Изобретение относится к области физиологически активных пептидов, а именно к способу получения нонапептидов, содержащих в молекуле остатки L-, D- или L- Nα-метил-аргинина формулы I-III:
R-Arg1-Lys2-Lys3-Tyr4-Lys5-Tyr6-Arg7-Xaa8-Lys9-NH2,
где
R = Н, Хаа = L-Arg (I)
R = Me, Хаа = L-Arg (II)
R = Н, Хаа = D-Arg (III)
Соединения (I-III) являются пептидными ингибиторами киназы легких цепей миозина (КЛЦМ) и обладают способностью регулировать изменение проницаемости эпителия и эндотелия сосудов [Секридова А.В., Сидорова М.В., Азьмуко А.А., Молокоедов А.С., Бушуев В.Н., Марченко А.В., Щербакова О.В., Ширинский В.П., Беспалова Ж.Д. Пептидные ингибиторы киназы легких цепей миозина, устойчивые к действию протеиназ. Биоорганическая химия. - 2010, 36 (4), с. 498-504]. Пептид (I) in vitro способен оказывать влияние на эпителиальную проницаемость кишечника [Clayburgh D.R., Barrett Т.Α., Tang Y., Meddings J.В., Van Eldik L.J., Watterson D.M., Clarke L.L., Mrsny R.J., Turner J.R. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates Τ cell activation-induced diarrhea in vivo. J. Clin. Invest. 115 (10): 2702-2715. 2005], пептид (II) [Патент РФ №2402565, МПК C07K 7/00, опубл. 27.10.2010 г.] и пептид (III) [Патент РФ №2493164, МПК C07K, опубл. 20.09.2013] обладают способностью предотвращать повышение проницаемости сосудистого эндотелия и могут найти применение в качестве средств снижения патологической гиперпроницаемости сосудистого эндотелия в различных областях медицины (в кардиологии, токсикологии, нейрохирургии, онкологии и др.).
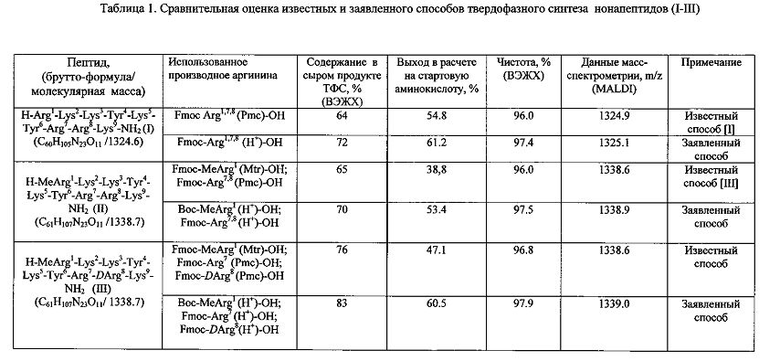
Известен способ получения пептидов формулы (I-III) твердофазным методом. В процессе твердофазного синтеза пептидов (ТФС) синтезируемая цепь полипептида ковалентно закрепляется на нерастворимой инертной полимерной матрице. Выделение целевого продукта на каждой стадии ТФС проводится путем соответствующих промывок и фильтрации пептидилполимера. Синтетический протокол включает в себя несколько последовательных химических превращений - стадий, которые повторяются в каждом цикле синтеза (цикл синтеза - присоединение одной аминокислоты). Стандартный цикл синтеза включает следующие основные стадии: 1) деблокирование - удаление Nα-защитной гуппы (Fmoc) обработкой пептидилполимера раствором вторичного амина (пиперидина, 4-метилпиперидина, 1,8-диазабицикло[5.4.0]ундец-7-ена и т.п.) в подходящем растворителе; 2) получение активированного производного присоединяемой аминокислоты; 3) конденсация - присоединение остатка Fmoc-защищенной аминокислоты к пептидилполимеру за счет образования амидной связи. Между основными стадиями проводятся промывки пептидилполимера органическим растворителем для удаления избытков соответствующих реагентов. Синтез пептидов (I-III) проведен путем ступенчатого наращивания пептидной цепи, начиная с С-концевой аминокислоты на полимерной матрице, с использованием 4-кратных избытков соответствующих защищенных Fmoc-аминокислот в присутствии конденсирующего агента с последующим деблокированием конечного пептидилполимера в 2 стадии: обработкой раствором вторичного основания (пиперидина или др.) для удаления Fmoc-защиты и затем трифторуксусной кислотой со специальными добавками в течение 16 часов с последующей очисткой полученного продукта с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). На Фиг. 1 показана схема синтеза нонапептидов, соответствующего известным способам. При ТФС пептидов (I-III) использована тактика максимальной защиты боковых функций аминокислот. При ТФС пептида (I) гуанидиновые группы остатков аргинина защищали с помощью 2,2,5,7,8-пентаметилхроман-6-сульфонильной (Pmc) группы. Суммарный выход пептида (в расчете на стартовую аминокислоту) составил 54.8%. При синтезе пептидов II и III гуанидиновые группы N-концевого Nα-метилзамещенного остатка Arg1 защищали с помощью 4-метокси-2,3,6-триметилбензолсульфонильной (Mtr) защитой, боковые группы остатков Arg7,8 блокировали Pmc-защитой. При этом получали пептиды формулы II и III с выходами 38.8 и 47.1% соответственно в расчете на стартовую аминокислоту, присоединенную к полимеру. В таблице 1 приведена сравнительная оценка известных и заявленного способов твердофазного синтеза нонапептидов (I-III).
Недостатками известных способов являются использование дорогих и труднодоступных производных аргинина, сложность отщепления защит аргинина по окончании синтеза (длительное время 16 ч и побочные реакции, связанные с отщеплением арилсульфонильных защит гуанидиновой группы остатков Arg), недостаточно высокий выход целевого продукта и многостадийность процесса.
Вышеуказанные недостатки делают известный способ малопригодным для крупномасштабного синтеза нонапептидов формулы (I-III).
Задачей изобретения является создание способа получения нонапептидов, который упрощает процедуру синтеза граммовых количеств нонапептидов формулы I-III, приводящую к: 1) отсутствию побочных реакций при удалении арилсульфонильных защит гуанидиновой функции остатков аргинина, 2) сокращению времени заключительного деблокирования пептидов, 3) удешевлению используемых производных аминокислот, 4) упрощению очистки сырого продукта.
Технический результат заключается в упрощении способа.
Технический результат достигается тем, что твердофазный синтез нонапептидов формулы I-III:
R-Arg1-Lys2-Lys3-Tyr4-Lys5-Tyr6-Arg7-Xaa8-Lys9-NH2,
где
R = Н, Хаа = L-Arg (I)
R = Me Xaa = L-Arg (II)
R = H, Хаа = D-Arg (III)
осуществляют путем последовательного наращивания пептидной цепи на полимерной матрице до получения соответствующего нонапептидилполимера с использованием производных аргинина с незащищенной гуанидиновой функцией.
Осуществление способа
При получении нонапептидов заявляемым способом iV-концевой остаток аргинина присоединяют в виде Nα- Вос(трет-бутилоксикарбонил)-Х-Arg-ОН, где X = H(I), X = СН3 (II), (III). Остатки Z-Arg7 и L-Arg8 (I) и (II) или D-Arg8 (III) вводят в пептидную цепь в виде соответствующих Fmoc-производных. На Фиг. 2 показана схема синтеза нонапептидов (II)-(III) в соответствии с заявляемым способом. Используемые производные аргинина являются недорогими коммерчески доступными продуктами, которые могут быть получены в одну стадию обычными методами органической химии в растворе. В ходе ТФС на стадии присоединения соответствующих остатков L-, D- и Nα-Ме-L-аргинина осуществляется временное блокирование его боковой гуанидиновой функции протонированием (солеобразованием).
Для протежирования гуанидиновой группы при синтезе пептидов в растворе используют хлор- и бромгидраты.
1-гидроксибензотриазол эффективно протонирует гуанидиновую группу с образованием солей, хорошо растворимых в применяемых для ТФС растворителях. В заявляемом способе присоединение Nα-Fmoc-L-Arg-OH или Nα-Fmoc-D-Arg-OH проводят в присутствии 2-3 эквивалентов HOBt по отношению к Fmoc-аминокислоте и эквивалентного количества Ν,Ν′-диизопропилкарбодиимида. Применение 1-гидроксибензотриазола (HOBt) в твердофазном синтезе (ТФС) пептидов широко известно. HOBt существенно увеличивает скорость реакции ацилирования с помощью карбодиимидов, эффективно подавляет рацемизацию и образование N-ацилмочевины.
Другим важным преимуществом предлагаемого способа является возможность использования конденсирующих агентов на основе солей фосфония/урония, таких как бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфат (ВОР), 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (HBTU) или тетрафторборат (TBTU) без применения органического основания. Обычно при использовании этих реагентов в ТФС для активации карбоксильной группы применяют 2-3 эквивалента органического основания (диизопропилэтиламина или N-метилморфолина), необходимого для связывания кислых противоионов - гексафторфосфата или тетрафторбората. Известно, что избытки оснований способны привести к рацемизации присоединяемой аминокислоты. Оказалось, что свободная гуанидиновая группа эффективно выполняет роль акцептора кислых противоионов и при этом происходит ее блокирование. Таким образом в случае активации карбоксильной группы Fmoc-Arg-OH с помощью солей фосфония/урония его гуанидиновая группа (рКа 12,5) играет роль органического основания. Дополнительным преимуществом заявленного способа является тот факт, что активированное производное аргинина получают in situ в условиях непрерывного технологического процесса, не требующего введения дополнительной стадии приготовления активированных производных аргинина.
Так как нонапептиды I-III содержат по три остатка аргинина в молекуле, в ходе деблокирования α-аминогруппы в аргининсодержащих пептидилполимерах, начиная с дипептидилполимера, освобождается не только α-аминогруппа, но происходит и депротонирование гуанидиновой функции уже имеющихся в растущей на полимере цепи остатков аргинина. В дальнейшем при присоединении следующей аминокислоты, независимо от выбранного способа конденсации, в реакцию ацилирования вступает не только α-аминогруппа, но и гуанидиновая группа аргинина. В результате образуются побочные продукты, которые не только понижают выход целевого вещества, но и существенно осложняют его очистку.
Для предотвращения побочной реакции ацилирования гуанидиновой функции остатков аргинина после каждой стадии деблокирования аргининсодержащих пептидилполимеров нужно проводить дополнительное протонирование аргинина. Дополнительная обработка пептидилполимера 5% раствором HOBt в Ν,Ν-диметилформамиде, Ν,Ν-диметилацетамиде или N-метипирролидоне в течение 5 мин перед конденсацией исчерпывающе защищает гуанидиновую группу остатков аргинина протонированием и при этом не препятствует ацилированию α-аминогруппы пептида, растущего на полимере. HOBt - нейтральная молекула, хорошо растворимая в органических растворителях, причем эти растворы устойчивы при хранении.
С практической точки зрения эта дополнительная промывка органично вписывается в технологический процесс ТФС, что важно при масштабировании синтеза для получения пептидов в укрупненных количествах.
По окончании синтеза полученный нонапептидилполимер обрабатывают деблокирующей смесью, в одну стадию отщепляют все защитные группы и полимерную матрицу и выделяют целевой продукт с помощью высокоэффективной жидкостной хроматографии. Чистоту полученных пептидов определяют с помощью ВЭЖХ на обращенной фазе, структуру пептидов подтверждают данными спектроскопии 1Н-ЯМР и масс-спектрометрии. Сравнительная оценка выходов и чистоты пептидов, полученных описанными и заявляемым способами, представлена графическими материалами. В таблице 1 представлена сравнительная оценка известных и заявленного способов твердофазного синтеза нонапептидов (I-III). На Фиг. 3 показан профиль аналитической ВЭЖХ сырого продукта твердофазного синтеза нонапептида (I) H-Arg-Lys-Lys-Tyr-Lys-Tyr-Arg-Arg-Lys-NH2 (а) - известным способом, (б) - заявленным способом. Содержание целевого пептида (I) составляет 55% (а) и 70% (б) соответственно. Колонка Kromasil C18 (4.6×250 мм), размер частиц 5 мкм, подвижная фаза: буфер А 0.05 M KH2PO4, рН 3, буфер Б 70% ацетонитрила + 30% буфера А, градиент Б от 0 до 60% за 30 мин, скорость элюции 1 мл/мин. На Фиг. 4 показан профиль аналитической ВЭЖХ сырого продукта твердофазного синтеза нонапептида (III) H-MeArg-Lys-Lys-Tyr-Lys-Tyr-Arg-DArg-Lys-NH2 (а) - известным способом, (б) - заявленным способом. Содержание пептида (III) составляет 76% (а) и 83% (б) соответственно. Колонка Kromasil C18 (4.6×250 мм), размер частиц 5 мкм, подвижная фаза: буфер А 01% TFA, буфер Б - 80% ацетонитрила + 20 буфера А, градиент Б от 0 до 60% за 30 мин, скорость элюции 1 мл/мин.
Способ иллюстрируется приведенными ниже примерами.
Пример 1. Синтез пептида H-Arg1-Lys2-Lys3-Tyr4-Lys5-Tyr6-Arg7-Arg8-Lys9-NH2 (I)
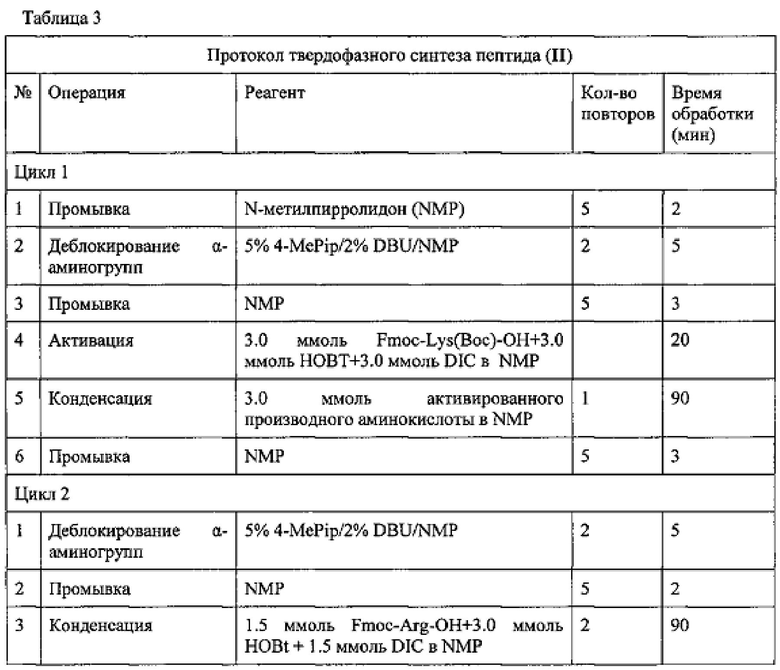
В работе использованы производные аминокислот (АА) и гексафторфосфат(бензотриазол-1-ил)окси-трис(диметиламино)фосфония (ВОР) NovaBiochem, Bachem, Швейцария), Ν,Ν′-диизопропилкарбодиимид (DIC), Ν,Ν-диизопропилэтиламин (DIPEA), гидроксибензотриазол (HOBt), триизобутилсилан (TIBS) компании Fluka, Швейцария, 4-метилпиперидин (4-MePip, Aldrich, США). Для синтеза применяли N-метилпирролидон, дихлорметан (DCM), трифторуксусную кислоту (TFA) компании Fluka, Швейцария, для хроматографии - ацетонитрил (Panreac, Испания). Аналитическую высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили на хроматографе (Gilson, Франция), использовали колонку Kromasil C18, 5 мкм, (4.6×250 мм) (Akzo Nobel, США), в качестве элюентов использовали буфер А - 0.05М KH2HO4, рН 3, буфер Б - 70% ацетонитрила в буфере А (условия 1) или буфер А - 0.1% TFA, буфер Б - 80% ацетонитрила в буфере А (условия 2), элюция проводилась градиентом концентрации буфера Б в буфере А от 0% до 60% за 30 мин. Скорость потока 1 мл/мин, детекция при 220 нм. Структура полученных пептидов доказана спектрами 1Н-ЯМР и данными масс-спектрометрии. 1Н-ЯМР-спектры снимали на спектрометре WM-500 (Bruker) 500 МГц (ФРГ) в дейтерированном диметилсульфоксиде (DMSO-d6) при 300 K, концентрация пептидов составляла 2-3 мг/мл. Химические (δ, м.д.) сдвиги измерялись относительно тетраметилсилана. Масс-спектры регистрировали на приборе UltraflexTOF/TOF (Bruker Daltonics, ФРГ) с времяпролетной базой методом MALDI. Для твердофазного синтеза использовали сополимер стирола с 1% дивинилбензола с 4-(2,4-диметоксифенил)-9-флуоренилметоксикарбонил-аминометилфенокси - якорной группой (Rink-amide-полимер) фирмы Nova BioChem, Швейцария, предназначенный для получения амидов пептидов, содержащий 0.64 ммоль/г аминогрупп. Синтез амида нонапептида проводили с С-конца, ступенчато (присоединяя по одной аминокислоте), исходя из 1.0 г (0.64 ммоль) Rink-amide-полимера в соответствии с протоколом. Для присоединения всех аминокислот, за исключением аргинина, использовали 4-кратные избытки ацилирующих агентов однократно. Гуанидиновые группы остатков Arg1,7,8 блокировали с помощью протонирования. В таблице 2 представлен протокол твердофазного синтеза пептида (I). При присоединении остатков аргинина конденсацию проводили дважды с использованием 2-кратных избытков ацилирующих агентов. Полноту протекания реакции контролировали с помощью теста на свободные аминогруппы в пептидилполимерах с нингидрином.
Заключительное деблокирование и отщепление нонапептида от полимера проводили в одну стадию путем обработки соответствующего нонапептидилполимера смесью 20 мл TFA, 0.5 мл деионизованной воды и 0.5 мл TIBS в течение 2 ч. Затем полимер отфильтровывали, промывали 2×5 мл деблокирующей смеси, фильтрат упаривали и к остатку прибавляли сухой эфир. Осадок отфильтровывали, промывали DCM (3×10 мл), эфиром (3×10 мл), сушили в вакуум-эксикаторе. Получено 0.82 г сырого продукта твердофазного синтеза с содержанием основного вещества 72%. Вещество очищали порциями с помощью препаративной ВЭЖХ на приборе Beckman (США), используя колонку Диасорб С16 130Т (25×250 мм), размер частиц сорбента 10 мкм. В качестве элюентов использовали буфер А - 0.01 M раствор ацетата аммония и буфер Б - 80% ацетонитрила в воде. Элюцию проводили градиентом 0.5% в минуту буфера Б в буфере А от 100% буфера А со скоростью 10 мл/мин. Пептиды детектировали при длине волны 220 нм. Фракции, содержащие целевой продукт, объединяли, ацетонитрил упаривали и лиофилизовали. В итоге получили 0.51 г (61.2% в расчете на стартовую аминокислоту) амида нонапептида (I). Гомогенность продукта, определенная с помощью аналитической ВЭЖХ, составляет 97.4%. Масс-спектр: 1325.1, вычислено 1324.6.
Пример 2. H-(N-Me)-Arg1-Lys-Lys-Tyr-Lys-Tyr-Arg7-Arg8-Lys-NH2 (II)
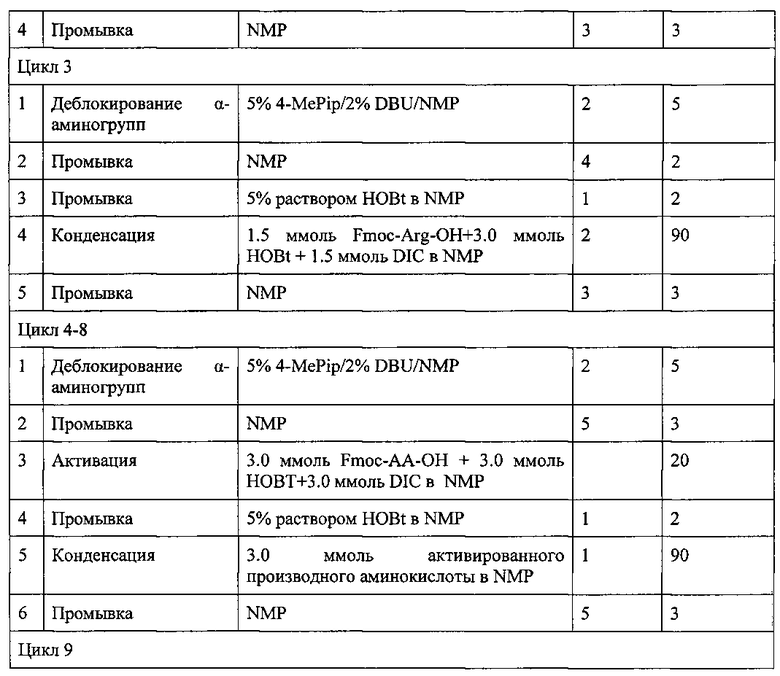
Синтез амида нонапептида (II) проводили аналогично синтезу пептида (I) по схеме 2, ступенчато (присоединяя по одной аминокислоте), исходя из 1.56 г (1.0 ммоль) Rink-amide-полимера. В таблице 3 представлен протокол твердофазного синтеза пептида (II). Для присоединения всех аминокислот, за исключением аргинина, использовали 3-кратные избытки ацилирующих агентов однократно. Гуанидиновые группы остатков Arg блокировали с помощью протонирования (см. протокол). При присоединении остатков аргинина конденсацию проводили дважды с использованием 1.5-кратных избытков ацилирующих агентов. Полноту протекания реакции контролировали с помощью теста на свободные аминогруппы в пептидилполимерах с нингидрином.
Заключительное деблокирование нонапептида (II) проводили в одну стадию путем обработки соответствующего нонапептидилполимера смесью 30 мл TFA, 0.75 мл деионизованной воды и 0.75 мл TIBS в течение 2 ч. Затем полимер отфильтровывали, промывали 2×7 мл деблокирующей смеси, фильтрат упаривали и к остатку прибавляли сухой эфир. Осадок отфильтровывали, промывали DCM (3×10 мл), эфиром (3×15 мл), сушили в вакуум-эксикаторе. 1.24 г сырого продукта твердофазного синтеза с содержанием основного вещества 70% очищали порциями с помощью препаративной ВЭЖХ, как описано в примере 1. В итоге получили 0.715 г (53.4% в расчете на стартовую аминокислоту) амида нонапептида (II). Гомогенность продукта, определенная с помощью аналитической ВЭЖХ в условиях 1 и 2, составляет 97.5%. Масс-спектр: 1338.9, вычислено 1338.7.
Пример 3. H-(N-Me)-Arg1-Lys-Lys-Tyr-Lys-Tyr-Arg7-D-Arg8-Lys9-NH2 (III)
Синтез амида нонапептида (III) проводили аналогично синтезу пептида (I) по схеме 2, ступенчато (присоединяя по одной аминокислоте), исходя из 3.12 г (2.0 ммоль) Rink-amide-полимера. В таблице 4 представлен протокол твердофазного синтеза пептида (III). Для присоединения всех аминокислот, за исключением аргинина, использовали 3-кратные избытки ацилирующих агентов однократно. Гуанидиновые группы остатков Arg блокировали с помощью протонирования (см. протокол). При присоединении остатков аргинина конденсацию проводили дважды с использованием 1.5-кратных избытков ацилирующих агентов. Полноту протекания реакции контролировали с помощью теста на свободные аминогруппы в пептидилполимерах с нингидрином.
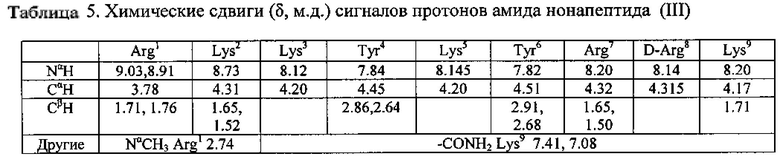
Заключительное деблокирование и отщепление нонапептида от полимера проводили в одну стадию путем обработки соответствующего нонапептидилполимера смесью 60 мл TFA, 1.5 мл деионизованной воды и 1.5 мл TIBS в течение 2 ч. Затем полимер отфильтровывали, промывали 2×15 мл деблокирующей смеси, фильтрат упаривали и к остатку прибавляли сухой эфир. Осадок отфильтровывали, промывали DCM (3×20 мл), эфиром (3×30 мл), сушили в вакуум-эксикаторе. Сырой продукт твердофазного синтеза (2.48 г) с содержанием основного вещества 83% очищали порциями. В итоге получили 1.62 г (60.5% в расчете на стартовую аминокислоту) амида нонапептида (III). Гомогенность продукта, определенная с помощью аналитической ВЭЖХ в условиях 1 и 2, составляет 97.9%. Масс-спектр: 1339.0, вычислено 1338.7. На Фиг. 5 приведены данные спектроскопии 1Н-ЯМР. В таблице 5 показаны химические сдвиги (δ, м.д.) сигналов протонов амида нонапептида (III).







| название | год | авторы | номер документа |
|---|---|---|---|
| АМИД НОНАПЕПТИДА, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ПРЕДОТВРАЩАТЬ ПОВЫШЕНИЕ ПРОНИЦАЕМОСТИ ЭНДОТЕЛИЯ СОСУДОВ | 2009 |
|
RU2402565C1 |
| ДОДЕКАПЕПТИДЫ, ОБЛАДАЮЩИЕ КАРДИОПРОТЕКТОРНЫМИ СВОЙСТВАМИ | 2010 |
|
RU2457216C1 |
| ЦИКЛИЧЕСКИЙ НОНАПЕПТИД, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ИНГИБИРОВАТЬ КИНАЗУ ЛЕГКИХ ЦЕПЕЙ МИОЗИНА | 2010 |
|
RU2443710C1 |
| АМИД НОНАПЕПТИДА, ПРЕПЯТСТВУЮЩИЙ ПОВЫШЕНИЮ ГИПЕРПРОНИЦАЕМОСТИ СОСУДИСТОГО ЭНДОТЕЛИЯ | 2012 |
|
RU2493164C1 |
| Пептид, обладающий способностью ингибировать миграцию клеток, стимулированную белком TARC | 2016 |
|
RU2629198C1 |
| СПОСОБ ИММУНОФЕРМЕНТНОГО АНАЛИЗА ДЛЯ ОПРЕДЕЛЕНИЯ АУТОАНТИТЕЛ К β-АДРЕНОРЕЦЕПТОРУ В ПЛАЗМЕ И СЫВОРОТКЕ КРОВИ ЧЕЛОВЕКА | 2011 |
|
RU2452964C1 |
| СИНТЕТИЧЕСКИЙ АНТИГЕН, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ СВЯЗЫВАТЬ АУТОАНТИТЕЛА К β-АДРЕНОРЕЦЕПТОРУ | 2007 |
|
RU2356576C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДОДЕКАПЕПТИДА И ТРИПЕПТИД ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2340626C1 |
| ТЕТРАДЕКАПЕПТИДЫ, УЛУЧШАЮЩИЕ ВОССТАНОВИТЕЛЬНУЮ ФУНКЦИЮ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ ПРИ ИШЕМИИ | 2017 |
|
RU2648846C1 |
| ПРОИЗВОДНЫЕ ГЕМИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2238950C2 |





Изобретение относится к твердофазному способу получения нонапептидов формулы I-III:

1. Способ получения нонапептидов формулы I-III:
R-Arg1-Lys2-Lys3-Tyr4-Lys5-Tyr6-Arg7-Xaa8-Lys9-NH2,
где
R = Н, Хаа = L-Arg (I)
R = Me, Хаа = L-Arg (II)
R = H, Хаа = D-Arg (III)
твердофазным методом путем последовательного наращивания пептидной цепи, начиная с С-концевой аминокислоты, ковалентно связанной с полимерной матрицей, последующей обработки полученных нонапептидилполимеров деблокирующим агентом для отщепления защитных групп и полимерной матрицы и выделения конечного продукта с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ), отличающийся тем, что для блокирования гуанидиновой функции остатков L-, D- и Me- L-аргинина применяют протонирование.
2. Способ по п. 1, отличающийся тем, что для протонирования гуанидиновой группы в пептидилполимерах применяют дополнительную промывку 5% раствором 1-гидроксибензотриазола.
3. Способ по п. 1, отличающийся тем, что конденсации остатков аргинина повторяют дважды с использованием половинного количества ацилирующих агентов, а в качестве конденсирующего агента применяют ВОР, HBTU или TBTU без использования органического основания.
4. Способ по п. 1, отличающийся тем, что конденсации остатков аргинина повторяют дважды с использованием 2-кратных избытков производных аминокислоты, 2-кратных избытков Ν,Ν′-диизопропилкарбодиимида и 4-6-кратных избытков HOBt по отношению к содержанию аминогрупп в пептидилполимерах.
| АМИД НОНАПЕПТИДА, ПРЕПЯТСТВУЮЩИЙ ПОВЫШЕНИЮ ГИПЕРПРОНИЦАЕМОСТИ СОСУДИСТОГО ЭНДОТЕЛИЯ | 2012 |
|
RU2493164C1 |
| ЦИКЛИЧЕСКИЙ НОНАПЕПТИД, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ИНГИБИРОВАТЬ КИНАЗУ ЛЕГКИХ ЦЕПЕЙ МИОЗИНА | 2010 |
|
RU2443710C1 |
| АМИД НОНАПЕПТИДА, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ПРЕДОТВРАЩАТЬ ПОВЫШЕНИЕ ПРОНИЦАЕМОСТИ ЭНДОТЕЛИЯ СОСУДОВ | 2009 |
|
RU2402565C1 |
| WO 2005108416 A2, 17.11.2005. | |||

