Уровень техники
Настоящее изобретение относится к фармацевтически приемлемым солям амидзамещенного индазола, которые являются ингибиторами фермента поли-АДФ-рибозополимеразы (PARP), ранее известной как поли-АДФ-рибозосинтаза и поли-АДФ-рибозилтрансфераза. Соединения по настоящему изобретению применимы как монотерапия при опухолях со специфическими дефектами в путях репарации ДНК и в качестве усилителей некоторых повреждающих ДНК средств, таких как противораковые средства и лучевая терапия. Кроме того, соединения по настоящему изобретению применимы для уменьшения клеточного некроза (при ударе и инфаркте миокарда), воспалении с отрицательной регуляцией и повреждении ткани, лечении ретровирусных инфекций и для защиты от токсичности химиотерапии.
Поли(АДФ-рибоза)полимераза (PARP) составляет суперсемейство из восемнадцати белков, содержащих каталитические домены PARP (Bioassays (2004), 26: 1148). Такие белки включают PARP-1, PARP-2, PARP-3, танкиразу-1, танкиразу-2, vaultPARP и TiPARP. Являющийся основой член семейства PARP-1 состоит из трех основных доменов: амино(N)-концевого ДНК-связывающего домена (DBD), содержащего два цинковых фингера, аутомодификационного домена и карбокси(С)-концевого каталитического домена.
PARP являются ядерными и цитоплазматическими ферментами, которые расщепляют NAD+ до никотинамида и АДФ-рибозы с образованием длинных и разветвленных полимеров АДФ-рибозы на белках-мишенях, включая топоизомеразы, гистоны и саму PARP (Biochem. Biophys. Res. Commun. (1998), 245: 1-10).
Поли-АДФ-рибозилирование вовлекается в некоторые биологические процессы, в том числе, репарацию ДНК, транскрипцию генов, развитие клеточного цикла, гибель клетки, функции хроматина и геномную устойчивость.
Показано, что каталитическая активность PARP-1 и PARP-2 быстро стимулируется разрывами цепи ДНК (см. Pharmacological Research (2005), 52: 25-33). В ответ на повреждение ДНК PARP-1 связывается с отдельными и двойными никами ДНК. При нормальных физиологических состояниях активность PARP минимальна, однако после повреждения ДНК происходит немедленная активация активности PARP до 500 крат. Как PARP-1, так и PARP-2 детектируют разрывы в цепи ДНК, действующие как ник-сенсоры, обеспечивая быструю передачу сигналов для начала транскрипции и рекрутинга ферментов, требуемых для репарации ДНК в месте повреждения. Так как лучевая терапия и многие химиотерапевтические подходы к лечению рака действуют посредством вызывания повреждений ДНК, ингибиторы PARP применимы в качестве хемо- и радиосенсибилизаторов для лечения рака. Сообщается, что ингибиторы PARP эффективны при радиосенсибилизации гипоксических опухолевых клеток (US 5032617, US 5215738 и US 5041653).
Большая часть биологических действий PARP относится к такому процессу поли-АДФ-рибозилирования, который влияет на свойства и функцию белков-мишеней; к олигомерам PAR, которые, когда отщепляются от поли-АДФ-рибозилированных белков, предоставляют различные клеточные действия; физической ассоциации PARP с ядерными белками с образованием функциональных комплексов; и снижению клеточного уровния его субстрата NAD+ (Nature Review (2005), 4: 421-440).
Помимо участия в репарации ДНК PARP также может действовать как медиатор гибели клеток. Ее избыточная активация при патологических состояниях, таких как ишемия и реперфузионное повреждение, может привести к существенному истощению внутриклеточного NAD+, которое может привести к ухудшению некоторых NAD+-зависимых путей метаболизма и, в результате, гибели клеток (см. Pharmacological Research (2005), 52: 44-59). В результате активации PARP уровни NAD+ существенно снижаются. Экстенсивная активация PARP ведет к серьезному истощению NAD+ в клетках, страдающих от массивного повреждения ДНК. Короткое время полужизни поли-АДФ-рибозы приводит к высокой скорости метаболизма, как только поли-АДФ-рибоза образуется, она быстро разрушается конститутивно активной поли-АДФ-рибозогликогидролазой (PARG). PARP и PARG образуют цикл, который превращает большое количество NAD+ в АДФ-рибозу, вызывая падение NAD+ и АТФ до уровня ниже 20% от нормального уровня. Такое развитие событий особенно пагубно во время ишемии, когда депривация кислорода уже однозначно поставила под угрозу выработку клеточной энергии. Предполагается, что существенное образование свободных радикалов во время реперфузии является основной причиной повреждения ткани. Частично падение АТФ, которое типично происходит во многих органах при ишемии и реперфузии, можно связать с истощением NAD+ из-за метаболизма поли-АДФ-рибозы. Таким образом, ожидается, что ингибирование PARP сохранит уровень клеточной энергии, причем посредством этого потенцируется выживание ишемических тканей после инсульта. Поэтому соединения, которые являются ингибиторами PARP, пригодны для лечения состояний, являющихся результатом опосредованной PARP гибели клеток, включая неврологические состояния, такие как удар, травма и болезнь Паркинсона.
Ингибиторы PARP описаны как применимые для специфического киллинга опухолей с дефицитом BRCA-1 и BRCA-2 (Nature (2005), 434: 913-916 и 917-921; и Cancer Biology & Therapy (2005), 4: 934-936).
Показано, что ингибиторы PARP повышают эффективность противораковых средств (Pharmacological Research (2005), 52: 25-33), в том числе, соединений платины, таких как цисплатин и карбоплатин (Cancer Chemother. Pharmacol. (1993), 33: 157-162, и Mol. Cancer Ther. (2003), 2: 371-382). Также показано, что ингибиторы PARP повышают противоопухолевую активность ингибиторов топоизомеразы I, таких как иринотекан и топотекан (Mol. Cancer Ther. (2003), 2: 371-382; и Clin. Cancer Res. (2000), 6: 2860-2867), и это показано in vivo на моделях (J. Natl. Cancer Inst. (2004), 96: 56-67).
Показано, что ингибиторы PARP восстанавливают восприимчивость к цитотоксическому и антипролиферативному действию темозоломида (TMZ) (см. Curr. Med. Chem. (2002), 9: 1285-1301; и Med. Chem. Rev. Online (2004), 1: 144-150). Это показано на ряде моделей in vitro (Br. J. Cancer (1995), 72: 849-856; Br. J. Cancer (1996), 74: 1030-1036; Mol. Pharmacol. (1997), 52: 249-258; Leukemia (1999), 13: 901-909; Glia (2002), 40: 44-54; и Clin. Cancer Res. (2000), 6: 2860-2867 и (2004), 10: 881-889); и на моделях in vivo (Blood (2002), 99: 2241-2244; Clin. Cancer Res. (2003), 9: 5370-5379, и J. Natl. Cancer Inst. (2004), 96: 56-67). Также показано, что ингибиторы PARP предотвращают появление некроза, вызываемого селективными N3-аденинметилирующими средствами, такими как MeOSO2(CH2)-лекситропсин (Me-Lex) (Pharmacological Research (2005), 52: 25-33).
Также показано, что ингибиторы PARP действуют как сенсибилизаторы облучения. Сообщается, что ингибиторы PARP эффективны при радиосенсибилизации (гипоксических) опухолевых клеток и эффективны для предотвращения восстановления опухолевых клеток после потенциально летального (Br. J. Cancer (1984), 49(Suppl. VI): 34-42; и Int. J. Radiat. Bioi. (1999), 75: 91-100) и сублетального (Clin. Oncol. (2004), 16(1): 29-30) повреждения ДНК после лучевой терапии, преимущественно, за счет их способности предотвращать повторное соединение разрыва цепи ДНК и путем воздействия на некоторые пути передачи сигналов повреждения ДНК.
Также показано, что ингибиторы PARP применимы для лечения острых и хронических болезней сердца (см. Pharmacological Research (2005), 52: 34-43). Например, показано, что однократные инъекции ингибиторов PARP уменьшают размер инфаркта, вызванного ишемией и реперфузией сердечной или скелетной мышцы у кроликов. В таких исследованиях однократная инъекция 3-аминобензамида (10 мг/кг) или за одну минуту до окклюзии или за одну минуту до реперфузии, вызывает схожее уменьшение размера инфаркта сердца (32-42%), в то время как другой ингибитор PARP 1,5-дигидроксиизохинолин (1 мг/кг) уменьшает размер инфаркта в сравнимой степени (38-48%). Такие результаты делают разумным предположение, что ингибиторы PARP могут заранее спасать от ишемии сердца или реперфузионного повреждения ткани скелетной мышцы (PNAS (1997), 94: 679-683). Также сообщается о подобных наблюдениях на свиньях (Eur. J. Pharmacol. (1998), 359: 143-150; и Ann. Thorac. Surg. (2002), 73: 575-581) и собаках (Shock. (2004), 21: 426-32).
Ингибиторы PARP показаны как применимые для лечения некоторых сосудистых заболеваний, септического шока, ишемического повреждения и нейротоксичности (Biochem. Biophys. Acta (1989), 1014: 1-7; J. Clin. Invest. (1997), 100: 723-735). Повреждение ДНК кислородными радикалами, которое приводит к разрушению цепи в ДНК, которое впоследствии распознается PARP, как показали исследования ингибиторов PARP, является главным фактором, вносящим вклад в такие болезненные состояния (J. Neurosci. Res. (1994), 39: 38-46, и PNAS (1996), 93: 4688-4692). Также показано, что PARP играет некую роль в патогенезе геморрагического шока (PNAS (2000), 97: 10203-10208).
Ингибиторы PARP показаны как применимые для лечения воспалительных заболеваний (см. Pharmacological Research (2005), 52: 72-82 и 83-92).
Также показано, что действительная вирусная инфекция клеток млекопитающих блокируется посредством ингибирования активности PARP. Показано, что такое ингибирование рекомбинантных ретровирусных инфекций происходит в различных типах клеток (J. Virology (1996), 70(6): 3992-4000). Также разработаны ингибиторы PARP для применения в противовирусной терапии и при лечении рака (WO 91/18591).
Эксперименты in vitro и in vivo показали, что ингибиторы PARP можно использовать для лечения или предупреждения аутоиммунных заболеваний, таких как диабет типа I и осложнения диабета (Pharmacological Research (2005), 52: 60-71).
Ингибирование PARP представляется как отсрочка появления признаков старения фибробластов человека (Biochem. Biophys. Res. Comm. (1994), 201(2): 665-672, и Pharmacological Research (2005), 52: 93-99). Это может быть связано с ролью, которую PARP играет в регуляции функции теломеров (Nature Gen, (1999), 23(1): 76-80).
Значительное большинство современных ингибиторов PARP взаимодействует с никотинамидсвязывающим доменом фермента и ведет себя как конкурентные ингибиторы в отношении NAD+ (Expert Opin. Ther. Patents (2004), 14: 1531-1551). Структурные аналоги никотинамида, такие как бензамид и производные, находятся среди первых соединений, исследованных как ингибиторы PARP. Однако такие молекулы имеют слабую ингибирующую активность и обладают другими действиями, не имеющими отношения к ингибированию PARP. Таким образом, существует потребность в обеспечении сильными ингибиторами фермента PARP.
Ранее описаны структурно родственные ингибиторы PARP. В WO 1999/59973 раскрываются амидзамещенные бензольные циклы, конденсированные с 5-членными гетероароматическими циклами; в WO 2001/85687 раскрываются амидзамещенные индолы; в WO 1997/04771, WO 2000/26192, WO 2000/32579, WO 2000/64878, WO 2000/68206, WO 2001/21615, WO 2002/068407, WO 2003/106430 и WO 2004/096793 раскрываются амидзамещенные бензоимидазолы; в WO 2000/29384 раскрываются амидзамещенные бензоимидазолы и индолы; и в ЕР 0879820 раскрываются амидзамещенные бензоксазолы. Структурно родственные индазолкарбоксамиды также раскрываются в WO 2007/113596 и WO 07/113596.
Теперь неожиданно обнаружено, что амидзамещенные индазолы по настоящему изобретению показывают особенно высокие уровни ингибирования активности поли-АДФ-рибозополимеразы (PARP). Так, соединения по настоящему изобретению особенно применимы в качестве ингибиторов PARP-1 и/или PARP-2. Они также показывают особенно хорошие уровни клеточной активности, демонстрируя хорошее антипролиферативное действие в клеточных линиях с дефицитом BRCA1 и BRCA2.
Настоящее изобретение относится к фармацевтически приемлемым солям соединения формулы I

2-{4-[(3S)-Пиперидин-3-ил]фенил}-2Н-индазол-7-карбоксамид
Особыми соединениями по настоящему изобретению являются
4-метилбензолсульфонат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния;
сульфат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния;
бензолсульфат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния;
фумарат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния;
сукцинат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния
и их стереоизомеры и таутомеры.
Особым соединением по настоящему изобретению является 4-метилбензолсульфонат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния или его стереоизомер или таутомер.
Особым соединением по настоящему изобретению является сульфат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния или его стереоизомер или таутомер.
Особым соединением по настоящему изобретению является бензолсульфат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния или его стереоизомер или таутомер.
Особым соединением по настоящему изобретению является фумарат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния или его стереоизомер или таутомер.
Особым соединением по настоящему изобретению является сукцинат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния или его стереоизомер или таутомер.
Настоящее изобретение также включает в свой объем N-оксиды соединений. Вообще, такие N-оксиды могут образовываться по любому доступному атому азота. N-Оксиды можно получить обычными способами, такими как взаимодействие соединений с оксоном в присутствии влажного оксида алюминия.
Настоящее изобретение включает в свой объем пролекарства соединений. Вообще, такие пролекарства будут представлять собой функциональные производные соединений, которые легко превращаются in vivo в требуемые соединения. Удобные процедуры отбора и получения подходящих пролекарственных производных описаны, например, в «Design of Prodrugs», ed. H. Bundgaard, Elsevier, 1985.
Пролекарство может представлять собой фармакологически неактивное производное биологически активного вещества («исходного лекарственного средства» или «исходной молекулы»), которое требует трансформации в организме для того, чтобы высвободилось активное лекарственное средство, и которое имеет улучшенные свойства для доставки по сравнению с исходной молекулой лекарственного средства. Трансформация in vivo может являться, например, результатом определенного метаболического процесса, такого как химический или ферментативный гидролиз эфира карбоновой, фосфорной или серной кислоты, или восстановление или окисление чувствительной функциональной группы.
Настоящее изобретение включает в свой объем сольваты соединений, например, гидраты.
Соединения по настоящему изобретению могут иметь асимметричные центры, хиральные оси и хиральные плоскости (описанные в E.L. Eliel and S.H. Wilen, Stereochemistry of Carbon Compounds, John Wiley & Sons. New York, 1994, pages 1119-1190) и встречаться в виде рацематов, рацемических смесей и в виде отдельных диастереомеров, всех возможных изомеров и их смесей, в том числе, оптических изомеров, причем все такие стереоизомеры включены в настоящее изобретение. Кроме того, соединения, раскрытые в данном описании, могут существовать в виде таутомеров, и обе таутомерные формы включены и охватываются объемом изобретеня, даже если отображена только одна таутомерная структура.
Соединения могут существовать в различных изомерных формах, которые все охватываются настоящим изобретением.
Соединения могут существовать в ряде различных полиморфных форм.
Свободное основание соединения по настоящему изобретению может быть протонировано по атому(ам) N аминогруппы и/или N-содержащей гетероциклической группы с образованием соли. Термин «свободное основание» относится к аминам в несолевой форме. Свободную форму определенной соли описанных соединений можно выделить с использованием методов, известных в технике. Например, свободную форму можно регенерировать обработкой соли подходящим разбавленным водным раствором основания, таким как рабавленный водный раствор NaOH, карбоната калия, аммиака и бикарбоната натрия. Свободные формы иногда могут отличаться от их соответствующих солевых форм по некоторым физическим свойствам, таким как растворимость в полярных растворителях, но соли кислот и оснований в других отношениях фармацевтически эквивалентны их соответствующим свободным формам для целей изобретения.
Фармацевтически приемлемые соли можно синтезировать из соединения формулы I, которые содержат основные группы, обычными химическими способами. Как правило, соли основных соединений получают или ионообменной хроматографией или взаимодействием свободного основания со стехиометрическим количеством или с избытком желательной солеобразующей неорганической или органической кислоты в подходящем растворителе или различных комбинациях растворителей.
Фармацевтически приемлемые соли по настоящему изобретению можно получить из соединений формулы I взаимодействием с неорганической, органической или полимерной кислотой. Например, обычные нетоксичные соли включают соли, образованные с такими кислотами, как толуолсульфоновая кислота, серная кислота, бензолсульфоновая кислота, фумаровая кислота или янтарная кислота, в особенности, с толуолсульфоновой кислотой.
Предпочтительно, фармацевтически приемлемая соль по данному изобретению содержит 1 эквивалент соединения формулы (I) и 1, 2 или 3 эквивалента кислоты. В одном воплощении фармацевтически приемлемая соль по данному изобретению содержит 2 эквивалента соединения формулы (I) и 1 эквивалент кислоты.
Термин «толуолсульфоновая кислота» может быть использован как взаимозаменяющий термина «4-метилбензолсульфоновая кислота», и толуолсульфонаты также могут называться солями тозилатами.
Получение фармацевтически приемлемых солей, описанных выше, и других типичных фармацевтически приемлемых солей полнее описано в Berg et al. (1977), J. Pharm. Sci., «Pharmaceutical Salts», 66: 1-19.
Также следует отметить, что соединения по настоящему изобретению являются потенциально внутренними солями или цвиттер-ионами, так как в физиологических условиях депротонированная кислотная группа в соединении, такая как карбоксильная группа, может представлять собой анион, и такой электронный заряд затем может быть разбалансирован внутри против катионного заряда протонированной или алкилированной щелочной группы, такой как четвертичный атом азота.
Соединения по изобретению можно использовать в способе лечения организма человека или животного методами терапии.
Изобретение относится к соединениям для применения при лечении или предупреждении состояний, которые можно облегчить посредством ингибирования поли-АДФ-рибозополимеразы (PARP) (см., например, Nature Review Drug Discovery (2005), 4: 421-440).
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения состояний, которые можно облегчить посредством ингибирования поли-АДФ-рибозополимеразы (PARP).
Настоящее изобретение также относится к способу лечения или предупреждения состояний, которые можно облегчить посредством ингибирования поли-АДФ-рибозополимеразы (PARP), включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Ингибиторы PARP по настоящему изобретению применимы для лечения заболеваний, описанных в WO 2005/082368.
Соединения по изобретению применимы для лечения воспалительных заболеваний, включая состояния, являющиеся, например, результатом отторжения трансплантатов органов; хронические заболевания суставов, включая артрит, ревматоидный артрит, остеоартрит и заболевания костной ткани, связанные с повышенной резорбцией костей; воспалительные заболевания кишечника, такие как илеит, специфический язвенный колит, синдром Барретта и болезнь Крона; воспалительные заболевания легких, такие как астма, респираторный дистерсс-синдром взрослых и хроническая обструктивная болезнь дыхательных путей; воспалительные заболевания глаз, включая дистрофию роговицы, трахому, онхоцеркоз, увеит, симпатический офтальмит и эндофтальмит; хронические заболевания десен, включая гингивит и периодонтит; туберкулез; лепру; воспалительные заболевания почек, включая уремические осложнения, гломерулонефрит и нефроз; воспалительные заболевания кожи, включая склеродермию, псориаз и экзему; воспалительные заболевания центральной нервной системы, включая хронические демиелинизирующие заболевания нервной системы, рассеяный склероз, нейродегенерацию, связанную со СПИДом, и болезнь Альцгеймера, инфекционный менингит, энцефаломиелит, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз и вирусный, и аутоиммунный энцефалит; диабетические осложнения, включая, но не ограничиваясь перечисленным, иммуннокомплексный васкулит, системную красную волчанку (SLE); воспалительные заболевания сердца, такие как кардиомиопатия, ишемическая болезнь сердца, гиперхолистеринемия и атеросклероз; а также различные другие заболевания, которые могут иметь воспалительные компоненты, включая преэклампсию, хроническую печеночную недостаточность, травму головного и спинного мозга и синдром множественной дисфункции органов (MODS) (множественная недостаточность органов (MOF)). Воспалительное заболевание также может представлять собой системное воспаление организма, примерами которого являются шок, вызванный грам-положительными или грам-отрицательными бактериями, геморрагический или анафилактический шок или шок, вызванный противораковой химиотерапией в ответ на провоспалительные цитокины, например, шок, связанный с провоспалительными цитокинами. Такой шок может быть вызван, например, химиотерапевтическим средством, которое вводят как лечение в случае рака.
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения воспалительных заболеваний.
Настоящее изобретение также относится к способу лечения или предупреждения воспалительных заболеваний, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Соединения по изобретению также могут применяться для лечения или предупреждения реперфузионных повреждений, являющихся результатом естественно встречающихся эпизодов и в ходе хирургической процедуры, таких как интестинальное реперфузионное повреждение; миокардиальное реперфузионное повреждение; реперфузионного повреждения, являющегося результатом операции с аппаратом искусственного кровообращения, операции по репарации аневризмы аорты, операции каротидной эндартерэктомии или геморрагического шока; и повреждения из-за реоксигенации в результате трансплантации органов, таких как сердце, легкое, печень, почка, поджелудочная железа, кишка и роговица.
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения реперфузионных повреждений.
Настоящее изобретение также относится к способу лечения или предупреждения реперфузионных повреждений, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Соединения по изобретению также могут применяться для лечения или предупреждения ишемических состояний, включая состояния, являющиеся результатом пересадки органов, такие как стабильная стенокардия, нестабильная стенокардия, ишемия сердца, ишемия печени, ишемия брыжеечной артерии, ишемия кишечника, тяжелая ишемия конечностей, хроническая тяжелая ишемия конечностей, церебральная ишемия, острая ишемия сердца, ишемическая болезнь почек, септический шок и ишемическая болезнь центральной нервной системы, такая как удар или церебральная ишемия.
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения ишемических состояний.
Настоящее изобретение также относится к способу лечения или предупреждения ишемических состояний, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения удара.
Настоящее изобретение также относится к способу лечения или предупреждения удара, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Соединения по настоящему изобретению также могут применяться для лечения или предупреждения хронической или острой почечной недостаточности.
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения почечной недостаточности.
Настоящее изобретение также относится к способу лечения или предупреждения почечной недостаточности, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Соединения по настоящему изобретению также могут применяться для лечения или предупреждения сосудистых заболеваний иных, чем сердечно-сосудистые заболевания, таких как периферическая окклюзия артерии, облетерирующий тромбангиит, болезнь и феномен Рейно, акроцианоз, эритромелалгия, варикозное расширение вен, артериовенозная фистула, лимфедема и жировой отек.
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения сосудистых заболеваний иных, чем сердечно-сосудистые заболевания.
Настоящее изобретение также относится к способу лечения или предупреждения сосудистых заболеваний иных, чем сердечно-сосудистые заболевания, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Соединения по настоящему изобретению также могут применяться для лечения или предупреждения сердечно-сосудистых заболеваний, таких как хроническая сердечная недостаточность, атеросклероз, застойная сердечная недостаточность, циркуляторный шок, кардиомиопатия, пересадка сердца, инфаркт миокарда и сердечная аритмия, такая как фибрилляция предсердий, суправентрикулярная тахикардия, трепетание предсердий и пароксизмальная предсердная тахикардия.
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения сердечно-сосудистых заболеваний.
Настоящее изобретение также относится к способу лечения или предупреждения сердечно-сосудистых заболеваний, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Соединения по настоящему изобретению также могут применяться для лечения или предупреждения сахарного диабета, включая диабет типа I (инсулинзависимый сахарный диабет), типа II (инсулиннезависимый сахарный диабет), гестационный диабет, аутоиммунный диабет, инсулинопатии, диабет из-за заболевания поджелудочной железы, диабет, ассоциированный с другими эндокринными заболеваниями (такими как синдром Кушинга, акромегалия, феохромоцитома, глюкагонома, первичный альдостеронизм или соматостатинома), синдром инсулинорезистентности типа А, синдром инсулинорезистентности типа В, липатрофный диабет и диабет, вызванный клеточными токсинами. Соединения по данному изобретению также могут применяться для лечения или предупреждения диабетических осложнений, таких как диабетическая катаракта, глаукома, ретинопатия, нефропатия (такая как микроальбуминурия и прогрессирующая диабетическая нефропатия), полиневропатия, гангрена ног, атеросклеротическая болезнь коронарной артерии, периферическая артериальная болезнь, некетозная гипергликемическая-гиперосмотическая кома, мононевропатии, вегетативная невропатия, язвы ног, проблемы с суставами и кожные осложнения или осложнения слизистых оболочек (такие как инфекция, лишай, заражение грибами Candida или necrobiosis lipoidica diabeticorumpbesity), гиперлипидемия, гипертензия, синдром инсулинорезистентности, коронарная артериальная болезнь, ретинопатия, диабетическая невропатия, грибковая инфекция, бактериальная инфекция и кардиомиопатия.
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения диабета.
Настоящее изобретение также относится к способу лечения или предупреждения диабета, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Соединения по настоящему изобретению также могут применяться для лечения или предупреждения рака, включая солидные опухоли, такие как фибросаркома, миксосаркома, липосаркома, хондросаркома, остеогенная саркома, хордома, ангиосаркома, эндотелиосаркома, лимфангиосаркома, лимфангиоэндотелиосаркома, синовиома, мезотелиома, опухоль Юинга, лейомиосаркома, рабдамиосаркома, рак ободочной кисшки, колоректальный рак, рак почек, рак поджелудочной железы, костный рак, рак молочной железы, рак яичников, рак предстательной железы, рак пищевода, рак желудка, рак ротовой полости, назальный рак, рак гортани, плоскоклеточная карцинома, безально-клеточный рак, аденокарцинома, карцинома потовых желез, карцинома сальных желез, папиллярная карцинома, папиллярная аденокарцинома, цистаденокарцинома, медуллярная карцинома, бронхогенный рак, почечно-клеточный рак, гепатома, карцинома желчных протоков, хориокарцинома, семинома, эмбриональная карцинома, опухоль Вильмса, цервикальный рак, рак яичка, мелкоклеточная карцинома легких, карцинома мочевого пузыря, рак легких, эпителиальная карцинома, рак кожи, меланома, нейробластома и ретинобластома; онкозаболевания кровеносной системы, такие как острый лимфобластный лейкоз («ALL»), острый лимфобластный В-клеточный лейкоз, острый лимфобластный Т-клеточный лейкоз, острый миелобластный лейкоз («AML»), острый промиелоидный лейкоз («APL»), острый монобластный лейкоз, острый эритролейкемический лейкоз, острый мегакариобластный лейкоз, острый миеломоноцитарный лейкоз, острый не-лимфоцитарный лейкоз, острый недифференцированный лейкоз, хронический миелоидный лейкоз («CML»), хронический лимфоцитарный лейкоз («CLL»), лейкемический ретикулез и множественная миелома; острые и хронические лейкозы, такие как лимфобластный, миелоидный, лимфоцитарный, миелоцитарный лейкозы; лимфомы, такие как болезнь Ходжкина, не-ходжкинская лимфома, множественная миелома, макроглобулинемия Вальденстрема, болезнь тяжелых цепей и истинная полицитемия; онкозаболевания ЦНС и головного мозга, такие как глиома, пилоидная астроцитома, астроцитома, анапластическая астроцитома, мультиформная глиобластома, медуллобластома, краниофарингиома, эпендимома, пинеалома, гемангиобластома, невринома слухового нерва, олигодендроглиома, менингиома, вестибулярная шваннома, адкнома, метастазирующая опухоль головного мозга и спинномозговая опухоль.
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения рака.
Настоящее изобретение также относится к способу лечения или предупреждения рака, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Соединения по настоящему изобретению также могут применяться для лечения или предупреждения рака, дефицитного в отношении активности репарации DSB ДНК, зависимой от гомологической рекомбинации (HR) (см. WO 2006/021801).
Путь HR-зависимой репарации DSB ДНК восстанавливает двухцепочечные разрывы (DSB) в ДНК через гомологические механизмы реформирования непрерывной спирали ДНК (Nat. Genet. (2001), 27(3): 247-254). Компоненты пути HR-зависимой репарации DSB ДНК включают, но не ограничиваются перечисленным, АТМ (NM-000051), RAD51 (NM-002875), RAD51 L1 (NM-002877), RAD51 C (NM-002876), RAD51L3 (NM-002878), DMC1 (NM-007068), XRCC2 (NM7005431), XRCC3 (NM-005432), RAD52 (NM-002879), RAD54L (NM-003579), RAD54B (NM-012415), BRCA-1 (NM-007295), BRCA-2 (NM-000059), RAD50 (NM-005732), MREI 1A (NM-005590), NBS1 (NM-002485), ADPRT (PARP-1), ADPRTL2, (PARP02) CTPS, RPA, RPA1, RPA2, RPA3, XPD, ERCC1, XPF, MMS19, RAD51, RAD51p, RAD51C, RAD51D, DMC1, XRCCR, XRCC3, BRCA1, BRCA2, RAD52, RAD54, RAD50, MRE11, NB51, WRN, BLMKU70, RU80, ATM, ATRCHK1, CHK2, FANCA, FANCB, FANCC, FANCD1, FANCE, FANCF, FANCG, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, RAD1 и RAD9. Другие белки, вовлеченные в путь HR-зависимой репарации DSB ДНК, включают регуляторные факторы, такие как EMSY (Cell (2003), 115: 523-535).
Рак, дефицитный в отношении HR-зависимой репарации DSB ДНК, может включать или содержать одну или несколько раковых клеток, которые имеют уменьшенную или аннулированную способность восстанавливать DSB ДНК по такому пути относительно нормальных клеток, т.е. путь активности HR-зависимой репарации DSB ДНК может быть урезан или отменен в одной или нескольких раковых клетках.
Активность одного или нескольких компонентов пути HR-зависимой репарации DSB ДНК может быть отменена в одной или нескольких раковых клетках индивидуума, имеющего рак, дефицитный в отношении HR-зависимой репарации DSB ДНК. Компоненты пути HR-зависимой репарации DSB ДНК хорошо охарактеризованы в технике (см., например, Science (2001), 291: 1284-1289) и включают компоненты, перечисленные выше.
Настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения рака, дефицитного в отношении активности HR-зависимой репарации DSB ДНК.
Настоящее изобретение также относится к способу лечения или предупреждения рака, дефицитного в отношении активности HR-зависимой репарации DSB ДНК, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
В одном воплощении раковые клетки являются дефицитными в отношении активности HR-зависимой репарации DSB ДНК одного или нескольких фенотипов, выбранных из АТМ (NM-000051), RAD51 (NM-002875), RAD51 L1 (NM-002877), RAD51 C (NM-002876), RAD51L3 (NM-002878), DMC1 (NM-007068), XRCC2 (NM7005431), XRCC3 (NM-005432), RAD52 (NM-002879), RAD54L (NM-003579), RAD54B (NM-012415), BRCA-1 (NM-007295), BRCA-2 (NM-000059), RAD50 (NM-005732), MREI 1A (NM-005590), NBS1 (NM-002485), ADPRT (PARP-1), ADPRTL2, (PARP02) CTPS, RPA, RPA1, RPA2, RPA3, XPD, ERCC1, XPF, MMS19, RAD51, RAD51p, RAD51C, RAD51D, DMC1, XRCCR, XRCC3, BRCA1, BRCA2, RAD52, RAD54, RAD50, MRE11, NB51, WRN, BLMKU70, RU80, ATM, ATRCHK1, CHK2, FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, RAD1 и RAD9.
В другом воплощении раковые клетки имеют фенотип, дефицитный в отношении BRCA1 и/или BRCA2. Раковыке клетки с таким фенотипом могут быть дефицитными в отношении BRCA1 и/или BRCA2, т.е., экспрессия и/или активность BRCA1 и/или BRCA2 в раковых клетках может быть снижена или отменена, например, посредством мутации или полиморфизма в кодирующей нуклеиновой кислоте, или посредством амплификации, мутации или полиморфизма в гене, кодирующем регуляторный фактор, например, гене EMSY, который кодирует регуляторный фактор BRCA2 (Cell (2003), 115: 523-535).
BRCA1 и BRCA2 являются известными опухолевыми супрессорами, чьи аллели дикого типа часто утрачены в опухолях гетерозиготных носителей (Oncogene (2002), 21(58): 8981-93; Trends Mol. Med. (2002), 8(12): 571-6). Ассоциация мутаций BRCA1 и/или BRCA2 с раком молочной железы хорошо охарактеризована (Exp. Clin. Cancer Res. (2002), 21 (3 Suppl): 9-12). Также известно, что амплификация гена EMSY, который кодирует фактор связывания BRCA2, ассоциируется с раком молочной железы и раком яичников. Носители мутаций BRCA1 и/или BRCA2 также находятся в зоне повышенного риска появления рака яичников, предстательной железы и поджелудочной железы. Детекция вариации BRCA1 и BRCA2 хорошо известна в технике и описана, например, в ЕР 699754, ЕР 705903, Genet. Test (1992), 1:75-83; Cancer Treat. Res. (2002), 107: 29-59; Neoplasm (2003), 50(4): 246-50; Ceska Gynecol. (2003), 68(1): 11-16). Определение амплификации фактора связывания BRCA2 EMSY описано в Cell, 115: 523-535. Ингибиторы PARP показаны как применимые для специфического киллинга опухолей с дефицитом BRCA1 и BRCA2 (Nature (2005), 434: 913-916 и 917-920).
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения опухолей с дефицитом BRCA1 и BRCA2.
Настоящее изобретение также относится к способу лечения или предупреждения опухолей с дефицитом BRCA1 и BRCA2, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
В одном воплощении ингибиторы PARP по настоящему изобретению можно использовать в профилактическом лечении для устранения клеток с дефицитом BRCA2 (см. Cancer Res. (2005), 65: 10145).
Соединения по данному изобретению могут применяться для лечения или предупреждения нейродегенеративных заболеваний, включая нейродегенерацию, связанную с экспансией полиглутамина, болезнь Гентингтона, болезнь Кеннеди, спиноцеребральную атаксию, dentatorubral-pallidoluysian атрофию (DPRLA), нейродегенерацию, связанную с агрегацией белков, болезнь Мачадо-Иозефа, болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз, губчатую энцефалопатию, болезнь, связанную с прионами, и рассеяный склероз (MS).
Таким образом, настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для лечения или предупреждения нейродегенеративных заболеваний.
Настоящее изобретение также относится к способу лечения или предупреждения нейродегенеративных заболеваний, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль.
Соединения по данному изобретению также могут применяться для лечения или предупреждения ретровирусной инфекции (US 5652260), повреждения сетчатки (Curr. Eye Res. (2004), 29: 403), физиологического старения кожи и повреждения кожи, вызванного УФ (US 5589483 и Biochem. Pharmacol. (2002), 63: 921).
Соединения по изобретению применимы для лечения или предупреждения преждевременного старения и отсрочки появления связанной с возрастом клеточной дисфункции (Pharmacological Research (2005), 52: 93-99).
Соединения по данному изобретению можно вводить млекопитающим, предпочтительно, людям, или одни или в комбинации с фармацевтически приемлемыми носителями, эксципиентами, разбавителями, адъювантами, наполнителями, буферами, стабилизиторами, консервантами, смазывающими веществами в фармацевтической композиции согласно стандартной фармацевтической практике.
Соединения по данному изобретению можно вводить субъекту любым обычным способом введения, или системно/периферически, или в место желательного действия, в том числе, но без ограничения, перорально (например, проглатыванием); местно (включая, например, трансдермальный, интраназальный, глазной, трансбуккальный и сублингвальный способ); легочно (например, ингаляцией или инсуффляцией с использованием, например, аэрозоля, например, через рот или нос); ректально; вагинально; парентерально (например, инъекцией, в том числе, подкожной интрадермальной, внутримышечной, внутривенной, интраартериальной, интракардиальной, интратекальной, интраспинальной, интракапсулярной, субкапсулярной, интраорбитальной, интраперитонеальной, интратрахеальной, субкутикулярной, интраартикулярной и интрастернальной) и с помощью имплантата депо (например, подкожного или внутримышечного).
Субъект может представлять собой эукариота, животное, позвоночное животное, млекопитающее, грызуна (например, морскую свинку, хомяка, крысу, мышь), представителя мышиных (мышь), представителя собачьих (например, собаку), представителя кошачьих (например, кошку), представителя лошадиных (например, лошадь), примата, обезьяну (например, мартышку или человекообразную обезьяну), мартышку (например, мармозетку, бабуина), человекообразную обезьяну (например, гориллу, шимпанзе, орангутана, гиббона) или человека.
Изобретение также относится к фармацевтическим композициям, включающим одно или несколько соединений по данному изобретению и фармацевтически приемлемый носитель. Фармацевтические композиции, содержащие активный ингредиент, могут находиться в форме, подходящей для перорального применения, например, таблеток, пастилок, лепешек, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального применения, можно получить согласно любому способу, известному для получения фармацевтических композиций, и такие композиции могут содержать одно или несколько веществ, выбранных из группы, включающей подслащивающие вещества, корригенты, красители и консерванты, для того, чтобы обеспечить фармацевтически элегантные и приятные препараты. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые подходят для получения таблеток. Такими эксципиентами могут являться, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; вещества, способствующие грануляции и рассыпанию, например, микрокристаллическая целлюлоза, натрийкросскармелоза, кукурузный крахмал или альгиновая кислота; связующие вещества, например, крахмал, желатин, поливинилпирролидон или аравийская камедь, и смазывающие вещества, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия, или на них может быть нанесено покрытие известными методами для маскирования неприятного вкуса лекарственного средства или отсрочки рассыпания и поглощения в желудочно-кишечном тракте, и посредством этого обеспечивается пролонгированное действие в течение длительного периода. Например, может быть использован водорастворимый материал, маскирующий вкус, такой как гидроксипропилметилцеллюлоза или гидроксипропилцеллюлоза, или материал, задерживающий высвобождение, такой как этилцеллюлоза или ацетатбутират целлюлозы.
Композиции для перорального применения также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или мягких желатиновых капсул, в которых активный ингредиент смешан с водорастворимым носителем, таким как полиэтиленгликоль, или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии содержат активный материал в смеси с эксципиентами, подходящими для получения водных суспензий. Такими эксципиентами являются суспендирующие вещества, например, натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующие или смачивающие вещества могут представлять собой встречающиеся в природе фосфатиды, например, лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например, полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепными алифатическими спиртами, например, гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и гексита, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и ангидридов гексита, например, моноолеат полиоксиэтиленсорбитана. Водные суспензии также могут содержать один или несколько консервантов, например этил- или н-пропил-п-гидроксибензоат, один или несколько красителей, один или несколько корригентов и одно или несколько подслащивающих веществ, таких как сахароза, сахарин или аспартам.
Масляные суспензии могут быть получены суспендированием активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, сезамовом масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Могут быть добавлены подслащивающие вещества, такие, какие указаны выше, и корригенты для получения приятного перорального препарата. Такие композиции могут быть сохранены за счет добавления антиоксидантов, таких как бутилированный гидроксианизол или альфа-токоферол.
Диспергируемые порошки и гранулы, подходящие для получения водной суспензии посредством добавления воды, предоставляют активный ингредиент в смеси с диспергирующим или смачивающим веществом, суспендирующим веществом и одним или несколькими консервантами. Примерами подходящих диспергирующих или смачивающих веществ и суспендирующих веществ являются вещества, уже указанные выше. Также могут присутствовать другие эксципиенты, например, подслащивающие вещества и красители. Такие композиции можно сохранить, добавляя антиоксидант, такой как аскорбиновая кислота.
Фармацевтические композиции по настоящему изобретению также могут находиться в форме эмульсий масло в воде. Масляная фаза может представлять собой растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин или их смесь. Подходящими эмульгаторами могут являться встречающиеся в природе фосфатиды, например лецитин соевых бобов, и эфиры или неполные эфиры, образованные жирными кислотами и ангидридами гексита, например, моноолеат сорбитана, и продукты конденсации указанных неполных эфиров с этиленоксидом, например, моноолеат полиоксиэтиленсорбитана. Эмульсии также могут содержать подслащивающие вещества, корригенты, консерванты и антиоксиданты.
Сиропы и эликсиры могут быть получены с подслащивающими веществами, например глицерином, пропиленгликолем, сорбитом или сахарозой. Такие композиции также могут содержать консерванты, подслащивающие вещества, красители и антиоксидант.
Фармацевтические композиции могут находиться в форме стерильных водных растворов для инъекции. К числу приемлемых сред и растворителей, которые можно использовать, относятся вода, раствор Рингера и изотонический раствор хлорида натрия.
Стерильный препарат для инъекции также может находиться в форме микроэмульсии масло в воде, где активный ингредиент растворен в масляной фазе. Например, активный ингредиент можно сначала растворить в смеси соевого масла и лецитина. Затем масляный раствор вводят в смесь воды и глицерина и обрабатывают с образованием микроэмульсии.
Растворы или микроэмульсии для инъекции можно вводить в кровоток пациента местной болюсной инъекцией. С другой стороны, может быть выгодным введение раствора или микроэмульсии таким путем, чтобы поддерживалась постоянная концентрация данного соединения в кровотоке. Для того чтобы поддерживать такую постоянную концентрацию, можно использовать устройство для постоянной внутривенной доставки. Примером такого устройства является внутривенный насос Deltec CADD-PLUS™, модель 5400.
Фармацевтические композиции могут находиться в форме стерильной водной или масляной суспензии для внутримышечного и подкожного введения. Такую суспензию можно получить согласно способам, известным в технике, с использованием таких подходящих диспергирующих или смачивающих веществ, которые указаны выше. Стерильный препарат для инъекции также может представлять собой стерильный раствор или суспензию для инъекции в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Кроме того, в качестве растворителя или суспендирующей среды удобно использовать нелетучие масла. Для такой цели можно использовать любое успокаивающее нелетучее масло, в том числе, синтетические моно- или диглицериды. Кроме того, при получении препаратов для инъекции находят применение жирные кислоты, такие как олеиновая кислота.
Соединения по настоящему изобретению также можно вводить в форме суппозиториев для ректального введения лекарственного средства. Такие композиции можно получить смешиванием лекарственного средства с подходящим не вызывающим раздражение эксципиентом, который является твердым при обычных температурах, но жидким при ректальной температуре, и поэтому будет плавиться в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао, глицеринированный желатин, гидрогенизированные растительные масла, смеси полиэтиленгликолей различной молекулярной массы и эфиры жирных кислот и полиэтиленгликоля.
Для местного применения используют кремы, мази, желе, растворы или суспензии и т.д. содержащие соединения по настоящему изобретению. (Для целей данной заявки местное применение будет включать жидкости для полоскания рта и горла.)
В случае настоящего изобретения соединения можно вводить в интраназальной форме путем местного применения подходящих интраназальных сред и устройств для доставки или трансдермальными путями с использованием форм трансдермальных пэтчей, хорошо известных специалистам в данной области техники. Для введения в форме системы трансдермальной доставки вводимая дозировка на протяжении схемы приема будет, конечно, скорее непрерывной, а не периодической. Соединения по настоящему изобретению также можно доставлять в виде суппозитория с использованием таких основ, как масло какао, глицеринированный желатин, гидрогенизированные растительные масла, смеси полиэтиленгликолей различной молекулярной массы и эфиры жирных кислот и полиэтиленгликоля.
Когда соединение по настоящему изобретению вводят субъекту, выбранный уровень дозировки будет зависеть от различных факторов, включая, но не ограничиваясь перечисленным, активность определенного соединения, тяжесть симптомов у индивидуума, способ введения, время введения, скорость экскреции соединения, длительность лечения, другие лекарственные средства, соединения и/или материалы, используемые в комбинации, и возраст, пол, массу, состояние, общее здоровье и предыдущую историю болезни пациента. Количество соединения и способ введения в конечном счете будут предписываться лечащим врачом, хотя, как правило, дозировка должна давать локальные концентрации в месте действия, которые достигают нужного эффекта, не вызывая существенного вредного или пагубного побочного действия.
Введение in vivo может быть осуществлено в одной дозе, непрерывно или периодически (например, в раздельных дозах через определенные интервалы) на протяжении лечения. Способы определения наиболее эффективных способов и доз для введения хорошо известны специалистам в данной области техники и будут изменяться с композицией, используемой для лечения, целью лечения, клетками-мишенями для лечения и субъектом, которого лечат. Можно осуществлять однократное или многократное введение, причем уровень дозы и схема выбираются лечащим врачом.
Как правило, подходящая доза активного соединения находится в интервале от примерно 100 мкг до примерно 250 мг на килограмм массы тела субъекта в сутки. Когда активное соединение представляет собой соль, эфир, пролекарство или подобное производное, вводимое количество рассчитывают на исходное соединение, и таким образом фактическая используемая масса пропорционально увеличивается.
Соединения по настоящему изобретению также применимы в комбинации с противораковыми средствами или химиотерапевтическими средствами.
Соединения по данному изобретению могут применяться в качестве хемо- и радиосенсибилизаторов при лечении рака. Они применимы для лечения млекопитающих, которые подвергались ранее или подвергаются в настоящее время лечению от рака. Такое предварительное лечение включает предшествующую химиотерапию, лучевую терапию, операцию или иммунотерапию, такую как противораковые вакцины.
Таким образом, настоящее изобретение относится к комбинации фармацевтически приемлемой соли соединения формулы I и противоракового средства для одновременного, раздельного или последовательного введения.
Настоящее изобретение также относится к комбинации фармацевтически приемлемой соли соединения формулы I, лучевой терапии и другого химиотерапевтического средства для одновременного, раздельного или последовательного введения.
Настоящее изобретение также относится к фармацевтически приемлемой соли соединения формулы I для применения при получении лекарственного средства для применения в качестве вспомогательного средства в противораковой терапии или для потенциации опухолевых клеток путем комбинации с ионизирующим излучением или химиотерапевтическими средствами.
Настоящее изобретение также относится к применению фармацевтически приемлемой соли соединения формулы I при получении лекарственного средства для применения в качестве вспомогательного средства в противораковой терапии или для потенциации опухолевых клеток путем комбинации с ионизирующим излучением и другими химиотерапевтическими средствами. Соединения также можно использовать в комбинации с ионизирующим излучением и другими химиотерапевтическими средствами.
Настоящее изобретение также относится к способу химиотерапии или лучевой терапии, включающему введение пациенту, нуждающемуся в этом, эффективного количества фармацевтически приемлемой соли соединения формулы I или композиции, включающей такую соль, в комбинации с ионизирующим излучением или химиотерапевтическими средствами. Соединения также можно вводить в комбинации с ионизирующим излучением и другими химиотерапевтическими средствами.
В комбинированной терапии соединения по данному изобретению можно вводить субъекту, нуждающемуся в этом, ранее (например, за 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часа, 96 часов, 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель или 12 недель), одновременно или после (например, через 5 минут, 15 минут, 30 минут, 45 минут, 1 час, 2 часа, 4 часа, 6 часов, 12 часов, 24 часа, 48 часов, 72 часа, 96 часов, 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 8 недель или 12 недель) введения другого протворакового средства. В различных воплощениях соединения по настоящему изобретению и другое противораковое средство вводят раздельно с перерывом в 1 минуту, с перерывом в 10 минут, с перерывом в 30 минут, с перерывом менее 1 часа, с перерывом от 1 часа до 2 часов, с перерывом от 2 часов до 3 часов, с перерывом от 3 часов до 4 часов, с перерывом от 4 часов до 5 часов, с перерывом от 5 часов до 6 часов, с перерывом от 6 часов до 7 часов, с перерывом от 7 часов до 8 часов, с перерывом от 8 часов до 9 часов, с перерывом от 9 часов до 10 часов, с перерывом от 10 часов до 11 часов, с перерывом от 11 часов до 12 часов, но с перерывом не более 24 часов или с перерывом не более 48 часов.
Соединения по данному изобретению и другое противораковое средство могут действовать аддитивно или синергически. Синергическая комбинация соединений по настоящему изобретению и другого противоракового средства может позволить использовать меньшие дозировки одного или обоих таких средств и/или менее частые дозировки одного или обоих средств соединений по настоящему изобретению и другого противоракового средства, и/или менее частое введение может уменьшить любую токсичность, связанную с введением средств субъекту, без уменьшения эффективности средств при лечении рака. Кроме того, синергическое действие может привести к повышенной эффективности таких средств при лечении рака и/или уменьшению любого вредного или нежелательного побочного действия, связанного с применением любого одного средства.
Примеры противораковых средств или химиотерапевтических средств для использования в комбинации с соединениями по настоящему изобретению можно найти в Cancer Principles and Practice of Oncology, V.T. Devita and S. Hellman (editors), 6th edition (February 15, 2001), Lippincott Williams & Wilkins Publishers. Специалист в данной области техники сможет понять, какие комбинации средств можно применить, основываясь на определенных характеристиках лекарственных средств и онкозаболевании. Такие противораковые средства включают, но не ограничиваются перечисленным, следующие средства: ингибиторы HDAC, модуляторы эстрогеновых рецепторов, модуляторы андрогеновых рецепторов, модуляторы ретиноидных рецепторов, цитотоксические/цитостатические средства, антипролиферативные средства, ингибиторы пренилпротеинтрансферазы, ингибиторы HMG-CoA-редуктазы, ингибиторы ВИЧ-протеазы, ингибиторы боратной транскриптазы и другие ингибиторы ангиогенеза, ингибиторы клеточной пролиферации и передачи сигналов выживания, средства, индуцирующие апоптоз, и средства, которые влияют на контрольные точки клеточного цикла. Соединения по настоящему изобретению особенно применимы для введения вместе с лучевой терапией.
Примеры «ингибиторов HDAC» включают субероиланилидгидроксамовую кислоту (SAHA), LAQ824, LBH589, PXD101, MS275, FK228, вальпроевую кислоту, масляную кислоту и CI-994.
«Модуляторы эстрогеновых рецепторов» относятся к соединениям, которые препятствуют или ингибируют связывание эстрогена с рецептором, независимо от механизма. Примеры модуляторов эстрогеновых рецепторов включают, но не ограничиваются перечисленным, тамоксифен, ралоксифен, идоксифен, LY353381, LY117081, торемифен, фулвестрант, 4-[7-(2,2-диметил-1-оксопропокси-4-метил-2-[4-[2-(1-пиперидинил)этокси]фенил]-2Н-1-бензопиран-3-ил]фенил-2,2-диметилпропаноат, 4,4'-дигидроксибензофенон-2,4-динитрофенилгидразон и SH646.
«Модуляторы андрогеновых рецепторов» относятся к соединениям, которые препятствуют или ингибируют связывание андрогенов с рецептором, независимо от механизма. Примеры модуляторов андрогеновых рецепторов включают финастерид и другие ингибиторы 5α-редуктазы, нилутамид, флутамид, бикалутамид, лиарозол и ацетат абиратерона.
«Модуляторы ретиноидных рецепторов» относятся к соеединениям, которые препятствуют или ингибируют связывание ретиноидов с рецептором, независимо от механизма. Примеры модуляторов ретиноидных рецепторов включают бексаротен, третиноин, 13-цис-ретиноевую кислоту, 9-цис-ретиноевую кислоту, α-дифторметилорнитин, ILX23-7553, транс-N-(4'-гидроксифенил)ретинамид и N-4-карбоксифенилретинамид.
«Цитотоксические/цитостатические средства» относятся к соединениям, которые вызывают гибель клетки или ингибируют пролиферацию клетки перимущественно путем прямого препятствования функционированию клетки, или ингибируют или препятствуют митозу клетки, включая алкилирующие агенты, факторы некроза опухоли, интеркаляторы, соединения, способные активировать гипоксию, ингибиторы образования микротрубочек/стабилизаторы образования микротрубочек, ингибиторы митоточеских кинезинов, ингибиторы киназ, вовлеченных в развитие митоза, антиметаболиты, модификаторы биологических реакций; гормональные/антигормональные терапевтические средства, гемопоэтические факторы роста терапевтические средства нацеленные моноклональные антитела, ингибиторы топоизомеразы, ингибиторы протеасом и ингибиторы убиквитинлигазы.
Примеры цитотоксических средств включают, но не ограничиваются перечисленным, циклофосфамид, хлорамбуцилкармустин (BCNU), ломустин (CCNU), бусульфан, треосульфан, сертенеф, кахектин, ифосфамид, тазонермин, лонидамин, карбоплатин, альтретамин, преднимустин, дибромдульцитол, ранимустин, фотемустин, недаплатин, ароплптин, оксалиплатин, темозоломид, метилметансульфонат, прокарбазин, декарбазин, гептаплатин, эстрамустин, тозилат импросульфана, трофосфамид, нимустин, хлорид диброспидия, пумитепу, лобаплатин, сатраплатин, профиромицин, цисплатин, ирофульвен, дексифосфамид, цис-аминдихлор(2-метилпиридин)платину, бензилгуанин, глуфосфамид, GPX100 (транс, транс, транс)-бис-мю-(гексан-1,6-диамин)-мю-[диаминплатина(II)]бис[диамин(хлор)платина(II)]тетрахлорид, диаризидинилспермин, триоксид мышьяка, 1-(11-додециламино-10-гидроксиундецил)-3,7-диметилксантин, зорубицин, идарубицин, даунорубицин, бизантрен, митоксантрон, пирарубицин, пинафид, валрубицин, амрубицин, доксорубицин, эпирубицин, пирарубицин, антинеопластон, 3'-дезамино-3'-морфолино-13-дезоксо-10-гидроксикарминомицин, аннамицин, галарубицин, элинафид, MEN10755 и 4-деметокси-3-дезамино-3-азиридинил-4-метилсульфонилдаунорубицин (см. WO 00/50032). Другие примеры включают ингибиторы Raf-киназы (такие как Вау43-9006) и ингибиторы mTOR (такие как Wyeth's CCI-779 и Ariad AP23573). Другими примерами являются ингибиторы Р13К (например, LY294002).
В одном воплощении соединения по данному изобретению можно использовать в комбинации с алкилирующими агентами.
Примеры алкилирующих агентов включают, но не ограничиваются перечисленным, азотные аналоги горчичного газа: циклофосфамид, ифосфамид, трофосфамид и хлорамбуцил; нитрозомочевины: кармустин (BCNU) и ломустин (CCNU); алкилсульфонаты: бусульфан и треосульфан; триазены: дакарбазин, прокарбазин и темозоломид; комплесы, содержащие платину: цисплатин, карбоплатин, ароплатин и оксалиплатин.
В одном воплощении алкилирующим агентом является дакарбазин. Дакарбазин можно вводить субъекту в дозировках, колеблющихся от примерно 150 мг/м2 (площади поверхности тела субъекта) до примерно 250 мг/м2. В другом воплощении дакарбазин вводят субъекту внутривенно один раз в сутки в течение пяти дней подряд в дозе, колеблющейся от примерно 150 мг/м2 до примерно 250 мг/м2.
В одном воплощении алкилирующим агентом является прокарбазин. Прокарбазин можно вводить субъекту в дозировках, колеблющихся от примерно 50 мг/м2 (площади поверхности тела субъекта) до примерно 100 мг/м2. В другом воплощении прокарбазин вводят субъекту внутривенно один раз в сутки в течение пяти дней подряд в дозе, колеблющейся от примерно 50 мг/м2 до примерно 100 мг/м2.
В одном воплощении алкилирующим агентом является темозоламид. Темозоламид можно вводить субъекту в дозировках, колеблющихся от примерно 150 мг/м2 (площади поверхности тела субъекта) до примерно 200 мг/м2. В другом воплощении темозоламид вводят субъекту внутривенно один раз в сутки в течение пяти дней подряд в дозе, колеблющейся от примерно 150 мг/м2 до примерно 200 мг/м2.
Примеры антимитотических средств включают аллоколхицин, халихондрин В, колхицин, производное колхицина, долстатин 10, мейтанзин, ризоксин, тиоколхицин и тритилцистеин.
Примером соединения, способного активировать гипоксию, является тирапазамин.
Примеры ингибиторов протеасом включают, но не ограничиваются перечисленным, лактацистин, бортезомиб, эпоксомицин и пептидальдегиды, такие как MG 132, MG 115 и PSI.
Примеры ингибиторов образования микротрубочек/стабилизаторов образования микротрубочек включают паклитаксел, сульфат виндезина, винкристин, винбластин, винорелбин, 3',4'-дидегидро-4'-дезокси-8'-норвинкалейкобластин, доцетаксол, ризоксин, доластатин, изэтионат мивобулина, ауристатин, цемадотин, RPR109881, BMS184476, винфлунин, криптофицин, 2,3,4,5,6-пентафтор-N-(3-фтор-4-метоксифенил)бензолсульфонамид, ангидровинбластин, N,N-диметил-L-валил-L-валил-N-метил-L-валил-L-пролил-L-пролин-трет-бутиламид, TDX258, эпотилоны (см., например, пат. США №№ 6284781 и 6288237) и BMS188797.
Некоторыми примерами ингибиторов топоизомераз являются топотекан, гикаптамин, иринотекан, рубитекан, экзатекан, гемитекан, дифломотекан, силил-камптотецины, 9-аминокамптотецин, камптотецин, криснатол, митомицин С, 6-этоксипропионил-3',4'-O-экзобензилиденшартрейзин, 9-метокси-N,N-диметил-5-нитропиразоло[3,4,5-kl]акридин-2-(6Н)пропанамин, 1-амино-9-этил-5-фтор-2,3-дигидро-9-гидрокси-4-метил-1Н,12Н-бензо[de]пирано[3',4':b,7]индолизино[1,2b]хинолин-10,13)9Н,15Н)дион, луртотекан, 7-[2-(N-изопропиламино)этил]-(20S)камптотецин, BNP1350, BNPI1100, BN80915, BN80942, фосфат этопозида, тенипозид, собузоксан, 2'-диметиламино-2'-дезоксиэтопозид, GL331, N-[2-(диметиламино)этил]-9-гидрокси-5,6-диметил-6Н-пиридо[4,3-b]карбазол-1-карбоксамид, азулакрин, (5а,5аВ,8аа,9b)-9-[2-(N-[2-(диметиламино)этил]-N-метиламино]этил]-5-[4-гидрокси-3,5-диметоксифенил]-5,5a,6,8,8a,9-гексогидрофуро(3',4':6,7)нафто(2,3-d)-1,3-диоксол-6-он, 2,3-(метилендиокси)-5-метил-7-гидрокси-8-метоксибензо[c]фенантридиний, 6,9-бис[(2-аминоэтил)амино]бензо[g]изогуинолин-5,10-дион, 5-(3-аминопропиламино)-7,10-дигидрокси-2-(2-гидроксиэтиламинометил)-6Н-пиразоло[4,5,1-de]акридин-6-он, N-[1-[2-(диэтиламино)этиламино]-7-метокси-9-оксо-9Н-тиоксантен-4-илметил]формамид, N-(2-(диметлиамино)этил)акридин-4-карбоксамид, 6-[[2-(диметиламино)этил]амино]-3-гидрокси-7Н-индено[2,1-c]хинолин-7-он и димесна; не-камтотециновые ингибиторы топоизомеразы-1, такие как индолокарбазолы; и двойные ингибиторы топоизомеразы 1 и II, такие как бензофеназины, XR20 115761MLN 576 и бензопиридоиндолы.
В одном воплощении ингибитором топоизомеразы является иринотекан. Иринотекан можно вводить субъекту в дозировках, колеблющихся от примерно 50 мг/м2 (площади поверхности тела субъекта) до примерно 150 мг/м2. В другом воплощении иринотекан вводят субъекту внутривенно один раз в сутки в течение пяти дней подряд в дозе, колеблющейся от примерно 50 мг/м2 до примерно 150 мг/м2 в течение 1-5 дней, затем снова внутривенно один раз в сутки в течение пяти дней подряд в дни 28-32 в дозе, колеблющейся от примерно 50 мг/м2 до примерно 150 мг/м2, затем снова внутривенно один раз в сутки в течение пяти дней подряд в дни 55-59 в дозе, колеблющейся от примерно 50 мг/м2 до примерно 150 мг/м2.
Примеры ингибиторов митотических кинезинов и, в частности, митотических кинезинов человека KSP, описаны в публикациях РСТ WO 01/30768, WO 01/98278, WO 02/056880, WO 03/050064, WO 03/050122, WO 03/049527, WO 03/049679, WO 03/049678, WO 03/039460, WO 03/079973, WO 03/099211, WO 2004/039774, WO 03/105855, WO 03/106417, WO 2004/087050, WO 2004/058700, WO 2004/058148 и WO 2004/03711, и в публикациях заявок на патент США US 2004/132830 и US 2004/132719. В одном воплощении ингибиторы митотических кинезинов включают, но не ограничиваются перечисленным, ингибиторы KSP, ингибиторы MKLP1, ингибиторы CENP-E, ингибиторы MCAK, ингибиторы Kif14, ингибиторы Mphosph1 и ингибиторы Rab6-KIFL.
«Ингибиторы киназ, вовлеченных в развитие митоза» включают, но не ограничиваются перечисленным, ингибиторы aurora-киназ, ингибиторы Polo-like киназ (PLK) (в частности, ингибиторы PLK-1), ингибиторы bub-1 и ингибиторы bub-R1.
«Антипролиферативные средства» включают антисмысловые олигонуклеотиды РНК и ДНК, такие как G3139, ODN698, RVASKRAS, GEM231 и INX3001, и антиметаболиты, такие как эноцитабин, кармофур, тегафур, пентостатин, доксифлуридин, триметрексат, флударабин, капецитабин, галоцитабин, цитарабин, окфосфат, гидрат фостеабина натрия, ралтитрексед, палтитрексид, эмитефур, тиазофурин, децитабин, нолатрексед, пеметрексед, нелзарабин, 2'-дезокси-2'-метилиденцитидин, 2'-фторометилен-2'-дезоксицитидин, N-[5-(2,3-дигидробензофурил)сульфонил]-N'-(3,4-дихлорфенил)мочевину, N6-[4-дезокси-4-[N2-[2(E),4(E)-тетрадекадиеноил]глициламино]-L-глицеро-В-L-манногептопиранозил]аденин, аплидин, эктеинасцидин, троксацитабин, 4-[2-амино-4-оксо-4,6,7,8-тетрагидро-3Н-пиримидино[5,4-b][1,4]тиазин-6-ил-(S)-этил]-2,5-тиеноил-L-глутаминовую кислоту, аминоптерин, 5-фторурацил, аланозин, эфир 11-ацетил-8-(карбамоилоксиметил)-4-формил-6-метокси-14-окса-1,11-диазатетрацикло(7.4.1.0.0)тетрадека-2,4,6-триен-9-илуксусной кислоты, швейнсонин, лометрексол, дексразоксан, метиониназу, 2'-циано-2'-дезокси-N4-пальмитоил-1-В-D-арабинофуранозилцитозин и тиосемикарбазон 3-аминопиридин-2-карбоксальдегида.
Примеры направленных терапевтических средств на основе моноклональных антител включают такие терапевтические средства, которые имеют цитотоксические средства или радиоизотопы, присоединенные к моноклональным антителам, специфическим против раковых клеток или специфическим против клеток-мишеней. Примеры включают бекссар.
«Ингибиторы HMG-CoA-редуктазы» относятся к ингибиторам 3-гидрокси-3-метилглутарил-CoA-редуктазы. Примеры ингибиторов HMG-CoA-редуктазы, которые можно использовать, включают, но не ограничиваются перечисленным, ловастатин (мевакор®; см. пат. США №№ 4231938, 4294926 и 4319039), симвастатин (зокор®; см. пат. США №№ 4444784, 4820850 и 4916239), правастатин (правахол®; см. пат. США №№ 4346227, 4537850, 4410629, 5030447 и 5180589), флувастатин (лескол®; см. пат. США №№ 5354772, 4911165, 4929437, 5189164, 5118853, 5290946 и 5356896) и аторвастатин (липитор®; см. пат. США №№ 5273995, 4681893, 5489691 и 5342952). Структурные формулы указанных и других ингибиторов HMG-CoA-редуктазы, которые можно использовать в данных способах, приводятся на странице 87 работы M. Yalpani «Cholesterol Lowering Drugs», Chemistry & Industry, pp. 85-89 (5 February 1996), и в патентах США №№ 4782084 и 4885314. Термин «ингибитор HMG-CoA-редуктазы», используемый в данном описании, включает все фармацевтически приемлемые лактоновые и открытые кислотные формы (т.е., где лактоновый цикл раскрывается с образованием свободной кислоты), а также солевые и эфирные формы соединений, которые обладают активностью ингибирования HMG-CoA-редуктазы, и, следовательно, применение таких солевых, эфирных, открытых кислотных и лактоновых форм включается в объем данного изобретения.
«Ингибитор пренилпротеинтрансферазы» относится к соединению, которое ингибирует любой один или любое сочетание ферментов пренилпротеинтрансфераз, включая фарнезилпротеинтрансферазу (FPTase), геранилгеранилпротеинтрансферазу типа I (GGPTase-I) и геранилгеранилпротеинтрансферазу типа II (GGPTase-II, также называемую Rab GGPTase).
Примеры ингибиторов пренилпротеинтрансфераз можно найти в следующих публикацих и патентах: WO 96/30343, WO 97/18813, WO 97/21701, WO 97/23478, WO 97/38665, WO 98/28980, WO 98/29119, WO 95/32987, пат. США № 5420245, пат. США № 5523430, пат. США № 5532359, пат. США № 5510510, пат. США № 5589485, пат. США № 5602098, публ. европейского патента 0618221, публ. европейского патента 0675112, публ. европейского патента 0604181, публ. европейского патента 0696593, WO 94/19357, WO 95/08542, WO 95/11917, WO 95/12612, WO 95/12572, WO 95/10514, пат. США № 5661152, WO 95/10515, WO 95/10516, WO 95/24612, WO 95/34535, WO 95/25086, WO 96/05529, WO 96/06138, WO 96/06193, WO 96/16443, WO 96/21701, WO 96/21456, WO 96/22278, WO 96/24611, WO 96/24612, WO 96/05168, WO 96/05169, WO 96/00736, пат. США № 5571792, WO 96/17861, WO 96/33159, WO 96/34850, WO 96/34851, WO 96/30017, WO 96/33018, WO 96/30362, WO 96/30363, WO 96/31111, WO 96/31477, WO 96/31478, WO 96/31501, WO 97/00252, WO 97/03047, WO 97/03050, WO 97/04785, WO 97/02920, WO 97/170070, WO 97/23478, WO 97/26246, WO 97/30053, WO 97/44350, WO 98/02436 и пат. США № 5532359. Пример, касающийся роли ингибитора пренилпротеинтрансферазы при ангиогенезе, см. в European. J. of Cancer (1999), 35(9): 1394-1401.
«Ингибиторы ангиогенеза» относятся к соединениям, которые ингибируют образование новых кровеносных сосудов, независимо от механизма. Примеры ингибиторов ангиогенеза включают, но не огириничиваются перечисленным, ингибиторы тирозинкиназ, такие как ингибиторы тирозинкиназовых рецепторов Flt-1 (VEGFR1) и Flk-1/KDR (VEGFR2), ингибиторы эпидермального, фибробластного или тромбоцитарного факторов роста, ингибиторы ММР (металлопротеаз матрикса), блокаторы интегрина, интерферон-α, интерлейкин-12, пентозанполисульфат, ингибиторы циклооксигеназы, в том числе, нестероидные противовоспалительные средства (NSAID), подобные аспирину и ибупрофену, а также селективные ингибиторы циклооксигеназы-2, подобные целекоксибу и рофекоксибу (PNAS (1992), 89: 7384; JNCI (1982), 69: 475; Arch. Ophtalmol. (1990), 108: 573; Anat. Rec. (1994), 238: 68; FEBS Letters (1995), 372: 83; Clin. Orthop. (1995), 313: 76; J. Mol. Endocrinol. (1996), 16: 107; Jpn. J. Pharmacol. (1997), 75: 105; Cancer Res. (1997), 57: 1625 (1997); Cell (1998), 93: 705; Intl. j. Mol. Med. (1998), 2: 715; J. Biol. Chem. (1999), 274: 9116), стероидные противовоспалительные средства (такие как кортикостероиды, минералокортикоиды, дексаметазон, преднизон, преднизолон, метилпред, бетаметазон), карбоксиамидотриазол, комбрестатин А-4, скваламин, 6-О-хлорацетилкарбонил)фумагиллол, талидомид, ангиостатин, тропонин-1, антагонисты ангиотензина II (см. J. Lab. Clin. Med. (1985), 105: 141-145) и антитела к VEGF (см. Nature Biotechnology (1999), 17: 963-968; Kim et al. (1993), Nature, 362: 841-844; WO 00/44777 и WO 00/61186).
Другие терапевтические средства, которые модулируют или ингибируют ангиогенез и также могут использоваться в сочетании с соединениями по настоящему изобретению, включают средства, которые модулируют или ингибируют системы коагуляции и фибринолиза (см. обзор в Clin. Chem. La. Med. (2000), 38: 679-692). Примеры таких средств, которые модулируют или ингибируют пути коагуляции и фибринолиза, включают, но не ограничиваются перечисленным, гепарин (см. Thromb. Haemost. (1998), 80: 10-23), низкомолекулярные гепарины и ингибиторы карбоксипептидазы U (также известные как ингибиторы тромбинактивируемого фибринолиза [TAFIa]) (см. Thrombosis Res. (2001), 101: 329-354). Ингибиторы TAFIa описаны в публикации РСТ WO 03/013526 и US, рег. № 60/349925 (зарегистрированной 18 января 2002).
«Средства, которые влияют на контрольные точки клеточного цикла» относятся к соединениям, которые ингибируют протеинкиназы, трансдуцирующие сигналы контрольных точек клеточного цикла, причем посредством этого раковая клетка сенсибилизируется к средствам, повреждающим ДНК. Такие средства включают ингибиторы ATR, ATM, киназ Chk1 и Chk2 и ингибиторы киназ cdk и cdc, и характерными примерами являются 7-гидроксистауроспорин, стауроспорин, флавопиридол, CYC202 (циклацел) и BMS-387032.
«Ингибиторы клеточной пролиферации и путей передачи сигналов выживания» относятся к фармацевтическим средствам, которые ингибируют рецепторы клеточной поверхности и каскады сигнальной трансдукции в прямом направлении таких поверхностых рецепторов. Такие средства включают ингибиторы EGFR (например, гефитиниб и эрлотиниб), ингибиторы ERB-2 (например, трастузумаб), ингибиторы IGFR (например, ингибиторы, описанные в WO 03/059951), ингибиторы цитокиновых рецепторов, ингибиторы МЕТ, ингибиторы PI3K (например, LY294002), ингибиторы серин/треонинкиназ (включая, но не ограничиваясь перечисленным, ингибиторы Akt, такие какие описаны в WO 03/086404, WO 03/68403, WO 03/086394, W) 03/086279, WO 02/083675, WO 02/083139, WO 02/083140 и WO 02/083138), ингибиторы Raf-киназ (например, BAY-43-9006), ингибиторы МЕК (например, CI-1040 и PD-098059) и ингибиторы mTOR (например, Wyeth CCI-779 и Ariad AP23573). Такие средства включают низкомолекулярные соединения-ингибиторы и антагонисты антител.
«Средства, индуцирующие апоптоз», включают активаторы членов семейств рецепторов TNF (включая рецепторы TRAIL).
В одном воплощении соединения по настоящему изобретению применимы для лечения рака в комбинации с одним или несколькими, в частности, одним, двумя или тремя средствами, выбранными из темозоломида, цисплатина, карбоплатина, оксалиплатина, иринотекана и топотекана.
Соединения по настоящему изобретению также можно применять для лечения рака в комбинации с одним или несколькими из следующих терапевтических средств: абареликс (пленаксис-депо®); альдеслейкин (прокин®); альдеслейкин (пролейкин®); алемтузумабб (кампат®); алитретиноин (панретин®); аллопуринол (зилоприм®); альтретамин (гексален®); амифостин (этиол®); анастрозол (аримидекс®); триоксид мышьяка (тризенокс®); аспарагиназа (элспар®); азацитидин (видаза®); бевакузимаб (авастин®); капсулы бексаротена (таргретин®); гель бексаротена (таргретин®); блеомицин (бленоксан®); бортезомиб (велкад®); бусульфан, внутривенное (бусульфекс®); бусульфан, пероральный (милеран®); калустерон (метосарб®); капецитабин (кселода®); карбоплатин (параплатин®); кармустин (BCNU®, BiCNU®); кармустин (глиадел®); имплантат кармустина с полифепрозаном 20 (Gliadel Wafer®); целекоксиб (целебрекс®); цетуксимаб (эрбитукс®); хлорамбуцил (лейкеран®); цисплатин (платинол®); кладрибин (лейстатин®, 2-CdA®); клофарибин (клолар®); циклофосфамид (цитоксан®, неозар®); циклофосфамид (цитоксан для инъекций®); циклофосфамид (цитоксан в таблетках®); цитарабин (цитозар-U®); цитарабин липосомный (DepoCyt®); дакарбазин (DTIC-Dome®); дактиномицин, актиномицин D (космеген®); дарбпоэтин-альфа (аранесп®); даунорубицин липосомный (DanuoXome®); даунорубицин, дауномицин (даунорубицин®); даунорубицин, дауномицин (церубидин®); денилейкиндифитокс (онтак®); дексразоксан (зинекард®); доцетаксел (таксотер®); доксорубицин (адриамицин PES®); доксорубицин (адриамицин®, рубекс®); доксорубицин (адриамицин PES для инъекций®); доксорубицин липосомный (доксил®); пропионат дромостанолона (дромостанолон®); пропионат дромостанолона (мастерон для инъекций®); раствор Эллиотта В (раствор Эллиотта В®); эпирубицин (элленс®); эпоэтин-альфа (эпоген®); эрлотиниб (тарцева®); эстрамустин (эмцит®); фосфат этопозида (этопофос®); этопозид VP-16 (вепезид®); эксеместан (аромазин®); филграстим (нейпоген®); флоксуридин (интраартериальный) (FUDR®); флударабин (флудара®); фторурацил, 5-FU (адруцил®); фулвестрант (фаслодекс®); гефитиниб (иресса®); гемцитабин (гемсар®); гемтузумаба озогамицин (милотарг®); ацетат гозерелина (золадекс имплантат®); ацетат гозерелина (золадекс®); ацетат гистрелина (гистрелин имплантат®); гидроксимочевина (гидреа®); ибритумомабтиуксетан (зевалин®); идарубицин (идамицин®); ифосфамид (IFEX®); мезилат иматиниба (гливек®); интерферон-альфа 2а (роферон А); интерферон-альфа 2b (интрон А); иринотекан (камптосар®); леналидомид (ревлимид®); летрозол (фемара®); лейковорин (веллковорин®, лейковорин®); ацетат лейпролида (элигард®); левамизол (эргамизол®); ломустин, CCNU (CeeBU®); меклоретамин, азотный аналог горчичного газа (мустарген®); ацетат мегестрола (мегац®); мелфалан, L-РАМ (алькеран®); меркаптопурин, 6-МР (пуринетол®); месна (меснекс®); месна (меснекс, табл.®); метотрексат (метотрексат®); метоксален (увадекс®); митомицин С (митомицин®); митотан (лизодрен®); митоксантрон (новантрон®); фенпропионат нандролона (дураболин-50®); неларабин (арранон®); нофетумомаб (верлума®); опрелвекин (неумега®); оксалиплатин (элоксатин®); паклитаксел (паксен®); паклитаксел (таксол®); паклитаксел, протеинсвязанные частицы (абраксан®); палифермин (кепиванс®); памидронат (аредиа®); пегадемаз (адаген (коровий пегадемаз)®); пегаспаргаза (онкаспар®); пегфилграстим (нейласта®); пеметрексед динатрий (алимта®); пентостатин (нипент®); пипоброман (верцит®); пликамицин, митрамицин (митрацин®); порфимернатрий (фотофрин®); прокарбазин (матулан®); хинакрин (атабрин®); расбуриказа (элитек®); ритуксимаб (ритуксан®); сарграмостим (лейкин®); сарграмостим (прокин®); сорафениб (нексавар®); стрептозоцин (занозар®); малеат сунитиниба (сутент®); тальк (склерозол®); тамоксифен (нолвадекс®); темозоломид (темодар®); тенипозид, VM-26 (вумон®); тестолактон (теслак®); тиогуанин, 6-TG (тиогуанин®); тиотепа (тиоплекс®); топотекан (гикамтин®); торемифен (фарестон®); тозитумомаб (бекссар®); тозитумомаб/I-131 тозитумомаб (бекссар®); трестазумаб (герцептин®); третиноин, ATRA (везаноид®); урацил-мустард (урацил-мустард, капсулы®); валрубицин (валстар®); винбластин (велбан®); винкристин (онковин®); винорелбин (навелбин®); вориностат (золинза®); золедронат (зомета®); нилотиниб (тазигна®) и дазатиниб (сприцел®).
Изобретение также охватывает комбинации с NSAID, которые являются селективными ингибиторами СОХ-2. Для целей данного описания NSAID, которые являются селективными ингибиторами СОХ-2, определяются как средства, которые обладают специфичностью ингибирования СОХ-2 относительно СОХ-1, по меньшей мере, 100 крат, измеренной как отношение IC50 для СОХ-2 относительно IC50 для СОХ-1, оцененные анализом клеток или липосом. Такие соединения включают, но не ограничиваются ими, соединения, раскрытые в пат. США № 5474995, пат. США № 5861419, пат. США № 6001843, пат. США № 60020343, пат. США № 5409944, пат. США № 5436265, пат. США № 5536752, пат. США № 5550142, пат. США № 5604260, пат. США № 5698584, пат. США № 5710140, WO 94/15932, пат. США № 5344991, пат. США № 5134142, пат. США № 5380738, пат. США № 5393790, пат. США № 5466823, пат. США № 5633272 и пат. США № 5932598, которые все включены в данное описание в качестве ссылок.
Ингибиторами СОХ-2, которые особенно применимы в способе лечения по изобретению, являются 5-хлор-3-(4-метилсульфонил)фенил-2-(2-метил-5-пиридинил)пиридин или его фармацевтически приемлемые соли.
Соединения, которые описаны как специфические ингибиторы СОХ-2, и, следовательно, особенно применимы в настоящем изобретении, включают, но не ограничиваются перечисленным, парекоксиб, целебрекс® и бекстра® или их фармацевтически приемлемые соли.
Другие примеры ингибиторов ангиогенеза включают, но не ограничиваются перечисленным, эндостатин, украин, ранприназу, IM862, 5-метокси-4-[2-метил-3-(3-метил-2-бутенил)оксиранил]-1-оксаспиро[2,5]окт-6-ил(хлорацетил)карбамат, ацетилдинаналин, 5-амино-[[3,5-дихлор-4-(4-хлорбензоил)фенил]метил]-1Н-1,2,3-триазол-4-карбоксамид, СМ101, скваламин, комбрестатин, RPI4610, NX31838, сульфированный фосфат маннопентаозы, 7,7-(карбонилбис[имино-N-метил-4,2-пирролокарбонилимино[N-метил-4,2-пиррол]карбонилимино]бис(1,3-нафталина дисульфонат) и 3-[(2,4-диметилпиррол-5-ил)метилен]-2-индолинон (SU5416).
Используемый выше термин «блокаторы интегрина» относится к соединениям, которые являются селективными антагонистами, ингибируют или противодействуют связыванию физиологического лиганда с интегрином αvβ3, к соединениям, которые являются селективными антагонистами, ингибируют или противодействуют связыванию физиологического лиганда с интегрином αvβ5, к соединениям, которые являются селективными антагонистами, ингибируют или противодействуют связыванию физиологического лиганда как с интегрином αvβ3, так и с интегрином αvβ5, и к соединениям, которые являются селективными антагонистами, ингибируют или противодействуют активности определнного(ых) интегрина(ов), экспрессированного(ых) на эндотелиальных клетках капилляров. Термин также относится к антагонистам интегринов αvβ6, αvβ8, α1β1, α2β1, α5β1, α6β1 и α6β4. Термин также относится к антоганистам любой комбинации интегринов v3, v5, v6, v8, 11, 21, 51, 61 и 64.
Некоторые конкретные примеры ингибиторов тирозинкиназ включают N-(трифторметилфенил)-5-метилизоксазол-4-карбоксамид, 3-[(2,4-диметилпиррол-5-ил)метилиденил]индолин-2-он, 17-(аллиламино)-17-деметоксигелданамицин, 4-(3-хлор-4-фторфениламино)-7-метокси-6-[3-(4-мофролинил)пропоксил]хиназолин, N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин, BIBX1382, 2,3,9,10,11,12-гексагидро-10-(гидроксиметил)-10-гидрокси-9-метил-9,12-эпокси-1Н-дииндоло[1,2,3-fg:3',2',1'-kl]пирроло[3,4-i][1,6]бензодиазоцин-1-он, SH268, генистеин, STI571, CEP2563, сульфонат 4-(3-хлорфениламино)-5,6-диметил-7Н-пирроло[2,3-d]пиримидинметан, 4-(3-бром-4-гидроксифенил)амино-6,7-диметоксихиназолин, 4-(4'-гидроксифенил)амино-6,7-диметоксихиназолин, SU6668, STI571A, N-4-хлорфенил-4-(4-пиридилметил)-1-фталазинамин и EMD121974.
В одном воплощении соединения по настоящему изобретению применимы для лечения или предупреждения появления некроза, вызываемого селективными N3-аденинметилирующими средствами, такими как MeOSO2(CH2)-лекситропсин (Me-Lex).
Комбинации с соединениями иными, чем противораковые соединения, также охватываются способами по изобретению. Например, комбинации соединений, заявленных в данном описании, с агонистами PPAR-α (т.е., PPAR-гамма) и агонистами PPAR-β (т.е., PPAR-дельта) применимы при лечении некоторых злокачественностей. PPAR-α и PPAR-β являются ядерными, активированными пероксисомными пролифераторами рецепторами α и αvβ5. Об экспрессии PPAR-α на эндотелиальных клетках и вовлечении его в ангиогенез сообщается в литературе (см. J. Cardiovasc. Pharmacol. (1998), 31: 909-913; J. Biol. Chem. (1999), 274: 9116-9121; Invest. Ophthalmol Vis. Sci. (2000), 41: 2309-2317). Позднее показано, что агонисты PPAR-α ингибируют ангиогенную реакцию на VEGF in vitro; как троглитазон, так и малеат розиглитазона ингибирует развитие неоваскуляризации сетчатки у мышей (Arch. Ophthalmol. (2001), 119: 709-717). Примеры агонистов PPAR-α и агонистов PPAR-α/β включают, но не ограничиваются перечисленным, тиазолидиндионы (такие как DRF2725, CS-011, троглитазон, розиглитазон и пиоглитазон), фенофибрат, гемифиброзил, клофибрат, GW2570, SB219994, AR-H039242, JTT-501, MCC-555, GW2331, GW409544, NN2344, KRP297, NP0110, DRF4158, NN622, GI262570, PNU181716, DRF552926, 2-[(5,7-дипропил-3-трифторметил-1,2-бензизоксазол-6-ил)окси]-2-метилпропионовую кислоту (раскрытую в USSN 09/782856) и 2(R)-7-(3-(2-хлор-4-(4-фторфенокси)фенокси)пропокси)-2-этилхроман-2-карбоновую кислоту (раскрытую в USSN 60/235708 и 60/244697).
Другим воплощением настоящего изобретения является применение раскрытых в данном описании соединений в комбинации с противовирусными средствами (такими как аналоги нуклеозидов, включая ганцикловир) для лечения рака. См. WO 98/04290.
Другим воплощением настоящего изобретения является применение раскрытых в данном описании соединений в комбинации с генной терапией для лечения рака. Обзор по генетическим стратегиям для лечения рака см. в Hall et al. (Am. J. Hum. Genet. (1997), 61: 785-789), и в Kufe et al. (Cancer Medicine, 5th Ed., pp. 876-889, B.C. Decker, Hamilton 2000). Генную терапию можно использовать для доставки любого гена, подавляющего опухоль. Примеры таких генов включают, но не ограничиваются перечисленным, р53, который можно доставлять через перенос гена, опосредуемый рекомбинантным вирусом (см. пат. США № 6069134, например), антагонист uPA/uPAR («Adenovirus-Mediated Delivery of a uPA/uPAR Antagonist Suppresses Angiogenesis-Dependent Tumor Growth and Dissemination in Mice», Gene Therapy, August (1998), 5(8): 1105-13) и интерферон-гамма (J. Immunol. (2000), 164: 217-222).
Соединения по данному изобретению также можно вводить в комбинации с ингибитором врожденной устойчивости к нескольким лекарственным средствам (MDR), в частности, MDR, связанной с высокими уровнями экспрессии белков-переносчиков. Такие ингибиторы MDR включают ингибиторы р-гликопротеина (P-gp), такие как LY335979, XR9576, OC144-093, R101922, VX853, верапамил и PSC833 (валсподар).
Соединение по настоящему изобретению можно использовать в сочетании с противорвотными средствами для лечения тошноты или рвоты, включая острую, задержанную, позднюю и преждевременную рвоту, которая может являться результатом применения соединения по настоящему изобретению, одного или с лучевой терапией. Для предупреждения или лечения рвоты соединение по настоящему изобретению можно использовать в сочетании с другими противорвотными средствами, в особенности, антагонистами рецепторов нейрокинина-1, антагонистами рецепторов 5НТ3, такими как ондансетрон, гранисетрон, трописетрон и затисетрон, агонистами рецепторов GABAB, такими как баклофен, кортикостероидом, таким как декадрон (дексаметазон), кеналог, аристокорт, назалид, преферид, бенкортен или другими, описанными в патентах США №№ 2789118, 2990401, 3048581, 3126375, 3929768, 3996359, 3928326 и 3749712, антидофаминергическими средствами, такими как фенотиазины (например, прохлорперазин, флуфеназин, тиоридазин и мезоридазин), метоклопрамидом или дронабинолом. В одном воплощении противорвотное средство, выбранное из антагонистов рецепторов нейрокинина-1, антагонистов рецепторов 5НТ3 и кортикостероидов, вводят как адъювант при лечении или предупреждении рвоты, которая может появиться после введения соединений по изобретению.
Антагонисты рецепторов нейрокинина-1, применеяемые в сочетании с соединениями по настоящему изобретению, хорошо описаны, например, в пат. США №№ 5162339, 5232929, 5242930, 5373003, 5387595, 5459270, 5494926, 5496833, 5637699, 5719147; европейских патентных публикациях №№ ЕР 0360390, 0394989, 0428434, 0429366, 0430771, 0436334, 0443132, 0482539, 0498069, 0499313, 0512901, 0512902, 0514273, 0514274, 0514275, 0514276, 0515681, 0517589, 0520555, 0522808, 0528495, 0532456, 0533280, 0536817, 0545478, 0558156, 0577394, 0585913, 0590152, 0599538, 0610793, 0634402, 0686629, 0693489, 0694535, 0699655, 0699674, 0707006, 0708101, 0709375, 0709376, 0714891, 0723959, 0733632 и 0776893; публикациях РСТ №№ WO 90/05525, 90/05729, 91/09844, 91/18899, 92/01688, 92/06079, 92/12151, 92/15585, 92/17449, 92/20661, 92/20676, 92/21677, 92/22569, 93/00330, 93/00331, 93/01159, 93/01165, 93/01169, 93/01170, 93/06099, 93/09116, 93/10073, 93/14084, 93/14113, 93/18023, 93/19064, 93/21155, 93/21181, 93/23380, 93/24465, 94/00440, 94/01402, 94/02461, 94/02595, 94/03429, 94/03445, 94/04494, 94/04496, 94/05625, 94/07843, 94/08997, 94/10165, 94/10167, 94/10168, 94/10170, 94/11368, 94/13639, 94/13663, 94/14767, 94/15903, 94/19320, 94/19323, 94/20500, 94/26735, 94/26740, 94/29309, 95/02595, 95/04040, 95/04042, 95/06645, 95/07886, 95/07908, 95/08549, 95/11880, 95/14017, 95/15311, 95/16679, 95/17382, 95/18124, 95/18129, 95/19344, 95/20575, 95/21819, 95/22525, 95/23798, 95/26338, 95/28418, 95/30674, 95/30687, 95/33744, 96/05181, 96/05193, 96/05203, 96/06094, 96/07649, 96/10562, 96/16939, 96/18643, 96/20197, 96/21661, 96/29304, 96/29317, 96/29326, 96/29328, 96/31214, 96/32385, 96/37489, 97/01553, 97/01554, 97/03066, 97/08144, 97/14671, 97/17362, 97/18206, 97/19084, 97/19942 и 97/210702; и в Британских публикациях патентов №№ 2266529, 2268931, 2269170, 2269590, 2271774, 2292144, 2293168, 2293169 и 2302689. Получение таких соединений хорошо описано в указанных выше патентах и публикациях, которые включены в данное описание в качестве ссылок.
В одном воплощении антагонист рецепторов нейрокинина-1 для применения в сочетании с соединениями по настоящему изобретению выбирают из 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фторфенил)-4-(3-(5-оксо-1Н,4Н-1,2,4-триазоло)метил)морфолина или его фармацевтически приемлемой соли, которые описаны в пат. США № 5719147.
Соединение по настоящему изобретению также можно вводить со средством, применимым при лечении анемии. Такое лечебное средство против анемии представляет собой, например, активатор рецепторов непрерывного эритропоэза (такой как эпоэтин-альфа).
Соединение по настоящему изобретению также можно вводить со средством, применимым при лечении нейтропении. Такое лечебное средство против нейтропении представляет собой, например, гемопоэтический фактор роста, который регулирует выработку и функцию нейтрофилов, такой как гранулоцитарный колониестимулирующий фактор (G-CSF) человека. Примеры G-CSF включают фиграстим.
Соединение по настоящему изобретению также можно вводить с иммунологически усиливающим лекарственным средством, таким как левамизол, изопринозин и задаксин.
Соединение по настоящему изобретению также может быть применимо для лечения или предупреждения рака, в том числе, костного рака, в комбинации с бисфосфонатами (термин включает бисфосфонаты, дифосфонаты, бисфосфоновые кислоты и дифосфоновые кислоты). Примеры бисфосфонатов включают, но не ограничиваются перечисленным, этидронат (дидронел), памидронат (аредиа), алендронат (фозамакс), ризедронат (актонель), золедронат (зомета), ибандронат (бонива), инкадронат или цимадронат, клодронат, ЕВ-1053, минодронат, неридронат, пиридронат и тилудронат, в том числе, их любые фармацевтически приемлемые соли, производные, гидраты и их смеси.
Таким образом, объем настоящего изобретения охватывает применение заявляемых в данном случае соединений в сочетании с ионизирующим излучением и/или в комбинации с другим соединением, выбранным из ингибиторов HDAC, модулятора эстрогеновых рецепторов, модулятора андрогеновых рецепторов, модулятора ретиноидных рецепторов, цитотоксического/цитостатического средства, антипролиферативного средства, ингибитора пренилпротеинтрансферазы, ингибитора HMG-CoA-редуктазы, ингибитора ангиогенеза, агониста PPAR-, агониста PPAR-, противовирусного средства, ингибитора врожденной устойчивости к нескольким лекарственным средствам, противорвотного средства, средства, применимого при лечении анемии, средства, применимого при лечении нейтропении, иммунологически усиливающего лекарственного средства, ингибитора пролиферации клеток и передачи сигналов выживания, средства, которое влияет на контрольные точки клеточного цикла, средства, индуцирующего апоптоз, и бисфосфоната.
Термин «введение» и его варианты (например, «введение» соединения) в отношении к соединению по изобретению обозначает введение соединения или пролекарства соединения в систему животного, нуждающегося в лечении. Когда соединение по изобретению или его пролекарство предоставляют в комбинации с одним или несколькими другими активными средствами (например, цитотоксическим средством и т.д.), «введение» и его варианты понимаются, каждый, как включающие одновременное и последовательное введение соединения или его пролекарства и других средств.
Используемый в данном описании термин «композиция» предполагает охват продукта, включающего определенные ингредиенты в определенных количествах, а также любого продукта, который прямо или косвенно является результатом сочетания определенных ингредиентов в определенных количествах.
Термин «терапевтически эффективное количество», используемый в данном описании, обозначает такое количество активного соединения или фармацевтического средства, которое выявляет биологическую или медицинскую реакцию в ткани, системе, у животного или человека, которой добивается исследователь, ветеринар, лечащий врач или другой врач.
Термин «лечение» относится к лечению млекопитающего, пораженного патологией, и относится к действию, которое облегчает состояние путем уничтожения раковых клеток, но также к действию, которое приводит к ингибированию развития состояния, и включает снижение скорости развития, приостановку развития, облегчение состояния и излечивание от состояния. Также включается лечение как профилактическая мера (т.е., профилактика).
Термин «фармацевтически приемлемый», используемый в данном описании, относится к соединениям, материалам, композициям и/или лекарственным формам, которые, в рамках здравого смысла, подходят для применения в контакте с тканями субъекта (например, человека) без излишней токсичности, раздражения, аллергической реакции или других проблем или осложнений, соответствуют разумному соотношению польза/риск. Каждый носитель, эксципиент и т.д. должен быть «приемлемым» в смысле совместимости с другими ингредиентами препарата.
Термин «вспомогательное средство» относится к применению соединений в сочетании с известными терапевтическими средствами. Такие средства включают цитотоксические схемы приема лекарственных средств и/или ионизирующего излучения, которые используются при лечении различных типов рака. В частности, известны активные соединения для потенцирования действия ряда противораковых химиотерапевтических способов лечения, которые включают класс топоизомеразных ядов (например, топотекан, иринотекан, рубитекан), большинство известных алкилирующих средств (например, DTIC, темозоламид) и лекарственные средства на основе платины (например, карбоплатин, цисплатин), используемые при лечении рака.
В объем формулы изобретения также включен способ лечения рака, включающий введение терапевтически эффективного количества фармацевтически приемлемой соли соединения формулы I в сочетании с лучевой терапией и/или в комбинации с соединением, выбранным из ингибиторов HDAC, модулятора эстрогеновых рецепторов, модулятора андрогеновых рецепторов, модулятора ретиноидных рецепторов, цитотоксического/цитостатического средства, антипролиферативного средства, ингибитора пренилпротеинтрансферазы, ингибитора HMG-CoA-редуктазы, ингибитора ангиогенеза, агониста PPAR-, агониста PPAR-, противовирусного средства, ингибитора врожденной устойчивости к нескольким лекарственным средствам, противорвотного средства, средства, применимого при лечении анемии, средства, применимого при лечении нейтропении, иммунологически усиливающего лекарственного средства, ингибитора пролиферации клеток и передачи сигнала выживания, средства, которое влияет на контрольные точки клеточного цикла, средства, индуцирующего апоптоз, и бисфосфоната.
Указанные и другие аспекты изобретения будут очевидны из указаний, содержащихся в данном описании.
В описании химии и в примерах, приведенных далее, используются следующие аббревиатуры:
AcCl (ацетилхлорид); (BzO)2 (пероксид бензоила; CDCl3 (дейтерированный хлороформ); DCM (дихлорметан); ДМФА (диметилформамид); ДМСО (диметилсульфоксид); экв. (эквивалент); ES (электронное распыление); EtOAc (этилацетат); EtOH (этанол); мол. с. (молекулярные сита); MeCN (ацетонитрил); МеОН (метанол); МС (масс-спектрометрия); MW (микроволновое излучение); NBS (N-бромсукцинимид); NMMO (N-метилморфолин-N-оксид); ЯМР (ядерный магнитный резонанс); Pкол (давление в колонке); изо-PrOH (изопропанол); RH (относительная влажность); КТ (комнатная температура); насыщ. водн. (насыщенный водный); SiO2 (силикагель) и ТГФ (тетрагидрофуран); колонка IST ISOLUTE® SPE SCX (колонка International Sorbent Technology для твердофазной экстракции с катионообменной смолой ISOLUTE®; SFC (сверхкритическая флюидная хроматография); TBTU - тетрафторборат O-(1Н-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония; Tкол (температура колонки); ТФК (трифторуксусная кислота) и ТСХ (тонкослойная хроматография).
Когда синтез промежуточных и исходных соединений не описывается, такие соединения являются коммерчески доступными или могут быть получены из коммерчески доступных соединений стандартными способами или при обращении к синтезу, описанному выше, схемам и примерам в данном описании.
Во время любой из последовательностей операций синтеза, описанной в данном описании, может стать необходимой или желательной защита чувствительных или реакционноспособных групп в любой из соответствующих молекул. Этого можно достичь с помощью обычных защитных групп, таких как группы, описанные в Protecting Groups in Organic Synthesis, 3rd Edition, Greene T.W. and Wuts P.G.M.; Wiley Interscience, 1999, и в Kocienski P.J., Protecting Groups, Thieme, 1994. Защитные группы можно удалить на удобной последующей стадии с использованием способов, известных в технике. Например, когда присутствует защитная группа Вос (трет-бутоксикарбонильная) или бензилкарбонильная защитная группа, такую группу можно удалить, добавляя растворители, такие как ТФК, DCM и/или MeCN, при примерно комнатной температуре. Соединение также можно гидрировать с использованием стандартных способов, таких как обработка в атмосфере водорода в присутствии катализатора, такого как Pd/C, в растворителе, таком как метанол. EtOAc в присутствии HCl и 1,4-диоксана также можно добавлять для удаления Вос или бензилкарбонильной защитной группы при примерно комнатной температуре.
Когда соединения по настоящему изобретению имеют хиральные центры, из рацемической смеси можно выделить энантиомеры стандартными способами разделения, такими как использование SFC, хиральной ВЭЖХ или расщепления хиральными кислотами. Разделение можно осуществить на любой стадии процесса получения соединений формулы I. Так, разделение можно осуществить на конечной стадии, или, с другой стороны, можно разделить промежуточные соединения и затем использовать определенные энантиомеры в последующих реакциях для получения нужного продукта.
Анализ PARP-1 SPA
В данном анализе испытывают соединения, описанные в данном описании в качестве примеров, и находят, что они имеют величины IC50 менее 5 мкМ, в частности, менее 50 нМ.
Рабочие реагенты
Буфер для анализа: 100 мМ трис, рН 8, 4 мМ MgCl2, 4 мМ спермина, 200 мМ KCl, 0,04% нонидета Р-40.
Смесь с ферментом: буфер для анализа (12,5 мкл), 10 мМ DTT (0,5 мкл), PARP-1 (5 нМ, Trevigen 4668-500-01), Н2О (до 35 мкл).
Смесь динуклеотида никотинамидаденина (NAD)/ДНК: [3H-NAD] (250 мкКи/мл, 0,4 мкл, Perkin-Elmer NET-443H), NAD (1,5 мМ, 0,05 мкл, Sigma N-1511), биотинилированный NAD (150 мкМ, 0,03 мкл, Trevigen 4670-500-01), активированный тимус теленка (1 мг/мл, 0,05 мкл, Amerscham Biosciences 27-4575), Н2О (до 10 мкл).
Смесь для проявления: гранулы SPA со стрептавидином (5 мг/мл, Amerscham Biosciences RPNQ 0007), растворенные в 500 мМ ЭДТК.
Выполнение эксперимента
Реакцию осуществляют в 96-луночном микропланшете в конечном объеме 50 мкл/лунка. Добавляют 5 мкл раствора 5% ДМСО/соединение, добавляют смесь с ферментом (35 мкл), начинают реакцию, добавляя смесь NAD/ДНК (10 мкл) и инкубируют в течение 2 час при КТ. Реакцию останавливают, добавляя смесь для проявления (25 мкл), и инкубируют 15 мин при КТ. Проводят замеры с использованием прибора Packard TOP COUNT.
Анализ пролиферации в клетках HeLa, заглушенных BRCA-1
Аббревиатуры
IMDM (среда Дульбекко, модифицированная по способу Исков); RPMI (среда от Roswell Park Memorial Institute); MOI (множественность заражения); GFP (зеленый белок флуоресценции); PBS (забуференный фосфатом физиологический раствор); FCS (фетальная телячья сыворотка) и DMEM (среда Игла, модифицированная по способу Дульбекко).
Соединения по настоящему изобретению также испытывают в антипролиферативном анализе в соответствующих парах клеток HeLa с BRCA1 wt и BRCA1-(shRNA). Анализ показывает, что ингибиторы PARP способны показывать селективность в отношении ингибирования роста клеток с дефицитом BRCA. Соединения показывают СС50 менее 5 мкМ в клетках с дефицитом BRCA1 и более чем 10-кратную селективность относительно BRCA-совершенных клеток.
Анализ основан на способности живых клеток превращать окислительно-восстановительный краситель (резазурин) в конечный продукт флуоресценции (резофурин). Количество образовавшегося резофурина прямо пропорционально числу клеток.
Клеточные линии
HeLa shBRCA1-GFP. Такие клетки представляют собой клетки HeLa, трансдуцированные при MOI 100 лентивирусом, содержащим shRNA против BRCA-1 и экспрессирующую кассету для GFP. Сайленсинг BRCA-1 превышает 80% при оценке анализом Taqman, и клетки устойчиво экспрессируют GFP.
HeLa THM-GFP. Такие клетки представляют собой клетки HeLa, трансдуцированные при MOI 100 контрольным вектором, неэкспрессирующим какую-либо shRNA.
Протокол
- Высевают 300 клеток/лунку в 96-луночный темный микропланшет в 90 мкл культуральной среды*
- Инкубируют 4 часа при 37оС, 5% СО2
- Добавляют 10 мкл/лунку 10× соединения (5% ДМСО в Н2О);
- Инкубируют 168 часов при 37оС, 5% СО2
- Добавляют 10 мкл раствора Celltiter Blue (Promega, G8081), предварительно разбавленного 1:1 в PBS1×
- Инкубируют смесь в течение 45' при 37оС, 5% СО2
- Инкубируют 15' при КТ в темноте
- Считывают планшет на флюориметре, экст. 550 нм, исп. 590 нм
*Культуральная среда: DMEM (Gibco, 4196-029), 10% FCS (Gibco, 10106-169), 0,1 мг/мл пенициллина-стрептомицина (Gibco, 15140-114), 2 мМ L-глутамина (Gibco, 3042190)
Анализ пролиферации в клеточных линиях с естественным дефицитом BRCA
Соединения по настоящему изобретению также показывают ингибирование пролиферации клеточных линий с естественным дефицитом BRCA-1 (MDA-MB-436) и BRCA-2 (CAPAN-1) с СС50 менее 5 микромоль.
Анализ пролиферации
Клетки высевают в 96-луночный микропланшет при 700 клеток/лунку в 100 мкл соответствующей культуральной среды*.
На другой день добавляют серийные разведения соединения в конечном объеме 200 мкл/лунку. Каждое разведение анализируют при трехкратном повторе.
Через шесть дней оценивают жизнеспособность клеток с использованием анализа жизнеспособности клеток с CellTiter Blue согласно инструкциям изготовителя (Promega). Планшеты считывают на приборе для прочтения микропланшетов Fusion Alpha (Packard Bioscience).
В случае клеточных линий со слабой пролиферацией (т.е., CAPAN-1) пролиферацию оценивают через 14 дней после добавления соединений и изменением среды один раз в день 7 (170 мкл среды на лунку аспирируют и заменяют 170 мкл свежей среды, содержащей соединения).
* Культуральная среда:
MDA-MB-436: RPMI (Gibco), 10% FBS (5% СО2)
CAPAN-1: IMDM (Gibco), 20% FBS (5% СО2)
Соединения, испытанные в онкологии на модели in vivo, показывают значительный уровень активности.
Примеры получения
Пример А
2-Фенил-2Н-индазол-7-карбоксамид (А6)
Стадия 1. Метил-3-метил-2-нитробензоат (А1)
К суспензии 3-метил-2-нитробензойной кислоты (1,0 экв.) в МеОН (0,4 М) при 0оС добавляют по каплям AcCl (3,0 экв.). Реакционную смесь перемешивают в течение 20 час при кипячении с обратным холодильником. Растворитель удаляют в вакууме, остаток растворяют в EtOAc и промывают несколько раз насыщ. водн. раствором NaHCO3 и рассолом, и сушат (Na2SO4). Выпаривание растворителя дает (А1) в виде белого твердого вещества, которое используют на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3, 300K) 
Стадия 2. Метил-3-(бромметил)-2-нитробензоат (А2)
Смесь (А1) (1,0 экв.), (BzO)2 (0,06 экв.) и NBS (1,18 экв.) в CCl4 (0,2 М относительно А1) кипятят с обратным холодильником в атмосфере N2 в течение 12 час. Смесь охлаждают до КТ, разбавляют DCM и концентрируют при пониженном давлении после сушки над SiO2. Остаток очищают колоночной флэш-хроматографией на SiO2 с использованием смеси EtOAc/петролейный эфир, 10:90, и получают нужный А2 в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3, 300K) 
Стадия 3. Метил-3-формил-2-нитробензоат (А3)
К смеси (А2) (1,0 экв.) и мол. сит 4Ǻ в MeCN (0,2 М) при КТ добавляют NMMO (2,0 экв.), и реакционную смесь перемешивают в течение 1,5 час в атмосфере N2. Затем смесь разбавляют EtOAc, фильтруют, и фильтрат промывают Н2О, 1 н HCl и рассолом и сушат (Na2SO4). Выпаривание растворителя дает (А3) в виде белого твердого вещества, которое используют на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3, 300K)
Стадия 4. Метил-2-нитро-3-[(фениламино)метил]бензоат (А4)
Смесь (А3) (1,0 экв.) и анилина (1,05 экв.) в EtOH (0,2М) перемешивают при кипячении с обратным холодильником в атмосфере N2 в течение 2 час, пока ТСХ не покажет завершение реакции (гексан/EtOAc=75:25). Выпаривание растворителя дает (А4) в виде белого твердого вещества, которое используют на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3, 300K)
Стадия 5. Метил-2-фенил-2Н-индазол-7-карбоксилат (А5)
Смесь (А4) (1,0 экв.) и NaN3 (1,05 экв.) в сухом ДМФА (0,3 М) перемешивают при 90оС в течение ночи в атмосфере N2. Сырую смесь упаривают в вакууме, и остаток очищают колоночной флэш-хроматографией на силикагеле с использованием градиента смеси EtOAc/петролейный эфир от 10:90 до 40:60, и получают нужный (А5) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3, 300K)
Стадия 6. 2-Фенил-2Н-индазол-7-карбоксамид (А6)
Эфир (А5) греют в смеси ТГФ и 32% водн. раствора NH3 при 70оС в течение ночи в запаяной трубке. Растворители удаляют в вакууме, остаток очищают колоночной флэш-хроматографией на силикагеле с использованием градиента смеси EtOAc/петролейный эфир от 30:70 до 50:50, и получают нужный (А6) в виде белого твердого вещества. 1H ЯМР (400 МГц, ДМСО, 300K)
Пример В
Хлорид 3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния (В4)
Стадия 1. трет-Бутил-3-[4-({[3-(метоксикарбонил)-2-нитрофенил]метилен}амино)фенил]пиперидин-1-карбоксилат (В1)
(В1) получают, следуя общей процедуре, описанной в примере А, стадия 4, с использованием А3 и трет-бутил-3-(4-аминофенил)пиперидин-1-карбоксилата до тех пор, пока ТСХ не покажет завершение реакции (петролейный эфир:EtOAc=4:1), и используют на следующей стадии без дополнительной очистки.
Стадия 2. Метил-2-{4-[1-(трет-бутоксикарбонил)пиперидин-3-ил]фенил}-2Н-индазол-7-карбоксилат (В2)
(В2) получают, следуя общей процедуре, описанной в примере А, стадия 5, и сырой продукт очищают колоночной флэш-хроматографией на силикагеле с использованием градиента 20-40% EtOAc/петролейный эфир, и получают нужный (В2) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3, 300K)
Стадия 3. трет-Бутил-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидин-1-карбоксилат (В3)
(В2) греют в 7 н растворе NH3 в МеОН (0,1М) в запаяной трубке в течение 2 суток при 60оС. Растворители удаляют в вакууме, сырой продукт реакции очищают, растирая в Et2O, и получают нужный (В3) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3, 300K)
Стадия 4. Хлорид 3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния (В4)
К раствору (В3) (1,0 экв.) в EtOAc (0,2 М) при перемешивании добавляют 4 н раствор HCl в диоксане (10,0 экв.), и реакционную смесь перемешивают при КТ в течение 3 час. Растворитель выпаривают при пониженном давлении, сырой продукт реакции очищают, растирая в Et2O, и получают нужный (В4) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-д6, 300K) 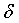
Пример С
2-{4-[(3R)-Пиперидин-3-ил]фенил}-2Н-индазол-7-карбоксамид (С1) и 2-{4-[(3S)-пиперидин-3-ил]фенил}-2Н-индазол-7-карбоксамид (С2)
Полученный в примере В В4 разделяют хиральной SFC (колонка Chiralpak AS-H, 1×25 мм, поток 10 мл/мин, Ткол 35оС, Ркол 10000 кПа (100 бар), модификатор 55% (изо-PrOH+4% Et2NH), с использованием СО2 в качестве сверхкритического элюента, и получают оба чистых энантиомера.
Энантиомер (С1), элюированный первым, время удерживания (SFC) 4,80 мин, получают в виде белого порошка. 1H ЯМР (400 МГц, ДМСО-д6, 300K) 
Энантиомер (С2), элюированный вторым, время удерживания (SFC) 6,51 мин, получают в виде белого порошка. 1H ЯМР (400 МГц, ДМСО-д6, 300K)
Примеры получения характерных соединений
Пример 1
4-Метилбензолсульфонат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния (D4)
Стадия 1. трет-Бутил(3S)-3-[4-({(IE)-[3-(метоксикарбонил)-2-нитрофенил]метилен}амино)фенил]пиперидин-1-карбоксилат (D1)
(D1) получают из соединения А3 примера А и трет-бутил(3S)-3-(4-аминофенил)пиперидин-1-карбоксилата (полученного расщеплением 3-(4-аминофенил)пиперидина 2 эквивалентами L-дибензоилвинной кислоты в МеОН и последующим введением защитной группы Вос), как описано в примере В для В1.
Стадия 2. 2-{4-[(3S)-1-(трет-Бутоксикарбонил)пиперидин-3-ил]фенил}-2Н-индазол-7-карбоновая кислота (D2)
(D1) (1 экв.) и азид натрия (1 экв.) перемешивают в ДМФА (0,25М), создают инертную атмосферу, и добавляют 2,6-лутидин (1,0 экв.). Смесь греют при внутренней температуре 110оС в течение 20 часов. Поолученный коричневый раствор охлаждают до 20оС, и добавляют ТГФ и 25 мас.% водный раствор LiCl. Фазы разделяют, и органическую фазу промывают еще три раза 25 %мас. водным раствором LiCl. К полученному выше органическому раствору добавляют 2,0 н раствор NaOH (10 экв.), смесь греют при 35оС в течение 20 часов и затем охлаждают до 20оС, и фазы разделяют. Органический слой промывают смесью 2,0 н соляной кислоты и рассола, слои разделяют, органический слой промывают дополнительно рассолом и концентрируют, и получают (D2), который дополнительно не очищают.
Стадия 3. трет-Бутил(3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидин-1-карбоксилат (D3)
(D2) растворяют в DCM (0,35 М), и при КТ добавляют ди-трет-бутилкарбонат (1,3 экв.) и пиридин (1,0 экв.). Через 30 минут добавляют бикарбонат аммония (1,3 экв.), и продолжают перемешивание в течение 20 часов. Добавляют 1 н HCl (5 мл/г), фазы разделяют, и органический слой промывают дважды водой и концентрируют до небольшого объема. Сырое соединение (D3) фильтруют через слой диоксида кремния и затем кристаллизуют из метил-трет-бутилового эфира.
Стадия 4. 4-Метилбензолсульфонат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния (D4)
(D3) растворяют в ТГФ (0,15 М), и добавляют воду (5% относительно ТГФ), добавляют моногидрат пара-толуолсульфоновой кислоты (2,2 экв.), и смесь греют при 66оС и перемешивают в течение ночи. После охлаждения нужную твердую соль извлекают фильтрацией и подтверждают, что она является моногидратом (D4). 1H ЯМР (400 МГц, ДМСО, 300K)
Пример 2
4-Метилбензолсульфонат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния
Вводят во взаимодействие 50 мг трет-бутил(3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидин-1-карбоксилата (пример 1, D3) в виде концентрированного раствора с 1 молярным эквивалентом п-толуолсульфоновой кислоты, добавленной в виде 1,0 М раствора в этаноле. Избыток растворителя удаляют центрифужным выпариванием, и добавляют 1 мл этанола при температуре помещения (~25оС). Полученный раствор перемешивают при 40оС в течение 24 часов, когда из раствора выпадает в осадок бесцветное твердое вещество, которое собирают фильтрацией. Полученное твердое вещество анализируют с использованием порошковой рентгенографии, протонного ЯМР, DSC и ТГА для подтверждения образования соли, названной в заголовке.
Пример 3
Бензолсульфат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния
Вводят во взаимодействие 50 мг трет-бутил(3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидин-1-карбоксилата (пример 1, D3) в виде концентрированного раствора с 1 молярным эквивалентом бензолсульфоновой кислоты, добавленной в виде 1,0 М раствора в ТГФ. Избыток растворителя удаляют центрифужным выпариванием, и добавляют 1 мл ТГФ при температуре помещения (~25оС). Полученный раствор перемешивают при 40оС в течение 24 часов, когда из раствора выпадает в осадок бесцветное твердое вещество, которое собирают фильтрацией. Полученное твердое вещество анализируют с использованием порошковой рентгенографии, протонного ЯМР, DSC и ТГА для подтверждения образования соли, названной в заголовке.
Пример 4
Фумарат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния
Вводят во взаимодействие 50 мг трет-бутил(3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидин-1-карбоксилата (пример 1, D3) в виде концентрированного раствора с 1 молярным эквивалентом фумаровой кислоты, добавленной в виде 0,405 М раствора в этаноле. Избыток растворителя удаляют центрифужным выпариванием, и добавляют 1 мл этанола при температуре помещения (~25оС). Полученный раствор перемешивают при 40оС в течение 24 часов. Смесь оставляют для постепенного выпаривания при атмосферых условиях, когда наблюдают бесцветное твердое вещество. Твердое вещество анализируют с использованием порошковой рентгенографии, протонного ЯМР, DSC и ТГА для подтверждения образования соли, названной в заголовке.
Пример 5
Сульфат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния
Вводят во взаимодействие 50 мг трет-бутил(3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидин-1-карбоксилата (пример 1, D3) в виде концентрированного раствора с 1 молярным эквивалентом серной кислоты, добавленной в виде 1 М раствора в смеси метанол:вода, 1:1 (об./об.). Избыток растворителя удаляют центрифужным выпариванием, и добавляют 1 мл смеси метанол:вода при температуре помещения (~25оС). Полученный раствор перемешивают при 40оС в течение 24 часов. Смесь оставляют для постепенного выпаривания при атмосферных условиях, когда наблюдают бесцветное твердое вещество.
В приведенной далее таблице сравниваются физико-химические свойства соединения примера 1 и соответствующего хлорида.
Сравнение физико-химических свойств
Хлорид = хлорид (3S)-3-[4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил]пиперидиния.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
| ПИРИДАЗИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2008 |
|
RU2490265C2 |
| ПРОИЗВОДНЫЕ (АЗА-)ИЗОХИНОЛИНОНА | 2013 |
|
RU2654216C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛИ | 2013 |
|
RU2660349C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2013 |
|
RU2650107C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПОЛИ(ADP-РИБОЗА) ПОЛИМЕРАЗЫ (PARP) | 2007 |
|
RU2472782C2 |
| ПИРИДИЛПИПЕРИДИНОВЫЕ АНТАГОНИСТЫ РЕЦЕПТОРА ОРЕКСИНОВ | 2008 |
|
RU2470021C2 |
| 3-АМИНОЦИКЛОПЕНТАНКАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ | 2013 |
|
RU2641913C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНМОЧЕВИНЫ | 2014 |
|
RU2666894C2 |
| ИНГИБИТОРЫ АКТИВНОСТИ АКТ | 2006 |
|
RU2421454C2 |
Настоящее изобретение относится к фармацевтически приемлемым солям амидзамещенного индазола, а именно 4-Метилбензолсульфонату (3S)-3-{4-[7-(аминокарбонил)-2H-индазол-2-ил]фенил}пиперидиния; бензолсульфату (3S)-3-{4-[7-(аминокарбонил)-2H-индазол-2-ил] фенил}пиперидиния и моногидрату 4-метилбензолсульфоната (3S)-3-{4-[7-(аминокарбонил)-2H-индазол-2-ил]фенил}пиперидиния, а также к их стереоизомерам и таутомерам. Кроме того, изобретение относится к фармацевтической композиции для лечения или предупреждения состояний, которые можно облегчить ингибированием поли-АДФ-рибозополимеразы (PARP), на основе данных солей, к комбинации солей и противоракового средства, к их применению и способу лечения или предупреждения рака, путем введения этих солей нуждающемуся в этом пациенту. Технический результат: получены и описаны новые соли амидзамещенного индазола, которые являются ингибиторами фермента поли-АДФ-рибозополимеразы (PARP), ранее известной как поли-АДФ-рибозосинтаза и поли-АДФ-рибозилтрансфераза. 6 н. и 6 з.п. ф-лы, 5 пр., 1 табл.
1. 4-Метилбензолсульфонат (3S)-3-{4-[7-(аминокарбонил)-2H-индазол-2-ил] фенил}пиперидиния;
бензолсульфат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния;
моногидрат 4-метилбензолсульфоната (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил] фенил}пиперидиния;
и их стереоизомеры и таутомеры.
2. Соединение по п.1, представляющее собой 4-метилбензолсульфонат (3S)-3-{4-[7-(аминокарбонил)-2H-индазол-2-ил]фенил}пиперидиния или его стереоизомер или таутомер.
3. Соединение по п.1, представляющее собой бензолсульфат (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния или его стереоизомер или таутомер.
4. Соединение по п.1, представляющее собой моногидрат 4-метилбензолсульфоната (3S)-3-{4-[7-(аминокарбонил)-2Н-индазол-2-ил]фенил}пиперидиния или его стереоизомер или таутомер.
5. Фармацевтическая композиция для лечения или предупреждения состояний, которые можно облегчить ингибированием поли-АДФ-рибозополимеразы (PARP), включающая соединение по любому из предшествующих пунктов или его стереоизомер или таутомер в сочетании с фармацевтически приемлемым носителем.
6. Комбинация соединения по любому из пп.1-4 или его стереоизомера, или таутомера и противоракового средства для одновременного, раздельного или последовательного введения.
7. Соединение по любому из пп.1-4 или его стереоизомер, или таутомер для применения в лечении или предупреждении состояний, которые можно облегчить ингибированием поли-АДФ-рибозополимеразы (PARP).
8. Применение соединения по любому из пп.1-4 или его стереоизомера, или таутомера для получения лекарственного средства для лечения или предупреждения состояний, которые можно облегчить ингибированием поли-АДФ-рибозополимеразы (PARP).
9. Применение по п.8 для получения лекарственного средства для лечения или предупреждения рака.
10. Соединение по п.7 или его стереоизомер, или таутомер для применения при лечении или предупреждении состояний, которые можно облегчить ингибированием поли-АДФ-рибозополимеразы (PARP), в частности рака.
11. Применение соединения по любому из пп.1-4 или его стереоизомера, или таутомера в качестве хемо- и/или радиосенсибилизатора в случае лечения рака.
12. Способ лечения или предупреждения рака, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения по п.1 или композиции по п.5.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Составной деревянный вал для текстильных машин | 1925 |
|
SU7430A1 |

