ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения производных R-бета-аминофенилмасляной кислоты формулы (I)

путем химического синтеза, включающему процесс разделения. Соединения формулы (I), которые получают в соответствии со способом по настоящему изобретению, можно использовать для синтеза ряда хиральных лекарств.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
С разработкой синтеза лекарств все больше хиральных лекарств синтезируют в виде отдельных энантиомеров. Производные R-бета-аминофенилмасляной кислоты, которые являются важными хиральными фармацевтическими промежуточными соединениями, обычно могут быть получены путем реакций восстановления, катализируемых хиральным катализатором. Этот способ описан в нескольких ссылках. Например, путь синтеза вышеупомянутого продукта раскрыт в J. Am. Chem. Soc, 1987, 5856. 2,4,5-Трифторфенилацетилацетоацетат используют в качестве исходного вещества, и Ru-(s)-BINAP используют в качестве хирального катализатора, а затем получают ацетат бета-гидрокси-2,4,5-трифторфенилмасляной кислоты. Затем ацетат R-бета-аминофенилмасляной кислоты может быть получен путем аминирования ацетата бета-гидрокси-2,4,5-трифторфенилмасляной кислоты. Способ получения хиральных производных R-бета-аминофенилмасляной кислоты раскрыт в J. Am. Chem. Soc, 1986, 7117 с использованием различных лигандов в качестве катализаторов восстановления. В публикации WO 2004085661 также раскрыт путь синтеза производных R-бета-аминофенилмасляной кислоты. В этой публикации раскрыт способ получения вышеупомянутых хиральных производных, как описано ниже: амид S-альфа-фенилглицина подвергают взаимодействию с 2,4,5-трифторфенилацетиламидом с получением производных альфа,бета-ненасыщенной бета-амино-2,4,5-трифторфенилмасляной кислоты, содержащих хиральный центр, а затем производные альфа, бета-ненасыщенной бета-амино-2,4,5-трифторфенилмасляной кислоты восстанавливают в присутствии катализатора оксида платины (PtO2) с получением хиральных производных бета-аминофенилмасляной кислоты. В публикации WO 2005020920 раскрыт способ получения соединений формулы (I) путем восстановления производных альфа, бета-ненасыщенной бета-амино-2,4,5-трифторфенилмасляной кислоты, с использованием димера хлор(1,5-циклооктадиен)родия(I) ([Rh(cod)Cl]2) и (R,S)-трет-бутил Josiphos в качестве катализаторов.
Получение производных бета-аминофенилмасляной кислоты путем хиральных реакций восстановления описано в нескольких ссылках, но результаты неудовлетворительны. Во-первых, катализаторы хирального восстановления, используемые в этих способах, обычно являются дорогостоящими, что по существу приводит к высоким затратам. На практике гомогенный катализ, вероятно, производит целевой продукт с высокой оптической чистотой. Однако рециклинг гомогенного катализатора затруднителен, что часто приводит к высоким затратам и делает этот путь синтеза непригодным для промышленных способов получения. Во-вторых, условия хирального восстановления обычно являются жесткими, хиральные катализаторы трудно получить, и способ является относительно сложным. В-третьих, поскольку селективность хиральных катализаторов часто низка, оптическая чистота продукта неудовлетворительна. Чтобы получить желаемый продукт, необходимо несколько стадий перекристаллизации, и способ непригоден для промышленных получений. Напротив, способ получения отдельных энантиомеров целевых продуктов, используя разделяющие агенты, демонстрирует преимущества во всех вышеописанных аспектах.
До настоящего времени получение производных R-бета-аминофенилмасляной кислоты путем использования разделяющих агентов не описано в ссылках. В свете фармацевтической ценности производных бета-аминофенилмасляной кислоты необходимо найти эффективный способ разделения для получения R-конфигурации производных бета-аминофенилмасляной кислоты, как упомянуто выше, с высокой оптической чистотой, с высокой эффективностью и высокими выходами.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
С целью преодоления недостатков предшествующего уровня техники задачей настоящего изобретения является предложение способа получения производных R-бета-аминофенилмасляной кислоты формулы (I):
где
Ar представляет собой незамещенный фенил или фенил, замещенный заместителями в количестве от одного до пяти, выбранными из группы, состоящей из атома фтора, метила, трифторметила и трифторметокси,
R1 представляет собой атом водорода или C1-6 алкил,
R2 представляет собой атом водорода или амино-защитную группу, включающую алкоксикарбонил и ацил, где алкоксикарбонил выбран из группы, состоящей из метоксилкарбонила, этоксилкарбонила и трет-бутоксилкарбонила, а ацил выбран из группы, состоящей из формацила, ацетила, хлорацетила, трихлорацетила, бензоила и фенилацила.
Способ включает приведенные ниже стадии:
(1) взаимодействие формиата аммония с незамещенным или замещенным фенилэтилацетоацетатом с получением имина, а затем взаимодействие имина с восстанавливающим агентом с получением рацемата бета-аминофенилмасляной кислоты эфира;
(2) взаимодействие рацемата бета-аминофенилмасляной кислоты эфира и разделяющего агента с образованием соли R-формы в спиртовом растворителе или в водном растворе спирта и кристаллизация этой соли; и
(3) гидролиз соли R-формы, образованной из бета-аминофенилмасляной кислоты эфира и разделяющего агента, или защита аминной группы бета-аминофенилмасляной кислоты эфира с получением производного R-бета-аминофенилмасляной кислоты формулы (I).
Одна форма осуществления настоящего изобретения дополнительно включает, что взаимодействие производных R-бета-аминофенилмасляной кислоты формулы (I), полученных на стадии (3), с соляной кислотой с получением соли соляной кислоты.
Хиральные фармацевтически промежуточные соединения, производные R-бета-аминофенилмасляной кислоты формулы (I), раскрытые в настоящем изобретении, могут быть получены, как изображено на приведенной ниже схеме:
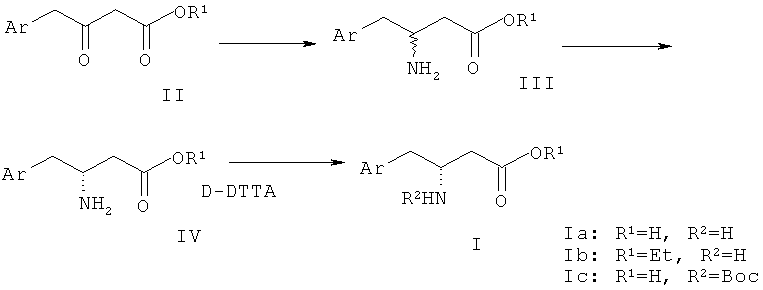
Предпочтительно в первом продукте (Ia): Ar представляет собой 2,4,5-трифторфенил, R1 и R2 представляют собой атом водорода.
Предпочтительно во втором продукте (Ib): Ar представляет собой 2,4,5-трифторфенил, R1 представляет собой этил и R2 представляет собой атом водорода.
Предпочтительно в третьем продукте (Ic): Ar представляет собой 2,4,5-трифторфенил, R1 представляет собой атом водорода, и R2 представляет собой трет-бутоксилкарбонил.
Чтобы лучше проиллюстрировать сущность изобретения, рассматривая репрезентативный способ получения хиральных фармацевтических промежуточных соединений, предпочтительно R-бета-амино-2,4,5-трифторфенилмасляной кислоты или ее фармацевтически приемлемых солей в качестве примеров, настоящее изобретение изложено далее по стадиям.
Способ получения R-бета-амино-2,4,5-трифторфенилмасляной кислоты включает приведенные ниже стадии.
Во-первых, формиат аммония подвергают взаимодействию с исходным веществом, 2,4,5-трифторфенилэтилацетоацетатом, с получением имина. Затем имин восстанавливают цианоборгидридом натрия с получением рацемата бета-аминофенилмасляной кислоты эфира. Во-вторых, R-бета-аминофенилмасляной кислоты эфир и разделяющий агент образуют соль R-формы в спиртовом растворителе или в водном растворе спирта. Соль R-бета-аминофенилмасляной кислоты эфира кристаллизуют. В-третьих, эту соль подвергают гидролизу или защите аминной группы с получением производных R-бета-аминофенилмасляной кислоты формулы (I) или их фармацевтически приемлемых солей.
В одной форме осуществления настоящего изобретения восстанавливающий агент, используемый на стадии (1), представляет собой цианоборгидрид натрия. Хиральный разделяющий агент, используемый на стадии (2), представляет собой хиральную диацилированную винную кислоту, включающую: дибензоил-D-винную кислоту, дибензоил-L-винную кислоту, ди-пара-толуоил-D-винную кислоту или ди-пара-толуоил-L-винную кислоту.
Предпочтительно хиральный разделяющий агент представляет собой ди-пара-толуоил-D-винную кислоту или ди-пара-толуоил-L-винную кислоту.
Разделяющий агент, ди-пара-толуоил-L-винную кислоту или ди-пара-толуоил-D-винную кислоту, используемую в способе по настоящему изобретению, можно использовать отдельно или вместе.
Кроме того, в способе получения производных R-бета-аминофенилмасляной кислоты спиртовой растворитель, используемый на стадии (2), представляет собой короткоцепочечный спирт из трех или менее чем трех атомов углерода. Предпочтительно он представляет собой метанол.
В одной форме осуществления настоящего изобретения водный раствор спирта, используемый на стадии (2), представляет собой водный раствор короткоцепочечного спирта из трех или менее чем трех атомов углерода.
В кратком изложении, задача, которая должна быть решена настоящим изобретением, состоит в получении отдельных энантиомеров формулы (I) путем химического синтеза. Способ включает разделение рацемата производных бета-аминофенилмасляной кислоты разделяющим агентом.
В вышеупомянутой схеме реакций соединение (II) (R1 представляет собой C1-6 алкил или атом водорода) может быть получено в соответствии с патентом США №5296482. 2,4,5-Трифторбромбензол, используемый в качестве исходного вещества, алкилируют диэтилмалонатом. Затем алкилированный продукт подвергают гидролизу и декарбоксилируют с получением 2,4,5-трифторуксусной кислоты. Эту кислоту конденсируют с кислотой Мелдрама. Затем продукт конденсации обрабатывают спиртом и декарбоксилируют путем нагревания реакционной смеси с получением 2,4,5-трифторфенилэтилацетоацетата, который можно использовать в качестве исходного вещества для получения продукта формулы (I).
2,4,5-Трифторфенилацетилацетоацетат (II) (R1 представляет собой этил) подвергают взаимодействию с формиатом аммония с получением имина. Затем имин восстанавливают цианоборгидридом натрия с получением соединения (III) (R1 представляет собой этил). Соединение (III) разделяют разделяющим агентом с получением соединения (IV) (R1 представляет собой этил). Затем соединение (IV) подвергают гидролизу, или аминную группу соединения (IV) защищают с получением продукта (I). Когда R1 и R2 представляют собой различные группы, как показано на приведенной выше схеме, продукт (I), содержащий различные заместители, может представлять собой определенное соединение на различных стадиях, такое как (Ia), (Ib) и (Ic), как проиллюстрировано выше.
После тщательного исследования авторы изобретения обнаружили, что различные общепринятые кислотные разделяющие агенты по существу неэффективны для разделения рацемата формулы (III) в вышеописанном процессе разделения, за исключением R-камфорсульфоновой кислоты, обладающей определенной селективностью. Некоторые кислотные разделяющие агенты не могут взаимодействовать с рацематом формулы (III) таким образом, чтобы эффективно образовывать кристаллические осадки в растворителях. Некоторые кислотные разделяющие агенты могут взаимодействовать с рацематом формулы (III) с образованием кристаллических осадков в растворителях, но отсутствует селективность, и полученные в результате осадки все еще представляют собой рацемическую смесь. После проведения некоторых исследований авторы изобретения идентифицировали, что неэффективные разделяющие агенты включают L-винную кислоту, R-миндальную кислоту, N-ацетил-L-глутаминовую кислоту, L-лейцин и тому подобное.
Далее, авторы изобретения обнаружили, что среди большого числа общепринятых кислотных разделяющих агентов, которые были протестированы, только винные кислоты, диацилированные бензоилом или замещенным бензоилом, такие как дибензоил-L-винная кислота (L-DBTA), дибензоил-D-винная кислота (D-DBTA), ди-пара-толуоил-L-винная кислота (L-DTTA) или ди-пара-толуоил-О-винная кислота (D-DTTA), могут эффективно разделять производные бета-аминофенилмасляной кислоты (R) конфигурации и (S) конфигурации.
В целом настоящее изобретение относится к способу получения производных R-бета-аминофенилмасляной кислоты (I). Этот способ включает не только стадию химического получения рацемата формулы (III), но также включает стадию взаимодействия разделяющего агента с рацематом формулы (III) с получением соответствующей соли в спиртовом растворителе или в водном растворе спирта и кристаллизации соответствующей соли с получением производных R-бета-аминофенилмасляной кислоты формулы (I) или соответствующих S-производных бета-аминофенилмасляной кислоты. Разделяющий агент представляет собой дибензоил-L-винную кислоту (L-DBTA), дибензоил-D-винную кислоту (D-DBTA), ди-пара-толуоил-L-винную кислоту (L-DTTA) или ди-пара-толуоил-D-винную кислоту (D-DTTA), и предпочтительно ди-пара-толуоил-L-винную кислоту или ди-пара-толуоил-D-винную кислоту.
Чтобы получить отдельные энантиомеры соединений формулы (I), такие как R-конфигурация (Ib), 1 моль D-DTTA подвергают взаимодействию с 2 моль рацемата формулы (III) (R1=этил) в метаноле с получением соответствующей соли. Соответствующую соль кристаллизуют с получением кристаллов с R-конфигурацией (IV) (R1=этил). R-конфигурацию соединения (Ib) получают из кристаллов. Напротив, когда L-DTTA используют в качестве разделяющего агента, получают продукт S-конфигурации.
Далее процесс разделения по настоящему изобретению включает стадию перекристаллизации после стадий образования и кристаллизации соли. Разделяющий агент, ди-пара-толуоил-L-винную кислоту (L-DTTA) и ди-пара-толуоил-D-винную кислоту (D-DTTA), используемую в настоящем изобретении, можно использовать отдельно или вместе. Конкретно настоящее изобретение относится к способу получения и разделения промежуточных соединений, производных бета-аминофенилмасляной кислоты формулы (I). Задача, которая должна быть решена настоящим изобретением, состоит в получении вышеописанного фармацевтически приемлемого, оптически чистого соединения формулы (I) R-конфигурации с хорошим выходом путем использования ди-пара-толуоил-L-винной кислоты. Этот способ характеризуется тем, что рацемат формулы (III) подвергают взаимодействию с кислотным разделяющим агентом в конкретном растворителе с получением соответствующей соли и селективно осаждают кристаллы солей желаемого хирального промежуточного соединения, производных бета-аминофенилмасляной кислоты.
Способ разделения промежуточного амина формулы (III) включает способ взаимодействия промежуточного амина формулы (III) с хиральным разделяющим агентом с получением соответствующей соли, перекристаллизации соответствующей соли с образованием кристаллических осадков и экстракции перекристаллизованных осадков с получением промежуточного амина формулы (Ib). Способ разделения может дополнительно включать стадию гидролиза (Ib) с получением (Ia) или защиты аминной группы с получением (Ic). Все хиральные фармацевтические промежуточные соединения можно использовать для синтеза ряда активных фармацевтических соединений.
Что касается количества разделяющего агента, в теории, поскольку для реакции кислотно-основной нейтрализации необходимы равные числа моль кислоты и основания, молярное отношение аминов к разделяющему агенту может составлять 2:1. Если желательна соль с определенной конфигурацией, молярное отношение может составлять 4:1. Если желательны соли присоединения кислоты с равными молярными количествами кислоты и основания, молярное отношение может составлять 1:1. Однако после проведения некоторых исследований авторы изобретения обнаружили, что более высокая доля разделяющего агента дает более удовлетворительные выходы продукта разделения с высокой хиральной чистотой. Вообще говоря, подходящее молярное отношение аминных промежуточных соединений к разделяющему агенту может составлять от 4:1 до 1:1, предпочтительное молярное отношение составляет от 2:1 до 1:1. Избыточное количество разделяющего агента не улучшает разделение.
Способ разделения рацемата формулы (III) можно осуществлять в общепринятом растворителе. Предпочтительно этот способ осуществляют в органическом растворителе, более предпочтительно в спиртовом растворителе. Спиртовой растворитель можно использовать отдельно или в комбинации с другими органическими растворителями. Спиртовые растворители, используемые в настоящем изобретении, включают спиртовые растворители, используемые отдельно, а также смешанные растворители спирта и основания. Спиртовой растворитель может представлять собой короткоцепочечный спирт из 3 или менее трех атомов углерода. Предпочтительно растворитель представляет собой метанол. Водный раствор спирта может представлять собой водный раствор вышеупомянутого короткоцепочечного спирта.
С целью улучшения хиральной чистоты аминов формулы (I) иногда необходимо перекристаллизовать полученную разделенную соль. Способ разделения в целом можно осуществлять при комнатной температуре; при необходимости в условиях нагревания. Как правило, стадию перекристаллизации осуществляют в условиях нагревания. Во-первых, соль, полученную в результате разделения, растворяют в конкретном растворителе, а затем медленно осуществляют перекристаллизацию при комнатной температуре. Как правило, после перекристаллизации дважды хиральная чистота часто удовлетворительна, и значение эй, как правило, выше 99%.
Способ получения свободного промежуточного соединения является общепринятым, где используемое основание предпочтительно представляет собой бикарбонат натрия. Экстрагирующий растворитель может представлять собой гидрофобный органический растворитель, используемый при общепринятых экстракциях, такой как этилацетат, метиленхлорид и хлороформ, и т.д., предпочтительно этилацетат и хлороформ. Способ гидролиза соединения формулы (I) также является общепринятым, и используемое основание предпочтительно представляет собой гидроксид натрия. Кислота, используемая при образовании соли, предпочтительно представляет собой соляную кислоту. Способ образования соли является общепринятым. Его может легко осуществить обычный специалист в данной области техники.
Оптическая чистота эфира или кислоты соединения формулы (I) в соответствии с настоящим изобретением составляет более чем 99%, что особенно пригодно в качестве промежуточного соединения синтеза хиральных лекарств.
ПРЕДПОЧТИТЕЛЬНЫЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение подробно проиллюстрировано приведенными ниже примерами, которые не следует рассматривать как ограничивающие объем настоящего изобретения.
ПРИМЕР ПОЛУЧЕНИЯ 1
114 г (0,60 моль) 2,4,5-трифторфенилуксусной кислоты растворяли в 600 мл ТГФ. К этой смеси добавляли 107 г (0,66 моль) карбонилдиимидазола при перемешивании (когда добавляли часть карбонилдиимидазола, образовывалось большое количество твердого вещества; затем это твердое вещество в результате растворялось в растворе при дальнейшем добавлении). После завершения добавления реакционную смесь нагревали до 50°С. Добавляли 95,1 г (0,66 моль) кислоты Мелдрама, и смесь выдерживали в течение 3 часов при 50°C. Смесь концентрировали для удаления ТГФ, и остаток растворяли в воде (600 мл) и дихлорметане (800 мл), а затем значение рН доводили до 2. Водную фазу отделяли, и органическую фазу промывали 0,1 н. HCl и водой (600 мл) соответственно. Органическую фазу высушивали и концентрировали с получением 182 г конденсата, 5-[2-(2,4,5-трифторфенил)-ацетил]-2,2-диметил-1,3-диоксан-4,6-диона, в виде твердого вещества (перекристаллизацию можно осуществить в этилацетате с получением белого твердого вещества). Точка плавления: 101,5-103,5°С, Выход: 96%.
ПРИМЕР 1
60 г конденсата (0,190 моль), полученного из примера получения 1, растворяли в этаноле (600 мл). Смесь перемешивали при 70°С в течение 3 часов и к смеси добавляли раствор 2,4,5-трифторфенилэтилацетоацетата в этаноле. К смеси добавляли 70 г формиата аммония (1,11 моль), и реакционную смесь нагревали до образования флегмы в течение 3 часов. После охлаждения до 40°С к реакционной смеси медленно добавляли 15 г цианоборгидрида натрия (0,239 моль), и реакционную смесь нагревали до образования флегмы в течение 2 часов. После охлаждения смесь концентрировали для удаления этанола, и остаток растворяли в воде, значение рН доводили до 9. Смесь экстрагировали дихлорметаном и промывали небольшим количеством воды. Органическую фазу высушивали и концентрировали с получением 45 г бета-аминофенилмасляной кислоты этилового эфира в виде коричневого масла. Выход: 90,5%.
ПРИМЕР 2
5,18 г (20 ммоль) рацемата бета-аминофенилмасляной кислоты этилового эфира растворяли в метаноле (60 мл) и добавляли 3,86 г (10 ммоль) D-DTTA при перемешивании. Из реакционного раствора быстро выпадало в осадок большое количество белого твердого вещества. Смесь нагревали до образования флегмы в течение 1-2 часов (твердое вещество не полностью растворялось в растворе). После охлаждения до температуры ниже 10°С полученные в результате осадки собирали фильтрованием и промывали небольшим количеством метанола, а затем осуществляли перекристаллизацию в метаноле. После перекристаллизации дважды получили 3,37 г белого порошка. Точка плавления: 187,0-188,0°С, [а]D 25=+96,7∈ (С1, 0,1 М NaOH). 3,0 г белого твердого вещества обрабатывали основанием с получением 1,20 г R-бета-аминофенилмасляной кислоты этилового эфира (Ib). Оптическая чистота (Ib) составляла более 99,7%, и выход первого разделения составлял 52,2%.
Остаточные растворы, полученные в результате во время вышеописанного процесса разделения и двух процессов перекристаллизации, объединяли, а затем концентрировали до сухости с получением сырого продукта. Этот сырой продукт обрабатывали насыщенным бикарбонатом натрия с получением свободного амина. Раствор экстрагировали хлороформом с получением 4,7 г рацемата, в основном, состоящего из S-конфигурации. Анализ ВЭЖХ показал 71,3% S-конфигурации. Рацемат растворяли в метаноле (60 мл) и добавляли 3,86 г L-DTTA (10 ммоль) для обратного разделения. Смесь нагревали до образования флегмы до получения прозрачного раствора. После охлаждения кристаллы выпадали в осадок из реакционного раствора. Полученные в результате осадки собирали фильтрованием, а затем высушивали с получением сырого продукта. Анализ ВЭЖХ показал 95,6% S-конфигурации. Сырой продукт S-конфигурации растворяли в 60 мл метанола, и смесь нагревали до образования флегмы до получения прозрачного раствора. После охлаждения кристаллы выпадали в осадок из раствора. Полученные в результате осадки собирали фильтрованием, а затем высушивали с получением 3,44 г соли L-DTTA S-конфигурации. Точка плавления: 182,0-183,5°С, [а]D 25=-90,3∈ (С1, 0,1 М NaOH). Выход обратного разделения составлял 53,3%. Анализ ВЭЖХ показал 98,4% S-конфигурации.
Остаточный раствор, полученный во время вышеописанного процесса обратного разделения и процесса перекристаллизации, объединяли, а затем концентрировали до сухости с получением сырого продукта. Этот сырой продукт обрабатывали насыщенным бикарбонатом натрия с получением свободного амина. Раствор экстрагировали хлороформом с получением 1,9 г рацемата, состоящего, в основном, из R-конфигурации. Анализ ВЭЖХ показал 67,4% R-конфигурации. Рацемат растворяли в 20 мл метанола и добавляли 1,5 г D-DTTA. Смесь нагревали до образования флегмы до получения прозрачного раствора. После охлаждения кристаллы выпадали в осадок в результате разделения. Осадок собирали фильтрованием и высушивали с получением 0,92 г соли. Анализ ВЭЖХ показал 99,30% R-конфигурации. Полученную в результате соль обрабатывали насыщенным бикарбонатом натрия с получением свободного амина. Раствор экстрагировали хлороформом с получением 0,5 г промежуточного амина (Ib) R-конфигурации. Выход: 19,5%. Анализ ВЭЖХ показал 99,3% R-конфигурации. Суммарный выход разделения составлял 71,4%.
ПРИМЕР 3
5,18 г (20 ммоль) рацемата бета-аминофенилмасляной кислоты этилового эфира растворяли в метаноле (60 мл) и добавляли 3,86 г (10 ммоль) L-DTTA при перемешивании. Из реакционного раствора быстро выпадало в осадок большое количество белого твердого вещества. Смесь нагревали до образования флегмы в течение 1-2 часов (твердое вещество не полностью растворялось в растворе). После охлаждения до температуры ниже 10°С полученные в результате осадки собирали фильтрованием и промывали небольшим количеством метанола. Остаточный раствор концентрировали до сухости и добавляли 70 мл воды, значение рН доводили до 8 насыщенным раствором бикарбоната натрия. Смесь экстрагировали дихлорметаном. Органическую фазу промывали водой и концентрировали с получением продукта в виде масла.
Этот продукт в виде масла растворяли в метаноле (60 мл) и добавляли 3,86 г D-DTTA (10 ммоль) при перемешивании. Из реакционного раствора быстро выпадало в осадок большое количество белого твердого вещества. Смесь нагревали до образования флегмы в течение 1-2 часов (твердое вещество не полностью растворялось в растворе). После охлаждения до температуры ниже 10°С кристаллы выпадали в осадок из раствора. Полученные в результате осадки собирали фильтрованием и промывали небольшим количеством метанола, а затем осуществляли перекристаллизацию в метаноле. После перекристаллизации получили 4,17 г белого порошка. Точка плавления: 185,0-186,5°С, [а]D 25=+95,8∈ (С1, 0,1 М NaOH). 4,0 г белого твердого вещества обрабатывали основанием с получением 1,61 г R-бета-аминофенилмасляной кислоты этилового эфира (Ib). Оптическая чистота Ib составляла более 99,7%, и полученный в результате выход составлял 64,8%.
ПРИМЕР 4
5,18 г (20 ммоль) рацемата бета-аминофенилмасляной кислоты этилового эфира растворяли в этаноле (120 мл) и добавляли 3,86 г (10 ммоль) D-DTTA при перемешивании. Смесь нагревали до образования флегмы до получения прозрачного раствора. После охлаждения кристаллы выпадали в осадок из раствора. Полученные в результате осадки собирали фильтрованием и высушивали с получением сырого продукта. Анализ ВЭЖХ показал 89,4% R-конфигурации.
Перекристаллизацию сырого продукта осуществляли в этаноле (120 мл). После перекристаллизации дважды получили 2,82 г белого твердого вещества. Точка плавления: 186,0-187,0°С, [а]D 25=+96,4∈ (С1, 0,1 М NaOH). Анализ ВЭЖХ показал 99,1% R-конфигурации. Белое твердое вещество растворяли в 20 мл воды, и значение рН доводили до 8-9 безводным карбонатом натрия. Смесь дважды экстрагировали дихлорметаном (10 млх2). Органическую фазу объединяли и промывали водой, органическую фазу концентрировали до сухости с получением 1,13 г R-бета-амино-2,4,5-трифторфенилмасляной кислоты этилового эфира (Ib). [а]D 25=-2,6∈(С=0,8, метанол). Выход разделения составлял 43,6%.
ПРИМЕР 5
5,18 г (20 ммоль) рацемата бета-аминофенилмасляной кислоты этилового эфира растворяли в этаноле (100 мл) и добавляли 3,58 г (10 ммоль) D-DTTA при перемешивании. Смесь нагревали до образования флегмы до получения прозрачного раствора. После охлаждения кристаллы выпадали в осадок из раствора. Полученные в результате осадки собирали фильтрованием и высушивали с получением сырого продукта. Анализ ВЭЖХ показал 83,37% R-конфигурации.
Перекристаллизацию сырого продукта осуществляли в этаноле (100 мл). После перекристаллизации дважды получили 2,59 г белого твердого вещества. Анализ ВЭЖХ показал 99,2% R-конфигурации. Белое твердое вещество растворяли в 18 мл воды, и значение рН доводили до 8-9 безводным карбонатом натрия. Смесь дважды экстрагировали дихлорметаном (10 млх2). Органическую фазу объединяли и промывали водой, органическую фазу концентрировали до сухости с получением 1,03 г R-бета-амино-2,4,5-трифторфенилмасляной кислоты этилового эфира (Ib). [а]D 25=-2,7∈(С=0,8, метанол). Выход разделения составлял 39,8%.
ПРИМЕР 6
1 г (3,84 ммоль) R-бета-амино-2,4,5-трифторфенилмасляной кислоты этилового эфира (1b) добавляли к смеси метанола (10 мл) и водного раствора карбоната натрия (10 мл), в котором значение рН было равно 10, а затем добавляли 1,0 г (ВОС)2О. Реакционную смесь подвергали взаимодействию при 30°С в течение 3 часов. После завершения реакции к смеси добавляли 4 М NaOH (8 мл). Гидролиз проводили при 40-45°С. Через 2 часа реакцию определяли по ТСХ. Растворитель выпаривали, и рН медленно доводили до 3. Смесь экстрагировали этилацетатом и промывали подкисленной водой. Органическую фазу высушивали и концентрировали, а затем осаждали кристаллы с получением 1,14 г R-бета-трет-бутоксилкарбониламино-2,4,5-трифторфенилмасляной кислоты (Ic). Точка плавления: 127-128°С. [а]D 25=14,2∈ (С=1, метанол). Выход: 89,1%.
ПРИМЕР 7
1,0 г (3,0 ммоль) R-бета-трет-бутоксилкарбониламино-2,4,5-трифторфенилмасляной кислоты (Ic) добавляли к 20 мл смеси этилацетата и HCl (2 М). Смесь перемешивали в течение 4 часов при комнатной температуре. Раствор концентрировали до половины его объема при низкой температуре, и кристаллы выпадали в осадок из раствора. Полученные в результате осадки собирали фильтрованием и высушивали с получением 0,67 г соли гидрохлорид R-бета-амино-2,4,5-трифторфенилмасляной кислоты (Ia). Точка плавления: 204,5-207,5°С. [а]D 25=-6,8∈(С=0,8, метанол). Выход: 82,8%.
ПРИМЕР 8
1,0 г R-бета-амино-2,4,5-трифторфенилмасляной кислоты этилового эфира (Ib) (3,84 ммоль) растворяли в 10 мл метанола и добавляли 4 М гидроксид натрия (6 мл). Гидролиз проводили при 40°С. Через 2 часа реакцию определяли по ТСХ. Значение рН доводили до 3, и растворитель концентрировали до сухости. Остаток растворяли в хлороформе и метаноле (4:1). Не растворенные соединения удаляли фильтрованием, и фильтрат наносили на колонку силикагеля. Основную фракцию собирали и концентрировали до сухости. К остатку добавляли 16 мл этилацетата, и смесь перемешивали в течение 2 часов при комнатной температуре. Кристаллы осаждали из раствора и высушивали с получением 0,90 г соли гидрохлорид R-бета-амино-2,4,5-трифторфенилмасляной кислоты (Ia). Точка плавления: 203,0-206,0°С. [а]D 25=-6,4∈ (С=0,8, метанол). Выход: 87,1%.
Изобретение относится к способу получения производных R-бета-аминофенилмасляной кислоты формулы  , где Ar представляет собой незамещенный фенил или фенил, замещенный заместителями в количестве от одного до пяти, выбранными из группы, состоящей из атома фтора, метил, трифторметил и трифторметокси; R1 представляет собой атом водорода или C1-6алкил; и R2 представляет собой атом водорода или аминозащитную группу, включающую алкоксикарбонил и ацил, которые могут найти применение в синтезе ряда хиральных лекарств. Способ включает (1) взаимодействие формиата аммония с незамещенным или замещенным фенилэтилацетоацетатом с получением имина, затем взаимодействие имина с восстанавливающим агентом с получением рацемата бета-аминофенилмасляной кислоты эфира; (2) взаимодействие рацемата бета-аминофенилмасляной кислоты эфира и разделяющего агента, который представляет собой D-винные кислоты, диацилированные бензоилом или замещенным бензоилом, с образованием соли в спиртовом растворителе или в водном растворе спирта, и кристаллизация соли; и (3) гидролиз соли, образованной из R-бета-аминофенилмасляной кислоты эфира и разделяющего агента, с возможной защитой аминной группы с получением производных R-бета-аминофенилмасляной кислоты формулы (I). Способ позволяет получать соединения формулы (I) с высокой оптической чистотой и высокими выходами. 11 з.п. ф-лы, 8 пр.
, где Ar представляет собой незамещенный фенил или фенил, замещенный заместителями в количестве от одного до пяти, выбранными из группы, состоящей из атома фтора, метил, трифторметил и трифторметокси; R1 представляет собой атом водорода или C1-6алкил; и R2 представляет собой атом водорода или аминозащитную группу, включающую алкоксикарбонил и ацил, которые могут найти применение в синтезе ряда хиральных лекарств. Способ включает (1) взаимодействие формиата аммония с незамещенным или замещенным фенилэтилацетоацетатом с получением имина, затем взаимодействие имина с восстанавливающим агентом с получением рацемата бета-аминофенилмасляной кислоты эфира; (2) взаимодействие рацемата бета-аминофенилмасляной кислоты эфира и разделяющего агента, который представляет собой D-винные кислоты, диацилированные бензоилом или замещенным бензоилом, с образованием соли в спиртовом растворителе или в водном растворе спирта, и кристаллизация соли; и (3) гидролиз соли, образованной из R-бета-аминофенилмасляной кислоты эфира и разделяющего агента, с возможной защитой аминной группы с получением производных R-бета-аминофенилмасляной кислоты формулы (I). Способ позволяет получать соединения формулы (I) с высокой оптической чистотой и высокими выходами. 11 з.п. ф-лы, 8 пр.
1. Способ получения производных R-бета-аминофенилмасляной кислоты формулы (I),

где Ar представляет собой незамещенный фенил или фенил, замещенный заместителями в количестве от одного до пяти, выбранными из группы, состоящей из атома фтора, метил, трифторметил и трифторметокси; R1 представляет собой атом водорода или C1-6алкил; и R2 представляет собой атом водорода или аминозащитную группу, включающую алкоксикарбонил и ацил, где алкоксикарбонил выбран из группы, состоящей из метоксилкарбонила, этоксилкарбонила и трет-бутоксилкарбонила, ацил выбран из группы, состоящей из формацила, ацетила, хлорацетила, трихлорацетила, бензоила и фенилацила,
включающий следующие стадии:
(1) взаимодействие формиата аммония с незамещенным или замещенным фенилэтилацетоацетатом с получением имина, затем взаимодействие имина с восстанавливающим агентом с получением рацемата бета-аминофенилмасляной кислоты эфира;
(2) взаимодействие рацемата бета-аминофенилмасляной кислоты эфира и разделяющего агента, который представляет собой D-винные кислоты, диацилированные бензоилом или замещенным бензоилом, с образованием соли в спиртовом растворителе или в водном растворе спирта, и кристаллизация соли; и
(3) гидролиз соли, образованной из R-бета-аминофенилмасляной кислоты эфира и разделяющего агента, с возможной защитой аминной группы с получением производных R-бета-аминофенилмасляной кислоты формулы (I).
2. Способ по п.1, характеризующийся тем, что Ar представляет собой 2,4,5-трифторфенил, и R1 и R2 представляют собой атом водорода.
3. Способ по п.1, характеризующийся тем, что Ar представляет собой 2,4,5-трифторфенил, R1 представляет собой этил, и R2 представляет собой атом водорода.
4. Способ по п.1, характеризующийся тем, что Ar представляет собой 2,4,5-трифторфенил, R1 представляет собой атом водорода, и R2 представляет собой трет-бутоксилкарбонил.
5. Способ по п.1, где хиральные фармацевтически промежуточные соединения, производные R-бета-аминофенилмасляной кислоты формулы (I), полученные на стадии (3), дополнительно подвергают взаимодействию с соляной кислотой с получением соли соляной кислоты.
6. Способ по любому из пп.1-4, характеризующийся тем, что восстанавливающий агент, используемый на стадии (1), представляет собой цианоборгидрид натрия.
7. Способ по любому из пп.1-4, характеризующийся тем, что разделяющий агент, используемый на стадии (2), представляет собой дибензоил-D-винную кислоту или ди-пара-толуоил-D-винную кислоту.
8. Способ по любому из пп.1-4, характеризующийся тем, что разделяющий агент представляет собой ди-пара-толуоил-D-винную кислоту.
9. Способ по п.7, характеризующийся тем, что используют одну ди-пара-толуоил-D-винную кислоту.
10. Способ по любому из пп.1-4, характеризующийся тем, что спиртовой растворитель, используемый на стадии (2), представляет собой короткоцепочечный спирт из трех или менее атомов углерода.
11. Способ по любому из пп.1-4, характеризующийся тем, что спиртовой растворитель представляет собой метанол.
12. Способ по любому из пп.1-4, характеризующийся тем, что водный раствор спирта, используемый на стадии (2), представляет собой раствор спирта из трех или менее чем трех атомов углерода.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 7385080 B2, 10.06.2008 | |||
| ПРОИЗВОДНЫЕ АМИНОУКСУСНОЙ КИСЛОТЫ В ВИДЕ РАЦЕМАТА ИЛИ D- И L-ЭНАНТИОМЕРОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ D- И L-ЭНАНТИОМЕРОВ ПРОИЗВОДНЫХ АМИНОУКСУСНОЙ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ И D- И L-ЭНАНТИОМЕРЫ АМИНОУКСУСНОЙ КИСЛОТЫ В ВИДЕ ЦИНХОНИДИНОВЫХ СОЛЕЙ | 1993 |
|
RU2111206C1 |

