ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям, проявляющим ингибирующее действие в отношении фосфодиэстераз, а также к их применению в качестве терапевтических средств при лечении воспалительных заболеваний и состояний.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Фосфодиэстеразы являются ферментами, которые катализируют гидролиз циклического AMP и/или циклического GMP в клетках до 5'-AMP и 5-GMP, соответственно, и, вследствие этого, они имеют решающее значение для клеточного регулирования уровней cAMP и cGMP. Из 11 фосфодиэстераз, идентифицированных до настоящего времени, фосфодиэстераза (PDE) 4, PDE7 и PDE8 проявляют селективность в отношении cAMP. PDE4 является наиболее важным регулятором cAMP, экспрессируемого в иммунных и воспалительных клетках, например, нейтрофилах, макрофагах и T-лимфоцитах (Z. Huang and J.A. Mancini, Current Med. Chem. 13, 2006, pp.3253-3262). Поскольку cAMP является ключевым вторичным мессенджером в модулировании воспалительных реакций, PDE4, как было обнаружено, регулирует воспалительные ответы клеток воспаления за счет модулирования провоспалительных цитокинов, таких как TNFα, IL-2, IFN-γ, GM-CSF и LTB4. Поэтому ингибирование PDE4 становится привлекательной целью при лечении воспалительных заболеваний, таких как астма, хронические обструктивные болезни легких (COPD), ревматоидный артрит, атопический дерматит, болезнь Крона и т.д. (M.D.Houslay et al., Drug Discovery Today 10 (22), 2005, pp.1503-1519). Поскольку у пациентов с атопическим дерматитом (AD) наблюдается повышенная активность PDE, ингибирование PDE4 также, по-видимому, могло бы стать продуктивным способом лечения AD (Journal of Investigative Dermatology (1986), 87(3), 372-6).
Семейство генов PDE4 состоит как минимум из четырех генов, а именно A, B, C и D, которые обладают высокой степенью гомологии (V. Boswell Smith and D. Spina, Curr. Opinion Investig. Drugs 6(11), 2006, pp. 1136-1141). Четыре изоформы PDE4 дифференцированно экспрессируются в различных тканях и типах клеток. Так, например, PDE4B преимущественно экспрессируется в моноцитах и нейтрофилах, но не в коре головного мозга и эпителиальных клетках, тогда как PDE4D экспресируется в легких, коре головного мозга, мозжечке и T-клетках (C. Kroegel and M. Foerster, Exp. Opinion Investig. Drugs 16(1), 2007, pp.109-124). Было выдвинуто предположение, что ингибирование PDE4D в мозге связано с неблагоприятными эффектами, обнаруженными при клиническом введении ингибиторов PDE4, в первую очередь тошнотой и рвотой, в то время как ингибирование PDE4B связано с противовоспалительными эффектами (B. Lipworth, Lancet 365, 2005, pp. 167-175). Однако разработанные до настоящего времени ингибиторы PDE, по-видимому, не являются специфичными для какой-либо из четырех изоформ PDE4.
Многочисленные ингибиторы PDE4 были исследованы с точки зрения их терапевтического действия при воспалительных заболеваниях, в первую очередь астме, воспалительных заболеваниях кишечника и COPD. Первый из них, а именно теофиллин, является слабым, неселективным ингибитором фосфодиэстеразы, применяемым при лечении респираторных заболеваний, таких как астма и COPD. Тем не менее, лечение теофиллином приводит к появлению как легких, так и тяжелых побочных эффектов, например, аритмии и конвульсий, ограничивающих клиническое применение теофиллина (Kroegel and Foerster, смотрите выше). Поскольку фосфодиэстераза остается привлекательной мишенью для противовоспалительной терапии, было разработано и исследовано в клинических условиях несколько других, более селективных ингибиторов PDE4. Клиническая разработка многих ингибиторов PDE4 первого поколения, например, ролипрама, была прекращена из-за ограничивающих дозы побочных эффектов, в первую очередь тошноты и рвоты. В настоящее время проходят клинические испытания ингибиторов PDE4 второго поколения, обладающих значительно менее выраженными нежелательными эффектами (Houslay, смотрите выше).
Разработанные в последнее время ингибиторы PDE4 раскрыты, например, в EP 0771794 и EP 0943613. В WO 96/31476 раскрыты структурно различные 4-замещенные-3,5-дихлорпиридины, которые являются ингибиторами фосфодиэстеразы циклического AMP.
Существует постоянная потребность в разработке новых ингибиторов PDE4, которые имеют более благоприятное терапевтическое окно, т.е. меньшие неблагоприятные эффекты, при сохранении их терапевтического противовоспалительного действия. Обзор доклинических и клинических испытаний селективных ингибиторов PDE4, в том числе ингибиторов, предназначенных для лечения атопического дерматита и псориаза, был приведен недавно в Inflammation & Allergy: Drug Targets, 2007, 6(1), 17-26.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы изобретения неожиданно обнаружили, что новые соединения по настоящему изобретению проявляют ингибирующую активность в отношении PDE4 и могут применяться в качестве терапевтических средств для лечения воспалительных аллергических заболеваний, таких как бронхиальная астма, аллергический ринит и нефрит; аутоиммунных заболеваний, таких как ревматоидный артрит, рассеянный склероз, болезнь Крона и системная красная волчанка; заболеваний центральной нервной системы, таких как депрессия, амнезия и деменция; органопатии, связанной с ишемическим рефлюксом, вызванным сердечной недостаточностью, шоком и цереброваскулярными заболеваниями, и т.п.; инсулин-резистентного диабета; язв; СПИДа и т.п.
Соединения по настоящему изобретению могут также оказаться полезными при профилактике, лечении и облегчении течения ряда заболеваний, например, кожных заболеваний или состояний, таких как пролиферативные и воспалительные кожные расстройства и, в частности, псориаз, воспаление эпидермиса, выпадение волос, атрофия кожи, атрофия кожи, вызванная стероидами, старение кожи, старение кожи под действием света, акне, дерматит, атопический дерматит, себорейный дерматит, контактный дерматит, аллергическая сыпь, зуд и экзема.
Соответственно, настоящее изобретение относится к соединениям, представленным формулой (I),

где m и n независимо представляют собой 0, 1, 2, 3, 4, 5, 6 или 7;
где G и E независимо представляют собой серу, кислород, -N=, -N(R5)- или -N(R5)C(O)-, и
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют ненасыщенный карбоцикл или гетероцикл, включающий один или два гетероатома, выбранных из кислорода, серы, -S(O)-, -S(O)2-, -N(R5)-, причем один или более атомов углерода в указанном ненасыщенном карбоцикле или гетероцикле необязательно замещены одним или более одинаковыми или различными заместителями, выбранными из числа заместителей R4; или
где G и E независимо представляют собой серу, кислород, -N=, -N(R5) или -N(R5)C(O)-, и
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют насыщенный карбоцикл, причем один или более атомов углерода в указанном насыщенном карбоцикле необязательно замещены одним или более одинаковыми или различными заместителями, выбранными из заместителей R4, при условии, что если G представляет собой кислород, оба коэффициента m и n не равны нулю, и при дополнительном условии, что если оба фрагмента G и E представляют собой атомы кислорода, сумма m и n равна шести или более;
R3 представляет собой галоген, гидрокси, алкил, алкенил, алкинил, галогеналкил, алкокси, галогеналкокси, алкилтио, формил, алкоксикарбонил, алкилкарбонил или аминокарбонил;
R4 представляет собой водород, амино, тиоксо, алкил, галогеналкил, гидроксиалкил, алкокси, галоген, оксо, тиа или гидрокси;
R5 представляет собой водород, алкил, галогеналкил, алкилкарбонил, гидроксиалкил, алкоксикарбонил, алкилсульфонил, алкиламиносульфонил или аминосульфонил;
X представляет собой связь, -CH2- или -NH-;
A представляет собой арил, циклоалкил, циклоалкенил, арилалкил, гетероарил, гетероарилалкил, гетероциклоалкил или гетероциклоалкенил, необязательно замещенный одним или более одинаковыми или различными заместителями, выбранными из числа заместителей R4;
а также их фармацевтически приемлемым солям, гидратам, N-оксидам или сольватам.
В другом аспекте изобретение относится к фармацевтической композиции, включающей соединение общей формулы (I) согласно данному выше определению в сочетании с фармацевтически приемлемым наполнителем или фармацевтически приемлемым носителем (носителями), необязательно, совместно с одним или более другими терапевтически активными соединениями (соединением).
В еще одном аспекте изобретение относится к соединению, формулы (I) согласно данному выше определению, а также его фармацевтически приемлемым солям, гидратам, N-оксидам или сольватам, для применения в профилактике, лечении или облегчении течения кожных заболеваний или состояний, или же острых или хронических кожных язвенных расстройств.
В еще одном аспекте изобретение относится к способу профилактики, лечения или облегчения течения кожных заболеваний или состояний, или же острых или хронических кожных язвенных расстройств, где способ включает введение пациенту, страдающему как минимум одним из указанных расстройств, эффективного количества одного или более соединений формулы (I), согласно данному выше определению, а также их фармацевтически приемлемых солей, гидратов, N-оксидов или сольватов; необязательно в сочетании с фармацевтически приемлемым носителем или одним или более наполнителями и, необязательно, в комбинации с другими терапевтически активными соединениями.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Имеется в виду, что термин «углеводородный радикал» означает радикал, содержащий только атомы водорода и углерода, он может содержать одну или более двойных и/или тройных углерод-углеродных связей, кроме того, он может включать циклические фрагменты в сочетании с разветвленными или линейными фрагментами. Упомянутые углеводороды включают 1-20 атомов углерода, и предпочтительно содержат 1-12, например, 1-6, например, 1-4, например, 1-3, например, 1-2, атомов углерода. Данный термин охватывает алкил, алкенил, циклоалкил, циклоалкенил, алкинил, а также арил, арилалкил, которые определены ниже.
Имеется в виду, что термин «арил» означает остаток ароматических карбоциклов, включающих 6-20 атомов углерода, например, 6-14 атомов углерода, предпочтительно 6-10 атомов углерода, в частности, 5- или 6-членных циклов, необязательно конденсированных карбоциклов, включающих как минимум один ароматический цикл, например, фенил, нафтил, инденил и инданил.
Имеется в виду, что термин «гетероарил» означает остаток ароматического гетероцикла, включающего 1-6 гетероатомов (выбранных из O, S и N) и 1-20 атомов углерода, например, 1-5 гетероатомов и 1-10 атомов углерода, например, 1-5 гетероатомов и 1-6 атомов углерода, например, 1-5 гетероатомов и 1-3 атома углерода, в частности, 5- или 6-членные циклы с 1-4 гетероатомами, выбранными из O, S и N, или необязательно конденсированные бициклические системы с 1-4 гетероатомами, в которых как минимум один цикл является ароматическим, например, пиридил, хинолил, изохинолил, индолил, тетразолил, тиазолил, имидазолил, пиразолил, оксазолил, изоксазолил, тиенил, пиразинил, изотиазолил, бензимидазолил и бензофуранил.
В контексте настоящего изобретения имеется в виду, что термин «алкил» означает радикал, полученный при удалении одного атома водорода из углеводорода. Упомянутый алкил включает 1-20, предпочтительно 1-12, например, 1-6, например, 1-4 атомов углерода. Этот термин включает подкласс нормальных алкилов (н-алкилов), вторичных и третичных алкилов, таких как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, гексил и изогексил.
Подразумевается, что термин «циклоалкил» означает остаток насыщенного циклоалкана, включающий 3-20 атомов углерода, предпочтительно 3-10 атомов углерода, в частности, 3-8 атомов углерода, например, 3-6 атомов углерода, включая конденсированные бициклические системы, например, циклопропил, циклобутил, циклопентил, циклогексил или циклогептил.
Имеется в виду, что термин «гетероциклоалкил» означает описанный выше остаток циклоалкана, в котором один или более атомов углерода заменены гетероатомами, включающий 1-19 атомов углерода, например, 2-4 атома углерода, содержащий помимо этого 1-6 гетероатомов, предпочтительно, 1, 2 или 3 гетероатома, выбранных из O, N или S, которые могут быть необязательно связаны с одним или двумя атомами кислорода, например, [1,3]диоксол, оксетан, [1,3]диоксолан, [1,3]диоксан, тетрагидротиопиран, тетрагидротиопиран-1,1-диоксид, тетрагидротиопиран-1-оксид, пиперидин, тетрагидротиофен, [1,3]дитиан, тиетан, [1,3]дитиан-1,3-диоксид или тиетан-1-оксид, или включающий конденсированную бициклическую систему с 1-4 гетероатомами, в которой как минимум один цикл содержит гетероатом, и другой цикл может являться, например, карбоциклом, например, изоиндолилом.
Имеется в виду, что термин «алкенил» означает моно-, ди-, три-, тетра- или пентаненасыщенный углеводородный радикал, содержащий 2-10 атомов углерода, в частности, 2-6 атомов углерода, например, 2-4 атома углерода, например, этенил, пропенил, бутенил, пентенил или гексенил.
Имеется в виду, что термин «циклоалкенил» означает моно-, ди-, три- или тетраненасыщенные неароматические циклические углеводородные радикалы, содержащие 3-20 атомов углерода, включая конденсированные бициклические системы, как правило содержащие 3-10 атомов углерода, как, например, 3, 4 или 6 атомов углерода, например, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил.
Имеется в виду, что термин «гетероциклоалкенил» означает описанный выше остаток циклоалкена, в котором один или более атомов углерода заменены на гетроатомы, содержащий 1-19 атомов углерода, например, 2-4 атома углерода, включающий помимо этого 1-6 гетероатомов, предпочтительно 1, 2 или 3 гетероатома, выбранных из O, N или S, включая конденсированные бициклические системы с 1-4 гетероатомами, в которых как минимум один цикл содержит гетероатом, и другой цикл может являться, например, карбоциклом, например, дигидрофуранилом или 2,5-дигидро-1H-пирролилом.
Имеется в виду, что термин «арилалкил» означает определенный выше арильный радикал, ковалентно присоединенный к алкильной группе, например, бензил.
Имеется в виду, что термин «гетероарилалкил» означает определенный выше гетероарильный радикал, ковалентно связанный с алкильной группой.
Имеется в виду, что термин «алкинил» означает углеводородный радикал, содержащий 1-5 тройных связей C-C и 2-20 атомов углерода, как правило включающий 2-10 атомов углерода, в частности, 2-6 атомов углерода, например, 2-4 атома углерода, например, этинил, пропинил, бутинил, пентинил или гексинил.
Имеется в виду, что термин «галоген» означает заместитель, относящийся к главной подгруппе 7-й группы периодической системы, например, фтор, хлор, бром и йод.
Имеется в виду, что термин «галогеналкил» означает определенную выше алкильную группу, замещенную одним или более определенными выше атомами галогенов, например, дифторметил.
Имеется в виду, что термин «гидроксиалкил» означает определенную выше алкильную группу, замещенную одной или более гидроксигруппами, например, гидроксиметил, гидроксиэтил, гидроксипропил.
Имеется в виду, что термин «алкокси» означает радикал формулы -OR', в котором R' представляет собой алкил согласно данному выше определению, например, метокси, этокси, н-пропокси, изопропокси, бутокси и т.д.
Имеется в виду, что термин «алкоксикарбонил» означает радикал формулы -C(O)-O-R', где R' представляет собой алкил согласно данному выше определению, например, метоксикарбонил, этоксикарбонил, н-пропоксикарбонил, изопропоксикарбонил и т.д.
Имеется в виду, что термин «алкилкарбонил» означает радикал формулы -C(O)-R', где R' представляет собой алкил согласно данному выше определению, например, этаноил, ацетил.
Имеется в виду, что термин «аминосульфонил» означает радикал формулы -S(O)2-NR”, где R” соответствует данному выше определению, например, -SO2Me.
Имеется в виду, что термин «гетероцикл» включает в себя приведенные выше определения определения гетероарила, гетероциклоалкила и гетероциклоалкенила, дополнительно включая аннелированные циклические системы, образованные с другими гетероциклами или с циклическими углеводородами, например, 2,5-дигидробензо(b)диоксоцин, 2,3,5,8-тетрагидро-[1,4]диоксоцин, 5,8-дигидро-[1,4]диоксоцин.
Имеется в виду, что термин «фармацевтически приемлемая соль» означает соли, полученные взаимодействием соединения формулы (I) с подходящей неорганической или органической кислотой, такой как хлористоводородная, бромистоводородная, йодистоводородная, серная, азотная, фосфорная, муравьиная, уксусная, 2,2-дихлоруксусная, адипиновая, аскорбиновая, L-аспарагиновая, L-глутаминовая, галактаровая, молочная, малеиновая, L-яблочная, фталевая, лимонная, пропионовая, бензойная, глутаровая, глюконовая, D-глюкуроновая, метансульфоновая, салициловая, янтарная, малоновая, винная, бензолсульфоновая, этан-1,2-дисульфоновая, 2-гидроксиэтансульфоновая, толуолсульфоновая, сульфамовая или фумаровая кислота. Фармацевтически приемлемые соли соединений формулы (I) также могут быть получены взаимодействием с подходящим основанием, таким как гидроксид натрия, гидроксид калия, гидроксид магния, гидроксид кальция, гидроксид серебра, аммиак и т.п. или подходящими нетоксичными аминами, такими как низшие алкиламины, например, триэтиламин, гидрокси производные низших алкиламинов, например, 2-гидроксиэтиламин, бис-(2-гидроксиэтил)амин, циклоалкиламины, например, дициклогексиламин или бензиламины, например, N,N'-дибензилэтилендиамин, а также дибензиламин или L-аргинин, или L-лизином. Соли, полученные реакцией с подходящим основанием включают, не ограничиваясь указанными, соли натрия, соли холина, соли 2-(диметиламино)этанола, соли 4-(2-гидроксиэтил)морфолина, соли L-лизина, соли N-(2-гидроксиэтил)пирролидина, соли этаноламина, соли калия, соли тетрабутиламмония, соли бензилтриметиламмония, соли цетилтриметиламмония, соли тетраметиламмония, соли тетрапропиламмония, соли трис(гидроксиметил)аминометана, соли N-метил-D-глюкамина, соли серебра, соли бензетония и соли триэтаноламина.
Имеется в виду, что термин «сольват» означает соединения, образованные взаимодействием между соединением, например, соединением формулы (I), и растворителем, например, спиртом, глицерином или водой, где указанные соединения существуют в твердой форме. Если растворителем является вода, указанные соединения именуют гидратами.
Варианты осуществления настоящего изобретения
В одном или нескольких вариантах осуществления настоящего изобретения оба фрагмента E и G представляют собой кислород.
В одном или нескольких вариантах осуществления настоящего изобретения оба коэффициента m и n равны единице.
В одном или нескольких вариантах осуществления настоящего изобретения оба коэффициента m и n равны нулю.
В одном или нескольких вариантах осуществления настоящего изобретения R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют гетероциклический фрагмент, включающий один или два гетероатома, выбранных из группы, состоящей из -O-, -S-, -S(O)-, -S(O)2-, -N= и -N(R5)-; причем один или более атомов углерода гетероциклического фрагмента необязательно замещены одним или более одинаковыми или различными заместителями, выбранными из числа заместителей R4.
В одном или нескольких вариантах осуществления настоящего изобретения R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют гетероциклический фрагмент, включающий один или два гетероатома, выбранных из группы, состоящей из -O-, -S-, -S(O)-, -S(O)2- и -N(R5)-; причем один или более атомов углерода гетероциклического фрагмента необязательно замещены одним или более одинаковыми или различными заместителями, выбранными из числа заместителей R4.
В одном или нескольких вариантах осуществления настоящего изобретения R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют 4-, 5- или 6-членный гетероциклический фрагмент, в частности, 6-членный гетероциклический фрагмент.
В одном или нескольких вариантах осуществления настоящего изобретения, указанный гетероциклический фрагмент представляет собой тетрагидропиран, оксетан, [1,3]диоксолан, [1,3]диоксан, тетрагидротиопиран, тетрагидротиопиран-1,1-диоксид, тетрагидротиопиран-1-оксид, пиперидин, тетрагидротиофен, [1,3]-дитиан, тиетан, [1,3]-дитиан-1,3-диоксид, тиетан-1-оксид или тиетан-1,1-диоксид.
В одном или нескольких вариантах осуществления настоящего изобретения гетероциклический фрагмент, образованный заместителями R1 и R2, а также атомом углерода, к которому они присоединены, включает один гетероатом или два гетероатома.
В одном или нескольких вариантах осуществления настоящего изобретения, гетероатом находится в положении 4 гетероциклического фрагмента. Этот гетероатом может являться, например, кислородом.
В одном или нескольких вариантах осуществления настоящего изобретения гетероатом (гетероатомы) представляет/представляют собой кислород, серу, -S(O)- или -S(O)2-.
В одном или нескольких вариантах осуществления настоящего изобретения фрагмент A представляет собой гетероарил или гетероарилалкил.
В одном или нескольких вариантах осуществления настоящего изобретения фрагмент A представляет собой пиридил, пиразинил или хинолил.
В других вариантах осуществления A может представлять собой фенил.
В одном или нескольких вариантах осуществления настоящего изобретения, фрагмент A замещен галогеном, в частности, хлором, фтором, бромом или йодом.
В одном или нескольких вариантах осуществления настоящего изобретения R3 представляет собой C1-6 алкокси, C1-6 галогеналкил или галоген.
В одном или нескольких вариантах осуществления настоящего изобретения R3 представляет собой метокси или этокси.
В одном или нескольких вариантах осуществления настоящего изобретения X представляет собой -CH2- или -NH-.
В одном или нескольких вариантах осуществления настоящего изобретения A представляет собой 4-(3,5-дихлорпиридил).
В одном или нескольких вариантах осуществления настоящего изобретения, соединение формулы (I) представлено формулой Ia или Ib
 .
.
где X, A, G, E, R1, R2, R3, R4, R5, m и n соответствуют данным выше определениям.
В отдельных вариантах осуществления настоящего изобретения X представляет собой -NH-, где R3 представляет собой C1-6 алкокси.
Настоящее изобретение охватывает все варианты осуществления, в которых X, A, G, E, R1, R2, R3, R4, R5 скомбинированы любым образом, описанным в настоящей заявке.
В частности, соединения формулы (I) могут быть выбраны из одного из следующих соединений:
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-ил)этанона (соединение 101),
N-(3,5-дихлорпиридин-4-ил)-7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоксамида (соединение 102),
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-ил)этанона (соединение 103),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-4',5'-дигидроспиро[1,3-бензодиоксол-2,3'-(2H)-тиофен]-4-ил)этанона (соединение 104),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 105),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-[метоксикарбонил]спиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 106),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-[метилсульфонил]спиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 107),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-ацетилспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 108),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-метилспиро[1,5-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 109),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 110),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1'-оксид]-4-ил)этанона (соединение 111),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1',1'-диоксид]-4-ил)этанона (соединение 112),
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1',1'-диоксид]-4-ил)этанона (соединение 113),
2-(3-бромпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 114),
2-(3-бромпиразин-2-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 115),
2-(пиразин-2-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 116),
2-(пиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 117),
2-(хинолин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 118),
2-(2,6-дихлорфенил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 119),
2-(2-хлорфенил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 120),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-оксетан]-6-ил}этанона (соединение 121),
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-оксетан]-6-ил}этанона (соединение 122),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан]-6-ил}этанона (соединение 123),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан-1',1'-диоксид]-6-ил}этанона (соединение 124),
2-(3,5-дихлорпиридин-1-оксидо-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан-1',1'-диоксид]-6-ил}этанона (соединение 125),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),2'-(1,3-диоксолан)]-6-ил}этанона (соединение 126),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),4'-тетрагидропиран]-6-ил}этанона (соединение 127),
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),4'-тетрагидропиран]-6-ил}этанона (соединение 128),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метокси-2',2'-диметилспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]диоксан]-6-ил}этанона (соединение 129),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]диоксан]-6-ил}этанона (соединение 130),
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]диоксан]-6-ил}этанона (соединение 131) и
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]дитиан]-6-ил}этанона (соединение 132),
а также их фармацевтически приемлемых солей, гидратов, N-оксидов или сольватов.
В одном или нескольких вариантах осуществления настоящего изобретения, соединения общей формулы (I) имеют молекулярную массу менее 800 Дальтон, например, менее 750 Дальтон, например, менее 700 Дальтон или менее 650, 600, 550 или 500 Дальтон.
В одном или нескольких вариантах осуществления настоящего изобретения, соединения формулы (I), соответствующие данному выше определению, применимы в терапии.
Соединения формулы (I) могут быть получены в кристаллической форме либо непосредственно путем концентрирования раствора в органическом растворителе, либо путем кристаллизации или перекристаллизации из органического растворителя или смеси указанного растворителя и сорастворителя, который может быть органическим или неорганическим, например, водой. Эти кристаллы могут быть выделены в форме, преимущественно свободной от растворителя, или в виде сольвата, например, гидрата. Настоящее изобретение охватывает все кристаллические модификации и формы, а также их смеси.
Соединения формулы (I) могут включать или не включать асимметрически замещенные (хиральные) атомы углерода, которые обеспечивают существование изомерных форм, например, энантиомеров и, возможно, диастереомеров. Настоящее изобретение относится ко всем таким изомерам, либо в виде чистых форм, либо в виде смесей чистых форм (например, рацематов). Чистые стереоизомерные формы соединений и промежуточные соединения по настоящему изобретению могут быть получены при использовании методик, известных в технике. Различные изомерные формы могут быть разделены с помощью физических способов разделения, например, селективной кристаллизации и хроматографических методик, например, жидкостной хроматографии с применением хиральных неподвижных фаз. Энантиомеры могут быть разделены друг от друга с помощью селективной кристаллизации их диастереомерных солей с оптически активными аминами, например, l-эфедрином. В качестве альтернативы, энантиомеры могут быть разделены посредством хроматографических методик с применением хиральных неподвижных фаз. Указанные чистые стереоизомерные формы могут также быть получены из соответствующих чистых стереоизомерных форм подходящих исходных соединений, при условии, что реакции проходят стереоселективно или стереоспецифично. Предпочтительно, если желательно получить конкретный стереоизомер, указанное соединение следует синтезировать с помощью стереоселективных или стереоспецифичных методик получения. В этих методиках преимущественно должны применяться хирально чистые исходные соединения.
Соединения по настоящему изобретению, необязательно в комбинации с другими действующими соединениями, могут применяться для лечения кожных заболеваний или состояний, или острых или хронических кожных язвенных расстройств, в частности, для лечения пролиферативных и воспалительных кожных расстройств, псориаза, рака, воспаления эпидермиса, выпадения волос, атрофии кожи, атрофии кожи, вызванной стероидами, старения кожи, старения кожи под действием света, акне, дерматита, атопического дерматита, себорейного дерматита, контактного дерматита, аллергической сыпи, зуда и экземы.
Помимо того, что соединения по настоящему изобретению применимы в лечении людей, их можно также применять для лечения животных, включая млекопитающих, например, лошадей, крупный рогатый скот, овец, свиней, собак и кошек.
Для применения в терапии, соединения по настоящему изобретению, как правило, применяются в форме фармацевтических композиций. Поэтому настоящее изобретение относится к фармацевтической композиции, включающей соединение формулы (I), необязательно вместе с одним или более другими терапевтически активными соединениями, в сочетании с фармацевтически приемлемым наполнителем или носителем. Этот носитель должен быть «приемлемым» в смысле совместимости с другими ингредиентами композиции, и не должен являться вредным для пациента, получающего композицию.
Как правило, действующий ингредиент составляет 0,05-99,9% по массе от массы состава.
В виде дозированной лекарственной формы соединение можно вводить один или более раз в день через соответствующие интервалы, однако, в любом случае в зависимости от состояния пациента и в соответствии с предписанием, сделанным практикующим медиком. Как правило, единица дозированной лекарственной формы содержит от 0,1 мг до 1000 мг, предпочтительно от 1 мг до 100 мг, например, 5-50 мг соединения формулы (I).
Подходящая дозировка соединения по настоящему изобретению будет зависеть, в числе прочего, от возраста и состояния пациента, тяжести заболевания, подвергаемого лечению, а также других факторов, хорошо известных практикующему врачу. Соединение по настоящему изобретению можно вводить перорально, парентерально или местно в зависимости от различного режима дозирования, например, дневного или с недельными интервалами. В основном разовая доза должна находиться в пределах от 0,01 до 400 мг/кг массы тела. Соединение можно вводить в форме болюсной дозы (т.е. всю дневную дозу вводят за один раз) или в виде отдельных доз два или более раз в день.
Что касается местного введения, возможно, более правильно использовать термин «единица применения», который означает разовую дозу, которую можно ввести пациенту и которая может быть легко обработана и упакована, сохраняясь при этом физически и химически стабильной дозировочной единицей, включающей либо действующее соединение как таковое, либо смесь этого соединения с твердыми или жидкими фармацевтическими разбавителями или носителями.
Термин «единица применения» в связи с местным применением означает единичную, т.е. одну дозу, допускающую при местном введении пациенту нанесение на квадратный сантиметр инфицированной поверхности от 0,1 мг до 10 мг и предпочтительно от 0,2 мг до 1 мг рассматриваемого действующего ингредиента.
Кроме того предусматривается, что в определенных режимах лечения, может оказаться применимым введение с более продолжительными интервалами, например, через день, раз в неделю или даже с еще более продолжительными интервалами.
Если лечение включает введение другого терапевтически активного соединения рекомендуется обратиться за справкой к Goodman & Gilman's The Pharmacological Basis of Therapeutics, 9th Ed., J.G. Hardman and L.E. Limbird (Eds.), McGraw-Hill 1995, для выяснения применимых дозировок указанных соединений.
Введение соединения по настоящему изобретению совместно с одним или более другими действующими соединениями может осуществляться либо одновременно либо последовательно.
Фармацевтические составы включают, например, составы в форме, подходящей для перорального (включая составы с отсроченным или долговременным высвобождением), ректального, парентерального (включая подкожное, интраперитонеальное, внутримышечное, внутрисуставное и внутривенное), трансдермального, глазного, местного, кожного, назального или буккального введения. Особенно предпочтительно местное введение заявленных составов.
Составы, как правило, могут быть представлены в виде дозированных лекарственных форм и их можно получать любым из способов, хорошо известных в фармацевтической технике, например, раскрытых в Remington, The Science and Practice of Pharmacy, 20th ed., 2000. Все методики включают стадию смешивания действующего ингредиента с носителем, который содержит один или более необходимых ингредиентов. В основном составы получают путем однородного и тщательного смешивания действующих ингредиентов с жидким носителем или тонкоизмельченным твердым носителем или носителями обоих этих типов и затем, если это необходимо, придания продукту желаемой формы.
Составы по настоящему изобретению, подходящие для перорального введения, могут иметь форму дискретных единиц, таких как капсулы, саше, таблетки или пастилки, каждая из которых содержит предусмотренное количество действующего ингредиента; форму порошка или гранул; форму раствора или суспензии в водной или неводной жидкости, например, этаноле или глицерине; или форму эмульсии масло-в-воде или вода-в-масле. Эти масла могут являться пищевыми маслами, как, например, хлопковым маслом, кунжутным маслом, кокосовым маслом или арахисовым маслом. Подходящие диспергирующие или суспендирующие средства для водных суспензий включают синтетические или натуральные камеди, такие как трагакант, альгинаты, гуммиарабик, декстран, натрий карбоксиметилцеллюлозу, желатин, метилцеллюлозу, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, карбомеры и поливинилпирролидон. Действующие ингредиенты могут вводиться также в форме болюсов, лекарственной кашки или пасты.
Таблетка может быть изготовлена прессованием или формованием действующего ингредиента необязательно с одним или более дополнительными ингредиентами. Прессованные таблетки могут быть получены путем прессования в подходящей машине для таблетирования действующего ингредиента(ов) в сыпучей форме, например, порошке или гранулах, необязательно смешанного со связующими веществами, как, например, лактозой, глюкозой, крахмалом, желатином, гуммиарабиком, смолой трагаканта, альгинатом натрия, карбоксиметилцеллюлозой, метилцеллюлозой, гидроксипропилметилцеллюлозой, полиэтиленгликолем, восками или подобными веществами; смазывающими средствами, как, например, олеатом натрия, стеаратом натрия, стеаратом магния, бензоатом натрия, ацетатом натрия, хлоридом натрия и т.п.; дезинтегрирующими средствами, как, например, крахмалом, метилцеллюлозой, агаром, бентонитом, кроскармелозой натрия, крахмал гликолятом натрия, кросповидоном и т.п., или дезинтегрирующим средством, как, например, полисорбатом 80. Формованные таблетки могут быть изготовлены путем формования в подходящем агрегате смеси порошкообразного действующего ингредиента и подходящего носителя, смоченного инертным жидким разбавителем.
Составы для ректального введения могут иметь форму суппозиториев, в которых соединение по настоящему изобретению смешано с легкоплавкими растворимыми или не растворимыми в воде твердыми веществами, например, маслом какао, гидрированными растительными маслами, полиэтиленгликолем или эфирами жирных кислот и полиэтиленгликолей, тогда как эликсиры могут быть приготовлены с применением миристилпальмитата.
Составы, пригодные для парентерального введения, обычно включают стерильные масляные или водные препараты действующего ингредиента, которые предпочтительно изотоничны крови пациента, получающего препарат, например, изотонический солевой раствор, изотонический раствор глюкозы или буферный раствор. Состав, как правило, можно стерилизовать, например, фильтрованием через фильтр, задерживающий бактерии, добавлением к составу стерилизующего средства, облучением или нагреванием состава. Липосомальные составы, описанные, например, в Encyclopedia of Pharmaceutical Technology, vol.9, 1994, также подходят для парентерального введения.
В качестве альтернативы, соединения формулы (I) могут входить в состав стерильных твердых препаратов, например, лиофилизованных порошков, которые легко растворяются в стерильном растворителе непосредственно перед применением.
Трансдермальные составы могут иметь форму пластыря или повязки.
Составы, подходящие для глазного введения, могут иметь форму стерильного водного препарата действующего ингредиента, который может присутствовать в микрокристаллической форме, например, в форме водной суспензии микрокристаллов. Липосомальные составы или биоразрушаемые полимерные системы, например, описанные в Encyclopedia of Pharmaceutical Technology, vol.2, 1989, также могут применяться для получения препаратов действующих ингредиентов, предназначенных для глазного введения.
Составы, пригодные для местного или глазного введения, включают жидкие или полужидкие препараты, такие как линименты, лосьоны, гели, спреи, пены, эмульсии масло-в-воде или вода-в-масле, такие как кремы, мази или пасты; или же растворы или суспензии, например, капли. Композиции для лечения глаз предпочтительно могут дополнительно содержать циклодекстрин.
Для местного введения, соединения формулы (I), как правило, могут присутствовать в количестве от 0,01 до 20% от массы композиции, как, например, от 0,1% до примерно 10%, но, кроме того, они могут присутствовать в количестве до примерно 50% от массы композиции.
Составы, применимые для назального или буккального введения, включают порошки, самораспыляющиеся составы и спреи, например, аэрозоли и распылители. Такие составы чрезвычайно подробно раскрыты, например, Modern Pharmaceutics, 2nd ed., G.S. Banker and C.T. Rhodes (Eds.), page 427-432, Marcel Dekker, New York; Modern Pharmaceutics, 3th ed., G.S. Banker and C.T. Rhodes (Eds.), page 618-619 and 718-721, Marcel Dekker, New York and Encyclopedia of Pharmaceutical Technology, vol. 10, J. Swarbrick and J.C. Boylan (Eds), page 191-221, Marcel Dekker, New York.
Помимо упомянутых выше ингредиентов, фармацевтические составы соединений формулы (I) могут включать один или более дополнительных ингредиентов, например, разбавителей, буферов, вкусоароматических средств, красителей, ПАВ, загустителей, консервантов, например, метил гидроксибензоата (включая антиоксиданты), эмульгирующих средств и т.п.
Если действующий ингредиент вводят в форме солей с фармацевтически приемлемыми нетоксичными кислотами или основаниями, предпочтительными солями являются, например, легко растворимые в воде или незначительно растворимые в воде, для достижения определенной и подходящей скорости абсорбции.
Фармацевтическая композиция может дополнительно включать один или более других действующих компонентов, обычно применяемых при лечении кожных заболеваний или состояний, например, выбранных из группы, состоящей из глюкокортикоидов, витамина D и аналогов витамина D, антигистаминных средств, антагонистов фактора активации тромбоцитов (PAF), антихолинергических средств, метилксантинов, β-адренергических средств, ингибиторов COX-2, салицилатов, индометацина, флуфенамата, напроксена, тимегадина, солей золота, пенициламина, средств для снижения уровней холестерина в сыворотке, ретиноидов, солей цинка, салицилазосульфапиридина и ингибиторов кальциневрина.
Изобретение более подробно описано в следующих примерах, которые никоим образом не ограничивают заявленный объем изобретения.
СПОСОБЫ ПОЛУЧЕНИЯ
Соединения по настоящему изобретению могут быть получены целым рядом способов, хорошо известных специалистам в области органического синтеза. Соединения формулы (I) могут быть получены, например, с применением реакций и методик, описанных ниже, в сочетании со способами, известными в технике синтетической органической химии, или их вариантами согласно оценке специалистов в данной области. Предпочтительные способы включают, не ограничиваясь этим, способы, описанные ниже. Реакции проводят в растворителях, которые подходят для применяемых реагентов и веществ и пригодны для осуществления выполняемых химических превращений. Кроме того, следует понимать, что в описанных ниже синтетических методиках все предложенные условия реакций, включая выбор растворителя, атмосферу реакции, температуру реакции, продолжительность эксперимента и способы обработки реакционной смеси, выбраны таким образом, чтобы являться стандартными условиями для данной реакции, которые должны быть полностью признаны специалистом в данной области. Не все соединения, относящиеся к данному классу, могут оказаться совместимыми с некоторыми из условий реакции, которые необходимы для некоторых из описанных способов. Эти ограничения на заместители, которые совместимы с условиями реакции, должны быть легко понятны специалисту в данной области техники, и могут применяться альтернативные способы.
Исходные вещества являются либо известными соединениями, которые имеются в продаже, либо их можно получить с помощью обычных синтетических методик, хорошо известных специалисту в данной области.
Соединения по настоящему изобретению или любые промежуточные соединения при необходимости могут быть очищены с применением стандартных методик, хорошо известных специалисту по синтетической органической химии, например, методик, описанных в “Purification of Laboratory Chemicals”, 5th ed.2003. Исходные соединения являются либо известными веществами, которые имеются в продаже, либо их можно получить стандартными способами, хороши известными специалистам в данной области.
ОБЩИЕ МЕТОДИКИ, ПОЛУЧЕНИЕ СОЕДИНЕНИЙ И ПРИМЕРЫ
Спектры ядерного магнитного резонанса (ЯМР) 1H регистрировали при частоте 300 МГц и спектры 13C-ЯМР при частоте 75,6 МГц. Значения химических сдвигов (δ, в м.д.) приведены для указанного растворителя по отношению к внутренним стандартам тетраметилсилану (δ=0,00), хлороформу (δ=7,25) или дейтерохлороформу (δ=76,81 для ЯМР 13C). Значения химического сдвига для мультиплета, либо указанного конкретно (дублет (д), триплет (т), квартет (кв)), либо нет (м) дано приблизительно для средней точки, если не указан диапазон. Сокращение ушир.с. указывает на уширенный синглет. Примененные органические растворители как правило являлись безводными.
Хроматографию осуществляли на силикагеле Merck silica gel 60 (0,040-0,063 мм). Соотношения растворителей приведены в виде соотношения объем:объем, если не отмечено иное.
В тексте заявки использованы следующие сокращения:
Препаративная ВЭЖХ/МС
Препаративную ВЭЖХ/МС проводили на системе Dionex APS с двумя препаративными насосами Shimadzu PP150 и масс-спектрометром Thermo MSQ Plus.
Колонка: Waters XTerra C-18, 150×19 мм, 5 мкм; система растворителей: A=вода (0,1% муравьиная кислота) и B=ацетонитрил (0,1% муравьиная кислота); скорость потока=18 мл/мин; методика (10 мин): методика линейного градиента, начиная с 10% B до 100% B в течение 6 минут и сохранение 100% B в течение еще 2 минут. Фракции собирали, основываясь на следовых количествах соответствующих ионов и сигнале PDA (240-400 нм).
Аналитическая ВЭЖХ/МС
Методика A: Аналитическую ВЭЖХ/МС проводили на системе Dionex APS с аналитическим насосом P680A и масс-спектрометром Thermo MSQ Plus. Колонка: Waters XTerra C-18, 150×4,6 мм, 5 мкм; система растворителей: A=вода (0,1% муравьиная кислота) и B=ацетонитрил (0,1% муравьиная кислота); скорость потока=1,0 мл/мин; методика (10 мин): методика линейного градиента, начиная с 10% B до 100% B в течение 6,6 минут и сохранение 100% B в течение еще 1,5 минут.
Методика B: Аналитическую ВЭЖХ/МС проводили на системе, состоящей из Waters 2795 HPLC, масс-спектрометра Micromass ZQ, Waters 996 PDA. Колонка: Waters XTerra C-18, 50×3,0 мм, 5 мкм; система растворителей: A=вода:ацетонитрил 95:5 (0,05% муравьиная кислота) и B=ацетонитрил (0,05% муравьиная кислота); скорость потока=1,0 мл/мин; методика (8 мин): методика линейного градиента, начиная с 10% B до 100% B в течение 6,0 минут и сохранение 100% B в течение 1 минуты.
Общая методика получения
Соединения по настоящему изобретению могут быть получены, например, следующими основными способами:
Соединения общей формулы Ia, в которых R1, R2 и R3 соответствуют данным выше определениям, могут быть получены следующим способом:
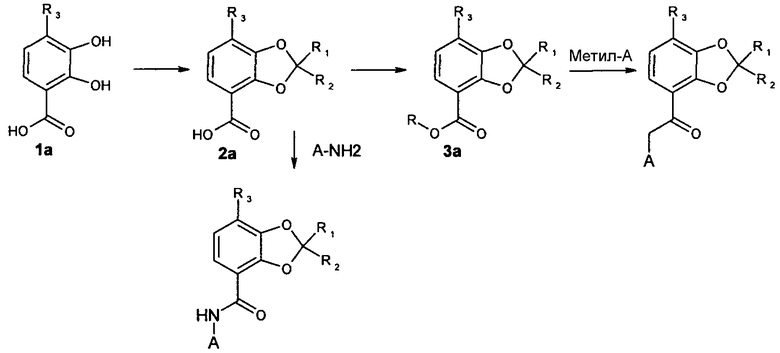
Исходные соединения формулы 1a получают по стандартным методикам, известным специалистам в области химии. 2,3,4-триметоксибензойную кислоту подвергают селективному ди-деметилированию по положениям 2- и 3-, используя BCl3, согласно работе Kaisalo et al. Synth. Commun, (1986), 16, 645-48.
Последующая реакция этого соединения, у которого снята защита, с чистыми (без примесей) кетонами, эфирами енолов, кеталями или смесями перечисленных реагентов с дополнительными катализаторами, такими как пара-толуолсульфоновая кислота или кислоты Льюиса или без них при температуре от комнатной до 180°C с использованием микроволнового излучения или обычного нагревания приводит к получению соединений 2a.
Взаимодействие соединений формулы 2a с MeI (или диметилсульфатом) в присутствии подходящего основания, например, K2CO3, KHCO3 или Et3N, в подходящем растворителе, таком как ДМФА, ацетон, ТГФ или DCM при температуре от комнатной до 100°C приводит к получению соединений формулы 3a.
Сложный эфир 3a может быть получен также по классической методике этерификации с использованием спирта и подходящей кислоты, например, H2SO4.
Соединения формулы Ia (X=CH2) получали конденсацией синтезированного метилового эфира с литиевыми производными карбанионов, полученными из соединений формулы A-метил, где A соответствует данным выше определениям, и подходящего основания, например, LDA или LiHMDS в подходящем растворителе, таком как ТГФ при температурах от -78°C до комнатной температуры. В качестве альтернативы литиевым производным карбанионов могут применяться реактивы Гриньяра.
Соединения формулы Ia (X=NH) получали взаимодействием соединений формулы 2a с (COCl)2, SOCl2 или PCl5 в подходящем растворителе, например, DCM или толуоле в присутствии или в отсутствие каталитического количества ДМФА при температурах от 0°C до 70°C, что приводило к образованию хлорангидрида соответствующей кислоты. После выпаривания растворителя в вакууме осуществлялась последующая конденсация полученного хлорангидрида кислоты с азотсодержащими анионами, генерированными добавлением подходящего основания, например, NaH, LDA или LiHMDS в подходящем растворителе, например, ТГФ, при температурах от -78°C до комнатной температуры к A-NH2, где A соответствует данному выше определению.
Соединения общей формулы Ib, где R1, R2 и R3 соответствуют данному выше определению, могут быть получены следующим способом:

Этерификация соединения 1b с использованием стандартных методик, например, MeOH и H2SO4, приводит к получению сложного эфира 2b.
Алкилирование 2b соединением 3b (X=Br, I, OTs) в присутствии подходящего основания, например, K2CO3 в подходящем растворителе, например, ДМСО, при температурах от комнатной до 120°C приводит к получению соединений формулы 4b.
Соединения формулы Ib (X=CH2) получали конденсацией синтезированного метилового эфира с литиевыми производными карбанионов, генерированными из соединений формулы A-метил, где A соответствует данному выше определению, и подходящего основания, например, LDA или LiHMDS в подходящем растворителе, например, ТГФ, при температурах от -78°C до комнатной. В качестве альтернативы литиевым производным карбанионов могут применяться реактивы Гриньяра.
Ожидается, что гидролиз сложных эфиров в стандартных условиях (кислотных или основных), приведет к образованию карбоновой кислоты 7b, которую можно превратить в хлорангидрид карбоновой кислоты и затем ввести в реакцию с азотсодержащим анионом (генерированным из A-NH2), как описано для синтеза Ia (X=NH).
Методика синтеза 1
7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоновая кислота (соединение 501)

Суспензию 2,3-дигидрокси-4-метоксибензойной кислоты (6,04 г, 32,8 ммоль) в 5,6-дигидро-4-метокси-2H-пиране (20 мл, 152 ммоль) выдерживали при 140°C в течение трех дней. При комнатной температуре добавляли этилацетат (200 мл) и органическую фазу экстрагировали насыщенным водным раствором NaHCO3 (2×50 мл). Водную фазу промывали Et2O (2х40 мл), подкисляли до pH=1 концентрированной HCl и экстрагировали дихлорметаном (2×50 мл). Органическую фазу высушивали над MgSO4. После упаривания при пониженном давлении получали следовые количества 2,3-дигидрокси-4-метоксибензойной кислоты наряду с 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоновой кислотой (1,23 г, 14%). 13C-ЯМР (ДМСО) δ 164,9, 148,2, 146,6, 134,5, 123,7, 117,0, 107,1, 106,8, 64,4, 56,0, 35,3.
Методика синтеза 2
Метил 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоксилат (соединение 502)

Суспензию неочищенной 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоновой кислоты (2,17 г, 8,15 ммоль), KHCO3 (2,58 г, 26,0 ммоль) и диметилсульфата (1,58 мл, 16,7 ммоль) в ацетоне (62 мл) перемешивали при комнатной температуре в течение двух дней, после чего выпаривали досуха при пониженном давлении. Добавляли этилацетат (100 мл). Органическую фазу промывали 0,5 М водным раствором NaOH (6×30 мл) и упаривали досуха при пониженном давлении. Неочищенный продукт вновь растворяли в дихлорметане (75 мл), высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная колоночная хроматография на силикагеле позволяла получить метил 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоксилат (1,87 г, 79%). 13C-ЯМР (CDCl3) δ 164,9, 149,1, 147,2, 135,2, 124,0, 117,5, 107,1, 106,5 65,2, 56,4, 51,8, 35,9.
Стандартная методика A
Пример 1
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-ил)этанон (соединение 101)
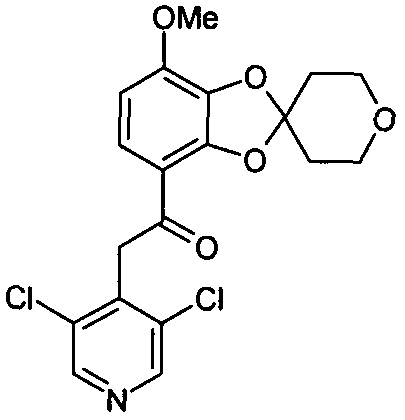
Раствор метил 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоксилата (1,80 г, 6,42 ммоль) и 3,5-дихлор-4-пиколина (1,46 г, 8,99 ммоль) в тетрагидрофуране (33 мл) охлаждали до 0°C. Добавляли 1,0 М раствор бис(триметилсилил)амида лития в тетрагидрофуране (19,3 мл, 19,3 ммоль) и давали подняться температуре реакционной смеси до комнатной в течение ночи. Добавляли насыщенный водный раствор NH4Cl (70 мл). Водную фазу экстрагировали дихлорметаном (3×100 мл). Объединенные органические фазы промывали водой (50 мл), высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная колоночная хроматография на силикагеле с последующей перекристаллизацией из изопропанола позволяла получить 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-ил)этанон (1,90 г, 71%). 13C-ЯМР (ДМСО) δ 189,1, 148,2, 147,7, 147,0, 141,2, 134,5, 132,8, 122,0, 118,0, 113,0, 107,8, 64,4, 56,3, 43,5, 35,2.
Пример 2
N-(3,5-дихлорпиридин-4-ил)-7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоксамид (соединение 102)

Оксалилхлорид (92 мкл, 1,1 ммоль) и каталитическое количество N,N-диметилформамида добавляли к суспензии 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоновой кислоты (48 мг, 0,18 ммоль) в дихлорметане (2 мл). После перемешивания в течение одного часа при комнатной температуре, растворитель удаляли при пониженном давлении и неочищенный хлорангидрид кислоты вновь растворяли в тетрагидрофуране (2 мл). Суспензию 3,5-дихлорпиридин-4-амина (67 мг, 0,40 ммоль) и NaH (60% дисперсия в минеральном масле, 16 мг, 0,40 ммоль) в тетрагидрофуране (1 мл) перемешивали в течение трех часов при комнатной температуре и затем при комнатной температуре по каплям добавляли к тетрагидрофурановому раствору, содержащему неочищенный хлорангидрид кислоты. После перемешивания при комнатной температуре в течение ночи, реакционную смесь разбавляли диэтиловым эфиром (30 мл) и органическую фазу промывали 0,5 М водным раствором NaOH (3×10 мл). Органическую фазу высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить N-(3,5-дихлорпиридин-4-ил)-7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоксамид (14 мг, 19%). 13C-ЯМР (ДМСО) δ 160,8, 148,0, 146,5, 146,2, 141,1, 134,1, 130,5, 122,5, 118,2, 108,3, 107,6, 64,2, 56,2, 35,2.
Пример 3
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-ил)этанон (соединение 103)

К раствору 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-ил)этанона (41 мг, 0,99 ммоль) в дихлорметане (0,5 мл) добавляли 30% H2O2 (25 мкл) и метилтриоксорений(VII) (3 мг). Полученную смесь перемешивали при комнатной температуре в течение ночи, добавляли MnO2 (3 мг) и перемешивали еще в течение часа. Добавляли воду (10 мл) и водную фазу экстрагировали дихлорметаном (3×10 мл). Объединенные органические фазы высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ приводила к получению соединения 103 (8 мг, 19%). ЖХ/МС (методика B): (m/z) 426,1; 428,1 (MH+); RT=2,98 мин; чистота (УФ)=100%.
Методика синтеза 3
Тетрагидро-3,3-диметокситиофен (соединение 503)

Раствор тетрагидротиофен-3-она (10,0 г, 97,9 ммоль), метилортоформиата (21,4 мл, 196 ммоль) и моногидрата пара-толуолсульфоновой кислоты (50 мг, 0,29 ммоль) в сухом метаноле (25 мл) кипятили с обратным холодильником в течение одного часа. Затем добавляли 1,0 М раствор NaOMe в метаноле (0,30 мл, 0,30 ммоль) и избыток метанола и триметилортоформиата удаляли отгонкой (при атмосферном давлении). Дальнейшая отгонка при пониженном давлении приводила к получению смеси тетрагидротиофен-3-она (~0,67 г, 7%) и тетрагидро-3,3-диметокситиофена (~9,8 г, 67%). 13C-ЯМР (MeOH) δ 113,01, 50,11, 36,90, 36,11, 27,72.
Методика синтеза 4
7-метокси-4',5'-дигидроспиро[1,3-бензодиоксол-2,3'-(2H)-тиофен]-4-карбоновая кислота (соединение 504)
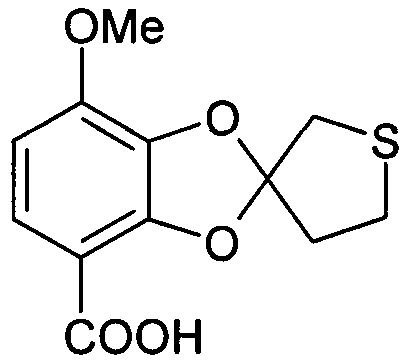
П-толуолсульфоновую кислоту (54 мг, 0,28 ммоль) добавляли к смеси тетрагидро-3,3-диметокситиофена (~9,8 г, 66 ммоль) и тетрагидротиофен-3-она (~0,67 г, 6,6 ммоль). Масляную баню нагревали до 145°C и отгоняли приблизительно один эквивалент метанола (2,7 мл, 67 ммоль). Понижали температуру, и в результате отгонки при пониженном давлении получали 7,04 г масла, к которому добавляли 2,3-дигидрокси-4-метоксибензойную кислоту (1,00 г, 5,43 ммоль). Полученную суспензию нагревали при действии микроволнового излучения (180°C, один час) в запаянном реакционном сосуде. Фильтрование с последующей стандартной очисткой ВЭЖХ приводило к получению соединения 504 (164 мг, 11%). ЖХ/МС (методика B): (m/z) 267,2 (M-1); RT=2,79 мин; чистота (УФ)=100%.
Методика синтеза 5
Метил 7-метокси-4',5'-дигидроспиро[1,3-бензодиоксол-2,3'-(2H)-тиофен]-4-карбоксилат (соединение 505)

Суспензию 7-метокси-4',5'-дигидроспиро[1,3-бензодиоксол-2,3'-(2H)-тиофен]-4-карбоновой кислоты (161 мг, 0,600 ммоль), K2CO3 (166 мг, 1,20 ммоль) и диметилсульфата (74 мкл, 0,78 ммоль) в ацетоне (1 мл) выдерживали при 50°C в течение ночи. При комнатной температуре добавляли воду и экстрагировали водную фазу этилацетатом (2×20 мл). Органическую фазу высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить соединение 505 (24 мг, 14%). 1H-ЯМР (CDCl3) δ 7,44 (д, 1Н), 6,56 (д, 1Н), 3,94 (с, 3Н), 3,88 (с, 3Н), 3,32 (д, 1Н), 3,24 )д, 1Н), 3,05 (т, 2Н), 2,49 (тд, 2Н).
Пример 4
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-4',5'-дигидроспиро[1,3-бензодиоксол-2,3'-(2H)-тиофен]-4-ил)этанон (соединение 104)

Раствор метил 7-метокси-4',5'-дигидроспиро[1,3-бензодиоксол-2,3'-(2H)-тиофен]-4-карбоксилата (24 мг, 85 мкмоль) и 3,5-дихлор-4-пиколина (21 мг, 0,13 ммоль) в тетрагидрофуране (1 мл) охлаждали до 0°C. Добавляли 1,0 М раствор бис(триметилсилил)амида лития в тетрагидрофуране (0,26 мл, 0,26 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Добавляли насыщенный водный раствор NH4Cl (10 мл). Водную фазу экстрагировали дихлорметаном (3×10 мл). Объединенные органические фазы промывали водой (20 мл), высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить указанное в заглавии соединение (12 мг, 34%). 13C-ЯМР (ДМСО) δ 189,02, 148,09, 147,51, 147,05, 140,98, 134,48, 132,69, 127,57, 122,31, 112,81, 107,81, 56,35, 43,26, 37,54, 36,54, 25,70.
Методика синтеза 6
1-ацетил-4,4-диметоксипиперидин (соединение 506)

Раствор 1-ацетил-4-пиперидона (17,0 г, 121 ммоль), триметилортоформиата (26,4 мл, 241 ммоль) и моногидрата пара-толуолсульфоновой кислоты (80 мг, 0,42 ммоль) в сухом метаноле (34 мл) кипятили с обратным холодильником в течение одного часа. Затем добавляли 1,0 М раствор NaOMe в метаноле (0,42 мл, 0,42 ммоль) и избыток метанола и триметилортоформиата удаляли отгонкой (при атмосферном давлении). Дополнительная отгонка при пониженном давлении позволила получить 1-ацетил-4,4-диметоксипиперидин (20,2 г, 89%). 1H-ЯМР (ДМСО) δ 3,45-3,32 (м, 4Н), 3,10 (с, 6Н), 1,99 (с, 3Н), 1,72-1,62 (м, 2Н), 1,61-1,52 (м, 2Н).
Методика синтеза 7
1-ацетил-1,2,3,6-тетрагидро-4-метоксипиридин (соединение 507)

К 1-ацетил-4,4-диметоксипиперидину (20,2 г, 108 ммоль) добавляли моногидрат пара-толуолсульфоновой кислоты (80 мг, 0,42 ммоль). Полученную смесь нагревали до 160°C и отгоняли примерно один эквивалент метанола (4,38 мл, 108 ммоль). Понижали температуру и в результате отгонки при пониженном давлении получали смесь 1-ацетил-4,4-диметоксипиперидина (1,4 г, 7%) и 1-ацетил-1,2,3,6-тетрагидро-4-метоксипиридина (14,2 г, 85%). 1H-ЯМР (ДМСО) δ 4,68-4,62 (м, 1Н), 4,00-3,88 (м, 2Н), 3,59-3,49 (м, 2Н), 3,49-3,45 (м, 3Н), 2,19-2,12 (м, 1Н), 2,09-2,03 (м, 1Н), 2,03-1,96 (м, 3Н).
Методика синтеза 8
7-метокси-1'-ацетилспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-карбоновая кислота (соединение 508)

Смесь 2,3-дигидрокси-4-метоксибензойной кислоты (1,23 г, 6,67 ммоль), 1-ацетил-4,4-диметоксипиперидина (1,4 г, 7,6 ммоль) и 1-ацетил-1,2,3,6-тетрагидро-4-метоксипиридина (14,2 г, 91,5 ммоль) нагревали под действием микроволнового излучения (180°C, один час) в запаянном реакционном сосуде. Фильтрование и последующая стандартная очистка ВЭЖХ позволяли получить соединение 508 (0,54 г, 26%). ЖХ/МС (методика B): (m/z) 308,2 (MH+); RT=2,27 мин; чистота (УФ)=95%.
Методика синтеза 9
Метил 7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-карбоксилат (соединение 509)

Раствор 7-метокси-1'-ацетилспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-карбоновой кислоты (143 мг, 0,467 ммоль) и LiOH (224 мг, 9,34 ммоль) в воде (3 мл) и MeOH (3 мл) нагревали до 75°C в течение пяти часов. При комнатной температуре смесь нейтрализовали 2 М HCl и упаривали досуха при пониженном давлении. Неочищенную 7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-карбоновую кислоту [ЖХ/МС (методика B): (m/z) 266,2 (MH+); RT=1,57 мин; чистота (УФ)=82%] кипятили с обратным холодильником в течение ночи в 1,7 М растворе HCl в метаноле (5 мл). При комнатной температуре добавляли воду (20 мл). Водную фазу промывали Et2O (10 мл), подщелачивали добавлением Na2CO3 и экстрагировали дихлорметаном (3×10 мл). Органическую фазу высушивали над MgSO4 и после упаривания при пониженном давлении получали метил 7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-карбоксилат (75 мг, 57%). 1H-ЯМР (ДМСО) δ 7,31 (д, 1Н), 8,72 (д, 1Н), 3,87 (с, 3Н), 3,78 (с, 3Н), 2,96-2,77 (м, 4Н), 1,94-1,83 (м, 4Н).
Пример 5
2-(3,5-дихлорпиридин-4-ил)-1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанон (соединение 105)
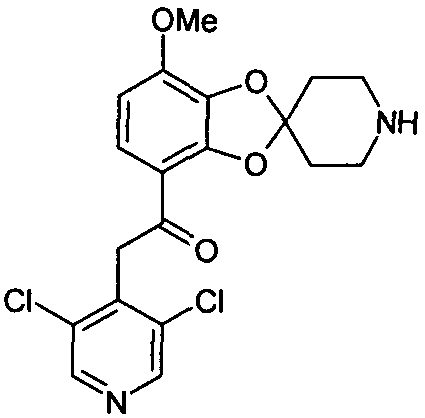
Раствор метил 7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-карбоксилата (75 мг, 0,268 моль) и 3,5-дихлор-4-пиколина (65 мг, 0,40 ммоль) в тетрагидрофуране (2,5 мл) охлаждали до 0°C. Добавляли 1,0 М раствор бис(триметилсилил)амида лития в тетрагидрофуране (0,80 мл, 0,80 ммоль) и температуре реакционной смеси давали подняться до комнатной в течение ночи. Добавляли насыщенный водный раствор NH4Cl (10 мл). Водную фазу экстрагировали дихлорметаном (3×10 мл). Объединенные органические фазы высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить соединение 105 (58 мг, 53%). 1H-ЯМР (ДМСО) δ 8,66 (с, 2H), 7,38-7,36 (м, 1H), 6,83-6,80 (м, 1H), 4,62 (с, 2H), 3,91 (с, 3H), 3,02-2,91 (м, 4H), 2,12-1,93 (м, 4H).
Пример 6
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-[метоксикарбонил]спиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанон (соединение 106)

Раствор 2-(3,5-дихлорпиридин-4-ил)-1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (10 мг, 24 мкмоль), триэтиламина (24 мкл, 171 мкмоль) и метилхлорформиата (10 мкл, 122 мкмоль) в дихлорметане (200 мкл) выдерживали при комнатной температуре в течение ночи. Добавляли вводу (500 мкл) и водную фазу экстрагировали дихлорметаном (3×500 мкл). Органическую фазу высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить соединение 106 (2,5 мг, 22%). 1H-ЯМР (ДМСО) δ 8,65 (с, 2Н), 7,39 (д, 1Н), 6,84 (д, 1Н), 4,62 (с, 2Н), 3,91 (с, 3Н), 3,80-3,66 (м, 2Н), 3,62 (с, 3Н), 3,58-3,46 (м, 2Н) 2,18-1,97 (м, 4Н).
Пример 7
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-[метилсульфонил]спиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанон (соединение 107)

Раствор 2-(3,5-дихлорпиридин-4-ил)-1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (10 мг, 24 мкмоль), триэтиламина (24 мкл, 171 мкмоль) и мезилхлорида (10 мкл, 122 мкмоль) в дихлорметане (200 мкл) выдерживали при комнатной температуре в течение ночи. Добавляли воду (500 мкл) и водную фазу экстрагировали дихлорметаном (3×500 мкл). Органическую фазу высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить соединение 107 (1,8 мг, 15%). 1H-ЯМР (ДМСО) δ 8,66 (с, 2Н), 7,45-7,38 (м, 1Н), 6,88-6,81 (м, 1Н), 4,62 (с, 2Н), 3,92 (с, 3Н), 3,50-3,36 (м, 4Н), 2,98 (с, 3Н), 2,29-2,11 (м, 4Н).
Пример 8
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-ацетил-спиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанон (соединение 108)
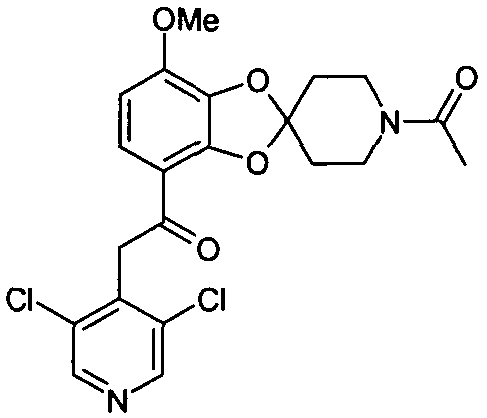
Раствор 2-(3,5-дихлорпиридин-4-ил)-1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (10 мг, 24 мкмоль), триэтиламина (24 мкл, 171 мкмоль) и уксусного ангидрида (12 мкл, 122 мкмоль) в дихлорметане (200 мкл) выдерживали при комнатной температуре в течение ночи. Добавляли вводу (500 мкл) и водную фазу экстрагировали дихлорметаном (3×500 мкл). Органическую фазу высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить соединение 108 (7,2 мг, 65%). 1H-ЯМР (ДМСО) δ 8,66 (с, 2Н), 7,40 (д, 1Н), 6,84 (д, 1Н), 4,63 (с, 2Н), 3,92 (с, 3Н), 3,90-3,84 (м, 1Н), 3,75-3,68 (м, 1Н), 3,64-3,58 (м, 1Н), 3,55-3,49 (м, 1Н), 2,21-2,15 (м, 1Н), 2,15-2,05 (м, 5Н), 2,01-1,94 (м, 1Н).
Методика синтеза 10
4,4-диметокситетрагидро-(4H)-тиопиран (соединение 510)

Смесь тетрагидро-(4H)-тиопиран-4-она (15,0 г, 129 ммоль), триметилортоформиата (28,3 мл, 258 ммоль) и моногидрата пара-толуолсульфоновой кислоты (67 мг, 0,35 ммоль) в метаноле (40 мл) кипятили с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, добавляли 1 М NaOMe (0,35 мл, 0,35 ммоль), и избыток метанола и триметилортоформиата удаляли отгонкой (при атмосферном давлении). Дополнительная отгонка при пониженном давлении позволяла получить 4,4-диметокситетрагидро-(4H)-тиопиран (20,7 г, 99%). 1H-ЯМР (ДМСО) δ 3,07 (с, 6Н), 2,56 (м, 4Н), 1,84 (м, 4Н).
Методика синтеза 11
7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-карбоновая кислота (соединение 511)
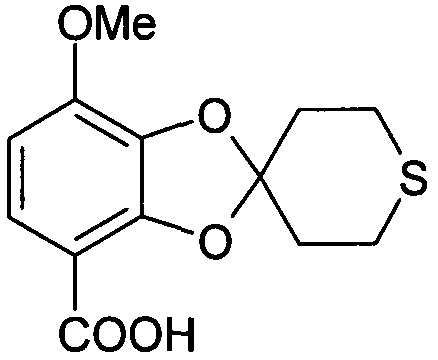
П-толуолсульфоновую кислоту (97 мг, 0,51 ммоль) добавляли к 4,4-диметокситетрагидро-(4H)-тиопирану (20,7 г, 128 ммоль), смесь нагревали до 145°C и выдерживали при этой температуре до отгонки приблизительно одного эквивалента метанола (5,17 мл, 128 ммоль). Затем смесь охлаждали до 130°C и в результате отгонки при пониженном давлении получали 10,1 г смеси 5:3 5,6-дигидро-4-метокси-(2H)-тиопирана [1H-ЯМР (ДМСО) δ 4,87 (м, 1Н), 3,44 (с, 3Н), 3,15 (дт, 2Н), 2,72 (т, 2Н), 2,22 (м, 2Н)] и 4,4-диметокситетрагидро-(4H)-тиопирана. Без дополнительной очистки эту смесь добавляли к 2,3-дигидрокси-4-метоксибензойной кислоте (2,00 г, 10,9 ммоль) и суспензию нагревали действием микроволнового излучения (180°C, один час) в запаянном реакционном сосуде. Добавляли этилацетат (100 мл) и органическую фазу, во-первых, промывали 0,5 М HCl (40 мл) и затем экстрагировали насыщенным водным раствором NaHCO3 (2×30 мл). Водную фазу промывали Et2O (2×40 мл), подкисляли до pH=1 концентрированной HCl и экстрагировали дихлорметаном (2×30 мл). Органическую фазу высушивали над MgSO4. Упаривание при пониженном давлении приводило к получению 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-карбоновой кислоты (1,86 г, 61%). 13C-ЯМР (ДМСО) δ 164,9, 148,2, 146,6, 134,5, 123,8, 118,0, 107,2, 106,9, 56,1, 35,9, 25,4.
Методика синтеза 12
Метил 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-карбоксилат (соединение 512)
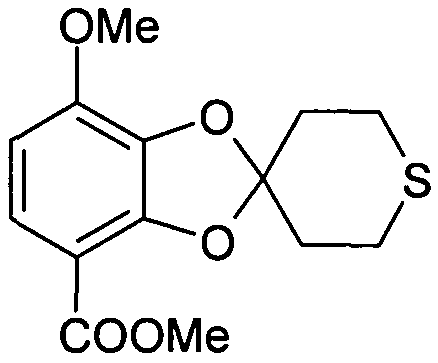
Суспензию 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-карбоновой кислоты (570 мг, 2,02 ммоль), K2CO3 (558 мг, 4,04 ммоль) и диметилсульфата (0,25 мл, 2,62 ммоль) в ацетоне (14 мл) перемешивали при 50°C в течение ночи. При комнатной температуре добавляли воду (30 мл). Водную фазу экстрагировали дихлорметаном (3×15 мл). Объединенные органические фазы высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная колоночная хроматография на силикагеле позволяла получить метил 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-карбоксилат (407 мг, 68%). 13C-ЯМР (ДМСО) δ 163,8, 148,1, 146,9, 134,6, 123,4, 118,3, 107,1, 105,9, 56,1, 51,6, 35,9, 25,4.
Пример 9
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанон (соединение 110)

Раствор метил 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-карбоксилата (40 мг, 0,14 ммоль) и 3,5-дихлор-4-пиколина (33 мг, 0,20 ммоль) в тетрагидрофуране (1,1 мл) охлаждали до 0°C. Добавляли 1,0 М раствор бис(триметилсилил)амида лития в тетрагидрофуране (0,41 мл, 0,41 ммоль), и реакционной смеси давали нагреться до комнатной температуры в течение ночи. Добавляли насыщенный водный раствор NH4Cl (20 мл). Водную фазу экстрагировали дихлорметаном (3×15 мл). Объединенные органические фазы высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанон (38 мг, 67%). 13C-ЯМР (ДМСО) δ 189,1, 148,2, 147,7, 147,0, 141,3, 134,5, 132,8, 122,0, 119,1, 113,0, 107,9, 56,3, 43,6, 35,9, 25,5.
Пример 10
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1'-оксид]-4-ил)этанон (соединение 111)

К раствору 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (17 мг, 40 мкмоль) в дихлорметане (0,5 мл) добавляли, во-первых, 0,25 М раствор H2O2 в этаноле (128 мкл, 32 мкмоль) и, во-вторых, метилтриоксорений(VII) (1 мг, 4 мкмоль). Смесь перемешивали при комнатной температуре в течение двух дней и выпаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1'-оксид]-4-ил)этанон (7 мг, 40%). 1H-ЯМР (ДМСО) δ 8,66 (с, 2Н), 7,42 (д, 1Н), 6,85 (д, 1Н), 4,63 (с, 2Н), 3,93 (с, 3Н), 3,17-2,94 (м, 4Н), 2,69-2,55 (м, 2Н), 2,36-2,24 (м, 2Н).
Пример 11
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1',1'-диоксид]-4-ил)этанон (соединение 112)

К раствору 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (11 мг, 26 мкмоль) в дихлорметане (0,25 мл) добавляли мета-хлорпербензойную кислоту (10 мг, 58 мкмоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный водный раствор NaHCO3 (1 мл) и водную фазу экстрагировали дихлорметаном (2×2 мл). Объединенные органические фазы высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1',1'-диоксид]-4-ил)этанон (5 мг, 42%). 1H-ЯМР (CDCl3) δ 8,52 (с, 2Н), 7,55 (д, 1Н), 6,68 (д, 1Н), 4,55 (с, 2Н), 3,99 (с, 3Н), 3,45-3,37 (м, 2Н), 3,33-3,25 (м, 2Н), 2,79-2,66 (м, 4Н).
Пример 12
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1',1'-диоксид]-4-ил)этанон (соединение 113)
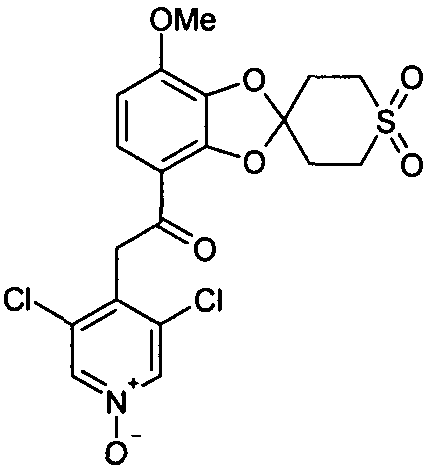
К раствору 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (10 мг, 23 мкмоль) в абсолютном этаноле (2 мл), во-первых, добавляли H2O2 (100 мкл, 0,97 ммоль) и затем, во-вторых, метилтриоксорений(VII) (2 мг, 8 мкмоль). Полученную смесь перемешивали при 40°C в течение ночи и затем добавляли 5% масс/объем водный раствор NaHSO3 (10 мл). Водную фазу экстрагировали дихлорметаном (3×20 мл). Объединенные органические фазы высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить 2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1',1'-диоксид]-4-ил)этанон (2,5 мг, 23%). 1H-ЯМР (ДМСО) δ 8,64 (с, 2Н), 7,42 (д, 1Н), 6,85 (д, 1Н), 4,59 (с, 2Н), 3,92 (с, 3Н), 3,51 (м, 2Н), 3,31 (м, 2Н), 2,59 (м, 4Н).
Общая методика A
LiHMDS (1 М в ТГФ, 3,0 экв.) по каплям добавляли к ледяному раствору сложного эфира 512 (1 экв.) и соединения формулы A-метил (1,3 экв.) в безводном ТГФ. Реакционную смесь перемешивали при к.т. в течение 12 часов, добавляли H2O (10 мл) и насыщенный водный раствор NH4Cl (20 мл), и затем экстрагировали EtOAc (3×50 мл). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной флэш-хроматографией, получая кетон.

Пример 13
2-(3-бромпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанон (соединение 114)
Указанное в заглавии соединение получали согласно общей методике A, используя 18 мг 4-бром-3-метилпиридина (выход: 40%)

ЖХ/МС (методика B): (m/z) 436,2 (MH+); RT=4,17 мин; чистота (УФ)=100%.
Пример 14
2-(3-бромпиразин-2-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанон (соединение 115)
Указанное в заглавии соединение получали согласно общей методике A, используя 17 мг 2-бром-3-метилпиразина (выход: 9%)
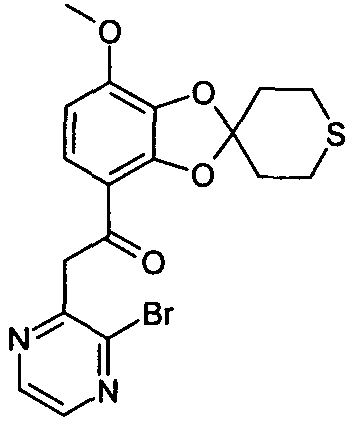
ЖХ/МС (методика B): (m/z) 437,2; 439,22 (MH+); RT=4,22 мин; чистота (УФ)=100%.
Пример 15
2-(пиразин-2-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанон (соединение 116)
Указанное в заглавии соединение получали согласно общей методике A, используя 16 мг 2-метилпиразина (выход: 52%)
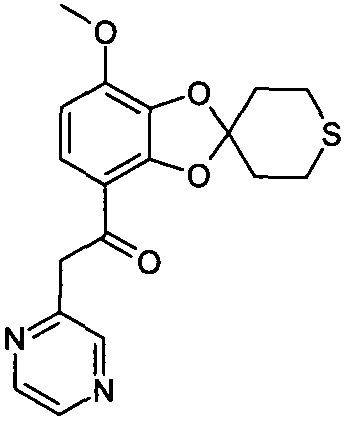
ЖХ/МС (методика B): (m/z) 359,3 (MH+); RT=3,33 мин; чистота (УФ)=100%.
Пример 16
2-(пиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанон (соединение 117)
Указанное в заглавии соединение получали согласно общей методике A, используя 18 мг 4-метилпиридина (выход: 13%)

ЖХ/МС (методика B): (m/z) 358,3 (MH+); RT=2,50 мин; чистота (УФ)=100%.
Пример 17
2-(хинолин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанон (соединение 118)
Указанное в заглавии соединение получали согласно общей методике A, используя 16 мг 4-метилхинолина (выход: 12%)
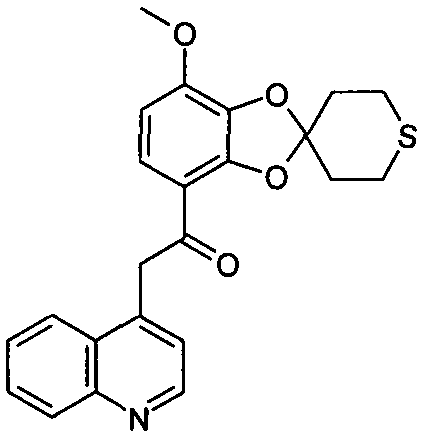
ЖХ/МС (методика B): (m/z) 408,3 (MH+); RT=3,33 мин; чистота (УФ)=100%.
Пример 18
2-(2,6-дихлорфенил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанон (соединение 119)
Указанное в заглавии соединение получали согласно общей методике A, используя 15 мг 2,6-дихлортолуола (выход: 8%)
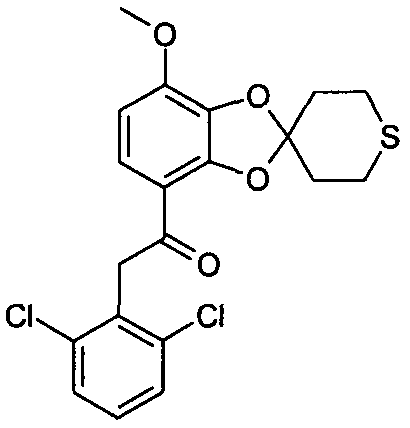
ЖХ/МС (методика B): (m/z) 425,24 (MH+); RT=5,28 мин; чистота (УФ)=100%.
Пример 19
2-(2-хлорфенил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанон (соединение 120)
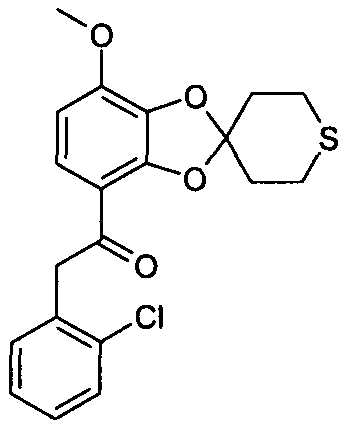
Раствор метил 7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-карбоксилата (14 мг) в сухом ТГФ (500 мл) охлаждали до 0°C в атмосфере Ar. Добавляли раствор 2-хлорбензилмагнийхлорида в диэтиловом эфире (0,25 М раствор, 189 мкл) и прекращали охлаждение. Через 2 часа при комнатной температуре добавляли дополнительную порцию 2-хлорбензилмагнийхлорида в диэтиловом эфире (0,25 М раствор, 189 мкл). Смесь перемешивали в течение 18 часов, добавляли воду и экстрагировали этилацетатом. Объединенные органические фазы высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить соединение 120 (9,2%).
ЖХ/МС (методика B): (m/z) 391,22 (MH+); RT=4,90 мин; чистота (УФ)=100%.
Методика синтеза 13
Метил 2,3-дигидрокси-4-метоксибензоат (соединение 513)

Раствор коммерчески доступной 2,3-дигидрокси-4-метоксибензойной кислоты (11,6 г, 63 ммоль) в безводном MeOH (150 мл) охлаждали на ледяной бане и по каплям добавляли концентрированную H2SO4 (8 мл). Реакционную смесь кипятили с обратным холодильником в течение 12 часов, затем охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Добавляли H2O (100 мл) и насыщенный водный раствор NaHCO3 (50 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали в вакууме, получая соединение 513 в виде бледно-желтого твердого вещества, которое применяли в следующей стадии без дополнительной очистки. ЖХ-МС: RT=2,31 мин; m/z 197,3 (M-H)-. 1H-ЯМР (CDCl3) δ 10,83 (1Н, с), 7,41 (1Н, д, J=9,0), 6,50 (1Н, д, J=8,9), 5,45 (1Н, с), 3,94 (3Н, с), 3,93 (3Н, с).
Общая методика B
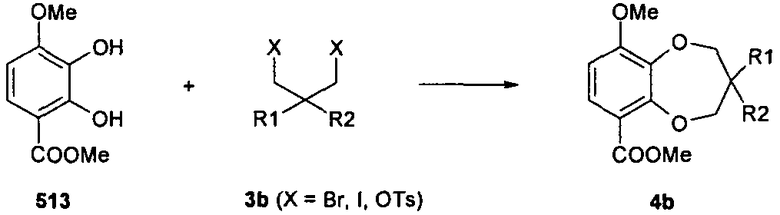
К перемешиваемому раствору соединений 513 и 3b (1,1 экв.) в безводном ДМСО добавляли K2CO3 (2,5 экв.) и смесь перемешивали при 100°C в течение 4-12 часов в атмосфере инертного газа. После охлаждения до комнатной температуры добавляли смесь воды и льда, перемешивали в течение 15 минут и затем экстрагировали EtOAc (3×50 мл). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной флэш-хроматографией.
С применением общей методики B были получены следующие соединения:
Методика синтеза 14
Метиловый эфир 9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-оксетан]-6-карбоновой кислоты (соединение 514)

Диалкилирование соединения 513 (198 мг, 1 ммоль) коммерчески доступным 3,3-бис(йодметил)оксетаном (372 мг, 1,1 ммоль) в ДМСО (5 мл) в присутствии K2CO3 (345 мг, 2,5 ммоль), выполненное в соответствии с общей методикой, позволяло получить соединение 514 в виде белого твердого вещества после очистки колоночной хроматографией (50-65% EtOAc в петролейном эфире). ЖХ-МС: RT=2,40 мин; m/z 281,26 (M+H)+. 1H-ЯМР (CDCl3) δ 7,49 (1Н, д, J=8,8), 6,62 (1Н, д, J=8,8), 4,61 (2Н, д, J=6,8), 4,58 (2Н, д, J=6,8), 4,48 (4Н, с), 3,90 (3Н, с), 3,87 (3Н, с).
Методика синтеза 15
Метиловый эфир 9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан]-6-карбоновой кислоты (соединение 516)
Стадия A
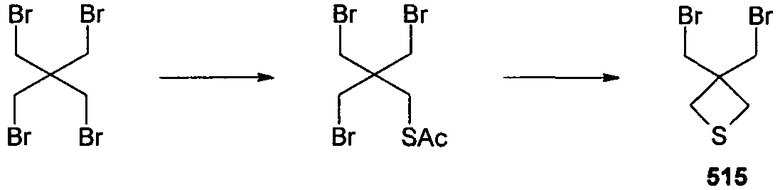
3,3-бис(бромметил)тиетан 515 получали по двухстадийной методике из 1,3-дибром-2,2-бис(бромметил)пропана, следуя описанному в литературе способу (Petrukhina, M.A.; Henck, C; Li, B.; Block, E.; Jin, J.; Zhang, S-Z.; Clerac, R. Inorg. Chem. 2005, 44, 77-84). Соответственно, смесь 1,3-дибром-2,2-бис(бромметил)пропана (7,76 г, 20 ммоль) и KSAc (2,28 г, 20 ммоль) в безводном ТГФ (30 мл) кипятили с обратным холодильником в течение 30 часов. Осадок отделяли фильтрованием, фильтрат концентрировали и полученный остаток очищали колоночной флэш-хроматографией (10-25% EtOAc в петролейном эфире), получая (2,2-(бисбромметил)-3-бромпропиловый)эфир тиоуксусной кислоты в виде бледно-желтого твердого вещества. Смесь (2,2-(бисбромметил)-3-бромпропилового)эфира тиоуксусной кислоты (1,53 г, 4 ммоль) и NaOMe (324 мг, 6 ммоль) в безводном MeOH (10 мл) перемешивали при 0°C в течение 2 часов. MeOH удаляли в вакууме, выпаривая совместно с толуолом (2×2 мл), и полученный остаток фильтровали через тонкий слой силикагеля, получая 3,3-бис(бромметил)тиетан 515 в виде густого масла, которое использовали без дополнительной очистки.
Стадия B
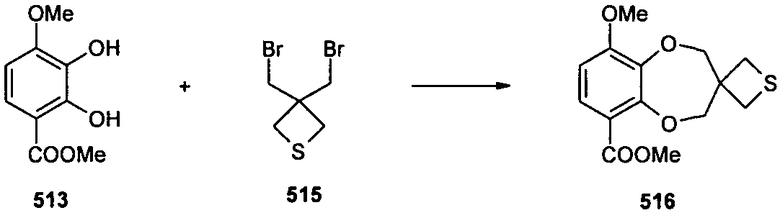
Диалкилирование соединения 513 (665 мг, 3,36 ммоль) соединением 515 (962 мг, 3,7 ммоль) в ДМСО (15 мл) в присутствии K2CO3 (1,16 г, 8,4 ммоль), выполненное в соответствии с общей методикой, позволяло получить соединение 516 в виде белого твердого вещества после очистки колоночной хроматографией (40-60% EtOAc в петролейном эфире). ЖХ-МС: RT=3,17 мин; m/z 297,19 (M+H)+. 1H-ЯМР (CDCl3) δ 7,49 (1Н, д, J=8,8), 6,63 (1Н, д, J=8,8), 4,30 (2Н, с), 4,28 (2Н, с), 3,90 (3Н, с), 3,87 (3Н, с), 3,11 (4Н, с).
Методика синтеза 16
Метиловый эфир 9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),2'-(1,3-диоксолан)]-6-карбоновой кислоты (соединение 518)
Стадия A

2,2-бис(бромметил)-1,3-диоксолан 517 получали из дибромацетона по описанной в литературе методике (Valentin, M-L.; Bolte, J. Bull. Soc. Chim. Fr., 1995, 132, 1167-71). В соответствии с методикой раствор дибромацетона (4,04 г, 18,7 ммоль), этиленгликоля (2,32 г, 37,4 ммоль) и п-TsOH (25 мг) в бензоле (70 мл) кипятили в течение 12 часов с азеотропной отгонкой воды. Реакционную смесь концентрировали при пониженном давлении, добавляли Et2O (50 мл) и органический слой промывали H2O (2×50 мл), высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной флэш-хроматографией (7-10 EtOAc в петролейном эфире), получая 2,2-бис(бромметил)-1,3-диоксолан (517) в виде бесцветной жидкости.
Стадия B

Диалкилирование соединения 513 (396 мг, 2 ммоль) соединением 517 (572 мг, 2,2 ммоль) в ДМСО (10 мл) в присутствии K2CO3 (690 мг, 5 ммоль), выполненное в соответствии с общей методикой, позволяло получить соединение 518 в виде белого твердого вещества после очистки колоночной хроматографией (40-60% EtOAc в петролейном эфире). ЖХ-МС: RT=2,70 мин; m/z 297,18 (M+H)+, 319,16 (M+Na)+. 1H-ЯМР (ДМСО-d6) δ 7,34 (1Н, д, J=8,8), 6,76 (1Н, д, J=8,8), 4,11 (2Н, с), 4,09 (2Н, с), 3,94 (4Н, с), 3,80 (3Н, с), 3,75 (3Н, с).
Методика синтеза 17
Метиловый эфир 9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),4'тетрагидропиран]-6-карбоновой кислоты (соединение 520)
Стадия A
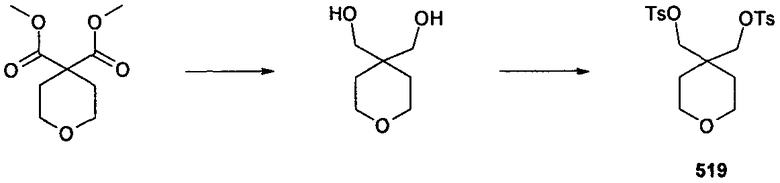
4,4-бис(п-толуолсульфонилоксиметил)тетрагидропиран (519) получали по двухстадийной методике из имеющегося в продаже диметилового эфира тетрагидропиран-4,4-дикарбоновой кислоты. В соответствии с методикой к ледяному раствору указанного диэфира (3,03 г, 15 ммоль) в безводном толуоле (45 мл) добавляли по каплям Synhydride (70% в толуоле, 19,5 мл, 66 ммоль) и полученную смесь перемешивали при 120°C в течение 3 часов. Смесь охлаждали до комнатной температуры, медленно добавляли H2O (50 мл) и концентрировали над силикагелем (35 г). Быстрая колоночная хроматография с линейным градиентом MeOH в CH2Cl2 позволяла получить 4,4-(бисгидроксиметил)тетрагидропиран в виде белого твердого вещества. Смесь диола (1,46 г, 10 ммоль) и TsCl (4,77 г, 25 ммоль) в безводном пиридине (25 мл) перемешивали при комнатной температуре в течение 48 часов. Растворитель удаляли в вакууме и осуществляли совместное выпаривание в толуоле (3×10 мл). Добавляли CH2Cl2 (100 мл) и насыщенный водный раствор NaHCO3 (100 мл), разделяли фазы и органическую фазу высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали флэш-хроматографией (35-45% EtOAc в петролейном эфире), получая 4,4-бис(п-толуолсульфонилоксиметил)тетрагидропиран (519) в виде белого твердого вещества.
Стадия B

Диалкилирование соединения 513 (95 мг, 0,48 ммоль) соединением 519 (240 мг, 0,53 ммоль) в ДМСО (3 мл) в присутствии K2CO3 (166 мг, 1,2 ммоль), выполненное в соответствии с общей методикой, позволяло получить соединение 520 в виде белого твердого вещества после очистки колоночной хроматографией (45-65% EtOAc в петролейном эфире). ЖХ-МС: RT=2,40 мин; 1H-ЯМР (ДМСО-d6) δ 7,34 (1Н, д, J=8,8), 6,78 (1Н, д, J=8,8), 3,99 (4Н, с), 3,80 (3Н, с), 3,76 (3Н, с), 3,61 (4Н, т, J=5,5), 1,54 (4Н, т, J=5,5).
Методика синтеза 18
Метиловый эфир 9-метокси-2',2'-диметилспиро[2H-1,5-бензодиоксепин-3(4H),5'[1,3]диоксан]-6-карбоновой кислоты (соединение 522)
Стадия A

Раствор 2,2-бис(бромметил)-1,3-пропандиол (5,0 г, 19,1 ммоль), безводного ацетона (20 мл) и п-TsOH (100 мг) в бензоле (75 мл) кипятили с обратным холодильником в течение 12 часов с азеотропной отгонкой воды. Реакционную смесь концентрировали при пониженном давлении, добавляли EtOAc (100 мл), и органический слой последовательно промывали H2O (3×30 мл) и насыщенным раствором соли (30 мл), высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученное белое вещество растирали с н-пентаном (20 мл), получая 5,5-бис(бромметил)-2,2-диметил-[1,3]диоксан (521) в виде бесцветных кристаллов.
Стадия B
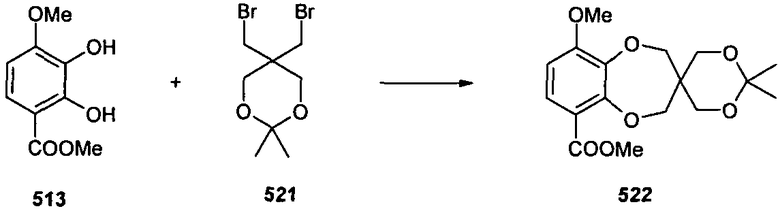
Диалкилирование соединения 513 (1,19 г, 6 ммоль) соединением 521 (2,0 г, 6,62 ммоль) в ДМСО (30 мл) в присутствии K2CO3 (2,07 г, 15 ммоль), выполненное в соответствии с общей методикой, позволяло получить соединение 522 в виде белого твердого вещества после очистки колоночной хроматографией (30-40% EtOAc в петролейном эфире). ЖХ-МС: RT=2,99 мин; m/z 339,31 (M+H)+, 361,25 (M+Na)+. 1H-ЯМР (CDCl3) δ 7,45 (1Н, д, J=8,8), 6,59 (1Н, д, J=8,8), 4,22 (2Н, с), 4,18 (2Н, с), 3,89 (3Н, с), 3,86 (7Н, с), 1,43 (6Н, с).
Методика синтеза 19
Метиловый эфир 9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'[1,3]диоксан]-6-карбоновой кислоты (соединение 524)
Стадия A

5,5-бис(бромметил)-[1,3]диоксан (523) получали из 2,2-бис(бромметил)-1,3-пропандиола, следуя известной из литературы методике (Bitha, P.; Carvajal, S.G.; Citarella, R.V.; Delos Santos, E.F.; Durr, F.E.; Hlavka, J.J.; Lang, S.A., Jr.; Lindsay, H.L.; Thomas, J.P.; Wallace, R.E.; Yang-I,L. J. Med. Chem. 1989, 32(9), 2063-7 и Mitkin, O.D.; Wan, Y.; Kurchan, A.N.; Kutateladze, A.G. Synthesis, 2001 (8), 1133-42). В соответствии с методикой, раствор 2,2-бис(бромметил)-1,3-пропандиола (2,5 г, 9,55 ммоль), формальдегида (37% водный раствор, 3,5 мл) и концентрированную HCl (2,0 мл) кипятили с обратным холодильником в течение 12 часов. После охлаждения до к.т. к реакционной смеси добавляли H2O (25 мл) и экстрагировали CH2Cl2 (2×25 мл). Объединенный органический слой последовательно промывали насыщенным водным раствором Na2CO3 (25 мл) и H2O (25 мл), высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении. С помощью 1H-ЯМР было установлено, что полученная жидкость имеет чистоту >95% и ее использовали без дальнейшей очистки.
Стадия B
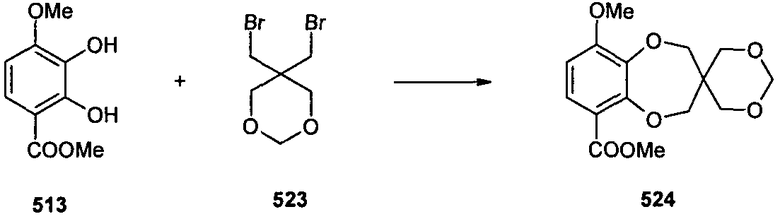
Диалкилирование соединения 513 (198 мг, 1 ммоль) соединением 523 (301 мг, 1,1 ммоль) в ДМСО (5 мл) в присутствии K2CO3 (345 мг, 2,5 ммоль), выполненное в соответствии с общей методикой, позволяло получить соединение 524 в виде белого твердого вещества после очистки колоночной хроматографией (50-65% EtOAc в петролейном эфире). ЖХ-МС: RT=2,65 мин; m/z 311,23 (M+H)+. 1H-ЯМР (ДМСО-d6) δ 7,37 (1Н, д, J=8,8), 6,81 (1Н, д, J=8,8), 4,80 (1Н, д, J=6,4), 4,78 (1Н, д, J=6,4), 4,07 (2Н, с), 4,01 (2Н, с), 3,81 (7Н, с), 3,76 (3Н, с).
Методика синтеза 20
Метиловый эфир 9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'[1,3]дитиан]-6-карбоновой кислоты (соединение 526)
Стадия A

4,4-бис(гидроксиметил)-[1,3]дитиан получали по двухстадийному способу из соединения 523, следуя известной из литературы методике (Mitkin, O.D.; Wan, Y.; Kurchan, A.N.; Kutateladze, A.G. Synthesis, 2001 (8), 1133-42). В соответствии с методикой смесь соединения 523 (1,36 г, 5 ммоль) и KSAc (1,71 г, 15 ммоль) в безводном ДМФА (10 мл) перемешивали при комнатной температуре в течение 30 часов. Удаляли растворитель при пониженном давлении и подвергали совместному выпариванию с толуолом (3×5 мл). Добавляли смесь воды со льдом и затем осуществляли экстракцию диизопропиловым эфиром (2×25 мл). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали, получая 5,5-бис(ацетилтиометил)-[1,3]диоксан (1,32 г, количественный выход) в виде бледно-желтого вязкого масла. С помощью 1H-ЯМР обнаружили, что чистота полученного вещества >95%, и использовали его без дальнейшей очистки.
Раствор 5,5-бис(ацетилтиометил)-[1,3]диоксана (1,32 г, 5 ммоль) в водном HCl (2 н., 25 мл) кипятили с обратным холодильником в течение 16 часов и затем охлаждали до комнатной температуры. Полученную смесь подщелачивали, добавляя по каплям водный раствор Na2CO3 (2 М), и затем экстрагировали CH2Cl2 (3×40 мл). Объединенные органические слои высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении, получая твердое белое вещество, которое растирали с горячей смесью н-гексан-диизопропиловый эфир (2:1, 15 мл). Белое твердое вещество отфильтровывали, получая 4,4-бис(гидроксиметил)-[1,3]дитиан, который использовали без дальнейшей очистки.
Смесь 4,4-бис(гидроксиметил)-[1,3]дитиана (405 мг, 2,25 ммоль) и TsCl (1,29 г, 6,75 ммоль) в безводном пиридине (4 мл) перемешивали при комнатной температуре в течение 48 часов. Растворитель удаляли в вакууме и выпаривали совместно с толуолом (3×3 мл). Добавляли CH2Cl2 (40 мл) и насыщенный водный раствор NaHCO3 (40 мл), разделяли фазы и органическую фазу высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали флэш-хроматографией (35-55% EtOAc в петролейном эфире), получая 4,4-бис(п-толуолсульфонилоксиметил)-[1,3]дитиан (525) в виде белого твердого вещества.
Стадия B

Диалкилирование соединения 513 (297 мг, 1,5 ммоль) соединением 525 (806 мг, 1,65 ммоль) в ДМСО (7,5 мл) в присутствии K2CO3 (518 мг, 3,75 ммоль), выполненное в соответствии с общей методикой, позволяло получить соединение 526 в виде белого твердого вещества после очистки колоночной хроматографией (40-50% EtOAc в петролейном эфире). ЖХ-МС: RT=3,53 мин; m/z 343,14 (M+H)+, 365,12 (M+Na)+, 327,28 (M-CH3)-. 1H-ЯМР (ДМСО-d6) δ 7,36 (1Н, д, J=8,8), 6,81 (1Н, д, J=8,8), 4,20 (2Н, с), 4,41 (2Н, с), 3,83 (2Н, с), 3,81 (3Н, с), 3,77 (3Н, с), 2,85 (4Н, с).

Общая методика C
LiHMDS (1 М в ТГФ, 3,0 экв.) по каплям добавляли к ледяному раствору сложного эфира 4b и коммерчески доступного 3,5-дихлор-4-метилпиридина (1,3 экв.) в безводном ТГФ. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, добавляли H2O (10 мл) и насыщенный водный раствор NH4Cl (20 мл), и смесь экстрагировали EtOAc (3×50 мл). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной флэш-хроматографией, получая кетон Ib.
Применяя общую методику C, получали следующие соединения:
Пример 20
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-оксетан]-6-ил}этанон (соединение 121)

Следуя общей методике, проводили конденсацию 3,5-дихлор-4-метилпиридина (169 мг, 1,04 ммоль) с соединением 514 (224 мг, 0,8 ммоль) в ТГФ (4 мл) в присутствии LiHMDS (2,4 мл, 2,4 ммоль), получая соединение 121 в виде белого твердого вещества после очистки колоночной хроматографией (65-80% EtOAc в петролейном эфире). ЖХ-МС: RT=3,24 мин; m/z 410,07 (M+H)+, 408,19, 410,22 (M-H)-. 1H-ЯМР (ДМСО-d6) δ 8,66 (2Н, с) 7,45 (1Н, д, J=8,9), 6,91 (1Н, д, J=9,0), 4,66 (2Н, с), 4,57 (2Н, с), 4,50 (2Н, д, J=6,5), 4,46 (2Н, д, J=6,5), 4,36 (2Н, с), 3,85 (3Н, с).
Пример 21
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-оксетан]-6-ил}этанон (соединение 122)
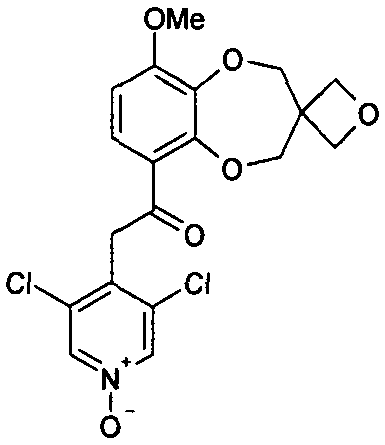
К раствору 2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-оксетан]-6-ил}этанона [121] (20,5 мг, 50 мкмоль) в CH2Cl2 (1 мл) добавляли 30% H2O2 (15 мкл) и метилтриоксорений(VII) (5 мг). Смесь перемешивали в течение 18 часов, добавляли MnO2 (5 мг) и перемешивали еще час. Добавляли CH2Cl2 (10 мл) и промывали органическую фазу водой. Объединенные органические фазы высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить 14 мг продукта.
ЖХ/МС (методика B): (m/z) 426,18; 428,20 (MH+); RT=2,42 мин; чистота (УФ)=100%.
Пример 22
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан]-6-ил}этанон (соединение 123)

Следуя общей методике, проводили конденсацию 3,5-дихлор-4-метилпиридина (366 мг, 2,26 ммоль) с соединением 516 (516 мг, 1,74 ммоль) в ТГФ (10 мл) в присутствии LiHMDS (5,2 мл, 5,2 ммоль), получая соединение 123 в виде белого твердого вещества после очистки колоночной хроматографией (55-65% EtOAc в петролейном эфире). ЖХ-МС: RT=4,23 мин; m/z 426,24, 428,25 (M+H)+, 424,23 (M-H)-. 1H-ЯМР (CDCl3) δ 8,51 (2Н, с), 7,57 (1Н, д, J=9,2), 6,71 (1Н, д, J=9,2), 4,64 (2Н, с), 4,46 (2Н, с), 4,33 (2Н, с), 3,93 (3Н, с), 3,20 (2Н, д, J=9,4), 3,12 (2Н, д, J=9,9).
Пример 23
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан-1',1'-диоксид]-6-ил}этанон (соединение 124)

К раствору 2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан]-6-ил}этанона [123] (42,6 мг) в CH2Cl2 (1 мл) добавляли 30% H2O2 (38 мкл) и метилтриоксорений(VII) (5 мг). Смесь перемешивали в течение 18 часов и затем промывали водой. Органическую фазу высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить 6 мг продукта.
ЖХ/МС (методика B): (m/z) 458,13, 460,10, 462,14 (MH+); RT=3,30 мин; чистота (УФ)=100%.
Пример 24
2-(3,5-дихлорпиридин-1-оксидо-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан-1',1'-диоксид]-6-ил}этанон (соединение 125)

К раствору 2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан]-6-ил}этанона [123] (39,6 мг) в CH2Cl2 (1 мл) добавляли 30% H2O2 (76 мкл) и метилтриоксорений(VII) (5 мг). Смесь перемешивали в течение 18 ч и затем добавляли воду. Органическую фазу высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить 8,4 мг продукта.
ЖХ/МС (методика B): (m/z) 474,17, 476,16, 478,18 (MH+); RT=2,39 мин; чистота (УФ)=100%.
Пример 25
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),2'-(1,3-диоксолан)]-6-ил}этанон (соединение 126)
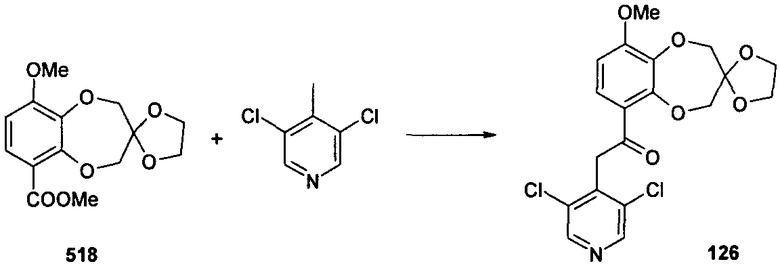
Следуя общей методике, проводили конденсацию 3,5-дихлор-4-метилпиридина (53 мг, 0,33 ммоль) с соединением 518 (74 мг, 0,25 ммоль) в ТГФ (1,5 мл) в присутствии LiHMDS (0,75 мл, 0,75 ммоль), получая соединение 126 в виде белого твердого вещества после очистки колоночной хроматографией (60-80% EtOAc в петролейном эфире). ЖХ-МС: RT=3,63 мин; m/z 426,18, 428,16 (M+H)+. 1H-ЯМР (ДМСО-d6) δ 8,64 (2Н, с), 7,39 (1Н, д, J=8,8), 6,86 (1Н, д, J=8,9), 4,60 (2Н, с), 4,34 (2Н, с), 4,17 (2Н, с), 3,97 (4Н, с), 3,84 (3Н, с).
Пример 26
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),4'-тетрагидропиран]-6-ил}этанон (соединение 127)
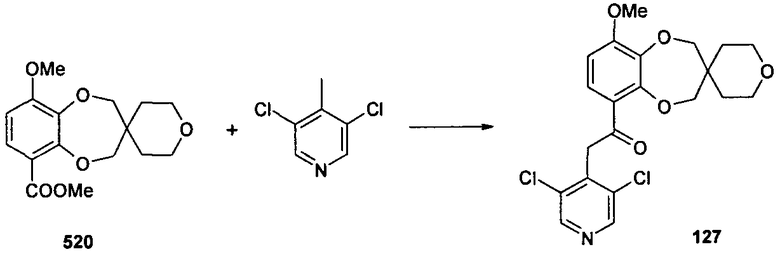
Следуя общей методике, проводили конденсацию 3,5-дихлор-4-метилпиридина (59 мг, 0,36 ммоль) с соединением 520 (85 мг, 0,28 ммоль) в ТГФ (1,5 мл) в присутствии LiHMDS (0,85 мл, 0,85 ммоль), получая соединение 127 в виде белого твердого вещества после очистки колоночной хроматографией (60-70% EtOAc в петролейном эфире). ЖХ-МС: RT=3,70 мин; m/z 438,21, 440,21 (M+H)+, 436,30, 438,27 (M-H)-. 1H-ЯМР (ДМСО-d6) δ 8,65 (2Н, с), 7,42 (1Н, д, J=8,9), 6,87 (1Н, д, J=9,0), 4,63 (2Н, с), 4,25 (2Н, с), 4,07 (2Н, с), 3,84 (3Н, с), 3,64 (4Н, т, J=5,3), 1,60 (4Н, т, J=5,3).
Пример 27
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),4'-тетрагидропиран]-6-ил}этанон (соединение 128)

К раствору 2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),4'-тетрагидропиран]-6-ил}этанона [127] (66 мг, 150 мкмоль) в CH2Cl2 (2 мл) добавляли 30% H2O2 (45 мкл) и метилтриоксорений(VII) (10 мг). Смесь перемешивали в течение 18 часов, добавляли MnO2 (10 мг) и перемешивали еще час. Добавляли CH2Cl2 (10 мкл) и органическую фазу промывали водой. Объединенные органические фазы высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить 5,6 мг продукта.
ЖХ/МС (методика B): (m/z) 454,32, 456,32 (MH+); RT=2,80 мин; чистота (УФ)=100%.
Пример 28
2-(3,5-дихлорпиридин-4-ил)-1-{9-метокси-2',2'-диметилспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]диоксан]-6-ил}этанон (соединение 129)
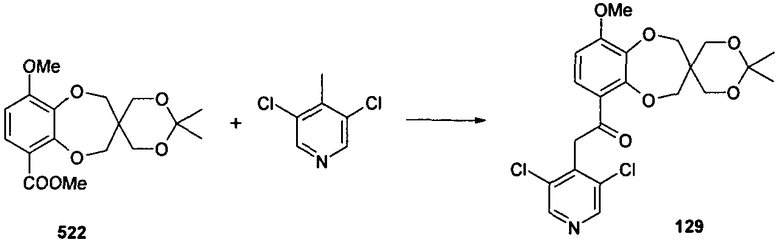
Следуя общей методике, проводили конденсацию 3,5-дихлор-4-метилпиридина (93 мг, 0,58 ммоль) с соединением 522 (150 мг, 0,44 ммоль) в ТГФ (2 мл) в присутствии LiHMDS (1,3 мл, 1,3 ммоль), получая соединение 129 в виде белого твердого вещества после очистки колоночной хроматографией (55-60% EtOAc в петролейном эфире). ЖХ-МС: RT=3,95 мин; m/z 468,19, 470,23 (M+H)+, 466,36, 468,33 (M-H)-. 1H-ЯМР (ДМСО-d6) δ 8,65 (2Н, с), 7,42 (1Н, д, J=8,8), 6,89 (1Н, д, J=9,1), 4,63 (2Н, с), 4,33 (2Н, с), 4,08 (2Н, с), 3,84 (3Н, с), 3,82 (4Н, с), 1,39 (3Н, с), 1,37 (3Н, с).
Пример 29
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]диоксан]-6-ил}этанон (соединение 130)

Следуя общей методике, проводили конденсацию 3,5-дихлор-4-метилпиридина (211 мг, 1,3 ммоль) с соединением 524 (310 мг, 1,0 ммоль) в ТГФ (6 мл) в присутствии LiHMDS (3 мл, 3,0 ммоль), получая соединение 130 в виде белого твердого вещества после очистки колоночной хроматографией (60-80% EtOAc в петролейном эфире). ЖХ-МС: RT=3,64 мин; m/z 440,16, 442,17 (M+H)+, 438,27, 440,29 (M-H)-. 1H-ЯМР (ДМСО-d6) δ 8,65 (2Н, с), 7,43 (1Н, д, J=8,9), 6,89 (1Н, д, J=8,9), 4,84 (1Н, д, J=6,1), 4,79 (1Н, д, J=6,1), 4,64 (2Н, с), 4,34 (2Н, с), 4,08 (2Н, с), 3,90 (2Н, д, J=11,4), 3,85 (3Н, с), 3,82 (2Н, д, J=11,2).
Пример 30
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]диоксан]-6-ил}этанон (соединение 131)
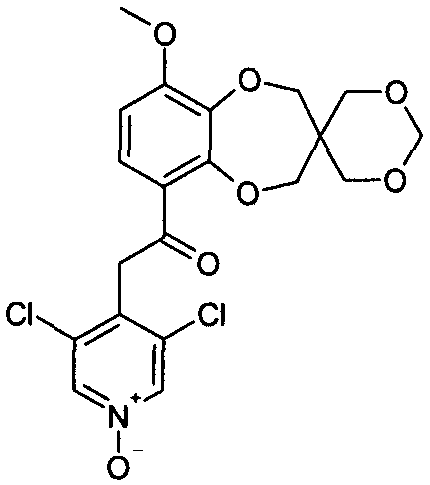
К раствору 2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]диоксан]-6-ил}этанона [130] (100 мг) в CH2Cl2 (2 мл) добавляли 30% H2O2 (120 мкл) и метилтриоксорений(VII) (5 мг). Смесь перемешивали в течение 18 часов и затем промывали водой. Органическую фазу высушивали над MgSO4 и упаривали досуха при пониженном давлении. Стандартная очистка ВЭЖХ позволяла получить 85 мг продукта.
ЖХ/МС (методика B): (m/z) 456,23 (MH+); RT=2,55 мин; чистота (УФ)=95%.
Пример 31
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]дитиан]-6-ил}этанон (соединение 132)

Следуя общей методике, проводили конденсацию 3,5-дихлор-4-метилпиридина (154 мг, 0,95 ммоль) с соединением 526 (250 мг, 0,73 ммоль) в ТГФ (4 мл) в присутствии LiHMDS (2,2 мл, 2,2 ммоль), получая соединение 132 в виде белого твердого вещества после очистки колоночной хроматографией (45-55% EtOAc в петролейном эфире). ЖХ-МС: RT=4,39 мин; m/z 472,15, 474,14 (M+H)+. 1H-ЯМР (ДМСО-d6) δ 8,65 (2Н, с), 7,42 (1Н, д, J=8,9), 6,88 (1Н, д, J=8,9), 4,64 (2Н, с), 4,43 (2Н, с), 4,24 (2Н, с), 3,85 (3Н, с), 3,84 (2Н, с), 2,91 (4Н, с).
Пример 32
Исследование ингибирования PDE4
Рекомбинантную человеческую PDE4 (инвентарный номер Genbank no NM_006203) инкубировали в течение 1 часа с тестируемыми соединениями в концентрациях до 10 мкМ с cAMP (1×10-5 М) и с незначительным количеством (0,021 МБк) радиоактивно меченного cAMP. В конце инкубирования расщепление субстрата оценивали по связыванию продукта AMP гранулами SPA, которые генерировали хемолюминисценцию при связывании с радиоактивной меткой. Продукт AMP ингибировал связывание радиоактивной метки с гранулами, что уменьшало люминисцентный сигнал. Результаты представляли в виде молярных концентраций, приводящих к 50% ингибированию расщепления субстрата по сравнению с контрольными образцами и выражали как IC50 (M).
Полученные результаты показаны ниже в таблице 1.
Пример 33
Высвобождение TNF-α
Мононуклеарные клетки периферической крови человека (PBMC) выделяли из лейкоцитарной пленки. Кровь смешивали с солевым раствором в соотношении 1:1 и выделяли PBMC, используя Lymphoprep tubesTM (Nycomed, Norway). PBMC суспендировали в RPMI1640 с 2% сывороткой эмбрионов телят (FCS), пенициллином/стрептоцидом и 2 мМ L-глутамином в концентрации 5×105 клеток/мл. Клетки преинкубировали в течение 30 минут с тестируемыми соединениями в 96-луночных планшетах для культур тканей, и в течение 18 часов стимулировали липополисахаридом 1 мг/мл (Sigma). Уровни TNF-α измеряли в культуральном супернатанте с помощью ферментного иммуноанализа, используя первичные и вторичные биотинилированные антитела систем R&D. Результаты выражали в виде значений IC50, рассчитанных на основании кривых ингибирования, с использованием в качестве положительного контроля секрецию в лунках, стимулированных LPS, и в качестве отрицательного контроля секрецию не стимулированных клеток.
Результаты показаны ниже в таблице 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОДИОКСОЛА ИЛИ БЕНЗОДИОКСЕПИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗ | 2011 |
|
RU2583787C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 1, 3-БЕНЗОДИОКСОЛЬНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2015 |
|
RU2697251C2 |
| ЗАМЕЩЕННЫЕ МЕТИЛФЕНИЛКЕТОНЫ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2007 |
|
RU2493149C2 |
| ЗАМЕЩЕННЫЕ ХРОМАНЫ | 2015 |
|
RU2718060C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 1,3-БЕНЗОДИОКСОЛЬНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2018 |
|
RU2814175C2 |
| НОВЫЕ БЕНЗОДИОКСОЛЫ | 2003 |
|
RU2304580C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ИХ ПРИМЕНЕНИЕ ПРОТИВ МИКОБАКТЕРИЙ | 2015 |
|
RU2664587C1 |
| ЗАМЕЩЕННЫЕ АРИЛАМИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-HT-РЕЦЕПТОРА | 2006 |
|
RU2440996C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ СИГМА РЕЦЕПТОРОВ | 2011 |
|
RU2582338C2 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
Настоящее изобретение относится к соединению общей формулы (I):

где m и n независимо представляют собой 0 или 1; G и E представляют собой кислород, R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют гетероцикл, включающий один или два гетероатома, выбранных из кислорода, серы, -S(O)-, -S(O)2-, -N=, -N(R5)-, причем один или более атомов углерода в указанном гетероцикле необязательно замещены одним или более одинаковыми или различными заместителями, выбранными из числа заместителей R4;
R3 представляет собой алкокси или галогеналкокси; R4 представляет собой водород, алкил или галоген; R5 представляет собой водород, алкилкарбонил, алкоксикарбонил или алкилсульфонил; X представляет собой связь, -CH2- или -NH-; A представляет собой фенил, пиридил, пиразинил или хинолил, необязательно замещенный одним или более одинаковыми или различными заместителями, выбранными из числа заместителей R4; или его фармацевтически приемлемым солям, гидратам, N-оксидам или сольватам. Изобретение также относится к фармацевтическим композициям на основе этих соединений в качестве ингибитора фосфодиэстеразы PDE4 для лечения, например, кожных заболеваний. 2 н. и 17 з.п. ф-лы, 2 табл., 32 пр.
1. Соединение общей формулы (I):

где m и n независимо представляют собой 0 или 1;
G и E представляют собой кислород,
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют гетероцикл, включающий один или два гетероатома, выбранных из кислорода, серы, -S(O)-, -S(O)2-, -N=, -N(R5)-, причем один или более атомов углерода в указанном гетероцикле необязательно замещены одним или более одинаковыми или различными заместителями, выбранными из числа заместителей R4;
R3 представляет собой алкокси или галогеналкокси;
R4 представляет собой водород, алкил или галоген;
R5 представляет собой водород, алкилкарбонил, алкоксикарбонил или алкилсульфонил;
X представляет собой связь, -CH2- или -NH-;
A представляет собой фенил, пиридил, пиразинил или хинолил, необязательно замещенный одним или более одинаковыми или различными заместителями, выбранными из числа заместителей R4;
и его фармацевтически приемлемые соли, гидраты, N-оксиды или соль ваты.
2. Соединение по п.1, где оба коэффициента m и n равны единице.
3. Соединение по п.1, где оба коэффициента m и n равны нулю.
4. Соединение по п.1, в котором R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл.
5. Соединение по п.4, в котором указанный гетероцикл представляет собой тетрагидропиран, оксетан, [1,3]диоксолан, [1,3]диоксан, тетрагидротиопиран, тетрагидротиопиран-1,1-диоксид, тетрагидротиопиран-1-оксид, пиперидин, тетрагидротиофен, [1,3]-дитиан, тиетан, [1,3]-дитиан-1,3-диоксид, тиетан-1-оксид или тиетан-1,1-диоксид.
6. Соединение по п.4, где гетероцикл включает один гетероатом.
7. Соединение по п.4, где гетероцикл включает два гетероатома.
8. Соединение по п.4, где гетероатом (гетероатомы) представляет/представляют собой кислород.
9. Соединение по п.4, где гетероатом (гетероатомы) представляет/представляют собой серу, -S(O)- или -S(O)2-.
10. Соединение по п.1, где A представляет собой фенил.
11. Соединение по п.1, где фрагмент A замещен галогеном.
12. Соединение по 11, где R3 представляет собой метокси или этокси.
13. Соединение по п.1, где X представляет собой -CH2-.
14. Соединение по п.1, где Х представляет собой -NH-.
15. Соединение по п.1, где A представляет собой 4-(3,5-дихлорпиридил).
16. Соединение по п.1, где R4 представляет собой водород.
17. Соединение по п.1, выбранное из группы, состоящей из 2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-ил)этанона (соединение 101),
N-(3,5-дихлорпиридин-4-ил)-7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-карбоксамида (соединение 102), 2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-пиран]-4-ил)этанона (соединение 103),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-4',5'-дигидроспиро[1,3-бензодиоксол-2,3'-(2H)-тиофен]-4-ил)этанона (соединение 104), 2-(3,5-дихлорпиридин-4-ил)-1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 105),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-[метоксикарбонил]спиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 106),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-[метилсульфонил]спиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 107),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-ацетилспиро[1,3-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 108),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-1'-метилспиро[1,5-бензодиоксол-2,4'-пиперидин]-4-ил)этанона (соединение 109),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4Н)-тиопиран]-4-ил)этанона (соединение 110),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1'-оксид]-4-ил)этанона (соединение 111),
2-(3,5-дихлорпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1',1'-диоксид]-4-ил)этанона (соединение 112),
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран-1',1'-диоксид]-4-ил)этанона (соединение 113),
2-(3-бромпиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 114),
2-(3-бромпиразин-2-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 115),
2-(пиразин-2-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4Н)-тиопиран]-4-ил)этанона (соединение 116),
2-(пиридин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4Н)-тиопиран]-4-ил)этанона (соединение 117),
2-(хинолин-4-ил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро [1,3-бензодиоксол-2,4'-(4Н)-тиопиран]-4-ил)этанона (соединение 118),
2-(2,6-дихлорфенил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4H)-тиопиран]-4-ил)этанона (соединение 119),
2-(2-хлорфенил)-1-(7-метокси-2',3',5',6'-тетрагидроспиро[1,3-бензодиоксол-2,4'-(4Н)-тиопиран]-4-ил)этанона (соединение 120),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-оксетан]-6-ил}этанона (соединение 121),
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-{9-метоксиспиро[2Н-1,5-бензодиоксепин-3(4Н),3'-оксетан]-6-ил}этанона (соединение 122),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2Н-1,5-бензодиоксепин-3(4Н),3'-тиетан]-6-ил}этанона (соединение 123),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2Н-1,5-бензодиоксепин-3(4Н),3'-тиетан-1',1'-диоксид]-6-ил}этанона (соединение 124),
2-(3,5-дихлорпиридин-1-оксидо-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),3'-тиетан-1',1'-диоксид]-6-ил}этанона (соединение 125),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2Н-1,5-бензодиоксепин-3(4Н),2'-(1,3-диоксолан)]-6-ил}этанона (соединение 126),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2Н-1,5-бензодиоксепин-3(4Н),4'-тетрагидропиран]-6-ил}этанона (соединение 127),
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-{9-метоксиспиро[2Н-1,5-бензодиоксепин-3(4Н),4'-тетрагидропиран]-6-ил}этанона (соединение 128),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метокси-2',2'-диметилспиро[2Н-1,5-бензодиоксепин-3(4H),5'-[1,3]диоксан]-6-ил}этанона (соединение 129),
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2Н-1,5-бензодиоксепин-3(4H), 5'-[1,3]диоксан]-6-ил}этанона (соединение 130),
2-(3,5-дихлор-1-оксидопиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]диоксан]-6-ил}этанона (соединение 131) и
2-(3,5-дихлорпиридин-4-ил)-1-{9-метоксиспиро[2H-1,5-бензодиоксепин-3(4H),5'-[1,3]дитиан]-6-ил}этанона (соединение 132),
и их фармацевтически приемлемых солей, гидратов, N-оксидов или сольватов.
18. Соединение по п.1 для применения в качестве ингибитора фосфодиэстеразы PDE4.
19. Фармацевтическая композиция для применения в качестве ингибитора фосфодиэстеразы PDE4, включающая соединение по п.1 в сочетании с фармацевтически приемлемым наполнителем или разбавителем или фармацевтически приемлемым носителем (носителями).
| US 20020128290 A1, 12.09.2002 | |||
| WO 9744337 A1, 27.11.1997 | |||
| Устройство для измерения постоянной времени вихревых токов | 1980 |
|
SU943613A1 |
| Машина для посадки лука-севка | 1982 |
|
SU1029860A1 |
| RU 99106587 A, 27.12.2000. | |||

