Предлагаемое изобретение относится к лекарственным средствам для лечения нарушений внеклеточной жидкости, а именно к биологически активным пептидам, способным препятствовать повышению проницаемости сосудистого эндотелия. Благодаря этому свойству данный пептид может найти применение в медицине в качестве противоотечного средства, а также средства лечения и профилактики других состояний, связанных с патологическим увеличением проницаемости сосудистого эндотелия.
Нарушение барьерной функции эндотелия сопровождается повышением проницаемости сосудистой стенки и развитием отека ткани, последствия которого могут варьировать от незначительных до катастрофических в том случае, если поражаются легкие, мозг и другие жизненно важные органы. Такая проблема нередко наблюдается при ряде патологических состояний, таких как острая сердечная недостаточность, патология клапанов сердца, почечная недостаточность, цирроз печени, онкологические заболевания, воздействие токсических веществ и др., и, несмотря на достижения современной медицины, продолжает оставаться причиной высокой смертности, даже в условиях стационарного лечения. Для борьбы с отеком используются диуретики, кристаллоиды и стероидные гормоны. Эти соединения улучшают выведение жидкости из организма, но не воздействуют на молекулярные механизмы развития отека - повышение проницаемости эндотелиальной выстилки микрососудов, возникающей вследствие сократительной активности эндотелиальных клеток.
Одним из ключевых регуляторов сократительной активности клеток, в том числе и эндотелиальных, является киназа легких цепей миозина (КЛЦМ). Этот фермент активируется при действии многих эдемагенных факторов и запускает сокращение эндотелия микрососудов, что приводит к нарушению его барьерной функции и развитию отека ткани. В связи с этим, очевидно, перспективным подходом к снижению гиперпроницаемости сосудов микроциркулярного русла в стрессовых ситуациях является подавление сократительной реакции эндотелия путем ингибирования эндотелиальной КЛЦМ.
Однако большинство существующих ингибиторов КЛЦМ не подходит для применения у человека по причине серьезных побочных эффектов. В этой связи актуален поиск ингибиторов КЛЦМ, характеризующихся фармакологически привилегированной структурой и высокой специфичностью. Одним из таких соединений является низкомолекулярный ингибитор, созданный на основе аминопиридазина [1. Behanna H.А., Watterson D.M., Ranaivo H.R. Development of a novel bioavailable inhibitor of the calmodulin-regulated protein kinase MLCK: a lead compound that attenuates vascular leak. Biochim. Biophys. Acta. 1763 (11), 1266-1274, 2006. 2. Rossi J.L., Velentza A.V., Steinhom D.M., Watterson D.M., Wainwright M.S. MLCK210 gene knockout or kinase inhibition preserves lung function following endotoxin-induced lung injury in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 292 (6), L1327-1334, 2007. 3. Wainwright M.S., Rossi J., Schavocky J., Crawford S., Steinhom D., Velentza A.V., Zasadzki M., Shirinsky V., Jia Y., Haiech J., Van Eldik L.J., Watterson D.M. Protein kinase involved in lung injury susceptibility: evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc. Natl. Acad. Sci. USA. 100 (10), 6233-6238, 20031-3]. В последние годы в мире разрабатываются пептидные ингибиторы КЛЦМ, для которых было показано влияние на проницаемость кишечного эпителия в эксперименте при отсутствии выраженной токсичности [Clay burgh D.R., Barrett Т.A., Tang Y., Meddings J.В., Van Eldik L.J., Watterson D.M., Clarke L. L., Mrsny R.J., Turner J.R. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates Т cell activation-induced diarrhea in vivo. J. Clin. Invest. 115 (10), 2702-2715, 2005.]. Нонапептид H-Arg-Lys-Lys-Tyr-Lys-Tyr-Arg-Arg-Lys-NH2 (L-ПИК), созданный на основе автоингибиторного участка КЛЦМ [Lukas T.J., Mirzoeva S., Slomczynska U., Watterson D.M. Identification of Novel Classes of Protein Kinase Inhibitors Using Combinatorial Peptide Chemistry Based on Functional Genomics Knowledge. J. Med. Chem. V.42., p.910-919, 1999.], способен проникать через плазматическую мембрану и влияет на эпителиальную проницаемость in vitro [Clayburgh D.R., Barrett T.A., Tang Y., Meddings J.В., Van Eldik L.J., Watterson D.M., Clarke L.L., Mrsny R.J., Turner J.R. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J. Clin. Invest. 115 (10), 2702-2715, 2005.], однако в силу особенностей его химической структуры чрезвычайно подвержен действию протеолитических ферментов [Owens S.E., Graham W.V., Siccardi D., Turmer J.R., Mrsny R.J., A Strategy to Identify Stable Membrane-Permeant Peptide Inhibitors of Myosin Light Chain Kinase. Pharm. Res. 22 (5), 703-709, 2005]. Это ставит под сомнение возможность его применения в практической медицине. Модификация пептида L-ПИК путем замены входящих в его состав L-аминокислот на D-конфигурацию, как и следовало ожидать, приводит к повышению устойчивости пептида к протеолитической деградации [Clayburgh D.R., Barrett T.A., Tang Y., Meddings J.В., Van Eldik L.J., Watterson D.M., Clarke L.L., Mrsny R.J., Turner J.R. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates Т cell activation-induced diarrhea in vivo. J. Clin. Invest. 115 (10), 2702-2715, 2005]. Однако ингибиторная активность D-ПИК значительно уступала таковой L-ПИК, по-видимому, вследствие того, что D-ПИК является полным энантиомером L-ПИК и обладает иной пространственной структурой [Секридова А.В., Сидорова M.В., Азьмуко А.А., Молокоедов А.С., Бушуев В.Н., Марченко А.В., Щербакова О.В., Ширинский В.П., Беспалова Ж.Д. Пептидные ингибиторы киназы легких цепей миозина, устойчивые к действию протеиназ. Биоорганическая химия 36 (4), 498-504, 2010]. Поскольку в ряду пептидных аналогов не существует четкой взаимосвязи между структурой и биологической активностью, даже небольшие модификации пептида могут кардинально сказаться на его ингибиторных свойствах.
Наиболее близким к заявляемому пептиду является пептид ПИК1 формулы H-(W-Me)-Arg-Lys-Lys-Tyr-Lys-Tyr-Arg-Arg-Lys-NH2 (или [NMe-Arg1]-ПИК). При сравнении пептидных ингибиторов КЛЦМ с различными вариантами модификации структуры исходного L-ПИК (D-ПИК, [NMe-Arg1]-ПИК, [εАса1]-ПИК, [Cit1]-ПИК, [Cit1,Orn3-ПИК) было показано, что активностью in vitro, сопоставимой с исходным L-ПИК, обладает только [NMe-Arg1]-ПИК (ПИК1) с модификацией N-концевой аминокислоты исходного пептида L-ПИК [Секридова А.В., Сидорова M.В., Азьмуко А.А., Молокоедов А.С., Бушуев В.Н., Марченко А.В., Щербакова О.В., Ширинский В.П., Беспалова Ж.Д. Пептидные ингибиторы киназы легких цепей миозина, устойчивые к действию протеиназ. Биоорганическая химия 36 (4), 498-504, 2010]. Время жизни ПИК1 в плазме крови т vitro почти в 3 раза превышало время жизни L-ПИК, и в концентрации 100 мкМ ПИК1 снижал индуцированное тромбином повышение проницаемости монослоя эндотелиальных клеток [Марченко А.В., Степанова Е.О., Секридова А.В., Сидорова М.В., Бушуев В.Н., Беспалова Ж.Д., Ширинский В.П. Новые пептидные ингибиторы киназы легких цепей миозина подавляют гиперпроницаемость эндотелия сосудов. Биофизика, 55, 1008-1013, 2010]. Однако эффект ПИК1 наблюдается в относительно высоких диапазонах концентраций (около 100 мкМ), что с фармакологической точки зрения делает его менее привлекательным для применения in vivo.
Задачей изобретения является обеспечение эффективного подавления гиперпроницаемости сосудистого эндотелия, и создание фармацевтической композиции для инъекций для борьбы с острым отеком жизненно важных органов. Актуальность этой задачи обусловлена необходимостью обеспечения практической медицины эффективными препаратами, подавляющими гиперпроницаемость сосудистого эндотелия, в частности для борьбы с острым отеком легких в критических для пациента ситуациях.
Технический результат заключается в получении пептида, обладающего усиленной активностью в отношении подавления гиперпроницаемости эндотелия сосудов и повышенным временем жизни в плазме крови.
Поставленная задача решается путем синтеза метилированного (Me) по аминоконцевой группе амида нонапептида формулы H-(W-Me)-Arg-Lys-Lys-Tyr-Lys-Tyr-Arg-D-Arg8-Lys-NH2 (ПИК2) и его фармацевтически приемлемых нетоксичных солей. Поставленная задача решается также созданием фармацевтической композиции для инъекций, содержащей терапевтически эффективное количество ПИК2 и фармацевтически приемлемый носитель. А также поставленная задача решается разработкой способа снижения гиперпроницаемости сосудистого эндотелия у реципиента посредством инъекции терапевтически эффективного количества указанной фармацевтической композиции.
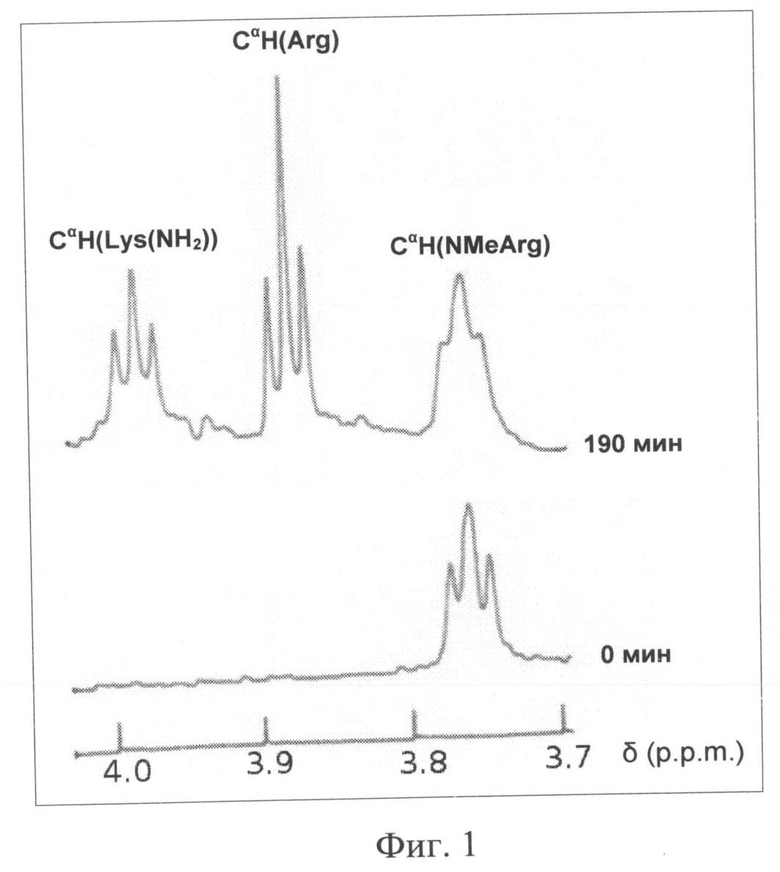
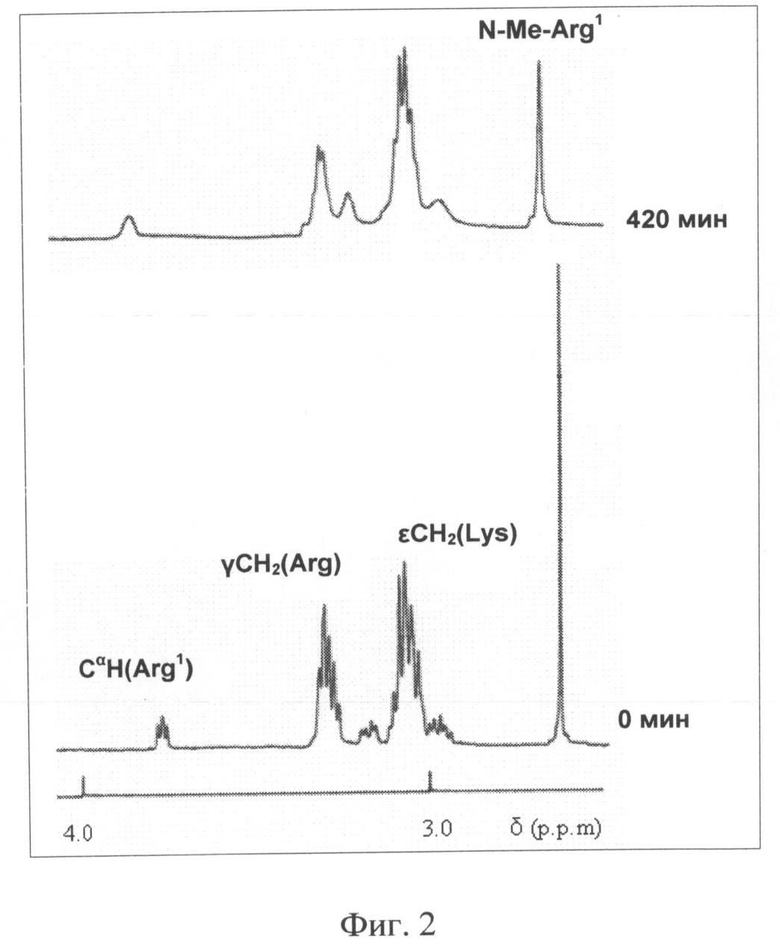
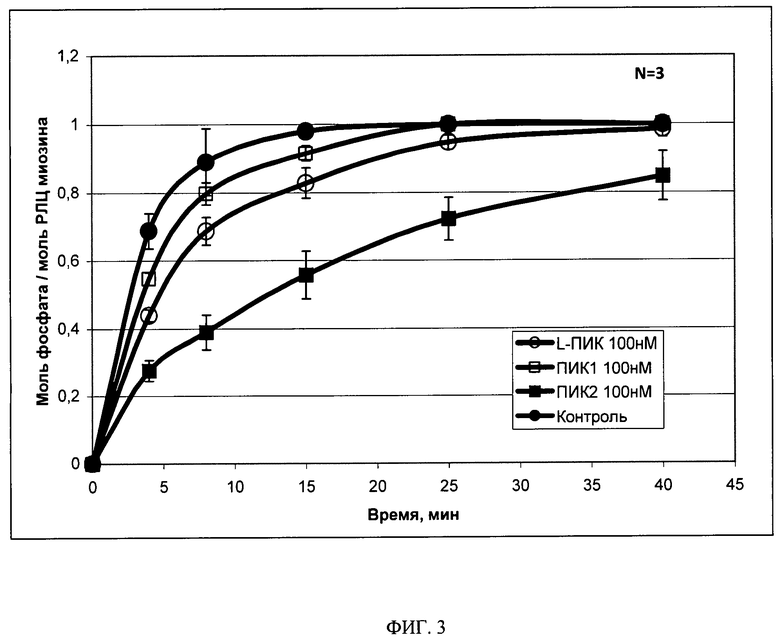
Изобретение поясняется 4 (четырьмя) иллюстрациями, где на Фиг.1 и Фиг.2, соответственно, представлены фрагменты 1N-ЯМР спектров прототипа (ПИК1) и заявляемого пептида (ПИК2) в растворе и в плазме крови человека (за вычетом спектра плазмы крови) с указанием сигналов соответствующих групп свободных аминокислот. На Фиг.3 представлена динамика фосфорилирования регуляторных легких цепей миозина под действием КЛЦМ in vitro в присутствии заявляемого пептида ПИК2 и пептидов-прототипов L-ПИК и ПИК1 в концентрации 100 нМ. На Фиг.4 представлено влияние заявляемого пептида (ПИК 2) и прототипа (ПИК 1) на диффузию альбумина, конъюгированного с флуоресцеинизотиоцианатом через монослой эндотелиальных клеток линии EA.hy926. Представлена относительная проницаемость эндотелия через 4 часа после стимуляции 100 нМ тромбином в присутствии 10 мкМ (белые столбцы), 20 мкМ (черные столбцы) и 100 мкМ (заштрихованные столбцы) заявляемого пептида (справа) и прототипа (слева). Проницаемость контрольных эндотелиальных клеток после стимуляции тромбином принята за 100% (показано пунктирной линией).
Заявляемый пептид был получен твердофазным методом пептидного синтеза с использованием 9-флуоренилметоксикарбонил (Fmoc)-технологии, описанным ниже в примере 1.
Фармацевтически приемлемые соли пептида изобретения включают соли, образованные с неорганическими или карбоновыми кислотами. Примеры неорганических кислот, образующих соли, включают галогенводородные кислоты, такие как соляная, бромистоводородная кислоты, фосфорная кислота, серная кислота. Соли карбоновых кислот образуются с такими кислотами, как уксусная, пропионовая, малоновая, малеиновая, лимонная, янтарная, яблочная, бензойная, фумаровая и другие подобные карбоновые кислоты. Соли с кислотами получают обычными методами, например, с помощью нейтрализации пептида ПИК2 в форме свободного основания с кислотой. Предпочтительными солями являются сульфат и гидрохлорид. Как указано выше, настоящее изобретение включает сольваты соединений этого изобретения и их фармацевтически приемлемые соли. Заявляемый пептид ПИК2 или его фармацевтически приемлемые соли могут образовывать сольваты с водой или обычными органическими растворителями. Такие сольваты включаются в область настоящего изобретения.
Термин «фармацевтически приемлемый» означает носитель, совместимый с другими ингредиентами фармакологической композиции и не оказывающий токсического или неблагоприятного действия на пациентов. Поскольку заявляемый пептид хорошо растворяется в водных растворах, в качестве носителей для ПИК2 могут быть использованы, например, изотонический раствор натрия хлорида 0,25%, стерильная вода для инъекций, раствор Рингера для инъекций, кровезаменители, раствор глюкозы для инфузий 5%, растворы других активных препаратов для внутривенного или внутримышечного введения при условии отсутствия неблагоприятных межлекарственных взаимодействий между ПИК2 или его фармацевтически приемлемыми солями и вторым активным веществом (активными веществами).
Терапевтически эффективное количество ПИК2 в качестве активного начала определяется как количество, необходимое для достижения желаемого физиологического ответа у реципиента, и может зависеть от массы тела реципиента, тяжести состояния реципиента, индивидуальных физиологических особенностей реципиента, проводимой сопутствующей терапией и др. Количество ПИК2 в фармакологической композиции будет зависеть от формы его выпуска - лиофилизат для приготовления раствора для инъекций или раствор для инъекций. Эффективные разовые дозы ПИК2 для внутривенного введения могут составить от 0,1 до 1,5 мг/кг веса тела реципиента. При введении препарата внутримышечно доза, необходимая для эффективного снижения гиперпроницаемости сосудистого эндотелия, будет выше и может составить до 5 мг/кг веса тела реципиента.
Лекарственные препараты с заявляемым пептидом или его фармацевтически приемлемьми солями в качестве активного вещества могут быть представлены в виде лиофилизата для приготовления раствора для инъекций или в виде готового раствора для инъекций. Предпочтительной является лиофилизованная форма для растворения.
Способы введения лекарственного препарата с заявляемым пептидом или его фармацевтически приемлемыми солями в качестве активного компонента включают внутрисосудистое болюсное введение, введение в виде инфузии в составе других растворов для введения, с компонентами которых заявляемый пептид ПИК2 или его фармацевтически приемлемые соли не образуют нежелательных взаимодействий, введение внутримышечно. Предпочтительным способом введения является болюсная внутрисосудистая инъекция, позволяющая быстро достичь терапевтически эффективных концентраций активного вещества в крови реципиента. Показаниями к применению заявляемого пептида или его фармацевтически приемлемых солей следует рассматривать появление клинических симптомов или признаков развития гиперпроницаемости сосудистого эндотелия, например, при остром отеке легких такими симптомами выступают цианоз, бледность, профузный пот, альтернация пульса, акцент II тона над легочной артерией, протодиастолический ритм галопа. Введение ПИК2 для симптоматического лечения следует осуществлять незамедлительно при появлении признаков развития острого отека.
Пример 1. Синтез заявляемого пептида (ПИК2)
H-(N-Me)-Arg1-Lys-Lys-Tyr-Lys-Tyr-Arg7-D-Arg8-Lys-NH2
В работе использованы производные аминокислот (АА) и гексафторфосфат (бензотриазол-1-ил)окси-трис(диметиламино)фосфония (ВОР, NovaBiochem, Швейцария), N,N'-диизопропилкарбодиимид (DIC), N,N-диизопропилэтиламин (DIPEA), гидроксибензотриазол (HOBt), триизобутилсилан (TIBS) компании Fluka, Швейцария, 4-метилпиперидин (4-MePip, Aldrich, США). Для синтеза применяли N-метилпирролидон, дихлорметан (DCM), трифторуксусная кислоту (TFA) компании Fluka, Швейцария, для хроматографии - ацетонитрил (Panreac, Испания). Аналитическую высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили на хроматографе (Gilson, Франция), использовали колонку Nucleosil 100 С18, 5 мкм, (4.6×250 мм) (Sigma, США) в качестве элюентов использовали буфер А - 0.1% TFA, буфер Б - 80% ацетонитрила в буфере А, элюция градиентом концентрации буфера Б в буфере А от 0% до 60% за 30 мин. Скорость потока 1 мл/мин, детекция при 220 нм. Структура полученных пептидов доказана спектрами 1H-ЯМР и данными масс-спектрометрии. 1H-ЯМР-спектры снимали на спектрометре WM-500 (Bruker) 500 МГц (ФРГ) в дейтерированном диметилсульфоксиде (DMSO-d6) при 300 К, концентрация пептидов составляла 2-3 мг/мл. Химические сдвиги измерялись относительно тетраметилсилана. Масс-спектры регистрировали на приборе PC-Kompact MALDI (Kratos, Англия).
Для твердофазного синтеза использовали сополимер стирола с 1% дивинилбензола с 4-(2,4-диметоксифенил)-9-флуоренилметоксикарбонил-аминометилфенокси - якорной группой (Rink-amide-полимер) фирмы Nova BoiChem, Швейцария, предназначенный для получения амидов пептидов, содержащий 0.64 ммоль/г аминогрупп. Синтез амида нонапептида проводили с С-конца, ступенчато (присоединяя по одной аминокислоте), исходя из 0.40 г (0.25 ммоль) Rink-amide-полимера. Первые 8 циклов синтеза проводили в автоматическом режиме на пептидном синтезаторе Applied Biosystems 431 А по стандартной программе для однократной конденсации Fmoc-аминокислот. Гуанидиновые группы остатков Arg и Arg блокировали с помощью 2,2,5,7,8-пентаметилхроман-6-сульфонила (Pmc) (см. протокол твердофазного синтеза). Для присоединения N-концевого, N-метилзамещенного остатка Arg1 гуанидиновую функцию защищали с помощью 4-метокси-2,3,6-триметилбензолсульфонила (Mtr), а для создания амидной связи применяли высокоэффективный реагент Кастро - ВОР [Castro В., Dormoy J.R., Evin G., Selve С. Peptide coupling reaction with benzotriazol-l-yl-tris(dimethylammo)phosphonium hexafluorophosphate. Tetrahedron Lett. V.14. P.1219-1222, 1975].
Заключительное деблокирование и отщепление нонапептида от полимера проводили в одну стадию путем обработки соответствующего нонапептидилполимера смесью 10 мл TFA, 0.25 мл деионизованной воды и 0.25 мл TIBS в течение 16 ч. Затем полимер отфильтровывали, промывали 2×2 мл деблокирующей смеси, фильтрат упаривали и к остатку прибавляли сухой эфир. Осадок отфильтровывали, промывали DCM (3×3 мл), эфиром (3×5 мл), сушили в вакуум-эксикаторе. 0.32 г сырого продукта твердофазного синтеза с содержанием основного вещества 76% очищали с помощью препаративной ВЭЖХ на приборе Beckman (США), используя колонку Диасорб С16 13 ОТ (25×250 мм), размер частиц сорбента 10 мкм. В качестве элюентов использовали буфер А - 0.01 М раствор ацетата аммония и буфер Б - 80% ацетонитрила в воде. Элюцию проводили градиентом 0.5% в минуту буфера Б в буфере А от 100% буфера А со скоростью 10 мл/мин. Пептиды детектировали при длине волны 220 нм. Фракции, содержащие целевой продукт, объединяли, ацетонитрил упаривали и лиофилизовали. В итоге получили 0.16 г (47.1% в расчете на стартовую аминокислоту, присоединенную к полимеру) амида нонапептида ПИК2. Гомогенность продукта, определенная с помощью аналитической ВЭЖХ, составляет 96.8%. Масс-спектр: 1338.6, вычислено 1338.7.
Времена жизни заявляемого пептида (ПИК2) и пептида-прототипа (ПИК1) в плазме крови сравнивали методом 1H-ЯМР, как описано в примере 2. Результаты сравнения заявляемого пептида (I) и прототипа представлены на Фиг.1 и Фиг.2, соответственно.
Пример 2. Время жизни пептидов в плазме крови in vitro
В асептических условиях у здоровых доноров забирали по 100 мл цельной крови в 50-мл пробирки с 5 мл консерванта (130 мМ цитрат на фосфатно-солевом буфере (ФСБ), рН 7,4). Кровь выдерживали 15-20 мин при комнатной температуре и центрифугировали при 1500 g в течение 15 мин при 18°С. Надосадочную жидкость переносили в новые пробирки и центрифугировали повторно в тех же условиях. Измеряли объем плазмы крови и добавляли сухой NaBr из расчета 280 мг NaBr на 1 мл плазмы. После растворения NaBr плазму (приблизительно по 30 мл) разливали по центрифужным стаканам (для ротора Type 45Ti, Beckman, США) и наслаивали сверху по 20 мл раствора с плотностью 1,216 г/л следующего состава: 11,42 г NaCl и 100 мг этилендиаминтетрауксусной кислоты (ЭДТА) растворяли в 1 л дистиллированной воды, рН 7,4, затем добавляли 281,44 г NaBr. Центрифугировали при 40000 об/мин в течение 48 ч. После центрифугирования отбирали верхний темно-желтый слой жидкости, содержащий липопротеиды, и бесцветный слой буфера, оставляя буфер на высоту приблизительно 1 см до видимой границы плазмы. Плазму, полученную от одного донора, объединяли и диализовали в течение 36 ч против 4 л ФСБ, свободного от ионов Са2+ и Mg2+ (MP Biomedicals, США) в диализных мешках с размером пор 12-14 кДа (Medicell, Великобритания). Процедуру диализа повторяли два раза по 24 ч против свежего буфера. После диализа плазму подвергали центрифугированию при 25000 об/мин в роторе Type 45Ti (Beckman, США) в течение 15 мин при 4°С. Надосадочную жидкость отбирали и лиофилизовали. Сухой остаток растворяли в D2O (в объеме, равном объему плазмы до внесения NaBr) и снова лиофилизовали. Затем растворяли в таком же объеме и до использования хранили аликвотами по 1 мл при -20°С.
Спектры 1H-ЯМР были получены на спектрометре WM-500 (Bruker, США) при 37°С. Химические сдвиги сигналов в спектрах измеряли относительно внутреннего стандарта - натриевой соли 2,2-диметил-2-силапентан-5-сульфаната. Отнесение сигналов выполняли с помощью метода двойного резонанса. Для выделения сигналов, принадлежащих протонам пептида, применяли разностную спектроскопию: из каждого спектра, полученного после добавления пептида в плазму крови, вычитали спектр свободной плазмы. Концентрация пептида в образце составляла 3 мг/мл.
Как видно из Фиг.1, в случае пептида-прототипа ПИК1 через 190 мин инкубации в плазме крови человека in vitro, хорошо выражены сигналы, соответствующие СαN-группам свободного аргинина и лизинамида. Это указывает на деградацию С-концевой части пептида, приводящую к образованию свободных аминокислот. При этом время полужизни ПИК1 в плазме крови составляет около 120 мин [Секридова А.В., Сидорова М.В., Азьмуко А.А., Молокоедов А.С., Бушуев В.Н., Марченко А.В., Щербакова О.В., Ширинский В.П., Беспалова Ж.Д. Пептидные ингибиторы киназы легких цепей миозина, устойчивые к действию протеиназ. Биоорганическая химия 36 (4), 498-504, 2010]. В то же время, в случае пептида ПИК2 (Фиг.2) исходный спектр не претерпевает существенных изменений даже через 420 мин инкубации в плазме крови, указывая на высокую стабильность ПИК2 в плазме крови с временем полужизни >420 мин. Таким образом, заявляемый пептид характеризуется повышенным временем жизни в плазме крови. Это позволяет поддерживать его эффективную концентрацию, необходимую для снижения гиперпроницаемости сосудистого эндотелия в критических состояниях. Повышенное время жизни пептида позволяет также использовать более низкие, при сравнении с гомологами, концентрации (5-30 мкМ в циркулирующей крови) при сохранении терапевтического эффекта.
Увеличение времени жизни пептида при замене природной L-конфигурации а-углеродного атома остатка Arg8 на D-конфигурацию в структуре заявляемого пептида ПИК2 следовало ожидать, поскольку известно, что пептидные связи, образованные D-аминокислотами более устойчивы, чем связи L-аминокислотных аналогов, так как не распознаются протеолитическими ферментами [Hruby VJ, Qian X. Approaches to the asymmetric synthesis of unusual amino acids. Methods Mol. Biol. 35, 249-286, 1994]. Однако наряду с увеличением времени жизни внесение модификаций может отрицательно сказаться на ингибиторной активности пептида. Подтверждением этому служит проведенное нами ранее сравнительное исследование активности пептидных ингибиторов D-ПИК, [NMe-Arg1]-ПИК, [εАса1]-ПИК, [Cit1]-ПИК и [Cit1,Orn3]-ПИК, из которых только [NMe-Arg1]-ПИК, (ПИК1) обладал активностью in vitro, сопоставимой с пептидом L-ПИК [Секридова А. В., Сидорова М.В., Азьмуко А.А., Молокоедов А.С., Бушуев В.П., Марченко А.В., Щербакова О.В., Ширинский В.П., Беспалова Ж.Д. Пептидные ингибиторы киназы легких цепей миозина, устойчивые к действию протеиназ. Биоорганическая химия 36 (4), 498-504, 2010]. Наши последующие исследования, направленные на решение задач данного изобретения, показали, что пептиды H-DLys-DArg-DArg-DTyr-DLys-DTyr-DLys-DLys-DArg-NH2 (ретроэнантио-ПИК), H-Arg(NO2)-Lys-Lys-Tyr-Lys-Tyr-Arg-Arg-Lys-NH2 (с защитой N-концевой части пептида с помощью введения нитро-группы в гуанидиновую группу остатка Arg1), H-Argψ[CH2NH]-Lys-Lys-Tyr-Lys-Tyr-Arg-DArg-Lys-NH2 (с защитой N-концевой части пептида с помощью введения псевдопептидной связи в первое положение), H-(NαMe)Arg-Lys-Lys-Tyr-Lys-Tyr-Arg-Arg-Lys-Pro-Gly-Pro-OH (с защитой N-концевой части пептида с помощью введения метильной группы и защитой С-концевой части с помощью трипептида Pro-Gly-Pro), и пептида H-Argψ[CH2NH]-Lys-Lys-Tyr-Lys-Tyr-Arg-Arg-Lys-Pro-Gly-Pro-OH (с защитой N-концевой части пептида с помощью введения псевдопептидной связи в первое положение и защитой С-концевой части с помощью трипептида Pro-Gly-Pro) также значительно уступали заявляемому пептиду ПИК2 по ингибиторной активности in vitro в отношении очищенной КЛЦМ и в модели подавления проницаемости эндотелиального монослоя in vitro.
Ингибиторную активность заявляемого пептида (ПИК2) и прототипов (ПИК1 и L-ПИК), являющихся наиболее активными из известных пептидов семейства ПИК, сравнивали по их влиянию на фосфорилирование регуляторных легких цепей миозина in vitro, как описано в примере 3. Результаты анализа представлены на Фиг.3.
Пример 3. Фосфорилирование регуляторных легких цепей миозина in vitro
Регуляторные легкие цепи (РЛЦ) миозина в конечной концентрации 20 мкМ фосфорилировали КЛЦМ из желудков индейки (9 нМ) при 30°С в буфере, содержащем 10 мМ 3-рМ-Морфолино]пропансульфоновая кислота (MOPS), pH 7.0, 100 мМ NaCl, 0.5 мМ CaCl2, 1 мМ MgCl2, 0.5 мМ Mg-АТФ, 0.3 мкМ кальмодулин, 1 мМ дитиотреитол (ДТТ) в присутствии 100 нМ заявляемого пептида или пептидов-прототипов L-ПИК или ПИК1. Реакцию инициировали добавлением КЛЦМ. Через определенные промежутки времени (0-40 мин) аликвоты реакционной смеси (5 мкл) отбирали и растворяли буфере для образцов. Полноту прохождения реакции анализировали с помощью электрофореза в присутствии мочевины и глицерина, позволяющем разделять белки на основании величины их заряда. Электрофорез проводили согласно [Persechini A, Kamm KE, Stull JT. Different phosphorylated forms of myosin in contracting tracheal smooth muscle. JBC, 261, 6293-6299, 1986] с модификациями. Белковые препараты растворяли в буфере для образцов (20 мМ трис-HCl, рН 6,8, 9 М мочевина, 10 мМ ДТТ, 0,05% бромфеноловый синий). Использовали 5,2% концентрирующий гель, приготовленный на 20 мМ трис-HCl буфере рН 6,8, содержащем 16,5% глицерол и 3,25 М мочевину; и 12% разделяющий гель, приготовленный на 20 мМ трис-23 мМ глициновом буфере рН 8,6, содержащем 40% глицерин. Электрофорез проводили в 20 мМ трис-23 мМ глициновом буфере, рН 8,6, содержащем 0,2% β-меркаптоэтанол при 330 В 20 мин и 400 В в течение 180 мин. Гели фиксировали и окрашивали Кумасси R-250. Эксперименты повторяли не менее 3 раз. Электрофоретическую подвижность белковых полос, соответствующих РЛЦ и фосфо-РЛЦ, устанавливали по подвижности соответствующих стандартов. Гели сканировали и количественный денситометрический анализ полученных электрофореграмм проводили с помощью программы Image J, версия 1.45. Степень фосфорилирования РЛЦ миозина рассчитывали как отношение фосфорилированных РЛЦ к общему количеству РЛЦ. Полученные данные обрабатывали в программе Excel и строили графики зависимости степени фосфорилирования РЛЦ миозина (моль фосфата/моль РЛЦ) от времени в присутствии разных концентраций пептида. Данные выражали как среднее + станд. откл.
Из приведенного примера видно, что в данных условиях протекания реакции в отсутствие ингибитора полное фосфорилирование РЛЦ миозина достигается через 15 мин. За то же время в присутствии пептидов-прототипов L-ПИК и ПИК1 реакция протекает на 90-95%, при этом в присутствии заявляемого пептида (ПИК2) реакция протекает только на 50-60%. Эти данные свидетельствуют о более сильных ингибиторных свойствах заявляемого пептида ПИК2 по сравнению с пептидами-прототипами L-ПИК и ПИК1. Следует отметить, что в данных условиях время жизни пептидов не влияет на их ингибиторную активность, поскольку реакция проводится в отсутствие протеолитических ферментов.
Влияние заявляемого пептида (ПИК2) и прототипа (ПИК1) на проницаемость эндотелия оценивали по скорости диффузии ФИТЦ-альбумина через монослой эндотелиальных клеток EA.hy926, как описано в примере 4. Результаты сравнения заявляемого пептида (ПИК2) и прототипа ПИК1 представлены на Фиг.4.
Пример 4. Проницаемость эндотелиального монослоя in vitro
Влияние пептидов на проницаемость эндотелия оценивали по скорости диффузии меченого флуоресцеинизотиоцианатом (ФИТЦ) альбумина через монослой эндотелиальных клеток EA.hy926 (рис.3). На мембраны вкладышей ThinCerts (Greiner Bio One, Австрия) с диаметром пор 0,4 мкм и плотностью пор 1×108/см2 высаживали 1×105 эндотелиальных клеток линии EA.hy926 в 200 мкл питательной среды Игла в модификации Дульбекко (ДМЕМ) с высоким содержанием глюкозы в присутствии 20% эмбриональной телячьей сыворотки. Во внешнюю камеру вносили 800 мкл той же среды без клеток. Клетки культивировали в течение 4 дней. За это время клетки достигали монослоя. Среду роста меняли на ДМЕМ без эмбриональной сыворотки и проводили депривацию клеток в течение 30 минут. После этого в нижнюю и верхнюю камеры добавляли исследуемый пептид в концентрациях 10-100 мкМ и инкубировали 30 минут. Затем в верхнюю камеру добавляли ФИТЦ-альбумин до конечной концентрации 0,5 мг/мл и каждые 30 мин отбирали пробы из нижней камеры для расчета базальной скорости диффузии ФИТЦ-альбумина через эндотелиальный монослой (относительные единицы интенсивности флуоресценции/час). Изменение проницаемости эндотелия индуцировали добавлением 100 нМ тромбина в верхнюю камеру и с интервалами по 30 мин продолжали отбор проб среды из нижней камеры для анализа содержания в ней ФИТЦ-альбумина. Содержание ФИТЦ-альбумина в пробах измеряли с помощью многофункционального планшетного анализатора Victor X3 (PerkinElmer, США). Результаты представляли как среднее±стандартное отклонение (Фиг.4). Достоверность отличий определяли по t-критерию Стьюдента.
Из приведенного примера видно, что заявляемый пептид (ПИК2) снижает индуцированную тромбином проницаемость монослоя эндотелиальных клеток более эффективно, чем прототип (ПИК1). Так, после преинкубации клеток с 10 мкМ прототипа ответ эндотелиальных клеток на тромбин снижается всего на 10% и составляет 90% от ответа клеток в отсутствие ингибиторов. В то же время после преинкубации с 10 мкМ заявляемым пептидом (ПИК2) ответ на тромбин снижается на 80%, составляя 20% от ответа клеток в отсутствие ингибиторов. В концентрации 20 мкМ прототип подавляет ответ на тромбин на 40%, тогда как заявляемый пептид (ПИК2) в той же концентрации полностью отменяет увеличение проницаемости монослоя. При концентрации прототипа, равной 100 мкМ, достигается существенное (90%) подавление реакции на тромбин. В то же время после преинкубации с 100 мкМ пептидом (ПИК2) усиливалась барьерная функция монослоя и проницаемость становилась на 30% ниже базовой проницаемости контрольных клеток в отсутствие стимуляции тромбином.
Применимость и эффективность лекарственных препаратов, включающих заявляемый пептид ПИК2 или его фармацевтически приемлемые соли, для снижения гиперпроницаемости сосудистого эндотелия in vivo демонстрируют эксперименты с моделированием острого отека легких, вызванного бактериальным липосахаридом. Конечным функциональным звеном при развитии отека тканей выступает усиление пара- и трансцеллюлярного транспорта веществ через клетки сосудистого эндотелия. Эти процессы сопряжены с активностью эндотелиальной КЛЦМ. Поэтому использование бактериального липосахарида позволяет смоделировать развитие острого отека легких различной этиологии.
Пример 5. Модель отека легких in vivo.
Самцам крыс линии Wistar весом 300-340 г под кетаминовым наркозом (100 мг/кг) вставляли катетеры в сонную артерию и яремную вену для регистрации среднего артериального давления (АД), частоты сердечных сокращений (ЧСС) и для введения веществ. Через 10 мин после регистрации исходных параметров крысам вводили липополисахарид (ЛПС) S. typhimurium в концентрации 8 мг/кг в физиологическом растворе. Через 20 мин крысам вводили заявляемый пептид (ПИК2) в концентрации 1,5 мг/кг внутривенно, болюсно в 0,5 мл физиологического раствора. Через 24 ч после эксперимента крыс забивали передозировкой уретана. Грудную клетку вскрывали, отсасывали и измеряли количество жидкости из плевральных полостей, вынимали и взвешивали легкие. Легкие сушили 3 суток при температуре 54°С до постоянного веса. Общее содержание воды в легких определяли как соотношение сырого веса легких плюс плеврального выпота к сухому весу легких. В контрольной группе животных определяли эти параметры без введения ЛПС и ПИК2. Результаты представляли как среднее ± ошибка среднего (табл.). Достоверность отличий определяли по t-критерию Стьюдента.
Как видно из табл., введение ЛПС приводит к значительному повышению массы легких, обусловленной поступлением воды и низкомолекулярных веществ, а также высокомолекулярных соединений и форменных элементов крови. При введении заявляемого пептида (ПИЮ) прирост количества жидкости в легких в ответ на воздействие ЛПС снижается в 2 раза. Таким образом, заявляемый пептид (ПИК2), введенный в дозе 1,5 мг/кг (концентрация в циркулирующей крови около 30 мкМ), способен ослаблять острый отек легких, вызванный воздействием бактериального эндотоксина.
Поскольку механизмы повышения проницаемости сосудистого эндотелия вовлечены в развитие острого отека легких различной этиологии, ПИК2 при введении внутривенно в физиологическом растворе или иных фармацевтически приемлемых носителях может выступать эффективным средством борьбы с острыми критическими состояниями, связанными с патологическим повышением проницаемости сосудистого эндотелия. Наиболее актуальной областью применения заявляемой фармацевтической композиции представляется борьба с острым отеком легких, являющимся причиной высокой смертности пациентов. Острый отек легких может возникать при проведении искусственной вентиляции легких при хирургических операциях, при сепсисе, интоксикациях, воспалительных и аллергических реакциях, травмах (ожогах, сдавливании), ишемии-реперфузии, сердечной недостаточности и ряде других нарушений.
| название | год | авторы | номер документа |
|---|---|---|---|
| Протеолитически устойчивый нонапептид, обладающий способностью предотвращать повышение гиперпроницаемости сосудистого эндотелия | 2018 |
|
RU2682878C1 |
| ЦИКЛИЧЕСКИЙ НОНАПЕПТИД, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ИНГИБИРОВАТЬ КИНАЗУ ЛЕГКИХ ЦЕПЕЙ МИОЗИНА | 2010 |
|
RU2443710C1 |
| АМИД НОНАПЕПТИДА, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ПРЕДОТВРАЩАТЬ ПОВЫШЕНИЕ ПРОНИЦАЕМОСТИ ЭНДОТЕЛИЯ СОСУДОВ | 2009 |
|
RU2402565C1 |
| СПОСОБ ПОЛУЧЕНИЯ НОНАПЕПТИДОВ | 2015 |
|
RU2592282C1 |
| СИНТЕТИЧЕСКИЙ АНТИГЕН, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ СВЯЗЫВАТЬ АУТОАНТИТЕЛА К β-АДРЕНОРЕЦЕПТОРУ | 2007 |
|
RU2356576C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДОТВРАЩЕНИЯ И ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ГЛАЗ, СОДЕРЖАЩАЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА БЕЛОК СЛИЯНИЯ, В КОТОРОМ СЛИТЫ ПРОНИКАЮЩИЙ В ТКАНЬ ПЕПТИД И ПРЕПАРАТ ПРОТИВ ФАКТОРА РОСТА ЭНДОТЕЛИЯ СОСУДОВ | 2016 |
|
RU2712623C2 |
| АНТИТЕЛО ПРОТИВ АДРЕНОМЕДУЛЛИНА (ADM), ИЛИ ФРАГМЕНТ АНТИ-ADM АНТИТЕЛА, ИЛИ АНТИ-ADM НЕ-IG КАРКАС ДЛЯ ПРИМЕНЕНИЯ ПРИ ВМЕШАТЕЛЬСТВЕ И ТЕРАПИИ ГИПЕРЕМИИ У ПАЦИЕНТА | 2017 |
|
RU2762059C2 |
| СПОСОБ ИММУНОФЕРМЕНТНОГО АНАЛИЗА ДЛЯ ОПРЕДЕЛЕНИЯ АУТОАНТИТЕЛ К β-АДРЕНОРЕЦЕПТОРУ В ПЛАЗМЕ И СЫВОРОТКЕ КРОВИ ЧЕЛОВЕКА | 2011 |
|
RU2452964C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА СЛИТЫЙ БЕЛОК, В КОТОРОМ СЛИТЫ ПРОНИКАЮЩИЙ В ОПУХОЛЬ ПЕПТИД И АНТИАНГИОГЕННОЕ СРЕДСТВО, ДЛЯ ПРЕДУПРЕЖДЕНИЯ И ЛЕЧЕНИЯ РАКА ИЛИ СВЯЗАННЫХ С АНГИОГЕНЕЗОМ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2727238C2 |
| ЛЕЧЕНИЕ СОСУДИСТЫХ ОСЛОЖНЕНИЙ ДИАБЕТА | 2010 |
|
RU2545718C2 |

Изобретение относится к биологически активным пептидам, способным предотвращать острое повышение проницаемости сосудистого эндотелия при введении реципиенту. Предложен пептид формулы H-(N-Me)-Arg-Lys-Lys-Tyr-Lys-Tyr-Arg-D-Arg-Lys-NH2. Заявленный пептид может найти применение в качестве средства снижения патологической гиперпроницаемости сосудистого эндотелия в различных областях медицины (в кардиологии, токсикологии, нейрохирургии, онкологии и др.). 3 н.п. ф-лы, 4 ил., 1 табл., 5 пр.
1. Пептид, обладающий усиленной активностью в отношении подавления гиперпроницаемости эндотелия сосудов и повышенным временем жизни в плазме крови, имеющий формулу H-(N-Me)-Arg-Lys-Lys-Tyr-Lys-Tyr-Arg-D-Arg-Lys-NH2.
2. Фармацевтическая композиция, обладающая способностью подавлять гиперпроницаемость эндотелия сосудов, содержащая терапевтически эффективное количество пептида по п.1 и фармацевтически приемлемый носитель для инъекций.
3. Способ подавления гиперпроницаемости эндотелия сосудов у реципиента посредством инъекции терапевтически эффективного количества фармацевтической композиции по п.2.
| АМИД НОНАПЕПТИДА, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ПРЕДОТВРАЩАТЬ ПОВЫШЕНИЕ ПРОНИЦАЕМОСТИ ЭНДОТЕЛИЯ СОСУДОВ | 2009 |
|
RU2402565C1 |
| US 2008081078 A1, 03.04.2008 | |||
| Секридова А.В | |||
| и др | |||
| Пептидные ингибиторы киназы легких цепей миозина, устойчивые к действию протеиназ | |||
| - Биоорганическая химия, 2010, т.36 (4), с.498-504. | |||

