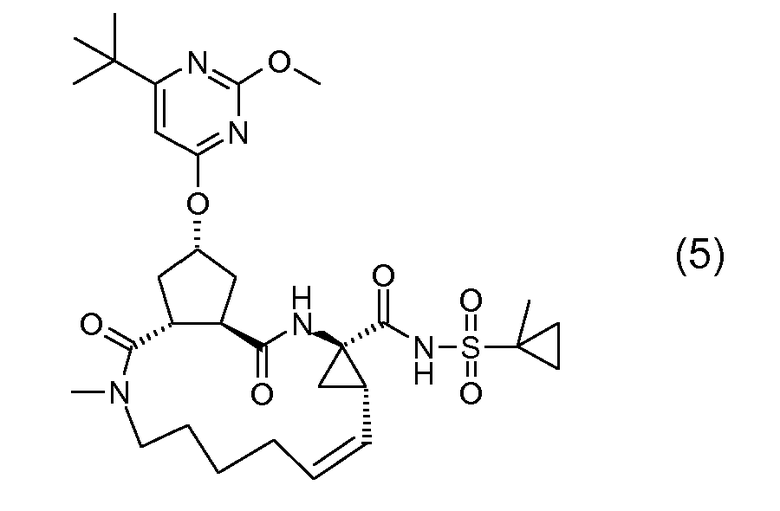
[17-(6-трет-Бутил-2-метокси-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид 1-метил-циклопропансульфоновой кислоты (5)
Соединение 3j (120 мг, 0,233 ммоль) растворяют в DCM (10 мл). Добавляют EDAC (54 мг, 0,28 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 2,5 час. Добавляют амид метилциклопропансульфоновой кислоты (35 мг, 0,256 ммоль) и DBU (73 мкл, 0,489 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют 5% лимонную кислоту, и органический слой отделяют и промывают насыщенным раствором соли, сушат, фильтруют и выпаривают. Остаток очищают с помощью препаративной ВЭЖХ, что дает чистое указанное в заголовке соединение (10,2 мг, 21%), MS (M+H)+ 632.
Пример 6
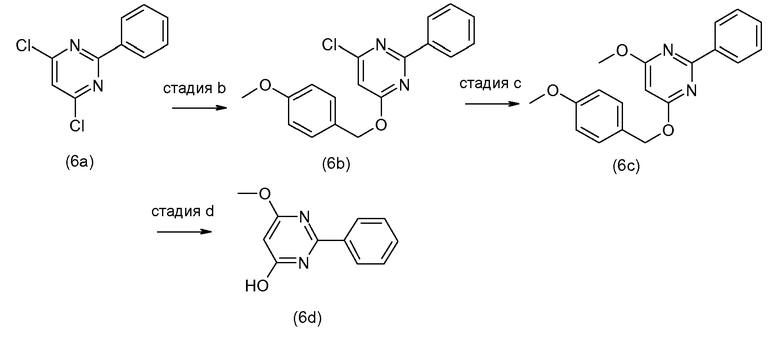
Стадия a: 4,6-Дихлор-2-фенил-пиримидин (6a)
К смеси 2-фенилпиримидин-4,6-диола (7 г, 0,037 моль) в POCl3 (26 мл, 0,279 моль) медленно добавляют N,N-диэтиламин (11,8 мл, 0,074 моль). Реакционную смесь нагревают с обратным холодильником в течение 3 час. Некоторую часть POCl3 выпаривают, и остаток выливают на лед с последующим экстрагированием EtOAc. Органический слой промывают насыщенным раствором соли, сушат (MgSO4), фильтруют и выпаривают, с получением указанного в заголовке соединения (5,16 г, 62%), MS (M+H)+ 226.
Стадия b: 4-Хлор-6-(4-метоксибензилокси)-2-фенилпиримидин (6b)
NaH (60%) (469 мг, 11,73 ммоль) добавляют порциями к перемешиваемому раствору 4,6-дихлор-2-фенил-пиримидина (2,2 г, 9,78 ммоль) и 4-метоксибензилового спирта (1,62 мг, 11,73 ммоль) в сухом ТГФ (55 мл) при 0°C. Через 1,5 час, добавляют NaHCO3 (водн.). Некоторую часть растворителя выпаривают, и остаток экстрагируют DCM, сушат (MgSO4), фильтруют и выпаривают, с получением указанного в заголовке соединения, (3,19 г, 100%), MS (M+H)+ 327.
Стадия c: 4-Метокси-6-(4-метокси-бензилокси)-2-фенил-пиримидин (6c)
NaOCH3 (2,64 г, 0,049 моль) растворяют в MeOH (180 мл). Добавляют 4-хлор-6-(4-метоксибензилокси)-2-фенилпиримидин (3,19 г, 9,78 ммоль), и реакционную смесь перемешивают при 0°C в течение 1 час. Затем смесь нагревают с обратным холодильником в течение 7 час. Растворитель выпаривают, и соединение очищают с помощью колоночной хроматографии (гептан → гептан/EtOAc 9/1), с получением указанного в заголовке соединения, (2 г, 64%), MS (M+H)+ 323.
Стадия d: 6-Метокси-2-фенилпиримидин-4-ол (6d)
4-Метокси-6-(4-метоксибензилокси)-2-фенилпиримидин растворяют в смеси TFA:DCM (1:2, 30 мл). Смесь перемешивают при комнатной температуре в течение 2 час. Добавляют NaHCO3 и DCM, и органический слой отделяют, сушат, фильтруют и выпаривают, с получением указанного в заголовке соединения (1,04 г, 83%), MS (M+H)+ 203.
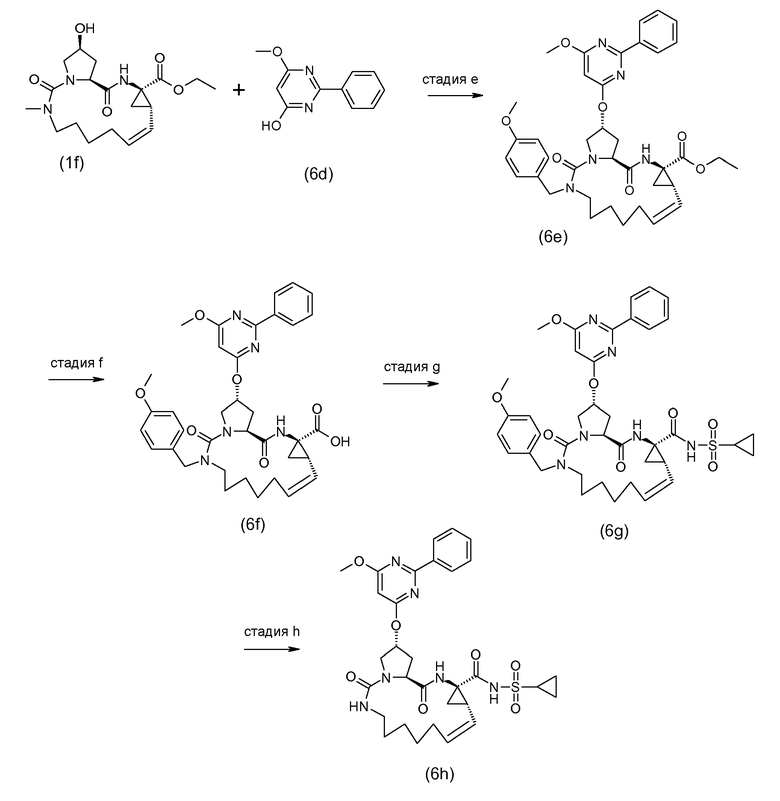
Стадия e: Сложный этиловый эфир 14-(4-метокси-бензил)-18-(6-метокси-2-фенил-пиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло[14.3.0.0*4.6*]нонадек-7-ен-4-карбоновой кислоты (6e)
Соединение 1f (500 мг, 1 ммоль), 6-метокси-2-фенилпиримидин-4-ол (243 мг, 1,2 ммоль), PPh3 (656 мг, 2,5 ммоль) и DIAD (0,49 мл, 2,5 ммоль) растворяют в ТГФ (40 мл) и ДМФ (3 мл). Реакционной смеси позволяют перемешиваться при комнатной температуре в течение ночи. Растворитель выпаривают, добавляют простой эфир, и PPh3O отфильтровывают. Очистка с помощью колоночной хроматографии, толуол/EtOAc, 9:1, дает указанное в заголовке соединение (410 мг, 60%), MS (M+H)+ 684.
Стадия f: 14-(4-Метоксибензил)-18-(6-метокси-2-фенилпиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло[14.3.0.0*4.6*]нонадек-7-ен-4-карбоновая кислота (6f)
Соединение 6e (410 мг, 0,60 ммоль) растворяют в смеси ТГФ:MeOH:H2O 2:1:1 (144 мл). Добавляют LiOH (1 M, 6 мл), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют 5% лимонную кислоту, а затем DCM. Органический слой сушат (MgSO4), фильтруют и выпаривают, что дает указанное в заголовке соединение (305 мг, 78%), MS (M+H)+ 656.
Стадия g: [14-(4-Метокси-бензил)-18-(6-метокси-2-фенил-пиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло[14.3.0.0*4.6*]нонадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (6g)
Соединение 6f (250 мг, 0,38 ммоль) растворяют в DCM (10 мл). Добавляют EDAC (88 мг, 0,46 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют амид циклопропансульфоновой кислоты (51 мг, 0,42 ммоль) и DBU (120 мкл, 0,80 ммоль), и перемешивают при комнатной температуре в течение 2 час. Добавляют лимонную кислоту, с последующим отделением органического слоя, который промывают насыщенным раствором соли, сушат, фильтруют и выпаривают. Этот материал используют непосредственно на следующей стадии без дополнительной очистки.
Стадия h: [18-(6-Метокси-2-фенилпиримидин-4-илокси)-2,15-диоксо-3,14,16-триаза-трицикло[14.3.0.0*4.6*]нонадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (6h)
Соединение 6g растворяют в смеси TFA:DCM, 1:2 (24 мл) и перемешивают при комнатной температуре в течение 1 часа. Добавляют NaHCO3, и органический слой отделяют, сушат, фильтруют и выпаривают. Остаток очищают с помощью препаративной ВЭЖХ, что дает чистое указанное в заголовке соединение (19 мг, 8% после двух стадий стадии), MS (M+H)+ 639.
Пример 7
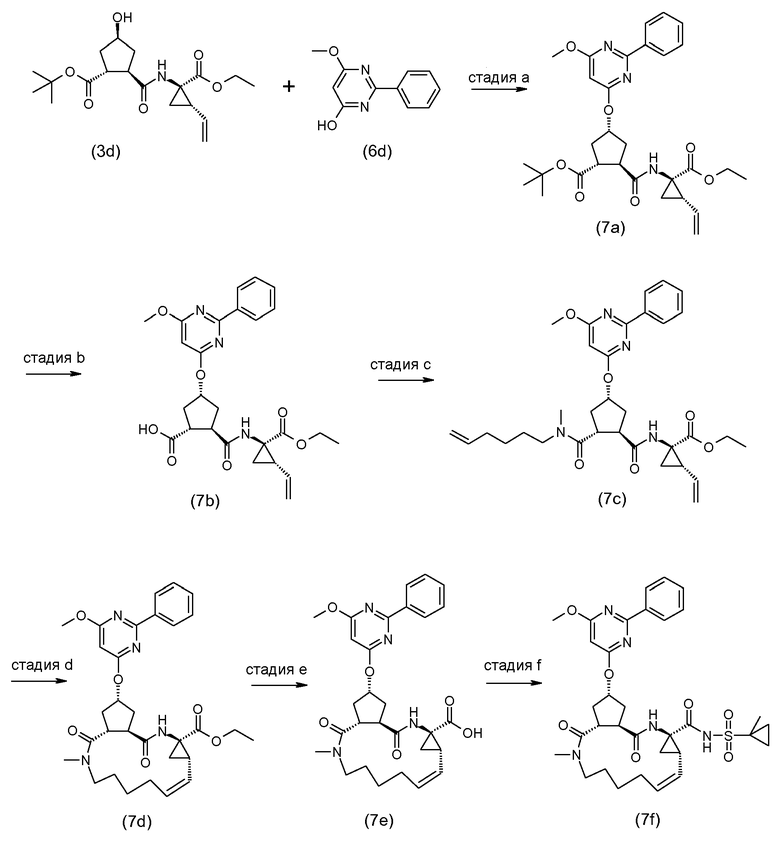
Стадия a: Сложный трет-бутиловый эфир 2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-4-(6-метокси-2-фенил-пиримидин-4-илокси)циклопентанкарбоновой кислоты (7a)
Соединение 6d (730 мг, 3,5 ммоль), соединение 3d (1,1 г, 3 ммоль) и PPh3 (1,97 г, 7,5 ммоль) суспендируют в ТГФ (80 мл), и колбу помещают на ледяную баню. Добавляют DIAD (1,5 мл, 7,5 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель выпаривают, и остаток растворяют в простом эфире. PPh3O отфильтровывают, и дополнительная очистка с помощью колоночной хроматографии (гептан/EtOAc, 4/1) дает указанное в заголовке соединение (1,47 г, 89%), MS (M+H)+ 552.
Стадия b: 2-(1-Этоксикарбонил-2-винил-циклопропилкарбамоил)-4-(6-метокси-2-фенил-пиримидин-4-илокси)циклопентанкарбоновая кислота (7b)
Соединение 7a (1,48 г, 2,68 ммоль) растворяют в 35 мл DCM. Добавляют триэтилсилан (1,07 мл, 6,7 ммоль) и 35 мл TFA, и смесь перемешивают при комнатной температуре в течение 45 минут. Растворитель выпаривают и совместно выпаривают с толуолом, что дает указанный в заголовке продукт (1,32 г, 99%), MS (M+H)+ 496.
Стадия c: Сложный этиловый эфир 1-{[2-(гекс-5-енил-метил-карбамоил)-4-(6-метокси-2-фенилпиримидин-4-илокси) циклопентанкарбонил]амино|-2-винил-циклопропанкарбоновой кислоты (7c)
Соединение 7b (1,32 г, 2,67 ммоль) растворяют в 30 мл сухого ДМФ. Добавляют DIEA (2,1 мл, 12,0 ммоль), N-метилгексенамин HCl (440 мг, 3,9 ммоль) и HATU (1,48 г, 3,9 ммоль) при 0°C, и реакционную смесь перемешивают при комнатной температуре в течение ночи. ДМФ выпаривают, и остаток растворяют в EtOAc, промывают насыщенным раствором NaHCO3 (водн.), H2O и насыщенным раствором соли. Органический слой сушат (MgSO4), фильтруют и выпаривают, с последующей колоночной хроматографией (гептан/ EtOAc), с получением указанного в заголовке соединения. (1,1 г, 70%), MS (M+H)+ 591.
Стадия d: Сложный этиловый эфир 17-(6-метокси-2-фенилпиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновой кислоты (7d)
Соединение 7c (1 г, 1,69 ммоль) и катализатор Ховейды-Граббса, 2-ое поколение (100 мг), растворяют в дегазированном и сухом DCE (1000 мл). Смесь нагревают с обратным холодильником в течение ночи в атмосфере N2. Материал перемешивают с окисью кремния, и растворитель выпаривают. Очистка с помощью колоночной хроматографии, EtOAc/гептан, 30:70 → 50:50, дает указанное в заголовке соединение (362 мг, 38%), MS (M+H)+ 563.
Стадия e: 17-(6-Метокси-2-фенилпиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновая кислота (7e)
Соединение 7d (362 мг, 0,644 ммоль) растворяют в смеси ТГФ:MeOH:H2O, 2:1:1 (100 мл). Добавляют LiOH (1 M, 6,5 мл), и реакционную смесь перемешивают при 60°C в течение 72 час. Реакционную смесь подкисляют посредством добавления 5% лимонной кислоты, после чего добавляют DCM. Органический слой отделяют, сушат (MgSO4), фильтруют и выпаривают, что дает указанное в заголовке соединение (344 мг, 100%), MS (M+H)+ 535.
Стадия f: [17-(6-Метокси-2-фенилпиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид 1-метил-циклопропансульфоновой кислоты (7f)
Соединение 7e (100 мг, 0,187 ммоль) растворяют в DCM (5 мл), и добавляют EDAC (43 мг, 0,224 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют амид метилциклопропансульфоновой кислоты (28 мг, 0,206 ммоль) и DBU (59 мкл, 0,393 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют лимонную кислоту, и органический слой отделяют, промывают насыщенным раствором соли, сушат, фильтруют и выпаривают. Очистка остатка с помощью препаративной ВЭЖХ дает чистое указанное в заголовке соединение (110 мг, 90%), MS (M+H)+ 652.
Пример 8
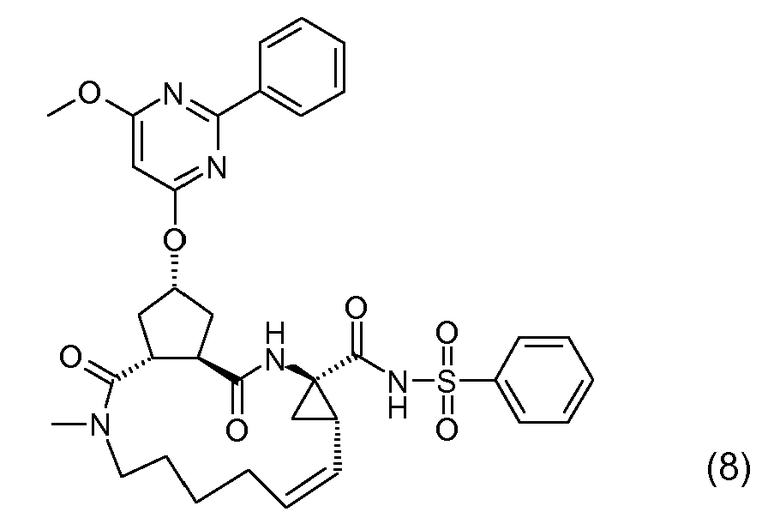
N-[17-(6-Метокси-2-фенилпиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]бензолсульфонамид (8)
Соединение 7e (60 мг, 0,112 ммоль) растворяют в DCM (5 мл). Добавляют EDAC (26 мг, 0,135 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют бензолсульфонамид (19 мг, 0,123 ммоль) и DBU (35 мкл, 0,235 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 2 час. Добавляют лимонную кислоту (5%), органический слой отделяют, промывают насыщенным раствором соли, сушат, фильтруют и выпаривают. Остаток очищают с помощью препаративной ВЭЖХ, что дает чистое указанное в заголовке соединение. (36 мг, 48%), MS (M+H)+ 674.
Пример 9
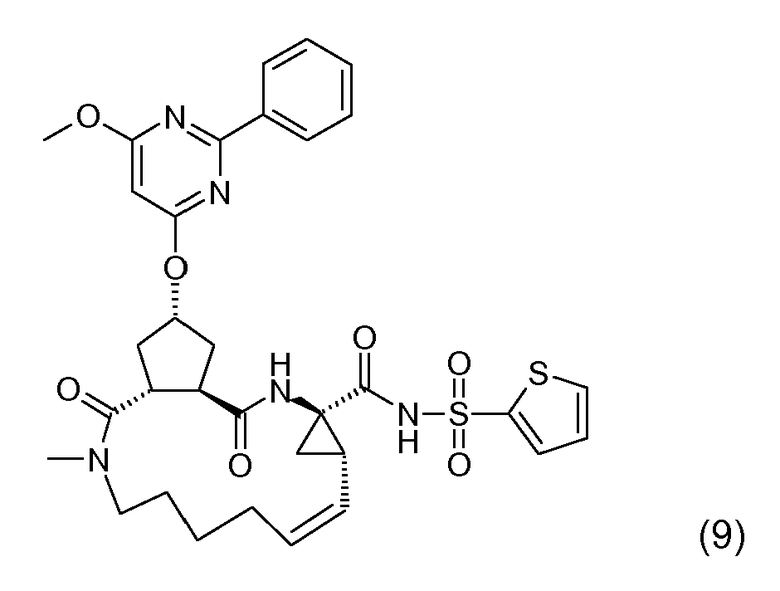
[17-(6-метокси-2-фенилпиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид тиофен-2-сульфоновой кислоты (9)
Соединение 7e (27 мг, 0,05 ммоль) растворяют в DCM (3 мл). Добавляют EDAC (12 мг, 0,06 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 1 час. Добавляют амид тиофен-2-сульфоноой кислоты (9 мг, 0,055 ммоль) и DBU (16 мкл, 0,105 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 2 час. Добавляют лимонную кислоту (5%), органический слой отделяют, промывают насыщенным раствором соли, сушат, фильтруют и выпаривают. Остаток очищают с помощью препаративной ВЭЖХ, что дает чистое указанное в заголовке соединение. (30 мг, 88%), MS (M+H)+ 680.
Пример 10
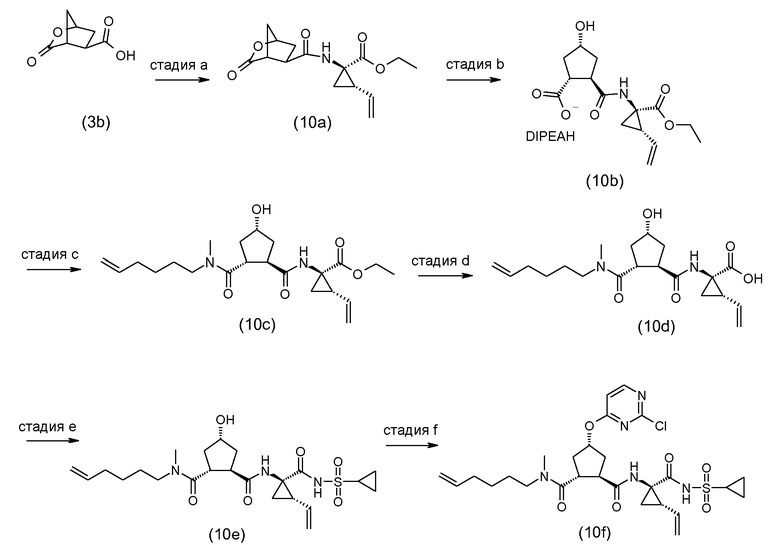
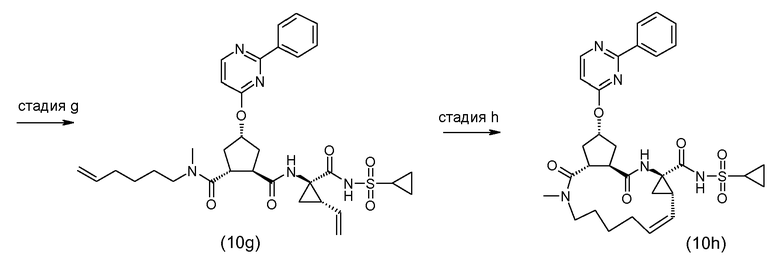
Стадия a: Сложный этиловый эфир 1-[(3-оксо-2-окса-бицикло[2,2,1]гептан-5-карбонил)амино]-2-винил-циклопропанкарбоновой кислоты (10a)
К раствору соединения 3b (857 мг, 5,5 ммоль) в ДМФ (14 мл) и DCM (25 мл) при комнатной температуре добавляют гидрохлорид сложного этилового эфира 1-амино-2-винил-циклопропанкарбоновой кислоты, полученный, как описано в заявке на Международный патент WO03/099274, (1,15 г, 6,0 ммоль), HATU (2,29 г, 6,0 ммоль) и DIPEA (3,82 мл, 22 ммоль). Реакционную смесь перемешивают в атмосфере N2 при температуре окружающей среды в течение 1 час. Анализ ЖХ/МС показывает полное преобразование, и реакционную смесь концентрируют в вакууме. Остаток повторно растворяют в DCM (100 мл) и 0,1 M HCl (водн.), и фазы разделяют. Органическую фазу промывают NaHCO3 (водн.) и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Удаление растворителя в вакууме дает целевое соединение (1,6 г, 99%). ЖХ/МС >95%, m/z (ESI+)= 294 (MH+)
Стадия b: Диизопропилэтиламиновая соль 2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-4-гидрокси-циклопентанкарбоновой кислоты (10b)
К раствору соединения 10a (800 мг, 2,73 ммоль) в воде (15 мл) в 20-мл реакционной емкости для микроволновой печи добавляют DIPEA (1,2 мл, 6,8 ммоль) и магнитную мешалку. Реакционную емкость герметизируют и не смешивающуюся суспензию энергично встряхивают перед помещением в полость микроволновой печи. После 1 мин предварительного перемешивания реакционную смесь облучают в течение 40 мин для достижения заданной температуры 100°C. После охлаждения до 40°C, прозрачный раствор концентрируют в вакууме, и оставшееся коричневое масло совместно выпаривают 3x с MeCN для удаления любой остаточной воды. Сырое указанное в заголовке соединение, в форме соли DIPEA, непосредственно отбирают для следующей стадии. ЖХ/МС >95%, m/z (ESI+)= 312 (MH+).
Стадия c: Сложный этиловый эфир 1-([2-(гекс-5-енил-метил-карбамоил)-4-гидрокси-циклопентанкарбонил]-амино}-2-винил-циклопропанкарбоновой кислоты (10c)
Сырое соединение 10b (5,5 моль) растворяют в DCM (50 мл) и ДМФ (14 мл), с последующим добавлением HATU (2,09 г, 5,5 ммоль), N-метил-N-гекс-5-ениламина (678 мг, 6,0 ммоль) и DIPEA (3,08 мл, 17,5 ммоль) при комнатной температуре. Реакционную смесь перемешивают при температуре окружающей среды в течение 1 час. Анализ ЖХ/МС показывает полное преобразование исходных материалов, и реакционную смесь концентрируют в вакууме. Остаток повторно растворяют в EtOAc (100 мл), и органическую фазу промывают 0,1 M HCl (водн.), K2CO3 (водн.) и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Удаление растворителя в вакууме дает масло, которое очищают с помощью флэш-хроматографии (окись кремния, EtOAc:MeOH), с получением указанного в заголовке соединения (1,65 г, 74%). ТСХ (окись кремния): MeOH:EtOAc 5:95, Rf=0,5; ЖХ/МС >95%, m/z (ESI+)= 407 (MH+).
Стадия d: 1-{[2-(Гекс-5-енил-метил-карбамоил)-4-гидрокси-циклопентанкарбонил]-амино}-2-винил-циклопропанкарбоновая кислота (10d)
Соединение 10c (493 мг, 1,21 ммоль) растворяют в ДМФ (1 мл) и переносят в 20-мл реакционную емкость для микроволновой печи, снабженную бруском магнитной мешалки, и добавляют водный раствор LiOH (2 M, 10,5 мл). Реакционную емкость герметизируют и несмешивающуюся суспензию энергично встряхивают перед помещением в полость микроволновой печи. Реакционную смесь облучают в течение 30 мин для достижения 130°C. Реакционную смесь охлаждают до 40°C, и прозрачный раствор подкисляют до pH 2 с помощью водного раствора HCl (1 M, 24 мл) и экстрагируют EtOAc (3×20 мл). Объединенные органические фазы промывают насыщенным раствором соли, сушат (MgSO4) и фильтруют. Растворитель удаляют в вакууме, с получением указанного в заголовке соединения (410 мг, 90%). ЖХ/МС >95%, m/z (ESI+)= 379 (MH+).
Стадия e: N-(1-Циклопропансульфониламинокарбонил-2-винил-циклопропил)-2-(гекс-5-енил-метил-амино-карбонил)-4-гидрокси-циклопентанкарбоксамид (10e)
Сырую кислоту 10d (410 мг, 1,09 ммоль) растворяют в ДМФ (1,5 мл) и DCM (4,5 мл) с последующим добавлением EDAC (417 мг, 2,18 ммоль) при комнатной температуре. Смеси дают возможность для инкубирования при перемешивании при комнатной температуре. Через 10 мин добавляют DMAP (133 мг, 1,09 ммоль), а затем следуют еще 20 мин инкубирования при комнатной температуре. Впоследствии добавляют предварительно перемешанный раствор амида циклопропансульфоновой кислоты (527 мг, 4,36 ммоль) и DBU (663 мг, 4,36 ммоль) в ДМФ (2 мл) и DCM (2 мл), с последующим нагревом в микроволновой печи для достижения 100°C в течение 30 мин. Полученный красный раствор концентрируют в вакууме и повторно растворяют в EtOAc (20 мл). Органическую фазу промывают 1 M HCl (водн.) (3×10 мл) и насыщенным раствором соли (10 мл), сушат (MgSO4) и фильтруют. Растворитель выпаривают в вакууме, с получением сырого сульфонамида, который дополнительно очищают с помощью хроматографии (окись кремния, EtOAc:MeOH, 97,5:2,5), что дает указанное в заголовке соединение (403 мг, 77%); ЖХ/МС >95%, m/z (ESI+)= 482 (MH+).
Стадия f: N-(1-Циклопропансульфониламинометил-2-винил-циклопропил)-2-(гекс-5-енил-метил-амино-карбонил)-4-(2-хлорпиримидин-4-илокси)циклопентанкарбоксамид (10f)
Соединение 10e (43 мг, 89,3 ммоль) растворяют в ДМФ (2 мл), и раствор охлаждают до 0°C и добавляют NaH (11 мг, 0,27 ммоль). Через 0,5 час добавляют 2,4-дихлорпиримидин. Реакционную смесь перемешивают при 0°C в течение 2 час, а затем гасят посредством добавления лимонной кислоты. Реакционную смесь экстрагируют DCM (3×10 мл), и объединенные органические фазы промывают лимонной кислотой, водой и насыщенным раствором соли. Органическую фазу сушат над MgSO4 и фильтруют. Растворитель удаляют в вакууме, с получением сырого целевого соединения (43 мг, 81%); ЖХ/МС m/z (ESI+)= 595 (MH+).
Стадия g: N-(1-Циклопропансульфониламинокарбонил-2-винил-циклопропил)-2-(гекс-5-енил-метил-амино-карбонил)-4-(2-фенилпиримидин-4-илокси)циклопентанкарбоксамид (10g)
Соединение 10f (43 мг, 72,4 ммоль), фенилбориновую кислоту (13 мг, 109 мкмоль) и (PPh3)2PdCl2 (5 мг, 7,2 мкмоль) смешивают в реакционном флаконе для микроволновой печи и нагревают в микроволновой печи при 120°C в течение 20 мин. Затем реакционную смесь гасят посредством добавления лимонной кислоты (20 мл) и экстрагируют 2 раза DCM (10 мл). Объединенные органические фазы промывают водой (2×10 мл) и насыщенным раствором соли (10 мл), сушат над MgSO4 и фильтруют. Растворитель удаляют в вакууме, и сырой продукт очищают с помощью препаративной ЖХ-МС, что дает указанное в заголовке соединение (5,9 мг, 13%), (ESI+)= 636 (MH+).
Стадия h: [13-Метил-2,14-диоксо-17-(2-фенилпиримидин-4-илокси)-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (10h)
Катализатор Ховейды-Граббса (2-ое поколение) (1,5 мг, 2,3 мкмоль) взвешивают в сухом реакционном флаконе для микроволновой печи, с последующим закрыванием флакона. Дегазированный раствор соединения 10g добавляют во флакон с помощью шприца, и реакционную смесь снова дегазируют с помощью N2. Реакционную смесь нагревают в микроволновой печи при 150°C в течение 10 минут. Растворитель удаляют в вакууме, и сырой продукт очищают с помощью препаративной ЖХ-MC, что дает указанное в заголовке соединение (0,9 мг, 16%), (ESI+)= 608 (MH+).
Пример 11
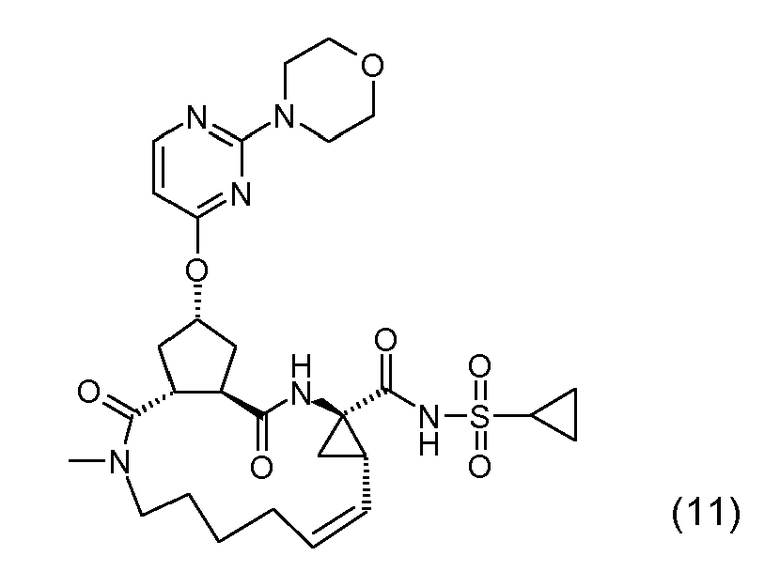
[13-Метил-17-(2-морфолин-4-ил-пиримидин-4-илокси)-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (11)
Соединение 10f (31 мг, 0,052 ммоль) и морфолин (0,1 мл) в ТГФ оставляют при температуре окружающей среды в течение 20 часов, летучие вещества удаляют, и остаток растворяют в DCE (15 мл), к нему добавляют катализатор Ховейды-Граббса II (7 мг). Смесь нагревают в микроволновой печи при 150°C в течение 10 мин в атмосфере азота, затем концентрируют досуха и очищают с помощью колоночной хроматографии на силикагеле, элюируя DCM-MeOH 2% с последующей препаративной ВЭЖХ-МС-УФ, с получением чистого указанного в заголовке соединения, (4,7 мг), (M+H)+ 616,2.
Пример 12

[17-(2-изопропиламинопиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (12)
Соединение 10f (30 мг, 0,05 ммоль) и изопропиламин (избыток) в ТГФ (10 мл) нагревают в микроволновой печи при 110°C в течение 30 мин, а затем оставляют при температуре окружающей среды в течение 16 час. Растворитель удаляют, и остаток повторно растворяют в DCE (15 мл) и дегазируют с помощью азота. Добавляют катализатор Ховейды-Граббса II (7 мг), и смесь нагревают в микроволновой печи при 150°C в течение 10 мин. Смесь концентрируют досуха и очищают с помощью препаративной ВЭЖХ-МС-УФ, что дает чистое указанное в заголовке соединение (3,1 мг), (M+H)+ = 589,2.
Пример 13
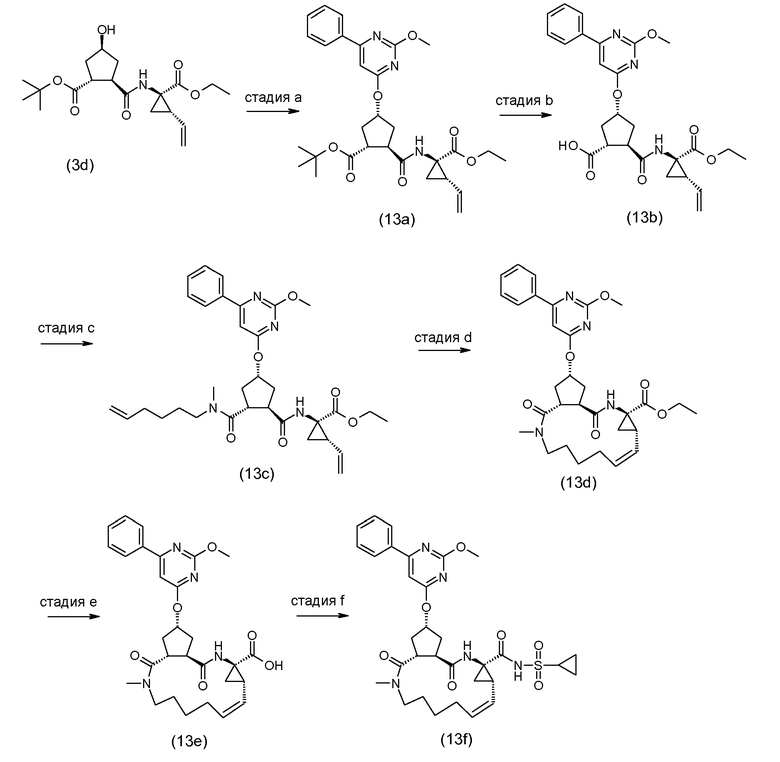
Стадия a: Сложный трет-бутиловый эфир 2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-4-(2-метокси-6-фенилпиримидин-4-илокси)циклопентанкарбоновой кислоты (13a)
К охлажденной суспензии спирта 3d (0,74 г, 2 ммоль), 2-метокси-6-фенил-пиримидин-4-ола (0,49 г, 2,4 ммоль) и трифенилфосфина (1,31 г, 5 ммоль) в сухом ТГФ (50 мл) добавляют DIAD (1,0 г, 5 ммоль), и смесь перемешивают при комнатной температуре в течение ночи. Смесь выпаривают, и указанное в заголовке соединение выделяют с помощью хроматографии на силикагеле, элюируя гексаном-этилацетатом, (1,0 г, 90%), (M+H)+ 552.
Стадия b: 2-(1-Этоксикарбонил-2-винил-циклопропилкарбамоил)-4-(2-метокси-6-фенил-пиримидин-4-илокси)циклопентанкарбоновая кислота (13b)
К раствору соединения 13a (1,0 г, 1,81 ммоль) в DCM (30 мл) добавляют триэтилсилан (0,53 г, 4,53 ммоль) и TFA (20 мл), и смесь перемешивают при комнатной температуре в течение 1 часа. Раствор выпаривают при пониженном давлении и совместно выпаривают два раза с толуолом, что дает указанное в заголовке соединение (1,2 г), (M+H)+ 496.
Стадия c: Сложный этиловый эфир 1-{[2-(Гекс-5-енил-метил-карбамоил-4-(2-метокси-6-фенил-пиримидин-4-илокси)-циклопентанкарбонил]амино}-2-винил-циклопропанкарбоновой кислоты (13c)
К охлажденному на льду раствору сырой кислоты 13b (1,2 г, 1,81 ммоль), N-метилгексенамина гидрохлорида (0,404 г, 2,7 ммоль) и DIEA (1,2 г, 9,1 ммоль) добавляют HATU (1,02 г, 2,7 ммоль), и смесь перемешивают в течение двадцати минут на ледяной бане и в течение двух часов при комнатной температуре. Добавляют насыщенный раствор гидрокарбоната натрия, и смесь экстрагируют три раза этилацетатом. Объединенные органические фазы промывают насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли и сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Продукт выделяют с помощью хроматографии на силикагеле с помощью гексана-этилацетата, (0,8 г, 74%), (M+H)+ 591.
Стадия d: Сложный этиловый эфир 17-(2-метокси-6-фенил-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновой кислоты (13d)
К раствору диолефина 13c (760 мг, 1,28 ммоль) в DCE (700 мл) в атмосфере аргона (три раза выпаривают, три раза заполняют аргоном) добавляют катализатор Ховейды Граббса, 2-ое поколение (80 мг), и смесь нагревают с обратным холодильником в течение ночи. Растворитель выпаривают при пониженном давление, и остаток очищают с помощью хроматографии на силикагеле, гексан-этилацетат, что дает указанное в заголовке соединение (0,52 г, 70%), (M+H)+ 563.
Стадия e: 17-(2-Метокси-6-фенилпиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновая кислота (13e)
К раствору соединения 13d (470 мг, 0,83 ммоль) в смеси ТГФ метанол, 1:1 (30 мл) добавляют раствор 1 M LiOH (10 мл), и смесь перемешивают в течение четырех дней при комнатной температуре. Добавляют 5% раствор лимонной кислоты, и смесь экстрагируют три раза этилацетатом. Органическую фазу сушат над сульфатом натрия и выпаривают на силикагеле. Продукт выделяют с помощью хроматографии на силикагеле, элюируя DCM-метанолом, (400 мг, 88%), (M+H)+ 535.
Стадия f: [17-(2-метокси-6-фенилпиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (13f)
Раствор кислоты 13e (150 мг, 0,28 ммоль) и EDAC (65 мг, 0,34 ммоль) в сухом DCM (3 мл) перемешивают в течение ночи. Добавляют амид циклопропансульфоновой кислоты (72,7 мг, 0,6 ммоль) и DBU (120 мг, 0,8 ммоль), и смесь перемешивают в течение ночи при комнатной температуре. Добавляют 5% лимонную кислоту, и смесь экстрагируют этилацетатом. Органическую фазу промывают два раза 5% лимонной кислотой и водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Очистка с помощью ВЭЖХ дает указанное в заголовке соединение, (100 мг, 56%), (M+H)+ 638.
Пример 14
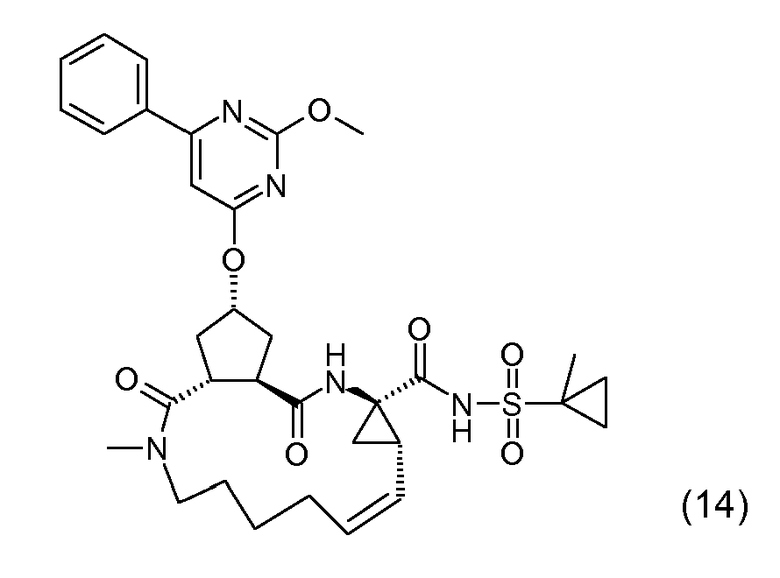
[17-(2-Метокси-6-фенил-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид 1-метил-циклопропансульфоновой кислоты (14)
Раствор кислоты 13e (250 мг, 0,46 ммоль) и EDAC (150 мг, 0,6 ммоль) в сухом DCM (4 мл) перемешивают в течение ночи при комнатной температуре. Добавляют амид метилциклопропансульфоновой кислоты (78 мг, 0,58 ммоль) и DBU (182 мг, 1,2 ммоль), и смесь перемешивают в течение дополнительных 6 часов при комнатной температуре. Добавляют 5% лимонную кислоту и смесь экстрагируют этилацетатом. Органическую фазу промывают два раза 5% лимонной кислотой и водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Очистка с помощью колоночной хроматографии на силикагеле, с элюированием простым диэтиловым эфиром - этилацетатом дает чистое указанное в заголовке соединение, (130 мг, 44%), (M+H)+ 652.
Пример 15
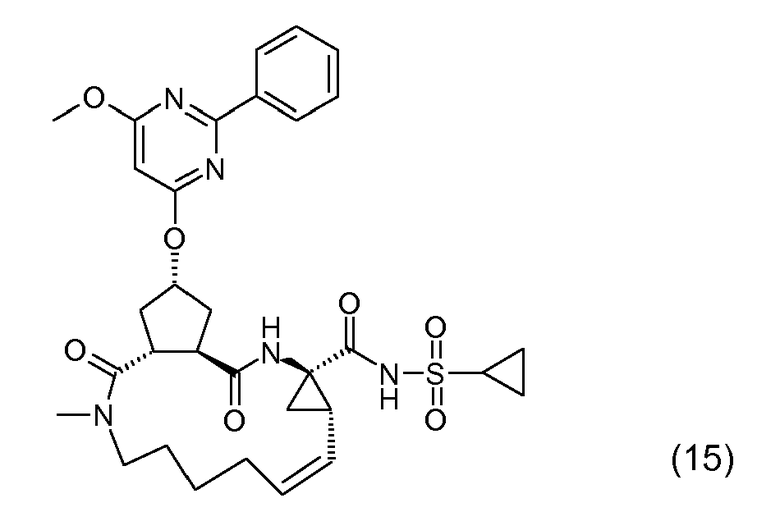
[17-(6-Метокси-2-фенилпиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (15)
Следуют процедуре, описанной в Примере 7, стадия f, но используя амид циклопропансульфоновой кислоты вместо амида метилциклопропансульфоновой кислоты, что дает указанное в заголовке соединение, (200 мг, 60%), (M+H)+ 638.
Пример 16
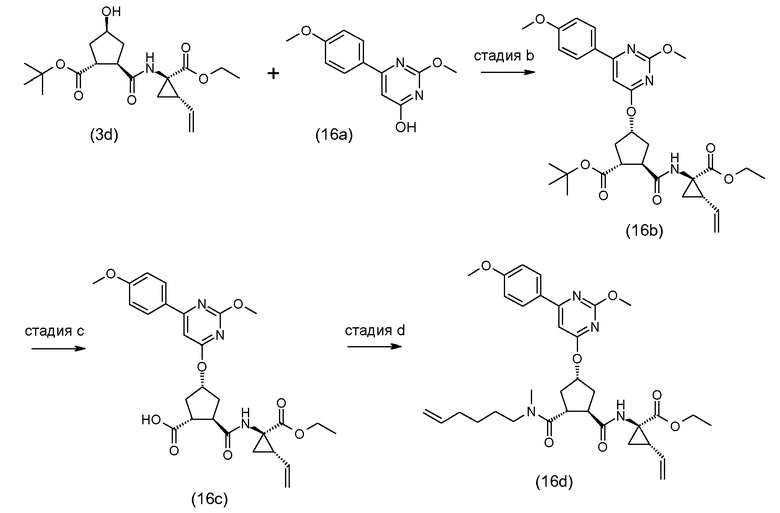
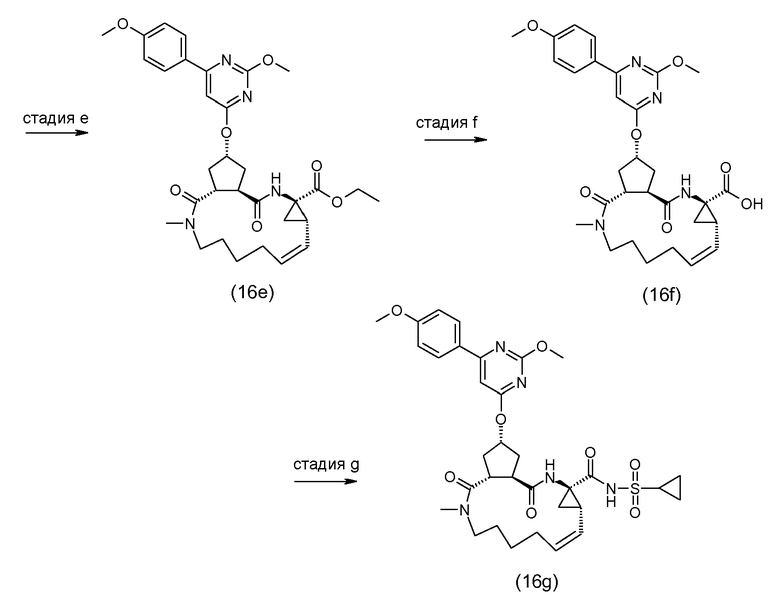
Стадия a: 2-Метокси-6-(4-метоксифенил)пиримидин-4-ол (16a)
К охлаждаемому на льду раствору метоксида натрия (5,8 г, 105 ммоль) в метаноле (60 мл) добавляют гидрохлорид O-метилизомочевины (6,1 г, 55 ммоль) и этил 4-метокси-бензоилацетат (11,11 г, 50 ммоль). Смесь перемешивают при комнатной температуре в течение ночи, а затем четыре часа при 60°С. Метанол удаляют, и смесь подкисляют 5% лимонной кислотой и позволяют стоять в течение ночи. Твердый продукт фильтруют, промывают водой и суспендируют в теплом этаноле. Смесь охлаждают, и указанное в заголовке соединение отфильтровывают и собирают в виде твердого продукта, (2,0 г, 7%), (M+H)+ 233.
Стадия b: Сложный трет-бутиловый эфир 2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-4-[2-метокси-6-(4-метоксифенил)-пиримидин-4'-илокси]циклопентанкарбоновой кислоты (16b)
Спирт 3d (0,74 г, 2 ммоль) взаимодействует с 2-метокси-6-(4-метоксифенил)пиримидин-4-олом (0,58 г, 2,5 ммоль) в присутствии трифенилфосфина (1,31 г, 5 ммоль) и DIAD (1,0 г, 5 ммоль) в сухом ТГФ (50 мл) в соответствии с процедурой, описанной в Примере 13, стадия a. Очистка с помощью колоночной хроматографии на силикагеле с элюированием этилацетатом-гексаном дает указанное в заголовке соединение (1,12 г, 95%), (M+H)+ 582.
Стадия c: 2-(1-Этоксикарбонил-2-винил-циклопропилкарбамоил)-4-[2-метокси-6-(4-метоксифенил)пиримидин-4-илокси]-циклопентанкарбоновая кислота (16c)
Соединение 16b (1,11 г, 1,9 ммоль) обрабатывают триэтилсиланом (0,55 г, 4,75 ммоль) и TFA (30 мл) в соответствии с процедурой, описанной в Примере 13, стадия b, что дает сырое указанное в заголовке соединение, (1,3 г), (M+H)+ 526.
Стадия d: Сложный этиловый эфир 1-((2-(гекс-5-енил-метил-карбамоил)-4-[2-метокси-6-(4-метоксифенил)пиримидин-4-илокси]-циклопентанкарбонил)амино)-2-винил-циклопропанкарбоновой кислоты (16d)
Кислота 16c (1,2 г, 2 ммоль) взаимодействует с N-метилгексенамином (340 мг, 3,0 ммоль) и DIEA (1,29 г, 10,0 ммоль) в сухом ДМФ (30 мл) в соответствии с процедурой, описанной в Примере 13, стадия c. Очистка с помощью колоночной хроматографии на силикагеле дает указанное в заголовке соединение, (1,02 г, 82%), (M+H)+ 621.
Стадия e: Сложный этиловый эфир 17-[2-метокси-6-(4-метокси-фенил)пиримидин-4-илокси]-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновой кислоты (16e)
Диолефин 16d (0,95 г, 1,53 ммоль) взаимодействует с катализатором Ховейды-Граббса, 2-ое поколение (140 мг), в DCE (900 мл) в соответствии с процедурой, описанной в Примере 13, стадия d, что дает сырое указанное в заголовке соединение, (560 мг, 62%), (M+H)+ 565.
Стадия f: 17-[2-Метокси-6-(4-метокси-фенил)-пиримидин-4-илокси]-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]-октадек-7-ен-4-карбоновая кислота (16f)
Раствор сложного этилового эфира 16e (1,2 г, 2 ммоль) в ТГФ/MeOH, 1:1 (30 мл), обрабатывают 1M раствором LiOH, как описано в Примере 13, стадия d. Очистка с помощью колоночной хроматографии на силикагеле дает сырое указанное в заголовке соединение, (400 мг, 77%), (M+H)+ 565.
Стадия g: {17-[2-метокси-6-(4-метоксифенил)пиримидин-4-илокси]-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]-октадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (16g)
Кислота 16f (195 мг, 0,34 ммоль) взаимодействует с амидом циклопропансульфоновой кислоты (53 мг, 0,44 ммоль), EDAC (85 мг, 0,44 ммоль) и DBU (137 мг, 0,9 ммоль) в сухом DCM (3 мл) в соответствии с процедурой, описанной в Примере 13, стадия f. Очистка с помощью колоночной хроматографии на силикагеле, с элюированием простым диэтиловым эфиром - этилацетатом дает указанное в заголовке соединение, (100 мг, 45%), (M+H)+ 668.
Пример 17
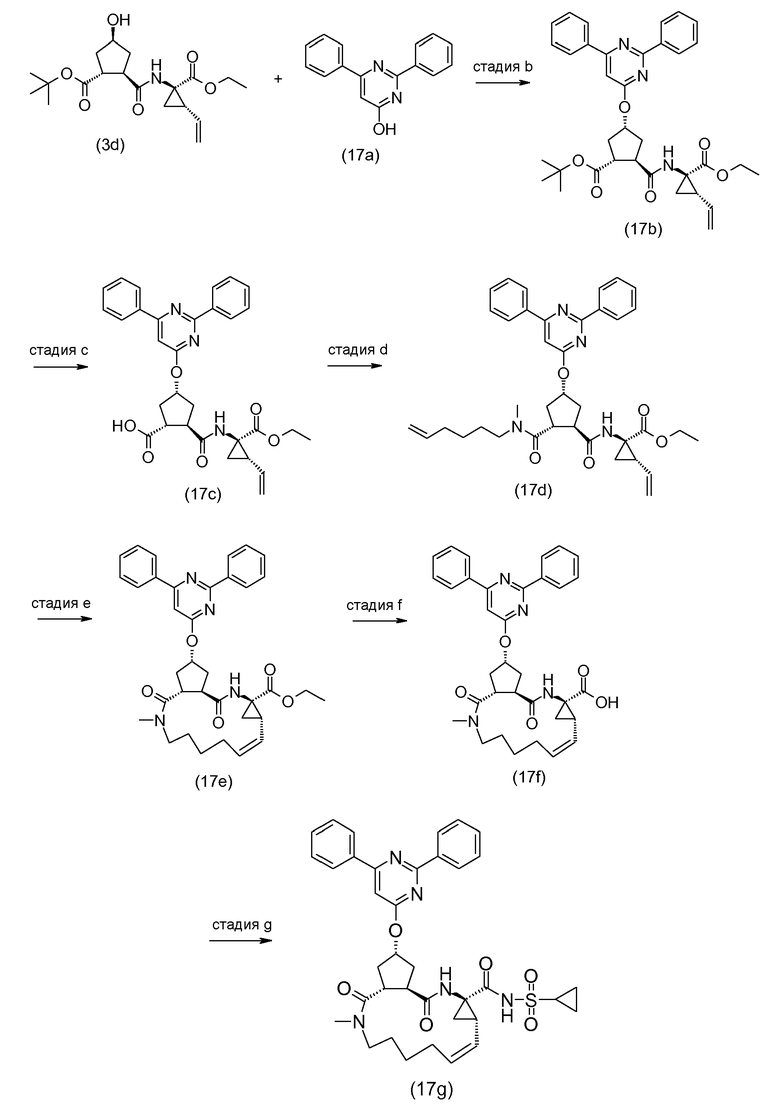
Стадия a: 2,6-Дифенил-пиримидин-4-ол (17a)
К охлаждаемому на льду раствору гидроксида натрия (2,4 г, 60 ммоль) в воде (30 мл) добавляют бензамидин гидрохлорид гидрат (9,4 г, 60 ммоль) и этилбензоилацетат (12,1 г, 63 ммоль). Добавляют этанол (примерно 30 мл), и смесь перемешивают при комнатной температуре в течение ночи. Твердый продукт отфильтровывают, промывают водой и простым диэтиловым эфиром и сушат, что дает указанное в заголовке соединение (9,0 г, 60%), (M+H)+ 1 249.
Стадия b: Сложный трет-бутиловый эфир 4-(2,6-дифенил-пиримидин-4-илокси)-2-(1-этоксикарбонил-2-винил-циклопропил-карбамоил)циклопентанкарбоновой кислоты (17b)
Спирт 3d (0,74 г, 2 ммоль) взаимодействует с 2,6-дифенилпиримидин-4-олом (0,62 г, 2,5 ммоль) в присутствии трифенилфосфина (1,31 г, 5 ммоль) и DIAD (1,0 г, 5 ммоль) в сухом ТГФ (50 мл) в соответствии с процедурой, описанной в Примере 13, стадия a. Очистка с помощью колоночной хроматографии на силикагеле с элюированием этилацетатом-гексаном дает указанное в заголовке соединение (1,2 г, 100%), (M+H)+ 598.
Стадия c: 4-(2,6-Дифенил-пиримидин-4-илокси)-2-(1-этоксикарбонил-2-винил-циклопропилкарбамоил)-циклопентанкарбоновая кислота (17c)
Соединение 17b (1,2 г, 2 ммоль) обрабатывают триэтилсиланом (0,58 г, 5,0 ммоль) и TFA (25 мл) в соответствии с процедурой, описанной в Примере 13, стадия b, что дает сырое указанное в заголовке соединение, (1,2 г), (M+H)+ 542.
Стадия d: Сложный этиловый эфир 1-{[4-(2,6-дифенил-пиримидин-4-илокси)-2-(гекс-5-енил-метил-карбамоил)-циклопентанкарбонил]амино}-2-винил-циклопропанкарбоновой кислоты (17d)
Кислота 17c (1,2 г, 2 ммоль) взаимодействует с N-метилгексенамином (340 мг, 3,0 ммоль) DTEA (1,29 г, 10,0 ммоль) и HATU (1,1 г, 3 ммоль) в сухом ДМФ (30 мл) в соответствии с процедурой, описанной в Примере 13, стадия c. Очистка с помощью колоночной хроматографии на силикагеле дает указанное в заголовке соединение (1,2 г, 93%), (M+H)+ 637.
Стадия e: Сложный этиловый эфир 17-(2,6-дифенил-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]-октадек-7-ен-4-карбоновой кислоты (17e)
Диолефин 17d (0,95 г, 1,49 ммоль) взаимодействует с катализатором Ховейды-Граббса, 2-ое поколение (135 мг), в DCE (900 мл) в соответствии с процедурой, описанной в Примере 13, стадия d, что дает указанное в заголовке соединение, (0,69 г, 76%), (M+H)+ 609.
Стадия f: 17-(2,6-Дифенил-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновая кислота (17f)
Раствор сложного этилового эфира 17e (0,67 г, 1,1 ммоль) в ТГФ/MeOH 1:1 (30 мл) обрабатывают 1M раствором LiOH (15 мл), как описано в Примере 13, стадия d, что дает указанное в заголовке соединение, (0,49 г, 76%), (M+H)+ 581.
Стадия g: [17-(2,6-дифенил-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (17g)
Кислота 17f (240 мг, 0,41 ммоль) взаимодействует с амидом циклопропансульфоновой кислоты (63 мг, 0,52 ммоль), EDAC (105 мг, 0,55 ммоль) и DBU (182 мг, 1,2 ммоль) в сухом DCM в соответствии с процедурой, описанной в Примере 13, стадия f. Очистка с помощью колоночной хроматографии на силикагеле, с элюированием простым диэтиловым эфиром - этилацетатом, дает указанное в заголовке соединение, (175 мг, 62%), (M+H)+ 684.
Пример 18
Стадия a: Синтез 4-хлор-2,6-диметоксипиримидина (18a)
2,4,6-Трихлорпиримидин (5,5 г, 30 ммоль) добавляют за один раз к охлаждаемому на льду раствору метоксида натрия (3,3 г, 60 ммоль) в метаноле (120 мл). Смесь перемешивают в течение одного часа примерно при 5°C и в течение двух часов при комнатной температуре. Растворитель удаляют при пониженном давлении, добавляют воду, и продукт экстрагируют три раза этилацетатом. Объединенные органические фазы промывают водой, сушат над сульфатом натрия и выпаривают, что дает указанное в заголовке соединение (5,0 г, 95%), чистота примерно 85%.
Стадия b: 2,6-Диметоксипиримидин-4-ол (18b)
Суспензию 4-хлор-2,6-диметокси-пиримидина (4,9 г, 28 ммоль), DABCO (6,4 г, 57 ммоль) и карбоната калия (13,8 г, 100 ммоль) нагревают с обратным холодильником в воде (150 мл) в течение одного часа. Смесь охлаждают, подкисляют 5% лимонной кислотой и экстрагируют три раза этилацетатом и три раза DCM вместе примерно с 105 ТГФ и 10% MeOH. Объединенные органические фазы сушат и выпаривают, что дает указанное в заголовке соединение (1,5 г, 34%), (M+H)+ 1 157.
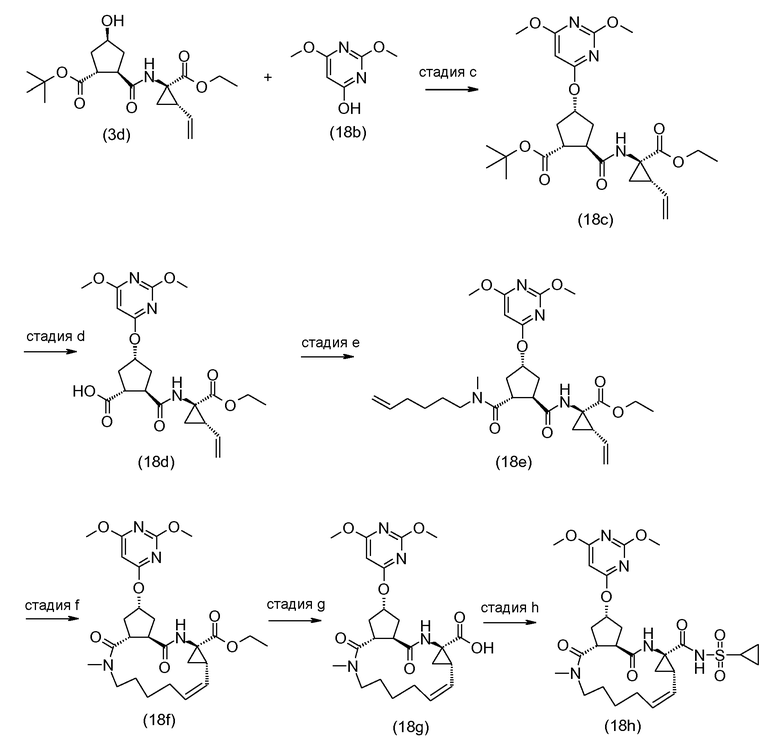
Стадия c: Сложный трет-бутиловый эфир 4-(2,6-диметокси-пиримидин-4-илокси)-2-(1-этоксикарбонил-2-винилцикло-пропилкарбамоил)циклопентанкарбоновой кислоты (18c)
Спирт 3d (0,74 г, 2 ммоль) взаимодействует с 2,6-диметоксипиримидин-4-олом (0,47 г, 3,0 ммоль) в присутствии трифенилфосфина (1,31 г, 5 ммоль) и DIAD (1,0 г, 5 ммоль) в сухом ТГФ (50 мл) в соответствии с процедурой, описанной в Примере 13, стадия a. Очистка с помощью колоночной хроматографии на силикагеле, с элюированием этилацетатом-гексаном дает указанное в заголовке соединение (0,93 г, 93%), (M+H)+ 506.
Стадия d: 4-(2,6-Диметокси-пиримидин-4-илокси)-2-(1-этоксикарбонил-2-винилцикло-пропилкарбамоил)-циклопентанкарбоновая кислота (18d)
Соединение 18c (0,92 г, 1,83 ммоль) обрабатывают триэтилсиланом (0,58 г, 5,0 ммоль) и TFA (25 мл) в DCM (25 мл) в соответствии с процедурой, описанной в Примере 13, стадия b, что дает указанное в заголовке соединение, (0,83 г), (M+H)+ 450.
Стадия c: Сложный этиловый эфир 1-{[4-(2,6-Диметокси-пиримидин-4-илокси)-2-(гекс-5-енил-метил-карбамоил)-циклопентанкарбонил]амино}-2-винил-циклопропанкарбоновой кислоты (18e)
Кислота 18d (1,2 г, 2 ммоль) взаимодействует с N-метилгексенамином (0,34 г, 3,0 ммоль), DIEA (1,29 г, 10,0 ммоль) и HATU (1,1 г, 3 ммоль) в сухом ДМФ (30 мл) в соответствии с процедурой, описанной в Примере 13, стадия c. Очистка с помощью колоночной хроматографии на силикагеле дает указанное в заголовке соединение (0,74 г, 74%), (M+H)+ 545.
Стадия f: Сложный этиловый эфир 17-(2,6-диметоксипиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло-[13.3.0.0*4.6*]октадек-7-ен-4-карбоновой кислоты (18f)
Диолефин 18e (0,73 г, 1,34 ммоль) взаимодействует с катализатором Ховейды-Граббса, 2-е поколение (100 мг), в DCE (700 мл) в соответствии с процедурой, описанной в Примере 13, стадия d. Очистка с помощью колоночной хроматографии на силикагеле дает указанное в заголовке соединение, (0,34 г, 49%), (M+H)+ 517.
Стадия g: 17-(2,6-Диметокси-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновая кислота (18g)
Раствор сложного этилового эфира 18f (0,33 г, 0,64 ммоль) в ТГФ/MeOH 1:1 (20 мл) обрабатывают 1M раствором LiOH (10 мл), как описано в Примере 13, стадия d. Очистка с помощью колоночной хроматографии на силикагеле, с элюированием DCM-MeOH, дает указанное в заголовке соединение, (0,31 г, 99%), (M+H)+ 489.
Стадия h: [17-(2,6-Диметокси-пиримидин-4-илокси)-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил]амид циклопропансульфоновой кислоты (18h)
Кислота 18g (1,2 г, 2 ммоль) взаимодействует с амидом циклопропансульфоновой кислоты (73 мг, 0,6 ммоль), EDAC (115 мг, 0,6 ммоль) и DBU (215 мг, 1,4 ммоль) в сухом DCM (3 мл) в соответствии с процедурой, описанной в Примере 13, стадия f. Очистка с помощью колоночной хроматографии на силикагеле, с элюированием простым диэтиловым эфиром - этилацетатом дает указанное в заголовке соединение, (185 мг, 70%), (M+H)+ 592.
Пример 19
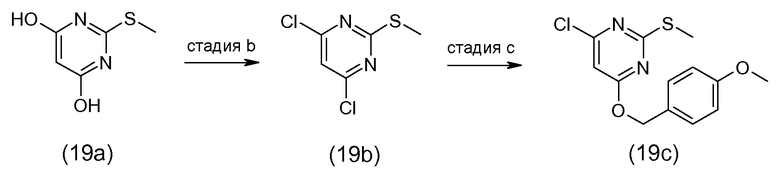
Стадия a: 2-Метилсульфанил-пиримидин-4,6-диол (19a)
Метилйодид (32,64 г, 220 ммоль) добавляют по каплям к суспензии тиобарбитуровой кислоты (29 г, 200 ммоль) в EtOH (300 мл) и в растворе 2M NaOH в воде (110 мл) при комнатной температуре. Смесь перемешивают при комнатной температуре в течение ночи, а затем перемешивают в течение двух часов при 60°C. Этанол удаляют, добавляют воду, и смеси позволяют стоять в течение 2 часов на ледяной бане. Твердый продукт указанного в заголовке соединения отфильтровывают, промывают водой со льдом и сушат (30 г, 95%).
Стадия b: 4,6-Дихлор-2-метилсульфанилпиримидин (19b)
2-Метилсульфанилпиримидин-4,6-диол (30,0 г, 189 ммоль) медленно добавляют к оксихлориду фосфора (350 мл), в то же время, охлаждая на льду, затем добавляют N,N-диэтиланилин (52,5 мл), в то же время, медленно охлаждая. Смесь медленно нагревают до появления флегмы и нагревают с обратным холодильником в течение 2,5 часов. Смесь выпаривают и добавляют к колотому льду. Смесь экстрагируют три раза этилацетатом, и объединенные органические слои промывают три раза водой, один раз насыщенным раствором соли и концентрируют. Очистка с помощью колоночной хроматографии на силикагеле, с элюированием гексаном-этилацетатом дает указанное в заголовке соединение (36 г, 97%).
Стадия c: 4-Хлор-6-(4-метоксибензилокси)-2-метилсульфанил-пиримидин (19c)
К раствору 4-метоксибензилового спирта (8,9 г, 65 ммоль) в сухом ДМФ (100 мл) порциями добавляют 60% суспензия гидрида натрия (2,2 г, 55 ммоль), и смесь перемешивают в течение двух часов при комнатной температуре. Смесь охлаждают на ледяной бане, и добавляют 4,6-дихлор-2-метилсульфанилпиримидин (9,8 г, 50 ммоль) в сухом ТГФ (20 мл). Затем смесь перемешивают в течение 72 час при комнатной температуре. Добавляют воду, и смесь дважды экстрагируют этилацетатом. Органическую фазу промывают водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Остаток очищают с помощью хроматографии на силикагеле, элюируя гексаном-этилацетатом, что дает указанное в заголовке соединение (9,1 г, 61%), (M+H)+ 296.
1H-ЯМР CDCl3 δ 2,59 (с, 3H), 3,80 (с, 3H), 5,38 (с, 2H), 6,40 (с, 1H) 6,90 (д, 2H), 7,38 (дд, 2H).
Пример 20
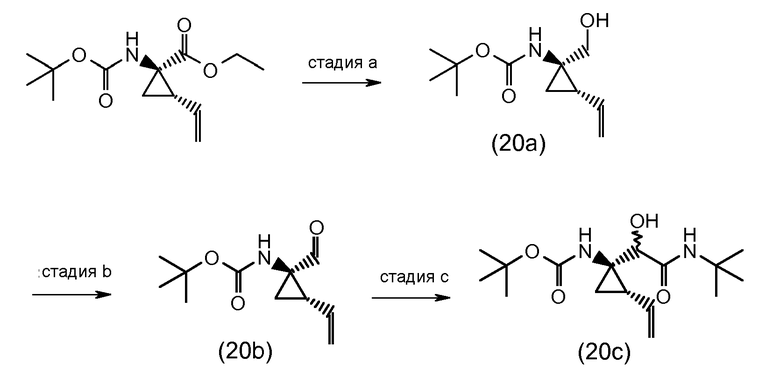
Стадия a: Сложный трет-бутиловый эфир (1-гидроксиметил-2-винилциклопропил)карбаминовой кислоты (20a)
К раствору сложного этилового эфира 1-трет-бутоксикарбониламино-2-винил-циклопропанкарбоновой кислоты (0,51 г, 2,0 ммоль) в ТГФ (10 мл) при 0°C добавляют 2M раствор боргидрида лития (4 мл, 8 ммоль). Реакционную смесь отслеживают с помощью ТСХ (7:3, гексан-этилацетат, окрашивают с использованием молибдата аммония - сульфата церия в водном растворе 10% серной кислоты), и после перемешивания в течение ночи при комнатной температуре реакцию осторожно гасят с использованием водного раствора 10% лимонной кислоты (25 мл, добавление по каплям при 0°C). Полученную смесь промывают дихлорметаном (3×10 мл), и объединенные органические слои сушат (Na2SO4), фильтруют и концентрируют. Флэш-хроматография остатка с использованием гексана-этилацетата 1:1 в качестве элюента, с последующим концентрированием соответствующих фракций и сушкой остатка в вакууме в течение ночи, дает продукт в виде бесцветного сиропа (0,407 г, 1,91 ммоль, 96%).
Данные ЯМР (400 МГц, CDCl3): 1H, δ 0,98 (м, 1H), 1,15 (м, 1H), 1,44 (с, 9H), 1,84 (м, 1H), 3,20 (ушир.с, 1H), 3,60 (дд, 1H), 3,78 (ушир.м, 1H), 5,10-5,26 (м, 3H), 5,70 (м, 1H).
Стадия b: Сложный трет-бутиловый эфир (1-формил-2-винил-циклопропил)карбаминовой кислоты (20b)
К перемешиваемому раствору спирта 20a (0,152 г, 0,71 ммоль) в дихлорметане (5 мл) добавляют периодинан Десса-Мартина (0,33 г, 0,78 ммоль) при комнатной температуре. Реакцию отслеживают с помощью ТСХ (гексан-этилацетат 3:2, УФ-мониторинг и окрашивание с использованием молибдата аммония - сульфата церия в водном растворе 10% серной кислоты). Окрашивание показывает довольно чистую реакционную смесь, но УФ-мониторинг показывает несколько побочных продуктов. Через 1 час, полученный желто-красный раствор разбавляют дихлорметаном (20 мл), затем промывают водным раствором 10% тиосульфата натрия/водным насыщенным раствором гидрокарбоната натрия (3×20 мл), 1:1, затем сушат (Na2SO4), фильтруют и концентрируют. Флэш-хроматография остатка с использованием элюирования в ступенчатом градиенте (этилацетат в гексане 20-30%) с последующим концентрированием соответствующих фракций и сушкой остатка в вакууме в течение ночи дает указанное в заголовке соединение в виде бесцветного масла (0,054 г, 0,255 ммоль, 36%).
Стадия c: Сложный трет-бутиловый эфир [1-(трет-бутилкарбамоил-гидрокси-метил)-2-винил-циклопропил]-карбаминовой кислоты (20c)
К раствору альдегида 20b (0,054 г, 0,255 ммоль) и трет-бутилизонитрила (0,043 мл, 0,38 ммоль) в дихлорметане (1 мл) и пиридине (0,083 мл, 1,02 ммоль) в атмосфере азота добавляют трифторуксусную кислоту (0,039 мл, 0,51 ммоль). Через 30 мин при комнатной температуре, реакционной смеси позволяют достичь комнатной температуры и перемешивают в течение следующих 2 дней. Затем ТСХ (7:3, гексан-этилацетат) и ЖХ-МС мониторинг показывает приблизительно 60% преобразование, и реакционную смесь разбавляют этилацетатом (10 мл). Раствор промывают последовательно водным раствором 10% лимонной кислоты (3×5 мл) и водным насыщенным раствором гидрокарбоната натрия (3×5 мл)), затем сушат (Na2SO4), фильтруют и концентрируют. Затем остаток обрабатывают 1M LiOH/ТГФ/MeOH (1,5 мл), водн., 1:1:1, в течение 10 мин при комнатной температуре, затем разбавляют водным раствором 10% лимонной кислоты и извлекают в этилацетате, затем сушат (Na2SO4), фильтруют и концентрируют. Колоночная хроматография остатка с использованием гексана-этилацетата, 7:3, в качестве элюента, с последующим концентрированием соответствующих фракций и сушкой остатка в вакууме в течение ночи, дает продукт в виде бесцветного твердого продукта (0,027 г, 0,086 ммоль, 34%).
ЯМР (400 МГц, CDCl3): 1H, δ 1,24 (м, 1H), 1,33-1,40 (м, 10 H), 1,44 (с, 9H), 1,87 (м, 1H), 3,65 (д, 1H), 5,21 (м, 3H), 5,50 (д, 1H), 5,89 (м, 1H), 7,03 (ушир.с, 1H).
α-Гидроксиамидные производные ингибиторов по настоящему изобретению получают затем посредством удаления группы N-boc из указанного в заголовке соединения, с последующим связыванием полученного амина с кислотой, такой как кислота 1i, в соответствии с процедурой, описанной в Примере 1, стадия j.
Общая процедура оксиления α-гидроксиамидов до α-кетоамидов:
Как правило, α-гидроксиамид растворяют в дихлорметане (20-30 мл/г) при комнатной температуре, затем добавляют периодинан Десса-Мартина (1,1 эквивалента), и реакционную смесь отслеживают с помощью ТСХ и ЖХ-МС. После завершения или примерного завершения реакции, реакционную смесь разбавляют дихлорметаном, а затем промывают водным раствором 10% тиосульфата натрия/водным насыщенным раствором гидрокарбоната натрия (3 раза), 1:1, затем сушат (Na2SO4), фильтруют и концентрируют. Остаток очищают с помощью колоночной хроматографии или препаративной ЖХ.
Пример 21
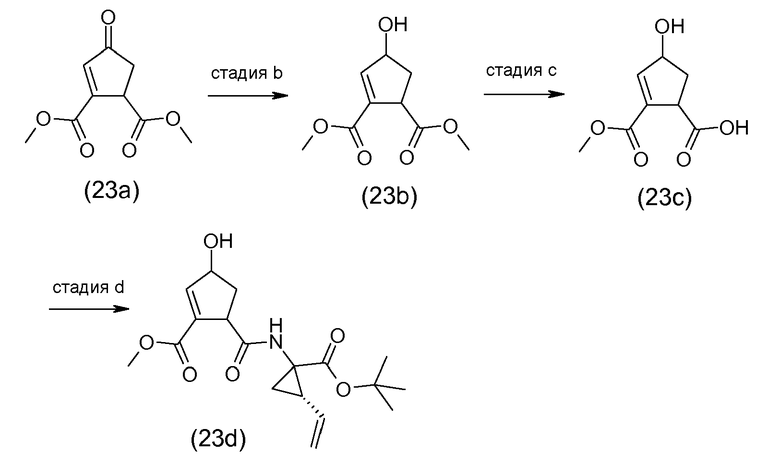
Стадия a: Сложный диметиловый эфир 4-оксоциклопент-2-ен-1,2-дикарбоновой кислоты (23a)
Сложный диметиловый эфир (1R,2S)-4-оксо-циклопентан-l,2-дикарбоновой кислоты (4,8 г, 23,8 ммоль) и CuBr2 (11,9 г, 53,2 ммоль) растворяют в сухом ТГФ (70 мл) и смесь нагревают с обратным холодильником в течение двух часов при 90°C. Образовавшийся CuBr отфильтровывают, и органическую фазу концентрируют. Добавляют CaCO3 (2,7 г, 27,2 ммоль) и ДМФ (70 мл), и смесь выдерживают при 100°C в течение одного часа. Темно-коричневую смесь выливают на лед (35 г), и образовавшийся преципитат отфильтровывают. Водный слой экстрагируют этилацетатом (1×300 мл + 3×150 мл). Органические фазы сушат, фильтруют и концентрируют. Очистка с помощью флэш-хроматографии (толуол/EtOAc 9:1) дает указанное в заголовке соединение (2,1 г, 45%) в виде желтых кристаллов.
Стадия b: Сложный диметиловый эфир 4-гидрокси-циклопент-2-ен-1,2-дикарбоновой кислоты (23b)
NaBH4 (0,66 г, 17,5 ммоль), растворенный в MeOH (23 мл), добавляют к холодному раствору (-30°C) кетона 23a (3,18 г, 16,1 ммоль). Через девять минут избыток NaBH4 разрушают посредством добавления насыщенного солевого раствора (80 мл). Смесь концентрируют и экстрагируют этилацетатом (4×80 мл). Органические фазы сушат, фильтруют и концентрируют, что дает указанное в заголовке соединение (3,0 г, 92%) в виде желтого масла.
Стадия c: Сложный 2-метиловый эфир 4-гидрокси-циклопент-2-ен-1,2-дикарбоновой кислоты (23c)
LiOH (0,52 г, 22 ммоль) добавляют к охлажденному на льду раствору спирта 23b (3,4 г, 22 ммоль), растворенному в диоксане и воде (1:1, 110 мл). Через два с половиной часа смесь совместно выпаривают с толуолом и метанолом. Очистка с помощью флэш-хроматографии (толуол/этилацетат, 3:1 + 1% HOAc) дает указанное в заголовке соединение (1,0 г, 27%) в виде желто-белых кристаллов.
1H-ЯМР (300 МГц, CD3OD): δ 1,78-1,89 (м, 1H), 2,70-2,84 (м, 1H), 3,56-3,71 (м, 1H), 3,76 (с, 3H), 4,81-4,90 (м, 1H), 6,76-6,81 (м, 1H); 13C-ЯМР (75,5 МГц, CDCl3): δ 38,0, 48,0, 52,4, 75,7, 137,0, 146,2, 165,0, 178,4.
Стадия d: Сложный метиловый эфир 5-(1-трет-бутоксикарбонил-2-винил-циклопропилкарбамоил)-3-гидрокси-циклопент-1-енкарбоновой кислоты (23d)
Взаимодействие соединения кислоты 23c (50 мг, 37 ммоль) со сложным трет-бутиловым эфиром (1R,2S)-1-амино-2-винил-циклопропанкарбоновой кислоты в соответствии со способом, описанным в Примере 1, стадия j, дает указанное в заголовке соединение в виде чуть желтоватого масла (50 мг, 38%). 1H-ЯМР (300 МГц, CDCl3): δ [(1,38 и 1,42) с, 9H], 1,75-1,83 (м, 1H), 2,00-2,21 (м, 3H), 3,55-3,63 (м, 1H), [(3,77 и 3,82) с, 3H], 4,20-4,38 (м, 1H), 4,65-4,80 (м, 1H), 5,13-5,20 (м, 1H), 5,22-5,38 (м, 1H), 5,60-5,82 (м, 1H), 6,95-6,96 (м, 2H).
Ингибиторы по настоящему изобретению получают из указанного в заголовке соединения посредством связывания полученного циклопентенольного производного с желаемым пиримидинолом, например, как описано в Примере 3, стадия f, с последующим гидролизом сложного метилового эфира с использованием реагента, подобного LiOH; связывания желаемого алкениламина и макроциклизации, как описано в Примере 3, стадия h и i; гидролиза сложного трет-бутилового эфира посредством обработки, например, TFA, и наконец, связывания желаемого сульфонамидного производного, например, как описано в Примере 3, стадия k.
Пример 22
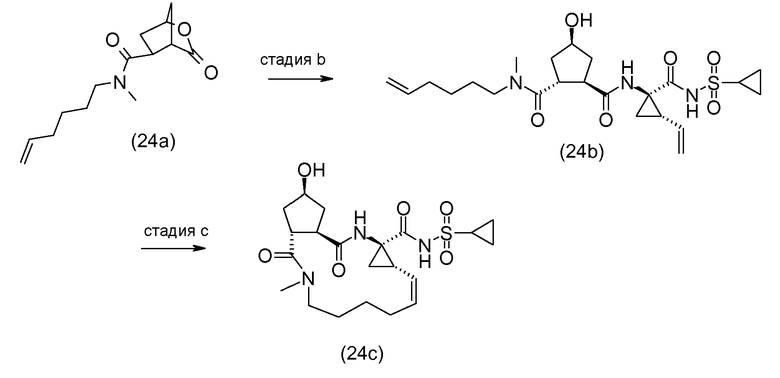
Стадия a: Гекс-5-енил-метиламид 3-оксо-2-окса-бицикло[2,2,1]гептан-5-карбоновой кислоты (24a)
К HATU (2,17 г, 5,7 ммоль) и N-метилгекс-5-ениламин гидрохлориду (6,47 ммоль) в 5 мл ДМФ, в атмосфере аргона на ледяной бане, добавляют 1R,4R,5R-3-оксо-2-окса-бицикло[2,2,1]гептан-5-карбоновую кислоту (835,6 мг, 5,35 ммоль) в 11 мл ДМФ, а затем DIEA (2,80 мл, 16 ммоль). После перемешивания в течение 40 мин, смесь перемешивают при комнатной температуре в течение 5 час. Растворитель выпаривают, остаток растворяют в EtOAc (70 мл) и промывают насыщенным раствором NaHCO3 (10 мл). Водную фазу экстрагируют EtOAc (2×25 мл). Органические фазы объединяют, промывают насыщенным раствором NaCl (20 мл), сушат над Na2SO4 и выпаривают. Колоночная флэш-хроматография (150 г силикагеля, 2/1, EtOAc - петролейный эфир (PE), детектирование ТСХ с помощью водного раствора KMnO4, Rf 0,55 в 4/1 EtOAc - PE) дает указанное в заголовке соединение в виде желтого масла (1,01 г, 75%).
Стадия b: N-(1-Циклопропансульфониламинокарбонил-2-винил-циклопропил)-2-(гекс-5-енил-метил-амино-карбонил)-4-гидрокси-циклопентан-карбоксамид (24b)
Раствор LiOH (0,15M, 53 мл, 8 ммоль) добавляют к лактонамиду 24a (996 мг, 3,96 ммоль) на ледяной бане и перемешивают в течение 1 час. Смесь подкисляют до pH 2 - 3 с помощью 1 н. HCl и выпаривают, совместно выпаривают с толуолом несколько раз и сушат в вакууме в течение ночи. Добавляют гидрохлорид (1-амино-2-винил-циклопропанкарбонил)амида (1R,2S)-циклопропансульфоновой кислоты (4,21 ммоль) и HATU (1,78 г, 4,68 ммоль). Смесь охлаждают на ледяной бане в атмосфере аргона, добавляют ДМФ (25 мл), а затем DIEA (2,0 мл, 11,5 ммоль). После перемешивания в течение 30 мин смесь перемешивают при комнатной температуре в течение 3 час. После выпаривания растворителя остаток растворяют в EtOAc (120 мл), промывают последовательно 0,5 н. HCl (20 мл) и насыщенным раствором NaCl (2×20 мл) и сушат над Na2SO4. Колоночная флэш-хроматография (200 г силикагеля YMC, 2-4% MeOH в CH2Cl2) дает белый твердый продукт (1,25 г, 66%).
Стадия c: (17-Гидрокси-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил)амид циклопропансульфоновой кислоты (24c)
Циклопентанол 24b (52,0 мг, 0,108 ммоль) растворяют в 19 мл 1,2-дихлор-этана (барботируют аргоном перед использованием). Катализатор Ховейды-Граббса 2-е поколение (6,62 мг, 10% моль) растворяют в DCE (2×0,5 мл) и добавляют. Зеленый раствор барботируют Ar в течение 1 мин. Аликвоты (4 мл, каждая) переносят в пять 2-5 - мл пробирок для микроволновой печи. В последнюю пробирку добавляют 0,8 мл промывания с растворителем. Каждую пробирку нагревают с помощью микроволновой печи (от комнатной температуры до 160°C через 5 мин). Все аликвоты объединяют, и растворитель выпаривают. Колоночная флэш-хроматография (силикагель, 3 → 7% MeOH в CH2Cl2) дает 24,39 мг твердых продуктов (Rf 0,28 в 10% MeOH-CH2Cl2 с двумя фракциями). Твердые продукты объединяют с 9,66-мг образцом и подвергают вторично хроматографии (2 → 8% MeOH в EtOAc) с получением кремообразных твердых продуктов (23 мг) с 80% желаемого соединения (выход 26%).
Ингибиторы по настоящему изобретению получают из указанного в заголовке соединения посредством связывания полученного циклопентенольного производного с желаемым пиримидинолом, например, как описано в Пример 1, стадия h.
Пример 23
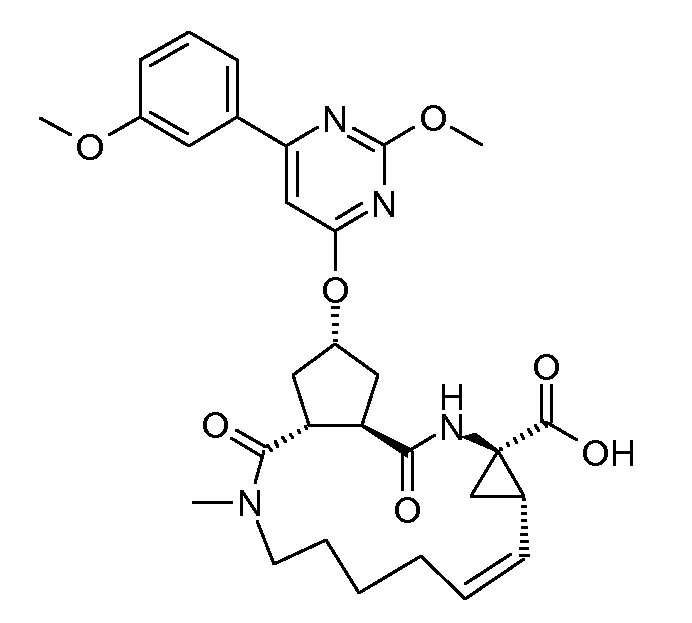
17-[2-Метокси-6-(3-метоксифенил)пиримидин-4-илокси]-13-метил-2,14-диоксо-3,13-диазатрицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбоновая кислота (25)
Следуют процедуре, описанной в примере 16, стадии a-f, но используют этил 3-метоксибензоилацетат (9 г, 40 ммоль) вместо этил 4-метоксибензоилацетата со стадии a, что дает указанное в заголовке соединение (305 мг), MS.
Пример 24
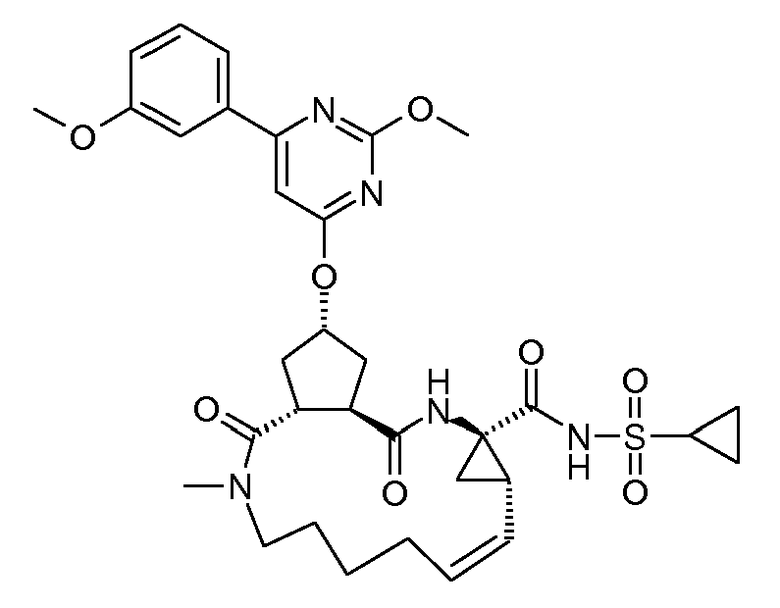
{17-[2-Метокси-6-(3-метокси-фенил)пиримидин-4-илокси]-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил}амид циклопропансульфоновой кислоты (26)
Кислота 25 (150 мг, 0,26 ммоль) взаимодействует с амидом циклопропансульфоновой кислоты (64 мг, 0,53 ммоль) в соответствии с процедурой, описанной в Примере 16, стадия g, что дает указанное в заголовке соединение (110 мг, 62%), MS [M+1] 668.
Пример 25
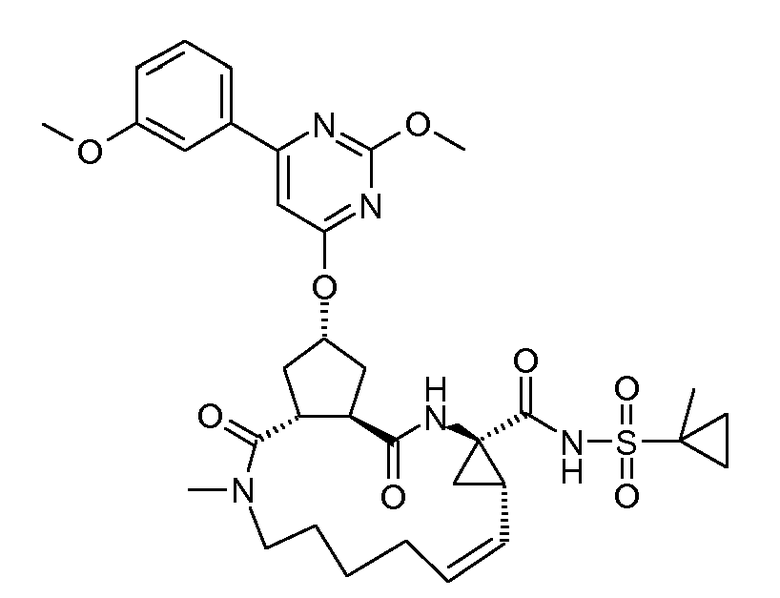
{17-[2-Метокси-6-(3-метоксифенил)пиримидин-4-илокси]-13-метил-2,14-диоксо-3,13-диаза-трицикло[13.3.0.0*4.6*]октадек-7-ен-4-карбонил}амид 1-метил-циклопропансульфоновой кислоты (27)
Кислота 25 (150 мг, 0,26 ммоль) взаимодействует с амидом метилциклопропансульфоновой кислоты (72 мг, 0,53 ммоль) в соответствии с процедурой, описанной в Примере 16, стадия g, что дает указанное в заголовке соединение (90 мг, 50%), MS [M+1] 682.
Активность соединений формулы (I)
Анализ с помощью репликонов
Соединения формулы (I) исследуют на активность при ингибировании репликации РНК HCV в клеточном анализе. Анализ демонстрирует, что соединения формулы (I) демонстрируют активность против репликонов HCV, функционирующих в культуре клеток. Клеточный анализ основывается на бицистронном конструкте экспрессии, как описано в Lohmann et al. (1999) Science, vol. 285 pp. 110-113, с модификациями, описанными Krieger et al. (2001) Journal of Virology 75: 4614-4624, с многоцелевой стратегией скрининга. В основном, способ представляет собой следующее.
Анализ использует стабильно трансфицированную линию клеток Huh-7 luc/neo (далее упоминаемую как Huh-Luc). Эта линия клеток содержит РНК, кодирующую бицистронный конструкт экспрессии, содержащий области NS3-NS5B дикого типа HCV типа 1b, транслированные из внутренного сайта входа в рибосому (IRES) от вируса энцефаломиокардита (EMCV), перед которым идет репортерная часть (FfL-люцифераза) и часть селектируемого маркера (neoR, неомицинфосфотрансфераза). Конструкт ограничивается NTR (нетранслируемыми областями) 5' и 3' от HCV типа 1b. Непрерывная культура клеток с репликоном в присутствии G418 (neoR) зависит от репликации РНК из HCV. Стабильно транфицированные клетки с репликонами, которые экспрессируют РНК из HCV, которая реплицируется автономно и при высоких уровнях, кодируя среди прочего люциферазу, используют для скрининга противовирусных соединений.
Клетки с репликонами помещают в 384-луночные планшеты в присутствии исследуемых и контрольных соединений, которые добавляют при различных концентрациях. После трех дней инкубирования, репликацию HCV измеряют посредством анализа активности люциферазы (используя стандартные субстраты и реагенты для анализа люциферазы и устройство для считывания микропланшетов Perkin Elmer ViewLuxTm ultraHTS). Клетки с репликонами в контрольных культурах имеют высокую экспрессию люциферазы в отсутствие любого ингибитора. Ингибиторную активность соединения по отношению к активности люциферазы отслеживают на клетках Huh-Luc, что дает возможность для получения кривых доза-реакция для каждого исследуемого соединения. Затем вычисляют значения EC50, это значение представляет собой количество соединения, необходимое для понижения на 50% уровня детектируемой активности люциферазы, или более конкретно, способности генетически связанного РНК репликона из HCV к реплицированию.
Анализ ингибирования
Цель этого анализа in vitro заключается в измерении ингибирования комплексов протеаза NS3/4A из HCV с помощью соединений по настоящему изобретению. Этот анализ показывает, насколько эффективными были бы соединения по настоящему изобретению при ингибировании протеолитической активности NS3/4A из HCV.
Ингибирование полноразмерного фермента протеазы NS3 гепатита C измеряют в основном, как описано в Poliakov, 2002 Prot Expression @ Purification 25 363 371. Вкратце, гидролиз депсипептидного субстрата Ac-DED(Edans)EEAbuψ[COO]ASK(Dabcyl)-NH2 (AnaSpec, San Jose, USA) измеряется спектрофлюорометрически в присутствии пептидного кофактора, KKGSVVIVGRIVLSGK (Ake Engstrom, Department of Medical Biochemistry and Microbiology, Uppsala University, Sweden). [Landro, 1997 #Biochem 36 9340-9348]. Фермент (1 нМ) инкубируют в 50 мМ HEPES, pH 7,5, 10 мМ DTT, 40% глицерине, 0,1% н-октил-D-глюкозиде, вместе с 25 мкМ кофактора NS4A и ингибитора при 30°C в течение 10 мин, при этом реакцию инициируют посредством добавления 0,5 мкМ субстрата. Ингибиторы растворяют в ДМСО, сонифицируют в течение 30 сек и перемешивают. Растворы хранят при -20°C между измерениями.
Конечная концентрация ДМСО в анализируемом образце доводится до 3,3%. Скорость гидролиза корректируется на внутренние эффекты фильтра в соответствии с опубликованными процедурами [Liu, 1999 Analytical Biochemistry 267 331-335]. Значения Ki оценивают с помощью нелинейного регрессионного анализа (GraFit, Erithacus Software, Staines, MX, UK), с использованием модели конкурентного ингибирования и фиксированного значения для Km (0,15 мкМ). Все измерения повторяют минимум по два раза.
Соединения по настоящему изобретению являются предпочтительно сильнодействующими против вируса дикого типа и мутантного вируса HCV, в частности, против вируса, содержащего мутации, вызывающие появление ускользнувших мутантов. Мутации, вызывающие появление ускользнувших мутантов, представляют собой такие, которые возникают у пациентов из-за селективного давления противовирусного препарата, известного из литературы, и которые придают повышенную резистентность к этому противовирусному препарату.
Ингибирование определенных мутантных HCV, демонстрируемое соединениями по настоящему изобретению, может определяться, как описано в заявке на Международный патент WO2004/039970.
A156T и D168V представляют собой особенно важные ускользнувшие мутанты в контексте терапии HCV с использованием ингибиторов протеазы NS3, и соединения по настоящему изобретению предпочтительно имеют низкие значения Ki против этих мутантов.
Следующая далее таблица перечисляет репрезентативные соединения, которые получены в соответствии с приведенными выше Примерами. Активности исследуемых соединений также изображены в таблице. Описание значений A, B, C, D, E и F является следующим:
- значение соответствует EC50 > 10 мкМ;
- значение B соответствует EC50 в пределах между 10 мкМ и 1 мкМ;
- значение C соответствует EC50 в пределах между 0,99 мкМ и 200 нМ;
- значение D соответствует EC50 в пределах между 199 нМ и 0,5 нМ.
- значение E соответствует Ki > 1 мкМ;
- значение F соответствует Ki в пределах между 1 мкМ и 100 нМ;
- значение G соответствует Ki в пределах между 99,9 нМ и 5 нМ;
- значение H соответствует Ki в пределах между 4,9 нМ и 0,1 нМ.
анализ с помощью репликонов
ферментативный анализ
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОЦИКЛИЧЕСКИЕ ФЕНИЛКАРБАМАТЫ, ИНГИБИРУЮЩИЕ HCV | 2008 |
|
RU2490261C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2437886C2 |
| ПРОИЗВОДНЫЕ ТИОКСАНТИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МИЕЛОПЕРОКСИДАЗЫ | 2003 |
|
RU2323219C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛО[3,2-d]ПИРИМИДИН-4-ОНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2005 |
|
RU2577858C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2419619C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ТИЕНО[3,2-b]ПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ IRAK-4 | 2011 |
|
RU2604062C2 |
| 1-(2-ИЗОПРОПОКСИЭТИЛ)-2-ТИОКСО-1,2,3,5-ТЕТРАГИДРО-ПИРРОЛО[3,2-d]ПИРИМИДИН-4-ОН И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 2005 |
|
RU2409578C2 |
| ИМИДАЗОЛО-5-ИЛ-2-АНИЛИНОПИРИМИДИНЫ КАК АГЕНТЫ ДЛЯ ИНГИБИРОВАНИЯ ПРОЛИФЕРАЦИИ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОДУЦИРОВАНИЯ | 2001 |
|
RU2284327C2 |
| СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ СНИЖЕНИЯ АКТИВНОСТИ ГОРМОН-ЧУВСТВИТЕЛЬНОЙ ЛИПАЗЫ | 2002 |
|
RU2317981C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ИНГИБИРОВАНИЯ ТИРОЗИНКИНАЗНОЙ АКТИВНОСТИ | 2011 |
|
RU2598852C2 |
Описываются новые пиримидин-замещенные макроциклические соединения общей формулы (I) 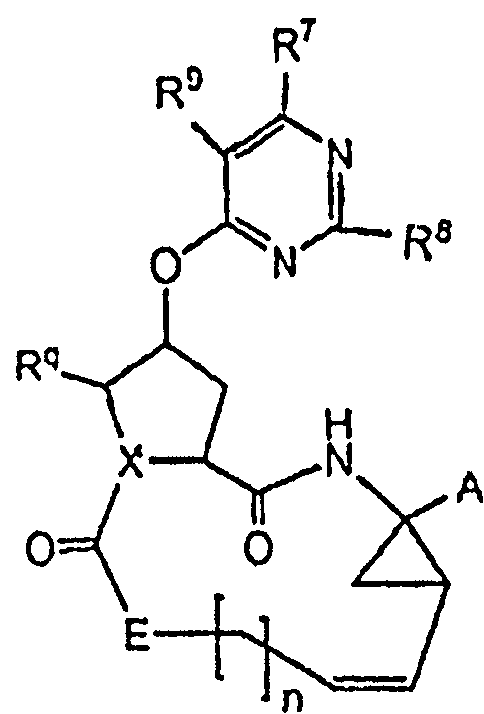 где
где
А=-C(=O)OR1 или -C(=O)-NH-SO2-R2; R1=Н или С1-6алкил; R2 = фенил, тиенил, С3-7циклоалкил, возможно замещенный С1-6алкилом; Х=N или СН; Е=NR5; R5=Н или С1-6алкил; n=4 или 5; R7=H, C1-6алкил, С1-6алкокси, фенил, возможно замещенный С1-6алкокси; R8=С1-6алкокси, фенил, возможно замещенный С1-6алкокси, морфолино или -NRaRb, где Ra и Rb независимо означают Н или C1-6алкил; R9=Rq=Н; или его фармацевтически приемлемые аддитивные соли, или стереоизомеры, и фармацевтические композиции, их содержащие. Данные соединения являются ингибиторами по отношению к серин-протеазе NS3 из HCV и могут найти применение при лечении хронических заболеваний печени, в частности хронического гепатита. 4 н. и 6 з.п. ф-лы, 1 табл., 25 прим.
1. Соединение формулы I:
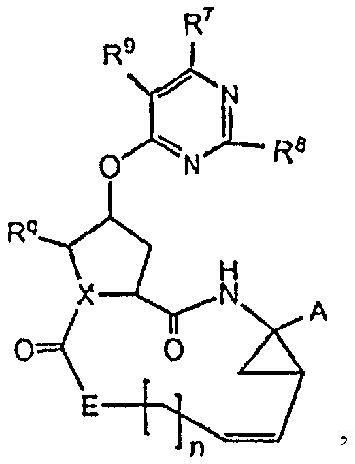
включая его стереоизомер , где
А представляет собой -C(=O)OR1, -C(=O)-NH-SO2-R2, где
R1 представляет собой водород или С1-6алкил;
R2 представляет собой фенил, тиенил, С3-7циклоалкил, необязательно замещенный С1-6алкилом;
Х представляет собой N, СН;
Rq представляет собой водород;
Е представляет собой NR5;
R5 представляет собой водород, С1-6алкил;
n равно 4 или 5;
R7 представляет собой водород, C1-6алкил, С1-6алкокси, фенил, возможно замещенный С1-6алкокси;
R8 представляет собой С1-6алкокси, фенил, возможно замещенный С1-6алкокси, морфолино или -NRaRb, где Ra и Rb независимо представляют собой Н или C1-6алкил;
R9 представляет собой водород;
или его фармацевтически приемлемая аддитивная соль.
2. Соединение по п.1, в котором Х представляет собой N.
3. Соединение по п.1, в котором пиримидинильный радикал (а) или (b) представляет собой:
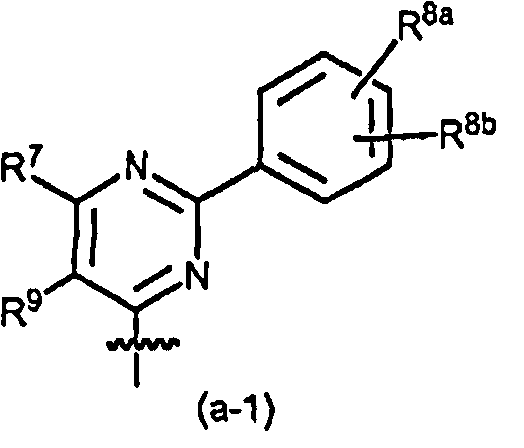
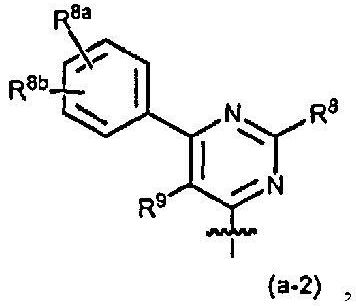
где R7 и R8 независимо представляют собой фенил или С1-6алкокси, например, метокси;
R9 представляет собой водород; R8a или R8b представляет собой водород, С1-6алкокси, например, метокси.
4. Соединение по п.1, в котором А представляет собой -C(=O)-NH-SO2R2, в частности, где R2 представляет собой С3-7циклоалкил или фенил, где С3-7циклоалкил необязательно замещен С1-6алкилом.
5. Соединение по п.1, в котором А представляет собой C(=O)OR1, где R1 представляет собой водород или C1-С6алкил, такой как метил.
6. Фармацевтическая композиция, обладающая HCV ингибирующей активностью, содержащая эффективное количество соединения по любому из пп.1-5 в качестве активного ингредиента и носитель.
7. Соединение по любому из пп.1-5 для применения в качестве лекарственного средства.
8. Применение соединения по любому из пп.1-5 для получения фармацевтической композиции для ингибирования репликации HCV.
9. Комбинация, обладающая HCV ингибирующей активностью, содержащая соединение по любому из пп.1-5 и другое противовирусное соединение.
10. Комбинация по п.9, в которой другое противовирусное соединение представляет собой анти-HCV соединение.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| WO 00/59929 A1 12.10.2000 | |||
| МАКРОЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ГЕПАТИТА C | 2000 |
|
RU2247126C2 |

